
癌の処置において使用するための多キナーゼインヒビター
本発明は、特定の癌を処置するための化合物、薬学的組成物および方法を提供する。このような組成物は、一般に、R1〜R3が本明細書中で定義されるとおりである式(I):

の化合物、またはその薬学的に受容可能な塩もしくはエステル;および薬学的に受容可能なキャリアを含有し得る。いくつかの局面において、本発明は、SrcTK/MEKに関連する癌の処置を必要とする被験体において、SrcTK/MEKに関連する癌を処置する方法を提供する。
の化合物、またはその薬学的に受容可能な塩もしくはエステル;および薬学的に受容可能なキャリアを含有し得る。いくつかの局面において、本発明は、SrcTK/MEKに関連する癌の処置を必要とする被験体において、SrcTK/MEKに関連する癌を処置する方法を提供する。
Notice: Undefined index: DEJ in /mnt/www/gzt_disp.php on line 298
【特許請求の範囲】
【請求項1】
SrcTK/MEKに関連する癌の処置を必要とする被験体において、SrcTK/MEKに関連する癌を処置する方法であって、該方法は、該被験体に、SrcTK/MEKに関連する癌を処置するための有効量のSrcTK/MEKインヒビターを含有する組成物を投与する工程を包含する、方法。
【請求項2】
前記SrcTK/MEKインヒビターが、約0.1μMの濃度で、Srcチロシンキナーゼファミリーの少なくとも1つのメンバーの活性を少なくとも約75%阻害する、請求項1に記載の方法。
【請求項3】
前記SrcTK/MEKインヒビターが、約0.1μMの濃度で、Srcチロシンキナーゼファミリーの少なくとも5つのメンバーの活性を少なくとも約50%阻害する、請求項1〜2のいずれか1項に記載の方法。
【請求項4】
前記Srcチロシンキナーゼファミリーの前記メンバーが、cSrc、Fyn、Lyn、LckおよびYesを含む、請求項3に記載の方法。
【請求項5】
前記SrcTK/MEKインヒビターが、約0.1μMの濃度で、MEK1の活性を少なくとも約50%阻害する、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記SrcTK/MEKインヒビターが式(I):
【化110】
の化合物、またはその薬学的に受容可能な塩もしくはエステルであり、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化111】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化112】
からなる群より選択されるヘテロシクリル部分を形成する;
請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記SrcTK/MEKに関連する癌が、慢性骨髄性白血病および結腸直腸癌からなる群より選択される癌である、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
前記癌がメシル酸イマチニブに耐性である、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
前記SrcTK/MEKインヒビターがまた、Bcl−Ablの活性を阻害し、そして前記SrcTK/MEKに関連する癌が白血病である、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
前記SrcTK/MEKインヒビターがまた、TrkBの活性を阻害する、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
