
癌治療用分子複合体
【課題】癌治療用の、トランスフェリン−薬剤複合体組成物、トランスフェリン−薬剤複合物などの癌治療に使用される分子複合体を形成する方法、および、そのために用いる中間化合物の提供。
【解決手段】式(I)を有する分子複合体を提供する。式中、nは複合数であり、Pはタンパク質などの担体分子の部分であり、R1は生物学的活性分子又はその類似体、誘導体、塩若しくは第二級アミンの部分であり、Zは−O−又はNH−であり、Yは、任意に1個又は複数のフェニルで置換され、1〜20個の炭素原子を有する直鎖状又は分岐状のアルキル基、任意に1個又は複数のアルキル又はフェニルで置換されたシクロアルキル基、或いは任意に1個又は複数のアルキル、電子吸引若しくは電子供与の基で置換された芳香族基である。分子複合体を適切な投与量で患者に投与することを含む、患者の目的とする標的細胞に生物学的活性分子を集中させる方法。

【解決手段】式(I)を有する分子複合体を提供する。式中、nは複合数であり、Pはタンパク質などの担体分子の部分であり、R1は生物学的活性分子又はその類似体、誘導体、塩若しくは第二級アミンの部分であり、Zは−O−又はNH−であり、Yは、任意に1個又は複数のフェニルで置換され、1〜20個の炭素原子を有する直鎖状又は分岐状のアルキル基、任意に1個又は複数のアルキル又はフェニルで置換されたシクロアルキル基、或いは任意に1個又は複数のアルキル、電子吸引若しくは電子供与の基で置換された芳香族基である。分子複合体を適切な投与量で患者に投与することを含む、患者の目的とする標的細胞に生物学的活性分子を集中させる方法。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2001年3月23日提出の米国仮出願第60/278,243号の利益を請求する。
【0002】
本発明は、一般に、患者の治療に使用する化合物及び方法に関する。より詳細には、本発明は癌治療用分子複合体に関する。特に、本発明は、トランスフェリン−薬剤複合体、その形成に有用な方法及び中間体、並びにその複合体を使用して患者を治療する方法に関する。
【背景技術】
【0003】
現在、数多くの抗癌剤が、様々な癌の治療において臨床的に使用されている。例えば、パクリタキセル(paclitaxel)及びタキソテール(taxotere)は、乳癌及び卵巣癌の治療に使用される2つの有望な抗癌剤であるが、皮膚、肺、頭部及び頚部の癌腫など他の癌の治療においても有望視されている。この他にも、これらの癌及び他の癌を治療するために有望な化学療法剤が開発され、試験されている。パクリタキセル、タキソテール、並びに他のタキサン類(taxanes)、カンプトテシン類(camptothecins)類、エポシロン類(epothilones)及びクアシノイド類(quassinoids)などの化合物、更に癌治療で有効性を示すその他の化合物が大きな関心を呼んでいる。特に、インビトロ及びインビボで抗癌活性を示す天然物薬剤が大きな関心を呼んでいる。このような化合物は、例えば、潜在的に再生可能な資源から入手できるという点で望ましいものである。
【0004】
しかし、確認されている多くの抗癌性化合物には、化学療法で使用する上での幾つかの問題点がある。その特有の問題の一つは、多くの抗癌性化合物が水に不溶であること関し、それが、化学療法に役立つ適切な医薬製剤を開発する上で大きな問題が生じさせていることである。これらの薬剤の水溶性を増大させる試みにおいて、これら薬剤は、しばしば、様々な担体化合物と共に製剤されている。しかし、これらの担体化合物は、その製剤を使用して治療した患者に、様々な副作用を引き起こすことがある。例えば、パクリタキセル、カンプトテシン及びこれらの類似体は、一般に、ポリエトキシレートひまし油およびエタノールの混合物と製剤されるが、これらの混合物は、臨床試験において好中球減少症、粘膜性、心性及び神経性の毒性、過敏症、ヒスタミン放出及び重症アレルギー反応を含む副作用をもたらすことが報告されている。
【0005】
癌治療でこれらの化学療法剤を使用する際のもう一つの問題は、正常健常細胞に有害な影響を及ぼさないで、癌細胞を攻撃することの難しさである。例えば、パクリタキセルは、正常細胞よりも癌細胞でより頻度が高い、有糸分裂及び細胞分裂過程を妨害することによってその抗癌活性を発揮する。しかし、化学療法を受けている患者は、正常健常細胞内での有糸分裂の妨害に関連する様々な副作用を体験することがある。
【0006】
従って、癌の治療法において、化学療法剤で癌細胞を直接的に攻撃するための化合物及びその使用法の開発が、極めて望まれている。これが可能になれば、担体化合物による有毒な副作用の軽減又は排除、薬剤の標的部位への効率的な送達、並びに投与薬剤の投与量の削減、及びその結果としての、健常細胞に対する毒性や化学療法における投与費用の低減に繋げることができる。
【0007】
その興味ある具体的手法の一つとして、腫瘍認識分子と複合させた抗癌剤部分を利用する方法がある。例えば、米国特許第6,191,290号(Safavy)には、腫瘍細胞表面の受容体と結合できる受容体リガンドペプチドと複合させたタキサン部分の形成及びその使用について考察がなされている。Safavyは特に、このような受容体リガンドペプチドを、BBN/GRP受容体認識ペプチド、ソマトスタチン受容体認識ペプチド、表皮成長因子受容体認識ペプチド、モノクローナル抗体又は受容体認識炭水化物とすることができることを指摘している。
【0008】
特に興味ある腫瘍認識分子の一つとしては、ヒトのタンパク質であるトランスフェリンがある。トランスフェリンは、分子量が約79550の血清糖タンパク質であり、ヘモグロビン合成のために成熟途上の赤血球に対する鉄の輸送に関与している。トランスフェリンは、鉄イオンに対する結合親和性が極めて高く、それによって、血漿中には、鉄の強毒性形態である遊離の鉄イオンが、本質的に存在しない。さらに、成熟過程にある細胞に必要な鉄分は、ジフェリック・トランスフェリン(各タンパク質分子が特異的に2個のFe3+イオンと結合してサーモンピンク色の錯体を形成したもの)によって供給される。すなわち、鉄が結合したトランスフェリンは、細胞膜上の受容体と結合し、そのトランスフェリン−受容体複合体が細胞膜内に取込まれ、次いで細胞質に鉄が放出されて、アポトランスフェリン−受容体複合体は細胞表面に戻り、アポトランスフェリンが受容体から放出されることになる。成熟過程にある細胞は、その細胞表面にトランスフェリン受容体を有しているが、静止期にある細胞ではトランスフェリン受容体が存在しないか、あっても極少数であることが明らかになっている。さらに、癌細胞には多くのトランスフェリン受容体が存在し、興味いことには、薬剤耐性癌細胞にはさらに多くのトランスフェリン受容体が存在することが明らかにされている。癌細胞上にはトランスフェリン受容体が存在し、正常細胞上には存在しないと言うことは、トランスフェリン複合体が癌細胞に対する標的薬剤としての一つの選択肢となり得る可能性を示唆している。例えば、Yeh他の論文「Killing of Human Tumer Cells in Culture with Adriamycin Conjugates of Human Transferrin」、Clin.Immunol.Immunopathol.32巻、1−11頁(1984年)、及びSizensky他の論文「Characterization of the Anti−Cancer Activity of Transferrin−Adriamycin Conjugates」、Am.J.Reprod.Immunol.27巻、163−166頁(1992年)で報告されているように、癌治療におけるトランスフェリン−アドリアマイシン複合体の治療指数はフリーのアドリアマイシンよりも大きい。
【0009】
その他の研究でも、ドコルビシン(Docorubicin)(Kratz他の論文「Transferrin conjugates of Docorubicin:Synthesis,Characterization,Cellular Uptake,and in vitro Efficacy」、J.Pharm Sci.、87巻、338−346頁(1998年))及びマイトマイシン(Mytomycin)C(Tanaka他の論文「Synthesis of Transferrin−Mitomycin C Conjugate as a Recepter−Mediated Drug Targeting System」、Biol.Pharm.Bull.、19巻、774−777頁(1996年))などの各種化学療法剤と複合させたトランスフェリンを利用する、癌治療に対する有望な方法が提案されている。
【0010】
有効なトランスフェリン−パクリタキセル複合体を目指した試みが、Bicamumpakaらの論文「In Vitro Cytotoxicity of Paclitaxel−Transferrin Conjugate on H69 Cells」、Oncol.Rep.、5巻、1381−1383頁(1998年)で報告されている。特に、Bicamumpakaらは、2’−グルタリル−ヘキサンジアミンパクリタキセルを合成し、次いで、それを、グルタルアルデヒドリンカーを使用して、2’−グルタリル−ヘキサンジアミン基のアミンにより、トランスフェリンと結合させた。しかし、Bicamumpakaは、得られたトランスフェリン−パクリタキセル複合体におけるH69細胞の増殖を阻止する能力が、元のパクリタキセル薬剤の5.4分の1未満であったと報告している。
【0011】
従って、トランスフェリン、Safavyが認知した受容体リガンドペプチド、又は他のタンパク質、抗体、レクチン、或いは細胞表面に付着できるその他の物質などの各種分子に、化学療法剤を結合するための、新規な化合物を提供することに対する要求が理解されよう。また、そのような化合物の形成方法を提供することに対する要求も理解される。さらに、癌治療に使用するための新規な分子複合体、特に、新規なトランスフェリン−薬剤複合体に対する要求も理解される。最後に、改善されたトランスフェリン−薬剤複合体などの分子複合体の使用などによって、患者に化学療法用薬剤製剤を投与する、癌治療法用の新規な方法に対する要求も理解される。本発明は、これらの要求に応じるものである。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】米国特許第6,191,290号明細書
【非特許文献】
【0013】
【非特許文献1】Yeh等、「Killing of Human Tumer Cells in Culture with Adriamycin Conjugates of Human Transferrin」、Clin.Immunol.Immunopathol.32巻、1−11頁(1984年)
【非特許文献2】Sizensky等、「Characterization of the Anti−Cancer Activity of Transferrin−Adriamycin Conjugates」、Am.J.Reprod.Immunol.27巻、163−166頁(1992年)
【非特許文献3】Kratz等、「Transferrin conjugates of Docorubicin:Synthesis,Characterization,Cellular Uptake,and in vitro Efficacy」、J.Pharm Sci.、87巻、338−346頁(1998年)
【非特許文献4】マイトマイシン(Mytomycin)C(Tanaka等、「Synthesis of Transferrin−Mitomycin C Conjugate as a Recepter−Mediated Drug Targeting System」、Biol.Pharm.Bull.、19巻、774−777頁(1996年)
【非特許文献5】Bicamumpaka等、「In Vitro Cytotoxicity of Paclitaxel−Transferrin Conjugate on H69 Cells」、Oncol.Rep.、5巻、1381−1383頁(1998年)
【発明の概要】
【発明が解決しようとする課題】
【0014】
本発明の目的は、ヒドロキシル基又はアミノ基を有する薬剤の新規かつ有用な分子複合体組成物を提供することである。
【0015】
本発明のさらなる目的は、癌治療用の、トランスフェリン−薬剤複合体組成物を提供することである。
【0016】
本発明の他の目的は、トランスフェリン−薬剤複合物などの、癌治療に使用される分子複合体を形成するために用いる中間化合物を提供することである。
【0017】
本発明の他の目的は、分子複合体、特にトランスフェリン−薬剤複合体を形成する効率的な方法を提供することである。
【0018】
本発明の更に他の目的は、癌患者が従来経験していた副作用を軽減又は排除する、患者に新規かつ有用な化学療法剤を投与する方法を提供することである。
【0019】
本発明の更に他の目的は、患者の癌細胞に化学療法剤を効率的に集中させるための方法を提供することである。
【課題を解決するための手段】
【0020】
本発明によれば、次の式を有する分子複合体が得られる。
【0021】
【化1】
「式中、nは分子複合体の複合数(例えば1〜5の整数)であり、Pはトランスフェリンタンパク質などの、少なくともn個の接近可能なアミノ官能基を有する分子の脱アミノ部分であり、R1はヒドロキシル基又はアミノ基を有する生物学的活性分子又はその類似体もしくは誘導体の、それぞれ、脱ヒドロキシル又は脱アミノの部分であり、Zはそれぞれ−O−又はNH−であり、Yは、任意に1個又は複数のフェニルで置換された、1〜20個の炭素原子を有する直鎖状又は分岐状のアルキル基、任意に1個又は複数のアルキル又はフェニルで置換されたシクロアルキル基、或いは任意に1個又は複数のアルキル基、電子吸引基もしくは電子供与基で置換された芳香族基である。」
Pは、好ましくはトランスフェリンなどのタンパク質であり、そのタンパク質は、生物学的活性分子と結合構造を通じて結合している。生物学的活性分子は、癌治療に有用であるような天然物薬剤でよく、各種タキサン類、カンプトテシン類、エポシロン類、ククルビタシン類、クアシノイド類、アントラサイクリン類、並びにその類似体及び誘導体などが挙げられる。
【0022】
本発明はまた、トランスフェリン−薬剤複合体などの分子複合体の形成に有用な化合物に関する。その化合物は次の一般式を有している。
【0023】
【化2】
「式中、R1、Y及びZは前記と同様であり、R2は−CH=CH(W)、−CH(OH)CH(OH)W、又は−C(O)Hである{Wは水素、任意に1個又は複数のフェニルで置換される1〜20個の炭素原子を有する直鎖状又は分岐状のアルキル、任意に1個又は複数のアルキル又はフェニルで置換されたシクロアルキル、又は任意に1個又は複数のアルキル基、電子吸引基もしくは電子供与基で置換された芳香族基である。}。」
【0024】
本発明はさらに、本発明による分子複合体の製造方法、特に癌治療に使用するためのトランスフェリン−薬剤複合体の製造方法に関する。この方法は、ヒドロキシル基又はアミノ基のいずれかを有する生物学的活性分子並びにその類似体及び誘導体(及び塩又は第二級アミン)からなる第1化合物を、次式の何れかの第2化合物と反応させて、
【0025】
【化3】
次式の第3化合物を形成する段階と、
【0026】
【化4】
第3化合物を次式の第4化合物に変換する段階と、
【0027】
【化5】
第4化合物を少なくともn個の接近可能なアミノ官能基を有する分子と結合させて、次式の分子複合体を形成する段階とを含んでいる。
【0028】
【化6】
「上記各式中、Xはハロゲンであり、R1、Z、W、Y、n及びPは前記と同様である。」
Wは、好ましくは水素であり、その結果第2化合物は末端オレフィンを有する。
第3化合物を第4化合物へ転換する段階は、第3化合物の酸化によって対応する次式のジオール中間体を介し、
【0029】
【化7】
その後に、当該ジオールを酸化して第4化合物にすることによって行われる。
【0030】
本発明はさらに、トランスフェリン−7−パクリタキセル複合体、トランスフェリン−2’−パクリタキセル複合体、トランスフェリン−3’−パクリタキセル複合体、及びトランスフェリン−20−カンプトテシン複合体の製造方法を提供する。本発明はまた、トランスフェリン−ローダミン123化合物、並びにその製造に用いる中間体及び方法を提供する。
【0031】
最後に、本発明は、本発明の複合化合物を利用して患者の選択された標的細胞に生物学的活性分子を、選択的に集中させる方法に関し、特に、トランスフェリン−薬剤複合体など本発明による分子複合体を、投与量を精選して、患者に投与することを含む方法に関するものである。
【0032】
本発明のこれらの目的及び他の目的については、本発明の例示的実施形態に関する以下の詳細な説明を添付の図面と併せ検討することによって容易に把握し、理解することができる。
【図面の簡単な説明】
【0033】
【図1】トランスフェリン−7−パクリタキセル複合体の形成で使用される7−アシル−ペンタナールパクリタキセルリンカー化合物を形成するための化学反応式を示す図である。
【図2】3−アシル−ペンタナールコレステロールリンカー化合物をトランスフェリンと結合させ、トランスフェリン−3−コレステロール複合体を形成するための化学反応式を示す図である。
【図3】20−アシル−ペンタナールカンプトテシンリンカー化合物をトランスフェリンと結合させ、トランスフェリン−20−カンプトテシン複合体を形成するための化学反応式を示す図である。
【図4(a)】トランスフェリン−2’−パクリタキセル複合体の形成で使用される2’−アシル−ペンタナールパクリタキセルリンカー化合物を形成するための化学反応式を示す図である。
【図4(b)】トランスフェリン−2’−パクリタキセル複合体の形成で使用される2’−アシル−ヘキサナールパクリタキセルリンカー化合物を形成するための化学反応式を示す図である。
【図4(c)】トランスフェリン−2’−パクリタキセル複合体の形成で使用される2’−アシル−ノナナールパクリタキセルリンカー化合物を形成するための化学反応式を示す図である。
【図5】プロトコルAにおける異なる濃度での、トランスフェリン−3−コレステロール複合体のKB細胞に対する細胞毒性を示すグラフである。
【図6】プロトコルAにおける異なる濃度での、トランスフェリン−ローダミン123複合体のKB細胞に対する細胞毒性を示すグラフである。
【図7】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のKB細胞に対する細胞毒性を示すグラフである。
【図8】プロトコルAにおける異なる濃度での、トランスフェリン−3−コレステロール複合体のLu細胞に対する細胞毒性を示すグラフである。
【図9】プロトコルAにおける異なる濃度での、トランスフェリン−ローダミン123複合体のLu細胞に対する細胞毒性を示すグラフである。
【図10】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のLu細胞に対する細胞毒性を示すグラフである。
【図11】プロトコルAにおける異なる濃度での、トランスフェリン−3−コレステロール複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【図12】プロトコルAにおける異なる濃度での、トランスフェリン−ローダミン123複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【図13】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【図14】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のKB細胞に対する細胞毒性を示すグラフである。
【図15】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のLu細胞に対する細胞毒性を示すグラフである。
【図16】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【図17】プロトコルBにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のKB細胞に対する細胞毒性を示すグラフである。
【図18】プロトコルBにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のLu細胞に対する細胞毒性を示すグラフである。
【図19】プロトコルBにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【発明を実施するための形態】
【0034】
本発明は、新規な分子複合体、特に患者の癌治療で使用する新規なトランスフェリン−薬剤複合体を提供する。さらに、本発明は生物学的活性分子をトランスフェリン又はその他の分子などの担体分子と結合するために用いる新規な中間体化合物に関するものである。特に、本発明は、癌治療薬並びにその類似体及び誘導体、並びにその前駆体など、それぞれ、ヒドロキシル基及びアミノ基を有する生物学的活性分子のアルデヒドエステル誘導体及びアルデヒドアミド誘導体を提供する。これらの誘導体は、そのエステル結合又はアミド結合しているアルデヒド官能基とトランスフェリン分子又は他のタンパク質の各種アミノ官能基との間でシッフ塩基を形成することによって、ヒト・トランスフェリンタンパク質などの担体分子と結合することができる。
【0035】
本発明はまた、ヒドロキシル基又はアミノ基を有する様々な生物学的活性化合物のトランスフェリン複合体、又は他の分子複合体、並びにその中間体の効率的な合成プロトコルを提供する。一般化した方法では、ヒドロキシル基又はアミノ基を含有する生物学的活性分子と、二重結合好ましくは末端オレフィンを有するカルボン酸又は酸ハロゲン化物などの適当なアシル化剤とを結合させることを含む。四酸化オスミウム触媒を用いて末端オレフィン部位を迅速かつ高効率で酸化し、次いで、得られたジオールを開裂してアルデヒドにすることによって、トランスフェリン複合体又は他の分子複合体の合成に適した前駆体が得られる。これら付加生成物の合成経路における最終段階は、アルデヒドを血液タンパク質であるトランスフェリンなどの担体分子と処理し、トランスフェリン単量体に生物学的活性分子を生成させることである。これらの生物学的活性分子では生物学的活性が増大していることが見出されている。本発明は、本明細書で開示するように、担体分子には、トランスフェリンの他、生物学的活性分子のエステルリンカー化合物及びアミドリンカー化合物のアルデヒド官能基と、シッフ塩基を形成することができる、少なくとも1個の接近可能なアミノ官能基を有する任意の分子を含めるように広く想定している。
