
癌の治療のための単純ヘルペスウイルス2型変異体の使用
本発明は、癌の治療における薬剤としての改変単純ヘルペスウイルス2型(HSV−2)の組成物および使用に関する。本改変HSV−2は、融合性活性を有する。本改変HSV−2は、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くポリペプチドをコードする改変/変異ICP10ポリヌクレオチドを含む。特定の態様では、このウイルスは、悪性細胞の治療に有用である。特定の実施態様では、このウイルスは、腫瘍細胞中で選択的に複製する。さらに別の特定の実施態様では、このウイルスは、細胞膜融合を起させて少なくともいくつかの望ましくない細胞の培養物、組織または生物を駆逐する。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願への相互参照)
この出願は、2005年6月23日に出願された仮出願第60/693,157号(この全体が、参考として本明細書に援用される)への優先権を主張する。
【0002】
(連邦の援助を受けた研究または開発においてなされた発明の権利に関する声明)
本発明は、少なくとも部分的に、NIH補助金第RO1 CA106671−01に従って米国政府が提供した資金を用いて開発された。米国政府は、本発明において特定の権利を有することがある。
【0003】
(発明の分野)
本発明は、ウイルス学、癌生物学、細胞生物学、分子生物学、および癌治療薬を含む薬剤の分野を目的とする。詳しくは、本発明は、ICP10遺伝子の改変形を含む変異体単純ヘルペスウイルス2(HSV−2)と、この変異体HSV−2の悪性疾患の治療への使用を提供する。
【背景技術】
【0004】
(発明の背景)
複製選択的殺腫瘍ウイルスは、固形腫瘍に対する抗腫瘍因子として大きな将来性を示した。これらのウイルスは、腫瘍細胞中では優先的に複製することができるが、正常細胞中では複製する能力が制限される。殺腫瘍ウイルスの基本的な抗腫瘍機序は、ウイルスが、最初に感染した腫瘍細胞から周囲の腫瘍細胞へ伝播し、広がるときの直接細胞変性効果によるものであり、大きな体積の分布と抗癌効果とを実現する。殺腫瘍を目的として、単純ヘルペスウイルス(HSV)は、最も普通には、正常細胞(非分裂型)中での効率的な複製に必要なウイルス遺伝子は削除するが、腫瘍細胞に関しては削除しないことによって改変されてきた。改変は、ウイルスγ34.5遺伝子またはICP6遺伝子の削除を含む。ウイルスγ34.5遺伝子は、HSV感染時に神経毒性因子として機能する(非特許文献1)。この遺伝子を除くと、非分裂細胞中でのウイルス複製が妨げられる(McKieら、(1996) Br J Cancer 74(5): 745−52)。ウイルスICP6遺伝子は、リボヌクレオチド1レダクターゼの大型サブユニットをコードする。この大型サブユニットは、ウイルスDNAが効率的に複製するのに十分なdNTPプールを作り出し、腫瘍細胞中では大量に発現するが、非分裂細胞中では発現しない。従って、この遺伝子を変異させたウイルスは、腫瘍細胞中で選択的に複製し、腫瘍細胞を殺傷することができる。動物研究段階で広範に試験され、現在は臨床試験段階にある殺腫瘍HSV G207は、γ34.5遺伝子座とICP6遺伝子中の挿入変異との両方の複製体において、大腸菌lacZ遺伝子による削除を受ける(Walkerら、(1999) Human Gene Ther. 10(13): 2237−2243)。あるいは、腫瘍特異性プロモーターを用いてHSV複製に必須のγ34.5または他の遺伝子を駆動することによって、殺腫瘍1型HSVを構築することができる(Chungら、(1999) J Virol 73(9): 7556−64)。
【0005】
殺腫瘍単純ヘルペスウイルス(HSV)は、当初、脳腫瘍の治療を目的として設計され、構築された(Andreanskyら、(1996) Proc Natl Acad. Sci.93(21): 11313−11318)。その後、それらは、乳腺(Toda, et at, (1998) Human Gene Ther. 9(15): 2177−2185)、前立腺(Walkerら、(1999) Harman Gene Ther. 10(13): 2237−2243)、肺(Toyoizumiら、(1999) Human Gene Ther. 10(18): 3013−3029)、卵巣(Coukosら、(1999) Clin. Cancer Res.5(6): 1523−1527)、結腸および肝臓(Pawlikら、(2000) Cancer Res. 61(11): 2790−2795)を含むさまざまなその他のヒト固形腫瘍において有効であることが見いだされた。殺腫瘍HSVの安全性も、マウス(Sundaresanら、(2000) J. Virol. 74(8): 3832−3841)および霊長類(ヨザル属)において検証された。後者は、HSV感染症に極めて敏感である(Todoら、(2000) Cancer Gene Ther. 7(6): 939−946)。これらの研究によって、殺腫瘍HSVはインビボ投与に極めて安全であることが確認された。
【0006】
殺腫瘍HSVは、もっぱらHSV−1から構築されてきた。HSV−2は、殺腫瘍ウイルスを構築する目的で検討されたことがない。しかし、HSV−2は、腫瘍溶解剤としての可能性を高める独特の特徴をいくつか有する。例えば、HSV−2は、HSV−1と異なり、好中球、単核細胞およびNK細胞の機能に影響を及ぼす糖蛋白質G(gG)の分泌形をコードすることが報告された(非特許文献2)。そのような性質は、HSV−2から誘導された殺腫瘍ウイルスに、体の先天性免疫の阻害効果に抵抗する能力を提供することがある。先天性免疫は、侵入する微生物に対する宿主の高速応答であり、インビボのHSV複製を制限する主要な因子であることが見いだされた(Dalloulら、(2004) J Clin Virol 30(4): 329−36; Wakimotoら、(2003) Gene Ther 10(11): 983−90)。従って、HSV−2から誘導された殺腫瘍ウイルスは、患者の体が抗HSV先天性免疫を発展させても、自己複製し、広がるはずである。
【0007】
前臨床研究は有望であったが、初期臨床試験の結果によると、現状の殺腫瘍ウイルスは、安全ではあるものの単独では限られた抗腫瘍活性しか有しないことが示唆された(Nemunaitisら、(2001) J. Clin Oncol. 19(2): 289−298)。本発明者らの研究によると、殺腫瘍HSVに細胞膜融合活性を組み込むと、ウイルスの抗腫瘍力を劇的に高めることができることが実証された(Fuら、(2002) Mot Ther. 7(6): 748−754; Fuら、(2003) Cancer Res. 62: 2306−2312。そのような融合殺腫瘍ウイルスは、腫瘍中の合胞体形成をもたらし、ウイルスの破壊力を直接高め、その腫瘍内の広がりを促進する(Fuら、(2003) Cancer Res. 62: 2306−2312)。合胞体形成の腫瘍崩壊機序と融合殺腫瘍HSVによる直接細胞溶解とを独自に組み合わせると、インサイチュ腫瘍抗原提示が促進され、強力な抗腫瘍免疫応答が得られる(Nakamoriら、(2004) Mol. Ther. 9(5): 658−665)。さらに、合胞体形成による融合殺腫瘍HSVの広がりによって、ホストの中の中和抗ウイルス抗体の存在下でも、抗腫瘍活性を維持することが可能になる。ウイルスは、生細胞の中でしか複製することができず、ウイルスの複製には、通常、特定の細胞信号伝達経路の活性化が必要である。多くのウイルスは、これらの信号伝達経路を活性化して複製に役立てるさまざまな戦略を進化の間に取得した。単純ヘルペスウイルス2型(HSV−2)リボヌクレオチドレダクターゼ(ICP10またはRRI)の大型サブユニットは、セリン/スレオニンプロテインキナーゼ(PK)活性を有する固有のアミノ末端ドメインを含む。このPK活性は、細胞Ras/MEK/MAPK経路を活性化することが見いだされた(Smithら、(2000) J Virol 74(22): 10417−29)。
【0008】
LuoとAurelianとは、HSV−2の中のICP10遺伝子のさまざまな削除を含むさまざまなベクターを記載して、特定のモチーフと特定の活性との間の関係を実証している (Luo and Aurelian, (1992) J Biol Chem 267(14): 9645−53)。HSV−2、ICP10遺伝子の改変構築体および削除構築体を用いて、リボヌクレオチドレダクターゼドメインの特定の特性が実証された(Peng et al. (1996) Virology 216(1): 184−96)。
【0009】
リボヌクレオチドレダクターゼ遺伝子からPKドメイン(ICP10 PK)を削除すると、活性化Ras信号伝達経路を予め存在させていない細胞中では、複製するウイルスの能力がひどく低下する(Smith et al (1998) J. Virol. 72(11): 9131−9141)。
【0010】
特許文献1は、HSV−2による攻撃からの保護を提供するワクチンを目的とする。ICP10のプロテインキナーゼドメインを削除したところ、細胞に感染し、細胞を変換させるHSV−2の能力に対して有害な効果が表れる。
【0011】
本発明は、改変されたHSV−2を利用して癌の治療のための新規な治療薬を提供することによって、当分野における必要を満たす。
【特許文献1】米国特許第6,013,265号明細書
【非特許文献1】Chouら、Science(1990)250:1262−1266
【非特許文献2】Bellnerら、J Immunol(2005)174(4): 2235−41
【発明の開示】
【課題を解決するための手段】
【0012】
(発明の要旨)
本発明は、腫瘍崩壊特性を有する強力な改変単純ヘルペスウイルス2型(HSV−2)を提供することによって、当分野の長年の求めに対する答を示す。本発明の特定の実施態様では、ウイルスは、改変されたICP10ポリヌクレオチドを有する。ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するが、プロテインキナーゼ活性の欠失するICP10ポリペプチドをコードする。特定の態様では、ウイルスは、悪性細胞の治療に有用である。特定の実施態様では、ウイルスは、腫瘍細胞中で選択的に複製する。さらに別の特定の実施態様では、ウイルスは、細胞膜融合を起させて少なくともいくつかの望ましくない細胞の培養物、組織または生物を駆逐する。さらに別の特定の実施態様では、ウイルスは、少なくともいくつかの望ましくない細胞の増殖を阻害し、および/または少なくともいくつかの望ましい細胞のアポトーシスを誘導し、および/または強力な抗腫瘍免疫応答を誘導し、および/またはそれらを組み合わせる。
【0013】
野生型HSV−2ウイルスは、セリン/スレオニンプロテインキナーゼ活性などのプロテインキナーゼ(PK)活性を有するアミノ末端ドメインと、リボヌクレオチドレダクターゼ活性を有するc−末端ドメインとを有するポリペプチドをコードするICP10ポリヌクレオチド(RR1ポリヌクレオチドとも呼ばれることがある)を含む。本発明の特定の態様では、ウイルスが、腫瘍細胞中で選択的複製活性(従って、腫瘍細胞を崩壊させる活性)および/またはウイルスを融合型にする活性を含み、あるいは膜融合(合胞体形成)活性を含むという意味で融合型活性を高めるように、内因性PKドメインを改変する。いくつかの実施態様では、プロテインキナーゼドメインをコードする内因性配列の少なくとも一部を削除することによって、コード化されるポリペプチドのプロテインキナーゼ活性が欠失するように、ICP10ポリヌクレオチドを改変する。
【0014】
本発明の別の実施態様では、プロテインキナーゼドメインの少なくとも一部をコードする内因性ICP10ポリヌクレオチドの少なくとも一部を第2のポリヌクレオチドが置換する。さらにその他の実施態様では、置換されなかったICP10配列は、RRドメイン全体を含む。内因性ICP10ポリヌクレオチドの少なくとも一部の置換は、例えば、相同組み換え、またはPCRおよび当業者に公知のその他の方法論の使用を含むその他の適当な遺伝子工学的方法によるなど、任意の適当な方法によって行ってよい。
【0015】
本発明の追加の態様では、ICP10の内因性PKドメインの少なくとも一部を置換するポリヌクレオチドは、任意の適当な配列であってよい。例えば、PKドメインを置換するポリヌクレオチドは、レポーター遺伝子産物または治療遺伝子産物をコードしてよい。第2のポリヌクレオチド(PKドメインの少なくとも一部を置換した)を含む改変ICP10ポリヌクレオチドは、置換ポリヌクレオチドと、残るICP10遺伝子の非置換部分とで構成される融合蛋白質をコードする。本発明で用いるのに適するレポーター遺伝子の非限定的な例は、緑色蛍光蛋白質(SEQ ID.NO:16、GenBank Accession No. U55761)、β−ガラクトシダーゼ、ルシフェラーゼおよび単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)を含む。治療ポリヌクレオチドの非限定例は、単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)、シトシンデアミナーゼ、カスパーゼ−3および野生型p53を含んでよい。
【0016】
本発明のさらにその他の実施態様では、ICP10の内因性PKドメインの少なくとも一部を置換するポリヌクレオチドは、免疫調節遺伝子、または融合膜糖蛋白質(FMG)をコードするポリヌクレオチドであってよい。本発明で用いるのに適する免疫調節遺伝子の非限定的な例は、IL−2、IL−12またはGM−CSF、およびその他のサイトカイン類、F42Kおよびその他のサイトカイン類縁体、あるいはMIP−1、MIP−1ベータ、MCP−1、RANTESおよびその他のケモカインを含む。本発明で用いるのに適する融合膜糖蛋白質をコードするポリヌクレオチドの非限定的な例は、パラミクソウイルスF蛋白質、HIV gp160蛋白質、SIV gp160蛋白質、レトロウイルスEnv蛋白質、エボラウイルスGpまたはインフルエンザウイルスヘムアグルチニン、テナガザル白血病ウイルス(GALV)由来の膜糖蛋白質またはテナガザル白血病ウイルスエンベロープ糖蛋白質C−末端切除形(GALV.fus)を含む。
【0017】
本発明のその他の実施態様では、改変ICP10ポリヌクレオチドは、構成的プロモーターに作動可能結合される。本発明で用いるのに適する構成的プロモーターの非限定的な例は、即時早期サイトメガロウイルス(CMV)プロモーター、SV40早期プロモーター、RSV LTR、ベータチキンアクチンプロモーターおよびHSV0−TKプロモーターを含む。本発明のその他の実施態様では、内因性PKドメイン(またはTMドメイン)の少なくとも一部を置換するポリヌクレオチドは、それに作動可能結合した調節配列を含む。特定の実施態様では、調節配列は、真核細胞中で作動可能であり、さらに別の態様では、癌細胞中で作動可能である。本明細書に記載されている方法および組成物を実際に使用するために有用なプロモーターの非限定的な例は、腫瘍特異性プロモーターおよび/または組織特異性プロモーター、例えば前立腺特異性抗原(PSA)プロモーター、カリクレイン2プロモーターおよびプロベイシンプロモーター(前立腺癌の場合)、L−プラスチンプロモーター(乳腺、卵巣および結腸の癌の場合)、サイログロブリンコアプロモーター(甲状腺癌の場合)、ミッドカインおよびシクロオキシゲナーゼ−2プロモーター(すい臓癌の場合)および大多数の腫瘍の場合のヒトテロメラーゼプロモーター(hTERT)を含んでよい。
【0018】
さらに別の実施態様では、第1の細胞と第2の細胞との間の融合を生成する方法であって、本発明の組成物を第1の細胞に導入することによって第2の細胞膜を第1の細胞膜と融合させる工程を含む方法が提供される。特定の実施態様では、第1の細胞、第2の細胞、または第1の細胞と第2の細胞との両方は、固形腫瘍における悪性細胞などの悪性細胞である。本明細書に記載されている方法および組成物を実際に使用する際に用いるのに適する悪性細胞の非限定的な例は、乳癌細胞、肺癌細胞、皮膚癌細胞、前立腺癌細胞、すい臓癌細胞、結腸癌細胞、脳癌細胞、肝臓癌細胞、甲状腺癌細胞、卵巣癌細胞、腎臓癌細胞、脾臓癌細胞、白血病細胞または骨癌細胞を含んでよい。
【0019】
特定の実施態様では、導入する工程は、さらに、ウイルスをヒトに全身供給することによるなど、ウイルスをヒトに供給する工程と定義される。投与の非限定的な経路は、本明細書に記載されている組成物を、静脈内投与、腫瘍内投与、腹腔内投与またはそれらの任意の組み合わせによって投与することを含んでよい。特定の実施態様では、本組成物を複数の細胞に導入する。
【0020】
追加の実施態様では、ヒトの悪性細胞などの悪性細胞を崩壊させる方法が提供される。この方法は、本発明の組成物を細胞に導入する工程を含み、この導入の後、悪性細胞の膜は別の細胞膜と融合する。
【0021】
別の実施態様では、本発明の組成物を含む哺乳類細胞がある。この哺乳類細胞は、正常リンパ球、マクロファージ、天然キラー細胞または本発明の組成物を腫瘍細胞に送るキャリアとして機能する任意のその他の種類の細胞であってよい。
【0022】
本発明のさらに別の実施態様では、本明細書に記載されている改変HSV−2ウイルスまたはウイルスベクターは、ウイルスに感染した癌細胞中にアポトーシスを誘導する。さらに別の実施態様では、ウイルスに感染していないが、本明細書に記載されている改変HSV−2ウイルスに感染した細胞の周囲の傍観者細胞中にアポトーシスが誘導される。
【0023】
本発明のさらに別の実施態様では、本明細書に記載されているウイルスまたはウイルスベクターは、細胞を溶解させるためのウイルスの効力およびまたは合胞体形成を評価するためのシステムの部分を含む。このシステムは、本明細書に記載されているウイルスまたはベクターと接触した細胞を含む。いくつかの実施態様では、細胞は、原発癌細胞または癌細胞系統由来の細胞などの真核細胞であってよい。その他の実施態様では、細胞は、本明細書に記載されているウイルスまたはベクターにとってホストとして働く原核細胞であってよい。本発明のさらにその他の実施態様では、ウイルスまたはウイルスベクターをさらに含む細胞をインビトロに保持してよい。本発明のさらにその他の実施態様では、ウイルスまたはベクターをさらに含む細胞をマウスなどの動物の中に配置する。本発明のさらにその他の実施態様では、癌細胞をウイルスまたはベクターと接触させて配置する前に、動物中に移植してよい。
【0024】
以上は、以下の本発明の詳細な説明をより良く理解することができるように、本発明の特徴および技術的利点の概要を広く示した。本発明の同じ目的を実行するために他の構造体を改変し、または設計するための基礎として、開示する概念および特定の実施態様を容易に利用し得ることは、当業者には自明である。そのような均等物な構築体は、添付の請求項に示される本発明の技術思想および範囲から逸脱しないことも、当業者には自明である。構成と、動作方法との両方に関して、本発明の特徴であると考えられる新規な特徴、その他の目的および利点とともに、添付の実施例および図と関連させて考慮すれば、以下の説明からよりよく理解されよう。しかし、実施例および図は、それぞれ、例を示し説明するために提供されるにすぎず、本発明の限定の定義としてではないことは明白に理解するべきである。
【発明を実施するための最良の形態】
【0025】
(発明の詳細な説明)
実施例1に記載するHSV−2ウイルス組成物を2006年6月8日にAmerican Type Culture Collection(ATCC)10801 University Blvd. Manassas, VA 20110−2209 USAに寄託した。ATCCは、ブダペスト条約にもとづいて設立された国際的な寄託機関(IDA)である。寄託証明書番号は である。
【0026】
I.定義
本明細書で用いられる用語「単純ヘルペスウイルス」または「HSV」は、ヒトを含む哺乳類に感染する被膜、二十面体、二本鎖DNAウイルスを指す。野生型HSVは、最終分化非分裂細胞と分裂細胞との両方に感染し、内部で複製する。「HSV−2」は、ICP10遺伝子を含むHSV系のメンバーを指す。本明細書で用いられる用語「FusOn−H2」は、本明細書で記載される、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性の欠失したポリペプチドをコードする改変ICP10ポリヌクレオチドを有するHSV−2変異体を指す。
【0027】
本明細書で用いられる用語「細胞膜融合」は、例えば2つの隣接する細胞など、少なくとも2つの細胞の外部膜の融合を指す。
【0028】
本明細書で用いられる用語「融合活性の増強」は、細胞膜融合の増強、増加、強化、増加、拡大またはそれらの組み合わせを指す。
【0029】
本明細書で用いられる用語「殺腫瘍」は、直接的または間接的に、結果として悪性細胞を崩壊させることができる剤の性質を指す。特定の実施態様では、この性質は、悪性細胞膜の別の膜と融合を引き起こすことを含む。
【0030】
本明細書で用いられる用語「選択複製」または「条件付き複製」は、特定の組織(例えば腫瘍)の中で選択的に成長する殺腫瘍ウイルスの能力を指す。
【0031】
本明細書で用いられる用語「合胞体」は、著しく大きな数の融合細胞を含む複数核巨大細胞形成を指す。
【0032】
本明細書で用いられる用語「ベクター」は、細胞の中に導入して自己複製させることができるようにするために、核酸配列を挿入することができるキャリア核酸分子を指す。挿入された核酸配列は、ベクターを導入した細胞に対して異物であるか、または細胞中の配列と相同であるが配列が本来見いだされない宿主細胞核酸中の位置にあるか、どちらの場合にも、「外因性」と呼ばれる。ベクターは、非ウイルスDNAベクターまたはウイルスベクターのどちらであってもよい。ウイルスベクターは、ウイルス蛋白質中にカプセル化され、細胞を感染させることができる。ベクターの非限定的な例は、ウイルスベクター、非ウイルスベクター、裸のDNA発現ベクター、プラスミド、コスミド、人工染色体(例えばYAC)、ファージベクター、陽イオン縮合剤と会合したDNA発現ベクター、リポソーム中にカプセル化されたDNA発現ベクターまたは特定の真核細胞、例えばプロデューサー細胞を含む。特に断らない限り、本明細書で用いられる「ベクター」は、DNAベクターおよびウイルスベクターを指す。当業者は、標準的な組み換え技法によってベクターを構築することが十分にできる。一般的に、これらは、Sambrook et al., Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press (1989)およびその中の引用文献を含む。ウイルス学的考察は、Coen D. M, Molecular Genetics of Animal Viruses in Virology, 2”d Edition, B. N. Fields (editor), Raven Press, N.Y. (1990)およびその中の引用文献にも概説されている。
【0033】
用語「発現ベクター」は、転写することができるRNAをコードする核酸を含む任意の種類の遺伝子の構築体を指す。ある場合には、RNA分子は、次に、蛋白質、ポリペプチドまたはペプチドに翻訳される。他の場合には、例えば、アンチセンス分子またはリボザイムの産生では、これらの配列は翻訳されない。発現ベクターは、特定の宿主細胞中に作動可能結合した暗号配列の転写およびおそらく翻訳に必要な核酸配列を参照するさまざまな「制御配列」を含んでよい。転写および翻訳を支配する制御配列に加えて、ベクターおよび発現ベクターは、他の機能を果し、ならびに下記に記載されている核酸配列を含んでよい。
【0034】
「プロモーター」は、転写の開始および転写の速度が制御される核酸配列の領域となる制御配列である。それは、核酸配列の時間的および空間的転写を開始するかまたは調節するRNAポリメラーゼおよびその他の転写因子などの調節蛋白質および分子が結合する遺伝要素を含んでよい。句「動作有効配置される」、「作動可能結合される」、「制御される」および「転写調節される」は、核酸配列に対してプロモーターがその配列の転写開始および/または発現を制御する正しい機能位置にありおよび/または方向を有することを意味する。非限定的なプロモーターの例は、構成的プロモーター、組織特異性プロモーター、腫瘍特異性プロモーターまたは外因性誘導要素に制御される内因性プロモーターを含む。
【0035】
本明細書で用いられる用語「構成的プロモーター」は、細胞周期全体を通じて時間的に連続して遺伝子またはポリヌクレオチドの発現を推進するプロモーターを指す。構成的プロモーターは、細胞周期全体を通して連続して動作してそれが関連する遺伝子またはポリヌクレオチドの発現を推進する限り、細胞型特異性または組織型特異性であってよい。非限定的な構成的プロモーターの例は、即時早期サイトメガロウイルス(CMV)プロモーター、SV40早期プロモーター、RSV LTR、ベータチキンアクチンプロモーターおよびHSV TKプロモーターを含む。
【0036】
用語「エンハンサー」は、核酸配列の転写活性化の制御に関与するシス活性調節配列を指す。
【0037】
本明細書では、用語「接触する」および「曝露される」は、細胞に適用されると、ウイルス、ウイルスベクター、非ウイルスベクター、DNAベクターまたは任意のその他の治療剤が、単独または組み合わされて、標的細胞に投与されるかまたは標的細胞と直接共存して配置されるプロセスを記述するために用いられる。
【0038】
句「改変ICP10ポリヌクレオチド」は、リボヌクレオチドレダクターゼ(RR)活性を有するがプロテインキナーゼ活性の欠失したICP10ポリペプチドをコードするICP10ポリヌクレオチドを指す。
【0039】
句「リボヌクレオチドレダクターゼ活性」は、ICP10ポリヌクレオチドによってコード化されたポリペプチドのC末端のドメインの、ウイルス複製に必要なデオキシヌクレオチド三リン酸(dNTP)を十分に発生させる能力を指す。
【0040】
句「プロテインキナーゼ活性」は、ICP10ポリヌクレオチドによってコード化されたポリペプチドのアミノ末端ドメインの、Ras/MEK/MAPK経路を活性化することができるセリンおよびスレオニン残基をリン酸化する能力を指す。
【0041】
本明細書で用いられる用語「傍観者腫瘍細胞」は、本明細書に記載されている改変HSV−2ウイルスに感染していないが、本明細書に記載されているウイルスまたはベクターに感染した腫瘍細胞に隣接するかまたは近傍にある腫瘍細胞を指す。
【0042】
本明細書で用いられる用語「抗癌剤」は、例えば、癌細胞を殺し、癌細胞中にアポトーシスを誘導し、癌細胞の成長速度を低下させ、転移の発生率または数を減らし、腫瘍サイズを小さくし、腫瘍成長を阻害し、腫瘍または癌細胞への血液の供給を減らし、癌細胞または腫瘍に対する免疫応答を促進し、癌の進行を妨げるかまたは阻害し、あるいは癌を有する被験者の寿命を伸ばすことによって、被験者の体内の癌に負の影響を及ぼすことができる剤を指す。
【0043】
本明細書で用いられる句「医薬品として」または「薬理学的に許容される」は、動物またはヒトに適切に投与されると、拒否反応、アレルギー反応またはその他の有害な反応を生じさせない分子実体および組成物を指す。句「医薬品として許容されるキャリア」は、任意のおよびすべての溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張剤および吸収遅延剤ならびに類似物を含む。
【0044】
用語「単位用量」は、被験者に用いるのに適する物理的に離散した単位を指す。各単位は、その投与に伴って所望の応答を生み出すと計算された、予め定められた量の治療組成物、すなわち適切な経路および治療計画を指す。
【0045】
本明細書で用いられる用語「有効な」または「治療として有効な」は、症状の悪化を阻むこと、疾患の発症を防ぐこと、疾患の広がりを防ぐこと、疾患の少なくとも1つの症状の回復、あるいはそれらの組み合わせを指す。
【0046】
II.導入
ウイルスは、生細胞の中でしか複製することができず、ウイルスの複製は、通常、特定の細胞信号伝達経路の活性化を必要とする。多くのウイルスは、これらの信号伝達経路を活性化して複製に利用するさまざまな戦略を進化の間に取得した。単純ヘルペスウイルス2型(HSV−2)リボヌクレオチドレダクターゼ(ICP10またはRR1)の大型サブユニットは、セリン/スレオニンプロテインキナーゼ(PK)活性を有する固有アミノ末端ドメインを含む。このPK活性は、細胞Ras/MEK/MAPK経路を活性化することが見いだされた(Smith, et at, (2000) J Virol 74(22): 10417−29)。その結果、このPKドメイン(ICP10 PK)をリボヌクレオチドレダクターゼ遺伝子から削除すると、予め存在する活性化Ras信号伝達経路がない細胞などの、細胞中で自己複製するウイルスの能力がひどく低下する報告された(Smith, et at, (1998) J. Virol. 72(11): 9131−9141)。
【0047】
ここで、本発明者らは、HSV−2のPKドメインを置換および/または改変し、その結果、改変ICP10遺伝子がコードする蛋白質にリボヌクレオチドレダクターゼ活性を持たせるがプロテインキナーゼ活性を欠失させると、ウイルスは、腫瘍細胞(少なくともRas信号伝達経路が腫瘍形成によって構成的に活性化された腫瘍細胞)の中で選択的に複製し、腫瘍細胞を崩壊させることを示す。さらに、本明細書に記載されているICP10ポリヌクレオチドの改変によって、ウイルスは固有融合型になる。すなわち、腫瘍細胞をこのウイルスに感染させると、広範な細胞膜融合(合胞体形成)が誘導される。この性質は、腫瘍細胞に対するウイルスの崩壊力を増大させる。さらに、インビボ研究によると、このウイルスは、局所投与にも全身投与にも極めて安全である。
【0048】
本発明のいくつかの実施態様では、PKドメインの改変は、緑色蛍光遺伝子を発現する遺伝子などのレポーター遺伝子の挿入、および/または即時早期サイトメガロウイルスプロモーターなどの構成的プロモーターによる野生型プロモーター遺伝子の置換を含む。
【0049】
いくつかの実施態様では、ICP10遺伝子のプロテインキナーゼ活性ドメインをコードするポリヌクレオチドに第2のポリヌクレオチドを挿入するか、またはプロテインキナーゼドメインの一部を第2のポリヌクレオチドで置換するかのどちらかによって、HSV−2を遺伝子操作し、これによって、改変ポリヌクレオチドがコードするポリペプチドに、リボヌクレオチドレダクターゼ活性を持たせるがプロテインキナーゼ活性を欠失させる。例えば、第2のポリヌクレオチドは、融合膜糖蛋白質などの糖蛋白質をコードしてよい。本発明の範囲内で用いられる好ましい糖蛋白質は、テナガザル白血病ウイルスエンベロープ融合膜糖蛋白質(GALV.fus)の切断型である。本発明の特定の態様では、本発明の殺腫瘍ウイルスとしてGALV.fusを発現させると、ウイルスの抗癌効果は顕著に増強される。
【0050】
いくつかの実施態様では、本発明の改変HSV−2は、削除などの、ICP10の中の変異を含む。変異によって、ウイルスは細胞融合特性を得る。そのような変異は、ウイルススクリーニング時にランダムに発生させても自然から得てもよく、次に、本明細書に記載され、および/または当分野で既知の手段によって、細胞融合特性を有する潜在的な候補のプールの機能を評価する。融合性表現型をもたらす変異は、点変異、フレームシフト、反転、削除、スプライシング誤変異、転写後処理変異、特定のウイルス糖蛋白質の過剰発現、それらの組み合わせ等であってよい。変異は、特定のHSV−2の配列を解析し、それを既知の野生型配列と比較することによって特定してよい。
【0051】
本発明の改変HSV−2は、例えば、悪性細胞の広がりを阻み、分裂を減少させまたは阻害し、根絶し、発生または増殖を妨げ、またはそれらを組み合わせるなど、悪性細胞の治療に有用である。悪性細胞は、固形腫瘍などの任意の形の癌由来であってよいが、その他の形も治療可能である。本発明の改変HSV−2は、肺、肝臓、前立腺、卵巣、胸、脳、すい臓、睾丸、結腸、頭および頸、黒色腫およびその他の種類の悪性腫瘍の治療に有用である。本発明は、転移段階を含む、癌疾患の任意の段階で悪性細胞を治療するのに有用である。本発明は、単独治療法として、または化学療法、手術、放射線治療および類似法を含む別の治療手段とともに利用してよい。
【0052】
III.改変ICP10ポリヌクレオチド
本発明は、改変ICP10ポリヌクレオチドを有するHSV−2変異体を記載する。改変ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ(PK)活性の欠失したポリペプチドをコードする。ICP10ポリヌクレオチドは、機能PKドメインをコードするために必要な配列の少なくとも1部を削除するか、またはPKドメインをコードする配列の少なくとも1部を第2のポリヌクレオチドで置換するかのどちらかによって改変してよい。突然変異生成、ポリメラーゼ連鎖反応(PCR)、相同組み換え、または当業者に既知の任意のその他の遺伝子操作技法を含む任意の適当な方法を用いて改変ICP10ポリヌクレオチドを生成させることができることは、当業者には自明である。
【0053】
A.突然変異生成
本発明の特定の実施態様では、例えば、さまざまな標準的な突然変異誘発手順のうち任意のものを用いる削除によって、HSV−2ウイルスのICP10配列を変異させる。変異は、ヌクレオチド配列、単一遺伝子または遺伝子のブロックの改変を含んでよい。変異は、単一ヌクレオチドを含んでよく(DNA配列内の単一ヌクレオチド塩基の除去、追加または置換を含む点変異など)、あるいは、多数のヌクレオチドの挿入または削除を含んでよい。変異は、DNA複製の忠実さの誤差などの出来事の結果として自発的に起きてよく、あるいは、化学的または物理的突然変異原への曝露に続いて誘起されてよい。当業者に公知の特定の標的設定法を用いて、変異に部位指向性も付与してよい。