TrkB/MEKに関連する癌の処置を必要とする被験体において、TrkB/MEKに関連する癌を処置する方法であって、該方法は、該被験体に、TrkB/MEKに関連する癌を処置するための有効量のTrkB/MEKインヒビターを含有する組成物を投与する工程を包含する、方法。
【請求項12】
前記TrkB/MEKインヒビターが、約0.1μMの濃度で、TrkBの活性を少なくとも約75%阻害する、請求項11に記載の方法。
【請求項13】
前記TrkB/MEKインヒビターが、約0.1μMの濃度で、MEK1の活性を少なくとも約55%阻害する、請求項11〜12のいずれか1項に記載の方法。
【請求項14】
前記TrkB/MEKインヒビターが、式(I):
【化113】
の化合物、またはその薬学的に受容可能な塩もしくはエステルであり、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化114】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化115】
からなる群より選択されるヘテロシクリル部分を形成する;
請求項11〜13のいずれか1項に記載の方法。
【請求項15】
前記TrkB/MEKに関連する癌が、膵臓癌、神経癌、神経膠腫、神経芽細胞腫および網膜芽細胞腫からなる群より選択される癌である、請求項10〜14のいずれか1項に記載の方法。
【請求項16】
前記TrkB/MEKインヒビターがまた、Srcチロシンキナーゼファミリーの少なくとも1つのメンバーの活性を阻害する、請求項10〜15のいずれか1項に記載の方法。
【請求項17】
転移プロセスの阻害を必要とする被験体において、転移プロセスを阻害する方法であって、該方法は、該被験体に、該転移プロセスを阻害するための有効量の式(I):
【化116】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化117】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化118】
からなる群より選択されるヘテロシクリル部分を形成する、
方法。
【請求項18】
B−RAF変異を有する癌の処置を必要とする被験体において、B−RAF変異を有する癌を処置する方法であって、該方法は、該被験体に、該B−RAF変異を有する癌を処置するための有効量の、少なくとも1つの式(I):
【化119】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化120】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化121】
からなる群より選択されるヘテロシクリル部分を形成する、
方法。
【請求項19】
前記B−RAF変異を有する癌が、B−RAF V600E変異を有する癌である、請求項18に記載の方法。
【請求項20】
前記B−RAF変異を有する癌が、卵巣癌、甲状腺癌、結腸直腸癌および黒色腫からなる群より選択される癌である、請求項18〜19のいずれか1項に記載の方法。
【請求項21】
FLT3変異を有する癌の処置を必要とする被験体において、FLT3変異を有する癌を処置する方法であって、該方法は、該被験体に、該FLT3変異を有する癌を処置するための有効量の、少なくとも1つの式(I):
【化122】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化123】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化124】
からなる群より選択されるヘテロシクリル部分を形成する、
方法。
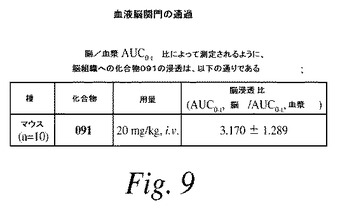