【0036】
また、本発明において、用語「生物学的活性分子」は、細胞、組織、脈管内などでの1種又は複数の生物学的過程に一般的に影響を与え、又は関与する任意の分子を含むものとして、広く解釈して理解されるべきである。このような生物学的活性分子には、薬剤、抗体、抗原、レクチン、色素、染料、トレーサ又は他の類似分子が含まれる。特に、本発明での使用を企図しているヒドロキシル基含有分子又はアミノ基含有分子には、パクリタキセル、ドセタキセル(docetaxel)及び他のタキサン類、コレステロール、ローダミン123、カンプトテシン類、エポシロンBなどのエポシロン類、ククルビタシン類、グローカルボロン(glaucarubolone)、ブルサトール(brusatol)及びブルシアンチン(bruceantin)などのクアシノイド類、アドリアマイシン、ダウノルビシン(daunorubicin)などのアントラサイクリン類、並びにこれらの類似体及び誘導体、並びにその他の化合物が含まれる。
用語「分子複合体」は、例えば本明細書で開示されるエステル及びアミドのシッフ塩基結合を通して本発明の担体分子に結合している生物学的活性分子を含む任意の化合物を広く包含するものとして理解されるべきである。
【0037】
本研究の中心は癌治療であるが、本願がさらに、各種のタンパク質又はその他の担体分子を、本発明により、他の応用分野を対象とした生物学的活性分子と複合させることを企図していることを理解されるべきである。
【0038】
(I.トランスフェリン−7−パクリタキセル複合体)
本発明によってトランスフェリン−7−パクリタキセル複合体を形成することができる。図1に示すように、先ず、パクリタキセルを様々な中間化合物を経由して7−パクリタキセルアルデヒドエステルに変換する。次いで、このアルデヒドエステルをトランスフェリンと結合させてトランスフェリン−7−パクリタキセル複合体を形成する。
【0039】
A.2’TBDMSパクリタキセルの調製
【0040】
【化8】
【0041】
先ず、パクリタキセルの2’−ヒドロキシル基をTBDMSで保護して2’−TBDMSパクリタキセルを形成する。反応例としてはTBDMSを示したが、TBDMSの代わりにTROC,BOM,CBZ,ベンジル、TES,EEなど他の保護基も使用可能であることを認識されたい。
【0042】
この材料は、Tetrahedron Letters、35巻、8931−8934頁,1994年でProf Gunda Georg他が述べている方法によって調製され、それに従って特徴付けられる。
【0043】
パクリタキセル(20.0g、23.45mM)のジメチルホルムアミド(150mL)溶液に、窒素雰囲気下でイミダゾール(23.95g、351.7mM)を添加し、続いてTBDMSCl(49.5g、328.3mM)を添加した。得られた溶液を、窒素雰囲気下、周囲温度で16時間攪拌した。この段階でTLC分析を行い、出発物質が完全に消費されていることを確認し、水(200mL)及び酢酸エチル(200mL)を添加して反応を終了した。有機層を分離し、水(100mL×2回)、塩水(50mL)で洗浄し、硫酸マグネシウム上で乾燥し、その後濾過、留去して残渣を取り出し、真空オーブン中で乾燥し、さらに精製することなしに次の反応に使用した。
【0044】
B.2’保護パクリタキセルの7−ヘキセン酸エステルの調製
【0045】
【化9】
【0046】
次に、好ましくは末端オレフィンを有する酸と反応させて、2’−保護パクリタキセルの7−ヘキセン酸エステルを形成した。本明細書の実施例では、5−ヘキセン酸を使用するが、本発明は、鎖末端から離れた位置にある二重結合を有するオレフィンアシル化剤をも想定していることを理解されたい。もっとも、好ましくは末端オレフィンを有する他の適当なアシル化剤を企図する。
例えば、本発明は次式の酸、
【0047】
【化10】
又は次式の酸ハロゲン化物の使用を企図する。
【0048】
【化11】
「上記各式中、Xはハロゲンであり、Yは、任意に1個若しくは複数のフェニル基で置換され1から20個の炭素原子を有する直鎖状若しくは分岐状のアルキル基、任意に1個若しくは複数のアルキル基またはフェニル基で置換されたシクロアルキル基、あるいは任意に1個若しくは複数のアルキル、電子吸引基または電子供与基で置換された芳香族基である。Wは、水素、任意に1個若しくは複数のフェニル基で置換され1から20個の炭素原子を有する直鎖状若しくは分岐状のアルキル基、任意に1個若しくは複数のアルキル基またはフェニル基で置換されたシクロアルキル基、あるいは任意に1個若しくは複数のアルキル、電子吸引基または電子供与基で置換された芳香族基である。」
【0049】
2’TBDMSパクリタキセル(2.0g、2.07mM)塩化メチレン(30mL)溶液に、窒素雰囲気下で、5−ヘキセン酸(0.49mL、4.14mM)を添加し、その後更にDIPC(0.81mL、5.18mM)及び4−PP(0.095g、0.64mM)を添加した。得られた反応混合物を、4時間攪拌し、TLC分析により完全に反応していることを判断した。水(50mL)及び酢酸エチル(90mL)を添加して反応を終了させ、有機層を分離し、水(50mL)、塩水(50mL)で洗浄し、硫酸マグネシウム上で乾燥した。得られた生成物を濾過し、溶媒を留去して残渣を取り出し、精製すること無しに次の反応に供した。
【0050】
C.2’保護パクリタキセルの7−アルデヒド誘導体の調製
【0051】
【化12】
【0052】
末端オレフィン部位を酸化してジオールに変え、続いて末端炭素を開裂するとパクリタキセルの7−アルデヒド2’保護誘導体が得られる。二重結合が前に考察したように鎖末端から移動している(すなわち、前記式中のWが水素でない)アシル化剤を使用する場合には、この反応中に起こる二重結合の開裂によって、二重結合を越えた鎖の部分が除去されることを理解されたい。また、例示した工程では単一段階として示されているが、次式のジオール、
【0053】
【化13】
(及び、本明細書に記載された化合物に対応するジオール)は、NaIO4無しでこの段階を実行することによって単離可能であることを認識されたい。NaIO4で処理してこのジオールを酸化開裂すると末端アルデヒドが得られる。
【0054】
7−ヘキセノイル、2’TBDMSパクリタキセル(2.2g、2.07mM)のACNとTHF(各20mL)の混合溶媒溶液に、水(20mL)を加え、続いて窒素雰囲気下で、NMO(0.49g、4.14mM)、NaIO4(0.89g、4.14mM)及びOsO4(13.15mg、0.052mM)のt−BuOH溶液を添加した。得られた反応混合物を周囲温度で2時間攪拌し、酢酸エチル及び水(各100mL)を加え反応終了とした。分離した有機層を塩水(20mL)で洗浄し、硫酸マグネシウム及びハイドロサルファイトナトリウムを通して濾過した。濾液を蒸発乾固して、粗生成物を精製すること無しに脱シリル化反応に供した。
【0055】
D.パクリタキセルの7−アルデヒド誘導体の調製
【0056】
【化14】
【0057】
2’位を次のように脱保護した。2’保護したパクリタキセルの7−アルデヒド誘導体(2.25g、2.07mM)THF(50mL)溶液に、窒素雰囲気下、周囲温度でTBAFのTHF溶液(1.0M溶液を3.11mL、3.11mM)を添加した。得られた反応混合物を2時間攪拌した。TLC分析によればこの時点で出発物質はなかった。酢酸エチル(200mL)及び0.5NHCl(100mL)を加え混合物の反応を終了し、分離した有機層を水(200mL)、塩水(100mL)で洗浄し、硫酸マグネシウム上で乾燥した。有機層を濾過、蒸発乾固し、次いで酢酸エチル及びヘプタンを用いたカラムクロマトグラフィーによって精製し、60%の総収率で精製物質を得た。化合物はMS及び1H NMRによって解析した。
【0058】
E.トランスフェリン−7−パクリタキセル複合体の調製
次にアルデヒドエステル誘導体をトランスフェリンと結合させて、複合数n(トランスフェリン1分子当たりのパクリタキセルの分子数)を有するトランスフェリン−7−パクリタキセル複合体を形成させることができる。複合数は3であったが、条件を変更すると複合数nが1から5のトランスフェリン−7−パクリタキセル複合体が生成する可能性も考えられる。本発明は、トランスフェリンの代わりに、接近可能なアミノ官能基を有する他のタンパク質などの担体分子を使用することが可能であり、得られる分子複合体の複合数もそれに応じて変動可能であることを当然考慮に入れている。
【0059】
【化15】
【0060】
80mg(1μmol)のトランスフェリンを含むPBS緩衝液/DMSO溶液に、19.04mg(20μmol)の7−アシル−ペンタナルパクリタキセルを含むDMSO溶液2mlを滴下した。トランスフェリンPBS緩衝液/DMSO溶液は、トランスフェリンを4mlのPBS(50mmol、pH8.0)に溶解し、さらにそのトランスフェリンPBS溶液に、0℃で、2mlのDMSOを加えて調製した。反応混合物を、C24振とう恒温器(New Brunswick Scientific classic series、Edison、アメリカ)を使用して、37℃で8時間振とうした。反応混合物を5.0μmの濾過装置で濾過し、澄明な濾液を、superdexHR200カラム(2.0×30cm)に20mMのTris−HCl(pH8.0)を0.5ml/分の流速で流すFPLCによって精製した。トランスフェリンに対応する分画を集めて凍結乾燥した。
【0061】
本明細書では例示しないが、7−パクリタキセル複合体と同様に10−デアセチルパクリタキセルの分子複合体が形成されることを理解されたい。例えば、出発化合物として次式の10−デアセチルパクリタキセルを用い、
【0062】
【化16】
アシル化剤として6−ヘプテン酸を用い、上述の方法と同様にしてトランスフェリン−10−アシル−ヘキサナールパクリタキセル複合体が形成される。当技術分野で知られているように、2’位及び7位の適切な保護及び脱保護を利用することができる。
【0063】
(II.トランスフェリン−3−コレステロール複合体)
トランスフェリンに非細胞毒性分子を結合した場合の結果を調べるためにトランスフェリン−3−コレステロール複合体を調製した。以下で考察するように、これらの結果は、本発明による癌治療用トランスフェリン−薬剤複合体は、インビトロ又はインビボで細胞毒性活性を示す癌治療剤を使用して形成されるべきことを示唆する。例えば癌治療用分子以外の分野での応用を研究するために、3−コレステロールと別のタンパク質との様々な複合体を同様にして調製して、そのようなタンパク質の複合物と他の生物学的活性化合物と比較することが可能であろう。
【0064】
図2に示すように、先ず、コレステロールを、各種中間化合物を経由して3−コレステロールアルデヒドエステルに変換し、次いで、それをトランスフェリンと結合させトランスフェリン−3−コレステロール複合体を形成する。前に考察したように、本明細書の実施例で使用する5−ヘキセン酸を、前に述べたようなその他各種アシル化剤で代替することができる。
【0065】
A.3−ヘキセノイルコレステロールの調製
【0066】
【化17】
【0067】
コレステロ−ル(2.0g、5.17mM)塩化メチレン(20mL)溶液に、5−ヘキセン酸(0.68mL、5.69mM)を添加し、続いて窒素雰囲気下で、DCC(1.60g、7.76mM)及び4−PP(0.115g、0.78mM)を加えた。得られた反応混合物を、周囲温度で1時間攪拌し、メチルt−ブチルエーテル(60mL)を加え反応終了とした。尿素を濾過により除去し、生成物を分液ロートに移し、1N HCl(10mL)、水(30mL)及び塩水(30mL)で洗浄した。生成物をMgSO4上で乾燥後、濾過し、溶媒を留去して残渣を得た。95%以上の収率で得られた生成物を1H NMRで解析した。
【0068】
B.コレステロールの3−アシル−ペンタナールエステルの調製
【0069】
【化18】
【0070】
コレステロール3−ヘキセン酸エステル(2.0g、4.14mM)のTHFとt−BuOHの混合溶媒(各10mL)溶液に、水(5mL)を加え、引き続き窒素雰囲気下で、NMO(0.97g、8.3mM)、NaIO4(1.78g、8.3mM)及びOsO4(21.3mg、0.083mM)のt−BuOH溶液を添加した。得られた反応混合物を、TLC分析で調べて変換が完全になるまで16時間攪拌した。反応混合物に珪藻土(1.6g)を加えて濾過した。フィルタケーキを酢酸エチル(100mL)で洗浄し、濾液を分液ロートに移し、1N HCl(15mL)、水(25mL)及び塩水(15mL)で洗浄し、次いでMgSO4上で乾燥した。濾過した溶液を蒸発乾固し、続いてカラムクロマトグラフィーで精製して85%の収率で生成物を得た。生成物を1H NMRで解析した。NMR分析によればコレステロール系の内部二重結合は酸化条件でも変化していないことがわかった。
【0071】
C.トランスフェリン−3−コレステロール複合体の調製
次に、アルデヒドエステル誘導体をトランスフェリンと結合させ、複合数がnであるトランスフェリン−3−コレステロール複合体を形成できる。
【0072】
【化19】
【0073】
20mg(0.5μmol)のトランスフェリンを含むPBS−緩衝液(50mmol、pH7.0)2mLに、1.21mg(2.5μmol)の3−アシル−ペンタナールコレステロールを含むDMSO溶液1mLを滴加した。反応混合物を、C24振とう恒温器(New Brunswick Scientific classic series、Edison、アメリカ)を用いて、37℃で30分間振とうした。この混合物に反応停止剤として1.527mg(25mmol)のエタノールアミンを含むPBS溶液0.5mLを添加して反応を停止させた。次に、濁った混合物を4℃、1000gで10分遠沈し、上澄み液を、superdexHR200カラム(2.0×30cm)を用いて20mMのTris−HCl(pH8.0)を0.5ml/分の流速で流すFPLCで精製した。トランスフェリンに対応する分画を集め、凍結乾燥した。
【0074】
III.トランスフェリン−20−カンプトテシン複合体
図3に示すように、先ず、カンプトテシンを様々な中間化合物を経由して20−カンプトテシンアルデヒドエステルに変換し、次いで、それをトランスフェリンと結合させて、トランスフェリン−20−カンプトテシン複合体を形成する。本発明においては、トランスフェリンを、接近可能なアミノ官能基を有する他のタンパク質などの担体分子で代替可能であり、5−ヘキセン酸を、前に述べたような他のアシル化剤で代替可能である。
【0075】
A.20−ヘキセノイルカンプトテシンの調製
【0076】
【化20】
【0077】
DMF(40mL)中にカンプトテシン(2.0g,5.74mM)及び5−ヘキセン酸(0.75mL、6.31mM)を含む混合物に、窒素雰囲気下で、DIPC(0.99ml,6,31mM)及び4−PP(0.13g、0.86mM)を添加した。得られた反応混合物を、周囲温度で24時間攪拌した。TLC分析でカンプトテシンが完全に消費されていることを確認した後、塩化メチレン及び水(各200mL)を用いながら反応混合物を分液ロートに移した。分離した有機層を塩水で洗浄し、MgSO4上で乾燥し、濾過して蒸発乾固した。固体残渣を塩化メチレン及び酢酸エチルを用いて結晶化した。結晶を濾過して純度98%以上の材料を70%の収率で得た。結晶をMS及び1H NMRデータで解析した。
【0078】
B.カンプトテシンの20−アシル−ペンタナールエステルの調製
【0079】
【化21】
【0080】
カンプトテシン20−ヘキセン酸エステル(1.0g、2.25mM)のTHF、アセトン及びACN混合溶媒(各15mL)溶液に、水(15mL)を加え、引き続き窒素雰囲気下で、NMO(0.53g、4.5mM)、NaIO4(0.96g、4.5mM)及びOsO4(15.3mg、0.06mM)のt−BuOH溶液を添加した。得られた反応混合物を、周囲温度で4時間攪拌し、ヘキセン酸エステルからカンプトテシンペンタナール誘導体への変換を完結させた。反応混合物を、塩化メチレン及び水(各200mL)に分配させた。有機層を分離し、1N HCl(10mL)、水(50mL)、塩水(50mL)で洗浄し、MgSO4上で乾燥し、濾過後、蒸発乾固した。残渣をカラムクロマトグラフィーで精製し、精製物をMS及び1H NMRデータにより解析した。
【0081】
C.トランスフェリン−20−カンプトテシン複合体の調製
次に、アルデヒドエステル誘導体をトランスフェリンと結合させ、複合数がnであるトランスフェリン−20−カンプトテシン複合体を形成する。
【0082】
【化22】
【0083】
トランスフェリン−20−カンプトテシン複合体を形成する方法は、先に述べた、トランスフェリン−7−パクリタキセル複合体及びトランスフェリン−3−コレステロール複合体を形成する方法と同様であり、DMSO及びトランスフェリンPBS溶液を利用する。得られたトランスフェリン−20−カンプトテシンのマススペクトル分析によれば、結合比はトランスフェリン1分子に対してカンプトテシンが3分子であった(すなわち、n=3)。トランスフェリンとトランスフェリン−20−カンプトテシン複合体の円偏光二色性(CD)スペクトルが相違しており、全体としての立体配座が変化したことを示唆する。
【0084】
(IV.トランスフェリン−ローダミン123複合体)
検出及び視覚化のためにローダミン123の蛍光を利用する目的で、トランスフェリン−ローダミン123複合体を調製した。ローダミン123及びトランスフェリンのそれぞれの遊離アミノ基の間を繋ぐリンカーとしてグルタルアルデヒドを用いて、1段階でトランスフェリン−ローダミン123複合体を調製した。この反応では中間体として次式の活性アルデヒド化合物が形成されると考えられる。
【0085】
【化23】
【0086】
A.トランスフェリン−ローダミン123複合体の調整
【0087】
【化24】
【0088】
トランスフェリン40mg(0.5μmol)を含むHeps緩衝食塩水(HBS、150mmolNaCl,10mmol/L Heps、pH7.4)溶液1mlを、1mlのローダミン123(5μmol)と4分間かき混ぜて混合した。1mlのグルタルアルデヒド(12.5mmol、HBS溶液)を、室温でかき混ぜながら4分間で滴加した。反応停止剤として0.5mlの25mmolエタノールアミンHBS溶液を添加し、4分間攪拌して結合反応を停止させた。混合物を透析チューブに移し、暗黒下に4℃で8時間、1LのHBSに対して透析した。次に、濁った混合物を4℃で10分間、1000gで遠沈し、澄明上澄み液に対して、superdexHR200カラム(2.0×30cm)を用い0.5ml/分の流速で20mMのTris−HCl(pH8.0)を流してクロマトグラフを行った。トランスフェリンに対応する分画を集めて凍結乾燥した。
【0089】
(V.2’−パクリタキセル化合物)
図4(a)、4(b)、4(c)に示すように、各種2’−パクリタキセル中間化合物を形成したが、アルキル鎖の短いアルデヒド誘導体(図4(a)に示した方法で5−ヘキセン酸を使用した場合など)では、トランスフェリン−2’−パクリタキセル複合体の形成に問題があった。おそらく、2’位が半球状タキサン骨格のC−13の周囲にある凹状領域に位置することに由来する障害によるものであろう。従って、この結果から、ヘキセン酸よりも鎖が長い結合用化合物を用いて形成したアルデヒド誘導体など、より長いアルキル鎖をもったアルデヒド誘導体を利用して、本発明によるトランスフェリン−2’−パクリタキセル複合体を形成できると考えられる。さらなる実験により、それぞれ図4(b)及び4(c)に示すように、同様な化学的方法を使用して2’−ヘプタナール トランスフェリン複合体及び2’−ノナナールトランスフェリン複合体をより容易に形成できることがわかった。2’−ノナナールパクリタキセル化合物の形成には、図4(c)に示すようにオレイン酸(9,10位に二重結合を有する18炭素鎖の酸)を使用したことに留意されたい。さらに、トランスフェリンが比較的大きなタンパクであることを考えれば、この結果から、前に述べたSafavyによって確認されたタンパク質などのより小さな担体分子は、パクリタキセルの凹状構造による障害が少なく、前記のアシル化剤の式においてY<4であるようなアルキル鎖の短い2’−パクリタキセルアルデヒド誘導体とより容易に複合体を形成できると考えられる。
【0090】
A.パクリタキセル2’−ヘキセン酸エステルの調製
【0091】
【化25】
【0092】
パクリタキセル(2.0g、2.34mM)及び5−ヘキセン酸(0.31mL、2.58mM)の塩化メチレン(25mL)溶液に、窒素雰囲気下、0℃でDCC(0.72g、3.51mM)を添加し、更にその後4−PP(0.17g、0.5mM)を添加した。得られた反応混合物を2時間攪拌し、その間に、反応混合物の温度は周囲温度に戻した。TLCで反応を調べ、2時間後に反応が完結していることを確認した。水及び酢酸エチルを各30mL添加して反応を終えた。混合物を分液ロートに移し、有機層を1N HCl(10mL)、水(30mL)及び塩水(20mL)で洗浄し、硫酸マグネシウム上で乾燥した。乾燥した濾液を蒸発乾固し、メチルt−ブチルエーテルで結晶化した。得られた化合物について、HPLCにより純度95%以上であることを確認した。また、1H NMR分析によりパクリタキセルの7位のヒドロキシル基には影響がなく2位のヒドロキシル基でエステル化されていることを確認した。収率は98%であった。
【0093】
B.パクリタキセル2’アルデヒド誘導体の調製
【0094】
【化26】
【0095】
パクリタキセル2’−ヘキセン酸エステル(0.475g、0.5mM)のt−BuOH、アセトン及び水(各2mL)の混合溶液に、窒素雰囲気下で、NMO(0.118g、1mM)NaIO4(0.214g、1mM)及びOsO4(2.54mg、0.01mM)のt−BuOH溶液を添加した。得られた混合物を周囲温度で2時間攪拌した。TLCによればこの時点で反応は完結していた。混合物に水及び酢酸エチル(各20mL)を添加して反応を終え、有機層を分離し、1N HCl(10mL)、水(10mL)及び塩水(10mL)で洗浄した。有機層を硫酸マグネシウム及びハイドロサルファイトナトリウムを通して濾過し、蒸発乾固した。粗化合物を酢酸エチル及びヘプタンを使用したカラムクロマトグラフィーで精製し、MS及び1H NMRで解析した。収率85%。
【0096】
C.パクリタキセル2’ジオール誘導体の調製
図4(a)に示したように、上記反応でNaIO4を使用しない場合には、次のようにジオールが単離される。
【0097】
【化27】
【0098】