【0054】
B.遺伝子組み換え
本発明の他の実施態様では、遺伝子組換え技法を用いてICP10ポリヌクレオチドを改変し、PKドメインをコードする配列の少なくとも一部を削除または置換する。削除/置換されるPKドメインの領域は、改変ICP10ポリヌクレオチドがコードするポリペプチドに、リボヌクレオチドレダクターゼ活性を維持させるがプロテインキナーゼ活性を欠失させる限り、任意の適当な領域であってよい。ただし、特定の実施態様では、PKドメインを改変すると、8つのPK触媒モチーフ(アミノ酸残基106〜445であるが、PK活性は、アミノ酸残基1〜445と考えられている)の1つ以上、および/または膜貫通(TM)領域、および/または不変のLys(Lys176)に影響を及ぼす。野生型ICP10ポリペプチド配列の例は、SEQ ID No:15(National Center for Biotechnology Information’s GenBank database Accession No.1813262A)に提供される。ICP10ポリペプチドをコードする野生型ポリヌクレオチドの例は、SEQ ID No:17に提供される。
【0055】
特定の実施態様では、PK活性に必要なPKドメインをコードする配列の一部を単に削除することによって、ICP10ポリヌクレオチドを改変する。PKドメインをコードする配列の少なくともいくつかが欠失したICP10ポリヌクレオチドの例は、SEQ ID No:18に提供される。別の実施態様の例では、SEQ ID:19に提供されるように、PKドメイン全体が削除されるように、ICP10ポリヌクレオチドを改変する。SEQ ID No:18とSEQ ID No:19との両方が、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くポリペプチドをコードするので、両方が、本明細書に記載されているHSV−2変異体を発生させるため用いるのに適する。特定の本発明の実施態様では、SEQ ID No:18またはSEQ ID No:19に開示されている改変ICP10ポリヌクレオチドは、内因性HSV−2プロモーターに制御されてよく、あるいは、SEQ ID No:20に記載されている即時早期サイトメガロウイルスプロモーターなどの構成的プロモーターに作動可能結合されてよい。
【0056】
本発明のさらに別の実施態様では、PKドメインをコードする配列の少なくとも一部を、緑色蛍光蛋白質など、ICP10ポリヌクレオチドのRRドメインをコードする配列のフレーム内に配置された、第2のポリヌクレオチドで置換することによって、ICP10ポリヌクレオチドを改変する。この構築体は、内因性HSV−2プロモーターに制御されてもよく、CMVプロモーター(SEQ ID NO:20)などの構成的プロモーターに制御されてもよい。実施例1に、この後者の構築体(GFP置換ポリヌクレオチドおよびCMVプロモーターを含む)をさらに詳しく記載する。
【0057】
本発明の別の態様では、HSV−2の内因性ICP10のプロテインキナーゼ活性ドメインの少なくとも一部を置換するポリヌクレオチドは、ウイルス融合膜糖蛋白質(FMG)などの細胞膜融合誘導性ポリペプチドの少なくとも融合性部分をコードすることができる。このポリペプチドは、好ましくは、例えば、実質的に中性pH(pH約6〜8など)で細胞膜融合を誘導することができる。
【0058】
特定の実施態様では、FMGは、MLV(例としてはSEQ ID NO:6)またはGALV(例としてはSEQ ID NO:5)などの、C−型レトロウイルスエンベロープ蛋白質由来の少なくとも融合性ドメインを含む。細胞質ドメインの一部、ほとんど、またはすべてを削除したレトロウイルスエンベロープ蛋白質は、ヒト細胞の場合、そのような操作が超融合活性を生む結果となるので有用である。いくつかの実施態様では、特定の改変をウイルス膜糖蛋白質中に導入して細胞膜融合を誘導する機能を増強する。例えば、複数のレトロウイルスおよびヘルペスウイルス糖蛋白質の細胞質ドメインを切断すると、融合活性が増加し、ときには、同時にウイルス粒子に組み込まれる効率が低下することが示された(Rein et al., (1994) J Virol 68(3): 1773−81)。
【0059】
細胞膜融合ポリペプチドのいくつかの例は、はしかウイルス融合蛋白質(SEQ ID NO:7)、HIV gp160(SEQ ID NO:8)およびSIV gp160(SEQ ID NO:9)蛋白質、レトロウイルスEnv蛋白質(SEQ ID NO:10)、エボラウイルスGp(SEQ ID NO:11)およびインフルエンザウイルスヘムアグルチニン(SEQ ID NO:12)を含む。
【0060】
その他の実施態様では、第2の機能ポリヌクレオチドをPKドメインの中に挿入するか、または用いてPKドメインの一部または全体を置換してよい。この第2の機能ポリヌクレオチドは、免疫調節剤または他の治療剤をコードしてよい。これらの追加の剤は、細胞表面受容体およびGAP接合の上方調節、細胞増殖抑制剤および分化剤に影響を及ぼし、細胞接着を阻害し、または、悪性細胞のアポトーシス感受性を増加させると考えられる。免疫調節性または他の治療薬剤をコードするポリヌクレオチドの非限定的な例は、腫瘍壊死因子;インターフェロン、アルファ、ベータ、ガンマ;インターロイキン−2(IL−2)、IL−12、顆粒球マクロファージコロニー刺激因子(GM−CSF)、F42K、MIP−1、MIP−1β、MCP−1、RANTES、単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)、シトシンデアミナーゼおよびカスパーゼ−3を含む。
【0061】
本発明のさらに別の実施態様では、レポーター蛋白質をコードするポリヌクレオチドの挿入によってICP10ポリヌクレオチドを改変する。非限定的なレポーター蛋白質をコードするポリヌクレオチドの例は、緑色蛍光蛋白質、強化緑色蛍光蛋白質、β−ガラクトシダーゼ、ルシフェラーゼおよびHSV−tkを含む。
【0062】
C.リボヌクレオチドレダクターゼ活性アッセイ
RRの生物学的活性は、既に説明した方法(Averettら、J. Biol. Chem. 258:9831−9838 (1983)およびSmith et al., J. Virol. 72:9131−9141 (1998))に以下の変更を施して検出することができる。BHK細胞を、最初、完全GMEM(10%FBSを含む)中で密生させて成長させた後、0.5%FBS EMEM中で3日間培養し、続いて、20pfuの野生型HSV、HSV−2変異体に感染させ、または偽感染させた。感染の20時間後に細胞を集め、500μlのHD緩衝液[100mMのHEPES緩衝液(pH7.6)、2mMのジチオスレイトール(DTT)]に再び懸濁させ、氷の上で15分間培養した後、30秒間超音波処理した。遠心分離(16,000g、20分間、4℃)によって細胞破片を除去し、上清を45%飽和の結晶硫酸アンモニウム(0.258g/ml)で沈殿させる。2回目の遠心分離(16,000g、30分間)の後、ペレットを100μlのHF緩衝液に溶解させ、50μlを取って等しい体積の2×反応緩衝液(400mMのHEPES緩衝液(pH8.0)、20mMのDTTおよび0.02mM[3H]−CDP(24Ci/mmol、Amersham, Chicago, Il)と混合する。100mMのヒドロキシ尿素と10mMのEDTA(pH8.0)とを加え、3分間沸騰させて反応を停止させる。次に1mlのCrotalux atrox毒(Sigma, St. Louis, MO)を加え、37℃で30分間培養した後、さらに3間沸騰させる。次に、この溶液を0.5mlのダウエックス−1ホウ酸塩カラムに通し、2mlの水で試料を溶出させ、4つの溶出画分に集めてバイオフルオール(Biofluor)(New England Nuclear, Boston, MA)と混合した後、シンチレーション計測する。リボヌクレオチドレダクターゼ活性を単位/mg蛋白質で表す。1単位は、1nmol[3H]CDPのdCDP/hr/mg蛋白質への変換を表す。
【0063】
D.プロテインキナーゼ活性アッセイ
改変ICP10ポリヌクレオチドがプロテインキナーゼ活性の欠失したポリペプチドをコードするか測定するために、改変ICP10ポリヌクレオチドまたは野生型HSV−2を有するHSV−2(moi=200、感染後16時間)に感染させた細胞の抽出分を抗LA−1抗体で免疫沈降させ、Chung et al. J. Virol. 63:3389−3398, 1998および米国特許第6,013,265号に記載されているPKアッセイに付す。通常、BCA蛋白質アッセイキット(PIERCE, Rockford IL.)を用いて細胞抽出物の免疫沈降物の蛋白質濃度を規格化し、20mMのTrid−HCl(pH7.4)、0.15MのNaClを含むTS緩衝液で洗浄し、20mMのTris−HCl(pH7.4)、5mMのMgCl2、2mMのMnCl2、10μCiの[32P]ATP(3000Ci/ミリモル、DuPont, New England Research Prod.)からなる50μlのキナーゼ反応緩衝液に懸濁させ、30℃で15分間培養する。ビーズを1mlのTS緩衝液で1回洗浄し、100μlの変性溶液に再懸濁させ、5分間沸騰させる。次に、7%ポリアクリルアミドゲルの上のSDS−PAGEによって蛋白質を分割する。次に、既に記載した(Aurelian et. al., Cancer Cells 7:187−191 1989)ように、蛋白質をニトロセルロース膜上に電気移動させ、特異抗体を用いるンキュベーションによって免疫ブロットした後、室温で1時間蛋白質A−ペルオキシダーゼ(Sigma, St. Louis, MO)処理した。Smith et al., Virol. 200:598−612(1994)に記載されているECL試薬(Amersham, Chicago, IL)によって検出を行ってよい。
【0064】
IV.ベクター構築。
【0065】
本発明は、ICP10配列の少なくとも一部を置換または削除し、これによって、改変ICP10ポリヌクレオチドによってコード化される蛋白質に、リボヌクレオチドレダクターゼ活性を持たせるがプロテインキナーゼ活性を欠失させ、特定の実施態様では、構成的プロモーターなどの調節配列をさらに含む、HSV−2ベクターを目的とする。いくつかの実施態様では、組成物は、改変ICP10遺伝子を含む裸のDNAベクター(非ウイルス)であり、他の実施態様では、組成物は、改変ICP10遺伝子を有する組み換えHSV−2である。裸のDNAベクターと組み換えウイルスとの両方は、以下の成分のいくつかまたはすべてをさらに含んでよい。
【0066】
A.ベクター
上記で定義したように、ベクターは、プラスミド、コスミド、ウイルス(バクテリオファージ、動物ウイルスおよび植物ウイルス)ならびに人工染色体(例えばYACs)を含むがそれらに限定されない。当分野では、設計されたウイルスおよびDNAベクターを構築するための方法が知られている。通常、これらは、Sambrooet al., Molecular Cloning:A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press(1989)およびその引用文献である。ウイルス学的考察は、Coen D. M., Molecular Genetics Animal Viruses in Virology, 2.sup.nd Edition, B. N. Fields (Editor), Raven Press, N.Y. (1990)およびその引用文献でも概説されている。
【0067】
発現ベクターは、さまざまな「制御配列」を含んでよい。制御配列は、特定の宿主細胞中に作動可能結合した暗号配列の転写およびおそらく翻訳に必要な核酸配列を参照する。転写および翻訳を支配する制御配列に加えて、DNAベクター、発現ベクターおよびウイルスは、他の機能を果し、ならびに下記に記載されている核酸配列を含んでよい。
【0068】
1.プロモーターおよびエンハンサー
一般に、プロモーターは、RNA合成の開始部位の位置を決めるために機能する配列を含む。これの最もよく知られた例は、TATAボックスであるが、TATAボックスが欠失したいくつかのプロモーター(例えば哺乳類の末端デオキシヌクレオチジルトランスフェラーゼ遺伝子のプロモーターおよびSB40後期遺伝子のプロモーター)では、開始部位自体の上にある離散的な要素が、開始の位置を決めるのを助ける。追加のプロモーター要素が転写開始の頻度を調節する。一般に、これらは、開始部位の上流の領域30から110に配置されるが、複数のプロモーターが開始部位の下流にも機能要素を含んでいることが示された。コード化配列をプロモーターに「制御される」ようにするために、転写解読フレームの転写開始部位の5’−末端を、選ばれたプロモーターの「下流」(すなわち3’−側)に配置する。「上流」のプロモーターは、DNAの転写を刺激し、コード化されたRNAの発現を促進する。
【0069】
プロモーター要素の間の間隔は、頻繁に柔軟であり、その結果、要素が反転しても、互いに移動してもプロモーター機能は保たれる。tkプロモーターにおいて、プロモーター要素の間の間隔は、50bpの間隔まで増加させないと活性は低下し始めない。プロモーターによって、個々の要素は、協力してまたは独立に機能して、転写を活性化することができるようである。プロモーターは、エンハンサーとともに用いてもよくてまたは用いなくてよい。
【0070】
プロモーターは、コード化セグメントおよび/またはエキソンの上流に位置する5’非コード化配列を単離することによって得られるように、核酸配列と自然に会合するものであってよい。同様に、エンハンサーは、その配列の下流または上流のどちらかに位置する核酸配列と自然に会合するものであってよい。あるいは、コード化核酸セグメントを組み換えプロモーターまたは非相同プロモーターに制御される位置に配置することによって、特定の利点が得られる。非相同プロモーターは、その自然環境では、通常、核酸配列と会合していないプロモーターを指す。組み換えエンハンサーまたは非相同エンハンサーは、その自然環境では正常な核酸配列と会合していないエンハンサーを指す。そのようなプロモーターまたはエンハンサーは、他の遺伝子のプロモーターまたはエンハンサー、任意の他のウイルス、原核生物細胞または真核生物細胞から単離されたプロモーターまたはエンハンサー、および「天然」でない、すなわち、さまざまな転写調節領域のさまざまな要素および/または発現を変える変異を含むプロモーターまたはエンハンサーを含む。例えば、組み換えDNA構築に最も普通に用いられるプロモーターは、βラクタマーゼ(ペニシリナーゼ)、ラクトースおよびトリプトファン(trp)プロモーター系を含む。プロモーターおよびエンハンサーの核酸配列を合成によって作り出すことに加えて、PCRを含む組み換えクローン形成および/または核酸増幅技術を本明細書に開示されている組成物とともに用いて配列を作り出してよい(米国特許第4,683,202号および第5,928,906号を参照のこと)。さらに、ミトコンドリア、クロロプラストおよび類似物などの非核オルガネラ中の配列の転写および/または発現を指令す制御配列も使用することができると考えられる。
【0071】
もちろん、発現のために選ばれたオルガネラ、細胞型、組織、器官または生物中のDNAセグメントの発現を効果的に導くプロモーターおよび/またはエンハンサーを使用することは重要になる。使用されるプロモーターは、導入されたDNAセグメントの高レベル発現を導く適切な条件下で、組み換え蛋白質および/またはペプチドの大量産生に有利であるように、構成的、組織特異的、誘導可能、および/または有用であってよい。プロモーターは、非相同性であってよく、または内因性であってよい。
【0072】
さらに、任意のプロモーター/エンハンサーの組み合わせを用いて発現を推進してよい。T3、T7またはSP6細胞質発現系を用いることは、別の可能な実施態様である。真核細胞は、適切な細菌ポリメラーゼが、投与複合体の一部または追加の遺伝子発現構築体として提供されるなら、特定の細菌プロモーターからの細胞質転写を支援することができる。
【0073】
組織特異性プロモーターまたは要素の同定、ならびに活性の特性を把握するアッセイは、当業者に公知である。そのような領域の非限定的な例は、ヒトLIMK2遺伝子(Nomoto et al. (1999) Gene 236(2): 259−271)、ソマトスタチン受容体−2遺伝子(Kraus et al., (1998) FEBS Lett. 428(3): 165−170)、マウス精巣上体レチノイン酸結合遺伝子(Lareyre et al., (1999) J. Biol. Chem. 274(12): 8282−8290)、ヒトCD4 (Zhao−Emonet et al., (1998) Biochem. Biophys. Aeta,1442(2−3): 109−119)、マウスα−2(XI)コラーゲン(Tsumaki, et at, (1998), J. Biol. Chem.273(36): 22861−4)、DIAドーパミン受容体遺伝子(Lee, et at, (1997), DNA Cell Biol. 16(11): 1267−1275)、インスリン様増殖因子II (Wu et at, (1997) Biophys Biochem Res. Comm. 233(1): 221−226)およびヒト小板内皮細胞接着分子−1(Almendro et at, (1996) J. Immunol. 157(12): 5411−5421)を含む。
【0074】
2.開始信号および内部リボソーム結合部位
コード化配列の効率的な翻訳には、特定の開始信号も必要になることがある。これらの信号は、ATG開始コドンまたは隣接配列を含む。ATG開始コドンを含む外因性翻訳制御信号を提供する必要がある。当業者ならこれを測定し、必要な信号を提供することは容易にできる。開始コドンは、挿入部分全体の翻訳を確実にするために、所望の暗号配列の解読フレームの「フレーム内」になければならないことは周知である。外因性翻訳制御信号および開始コドンは、天然であっても合成であってもよい。適切な転写エンハンサー要素をふくむことによって、発現の効率が増強されることがある。
【0075】
本発明の特定の実施態様では、内部リボソーム進入部位(IRES)要素を用いて、多重遺伝子メッセージ、または多シストロン性メッセージを作り出す。IRES要素は、5’メチル化キャップ依存性翻訳のリボソーム走査モデルを省略し、内部部位で翻訳を開始することができる。IRES要素は、非相同性開放読み取りフレームに結合することができる。IRESによってそれぞれ分離された複数の開放読み取りフレームを一緒に転写して、多シストロン性メッセージを作り出すことができる。IRES要素のおかげで、各開放読み取りフレームは、効率的な翻訳のためにリボソームにアクセスすることができる。単一のメッセージを転写する単一のプロモーター/エンハンサーを用いて、複数の遺伝子を効率的に発現することができる(米国特許第5,925,565号および第5,935,819号を参照すること)。
【0076】
3.終止シグナル
本発明のベクターまたは構築体は、通常、少なくとも1つの終止シグナルを含む。「終止シグナル」または「ターミネーター」は、RNAポリメラーゼによるRNA転写物の特定の終了に関与するDNA配列で構成される。従って、特定の実施態様では、RNA転写体の産生を終了する終止シグナルを包含する。望ましいメッセージレベルを実現するために、in vivoでターミネーターが必要になることがある。
【0077】
真核生物系では、ターミネーター領域は、ポリアデニル化部位を露出させるように新しい転写産物を部位特異的に開裂させることができる特定のDNA配列も含んでよい。これによって、専用の内因性ポリメラーゼに、転写体の3’末端に約200A残基の並び(ポリA)を加えるように信号を伝達する。このポリA尾部で改変されたRNA分子は、安定性が増し、より効率的に変換されるようである。従って、真核生物が関与する他の実施態様では、ターミネーターは、RNAの開裂のための信号を含み、ターミネーター信号は、メッセージのポリアデニル化を促進すると考えられる。ターミネーターおよび/またはポリアデニル化部位要素は、メッセージレベルを改良し、カセットから他の配列への読み通しを最小限にするように働くことができる。
【0078】
本発明において用いられるターミネーターは、本明細書に記載されている任意の既知の転写ターミネーター、または当業者に既知の転写ターミネーターを含み、例えばウシ成長ホルモンターミネーターまたは例えばSV40ターミネーターなどのウイルス終止配列などの遺伝子の終結配列を例えば含むが、それらに限定されない。特定の実施態様では、終止シグナルは、配列の切断によるなどの転写可能配列または翻訳可能配列の欠失であってよい。
【0079】
4.ポリアデニル化シグナル
発現、特に真核生物の発現では、一般に、転写体の適切なポリアデニル化を実現するために、ポリアデニル化シグナルを含む。ポリアデニル化シグナルの性質は、本発明の実施を成功させる上で不可欠とは考えられず、任意のそのような配列を使用してよい。好ましい実施態様は、SV40ポリアデニル化シグナルまたはウシ成長ホルモンポリアデニル化シグナルを含む。これらはともに簡便であり、さまざまな標的細胞中で良好に機能することが知られている。ポリアデニル化は、転写体の安定性を増し、または細胞質輸送を促進することがある。
【0080】
5.選択可能および検索可能マーカー
本発明の特定の実施態様では、本発明の核酸構築体を含む細胞は、発現ベクターにマーカーを含ませることによってインビボまたはインビトロで特定してよい。そのようなマーカーによって、特定することができる変化を細胞に付与し、発現ベクターを含む細胞を容易に特定することが可能になる。通常、選択可能なマーカーは、選抜を可能にする属性を付与するものである。正の選択可能マーカーは、マーカーの存在がその選択を可能にするものであり、負の選択可能マーカーは、その存在がその選択を妨げるものである。正の選択可能マーカーの1例は、薬剤耐性マーカーである。
【0081】
通常、薬物選択マーカーを含むと、クローン形成および変換体の特定が容易になる。例えば、ネオマイシン、ピューロマイシン、ハイグロマイシン、DHFR、GPT、ゼオシンおよびヒスチジノールへの抵抗性を付与する遺伝子は、有用な選択可能マーカーである。諸条件の実体化によって変換体の差別化を可能にする表現型を付与するマーカーに加えて、比色分析を土台とするGFPなどの選抜可能マーカーを含むその他の種類のマーカーも包含される。あるいは、単純ヘルペスウイルスチミジンキナーゼ(tk)またはクロラムフェニコールアセチルトランスフェラーゼ(CAT)などの選抜可能酵素を利用してよい。当業者には、どのように免疫学的マーカーを、おそらく蛍光活性化細胞分類(FACS)分析とともに、使用するかも既知と考えられる。遺伝子産物をコードする核酸と同時に発現することができる限り、用いられるマーカーは重要ではないと考えられる。選択可能マーカーおよび選抜可能マーカーのさらに別の例が当業者に公知である。
【0082】
ベクターは、適当な方法によって、初期感染細胞に導入される。本発明において用いられる、オルガネラ、細胞、組織または生物を変換するために核酸を供給するためのそのような方法は、事実上、本明細書に記載されているかまたは当業者に既知の、核酸(例えばHSVベクター)をオルガネラ、細胞、組織または生物中に導入することができる任意の方法を含むと考えられる。非限定的な方法の例は、エクスビボの形質移入によるDNAの直接投与、注射(米国特許第5,994,624号、第5,981,274号、第5,945,100号、第5,780,448号、第5,736,524号、第5,702,932号、第5,656,610号、第5,589,466号および第5,580,859号)、マイクロインジェクション(米国特許第5,789,215号)、エレクトロポレーション(米国特許第5,384,253号)、リン酸カルシウム沈殿、DEAEデキストランおよびその後のポリエチレングリコール、直接超音波投与、リポソーム媒介形質移入、受容体媒介形質移入、微粒子銃(国際特許出願第94/09699号および第95/06128号、米国特許第5,610,042号、第5,322,783号、第5,563,055号、第5,550,318号、第5,538,877号および第5,538,880号)、炭化ケイ素繊維による撹拌(米国特許第5,302,523号および第5,464,765号)、アグロバクテリウム媒介変換(米国特許番号5,591,616号および第5,563,055号)、プロトプラストのPEG媒介変換(米国特許第4,684,611号および第4,952,500号)、除湿/阻害媒介DNA取り込み、およびこれらの方法の任意の組み合わせ、または当業者に既知のその他の方法を含む。組成物は、医薬品として許容される賦形剤中で、静注などの全身投与法によって、哺乳類の細胞に供給してもよい。
【0083】
B.細胞へのDNAベクター供給の方法
1.エクスビボ変換
生物から取り出した細胞および組織にエクスビボ環境で形質移入するための方法が当業者に知られている。従って、本発明において、細胞または組織を取り出し、本明細書に記載されている核酸および組成物を用いてエクスビボで形質移入してよいとみなされる。特定の態様では、形質移入された細胞または組織を生物中に配置してよい。いくつかの実施態様では、形質移入された細胞または組織の中で核酸を発現させる。
【0084】
2.注射
特定の実施態様では、核酸は、例えば、皮下、皮膚内、筋肉内、静脈内、腹腔内等などの1回以上の注射(すなわち針注射)によって、オルガネラ、細胞、組織または生物に投与してよい。注射の方法は、当業者に公知である(例えば食塩水を含む組成物の注射)。本発明のさらに別の実施態様は、直接マイクロインジェクションによる核酸の導入を含む。用いられる本発明の組成物の量は、影響を受ける細胞、組織または生物の性質によって変化させてよい。
【0085】
3.エレクトロポレーション
本発明の特定の実施態様では、エレクトロポレーションによってオルガネラ、細胞、組織または生物の中に核酸を導入する。エレクトロポレーションは、細胞およびDNAの懸濁液の高電圧放電への曝露を含む。この方法のいくつかの変化形では、ペクチン分解酵素などの特定の細胞壁分解酵素を使用して受け入れ標的細胞を非処理細胞よりエレクトロポレーションによる変換に対して敏感にする(米国特許第5,384,253号)。あるいは、受け入れ細胞を機械的損傷による変換に対してより敏感にしてよい。
【0086】
4.リポソーム媒介トランスフェクション
本発明のさらに別の実施態様では、改変ICP10ポリヌクレオチドを有するベクターなどの本明細書に記載されている組成物を、例えばリポソームなどの脂質複合体の中に捕捉してよい。リポソームは、リン脂質2分子膜と内部の水性媒質とを特徴とするベシクル構造体である。多重ラメラリポソームは、水性媒質で分離された複数の脂質層を有する。それらは、リン脂質を過剰の水溶液の中に懸濁させると自発的に形成される。脂質成分は、自己再配置を行ってから閉鎖構造体を形成し、水および溶解した溶質を脂質2重層の間に捕捉する。リポフェクタミン(Gibco BRL)またはスーパーフェクト(Qiagen)と複合体を形成した核酸も包含される。
【0087】
本発明の特定の実施態様では、リポソームは、ヘマグルチナチンウイルス(HVJ)と複合体を形成させてよい。こうすると、細胞膜との融合が促進され、リポソームカプセル化DNAの細胞への進入が促進されることが示された(Kaneda et al., (1989) Science 20;243(4889): 375−8)。その他の実施態様では、リポソームを核非ヒストン染色体蛋白質(HMG1)と複合体形成させるかまたはともに使用してよい(Kato et al., (1991) JBiol Chem. (1991) Feb 25;266(6): 3361−4)。さらにまた別の実施態様では、リポソームをHVJとHMG1との両方と複合体形成させてよく、またはHVJとHMG1との両方とともに使用してよい。その他の実施態様では、投与用ベヒクルは、リガンドおよびリポソームを含んでよい。
【0088】
5.受容体媒介形質移入
核酸は、受容体媒介投与ベヒクルによって標的細胞に投与してよい。この手法は、受容体媒介エンドサイトーシスによる巨大分子の選択的取り込みを利用する。さまざまな受容体の細胞型特異的分布からみると、この投与方法は、別の度合いの特異性を本発明に加える。
【0089】
特定の実施態様では、受容体媒介遺伝子標的化ベヒクルは、受容体特異性リガンドおよび核酸結合剤を含む。他の実施態様は、投与される核酸を動作有効結合した受容体特異性リガンドを含む。欧州特許EPO 0273 085に記載されている、遺伝子をへん平上皮がん細胞に供給するために用いられた上皮細胞成長因子(EGF)を含むいくつかのリガンドが、受容体媒介遺伝子移入のために、用いられてきた。
【0090】
その他の実施態様では、細胞特異性核酸標的化ベヒクルの核酸投与ベヒクル成分は、リポソームと組み合わされた特異的結合リガンドを含んでよい。投与される核酸(単数または複数)をリポソームの中に収容し、特異的結合リガンドをリポソーム膜の中に官能基によって組み込む。従って、リポソームは、標的細胞の受容体(単数または複数)に特異的に結合し、内容物を細胞に供給する。
【0091】
さらに別の実施態様では、標的化投与ベヒクルの核酸供給ベヒクル成分は、リポソーム自体であってよく、好ましくは、細胞特異的結合を導く1つ以上の脂質または糖蛋白質を含む。例えば、ガラクトース末端アシアルガングリオシドであるラクトシルセラミドをリポソームの中に組み込み、肝細胞によるインシュリン遺伝子の取り込みの増加が観測された(Nicolau et al., (1987) Methods Enzymol. 149:157−76)。本発明の組織特異的変換構築体は、同様な方法で標的細胞の中に特異的に投与することができると考えられる。
【0092】
6.微粒子銃
微粒子銃技法を用いて、核酸を少なくとも1つのオルガネラ、細胞、組織または生物中に導入することができる(米国特許第5,550,318号、米国特許第5,538,880号、米国特許第5,610,042号、および国際特許出願第94/09699号)。この方法は、DNAで被覆されるかまたはDNAを含むマイクロ発射体を、細胞を死滅させないで細胞膜を突き通し、細胞に入ることができる高速度に加速する能力に依拠する。マイクロ発射体は、タングステン、白金または金など、任意の生物学的に不活性な物質で構成してよい。爆撃のために、懸濁液の中の細胞は、フィルタまたは固体培地上で濃縮される。あるいは、未成熟胚またはその他の標的細胞を、固体培地上に配置してよい。爆撃される細胞は、固定プレート上の微粒子銃デバイスの下の適切な距離に配置する。本発明の実施に有用な非常に広範な微粒子銃技法が当業者に既知である。
【0093】
C.宿主細胞
本明細書で用いられる用語「細胞」、「細胞系統」および「細胞培養物」は、区別なく用いてよい。これらの用語はすべて、それらの子孫を含む。子孫は、任意のおよびすべての後継世代である。すべての子孫は、故意または不慮の変異によって、同一でないことがあると理解される。非相同核酸配列を発現する場合、「宿主細胞」は、原核生物または真核生物の細胞を指し、ベクターを自己複製し、および/またはベクターによってコード化される非相同遺伝子を発現することができる任意の変換可能な生物を含む。宿主細胞は、ベクターの受け入れ体として用いることができ、用いられてきた。宿主細胞は「形質移入され」または「変換され」てよい。これは、外因性核酸が宿主細胞の中に移入または導入されるプロセスを指す。変換細胞は、被験者一次細胞およびその子孫を含む。本明細書で用いられる用語「設計された」および「組み換え」細胞または宿主細胞は、例えばベクターなどの外因性核酸配列が導入された細胞を指すものとする。従って、組み換え細胞は、組み替えによって導入された核酸を含まない天然細胞と区別することができる。
【0094】
組織は、細胞膜融合発生HSV−2変異体によって変換される単数または複数の宿主細胞を含んでよい。組織は、生物の一部分であってよく、または生物から分離されてよい。特定の実施態様では、組織は、脂肪細胞、歯槽、エナメル芽細胞、神経、基底細胞、血液(例えばリンパ球)、血管、骨、骨髄、グリア細胞、胸、軟骨、頸部、結腸、角膜、胎生期、子宮内膜、内皮、上皮、食道、色帯、繊維芽細胞、小胞、神経節細胞、グリア細胞、杯状細胞、腎臓、肝臓、肺、リンパ節、筋肉、ニューロン、卵巣、すい臓、末しょう血液、前立腺、皮膚、小腸、脾臓、幹細胞、胃、睾丸、およびそれらのすべての癌を含んでよいが、それらに限定されない。
【0095】
特定の実施態様では、宿主細胞または組織は、少なくとも1つの生物の一部であってよい。特定の実施態様では、生物は、当業者には自明のように、原核生物(例えば真正細菌、古細菌)または真核生物であってよいが、それらに限定されない。
【0096】
多数の細胞系統および培養物が宿主細胞として用いるために利用可能であり、American Type Culture Collection(ATCC)などの組織によって市販されている。当業者は、ベクター主鎖および所望の結果によって適切なホストを決定することができる。ベクター複製および/または発現に利用できる細胞型の非限定的な例は、大腸菌(例えば大腸菌株RR1、LE392、B、X1776(ATCC第31537号)、W3110、F、ラムダ、DH5α、JM109およびKC8)、桿菌、例えば枯草菌などの細菌、その他の腸内細菌科、例えばサルモネラチフィムリウム、セラチアマルセッセンス、ならびに複数の市販の細菌宿主およびSURE(登録商標)コンピテント細胞およびソロパック(登録商標)(SOLOPACKT)ゴールド細胞(Gold Cell)(ストラタジーン(登録商標)(STRATAGENE, La Jolla, CA)などのコンピテント細胞を含む。ベクターの複製および/または発現のための真核生物宿主細胞の非限定的な例は、HeLa、NIH3T3、Jurkat、293、Cos、CHO、SaosおよびPC12を含む。