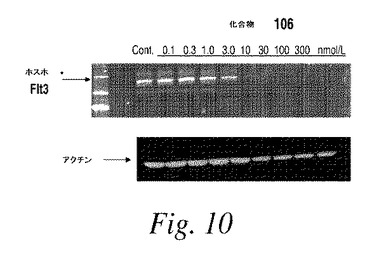
【請求項22】
前記FLT3変異を有する癌が、FLT3 D835Y、FLT3 Y842C、FLT3 K663QまたはFLT3 V592A変異を有する癌である、請求項21に記載の方法。
【請求項23】
前記FLT3変異を有する癌が白血病である、請求項21〜22のいずれか1項に記載の方法。
【請求項24】
中枢神経系腫瘍の処置を必要とする被験体において、中枢神経系腫瘍を処置する方法であって、該方法は、該被験体に、該腫瘍を処置するための有効量の、式(I):
【化125】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化126】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化127】
からなる群より選択されるヘテロシクリル部分を形成し、
該式(I)の化合物は血液脳関門を通過し得る、
方法。
【請求項25】
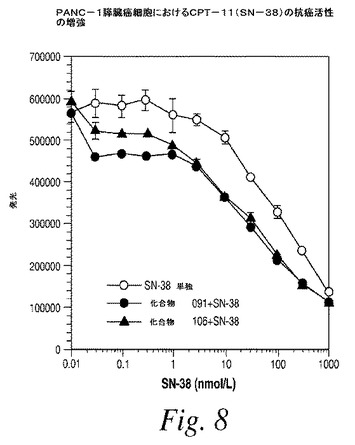
動物の脳内の式(I)の化合物の量対血漿中の式(I)の化合物の量の比に基づいて、該式(I)の化合物の少なくとも約20%が前記血液脳関門を通過する、請求項24に記載の方法。
【請求項26】
動物の脳内の式(I)の化合物の量対血漿中の式(I)の化合物の量の比に基づいて、該式(I)の化合物の少なくとも約50%が血液脳関門を通過する、請求項24に記載の方法。
【請求項27】
前記中枢神経系腫瘍が、脳腫瘍、神経膠腫および神経芽細胞腫からなる群より選択される腫瘍である、請求項24〜26のいずれか1項に記載の方法。
【請求項28】
慢性骨髄性白血病、膵臓癌、神経癌、神経膠腫、神経芽細胞腫、網膜芽細胞腫、卵巣癌、甲状腺癌、結腸直腸癌および黒色腫からなる群より選択される少なくとも1つの癌の処置を必要とする被験体において、該癌を処置する方法であって、該方法は、該被験体に、該癌を処置するための有効量の、式(I):
【化128】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化129】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化130】
からなる群より選択されるヘテロシクリル部分を形成する、
方法。
【請求項29】
【化131】
が二重結合を表す、請求項6〜10または14〜28のいずれか1項に記載の方法。
【請求項30】
R3が−NRbRcであり、ここでRcがHであり、そしてRbが非置換C1〜4アルキルである、請求項6〜10または14〜28のいずれか1項に記載の方法。
【請求項31】
RcがHであり、そしてRbがメチルまたはエチルである、請求項6〜10または14〜28のいずれか1項に記載の方法。
【請求項32】
前記化合物が:
【化132】
、ならびにその薬学的に受容可能な塩およびエステルからなる群より選択される少なくとも1つの化合物である、請求項1〜31のいずれか1項に記載の方法。
【請求項33】
前記式(I)の化合物が、式(II):
【化133】
の化合物、またはその薬学的に受容可能な塩もしくはエステルであり、式(II)において、
R3は−NHRbであり、そしてRbは、0、1個または2個のヒドロキシル基で置換されたC1〜C3アルキルである、
請求項6〜10または14〜28のいずれか1項に記載の方法。
【請求項34】
R3が非置換C1〜3アルキルアミノである、請求項33に記載の方法。
【請求項35】
R3が、メチルアミノおよびエチルアミノからなる群より選択される基である、請求項34に記載の方法。
【請求項36】
R3が、2−ヒドロキシエチルアミノおよび2,3−ジヒドロキシプロピルアミノからなる群より選択される基である、請求項33に記載の方法。
