
癌治療のための組成物及び方法
本発明は、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌を、mTOR阻害剤及び抗IGF−1R抗体を用いて治療する方法であって、ここで、mTOR阻害剤が、リダフォロリムス、エベロリムス、テムシロリムス、又はそれらの組合せである方法を提供する。

【発明の詳細な説明】
【背景技術】
【0001】
ホスファチジルイノシトール−3−キナーゼ(PI3K)シグナリング経路は、多くの様々なタイプのヒトの悪性腫瘍において、癌細胞の増殖及び生存に重要である。グランビル(Granville CA)ら著、「Handicapping the Race to Develop Inhibitors of the Phosphoinositide 4−Kinase/Akt/Mammalian Target of Rapamaycin Pathway(ホスホイノシチド−4−キナーゼ/Akt/哺乳類ラパマイシン経路標的タンパク質の阻害剤開発競争のハンディキャッピング)、Clin Cancer Res」、2006年、第12巻、第3号、p.679−89参照。この経路は、上皮成長因子受容体及びインスリン様成長因子受容体などの、リガンド受容体相互作用からの上流のインプットを受け、哺乳類のラパマイシン標的タンパク質(mTOR)などの、下流のエフェクターを介してシグナリングする。mTORは、細胞周期進行及び多くの他の重要な細胞増殖プロセスにとり決定的なタンパク質の産生を調節する、極めて重要な下流エフェクター分子である。アブラハム(Abraham RT)及びギボンス(Gibbons,JJ)著、「The mammalian target of rapamycin signaling pathway:twists and turns in the road to cancer therapy(哺乳類のラパマイシンシグナリング経路標的タンパク質:癌療法への道における紆余曲折)、Clin Cancer Res」、2007年、第13巻、第11号、p.3109−14参照。
【0002】
PI3キナーゼ軸(PI3 Kinase axis)の調節異常は、ヒトの癌に共通しており、それらは、過活動性の成長因子受容体シグナリング、PI3Kの突然変異の活性化、PTEN腫瘍抑制因子の機能喪失、及びmTORキナーゼ活性の活性化をもたらす他のいくつかのメカニズムに起因する。臨床的には、PI3K軸の首尾よい薬理学的阻害は、PI3キナーゼの、上流の成長因子受容体と、下流エフェクター(例えばmTOR)とにフォーカスされてきた。現在、mTOR阻害剤が、進行性悪性腫瘍の患者に臨床上の利益を与え得ることを示す実質的な臨床的証拠がある。
【0003】
インスリン受容体ファミリーのチロシンキナーゼ受容体である、インスリン様成長因子1型受容体(IGF−1R)は、細胞増殖、分化に関与し、多くのタイプの癌において、悪性細胞の形質転換及び維持に重要な役割を果たす。バセルガ(Baserga,R)ら著、「Mini Review:The IGF−1R receptor in cancer biology(ミニレビュー:癌生物学におけるIGF−1R受容体)、Int.J.Cancer」、2003年、第107巻、p.873−77参照。IGF−1R及びそのリガンドIGF−2は、多くのタイプの進行癌において過剰発現され、リガンド刺激による受容体シグナリングが、癌細胞の増殖をインビトロにおいて促進する。重要なことに、IGF−1Rシグナリングは、PI3K軸に密に関連している。IGF−1R阻害は、強力な抗癌効果を前臨床研究において示しており、現在いくつかのIGF−1R阻害剤が臨床開発中である。
【0004】
mTORとIGF−1R阻害剤との組合せは、PI3K軸における上流及び下流の双方の分子標的を阻害することにより、相乗効果を提供し得る。mTORの阻害は、Akt発癌遺伝子を活性化するフィードバックループの活性化につながり得るものであり、このことは、インビトロの及びmTOR阻害剤を処置された患者から採取された腫瘍生検からの腫瘍細胞において、ホスホ−Aktのレベルの上昇として具現化する。サン(Sun,S−Y)ら著、「Priority Report:Activation of Akt and eIF4E survival pathways by rapamycin−mediated mammalian target of rapamycin inhibition(プライオリティレポート:ラパマイシン媒介性哺乳類ラパマイシン標的タンパク質阻害によるAkt及びeIF4E生存経路の活性化)、CANCER RES」、2005年、第65巻、第16号、p.7052−58;及びガードナー(Gardner,H)ら著、「Biomarker analysis of a phase II double−blind randomized trial of daily oral RAD001(everolimus)plus letrozole or placebo plus letrozole as neoadjuvant therapy for patients with estrogen receptor positive breast cancer(エストロゲン受容体陽性乳癌患者のためのネオアジュバント療法としての、RAD001(エベロリムス)プラスレトロゾール、又はプラセボプラスレトロゾールの、毎日の経口投与に関する第II相無作為化二重盲検試験のバイオマーカー分析)、San Antonio Breast Cancer Symposium(サンアントニオ乳癌シンポジウム)」、2007年、12月13−16日、San Antonio、TX、参照。このフィードバックループは、IGF−1R及びインスリン受容体基質を介したシグナリングを含み得るものであり、IGF−1R阻害剤によって阻害される。結果として、前臨床試験は、IGF−1R阻害剤とmTOR阻害剤との組合せが、相加的又は相乗的な抗腫瘍活性をインビトロでもたらすことを示してきた。最近、2つのグループが独自に、ヒト肉腫の異種移植モデルで、ラパマイシンと抗IGF−1R抗体とを組合せた結果を報告した。クルマシェヴァ(Kurmasheva RT)ら、ポスター:「Combination of CP−751871,a human monoclonal antibody against the IGF−1 receptor with rapamycin results in highly effective therapy for xenografts derived from childhood sarcomas(CP−751871(IGF−1受容体に対するヒトモノクローナル抗体)とラパマイシンとの併用は小児肉腫由来の異種移植に高度に有効な療法をもたらす)」、EORTC 2007、及びダルコ(Darko,IA)ら著、要約:「Evaluation of combined insulin−like growth factor receptor type I(IGF−1R) and mTOR pathway blockade in sarcoma xenograft models(肉腫異種移植モデルにおけるインスリン様成長因子1型受容体(IGF−1R)とmTOR経路遮断との併用の評価)」、AACR Annual Meeting(AACR年回)、2007年、p.4760を参照。これらの研究の1つにおいては、確立されたユーイング及び骨肉腫異種移植片の完全な抑制が観察されたが、別の研究では、併用することで少なくとも相加的な利益を伴う強力な抗腫瘍活性が観察された。
【発明の概要】
【0005】
発明の要旨
本発明は、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌を、mTOR阻害剤及び抗IGF−1R抗体を用いて治療する方法であって、ここで、mTOR阻害剤が、リダフォロリムス、エベロリムス、テムシロリムス、ラパマイシン類似体、又はそれらの組合せであり、抗IGF−1R抗体が、ダロツズマブ(dalotuzumab)、フィギツムマブ、シクスツムマブ、SHC717454、ロシュ(Roche)R1507、EM164、又はアムジェン(Amgen)AMG479である方法を提供する。
【図面の簡単な説明】
【0006】
【図1】リダフォロリムス+MK−0646の併用による発癌性PI3Kシグナリングの改善されたターゲティングを示す図である。MK−0646&リダフォロリムスによる併用療法は、PI3K経路の阻害を増大し、癌細胞増殖を阻止する。(A)ネガティブフィードバックループを説明するPI3キナーゼシグナリング経路;(B)mTOR阻害が患者の腫瘍でAkt−Pの上昇をもたらすことを示す公表データ;(C)インビトロのH2122細胞におけるリダフォロリムス(10nM)、MK−0646(10ug/ml)、又はそれらの組合せに応答した経路シグナリング;(D)Cに用いた濃度で示した処置で24時間処理された細胞における、細胞周期分布及び細胞死を示すFACSプロフィールデータ。
【図2】リダフォロリムス&MK−0646の併用がインビトロの効果を増大することを示す図である。肺癌細胞系を、軟寒天中で、MK−0646若しくはリダフォロリムス又はそれらの組合せの存在下で培養した。軟寒天コロニー形成は、蛍光色素(LavaCell)を用いて定量化し、コロニーの面積及び数は、画像入力及び解析プラットフォーム(Isocyte)を使用して計数した。A)96ウエルプレートにおける相対コロニー面積がプロットされている。併用は、A549&H2122細胞系において、有意に増殖阻害を増強した(p<0.02)。P−RTKアレイにより測定されるRTKの活性化状態、及びKRASにおいて活性化する突然変異を下に示す。B)9NSCLC細胞系における、リダフォロリムス(10nM)若しくはMK−0646(10ug/ml)又はそれらの組合せのいずれかに応答した相対的軟寒天コロニー形成がプロットされている。細胞系は、KRAS中の活性化突然変異に基づき分類した。
【図3】突然変異体−KRAS異種移植腫瘍におけるリダフォロリムス&MK−0646併用の抗腫瘍活性を示す図である。MK−0646&リダフォロリムスによる併用処置は、A549異種移植片増殖の阻止において有効である。有意な腫瘍増殖阻害が、多様な濃度のMK−0646&リダフォロリムスを併用した併用処置群で、二元配置分散(2way ANOVA)分析により評価した場合に観察された。
【図4】リダフォロリムスと併用した高濃度のMK−0646により統計的に低減された腫瘍重量を示す図である。マウスに、MK−0646(20mpk)を単独で又はリダフォロリムス(0.1mpk)と併用でのいずれかにより、3週間にわたり投薬処置した。腫瘍重量(上記参照)は、リダフォロリムスと併用した、より高用量のMK−0646で、統計的に小さく、腫瘍の退縮が強調された。
【図5】A549異種移植片モデルがエルロチニブ処置に抵抗性であることを示す図である。ビヒクルに比較して、エルロチニブ処置群では何ら有意な増殖阻害が観察されなかった。
【図6】IGF1受容体シグナル伝達を示す図である。ホスファチジルイノシトール3−キナーゼ(PIK3CA)経路及びRAS経路を標的とするスクリーニングヒットが、それぞれ赤又は青で囲まれている。
【図7】コロニーアッセイによるヒットの検証により、PI3K経路のレギュレータを標的とするshRNAがMK−0646の効力に強く影響を及ぼすことを実証している図である。コロニー数(A)及びコロニー面積(B)は、アルファ・イメージャー(Alpha Imager)でプレートをスキャンすることにより測定した。MK−0646の上位3つの強力なエンハンサーは、PI3K経路のエフェクター(赤色で強調)をサイレンスさせたshRNAであり、一方PTENを標的とするshRNAは、薬剤に対する抵抗性を伝達した。
【図8】HT29結腸癌細胞におけるPTENの安定なサイレンシングが、MK−0646に対する抵抗性を伝達することを示す図である。PTEN又はCSK又はMAP3Kを標的とする種々のshRNAを発現する安定なHT29細胞を生成し、MK−0646に対する感受性をコロニー形成アッセイにおいて試験した。PTENのサイレンシングにより、MK−0646による増殖阻害の低減が示された。
【図9】PI3Kのサイレンシングは、PTEN欠失HT29細胞をMK−0646の効果に対し再感受性化するが、PDPK1又はMELKは再感受性化しないことを示す図である。PTENのRNAi媒介ノックダウンは、MK−0646媒介増殖阻害に対し抵抗性を付与する(図7&8参照)。PTENノックダウン細胞の増殖は、MK−0646&PI3K RNAiによる、IGF−1R及びPIK3CAの併用阻害により阻止し得る。
【図10】PI3K及びRas経路キナーゼが、MK−0646エンハンサースクリーンからの上位のコンセンサスヒットのうちで傑出していることを示す図である。PI3K及びRas経路の標準的な及び推定上のキナーゼレギュレータが、各々赤及び青で強調されている。定量的PCR分析を行なって、標的サイレンシング効率を評価した。ヒットの確認は、図7及び8に記載した通りのコロニー形成アッセイ又は短期間の増殖アッセイを用いて実施した。
【図11】確認されたMK−0646感受性化ヒットを示す図である。ある用量設定の薬剤(titration of drug)への暴露に続く、コロニーアウトグロースアッセイ(>2倍)又は72時間のアラマー(Alamar)アッセイ(p<0.05)のいずれかにおいて、MK−0646に対する腫瘍細胞の感受性を増大させたスクリーニングヒットを下に示す。あるベクターは、薬剤の不在下での毒性のため、いずれのアッセイでも検査不能であったことに注目されたい。色分けは、図10に定義した通りである。
【図12】リダフォロリムス−ダロツズマブ併用に応答している患者のCT画像を示す図である。画像は、肝臓(図に示した通り)及び他の部位に転移した、エストロゲン受容体陽性乳癌をもつ56歳の女性からである。患者は、多数回の事前の化学療法及びホルモン療法の後に進行した。上部パネルは、肝臓の左葉中の大きい腫瘍を示しており、下部パネルは、ダロツズマブと併用したリダフォロリムスの第I相臨床試験による2サイクルの処置後の、著しくサイズの縮小した同腫瘍を示す。患者は部分寛解を達成しており、これは8ヵ月を超える実験的治療後に継続していた。
【0007】
発明の詳細な記載
絶え間ない研究の結果、本発明者らは、mTOR阻害剤又はその薬学的に許容される塩を抗IGF−1R抗体と組合せて使用することにより、相乗的に優れた抗癌活性を達成し得ることを見出しており、ここで、mTOR阻害剤は、リダフォロリムス、エベロリムス、テムシロリムス、ラパマイシン類似体、又はそれらの組合せであり、抗IGF−1R抗体は、ダロツズマブ、フィギツムマブ、シクスツムマブ、SHC717454、ロシュR1507、EM164、又はアムジェンAMG479である。本発明は特に、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌の治療に有用である。しかしながら、本発明は、様々な他の癌、例えば脳腫瘍、頚大脳癌(cervicocerebral cancer)、食道癌、甲状腺癌、小細胞肺癌、肺癌、胃癌、胆嚢/胆管癌、肝臓癌、膵臓癌、卵巣癌、絨毛上皮癌、子宮体癌、子宮頚癌、腎盂/尿管癌、膀胱癌、前立腺癌、陰茎癌、精巣癌、胎児性癌、ウィルムス癌、皮膚癌、悪性黒色腫、神経芽細胞腫、骨肉腫、ユーイング腫瘍、軟部肉腫、急性白血病、慢性リンパ球性白血病、慢性骨髄性白血病、及びホジキンリンパ腫の治療に有用であることも立証し得た。
【0008】
したがって、本発明は、mTOR阻害剤及び抗IGF−1R抗体を用いて、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌を治療する方法であって、ここで、mTOR阻害剤が、リダフォロリムス、エベロリムス、テムシロリムス、ラパマイシン類似体、又はそれらの組合せであり、抗IGF−1R抗体が、ダロツズマブ、フィギツムマブ、シクスツムマブ、SHC717454、ロシュR1507、EM164、又はアムジェンAMG479である、該方法に関する。
【0009】
本発明の1つの実施態様においては、mTOR阻害剤は、リダフォロリムスである。
【0010】
本発明の別の実施態様においては、抗IGF−1R抗体は、少なくとも1つの非ヒト由来の重鎖相補性決定領域(CDR)と、少なくとも1つの非ヒト由来の軽鎖相補性決定領域(CDR)とを含んでなり、ここで、IGF−1Rに結合する前記抗体は、以下:(a)IGF−1Rには結合するが、IRには結合しない;(b)インスリン受容体とインスリン成長因子受容体とを含んでなるハイブリッド受容体(IR/IGF−1Rハイブリッド−R)には結合するが、単独のIRには結合しない;(c)ヒトIGF−1Rと、IGF−1及び/又はIGF−2との間の結合を阻害する;(d)前記ハイブリッド−R及びその天然のリガンド(好ましくは本明細書ではIGF1及び/又はIGF2及び/又はインスリンとして示されている)と、100nM未満の阻害定数及び/又はIC50で結合する;(e)前記IGF−1Rのチロシンキナーゼ活性を特異的に阻害する;(f)前記ハイブリッド−Rのチロシンキナーゼ活性を特異的に阻害する;(g)前記ハイブリッド−Rについて、10nM以下の結合親和性を有する;(h)IGF−1R発現をダウンレギュレートする;(i)ハイブリッド−R発現をダウンレギュレートする;(j)インビボの腫瘍増殖を阻害する、からなる群より選択される特性の少なくとも1つを有する。
【0011】
本発明の1つのクラスにおいては、重鎖CDRは、配列番号4、5、又は6からなる群より選択されるアミノ酸配列を含んでなり、かつ軽鎖CDRは、配列番号1、2、又は3からなる群より選択されるアミノ酸配列を含んでなる。
【0012】
本発明の別のクラスにおいては、ヒト化抗体、又はその機能性フラグメントの1つは、配列番号7又は8からなる群より選択されるアミノ酸配列を含んでなる軽鎖か、或いは配列番号9、10、又は11からなる群より選択されるアミノ酸配列を含んでなる重鎖を含んでなる。
【0013】
本発明の別のクラスにおいては、抗IGF−1R抗体は、ダロツズマブである。
【0014】
本発明の別の実施態様においては、mTOR阻害剤は、リダフォロリムスであり、かつ抗IGF−1R抗体は、ダロツズマブである。
【0015】
本発明の別の実施態様においては、mTOR阻害剤は、10mgないし40mgの用量で投与される。本発明の1つのクラスにおいては、リダフォロリムスは、1週間に5回投与される。
【0016】
本発明の別の実施態様においては、抗IGF−1R抗体は、10mg/kgの用量で静脈内投与される。本発明の1つのクラスにおいては、抗IGF−1R抗体は、週1回投与される。本発明の別のクラスにおいては、抗IGF−1R抗体は、2週間に1回投与される。
【0017】
mTOR阻害剤及び抗IGF−1R抗体は、同時に、別々に、又は逐次に投与するために調製し得る。
【0018】
上記に示した好ましい実施態様についての記載は、別に指定されない限り、特定の及び好ましい群の全ての組合せを含むものとする。本明細書において使用される用語の意味は、以下に記載されており、本発明は以下にさらに詳細に記載される。
【0019】
用語「同時」は、本明細書で言及する場合、本発明の医薬品が、時間的に同時に投与されることを意味する。
【0020】
用語「別々」は、本明細書で言及する場合、本発明の医薬品が、共通の治療スケジュールの過程の間の別々の時間に投与されることを意味する。
【0021】
用語「逐次」は、本明細書で言及する場合、1つの医薬品の投与に続いて、別の医薬品の投与がなされることを意味し;第2の医薬品は、1つの医薬品の投与後に、第1の医薬品の実質的に直後に投与してもよく、又は第2の医薬品を、第1の医薬品の有効期間の後に投与してもよく;また有効期間は、第1の医薬品の投与からの最大利益の実現のために与えられる時間の量である。
【0022】
用語「癌」は、本明細書で言及する場合、種々の肉腫及び癌腫を包含し、かつ固形癌及び造血器癌を包含する。固形癌は、本明細書で言及する場合、例えば、脳腫瘍、頚大脳癌、食道癌、甲状腺癌、小細胞肺癌、非小細胞肺癌、乳癌、子宮内膜癌、肺癌、胃癌、胆嚢/胆管癌、肝臓癌、膵臓癌、結腸癌、直腸癌、卵巣癌、絨毛上皮癌、子宮体癌、子宮頚癌、腎盂/尿管癌、膀胱癌、前立腺癌、陰茎癌、精巣癌、胎児性癌、ウィルムス腫瘍、皮膚癌、悪性黒色腫、神経芽細胞腫、骨肉腫、ユーイング腫瘍、軟部肉腫を包含する。一方、造血器癌は、例えば、急性白血病、慢性リンパ球性白血病、慢性骨髄性白血病、赤血球増加症、悪性リンパ腫、多発性骨髄腫、及びホジキンリンパ腫、非ホジキンリンパ腫を包含する。
【0023】
用語「癌の治療」は、本明細書で言及する場合、癌の症例に抗癌剤を投与して、その症例の癌細胞の増殖を阻害するようにすることを意味する。好ましくは、治療の結果として、癌増殖の退縮を生じるか、又はすなわち、それにより検出可能な癌のサイズが縮小される。さらに好ましくは、治療は癌の完全な消失をもたらす。
【0024】
mTOR阻害剤
現在臨床開発中のmTOR阻害剤は、ラパマイシンの構造類似体である。本発明のmTOR阻害剤は、リダフォロリムス、テムシロリムス、エベロリムス、ラパマイシン類似体、及びそれらの組合せを包含する。
【0025】
リダフォロリムスは、AP 23573、MK−8669、及びデフォロリムスとしても知られ、インビトロの広範囲のヒト腫瘍細胞系において、またヒト腫瘍細胞系を利用したマウス腫瘍異種移植片モデルにおいて抗増殖活性をもつ、ラパマイシンの独特の非プロドラッグ類似体である。リダフォロリムスは、進行癌患者に投与されてきており、現在、進行した軟部組織又は骨肉腫の患者における研究を含め、様々な進行性悪性腫瘍について臨床開発中である。今までのところ、これらの試験は、リダフォロリムスが、予測可能かつ管理可能な有害事象プロフィールをもち、全般的に充分に許容されること、及び広範囲の癌において抗腫瘍活性をもつことを証明してきた。リダフォロリムスの記載及び調製法は、アリアド・ジーン・セラピューティクス・インク(Ariad Gene Therapeutics,Inc.)に対する米国特許第7,091,213号に記載されており、これは、その記載全体が本明細書に援用される。
【0026】
テムシロリムスは、トリセル(Torisel)(登録商標)としても知られ、現在、腎細胞癌の治療用に市販されている。テムシロリムスの記載及び調製法は、アメリカン・ホーム・プロダクツ・コーポレーション(American Home Products Corporation)に対する米国特許第5,362,718号に記載されており、これは、その記載全体が本明細書援用される。
【0027】
エベロリムスは、サーティカン(Certican)(登録商標)又はRAD001としても知られ、ノバルティス(Novartis)から市販されており、ラパマイシン(シロリムス)と比較して、有機溶媒中でより優れた安定性及び増強された溶解性を有するとともに、副作用が少なくより好ましい薬物動態を有する。エベロリムスは、免疫抑制レジメンの効力を高めるため、シクロスポリンのマイクロエマルジョン製剤(ネオラール(Neoral)(登録商標)、ノバルティス)と併用されてきた。
【0028】
本発明のmTOR阻害剤はまた、種々の結晶、アモルファス物質、薬学的に許容される塩、水和物、及び溶媒和物として存在してもよい。さらに、本発明のmTOR阻害剤は、プロドラッグとして提供してもよい。一般に、かかるプロドラッグは、本発明のmTOR阻害剤の機能性の誘導体であり、これは、生体によって必要とされる化合物へ容易に変換し得る。したがって、本発明の種々の癌の治療法においては、用語「投与」は、特定の化合物の投与のみならず、患者への投与後に生体内で特定の化合物に変換し得る化合物の投与も包含する。適切なプロドラッグ誘導体の選択及び製造のための通常の方法は、例えば、バンガード(H.Bumdgaard)編、「Design of Prodrugs(プロドラッグの設計)」、Elsevier、1985年、に記載されており、これは、本明細書において参照され、かつその記載全体が本明細書の一部として本明細書に含まれる。化合物の代謝産物は、該化合物を生物学的環境内に置くことにより産生される活性化合物を包含してもよく、これらは本発明の化合物の範囲内にある。
【0029】
抗IGF−1R抗体
本発明の抗IGF−1R抗体は、単離された抗体か、又はその機能性のフラグメントであり、ここで、前記抗体又はその前記フラグメントは、ヒトインスリン様増殖因子I受容体に特異的に結合可能であり、かつ必要であれば、好ましくはさらに、リガンドであるIGF1及び/又はIGF2が、IGF−1Rへ結合するのを阻害することができ、及び/又は少なくとも1つのリガンドが、前記IGF−1R受容体へ結合するのに付随するシグナリングカスケードを特異的に阻害し得る。本発明のIGF−1R抗体は、特異的にIGF−1Rに結合し得る、モノクローナル及び/又はポリクローナル抗体を包含する。本発明の抗IGF−1R抗体は、ダロツズマブ、フィギツムマブ、シクスツムマブ、SHC717454、ロシュR1507、EM164、及びアムジェンAMG479を包含する。
【0030】
ダロツズマブは、配列番号1、2、又は3のアミノ酸配列のCDRから選択される少なくとも1つの相補性決定領域CDRか、又はその配列が、最適アラインメントの後に、配列番号1、2、又は3の配列と、少なくとも80%、好ましくは85%、90%、95%、及び98%の同一性をもつ少なくとも1つのCDRを含んでなる、軽鎖を含んでなること、或いは、配列番号4、5、及び6のアミノ酸配列のCDRから選択される少なくとも1つのCDRか、又はその配列が、最適アラインメントの後に、配列番号4、5、及び6の配列と、少なくとも80%、好ましくは85%、90%、95%、及び98%の同一性をもつ少なくとも1つのCDRを含んでなる、重鎖を含んでなること、により特徴づけられる。前記抗IGF−1R抗体を製造及び使用する方法は、米国特許第7,214,444号に記載されており、その記載全体が本明細書に援用される。
【0031】
本明細書においては、用語「結合すること(to bind)」及び「結合すること(to attach)」は、同じ意味をもち、互に交換可能である。
【0032】
本明細書においては、抗体化合物又はその配列に結合した、用語であるポリペプチド、ポリペプチド配列、ペプチド、及びタンパク質は、互に交換可能である。
【0033】
本発明は、天然型の抗体には関係しないこと、すなわち、それらは、その天然の環境中にあるのはなく、天然の供給源から精製により単離又は入手し得たものであるか、或いは遺伝的組み換えによるか、又は化学合成によって得られたものであること、及びしたがって、これより先に記載される、非天然のアミノ酸を含有し得ることが理解されるべきである。
【0034】
CDR領域又はCDRにより、カバット(Kabat)らにより定義された、免疫グロブリンの重及び軽鎖の超可変領域を示すことが意図されている(Kabat et al、「Sequences of proteins of immunological interest(免疫学的に興味深いタンパク質の配列)、第5版」、米国保健社会福祉省、NIH、1991年及びその後の版)。3つの重鎖CDR及び3つの軽鎖CDRが存在する。用語CDR又は複数のCDR(CDRs)は、本明細書では、抗体の抗原若しくはそれを認識するエピトープへの、親和性による結合に役割を果たしているアミノ酸残基の大半を含有するこれらの領域の、場合により、1つ若しくはいくつか、又はさらに全てを示すために使用する。
【0035】
本発明の意味においては、2つの核酸又はアミノ酸配列の間の「同一性のパーセント」により、最良のアラインメント(最適アラインメント)後に得られた、比較されるべき2つの配列間のヌクレオチド又は同一のアミノ酸残基のパーセントを示すことが意図され、このパーセントは、純粋に統計的であり、2つの配列の間の差異は、ランダムに、かつその全長にわたり分布している。2つの核酸又はアミノ酸配列間の比較は、慣例的に、それらを最適な方法で整列させた後、これらの配列を比較することにより実施され、前記比較は、セグメントごとに、又は「比較ウィンドウ」ごとに行ない得る。比較のための配列の最適アラインメントは、手作業によるものに加えて、スミス(Smith)及びウォーターマン(Waterman)の局所ホモロジーアルゴリズム[「Ad.App.Math.」、1981年、第2巻、p.482]によるか、ネドルマン(Neddleman)及びヴンシュ(Wunsch)の局所ホモロジーアルゴリズム[「J.Mol.Biol.」、1970年、第48巻、p.443]によるか、ピアソン(Pearson)及びリプマン(Lipman)の同様の検索法[「Proc.Natl.Acad.Sci.USA」、1988年、第85巻、p.2444]によるか、又はこれらのアルゴリズムを用いたコンピューターソフトウェア(ウィスコンシン・ジェネティクス・ソフトウェア・パッケージ(Wisconsin Genetics Software Package)、ジェネティクス・コンピュータ・グループ(Genetics Computer Group)、575サイエンス(Science)Dr.、Madison、WI)のGAP、BESTFIT、FASTA、及びTFASTA又はBLASTN若しくはBLASTP比較ソフトウェア)により実行し得る。
【0036】
2つの核酸又はアミノ酸配列の間の同一性のパーセントは、最適な方法で整列されたこれら2つの配列を比較することにより決定し、これにおいて、比較されるべき核酸又はアミノ酸配列は、これら2つの配列間の最適アラインメント用の参照配列に比較して、付加又は欠失を含んでいてもよい。同一性のパーセントは、その位置について核酸又はアミノ酸残基が2つの配列間で同一である、同一位置の数を測定すること、同一位置数を比較ウィンドウ内の全位置数で割ること、及び得られた結果を100倍して、これら2つの配列間の同一性のパーセントを得ることにより計算する。
【0037】
例えば、http://www.ncbi.nlm.nih.gov/gorf/bl2.htmlのサイトにおいて利用可能なBLASTプログラム、「BLAST2sequences」(タツソヴァ(Tatusova)ら、「Blast 2 sequences−a new tool for comparing protein and nucleotide sequences(Blast2配列−タンパク質及びヌクレオチド配列比較のための新規なツール)」FEMS Microbiol Lett.第174巻、p.247−250)を使用可能であり、用いたパラメータは、デフォルトによって与えられたものであり(特に、パラメータである、「オープンギャップペナルティ」:5、及び「伸長ギャップペナルティ」:2について、選択されたマトリックスは、例えば、プログラムによって推奨されたマトリックス「BLOSUM 62」である)、比較されるべき2つの配列間の同一性のパーセントは、このプログラムにより直接計算される。
【0038】
参照アミノ酸配列と少なくとも80%、好ましくは85%、90%、95%、及び98%の同一性をもつアミノ酸配列により、参照配列に比較して、ある修飾、特に、少なくとも1つのアミノ酸の欠失、付加、若しくは置換、トランケーション(先端の切断)、又は伸長を有するものが好ましい。1つ以上の連続又は不連続のアミノ酸の置換の場合、置換は、好ましくは、置換されたアミノ酸が「同等(equivalent)」のアミノ酸に置き換えられる。「同等のアミノ酸(equivalent amino acids)」という表現は、本明細書では、基本構造をもつアミノ酸の1つによって置換され得るが、しかしながら、対応する抗体の生物活性を本質的に修飾することはない、任意のアミノ酸を示すことを意図しており、例えば、後に特に実施例において定義されるものなどである。
【0039】
これらの同等のアミノ酸は、それらが置換するアミノ酸との、その構造上の相同性か、又は実行可能な種々の抗体間の生物活性の比較試験の結果のいずれかに依存して決定し得る。
【0040】
単なる例としては、対応する修飾された抗体の生物活性の大きな修飾をもたらすことなく実行し得る置換の可能性が挙げられる。それ故、ロイシンをバリン又はイソロイシンで、アスパラギン酸をグルタミン酸で、グルタミンをアスパラギンで、アルギニンをリジンで、などの置換が可能であり、逆の置換が、同じ条件下で必然的に予想される。
【0041】
本発明の抗体は、好ましくは、特異的なモノクローナル抗体であり、特に、マウス、キメラ、又はヒト化起源のものであり、これらは当業者に周知の標準的な方法により取得し得る。
【0042】
一般に、モノクローナル抗体又はその機能性フラグメント、特にマウス起源のものの調製には、特に、マニュアル「Antibodies(抗体)」(ハーロー(Harlow)及びレーン(Lane)著、「Antibodies :A Laboratory Manual(抗体:実験室マニュアル)」、Cold Spring Harbor Laboratory、Cold Spring Harbor NY、1988年、p.726)に記載されている技術、又はコーラー(Kohler)及びミルスタイン(Milstein)(Nature、1975年、第256巻、p.495−497)により記載されたハイブリドーマからの調製技術を参照し得る。
【0043】
本発明のモノクローナル抗体は、例えば、IGF−1R受容体か又は本発明の前記モノクローナル抗体によって特異的に認識されるエピトープを含有するそのフラグメントの1つに対し免疫化された、動物細胞から得られる。前記IGF−1R受容体、又はその前記フラグメントの1つは、通常の実施の方法に従い、IGF−1R受容体をコードするcDNA配列中に含まれる核酸配列から出発する遺伝的組換えによるか、又はIGF−1R受容体のペプチド配列中に含まれるアミノ酸配列から出発するペプチド合成により、特別に合成し得る。
【0044】
本発明のモノクローナル抗体は、例えば、IGF−1R受容体か又は本発明の前記モノクローナル抗体によって特異的に認識されるエピトープを含有するそのフラグメントの1つがその上に予め固定されている、アフィニティカラム上で精製し得る。さらに具体的には、前記モノクローナル抗体は、プロテインA及び/又はG上でのクロマトグラフィー後に、残留するタンパク質不純物、並びにDNA及びLPSを除去する目的で、イオン交換クロマトグラフィーを続けるか、又は続けずに精製してもよく、これに、二量体又は他の多量体の存在による潜在的な凝集体を除去する目的で、セファロース(Sepharose)ゲル上での排除クロマトグラフィーを続けるか、又は続けなくてもよい。なおさらに好ましい方法では、これらの技術の全てを同時に、又は逐次に使用し得る。
【0045】
キメラ又はヒト化された抗体は、同様に本発明の抗体に包含される。
【0046】
キメラ抗体により、所与の種の抗体に由来する天然の可変(軽鎖及び重鎖)領域を、前記所与の種に対して異種の抗体の軽鎖及び重鎖定常領域と組合せて含有する抗体を示すことが意図されている。
【0047】
本発明の抗体又はそのキメラ型のフラグメントは、遺伝的組換えの技術を用いることにより調製し得る。例えば、キメラ抗体は、プロモータと、本発明の非ヒトの、特にマウスのモノクローナル抗体の可変領域をコードする配列と、及びヒト抗体の定常領域をコードする配列とを含有する、組換えDNAをクローニングすることにより産生し得る。かかる組換え遺伝子によりコードされた本発明のキメラ抗体は、例えば、マウス−ヒトキメラであり、この抗体の特異性は、マウスDNA由来の可変領域により決定され、そのアイソタイプは、ヒトDNA由来の定常領域によって決定される。キメラ抗体の調製法については、例えば、ヴァーホイエン(Verhoeyn)らの文献(「BioEssays」、1988年、第8巻、p.74)を参照し得る。
【0048】
ヒト化抗体により、非ヒト起源の抗体に由来するCDR領域を含有する抗体を示すことが意図され、抗体分子の他の部分は、一つの(又は数個の)ヒトの抗体に由来する。さらに、骨格のセグメント(FRと称する)のいくつかの残基を、結合の親和性を保持する目的で、修飾してもよい(ジョーンズ(Jones)ら著、「Nature」、1986年、第321巻、p.522−525;ヴァーホイエン(Verhoeyn)ら著、「Science」、1988年、第239巻、p.1534−1536;リーチマン(Riechman)ら著、「Nature」、1988年、第332巻、p.323−327)。
【0049】
本発明のヒト化抗体又はそのフラグメントは、当業者に周知の技術により調製し得る(例えば、シンガー(Singer)ら著、「J.Immun.」、1992年、第150巻、p.2844−2857;マウンテン(Mountain)ら著、「Biotechnol.Genet.Eng.Rev.」、1992年、第10巻、p.1−142;又はベビントン(Bebbington)ら著、「Bio/Technology」、1992年、第10巻、p.169−175、などの文献に記載されたもの)。本発明のかかるヒト化抗体は、好ましくは、インビトロの診断法、又はインビボの予防的及び/又は治療的処置における使用が好ましい。
【0050】
本発明の抗体の機能性フラグメントにより、特に抗体フラグメント、例えば、Fv、scFv(scは単鎖)、Fab、F(ab’)2、Fab’、scFv−Fcフラグメント、又はダイアボディ、或いは、例えば、ポリ(エチレン)グリコール(「PEG化」)(Fv−PEG、scFv−PEG、Fab−PEG、F(ab’)2−PEG、又はFab’−PEGと呼ばれるフラグメント)(「PEG」はポリ(エチレン)グリコール)などのポリ(アルキレン)グリコールの付加による化学修飾によるか、又はリポソーム内への取込みによって、半減期が増大しているであろう、任意のフラグメントであって、前記フラグメントは、本発明の配列番号1、2、3、4、5、又は6の特徴的なCDRの少なくとも1つを有し、かつ特に、抗体IGF−1R受容体を認識及び結合し、必要であればIGF−1R受容体の活性を阻害する能力など、それが由来する抗体のたとえ部分的な活性であっても、一般的な方法で及ぼすことができる前記フラグメント、を示すものとする。
【0051】
好ましくは、前記機能性フラグメントは、それらが由来する抗体の重又は軽可変鎖の、部分的な配列からなるか、又は含んでなり、前記部分的な配列は、IGF−1R受容体について、それが由来する抗体と同様の結合特異性及び、十分な親和性(好ましくは、それが由来する抗体の親和性の少なくとも1/100、さらに好ましい態様では、少なくとも1/10に等しい充分な親和性)を保持するのに充分である。
【0052】
かかる機能性フラグメントは、それが由来する抗体の配列の、最低5個のアミノ酸、好ましくは10、15、25、50、及び100個の連続したアミノ酸を含有することができる。
【0053】
好ましくは、これらの機能性フラグメントは、Fv、scFv、Fab、F(ab’)2、Fab’、scFv−Fc型、又はダイアボディのフラグメントであり、これらは一般に、それらが由来する抗体と同じ結合特性を有する。本発明によれば、本発明の抗体フラグメントは、例えば、ペプシン又はパパインなどの酵素による消化等の方法により、及び/又は化学的還元によるジスルフィド結合の切断により、上記記載の抗体などから出発して取得し得る。別の方法では、本発明に含まれる抗体フラグメントは、同様に当業者に周知の遺伝的組換え技術により、又はさもなければ、例えば、アプライド・バイオシステムズ(Applied Biosystems)社等により供給されるものなどの自動ペプチド合成装置によるペプチド合成によって取得し得る。
【0054】
さらに好ましい態様においては、本発明は、本発明の抗体、又はその機能性フラグメント、特に、遺伝的組換えにより、又は化学合成により取得された、キメラ又はヒト化抗体を含んでなる。
【0055】
好ましい実施態様においては、本発明の主題は、配列番号6の少なくとも1つのCDR、又は配列番号6の配列との最適アラインメント後に少なくとも80%の同一性を有する配列を含んでなることを特徴とする、本発明の抗体又はその機能性フラグメントの1つである。
【0056】
6つの短いCDR配列の中で、重鎖の3番目のCDR(CDRH3)は、サイズの変異性が大きい(高い多様性は、本質的に、それを生じるもとである遺伝子の配列のメカニズムに起因する)。これは、2アミノ酸程の短さであってもよいが、既知の最長のサイズは26である。機能上は、CDRH3は、一部は、抗体の特異性の決定に役割を果たしている(セガル(Segal)ら著、「PNAS」、1974年、第71巻、p.4298−4302;アミット(Amit)ら著、「Science」、1986年、第233巻、p.747−753;チョシア(Chothia)ら著、「J.Mol.Biol.」、1987年、第196巻、p.901−917;チョシア(Chothia)ら著、「Nature」、1989年、第342巻、p.877−883;ケイトン(Caton)ら著、「J.Immunol.」、1990年、第144巻、p.1965−1968;シャロン(Sharon)ら著、「PNAS」、1990年、第87巻、p.4814−4817;シャロン(Sharon)ら著、「J.Immunol.」、1990年、第144巻、p.4863−4869;カバット(Kabat)ら著、「J.Immunol.」、1991年、第147巻、p.1709−1719)。
【0057】
CDRのアミノ酸のうちのわずかなパーセントのものだけが、抗体の結合部位の構造に寄与することが知られているが、これらの残基は、非常に特異的な三次元的立体配座において維持されているはずである。
【0058】
さらに好ましい態様においては、本発明は、3つのCDR又は配列番号4、5、及び6の配列の3つのCDRの、少なくとも2つか、或いは3つのCDR又は配列番号4、5、及び6の配列との最適アラインメント後に各々少なくとも80%の同一性を有する配列の3つのCDRの、少なくとも2つを含んでなる、重鎖を含んでなることを特徴とする、本発明の抗体又はその機能性フラグメントの1つに関する。
【0059】
同様の好ましい実施態様においては、本発明の主題は、配列番号1、2、又は3の配列のCDRから選択される少なくとも1つのCDR、又は、その配列が、配列番号1、2、又は3の配列との最適アラインメント後に少なくとも80%の同一性を有するCDRを含んでなる、軽鎖を含んでなることを特徴とする、本発明の抗体又はその機能性フラグメントの1つである。
