
癌細胞を選択的に死滅させるRasGAP由来ペプチド
本発明は、選択的に細胞を死滅させる薬物の能力を増強する、RasGAPタンパク質のN2配列、その断片、もしくはその変異体であるペプチドに関する。さらに、本発明は、活性物質として、製薬上有効な量の前記ペプチドを含んでなる、医薬組成物に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、癌細胞において薬物が選択的に細胞を死滅させる能力を強めるペプチドに関する。さらに、本発明は、活性成分として、製薬上有効な量の前記ペプチドを少なくとも1つ含んでなる、医薬組成物に関する。
【背景技術】
【0002】
腫瘍は多様で不均質であるが、いずれも際限なく増殖する能力を共有する。抑制されたアポトーシス感受性と結びついた、脱制御された細胞増殖が、腫瘍の進展を生じる最低限の必要条件となる。
【0003】
アポトーシスは、細胞がプログラム細胞死に入るプロセスであり、発生時に生じる生命維持に必要な現象であって、恒常性の維持に欠かせない。不可逆的に細胞をアポトーシスに委ねると考えられている生化学的事象は、カスパーゼ(アスパラギン酸残基の後で切断するシステインプロテアーゼ)の活性化である。アポトーシスを受ける細胞は、特徴的な形態的変化および生化学的変化を示すが、これは膜のブレッビング、細胞が丸くなること、クロマチン凝縮、DNA切断、細胞表面でのアポトーシスマーカーの発現、および抗アポトーシスシグナル伝達経路の阻害を包含する。これらすべての事象は、特異的カスパーゼ阻害剤によって阻止することができる。したがって、アポトーシスの間に観察される特徴的な変化のすべてとはいえないまでもほとんどの原因となるのは、カスパーゼ基質の切断である。
【0004】
アポトーシスの実行段階は、細胞内のカスパーゼ基質が切断されたときに、誘発される。数十ものカスパーゼ基質が同定され、そのリストは確実に増え続けている(Earnshaw W. C. etal.,"Mammalian caspases: structure, activation, substrates, and functions during apoptosis"Annu. Rev.Biochem. 68,383, 1999)。ひとたび切断されると、カスパーゼ基質は、カスパーゼ活性の増幅、DNAの断片化、核の分解などといった、アポトーシスの際に観察される生化学的および形態学的事象を媒介する。
【0005】
さらに、マイトジェン活性化プロテインキナーゼ(MAPK)経路がアポトーシスを正または負に調節することが明らかになった(Jarpe M. B. et al., "Anti-apoptotic versus pro-apoptotic signal transduction : checkpoints and stop signs along the road to death" Oncogene, 17,1475, 1998; Widmann C. et al., "Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human" Physiol. Rev. 79,143,1999)。このことから、アポトーシス性カスパーゼが、MAPKを調節する、および/またはMAPK経路の成分である、シグナル伝達タンパク質の一部を標的とする理由が説明できると思われる(Widmann C. et al., "Caspase- dependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti- apoptotic signals" J. Biol. Chem. 273, 7141, 1998)。これらのタンパク質にはMEKK1, PAK2, Mst1およびRasGAPがある。
【0006】
最近、Yang and Widmann、(Yang J. -Y. and Widmann C., "Antiapoptotic signaling generated by caspase-induced cleavage of RasGAP" Mol. Cell. Biol. , 21,5346,2001 ; "A subset of caspase substrates functions as the Jekylland Hyde of apoptosis "Eur. Cytokine Netw., 13,387, 2002a ; "The RasGAP N-terminal fragment generated by caspase cleavage protects cells in aRas/PI3K/Akt-dependent manner that does not rely on NFkappa B"J. Biol. Chem., 277,14641,2002b)は、Ras およびRho GTP結合タンパク質の制御因子であるRasGAPが、カスパーゼによる切断の程度に応じて、抗アポトーシス性シグナルおよびアポトーシス促進性シグナルをいずれも誘導することができるため、異例のカスパーゼ基質であることを示した。かれらは、カスパーゼ活性が低レベルである場合、RasGAPは455位で切断され、N末端配列(配列N)およびC末端配列(配列C)を生成することを示した。
【0007】
配列Cは全長RasGAPではないが、核濃縮の出現、カスパーゼ3の活性化およびPARPの切断を引き起こす能力から評価されるように、Hela細胞において強力なアポトーシス反応を引き起こした。
【0008】
同じ研究において、著者らはまた、配列Nが細胞死を促進するのではなく、むしろカスパーゼ活性化の下流でアポトーシスの全般的なブロッカーとなるように見えることを示した。カスパーゼ活性が高レベルの場合、配列Nがアポトーシスを妨げる能力は、配列Nが157位で切断されると抑制される。この後者の切断事象は、配列Nとは対照的に細胞を感作することが示されているN1およびN2の2つの配列を生成するが、これらは、癌治療の化学療法で使用される薬物、シスプラチンによって誘導されるアポトーシスに向けて高いカスパーゼ活性を生じることができる。
【0009】
しかしながら、Leblancら(Leblanc V. et al., "Ras-GTPase activating protein inhibition specifically induces apoptosis of tumour cells" Oncogene, 18, 4884,1999)が明らかにしたように、RasGAPのN2配列のSH3ドメインに対するモノクローナル抗体を、このタンパク質を特異的に阻害するために注入すると、癌細胞においてアポトーシスを誘導する。特許出願WO99/65947(Psrlerら)から、RasGAP SH3ドメイン結合タンパク質、G3BPに対するモノクローナル抗体が、G3BPが特異的に過剰発現している癌細胞において、アポトーシスを誘導することも知られている。
【0010】
これらの結果は、RasGAP SH3ドメインを介して増殖を制御するRasGAP経路が、ある種の癌細胞の生存に必須であること示すと思われる。これらの発見は、YangおよびWidmannにより得られた結果とは対照的であると思われるので、RasGAP SH3ドメインは細胞におけるアポトーシスの誘導と制御において極めて相反する二面的機能を有するという結論をもたらす。
【0011】
化学療法は、単独で、または他の治療法(たとえば、放射線療法)と併用して、現在、最も一般的で有効な癌の治療手段である。癌を治療する化学療法で使用される薬物の有効性は、癌細胞を死滅させる能力に依存する。しかしながら、このような薬物の使用には限界があり、その限界は薬物が癌細胞ではない正常細胞にも悪影響を及ぼす可能性があるという事実によってもたらされるが、それは、こうした薬物が癌細胞のみならず、癌細胞でない正常細胞においても、特に迅速に分裂する細胞において、カスパーゼへの強い刺激を引き起こすためである。
【0012】
したがって、臨床医の課題は、腫瘍を取り除くには十分に高用量であるが、患者に脱毛、悪心嘔吐、心臓毒性および二次癌といった深刻な副作用を引き起こすほどには高くない薬物投与量を選択することである。
【0013】
薬物の癌細胞に対する選択性を向上させることは、明らかに化学療法処置の有効性を高め、それによって薬物投与量を低下させることができる。このことはまた、上記の深刻な副作用を可能な限り減少させる結果につながることになる。
【0014】
したがって、本発明の目的は、薬物と併用して、癌の治療もしくは予防を目的とする、上記の欠点のない改善されたアプローチを提供することである。
【0015】
発明の概要
上記目的は、選択的に癌細胞を死滅させる薬物の能力を高める、基本的にRasGAPタンパク質のN2配列、その断片、またはその変異体からなるペプチドを提供することによって達成された。
【0016】
さらに、本発明は、ペプチドをコードする精製単離された核酸配列、少なくとも1コピーの精製単離された核酸配列を含んでなる発現ベクター、ならびにペプチド、単離精製された核酸配列および/または発現ベクターを含有する、真核もしくは原核宿主細胞を提供する。
【0017】
本発明はさらに、活性成分として、少なくとも1つの本発明のペプチドを、製薬上有効な量含んでなる医薬組成物を提供する。
【0018】
発明の詳細な説明
本明細書で使用される場合、「ペプチド」、「タンパク質」、「ポリペプチド」、「ポリペプチド性」、および「ペプチド性」という用語は相互に交換可能なものとして使用され、隣接するアミノ酸残基のαアミノ基とカルボキシル基の間のペプチド結合によって他のアミノ酸と結合した一連のアミノ酸残基を示す。
【0019】
Ras およびRho GTP結合タンパク質の制御因子であるRasGAPは、カスパーゼによる切断の程度に応じて、抗アポトーシス性シグナルおよびアポトーシス促進性シグナルをいずれも誘導することができるため、異例のカスパーゼ基質である。カスパーゼ活性が低レベルである場合、RasGAPは455位で切断され、N末端断片(断片N、約56kD)およびC末端断片(断片C、約64kD)を生成する。断片Nはカスパーゼ活性化の下流でアポトーシスの一般的なブロッカーとなるように思われる(Yang J. -Y. and Widmann C., Mol. Cell. Biol., 21,5346, 2001およびJ. Biol. Chem., 277,14641, 2002b)。カスパーゼ活性が高レベルの場合、断片Nは157位でさらに切断されて、2つの断片、N1(アミノ酸1〜157)およびN2(アミノ酸158-455)を生成する。
【0020】
「癌細胞」は、動物においてin vivoに生じる細胞であって、望ましくない無秩序な細胞増殖、または組織の異常な持続性もしくは異常な侵入をなしうる細胞を意味する。in vitroで、この用語はまた、永久に不死化された樹立細胞培養系である細胞株を指し、これは適当な新培地および空間を与えると無期限に、しかも無秩序に増殖する。
【0021】
「薬物」という用語は、哺乳動物細胞、好ましくはヒト細胞を死滅させることができる薬物を指す。さまざまな起源の、さまざまな作用機序をもつ、いくつかに分類される薬物がある。
【0022】
本発明の薬物は、宿主の生物学的プロセスに由来する、またはそのプロセスを有益に調節する作用物質に関わる。インターフェロン、腫瘍増殖因子、腫瘍壊死因子、GM-CSFおよびG-CSFといった増殖因子、ならびにインターロイキン2,インターロイキン6、インターロイキン7およびインターロイキン12といったインターロイキン類が、癌治療に現在使用されている、上記のような生物学的薬物の例である。
【0023】
本発明の薬物は、さらに、DNAを損傷し、および/または、細胞が増殖できないようにする、遺伝毒性物質のような作用物質にも関係すると考えられる。遺伝毒性物質は、アルキル化剤、代謝拮抗物質、DNA切断物質、DNA結合物質、トポイソメラーゼ毒および紡錘体毒を含んでなる一群から選択することができる。
【0024】
アルキル化剤の例としては、ロムスチン、カルムスチン、ストレプトゾシン、メクロレタミン、メルファラン、ウラシルナイトロジェンマスタード、クロラムブシル、シクロスファミド、イホスファミド、シスプラチン、カルボプラチン、マイトマイシン、チオテパ、ダカルバジン、プロカルバジン、ヘキサメチルメラミン、トリエチレンメラミン、ブスルファン、ピポブロマン、ミトタンおよび他のプラチナ誘導体がある。
【0025】
DNA切断物質はたとえばブレオマイシンである。
【0026】
トポイソメラーゼ毒は、トポテカン、イリノテカン、カンプトテシン・ナトリウム塩、ダウノルビシン、ドキソルビシン、イダルビシン、ミトキサントロン、テニポシド、アドリアマイシンおよびエトポシドを含んでなる一群から選択することができる。
【0027】
DNA結合物質の例はダクチノマイシンおよびミトラマイシンであるが、紡錘体毒は、ビンブラスチン、ビンクリスチン、ナベルビン、パクリタキセルおよびドセタキセルを含んでなる一群から選択することができる。
【0028】
薬物として代謝拮抗剤も使用することができるが、これは、下記の化合物の中から選択することができる:メトトレキサート、トリメトレキサート、ペントスタチン、シタラビン、ara-CMP、リン酸フルダラビン、ヒドロキシ尿素、フルオロウラシル、フロクスウリジン、クロロデオキシアデノシン、ゲムシタビン、チオグアニンおよび6-メルカプトプリン。
【0029】
好ましくは遺伝毒性物質、より好ましくはシスプラチン、ミトキサントロン、およびアドリアマイシンが、本発明において薬物として使用される。
【0030】
これらの薬物は、単独で、または相互に併用して使用することができる。2つ以上の薬物を使用する場合には、薬物の有用な組み合わせを決定することは、十分に当業者の能力の範囲内であって、たとえば死滅させるべき癌細胞によって決まることになるだろう。
【0031】
「増強する」という用語は、細胞を死滅させることができる薬物の効果を増大させるペプチドの能力を指す。この能力は、in vitroで、たとえば、核濃縮(アポトーシス細胞のマーカー)を示す細胞の数を記録することによって、ペプチドを含有し少なくとも1つの薬物で処理された細胞のアポトーシスの割合を測定することにより、評価することができる。典型的には、その結果を、前記ペプチドを含有しない薬物処理細胞から得られた結果と比較する。所定の濃度での細胞のアポトーシスを2倍以上増加させるペプチド、または一定のアポトーシス反応を誘導する薬物の用量を1/2以下に減少させるペプチドは、薬物が細胞を死滅させる能力を増強すると見なすことができるであろう。
【0032】
本明細書で使用される場合、「選択的に」という用語は、本発明のペプチドが、所定の濃度で細胞を死滅させる薬物の能力を、癌細胞においては特異的に増強するが、非癌細胞では驚くべきことに増強しないことを意味する。
【0033】
ペプチドが薬物の細胞死滅能力を癌細胞において特異的に増強するin vitroでの薬物の濃度範囲は、通常、使用される薬物によって決まる。たとえば、遺伝毒性物質を使用する場合、通常、in vitroの薬物濃度は、0.1から100μMであり、好ましくは0.15から30μMである。
【0034】
PasGAPタンパク質のN2配列は、好ましくはヒト由来であって、図2Aに示すように2つのSH2ドメインおよび1つのSH3ドメインを含む297アミノ酸からなる36kDタンパク質を指す。
【0035】
一般に、Srcホモロジー2(SH2)ドメインは、リン酸化チロシンの認識に関わるのに対して、Srcホモロジー3(SH3)ドメインは、細胞骨格の組織化に関係するシグナル伝達に関わるタンパク質をしばしば示唆する。
【0036】
「断片」は、RasGAPタンパク質のN2配列よりも長さとして少ないアミノ酸を含有する配列を指す。この配列は、その元になった天然配列と同じ性質を示す限り使用することができる。こうした配列は、RasGAPタンパク質のそれぞれのN2配列よりも長さが90%未満、好ましくは60%未満、特に30%未満のアミノ酸を含有することが好ましい。
【0037】
本発明はまた、RasGAPタンパク質のN2配列の変異体も包含する。「変異体」という用語は、天然配列のペプチドとはある程度異なるアミノ酸配列を有するペプチドを指すが、このアミノ酸配列は、保存的アミノ酸置換によって天然配列から変化したアミノ酸配列であって、保存的置換によって1つまたは複数のアミノ酸が同じ性質および同じ高次構造上の役割を有する別のアミノ酸によって置換される。アミノ酸配列変異体は、天然アミノ酸配列のアミノ酸配列内のある位置に、置換、欠失および/または挿入を有する。保存的アミノ酸置換は、本明細書では、下記の5群のうちの1群の範囲内での交換と定義される:
I.小、脂肪族、非極性もしくはわずかに極性の残基:Ala, Ser, Thr, Pro, Gly
II.極性、正電荷を有する残基:His, Arg, Lys
III.極性、負電荷を有する残基:およびそのアミド:Asp, Asn, Glu, Gln
IV.大、芳香族残基:Phe, Tyr, Trp
V.大、脂肪族、非極性残基:Met. Leu. Ile, Val, Cys。
【0038】
N2配列、ならびにその断片および変異体は、当技術分野で知られている様々な方法および技術、たとえば、化学合成、もしくはManiatis et al. 1982, Molecular Cloning, A laboratory Manual, Cold Spring Harbor Laboratoryに記載の遺伝子組換え技術によって調製することができる。
【0039】
好ましくは、RasGAPタンパク質のN2配列の断片は、N2配列のSH3ドメイン、その一部もしくはその変異体のアミノ酸配列を含んでなる。
【0040】
出願人らは、驚くべきことに、RasGAPタンパク質のN2配列の、さらに短い配列の特徴を明らかにしたが、その配列は、短くても、選択的に癌細胞を死滅させる薬物の能力を増強し、しかもより容易に合成できるという利点も有するものである。このような部分配列を分離できるかどうかを評価するために、出願人らは断片Nの一連のトランケート型を図2Aに示すように生成し、癌細胞株において薬物が誘導するアポトーシスを増強するそれらの能力を測定した(図2B)。N2配列からのこれらの部分配列をベクターにクローニングして癌細胞株(HeLa)にトランスフェクトした。
【0041】
本出願人らは、空の構築物またはN2のSH2ドメインのみをコードする構築物でトランスフェクトされたHeLa細胞が、シスプラチン誘導細胞死を増強しないことを明らかにした。これに対して、SH3ドメインを含有する構築物を発現する細胞は、図2Bに示すように、シスプラチン誘導アポトーシスのさらなる増強を示した。
【0042】
そこで本出願人らは、最小の増強ペプチドを同定する目的で、SH3ドメインにおいてトランケーションを次々に進めた。これらの構築物のすべて、またはN2配列の部分配列(図2A)は、10アミノ酸の長さのペプチドをコードする最も短い配列(317-326)を含めて、シスプラチンのHeLa細胞死滅能力を増強した(図2B)。断片N2の細胞死増強特性は完全なSH3ドメインを必要とせず、短いペプチド性配列のようなSH3ドメインの部分配列によって媒介されることを、これらの結果は示している。
【0043】
SH3ドメインの一部分、またはその変異体は、SH3ドメインのアミノ酸配列のうち、好ましくは70個以下の、より好ましくは30個以下の、もっとも好ましくは10個以下のアミノ酸を含有する。
【0044】
詳細には、本発明に含まれるのは、表1のDNA配列によってコードされるアミノ酸配列である、SH3ドメインの部分配列である。
【表1】
【0045】
N2配列のSH3ドメインの部分配列が配列番号4(RasGAP317-326)である場合、ヒトにおいて前記配列番号4によりコードされる、結果として得られるアミノ酸配列はWMWVTNLRTDである。異なる種との比較から、異なるアミノ酸が存在することが明らかになったが、それは表2に示すように種の間で保存されている。
【表2】
【0046】
種の間で保存されているアミノ酸配列は、太字下線付きの活字の残基として表わされるのに対して、Xは、N2のSH3ドメインのこの10アミノ酸部分の発明的特性を損なうことなく、保存的もしくは非保存的アミノ酸置換によって変えることができるアミノ酸残基に対応する。
【0047】