【0097】
いくつかのベクターは、原核生物細胞と真核生物細胞との両方の中で複製および/または発現することを可能にする制御配列を使用してよい。さらに、上記に記載した宿主細胞をすべて培養して維持し、ベクターの複製を可能にする条件は、当業者には自明と考えられる。ベクターの大量産生、ならびにベクターによってコード化される核酸および同種ポリペプチド、蛋白質またはペプチドの産生を可能にする技法および条件も自明であり、既知である。
【0098】
D.ウイルスベクターパッケージングおよび増殖。
【0099】
1.ウイルスパッケージング
本発明の特定の実施態様では、ICP10遺伝子を改変した後、相同組み換えによってウイルスに挿入する。通常、改変ICP10遺伝子を含むプラスミドDNAと精製HSV−2ゲノムDNAとをリポフェクタミンを用いてベロ細胞に共移入することによってこれを実行する。次に、組み換えウイルスを特定し(通常、ウイルスプラークを検索して選択可能マーカーの有無を調べて)、改変ICP10ポリヌクレオチドを含むプラークを選択する。次に、組み換えウイルス選択したら、インビトロでキャラクタリゼーションして改変ICP10遺伝子がHSV−2ゲノム中に正しく挿入され、元のICP10遺伝子を置換したことを確認する。実施例1および2に、ウイルスパッケージングおよびインビトロのキャラクタリゼーションをさらに詳細に記載する。
【0100】
2.ウイルス原料の調製
組み換えHSV−2変異体ウイルスを選択したら、以下のようにウイルス原料を調製する。10%ウシ胎児血清(FBS)中でベロ細胞を成長させ、細胞あたり0.01プラーク形成単位(pfu)に感染させる。次に、2日後、凍結、解凍および超音波処理を繰り返して、細胞からウイルスを集める。ウイルスを集めたら、記載された(Nakamoriら、(2003) Clinical Cancer Res. 9(7): 2727−2733)ように精製する。ウイルスを精製したら、滴定(実施例10に記載されている)し、等分し、使用するまで−80℃で保管する。
【0101】
E.蛋白質発現系
例えば、本発明のDNAベクター組成物の発生において蛋白質発現系を利用して、改変ICP10ポリヌクレオチドによってコード化されたポリペプチドを機能研究のために発現させてよい。上記で考察した組成物の少なくとも一部または全体を含む多数の発現系が存在する。原核生物および/または真核生物による系を本発明とともに使用して核酸配列または同種ポリペプチド、蛋白質およびペプチドを産生させてよい。多くのそのような系が市販され、広く利用可能である。
【0102】
米国特許第5,871,986号および第4,879,236号に記載され、市販されている(例えば、CLONTECH, Inc. Mountain View, CA)ものなどの昆虫細胞/バキュロウイルス系は、高水準の非相同核酸セグメントの蛋白質発現を行うことができる。
【0103】
市販発現系のその他の例は、合成エクジソン誘導性受容体を含む誘導性哺乳類発現系、pET発現系または大腸菌発現系(STRATAGENE, LaJolla, CA);テトラサイクリン調節発現系、全長CMVプロモーターまたはメチロトローフ酵母ピキアメタノーリカ(INVITROGEN, Carlsbad, CA)中の高レベルの組み換え蛋白質の産生用に設計された酵母発現系を用いる誘導性哺乳類発現系を含む。
【0104】
本発明の方法によって産生される蛋白質、ポリペプチドまたはペプチドは、「過剰発現」、すなわち、細胞中での天然の発現と比較すると増加したレベルで発現されてよいと考えられる。そのような過剰発現は、放射能標識および/または蛋白質精製を含むさまざまな方法によって評価してよい。しかし、簡単で直接的な方法、例えばSDS/PAGEおよび蛋白質染色またはウェスタンブロッティングとそれに続く、結果として得られたゲルまたはブロットの密度測定掃引などの定量分析法を含むものが好ましい。特定の蛋白質、ポリペプチドまたはペプチドが宿主細胞によって産生される他の蛋白質と比べると相対的に多くなるように、天然細胞中のレベルと比較した組み換え蛋白質、ポリペプチドまたはペプチドのレベルの特定の増加は過剰発現を示し、例えば、ゲル上で目に見える。
【0105】
V.HSV−2変異体の機能役割
本明細書に記載されているHSV−2変異体は、殺腫瘍剤としての複数の機能役割を示す。例えば、本ウイルスは、感染細胞ならびに傍観者細胞の両方において、溶菌ならびに合胞体形成およびアポトーシス誘導によって、腫瘍細胞を崩壊させることができる。さらに、HSV−2変異体によって腫瘍を崩壊させると、強力な抗腫瘍免疫応答を誘導し、抗腫瘍免疫応答が、悪性疾患治療用の殺腫瘍剤としての本変異体ウイルスの治療効力にさらに貢献する。
【0106】
本HSV−2変異体ウイルスは、周期活動中の細胞中で選択複製を示すが、非サイクリング細胞中では示さない。実施例4にさらに詳細に記載するように、本変異体HSV−2は、プロテインキナーゼ活性が欠失しているので、周期活動中の細胞中での成長と比較して周期活動中でない細胞中での成長は少なくとも40分の1に低下する。これとは対照的に、野生型HSV−2の周期活動中の細胞と周期活動中でない細胞との間の増殖特性はわずかに影響を受けるだけである。従って、本明細書に記載の本HSV−2変異体は、腫瘍細胞など、活性化Ras経路を有するサイクリング細胞における殺腫瘍剤として用いるのに優れて適する。
【0107】
本明細書に記載の本改変HSV−2は、他の殺腫瘍ウイルスおよび野生型HSV−2と比較すると、優れた殺腫瘍細胞能力を有する。実施例5に記載するインビトロアッセイを用いて、さまざまな組織起源のヒト腫瘍細胞に対するFusOn−H2の殺細胞能力が、米国特許出願第10/397,635号に記載され、および/または従来検証された殺腫瘍HSV−1の殺細胞能力より有意に強く、親の野生型HSV−2の殺細胞能力を超えることを示す。さらに、実施例6に記載するように、本発明のウイルスの穏やかな投与量(1×106プラーク形成単位)を1回注射したところ、局所塗布によってヌードマウスに確立した胸腺腫瘍が、この動物の100%(n=8)で完全に消失した。一方、殺腫瘍HSV−1を同じ投与量で投与したが、30%より少ないマウスで腫瘍が縮小しただけであった。
【0108】
溶解活性および融合活性に加えて、本HSV−2変異体は、強力なアポトーシス誘導活性も有し、強力な抗腫瘍免疫応答も誘導することができる。本HSV−2変異体は、インビトロ環境で、ウイルスに感染した細胞ならびに感染細胞の周りの非感染傍観者細胞中にアポトーシスを誘導することができる。さらに、本HSV−2変異体は、インビボで、腫瘍細胞のアポトーシスを誘導する効果を有する。実施例8にこれをさらに詳しく記載する。本明細書に記載の組成物は、他の殺腫瘍ウイルスより腫瘍細胞を殺傷する上で有効であるだけでなく、本HSV−2変異体は、強い抗腫瘍免疫応答の誘導によって、原発腫瘍および転移腫瘍に対してインビボで強い治療効果を示す。実施例9に記載するように、FusOn−H2処理マウス由来の養子伝達CTLは、元の腫瘍の成長を阻害し、転移の発達を効果的に防ぐことができる。
【0109】
アポトーシスまたはプログラム化細胞死は、正常な胚発育の必須過程であり、成体組織中の恒常状態を維持し、発癌を抑制する。本発明のいくつかの実施態様では、本改変HSV−2は、本ウイルスに感染した腫瘍細胞と、非感染傍観者腫瘍細胞とのアポトーシスの強力な誘導因子である。例えば、特定の実施態様では、腫瘍細胞を、ICP10遺伝子のプロテインキナーゼドメインの一部を緑色蛍光蛋白質(GFP)コード化遺伝子で置換したHSV−2構築体に感染させた。蛍光顕微鏡下でGFPを可視化することによって感染細胞を識別することができ、クロマチン凝集によって明らかにして、アポトーシスを行う細胞を識別した。GFP発現に対してクロマチン凝集を示す細胞の比は、2.6:1であり、本改変HSV−2に感染しなかった腫瘍細胞のうちのかなりの数がアポトーシスを行うことを示唆した。実施例8に、本発明の殺腫瘍ウイルスのアポトーシスを誘導する能力をさらに詳細に記載する。
【0110】
悪性疾患と戦う際には、強い抗腫瘍免疫応答が有用である。本明細書に記載の本HSV−2変異体は、原発腫瘍および転移腫瘍に対してインビボで強力な抗腫瘍免疫応答を誘導することができる。特定の実施態様では、本変異体HSV−2(FusOn−H2)は、4T1マウス乳腺腫瘍細胞系統を用いるマウス乳腺腫瘍モデル中で選択複製し、腫瘍細胞を溶解させ、強い抗腫瘍免疫応答の誘導によって、インビボで原発腫瘍および転移腫瘍に対する強い治療効果を示した。詳しくは、FusOn−H2で治療したマウス由来の養子伝達細胞毒性リンパ球(CTL)は、元の腫瘍の成長を阻害し、FusOn−H2による治療を受けていないマウスの転移を効果的に防ぐことができる。実施例9に、これをさらに詳細に記載する。
【0111】
VI.医薬品組成物および投与経路
A.総合的考察
本発明の組成物は、改変ICP10遺伝子を有する組み換えHSV−2変異体か、本明細書に記載の改変ICP10遺伝子を有する裸の(非ウイルス)DNAベクターのどちらかを含む医薬品組成物として投与することができる。本発明の組成物は、通常の医薬品調製物を含む。通常、本発明の組成物は、本組成物を医薬品として許容されるキャリアまたは水性媒質に溶解または分散させて、薬学的剤として投与することができる。医薬活性物質のためのそのような媒質および剤の使用は、当分野で公知である。任意の通常の媒質または剤が本発明の組成物と適合する限り、治療組成物中のその使用は考慮範囲内である。他の抗疾患剤などの補助活性成分も、医薬品組成物中に組み込んでよい。本組成物の投与は、その経路によって標的細胞を利用できる限り、任意の普通の経路によってよい。投与経路の例は、口、鼻、舌下、直腸、膣または皮膚を含む。あるいは、同所注射、皮内注射、皮下注射、筋肉内注射、腹腔内注射、静脈注射、または、直接腫瘍内注射によって投与してよい。本発明の組成物のための医薬組成物、用量および投与経路を下に記載する。
【0112】
B.HSV−2変異体の医薬品製剤
本発明の変異体ウイルス組成物は、薬理学的に許容される製剤として調製することができる。通常、変異体ウイルスを、医薬品として許容され、ウイルスと適合する賦形剤と混合する。適当な賦形剤は、例えば、水、食塩水、デキストロース、グリセロール、エタノールまたは類似物およびそれらの組み合わせである。さらに、望むなら、調製物は、湿潤剤または乳化剤、pH緩衝剤、アジュバントまたは免疫強化物質など、少量の補助物質を含んでよい。これらの補助物質は、本ウイルス変異体の有効性を高める(Remington’s Pharmaceutical Sciences, Gennaro, A. R. et al., eds., Mack Publishing Co., pub., 18th ed., 1990を参照すること)。例えば、注射用に医薬品として許容される一般的なキャリアは、リン酸緩衝生理食塩水のミリリットルあたり50mgから最大約100mgのヒト血清アルブミンを含んでよい。薬理学的に許容される組成物の製剤に用いるのに適する非水溶溶媒の別の非限定的な例は、プロピレングリコール、ポリエチレングリコール、植物油、ゴマ油、落花生油およびオレイン酸エチルなどの注射可能な有機エステルを含む。水系キャリアの非限定的な例は、水、水溶液、食塩水、塩化ナトリウムなどの非経口ベヒクル、リンゲルのデキストロース等を含む。静脈内ベヒクルは、流体および栄養補液を含む。医薬品組成物のさまざまな成分のpHおよび正確な濃度を測定することは、定型的であり、当業者の知識の範囲内にある(Goodman and Gilman’s The Pharmacological Basis for Therapeutics, Gilman, A. G. et al., eds., Pergamon Press, pub., 8th ed., 1990を参照すること)。
【0113】
必要な、上記に記載したさまざまなその他の無菌成分とともに適切な溶媒の中に必要な量の活性化合物を組み込んで、無菌の注射可能溶液を調製する。通常、上記に記載した基本的な分散媒質と必要なその他の成分とを含む無菌ベヒクルにさまざまな殺菌された活性成分を組み込むことによって、分散液を調製する。
【0114】
C.HSV−2変異体の投与経路および用量
本変異体ウイルス組成物は、標的組織へのアクセスを提供する任意の経路によって投与してよい。投与経路の非限定的な例は、口、鼻、舌下、直腸、膣、塗布を含んでよく、あるいは注射(同所注射、皮内注射、皮下注射、筋肉内注射、腹腔内注射、静脈内注射または直接腫瘍内注入を含む)によってであってよい。通常、本ウイルス変異体は、液体溶液または懸濁液としての注射可能薬物として調製してよく、注入前に、液体への溶解または液体への懸濁に適する固体も調製してよい。調製物は、乳化もさせてよい。
【0115】
例えば、水溶液で非経口投与する場合、必要なら溶液を適当に緩衝性とするひつようがある。まず、十分な食塩水またはグルコースで液体希釈剤を等張性にする。これらの特定の水溶液は静脈投与、筋肉内投与、皮下投与および腹腔内投与に、特に適する。これに関連して、使用することができる無菌水性媒質は、本開示によって、当業者に既知である。例えば、1用量を1mlの等張性NaCl溶液に溶解させ、1000mlの皮下注入流体に加えるか、または提案された注入部位に注入するかのどちらかを行う(例えば、”Remington’s Pharmaceutical Sciences” 15th Edition, pages 1035−1038 and 1570−1580を参照すること)。治療を受ける被験者の状態によって、若干の用量の変化が必然的に起こる。いずれにしても、投与の責任者は、個々の被験者に適切な用量を決定する。
【0116】
本発明の組成物を用いて治療を提供するための最善の治療計画は、簡明直截的に決定することができることは、当業者には自明であろう。これは、実験の問題ではなく、最適化の問題であり、医術では定型的に行われている。例えば、マウスでインビボに調べ、用量および投与計画を最適化し始める出発点を得る。注射の頻度は、最初、週に一度であってよい。しかし、この頻度は、初期臨床試験から得られる結果および特定の患者の必要性によって、1日から2週間ごと、1月ごとまで、最適に調節する可能性がある。最初、マウスで用いた組成物の量から外挿してヒト投与量を決定してよい。
【0117】
1.用量
投与するウイルスベクターの量は、治療の回数、治療を受ける被験者、抗ウイルス抗体を合成する被験者免疫系の容量、崩壊させる標的組織、および所望の保護の程度を含むいくつかの因子に依存する。投与されるウイルス組成物の正確な量は、担当医の判断によって決まり、各個人独自である。しかし、適当な用量は、105プラーク形成単位(pfu)から1010pfuの範囲にある。特定の実施態様では、ウイルスDNAの用量は、約105、106、107、108、109、最大1010pfu以内であってよい。
【0118】
D.非ウイルスDNAベクター製剤
上記に記載したウイルス医薬品調合物のための調合物に加えて、非ウイルスDNAベクターも薬理学的に許容される無菌溶液の調製のための無菌粉体として調製することができる。無菌粉体の調製のための一般的な方法は、活性成分と、無菌ろ過済み溶液からの任意の別の所望の成分との粉体を生じる真空乾燥および凍結乾燥技法を含む。
【0119】
E.非ウイルスDNAベクターの投与経路および用量
本発明のポリヌクレオチドを哺乳類細胞に移入するための非ウイルスベクターを供給するためのいくつかの方法が考慮範囲内にある。これらは、リン酸カルシウム沈殿、DEAE−デキストラン、エレクトロポレーション、直接マイクロインジェクション、DNA保持リポソームおよびリポフェクタミン−DNA複合体、細胞超音波処理、高速マイクロ発射体を用いる遺伝子衝撃、既に考察した受容体媒介形質移入を含む。これらの技法のいくつかを、インビボまたはエクスビボ使用のために適応させることができる。
【0120】
本発明のいくつかの実施態様では、発現ベクターは、単に、ポリヌクレオチドを含む裸の組み換えDNAまたはプラスミドからなっていてよい。構築体の移入は、物理的または化学的に細胞膜を透過する本明細書に言及の任意の方法によって実行してよい。これは、特にインビトロ移入に適用可能であるが、インビボ使用にも適用してよい。
【0121】
その他の実施態様では、投与用ベヒクルは、リガンドおよびリポソームを含んでよい。例えば、Nicolauらは、リポソームに組み込まれたガラクトース末端アシアルガングリオシドであるラクトシルセラミドを使用し、肝細胞によるインシュリン遺伝子の取り込みの増加を観測した(Nicolau et at, (1987) Methods Enzyrnol. 149:157−76)。従って、リポソームの有無にかかわらず、任意の数の受容体−配位子システムによって、特定の遺伝子をコードする核酸を細胞型に特異的に供給することも実行できる。例えば、EGF受容体の上方調節(欧州特許第0 273 085号に記載されている)を示す細胞へ核酸を媒介投与するための受容体として上皮細胞成長因子(EGF)を用いてよく、マンノースを用いて肝臓細胞上のマンノース受容体を標的化してよい。
【0122】
特定の実施態様では、エクスビボ条件下での方が、DNA移入を容易に実行することができる。エクスビボ遺伝子治療は、動物からの細胞を取り出し、核酸をインビトロで細胞に投与した後、改変細胞を動物へ戻すことを指す。これは、動物由来の組織/器官の外科的取り出し、または細胞および組織の初代培養を含んでよい。
【0123】
1.用量
特定の実施態様では、用量を約103pfu/kg体重から約108pfu/kg体重の間で変化させることは想定の範囲内である。特定の実施態様では、用量は、約103、104、105、106、107から108pfu/kg体重以内であってよい。もちろん、通常そのような治療プロトコルで実行するように、初期臨床試験の結果および特定の患者の必要性に依存して、この用量を、上下に調節してよい。
【0124】
VII.併用療法
本発明の方法および組成物の有効性を増加させるために、本明細書に開示される方法および組成物を他の抗癌剤と組み合わせることが望ましいことがある。このプロセスは、癌細胞を少なくとも1つの他の抗癌剤と組み合わせた本発明の組成物と接触させることを含んでよい。これは、細胞を、両方の剤を含む単一の組成物または薬理学的製剤と接触させることによって、または細胞を2つの別個の組成物または製剤と接触させることによって、実現してよい。2つの別個の製剤を用いる場合、癌細胞を両方の製剤と同時に接触してもよく、他の製剤より先に1つの製剤と接触させてもよく、それらの任意の組み合わせまたは繰り返しサイクルであってもよい(例えば、本発明の組成物を、別の抗癌剤の投与の前または後に投与する)。本発明の組成物と他の剤とを別個に投与する実施態様では、通常、本発明の組成物と他の剤とが、依然として癌細胞に併用効果を有利に及ぼすことができるよう、各供与時間の間にあまり時間を置かないように注意する。2つの製剤の投与の間のこの時間間隔は、分から週の範囲にあってよい。
【0125】
本発明の組成物または方法とともに用いてよい抗癌剤の非限定的な例は、化学療法薬(例えばシスプラチン(CDDP)、カルボプラチン、プロカルバジン、メクロレタミン、シクロホスファミド、カンプトセシン、イホスファミド、メルファラン、クロラムブシル、ブスルファン、ニトロソ尿素、ダクチノマイシン、ダウノルビシン、ドキソルビシン、ブレオマイシン、ピコマイシン、マイトマイシン、エトポシド(VP16)、タモキシフェン、ラロキシフェン、エストロゲン受容体結合剤、タキソール、ジェムシタビエン、ナベルビン、ファルネシル−蛋白質トランスフェラーゼ阻害剤、トランス白金、5−フルオロウラシル、ビンクリスチン、ビンブラスチンおよびメトトレキサン、またはそれらの任意の類縁体または誘導体変化形)、放射線治療薬剤(例えば腫瘍細胞へのγ線、X線、マイクロ波およびUV照射および/またはラジオアイソトープの指向投与)、免疫療法剤および免疫調節剤、遺伝子治療薬剤、アポトーシス促進剤および当業者に公知のその他の細胞周期調節剤を含んでよい。
【0126】
免疫療法を本明細書に記載の組成物および方法とともに用いて、悪性疾患の治療のための併用療法としてもよい。通常、免疫療法は、癌細胞を標的とし、崩壊させる免疫エフェクター細胞と分子との使用に依拠する。免疫エフェクターは、例えば、腫瘍細胞の表面のマーカーに特異的な抗体であってよい。この抗体は、単独で治療のエフェクターとして働いてよく、あるいは、他の細胞(例えば細胞毒性T細胞またはNK細胞)を募って殺細胞を実際に実行してよい。この抗体は、薬物またはトキシン(例えば化学療法剤、放射性核種、リシンA鎖、コレラトキシン、百日咳毒素等)と結合し、単に標的化剤として働いてよい。いくつかの実施態様では、エフェクターは、腫瘍細胞標的と直接的または間接的に相互作用する表面分子を有するリンパ球であってよい。その他の実施態様では、腫瘍細胞は、標的化の対象となるなんらかのマーカーを持っていなければならない。標的化に適する腫瘍マーカーの非限定的な例は、癌胎児性抗原(CEA)、前立腺特異性抗原、尿腫瘍関連抗原、胎児抗原、チロシナーゼ(p97)、gp68、TAG−72、HMFG、シアリルルイス抗原、MucA、MucB、PLAP、エストロゲン受容体、ラミニン受容器、erbBおよびp155を含んでよい。
【0127】
悪性疾患の治療のための複合治療法として本明細書に記載の組成物および方法とともに、遺伝子治療を併用してよい。併用療法としての遺伝子治療は、本明細書に記載の変異体HSV−2とは別の治療遺伝子の投与および発現に依拠する。この遺伝子治療は、本明細書に記載のHSV−2変異体の前、後、または同時に起用してよい。遺伝子治療の非限定的な標的の例は、免疫調節剤、細胞表面受容体およびGAP接合の上方調節に影響を及ぼす剤、細胞静止剤および分化剤、細胞接着の阻害剤、または標的細胞のアポトーシス感受性を誘導し、または増加させる剤を含む。本発明と組み合わせて遺伝子治療の一部として用いることができる免疫調節遺伝子の非限定的な例は、腫瘍壊死因子;インターフェロンアルファ、ベータおよびガンマ;IL−2およびその他のサイトカイン;F42Kおよびその他のサイトカイン類縁体;またはMIP−1、MIP−1ベータ、MCP−1、RANTESおよびその他のケモカインを含む。
【0128】
細胞増殖の阻害剤の例は、p16である。サイクリン依存性キナーゼ、すなわちCDKによって真核生物の細胞周期の主要遷移を引き起こす。1つのCDK、すなわちサイクリン依存性キナーゼ4(CDK4)は、G1期を通して進行を調節する。この酵素の活性は、G1後期にRbをリン酸化することであってよい。CDK4の活性は、活性化サブユニット、すなわちD−型サイクリンによって、および阻害サブユニット、すなわちCDK4に特異的に結合し、阻害し、従って、Rbリン酸化を調節してよいp16INK4によって制御される。p16INK4遺伝子は、新しく記載されたCDK阻害性蛋白質の種類に属し、この種類は、p16B、p19、p21WAF1およびp27KI1も含む。p16INK4遺伝子の同型接合削除および変異は、ヒト腫瘍細胞系統では頻繁である。p16INK4蛋白質はCDK4阻害剤なので、この遺伝子を削除すると、CDK4の活性が増加し、その結果、Rb蛋白質が高リン酸化される。遺伝子治療に使用して細胞増殖を阻害してよい他の遺伝子は、Rb、APC、DCC、NF−1、NF−2、WT−1、MEN−I、MEN−II、zac1、p73、VHL、MMAC1/PTEN、DBCCR−1、FCC、rsk−3、p27、p27/p16融合体、p21/p27融合体、抗血栓遺伝子(例えばCOX−1、TFPI)、PGS、Dp、E2F、ras、myc、neu、raf、erb、fms、trk、ret、gsp、hst、abl、E1A、p300、血管新生に関与する遺伝子(例えばVEGF、FGF、トロンボスポンジン、BAI−1、GDAIFまたはそれらの受容体)およびMCCを含む。
【0129】
細胞表面受容体またはFas/Fasリガンド、DR4またはDR5/TRAILなどのリガンドの上方調節は、過剰増殖性細胞に対する自己分泌効果または傍分泌効果を確立し、本発明のアポトーシス誘導能力を強化することも考慮の範囲内である。GAP接合の数を上げて細胞間の信号伝達を増加させると、隣接する過剰増殖性細胞ポピュレーションに対する抗過剰増殖性効果が増加する。他の実施態様では、細胞増殖抑制剤または分化剤を本発明で用いて、治療法の抗過剰増殖性抗力を高めることができる。細胞接着の阻害剤は、本発明の効力を高めるとみなされる。細胞接着阻害剤の例は、局所接着キナーゼ(FAK)阻害剤およびロバスタチンである。さらに、抗体c225などの過剰増殖性細胞のアポトーシス感受性を増加させる他の剤を本発明と組み合わせて用いて治療効果を高めることは、考慮の範囲内である。
【0130】
本発明とともにホルモン療法も用いてよい。乳癌、前立腺癌、卵巣癌または子宮頸部癌などの特定の癌の治療においてホルモンを使用してテストステロンまたはエストロゲンなどの特定のホルモンの効果のレベルを下げ、あるいは遮断してよい。この治療は、治療オプションとして、または転移の危険性を小さくするために、多くの場合に少なくとも1つの他の癌療法と組み合わせて用いられる。
【0131】
本願において引用するすべての特許、特許出願およびその他の刊行物は、公開されたアミノ酸またはポリヌクレオチド配列を含めて、参照によって全体としてあらゆる目的について組み込まれる。
【実施例】
【0132】
以下の実施例を掲載して本発明の好ましい実施態様を示す。当業者は、以下の実施例に開示する技法は、本発明の実施において良好に機能することを本発明者が発見した技法を表し、従って本発明の実施にとって好ましいモードを構成するとみなしてよいと理解するべきである。しかし、当業者は、本開示を参照し、開示された特定の実施態様において多くの変化を施してもなお本発明の技術思想および範囲から逸脱せずに同様な類似の結果を得ることができることを理解するべきである。
【0133】
(実施例1)
FusOn−H2の構築
図1に、FusOn−H2の例の構築の例を示す。最初に、以下のプライマー対の例、すなわち5’−TTGGTCTTCACCTACCGACA(SEQ ID NO:1)と3’−GACGCGATGAACGGAAAC(SEQ ID NO:2)とを用いて、ICP10左側(left−flanking)領域を含むHSVゲノム領域(HSV−2ゲノムのヌクレオチド番号85994〜86999と等価)を増幅させた。以下のプライマー対の例、すなわち、5’−ACACGCCCTATCATCTGAGG(SEQ ID NO:13)と5’−AACATGATGAAGGGGCTTCC(SEQ ID NO:14)とを用いて、RRドメインおよび右側(right−flank)領域(HSV−2ゲノムのヌクレオチド配列番号88228〜89347と等価)を増幅させた。これらの2つのPCR産物を、EcoRI−NotI−XbaIライゲーションによってpNeb193の中にクローン化し、pNeb−ICP10−デルタPKを発生させた。次に、以下のプライマー対の例、すなわち、5’−ATGGTGAGCAAGGGCGAG(SEQ ID NO:3)と3’−CTTGTACAGCTCGTCCATGC(SEQ ID NO:4)とを用いて、CMVプロモーター−EGFP遺伝子を含むDNA配列を、pSZ−EGFPからPCR増幅させた。次に、BgIIIおよびNotIライゲーションによって、PCR増幅DNAをpNeb−ICP10−デルタPKの削除PK遺伝子座にクローン化してpNeb−PKF−2を発生させた。PCR増幅戦略を設計する際には、EGFP遺伝子がICP10遺伝子の残るRRドメインのフレーム内に融合し、その結果、この融合遺伝子の新しい蛋白質産物は、以下の実験段階において組み換えウイルスの選択を容易にする無傷の機能EGFPを含むように、プライマーを設計した。
【0134】
pNeb−PKF−2プラスミドDNAと、精製したHSV−2ゲノムDNA(186株)とをリポフェクタミンによってベロ細胞に共形質移入する相同組み換えによって、改変ICP10遺伝子をウイルス中に挿入した。GFP陽性ウイルスプラークを検索して組み換えウイルスを選抜し、特定した。検索プロセスの間に、GFP陽性プラークがすべて、明らかな感染細胞の合胞体形成を示すことに気がついた。これは、本発明の特定の実施態様では、この改変ウイルスが広範囲な細胞膜融合を誘導することを示している。合計6つのプラークを選抜した。さらにキャラクタリゼーションするため、およびその後のすべての実験のためにそれらの1つを選択した。これをFusOn−H2と呼ぶ。
【0135】
実施例2
インビトロキャラクタリゼーション
FusOn−H2ベクターの例を、当分野の標準的な分析法によってキャラクタリゼーションした。
【0136】
サザンブロット分析。
【0137】
改変ICP10遺伝子がHSV−2ゲノムに正しく挿入され、元のICP10遺伝子を置換したことを確認するために、精製したFusOn−H2ウイルス原料からウイルスDNAを抽出した。対照として、同じ手順によって、親の野生型HSV−2からウイルスDNAを抽出した。BamHIを用いてウイルスDNAを消化し、0.8%アガロースゲル中で電気泳動した。BamHIによって消化すると、野生型HSV−2ゲノムから、ICP10遺伝子全体およびその左側領域および右側領域を含む7390bpのDNA断片が生じる。しかし、FusOn−H2ゲノムを同じ酵素で消化すると、ICP10遺伝子座から2つのDNA断片、すなわち、1)左側領域とCMVプロモーター配列とを含む4830bp断片、および2)GFP、RRおよび右側領域を含む3034bp配列、が生じる。DNAをナイロン膜に移し、1)DNA配列の左側領域、2)PK領域全体、3)PKの右側領域、および4)GFP遺伝子、から調製した4つのプローブとハイブリッド化させる。結果(図2)によると、GFP遺伝子から作った1つを除くすべてのプローブが7390bpDNAバンドにハイブリッド化した。左側領域プローブは、GFPおよび右側領域DNA配列から調製したプローブによって認識されるDNAバンドにハイブリッド化した。PKドメイン配列から作ったプローブは、DNA断片のどれにもハイブリッド化することができず、PKドメインがFusOn−H2のゲノムから完全に除去されたことを示した。
【0138】
ウェスタンブロットハイブリダイゼーション。
【0139】
FusOn−H2のゲノムの中の改変ICP10遺伝子の正しさをさらに確認するために、FusOn−H2または親の野生型HSV−2のどちらかに感染させたベロ細胞から、またはpSZ−EGFPプラスミドDNAで形質移入された細胞から蛋白質を抽出した。12%SDS−PAGEゲル上で蛋白質を分離し、ハイボンド−C膜に移した。次に、抗GFPモノクローナル抗体(抗GFP#Ab290、ABCAM Inc., Cambridge, MA)で膜をブロットした。この抗GFP抗体は、pSZ−EGP形質移入細胞から発現された小型のGFP蛋白質(約28kD)を選択した。同じ抗体は、著しく大きな蛋白質バンド(融合蛋白質のサイズは、約120kDと予想される)も認識した。しかし、この抗体は、野生型HSV−2に感染した細胞由来のどの蛋白質産物とも反応することができず、この抗体の特異性が確認された。これらの結果は、GFP遺伝子がFusOn−H2ゲノム中のICP10遺伝子の残っているRRドメインと正しく融合したことをさらに確認する。
【0140】
(実施例3)
インビトロFusOn−H2表現型キャラクタリゼーション
FusOn−H2の表現型を決定するために、本発明者らは、ベロ細胞を野生型HSV−2またはFusOn−H2のどちらかに感染させ、あるいは、細胞を感染させずにおいた。感染の24時間後、FusOn−H2に感染させた細胞単一層中に明らかな合胞体形成が見えた。非感染細胞にも、野生型HSV−2に感染させた細胞にも、合胞体は見られなかった。さまざまな組織起源のヒト腫瘍細胞中でも同様な合胞体形成が観測された。いくつかの腫瘍細胞では、野生型HSV−2に感染させても、ある程度の合胞体形成が誘導された。しかし、これらの細胞上でFusOn−H2によって誘導される合胞体形成は、通常、有意に大規模であった。従って、この場合、FusOn−H2は、親の野生型HSV−2と比較すると、融合活性の増強を示す。これらの結果は、FusOn−H2は、その感染が広範な合胞体形成を誘導するか、または腫瘍細胞中の合胞体形成の強度を強める点で、表現型として親のウイルスと異なることを示している。PKドメインも、ICP10遺伝子全体も、これまで細胞膜融合といかなる機能関連を有すると報告されたことがない(Smith et al., (1998) J Virol. 72(11): 9131−9141; Smith et al., (1994) Virol. 200(2): 598−612; Smith et al., (1992) J. Gen. Virol. 73(pt6): 1417−1428)。いくつかの実施態様では、GFP遺伝子を加え、および/またはICP10の天然プロモーターを強力なCMVプロモーターで置換すると、このウイルスのこの表現型変化に効果があった。1型殺腫瘍HSVによって合胞体形成が誘導されると、例えば、ヒト腫瘍細胞に対するウイルスの殺細胞能力が著しく増加することが示されている(2003年3月26日出願の米国特許出願第10/397,635号)ので、殺腫瘍を目的とする用途では、FusOn−H2の融合性表現型は重要である。
【0141】
(実施例4)
増殖活動中の細胞および休止状態の細胞中のFusOn−H2の成長曲線