【0060】
さらに好ましい実施態様においては、本発明の主題は、3つのCDR又は配列番号1、2、及び3の配列の3つのCDRの、少なくとも2つか、或いは3つのCDR又は配列番号1、2、及び3の配列との最適アラインメント後に各々少なくとも80%の同一性を有する配列の3つのCDRの、少なくとも2つを含んでなる、軽鎖を含んでなることを特徴とする、本発明の抗体又はその機能性フラグメントである。
【0061】
さらに好ましい態様においては、本発明の抗体又はその機能性フラグメントの1つは、それが、配列番号4、5、及び6の配列の3つのCDR、又は配列番号4、5、及び6の配列との最適アラインメント後に各々少なくとも80%の同一性を有する配列の3つのCDRを含んでなる、重鎖を含んでなること、及びさらに、配列番号1、2、及び3の配列の3つのCDR、又は配列番号1、2、及び3の配列との最適アラインメント後に各々少なくとも80%の同一性を有する配列の3つのCDRを含んでなる、軽鎖を含んでなることを特徴とする。
【0062】
別の態様によれば、本発明の主題は、ヒトインスリン受容体IRに結合しないか、又は有意な態様では結合しないことを特徴とする、本発明の抗体又はその機能性フラグメントの1つである。
【0063】
好ましい態様においては、本発明の前記機能性フラグメントは、フラグメントであるFv、scFv、Fab、F(ab’)2、Fab’、scFv−Fc、又はダイアボディ、或いは、化学修飾、特にPEG化によるか、又はリポソーム内への取込みによって半減期が増大しているであろう、任意のフラグメントから選択される。
【0064】
別の態様によれば、本発明は、本発明のモノクローナル抗体を分泌することが可能なマウスハイブリドーマ、特に、2001年9月19日に、Centre National de Culture De Microorganisme(CNCM、国立微生物培養センター)(パスツール研究所、パリ、フランス)に、番号I−2717で寄託されたマウス由来のハイブリドーマに関する。
【0065】
抗体が、2001年9月19日に、CNCMに、番号I−2717で寄託されたハイブリドーマにより分泌されることを特徴とする、本明細書で7C10と称されるモノクローナル抗体、又はその機能性フラグメントの1つは、もちろん本発明の一部である。
【0066】
「ダロツズマブ」、「h7C10」、「MK−0646」、又は「F50035」は、IGF−1Rに結合すること、並びにIR/IGF−1ハイブリッド受容体に結合することを特徴とするヒト化抗体を記載するべく区別なく使用される。かかる抗体は、例えば、米国特許出願第10/735,916号(US20050084906)(これは、PCT/FR03/00178及び/又はUS20050249730の一部継続出願である)において記載された抗体を包含してもよく、ここで前記のものは、ヒト化抗体又はそのフラグメントであり、かつ軽鎖及び/又は重鎖を含んでなり、これにおいて前記軽鎖及び/又は重鎖の骨格セグメントFR1ないしFR4は、それぞれヒト抗体の軽鎖及び/又は重鎖の骨格セグメントFR1ないしFR4に由来する。ヒト化抗体は、非ヒト由来であり、かつ配列番号1、2、又は3からなる群より選択されるアミノ酸配列を有する少なくとも1つ以上の相補性決定領域を含んでなる、少なくとも1つの軽鎖と、配列番号4、5、又は6からなる群より選択されるアミノ酸配列を有する少なくとも1つ以上の相補性決定領域を含んでなる、少なくとも1つの重鎖とを含んでなってもよい。軽鎖は、配列番号7又は8のうちの1つに示した1つ以上のアミノ酸配列か、又は配列番号7又は8との最適アラインメントの後に少なくとも80%の同一性を有する配列を含んでなってもよい。同様に、重鎖は、配列番号9、10、又は11のうちの1つに示した1つ以上のアミノ酸配列か、又は配列番号9、10、又は11との最適アラインメントの後に少なくとも80%の同一性を有する配列を含んでなってもよい。
【0067】
特定の実施態様においては、本発明は、本発明のマウス抗体又はその機能性フラグメントの1つに関し、前記抗体は、配列番号12のアミノ酸配列、又は配列番号12の配列との最適アラインメントの後に少なくとも80%の同一性を有する配列を含んでなる配列の軽鎖を含んでなること、及び/又は、配列番号13のアミノ酸配列、又は配列番号13の配列との最適アラインメントの後に少なくとも80%の同一性を有する配列を含んでなる配列の重鎖を含んでなることを特徴とする。
【0068】
フィギツムマブは、CP751,871としても知られ、ファイザー(Pfizer)による研究下にある完全ヒト抗体である。フィギツムマブの記載及び調製法は、アブジェニックス・インク(Abgenix,Inc)及びファイザー・インク(Phizer,Inc.)に対する米国特許第7,037,498号に記載されており、これはその記載全体が本明細書に援用される。
【0069】
シクスツムマブは、IMC A−12としても知られ、イムクローン(ImClone)による研究下にある完全ヒト抗体である。シクスツムマブの記載及び調製法は、米国特許公報第US2008/0025990号に記載されており、これはその記載全体が本明細書に援用される。
【0070】
SHC717454は、CP 751,871としても知られ、シェーリング・プラウ(Schering Plough)(現在はメルク・アンド・カンパニー・インク(Merck&Co.,Inc.))による研究下にある完全ヒト抗体である。SHC717454の記載及び調製は、シェーリング・コーポレーションに対する米国特許第7,217,796号に記載されており、これはその記載全体が本明細書に援用される。
【0071】
ロシュR1507は、ホフマン−ラロシュ(Hoffmann−LaRoche)による研究下にある。ロシュR1507の記載及び調製は、ホフマン−ラロシュに対する米国特許第7,378,503号に記載されており、これはその記載全体が本明細書に援用される。
【0072】
EM164は、イムノジェン(Immunogen)による研究下にある。EM164の記載及び調製は、イムノジェン・インクに対する国際特許公開 WO03/106621に記載されており、これはその記載全体が本明細書に援用される。
【0073】
アムジェンAMG479は、アムジェン・インクによる研究下にある。アムジェンAMG479の記載及び調製は、アムジェン・インク対する国際特許公開 WO06/069202に記載されており、これはその記載全体が本明細書に援用される。
【0074】
同様の特定の態様によれば、本発明は、本発明のキメラ抗体又はその機能性フラグメントの1つに関係し、前記抗体はさらに、マウスに対して異種の、特にヒトの抗体に由来する軽鎖及び重鎖定常領域含んでなること、及び好ましい態様においては、ヒト抗体に由来する軽鎖及び重鎖定常領域は、各々カッパ及びガンマ−1、ガンマ−2、又はガンマ−4領域であることを特徴とする。
【0075】
同様の特定の態様によれば、本発明は、本発明のヒト化抗体又はその機能性フラグメントの1つに関し、前記抗体は、軽鎖及び/又は重鎖を含んでなり、これにおいて前記軽鎖及び/又は重鎖の骨格セグメントFR1ないしFR4(以下の実施例12及び13、表5及び6において定義された通り)は、それぞれヒト抗体の軽鎖及び/又は重鎖の骨格セグメントFR1ないしFR4に由来することを特徴とする。
【0076】
好ましくは、本発明のヒト化抗体又はその機能性フラグメントの1つは、前記ヒト化抗体が、配列番号8のアミノ酸配列を含んでなる軽鎖を含んでなること、及び配列番号10又は11の、好ましくは配列番号11のアミノ酸配列を含んでなる重鎖を含んでなることを特徴とする。
【0077】
本発明において区別なく使用される用語である、核酸、核又は核酸配列、ポリヌクレオチド、オリゴヌクレオチド、ポリヌクレオチド配列、ヌクレオチド配列により、ヌクレオチドの厳密な結合を示すことが意図されており、これらは修飾されているか又は未修飾であり、フラグメント又は定義されるべき核酸の領域を与え、非天然のヌクレオチドを含有するか又は含有せず、また二本鎖DNA、一本鎖DNAにも、前記DNAの転写産物と同様に対応し得るものである。
【0078】
また本明細書では、本発明が、その天然の染色体環境、すなわち、天然の状態にあるヌクレオチド配列には関与しないことも理解されるべきである。本発明は、単離及び/又は精製された配列に関するものであり、すなわち、それらは、直接又は間接的に、例えばコピーにより選択されたものであって、その環境は、少なくとも部分的に修飾されたものである。それ故それは、同様に、本明細書では、例えば、宿主細胞による遺伝的組換えによって得られるか、又は化学合成によって得られる、単離した核酸を示すことが意図されている。
【0079】
好ましい配列との最適アラインメントの後に、少なくとも80%、好ましくは85%、90%、95%、及び98%の同一性を有する核酸配列により、該核酸配列が、参照核酸配列と比較して、ある種の修飾、特に、欠失、トランケーション、伸長、キメラ融合、及び/又は置換、とりわけ点置換などを有する核酸配列を示すことが意図されている。好ましくは、本発明は、その配列が参照配列と同じアミノ酸配列をコードしている配列に関与しており、このことは、遺伝コードの縮重か、又は、好ましくは、特に以下に定義される高ストリンジェント条件下で、参照配列と特異的にハイブリダイズし得る相補的配列につながる。
【0080】
高ストリンジェント条件下のハイブリダイゼーションは、温度条件及びイオン強度条件が、それらが相補的なDNAの2つのフラグメント間のハイブリダイゼーションを維持させるように選択されることを意味する。単なる例証として、上記記載のポリヌクレオチドフラグメントを定義することを目的とする高ストリンジェントなハイブリダイゼーション工程の条件は、好ましくは以下の通りである。
【0081】
DNA−DNA、又はDNA−RNAハイブリダイゼーションは、2工程で行なわれる:(1)5xSSC(1xSSCは、0.15M NaCl+0.015M クエン酸ナトリウム溶液に相当)、50% ホルムアミド、7% ドデシル硫酸ナトリウム(SDS)、10x デンハート(denhardt’s)、5% 硫酸デキストラン、及び1% サケ精子DNAを含有するリン酸バッファ(20mM、pH7.5)中で、42℃で3時間のプレハイブリダイゼーション;(2)プローブのサイズに依存した温度(すなわち、プローブサイズ>100ヌクレオチドに対し、42℃)において、20時間の実際のハイブリダイゼーションの後に、2xSSC+2% SDS中での、20℃で20分間の2回の洗浄、0.1xSSC+0.1% SDS中での、20℃で20分間の1回の洗浄。最後の洗浄は、プローブサイズ>100ヌクレオチドに対し、0.1xSSC+0.1% SDS中で、60℃で30分間行なった。上記記載のストリンジェンシーの高いハイブリダイゼーション条件は、ある規定されたサイズのポリヌクレオチドについては、当業者により、サンブルック(Sambrook)らの教示(「Molcular Cloning:a Laboratory manual(分子クローニング:実験室マニュアル)」、第2版、Cold Spring Harbor、1989年)に従って、より大きい又はより小さいサイズのオリゴヌクレオチドに適用し得る。
【0082】
投薬及び投与経路
本発明のmTOR阻害剤及び抗IGF−1R抗体については、様々な製剤形態を選択でき、その例は、錠剤、カプセル、粉末、顆粒又は液体などの経口製剤、溶液又は懸濁液などの無菌の液体製剤、坐剤、軟膏などを包含する。mTOR阻害剤は、薬学的に許容される塩として利用し得る。本発明のmTOR阻害剤及び抗IGF−1R抗体は、薬学的に許容される担体又は希釈剤と共に調製される。
【0083】
用語「薬学的に許容される塩」は、本明細書で言及される場合、通常の薬学的に許容される塩を意味する。例えば、化合物がヒドロキシル基か、又はカルボキシル基及びテトラゾリル基などの酸性基をもつ場合、ヒドロキシル基又は酸性基において塩基付加塩を形成してもよく;或いは化合物がアミノ基又は塩基性の複素環基をもつ場合には、アミノ基又は塩基性複素環基において酸付加塩を形成してもよい。
【0084】
塩基付加塩は、例えば、ナトリウム塩、カリウム塩などのアルカリ金属塩;カルシウム塩、マグネシウム塩などのアルカリ土類金属塩;アンモニウム塩;及び有機アミン塩、例えばトリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、プロカイン塩、N,N’−ジベンジルエチレンジアミン塩を包含する。
【0085】
酸付加塩は、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、過塩素酸塩などの無機酸塩;マレイン酸塩、フマル酸塩、酒石酸塩、クエン酸塩、アスコルビン酸塩、トリフルオロ酢酸塩などの有機酸塩;及びメタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩などのスルホン酸塩を包含する。
【0086】
用語「薬学的に許容される担体又は希釈剤」は、賦形剤[例えば、脂肪、蜜蝋、半固体及び液体ポリオール、天然又は硬化油など];水(例えば、蒸留水、特に注射用蒸留水など)、生理食塩水、アルコール(例えば、エタノール)、グリセロール、ポリオール、グルコース水溶液、マンニトール、植物油など);添加剤[例えば、増量剤、崩壊剤、結合剤、滑沢剤、湿潤剤、安定化剤、乳化剤、分散剤、保存剤、甘味剤、着色剤、調味剤又は芳香剤、濃縮剤、希釈剤、緩衝剤、溶媒又は可溶化剤、貯蔵効果を達成するための化学物質、浸透圧を調整するための塩、コーティング剤、又は酸化防止剤]などを指す。
【0087】
固形製剤は、錠剤、カプセル、顆粒、及び粉末の形態で、何ら添加剤を用いずに、又は適当な担体(添加剤)を用いて調製し得る。かかる担体(添加剤)の例には、ラクトース又はグルコースなどの糖類;トウモロコシ、コムギ、又はコメのデンプン;ステアリン酸などの脂肪酸;メタケイ酸アルミン酸マグネシウム又は無水リン酸カルシウムなどの無機塩;ポリビニルピロリドン又はポリアルキレングリコールなどの合成ポリマー;ステアリルアルコール、ベンジルアルコールなどのアルコール;メチルセルロース、カルボキシメチルセルロース、エチルセルロース、又はヒドロキシプロピルメチルセルロースなどの合成セルロース誘導体;及び他の通常使用される添加剤、例えばゼラチン、タルク、植物油、及びアラビアゴムが挙げられてよい。
【0088】
錠剤、カプセル、顆粒、及び粉末などのこれらの固形製剤は、各製剤の全重量に基づき、一般に、例えば、0.1ないし100重量%、及び好ましくは、5ないし98重量%の、mTOR阻害剤を含有してもよい。
【0089】
液体製剤は、懸濁液、シロップ、注射液、及び点滴剤(静脈内輸液)の形態で、水、アルコール、又は植物由来の油、例えば大豆油、ラッカセイ油、及びゴマ油などの、液体製剤において通常用いられる適当な添加剤を使用して製造される。
【0090】
特に、製剤が、筋肉内注射、静脈内注射、又は皮下注射の形態で、非経口的に投与される場合、適当な溶媒又は希釈剤は、注射用の蒸留水、塩酸リドカインの水溶液(筋肉内注射用)、生理食塩水、グルコース水溶液、エタノール、ポリエチレングリコール、プロピレングリコール、静脈内注射用の液体(例えば、クエン酸、又はクエン酸ナトリウムなどの水溶液)、又は電解質溶液(静脈内点滴輸液及び静脈内注射用)、又はそれらの混合溶液に例示される。
【0091】
かかる注射剤は、予め溶解された溶液の形態か、或いは、使用時に溶解される、粉末自体、又は適当な担体(添加剤)と混合された粉末の形態でよい。注射液は、各製剤の全重量に基づき、例えば、0.1ないし10重量%の活性成分を含有してもよい。
【0092】
経口投与用の懸濁液又はシロップなどの液体製剤は、各製剤の全重量に基づき、例えば、0.1ないし10重量%の活性成分を含有してもよい。
【0093】
本発明の各製剤は、当業者により、通常の方法又は一般的な技術によって調製し得る。例えば、調製は、製剤が経口用製剤である場合には、例えば、適切な量の本発明の化合物を、適切な量のラクトースと混合すること、この混合物を経口投与に適した硬ゼラチンカプセル内に充填することによって実施し得る。一方、本発明の化合物を含有する製剤が注射剤の場合には、適切な量の本発明の化合物を、適切な量の0.9%生理食塩水と混合すること、及びこの混合物を注射用のバイアル内に充填することにより実施し得る。
【0094】
本発明の成分(component)は、ヒトを含む哺乳類へ、単独で、又は薬学的に許容される担体、賦形剤、又は希釈剤と組合せて、医薬組成物の状態で、標準的な薬学のプラクティスに従って投与してもよい。かかる成分は、経口的に、又は静脈内、筋肉内、腹腔内、皮下、直腸内、及び局所の投与経路を含め、非経口的に投与し得る。
【0095】
適切な投薬量は、医師にとり既知であり、当然のことながら、特定の疾病状態、投与される組成物の比活性、及び治療を受ける個々の患者に依存するであろう。ある場合には、所望の治療量を達成するため、反復した投与を提供すること、すなわち特定のモニター又は計測された用量の個々の投与を反復することが必要であってもよく、この場合、個々の投与は所望の1日用量又は効果が達成されるまで反復される。適切な投薬量についてのさらなる情報は、以下に提供される。
【0096】
用語「投与」及びその変形(例えば、化合物を「投与すること」)は、本発明の成分に関して、かかる成分又は該成分のプロドラッグを、治療を要する動物の系内に導入することを意味する。本発明の成分又はそのプロドラッグが、1つ以上の他の活性薬剤(例えば、mTOR阻害剤)と組合せて与えられる場合、「投与」及びその変形は、各々、かかる成分又はそのプロドラッグと他の薬剤との、同時の、及び逐次の導入を包含するものと理解される。
【0097】
本明細書で用いる場合、用語「組成物」は、指定された量の指定された成分を含んでなる生成物、並びに指定された量の指定された成分の組合せから、直接又は間接的にもたらされる任意の生成物を包含することが意図されている。
【0098】
用語「治療有効量」は、本明細書で用いるとき、研究者、獣医師、医師、又は他の臨床家によって求められている、組織、系、動物、又はヒトにおける生物学的又は医学的応答を示す活性化合物又は医薬の量を意味する。
【0099】
mTOR阻害剤の適切な量が、癌の治療を受けている患者に投与される。1つの実施態様においては、mTOR阻害剤は、1日当たり約10mgないし40mgの用量で投与される。本発明の1つの実施態様においては、mTOR阻害剤は、1日当たり10mgの用量で投与される。本発明の別の実施態様においては、mTOR阻害剤は、1日当たり20mgの用量で投与される。本発明の別の実施態様においては、mTOR阻害剤は、1日当たり30mgの用量で投与される。本発明の別の実施態様においては、mTOR阻害剤は、1日当たり40mgの用量で投与される。
【0100】
本発明の1つの実施態様においては、mTOR阻害剤は、1週間当たり5回投与される。例えば、リダフォロリムスは、1日目に開始され、指定された用量レベルで連続して5日間継続され、これにリダフォロリムスの処置のない2日間が続けられる。リダフォロリムスは次に、この毎週5回のスケジュールで継続される。
【0101】
本発明の抗IGF−1R抗体とmTOR阻害剤とを含んでなる併用療法は、ボーラスとしての静脈内投与、又は一定期間の連続した輸液によるか、筋肉内、腹腔内、脳脊髄内、皮下、関節内、滑液包内、髄腔内、経口、局所、又は吸入経路によるなどの既知の方法に従って、ヒト患者に投与される。抗体の静脈内又は皮下投与が好ましい。3つの異なる送達アプローチが、本発明の抗体の送達に有用であることが期待される。通常の静脈内送達は、おそらく大多数の腫瘍のための標準的な送達技術であろう。しかしながら、卵巣、胆管、他の管系などの腫瘍に代表される腹腔内のものなどのいくつかの腫瘍に関しては、腫瘍において高線量の抗体を得るために、また抗体クリアランスを最少化するために、腹腔内投与が好適であることを示すことがある。同様に、ある固体腫瘍は血管系を有しており、これは局所灌流に適する。局所灌流により、腫瘍部位において高線量の抗体を得ることができ、また短時間の抗体クリアランスを最小にすることができる。
【0102】
任意のタンパク質又は抗体の輸液をベースとする治療と同様に、安全性の懸念は主として(i)サイトカイン放出症候群、すなわち、低血圧、発熱、震え、悪寒、(ii)物質に対する免疫応答の発生(すなわち、患者による治療抗体に対するヒト抗体の発生、又はHAHA若しくはHACA応答)、及び(iii)EGF受容体を発現する正常細胞、例えば、EGFR及び/又はIGF−1Rを発現する肝細胞に対する毒性、に関連する。標準的試験及び追跡調査を利用して、これらの安全性懸念の各々がモニターされる。特に、肝機能は、もしあれば肝臓に対する損傷を評価する目的で、臨床試験の間頻繁にモニターされる。
【0103】
疾患の予防又は治療用には、抗体の適切な用量は、上記に定義した通りの、治療されるべき疾患のタイプ、疾患の重症度及び経過、抗体が予防又は治療目的で投与されるのかどうか、以前の療法、患者の病歴及び抗体に対する応答、及び主治医の自由裁量に依存するであろう。抗体は、一度に、又は一連の治療にわたり、適宜患者に投与される。併用療法レジメンでは、本発明の組成物は、治療上の有効量又は相乗的な量で投与される。本明細書で用いるとき、治療有効量は、抗IGF−1R抗体及びmTOR阻害剤の同時投与、又は本発明の組成物の投与が、標的とする疾患又は症状の低減又は阻害をもたらす量である。治療上の相乗的な量は、特定の疾患に伴う症状又は症候群を、相乗的に又は有意に、緩和又は除去するのに必要な、抗IGF−1R抗体及びmTOR阻害剤の量である。
【0104】
広い実施態様においては、本発明の治療は、抗IGF−1R抗体とmTOR阻害剤との
併用投与を包含する。併用投与は、別々の製剤又は単一の医薬製剤を用いた同時投与、及びいずれかの順序での逐次的な投与を包含し、これにおいて、その間に双方の(又は全ての)活性薬剤が同時にその生物活性を及ぼす期間があることが好ましい。かかる化学療法剤のための製剤及び投薬スケジュールは、製造業者の指示に従って、又は熟練の開業医により経験的に決定された通りに使用してもよい。化学療法のための製剤及び投薬スケジュールはまた、ペリー(M.C.Perry)編、「Chemotherapy(化学療法)、Service Ed」、Williams & Wilkins、Baltimore、Md.、1992年に記載されている。mTOR阻害剤は、抗体の投与に先行するか、又は後続して投与してもよく、或いはそれらを同時に投与してもよい。本発明の治療的併用での臨床的投薬は、副作用の程度によって制限されることが見込まれる。
【0105】
例えば、1つ以上の別々の投与によるものであろうと、連続した輸液によるものであろうと、疾患のタイプ及び重症度に依存して、約1μg/kgないし50mg/kg(例えば、0.1−20mg/kg)の抗体が、患者への投与の最初の候補投薬量である。典型的な一日用量は、上記の因子に依存して、約1μg/kgないし約100mg/kg以上の範囲内である。数日間以上にわたる反復投与では、症状に依存して、疾患症状に所望の抑制が生じるまで継続される。しかしながら、他の用量用法を使用してもよい。
【0106】
1つの態様においては、本発明の抗体は、約5mg/kgないし約15mg/kgの用量範囲で、毎週投与されるか、又は2週間ないし3週間ごとに投与してもよい。ある態様においては、化学療法レジメンは、従来の高用量の間欠的投与を包含する。別の態様においては、化学療法剤は、計画的中断なしに、より少量で頻繁な投薬を用いて投与される(「メトロノミック・ケモセラピー」)。本発明の療法の進行は、通常の技術及びアッセイによりによりモニターし得る。
【0107】
1つの実施態様においては、投薬順序は、mTOR阻害剤をIGF−1R抗体と同時に投与することを含んでなる−例えば、リダフォロリムスは毎日投与され、一方IGF−1R抗体(ダロツズマブ)は毎週投与される。特に、ダロツズマブは10mg/kgの用量で、毎週静脈内投与され、一方リダフォロリムスは毎日10mgのスケジュールで投与される。
【0108】
別の実施態様においては、投薬順序は、mTOR阻害剤をIGF−1R抗体と同時に投与することを含んでなる−例えば、リダフォロリムスは毎日投与され、一方IGF−1R抗体(ダロツズマブ)は毎週投与される。特に、ダロツズマブは10mg/kgの用量で、毎週静脈内投与され、一方リダフォロリムスは毎日20mgのスケジュールで投与される。
【0109】
1つの実施態様においては、投薬順序は、mTOR阻害剤をIGF−1R抗体と同時に投与することを含んでなる−例えば、リダフォロリムスは毎日投与され、一方IGF−1R抗体(ダロツズマブ)は毎週投与される。特に、ダロツズマブは10mg/kgの用量で、毎週静脈内投与され、一方リダフォロリムスは毎日30mgのスケジュールで投与される。
【0110】
1つの実施態様においては、投薬順序は、mTOR阻害剤をIGF−1R抗体と同時に投与することを含んでなる−例えば、リダフォロリムスは毎日投与され、一方IGF−1R抗体(ダロツズマブ)は毎週投与される。特に、ダロツズマブは10mg/kgの用量で、毎週静脈内投与され、一方リダフォロリムスは毎日40mgのスケジュールで投与される。
【0111】
IGF−1R抗体に関する別の投薬レジメンは、以下の通りである:(a)15mg/kg(loading)を、続いて毎週7.5mg/kgを投入する;(b)一週間当たり7.5mg/kg;(c)一週間当たり10.0mg/kg;(d)隔週ごとに7.5mg/kg;(e)隔週ごとに10.0mg/kg;(f)隔週ごとに20.0mg/kg;(g)3週間ごとに30.0mg/kg。
【0112】
併用のためのサンプル投薬レジメンは、以下の通りである:
【0113】
【化1−1】
【0114】
【化1−2】
【0115】
効能追加
非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌の治療に加えて、mTOR阻害剤及び抗IGF−1R抗体の併用はまた、以下の癌の治療にも有用であってよい:心臓:肉腫(血管肉腫、線維肉腫、横紋筋肉腫、脂肪肉腫)、粘液腫、横紋筋腫、線維腫、脂肪腫、及び奇形腫;肺:気管支原性癌(扁平上皮細胞、未分化小細胞、未分化大細胞、腺癌)、肺胞(細気管支)癌、気管支腺腫、肉腫、リンパ腫、軟骨腫様過誤腫、中皮腫;胃腸:食道(扁平上皮細胞癌、腺癌、平滑筋肉腫、リンパ腫)、胃(癌腫、リンパ腫、平滑筋肉腫)、膵臓(腺管腺癌、インスリノーマ、グルカゴノーマ、ガストリノーマ、カルチノイド腫瘍、ビポーマ)、小腸(腺癌、リンパ腫、カルチノイド腫瘍、カポジ肉腫、平滑筋腫、血管腫、脂肪腫、神経線維腫、線維腫)、大腸(腺癌、管状腺腫、絨毛腺腫、過誤腫、平滑筋腫)、結腸、結腸直腸、直腸;尿生殖路:腎臓(腺癌、ウィルムス腫瘍[腎芽細胞腫]、リンパ腫、白血病)、膀胱及び尿道(扁平上皮細胞癌、移行上皮癌、腺癌)、前立腺(腺癌、肉腫)、精巣(精上皮腫、奇形腫、胎児性癌、奇形癌、絨毛上皮腫、肉腫、間質細胞癌、線維腫、線維腺腫、類腺腫瘍、脂肪腫);肝臓:肝癌(肝細胞癌)、胆管癌、肝芽細胞腫、血管肉腫、肝細胞腺腫、血管腫;骨:骨原性肉腫(骨肉腫)、線維肉腫、悪性線維性組織球腫、軟骨肉腫、ユーイング肉腫、悪性リンパ腫(細網肉腫)、多発性骨髄種、悪性巨細胞腫軟骨腫、骨軟骨腫(骨軟骨性外骨症)、良性軟骨腫、軟骨芽細胞腫、軟骨粘液線維腫、類骨骨腫及び、巨細胞腫;神経系:頭蓋(骨腫、血管腫、肉芽腫、黄色腫、変形性骨炎)、髄膜(髄膜腫、髄膜肉腫、神経膠腫症)、脳(星状細胞腫、髄芽細胞腫、神経膠腫、上衣細胞腫、胚細胞腫[松果体腫]、多形性神経膠芽腫、乏突起神経膠腫、神経鞘種、網膜芽細胞腫、先天性腫瘍)、脊髄神経線維腫、髄膜腫、神経膠腫、肉腫);婦人科系:子宮(子宮内膜癌)、子宮頚(子宮頚癌、前腫瘍性子宮頚部異形成)、卵巣(卵巣癌[漿液性嚢胞腺癌、粘液性嚢胞腺癌、分類不能癌腫]、顆粒膜卵胞膜細胞腫、セルトリ−ライディッヒ細胞腫、未分化胚細胞腫、悪性奇形腫)、外陰(扁平上皮細胞癌、上皮内癌、腺癌、線維肉腫、黒色腫)、膣(明細胞癌、扁平上皮細胞癌、ブドウ状肉腫(胎児性横紋筋肉腫)、卵管(癌腫);血液系:血液(骨髄性白血病[急性及び慢性]、急性リンパ芽球生白血病、慢性リンパ球性白血病、骨髄増殖性疾患、多発性骨髄腫、脊髄異形成症候群)、ホジキン病、非ホジキンリンパ腫[悪性リンパ腫];皮膚:悪性黒色腫、基底細胞癌、扁平上皮細胞癌、カポジ肉腫、色素性異形成毋斑、脂肪腫、血管腫、皮膚線維種;及び副腎:神経芽細胞腫を包含する。したがって、本明細書に提供された、用語「癌性細胞」は、上記に定義された症状の任意の1つに苦しめられた細胞を包含する。
【0116】
本発明のmTOR阻害剤及び抗IGF−1R抗体の併用はまた、以下の疾患症状の治療においても有用であってよい:ケロイド及び乾癬。
【0117】
本発明の範囲内にさらに包含されるのは、血管新生が関与する疾患を治療又は予防する方法であり、これは、かかる治療を必要とする哺乳類に対し、治療有効量の本発明の組合せを投与することを含んでなる。眼の血管新生疾患は、結果として生じる組織損傷の大部分を眼の血管の異常な浸潤に帰し得る症状の一例である(2000年6月2日公開のWO 00/30651を参照)。望ましくない浸潤は、虚血性網膜症、例えば、糖尿病性網膜症、未熟児網膜症、網膜静脈閉塞その他から結果として生じるものにより、又は、神経変性疾患、例えば、加齢関連黄班変性症において観察される脈絡膜血管新生により誘発され得る。本発明化合物の投与による血管増殖の阻害は、それ故、血管の浸潤を防止し、かつ、血管新生が関与する疾患、例えば、網膜血管新生、糖尿病性網膜症、加齢関連黄班変性症などのような眼疾患を予防又は治療するであろう。
【0118】
本発明の範囲内にさらに包含されるのは、限定されないが:眼疾患(例えば、網膜血管新生、糖尿病性網膜症、及び加齢関連黄班変性症)、アテローム性動脈硬化症、関節炎、乾癬、肥満、及びアルツハイマー病を含む、血管新生が関与する非悪性疾患(ドレッジ(Dredge)ら、Expert Opin.Biol.Ther.、2002年、第2巻、第8号、p.953−966)を、治療又は予防する方法である。別の実施態様においては、血管新生が関与する疾患を治療又は予防する方法は、眼疾患(例えば、網膜血管新生、糖尿病性網膜症、及び加齢関連黄班変性症)、アテローム性動脈硬化症、関節炎、及び乾癬を包含する。
【0119】
本発明の範囲内にさらに包含されるのは、再狭窄、炎症、自己免疫疾患、及びアレルギー/喘息といった、過増殖性疾患を治療する方法である。
【0120】
本発明の範囲内にさらに包含されるのは、ステントを被覆するための本発明の組合せの使用であり、かつそれ故に、再狭窄の治療及び/又は予防用の被覆ステント(WO 03/032809)に対する本化合物の使用である。
【0121】
本発明の範囲内にさらに包含されるのは、変形性関節症の治療及び/又は予防(WO 03/035048)のための本発明の組合せの使用である。
【0122】
本発明の範囲内にさらに包含されるのは、低インスリン症を治療する方法である。
【0123】
本発明を例示するものは、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌の治療及び/又は予防用医薬の調製における、上記記載のmTOR阻害剤及び抗IGF−1R抗体の併用使用である。
【0124】
追加の抗癌剤
本発明のmTOR阻害剤及び抗IGF−1R抗体の併用はまた、さらなる治療剤、化学療法剤、及び抗癌剤との併用においても有用である。本発明のmTOR阻害剤及び抗IGF−1R抗体の併用を、治療剤、化学療法剤、及び抗癌剤とさらに併用することは、本発明の範囲内である。かかる薬剤の例は、デヴィータ(V.T.Devita)及びヘルマン(S.Hellman)(共編)“Cancer Principles and Practice of Oncology)(癌の原理及び腫瘍学のプラクティス)”、第6版、2001年2月15日、Lippincott Williams&Wilkins Publishers、において見出すことができる。当業者は、薬物及び関係する癌の、特定の性質に基づき、どの薬剤の組合せが有用であるかを識別することができるであろう。かかるさらなる抗癌剤は、限定されないが以下を包含する:エストロゲンレセプターモジュレータ、アンドロゲンレセプターモジュレータ、レチノイドレセプターモジュレータ、細胞傷害剤/細胞増殖抑制剤、抗増殖剤、プレニル−タンパク質トランスフェラーゼ阻害剤、HMG−CoAレダクターゼ阻害剤及び他の血管新生インヒビター、HIVプロテアーゼ阻害剤、逆転写酵素阻害剤、細胞増殖及び生存シグナリングの阻害剤、ビスホスホネート、アロマターゼ阻害剤、siRNA治療、γ−セクレターゼ阻害剤、受容体チロシンキナーゼ(RTK)を妨害する薬剤、及び細胞周期チェックポイントを妨害する薬剤。本発明のmTOR阻害剤及び抗IGF−1R抗体の併用は、放射線療法と同時投与された場合、特に有用でありえる。
【0125】
「エストロゲンレセプターモジュレータ」は、メカニズムにかかわらず、エストロゲンの受容体への結合を妨害又は阻害する化合物を指す。エストロゲンレセプターモジュレータの例は、限定されないが、タモキシフェン、ラロキシフェン、ヨードキシフェン、LY353381、LY117081、トレミフェン、フルベストラント、4−[7−(2,2−ジメチル−1−オキソプロポキシ−4−メチル−2−[4−[2−(1−ピペリジニル)エトキシ]フェニル]−2H−1−ベンゾピラン−3−イル]−フェニル−2,2−ジメチルプロパノアート、4,4’−ジヒドロキシベンゾフェノン−2,4−ジニトロフェニル−ヒドラゾン、及びSH646を包含する。
【0126】
「アンドロゲンレセプターモジュレータ」は、メカニズムにかかわらず、アンドロゲンの受容体への結合を妨害又は阻害する化合物を指す。アンドロゲンレセプターモジュレータの例は、フィナステリド及び他の5α−レダクターゼ阻害剤、ニルタミド、フルタミド、ビカルタミド、リアロゾール、及びアビラテロンアセタートを包含する。
【0127】
「レチノイドレセプターモジュレータ」は、メカニズムにかかわらず、受容体へのレチノイドの結合を妨害又は阻害する化合物を指す。かかるレチノイドレセプターモジュレータの例は、ベキサロテン、トレチノイン、13−シス−レチノイン酸、9−シス−レチノイン酸、α−ジフルオロメチルオルニチン、ILX23−7553、トランス−N−(4’−ヒドロキシフェニル)レチンアミド、及びN−4−カルボキシフェニルレチンアミドを包含する。
【0128】
「細胞傷害剤/細胞増殖抑制剤」は、主として細胞の機能化を直接妨害するか又は細胞有糸分裂を阻害又は妨害することにより、細胞死を引き起こすか又は細胞増殖を阻害する化合物を指し、アルキル化剤、腫瘍壊死因子、インターカレーター、低酸素活性化化合物、微小管阻害剤/微小管安定化剤、有糸分裂キネシンの阻害剤、ヒストン脱アセチル化酵素阻害剤、有糸分裂の進行に関与するキナーゼの阻害剤、成長因子及びサイトカインシグナル伝達経路に関与するキナーゼの阻害剤、代謝拮抗物質、生物応答調節剤;ホルモン/抗ホルモン治療剤、造血成長因子、モノクローナル抗体標的化治療剤、トポイソメラーゼ阻害剤、プロテオソーム阻害剤、ユビキチンリガーゼ阻害剤、及びオーロラキナーゼ阻害剤を包含する。
【0129】
細胞傷害剤/細胞増殖抑制剤の例は、制限なされることなく、セルテネフ、カケクチン、イフォスファミド、タソネルミン、ロニダミン、カルボプラチン、アルトレタミン、プレドニムスチン、ジブロモダルシトール、ラニムスチン、フォテムスチン、ネダプラチン、オキザリプラチン、テモゾロミド、ヘプタプラチン、エストラムスチン、インプロスルファントシラート、トロホスファミド、ニムスチン、塩化ジブロスピジウム、プミテパ、ロバプラチン、サトラプラチン、プロフィロマイシン、シスプラチン、イロフルベン、デキシホスファミド、シス−アミンジクロロ(2−メチル−ピリジン)白金、ベンジルグアニン、グルホスファミド、GPX100、(トランス、トランス、トランス)−ビス−ミュー−(ヘキサン−1,6−ジアミン)−ミュー−[ジアミン−白金(II)]ビス[ジアミン(クロロ)白金(II)]テトラクロリド、ジアリジジニル(diarizidinyl)スペルミン、三酸化ヒ素、1−(11−ドデシルアミノ−10−ヒドロキシウンデシル)−3,7−ジメチルキサンチン、ゾルビシン、イダルビシン、ダウノルビシン、ビサントレン、ミトキサントロン、ピラルビシン、ピナフィド、バルルビシン、アムルビシン、アンチネオプラストン、3’−デアミノ−3’−モルホリノ−13−デオキソ−10−ヒドロキシカルミノマイシン、アナマイシン、ガラルビシン、エリナフィド、MEN10755、及び4−デメトキシ−3−デアミノ−3−アジリジニル−4−メチルスルホニル−ダウノルビシン(WO00/50032参照)、Rafキナーゼ阻害剤(例えば、Bay43−9006)、及びさらなるmTOR阻害剤を包含する。
【0130】
低酸素活性化化合物の1つの例は、チラパザミンである。
【0131】
プロテオソーム阻害剤の例は、限定されないが、ラクタシスチン及びMLN−341(ベルケード(Velcade))を包含する。
【0132】
微小管阻害剤/微小管安定化剤の例は、パクリタキセル、硫酸ビンデシン、3’,4’−ジデヒドロ−4’−デオキシ−8’−ノルビンカロイコブラスチン、ドセタキソール、リゾキシン、ドラスタチン、ミボブリンイセチオナート、アウリスタチン、セマドチン、RPR109881、BMS184476、ビンフルニン、クリプトフィシン、2,3,4,5,6−ペンタフルオロ−N−(3−フルオロ−4−メトキシフェニル)ベンゼンスルホンアミド、アンヒドロビンブラスチン、N,N−ジメチル−L−バリル−L−バリル−N−メチル−L−バリル−L−プロリル−L−プロリン−t−ブチルアミド、TDX258、エポチロン(例えば米国特許第6,284,781及び6,288,237号参照)、及び、BMS188797を包含する。1つの実施態様においては、エポチロンは微小管阻害剤/微小管安定化剤に包含されない。
【0133】
トポイソメラーゼラーゼ阻害剤のいくつかの例は、トポテカン、ハイカプタミン(hycaptamine)、イリノテカン、ルビテカン、6−エトキシプロピオニル−3’,4’−O−エキソ−ベンジリデン−シャールトルーシン、9−メトキシ−N,N−ジメチル−5−ニトロピラゾロ[3,4,5−kl]アクリジン−2−(6H)プロパンアミン、1−アミノ−9−エチル−5−フルオロ−2,3−ジヒドロ−9−ヒドロキシ−4−メチル−1H,12H−ベンゾ[de]ピラノ[3’,4’:b,7]−インドリジノ[1,2b]キノリン−10,13(9H,15H)ジオン、ルルトテカン(lurtotecan)、7−[2−(N−イソプロピルアミノ)エチル]−(20S)カンプトテシン、BNP1350、BNPI1100、BN80915、BN80942、エトポシドホスファート、テニポシド、ソブゾキサン、2’−ジメチルアミノ−2’−デオキシ−エトポシド、GL331、N−[2−(ジメチルアミノ)エチル]−9−ヒドロキシ−5,6−ジメチル−6H−ピリド[4,3−b]カルバゾール−1−カルボキサミド、アスラクライン(asulacrine)、(5a,5aB,8aa,9b)−9−[2−[N−[2−(ジメチルアミノ)エチル]−N−メチルアミノ]エチル]−5−[4−ヒドロキシ−3,5−ジメトキシフェニル]−5,5a,6,8,8a,9−ヘキソヒドロフロ(3’,4’:6,7)ナフト(2,3−d)−1,3−ジオキソール−6−オン、2,3−(メチレンジオキシ)−5−メチル−7−ヒドロキシ−8−メトキシベンゾ[c]−フェナントリジニウム、6,9−ビス[(2−アミノエチル)アミノ]ベンゾ[g]イソキノリン−5,10−ジオン、5−(3−アミノプロピルアミノ)−7,10−ジヒドロキシ−2−(2−ヒドロキシエチルアミノメチル)−6H−ピラゾロ[4,5,1−de]アクリジン−6−オン、N−[1−[2(ジエチルアミノ)エチルアミノ]−7−メトキシ−9−オキソ−9H−チオキサンテン−4−イルメチル]ホルムアミド、N−(2−(ジメチルアミノ)エチル)アクリジン−4−カルボキサミド、6−[[2−(ジメチルアミノ)エチル]アミノ]−3−ヒドロキシ−7H−インデノ[2,1−c]キノリン−7−オン、及び、ジメスナである。
【0134】
有糸分裂キネシン、及び特にヒト有糸分裂キネシンKSPの阻害剤の例は、国際公開WO 03/039460、WO 03/050064、WO 03/050122、WO 03/049527、WO03/049679、WO 03/049678、WO 04/039774、WO 03/079973、WO 03/099211、WO 03/015855、WO 03/106417、WO 04/037171、WO 04/058148、WO 04/058700、WO 04/126699、WO05/018638、WO05/019206、WO05/019205、WO05/018547、WO05/017190、US2005/0176776に記載されている。1つの実施態様においては、有糸分裂キネシンの阻害剤は、限定されないが、KSPの阻害剤、MKLP1の阻害剤、CENP−Eの阻害剤、MCAKの阻害剤、及びRab6−KIFLの阻害剤を包含する。
【0135】
「ヒストン脱アセチル化酵素阻害剤」の例は、限定されないが、SAHA、TSA、オキサムフラチン、PXD101、MG98、及びスクリプタイドを包含する。他のヒストン脱アセチル化酵素阻害剤に関するさらなる参考文献は、以下の文書に見出されてよい;ミラー(Miller,T.A)ら、J.Med.Chem.、2003年、第46巻、第24号、p.5097−5116。
【0136】
「有糸分裂の進行に関与するキナーゼの阻害剤」は、限定されないが、オーロラキナーゼの阻害剤、ポロ様キナーゼの阻害剤(PLK;特にPLK−1の阻害剤)、bub−1の阻害剤、及びbub−R1の阻害剤を包含する。「オーロラキナーゼ阻害剤」の一例は、VX−680である。
【0137】