N2のヒトSH3ドメインのこの10アミノ酸部分からなる、これらのペプチド性変異体、および特にアラインメント配列WXWVTXXRTXもまた、本発明の範囲に含まれるが、それらは、ある程度、天然型配列のペプチドとは異なるアミノ酸配列を有するペプチドを表し、それは、保存的もしくは非保存的アミノ酸置換によって天然型配列WMWVTNLRTDから変化したアミノ酸配列であって、それにより1つもしくは複数のアミノ酸残基が同じ特性および高次構造上の役割を有する別の残基によって置換されている。
【0048】
通常、基本的にRasGAPタンパク質のN2配列、その断片、またはその変異体からなるペプチドは、本発明で明らかにされるように、細胞内のペプチドの蓄積を増加させる薬剤と結合している。
【0049】
こうした薬剤は、たとえば、治療薬と結合したトランスフェリンの、膜トランスフェリン受容体を介したエンドサイトーシスのような、受容体を介したエンドサイトーシスを引き起こす化合物(Qian Z. M. et al.,"Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway "Pharmacological Reviews, 54,561, 2002)とすることができ、または、たとえば、プロテインキナーゼC(Ioannides C. G. et al., "Inhibition of IL-2 receptor induction andIL-2 production in the human leukemic cell line Jurkat by a novel peptide inhibitor of proteinkinase C" Cell Immunol., 131,242, 1990)およびプロテインチロシンホスファターゼ(Kole H. K. et al., "A peptide-based protein-tyrosine phosphatase inhibitor specifically enhances insulin receptor function in intact cells" J. Biol. Chem. 271,14302, 1996)のペプチドインヒビターの細胞内デリバリーにすでに使用されている、デカン酸、ミリスチン酸およびステアリン酸といった脂肪酸の一群から、もしくはペプチドから選択することができる細胞膜透過性キャリアとすることができる。
【0050】
細胞膜透過性キャリアがペプチドである場合、アルギニンリッチのペプチドであることが好ましい。Futakiら(Futaki S. et al., "Arginine- rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery" J. Biol. Chem., 276, 5836, 2001)において、細胞膜透過性キャリアペプチド中のアルギニン残基数がインターナリゼーションの方式に相当の影響を及ぼすこと、ならびにインターナリゼーションに最適なアルギニン残基数が存在すると思われ、それは好ましくはアルギニン7残基以上であることが、最近明らかになった。
【0051】
本発明のペプチドは、通常、スペーサーによって細胞膜透過性キャリアと結合する。この場合、細胞膜透過性キャリアはペプチドであることが好ましい。
【0052】
通常、アルギニンリッチペプチドは、HIV-TAT48-57ペプチド、FHV-coat35-49ペプチド、HTLV-II Rex4-16ペプチドおよびBMV gag7-25ペプチドを含んでなる一群から選択される。好ましくは、アルギニンリッチペプチドはHIV-TAT48-57ペプチドとする。
【0053】
HIV-TAT48-57ペプチドが、たとえばRasGAP317-326のよなRasGAP配列と結合する場合、2つのグリシン残基をTATとRasGAP配列の間にスペーサーとして挿入して柔軟性を与える。
【0054】
天然ペプチド(L型)に固有の問題は天然プロテアーゼによる分解であるので、ペプチドのD型および/または「レトロ‐インベルソ(retro-inverso)型異性体」を含めるように本発明のペプチドを調製してもよい。
【0055】
この場合、本発明のペプチドの短い断片および変異体のレトロ‐インベルソ型異性体を調製する。
【0056】
したがって、ペプチドを自然のタンパク質加水分解から保護することは、特異的なヘテロ二価もしくはヘテロ多価化合物の有効性を高めるはずである。天然プロテアーゼによる分解から保護されるので、非レトロ‐インベルソ含有アナログより高い生物学的活性がレトロ‐インベルソ含有ペプチドについて予測される。さらに、これらのペプチドは高い安定性ならびに低い免疫原性を示すことが明らかになった(Sela M. and Zisman E. ,"Different roles of D-amino acids in immune phenomena "FASEB J. 11,449, 1997)。
【0057】
レトロ‐インベルソペプチドは、たとえばSela and Zisman, (1997)に記載の既知の配列を有するペプチドについて調製される。
【0058】
「レトロ‐インベルソ型異性体」は、配列の方向を逆転し、各アミノ酸のキラリティーも反転させた、直鎖状ペプチドの異性体を意味する;したがって末端鎖相補性は存在し得ない。
【0059】
また本発明は、ペプチドの修飾(通常は一次配列を変更しない)も包含するが、そうした修飾には、in vivo もしくはin vitroでのペプチドの化学的な誘導体化、たとえば、アシル化もしくはカルボキシル化がある。グリコシル化修飾も含まれるが、たとえばこうした修飾は、合成およびプロセシングのときに、または追加のプロセッシングステップにおいて、グリコシル化パターンを変更することによってなされ、それは、たとえば、ペプチドをグリコシル化に影響を与える酵素(たとえば、哺乳類のグリコシル化酵素もしくは脱グリコシル化酵素)と接触させることによる。リン酸化されたアミノ酸残基、たとえばホスホチロシン、ホスホセリン、もしくはホスホスレオニンを有する配列も含まれる。
【0060】
本発明はまた、1つもしくは複数のペプチド結合が、ペプチダーゼによる切断を受けにくい、代替型の共有結合で置換されたアナログ(ペプチドミメティック)を包含する。被験体に注入した後のペプチドのタンパク質分解が問題となる場合、特に感受性のペプチド結合を切断されないペプチドミメティックで置換することによって、その結果得られたペプチドはいっそう安定となり、したがって活性物質としてより有用なものとなる。このようなミメティック、およびそれをペプチドに取り入れる方法は、当技術分野においてよく知られている。
【0061】
アミノ末端保護基も有用であって、たとえば、t-ブチルオキシカルボニル, アセチル, エチル, スクシニル,メトキシスクシニル, スベリル, アジピル, アゼライル(azelayl), ダンシル, ベンジルオキシカルボニル, フルオレニルメトキシカルボニル,メトキシアゼライル(methoxyazelayl), メトキシアジピル, メトキシスベリルおよび2,4-ジニトロフェニルがある。ペプチドの、荷電したアミノおよびカルボキシ末端を保護することには、疎水性の細胞膜を通って細胞内に入るペプチドの通過を向上させるという追加の利点もある。
【0062】
本発明に基づいて、組換え技法を用いて、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドを調製する場合には、そのポリペプチドをコードする核酸配列を使用することが好ましい。組換え技法を実施する方法に関しては、たとえば、Maniatis et al. 1982, Molecular Cloning, A laboratory Manual, Cold Spring Harbor Laboratoryおよび商業的に利用可能な方法を参照されたい。
【0063】
したがって、本発明はまた、上記の、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする、精製単離された核酸配列に関する。
【0064】
「精製および単離された核酸もしくは核酸配列」は、本発明のペプチドをコードする核酸配列、または基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする核酸が本発明に従って存在する状態を表す。
【0065】
本発明に含まれる精製および単離された核酸もしくは核酸配列は、DNA, RNA, またはDNA/RNAハイブリッドとすることができる。
【0066】
本明細書で使用されるDNAは任意のポリデオキシヌクレオチド配列であって、たとえば、二本鎖DNA、一本鎖DNA、二本鎖DNAであってその一本鎖もしくは二本鎖ともが2つ以上の断片で構成されるDNA、二本鎖DNAであってその一本鎖もしくは二本鎖ともが中断されていないホスホジエステル骨格を有するDNA、1つもしくは複数の一本鎖部分および1つもしくは複数の二本鎖部分を含有するDNA、二本鎖DNAであってそのDNA鎖が完全に相補的であるDNA、二本鎖DNAであってそのDNA鎖が一部分だけ相補的であるDNA、環状DNA、共有結合DNA、直鎖状DNA、共有架橋結合DNA、cDNA、化学合成されたDNA、半合成DNA、生合成DNA、自然分離されたDNA、酵素消化されたDNA、剪断されたDNA、標識されたDNA、たとえば放射標識DNAおよび蛍光色素標識DNA、1つもしくは複数の天然に存在しないタイプの核酸を含有するDNAを含める。
【0067】
基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードするDNA配列は、標準的な化学技術、たとえば、ホスホトリエステル法によって、または自動化合成法およびPCR法によって合成することができる。
【0068】
本発明により、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする精製および単離されたDNA配列は、酵素による技法によって作製することもできる。したがって、制限酵素は所定の認識配列の位置で核酸分子を切断するが、その制限酵素を用いて核酸配列を、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードするDNA(またはRNA)のような、核酸配列を含有するより大きい核酸分子から単離することができる。
【0069】
本発明はまた、ポリリボヌクレオチド(RNA)の形の核酸も包含し、たとえば、一本鎖RNA、cRNA、二本鎖RNA、二本鎖RNAであってその一本鎖もしくは二本鎖ともが2つ以上の断片で構成されるRNA、二本鎖RNAであってその一本鎖もしくは二本鎖ともが中断されていないホスホジエステル骨格を有するRNA、1つもしくは複数の一本鎖部分および1つもしくは複数の二本鎖部分を含有するRNA、二本鎖RNAであってそのRNA鎖が完全に相補的であるRNA、二本鎖RNAであってそのRNA鎖が一部分だけ相補的であるRNA、共有架橋結合RNA、酵素消化されたRNA、剪断されたRNA、mRNA、化学合成されたRNA、半合成RNA、生合成RNA、自然分離されたRNA、標識されたRNA、たとえば放射標識RNAおよび蛍光色素標識RNA、1つもしくは複数の天然に存在しないタイプの核酸を含有するRNAを含める。
【0070】
核酸として使用するのに好ましいのは、配列番号1、配列番号2、配列番号3、または配列番号4からなる一群から選択される、精製および単離されたDNA配列である。
【0071】
本発明は、前記配列の変異体も包含し、それは保存的ヌクレオチド置換によって基準配列から変化したヌクレオチド配列であるが、その置換によって1つもしくは複数のヌクレオチドが同様の性質を持つ別のヌクレオチドによって置換されている。
【0072】
本発明はまた、開示された精製および単離された核酸配列の対立遺伝子変異体を包含する;前記変異体は、すなわち、単離精製された核酸の、天然に存在する代替形態であり、それらは精製単離された核酸配列によってコードされるペプチドと同一、相同、もしくは関連するペプチドをコードする。あるいはまた、天然に存在しない変異体を変異誘導法もしくは直接合成によって作製することができる。
【0073】
基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする前記の精製および単離された核酸配列は、さらに、細胞膜透過性キャリアペプチドをコードするヌクレオチド配列を含んでなることができる。
【0074】
さらにもう一つの本発明の関心事は、上記の、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする、単離および精製された核酸配列を少なくとも1コピー含んでなる発現ベクターを提供することである。好ましくは、本発明のペプチドをコードする単離および精製された核酸配列は、DNAである。
【0075】
本明細書で使用される場合、「ベクター」、「プラスミド」、および「発現ベクター」は相互に交換可能なものとして使用されるが、これは、最も広く使用されるベクターの形がプラスミドであるためである。
【0076】
ベクターはさらに、本発明の細胞膜透過性キャリアペプチドをコードするヌクレオチド配列も含んでなることができる。発現ベクターの選択は、当技術分野でよく知られているように、まさに、求められる機能上の特性次第であり、たとえば、ペプチドの発現、ならびに形質転換されるべき、もしくはトランスフェクトされるべき宿主細胞によって決まる。
【0077】
それに加えて、発現ベクターはさらに、精製および単離されたDNA配列に機能しうるように連結されたプロモーターを含んでなることができる。このことは、連結された、本発明のペプチドをコードする単離および精製されたDNA配列が、適当な制御配列のコントロール下にあり、その制御配列によって、挿入された単離精製DNA配列の発現、すなわち転写および翻訳が可能となることを意味する。
【0078】
本明細書で使用される場合、「プロモーター」という用語は、当技術分野で知られている任意の追加的な制御配列、たとえば、ポリペプチドの発現のために通常使用されるプロモーターおよび/またはエンハンサー、ポリアデニル化部位、ならびにスプライス部位を表すが、あるいは追加して1つもしくは複数の別個のターゲティング配列を包含してもよく、状況に応じて選択マーカーをコードしていてもよい。使用可能なプロモーターは、そうしたプロモーターが宿主細胞に適合するならば、たとえば、ポリオーマウイルス、アデノウイルス(たとえばアデノウイルス2)、パピローマウイルス(たとえばウシパピローマウイルス)、トリ肉腫ウイルス、サイトメガロウイルス(たとえばマウスもしくはヒトサイトメガロウイルス最初期プロモーター)、レトロウイルス、B型肝炎ウイルスおよびサルウイルス40(たとえばSV40初期および後期プロモーター)といったウイルスのゲノム由来のプロモーター、またはアクチンプロモーター、免疫グロブリンプロモーターもしくは熱ショックプロモーターといった異種哺乳類プロモーターから得られるプロモーターがある。
【0079】
使用可能なエンハンサーは、たとえば、哺乳類遺伝子由来の既知のエンハンサー配列(グロビン、エラスターゼ、アルブミン、αフェトプロテインおよびインスリン)または真核細胞ウイルス由来のエンハンサー、たとえば、SV40エンハンサー、サイトメガロウイルス初期プロモーターエンハンサー、ポリオーマおよびアデノウイルスエンハンサーである。
【0080】
本発明のDNA配列の発現には、さまざまな宿主/発現ベクターの組み合わせを用いることができる。有用な発現ベクターは、たとえば、染色体性、非染色体性および合成DNA配列のセグメントからなっていてもよい。適当なベクターとしては、SV40から作製されたベクター、ならびに既知の細菌プラスミド、たとえば、大腸菌(E. coli)プラスミドcol E1、pCR1、pBR322、pcDNA3、pMB9およびそれらから作られたプラスミド、RP4のようなプラスミド;ファージDNA、たとえば、ファージXから作られた多数のDNA、たとえばNM989、および他のファージDNA、たとえばM13および繊維状一本鎖ファージDNA;酵母プラスミド、たとえば、2μプラスミドもしくはそれから作られたプラスミド;真核細胞において有用なベクター、たとえば、昆虫もしくは哺乳類細胞において有用なベクター;プラスミドおよびファージDNAの組み合わせから作られたベクター、たとえば、ファージDNAもしくは他の発現制御配列を用いるように改変されたプラスミド;などがある。
【0081】
もっとも好ましくは、発現ベクターはpcDNA3である。
【0082】
本発明のもう一つの関心事は、本発明のペプチド、単離精製された本発明の核酸配列、および/または本明細書に記載の発現ベクターを含有する真核もしくは原核宿主細胞を提供することである。
【0083】
精製単離された本発明のDNA配列を含んでなる発現ベクターによる、適当な真核もしくは原核宿主細胞の形質転換またはトランスフェクションは、概して使用するベクターの種類によって決まる公知の方法によって達成される。こうした方法に関しては、たとえば、Maniatis et al. 1982, Molecular Cloning, A laboratory Manual, Cold Spring Harbor Laboratoryおよび商業的に利用可能な方法を参照されたい。「トランスフェクトされた細胞」、「形質転換された細胞」もしくは「トランスフェクト/形質転換された細胞」という用語は、細胞外DNAがその中に導入され、したがって細胞外DNAを保有する細胞を意味する。核酸が染色体組み込み体として、または染色体外エレメントとして複製可能であるように、DNAを細胞内に導入することができる。
【0084】
基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドは、状況に応じて、本明細書に記載のように細胞内でのペプチドの蓄積を増加させる作用物質と結合しているが、そのペプチドは、細胞発現系において、好ましくは組換えによって作製される。
【0085】
さまざまな単細胞の宿主細胞が、本発明のDNA配列の発現に有用である。これらの宿主には、既知の真核および原核宿主、たとえば、大腸菌、Pseudomonas, Bacillus, Streptomyces、酵母のような真菌、および動物細胞、たとえば、CHO、YB/20、NSO、SP2/0、Rl.1、B-WおよびL-M細胞、アフリカミドリザル腎細胞(例、COS1、COS 7、BSC1、BSC40およびBMT10)、昆虫細胞(例、Sf9)、およびヒト細胞、ならびに組織培養植物細胞を含めることができる。好ましくは、宿主細胞は細菌細胞であって、大腸菌細胞がさらに好ましい。
【0086】
本発明はまた、活性成分として、製薬上有効な量の少なくとも1つの記載されたペプチドを、状況に応じて製薬上許容される担体、希釈剤および補助剤と組み合わせて含んでなる、医薬組成物に関する。
【0087】
「製薬上有効な量」は、ヒトもしくは動物に投与したときに検出可能な薬理学的および/または生理学的な効果を生じる化学物質もしくは化合物を指す。
【0088】
それぞれの製薬上有効な量は、治療すべき個別の患者、治療すべき疾患および投与方法によって決まる。さらに、製薬上有効な量は、使用される具体的なペプチド次第であって、特にペプチドが記載のように薬物を追加して含有するか否かによる。治療は通常、数時間、数日もしくは数週間の間隔をおいた、医薬組成物の複数回投与を含んでなる。ポリペプチドの製薬上有効な投薬量単位は、治療を受ける患者の体重kg当たり0.001ngから100μgの範囲である。
【0089】
好ましくは、医薬組成物は、少なくとも1つの本明細書に記載のペプチドに加えて、1つもしくは複数の製薬上許容される担体、希釈剤および補助剤を含有することができる。
【0090】
許容される担体、希釈剤および補助剤は、医薬として使用できる製剤への活性化合物の加工を助けるが、これらは使用される投与量および濃度で受容者に対して無毒であって、バッファー、たとえばリン酸、クエン酸および他の有機酸;アスコルビン酸およびメチオニンを含めた抗酸化剤;防腐剤(たとえば、塩化オクタデシルジメチルベンジルアンモニウム;塩化ヘキサメトニウム;塩化ベンザルコニウム、塩化ベンゼトニウム;フェノール、ブチルもしくはベンジルアルコール;メチルもしくはプロピルパラベンといったアルキルパラベン類;カテコール;レゾルシノール;シクロヘキサノール;3-ペンタノール;およびm-クレゾール);低分子量(約10残基未満)ポリペプチド;タンパク質、たとえば、血清アルブミン、ゼラチン、もしくは免疫グロブリン;親水性ポリマー、たとえばポリビニルピロリドン;アミノ酸、たとえばグリシン、グルタミン、アスパラギン、ヒスチジン、アルギニンもしくはリジン;単糖類、二糖類、および他の炭水化物、たとえばグルコース、マンノースもしくはデキストリン;キレート剤、たとえばEDTA;糖類、たとえばショ糖、マンニトール、トレハロースもしくはソルビトール;塩形成対イオン、たとえばナトリウム;金属錯体(たとえば亜鉛タンパク質錯体);および/または非イオン性界面活性剤、たとえばTWEEN(登録商標)、PLURONICS(登録商標)、もしくはポリエチレングリコール(PEG)を包含する。