【請求項37】
前記化合物が:
【化134】
、ならびにその薬学的に受容可能な塩およびエステルからなる群より選択される少なくとも1つの化合物である、請求項33に記載の方法。
【請求項38】
前記方法が、第二の化学療法薬物を投与する工程をさらに包含する、請求項1〜37のいずれか1項に記載の方法。
【請求項39】
前記式(I)の化合物を含有する組成物が静脈内投与される、請求項1〜38のいずれか1項に記載の方法。
【請求項40】
前記式(I)の化合物を含有する組成物が、体重1kgあたり約0.10mg〜約25mgの投薬量で投与される、請求項1〜39のいずれか1項に記載の方法。
【請求項41】
以下に列挙される化合物:
【化135】
【化136】
【化137】
【化138】
、ならびにその薬学的に受容可能な塩およびエステルから選択される、化合物。
【請求項1】
SrcTK/MEKに関連する癌の処置を必要とする被験体において、SrcTK/MEKに関連する癌を処置する方法であって、該方法は、該被験体に、SrcTK/MEKに関連する癌を処置するための有効量のSrcTK/MEKインヒビターを含有する組成物を投与する工程を包含する、方法。
【請求項2】
前記SrcTK/MEKインヒビターが、約0.1μMの濃度で、Srcチロシンキナーゼファミリーの少なくとも1つのメンバーの活性を少なくとも約75%阻害する、請求項1に記載の方法。
【請求項3】
前記SrcTK/MEKインヒビターが、約0.1μMの濃度で、Srcチロシンキナーゼファミリーの少なくとも5つのメンバーの活性を少なくとも約50%阻害する、請求項1〜2のいずれか1項に記載の方法。
【請求項4】
前記Srcチロシンキナーゼファミリーの前記メンバーが、cSrc、Fyn、Lyn、LckおよびYesを含む、請求項3に記載の方法。
【請求項5】
前記SrcTK/MEKインヒビターが、約0.1μMの濃度で、MEK1の活性を少なくとも約50%阻害する、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記SrcTK/MEKインヒビターが式(I):
【化110】
の化合物、またはその薬学的に受容可能な塩もしくはエステルであり、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化111】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化112】
からなる群より選択されるヘテロシクリル部分を形成する;
請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記SrcTK/MEKに関連する癌が、慢性骨髄性白血病および結腸直腸癌からなる群より選択される癌である、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
前記癌がメシル酸イマチニブに耐性である、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
前記SrcTK/MEKインヒビターがまた、Bcl−Ablの活性を阻害し、そして前記SrcTK/MEKに関連する癌が白血病である、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
前記SrcTK/MEKインヒビターがまた、TrkBの活性を阻害する、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
TrkB/MEKに関連する癌の処置を必要とする被験体において、TrkB/MEKに関連する癌を処置する方法であって、該方法は、該被験体に、TrkB/MEKに関連する癌を処置するための有効量のTrkB/MEKインヒビターを含有する組成物を投与する工程を包含する、方法。