「抗増殖剤」は、アンチセンスRNA及びDNAオリゴヌクレオチド、例えばG3139、ODN698、RVASKRAS、GEM231、及びINX3001;及び、代謝拮抗物質、例えばエノシタビン、カルモフール、テガフール、ペントスタチン、ドキシフルリジン、トリメトレキセート、フルダラビン、カペシタビン、ガロシタビン、シタラビンオクフォスファート、フォステアビン(fosteabine)ナトリウム水和物、ラルチトレキセド、パルチトレキシド、エミテフール、チアゾフリン、デシタビン、ノラトレキセド、ペメトレキセド、ネルザラビン、2’−デオキシ−2’−メチリデンシチジン、2’−フルオロメチレン−2’−デオキシシチジンン、N−[5−(2,3−ジヒドロ−ベンゾフリル)スルホニル]−N’−(3,4−ジクロロフェニル)尿素、N6−[4−デオキシ−4−[N2−[2(E),4(E)−テトラデカジエノイル]グリシルアミノ]−L−グリセロ−B−L−マンノ−ヘプトピラノシル]アデニン、アプリジン、エクチナサイジン、トロキサシタビン、4−[2−アミノ−4−オキソ−4,6,7,8−テトラヒドロ−3H−ピリミジノ[5,4−b][1,4]チアジン−6−イル−(S)−エチル]−2,5−チエノイル−L−グルタミン酸、アミノプテリン、5−フルオロウラシル、アラノシン、11−アセチル−8−(カルバモイルオキシメチル)−4−ホルミル−6−メトキシ−14−オキサ−1,11−ジアザテトラシクロ(7.4.1.0.0)−テトラデカ−2,4,6−トリエン−9−イル酢酸エステル、スワインソニン、ロメトレキソール、デキシラゾキサン(dexrazoxane)、メチオニナーゼ、2’−シアノ−2’−デオキシ−N4−パルミトイル−1−B−D−アラビノフラノシルシトシン、3−アミノピリジン−2−カルボキシアルデヒドチオセミカルバゾン、及びトラツズマブを包含する。
【0138】
モノクローナル抗体標的化治療剤の例は、癌細胞特異又は標的細胞特異モノクローナル抗体へ結合した、細胞傷害剤又は放射性同位元素を有する治療剤を包含する。例は、ベキサール(Bexxar)を包含する。
【0139】
「HMG−CoAレダクターゼ阻害剤」は、3−ヒドロキシ−3−メチルグルタリル−CoAレダクターゼの阻害剤を指す。使用してもよいHMG−CoAレダクターゼ阻害剤の例は、限定されないが、ロバスタチン(メバコール(MEVACOR)(登録商標);米国特許第4,231,938、4,294,926、及び4,319,039号参照)、シンバスタチン(ゾコール(ZOCOR)(登録商標);米国特許第4,444,784、4,820,850、及び4,916,239号参照)、プラバスタチン(プラバコール(PRAVACHOL)(登録商標);米国特許第4,346,227、4,537,859、4,410,629、5,030,447、及び5,180,589号参照)、フルバスタチン(レスコール(LESCOL)(登録商標);米国特許第5,354,772、4,911,165、4,929,437、5,189,164、5,118,853、5,290,946、及び5,356,896号参照)、アトルバスタチン(リピトール(LIPITOR)(登録商標);米国特許第5,273,995、4,681,893、5,489,691、及び5,342,952号参照)、及びセリバスタチン(リバスタチン及びバイコール(BAYCHOL)(登録商標)としても知られる;米国特許第5,177,080号参照)を包含する。これらの、及び、本方法において使用してもよい追加のHMG−CoAレダクターゼ阻害剤の構造式は、ヤルパーニ(M.Yalpani)、“Cholesterol Lowering Drugs(コレステロール低下薬)”,Chemistry & Industry,1996年2月5日、p.85−89の第87頁、及び、米国特許第4,782,084及び4,885,314号に記載されている。本明細書で使用される場合、用語、HMG−CoAレダクターゼ阻害剤は、全ての薬学的に許容されるラクトン及びオープン酸型(すなわち、ラクトン環が開裂されて遊離酸を形成する場合)、並びに、HMG−CoAレダクターゼ阻害活性を有する化合物の塩及びエステル型であって、それ故、かかる塩、エステル、オープン酸、及びラクトン型の使用は、本発明の範囲内に包含される。
【0140】
「プレニル−タンパク質トランスフェラーゼ阻害剤」は、プレニルタンパク質トランスフェラーゼ酵素の任意の1つ又は任意の組合せを阻害する化合物を指し、ファルネシル−タンパク質トランスフェラーゼ(FPTアーゼ)、ゲラニルゲラニル−タンパク質トランスフェラーゼI型(GGPTアーゼ−I)、及び、ゲラニルゲラニル−タンパク質トランスフェラーゼII型(GGPTアーゼ−II、またRab GGPTアーゼとも呼ばれる)を包含する。
【0141】
プレニル−タンパク質トランスフェラーゼ阻害剤の例は、以下の出版物及び特許に見出すことができる:WO 96/30343、WO 97/18813、WO 97/21701、WO 97/23478、WO 97/38665、WO 98/28980、WO 98/29119、WO95/32987、米国特許第5,420,245号、米国特許第5,523,430号、米国特許第5,532,359号、米国特許第5,510,510号、米国特許第5,589,485号、米国特許第5,602,098号、欧州特許公開0 618 221、欧州特許公開0 675 112、欧州特許公開0 604 181、欧州特許公開第0 696 593、WO 94/19357、WO 95/08542、WO 95/11917、WO 95/12612、WO 95/12572、WO 95/10514、米国特許第5,661,152号、WO 95/10515、WO 95/10516、WO 95/24612、WO 95/34535、WO 95/25086、WO 96/05529、WO 96/06138、WO 96/06193、WO 96/16443、WO 96/21701、WO 96/21456、WO 96/22278、WO 96/24611、WO 96/24612、WO 96/05168、WO 96/05169、WO 96/00736、米国特許第5,571,792号、WO 96/17861、WO 96/33159、WO 96/34850、WO 96/34851、WO 96/30017、WO 96/30018、WO 96/30362、WO 96/30363、WO 96/31111、WO 96/31477、WO 96/31478、WO 96/31501、WO 97/00252、WO 97/03047、WO 97/03050、WO 97/04785、WO 97/02920、WO 97/17070、WO 97/23478、WO 97/26246、WO 97/30053、WO 97/44350、WO 98/02436、及び米国特許第5,532,359号。血管新生に対するプレニル−タンパク質トランスフェラーゼ阻害剤の役割の1つの例については、European J.of Cancer、1999年、第35巻、第9号、p.1394−1401参照。
【0142】
「血管新生インヒビター」は、メカニズムにかかわらず、新たな血管の形成を阻害する化合物を指す。血管新生インヒビターの例は、限定されないが、チロシンキナーゼ阻害剤、例えば、チロシンキナーゼ受容体Flt−1(VEGFR1)及びFlk−1/KDR(VEGFR2)の阻害剤、上皮由来、線維芽細胞由来、又は血小板由来の成長因子の阻害剤、MMP(マトリックスメタロプロテアーゼ)阻害剤、インテグリンブロッカー、インターフェロン−α、インターロイキン−12、ペントサン多硫酸塩、アスピリン及びイブプロフェンのような非ステロイド系抗炎症剤(NSAID)並びにセレコキシブ及びロフェコキシブのような選択的シクロオキシ−ゲナーゼ−2阻害剤を含む、シクロオキシゲナーゼ阻害剤(PNAS、1992年、第89巻、p.7384;JNCI、1982年、第69巻、p.475;Arch.Opthalmol.1990年、第108巻、p.573;Anat.Rec.1994年、第238巻、p.68;FEBS Letters、1995年、第372巻、p.83;Clin.Orthop.1995年、第313巻、p.76;J.Mol.Endocrinol.1996年、第16巻、p.107;Jpn.J.Pharmacol.1997年、第75巻、p.105;Cancer Res.1997年、第57巻、p.1625;Cell、1998年、第93巻、p.705;Intl.J.Mol.Med.1998年、第2巻、p.715;J.Biol.Chem.1999年、第274巻、p.9116、ステロイド系抗炎症剤(例えばコルチコステロイド、ミネラルコルチコイド、デキサメタゾン、プレドニソン、プレドニソロン、メチルプレド、ベタメタゾン)、カルボキサミドトリアゾール、コンブレタスタチンA−4、スクアラミン、6−O−クロロアセチル−カルボニル)−フマギロール、サリドマイド、アンギオスタチン、トロポニン−1、アンギオテンシンIIアンタゴニスト(例えば、フェルナンデス(Fernandez)ら、J.Lab.Clin.Med.1985年、第105巻、p.141−145を参照)、及びVEGFに対する抗体(Nature Biotechnology、1999年10月、第17巻、p.963−968;キム(Kim)ら、Nature、1993年、第362巻、p.841−844;WO 00/44777;及び、WO 00/61186参照)を包含する。
【0143】
血管新生を調節又は阻害し、かつ本発明化合物と組合せて使用されてもよい他の治療薬は、凝固及び線溶系を調節又は阻害する薬剤を包含する(Clin.Chem.La.Med.2000年、第38巻、p.679−692の総説を参照)。凝固及び線溶経路を調節又は阻害する、かかる薬剤の例は、限定されないが、ヘパリン(Thromb.Haemost.1998年、第80巻、p.10−23を参照)、低分子量ヘパリン、及びカルボキシペプチダーゼU阻害剤(活性型のトロンビン活性化線溶抑制因子[TAFIa]の阻害剤としても公知)(Thrombosis Res.2001年、第101巻、p.329−354参照)を包含する。TAFIa阻害剤は、米国特許出願番号60/310,927(2001年8月8日出願)及び60/349,925号(2002年1月18日出願)に記載されている。
【0144】
「細胞周期チェックポイントを妨害する薬剤」は、細胞周期チェックポイントシグナルを伝達するプロテインキナーゼを阻害し、それにより癌細胞をDNA損傷剤に対し感受性化する化合物を指す。かかる薬剤は、ATR、ATM、CHK11及びCHK12キナーゼの阻害剤、及び、cdk及びcdcキナーゼ阻害剤を包含し、かつ特に、7−ヒドロキシスタウロスポリン、フラボピリドール、CYC202(サイクラセル(Cyclacel))、及びBMS−387032に例示される。
【0145】
「受容体チロシンキナーゼ(RTK)を妨害する薬剤」は、RTKを、及びそれ故に、発癌及び腫瘍進行に関与するメカニズムを阻害する化合物を指す。かかる薬剤は、c−Kit、Eph、PDGF、Flt3、及びc−Metの阻害剤を包含する。さらなる薬剤は、ブーム・ジェンセン(Bume−Jensen)及びハンター(Hunter)、Nature、2001年、第411巻、p.355−365により記述された、RTKの阻害剤を包含する。
【0146】
「細胞増殖及び生存シグナリング経路の阻害剤」は、細胞表面受容体の下流のシグナル伝達カスケードを阻害する化合物を指す。かかる薬剤は、セリン/スレオニンキナーゼ(限定されないが、Aktの阻害剤、例えばWO 02/083064、WO 02/083139、WO 02/083140、US2004−0116432、WO 02/083138、US2004−0102360、WO 03/086404、WO 03/086279、WO 03/086394、WO 03/084473、WO 03/086403、WO 2004/041162、WO 2004/096131、WO 2004/096129、WO 2004/096135、WO 2004/096130、WO 2005/100356、WO 2005/100344、US2005/029941、US2005/44294、US2005/43361、60/734188、60/652737、60/670469に開示されたものを包含する)、Rafキナーゼの阻害剤(例えばBAY−43−9006)、MEKの阻害剤(例えば、CI−1040及びPD−098059)、mTORの阻害剤(例えばワイス(Wyeth)CCI−779)、及びPI3Kの阻害剤(例えば、LY294002)を包含する。
【0147】
上記記載のように、NSAIDとの併用は、強力なCOX−2阻害剤であるNSAIDの使用に向けたものである。本明細書では、NSAIDは、細胞又はミクロソームアッセイにより測定されるとき、COX−2の阻害について1μM以下のIC50をもつ場合に効力がある。
【0148】
本発明はまた、選択的なCOX−2阻害剤である、NSAIDとの併用も包含する。本明細書では、選択的COX−2阻害剤であるNSAIDは、細胞又はミクロソームアッセイにより評価される、COX−1のIC50に対するCOX−2のIC50の比率によって測定されるとき、少なくとも100倍の、COX−1に対するCOX−2阻害特異性を有するものとして定義される。かかる化合物は、限定されないが、米国特許5,474,995、米国特許5,861,419、米国特許6,001,843、米国特許6,020,343、米国特許5,409,944、米国特許5,436,265、米国特許5,536,752、米国特許5,550,142、米国特許5,604,260、米国特許5,698,584、米国特許5,710,140、WO 94/15932、米国特許5,344,991、米国特許5,134,142、米国特許5,380,738、米国特許5,393,790、米国特許5,466,823、米国特許5,633,272、及び米国特許5,932,598に開示されているものを包含し、これらは全て参考として本明細書に含まれる。
【0149】
本治療法において特に有用であるCOX−2の阻害剤は、3−フェニル−4−(4−(メチルスルホニル)フェニル)−2−(5H)−フラノン;及び5−クロロ−3−(4−メチルスルホニル)フェニル−2−(2−メチル−5−ピリジニル)ピリジン;又はその薬学的に許容される塩である。
【0150】
COX−2の特異的阻害剤として記載されてきており、かつそれ故、本発明において有用である化合物は、限定されないが以下を包含する:パレコキシブ、ベクストラ(BEXTRA(登録商標))、セレブレックス(CELEBREX(登録商標))、又はその薬学的に許容される塩。
【0151】
血管新生インヒビターの他の例は、限定されないが、エンドスタチン、ウクライン、ランピルナーゼ、IM862、5−メトキシ−4−[2−メチル−3−(3−メチル−2−ブテニル)オキシラニル]−1−オキサスピロ[2,5]オクト−6−イル(クロロアセチル)カルバメート、アセチルジナナリン(acetyldinanaline)、5−アミノ−1−[[3,5−ジクロロ−4−(4−クロロベンゾイル)フェニル]メチル]−1H−1,2,3−トリアゾール−4−カルボキサミド、CM101、スクアラミン、コンブレタスタチン、RPI4610、NX31838、硫酸化マンノペンタオースホスファート、7,7−(カルボニル−ビス[イミノ−N−メチル−4,2−ピロロカルボニルイミノ[N−メチル−4,2−ピロール]−カルボニルイミノ]−ビス−(1,3−ナフタレンジスルホナート)、及び、3−[(2,4−ジメチルピロール−5−イル)メチレン]−2−インドリノン(SU5416)を包含する。
【0152】
前記に使用したように、「インテグリンブロッカー」は、生理的リガンドの、αvβ3インテグリンへの結合を選択的に拮抗するか、阻害するか、又は反対に作用する化合物、生理的リガンドの、αvβ5インテグリンへの結合を選択的に拮抗するか、阻害するか、又は反対に作用する化合物、生理的リガンドの、αvβ3インテグリン及びαvβ5インテグリン双方への結合を拮抗するか、阻害するか、又は反対に作用する化合物、及び、毛細血管内皮細胞上に発現された特定のインテグリンの活性を拮抗するか、阻害するか、又は反対に作用する化合物を指す。該用語はまた、αvβ6、αvβ8、α1β1、α2β1、α5β1、α6β1、及びα6β4インテグリンのアンタゴニストも指す。該用語はまた、αvβ3、αvβ5、αvβ6、αvβ8、α1β1、α2β1、β5α1、α6β1、及びα6β4インテグリンの任意の組合せのアンタゴニストも指す。
【0153】
チロシンキナーゼ阻害剤のいくつかの具体的な例は、N−(トリフルオロメチルフェニル)−5−メチルイソオキサゾール−4−カルボキサミド、3−[(2,4−ジメチルピロール−5−イル)メチリデニル]インドリン−2−オン、17−(アリルアミノ)−17−デメトキシゲルダナマイシン、4−(3−クロロ−4−フルオロフェニルアミノ)−7−メトキシ−6−[3−(4−モルホリニル)プロポキシル]キナゾリン、N−(3−エチニルフェニル)−6,7−ビス(2−メトキシエトキシ)−4−キナゾリンアミン、BIBX1382、2,3,9,10,11,12−ヘキサヒドロ−10−(ヒドロキシメチル)−10−ヒドロキシ−9−メチル−9,12−エポキシ−1H−ジインドロ[1,2,3−fg:3’,2’,1’−kl]ピロロ[3,4−i][1,6]ベンゾジアゾシン−1−オン、SH268、ゲニステイン、STI571、CEP2563、4−(3−クロロフェニルアミノ)−5,6−ジメチル−7H−ピロロ[2,3−d]ピリミジンメタンスルホナート、4−(3−ブロモ−4−ヒドロキシフェニル)アミノ−6,7−ジメトキシキナゾリン、4−(4’−ヒドロキシフェニル)アミノ−6,7−ジメトキシキナゾリン、SU6668、STI571A、N−4−クロロフェニル−4−(4−ピリジルメチル)−1−フタラジンアミン、及びEMD121974を包含する。
【0154】
抗癌化合物以外の化合物との併用もまた、本方法に包含される。例えば、本発明のmTOR阻害剤及び抗IGF−1R抗体の併用と、PPAR−γ(すなわち、PPAR−ガンマ)アゴニスト及びPPAR−δ(すなわち、PPAR−デルタ)アゴニストとの組合せは、いくつかの悪性疾患の治療において有用である。PPAR−γ及びPPAR−δは、核のペルオキシソーム増殖剤活性化受容体γ及びδである。PPAR−γの、内皮細胞上での発現及び、血管新生におけるその関与は、文献に報告されてきた(J.Cardiovasc.Pharmacol.1998年、第31巻、p.909−913;J.Biol.Chem.1999年、第274巻、p.9116−9121;Invest.Ophthalmol Vis.Sci.2000年、第41巻、p.2309−2317参照)。より最近では、PPAR−γアゴニストが、インビトロでVEGFに対する血管新生応答を阻害することが示された;トログリタゾン及びマレイン酸ロシグリタゾンの双方は、マウスにおいて、網膜の血管新生の発生を阻害する(Arch.Opthamol.2001年、第119巻、p.709−717)。PPAR−γアゴニスト及びPPAR−γ/αアゴニストの例は、限定されないが、チアゾリジンジオン(例えばDRF2725、CS−011、トログリタゾン、ロシグリタゾン、及びピオグリタゾン)、フェノフィブラート、ゲンフィブロジル、クロフィブラート、GW2570、SB219994、AR−H039242、JTT−501、MCC−555、GW2331、GW409544、NN2344、KRP297、NP0110、DRF4158、NN622、GI262570、PNU182716、DRF552926、2−[(5,7−ジプロピル−3−トリフルオロメチル−1,2−ベンゾイソオキサゾール−6−イル)オキシ]−2−メチルプロピオン酸(USSN 09/782,856に開示)、及び2(R)−7−(3−(2−クロロ−4−(4−フルオロフェノキシ)フェノキシ)プロポキシ)−2−エチルクロマン−2−カルボン酸(USSN 60/235,708及び60/244,697に開示)を包含する。
【0155】
本発明の別の実施態様は、本開示化合物の、癌の治療のための遺伝子療法と組合せた使用である。癌を治療するための遺伝学的戦略の概要に関しては、ホール(Hall)ら、(Am.J.Hum,Genet.1997年、第61巻、p.785−789)、及びクーフェ(Kufe)ら(「Cancer Medicine(癌医療)」、第5版、BC Decker、ハミルトン、2000年、p.876−889)を参照のこと。遺伝子療法を用いて、任意の腫瘍抑制遺伝子を送達することができる。かかる遺伝子の例は、限定されないが、組換えウイルス媒介性の遺伝子導入により送達可能であるp53(例えば、米国特許第6,069,134号参照)、uPA/uPARアンタゴニスト(「Adenovirus−Mediated Delivery of a uPA/uPAR Antagonist Suppresses Angiogenesis−Dependent Tumor Growth and Dissemination in Mice(uPA/uPARアンタゴニストのアデノウイルス媒介送達は、血管新生依存性の腫瘍増殖及び播種をマウスにおいて抑制する)」、Gene Therapy、1998年8月、第5巻、第8号、p.1105−13)、及びインターフェロンガンマ(J.Immunol.2000年、第164巻、p.217−222)を包含する。
【0156】
本発明化合物はまた、生来多剤耐性(MDR)、特に、高レベルのトランスポータ−タンパク質の発現に関与しているMDRの阻害剤と併用して投与されてもよい。かかるMDR阻害剤は、p−糖タンパク質(P−gp)の阻害剤、例えばLY335979、XR9576、OC144−093、R101922、VX853、及びPSC833(バルスポダール)を包含する。
【0157】
本発明化合物は、本発明化合物の、単独の又は放射線療法との使用の結果として生じるかもしれない急性、遅発性、遅延相、及び予測性嘔吐を含めた、悪心又は嘔吐を治療するべく、抗嘔吐薬と一緒に用いてもよい。嘔吐の予防又は治療のためには、本発明化合物は、他の抗嘔吐薬、特にニューロキニン−1受容体アンタゴニスト;5HT3受容体アンタゴニスト、例えばオンダンセトロン、グラニセトロン、トロピセトロン、及びザチセトロン;GABAB受容体アゴニスト、例えばバクロフェン;コルチコステロイド、例えばデカドロン(デキサメタゾン)、ケナログ(Kenalog)、アリストコート(Aristocort)、ナサライド(Nasalide)、プレフェリド(Preferid)、ベネコルテン(Benecorten)、又は他の、米国特許第2,789,118、2,990,401、3,048,581、3,126,375、3,929,768、3,996,359、3,928,326、及び3,749,712号に開示されたもの;抗ドーパミン作動薬、例えばフェノチアジン類(例えばプロクロペラジン、フルフェナジン、チオリダジン、及びメソリダジン)、メトクロプラマイド、又はドロナビノールと一緒に使用してもよい。別の実施態様においては、ニューロキニン−1受容体アンタゴニスト、5HT3受容体アンタゴニスト、及びコルチコステロイドから選ばれる抗嘔吐薬との併用療法が、本化合物の投与の結果として生じるかもしれない嘔吐の治療又は予防用に開示されている。
【0158】
本発明化合物との併用において役立つニューロキニン−1受容体アンタゴニストは、例えば、米国特許第5,162,339、5,232,929、5,242,930、5,373,003、5,387,595、5,459,270、5,494,926、5,496,833、5,637,699、5,719,147号;欧州特許公開番号EP 0 360 390、0 394 989、0 428 434、0 429 366、0 430 771、0 436 334、0 443 132、0 482 539、0 498 069、0 499 313、0 512 901、0 512 902、0 514 273、0 514 274、0 514 275、0 514 276、0 515 681、0 517 589、0 520 555、0 522 808、0 528 495、0 532 456、0 533 280、0 536 817、0 545 478、0 558 156、0 577 394、0 585 913、0 590 152、0 599 538、0 610 793、0 634 402、0 686 629、0 693 489、0 694 535、0 699 655、0 699 674、0 707 006、0 708 101、0 709 375、0 709 376、0 714 891、0 723 959、0 733 632、及び0 776 893;PCT国際特許公開番号WO 90/05525、90/05729、91/09844、91/18899、92/01688、92/06079、92/12151、92/15585、92/17449、92/20661、92/20676、92/21677、92/22569、93/00330、93/00331、93/01159、93/01165、93/01169、93/01170、93/06099、93/09116、93/10073、93/14084、93/14113、93/18023、93/19064、93/21155、93/21181、93/23380、93/24465、94/00440、94/01402、94/02461、94/02595、94/03429、94/03445、94/04494、94/04496、94/05625、94/07843、94/08997、94/10165、94/10167、94/10168、94/10170、94/11368、94/13639、94/13663、94/14767、94/15903、94/19320、94/19323、94/20500、94/26735、94/26740、94/29309、95/02595、95/04040、95/04042、95/06645、95/07886、95/07908、95/08549、95/11880、95/14017、95/15311、95/16679、95/17382、95/18124、95/18129、95/19344、95/20575、95/21819、95/22525、95/23798、95/26338、95/28418、95/30674、95/30687、95/33744、96/05181、96/05193、96/05203、96/06094、96/07649、96/10562、96/16939、96/18643、96/20197、96/21661、96/29304、96/29317、96/29326、96/29328、96/31214、96/32385、96/37489、97/01553、97/01554、97/03066、97/08144、97/14671、97/17362、97/18206、97/19084、97/19942、及び97/21702;及び英国特許公開番号2 266 529、2 268 931、2 269 170、2 269 590、2 271 774、2 292 144、2 293 168、2 293 169、及び2 302 689において充分に記載されている。かかる化合物の調製は、上述の特許及び刊行物において充分に記載されており、それらは参考として本明細書に含まれている。
【0159】
1つの実施態様においては、本発明化合物との併用使用のためのニューロキニン−1受容体アンタゴニストは:2−(R)−(1−(R)−(3,5−ビス(トリフルオロメチル)フェニル)エトキシ)−3−(S)−(4−フルオロフェニル)−4−(3−(5−オキソ−1H,4H−1,2,4−トリアゾロ)メチル)モルホリン、又は、その薬学的に許容される塩から選ばれ、それは、米国特許第5,719,147号に記載されている。
【0160】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、貧血症の治療において有用な薬剤とともに投与してもよい。かかる貧血症治療薬は、例えば、持続性の赤血球形成受容体活性化剤(例えばエポエチン・アルファ)である。
【0161】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、好中球減少症の治療において有用な薬剤とともに投与されてよい。かかる好中球減少症治療薬は、例えば、ヒト顆粒球コロニー刺激因子(G−CSF)のような、好中球の産生及び機能を調節する造血成長因子である。G−CSFの例は、フィルグラスチムを包含する。
【0162】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、免疫強化剤、例えば、レバミソール、イソプリノシン、及びザダキシン(Zadaxin)とともに投与してもよい。
【0163】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、ビスホスホネート(ビスホスホナート、ジホスホナート、ビスホスホン酸、及びジホスホン酸を包含するべく理解される)と併用して、骨癌を含む癌の治療又は予防に有用であってよい。ビホスホネートの例は、限定されないが:エチドロネート(ダイドロネル(Didronel))、パミドロネート(アレディア(Aredia))、アレンドロネート(フォサマックス(Fosamax))、リセドロネート(アクトネル(Actonel))、ゾレドロネート(ゾメタ(Zometa))、イバンドロネート(ボニバ(Boniva))、インカドロネート又はシマドロネート、クロドロネート、EB−1053、ミノドロネート、ネリドロネート、ピリドロネート(piridronate)、及びチルドロネートを包含し、任意の及び全ての、薬学的に許容される塩、誘導体、水和物、及びそれらの混合物を包含する。
【0164】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、アロマターゼ阻害剤と組合せて、乳癌の治療又は予防に有用でありえる。アロマターゼ阻害剤の例は、限定されないが、アナストロゾール、レトロゾール、及びエキセメスタンを包含する。
【0165】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、siRNA治療薬との併用において、癌の治療又は予防に有用でありえる。
【0166】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、γ−セクレターゼ阻害剤及び/又はNOTCHシグナリング阻害剤と併用して投与してもよい。かかる阻害剤は、WO 01/90084、WO 02/30912、WO 01/70677、WO 03/013506、WO 02/36555、WO 03/093252、WO 03/093264、WO 03/093251、WO 03/093253、WO 2004/039800、WO 2004/039370、WO 2005/030731、WO 2005/014553、USSN 10/957,251、WO 2004/089911、WO 02/081435、WO 02/081433、WO 03/018543、WO 2004/031137、WO 2004/031139、WO 2004/031138、WO 2004/101538、WO 2004/101539、及びWO 02/47671(LY−450139を含む)。
【0167】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、Aktの阻害剤との併用において、癌の治療又は予防に有用でありえる。かかる阻害剤は、限定されないが、以下の出版物に記載された化合物を包含する:WO 02/083064、WO 02/083139、WO 02/083140、US2004−0116432、WO 02/083138、US2004−0102360、WO 03/086404、WO 03/086279、WO 03/086394、WO 03/084473、WO 03/086403、WO 2004/041162、WO 2004/096131、WO 2004/096129、WO 2004/096135、WO 2004/096130、WO 2005/100356、WO 2005/100344、US2005/029941、US2005/44294、US2005/43361、60/734188、60/652737、60/670469。
【0168】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、PARP阻害剤との併用において、癌の治療又は予防に有用でありえる。
【0169】
放射線療法自体は、癌治療の分野では通常の方法を意味する。放射線療法には、X線、γ線、中性子線、電子線、陽子線などの種々の放射線;及び放射線源を使用し得る。最も一般的な放射線療法においては、外部放射線、γ線による照射にリニア加速器が使用される。
【0170】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、以下の治療薬との併用において、癌の治療に有用でありえる:アバレリックス(プレナキシス・デポ(Plenaxis depot)(登録商標));アルデスロイキン(プロカイン(Prokine)(登録商標));アルデスロイキン(プロロイキン(Proleukin)(登録商標));アレムツズマブ(キャンパス(Campath)(登録商標));アリトレチノイン(パンレチン(Panretin)(登録商標));アロプリノール(ザイロプリム(Zyloprim)(登録商標));アルトレタミン(ヘキサレン(Hexalen)(登録商標));アミフォスチン(エチヨル(Ethyol)(登録商標));アナストロゾール(アリミデックス(Arimidex)(登録商標));三酸化ヒ素(トリセノックス(Trisenox)(登録商標));アスパラギナーゼ(エルスパル(Elspar)(登録商標));アザシチジン(ビダザ(Vidaza)(登録商標));ベバシズマブ(アバスチン(Avastin)(登録商標));ベキサロテン・カプセル(タルグレチン(Targretin)(登録商標));ベキサロテン・ゲル(タルグレチン(Targretin)(登録商標));ブレオマイシン(ブレノキサン(Blenoxane)(登録商標));ボルテゾミブ(ベルケード(Velcade)(登録商標));ブスルファン・静脈内(ブスルフェックス(Busulfex)(登録商標));ブスルファン・経口(マイレラン(Myleran)(登録商標));カルステロン(メトサーブ(Methosarb)(登録商標));カペシタビン(ゼローダ(Xeloda)(登録商標));カルボプラチン(パラプラチン(Paraplatin)(登録商標));カルムスチン(BCNU(登録商標)、BiCNU(登録商標));カルムスチン(グリアデル(Gliadel)(登録商標));インプラント型ポリフェプロサン20カルムスチン(グリアデル・ウエファー(Gliadel Wafer)(登録商標));セレコキシブ(セレブレックス((Celebex)(登録商標));セツキシマブ(アービタックス(Erbitux)(登録商標));クロランブシル(ロイケラン(Leukeran)(登録商標));シスプラチン(プラチノール(Platinol)(登録商標));クラドリビン(ロイスタチン(Leustatin)(登録商標)、2−CdA(登録商標));クロファラビン(クロラール(Clolar)(登録商標));シクロホスファミド(サイトキサン(Cytoxan)(登録商標)、ネオサール(Neosar)(登録商標));シクロホスファミド(サイトキサン注射薬(Cytoxan Injection)(登録商標));シクロホスファミド(サイトキサン・錠剤(Cytoxan Tablet)(登録商標));シタラビン(サイトサール−U(Cytosar−U)(登録商標));リポソーム化シタラビン(DepoCyt(登録商標));デカルバジン(DTIC−Dome(登録商標));ダクチノマイシン、アクチノマイシンD(コスメゲン(Cosmegen)(登録商標));ダルベポエチン・アルファ(アラネスプ(Aranesp)(登録商標));リポソーム化ダウノルビシン(DanuoXome(登録商標));ダウノルビシン、ダウノマイシン(ダウノルビシン(Daunorubicin)(登録商標));ダウノルビシン、ダウノマイシン(セルビジン(Cerbidine)(登録商標));デニロイキン・diftitox(オンタック(Ontak)(登録商標));デクスラゾキサン(ザインカード(Zinecard)(登録商標));ドセタキセル(タキソテール(Taxotere)(登録商標));ドキソルビシン(アドリアマイシンPFS(Adriamycin PFS)(登録商標));ドキソルビシン(アドリアマイシン(Adriamycin)(登録商標)、ルベックス(Rubex)(登録商標));ドキソルビシン(アドリアマイシンPFS注射薬(Adriamycin PFS Injection)(登録商標));リポソーム化ドキソルビシン(ドキシル(Doxil)(登録商標));プロピオン酸ドロモスタノロン(ドロモスタノロン(Dromostanolone)(登録商標));プロピオン酸ドロモスタノロン(マステロン注射薬(Masterone Injection)(登録商標));エリオットのB溶液(エリオットのB溶液(Elliott’s B Solution)(登録商標));エピルビシン(エレンス(Ellence)(登録商標));エポエチン・アルファ(エポジェン(epogen)(登録商標));エルロチニブ(タルセバ(Tarceva)(登録商標));エストラムスチン(Emcyt(登録商標));エトポシドリン酸塩(エトポフォス(Etopophos)(登録商標));エトポシド、VP−16(ベペシド(Vepesid)(登録商標));エキセメスタン(アロマシン(Aromasin)(登録商標));フィルグラスチム(ノイポゲン(Neupogen)(登録商標));フロクスウリジン(動脈内)(FUDR(登録商標));フルダラビン(フルダラ(Fludara)(登録商標));フルオロウラシル、5−FU(アドルシル(Adrucil)(登録商標));フルベストラント(ファスロデックス(Faslodex)(登録商標));ゲフィチニブ(イレッサ(Iressa)(登録商標));ゲムシタビン(ゲムザール(Gemzar)(登録商標));ゲムツズマブ・オゾガマイシン(マイロターグ(Mylotarg)(登録商標));ゴセレリン酢酸塩(ゾラデックス・インプラント(Zoladex Implant)(登録商標));ゴセレリン酢酸塩(ゾラデックス(Zoladex)(登録商標));ヒストレリン酢酸塩(ヒストレリン・インプラント(Histrelin implant)(登録商標));ヒドロキシウレア(ハイドレア(Hydrea)(登録商標));イブリツモマブ・チウキセタン(ゼヴァリン(Zevalin)(登録商標));イダルビシン(イダマイシン(Idamycin)(登録商標));イフォスファミド(IFEX(登録商標));イマチニブメシル酸塩(グリベック(Gleevec)(登録商標));インターフェロンアルファ2a(ロフェロンA(Roferon A)(登録商標));インターフェロンアルファ−2b(イントロンA(Intron A)(登録商標));イリノテカン(カンプトサール(Camptosar)(登録商標));レナリドマイド(レブリミド(Revlimid)(登録商標));レトロゾール(フェマーラ(Femara)(登録商標));ロイコボリン(ウェルコボリン(Wellcovorin)(登録商標)、ロイコボリン(Leucovorin)(登録商標));酢酸ロイプロリド(エリガード(Eligard)(登録商標));レバミソール(エルガミゾール(Ergamisol)(登録商標));ロムスチン、CCNU(CeeBU(登録商標));メクロレタミン・ナイトロジェンマスタード(マスタルゲン(Mustargen)(登録商標));酢酸メゲストロール(メゲース(Megace)(登録商標));メルファラン、L−PAM(アルケラン(Alkeran)(登録商標));メルカプトプリン、6−MP(プリネトール(Purinethol)(登録商標));メスナ(メスネックス(Mesnex)(登録商標));メスナ(メスネックス錠(Mesnex tabs)(登録商標));メトトレキサート(メトトレキセート(Methotrexate)(登録商標));メトキサレン(ウヴァデクス(Uvadex)(登録商標));マイトマイシンC(ミュータマイシン(Mutamycin)(登録商標));ミトタン(リソドレン(Lysodren)(登録商標));ミトキサントロン(ノバントロン(Novantrone)(登録商標));フェンプロピオン酸ナンドロロン(デュラボリン−50(Durabolin−50)(登録商標));ネララビン(アラノン(Arranon)(登録商標));ノフェツモマブ(ヴァールマ(Verluma)(登録商標));オプレルベキン(ニューメガ(Neumega)(登録商標));オキサリプラチン(エロキサチン(Eloxatin)(登録商標));パクリタキセル(パクセン(Paxene)(登録商標));パクリタキセル(タキソール(Taxol)(登録商標));パクリタキセル・タンパク質結合粒子(アブラキサン(Abraxane)(登録商標));パリフェルミン(ケピバンス(Kepivance)(登録商標));パミドロネート(アレディア(Aredia)(登録商標));ペグアデマーゼ(アダジェン(Adagen)(ウシ・ペグアデマーゼ(Pegademase Bovine))(登録商標));ペグアスパラガーゼ(オンキャスパー(Oncaspar)(登録商標));ペグフィルグラスチム(ニューラスタ(Neulasta)(登録商標));ペメトレキセド2ナトリウム(アリムタ(Alimta)(登録商標));ペントスタチン(ナイペント(Nipent)(登録商標));ピポブロマン(バーサイト(Vercyte)(登録商標));プリカマイシン、ミトラマイシン(ミトラシン(Mithracin)(登録商標));ポルフィマー・ナトリウム(フォトフリン(Photofrin)(登録商標));プロカルバジン(マツラン(Matulane)(登録商標));キナクリン(アタブリン(Atabrine)(登録商標));ラスブリカーゼ(エリテック(Elitek)(登録商標));リツキシマブ(リツキサン(Rituxan)(登録商標));サルグラモスチン(リューカイン(Leukine)(登録商標));サルグラモスチン(プロカイン(Prokine)(登録商標));ソラフェニブ(ネクサバール(Nexavar)(登録商標));ストレプトゾシン(ザノサール(Zanosar)(登録商標));スニチニブ・マレイン酸塩(スーテント(Sutent)(登録商標));タルク(スクレロゾール(Sclerosol)(登録商標));タモキシフェン(ノルバデックス(Nolvadex)(登録商標));テモゾロミド(テモダール(Temodar)(登録商標));テニポシド、VM−26(ヴァモン(Vumon)(登録商標));テストラクトン(テスラク(Teslac)(登録商標));チオグアニン、6−TG(チオグアニン(Thioguanine)(登録商標));チオテパ(チオプレックス(Thioplex)(登録商標));トポテカン(ハイカムチン(Hycamtin)(登録商標));トレミフェン(フェアストン(Fareston)(登録商標));トシツモマブ(ベキサール(Bexxar)(登録商標));トシツモマブ/I−131 トシツモマブ(ベキサール(Bexxar)(登録商標));トラツズマブ(ハーセプチン(Herceptin)(登録商標));トレチノイン、ATRA(ベサノイド(Vesanoid)(登録商標));ウラシル・マスタード(ウラシル・マスタード・カプセル(Uracil Mustard Capsules)(登録商標));バルルビシン(バルスター(Valstar)(登録商標));ビンブラスチン(ベルバン(Velban)(登録商標));ビンクリスチン(オンコビン(Oncovin)(登録商標));ビノレルビン(ナベルビン(Navelbine)(登録商標));及びゾレドロネート(ゾメタ(Zometa)(登録商標))。