【0091】
医薬組成物の投与法は、全身性または局所性とすることができる。たとえば、このような組成物の投与は、さまざまな非経口経路、たとえば皮下、静脈内、皮内、筋肉内、腹腔内、鼻腔内、経皮、頬側経路、または埋め込み装置によることができるが、蠕動によって送達することもできる。
【0092】
本明細書に記載のペプチドを活性成分として含んでなる医薬組成物を、生体吸収性マトリックスに組み入れ、またはそれに浸透させることもできるが、そのマトリックスはマトリックスの懸濁液、ゲルもしくは固体の形で投与される。加えて、マトリクスはバイオポリマーから構成されていてもよい。
【0093】
徐放性製剤を調製することができる。徐放性製剤の適当な例としては、抗体を含有する固体疎水性ポリマーの半浸透性マトリックスがあり、このマトリックスはフィルムもしくはマイクロカプセルといった成型品の形をとる。徐放性マトリックスの例には、ポリエステル、ハイドロゲル(たとえば、ポリ(2-ヒドロキシエチル-メタクリレート)もしくはポリ(ビニルアルコール))、ポリラクチド(米国特許第3,773,919号)、L-グルタミン酸およびγ-エチル-L-グルタメートのコポリマー、非分解性エチレン−ビニルアセテート、分解性乳酸-グリコール酸コポリマー、たとえばLUPRON DEPOT(商標名)(乳酸-グリコール酸コポリマーおよび酢酸ロイプロリドからなる注射用マイクロスフェア)、ならびにポリ-D-(-)-3-ヒドロキシ酪酸がある。
【0094】
in vivo投与に使用される製剤は無菌でなければならない。これは、たとえば、無菌濾過膜を通す濾過によって容易に達成される。
【0095】
当然のことながら、本発明のペプチドの適切な投与量は、投与を受ける人の年齢、性別、健康状態および体重、もしあれば併用治療の種類、ならびに求める効果の内容によって左右される。
【0096】
適当な剤形は、疾患、ペプチド、および投与法によって決まる;可能性としては、錠剤、カプセル剤、トローチ剤、歯科用ペースト、座剤、吸入剤、溶液、軟膏剤および非経口デポー製剤が含まれる。
【0097】
ペプチドのアミノ酸のアミノ酸修飾も本発明に含まれるので、このことは、本発明のペプチドを水に不溶性のマトリックスもしくは他の巨大分子基剤に架橋結合するために、または溶解性、吸着性および血液脳関門を越える透過性を改善するために、有用であると考えられる。このような修飾は当技術分野ではよく知られており、あるいはまた、起こりうる望ましくないペプチドの副作用などを取り除き、もしくは弱めることができる。
【0098】
本発明の好ましい医薬組成物は、活性成分としてペプチドを含んでなるが、また別の医薬組成物は、本明細書に記載のペプチドをコードする精製単離された核酸配列を、活性成分として含有することができる。この医薬組成物は、単一の精製単離されたDNA配列、前記の精製単離されたDNA配列を含んでなる発現ベクター、または本明細書に記載の発現ベクターであらかじめトランスフェクトされ、もしくは形質転換された宿主細胞のいずれかを包含することができる。この後者の例では、宿主細胞は、抗原性の問題を回避するために、好ましくは治療を受ける患者から分離されることになる。これらの遺伝子治療および細胞治療のアプローチは、特に、医薬組成物の反復投与を必要とする患者に非常に適しているが、それは、前記の精製単離されたDNA配列、発現ベクター、または発現ベクターであらかじめトランスフェクトされ、もしくは形質転換された宿主細胞を、その後内生的にタンパク質を産生する患者の細胞に、組み入れることができるためである。
【0099】
通常、本明細書に記載の医薬組成物は、癌の治療もしくは予防に使用される。
【0100】
本発明はまた、癌の治療用もしくは予防のための薬物を製剤するための、本発明の医薬組成物の使用を包含する。
【0101】
「癌」という用語は、基本的に無秩序な細胞増殖によって特徴付けられる、哺乳類における生理的状態を指し、または表現するものである。
【0102】
通常、治療もしくは予防すべき癌は、癌腫、リンパ腫、芽細胞種、肉腫、脂肪肉腫、神経内分泌腫瘍、中皮腫、神経鞘腫、髄膜腫、腺癌、黒色腫、白血病、悪性リンパ腫、扁平上皮癌、上皮性扁平上皮癌、肺癌、小細胞肺癌、非小細胞肺癌、肺腺癌、肺扁平上皮癌、腹膜癌、肝細胞癌、胃癌、消化器癌、膵臓癌、神経膠芽腫、子宮頸癌、卵巣癌、肝臓癌、膀胱癌、肝細胞腫、乳癌、大腸癌、直腸癌、直腸結腸癌、子宮内膜癌もしくは子宮癌、唾液腺癌、腎臓癌、前立腺癌、外陰癌、甲状腺癌、肝癌、肛門癌、陰茎癌、睾丸癌、食道癌、胆道腫瘍、および頭頸部癌からなる一群から選択されることとなる。
【0103】
好ましくは、癌は、中皮種、睾丸癌もしくは膵臓癌である。
【0104】
本発明のペプチドは、概して、意図した目的を達成する量で使用される。癌の治療もしくは予防に使用するために、ペプチドもしくはその医薬組成物は、治療上有効な量投与され、または適用される。「治療上有効な量」は、症状を改善もしくは予防するのに有効な量、または治療を受ける被験体の生存を延長するのに有効な量である。治療上有効な量の決定は、特に、本明細書で提示される詳細な開示に照らして、十分、当業者に可能な範囲である。
【0105】
全身投与のために、治療上有効な量または用量は、初めにin vitroアッセイから見積もることができる。たとえば、細胞培養で決定されるIC50を含めた循環濃度範囲を達成するように、動物モデルにおいて用量を定式化することができる。そうした情報を用いて、より正確に、ヒトにおいて有効な用量を決定することができる。
【0106】
初回投与量も、たとえば動物モデルのin vivoデータから、当技術分野でよく知られた技法によって、見積もることができる。当業者は、動物データに基づいて容易にヒトに対する投与を最適化することができるであろうが、当然、治療を受ける被験体、被験体の体重、疾患の重症度、投与の方法、および処方医の判断に依存する。
【0107】
本明細書はまた、癌の治療もしくは予防を必要とする被験体に本明細書に記載の医薬組成物を治療上有効な量投与することを含んでなる、前記被験体において癌を治療もしくは予防する方法を提供する。
【0108】
治療もしくは予防することができる癌の例は、上記に記載した。好ましくは、癌は中皮種、睾丸癌もしくは膵臓癌である。
【0109】
好ましい方法において、被験体はヒト患者であり、投与されるペプチドは、癌細胞を死滅させる少なくとも1つの薬物の能力を選択的に増強するものであって、TAT-RasGAP317-326ペプチドである。
【0110】
癌細胞において選択的にアポトーシスを強める方法もまた本発明の範囲に含まれるが、その方法は、癌細胞を、本発明の少なくとも1つのペプチドおよび薬物と接触させることを含んでなる。
【0111】
癌細胞を本発明の少なくとも1つのペプチドおよび薬物と接触させることを含んでなる、選択的に癌細胞を死滅させる方法も想定される。
【0112】
癌細胞において選択的に細胞を死滅させる薬物の能力を増強するために、本明細書に記載のペプチドを使用することも想定される。
【0113】
本発明のもう一つの目的は、被験体において癌を治療もしくは予防するキットを提供することであって、該キットは、少なくとも1つの本明細書に記載のペプチドを、状況に応じて試薬および/または使用説明書と共に含んでなる。
【0114】
概して、キットは、容器、およびその容器の上もしくは容器に付随してラベルまたは添付文書を含んでなる。適当な容器には、ビン、バイアル、シリンジなどがある。容器はガラスやプラスチックといった、さまざまな材料から作製することができる。容器は、癌の治療に有効な組成物が入っており、無菌のアクセスポートを有していてもよい(たとえば、容器は静脈注射用溶液バッグ、または皮下注射針で突き刺すことができるストッパー付きバイアルとすることができる)。ラベルもしくは添付文書は、その組成物が好みの癌の治療用に使用されることを示す。
【0115】
状況に応じて、キットはさらに、上記の薬物からなる一群から選択される追加の抗ガン剤、抗-上皮成長因子受容体抗体、放射免疫治療薬、およびそれらの組み合わせを含んでなる、別個の医薬剤形を含んでなる。
【0116】
実施例
【実施例1】
【0117】
細胞およびトランスフェクション
HeLaおよびMCF-7細胞を、10%ウシ新生仔血清(Sigma ; カタログ番号N4637)を含有するRPMI 1640 (Sigma; カタログ番号8758)中で、37℃、5% CO2にて維持した。 U20S細胞は、 15% ウシ胎仔血清(Sigma; カタログ番号F7524) を含有するDMEM (Sigma ; カタログ番号5796)中で、37℃、5% CO2にて維持した。H-Meso-1細胞は10% ウシ胎仔血清を含有するRPMI 1640中で37℃、5%CO2にて維持した。HUV-EC-C細胞は、10% ウシ胎仔血清、20 ng/ml塩基性線維芽細胞成長因子(Gibco; カタログ番号13256-029)、10 ng/ml 上皮成長因子 (Gibco; カタログ番号13247-051)、10, ug/ml フィブロネクチン(Gibco; カタログ番号33016-015)を添加したヒト内皮SFM培地(Gibco; カタログ番号l1111-044)中で37℃、5%CO2にて維持した。HaCat細胞は、上皮成長因子1-53およびウシ脳下垂体抽出物(Gibco ; カタログ番号17005-075)を含有するケラチノサイトSFM培地中で37℃、5%CO2にて維持した。HeLa細胞は、既述のようにトランスフェクトした (Yang J. -Y. and Widmann C. , Mol. Cell. Biol. , 21,5346, 2001)。遺伝毒性物質処理は、6-ウェルプレートで行った。2.5x105細胞/ウェルの濃度で、処理の前日に細胞を分裂した。U20S細胞は6ウェルプレートで、カルシウム/リン酸沈澱法によってトランスフェクトした(Jordan M. et al., "Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation" Nucleic Acids Res., 24, 596, 1996)。簡単に述べると、プラスミドを90μl H20中に希釈し、10μl CaCl2 2.5 M と混合して、室温にて10分間インキュベートした。その後、100μlのHEP溶液(280 mM NaCl、10 mM KCl、1.5 mM Na2HP04、12 mM D-グルコース、50 mM HEPES)を急速に上記DNA溶液と混合し、室温にて厳密に1分間インキュベートし、最後に、細胞培養培地に移した。37℃、5%CO2にて8時間後、培地を新培地と交換し、細胞をさらに16-24時間インキュベートした後、分析した。
【0118】
化学薬品
シスプラチンおよびミトキサントロンはSigma (カタログ番号はそれぞれP4394およびM6545)から入手した。シスプラチンは終濃度100 mMとなるようにDMSO中に希釈し、-20℃で保存した。ミトキサントロンは終濃度10 mMとなるように100%エタノール中に希釈し、-80℃で保存した。アドリアマイシンはCalbiochem (カタログ番号324380)から入手した。これは水で終濃度10mMとなるように希釈し、-20℃で保存した。ヘキスト(Hoechst)33342はRoche(カタログ番号H-1399)から入手した。これは水で終濃度10mg/mlとなるように希釈して4℃にて暗所に保存した。
【0119】
ペプチド合成および標識化
HIV-TAT48-57(GRKKRRQRRR)およびTAT-RasGAP317-326(GRKKRRQRRRGGWMWVTNLRTD)ペプチドは、スイス、ローザンヌ大学生化学研究所で、FMOC法を用いて合成し、HPLCで精製して、質量分析法で検査した。
【0120】
フルオレセインイソチオシアネート(FITC)標識は、β-アラニン- GRKKRRQRRRGGWMWVTNLRTD配列について行ったが、その側鎖Fmoc保護アミノ酸は、Arg(bpf), Lys (Boc), Gln (Trt), Trp (Boc), Thr (tBu), Asn (Trt)、およびAsp (OtBu)であった。このペプチドは、0.2 mmol Rink Amide AM樹脂上でFmoc法によって段階的に合成された。ニンヒドリン試験によって合成をモニターした。β-アラニンを結合した後、ジメチルホルムアミド(DMF)中20% ピペリジンを用いてFmoc基を除去した。この段階で、FITC(4ml DMFおよび1ml N-エチルジイソプロピルアミン中、樹脂を置換する5倍過剰量)を用いてフルオレセイン基をペプチドのN末端に結合し、フルオレセイン誘導体化ペプチドを生成した。
【0121】
ペプチドは脱イオン水に終濃度1mMとなるように溶解し、追って使用するまで−20℃にて保存した。
【0122】
プラスミド
プラスミドの名称に付け加えられたdn3は、そのバックボーンプラスミドが発現ベクターpcDNA3(Invitrogen)であることを示す。構築物はすべてNアミノ末端でHA配列(MGYPYDVPDYAS)によってタグを付けた。プラスミドN2.dn3は、ヒトRasGAP断片N2をコードし、プラスミドSH2-SH3.dn3はヒトRasGAPアミノ酸158-361をコードし、プラスミドSH2.dn3はヒトRasGAPアミノ酸158-277をコードし、プラスミドSH3.dn3はヒトRasGAPアミノ酸279-361をコードする。プラスミドIκBαΔN2はNFκBの活性化を阻止するある種のIκBαをコードする (YangおよびWidmann, 2002b)。GFPタンパク質をコードするプラスミドpEGFP-Clは、Clontechから入手した。pRL-TKは、ウミシイタケ(Renilla renifornis)ルシフェラーゼをコードするベクターであるが、Promegaより入手した。prLUC は、NFκB-応答性エレメントの制御下でホタルルシフェラーゼcDNAを有するレポータープラスミドである(Yang J.-Y. and Widmann C., Mol. Cell. Biol., 21, 5346, 2001)。
【0123】
アポトーシス測定
アポトーシスは、核濃縮を示す細胞の数を記録することによって測定した。生きた細胞の核をヘキスト33342(終濃度10μg/ml)で約5分間標識し、次にその細胞を、蛍光および透過光光学系を装備したLeica DMIRB倒立顕微鏡を用いて分析した(少なくとも条件当り細胞400個)。
【0124】
細胞をトランスフェクトもしくは処理した1日後にアポトーシスの評価を行った。トランスフェクトされた細胞に関わる実験において、トランスフェクション溶液にpEDFP-C1を入れ、トランスフェクト細胞をGFPで標識した。この場合、アポトーシスの程度はトランスフェクト細胞のみについて評価した。
【0125】
ルシフェラーゼレポーターアッセイ
ルシフェラーゼアッセイはPromega製Dual-Luciferase(登録商標)Reporter Assay(カタログ番号E1910)を用いて行った。Promegaのキットから供給されたPLB溶解バッファー100μlを用いて6ウェルプレートから細胞を溶解し、氷上で30分間インキュベートした。次に溶解液を16,000gで15分間遠心して透明化した。この溶解液20μlをLARII試薬25μlと混合してホタルルシフェラーゼ活性を記録し、前記混合物にStop & Glo 試薬25μlを加えてウミシイタケルシフェラーゼ活性を記録した。それぞれの測定について、Lumat LB 9501ルミノメーター(Berthold Technologies、チューリッヒ、スイス)を用いて12秒間、発光を数量化した。
【0126】
ウェスタンブロット分析
細胞を溶解バッファー(25 mM Hepes、300mM NaCl、1.5mM MgCl2、0.2mM EDTA、0.1mM Na3V04、1% Triton X100、コンプリートEDTAフリープロテアーゼインヒビターカクテル錠(Roche;カタログ番号1873580))中で溶解した。SDS-PAGEでタンパク質を分離し、ニトロセルロース膜(BioRad;カタログ番号162-0115)にブロットした。その後、室温にて30分間、TBS (18 mM HC1、130 mM NaCl、20 mM Tris)、5%脱脂粉乳で膜をブロックし、適当な一次抗体とともに一晩インキュベートした。これらの抗体を、TBS、5%脱脂粉乳で1:2500に希釈したAlexa Fluor 680結合二次抗体(Molecular Probes;カタログ番号A21109)によって検出し、続いてOdysseyインフラレッドイメージングシステム(Licor、ホンブルク、ドイツ)によって可視化した。phospho-p38に対する一次抗体(Cell Signaling Technology;カタログ番号9211L)をTBS中5%BSAで1:500に希釈した。phospho-JNKに対する一次抗体(Cell Signaling Technology;カタログ番号9551L)は、TBS中5%BSAで1:1000に希釈した。Odysseyインフラレッドイメージングソフトウェアを用いて数量化した。
【0127】
統計分析
すべての統計分析は、スチューデントtテストによりMicrosoft Excel (XP edition)で行った。
【実施例2】
【0128】
RasGAP断片N2はさまざまな遺伝毒性物質によって引き起こされるアポトーシス反応を増強する。
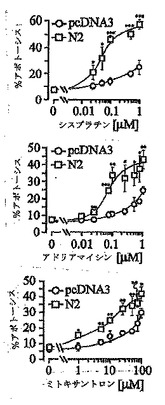
最近、本出願人らは、断片N2がHeLa腫瘍細胞株を死滅させるシスプラチンの能力を増強することを明らかにした(Yang J.-Y. and Widmann C., Mol. Cell. Biol., 21,5346, 2001)。断片N2が、他の遺伝毒性物質によって引き起こされるアポトーシス反応を増強することができるかどうかを評価するために、断片N2を発現するHeLa細胞もしくは発現しない同細胞を、アドリアマイシンおよびミトキサントロン(および対照としてシスプラチン)の濃度を増加させてその影響下においた。図1は、断片N2の存在が、さまざまな薬物に対するHeLa細胞の感受性を対照細胞より少なくとも10倍高めることを示す。この結果は、断片N2が広域性の遺伝毒性物質増感剤であることを示す。
【0129】
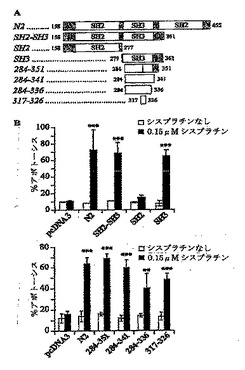
癌細胞を死滅させる薬物の能力を増強する断片Nの中で最小の配列の同定
断片N2は36 kDaのタンパク質であり、そのため化学的な合成は困難であると考えられる。短くてもなお遺伝毒性物質増感能力を有する配列の特徴を示すことは、断片N2から治療手段を開発するプロセスにおいて重要なステップである。そういった短い配列が分離できるかどうかを評価するために、断片N2の一連のトランケート型を作製し(図2A)、HeLa細胞においてシスプラチンが引き起こすアポトーシスを増強する能力を測定した(図2B)。断片N2は2つのSH2ドメインおよび1つのSH3ドメインを含有する(図2A)。これらのドメインのうちどれが断片N2のアポトーシス促進活性を有するかをまず判定した。HeLa細胞を、断片N2を発現する細胞と発現しない細胞で薬物に対する感受性の差が最大となる濃度である、0.15μMのシスプラチン存在下もしくは非存在下で、さまざまなSHドメインをコードするプラスミドによりトランスフェクトした(図1を参照されたい)。何もコードしていない構築物またはSH2ドメインをコードする構築物は、これらの構築物でトランスフェクトされたHaLa細胞を死滅させるシスプラチンの能力を増強しなかった。これに対して、HS3ドメインを含有する構築物は、アポトーシスによってシスプラチンが細胞を死滅させる能力を増強する(図2B)。ウェスタンブロット分析から、SH2ドメインが他の構築物と同じく効率的に発現されていることはあきらかなので、SH2ドメイン単独でHeLa細胞において細胞死を増強することができないのは、タンパク質発現が低下した結果ではない(データは示さない)。
【0130】
