
癌転移および癌転移に関連する骨量減少を予防および処置するための方法
【課題】癌転移および癌転移に関連する骨量減少を予防および処置するための方法の提供。
【解決手段】治療薬と組み合わされたM−CSFアンタゴニストを対象に投与することにより骨溶解、癌転移および癌転移に関連する骨量減少を防御および処置するための方法が提供される。本発明の一つの実施態様では、骨溶解性障害を患うかまたはその危険性を有する対象に単剤治療の量のM−CSFアンタゴニストおよび単剤治療上有効な量の第二の抗破骨細胞薬を約1日から1年の移行期間の間投与し、その間M−CSFアンタゴニストは活性破骨細胞の数を治療上望ましいレベルまで低下させることを含む、その対象を処置するための方法が提供される。M−CSFアンタゴニストの実例には、M−CSF抗体が挙げられ、そして第二の抗破骨細胞薬の実例には、ビスホスホネート系および抗RANKL抗体を含むRANKL阻害剤が挙げられる。
【解決手段】治療薬と組み合わされたM−CSFアンタゴニストを対象に投与することにより骨溶解、癌転移および癌転移に関連する骨量減少を防御および処置するための方法が提供される。本発明の一つの実施態様では、骨溶解性障害を患うかまたはその危険性を有する対象に単剤治療の量のM−CSFアンタゴニストおよび単剤治療上有効な量の第二の抗破骨細胞薬を約1日から1年の移行期間の間投与し、その間M−CSFアンタゴニストは活性破骨細胞の数を治療上望ましいレベルまで低下させることを含む、その対象を処置するための方法が提供される。M−CSFアンタゴニストの実例には、M−CSF抗体が挙げられ、そして第二の抗破骨細胞薬の実例には、ビスホスホネート系および抗RANKL抗体を含むRANKL阻害剤が挙げられる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は別の治療薬と組み合わされたM−CSFアンタゴニストを対象に投与することにより、癌転移および癌転移に関連する骨量減少(bone loss)を含む骨溶解性疾患を防御および処置するための方法に関する。
【背景技術】
【0002】
(発明の背景)
骨吸収を媒介する破骨細胞は骨溶解性疾患を含む正常および異常な骨リモデリング過程に関与する。破骨細胞は造血細胞から分化する多核細胞である。破骨細胞が不完全な細胞分裂よりもむしろ、骨髄の造血幹細胞に由来する単核前駆体の融合により形成されることは一般的に知られている(Chambers、Bone and Mineral Research 6:1−25(1989);Gothlingら、Clin Orthop Relat R.120:201−228(1976);Kahnら、Nature
258:325−327(1975)、Sudaら、Endocr Rev 13:66−80、(1992);Walker、Science 180:875(1973);Walker、Science 190:785−787(1975);Walker、Science 190:784−785(1975))。これらは単球−マクロファージ細胞系と共通の幹細胞を共有する(Ashら、Nature 283:669−670(1980)、Kerbyら、J.Bone Miner Res 7:353−62(1992))。破骨細胞前駆体の成熟多核破骨細胞への分化はホルモン性および局部刺激を含む様々な因子を必要とし(Athanasouら、Bone Miner 3:317−333(1988);Feldmanら、Endocrinology 107:1137−1143(1980);Walker、Science 190:784−785(1975);Zhengら、Histochem J 23:180−188(1991))、そして生体骨および骨細胞は破骨細胞発達において非常に重要な役割を果たすことが示されている(Hagenaarsら、Bone Miner 6:179−189(1989))。骨芽または骨髄間質細胞もまた破骨細胞分化に必要とされる。破骨細胞形成を支持するこれらの細胞により生成される因子の一つはマクロファージコロニー刺激因子、M−CSFである(Wiktor−Jedrzejczakら、Proc Natl Acad Sci USA 87:4828−4832(1990);Yoshidaら、Nature 345:442−444(1990))。NF−κBリガンドに関する受容体アクチベーター(RANKL、TRANCE、ODFおよびOPGLとしても公知)は別のシグナルであり(Sudaら、Endocr Rev 13:66−80(1992))、それにより骨芽/間質細胞が破骨細胞および破骨細胞前駆体に位置する受容体、RANK(TRANCER)を介する破骨細胞形成および吸収を刺激する(Laceyら、Cell 93:165−176(1998);Tsudaら、Biochem Biophys Res Co 234:137−142(1997);Wongら、J Exp Med 186:2075−2080(1997);Wongら、J Biol.Chem 272:25190−25194(1997);Yasudaら、Endocrinology 139:1329−1337(1998);Yasudaら、Proc Natl Acad Sci US 95:3597−3602(1998))。骨芽細胞はまたオステオプロテジェリン(OPG、OCIFとしても公知)と称される、破骨細胞形成を強く阻止するタンパク質を分泌し、それはRANKLに関するデコイ受容体として作用し、したがってRANKおよびRANKLを介する破骨細胞と骨芽細胞との間の陽性シグナルを阻止する。
【0003】
破骨細胞はミネラルおよび有機骨マトリックスの双方の溶解に寄与する(Blairら、J Cell Biol 102:1164−1172(1986))。破骨細胞は特殊な膜部分ならびに、酒石酸抵抗性酸ホスファターゼ(TRAP)(Andersonら、1979)、炭酸脱水酵素II(Vaananenら、Histochemistry
78:481−485(1983))、カルシトニン受容体(Warshafskyら、Bone 6:179−185(1985))およびビトロネクチン受容体(Daviesら、J Cell Biol 109:1817−1826(1989))のようないくつかの膜および細胞質マーカーを伴う独特な極性形態学を表現する高分化型細胞を表す。多核破骨細胞は通常10個未満の核を含有するが、それらは直径10μmと100μmの間である100個までの核を含有し得る(Gothlingら、Clin Orthop Relat R 120:201−228(1976))。これによりそれらは光学顕微鏡により比較的容易に同定されることになる。それらは活性化状態にあるときに高度に空胞化され、そしてまた高い代謝速度の指標である多くのミトコンドリアを含有する(Mundy、Primer on the metabolic bone diseases and disorders of mineral metabolism、18−22頁(1990))。破骨細胞は破骨性骨転移において主要な役割を果たすので、当分野では破骨細胞刺激および機能を防御するための新しい薬剤および方法が必要とされる。
【0004】
癌転移は癌患者における術後または治療後の再発の主な原因である。処置を開発するための集中的な努力にもかかわらず、癌転移は治療に対して依然実質的に不応性である。骨は種々の型のヒト癌(乳腺、肺、前立腺および甲状腺癌)の転移の最も一般的な部位の一つである。破骨性骨転移の発症は難治性の疼痛、高度な骨折し易さ、神経圧迫および高カルシウム血症による深刻な病的状態を引き起こす。これらの臨床上の問題にもかかわらず、癌転移に関連する骨量減少に利用可能な処置はわずかしかない。
【0005】
骨溶解性疾患を標的とするいくつかの治療計画が現在用いられているか、または開発中であり、ここでは主に破骨細胞の形成または活性を阻止することにより骨吸収を遮断する薬物の開発に尽力している。ビスホスホネート系(BPs)、骨に集中するピロリン酸類似体は今日までの最も有効な骨吸収の阻害剤である。BPsは破骨細胞に取り込まれ、その活性を阻止し、そして細胞にアポトーシスを被らせ、それにより骨吸収を阻止する。アレンドロネートは脊椎/股関節骨折の有意な減少を示す最初の骨吸収のBP阻害剤であり、そして骨粗鬆症の処置のために承認されている。最新世代のBPであるZometaは高カルシウム血症ならびに固形腫瘍および多発性骨髄腫における骨疾患の処置のために承認されており、そしてパジェット病ならびに固形腫瘍および多発性骨髄腫の結果である骨転移の処置の可能性に関して調査中である。Zometaは非常に低用量で作用し、そして月1回の15分間静脈内注入として与えられるが、骨芽細胞にも影響し、そして腎毒性および顎の骨壊死のような副作用を引き起こし得る(非特許文献1;非特許文献2;非特許文献3;非特許文献4;非特許文献5;非特許文献6;非特許文献7;非特許文献8)。したがって骨溶解性疾患および/または骨溶解性骨転移を含む癌転移を防御または処置するための新しい薬剤および方法を同定することが当分野では依然必要とされている。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】FromigueおよびBrody、J.Endocrinol.Invest.(2002)25:39−46
【非特許文献2】Ibrahim,A.ら、Clin.Canc.Res.(2003)9:2394−99
【非特許文献3】Body,J.J.、The Breast.(2003)S2:S37−44
【非特許文献4】Yaccoby,S.ら、Brit.J.Hemat.(2002)116:278−80
【非特許文献5】Corey,E.ら、Clin.Canc.Res.(2003)9:295−306
【非特許文献6】Coleman,R.E.、Sem.Oncol.,(2002)29(6):43−49
【非特許文献7】Coleman,R.E.、Eur.Soc.Med.Oncol.(2005)16:687−95
【非特許文献8】Bamiasら、J Clin Oncol(2005)13:8580−8587
【発明の概要】
【課題を解決するための手段】
【0007】
(発明の要旨)
本発明の組成物および方法は当分野において前記された、およびその他の関連する必要性を満足する。本発明の一つの実施態様では、骨溶解性障害を患うかまたはその危険性を有する対象に単剤治療の量のM−CSFアンタゴニストおよび単剤治療上有効な量の第二の抗破骨細胞薬を約1日から1年の移行期間(transition period)の間投与し、その間M−CSFアンタゴニストは活性破骨細胞の数を治療上望ましいレベルまで低下させることを含む、その対象を処置するための方法が提供される。M−CSFアンタゴニストの実例には、M−CSF抗体が挙げられ、そして第二の抗破骨細胞薬の実例には、ビスホスホネート系および抗RANKL抗体を含むRANKL阻害剤が挙げられる。抗M−CSF抗体および破骨細胞阻害剤を伴う方法および/または使用は本明細書では場合によっては国際公開番号第WO2005/068503号に開示されるRX1、5H4、MCIおよびMC3由来の抗体の使用を排除する。移行期間の長さは、例えば少なくとも1日から1年まででよく、そして例えば破骨細胞成長または活性の関連マーカーによりモニタリングすることができる。これに代えて、それらを同時に与えることができる。
【0008】
実例として骨形成のマーカーには、限定するものではないがカルシウム、ならびに全および骨特異的アルカリ性ホスファターゼ(BAP)、オステオカルシン(OC、骨glaタンパク質)、IC型プロコラーゲンプロペプチド(PICP)、IN型プロコラーゲンプロペプチド(PINP)が挙げられ、そして骨吸収のマーカーには、限定するものではないがNTX(骨コラーゲンのN末端架橋テロペプチド)およびCTX(骨コラーゲンのC末端架橋テロペプチド)、ピリジニウム架橋(ピリジノリンおよびデオキシピリジノリン(DPD))および関連ペプチド、骨のI型コラーゲン分解産物、ヒドロキシプロリンおよびヒドロキシリジングリコシド、酒石酸抵抗性酸ホスファターゼ(TRACP)、ならびに骨シアロタンパク質(BSP)が挙げられる。Fohrら、J.Clin.Endocrinol.Metab.,88(11):5059−5075(2003年11月)を参照のこと。
【0009】
関連する実施態様では、第二の抗破骨細胞薬が移行期間の後に中断される前記された方法が提供される。その他の関連する実施態様では、第二の抗破骨細胞薬を移行期間の後に減少させる前記された方法が提供される。さらなる関連する実施態様では、M−CSFアンタゴニストの量を移行期間の後に減少させる前記された方法が提供される。
【0010】
本発明の方法がM−CSFとその受容体(M−CSFR)との間の相互作用を阻止することによりその治療可能性を達成すると企図される。M−CSF/M−CSFR相互作用の阻止が破骨細胞増殖および/または分化を阻止することがさらに企図される。本発明のいずれかの方法または組成物では、M−CSFアンタゴニストは抗M−CSF抗体を含むポリペプチド;抗M−CSFR抗体を含むポリペプチド;M−CSFムテインもしくはその誘導体を含む可溶性ポリペプチド;またはM−CSFRムテインもしくはその誘導体を含む可溶性ポリペプチド;またはM−CSFもしくはM−CSFRの発現を阻止する核酸分子でよい。種々のM−CSFアンタゴニストの同定、生成および修飾は国際公開番号第WO2005/068503号に記載され、その全てを出典明示により本明細書の一部とする。
【0011】
M−CSF抗体はポリクローナル抗体;モノクローナル抗体;ヒト化抗体;ヒト抗体;ヒト改変抗体;キメラ抗体;Fab、F(ab’)2もしくはFv抗体フラグメント;または前記された抗体のいずれか一つの抗体のムテインでよい。
【0012】
骨溶解を阻止する本発明のM−CSF抗体は国際公開番号第WO2005/068503号(M−CSF抗体に関するその教示に関してその全てを出典明示により本明細書の一部とする)に記載されている。
【0013】
本発明の一つの実施態様では、図1、3および4の各々に示されるアミノ酸配列を有するネズミモノクローナル抗体RX1、MC1またはMC3のいずれか一つと同一のM−CSFのエピトープに特異的に結合する機能的フラグメントを含む非ネズミモノクローナル抗体が提供される。関連する実施態様では抗体がポリクローナル抗体;Human Engineered(商標)抗体を含むモノクローナル抗体;ヒト化抗体;ヒト抗体;;キメラ抗体;Fab、F(ab’)2;Fv;Sc FvまたはSCA抗体フラグメント;ダイアボディー;直鎖状抗体;または好ましくは少なくとも10−7、10−8もしくは10−9またはそれより高い結合親和性を保持するこれらの抗体のいずれか一つのムテイン;からなる群から選択される前記された抗体が提供される。M−CSFへの結合に関して、図1に示されるアミノ酸配列を有するモノクローナル抗体RX1、MC1および/またはMC3と75%を超えて競合する機能的フラグメントを含む非ネズミモノクローナル抗体もまた企図される。
【0014】
別の実施態様では、非ネズミモノクローナル抗体またはその機能的フラグメントが図7のアミノ酸98−105の少なくとも4、5、6、7または8個の近接する残基を含むM−CSFのエピトープに結合する機能的フラグメントを含む該非ネズミモノクローナル抗体が提供される。
【0015】
別の実施態様では、本発明は非ネズミモノクローナル抗体またはその機能的フラグメントが図7のアミノ酸65−73または138−144の少なくとも4、5、6、7または8個の近接する残基を含むM−CSFのエピトープ(5H4またはMC3により認識されるM−CSFエピトープに相当する)に結合する機能的フラグメントを含む該非ネズミモノクローナル抗体を提供する。
【0016】
さらに別の実施態様では、図7のアミノ酸98−105を含むM−CSFのエピトープに結合する前記された抗体またはフラグメントが提供される。関連する実施態様では、図1AのCDR3を含む前記された抗体が提供される。別の実施態様では、図1Aで示されるネズミ抗体RX1の少なくとも1、2、3、4、5または6個のCDRを含む抗体が提供される。ネズミ抗体RX1の少なくとも1、2、3、4または5個のCDRを含むかかる抗体はまた図8A−Bで示される抗体5H4の6個のCDRのいずれかの少なくとも1、2、3、4または5個のCDRを含んでなってよい。これに代えてネズミ抗体RX1の少なくとも1、2、3、4または5個のCDRを含む抗体はまた図8A−Bで示される抗体MC1の6個のCDRのいずれかの少なくとも1、2、3、4または5個のCDRを含んでなってよい。さらに別の代替えでは、前記された抗体はまた図8A−Bで示される抗体MC3の6個のCDRのいずれかの少なくとも1、2、3、4または5個のCDRを含んでなってよい。関連する実施態様では、ネズミ抗体RX1の少なくとも1、2、3、4または5個のCDRを含む抗体は提供される図8A−Bで示されるコンセンサスCDRの少なくとも1、2、3、4または5個のCDRを含んでなってよい。さらに別の関連する実施態様では、前記された抗体では(複数の)コンセンサスCDRの一つまたはそれより多い残基はネズミ抗体RX1、5H4、MC1またはMC3のCDRのいずれかの対応する残基により置換されている。抗体の一つまたはそれより多いアミノ酸が例えばCDRの保存置換、および/または低および中リスク残基における保存もしくは非保存変化により変異していたとしても、望ましい結合親和性を保持することができる。