【0171】
全ての特許、刊行物、及び確認された係属中の特許は、参考として本明細書に含まれる。
【0172】
本明細書において使用された略号は、以下の表に示した意味を有する。以下の表に記載されていない略号は、別に特に述べない限り、一般に使用される意味を有する。
【0173】
【化2】
【0174】
本発明のmTOR阻害剤及び抗IGF−1R抗体は、適当な材料を使用して、以下の一般的なスキームに従って調製してよく、かつその後の具体的な実施例によってさらに例示される。しかしながら、実施例に例示された具体的な抗癌剤が、本発明とみなされる唯一のジーナス(Genus)を形成するものと解釈されるべきではない。それ故、以下の例示的な実施例は、リストされた抗癌剤によって、又は例示を目的として使用された特定の物質によって、限定されるものではない。当業者は、これらの化合物を調整するため、以下の調製法の条件及びプロセスについて既知の変形を用いてもよいことを容易に理解するであろう。別に記載のない限り、すべての温度は摂氏である。
【0175】
合成法
【0176】
【化3】
【0177】
本発明は、ここに、以下の非限定的例において例示され、ここで、別段の指定のない限り:
1.全ての最終生成物は、NMR、LCMSにより分析した。
2.中間体は、NMR及び/又はTLC及び/又はLCMSにより分析した。
3.ほとんどの化合物は、シリカゲル上でのフラッシュクロマトグラフィー、逆相HPLC、再結晶化、及び/又はスウィッシュ(溶媒中に懸濁した後、固体を濾過する)により精製した。
4.反応の経過は、薄層クロマトグラフィー(TLC)及び/又はLCMSにより追跡し、また反応時間は例示のみのために示されている。
【0178】
実施例1
ジメチル−ホスフィン酸C−43ラパマイシンエステル
【0179】
【化4】
【0180】
ジクロロメタン 1.8mL中のラパマイシン(0.1g、0.109mmol)の冷却された(0℃)溶液に、2,6−ジ−t−ブチル−4−メチルピリジン 0.168g(0.82mmol)を、N2流下で添加し、直後にジクロロメタン 0.2mL中のジメチルホスフィン酸クロリド(0.062g、0.547mmol)の溶液を添加した。わずかに黄色の反応溶液を、0℃で、N2雰囲気下で3.5時間攪拌した(反応はTLCによりモニターした)。冷却した(0℃)反応溶液を、EtOAc 約20mLで希釈し、次にEtOAc(150mL)及び飽和NaHCO3(100mL)を含有する分液漏斗に移した。水層を除去し、有機層を氷冷した1N 塩酸(1x100mL)、飽和NaHCO3(1x100mL)、及び食塩水(1x100mL)で順次洗浄し、次にMgSO4上で乾燥し、濃縮した。粗生成物をシリカゲルフラッシュクロマトグラフィー(1:10:3:3 MeOH/DCM/EtOAc/ヘキサンで溶出)により精製して、白色固体 0.092gを得た:
1H NMR(300MHz,CDCl3)δ4.18(m,1H)、4.10(m,1H)、3.05(m,1H)、1.51(m,6H);31P NMR(121MHz,CDCl3)δ53.6;1013m/z(M+Na)。
【0181】
実施例2
ジメチルホスフィン酸C−43ラパマイシンエステル、別の合成法
ラパマイシン及びジクロロメタンを、窒素でパージした反応フラスコに装入する。攪拌した溶液を、約0℃に冷却する(−5±5℃の外部温度を反応の間中、維持する)。次に、ジクロロメタン中のクロロジメチルホスフィン酸クロリド(2.0モル当量)の溶液を、約8−13分間にわたり添加する。この直後に、ジクロロメタン中の3,5−ルチジン(2.2モル当量)の溶液を、約15−20分間にわたり添加する。双方の添加の間中、反応は0℃未満にとどまる。冷却した反応溶液を1時間攪拌し、次に、まだ冷たいうちに、飽和NaHCO3水溶液及びメチル−t−ブチルエーテル(MTBE)、酢酸エチル、又はジエチルエーテルを含有する抽出器に移す。製造過程のサンプルは、30及び60分の時点で取出す。サンプルは、反応ワークアップに記載したものと同様の方法で調製する。反応の進行は、TLC(1:10:3:3 MeOH/DCM/EtOAc/ヘキサン)及び逆相HPLC分析によりモニターする。単離した有機層を、氷冷した1N 塩酸、飽和NaHCO3(2x)、飽和NaCl水溶液で順次洗浄し、硫酸ナトリウム上で乾燥する。濾過及び溶媒除去し、残渣をアセトンで溶媒交換し、続いて真空中で濃縮して粗生成物を得て、これを順相及び逆相HPLCにより、純度について分析してもよい。
【0182】
実施例3
マウスモノクローナル抗体(MAB)の生成及び選択
IGF−1Rに対し特異的に向けられ、かつIRは認識しない、MAbの生成を目的として、6工程のスクリーニングを含んでなるプロトコールについて考察した。
【0183】
それは以下のものから構成される:
−ハイブリドーマを生成する目的で、マウスを組換えIGF−1Rで免疫化すること、
−培養上清を、免疫化に寄与した組換えタンパク質について、ELISAによりスクリーニングすること、
−陽性のハイブリドーマの全ての上清を、MCF−7腫瘍細胞の表面上で過剰発現している天然の受容体について、ELISAにより試験すること、
−ハイブリドーマ陽性の上清を、IGF−1R又はIRを各々発現するバキュロウイルスで感染された昆虫細胞上での、IGF−1R及びIRの特異的認識の観点から、2つの最初のスクリーニングで評価すること、
−この工程で選択された抗体が、MCF−7細胞の誘導されたIGF1増殖を、インビトロで阻害し得ることを検証すること、
−腫瘍MCF−7の増殖に対するインパクトという観点から保持された候補の、インビボの活性をヌードマウスにおいて確認すること。
【0184】
これらの個別の工程及び得られた結果の全てを、以下の実施例1において簡単に記載する。
【0185】
免疫化の工程では、マウスに8μgの組換えIGF−1Rを、皮下経路により2回注射した。雌ラットの細胞が、マウス骨髄腫Sp2OAg14の細胞と融合する3日前に、3μgの組換え受容体の静脈内注射により、マウスを刺激した。融合の14日後、ハイブリドーマ上清を、組換えIGF−1Rによって感作したプレート上で、ELISAによってスクリーンした。その上清が陽性であると判明したハイブリドーマを保持し、増殖させた後、FACScanで試験して、産生した抗体が同様に天然のIGF−1Rを認識し得ることを確認した。これを行なうため、IGF−1Rを過剰発現するエストロゲン依存性の乳房腫瘍由来のMCF−7細胞を、ELISAで選択されたハイブリドーマにより産生し各培養上清とともにインキュベートした。細胞表面上の天然/MAb受容体複合体は、蛍光色素へ結合した二次抗種抗体(secondary anti−species antibody)によって示された。図3Aないし3Cは、ハイブリドーマ7C10の上清で得られたヒストグラムタイプ(図3C)を、細胞標識単独+二次抗体(図3A)、又はコントロールアイソタイプを利用した標識(図3B)と比較して示す。
【0186】
選択のこの工程において、MAbを分泌すると同時に組換え受容体及び天然受容体を認識するハイブリドーマのみを選択し、クローニングした。これらのハイブリドーマによって分泌されるMAbを産生し、次に精製した後、2つの受容体を同時に認識するハイブリドーマを除去する目的で、IGF−1R又はIRを発現するSf9昆虫細胞上で、上記記載の方法に従ってFACScanで試験した。図4Aは、未感染細胞+二次抗体(1)、αIR3で標識された未感染細胞+二次抗体(2)、及び抗IGF−1R抗体で標識された未感染細胞+二次抗体(3)にそれぞれ対応するヒストグラム1、2、3の全回収率を示す。この最初の結果は、これらの未感染昆虫細胞の表面上には、検出可能なIGF−1R及びIRが存在しないことを充分に示している。図4Bは、IGF−1Rを発現するバキュロウイルスに感染された細胞の標識を示す。この2番目の図では、陽性コントロールとして用いたαIR3が、予想通り充分に細胞を標識するが(ピーク2)、一方、抗IR(ピーク3)は、単一細胞のピークに重なっている。最後に、図4Cでは、抗IRが、予想通り充分にIRを発現するSf9細胞を標識するが(ピーク3)、予想外なことに、文献ではIGF−1Rに特異的であると記載されているαIR3が、IRを認識するように見られる(ピーク2)ことが示されている。
【0187】
この3番目のスクリーニング系で得られた結果は、表1に要約されており、IGF−1Rを認識しかつIRを認識しないという基準を満たすMAb:7C10の生成を示している。Mab 7C10のアイソタイピングにより、それがIgG1を含むことが示されている。
【0188】
【表1】
【0189】
MAbの選択用に提供された最後の2つのスクリーニングは、後者が、細胞系MCF−7においてIGF−Iによって誘導される細胞増殖を、インビボ及びインビトロにおいて大いに阻害し得ることを証明するものであった。
【0190】
インビトロの選択では、MCF−7細胞を接種し、ウシ胎児血清を除去し、次に漸増濃度のIGF−I(1ないし50ng/ml)の存在下で、最終濃度10μg/mlまで添加された、試験されるべき7C10抗体の存在下又は非存在下でインキュベートした。この実験では、市販のαIR3 MAbを陽性コントロールとし、7G3 MAb(7C10と同時に単離され、天然の受容体を弱く認識する(FACSによるMFIは、MAb 7C10の200に比較して50))を、コントロールアイソタイプとして添加した。細胞増殖は、細胞によるトリチウムチミジンの取込みをβカウンターで追跡することにより推定する。結果は、増殖指数として表わす。図5に提示したデータは、IGF1がMCF−7細胞の増殖を用量依存的に刺激し得ることを示している。陽性コントロールとして使用したMAb αIR3は、IGF−Iによって誘導されるMCF−7細胞の増殖を完全に阻害する。同様に、MAb 7C10は、IGF−Iによって誘導されたMCF−7細胞の増殖を有意に阻害する。最後に、アイソタイプコントロールとして用いたMAb 7G3は、予想通りに、MCF−7細胞のインビトロの腫瘍細胞増殖に影響なしにうまくいく。
【0191】
インビボの選択は、確立された腫瘍モデルにおいて実施した。これを行なうため、ヌードマウスは、マウスモデルにおいて腫瘍を得るために不可欠な徐放性エストロゲンの皮下移植を受けた。エストロゲンの移植の24時間後、5.106のMCF−7細胞を、マウスの右側腹部の皮下に移植する。この細胞移植の5日後には、腫瘍は測定可能であり、6匹のマウスからなるバッチがランダムに形成される。マウスの処置は、週当たり2回、5ないし6週間にわたり、250μg/1回用量/マウスの用量で実施する。コントロール群では、マウスを、マウスコントロールアイソタイプと同様に処理する。図6Aに提示した結果は、抗体7C10により誘導される、非常に有意な腫瘍増殖阻害を示している。通常はIGF1の受容体のドメインの参照として用いられ、エストロゲン依存性の腫瘍の増殖に対し何らインビボで活性をもたないことが知られる、αIR3について得られるデータを参照した場合、この活性は特に予想外である(図6B参照)。同様に、マウスMAb 1H7由来の組換え抗体scFv−Fcで得られた結果に比較して(図6C参照)、MAb 7C10は、MCF−7細胞の増殖のインビボでの阻害においてはるかに効力がある。
【0192】
実施例4
ヒト肺癌細胞系におけるMK−0646及びリダフォロリムスの効果
要約:併用が提案された根拠は、MK−0646及びリダフォロリムスの各々が、組合された場合に、PI3キナーゼシグナリング経路を介して発癌シグナリングを阻害することにより作用すること、及び2つが組合された状態では、いずれかの薬剤が単独で作用するよりも有効な経路の阻害をもたらすことを示唆する観察に基づいている。また、カオ(Cao)ら著、「Cancer Research」、2008年も参照。
【0193】
手短に言えば、MK−0646(これは、IGF1Rを標的とするモノクローナル抗体である)、及び、リダフォロリムス(これは、mTOR阻害剤であり、またラパマイシン類似体である)は、各々、肺癌の治療用に現在開発中である。ラパマイシン類似体による治療は、AKTのリン酸化により測定される、AKTシグナリングのアップレギュレーションをもたらす。リダフォロリムスによるmTORの阻害は、腫瘍増殖停止を誘導し得るが、IRS−1により媒介されるネガティブフィードバックループを無効にし、結果としてAKTを活性化し、このことがその抗腫瘍活性の低減に関連づけられてきた。AKTの、このフィードバック活性化は、IGF1Rシグナリング経路によって媒介される。最近の臨床研究は、このフィードバックメカニズムによるAKTの活性化が、ラパマイシンを処置された患者におけるより短い進行期間(shorter time to progression)に関連づけ得ることを示唆している(クラフェシー(Cloughesy)ら著、「PLoS Medicine」、2008年)。さらに、mTORは、MK−0646の効力の重要なエンハンサーであると考えられ、したがって、リダフォロリムスをMK−0646と併用することでAKTのこのフィードバック活性化を阻止することが、PI3K経路の阻害、並びにMK0646の抗腫瘍活性を増強するために有益り得る。併用が提案された根拠は、一部は、上記の観察に基づいている。この可能性を研究するため、本発明者らは、提案された組合せを、一群の肺癌細胞系において検査した。本明細書において以下に詳述されるのは、MK−0646とリダフォロリムスとを含んでなる併用治療がPI3K経路の阻害及び細胞増殖を有意に増強するという仮説を支持するものである。同様に、提案された組合せは、エルロチニブ抵抗性、KRAS突然変異肺癌異種移植モデルにおいて、抗腫瘍活性の増強を実証した。
【0194】
(A)MK−0646+リダフォロリムス併用は、PI3K経路標的化を増強する:
方法:すべてのNSCLC細胞系は、ATCCから入手し、10% ウシ胎児血清(インビトロジェン(Invitrogen))と共に、RPMI 1640中に維持した。ウエスタンブロット分析では、細胞からの全タンパク質溶解物を、6ウエルプレート中で培養し、リダフォロリムス(10nM)若しくはMK−0646(10ng/ml)のいずれかで、又は組合せて4時間処置し、SDSゲルローディング色素(インビトロジェン)中に採取した。サンプルは、指示された全抗体又はホスホ特異抗体で、次に二次抗体(Cell Signaling Technology, CST)でウエスタンブロットし、次にスーパーシグナル(SuperSignal)化学発光基質(Pierce)とインキュベートした。次いでブロットを、コダック バイオマックス ライト フィルム(Kodak Biomax Light Film)に暴露させた。ERK、p−ERK(Thr202/Tyr204)、AKT及びp−AKT(Ser473)、IGF1R S6K&P−S6K(T389)、IRS1&P−IRS1(S302)、及びアクチンに対する抗体は、CSTから入手した.細胞周期分析は、H2122細胞にて、指示された化合物(上記参照)による24時間処理の後に実施した。百万個の細胞を、透過性化し、ヨウ化プロピジウム(PI)で、製造業者による記載の通り染色した(BD Pharmingen #550825)。PIは、DNA及びRNAの双方に結合するため、後者をリボヌクレアーゼ(RNアーゼA)による消化により除去した。フローサイトメトリーにより測定されるDNA含有量は、細胞周期について有用な情報を示し得る。細胞周期のG2及びM期にある細胞は、G0及びG1期にあるものの2倍のDNA含有量をもつ。S期にある細胞は、これらの両極端の間にあるDNA含有量をもつ。PIは、フローサイトメーターを使用し、562−588nmバンドパスフィルタを用いて、橙色の範囲内で検出される。
【0195】
分析:リダフォロリムスとMK−0646との併用は、ヒト肺癌細胞系で評価されてきた。双方の化合物は、PI3キナーゼシグナリング経路を介して発癌シグナリングを阻害することにより作用する。本明細書で実施された研究は、2つの薬剤がいずれかの薬剤単独よりもさらに有効な経路阻害をもたらすという他の報告を確認した。mTOR(TORC1複合体)の阻害が、二次的効果をもたらし、この結果、活性のあるリン酸化型のAKTが生じ、これにより腫瘍細胞の生存が促進されることが先に報告されている。重要なことには、MK−0646によるIGF1Rの阻害は、この効果を阻止する上で有効である。図1は、フィードバックループ現象を例示しており、2つの薬剤の併用が、この効果を打ち消し、発癌性PI3K経路の標的化を改善することを示している。mTOR阻害剤は、S6キナーゼの、及びしたがってタンパク質翻訳の有効な阻害剤である。しかしながら、S6キナーゼ阻害はまた、IGF1Rシグナリングを抑制するS6キナーゼ(S6K)により媒介されるネガティブフィードバックの廃止につながる。このフィードバックループを阻止することの効果は、IGF1Rシグナリングが高められることであり、活性のあるリン酸化型のAKT(AKT−P)のレベルの上昇につながり、これにより腫瘍生存が駆動される。リダフォロリムスに類似しているラパログ(rapalogue)RAD001による患者の治療は、結果としてAKT−Pの上昇をもたらす(オーライリー(O’Reilly,K.E.)ら著、「Cancer Research」、2006年、図1B)。ある研究者らは、このことが、ある状況において単一の薬剤として使用した場合の化合物の有効性を限定し得ることを示唆してきた(クラフェシー(Cloughesy)ら著、「PLoS Medicine」、2008年)。この効果がIGF1R活性に依存すると考えられることから、本発明者らは、mTORを、IGF−1R阻害剤であるMK−0646と組合せて使用することを提案した。今日までのところ、収集されたデータは先の知見と一致する。データは、非小細胞肺癌細胞系H2122を、リダフォロリムスとMK−0646との組合せと共に同時治療することが、シグナリングの阻止において、いずれかの薬剤単独よりもさらに有効であり(図1C、右)、S6Kのリン酸化(リダフォロリムスによる)及びAKTリン酸化(MK−0646による)の双方を妨げることを示唆している。併用はまた、細胞周期阻害及びこれらの腫瘍細胞のインビトロの生存の阻害においても、単一の薬剤よりもさらに有効である。併用によるH2122腫瘍細胞系の治療は、活発に分裂している細胞のパーセントの低減、及び死んだか又は死にかけている細胞の増加をもたらした(図1D)。
【0196】
(B)リダフォロリムス+MK−0646併用は、インビトロの効力を増大する。
MK−0646若しくはリダフォロリムス又は組合せの効力を評価するため、本発明者らは、軟寒天アッセイを用いて、一群中の肺腫瘍細胞系において、これらの阻害剤の存在下でアンカレッジ(anchorage)非依存性増殖を評価した。コロニー形成アッセイは、補充IGFの不在下で実施した。MK−0646若しくはリダフォロリムス又は組合せの効果は、9つの細胞系(5つの突然変異−KRAS;4つのwt−KRAS)で評価した。MK−0646単一薬剤の感受性は、1つのKRAS突然変異細胞系(H23)及び3つのKRAS野生型細胞系において観察された(図2)。IGF1Rの発現が低いH1703は、MK−0646には抵抗性であった。高レベルのIGF−1Rをもつ2つのKRAS突然変異細胞系(A549&H2122)では、単一薬剤の活性は限られたものにすぎなかったのに対し、併用は有意な増殖阻害を示した。一群中のほぼ全体の細胞系が、併用の利点を示した。KRAS状態と、併用に対する応答との有意な相互関係は何ら観察されなかった。
【0197】
方法:軟寒天アッセイは、96ウエルガラス底プレート(MatriCal)において行なった。細胞は、14%FBS及び0.3%(w/v)シープラーク(SeaPlaque)アガロース(Lonza Rockland,Inc)を補充したRMPI1640 100μL中で、各ウエル当たり3,000−9,000細胞の濃度で、0.8%アガロースを補充した同じ培地からなるボトムレイヤーの上に播種した。化合物は、アガロースを固化させた後に、補充した培地100μl中で添加した。細胞は、7−14日間インキュベートした後、ラバセル(LavaCell)(Active Motif)で一晩染色した。コロニーは、Isocyte(登録商標)レーザースキャニングサイトメータを用いて定量化した。MK−0646の、単独又は標準的な治療薬剤と組合せての、アンカレッジ非依存性増殖阻害能を、軟寒天コロニー形成アッセイにおいて評価した。RTK状態は、全タンパク質溶解物中で、P−RTKアレイ(R&D biosciences)を用いて、製造業者による記載の通り評価した。KRASにおける活性化突然変異は、公開された癌ゲノムデータベース(Sanger)から同定した。
【0198】
(C)A549肺異種移植モデルの組合せにおける、MK−0646及びリダフォロリムスの併用の利点。
リダフォロリムス及びMK−0646の組合せを、A549突然変異−KRAS異種移植モデル(A549 mutant−KRAS xenograft model)において評価した。MK−0646が、0.2又は2mpkのいずれかで、リダフォロリムス(0.1mpk)と組合せて投薬された場合、有意な抗腫瘍活性を示した。もっとも、MK−0646を20mpkで、リダフォロリムス(0.1mpk)と組合せて投薬すると、いずれかの薬剤単独に対し、統計的な有意性をもって腫瘍退縮を示した(図3)。同様の結果は、腫瘍重量を群間で比較した場合に観察された。MK−0646(20mpk)をリダフォロリムス(0.1mpk)と組合せて投薬されたマウスでの腫瘍重量には、いずれかの薬剤単独に対し、統計的に有意な減少があった(図3)。これらのデータを合わせれば、リダフォロリムスと組合された低い用量のMK−0646は腫瘍静止状態を誘導するのに対し、リダフォロリムスと組合された高用量のMK−0646(20mpk)は、腫瘍退縮を引き起こし得ることの、強力なインビボの証拠を提供する。また、腫瘍重量の有意な減少も、この組合せにおいて観察された(図4)。このモデルにおけるエルロチニブによる処置は、結果として何ら容易に評価し得る腫瘍増殖抑制を生じなかった(図5)。このモデルでのエルロチニブによる効力の欠如は、KRASにおける活性化突然変異がこのモデルに存在するとすれば、驚くべきことではない。結果として、データは、エルロチニブ耐性のNSCLCモデルにおける、MK−0646&リダフォロリムス併用の利益を証明している。
【0199】
方法:2.5x106のA549ヒトNSCLC細胞を、4−6週齢の無胸腺Nude−Foxnlnuマウス(Charles River Laboratories)の右側腹部の皮下に注射した。腫瘍が約300mm3のサイズ(長さ*幅*幅*0.5)に達した時、マウスを処置群に無作為化した。マウス(n=8/群)に、ビヒクルを週1回3週間にわたり(qwkx3)(20mM L−ヒスチジン、150mM NaCl、0.5% PS80 pH=6)、又は0.2若しくは2若しくは20mpkのMK−0646を腹腔内に、qwkx3で、或いはリダフォロリムス(0.1mg/kg)を、又はMK−0646と組合せて、3週間にわたり投薬した。動物を秤量し、腫瘍体積を、実験の間週2回、及び終了時に、キャリバーで測定した。腫瘍重量は、終了時に測定した。21日目、動物をCO2窒息により犠牲にした。マウスは、最後の投薬の24時間後に犠牲にした。犠牲にする時点で、組織サンプルを収集し、加工した。
【0200】
処理の終了時の相対腫瘍体積を、以下の表に表わす。負の値は、腫瘍の退縮を表わす。単一薬剤に比較して、有意な腫瘍増殖阻害が、全てのMK−0646&リダフォロリムスの併用群において観察された。
【0201】
【表2】
【0202】
実施例5
MK−0646エンハンサースクリーン
要約:多くの一連の証拠が、IGF1Rシグナリングの過剰活性化が、腫瘍進行と関連することを示唆している。IGF1R及びそのリガンドIGF1の双方は、しばしば、ヒト癌において過剰発現されており、不良な予後に関係づけられている(ミラー(Miller)及びイー(Yee)、2005年)。さらに、IGF1又はIGF1Rのいずれかの強制された過剰発現は、動物組織において、自然発生腫瘍の形成をもたらす(ジョーンズ(Jones)ら、2006年)。対照的に、IGF1Rノックアウトマウスから単離された線維芽細胞腫が、発癌遺伝子の過剰発現による形質転換に抵抗性であることから、低減したIGF1R機能は、腫瘍発生を防止し得る(セル(Sell)ら、1994年)。
【0203】
IGF−1R特異的抗体(MK−0646)を用いて、併用療法プロトコールにおいての有効性を増強し得るかどうかを確認するため、レンチウイルス媒介RNAiスクリーンを使用した。スクリーンの結果、37の標的が同定され、そのサイレンシングは、MK−0646に対する腫瘍細胞の感受性を有意に増強した。これらのエンハンサーの中には、PI3キナーゼシグナリングカスケードの4つの別個のポジティブレギュレータ(PI3KCA、PDPK1、AKT2、及びFRAP1/mTOR)を標的とするshRNAがあった。対照的に、その経路のネガティブレギュレータであるPTENを標的とするshRNAは、MK−0646に対する抵抗性を示した。これらのデータを合わせれば、PTENについてネガティブな腫瘍は、MK−0646に対し抵抗性を示し得る。さらに、データは、mTORi又はAKTi又はPI3KiなどのPI3K経路を標的とする阻害剤を、MK−0646と組み合せることが、MK−0646応答を実際に強化し得ることを示唆する仮説を支持している。
【0204】
結果:本発明者らは、レンチウイルス媒介RNAiスクリーニングを適用して、インスリン様増殖因子1受容体(IGF1R)mab、特にMK−0646に対する腫瘍細胞感受性のキナーゼレギュレータを同定した。この目的に向けて、480の別個のヒトキナーゼを標的とする1439のレンチウイルスshRNAベクターをスクリーンして、MK−0646の抗腫瘍細胞活性の強力なエンハンサーを同定した。
【0205】
種々の濃度のMK−0646(200μg/mLないし300ng/mL)を用いた9つの別個のスクリーンを、MK−0646及びキナーゼ標的レンチウイルスshRNAベクターを用いて実施した。スクリーンから、MK−0646エンハンサーの上位3%を代表する37のコンセンサスヒットのリストが同定された。図10参照。これらのヒットのうち顕著なものは、2つの別個のベクター、PDPK1,AKT2、及びFRAP1/mTORによる、標準ホスファチジルイノシトール3キナーゼ(PI3K)経路−PIK3CAの多数のメンバーを標的とするshRNAベクターであった。スクリーンからの2つの別々のコンセンサスヒット(CCRK及びNEK8)は、PI3K/AKT経路のレギュレータとして、既にsiRNAスクリーンにおいて同定されていた(ブレイス(Brace)ら、2006年参照)。またMK−0646エンハンサーのリストの中で注目すべきであるのは、Rasシグナリングカスケードの2つのメンバーを標的とするshRNAであった(2つの別個のベクターによるB−Raf及び2つの別個のベクターによるMAP2K1)。全てにおいて、上位37のshRNAヒットのうちの11は、IGF1R活性化に続いて活性化されるものと仮定された、2つの確立されたシグナリングカスケードのメンバーであるキナーゼを標的とする。図6参照。
【0206】
HT29結腸癌細胞によるコロニー形成に基づく、独立した増殖アッセイを実施して、上記で言及されたスクリーニングヒットを確認した。
【0207】
方法/分析−図6:新鮮なベクターによる感染に続き、細胞を5用量力価のMK−0646に暴露し、次に9日間にわたりコロニー形成させた。37のshRNAヒットの各々について、MK−0646に対する感受性の増強能を、空のベクターコントロール、並びにshRNAキナーゼベクターセットから無作為に選択された3つの「非ヒット」(”non−hit”)shRNAベクターと比較した。さらに、PI3K経路の多くのポジティブエフェクターがヒットとしてスコアされたことから、脂質ホスファターゼPTEN(PI3Kシグナリングの主要なネガティブレギュレータ)を効率的にサイレンスさせる別のベクターもまた試験した。PTENは、確立した腫瘍サプレッサーであり、PI3K活性の生成物を脱リン酸化することによって作用し、それ故下流キナーゼの活性化を妨げる。ベクターなし(MK−0646単独)及び処置なし(ウイルスなし及び薬剤なし)のコントロールもまた実施した。
【0208】
方法:480の個別の遺伝子、主としてキナーゼ、を標的とするレンチウイルスは、1439のレンチウイルスshRNAベクター(Kalypsis library,GNF)を、293T細胞において、インビトロジェン・レンチウイルスパッケージングシステム(Virapower packaging system,Invtrogen)により推奨されるパッケージングベクターと同時トランスフェクトすることにより、パッケージし生成した。HT−29細胞(n=500)を、384ウエルプレートにて、10% FBS(DMEM中)の存在下で培養した。翌日、細胞をパッケージされたウイルス10uLで感染させた。3日目、培地を除去し、細胞を1日回復させた。4日目、細胞をMK−0646(200μg/mLないし300ng/mL)で処置した。8日目、細胞増殖の量を、既に記載された通りにアラマーブルーで染色することにより評価した(クリングホッファー(Klinghoffer)ら、「Assay and Drug Development Technologies」、2008年)。
【0209】
方法−図7:HT29細胞は、1500細胞/ウエルで、6ウエルプレートに播種し、ウイルスに16時間暴露した。ウイルスを除去し、薬剤を含有しないか又はMK−0646の希釈物(0.05、0.1、0.5、1、又は10ug/ml)を含有する新鮮な培地で置き換えた。MK−0646は、アッセイの過程にわたり3日ごとに交換した。プレートは、薬剤添加の9日後にクリスタル紫で染色した。コロニー数及び面積は、AlphaImagerを用いたイメージングにより計算した。
【0210】
無作為に選ばれた3つのshRNAベクターの、空のベクター及びベクターなしのコントロールとの比較は、腫瘍細胞のMK−0646に対する感受性に何ら差異を示さなかった。対照的に、このアッセイにおいて評価し得た20スクリーニングのうち11は、腫瘍細胞増殖のMK−0646阻害に2倍以上の増強を生じる結果となった。図7参照。
【0211】
定量的リアルタイムPCR分析は、これらのベクターの10/11が、その標的を>50%、また7/11が>70%サイレンスさせたことを示し、観察された増強が意図されたサイレンシングによるものであったことを示唆している−図10。同様に、このアッセイの使用により、37のshRNAヒットのうち17が、薬剤の不在下で細胞毒性を生じる結果となり、MK−0646感受性化の評価が妨げられた。これらの毒性shRNAは、細胞が感染に続き、10用量力価のMK−0646に暴露されたことを除いて、プライマリースクリーンに類似の短時間(72時間)のアラマーブルーアッセイを用いて分析した。このアッセイの結果として、さらなる8つのshRNAが同定され、MK−0646に対する腫瘍細胞感受性の有意な(p<0.05)増強を実証した。図11参照。
【0212】
図7及び10を参照すれば、IGF媒介性の腫瘍増殖におけるPI3K経路シグナリングの主要な役割と一致して、コロニーアッセイによる上位3つのMK−0646エンハンサーは、PI3K、FRAP1/mTOR、及びNEK8を標的とするベクターであった(これら3つのベクターは、それぞれ86%、80%、及び70%の標的サイレンシングをもたらした)。
【0213】
さらに、72時間のアラマーブルーアッセイによる最強のエンハンサーは、推定上のPI3K経路レギュレータCCRKを標的とするベクターであった(64%の標的サイレンシング:示さず)。反対に、PTENを標的とするベクターに暴露された細胞は、MK−0646の抗増殖効果に対し抵抗性であり、処置されなかった細胞と同様の増殖を示した(図6)。Ras経路のメンバーを標的とするベクター、MAP2K1及びB−raf(それぞれ97%及び75%のサイレンシング)もまた確認された。図10参照。PI3K及びRASシグナリングカスケードのメンバーの間に広範囲のクロスレギュレーションが存在するとすれば、MAP2K1及びB−raf shRNAの感受性効果は、一部は、PI3Kシグナリングのクエンチングによることが仮定される。
【0214】
今日までのところ、同じ遺伝子を標的とする2つのshRNAが、プライマリースクリーンで増強を証明する場合(PIK3CA、B−raf、MAP2K1、BUB1、及びCSNK1A1)、2つのshRNAの一方は確認されるのに対し、他方は薬剤の不在下で実証された毒性をもつように見える。第2のshRNAの確認には、より短いアッセイフォーマットが必要であってよい。
【0215】
PTENの結果をさらに支持するため、アッセイを、PTEN shRNAの安定なインテグレーションを内部にもつ細胞上で実施した。これらを、ベクターコントロール、又はCSK及びMAP4K5を標的とするshRNA、を安定に発現する細胞に対し比較することを開始した。CSK及びMAP4K5を標的とするベクターは、プライマリースクリーンからのコンセンサスヒットであったが、MAP4K5のみをコロニーアッセイで確認した。新鮮なウイルスの適用の結果と同様に、PTEN shRNAを発現する細胞系は、コロニーアッセイによってMK−0646抵抗性を示したのに対し、CSK shRNAを発現する系は、ベクターコントロールと同様に応答し、MAP4K5 shRNAを発現する細胞は、薬剤に対し感受性化された。図8参照。
【0216】
方法−図8:PTEN、CSK、及びMAP4K5を標的とする安定なインテグレーションshRNAを内部にもつHT29細胞を、1500細胞/ウエルで、6ウエルプレートに播種した。翌日、何ら薬剤を含有しないか、又は0.01、0.1、1、10、又は100ug/mlのMK−0646を含有する新鮮な培地を、細胞に与えた。MK−0646は、アッセイの過程にわたり3日ごとに交換した。プレートは、薬剤添加の9日後にクリスタル紫で染色した。コロニー増殖は、上記記載の通り定量化した。MK−0646に対する応答は、上記で分析された各レンチウイルスベクターへの急性暴露に続いて観察されたものと同様であった。
【0217】
データ及び付随する観察から、PTENに機能喪失型突然変異のある腫瘍をもつ患者は、MK−0646による処置に対し不十分に応答し得ることが示唆される。かかる突然変異は癌において一般的であることから、本発明者らは、PI3Kを阻害することによりMK−0646抵抗性細胞が薬剤に対し再感受性化され得るかどうか判定することを試みて、追加のアッセイを実施した。
【0218】
方法−図9:ベクターコントロールの安定な発現があるか(赤色の線)(MK−0646に感受性)、又はPTEN shRNAの安定な発現がある(青色の線)(MK−0646に抵抗性)、HT29細胞を、PI3K、PDPK1、又はMELKを標的とするshRNAベクターで感染させた。(A)図6に示した本発明者らのアッセイと同様に、PI3K又はMELKを標的とするshRNAによる、ベクターコントロールHT29細胞の感染は、結果としてMK−0646に対し、各々強い及び中程度の感受性化を生じるが、一方PDPKlを標的とするshRNAは何ら効果をもたない。(B)MELK又はPDPK1による安定なPTEN、shRNA HT29細胞の感染は、PTEN欠失により引き起こされるMK−0646抵抗性の逆転には、何ら影響をもたない。対照的に、PI3K shRNAベクターによる感染は、この抵抗性を完全に逆転させる。
【0219】
分析:本発明者らの先の結果に一致して、PTEN shRNAを発現する細胞は、ベクターコントロールを発現する細胞に比較して、MK−0646に抵抗性であった。図9Aを参照すれば、本発明者らの先の実験と一致して、PDPK1ではないがPI3K又はMELKのいずれかを標的とするベクターによるコントロール細胞の感染は、細胞をMK−0646に対し感受性化した。PTEN欠失細胞では、MELK shRNAによる感染は、これらの細胞がMK−0646抵抗性のままであるため、何ら強い影響はなかった。PI3Kを標的とするshRNAによる感染は、しかしながら、MK−0646の効果に対し、PTEN shRNA系を再感受性化し得た−図9B。したがって、PI3Kを介したシグナリングが、IGF1R媒介性の腫瘍細胞増殖にとり律速となるように考えられる。この観察は、過剰活性PI3Kシグナリングをもつ患者は、抗IGF−1R薬剤による単一療法に対し不応答性でありえる、mTORなどのPI3K経路のメンバーの阻害剤による併用療法は、実際に有効であり得ることを示唆している。
【0220】
本発明の多数の実施態様を記載してきたが、基本的な例は、本発明によって包含される別の実施態様を提供するべく、変更されてもよい。それ故、本発明の範囲は、単なる例として記述された具体的な実施態様よりもむしろ、添付の特許請求の範囲によって提起されるべきであることが理解されるべきである。
【図1−A】
【図1−B】
【図1−C】
【図1−D】
【図2−A】
【図2−B】
【図3−A】
【図3−B】
【背景技術】
【0001】
ホスファチジルイノシトール−3−キナーゼ(PI3K)シグナリング経路は、多くの様々なタイプのヒトの悪性腫瘍において、癌細胞の増殖及び生存に重要である。グランビル(Granville CA)ら著、「Handicapping the Race to Develop Inhibitors of the Phosphoinositide 4−Kinase/Akt/Mammalian Target of Rapamaycin Pathway(ホスホイノシチド−4−キナーゼ/Akt/哺乳類ラパマイシン経路標的タンパク質の阻害剤開発競争のハンディキャッピング)、Clin Cancer Res」、2006年、第12巻、第3号、p.679−89参照。この経路は、上皮成長因子受容体及びインスリン様成長因子受容体などの、リガンド受容体相互作用からの上流のインプットを受け、哺乳類のラパマイシン標的タンパク質(mTOR)などの、下流のエフェクターを介してシグナリングする。mTORは、細胞周期進行及び多くの他の重要な細胞増殖プロセスにとり決定的なタンパク質の産生を調節する、極めて重要な下流エフェクター分子である。アブラハム(Abraham RT)及びギボンス(Gibbons,JJ)著、「The mammalian target of rapamycin signaling pathway:twists and turns in the road to cancer therapy(哺乳類のラパマイシンシグナリング経路標的タンパク質:癌療法への道における紆余曲折)、Clin Cancer Res」、2007年、第13巻、第11号、p.3109−14参照。
【0002】
PI3キナーゼ軸(PI3 Kinase axis)の調節異常は、ヒトの癌に共通しており、それらは、過活動性の成長因子受容体シグナリング、PI3Kの突然変異の活性化、PTEN腫瘍抑制因子の機能喪失、及びmTORキナーゼ活性の活性化をもたらす他のいくつかのメカニズムに起因する。臨床的には、PI3K軸の首尾よい薬理学的阻害は、PI3キナーゼの、上流の成長因子受容体と、下流エフェクター(例えばmTOR)とにフォーカスされてきた。現在、mTOR阻害剤が、進行性悪性腫瘍の患者に臨床上の利益を与え得ることを示す実質的な臨床的証拠がある。
【0003】
インスリン受容体ファミリーのチロシンキナーゼ受容体である、インスリン様成長因子1型受容体(IGF−1R)は、細胞増殖、分化に関与し、多くのタイプの癌において、悪性細胞の形質転換及び維持に重要な役割を果たす。バセルガ(Baserga,R)ら著、「Mini Review:The IGF−1R receptor in cancer biology(ミニレビュー:癌生物学におけるIGF−1R受容体)、Int.J.Cancer」、2003年、第107巻、p.873−77参照。IGF−1R及びそのリガンドIGF−2は、多くのタイプの進行癌において過剰発現され、リガンド刺激による受容体シグナリングが、癌細胞の増殖をインビトロにおいて促進する。重要なことに、IGF−1Rシグナリングは、PI3K軸に密に関連している。IGF−1R阻害は、強力な抗癌効果を前臨床研究において示しており、現在いくつかのIGF−1R阻害剤が臨床開発中である。
【0004】