本出願人らは次に、最小の遺伝毒性物質感作配列を同定する目的で、SH3ドメインのトランケーションを進めた。これらの構築物(図2A参照)はすべて、10アミノ酸の長さのペプチドをコードする最も短いもの(317-326)を含めて、シスプラチンがHeLa細胞を死滅させる能力を増強した(図2B)。こうした結果は、断片N2の細胞死感作特性が、完全なSHドメインを必要とせず、非常に短いペプチド配列によって媒介されることを示唆する。
【0131】
317-326 RasGAP配列と結合したHIV-TAT48-57ペプチドは細胞透過性遺伝毒性物質増感ペプチドとして機能する。
【0132】
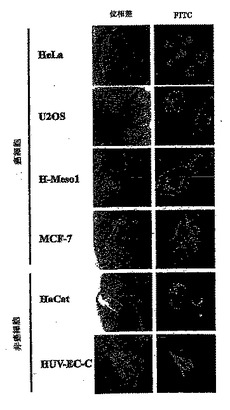
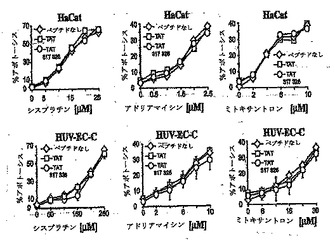
RasGAPのアミノ酸317-326をコードするプラスミドに、薬物が癌細胞を死滅させる能力を強める力があるならば、316-326に相当する合成ペプチドも細胞に浸透することができればこうした活性を示すはずである。HIV TATタンパク質に由来する短い配列(HIV-TAT48-57)を付加することによって、ポリペプチドの細胞内での効率的な蓄積が可能になることが実証されている(Schwarze S. R. et al.,"n vivo protein transduction: delivery of a biologically active protein into the mouse" Science, 285,1569, 1999)。したがって、本出願人らは、アミノ酸キャリアペプチドHIV-TAT48-57と共有結合したRasGAPのアミノ酸317-326を含有するペプチドを合成した。TATとRasGAP配列の間にスペーサーとして2つのグリシン残基を挿入し、柔軟性を与えた。このペプチド(以後TAT-RasGAP317-326ペプチドと称する)の細胞取り込みを評価する手段として、そのペプチドをFITCフルオロフォアで標識した。標識したペプチドを4つの異なる腫瘍細胞株とともにインキュベートした:子宮頸部由来ヒト腺癌(HeLa細胞)、ヒト骨肉腫(U2OS細胞)、乳癌細胞株(MCF-7細胞)およびヒト悪性中皮種(H-Meso-1細胞)、ならびに2つの非癌細胞株(HaCatヒト皮膚ケラチノサイト細胞株およびHUV-EC-Cヒト臍静脈内皮細胞)。図3に示すように、TAT-RasGAP317-326ペプチドは、これらすべての細胞株の中に効率よく入った。非癌細胞と癌細胞の間でペプチドの移動に差は認められなかった。最初に、3つの遺伝毒性物質に対する癌細胞株の感受性を、毒性物質の濃度を上げて評価した(データは示さない)。これによって、テストした各細胞株についての遺伝毒性物質の亜致死濃度を測定することができた。驚くべきことに、TAT-RasGAP317-326ペプチドは、テストした癌細胞株を死滅させるシスプラチン、アドリアマイシンおよびミトキサントロンの能力を増強したがRasGAP配列を欠いた対照のHIV-TAT48-57ペプチドは増強しなかったこと、および対照条件においてアポトーシスは誘導されず、もしくはごくわずかしか誘導されないことが明らかになった。これに対して、2つの非癌細胞株においてシスプラチン、アドリアマイシンおよびミトキサントロンによって誘導されるアポトーシス反応は、ペプチドの存在によって影響を受けなかった(図4B)。したがって、本出願人らは、特異的に細胞に入り、癌細胞を死滅させる薬物の能力を増強することができる最小の合成ペプチドを同定した。
【0133】
NFκBおよびSAPK経路はTAT-RasGAP317-326の特性に関与しない。
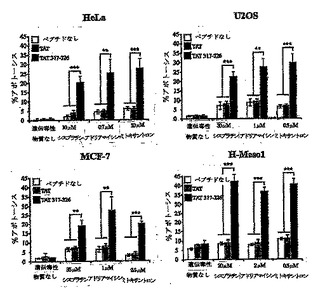
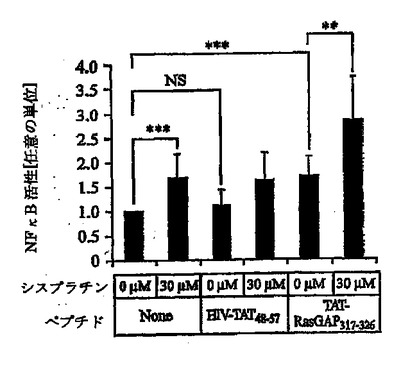
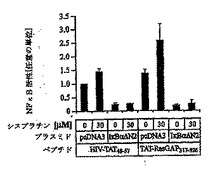
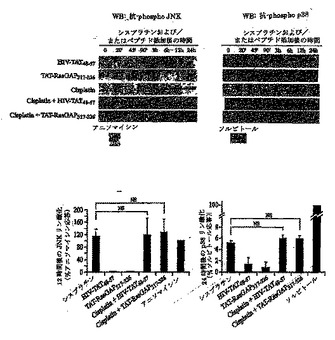
Rasモジュレータとして、RasGAPは、細胞死を制御するRas依存性経路の一部、たとえばRas-P13K-Akt-NFκB経路に影響を及ぼすと考えられる(Datta S. R. et al., "Cellular survival: a play in three Akts" Genes Dev., 13, 2905, 1999)。したがって、NFκB活性のモジュレーションが、TAT-RasGAP317-326による能力の増強に関与するかどうかを評価した。図5Aに示すように、NFκBはU2OS細胞において、シスプラチン処理もしくはTAT-RasGAP317-326による処理の後、活性化されたが、対照ペプチドHIV-TAT48-57による刺激では活性化されなかった。シスプラチンおよびTAT-RasGAP317-326の両者とともに細胞をインキュベートすると、結果としてNFκBのさらなる活性化を生じた(図5A)。NFκB経路は、多くの種類の細胞において細胞生存反応の誘導に関与する(Van Antwerp D. J. etal., "Suppression of TNF-alpha- induced apoptosis by NF-kappaB" Science, 274,787, 1996; Beg A. A. and Baltimore D., "An essential role for NF-kappaB in preventing TNF-alpha-induced cell death" Science, 274,782, 1996)が、ある種の状況においてはアポトーシス反応の誘導にも必要とされる可能性がある(Ryan K. M. et al., "Role of NF-kappaB in p53-mediated programmed cell death" Nature, 404, 892,2000)。TAT-RasGAP317-326によるNFκBの活性化が増強する能力機能に必要であるかを判断するために、非分解型IκB(IκBαΔN2)を細胞において発現させた。予想されるように、この構築物は効率的にシスプラチンおよびTAT-RasGAP317-326によるNFκBの活性化を阻止した(図5B)。IκBαΔN2は、わずかにシスプラチンによるアポトーシス反応を増大させた(図5C)。しかしながら、アポトーシスによってU2OS細胞を死滅させるシスプラチンの能力を増強するTAT-RasGAP317-326の効力は、NFκBインヒビターの影響を受けなかった(図5C)。これらの結果は、NFκB経路の活性化がTAT-RasGAP317-326による細胞死増強能力の効果に関与しないことを示す。
【0134】
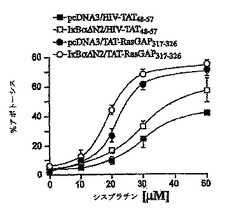
ストレス活性化プロテインキナーゼ(SAPK)-JNKおよびp38 MAPK-は、さまざまな刺激によって引き起こされるアポトーシス反応に関与していた(Jarpe M. B. et al., Oncogene, 17, 1475, 1998)。図6は、対照HIV-TAT48-57ペプチドもTAT-RasGAP317-326ペプチドもいずれもこれらのMAPK経路を活性化しなかったことを示す。これらのペプチドはまた、シスプラチンがJNKもしくはp38 MAPKを刺激する能力を高めることはできなかった(図6)。これらの結果は、ストレス活性化MAPK経路が、癌細胞を死滅させる遺伝毒性物質の能力を増強する、TAT-RasGAP317-326の機能に関与しないことを示す。
【図面の簡単な説明】
【0135】
【図1】図1は、断片N2もしくは空のpcDNAプラスミドでトランスフェクトされたHeLa細胞における、さまざまな薬物によるアポトーシス誘導のパーセンテージを示す。 HeLa細胞(2x106)を直径10cmのペトリ皿に播き、空のpcDNA3プラスミド2μg、または断片N2をコードするpcDNA3ベクター2μgとともに、GFP発現プラスミド1μg(トランスフェクト細胞を標識するため)でトランスフェクトした。トランスフェクションの1日後、細胞を24時間、指示された濃度のシスプラチン、アドリアマイシン、もしくはミトキサントロンとともにインキュベートした。その後核濃縮を示すGFP陽性細胞の数を記録した。 結果は、3回の独立した測定の平均値±標準偏差に対応する。星印は対照細胞とシスプラチン処理細胞の間の有意な相違を示す(*,p < 0. 05 ; **, p < 0.01 ; ***, p < 0.001)。
【図2】図2Aは本研究に用いられたさまざまな構築物の概要図である。 SHはSrc相同ドメインを表す。図2Bは、図2Aに記載の構築物をコードするプラスミドでトランスフェクトされたHeLa細胞における、シスプラチンによるアポトーシス誘導のパーセンテージを示す。 HeLa細胞を、図2Aに記載の構築物をコードしたプラスミドで、図1に記載のようにトランスフェクトした。その後、HeLa細胞を0.15μMシスプラチンで処理し、または処理せずに、アポトーシスの程度を20時間後に測定した。結果は、3つの独立した測定の平均値±標準偏差に対応する。星印は0.15μMシスプラチン処理細胞と非処理細胞の間の有意な相違を示す(**, p < 0.01 ; ***, p < 0.001)。
【図3】図3は、FITC標識TAT-RasGAP317-326ペプチドとともにインキュベートした生きた細胞の位相差および落射蛍光画像を示す。 次の細胞株(HeLa、U20S、H-Mesol、MCF-7、HaCatおよびHUV-EC-C)を、FITC標識TAT-RasGAP317-326ペプチド20μMを含む培地中で37℃、5% CO2にて3時間インキュベートした後、培地で3回洗浄した。
【図4A】図4Aは、TAT-RASGAP317-326で処理された、または処理されない、2つの非癌細胞における、さまざまな薬物によるアポトーシス誘導のパーセンテージを示す。 2つの非癌細胞株(HaCatおよびHUV-EC-C)を、20μM TAT-RasGAP317-326 の存在下もしくは非存在下で、シスプラチン、アドリアマイシンおよびミトキサントロンとともに、それらの濃度を増加させてインキュベートした。アポトーシスの程度を20時間後に記録した。
【図4B】図4Bは、TAT-RASGAP317-326で処理された、または処理されない、4つの癌細胞株における、さまざまな薬物によるアポトーシス誘導のパーセンテージを示す。 4つの癌細胞株(HeLa、U20S、MCF-7およびH-Mesol)を6ウェルプレートに播き、20μMHIV-TAT48-57もしくはTAT-RasGAP317-326ペプチドのいずれか一方の存在下、またはいずれも存在しない条件下で、指示された濃度のシスプラチン、アドリアマイシンおよびミトキサントロンで20時間処理した。次に核濃縮を示す細胞の数を記録した。星印は、TAT-RasGAP317-326とともにインキュベートした遺伝毒性物質処理細胞と、未処理のままの細胞もしくはHIV-TAT48-57ペプチドとともにインキュベートした細胞との間の有意な相違を示す(**, p < 0.01 ; ***, p < 0.001)。
【図5A】図5Aは、シスプラチンで処理したU2OS細胞における、HIV-TAT48-57もしくはTAT-RasGAP317-326ペプチド存在下でのNFκB活性を示す。 U2OS細胞(1x105)を6ウェルプレートに播き、NFκB活性のためのホタルルシフェラーゼレポータープラスミド1μgおよびウミシイタケルシフェラーゼをコードするプラスミド0.1μgでトランスフェクトした。1日後に、20μM HIV-TAT48-57もしくはTAT-RasGAP317-326ペプチドの存在下または非存在下で、20時間、細胞を指示された濃度のシスプラチンで処理した。データはウミシイタケルシフェラーゼ活性に対して標準化されたホタルルシフェラーゼ活性を表し、対照の非処理細胞で得られた基底NFκB活性からの増加倍数として示した。結果は、3つの独立した測定の平均値±標準偏差に対応する。星印は指示された条件の間の有意な相違を示す(**, p < 0.01 ; ***, p < 0.001)。
【図5B】図5Bは、シスプラチンで処理したU2OS細胞における、HIV-TAT48-57ペプチドおよびIκBαΔN2、またはTAT-RasGAP317-326ペプチドおよびIκBαΔN2の存在下でのNFκB活性を示す。 U2OS細胞を、NFκB活性のためのホタルルシフェラーゼレポータープラスミド1μg、ウミシイタケルシフェラーゼをコードするプラスミド0.1μg、GFP発現プラスミド(トランスフェクト細胞を標識するため)0.5μg、NFκB経路を阻害するIκBαΔN2をコードするプラスミド1μgもしくは空のpcDNA3ベクター1μgでトランスフェクトした。1日後に細胞を、さらに20時間、20μM HIV-TAT48-57または20μM TAT-RasGAP317-326の存在下で、シスプラチンとともに、シスプラチンの濃度を増加させてインキュベートした。次に、細胞を溶解し、パネルAに記載のようにNFκB活性を測定したが、ただし結果は、対照のHIV-TAT48-57ペプチドとともにインキュベートした細胞において検出されたNFκB活性からの増加倍数として表現した。
【図5C】図5Cは、図5AおよびBのトランスフェクト細胞のアポトーシスのパーセンテージを示す。 代わりになるべきものとして、核濃縮を示すGFP陽性細胞の数を測定した。結果は、3つの独立した測定の平均値±標準偏差に対応する。
【図6】図6は、HIV-TAT48-57もしくはTAT-RasGAP317-326ペプチドのいずれかでトランスフェクトされ、およびシスプラチンで処理された、U2OS細胞のJNKリン酸化のウェスタンブロットおよびパーセンテージを表す。 U2OS細胞(2x105)を6ウェルプレートに播き、HIV-TAT48-57もしくはTAT-RasGAP317-326ペプチド(濃度20μM)およびシスプラチン(濃度30μM)の指示された組み合わせによって、指示された時間、処理した。JNKおよびp38活性化に対する陽性対照は、それぞれ、1μg/mlアニソマイシンで3時間、および0.5Mソルビトールで30分間、細胞を刺激した後、得られた。ウェスタンブロットに基づいて示される定量化は12時間のバンドについて行われ、陽性対照に対して標準化された。結果は、3つの独立した測定の平均値±標準偏差に対応する。
【技術分野】
【0001】
本発明は、癌細胞において薬物が選択的に細胞を死滅させる能力を強めるペプチドに関する。さらに、本発明は、活性成分として、製薬上有効な量の前記ペプチドを少なくとも1つ含んでなる、医薬組成物に関する。
【背景技術】
【0002】
腫瘍は多様で不均質であるが、いずれも際限なく増殖する能力を共有する。抑制されたアポトーシス感受性と結びついた、脱制御された細胞増殖が、腫瘍の進展を生じる最低限の必要条件となる。
【0003】
アポトーシスは、細胞がプログラム細胞死に入るプロセスであり、発生時に生じる生命維持に必要な現象であって、恒常性の維持に欠かせない。不可逆的に細胞をアポトーシスに委ねると考えられている生化学的事象は、カスパーゼ(アスパラギン酸残基の後で切断するシステインプロテアーゼ)の活性化である。アポトーシスを受ける細胞は、特徴的な形態的変化および生化学的変化を示すが、これは膜のブレッビング、細胞が丸くなること、クロマチン凝縮、DNA切断、細胞表面でのアポトーシスマーカーの発現、および抗アポトーシスシグナル伝達経路の阻害を包含する。これらすべての事象は、特異的カスパーゼ阻害剤によって阻止することができる。したがって、アポトーシスの間に観察される特徴的な変化のすべてとはいえないまでもほとんどの原因となるのは、カスパーゼ基質の切断である。
【0004】
アポトーシスの実行段階は、細胞内のカスパーゼ基質が切断されたときに、誘発される。数十ものカスパーゼ基質が同定され、そのリストは確実に増え続けている(Earnshaw W. C. etal.,"Mammalian caspases: structure, activation, substrates, and functions during apoptosis"Annu. Rev.Biochem. 68,383, 1999)。ひとたび切断されると、カスパーゼ基質は、カスパーゼ活性の増幅、DNAの断片化、核の分解などといった、アポトーシスの際に観察される生化学的および形態学的事象を媒介する。
【0005】
さらに、マイトジェン活性化プロテインキナーゼ(MAPK)経路がアポトーシスを正または負に調節することが明らかになった(Jarpe M. B. et al., "Anti-apoptotic versus pro-apoptotic signal transduction : checkpoints and stop signs along the road to death" Oncogene, 17,1475, 1998; Widmann C. et al., "Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human" Physiol. Rev. 79,143,1999)。このことから、アポトーシス性カスパーゼが、MAPKを調節する、および/またはMAPK経路の成分である、シグナル伝達タンパク質の一部を標的とする理由が説明できると思われる(Widmann C. et al., "Caspase- dependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti- apoptotic signals" J. Biol. Chem. 273, 7141, 1998)。これらのタンパク質にはMEKK1, PAK2, Mst1およびRasGAPがある。
【0006】
最近、Yang and Widmann、(Yang J. -Y. and Widmann C., "Antiapoptotic signaling generated by caspase-induced cleavage of RasGAP" Mol. Cell. Biol. , 21,5346,2001 ; "A subset of caspase substrates functions as the Jekylland Hyde of apoptosis "Eur. Cytokine Netw., 13,387, 2002a ; "The RasGAP N-terminal fragment generated by caspase cleavage protects cells in aRas/PI3K/Akt-dependent manner that does not rely on NFkappa B"J. Biol. Chem., 277,14641,2002b)は、Ras およびRho GTP結合タンパク質の制御因子であるRasGAPが、カスパーゼによる切断の程度に応じて、抗アポトーシス性シグナルおよびアポトーシス促進性シグナルをいずれも誘導することができるため、異例のカスパーゼ基質であることを示した。かれらは、カスパーゼ活性が低レベルである場合、RasGAPは455位で切断され、N末端配列(配列N)およびC末端配列(配列C)を生成することを示した。
【0007】
配列Cは全長RasGAPではないが、核濃縮の出現、カスパーゼ3の活性化およびPARPの切断を引き起こす能力から評価されるように、Hela細胞において強力なアポトーシス反応を引き起こした。
【0008】
同じ研究において、著者らはまた、配列Nが細胞死を促進するのではなく、むしろカスパーゼ活性化の下流でアポトーシスの全般的なブロッカーとなるように見えることを示した。カスパーゼ活性が高レベルの場合、配列Nがアポトーシスを妨げる能力は、配列Nが157位で切断されると抑制される。この後者の切断事象は、配列Nとは対照的に細胞を感作することが示されているN1およびN2の2つの配列を生成するが、これらは、癌治療の化学療法で使用される薬物、シスプラチンによって誘導されるアポトーシスに向けて高いカスパーゼ活性を生じることができる。
【0009】
しかしながら、Leblancら(Leblanc V. et al., "Ras-GTPase activating protein inhibition specifically induces apoptosis of tumour cells" Oncogene, 18, 4884,1999)が明らかにしたように、RasGAPのN2配列のSH3ドメインに対するモノクローナル抗体を、このタンパク質を特異的に阻害するために注入すると、癌細胞においてアポトーシスを誘導する。特許出願WO99/65947(Psrlerら)から、RasGAP SH3ドメイン結合タンパク質、G3BPに対するモノクローナル抗体が、G3BPが特異的に過剰発現している癌細胞において、アポトーシスを誘導することも知られている。
