
癌の処置用のポリ−ヘテロアリール誘導体
本発明は、それぞれ式(I)、(II)、(III)及び(IV)の複素環1(Het−1)a及び/又は複素環2(Het−2)b及び/又は複素環3(Het−3)c及び/又は複素環4(Het−4)dの組合せ、N−オキシド、薬学的に許容しうる付加塩を含む、ペンタ−、ヘキサ−、ヘプタ−、オクタ−、ノナ−及びデカ−ヘテロアリール誘導体に関する。本特許請求化合物は、癌の処置に適している。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、四重鎖DNAに特異的に結合してこれを安定化する、ポリ−ヘテロアリール誘導体に関する。本発明はまた、このような化合物の薬学的に許容しうる塩、これらを含有する医薬組成物の製造方法、並びに癌及び感染症の処置におけるこのような化合物の使用に関する。
【背景技術】
【0002】
「ポリヘテロアリール」は、本明細書及び請求の範囲において使用されるとき、場合によりフェニレン部分を包含するか、かつ/又は末端フェニル基を有する、芳香族複素環部分よりなる配列/鎖を意味する。
【0003】
G−四重鎖DNAは、現在のところ高G含量配列の機能を調節できる構造要素と考えられている。詳細には、多くの証拠から、テロメアにおける、又はc−Myc、Bcl−2、VEGF、Hif−1a、Ret、c−Kit、PDGF−A、KRAS及びc−Myb(1)を包含する種々のヒト腫瘍遺伝子中の特異的遺伝子プロモーターにおける、この特有のDNA構造の形成が、癌細胞増殖を阻害することが示唆されている。
【0004】
ヒト細胞のテロメアDNAは、配列:5'-TTAGGG-3'の縦列反復を含むが、これはその3’側で、インビトロでG−四重鎖中に折り畳まれることが証明されている、一本鎖オーバーハングで終わる。インビボでのこのような四重鎖構造の形成は、テロメアと通常結合した保護タンパク質(シェルタリン(shelterin)複合体)の変位をもたらし、このためテロメア構造が崩壊してゲノムが不安定になると仮定されている。
【0005】
腫瘍遺伝子プロモーターでは、四重鎖の安定化は、遺伝子発現を調節するタンパク質因子の局所環境を崩壊させることが予想されるが、これはc−mycに関しては充分な証拠がある(1)。よって四重鎖DNA形成を誘導又は安定化することができる化学的介入は、低分子四重鎖結合剤を介して、特に癌細胞においてDNA関連機能(テロメア伸長、腫瘍遺伝子転写)を制御することを目的として、徹底調査されている。
【0006】
G−四重鎖DNAと相互作用する化合物は、現在これらに傾けられた最近の総説(2〜9)の数に例証されるとおり、文献上非常に多い。しかし、二重鎖DNAよりも四重鎖DNAに対して高度の選択性を示す化合物は少なく;更には、これらの化合物の少数のものだけが、抗癌剤として徹底調査されている。このファミリーのリード化合物は、5個のオキサゾール環、2個のメチルオキサゾール環及び1個のチアゾリン環からなる、天然の大環状物質(Streptomyces anulatus 3533-SV4から単離)である、テロメスタチン(telomestatin)である。テロメスタチンは、四重鎖選択的相互作用に関して1つのパラダイムを表し、そしてインビトロで並外れたテロメラーゼ阻害特性を示す(TRAP又は直接アッセイのいずれかにより評価、IC50−TRAP=0.6nM及びIC50−直接アッセイ=58nM)(10)。更に、テロメスタチンはまた、その細胞効果についても深く調査され、そして種々の腫瘍細胞株において抗増殖及びアポトーシス特性を呈した(11及び12)。テロメスタチンは、テロメアの完全性を変え(13)、テロメアでのDNA損傷応答を引き起こす(14)。テロメスタチンは、インビトロでPOT1脱キャップ形成(uncapping)を誘導し(IC50−POT1=500nM)、そして腫瘍細胞でテロメアからGFP−POT1を除去する(15)。テロメスタチンの厳しい合成経路を考慮して(16)、興味深い四重鎖相互作用特性を呈する類似体が報告されている(17、18及び19)。
【0007】
しかし、これらの化合物のどれもが、テロメスタチンの並外れた特性を示していない。更に、テロメスタチン自体を包含するこれらの化合物のどれもが、その蛍光特性について調査されていない。テロメスタチンの化学的不安定性、難水溶性及び特に困難な合成を前提とすると、それの大規模な治療的使用には問題が多い。
【0008】
このような状況において、本発明者らは、新しい非大環状ポリヘテロアリール誘導体が、二重鎖DNAよりも四重鎖DNAに対して優れた選択性を示すこと、そしてこれらの誘導体のそのDNA標的との結合が、i)その分光学的特性の大きな変化をもたらし、ii)テロメアG−オーバーハングからのヒトPOT1(テロメアの保護1)タンパク質を脱キャップし、iii)ヒト腫瘍細胞株の細胞増殖を阻害することを見い出した。この新しいファミリーの化合物の生理学的液体中での安定性及び可溶性は、治療処置におけるその使用に適合する。更には、本発明者らは、これらの化合物を入手するための効率的かつ独創的な製造法を見い出した。
【0009】
次に本発明の1つの目的は、新しい製品としてこのような非大環状ポリヘテロアリール誘導体を提供することである。
【0010】
また、これらの合成方法にも関する。
【0011】
別の目的により、本発明は、該化合物の四重鎖相互作用特性を利用して、更に四重鎖特異的プローブとしての該誘導体の使用に関する。詳細には、開示化合物の高蛍光量子収率量子を考慮して、このような化合物は、四重鎖DNA又は関連核酸構造(特に限定されないが四重鎖RNAを包含する)の検出及び/又は精製用の蛍光プローブとして使用することができよう。
【0012】
更に別の目的では、本発明は、活性成分として該誘導体を含有する医薬組成物を提供し、また広範な疾患用の薬物、特に例えば、癌又はマラリアのような感染症に罹患している患者の処置用の薬物の製造における該誘導体の使用に関する。
【0013】
本発明はまた、癌及び感染症の処置方法であって、有効量の該誘導体をこれを必要とする患者に投与することを含む方法に関する。
【図面の簡単な説明】
【0014】
【図1】図1は、種々の条件での本発明の誘導体の吸収スペクトルを表す。
【図2】図2は、種々の滴定の要約を表す。
【図3】図3は、ヒト又はマラリア原虫テロメア配列を模倣するオリゴヌクレオチド及びFRETパートナーによるFRET融解試験法の結果を表す。
【図4】図4は、本発明の誘導体を伴うか又は伴わないG−四重鎖の蛍光融解の結果を表す。
【図5】図5は、G−四重鎖単独又は本発明の誘導体の存在下での蛍光融解の結果を表す。
【図6】図6は、種々のFRET融解実験の要約を表す。
【図7】図7は、本発明の誘導体を加えた四重鎖DNAでの蛍光融解の結果を表す。
【図8】図8は、本発明の誘導体を加えた四重鎖又は短い二重鎖DNAを用いた蛍光滴定の結果を表す。
【図9】図9は、本発明の誘導体を加えた種々の四重鎖DNA構造を用いた蛍光滴定の結果を表す。
【図10】図10は、種々の蛍光滴定の要約のグラフ表示である。
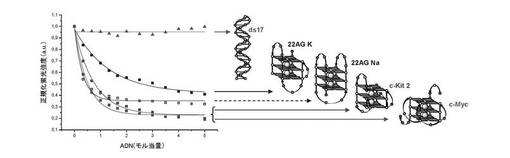
【図11】図11は、本発明の誘導体を加えた四重鎖DNAの円偏光二色性の結果を表す。
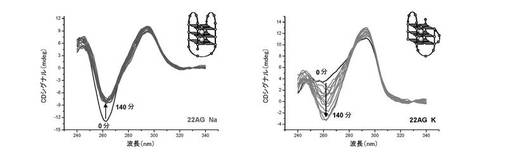
【図12】図12は、本発明の誘導体を加えた四重鎖DNAの円偏光二色性の結果を表す。
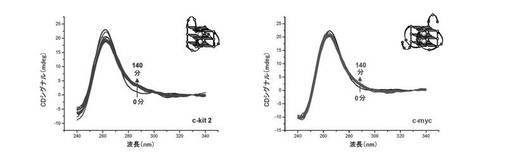
【図13】図13は、四重鎖構造の誘導に関する結果を表す。
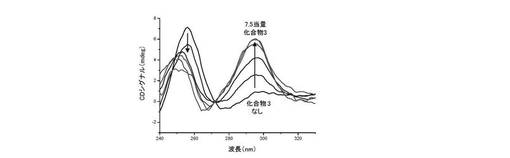
【図14】図14は、本発明の誘導体によるインビトロでのテロメア配列へのPOT1結合の阻害に関する結果を表す。
【図15】図15は、本発明の誘導体による細胞増殖の阻害に関する結果を表す。
【図16】図16は、四重鎖DNAの安定化、及び本発明の誘導体の二重鎖に対する四重鎖DNAの選択性に関する結果を表す。
【0015】
本発明のポリヘテロアリール誘導体は、ペンタ−、ヘキサ−、ヘプタ−、オクタ−、ノナ−及びデカ−ヘテロアリール誘導体(以降ペンタ−〜デカ−ヘテロアリールと略す)であって、それぞれ式(I)、(II)、(III)及び(IV)の複素環1(Het−1)a及び/又は複素環2(Het−2)b及び/又は複素環3(Het−3)c及び/又は複素環4(Het−4)d:
【0016】
【化1】

の組合せ、N−オキシド、薬学的に許容しうる付加塩[ここで、
− 該組合せは、少なくとも2種の異なる複素環部分を含み、そして場合により式(V)の(Aryl−5)e部分を含み、
− a、b及びeは、0〜6の整数であり、c及びdは、0〜2の整数であり、a+b+c+d+eの合計は、≦10であり、
− Yは、O又はSであり;
− Het−1は、2,6−ピリジン−ジイル又は2,4−ピリミジン−ジイル又は3,5−ピラジン−ジイル又は2,4−(1,3,5−トリアジン)ジイル又は3,5−(1,2,4−トリアジン)ジイル又は2,4−オキサゾリン−ジイル又は2,4−チアゾリン−ジイルを含む群において選択される窒素複素環−ジイル環のクラスであり;
− Het−2は、2,5−オキサゾリン−ジイル又は2,5−チアゾリン−ジイル又は2,5−チオフェン−ジイル又は2,5−フラン−ジイルを含む群において選択される5員複素環−ジイル環のクラスであり;
− Het−3及び/又はHet−4は、該ペンタ−、ヘキサ−、ヘプタ−、オクタ−、ノナ−及びデカ−ヘテロアリール誘導体の終点を構成するが、ここで、
− Het−3は、2−ピリジル又は2−ピリミジル又は4−ピリミジル又は2−ピラジル又は2−(1,3,5)トリアジル又は3−(1,2,4)トリアジル又は5−(1,2,4)トリアジル又は4−オキサゾリル又は4−チアゾリルから選択される窒素複素環−イル環のクラスであり;
− Het−4は、2−オキサゾリル又は5−オキサゾリル又は2−チアゾリル又は5−チアゾリル又は2−チエニル又は2−フラニルから選択される5員複素環−イルのクラスであり;
− R1及びR2は、それぞれ独立に、水素;C1−6アルキル;C1−4アルキルオキシ;ハロ;ヒドロキシ;ヒドロキシメチル;ニトロ;アミノ;モノ−又はジ(C1−4アルキル)アミノ;C1−4アルキルメチルアミノ;モノ−又はジ(C1−4アルキル)アミノ(C2−4アルキル)アミノメチル;モルホリン−4−イルC2−4アルキルオキシ;ピペラジン−1−イルC2−4アルキルオキシ;4−C1−4アルキルピペラジン−1−イル;フェニル、ベンジル、ナフチルから選択される単環又は二環から選択される]を含む誘導体である。
【0017】
本発明のある実施態様では、a、b、c及びdは、0〜3の整数であり、e=0であり、そしてa+b+c+dの合計は、≦10である。
【0018】
本発明のある実施態様では、1個又は数個の上記部分は、それぞれ独立にC1−6アルキル;C1−4アルキルオキシ;ハロ;ヒドロキシ;ニトロ;アミノ;モノ−又はジ(C1−4アルキル)アミノ;C1−4アルキルメチルアミノ;モルホリン−4−イルC2−4アルキルオキシ;ピペラジン−1−イルC2−4アルキルオキシ;4−C1−4アルキルピペラジン−1−イルC2−4アルキルオキシ;フェニル、ベンジル、ナフチルから選択される単環式又は二環式環から選択される、1、2又は3個の置換基で置換されている。
【0019】
本発明は、詳細には少なくとも5個、少なくとも7個、少なくとも8個、少なくとも9個又は10個の複素環部分を含む、上記と同義の誘導体に関する。
【0020】
別の態様では、本発明はまた、詳細には1個又は2、3若しくは4個のような数個の1,3−フェニレン−ジイル部分を更に含む、上記と同義の誘導体に関する。
【0021】
本発明の特に好ましい誘導体は、下記構造の1つを有する:
−(R2により置換されたHet−4)−(Het−1)−(R2により置換されたHet−4)
−(R1により置換されたHet−3)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−1,3−フェニレン−ジイル−(Het−2)−(R1により置換されたHet−3)
−(R1又はR2により置換された1,3−フェニレン−ジイル)−(Het−2)−(Het−1)−(Het−2)−(R1又はR2により置換された1,3−フェニレン−ジイル)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−1)−(Het−2)−(R1により置換されたHet−3)。
【0022】
好ましい誘導体において、
・ R1及びR2は、オキサゾリン−若しくはピリジン−ジイル基であるか、かつ/又は
・ Het−2は、オキサゾリン−ジイル基であるか、かつ/又は
・ Het−1は、2,6−ピリジン−ジイル、2,6−ピリミジン−ジイル、2,6−ピラジン−ジイル、若しくは1,3−フェニレン−ジイル基である。
【0023】
有利な群では、誘導体のG−四重鎖相互作用特性を考慮して、Het−1は、2個のHet−2に結合している。
【0024】
該群の好ましい誘導体において、Het−1は、ピリジン−ジイルであり、そしてHet−4は、ピリジル又はピリジル−オキサゾリルにより置換されている、オキサゾリルである。
【0025】
第2の有利な群では、Het−2は、2個のHet−1に結合している。
【0026】
好ましい誘導体において、Het−2は、オキサゾリルであり、そしてHet−1は、ピリジル−オキサゾリルである。
【0027】
本発明の特定の好ましい化合物は、後述の実施例の項にリストされているもの、特に化合物(3)及び(4)である。
【0028】
上で定義された誘導体の薬学的に許容しうる付加塩は、その酸付加塩及び塩基塩(二塩を包含する)を包含する。
【0029】
適切な酸付加塩は、非毒性塩を形成する酸から形成される。例は、酢酸、アスパラギン酸、安息香酸、ベシル酸、重炭酸/炭酸、重硫酸/硫酸、ホウ酸、カンシル酸、クエン酸、エジシル酸、エシル酸、ギ酸、フマル酸、グルセプト酸、グルコン酸、グルクロン酸、ヘキサフルオロリン酸、ヒベンズ酸、塩酸/塩化物、臭化水素酸/臭化物、ヨウ化水素酸/ヨウ化物、イセチオン酸、乳酸、リンゴ酸、マレイン酸、マロン酸、メシル酸、メチル硫酸、ナフチル酸、2−ナプシル酸、ニコチン酸、硝酸、オロチン酸、シュウ酸、パルミチン酸、パモ酸、リン酸/リン酸水素/リン酸二水素、サッカラート、ステアリン酸、コハク酸、酒石酸、トシル酸及びトリフルオロ酢酸塩を包含する。
【0030】
適切な塩基塩は、非毒性塩を形成する塩基から形成される。例は、アルミニウム、アルギニン、ベンザチン、カルシウム、コリン、ジエチルアミン、ジオラミン、グリシン、リシン、マグネシウム、メグルミン、オラミン、カリウム、ナトリウム、トロメタミン及び亜鉛塩を包含する。
【0031】
上で定義された誘導体のこのような薬学的に許容しうる塩は、該誘導体及び所望の酸又は塩基の溶液を必要に応じて一緒に混合することにより、容易に調製できる。塩は、溶液から沈殿するため、これを濾過により収集するか、又は溶媒の留去により回収することができる。塩におけるイオン化の程度は、完全にイオン化からほぼイオン化がない状態まで変化しうる。
【0032】
本発明はまた、上記と同義の同位体標識誘導体、好ましくは薬学的に許容しうる同位体標識誘導体を包含するが、この誘導体では1個以上の原子が、同一の原子番号を有するが、自然界で通常見られる原子質量又は質量数とは異なる原子質量又は質量数を有する原子により置換されている。
【0033】
本発明の化合物に含めるのに適切な同位体の例は、2H及び3Hのような水素、11C、13C及び14Cのような炭素、36Clのような塩素、18Fのようなフッ素、123I及び125Iのようなヨウ素、13N及び15Nのような窒素、15O、17O及び18Oのような酸素、32Pのようなリン、並びに35Sのような硫黄の同位体を包含する。
【0034】
本発明のある種の同位体標識誘導体、例えば、放射性同位体を取り込んだ誘導体は、薬物及び/又は基質の組織分布研究において有用である。放射性同位体のトリチウム、即ち、3H、及び炭素14、即ち、14Cは、その取り込みの容易さ及び即時の検出手段を考慮して、この目的に特に有用である。
【0035】
ジュウテリウム、即ち、2Hのような重い同位体での置換では、代謝安定性の高さに起因するある種の治療上の利点、例えば、インビボ半減期の延長又は必要用量の減少が得られ、このため幾つかの状況では好ましい。
【0036】
11C、18F、15O及び13Nのような陽電子放出同位体での置換では、基質の受容体占有率を検査するための陽電子放出断層撮影法(PET)研究において有用でありうる。
【0037】
後述の実施例に示されるように、本発明の誘導体は、四重鎖DNAに特異的に結合して安定化するが、このため四重鎖特異的プローブとして大いに興味深い。四重鎖DNAに結合する該誘導体の能力、更にはその選択性は、後述の実施例の項に記述される試験法を包含する、当業者には既知の試験法を用いて測定することができる。
【0038】
これらはまた、種々の疾患の処置のために大いに価値がある。
【0039】
よって本発明は、上記誘導体の薬物としての使用に関する。
【0040】
本発明はまた、有効量の少なくとも1つの上記のような誘導体を薬学的に許容しうる担体と組合せて含む医薬組成物に関する。
【0041】
本明細書において使用されるとき、「治療有効量」又は「有効量」は、疾患の処置を構成するのに充分な、患者に投与される上記誘導体のような治療剤の量を意味する。
【0042】
本明細書において使用されるとき、疾患の「処置」という用語は、(1)このような用語が適用される病態又は病状の症状の進行、増悪、再発、播種又は悪化を遅延又は停止させること;(2)このような用語が適用される病態又は病状の症状を軽減させるか又は改善をもたらすこと;及び/あるいは(3)このような用語が適用される病態又は病状を回復させるか又は治癒することを目指した任意の活動のことをいう。
【0043】
医薬用途の本発明の誘導体は、実際に単独で、又は1種以上の他の本発明の誘導体と組合せて、又は1種以上の他の薬物、特に抗癌薬若しくは抗感染症薬と組合せて(又はその任意の組合せとして)投与することができる。一般に、これらは1種以上の薬学的に許容しうる担体と結び付けた製剤として投与されよう。「担体」という用語は、本明細書において本発明の誘導体以外の任意の成分を記述するために使用される。担体の選択は、大体において、特定の投与様式、溶解度及び安定性に及ぼす担体の効果、並びに投与剤形の性質のような要因に依存する。
【0044】