mTORとIGF−1R阻害剤との組合せは、PI3K軸における上流及び下流の双方の分子標的を阻害することにより、相乗効果を提供し得る。mTORの阻害は、Akt発癌遺伝子を活性化するフィードバックループの活性化につながり得るものであり、このことは、インビトロの及びmTOR阻害剤を処置された患者から採取された腫瘍生検からの腫瘍細胞において、ホスホ−Aktのレベルの上昇として具現化する。サン(Sun,S−Y)ら著、「Priority Report:Activation of Akt and eIF4E survival pathways by rapamycin−mediated mammalian target of rapamycin inhibition(プライオリティレポート:ラパマイシン媒介性哺乳類ラパマイシン標的タンパク質阻害によるAkt及びeIF4E生存経路の活性化)、CANCER RES」、2005年、第65巻、第16号、p.7052−58;及びガードナー(Gardner,H)ら著、「Biomarker analysis of a phase II double−blind randomized trial of daily oral RAD001(everolimus)plus letrozole or placebo plus letrozole as neoadjuvant therapy for patients with estrogen receptor positive breast cancer(エストロゲン受容体陽性乳癌患者のためのネオアジュバント療法としての、RAD001(エベロリムス)プラスレトロゾール、又はプラセボプラスレトロゾールの、毎日の経口投与に関する第II相無作為化二重盲検試験のバイオマーカー分析)、San Antonio Breast Cancer Symposium(サンアントニオ乳癌シンポジウム)」、2007年、12月13−16日、San Antonio、TX、参照。このフィードバックループは、IGF−1R及びインスリン受容体基質を介したシグナリングを含み得るものであり、IGF−1R阻害剤によって阻害される。結果として、前臨床試験は、IGF−1R阻害剤とmTOR阻害剤との組合せが、相加的又は相乗的な抗腫瘍活性をインビトロでもたらすことを示してきた。最近、2つのグループが独自に、ヒト肉腫の異種移植モデルで、ラパマイシンと抗IGF−1R抗体とを組合せた結果を報告した。クルマシェヴァ(Kurmasheva RT)ら、ポスター:「Combination of CP−751871,a human monoclonal antibody against the IGF−1 receptor with rapamycin results in highly effective therapy for xenografts derived from childhood sarcomas(CP−751871(IGF−1受容体に対するヒトモノクローナル抗体)とラパマイシンとの併用は小児肉腫由来の異種移植に高度に有効な療法をもたらす)」、EORTC 2007、及びダルコ(Darko,IA)ら著、要約:「Evaluation of combined insulin−like growth factor receptor type I(IGF−1R) and mTOR pathway blockade in sarcoma xenograft models(肉腫異種移植モデルにおけるインスリン様成長因子1型受容体(IGF−1R)とmTOR経路遮断との併用の評価)」、AACR Annual Meeting(AACR年回)、2007年、p.4760を参照。これらの研究の1つにおいては、確立されたユーイング及び骨肉腫異種移植片の完全な抑制が観察されたが、別の研究では、併用することで少なくとも相加的な利益を伴う強力な抗腫瘍活性が観察された。
【発明の概要】
【0005】
発明の要旨
本発明は、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌を、mTOR阻害剤及び抗IGF−1R抗体を用いて治療する方法であって、ここで、mTOR阻害剤が、リダフォロリムス、エベロリムス、テムシロリムス、ラパマイシン類似体、又はそれらの組合せであり、抗IGF−1R抗体が、ダロツズマブ(dalotuzumab)、フィギツムマブ、シクスツムマブ、SHC717454、ロシュ(Roche)R1507、EM164、又はアムジェン(Amgen)AMG479である方法を提供する。
【図面の簡単な説明】
【0006】
【図1】リダフォロリムス+MK−0646の併用による発癌性PI3Kシグナリングの改善されたターゲティングを示す図である。MK−0646&リダフォロリムスによる併用療法は、PI3K経路の阻害を増大し、癌細胞増殖を阻止する。(A)ネガティブフィードバックループを説明するPI3キナーゼシグナリング経路;(B)mTOR阻害が患者の腫瘍でAkt−Pの上昇をもたらすことを示す公表データ;(C)インビトロのH2122細胞におけるリダフォロリムス(10nM)、MK−0646(10ug/ml)、又はそれらの組合せに応答した経路シグナリング;(D)Cに用いた濃度で示した処置で24時間処理された細胞における、細胞周期分布及び細胞死を示すFACSプロフィールデータ。
【図2】リダフォロリムス&MK−0646の併用がインビトロの効果を増大することを示す図である。肺癌細胞系を、軟寒天中で、MK−0646若しくはリダフォロリムス又はそれらの組合せの存在下で培養した。軟寒天コロニー形成は、蛍光色素(LavaCell)を用いて定量化し、コロニーの面積及び数は、画像入力及び解析プラットフォーム(Isocyte)を使用して計数した。A)96ウエルプレートにおける相対コロニー面積がプロットされている。併用は、A549&H2122細胞系において、有意に増殖阻害を増強した(p<0.02)。P−RTKアレイにより測定されるRTKの活性化状態、及びKRASにおいて活性化する突然変異を下に示す。B)9NSCLC細胞系における、リダフォロリムス(10nM)若しくはMK−0646(10ug/ml)又はそれらの組合せのいずれかに応答した相対的軟寒天コロニー形成がプロットされている。細胞系は、KRAS中の活性化突然変異に基づき分類した。
【図3】突然変異体−KRAS異種移植腫瘍におけるリダフォロリムス&MK−0646併用の抗腫瘍活性を示す図である。MK−0646&リダフォロリムスによる併用処置は、A549異種移植片増殖の阻止において有効である。有意な腫瘍増殖阻害が、多様な濃度のMK−0646&リダフォロリムスを併用した併用処置群で、二元配置分散(2way ANOVA)分析により評価した場合に観察された。
【図4】リダフォロリムスと併用した高濃度のMK−0646により統計的に低減された腫瘍重量を示す図である。マウスに、MK−0646(20mpk)を単独で又はリダフォロリムス(0.1mpk)と併用でのいずれかにより、3週間にわたり投薬処置した。腫瘍重量(上記参照)は、リダフォロリムスと併用した、より高用量のMK−0646で、統計的に小さく、腫瘍の退縮が強調された。
【図5】A549異種移植片モデルがエルロチニブ処置に抵抗性であることを示す図である。ビヒクルに比較して、エルロチニブ処置群では何ら有意な増殖阻害が観察されなかった。
【図6】IGF1受容体シグナル伝達を示す図である。ホスファチジルイノシトール3−キナーゼ(PIK3CA)経路及びRAS経路を標的とするスクリーニングヒットが、それぞれ赤又は青で囲まれている。
【図7】コロニーアッセイによるヒットの検証により、PI3K経路のレギュレータを標的とするshRNAがMK−0646の効力に強く影響を及ぼすことを実証している図である。コロニー数(A)及びコロニー面積(B)は、アルファ・イメージャー(Alpha Imager)でプレートをスキャンすることにより測定した。MK−0646の上位3つの強力なエンハンサーは、PI3K経路のエフェクター(赤色で強調)をサイレンスさせたshRNAであり、一方PTENを標的とするshRNAは、薬剤に対する抵抗性を伝達した。
【図8】HT29結腸癌細胞におけるPTENの安定なサイレンシングが、MK−0646に対する抵抗性を伝達することを示す図である。PTEN又はCSK又はMAP3Kを標的とする種々のshRNAを発現する安定なHT29細胞を生成し、MK−0646に対する感受性をコロニー形成アッセイにおいて試験した。PTENのサイレンシングにより、MK−0646による増殖阻害の低減が示された。
【図9】PI3Kのサイレンシングは、PTEN欠失HT29細胞をMK−0646の効果に対し再感受性化するが、PDPK1又はMELKは再感受性化しないことを示す図である。PTENのRNAi媒介ノックダウンは、MK−0646媒介増殖阻害に対し抵抗性を付与する(図7&8参照)。PTENノックダウン細胞の増殖は、MK−0646&PI3K RNAiによる、IGF−1R及びPIK3CAの併用阻害により阻止し得る。
【図10】PI3K及びRas経路キナーゼが、MK−0646エンハンサースクリーンからの上位のコンセンサスヒットのうちで傑出していることを示す図である。PI3K及びRas経路の標準的な及び推定上のキナーゼレギュレータが、各々赤及び青で強調されている。定量的PCR分析を行なって、標的サイレンシング効率を評価した。ヒットの確認は、図7及び8に記載した通りのコロニー形成アッセイ又は短期間の増殖アッセイを用いて実施した。
【図11】確認されたMK−0646感受性化ヒットを示す図である。ある用量設定の薬剤(titration of drug)への暴露に続く、コロニーアウトグロースアッセイ(>2倍)又は72時間のアラマー(Alamar)アッセイ(p<0.05)のいずれかにおいて、MK−0646に対する腫瘍細胞の感受性を増大させたスクリーニングヒットを下に示す。あるベクターは、薬剤の不在下での毒性のため、いずれのアッセイでも検査不能であったことに注目されたい。色分けは、図10に定義した通りである。
【図12】リダフォロリムス−ダロツズマブ併用に応答している患者のCT画像を示す図である。画像は、肝臓(図に示した通り)及び他の部位に転移した、エストロゲン受容体陽性乳癌をもつ56歳の女性からである。患者は、多数回の事前の化学療法及びホルモン療法の後に進行した。上部パネルは、肝臓の左葉中の大きい腫瘍を示しており、下部パネルは、ダロツズマブと併用したリダフォロリムスの第I相臨床試験による2サイクルの処置後の、著しくサイズの縮小した同腫瘍を示す。患者は部分寛解を達成しており、これは8ヵ月を超える実験的治療後に継続していた。
【0007】
発明の詳細な記載
絶え間ない研究の結果、本発明者らは、mTOR阻害剤又はその薬学的に許容される塩を抗IGF−1R抗体と組合せて使用することにより、相乗的に優れた抗癌活性を達成し得ることを見出しており、ここで、mTOR阻害剤は、リダフォロリムス、エベロリムス、テムシロリムス、ラパマイシン類似体、又はそれらの組合せであり、抗IGF−1R抗体は、ダロツズマブ、フィギツムマブ、シクスツムマブ、SHC717454、ロシュR1507、EM164、又はアムジェンAMG479である。本発明は特に、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌の治療に有用である。しかしながら、本発明は、様々な他の癌、例えば脳腫瘍、頚大脳癌(cervicocerebral cancer)、食道癌、甲状腺癌、小細胞肺癌、肺癌、胃癌、胆嚢/胆管癌、肝臓癌、膵臓癌、卵巣癌、絨毛上皮癌、子宮体癌、子宮頚癌、腎盂/尿管癌、膀胱癌、前立腺癌、陰茎癌、精巣癌、胎児性癌、ウィルムス癌、皮膚癌、悪性黒色腫、神経芽細胞腫、骨肉腫、ユーイング腫瘍、軟部肉腫、急性白血病、慢性リンパ球性白血病、慢性骨髄性白血病、及びホジキンリンパ腫の治療に有用であることも立証し得た。
【0008】
したがって、本発明は、mTOR阻害剤及び抗IGF−1R抗体を用いて、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌を治療する方法であって、ここで、mTOR阻害剤が、リダフォロリムス、エベロリムス、テムシロリムス、ラパマイシン類似体、又はそれらの組合せであり、抗IGF−1R抗体が、ダロツズマブ、フィギツムマブ、シクスツムマブ、SHC717454、ロシュR1507、EM164、又はアムジェンAMG479である、該方法に関する。
【0009】
本発明の1つの実施態様においては、mTOR阻害剤は、リダフォロリムスである。
【0010】
本発明の別の実施態様においては、抗IGF−1R抗体は、少なくとも1つの非ヒト由来の重鎖相補性決定領域(CDR)と、少なくとも1つの非ヒト由来の軽鎖相補性決定領域(CDR)とを含んでなり、ここで、IGF−1Rに結合する前記抗体は、以下:(a)IGF−1Rには結合するが、IRには結合しない;(b)インスリン受容体とインスリン成長因子受容体とを含んでなるハイブリッド受容体(IR/IGF−1Rハイブリッド−R)には結合するが、単独のIRには結合しない;(c)ヒトIGF−1Rと、IGF−1及び/又はIGF−2との間の結合を阻害する;(d)前記ハイブリッド−R及びその天然のリガンド(好ましくは本明細書ではIGF1及び/又はIGF2及び/又はインスリンとして示されている)と、100nM未満の阻害定数及び/又はIC50で結合する;(e)前記IGF−1Rのチロシンキナーゼ活性を特異的に阻害する;(f)前記ハイブリッド−Rのチロシンキナーゼ活性を特異的に阻害する;(g)前記ハイブリッド−Rについて、10nM以下の結合親和性を有する;(h)IGF−1R発現をダウンレギュレートする;(i)ハイブリッド−R発現をダウンレギュレートする;(j)インビボの腫瘍増殖を阻害する、からなる群より選択される特性の少なくとも1つを有する。
【0011】
本発明の1つのクラスにおいては、重鎖CDRは、配列番号4、5、又は6からなる群より選択されるアミノ酸配列を含んでなり、かつ軽鎖CDRは、配列番号1、2、又は3からなる群より選択されるアミノ酸配列を含んでなる。
【0012】
本発明の別のクラスにおいては、ヒト化抗体、又はその機能性フラグメントの1つは、配列番号7又は8からなる群より選択されるアミノ酸配列を含んでなる軽鎖か、或いは配列番号9、10、又は11からなる群より選択されるアミノ酸配列を含んでなる重鎖を含んでなる。
【0013】
本発明の別のクラスにおいては、抗IGF−1R抗体は、ダロツズマブである。
【0014】
本発明の別の実施態様においては、mTOR阻害剤は、リダフォロリムスであり、かつ抗IGF−1R抗体は、ダロツズマブである。
【0015】
本発明の別の実施態様においては、mTOR阻害剤は、10mgないし40mgの用量で投与される。本発明の1つのクラスにおいては、リダフォロリムスは、1週間に5回投与される。
【0016】
本発明の別の実施態様においては、抗IGF−1R抗体は、10mg/kgの用量で静脈内投与される。本発明の1つのクラスにおいては、抗IGF−1R抗体は、週1回投与される。本発明の別のクラスにおいては、抗IGF−1R抗体は、2週間に1回投与される。
【0017】
mTOR阻害剤及び抗IGF−1R抗体は、同時に、別々に、又は逐次に投与するために調製し得る。
【0018】
上記に示した好ましい実施態様についての記載は、別に指定されない限り、特定の及び好ましい群の全ての組合せを含むものとする。本明細書において使用される用語の意味は、以下に記載されており、本発明は以下にさらに詳細に記載される。
【0019】
用語「同時」は、本明細書で言及する場合、本発明の医薬品が、時間的に同時に投与されることを意味する。
【0020】
用語「別々」は、本明細書で言及する場合、本発明の医薬品が、共通の治療スケジュールの過程の間の別々の時間に投与されることを意味する。
【0021】
用語「逐次」は、本明細書で言及する場合、1つの医薬品の投与に続いて、別の医薬品の投与がなされることを意味し;第2の医薬品は、1つの医薬品の投与後に、第1の医薬品の実質的に直後に投与してもよく、又は第2の医薬品を、第1の医薬品の有効期間の後に投与してもよく;また有効期間は、第1の医薬品の投与からの最大利益の実現のために与えられる時間の量である。
【0022】
用語「癌」は、本明細書で言及する場合、種々の肉腫及び癌腫を包含し、かつ固形癌及び造血器癌を包含する。固形癌は、本明細書で言及する場合、例えば、脳腫瘍、頚大脳癌、食道癌、甲状腺癌、小細胞肺癌、非小細胞肺癌、乳癌、子宮内膜癌、肺癌、胃癌、胆嚢/胆管癌、肝臓癌、膵臓癌、結腸癌、直腸癌、卵巣癌、絨毛上皮癌、子宮体癌、子宮頚癌、腎盂/尿管癌、膀胱癌、前立腺癌、陰茎癌、精巣癌、胎児性癌、ウィルムス腫瘍、皮膚癌、悪性黒色腫、神経芽細胞腫、骨肉腫、ユーイング腫瘍、軟部肉腫を包含する。一方、造血器癌は、例えば、急性白血病、慢性リンパ球性白血病、慢性骨髄性白血病、赤血球増加症、悪性リンパ腫、多発性骨髄腫、及びホジキンリンパ腫、非ホジキンリンパ腫を包含する。
【0023】
用語「癌の治療」は、本明細書で言及する場合、癌の症例に抗癌剤を投与して、その症例の癌細胞の増殖を阻害するようにすることを意味する。好ましくは、治療の結果として、癌増殖の退縮を生じるか、又はすなわち、それにより検出可能な癌のサイズが縮小される。さらに好ましくは、治療は癌の完全な消失をもたらす。
【0024】
mTOR阻害剤
現在臨床開発中のmTOR阻害剤は、ラパマイシンの構造類似体である。本発明のmTOR阻害剤は、リダフォロリムス、テムシロリムス、エベロリムス、ラパマイシン類似体、及びそれらの組合せを包含する。
【0025】
リダフォロリムスは、AP 23573、MK−8669、及びデフォロリムスとしても知られ、インビトロの広範囲のヒト腫瘍細胞系において、またヒト腫瘍細胞系を利用したマウス腫瘍異種移植片モデルにおいて抗増殖活性をもつ、ラパマイシンの独特の非プロドラッグ類似体である。リダフォロリムスは、進行癌患者に投与されてきており、現在、進行した軟部組織又は骨肉腫の患者における研究を含め、様々な進行性悪性腫瘍について臨床開発中である。今までのところ、これらの試験は、リダフォロリムスが、予測可能かつ管理可能な有害事象プロフィールをもち、全般的に充分に許容されること、及び広範囲の癌において抗腫瘍活性をもつことを証明してきた。リダフォロリムスの記載及び調製法は、アリアド・ジーン・セラピューティクス・インク(Ariad Gene Therapeutics,Inc.)に対する米国特許第7,091,213号に記載されており、これは、その記載全体が本明細書に援用される。
【0026】
テムシロリムスは、トリセル(Torisel)(登録商標)としても知られ、現在、腎細胞癌の治療用に市販されている。テムシロリムスの記載及び調製法は、アメリカン・ホーム・プロダクツ・コーポレーション(American Home Products Corporation)に対する米国特許第5,362,718号に記載されており、これは、その記載全体が本明細書援用される。
【0027】
エベロリムスは、サーティカン(Certican)(登録商標)又はRAD001としても知られ、ノバルティス(Novartis)から市販されており、ラパマイシン(シロリムス)と比較して、有機溶媒中でより優れた安定性及び増強された溶解性を有するとともに、副作用が少なくより好ましい薬物動態を有する。エベロリムスは、免疫抑制レジメンの効力を高めるため、シクロスポリンのマイクロエマルジョン製剤(ネオラール(Neoral)(登録商標)、ノバルティス)と併用されてきた。
【0028】
本発明のmTOR阻害剤はまた、種々の結晶、アモルファス物質、薬学的に許容される塩、水和物、及び溶媒和物として存在してもよい。さらに、本発明のmTOR阻害剤は、プロドラッグとして提供してもよい。一般に、かかるプロドラッグは、本発明のmTOR阻害剤の機能性の誘導体であり、これは、生体によって必要とされる化合物へ容易に変換し得る。したがって、本発明の種々の癌の治療法においては、用語「投与」は、特定の化合物の投与のみならず、患者への投与後に生体内で特定の化合物に変換し得る化合物の投与も包含する。適切なプロドラッグ誘導体の選択及び製造のための通常の方法は、例えば、バンガード(H.Bumdgaard)編、「Design of Prodrugs(プロドラッグの設計)」、Elsevier、1985年、に記載されており、これは、本明細書において参照され、かつその記載全体が本明細書の一部として本明細書に含まれる。化合物の代謝産物は、該化合物を生物学的環境内に置くことにより産生される活性化合物を包含してもよく、これらは本発明の化合物の範囲内にある。
【0029】
抗IGF−1R抗体
本発明の抗IGF−1R抗体は、単離された抗体か、又はその機能性のフラグメントであり、ここで、前記抗体又はその前記フラグメントは、ヒトインスリン様増殖因子I受容体に特異的に結合可能であり、かつ必要であれば、好ましくはさらに、リガンドであるIGF1及び/又はIGF2が、IGF−1Rへ結合するのを阻害することができ、及び/又は少なくとも1つのリガンドが、前記IGF−1R受容体へ結合するのに付随するシグナリングカスケードを特異的に阻害し得る。本発明のIGF−1R抗体は、特異的にIGF−1Rに結合し得る、モノクローナル及び/又はポリクローナル抗体を包含する。本発明の抗IGF−1R抗体は、ダロツズマブ、フィギツムマブ、シクスツムマブ、SHC717454、ロシュR1507、EM164、及びアムジェンAMG479を包含する。
【0030】
ダロツズマブは、配列番号1、2、又は3のアミノ酸配列のCDRから選択される少なくとも1つの相補性決定領域CDRか、又はその配列が、最適アラインメントの後に、配列番号1、2、又は3の配列と、少なくとも80%、好ましくは85%、90%、95%、及び98%の同一性をもつ少なくとも1つのCDRを含んでなる、軽鎖を含んでなること、或いは、配列番号4、5、及び6のアミノ酸配列のCDRから選択される少なくとも1つのCDRか、又はその配列が、最適アラインメントの後に、配列番号4、5、及び6の配列と、少なくとも80%、好ましくは85%、90%、95%、及び98%の同一性をもつ少なくとも1つのCDRを含んでなる、重鎖を含んでなること、により特徴づけられる。前記抗IGF−1R抗体を製造及び使用する方法は、米国特許第7,214,444号に記載されており、その記載全体が本明細書に援用される。
【0031】
本明細書においては、用語「結合すること(to bind)」及び「結合すること(to attach)」は、同じ意味をもち、互に交換可能である。
【0032】
本明細書においては、抗体化合物又はその配列に結合した、用語であるポリペプチド、ポリペプチド配列、ペプチド、及びタンパク質は、互に交換可能である。
【0033】
本発明は、天然型の抗体には関係しないこと、すなわち、それらは、その天然の環境中にあるのはなく、天然の供給源から精製により単離又は入手し得たものであるか、或いは遺伝的組み換えによるか、又は化学合成によって得られたものであること、及びしたがって、これより先に記載される、非天然のアミノ酸を含有し得ることが理解されるべきである。
【0034】
CDR領域又はCDRにより、カバット(Kabat)らにより定義された、免疫グロブリンの重及び軽鎖の超可変領域を示すことが意図されている(Kabat et al、「Sequences of proteins of immunological interest(免疫学的に興味深いタンパク質の配列)、第5版」、米国保健社会福祉省、NIH、1991年及びその後の版)。3つの重鎖CDR及び3つの軽鎖CDRが存在する。用語CDR又は複数のCDR(CDRs)は、本明細書では、抗体の抗原若しくはそれを認識するエピトープへの、親和性による結合に役割を果たしているアミノ酸残基の大半を含有するこれらの領域の、場合により、1つ若しくはいくつか、又はさらに全てを示すために使用する。
【0035】
本発明の意味においては、2つの核酸又はアミノ酸配列の間の「同一性のパーセント」により、最良のアラインメント(最適アラインメント)後に得られた、比較されるべき2つの配列間のヌクレオチド又は同一のアミノ酸残基のパーセントを示すことが意図され、このパーセントは、純粋に統計的であり、2つの配列の間の差異は、ランダムに、かつその全長にわたり分布している。2つの核酸又はアミノ酸配列間の比較は、慣例的に、それらを最適な方法で整列させた後、これらの配列を比較することにより実施され、前記比較は、セグメントごとに、又は「比較ウィンドウ」ごとに行ない得る。比較のための配列の最適アラインメントは、手作業によるものに加えて、スミス(Smith)及びウォーターマン(Waterman)の局所ホモロジーアルゴリズム[「Ad.App.Math.」、1981年、第2巻、p.482]によるか、ネドルマン(Neddleman)及びヴンシュ(Wunsch)の局所ホモロジーアルゴリズム[「J.Mol.Biol.」、1970年、第48巻、p.443]によるか、ピアソン(Pearson)及びリプマン(Lipman)の同様の検索法[「Proc.Natl.Acad.Sci.USA」、1988年、第85巻、p.2444]によるか、又はこれらのアルゴリズムを用いたコンピューターソフトウェア(ウィスコンシン・ジェネティクス・ソフトウェア・パッケージ(Wisconsin Genetics Software Package)、ジェネティクス・コンピュータ・グループ(Genetics Computer Group)、575サイエンス(Science)Dr.、Madison、WI)のGAP、BESTFIT、FASTA、及びTFASTA又はBLASTN若しくはBLASTP比較ソフトウェア)により実行し得る。
【0036】
2つの核酸又はアミノ酸配列の間の同一性のパーセントは、最適な方法で整列されたこれら2つの配列を比較することにより決定し、これにおいて、比較されるべき核酸又はアミノ酸配列は、これら2つの配列間の最適アラインメント用の参照配列に比較して、付加又は欠失を含んでいてもよい。同一性のパーセントは、その位置について核酸又はアミノ酸残基が2つの配列間で同一である、同一位置の数を測定すること、同一位置数を比較ウィンドウ内の全位置数で割ること、及び得られた結果を100倍して、これら2つの配列間の同一性のパーセントを得ることにより計算する。
【0037】
例えば、http://www.ncbi.nlm.nih.gov/gorf/bl2.htmlのサイトにおいて利用可能なBLASTプログラム、「BLAST2sequences」(タツソヴァ(Tatusova)ら、「Blast 2 sequences−a new tool for comparing protein and nucleotide sequences(Blast2配列−タンパク質及びヌクレオチド配列比較のための新規なツール)」FEMS Microbiol Lett.第174巻、p.247−250)を使用可能であり、用いたパラメータは、デフォルトによって与えられたものであり(特に、パラメータである、「オープンギャップペナルティ」:5、及び「伸長ギャップペナルティ」:2について、選択されたマトリックスは、例えば、プログラムによって推奨されたマトリックス「BLOSUM 62」である)、比較されるべき2つの配列間の同一性のパーセントは、このプログラムにより直接計算される。
【0038】
参照アミノ酸配列と少なくとも80%、好ましくは85%、90%、95%、及び98%の同一性をもつアミノ酸配列により、参照配列に比較して、ある修飾、特に、少なくとも1つのアミノ酸の欠失、付加、若しくは置換、トランケーション(先端の切断)、又は伸長を有するものが好ましい。1つ以上の連続又は不連続のアミノ酸の置換の場合、置換は、好ましくは、置換されたアミノ酸が「同等(equivalent)」のアミノ酸に置き換えられる。「同等のアミノ酸(equivalent amino acids)」という表現は、本明細書では、基本構造をもつアミノ酸の1つによって置換され得るが、しかしながら、対応する抗体の生物活性を本質的に修飾することはない、任意のアミノ酸を示すことを意図しており、例えば、後に特に実施例において定義されるものなどである。
【0039】
これらの同等のアミノ酸は、それらが置換するアミノ酸との、その構造上の相同性か、又は実行可能な種々の抗体間の生物活性の比較試験の結果のいずれかに依存して決定し得る。
【0040】
単なる例としては、対応する修飾された抗体の生物活性の大きな修飾をもたらすことなく実行し得る置換の可能性が挙げられる。それ故、ロイシンをバリン又はイソロイシンで、アスパラギン酸をグルタミン酸で、グルタミンをアスパラギンで、アルギニンをリジンで、などの置換が可能であり、逆の置換が、同じ条件下で必然的に予想される。
【0041】
本発明の抗体は、好ましくは、特異的なモノクローナル抗体であり、特に、マウス、キメラ、又はヒト化起源のものであり、これらは当業者に周知の標準的な方法により取得し得る。
【0042】
一般に、モノクローナル抗体又はその機能性フラグメント、特にマウス起源のものの調製には、特に、マニュアル「Antibodies(抗体)」(ハーロー(Harlow)及びレーン(Lane)著、「Antibodies :A Laboratory Manual(抗体:実験室マニュアル)」、Cold Spring Harbor Laboratory、Cold Spring Harbor NY、1988年、p.726)に記載されている技術、又はコーラー(Kohler)及びミルスタイン(Milstein)(Nature、1975年、第256巻、p.495−497)により記載されたハイブリドーマからの調製技術を参照し得る。
【0043】
本発明のモノクローナル抗体は、例えば、IGF−1R受容体か又は本発明の前記モノクローナル抗体によって特異的に認識されるエピトープを含有するそのフラグメントの1つに対し免疫化された、動物細胞から得られる。前記IGF−1R受容体、又はその前記フラグメントの1つは、通常の実施の方法に従い、IGF−1R受容体をコードするcDNA配列中に含まれる核酸配列から出発する遺伝的組換えによるか、又はIGF−1R受容体のペプチド配列中に含まれるアミノ酸配列から出発するペプチド合成により、特別に合成し得る。
【0044】
本発明のモノクローナル抗体は、例えば、IGF−1R受容体か又は本発明の前記モノクローナル抗体によって特異的に認識されるエピトープを含有するそのフラグメントの1つがその上に予め固定されている、アフィニティカラム上で精製し得る。さらに具体的には、前記モノクローナル抗体は、プロテインA及び/又はG上でのクロマトグラフィー後に、残留するタンパク質不純物、並びにDNA及びLPSを除去する目的で、イオン交換クロマトグラフィーを続けるか、又は続けずに精製してもよく、これに、二量体又は他の多量体の存在による潜在的な凝集体を除去する目的で、セファロース(Sepharose)ゲル上での排除クロマトグラフィーを続けるか、又は続けなくてもよい。なおさらに好ましい方法では、これらの技術の全てを同時に、又は逐次に使用し得る。
【0045】
キメラ又はヒト化された抗体は、同様に本発明の抗体に包含される。
【0046】
キメラ抗体により、所与の種の抗体に由来する天然の可変(軽鎖及び重鎖)領域を、前記所与の種に対して異種の抗体の軽鎖及び重鎖定常領域と組合せて含有する抗体を示すことが意図されている。
【0047】
本発明の抗体又はそのキメラ型のフラグメントは、遺伝的組換えの技術を用いることにより調製し得る。例えば、キメラ抗体は、プロモータと、本発明の非ヒトの、特にマウスのモノクローナル抗体の可変領域をコードする配列と、及びヒト抗体の定常領域をコードする配列とを含有する、組換えDNAをクローニングすることにより産生し得る。かかる組換え遺伝子によりコードされた本発明のキメラ抗体は、例えば、マウス−ヒトキメラであり、この抗体の特異性は、マウスDNA由来の可変領域により決定され、そのアイソタイプは、ヒトDNA由来の定常領域によって決定される。キメラ抗体の調製法については、例えば、ヴァーホイエン(Verhoeyn)らの文献(「BioEssays」、1988年、第8巻、p.74)を参照し得る。
【0048】
ヒト化抗体により、非ヒト起源の抗体に由来するCDR領域を含有する抗体を示すことが意図され、抗体分子の他の部分は、一つの(又は数個の)ヒトの抗体に由来する。さらに、骨格のセグメント(FRと称する)のいくつかの残基を、結合の親和性を保持する目的で、修飾してもよい(ジョーンズ(Jones)ら著、「Nature」、1986年、第321巻、p.522−525;ヴァーホイエン(Verhoeyn)ら著、「Science」、1988年、第239巻、p.1534−1536;リーチマン(Riechman)ら著、「Nature」、1988年、第332巻、p.323−327)。
【0049】
本発明のヒト化抗体又はそのフラグメントは、当業者に周知の技術により調製し得る(例えば、シンガー(Singer)ら著、「J.Immun.」、1992年、第150巻、p.2844−2857;マウンテン(Mountain)ら著、「Biotechnol.Genet.Eng.Rev.」、1992年、第10巻、p.1−142;又はベビントン(Bebbington)ら著、「Bio/Technology」、1992年、第10巻、p.169−175、などの文献に記載されたもの)。本発明のかかるヒト化抗体は、好ましくは、インビトロの診断法、又はインビボの予防的及び/又は治療的処置における使用が好ましい。
【0050】
本発明の抗体の機能性フラグメントにより、特に抗体フラグメント、例えば、Fv、scFv(scは単鎖)、Fab、F(ab’)2、Fab’、scFv−Fcフラグメント、又はダイアボディ、或いは、例えば、ポリ(エチレン)グリコール(「PEG化」)(Fv−PEG、scFv−PEG、Fab−PEG、F(ab’)2−PEG、又はFab’−PEGと呼ばれるフラグメント)(「PEG」はポリ(エチレン)グリコール)などのポリ(アルキレン)グリコールの付加による化学修飾によるか、又はリポソーム内への取込みによって、半減期が増大しているであろう、任意のフラグメントであって、前記フラグメントは、本発明の配列番号1、2、3、4、5、又は6の特徴的なCDRの少なくとも1つを有し、かつ特に、抗体IGF−1R受容体を認識及び結合し、必要であればIGF−1R受容体の活性を阻害する能力など、それが由来する抗体のたとえ部分的な活性であっても、一般的な方法で及ぼすことができる前記フラグメント、を示すものとする。
【0051】
好ましくは、前記機能性フラグメントは、それらが由来する抗体の重又は軽可変鎖の、部分的な配列からなるか、又は含んでなり、前記部分的な配列は、IGF−1R受容体について、それが由来する抗体と同様の結合特異性及び、十分な親和性(好ましくは、それが由来する抗体の親和性の少なくとも1/100、さらに好ましい態様では、少なくとも1/10に等しい充分な親和性)を保持するのに充分である。
【0052】
かかる機能性フラグメントは、それが由来する抗体の配列の、最低5個のアミノ酸、好ましくは10、15、25、50、及び100個の連続したアミノ酸を含有することができる。
【0053】
好ましくは、これらの機能性フラグメントは、Fv、scFv、Fab、F(ab’)2、Fab’、scFv−Fc型、又はダイアボディのフラグメントであり、これらは一般に、それらが由来する抗体と同じ結合特性を有する。本発明によれば、本発明の抗体フラグメントは、例えば、ペプシン又はパパインなどの酵素による消化等の方法により、及び/又は化学的還元によるジスルフィド結合の切断により、上記記載の抗体などから出発して取得し得る。別の方法では、本発明に含まれる抗体フラグメントは、同様に当業者に周知の遺伝的組換え技術により、又はさもなければ、例えば、アプライド・バイオシステムズ(Applied Biosystems)社等により供給されるものなどの自動ペプチド合成装置によるペプチド合成によって取得し得る。
【0054】
さらに好ましい態様においては、本発明は、本発明の抗体、又はその機能性フラグメント、特に、遺伝的組換えにより、又は化学合成により取得された、キメラ又はヒト化抗体を含んでなる。
【0055】
好ましい実施態様においては、本発明の主題は、配列番号6の少なくとも1つのCDR、又は配列番号6の配列との最適アラインメント後に少なくとも80%の同一性を有する配列を含んでなることを特徴とする、本発明の抗体又はその機能性フラグメントの1つである。
【0056】
6つの短いCDR配列の中で、重鎖の3番目のCDR(CDRH3)は、サイズの変異性が大きい(高い多様性は、本質的に、それを生じるもとである遺伝子の配列のメカニズムに起因する)。これは、2アミノ酸程の短さであってもよいが、既知の最長のサイズは26である。機能上は、CDRH3は、一部は、抗体の特異性の決定に役割を果たしている(セガル(Segal)ら著、「PNAS」、1974年、第71巻、p.4298−4302;アミット(Amit)ら著、「Science」、1986年、第233巻、p.747−753;チョシア(Chothia)ら著、「J.Mol.Biol.」、1987年、第196巻、p.901−917;チョシア(Chothia)ら著、「Nature」、1989年、第342巻、p.877−883;ケイトン(Caton)ら著、「J.Immunol.」、1990年、第144巻、p.1965−1968;シャロン(Sharon)ら著、「PNAS」、1990年、第87巻、p.4814−4817;シャロン(Sharon)ら著、「J.Immunol.」、1990年、第144巻、p.4863−4869;カバット(Kabat)ら著、「J.Immunol.」、1991年、第147巻、p.1709−1719)。
【0057】
CDRのアミノ酸のうちのわずかなパーセントのものだけが、抗体の結合部位の構造に寄与することが知られているが、これらの残基は、非常に特異的な三次元的立体配座において維持されているはずである。
【0058】
さらに好ましい態様においては、本発明は、3つのCDR又は配列番号4、5、及び6の配列の3つのCDRの、少なくとも2つか、或いは3つのCDR又は配列番号4、5、及び6の配列との最適アラインメント後に各々少なくとも80%の同一性を有する配列の3つのCDRの、少なくとも2つを含んでなる、重鎖を含んでなることを特徴とする、本発明の抗体又はその機能性フラグメントの1つに関する。
【0059】
同様の好ましい実施態様においては、本発明の主題は、配列番号1、2、又は3の配列のCDRから選択される少なくとも1つのCDR、又は、その配列が、配列番号1、2、又は3の配列との最適アラインメント後に少なくとも80%の同一性を有するCDRを含んでなる、軽鎖を含んでなることを特徴とする、本発明の抗体又はその機能性フラグメントの1つである。
【0060】
さらに好ましい実施態様においては、本発明の主題は、3つのCDR又は配列番号1、2、及び3の配列の3つのCDRの、少なくとも2つか、或いは3つのCDR又は配列番号1、2、及び3の配列との最適アラインメント後に各々少なくとも80%の同一性を有する配列の3つのCDRの、少なくとも2つを含んでなる、軽鎖を含んでなることを特徴とする、本発明の抗体又はその機能性フラグメントである。
【0061】
さらに好ましい態様においては、本発明の抗体又はその機能性フラグメントの1つは、それが、配列番号4、5、及び6の配列の3つのCDR、又は配列番号4、5、及び6の配列との最適アラインメント後に各々少なくとも80%の同一性を有する配列の3つのCDRを含んでなる、重鎖を含んでなること、及びさらに、配列番号1、2、及び3の配列の3つのCDR、又は配列番号1、2、及び3の配列との最適アラインメント後に各々少なくとも80%の同一性を有する配列の3つのCDRを含んでなる、軽鎖を含んでなることを特徴とする。
【0062】
別の態様によれば、本発明の主題は、ヒトインスリン受容体IRに結合しないか、又は有意な態様では結合しないことを特徴とする、本発明の抗体又はその機能性フラグメントの1つである。
【0063】
好ましい態様においては、本発明の前記機能性フラグメントは、フラグメントであるFv、scFv、Fab、F(ab’)2、Fab’、scFv−Fc、又はダイアボディ、或いは、化学修飾、特にPEG化によるか、又はリポソーム内への取込みによって半減期が増大しているであろう、任意のフラグメントから選択される。
【0064】
別の態様によれば、本発明は、本発明のモノクローナル抗体を分泌することが可能なマウスハイブリドーマ、特に、2001年9月19日に、Centre National de Culture De Microorganisme(CNCM、国立微生物培養センター)(パスツール研究所、パリ、フランス)に、番号I−2717で寄託されたマウス由来のハイブリドーマに関する。
【0065】
抗体が、2001年9月19日に、CNCMに、番号I−2717で寄託されたハイブリドーマにより分泌されることを特徴とする、本明細書で7C10と称されるモノクローナル抗体、又はその機能性フラグメントの1つは、もちろん本発明の一部である。
【0066】
「ダロツズマブ」、「h7C10」、「MK−0646」、又は「F50035」は、IGF−1Rに結合すること、並びにIR/IGF−1ハイブリッド受容体に結合することを特徴とするヒト化抗体を記載するべく区別なく使用される。かかる抗体は、例えば、米国特許出願第10/735,916号(US20050084906)(これは、PCT/FR03/00178及び/又はUS20050249730の一部継続出願である)において記載された抗体を包含してもよく、ここで前記のものは、ヒト化抗体又はそのフラグメントであり、かつ軽鎖及び/又は重鎖を含んでなり、これにおいて前記軽鎖及び/又は重鎖の骨格セグメントFR1ないしFR4は、それぞれヒト抗体の軽鎖及び/又は重鎖の骨格セグメントFR1ないしFR4に由来する。ヒト化抗体は、非ヒト由来であり、かつ配列番号1、2、又は3からなる群より選択されるアミノ酸配列を有する少なくとも1つ以上の相補性決定領域を含んでなる、少なくとも1つの軽鎖と、配列番号4、5、又は6からなる群より選択されるアミノ酸配列を有する少なくとも1つ以上の相補性決定領域を含んでなる、少なくとも1つの重鎖とを含んでなってもよい。軽鎖は、配列番号7又は8のうちの1つに示した1つ以上のアミノ酸配列か、又は配列番号7又は8との最適アラインメントの後に少なくとも80%の同一性を有する配列を含んでなってもよい。同様に、重鎖は、配列番号9、10、又は11のうちの1つに示した1つ以上のアミノ酸配列か、又は配列番号9、10、又は11との最適アラインメントの後に少なくとも80%の同一性を有する配列を含んでなってもよい。
【0067】
特定の実施態様においては、本発明は、本発明のマウス抗体又はその機能性フラグメントの1つに関し、前記抗体は、配列番号12のアミノ酸配列、又は配列番号12の配列との最適アラインメントの後に少なくとも80%の同一性を有する配列を含んでなる配列の軽鎖を含んでなること、及び/又は、配列番号13のアミノ酸配列、又は配列番号13の配列との最適アラインメントの後に少なくとも80%の同一性を有する配列を含んでなる配列の重鎖を含んでなることを特徴とする。
【0068】