【請求項12】
前記TrkB/MEKインヒビターが、約0.1μMの濃度で、TrkBの活性を少なくとも約75%阻害する、請求項11に記載の方法。
【請求項13】
前記TrkB/MEKインヒビターが、約0.1μMの濃度で、MEK1の活性を少なくとも約55%阻害する、請求項11〜12のいずれか1項に記載の方法。
【請求項14】
前記TrkB/MEKインヒビターが、式(I):
【化113】
の化合物、またはその薬学的に受容可能な塩もしくはエステルであり、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化114】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化115】
からなる群より選択されるヘテロシクリル部分を形成する;
請求項11〜13のいずれか1項に記載の方法。
【請求項15】
前記TrkB/MEKに関連する癌が、膵臓癌、神経癌、神経膠腫、神経芽細胞腫および網膜芽細胞腫からなる群より選択される癌である、請求項10〜14のいずれか1項に記載の方法。
【請求項16】
前記TrkB/MEKインヒビターがまた、Srcチロシンキナーゼファミリーの少なくとも1つのメンバーの活性を阻害する、請求項10〜15のいずれか1項に記載の方法。
【請求項17】
転移プロセスの阻害を必要とする被験体において、転移プロセスを阻害する方法であって、該方法は、該被験体に、該転移プロセスを阻害するための有効量の式(I):
【化116】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化117】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化118】
からなる群より選択されるヘテロシクリル部分を形成する、
方法。
【請求項18】
B−RAF変異を有する癌の処置を必要とする被験体において、B−RAF変異を有する癌を処置する方法であって、該方法は、該被験体に、該B−RAF変異を有する癌を処置するための有効量の、少なくとも1つの式(I):
【化119】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化120】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化121】
からなる群より選択されるヘテロシクリル部分を形成する、
方法。
【請求項19】
前記B−RAF変異を有する癌が、B−RAF V600E変異を有する癌である、請求項18に記載の方法。
【請求項20】
前記B−RAF変異を有する癌が、卵巣癌、甲状腺癌、結腸直腸癌および黒色腫からなる群より選択される癌である、請求項18〜19のいずれか1項に記載の方法。
【請求項21】
FLT3変異を有する癌の処置を必要とする被験体において、FLT3変異を有する癌を処置する方法であって、該方法は、該被験体に、該FLT3変異を有する癌を処置するための有効量の、少なくとも1つの式(I):
【化122】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化123】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化124】
からなる群より選択されるヘテロシクリル部分を形成する、
方法。
【請求項22】
前記FLT3変異を有する癌が、FLT3 D835Y、FLT3 Y842C、FLT3 K663QまたはFLT3 V592A変異を有する癌である、請求項21に記載の方法。
【請求項23】