【0010】
これらの結果は、RasGAP SH3ドメインを介して増殖を制御するRasGAP経路が、ある種の癌細胞の生存に必須であること示すと思われる。これらの発見は、YangおよびWidmannにより得られた結果とは対照的であると思われるので、RasGAP SH3ドメインは細胞におけるアポトーシスの誘導と制御において極めて相反する二面的機能を有するという結論をもたらす。
【0011】
化学療法は、単独で、または他の治療法(たとえば、放射線療法)と併用して、現在、最も一般的で有効な癌の治療手段である。癌を治療する化学療法で使用される薬物の有効性は、癌細胞を死滅させる能力に依存する。しかしながら、このような薬物の使用には限界があり、その限界は薬物が癌細胞ではない正常細胞にも悪影響を及ぼす可能性があるという事実によってもたらされるが、それは、こうした薬物が癌細胞のみならず、癌細胞でない正常細胞においても、特に迅速に分裂する細胞において、カスパーゼへの強い刺激を引き起こすためである。
【0012】
したがって、臨床医の課題は、腫瘍を取り除くには十分に高用量であるが、患者に脱毛、悪心嘔吐、心臓毒性および二次癌といった深刻な副作用を引き起こすほどには高くない薬物投与量を選択することである。
【0013】
薬物の癌細胞に対する選択性を向上させることは、明らかに化学療法処置の有効性を高め、それによって薬物投与量を低下させることができる。このことはまた、上記の深刻な副作用を可能な限り減少させる結果につながることになる。
【0014】
したがって、本発明の目的は、薬物と併用して、癌の治療もしくは予防を目的とする、上記の欠点のない改善されたアプローチを提供することである。
【0015】
発明の概要
上記目的は、選択的に癌細胞を死滅させる薬物の能力を高める、基本的にRasGAPタンパク質のN2配列、その断片、またはその変異体からなるペプチドを提供することによって達成された。
【0016】
さらに、本発明は、ペプチドをコードする精製単離された核酸配列、少なくとも1コピーの精製単離された核酸配列を含んでなる発現ベクター、ならびにペプチド、単離精製された核酸配列および/または発現ベクターを含有する、真核もしくは原核宿主細胞を提供する。
【0017】
本発明はさらに、活性成分として、少なくとも1つの本発明のペプチドを、製薬上有効な量含んでなる医薬組成物を提供する。
【0018】
発明の詳細な説明
本明細書で使用される場合、「ペプチド」、「タンパク質」、「ポリペプチド」、「ポリペプチド性」、および「ペプチド性」という用語は相互に交換可能なものとして使用され、隣接するアミノ酸残基のαアミノ基とカルボキシル基の間のペプチド結合によって他のアミノ酸と結合した一連のアミノ酸残基を示す。
【0019】
Ras およびRho GTP結合タンパク質の制御因子であるRasGAPは、カスパーゼによる切断の程度に応じて、抗アポトーシス性シグナルおよびアポトーシス促進性シグナルをいずれも誘導することができるため、異例のカスパーゼ基質である。カスパーゼ活性が低レベルである場合、RasGAPは455位で切断され、N末端断片(断片N、約56kD)およびC末端断片(断片C、約64kD)を生成する。断片Nはカスパーゼ活性化の下流でアポトーシスの一般的なブロッカーとなるように思われる(Yang J. -Y. and Widmann C., Mol. Cell. Biol., 21,5346, 2001およびJ. Biol. Chem., 277,14641, 2002b)。カスパーゼ活性が高レベルの場合、断片Nは157位でさらに切断されて、2つの断片、N1(アミノ酸1〜157)およびN2(アミノ酸158-455)を生成する。
【0020】
「癌細胞」は、動物においてin vivoに生じる細胞であって、望ましくない無秩序な細胞増殖、または組織の異常な持続性もしくは異常な侵入をなしうる細胞を意味する。in vitroで、この用語はまた、永久に不死化された樹立細胞培養系である細胞株を指し、これは適当な新培地および空間を与えると無期限に、しかも無秩序に増殖する。
【0021】
「薬物」という用語は、哺乳動物細胞、好ましくはヒト細胞を死滅させることができる薬物を指す。さまざまな起源の、さまざまな作用機序をもつ、いくつかに分類される薬物がある。
【0022】
本発明の薬物は、宿主の生物学的プロセスに由来する、またはそのプロセスを有益に調節する作用物質に関わる。インターフェロン、腫瘍増殖因子、腫瘍壊死因子、GM-CSFおよびG-CSFといった増殖因子、ならびにインターロイキン2,インターロイキン6、インターロイキン7およびインターロイキン12といったインターロイキン類が、癌治療に現在使用されている、上記のような生物学的薬物の例である。
【0023】
本発明の薬物は、さらに、DNAを損傷し、および/または、細胞が増殖できないようにする、遺伝毒性物質のような作用物質にも関係すると考えられる。遺伝毒性物質は、アルキル化剤、代謝拮抗物質、DNA切断物質、DNA結合物質、トポイソメラーゼ毒および紡錘体毒を含んでなる一群から選択することができる。
【0024】
アルキル化剤の例としては、ロムスチン、カルムスチン、ストレプトゾシン、メクロレタミン、メルファラン、ウラシルナイトロジェンマスタード、クロラムブシル、シクロスファミド、イホスファミド、シスプラチン、カルボプラチン、マイトマイシン、チオテパ、ダカルバジン、プロカルバジン、ヘキサメチルメラミン、トリエチレンメラミン、ブスルファン、ピポブロマン、ミトタンおよび他のプラチナ誘導体がある。
【0025】
DNA切断物質はたとえばブレオマイシンである。
【0026】
トポイソメラーゼ毒は、トポテカン、イリノテカン、カンプトテシン・ナトリウム塩、ダウノルビシン、ドキソルビシン、イダルビシン、ミトキサントロン、テニポシド、アドリアマイシンおよびエトポシドを含んでなる一群から選択することができる。
【0027】
DNA結合物質の例はダクチノマイシンおよびミトラマイシンであるが、紡錘体毒は、ビンブラスチン、ビンクリスチン、ナベルビン、パクリタキセルおよびドセタキセルを含んでなる一群から選択することができる。
【0028】
薬物として代謝拮抗剤も使用することができるが、これは、下記の化合物の中から選択することができる:メトトレキサート、トリメトレキサート、ペントスタチン、シタラビン、ara-CMP、リン酸フルダラビン、ヒドロキシ尿素、フルオロウラシル、フロクスウリジン、クロロデオキシアデノシン、ゲムシタビン、チオグアニンおよび6-メルカプトプリン。
【0029】
好ましくは遺伝毒性物質、より好ましくはシスプラチン、ミトキサントロン、およびアドリアマイシンが、本発明において薬物として使用される。
【0030】
これらの薬物は、単独で、または相互に併用して使用することができる。2つ以上の薬物を使用する場合には、薬物の有用な組み合わせを決定することは、十分に当業者の能力の範囲内であって、たとえば死滅させるべき癌細胞によって決まることになるだろう。
【0031】
「増強する」という用語は、細胞を死滅させることができる薬物の効果を増大させるペプチドの能力を指す。この能力は、in vitroで、たとえば、核濃縮(アポトーシス細胞のマーカー)を示す細胞の数を記録することによって、ペプチドを含有し少なくとも1つの薬物で処理された細胞のアポトーシスの割合を測定することにより、評価することができる。典型的には、その結果を、前記ペプチドを含有しない薬物処理細胞から得られた結果と比較する。所定の濃度での細胞のアポトーシスを2倍以上増加させるペプチド、または一定のアポトーシス反応を誘導する薬物の用量を1/2以下に減少させるペプチドは、薬物が細胞を死滅させる能力を増強すると見なすことができるであろう。
【0032】
本明細書で使用される場合、「選択的に」という用語は、本発明のペプチドが、所定の濃度で細胞を死滅させる薬物の能力を、癌細胞においては特異的に増強するが、非癌細胞では驚くべきことに増強しないことを意味する。
【0033】
ペプチドが薬物の細胞死滅能力を癌細胞において特異的に増強するin vitroでの薬物の濃度範囲は、通常、使用される薬物によって決まる。たとえば、遺伝毒性物質を使用する場合、通常、in vitroの薬物濃度は、0.1から100μMであり、好ましくは0.15から30μMである。
【0034】
PasGAPタンパク質のN2配列は、好ましくはヒト由来であって、図2Aに示すように2つのSH2ドメインおよび1つのSH3ドメインを含む297アミノ酸からなる36kDタンパク質を指す。
【0035】
一般に、Srcホモロジー2(SH2)ドメインは、リン酸化チロシンの認識に関わるのに対して、Srcホモロジー3(SH3)ドメインは、細胞骨格の組織化に関係するシグナル伝達に関わるタンパク質をしばしば示唆する。
【0036】
「断片」は、RasGAPタンパク質のN2配列よりも長さとして少ないアミノ酸を含有する配列を指す。この配列は、その元になった天然配列と同じ性質を示す限り使用することができる。こうした配列は、RasGAPタンパク質のそれぞれのN2配列よりも長さが90%未満、好ましくは60%未満、特に30%未満のアミノ酸を含有することが好ましい。
【0037】
本発明はまた、RasGAPタンパク質のN2配列の変異体も包含する。「変異体」という用語は、天然配列のペプチドとはある程度異なるアミノ酸配列を有するペプチドを指すが、このアミノ酸配列は、保存的アミノ酸置換によって天然配列から変化したアミノ酸配列であって、保存的置換によって1つまたは複数のアミノ酸が同じ性質および同じ高次構造上の役割を有する別のアミノ酸によって置換される。アミノ酸配列変異体は、天然アミノ酸配列のアミノ酸配列内のある位置に、置換、欠失および/または挿入を有する。保存的アミノ酸置換は、本明細書では、下記の5群のうちの1群の範囲内での交換と定義される:
I.小、脂肪族、非極性もしくはわずかに極性の残基:Ala, Ser, Thr, Pro, Gly
II.極性、正電荷を有する残基:His, Arg, Lys
III.極性、負電荷を有する残基:およびそのアミド:Asp, Asn, Glu, Gln
IV.大、芳香族残基:Phe, Tyr, Trp
V.大、脂肪族、非極性残基:Met. Leu. Ile, Val, Cys。
【0038】
N2配列、ならびにその断片および変異体は、当技術分野で知られている様々な方法および技術、たとえば、化学合成、もしくはManiatis et al. 1982, Molecular Cloning, A laboratory Manual, Cold Spring Harbor Laboratoryに記載の遺伝子組換え技術によって調製することができる。
【0039】
好ましくは、RasGAPタンパク質のN2配列の断片は、N2配列のSH3ドメイン、その一部もしくはその変異体のアミノ酸配列を含んでなる。
【0040】
出願人らは、驚くべきことに、RasGAPタンパク質のN2配列の、さらに短い配列の特徴を明らかにしたが、その配列は、短くても、選択的に癌細胞を死滅させる薬物の能力を増強し、しかもより容易に合成できるという利点も有するものである。このような部分配列を分離できるかどうかを評価するために、出願人らは断片Nの一連のトランケート型を図2Aに示すように生成し、癌細胞株において薬物が誘導するアポトーシスを増強するそれらの能力を測定した(図2B)。N2配列からのこれらの部分配列をベクターにクローニングして癌細胞株(HeLa)にトランスフェクトした。
【0041】
本出願人らは、空の構築物またはN2のSH2ドメインのみをコードする構築物でトランスフェクトされたHeLa細胞が、シスプラチン誘導細胞死を増強しないことを明らかにした。これに対して、SH3ドメインを含有する構築物を発現する細胞は、図2Bに示すように、シスプラチン誘導アポトーシスのさらなる増強を示した。
【0042】
そこで本出願人らは、最小の増強ペプチドを同定する目的で、SH3ドメインにおいてトランケーションを次々に進めた。これらの構築物のすべて、またはN2配列の部分配列(図2A)は、10アミノ酸の長さのペプチドをコードする最も短い配列(317-326)を含めて、シスプラチンのHeLa細胞死滅能力を増強した(図2B)。断片N2の細胞死増強特性は完全なSH3ドメインを必要とせず、短いペプチド性配列のようなSH3ドメインの部分配列によって媒介されることを、これらの結果は示している。
【0043】
SH3ドメインの一部分、またはその変異体は、SH3ドメインのアミノ酸配列のうち、好ましくは70個以下の、より好ましくは30個以下の、もっとも好ましくは10個以下のアミノ酸を含有する。
【0044】
詳細には、本発明に含まれるのは、表1のDNA配列によってコードされるアミノ酸配列である、SH3ドメインの部分配列である。
【表1】
【0045】
N2配列のSH3ドメインの部分配列が配列番号4(RasGAP317-326)である場合、ヒトにおいて前記配列番号4によりコードされる、結果として得られるアミノ酸配列はWMWVTNLRTDである。異なる種との比較から、異なるアミノ酸が存在することが明らかになったが、それは表2に示すように種の間で保存されている。
【表2】
【0046】
種の間で保存されているアミノ酸配列は、太字下線付きの活字の残基として表わされるのに対して、Xは、N2のSH3ドメインのこの10アミノ酸部分の発明的特性を損なうことなく、保存的もしくは非保存的アミノ酸置換によって変えることができるアミノ酸残基に対応する。
【0047】
N2のヒトSH3ドメインのこの10アミノ酸部分からなる、これらのペプチド性変異体、および特にアラインメント配列WXWVTXXRTXもまた、本発明の範囲に含まれるが、それらは、ある程度、天然型配列のペプチドとは異なるアミノ酸配列を有するペプチドを表し、それは、保存的もしくは非保存的アミノ酸置換によって天然型配列WMWVTNLRTDから変化したアミノ酸配列であって、それにより1つもしくは複数のアミノ酸残基が同じ特性および高次構造上の役割を有する別の残基によって置換されている。
【0048】
通常、基本的にRasGAPタンパク質のN2配列、その断片、またはその変異体からなるペプチドは、本発明で明らかにされるように、細胞内のペプチドの蓄積を増加させる薬剤と結合している。
【0049】
こうした薬剤は、たとえば、治療薬と結合したトランスフェリンの、膜トランスフェリン受容体を介したエンドサイトーシスのような、受容体を介したエンドサイトーシスを引き起こす化合物(Qian Z. M. et al.,"Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway "Pharmacological Reviews, 54,561, 2002)とすることができ、または、たとえば、プロテインキナーゼC(Ioannides C. G. et al., "Inhibition of IL-2 receptor induction andIL-2 production in the human leukemic cell line Jurkat by a novel peptide inhibitor of proteinkinase C" Cell Immunol., 131,242, 1990)およびプロテインチロシンホスファターゼ(Kole H. K. et al., "A peptide-based protein-tyrosine phosphatase inhibitor specifically enhances insulin receptor function in intact cells" J. Biol. Chem. 271,14302, 1996)のペプチドインヒビターの細胞内デリバリーにすでに使用されている、デカン酸、ミリスチン酸およびステアリン酸といった脂肪酸の一群から、もしくはペプチドから選択することができる細胞膜透過性キャリアとすることができる。
【0050】
細胞膜透過性キャリアがペプチドである場合、アルギニンリッチのペプチドであることが好ましい。Futakiら(Futaki S. et al., "Arginine- rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery" J. Biol. Chem., 276, 5836, 2001)において、細胞膜透過性キャリアペプチド中のアルギニン残基数がインターナリゼーションの方式に相当の影響を及ぼすこと、ならびにインターナリゼーションに最適なアルギニン残基数が存在すると思われ、それは好ましくはアルギニン7残基以上であることが、最近明らかになった。
【0051】
本発明のペプチドは、通常、スペーサーによって細胞膜透過性キャリアと結合する。この場合、細胞膜透過性キャリアはペプチドであることが好ましい。
【0052】
通常、アルギニンリッチペプチドは、HIV-TAT48-57ペプチド、FHV-coat35-49ペプチド、HTLV-II Rex4-16ペプチドおよびBMV gag7-25ペプチドを含んでなる一群から選択される。好ましくは、アルギニンリッチペプチドはHIV-TAT48-57ペプチドとする。
【0053】
HIV-TAT48-57ペプチドが、たとえばRasGAP317-326のよなRasGAP配列と結合する場合、2つのグリシン残基をTATとRasGAP配列の間にスペーサーとして挿入して柔軟性を与える。
【0054】
天然ペプチド(L型)に固有の問題は天然プロテアーゼによる分解であるので、ペプチドのD型および/または「レトロ‐インベルソ(retro-inverso)型異性体」を含めるように本発明のペプチドを調製してもよい。
【0055】
この場合、本発明のペプチドの短い断片および変異体のレトロ‐インベルソ型異性体を調製する。
【0056】
したがって、ペプチドを自然のタンパク質加水分解から保護することは、特異的なヘテロ二価もしくはヘテロ多価化合物の有効性を高めるはずである。天然プロテアーゼによる分解から保護されるので、非レトロ‐インベルソ含有アナログより高い生物学的活性がレトロ‐インベルソ含有ペプチドについて予測される。さらに、これらのペプチドは高い安定性ならびに低い免疫原性を示すことが明らかになった(Sela M. and Zisman E. ,"Different roles of D-amino acids in immune phenomena "FASEB J. 11,449, 1997)。
【0057】
レトロ‐インベルソペプチドは、たとえばSela and Zisman, (1997)に記載の既知の配列を有するペプチドについて調製される。
【0058】
「レトロ‐インベルソ型異性体」は、配列の方向を逆転し、各アミノ酸のキラリティーも反転させた、直鎖状ペプチドの異性体を意味する;したがって末端鎖相補性は存在し得ない。
【0059】
また本発明は、ペプチドの修飾(通常は一次配列を変更しない)も包含するが、そうした修飾には、in vivo もしくはin vitroでのペプチドの化学的な誘導体化、たとえば、アシル化もしくはカルボキシル化がある。グリコシル化修飾も含まれるが、たとえばこうした修飾は、合成およびプロセシングのときに、または追加のプロセッシングステップにおいて、グリコシル化パターンを変更することによってなされ、それは、たとえば、ペプチドをグリコシル化に影響を与える酵素(たとえば、哺乳類のグリコシル化酵素もしくは脱グリコシル化酵素)と接触させることによる。リン酸化されたアミノ酸残基、たとえばホスホチロシン、ホスホセリン、もしくはホスホスレオニンを有する配列も含まれる。
【0060】
本発明はまた、1つもしくは複数のペプチド結合が、ペプチダーゼによる切断を受けにくい、代替型の共有結合で置換されたアナログ(ペプチドミメティック)を包含する。被験体に注入した後のペプチドのタンパク質分解が問題となる場合、特に感受性のペプチド結合を切断されないペプチドミメティックで置換することによって、その結果得られたペプチドはいっそう安定となり、したがって活性物質としてより有用なものとなる。このようなミメティック、およびそれをペプチドに取り入れる方法は、当技術分野においてよく知られている。
【0061】