上記の誘導体の送達に適切な医薬組成物及びその製造方法は、当業者には容易に明らかとなるだろう。このような組成物及びその製造方法は、例えば、'Remington’s Pharmaceutical Sciences', 19th Edition (Mack Publishing Company, 1995)に見い出すことができる。
【0045】
本医薬組成物は、例えば、錠剤、カプセル剤、丸剤、粉剤、徐放製剤、液剤、懸濁剤として経口投与に;液剤、懸濁剤又は乳剤として非経口投与に;軟膏剤又はクリーム剤として局所投与に;あるいは坐剤として直腸内投与に適切な剤形にしてもよい。本医薬組成物は、正確な用量の単回投与用に適切な単位投与剤形にしてもよい。
【0046】
適切な製剤担体は、不活性希釈剤又は増量剤、水及び種々の有機溶媒を包含する。本医薬組成物は、必要に応じて、香味料、結合剤、賦形剤などのような追加の成分を含有してもよい。よって経口投与には、クエン酸のような種々の賦形剤を含有する錠剤を、デンプン、アルギン酸及びある種のケイ酸複合体のような種々の崩壊剤と、並びにショ糖、ゼラチン及びアカシアゴムのような結合剤と一緒にして利用してもよい。更には、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム及びタルクのような滑沢剤は、錠剤化目的にしばしば有用である。同様のタイプの固体組成物はまた、軟及び硬充填ゼラチンカプセル剤にして利用することができる。このために好ましい材料は、ラクトース、即ち乳糖及び高分子量ポリエチレングリコール類を包含する。経口投与に水性懸濁剤又はエリキシル剤が必要であれば、上記誘導体は、種々の甘味剤又は着香剤、着色料又は色素、及び必要に応じて乳化剤又は懸濁剤と、水、エタノール、プロピレングリコール、グリセリン、又はこれらの組合せと一緒にして合せることができる。
【0047】
本明細書において使用されるとき、医薬組成物の「非経口投与」は、対象の組織の物理的突破を特徴とする投与の任意の経路、及び組織での突破による医薬組成物の投与を包含する。よって非経口投与は、特に限定されないが、組成物の注射による、外科的切開を介した組成物の適用による、組織浸透性の非外科的創傷を介した組成物の適用によるなどの医薬組成物の投与を包含する。特に、非経口投与は、特に限定されないが、皮下、静脈内、皮内、腹腔内、筋肉内、胸骨内注射、及び腎臓透析点滴法を包含するものである。
【0048】
非経口投与に適切な医薬組成物の製剤は、活性成分を無菌水又は無菌等張性食塩水のような薬学的に許容しうる担体と合わせて含む。このような製剤は、ボーラス投与又は連続投与に適した剤形で調製、包装、又は販売することができる。非経口投与用の製剤は、特に限定されないが、後述のような懸濁剤、液剤、油性又は水性ビヒクル中の乳剤、ペースト、及び埋め込み型徐放性又は生分解性製剤を包含する。このような製剤は更に、特に限定されないが、懸濁剤、安定化剤、又は分散剤を包含する1種以上の追加の成分を含んでいてもよい。非経口投与用製剤の1つの実施態様において、活性成分は、復元組成物の非経口投与に先立って適切なビヒクル(例えば、無菌の発熱性物質除去水)で復元するための、乾燥(即ち、粉末又は顆粒)製剤として提供される。
【0049】
本発明の医薬組成物は、インプラント、経皮パッチ、及びマイクロカプセル化送達系を包含する放出制御製剤のように、本化合物を急速放出に対して保護する担体と共に調製することができる。エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル類、及びポリ乳酸のような、生分解性の生体適合性ポリマーを使用することができる。
【0050】
治療用途には、組成物中の活性成分の有効な1日量は、一般に、0.01mg/kg〜50mg/kg体重、更に好ましくは0.1mg/kg〜10mg/kg体重であろう。当然ながら、これは使用する誘導体、投与様式、必要な処置、適応疾患、更には患者の生理学的データ(例えば、年齢、サイズ、及び体重)により変化する。この1日総用量は、単回又は分割用量として投与することができる。治療剤の投与に適切な用量及び用法の決定は、当該分野において周知であり、当業者であれば含まれることを理解するであろう。
【0051】
上記の誘導体及びその医薬組成物は、癌及びマラリアのような感染症を処置するのに特に有用である。
【0052】
「癌」という用語は、本明細書において使用されるとき、無制限増殖、不死性、転移能、及びある種の特徴的な形態学的状態のような、発癌性細胞に典型的な特徴を持つ細胞の存在のことをいう。この用語は、任意のタイプの悪性腫瘍(原発性又は転移性)のことをいう。
【0053】
「感染症」という用語は、本明細書において使用されるとき、細菌、寄生虫、酵母、真菌のような微生物による哺乳動物(ヒト又は動物)の感染に起因する疾患のことをいう。ウイルスによる感染もまた包含される。
【0054】
癌又はマラリアのような感染症の処置用薬物を製造するための上記誘導体及びその医薬組成物の使用は、本発明の範囲の一部になる。
【0055】
本発明はまた、癌又は感染症の処置方法であって、有効量の上記の誘導体又はその医薬組成物をこれを必要とする患者に投与することを含む方法を包含する。
【0056】
本発明はまた、上記の誘導体の製造方法を含める。
【0057】
該方法は、
・ X1−A−X2を
・ B−X3と、又はX4−B−X3と、
所望のポリヘテロアリール誘導体を与える条件下で反応させること[ここで、
− Aは、ジ−又はトリ−又はテトラ−ヘテロアリール−1,4鎖であり、そしてBは、(モノ−又はジ−又はトリ−又はテトラ−ヘテロアリール−1,4)−であり(ここで、ヘテロアリール部分は、場合により1個、2個、3個又は4個のAryl−5部分又はAryl−5末端部分を含み、ヘテロアリール−1,4は、上記と同義のHet−1及び/又はHet−2及び/又はHet−3及び/又はHet−4の組合せである)、
− X1及びX2は、同一であるか又は異なって、水素又はハロゲン又はトリフラート基を表し、
− X3は、ハロゲン又はハロゲン化亜鉛又はトリアルキルスズ又はボロン酸基を表し、
− X4は、カルボキサルデヒド基を表す]を含む。
【0058】
本発明の他の特徴及び利点は、図1〜図16を参照する以下の実施例に与えられるが、ここで、
− 図1A〜図1Dは、種々の条件での本発明の誘導体の吸収スペクトルを表し;
− 図2、種々の滴定の要約;
− 図3A〜図3C、ヒト又はマラリア原虫テロメア配列を模倣するオリゴヌクレオチド及びFRETパートナーによるFRET融解試験法の結果;
− 図4A〜図4b及び図5、本発明の誘導体を伴うか又は伴わないG−四重鎖の蛍光融解の結果;
− 図5、G−四重鎖単独又は本発明の誘導体の存在下での蛍光融解の結果(過剰の四重鎖DNA競合物質を伴うか又は伴わない);
− 図6、種々のFRET融解実験の要約;
− 図7A及び図7B、本発明の誘導体を加えた四重鎖DNAでの蛍光融解の結果;
− 図8、本発明の誘導体を加えた四重鎖又は短い二重鎖DNAを用いた蛍光滴定の結果;
− 図9、本発明の誘導体を加えた種々の四重鎖DNA構造を用いた蛍光滴定の結果;
− 図10、種々の蛍光滴定の要約のグラフ表示;
− 図11A及び図11b及び図12、本発明の誘導体を加えた四重鎖DNAの円偏光二色性の結果;
− 図13、四重鎖構造の誘導に関する結果;
− 図14、本発明の誘導体によるインビトロでのテロメア配列へのPOT1結合の阻害に関する結果;
− 図15、本発明の誘導体による細胞増殖の阻害に関する結果;並びに
− 図16、四重鎖DNAの安定化、及び本発明の誘導体の二重鎖に対する四重鎖DNAの選択性に関する結果。
【0059】
I − ポリ−ヘテロアリール誘導体の合成
以下の実施例は、本発明を説明するものである。本化合物はまた、当業者が既知の他の製造法によっても得られる。出発物質として使用される、2,6−ピリジンジカルボキサルデヒド、6−ブロモピリジン−2−カルバルデヒド、イソシアン化p−トルエンスルホニルメチル(TosMIC)、2−ピリジル亜鉛ブロミド、1,3−ベンゼンジカルボキサルデヒド、6−ブロモピリジン−2−カルボキサルデヒド、3−ブロモベンズアルデヒド及び5−ブロモ−2−フルアルデヒドは、市販の製品である。
【0060】
1.:実施例1: 2,6−ビス[2−(ピリジン−2−イル)オキサゾール−5−イル]ピリジン(化合物(1))
【0061】
【化2】
【0062】
1.1.: 2,6−ビス(オキサゾール−5−イル)ピリジン(中間体(1))
【0063】
【化3】
メタノール100mL中の2,6−ピリジンジカルボキサルデヒド(4.0g;29.2mmol)、TosMIC(11.4g;58.3mmol)及び炭酸カリウム(16.3g;117.8mmol)を3時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタン(4×150mL)で抽出した。合わせた有機層を硫酸マグネシウムで乾燥して、真空で濃縮した。残渣は、ジクロロメタン:エタノール(95:5)を溶離液とするシリカゲルカラムのフラッシュクロマトグラフィーにより精製して、標題化合物を黄色の固体として得た(5.6g、90%)、融点=190〜191℃;1H NMR (300 MHz, CDCl3) 7.56 (d ; 2H ; J= 8.0) ; 7.74 (s ; 2H ) ; 7.80 (t ; 1H ; J= 8.0 Hz) ; 7.96 (s ; 2H) ; 13C NMR (75 MHz, CDCl3) 118.7, 125.7, 138.0, 147.4, 150.8, 151.0; SM m/z 214.1 (M+1) ; 元素分析、C11H7N3O2の理論値: C, 91.97 ; H, 3.31 ; N, 19.71. 実測値: C, 61.51, H, 3.43, 19.48.
【0064】
1.2.: 2,6−ビス−(2−ヨードオキサゾール−5−イル)ピリジン(中間体(2))
【0065】
【化4】
アルゴン雰囲気下、中間体(1)(0.1g;0.5mmol)及びTMEDA(0.2mL;1.0mmol)を無水THF 5mLに溶解して、−78℃で冷却した。THF中のLiHMDS 1Mの溶液(0.5mL;0.5mmol)を滴下により加え、−78℃で30分間及び−40℃で1時間撹拌した。この混合物を再度−78℃で冷却して、1,2−ジヨードエタン(0.5g;1.9mmol)を加えた。室温で一晩撹拌後、この混合物をチオ硫酸ナトリウム溶液中に注ぎ入れ、酢酸エチルで抽出した(3×10mL)。合わせた有機層を硫酸マグネシウムで乾燥して、真空で濃縮した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、中間体(2)をベージュ色の固体として得た(117mg、50%)、1H NMR (300 MHz, CDCl3) 7.58 (d ; 2H ; J= 8 Hz) ; 7.69 (s ; 2H) ; 7.85 (t ; 1H ; J= 8 Hz ) ; 13C NMR (75 MHz, CDCl3) 119.5, 129.7, 138.7, 147.0, 157.2, 151.5; SM m/z 465.8 (M+1), 487.8 (M+23).
【0066】
1.3.: 2,6−ビス[2−(ピリジン−2−イル)オキサゾール−5−イル]ピリジン(化合物(1))
方法1: 中間体(2)(65mg;0.14mmol)、2−ピリジル亜鉛ブロミド(1.2mL;0.60mmol)及びPd(PPh3)4(12mg;0.01mmol)の混合物を無水THF 2mL中で4時間加熱還流した。水を加えて、この混合物を酢酸エチルで抽出した(3×10mL)。合わせた有機層を硫酸マグネシウムで乾燥し、濾過して真空で濃縮した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、目的化合物(1)をベージュ色の固体として得た(13mg、25%)、融点=242〜245℃;1H NMR (300 MHz, CDCl3) 7.40 (m ; 2H) ; 7.83-7.88 (m ; 5H) ; 7.97 (s ; 2H) ; 8.23 (d ; 2H ; J= 7.9 Hz) ; 8.77 (d ; 2H ; J= 4.5 Hz) ; 13C NMR (75 MHz, CDCl3) 119.7, 123.3, 125.6, 128.6, 137.8, 138.5, 146.6, 147.9, 150.9,152.3, 161.5; SM m/z 368.0 (M+1), 390.0 (M+23).
【0067】
方法2: 中間体(1)(0.1g;0.5mmol)、2−ブロモピリジン(91μL;0.9mmol)、二酢酸パラジウム(11mg;0.05mmol;10mol%)、PCy3.HBF4(35mg;0.1mmol;20mol%)、ヨウ化銅(I)(0.18g;0.9mmol)、炭酸セシウム(0.61mg;1.88mmol)及び無水トルエン1.3mLの混合物をマイクロ波照射条件(130℃、150W)下に4時間置いた。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、化合物(1)(8mg、33%)を得たが、これは上の方法1に記述されたものと同一である。
【0068】
2.実施例2: 2,5−ビス−[6−(オキサゾール−5−イル)]ピリジン−2−イルオキサゾール(化合物(2))
【0069】
【化5】
【0070】
2.1: 5−(2−ブロモピリジン−2−イル)オキサゾール(中間体(3))
【0071】
【化6】
6−ブロモピリジン−2−カルバルデヒド(4.0g;21.5mmol)、TosMIC(4.2g;21.5mmol)及び炭酸カリウム(6.0g;43.4mmol)をメタノール75mL中で3時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタンで抽出した(4×100mL)。合わせた有機層を硫酸マグネシウムで乾燥して真空で濃縮した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、中間体(3)を黄色の固体として得た(2.9g、60%);融点=90〜91℃;1H NMR (300 MHz, CDCl3) 7.38 (dd ; 1H ; J= 3.5 & 5.3 Hz) ; 7.57 (m ; 2H ) ; 7.73 (s ; 1H) ; 7.97 (s ; 1H) ; 13C NMR (75 MHz, CDCl3) 118.1, 126.4, 127.6, 139.3, 142.4, 148.0, 151.6 ; SM m/z 225.0 and 227.1 (M+1).
【0072】
2.2: 2,5−ビス−[6−(オキサゾール−5−イル)]ピリジン−2−イルオキサゾール(化合物(2))
中間体(1)(0.10g;0.5mmol)、中間体(3)(0.10g;0.5mmol)、二酢酸パラジウム(11mg;0.05mmol;10mol%)、PCy3.HBF4(35mg;0.09mmol;20mol%)、ヨウ化銅(I)(0.1g;0.5mmol)、炭酸セシウム(0.3mg;0.9mmol)及び無水トルエン1.3mLの混合物をマイクロ波照射条件(130℃、150W)下に6時間置いた。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、目的化合物(2)をベージュ色の固体として得た(60mg、36%);1H NMR (300 MHz, CDCl3) 7.70 (d ; 2H ; J= 7.5 Hz) ; 7.80 (d ; 1H; J= 7.5Hz) ; 7.85 (d ; 2H ; J= 7.5 Hz ) ; 7.90 (s ; 1H) ; 7.93 (d; 2H ; J= 5 Hz) ; 8.00 (s ; 1H) ; 8.04 (d ; 1H ; J= 5 Hz) ; 8.20 (d ; 1H ; J= 7.5 Hz); SM m/z358.1 (M+1).
【0073】
3.実施例3: 2,6−ビス{2−[6−(オキサゾール−5−イル)ピリジン−2−イル]オキサゾール−5−イル}ピリジン(化合物(3))
【0074】
【化7】
中間体(1)(0.10g;0.5mmol)、中間体(3)(0.21g;0.9mmol)、二酢酸パラジウム(11mg;0.05mmol;10mol%)、PCy3.HBF4(35mg;0.09mmol;20mol%)、ヨウ化銅(I)(0.18g;0.9mmol)、炭酸セシウム(0.61mg;1.9mmol)及び無水トルエン1.3mLの混合物をマイクロ波照射条件(130℃、150W)下に6時間置いた。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、目的化合物(3)をベージュ色の固体として得た(52mg、22%);融点>250℃;1H NMR (300 MHz, CDCl3) 7.65 (m; 1H) ; 7.75 (m; 2H); 7.90-8.05 (m; 9H); 8.10 (s; 1H); 8.20 (m; 2H); SM m/z 524.2 (M+23).
【0075】
4.実施例4: 1,3−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)ベンゼン(化合物(4))
【0076】
【化8】
【0077】
4.1: 6,6’−(5,5’−(1,3−フェニレン)ビス(オキサゾール−5,2−ジイル))ジピリジン−2−カルバルデヒド(中間体(4))
【0078】
【化9】
Sambavisarao及びVrajesh(Synthesis, 2007, 3653)に報告されたように得られた1,3−ジ(オキサゾール−5−イル)ベンゼン(0.21g;1mmol)、6−ブロモピリジン−2−カルボキサルデヒド(0.24g;1.3mmol)、二酢酸パラジウム(64mg;0.28mmol、28mol%)、PCy3.HBF4(62mg;0.17mmol;17mol%)、ヨウ化銅(I)(0.43g;2.3mmol)、炭酸セシウム(1.4g;4.3mmol)及び無水ジオキサン4mLの混合物を封管中で130℃で2時間加熱した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、中間体(4)をベージュ色の固体として得た(83mg、30%);1H NMR (300 MHz, CDCl3) 10.27 (s ; 1H); 8.45-8.40 (m ; 1H) ; 8.20 (s ; 0.5H) ; 8.10-8.05 (m ; 2H) ; 7.83 (dd ; 1H; J= 7.8 & 1.6 Hz) ; 7.71 (s; 1H) ; 7.61 (t ; 0.5H; J= 7.93 Hz); SM m/z 423.1 (M+1).