パクリタキセル2’−ヘキセン酸エステル(9.0g、9.47mM)アセトン(135mL)溶液に、水(50mL)を添加した。得られた溶液に、窒素雰囲気下で、NMO(2.22g、18.94mM)を添加し、続いてOsO4(48.2mg、0.19mM)t−BuOH溶液を添加して16時間攪拌を続けた。TLCで変換の完結を確認した後、反応混合物に珪藻土(15g)を加えて濾過した。濾液をロータリー蒸留器でアセトンが無くなるまで濃縮し、引き続き水層を固形食塩で飽和させた後、酢酸エチル(200mL)による抽出操作に付した。得られた有機層を水(10mL)、1N HCl(100mL)、水(10mL)及び塩水(100mL)で洗浄し、MgSO4を通して濾過した。溶媒を留去し、残渣をカラムクロマトグラフィーで精製し、収率65%で純粋なジオールを得た。先に考察した7−パクリタキセル誘導体の対応するジオールの場合と同様、ジオールをNaIO4で処理する酸化的開裂によって末端アルデヒドが得られる。
【0099】
D.トランスフェリン−2’−アシル−ヘキサナルパクリタキセル複合体の調製
図4(b)に示すように、まず、パクリタキセルを、各種中間体を経由して2’−アシル−ヘキサナールパクリタキセル化合物に変換した。2’−アシル−ヘキサナールパクリタキセルアルデヒドエステル誘導体を形成するための化学過程は、5−ヘキセン酸の代わりに6−ヘプテン酸を使用すること以外は2’−アシル−ペンタナールパクリタキセル誘導体の形成について既に述べた化学過程と同様である。
【0100】
次に、アルデヒドエステルをトランスフェリンに結合させ、以下に示すような複合数がnであるトランスフェリン−2’−パクリタキセル複合体を形成する。
【0101】
【化28】
この手順は、トランスフェリン−7−パクリタキセル複合体の調製について既に述べたものと同様である。マススペクトルによれば、各トランスフェリンに2分子のパクリタキセルが複合したもの(すなわち、n=2)も検出されたが、主なトランスフェリン−2’−パクリタキセル複合生成物の結合比はトランスフェリン1分子に対しパクリタキセル1分子(すなわち、n=1)であった。遠紫外領域でのトランスフェリンとトランスフェリン−2’−パクリタキセル複合体との円偏光二色性スペクトルが相違しており、2’−パクリタキセル複合体の全体としての立体配座の変化が示唆される。
【0102】
E.トランスフェリン−2’−アシル−ノナナールパクリタキセル複合体の調製
図4(c)に示すように、パクリタキセルを、各種中間体を経由して先ず2’−アシル−ノナナールパクリタキセルアルデヒドエステル化合物に変換することによって、2’−パクリタキセルのトランスフェリン複合体を形成した。化学過程は2’−アシル−ペンタナールパクリタキセル誘導体について既に述べたものとやはり同様であるが、図4(c)で示したように、アシル化剤としてオレイン酸(cis−9−オクタデセン酸、CH3(CH2)7CH=CH(CH2)7CO2H)を使用したことに留意されたい。従って、鎖を開裂してアルデヒドを形成させるに際しては、9,10位の二重結合を越えて延びている鎖の部分が除去される。
【0103】
2’−アシル−ノナナールパクリタキセルとトランスフェリンを複合させて次式のトランスフェリン−2’−パクリタキセル複合体を形成させることは、トランスフェリン−7−パクリタキセル複合体について既に述べた複合の場合と同様である。
【0104】
【化29】
【0105】
(VI.アミド誘導体)
本発明はまた、アミノ基含有化合物のアミド誘導体の形成をも想定している。例えば、次式
【0106】
【化30】
のパクリタキセル類似体を、適切なアシル化剤、好ましくは末端オレフィンを有するアシル化剤と結合させ、その末端オレフィンによって、トランスフェリン又はその他の担体分子/タンパク質と共に使用する、アルデヒドリンカーに変換可能なパクリタキセルアミド誘導体を形成することができる。
上記のアミノ基含有パクリタキセル類似体をヘキセン酸と結合させることによって形成される中間体の例を次式に示す。
【0107】
【化31】
このような中間体は、例えば7−パクリタキセル及び2−パクリタキセル誘導体について既に開示した方法によって、対応するジオール、
【0108】
【化32】
及びアルデヒド
【0109】
【化33】
に変換することができる。従って、次式のトランスフェリン複合体を企図する。
【0110】
【化34】
半球状タキサン骨格のC−13位周囲の凹状領域に側鎖が存在することに由来する障害に基づく、トランスフェリン複合体を形成する困難さに対処するためには、ヘキセン酸よりも鎖長がより長い結合用化合物を使用することが考えられる。例えば、ヘプテン酸を使用すると次式のトランスフェリン複合体が形成される。
【0111】
【化35】
ここでは、保護したタキサンを、先ず、次のように対応するアミン−塩酸塩に変換する。
【0112】
【化36】
上式で示した7−O,3’−N−ジ−(CBZ)−2’−O−BOMパクリタキセルなどの保護されたタキサンの形成は、当技術分野で周知であり、例えば米国特許第5,750,737号、5,973,170号、6,133,462号、6,066,749号、6,048,990号、6,136,999号、及び6,143,902号に開示されており、その教示を参照として本明細書に組込む。上記反応式に示したアミン−塩酸塩の形成は、米国特許出願第09/843,284号により完全に記載されており、その教示を参照として本明細書に組込む。
【0113】
例えば、反応例を挙げれば、5.05gの7−O,3’−ジ−(CBZ)−2’−O−BOMパクリタキセルを、磁気攪拌子を備えた0.5L丸底フラスコ内の90.0mLのTHFに溶解し、フラスコに6.01mLの3.62M塩酸(22.08mmol)及び8.10g(湿分50%)の10%Pd/Cを添加した。反応容器を窒素で3回、水素で2回流洗し、反応混合物を、水素を充填したバルーンから供給される水素雰囲気下、室温下で、約1時間激しく攪拌した。この操作により上記反応式に示したアミン塩酸塩が生成した。上記の工程で使用した塩酸に代わって、他の鉱酸並びに有機酸を使用できることを認識されたい。
【0114】
その他にもタクサンのアンモニウム塩を形成する方法が知られている。例えば、トリフルオロ酢酸を使用して対応するアンモニウムトリフルオロ酢酸(TFA)塩を形成する方法が、例えば米国特許第5,675,025号、5,684,175号、5,770,745号、5,939,566号、5,948,919号、6,048,990号、6,066,749号、6,072,060号、6,136,999号、6,143,902号、6,262,281号、及び6,307,088号に開示されており、その教示を参照として本明細書に組込む。
【0115】
アミン−塩酸塩を形成したら、次に、以下に示すように、その塩酸塩をヘプテン酸と反応させて、対応するヘプテン酸エステルを形成する。
【0116】
【化37】
この反応は、トリエチルアミン(TEA)を加えて、アミン塩を対応する遊離のアミンに解離させていることを理解すべき以外は、7−パクリタキセル誘導体又は2’−パクリタキセル誘導体に関して既に開示している反応とほぼ同様である。次に、以下に示すように、得られたヘプテン酸エステルを対応する3’−アミドヘキサナールパクリタキセルに変換した。
【0117】
【化38】
この反応もやはり前に他の誘導体に関して述べた反応と同様である。次に、得られたアルデヒドを、トランスフェリン−7−パクリタキセル複合体の形成について前に述べた方法と同様の方法を用いて、トランスフェリンと結合させ、トランスフェリン−3’−アミド−ヘキサナールパクリタキセル複合体を得た。トランスフェリンとトランスフェリン−3’−アミド−ヘキサナールパクリタキセル複合体の円偏光二色性スペクトルは類似しており、双方の全体的立体配座が類似していることを示す。
【0118】
前述したことから、アミン塩の代わりに対応する遊離のアミンそれ自体を使用できることを認識されたい。タキサンの遊離アミンの形成は、例えば米国特許第5,688,977号、5,770,745号、5,939,566号、6,048,990号、6,066,749号、6,072,060号、6,107,497号、6,262,281号、及び6,307,088号に開示されており、その教示を参照として本明細書に組込む。
従って、本発明は、上述の実施例のアミン塩酸塩を、次の一般式で表される化合物並びにその類似体及び誘導体で代替することを企図する。
【0119】
【化39】
(式中、R3はNH2又はNH2HAでよくHAは有機酸又は鉱酸である)
【0120】
(VII.一般的方法及び化合物)
これまでの考察から明らかなように、本発明は、タンパク質−薬剤複合体を形成するための一般的方法を提供するものである。先ず、ヒドロキシル基又はアミノ基を有する、式R1−NH2又はR1−OHで表される生物学的活性化合物、あるいはその類似体又は誘導体を準備するが、場合によっては、それらの化合物の別の位置を、当技術分野で周知の範囲で1種又は複数の保護基で保護することができる。また、本発明がアミノ基を有する生物学的活性分子の第二級アミン(すなわち、R4が当技術分野で周知の適当なラジカルである式R1R4NHで表される分子)も想定していることを認識されたい。
【0121】
生物学的活性化合物を、次式(W、X及びYは前記と同様である)
【0122】
【化40】
から選択される化合物と反応させることによって、次式の構造を有する化合物を形成する。
【0123】
【化41】
「式中、Zは、生物学的活性化合物が式R1−OHを有する場合には−O−、生物学的活性化合物が式R1−NH2を有する場合には−NH−であり、W及びYは前記と同様である。」
この化合物を酸化して次式のアルデヒドにする。
【0124】
【化42】
別法としては、中間体として次式の対応するジオールを形成し、それを開裂してアルデヒドとすることもできる。
【0125】
【化43】
アルデヒドを、接近可能なアミノ官能基を含有するタンパク質又は他の担体分子、例えば式中、mが整数、Pがタンパク質又は他の担体分子、(NH2)mがその接近可能なアミノ官能基である次の一般式を有する化合物などと結合させて、
【0126】
【化44】
次式の複合体を形成する。
【0127】
【化45】
「式中、nは分子複合体の複合数であり、単一の担体分子に結合している特定の薬剤の分子数を表し、反応条件や特定の薬剤をトランスフェリンタンパク質などの担体分子に結合させるために使用される基礎となる中間化合物によって異なる。
【0128】
本発明で使用を想定している担体分子には、トランスフェリンなどのタンパク質、Safavyによって明らかにされた受容体リガンドペプチド、又はその他のタンパク質、抗体、レクチン又は細胞表面に付着できるその他の物質が含まれる。
【0129】
本発明の一般的方法は、次の一般式で表されるオレフィン、ジオール、及びアルデヒドなど、各種中間化合物の形成を想定していることを認識されたい。
【0130】
【化46】
「式中、R1、Z、及びYは前記と同様であり、R2は−CH=CH(W)、−CH(OH)CH(OH)W、又は−C(O)Hであり、Wは前記と同様である。」
R1には、必要であれば、例えばパクリタキセルの場合であればTBDMS、TROC、BOM、ベンジル、TES、又はEEなどの1個又は複数の保護基を含めることができる。その場合必要であれば、本発明の方法に1個又は複数の保護基でR1を保護する段階及び脱保護する段階を含めることができる。例えば、パクリタキセルをアシル化剤と結合させる段階に先立って、パクリタキセルの2’位をTBDMS、TROC、BOM、ベンジル、TES、又はEEなどで保護し、その後、その化合物を対応するアルデヒドに変換する段階の後に2’位を脱保護することができる。
【0131】
(VIII.分析/解析)
上記の方法で形成されたトランスフェリン−薬剤複合生成物を、マススペクトル及びFPLCで解析した。200nmから800nmのUVスペクトルを測定した。トランスフェリン/トランスフェリン複合体の測定濃度は0.5mg/mlとした。測定サンプルはPBS緩衝液(pH=7.4)で調製した。円偏光二色性の測定は、光路長1mmの円筒状水晶セルを用い、240nmから190nmまでのスペクトルを測定した。トランスフェリン/トランスフェリン複合体の測定濃度は1mg/mlとした。測定サンプルは水又はPBS緩衝液(pH=7.4)で調製した。
【0132】
蛍光分光光度計を用いてトランスフェリン、ローダミン123、及びトランスフェリン−ローダミン123複合体の蛍光を測定した。トランスフェリン(0.25mg/ml)、ローダミン123(10ng/ml)及びトランスフェリン−ローダミン123複合体(0.25mg/ml)は、リン酸塩緩衝液(pH7.0)で溶解した。励起波長はトランスフェリン及びトランスフェリン−ローダミン123複合体では280nm、ローダミン123では500nmとし、300から700nmの範囲の発光スペクトルを記録した。
【0133】
Laemmliゲル電気泳動法(「Cleavage of Structural Proteins During the Assembly of the Head of Bacteriophage T4」、Nature、227巻、680−685頁(1970年))を用いて、トランスフェリン複合体の純度を評価した。12%のアクリルアミドからなる垂直スラブゲル(NuPAGE電気泳動装置、NOVEX(登録商標))及びE1900−XCELL(登録商標)Mini Cell装置(Novex、サンジエゴ、カリフォルニア州)を用いて、複合体カラム分画のドデシル硫酸ナトリウム(SDS)ポリアクリルアミドゲル電気泳動を行った。サンプルを調製し、100℃で3分間加熱後、ゲル上に載せた。トランスフェリン複合体(20μl)を、ゲル上に約0.03mg/mlのタンパク質濃度で添加した。電気泳動が終了した後、ゲルをクマシーブルー染色液で30分間染色し、次いで5〜12時間脱色した。トランスフェリン−7−パクリタキセル複合体については、2−メルカプトエタノールの存在下(1μl、還元剤)及び非存在下でサンプルを分析した。
【0134】
既知量のパクリタキセルを注入し、そのピーク面積をパクリタキセル濃度に対してプロットすることにより、HPLCによるパクリタキセルの検量線を作成した。サンプルは、C−4カラム(5μm、300Å、25cm×4.6mm内径、流速1ml/分)を用い、溶媒A(80%水、20%ACN、0.1%TFA)と溶媒B(80%ACN、20%水、0.1%TFA)とのグラジエントで溶離する分析用逆相HPLCによって分析した。HPLC分析に要する時間は全部で24分であった。分析用HPLCに用いたグラジエント法では、溶媒Bを0%から始めて、20分間で100%まで増やし、次いで溶媒B100%で2分間溶離し、最後に2分間で溶媒Bを0%に徐々に戻した。
【0135】
パクリタキセルの原液(1mg/ml)は、パクリタキセルを酢酸エチルに溶解して調製した。パクリタキセルの最終濃度は、25、50、75、100μg/mlとした。各濃度について、サンプルをHPLCに注入し、2回繰返して得られたピーク面積の平均を、パクリタキセルの濃度に対してプロットし、パクリタキセルの検量線を作成した。
【0136】
トランスフェリン−7−パクリタキセル複合体の結合比は、複合体を酸加水解し、続いてHPLCでパクリタキセルの量を測定した後に判断した。1mgのトランスフェリン−パクリタキセルを、0.4mlのPBS緩衝液に溶解し、酢酸を加えてpHを4に調整後、反応混合物を室温で10分間攪拌した。反応混合物に酢酸エチルを0.2ml添加して2分間かき混ぜ、パクリタキセルを単離した。次に濁った混合物を、4℃で10分間、1000gで遠沈し、パクリタキセルを含む澄明な上澄液20μlをHPLCに注入した。パクリタキセルの検量線によれば、結合比はトランスフェリン1個に対しパクリタキセルが3個であった。
【0137】
(IX.トランスフェリン複合体の安定性)
A.熱安定性
トランスフェリン及びトランスフェリン複合体を、PBS緩衝液(pH7.0、0.05モル)に溶解(1mg/ml)して、トランスフェリン及びトランスフェリン複合体の貯蔵液を調製した。封管中に0.1mlずつ等分し、37℃に保った。適当な時間間隔で3本の封管を取り出し、直ちにドライアイスで凍結してCD及びSDS−PAGE電気泳動による分析を行うまで−70℃で保存した。分析直前に該当するサンプルを急速解凍した。
【0138】
B.pH依存安定性
トランスフェリン及びトランスフェリン複合体を、水に溶解してそれぞれの貯蔵液(1mg/ml)を調製した。さらに、貯蔵液を様々なpHの緩衝液(0.05モル)で希釈して最終濃度を0.1mg/mlとした。このサンプルを、室温に2時間、又は37℃に2時間保ち、CD、蛍光及びUV分光法で分析した。
【0139】
(X.細胞毒性データ)
哺乳動物の培養細胞を用いて、トランスフェリン−3−コレステロール複合体、トランスフェリン−ローダミン123複合体及びトランスフェリン−7−パクリタキセル複合体の増殖抑制能を調べた。以下で考察するように、ローダミン123及びコレステロールのトランスフェリン複合体は、腫瘍細胞及び正常細胞のいずれに対しても大きな有害作用を示さなかった。このことから、物質をトランスフェリンに複合させること自体が細胞の増殖に大きな影響を及ぼしている訳ではないことがわかる。しかし、パクリタキセルの複合体は、癌に対して有効性を示す化合物であり、正常細胞に有害な影響を与えないで癌細胞を攻撃できる可能性のあることがわかった。従って、本発明は、できれば最適投与量下で正常細胞を傷害することなしに、癌治療で効果を示す化合物で癌細胞を特異的に攻撃するための有望な方法を提案するものである。
【0140】
細胞系としては、KB(ヒト口腔類表皮癌種、ATCC#CCL−17)、Lu−1(ヒト肺癌細胞系、イリノイ大学医学部、腫瘍外科部門より入手)、及びhTERT(テロメラーゼ−不朽化正常上皮細胞系、Clontech#C4000−1)を選択した。二つの治療プロトコルで、化合物の投与量を変えて評価した。プロトコルAでは、KB、Lu、及びhTERT細胞の各培養物を、培養日以降、3種の複合化合物の投与量を様々に変えて処理した。プロトコルBでは、KB、Lu、及びhTERT細胞の各培養物を集密するまで(9日間)増殖させ、次いで、試験の期間中、トランスフェリン−7−パクリタキセル複合体の様々な投与量で処理した。
【0141】
KBは、10%ウシ胎児血清(FBS)、100単位/mlペニシリンG、100μg/mlストレプトマイシン硫酸塩、0.25μg/mlアンフォテリシンB(Fungizone)(PSF)(GIBCO)及び1%可欠アミノ酸(NAA)(Sigma)を追加したDMEM(GIBCO)中で培養した。Luは、10%FBS、PSF、及び1%NAAを追加したMEME(GIBCO)中に保持した。hTERT−RPE1は、10%FBS+PSFを追加したDMEM/F−12(GIBCO)中に保持した。すべての細胞系を空気中5%CO2雰囲気下、37℃、湿度100%で培養した。
【0142】
全般的な手順は、Skehan他による論文「New colorimetric cytotoxicity assay for anticancer−drug screening」、J.Natl.Cancer Inst.B2、1107−1112頁、1990年、及びLikhitwitayawuid他による論文「Cytotoxic and antimalarial bisbenzylisoquinoline alkaloids from Stephania erecta」、J.Nat.Prod.、56巻30−38頁1993年、に記載されているものに従った。典型的には細胞を60%から70%集密するまで増殖させ、培地を交換し、1日後にその細胞を試験に供した。最初に試験サンプルを滅菌したPBSに溶解した。PBS溶媒を用いて順次希釈して、その10μlを3つのウェルに添加した。対照群にはPBS(10μl)を添加した。プレートを調製した後、トリプシンで処理して組織培養フラスコから細胞を取り出し、計数し,新鮮培地で希釈した。KB細胞(3×104細胞/ml)、Lu細胞(5×104細胞/ml)及びhTERT−RPE1細胞(4×104細胞/ml)(培地190μl)を96穴プレートに添加した。プレートを5%CO2存在下で37℃で培養し、20%冷トリクロロ酢酸100μlを添加して細胞を固定し、4℃に30分間保った。プレートを水道水で洗浄(3回)し、一夜乾燥した。1%酢酸に溶かした0.4%(w/v)スルホローダミンBを100μl添加して固定した細胞を30分間染色した。プレートを1%酢酸で洗浄(3回)し、乾燥させた。10mM非緩衝Tris塩基(pH10)を添加(200μl/ウェル)して結合している染料を可溶化した。プレートを5分間シェイカー上に保持し、ELISAプレートリーダーを用いて515nmでの吸収を測定した。いずれの場合も、96穴プレートの幾つかのウェルに同数の細胞を添加し、37℃で30分間培養して零日対照とした。次いで、トリクロロ酢酸で細胞を固定し、上記同様に処理した。
【0143】
始めに、プロトコルAに従って、トランスフェリン−3−コレステロール複合体、トランスフェリン−ローダミン123複合体及びトランスフェリン−7−パクリタキセル複合体を用いて一連の実験を行った。プロトコルAでは、プレート作製日に試験化合物を62.5〜250μg/mlの範囲の様々な濃度で添加し、3日毎に培地を補充し、試験化合物の濃度を同一に維持した。時間に対する吸光度のプロットで示されるように、トランスフェリン−3−コレステロール複合体及びトランスフェリン−ローダミン123複合体はいずれの細胞系に対しても活性を示さなかった(図5、6、8、9、11、12)。これに対してトランスフェリン−7−パクリタキセル複合体では、試験したその全ての濃度で細胞増殖の完全な抑制が観察された(図7、10、13)。
【0144】
そこで、さらに低濃度のトランスフェリン−7−パクリタキセル複合体を使用して再度試験した。図14に示すように、KB細胞をプロトコルAで試験してみると、5又は50μg/mlの濃度では完全な増殖抑制が観察されたが、0.5μg/mlの濃度では増殖に対する影響は認められなかった。それぞれ図15及び16に示すように、Lu及びhTERT細胞についても同様な応答が観察された。
【0145】
プロトコルBによるKB細胞についての試験でも類似の結果が観察された。プロトコルBでは、9日目まで培地を取り替えることなしに細胞を増殖させ、9日目に様々な濃度の試験化合物を添加し、次いで、3日毎に化合物及び培地を補充した。50又は5μg/mlの濃度では細胞数のかなりの減少が観察されたが、0.5μg/mlの濃度では細胞数は対照と同程度であった(図17)。
【0146】
Lu細胞は抵抗性が若干大であった。50μg/mlでは細胞数のかなりの減少が観察されたが、5μg/mlの濃度ではその減少の程度が小さくなり、0.5μg/mlの濃度では細胞数が減少しなかった(図18)。
【0147】