前記FLT3変異を有する癌が白血病である、請求項21〜22のいずれか1項に記載の方法。
【請求項24】
中枢神経系腫瘍の処置を必要とする被験体において、中枢神経系腫瘍を処置する方法であって、該方法は、該被験体に、該腫瘍を処置するための有効量の、式(I):
【化125】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化126】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化127】
からなる群より選択されるヘテロシクリル部分を形成し、
該式(I)の化合物は血液脳関門を通過し得る、
方法。
【請求項25】
動物の脳内の式(I)の化合物の量対血漿中の式(I)の化合物の量の比に基づいて、該式(I)の化合物の少なくとも約20%が前記血液脳関門を通過する、請求項24に記載の方法。
【請求項26】
動物の脳内の式(I)の化合物の量対血漿中の式(I)の化合物の量の比に基づいて、該式(I)の化合物の少なくとも約50%が血液脳関門を通過する、請求項24に記載の方法。
【請求項27】
前記中枢神経系腫瘍が、脳腫瘍、神経膠腫および神経芽細胞腫からなる群より選択される腫瘍である、請求項24〜26のいずれか1項に記載の方法。
【請求項28】
慢性骨髄性白血病、膵臓癌、神経癌、神経膠腫、神経芽細胞腫、網膜芽細胞腫、卵巣癌、甲状腺癌、結腸直腸癌および黒色腫からなる群より選択される少なくとも1つの癌の処置を必要とする被験体において、該癌を処置する方法であって、該方法は、該被験体に、該癌を処置するための有効量の、式(I):
【化128】
の化合物、またはその薬学的に受容可能な塩もしくはエステルを含有する組成物を投与する工程を包含し、式(I)において:
R1はHであり、
R2は、Hおよびトリフルオロメチルカルボニルからなる群より選択される部分であるか;またはR1およびR2は、コア構造と一緒になって、式(a):
【化129】
のヘテロシクリルジイル部分を形成し;
R3は、−ORaおよび−NRbRcからなる群より選択される部分であり;
Raは、イミダゾリルで必要に応じて置換されたC1〜4アルキル基であり;
Rbは、C1〜4アルキル基およびC1〜4アルコキシ基からなる群より選択される部分であり、ここでRbは、0、1個もしくは2個の基で置換されており、該基は各々独立して、−OCH3、−C(O)OH、−C(O)NR’R”、−NH(C1〜3アルキル)、nが2〜4である−NH(CH2CH2O)nCH3、ピペラジニル、N−メチルピペラジニル、ピペリジニル、N−メチルピペリジニル、N−モルホリニル、イミダゾリル、ピロリジニル、−OPO3H2およびヒドロキシルからなる群より選択され;該−NH(C1〜3アルキル)基は、0、1個もしくは2個のヒドロキシル部分で置換されており、そしてR’およびR”は各々独立して、−Hまたは−CH3から選択され;そして
RcはHであるか、またはRbおよびRcは、これらが結合している窒素と一緒になって、式(b)および式(c):
【化130】
からなる群より選択されるヘテロシクリル部分を形成する、
方法。
【請求項29】
【化131】
が二重結合を表す、請求項6〜10または14〜28のいずれか1項に記載の方法。
【請求項30】
R3が−NRbRcであり、ここでRcがHであり、そしてRbが非置換C1〜4アルキルである、請求項6〜10または14〜28のいずれか1項に記載の方法。
【請求項31】
RcがHであり、そしてRbがメチルまたはエチルである、請求項6〜10または14〜28のいずれか1項に記載の方法。
【請求項32】
前記化合物が:
【化132】
、ならびにその薬学的に受容可能な塩およびエステルからなる群より選択される少なくとも1つの化合物である、請求項1〜31のいずれか1項に記載の方法。