【0017】
本発明の別の実施態様では、図1A、2、3または4で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変重鎖アミノ酸配列を含む前記された抗体のバリアントが提供される。関連する実施態様では、抗体は図1A、2、3または4で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変軽鎖アミノ酸配列を含む。
【0018】
さらに別の実施態様では、抗体はヒト抗体配列の定常領域ならびに一つまたはそれより多い重および軽鎖可変フレームワーク領域を含む。関連する実施態様では、抗体はヒトIgG1、IgG2、IgG3またはIgG4の修飾または未修飾定常領域を含む。好ましい実施態様では、定常領域はヒトIgG1またはIgG4であり、それは場合によっては特定の特性を強化または低下させるために修飾され得る。IgG1の場合、定常領域、特にヒンジまたはCH2領域に対する修飾は、ADCCおよび/またはCDC活性を含むエフェクター機能を上昇または低下させ得る。その他の実施態様では、IgG2定常領域を修飾して抗体−抗原凝集形成を低下させる。IgG4の場合、定常領域、特にヒンジ領域に対する修飾は、半抗体の形成を低下させ得る。
【0019】
本発明の一つの実施態様では、国際公開番号第WO2005/068503号に記載されるようなネズミ抗体RX1、5H4、MC1もしくはMC3のいずれか一つと同一のM−CSFのエピトープに特異的に結合するか、またはM−CSFに対する結合に関して10%を超えて、さらに好ましくは25%を超えて、なおさらに好ましくは50%を超えて、もっとさらに好ましくは75%を超えて、そして最も好ましくは90%を超えて前記されたネズミ抗体のいずれか一つと拮抗する非ネズミモノクローナル抗体が提供される。キメラ、ヒト、ヒト化、ヒト改変抗体またはそれのフラグメントもしくはムテインもしくは化学的に誘導体化されたものを含むかかるネズミ抗体の配列由来の抗体が第WO2005/068503号に記載されている。
【0020】
M−CSFに結合する能力を保持する「RX1由来の抗体」には以下のいずれか一つが含まれる:
1)相同性決定に関する類似アミノ酸を考慮して、図1で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変重鎖アミノ酸配列を含む、および/または図1で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変軽鎖アミノ酸配列を含むバリアントを含む、図1に示されるアミノ酸配列を有するネズミ抗体RX1のアミノ酸バリアント;
2)図1で示されるアミノ酸配列を有するネズミ抗体RX1の一つまたはそれより多い相補性決定領域(CDR)を含んでなり、好ましくはRX1重鎖の少なくともCDR3を含んでなり、そして好ましくは二つもしくはそれより多い、または三つもしくはそれより多い、または四つもしくはそれより多い、または五つもしくはそれより多い、または六つ全部のCDRを含むM−CSF結合ポリペプチド(ネズミ抗体RX1を除く);
3)図9Bから12Bで示される重および軽鎖アミノ酸配列を有するHuman Engineered(商標)抗体または図9Bから12Bの元来のHuman Engineered(商標)重もしくは軽鎖と少なくとも60%のアミノ酸配列同一性、さらに好ましくは少なくとも80%、さらに好ましくは少なくとも85%、さらに好ましくは少なくとも90%、そして最も好ましくは例えば65%、70%、75%、80%、81%、82%、83%、84%、85%。86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%および100%を含む少なくとも95%の同一性を有する重もしくは軽鎖を含むそのバリアント;
4)図9Bから12BのHuman Engineered(商標)抗体の一つまたはそれより多いCDRの高リスク残基を含んでなり、そして好ましくは二つもしくはそれより多い、または三つもしくはそれより多い、または四つもしくはそれより多い、または五つもしくはそれより多い、または六つ全部のCDRの高リスク残基を含むM−CSF結合ポリペプチド(ネズミ抗体RX1を除く);
5)図1Bで示される高リスクアミノ酸残基を保持し、そして図1Bで示される低または中リスク残基の一つまたはそれより多い変化を含む;
例えば図1Bで示される低リスク残基での一つまたはそれより多い変化および中リスク残基での保存置換を含むか;または
例えば図1Bで示される中および高リスクアミノ酸残基を保持し、そして低リスク残基で一つまたはそれより多い変化を含んでなり;
ここで変化は挿入、欠失または置換を含み、そして保存置換でよいか、または改変抗体の配列をヒト軽鎖もしくは重鎖配列、ヒト生殖系軽鎖もしくは重鎖配列、コンセンサスヒト軽鎖もしくは重鎖配列、またはコンセンサスヒト生殖系軽鎖もしくは重鎖配列に近づけることができる;
Human Engineered(商標)抗体またはバリアント。かかる抗体は好ましくは少なくとも10−7、10−8もしくは10−9またはそれより高い親和性を伴ってM−CSFに結合し、そして好ましくはM−CSFの活性を誘起する破骨細胞形成を中和する。
【0021】
同様にM−CSFに結合する能力を保持する「MC3由来の抗体」には以下のいずれか一つが含まれる:
1)相同性決定に関する類似アミノ酸を考慮して、図4で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変重鎖アミノ酸配列を含む、および/または図4で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変軽鎖アミノ酸配列を含むバリアントを含む、図4に示されるアミノ酸配列を有するネズミ抗体MC3のアミノ酸バリアント;
2)図4で示されるアミノ酸配列を有するネズミ抗体MC3の一つまたはそれより多い相補性決定領域(CDR)を含んでなり、好ましくはMC3重鎖の少なくともCDR3を含んでなり、そして好ましくは二つもしくはそれより多い、または三つもしくはそれより多い、または四つもしくはそれより多い、または五つもしくはそれより多い、または六つ全部のCDRを含むM−CSF結合ポリペプチド(場合によってはネズミ抗体MC3を含むかまたは除く);
3)Studnickaら、米国特許第5,766,886号および本明細書の実施例4Aに示される方法に従って、図13C−13Eに示されるKabat番号付けを用いて、低、中および高リスク残基を同定するためにネズミ配列を変化させることにより作成されたHuman Engineered(商標)抗体;かかる抗体は少なくとも一つの以下の重鎖および少なくとも一つの以下の軽鎖を含む:(a)低リスク残基の全てが修飾されており、必要によりヒト参照免疫グロブリン配列と同一の残基になる重鎖、または(b)低および中リスク残基の全てが修飾されており、必要によりヒト参照免疫グロブリン配列と同一の残基になる重鎖、(c)低リスク残基の全てが修飾されており、必要によりヒト参照免疫グロブリン配列と同一の残基になる軽鎖、または(d)低および中リスク残基の全てが修飾されており、必要によりヒト参照免疫グロブリン配列と同一の残基になる軽鎖;
4)元来のHuman Engineered(商標)重または軽鎖と少なくとも60%のアミノ酸配列同一性、さらに好ましくは少なくとも80%、さらに好ましくは少なくとも85%、さらに好ましくは少なくとも90%、そして最も好ましくは例えば65%、70%、75%、80%、81%、82%、83%、84%、85%。86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%および100%を含む少なくとも95%の同一性を有する重または軽鎖を含む前記のパラグラフ(3)において前記された抗体のバリアント;
5)図4のネズミMC3抗体の一つまたはそれより多いCDRの高リスク残基を含んでなり、そして好ましくは二つもしくはそれより多い、または三つもしくはそれより多い、または四つもしくはそれより多い、または五つもしくはそれより多い、または六つ全部のCDRの高リスク残基を含むM−CSF結合ポリペプチド(場合によってはネズミ抗体MC3を含むかまたは除く);
6)ネズミMC3抗体の高リスクアミノ酸残基を保持し、そして低または中リスク残基で一つまたはそれより多い変化を含む;
例えば低リスク残基での一つまたはそれより多い変化および中リスク残基での保存置換を含むか;または
例えば中および高リスクアミノ酸残基を保持し、そして低リスク残基で一つまたはそれより多い変化を含んでなり;
ここで変化は挿入、欠失または置換を含み、そして保存置換でよいか、または改変抗体の配列をヒト軽鎖もしくは重鎖配列、ヒト生殖系軽鎖もしくは重鎖配列、コンセンサスヒト軽鎖もしくは重鎖配列、またはコンセンサスヒト生殖系軽鎖もしくは重鎖配列に近づけることができる;
Human Engineered(商標)抗体またはバリアント。かかる抗体は好ましくは少なくとも10−7、10−8もしくは10−9またはそれより高い親和性を伴ってM−CSFに結合し、そして好ましくはM−CSFの活性を誘起する破骨細胞形成を中和する。
【0022】
「5H4由来の抗体」または「MC1由来の抗体」なる用語は前記の記載に従って同様に定義される。
【0023】
本明細書にて詳細に記載されるように、Human Engineered(商標)抗体またはバリアントを含むRX1、5H4、MC1またはMC3由来の抗体は、IgG、IgA、IgMまたはIgEのような異なるアイソタイプのものでよい。IgGクラスの抗体は異なる定常領域を含むことができ、例えばIgG2抗体をIgG1またはIgG4定常領域を表示するように修飾することができる。好ましい実施態様では、本発明は修飾または未修飾IgG1またはIgG4定常領域を含むHuman Engineered(商標)抗体またはバリアントを提供する。IgG1の場合、定常領域、特にヒンジまたはCH2領域に対する修飾は、ADCCおよび/またはCDC活性を含むエフェクター機能を上昇または低下させることができる。その他の実施態様では、IgG2定常領域を修飾して抗体−抗原凝集形成を低下させる。IgG4の場合、定常領域、特にヒンジ領域に対する修飾は半抗体の形成を低下させることができる。具体的な例示的な実施態様では、IgG4ヒンジ配列Cys−Pro−Ser−CysをIgG1ヒンジ配列Cys−Pro−Pro−Cysに変異させることが提供される。
【0024】
前記されたM−CSFアンタゴニストまたはM−CSF抗体および薬学的に許容される担体、賦形剤または希釈剤を含む医薬組成物を本発明に従って投与することができる。
【0025】
二つまたはそれより多いM−CSFアンタゴニストを一緒に混合するか、またはM−CSFアンタゴニストおよび第二の抗破骨細胞薬を同時投与して、癌転移および/または癌転移に関連する骨量減少を含む本発明の骨溶解性疾患に対して改善された効果を提供することはさらに有利であり得る。
【0026】
本発明の例示的な実施態様では、第二の抗破骨細胞薬がビスホスホネートである前記された方法が提供される。さらなる実施態様では、ビスホスホネートはゾレドロネート(zoledronate)、パミドロネート(pamidronate)、クロドロネート(clodronate)、エチドロネート(etidronate)、チルドロネート(tiludronate)、アレンドロネート(alendronate)、イバンドロナート(ibandronate)またはリセドロネート(risedronate)である。その他の抗破骨細胞薬の実例にはビスホスホネート系、PTHrP中和剤(例えば抗体、アンチセンス、siRNA)、カテプシンK阻害剤、MIP−1−αアンタゴニスト、RANK/RANKL中和剤(例えば抗RANK抗体、抗RANKL抗体またはアンチセンス、可溶性RANKL受容体またはそのムテイン)、RANKLワクチン、オステオプロテグリン(OPG)、血小板由来成長因子(PDGF)、srcキナーゼ阻害剤、マロン酸ガリウムおよびマトリックスメタロプロテイナーゼ(MMP)阻害剤が挙げられる。
【0027】
本発明の治療方法をさらに癌化学療法剤のような第三の治療薬と、または放射線治療もしくは外科的手術と組み合わせることができる。癌化学療法剤には、限定するものではないがカルボプラチンおよびシスプラチンのようなアルキル化剤;ナイトロジェンマスタードアルキル化剤;カルムスチン(BCNU)のようなニトロソウレアアルキル化剤;メトトレキサートのような代謝拮抗剤;プリン類似体代謝拮抗剤、メルカプトプリン;フルオロウラシル(5−FU)およびゲムシタビンのようなピリミジン類似体代謝拮抗剤;ゴセレリン、リュープロリドおよびタモキシフェンのような抗腫瘍性ホルモン薬;アルデスロイキン、インターロイキン−2、ドセタキセル、エトポシド(VP−16)、インターフェロンアルファ、パクリタキセルおよびトレチノイン(ATRA)のような天然抗悪性腫瘍薬;ブレオマイシン、ダクチノマイシン、ダウノルビシン、ドキソルビシンおよびマイトマイシンのような抗菌性天然抗悪性腫瘍薬;ならびにビンブラスチン、ビンクリスチン、ビンデシンのようなビンカアルカロイド天然抗悪性腫瘍薬;ヒドロキシウレア;アセグラトン、アドリアマイシン、イフォスファミド、エノシタビン、エピチオスタノール、アクラルビシン、アンシタビン、ニムスチン、塩酸プロカルバジン、カルボコン、カルボプラチン、カルモフール、クロモマイシンA3、抗腫瘍性多糖類、抗腫瘍性血小板因子、シクロホスファミド、シゾフィラン、シタラビン、ダカルバジン、チオイノシン、チオテパ、テガフル、ネオカルチノスタチン、OK−432、ブレオマイシン、フルツロン、ブロクスウリジン、ブスルファン、ホンバン、ペプロマイシン、ベスタチン(ウベニメクス)、インターフェロン−β、メピチオスタン、ミトブロニトール、メルファラン、ラミニンペプチド、レンチナン,カワラタケ(Coriolus versicolor)抽出物、テガフル/ウラシル、エストラムスチン(エストロゲン/メクロレタミン)が含まれる。
【0028】
さらに、癌患者の補助的療法として用いられるさらなる薬剤には、EPO、G−CSF、ガンシクロビル;抗生物質、ロイプロリド;メペリジン;ジドブジン(AZT);変異体および類似体を含むインターロイキン1から18;インターフェロンα、βおよびγのようなインターフェロン系またはサイトカイン系;黄体形成ホルモン放出ホルモン(LHRH)および類似体、および生殖腺刺激ホルモン放出ホルモン(GnRH)のようなホルモン系;形質転換成長因子β(TGF−β)、線維芽細胞成長因子(FGF)、神経成長因子(NGF)、成長ホルモン放出因子(GHRF)、上皮細胞成長因子(EGF)、線維芽細胞成長因子相同因子(FGFHF)、肝細胞成長因子(HGF)およびインスリン成長因子(IGF)のような成長因子;腫瘍壊死因子α&β(TNF−α&β);浸潤阻止因子(invasion inhibiting factor)2(IIF−2);骨形成タンパク質1−7(BMP1−7);ソマトスタチン;チモシンα−1;γグロブリン;スーパーオキシドジスムターゼ(SOD);補体因子;抗血管形成因子;抗原性物質;プロドラッグ;成長因子受容体キナーゼ阻害剤;抗Her2抗体;ならびにVEGF中和抗体が含まれる。
【0029】
移行期間に続いて、治療効果を達成するために必要とされるM−CSFアンタゴニストの量または第二の抗破骨細胞薬の量を減少させることができる。したがってかかる期間の後、M−CSFアンタゴニストが第二の抗破骨細胞薬の効果を改善するか、または第二の抗破骨細胞薬の投与に随伴される副作用を低減するか、または第二の抗破骨細胞薬の安全性を改善することができる。M−CSFアンタゴニストはまた効果を改善するか、癌化学療法剤、その他の補助的療法、外科的手術または放射線治療のような第三の治療法の副作用を低減するか、または安全性を改善することができる。本発明の別の実施態様では、M−CSFアンタゴニストを含む医薬品、ならびにその医薬品が第二および/もしくは第三の治療薬との、および/または外科的手術もしくは放射線治療との組み合わせで用いられるべきであるとする説明書を含む包装、バイアルまたは容器が提供される。
【0030】
多くの骨溶解性障害は本発明による処置に適していると企図される。本明細書にて使用される際には、「骨溶解性障害」は破骨細胞活性の上昇の結果である任意の症状である。骨溶解性障害の危険性を有する対象は、骨溶解性障害を発達させる傾向がある群の対象、または骨破壊活性の上昇を引き起こすかもしくはそれに寄与する疾患を患う対象でよい。本発明の例示的な実施態様では骨溶解性障害は、内分泌疾患(副腎皮質ホルモン過剰症、性腺機能低下症、原発性または二次性副甲状腺機能亢進症、甲状腺機能亢進症)、高カルシウム血症、欠乏状態(クル病/骨軟化症、壊血病、栄養障害)、慢性疾患(吸収不良症候群、慢性腎不全(腎性骨異栄養症)、慢性肝疾患(肝性骨異栄養症))、薬物(糖質コルチコイド(糖質コルチコイド誘起骨粗鬆症)、ヘパリン、アルコール)または遺伝性疾患(骨形成不全症、ホモシスチン尿症)、癌、骨粗鬆症、大理石骨病、関節炎および関節リウマチに関連する骨の炎症、歯周病、線維性骨異形成症および/またはパジェット病を含む破骨細胞活性の相対的な上昇に関連する代謝性骨疾患でよい。