フィギツムマブは、CP751,871としても知られ、ファイザー(Pfizer)による研究下にある完全ヒト抗体である。フィギツムマブの記載及び調製法は、アブジェニックス・インク(Abgenix,Inc)及びファイザー・インク(Phizer,Inc.)に対する米国特許第7,037,498号に記載されており、これはその記載全体が本明細書に援用される。
【0069】
シクスツムマブは、IMC A−12としても知られ、イムクローン(ImClone)による研究下にある完全ヒト抗体である。シクスツムマブの記載及び調製法は、米国特許公報第US2008/0025990号に記載されており、これはその記載全体が本明細書に援用される。
【0070】
SHC717454は、CP 751,871としても知られ、シェーリング・プラウ(Schering Plough)(現在はメルク・アンド・カンパニー・インク(Merck&Co.,Inc.))による研究下にある完全ヒト抗体である。SHC717454の記載及び調製は、シェーリング・コーポレーションに対する米国特許第7,217,796号に記載されており、これはその記載全体が本明細書に援用される。
【0071】
ロシュR1507は、ホフマン−ラロシュ(Hoffmann−LaRoche)による研究下にある。ロシュR1507の記載及び調製は、ホフマン−ラロシュに対する米国特許第7,378,503号に記載されており、これはその記載全体が本明細書に援用される。
【0072】
EM164は、イムノジェン(Immunogen)による研究下にある。EM164の記載及び調製は、イムノジェン・インクに対する国際特許公開 WO03/106621に記載されており、これはその記載全体が本明細書に援用される。
【0073】
アムジェンAMG479は、アムジェン・インクによる研究下にある。アムジェンAMG479の記載及び調製は、アムジェン・インク対する国際特許公開 WO06/069202に記載されており、これはその記載全体が本明細書に援用される。
【0074】
同様の特定の態様によれば、本発明は、本発明のキメラ抗体又はその機能性フラグメントの1つに関係し、前記抗体はさらに、マウスに対して異種の、特にヒトの抗体に由来する軽鎖及び重鎖定常領域含んでなること、及び好ましい態様においては、ヒト抗体に由来する軽鎖及び重鎖定常領域は、各々カッパ及びガンマ−1、ガンマ−2、又はガンマ−4領域であることを特徴とする。
【0075】
同様の特定の態様によれば、本発明は、本発明のヒト化抗体又はその機能性フラグメントの1つに関し、前記抗体は、軽鎖及び/又は重鎖を含んでなり、これにおいて前記軽鎖及び/又は重鎖の骨格セグメントFR1ないしFR4(以下の実施例12及び13、表5及び6において定義された通り)は、それぞれヒト抗体の軽鎖及び/又は重鎖の骨格セグメントFR1ないしFR4に由来することを特徴とする。
【0076】
好ましくは、本発明のヒト化抗体又はその機能性フラグメントの1つは、前記ヒト化抗体が、配列番号8のアミノ酸配列を含んでなる軽鎖を含んでなること、及び配列番号10又は11の、好ましくは配列番号11のアミノ酸配列を含んでなる重鎖を含んでなることを特徴とする。
【0077】
本発明において区別なく使用される用語である、核酸、核又は核酸配列、ポリヌクレオチド、オリゴヌクレオチド、ポリヌクレオチド配列、ヌクレオチド配列により、ヌクレオチドの厳密な結合を示すことが意図されており、これらは修飾されているか又は未修飾であり、フラグメント又は定義されるべき核酸の領域を与え、非天然のヌクレオチドを含有するか又は含有せず、また二本鎖DNA、一本鎖DNAにも、前記DNAの転写産物と同様に対応し得るものである。
【0078】
また本明細書では、本発明が、その天然の染色体環境、すなわち、天然の状態にあるヌクレオチド配列には関与しないことも理解されるべきである。本発明は、単離及び/又は精製された配列に関するものであり、すなわち、それらは、直接又は間接的に、例えばコピーにより選択されたものであって、その環境は、少なくとも部分的に修飾されたものである。それ故それは、同様に、本明細書では、例えば、宿主細胞による遺伝的組換えによって得られるか、又は化学合成によって得られる、単離した核酸を示すことが意図されている。
【0079】
好ましい配列との最適アラインメントの後に、少なくとも80%、好ましくは85%、90%、95%、及び98%の同一性を有する核酸配列により、該核酸配列が、参照核酸配列と比較して、ある種の修飾、特に、欠失、トランケーション、伸長、キメラ融合、及び/又は置換、とりわけ点置換などを有する核酸配列を示すことが意図されている。好ましくは、本発明は、その配列が参照配列と同じアミノ酸配列をコードしている配列に関与しており、このことは、遺伝コードの縮重か、又は、好ましくは、特に以下に定義される高ストリンジェント条件下で、参照配列と特異的にハイブリダイズし得る相補的配列につながる。
【0080】
高ストリンジェント条件下のハイブリダイゼーションは、温度条件及びイオン強度条件が、それらが相補的なDNAの2つのフラグメント間のハイブリダイゼーションを維持させるように選択されることを意味する。単なる例証として、上記記載のポリヌクレオチドフラグメントを定義することを目的とする高ストリンジェントなハイブリダイゼーション工程の条件は、好ましくは以下の通りである。
【0081】
DNA−DNA、又はDNA−RNAハイブリダイゼーションは、2工程で行なわれる:(1)5xSSC(1xSSCは、0.15M NaCl+0.015M クエン酸ナトリウム溶液に相当)、50% ホルムアミド、7% ドデシル硫酸ナトリウム(SDS)、10x デンハート(denhardt’s)、5% 硫酸デキストラン、及び1% サケ精子DNAを含有するリン酸バッファ(20mM、pH7.5)中で、42℃で3時間のプレハイブリダイゼーション;(2)プローブのサイズに依存した温度(すなわち、プローブサイズ>100ヌクレオチドに対し、42℃)において、20時間の実際のハイブリダイゼーションの後に、2xSSC+2% SDS中での、20℃で20分間の2回の洗浄、0.1xSSC+0.1% SDS中での、20℃で20分間の1回の洗浄。最後の洗浄は、プローブサイズ>100ヌクレオチドに対し、0.1xSSC+0.1% SDS中で、60℃で30分間行なった。上記記載のストリンジェンシーの高いハイブリダイゼーション条件は、ある規定されたサイズのポリヌクレオチドについては、当業者により、サンブルック(Sambrook)らの教示(「Molcular Cloning:a Laboratory manual(分子クローニング:実験室マニュアル)」、第2版、Cold Spring Harbor、1989年)に従って、より大きい又はより小さいサイズのオリゴヌクレオチドに適用し得る。
【0082】
投薬及び投与経路
本発明のmTOR阻害剤及び抗IGF−1R抗体については、様々な製剤形態を選択でき、その例は、錠剤、カプセル、粉末、顆粒又は液体などの経口製剤、溶液又は懸濁液などの無菌の液体製剤、坐剤、軟膏などを包含する。mTOR阻害剤は、薬学的に許容される塩として利用し得る。本発明のmTOR阻害剤及び抗IGF−1R抗体は、薬学的に許容される担体又は希釈剤と共に調製される。
【0083】
用語「薬学的に許容される塩」は、本明細書で言及される場合、通常の薬学的に許容される塩を意味する。例えば、化合物がヒドロキシル基か、又はカルボキシル基及びテトラゾリル基などの酸性基をもつ場合、ヒドロキシル基又は酸性基において塩基付加塩を形成してもよく;或いは化合物がアミノ基又は塩基性の複素環基をもつ場合には、アミノ基又は塩基性複素環基において酸付加塩を形成してもよい。
【0084】
塩基付加塩は、例えば、ナトリウム塩、カリウム塩などのアルカリ金属塩;カルシウム塩、マグネシウム塩などのアルカリ土類金属塩;アンモニウム塩;及び有機アミン塩、例えばトリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、プロカイン塩、N,N’−ジベンジルエチレンジアミン塩を包含する。
【0085】
酸付加塩は、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、過塩素酸塩などの無機酸塩;マレイン酸塩、フマル酸塩、酒石酸塩、クエン酸塩、アスコルビン酸塩、トリフルオロ酢酸塩などの有機酸塩;及びメタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩などのスルホン酸塩を包含する。
【0086】
用語「薬学的に許容される担体又は希釈剤」は、賦形剤[例えば、脂肪、蜜蝋、半固体及び液体ポリオール、天然又は硬化油など];水(例えば、蒸留水、特に注射用蒸留水など)、生理食塩水、アルコール(例えば、エタノール)、グリセロール、ポリオール、グルコース水溶液、マンニトール、植物油など);添加剤[例えば、増量剤、崩壊剤、結合剤、滑沢剤、湿潤剤、安定化剤、乳化剤、分散剤、保存剤、甘味剤、着色剤、調味剤又は芳香剤、濃縮剤、希釈剤、緩衝剤、溶媒又は可溶化剤、貯蔵効果を達成するための化学物質、浸透圧を調整するための塩、コーティング剤、又は酸化防止剤]などを指す。
【0087】
固形製剤は、錠剤、カプセル、顆粒、及び粉末の形態で、何ら添加剤を用いずに、又は適当な担体(添加剤)を用いて調製し得る。かかる担体(添加剤)の例には、ラクトース又はグルコースなどの糖類;トウモロコシ、コムギ、又はコメのデンプン;ステアリン酸などの脂肪酸;メタケイ酸アルミン酸マグネシウム又は無水リン酸カルシウムなどの無機塩;ポリビニルピロリドン又はポリアルキレングリコールなどの合成ポリマー;ステアリルアルコール、ベンジルアルコールなどのアルコール;メチルセルロース、カルボキシメチルセルロース、エチルセルロース、又はヒドロキシプロピルメチルセルロースなどの合成セルロース誘導体;及び他の通常使用される添加剤、例えばゼラチン、タルク、植物油、及びアラビアゴムが挙げられてよい。
【0088】
錠剤、カプセル、顆粒、及び粉末などのこれらの固形製剤は、各製剤の全重量に基づき、一般に、例えば、0.1ないし100重量%、及び好ましくは、5ないし98重量%の、mTOR阻害剤を含有してもよい。
【0089】
液体製剤は、懸濁液、シロップ、注射液、及び点滴剤(静脈内輸液)の形態で、水、アルコール、又は植物由来の油、例えば大豆油、ラッカセイ油、及びゴマ油などの、液体製剤において通常用いられる適当な添加剤を使用して製造される。
【0090】
特に、製剤が、筋肉内注射、静脈内注射、又は皮下注射の形態で、非経口的に投与される場合、適当な溶媒又は希釈剤は、注射用の蒸留水、塩酸リドカインの水溶液(筋肉内注射用)、生理食塩水、グルコース水溶液、エタノール、ポリエチレングリコール、プロピレングリコール、静脈内注射用の液体(例えば、クエン酸、又はクエン酸ナトリウムなどの水溶液)、又は電解質溶液(静脈内点滴輸液及び静脈内注射用)、又はそれらの混合溶液に例示される。
【0091】
かかる注射剤は、予め溶解された溶液の形態か、或いは、使用時に溶解される、粉末自体、又は適当な担体(添加剤)と混合された粉末の形態でよい。注射液は、各製剤の全重量に基づき、例えば、0.1ないし10重量%の活性成分を含有してもよい。
【0092】
経口投与用の懸濁液又はシロップなどの液体製剤は、各製剤の全重量に基づき、例えば、0.1ないし10重量%の活性成分を含有してもよい。
【0093】
本発明の各製剤は、当業者により、通常の方法又は一般的な技術によって調製し得る。例えば、調製は、製剤が経口用製剤である場合には、例えば、適切な量の本発明の化合物を、適切な量のラクトースと混合すること、この混合物を経口投与に適した硬ゼラチンカプセル内に充填することによって実施し得る。一方、本発明の化合物を含有する製剤が注射剤の場合には、適切な量の本発明の化合物を、適切な量の0.9%生理食塩水と混合すること、及びこの混合物を注射用のバイアル内に充填することにより実施し得る。
【0094】
本発明の成分(component)は、ヒトを含む哺乳類へ、単独で、又は薬学的に許容される担体、賦形剤、又は希釈剤と組合せて、医薬組成物の状態で、標準的な薬学のプラクティスに従って投与してもよい。かかる成分は、経口的に、又は静脈内、筋肉内、腹腔内、皮下、直腸内、及び局所の投与経路を含め、非経口的に投与し得る。
【0095】
適切な投薬量は、医師にとり既知であり、当然のことながら、特定の疾病状態、投与される組成物の比活性、及び治療を受ける個々の患者に依存するであろう。ある場合には、所望の治療量を達成するため、反復した投与を提供すること、すなわち特定のモニター又は計測された用量の個々の投与を反復することが必要であってもよく、この場合、個々の投与は所望の1日用量又は効果が達成されるまで反復される。適切な投薬量についてのさらなる情報は、以下に提供される。
【0096】
用語「投与」及びその変形(例えば、化合物を「投与すること」)は、本発明の成分に関して、かかる成分又は該成分のプロドラッグを、治療を要する動物の系内に導入することを意味する。本発明の成分又はそのプロドラッグが、1つ以上の他の活性薬剤(例えば、mTOR阻害剤)と組合せて与えられる場合、「投与」及びその変形は、各々、かかる成分又はそのプロドラッグと他の薬剤との、同時の、及び逐次の導入を包含するものと理解される。
【0097】
本明細書で用いる場合、用語「組成物」は、指定された量の指定された成分を含んでなる生成物、並びに指定された量の指定された成分の組合せから、直接又は間接的にもたらされる任意の生成物を包含することが意図されている。
【0098】
用語「治療有効量」は、本明細書で用いるとき、研究者、獣医師、医師、又は他の臨床家によって求められている、組織、系、動物、又はヒトにおける生物学的又は医学的応答を示す活性化合物又は医薬の量を意味する。
【0099】
mTOR阻害剤の適切な量が、癌の治療を受けている患者に投与される。1つの実施態様においては、mTOR阻害剤は、1日当たり約10mgないし40mgの用量で投与される。本発明の1つの実施態様においては、mTOR阻害剤は、1日当たり10mgの用量で投与される。本発明の別の実施態様においては、mTOR阻害剤は、1日当たり20mgの用量で投与される。本発明の別の実施態様においては、mTOR阻害剤は、1日当たり30mgの用量で投与される。本発明の別の実施態様においては、mTOR阻害剤は、1日当たり40mgの用量で投与される。
【0100】
本発明の1つの実施態様においては、mTOR阻害剤は、1週間当たり5回投与される。例えば、リダフォロリムスは、1日目に開始され、指定された用量レベルで連続して5日間継続され、これにリダフォロリムスの処置のない2日間が続けられる。リダフォロリムスは次に、この毎週5回のスケジュールで継続される。
【0101】
本発明の抗IGF−1R抗体とmTOR阻害剤とを含んでなる併用療法は、ボーラスとしての静脈内投与、又は一定期間の連続した輸液によるか、筋肉内、腹腔内、脳脊髄内、皮下、関節内、滑液包内、髄腔内、経口、局所、又は吸入経路によるなどの既知の方法に従って、ヒト患者に投与される。抗体の静脈内又は皮下投与が好ましい。3つの異なる送達アプローチが、本発明の抗体の送達に有用であることが期待される。通常の静脈内送達は、おそらく大多数の腫瘍のための標準的な送達技術であろう。しかしながら、卵巣、胆管、他の管系などの腫瘍に代表される腹腔内のものなどのいくつかの腫瘍に関しては、腫瘍において高線量の抗体を得るために、また抗体クリアランスを最少化するために、腹腔内投与が好適であることを示すことがある。同様に、ある固体腫瘍は血管系を有しており、これは局所灌流に適する。局所灌流により、腫瘍部位において高線量の抗体を得ることができ、また短時間の抗体クリアランスを最小にすることができる。
【0102】
任意のタンパク質又は抗体の輸液をベースとする治療と同様に、安全性の懸念は主として(i)サイトカイン放出症候群、すなわち、低血圧、発熱、震え、悪寒、(ii)物質に対する免疫応答の発生(すなわち、患者による治療抗体に対するヒト抗体の発生、又はHAHA若しくはHACA応答)、及び(iii)EGF受容体を発現する正常細胞、例えば、EGFR及び/又はIGF−1Rを発現する肝細胞に対する毒性、に関連する。標準的試験及び追跡調査を利用して、これらの安全性懸念の各々がモニターされる。特に、肝機能は、もしあれば肝臓に対する損傷を評価する目的で、臨床試験の間頻繁にモニターされる。
【0103】
疾患の予防又は治療用には、抗体の適切な用量は、上記に定義した通りの、治療されるべき疾患のタイプ、疾患の重症度及び経過、抗体が予防又は治療目的で投与されるのかどうか、以前の療法、患者の病歴及び抗体に対する応答、及び主治医の自由裁量に依存するであろう。抗体は、一度に、又は一連の治療にわたり、適宜患者に投与される。併用療法レジメンでは、本発明の組成物は、治療上の有効量又は相乗的な量で投与される。本明細書で用いるとき、治療有効量は、抗IGF−1R抗体及びmTOR阻害剤の同時投与、又は本発明の組成物の投与が、標的とする疾患又は症状の低減又は阻害をもたらす量である。治療上の相乗的な量は、特定の疾患に伴う症状又は症候群を、相乗的に又は有意に、緩和又は除去するのに必要な、抗IGF−1R抗体及びmTOR阻害剤の量である。
【0104】
広い実施態様においては、本発明の治療は、抗IGF−1R抗体とmTOR阻害剤との
併用投与を包含する。併用投与は、別々の製剤又は単一の医薬製剤を用いた同時投与、及びいずれかの順序での逐次的な投与を包含し、これにおいて、その間に双方の(又は全ての)活性薬剤が同時にその生物活性を及ぼす期間があることが好ましい。かかる化学療法剤のための製剤及び投薬スケジュールは、製造業者の指示に従って、又は熟練の開業医により経験的に決定された通りに使用してもよい。化学療法のための製剤及び投薬スケジュールはまた、ペリー(M.C.Perry)編、「Chemotherapy(化学療法)、Service Ed」、Williams & Wilkins、Baltimore、Md.、1992年に記載されている。mTOR阻害剤は、抗体の投与に先行するか、又は後続して投与してもよく、或いはそれらを同時に投与してもよい。本発明の治療的併用での臨床的投薬は、副作用の程度によって制限されることが見込まれる。
【0105】
例えば、1つ以上の別々の投与によるものであろうと、連続した輸液によるものであろうと、疾患のタイプ及び重症度に依存して、約1μg/kgないし50mg/kg(例えば、0.1−20mg/kg)の抗体が、患者への投与の最初の候補投薬量である。典型的な一日用量は、上記の因子に依存して、約1μg/kgないし約100mg/kg以上の範囲内である。数日間以上にわたる反復投与では、症状に依存して、疾患症状に所望の抑制が生じるまで継続される。しかしながら、他の用量用法を使用してもよい。
【0106】
1つの態様においては、本発明の抗体は、約5mg/kgないし約15mg/kgの用量範囲で、毎週投与されるか、又は2週間ないし3週間ごとに投与してもよい。ある態様においては、化学療法レジメンは、従来の高用量の間欠的投与を包含する。別の態様においては、化学療法剤は、計画的中断なしに、より少量で頻繁な投薬を用いて投与される(「メトロノミック・ケモセラピー」)。本発明の療法の進行は、通常の技術及びアッセイによりによりモニターし得る。
【0107】
1つの実施態様においては、投薬順序は、mTOR阻害剤をIGF−1R抗体と同時に投与することを含んでなる−例えば、リダフォロリムスは毎日投与され、一方IGF−1R抗体(ダロツズマブ)は毎週投与される。特に、ダロツズマブは10mg/kgの用量で、毎週静脈内投与され、一方リダフォロリムスは毎日10mgのスケジュールで投与される。
【0108】
別の実施態様においては、投薬順序は、mTOR阻害剤をIGF−1R抗体と同時に投与することを含んでなる−例えば、リダフォロリムスは毎日投与され、一方IGF−1R抗体(ダロツズマブ)は毎週投与される。特に、ダロツズマブは10mg/kgの用量で、毎週静脈内投与され、一方リダフォロリムスは毎日20mgのスケジュールで投与される。
【0109】
1つの実施態様においては、投薬順序は、mTOR阻害剤をIGF−1R抗体と同時に投与することを含んでなる−例えば、リダフォロリムスは毎日投与され、一方IGF−1R抗体(ダロツズマブ)は毎週投与される。特に、ダロツズマブは10mg/kgの用量で、毎週静脈内投与され、一方リダフォロリムスは毎日30mgのスケジュールで投与される。
【0110】
1つの実施態様においては、投薬順序は、mTOR阻害剤をIGF−1R抗体と同時に投与することを含んでなる−例えば、リダフォロリムスは毎日投与され、一方IGF−1R抗体(ダロツズマブ)は毎週投与される。特に、ダロツズマブは10mg/kgの用量で、毎週静脈内投与され、一方リダフォロリムスは毎日40mgのスケジュールで投与される。
【0111】
IGF−1R抗体に関する別の投薬レジメンは、以下の通りである:(a)15mg/kg(loading)を、続いて毎週7.5mg/kgを投入する;(b)一週間当たり7.5mg/kg;(c)一週間当たり10.0mg/kg;(d)隔週ごとに7.5mg/kg;(e)隔週ごとに10.0mg/kg;(f)隔週ごとに20.0mg/kg;(g)3週間ごとに30.0mg/kg。
【0112】
併用のためのサンプル投薬レジメンは、以下の通りである:
【0113】
【化1−1】
【0114】
【化1−2】
【0115】
効能追加
非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌の治療に加えて、mTOR阻害剤及び抗IGF−1R抗体の併用はまた、以下の癌の治療にも有用であってよい:心臓:肉腫(血管肉腫、線維肉腫、横紋筋肉腫、脂肪肉腫)、粘液腫、横紋筋腫、線維腫、脂肪腫、及び奇形腫;肺:気管支原性癌(扁平上皮細胞、未分化小細胞、未分化大細胞、腺癌)、肺胞(細気管支)癌、気管支腺腫、肉腫、リンパ腫、軟骨腫様過誤腫、中皮腫;胃腸:食道(扁平上皮細胞癌、腺癌、平滑筋肉腫、リンパ腫)、胃(癌腫、リンパ腫、平滑筋肉腫)、膵臓(腺管腺癌、インスリノーマ、グルカゴノーマ、ガストリノーマ、カルチノイド腫瘍、ビポーマ)、小腸(腺癌、リンパ腫、カルチノイド腫瘍、カポジ肉腫、平滑筋腫、血管腫、脂肪腫、神経線維腫、線維腫)、大腸(腺癌、管状腺腫、絨毛腺腫、過誤腫、平滑筋腫)、結腸、結腸直腸、直腸;尿生殖路:腎臓(腺癌、ウィルムス腫瘍[腎芽細胞腫]、リンパ腫、白血病)、膀胱及び尿道(扁平上皮細胞癌、移行上皮癌、腺癌)、前立腺(腺癌、肉腫)、精巣(精上皮腫、奇形腫、胎児性癌、奇形癌、絨毛上皮腫、肉腫、間質細胞癌、線維腫、線維腺腫、類腺腫瘍、脂肪腫);肝臓:肝癌(肝細胞癌)、胆管癌、肝芽細胞腫、血管肉腫、肝細胞腺腫、血管腫;骨:骨原性肉腫(骨肉腫)、線維肉腫、悪性線維性組織球腫、軟骨肉腫、ユーイング肉腫、悪性リンパ腫(細網肉腫)、多発性骨髄種、悪性巨細胞腫軟骨腫、骨軟骨腫(骨軟骨性外骨症)、良性軟骨腫、軟骨芽細胞腫、軟骨粘液線維腫、類骨骨腫及び、巨細胞腫;神経系:頭蓋(骨腫、血管腫、肉芽腫、黄色腫、変形性骨炎)、髄膜(髄膜腫、髄膜肉腫、神経膠腫症)、脳(星状細胞腫、髄芽細胞腫、神経膠腫、上衣細胞腫、胚細胞腫[松果体腫]、多形性神経膠芽腫、乏突起神経膠腫、神経鞘種、網膜芽細胞腫、先天性腫瘍)、脊髄神経線維腫、髄膜腫、神経膠腫、肉腫);婦人科系:子宮(子宮内膜癌)、子宮頚(子宮頚癌、前腫瘍性子宮頚部異形成)、卵巣(卵巣癌[漿液性嚢胞腺癌、粘液性嚢胞腺癌、分類不能癌腫]、顆粒膜卵胞膜細胞腫、セルトリ−ライディッヒ細胞腫、未分化胚細胞腫、悪性奇形腫)、外陰(扁平上皮細胞癌、上皮内癌、腺癌、線維肉腫、黒色腫)、膣(明細胞癌、扁平上皮細胞癌、ブドウ状肉腫(胎児性横紋筋肉腫)、卵管(癌腫);血液系:血液(骨髄性白血病[急性及び慢性]、急性リンパ芽球生白血病、慢性リンパ球性白血病、骨髄増殖性疾患、多発性骨髄腫、脊髄異形成症候群)、ホジキン病、非ホジキンリンパ腫[悪性リンパ腫];皮膚:悪性黒色腫、基底細胞癌、扁平上皮細胞癌、カポジ肉腫、色素性異形成毋斑、脂肪腫、血管腫、皮膚線維種;及び副腎:神経芽細胞腫を包含する。したがって、本明細書に提供された、用語「癌性細胞」は、上記に定義された症状の任意の1つに苦しめられた細胞を包含する。
【0116】
本発明のmTOR阻害剤及び抗IGF−1R抗体の併用はまた、以下の疾患症状の治療においても有用であってよい:ケロイド及び乾癬。
【0117】
本発明の範囲内にさらに包含されるのは、血管新生が関与する疾患を治療又は予防する方法であり、これは、かかる治療を必要とする哺乳類に対し、治療有効量の本発明の組合せを投与することを含んでなる。眼の血管新生疾患は、結果として生じる組織損傷の大部分を眼の血管の異常な浸潤に帰し得る症状の一例である(2000年6月2日公開のWO 00/30651を参照)。望ましくない浸潤は、虚血性網膜症、例えば、糖尿病性網膜症、未熟児網膜症、網膜静脈閉塞その他から結果として生じるものにより、又は、神経変性疾患、例えば、加齢関連黄班変性症において観察される脈絡膜血管新生により誘発され得る。本発明化合物の投与による血管増殖の阻害は、それ故、血管の浸潤を防止し、かつ、血管新生が関与する疾患、例えば、網膜血管新生、糖尿病性網膜症、加齢関連黄班変性症などのような眼疾患を予防又は治療するであろう。
【0118】
本発明の範囲内にさらに包含されるのは、限定されないが:眼疾患(例えば、網膜血管新生、糖尿病性網膜症、及び加齢関連黄班変性症)、アテローム性動脈硬化症、関節炎、乾癬、肥満、及びアルツハイマー病を含む、血管新生が関与する非悪性疾患(ドレッジ(Dredge)ら、Expert Opin.Biol.Ther.、2002年、第2巻、第8号、p.953−966)を、治療又は予防する方法である。別の実施態様においては、血管新生が関与する疾患を治療又は予防する方法は、眼疾患(例えば、網膜血管新生、糖尿病性網膜症、及び加齢関連黄班変性症)、アテローム性動脈硬化症、関節炎、及び乾癬を包含する。
【0119】
本発明の範囲内にさらに包含されるのは、再狭窄、炎症、自己免疫疾患、及びアレルギー/喘息といった、過増殖性疾患を治療する方法である。
【0120】
本発明の範囲内にさらに包含されるのは、ステントを被覆するための本発明の組合せの使用であり、かつそれ故に、再狭窄の治療及び/又は予防用の被覆ステント(WO 03/032809)に対する本化合物の使用である。
【0121】
本発明の範囲内にさらに包含されるのは、変形性関節症の治療及び/又は予防(WO 03/035048)のための本発明の組合せの使用である。
【0122】
本発明の範囲内にさらに包含されるのは、低インスリン症を治療する方法である。
【0123】
本発明を例示するものは、非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌の治療及び/又は予防用医薬の調製における、上記記載のmTOR阻害剤及び抗IGF−1R抗体の併用使用である。
【0124】
追加の抗癌剤
本発明のmTOR阻害剤及び抗IGF−1R抗体の併用はまた、さらなる治療剤、化学療法剤、及び抗癌剤との併用においても有用である。本発明のmTOR阻害剤及び抗IGF−1R抗体の併用を、治療剤、化学療法剤、及び抗癌剤とさらに併用することは、本発明の範囲内である。かかる薬剤の例は、デヴィータ(V.T.Devita)及びヘルマン(S.Hellman)(共編)“Cancer Principles and Practice of Oncology)(癌の原理及び腫瘍学のプラクティス)”、第6版、2001年2月15日、Lippincott Williams&Wilkins Publishers、において見出すことができる。当業者は、薬物及び関係する癌の、特定の性質に基づき、どの薬剤の組合せが有用であるかを識別することができるであろう。かかるさらなる抗癌剤は、限定されないが以下を包含する:エストロゲンレセプターモジュレータ、アンドロゲンレセプターモジュレータ、レチノイドレセプターモジュレータ、細胞傷害剤/細胞増殖抑制剤、抗増殖剤、プレニル−タンパク質トランスフェラーゼ阻害剤、HMG−CoAレダクターゼ阻害剤及び他の血管新生インヒビター、HIVプロテアーゼ阻害剤、逆転写酵素阻害剤、細胞増殖及び生存シグナリングの阻害剤、ビスホスホネート、アロマターゼ阻害剤、siRNA治療、γ−セクレターゼ阻害剤、受容体チロシンキナーゼ(RTK)を妨害する薬剤、及び細胞周期チェックポイントを妨害する薬剤。本発明のmTOR阻害剤及び抗IGF−1R抗体の併用は、放射線療法と同時投与された場合、特に有用でありえる。
【0125】
「エストロゲンレセプターモジュレータ」は、メカニズムにかかわらず、エストロゲンの受容体への結合を妨害又は阻害する化合物を指す。エストロゲンレセプターモジュレータの例は、限定されないが、タモキシフェン、ラロキシフェン、ヨードキシフェン、LY353381、LY117081、トレミフェン、フルベストラント、4−[7−(2,2−ジメチル−1−オキソプロポキシ−4−メチル−2−[4−[2−(1−ピペリジニル)エトキシ]フェニル]−2H−1−ベンゾピラン−3−イル]−フェニル−2,2−ジメチルプロパノアート、4,4’−ジヒドロキシベンゾフェノン−2,4−ジニトロフェニル−ヒドラゾン、及びSH646を包含する。
【0126】
「アンドロゲンレセプターモジュレータ」は、メカニズムにかかわらず、アンドロゲンの受容体への結合を妨害又は阻害する化合物を指す。アンドロゲンレセプターモジュレータの例は、フィナステリド及び他の5α−レダクターゼ阻害剤、ニルタミド、フルタミド、ビカルタミド、リアロゾール、及びアビラテロンアセタートを包含する。
【0127】
「レチノイドレセプターモジュレータ」は、メカニズムにかかわらず、受容体へのレチノイドの結合を妨害又は阻害する化合物を指す。かかるレチノイドレセプターモジュレータの例は、ベキサロテン、トレチノイン、13−シス−レチノイン酸、9−シス−レチノイン酸、α−ジフルオロメチルオルニチン、ILX23−7553、トランス−N−(4’−ヒドロキシフェニル)レチンアミド、及びN−4−カルボキシフェニルレチンアミドを包含する。
【0128】
「細胞傷害剤/細胞増殖抑制剤」は、主として細胞の機能化を直接妨害するか又は細胞有糸分裂を阻害又は妨害することにより、細胞死を引き起こすか又は細胞増殖を阻害する化合物を指し、アルキル化剤、腫瘍壊死因子、インターカレーター、低酸素活性化化合物、微小管阻害剤/微小管安定化剤、有糸分裂キネシンの阻害剤、ヒストン脱アセチル化酵素阻害剤、有糸分裂の進行に関与するキナーゼの阻害剤、成長因子及びサイトカインシグナル伝達経路に関与するキナーゼの阻害剤、代謝拮抗物質、生物応答調節剤;ホルモン/抗ホルモン治療剤、造血成長因子、モノクローナル抗体標的化治療剤、トポイソメラーゼ阻害剤、プロテオソーム阻害剤、ユビキチンリガーゼ阻害剤、及びオーロラキナーゼ阻害剤を包含する。
【0129】
細胞傷害剤/細胞増殖抑制剤の例は、制限なされることなく、セルテネフ、カケクチン、イフォスファミド、タソネルミン、ロニダミン、カルボプラチン、アルトレタミン、プレドニムスチン、ジブロモダルシトール、ラニムスチン、フォテムスチン、ネダプラチン、オキザリプラチン、テモゾロミド、ヘプタプラチン、エストラムスチン、インプロスルファントシラート、トロホスファミド、ニムスチン、塩化ジブロスピジウム、プミテパ、ロバプラチン、サトラプラチン、プロフィロマイシン、シスプラチン、イロフルベン、デキシホスファミド、シス−アミンジクロロ(2−メチル−ピリジン)白金、ベンジルグアニン、グルホスファミド、GPX100、(トランス、トランス、トランス)−ビス−ミュー−(ヘキサン−1,6−ジアミン)−ミュー−[ジアミン−白金(II)]ビス[ジアミン(クロロ)白金(II)]テトラクロリド、ジアリジジニル(diarizidinyl)スペルミン、三酸化ヒ素、1−(11−ドデシルアミノ−10−ヒドロキシウンデシル)−3,7−ジメチルキサンチン、ゾルビシン、イダルビシン、ダウノルビシン、ビサントレン、ミトキサントロン、ピラルビシン、ピナフィド、バルルビシン、アムルビシン、アンチネオプラストン、3’−デアミノ−3’−モルホリノ−13−デオキソ−10−ヒドロキシカルミノマイシン、アナマイシン、ガラルビシン、エリナフィド、MEN10755、及び4−デメトキシ−3−デアミノ−3−アジリジニル−4−メチルスルホニル−ダウノルビシン(WO00/50032参照)、Rafキナーゼ阻害剤(例えば、Bay43−9006)、及びさらなるmTOR阻害剤を包含する。
【0130】
低酸素活性化化合物の1つの例は、チラパザミンである。
【0131】
プロテオソーム阻害剤の例は、限定されないが、ラクタシスチン及びMLN−341(ベルケード(Velcade))を包含する。
【0132】
微小管阻害剤/微小管安定化剤の例は、パクリタキセル、硫酸ビンデシン、3’,4’−ジデヒドロ−4’−デオキシ−8’−ノルビンカロイコブラスチン、ドセタキソール、リゾキシン、ドラスタチン、ミボブリンイセチオナート、アウリスタチン、セマドチン、RPR109881、BMS184476、ビンフルニン、クリプトフィシン、2,3,4,5,6−ペンタフルオロ−N−(3−フルオロ−4−メトキシフェニル)ベンゼンスルホンアミド、アンヒドロビンブラスチン、N,N−ジメチル−L−バリル−L−バリル−N−メチル−L−バリル−L−プロリル−L−プロリン−t−ブチルアミド、TDX258、エポチロン(例えば米国特許第6,284,781及び6,288,237号参照)、及び、BMS188797を包含する。1つの実施態様においては、エポチロンは微小管阻害剤/微小管安定化剤に包含されない。
【0133】
トポイソメラーゼラーゼ阻害剤のいくつかの例は、トポテカン、ハイカプタミン(hycaptamine)、イリノテカン、ルビテカン、6−エトキシプロピオニル−3’,4’−O−エキソ−ベンジリデン−シャールトルーシン、9−メトキシ−N,N−ジメチル−5−ニトロピラゾロ[3,4,5−kl]アクリジン−2−(6H)プロパンアミン、1−アミノ−9−エチル−5−フルオロ−2,3−ジヒドロ−9−ヒドロキシ−4−メチル−1H,12H−ベンゾ[de]ピラノ[3’,4’:b,7]−インドリジノ[1,2b]キノリン−10,13(9H,15H)ジオン、ルルトテカン(lurtotecan)、7−[2−(N−イソプロピルアミノ)エチル]−(20S)カンプトテシン、BNP1350、BNPI1100、BN80915、BN80942、エトポシドホスファート、テニポシド、ソブゾキサン、2’−ジメチルアミノ−2’−デオキシ−エトポシド、GL331、N−[2−(ジメチルアミノ)エチル]−9−ヒドロキシ−5,6−ジメチル−6H−ピリド[4,3−b]カルバゾール−1−カルボキサミド、アスラクライン(asulacrine)、(5a,5aB,8aa,9b)−9−[2−[N−[2−(ジメチルアミノ)エチル]−N−メチルアミノ]エチル]−5−[4−ヒドロキシ−3,5−ジメトキシフェニル]−5,5a,6,8,8a,9−ヘキソヒドロフロ(3’,4’:6,7)ナフト(2,3−d)−1,3−ジオキソール−6−オン、2,3−(メチレンジオキシ)−5−メチル−7−ヒドロキシ−8−メトキシベンゾ[c]−フェナントリジニウム、6,9−ビス[(2−アミノエチル)アミノ]ベンゾ[g]イソキノリン−5,10−ジオン、5−(3−アミノプロピルアミノ)−7,10−ジヒドロキシ−2−(2−ヒドロキシエチルアミノメチル)−6H−ピラゾロ[4,5,1−de]アクリジン−6−オン、N−[1−[2(ジエチルアミノ)エチルアミノ]−7−メトキシ−9−オキソ−9H−チオキサンテン−4−イルメチル]ホルムアミド、N−(2−(ジメチルアミノ)エチル)アクリジン−4−カルボキサミド、6−[[2−(ジメチルアミノ)エチル]アミノ]−3−ヒドロキシ−7H−インデノ[2,1−c]キノリン−7−オン、及び、ジメスナである。
【0134】
有糸分裂キネシン、及び特にヒト有糸分裂キネシンKSPの阻害剤の例は、国際公開WO 03/039460、WO 03/050064、WO 03/050122、WO 03/049527、WO03/049679、WO 03/049678、WO 04/039774、WO 03/079973、WO 03/099211、WO 03/015855、WO 03/106417、WO 04/037171、WO 04/058148、WO 04/058700、WO 04/126699、WO05/018638、WO05/019206、WO05/019205、WO05/018547、WO05/017190、US2005/0176776に記載されている。1つの実施態様においては、有糸分裂キネシンの阻害剤は、限定されないが、KSPの阻害剤、MKLP1の阻害剤、CENP−Eの阻害剤、MCAKの阻害剤、及びRab6−KIFLの阻害剤を包含する。
【0135】
「ヒストン脱アセチル化酵素阻害剤」の例は、限定されないが、SAHA、TSA、オキサムフラチン、PXD101、MG98、及びスクリプタイドを包含する。他のヒストン脱アセチル化酵素阻害剤に関するさらなる参考文献は、以下の文書に見出されてよい;ミラー(Miller,T.A)ら、J.Med.Chem.、2003年、第46巻、第24号、p.5097−5116。
【0136】
「有糸分裂の進行に関与するキナーゼの阻害剤」は、限定されないが、オーロラキナーゼの阻害剤、ポロ様キナーゼの阻害剤(PLK;特にPLK−1の阻害剤)、bub−1の阻害剤、及びbub−R1の阻害剤を包含する。「オーロラキナーゼ阻害剤」の一例は、VX−680である。
【0137】
「抗増殖剤」は、アンチセンスRNA及びDNAオリゴヌクレオチド、例えばG3139、ODN698、RVASKRAS、GEM231、及びINX3001;及び、代謝拮抗物質、例えばエノシタビン、カルモフール、テガフール、ペントスタチン、ドキシフルリジン、トリメトレキセート、フルダラビン、カペシタビン、ガロシタビン、シタラビンオクフォスファート、フォステアビン(fosteabine)ナトリウム水和物、ラルチトレキセド、パルチトレキシド、エミテフール、チアゾフリン、デシタビン、ノラトレキセド、ペメトレキセド、ネルザラビン、2’−デオキシ−2’−メチリデンシチジン、2’−フルオロメチレン−2’−デオキシシチジンン、N−[5−(2,3−ジヒドロ−ベンゾフリル)スルホニル]−N’−(3,4−ジクロロフェニル)尿素、N6−[4−デオキシ−4−[N2−[2(E),4(E)−テトラデカジエノイル]グリシルアミノ]−L−グリセロ−B−L−マンノ−ヘプトピラノシル]アデニン、アプリジン、エクチナサイジン、トロキサシタビン、4−[2−アミノ−4−オキソ−4,6,7,8−テトラヒドロ−3H−ピリミジノ[5,4−b][1,4]チアジン−6−イル−(S)−エチル]−2,5−チエノイル−L−グルタミン酸、アミノプテリン、5−フルオロウラシル、アラノシン、11−アセチル−8−(カルバモイルオキシメチル)−4−ホルミル−6−メトキシ−14−オキサ−1,11−ジアザテトラシクロ(7.4.1.0.0)−テトラデカ−2,4,6−トリエン−9−イル酢酸エステル、スワインソニン、ロメトレキソール、デキシラゾキサン(dexrazoxane)、メチオニナーゼ、2’−シアノ−2’−デオキシ−N4−パルミトイル−1−B−D−アラビノフラノシルシトシン、3−アミノピリジン−2−カルボキシアルデヒドチオセミカルバゾン、及びトラツズマブを包含する。
【0138】
モノクローナル抗体標的化治療剤の例は、癌細胞特異又は標的細胞特異モノクローナル抗体へ結合した、細胞傷害剤又は放射性同位元素を有する治療剤を包含する。例は、ベキサール(Bexxar)を包含する。
【0139】
「HMG−CoAレダクターゼ阻害剤」は、3−ヒドロキシ−3−メチルグルタリル−CoAレダクターゼの阻害剤を指す。使用してもよいHMG−CoAレダクターゼ阻害剤の例は、限定されないが、ロバスタチン(メバコール(MEVACOR)(登録商標);米国特許第4,231,938、4,294,926、及び4,319,039号参照)、シンバスタチン(ゾコール(ZOCOR)(登録商標);米国特許第4,444,784、4,820,850、及び4,916,239号参照)、プラバスタチン(プラバコール(PRAVACHOL)(登録商標);米国特許第4,346,227、4,537,859、4,410,629、5,030,447、及び5,180,589号参照)、フルバスタチン(レスコール(LESCOL)(登録商標);米国特許第5,354,772、4,911,165、4,929,437、5,189,164、5,118,853、5,290,946、及び5,356,896号参照)、アトルバスタチン(リピトール(LIPITOR)(登録商標);米国特許第5,273,995、4,681,893、5,489,691、及び5,342,952号参照)、及びセリバスタチン(リバスタチン及びバイコール(BAYCHOL)(登録商標)としても知られる;米国特許第5,177,080号参照)を包含する。これらの、及び、本方法において使用してもよい追加のHMG−CoAレダクターゼ阻害剤の構造式は、ヤルパーニ(M.Yalpani)、“Cholesterol Lowering Drugs(コレステロール低下薬)”,Chemistry & Industry,1996年2月5日、p.85−89の第87頁、及び、米国特許第4,782,084及び4,885,314号に記載されている。本明細書で使用される場合、用語、HMG−CoAレダクターゼ阻害剤は、全ての薬学的に許容されるラクトン及びオープン酸型(すなわち、ラクトン環が開裂されて遊離酸を形成する場合)、並びに、HMG−CoAレダクターゼ阻害活性を有する化合物の塩及びエステル型であって、それ故、かかる塩、エステル、オープン酸、及びラクトン型の使用は、本発明の範囲内に包含される。
【0140】
「プレニル−タンパク質トランスフェラーゼ阻害剤」は、プレニルタンパク質トランスフェラーゼ酵素の任意の1つ又は任意の組合せを阻害する化合物を指し、ファルネシル−タンパク質トランスフェラーゼ(FPTアーゼ)、ゲラニルゲラニル−タンパク質トランスフェラーゼI型(GGPTアーゼ−I)、及び、ゲラニルゲラニル−タンパク質トランスフェラーゼII型(GGPTアーゼ−II、またRab GGPTアーゼとも呼ばれる)を包含する。
【0141】