分裂(腫瘍)細胞中のFusOn−H2の選択複製の性質を求めるために、本発明者らは、ベロ細胞を感染させ、10%ウシ胎児血清(FBS)(十分に増殖状態にある細胞)中、またはFBSを含まない媒地(休止状態の細胞)中のどちらかで、野生型HSV−2またはFusOn−H2のどちらかとともに培養した。感染後、異なる時点でウイルスを集め、ベロ細胞で滴定した。細胞を休止状態にしても、野生型ウイルスの成長は、わずかな影響しか受けなかった(2分の1まで減少しない)。これとは対照的に、休止状態にある細胞中のFusOn−H2の成長は、増殖状態にある細胞からのウイルス収量と比較すると、劇的に減少した(40分の1より小さくなる)。これらの結果(図5)は、FusOn−H2は、腫瘍細胞中で十分複製能力があるが、体の中の正常な体細胞を表す休止状態の細胞中では、通常、最小限の複製能力しかなく、従って、腫瘍細胞中でのFusOn−H2の選択複製能力が得られることを示した。
【0142】
(実施例5)
ヒト腫瘍細胞に対するFusOn−H2のインビトロ殺細胞アッセイ
次に、本発明者らは、直接、FusOn−H2とその親の野生型HSV−2とのインビトロ殺腫瘍効果、あるいは1型HSV(HSV−1)から構築した殺腫瘍ウイルスを比較した。ヒト卵巣がん細胞系統Skov−3またはヒト乳癌細胞系統MDA−MB−435の例となる細胞を0.01または0.1pfu/細胞のどちらかで本ウイルスに感染させ、ウイルス感染の24時間後または48時間後のどちらかの時点で、例えば比色法乳酸デヒドロゲナーゼ(LDH)アッセイによって細胞生存率を測定した。結果(図6)によると、試験した殺腫瘍HSVの中で、FusOn−H2が両方のヒト腫瘍細胞系統に対して最も高い殺細胞能力を有する。その殺細胞能力は、腫瘍細胞中に合胞体形成を誘導するその能力によって、その親の野生株ウイルスと比較しても有意に高かった。
【0143】
(実施例6)
FusOn−H2のインビボ治療評価
インビボ条件下で、例となるFusOn−H2ウイルスをキャラクタリゼーションした。
【0144】
ヒト乳癌異種移植部に対して
FusOn−H2のインビボ抗腫瘍効果を評価するために、本発明者らは、非常におだやかな用量(1×106pfu)のウイルスを、ヒト乳癌(乳腺脂肪層中のMDA−MB−435細胞の注入から)の確立された異種移植部(直径約5〜8mm)に直接注入した。比較のため、本発明者らは、HSV−1由来の殺腫瘍HSV(Baco−1)をFusOn−H2と同じ用量で同時に評価した。Baco−1は、2003年3月26日出願の米国特許出願第10/397,635号に記載されている。4週間にわたり、毎週腫瘍サイズを測定した。PBS対照と比較すると、どちらのウイルスの単回注射も腫瘍成長(図7)に対して即時効果を示した。ウイルス注入後1週間以内に、殺腫瘍ウイルスのどちらで治療したマウスの腫瘍も、PBSを注射した腫瘍より著しく小さかった(P<0.001)。しかし、第2週から第4週では、FusOn−H2は、Baco−1より有意に大きな抗癌効果(P<0.01)を生じた。FusOn−H2投与後、第2週には、すべての動物(8匹中8匹)の腫瘍が消えた。これに対して、Baco−1を注入した群では、2匹のマウスの腫瘍しか消えなかった。他の6匹のマウスでは、腫瘍は最初に縮んだが、ウイルス注入後第3週には再び成長し始めた。これらの結果は、FusOn−H2がヒト乳癌に対する強力な抗腫瘍剤であり、HSV−1から構築した融合性殺腫瘍HSVより有意に効果が大きいことを示している。
【0145】
ヒト卵巣がん異種移植部に対して
卵巣がんの腹腔内侵入は、よくある重大な臨床問題である。後期卵巣がん患者の約70%が腹腔空洞内に転移性疾患を有すると報告された。従って、本発明者らは、例えば、ヒト卵巣がんに対するFusOn−H2の有効性を試験する手段として、腹腔内転移モデル(異物移植Skov−3細胞)を選んだ。集めたばかりのSkov−3細胞を3X106細胞/マウスの用量でヌードマウスの腹腔内に接種した。2週間後、マウスの腫瘍細胞注射部位から遠く離れた部位に、3×106pfuのBaco−1、FusOn−H2またはPBS(対照)のどれかを単回腹腔内(i.p.)注射した。1週間後、この治療注射を繰り返した。最初の治療注射の4週間後、マウスを安楽死させ、腹腔空洞内の腫瘍成長を評価した。これらの治療群の各動物の複数の空洞内腫瘍小結節を観察すると明らかであったが、PBS−またはBaco−1治療群のどちらにも明らかな腫瘍のi.p.転移があった(図8および表1)。
【0146】
表1 ヒト卵巣癌異種移植部の殺腫瘍治療後の腹腔内腫瘍小結節の数および重量
【0147】
【表1】
PBSと比較すると、Baco−1治療は、確立された卵巣がんに対して一定の治療効果を示した。1匹のマウスの腫瘍は完全に消え、1匹の腫瘍小結節は著しく小さくなった(1つの腫瘍小結節しか見られなかった)。しかし、FusOn−H2の治療効果は明らかに大規模であった。実験の終りまでに、FusOn−H2治療を受けた群の8匹のマウスのうち7匹の腫瘍が消えた(表1および図8)。腫瘍がなくならなかった唯一のマウスの腫瘍小結節は1つしかなかったが、小結節は、Baco−1またはPBSの治療を受けたマウスの小結節よりはるかに小さかった。これらの結果は、FusOn−H2が、比較的大きな空洞の中に確立したヒト固形腫瘍を治療する上で、ウイルスを非常におだやかな用量で投与したときでも、極めて有効であることを明らかに実証している。
【0148】
(実施例7)
FusOn−H2のインビボ毒性評価
FusOn−H2の毒性を評価するための第一段階として、本発明者らは、5×106pfuの野生型HSV−1、HSV−2またはFusOn−H2のどれかをC57/ブラックマウス(N=5)に皮下注射した。ウイルス投与の5日後に、野生型HSV−lを受けた群の5匹のマウスのうち4匹が死んだ。野生型HSV−2を受けた群では1匹のマウスが死んだ。しかし、FusOn−H2を注射した群ではマウスは1匹も死ななかった。これらの結果は、FusOn−H2が腫瘍細胞を殺傷することには極めて強力であるが、受け入れ宿主に対する毒性は親の野生型HSVより著しく低く、特定の実施態様では、臨床応用で安全なことを示している。
【0149】
(実施例8)
アポトーシスを誘導するFusOn−H2の能力
本実施例によって、本発明に記載の改変HSV−2ウイルス(FusOn−H2)は、感染腫瘍細胞および周囲の腫瘍細胞においてアポトーシスを効率的に誘導することができ、別の腫瘍破壊機序を提供することを示す。
【0150】
アフリカミドリザル腎臓(ベロ)細胞、SW403およびSW480細胞(ヒト結腸癌細胞系統)、およびA549細胞(ヒト肺癌細胞系統)は、American Type Culture Collection (Rockville, MD)から入手した。ヒト食道癌細胞系統のEC9706は、Mingrong Wang博士(Chinese Academy of Medical Sciences)の提供である。ヒト卵巣がん細胞系統のSKOV3細胞は、Robert Bast博士(the M. D. Anderson Cancer Center)の提供である。ヒト骨肉腫系統のU20S細胞は、Dr. Lawrence Donehowerの提供である。細胞はすべて、10%ウシ胎児血清(FBS)を含むDMEM中で培養した。
【0151】
野生型HSV−2株186(wt186)からFusOn−H2を誘導した。その構築は実施例1に記載した。HSV−1系殺腫瘍ウイルスBaco−1の構築は、米国特許出願第10/397,635号に記載されている。ウイルス原料は、細胞あたり0.01プラーク形成単位(pfu)のベロ細胞を感染させて調製した。2日後、ウイルスを集め、記載されている(Nakamori et al., (2003) Clinical Cancer Res. 9(7): 2727−2733)ように精製した。純化ウイルスを滴定し、等分し、使用するまで−80℃で保管した。
【0152】
ウイルス成長キャラクタリゼーション
24ウェルプレートに3つ1組で、50%密度で細胞を接種した。次の日、細胞を1pfu/細胞のウイルスに1時間感染させた。細胞をPBSで1度洗浄し、吸収されなかったウイルスと取り込まれなかったウイルスとを除去した後、新鮮な培地を加えた。感染後24時間で細胞を集めた。凍結および解凍ならびに超音波処理を繰り返して、ウイルスを放出させた。プラークアッセイによってベロ細胞基準のウイルス力価を測定した。
【0153】
感染細胞のヘキスト(Hoechst)染料染色およびクロマチン凝集の定量
次の日、24ウェルプレートに接種した細胞を、10pfu/細胞のFusOn−H2、wt186またはBaco−1に感染させ、あるいは偽感染させた。感染の24時間後、細胞を37℃で1μg/mlの最終濃度のヘキスト染料33358(Sigma−Aldrich, MO)で30分間染色してから、蛍光顕微鏡下で顕微写真を撮影した。
【0154】
DNAラダーリングアッセイ
細胞を、6ウェルプレートに70%密度で接種した。次の日、細胞を10pfu/細胞のウイルスに感染させた。ウイルス感染の24時間後に、細胞を集め、DNAzo1試薬(Invitrogen, CA)を用いてDNAを細胞から抽出した。抽出したDNAをRNアーゼ(100μg/ml)で処理した後、フェノール:クロロホルム抽出およびエタノール沈殿に付した。次に、DNAを、1%アガロースゲルに載せ、電気泳動を行い、臭化エチジウムで染色した後、UV照明下で視覚化した。
【0155】
EGFPの発現は、クロマチン凝集に対応する
次の日、12ウェルプレートに接種した細胞を、1pfu/細胞のFusOn−H2に感染させた。クロマチン凝集のためのヘキスト染料染色を、上記で説明したように実行した。Spot Image Software(Diagnostic Instrument, Inc, IL)を用いて、別々の蛍光灯による同じ画像域からの顕微鏡写真の重ね合せを実行した。同じ区域の中のGFP陽性アポトーシス細胞とGFP陰性アポトーシス細胞とを別々に計数した。各区域で約100のアポトーシス細胞を数えた。全部で3つの区域を計算して、FusOn−H2感染細胞によって誘導された傍観者効果を証明した。
【0156】
末端デオキシヌクレオチジルトランスフェラーゼ媒介ニック端標識化(Tunel)アッセイ
雌のHsd無胸腺(nu/nu)マウス(Harlan, Indianapolis, Indianaから入手した)を特定の病原体感染防止条件下に保ち、5から6週齢に達したとき実験に用いた。0.25%のトリプシンおよび0.05%のEDTAに短時間曝露した亜集密培養からEC9706細胞を集めした。10%FBSを含む培地でトリプシン処理を停止させた後、無血清培地中で細胞を1回洗浄し、PBS中に再懸濁した。第0日に、ヌードマウスの右横腹に5×106のEC9706細胞を接種した。腫瘍細胞注入の2週間後、腫瘍の直径が約5mmに到達したとき、体積100μl中3×106pfuのFusOn−H2またはBaco−1、あるいは同じ体積のPBSをマウスに単回腫瘍内注射した。腫瘍を毎週測定し、式:腫瘍体積[mm3]=(長さ[mm])×(幅[mm])2×0.52によって腫瘍の体積を求めた。Tunelアッセイでは、1×107pfuのFusOn−H2またはBaco−1ウイルスを腫瘍内注入して3日後、CO2に曝露してマウスを安楽死させた。腫瘍組織を外植し、Tunel染色に備えて区分けした。
【0157】
FusOn−H2は、さまざまな組織起源のヒト腫瘍細胞中にアポトーシスを誘導する
シクロヘキシミドなどの翻訳阻害剤によってウイルス蛋白質合成を遮断しなければ、特定のHSV−2遺伝子産物の抗アポトーシス活性によって、HSV−2に感染させても、通常、アポトーシスを誘導しない(Aubert et aG, (1999) J Virol 73(12): 10359−70)。HSV−2由来のICP10遺伝子のPKドメインは、抗アポトーシス機能を有するウイルス遺伝子産物の1つとして認識され、ウイルスゲノムから削除すると、ある種の体細胞のアポトーシス死を誘導する能力をウイルスに与えることが記載された(Perkinsら、(2002) J Virol76(3): 1435−49)。
【0158】
FusOn−H2が腫瘍細胞のアポトーシス死を誘導するか定めるために、さまざまな組織起源のヒト腫瘍細胞のパネルを10m.o.i.のウイルスに感染させた。HSV−1、Baco−1から誘導した殺腫瘍ウイルスを対照として評価に含めた。腫瘍細胞の中で、EC9706はヒト食道癌細胞系統であり、SKOV3はヒト卵巣がん細胞系統であり、SW403およびSW480はヒト大腸癌細胞系統である。細胞を6ウェルプレートに接種し、翌日、ウイルスに感染させた。感染の24時間後、細胞をヘキスト染料33358で染色した。FusOn−H2による腫瘍細胞の感染は、大量のクロマチン凝集を誘導し、アポトーシスを示した。これは、FusOn−H2感染細胞中に強い小さな青色の核染色が出現したことによって明白だった。全体として、FusOn−H2に感染した腫瘍細胞の80%超がクロマチン凝集を示した。非感染腫瘍細胞は、そのようなアポトーシスの特徴を非常にわずかに、またはまったく示さなかった。これらの腫瘍細胞を親の野生型HSV−2(wt186)またはBaco−1に感染させても、クロマチン凝集の青色蛍光染色の背景レベルはあまり増加しなかった。
【0159】
腫瘍細胞中にアポトーシスを誘導するFusOn−H2の能力をさらに検証するために、DNA断片化を解析した。前の実験に用いた3つの腫瘍細胞を10pfu/細胞のウイルスに感染させるかまたは偽感染させた。感染の24時間後、細胞を集めた。DNAを細胞から抽出し、1%アガロースゲル中で分離した。FusOn−H2感染物質が載っているウェルには明らかなラダーリングがあった。このラダーリングは、wt186またはBaco−1感染細胞のどちらか由来のDNA試料が載っているウェルの中で検出され、従って、上記で提示したクロマチン凝集の結果を再確認した。これらの結果を一緒にすると、FusOn−H2に感染させるとこれらのヒト腫瘍細胞中にアポトーシスが効率的に誘導されるが、親の野生型HSV−2にもHSV−1系殺腫瘍ウイルスにもそのような特性はない。
【0160】
FusOn−H2に感染すると、傍観者細胞のアポトーシス死も誘導される
FusOn−H2には強化された緑色蛍光蛋白質をコードする遺伝子があるので、その感染力は、蛍光顕微鏡下で容易に測定することができた。クロマチン凝集の蛍光性検出と感染力とを相互検証しているとき、青色の蛍光性クロマチン凝集を示す細胞と、GFP染色を示す細胞との百分率の間の明らかな不一致を認めた。クロマチン凝集を示す腫瘍細胞とGFP発現を示す腫瘍細胞との絶対数を数えると、比は、約2.6:1であった。この結果によると、FusOn−H2感染には、周囲の腫瘍細胞に対するかなりの傍観者アポトーシス効果があった。
【0161】
FusOn−H2誘導アポトーシス死は、腫瘍細胞死を加速し、腫瘍細胞内のウイルス複製を弱める
FusOn−H2に感染した腫瘍細胞と、殺腫瘍ウイルス誘導HSV−1に感染した腫瘍細胞との間で、細胞が細胞変性効果(CPE)を示した時間にも明らかな差異を認めた。1pfu/細胞の用量のFusOn−H2に感染した腫瘍細胞は、通常、24時間以内に十分なCPEを示したが、同じ用量のBaco−1に感染した腫瘍細胞は、形態学的にほとんど正常に見えた。それらは、通常、感染後72時間を超えるまで明らかなCPEの徴候を示さなかった。FusOn−H2に感染させたウェルの中では、感染後24時間で細胞の集団化と相互離脱を含む一般的なCPEを容易に見ることができたが、Baco−1に感染させた細胞は、感染後48時間でも基本的に偽感染細胞と同じに見えた。これらの結果によると、FusOn−H2によって誘導されるアポトーシス細胞死は、ウイルス感染後すぐに起こったが、ウイルス複製の殺腫瘍効果が起こるにはもっと長い時間を要した。
【0162】
アポトーシス腫瘍細胞死は重要なウイルスのインビボ抗腫瘍機序である
既に記載した実験で用いた腫瘍細胞の1つから確立した腫瘍異種移植物に対するFusOn−H2のインビボ抗腫瘍活性を評価した。Baco−1をこの実験に含め、これらの2つのウイルスの治療効果を直接比較することができるようにした。新たに調製した5×106のEC9706細胞の皮下注射によって、腫瘍異種移植物を右横腹に確立した。腫瘍サイズが直径約5mmに達したとき、マウスにどちらかのウイルス(FusOn−H2またはBaco−1)、あるいは対照としてのPBSを、3×106pfuの用量で単回腫瘍内注射した。6週にわたって腫瘍を定期的に測定し、治療前の腫瘍体積を治療後の異なる時点の腫瘍体積で除することによって、腫瘍成長比を求めた。FusOn−H2を治療投与すると、基本的に、腫瘍成長は1週間以内に停止した。その後、腫瘍は縮み始め、実験終了までに平均腫瘍サイズはビボ治療前のサイズの約2分の1でしかなくなり、半数を超えるマウスの腫瘍は完全に消えた。対照のPBSと比べると、Baco−1を投与しても、3週までいかなる治療効果も示さなかった。しかし、腫瘍は第35日には再び成長し始めたので、腫瘍収縮は一時的なようであった。この腫瘍細胞中ではアポトーシスの誘導によって複製能力が制約されるという事実にもかかわらず、全体として、FusOn−H2の治療効果は、評価を行ったすべての時点でBaco−1の治療効果より有意に強かった(p<0.05)。これらの結果によると、このインビボ研究では、FusOn−H2によって誘導されたアポトーシス死と、同時に起こる傍観者効果とが主要抗腫瘍機序であった可能性が高い。
【0163】
(実施例9)
FusOn−H2による腫瘍破壊は強力な抗腫瘍免疫を誘導する
2つの純系腫瘍モデル、すなわちマウス乳腺腫瘍(4T1細胞)とマウス神経芽細胞腫(ニューロ2A細胞)におけるFusOn−H2の抗腫瘍活性を評価した。どちらの場合にも、FusOn−H2は、統計的に有意な抗腫瘍効果を示し、同時に堅固な腫瘍特異性免疫応答も示した。下記に提示するのは、乳腺腫瘍モデルにおける研究からの一般的なデータである。
【0164】
この評価には、4T1細胞を利用した。これは、非免疫原性であり、非常に悪性であり、純系BALB/cマウスにおいて高度に転移性である(Aslakson and Miller (1992) Cancer Res 52(6): 1399−405; Pulaski and Ostrand−Rosenberg (1998) Cancer Res. 58(7): 1486−93)。4T1細胞(1×105)を免疫能力のあるBALB/cマウスの乳腺脂肪層に同所投与注射して同所性腫瘍を確立した。マウスを10日間飼育したところ、群の90%を超える個体で肺転位を検知することができた。次に、腫瘍を持つマウスを3つの群(それぞれn=10)に分け、FusOn−H2か、あるいは、このモデルにおいて有効な抗腫瘍免疫を誘導することが既に示されている二重融合型Synco−2D(Nakamori, Fu et al., (2004) Mol. Ther. 9(5): 658−665)を含むHSV−1から誘導した他の殺腫瘍HSVか、のどちらかの1×107pfuを腫瘍内注射した。2週間にわたって、正所性部位の腫瘍質量を毎週測定した後、マウスを死なせ、免疫学的アッセイおよび肺転位の評価を行った(墨汁灌流後、解剖顕微鏡下で計測)。免疫学的アッセイでは、外植された脾臓から脾細胞を調製し、照射4T1細胞で5日間インビトロ刺激してから、以下のアッセイ、すなわち、1)51Cr放出アッセイによる腫瘍特異性CTL活性(4T1細胞または純系肉腫細胞系統Meth−Aのどちらかを標的細胞とする)、2)BD Biosciences社から購入した検出キットを用いるマウスIFN−γ分泌細胞のElispot分析、3)サイトカイン分泌の定量(インターフェロン−γとIL−10との両方について)を用いた。結果によると、FusOn−H2を局所腫瘍内投与すると、正所性腫瘍だけででなく遠隔肺転位に対しても他のウイルスより有意に良好な治療効果が認められた。Synco−2Dは、Baco−1と比較すると、正所性腫瘍および転移性腫瘍の成長を阻害することができた。この結果は、本発明者らの既報の観察結果(Nakamori, Fu et al., (2004) Mol. Ther. 9(5): 658−665)と類似していた。しかし、FusOn−H2は、この腫瘍を治療する上でSynco−2Dより有効でさえあるように見えた。FusOn−H2によって誘導される同時発生抗腫瘍免疫応答も、腫瘍特異性CTL活性および頻度ならびにサイトカイン放出を含め、Synco−2Dの腫瘍免疫応答より明らかであり、局所腫瘍および転移性腫瘍の除去に対するそれらの寄与を示した。
【0165】
(実施例10)
ウイルス力価を測定するためのプラーク形成アッセイ
ウイルス原料を調製した後、既に記載したプラーク形成アッセイ(Lancz GJ. (1974). Arch Virol., 46, 36−43を参照すること)を用いてウイルス力価を測定した。ベロ細胞をトリプシン処理し、計数し、ウェルあたり4×105細胞で6ウェルプレートに塗布し、37℃、5%CO2および90%湿度で温置し、24時間培養する。次の日、ウイルスを、1×最小必須培地(MEM)の中で次々に1:10に希釈して10−3〜10−8の6つの濃度にする。次に、培地をウェルから吸い取り、0.5mlのウイルス希釈物を3つ1組で各ウェルに加える。次に、15分ごとに振盪してプレートを1時間培養する。培養期間の後、ウイルス溶液を吸い取り、1%アガロースを含む2mlのMEMを各ウェルに加え、プレートを3日間培養した後、0.1%クリスタルバイオレットと20%エタノールとを含む溶液で細胞を染色する。30分間の培養期間の終わりに、染色物を吸い取り、10倍の倍率の立体顕微鏡を用いてプラークを計数する。次に、ウイルス力価をmlあたりプラーク形成単位として表す。
【図面の簡単な説明】
【0166】
【図1】図1A〜1Cは、FusOn−H2構築のための戦略を示す。図1A。HSV−2ゲノムの概略図。ゲノムは灰色の横棒によって表され、末端繰り返し単位(TR)および内部繰り返し単位(IR)は灰色の箱として示される。ICP10遺伝子の位置も示されている。図1B。ICP10遺伝子の拡大図であり、PKドメインおよびRR1ドメイン、ならびに天然プロモーターの位置を示す。図1C。続いてウイルスゲノムに挿入されてFusOn−H2を構築した改変ICP10遺伝子。図に示したように、PKドメインは、EGFP遺伝子(RR遺伝子との枠組の中)で置換され、遺伝子の元のプロモーターは、最も強い哺乳類遺伝子プロモーターの1つであるサイトメガロウイルスの即時早期プロモーターで置換された。非改変ICP10遺伝子座および改変ICP10遺伝子座のBamHI制限部位を標識化する。PKL、PK、GFPおよびPKRと印を付けたボックスは、図2のサザンハイブリダイゼーションにおいて用いられる4つのプローブがハイブリッド形成する位置を示す。
【図2】図2は、FusOn−H2のサザンブロット分析を示す。サザンブロットハイブリダイゼーションは、親の野生型HSV−2(w)またはFusOn−H2(m)のどちらか由来のBamHI溶解ウイルスDNAを示す。サザンハイブリダイゼーションに用いられる4つのプローブは、左側(left−flank)領域から作られたPKL、PKドメイン領域から作られたPK、右側(right−flank)領域から作られたPKR、EGFP遺伝子から調製されたGFPであった。
【図3】図3は、抗GFPmAbを用いるFusOn−H2のウェスタンブロット分析を示す。FusOn−H2(m)またはその親の野生型HSV−2(w)のどちらかに感染したベロ細胞から、あるいはpSZ−EGFPプラスミドDNA(p)で形質移入したベロ細胞から、細胞溶解物を調製した。
【図4】図4は、培養細胞中のFusOn−H2の表現型キャラクタリゼーションを示す。細胞は、0.01pfu/細胞の図に示したウイルスに感染させるか、または感染させなかった。感染の24時間後に顕微鏡写真を撮影した。白色の矢印によって合胞体を示す。被検細胞の中で、MDA−MB−435はヒト乳癌系統であり、MPans−96はヒトすい臓癌系統であり、SKOV3はヒト卵巣がん系統である。元の倍率は200倍である。
【図5】図5A〜Cは、FusOn−H2の選択的複製を示す。図5A。ベロ細胞を完全循環状態(10%FBS)に保持するか、または24時間血清飢餓状態に保持してから、1pfu/細胞のウイルスに感染させた。図に示した時点で細胞を集め、ベロ細胞単一層上のプラークアッセイによってウイルス収量を定量した。図5B。ウイルス感染の間、単独または50μMのPD98059共存下、低い百分率の血清(2%)を含む培地中でベロ細胞を培養した。感染の24時間および48時間後に細胞を集め、PD98059非存在下でのウェル中の全ウイルス収量を、薬物を含むウェルからの収量で除することによって、ウイルス複製における倍数減少を計算した。図5C。インビトロ培養した一次肝細胞を1pfu/細胞の図に示したウイルスに感染させた。図に示した感染後の時点でウイルスを集め、ベロ細胞単一層上のプラークアッセイによって定量した。*p<0.01、wt186と比較したFusOn−H2(スチューデントt検定)。
【図6】図6AおよびB。殺腫瘍HSVによるヒト癌細胞のインビトロ殺細胞能力。0.01pfu/細胞(A)または0.1pfu/細胞(B)のどちらかで細胞をウイルスに感染させた。図に示した時点のLDHアッセイによって細胞生存率を求めた。ウイルス感染細胞から放出されたLDHを非感染細胞からのそれで除することによって殺細胞百分率を計算した。p<0.01、wt186またはBaco−1と比較したFusOn−H2、Ψp<0.01、wt186と比較したFusOn−H2(スチューデントt検定)。
【図7】図7AおよびB。異種移植型ヒト乳癌に対するFusOn−H2のインビボ抗腫瘍活性。図7A。腫瘍内投与後の治療効果。MDA−MB−435細胞を第2乳腺の脂肪部分に注入することによってヒト乳腺腫瘍異種移植を実施した。腫瘍が直径約5mmに到達したとき、1×106pfuの投与量のウイルスを腫瘍内注射した。治療群は、FusOn−H2、Baco−1またはPBSを含む。ウイルス注入してから図に示した週後に測定した腫瘍体積を治療前の腫瘍体積で除することによって腫瘍成長比を求めた(群あたりn=8のマウス)。図7B。大型乳腺腫瘍異種移植に対する治療効果。腫瘍内注射群および静脈内注射群(それぞれn=5)について、腫瘍はそれぞれ直径10および10〜15mmであった。腫瘍内注射および静脈内注射のそれぞれで、3×106pfuおよび1.5×107pfuの投与量のウイルスを投与した。図6Aの場合と同じ方法で腫瘍成長比を計算した。Ψp<0.05、Baco−1と比較したFusOn−H2、*p<0.01、Synco−2Dと比較したFusOn−H2(スチューデントt検定)。
【図8】図8。ヌードマウスの腹腔内に確立した転移性ヒト卵巣癌異種移植に対するFusOn−H2の治療効果。腹腔への2×106のSKOV3細胞の腹腔内接種によってヒト卵巣がん異種移植を確立した(治療群あたりn=8のマウス)。腫瘍細胞接種の8日後および15日後、腫瘍着床部位から遠く離れた部位への3×106pfuの投与量の殺腫瘍HSVの腹腔内投与をマウスに施した。最初のウイルス注入の4週間後(すなわち腫瘍細胞注射の5週間後)に、マウスを安楽死させた。この図に腫瘍小結節の概観を示し、表1に各動物からの腫瘍小結節の数および腫瘍重量を示す。
【技術分野】
【0001】
(関連出願への相互参照)
この出願は、2005年6月23日に出願された仮出願第60/693,157号(この全体が、参考として本明細書に援用される)への優先権を主張する。
【0002】
(連邦の援助を受けた研究または開発においてなされた発明の権利に関する声明)
本発明は、少なくとも部分的に、NIH補助金第RO1 CA106671−01に従って米国政府が提供した資金を用いて開発された。米国政府は、本発明において特定の権利を有することがある。
【0003】
(発明の分野)
本発明は、ウイルス学、癌生物学、細胞生物学、分子生物学、および癌治療薬を含む薬剤の分野を目的とする。詳しくは、本発明は、ICP10遺伝子の改変形を含む変異体単純ヘルペスウイルス2(HSV−2)と、この変異体HSV−2の悪性疾患の治療への使用を提供する。
【背景技術】
【0004】
(発明の背景)
複製選択的殺腫瘍ウイルスは、固形腫瘍に対する抗腫瘍因子として大きな将来性を示した。これらのウイルスは、腫瘍細胞中では優先的に複製することができるが、正常細胞中では複製する能力が制限される。殺腫瘍ウイルスの基本的な抗腫瘍機序は、ウイルスが、最初に感染した腫瘍細胞から周囲の腫瘍細胞へ伝播し、広がるときの直接細胞変性効果によるものであり、大きな体積の分布と抗癌効果とを実現する。殺腫瘍を目的として、単純ヘルペスウイルス(HSV)は、最も普通には、正常細胞(非分裂型)中での効率的な複製に必要なウイルス遺伝子は削除するが、腫瘍細胞に関しては削除しないことによって改変されてきた。改変は、ウイルスγ34.5遺伝子またはICP6遺伝子の削除を含む。ウイルスγ34.5遺伝子は、HSV感染時に神経毒性因子として機能する(非特許文献1)。この遺伝子を除くと、非分裂細胞中でのウイルス複製が妨げられる(McKieら、(1996) Br J Cancer 74(5): 745−52)。ウイルスICP6遺伝子は、リボヌクレオチド1レダクターゼの大型サブユニットをコードする。この大型サブユニットは、ウイルスDNAが効率的に複製するのに十分なdNTPプールを作り出し、腫瘍細胞中では大量に発現するが、非分裂細胞中では発現しない。従って、この遺伝子を変異させたウイルスは、腫瘍細胞中で選択的に複製し、腫瘍細胞を殺傷することができる。動物研究段階で広範に試験され、現在は臨床試験段階にある殺腫瘍HSV G207は、γ34.5遺伝子座とICP6遺伝子中の挿入変異との両方の複製体において、大腸菌lacZ遺伝子による削除を受ける(Walkerら、(1999) Human Gene Ther. 10(13): 2237−2243)。あるいは、腫瘍特異性プロモーターを用いてHSV複製に必須のγ34.5または他の遺伝子を駆動することによって、殺腫瘍1型HSVを構築することができる(Chungら、(1999) J Virol 73(9): 7556−64)。
【0005】
殺腫瘍単純ヘルペスウイルス(HSV)は、当初、脳腫瘍の治療を目的として設計され、構築された(Andreanskyら、(1996) Proc Natl Acad. Sci.93(21): 11313−11318)。その後、それらは、乳腺(Toda, et at, (1998) Human Gene Ther. 9(15): 2177−2185)、前立腺(Walkerら、(1999) Harman Gene Ther. 10(13): 2237−2243)、肺(Toyoizumiら、(1999) Human Gene Ther. 10(18): 3013−3029)、卵巣(Coukosら、(1999) Clin. Cancer Res.5(6): 1523−1527)、結腸および肝臓(Pawlikら、(2000) Cancer Res. 61(11): 2790−2795)を含むさまざまなその他のヒト固形腫瘍において有効であることが見いだされた。殺腫瘍HSVの安全性も、マウス(Sundaresanら、(2000) J. Virol. 74(8): 3832−3841)および霊長類(ヨザル属)において検証された。後者は、HSV感染症に極めて敏感である(Todoら、(2000) Cancer Gene Ther. 7(6): 939−946)。これらの研究によって、殺腫瘍HSVはインビボ投与に極めて安全であることが確認された。
【0006】
殺腫瘍HSVは、もっぱらHSV−1から構築されてきた。HSV−2は、殺腫瘍ウイルスを構築する目的で検討されたことがない。しかし、HSV−2は、腫瘍溶解剤としての可能性を高める独特の特徴をいくつか有する。例えば、HSV−2は、HSV−1と異なり、好中球、単核細胞およびNK細胞の機能に影響を及ぼす糖蛋白質G(gG)の分泌形をコードすることが報告された(非特許文献2)。そのような性質は、HSV−2から誘導された殺腫瘍ウイルスに、体の先天性免疫の阻害効果に抵抗する能力を提供することがある。先天性免疫は、侵入する微生物に対する宿主の高速応答であり、インビボのHSV複製を制限する主要な因子であることが見いだされた(Dalloulら、(2004) J Clin Virol 30(4): 329−36; Wakimotoら、(2003) Gene Ther 10(11): 983−90)。従って、HSV−2から誘導された殺腫瘍ウイルスは、患者の体が抗HSV先天性免疫を発展させても、自己複製し、広がるはずである。
【0007】
前臨床研究は有望であったが、初期臨床試験の結果によると、現状の殺腫瘍ウイルスは、安全ではあるものの単独では限られた抗腫瘍活性しか有しないことが示唆された(Nemunaitisら、(2001) J. Clin Oncol. 19(2): 289−298)。本発明者らの研究によると、殺腫瘍HSVに細胞膜融合活性を組み込むと、ウイルスの抗腫瘍力を劇的に高めることができることが実証された(Fuら、(2002) Mot Ther. 7(6): 748−754; Fuら、(2003) Cancer Res. 62: 2306−2312。そのような融合殺腫瘍ウイルスは、腫瘍中の合胞体形成をもたらし、ウイルスの破壊力を直接高め、その腫瘍内の広がりを促進する(Fuら、(2003) Cancer Res. 62: 2306−2312)。合胞体形成の腫瘍崩壊機序と融合殺腫瘍HSVによる直接細胞溶解とを独自に組み合わせると、インサイチュ腫瘍抗原提示が促進され、強力な抗腫瘍免疫応答が得られる(Nakamoriら、(2004) Mol. Ther. 9(5): 658−665)。さらに、合胞体形成による融合殺腫瘍HSVの広がりによって、ホストの中の中和抗ウイルス抗体の存在下でも、抗腫瘍活性を維持することが可能になる。ウイルスは、生細胞の中でしか複製することができず、ウイルスの複製には、通常、特定の細胞信号伝達経路の活性化が必要である。多くのウイルスは、これらの信号伝達経路を活性化して複製に役立てるさまざまな戦略を進化の間に取得した。単純ヘルペスウイルス2型(HSV−2)リボヌクレオチドレダクターゼ(ICP10またはRRI)の大型サブユニットは、セリン/スレオニンプロテインキナーゼ(PK)活性を有する固有のアミノ末端ドメインを含む。このPK活性は、細胞Ras/MEK/MAPK経路を活性化することが見いだされた(Smithら、(2000) J Virol 74(22): 10417−29)。