【請求項33】
前記式(I)の化合物が、式(II):
【化133】
の化合物、またはその薬学的に受容可能な塩もしくはエステルであり、式(II)において、
R3は−NHRbであり、そしてRbは、0、1個または2個のヒドロキシル基で置換されたC1〜C3アルキルである、
請求項6〜10または14〜28のいずれか1項に記載の方法。
【請求項34】
R3が非置換C1〜3アルキルアミノである、請求項33に記載の方法。
【請求項35】
R3が、メチルアミノおよびエチルアミノからなる群より選択される基である、請求項34に記載の方法。
【請求項36】
R3が、2−ヒドロキシエチルアミノおよび2,3−ジヒドロキシプロピルアミノからなる群より選択される基である、請求項33に記載の方法。
【請求項37】
前記化合物が:
【化134】
、ならびにその薬学的に受容可能な塩およびエステルからなる群より選択される少なくとも1つの化合物である、請求項33に記載の方法。
【請求項38】
前記方法が、第二の化学療法薬物を投与する工程をさらに包含する、請求項1〜37のいずれか1項に記載の方法。
【請求項39】
前記式(I)の化合物を含有する組成物が静脈内投与される、請求項1〜38のいずれか1項に記載の方法。
【請求項40】
前記式(I)の化合物を含有する組成物が、体重1kgあたり約0.10mg〜約25mgの投薬量で投与される、請求項1〜39のいずれか1項に記載の方法。
【請求項41】
以下に列挙される化合物:
【化135】
【化136】
【化137】
【化138】
、ならびにその薬学的に受容可能な塩およびエステルから選択される、化合物。
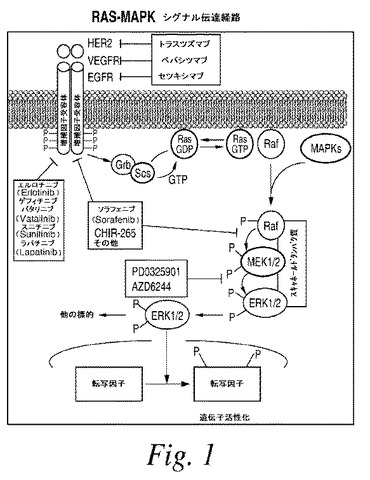
【図1】

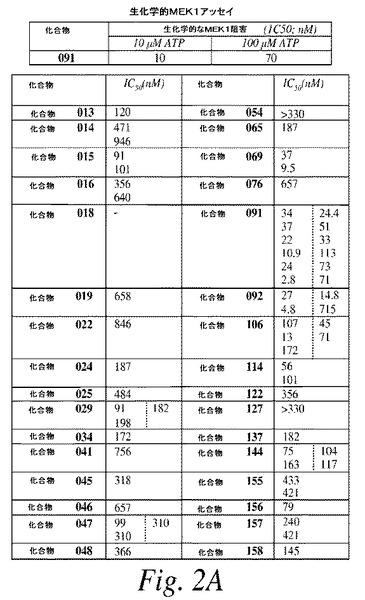
【図2A】
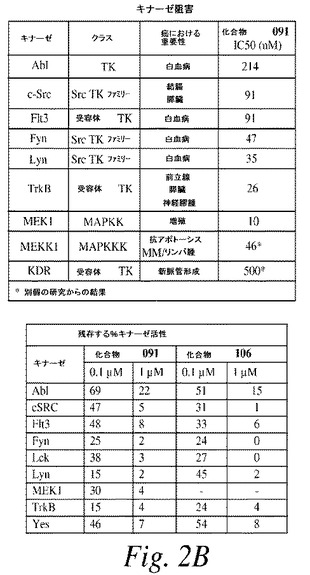
【図2B】
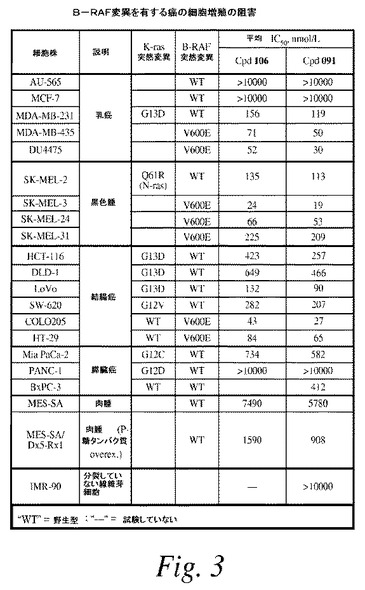
【図3】
【図4】
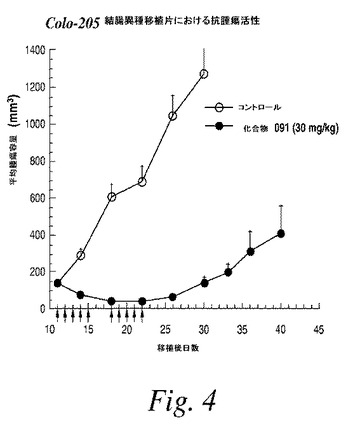
【図5】
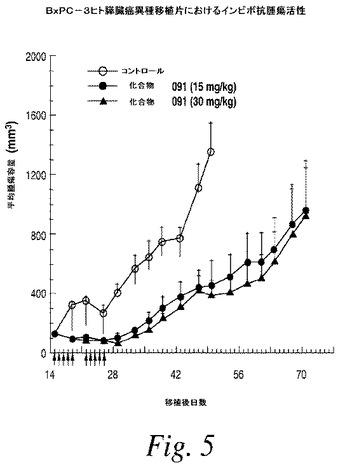
【図6】
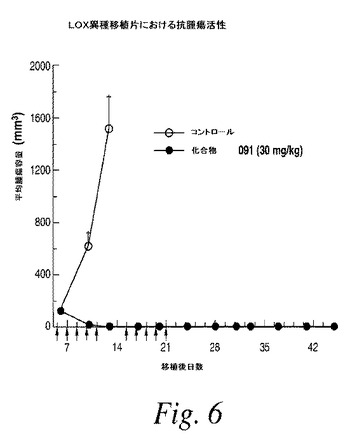
【図7】
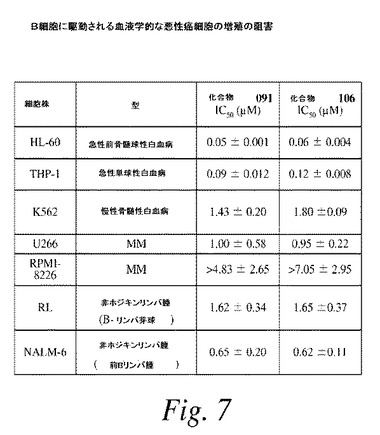
【図8】
【図9】
【図10】
【図11】
【図2A】
【図2B】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2010−534682(P2010−534682A)
【公表日】平成22年11月11日(2010.11.11)
【国際特許分類】
【出願番号】特願2010−518424(P2010−518424)
【出願日】平成20年7月25日(2008.7.25)
【国際出願番号】PCT/US2008/071256
【国際公開番号】WO2009/015368
【国際公開日】平成21年1月29日(2009.1.29)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
【公表日】平成22年11月11日(2010.11.11)
【国際特許分類】
【出願日】平成20年7月25日(2008.7.25)
【国際出願番号】PCT/US2008/071256
【国際公開番号】WO2009/015368
【国際公開日】平成21年1月29日(2009.1.29)
【出願人】(506137147)エーザイ・アール・アンド・ディー・マネジメント株式会社 (215)
【Fターム(参考)】
[ Back to top ]