プレニル−タンパク質トランスフェラーゼ阻害剤の例は、以下の出版物及び特許に見出すことができる:WO 96/30343、WO 97/18813、WO 97/21701、WO 97/23478、WO 97/38665、WO 98/28980、WO 98/29119、WO95/32987、米国特許第5,420,245号、米国特許第5,523,430号、米国特許第5,532,359号、米国特許第5,510,510号、米国特許第5,589,485号、米国特許第5,602,098号、欧州特許公開0 618 221、欧州特許公開0 675 112、欧州特許公開0 604 181、欧州特許公開第0 696 593、WO 94/19357、WO 95/08542、WO 95/11917、WO 95/12612、WO 95/12572、WO 95/10514、米国特許第5,661,152号、WO 95/10515、WO 95/10516、WO 95/24612、WO 95/34535、WO 95/25086、WO 96/05529、WO 96/06138、WO 96/06193、WO 96/16443、WO 96/21701、WO 96/21456、WO 96/22278、WO 96/24611、WO 96/24612、WO 96/05168、WO 96/05169、WO 96/00736、米国特許第5,571,792号、WO 96/17861、WO 96/33159、WO 96/34850、WO 96/34851、WO 96/30017、WO 96/30018、WO 96/30362、WO 96/30363、WO 96/31111、WO 96/31477、WO 96/31478、WO 96/31501、WO 97/00252、WO 97/03047、WO 97/03050、WO 97/04785、WO 97/02920、WO 97/17070、WO 97/23478、WO 97/26246、WO 97/30053、WO 97/44350、WO 98/02436、及び米国特許第5,532,359号。血管新生に対するプレニル−タンパク質トランスフェラーゼ阻害剤の役割の1つの例については、European J.of Cancer、1999年、第35巻、第9号、p.1394−1401参照。
【0142】
「血管新生インヒビター」は、メカニズムにかかわらず、新たな血管の形成を阻害する化合物を指す。血管新生インヒビターの例は、限定されないが、チロシンキナーゼ阻害剤、例えば、チロシンキナーゼ受容体Flt−1(VEGFR1)及びFlk−1/KDR(VEGFR2)の阻害剤、上皮由来、線維芽細胞由来、又は血小板由来の成長因子の阻害剤、MMP(マトリックスメタロプロテアーゼ)阻害剤、インテグリンブロッカー、インターフェロン−α、インターロイキン−12、ペントサン多硫酸塩、アスピリン及びイブプロフェンのような非ステロイド系抗炎症剤(NSAID)並びにセレコキシブ及びロフェコキシブのような選択的シクロオキシ−ゲナーゼ−2阻害剤を含む、シクロオキシゲナーゼ阻害剤(PNAS、1992年、第89巻、p.7384;JNCI、1982年、第69巻、p.475;Arch.Opthalmol.1990年、第108巻、p.573;Anat.Rec.1994年、第238巻、p.68;FEBS Letters、1995年、第372巻、p.83;Clin.Orthop.1995年、第313巻、p.76;J.Mol.Endocrinol.1996年、第16巻、p.107;Jpn.J.Pharmacol.1997年、第75巻、p.105;Cancer Res.1997年、第57巻、p.1625;Cell、1998年、第93巻、p.705;Intl.J.Mol.Med.1998年、第2巻、p.715;J.Biol.Chem.1999年、第274巻、p.9116、ステロイド系抗炎症剤(例えばコルチコステロイド、ミネラルコルチコイド、デキサメタゾン、プレドニソン、プレドニソロン、メチルプレド、ベタメタゾン)、カルボキサミドトリアゾール、コンブレタスタチンA−4、スクアラミン、6−O−クロロアセチル−カルボニル)−フマギロール、サリドマイド、アンギオスタチン、トロポニン−1、アンギオテンシンIIアンタゴニスト(例えば、フェルナンデス(Fernandez)ら、J.Lab.Clin.Med.1985年、第105巻、p.141−145を参照)、及びVEGFに対する抗体(Nature Biotechnology、1999年10月、第17巻、p.963−968;キム(Kim)ら、Nature、1993年、第362巻、p.841−844;WO 00/44777;及び、WO 00/61186参照)を包含する。
【0143】
血管新生を調節又は阻害し、かつ本発明化合物と組合せて使用されてもよい他の治療薬は、凝固及び線溶系を調節又は阻害する薬剤を包含する(Clin.Chem.La.Med.2000年、第38巻、p.679−692の総説を参照)。凝固及び線溶経路を調節又は阻害する、かかる薬剤の例は、限定されないが、ヘパリン(Thromb.Haemost.1998年、第80巻、p.10−23を参照)、低分子量ヘパリン、及びカルボキシペプチダーゼU阻害剤(活性型のトロンビン活性化線溶抑制因子[TAFIa]の阻害剤としても公知)(Thrombosis Res.2001年、第101巻、p.329−354参照)を包含する。TAFIa阻害剤は、米国特許出願番号60/310,927(2001年8月8日出願)及び60/349,925号(2002年1月18日出願)に記載されている。
【0144】
「細胞周期チェックポイントを妨害する薬剤」は、細胞周期チェックポイントシグナルを伝達するプロテインキナーゼを阻害し、それにより癌細胞をDNA損傷剤に対し感受性化する化合物を指す。かかる薬剤は、ATR、ATM、CHK11及びCHK12キナーゼの阻害剤、及び、cdk及びcdcキナーゼ阻害剤を包含し、かつ特に、7−ヒドロキシスタウロスポリン、フラボピリドール、CYC202(サイクラセル(Cyclacel))、及びBMS−387032に例示される。
【0145】
「受容体チロシンキナーゼ(RTK)を妨害する薬剤」は、RTKを、及びそれ故に、発癌及び腫瘍進行に関与するメカニズムを阻害する化合物を指す。かかる薬剤は、c−Kit、Eph、PDGF、Flt3、及びc−Metの阻害剤を包含する。さらなる薬剤は、ブーム・ジェンセン(Bume−Jensen)及びハンター(Hunter)、Nature、2001年、第411巻、p.355−365により記述された、RTKの阻害剤を包含する。
【0146】
「細胞増殖及び生存シグナリング経路の阻害剤」は、細胞表面受容体の下流のシグナル伝達カスケードを阻害する化合物を指す。かかる薬剤は、セリン/スレオニンキナーゼ(限定されないが、Aktの阻害剤、例えばWO 02/083064、WO 02/083139、WO 02/083140、US2004−0116432、WO 02/083138、US2004−0102360、WO 03/086404、WO 03/086279、WO 03/086394、WO 03/084473、WO 03/086403、WO 2004/041162、WO 2004/096131、WO 2004/096129、WO 2004/096135、WO 2004/096130、WO 2005/100356、WO 2005/100344、US2005/029941、US2005/44294、US2005/43361、60/734188、60/652737、60/670469に開示されたものを包含する)、Rafキナーゼの阻害剤(例えばBAY−43−9006)、MEKの阻害剤(例えば、CI−1040及びPD−098059)、mTORの阻害剤(例えばワイス(Wyeth)CCI−779)、及びPI3Kの阻害剤(例えば、LY294002)を包含する。
【0147】
上記記載のように、NSAIDとの併用は、強力なCOX−2阻害剤であるNSAIDの使用に向けたものである。本明細書では、NSAIDは、細胞又はミクロソームアッセイにより測定されるとき、COX−2の阻害について1μM以下のIC50をもつ場合に効力がある。
【0148】
本発明はまた、選択的なCOX−2阻害剤である、NSAIDとの併用も包含する。本明細書では、選択的COX−2阻害剤であるNSAIDは、細胞又はミクロソームアッセイにより評価される、COX−1のIC50に対するCOX−2のIC50の比率によって測定されるとき、少なくとも100倍の、COX−1に対するCOX−2阻害特異性を有するものとして定義される。かかる化合物は、限定されないが、米国特許5,474,995、米国特許5,861,419、米国特許6,001,843、米国特許6,020,343、米国特許5,409,944、米国特許5,436,265、米国特許5,536,752、米国特許5,550,142、米国特許5,604,260、米国特許5,698,584、米国特許5,710,140、WO 94/15932、米国特許5,344,991、米国特許5,134,142、米国特許5,380,738、米国特許5,393,790、米国特許5,466,823、米国特許5,633,272、及び米国特許5,932,598に開示されているものを包含し、これらは全て参考として本明細書に含まれる。
【0149】
本治療法において特に有用であるCOX−2の阻害剤は、3−フェニル−4−(4−(メチルスルホニル)フェニル)−2−(5H)−フラノン;及び5−クロロ−3−(4−メチルスルホニル)フェニル−2−(2−メチル−5−ピリジニル)ピリジン;又はその薬学的に許容される塩である。
【0150】
COX−2の特異的阻害剤として記載されてきており、かつそれ故、本発明において有用である化合物は、限定されないが以下を包含する:パレコキシブ、ベクストラ(BEXTRA(登録商標))、セレブレックス(CELEBREX(登録商標))、又はその薬学的に許容される塩。
【0151】
血管新生インヒビターの他の例は、限定されないが、エンドスタチン、ウクライン、ランピルナーゼ、IM862、5−メトキシ−4−[2−メチル−3−(3−メチル−2−ブテニル)オキシラニル]−1−オキサスピロ[2,5]オクト−6−イル(クロロアセチル)カルバメート、アセチルジナナリン(acetyldinanaline)、5−アミノ−1−[[3,5−ジクロロ−4−(4−クロロベンゾイル)フェニル]メチル]−1H−1,2,3−トリアゾール−4−カルボキサミド、CM101、スクアラミン、コンブレタスタチン、RPI4610、NX31838、硫酸化マンノペンタオースホスファート、7,7−(カルボニル−ビス[イミノ−N−メチル−4,2−ピロロカルボニルイミノ[N−メチル−4,2−ピロール]−カルボニルイミノ]−ビス−(1,3−ナフタレンジスルホナート)、及び、3−[(2,4−ジメチルピロール−5−イル)メチレン]−2−インドリノン(SU5416)を包含する。
【0152】
前記に使用したように、「インテグリンブロッカー」は、生理的リガンドの、αvβ3インテグリンへの結合を選択的に拮抗するか、阻害するか、又は反対に作用する化合物、生理的リガンドの、αvβ5インテグリンへの結合を選択的に拮抗するか、阻害するか、又は反対に作用する化合物、生理的リガンドの、αvβ3インテグリン及びαvβ5インテグリン双方への結合を拮抗するか、阻害するか、又は反対に作用する化合物、及び、毛細血管内皮細胞上に発現された特定のインテグリンの活性を拮抗するか、阻害するか、又は反対に作用する化合物を指す。該用語はまた、αvβ6、αvβ8、α1β1、α2β1、α5β1、α6β1、及びα6β4インテグリンのアンタゴニストも指す。該用語はまた、αvβ3、αvβ5、αvβ6、αvβ8、α1β1、α2β1、β5α1、α6β1、及びα6β4インテグリンの任意の組合せのアンタゴニストも指す。
【0153】
チロシンキナーゼ阻害剤のいくつかの具体的な例は、N−(トリフルオロメチルフェニル)−5−メチルイソオキサゾール−4−カルボキサミド、3−[(2,4−ジメチルピロール−5−イル)メチリデニル]インドリン−2−オン、17−(アリルアミノ)−17−デメトキシゲルダナマイシン、4−(3−クロロ−4−フルオロフェニルアミノ)−7−メトキシ−6−[3−(4−モルホリニル)プロポキシル]キナゾリン、N−(3−エチニルフェニル)−6,7−ビス(2−メトキシエトキシ)−4−キナゾリンアミン、BIBX1382、2,3,9,10,11,12−ヘキサヒドロ−10−(ヒドロキシメチル)−10−ヒドロキシ−9−メチル−9,12−エポキシ−1H−ジインドロ[1,2,3−fg:3’,2’,1’−kl]ピロロ[3,4−i][1,6]ベンゾジアゾシン−1−オン、SH268、ゲニステイン、STI571、CEP2563、4−(3−クロロフェニルアミノ)−5,6−ジメチル−7H−ピロロ[2,3−d]ピリミジンメタンスルホナート、4−(3−ブロモ−4−ヒドロキシフェニル)アミノ−6,7−ジメトキシキナゾリン、4−(4’−ヒドロキシフェニル)アミノ−6,7−ジメトキシキナゾリン、SU6668、STI571A、N−4−クロロフェニル−4−(4−ピリジルメチル)−1−フタラジンアミン、及びEMD121974を包含する。
【0154】
抗癌化合物以外の化合物との併用もまた、本方法に包含される。例えば、本発明のmTOR阻害剤及び抗IGF−1R抗体の併用と、PPAR−γ(すなわち、PPAR−ガンマ)アゴニスト及びPPAR−δ(すなわち、PPAR−デルタ)アゴニストとの組合せは、いくつかの悪性疾患の治療において有用である。PPAR−γ及びPPAR−δは、核のペルオキシソーム増殖剤活性化受容体γ及びδである。PPAR−γの、内皮細胞上での発現及び、血管新生におけるその関与は、文献に報告されてきた(J.Cardiovasc.Pharmacol.1998年、第31巻、p.909−913;J.Biol.Chem.1999年、第274巻、p.9116−9121;Invest.Ophthalmol Vis.Sci.2000年、第41巻、p.2309−2317参照)。より最近では、PPAR−γアゴニストが、インビトロでVEGFに対する血管新生応答を阻害することが示された;トログリタゾン及びマレイン酸ロシグリタゾンの双方は、マウスにおいて、網膜の血管新生の発生を阻害する(Arch.Opthamol.2001年、第119巻、p.709−717)。PPAR−γアゴニスト及びPPAR−γ/αアゴニストの例は、限定されないが、チアゾリジンジオン(例えばDRF2725、CS−011、トログリタゾン、ロシグリタゾン、及びピオグリタゾン)、フェノフィブラート、ゲンフィブロジル、クロフィブラート、GW2570、SB219994、AR−H039242、JTT−501、MCC−555、GW2331、GW409544、NN2344、KRP297、NP0110、DRF4158、NN622、GI262570、PNU182716、DRF552926、2−[(5,7−ジプロピル−3−トリフルオロメチル−1,2−ベンゾイソオキサゾール−6−イル)オキシ]−2−メチルプロピオン酸(USSN 09/782,856に開示)、及び2(R)−7−(3−(2−クロロ−4−(4−フルオロフェノキシ)フェノキシ)プロポキシ)−2−エチルクロマン−2−カルボン酸(USSN 60/235,708及び60/244,697に開示)を包含する。
【0155】
本発明の別の実施態様は、本開示化合物の、癌の治療のための遺伝子療法と組合せた使用である。癌を治療するための遺伝学的戦略の概要に関しては、ホール(Hall)ら、(Am.J.Hum,Genet.1997年、第61巻、p.785−789)、及びクーフェ(Kufe)ら(「Cancer Medicine(癌医療)」、第5版、BC Decker、ハミルトン、2000年、p.876−889)を参照のこと。遺伝子療法を用いて、任意の腫瘍抑制遺伝子を送達することができる。かかる遺伝子の例は、限定されないが、組換えウイルス媒介性の遺伝子導入により送達可能であるp53(例えば、米国特許第6,069,134号参照)、uPA/uPARアンタゴニスト(「Adenovirus−Mediated Delivery of a uPA/uPAR Antagonist Suppresses Angiogenesis−Dependent Tumor Growth and Dissemination in Mice(uPA/uPARアンタゴニストのアデノウイルス媒介送達は、血管新生依存性の腫瘍増殖及び播種をマウスにおいて抑制する)」、Gene Therapy、1998年8月、第5巻、第8号、p.1105−13)、及びインターフェロンガンマ(J.Immunol.2000年、第164巻、p.217−222)を包含する。
【0156】
本発明化合物はまた、生来多剤耐性(MDR)、特に、高レベルのトランスポータ−タンパク質の発現に関与しているMDRの阻害剤と併用して投与されてもよい。かかるMDR阻害剤は、p−糖タンパク質(P−gp)の阻害剤、例えばLY335979、XR9576、OC144−093、R101922、VX853、及びPSC833(バルスポダール)を包含する。
【0157】
本発明化合物は、本発明化合物の、単独の又は放射線療法との使用の結果として生じるかもしれない急性、遅発性、遅延相、及び予測性嘔吐を含めた、悪心又は嘔吐を治療するべく、抗嘔吐薬と一緒に用いてもよい。嘔吐の予防又は治療のためには、本発明化合物は、他の抗嘔吐薬、特にニューロキニン−1受容体アンタゴニスト;5HT3受容体アンタゴニスト、例えばオンダンセトロン、グラニセトロン、トロピセトロン、及びザチセトロン;GABAB受容体アゴニスト、例えばバクロフェン;コルチコステロイド、例えばデカドロン(デキサメタゾン)、ケナログ(Kenalog)、アリストコート(Aristocort)、ナサライド(Nasalide)、プレフェリド(Preferid)、ベネコルテン(Benecorten)、又は他の、米国特許第2,789,118、2,990,401、3,048,581、3,126,375、3,929,768、3,996,359、3,928,326、及び3,749,712号に開示されたもの;抗ドーパミン作動薬、例えばフェノチアジン類(例えばプロクロペラジン、フルフェナジン、チオリダジン、及びメソリダジン)、メトクロプラマイド、又はドロナビノールと一緒に使用してもよい。別の実施態様においては、ニューロキニン−1受容体アンタゴニスト、5HT3受容体アンタゴニスト、及びコルチコステロイドから選ばれる抗嘔吐薬との併用療法が、本化合物の投与の結果として生じるかもしれない嘔吐の治療又は予防用に開示されている。
【0158】
本発明化合物との併用において役立つニューロキニン−1受容体アンタゴニストは、例えば、米国特許第5,162,339、5,232,929、5,242,930、5,373,003、5,387,595、5,459,270、5,494,926、5,496,833、5,637,699、5,719,147号;欧州特許公開番号EP 0 360 390、0 394 989、0 428 434、0 429 366、0 430 771、0 436 334、0 443 132、0 482 539、0 498 069、0 499 313、0 512 901、0 512 902、0 514 273、0 514 274、0 514 275、0 514 276、0 515 681、0 517 589、0 520 555、0 522 808、0 528 495、0 532 456、0 533 280、0 536 817、0 545 478、0 558 156、0 577 394、0 585 913、0 590 152、0 599 538、0 610 793、0 634 402、0 686 629、0 693 489、0 694 535、0 699 655、0 699 674、0 707 006、0 708 101、0 709 375、0 709 376、0 714 891、0 723 959、0 733 632、及び0 776 893;PCT国際特許公開番号WO 90/05525、90/05729、91/09844、91/18899、92/01688、92/06079、92/12151、92/15585、92/17449、92/20661、92/20676、92/21677、92/22569、93/00330、93/00331、93/01159、93/01165、93/01169、93/01170、93/06099、93/09116、93/10073、93/14084、93/14113、93/18023、93/19064、93/21155、93/21181、93/23380、93/24465、94/00440、94/01402、94/02461、94/02595、94/03429、94/03445、94/04494、94/04496、94/05625、94/07843、94/08997、94/10165、94/10167、94/10168、94/10170、94/11368、94/13639、94/13663、94/14767、94/15903、94/19320、94/19323、94/20500、94/26735、94/26740、94/29309、95/02595、95/04040、95/04042、95/06645、95/07886、95/07908、95/08549、95/11880、95/14017、95/15311、95/16679、95/17382、95/18124、95/18129、95/19344、95/20575、95/21819、95/22525、95/23798、95/26338、95/28418、95/30674、95/30687、95/33744、96/05181、96/05193、96/05203、96/06094、96/07649、96/10562、96/16939、96/18643、96/20197、96/21661、96/29304、96/29317、96/29326、96/29328、96/31214、96/32385、96/37489、97/01553、97/01554、97/03066、97/08144、97/14671、97/17362、97/18206、97/19084、97/19942、及び97/21702;及び英国特許公開番号2 266 529、2 268 931、2 269 170、2 269 590、2 271 774、2 292 144、2 293 168、2 293 169、及び2 302 689において充分に記載されている。かかる化合物の調製は、上述の特許及び刊行物において充分に記載されており、それらは参考として本明細書に含まれている。
【0159】
1つの実施態様においては、本発明化合物との併用使用のためのニューロキニン−1受容体アンタゴニストは:2−(R)−(1−(R)−(3,5−ビス(トリフルオロメチル)フェニル)エトキシ)−3−(S)−(4−フルオロフェニル)−4−(3−(5−オキソ−1H,4H−1,2,4−トリアゾロ)メチル)モルホリン、又は、その薬学的に許容される塩から選ばれ、それは、米国特許第5,719,147号に記載されている。
【0160】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、貧血症の治療において有用な薬剤とともに投与してもよい。かかる貧血症治療薬は、例えば、持続性の赤血球形成受容体活性化剤(例えばエポエチン・アルファ)である。
【0161】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、好中球減少症の治療において有用な薬剤とともに投与されてよい。かかる好中球減少症治療薬は、例えば、ヒト顆粒球コロニー刺激因子(G−CSF)のような、好中球の産生及び機能を調節する造血成長因子である。G−CSFの例は、フィルグラスチムを包含する。
【0162】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、免疫強化剤、例えば、レバミソール、イソプリノシン、及びザダキシン(Zadaxin)とともに投与してもよい。
【0163】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、ビスホスホネート(ビスホスホナート、ジホスホナート、ビスホスホン酸、及びジホスホン酸を包含するべく理解される)と併用して、骨癌を含む癌の治療又は予防に有用であってよい。ビホスホネートの例は、限定されないが:エチドロネート(ダイドロネル(Didronel))、パミドロネート(アレディア(Aredia))、アレンドロネート(フォサマックス(Fosamax))、リセドロネート(アクトネル(Actonel))、ゾレドロネート(ゾメタ(Zometa))、イバンドロネート(ボニバ(Boniva))、インカドロネート又はシマドロネート、クロドロネート、EB−1053、ミノドロネート、ネリドロネート、ピリドロネート(piridronate)、及びチルドロネートを包含し、任意の及び全ての、薬学的に許容される塩、誘導体、水和物、及びそれらの混合物を包含する。
【0164】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、アロマターゼ阻害剤と組合せて、乳癌の治療又は予防に有用でありえる。アロマターゼ阻害剤の例は、限定されないが、アナストロゾール、レトロゾール、及びエキセメスタンを包含する。
【0165】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、siRNA治療薬との併用において、癌の治療又は予防に有用でありえる。
【0166】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、γ−セクレターゼ阻害剤及び/又はNOTCHシグナリング阻害剤と併用して投与してもよい。かかる阻害剤は、WO 01/90084、WO 02/30912、WO 01/70677、WO 03/013506、WO 02/36555、WO 03/093252、WO 03/093264、WO 03/093251、WO 03/093253、WO 2004/039800、WO 2004/039370、WO 2005/030731、WO 2005/014553、USSN 10/957,251、WO 2004/089911、WO 02/081435、WO 02/081433、WO 03/018543、WO 2004/031137、WO 2004/031139、WO 2004/031138、WO 2004/101538、WO 2004/101539、及びWO 02/47671(LY−450139を含む)。
【0167】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、Aktの阻害剤との併用において、癌の治療又は予防に有用でありえる。かかる阻害剤は、限定されないが、以下の出版物に記載された化合物を包含する:WO 02/083064、WO 02/083139、WO 02/083140、US2004−0116432、WO 02/083138、US2004−0102360、WO 03/086404、WO 03/086279、WO 03/086394、WO 03/084473、WO 03/086403、WO 2004/041162、WO 2004/096131、WO 2004/096129、WO 2004/096135、WO 2004/096130、WO 2005/100356、WO 2005/100344、US2005/029941、US2005/44294、US2005/43361、60/734188、60/652737、60/670469。
【0168】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、PARP阻害剤との併用において、癌の治療又は予防に有用でありえる。
【0169】
放射線療法自体は、癌治療の分野では通常の方法を意味する。放射線療法には、X線、γ線、中性子線、電子線、陽子線などの種々の放射線;及び放射線源を使用し得る。最も一般的な放射線療法においては、外部放射線、γ線による照射にリニア加速器が使用される。
【0170】
本発明のmTOR阻害剤及び抗IGF−1R抗体の組合せはまた、以下の治療薬との併用において、癌の治療に有用でありえる:アバレリックス(プレナキシス・デポ(Plenaxis depot)(登録商標));アルデスロイキン(プロカイン(Prokine)(登録商標));アルデスロイキン(プロロイキン(Proleukin)(登録商標));アレムツズマブ(キャンパス(Campath)(登録商標));アリトレチノイン(パンレチン(Panretin)(登録商標));アロプリノール(ザイロプリム(Zyloprim)(登録商標));アルトレタミン(ヘキサレン(Hexalen)(登録商標));アミフォスチン(エチヨル(Ethyol)(登録商標));アナストロゾール(アリミデックス(Arimidex)(登録商標));三酸化ヒ素(トリセノックス(Trisenox)(登録商標));アスパラギナーゼ(エルスパル(Elspar)(登録商標));アザシチジン(ビダザ(Vidaza)(登録商標));ベバシズマブ(アバスチン(Avastin)(登録商標));ベキサロテン・カプセル(タルグレチン(Targretin)(登録商標));ベキサロテン・ゲル(タルグレチン(Targretin)(登録商標));ブレオマイシン(ブレノキサン(Blenoxane)(登録商標));ボルテゾミブ(ベルケード(Velcade)(登録商標));ブスルファン・静脈内(ブスルフェックス(Busulfex)(登録商標));ブスルファン・経口(マイレラン(Myleran)(登録商標));カルステロン(メトサーブ(Methosarb)(登録商標));カペシタビン(ゼローダ(Xeloda)(登録商標));カルボプラチン(パラプラチン(Paraplatin)(登録商標));カルムスチン(BCNU(登録商標)、BiCNU(登録商標));カルムスチン(グリアデル(Gliadel)(登録商標));インプラント型ポリフェプロサン20カルムスチン(グリアデル・ウエファー(Gliadel Wafer)(登録商標));セレコキシブ(セレブレックス((Celebex)(登録商標));セツキシマブ(アービタックス(Erbitux)(登録商標));クロランブシル(ロイケラン(Leukeran)(登録商標));シスプラチン(プラチノール(Platinol)(登録商標));クラドリビン(ロイスタチン(Leustatin)(登録商標)、2−CdA(登録商標));クロファラビン(クロラール(Clolar)(登録商標));シクロホスファミド(サイトキサン(Cytoxan)(登録商標)、ネオサール(Neosar)(登録商標));シクロホスファミド(サイトキサン注射薬(Cytoxan Injection)(登録商標));シクロホスファミド(サイトキサン・錠剤(Cytoxan Tablet)(登録商標));シタラビン(サイトサール−U(Cytosar−U)(登録商標));リポソーム化シタラビン(DepoCyt(登録商標));デカルバジン(DTIC−Dome(登録商標));ダクチノマイシン、アクチノマイシンD(コスメゲン(Cosmegen)(登録商標));ダルベポエチン・アルファ(アラネスプ(Aranesp)(登録商標));リポソーム化ダウノルビシン(DanuoXome(登録商標));ダウノルビシン、ダウノマイシン(ダウノルビシン(Daunorubicin)(登録商標));ダウノルビシン、ダウノマイシン(セルビジン(Cerbidine)(登録商標));デニロイキン・diftitox(オンタック(Ontak)(登録商標));デクスラゾキサン(ザインカード(Zinecard)(登録商標));ドセタキセル(タキソテール(Taxotere)(登録商標));ドキソルビシン(アドリアマイシンPFS(Adriamycin PFS)(登録商標));ドキソルビシン(アドリアマイシン(Adriamycin)(登録商標)、ルベックス(Rubex)(登録商標));ドキソルビシン(アドリアマイシンPFS注射薬(Adriamycin PFS Injection)(登録商標));リポソーム化ドキソルビシン(ドキシル(Doxil)(登録商標));プロピオン酸ドロモスタノロン(ドロモスタノロン(Dromostanolone)(登録商標));プロピオン酸ドロモスタノロン(マステロン注射薬(Masterone Injection)(登録商標));エリオットのB溶液(エリオットのB溶液(Elliott’s B Solution)(登録商標));エピルビシン(エレンス(Ellence)(登録商標));エポエチン・アルファ(エポジェン(epogen)(登録商標));エルロチニブ(タルセバ(Tarceva)(登録商標));エストラムスチン(Emcyt(登録商標));エトポシドリン酸塩(エトポフォス(Etopophos)(登録商標));エトポシド、VP−16(ベペシド(Vepesid)(登録商標));エキセメスタン(アロマシン(Aromasin)(登録商標));フィルグラスチム(ノイポゲン(Neupogen)(登録商標));フロクスウリジン(動脈内)(FUDR(登録商標));フルダラビン(フルダラ(Fludara)(登録商標));フルオロウラシル、5−FU(アドルシル(Adrucil)(登録商標));フルベストラント(ファスロデックス(Faslodex)(登録商標));ゲフィチニブ(イレッサ(Iressa)(登録商標));ゲムシタビン(ゲムザール(Gemzar)(登録商標));ゲムツズマブ・オゾガマイシン(マイロターグ(Mylotarg)(登録商標));ゴセレリン酢酸塩(ゾラデックス・インプラント(Zoladex Implant)(登録商標));ゴセレリン酢酸塩(ゾラデックス(Zoladex)(登録商標));ヒストレリン酢酸塩(ヒストレリン・インプラント(Histrelin implant)(登録商標));ヒドロキシウレア(ハイドレア(Hydrea)(登録商標));イブリツモマブ・チウキセタン(ゼヴァリン(Zevalin)(登録商標));イダルビシン(イダマイシン(Idamycin)(登録商標));イフォスファミド(IFEX(登録商標));イマチニブメシル酸塩(グリベック(Gleevec)(登録商標));インターフェロンアルファ2a(ロフェロンA(Roferon A)(登録商標));インターフェロンアルファ−2b(イントロンA(Intron A)(登録商標));イリノテカン(カンプトサール(Camptosar)(登録商標));レナリドマイド(レブリミド(Revlimid)(登録商標));レトロゾール(フェマーラ(Femara)(登録商標));ロイコボリン(ウェルコボリン(Wellcovorin)(登録商標)、ロイコボリン(Leucovorin)(登録商標));酢酸ロイプロリド(エリガード(Eligard)(登録商標));レバミソール(エルガミゾール(Ergamisol)(登録商標));ロムスチン、CCNU(CeeBU(登録商標));メクロレタミン・ナイトロジェンマスタード(マスタルゲン(Mustargen)(登録商標));酢酸メゲストロール(メゲース(Megace)(登録商標));メルファラン、L−PAM(アルケラン(Alkeran)(登録商標));メルカプトプリン、6−MP(プリネトール(Purinethol)(登録商標));メスナ(メスネックス(Mesnex)(登録商標));メスナ(メスネックス錠(Mesnex tabs)(登録商標));メトトレキサート(メトトレキセート(Methotrexate)(登録商標));メトキサレン(ウヴァデクス(Uvadex)(登録商標));マイトマイシンC(ミュータマイシン(Mutamycin)(登録商標));ミトタン(リソドレン(Lysodren)(登録商標));ミトキサントロン(ノバントロン(Novantrone)(登録商標));フェンプロピオン酸ナンドロロン(デュラボリン−50(Durabolin−50)(登録商標));ネララビン(アラノン(Arranon)(登録商標));ノフェツモマブ(ヴァールマ(Verluma)(登録商標));オプレルベキン(ニューメガ(Neumega)(登録商標));オキサリプラチン(エロキサチン(Eloxatin)(登録商標));パクリタキセル(パクセン(Paxene)(登録商標));パクリタキセル(タキソール(Taxol)(登録商標));パクリタキセル・タンパク質結合粒子(アブラキサン(Abraxane)(登録商標));パリフェルミン(ケピバンス(Kepivance)(登録商標));パミドロネート(アレディア(Aredia)(登録商標));ペグアデマーゼ(アダジェン(Adagen)(ウシ・ペグアデマーゼ(Pegademase Bovine))(登録商標));ペグアスパラガーゼ(オンキャスパー(Oncaspar)(登録商標));ペグフィルグラスチム(ニューラスタ(Neulasta)(登録商標));ペメトレキセド2ナトリウム(アリムタ(Alimta)(登録商標));ペントスタチン(ナイペント(Nipent)(登録商標));ピポブロマン(バーサイト(Vercyte)(登録商標));プリカマイシン、ミトラマイシン(ミトラシン(Mithracin)(登録商標));ポルフィマー・ナトリウム(フォトフリン(Photofrin)(登録商標));プロカルバジン(マツラン(Matulane)(登録商標));キナクリン(アタブリン(Atabrine)(登録商標));ラスブリカーゼ(エリテック(Elitek)(登録商標));リツキシマブ(リツキサン(Rituxan)(登録商標));サルグラモスチン(リューカイン(Leukine)(登録商標));サルグラモスチン(プロカイン(Prokine)(登録商標));ソラフェニブ(ネクサバール(Nexavar)(登録商標));ストレプトゾシン(ザノサール(Zanosar)(登録商標));スニチニブ・マレイン酸塩(スーテント(Sutent)(登録商標));タルク(スクレロゾール(Sclerosol)(登録商標));タモキシフェン(ノルバデックス(Nolvadex)(登録商標));テモゾロミド(テモダール(Temodar)(登録商標));テニポシド、VM−26(ヴァモン(Vumon)(登録商標));テストラクトン(テスラク(Teslac)(登録商標));チオグアニン、6−TG(チオグアニン(Thioguanine)(登録商標));チオテパ(チオプレックス(Thioplex)(登録商標));トポテカン(ハイカムチン(Hycamtin)(登録商標));トレミフェン(フェアストン(Fareston)(登録商標));トシツモマブ(ベキサール(Bexxar)(登録商標));トシツモマブ/I−131 トシツモマブ(ベキサール(Bexxar)(登録商標));トラツズマブ(ハーセプチン(Herceptin)(登録商標));トレチノイン、ATRA(ベサノイド(Vesanoid)(登録商標));ウラシル・マスタード(ウラシル・マスタード・カプセル(Uracil Mustard Capsules)(登録商標));バルルビシン(バルスター(Valstar)(登録商標));ビンブラスチン(ベルバン(Velban)(登録商標));ビンクリスチン(オンコビン(Oncovin)(登録商標));ビノレルビン(ナベルビン(Navelbine)(登録商標));及びゾレドロネート(ゾメタ(Zometa)(登録商標))。
【0171】
全ての特許、刊行物、及び確認された係属中の特許は、参考として本明細書に含まれる。
【0172】
本明細書において使用された略号は、以下の表に示した意味を有する。以下の表に記載されていない略号は、別に特に述べない限り、一般に使用される意味を有する。
【0173】
【化2】
【0174】
本発明のmTOR阻害剤及び抗IGF−1R抗体は、適当な材料を使用して、以下の一般的なスキームに従って調製してよく、かつその後の具体的な実施例によってさらに例示される。しかしながら、実施例に例示された具体的な抗癌剤が、本発明とみなされる唯一のジーナス(Genus)を形成するものと解釈されるべきではない。それ故、以下の例示的な実施例は、リストされた抗癌剤によって、又は例示を目的として使用された特定の物質によって、限定されるものではない。当業者は、これらの化合物を調整するため、以下の調製法の条件及びプロセスについて既知の変形を用いてもよいことを容易に理解するであろう。別に記載のない限り、すべての温度は摂氏である。
【0175】
合成法
【0176】
【化3】
【0177】
本発明は、ここに、以下の非限定的例において例示され、ここで、別段の指定のない限り:
1.全ての最終生成物は、NMR、LCMSにより分析した。
2.中間体は、NMR及び/又はTLC及び/又はLCMSにより分析した。
3.ほとんどの化合物は、シリカゲル上でのフラッシュクロマトグラフィー、逆相HPLC、再結晶化、及び/又はスウィッシュ(溶媒中に懸濁した後、固体を濾過する)により精製した。
4.反応の経過は、薄層クロマトグラフィー(TLC)及び/又はLCMSにより追跡し、また反応時間は例示のみのために示されている。
【0178】
実施例1
ジメチル−ホスフィン酸C−43ラパマイシンエステル
【0179】
【化4】
【0180】
ジクロロメタン 1.8mL中のラパマイシン(0.1g、0.109mmol)の冷却された(0℃)溶液に、2,6−ジ−t−ブチル−4−メチルピリジン 0.168g(0.82mmol)を、N2流下で添加し、直後にジクロロメタン 0.2mL中のジメチルホスフィン酸クロリド(0.062g、0.547mmol)の溶液を添加した。わずかに黄色の反応溶液を、0℃で、N2雰囲気下で3.5時間攪拌した(反応はTLCによりモニターした)。冷却した(0℃)反応溶液を、EtOAc 約20mLで希釈し、次にEtOAc(150mL)及び飽和NaHCO3(100mL)を含有する分液漏斗に移した。水層を除去し、有機層を氷冷した1N 塩酸(1x100mL)、飽和NaHCO3(1x100mL)、及び食塩水(1x100mL)で順次洗浄し、次にMgSO4上で乾燥し、濃縮した。粗生成物をシリカゲルフラッシュクロマトグラフィー(1:10:3:3 MeOH/DCM/EtOAc/ヘキサンで溶出)により精製して、白色固体 0.092gを得た:
1H NMR(300MHz,CDCl3)δ4.18(m,1H)、4.10(m,1H)、3.05(m,1H)、1.51(m,6H);31P NMR(121MHz,CDCl3)δ53.6;1013m/z(M+Na)。
【0181】
実施例2
ジメチルホスフィン酸C−43ラパマイシンエステル、別の合成法