【0031】
その他の例示的な実施態様では、骨溶解性障害は骨への転移性癌でよく、ここで転移性癌は乳腺、肺、腎臓、多発性骨髄腫、甲状腺、前立腺、腺癌、白血病およびリンパ腫を含む血液細胞悪性腫瘍;頭頸部癌;食道癌、胃癌、結腸癌、腸癌、結腸直腸癌、直腸癌、膵臓癌、肝臓癌、胆管もしくは胆嚢の癌を含む胃腸管系癌;卵巣癌腫、子宮内膜癌、膣癌もしくは子宮頸癌を含む女性生殖器の悪性腫瘍;膀胱癌;神経芽細胞腫を含む脳癌;肉腫、骨肉腫;または悪性メラノーマもしくは扁平上皮癌を含む皮膚癌である。
【0032】
本発明の例示的な実施態様では、前記の方法のいずれかは骨量減少を防御するかまたは低減させることができるか、または疾患に関連する骨転移もしくは骨量減少の重篤度を防御もしくは低減させる。
【0033】
本発明に従って投与されるM−CSF抗体を約2μg/kgから30mg/kg、0.1mg/kgから30mg/kg、または0.1mg/kgから10mg/kg体重の間の用量で与えることができる。
例えば、本願発明は以下の項目を提供する。
(項目1)
単剤治療上有効な用量の抗M−CSF抗体および単剤治療上有効な用量の破骨細胞阻害剤を骨溶解性障害を患う患者に投与する工程を含む、該患者を処置する方法。
(項目2)
単剤治療上有効な用量の抗M−CSF抗体および単剤治療上有効な用量のビスホスホネートを骨溶解性障害を患う患者に投与する工程を含む該患者を処置する方法。
(項目3)
前記破骨細胞阻害剤がRANKL阻害剤である項目1に記載の方法。
(項目4)
前記抗MCSF抗体がRX1由来の抗体、MC1由来の抗体、MC3由来の抗体または5H4由来の抗体である項目2に記載の方法。
(項目5)
前記抗MCSF抗体がheRX1である項目2に記載の方法。
(項目6)
前記骨溶解性障害が骨粗鬆症、癌転移に関連する骨量減少、パジェット病またはプロテーゼ周囲骨量減少である項目1から5のいずれかに記載の方法。
(項目7)
前記破骨細胞阻害剤および前記MCSF抗体が同時に投与される項目1から6のいずれかに記載の方法。
(項目8)
前記RANKL阻害剤が抗RANKL抗体、可溶性RANKL受容体およびRANKLワクチンからなる群から選択される項目に記載の方法。
(項目9)
前記抗RANKL抗体がAMG−162である項目3、4、6および7に記載の方法。
(項目10)
前記ビスホスホネートがゾレドロネート、パミドロネート、クロドロネート、エチドロネート、チルドロネート、アレンドロネート、イバンドロナートおよびリセドロネートからなる群から選択される項目2、4、6および7に記載の方法。
(項目11)
前記ビスホスホネートがゾレドロネートである項目10に記載の方法。
(項目12)
移行期間に単剤治療上有効な用量の抗M−CSF抗体および単剤治療上有効な用量のビスホスホネートを骨溶解性障害を患う患者に投与する工程を含む、該患者を処置する方法。
(項目13)
前記破骨細胞阻害剤がRANKL阻害剤である項目12に記載の方法。
(項目14)
前記移行期間がおよそ1−7日である項目13に記載の方法。
(項目15)
前記移行期間が1週間から1か月である項目13に記載の方法。
(項目16)
前記移行期間が1か月から3か月である項目13に記載の方法。
(項目17)
前記移行期間が3から6か月である項目13に記載の方法。
(項目18)
前記移行期間が6から12か月である項目13に記載の方法。
(項目19)
前記移行期間後に前記ビスホスホネート治療を中断する工程をさらに含む項目13に記載の方法。
(項目20)
前記移行期間後に前記ビスホスホネートの用量を減少させる工程をさらに含む項目13に記載の方法。
(項目21)
前記移行期間の直後にビスホスホネートの用量を減少させる項目13に記載の方法。
(項目22)
前記移行期間後に抗MCSF抗体の用量を減少させる工程をさらに含む項目13に記載の方法。
(項目24)
前記移行期間の直後に抗MCSF抗体の用量を減少させる項目13に記載の方法。
(項目25)
前記移行期間後に前記ビスホスホネートが少なくとも1回投与される項目13に記載の方法。
(項目26)
前記抗MCSF抗体が配列番号5および配列番号6のネズミRX1抗体からの少なくとも二つのCDRを含む項目1から25のいずれかに記載の方法。
(項目27)
前記抗MCSF抗体が配列番号5および配列番号6のネズミRX1抗体からの少なくとも三つのCDRを含む項目1から26のいずれかに記載の方法。
(項目28)
前記抗MCSF抗体が配列番号5および配列番号6のM−CSFに対する結合に関して少なくとも75%、ネズミRX1抗体と競合する項目1から27のいずれかに記載の方法。
(項目29)
前記破骨細胞阻害剤がゾレドロネートであり、そして前記MCSF抗体がRX1由来の抗体である項目6、13および20に記載の方法。
(項目30)
前記ゾレドロネートが0.5mgと4mgの間の用量で投与される項目29に記載の方法。
(項目31)
前記抗MCSF抗体がRX1、MC1、MC3または5H4抗体と同一のエピトープに結合する項目4、6、8、10、11または12に記載の方法。
【図面の簡単な説明】
【0034】
【図1A】M−CSF特異的ネズミ抗体RX1のアミノ酸配列(配列番号2および4)(アメリカン・タイプ・カルチャー・コレクション(米国、バージニア州、マナサス)でATCC寄託番号PTA−6113の下に寄託されたプラスミドのcDNAインサートによりコードされる)および対応する核酸配列(配列番号1および3)を示す。CDR領域は番号付けされ、そして太字で示される。
【図1B】各々M−CSF特異的ネズミ抗体RX1軽鎖(配列番号5)および重鎖(配列番号6)のアミノ酸配列を示し、Studnickaら、第WO93/11794号に従って同定された高リスク(太字)、中リスク(下線)および低リスク残基を伴う。
【図1C】各々M−CSF特異的ネズミ抗体RX1軽鎖(配列番号5)および重鎖(配列番号6)のアミノ酸配列を示し、Studnickaら、第WO93/11794号に従って同定された高リスク(太字)、中リスク(下線)および低リスク残基を伴う。
【図2】各々M−CSF特異的ネズミ抗体5H4(配列番号10および11)、MC1(配列番号12および13)(ATCC寄託番号PTA−6263の下に寄託されたハイブリドーマにより生成される)およびMC3(配列番号14および15)(ATCC寄託番号PTA−6264の下に寄託されたハイブリドーマにより生成される)のアミノ酸配列を示す。
【図3】各々M−CSF特異的ネズミ抗体5H4(配列番号10および11)、MC1(配列番号12および13)(ATCC寄託番号PTA−6263の下に寄託されたハイブリドーマにより生成される)およびMC3(配列番号14および15)(ATCC寄託番号PTA−6264の下に寄託されたハイブリドーマにより生成される)のアミノ酸配列を示す。
【図4】各々M−CSF特異的ネズミ抗体5H4(配列番号10および11)、MC1(配列番号12および13)(ATCC寄託番号PTA−6263の下に寄託されたハイブリドーマにより生成される)およびMC3(配列番号14および15)(ATCC寄託番号PTA−6264の下に寄託されたハイブリドーマにより生成される)のアミノ酸配列を示す。
【図5】M−CSFαのアミノ酸配列(配列番号7)である。
【図6】M−CSFβのアミノ酸配列(配列番号8)である。
【図7】M−CSFγのアミノ酸配列(配列番号9)である。DNA配列における多くの多型はアミノ酸の差異を招き得る。例えば共通の多型は104位でProよりむしろAlaを提供する。
【図8A】ヒトM−CSF特異的ネズミ抗体RX1;5H4;MC1;およびMC3の重および軽鎖アミノ酸配列(配列番号16−38)のCDR領域のアラインメントである。
【図8B】ヒトM−CSF特異的ネズミ抗体RX1;5H4;MC1;およびMC3の重および軽鎖アミノ酸配列(配列番号16−38)のCDR領域のアラインメントである。
【図9A】(a)ネズミRX1重鎖のリスクライン(H=高リスク、M=中リスク、L=低リスク)、(b)RX1重鎖アミノ酸配列(配列番号6)、(c)RX1とアラインメントされた最も近いヒトコンセンサス配列、KabatVh2コンセンサスのアミノ酸配列(配列番号39)および(d)二つのHuman Engineered(商標)配列(配列番号41および43)の実例を生成させた変化を示す。
【図9B】「低リスク」および「低+中リスク」と称される二つの重鎖Human Engineered(商標)配列の実例のアミノ酸配列(配列番号41および43)ならびに対応する核酸配列(配列番号40および42)を示す。
【図10A】(a)ネズミRX1軽鎖のリスクライン(H=高リスク、M=中リスク、L=低リスク)、(b)RX1軽鎖アミノ酸配列(配列番号5)、(c)RX1とアラインメントされた最も近いヒトコンセンサス配列、KabatVk3コンセンサスのアミノ酸配列(配列番号49)および(d)二つのHuman Engineered(商標)配列(配列番号45および47)の実例を生成させた変化を示す。
【図10B】「低リスク」および「低+中リスク」と称される二つの軽鎖Human Engineered(商標)配列の実例のアミノ酸配列(配列番号45および47)ならびに対応する核酸配列(配列番号44および46)を示す。
【図11A】(a)ネズミRX1軽鎖のリスクライン(H=高リスク、M=中リスク、L=低リスク)、(b)RX1軽鎖アミノ酸配列(配列番号5)、(c)RX1とアラインメントされた最も近いヒトコンセンサス配列、KabatVk3コンセンサスのアミノ酸配列(配列番号49)および(d)54−56位が変化しなかった(すなわちネズミ配列のままである)代替えのアミノ酸配列の実例(配列番号48)を示す。
【図11B】二つの代替えの軽鎖Human Engineered(商標)配列の実例のアミノ酸配列(配列番号48、87)ならびに対応する核酸配列(配列番号88および86)を示す。
【図12A】(a)ネズミRX1軽鎖のリスクライン(H=高リスク、M=中リスク、L=低リスク)、(b)RX1軽鎖アミノ酸配列(配列番号5)、(c)RX1とアラインメントされた最も近いヒトコンセンサス生殖系配列、Vk6サブグループ2−1−(1)A14のアミノ酸配列(配列番号50)および(d)二つのHuman Engineered(商標)配列(配列番号51および53)の実例を生成させた変化を示す。
【図12B】「低リスク」および「低+中リスク」と称される二つの軽鎖Human Engineered(商標)配列の実例のアミノ酸配列(配列番号51および53)ならびに対応する核酸配列(配列番号52)を示す。
【図13A】Kabat番号付けシステム(「POS」と称される一直線で示されるアミノ酸番号付け)を用いて種々のヒトコンセンサスおよびヒト生殖系コンセンサス配列を伴うネズミRX1重鎖アミノ酸配列(配列番号54)のアラインメント(配列番号55−83)を示す。
【図13B】Kabat番号付けシステム(「POS」と称される一直線で示されるアミノ酸番号付け)を用いて種々のヒトコンセンサスおよびヒト生殖系コンセンサス配列を伴うネズミRX1重鎖アミノ酸配列(配列番号54)のアラインメント(配列番号55−83)を示す。
【図13C】Kabat番号付けシステムに対応する抗体5H4、MC1およびMC3のアミノ酸残基(各々配列番号10および11;配列番号12および13;配列番号14および15)がどのようになっているかを示す。
【図13D】Kabat番号付けシステムに対応する抗体5H4、MC1およびMC3のアミノ酸残基(各々配列番号10および11;配列番号12および13;配列番号14および15)がどのようになっているかを示す。
【図13E】Kabat番号付けシステムに対応する抗体5H4、MC1およびMC3のアミノ酸残基(各々配列番号10および11;配列番号12および13;配列番号14および15)がどのようになっているかを示す。
【図14】動物モデルにおけるZometaの抗吸収効果を示す。
【図15】検出可能な骨溶解を伴う各群の動物のパーセントを示す。
【図16】試験の最後の日のX線画像分析に基づく平均骨溶解スコアを示す。
【図17】試験の最終日の脛骨(腫瘍接種部位)の代表的なFaxitron X線画像を示す。
【図18】破骨細胞活性に及ぼすRX1の効果を示す。
【図19】Zometaによる破骨細胞活性の阻止を示す。
【図20】霊長類におけるRX1での薬物動態試験の結果を示す。
【図21】霊長類におけるRX1での薬物動態試験の結果を示す。
【発明を実施するための形態】
【0035】
(詳細な説明)
マクロファージコロニー刺激因子(M−CSF)としても公知のコロニー刺激因子(CSF−1)は破骨細胞形成に重要であることが分かっている。加えて、M−CSFは成熟破骨細胞の骨破壊機能、その他の可溶性因子と協働してその遊走およびその生存性、ならびに骨芽細胞および線維芽細胞により提供される細胞間相互作用を変調することが示されている(FixeおよびPraloran、Cytokine 10:3−7(1998);Martinら、Critical Rev.in Eukaryotic Gene Expression 8:107−23(1998))。
【0036】
全長ヒトM−CSF mRNAは554個のアミノ酸の前駆体タンパク質をコードする。選択的mRNAスプライシングおよび差次的翻訳後タンパク質プロセシングにより、M−CSFは糖タンパク質もしくは硫酸コンドロイチン含有プロテオグリカンとして循環中に分泌されるか、またはM−CSF産生細胞の表面の膜を貫通する糖タンパク質として発現されるかのいずれかになり得る。完全なインビトロ生物学的活性に必要とされる最小配列であるヒトM−CSFの細菌発現されたアミノ末端の150個のアミノ酸の三次元構造は、このタンパク質は各単量体が四つのアルファヘリックス束および逆平行ベータシートからなる、ジスルフィド連結された二量体であることを示している(Panditら、Science 258:1358−62(1992))。三つの異なるM−CSF種は選択的mRNAスプライシングにより生成される。三つのポリペプチド前駆体は256個のアミノ酸のM−CFSα、554個のアミノ酸のM−CSFβ、および438個のアミノ酸のM−CSFγである。M−CSFβは膜結合形態では生じない分泌タンパク質である。M−CSFαはタンパク質切断により緩徐に放出される膜内在性タンパク質として発現される。M−CSFαは図5で示される配列のアミノ酸191−197で切断されている。M−CSFの膜結合形態は近くの細胞の受容体と相互作用することができ、そしてしたがって特異的細胞間接触を媒介する。「M−CSF」なる用語はまた図7のアミノ酸36−438をも含み得る。
【0037】
種々の形態のM−CSFは標的細胞でその受容体M−CSFRに結合することにより機能する。M−CSFRは五つの細胞外免疫グロブリン様ドメイン、膜貫通ドメインおよび細胞内分断Src関連チロシンキナーゼドメインを有する膜を貫通する分子である。M−CSFRはc−fms癌原遺伝子によりコードされる。M−CSFRの細胞外ドメインへのM−CSFの結合により、受容体の二量体化に至り、それは細胞質キナーゼドメインを活性化し、自己リン酸化およびその他の細胞タンパク質のリン酸化に至る(Hamilton J.A.、J Leukoc Biol.62(2):145−55(1997);Hamilton J.A.、Immuno Today.,18(7):313−7(1997))。
【0038】
リン酸化された細胞タンパク質は細胞応答に至る生化学的事象のカスケード:有糸分裂、サイトカインの分泌、膜ラフリングおよびその独自の受容体の転写の調節を誘起する(FixeおよびPraloran、Cytokine 10:32−37(1998))。
【0039】
「腫瘍」とは本明細書にて使用される際には、悪性であろうと良性であろうと、全ての新生物の細胞成長および増殖、ならびに全ての前癌性および癌性細胞および組織を指す。
【0040】
「癌」および「癌性」なる用語は、典型的には無秩序な細胞成長を特徴とする哺乳動物における生理学的症状を指すかまたは記載する。癌の実例には、限定するものではないが癌腫;リンパ腫、芽細胞腫、肉腫;および白血病が挙げられる。かかる癌のさらに特定の実例には、乳癌、前立腺癌、結腸癌、扁平上皮癌、小細胞肺癌、非小細胞肺癌、胃腸管系の癌、膵臓癌、神経膠芽腫、子宮頸癌、卵巣癌、肝臓癌、膀胱癌、肝細胞腫、結腸直腸癌、子宮内膜癌腫、唾液腺癌腫;腎臓癌、肝臓癌、外陰癌、甲状腺癌、肝細胞癌腫および種々の型の頭頸部癌が挙げられる。
【0041】
「処置」は障害の発達を防御するか、または病理を変化させる意図を伴って実施される介入である。したがって「処置」は治療的処置および予防的または防御的手段の双方を指す。処置を必要とするものには既に障害を有するもの、および障害が防御されるべきものが含まれる。腫瘍(例えば癌)処置において、治療薬は腫瘍細胞の病理を直接低減させるか、またはその他の治療薬、例えば放射線および/もしくは化学療法による処置に対する腫瘍細胞の感受性をさらに高めることができる。癌の「病理」には患者の健康を危険にさらす全ての現象が含まれる。これには限定するものではないが、異常なまたは制御できない細胞成長、転移、近隣細胞の正常な機能の妨害、サイトカインまたはその他の分泌性生成物の異常レベルでの放出、炎症性または免疫学的応答の抑制または激化等が含まれる。