【0008】
LuoとAurelianとは、HSV−2の中のICP10遺伝子のさまざまな削除を含むさまざまなベクターを記載して、特定のモチーフと特定の活性との間の関係を実証している (Luo and Aurelian, (1992) J Biol Chem 267(14): 9645−53)。HSV−2、ICP10遺伝子の改変構築体および削除構築体を用いて、リボヌクレオチドレダクターゼドメインの特定の特性が実証された(Peng et al. (1996) Virology 216(1): 184−96)。
【0009】
リボヌクレオチドレダクターゼ遺伝子からPKドメイン(ICP10 PK)を削除すると、活性化Ras信号伝達経路を予め存在させていない細胞中では、複製するウイルスの能力がひどく低下する(Smith et al (1998) J. Virol. 72(11): 9131−9141)。
【0010】
特許文献1は、HSV−2による攻撃からの保護を提供するワクチンを目的とする。ICP10のプロテインキナーゼドメインを削除したところ、細胞に感染し、細胞を変換させるHSV−2の能力に対して有害な効果が表れる。
【0011】
本発明は、改変されたHSV−2を利用して癌の治療のための新規な治療薬を提供することによって、当分野における必要を満たす。
【特許文献1】米国特許第6,013,265号明細書
【非特許文献1】Chouら、Science(1990)250:1262−1266
【非特許文献2】Bellnerら、J Immunol(2005)174(4): 2235−41
【発明の開示】
【課題を解決するための手段】
【0012】
(発明の要旨)
本発明は、腫瘍崩壊特性を有する強力な改変単純ヘルペスウイルス2型(HSV−2)を提供することによって、当分野の長年の求めに対する答を示す。本発明の特定の実施態様では、ウイルスは、改変されたICP10ポリヌクレオチドを有する。ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するが、プロテインキナーゼ活性の欠失するICP10ポリペプチドをコードする。特定の態様では、ウイルスは、悪性細胞の治療に有用である。特定の実施態様では、ウイルスは、腫瘍細胞中で選択的に複製する。さらに別の特定の実施態様では、ウイルスは、細胞膜融合を起させて少なくともいくつかの望ましくない細胞の培養物、組織または生物を駆逐する。さらに別の特定の実施態様では、ウイルスは、少なくともいくつかの望ましくない細胞の増殖を阻害し、および/または少なくともいくつかの望ましい細胞のアポトーシスを誘導し、および/または強力な抗腫瘍免疫応答を誘導し、および/またはそれらを組み合わせる。
【0013】
野生型HSV−2ウイルスは、セリン/スレオニンプロテインキナーゼ活性などのプロテインキナーゼ(PK)活性を有するアミノ末端ドメインと、リボヌクレオチドレダクターゼ活性を有するc−末端ドメインとを有するポリペプチドをコードするICP10ポリヌクレオチド(RR1ポリヌクレオチドとも呼ばれることがある)を含む。本発明の特定の態様では、ウイルスが、腫瘍細胞中で選択的複製活性(従って、腫瘍細胞を崩壊させる活性)および/またはウイルスを融合型にする活性を含み、あるいは膜融合(合胞体形成)活性を含むという意味で融合型活性を高めるように、内因性PKドメインを改変する。いくつかの実施態様では、プロテインキナーゼドメインをコードする内因性配列の少なくとも一部を削除することによって、コード化されるポリペプチドのプロテインキナーゼ活性が欠失するように、ICP10ポリヌクレオチドを改変する。
【0014】
本発明の別の実施態様では、プロテインキナーゼドメインの少なくとも一部をコードする内因性ICP10ポリヌクレオチドの少なくとも一部を第2のポリヌクレオチドが置換する。さらにその他の実施態様では、置換されなかったICP10配列は、RRドメイン全体を含む。内因性ICP10ポリヌクレオチドの少なくとも一部の置換は、例えば、相同組み換え、またはPCRおよび当業者に公知のその他の方法論の使用を含むその他の適当な遺伝子工学的方法によるなど、任意の適当な方法によって行ってよい。
【0015】
本発明の追加の態様では、ICP10の内因性PKドメインの少なくとも一部を置換するポリヌクレオチドは、任意の適当な配列であってよい。例えば、PKドメインを置換するポリヌクレオチドは、レポーター遺伝子産物または治療遺伝子産物をコードしてよい。第2のポリヌクレオチド(PKドメインの少なくとも一部を置換した)を含む改変ICP10ポリヌクレオチドは、置換ポリヌクレオチドと、残るICP10遺伝子の非置換部分とで構成される融合蛋白質をコードする。本発明で用いるのに適するレポーター遺伝子の非限定的な例は、緑色蛍光蛋白質(SEQ ID.NO:16、GenBank Accession No. U55761)、β−ガラクトシダーゼ、ルシフェラーゼおよび単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)を含む。治療ポリヌクレオチドの非限定例は、単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)、シトシンデアミナーゼ、カスパーゼ−3および野生型p53を含んでよい。
【0016】
本発明のさらにその他の実施態様では、ICP10の内因性PKドメインの少なくとも一部を置換するポリヌクレオチドは、免疫調節遺伝子、または融合膜糖蛋白質(FMG)をコードするポリヌクレオチドであってよい。本発明で用いるのに適する免疫調節遺伝子の非限定的な例は、IL−2、IL−12またはGM−CSF、およびその他のサイトカイン類、F42Kおよびその他のサイトカイン類縁体、あるいはMIP−1、MIP−1ベータ、MCP−1、RANTESおよびその他のケモカインを含む。本発明で用いるのに適する融合膜糖蛋白質をコードするポリヌクレオチドの非限定的な例は、パラミクソウイルスF蛋白質、HIV gp160蛋白質、SIV gp160蛋白質、レトロウイルスEnv蛋白質、エボラウイルスGpまたはインフルエンザウイルスヘムアグルチニン、テナガザル白血病ウイルス(GALV)由来の膜糖蛋白質またはテナガザル白血病ウイルスエンベロープ糖蛋白質C−末端切除形(GALV.fus)を含む。
【0017】
本発明のその他の実施態様では、改変ICP10ポリヌクレオチドは、構成的プロモーターに作動可能結合される。本発明で用いるのに適する構成的プロモーターの非限定的な例は、即時早期サイトメガロウイルス(CMV)プロモーター、SV40早期プロモーター、RSV LTR、ベータチキンアクチンプロモーターおよびHSV0−TKプロモーターを含む。本発明のその他の実施態様では、内因性PKドメイン(またはTMドメイン)の少なくとも一部を置換するポリヌクレオチドは、それに作動可能結合した調節配列を含む。特定の実施態様では、調節配列は、真核細胞中で作動可能であり、さらに別の態様では、癌細胞中で作動可能である。本明細書に記載されている方法および組成物を実際に使用するために有用なプロモーターの非限定的な例は、腫瘍特異性プロモーターおよび/または組織特異性プロモーター、例えば前立腺特異性抗原(PSA)プロモーター、カリクレイン2プロモーターおよびプロベイシンプロモーター(前立腺癌の場合)、L−プラスチンプロモーター(乳腺、卵巣および結腸の癌の場合)、サイログロブリンコアプロモーター(甲状腺癌の場合)、ミッドカインおよびシクロオキシゲナーゼ−2プロモーター(すい臓癌の場合)および大多数の腫瘍の場合のヒトテロメラーゼプロモーター(hTERT)を含んでよい。
【0018】
さらに別の実施態様では、第1の細胞と第2の細胞との間の融合を生成する方法であって、本発明の組成物を第1の細胞に導入することによって第2の細胞膜を第1の細胞膜と融合させる工程を含む方法が提供される。特定の実施態様では、第1の細胞、第2の細胞、または第1の細胞と第2の細胞との両方は、固形腫瘍における悪性細胞などの悪性細胞である。本明細書に記載されている方法および組成物を実際に使用する際に用いるのに適する悪性細胞の非限定的な例は、乳癌細胞、肺癌細胞、皮膚癌細胞、前立腺癌細胞、すい臓癌細胞、結腸癌細胞、脳癌細胞、肝臓癌細胞、甲状腺癌細胞、卵巣癌細胞、腎臓癌細胞、脾臓癌細胞、白血病細胞または骨癌細胞を含んでよい。
【0019】
特定の実施態様では、導入する工程は、さらに、ウイルスをヒトに全身供給することによるなど、ウイルスをヒトに供給する工程と定義される。投与の非限定的な経路は、本明細書に記載されている組成物を、静脈内投与、腫瘍内投与、腹腔内投与またはそれらの任意の組み合わせによって投与することを含んでよい。特定の実施態様では、本組成物を複数の細胞に導入する。
【0020】
追加の実施態様では、ヒトの悪性細胞などの悪性細胞を崩壊させる方法が提供される。この方法は、本発明の組成物を細胞に導入する工程を含み、この導入の後、悪性細胞の膜は別の細胞膜と融合する。
【0021】
別の実施態様では、本発明の組成物を含む哺乳類細胞がある。この哺乳類細胞は、正常リンパ球、マクロファージ、天然キラー細胞または本発明の組成物を腫瘍細胞に送るキャリアとして機能する任意のその他の種類の細胞であってよい。
【0022】
本発明のさらに別の実施態様では、本明細書に記載されている改変HSV−2ウイルスまたはウイルスベクターは、ウイルスに感染した癌細胞中にアポトーシスを誘導する。さらに別の実施態様では、ウイルスに感染していないが、本明細書に記載されている改変HSV−2ウイルスに感染した細胞の周囲の傍観者細胞中にアポトーシスが誘導される。
【0023】
本発明のさらに別の実施態様では、本明細書に記載されているウイルスまたはウイルスベクターは、細胞を溶解させるためのウイルスの効力およびまたは合胞体形成を評価するためのシステムの部分を含む。このシステムは、本明細書に記載されているウイルスまたはベクターと接触した細胞を含む。いくつかの実施態様では、細胞は、原発癌細胞または癌細胞系統由来の細胞などの真核細胞であってよい。その他の実施態様では、細胞は、本明細書に記載されているウイルスまたはベクターにとってホストとして働く原核細胞であってよい。本発明のさらにその他の実施態様では、ウイルスまたはウイルスベクターをさらに含む細胞をインビトロに保持してよい。本発明のさらにその他の実施態様では、ウイルスまたはベクターをさらに含む細胞をマウスなどの動物の中に配置する。本発明のさらにその他の実施態様では、癌細胞をウイルスまたはベクターと接触させて配置する前に、動物中に移植してよい。
【0024】
以上は、以下の本発明の詳細な説明をより良く理解することができるように、本発明の特徴および技術的利点の概要を広く示した。本発明の同じ目的を実行するために他の構造体を改変し、または設計するための基礎として、開示する概念および特定の実施態様を容易に利用し得ることは、当業者には自明である。そのような均等物な構築体は、添付の請求項に示される本発明の技術思想および範囲から逸脱しないことも、当業者には自明である。構成と、動作方法との両方に関して、本発明の特徴であると考えられる新規な特徴、その他の目的および利点とともに、添付の実施例および図と関連させて考慮すれば、以下の説明からよりよく理解されよう。しかし、実施例および図は、それぞれ、例を示し説明するために提供されるにすぎず、本発明の限定の定義としてではないことは明白に理解するべきである。
【発明を実施するための最良の形態】
【0025】
(発明の詳細な説明)
実施例1に記載するHSV−2ウイルス組成物を2006年6月8日にAmerican Type Culture Collection(ATCC)10801 University Blvd. Manassas, VA 20110−2209 USAに寄託した。ATCCは、ブダペスト条約にもとづいて設立された国際的な寄託機関(IDA)である。寄託証明書番号は である。
【0026】
I.定義
本明細書で用いられる用語「単純ヘルペスウイルス」または「HSV」は、ヒトを含む哺乳類に感染する被膜、二十面体、二本鎖DNAウイルスを指す。野生型HSVは、最終分化非分裂細胞と分裂細胞との両方に感染し、内部で複製する。「HSV−2」は、ICP10遺伝子を含むHSV系のメンバーを指す。本明細書で用いられる用語「FusOn−H2」は、本明細書で記載される、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性の欠失したポリペプチドをコードする改変ICP10ポリヌクレオチドを有するHSV−2変異体を指す。
【0027】
本明細書で用いられる用語「細胞膜融合」は、例えば2つの隣接する細胞など、少なくとも2つの細胞の外部膜の融合を指す。
【0028】
本明細書で用いられる用語「融合活性の増強」は、細胞膜融合の増強、増加、強化、増加、拡大またはそれらの組み合わせを指す。
【0029】
本明細書で用いられる用語「殺腫瘍」は、直接的または間接的に、結果として悪性細胞を崩壊させることができる剤の性質を指す。特定の実施態様では、この性質は、悪性細胞膜の別の膜と融合を引き起こすことを含む。
【0030】
本明細書で用いられる用語「選択複製」または「条件付き複製」は、特定の組織(例えば腫瘍)の中で選択的に成長する殺腫瘍ウイルスの能力を指す。
【0031】
本明細書で用いられる用語「合胞体」は、著しく大きな数の融合細胞を含む複数核巨大細胞形成を指す。
【0032】
本明細書で用いられる用語「ベクター」は、細胞の中に導入して自己複製させることができるようにするために、核酸配列を挿入することができるキャリア核酸分子を指す。挿入された核酸配列は、ベクターを導入した細胞に対して異物であるか、または細胞中の配列と相同であるが配列が本来見いだされない宿主細胞核酸中の位置にあるか、どちらの場合にも、「外因性」と呼ばれる。ベクターは、非ウイルスDNAベクターまたはウイルスベクターのどちらであってもよい。ウイルスベクターは、ウイルス蛋白質中にカプセル化され、細胞を感染させることができる。ベクターの非限定的な例は、ウイルスベクター、非ウイルスベクター、裸のDNA発現ベクター、プラスミド、コスミド、人工染色体(例えばYAC)、ファージベクター、陽イオン縮合剤と会合したDNA発現ベクター、リポソーム中にカプセル化されたDNA発現ベクターまたは特定の真核細胞、例えばプロデューサー細胞を含む。特に断らない限り、本明細書で用いられる「ベクター」は、DNAベクターおよびウイルスベクターを指す。当業者は、標準的な組み換え技法によってベクターを構築することが十分にできる。一般的に、これらは、Sambrook et al., Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press (1989)およびその中の引用文献を含む。ウイルス学的考察は、Coen D. M, Molecular Genetics of Animal Viruses in Virology, 2”d Edition, B. N. Fields (editor), Raven Press, N.Y. (1990)およびその中の引用文献にも概説されている。
【0033】
用語「発現ベクター」は、転写することができるRNAをコードする核酸を含む任意の種類の遺伝子の構築体を指す。ある場合には、RNA分子は、次に、蛋白質、ポリペプチドまたはペプチドに翻訳される。他の場合には、例えば、アンチセンス分子またはリボザイムの産生では、これらの配列は翻訳されない。発現ベクターは、特定の宿主細胞中に作動可能結合した暗号配列の転写およびおそらく翻訳に必要な核酸配列を参照するさまざまな「制御配列」を含んでよい。転写および翻訳を支配する制御配列に加えて、ベクターおよび発現ベクターは、他の機能を果し、ならびに下記に記載されている核酸配列を含んでよい。
【0034】
「プロモーター」は、転写の開始および転写の速度が制御される核酸配列の領域となる制御配列である。それは、核酸配列の時間的および空間的転写を開始するかまたは調節するRNAポリメラーゼおよびその他の転写因子などの調節蛋白質および分子が結合する遺伝要素を含んでよい。句「動作有効配置される」、「作動可能結合される」、「制御される」および「転写調節される」は、核酸配列に対してプロモーターがその配列の転写開始および/または発現を制御する正しい機能位置にありおよび/または方向を有することを意味する。非限定的なプロモーターの例は、構成的プロモーター、組織特異性プロモーター、腫瘍特異性プロモーターまたは外因性誘導要素に制御される内因性プロモーターを含む。
【0035】
本明細書で用いられる用語「構成的プロモーター」は、細胞周期全体を通じて時間的に連続して遺伝子またはポリヌクレオチドの発現を推進するプロモーターを指す。構成的プロモーターは、細胞周期全体を通して連続して動作してそれが関連する遺伝子またはポリヌクレオチドの発現を推進する限り、細胞型特異性または組織型特異性であってよい。非限定的な構成的プロモーターの例は、即時早期サイトメガロウイルス(CMV)プロモーター、SV40早期プロモーター、RSV LTR、ベータチキンアクチンプロモーターおよびHSV TKプロモーターを含む。
【0036】
用語「エンハンサー」は、核酸配列の転写活性化の制御に関与するシス活性調節配列を指す。
【0037】
本明細書では、用語「接触する」および「曝露される」は、細胞に適用されると、ウイルス、ウイルスベクター、非ウイルスベクター、DNAベクターまたは任意のその他の治療剤が、単独または組み合わされて、標的細胞に投与されるかまたは標的細胞と直接共存して配置されるプロセスを記述するために用いられる。
【0038】
句「改変ICP10ポリヌクレオチド」は、リボヌクレオチドレダクターゼ(RR)活性を有するがプロテインキナーゼ活性の欠失したICP10ポリペプチドをコードするICP10ポリヌクレオチドを指す。
【0039】
句「リボヌクレオチドレダクターゼ活性」は、ICP10ポリヌクレオチドによってコード化されたポリペプチドのC末端のドメインの、ウイルス複製に必要なデオキシヌクレオチド三リン酸(dNTP)を十分に発生させる能力を指す。
【0040】
句「プロテインキナーゼ活性」は、ICP10ポリヌクレオチドによってコード化されたポリペプチドのアミノ末端ドメインの、Ras/MEK/MAPK経路を活性化することができるセリンおよびスレオニン残基をリン酸化する能力を指す。
【0041】
本明細書で用いられる用語「傍観者腫瘍細胞」は、本明細書に記載されている改変HSV−2ウイルスに感染していないが、本明細書に記載されているウイルスまたはベクターに感染した腫瘍細胞に隣接するかまたは近傍にある腫瘍細胞を指す。
【0042】
本明細書で用いられる用語「抗癌剤」は、例えば、癌細胞を殺し、癌細胞中にアポトーシスを誘導し、癌細胞の成長速度を低下させ、転移の発生率または数を減らし、腫瘍サイズを小さくし、腫瘍成長を阻害し、腫瘍または癌細胞への血液の供給を減らし、癌細胞または腫瘍に対する免疫応答を促進し、癌の進行を妨げるかまたは阻害し、あるいは癌を有する被験者の寿命を伸ばすことによって、被験者の体内の癌に負の影響を及ぼすことができる剤を指す。
【0043】
本明細書で用いられる句「医薬品として」または「薬理学的に許容される」は、動物またはヒトに適切に投与されると、拒否反応、アレルギー反応またはその他の有害な反応を生じさせない分子実体および組成物を指す。句「医薬品として許容されるキャリア」は、任意のおよびすべての溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張剤および吸収遅延剤ならびに類似物を含む。
【0044】
用語「単位用量」は、被験者に用いるのに適する物理的に離散した単位を指す。各単位は、その投与に伴って所望の応答を生み出すと計算された、予め定められた量の治療組成物、すなわち適切な経路および治療計画を指す。
【0045】
本明細書で用いられる用語「有効な」または「治療として有効な」は、症状の悪化を阻むこと、疾患の発症を防ぐこと、疾患の広がりを防ぐこと、疾患の少なくとも1つの症状の回復、あるいはそれらの組み合わせを指す。
【0046】
II.導入
ウイルスは、生細胞の中でしか複製することができず、ウイルスの複製は、通常、特定の細胞信号伝達経路の活性化を必要とする。多くのウイルスは、これらの信号伝達経路を活性化して複製に利用するさまざまな戦略を進化の間に取得した。単純ヘルペスウイルス2型(HSV−2)リボヌクレオチドレダクターゼ(ICP10またはRR1)の大型サブユニットは、セリン/スレオニンプロテインキナーゼ(PK)活性を有する固有アミノ末端ドメインを含む。このPK活性は、細胞Ras/MEK/MAPK経路を活性化することが見いだされた(Smith, et at, (2000) J Virol 74(22): 10417−29)。その結果、このPKドメイン(ICP10 PK)をリボヌクレオチドレダクターゼ遺伝子から削除すると、予め存在する活性化Ras信号伝達経路がない細胞などの、細胞中で自己複製するウイルスの能力がひどく低下する報告された(Smith, et at, (1998) J. Virol. 72(11): 9131−9141)。
【0047】
ここで、本発明者らは、HSV−2のPKドメインを置換および/または改変し、その結果、改変ICP10遺伝子がコードする蛋白質にリボヌクレオチドレダクターゼ活性を持たせるがプロテインキナーゼ活性を欠失させると、ウイルスは、腫瘍細胞(少なくともRas信号伝達経路が腫瘍形成によって構成的に活性化された腫瘍細胞)の中で選択的に複製し、腫瘍細胞を崩壊させることを示す。さらに、本明細書に記載されているICP10ポリヌクレオチドの改変によって、ウイルスは固有融合型になる。すなわち、腫瘍細胞をこのウイルスに感染させると、広範な細胞膜融合(合胞体形成)が誘導される。この性質は、腫瘍細胞に対するウイルスの崩壊力を増大させる。さらに、インビボ研究によると、このウイルスは、局所投与にも全身投与にも極めて安全である。
【0048】
本発明のいくつかの実施態様では、PKドメインの改変は、緑色蛍光遺伝子を発現する遺伝子などのレポーター遺伝子の挿入、および/または即時早期サイトメガロウイルスプロモーターなどの構成的プロモーターによる野生型プロモーター遺伝子の置換を含む。
【0049】
いくつかの実施態様では、ICP10遺伝子のプロテインキナーゼ活性ドメインをコードするポリヌクレオチドに第2のポリヌクレオチドを挿入するか、またはプロテインキナーゼドメインの一部を第2のポリヌクレオチドで置換するかのどちらかによって、HSV−2を遺伝子操作し、これによって、改変ポリヌクレオチドがコードするポリペプチドに、リボヌクレオチドレダクターゼ活性を持たせるがプロテインキナーゼ活性を欠失させる。例えば、第2のポリヌクレオチドは、融合膜糖蛋白質などの糖蛋白質をコードしてよい。本発明の範囲内で用いられる好ましい糖蛋白質は、テナガザル白血病ウイルスエンベロープ融合膜糖蛋白質(GALV.fus)の切断型である。本発明の特定の態様では、本発明の殺腫瘍ウイルスとしてGALV.fusを発現させると、ウイルスの抗癌効果は顕著に増強される。
【0050】
いくつかの実施態様では、本発明の改変HSV−2は、削除などの、ICP10の中の変異を含む。変異によって、ウイルスは細胞融合特性を得る。そのような変異は、ウイルススクリーニング時にランダムに発生させても自然から得てもよく、次に、本明細書に記載され、および/または当分野で既知の手段によって、細胞融合特性を有する潜在的な候補のプールの機能を評価する。融合性表現型をもたらす変異は、点変異、フレームシフト、反転、削除、スプライシング誤変異、転写後処理変異、特定のウイルス糖蛋白質の過剰発現、それらの組み合わせ等であってよい。変異は、特定のHSV−2の配列を解析し、それを既知の野生型配列と比較することによって特定してよい。
【0051】
本発明の改変HSV−2は、例えば、悪性細胞の広がりを阻み、分裂を減少させまたは阻害し、根絶し、発生または増殖を妨げ、またはそれらを組み合わせるなど、悪性細胞の治療に有用である。悪性細胞は、固形腫瘍などの任意の形の癌由来であってよいが、その他の形も治療可能である。本発明の改変HSV−2は、肺、肝臓、前立腺、卵巣、胸、脳、すい臓、睾丸、結腸、頭および頸、黒色腫およびその他の種類の悪性腫瘍の治療に有用である。本発明は、転移段階を含む、癌疾患の任意の段階で悪性細胞を治療するのに有用である。本発明は、単独治療法として、または化学療法、手術、放射線治療および類似法を含む別の治療手段とともに利用してよい。
【0052】
III.改変ICP10ポリヌクレオチド
本発明は、改変ICP10ポリヌクレオチドを有するHSV−2変異体を記載する。改変ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ(PK)活性の欠失したポリペプチドをコードする。ICP10ポリヌクレオチドは、機能PKドメインをコードするために必要な配列の少なくとも1部を削除するか、またはPKドメインをコードする配列の少なくとも1部を第2のポリヌクレオチドで置換するかのどちらかによって改変してよい。突然変異生成、ポリメラーゼ連鎖反応(PCR)、相同組み換え、または当業者に既知の任意のその他の遺伝子操作技法を含む任意の適当な方法を用いて改変ICP10ポリヌクレオチドを生成させることができることは、当業者には自明である。
【0053】
A.突然変異生成
本発明の特定の実施態様では、例えば、さまざまな標準的な突然変異誘発手順のうち任意のものを用いる削除によって、HSV−2ウイルスのICP10配列を変異させる。変異は、ヌクレオチド配列、単一遺伝子または遺伝子のブロックの改変を含んでよい。変異は、単一ヌクレオチドを含んでよく(DNA配列内の単一ヌクレオチド塩基の除去、追加または置換を含む点変異など)、あるいは、多数のヌクレオチドの挿入または削除を含んでよい。変異は、DNA複製の忠実さの誤差などの出来事の結果として自発的に起きてよく、あるいは、化学的または物理的突然変異原への曝露に続いて誘起されてよい。当業者に公知の特定の標的設定法を用いて、変異に部位指向性も付与してよい。
【0054】
B.遺伝子組み換え
本発明の他の実施態様では、遺伝子組換え技法を用いてICP10ポリヌクレオチドを改変し、PKドメインをコードする配列の少なくとも一部を削除または置換する。削除/置換されるPKドメインの領域は、改変ICP10ポリヌクレオチドがコードするポリペプチドに、リボヌクレオチドレダクターゼ活性を維持させるがプロテインキナーゼ活性を欠失させる限り、任意の適当な領域であってよい。ただし、特定の実施態様では、PKドメインを改変すると、8つのPK触媒モチーフ(アミノ酸残基106〜445であるが、PK活性は、アミノ酸残基1〜445と考えられている)の1つ以上、および/または膜貫通(TM)領域、および/または不変のLys(Lys176)に影響を及ぼす。野生型ICP10ポリペプチド配列の例は、SEQ ID No:15(National Center for Biotechnology Information’s GenBank database Accession No.1813262A)に提供される。ICP10ポリペプチドをコードする野生型ポリヌクレオチドの例は、SEQ ID No:17に提供される。
【0055】
特定の実施態様では、PK活性に必要なPKドメインをコードする配列の一部を単に削除することによって、ICP10ポリヌクレオチドを改変する。PKドメインをコードする配列の少なくともいくつかが欠失したICP10ポリヌクレオチドの例は、SEQ ID No:18に提供される。別の実施態様の例では、SEQ ID:19に提供されるように、PKドメイン全体が削除されるように、ICP10ポリヌクレオチドを改変する。SEQ ID No:18とSEQ ID No:19との両方が、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くポリペプチドをコードするので、両方が、本明細書に記載されているHSV−2変異体を発生させるため用いるのに適する。特定の本発明の実施態様では、SEQ ID No:18またはSEQ ID No:19に開示されている改変ICP10ポリヌクレオチドは、内因性HSV−2プロモーターに制御されてよく、あるいは、SEQ ID No:20に記載されている即時早期サイトメガロウイルスプロモーターなどの構成的プロモーターに作動可能結合されてよい。
【0056】
本発明のさらに別の実施態様では、PKドメインをコードする配列の少なくとも一部を、緑色蛍光蛋白質など、ICP10ポリヌクレオチドのRRドメインをコードする配列のフレーム内に配置された、第2のポリヌクレオチドで置換することによって、ICP10ポリヌクレオチドを改変する。この構築体は、内因性HSV−2プロモーターに制御されてもよく、CMVプロモーター(SEQ ID NO:20)などの構成的プロモーターに制御されてもよい。実施例1に、この後者の構築体(GFP置換ポリヌクレオチドおよびCMVプロモーターを含む)をさらに詳しく記載する。
【0057】
本発明の別の態様では、HSV−2の内因性ICP10のプロテインキナーゼ活性ドメインの少なくとも一部を置換するポリヌクレオチドは、ウイルス融合膜糖蛋白質(FMG)などの細胞膜融合誘導性ポリペプチドの少なくとも融合性部分をコードすることができる。このポリペプチドは、好ましくは、例えば、実質的に中性pH(pH約6〜8など)で細胞膜融合を誘導することができる。
【0058】
特定の実施態様では、FMGは、MLV(例としてはSEQ ID NO:6)またはGALV(例としてはSEQ ID NO:5)などの、C−型レトロウイルスエンベロープ蛋白質由来の少なくとも融合性ドメインを含む。細胞質ドメインの一部、ほとんど、またはすべてを削除したレトロウイルスエンベロープ蛋白質は、ヒト細胞の場合、そのような操作が超融合活性を生む結果となるので有用である。いくつかの実施態様では、特定の改変をウイルス膜糖蛋白質中に導入して細胞膜融合を誘導する機能を増強する。例えば、複数のレトロウイルスおよびヘルペスウイルス糖蛋白質の細胞質ドメインを切断すると、融合活性が増加し、ときには、同時にウイルス粒子に組み込まれる効率が低下することが示された(Rein et al., (1994) J Virol 68(3): 1773−81)。
【0059】
細胞膜融合ポリペプチドのいくつかの例は、はしかウイルス融合蛋白質(SEQ ID NO:7)、HIV gp160(SEQ ID NO:8)およびSIV gp160(SEQ ID NO:9)蛋白質、レトロウイルスEnv蛋白質(SEQ ID NO:10)、エボラウイルスGp(SEQ ID NO:11)およびインフルエンザウイルスヘムアグルチニン(SEQ ID NO:12)を含む。
【0060】
その他の実施態様では、第2の機能ポリヌクレオチドをPKドメインの中に挿入するか、または用いてPKドメインの一部または全体を置換してよい。この第2の機能ポリヌクレオチドは、免疫調節剤または他の治療剤をコードしてよい。これらの追加の剤は、細胞表面受容体およびGAP接合の上方調節、細胞増殖抑制剤および分化剤に影響を及ぼし、細胞接着を阻害し、または、悪性細胞のアポトーシス感受性を増加させると考えられる。免疫調節性または他の治療薬剤をコードするポリヌクレオチドの非限定的な例は、腫瘍壊死因子;インターフェロン、アルファ、ベータ、ガンマ;インターロイキン−2(IL−2)、IL−12、顆粒球マクロファージコロニー刺激因子(GM−CSF)、F42K、MIP−1、MIP−1β、MCP−1、RANTES、単純ヘルペスウイルスチミジンキナーゼ(HSV−tk)、シトシンデアミナーゼおよびカスパーゼ−3を含む。
【0061】
本発明のさらに別の実施態様では、レポーター蛋白質をコードするポリヌクレオチドの挿入によってICP10ポリヌクレオチドを改変する。非限定的なレポーター蛋白質をコードするポリヌクレオチドの例は、緑色蛍光蛋白質、強化緑色蛍光蛋白質、β−ガラクトシダーゼ、ルシフェラーゼおよびHSV−tkを含む。
【0062】
C.リボヌクレオチドレダクターゼ活性アッセイ
RRの生物学的活性は、既に説明した方法(Averettら、J. Biol. Chem. 258:9831−9838 (1983)およびSmith et al., J. Virol. 72:9131−9141 (1998))に以下の変更を施して検出することができる。BHK細胞を、最初、完全GMEM(10%FBSを含む)中で密生させて成長させた後、0.5%FBS EMEM中で3日間培養し、続いて、20pfuの野生型HSV、HSV−2変異体に感染させ、または偽感染させた。感染の20時間後に細胞を集め、500μlのHD緩衝液[100mMのHEPES緩衝液(pH7.6)、2mMのジチオスレイトール(DTT)]に再び懸濁させ、氷の上で15分間培養した後、30秒間超音波処理した。遠心分離(16,000g、20分間、4℃)によって細胞破片を除去し、上清を45%飽和の結晶硫酸アンモニウム(0.258g/ml)で沈殿させる。2回目の遠心分離(16,000g、30分間)の後、ペレットを100μlのHF緩衝液に溶解させ、50μlを取って等しい体積の2×反応緩衝液(400mMのHEPES緩衝液(pH8.0)、20mMのDTTおよび0.02mM[3H]−CDP(24Ci/mmol、Amersham, Chicago, Il)と混合する。100mMのヒドロキシ尿素と10mMのEDTA(pH8.0)とを加え、3分間沸騰させて反応を停止させる。次に1mlのCrotalux atrox毒(Sigma, St. Louis, MO)を加え、37℃で30分間培養した後、さらに3間沸騰させる。次に、この溶液を0.5mlのダウエックス−1ホウ酸塩カラムに通し、2mlの水で試料を溶出させ、4つの溶出画分に集めてバイオフルオール(Biofluor)(New England Nuclear, Boston, MA)と混合した後、シンチレーション計測する。リボヌクレオチドレダクターゼ活性を単位/mg蛋白質で表す。1単位は、1nmol[3H]CDPのdCDP/hr/mg蛋白質への変換を表す。