【0079】
4.2: 1,3−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)ベンゼン(化合物(4))
無水エタノール8mL中のジカルボキサルデヒド中間体(4)(75mg;0.18mmol)、TosMIC(86mg;0.44mmol)及び炭酸カリウム(126mg;0.9mmol)の混合物を2時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタンで抽出した(3×50mL)。合わせた有機層を硫酸マグネシウムで乾燥して真空で濃縮した。残渣は、ジクロロメタン:エタノール(95:5)を溶離液とするシリカゲルカラムのフラッシュクロマトグラフィーにより精製して、標題化合物をオフホワイト色の固体として得た(55mg、62%)、融点>250℃;1H NMR (300 MHz, CDCl3) 8.22 (br s ; 0.5H) ; 8.17 (d ; 1H; J= 7.8 Hz) ; 8.03 (s; 1H) ; 7.99 (t ; 1H, J= 7.8 Hz), 7.93 (s; 1H) ; 7.85-7.76 (m; 2H); 7.68 (s; 1H); 7.64-7.57 (m; 0.5H); SM m/z 501.1 (M+1) ; 元素分析、C28H16N6O4. 0.75 H2Oの理論値: C, 65.43 ; H, 3.40 ; N, 16.36, 実測値: C, 65.92, H, 3.37, 15.92.
【0080】
5.実施例5: 2,6−ビス(2−(3−(オキサゾール−5−イル)フェニル)オキサゾール−5−イル)ピリジン(化合物(5))
【0081】
【化10】
【0082】
5.1: 3,3’−(5,5’−(ピリジン−2,6−ジイル)ビス(オキサゾール−5,2−ジイル))ジベンズアルデヒド(中間体(5))
【0083】
【化11】
中間体(1)(0.18g;0.84mmol)、3−ブロモベンズアルデヒド(0.50g;2.7mmol)、二酢酸パラジウム(80mg;0.36mmol;43mol%)、PCy3.HBF4(68mg;0.18mmol;20mol%)、ヨウ化銅(I)(0.37g;1.9mmol)、炭酸セシウム(1.2g;3.7mmol)及び無水ジオキサン3.5mLの混合物を封管中で130℃で18時間加熱した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、98:2)により精製して、中間体(5)を黄色の固体として得た(50mg、14%);1H NMR (300 MHz, CDCl3) 10.15 (s ; 1H); 8.66 (s ; 1H) ; 8.45 (d ; 1H; J= 7.7 Hz) ; 8.04 (d; 1H; J= 7.6 Hz) ; 7.98-7.92 (m ; 1.5H) ; 7.76 (d ; 1H; J= 7.9 Hz) ; 7.72 (t ; 1H; J= 7.7 Hz); SM m/z 444.0 (M+Na); 元素分析、C25H15N3O4. 0.25 H2Oの理論値: C, 70.50 ; H, 3.64 ; N, 9.87, 実測値: C, 70.11, H, 3.51, 9.75.
【0084】
5.2: 2,6−ビス(2−(3−(オキサゾール−5−イル)フェニル)オキサゾール−5−イル)ピリジン(化合物(5))
無水エタノール6mL中のジカルボキサルデヒド中間体(5)(50mg;0.12mmol)、TosMIC(50mg;0.25mmol)及び炭酸カリウム(70mg;0.5mmol)の混合物を2時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタンで抽出した(3×50mL)。合わせた有機層を硫酸マグネシウムで乾燥して、真空で濃縮した。残渣は、ジクロロメタン:エタノール(94:6)を溶離液とするシリカゲルカラムのフラッシュクロマトグラフィーにより精製して、標題化合物を淡黄色の固体として得た(40mg;67%)、融点254〜256℃;1H NMR (300 MHz, CDCl3) 8.45 (s ; 1H) ; 8.14 (d ; 1H; J= 7.9 Hz) ; 7.99 (s; 1H) ; 7.96-7.88 (m ; 1.5H), 7.79 (d ; 1H; J= 7.9 Hz); 7.74 (d ; 1H; J= 7.9 Hz); 7.60 (t ; 1H; J= 7.9 Hz); 7.51 (s; 1H); SM m/z 500.2 (M+1) ; 元素分析、C29H17N5O4. H2Oの理論値: C, 67.31 ; H, 3.67 ; N, 13.53, 実測値: C, 67.53, H, 3.82, 13.29.
【0085】
6.実施例6: 2,6−ビス(2−(5−(オキサゾール−5−イル)フラン−2−イル)オキサゾール−5−イル)ピリジン(化合物(6))
【0086】
【化12】
【0087】
6.1: 5,5’−(5,5’−(ピリジン−2,6−ジイル)ビス(オキサゾール−5,2−ジイル))ジフラン−2−カルバルデヒド(中間体(6))
【0088】
【化13】
中間体(1)(0.21g;1mmol)、5−ブロモ−2−フルアルデヒド(0.21g;1.2mmol)、二酢酸パラジウム(83mg;0.37mmol;37mol%)、PCy3.HBF4(72mg;0.2mmol;20mol%)、ヨウ化銅(I)(0.46g;2.4mmol)、炭酸セシウム(1.4g;4.3mmol)及び無水ジオキサン4mLの混合物を封管中で130℃で2時間加熱した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、98:2)により精製して、中間体(6)を黄色の固体として得た(62mg、26%);1H NMR (300 MHz, CDCl3) 9.83 (s ; 1H); 7.97 (s ; 1H) ; 7.95-7.90 (m ; 0.5H) ; 7.79 (d; 1H; J= 8.2 Hz) ; 7.39 (d; 1H; J= 3.8 Hz) ; 7.31 (d ; 1H; J= 3.8 Hz); SM m/z 402.1 (M+1);元素分析、C21H11N3O6. 1.5 H2Oの理論値: C, 58.87 ; H, 3.27 ; N, 9.81, 実測値: C, 58.77, H, 2.85, 9.83.
【0089】
6.2: 2,6−ビス(2−(5−(オキサゾール−5−イル)フラン−2−イル)オキサゾール−5−イル)ピリジン(化合物(6))
無水エタノール3mL中のジカルボキサルデヒド中間体(6)(30mg;0.07mmol)、TosMIC(43mg;0.22mmol)及び炭酸カリウム(63mg;0.45mmol)の混合物を2時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタンで抽出した(3×50mL)。合わせた有機層を硫酸マグネシウムで乾燥して、真空で濃縮した。残渣は、ジクロロメタン:エタノール(96:4)を溶離液とするシリカゲルカラムのフラッシュクロマトグラフィーにより精製して、標題化合物を淡黄色の固体として得た(13mg;36%)、融点>250℃;1H NMR (300 MHz, CDCl3) 7.95-7.91 (m ; 2.5H) ; 7.71 (d ; 1H; J= 8.1 Hz) ; 7.53 (s; 1H) ; 7.34-7.29 (m ; 2H); SM m/z 480.1 (M+1).
【0090】
7.: 2,6−ビス(2−(2,2’−ビピリジン−6−イル)オキサゾール−5−イル)ピリジン(化合物(7))
【0091】
【化14】
中間体(1)から出発し、そして中間体(3)の代わりに6−ブロモ−2,2’−ビピリジン(U. Lehmann and A.D. Schluter, Eur. J. Org. Chem. 2000, 3483-3487)を用いて、化合物(3)の合成に関して上記されたプロトコールを適用することにより、標題化合物(7)を得た。
【0092】
8.: 2,6−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)ピラジン(化合物(8))
【0093】
【化15】
【0094】
8.1: 2,6−ジ(オキサゾール−5−イル)ピラジン(中間体(7))
【0095】
【化16】
2,6−ピリジンジカルボキサルデヒドの代わりにピラジン−2,6−ジカルバルデヒド(H. Schumann and H.-K. Luo, Zeitschrift fuer Naturforschung, B : Chemical Sciences, 2005, 60(1), 22-24)から出発して、中間体(1)の合成に関して上記されたプロトコールを適用することにより、標題中間体(7)を得た。
【0096】
8.2: 6,6’−(5,5’−(ピラジン−2,6−ジイル)ビス(オキサゾール−5,2−ジイル))ジピリジン−2−カルバルデヒド(中間体(8))
【0097】
【化17】
中間体(1)及び3−ブロモベンズアルデヒドの代わりに中間体(7)及び6−ブロモピリジン−2−カルバルデヒドから出発して、中間体(5)の合成に関して上記されたプロトコールを適用することにより、標題中間体(8)を得た。
【0098】
8.3: 2,6−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)ピラジン(化合物(8))
中間体(5)の代わりに中間体(8)から出発して、化合物(5)の合成に関して上記されたプロトコールを適用することにより、標題化合物(8)を得た。
【0099】
9.: 6,6’−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)−2,2’−ビピリジン(化合物(9))
【0100】
【化18】
【0101】
9.1: 6,6’−ジ(オキサゾール−5−イル)−2,2’−ビピリジン(中間体(10))
【0102】
【化19】
2,6−ピリジンジカルボキサルデヒドの代わりに2,2’−ビピリジン−6,6’−ジカルバルデヒド(G. R. Newkome and H.-W. Lee; J. Am. Chem. Soc. 1983, 105(18), 5956-5957)から出発して、中間体(1)の合成に関して上記されたプロトコールを適用することにより、標題中間体(10)を得た。
【0103】
9.2: 6,6’−(5,5’−(2,2’−ビピリジン−6,6’−ジイル)ビス(オキサゾール−5,2−ジイル))ジピリジン−2−カルバルデヒド(中間体(11))
【0104】
【化20】
中間体(1)及び3−ブロモベンズアルデヒドの代わりに中間体(10)及び6−ブロモピリジン−2−カルバルデヒドから出発して、中間体(5)の合成に関して上記されたプロトコールを適用することにより、標題中間体(11)を得た。
【0105】
9.3: 6,6’−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)−2,2’−ビピリジン(化合物(9))
中間体(5)の代わりに中間体(11)から出発して、化合物(5)の合成に関して上記されたプロトコールを適用することにより、標題化合物(9)を得た。
【0106】
II − 生物物理学的方法及びデータ:
試験II−1: 可溶性を調査するための紫外可視分光光度法
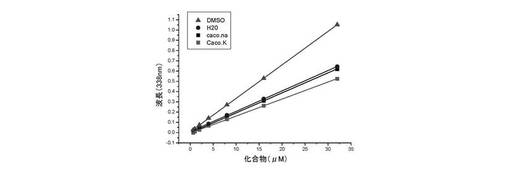
a) 紫外可視滴定:
本試験において詳述されるピリジン系ポリ芳香族複素環化合物の紫外可視特性の試験によって、種々の条件でその可溶性の測定が可能になる(DMSO、H2O及びカコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM NaCl(Caco.Na用)又はKCl(Caco.K用)):このための最も簡便な方法は、種々の濃度(0〜32μM)で化合物(3)の吸収スペクトルを測定することである;こうして化合物(3)の可溶性は、化合物(3)の濃度の関数としての所定の波長(ここでは338nm(A338))でのその吸光度の報告とBeer-Lambertの法則の適用とによって評価する。結果は、図1に与えられる。
【0107】
b) 要約
種々の滴定は、図2に要約する。
これらのデータは、化合物(3)が、
− ここで使用した全ての溶媒系に可溶性であること(DMSO、H2O及びカコジル酸水性緩衝液(高Na+又は高K+の両方);
− ここで使用した全ての溶媒中で、0〜32μM濃度範囲内で自己凝集しないこと;
を示した。
【0108】
試験II−2: 四重鎖DNAの安定化及び二重鎖に対する四重鎖DNAの選択性
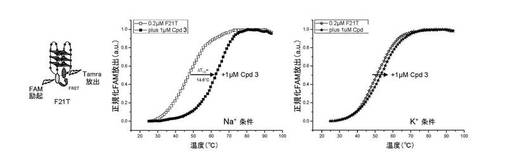
四重鎖構造を持つ化合物の安定化、ひいては相互作用は、二重鎖に対する四重鎖DNA選択性、更には四重鎖内選択性の測定をも可能にするバージョンで、FRET融解試験法によりモニターする(17)。
【0109】
FRET試験法は、ヒト又はマラリア原虫テロメア配列を模倣するオリゴヌクレオチドで実施し、そしてそれぞれの先端にFRETパートナーを取り付ける:F21T(FAM-G3[T2AG3]3-Tamra)、FPf1T(FAM-G3[T3AG3]3-Tamra)及びFPf8T(FAM-G3[T2CAG3]3-Tamra)(ここで、FAM:6−カルボキシフルオレセインであり、そしてTamra:6−カルボキシ−テトラメチルローダミンである)。測定は492nm励起及び516nm検出で行った。
【0110】
a) 四重鎖安定化:
蛍光融解を実施した。結果は、図3A〜図3Cに与えられる。本実験は、10mMカコジル酸リチウム(pH7.2)及び100mM NaCl又はKClを含有する緩衝液中の0.2μMのF21T、FPf1T又はFPf8Tで実施した(後述のF21Tの例を参照のこと、NaCl(左)及びKCl(右));四重鎖DNAの融解は、単独及び1μM(5当量)の化合物(3)の存在下でモニターした。
【0111】
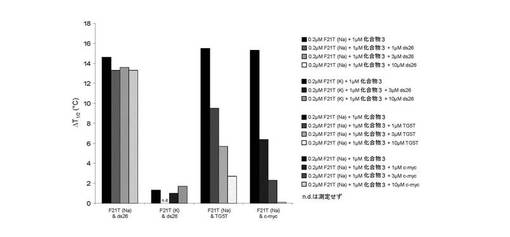
3種のオリゴヌクレオチドでの化合物(3)により誘導される安定化効果は、融解温度(ΔT1/2)の上昇により定量する。値は以下の表に示す。
【0112】
【表1】
【0113】
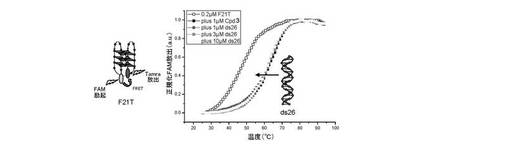
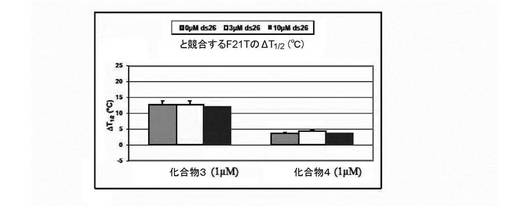
b) 二重鎖に対する四重鎖DNAの選択性:
蛍光融解は、10mMカコジル酸リチウム(pH7.2)及び100mM NaClを含有する緩衝液中の0.2μMのF21Tで実施した;G−四重鎖の融解は、単独及び1μMの化合物(3)(過剰(1、3及び10μM)の二重鎖DNA競合物質ds26(自己相補的配列[5’-CAATCGGATCGAATTCGATCCGATTG-3’]よりなる26塩基対の二重鎖DNA)を含むか又は含まない)の存在下でモニターした。結果は図4に与えられる。
【0114】
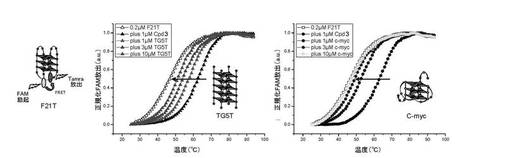
c) 四重鎖内選択性:
蛍光融解実験は、10mMカコジル酸リチウム(pH7.2)及び100mM NaClを含有する緩衝液中の0.2μMのF21Tで実施した;G−四重鎖の融解は、単独及び1μMの化合物(3)(過剰(1、3及び10μM)の四重鎖DNA競合物質(両方とも分子内の四重鎖構造中に折り畳まれることが強く疑われる、TG5T([(5’-TG5T-3’)4]、以前の研究(21)において使用した4分子四重鎖DNA)又はc−myc([5'-GAGGGTGGGGAGGGTGGGGAAG-3']、腫瘍遺伝子c−mycのプロモーター領域に存在する配列))を含むか又は含まない)の存在下でモニターした(22〜25)。結果は図5A〜図5Bに与えられる。
【0115】
d) 要約
種々のFRET融解実験は、図6の棒グラフ表示に要約する。
これらのデータは、化合物(3)が、
− ナトリウム緩衝液中でテロメア四重鎖F21T(ヒト)、FPf1T及びFPf8T(Plasmodium falciparum)と効率的に相互作用するが、一方カリウム条件ではこれらのオリゴヌクレオチドを安定化しないこと(表を参照のこと)(試験した3つのケースでは、化合物(3)が、カチオンに基づく四重鎖内選択性を提示することを意味する);
− 二重鎖DNAから四重鎖DNAを非常に効率的に識別すること(F21T安定化(ナトリウム緩衝液中)が、1、3又は10μMのds26の存在により限定的に影響を受けるに過ぎないため(安定化は、全てのケースで>91%維持される));
− ほぼ確実にスタッキング相互作用を介して四重鎖構造の外部G−カルテットと相互作用すること(F21T安定化が、非ループ4分子四重鎖DNA(TG5T、F21T安定化は、1、3及び10μMのTG5Tの存在下でそれぞれ61、37及び17%維持されている)又は二本鎖のみ反転ループ四重鎖DNA(c−myc、F21T安定化は、1、3及び10μMのc−mycの存在下でそれぞれ42、15及び1%維持されている)の競合に対して高感受性であるため)
を示す。
Na+及びK+条件の間で観測される劇的な違いを考えると、4分子及びループ両方との本化合物の相互作用もまた仮定することができる。
【0116】
試験II−2の2: 図16に、a)及びb)に上記されたのと同じナトリウム条件を用いた、化合物(3)及び(4)でのFRET融解で得られた結果を与える。蛍光標識四重鎖F21Tの融解は、単独及び1μMの化合物(3)又は化合物(4)(過剰(1、3及び10μM)の二重鎖DNA競合物質ds26(上記の自己相補的配列[5’-CAATCGGATCGAATTCGATCCGATTG-3’]よりなる26塩基対の二重鎖DNA)を含むか又は含まない)の存在下でモニターした。
【0117】
ds26の存在によりほとんど影響されない融解温度の上昇が、両方の化合物について得られる。
【0118】
試験II−3: 蛍光試験によりモニターされるDNAとの相互作用
本試験に詳述されるピリジン系ポリ芳香族複素環化合物は、強い蛍光を特徴とする。注目すべきは、量子収率が、純粋な有機(例えば、DMSO)から生理学的条件(例えば、緩衝液:10mMカコジル酸ナトリウム+100mM KCl(pH7.2)、表を参照のこと)までの使用される溶媒の性質に影響されないことである。DNAとの相互作用に及ぼす試験化合物の蛍光特性の変化により、生物学的に関連する幾つかの四重鎖に対する、これらの見かけの結合親和性を比較することができる(27)。
【0119】
【表2】
注意:化合物(3)の量子収率(及び標準偏差(s.d.))は、エタノール中のアントラセンを参照として、CH2Cl2(DCM)及び水中で測定した。
【0120】
a) 四重鎖相互作用:
ここで使用した四重鎖DNAは22AGである:これは、ヒトテロメア配列を模倣する22ntオリゴヌクレオチドの折り畳みに由来する:22AGは[5’-AG3(T2AG3)3-3’]である。22AGからの四重鎖構造は、対応するオリゴヌクレオチドを90℃で5分間、10mMカコジル酸ナトリウム緩衝液(pH7.2)、100mM NaCl(22AG Na用)又はKCl(22AG K用)中で加熱し、そして氷中で冷却して速度論的トラップにより分子内折り畳みを助けることによって調製する。濃度は、使用前に紫外可視測定(85℃で5分の熱変性後)により260nmで測定する(図7A及び図7Bを参照のこと)。
【0121】
増加量の22AG Na(左)又は22AG K(右)をカコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM NaCl(右)又はKCl(左))中の化合物(3)の0.25μM溶液に加えると、化合物(3)の蛍光の漸進的消光が起こる(λex=340nm)。
【0122】
b) 二重鎖に対する四重鎖DNAの選択性:
二重鎖に対する四重鎖選択性は、22AG(上記参照)及び短い二重鎖DNA:ds17(以前の研究に使用した生物学的配列を表す17塩基対の二重鎖DNAであり(28);2本の相補鎖の配列は、以下のとおりである:[5’-CCAGTTCGTAGTAACCC-3’]/[5’-GGGTTACTACGAACTGG-3’])で実施した実験の比較による蛍光滴定によって評価する。二重鎖構造は、2本の対応する相補鎖を90℃で5分間、10mMカコジル酸ナトリウム緩衝液(pH7.3)、100mM KCl中で加熱し、次に6時間ゆっくり冷却することによって調製する。濃度は、使用前に紫外可視測定(85℃で5分の熱変性後)により260nmで測定する。結果は図8に与えられる。
【0123】
増加量のds17をカコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM KCl)中の化合物(3)の0.25μM溶液に加えても、化合物(3)の蛍光の漸進的消光は起こらない(λex=340nm)。
【0124】
c) 四重鎖内選択性:
四重鎖内選択性は、22AG(上記参照)及び2種の他の四重鎖構造:c−myc及びc−kit2で実施した実験の比較による蛍光滴定によって評価する。これら2種の四重鎖DNAの形成は、目下c−myc(上記参照)及びc−kit(22;29及び30)腫瘍遺伝子のプロモーター領域で起こることが強く疑われる。この配列は以下のとおりである:c−myc:[5’-TGAGGGTGGGTAGGGTGGGTAA-3’]及びc−kit2:[(5’-CGGGCGGGCGCGAGGGAGGGG-3’]。四重鎖構造は、対応するオリゴヌクレオチドを90℃で5分間、10mMカコジル酸ナトリウム緩衝液(pH7.2)、100mM KCl中で加熱し、そして氷中で冷却して速度論的トラップにより分子内折り畳みを助けることによって調製する。濃度は、使用前に紫外可視測定(85℃で5分の熱変性後)により260nmで測定する。
結果は、図9A及び図9Bに与えられる。
【0125】
増加量のc−myc(左)又はc−kit2(右)をカコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM KCl)中の化合物(3)の0.25μM溶液に加えると、化合物(3)の蛍光の漸進的消光が起こる(λex=340nm)。
【0126】
d) 要約:
種々の蛍光滴定(λex=340nm)は、グラフ表示を与える図10に要約する:
これらのデータは、化合物(3)が、
− 二重鎖DNAマトリックス(ds17)から四重鎖DNA(22AG K、22AG Na、c−kit2及びc−myc)を非常に効率的に識別し、よってFRET融解の結果を裏付けること;
− 更に効率的かつ迅速に22AG Na、c−kit2及びc−mycと相互作用するが、一方22AG Kとのその相互作用は弱いこと;この結果は、FRET融解実験を介して観測される結果に一致し、そして四重鎖内選択性において有望な候補であること
を示した。
【0127】
試験II−4: 円偏光二色性試験
円偏光二色性(CD)により、リガンドの結合時のDNA構造の修飾の深い研究が可能である;よってこれは、リガンドのその標的に対する親和性を反映し、またその結合様式への洞察をも提供しうる(Paramasivan et al, Methods, 2007, 43, 324)。
【0128】
a) 四重鎖相互作用:
ここで使用した四重鎖DNAは、22AG(上記参照)であり、両方とも10mMカコジル酸ナトリウム緩衝液(pH7.2)、100mM NaCl(22AG Na用)又はKCl(22AG K用)中でアニーリングされている。両方の緩衝液中の22AGの3μM溶液に、過剰の化合物(3)(30μM、10当量)を加える。結果は図11に与えられる。四重鎖との化合物(3)の相互作用は、時間の関数としてモニターする:これにより22AG NaのCDシグナルは大きさが変化する(左、特に263nmで)が、一方22AG Kの構造は完全に再編成する(右、特に263nmで)。
【0129】
b) 四重鎖内選択性:
四重鎖内選択性は、22AG(上記参照)及び2種の他の四重鎖構造:c−myc及びc−kit2(上記参照)で実施した実験の比較によるCDによって評価する。結果は図11A及び図11Bに与えられる。カコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM KCl)中のc−kit2(左)及びc−myc(右)両方の3μM溶液に、過剰の化合物(3)(30μM、10当量)を加える。四重鎖との化合物(3)の相互作用は、時間の関数としてモニターする:これによりc−kit2のCDシグナルの大きさが変化し(特に285nmで)、これより少ないがc−mycでも変化する(285nm)。
【0130】
c) 四重鎖構造の誘導:
22AGオリゴヌクレオチドの折り畳まれた形と折り畳まれていない形の間の平衡は、入れられる緩衝液の存在及び性質に高感受性である。対応する結果は図12A及び図12Bに与えられる。10mMトリスHCl(pH7.2)のような低カチオン性緩衝液では、22AGは大部分折り畳まれていない(ランダムコイル状);よって化合物(3)を加えることにより折り畳まれた形<−>折り畳まれていない形の平衡に影響を及ぼすことができる。即ち、トリスHCl緩衝液中のランダムコイル状22AGの3μM溶液に、増加量の化合物(3)(0〜7.5当量)を加える;これにより22AGのCDシグナルは大きく変化し、ランダムコイルに典型的な257nmシグナルの消失と、2つの新規な極大[一方は正(290nm付近)であり他方は負(265nm付近)であり、両方とも「逆平行」四重鎖DNA構造(22AG Naとして、上記参照)のCDシグナルに典型的である]の獲得に至る。
【0131】
d) 要約:
これらのデータは、化合物(3)が、
− 効率的にかつほぼ自発的に22AG Naと相互作用するが、22AG Kとは効率的ではあるがゆっくりと反応すること(これは蛍光滴定及びFRET融解実験結果と一致する);
− c−myc及びc−kit2と相互作用するが、その相互作用は、これら2つの四重鎖の構造を修飾しないこと;
− 22AGの折り畳まれていない形と折り畳まれた形の間の平衡を折り畳まれた形へとシフトさせることができること
を示した。
【0132】
III − 生物学的方法及びデータ:
試験III−1: 化合物(3)によるインビトロでのテロメア配列へのPOT1結合の阻害
hPOT1を用いる電気泳動移動度シフトアッセイをテロメア22AGオリゴヌクレオチド(上記参照)に実行した。22AGは、T4ポリヌクレオチドキナーゼを用いて5’末端を[γ−32P]−ATPで標識した。精製組換えhPOT1は、バキュロウイルス発現系で産生させた。POT1/22AG結合アッセイは、50mMヘペス(pH7.9)、100mM NaCl、0.1mM EDTA、4%w/vショ糖、2%v/vグリセロール、0.1mg/ml BSA、0.02%w/vブロモフェノールブルー、30nM hPOT1、20nM[α−32P]−22AGを含有する総容量10μl中で実行した。様々な濃度の化合物(3)(10、1、0.1及び0.01μM)をhPOT1と共にこの溶液に加え、この混合物を室温で30分間インキュベートした。それぞれ個々の試料は、0.5×トリス−ホウ酸−EDTA緩衝液中で1%アガロースゲル上で電気泳動により分離した。ゲルは80Vで35分間流して、ワットマンDE81紙上で乾燥して、放射活性はホスファイメージャー(phosphorimager)(Typhoon 9210, Amersham)により可視化した。データの解析は、ImageQuantソフトウェア(Amersham)により実施して、結果は未処理対照(100%と定義)で得られるPOT1−22AG複合体の百分率として表した(図14を参照のこと)。
【0133】
これらのデータは、化合物(3)が、テロメア22AG配列へのhPOT1の結合を用量依存的に阻害することを示した。後述の実験において、化合物(3)のIC50−POT1は300nMに等しい。
【0134】
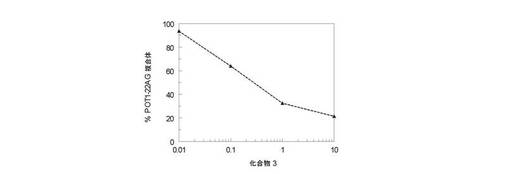
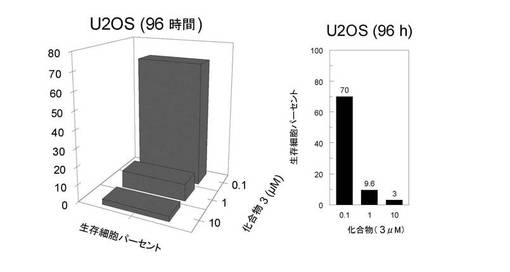
試験III−2: 化合物(3)による細胞増殖の阻害
pEGFP−POT1ベクターで安定にトランスフェクトしたHT1080ヒト線維肉腫細胞株(HT1080GFP−POT1)は以前に報告されており(32)、U20Sヒト骨肉腫はAmerican Type Culture Collection(Rockville, 米国)から得た。細胞は、HT1080GFP−POT1用に10%v/vウシ胎仔血清及び400μg/mlジェネティシンを補足したダルベッコー修飾イーグル培地(Invitrogen)で増殖させた。細胞は、様々な濃度の化合物(3)(10、3、1、0.3及び0.1μM)の存在下で各濃度二重反復で、0日目に4.5×104細胞/ウェルで6ウェル培養プレートに蒔き、更に72時間培養した。3日目に、細胞を1×PBSで洗浄してトリプシン処理した。各処理細胞試料について、生存細胞の数をトリパンブルーの存在下で測定した。結果は図15A及び図15Bに与えられる。これらは、100%と定義した未処理対照細胞に比較した相対細胞数(パーセント)に相当する。
【0135】
これらのデータは、化合物(3)が、癌細胞の増殖を0.1〜1μMの間であるIC50で妨害することを示す。
【0136】
特に限定されないが、特許公報、特許出願、本及び学術論文を包含する、本出願に引用される全ての刊行物は、それぞれ引用例としてその全体が本明細書に取り込まれる。本発明は開示された実施態様を参照して上述されているが、当業者には、詳述された特定の実験は、単に本発明の例証であることが容易に理解されよう。当然のことながら、本発明の本質を逸することなく、ここに種々の変更を加えることができる。したがって、本発明は以下の請求の範囲によってのみ限定される。
【0137】
【表3】
【技術分野】
【0001】
本発明は、四重鎖DNAに特異的に結合してこれを安定化する、ポリ−ヘテロアリール誘導体に関する。本発明はまた、このような化合物の薬学的に許容しうる塩、これらを含有する医薬組成物の製造方法、並びに癌及び感染症の処置におけるこのような化合物の使用に関する。
【背景技術】
【0002】
「ポリヘテロアリール」は、本明細書及び請求の範囲において使用されるとき、場合によりフェニレン部分を包含するか、かつ/又は末端フェニル基を有する、芳香族複素環部分よりなる配列/鎖を意味する。
【0003】
G−四重鎖DNAは、現在のところ高G含量配列の機能を調節できる構造要素と考えられている。詳細には、多くの証拠から、テロメアにおける、又はc−Myc、Bcl−2、VEGF、Hif−1a、Ret、c−Kit、PDGF−A、KRAS及びc−Myb(1)を包含する種々のヒト腫瘍遺伝子中の特異的遺伝子プロモーターにおける、この特有のDNA構造の形成が、癌細胞増殖を阻害することが示唆されている。
【0004】
ヒト細胞のテロメアDNAは、配列:5'-TTAGGG-3'の縦列反復を含むが、これはその3’側で、インビトロでG−四重鎖中に折り畳まれることが証明されている、一本鎖オーバーハングで終わる。インビボでのこのような四重鎖構造の形成は、テロメアと通常結合した保護タンパク質(シェルタリン(shelterin)複合体)の変位をもたらし、このためテロメア構造が崩壊してゲノムが不安定になると仮定されている。
【0005】
腫瘍遺伝子プロモーターでは、四重鎖の安定化は、遺伝子発現を調節するタンパク質因子の局所環境を崩壊させることが予想されるが、これはc−mycに関しては充分な証拠がある(1)。よって四重鎖DNA形成を誘導又は安定化することができる化学的介入は、低分子四重鎖結合剤を介して、特に癌細胞においてDNA関連機能(テロメア伸長、腫瘍遺伝子転写)を制御することを目的として、徹底調査されている。
【0006】
G−四重鎖DNAと相互作用する化合物は、現在これらに傾けられた最近の総説(2〜9)の数に例証されるとおり、文献上非常に多い。しかし、二重鎖DNAよりも四重鎖DNAに対して高度の選択性を示す化合物は少なく;更には、これらの化合物の少数のものだけが、抗癌剤として徹底調査されている。このファミリーのリード化合物は、5個のオキサゾール環、2個のメチルオキサゾール環及び1個のチアゾリン環からなる、天然の大環状物質(Streptomyces anulatus 3533-SV4から単離)である、テロメスタチン(telomestatin)である。テロメスタチンは、四重鎖選択的相互作用に関して1つのパラダイムを表し、そしてインビトロで並外れたテロメラーゼ阻害特性を示す(TRAP又は直接アッセイのいずれかにより評価、IC50−TRAP=0.6nM及びIC50−直接アッセイ=58nM)(10)。更に、テロメスタチンはまた、その細胞効果についても深く調査され、そして種々の腫瘍細胞株において抗増殖及びアポトーシス特性を呈した(11及び12)。テロメスタチンは、テロメアの完全性を変え(13)、テロメアでのDNA損傷応答を引き起こす(14)。テロメスタチンは、インビトロでPOT1脱キャップ形成(uncapping)を誘導し(IC50−POT1=500nM)、そして腫瘍細胞でテロメアからGFP−POT1を除去する(15)。テロメスタチンの厳しい合成経路を考慮して(16)、興味深い四重鎖相互作用特性を呈する類似体が報告されている(17、18及び19)。
【0007】
しかし、これらの化合物のどれもが、テロメスタチンの並外れた特性を示していない。更に、テロメスタチン自体を包含するこれらの化合物のどれもが、その蛍光特性について調査されていない。テロメスタチンの化学的不安定性、難水溶性及び特に困難な合成を前提とすると、それの大規模な治療的使用には問題が多い。
【0008】
このような状況において、本発明者らは、新しい非大環状ポリヘテロアリール誘導体が、二重鎖DNAよりも四重鎖DNAに対して優れた選択性を示すこと、そしてこれらの誘導体のそのDNA標的との結合が、i)その分光学的特性の大きな変化をもたらし、ii)テロメアG−オーバーハングからのヒトPOT1(テロメアの保護1)タンパク質を脱キャップし、iii)ヒト腫瘍細胞株の細胞増殖を阻害することを見い出した。この新しいファミリーの化合物の生理学的液体中での安定性及び可溶性は、治療処置におけるその使用に適合する。更には、本発明者らは、これらの化合物を入手するための効率的かつ独創的な製造法を見い出した。
【0009】
次に本発明の1つの目的は、新しい製品としてこのような非大環状ポリヘテロアリール誘導体を提供することである。
【0010】
また、これらの合成方法にも関する。
【0011】
別の目的により、本発明は、該化合物の四重鎖相互作用特性を利用して、更に四重鎖特異的プローブとしての該誘導体の使用に関する。詳細には、開示化合物の高蛍光量子収率量子を考慮して、このような化合物は、四重鎖DNA又は関連核酸構造(特に限定されないが四重鎖RNAを包含する)の検出及び/又は精製用の蛍光プローブとして使用することができよう。
【0012】
更に別の目的では、本発明は、活性成分として該誘導体を含有する医薬組成物を提供し、また広範な疾患用の薬物、特に例えば、癌又はマラリアのような感染症に罹患している患者の処置用の薬物の製造における該誘導体の使用に関する。
【0013】
本発明はまた、癌及び感染症の処置方法であって、有効量の該誘導体をこれを必要とする患者に投与することを含む方法に関する。
【図面の簡単な説明】
【0014】
【図1】図1は、種々の条件での本発明の誘導体の吸収スペクトルを表す。
【図2】図2は、種々の滴定の要約を表す。
【図3】図3は、ヒト又はマラリア原虫テロメア配列を模倣するオリゴヌクレオチド及びFRETパートナーによるFRET融解試験法の結果を表す。
【図4】図4は、本発明の誘導体を伴うか又は伴わないG−四重鎖の蛍光融解の結果を表す。
【図5】図5は、G−四重鎖単独又は本発明の誘導体の存在下での蛍光融解の結果を表す。
【図6】図6は、種々のFRET融解実験の要約を表す。
【図7】図7は、本発明の誘導体を加えた四重鎖DNAでの蛍光融解の結果を表す。
【図8】図8は、本発明の誘導体を加えた四重鎖又は短い二重鎖DNAを用いた蛍光滴定の結果を表す。
【図9】図9は、本発明の誘導体を加えた種々の四重鎖DNA構造を用いた蛍光滴定の結果を表す。
【図10】図10は、種々の蛍光滴定の要約のグラフ表示である。
【図11】図11は、本発明の誘導体を加えた四重鎖DNAの円偏光二色性の結果を表す。
【図12】図12は、本発明の誘導体を加えた四重鎖DNAの円偏光二色性の結果を表す。
【図13】図13は、四重鎖構造の誘導に関する結果を表す。
【図14】図14は、本発明の誘導体によるインビトロでのテロメア配列へのPOT1結合の阻害に関する結果を表す。
【図15】図15は、本発明の誘導体による細胞増殖の阻害に関する結果を表す。
【図16】図16は、四重鎖DNAの安定化、及び本発明の誘導体の二重鎖に対する四重鎖DNAの選択性に関する結果を表す。
【0015】
本発明のポリヘテロアリール誘導体は、ペンタ−、ヘキサ−、ヘプタ−、オクタ−、ノナ−及びデカ−ヘテロアリール誘導体(以降ペンタ−〜デカ−ヘテロアリールと略す)であって、それぞれ式(I)、(II)、(III)及び(IV)の複素環1(Het−1)a及び/又は複素環2(Het−2)b及び/又は複素環3(Het−3)c及び/又は複素環4(Het−4)d:
【0016】
【化1】
の組合せ、N−オキシド、薬学的に許容しうる付加塩[ここで、
− 該組合せは、少なくとも2種の異なる複素環部分を含み、そして場合により式(V)の(Aryl−5)e部分を含み、
− a、b及びeは、0〜6の整数であり、c及びdは、0〜2の整数であり、a+b+c+d+eの合計は、≦10であり、
− Yは、O又はSであり;
− Het−1は、2,6−ピリジン−ジイル又は2,4−ピリミジン−ジイル又は3,5−ピラジン−ジイル又は2,4−(1,3,5−トリアジン)ジイル又は3,5−(1,2,4−トリアジン)ジイル又は2,4−オキサゾリン−ジイル又は2,4−チアゾリン−ジイルを含む群において選択される窒素複素環−ジイル環のクラスであり;