【0042】
処置の目的のための「哺乳動物」とはヒト、家庭用および農場用の動物、ならびに動物園、スポーツまたはペット用動物、例えばイヌ、ウマ、ネコ、ウシ、等を含む哺乳動物として分類される任意の動物を指す。好ましくは、哺乳動物はヒトである。
【0043】
本明細書にて使用される際には、「転移性癌」なる語句は身体のその他の部分、特に骨にまで広がる可能性を有する癌として定義される。種々の癌が骨に転移することができるが、最も一般的な転移する癌は乳腺、肺、腎臓、多発性骨髄腫、甲状腺および前立腺である。実例としては、骨に転移する可能性を有するその他の癌には、限定するものではないが腺癌、白血病およびリンパ腫を含む血液細胞悪性腫瘍;頭頸部癌;食道癌、胃癌、結腸癌、腸癌、結腸直腸癌、直腸癌、膵臓癌、肝臓癌、胆管もしくは胆嚢の癌を含む胃腸管系癌;卵巣癌腫、子宮内膜癌、膣癌および子宮頸癌を含む女性生殖器の悪性腫瘍;膀胱癌;神経芽細胞腫を含む脳癌;肉腫、骨肉腫;ならびに悪性メラノーマおよび扁平上皮癌を含む皮膚癌が挙げられる。本発明は特に骨における腫瘍誘起の骨溶解性病変の防御および処置を企図する。
【0044】
本明細書にて使用される際には、「治療上有効な量」なる語句は、望ましい処置レジメンに従って投与された場合、望ましい治療的または予防的効果または応答を引き出すM−CSF抗体のような本発明の実施態様に適切であろう、治療用または予防用のM−CSFアンタゴニストの量を指すと意図されることを指す。
【0045】
ヒト「M−CSF」とは、本明細書にて使用される際には、Kawasakiら、Science 230:291(1985)、Cerrettiら、Molecular Immunology,25:761(1988)、またはLadnerら、EMBO Journal 6:2693(1987)(その各々を出典明示により本明細書の一部とする)に記載される成熟ヒトM−CSFα、M−CSFβまたはM−CSFγポリペプチドと実質的に同一のアミノ酸配列を有するヒトポリペプチドを指す。かかる用語法は、三つの成熟M−CSFが前記で記載されたように異なるアミノ酸配列を有し、そしてM−CSFの活性形態はジスルフィド結合した二量体であることの理解を反映しており;したがって「M−CSF」なる用語が生物学的に活性な形態を指す場合、二量体形態が意図される。「M−CSF二量体」とは二量体化している二つのM−CSFポリペプチド単量体を指し、そして双方のホモ二量体(同一の型のM−CSF単量体の二つからなる)およびヘテロ二量体(二つの異なる単量体からなる)を含む。米国特許第4,929,700号(出典明示により本明細書の一部とする)に記載されるように、M−CSF単量体をインビトロでM−CSF二量体に変換することができる。
【0046】
1.アンタゴニスト
本明細書にて使用される際には、「アンタゴニスト」なる用語は一般的に、例えば一つの分子のと別の分子との結合を妨害するための分子、化合物もしくはその他の薬剤の特性、または立体障害、立体構造の変化もしくはその他の生化学的メカニズムのいずれかによる一つ細胞の別の細胞による刺激を指す。一つの点については、アンタゴニストなる用語は、受容体のそのリガンドに対する結合、例えばそれによりM−CSFが誘因となるシグナル伝達経路を阻止するM−CSFのM−CSFRとの結合を防御する薬剤の特性を指す。アンタゴニストなる用語は任意の具体的な作用メカニズムにより限定されないが、むしろ一般的に現在定義されている機能的特性を指す。本発明のアンタゴニストには、限定するものではないが:M−CSF抗体およびそのフラグメントおよびムテインおよび修飾体、可溶性M−CSFおよびそのフラグメントおよびムテインおよび修飾体、M−CSFR抗体およびそのフラグメントおよびムテインおよび修飾体、可溶性M−CSFRおよびそのフラグメントおよびムテインおよび修飾体、ならびにM−CSFまたはM−CSFRに結合するペプチドおよびその他の化学的化合物および分子、ならびにM−CSFおよびM−CSFRの発現を阻止するアンチセンスまたはRNAiのような核酸分子が含まれる。本発明のアンタゴニストのいずれかを当分野において公知の任意の様式で投与することができる。例えばM−CSFムテイン、M−CSFRムテインまたはM−CSFもしくはM−CSFRに結合する抗体フラグメントを、遺伝子治療を介して投与することができる。
【0047】
本発明のM−CSFアンタゴニストには、適用可能な場合、機能的均等物が含まれる。例えば分子は長さ、構造、構成成分等で異なり得るが、依然として一つまたはそれより多い、定義された機能を保持し得る。さらに特に、本発明の抗体、抗体フラグメントまたはペプチドの機能的均等物には、擬似化合物、すなわち抗原結合に関して適切な立体構造および/または配向を擬似するように設計された構築物が含まれ得る。
【0048】
好ましいM−CSFアンタゴニストを、場合によっては側鎖の付加等により、例えばアミノ末端アシル化、カルボキシ末端アミド化またはアミノ酸側鎖へのさらなる基の結合により修飾することができる。アンタゴニストはまた一つまたはそれより多い保存アミノ酸置換を含むことができる。「保存アミノ酸置換」とは全体的な電荷、疎水性/親水性および/または置換されたアミノ酸の立体容積を保存する、アミノ酸配列におけるこれらの変化を意味する。例えば以下の基の間の置換は保存的である:Gly/Ala、Val/Ile/Leu、Asp/Glu、Lys/Arg、Asn/Gln、Ser/Cys/ThrおよびPhe/Trp/Tyr。かかる修飾は実質的にはM−CSFアンタゴニストの効果を減じることはなく、そして例えばインビボで半減期の増加または毒性の低下のような望ましい特性を付与し得る。
【0049】
本発明はまたアミノ酸残基の挿入、欠失または置換以外の修飾を担持するポリペプチドを含むことも意図される。実例としては、修飾は天然の共有結合性でよく、そして例えば重合体、脂質、その他の有機および無機部分との化学的結合を含み得る。かかる誘導体を調製して、ポリペプチドの循環半減期を増すことができるか、またはポリペプチドが望ましい細胞、組織もしくは器官を標的とする能力を改善するように設計することができる。同様に、本発明はさらにポリエチレングリコール、ポリオキシエチレングリコールまたはポリプロピレングリコールのような一つまたはそれより多い水溶性重合体連結を含むように共有結合的に修飾されているM−CSFまたはM−CSFRポリペプチドを包含する。
【0050】
A.M−CSF抗体
「抗体」なる用語は最も広義で用いられ、そして完全に組み立てられた抗体、モノクローナル抗体、ポリクローナル抗体、多重特異性抗体(例えば二重特異性抗体)、抗原に結合できる抗体フラグメント(例えばFab’、F’(ab)2、Fv、一本鎖抗体、ダイアボディー)および望ましい生物学的活性を呈する限り前記のものを含む組換えペプチドを含む。
【0051】
「モノクローナル抗体」なる用語は本明細書にて使用される際には、実質的に均一な抗体の集団から得られる抗体を指し、すなわち集団を含む個々の抗体は少量で存在し得る可能な天然発生変異を除いて同一である。モノクローナル抗体は高度に特異的であり、単一の抗原部位に対して指向する。さらに典型的には異なる決定基(エピトープ)に対して指向する異なる抗体を含む従来の(ポリクローナル)抗体調製物とは対照的に、各モノクローナル抗体は抗原上の単一の決定基に対して指向する。その特異性に加えて、モノクローナル抗体は均一な培養により合成され、異なる特異性および特徴を有するその他の免疫グロブリンにより汚染されていないという点で有利である。
【0052】
「モノクローナル」なる修飾語句は抗体の実質的に均一な集団から得られるような抗体の性状を示し、そして任意の特定の方法による抗体の産生を必要とすると解釈されるべきではない。例えば本発明に従って用いられるモノクローナル抗体を、Kohlerら、Nature,256:495(1975)により最初に記載されたハイブリドーマ法により作成することができるか、または組換えDNA法(例えば米国特許第4,816,567号参照)により作成することができる。「モノクローナル抗体」を例えばClacksonら、Nature,352:624628(1991)およびMarksら、J.Mol.Biol.,222:581−597(1991)に記載される技術を用いてファージ抗体ライブラリーから単離することもできる。
【0053】
その重鎖の定常ドメインのアミノ酸配列に依存して、免疫グロブリンを異なるクラスに割り当てることができる。IgA、IgD、IgE、IgGおよびIgMの五つの主要なクラスがあり、そしてこれらのうちのいくつかをさらにサブクラスまたはアイソタイプ、例えばIgG1、IgG2、IgG3、IgG4、IgA1およびIgA2に分けることができる。免疫グロブリンの異なるクラスに相当する重鎖定常ドメインを各々アルファ、デルタ、イプシロン、ガンマおよびミューと称する。免疫グロブリンの異なるクラスのサブユニット構造および三次元立体構造は周知である。異なるアイソタイプは異なるエフェクター機能を有し、例えばIgG1およびIgG3アイソタイプはADCC活性を有する。
【0054】
「抗体フラグメント」はインタクトな全長抗体の一部、好ましくはインタクトな抗体の抗原結合または可変領域を含む。抗体フラグメントの実例には、Fab、Fab’、F(ab’)2およびFvフラグメント;ダイアボディー;直鎖状抗体(Zapataら、Protein Eng.,8(10):1057−1062(1995));一本鎖抗体分子;および抗体フラグメントから形成された多重特異性抗体が挙げられる。抗体のパパイン消化により「Fab」フラグメントと称される二つの同一の抗原結合フラグメントが生成され、各々単一の抗原結合部位および、残りの「Fc」フラグメントを有し、「Fc」フラグメントの名前は35を容易に結晶化するその能力を反映する。ペプシン処理により、抗体のVHおよびVLドメイン(これらのドメインは単一のポリペプチド鎖に存在する)を含む二つの「一本鎖Fv」すなわち「sFv」抗体フラグメントを有するF(ab’)2フラグメントを生じる。好ましくは、FvポリペプチドはさらにVHおよびVLドメイン間でポリペプチドリンカーを含んでなり、それはFvが抗原結合に関して望ましい構造を形成するのを可能にする。sFvの概説に関しては、Pluckthun、The
Pharmacology of Monoclonal Antibodies、113巻、RosenburgおよびMoore編、Springer−Verlag(ニューヨーク)、269−315頁(1994)を参照のこと。
【0055】
「超可変」領域なる用語は抗原結合に寄与する抗体のアミノ酸残基を指す。超可変領域は相補性決定領域、すなわちCDRからのアミノ酸残基(すなわちKabatら、Sequences of Proteins of Immunological Interest、第5版、公衆衛生局、国立衛生研究所、メリーランド州、ベセスダ(1991)に記載されるように、軽鎖可変ドメインの残基24−34(L1)、50−56(L2)および89−97(L3)ならびに重鎖可変ドメインの31−35(H1)、50−65(H2)および95−102(H3))および/または超可変ループからのこれらの残基(すなわちChothiaら、J.Mol.Biol.196:901−917(1987)に記載されるように、軽鎖可変ドメインの残基26−32(L1)、50−52(L2)および91−96(L3)ならびに重鎖可変ドメインの26−32(H1)、53−55(H2)および96−101(H3)を含む。
【0056】
「フレームワーク」またはFR残基は超可変領域残基以外のこれらの可変ドメイン残基である。
【0057】
「ダイアボディー」なる用語は二つの抗原結合部位を有する小型の抗体フラグメントを指し、そのフラグメントは同一のポリペプチド鎖(VH VL)で軽鎖可変ドメイン(VL)に連結された重鎖可変ドメイン(VH)を含む。非常に短くて同一鎖上の二つのドメイン間で対形成することができないリンカーを用いることにより、ドメインをその他の鎖の相補的ドメインと対形成させ、そして二つの抗原結合部位を創成する。ダイアボディーは例えば欧州特許第404,097号;第WO93/11161号;および30Hollingerら、Proc.Natl.Acad.Sci.USA,90:6444−6448(1993)にてさらに十分に記載されている。
【0058】
いくつかの実施態様では、少なくとも二つの異なるエピトープに関して結合特異性を有するモノクローナル、ヒト、ヒト化、Human Engineered(商標)またはバリアント抗M−CSF抗体を含む多重特異性(例えば二重特異性)モノクローナル抗体を作成するのが望ましい場合もある。二重特異性抗体の実例はM−CSFの二つの異なるエピトープに結合し得る。これに代えて、抗M−CSFアームをT細胞受容体分子(例えばCD2またはCD3)のような白血球上のトリガー分子、またはFcγRI(CD64)、FcγRII(CD32)およびFcγRIII(CD16)のようなIgGに関するFc受容体(FcγR)に結合するアームと組み合わせて、M−CSF発現細胞に対する細胞防衛メカニズムに集中させることができる。二重特異性抗体を用いてM−CSFを発現する細胞に細胞毒性薬を局在化させることもできる。これらの抗体はM−CSF結合アームおよび細胞毒性薬(例えばサポリン、抗インターフェロン60、ビンカアルカロイド、リシンA鎖、メトトレキサートまたは放射性同位元素ハプテン)に結合するアームを有する。二重特異性抗体を全長抗体または抗体フラグメント(例えばF(ab’).sub.2二重特異性抗体)として調製することができる。
【0059】
二重特異性抗体を作成するための別の研究法に従って、抗体分子の対の間のインターフェースを操作して組換え細胞培養から回収されるヘテロ二量体のパーセンテージを最大にすることができる。好ましいインターフェースは抗体定常ドメインのCH3ドメインの少なくとも一部を含む。この方法では、最初の抗体分子のインターフェースからの一つまたはそれより多い小型アミノ酸側鎖をより大型の側鎖(例えばチロシンまたはトリプトファン)と置き換える。大型のアミノ酸側鎖を小型のもの(例えばアラニンまたはスレオニン)と置き換えることにより、(複数の)大型側鎖と同一かまたは類似した大きさの代償性「空洞」が第二の抗体分子のインターフェースに創成される。これにより、ホモ二量体のようなその他の望ましくない最終生成物よりもヘテロ二量体の収量を増加させるためのメカニズムが提供される。1996年9月6日公開の第WO96/27011号を参照のこと。
【0060】
二重特異性抗体には架橋または「ヘテロ抱合」抗体が含まれる。例えばヘテロ抱合の抗体の一方をアビジンに結合させ、他方をビオチンに結合させることができる。任意の便宜的な架橋方法を用いてヘテロ抱合抗体を作成することができる。適当な架橋剤は当分野において周知であり、そして米国特許第4,676,980号にて、多くの架橋技術と一緒に開示されている。
【0061】
抗体フラグメントから二重特異性抗体を作成するための技術もまた文献に記載されている。例えば二重特異性抗体を、化学的連結を用いて調製することができる。Brennanら、Science 229:81(1985)は、インタクトな抗体がタンパク質分解により切断されてF(ab’)2フラグメントを作成する手順を記載している。これらのフラグメントはジチオール錯化剤、亜ヒ酸ナトリウムの存在下で還元されて隣接ジチオールを安定化し、そして分子間ジスルフィド形成を防御する。作成されたFab’フラグメントは次いでチオニトロ安息香酸(TNB)誘導体に変換される。Fab’−TNB誘導体の一つは次いでメルカプトエチルアミンでの還元によりFab’−チオールに再変換され、そして等モル濃度の量のその他のFab’−TNB誘導体と混合されて二重特異性抗体を形成する。生成された二重特異性抗体を酵素の選択的固定のための薬剤として用いることができる。なおさらなる実施態様では、大腸菌から直接回収されたFab’−SHフラグメントをインビトロで化学的に結合させて二重特異性抗体を形成することができる。(Shalabyら、J.Exp.Med.175:217−225(1992))。
【0062】
Shalabyら、J.Exp.Med.175:217−225(1992)は完全なヒト化二重特異性抗体F(ab’)2分子の生成を記載している。各Fab’フラグメントは大腸菌から別個に分泌され、そしてインビトロで指示された化学的結合に供されて二重特異性抗体を形成した。このように形成された二重特異性抗体はHER2受容体を過剰発現する細胞および正常なヒトT細胞に結合でき、そしてヒト乳腺腫瘍標的に対するヒト細胞毒性リンパ球の溶解活性の誘因となることができた。
【0063】
組換え細胞培養から直接二重特異性抗体フラグメントを作成および単離するための種々の技術もまた記載されている。例えばロイシンジッパーを用いて二重特異性抗体が生成されている(Kostelnyら、J.Immunol.148(5):1547−1553(1992))。FosおよびJunタンパク質からのロイシンジッパーペプチドは遺伝子融合により二つの異なる抗体のFab’部分に連結された。抗体ホモ二量体はヒンジ領域で還元されて単量体を形成し、そして次に再酸化されて抗体ヘテロ二量体を形成する。この方法を抗体ホモ二量体の生成に利用することもできる。Hollingerら、Proc.Natl.Acad.Sci.USA 90:6444−6448(1993)に記載される「ダイアボディー」テクノロジーは二重特異性抗体フラグメントを作成するための代替えメカニズムを提供している。