hTERT細胞に対しては効果が最も小さかった。対照培養物及び0.5又は5μg/mlのトランスフェリン−7−パクリタキセル複合体で処理した培養物で細胞増殖のわずかな増加が観察された。細胞を50μg/mlで処理した場合、この増殖は減少したが、それでも16日目で細胞数は9日目とほぼ同じであった(図19)。これらの結果は、トランスフェリン−7−パクリタキセル複合体が、主として癌細胞(KB及びLu)を攻撃し、正常細胞(hTERT)にはあまり影響を与えないことを示唆しており、本発明によるトランスフェリン−7−パクリタキセル複合体及び他のタンパク質−薬剤複合体によって、癌治療に対する有望な方法が得られることを示している。
【0148】
以上、本発明の例示的実施形態に則して本発明をある程度詳細に説明してきた。しかしながら、本発明は、先行技術を考慮して解釈される特許請求の範囲で定義されるものであって、本発明の例示的実施形態に、本明細書に含まれる発明の概念から逸脱することなしに修正又は変更を加えることができることを認識されたい。
【技術分野】
【0001】
本出願は、2001年3月23日提出の米国仮出願第60/278,243号の利益を請求する。
【0002】
本発明は、一般に、患者の治療に使用する化合物及び方法に関する。より詳細には、本発明は癌治療用分子複合体に関する。特に、本発明は、トランスフェリン−薬剤複合体、その形成に有用な方法及び中間体、並びにその複合体を使用して患者を治療する方法に関する。
【背景技術】
【0003】
現在、数多くの抗癌剤が、様々な癌の治療において臨床的に使用されている。例えば、パクリタキセル(paclitaxel)及びタキソテール(taxotere)は、乳癌及び卵巣癌の治療に使用される2つの有望な抗癌剤であるが、皮膚、肺、頭部及び頚部の癌腫など他の癌の治療においても有望視されている。この他にも、これらの癌及び他の癌を治療するために有望な化学療法剤が開発され、試験されている。パクリタキセル、タキソテール、並びに他のタキサン類(taxanes)、カンプトテシン類(camptothecins)類、エポシロン類(epothilones)及びクアシノイド類(quassinoids)などの化合物、更に癌治療で有効性を示すその他の化合物が大きな関心を呼んでいる。特に、インビトロ及びインビボで抗癌活性を示す天然物薬剤が大きな関心を呼んでいる。このような化合物は、例えば、潜在的に再生可能な資源から入手できるという点で望ましいものである。
【0004】
しかし、確認されている多くの抗癌性化合物には、化学療法で使用する上での幾つかの問題点がある。その特有の問題の一つは、多くの抗癌性化合物が水に不溶であること関し、それが、化学療法に役立つ適切な医薬製剤を開発する上で大きな問題が生じさせていることである。これらの薬剤の水溶性を増大させる試みにおいて、これら薬剤は、しばしば、様々な担体化合物と共に製剤されている。しかし、これらの担体化合物は、その製剤を使用して治療した患者に、様々な副作用を引き起こすことがある。例えば、パクリタキセル、カンプトテシン及びこれらの類似体は、一般に、ポリエトキシレートひまし油およびエタノールの混合物と製剤されるが、これらの混合物は、臨床試験において好中球減少症、粘膜性、心性及び神経性の毒性、過敏症、ヒスタミン放出及び重症アレルギー反応を含む副作用をもたらすことが報告されている。
【0005】
癌治療でこれらの化学療法剤を使用する際のもう一つの問題は、正常健常細胞に有害な影響を及ぼさないで、癌細胞を攻撃することの難しさである。例えば、パクリタキセルは、正常細胞よりも癌細胞でより頻度が高い、有糸分裂及び細胞分裂過程を妨害することによってその抗癌活性を発揮する。しかし、化学療法を受けている患者は、正常健常細胞内での有糸分裂の妨害に関連する様々な副作用を体験することがある。
【0006】
従って、癌の治療法において、化学療法剤で癌細胞を直接的に攻撃するための化合物及びその使用法の開発が、極めて望まれている。これが可能になれば、担体化合物による有毒な副作用の軽減又は排除、薬剤の標的部位への効率的な送達、並びに投与薬剤の投与量の削減、及びその結果としての、健常細胞に対する毒性や化学療法における投与費用の低減に繋げることができる。
【0007】
その興味ある具体的手法の一つとして、腫瘍認識分子と複合させた抗癌剤部分を利用する方法がある。例えば、米国特許第6,191,290号(Safavy)には、腫瘍細胞表面の受容体と結合できる受容体リガンドペプチドと複合させたタキサン部分の形成及びその使用について考察がなされている。Safavyは特に、このような受容体リガンドペプチドを、BBN/GRP受容体認識ペプチド、ソマトスタチン受容体認識ペプチド、表皮成長因子受容体認識ペプチド、モノクローナル抗体又は受容体認識炭水化物とすることができることを指摘している。
【0008】
特に興味ある腫瘍認識分子の一つとしては、ヒトのタンパク質であるトランスフェリンがある。トランスフェリンは、分子量が約79550の血清糖タンパク質であり、ヘモグロビン合成のために成熟途上の赤血球に対する鉄の輸送に関与している。トランスフェリンは、鉄イオンに対する結合親和性が極めて高く、それによって、血漿中には、鉄の強毒性形態である遊離の鉄イオンが、本質的に存在しない。さらに、成熟過程にある細胞に必要な鉄分は、ジフェリック・トランスフェリン(各タンパク質分子が特異的に2個のFe3+イオンと結合してサーモンピンク色の錯体を形成したもの)によって供給される。すなわち、鉄が結合したトランスフェリンは、細胞膜上の受容体と結合し、そのトランスフェリン−受容体複合体が細胞膜内に取込まれ、次いで細胞質に鉄が放出されて、アポトランスフェリン−受容体複合体は細胞表面に戻り、アポトランスフェリンが受容体から放出されることになる。成熟過程にある細胞は、その細胞表面にトランスフェリン受容体を有しているが、静止期にある細胞ではトランスフェリン受容体が存在しないか、あっても極少数であることが明らかになっている。さらに、癌細胞には多くのトランスフェリン受容体が存在し、興味いことには、薬剤耐性癌細胞にはさらに多くのトランスフェリン受容体が存在することが明らかにされている。癌細胞上にはトランスフェリン受容体が存在し、正常細胞上には存在しないと言うことは、トランスフェリン複合体が癌細胞に対する標的薬剤としての一つの選択肢となり得る可能性を示唆している。例えば、Yeh他の論文「Killing of Human Tumer Cells in Culture with Adriamycin Conjugates of Human Transferrin」、Clin.Immunol.Immunopathol.32巻、1−11頁(1984年)、及びSizensky他の論文「Characterization of the Anti−Cancer Activity of Transferrin−Adriamycin Conjugates」、Am.J.Reprod.Immunol.27巻、163−166頁(1992年)で報告されているように、癌治療におけるトランスフェリン−アドリアマイシン複合体の治療指数はフリーのアドリアマイシンよりも大きい。
【0009】
その他の研究でも、ドコルビシン(Docorubicin)(Kratz他の論文「Transferrin conjugates of Docorubicin:Synthesis,Characterization,Cellular Uptake,and in vitro Efficacy」、J.Pharm Sci.、87巻、338−346頁(1998年))及びマイトマイシン(Mytomycin)C(Tanaka他の論文「Synthesis of Transferrin−Mitomycin C Conjugate as a Recepter−Mediated Drug Targeting System」、Biol.Pharm.Bull.、19巻、774−777頁(1996年))などの各種化学療法剤と複合させたトランスフェリンを利用する、癌治療に対する有望な方法が提案されている。
【0010】
有効なトランスフェリン−パクリタキセル複合体を目指した試みが、Bicamumpakaらの論文「In Vitro Cytotoxicity of Paclitaxel−Transferrin Conjugate on H69 Cells」、Oncol.Rep.、5巻、1381−1383頁(1998年)で報告されている。特に、Bicamumpakaらは、2’−グルタリル−ヘキサンジアミンパクリタキセルを合成し、次いで、それを、グルタルアルデヒドリンカーを使用して、2’−グルタリル−ヘキサンジアミン基のアミンにより、トランスフェリンと結合させた。しかし、Bicamumpakaは、得られたトランスフェリン−パクリタキセル複合体におけるH69細胞の増殖を阻止する能力が、元のパクリタキセル薬剤の5.4分の1未満であったと報告している。
【0011】
従って、トランスフェリン、Safavyが認知した受容体リガンドペプチド、又は他のタンパク質、抗体、レクチン、或いは細胞表面に付着できるその他の物質などの各種分子に、化学療法剤を結合するための、新規な化合物を提供することに対する要求が理解されよう。また、そのような化合物の形成方法を提供することに対する要求も理解される。さらに、癌治療に使用するための新規な分子複合体、特に、新規なトランスフェリン−薬剤複合体に対する要求も理解される。最後に、改善されたトランスフェリン−薬剤複合体などの分子複合体の使用などによって、患者に化学療法用薬剤製剤を投与する、癌治療法用の新規な方法に対する要求も理解される。本発明は、これらの要求に応じるものである。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】米国特許第6,191,290号明細書
【非特許文献】
【0013】
【非特許文献1】Yeh等、「Killing of Human Tumer Cells in Culture with Adriamycin Conjugates of Human Transferrin」、Clin.Immunol.Immunopathol.32巻、1−11頁(1984年)
【非特許文献2】Sizensky等、「Characterization of the Anti−Cancer Activity of Transferrin−Adriamycin Conjugates」、Am.J.Reprod.Immunol.27巻、163−166頁(1992年)
【非特許文献3】Kratz等、「Transferrin conjugates of Docorubicin:Synthesis,Characterization,Cellular Uptake,and in vitro Efficacy」、J.Pharm Sci.、87巻、338−346頁(1998年)
【非特許文献4】マイトマイシン(Mytomycin)C(Tanaka等、「Synthesis of Transferrin−Mitomycin C Conjugate as a Recepter−Mediated Drug Targeting System」、Biol.Pharm.Bull.、19巻、774−777頁(1996年)
【非特許文献5】Bicamumpaka等、「In Vitro Cytotoxicity of Paclitaxel−Transferrin Conjugate on H69 Cells」、Oncol.Rep.、5巻、1381−1383頁(1998年)
【発明の概要】
【発明が解決しようとする課題】
【0014】
本発明の目的は、ヒドロキシル基又はアミノ基を有する薬剤の新規かつ有用な分子複合体組成物を提供することである。
【0015】
本発明のさらなる目的は、癌治療用の、トランスフェリン−薬剤複合体組成物を提供することである。
【0016】
本発明の他の目的は、トランスフェリン−薬剤複合物などの、癌治療に使用される分子複合体を形成するために用いる中間化合物を提供することである。
【0017】
本発明の他の目的は、分子複合体、特にトランスフェリン−薬剤複合体を形成する効率的な方法を提供することである。
【0018】
本発明の更に他の目的は、癌患者が従来経験していた副作用を軽減又は排除する、患者に新規かつ有用な化学療法剤を投与する方法を提供することである。
【0019】
本発明の更に他の目的は、患者の癌細胞に化学療法剤を効率的に集中させるための方法を提供することである。
【課題を解決するための手段】
【0020】
本発明によれば、次の式を有する分子複合体が得られる。
【0021】
【化1】
「式中、nは分子複合体の複合数(例えば1〜5の整数)であり、Pはトランスフェリンタンパク質などの、少なくともn個の接近可能なアミノ官能基を有する分子の脱アミノ部分であり、R1はヒドロキシル基又はアミノ基を有する生物学的活性分子又はその類似体もしくは誘導体の、それぞれ、脱ヒドロキシル又は脱アミノの部分であり、Zはそれぞれ−O−又はNH−であり、Yは、任意に1個又は複数のフェニルで置換された、1〜20個の炭素原子を有する直鎖状又は分岐状のアルキル基、任意に1個又は複数のアルキル又はフェニルで置換されたシクロアルキル基、或いは任意に1個又は複数のアルキル基、電子吸引基もしくは電子供与基で置換された芳香族基である。」
Pは、好ましくはトランスフェリンなどのタンパク質であり、そのタンパク質は、生物学的活性分子と結合構造を通じて結合している。生物学的活性分子は、癌治療に有用であるような天然物薬剤でよく、各種タキサン類、カンプトテシン類、エポシロン類、ククルビタシン類、クアシノイド類、アントラサイクリン類、並びにその類似体及び誘導体などが挙げられる。
【0022】
本発明はまた、トランスフェリン−薬剤複合体などの分子複合体の形成に有用な化合物に関する。その化合物は次の一般式を有している。
【0023】
【化2】
「式中、R1、Y及びZは前記と同様であり、R2は−CH=CH(W)、−CH(OH)CH(OH)W、又は−C(O)Hである{Wは水素、任意に1個又は複数のフェニルで置換される1〜20個の炭素原子を有する直鎖状又は分岐状のアルキル、任意に1個又は複数のアルキル又はフェニルで置換されたシクロアルキル、又は任意に1個又は複数のアルキル基、電子吸引基もしくは電子供与基で置換された芳香族基である。}。」
【0024】
本発明はさらに、本発明による分子複合体の製造方法、特に癌治療に使用するためのトランスフェリン−薬剤複合体の製造方法に関する。この方法は、ヒドロキシル基又はアミノ基のいずれかを有する生物学的活性分子並びにその類似体及び誘導体(及び塩又は第二級アミン)からなる第1化合物を、次式の何れかの第2化合物と反応させて、
【0025】
【化3】
次式の第3化合物を形成する段階と、
【0026】
【化4】
第3化合物を次式の第4化合物に変換する段階と、
【0027】
【化5】
第4化合物を少なくともn個の接近可能なアミノ官能基を有する分子と結合させて、次式の分子複合体を形成する段階とを含んでいる。
【0028】
【化6】
「上記各式中、Xはハロゲンであり、R1、Z、W、Y、n及びPは前記と同様である。」
Wは、好ましくは水素であり、その結果第2化合物は末端オレフィンを有する。
第3化合物を第4化合物へ転換する段階は、第3化合物の酸化によって対応する次式のジオール中間体を介し、
【0029】
【化7】
その後に、当該ジオールを酸化して第4化合物にすることによって行われる。
【0030】
本発明はさらに、トランスフェリン−7−パクリタキセル複合体、トランスフェリン−2’−パクリタキセル複合体、トランスフェリン−3’−パクリタキセル複合体、及びトランスフェリン−20−カンプトテシン複合体の製造方法を提供する。本発明はまた、トランスフェリン−ローダミン123化合物、並びにその製造に用いる中間体及び方法を提供する。
【0031】
最後に、本発明は、本発明の複合化合物を利用して患者の選択された標的細胞に生物学的活性分子を、選択的に集中させる方法に関し、特に、トランスフェリン−薬剤複合体など本発明による分子複合体を、投与量を精選して、患者に投与することを含む方法に関するものである。
【0032】
本発明のこれらの目的及び他の目的については、本発明の例示的実施形態に関する以下の詳細な説明を添付の図面と併せ検討することによって容易に把握し、理解することができる。
【図面の簡単な説明】
【0033】
【図1】トランスフェリン−7−パクリタキセル複合体の形成で使用される7−アシル−ペンタナールパクリタキセルリンカー化合物を形成するための化学反応式を示す図である。
【図2】3−アシル−ペンタナールコレステロールリンカー化合物をトランスフェリンと結合させ、トランスフェリン−3−コレステロール複合体を形成するための化学反応式を示す図である。
【図3】20−アシル−ペンタナールカンプトテシンリンカー化合物をトランスフェリンと結合させ、トランスフェリン−20−カンプトテシン複合体を形成するための化学反応式を示す図である。
【図4(a)】トランスフェリン−2’−パクリタキセル複合体の形成で使用される2’−アシル−ペンタナールパクリタキセルリンカー化合物を形成するための化学反応式を示す図である。
【図4(b)】トランスフェリン−2’−パクリタキセル複合体の形成で使用される2’−アシル−ヘキサナールパクリタキセルリンカー化合物を形成するための化学反応式を示す図である。
【図4(c)】トランスフェリン−2’−パクリタキセル複合体の形成で使用される2’−アシル−ノナナールパクリタキセルリンカー化合物を形成するための化学反応式を示す図である。
【図5】プロトコルAにおける異なる濃度での、トランスフェリン−3−コレステロール複合体のKB細胞に対する細胞毒性を示すグラフである。
【図6】プロトコルAにおける異なる濃度での、トランスフェリン−ローダミン123複合体のKB細胞に対する細胞毒性を示すグラフである。
【図7】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のKB細胞に対する細胞毒性を示すグラフである。
【図8】プロトコルAにおける異なる濃度での、トランスフェリン−3−コレステロール複合体のLu細胞に対する細胞毒性を示すグラフである。
【図9】プロトコルAにおける異なる濃度での、トランスフェリン−ローダミン123複合体のLu細胞に対する細胞毒性を示すグラフである。
【図10】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のLu細胞に対する細胞毒性を示すグラフである。
【図11】プロトコルAにおける異なる濃度での、トランスフェリン−3−コレステロール複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【図12】プロトコルAにおける異なる濃度での、トランスフェリン−ローダミン123複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【図13】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【図14】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のKB細胞に対する細胞毒性を示すグラフである。
【図15】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のLu細胞に対する細胞毒性を示すグラフである。
【図16】プロトコルAにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【図17】プロトコルBにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のKB細胞に対する細胞毒性を示すグラフである。
【図18】プロトコルBにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のLu細胞に対する細胞毒性を示すグラフである。
【図19】プロトコルBにおける異なる濃度での、トランスフェリン−7−パクリタキセル複合体のhTERT細胞に対する細胞毒性を示すグラフである。
【発明を実施するための形態】
【0034】
本発明は、新規な分子複合体、特に患者の癌治療で使用する新規なトランスフェリン−薬剤複合体を提供する。さらに、本発明は生物学的活性分子をトランスフェリン又はその他の分子などの担体分子と結合するために用いる新規な中間体化合物に関するものである。特に、本発明は、癌治療薬並びにその類似体及び誘導体、並びにその前駆体など、それぞれ、ヒドロキシル基及びアミノ基を有する生物学的活性分子のアルデヒドエステル誘導体及びアルデヒドアミド誘導体を提供する。これらの誘導体は、そのエステル結合又はアミド結合しているアルデヒド官能基とトランスフェリン分子又は他のタンパク質の各種アミノ官能基との間でシッフ塩基を形成することによって、ヒト・トランスフェリンタンパク質などの担体分子と結合することができる。
【0035】
本発明はまた、ヒドロキシル基又はアミノ基を有する様々な生物学的活性化合物のトランスフェリン複合体、又は他の分子複合体、並びにその中間体の効率的な合成プロトコルを提供する。一般化した方法では、ヒドロキシル基又はアミノ基を含有する生物学的活性分子と、二重結合好ましくは末端オレフィンを有するカルボン酸又は酸ハロゲン化物などの適当なアシル化剤とを結合させることを含む。四酸化オスミウム触媒を用いて末端オレフィン部位を迅速かつ高効率で酸化し、次いで、得られたジオールを開裂してアルデヒドにすることによって、トランスフェリン複合体又は他の分子複合体の合成に適した前駆体が得られる。これら付加生成物の合成経路における最終段階は、アルデヒドを血液タンパク質であるトランスフェリンなどの担体分子と処理し、トランスフェリン単量体に生物学的活性分子を生成させることである。これらの生物学的活性分子では生物学的活性が増大していることが見出されている。本発明は、本明細書で開示するように、担体分子には、トランスフェリンの他、生物学的活性分子のエステルリンカー化合物及びアミドリンカー化合物のアルデヒド官能基と、シッフ塩基を形成することができる、少なくとも1個の接近可能なアミノ官能基を有する任意の分子を含めるように広く想定している。
【0036】
また、本発明において、用語「生物学的活性分子」は、細胞、組織、脈管内などでの1種又は複数の生物学的過程に一般的に影響を与え、又は関与する任意の分子を含むものとして、広く解釈して理解されるべきである。このような生物学的活性分子には、薬剤、抗体、抗原、レクチン、色素、染料、トレーサ又は他の類似分子が含まれる。特に、本発明での使用を企図しているヒドロキシル基含有分子又はアミノ基含有分子には、パクリタキセル、ドセタキセル(docetaxel)及び他のタキサン類、コレステロール、ローダミン123、カンプトテシン類、エポシロンBなどのエポシロン類、ククルビタシン類、グローカルボロン(glaucarubolone)、ブルサトール(brusatol)及びブルシアンチン(bruceantin)などのクアシノイド類、アドリアマイシン、ダウノルビシン(daunorubicin)などのアントラサイクリン類、並びにこれらの類似体及び誘導体、並びにその他の化合物が含まれる。