アミノ末端保護基も有用であって、たとえば、t-ブチルオキシカルボニル, アセチル, エチル, スクシニル,メトキシスクシニル, スベリル, アジピル, アゼライル(azelayl), ダンシル, ベンジルオキシカルボニル, フルオレニルメトキシカルボニル,メトキシアゼライル(methoxyazelayl), メトキシアジピル, メトキシスベリルおよび2,4-ジニトロフェニルがある。ペプチドの、荷電したアミノおよびカルボキシ末端を保護することには、疎水性の細胞膜を通って細胞内に入るペプチドの通過を向上させるという追加の利点もある。
【0062】
本発明に基づいて、組換え技法を用いて、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドを調製する場合には、そのポリペプチドをコードする核酸配列を使用することが好ましい。組換え技法を実施する方法に関しては、たとえば、Maniatis et al. 1982, Molecular Cloning, A laboratory Manual, Cold Spring Harbor Laboratoryおよび商業的に利用可能な方法を参照されたい。
【0063】
したがって、本発明はまた、上記の、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする、精製単離された核酸配列に関する。
【0064】
「精製および単離された核酸もしくは核酸配列」は、本発明のペプチドをコードする核酸配列、または基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする核酸が本発明に従って存在する状態を表す。
【0065】
本発明に含まれる精製および単離された核酸もしくは核酸配列は、DNA, RNA, またはDNA/RNAハイブリッドとすることができる。
【0066】
本明細書で使用されるDNAは任意のポリデオキシヌクレオチド配列であって、たとえば、二本鎖DNA、一本鎖DNA、二本鎖DNAであってその一本鎖もしくは二本鎖ともが2つ以上の断片で構成されるDNA、二本鎖DNAであってその一本鎖もしくは二本鎖ともが中断されていないホスホジエステル骨格を有するDNA、1つもしくは複数の一本鎖部分および1つもしくは複数の二本鎖部分を含有するDNA、二本鎖DNAであってそのDNA鎖が完全に相補的であるDNA、二本鎖DNAであってそのDNA鎖が一部分だけ相補的であるDNA、環状DNA、共有結合DNA、直鎖状DNA、共有架橋結合DNA、cDNA、化学合成されたDNA、半合成DNA、生合成DNA、自然分離されたDNA、酵素消化されたDNA、剪断されたDNA、標識されたDNA、たとえば放射標識DNAおよび蛍光色素標識DNA、1つもしくは複数の天然に存在しないタイプの核酸を含有するDNAを含める。
【0067】
基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードするDNA配列は、標準的な化学技術、たとえば、ホスホトリエステル法によって、または自動化合成法およびPCR法によって合成することができる。
【0068】
本発明により、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする精製および単離されたDNA配列は、酵素による技法によって作製することもできる。したがって、制限酵素は所定の認識配列の位置で核酸分子を切断するが、その制限酵素を用いて核酸配列を、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードするDNA(またはRNA)のような、核酸配列を含有するより大きい核酸分子から単離することができる。
【0069】
本発明はまた、ポリリボヌクレオチド(RNA)の形の核酸も包含し、たとえば、一本鎖RNA、cRNA、二本鎖RNA、二本鎖RNAであってその一本鎖もしくは二本鎖ともが2つ以上の断片で構成されるRNA、二本鎖RNAであってその一本鎖もしくは二本鎖ともが中断されていないホスホジエステル骨格を有するRNA、1つもしくは複数の一本鎖部分および1つもしくは複数の二本鎖部分を含有するRNA、二本鎖RNAであってそのRNA鎖が完全に相補的であるRNA、二本鎖RNAであってそのRNA鎖が一部分だけ相補的であるRNA、共有架橋結合RNA、酵素消化されたRNA、剪断されたRNA、mRNA、化学合成されたRNA、半合成RNA、生合成RNA、自然分離されたRNA、標識されたRNA、たとえば放射標識RNAおよび蛍光色素標識RNA、1つもしくは複数の天然に存在しないタイプの核酸を含有するRNAを含める。
【0070】
核酸として使用するのに好ましいのは、配列番号1、配列番号2、配列番号3、または配列番号4からなる一群から選択される、精製および単離されたDNA配列である。
【0071】
本発明は、前記配列の変異体も包含し、それは保存的ヌクレオチド置換によって基準配列から変化したヌクレオチド配列であるが、その置換によって1つもしくは複数のヌクレオチドが同様の性質を持つ別のヌクレオチドによって置換されている。
【0072】
本発明はまた、開示された精製および単離された核酸配列の対立遺伝子変異体を包含する;前記変異体は、すなわち、単離精製された核酸の、天然に存在する代替形態であり、それらは精製単離された核酸配列によってコードされるペプチドと同一、相同、もしくは関連するペプチドをコードする。あるいはまた、天然に存在しない変異体を変異誘導法もしくは直接合成によって作製することができる。
【0073】
基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする前記の精製および単離された核酸配列は、さらに、細胞膜透過性キャリアペプチドをコードするヌクレオチド配列を含んでなることができる。
【0074】
さらにもう一つの本発明の関心事は、上記の、基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドをコードする、単離および精製された核酸配列を少なくとも1コピー含んでなる発現ベクターを提供することである。好ましくは、本発明のペプチドをコードする単離および精製された核酸配列は、DNAである。
【0075】
本明細書で使用される場合、「ベクター」、「プラスミド」、および「発現ベクター」は相互に交換可能なものとして使用されるが、これは、最も広く使用されるベクターの形がプラスミドであるためである。
【0076】
ベクターはさらに、本発明の細胞膜透過性キャリアペプチドをコードするヌクレオチド配列も含んでなることができる。発現ベクターの選択は、当技術分野でよく知られているように、まさに、求められる機能上の特性次第であり、たとえば、ペプチドの発現、ならびに形質転換されるべき、もしくはトランスフェクトされるべき宿主細胞によって決まる。
【0077】
それに加えて、発現ベクターはさらに、精製および単離されたDNA配列に機能しうるように連結されたプロモーターを含んでなることができる。このことは、連結された、本発明のペプチドをコードする単離および精製されたDNA配列が、適当な制御配列のコントロール下にあり、その制御配列によって、挿入された単離精製DNA配列の発現、すなわち転写および翻訳が可能となることを意味する。
【0078】
本明細書で使用される場合、「プロモーター」という用語は、当技術分野で知られている任意の追加的な制御配列、たとえば、ポリペプチドの発現のために通常使用されるプロモーターおよび/またはエンハンサー、ポリアデニル化部位、ならびにスプライス部位を表すが、あるいは追加して1つもしくは複数の別個のターゲティング配列を包含してもよく、状況に応じて選択マーカーをコードしていてもよい。使用可能なプロモーターは、そうしたプロモーターが宿主細胞に適合するならば、たとえば、ポリオーマウイルス、アデノウイルス(たとえばアデノウイルス2)、パピローマウイルス(たとえばウシパピローマウイルス)、トリ肉腫ウイルス、サイトメガロウイルス(たとえばマウスもしくはヒトサイトメガロウイルス最初期プロモーター)、レトロウイルス、B型肝炎ウイルスおよびサルウイルス40(たとえばSV40初期および後期プロモーター)といったウイルスのゲノム由来のプロモーター、またはアクチンプロモーター、免疫グロブリンプロモーターもしくは熱ショックプロモーターといった異種哺乳類プロモーターから得られるプロモーターがある。
【0079】
使用可能なエンハンサーは、たとえば、哺乳類遺伝子由来の既知のエンハンサー配列(グロビン、エラスターゼ、アルブミン、αフェトプロテインおよびインスリン)または真核細胞ウイルス由来のエンハンサー、たとえば、SV40エンハンサー、サイトメガロウイルス初期プロモーターエンハンサー、ポリオーマおよびアデノウイルスエンハンサーである。
【0080】
本発明のDNA配列の発現には、さまざまな宿主/発現ベクターの組み合わせを用いることができる。有用な発現ベクターは、たとえば、染色体性、非染色体性および合成DNA配列のセグメントからなっていてもよい。適当なベクターとしては、SV40から作製されたベクター、ならびに既知の細菌プラスミド、たとえば、大腸菌(E. coli)プラスミドcol E1、pCR1、pBR322、pcDNA3、pMB9およびそれらから作られたプラスミド、RP4のようなプラスミド;ファージDNA、たとえば、ファージXから作られた多数のDNA、たとえばNM989、および他のファージDNA、たとえばM13および繊維状一本鎖ファージDNA;酵母プラスミド、たとえば、2μプラスミドもしくはそれから作られたプラスミド;真核細胞において有用なベクター、たとえば、昆虫もしくは哺乳類細胞において有用なベクター;プラスミドおよびファージDNAの組み合わせから作られたベクター、たとえば、ファージDNAもしくは他の発現制御配列を用いるように改変されたプラスミド;などがある。
【0081】
もっとも好ましくは、発現ベクターはpcDNA3である。
【0082】
本発明のもう一つの関心事は、本発明のペプチド、単離精製された本発明の核酸配列、および/または本明細書に記載の発現ベクターを含有する真核もしくは原核宿主細胞を提供することである。
【0083】
精製単離された本発明のDNA配列を含んでなる発現ベクターによる、適当な真核もしくは原核宿主細胞の形質転換またはトランスフェクションは、概して使用するベクターの種類によって決まる公知の方法によって達成される。こうした方法に関しては、たとえば、Maniatis et al. 1982, Molecular Cloning, A laboratory Manual, Cold Spring Harbor Laboratoryおよび商業的に利用可能な方法を参照されたい。「トランスフェクトされた細胞」、「形質転換された細胞」もしくは「トランスフェクト/形質転換された細胞」という用語は、細胞外DNAがその中に導入され、したがって細胞外DNAを保有する細胞を意味する。核酸が染色体組み込み体として、または染色体外エレメントとして複製可能であるように、DNAを細胞内に導入することができる。
【0084】
基本的に、RasGAPタンパク質のN2配列、その断片もしくはその変異体からなるペプチドは、状況に応じて、本明細書に記載のように細胞内でのペプチドの蓄積を増加させる作用物質と結合しているが、そのペプチドは、細胞発現系において、好ましくは組換えによって作製される。
【0085】
さまざまな単細胞の宿主細胞が、本発明のDNA配列の発現に有用である。これらの宿主には、既知の真核および原核宿主、たとえば、大腸菌、Pseudomonas, Bacillus, Streptomyces、酵母のような真菌、および動物細胞、たとえば、CHO、YB/20、NSO、SP2/0、Rl.1、B-WおよびL-M細胞、アフリカミドリザル腎細胞(例、COS1、COS 7、BSC1、BSC40およびBMT10)、昆虫細胞(例、Sf9)、およびヒト細胞、ならびに組織培養植物細胞を含めることができる。好ましくは、宿主細胞は細菌細胞であって、大腸菌細胞がさらに好ましい。
【0086】
本発明はまた、活性成分として、製薬上有効な量の少なくとも1つの記載されたペプチドを、状況に応じて製薬上許容される担体、希釈剤および補助剤と組み合わせて含んでなる、医薬組成物に関する。
【0087】
「製薬上有効な量」は、ヒトもしくは動物に投与したときに検出可能な薬理学的および/または生理学的な効果を生じる化学物質もしくは化合物を指す。
【0088】
それぞれの製薬上有効な量は、治療すべき個別の患者、治療すべき疾患および投与方法によって決まる。さらに、製薬上有効な量は、使用される具体的なペプチド次第であって、特にペプチドが記載のように薬物を追加して含有するか否かによる。治療は通常、数時間、数日もしくは数週間の間隔をおいた、医薬組成物の複数回投与を含んでなる。ポリペプチドの製薬上有効な投薬量単位は、治療を受ける患者の体重kg当たり0.001ngから100μgの範囲である。
【0089】
好ましくは、医薬組成物は、少なくとも1つの本明細書に記載のペプチドに加えて、1つもしくは複数の製薬上許容される担体、希釈剤および補助剤を含有することができる。
【0090】
許容される担体、希釈剤および補助剤は、医薬として使用できる製剤への活性化合物の加工を助けるが、これらは使用される投与量および濃度で受容者に対して無毒であって、バッファー、たとえばリン酸、クエン酸および他の有機酸;アスコルビン酸およびメチオニンを含めた抗酸化剤;防腐剤(たとえば、塩化オクタデシルジメチルベンジルアンモニウム;塩化ヘキサメトニウム;塩化ベンザルコニウム、塩化ベンゼトニウム;フェノール、ブチルもしくはベンジルアルコール;メチルもしくはプロピルパラベンといったアルキルパラベン類;カテコール;レゾルシノール;シクロヘキサノール;3-ペンタノール;およびm-クレゾール);低分子量(約10残基未満)ポリペプチド;タンパク質、たとえば、血清アルブミン、ゼラチン、もしくは免疫グロブリン;親水性ポリマー、たとえばポリビニルピロリドン;アミノ酸、たとえばグリシン、グルタミン、アスパラギン、ヒスチジン、アルギニンもしくはリジン;単糖類、二糖類、および他の炭水化物、たとえばグルコース、マンノースもしくはデキストリン;キレート剤、たとえばEDTA;糖類、たとえばショ糖、マンニトール、トレハロースもしくはソルビトール;塩形成対イオン、たとえばナトリウム;金属錯体(たとえば亜鉛タンパク質錯体);および/または非イオン性界面活性剤、たとえばTWEEN(登録商標)、PLURONICS(登録商標)、もしくはポリエチレングリコール(PEG)を包含する。
【0091】
医薬組成物の投与法は、全身性または局所性とすることができる。たとえば、このような組成物の投与は、さまざまな非経口経路、たとえば皮下、静脈内、皮内、筋肉内、腹腔内、鼻腔内、経皮、頬側経路、または埋め込み装置によることができるが、蠕動によって送達することもできる。
【0092】
本明細書に記載のペプチドを活性成分として含んでなる医薬組成物を、生体吸収性マトリックスに組み入れ、またはそれに浸透させることもできるが、そのマトリックスはマトリックスの懸濁液、ゲルもしくは固体の形で投与される。加えて、マトリクスはバイオポリマーから構成されていてもよい。
【0093】
徐放性製剤を調製することができる。徐放性製剤の適当な例としては、抗体を含有する固体疎水性ポリマーの半浸透性マトリックスがあり、このマトリックスはフィルムもしくはマイクロカプセルといった成型品の形をとる。徐放性マトリックスの例には、ポリエステル、ハイドロゲル(たとえば、ポリ(2-ヒドロキシエチル-メタクリレート)もしくはポリ(ビニルアルコール))、ポリラクチド(米国特許第3,773,919号)、L-グルタミン酸およびγ-エチル-L-グルタメートのコポリマー、非分解性エチレン−ビニルアセテート、分解性乳酸-グリコール酸コポリマー、たとえばLUPRON DEPOT(商標名)(乳酸-グリコール酸コポリマーおよび酢酸ロイプロリドからなる注射用マイクロスフェア)、ならびにポリ-D-(-)-3-ヒドロキシ酪酸がある。
【0094】
in vivo投与に使用される製剤は無菌でなければならない。これは、たとえば、無菌濾過膜を通す濾過によって容易に達成される。
【0095】
当然のことながら、本発明のペプチドの適切な投与量は、投与を受ける人の年齢、性別、健康状態および体重、もしあれば併用治療の種類、ならびに求める効果の内容によって左右される。
【0096】
適当な剤形は、疾患、ペプチド、および投与法によって決まる;可能性としては、錠剤、カプセル剤、トローチ剤、歯科用ペースト、座剤、吸入剤、溶液、軟膏剤および非経口デポー製剤が含まれる。
【0097】
ペプチドのアミノ酸のアミノ酸修飾も本発明に含まれるので、このことは、本発明のペプチドを水に不溶性のマトリックスもしくは他の巨大分子基剤に架橋結合するために、または溶解性、吸着性および血液脳関門を越える透過性を改善するために、有用であると考えられる。このような修飾は当技術分野ではよく知られており、あるいはまた、起こりうる望ましくないペプチドの副作用などを取り除き、もしくは弱めることができる。
【0098】
本発明の好ましい医薬組成物は、活性成分としてペプチドを含んでなるが、また別の医薬組成物は、本明細書に記載のペプチドをコードする精製単離された核酸配列を、活性成分として含有することができる。この医薬組成物は、単一の精製単離されたDNA配列、前記の精製単離されたDNA配列を含んでなる発現ベクター、または本明細書に記載の発現ベクターであらかじめトランスフェクトされ、もしくは形質転換された宿主細胞のいずれかを包含することができる。この後者の例では、宿主細胞は、抗原性の問題を回避するために、好ましくは治療を受ける患者から分離されることになる。これらの遺伝子治療および細胞治療のアプローチは、特に、医薬組成物の反復投与を必要とする患者に非常に適しているが、それは、前記の精製単離されたDNA配列、発現ベクター、または発現ベクターであらかじめトランスフェクトされ、もしくは形質転換された宿主細胞を、その後内生的にタンパク質を産生する患者の細胞に、組み入れることができるためである。
【0099】
通常、本明細書に記載の医薬組成物は、癌の治療もしくは予防に使用される。
【0100】
本発明はまた、癌の治療用もしくは予防のための薬物を製剤するための、本発明の医薬組成物の使用を包含する。
【0101】
「癌」という用語は、基本的に無秩序な細胞増殖によって特徴付けられる、哺乳類における生理的状態を指し、または表現するものである。
【0102】
通常、治療もしくは予防すべき癌は、癌腫、リンパ腫、芽細胞種、肉腫、脂肪肉腫、神経内分泌腫瘍、中皮腫、神経鞘腫、髄膜腫、腺癌、黒色腫、白血病、悪性リンパ腫、扁平上皮癌、上皮性扁平上皮癌、肺癌、小細胞肺癌、非小細胞肺癌、肺腺癌、肺扁平上皮癌、腹膜癌、肝細胞癌、胃癌、消化器癌、膵臓癌、神経膠芽腫、子宮頸癌、卵巣癌、肝臓癌、膀胱癌、肝細胞腫、乳癌、大腸癌、直腸癌、直腸結腸癌、子宮内膜癌もしくは子宮癌、唾液腺癌、腎臓癌、前立腺癌、外陰癌、甲状腺癌、肝癌、肛門癌、陰茎癌、睾丸癌、食道癌、胆道腫瘍、および頭頸部癌からなる一群から選択されることとなる。
【0103】
好ましくは、癌は、中皮種、睾丸癌もしくは膵臓癌である。
【0104】
本発明のペプチドは、概して、意図した目的を達成する量で使用される。癌の治療もしくは予防に使用するために、ペプチドもしくはその医薬組成物は、治療上有効な量投与され、または適用される。「治療上有効な量」は、症状を改善もしくは予防するのに有効な量、または治療を受ける被験体の生存を延長するのに有効な量である。治療上有効な量の決定は、特に、本明細書で提示される詳細な開示に照らして、十分、当業者に可能な範囲である。
【0105】
全身投与のために、治療上有効な量または用量は、初めにin vitroアッセイから見積もることができる。たとえば、細胞培養で決定されるIC50を含めた循環濃度範囲を達成するように、動物モデルにおいて用量を定式化することができる。そうした情報を用いて、より正確に、ヒトにおいて有効な用量を決定することができる。
【0106】
初回投与量も、たとえば動物モデルのin vivoデータから、当技術分野でよく知られた技法によって、見積もることができる。当業者は、動物データに基づいて容易にヒトに対する投与を最適化することができるであろうが、当然、治療を受ける被験体、被験体の体重、疾患の重症度、投与の方法、および処方医の判断に依存する。
【0107】
本明細書はまた、癌の治療もしくは予防を必要とする被験体に本明細書に記載の医薬組成物を治療上有効な量投与することを含んでなる、前記被験体において癌を治療もしくは予防する方法を提供する。
【0108】
治療もしくは予防することができる癌の例は、上記に記載した。好ましくは、癌は中皮種、睾丸癌もしくは膵臓癌である。