【0064】
フラグメントは、短すぎて同一鎖上の二つのドメイン間で対形成できないリンカーにより、軽鎖可変領域(VL)に連結されている重鎖可変領域(VH)を含む。したがって、一つのフラグメントのVHおよびVLドメインは別のフラグメントの相補的VLおよびVHドメインと対形成を強いられ、それにより二つの抗原部位を形成する。一本鎖Fv(sFv)二量体の使用による二重特異性抗体フラグメントを作成するための別の計画もまた報告されている。Gruberら、J.Immunol.152:5368(1994)を参照のこと。
【0065】
これに代えて、二重特異性抗体はZapataら、Protein Eng.8(10):1057−1062(1995)に記載されるように生成された「直鎖状抗体」でよい。簡単には、これらの抗体は一対の抗原結合領域を形成する一対のタンデムFdセグメント(VH−CH1−VH−CH1)を含む。直鎖状抗体は二重特異性または単一特異性でよい。
【0066】
2価を超える抗体もまた企図される。例えば三重特異性抗体を調製することができる。(Tuffら、J.lmmunol.147:60(1991))
特定の実施態様では、モノクローナル、ヒト、ヒト化、Human Engineered(商標)またはバリアント抗M−CSF抗体はRX1、5H4、MC1またはMC3抗体フラグメントのような抗体フラグメントである。抗体フラグメントを生成するための種々の技術が開発されている。伝統的にはこれらのフラグメントはインタクトな抗体のタンパク質消化を介して誘導される(例えばMorimotoら、Journal of Biochemical and Biophysical Methods 24:107−117(1992)およびBrennanら、Science 229:81(1985)を参照のこと)。しかしながら、今やこれらのフラグメントを組換え宿主細胞により直接生成することができる。Betterら、Science 240:1041−1043(1988)は細菌からの機能的抗体フラグメントの分泌を開示している(例えばBetterら、Skerraら、Science 240:1038−1041(1988)を参照のこと)。例えばFab’−SHフラグメントを大腸菌から直接回収し、そして化学的に連結させてF(ab’)2フラグメントを形成することができる(Carterら、Bio/Technology 10:163−167(1992))。別の実施態様では、ロイシンジッパーGCN4を用いてF(ab’)2分子の組み立てを促進してF(ab’)2を形成する。別の研究法に従って、Fv、FabまたはF(ab’)2フラグメントを宿主細胞培養から直接単離することができる。抗体フラグメントの生成のためのその他の技術は当業者には明白であろう。
【0067】
「単離された」抗体は同定され、そしてその天然の環境の構成成分から分離されて、そして回収されているものである。その天然の環境の夾雑する構成成分は抗体の診断的または治療的使用を妨害する材料であり、そして酵素、ホルモンおよびその他のタンパク質性または非タンパク質性溶質を含み得る。好ましい実施態様では、抗体は(1)ローリー法により決定されるように、抗体の95重量%、そして最も好ましくは99重量%を超えるまで、(2)スピニングカップシークエネーターの使用により、N末端、もしくは内部のアミノ酸配列の少なくとも15残基を得るのに十分な程度まで、または(3)クーマシーブルーもしくは好ましくは銀染色を用いて還元もしくは非還元条件下でSDS−PAGEにより均一になるまで精製される。組換え細胞内で原位置の抗体は、抗体の天然の環境の少なくとも一つの構成成分が存在しないので、単離された抗体に含まれる。しかしながら通常単離された抗体は少なくとも一つの精製工程により調製される。
【0068】
抗体の構造および作成の詳細な記載に関してはRoth,D.B.およびCraig,N.L.、Cell,94:411−414(1998)および米国特許第6,255,458号(その全てを出典明示により本明細書の一部とする)を参照のこと。簡単には重および軽鎖免疫グロブリン遺伝子をコードするDNAを作成するための過程は一次的にはB細胞の発達の際に生じる。種々の免疫グロブリン遺伝子セグメントの再配列および結合の前に、V、D、Jおよび定常(C)遺伝子セグメントは一般的に単一の染色体上の比較的近接近して見出される。B細胞分化の間に、V、D、J(または軽鎖遺伝子の場合VおよびJのみ)遺伝子セグメントの適切なファミリーメンバーの各々の一つは組換えられて機能的に再配列された重および軽鎖免疫グロブリン遺伝子を形成する。この遺伝子セグメント再配列過程は逐次的であると思われる。最初に重鎖D−J結合、続いて重鎖V−DJ結合および軽鎖V−J結合が作られる。
【0069】
機能的重および軽鎖可変領域を形成する可変領域遺伝子セグメントの組換えは、組換え能力のあるV、DおよびJセグメントをフランキングする組換えシグナル配列(RSS’s)により媒介される。直接組換えに必要および十分なRSS’sは二分子対称七量体、ATリッチ九量体および12または23塩基対のいずれかの介在スペーサー領域を含む。これらのシグナルは異なる遺伝子座およびD−J(またはV−J)組換えを担持する種間で保存され、そして機能的に互換性である。Oettingerら、Science 248:1517−1523(1990)およびそこに引用される参照文献を参照のこと。七量体は配列CACAGTGまたはその類似体、続いて非保存配列のスペーサー、そして次に配列ACAAAAACCまたはその類似体を有する九量体を含む。これらの配列は各VおよびD遺伝子セグメントのJまたは下流側に見出される。生殖系列DおよびJセグメントの直ぐ先は再度二つの組換えシグナル配列、再度非保存配列により分けられた、最初に九量体、そして次に七量体がある。VL、VHまたはDセグメントに続く七量体および九量体配列は、それらが組換えるJL、DまたはJHセグメントに先行するものに相補的である。七量体配列と九量体配列の間のスペーサーは12塩基対の長さか、または22塩基対と24塩基対の間の長さのいずれかである。
【0070】
V、DおよびJセグメントの再配列に加えて、軽鎖のVおよびJセグメントが結合し、そして重鎖のDおよびJセグメントが結合する位置の可変組換えにより、免疫グロブリン重および軽鎖の一次的なレパートリーでさらなる多様性を生じる。軽鎖におけるかかる変化は典型的にはV遺伝子セグメントの最後のコドンおよびJセグメントの最初のコドン内で生じる。結合における類似の不正確さはDセグメントとJHセグメントの間の重鎖染色体上で生じ、そして10ヌクレオチドほどまで伸長し得る。さらに、DとJH遺伝子セグメントの間およびVHとD遺伝子セグメントの間で、ゲノムDNAによりコードされないいくつかのヌクレオチドが挿入され得る。これらのヌクレオチドの付加はN領域多様性として公知である。
【0071】
可変領域遺伝子セグメントにおけるかかる再配列およびかかる結合の間に生じ得る可変組換えの正味の効果は一次抗体レパートリーの生成である。
【0072】
「Fv」は完全抗原認識および結合部位を含有する最小抗体フラグメントである。この領域は緊密な、非共有結合性会合の一つの重鎖および一つの軽鎖可変ドメインの二量体からなる。各可変ドメインの三つのCDRが相互作用してVH V1二量体の表面で抗原結合部位を定義するのはこの立体構造においてである。六つのCDRが集団で抗体に対する抗原結合特異性に寄与する。しかしながら単一の可変ドメイン(または抗原に特異的な三つのCDRのみを含むFvの半分)でさえ抗原を認識し、そして結合する能力を有しているが、親和性は全結合部位よりも低い。
【0073】
Fabフラグメントもまた軽鎖の定常ドメインおよび重鎖の第一定常ドメイン(CH1)を含有する。Fabフラグメントは抗体ヒンジ領域からの一つまたはそれより多いシステインを含む重鎖CH1ドメインのカルボキシ末端における数個の残基の付加によりFab’フラグメントと異なる。本明細書ではFab’−SHは定常ドメインの(複数の)システイン残基が遊離チオール基を担持するFab’に関する名称である。F(ab’)2抗体フラグメントは元来それらの間にヒンジシステインを有するFab’フラグメントの対として生成された。
【0074】
「中和抗体」とは、それが結合する標的抗原のエフェクター機能を排除するかまたは有意に低減させることができる抗体分子を意味する。したがって抗標的「中和」抗体は酵素活性、リガンド結合または細胞内シグナリングのようなエフェクター機能を排除するかまたは有意に低減させることができる。
【0075】
本明細書にて提供されるような癌転移または癌転移に関連する骨量減少の処置のための組成物および方法は、望ましい効果を達成するために単独でまたはその他の治療と組み合わされて使用される一つまたはそれより多い抗体を利用できる。環境抗原との直接接触または抗原での免疫のいずれかの結果として、本発明による抗体を、抗体を産生する動物から単離することができる。これに代えて、当分野において周知の抗体発現系の一つを用いて組換えDNA方法論により抗体を産生することができる(例えばHarlowおよびLane、Antibodies:A Laboratory Manual、Cold Spring Harbor Laboratory(1988)を参照のこと)。かかる抗体は組換えIgG、免疫グロブリンから誘導される配列または「Human Engineered(商標)」抗体を有するキメラ融合タンパク質を含むことができ、それを全て本発明による癌転移および/または癌転移に関連する骨量減少の処置のために用いることができる。インタクトな全長分子に加えて、「抗体」なる用語はまたそのフラグメント(例えばscFv、Fv、Fd、Fab、Fab’およびF(ab)’2フラグメントのような)またはM−CSF(またはM−CSFR)に結合するインタクトな分子および/もしくはフラグメントの多量体もしくは凝集体をも指す。これらの抗体フラグメントは抗原に結合し、そして例えばガラクトース残基の組み込みによるクリアランスおよび取り込みを促進する構造的様相を呈するように誘導体化され得る。
【0076】
本発明の一つの実施態様では、本質的にHalenbeckら、米国特許第5,491,065号(1997)(出典明示により本明細書の一部とする)に記載されるようにM−CSFモノクローナル抗体を調製することができる。M−CSFモノクローナル抗体の実例としては、付随する生物学的活性の中和を伴う組換えまたは未変性二量体M−CSFに随伴される見かけの立体構造エピトープに結合するものが挙げられる。これらの抗体は単量体および化学的に誘導体化された二量体M−CSFを含む生物学的に不活性な形態のM−CSFとは実質的には反応しない。
【0077】
本発明のその他の実施態様では、Human Engineered(商標)抗M−CSFモノクローナル抗体が提供される。「Human Engineered(商標)抗体」なる語句は非ヒト抗体、典型的にはマウスモノクローナル抗体から誘導された抗体を指す。これに代えて、親、非ヒト抗体の抗原結合特性を保持するかまたは実質的に保持するが、ヒトに投与された場合、親抗体と比較して免疫原性の低下を呈するキメラ抗体からHuman Engineered(商標)抗体を誘導することができる。本明細書にて使用される際には、「キメラ抗体」なる語句は典型的には異なる種を起源とする二つの異なる抗体から誘導される配列を含有する抗体(例えば米国特許第4,816,567号を参照のこと)を指す。最も典型的には、キメラ抗体はヒトおよびネズミ抗体フラグメント、一般的にはヒト定常およびマウス可変領域を含む。
【0078】
「相補性決定領域」なる語句または「CDR」なる用語は未変性免疫グロブリン結合部位の天然Fv領域の結合親和性および特異性を一緒に定義するアミノ酸配列を指す(例えばChothiaら、J.Mol.Biol.196:901−917(1987);Kabatら、米国保健社会福祉省NIH出版第91 3242号(1991)を参照のこと)。「定常領域」なる語句はエフェクター機能を付与する抗体分子の部分を指す。本発明では、マウス定常領域はヒト定常領域により置換されるのが好ましい。対象抗体の定常領域はヒト免疫グロブリンから誘導される。重鎖定常領域を五つのアイソタイプ:アルファ、デルタ、イプシロン、ガンマまたはミューのいずれかから選択することができる。
【0079】
本発明の抗体は、約106M−1以上、好ましくは約107M−1以上、さらに好ましくは約108M−1以上、そして最も好ましくは約109M−1、1010M−1、1011M−1または1012M−1以上のKaで抗原に結合する場合、免疫特異的または特異的に結合するとされる。抗M−CSF抗体は、宿主/対象組織により発現されるもの、および腫瘍により発現されるものを含むM−CSFの様々な天然発生形態に結合し得る。RX1、5H4、MC1またはMC3抗体のような本明細書にて開示されるモノクローナル抗体はM−CSFに関して親和性を有し、そして少なくとも10−4M、好ましくは少なくとも約10−7Mから約10−8M、さらに好ましくは少なくとも約l0−8M、10−10M、l0−11Mまたは10−12Mの解離平衡定数(Kd)を特徴とする。平衡透析による;製造者により概説される一般的な手順を用いてBlAcore2000装置を使用することによる;125I標識M−CSFを用いるラジオイムノアッセイによる;または当業者に公知の別の方法によるような従来の技術を用いてかかる親和性を容易に決定することができる。例えばScatchardら、Ann N.Y.Acad.Sci.,51:660(1949)の方法により親和性データを分析することができる。このように好ましいM−CSF抗体はM−CSFに関して高度な特性を呈し、そしてその他の分子とは実質的に低い親和性で結合することは明白である。好ましい抗体は、図4のネズミRX1がM−CSFと結合するのと類似の親和性でM−CSFに結合し、低い免疫原性を呈し、そして転移性疾患動物モデルにおいて試験した場合、癌の転移を阻止する。その他の抗体の実例は図2、3または4のネズミ5H4、MC1またはMC3が各々M−CSFと結合するのと類似の親和性でM−CSFに結合する。
【0080】
抗体の生成に使用される抗原は例えば、場合によってはエピトープがその未変性立体構造を表示することを可能にする別のポリペプチドに融合された、望ましいエピトープを保持するインタクトなM−CSFまたはM−CSFのフラグメントでよい。これに代えて、その細胞表面でM−CSFを発現する細胞を用いて抗体を作成することができる。かかる細胞は形質転換されてM−CSFを発現できるか、またはM−CSFを発現するその他の天然発生細胞でよい。抗体の作成に有用なM−CSFのその他の形態は当業者には明白であろう。
【0081】
i.ポリクローナル抗体
ポリクローナル抗体を好ましくは動物において関連抗原およびアジュバントの複数回皮下(sc)または腹腔内(ip)注射により上昇させる。二機能性または誘導体化剤、例えばマレイミドベンゾイルスルホスクシンイミドエステル(システイン残基により抱合)、N−ヒドロキシスクシンイミド(リジン残基による)、グルタルアルデヒド、無水コハク酸または当分野において公知のその他の薬剤を用いて、関連抗原を免疫される種で免疫原性であるタンパク質、例えばキーホールリンペットヘモシアニン、血清アルブミン、ウシチログロブリン、大豆トリプシン阻害剤に抱合させることにより改善された抗体応答を得ることができる。
【0082】
例えば100μgまたは5μgのタンパク質または抱合体(各々ウサギまたはマウスに関して)をフロイント完全アジュバント3容量と組み合わせ、そして溶液を複数の部位に皮内注射することにより、動物を抗原、免疫原性抱合体または誘導体に対して免疫する。1か月後、フロイント完全アジュバント中1/5(分画(1/10))から元来の量のペプチドまたは包合体で複数の部位に皮下注射することにより動物をブーストする。ブースター注射後7−14日に動物を出血させ、そして血清を抗体力価に関して検定する。力価がプラトーになるまで動物をブーストする。好ましくは同一抗原であるが、異なるタンパク質に、および/または異なる架橋剤により抱合させた抱合体で動物をブーストする。組換え細胞培養でタンパク質融合体として抱合体を作成することもできる。またミョウバンのような凝集剤を用いて免疫応答を強化するのが適当である。
【0083】
ii.モノクローナル抗体
モノクローナル抗体を、Kohlerら、Nature,256:495(1975)により最初に記載されたハイブリドーマ法を用いて作成できるか、または組換えDNA法により作成できる。
【0084】
ハイブリドーマ法では、マウスまたはハムスターもしくはマカクザルのようなその他の適切な宿主動物を本明細書に記載されるように免疫して、免疫に用いられるタンパク質に特異的に結合する抗体を産生するか、または産生できるリンパ球を導く。これに代えてリンパ球をインビトロで免疫できる。次いでポリエチレングリコールのような適当な融合剤を用いてリンパ球を骨髄腫細胞と融合してハイブリドーマ細胞を形成する(Goding、Monoclonal Antibodies:Principles and Practice、59−103頁(Academic Press、1986))。