【0063】
D.プロテインキナーゼ活性アッセイ
改変ICP10ポリヌクレオチドがプロテインキナーゼ活性の欠失したポリペプチドをコードするか測定するために、改変ICP10ポリヌクレオチドまたは野生型HSV−2を有するHSV−2(moi=200、感染後16時間)に感染させた細胞の抽出分を抗LA−1抗体で免疫沈降させ、Chung et al. J. Virol. 63:3389−3398, 1998および米国特許第6,013,265号に記載されているPKアッセイに付す。通常、BCA蛋白質アッセイキット(PIERCE, Rockford IL.)を用いて細胞抽出物の免疫沈降物の蛋白質濃度を規格化し、20mMのTrid−HCl(pH7.4)、0.15MのNaClを含むTS緩衝液で洗浄し、20mMのTris−HCl(pH7.4)、5mMのMgCl2、2mMのMnCl2、10μCiの[32P]ATP(3000Ci/ミリモル、DuPont, New England Research Prod.)からなる50μlのキナーゼ反応緩衝液に懸濁させ、30℃で15分間培養する。ビーズを1mlのTS緩衝液で1回洗浄し、100μlの変性溶液に再懸濁させ、5分間沸騰させる。次に、7%ポリアクリルアミドゲルの上のSDS−PAGEによって蛋白質を分割する。次に、既に記載した(Aurelian et. al., Cancer Cells 7:187−191 1989)ように、蛋白質をニトロセルロース膜上に電気移動させ、特異抗体を用いるンキュベーションによって免疫ブロットした後、室温で1時間蛋白質A−ペルオキシダーゼ(Sigma, St. Louis, MO)処理した。Smith et al., Virol. 200:598−612(1994)に記載されているECL試薬(Amersham, Chicago, IL)によって検出を行ってよい。
【0064】
IV.ベクター構築。
【0065】
本発明は、ICP10配列の少なくとも一部を置換または削除し、これによって、改変ICP10ポリヌクレオチドによってコード化される蛋白質に、リボヌクレオチドレダクターゼ活性を持たせるがプロテインキナーゼ活性を欠失させ、特定の実施態様では、構成的プロモーターなどの調節配列をさらに含む、HSV−2ベクターを目的とする。いくつかの実施態様では、組成物は、改変ICP10遺伝子を含む裸のDNAベクター(非ウイルス)であり、他の実施態様では、組成物は、改変ICP10遺伝子を有する組み換えHSV−2である。裸のDNAベクターと組み換えウイルスとの両方は、以下の成分のいくつかまたはすべてをさらに含んでよい。
【0066】
A.ベクター
上記で定義したように、ベクターは、プラスミド、コスミド、ウイルス(バクテリオファージ、動物ウイルスおよび植物ウイルス)ならびに人工染色体(例えばYACs)を含むがそれらに限定されない。当分野では、設計されたウイルスおよびDNAベクターを構築するための方法が知られている。通常、これらは、Sambrooet al., Molecular Cloning:A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press(1989)およびその引用文献である。ウイルス学的考察は、Coen D. M., Molecular Genetics Animal Viruses in Virology, 2.sup.nd Edition, B. N. Fields (Editor), Raven Press, N.Y. (1990)およびその引用文献でも概説されている。
【0067】
発現ベクターは、さまざまな「制御配列」を含んでよい。制御配列は、特定の宿主細胞中に作動可能結合した暗号配列の転写およびおそらく翻訳に必要な核酸配列を参照する。転写および翻訳を支配する制御配列に加えて、DNAベクター、発現ベクターおよびウイルスは、他の機能を果し、ならびに下記に記載されている核酸配列を含んでよい。
【0068】
1.プロモーターおよびエンハンサー
一般に、プロモーターは、RNA合成の開始部位の位置を決めるために機能する配列を含む。これの最もよく知られた例は、TATAボックスであるが、TATAボックスが欠失したいくつかのプロモーター(例えば哺乳類の末端デオキシヌクレオチジルトランスフェラーゼ遺伝子のプロモーターおよびSB40後期遺伝子のプロモーター)では、開始部位自体の上にある離散的な要素が、開始の位置を決めるのを助ける。追加のプロモーター要素が転写開始の頻度を調節する。一般に、これらは、開始部位の上流の領域30から110に配置されるが、複数のプロモーターが開始部位の下流にも機能要素を含んでいることが示された。コード化配列をプロモーターに「制御される」ようにするために、転写解読フレームの転写開始部位の5’−末端を、選ばれたプロモーターの「下流」(すなわち3’−側)に配置する。「上流」のプロモーターは、DNAの転写を刺激し、コード化されたRNAの発現を促進する。
【0069】
プロモーター要素の間の間隔は、頻繁に柔軟であり、その結果、要素が反転しても、互いに移動してもプロモーター機能は保たれる。tkプロモーターにおいて、プロモーター要素の間の間隔は、50bpの間隔まで増加させないと活性は低下し始めない。プロモーターによって、個々の要素は、協力してまたは独立に機能して、転写を活性化することができるようである。プロモーターは、エンハンサーとともに用いてもよくてまたは用いなくてよい。
【0070】
プロモーターは、コード化セグメントおよび/またはエキソンの上流に位置する5’非コード化配列を単離することによって得られるように、核酸配列と自然に会合するものであってよい。同様に、エンハンサーは、その配列の下流または上流のどちらかに位置する核酸配列と自然に会合するものであってよい。あるいは、コード化核酸セグメントを組み換えプロモーターまたは非相同プロモーターに制御される位置に配置することによって、特定の利点が得られる。非相同プロモーターは、その自然環境では、通常、核酸配列と会合していないプロモーターを指す。組み換えエンハンサーまたは非相同エンハンサーは、その自然環境では正常な核酸配列と会合していないエンハンサーを指す。そのようなプロモーターまたはエンハンサーは、他の遺伝子のプロモーターまたはエンハンサー、任意の他のウイルス、原核生物細胞または真核生物細胞から単離されたプロモーターまたはエンハンサー、および「天然」でない、すなわち、さまざまな転写調節領域のさまざまな要素および/または発現を変える変異を含むプロモーターまたはエンハンサーを含む。例えば、組み換えDNA構築に最も普通に用いられるプロモーターは、βラクタマーゼ(ペニシリナーゼ)、ラクトースおよびトリプトファン(trp)プロモーター系を含む。プロモーターおよびエンハンサーの核酸配列を合成によって作り出すことに加えて、PCRを含む組み換えクローン形成および/または核酸増幅技術を本明細書に開示されている組成物とともに用いて配列を作り出してよい(米国特許第4,683,202号および第5,928,906号を参照のこと)。さらに、ミトコンドリア、クロロプラストおよび類似物などの非核オルガネラ中の配列の転写および/または発現を指令す制御配列も使用することができると考えられる。
【0071】
もちろん、発現のために選ばれたオルガネラ、細胞型、組織、器官または生物中のDNAセグメントの発現を効果的に導くプロモーターおよび/またはエンハンサーを使用することは重要になる。使用されるプロモーターは、導入されたDNAセグメントの高レベル発現を導く適切な条件下で、組み換え蛋白質および/またはペプチドの大量産生に有利であるように、構成的、組織特異的、誘導可能、および/または有用であってよい。プロモーターは、非相同性であってよく、または内因性であってよい。
【0072】
さらに、任意のプロモーター/エンハンサーの組み合わせを用いて発現を推進してよい。T3、T7またはSP6細胞質発現系を用いることは、別の可能な実施態様である。真核細胞は、適切な細菌ポリメラーゼが、投与複合体の一部または追加の遺伝子発現構築体として提供されるなら、特定の細菌プロモーターからの細胞質転写を支援することができる。
【0073】
組織特異性プロモーターまたは要素の同定、ならびに活性の特性を把握するアッセイは、当業者に公知である。そのような領域の非限定的な例は、ヒトLIMK2遺伝子(Nomoto et al. (1999) Gene 236(2): 259−271)、ソマトスタチン受容体−2遺伝子(Kraus et al., (1998) FEBS Lett. 428(3): 165−170)、マウス精巣上体レチノイン酸結合遺伝子(Lareyre et al., (1999) J. Biol. Chem. 274(12): 8282−8290)、ヒトCD4 (Zhao−Emonet et al., (1998) Biochem. Biophys. Aeta,1442(2−3): 109−119)、マウスα−2(XI)コラーゲン(Tsumaki, et at, (1998), J. Biol. Chem.273(36): 22861−4)、DIAドーパミン受容体遺伝子(Lee, et at, (1997), DNA Cell Biol. 16(11): 1267−1275)、インスリン様増殖因子II (Wu et at, (1997) Biophys Biochem Res. Comm. 233(1): 221−226)およびヒト小板内皮細胞接着分子−1(Almendro et at, (1996) J. Immunol. 157(12): 5411−5421)を含む。
【0074】
2.開始信号および内部リボソーム結合部位
コード化配列の効率的な翻訳には、特定の開始信号も必要になることがある。これらの信号は、ATG開始コドンまたは隣接配列を含む。ATG開始コドンを含む外因性翻訳制御信号を提供する必要がある。当業者ならこれを測定し、必要な信号を提供することは容易にできる。開始コドンは、挿入部分全体の翻訳を確実にするために、所望の暗号配列の解読フレームの「フレーム内」になければならないことは周知である。外因性翻訳制御信号および開始コドンは、天然であっても合成であってもよい。適切な転写エンハンサー要素をふくむことによって、発現の効率が増強されることがある。
【0075】
本発明の特定の実施態様では、内部リボソーム進入部位(IRES)要素を用いて、多重遺伝子メッセージ、または多シストロン性メッセージを作り出す。IRES要素は、5’メチル化キャップ依存性翻訳のリボソーム走査モデルを省略し、内部部位で翻訳を開始することができる。IRES要素は、非相同性開放読み取りフレームに結合することができる。IRESによってそれぞれ分離された複数の開放読み取りフレームを一緒に転写して、多シストロン性メッセージを作り出すことができる。IRES要素のおかげで、各開放読み取りフレームは、効率的な翻訳のためにリボソームにアクセスすることができる。単一のメッセージを転写する単一のプロモーター/エンハンサーを用いて、複数の遺伝子を効率的に発現することができる(米国特許第5,925,565号および第5,935,819号を参照すること)。
【0076】
3.終止シグナル
本発明のベクターまたは構築体は、通常、少なくとも1つの終止シグナルを含む。「終止シグナル」または「ターミネーター」は、RNAポリメラーゼによるRNA転写物の特定の終了に関与するDNA配列で構成される。従って、特定の実施態様では、RNA転写体の産生を終了する終止シグナルを包含する。望ましいメッセージレベルを実現するために、in vivoでターミネーターが必要になることがある。
【0077】
真核生物系では、ターミネーター領域は、ポリアデニル化部位を露出させるように新しい転写産物を部位特異的に開裂させることができる特定のDNA配列も含んでよい。これによって、専用の内因性ポリメラーゼに、転写体の3’末端に約200A残基の並び(ポリA)を加えるように信号を伝達する。このポリA尾部で改変されたRNA分子は、安定性が増し、より効率的に変換されるようである。従って、真核生物が関与する他の実施態様では、ターミネーターは、RNAの開裂のための信号を含み、ターミネーター信号は、メッセージのポリアデニル化を促進すると考えられる。ターミネーターおよび/またはポリアデニル化部位要素は、メッセージレベルを改良し、カセットから他の配列への読み通しを最小限にするように働くことができる。
【0078】
本発明において用いられるターミネーターは、本明細書に記載されている任意の既知の転写ターミネーター、または当業者に既知の転写ターミネーターを含み、例えばウシ成長ホルモンターミネーターまたは例えばSV40ターミネーターなどのウイルス終止配列などの遺伝子の終結配列を例えば含むが、それらに限定されない。特定の実施態様では、終止シグナルは、配列の切断によるなどの転写可能配列または翻訳可能配列の欠失であってよい。
【0079】
4.ポリアデニル化シグナル
発現、特に真核生物の発現では、一般に、転写体の適切なポリアデニル化を実現するために、ポリアデニル化シグナルを含む。ポリアデニル化シグナルの性質は、本発明の実施を成功させる上で不可欠とは考えられず、任意のそのような配列を使用してよい。好ましい実施態様は、SV40ポリアデニル化シグナルまたはウシ成長ホルモンポリアデニル化シグナルを含む。これらはともに簡便であり、さまざまな標的細胞中で良好に機能することが知られている。ポリアデニル化は、転写体の安定性を増し、または細胞質輸送を促進することがある。
【0080】
5.選択可能および検索可能マーカー
本発明の特定の実施態様では、本発明の核酸構築体を含む細胞は、発現ベクターにマーカーを含ませることによってインビボまたはインビトロで特定してよい。そのようなマーカーによって、特定することができる変化を細胞に付与し、発現ベクターを含む細胞を容易に特定することが可能になる。通常、選択可能なマーカーは、選抜を可能にする属性を付与するものである。正の選択可能マーカーは、マーカーの存在がその選択を可能にするものであり、負の選択可能マーカーは、その存在がその選択を妨げるものである。正の選択可能マーカーの1例は、薬剤耐性マーカーである。
【0081】
通常、薬物選択マーカーを含むと、クローン形成および変換体の特定が容易になる。例えば、ネオマイシン、ピューロマイシン、ハイグロマイシン、DHFR、GPT、ゼオシンおよびヒスチジノールへの抵抗性を付与する遺伝子は、有用な選択可能マーカーである。諸条件の実体化によって変換体の差別化を可能にする表現型を付与するマーカーに加えて、比色分析を土台とするGFPなどの選抜可能マーカーを含むその他の種類のマーカーも包含される。あるいは、単純ヘルペスウイルスチミジンキナーゼ(tk)またはクロラムフェニコールアセチルトランスフェラーゼ(CAT)などの選抜可能酵素を利用してよい。当業者には、どのように免疫学的マーカーを、おそらく蛍光活性化細胞分類(FACS)分析とともに、使用するかも既知と考えられる。遺伝子産物をコードする核酸と同時に発現することができる限り、用いられるマーカーは重要ではないと考えられる。選択可能マーカーおよび選抜可能マーカーのさらに別の例が当業者に公知である。
【0082】
ベクターは、適当な方法によって、初期感染細胞に導入される。本発明において用いられる、オルガネラ、細胞、組織または生物を変換するために核酸を供給するためのそのような方法は、事実上、本明細書に記載されているかまたは当業者に既知の、核酸(例えばHSVベクター)をオルガネラ、細胞、組織または生物中に導入することができる任意の方法を含むと考えられる。非限定的な方法の例は、エクスビボの形質移入によるDNAの直接投与、注射(米国特許第5,994,624号、第5,981,274号、第5,945,100号、第5,780,448号、第5,736,524号、第5,702,932号、第5,656,610号、第5,589,466号および第5,580,859号)、マイクロインジェクション(米国特許第5,789,215号)、エレクトロポレーション(米国特許第5,384,253号)、リン酸カルシウム沈殿、DEAEデキストランおよびその後のポリエチレングリコール、直接超音波投与、リポソーム媒介形質移入、受容体媒介形質移入、微粒子銃(国際特許出願第94/09699号および第95/06128号、米国特許第5,610,042号、第5,322,783号、第5,563,055号、第5,550,318号、第5,538,877号および第5,538,880号)、炭化ケイ素繊維による撹拌(米国特許第5,302,523号および第5,464,765号)、アグロバクテリウム媒介変換(米国特許番号5,591,616号および第5,563,055号)、プロトプラストのPEG媒介変換(米国特許第4,684,611号および第4,952,500号)、除湿/阻害媒介DNA取り込み、およびこれらの方法の任意の組み合わせ、または当業者に既知のその他の方法を含む。組成物は、医薬品として許容される賦形剤中で、静注などの全身投与法によって、哺乳類の細胞に供給してもよい。
【0083】
B.細胞へのDNAベクター供給の方法
1.エクスビボ変換
生物から取り出した細胞および組織にエクスビボ環境で形質移入するための方法が当業者に知られている。従って、本発明において、細胞または組織を取り出し、本明細書に記載されている核酸および組成物を用いてエクスビボで形質移入してよいとみなされる。特定の態様では、形質移入された細胞または組織を生物中に配置してよい。いくつかの実施態様では、形質移入された細胞または組織の中で核酸を発現させる。
【0084】
2.注射
特定の実施態様では、核酸は、例えば、皮下、皮膚内、筋肉内、静脈内、腹腔内等などの1回以上の注射(すなわち針注射)によって、オルガネラ、細胞、組織または生物に投与してよい。注射の方法は、当業者に公知である(例えば食塩水を含む組成物の注射)。本発明のさらに別の実施態様は、直接マイクロインジェクションによる核酸の導入を含む。用いられる本発明の組成物の量は、影響を受ける細胞、組織または生物の性質によって変化させてよい。
【0085】
3.エレクトロポレーション
本発明の特定の実施態様では、エレクトロポレーションによってオルガネラ、細胞、組織または生物の中に核酸を導入する。エレクトロポレーションは、細胞およびDNAの懸濁液の高電圧放電への曝露を含む。この方法のいくつかの変化形では、ペクチン分解酵素などの特定の細胞壁分解酵素を使用して受け入れ標的細胞を非処理細胞よりエレクトロポレーションによる変換に対して敏感にする(米国特許第5,384,253号)。あるいは、受け入れ細胞を機械的損傷による変換に対してより敏感にしてよい。
【0086】
4.リポソーム媒介トランスフェクション
本発明のさらに別の実施態様では、改変ICP10ポリヌクレオチドを有するベクターなどの本明細書に記載されている組成物を、例えばリポソームなどの脂質複合体の中に捕捉してよい。リポソームは、リン脂質2分子膜と内部の水性媒質とを特徴とするベシクル構造体である。多重ラメラリポソームは、水性媒質で分離された複数の脂質層を有する。それらは、リン脂質を過剰の水溶液の中に懸濁させると自発的に形成される。脂質成分は、自己再配置を行ってから閉鎖構造体を形成し、水および溶解した溶質を脂質2重層の間に捕捉する。リポフェクタミン(Gibco BRL)またはスーパーフェクト(Qiagen)と複合体を形成した核酸も包含される。
【0087】
本発明の特定の実施態様では、リポソームは、ヘマグルチナチンウイルス(HVJ)と複合体を形成させてよい。こうすると、細胞膜との融合が促進され、リポソームカプセル化DNAの細胞への進入が促進されることが示された(Kaneda et al., (1989) Science 20;243(4889): 375−8)。その他の実施態様では、リポソームを核非ヒストン染色体蛋白質(HMG1)と複合体形成させるかまたはともに使用してよい(Kato et al., (1991) JBiol Chem. (1991) Feb 25;266(6): 3361−4)。さらにまた別の実施態様では、リポソームをHVJとHMG1との両方と複合体形成させてよく、またはHVJとHMG1との両方とともに使用してよい。その他の実施態様では、投与用ベヒクルは、リガンドおよびリポソームを含んでよい。
【0088】
5.受容体媒介形質移入
核酸は、受容体媒介投与ベヒクルによって標的細胞に投与してよい。この手法は、受容体媒介エンドサイトーシスによる巨大分子の選択的取り込みを利用する。さまざまな受容体の細胞型特異的分布からみると、この投与方法は、別の度合いの特異性を本発明に加える。
【0089】
特定の実施態様では、受容体媒介遺伝子標的化ベヒクルは、受容体特異性リガンドおよび核酸結合剤を含む。他の実施態様は、投与される核酸を動作有効結合した受容体特異性リガンドを含む。欧州特許EPO 0273 085に記載されている、遺伝子をへん平上皮がん細胞に供給するために用いられた上皮細胞成長因子(EGF)を含むいくつかのリガンドが、受容体媒介遺伝子移入のために、用いられてきた。
【0090】
その他の実施態様では、細胞特異性核酸標的化ベヒクルの核酸投与ベヒクル成分は、リポソームと組み合わされた特異的結合リガンドを含んでよい。投与される核酸(単数または複数)をリポソームの中に収容し、特異的結合リガンドをリポソーム膜の中に官能基によって組み込む。従って、リポソームは、標的細胞の受容体(単数または複数)に特異的に結合し、内容物を細胞に供給する。
【0091】
さらに別の実施態様では、標的化投与ベヒクルの核酸供給ベヒクル成分は、リポソーム自体であってよく、好ましくは、細胞特異的結合を導く1つ以上の脂質または糖蛋白質を含む。例えば、ガラクトース末端アシアルガングリオシドであるラクトシルセラミドをリポソームの中に組み込み、肝細胞によるインシュリン遺伝子の取り込みの増加が観測された(Nicolau et al., (1987) Methods Enzymol. 149:157−76)。本発明の組織特異的変換構築体は、同様な方法で標的細胞の中に特異的に投与することができると考えられる。
【0092】
6.微粒子銃
微粒子銃技法を用いて、核酸を少なくとも1つのオルガネラ、細胞、組織または生物中に導入することができる(米国特許第5,550,318号、米国特許第5,538,880号、米国特許第5,610,042号、および国際特許出願第94/09699号)。この方法は、DNAで被覆されるかまたはDNAを含むマイクロ発射体を、細胞を死滅させないで細胞膜を突き通し、細胞に入ることができる高速度に加速する能力に依拠する。マイクロ発射体は、タングステン、白金または金など、任意の生物学的に不活性な物質で構成してよい。爆撃のために、懸濁液の中の細胞は、フィルタまたは固体培地上で濃縮される。あるいは、未成熟胚またはその他の標的細胞を、固体培地上に配置してよい。爆撃される細胞は、固定プレート上の微粒子銃デバイスの下の適切な距離に配置する。本発明の実施に有用な非常に広範な微粒子銃技法が当業者に既知である。
【0093】
C.宿主細胞
本明細書で用いられる用語「細胞」、「細胞系統」および「細胞培養物」は、区別なく用いてよい。これらの用語はすべて、それらの子孫を含む。子孫は、任意のおよびすべての後継世代である。すべての子孫は、故意または不慮の変異によって、同一でないことがあると理解される。非相同核酸配列を発現する場合、「宿主細胞」は、原核生物または真核生物の細胞を指し、ベクターを自己複製し、および/またはベクターによってコード化される非相同遺伝子を発現することができる任意の変換可能な生物を含む。宿主細胞は、ベクターの受け入れ体として用いることができ、用いられてきた。宿主細胞は「形質移入され」または「変換され」てよい。これは、外因性核酸が宿主細胞の中に移入または導入されるプロセスを指す。変換細胞は、被験者一次細胞およびその子孫を含む。本明細書で用いられる用語「設計された」および「組み換え」細胞または宿主細胞は、例えばベクターなどの外因性核酸配列が導入された細胞を指すものとする。従って、組み換え細胞は、組み替えによって導入された核酸を含まない天然細胞と区別することができる。
【0094】
組織は、細胞膜融合発生HSV−2変異体によって変換される単数または複数の宿主細胞を含んでよい。組織は、生物の一部分であってよく、または生物から分離されてよい。特定の実施態様では、組織は、脂肪細胞、歯槽、エナメル芽細胞、神経、基底細胞、血液(例えばリンパ球)、血管、骨、骨髄、グリア細胞、胸、軟骨、頸部、結腸、角膜、胎生期、子宮内膜、内皮、上皮、食道、色帯、繊維芽細胞、小胞、神経節細胞、グリア細胞、杯状細胞、腎臓、肝臓、肺、リンパ節、筋肉、ニューロン、卵巣、すい臓、末しょう血液、前立腺、皮膚、小腸、脾臓、幹細胞、胃、睾丸、およびそれらのすべての癌を含んでよいが、それらに限定されない。
【0095】
特定の実施態様では、宿主細胞または組織は、少なくとも1つの生物の一部であってよい。特定の実施態様では、生物は、当業者には自明のように、原核生物(例えば真正細菌、古細菌)または真核生物であってよいが、それらに限定されない。
【0096】
多数の細胞系統および培養物が宿主細胞として用いるために利用可能であり、American Type Culture Collection(ATCC)などの組織によって市販されている。当業者は、ベクター主鎖および所望の結果によって適切なホストを決定することができる。ベクター複製および/または発現に利用できる細胞型の非限定的な例は、大腸菌(例えば大腸菌株RR1、LE392、B、X1776(ATCC第31537号)、W3110、F、ラムダ、DH5α、JM109およびKC8)、桿菌、例えば枯草菌などの細菌、その他の腸内細菌科、例えばサルモネラチフィムリウム、セラチアマルセッセンス、ならびに複数の市販の細菌宿主およびSURE(登録商標)コンピテント細胞およびソロパック(登録商標)(SOLOPACKT)ゴールド細胞(Gold Cell)(ストラタジーン(登録商標)(STRATAGENE, La Jolla, CA)などのコンピテント細胞を含む。ベクターの複製および/または発現のための真核生物宿主細胞の非限定的な例は、HeLa、NIH3T3、Jurkat、293、Cos、CHO、SaosおよびPC12を含む。
【0097】
いくつかのベクターは、原核生物細胞と真核生物細胞との両方の中で複製および/または発現することを可能にする制御配列を使用してよい。さらに、上記に記載した宿主細胞をすべて培養して維持し、ベクターの複製を可能にする条件は、当業者には自明と考えられる。ベクターの大量産生、ならびにベクターによってコード化される核酸および同種ポリペプチド、蛋白質またはペプチドの産生を可能にする技法および条件も自明であり、既知である。
【0098】
D.ウイルスベクターパッケージングおよび増殖。
【0099】
1.ウイルスパッケージング
本発明の特定の実施態様では、ICP10遺伝子を改変した後、相同組み換えによってウイルスに挿入する。通常、改変ICP10遺伝子を含むプラスミドDNAと精製HSV−2ゲノムDNAとをリポフェクタミンを用いてベロ細胞に共移入することによってこれを実行する。次に、組み換えウイルスを特定し(通常、ウイルスプラークを検索して選択可能マーカーの有無を調べて)、改変ICP10ポリヌクレオチドを含むプラークを選択する。次に、組み換えウイルス選択したら、インビトロでキャラクタリゼーションして改変ICP10遺伝子がHSV−2ゲノム中に正しく挿入され、元のICP10遺伝子を置換したことを確認する。実施例1および2に、ウイルスパッケージングおよびインビトロのキャラクタリゼーションをさらに詳細に記載する。
【0100】
2.ウイルス原料の調製
組み換えHSV−2変異体ウイルスを選択したら、以下のようにウイルス原料を調製する。10%ウシ胎児血清(FBS)中でベロ細胞を成長させ、細胞あたり0.01プラーク形成単位(pfu)に感染させる。次に、2日後、凍結、解凍および超音波処理を繰り返して、細胞からウイルスを集める。ウイルスを集めたら、記載された(Nakamoriら、(2003) Clinical Cancer Res. 9(7): 2727−2733)ように精製する。ウイルスを精製したら、滴定(実施例10に記載されている)し、等分し、使用するまで−80℃で保管する。
【0101】
E.蛋白質発現系
例えば、本発明のDNAベクター組成物の発生において蛋白質発現系を利用して、改変ICP10ポリヌクレオチドによってコード化されたポリペプチドを機能研究のために発現させてよい。上記で考察した組成物の少なくとも一部または全体を含む多数の発現系が存在する。原核生物および/または真核生物による系を本発明とともに使用して核酸配列または同種ポリペプチド、蛋白質およびペプチドを産生させてよい。多くのそのような系が市販され、広く利用可能である。
【0102】
米国特許第5,871,986号および第4,879,236号に記載され、市販されている(例えば、CLONTECH, Inc. Mountain View, CA)ものなどの昆虫細胞/バキュロウイルス系は、高水準の非相同核酸セグメントの蛋白質発現を行うことができる。
【0103】
市販発現系のその他の例は、合成エクジソン誘導性受容体を含む誘導性哺乳類発現系、pET発現系または大腸菌発現系(STRATAGENE, LaJolla, CA);テトラサイクリン調節発現系、全長CMVプロモーターまたはメチロトローフ酵母ピキアメタノーリカ(INVITROGEN, Carlsbad, CA)中の高レベルの組み換え蛋白質の産生用に設計された酵母発現系を用いる誘導性哺乳類発現系を含む。
【0104】
本発明の方法によって産生される蛋白質、ポリペプチドまたはペプチドは、「過剰発現」、すなわち、細胞中での天然の発現と比較すると増加したレベルで発現されてよいと考えられる。そのような過剰発現は、放射能標識および/または蛋白質精製を含むさまざまな方法によって評価してよい。しかし、簡単で直接的な方法、例えばSDS/PAGEおよび蛋白質染色またはウェスタンブロッティングとそれに続く、結果として得られたゲルまたはブロットの密度測定掃引などの定量分析法を含むものが好ましい。特定の蛋白質、ポリペプチドまたはペプチドが宿主細胞によって産生される他の蛋白質と比べると相対的に多くなるように、天然細胞中のレベルと比較した組み換え蛋白質、ポリペプチドまたはペプチドのレベルの特定の増加は過剰発現を示し、例えば、ゲル上で目に見える。
【0105】
V.HSV−2変異体の機能役割
本明細書に記載されているHSV−2変異体は、殺腫瘍剤としての複数の機能役割を示す。例えば、本ウイルスは、感染細胞ならびに傍観者細胞の両方において、溶菌ならびに合胞体形成およびアポトーシス誘導によって、腫瘍細胞を崩壊させることができる。さらに、HSV−2変異体によって腫瘍を崩壊させると、強力な抗腫瘍免疫応答を誘導し、抗腫瘍免疫応答が、悪性疾患治療用の殺腫瘍剤としての本変異体ウイルスの治療効力にさらに貢献する。
【0106】
本HSV−2変異体ウイルスは、周期活動中の細胞中で選択複製を示すが、非サイクリング細胞中では示さない。実施例4にさらに詳細に記載するように、本変異体HSV−2は、プロテインキナーゼ活性が欠失しているので、周期活動中の細胞中での成長と比較して周期活動中でない細胞中での成長は少なくとも40分の1に低下する。これとは対照的に、野生型HSV−2の周期活動中の細胞と周期活動中でない細胞との間の増殖特性はわずかに影響を受けるだけである。従って、本明細書に記載の本HSV−2変異体は、腫瘍細胞など、活性化Ras経路を有するサイクリング細胞における殺腫瘍剤として用いるのに優れて適する。
【0107】
本明細書に記載の本改変HSV−2は、他の殺腫瘍ウイルスおよび野生型HSV−2と比較すると、優れた殺腫瘍細胞能力を有する。実施例5に記載するインビトロアッセイを用いて、さまざまな組織起源のヒト腫瘍細胞に対するFusOn−H2の殺細胞能力が、米国特許出願第10/397,635号に記載され、および/または従来検証された殺腫瘍HSV−1の殺細胞能力より有意に強く、親の野生型HSV−2の殺細胞能力を超えることを示す。さらに、実施例6に記載するように、本発明のウイルスの穏やかな投与量(1×106プラーク形成単位)を1回注射したところ、局所塗布によってヌードマウスに確立した胸腺腫瘍が、この動物の100%(n=8)で完全に消失した。一方、殺腫瘍HSV−1を同じ投与量で投与したが、30%より少ないマウスで腫瘍が縮小しただけであった。
【0108】
溶解活性および融合活性に加えて、本HSV−2変異体は、強力なアポトーシス誘導活性も有し、強力な抗腫瘍免疫応答も誘導することができる。本HSV−2変異体は、インビトロ環境で、ウイルスに感染した細胞ならびに感染細胞の周りの非感染傍観者細胞中にアポトーシスを誘導することができる。さらに、本HSV−2変異体は、インビボで、腫瘍細胞のアポトーシスを誘導する効果を有する。実施例8にこれをさらに詳しく記載する。本明細書に記載の組成物は、他の殺腫瘍ウイルスより腫瘍細胞を殺傷する上で有効であるだけでなく、本HSV−2変異体は、強い抗腫瘍免疫応答の誘導によって、原発腫瘍および転移腫瘍に対してインビボで強い治療効果を示す。実施例9に記載するように、FusOn−H2処理マウス由来の養子伝達CTLは、元の腫瘍の成長を阻害し、転移の発達を効果的に防ぐことができる。