− Het−2は、2,5−オキサゾリン−ジイル又は2,5−チアゾリン−ジイル又は2,5−チオフェン−ジイル又は2,5−フラン−ジイルを含む群において選択される5員複素環−ジイル環のクラスであり;
− Het−3及び/又はHet−4は、該ペンタ−、ヘキサ−、ヘプタ−、オクタ−、ノナ−及びデカ−ヘテロアリール誘導体の終点を構成するが、ここで、
− Het−3は、2−ピリジル又は2−ピリミジル又は4−ピリミジル又は2−ピラジル又は2−(1,3,5)トリアジル又は3−(1,2,4)トリアジル又は5−(1,2,4)トリアジル又は4−オキサゾリル又は4−チアゾリルから選択される窒素複素環−イル環のクラスであり;
− Het−4は、2−オキサゾリル又は5−オキサゾリル又は2−チアゾリル又は5−チアゾリル又は2−チエニル又は2−フラニルから選択される5員複素環−イルのクラスであり;
− R1及びR2は、それぞれ独立に、水素;C1−6アルキル;C1−4アルキルオキシ;ハロ;ヒドロキシ;ヒドロキシメチル;ニトロ;アミノ;モノ−又はジ(C1−4アルキル)アミノ;C1−4アルキルメチルアミノ;モノ−又はジ(C1−4アルキル)アミノ(C2−4アルキル)アミノメチル;モルホリン−4−イルC2−4アルキルオキシ;ピペラジン−1−イルC2−4アルキルオキシ;4−C1−4アルキルピペラジン−1−イル;フェニル、ベンジル、ナフチルから選択される単環又は二環から選択される]を含む誘導体である。
【0017】
本発明のある実施態様では、a、b、c及びdは、0〜3の整数であり、e=0であり、そしてa+b+c+dの合計は、≦10である。
【0018】
本発明のある実施態様では、1個又は数個の上記部分は、それぞれ独立にC1−6アルキル;C1−4アルキルオキシ;ハロ;ヒドロキシ;ニトロ;アミノ;モノ−又はジ(C1−4アルキル)アミノ;C1−4アルキルメチルアミノ;モルホリン−4−イルC2−4アルキルオキシ;ピペラジン−1−イルC2−4アルキルオキシ;4−C1−4アルキルピペラジン−1−イルC2−4アルキルオキシ;フェニル、ベンジル、ナフチルから選択される単環式又は二環式環から選択される、1、2又は3個の置換基で置換されている。
【0019】
本発明は、詳細には少なくとも5個、少なくとも7個、少なくとも8個、少なくとも9個又は10個の複素環部分を含む、上記と同義の誘導体に関する。
【0020】
別の態様では、本発明はまた、詳細には1個又は2、3若しくは4個のような数個の1,3−フェニレン−ジイル部分を更に含む、上記と同義の誘導体に関する。
【0021】
本発明の特に好ましい誘導体は、下記構造の1つを有する:
−(R2により置換されたHet−4)−(Het−1)−(R2により置換されたHet−4)
−(R1により置換されたHet−3)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−1,3−フェニレン−ジイル−(Het−2)−(R1により置換されたHet−3)
−(R1又はR2により置換された1,3−フェニレン−ジイル)−(Het−2)−(Het−1)−(Het−2)−(R1又はR2により置換された1,3−フェニレン−ジイル)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−1)−(Het−2)−(R1により置換されたHet−3)。
【0022】
好ましい誘導体において、
・ R1及びR2は、オキサゾリン−若しくはピリジン−ジイル基であるか、かつ/又は
・ Het−2は、オキサゾリン−ジイル基であるか、かつ/又は
・ Het−1は、2,6−ピリジン−ジイル、2,6−ピリミジン−ジイル、2,6−ピラジン−ジイル、若しくは1,3−フェニレン−ジイル基である。
【0023】
有利な群では、誘導体のG−四重鎖相互作用特性を考慮して、Het−1は、2個のHet−2に結合している。
【0024】
該群の好ましい誘導体において、Het−1は、ピリジン−ジイルであり、そしてHet−4は、ピリジル又はピリジル−オキサゾリルにより置換されている、オキサゾリルである。
【0025】
第2の有利な群では、Het−2は、2個のHet−1に結合している。
【0026】
好ましい誘導体において、Het−2は、オキサゾリルであり、そしてHet−1は、ピリジル−オキサゾリルである。
【0027】
本発明の特定の好ましい化合物は、後述の実施例の項にリストされているもの、特に化合物(3)及び(4)である。
【0028】
上で定義された誘導体の薬学的に許容しうる付加塩は、その酸付加塩及び塩基塩(二塩を包含する)を包含する。
【0029】
適切な酸付加塩は、非毒性塩を形成する酸から形成される。例は、酢酸、アスパラギン酸、安息香酸、ベシル酸、重炭酸/炭酸、重硫酸/硫酸、ホウ酸、カンシル酸、クエン酸、エジシル酸、エシル酸、ギ酸、フマル酸、グルセプト酸、グルコン酸、グルクロン酸、ヘキサフルオロリン酸、ヒベンズ酸、塩酸/塩化物、臭化水素酸/臭化物、ヨウ化水素酸/ヨウ化物、イセチオン酸、乳酸、リンゴ酸、マレイン酸、マロン酸、メシル酸、メチル硫酸、ナフチル酸、2−ナプシル酸、ニコチン酸、硝酸、オロチン酸、シュウ酸、パルミチン酸、パモ酸、リン酸/リン酸水素/リン酸二水素、サッカラート、ステアリン酸、コハク酸、酒石酸、トシル酸及びトリフルオロ酢酸塩を包含する。
【0030】
適切な塩基塩は、非毒性塩を形成する塩基から形成される。例は、アルミニウム、アルギニン、ベンザチン、カルシウム、コリン、ジエチルアミン、ジオラミン、グリシン、リシン、マグネシウム、メグルミン、オラミン、カリウム、ナトリウム、トロメタミン及び亜鉛塩を包含する。
【0031】
上で定義された誘導体のこのような薬学的に許容しうる塩は、該誘導体及び所望の酸又は塩基の溶液を必要に応じて一緒に混合することにより、容易に調製できる。塩は、溶液から沈殿するため、これを濾過により収集するか、又は溶媒の留去により回収することができる。塩におけるイオン化の程度は、完全にイオン化からほぼイオン化がない状態まで変化しうる。
【0032】
本発明はまた、上記と同義の同位体標識誘導体、好ましくは薬学的に許容しうる同位体標識誘導体を包含するが、この誘導体では1個以上の原子が、同一の原子番号を有するが、自然界で通常見られる原子質量又は質量数とは異なる原子質量又は質量数を有する原子により置換されている。
【0033】
本発明の化合物に含めるのに適切な同位体の例は、2H及び3Hのような水素、11C、13C及び14Cのような炭素、36Clのような塩素、18Fのようなフッ素、123I及び125Iのようなヨウ素、13N及び15Nのような窒素、15O、17O及び18Oのような酸素、32Pのようなリン、並びに35Sのような硫黄の同位体を包含する。
【0034】
本発明のある種の同位体標識誘導体、例えば、放射性同位体を取り込んだ誘導体は、薬物及び/又は基質の組織分布研究において有用である。放射性同位体のトリチウム、即ち、3H、及び炭素14、即ち、14Cは、その取り込みの容易さ及び即時の検出手段を考慮して、この目的に特に有用である。
【0035】
ジュウテリウム、即ち、2Hのような重い同位体での置換では、代謝安定性の高さに起因するある種の治療上の利点、例えば、インビボ半減期の延長又は必要用量の減少が得られ、このため幾つかの状況では好ましい。
【0036】
11C、18F、15O及び13Nのような陽電子放出同位体での置換では、基質の受容体占有率を検査するための陽電子放出断層撮影法(PET)研究において有用でありうる。
【0037】
後述の実施例に示されるように、本発明の誘導体は、四重鎖DNAに特異的に結合して安定化するが、このため四重鎖特異的プローブとして大いに興味深い。四重鎖DNAに結合する該誘導体の能力、更にはその選択性は、後述の実施例の項に記述される試験法を包含する、当業者には既知の試験法を用いて測定することができる。
【0038】
これらはまた、種々の疾患の処置のために大いに価値がある。
【0039】
よって本発明は、上記誘導体の薬物としての使用に関する。
【0040】
本発明はまた、有効量の少なくとも1つの上記のような誘導体を薬学的に許容しうる担体と組合せて含む医薬組成物に関する。
【0041】
本明細書において使用されるとき、「治療有効量」又は「有効量」は、疾患の処置を構成するのに充分な、患者に投与される上記誘導体のような治療剤の量を意味する。
【0042】
本明細書において使用されるとき、疾患の「処置」という用語は、(1)このような用語が適用される病態又は病状の症状の進行、増悪、再発、播種又は悪化を遅延又は停止させること;(2)このような用語が適用される病態又は病状の症状を軽減させるか又は改善をもたらすこと;及び/あるいは(3)このような用語が適用される病態又は病状を回復させるか又は治癒することを目指した任意の活動のことをいう。
【0043】
医薬用途の本発明の誘導体は、実際に単独で、又は1種以上の他の本発明の誘導体と組合せて、又は1種以上の他の薬物、特に抗癌薬若しくは抗感染症薬と組合せて(又はその任意の組合せとして)投与することができる。一般に、これらは1種以上の薬学的に許容しうる担体と結び付けた製剤として投与されよう。「担体」という用語は、本明細書において本発明の誘導体以外の任意の成分を記述するために使用される。担体の選択は、大体において、特定の投与様式、溶解度及び安定性に及ぼす担体の効果、並びに投与剤形の性質のような要因に依存する。
【0044】
上記の誘導体の送達に適切な医薬組成物及びその製造方法は、当業者には容易に明らかとなるだろう。このような組成物及びその製造方法は、例えば、'Remington’s Pharmaceutical Sciences', 19th Edition (Mack Publishing Company, 1995)に見い出すことができる。
【0045】
本医薬組成物は、例えば、錠剤、カプセル剤、丸剤、粉剤、徐放製剤、液剤、懸濁剤として経口投与に;液剤、懸濁剤又は乳剤として非経口投与に;軟膏剤又はクリーム剤として局所投与に;あるいは坐剤として直腸内投与に適切な剤形にしてもよい。本医薬組成物は、正確な用量の単回投与用に適切な単位投与剤形にしてもよい。
【0046】
適切な製剤担体は、不活性希釈剤又は増量剤、水及び種々の有機溶媒を包含する。本医薬組成物は、必要に応じて、香味料、結合剤、賦形剤などのような追加の成分を含有してもよい。よって経口投与には、クエン酸のような種々の賦形剤を含有する錠剤を、デンプン、アルギン酸及びある種のケイ酸複合体のような種々の崩壊剤と、並びにショ糖、ゼラチン及びアカシアゴムのような結合剤と一緒にして利用してもよい。更には、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム及びタルクのような滑沢剤は、錠剤化目的にしばしば有用である。同様のタイプの固体組成物はまた、軟及び硬充填ゼラチンカプセル剤にして利用することができる。このために好ましい材料は、ラクトース、即ち乳糖及び高分子量ポリエチレングリコール類を包含する。経口投与に水性懸濁剤又はエリキシル剤が必要であれば、上記誘導体は、種々の甘味剤又は着香剤、着色料又は色素、及び必要に応じて乳化剤又は懸濁剤と、水、エタノール、プロピレングリコール、グリセリン、又はこれらの組合せと一緒にして合せることができる。
【0047】
本明細書において使用されるとき、医薬組成物の「非経口投与」は、対象の組織の物理的突破を特徴とする投与の任意の経路、及び組織での突破による医薬組成物の投与を包含する。よって非経口投与は、特に限定されないが、組成物の注射による、外科的切開を介した組成物の適用による、組織浸透性の非外科的創傷を介した組成物の適用によるなどの医薬組成物の投与を包含する。特に、非経口投与は、特に限定されないが、皮下、静脈内、皮内、腹腔内、筋肉内、胸骨内注射、及び腎臓透析点滴法を包含するものである。
【0048】
非経口投与に適切な医薬組成物の製剤は、活性成分を無菌水又は無菌等張性食塩水のような薬学的に許容しうる担体と合わせて含む。このような製剤は、ボーラス投与又は連続投与に適した剤形で調製、包装、又は販売することができる。非経口投与用の製剤は、特に限定されないが、後述のような懸濁剤、液剤、油性又は水性ビヒクル中の乳剤、ペースト、及び埋め込み型徐放性又は生分解性製剤を包含する。このような製剤は更に、特に限定されないが、懸濁剤、安定化剤、又は分散剤を包含する1種以上の追加の成分を含んでいてもよい。非経口投与用製剤の1つの実施態様において、活性成分は、復元組成物の非経口投与に先立って適切なビヒクル(例えば、無菌の発熱性物質除去水)で復元するための、乾燥(即ち、粉末又は顆粒)製剤として提供される。
【0049】
本発明の医薬組成物は、インプラント、経皮パッチ、及びマイクロカプセル化送達系を包含する放出制御製剤のように、本化合物を急速放出に対して保護する担体と共に調製することができる。エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル類、及びポリ乳酸のような、生分解性の生体適合性ポリマーを使用することができる。
【0050】
治療用途には、組成物中の活性成分の有効な1日量は、一般に、0.01mg/kg〜50mg/kg体重、更に好ましくは0.1mg/kg〜10mg/kg体重であろう。当然ながら、これは使用する誘導体、投与様式、必要な処置、適応疾患、更には患者の生理学的データ(例えば、年齢、サイズ、及び体重)により変化する。この1日総用量は、単回又は分割用量として投与することができる。治療剤の投与に適切な用量及び用法の決定は、当該分野において周知であり、当業者であれば含まれることを理解するであろう。
【0051】
上記の誘導体及びその医薬組成物は、癌及びマラリアのような感染症を処置するのに特に有用である。
【0052】
「癌」という用語は、本明細書において使用されるとき、無制限増殖、不死性、転移能、及びある種の特徴的な形態学的状態のような、発癌性細胞に典型的な特徴を持つ細胞の存在のことをいう。この用語は、任意のタイプの悪性腫瘍(原発性又は転移性)のことをいう。
【0053】
「感染症」という用語は、本明細書において使用されるとき、細菌、寄生虫、酵母、真菌のような微生物による哺乳動物(ヒト又は動物)の感染に起因する疾患のことをいう。ウイルスによる感染もまた包含される。
【0054】
癌又はマラリアのような感染症の処置用薬物を製造するための上記誘導体及びその医薬組成物の使用は、本発明の範囲の一部になる。
【0055】
本発明はまた、癌又は感染症の処置方法であって、有効量の上記の誘導体又はその医薬組成物をこれを必要とする患者に投与することを含む方法を包含する。
【0056】
本発明はまた、上記の誘導体の製造方法を含める。
【0057】
該方法は、
・ X1−A−X2を
・ B−X3と、又はX4−B−X3と、
所望のポリヘテロアリール誘導体を与える条件下で反応させること[ここで、
− Aは、ジ−又はトリ−又はテトラ−ヘテロアリール−1,4鎖であり、そしてBは、(モノ−又はジ−又はトリ−又はテトラ−ヘテロアリール−1,4)−であり(ここで、ヘテロアリール部分は、場合により1個、2個、3個又は4個のAryl−5部分又はAryl−5末端部分を含み、ヘテロアリール−1,4は、上記と同義のHet−1及び/又はHet−2及び/又はHet−3及び/又はHet−4の組合せである)、
− X1及びX2は、同一であるか又は異なって、水素又はハロゲン又はトリフラート基を表し、
− X3は、ハロゲン又はハロゲン化亜鉛又はトリアルキルスズ又はボロン酸基を表し、
− X4は、カルボキサルデヒド基を表す]を含む。
【0058】
本発明の他の特徴及び利点は、図1〜図16を参照する以下の実施例に与えられるが、ここで、
− 図1A〜図1Dは、種々の条件での本発明の誘導体の吸収スペクトルを表し;
− 図2、種々の滴定の要約;
− 図3A〜図3C、ヒト又はマラリア原虫テロメア配列を模倣するオリゴヌクレオチド及びFRETパートナーによるFRET融解試験法の結果;
− 図4A〜図4b及び図5、本発明の誘導体を伴うか又は伴わないG−四重鎖の蛍光融解の結果;
− 図5、G−四重鎖単独又は本発明の誘導体の存在下での蛍光融解の結果(過剰の四重鎖DNA競合物質を伴うか又は伴わない);
− 図6、種々のFRET融解実験の要約;
− 図7A及び図7B、本発明の誘導体を加えた四重鎖DNAでの蛍光融解の結果;
− 図8、本発明の誘導体を加えた四重鎖又は短い二重鎖DNAを用いた蛍光滴定の結果;
− 図9、本発明の誘導体を加えた種々の四重鎖DNA構造を用いた蛍光滴定の結果;
− 図10、種々の蛍光滴定の要約のグラフ表示;
− 図11A及び図11b及び図12、本発明の誘導体を加えた四重鎖DNAの円偏光二色性の結果;
− 図13、四重鎖構造の誘導に関する結果;
− 図14、本発明の誘導体によるインビトロでのテロメア配列へのPOT1結合の阻害に関する結果;
− 図15、本発明の誘導体による細胞増殖の阻害に関する結果;並びに
− 図16、四重鎖DNAの安定化、及び本発明の誘導体の二重鎖に対する四重鎖DNAの選択性に関する結果。
【0059】
I − ポリ−ヘテロアリール誘導体の合成
以下の実施例は、本発明を説明するものである。本化合物はまた、当業者が既知の他の製造法によっても得られる。出発物質として使用される、2,6−ピリジンジカルボキサルデヒド、6−ブロモピリジン−2−カルバルデヒド、イソシアン化p−トルエンスルホニルメチル(TosMIC)、2−ピリジル亜鉛ブロミド、1,3−ベンゼンジカルボキサルデヒド、6−ブロモピリジン−2−カルボキサルデヒド、3−ブロモベンズアルデヒド及び5−ブロモ−2−フルアルデヒドは、市販の製品である。
【0060】
1.:実施例1: 2,6−ビス[2−(ピリジン−2−イル)オキサゾール−5−イル]ピリジン(化合物(1))
【0061】
【化2】
【0062】
1.1.: 2,6−ビス(オキサゾール−5−イル)ピリジン(中間体(1))
【0063】
【化3】
メタノール100mL中の2,6−ピリジンジカルボキサルデヒド(4.0g;29.2mmol)、TosMIC(11.4g;58.3mmol)及び炭酸カリウム(16.3g;117.8mmol)を3時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタン(4×150mL)で抽出した。合わせた有機層を硫酸マグネシウムで乾燥して、真空で濃縮した。残渣は、ジクロロメタン:エタノール(95:5)を溶離液とするシリカゲルカラムのフラッシュクロマトグラフィーにより精製して、標題化合物を黄色の固体として得た(5.6g、90%)、融点=190〜191℃;1H NMR (300 MHz, CDCl3) 7.56 (d ; 2H ; J= 8.0) ; 7.74 (s ; 2H ) ; 7.80 (t ; 1H ; J= 8.0 Hz) ; 7.96 (s ; 2H) ; 13C NMR (75 MHz, CDCl3) 118.7, 125.7, 138.0, 147.4, 150.8, 151.0; SM m/z 214.1 (M+1) ; 元素分析、C11H7N3O2の理論値: C, 91.97 ; H, 3.31 ; N, 19.71. 実測値: C, 61.51, H, 3.43, 19.48.