【0085】
このように調製されたハイブリドーマ細胞を、好ましくは融合されていない親骨髄腫細胞の成長または生存を阻止する一つまたはそれより多い物質を含有する適当な培養培地に播種し、そして成長させる。例えば親骨髄腫細胞が酵素ヒポキサンチングアニンホスホリボシルトランスフェラーゼ(HGPRTまたはHPRT)を欠く場合、ハイブリドーマのための培養培地は典型的にはヒポキサンチン、アミノプテリンおよびチミジンを含み(HAT培地)、その物質はHGPRT欠損細胞の成長を防御する。
【0086】
好ましい骨髄腫細胞は効率的に融合し、選択された抗体産生細胞による安定した高レベルの抗体の産生を支持し、そして培地に対して感受性があるものである。ヒト骨髄腫およびマウス−ヒトヘテロ骨髄腫細胞系はまたヒトモノクローナル抗体の産生に関して記載されている(Kozbor、J.Immunol.,133:3001(1984);Brodeurら、Monoclonal Antibody Production Techniques and Applications、51−63頁(Marcel Dekker社、ニューヨーク、1987))。ネズミ骨髄腫系の実例にはソーク研究所セル・ディストリビューション・センター(米国、カリフォルニア州、サンディエゴ)から入手可能なMOP−21およびM.C.−11マウス腫瘍、ならびにアメリカン・タイプ・カルチャー・コレクション(米国、メリーランド州、ロックビル)から入手可能なSP−2またはX63−Ag8−653細胞から誘導されるものが挙げられる。
【0087】
ハイブリドーマ細胞が成長している培養培地を、抗原に対して指向するモノクローナル抗体の産生に関して検定する。好ましくはハイブリドーマ細胞により産生されたモノクローナル抗体の結合特異性を免疫沈殿により、またはラジオイムノアッセイ(RIA)もしくは酵素結合免疫吸着アッセイ(ELISA)のようなインビトロ結合アッセイにより決定する。モノクローナル抗体の結合親和性を例えばスカッチャード分析により決定することができる(Munsonら、Anal.Biochem.,107:220(1980))。
【0088】
望ましい特異性、親和性および/または活性の抗体を産生するハイブリドーマ細胞を同定した後、限界希釈手順によりクローンをサブクローニングし、そして標準的な方法により成長させることができる(Goding、Monoclonal Antibodies:Principles and Practice、59−103頁(Academic Press、1986))。この目的のための適当な培養培地には、例えばD−MEMまたはRPMI−1640培地が含まれる。加えて、ハイブリドーマ細胞をインビボで動物の腹水腫瘍として成長させることができる。サブクローンにより分泌されるモノクローナル抗体を例えばプロテインAセファロース、ヒドロキシルアパタイトクロマトグラフィー、ゲル電気泳動、透析または親和性クロマトグラフィーのような従来の免疫グロブリン精製手順により培養培地、腹水または血清から分離するのが適当である。
【0089】
本発明の抗体は、約106M−1以上、好ましくは約107M−1以上、さらに好ましくは約108M−1以上、そして最も好ましくは約109M−1、1010M−1、1011M−1または1012M−1以上のKaで抗原に結合する場合、免疫特異的または特異的に結合するとされる。抗M−CSF抗体は、宿主/対象組織により発現されるもの、および腫瘍により発現されるものを含むM−CSFの様々な天然発生形態に結合し得る。RX1、5H4、MC1またはMC3抗体のような本明細書にて開示されるモノクローナル抗体はM−CSFに関して親和性を有し、そして少なくとも10−4M、好ましくは少なくとも約10−7Mから約10−8M、さらに好ましくは少なくとも約l0−8M、10−10M、l0−11Mまたは10−12Mの解離平衡定数(Kd)を特徴とする。平衡透析による;製造者により概説される一般的な手順を用いてBlAcore2000装置を使用することによる;125I標識M−CSFを用いるラジオイムノアッセイによる;または当業者に公知の別の方法によるような従来の技術を用いてかかる親和性を容易に決定することができる。例えばScatchardら、Ann N.Y.Acad.Sci.,51:660(1949)の方法により親和性データを分析することができる。このように好ましいM−CSF抗体はM−CSFに関して高度な特異性を呈し、そしてその他の分子とは実質的に低い親和性で結合する。好ましい抗体は、図1のネズミRX1がM−CSFと結合するのと類似の親和性でM−CSFに結合し、低い免疫原性を呈し、そして転移性疾患動物モデルにおいて試験した場合、癌の転移を阻止する。その他の抗体の実例は図2、3または4のネズミ5H4、MC1またはMC3が各々M−CSFと結合するのと類似の親和性でM−CSFに結合する。
【0090】
保存置換を表1に「好ましい置換」の見出しの下で示す。かかる置換が生物学的活性の変化を招く場合、次いで表1の「置換の実例」と表示される、またはアミノ酸クラスに関して以下にさらに記載されるようなさらに実質的な変化を導入し、そして生成物をスクリーニングすることができる。
【0091】
【表1】

抗体の生物学的特性における実質的な修飾は、(a)置換の部分のポリペプチドバックボーンの構造、例えばシートもしくはヘリックス構造として;(b)標的部位での分子の電荷もしくは疎水性;または(c)側鎖の容積;の維持に及ぼすその効果において有意に異なる置換基を選択することにより達成される。天然発生残基は共通の側鎖特性に基づいて群に分けられる:
(1)疎水性:ノルロイシン、met、ala、val、leu、ile;
(2)中性親水性:cys、ser、thr;
(3)酸性:asp、glu;
(4)塩基性:asn、gln、his、lys、arg;
(5)鎖配向に影響する残基:gly、pro;および
(6)芳香族:trp、tyr、phe。
【0092】
非保存置換はこれらのクラスの一つのメンバーを別のクラスのメンバーと置き換えることを伴う。
【0093】
ヒト化またはバリアント抗体の適切な立体構造の維持に関与しない任意のシステイン残基はまた、分子の酸化安定性の改善および異常架橋の防御のために一般的にセリンで置換され得る。逆にその安定性の改善のために(複数の)システイン結合を抗体に付加できる(特に抗体がFvフラグメントのような抗体フラグメントである場合)。
【0094】
B.M−CSFムテイン
本発明はさらに本発明の方法によるMCSFアンタゴニストとして用いることができるM−CSFムテインを提供する。
【0095】
「フラグメント」は本明細書にて使用される際には、インタクトな未変性分子の一部を意味し;例えばフラグメントポリペプチドは、N末端またはC末端のいずれかからの一つまたはそれより多いアミノ酸が欠失している未変性ポリペプチドのフラグメントである。
【0096】
「ムテイン」はポリペプチドに関して本明細書にて使用される際には、インタクトな未変性分子のバリアントまたは一つもしくはそれより多いアミノ酸が置換、挿入もしくは欠失している未変性分子のフラグメントのバリアントを意味する。かかる置換、挿入または欠失はN末端、C末端または分子の内部でよい。したがって「ムテイン」なる用語にはその範囲内に未変性分子のフラグメントが含まれる。挿入ムテインにはN末端またはC末端での融合、例えば半減期を増すための免疫グロブリンのFc部分への融合が含まれる。
【0097】
スミス・ウォーターマン相同性検索アルゴリズム(Meth.Mol.Biol.70:173−187(1997))により決定され、以下の検索パラメーター:ギャップオープンペナルティー12およびギャップ伸長ペナルティー1でアフィンギャップ検索を用いてMSPRCHプログラム(Oxford Molecular)で実行されるように、本発明による好ましいムテインは未変性ポリペプチドに対して少なくとも約65%、70%、75%、80%、85%、90%、95%、97%またはそれより高い配列同一性(相同性)を呈する。その他の周知の、および日常的に用いられる相同性/同一性走査アルゴリズムプログラムには、PearsonおよびLipman、PNAS USA,85:2444−2448(1988);LipmanおよびPearson、Science,222:1435(1985);Devereauxら、Nuc.Acids Res.,12:387−395(1984);またはAltschulら、Mol.Biol.,215:403−410(1990)のBLASTP、BLASTNまたはBLASTXアルゴリズムが含まれる。これらのアルゴリズムを用いるコンピュータープログラムもまた利用可能であり、そして限定するものではないが:GAP、BESTFIT、BLAST、FASTAおよびTFASTA(Genetics Computing Group(GCG)パッケージ、バージョン8(米国、ウィスコンシン州、マジソン)から市販により入手可能である);ならびにPC/GeneプログラムのCLUSTAL(Intellegenetics(カリフォルニア州マウンテンビュー)による)が含まれる。好ましくはプログラムにより決定されるデフォルトパラメーターを用いて配列同一性のパーセンテージを決定する。
【0098】
「修飾」は本明細書にて使用される際には、望ましい活性(アゴニストまたはアンタゴニスト)が保持されている限り、グリコシル化、リン酸化、重合体抱合(例えばポリエチレングリコールとの)またはその他の外来分子の付加のような未変性ポリペプチド、フラグメントまたはムテインの任意の修飾を意味する。
【0099】
米国特許第6,025,146号およびKoths、Mol.Reprod.Dev.46(1):31−38(1997年1月)(双方共にその全てを出典明示により本明細書の一部とする)はM−CSF単独およびMCSF−Rと複合化されたM−CSFの結晶化を記載し、そしてM−CSF三次元構造および受容体結合に関与する残基を特徴付けしている。米国特許第6,025,146号はまた、構造情報に基づいてM−CSFにおけるアミノ酸置換候補を選択するための方法を記載している。この形態のM−CSFの全体的なトポロジーは逆平行の四つのアルファヘリックス束のものであり、そこでヘリックスは、たいてい四つのヘリックス束の、より一般的に観察される上下上下連結とは異なり、上上下下と走る。長い交差接続がヘリックスAをヘリックスBに連結し、そして類似の接続がヘリックスCとDの間で見出される。ジスルフィド連結された二量体形態では、束は終端間で連結され、極めて平らな細長い構造を形成する(直径およそ85×35×25)。各単量体で三つの分子内ジスルフィド結合(Cys7−Cys90、Cys48−Cys139、Cys102−Cys146)があり、その全てが分子の遠位末端にある。一つの鎖間ジスルフィド結合(Cys31−−Cys31)は図2で示されるようにそれを通過する非結晶学的な2回対称軸を有する二量体インターフェースに位置する。変異実験によりこの形態のM−CSFでは全てのシステイン残基が完全な生物学的活性に必要であり得ることが示される。本明細書にて記載される構造は、その役割が受容体認識に関連しているというよりむしろ一次構造的であることを示唆している。米国特許第6,025,146号は、配列のアミノ酸残基のアルファ炭素位置により同定されるような、トランケートされた組換えM−CSFα二量体の三次元的構造を提供する。
【0100】
ヘリックスA、CおよびDの具体的な残基は受容体結合相互作用の特異性に関与するようである。M−CSFβがシステイン157および/または159を伴う鎖内ジスルフィド結合を有するので、M−CSFのC末端領域は構造の「後方」から伸びてM−CSFの膜結合形態に関する可変長「テザー」を提供する。したがってM−CSFの「正面」または受容体結合領域は分子の反対側にあり、未変性M−CSFの各々約6から26、71から90、および110から130の残基を含むヘリックスA、CおよびDの、またはそれの近くの溶媒接触可能残基からなる。部位特異的変異誘発によりこれらの領域における溶媒接触可能残基を変化させて受容体との側鎖相互作用を上昇または低下させることにより、M−CSFアゴニストまたはアンタゴニストを作成することができる。トリペプチドgly−x−glyの場合、接触可能なアミノ酸の表面積の正規化に基づいて、約0.25を超える、および好ましくは約0.4を超える溶媒接触可能表面積を有する残基が好ましい(Kabsch,W.ら、Biopolymers 22:2577(1983))。好ましくは、単量体の相対的配向を維持し、そしてタンパク質フォールディングの過程を乱すのを回避するために、二量体インターフェースのようなタンパク質のその他の部分と相互作用しない残基が選択される。場合によってさらに考えられることは、ヒトM−CSFとマウスM−CSF間で保存されない残基を選択することであり、それはヒトM−CSF受容体を認識しない。候補アミノ酸は好ましくは、MCSF−R残基との水素結合および/または疎水性相互作用を破壊するために、非保存アミノ酸での置換のために選択される。例えば一つまたはそれより多いヒスチジンを類似の大きさの非水素供与性アミノ酸に変えることにより、変化した受容体結合能力を有するM−CSFを創成できる。置換に好ましいアミノ酸には、限定するものではないが:H15;Q79;R86;E115;E41;K93;D99;L55;S18;Q20;175;V78;L85;D69;N70;H9;N63;およびT34が含まれる。受容体シグナリングに重要なM−CSF残基は、M−CSFの不連続領域から構成されると考えられる。投与された可能性のあるM−CSF基盤のタンパク質性薬物に対する抗体形成の可能性を最小限にするために、可能な限り溶媒接触可能な親M−CSF残基を保持するのが望ましい。
【0101】
N末端/Aヘリックス領域のアミノ酸H15およびH9の変異誘発は有意に低い生物学的活性および有意に低いMCSF−R結合能力を有するムテインに至った。この結果により、生物学的活性の低下は受容体結合親和性の低下によるものであったことが示され
;したがってこれらのヒスチジンアミノ酸は、M−CSF受容体結合親和性に重要である接触を表し、そして完全な受容体結合能力が望ましい場合、未変化にしておくべきである。Y6およびS13のような直ぐ近くの溶媒接触可能残基ならびにその他のものもまたM−CSF受容体コンタクト残基を表し得る。ヘリックスAおよびCの中央部分の溶媒接触可能残基の重要性を試験するために、M−CSFの二重変異体(Q20A、V78K)を構築した。この二重ムテインの生物学的活性はわずかに低く(8−10倍)、そしてしたがって受容体結合活性が低かった。残基Q17、R21、E115およびE119の変異誘発は目的の部分の溶媒接触可能アミノ酸の側鎖特性を変化させたが、生物学的特異活性には影響しなかったが、これはアンタゴニスト活性を有するように設計されたムテインでこれらの残基を変化させる必要がないことを示唆している。
【0102】
一つの実施態様では、本発明は受容体結合に関与するヘリックスAおよび/またはCおよび/またはDの残基(例えばアミノ酸6から26、71から90および/または110から130)が非保存的に変異しているM−CSFムテインの使用を企図する。かかるムテインは好ましくはヘリックスA、CまたはD内の未変性配列に対して少なくとも65%、70%、75%、80%、85%または90%の類似性を保持する(すなわち同一であるかまたは類似の特性を有するアミノ酸)が、残りのペプチドの未変性配列に対して高い類似性、例えば少なくとも95%、98%または99%の類似性を有する。加えて、受容体結合部位の三次元立体構造を支持する残基は非保存的に変異され得る。
【0103】
別の実施態様では、M−CSFムテインはM−CSFの単量体形態である。M−CSFの二量体形態は生物学的に活性な形態であり、そしてM−CSFの単量体形態は一般的に不活性である。単量体のジスルフィド結合はCys31−Cys31鎖間連結により生じるようである。したがってM−CSFの単量体形態がアンタゴニストとしての使用に適当であり得ることが企図される。かかる形態にはCys31および/もしくはその他のシステインのシステイン欠失および/もしくはシステイン置き換え(例えばシステインのアラニンへの置換)を含むムテイン、または(複数の)システイン、特にCys31が化学的に修飾されてジスルフィド結合に利用できなくなっているムテインが含まれる。
【0104】
さらに別の実施態様では、M−CSFムテインは一つもしくはそれより多いヘリックスA、CもしくはD、または受容体結合に関与するその部分を単独で、または適切な三次元立体構造でフラグメントの表示を可能にするその他のポリペプチドに融合されて含む。
【0105】
組換え生成または化学的合成を含む当分野において周知の技術を用いて、任意の望ましい保存および/または非保存ムテインを含有するムテインが容易に調製される。
【0106】
保存置換、特にリガンド−受容体結合に直接関与する領域の外側の置換はM−CSFムテイン(またはM−CSFRムテイン)の結合特性を有意に変化しないと予測される。物理学的特性ならびに二次および三次タンパク質構造に対する寄与に従ってアミノ酸を分類することができる。保存置換は一つのアミノ酸の、類似の特性を有する別のアミノ酸との置換として当分野において認識されている。保存置換の実例を直ぐ下の表2(1997年3月13日公開の第WO97/09433号、10頁より(PCT/GB96/02197、9/6/96出願)に示す。