【0109】
アポトーシスまたはプログラム化細胞死は、正常な胚発育の必須過程であり、成体組織中の恒常状態を維持し、発癌を抑制する。本発明のいくつかの実施態様では、本改変HSV−2は、本ウイルスに感染した腫瘍細胞と、非感染傍観者腫瘍細胞とのアポトーシスの強力な誘導因子である。例えば、特定の実施態様では、腫瘍細胞を、ICP10遺伝子のプロテインキナーゼドメインの一部を緑色蛍光蛋白質(GFP)コード化遺伝子で置換したHSV−2構築体に感染させた。蛍光顕微鏡下でGFPを可視化することによって感染細胞を識別することができ、クロマチン凝集によって明らかにして、アポトーシスを行う細胞を識別した。GFP発現に対してクロマチン凝集を示す細胞の比は、2.6:1であり、本改変HSV−2に感染しなかった腫瘍細胞のうちのかなりの数がアポトーシスを行うことを示唆した。実施例8に、本発明の殺腫瘍ウイルスのアポトーシスを誘導する能力をさらに詳細に記載する。
【0110】
悪性疾患と戦う際には、強い抗腫瘍免疫応答が有用である。本明細書に記載の本HSV−2変異体は、原発腫瘍および転移腫瘍に対してインビボで強力な抗腫瘍免疫応答を誘導することができる。特定の実施態様では、本変異体HSV−2(FusOn−H2)は、4T1マウス乳腺腫瘍細胞系統を用いるマウス乳腺腫瘍モデル中で選択複製し、腫瘍細胞を溶解させ、強い抗腫瘍免疫応答の誘導によって、インビボで原発腫瘍および転移腫瘍に対する強い治療効果を示した。詳しくは、FusOn−H2で治療したマウス由来の養子伝達細胞毒性リンパ球(CTL)は、元の腫瘍の成長を阻害し、FusOn−H2による治療を受けていないマウスの転移を効果的に防ぐことができる。実施例9に、これをさらに詳細に記載する。
【0111】
VI.医薬品組成物および投与経路
A.総合的考察
本発明の組成物は、改変ICP10遺伝子を有する組み換えHSV−2変異体か、本明細書に記載の改変ICP10遺伝子を有する裸の(非ウイルス)DNAベクターのどちらかを含む医薬品組成物として投与することができる。本発明の組成物は、通常の医薬品調製物を含む。通常、本発明の組成物は、本組成物を医薬品として許容されるキャリアまたは水性媒質に溶解または分散させて、薬学的剤として投与することができる。医薬活性物質のためのそのような媒質および剤の使用は、当分野で公知である。任意の通常の媒質または剤が本発明の組成物と適合する限り、治療組成物中のその使用は考慮範囲内である。他の抗疾患剤などの補助活性成分も、医薬品組成物中に組み込んでよい。本組成物の投与は、その経路によって標的細胞を利用できる限り、任意の普通の経路によってよい。投与経路の例は、口、鼻、舌下、直腸、膣または皮膚を含む。あるいは、同所注射、皮内注射、皮下注射、筋肉内注射、腹腔内注射、静脈注射、または、直接腫瘍内注射によって投与してよい。本発明の組成物のための医薬組成物、用量および投与経路を下に記載する。
【0112】
B.HSV−2変異体の医薬品製剤
本発明の変異体ウイルス組成物は、薬理学的に許容される製剤として調製することができる。通常、変異体ウイルスを、医薬品として許容され、ウイルスと適合する賦形剤と混合する。適当な賦形剤は、例えば、水、食塩水、デキストロース、グリセロール、エタノールまたは類似物およびそれらの組み合わせである。さらに、望むなら、調製物は、湿潤剤または乳化剤、pH緩衝剤、アジュバントまたは免疫強化物質など、少量の補助物質を含んでよい。これらの補助物質は、本ウイルス変異体の有効性を高める(Remington’s Pharmaceutical Sciences, Gennaro, A. R. et al., eds., Mack Publishing Co., pub., 18th ed., 1990を参照すること)。例えば、注射用に医薬品として許容される一般的なキャリアは、リン酸緩衝生理食塩水のミリリットルあたり50mgから最大約100mgのヒト血清アルブミンを含んでよい。薬理学的に許容される組成物の製剤に用いるのに適する非水溶溶媒の別の非限定的な例は、プロピレングリコール、ポリエチレングリコール、植物油、ゴマ油、落花生油およびオレイン酸エチルなどの注射可能な有機エステルを含む。水系キャリアの非限定的な例は、水、水溶液、食塩水、塩化ナトリウムなどの非経口ベヒクル、リンゲルのデキストロース等を含む。静脈内ベヒクルは、流体および栄養補液を含む。医薬品組成物のさまざまな成分のpHおよび正確な濃度を測定することは、定型的であり、当業者の知識の範囲内にある(Goodman and Gilman’s The Pharmacological Basis for Therapeutics, Gilman, A. G. et al., eds., Pergamon Press, pub., 8th ed., 1990を参照すること)。
【0113】
必要な、上記に記載したさまざまなその他の無菌成分とともに適切な溶媒の中に必要な量の活性化合物を組み込んで、無菌の注射可能溶液を調製する。通常、上記に記載した基本的な分散媒質と必要なその他の成分とを含む無菌ベヒクルにさまざまな殺菌された活性成分を組み込むことによって、分散液を調製する。
【0114】
C.HSV−2変異体の投与経路および用量
本変異体ウイルス組成物は、標的組織へのアクセスを提供する任意の経路によって投与してよい。投与経路の非限定的な例は、口、鼻、舌下、直腸、膣、塗布を含んでよく、あるいは注射(同所注射、皮内注射、皮下注射、筋肉内注射、腹腔内注射、静脈内注射または直接腫瘍内注入を含む)によってであってよい。通常、本ウイルス変異体は、液体溶液または懸濁液としての注射可能薬物として調製してよく、注入前に、液体への溶解または液体への懸濁に適する固体も調製してよい。調製物は、乳化もさせてよい。
【0115】
例えば、水溶液で非経口投与する場合、必要なら溶液を適当に緩衝性とするひつようがある。まず、十分な食塩水またはグルコースで液体希釈剤を等張性にする。これらの特定の水溶液は静脈投与、筋肉内投与、皮下投与および腹腔内投与に、特に適する。これに関連して、使用することができる無菌水性媒質は、本開示によって、当業者に既知である。例えば、1用量を1mlの等張性NaCl溶液に溶解させ、1000mlの皮下注入流体に加えるか、または提案された注入部位に注入するかのどちらかを行う(例えば、”Remington’s Pharmaceutical Sciences” 15th Edition, pages 1035−1038 and 1570−1580を参照すること)。治療を受ける被験者の状態によって、若干の用量の変化が必然的に起こる。いずれにしても、投与の責任者は、個々の被験者に適切な用量を決定する。
【0116】
本発明の組成物を用いて治療を提供するための最善の治療計画は、簡明直截的に決定することができることは、当業者には自明であろう。これは、実験の問題ではなく、最適化の問題であり、医術では定型的に行われている。例えば、マウスでインビボに調べ、用量および投与計画を最適化し始める出発点を得る。注射の頻度は、最初、週に一度であってよい。しかし、この頻度は、初期臨床試験から得られる結果および特定の患者の必要性によって、1日から2週間ごと、1月ごとまで、最適に調節する可能性がある。最初、マウスで用いた組成物の量から外挿してヒト投与量を決定してよい。
【0117】
1.用量
投与するウイルスベクターの量は、治療の回数、治療を受ける被験者、抗ウイルス抗体を合成する被験者免疫系の容量、崩壊させる標的組織、および所望の保護の程度を含むいくつかの因子に依存する。投与されるウイルス組成物の正確な量は、担当医の判断によって決まり、各個人独自である。しかし、適当な用量は、105プラーク形成単位(pfu)から1010pfuの範囲にある。特定の実施態様では、ウイルスDNAの用量は、約105、106、107、108、109、最大1010pfu以内であってよい。
【0118】
D.非ウイルスDNAベクター製剤
上記に記載したウイルス医薬品調合物のための調合物に加えて、非ウイルスDNAベクターも薬理学的に許容される無菌溶液の調製のための無菌粉体として調製することができる。無菌粉体の調製のための一般的な方法は、活性成分と、無菌ろ過済み溶液からの任意の別の所望の成分との粉体を生じる真空乾燥および凍結乾燥技法を含む。
【0119】
E.非ウイルスDNAベクターの投与経路および用量
本発明のポリヌクレオチドを哺乳類細胞に移入するための非ウイルスベクターを供給するためのいくつかの方法が考慮範囲内にある。これらは、リン酸カルシウム沈殿、DEAE−デキストラン、エレクトロポレーション、直接マイクロインジェクション、DNA保持リポソームおよびリポフェクタミン−DNA複合体、細胞超音波処理、高速マイクロ発射体を用いる遺伝子衝撃、既に考察した受容体媒介形質移入を含む。これらの技法のいくつかを、インビボまたはエクスビボ使用のために適応させることができる。
【0120】
本発明のいくつかの実施態様では、発現ベクターは、単に、ポリヌクレオチドを含む裸の組み換えDNAまたはプラスミドからなっていてよい。構築体の移入は、物理的または化学的に細胞膜を透過する本明細書に言及の任意の方法によって実行してよい。これは、特にインビトロ移入に適用可能であるが、インビボ使用にも適用してよい。
【0121】
その他の実施態様では、投与用ベヒクルは、リガンドおよびリポソームを含んでよい。例えば、Nicolauらは、リポソームに組み込まれたガラクトース末端アシアルガングリオシドであるラクトシルセラミドを使用し、肝細胞によるインシュリン遺伝子の取り込みの増加を観測した(Nicolau et at, (1987) Methods Enzyrnol. 149:157−76)。従って、リポソームの有無にかかわらず、任意の数の受容体−配位子システムによって、特定の遺伝子をコードする核酸を細胞型に特異的に供給することも実行できる。例えば、EGF受容体の上方調節(欧州特許第0 273 085号に記載されている)を示す細胞へ核酸を媒介投与するための受容体として上皮細胞成長因子(EGF)を用いてよく、マンノースを用いて肝臓細胞上のマンノース受容体を標的化してよい。
【0122】
特定の実施態様では、エクスビボ条件下での方が、DNA移入を容易に実行することができる。エクスビボ遺伝子治療は、動物からの細胞を取り出し、核酸をインビトロで細胞に投与した後、改変細胞を動物へ戻すことを指す。これは、動物由来の組織/器官の外科的取り出し、または細胞および組織の初代培養を含んでよい。
【0123】
1.用量
特定の実施態様では、用量を約103pfu/kg体重から約108pfu/kg体重の間で変化させることは想定の範囲内である。特定の実施態様では、用量は、約103、104、105、106、107から108pfu/kg体重以内であってよい。もちろん、通常そのような治療プロトコルで実行するように、初期臨床試験の結果および特定の患者の必要性に依存して、この用量を、上下に調節してよい。
【0124】
VII.併用療法
本発明の方法および組成物の有効性を増加させるために、本明細書に開示される方法および組成物を他の抗癌剤と組み合わせることが望ましいことがある。このプロセスは、癌細胞を少なくとも1つの他の抗癌剤と組み合わせた本発明の組成物と接触させることを含んでよい。これは、細胞を、両方の剤を含む単一の組成物または薬理学的製剤と接触させることによって、または細胞を2つの別個の組成物または製剤と接触させることによって、実現してよい。2つの別個の製剤を用いる場合、癌細胞を両方の製剤と同時に接触してもよく、他の製剤より先に1つの製剤と接触させてもよく、それらの任意の組み合わせまたは繰り返しサイクルであってもよい(例えば、本発明の組成物を、別の抗癌剤の投与の前または後に投与する)。本発明の組成物と他の剤とを別個に投与する実施態様では、通常、本発明の組成物と他の剤とが、依然として癌細胞に併用効果を有利に及ぼすことができるよう、各供与時間の間にあまり時間を置かないように注意する。2つの製剤の投与の間のこの時間間隔は、分から週の範囲にあってよい。
【0125】
本発明の組成物または方法とともに用いてよい抗癌剤の非限定的な例は、化学療法薬(例えばシスプラチン(CDDP)、カルボプラチン、プロカルバジン、メクロレタミン、シクロホスファミド、カンプトセシン、イホスファミド、メルファラン、クロラムブシル、ブスルファン、ニトロソ尿素、ダクチノマイシン、ダウノルビシン、ドキソルビシン、ブレオマイシン、ピコマイシン、マイトマイシン、エトポシド(VP16)、タモキシフェン、ラロキシフェン、エストロゲン受容体結合剤、タキソール、ジェムシタビエン、ナベルビン、ファルネシル−蛋白質トランスフェラーゼ阻害剤、トランス白金、5−フルオロウラシル、ビンクリスチン、ビンブラスチンおよびメトトレキサン、またはそれらの任意の類縁体または誘導体変化形)、放射線治療薬剤(例えば腫瘍細胞へのγ線、X線、マイクロ波およびUV照射および/またはラジオアイソトープの指向投与)、免疫療法剤および免疫調節剤、遺伝子治療薬剤、アポトーシス促進剤および当業者に公知のその他の細胞周期調節剤を含んでよい。
【0126】
免疫療法を本明細書に記載の組成物および方法とともに用いて、悪性疾患の治療のための併用療法としてもよい。通常、免疫療法は、癌細胞を標的とし、崩壊させる免疫エフェクター細胞と分子との使用に依拠する。免疫エフェクターは、例えば、腫瘍細胞の表面のマーカーに特異的な抗体であってよい。この抗体は、単独で治療のエフェクターとして働いてよく、あるいは、他の細胞(例えば細胞毒性T細胞またはNK細胞)を募って殺細胞を実際に実行してよい。この抗体は、薬物またはトキシン(例えば化学療法剤、放射性核種、リシンA鎖、コレラトキシン、百日咳毒素等)と結合し、単に標的化剤として働いてよい。いくつかの実施態様では、エフェクターは、腫瘍細胞標的と直接的または間接的に相互作用する表面分子を有するリンパ球であってよい。その他の実施態様では、腫瘍細胞は、標的化の対象となるなんらかのマーカーを持っていなければならない。標的化に適する腫瘍マーカーの非限定的な例は、癌胎児性抗原(CEA)、前立腺特異性抗原、尿腫瘍関連抗原、胎児抗原、チロシナーゼ(p97)、gp68、TAG−72、HMFG、シアリルルイス抗原、MucA、MucB、PLAP、エストロゲン受容体、ラミニン受容器、erbBおよびp155を含んでよい。
【0127】
悪性疾患の治療のための複合治療法として本明細書に記載の組成物および方法とともに、遺伝子治療を併用してよい。併用療法としての遺伝子治療は、本明細書に記載の変異体HSV−2とは別の治療遺伝子の投与および発現に依拠する。この遺伝子治療は、本明細書に記載のHSV−2変異体の前、後、または同時に起用してよい。遺伝子治療の非限定的な標的の例は、免疫調節剤、細胞表面受容体およびGAP接合の上方調節に影響を及ぼす剤、細胞静止剤および分化剤、細胞接着の阻害剤、または標的細胞のアポトーシス感受性を誘導し、または増加させる剤を含む。本発明と組み合わせて遺伝子治療の一部として用いることができる免疫調節遺伝子の非限定的な例は、腫瘍壊死因子;インターフェロンアルファ、ベータおよびガンマ;IL−2およびその他のサイトカイン;F42Kおよびその他のサイトカイン類縁体;またはMIP−1、MIP−1ベータ、MCP−1、RANTESおよびその他のケモカインを含む。
【0128】
細胞増殖の阻害剤の例は、p16である。サイクリン依存性キナーゼ、すなわちCDKによって真核生物の細胞周期の主要遷移を引き起こす。1つのCDK、すなわちサイクリン依存性キナーゼ4(CDK4)は、G1期を通して進行を調節する。この酵素の活性は、G1後期にRbをリン酸化することであってよい。CDK4の活性は、活性化サブユニット、すなわちD−型サイクリンによって、および阻害サブユニット、すなわちCDK4に特異的に結合し、阻害し、従って、Rbリン酸化を調節してよいp16INK4によって制御される。p16INK4遺伝子は、新しく記載されたCDK阻害性蛋白質の種類に属し、この種類は、p16B、p19、p21WAF1およびp27KI1も含む。p16INK4遺伝子の同型接合削除および変異は、ヒト腫瘍細胞系統では頻繁である。p16INK4蛋白質はCDK4阻害剤なので、この遺伝子を削除すると、CDK4の活性が増加し、その結果、Rb蛋白質が高リン酸化される。遺伝子治療に使用して細胞増殖を阻害してよい他の遺伝子は、Rb、APC、DCC、NF−1、NF−2、WT−1、MEN−I、MEN−II、zac1、p73、VHL、MMAC1/PTEN、DBCCR−1、FCC、rsk−3、p27、p27/p16融合体、p21/p27融合体、抗血栓遺伝子(例えばCOX−1、TFPI)、PGS、Dp、E2F、ras、myc、neu、raf、erb、fms、trk、ret、gsp、hst、abl、E1A、p300、血管新生に関与する遺伝子(例えばVEGF、FGF、トロンボスポンジン、BAI−1、GDAIFまたはそれらの受容体)およびMCCを含む。
【0129】
細胞表面受容体またはFas/Fasリガンド、DR4またはDR5/TRAILなどのリガンドの上方調節は、過剰増殖性細胞に対する自己分泌効果または傍分泌効果を確立し、本発明のアポトーシス誘導能力を強化することも考慮の範囲内である。GAP接合の数を上げて細胞間の信号伝達を増加させると、隣接する過剰増殖性細胞ポピュレーションに対する抗過剰増殖性効果が増加する。他の実施態様では、細胞増殖抑制剤または分化剤を本発明で用いて、治療法の抗過剰増殖性抗力を高めることができる。細胞接着の阻害剤は、本発明の効力を高めるとみなされる。細胞接着阻害剤の例は、局所接着キナーゼ(FAK)阻害剤およびロバスタチンである。さらに、抗体c225などの過剰増殖性細胞のアポトーシス感受性を増加させる他の剤を本発明と組み合わせて用いて治療効果を高めることは、考慮の範囲内である。
【0130】
本発明とともにホルモン療法も用いてよい。乳癌、前立腺癌、卵巣癌または子宮頸部癌などの特定の癌の治療においてホルモンを使用してテストステロンまたはエストロゲンなどの特定のホルモンの効果のレベルを下げ、あるいは遮断してよい。この治療は、治療オプションとして、または転移の危険性を小さくするために、多くの場合に少なくとも1つの他の癌療法と組み合わせて用いられる。
【0131】
本願において引用するすべての特許、特許出願およびその他の刊行物は、公開されたアミノ酸またはポリヌクレオチド配列を含めて、参照によって全体としてあらゆる目的について組み込まれる。
【実施例】
【0132】
以下の実施例を掲載して本発明の好ましい実施態様を示す。当業者は、以下の実施例に開示する技法は、本発明の実施において良好に機能することを本発明者が発見した技法を表し、従って本発明の実施にとって好ましいモードを構成するとみなしてよいと理解するべきである。しかし、当業者は、本開示を参照し、開示された特定の実施態様において多くの変化を施してもなお本発明の技術思想および範囲から逸脱せずに同様な類似の結果を得ることができることを理解するべきである。
【0133】
(実施例1)
FusOn−H2の構築
図1に、FusOn−H2の例の構築の例を示す。最初に、以下のプライマー対の例、すなわち5’−TTGGTCTTCACCTACCGACA(SEQ ID NO:1)と3’−GACGCGATGAACGGAAAC(SEQ ID NO:2)とを用いて、ICP10左側(left−flanking)領域を含むHSVゲノム領域(HSV−2ゲノムのヌクレオチド番号85994〜86999と等価)を増幅させた。以下のプライマー対の例、すなわち、5’−ACACGCCCTATCATCTGAGG(SEQ ID NO:13)と5’−AACATGATGAAGGGGCTTCC(SEQ ID NO:14)とを用いて、RRドメインおよび右側(right−flank)領域(HSV−2ゲノムのヌクレオチド配列番号88228〜89347と等価)を増幅させた。これらの2つのPCR産物を、EcoRI−NotI−XbaIライゲーションによってpNeb193の中にクローン化し、pNeb−ICP10−デルタPKを発生させた。次に、以下のプライマー対の例、すなわち、5’−ATGGTGAGCAAGGGCGAG(SEQ ID NO:3)と3’−CTTGTACAGCTCGTCCATGC(SEQ ID NO:4)とを用いて、CMVプロモーター−EGFP遺伝子を含むDNA配列を、pSZ−EGFPからPCR増幅させた。次に、BgIIIおよびNotIライゲーションによって、PCR増幅DNAをpNeb−ICP10−デルタPKの削除PK遺伝子座にクローン化してpNeb−PKF−2を発生させた。PCR増幅戦略を設計する際には、EGFP遺伝子がICP10遺伝子の残るRRドメインのフレーム内に融合し、その結果、この融合遺伝子の新しい蛋白質産物は、以下の実験段階において組み換えウイルスの選択を容易にする無傷の機能EGFPを含むように、プライマーを設計した。
【0134】
pNeb−PKF−2プラスミドDNAと、精製したHSV−2ゲノムDNA(186株)とをリポフェクタミンによってベロ細胞に共形質移入する相同組み換えによって、改変ICP10遺伝子をウイルス中に挿入した。GFP陽性ウイルスプラークを検索して組み換えウイルスを選抜し、特定した。検索プロセスの間に、GFP陽性プラークがすべて、明らかな感染細胞の合胞体形成を示すことに気がついた。これは、本発明の特定の実施態様では、この改変ウイルスが広範囲な細胞膜融合を誘導することを示している。合計6つのプラークを選抜した。さらにキャラクタリゼーションするため、およびその後のすべての実験のためにそれらの1つを選択した。これをFusOn−H2と呼ぶ。
【0135】
実施例2
インビトロキャラクタリゼーション
FusOn−H2ベクターの例を、当分野の標準的な分析法によってキャラクタリゼーションした。
【0136】
サザンブロット分析。
【0137】
改変ICP10遺伝子がHSV−2ゲノムに正しく挿入され、元のICP10遺伝子を置換したことを確認するために、精製したFusOn−H2ウイルス原料からウイルスDNAを抽出した。対照として、同じ手順によって、親の野生型HSV−2からウイルスDNAを抽出した。BamHIを用いてウイルスDNAを消化し、0.8%アガロースゲル中で電気泳動した。BamHIによって消化すると、野生型HSV−2ゲノムから、ICP10遺伝子全体およびその左側領域および右側領域を含む7390bpのDNA断片が生じる。しかし、FusOn−H2ゲノムを同じ酵素で消化すると、ICP10遺伝子座から2つのDNA断片、すなわち、1)左側領域とCMVプロモーター配列とを含む4830bp断片、および2)GFP、RRおよび右側領域を含む3034bp配列、が生じる。DNAをナイロン膜に移し、1)DNA配列の左側領域、2)PK領域全体、3)PKの右側領域、および4)GFP遺伝子、から調製した4つのプローブとハイブリッド化させる。結果(図2)によると、GFP遺伝子から作った1つを除くすべてのプローブが7390bpDNAバンドにハイブリッド化した。左側領域プローブは、GFPおよび右側領域DNA配列から調製したプローブによって認識されるDNAバンドにハイブリッド化した。PKドメイン配列から作ったプローブは、DNA断片のどれにもハイブリッド化することができず、PKドメインがFusOn−H2のゲノムから完全に除去されたことを示した。
【0138】
ウェスタンブロットハイブリダイゼーション。
【0139】
FusOn−H2のゲノムの中の改変ICP10遺伝子の正しさをさらに確認するために、FusOn−H2または親の野生型HSV−2のどちらかに感染させたベロ細胞から、またはpSZ−EGFPプラスミドDNAで形質移入された細胞から蛋白質を抽出した。12%SDS−PAGEゲル上で蛋白質を分離し、ハイボンド−C膜に移した。次に、抗GFPモノクローナル抗体(抗GFP#Ab290、ABCAM Inc., Cambridge, MA)で膜をブロットした。この抗GFP抗体は、pSZ−EGP形質移入細胞から発現された小型のGFP蛋白質(約28kD)を選択した。同じ抗体は、著しく大きな蛋白質バンド(融合蛋白質のサイズは、約120kDと予想される)も認識した。しかし、この抗体は、野生型HSV−2に感染した細胞由来のどの蛋白質産物とも反応することができず、この抗体の特異性が確認された。これらの結果は、GFP遺伝子がFusOn−H2ゲノム中のICP10遺伝子の残っているRRドメインと正しく融合したことをさらに確認する。
【0140】
(実施例3)
インビトロFusOn−H2表現型キャラクタリゼーション
FusOn−H2の表現型を決定するために、本発明者らは、ベロ細胞を野生型HSV−2またはFusOn−H2のどちらかに感染させ、あるいは、細胞を感染させずにおいた。感染の24時間後、FusOn−H2に感染させた細胞単一層中に明らかな合胞体形成が見えた。非感染細胞にも、野生型HSV−2に感染させた細胞にも、合胞体は見られなかった。さまざまな組織起源のヒト腫瘍細胞中でも同様な合胞体形成が観測された。いくつかの腫瘍細胞では、野生型HSV−2に感染させても、ある程度の合胞体形成が誘導された。しかし、これらの細胞上でFusOn−H2によって誘導される合胞体形成は、通常、有意に大規模であった。従って、この場合、FusOn−H2は、親の野生型HSV−2と比較すると、融合活性の増強を示す。これらの結果は、FusOn−H2は、その感染が広範な合胞体形成を誘導するか、または腫瘍細胞中の合胞体形成の強度を強める点で、表現型として親のウイルスと異なることを示している。PKドメインも、ICP10遺伝子全体も、これまで細胞膜融合といかなる機能関連を有すると報告されたことがない(Smith et al., (1998) J Virol. 72(11): 9131−9141; Smith et al., (1994) Virol. 200(2): 598−612; Smith et al., (1992) J. Gen. Virol. 73(pt6): 1417−1428)。いくつかの実施態様では、GFP遺伝子を加え、および/またはICP10の天然プロモーターを強力なCMVプロモーターで置換すると、このウイルスのこの表現型変化に効果があった。1型殺腫瘍HSVによって合胞体形成が誘導されると、例えば、ヒト腫瘍細胞に対するウイルスの殺細胞能力が著しく増加することが示されている(2003年3月26日出願の米国特許出願第10/397,635号)ので、殺腫瘍を目的とする用途では、FusOn−H2の融合性表現型は重要である。
【0141】
(実施例4)
増殖活動中の細胞および休止状態の細胞中のFusOn−H2の成長曲線
分裂(腫瘍)細胞中のFusOn−H2の選択複製の性質を求めるために、本発明者らは、ベロ細胞を感染させ、10%ウシ胎児血清(FBS)(十分に増殖状態にある細胞)中、またはFBSを含まない媒地(休止状態の細胞)中のどちらかで、野生型HSV−2またはFusOn−H2のどちらかとともに培養した。感染後、異なる時点でウイルスを集め、ベロ細胞で滴定した。細胞を休止状態にしても、野生型ウイルスの成長は、わずかな影響しか受けなかった(2分の1まで減少しない)。これとは対照的に、休止状態にある細胞中のFusOn−H2の成長は、増殖状態にある細胞からのウイルス収量と比較すると、劇的に減少した(40分の1より小さくなる)。これらの結果(図5)は、FusOn−H2は、腫瘍細胞中で十分複製能力があるが、体の中の正常な体細胞を表す休止状態の細胞中では、通常、最小限の複製能力しかなく、従って、腫瘍細胞中でのFusOn−H2の選択複製能力が得られることを示した。
【0142】
(実施例5)
ヒト腫瘍細胞に対するFusOn−H2のインビトロ殺細胞アッセイ
次に、本発明者らは、直接、FusOn−H2とその親の野生型HSV−2とのインビトロ殺腫瘍効果、あるいは1型HSV(HSV−1)から構築した殺腫瘍ウイルスを比較した。ヒト卵巣がん細胞系統Skov−3またはヒト乳癌細胞系統MDA−MB−435の例となる細胞を0.01または0.1pfu/細胞のどちらかで本ウイルスに感染させ、ウイルス感染の24時間後または48時間後のどちらかの時点で、例えば比色法乳酸デヒドロゲナーゼ(LDH)アッセイによって細胞生存率を測定した。結果(図6)によると、試験した殺腫瘍HSVの中で、FusOn−H2が両方のヒト腫瘍細胞系統に対して最も高い殺細胞能力を有する。その殺細胞能力は、腫瘍細胞中に合胞体形成を誘導するその能力によって、その親の野生株ウイルスと比較しても有意に高かった。
【0143】
(実施例6)
FusOn−H2のインビボ治療評価
インビボ条件下で、例となるFusOn−H2ウイルスをキャラクタリゼーションした。
【0144】
ヒト乳癌異種移植部に対して
FusOn−H2のインビボ抗腫瘍効果を評価するために、本発明者らは、非常におだやかな用量(1×106pfu)のウイルスを、ヒト乳癌(乳腺脂肪層中のMDA−MB−435細胞の注入から)の確立された異種移植部(直径約5〜8mm)に直接注入した。比較のため、本発明者らは、HSV−1由来の殺腫瘍HSV(Baco−1)をFusOn−H2と同じ用量で同時に評価した。Baco−1は、2003年3月26日出願の米国特許出願第10/397,635号に記載されている。4週間にわたり、毎週腫瘍サイズを測定した。PBS対照と比較すると、どちらのウイルスの単回注射も腫瘍成長(図7)に対して即時効果を示した。ウイルス注入後1週間以内に、殺腫瘍ウイルスのどちらで治療したマウスの腫瘍も、PBSを注射した腫瘍より著しく小さかった(P<0.001)。しかし、第2週から第4週では、FusOn−H2は、Baco−1より有意に大きな抗癌効果(P<0.01)を生じた。FusOn−H2投与後、第2週には、すべての動物(8匹中8匹)の腫瘍が消えた。これに対して、Baco−1を注入した群では、2匹のマウスの腫瘍しか消えなかった。他の6匹のマウスでは、腫瘍は最初に縮んだが、ウイルス注入後第3週には再び成長し始めた。これらの結果は、FusOn−H2がヒト乳癌に対する強力な抗腫瘍剤であり、HSV−1から構築した融合性殺腫瘍HSVより有意に効果が大きいことを示している。
【0145】
ヒト卵巣がん異種移植部に対して
卵巣がんの腹腔内侵入は、よくある重大な臨床問題である。後期卵巣がん患者の約70%が腹腔空洞内に転移性疾患を有すると報告された。従って、本発明者らは、例えば、ヒト卵巣がんに対するFusOn−H2の有効性を試験する手段として、腹腔内転移モデル(異物移植Skov−3細胞)を選んだ。集めたばかりのSkov−3細胞を3X106細胞/マウスの用量でヌードマウスの腹腔内に接種した。2週間後、マウスの腫瘍細胞注射部位から遠く離れた部位に、3×106pfuのBaco−1、FusOn−H2またはPBS(対照)のどれかを単回腹腔内(i.p.)注射した。1週間後、この治療注射を繰り返した。最初の治療注射の4週間後、マウスを安楽死させ、腹腔空洞内の腫瘍成長を評価した。これらの治療群の各動物の複数の空洞内腫瘍小結節を観察すると明らかであったが、PBS−またはBaco−1治療群のどちらにも明らかな腫瘍のi.p.転移があった(図8および表1)。
【0146】
表1 ヒト卵巣癌異種移植部の殺腫瘍治療後の腹腔内腫瘍小結節の数および重量
【0147】
【表1】
PBSと比較すると、Baco−1治療は、確立された卵巣がんに対して一定の治療効果を示した。1匹のマウスの腫瘍は完全に消え、1匹の腫瘍小結節は著しく小さくなった(1つの腫瘍小結節しか見られなかった)。しかし、FusOn−H2の治療効果は明らかに大規模であった。実験の終りまでに、FusOn−H2治療を受けた群の8匹のマウスのうち7匹の腫瘍が消えた(表1および図8)。腫瘍がなくならなかった唯一のマウスの腫瘍小結節は1つしかなかったが、小結節は、Baco−1またはPBSの治療を受けたマウスの小結節よりはるかに小さかった。これらの結果は、FusOn−H2が、比較的大きな空洞の中に確立したヒト固形腫瘍を治療する上で、ウイルスを非常におだやかな用量で投与したときでも、極めて有効であることを明らかに実証している。
【0148】
(実施例7)
FusOn−H2のインビボ毒性評価
FusOn−H2の毒性を評価するための第一段階として、本発明者らは、5×106pfuの野生型HSV−1、HSV−2またはFusOn−H2のどれかをC57/ブラックマウス(N=5)に皮下注射した。ウイルス投与の5日後に、野生型HSV−lを受けた群の5匹のマウスのうち4匹が死んだ。野生型HSV−2を受けた群では1匹のマウスが死んだ。しかし、FusOn−H2を注射した群ではマウスは1匹も死ななかった。これらの結果は、FusOn−H2が腫瘍細胞を殺傷することには極めて強力であるが、受け入れ宿主に対する毒性は親の野生型HSVより著しく低く、特定の実施態様では、臨床応用で安全なことを示している。
【0149】
(実施例8)
アポトーシスを誘導するFusOn−H2の能力
本実施例によって、本発明に記載の改変HSV−2ウイルス(FusOn−H2)は、感染腫瘍細胞および周囲の腫瘍細胞においてアポトーシスを効率的に誘導することができ、別の腫瘍破壊機序を提供することを示す。
【0150】
アフリカミドリザル腎臓(ベロ)細胞、SW403およびSW480細胞(ヒト結腸癌細胞系統)、およびA549細胞(ヒト肺癌細胞系統)は、American Type Culture Collection (Rockville, MD)から入手した。ヒト食道癌細胞系統のEC9706は、Mingrong Wang博士(Chinese Academy of Medical Sciences)の提供である。ヒト卵巣がん細胞系統のSKOV3細胞は、Robert Bast博士(the M. D. Anderson Cancer Center)の提供である。ヒト骨肉腫系統のU20S細胞は、Dr. Lawrence Donehowerの提供である。細胞はすべて、10%ウシ胎児血清(FBS)を含むDMEM中で培養した。
【0151】