【0064】
1.2.: 2,6−ビス−(2−ヨードオキサゾール−5−イル)ピリジン(中間体(2))
【0065】
【化4】
アルゴン雰囲気下、中間体(1)(0.1g;0.5mmol)及びTMEDA(0.2mL;1.0mmol)を無水THF 5mLに溶解して、−78℃で冷却した。THF中のLiHMDS 1Mの溶液(0.5mL;0.5mmol)を滴下により加え、−78℃で30分間及び−40℃で1時間撹拌した。この混合物を再度−78℃で冷却して、1,2−ジヨードエタン(0.5g;1.9mmol)を加えた。室温で一晩撹拌後、この混合物をチオ硫酸ナトリウム溶液中に注ぎ入れ、酢酸エチルで抽出した(3×10mL)。合わせた有機層を硫酸マグネシウムで乾燥して、真空で濃縮した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、中間体(2)をベージュ色の固体として得た(117mg、50%)、1H NMR (300 MHz, CDCl3) 7.58 (d ; 2H ; J= 8 Hz) ; 7.69 (s ; 2H) ; 7.85 (t ; 1H ; J= 8 Hz ) ; 13C NMR (75 MHz, CDCl3) 119.5, 129.7, 138.7, 147.0, 157.2, 151.5; SM m/z 465.8 (M+1), 487.8 (M+23).
【0066】
1.3.: 2,6−ビス[2−(ピリジン−2−イル)オキサゾール−5−イル]ピリジン(化合物(1))
方法1: 中間体(2)(65mg;0.14mmol)、2−ピリジル亜鉛ブロミド(1.2mL;0.60mmol)及びPd(PPh3)4(12mg;0.01mmol)の混合物を無水THF 2mL中で4時間加熱還流した。水を加えて、この混合物を酢酸エチルで抽出した(3×10mL)。合わせた有機層を硫酸マグネシウムで乾燥し、濾過して真空で濃縮した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、目的化合物(1)をベージュ色の固体として得た(13mg、25%)、融点=242〜245℃;1H NMR (300 MHz, CDCl3) 7.40 (m ; 2H) ; 7.83-7.88 (m ; 5H) ; 7.97 (s ; 2H) ; 8.23 (d ; 2H ; J= 7.9 Hz) ; 8.77 (d ; 2H ; J= 4.5 Hz) ; 13C NMR (75 MHz, CDCl3) 119.7, 123.3, 125.6, 128.6, 137.8, 138.5, 146.6, 147.9, 150.9,152.3, 161.5; SM m/z 368.0 (M+1), 390.0 (M+23).
【0067】
方法2: 中間体(1)(0.1g;0.5mmol)、2−ブロモピリジン(91μL;0.9mmol)、二酢酸パラジウム(11mg;0.05mmol;10mol%)、PCy3.HBF4(35mg;0.1mmol;20mol%)、ヨウ化銅(I)(0.18g;0.9mmol)、炭酸セシウム(0.61mg;1.88mmol)及び無水トルエン1.3mLの混合物をマイクロ波照射条件(130℃、150W)下に4時間置いた。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、化合物(1)(8mg、33%)を得たが、これは上の方法1に記述されたものと同一である。
【0068】
2.実施例2: 2,5−ビス−[6−(オキサゾール−5−イル)]ピリジン−2−イルオキサゾール(化合物(2))
【0069】
【化5】
【0070】
2.1: 5−(2−ブロモピリジン−2−イル)オキサゾール(中間体(3))
【0071】
【化6】
6−ブロモピリジン−2−カルバルデヒド(4.0g;21.5mmol)、TosMIC(4.2g;21.5mmol)及び炭酸カリウム(6.0g;43.4mmol)をメタノール75mL中で3時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタンで抽出した(4×100mL)。合わせた有機層を硫酸マグネシウムで乾燥して真空で濃縮した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、中間体(3)を黄色の固体として得た(2.9g、60%);融点=90〜91℃;1H NMR (300 MHz, CDCl3) 7.38 (dd ; 1H ; J= 3.5 & 5.3 Hz) ; 7.57 (m ; 2H ) ; 7.73 (s ; 1H) ; 7.97 (s ; 1H) ; 13C NMR (75 MHz, CDCl3) 118.1, 126.4, 127.6, 139.3, 142.4, 148.0, 151.6 ; SM m/z 225.0 and 227.1 (M+1).
【0072】
2.2: 2,5−ビス−[6−(オキサゾール−5−イル)]ピリジン−2−イルオキサゾール(化合物(2))
中間体(1)(0.10g;0.5mmol)、中間体(3)(0.10g;0.5mmol)、二酢酸パラジウム(11mg;0.05mmol;10mol%)、PCy3.HBF4(35mg;0.09mmol;20mol%)、ヨウ化銅(I)(0.1g;0.5mmol)、炭酸セシウム(0.3mg;0.9mmol)及び無水トルエン1.3mLの混合物をマイクロ波照射条件(130℃、150W)下に6時間置いた。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、目的化合物(2)をベージュ色の固体として得た(60mg、36%);1H NMR (300 MHz, CDCl3) 7.70 (d ; 2H ; J= 7.5 Hz) ; 7.80 (d ; 1H; J= 7.5Hz) ; 7.85 (d ; 2H ; J= 7.5 Hz ) ; 7.90 (s ; 1H) ; 7.93 (d; 2H ; J= 5 Hz) ; 8.00 (s ; 1H) ; 8.04 (d ; 1H ; J= 5 Hz) ; 8.20 (d ; 1H ; J= 7.5 Hz); SM m/z358.1 (M+1).
【0073】
3.実施例3: 2,6−ビス{2−[6−(オキサゾール−5−イル)ピリジン−2−イル]オキサゾール−5−イル}ピリジン(化合物(3))
【0074】
【化7】
中間体(1)(0.10g;0.5mmol)、中間体(3)(0.21g;0.9mmol)、二酢酸パラジウム(11mg;0.05mmol;10mol%)、PCy3.HBF4(35mg;0.09mmol;20mol%)、ヨウ化銅(I)(0.18g;0.9mmol)、炭酸セシウム(0.61mg;1.9mmol)及び無水トルエン1.3mLの混合物をマイクロ波照射条件(130℃、150W)下に6時間置いた。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、目的化合物(3)をベージュ色の固体として得た(52mg、22%);融点>250℃;1H NMR (300 MHz, CDCl3) 7.65 (m; 1H) ; 7.75 (m; 2H); 7.90-8.05 (m; 9H); 8.10 (s; 1H); 8.20 (m; 2H); SM m/z 524.2 (M+23).
【0075】
4.実施例4: 1,3−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)ベンゼン(化合物(4))
【0076】
【化8】
【0077】
4.1: 6,6’−(5,5’−(1,3−フェニレン)ビス(オキサゾール−5,2−ジイル))ジピリジン−2−カルバルデヒド(中間体(4))
【0078】
【化9】
Sambavisarao及びVrajesh(Synthesis, 2007, 3653)に報告されたように得られた1,3−ジ(オキサゾール−5−イル)ベンゼン(0.21g;1mmol)、6−ブロモピリジン−2−カルボキサルデヒド(0.24g;1.3mmol)、二酢酸パラジウム(64mg;0.28mmol、28mol%)、PCy3.HBF4(62mg;0.17mmol;17mol%)、ヨウ化銅(I)(0.43g;2.3mmol)、炭酸セシウム(1.4g;4.3mmol)及び無水ジオキサン4mLの混合物を封管中で130℃で2時間加熱した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、95:5)により精製して、中間体(4)をベージュ色の固体として得た(83mg、30%);1H NMR (300 MHz, CDCl3) 10.27 (s ; 1H); 8.45-8.40 (m ; 1H) ; 8.20 (s ; 0.5H) ; 8.10-8.05 (m ; 2H) ; 7.83 (dd ; 1H; J= 7.8 & 1.6 Hz) ; 7.71 (s; 1H) ; 7.61 (t ; 0.5H; J= 7.93 Hz); SM m/z 423.1 (M+1).
【0079】
4.2: 1,3−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)ベンゼン(化合物(4))
無水エタノール8mL中のジカルボキサルデヒド中間体(4)(75mg;0.18mmol)、TosMIC(86mg;0.44mmol)及び炭酸カリウム(126mg;0.9mmol)の混合物を2時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタンで抽出した(3×50mL)。合わせた有機層を硫酸マグネシウムで乾燥して真空で濃縮した。残渣は、ジクロロメタン:エタノール(95:5)を溶離液とするシリカゲルカラムのフラッシュクロマトグラフィーにより精製して、標題化合物をオフホワイト色の固体として得た(55mg、62%)、融点>250℃;1H NMR (300 MHz, CDCl3) 8.22 (br s ; 0.5H) ; 8.17 (d ; 1H; J= 7.8 Hz) ; 8.03 (s; 1H) ; 7.99 (t ; 1H, J= 7.8 Hz), 7.93 (s; 1H) ; 7.85-7.76 (m; 2H); 7.68 (s; 1H); 7.64-7.57 (m; 0.5H); SM m/z 501.1 (M+1) ; 元素分析、C28H16N6O4. 0.75 H2Oの理論値: C, 65.43 ; H, 3.40 ; N, 16.36, 実測値: C, 65.92, H, 3.37, 15.92.
【0080】
5.実施例5: 2,6−ビス(2−(3−(オキサゾール−5−イル)フェニル)オキサゾール−5−イル)ピリジン(化合物(5))
【0081】
【化10】
【0082】
5.1: 3,3’−(5,5’−(ピリジン−2,6−ジイル)ビス(オキサゾール−5,2−ジイル))ジベンズアルデヒド(中間体(5))
【0083】
【化11】
中間体(1)(0.18g;0.84mmol)、3−ブロモベンズアルデヒド(0.50g;2.7mmol)、二酢酸パラジウム(80mg;0.36mmol;43mol%)、PCy3.HBF4(68mg;0.18mmol;20mol%)、ヨウ化銅(I)(0.37g;1.9mmol)、炭酸セシウム(1.2g;3.7mmol)及び無水ジオキサン3.5mLの混合物を封管中で130℃で18時間加熱した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、98:2)により精製して、中間体(5)を黄色の固体として得た(50mg、14%);1H NMR (300 MHz, CDCl3) 10.15 (s ; 1H); 8.66 (s ; 1H) ; 8.45 (d ; 1H; J= 7.7 Hz) ; 8.04 (d; 1H; J= 7.6 Hz) ; 7.98-7.92 (m ; 1.5H) ; 7.76 (d ; 1H; J= 7.9 Hz) ; 7.72 (t ; 1H; J= 7.7 Hz); SM m/z 444.0 (M+Na); 元素分析、C25H15N3O4. 0.25 H2Oの理論値: C, 70.50 ; H, 3.64 ; N, 9.87, 実測値: C, 70.11, H, 3.51, 9.75.
【0084】
5.2: 2,6−ビス(2−(3−(オキサゾール−5−イル)フェニル)オキサゾール−5−イル)ピリジン(化合物(5))
無水エタノール6mL中のジカルボキサルデヒド中間体(5)(50mg;0.12mmol)、TosMIC(50mg;0.25mmol)及び炭酸カリウム(70mg;0.5mmol)の混合物を2時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタンで抽出した(3×50mL)。合わせた有機層を硫酸マグネシウムで乾燥して、真空で濃縮した。残渣は、ジクロロメタン:エタノール(94:6)を溶離液とするシリカゲルカラムのフラッシュクロマトグラフィーにより精製して、標題化合物を淡黄色の固体として得た(40mg;67%)、融点254〜256℃;1H NMR (300 MHz, CDCl3) 8.45 (s ; 1H) ; 8.14 (d ; 1H; J= 7.9 Hz) ; 7.99 (s; 1H) ; 7.96-7.88 (m ; 1.5H), 7.79 (d ; 1H; J= 7.9 Hz); 7.74 (d ; 1H; J= 7.9 Hz); 7.60 (t ; 1H; J= 7.9 Hz); 7.51 (s; 1H); SM m/z 500.2 (M+1) ; 元素分析、C29H17N5O4. H2Oの理論値: C, 67.31 ; H, 3.67 ; N, 13.53, 実測値: C, 67.53, H, 3.82, 13.29.
【0085】
6.実施例6: 2,6−ビス(2−(5−(オキサゾール−5−イル)フラン−2−イル)オキサゾール−5−イル)ピリジン(化合物(6))
【0086】
【化12】
【0087】
6.1: 5,5’−(5,5’−(ピリジン−2,6−ジイル)ビス(オキサゾール−5,2−ジイル))ジフラン−2−カルバルデヒド(中間体(6))
【0088】
【化13】
中間体(1)(0.21g;1mmol)、5−ブロモ−2−フルアルデヒド(0.21g;1.2mmol)、二酢酸パラジウム(83mg;0.37mmol;37mol%)、PCy3.HBF4(72mg;0.2mmol;20mol%)、ヨウ化銅(I)(0.46g;2.4mmol)、炭酸セシウム(1.4g;4.3mmol)及び無水ジオキサン4mLの混合物を封管中で130℃で2時間加熱した。残渣をフラッシュクロマトグラフィー(SiO2、ジクロロメタン:エタノール、98:2)により精製して、中間体(6)を黄色の固体として得た(62mg、26%);1H NMR (300 MHz, CDCl3) 9.83 (s ; 1H); 7.97 (s ; 1H) ; 7.95-7.90 (m ; 0.5H) ; 7.79 (d; 1H; J= 8.2 Hz) ; 7.39 (d; 1H; J= 3.8 Hz) ; 7.31 (d ; 1H; J= 3.8 Hz); SM m/z 402.1 (M+1);元素分析、C21H11N3O6. 1.5 H2Oの理論値: C, 58.87 ; H, 3.27 ; N, 9.81, 実測値: C, 58.77, H, 2.85, 9.83.
【0089】
6.2: 2,6−ビス(2−(5−(オキサゾール−5−イル)フラン−2−イル)オキサゾール−5−イル)ピリジン(化合物(6))
無水エタノール3mL中のジカルボキサルデヒド中間体(6)(30mg;0.07mmol)、TosMIC(43mg;0.22mmol)及び炭酸カリウム(63mg;0.45mmol)の混合物を2時間加熱還流した。真空で溶媒を留去して、残渣を食塩水中に注ぎ入れ、ジクロロメタンで抽出した(3×50mL)。合わせた有機層を硫酸マグネシウムで乾燥して、真空で濃縮した。残渣は、ジクロロメタン:エタノール(96:4)を溶離液とするシリカゲルカラムのフラッシュクロマトグラフィーにより精製して、標題化合物を淡黄色の固体として得た(13mg;36%)、融点>250℃;1H NMR (300 MHz, CDCl3) 7.95-7.91 (m ; 2.5H) ; 7.71 (d ; 1H; J= 8.1 Hz) ; 7.53 (s; 1H) ; 7.34-7.29 (m ; 2H); SM m/z 480.1 (M+1).