【0107】
【表2】
これに代えて保存アミノ酸をLehninger(Biochemistry、第2版;Worth Publishers社、ニューヨーク州、ニューヨーク(1975)、71−77頁)に記載されるように群分けすることができ、直ぐ下の表3に示す。
【0108】
【表3】
さらに別の代替えとして、保存置換の実例を直ぐ下の表4に示す。
【0109】
【表4】
M−CSFをコードするDNA配列の利用性により望ましいポリペプチドを生成するための種々の発現系の使用が可能になる。発現ベクターの構築および適切なDNA配列からの組換え体の生成は当分野において周知の方法により実施される。これらの技術および種々のその他の技術は一般的にSambrookら、Molecular Cloning−A Laboratory Manual、Cold Spring Harbor Laboratory、ニューヨーク州、コールドスプリングハーバー(1989)、およびKriegler,M.、Gene Transfer and Expression,A Laboratory Manual、Stockton Press、ニューヨーク(1990)に従って実施される(双方共に出典明示により本明細書の一部とする)。
【0110】
周知の組換えDNA技術に従って望ましい構造(例えばM−CSFの受容体結合能力)を破壊せずにDNA配列によりコードされるアミノ酸の欠失、付加または変更によりM−CSFの一次配列に対して特定の修飾を作ることができる。さらに、個々のアミノ酸を置換するかまたは酸化、還元もしくはその他の修飾により修飾することができ、そして活性結合部位および構造情報を保持するフラグメントを得るためにポリペプチドを切断することができることは当業者には理解されよう。かかる置換および変更により、「成熟M−CSFα(配列番号7)、M−CSFβ(配列番号8)およびM−CSFγ(配列番号9)ポリペプチドと実質的に同一のアミノ酸配列を有している」ポリペプチドの定義に入るアミノ酸配列を有するポリペプチドが導かれる。
【0111】
当分野において公知の化学的合成または組換え生成技術によりポリペプチドを生成することができる。
【0112】
タンパク質の関連性を、そのコード化核酸の関連性により特徴づけることもできる。ポリヌクレオチド配列の同一性および/または類似性を決定する方法は前記で記載される。加えて、穏やかにまたは高度にストリンジェントな条件下でハイブリダイズするその能力を試験することにより、ポリヌクレオチド配列の類似性を決定する方法を以下のように決定することができる。穏やかにストリンジェントな条件の実例は以下のとおりである:50%ホルムアミド、1%SDS、1M NaCl、10%硫酸デキストランを含むハイブリダイゼーション溶液中42℃でハイブリダイゼーション、そして0.1×SSCおよび1%SDSを含む洗浄溶液中60℃で30分間2回洗浄。高度にストリンジェントな条件には0.1×SSCおよび1%SDSを含む洗浄溶液中68℃での洗浄が含まれる。当分野において記載されるような(Ausubelら(編)、Protocols in Molecular Biology、John Wiley & Sons(1994)、6.0.3から6.4.10頁)温度およびバッファーまたは塩濃度の変化により均等なストリンジェンシーの条件を達成できることは当分野において理解されている。ハイブリダイゼーション条件の修飾を経験的に決定できるか、またはプローブのグアノシン/シトシン(GC)塩基対の長さおよびパーセンテージに基づいて正確に計算することができる。Sambrookら(編)、Molecular Cloning:A Laboratory Manual、Cold Spring Harbor Laboratory Press:ニューヨーク州、コールドスプリングハーバー(1989)、9.47から9.51頁に記載されるように、ハイブリダイゼーション条件を計算することができる。
【0113】
C.可溶性M−CSFR
本発明によるM−CSFRフラグメントの実例は一つもしくはそれより多い、または二つもしくはそれより多いM−CSF/受容体結合に関与するドメイン(ドメイン1、2および3と考えられる)を含んでなり得る。好ましいM−CSFRフラグメントはM−CSFRのドメイン1、2および3の三つ全てを含む。かかるフラグメントに対する、またはM−CSFRの細胞外ドメイン全体に対するさらなる変異および/または修飾が企図され、そして前記でM−CSFムテインに関するセクションで記載されるように生成され得る。
【0114】
M−CSFR(配列番号84および85)は五つの細胞外免疫グロブリン様ドメイン(そのドメイン1−3はリガンド−受容体結合に関与すると考えられる)、膜貫通ドメインおよび細胞内分断Src関連チロシンキナーゼドメインを有する、膜を貫通する分子である。配列番号85に関して、前記されたドメインは以下のように位置する:Igドメイン1:アミノ酸27−102;Igドメイン2:アミノ酸112−196;Igドメイン3:アミノ酸215−285;Igドメイン4:アミノ酸308−399;Igドメイン5:アミノ酸410−492;膜貫通ドメイン:アミノ酸515−537;およびキナーゼドメイン:アミノ酸582−910。「典型的な」免疫グロブリン様ドメインは通常各ループの先端で二つのシステインの間のジスルフィド結合により繋留されるループ構造を含有する。M−CSF−Rでは、Ig様ループを形成するこれらのシステインは以下のアミノ酸位置にある:ドメイン1:42、84;ドメイン2:127、177;ドメイン3:224、278;ドメイン4:関与するシステインなし;ドメイン5:419、485。
【0115】
M−CSFRのインタクトな細胞外部分または抗原性を保持するその任意のフラグメント、例えば一つまたはそれより多いIg様ループを用いて未変性受容体に結合する抗体を上昇させることができる。ポリクローナル、モノクローナル、キメラ、CDRグラフト、ヒト化、完全ヒト抗体およびその抗原結合フラグメントをM−CSFに対する抗体に関して前記されたように調製することができる。本明細書にて「スクリーニング方法」の表題のセクションに記載されるようなアッセイを用いて、または当分野において公知の任意の適当なアッセイを用いてMCSFアンタゴニストとしての活性に関して、および本発明の処置方法における適性に関して抗体産生物をスクリーニングすることができる。
【0116】
受容体の細胞外ドメイン内の一つまたはそれより多い前記されたIg様ループは、M−CSFとM−CSFRとの間の相互作用を阻止するのに十分であり得る。したがってM−CSFRおよびそのムテインの細胞外ドメインのフラグメントを当分野において周知の組換えまたは化学的合成手段を用いて容易に調製することができる。本明細書にて「スクリーニング方法」の表題のセクションに記載されるようなアッセイを用いて、または当分野において公知の任意の適当なアッセイを用いてMCSFアンタゴニストとしての活性に関して、および本発明の処置方法における適性に関して産生物をスクリーニングすることができる。
【0117】
D.遺伝子治療
治療用タンパク質の適切な細胞への分配を、物理的DNA移入法(例えばリポソームまたは化学的処理)の使用を含む当分野において公知の任意の適当な研究法の使用による、またはウイルスベクター(例えばアデノウイルス、アデノ随伴ウイルスまたはレトロウイルス)使用によるエキソビボ、インサイチュまたはインビボ遺伝子治療により行うことができる。アンチセンス化合物およびそれを用いる方法もまた本発明により提供される。遺伝子発現レベルを低下させるための周知のアンチセンス、遺伝子「ノックアウト」、リボザイム、三重ヘリックス、二重ヘリックスまたはRNAi法の使用によりM−CSFまたはM−CSFR活性のレベルを低下させることができる。かかる分子の生成および使用のための技術は当業者に周知である。
【0118】
本明細書にて使用される際には、「ペプチド擬似物質」なる用語は、ファルマコフォアの空間的配向が天然のペプチドの生理活性立体構造を実質的に擬似するように足場に支持された、アミノ酸側鎖の集合体またはファルマコフォア、またはその適当な誘導体を含む非ペプチド化合物である。例えばペプチド擬似物質はアミノ酸またはペプチド結合を欠くが、結合活性に必要である親ペプチドからのペプチド鎖群の特定の三次元配置を保持する。足場は二環式、三環式またはさらに高次の多環式炭素もしくはヘテロ原子骨格を含んでなり得るか、または一つもしくはそれより多い環構造(例えばピリジン、インダゾール等)もしくはアミド結合に基づき得る。この足場をコアの一方の末端でスペーサーにより酸性基(例えばカルボン酸官能基)に、および他方の末端で塩基性基(例えばアミジンまたはグアニジンのようなN含有部分)に連結させることができる。ペプチド擬似物質を合成するための技術の実例は2003年10月23日公開の米国特許出願番号第20030199531号、2003年7月24日公開の米国特許出願番号第20030139348号に記載されている。
【0119】
抗体およびその他のタンパク質に加えて、本発明はまた、限定するものではないがM−CSFとM−CSFRの間の相互作用またはM−CSFRの活性化を阻止するのにも有効であるペプチドまたは小型有機分子を含む代替えのM−CSFアンタゴニストをも企図する。
【0120】
II.組み合わせ治療
M−CSFアンタゴニストおよび第二の抗破骨細胞薬のような本発明による二つの治療薬の併用投与は、薬剤がその治療効果を奏する期間に重複が存在する限り、同時にまたは同一経路により薬剤が投与される必要はない。異なる日または週の投与のような同時または逐次的投与が企図される。
【0121】
M−CSF抗体(M−CSFアンタゴニストの実例)での処置の開始後、治療効果が観察される有意なタイムラグが見出されることから、移行期間の間の作用がより迅速に発現する第二の抗破骨細胞薬の同時投与が望ましくなる。移行期間の間、二つの薬剤は単剤治療で有効な量で投与されなければならない。移行期間に続いて、第二の抗破骨細胞薬を中止するか、または投薬量を減少させることができる。M−CSFアンタゴニストおよび第二の抗破骨細胞薬が相乗効果を奏する場合、移行期間後に一つまたは双方の用量を減らすことができる。
【0122】
本発明の組成物を、癌転移および/または癌転移に関連する骨量減少、または骨粗鬆症のようなその他の骨量減少に関連する疾患を含む骨溶解性障害を既に患っているか、またはその傾向がある哺乳動物に、かかる疾患の進行を防御または少なくとも部分的に停止させるのに十分な量で投与する。治療薬が単独で与えられる(第二の治療薬と組み合わされない)場合にこれを達成するのに十分な治療薬の量は「単剤治療上有効な用量」として定義される。
【0123】
本発明の組み合わせ治療では、M−CSF抗体のようなM−CSFアンタゴニストおよび第二の抗破骨細胞薬を同時にまたは異なる時間に投与することができる。二つの薬剤を例えば互いに8時間、1日、14日、30日、3か月、6か月、9か月または1年以内に投与することができる。
【0124】
第二の抗破骨細胞薬の実例には、限定するものではないがゾレドロネート、パミドロネート、クロドロネート、エチドロネート、チルドロネート、アレンドロネート、イバンドロナートまたはリセドロネートを含むビスホスホネート系が挙げられる。その他の抗破骨細胞薬の実例には、ビスホスホネート系、PTHrP中和剤(例えば抗体、アンチセンス、siRNA)、カテプシンK阻害剤、MIP−1−αアンタゴニスト、RANK/RANKL中和剤(例えばAMG−162のような抗RANK抗体、またはアンチセンス、可溶性RANKL受容体もしくはそのムテイン)、RANKLワクチン、オステオプロテグリン(OPG)、血小板由来成長因子(PDGF)、srcキナーゼ阻害剤、マロン酸ガリウムおよびマトリックスメタロプロテイナーゼ(MMP)阻害剤が挙げられる。
【0125】
ビスホスホネート系の用量の実例には、静脈内投与4mgが挙げられる。3.5mg、3.3mgまたは3.0mgを含むより少ない投薬量を投与することもできる。皮下および第WO02/087555号に記載されるようなものを含むその他の投与経路が可能である。M−CSF抗体の有効量は異なり、そして疾患の重篤度ならびに処置される患者の体重および一般状態に依存するが、一般的に約1.0mg/kgから約100mg/kg体重または約10mg/kgから約30mg/kgの範囲であり、適用あたり約0.1mg/kgから約10mg/kgまたは約1mg/kgから約10mg/kgの投薬量がさらに一般的に用いられる。例えば一つまたはそれより多い別個の投与によっても、または連続注入によっても、例えば約1011g/kgから5mg/kgまたは約30mg/kgから1mg/kgの抗体が患者への投与のための初期候補投薬量である。投与は毎日、隔日、毎週またはより少ない頻度であり、必要により疾患に対する応答性および患者の治療耐性に依存する。望ましい疾患病徴の抑制が生じるまで4、5、6、7、8、10もしくは12週またはそれより長いような長期間にわたる維持投薬量を必要とする場合もあり、そして必要により投薬量を調整することができる。この治療の進展は従来の技術およびアッセイにより容易にモニタリングされる。
【0126】
本発明の方法は癌の全段階で有用であり得るが、進行性または転移性癌において特に適切であり得る。化学療法剤処置を受けたことのない患者では、治療方法を化学療法または放射線レジメンと組み合わせるのが好ましいであろうが、一つまたはそれより多い化学療法剤を投与されている患者には本発明の治療方法での処置が指示され得る。加えて本発明の治療方法により、特に化学療法剤の毒性に対して十分な耐性のない患者では、併用化学療法の投薬量を減少させて使用することも可能になる。
【0127】
本発明の方法は単一の抗M−CSF抗体および異なる抗体の組み合わせまたは「カクテル」の投与を企図する。かかる抗体カクテルは異なるエフェクターメカニズムを引き出す抗体を含有するか、または細胞毒性抗体を免疫エフェクター機能性に依存する抗体と直接組み合わせるので、特定の利点を有し得る。組み合わされたかかる抗体は相乗的治療効果を奏し得る。
【0128】
本発明の方法を癌治療のようなさらにその他の治療方法と組み合わせて使用することができる。癌治療薬および/または手順の実例には、限定するものではないが種々の化学療法剤、アンドロゲン遮断薬および免疫モジュレーター(例えばIL−2、GM−CSF、SLC)、ビスホスホネート(系)、例えばアレディア(すなわちパミドロネート、パミドロン酸、パミドロン酸二ナトリウム、パミドロン酸二ナトリウム五水和物);Zometa(すなわちアクラスタ、ゾレドロン酸、ゾレドロネート);クロンドロネート(すなわちボネフォス、ロロン、クロドロン酸二ナトリウム、クロンドロン酸ナトリウム);フォサマックス(すなわちアレンドロネート、アレンドロン酸ナトリウム塩三水和物、アレンドロン酸);フォサバンス(すなわちビタミンDと共に処方されたフォサマックス);ボンドロナト(Bondronat)またはボンビバまたはボニバ(すなわちイバンドロナート、イバンドロン酸、イバンドロン酸ナトリウム);アクトネル(すなわちリセドロネート、リセドロン酸ナトリウム、リセドロン酸);ジドロネルまたはジドロカル(すなわちエチドロネート、エチドロン酸、エチドロン酸二ナトリウム);ネリキシア(Nerixia)(すなわちネリドロネート、ネリドロン酸);スケリッド(すなわちチルドロネート、チルドロン酸);ジメチル−APD(すなわちオルパドロネート、オルパドロン酸);およびメドロン酸またはメドロナート)、外科的手術、放射線、細胞毒性化学療法、ホルモン療法(例えばタモキシフェン;抗アンドロゲン療法)、抗体療法(例えばRANKL/RANK中和抗体;PTHrP中和、抗Her2、抗CD20、抗CD40、CD22、VEGF、IGFR−1、EphA2、HAAH、TMEFF2、CAIX抗体)、治療用タンパク質療法(例えば可溶性RANKL受容体;OPG、ならびにPDGFおよびMMP阻害剤)、小型分子薬物治療(例えばSrcキナーゼ阻害剤)、成長因子受容体のキナーゼ阻害剤、またはRANKL阻害剤、オリゴヌクレオチド療法(例えばRANKLまたはRANKまたはPTHrPアンチセンス)、遺伝子治療(例えば抗RANKL抗体のようなRANKLまたはRANK阻害剤)、ペプチド療法(例えばRANKLのムテイン)ならびに本明細書に記載されるこれれのタンパク質、ペプチド、化合物および小型分子が挙げられる。
【0129】