ラパマイシン及びジクロロメタンを、窒素でパージした反応フラスコに装入する。攪拌した溶液を、約0℃に冷却する(−5±5℃の外部温度を反応の間中、維持する)。次に、ジクロロメタン中のクロロジメチルホスフィン酸クロリド(2.0モル当量)の溶液を、約8−13分間にわたり添加する。この直後に、ジクロロメタン中の3,5−ルチジン(2.2モル当量)の溶液を、約15−20分間にわたり添加する。双方の添加の間中、反応は0℃未満にとどまる。冷却した反応溶液を1時間攪拌し、次に、まだ冷たいうちに、飽和NaHCO3水溶液及びメチル−t−ブチルエーテル(MTBE)、酢酸エチル、又はジエチルエーテルを含有する抽出器に移す。製造過程のサンプルは、30及び60分の時点で取出す。サンプルは、反応ワークアップに記載したものと同様の方法で調製する。反応の進行は、TLC(1:10:3:3 MeOH/DCM/EtOAc/ヘキサン)及び逆相HPLC分析によりモニターする。単離した有機層を、氷冷した1N 塩酸、飽和NaHCO3(2x)、飽和NaCl水溶液で順次洗浄し、硫酸ナトリウム上で乾燥する。濾過及び溶媒除去し、残渣をアセトンで溶媒交換し、続いて真空中で濃縮して粗生成物を得て、これを順相及び逆相HPLCにより、純度について分析してもよい。
【0182】
実施例3
マウスモノクローナル抗体(MAB)の生成及び選択
IGF−1Rに対し特異的に向けられ、かつIRは認識しない、MAbの生成を目的として、6工程のスクリーニングを含んでなるプロトコールについて考察した。
【0183】
それは以下のものから構成される:
−ハイブリドーマを生成する目的で、マウスを組換えIGF−1Rで免疫化すること、
−培養上清を、免疫化に寄与した組換えタンパク質について、ELISAによりスクリーニングすること、
−陽性のハイブリドーマの全ての上清を、MCF−7腫瘍細胞の表面上で過剰発現している天然の受容体について、ELISAにより試験すること、
−ハイブリドーマ陽性の上清を、IGF−1R又はIRを各々発現するバキュロウイルスで感染された昆虫細胞上での、IGF−1R及びIRの特異的認識の観点から、2つの最初のスクリーニングで評価すること、
−この工程で選択された抗体が、MCF−7細胞の誘導されたIGF1増殖を、インビトロで阻害し得ることを検証すること、
−腫瘍MCF−7の増殖に対するインパクトという観点から保持された候補の、インビボの活性をヌードマウスにおいて確認すること。
【0184】
これらの個別の工程及び得られた結果の全てを、以下の実施例1において簡単に記載する。
【0185】
免疫化の工程では、マウスに8μgの組換えIGF−1Rを、皮下経路により2回注射した。雌ラットの細胞が、マウス骨髄腫Sp2OAg14の細胞と融合する3日前に、3μgの組換え受容体の静脈内注射により、マウスを刺激した。融合の14日後、ハイブリドーマ上清を、組換えIGF−1Rによって感作したプレート上で、ELISAによってスクリーンした。その上清が陽性であると判明したハイブリドーマを保持し、増殖させた後、FACScanで試験して、産生した抗体が同様に天然のIGF−1Rを認識し得ることを確認した。これを行なうため、IGF−1Rを過剰発現するエストロゲン依存性の乳房腫瘍由来のMCF−7細胞を、ELISAで選択されたハイブリドーマにより産生し各培養上清とともにインキュベートした。細胞表面上の天然/MAb受容体複合体は、蛍光色素へ結合した二次抗種抗体(secondary anti−species antibody)によって示された。図3Aないし3Cは、ハイブリドーマ7C10の上清で得られたヒストグラムタイプ(図3C)を、細胞標識単独+二次抗体(図3A)、又はコントロールアイソタイプを利用した標識(図3B)と比較して示す。
【0186】
選択のこの工程において、MAbを分泌すると同時に組換え受容体及び天然受容体を認識するハイブリドーマのみを選択し、クローニングした。これらのハイブリドーマによって分泌されるMAbを産生し、次に精製した後、2つの受容体を同時に認識するハイブリドーマを除去する目的で、IGF−1R又はIRを発現するSf9昆虫細胞上で、上記記載の方法に従ってFACScanで試験した。図4Aは、未感染細胞+二次抗体(1)、αIR3で標識された未感染細胞+二次抗体(2)、及び抗IGF−1R抗体で標識された未感染細胞+二次抗体(3)にそれぞれ対応するヒストグラム1、2、3の全回収率を示す。この最初の結果は、これらの未感染昆虫細胞の表面上には、検出可能なIGF−1R及びIRが存在しないことを充分に示している。図4Bは、IGF−1Rを発現するバキュロウイルスに感染された細胞の標識を示す。この2番目の図では、陽性コントロールとして用いたαIR3が、予想通り充分に細胞を標識するが(ピーク2)、一方、抗IR(ピーク3)は、単一細胞のピークに重なっている。最後に、図4Cでは、抗IRが、予想通り充分にIRを発現するSf9細胞を標識するが(ピーク3)、予想外なことに、文献ではIGF−1Rに特異的であると記載されているαIR3が、IRを認識するように見られる(ピーク2)ことが示されている。
【0187】
この3番目のスクリーニング系で得られた結果は、表1に要約されており、IGF−1Rを認識しかつIRを認識しないという基準を満たすMAb:7C10の生成を示している。Mab 7C10のアイソタイピングにより、それがIgG1を含むことが示されている。
【0188】
【表1】
【0189】
MAbの選択用に提供された最後の2つのスクリーニングは、後者が、細胞系MCF−7においてIGF−Iによって誘導される細胞増殖を、インビボ及びインビトロにおいて大いに阻害し得ることを証明するものであった。
【0190】
インビトロの選択では、MCF−7細胞を接種し、ウシ胎児血清を除去し、次に漸増濃度のIGF−I(1ないし50ng/ml)の存在下で、最終濃度10μg/mlまで添加された、試験されるべき7C10抗体の存在下又は非存在下でインキュベートした。この実験では、市販のαIR3 MAbを陽性コントロールとし、7G3 MAb(7C10と同時に単離され、天然の受容体を弱く認識する(FACSによるMFIは、MAb 7C10の200に比較して50))を、コントロールアイソタイプとして添加した。細胞増殖は、細胞によるトリチウムチミジンの取込みをβカウンターで追跡することにより推定する。結果は、増殖指数として表わす。図5に提示したデータは、IGF1がMCF−7細胞の増殖を用量依存的に刺激し得ることを示している。陽性コントロールとして使用したMAb αIR3は、IGF−Iによって誘導されるMCF−7細胞の増殖を完全に阻害する。同様に、MAb 7C10は、IGF−Iによって誘導されたMCF−7細胞の増殖を有意に阻害する。最後に、アイソタイプコントロールとして用いたMAb 7G3は、予想通りに、MCF−7細胞のインビトロの腫瘍細胞増殖に影響なしにうまくいく。
【0191】
インビボの選択は、確立された腫瘍モデルにおいて実施した。これを行なうため、ヌードマウスは、マウスモデルにおいて腫瘍を得るために不可欠な徐放性エストロゲンの皮下移植を受けた。エストロゲンの移植の24時間後、5.106のMCF−7細胞を、マウスの右側腹部の皮下に移植する。この細胞移植の5日後には、腫瘍は測定可能であり、6匹のマウスからなるバッチがランダムに形成される。マウスの処置は、週当たり2回、5ないし6週間にわたり、250μg/1回用量/マウスの用量で実施する。コントロール群では、マウスを、マウスコントロールアイソタイプと同様に処理する。図6Aに提示した結果は、抗体7C10により誘導される、非常に有意な腫瘍増殖阻害を示している。通常はIGF1の受容体のドメインの参照として用いられ、エストロゲン依存性の腫瘍の増殖に対し何らインビボで活性をもたないことが知られる、αIR3について得られるデータを参照した場合、この活性は特に予想外である(図6B参照)。同様に、マウスMAb 1H7由来の組換え抗体scFv−Fcで得られた結果に比較して(図6C参照)、MAb 7C10は、MCF−7細胞の増殖のインビボでの阻害においてはるかに効力がある。
【0192】
実施例4
ヒト肺癌細胞系におけるMK−0646及びリダフォロリムスの効果
要約:併用が提案された根拠は、MK−0646及びリダフォロリムスの各々が、組合された場合に、PI3キナーゼシグナリング経路を介して発癌シグナリングを阻害することにより作用すること、及び2つが組合された状態では、いずれかの薬剤が単独で作用するよりも有効な経路の阻害をもたらすことを示唆する観察に基づいている。また、カオ(Cao)ら著、「Cancer Research」、2008年も参照。
【0193】
手短に言えば、MK−0646(これは、IGF1Rを標的とするモノクローナル抗体である)、及び、リダフォロリムス(これは、mTOR阻害剤であり、またラパマイシン類似体である)は、各々、肺癌の治療用に現在開発中である。ラパマイシン類似体による治療は、AKTのリン酸化により測定される、AKTシグナリングのアップレギュレーションをもたらす。リダフォロリムスによるmTORの阻害は、腫瘍増殖停止を誘導し得るが、IRS−1により媒介されるネガティブフィードバックループを無効にし、結果としてAKTを活性化し、このことがその抗腫瘍活性の低減に関連づけられてきた。AKTの、このフィードバック活性化は、IGF1Rシグナリング経路によって媒介される。最近の臨床研究は、このフィードバックメカニズムによるAKTの活性化が、ラパマイシンを処置された患者におけるより短い進行期間(shorter time to progression)に関連づけ得ることを示唆している(クラフェシー(Cloughesy)ら著、「PLoS Medicine」、2008年)。さらに、mTORは、MK−0646の効力の重要なエンハンサーであると考えられ、したがって、リダフォロリムスをMK−0646と併用することでAKTのこのフィードバック活性化を阻止することが、PI3K経路の阻害、並びにMK0646の抗腫瘍活性を増強するために有益り得る。併用が提案された根拠は、一部は、上記の観察に基づいている。この可能性を研究するため、本発明者らは、提案された組合せを、一群の肺癌細胞系において検査した。本明細書において以下に詳述されるのは、MK−0646とリダフォロリムスとを含んでなる併用治療がPI3K経路の阻害及び細胞増殖を有意に増強するという仮説を支持するものである。同様に、提案された組合せは、エルロチニブ抵抗性、KRAS突然変異肺癌異種移植モデルにおいて、抗腫瘍活性の増強を実証した。
【0194】
(A)MK−0646+リダフォロリムス併用は、PI3K経路標的化を増強する:
方法:すべてのNSCLC細胞系は、ATCCから入手し、10% ウシ胎児血清(インビトロジェン(Invitrogen))と共に、RPMI 1640中に維持した。ウエスタンブロット分析では、細胞からの全タンパク質溶解物を、6ウエルプレート中で培養し、リダフォロリムス(10nM)若しくはMK−0646(10ng/ml)のいずれかで、又は組合せて4時間処置し、SDSゲルローディング色素(インビトロジェン)中に採取した。サンプルは、指示された全抗体又はホスホ特異抗体で、次に二次抗体(Cell Signaling Technology, CST)でウエスタンブロットし、次にスーパーシグナル(SuperSignal)化学発光基質(Pierce)とインキュベートした。次いでブロットを、コダック バイオマックス ライト フィルム(Kodak Biomax Light Film)に暴露させた。ERK、p−ERK(Thr202/Tyr204)、AKT及びp−AKT(Ser473)、IGF1R S6K&P−S6K(T389)、IRS1&P−IRS1(S302)、及びアクチンに対する抗体は、CSTから入手した.細胞周期分析は、H2122細胞にて、指示された化合物(上記参照)による24時間処理の後に実施した。百万個の細胞を、透過性化し、ヨウ化プロピジウム(PI)で、製造業者による記載の通り染色した(BD Pharmingen #550825)。PIは、DNA及びRNAの双方に結合するため、後者をリボヌクレアーゼ(RNアーゼA)による消化により除去した。フローサイトメトリーにより測定されるDNA含有量は、細胞周期について有用な情報を示し得る。細胞周期のG2及びM期にある細胞は、G0及びG1期にあるものの2倍のDNA含有量をもつ。S期にある細胞は、これらの両極端の間にあるDNA含有量をもつ。PIは、フローサイトメーターを使用し、562−588nmバンドパスフィルタを用いて、橙色の範囲内で検出される。
【0195】
分析:リダフォロリムスとMK−0646との併用は、ヒト肺癌細胞系で評価されてきた。双方の化合物は、PI3キナーゼシグナリング経路を介して発癌シグナリングを阻害することにより作用する。本明細書で実施された研究は、2つの薬剤がいずれかの薬剤単独よりもさらに有効な経路阻害をもたらすという他の報告を確認した。mTOR(TORC1複合体)の阻害が、二次的効果をもたらし、この結果、活性のあるリン酸化型のAKTが生じ、これにより腫瘍細胞の生存が促進されることが先に報告されている。重要なことには、MK−0646によるIGF1Rの阻害は、この効果を阻止する上で有効である。図1は、フィードバックループ現象を例示しており、2つの薬剤の併用が、この効果を打ち消し、発癌性PI3K経路の標的化を改善することを示している。mTOR阻害剤は、S6キナーゼの、及びしたがってタンパク質翻訳の有効な阻害剤である。しかしながら、S6キナーゼ阻害はまた、IGF1Rシグナリングを抑制するS6キナーゼ(S6K)により媒介されるネガティブフィードバックの廃止につながる。このフィードバックループを阻止することの効果は、IGF1Rシグナリングが高められることであり、活性のあるリン酸化型のAKT(AKT−P)のレベルの上昇につながり、これにより腫瘍生存が駆動される。リダフォロリムスに類似しているラパログ(rapalogue)RAD001による患者の治療は、結果としてAKT−Pの上昇をもたらす(オーライリー(O’Reilly,K.E.)ら著、「Cancer Research」、2006年、図1B)。ある研究者らは、このことが、ある状況において単一の薬剤として使用した場合の化合物の有効性を限定し得ることを示唆してきた(クラフェシー(Cloughesy)ら著、「PLoS Medicine」、2008年)。この効果がIGF1R活性に依存すると考えられることから、本発明者らは、mTORを、IGF−1R阻害剤であるMK−0646と組合せて使用することを提案した。今日までのところ、収集されたデータは先の知見と一致する。データは、非小細胞肺癌細胞系H2122を、リダフォロリムスとMK−0646との組合せと共に同時治療することが、シグナリングの阻止において、いずれかの薬剤単独よりもさらに有効であり(図1C、右)、S6Kのリン酸化(リダフォロリムスによる)及びAKTリン酸化(MK−0646による)の双方を妨げることを示唆している。併用はまた、細胞周期阻害及びこれらの腫瘍細胞のインビトロの生存の阻害においても、単一の薬剤よりもさらに有効である。併用によるH2122腫瘍細胞系の治療は、活発に分裂している細胞のパーセントの低減、及び死んだか又は死にかけている細胞の増加をもたらした(図1D)。
【0196】
(B)リダフォロリムス+MK−0646併用は、インビトロの効力を増大する。
MK−0646若しくはリダフォロリムス又は組合せの効力を評価するため、本発明者らは、軟寒天アッセイを用いて、一群中の肺腫瘍細胞系において、これらの阻害剤の存在下でアンカレッジ(anchorage)非依存性増殖を評価した。コロニー形成アッセイは、補充IGFの不在下で実施した。MK−0646若しくはリダフォロリムス又は組合せの効果は、9つの細胞系(5つの突然変異−KRAS;4つのwt−KRAS)で評価した。MK−0646単一薬剤の感受性は、1つのKRAS突然変異細胞系(H23)及び3つのKRAS野生型細胞系において観察された(図2)。IGF1Rの発現が低いH1703は、MK−0646には抵抗性であった。高レベルのIGF−1Rをもつ2つのKRAS突然変異細胞系(A549&H2122)では、単一薬剤の活性は限られたものにすぎなかったのに対し、併用は有意な増殖阻害を示した。一群中のほぼ全体の細胞系が、併用の利点を示した。KRAS状態と、併用に対する応答との有意な相互関係は何ら観察されなかった。
【0197】
方法:軟寒天アッセイは、96ウエルガラス底プレート(MatriCal)において行なった。細胞は、14%FBS及び0.3%(w/v)シープラーク(SeaPlaque)アガロース(Lonza Rockland,Inc)を補充したRMPI1640 100μL中で、各ウエル当たり3,000−9,000細胞の濃度で、0.8%アガロースを補充した同じ培地からなるボトムレイヤーの上に播種した。化合物は、アガロースを固化させた後に、補充した培地100μl中で添加した。細胞は、7−14日間インキュベートした後、ラバセル(LavaCell)(Active Motif)で一晩染色した。コロニーは、Isocyte(登録商標)レーザースキャニングサイトメータを用いて定量化した。MK−0646の、単独又は標準的な治療薬剤と組合せての、アンカレッジ非依存性増殖阻害能を、軟寒天コロニー形成アッセイにおいて評価した。RTK状態は、全タンパク質溶解物中で、P−RTKアレイ(R&D biosciences)を用いて、製造業者による記載の通り評価した。KRASにおける活性化突然変異は、公開された癌ゲノムデータベース(Sanger)から同定した。
【0198】
(C)A549肺異種移植モデルの組合せにおける、MK−0646及びリダフォロリムスの併用の利点。
リダフォロリムス及びMK−0646の組合せを、A549突然変異−KRAS異種移植モデル(A549 mutant−KRAS xenograft model)において評価した。MK−0646が、0.2又は2mpkのいずれかで、リダフォロリムス(0.1mpk)と組合せて投薬された場合、有意な抗腫瘍活性を示した。もっとも、MK−0646を20mpkで、リダフォロリムス(0.1mpk)と組合せて投薬すると、いずれかの薬剤単独に対し、統計的な有意性をもって腫瘍退縮を示した(図3)。同様の結果は、腫瘍重量を群間で比較した場合に観察された。MK−0646(20mpk)をリダフォロリムス(0.1mpk)と組合せて投薬されたマウスでの腫瘍重量には、いずれかの薬剤単独に対し、統計的に有意な減少があった(図3)。これらのデータを合わせれば、リダフォロリムスと組合された低い用量のMK−0646は腫瘍静止状態を誘導するのに対し、リダフォロリムスと組合された高用量のMK−0646(20mpk)は、腫瘍退縮を引き起こし得ることの、強力なインビボの証拠を提供する。また、腫瘍重量の有意な減少も、この組合せにおいて観察された(図4)。このモデルにおけるエルロチニブによる処置は、結果として何ら容易に評価し得る腫瘍増殖抑制を生じなかった(図5)。このモデルでのエルロチニブによる効力の欠如は、KRASにおける活性化突然変異がこのモデルに存在するとすれば、驚くべきことではない。結果として、データは、エルロチニブ耐性のNSCLCモデルにおける、MK−0646&リダフォロリムス併用の利益を証明している。
【0199】
方法:2.5x106のA549ヒトNSCLC細胞を、4−6週齢の無胸腺Nude−Foxnlnuマウス(Charles River Laboratories)の右側腹部の皮下に注射した。腫瘍が約300mm3のサイズ(長さ*幅*幅*0.5)に達した時、マウスを処置群に無作為化した。マウス(n=8/群)に、ビヒクルを週1回3週間にわたり(qwkx3)(20mM L−ヒスチジン、150mM NaCl、0.5% PS80 pH=6)、又は0.2若しくは2若しくは20mpkのMK−0646を腹腔内に、qwkx3で、或いはリダフォロリムス(0.1mg/kg)を、又はMK−0646と組合せて、3週間にわたり投薬した。動物を秤量し、腫瘍体積を、実験の間週2回、及び終了時に、キャリバーで測定した。腫瘍重量は、終了時に測定した。21日目、動物をCO2窒息により犠牲にした。マウスは、最後の投薬の24時間後に犠牲にした。犠牲にする時点で、組織サンプルを収集し、加工した。
【0200】
処理の終了時の相対腫瘍体積を、以下の表に表わす。負の値は、腫瘍の退縮を表わす。単一薬剤に比較して、有意な腫瘍増殖阻害が、全てのMK−0646&リダフォロリムスの併用群において観察された。
【0201】
【表2】
【0202】
実施例5
MK−0646エンハンサースクリーン
要約:多くの一連の証拠が、IGF1Rシグナリングの過剰活性化が、腫瘍進行と関連することを示唆している。IGF1R及びそのリガンドIGF1の双方は、しばしば、ヒト癌において過剰発現されており、不良な予後に関係づけられている(ミラー(Miller)及びイー(Yee)、2005年)。さらに、IGF1又はIGF1Rのいずれかの強制された過剰発現は、動物組織において、自然発生腫瘍の形成をもたらす(ジョーンズ(Jones)ら、2006年)。対照的に、IGF1Rノックアウトマウスから単離された線維芽細胞腫が、発癌遺伝子の過剰発現による形質転換に抵抗性であることから、低減したIGF1R機能は、腫瘍発生を防止し得る(セル(Sell)ら、1994年)。
【0203】
IGF−1R特異的抗体(MK−0646)を用いて、併用療法プロトコールにおいての有効性を増強し得るかどうかを確認するため、レンチウイルス媒介RNAiスクリーンを使用した。スクリーンの結果、37の標的が同定され、そのサイレンシングは、MK−0646に対する腫瘍細胞の感受性を有意に増強した。これらのエンハンサーの中には、PI3キナーゼシグナリングカスケードの4つの別個のポジティブレギュレータ(PI3KCA、PDPK1、AKT2、及びFRAP1/mTOR)を標的とするshRNAがあった。対照的に、その経路のネガティブレギュレータであるPTENを標的とするshRNAは、MK−0646に対する抵抗性を示した。これらのデータを合わせれば、PTENについてネガティブな腫瘍は、MK−0646に対し抵抗性を示し得る。さらに、データは、mTORi又はAKTi又はPI3KiなどのPI3K経路を標的とする阻害剤を、MK−0646と組み合せることが、MK−0646応答を実際に強化し得ることを示唆する仮説を支持している。
【0204】
結果:本発明者らは、レンチウイルス媒介RNAiスクリーニングを適用して、インスリン様増殖因子1受容体(IGF1R)mab、特にMK−0646に対する腫瘍細胞感受性のキナーゼレギュレータを同定した。この目的に向けて、480の別個のヒトキナーゼを標的とする1439のレンチウイルスshRNAベクターをスクリーンして、MK−0646の抗腫瘍細胞活性の強力なエンハンサーを同定した。
【0205】
種々の濃度のMK−0646(200μg/mLないし300ng/mL)を用いた9つの別個のスクリーンを、MK−0646及びキナーゼ標的レンチウイルスshRNAベクターを用いて実施した。スクリーンから、MK−0646エンハンサーの上位3%を代表する37のコンセンサスヒットのリストが同定された。図10参照。これらのヒットのうち顕著なものは、2つの別個のベクター、PDPK1,AKT2、及びFRAP1/mTORによる、標準ホスファチジルイノシトール3キナーゼ(PI3K)経路−PIK3CAの多数のメンバーを標的とするshRNAベクターであった。スクリーンからの2つの別々のコンセンサスヒット(CCRK及びNEK8)は、PI3K/AKT経路のレギュレータとして、既にsiRNAスクリーンにおいて同定されていた(ブレイス(Brace)ら、2006年参照)。またMK−0646エンハンサーのリストの中で注目すべきであるのは、Rasシグナリングカスケードの2つのメンバーを標的とするshRNAであった(2つの別個のベクターによるB−Raf及び2つの別個のベクターによるMAP2K1)。全てにおいて、上位37のshRNAヒットのうちの11は、IGF1R活性化に続いて活性化されるものと仮定された、2つの確立されたシグナリングカスケードのメンバーであるキナーゼを標的とする。図6参照。
【0206】
HT29結腸癌細胞によるコロニー形成に基づく、独立した増殖アッセイを実施して、上記で言及されたスクリーニングヒットを確認した。
【0207】
方法/分析−図6:新鮮なベクターによる感染に続き、細胞を5用量力価のMK−0646に暴露し、次に9日間にわたりコロニー形成させた。37のshRNAヒットの各々について、MK−0646に対する感受性の増強能を、空のベクターコントロール、並びにshRNAキナーゼベクターセットから無作為に選択された3つの「非ヒット」(”non−hit”)shRNAベクターと比較した。さらに、PI3K経路の多くのポジティブエフェクターがヒットとしてスコアされたことから、脂質ホスファターゼPTEN(PI3Kシグナリングの主要なネガティブレギュレータ)を効率的にサイレンスさせる別のベクターもまた試験した。PTENは、確立した腫瘍サプレッサーであり、PI3K活性の生成物を脱リン酸化することによって作用し、それ故下流キナーゼの活性化を妨げる。ベクターなし(MK−0646単独)及び処置なし(ウイルスなし及び薬剤なし)のコントロールもまた実施した。
【0208】
方法:480の個別の遺伝子、主としてキナーゼ、を標的とするレンチウイルスは、1439のレンチウイルスshRNAベクター(Kalypsis library,GNF)を、293T細胞において、インビトロジェン・レンチウイルスパッケージングシステム(Virapower packaging system,Invtrogen)により推奨されるパッケージングベクターと同時トランスフェクトすることにより、パッケージし生成した。HT−29細胞(n=500)を、384ウエルプレートにて、10% FBS(DMEM中)の存在下で培養した。翌日、細胞をパッケージされたウイルス10uLで感染させた。3日目、培地を除去し、細胞を1日回復させた。4日目、細胞をMK−0646(200μg/mLないし300ng/mL)で処置した。8日目、細胞増殖の量を、既に記載された通りにアラマーブルーで染色することにより評価した(クリングホッファー(Klinghoffer)ら、「Assay and Drug Development Technologies」、2008年)。
【0209】
方法−図7:HT29細胞は、1500細胞/ウエルで、6ウエルプレートに播種し、ウイルスに16時間暴露した。ウイルスを除去し、薬剤を含有しないか又はMK−0646の希釈物(0.05、0.1、0.5、1、又は10ug/ml)を含有する新鮮な培地で置き換えた。MK−0646は、アッセイの過程にわたり3日ごとに交換した。プレートは、薬剤添加の9日後にクリスタル紫で染色した。コロニー数及び面積は、AlphaImagerを用いたイメージングにより計算した。
【0210】
無作為に選ばれた3つのshRNAベクターの、空のベクター及びベクターなしのコントロールとの比較は、腫瘍細胞のMK−0646に対する感受性に何ら差異を示さなかった。対照的に、このアッセイにおいて評価し得た20スクリーニングのうち11は、腫瘍細胞増殖のMK−0646阻害に2倍以上の増強を生じる結果となった。図7参照。
【0211】
定量的リアルタイムPCR分析は、これらのベクターの10/11が、その標的を>50%、また7/11が>70%サイレンスさせたことを示し、観察された増強が意図されたサイレンシングによるものであったことを示唆している−図10。同様に、このアッセイの使用により、37のshRNAヒットのうち17が、薬剤の不在下で細胞毒性を生じる結果となり、MK−0646感受性化の評価が妨げられた。これらの毒性shRNAは、細胞が感染に続き、10用量力価のMK−0646に暴露されたことを除いて、プライマリースクリーンに類似の短時間(72時間)のアラマーブルーアッセイを用いて分析した。このアッセイの結果として、さらなる8つのshRNAが同定され、MK−0646に対する腫瘍細胞感受性の有意な(p<0.05)増強を実証した。図11参照。
【0212】
図7及び10を参照すれば、IGF媒介性の腫瘍増殖におけるPI3K経路シグナリングの主要な役割と一致して、コロニーアッセイによる上位3つのMK−0646エンハンサーは、PI3K、FRAP1/mTOR、及びNEK8を標的とするベクターであった(これら3つのベクターは、それぞれ86%、80%、及び70%の標的サイレンシングをもたらした)。
【0213】
さらに、72時間のアラマーブルーアッセイによる最強のエンハンサーは、推定上のPI3K経路レギュレータCCRKを標的とするベクターであった(64%の標的サイレンシング:示さず)。反対に、PTENを標的とするベクターに暴露された細胞は、MK−0646の抗増殖効果に対し抵抗性であり、処置されなかった細胞と同様の増殖を示した(図6)。Ras経路のメンバーを標的とするベクター、MAP2K1及びB−raf(それぞれ97%及び75%のサイレンシング)もまた確認された。図10参照。PI3K及びRASシグナリングカスケードのメンバーの間に広範囲のクロスレギュレーションが存在するとすれば、MAP2K1及びB−raf shRNAの感受性効果は、一部は、PI3Kシグナリングのクエンチングによることが仮定される。
【0214】
今日までのところ、同じ遺伝子を標的とする2つのshRNAが、プライマリースクリーンで増強を証明する場合(PIK3CA、B−raf、MAP2K1、BUB1、及びCSNK1A1)、2つのshRNAの一方は確認されるのに対し、他方は薬剤の不在下で実証された毒性をもつように見える。第2のshRNAの確認には、より短いアッセイフォーマットが必要であってよい。
【0215】
PTENの結果をさらに支持するため、アッセイを、PTEN shRNAの安定なインテグレーションを内部にもつ細胞上で実施した。これらを、ベクターコントロール、又はCSK及びMAP4K5を標的とするshRNA、を安定に発現する細胞に対し比較することを開始した。CSK及びMAP4K5を標的とするベクターは、プライマリースクリーンからのコンセンサスヒットであったが、MAP4K5のみをコロニーアッセイで確認した。新鮮なウイルスの適用の結果と同様に、PTEN shRNAを発現する細胞系は、コロニーアッセイによってMK−0646抵抗性を示したのに対し、CSK shRNAを発現する系は、ベクターコントロールと同様に応答し、MAP4K5 shRNAを発現する細胞は、薬剤に対し感受性化された。図8参照。
【0216】
方法−図8:PTEN、CSK、及びMAP4K5を標的とする安定なインテグレーションshRNAを内部にもつHT29細胞を、1500細胞/ウエルで、6ウエルプレートに播種した。翌日、何ら薬剤を含有しないか、又は0.01、0.1、1、10、又は100ug/mlのMK−0646を含有する新鮮な培地を、細胞に与えた。MK−0646は、アッセイの過程にわたり3日ごとに交換した。プレートは、薬剤添加の9日後にクリスタル紫で染色した。コロニー増殖は、上記記載の通り定量化した。MK−0646に対する応答は、上記で分析された各レンチウイルスベクターへの急性暴露に続いて観察されたものと同様であった。
【0217】
データ及び付随する観察から、PTENに機能喪失型突然変異のある腫瘍をもつ患者は、MK−0646による処置に対し不十分に応答し得ることが示唆される。かかる突然変異は癌において一般的であることから、本発明者らは、PI3Kを阻害することによりMK−0646抵抗性細胞が薬剤に対し再感受性化され得るかどうか判定することを試みて、追加のアッセイを実施した。
【0218】
方法−図9:ベクターコントロールの安定な発現があるか(赤色の線)(MK−0646に感受性)、又はPTEN shRNAの安定な発現がある(青色の線)(MK−0646に抵抗性)、HT29細胞を、PI3K、PDPK1、又はMELKを標的とするshRNAベクターで感染させた。(A)図6に示した本発明者らのアッセイと同様に、PI3K又はMELKを標的とするshRNAによる、ベクターコントロールHT29細胞の感染は、結果としてMK−0646に対し、各々強い及び中程度の感受性化を生じるが、一方PDPKlを標的とするshRNAは何ら効果をもたない。(B)MELK又はPDPK1による安定なPTEN、shRNA HT29細胞の感染は、PTEN欠失により引き起こされるMK−0646抵抗性の逆転には、何ら影響をもたない。対照的に、PI3K shRNAベクターによる感染は、この抵抗性を完全に逆転させる。
【0219】
分析:本発明者らの先の結果に一致して、PTEN shRNAを発現する細胞は、ベクターコントロールを発現する細胞に比較して、MK−0646に抵抗性であった。図9Aを参照すれば、本発明者らの先の実験と一致して、PDPK1ではないがPI3K又はMELKのいずれかを標的とするベクターによるコントロール細胞の感染は、細胞をMK−0646に対し感受性化した。PTEN欠失細胞では、MELK shRNAによる感染は、これらの細胞がMK−0646抵抗性のままであるため、何ら強い影響はなかった。PI3Kを標的とするshRNAによる感染は、しかしながら、MK−0646の効果に対し、PTEN shRNA系を再感受性化し得た−図9B。したがって、PI3Kを介したシグナリングが、IGF1R媒介性の腫瘍細胞増殖にとり律速となるように考えられる。この観察は、過剰活性PI3Kシグナリングをもつ患者は、抗IGF−1R薬剤による単一療法に対し不応答性でありえる、mTORなどのPI3K経路のメンバーの阻害剤による併用療法は、実際に有効であり得ることを示唆している。
【0220】
本発明の多数の実施態様を記載してきたが、基本的な例は、本発明によって包含される別の実施態様を提供するべく、変更されてもよい。それ故、本発明の範囲は、単なる例として記述された具体的な実施態様よりもむしろ、添付の特許請求の範囲によって提起されるべきであることが理解されるべきである。
【図1−A】
【図1−B】
【図1−C】
【図1−D】
【図2−A】
【図2−B】
【図3−A】
【図3−B】
【特許請求の範囲】
【請求項1】
非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌を、mTOR阻害剤及び抗IGF−1R抗体を用いて治療する方法であって、ここで、mTOR阻害剤が、リダフォロリムス、エベロリムス、テムシロリムス、及びそれらの組合せからなる群より選択され、また抗IGF−1R抗体が、ダロツズマブ、フィギツムマブ、シクスツムマブ、SHC717454、ロシュ(Roche)R1507、及びアムジェン(Amgen)AMG479からなる群より選択される、該方法。
【請求項2】
前記mTOR阻害剤がリダフォロリムスである、請求項1に記載の方法。
【請求項3】
前記抗IGF−1R抗体が、少なくとも1つの非ヒト由来の重鎖相補性決定領域(CDR)と、少なくとも1つの非ヒト由来の軽鎖相補性決定領域(CDR)とを含んでなり、ここで、IGF−1Rに結合する前記抗体が、以下:(a)IGF−1Rには結合するが、IRには結合しない;(b)インスリン受容体とインスリン成長因子受容体とを含んでなるハイブリッド受容体(IR/IGF−1Rハイブリッド−R)には結合するが、単独のIRには結合しない;(c)ヒトIGF−1Rと、IGF−1及び/又はIGF−2との間の結合を阻害する;(d)前記ハイブリッド−R及びその天然のリガンド(好ましくは本明細書ではIGF1及び/又はIGF2及び/又はインスリンとして示されている)と、100nM未満の阻害定数及び/又はIC50で結合する;(e)前記IGF−1Rのチロシンキナーゼ活性を特異的に阻害する;(f)前記ハイブリッド−Rのチロシンキナーゼ活性を特異的に阻害する;(g)前記ハイブリッド−Rについて、10nM以下の結合親和性を有する;(h)IGF−1R発現をダウンレギュレートする;(i)ハイブリッド−R発現をダウンレギュレートする;(j)インビボの腫瘍増殖を阻害する、からなる群より選択される特性の少なくとも1つを有する、請求項2に記載の方法。
【請求項4】
前記重鎖CDRが、配列番号4、5、又は6からなる群より選択されるアミノ酸配列を含んでなり、かつ前記軽鎖CDRが、配列番号1、2、又は3からなる群より選択されるアミノ酸配列を含んでなる、請求項3に記載の方法。
【請求項5】
ヒト化抗体、又はその機能性フラグメントの1つが、配列番号7、若しくは8からなる群より選択されるアミノ酸配列を含んでなる軽鎖か、又は配列番号9、10、若しくは11からなる群より選択されるアミノ酸配列を含んでなる重鎖を含んでなる、請求項3に記載の方法。
【請求項6】
前記抗IGF−1R抗体が、ダロツズマブである、請求項3に記載の方法。
【請求項7】
前記mTOR阻害剤が、リダフォロリムスであり、かつ前記抗IGF−1R抗体が、ダロツズマブである、請求項1に記載の方法。
【請求項8】
リダフォロリムスが10mgないし40mgの用量で投与される、請求項7に記載の方法。
【請求項9】
リダフォロリムスが、週5回投与される、請求項8に記載の方法。
【請求項10】
ダロツズマブが10mg/kgの用量で静脈内投与される、請求項7に記載の方法。
【請求項11】
ダロツズマブが週1回投与される、請求項7に記載の方法。
【請求項12】
ダロツズマブが2週間に1回投与される、請求項7に記載の方法。
【請求項1】
非小細胞肺癌、乳癌、結腸直腸癌、軟部組織又は骨肉腫、及び子宮内膜癌からなる群より選択される癌を、mTOR阻害剤及び抗IGF−1R抗体を用いて治療する方法であって、ここで、mTOR阻害剤が、リダフォロリムス、エベロリムス、テムシロリムス、及びそれらの組合せからなる群より選択され、また抗IGF−1R抗体が、ダロツズマブ、フィギツムマブ、シクスツムマブ、SHC717454、ロシュ(Roche)R1507、及びアムジェン(Amgen)AMG479からなる群より選択される、該方法。
【請求項2】
前記mTOR阻害剤がリダフォロリムスである、請求項1に記載の方法。
【請求項3】
前記抗IGF−1R抗体が、少なくとも1つの非ヒト由来の重鎖相補性決定領域(CDR)と、少なくとも1つの非ヒト由来の軽鎖相補性決定領域(CDR)とを含んでなり、ここで、IGF−1Rに結合する前記抗体が、以下:(a)IGF−1Rには結合するが、IRには結合しない;(b)インスリン受容体とインスリン成長因子受容体とを含んでなるハイブリッド受容体(IR/IGF−1Rハイブリッド−R)には結合するが、単独のIRには結合しない;(c)ヒトIGF−1Rと、IGF−1及び/又はIGF−2との間の結合を阻害する;(d)前記ハイブリッド−R及びその天然のリガンド(好ましくは本明細書ではIGF1及び/又はIGF2及び/又はインスリンとして示されている)と、100nM未満の阻害定数及び/又はIC50で結合する;(e)前記IGF−1Rのチロシンキナーゼ活性を特異的に阻害する;(f)前記ハイブリッド−Rのチロシンキナーゼ活性を特異的に阻害する;(g)前記ハイブリッド−Rについて、10nM以下の結合親和性を有する;(h)IGF−1R発現をダウンレギュレートする;(i)ハイブリッド−R発現をダウンレギュレートする;(j)インビボの腫瘍増殖を阻害する、からなる群より選択される特性の少なくとも1つを有する、請求項2に記載の方法。
【請求項4】
前記重鎖CDRが、配列番号4、5、又は6からなる群より選択されるアミノ酸配列を含んでなり、かつ前記軽鎖CDRが、配列番号1、2、又は3からなる群より選択されるアミノ酸配列を含んでなる、請求項3に記載の方法。
【請求項5】
ヒト化抗体、又はその機能性フラグメントの1つが、配列番号7、若しくは8からなる群より選択されるアミノ酸配列を含んでなる軽鎖か、又は配列番号9、10、若しくは11からなる群より選択されるアミノ酸配列を含んでなる重鎖を含んでなる、請求項3に記載の方法。
【請求項6】
前記抗IGF−1R抗体が、ダロツズマブである、請求項3に記載の方法。
【請求項7】
前記mTOR阻害剤が、リダフォロリムスであり、かつ前記抗IGF−1R抗体が、ダロツズマブである、請求項1に記載の方法。
【請求項8】
リダフォロリムスが10mgないし40mgの用量で投与される、請求項7に記載の方法。
【請求項9】
リダフォロリムスが、週5回投与される、請求項8に記載の方法。
【請求項10】
ダロツズマブが10mg/kgの用量で静脈内投与される、請求項7に記載の方法。
【請求項11】
ダロツズマブが週1回投与される、請求項7に記載の方法。
【請求項12】
ダロツズマブが2週間に1回投与される、請求項7に記載の方法。
【図4】

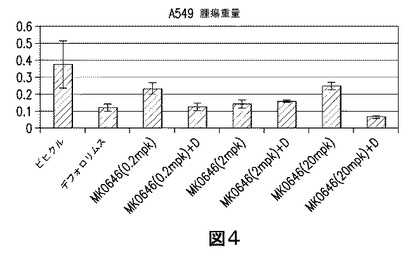
【図5】
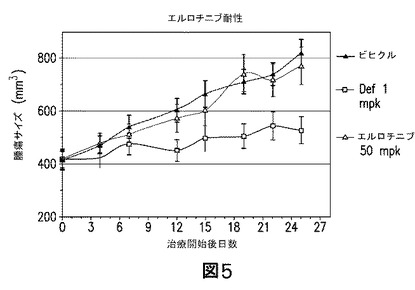
【図6】

【図7−A】
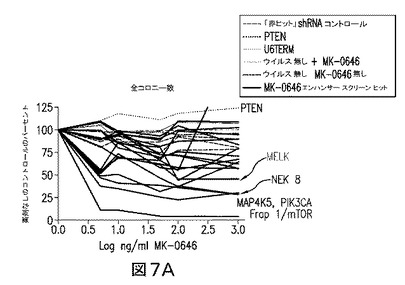
【図7−B】
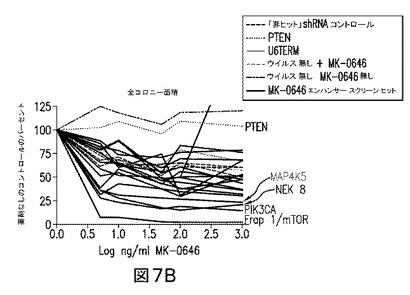
【図8】
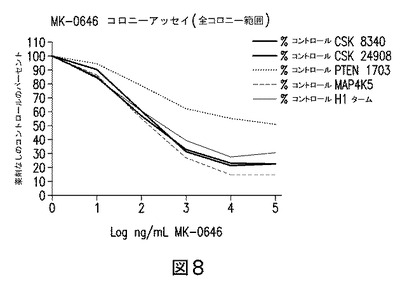
【図9−A】
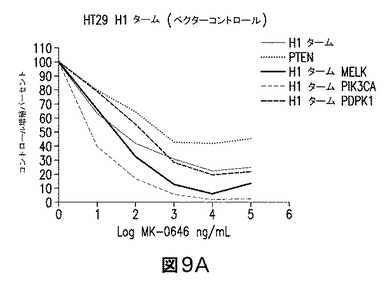
【図9−B】

【図10】
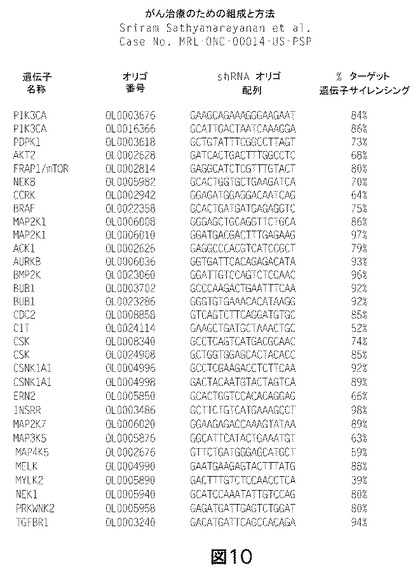
【図11】

【図12】
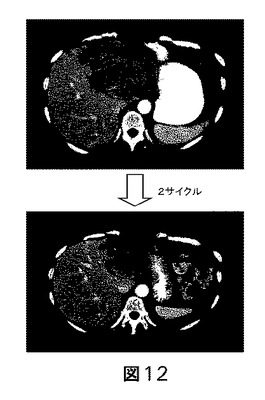
【図5】
【図6】
【図7−A】
【図7−B】
【図8】
【図9−A】
【図9−B】
【図10】
【図11】
【図12】
【公表番号】特表2012−524088(P2012−524088A)
【公表日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願番号】特願2012−506070(P2012−506070)
【出願日】平成22年4月6日(2010.4.6)
【国際出願番号】PCT/US2010/030074
【国際公開番号】WO2010/120599
【国際公開日】平成22年10月21日(2010.10.21)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
【公表日】平成24年10月11日(2012.10.11)
【国際特許分類】
【出願日】平成22年4月6日(2010.4.6)
【国際出願番号】PCT/US2010/030074
【国際公開番号】WO2010/120599
【国際公開日】平成22年10月21日(2010.10.21)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
[ Back to top ]