【0090】
7.: 2,6−ビス(2−(2,2’−ビピリジン−6−イル)オキサゾール−5−イル)ピリジン(化合物(7))
【0091】
【化14】
中間体(1)から出発し、そして中間体(3)の代わりに6−ブロモ−2,2’−ビピリジン(U. Lehmann and A.D. Schluter, Eur. J. Org. Chem. 2000, 3483-3487)を用いて、化合物(3)の合成に関して上記されたプロトコールを適用することにより、標題化合物(7)を得た。
【0092】
8.: 2,6−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)ピラジン(化合物(8))
【0093】
【化15】
【0094】
8.1: 2,6−ジ(オキサゾール−5−イル)ピラジン(中間体(7))
【0095】
【化16】
2,6−ピリジンジカルボキサルデヒドの代わりにピラジン−2,6−ジカルバルデヒド(H. Schumann and H.-K. Luo, Zeitschrift fuer Naturforschung, B : Chemical Sciences, 2005, 60(1), 22-24)から出発して、中間体(1)の合成に関して上記されたプロトコールを適用することにより、標題中間体(7)を得た。
【0096】
8.2: 6,6’−(5,5’−(ピラジン−2,6−ジイル)ビス(オキサゾール−5,2−ジイル))ジピリジン−2−カルバルデヒド(中間体(8))
【0097】
【化17】
中間体(1)及び3−ブロモベンズアルデヒドの代わりに中間体(7)及び6−ブロモピリジン−2−カルバルデヒドから出発して、中間体(5)の合成に関して上記されたプロトコールを適用することにより、標題中間体(8)を得た。
【0098】
8.3: 2,6−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)ピラジン(化合物(8))
中間体(5)の代わりに中間体(8)から出発して、化合物(5)の合成に関して上記されたプロトコールを適用することにより、標題化合物(8)を得た。
【0099】
9.: 6,6’−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)−2,2’−ビピリジン(化合物(9))
【0100】
【化18】
【0101】
9.1: 6,6’−ジ(オキサゾール−5−イル)−2,2’−ビピリジン(中間体(10))
【0102】
【化19】
2,6−ピリジンジカルボキサルデヒドの代わりに2,2’−ビピリジン−6,6’−ジカルバルデヒド(G. R. Newkome and H.-W. Lee; J. Am. Chem. Soc. 1983, 105(18), 5956-5957)から出発して、中間体(1)の合成に関して上記されたプロトコールを適用することにより、標題中間体(10)を得た。
【0103】
9.2: 6,6’−(5,5’−(2,2’−ビピリジン−6,6’−ジイル)ビス(オキサゾール−5,2−ジイル))ジピリジン−2−カルバルデヒド(中間体(11))
【0104】
【化20】
中間体(1)及び3−ブロモベンズアルデヒドの代わりに中間体(10)及び6−ブロモピリジン−2−カルバルデヒドから出発して、中間体(5)の合成に関して上記されたプロトコールを適用することにより、標題中間体(11)を得た。
【0105】
9.3: 6,6’−ビス(2−(6−(オキサゾール−5−イル)ピリジン−2−イル)オキサゾール−5−イル)−2,2’−ビピリジン(化合物(9))
中間体(5)の代わりに中間体(11)から出発して、化合物(5)の合成に関して上記されたプロトコールを適用することにより、標題化合物(9)を得た。
【0106】
II − 生物物理学的方法及びデータ:
試験II−1: 可溶性を調査するための紫外可視分光光度法
a) 紫外可視滴定:
本試験において詳述されるピリジン系ポリ芳香族複素環化合物の紫外可視特性の試験によって、種々の条件でその可溶性の測定が可能になる(DMSO、H2O及びカコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM NaCl(Caco.Na用)又はKCl(Caco.K用)):このための最も簡便な方法は、種々の濃度(0〜32μM)で化合物(3)の吸収スペクトルを測定することである;こうして化合物(3)の可溶性は、化合物(3)の濃度の関数としての所定の波長(ここでは338nm(A338))でのその吸光度の報告とBeer-Lambertの法則の適用とによって評価する。結果は、図1に与えられる。
【0107】
b) 要約
種々の滴定は、図2に要約する。
これらのデータは、化合物(3)が、
− ここで使用した全ての溶媒系に可溶性であること(DMSO、H2O及びカコジル酸水性緩衝液(高Na+又は高K+の両方);
− ここで使用した全ての溶媒中で、0〜32μM濃度範囲内で自己凝集しないこと;
を示した。
【0108】
試験II−2: 四重鎖DNAの安定化及び二重鎖に対する四重鎖DNAの選択性
四重鎖構造を持つ化合物の安定化、ひいては相互作用は、二重鎖に対する四重鎖DNA選択性、更には四重鎖内選択性の測定をも可能にするバージョンで、FRET融解試験法によりモニターする(17)。
【0109】
FRET試験法は、ヒト又はマラリア原虫テロメア配列を模倣するオリゴヌクレオチドで実施し、そしてそれぞれの先端にFRETパートナーを取り付ける:F21T(FAM-G3[T2AG3]3-Tamra)、FPf1T(FAM-G3[T3AG3]3-Tamra)及びFPf8T(FAM-G3[T2CAG3]3-Tamra)(ここで、FAM:6−カルボキシフルオレセインであり、そしてTamra:6−カルボキシ−テトラメチルローダミンである)。測定は492nm励起及び516nm検出で行った。
【0110】
a) 四重鎖安定化:
蛍光融解を実施した。結果は、図3A〜図3Cに与えられる。本実験は、10mMカコジル酸リチウム(pH7.2)及び100mM NaCl又はKClを含有する緩衝液中の0.2μMのF21T、FPf1T又はFPf8Tで実施した(後述のF21Tの例を参照のこと、NaCl(左)及びKCl(右));四重鎖DNAの融解は、単独及び1μM(5当量)の化合物(3)の存在下でモニターした。
【0111】
3種のオリゴヌクレオチドでの化合物(3)により誘導される安定化効果は、融解温度(ΔT1/2)の上昇により定量する。値は以下の表に示す。
【0112】
【表1】
【0113】
b) 二重鎖に対する四重鎖DNAの選択性:
蛍光融解は、10mMカコジル酸リチウム(pH7.2)及び100mM NaClを含有する緩衝液中の0.2μMのF21Tで実施した;G−四重鎖の融解は、単独及び1μMの化合物(3)(過剰(1、3及び10μM)の二重鎖DNA競合物質ds26(自己相補的配列[5’-CAATCGGATCGAATTCGATCCGATTG-3’]よりなる26塩基対の二重鎖DNA)を含むか又は含まない)の存在下でモニターした。結果は図4に与えられる。
【0114】
c) 四重鎖内選択性:
蛍光融解実験は、10mMカコジル酸リチウム(pH7.2)及び100mM NaClを含有する緩衝液中の0.2μMのF21Tで実施した;G−四重鎖の融解は、単独及び1μMの化合物(3)(過剰(1、3及び10μM)の四重鎖DNA競合物質(両方とも分子内の四重鎖構造中に折り畳まれることが強く疑われる、TG5T([(5’-TG5T-3’)4]、以前の研究(21)において使用した4分子四重鎖DNA)又はc−myc([5'-GAGGGTGGGGAGGGTGGGGAAG-3']、腫瘍遺伝子c−mycのプロモーター領域に存在する配列))を含むか又は含まない)の存在下でモニターした(22〜25)。結果は図5A〜図5Bに与えられる。
【0115】
d) 要約
種々のFRET融解実験は、図6の棒グラフ表示に要約する。
これらのデータは、化合物(3)が、
− ナトリウム緩衝液中でテロメア四重鎖F21T(ヒト)、FPf1T及びFPf8T(Plasmodium falciparum)と効率的に相互作用するが、一方カリウム条件ではこれらのオリゴヌクレオチドを安定化しないこと(表を参照のこと)(試験した3つのケースでは、化合物(3)が、カチオンに基づく四重鎖内選択性を提示することを意味する);
− 二重鎖DNAから四重鎖DNAを非常に効率的に識別すること(F21T安定化(ナトリウム緩衝液中)が、1、3又は10μMのds26の存在により限定的に影響を受けるに過ぎないため(安定化は、全てのケースで>91%維持される));
− ほぼ確実にスタッキング相互作用を介して四重鎖構造の外部G−カルテットと相互作用すること(F21T安定化が、非ループ4分子四重鎖DNA(TG5T、F21T安定化は、1、3及び10μMのTG5Tの存在下でそれぞれ61、37及び17%維持されている)又は二本鎖のみ反転ループ四重鎖DNA(c−myc、F21T安定化は、1、3及び10μMのc−mycの存在下でそれぞれ42、15及び1%維持されている)の競合に対して高感受性であるため)
を示す。
Na+及びK+条件の間で観測される劇的な違いを考えると、4分子及びループ両方との本化合物の相互作用もまた仮定することができる。
【0116】
試験II−2の2: 図16に、a)及びb)に上記されたのと同じナトリウム条件を用いた、化合物(3)及び(4)でのFRET融解で得られた結果を与える。蛍光標識四重鎖F21Tの融解は、単独及び1μMの化合物(3)又は化合物(4)(過剰(1、3及び10μM)の二重鎖DNA競合物質ds26(上記の自己相補的配列[5’-CAATCGGATCGAATTCGATCCGATTG-3’]よりなる26塩基対の二重鎖DNA)を含むか又は含まない)の存在下でモニターした。
【0117】
ds26の存在によりほとんど影響されない融解温度の上昇が、両方の化合物について得られる。
【0118】
試験II−3: 蛍光試験によりモニターされるDNAとの相互作用
本試験に詳述されるピリジン系ポリ芳香族複素環化合物は、強い蛍光を特徴とする。注目すべきは、量子収率が、純粋な有機(例えば、DMSO)から生理学的条件(例えば、緩衝液:10mMカコジル酸ナトリウム+100mM KCl(pH7.2)、表を参照のこと)までの使用される溶媒の性質に影響されないことである。DNAとの相互作用に及ぼす試験化合物の蛍光特性の変化により、生物学的に関連する幾つかの四重鎖に対する、これらの見かけの結合親和性を比較することができる(27)。
【0119】
【表2】
注意:化合物(3)の量子収率(及び標準偏差(s.d.))は、エタノール中のアントラセンを参照として、CH2Cl2(DCM)及び水中で測定した。
【0120】
a) 四重鎖相互作用:
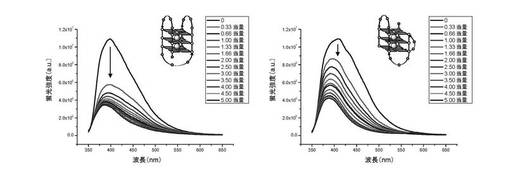
ここで使用した四重鎖DNAは22AGである:これは、ヒトテロメア配列を模倣する22ntオリゴヌクレオチドの折り畳みに由来する:22AGは[5’-AG3(T2AG3)3-3’]である。22AGからの四重鎖構造は、対応するオリゴヌクレオチドを90℃で5分間、10mMカコジル酸ナトリウム緩衝液(pH7.2)、100mM NaCl(22AG Na用)又はKCl(22AG K用)中で加熱し、そして氷中で冷却して速度論的トラップにより分子内折り畳みを助けることによって調製する。濃度は、使用前に紫外可視測定(85℃で5分の熱変性後)により260nmで測定する(図7A及び図7Bを参照のこと)。
【0121】
増加量の22AG Na(左)又は22AG K(右)をカコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM NaCl(右)又はKCl(左))中の化合物(3)の0.25μM溶液に加えると、化合物(3)の蛍光の漸進的消光が起こる(λex=340nm)。
【0122】
b) 二重鎖に対する四重鎖DNAの選択性:
二重鎖に対する四重鎖選択性は、22AG(上記参照)及び短い二重鎖DNA:ds17(以前の研究に使用した生物学的配列を表す17塩基対の二重鎖DNAであり(28);2本の相補鎖の配列は、以下のとおりである:[5’-CCAGTTCGTAGTAACCC-3’]/[5’-GGGTTACTACGAACTGG-3’])で実施した実験の比較による蛍光滴定によって評価する。二重鎖構造は、2本の対応する相補鎖を90℃で5分間、10mMカコジル酸ナトリウム緩衝液(pH7.3)、100mM KCl中で加熱し、次に6時間ゆっくり冷却することによって調製する。濃度は、使用前に紫外可視測定(85℃で5分の熱変性後)により260nmで測定する。結果は図8に与えられる。
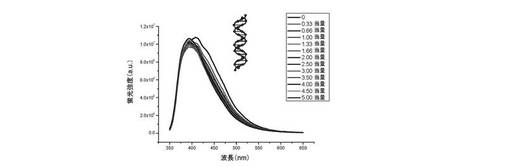
【0123】
増加量のds17をカコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM KCl)中の化合物(3)の0.25μM溶液に加えても、化合物(3)の蛍光の漸進的消光は起こらない(λex=340nm)。
【0124】
c) 四重鎖内選択性:
四重鎖内選択性は、22AG(上記参照)及び2種の他の四重鎖構造:c−myc及びc−kit2で実施した実験の比較による蛍光滴定によって評価する。これら2種の四重鎖DNAの形成は、目下c−myc(上記参照)及びc−kit(22;29及び30)腫瘍遺伝子のプロモーター領域で起こることが強く疑われる。この配列は以下のとおりである:c−myc:[5’-TGAGGGTGGGTAGGGTGGGTAA-3’]及びc−kit2:[(5’-CGGGCGGGCGCGAGGGAGGGG-3’]。四重鎖構造は、対応するオリゴヌクレオチドを90℃で5分間、10mMカコジル酸ナトリウム緩衝液(pH7.2)、100mM KCl中で加熱し、そして氷中で冷却して速度論的トラップにより分子内折り畳みを助けることによって調製する。濃度は、使用前に紫外可視測定(85℃で5分の熱変性後)により260nmで測定する。
結果は、図9A及び図9Bに与えられる。
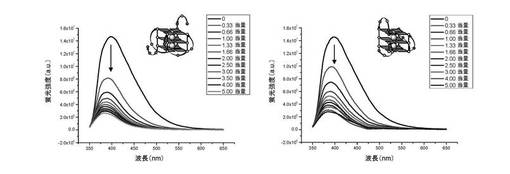
【0125】
増加量のc−myc(左)又はc−kit2(右)をカコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM KCl)中の化合物(3)の0.25μM溶液に加えると、化合物(3)の蛍光の漸進的消光が起こる(λex=340nm)。
【0126】
d) 要約:
種々の蛍光滴定(λex=340nm)は、グラフ表示を与える図10に要約する:
これらのデータは、化合物(3)が、
− 二重鎖DNAマトリックス(ds17)から四重鎖DNA(22AG K、22AG Na、c−kit2及びc−myc)を非常に効率的に識別し、よってFRET融解の結果を裏付けること;
− 更に効率的かつ迅速に22AG Na、c−kit2及びc−mycと相互作用するが、一方22AG Kとのその相互作用は弱いこと;この結果は、FRET融解実験を介して観測される結果に一致し、そして四重鎖内選択性において有望な候補であること
を示した。
【0127】
試験II−4: 円偏光二色性試験
円偏光二色性(CD)により、リガンドの結合時のDNA構造の修飾の深い研究が可能である;よってこれは、リガンドのその標的に対する親和性を反映し、またその結合様式への洞察をも提供しうる(Paramasivan et al, Methods, 2007, 43, 324)。
【0128】
a) 四重鎖相互作用:
ここで使用した四重鎖DNAは、22AG(上記参照)であり、両方とも10mMカコジル酸ナトリウム緩衝液(pH7.2)、100mM NaCl(22AG Na用)又はKCl(22AG K用)中でアニーリングされている。両方の緩衝液中の22AGの3μM溶液に、過剰の化合物(3)(30μM、10当量)を加える。結果は図11に与えられる。四重鎖との化合物(3)の相互作用は、時間の関数としてモニターする:これにより22AG NaのCDシグナルは大きさが変化する(左、特に263nmで)が、一方22AG Kの構造は完全に再編成する(右、特に263nmで)。
【0129】
b) 四重鎖内選択性:
四重鎖内選択性は、22AG(上記参照)及び2種の他の四重鎖構造:c−myc及びc−kit2(上記参照)で実施した実験の比較によるCDによって評価する。結果は図11A及び図11Bに与えられる。カコジル酸緩衝液(10mMカコジル酸ナトリウム+100mM KCl)中のc−kit2(左)及びc−myc(右)両方の3μM溶液に、過剰の化合物(3)(30μM、10当量)を加える。四重鎖との化合物(3)の相互作用は、時間の関数としてモニターする:これによりc−kit2のCDシグナルの大きさが変化し(特に285nmで)、これより少ないがc−mycでも変化する(285nm)。
【0130】
c) 四重鎖構造の誘導:
22AGオリゴヌクレオチドの折り畳まれた形と折り畳まれていない形の間の平衡は、入れられる緩衝液の存在及び性質に高感受性である。対応する結果は図12A及び図12Bに与えられる。10mMトリスHCl(pH7.2)のような低カチオン性緩衝液では、22AGは大部分折り畳まれていない(ランダムコイル状);よって化合物(3)を加えることにより折り畳まれた形<−>折り畳まれていない形の平衡に影響を及ぼすことができる。即ち、トリスHCl緩衝液中のランダムコイル状22AGの3μM溶液に、増加量の化合物(3)(0〜7.5当量)を加える;これにより22AGのCDシグナルは大きく変化し、ランダムコイルに典型的な257nmシグナルの消失と、2つの新規な極大[一方は正(290nm付近)であり他方は負(265nm付近)であり、両方とも「逆平行」四重鎖DNA構造(22AG Naとして、上記参照)のCDシグナルに典型的である]の獲得に至る。
【0131】
d) 要約:
これらのデータは、化合物(3)が、
− 効率的にかつほぼ自発的に22AG Naと相互作用するが、22AG Kとは効率的ではあるがゆっくりと反応すること(これは蛍光滴定及びFRET融解実験結果と一致する);
− c−myc及びc−kit2と相互作用するが、その相互作用は、これら2つの四重鎖の構造を修飾しないこと;
− 22AGの折り畳まれていない形と折り畳まれた形の間の平衡を折り畳まれた形へとシフトさせることができること
を示した。
【0132】
III − 生物学的方法及びデータ:
試験III−1: 化合物(3)によるインビトロでのテロメア配列へのPOT1結合の阻害