用語「分子複合体」は、例えば本明細書で開示されるエステル及びアミドのシッフ塩基結合を通して本発明の担体分子に結合している生物学的活性分子を含む任意の化合物を広く包含するものとして理解されるべきである。
【0037】
本研究の中心は癌治療であるが、本願がさらに、各種のタンパク質又はその他の担体分子を、本発明により、他の応用分野を対象とした生物学的活性分子と複合させることを企図していることを理解されるべきである。
【0038】
(I.トランスフェリン−7−パクリタキセル複合体)
本発明によってトランスフェリン−7−パクリタキセル複合体を形成することができる。図1に示すように、先ず、パクリタキセルを様々な中間化合物を経由して7−パクリタキセルアルデヒドエステルに変換する。次いで、このアルデヒドエステルをトランスフェリンと結合させてトランスフェリン−7−パクリタキセル複合体を形成する。
【0039】
A.2’TBDMSパクリタキセルの調製
【0040】
【化8】
【0041】
先ず、パクリタキセルの2’−ヒドロキシル基をTBDMSで保護して2’−TBDMSパクリタキセルを形成する。反応例としてはTBDMSを示したが、TBDMSの代わりにTROC,BOM,CBZ,ベンジル、TES,EEなど他の保護基も使用可能であることを認識されたい。
【0042】
この材料は、Tetrahedron Letters、35巻、8931−8934頁,1994年でProf Gunda Georg他が述べている方法によって調製され、それに従って特徴付けられる。
【0043】
パクリタキセル(20.0g、23.45mM)のジメチルホルムアミド(150mL)溶液に、窒素雰囲気下でイミダゾール(23.95g、351.7mM)を添加し、続いてTBDMSCl(49.5g、328.3mM)を添加した。得られた溶液を、窒素雰囲気下、周囲温度で16時間攪拌した。この段階でTLC分析を行い、出発物質が完全に消費されていることを確認し、水(200mL)及び酢酸エチル(200mL)を添加して反応を終了した。有機層を分離し、水(100mL×2回)、塩水(50mL)で洗浄し、硫酸マグネシウム上で乾燥し、その後濾過、留去して残渣を取り出し、真空オーブン中で乾燥し、さらに精製することなしに次の反応に使用した。
【0044】
B.2’保護パクリタキセルの7−ヘキセン酸エステルの調製
【0045】
【化9】
【0046】
次に、好ましくは末端オレフィンを有する酸と反応させて、2’−保護パクリタキセルの7−ヘキセン酸エステルを形成した。本明細書の実施例では、5−ヘキセン酸を使用するが、本発明は、鎖末端から離れた位置にある二重結合を有するオレフィンアシル化剤をも想定していることを理解されたい。もっとも、好ましくは末端オレフィンを有する他の適当なアシル化剤を企図する。
例えば、本発明は次式の酸、
【0047】
【化10】
又は次式の酸ハロゲン化物の使用を企図する。
【0048】
【化11】
「上記各式中、Xはハロゲンであり、Yは、任意に1個若しくは複数のフェニル基で置換され1から20個の炭素原子を有する直鎖状若しくは分岐状のアルキル基、任意に1個若しくは複数のアルキル基またはフェニル基で置換されたシクロアルキル基、あるいは任意に1個若しくは複数のアルキル、電子吸引基または電子供与基で置換された芳香族基である。Wは、水素、任意に1個若しくは複数のフェニル基で置換され1から20個の炭素原子を有する直鎖状若しくは分岐状のアルキル基、任意に1個若しくは複数のアルキル基またはフェニル基で置換されたシクロアルキル基、あるいは任意に1個若しくは複数のアルキル、電子吸引基または電子供与基で置換された芳香族基である。」
【0049】
2’TBDMSパクリタキセル(2.0g、2.07mM)塩化メチレン(30mL)溶液に、窒素雰囲気下で、5−ヘキセン酸(0.49mL、4.14mM)を添加し、その後更にDIPC(0.81mL、5.18mM)及び4−PP(0.095g、0.64mM)を添加した。得られた反応混合物を、4時間攪拌し、TLC分析により完全に反応していることを判断した。水(50mL)及び酢酸エチル(90mL)を添加して反応を終了させ、有機層を分離し、水(50mL)、塩水(50mL)で洗浄し、硫酸マグネシウム上で乾燥した。得られた生成物を濾過し、溶媒を留去して残渣を取り出し、精製すること無しに次の反応に供した。
【0050】
C.2’保護パクリタキセルの7−アルデヒド誘導体の調製
【0051】
【化12】
【0052】
末端オレフィン部位を酸化してジオールに変え、続いて末端炭素を開裂するとパクリタキセルの7−アルデヒド2’保護誘導体が得られる。二重結合が前に考察したように鎖末端から移動している(すなわち、前記式中のWが水素でない)アシル化剤を使用する場合には、この反応中に起こる二重結合の開裂によって、二重結合を越えた鎖の部分が除去されることを理解されたい。また、例示した工程では単一段階として示されているが、次式のジオール、
【0053】
【化13】
(及び、本明細書に記載された化合物に対応するジオール)は、NaIO4無しでこの段階を実行することによって単離可能であることを認識されたい。NaIO4で処理してこのジオールを酸化開裂すると末端アルデヒドが得られる。
【0054】
7−ヘキセノイル、2’TBDMSパクリタキセル(2.2g、2.07mM)のACNとTHF(各20mL)の混合溶媒溶液に、水(20mL)を加え、続いて窒素雰囲気下で、NMO(0.49g、4.14mM)、NaIO4(0.89g、4.14mM)及びOsO4(13.15mg、0.052mM)のt−BuOH溶液を添加した。得られた反応混合物を周囲温度で2時間攪拌し、酢酸エチル及び水(各100mL)を加え反応終了とした。分離した有機層を塩水(20mL)で洗浄し、硫酸マグネシウム及びハイドロサルファイトナトリウムを通して濾過した。濾液を蒸発乾固して、粗生成物を精製すること無しに脱シリル化反応に供した。
【0055】
D.パクリタキセルの7−アルデヒド誘導体の調製
【0056】
【化14】
【0057】
2’位を次のように脱保護した。2’保護したパクリタキセルの7−アルデヒド誘導体(2.25g、2.07mM)THF(50mL)溶液に、窒素雰囲気下、周囲温度でTBAFのTHF溶液(1.0M溶液を3.11mL、3.11mM)を添加した。得られた反応混合物を2時間攪拌した。TLC分析によればこの時点で出発物質はなかった。酢酸エチル(200mL)及び0.5NHCl(100mL)を加え混合物の反応を終了し、分離した有機層を水(200mL)、塩水(100mL)で洗浄し、硫酸マグネシウム上で乾燥した。有機層を濾過、蒸発乾固し、次いで酢酸エチル及びヘプタンを用いたカラムクロマトグラフィーによって精製し、60%の総収率で精製物質を得た。化合物はMS及び1H NMRによって解析した。
【0058】
E.トランスフェリン−7−パクリタキセル複合体の調製
次にアルデヒドエステル誘導体をトランスフェリンと結合させて、複合数n(トランスフェリン1分子当たりのパクリタキセルの分子数)を有するトランスフェリン−7−パクリタキセル複合体を形成させることができる。複合数は3であったが、条件を変更すると複合数nが1から5のトランスフェリン−7−パクリタキセル複合体が生成する可能性も考えられる。本発明は、トランスフェリンの代わりに、接近可能なアミノ官能基を有する他のタンパク質などの担体分子を使用することが可能であり、得られる分子複合体の複合数もそれに応じて変動可能であることを当然考慮に入れている。
【0059】
【化15】
【0060】
80mg(1μmol)のトランスフェリンを含むPBS緩衝液/DMSO溶液に、19.04mg(20μmol)の7−アシル−ペンタナルパクリタキセルを含むDMSO溶液2mlを滴下した。トランスフェリンPBS緩衝液/DMSO溶液は、トランスフェリンを4mlのPBS(50mmol、pH8.0)に溶解し、さらにそのトランスフェリンPBS溶液に、0℃で、2mlのDMSOを加えて調製した。反応混合物を、C24振とう恒温器(New Brunswick Scientific classic series、Edison、アメリカ)を使用して、37℃で8時間振とうした。反応混合物を5.0μmの濾過装置で濾過し、澄明な濾液を、superdexHR200カラム(2.0×30cm)に20mMのTris−HCl(pH8.0)を0.5ml/分の流速で流すFPLCによって精製した。トランスフェリンに対応する分画を集めて凍結乾燥した。
【0061】
本明細書では例示しないが、7−パクリタキセル複合体と同様に10−デアセチルパクリタキセルの分子複合体が形成されることを理解されたい。例えば、出発化合物として次式の10−デアセチルパクリタキセルを用い、
【0062】
【化16】
アシル化剤として6−ヘプテン酸を用い、上述の方法と同様にしてトランスフェリン−10−アシル−ヘキサナールパクリタキセル複合体が形成される。当技術分野で知られているように、2’位及び7位の適切な保護及び脱保護を利用することができる。
【0063】
(II.トランスフェリン−3−コレステロール複合体)
トランスフェリンに非細胞毒性分子を結合した場合の結果を調べるためにトランスフェリン−3−コレステロール複合体を調製した。以下で考察するように、これらの結果は、本発明による癌治療用トランスフェリン−薬剤複合体は、インビトロ又はインビボで細胞毒性活性を示す癌治療剤を使用して形成されるべきことを示唆する。例えば癌治療用分子以外の分野での応用を研究するために、3−コレステロールと別のタンパク質との様々な複合体を同様にして調製して、そのようなタンパク質の複合物と他の生物学的活性化合物と比較することが可能であろう。
【0064】
図2に示すように、先ず、コレステロールを、各種中間化合物を経由して3−コレステロールアルデヒドエステルに変換し、次いで、それをトランスフェリンと結合させトランスフェリン−3−コレステロール複合体を形成する。前に考察したように、本明細書の実施例で使用する5−ヘキセン酸を、前に述べたようなその他各種アシル化剤で代替することができる。
【0065】
A.3−ヘキセノイルコレステロールの調製
【0066】
【化17】
【0067】
コレステロ−ル(2.0g、5.17mM)塩化メチレン(20mL)溶液に、5−ヘキセン酸(0.68mL、5.69mM)を添加し、続いて窒素雰囲気下で、DCC(1.60g、7.76mM)及び4−PP(0.115g、0.78mM)を加えた。得られた反応混合物を、周囲温度で1時間攪拌し、メチルt−ブチルエーテル(60mL)を加え反応終了とした。尿素を濾過により除去し、生成物を分液ロートに移し、1N HCl(10mL)、水(30mL)及び塩水(30mL)で洗浄した。生成物をMgSO4上で乾燥後、濾過し、溶媒を留去して残渣を得た。95%以上の収率で得られた生成物を1H NMRで解析した。
【0068】
B.コレステロールの3−アシル−ペンタナールエステルの調製
【0069】
【化18】
【0070】
コレステロール3−ヘキセン酸エステル(2.0g、4.14mM)のTHFとt−BuOHの混合溶媒(各10mL)溶液に、水(5mL)を加え、引き続き窒素雰囲気下で、NMO(0.97g、8.3mM)、NaIO4(1.78g、8.3mM)及びOsO4(21.3mg、0.083mM)のt−BuOH溶液を添加した。得られた反応混合物を、TLC分析で調べて変換が完全になるまで16時間攪拌した。反応混合物に珪藻土(1.6g)を加えて濾過した。フィルタケーキを酢酸エチル(100mL)で洗浄し、濾液を分液ロートに移し、1N HCl(15mL)、水(25mL)及び塩水(15mL)で洗浄し、次いでMgSO4上で乾燥した。濾過した溶液を蒸発乾固し、続いてカラムクロマトグラフィーで精製して85%の収率で生成物を得た。生成物を1H NMRで解析した。NMR分析によればコレステロール系の内部二重結合は酸化条件でも変化していないことがわかった。
【0071】
C.トランスフェリン−3−コレステロール複合体の調製
次に、アルデヒドエステル誘導体をトランスフェリンと結合させ、複合数がnであるトランスフェリン−3−コレステロール複合体を形成できる。
【0072】
【化19】
【0073】
20mg(0.5μmol)のトランスフェリンを含むPBS−緩衝液(50mmol、pH7.0)2mLに、1.21mg(2.5μmol)の3−アシル−ペンタナールコレステロールを含むDMSO溶液1mLを滴加した。反応混合物を、C24振とう恒温器(New Brunswick Scientific classic series、Edison、アメリカ)を用いて、37℃で30分間振とうした。この混合物に反応停止剤として1.527mg(25mmol)のエタノールアミンを含むPBS溶液0.5mLを添加して反応を停止させた。次に、濁った混合物を4℃、1000gで10分遠沈し、上澄み液を、superdexHR200カラム(2.0×30cm)を用いて20mMのTris−HCl(pH8.0)を0.5ml/分の流速で流すFPLCで精製した。トランスフェリンに対応する分画を集め、凍結乾燥した。
【0074】
III.トランスフェリン−20−カンプトテシン複合体
図3に示すように、先ず、カンプトテシンを様々な中間化合物を経由して20−カンプトテシンアルデヒドエステルに変換し、次いで、それをトランスフェリンと結合させて、トランスフェリン−20−カンプトテシン複合体を形成する。本発明においては、トランスフェリンを、接近可能なアミノ官能基を有する他のタンパク質などの担体分子で代替可能であり、5−ヘキセン酸を、前に述べたような他のアシル化剤で代替可能である。
【0075】
A.20−ヘキセノイルカンプトテシンの調製
【0076】
【化20】
【0077】
DMF(40mL)中にカンプトテシン(2.0g,5.74mM)及び5−ヘキセン酸(0.75mL、6.31mM)を含む混合物に、窒素雰囲気下で、DIPC(0.99ml,6,31mM)及び4−PP(0.13g、0.86mM)を添加した。得られた反応混合物を、周囲温度で24時間攪拌した。TLC分析でカンプトテシンが完全に消費されていることを確認した後、塩化メチレン及び水(各200mL)を用いながら反応混合物を分液ロートに移した。分離した有機層を塩水で洗浄し、MgSO4上で乾燥し、濾過して蒸発乾固した。固体残渣を塩化メチレン及び酢酸エチルを用いて結晶化した。結晶を濾過して純度98%以上の材料を70%の収率で得た。結晶をMS及び1H NMRデータで解析した。
【0078】
B.カンプトテシンの20−アシル−ペンタナールエステルの調製
【0079】
【化21】
【0080】
カンプトテシン20−ヘキセン酸エステル(1.0g、2.25mM)のTHF、アセトン及びACN混合溶媒(各15mL)溶液に、水(15mL)を加え、引き続き窒素雰囲気下で、NMO(0.53g、4.5mM)、NaIO4(0.96g、4.5mM)及びOsO4(15.3mg、0.06mM)のt−BuOH溶液を添加した。得られた反応混合物を、周囲温度で4時間攪拌し、ヘキセン酸エステルからカンプトテシンペンタナール誘導体への変換を完結させた。反応混合物を、塩化メチレン及び水(各200mL)に分配させた。有機層を分離し、1N HCl(10mL)、水(50mL)、塩水(50mL)で洗浄し、MgSO4上で乾燥し、濾過後、蒸発乾固した。残渣をカラムクロマトグラフィーで精製し、精製物をMS及び1H NMRデータにより解析した。
【0081】
C.トランスフェリン−20−カンプトテシン複合体の調製
次に、アルデヒドエステル誘導体をトランスフェリンと結合させ、複合数がnであるトランスフェリン−20−カンプトテシン複合体を形成する。
【0082】
【化22】
【0083】
トランスフェリン−20−カンプトテシン複合体を形成する方法は、先に述べた、トランスフェリン−7−パクリタキセル複合体及びトランスフェリン−3−コレステロール複合体を形成する方法と同様であり、DMSO及びトランスフェリンPBS溶液を利用する。得られたトランスフェリン−20−カンプトテシンのマススペクトル分析によれば、結合比はトランスフェリン1分子に対してカンプトテシンが3分子であった(すなわち、n=3)。トランスフェリンとトランスフェリン−20−カンプトテシン複合体の円偏光二色性(CD)スペクトルが相違しており、全体としての立体配座が変化したことを示唆する。
【0084】
(IV.トランスフェリン−ローダミン123複合体)
検出及び視覚化のためにローダミン123の蛍光を利用する目的で、トランスフェリン−ローダミン123複合体を調製した。ローダミン123及びトランスフェリンのそれぞれの遊離アミノ基の間を繋ぐリンカーとしてグルタルアルデヒドを用いて、1段階でトランスフェリン−ローダミン123複合体を調製した。この反応では中間体として次式の活性アルデヒド化合物が形成されると考えられる。
【0085】
【化23】
【0086】
A.トランスフェリン−ローダミン123複合体の調整
【0087】
【化24】
【0088】
トランスフェリン40mg(0.5μmol)を含むHeps緩衝食塩水(HBS、150mmolNaCl,10mmol/L Heps、pH7.4)溶液1mlを、1mlのローダミン123(5μmol)と4分間かき混ぜて混合した。1mlのグルタルアルデヒド(12.5mmol、HBS溶液)を、室温でかき混ぜながら4分間で滴加した。反応停止剤として0.5mlの25mmolエタノールアミンHBS溶液を添加し、4分間攪拌して結合反応を停止させた。混合物を透析チューブに移し、暗黒下に4℃で8時間、1LのHBSに対して透析した。次に、濁った混合物を4℃で10分間、1000gで遠沈し、澄明上澄み液に対して、superdexHR200カラム(2.0×30cm)を用い0.5ml/分の流速で20mMのTris−HCl(pH8.0)を流してクロマトグラフを行った。トランスフェリンに対応する分画を集めて凍結乾燥した。
【0089】
(V.2’−パクリタキセル化合物)
図4(a)、4(b)、4(c)に示すように、各種2’−パクリタキセル中間化合物を形成したが、アルキル鎖の短いアルデヒド誘導体(図4(a)に示した方法で5−ヘキセン酸を使用した場合など)では、トランスフェリン−2’−パクリタキセル複合体の形成に問題があった。おそらく、2’位が半球状タキサン骨格のC−13の周囲にある凹状領域に位置することに由来する障害によるものであろう。従って、この結果から、ヘキセン酸よりも鎖が長い結合用化合物を用いて形成したアルデヒド誘導体など、より長いアルキル鎖をもったアルデヒド誘導体を利用して、本発明によるトランスフェリン−2’−パクリタキセル複合体を形成できると考えられる。さらなる実験により、それぞれ図4(b)及び4(c)に示すように、同様な化学的方法を使用して2’−ヘプタナール トランスフェリン複合体及び2’−ノナナールトランスフェリン複合体をより容易に形成できることがわかった。2’−ノナナールパクリタキセル化合物の形成には、図4(c)に示すようにオレイン酸(9,10位に二重結合を有する18炭素鎖の酸)を使用したことに留意されたい。さらに、トランスフェリンが比較的大きなタンパクであることを考えれば、この結果から、前に述べたSafavyによって確認されたタンパク質などのより小さな担体分子は、パクリタキセルの凹状構造による障害が少なく、前記のアシル化剤の式においてY<4であるようなアルキル鎖の短い2’−パクリタキセルアルデヒド誘導体とより容易に複合体を形成できると考えられる。
【0090】
A.パクリタキセル2’−ヘキセン酸エステルの調製
【0091】
【化25】
【0092】
パクリタキセル(2.0g、2.34mM)及び5−ヘキセン酸(0.31mL、2.58mM)の塩化メチレン(25mL)溶液に、窒素雰囲気下、0℃でDCC(0.72g、3.51mM)を添加し、更にその後4−PP(0.17g、0.5mM)を添加した。得られた反応混合物を2時間攪拌し、その間に、反応混合物の温度は周囲温度に戻した。TLCで反応を調べ、2時間後に反応が完結していることを確認した。水及び酢酸エチルを各30mL添加して反応を終えた。混合物を分液ロートに移し、有機層を1N HCl(10mL)、水(30mL)及び塩水(20mL)で洗浄し、硫酸マグネシウム上で乾燥した。乾燥した濾液を蒸発乾固し、メチルt−ブチルエーテルで結晶化した。得られた化合物について、HPLCにより純度95%以上であることを確認した。また、1H NMR分析によりパクリタキセルの7位のヒドロキシル基には影響がなく2位のヒドロキシル基でエステル化されていることを確認した。収率は98%であった。
【0093】
B.パクリタキセル2’アルデヒド誘導体の調製
【0094】
【化26】
【0095】
パクリタキセル2’−ヘキセン酸エステル(0.475g、0.5mM)のt−BuOH、アセトン及び水(各2mL)の混合溶液に、窒素雰囲気下で、NMO(0.118g、1mM)NaIO4(0.214g、1mM)及びOsO4(2.54mg、0.01mM)のt−BuOH溶液を添加した。得られた混合物を周囲温度で2時間攪拌した。TLCによればこの時点で反応は完結していた。混合物に水及び酢酸エチル(各20mL)を添加して反応を終え、有機層を分離し、1N HCl(10mL)、水(10mL)及び塩水(10mL)で洗浄した。有機層を硫酸マグネシウム及びハイドロサルファイトナトリウムを通して濾過し、蒸発乾固した。粗化合物を酢酸エチル及びヘプタンを使用したカラムクロマトグラフィーで精製し、MS及び1H NMRで解析した。収率85%。
【0096】
C.パクリタキセル2’ジオール誘導体の調製
図4(a)に示したように、上記反応でNaIO4を使用しない場合には、次のようにジオールが単離される。
【0097】
【化27】
【0098】
パクリタキセル2’−ヘキセン酸エステル(9.0g、9.47mM)アセトン(135mL)溶液に、水(50mL)を添加した。得られた溶液に、窒素雰囲気下で、NMO(2.22g、18.94mM)を添加し、続いてOsO4(48.2mg、0.19mM)t−BuOH溶液を添加して16時間攪拌を続けた。TLCで変換の完結を確認した後、反応混合物に珪藻土(15g)を加えて濾過した。濾液をロータリー蒸留器でアセトンが無くなるまで濃縮し、引き続き水層を固形食塩で飽和させた後、酢酸エチル(200mL)による抽出操作に付した。得られた有機層を水(10mL)、1N HCl(100mL)、水(10mL)及び塩水(100mL)で洗浄し、MgSO4を通して濾過した。溶媒を留去し、残渣をカラムクロマトグラフィーで精製し、収率65%で純粋なジオールを得た。先に考察した7−パクリタキセル誘導体の対応するジオールの場合と同様、ジオールをNaIO4で処理する酸化的開裂によって末端アルデヒドが得られる。