【0109】
好ましい方法において、被験体はヒト患者であり、投与されるペプチドは、癌細胞を死滅させる少なくとも1つの薬物の能力を選択的に増強するものであって、TAT-RasGAP317-326ペプチドである。
【0110】
癌細胞において選択的にアポトーシスを強める方法もまた本発明の範囲に含まれるが、その方法は、癌細胞を、本発明の少なくとも1つのペプチドおよび薬物と接触させることを含んでなる。
【0111】
癌細胞を本発明の少なくとも1つのペプチドおよび薬物と接触させることを含んでなる、選択的に癌細胞を死滅させる方法も想定される。
【0112】
癌細胞において選択的に細胞を死滅させる薬物の能力を増強するために、本明細書に記載のペプチドを使用することも想定される。
【0113】
本発明のもう一つの目的は、被験体において癌を治療もしくは予防するキットを提供することであって、該キットは、少なくとも1つの本明細書に記載のペプチドを、状況に応じて試薬および/または使用説明書と共に含んでなる。
【0114】
概して、キットは、容器、およびその容器の上もしくは容器に付随してラベルまたは添付文書を含んでなる。適当な容器には、ビン、バイアル、シリンジなどがある。容器はガラスやプラスチックといった、さまざまな材料から作製することができる。容器は、癌の治療に有効な組成物が入っており、無菌のアクセスポートを有していてもよい(たとえば、容器は静脈注射用溶液バッグ、または皮下注射針で突き刺すことができるストッパー付きバイアルとすることができる)。ラベルもしくは添付文書は、その組成物が好みの癌の治療用に使用されることを示す。
【0115】
状況に応じて、キットはさらに、上記の薬物からなる一群から選択される追加の抗ガン剤、抗-上皮成長因子受容体抗体、放射免疫治療薬、およびそれらの組み合わせを含んでなる、別個の医薬剤形を含んでなる。
【0116】
実施例
【実施例1】
【0117】
細胞およびトランスフェクション
HeLaおよびMCF-7細胞を、10%ウシ新生仔血清(Sigma ; カタログ番号N4637)を含有するRPMI 1640 (Sigma; カタログ番号8758)中で、37℃、5% CO2にて維持した。 U20S細胞は、 15% ウシ胎仔血清(Sigma; カタログ番号F7524) を含有するDMEM (Sigma ; カタログ番号5796)中で、37℃、5% CO2にて維持した。H-Meso-1細胞は10% ウシ胎仔血清を含有するRPMI 1640中で37℃、5%CO2にて維持した。HUV-EC-C細胞は、10% ウシ胎仔血清、20 ng/ml塩基性線維芽細胞成長因子(Gibco; カタログ番号13256-029)、10 ng/ml 上皮成長因子 (Gibco; カタログ番号13247-051)、10, ug/ml フィブロネクチン(Gibco; カタログ番号33016-015)を添加したヒト内皮SFM培地(Gibco; カタログ番号l1111-044)中で37℃、5%CO2にて維持した。HaCat細胞は、上皮成長因子1-53およびウシ脳下垂体抽出物(Gibco ; カタログ番号17005-075)を含有するケラチノサイトSFM培地中で37℃、5%CO2にて維持した。HeLa細胞は、既述のようにトランスフェクトした (Yang J. -Y. and Widmann C. , Mol. Cell. Biol. , 21,5346, 2001)。遺伝毒性物質処理は、6-ウェルプレートで行った。2.5x105細胞/ウェルの濃度で、処理の前日に細胞を分裂した。U20S細胞は6ウェルプレートで、カルシウム/リン酸沈澱法によってトランスフェクトした(Jordan M. et al., "Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation" Nucleic Acids Res., 24, 596, 1996)。簡単に述べると、プラスミドを90μl H20中に希釈し、10μl CaCl2 2.5 M と混合して、室温にて10分間インキュベートした。その後、100μlのHEP溶液(280 mM NaCl、10 mM KCl、1.5 mM Na2HP04、12 mM D-グルコース、50 mM HEPES)を急速に上記DNA溶液と混合し、室温にて厳密に1分間インキュベートし、最後に、細胞培養培地に移した。37℃、5%CO2にて8時間後、培地を新培地と交換し、細胞をさらに16-24時間インキュベートした後、分析した。
【0118】
化学薬品
シスプラチンおよびミトキサントロンはSigma (カタログ番号はそれぞれP4394およびM6545)から入手した。シスプラチンは終濃度100 mMとなるようにDMSO中に希釈し、-20℃で保存した。ミトキサントロンは終濃度10 mMとなるように100%エタノール中に希釈し、-80℃で保存した。アドリアマイシンはCalbiochem (カタログ番号324380)から入手した。これは水で終濃度10mMとなるように希釈し、-20℃で保存した。ヘキスト(Hoechst)33342はRoche(カタログ番号H-1399)から入手した。これは水で終濃度10mg/mlとなるように希釈して4℃にて暗所に保存した。
【0119】
ペプチド合成および標識化
HIV-TAT48-57(GRKKRRQRRR)およびTAT-RasGAP317-326(GRKKRRQRRRGGWMWVTNLRTD)ペプチドは、スイス、ローザンヌ大学生化学研究所で、FMOC法を用いて合成し、HPLCで精製して、質量分析法で検査した。
【0120】
フルオレセインイソチオシアネート(FITC)標識は、β-アラニン- GRKKRRQRRRGGWMWVTNLRTD配列について行ったが、その側鎖Fmoc保護アミノ酸は、Arg(bpf), Lys (Boc), Gln (Trt), Trp (Boc), Thr (tBu), Asn (Trt)、およびAsp (OtBu)であった。このペプチドは、0.2 mmol Rink Amide AM樹脂上でFmoc法によって段階的に合成された。ニンヒドリン試験によって合成をモニターした。β-アラニンを結合した後、ジメチルホルムアミド(DMF)中20% ピペリジンを用いてFmoc基を除去した。この段階で、FITC(4ml DMFおよび1ml N-エチルジイソプロピルアミン中、樹脂を置換する5倍過剰量)を用いてフルオレセイン基をペプチドのN末端に結合し、フルオレセイン誘導体化ペプチドを生成した。
【0121】
ペプチドは脱イオン水に終濃度1mMとなるように溶解し、追って使用するまで−20℃にて保存した。
【0122】
プラスミド
プラスミドの名称に付け加えられたdn3は、そのバックボーンプラスミドが発現ベクターpcDNA3(Invitrogen)であることを示す。構築物はすべてNアミノ末端でHA配列(MGYPYDVPDYAS)によってタグを付けた。プラスミドN2.dn3は、ヒトRasGAP断片N2をコードし、プラスミドSH2-SH3.dn3はヒトRasGAPアミノ酸158-361をコードし、プラスミドSH2.dn3はヒトRasGAPアミノ酸158-277をコードし、プラスミドSH3.dn3はヒトRasGAPアミノ酸279-361をコードする。プラスミドIκBαΔN2はNFκBの活性化を阻止するある種のIκBαをコードする (YangおよびWidmann, 2002b)。GFPタンパク質をコードするプラスミドpEGFP-Clは、Clontechから入手した。pRL-TKは、ウミシイタケ(Renilla renifornis)ルシフェラーゼをコードするベクターであるが、Promegaより入手した。prLUC は、NFκB-応答性エレメントの制御下でホタルルシフェラーゼcDNAを有するレポータープラスミドである(Yang J.-Y. and Widmann C., Mol. Cell. Biol., 21, 5346, 2001)。
【0123】
アポトーシス測定
アポトーシスは、核濃縮を示す細胞の数を記録することによって測定した。生きた細胞の核をヘキスト33342(終濃度10μg/ml)で約5分間標識し、次にその細胞を、蛍光および透過光光学系を装備したLeica DMIRB倒立顕微鏡を用いて分析した(少なくとも条件当り細胞400個)。
【0124】
細胞をトランスフェクトもしくは処理した1日後にアポトーシスの評価を行った。トランスフェクトされた細胞に関わる実験において、トランスフェクション溶液にpEDFP-C1を入れ、トランスフェクト細胞をGFPで標識した。この場合、アポトーシスの程度はトランスフェクト細胞のみについて評価した。
【0125】
ルシフェラーゼレポーターアッセイ
ルシフェラーゼアッセイはPromega製Dual-Luciferase(登録商標)Reporter Assay(カタログ番号E1910)を用いて行った。Promegaのキットから供給されたPLB溶解バッファー100μlを用いて6ウェルプレートから細胞を溶解し、氷上で30分間インキュベートした。次に溶解液を16,000gで15分間遠心して透明化した。この溶解液20μlをLARII試薬25μlと混合してホタルルシフェラーゼ活性を記録し、前記混合物にStop & Glo 試薬25μlを加えてウミシイタケルシフェラーゼ活性を記録した。それぞれの測定について、Lumat LB 9501ルミノメーター(Berthold Technologies、チューリッヒ、スイス)を用いて12秒間、発光を数量化した。
【0126】
ウェスタンブロット分析
細胞を溶解バッファー(25 mM Hepes、300mM NaCl、1.5mM MgCl2、0.2mM EDTA、0.1mM Na3V04、1% Triton X100、コンプリートEDTAフリープロテアーゼインヒビターカクテル錠(Roche;カタログ番号1873580))中で溶解した。SDS-PAGEでタンパク質を分離し、ニトロセルロース膜(BioRad;カタログ番号162-0115)にブロットした。その後、室温にて30分間、TBS (18 mM HC1、130 mM NaCl、20 mM Tris)、5%脱脂粉乳で膜をブロックし、適当な一次抗体とともに一晩インキュベートした。これらの抗体を、TBS、5%脱脂粉乳で1:2500に希釈したAlexa Fluor 680結合二次抗体(Molecular Probes;カタログ番号A21109)によって検出し、続いてOdysseyインフラレッドイメージングシステム(Licor、ホンブルク、ドイツ)によって可視化した。phospho-p38に対する一次抗体(Cell Signaling Technology;カタログ番号9211L)をTBS中5%BSAで1:500に希釈した。phospho-JNKに対する一次抗体(Cell Signaling Technology;カタログ番号9551L)は、TBS中5%BSAで1:1000に希釈した。Odysseyインフラレッドイメージングソフトウェアを用いて数量化した。
【0127】
統計分析
すべての統計分析は、スチューデントtテストによりMicrosoft Excel (XP edition)で行った。
【実施例2】
【0128】
RasGAP断片N2はさまざまな遺伝毒性物質によって引き起こされるアポトーシス反応を増強する。
最近、本出願人らは、断片N2がHeLa腫瘍細胞株を死滅させるシスプラチンの能力を増強することを明らかにした(Yang J.-Y. and Widmann C., Mol. Cell. Biol., 21,5346, 2001)。断片N2が、他の遺伝毒性物質によって引き起こされるアポトーシス反応を増強することができるかどうかを評価するために、断片N2を発現するHeLa細胞もしくは発現しない同細胞を、アドリアマイシンおよびミトキサントロン(および対照としてシスプラチン)の濃度を増加させてその影響下においた。図1は、断片N2の存在が、さまざまな薬物に対するHeLa細胞の感受性を対照細胞より少なくとも10倍高めることを示す。この結果は、断片N2が広域性の遺伝毒性物質増感剤であることを示す。
【0129】
癌細胞を死滅させる薬物の能力を増強する断片Nの中で最小の配列の同定
断片N2は36 kDaのタンパク質であり、そのため化学的な合成は困難であると考えられる。短くてもなお遺伝毒性物質増感能力を有する配列の特徴を示すことは、断片N2から治療手段を開発するプロセスにおいて重要なステップである。そういった短い配列が分離できるかどうかを評価するために、断片N2の一連のトランケート型を作製し(図2A)、HeLa細胞においてシスプラチンが引き起こすアポトーシスを増強する能力を測定した(図2B)。断片N2は2つのSH2ドメインおよび1つのSH3ドメインを含有する(図2A)。これらのドメインのうちどれが断片N2のアポトーシス促進活性を有するかをまず判定した。HeLa細胞を、断片N2を発現する細胞と発現しない細胞で薬物に対する感受性の差が最大となる濃度である、0.15μMのシスプラチン存在下もしくは非存在下で、さまざまなSHドメインをコードするプラスミドによりトランスフェクトした(図1を参照されたい)。何もコードしていない構築物またはSH2ドメインをコードする構築物は、これらの構築物でトランスフェクトされたHaLa細胞を死滅させるシスプラチンの能力を増強しなかった。これに対して、HS3ドメインを含有する構築物は、アポトーシスによってシスプラチンが細胞を死滅させる能力を増強する(図2B)。ウェスタンブロット分析から、SH2ドメインが他の構築物と同じく効率的に発現されていることはあきらかなので、SH2ドメイン単独でHeLa細胞において細胞死を増強することができないのは、タンパク質発現が低下した結果ではない(データは示さない)。
【0130】
本出願人らは次に、最小の遺伝毒性物質感作配列を同定する目的で、SH3ドメインのトランケーションを進めた。これらの構築物(図2A参照)はすべて、10アミノ酸の長さのペプチドをコードする最も短いもの(317-326)を含めて、シスプラチンがHeLa細胞を死滅させる能力を増強した(図2B)。こうした結果は、断片N2の細胞死感作特性が、完全なSHドメインを必要とせず、非常に短いペプチド配列によって媒介されることを示唆する。
【0131】
317-326 RasGAP配列と結合したHIV-TAT48-57ペプチドは細胞透過性遺伝毒性物質増感ペプチドとして機能する。
【0132】
RasGAPのアミノ酸317-326をコードするプラスミドに、薬物が癌細胞を死滅させる能力を強める力があるならば、316-326に相当する合成ペプチドも細胞に浸透することができればこうした活性を示すはずである。HIV TATタンパク質に由来する短い配列(HIV-TAT48-57)を付加することによって、ポリペプチドの細胞内での効率的な蓄積が可能になることが実証されている(Schwarze S. R. et al.,"n vivo protein transduction: delivery of a biologically active protein into the mouse" Science, 285,1569, 1999)。したがって、本出願人らは、アミノ酸キャリアペプチドHIV-TAT48-57と共有結合したRasGAPのアミノ酸317-326を含有するペプチドを合成した。TATとRasGAP配列の間にスペーサーとして2つのグリシン残基を挿入し、柔軟性を与えた。このペプチド(以後TAT-RasGAP317-326ペプチドと称する)の細胞取り込みを評価する手段として、そのペプチドをFITCフルオロフォアで標識した。標識したペプチドを4つの異なる腫瘍細胞株とともにインキュベートした:子宮頸部由来ヒト腺癌(HeLa細胞)、ヒト骨肉腫(U2OS細胞)、乳癌細胞株(MCF-7細胞)およびヒト悪性中皮種(H-Meso-1細胞)、ならびに2つの非癌細胞株(HaCatヒト皮膚ケラチノサイト細胞株およびHUV-EC-Cヒト臍静脈内皮細胞)。図3に示すように、TAT-RasGAP317-326ペプチドは、これらすべての細胞株の中に効率よく入った。非癌細胞と癌細胞の間でペプチドの移動に差は認められなかった。最初に、3つの遺伝毒性物質に対する癌細胞株の感受性を、毒性物質の濃度を上げて評価した(データは示さない)。これによって、テストした各細胞株についての遺伝毒性物質の亜致死濃度を測定することができた。驚くべきことに、TAT-RasGAP317-326ペプチドは、テストした癌細胞株を死滅させるシスプラチン、アドリアマイシンおよびミトキサントロンの能力を増強したがRasGAP配列を欠いた対照のHIV-TAT48-57ペプチドは増強しなかったこと、および対照条件においてアポトーシスは誘導されず、もしくはごくわずかしか誘導されないことが明らかになった。これに対して、2つの非癌細胞株においてシスプラチン、アドリアマイシンおよびミトキサントロンによって誘導されるアポトーシス反応は、ペプチドの存在によって影響を受けなかった(図4B)。したがって、本出願人らは、特異的に細胞に入り、癌細胞を死滅させる薬物の能力を増強することができる最小の合成ペプチドを同定した。
【0133】
NFκBおよびSAPK経路はTAT-RasGAP317-326の特性に関与しない。
Rasモジュレータとして、RasGAPは、細胞死を制御するRas依存性経路の一部、たとえばRas-P13K-Akt-NFκB経路に影響を及ぼすと考えられる(Datta S. R. et al., "Cellular survival: a play in three Akts" Genes Dev., 13, 2905, 1999)。したがって、NFκB活性のモジュレーションが、TAT-RasGAP317-326による能力の増強に関与するかどうかを評価した。図5Aに示すように、NFκBはU2OS細胞において、シスプラチン処理もしくはTAT-RasGAP317-326による処理の後、活性化されたが、対照ペプチドHIV-TAT48-57による刺激では活性化されなかった。シスプラチンおよびTAT-RasGAP317-326の両者とともに細胞をインキュベートすると、結果としてNFκBのさらなる活性化を生じた(図5A)。NFκB経路は、多くの種類の細胞において細胞生存反応の誘導に関与する(Van Antwerp D. J. etal., "Suppression of TNF-alpha- induced apoptosis by NF-kappaB" Science, 274,787, 1996; Beg A. A. and Baltimore D., "An essential role for NF-kappaB in preventing TNF-alpha-induced cell death" Science, 274,782, 1996)が、ある種の状況においてはアポトーシス反応の誘導にも必要とされる可能性がある(Ryan K. M. et al., "Role of NF-kappaB in p53-mediated programmed cell death" Nature, 404, 892,2000)。TAT-RasGAP317-326によるNFκBの活性化が増強する能力機能に必要であるかを判断するために、非分解型IκB(IκBαΔN2)を細胞において発現させた。予想されるように、この構築物は効率的にシスプラチンおよびTAT-RasGAP317-326によるNFκBの活性化を阻止した(図5B)。IκBαΔN2は、わずかにシスプラチンによるアポトーシス反応を増大させた(図5C)。しかしながら、アポトーシスによってU2OS細胞を死滅させるシスプラチンの能力を増強するTAT-RasGAP317-326の効力は、NFκBインヒビターの影響を受けなかった(図5C)。これらの結果は、NFκB経路の活性化がTAT-RasGAP317-326による細胞死増強能力の効果に関与しないことを示す。
【0134】
ストレス活性化プロテインキナーゼ(SAPK)-JNKおよびp38 MAPK-は、さまざまな刺激によって引き起こされるアポトーシス反応に関与していた(Jarpe M. B. et al., Oncogene, 17, 1475, 1998)。図6は、対照HIV-TAT48-57ペプチドもTAT-RasGAP317-326ペプチドもいずれもこれらのMAPK経路を活性化しなかったことを示す。これらのペプチドはまた、シスプラチンがJNKもしくはp38 MAPKを刺激する能力を高めることはできなかった(図6)。これらの結果は、ストレス活性化MAPK経路が、癌細胞を死滅させる遺伝毒性物質の能力を増強する、TAT-RasGAP317-326の機能に関与しないことを示す。
【図面の簡単な説明】
【0135】