野生型HSV−2株186(wt186)からFusOn−H2を誘導した。その構築は実施例1に記載した。HSV−1系殺腫瘍ウイルスBaco−1の構築は、米国特許出願第10/397,635号に記載されている。ウイルス原料は、細胞あたり0.01プラーク形成単位(pfu)のベロ細胞を感染させて調製した。2日後、ウイルスを集め、記載されている(Nakamori et al., (2003) Clinical Cancer Res. 9(7): 2727−2733)ように精製した。純化ウイルスを滴定し、等分し、使用するまで−80℃で保管した。
【0152】
ウイルス成長キャラクタリゼーション
24ウェルプレートに3つ1組で、50%密度で細胞を接種した。次の日、細胞を1pfu/細胞のウイルスに1時間感染させた。細胞をPBSで1度洗浄し、吸収されなかったウイルスと取り込まれなかったウイルスとを除去した後、新鮮な培地を加えた。感染後24時間で細胞を集めた。凍結および解凍ならびに超音波処理を繰り返して、ウイルスを放出させた。プラークアッセイによってベロ細胞基準のウイルス力価を測定した。
【0153】
感染細胞のヘキスト(Hoechst)染料染色およびクロマチン凝集の定量
次の日、24ウェルプレートに接種した細胞を、10pfu/細胞のFusOn−H2、wt186またはBaco−1に感染させ、あるいは偽感染させた。感染の24時間後、細胞を37℃で1μg/mlの最終濃度のヘキスト染料33358(Sigma−Aldrich, MO)で30分間染色してから、蛍光顕微鏡下で顕微写真を撮影した。
【0154】
DNAラダーリングアッセイ
細胞を、6ウェルプレートに70%密度で接種した。次の日、細胞を10pfu/細胞のウイルスに感染させた。ウイルス感染の24時間後に、細胞を集め、DNAzo1試薬(Invitrogen, CA)を用いてDNAを細胞から抽出した。抽出したDNAをRNアーゼ(100μg/ml)で処理した後、フェノール:クロロホルム抽出およびエタノール沈殿に付した。次に、DNAを、1%アガロースゲルに載せ、電気泳動を行い、臭化エチジウムで染色した後、UV照明下で視覚化した。
【0155】
EGFPの発現は、クロマチン凝集に対応する
次の日、12ウェルプレートに接種した細胞を、1pfu/細胞のFusOn−H2に感染させた。クロマチン凝集のためのヘキスト染料染色を、上記で説明したように実行した。Spot Image Software(Diagnostic Instrument, Inc, IL)を用いて、別々の蛍光灯による同じ画像域からの顕微鏡写真の重ね合せを実行した。同じ区域の中のGFP陽性アポトーシス細胞とGFP陰性アポトーシス細胞とを別々に計数した。各区域で約100のアポトーシス細胞を数えた。全部で3つの区域を計算して、FusOn−H2感染細胞によって誘導された傍観者効果を証明した。
【0156】
末端デオキシヌクレオチジルトランスフェラーゼ媒介ニック端標識化(Tunel)アッセイ
雌のHsd無胸腺(nu/nu)マウス(Harlan, Indianapolis, Indianaから入手した)を特定の病原体感染防止条件下に保ち、5から6週齢に達したとき実験に用いた。0.25%のトリプシンおよび0.05%のEDTAに短時間曝露した亜集密培養からEC9706細胞を集めした。10%FBSを含む培地でトリプシン処理を停止させた後、無血清培地中で細胞を1回洗浄し、PBS中に再懸濁した。第0日に、ヌードマウスの右横腹に5×106のEC9706細胞を接種した。腫瘍細胞注入の2週間後、腫瘍の直径が約5mmに到達したとき、体積100μl中3×106pfuのFusOn−H2またはBaco−1、あるいは同じ体積のPBSをマウスに単回腫瘍内注射した。腫瘍を毎週測定し、式:腫瘍体積[mm3]=(長さ[mm])×(幅[mm])2×0.52によって腫瘍の体積を求めた。Tunelアッセイでは、1×107pfuのFusOn−H2またはBaco−1ウイルスを腫瘍内注入して3日後、CO2に曝露してマウスを安楽死させた。腫瘍組織を外植し、Tunel染色に備えて区分けした。
【0157】
FusOn−H2は、さまざまな組織起源のヒト腫瘍細胞中にアポトーシスを誘導する
シクロヘキシミドなどの翻訳阻害剤によってウイルス蛋白質合成を遮断しなければ、特定のHSV−2遺伝子産物の抗アポトーシス活性によって、HSV−2に感染させても、通常、アポトーシスを誘導しない(Aubert et aG, (1999) J Virol 73(12): 10359−70)。HSV−2由来のICP10遺伝子のPKドメインは、抗アポトーシス機能を有するウイルス遺伝子産物の1つとして認識され、ウイルスゲノムから削除すると、ある種の体細胞のアポトーシス死を誘導する能力をウイルスに与えることが記載された(Perkinsら、(2002) J Virol76(3): 1435−49)。
【0158】
FusOn−H2が腫瘍細胞のアポトーシス死を誘導するか定めるために、さまざまな組織起源のヒト腫瘍細胞のパネルを10m.o.i.のウイルスに感染させた。HSV−1、Baco−1から誘導した殺腫瘍ウイルスを対照として評価に含めた。腫瘍細胞の中で、EC9706はヒト食道癌細胞系統であり、SKOV3はヒト卵巣がん細胞系統であり、SW403およびSW480はヒト大腸癌細胞系統である。細胞を6ウェルプレートに接種し、翌日、ウイルスに感染させた。感染の24時間後、細胞をヘキスト染料33358で染色した。FusOn−H2による腫瘍細胞の感染は、大量のクロマチン凝集を誘導し、アポトーシスを示した。これは、FusOn−H2感染細胞中に強い小さな青色の核染色が出現したことによって明白だった。全体として、FusOn−H2に感染した腫瘍細胞の80%超がクロマチン凝集を示した。非感染腫瘍細胞は、そのようなアポトーシスの特徴を非常にわずかに、またはまったく示さなかった。これらの腫瘍細胞を親の野生型HSV−2(wt186)またはBaco−1に感染させても、クロマチン凝集の青色蛍光染色の背景レベルはあまり増加しなかった。
【0159】
腫瘍細胞中にアポトーシスを誘導するFusOn−H2の能力をさらに検証するために、DNA断片化を解析した。前の実験に用いた3つの腫瘍細胞を10pfu/細胞のウイルスに感染させるかまたは偽感染させた。感染の24時間後、細胞を集めた。DNAを細胞から抽出し、1%アガロースゲル中で分離した。FusOn−H2感染物質が載っているウェルには明らかなラダーリングがあった。このラダーリングは、wt186またはBaco−1感染細胞のどちらか由来のDNA試料が載っているウェルの中で検出され、従って、上記で提示したクロマチン凝集の結果を再確認した。これらの結果を一緒にすると、FusOn−H2に感染させるとこれらのヒト腫瘍細胞中にアポトーシスが効率的に誘導されるが、親の野生型HSV−2にもHSV−1系殺腫瘍ウイルスにもそのような特性はない。
【0160】
FusOn−H2に感染すると、傍観者細胞のアポトーシス死も誘導される
FusOn−H2には強化された緑色蛍光蛋白質をコードする遺伝子があるので、その感染力は、蛍光顕微鏡下で容易に測定することができた。クロマチン凝集の蛍光性検出と感染力とを相互検証しているとき、青色の蛍光性クロマチン凝集を示す細胞と、GFP染色を示す細胞との百分率の間の明らかな不一致を認めた。クロマチン凝集を示す腫瘍細胞とGFP発現を示す腫瘍細胞との絶対数を数えると、比は、約2.6:1であった。この結果によると、FusOn−H2感染には、周囲の腫瘍細胞に対するかなりの傍観者アポトーシス効果があった。
【0161】
FusOn−H2誘導アポトーシス死は、腫瘍細胞死を加速し、腫瘍細胞内のウイルス複製を弱める
FusOn−H2に感染した腫瘍細胞と、殺腫瘍ウイルス誘導HSV−1に感染した腫瘍細胞との間で、細胞が細胞変性効果(CPE)を示した時間にも明らかな差異を認めた。1pfu/細胞の用量のFusOn−H2に感染した腫瘍細胞は、通常、24時間以内に十分なCPEを示したが、同じ用量のBaco−1に感染した腫瘍細胞は、形態学的にほとんど正常に見えた。それらは、通常、感染後72時間を超えるまで明らかなCPEの徴候を示さなかった。FusOn−H2に感染させたウェルの中では、感染後24時間で細胞の集団化と相互離脱を含む一般的なCPEを容易に見ることができたが、Baco−1に感染させた細胞は、感染後48時間でも基本的に偽感染細胞と同じに見えた。これらの結果によると、FusOn−H2によって誘導されるアポトーシス細胞死は、ウイルス感染後すぐに起こったが、ウイルス複製の殺腫瘍効果が起こるにはもっと長い時間を要した。
【0162】
アポトーシス腫瘍細胞死は重要なウイルスのインビボ抗腫瘍機序である
既に記載した実験で用いた腫瘍細胞の1つから確立した腫瘍異種移植物に対するFusOn−H2のインビボ抗腫瘍活性を評価した。Baco−1をこの実験に含め、これらの2つのウイルスの治療効果を直接比較することができるようにした。新たに調製した5×106のEC9706細胞の皮下注射によって、腫瘍異種移植物を右横腹に確立した。腫瘍サイズが直径約5mmに達したとき、マウスにどちらかのウイルス(FusOn−H2またはBaco−1)、あるいは対照としてのPBSを、3×106pfuの用量で単回腫瘍内注射した。6週にわたって腫瘍を定期的に測定し、治療前の腫瘍体積を治療後の異なる時点の腫瘍体積で除することによって、腫瘍成長比を求めた。FusOn−H2を治療投与すると、基本的に、腫瘍成長は1週間以内に停止した。その後、腫瘍は縮み始め、実験終了までに平均腫瘍サイズはビボ治療前のサイズの約2分の1でしかなくなり、半数を超えるマウスの腫瘍は完全に消えた。対照のPBSと比べると、Baco−1を投与しても、3週までいかなる治療効果も示さなかった。しかし、腫瘍は第35日には再び成長し始めたので、腫瘍収縮は一時的なようであった。この腫瘍細胞中ではアポトーシスの誘導によって複製能力が制約されるという事実にもかかわらず、全体として、FusOn−H2の治療効果は、評価を行ったすべての時点でBaco−1の治療効果より有意に強かった(p<0.05)。これらの結果によると、このインビボ研究では、FusOn−H2によって誘導されたアポトーシス死と、同時に起こる傍観者効果とが主要抗腫瘍機序であった可能性が高い。
【0163】
(実施例9)
FusOn−H2による腫瘍破壊は強力な抗腫瘍免疫を誘導する
2つの純系腫瘍モデル、すなわちマウス乳腺腫瘍(4T1細胞)とマウス神経芽細胞腫(ニューロ2A細胞)におけるFusOn−H2の抗腫瘍活性を評価した。どちらの場合にも、FusOn−H2は、統計的に有意な抗腫瘍効果を示し、同時に堅固な腫瘍特異性免疫応答も示した。下記に提示するのは、乳腺腫瘍モデルにおける研究からの一般的なデータである。
【0164】
この評価には、4T1細胞を利用した。これは、非免疫原性であり、非常に悪性であり、純系BALB/cマウスにおいて高度に転移性である(Aslakson and Miller (1992) Cancer Res 52(6): 1399−405; Pulaski and Ostrand−Rosenberg (1998) Cancer Res. 58(7): 1486−93)。4T1細胞(1×105)を免疫能力のあるBALB/cマウスの乳腺脂肪層に同所投与注射して同所性腫瘍を確立した。マウスを10日間飼育したところ、群の90%を超える個体で肺転位を検知することができた。次に、腫瘍を持つマウスを3つの群(それぞれn=10)に分け、FusOn−H2か、あるいは、このモデルにおいて有効な抗腫瘍免疫を誘導することが既に示されている二重融合型Synco−2D(Nakamori, Fu et al., (2004) Mol. Ther. 9(5): 658−665)を含むHSV−1から誘導した他の殺腫瘍HSVか、のどちらかの1×107pfuを腫瘍内注射した。2週間にわたって、正所性部位の腫瘍質量を毎週測定した後、マウスを死なせ、免疫学的アッセイおよび肺転位の評価を行った(墨汁灌流後、解剖顕微鏡下で計測)。免疫学的アッセイでは、外植された脾臓から脾細胞を調製し、照射4T1細胞で5日間インビトロ刺激してから、以下のアッセイ、すなわち、1)51Cr放出アッセイによる腫瘍特異性CTL活性(4T1細胞または純系肉腫細胞系統Meth−Aのどちらかを標的細胞とする)、2)BD Biosciences社から購入した検出キットを用いるマウスIFN−γ分泌細胞のElispot分析、3)サイトカイン分泌の定量(インターフェロン−γとIL−10との両方について)を用いた。結果によると、FusOn−H2を局所腫瘍内投与すると、正所性腫瘍だけででなく遠隔肺転位に対しても他のウイルスより有意に良好な治療効果が認められた。Synco−2Dは、Baco−1と比較すると、正所性腫瘍および転移性腫瘍の成長を阻害することができた。この結果は、本発明者らの既報の観察結果(Nakamori, Fu et al., (2004) Mol. Ther. 9(5): 658−665)と類似していた。しかし、FusOn−H2は、この腫瘍を治療する上でSynco−2Dより有効でさえあるように見えた。FusOn−H2によって誘導される同時発生抗腫瘍免疫応答も、腫瘍特異性CTL活性および頻度ならびにサイトカイン放出を含め、Synco−2Dの腫瘍免疫応答より明らかであり、局所腫瘍および転移性腫瘍の除去に対するそれらの寄与を示した。
【0165】
(実施例10)
ウイルス力価を測定するためのプラーク形成アッセイ
ウイルス原料を調製した後、既に記載したプラーク形成アッセイ(Lancz GJ. (1974). Arch Virol., 46, 36−43を参照すること)を用いてウイルス力価を測定した。ベロ細胞をトリプシン処理し、計数し、ウェルあたり4×105細胞で6ウェルプレートに塗布し、37℃、5%CO2および90%湿度で温置し、24時間培養する。次の日、ウイルスを、1×最小必須培地(MEM)の中で次々に1:10に希釈して10−3〜10−8の6つの濃度にする。次に、培地をウェルから吸い取り、0.5mlのウイルス希釈物を3つ1組で各ウェルに加える。次に、15分ごとに振盪してプレートを1時間培養する。培養期間の後、ウイルス溶液を吸い取り、1%アガロースを含む2mlのMEMを各ウェルに加え、プレートを3日間培養した後、0.1%クリスタルバイオレットと20%エタノールとを含む溶液で細胞を染色する。30分間の培養期間の終わりに、染色物を吸い取り、10倍の倍率の立体顕微鏡を用いてプラークを計数する。次に、ウイルス力価をmlあたりプラーク形成単位として表す。
【図面の簡単な説明】
【0166】
【図1】図1A〜1Cは、FusOn−H2構築のための戦略を示す。図1A。HSV−2ゲノムの概略図。ゲノムは灰色の横棒によって表され、末端繰り返し単位(TR)および内部繰り返し単位(IR)は灰色の箱として示される。ICP10遺伝子の位置も示されている。図1B。ICP10遺伝子の拡大図であり、PKドメインおよびRR1ドメイン、ならびに天然プロモーターの位置を示す。図1C。続いてウイルスゲノムに挿入されてFusOn−H2を構築した改変ICP10遺伝子。図に示したように、PKドメインは、EGFP遺伝子(RR遺伝子との枠組の中)で置換され、遺伝子の元のプロモーターは、最も強い哺乳類遺伝子プロモーターの1つであるサイトメガロウイルスの即時早期プロモーターで置換された。非改変ICP10遺伝子座および改変ICP10遺伝子座のBamHI制限部位を標識化する。PKL、PK、GFPおよびPKRと印を付けたボックスは、図2のサザンハイブリダイゼーションにおいて用いられる4つのプローブがハイブリッド形成する位置を示す。
【図2】図2は、FusOn−H2のサザンブロット分析を示す。サザンブロットハイブリダイゼーションは、親の野生型HSV−2(w)またはFusOn−H2(m)のどちらか由来のBamHI溶解ウイルスDNAを示す。サザンハイブリダイゼーションに用いられる4つのプローブは、左側(left−flank)領域から作られたPKL、PKドメイン領域から作られたPK、右側(right−flank)領域から作られたPKR、EGFP遺伝子から調製されたGFPであった。
【図3】図3は、抗GFPmAbを用いるFusOn−H2のウェスタンブロット分析を示す。FusOn−H2(m)またはその親の野生型HSV−2(w)のどちらかに感染したベロ細胞から、あるいはpSZ−EGFPプラスミドDNA(p)で形質移入したベロ細胞から、細胞溶解物を調製した。
【図4】図4は、培養細胞中のFusOn−H2の表現型キャラクタリゼーションを示す。細胞は、0.01pfu/細胞の図に示したウイルスに感染させるか、または感染させなかった。感染の24時間後に顕微鏡写真を撮影した。白色の矢印によって合胞体を示す。被検細胞の中で、MDA−MB−435はヒト乳癌系統であり、MPans−96はヒトすい臓癌系統であり、SKOV3はヒト卵巣がん系統である。元の倍率は200倍である。
【図5】図5A〜Cは、FusOn−H2の選択的複製を示す。図5A。ベロ細胞を完全循環状態(10%FBS)に保持するか、または24時間血清飢餓状態に保持してから、1pfu/細胞のウイルスに感染させた。図に示した時点で細胞を集め、ベロ細胞単一層上のプラークアッセイによってウイルス収量を定量した。図5B。ウイルス感染の間、単独または50μMのPD98059共存下、低い百分率の血清(2%)を含む培地中でベロ細胞を培養した。感染の24時間および48時間後に細胞を集め、PD98059非存在下でのウェル中の全ウイルス収量を、薬物を含むウェルからの収量で除することによって、ウイルス複製における倍数減少を計算した。図5C。インビトロ培養した一次肝細胞を1pfu/細胞の図に示したウイルスに感染させた。図に示した感染後の時点でウイルスを集め、ベロ細胞単一層上のプラークアッセイによって定量した。*p<0.01、wt186と比較したFusOn−H2(スチューデントt検定)。
【図6】図6AおよびB。殺腫瘍HSVによるヒト癌細胞のインビトロ殺細胞能力。0.01pfu/細胞(A)または0.1pfu/細胞(B)のどちらかで細胞をウイルスに感染させた。図に示した時点のLDHアッセイによって細胞生存率を求めた。ウイルス感染細胞から放出されたLDHを非感染細胞からのそれで除することによって殺細胞百分率を計算した。p<0.01、wt186またはBaco−1と比較したFusOn−H2、Ψp<0.01、wt186と比較したFusOn−H2(スチューデントt検定)。
【図7】図7AおよびB。異種移植型ヒト乳癌に対するFusOn−H2のインビボ抗腫瘍活性。図7A。腫瘍内投与後の治療効果。MDA−MB−435細胞を第2乳腺の脂肪部分に注入することによってヒト乳腺腫瘍異種移植を実施した。腫瘍が直径約5mmに到達したとき、1×106pfuの投与量のウイルスを腫瘍内注射した。治療群は、FusOn−H2、Baco−1またはPBSを含む。ウイルス注入してから図に示した週後に測定した腫瘍体積を治療前の腫瘍体積で除することによって腫瘍成長比を求めた(群あたりn=8のマウス)。図7B。大型乳腺腫瘍異種移植に対する治療効果。腫瘍内注射群および静脈内注射群(それぞれn=5)について、腫瘍はそれぞれ直径10および10〜15mmであった。腫瘍内注射および静脈内注射のそれぞれで、3×106pfuおよび1.5×107pfuの投与量のウイルスを投与した。図6Aの場合と同じ方法で腫瘍成長比を計算した。Ψp<0.05、Baco−1と比較したFusOn−H2、*p<0.01、Synco−2Dと比較したFusOn−H2(スチューデントt検定)。
【図8】図8。ヌードマウスの腹腔内に確立した転移性ヒト卵巣癌異種移植に対するFusOn−H2の治療効果。腹腔への2×106のSKOV3細胞の腹腔内接種によってヒト卵巣がん異種移植を確立した(治療群あたりn=8のマウス)。腫瘍細胞接種の8日後および15日後、腫瘍着床部位から遠く離れた部位への3×106pfuの投与量の殺腫瘍HSVの腹腔内投与をマウスに施した。最初のウイルス注入の4週間後(すなわち腫瘍細胞注射の5週間後)に、マウスを安楽死させた。この図に腫瘍小結節の概観を示し、表1に各動物からの腫瘍小結節の数および腫瘍重量を示す。
【特許請求の範囲】
【請求項1】
改変ICP10ポリヌクレオチドを内部に有する単純ヘルペスウイルス2型(HSV−2)を含む化合物の、宿主内の癌細胞を選択的に殺傷するための薬物の製造における使用であって、該改変ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くICP10ポリペプチドをコードする、使用。
【請求項2】
前記ICP10ポリヌクレオチドは、該ICP10ポリヌクレオチドのプロテインキナーゼドメインの一部を削除することによって改変されている、請求項1に記載の使用。
【請求項3】
前記ICP10ポリヌクレオチドは、緑色蛍光蛋白質、β−ガラクトシダーゼ、ルシフェラーゼおよび単純ヘルペスウイルスチミジンキナーゼ(HSVtk)からなる群より選択されるレポーター蛋白質をコードするポリヌクレオチドの挿入によって改変されている、請求項1に記載の使用。
【請求項4】
前記ICP10ポリヌクレオチドは、腫瘍壊死因子、インターフェロンアルファ、インターフェロンベータ、インターフェロンガンマ、インターロイキン−2、インターロイキン12、GM−CSF、F42K、MIP−1、MIP−1βおよびMCP−1からなる群より選択される免疫調節遺伝子をコードするポリヌクレオチドの挿入によって改変されている、請求項1に記載の使用。
【請求項5】
前記改変ICP10ポリヌクレオチドは、構成的プロモーターに作動可能に結合している、請求項1から4の任意の1項に記載の使用。
【請求項6】
前記ICP10ポリヌクレオチドは、前記内因性プロテインキナーゼ活性ドメインの少なくとも一部を融合性膜糖蛋白質をコードするポリヌクレオチドで置換することによってさらに改変されている、請求項1〜5のいずれか1項に記載の使用。
【請求項7】
前記融合性膜糖蛋白質は、テナガザル白血病ウイルスエンベロープ融合性膜糖蛋白質、マウス白血病ウイルスエンベロープ蛋白質、細胞質ドメインを欠くレトロウイルスエンベロープ蛋白質、はしかウイルス融合蛋白質、HIV gp160蛋白質、SIV gp160、レトロウイルスEnv蛋白質、エボラウイルスGpおよびインフルエンザウイルスヘムアグルチニンからなる群より選択される、請求項6に記載の使用。
【請求項8】
前記薬物は、癌細胞を、該癌細胞膜の融合(合胞体形成)によって選択的に殺傷する、請求項1に記載の使用。
【請求項9】
前記薬物は、感染腫瘍細胞および周囲の腫瘍細胞においてアポトーシスを誘導することによって、癌細胞を選択的に殺傷する、請求項1に記載の使用。
【請求項10】
前記薬物を腫瘍内投与する、請求項1に記載の使用。
【請求項11】
前記薬物を全身投与する、請求項1に記載の使用。
【請求項12】
前記薬物を腹腔内投与する、請求項1に記載の使用。
【請求項13】
腫瘍崩壊特性と融合特性とを有するHSV−2ウイルスを生成する方法であって、
ICP10ポリヌクレオチドを改変する工程であって、該改変ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くICP10ポリペプチドをコードする、工程、および
該改変ICP10ポリヌクレオチドを構成的に活性なプロモーターと作動可能に結合させる工程、
を含む方法。
【請求項14】
腫瘍崩壊特性および融合特性を有する単純ヘルペスウイルス2型(HSV−2)を含む組成物であって、該HSV−2は、改変ICP10ポリヌクレオチドを含み、該改変ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くICP10ポリペプチドをコードし、該改変ICP10ポリヌクレオチドは、構成的プロモーターに作動可能に結合された、組成物。
【請求項15】
前記HSV−2は、前記ICP10遺伝子のプロテインキナーゼ活性ドメインに挿入された、融合性膜糖蛋白質をコードするポリヌクレオチドをさらに含み、該融合性膜糖蛋白質は、テナガザル白血病ウイルスエンベロープ融合性膜糖蛋白質、該テナガザル白血病ウイルスエンベロープ糖蛋白質のC末端切除形(GALV.fus)、マウス白血病ウイルスエンベロープ蛋白質、細胞質ドメインを欠くレトロウイルスエンベロープ蛋白質、はしかウイルス融合蛋白質、HIV gp160蛋白質、SIV gp160、レトロウイルスEnv蛋白質、エボラウイルスGpおよびインフルエンザウイルスヘムアグルチニンからなる群より選択される、請求項14に記載の組成物。
【請求項16】
前記HSV−2は、前記ICP10遺伝子のプロテインキナーゼ活性ドメインに挿入されたポリヌクレオチドを含み、該ポリヌクレオチドは、緑色蛍光蛋白質、β−ガラクトシダーゼ、ルシフェラーゼおよび単純ヘルペスウイルスチミジンキナーゼ(HSVtk)からなる群より選択されるレポーター蛋白質をコードする、請求項14に記載の組成物。
【請求項17】
前記HSV−2は、前記ICP10遺伝子のプロテインキナーゼ活性ドメインに挿入されたポリヌクレオチドを含み、該ポリヌクレオチドは、腫瘍壊死因子、インターフェロンアルファ、インターフェロンベータ、インターフェロンガンマ、インターロイキン−2、インターロイキン12、GM−CSF、F42K、MIP−1、MIP−1βおよびMCP−1からなる群より選択される免疫調節遺伝子をコードする、請求項14に記載の組成物。
【請求項18】
前記HSV−2は、前記ICP10ポリヌクレオチドのプロテインキナーゼ活性ドメインに挿入された治療用ポリヌクレオチドをさらに含み、該治療用ポリヌクレオチドは、HSVtk、シトシンデアミナーゼおよびカスパーゼ−3からなる群より選択される、請求項14に記載の組成物。
【請求項19】
前記構成的に活性なプロモーターは、即時早期サイトメガロウイルスプロモーターである、請求項14に記載の組成物。
【請求項20】
リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くポリペプチドをコードする改変ICP10ポリヌクレオチドを含むHSV−2ウイルスベクターを含む組成物であって、該改変ポリヌクレオチドは、構成的プロモーターに作動可能に結合した、組成物。
【請求項1】
改変ICP10ポリヌクレオチドを内部に有する単純ヘルペスウイルス2型(HSV−2)を含む化合物の、宿主内の癌細胞を選択的に殺傷するための薬物の製造における使用であって、該改変ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くICP10ポリペプチドをコードする、使用。
【請求項2】
前記ICP10ポリヌクレオチドは、該ICP10ポリヌクレオチドのプロテインキナーゼドメインの一部を削除することによって改変されている、請求項1に記載の使用。
【請求項3】
前記ICP10ポリヌクレオチドは、緑色蛍光蛋白質、β−ガラクトシダーゼ、ルシフェラーゼおよび単純ヘルペスウイルスチミジンキナーゼ(HSVtk)からなる群より選択されるレポーター蛋白質をコードするポリヌクレオチドの挿入によって改変されている、請求項1に記載の使用。
【請求項4】
前記ICP10ポリヌクレオチドは、腫瘍壊死因子、インターフェロンアルファ、インターフェロンベータ、インターフェロンガンマ、インターロイキン−2、インターロイキン12、GM−CSF、F42K、MIP−1、MIP−1βおよびMCP−1からなる群より選択される免疫調節遺伝子をコードするポリヌクレオチドの挿入によって改変されている、請求項1に記載の使用。
【請求項5】
前記改変ICP10ポリヌクレオチドは、構成的プロモーターに作動可能に結合している、請求項1から4の任意の1項に記載の使用。
【請求項6】
前記ICP10ポリヌクレオチドは、前記内因性プロテインキナーゼ活性ドメインの少なくとも一部を融合性膜糖蛋白質をコードするポリヌクレオチドで置換することによってさらに改変されている、請求項1〜5のいずれか1項に記載の使用。
【請求項7】
前記融合性膜糖蛋白質は、テナガザル白血病ウイルスエンベロープ融合性膜糖蛋白質、マウス白血病ウイルスエンベロープ蛋白質、細胞質ドメインを欠くレトロウイルスエンベロープ蛋白質、はしかウイルス融合蛋白質、HIV gp160蛋白質、SIV gp160、レトロウイルスEnv蛋白質、エボラウイルスGpおよびインフルエンザウイルスヘムアグルチニンからなる群より選択される、請求項6に記載の使用。
【請求項8】
前記薬物は、癌細胞を、該癌細胞膜の融合(合胞体形成)によって選択的に殺傷する、請求項1に記載の使用。
【請求項9】
前記薬物は、感染腫瘍細胞および周囲の腫瘍細胞においてアポトーシスを誘導することによって、癌細胞を選択的に殺傷する、請求項1に記載の使用。
【請求項10】
前記薬物を腫瘍内投与する、請求項1に記載の使用。
【請求項11】
前記薬物を全身投与する、請求項1に記載の使用。
【請求項12】
前記薬物を腹腔内投与する、請求項1に記載の使用。
【請求項13】
腫瘍崩壊特性と融合特性とを有するHSV−2ウイルスを生成する方法であって、
ICP10ポリヌクレオチドを改変する工程であって、該改変ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くICP10ポリペプチドをコードする、工程、および
該改変ICP10ポリヌクレオチドを構成的に活性なプロモーターと作動可能に結合させる工程、
を含む方法。
【請求項14】
腫瘍崩壊特性および融合特性を有する単純ヘルペスウイルス2型(HSV−2)を含む組成物であって、該HSV−2は、改変ICP10ポリヌクレオチドを含み、該改変ICP10ポリヌクレオチドは、リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くICP10ポリペプチドをコードし、該改変ICP10ポリヌクレオチドは、構成的プロモーターに作動可能に結合された、組成物。
【請求項15】
前記HSV−2は、前記ICP10遺伝子のプロテインキナーゼ活性ドメインに挿入された、融合性膜糖蛋白質をコードするポリヌクレオチドをさらに含み、該融合性膜糖蛋白質は、テナガザル白血病ウイルスエンベロープ融合性膜糖蛋白質、該テナガザル白血病ウイルスエンベロープ糖蛋白質のC末端切除形(GALV.fus)、マウス白血病ウイルスエンベロープ蛋白質、細胞質ドメインを欠くレトロウイルスエンベロープ蛋白質、はしかウイルス融合蛋白質、HIV gp160蛋白質、SIV gp160、レトロウイルスEnv蛋白質、エボラウイルスGpおよびインフルエンザウイルスヘムアグルチニンからなる群より選択される、請求項14に記載の組成物。
【請求項16】
前記HSV−2は、前記ICP10遺伝子のプロテインキナーゼ活性ドメインに挿入されたポリヌクレオチドを含み、該ポリヌクレオチドは、緑色蛍光蛋白質、β−ガラクトシダーゼ、ルシフェラーゼおよび単純ヘルペスウイルスチミジンキナーゼ(HSVtk)からなる群より選択されるレポーター蛋白質をコードする、請求項14に記載の組成物。
【請求項17】
前記HSV−2は、前記ICP10遺伝子のプロテインキナーゼ活性ドメインに挿入されたポリヌクレオチドを含み、該ポリヌクレオチドは、腫瘍壊死因子、インターフェロンアルファ、インターフェロンベータ、インターフェロンガンマ、インターロイキン−2、インターロイキン12、GM−CSF、F42K、MIP−1、MIP−1βおよびMCP−1からなる群より選択される免疫調節遺伝子をコードする、請求項14に記載の組成物。
【請求項18】
前記HSV−2は、前記ICP10ポリヌクレオチドのプロテインキナーゼ活性ドメインに挿入された治療用ポリヌクレオチドをさらに含み、該治療用ポリヌクレオチドは、HSVtk、シトシンデアミナーゼおよびカスパーゼ−3からなる群より選択される、請求項14に記載の組成物。
【請求項19】
前記構成的に活性なプロモーターは、即時早期サイトメガロウイルスプロモーターである、請求項14に記載の組成物。
【請求項20】
リボヌクレオチドレダクターゼ活性を有するがプロテインキナーゼ活性を欠くポリペプチドをコードする改変ICP10ポリヌクレオチドを含むHSV−2ウイルスベクターを含む組成物であって、該改変ポリヌクレオチドは、構成的プロモーターに作動可能に結合した、組成物。
【図1】

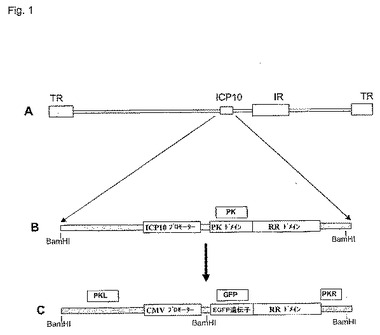
【図2】

【図3】
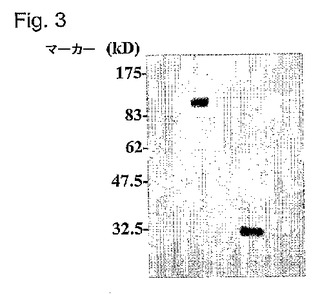
【図4】
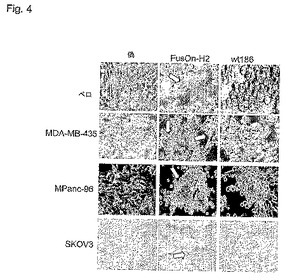
【図5】
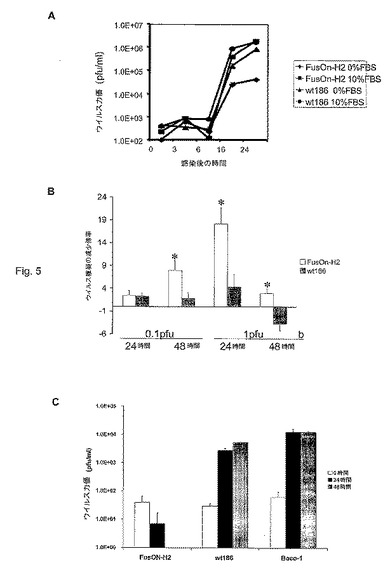
【図6】
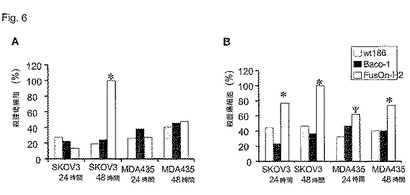
【図7】
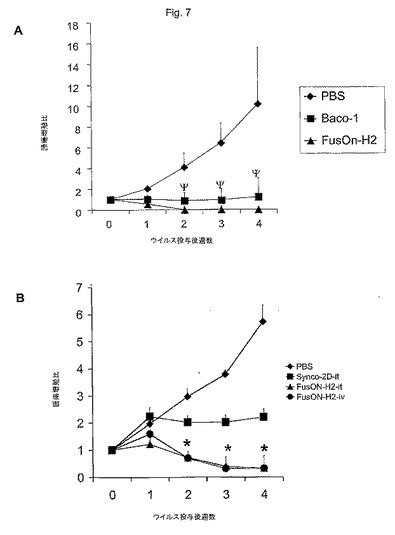
【図8】
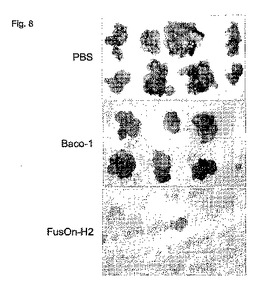
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2008−546801(P2008−546801A)
【公表日】平成20年12月25日(2008.12.25)
【国際特許分類】
【出願番号】特願2008−518422(P2008−518422)
【出願日】平成18年6月23日(2006.6.23)
【国際出願番号】PCT/US2006/024440
【国際公開番号】WO2007/002373
【国際公開日】平成19年1月4日(2007.1.4)
【出願人】(391058060)ベイラー カレッジ オブ メディスン (16)
【氏名又は名称原語表記】BAYLOR COLLEGE OF MEDICINE
【Fターム(参考)】
【公表日】平成20年12月25日(2008.12.25)
【国際特許分類】
【出願日】平成18年6月23日(2006.6.23)
【国際出願番号】PCT/US2006/024440
【国際公開番号】WO2007/002373
【国際公開日】平成19年1月4日(2007.1.4)
【出願人】(391058060)ベイラー カレッジ オブ メディスン (16)
【氏名又は名称原語表記】BAYLOR COLLEGE OF MEDICINE
【Fターム(参考)】
[ Back to top ]