【0099】
D.トランスフェリン−2’−アシル−ヘキサナルパクリタキセル複合体の調製
図4(b)に示すように、まず、パクリタキセルを、各種中間体を経由して2’−アシル−ヘキサナールパクリタキセル化合物に変換した。2’−アシル−ヘキサナールパクリタキセルアルデヒドエステル誘導体を形成するための化学過程は、5−ヘキセン酸の代わりに6−ヘプテン酸を使用すること以外は2’−アシル−ペンタナールパクリタキセル誘導体の形成について既に述べた化学過程と同様である。
【0100】
次に、アルデヒドエステルをトランスフェリンに結合させ、以下に示すような複合数がnであるトランスフェリン−2’−パクリタキセル複合体を形成する。
【0101】
【化28】
この手順は、トランスフェリン−7−パクリタキセル複合体の調製について既に述べたものと同様である。マススペクトルによれば、各トランスフェリンに2分子のパクリタキセルが複合したもの(すなわち、n=2)も検出されたが、主なトランスフェリン−2’−パクリタキセル複合生成物の結合比はトランスフェリン1分子に対しパクリタキセル1分子(すなわち、n=1)であった。遠紫外領域でのトランスフェリンとトランスフェリン−2’−パクリタキセル複合体との円偏光二色性スペクトルが相違しており、2’−パクリタキセル複合体の全体としての立体配座の変化が示唆される。
【0102】
E.トランスフェリン−2’−アシル−ノナナールパクリタキセル複合体の調製
図4(c)に示すように、パクリタキセルを、各種中間体を経由して先ず2’−アシル−ノナナールパクリタキセルアルデヒドエステル化合物に変換することによって、2’−パクリタキセルのトランスフェリン複合体を形成した。化学過程は2’−アシル−ペンタナールパクリタキセル誘導体について既に述べたものとやはり同様であるが、図4(c)で示したように、アシル化剤としてオレイン酸(cis−9−オクタデセン酸、CH3(CH2)7CH=CH(CH2)7CO2H)を使用したことに留意されたい。従って、鎖を開裂してアルデヒドを形成させるに際しては、9,10位の二重結合を越えて延びている鎖の部分が除去される。
【0103】
2’−アシル−ノナナールパクリタキセルとトランスフェリンを複合させて次式のトランスフェリン−2’−パクリタキセル複合体を形成させることは、トランスフェリン−7−パクリタキセル複合体について既に述べた複合の場合と同様である。
【0104】
【化29】
【0105】
(VI.アミド誘導体)
本発明はまた、アミノ基含有化合物のアミド誘導体の形成をも想定している。例えば、次式
【0106】
【化30】
のパクリタキセル類似体を、適切なアシル化剤、好ましくは末端オレフィンを有するアシル化剤と結合させ、その末端オレフィンによって、トランスフェリン又はその他の担体分子/タンパク質と共に使用する、アルデヒドリンカーに変換可能なパクリタキセルアミド誘導体を形成することができる。
上記のアミノ基含有パクリタキセル類似体をヘキセン酸と結合させることによって形成される中間体の例を次式に示す。
【0107】
【化31】
このような中間体は、例えば7−パクリタキセル及び2−パクリタキセル誘導体について既に開示した方法によって、対応するジオール、
【0108】
【化32】
及びアルデヒド
【0109】
【化33】
に変換することができる。従って、次式のトランスフェリン複合体を企図する。
【0110】
【化34】
半球状タキサン骨格のC−13位周囲の凹状領域に側鎖が存在することに由来する障害に基づく、トランスフェリン複合体を形成する困難さに対処するためには、ヘキセン酸よりも鎖長がより長い結合用化合物を使用することが考えられる。例えば、ヘプテン酸を使用すると次式のトランスフェリン複合体が形成される。
【0111】
【化35】
ここでは、保護したタキサンを、先ず、次のように対応するアミン−塩酸塩に変換する。
【0112】
【化36】
上式で示した7−O,3’−N−ジ−(CBZ)−2’−O−BOMパクリタキセルなどの保護されたタキサンの形成は、当技術分野で周知であり、例えば米国特許第5,750,737号、5,973,170号、6,133,462号、6,066,749号、6,048,990号、6,136,999号、及び6,143,902号に開示されており、その教示を参照として本明細書に組込む。上記反応式に示したアミン−塩酸塩の形成は、米国特許出願第09/843,284号により完全に記載されており、その教示を参照として本明細書に組込む。
【0113】
例えば、反応例を挙げれば、5.05gの7−O,3’−ジ−(CBZ)−2’−O−BOMパクリタキセルを、磁気攪拌子を備えた0.5L丸底フラスコ内の90.0mLのTHFに溶解し、フラスコに6.01mLの3.62M塩酸(22.08mmol)及び8.10g(湿分50%)の10%Pd/Cを添加した。反応容器を窒素で3回、水素で2回流洗し、反応混合物を、水素を充填したバルーンから供給される水素雰囲気下、室温下で、約1時間激しく攪拌した。この操作により上記反応式に示したアミン塩酸塩が生成した。上記の工程で使用した塩酸に代わって、他の鉱酸並びに有機酸を使用できることを認識されたい。
【0114】
その他にもタクサンのアンモニウム塩を形成する方法が知られている。例えば、トリフルオロ酢酸を使用して対応するアンモニウムトリフルオロ酢酸(TFA)塩を形成する方法が、例えば米国特許第5,675,025号、5,684,175号、5,770,745号、5,939,566号、5,948,919号、6,048,990号、6,066,749号、6,072,060号、6,136,999号、6,143,902号、6,262,281号、及び6,307,088号に開示されており、その教示を参照として本明細書に組込む。
【0115】
アミン−塩酸塩を形成したら、次に、以下に示すように、その塩酸塩をヘプテン酸と反応させて、対応するヘプテン酸エステルを形成する。
【0116】
【化37】
この反応は、トリエチルアミン(TEA)を加えて、アミン塩を対応する遊離のアミンに解離させていることを理解すべき以外は、7−パクリタキセル誘導体又は2’−パクリタキセル誘導体に関して既に開示している反応とほぼ同様である。次に、以下に示すように、得られたヘプテン酸エステルを対応する3’−アミドヘキサナールパクリタキセルに変換した。
【0117】
【化38】
この反応もやはり前に他の誘導体に関して述べた反応と同様である。次に、得られたアルデヒドを、トランスフェリン−7−パクリタキセル複合体の形成について前に述べた方法と同様の方法を用いて、トランスフェリンと結合させ、トランスフェリン−3’−アミド−ヘキサナールパクリタキセル複合体を得た。トランスフェリンとトランスフェリン−3’−アミド−ヘキサナールパクリタキセル複合体の円偏光二色性スペクトルは類似しており、双方の全体的立体配座が類似していることを示す。
【0118】
前述したことから、アミン塩の代わりに対応する遊離のアミンそれ自体を使用できることを認識されたい。タキサンの遊離アミンの形成は、例えば米国特許第5,688,977号、5,770,745号、5,939,566号、6,048,990号、6,066,749号、6,072,060号、6,107,497号、6,262,281号、及び6,307,088号に開示されており、その教示を参照として本明細書に組込む。
従って、本発明は、上述の実施例のアミン塩酸塩を、次の一般式で表される化合物並びにその類似体及び誘導体で代替することを企図する。
【0119】
【化39】
(式中、R3はNH2又はNH2HAでよくHAは有機酸又は鉱酸である)
【0120】
(VII.一般的方法及び化合物)
これまでの考察から明らかなように、本発明は、タンパク質−薬剤複合体を形成するための一般的方法を提供するものである。先ず、ヒドロキシル基又はアミノ基を有する、式R1−NH2又はR1−OHで表される生物学的活性化合物、あるいはその類似体又は誘導体を準備するが、場合によっては、それらの化合物の別の位置を、当技術分野で周知の範囲で1種又は複数の保護基で保護することができる。また、本発明がアミノ基を有する生物学的活性分子の第二級アミン(すなわち、R4が当技術分野で周知の適当なラジカルである式R1R4NHで表される分子)も想定していることを認識されたい。
【0121】
生物学的活性化合物を、次式(W、X及びYは前記と同様である)
【0122】
【化40】
から選択される化合物と反応させることによって、次式の構造を有する化合物を形成する。
【0123】
【化41】
「式中、Zは、生物学的活性化合物が式R1−OHを有する場合には−O−、生物学的活性化合物が式R1−NH2を有する場合には−NH−であり、W及びYは前記と同様である。」
この化合物を酸化して次式のアルデヒドにする。
【0124】
【化42】
別法としては、中間体として次式の対応するジオールを形成し、それを開裂してアルデヒドとすることもできる。
【0125】
【化43】
アルデヒドを、接近可能なアミノ官能基を含有するタンパク質又は他の担体分子、例えば式中、mが整数、Pがタンパク質又は他の担体分子、(NH2)mがその接近可能なアミノ官能基である次の一般式を有する化合物などと結合させて、
【0126】
【化44】
次式の複合体を形成する。
【0127】
【化45】
「式中、nは分子複合体の複合数であり、単一の担体分子に結合している特定の薬剤の分子数を表し、反応条件や特定の薬剤をトランスフェリンタンパク質などの担体分子に結合させるために使用される基礎となる中間化合物によって異なる。
【0128】
本発明で使用を想定している担体分子には、トランスフェリンなどのタンパク質、Safavyによって明らかにされた受容体リガンドペプチド、又はその他のタンパク質、抗体、レクチン又は細胞表面に付着できるその他の物質が含まれる。
【0129】
本発明の一般的方法は、次の一般式で表されるオレフィン、ジオール、及びアルデヒドなど、各種中間化合物の形成を想定していることを認識されたい。
【0130】
【化46】
「式中、R1、Z、及びYは前記と同様であり、R2は−CH=CH(W)、−CH(OH)CH(OH)W、又は−C(O)Hであり、Wは前記と同様である。」
R1には、必要であれば、例えばパクリタキセルの場合であればTBDMS、TROC、BOM、ベンジル、TES、又はEEなどの1個又は複数の保護基を含めることができる。その場合必要であれば、本発明の方法に1個又は複数の保護基でR1を保護する段階及び脱保護する段階を含めることができる。例えば、パクリタキセルをアシル化剤と結合させる段階に先立って、パクリタキセルの2’位をTBDMS、TROC、BOM、ベンジル、TES、又はEEなどで保護し、その後、その化合物を対応するアルデヒドに変換する段階の後に2’位を脱保護することができる。
【0131】
(VIII.分析/解析)
上記の方法で形成されたトランスフェリン−薬剤複合生成物を、マススペクトル及びFPLCで解析した。200nmから800nmのUVスペクトルを測定した。トランスフェリン/トランスフェリン複合体の測定濃度は0.5mg/mlとした。測定サンプルはPBS緩衝液(pH=7.4)で調製した。円偏光二色性の測定は、光路長1mmの円筒状水晶セルを用い、240nmから190nmまでのスペクトルを測定した。トランスフェリン/トランスフェリン複合体の測定濃度は1mg/mlとした。測定サンプルは水又はPBS緩衝液(pH=7.4)で調製した。
【0132】
蛍光分光光度計を用いてトランスフェリン、ローダミン123、及びトランスフェリン−ローダミン123複合体の蛍光を測定した。トランスフェリン(0.25mg/ml)、ローダミン123(10ng/ml)及びトランスフェリン−ローダミン123複合体(0.25mg/ml)は、リン酸塩緩衝液(pH7.0)で溶解した。励起波長はトランスフェリン及びトランスフェリン−ローダミン123複合体では280nm、ローダミン123では500nmとし、300から700nmの範囲の発光スペクトルを記録した。
【0133】
Laemmliゲル電気泳動法(「Cleavage of Structural Proteins During the Assembly of the Head of Bacteriophage T4」、Nature、227巻、680−685頁(1970年))を用いて、トランスフェリン複合体の純度を評価した。12%のアクリルアミドからなる垂直スラブゲル(NuPAGE電気泳動装置、NOVEX(登録商標))及びE1900−XCELL(登録商標)Mini Cell装置(Novex、サンジエゴ、カリフォルニア州)を用いて、複合体カラム分画のドデシル硫酸ナトリウム(SDS)ポリアクリルアミドゲル電気泳動を行った。サンプルを調製し、100℃で3分間加熱後、ゲル上に載せた。トランスフェリン複合体(20μl)を、ゲル上に約0.03mg/mlのタンパク質濃度で添加した。電気泳動が終了した後、ゲルをクマシーブルー染色液で30分間染色し、次いで5〜12時間脱色した。トランスフェリン−7−パクリタキセル複合体については、2−メルカプトエタノールの存在下(1μl、還元剤)及び非存在下でサンプルを分析した。
【0134】
既知量のパクリタキセルを注入し、そのピーク面積をパクリタキセル濃度に対してプロットすることにより、HPLCによるパクリタキセルの検量線を作成した。サンプルは、C−4カラム(5μm、300Å、25cm×4.6mm内径、流速1ml/分)を用い、溶媒A(80%水、20%ACN、0.1%TFA)と溶媒B(80%ACN、20%水、0.1%TFA)とのグラジエントで溶離する分析用逆相HPLCによって分析した。HPLC分析に要する時間は全部で24分であった。分析用HPLCに用いたグラジエント法では、溶媒Bを0%から始めて、20分間で100%まで増やし、次いで溶媒B100%で2分間溶離し、最後に2分間で溶媒Bを0%に徐々に戻した。
【0135】
パクリタキセルの原液(1mg/ml)は、パクリタキセルを酢酸エチルに溶解して調製した。パクリタキセルの最終濃度は、25、50、75、100μg/mlとした。各濃度について、サンプルをHPLCに注入し、2回繰返して得られたピーク面積の平均を、パクリタキセルの濃度に対してプロットし、パクリタキセルの検量線を作成した。
【0136】
トランスフェリン−7−パクリタキセル複合体の結合比は、複合体を酸加水解し、続いてHPLCでパクリタキセルの量を測定した後に判断した。1mgのトランスフェリン−パクリタキセルを、0.4mlのPBS緩衝液に溶解し、酢酸を加えてpHを4に調整後、反応混合物を室温で10分間攪拌した。反応混合物に酢酸エチルを0.2ml添加して2分間かき混ぜ、パクリタキセルを単離した。次に濁った混合物を、4℃で10分間、1000gで遠沈し、パクリタキセルを含む澄明な上澄液20μlをHPLCに注入した。パクリタキセルの検量線によれば、結合比はトランスフェリン1個に対しパクリタキセルが3個であった。
【0137】
(IX.トランスフェリン複合体の安定性)
A.熱安定性
トランスフェリン及びトランスフェリン複合体を、PBS緩衝液(pH7.0、0.05モル)に溶解(1mg/ml)して、トランスフェリン及びトランスフェリン複合体の貯蔵液を調製した。封管中に0.1mlずつ等分し、37℃に保った。適当な時間間隔で3本の封管を取り出し、直ちにドライアイスで凍結してCD及びSDS−PAGE電気泳動による分析を行うまで−70℃で保存した。分析直前に該当するサンプルを急速解凍した。
【0138】
B.pH依存安定性
トランスフェリン及びトランスフェリン複合体を、水に溶解してそれぞれの貯蔵液(1mg/ml)を調製した。さらに、貯蔵液を様々なpHの緩衝液(0.05モル)で希釈して最終濃度を0.1mg/mlとした。このサンプルを、室温に2時間、又は37℃に2時間保ち、CD、蛍光及びUV分光法で分析した。
【0139】
(X.細胞毒性データ)
哺乳動物の培養細胞を用いて、トランスフェリン−3−コレステロール複合体、トランスフェリン−ローダミン123複合体及びトランスフェリン−7−パクリタキセル複合体の増殖抑制能を調べた。以下で考察するように、ローダミン123及びコレステロールのトランスフェリン複合体は、腫瘍細胞及び正常細胞のいずれに対しても大きな有害作用を示さなかった。このことから、物質をトランスフェリンに複合させること自体が細胞の増殖に大きな影響を及ぼしている訳ではないことがわかる。しかし、パクリタキセルの複合体は、癌に対して有効性を示す化合物であり、正常細胞に有害な影響を与えないで癌細胞を攻撃できる可能性のあることがわかった。従って、本発明は、できれば最適投与量下で正常細胞を傷害することなしに、癌治療で効果を示す化合物で癌細胞を特異的に攻撃するための有望な方法を提案するものである。
【0140】
細胞系としては、KB(ヒト口腔類表皮癌種、ATCC#CCL−17)、Lu−1(ヒト肺癌細胞系、イリノイ大学医学部、腫瘍外科部門より入手)、及びhTERT(テロメラーゼ−不朽化正常上皮細胞系、Clontech#C4000−1)を選択した。二つの治療プロトコルで、化合物の投与量を変えて評価した。プロトコルAでは、KB、Lu、及びhTERT細胞の各培養物を、培養日以降、3種の複合化合物の投与量を様々に変えて処理した。プロトコルBでは、KB、Lu、及びhTERT細胞の各培養物を集密するまで(9日間)増殖させ、次いで、試験の期間中、トランスフェリン−7−パクリタキセル複合体の様々な投与量で処理した。
【0141】
KBは、10%ウシ胎児血清(FBS)、100単位/mlペニシリンG、100μg/mlストレプトマイシン硫酸塩、0.25μg/mlアンフォテリシンB(Fungizone)(PSF)(GIBCO)及び1%可欠アミノ酸(NAA)(Sigma)を追加したDMEM(GIBCO)中で培養した。Luは、10%FBS、PSF、及び1%NAAを追加したMEME(GIBCO)中に保持した。hTERT−RPE1は、10%FBS+PSFを追加したDMEM/F−12(GIBCO)中に保持した。すべての細胞系を空気中5%CO2雰囲気下、37℃、湿度100%で培養した。
【0142】
全般的な手順は、Skehan他による論文「New colorimetric cytotoxicity assay for anticancer−drug screening」、J.Natl.Cancer Inst.B2、1107−1112頁、1990年、及びLikhitwitayawuid他による論文「Cytotoxic and antimalarial bisbenzylisoquinoline alkaloids from Stephania erecta」、J.Nat.Prod.、56巻30−38頁1993年、に記載されているものに従った。典型的には細胞を60%から70%集密するまで増殖させ、培地を交換し、1日後にその細胞を試験に供した。最初に試験サンプルを滅菌したPBSに溶解した。PBS溶媒を用いて順次希釈して、その10μlを3つのウェルに添加した。対照群にはPBS(10μl)を添加した。プレートを調製した後、トリプシンで処理して組織培養フラスコから細胞を取り出し、計数し,新鮮培地で希釈した。KB細胞(3×104細胞/ml)、Lu細胞(5×104細胞/ml)及びhTERT−RPE1細胞(4×104細胞/ml)(培地190μl)を96穴プレートに添加した。プレートを5%CO2存在下で37℃で培養し、20%冷トリクロロ酢酸100μlを添加して細胞を固定し、4℃に30分間保った。プレートを水道水で洗浄(3回)し、一夜乾燥した。1%酢酸に溶かした0.4%(w/v)スルホローダミンBを100μl添加して固定した細胞を30分間染色した。プレートを1%酢酸で洗浄(3回)し、乾燥させた。10mM非緩衝Tris塩基(pH10)を添加(200μl/ウェル)して結合している染料を可溶化した。プレートを5分間シェイカー上に保持し、ELISAプレートリーダーを用いて515nmでの吸収を測定した。いずれの場合も、96穴プレートの幾つかのウェルに同数の細胞を添加し、37℃で30分間培養して零日対照とした。次いで、トリクロロ酢酸で細胞を固定し、上記同様に処理した。
【0143】
始めに、プロトコルAに従って、トランスフェリン−3−コレステロール複合体、トランスフェリン−ローダミン123複合体及びトランスフェリン−7−パクリタキセル複合体を用いて一連の実験を行った。プロトコルAでは、プレート作製日に試験化合物を62.5〜250μg/mlの範囲の様々な濃度で添加し、3日毎に培地を補充し、試験化合物の濃度を同一に維持した。時間に対する吸光度のプロットで示されるように、トランスフェリン−3−コレステロール複合体及びトランスフェリン−ローダミン123複合体はいずれの細胞系に対しても活性を示さなかった(図5、6、8、9、11、12)。これに対してトランスフェリン−7−パクリタキセル複合体では、試験したその全ての濃度で細胞増殖の完全な抑制が観察された(図7、10、13)。
【0144】
そこで、さらに低濃度のトランスフェリン−7−パクリタキセル複合体を使用して再度試験した。図14に示すように、KB細胞をプロトコルAで試験してみると、5又は50μg/mlの濃度では完全な増殖抑制が観察されたが、0.5μg/mlの濃度では増殖に対する影響は認められなかった。それぞれ図15及び16に示すように、Lu及びhTERT細胞についても同様な応答が観察された。
【0145】
プロトコルBによるKB細胞についての試験でも類似の結果が観察された。プロトコルBでは、9日目まで培地を取り替えることなしに細胞を増殖させ、9日目に様々な濃度の試験化合物を添加し、次いで、3日毎に化合物及び培地を補充した。50又は5μg/mlの濃度では細胞数のかなりの減少が観察されたが、0.5μg/mlの濃度では細胞数は対照と同程度であった(図17)。
【0146】
Lu細胞は抵抗性が若干大であった。50μg/mlでは細胞数のかなりの減少が観察されたが、5μg/mlの濃度ではその減少の程度が小さくなり、0.5μg/mlの濃度では細胞数が減少しなかった(図18)。
【0147】
hTERT細胞に対しては効果が最も小さかった。対照培養物及び0.5又は5μg/mlのトランスフェリン−7−パクリタキセル複合体で処理した培養物で細胞増殖のわずかな増加が観察された。細胞を50μg/mlで処理した場合、この増殖は減少したが、それでも16日目で細胞数は9日目とほぼ同じであった(図19)。これらの結果は、トランスフェリン−7−パクリタキセル複合体が、主として癌細胞(KB及びLu)を攻撃し、正常細胞(hTERT)にはあまり影響を与えないことを示唆しており、本発明によるトランスフェリン−7−パクリタキセル複合体及び他のタンパク質−薬剤複合体によって、癌治療に対する有望な方法が得られることを示している。
【0148】