hPOT1を用いる電気泳動移動度シフトアッセイをテロメア22AGオリゴヌクレオチド(上記参照)に実行した。22AGは、T4ポリヌクレオチドキナーゼを用いて5’末端を[γ−32P]−ATPで標識した。精製組換えhPOT1は、バキュロウイルス発現系で産生させた。POT1/22AG結合アッセイは、50mMヘペス(pH7.9)、100mM NaCl、0.1mM EDTA、4%w/vショ糖、2%v/vグリセロール、0.1mg/ml BSA、0.02%w/vブロモフェノールブルー、30nM hPOT1、20nM[α−32P]−22AGを含有する総容量10μl中で実行した。様々な濃度の化合物(3)(10、1、0.1及び0.01μM)をhPOT1と共にこの溶液に加え、この混合物を室温で30分間インキュベートした。それぞれ個々の試料は、0.5×トリス−ホウ酸−EDTA緩衝液中で1%アガロースゲル上で電気泳動により分離した。ゲルは80Vで35分間流して、ワットマンDE81紙上で乾燥して、放射活性はホスファイメージャー(phosphorimager)(Typhoon 9210, Amersham)により可視化した。データの解析は、ImageQuantソフトウェア(Amersham)により実施して、結果は未処理対照(100%と定義)で得られるPOT1−22AG複合体の百分率として表した(図14を参照のこと)。
【0133】
これらのデータは、化合物(3)が、テロメア22AG配列へのhPOT1の結合を用量依存的に阻害することを示した。後述の実験において、化合物(3)のIC50−POT1は300nMに等しい。
【0134】
試験III−2: 化合物(3)による細胞増殖の阻害
pEGFP−POT1ベクターで安定にトランスフェクトしたHT1080ヒト線維肉腫細胞株(HT1080GFP−POT1)は以前に報告されており(32)、U20Sヒト骨肉腫はAmerican Type Culture Collection(Rockville, 米国)から得た。細胞は、HT1080GFP−POT1用に10%v/vウシ胎仔血清及び400μg/mlジェネティシンを補足したダルベッコー修飾イーグル培地(Invitrogen)で増殖させた。細胞は、様々な濃度の化合物(3)(10、3、1、0.3及び0.1μM)の存在下で各濃度二重反復で、0日目に4.5×104細胞/ウェルで6ウェル培養プレートに蒔き、更に72時間培養した。3日目に、細胞を1×PBSで洗浄してトリプシン処理した。各処理細胞試料について、生存細胞の数をトリパンブルーの存在下で測定した。結果は図15A及び図15Bに与えられる。これらは、100%と定義した未処理対照細胞に比較した相対細胞数(パーセント)に相当する。
【0135】
これらのデータは、化合物(3)が、癌細胞の増殖を0.1〜1μMの間であるIC50で妨害することを示す。
【0136】
特に限定されないが、特許公報、特許出願、本及び学術論文を包含する、本出願に引用される全ての刊行物は、それぞれ引用例としてその全体が本明細書に取り込まれる。本発明は開示された実施態様を参照して上述されているが、当業者には、詳述された特定の実験は、単に本発明の例証であることが容易に理解されよう。当然のことながら、本発明の本質を逸することなく、ここに種々の変更を加えることができる。したがって、本発明は以下の請求の範囲によってのみ限定される。
【0137】
【表3】
【特許請求の範囲】
【請求項1】
ペンタ−、ヘキサ−、ヘプタ−、オクタ−、ノナ−及びデカ−ヘテロアリール誘導体であって、それぞれ式(I)、(II)、(III)及び(IV)の複素環1(Het−1)a及び/又は複素環2(Het−2)b及び/又は複素環3(Het−3)c及び/又は複素環4(Het−4)d:
【化21】
の組合せ、N−オキシド、薬学的に許容しうる付加塩[ここで、
− 該組合せは、少なくとも2種の異なる複素環部分を含み、そして場合により式(V)の(Aryl−5)e部分を含み、
− a、b及びeは、0〜6の整数であり、c及びdは、0〜2の整数であり、a+b+c+d+eの合計は、≦10であり、
− Yは、O又はSであり;
− Het−1は、2,6−ピリジン−ジイル又は2,4−ピリミジン−ジイル又は3,5−ピラジン−ジイル又は2,4−(1,3,5−トリアジン)ジイル又は3,5−(1,2,4−トリアジン)ジイル又は2,4−オキサゾリン−ジイル又は2,4−チアゾリン−ジイルを含む群において選択される窒素複素環−ジイル環のクラスであり;
− Het−2は、2,5−オキサゾリン−ジイル又は2,5−チアゾリン−ジイル又は2,5−チオフェン−ジイル又は2,5−フラン−ジイルを含む群において選択される5員複素環−ジイル環のクラスであり;
− Het−3及び/又はHet−4は、該ペンタ−〜デカ−ヘテロアリール誘導体の終点を構成するが、ここで、
− Het−3は、2−ピリジル又は2−ピリミジル又は4−ピリミジル又は2−ピラジル又は2−(1,3,5)トリアジル又は3−(1,2,4)トリアジル又は5−(1,2,4)トリアジル又は4−オキサゾリル又は4−チアゾリルから選択される窒素複素環−イル環のクラスであり;
− Het−4は、2−オキサゾリル又は5−オキサゾリル又は2−チアゾリル又は5−チアゾリル又は2−チエニル又は2−フラニルから選択される5員複素環−イルのクラスであり;
− R1及びR2は、それぞれ独立に、水素;C1−6アルキル;C1−4アルキルオキシ;ハロ;ヒドロキシ;ヒドロキシメチル;ニトロ;アミノ;モノ−又はジ(C1−4アルキル)アミノ;C1−4アルキルメチルアミノ;モノ−又はジ(C1−4アルキル)アミノ(C2−4アルキル)アミノメチル;モルホリン−4−イルC2−4アルキルオキシ;ピペラジン−1−イルC2−4アルキルオキシ;4−C1−4アルキルピペラジン−1−イル;フェニル、ベンジル、ナフチルから選択される単環又は二環から選択される]を含む誘導体。
【請求項2】
1個又は数個の該部分が、それぞれ独立にC1−6アルキル;C1−4アルキルオキシ;ハロ;ヒドロキシ;ニトロ;アミノ;モノ−又はジ(C1−4アルキル)アミノ;C1−4アルキルメチルアミノ;モルホリン−4−イルC2−4アルキルオキシ;ピペラジン−1−イルC2−4アルキルオキシ;4−C1−4アルキルピペラジン−1−イルC2−4アルキルオキシ;フェニル、ベンジル、ナフチルから選択される単環式又は二環式環から選択される、1、2又は3個の置換基で置換されている、請求項1に記載の誘導体。
【請求項3】
少なくとも5個、少なくとも7個、少なくとも8個、少なくとも9個又は10個の複素環部分を含む、請求項1又は2に記載の誘導体。
【請求項4】
1個又は数個の1,3−フェニレン−ジイル部分を更に含む、請求項3に記載の誘導体。
【請求項5】
下記構造:
−(R2により置換されたHet−4)−(Het−1)−(R2により置換されたHet−4)
−(R1により置換されたHet−3)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−1,3−フェニレン−ジイル−(Het−2)−(R1により置換されたHet−3)
−(R1又はR2により置換された1,3−フェニレン−ジイル)−(Het−2)−(Het−1)−(Het−2)−(R1又はR2により置換された1,3−フェニレン−ジイル)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
の1つを有する、請求項1〜4のいずれか1項に記載の誘導体。
【請求項6】
・ R1及びR2が、オキサゾリン−若しくはピリジン−ジイル基であるか、かつ/又は
・ Het−2が、オキサゾリン−ジイル基であるか、かつ/又は
・ Het−1が、2,6−ピリジン−ジイル、2,6−ピリミジン−ジイル、2,6−ピラジン−ジイル、若しくは1,3−フェニレン−ジイル基である、請求項1〜5のいずれか1項に記載の誘導体。
【請求項7】
1個以上の原子が、同一の原子番号を有するが、自然界で通常見られる原子質量又は質量数とは異なる原子質量又は質量数を有する原子により置換されている、請求項1〜6のいずれか1項に記載の誘導体。
【請求項8】
薬物としての請求項1〜7のいずれか1項に記載の誘導体の使用。
【請求項9】
有効量の請求項1〜7のいずれか1項に記載の少なくとも1つの誘導体を薬学的に許容しうる担体と組合せて含む医薬組成物。
【請求項10】
経口又は非経口経路による投与に適切な剤形下の、請求項9に記載の医薬組成物。
【請求項11】
癌及びマラリアのような感染症を処置するための、請求項9又は10に記載の医薬組成物。
【請求項12】
癌又は感染症の処置用薬物を製造するための、請求項1〜7のいずれか1項に記載の誘導体の使用。
【請求項13】
請求項1〜7のいずれか1項に記載のポリヘテロアリール誘導体の製造方法であって、 ・ X1−A−X2を
・ B−X3と、又はX4−B−X3と、
所望のポリヘテロアリール誘導体を与える条件下で反応させること[ここで、
− Aは、ジ−又はトリ−又はテトラ−ヘテロアリール−1,4鎖であり、そしてBは、(モノ−又はジ−又はトリ−又はテトラ−ヘテロアリール−1,4)−であり(ここで、ヘテロアリール部分は、場合により1個、2個、3個又は4個のAryl−5部分又はAryl−5末端部分を含み、ヘテロアリール−1,4は、上記と同義のHet−1及び/又はHet−2及び/又はHet−3及び/又はHet−4の組合せである)、
− X1及びX2は、同一であるか又は異なって、水素又はハロゲン又はトリフラート基を表し、
− X3は、ハロゲン又はハロゲン化亜鉛又はトリアルキルスズ又はボロン酸基を表し、
− X4は、カルボキサルデヒド基を表す]を含む方法。
【請求項14】
DNA四重鎖特異的プローブとしての、請求項1〜7のいずれか1項に記載の誘導体の使用。
【請求項15】
四重鎖DNA又は関連核酸構造を検出及び/又は精製するための、請求項1〜7のいずれか1項に記載の誘導体の使用。
【請求項1】
ペンタ−、ヘキサ−、ヘプタ−、オクタ−、ノナ−及びデカ−ヘテロアリール誘導体であって、それぞれ式(I)、(II)、(III)及び(IV)の複素環1(Het−1)a及び/又は複素環2(Het−2)b及び/又は複素環3(Het−3)c及び/又は複素環4(Het−4)d:
【化21】
の組合せ、N−オキシド、薬学的に許容しうる付加塩[ここで、
− 該組合せは、少なくとも2種の異なる複素環部分を含み、そして場合により式(V)の(Aryl−5)e部分を含み、
− a、b及びeは、0〜6の整数であり、c及びdは、0〜2の整数であり、a+b+c+d+eの合計は、≦10であり、
− Yは、O又はSであり;
− Het−1は、2,6−ピリジン−ジイル又は2,4−ピリミジン−ジイル又は3,5−ピラジン−ジイル又は2,4−(1,3,5−トリアジン)ジイル又は3,5−(1,2,4−トリアジン)ジイル又は2,4−オキサゾリン−ジイル又は2,4−チアゾリン−ジイルを含む群において選択される窒素複素環−ジイル環のクラスであり;
− Het−2は、2,5−オキサゾリン−ジイル又は2,5−チアゾリン−ジイル又は2,5−チオフェン−ジイル又は2,5−フラン−ジイルを含む群において選択される5員複素環−ジイル環のクラスであり;
− Het−3及び/又はHet−4は、該ペンタ−〜デカ−ヘテロアリール誘導体の終点を構成するが、ここで、
− Het−3は、2−ピリジル又は2−ピリミジル又は4−ピリミジル又は2−ピラジル又は2−(1,3,5)トリアジル又は3−(1,2,4)トリアジル又は5−(1,2,4)トリアジル又は4−オキサゾリル又は4−チアゾリルから選択される窒素複素環−イル環のクラスであり;
− Het−4は、2−オキサゾリル又は5−オキサゾリル又は2−チアゾリル又は5−チアゾリル又は2−チエニル又は2−フラニルから選択される5員複素環−イルのクラスであり;
− R1及びR2は、それぞれ独立に、水素;C1−6アルキル;C1−4アルキルオキシ;ハロ;ヒドロキシ;ヒドロキシメチル;ニトロ;アミノ;モノ−又はジ(C1−4アルキル)アミノ;C1−4アルキルメチルアミノ;モノ−又はジ(C1−4アルキル)アミノ(C2−4アルキル)アミノメチル;モルホリン−4−イルC2−4アルキルオキシ;ピペラジン−1−イルC2−4アルキルオキシ;4−C1−4アルキルピペラジン−1−イル;フェニル、ベンジル、ナフチルから選択される単環又は二環から選択される]を含む誘導体。
【請求項2】
1個又は数個の該部分が、それぞれ独立にC1−6アルキル;C1−4アルキルオキシ;ハロ;ヒドロキシ;ニトロ;アミノ;モノ−又はジ(C1−4アルキル)アミノ;C1−4アルキルメチルアミノ;モルホリン−4−イルC2−4アルキルオキシ;ピペラジン−1−イルC2−4アルキルオキシ;4−C1−4アルキルピペラジン−1−イルC2−4アルキルオキシ;フェニル、ベンジル、ナフチルから選択される単環式又は二環式環から選択される、1、2又は3個の置換基で置換されている、請求項1に記載の誘導体。
【請求項3】
少なくとも5個、少なくとも7個、少なくとも8個、少なくとも9個又は10個の複素環部分を含む、請求項1又は2に記載の誘導体。
【請求項4】
1個又は数個の1,3−フェニレン−ジイル部分を更に含む、請求項3に記載の誘導体。
【請求項5】
下記構造:
−(R2により置換されたHet−4)−(Het−1)−(R2により置換されたHet−4)
−(R1により置換されたHet−3)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−1,3−フェニレン−ジイル−(Het−2)−(R1により置換されたHet−3)
−(R1又はR2により置換された1,3−フェニレン−ジイル)−(Het−2)−(Het−1)−(Het−2)−(R1又はR2により置換された1,3−フェニレン−ジイル)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
−(R1により置換されたHet−3)−(Het−2)−(Het−1)−(Het−1)−(Het−2)−(R1により置換されたHet−3)
の1つを有する、請求項1〜4のいずれか1項に記載の誘導体。
【請求項6】
・ R1及びR2が、オキサゾリン−若しくはピリジン−ジイル基であるか、かつ/又は
・ Het−2が、オキサゾリン−ジイル基であるか、かつ/又は
・ Het−1が、2,6−ピリジン−ジイル、2,6−ピリミジン−ジイル、2,6−ピラジン−ジイル、若しくは1,3−フェニレン−ジイル基である、請求項1〜5のいずれか1項に記載の誘導体。
【請求項7】
1個以上の原子が、同一の原子番号を有するが、自然界で通常見られる原子質量又は質量数とは異なる原子質量又は質量数を有する原子により置換されている、請求項1〜6のいずれか1項に記載の誘導体。
【請求項8】
薬物としての請求項1〜7のいずれか1項に記載の誘導体の使用。
【請求項9】
有効量の請求項1〜7のいずれか1項に記載の少なくとも1つの誘導体を薬学的に許容しうる担体と組合せて含む医薬組成物。
【請求項10】
経口又は非経口経路による投与に適切な剤形下の、請求項9に記載の医薬組成物。
【請求項11】
癌及びマラリアのような感染症を処置するための、請求項9又は10に記載の医薬組成物。
【請求項12】
癌又は感染症の処置用薬物を製造するための、請求項1〜7のいずれか1項に記載の誘導体の使用。
【請求項13】
請求項1〜7のいずれか1項に記載のポリヘテロアリール誘導体の製造方法であって、 ・ X1−A−X2を
・ B−X3と、又はX4−B−X3と、
所望のポリヘテロアリール誘導体を与える条件下で反応させること[ここで、
− Aは、ジ−又はトリ−又はテトラ−ヘテロアリール−1,4鎖であり、そしてBは、(モノ−又はジ−又はトリ−又はテトラ−ヘテロアリール−1,4)−であり(ここで、ヘテロアリール部分は、場合により1個、2個、3個又は4個のAryl−5部分又はAryl−5末端部分を含み、ヘテロアリール−1,4は、上記と同義のHet−1及び/又はHet−2及び/又はHet−3及び/又はHet−4の組合せである)、
− X1及びX2は、同一であるか又は異なって、水素又はハロゲン又はトリフラート基を表し、
− X3は、ハロゲン又はハロゲン化亜鉛又はトリアルキルスズ又はボロン酸基を表し、
− X4は、カルボキサルデヒド基を表す]を含む方法。
【請求項14】
DNA四重鎖特異的プローブとしての、請求項1〜7のいずれか1項に記載の誘導体の使用。
【請求項15】
四重鎖DNA又は関連核酸構造を検出及び/又は精製するための、請求項1〜7のいずれか1項に記載の誘導体の使用。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公表番号】特表2012−507504(P2012−507504A)
【公表日】平成24年3月29日(2012.3.29)
【国際特許分類】
【出願番号】特願2011−533915(P2011−533915)
【出願日】平成21年11月2日(2009.11.2)
【国際出願番号】PCT/IB2009/054856
【国際公開番号】WO2010/049915
【国際公開日】平成22年5月6日(2010.5.6)
【出願人】(500026533)アンスティテュ・キュリ (20)
【氏名又は名称原語表記】INSTITUT CURIE
【出願人】(595040744)サントル・ナショナル・ドゥ・ラ・ルシェルシュ・シャンティフィク (88)
【氏名又は名称原語表記】CENTRE NATIONAL DE LA RECHERCHE SCIENTIFIQUE
【出願人】(591100596)アンスティチュ ナショナル ドゥ ラ サンテ エ ドゥ ラ ルシェルシュ メディカル (59)
【出願人】(506140550)ミュゼオム・ナショナル・ディストワール・ナチュレル (4)
【氏名又は名称原語表記】MUSEUM NATIONAL D’HISTOIRE NATURELLE
【Fターム(参考)】
【公表日】平成24年3月29日(2012.3.29)
【国際特許分類】
【出願日】平成21年11月2日(2009.11.2)
【国際出願番号】PCT/IB2009/054856
【国際公開番号】WO2010/049915
【国際公開日】平成22年5月6日(2010.5.6)
【出願人】(500026533)アンスティテュ・キュリ (20)
【氏名又は名称原語表記】INSTITUT CURIE
【出願人】(595040744)サントル・ナショナル・ドゥ・ラ・ルシェルシュ・シャンティフィク (88)
【氏名又は名称原語表記】CENTRE NATIONAL DE LA RECHERCHE SCIENTIFIQUE
【出願人】(591100596)アンスティチュ ナショナル ドゥ ラ サンテ エ ドゥ ラ ルシェルシュ メディカル (59)
【出願人】(506140550)ミュゼオム・ナショナル・ディストワール・ナチュレル (4)
【氏名又は名称原語表記】MUSEUM NATIONAL D’HISTOIRE NATURELLE
【Fターム(参考)】
[ Back to top ]