【図1】図1は、断片N2もしくは空のpcDNAプラスミドでトランスフェクトされたHeLa細胞における、さまざまな薬物によるアポトーシス誘導のパーセンテージを示す。 HeLa細胞(2x106)を直径10cmのペトリ皿に播き、空のpcDNA3プラスミド2μg、または断片N2をコードするpcDNA3ベクター2μgとともに、GFP発現プラスミド1μg(トランスフェクト細胞を標識するため)でトランスフェクトした。トランスフェクションの1日後、細胞を24時間、指示された濃度のシスプラチン、アドリアマイシン、もしくはミトキサントロンとともにインキュベートした。その後核濃縮を示すGFP陽性細胞の数を記録した。 結果は、3回の独立した測定の平均値±標準偏差に対応する。星印は対照細胞とシスプラチン処理細胞の間の有意な相違を示す(*,p < 0. 05 ; **, p < 0.01 ; ***, p < 0.001)。
【図2】図2Aは本研究に用いられたさまざまな構築物の概要図である。 SHはSrc相同ドメインを表す。図2Bは、図2Aに記載の構築物をコードするプラスミドでトランスフェクトされたHeLa細胞における、シスプラチンによるアポトーシス誘導のパーセンテージを示す。 HeLa細胞を、図2Aに記載の構築物をコードしたプラスミドで、図1に記載のようにトランスフェクトした。その後、HeLa細胞を0.15μMシスプラチンで処理し、または処理せずに、アポトーシスの程度を20時間後に測定した。結果は、3つの独立した測定の平均値±標準偏差に対応する。星印は0.15μMシスプラチン処理細胞と非処理細胞の間の有意な相違を示す(**, p < 0.01 ; ***, p < 0.001)。
【図3】図3は、FITC標識TAT-RasGAP317-326ペプチドとともにインキュベートした生きた細胞の位相差および落射蛍光画像を示す。 次の細胞株(HeLa、U20S、H-Mesol、MCF-7、HaCatおよびHUV-EC-C)を、FITC標識TAT-RasGAP317-326ペプチド20μMを含む培地中で37℃、5% CO2にて3時間インキュベートした後、培地で3回洗浄した。
【図4A】図4Aは、TAT-RASGAP317-326で処理された、または処理されない、2つの非癌細胞における、さまざまな薬物によるアポトーシス誘導のパーセンテージを示す。 2つの非癌細胞株(HaCatおよびHUV-EC-C)を、20μM TAT-RasGAP317-326 の存在下もしくは非存在下で、シスプラチン、アドリアマイシンおよびミトキサントロンとともに、それらの濃度を増加させてインキュベートした。アポトーシスの程度を20時間後に記録した。
【図4B】図4Bは、TAT-RASGAP317-326で処理された、または処理されない、4つの癌細胞株における、さまざまな薬物によるアポトーシス誘導のパーセンテージを示す。 4つの癌細胞株(HeLa、U20S、MCF-7およびH-Mesol)を6ウェルプレートに播き、20μMHIV-TAT48-57もしくはTAT-RasGAP317-326ペプチドのいずれか一方の存在下、またはいずれも存在しない条件下で、指示された濃度のシスプラチン、アドリアマイシンおよびミトキサントロンで20時間処理した。次に核濃縮を示す細胞の数を記録した。星印は、TAT-RasGAP317-326とともにインキュベートした遺伝毒性物質処理細胞と、未処理のままの細胞もしくはHIV-TAT48-57ペプチドとともにインキュベートした細胞との間の有意な相違を示す(**, p < 0.01 ; ***, p < 0.001)。
【図5A】図5Aは、シスプラチンで処理したU2OS細胞における、HIV-TAT48-57もしくはTAT-RasGAP317-326ペプチド存在下でのNFκB活性を示す。 U2OS細胞(1x105)を6ウェルプレートに播き、NFκB活性のためのホタルルシフェラーゼレポータープラスミド1μgおよびウミシイタケルシフェラーゼをコードするプラスミド0.1μgでトランスフェクトした。1日後に、20μM HIV-TAT48-57もしくはTAT-RasGAP317-326ペプチドの存在下または非存在下で、20時間、細胞を指示された濃度のシスプラチンで処理した。データはウミシイタケルシフェラーゼ活性に対して標準化されたホタルルシフェラーゼ活性を表し、対照の非処理細胞で得られた基底NFκB活性からの増加倍数として示した。結果は、3つの独立した測定の平均値±標準偏差に対応する。星印は指示された条件の間の有意な相違を示す(**, p < 0.01 ; ***, p < 0.001)。
【図5B】図5Bは、シスプラチンで処理したU2OS細胞における、HIV-TAT48-57ペプチドおよびIκBαΔN2、またはTAT-RasGAP317-326ペプチドおよびIκBαΔN2の存在下でのNFκB活性を示す。 U2OS細胞を、NFκB活性のためのホタルルシフェラーゼレポータープラスミド1μg、ウミシイタケルシフェラーゼをコードするプラスミド0.1μg、GFP発現プラスミド(トランスフェクト細胞を標識するため)0.5μg、NFκB経路を阻害するIκBαΔN2をコードするプラスミド1μgもしくは空のpcDNA3ベクター1μgでトランスフェクトした。1日後に細胞を、さらに20時間、20μM HIV-TAT48-57または20μM TAT-RasGAP317-326の存在下で、シスプラチンとともに、シスプラチンの濃度を増加させてインキュベートした。次に、細胞を溶解し、パネルAに記載のようにNFκB活性を測定したが、ただし結果は、対照のHIV-TAT48-57ペプチドとともにインキュベートした細胞において検出されたNFκB活性からの増加倍数として表現した。
【図5C】図5Cは、図5AおよびBのトランスフェクト細胞のアポトーシスのパーセンテージを示す。 代わりになるべきものとして、核濃縮を示すGFP陽性細胞の数を測定した。結果は、3つの独立した測定の平均値±標準偏差に対応する。
【図6】図6は、HIV-TAT48-57もしくはTAT-RasGAP317-326ペプチドのいずれかでトランスフェクトされ、およびシスプラチンで処理された、U2OS細胞のJNKリン酸化のウェスタンブロットおよびパーセンテージを表す。 U2OS細胞(2x105)を6ウェルプレートに播き、HIV-TAT48-57もしくはTAT-RasGAP317-326ペプチド(濃度20μM)およびシスプラチン(濃度30μM)の指示された組み合わせによって、指示された時間、処理した。JNKおよびp38活性化に対する陽性対照は、それぞれ、1μg/mlアニソマイシンで3時間、および0.5Mソルビトールで30分間、細胞を刺激した後、得られた。ウェスタンブロットに基づいて示される定量化は12時間のバンドについて行われ、陽性対照に対して標準化された。結果は、3つの独立した測定の平均値±標準偏差に対応する。
【特許請求の範囲】
【請求項1】
細胞を死滅させる薬物の能力を増強するペプチドであって、そのペプチドが基本的にRasGAPタンパク質のN2配列、その断片、もしくはその変異体からなり、それらが癌細胞において選択的に前記能力を増強することを特徴とする、前記ペプチド。
【請求項2】
RasGAPタンパク質のN2配列の断片がN2配列のSH3ドメインのアミノ酸配列、その一部、もしくはその変異体を含んでなることを特徴とする、請求項1に記載のペプチド。
【請求項3】
SH3ドメインの一部、もしくはその変異体が、SH3ドメインのアミノ酸配列のうち70個以下のアミノ酸を含有することを特徴とする、請求項2に記載のペプチド。
【請求項4】
SH3ドメインの一部がDNA配列、配列番号1、配列番号2、配列番号3、または配列番号4によりコードされるアミノ酸配列であることを特徴とする、請求項2に記載のペプチド。
【請求項5】
前記ペプチドが、細胞内で当該ペプチドの蓄積を増加させる薬剤と結合していることを特徴とする、請求項1〜4のいずれか1つに記載のペプチド。
【請求項6】
前記薬剤が細胞膜透過性キャリアであることを特徴とする、請求項5に記載のペプチド。
【請求項7】
前記細胞膜透過性キャリアがペプチドであることを特徴とする、請求項6に記載のペプチド。
【請求項8】
前記細胞膜透過性キャリアペプチドが、HIV-TAT48-57ペプチド、FHV-coat35-49ペプチド、HTLV-II Rex4-16ペプチド、およびBMVgag7-25ペプチドを含んでなる一群から選択される、アルギニンリッチペプチドであることを特徴とする、請求項7に記載のペプチド。
【請求項9】
アルギニンリッチペプチドがHIV-TAT48-57ペプチドであることを特徴とする、請求項8に記載のペプチド。
【請求項10】
薬物が遺伝毒性物質であることを特徴とする、請求項1〜9のいずれか1つに記載のペプチド。
【請求項11】
遺伝毒性物質がシスプラチン、ミトキサントロンおよびアドリアマイシンを含んでなる一群から選択されることを特徴とする、請求項10に記載のペプチド。
【請求項12】
請求項1〜4のいずれか1つに記載のペプチドをコードする核酸配列を含んでなる、単離および精製された核酸配列。
【請求項13】
細胞膜透過性キャリアペプチドをコードするヌクレオチド配列を追加して含んでなる、請求項12に記載の核酸配列。
【請求項14】
請求項12または13に記載の単離精製された核酸配列の少なくとも1コピーを含んでなる、発現ベクター。
【請求項15】
請求項1〜4のいずれか1つに記載のペプチド、請求項12もしくは13に記載の単離精製された核酸配列、または請求項14に記載の発現ベクターを含有する、原核または真核宿主細胞。
【請求項16】
活性成分として、製薬上有効な量の、請求項1〜9のいずれか1つに記載の少なくとも1つのペプチドを、状況に応じて製薬上許容される担体、希釈剤および補助剤と組み合わせて含んでなる、医薬組成物。
【請求項17】
癌の治療または予防を目的とする、請求項16に記載の医薬組成物。
【請求項18】
癌の治療または予防のための薬物を製剤するための、請求項16に記載の医薬組成物の使用。
【請求項19】
癌が、癌腫、リンパ腫、芽細胞種、肉腫、脂肪肉腫、神経内分泌腫瘍、中皮腫、神経鞘腫、髄膜腫、腺癌、黒色腫、白血病、悪性リンパ腫、扁平上皮癌、上皮性扁平上皮癌、肺癌、小細胞肺癌、非小細胞肺癌、肺腺癌、肺扁平上皮癌、腹膜癌、肝細胞癌、胃癌、消化器癌、膵臓癌、神経膠芽腫、子宮頸癌、卵巣癌、肝臓癌、膀胱癌、肝細胞腫、乳癌、大腸癌、直腸癌、直腸結腸癌、子宮内膜癌もしくは子宮癌、唾液腺癌、腎臓癌、前立腺癌、外陰癌、甲状腺癌、肝癌、肛門癌、陰茎癌、睾丸癌、食道癌、胆道腫瘍、および頭頸部癌からなる一群から選択されることを特徴とする、請求項18に記載の使用。
【請求項20】
癌が中皮種、睾丸癌もしくは膵臓癌であることを特徴とする、請求項19に記載の使用。
【請求項21】
治療上有効な量の請求項16に記載の医薬組成物を、それを必要とする被験体に投与することを含んでなる、癌腫、リンパ腫、芽細胞種、肉腫、脂肪肉腫、神経内分泌腫瘍、中皮腫、神経鞘腫、髄膜腫、腺癌、黒色腫、白血病、悪性リンパ腫、扁平上皮癌、上皮性扁平上皮癌、肺癌、小細胞肺癌、非小細胞肺癌、肺腺癌、肺扁平上皮癌、腹膜癌、肝細胞癌、胃癌、消化器癌、膵臓癌、神経膠芽腫、子宮頸癌、卵巣癌、肝臓癌、膀胱癌、肝細胞腫、乳癌、大腸癌、直腸癌、直腸結腸癌、子宮内膜癌もしくは子宮癌、唾液腺癌、腎臓癌、前立腺癌、外陰癌、甲状腺癌、肝癌、肛門癌、陰茎癌、睾丸癌、食道癌、胆道腫瘍、および頭頸部癌からなる一群から選択される癌を治療または予防する方法。
【請求項22】
癌が中皮種、睾丸癌もしくは膵臓癌であることを特徴とする、請求項21に記載の方法。
【請求項23】
請求項1〜11のいずれか1つに記載の、少なくとも1つのペプチドおよび薬物と癌細胞を接触させることを含んでなる、癌細胞において選択的にアポトーシスを増強する方法。
【請求項24】
薬物が遺伝毒性物質であることを特徴とする、請求項23に記載の方法。
【請求項25】
遺伝毒性物質がシスプラチン、ミトキサントロン、アドリアマイシンを含んでなる一群から選択されることを特徴とする、請求項24に記載の方法。
【請求項26】
請求項1〜11のいずれか1つに記載の、少なくとも1つのペプチドおよび薬物と癌細胞を接触させることを含んでなる、選択的に癌細胞を死滅させる方法。
【請求項27】
薬物が遺伝毒性物質であることを特徴とする、請求項26に記載の方法。
【請求項28】
癌細胞中で選択的に細胞を死滅させる遺伝毒性物質の能力を増強するための、請求項1〜11のいずれか1つに記載のペプチドの使用。
【請求項29】
被験体において癌を治療もしくは予防するためのキットであって、請求項1〜11のいずれか1つに記載の、少なくとも1つのペプチドを、状況に応じて試薬および/または使用説明書とともに含んでなる、前記キット。
【請求項30】
薬物、抗-上皮成長因子受容体抗体、放射免疫治療薬、およびそれらの組み合わせからなる一群から選択される、追加の抗ガン剤を含んでなる別の医薬剤形をさらに含んでなる、請求項29に記載のキット。
【請求項31】
一般配列:WXWVTXXRTXによって特徴付けられ、Xはアミノ酸残基である、請求項1〜9のいずれか1つに記載のペプチド。
【請求項1】
細胞を死滅させる薬物の能力を増強するペプチドであって、そのペプチドが基本的にRasGAPタンパク質のN2配列、その断片、もしくはその変異体からなり、それらが癌細胞において選択的に前記能力を増強することを特徴とする、前記ペプチド。
【請求項2】
RasGAPタンパク質のN2配列の断片がN2配列のSH3ドメインのアミノ酸配列、その一部、もしくはその変異体を含んでなることを特徴とする、請求項1に記載のペプチド。
【請求項3】
SH3ドメインの一部、もしくはその変異体が、SH3ドメインのアミノ酸配列のうち70個以下のアミノ酸を含有することを特徴とする、請求項2に記載のペプチド。
【請求項4】
SH3ドメインの一部がDNA配列、配列番号1、配列番号2、配列番号3、または配列番号4によりコードされるアミノ酸配列であることを特徴とする、請求項2に記載のペプチド。
【請求項5】
前記ペプチドが、細胞内で当該ペプチドの蓄積を増加させる薬剤と結合していることを特徴とする、請求項1〜4のいずれか1つに記載のペプチド。
【請求項6】
前記薬剤が細胞膜透過性キャリアであることを特徴とする、請求項5に記載のペプチド。
【請求項7】
前記細胞膜透過性キャリアがペプチドであることを特徴とする、請求項6に記載のペプチド。
【請求項8】
前記細胞膜透過性キャリアペプチドが、HIV-TAT48-57ペプチド、FHV-coat35-49ペプチド、HTLV-II Rex4-16ペプチド、およびBMVgag7-25ペプチドを含んでなる一群から選択される、アルギニンリッチペプチドであることを特徴とする、請求項7に記載のペプチド。
【請求項9】
アルギニンリッチペプチドがHIV-TAT48-57ペプチドであることを特徴とする、請求項8に記載のペプチド。
【請求項10】
薬物が遺伝毒性物質であることを特徴とする、請求項1〜9のいずれか1つに記載のペプチド。
【請求項11】
遺伝毒性物質がシスプラチン、ミトキサントロンおよびアドリアマイシンを含んでなる一群から選択されることを特徴とする、請求項10に記載のペプチド。
【請求項12】
請求項1〜4のいずれか1つに記載のペプチドをコードする核酸配列を含んでなる、単離および精製された核酸配列。
【請求項13】
細胞膜透過性キャリアペプチドをコードするヌクレオチド配列を追加して含んでなる、請求項12に記載の核酸配列。
【請求項14】
請求項12または13に記載の単離精製された核酸配列の少なくとも1コピーを含んでなる、発現ベクター。
【請求項15】
請求項1〜4のいずれか1つに記載のペプチド、請求項12もしくは13に記載の単離精製された核酸配列、または請求項14に記載の発現ベクターを含有する、原核または真核宿主細胞。
【請求項16】
活性成分として、製薬上有効な量の、請求項1〜9のいずれか1つに記載の少なくとも1つのペプチドを、状況に応じて製薬上許容される担体、希釈剤および補助剤と組み合わせて含んでなる、医薬組成物。
【請求項17】
癌の治療または予防を目的とする、請求項16に記載の医薬組成物。
【請求項18】
癌の治療または予防のための薬物を製剤するための、請求項16に記載の医薬組成物の使用。
【請求項19】
癌が、癌腫、リンパ腫、芽細胞種、肉腫、脂肪肉腫、神経内分泌腫瘍、中皮腫、神経鞘腫、髄膜腫、腺癌、黒色腫、白血病、悪性リンパ腫、扁平上皮癌、上皮性扁平上皮癌、肺癌、小細胞肺癌、非小細胞肺癌、肺腺癌、肺扁平上皮癌、腹膜癌、肝細胞癌、胃癌、消化器癌、膵臓癌、神経膠芽腫、子宮頸癌、卵巣癌、肝臓癌、膀胱癌、肝細胞腫、乳癌、大腸癌、直腸癌、直腸結腸癌、子宮内膜癌もしくは子宮癌、唾液腺癌、腎臓癌、前立腺癌、外陰癌、甲状腺癌、肝癌、肛門癌、陰茎癌、睾丸癌、食道癌、胆道腫瘍、および頭頸部癌からなる一群から選択されることを特徴とする、請求項18に記載の使用。
【請求項20】
癌が中皮種、睾丸癌もしくは膵臓癌であることを特徴とする、請求項19に記載の使用。
【請求項21】
治療上有効な量の請求項16に記載の医薬組成物を、それを必要とする被験体に投与することを含んでなる、癌腫、リンパ腫、芽細胞種、肉腫、脂肪肉腫、神経内分泌腫瘍、中皮腫、神経鞘腫、髄膜腫、腺癌、黒色腫、白血病、悪性リンパ腫、扁平上皮癌、上皮性扁平上皮癌、肺癌、小細胞肺癌、非小細胞肺癌、肺腺癌、肺扁平上皮癌、腹膜癌、肝細胞癌、胃癌、消化器癌、膵臓癌、神経膠芽腫、子宮頸癌、卵巣癌、肝臓癌、膀胱癌、肝細胞腫、乳癌、大腸癌、直腸癌、直腸結腸癌、子宮内膜癌もしくは子宮癌、唾液腺癌、腎臓癌、前立腺癌、外陰癌、甲状腺癌、肝癌、肛門癌、陰茎癌、睾丸癌、食道癌、胆道腫瘍、および頭頸部癌からなる一群から選択される癌を治療または予防する方法。
【請求項22】
癌が中皮種、睾丸癌もしくは膵臓癌であることを特徴とする、請求項21に記載の方法。
【請求項23】
請求項1〜11のいずれか1つに記載の、少なくとも1つのペプチドおよび薬物と癌細胞を接触させることを含んでなる、癌細胞において選択的にアポトーシスを増強する方法。
【請求項24】
薬物が遺伝毒性物質であることを特徴とする、請求項23に記載の方法。
【請求項25】
遺伝毒性物質がシスプラチン、ミトキサントロン、アドリアマイシンを含んでなる一群から選択されることを特徴とする、請求項24に記載の方法。
【請求項26】
請求項1〜11のいずれか1つに記載の、少なくとも1つのペプチドおよび薬物と癌細胞を接触させることを含んでなる、選択的に癌細胞を死滅させる方法。
【請求項27】
薬物が遺伝毒性物質であることを特徴とする、請求項26に記載の方法。
【請求項28】
癌細胞中で選択的に細胞を死滅させる遺伝毒性物質の能力を増強するための、請求項1〜11のいずれか1つに記載のペプチドの使用。
【請求項29】
被験体において癌を治療もしくは予防するためのキットであって、請求項1〜11のいずれか1つに記載の、少なくとも1つのペプチドを、状況に応じて試薬および/または使用説明書とともに含んでなる、前記キット。
【請求項30】
薬物、抗-上皮成長因子受容体抗体、放射免疫治療薬、およびそれらの組み合わせからなる一群から選択される、追加の抗ガン剤を含んでなる別の医薬剤形をさらに含んでなる、請求項29に記載のキット。
【請求項31】
一般配列:WXWVTXXRTXによって特徴付けられ、Xはアミノ酸残基である、請求項1〜9のいずれか1つに記載のペプチド。
【図1】

【図2】
【図3】
【図4A】
【図4B】
【図5A】
【図5B】
【図5C】
【図6】
【図2】
【図3】
【図4A】
【図4B】
【図5A】
【図5B】
【図5C】
【図6】
【公表番号】特表2007−527856(P2007−527856A)
【公表日】平成19年10月4日(2007.10.4)
【国際特許分類】
【出願番号】特願2006−516588(P2006−516588)
【出願日】平成16年6月30日(2004.6.30)
【国際出願番号】PCT/IB2004/002165
【国際公開番号】WO2005/000887
【国際公開日】平成17年1月6日(2005.1.6)
【出願人】(506000793)ユニベルシテ ド ローザンヌ (1)
【Fターム(参考)】
【公表日】平成19年10月4日(2007.10.4)
【国際特許分類】
【出願日】平成16年6月30日(2004.6.30)
【国際出願番号】PCT/IB2004/002165
【国際公開番号】WO2005/000887
【国際公開日】平成17年1月6日(2005.1.6)
【出願人】(506000793)ユニベルシテ ド ローザンヌ (1)
【Fターム(参考)】
[ Back to top ]