以上、本発明の例示的実施形態に則して本発明をある程度詳細に説明してきた。しかしながら、本発明は、先行技術を考慮して解釈される特許請求の範囲で定義されるものであって、本発明の例示的実施形態に、本明細書に含まれる発明の概念から逸脱することなしに修正又は変更を加えることができることを認識されたい。
【特許請求の範囲】
【請求項1】
次式の構造を有する分子複合体、
【化1】
[式中、
(a)nは、前記分子複合体の複合数であり、
(b)Pは、少なくともn個の接近可能なアミノ官能基を有する分子の脱アミノ部分であり、
(c)R1は、
(1)(i)式R1−OHで表される、ヒドロキシル基を有する生物学的活性分子並びにその類似体及び誘導体の脱ヒドロキシル部分、並びに
(2)(i)式R1−NH2で表される、アミノ基を有する生物学的活性分子並びにその類似体及び誘導体あるいはこれらの塩又は第二級アミンの脱アミノ部分、から選択され、
(d)Zは、R1が前記の脱ヒドロキシル部分である場合には−O−、R1が前記の脱アミノ部分である場合には−NH−であり、
(e)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個又は複数のアルキル、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項2】
Pがタンパク質である、請求項1に記載の分子複合体。
【請求項3】
前記タンパク質がトランスフェリンである、請求項2に記載の分子複合体。
【請求項4】
nが1から5の整数である、請求項1に記載の分子複合体。
【請求項5】
前記生物学的活性分子が癌治療に有効な薬剤である、請求項1に記載の分子複合体。
【請求項6】
前記薬剤が天然物癌治療薬である、請求項5に記載の分子複合体。
【請求項7】
前記薬剤が、タキサン、カンプトテシン、エポシロン、ククルビタシン、クアシノイド、アントラサイクリン、並びにこれらの類似体及び誘導体から選択される、請求項5に記載の分子複合体。
【請求項8】
R1部分が、7−デヒドロキシルパクリタキセル、10−デヒドロキシルパクリタキセル、2’−デヒドロキシルパクリタキセル、3’−デ−ベンズアミドパクリタキセル、3−デヒドロキシルコレステロール、及び20−デヒドロキシルカンプトテシン、並びにこれらの類似体及び誘導体から選択される、請求項1に記載の分子複合体。
【請求項9】
Yが−(CH2)r−(rは3から7の整数)である、請求項1に記載の分子複合体。
【請求項10】
R1が、7−デヒドロキシルパクリタキセル部分であり、Zが−O−であり、Yが−(CH2)3−である、請求項1に記載の分子複合体。
【請求項11】
Pがトランスフェリンであり、nが3である、請求項10に記載の分子複合体。
【請求項12】
R1が2’−デヒドロキシルパクリタキセル部分であり、Zが−O−であり、Yが−(CH2)4−である、請求項1に記載の分子複合体。
【請求項13】
Pがトランスフェリンであり、nが1及び2から選択される、請求項12に記載の分子複合体。
【請求項14】
分子複合体の形成に有用な、次式の構造を有する化合物。
【化2】
[式中、
(a)R1は、
(1)(i)式R1−OHで表される、ヒドロキシル基を有する生物学的活性分子並びにその類似体及び誘導体の脱ヒドロキシル部分、並びに
(2)(i)式R1−NH2で表される、アミノ基を有する生物学的活性分子並びにその類似体及び誘導体あるいはこれらの塩又は第二級アミンの脱アミノ部分、から選択され、
(c)Zは、R1が前記の脱ヒドロキシル部分である場合には−O−であり、R1が前記の脱アミノ部分である場合には−NH−であり、
(d)R2は、−CH=CH(W)、−CH(OH)CH(OH)W、及び−C(O)Hから選択され、
(e)Wは、
(1)水素、
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(f)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択される。]
【請求項15】
R2が、−CH=CH2、−CH(OH)CH2(OH)、及び−C(O)Hから選択される、請求項14に記載の化合物。
【請求項16】
前記生物学的活性分子が癌治療に有効な薬剤である、請求項14に記載の化合物。
【請求項17】
前記薬剤が天然物癌治療薬である、請求項16に記載の化合物。
【請求項18】
前記薬剤が、タキサン、カンプトテシン、エポシロン、ククルビタシン、クアシノイド、アントラサイクリン、並びにこれらの類似体及び誘導体から選択される、請求項16に記載の化合物。
【請求項19】
R1部分が、7−デヒドロキシルパクリタキセル、10−デヒドロキシルパクリタキセル、2’−デヒドロキシルパクリタキセル、3’−デ−ベンズアミドパクリタキセル、3−デヒドロキシルコレステロール、及び20−デヒドロキシルカンプトテシン、並びにこれらの類似体及び誘導体から選択される、請求項14に記載の化合物。
【請求項20】
Yが、−(CH2)r−(rは3から7の整数)である、請求項14に記載の化合物。
【請求項21】
R1が、7−デヒドロキシルパクリタキセル部分であり、Zが−O−であり、Yが−(CH2)3−である、請求項14に記載の化合物。
【請求項22】
R1が、2’−デヒドロキシルパクリタキセル部分であり、Zが−O−であり、Yが−(CH2)r−(rは4から7の整数)である、請求項14に記載の化合物。
【請求項23】
R1が、7−デヒドロキシル2’−保護パクリタキセル部分であり、R2が、−CH=CH2及び−CH(OH)CH2(OH)から選択される、請求項14に記載の化合物。
【請求項24】
R1が、7−デヒドロキシル−2’−ヒドロキシルパクリタキセル部分であり、R2が、−C(O)Hである、請求項14に記載の化合物。
【請求項25】
(A)
(1)式R1−OHで表される、ヒドロキシル基を有する生物学的活性分子、並びにその類似体及び誘導体、及び、
(2)式R1−NH2で表される、アミノ基を有する生物学的活性分子並びにその類似体及び誘導体、あるいはそれらの塩又は第二級アミン
から選択される第一化合物を、次式
【化3】
から選択される第二化合物と反応させて次式の第三化合物を生成する段階と、
【化4】
(B)前記第三化合物を、次式の第四化合物に変換する段階と、
【化5】
(C)前記第四化合物を、少なくともn個の接近可能なアミノ官能基を有する分子と複合させて次式の分子複合体を形成させる段階、
とを含む、分子複合体の製造方法。
【化6】
[上記各式で、
(a)Xはハロゲンであり、
(b)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(c)Wは、
(1)水素、
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(d)R1は、前記第一化合物の脱ヒドロキシ部分であり、Zは、前記第一化合物が前記のヒドロキシル基を有する生物学的活性分子並びにその類似体及び誘導体である場合には、−O−であり、
(e)R1は、前記第一化合物の脱アミノ部分であり、Zは、前記第一化合物が前記のアミノ基を有する生物学的活性分子並びにその類似体及び誘導体、あるいはそれらの塩又は第二級アミンである場合には、−NH−であり、
(f)nは、前記の分子複合体の複合数であり、及び
(g)Pは、少なくともn個の接近可能なアミノ官能基を有する前記分子である。]
【請求項26】
Wが水素である、請求項25に記載の方法。
【請求項27】
Pがタンパク質である、請求項25に記載の方法。
【請求項28】
前記タンパク質がトランスフェリンである、請求項27に記載の方法。
【請求項29】
nが1から5の整数である、請求項25に記載の方法。
【請求項30】
前記の生物学的活性分子が癌治療に有効な薬剤である、請求項25に記載の方法。
【請求項31】
前記薬剤が天然物癌治療薬である、請求項30に記載の方法。
【請求項32】
前記薬剤が、タキサン、カンプトテシン、エポシロン、ククルビタシン、クアシノイド、アントラサイクリン、並びにこれらの類似体及び誘導体から選択される、請求項30に記載の方法。
【請求項33】
R1部分が、7−デヒドロキシルパクリタキセル、10−デヒドロキシルパクリタキセル、2’−デヒドロキシルパクリタキセル、3’−デ−ベンズアミドパクリタキセル、3−デヒドロキシルコレステロール、及び20−デヒドロキシルカンプトテシン、並びにこれらの類似体及び誘導体から選択される、請求項25に記載の方法。
【請求項34】
Yが−(CH2)r−(rは3から7の整数)である、請求項25に記載の方法。
【請求項35】
前記第三化合物を前記第四化合物に変換する段階が、前記第三化合物を次式
【化7】
のジオールに酸化し、その後に前記ジオールを前記第四化合物に酸化することを含む、請求項25に記載の方法。
【請求項36】
少なくともn個の接近可能なアミノ官能基を有する前記分子がトランスフェリンタンパク質であり、前記第四化合物を前記分子と複合させる段階が、第四化合物、DMSO及びトランスフェリンPBS緩衝液を混合して分子複合体を含有する混合物を形成することを含む、請求範囲25に記載の方法。
【請求項37】
分子複合体を含有する前記混合物に反応停止剤を添加する段階を含む、請求項36に記載の方法。
【請求項38】
前記反応停止剤がエタノールアミンPBS溶液である、請求項37に記載の方法。
【請求項39】
R1が3−デヒドロキシルコレステロール部分であり、前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、コレステロール、塩化メチレン、5−ヘキセン酸、DCC及び4−PPを混合することを含む、請求項25に記載の方法。
【請求項40】
R1が、3−デヒドロキシルコレステロール部分であり、前記第三化合物を前記第四化合物に変換する段階が、窒素雰囲気下で、前記第三化合物、THF、t−BuOH、水、NMO、NaIO4、及びOsO4を混合することを含む、請求項25に記載の方法。
【請求項41】
R1が、20−デヒドロキシルカンプトテシン部分であり、前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、カンプトテシン、5−ヘキセン酸、DMF、DIPC及び4−PPを混合することを含む、請求項25に記載の方法。
【請求項42】
R1が、20−デヒドロキシルカンプトテシン部分であり、前記第三化合物を前記第四化合物に変換する段階が、窒素雰囲気下で、前記第三化合物、THF、アセトン、ACN、水、T−BuOH、NMO、NaIO4及びOsO4を混合することを含む、請求項25に記載の方法。
【請求項43】
(A)パクリタキセルの2’−ヒドロキシル位を保護して、次式の第一化合物を形成する段階と、
【化8】
(B)前記第一化合物を次式から選択される第二化合物と反応させて、
【化9】
次式の第三化合物を形成する段階と、
【化10】
(C)前記第三化合物を、次式の第四化合物に変換する段階と、
【化11】
(D)前記第四化合物の2’位を脱保護して、次式の第五化合物を形成する段階と、
【化12】
(E)前記第五化合物をトランスフェリンと複合させて、次式の構造を有する分子複合体を形成する段階と
【化13】
を含む、癌治療に使用するためのトランスフェリン−7−パクリタキセル複合体の製造方法。
[上記各式で、
(a)nは1から5の整数であり、
(b)P1はヒドロキシル保護基であり、
(c)Xはハロゲンであり、
(d)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直線は分岐状アルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項44】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、塩化メチレン、5−ヘキセン酸、DIPC及び4−PPを混合することを含む、請求項43に記載の方法。
【請求項45】
前記第三化合物を前記第四化合物に変換させる段階が、窒素雰囲気下で、前記第三化合物、ACN、THF、水、NMO、NaIO4、OsO4及びt−BuOHを混合することを含む、請求項43に記載の方法。
【請求項46】
前記第四化合物を脱保護する段階が、窒素雰囲気下で、前記第四化合物、TBAF及びTHFを混合することを含む、請求項43に記載の方法。
【請求項47】
前記第五化合物をトランスフェリンと複合させる段階が、前記第五化合物、DMSO及びトランスフェリンPBS緩衝液/DMSO溶液を混合することを含む、請求項43に記載の方法。
【請求項48】
(A)次式の第一化合物を、
【化14】
次式から選択される第二化合物と反応させて、
【化15】
次式の第三化合物を形成する段階と、
【化16】
(B)前記第三化合物を次式の第四化合物に変換する段階と、
【化17】
(C)前記第四化合物をトランスフェリンと複合させて、次式の構造を有する分子複合体を形成する段階と
【化18】
を含む、癌治療に使用するためのトランスフェリン−2’−パクリタキセル複合体の製造方法。
「上記各式で、
(a)nは1から5の整数であり
(b)Xはハロゲンであり
(c)Wは
(1)水素
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(d)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。」
【請求項49】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、塩化メチレン、5−ヘプテン酸、DIPC及び4−PPを混合することを含む、請求項48に記載の方法。
【請求項50】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、塩化メチレン、オレイン酸、DIPC及び4−PPを混合することを含む、請求項48に記載の方法。
【請求項51】
前記第三化合物を前記第四化合物に変換させる段階が、窒素雰囲気下で前記第三化合物、アセトン、水、NMO、NaIO4、OsO4及びt−BuOHを混合することを含む、請求項48に記載の方法。
【請求項52】
前記第四化合物をトランスフェリンと複合させる段階が、前記第四化合物、DMSO及びトランスフェリンPBS緩衝液/DMSO溶液を混合することを含む、請求項43に記載の方法。
【請求項53】
(A)次式の第一化合物を、
【化19】
次式から選択される第二化合物と反応させて、
【化20】
次式の第三化合物を形成する段階と、
【化21】
(B)前記第三化合物を次式の第四化合物に変換する段階と、
【化22】
(C)前記第四化合物をトランスフェリンと複合させて、次式の構造を有する分子複合体を形成する段階と
【化23】
を含む、癌治療に使用するためのトランスフェリン−3’−パクリタキセル複合体の製造方法。
[上記各式で、
(a)nは1から5の整数であり、
(b)R3は、
(1)NH2、及び
(2)NH2HA(ここでHAは(i)有機酸及び(ii)鉱酸から選択される)から選択され、
(c)Xはハロゲンであり、
(d)Wは、
(1)水素、
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(e)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項54】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、塩化メチレン、5−ヘプテン酸、DCC及び4−PPを混合することを含む、請求項53に記載の方法。
【請求項55】
前記第一化合物を前記第二化合物と反応させる段階が、最初に、前記第一化合物を、対応するアミンに遊離させることを含む、請求項53に記載の方法。
【請求項56】
前記第三化合物を前記第四化合物に変換させる段階が、窒素雰囲気下で、前記第三化合物、ACN、水、NMO、NaIO4、OsO4及びTHFを混合することを含む、請求項53に記載の方法。
【請求項57】
前記第四化合物をトランスフェリンと複合させる段階が、前記第四化合物、DMSO及びトランスフェリンPBS緩衝液/DMSO溶液を混合することを含む、請求項53に記載の方法。
【請求項58】
(A)次式の第一化合物を、
【化24】
次式から選択される第二化合物と反応させて、
【化25】
次式の第三化合物を形成する段階と、
【化26】
(B)前記第三化合物を次式の第四化合物に変換する段階と、
【化27】
(C)前記第四化合物をトランスフェリンと複合させて、次式の構造を有する分子複合体を形成する段階と
【化28】
を含む、癌治療に使用するためのトランスフェリン−20−カンプトテシン複合体の製造方法。
[上記各式で、
(a)nは1から5の整数であり、
(b)Xはハロゲンであり、
(c)Wは、
(1)水素、
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(d)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項59】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、5−ヘキセン酸、DMF、DIPC及び4−PPを混合することを含む、請求項58に記載の方法。
【請求項60】
前記第三化合物を前記第四化合物に変換させる段階が、窒素雰囲気下で、前記第三化合物、アセトン、ACN、THF、水、NMO、NaIO4、OsO4及びt−BuOHを混合することを含む、請求項58に記載の方法。
【請求項61】
前記第四化合物をトランスフェリンと複合させる段階が、前記第四化合物、DMSO及びトランスフェリンPBS緩衝液/DMSO溶液を混合することを含む、請求項58に記載の方法。
【請求項62】
トランスフェリン−ローダミン123複合体を形成するのに有用な、次式の構造を有する化合物。
【化29】
[上式で、Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項63】
Yが−(CH2)3−である、請求項62に記載の化合物。
【請求項64】
次式のアルデヒドを、
【化30】
トランスフェリン及び次式のローダミン123を含む溶液に添加して、
【化31】
次式のトランスフェリン−ローダミン123複合体を形成する
ことを含む、トランスフェリン−ローダミン123複合体の製造方法。
【化32】
[上記各式で
(a)nは1から5の整数であり、
(b)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項65】
トランスフェリン、Hepes緩衝塩、ローダミン123及びグルタルアルデヒドを混合して前記トランスフェリン−ローダミン123複合体を形成する、請求項64に記載の方法。
【請求項66】
前記溶液を反応停止剤で停止させることを含む、請求項65に記載の方法。
【請求項67】
前記反応停止剤がエタノールアミンHBS溶液である、請求項66に記載の方法。
【請求項68】
次式の構造を有する分子複合体の選択された量を患者に投与することを含む、患者の標的細胞に選択的に生物学的活性分子を集中させる方法。
【化33】
[式中、
(a)nは前記分子複合体の複合数であり、
(b)Pは少なくともn個の接近可能なアミノ官能基を有し前記選択される標的細胞を攻撃する分子の脱アミノ部分であり、
(c)R1は、
(1)(i)式R1−OHで表される、ヒドロキシル基を有する生物学的活性分子並びにその類似体及び誘導体の脱ヒドロキシル部分、並びに
(2)(i)式R1−NH2で表される、アミノ基を有する生物学的活性分子並びにその類似体及び誘導体、あるいはこれらの塩又は第二級アミンの脱アミノ部分から選択され、
(d)Zは、R1が前記の脱ヒドロキシル部分から選択される場合には−O−であり、R1が前記の脱アミノ部分から選択される場合には−NH−であり、
(e)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項69】
Pがタンパク質である、請求項68に記載の方法。
【請求項70】
前記タンパク質がトランスフェリンである、請求項69に記載の方法。
【請求項71】
nが1から5の整数である、請求項68に記載の方法。
【請求項72】
前記生物学的活性分子が癌治療に有効な薬剤である、請求項68に記載の方法。
【請求項73】
前記薬剤が天然物癌治療剤である、請求項72に記載の方法。
【請求項74】
前記薬剤がタキサン、カンプトテシン、エポシロン、ククルビタシン、クアシノイド、アントラサイクリン、並びにこれらの類似体及び誘導体から選択される、請求項72に記載の方法。
【請求項75】
R1部分が、7−デヒドロキシルパクリタキセル、10−デヒドロキシルパクリタキセル、2’−デヒドロキシルパクリタキセル、3’−デ−ベンズアミドパクリタキセル、3−デヒドロキシルコレステロール、及び20−デヒドロキシルカンプトテシン、並びにこれらの類似体及び誘導体から選択される、請求項68に記載の方法。
【請求項76】
Yが−(CH2)r−(rは3から7の整数)である、請求項68に記載の方法。
【請求項1】
次式の構造を有する分子複合体、
【化1】
[式中、
(a)nは、前記分子複合体の複合数であり、
(b)Pは、少なくともn個の接近可能なアミノ官能基を有する分子の脱アミノ部分であり、
(c)R1は、
(1)(i)式R1−OHで表される、ヒドロキシル基を有する生物学的活性分子並びにその類似体及び誘導体の脱ヒドロキシル部分、並びに
(2)(i)式R1−NH2で表される、アミノ基を有する生物学的活性分子並びにその類似体及び誘導体あるいはこれらの塩又は第二級アミンの脱アミノ部分、から選択され、
(d)Zは、R1が前記の脱ヒドロキシル部分である場合には−O−、R1が前記の脱アミノ部分である場合には−NH−であり、
(e)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個又は複数のアルキル、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項2】
Pがタンパク質である、請求項1に記載の分子複合体。
【請求項3】
前記タンパク質がトランスフェリンである、請求項2に記載の分子複合体。
【請求項4】
nが1から5の整数である、請求項1に記載の分子複合体。
【請求項5】
前記生物学的活性分子が癌治療に有効な薬剤である、請求項1に記載の分子複合体。
【請求項6】
前記薬剤が天然物癌治療薬である、請求項5に記載の分子複合体。
【請求項7】
前記薬剤が、タキサン、カンプトテシン、エポシロン、ククルビタシン、クアシノイド、アントラサイクリン、並びにこれらの類似体及び誘導体から選択される、請求項5に記載の分子複合体。
【請求項8】
R1部分が、7−デヒドロキシルパクリタキセル、10−デヒドロキシルパクリタキセル、2’−デヒドロキシルパクリタキセル、3’−デ−ベンズアミドパクリタキセル、3−デヒドロキシルコレステロール、及び20−デヒドロキシルカンプトテシン、並びにこれらの類似体及び誘導体から選択される、請求項1に記載の分子複合体。
【請求項9】
Yが−(CH2)r−(rは3から7の整数)である、請求項1に記載の分子複合体。
【請求項10】
R1が、7−デヒドロキシルパクリタキセル部分であり、Zが−O−であり、Yが−(CH2)3−である、請求項1に記載の分子複合体。
【請求項11】
Pがトランスフェリンであり、nが3である、請求項10に記載の分子複合体。
【請求項12】
R1が2’−デヒドロキシルパクリタキセル部分であり、Zが−O−であり、Yが−(CH2)4−である、請求項1に記載の分子複合体。
【請求項13】
Pがトランスフェリンであり、nが1及び2から選択される、請求項12に記載の分子複合体。
【請求項14】
分子複合体の形成に有用な、次式の構造を有する化合物。
【化2】
[式中、
(a)R1は、
(1)(i)式R1−OHで表される、ヒドロキシル基を有する生物学的活性分子並びにその類似体及び誘導体の脱ヒドロキシル部分、並びに
(2)(i)式R1−NH2で表される、アミノ基を有する生物学的活性分子並びにその類似体及び誘導体あるいはこれらの塩又は第二級アミンの脱アミノ部分、から選択され、
(c)Zは、R1が前記の脱ヒドロキシル部分である場合には−O−であり、R1が前記の脱アミノ部分である場合には−NH−であり、
(d)R2は、−CH=CH(W)、−CH(OH)CH(OH)W、及び−C(O)Hから選択され、
(e)Wは、
(1)水素、
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(f)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択される。]
【請求項15】
R2が、−CH=CH2、−CH(OH)CH2(OH)、及び−C(O)Hから選択される、請求項14に記載の化合物。
【請求項16】
前記生物学的活性分子が癌治療に有効な薬剤である、請求項14に記載の化合物。
【請求項17】
前記薬剤が天然物癌治療薬である、請求項16に記載の化合物。
【請求項18】
前記薬剤が、タキサン、カンプトテシン、エポシロン、ククルビタシン、クアシノイド、アントラサイクリン、並びにこれらの類似体及び誘導体から選択される、請求項16に記載の化合物。
【請求項19】
R1部分が、7−デヒドロキシルパクリタキセル、10−デヒドロキシルパクリタキセル、2’−デヒドロキシルパクリタキセル、3’−デ−ベンズアミドパクリタキセル、3−デヒドロキシルコレステロール、及び20−デヒドロキシルカンプトテシン、並びにこれらの類似体及び誘導体から選択される、請求項14に記載の化合物。
【請求項20】
Yが、−(CH2)r−(rは3から7の整数)である、請求項14に記載の化合物。
【請求項21】
R1が、7−デヒドロキシルパクリタキセル部分であり、Zが−O−であり、Yが−(CH2)3−である、請求項14に記載の化合物。
【請求項22】
R1が、2’−デヒドロキシルパクリタキセル部分であり、Zが−O−であり、Yが−(CH2)r−(rは4から7の整数)である、請求項14に記載の化合物。
【請求項23】
R1が、7−デヒドロキシル2’−保護パクリタキセル部分であり、R2が、−CH=CH2及び−CH(OH)CH2(OH)から選択される、請求項14に記載の化合物。
【請求項24】
R1が、7−デヒドロキシル−2’−ヒドロキシルパクリタキセル部分であり、R2が、−C(O)Hである、請求項14に記載の化合物。
【請求項25】
(A)
(1)式R1−OHで表される、ヒドロキシル基を有する生物学的活性分子、並びにその類似体及び誘導体、及び、
(2)式R1−NH2で表される、アミノ基を有する生物学的活性分子並びにその類似体及び誘導体、あるいはそれらの塩又は第二級アミン
から選択される第一化合物を、次式
【化3】
から選択される第二化合物と反応させて次式の第三化合物を生成する段階と、
【化4】
(B)前記第三化合物を、次式の第四化合物に変換する段階と、
【化5】
(C)前記第四化合物を、少なくともn個の接近可能なアミノ官能基を有する分子と複合させて次式の分子複合体を形成させる段階、
とを含む、分子複合体の製造方法。
【化6】
[上記各式で、
(a)Xはハロゲンであり、
(b)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(c)Wは、
(1)水素、
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(d)R1は、前記第一化合物の脱ヒドロキシ部分であり、Zは、前記第一化合物が前記のヒドロキシル基を有する生物学的活性分子並びにその類似体及び誘導体である場合には、−O−であり、
(e)R1は、前記第一化合物の脱アミノ部分であり、Zは、前記第一化合物が前記のアミノ基を有する生物学的活性分子並びにその類似体及び誘導体、あるいはそれらの塩又は第二級アミンである場合には、−NH−であり、
(f)nは、前記の分子複合体の複合数であり、及び
(g)Pは、少なくともn個の接近可能なアミノ官能基を有する前記分子である。]
【請求項26】
Wが水素である、請求項25に記載の方法。
【請求項27】
Pがタンパク質である、請求項25に記載の方法。
【請求項28】
前記タンパク質がトランスフェリンである、請求項27に記載の方法。
【請求項29】
nが1から5の整数である、請求項25に記載の方法。
【請求項30】
前記の生物学的活性分子が癌治療に有効な薬剤である、請求項25に記載の方法。
【請求項31】
前記薬剤が天然物癌治療薬である、請求項30に記載の方法。
【請求項32】
前記薬剤が、タキサン、カンプトテシン、エポシロン、ククルビタシン、クアシノイド、アントラサイクリン、並びにこれらの類似体及び誘導体から選択される、請求項30に記載の方法。
【請求項33】
R1部分が、7−デヒドロキシルパクリタキセル、10−デヒドロキシルパクリタキセル、2’−デヒドロキシルパクリタキセル、3’−デ−ベンズアミドパクリタキセル、3−デヒドロキシルコレステロール、及び20−デヒドロキシルカンプトテシン、並びにこれらの類似体及び誘導体から選択される、請求項25に記載の方法。
【請求項34】
Yが−(CH2)r−(rは3から7の整数)である、請求項25に記載の方法。
【請求項35】
前記第三化合物を前記第四化合物に変換する段階が、前記第三化合物を次式
【化7】
のジオールに酸化し、その後に前記ジオールを前記第四化合物に酸化することを含む、請求項25に記載の方法。
【請求項36】
少なくともn個の接近可能なアミノ官能基を有する前記分子がトランスフェリンタンパク質であり、前記第四化合物を前記分子と複合させる段階が、第四化合物、DMSO及びトランスフェリンPBS緩衝液を混合して分子複合体を含有する混合物を形成することを含む、請求範囲25に記載の方法。
【請求項37】
分子複合体を含有する前記混合物に反応停止剤を添加する段階を含む、請求項36に記載の方法。
【請求項38】
前記反応停止剤がエタノールアミンPBS溶液である、請求項37に記載の方法。
【請求項39】
R1が3−デヒドロキシルコレステロール部分であり、前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、コレステロール、塩化メチレン、5−ヘキセン酸、DCC及び4−PPを混合することを含む、請求項25に記載の方法。
【請求項40】
R1が、3−デヒドロキシルコレステロール部分であり、前記第三化合物を前記第四化合物に変換する段階が、窒素雰囲気下で、前記第三化合物、THF、t−BuOH、水、NMO、NaIO4、及びOsO4を混合することを含む、請求項25に記載の方法。
【請求項41】
R1が、20−デヒドロキシルカンプトテシン部分であり、前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、カンプトテシン、5−ヘキセン酸、DMF、DIPC及び4−PPを混合することを含む、請求項25に記載の方法。
【請求項42】
R1が、20−デヒドロキシルカンプトテシン部分であり、前記第三化合物を前記第四化合物に変換する段階が、窒素雰囲気下で、前記第三化合物、THF、アセトン、ACN、水、T−BuOH、NMO、NaIO4及びOsO4を混合することを含む、請求項25に記載の方法。
【請求項43】
(A)パクリタキセルの2’−ヒドロキシル位を保護して、次式の第一化合物を形成する段階と、
【化8】
(B)前記第一化合物を次式から選択される第二化合物と反応させて、
【化9】
次式の第三化合物を形成する段階と、
【化10】
(C)前記第三化合物を、次式の第四化合物に変換する段階と、
【化11】
(D)前記第四化合物の2’位を脱保護して、次式の第五化合物を形成する段階と、
【化12】
(E)前記第五化合物をトランスフェリンと複合させて、次式の構造を有する分子複合体を形成する段階と
【化13】
を含む、癌治療に使用するためのトランスフェリン−7−パクリタキセル複合体の製造方法。
[上記各式で、
(a)nは1から5の整数であり、
(b)P1はヒドロキシル保護基であり、
(c)Xはハロゲンであり、
(d)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直線は分岐状アルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項44】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、塩化メチレン、5−ヘキセン酸、DIPC及び4−PPを混合することを含む、請求項43に記載の方法。
【請求項45】
前記第三化合物を前記第四化合物に変換させる段階が、窒素雰囲気下で、前記第三化合物、ACN、THF、水、NMO、NaIO4、OsO4及びt−BuOHを混合することを含む、請求項43に記載の方法。
【請求項46】
前記第四化合物を脱保護する段階が、窒素雰囲気下で、前記第四化合物、TBAF及びTHFを混合することを含む、請求項43に記載の方法。
【請求項47】
前記第五化合物をトランスフェリンと複合させる段階が、前記第五化合物、DMSO及びトランスフェリンPBS緩衝液/DMSO溶液を混合することを含む、請求項43に記載の方法。
【請求項48】
(A)次式の第一化合物を、
【化14】
次式から選択される第二化合物と反応させて、
【化15】
次式の第三化合物を形成する段階と、
【化16】
(B)前記第三化合物を次式の第四化合物に変換する段階と、
【化17】
(C)前記第四化合物をトランスフェリンと複合させて、次式の構造を有する分子複合体を形成する段階と
【化18】
を含む、癌治療に使用するためのトランスフェリン−2’−パクリタキセル複合体の製造方法。
「上記各式で、
(a)nは1から5の整数であり
(b)Xはハロゲンであり
(c)Wは
(1)水素
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(d)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。」
【請求項49】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、塩化メチレン、5−ヘプテン酸、DIPC及び4−PPを混合することを含む、請求項48に記載の方法。
【請求項50】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、塩化メチレン、オレイン酸、DIPC及び4−PPを混合することを含む、請求項48に記載の方法。
【請求項51】
前記第三化合物を前記第四化合物に変換させる段階が、窒素雰囲気下で前記第三化合物、アセトン、水、NMO、NaIO4、OsO4及びt−BuOHを混合することを含む、請求項48に記載の方法。
【請求項52】
前記第四化合物をトランスフェリンと複合させる段階が、前記第四化合物、DMSO及びトランスフェリンPBS緩衝液/DMSO溶液を混合することを含む、請求項43に記載の方法。
【請求項53】
(A)次式の第一化合物を、
【化19】
次式から選択される第二化合物と反応させて、
【化20】
次式の第三化合物を形成する段階と、
【化21】
(B)前記第三化合物を次式の第四化合物に変換する段階と、
【化22】
(C)前記第四化合物をトランスフェリンと複合させて、次式の構造を有する分子複合体を形成する段階と
【化23】
を含む、癌治療に使用するためのトランスフェリン−3’−パクリタキセル複合体の製造方法。
[上記各式で、
(a)nは1から5の整数であり、
(b)R3は、
(1)NH2、及び
(2)NH2HA(ここでHAは(i)有機酸及び(ii)鉱酸から選択される)から選択され、
(c)Xはハロゲンであり、
(d)Wは、
(1)水素、
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(e)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項54】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、塩化メチレン、5−ヘプテン酸、DCC及び4−PPを混合することを含む、請求項53に記載の方法。
【請求項55】
前記第一化合物を前記第二化合物と反応させる段階が、最初に、前記第一化合物を、対応するアミンに遊離させることを含む、請求項53に記載の方法。
【請求項56】
前記第三化合物を前記第四化合物に変換させる段階が、窒素雰囲気下で、前記第三化合物、ACN、水、NMO、NaIO4、OsO4及びTHFを混合することを含む、請求項53に記載の方法。
【請求項57】
前記第四化合物をトランスフェリンと複合させる段階が、前記第四化合物、DMSO及びトランスフェリンPBS緩衝液/DMSO溶液を混合することを含む、請求項53に記載の方法。
【請求項58】
(A)次式の第一化合物を、
【化24】
次式から選択される第二化合物と反応させて、
【化25】
次式の第三化合物を形成する段階と、
【化26】
(B)前記第三化合物を次式の第四化合物に変換する段階と、
【化27】
(C)前記第四化合物をトランスフェリンと複合させて、次式の構造を有する分子複合体を形成する段階と
【化28】
を含む、癌治療に使用するためのトランスフェリン−20−カンプトテシン複合体の製造方法。
[上記各式で、
(a)nは1から5の整数であり、
(b)Xはハロゲンであり、
(c)Wは、
(1)水素、
(2)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(3)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(4)任意に、1個若しくは複数のアルキル基又は電子吸引若しくは電子供与の基で置換された芳香族基から選択され、
(d)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項59】
前記第一化合物を前記第二化合物と反応させる段階が、窒素雰囲気下で、前記第一化合物、5−ヘキセン酸、DMF、DIPC及び4−PPを混合することを含む、請求項58に記載の方法。
【請求項60】
前記第三化合物を前記第四化合物に変換させる段階が、窒素雰囲気下で、前記第三化合物、アセトン、ACN、THF、水、NMO、NaIO4、OsO4及びt−BuOHを混合することを含む、請求項58に記載の方法。
【請求項61】
前記第四化合物をトランスフェリンと複合させる段階が、前記第四化合物、DMSO及びトランスフェリンPBS緩衝液/DMSO溶液を混合することを含む、請求項58に記載の方法。
【請求項62】
トランスフェリン−ローダミン123複合体を形成するのに有用な、次式の構造を有する化合物。
【化29】
[上式で、Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項63】
Yが−(CH2)3−である、請求項62に記載の化合物。
【請求項64】
次式のアルデヒドを、
【化30】
トランスフェリン及び次式のローダミン123を含む溶液に添加して、
【化31】
次式のトランスフェリン−ローダミン123複合体を形成する
ことを含む、トランスフェリン−ローダミン123複合体の製造方法。
【化32】
[上記各式で
(a)nは1から5の整数であり、
(b)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項65】
トランスフェリン、Hepes緩衝塩、ローダミン123及びグルタルアルデヒドを混合して前記トランスフェリン−ローダミン123複合体を形成する、請求項64に記載の方法。
【請求項66】
前記溶液を反応停止剤で停止させることを含む、請求項65に記載の方法。
【請求項67】
前記反応停止剤がエタノールアミンHBS溶液である、請求項66に記載の方法。
【請求項68】
次式の構造を有する分子複合体の選択された量を患者に投与することを含む、患者の標的細胞に選択的に生物学的活性分子を集中させる方法。
【化33】
[式中、
(a)nは前記分子複合体の複合数であり、
(b)Pは少なくともn個の接近可能なアミノ官能基を有し前記選択される標的細胞を攻撃する分子の脱アミノ部分であり、
(c)R1は、
(1)(i)式R1−OHで表される、ヒドロキシル基を有する生物学的活性分子並びにその類似体及び誘導体の脱ヒドロキシル部分、並びに
(2)(i)式R1−NH2で表される、アミノ基を有する生物学的活性分子並びにその類似体及び誘導体、あるいはこれらの塩又は第二級アミンの脱アミノ部分から選択され、
(d)Zは、R1が前記の脱ヒドロキシル部分から選択される場合には−O−であり、R1が前記の脱アミノ部分から選択される場合には−NH−であり、
(e)Yは、
(1)任意に、1個又は複数のフェニル基で置換され、1から20個の炭素原子を有する直鎖状又は分岐状のアルキル基、
(2)任意に、1個若しくは複数のアルキル基又はフェニル基で置換されたシクロアルキル基、及び
(3)任意に、1個若しくは複数のアルキル基、電子吸引基又は電子供与基で置換された芳香族基から選択される。]
【請求項69】
Pがタンパク質である、請求項68に記載の方法。
【請求項70】
前記タンパク質がトランスフェリンである、請求項69に記載の方法。
【請求項71】
nが1から5の整数である、請求項68に記載の方法。
【請求項72】
前記生物学的活性分子が癌治療に有効な薬剤である、請求項68に記載の方法。
【請求項73】
前記薬剤が天然物癌治療剤である、請求項72に記載の方法。
【請求項74】
前記薬剤がタキサン、カンプトテシン、エポシロン、ククルビタシン、クアシノイド、アントラサイクリン、並びにこれらの類似体及び誘導体から選択される、請求項72に記載の方法。
【請求項75】
R1部分が、7−デヒドロキシルパクリタキセル、10−デヒドロキシルパクリタキセル、2’−デヒドロキシルパクリタキセル、3’−デ−ベンズアミドパクリタキセル、3−デヒドロキシルコレステロール、及び20−デヒドロキシルカンプトテシン、並びにこれらの類似体及び誘導体から選択される、請求項68に記載の方法。
【請求項76】
Yが−(CH2)r−(rは3から7の整数)である、請求項68に記載の方法。
【図1】

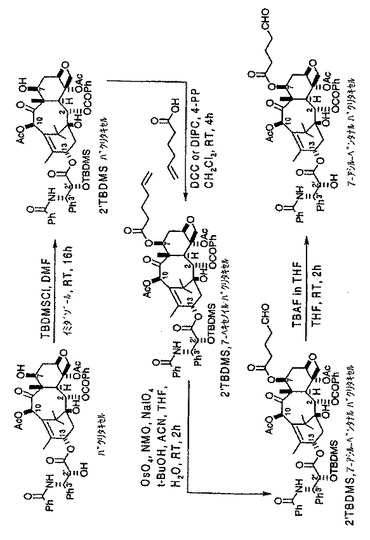
【図2】
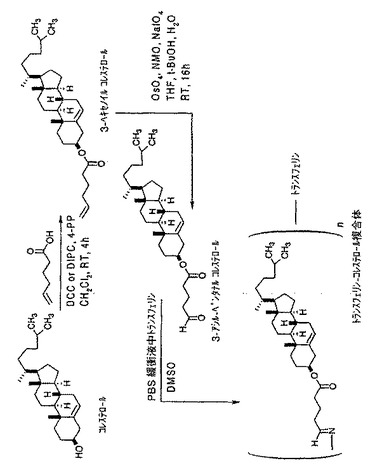
【図3】
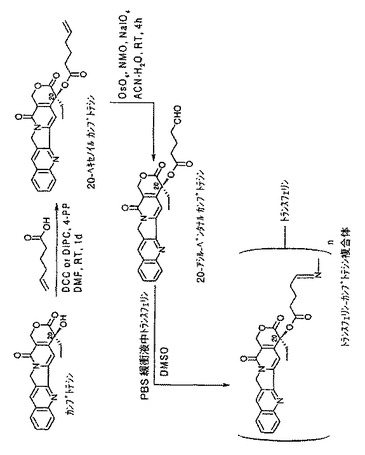
【図4(a)】
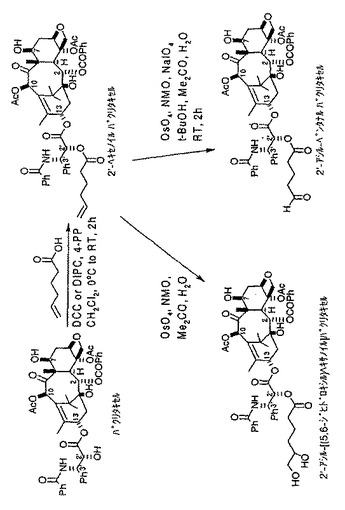
【図4(b)】
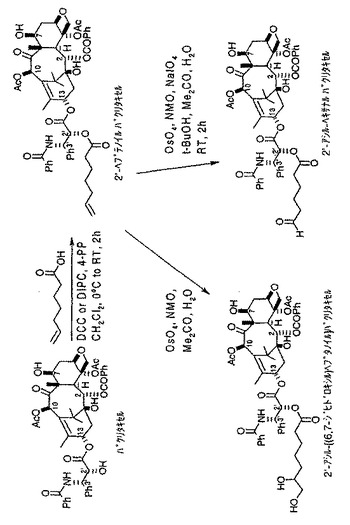
【図4(c)】
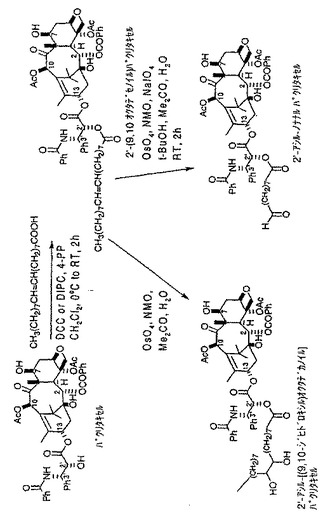
【図5】
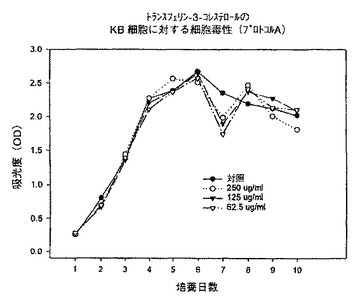
【図6】
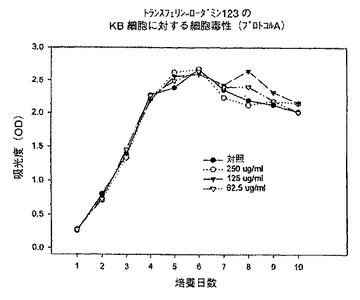
【図7】
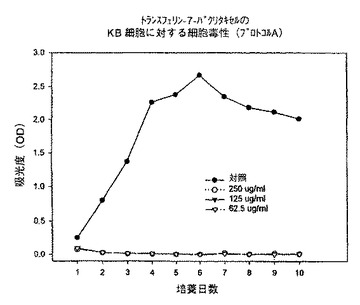
【図8】
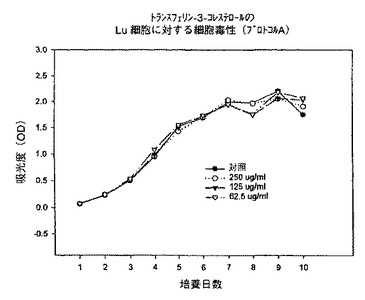
【図9】
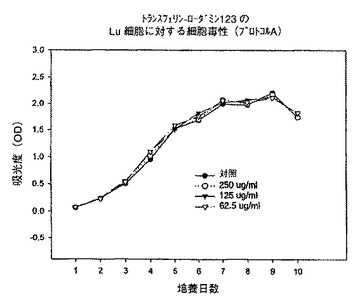
【図10】
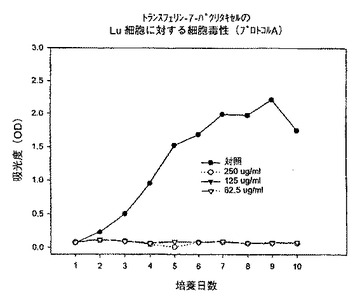
【図11】
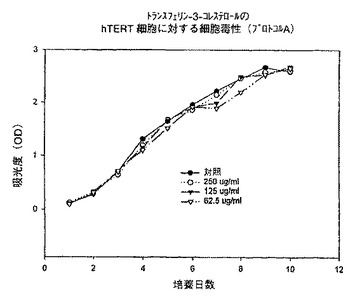
【図12】
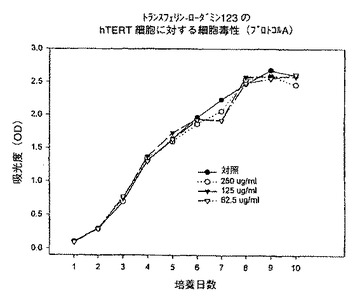
【図13】
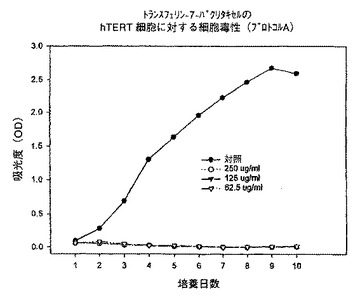
【図14】
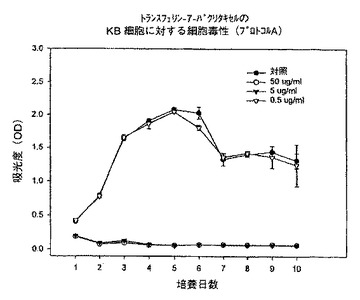
【図15】
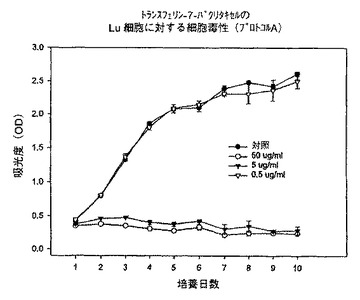
【図16】
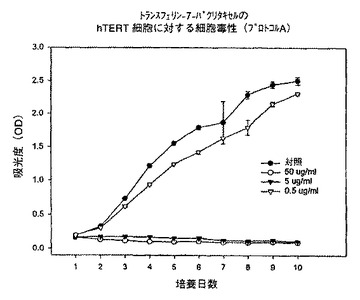
【図17】
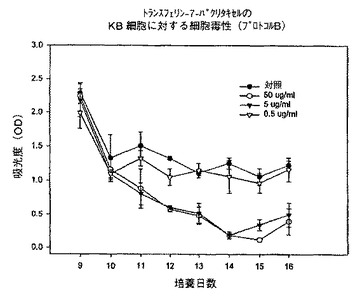
【図18】
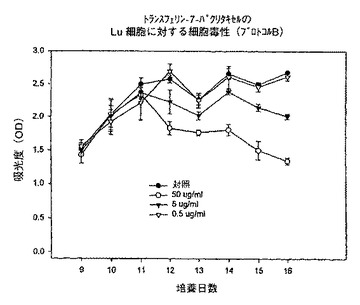
【図19】
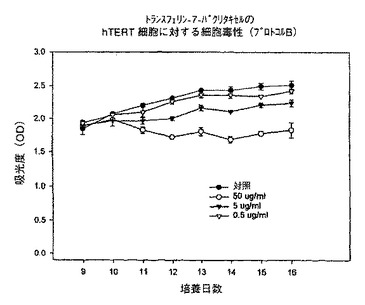
【図2】
【図3】
【図4(a)】
【図4(b)】
【図4(c)】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2010−254696(P2010−254696A)
【公開日】平成22年11月11日(2010.11.11)
【国際特許分類】
【出願番号】特願2010−109672(P2010−109672)
【出願日】平成22年4月19日(2010.4.19)
【分割の表示】特願2002−574963(P2002−574963)の分割
【原出願日】平成14年3月25日(2002.3.25)
【出願人】(503344861)ナプロ バイオセラピューティクス,インコーポレイテッド (1)
【Fターム(参考)】
【公開日】平成22年11月11日(2010.11.11)
【国際特許分類】
【出願日】平成22年4月19日(2010.4.19)
【分割の表示】特願2002−574963(P2002−574963)の分割
【原出願日】平成14年3月25日(2002.3.25)
【出願人】(503344861)ナプロ バイオセラピューティクス,インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]