癌化学療法剤には、限定するものではないがカルボプラチンおよびシスプラチンのようなアルキル化剤;ナイトロジェンマスタードアルキル化剤;カルムスチン(BCNU)のようなニトロソウレアアルキル化剤;メトトレキサートのような代謝拮抗剤;葉酸;プリン類似体代謝拮抗剤、メルカプトプリン;フルオロウラシル(5−FU)およびゲムシタビン(Gemzar(登録商標))のようなピリミジン類似体代謝拮抗剤;ゴセレリン、リュープロリドおよびタモキシフェンのような抗腫瘍性ホルモン薬;アルデスロイキン、インターロイキン−2、ドセタキセル、エトポシド(VP−16)、インターフェロンアルファ、パクリタキセル(Taxol(登録商標))およびトレチノイン(ATRA)のような天然抗悪性腫瘍薬;ブレオマイシン、ダクチノマイシン、ダウノルビシン、ドキソルビシン、ダウノマイシンおよびマイトマイシンCを含むマイトマイシンのような抗菌性天然抗悪性腫瘍薬;ならびにビンブラスチン、ビンクリスチン、ビンデシンのようなビンカアルカロイド天然抗悪性腫瘍薬;ヒドロキシウレア;アセグラトン、アドリアマイシン、イフォスファミド、エノシタビン、エピチオスタノール、アクラルビシン、アンシタビン、ニムスチン、塩酸プロカルバジン、カルボコン、カルボプラチン、カルモフール、クロモマイシンA3、抗腫瘍性多糖類、抗腫瘍性血小板因子、シクロホスファミド(Cytoxin(登録商標))、シゾフィラン、シタラビン(シトシンアラビノシド)、ダカルバジン、チオイノシン、チオテパ、テガフル、ドラスタチン系、アウリスタチンのようなドラスタチン類似体、CPT−11(イリノテカン)、ミトザントロン、ビノレルビン、テニポシド、アミノプテリン、カルミノマイシン、エスペラミシン系(例えば米国特許第4,675,187号参照)、ネオカルチノスタチン、OK−432、ブレオマイシン、フルツロン、ブロクスウリジン、ブスルファン、ホンバン、ペプロマイシン、ベスタチン(Ubenimex(登録商標))、インターフェロン−β、メピチオスタン、ミトブロニトール、メルファラン、ラミニンペプチド、レンチナン,カワラタケ(Coriolus versicolor)抽出物、テガフル/ウラシル、エストラムスチン(エストロゲン/メクロレタミン)が含まれる。
【0130】
さらに、癌患者のための治療として使用されるさらなる薬剤には、EPO、G−CSF、ガンシクロビル;抗生物質、ロイプロリド;メペリジン;ジドブジン(AZT);変異体および類似体を含むインターロイキン1から18;インターフェロンα、βおよびγのようなインターフェロン系またはサイトカイン系;黄体形成ホルモン放出ホルモン(LHRH)および類似体、および生殖腺刺激ホルモン放出ホルモン(GnRH)のようなホルモン系;形質転換成長因子β(TGF−β)、線維芽細胞成長因子(FGF)、神経成長因子(NGF)、成長ホルモン放出因子(GHRF)、上皮細胞成長因子(EGF)、線維芽細胞成長因子相同因子(FGFHF)、肝細胞成長因子(HGF)およびインスリン成長因子(IGF)のような成長因子;腫瘍壊死因子α&β(TNF−α&β);浸潤阻止因子(invasion inhibiting factor)2(IIF−2);骨形成タンパク質1−7(BMP1−7);ソマトスタチン;チモシンα−1;γグロブリン;スーパーオキシドジスムターゼ(SOD);補体因子;抗血管形成因子;抗原性物質;ならびにプロドラッグが含まれる。
【0131】
プロドラッグとは、親薬物と比較して腫瘍細胞に対して細胞毒性が弱いかまたは細胞無毒性であり、そして酵素により活性化されるかまたは活性なもしくはさらに活性な親形態に変換されることが可能である、医薬的に活性な物質の前駆体または誘導体形態を指す。例えばWilman、「Prodrugs in Cancer Chemotherapy」、Biochemical Society Transactions、14、375−382頁、第615回ベルファスト会議(1986)およびStellaら、「Prodrugs:A Chemical Approach to Targeted
Drug Deliver」、Directed Drug Delivery、Borchardtら(編)、247−267頁、Humana Press(1985)参照。プロドラッグには限定するものではないが、さらに活性な細胞毒性遊離薬物に変換することができるリン酸含有プロドラッグ、チオリン酸含有プロドラッグ、硫酸含有プロドラッグ、ペプチド含有プロドラッグ、Dアミノ酸修飾プロドラッグ、グリコシル化プロドラッグ、βラクタム含有プロドラッグ、場合によっては置換されたフェノキシアセトアミド含有プロドラッグまたは場合によっては置換されたフェニルアセトアミド含有プロドラッグ、5−フルオロシトシンおよびその他の5−フルオロウリジンプロドラッグが含まれる。本明細書における使用のためのプロドラッグ形態に誘導体化できる細胞毒性薬物の実例には、限定するものではないが前記されたこれらの化学療法剤が挙げられる。
【0132】
III.投与および調製
M−CSFアンタゴニストの有効量は異なり、そして疾患の重篤度ならびに処置される患者の体重および一般状態に依存するが、一般的に約1.0μg/kgから約100mg/kg体重の範囲であり、適用あたり約10μg/kgから約10mg/kgの投薬量がさらに一般的に用いられる。本発明の組成物の有効量の決定を当分野において周知である標準的な経験的な方法により達成することができる。例えば所定の投薬量のM−CSFアンタゴニストで処置された対象からの血清のインビボ中和活性を、Cenciら、J Clin.Invest.1055:1279−87(2000)に記載されるように、インビトロでM−CSF誘起のネズミ単球(M−CSFに対する受容体を高レベルに発現するCD11b+細胞、CD11細胞のサブセット)の増殖および生存を遮断する血清の能力を決定するアッセイを用いて評価することができる。
【0133】
投与は毎日、2日毎、3日毎、週2回、毎週またはより少ない頻度であり、必要により疾患に対する応答性および患者の治療耐性に依存する。長期間にわたる維持投薬量を必要とする場合もあり、そして必要により投薬量を調整することができる。
【0134】
組成物の単回または複数回投与を実施することができ、用量レベルおよびパターンは処置する医師により選択される。
【0135】
本発明の方法の実施において使用される抗M−CSF抗体を含むM−CSFアンタゴニストを望ましい分配方法に適当な担体を含む医薬組成物に処方することができる。適当な担体には、M−CSFアンタゴニストと組み合わされた場合にアンタゴニストの抗腫瘍機能を保持し、そして対象の免疫系とは非反応性である任意の材料が含まれる。実例には、限定するものではないが滅菌リン酸塩緩衝生理食塩水、静菌水等のようないずれかの多くの標準的な医薬用担体が挙げられる。種々の水性担体、例えば水、緩衝用水、0.4%生理食塩水、0.3%グリシン等が用いられ、そして穏やかな化学的修飾等に供された、アルブミン、リポタンパク質、グロブリン等のような安定性を強化するためのその他のタンパク質を含むことができる。
【0136】
望ましい純度を有するアンタゴニストを場合によっては生理学的に許容される担体、賦形剤または安定化剤(Remington’s Pharmaceutical Sciences、第16版、Osol,A.編(1980))と混合することにより、凍結乾燥処方または水溶液の形態の保存用のアンタゴニストの治療用処方を調製する。許容される担体、賦形剤または安定化剤は用いられると投薬量および濃度でレシピエントに対して無毒性であり、そしてリン酸塩、クエン酸塩およびその他の有機酸のようなバッファー;アスコルビン酸およびメチオニンを含む抗酸化剤;保存剤(例えば塩化オクタデシルジメチルベンジルアンモニウム;塩化ヘキサメトニウム;塩化ベンザルコニウム、塩化ベンゼトニウム;フェノール、ブチルまたはベンジルアルコール;メチルまたはプロピルパラベンのようなアルキルパラベン;カテコール;レソルシノール;シクロヘキサノール;3−ペンタノール;およびm−クレゾール);低分子量(約10残基未満)ポリペプチド;血清アルブミン、ゼラチンもしくは免疫グロブリンのようなタンパク質;ポリビニルピロリドンのような親水性重合体;グリシン、グルタミン、アスパラギン、ヒスチジン、アルギニンもしくはリジンのようなアミノ酸;グルコース、マンノースもしくはデキストリンを含む単糖類、二糖類およびその他の炭水化物;EDTAのようなキレート剤;スクロース、マンニトール、トレハロースもしくはソルビトールのような糖;ナトリウムのような塩形成対イオン;金属複合体(例えばZnタンパク質複合体);ならびに/またはTWEEN(商標)、PLURONICS(商標)もしくはポリエチレングリコール(PEG)のような非イオン性界面活性剤が含まれる。
【0137】
本明細書では処方はまた、処置される特定の適応症に関して必要により一つより多い活性化合物、好ましくは互いに悪影響を及ぼさない相補的活性を有するものを含有し得る。例えばさらに免疫抑制剤を提供するのが望ましい場合もある。かかる分子は意図される目的のために有効である量で組み合わされて存在するのが適当である。
【0138】
例えばコアセルベーション技術により、または界面重合により調製されたマイクロカプセル、例えばヒドロキシメチルセルロースまたはゼラチンマイクロカプセルおよびポリ−(メチルメタクリラート)マイクロカプセルに、各々コロイド薬物分配系(例えばリポソーム、アルブミンマイクロスフェア、マイクロエマルジョン、ナノ粒子およびナノカプセル)で、またはマクロエマルジョンで活性成分を封入することができる。かかる技術はRemington’s Pharmaceutical Sciences、第16版、Osol,A.編(1980)に開示されている。
【0139】
インビボ投与に用いられる処方は滅菌されていなければならない。これは滅菌ろ過膜を通してろ過することにより容易に達成される。
【0140】
非経口、皮下、腹腔内、肺内および鼻内を含む任意の適当な手段により、ならびに所望により局部処置、病巣内投与用にアンタゴニストが投与される。非経口注入には静脈内、動脈内、腹腔内、筋肉内、皮内または皮下投与が含まれる。加えてアンタゴニストをパルス注入により、特にアンタゴニストの漸減用量で投与するのが適当である。好ましくは投与が短期か長期かにある程度依存して、注射により、最も好ましくは静脈内または皮下注射により用量を与える。例えば望ましい部位に接近して配置されたカテーテルにより、局所、特に経皮、経粘膜、直腸、経口または局部投与を含むその他の投与方法が企図される。
【0141】
本発明の組成物を例えば顆粒、粉末、錠剤、カプセル、シロップ、坐剤、注射、エマルジョン、エリキシル、懸濁液または溶液の形態にできる。本組成物を例えば経口投与による、鼻内投与による、直腸投与、皮下注射、静脈内注射、筋肉内注射または腹腔内注射による種々の投与経路用に処方できる。
【0142】
注射用投薬形態には一般的に、適当な分散剤または湿潤剤および懸濁化剤を用いて調製できる水性懸濁液または油性懸濁液が含まれる。注射用形態は溶媒または希釈剤で調製される溶液相または懸濁液の形態でよい。許容される溶媒またはベヒクルには、滅菌水、リンガー溶液または等張水性生理食塩水が含まれる。これに代えて、滅菌油を溶媒または懸濁化剤として用いることができる。好ましくは油または脂肪酸は非揮発性であり、天然または合成油、脂肪酸、モノ、ジまたはトリグリセリドを含む。
【0143】
注射用の医薬用処方および/または医薬品は前記されたような適切な溶液での再構築に適当な粉末でよい。これの実例には限定するものではないが、凍結−乾燥、回転乾燥またはスプレー乾燥粉末、非晶質粉末、顆粒、沈殿物または微粒子が挙げられる。注射用の処方は場合によっては安定化剤、pH調整剤、界面活性剤、バイオアベイラビリティー調整剤およびこれらの組み合わせを含有し得る。
【0144】
徐放性調製物を調製することができる。徐放性調製物の適当な実例にはアンタゴニストを含有する固体疎水性重合体の半透性マトリックスが挙げられ、そのマトリックスは造形物、例えばフィルムまたはマイクロカプセルの形態である。徐放性マトリックスの実例には、ポリエステル、ヒドロゲル(例えばポリ(2−ヒドロキシエチル−メタクリラート)、またはポリ(ビニルアルコール))、ポリラクチド系(米国特許第3,773,919号)、L−グルタミン酸およびγエチル−L−グルタマートの共重合体、非分解性エチレン−ビニルアセタート、Lupron Depot(商標)(乳酸−グリコール酸共重合体および酢酸ロイプロリドから構成される注射用マイクロスフェア)のような分解性乳酸−グリコール酸共重合体およびポリ−D−(−)−3−ヒドロキシ酪酸が挙げられる。エチレン−ビニルアセタートおよび乳酸−グリコール酸のような重合体は100日間にわたる分子の放出を可能にするが、特定のヒドロゲルはタンパク質をより短い期間で放出する。カプセル化アンタゴニストは体内に長時間留まる場合、37℃で水分に暴露された結果、変性するかまたは凝集して生物学的活性の喪失および免疫原性の変化の可能性を招き得る。関与するメカニズムに依存して安定化のために合理的な計画を考案することができる。例えば凝集メカニズムがチオ−ジスルフィド交換による分子間S−S結合形成であることが見出された場合、スルフヒドリル残基を修飾し、酸性溶液から凍結乾燥し、水分含量を制御し、適切な添加剤を使用し、そして具体的な重合体マトリックス組成物を開発することにより安定化を達成することができる。
【0145】
本発明の処方を本明細書にて前記されたような短時間作用型、即時放出型、長時間作用型または徐放型になるように設計することができる。したがって医薬用処方を放出制御用または放出遅延用に処方することもできる。
【0146】
本組成物は例えばミセルもしくはリポソーム、またはその他の何らかのカプセル化形態を含むか、または放出延長形態で投与されて長時間保存および/または分配効果を提供することができる。したがって医薬用処方および医薬品をペレットまたはシリンダーに圧縮し、そしてデポー注射として、またはステントのようなインプラントとして筋肉内または皮下に埋め込むことができる。かかるインプラントはシリコンおよび生分解性重合体のような公知の不活性材料を用いることができる。
【0147】
前記したようなこれらの代表的な投薬形態に加えて、薬学的に許容される賦形剤および担体が一般的に当業者に公知であり、そしてしたがって本発明に含まれる。かかる賦形剤および担体は例えば「Remingtons Pharmaceutical Sciences」、Mack出版社、ニュージャージー州(1991)に記載されており、それを出典明示により本明細書の一部とする。
【0148】
具体的な投薬量を疾患の症状、対象の年齢、体重、一般健康状態、性別および食事、投薬間隔、投与経路、排泄速度、ならびに薬物の組み合わせに依存して調整することができる。有効量を含有する前記のいずれかの投薬形態は十分に通常の実験の範疇であり、そしてしたがって十分に本発明の範囲内である。
【0149】
本発明による治療薬として有用なM−CSFアンタゴニストまたは抗体はしばしばその他の天然発生免疫グロブリンまたはその他の生物学的分子を実質的に含まずに調製される。好ましいM−CSFアンタゴニストはまた癌転移および/または癌転移に関連する骨量減少を含む骨溶解性障害に苦しむか、またはそれを患う傾向がある哺乳動物に投与される場合、最小限の毒性しか呈さない。
【0150】
本発明の組成物を従来の周知の滅菌技術により滅菌することができる。得られた溶液を使用のために包装するかまたは無菌条件下でろ過し、そして凍結乾燥することができ、凍結乾燥された調製物を投与前に滅菌溶液と組み合わせる。組成物はpH調整剤および緩衝剤、等張化剤等、例えば酢酸ナトリウム、酪酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウムのような生理学的状態に近づけるのに必要とされるような薬学的に許容される補助物質ならびに安定化剤(例えば120%マルトース等)を含有し得る。
【0151】
また本発明のM−CSFアンタゴニストを、薬物(例えば本明細書に開示されるアンタゴニストおよび場合によっては化学療法剤)の分配に有用である種々の型の脂質および/またはリン脂質および/または界面活性剤から構成される小型ベシクルであるリポソームを介して投与することもできる。リポソームはエマルジョン、フォーム、ミセル、不溶性単層、リン脂質分散物、ラメラ層等を含み、そしてM−CSFアンタゴニストを特定の組織に標的化し、そして組成物の半減期を増すベヒクルとして提供され得る。リポソームを調製するために、例えば米国特許第4,837,028号および第5,019,369号(これらの特許を出典明示により本明細書の一部とする)に記載されるような種々の方法が利用可能である。
【0152】
アンタゴニストを含有するリポソームは、Epsteinら、Proc.Natl.Acad.Sci.USA 82:3688(1985);Hwangら、Proc.Natl Acad.Sci.USA 77:4030(1980);ならびに米国特許第4,485,045号および第4,544,545号に記載されるように当分野において公知の方法により調製される。循環時間が延長されたリポソームが米国特許第5,013,556号に開示されている。特に有用なリポソームをホスファチジルコリン、コレステロールおよびPEG誘導体化ホスファチジルエタノールアミン(PEG−PE)を含む脂質組成物を用いる逆相蒸発法により作成することができる。望ましい直径を有するリポソームを生じるために規定の孔径のフィルターを通してリポソームを押し出す。Martinら、J.Biol.Chem.257:286−288(1982)に記載されるように、ジスルフィド交換反応により本発明のM−CSF抗体のFab’フラグメントをリポソームに抱合させることができる。化学療法剤(例えばドキソルビシン)は場合によってはリポソームに含有される(例えばGabizonら、J.National Cancer Inst.81(19):1484(1989)参照)。
【0153】
これらの組成物中のM−CSFアンタゴニストの濃度は大きく、すなわち重量で約10%未満、通常少なくとも約25%から75%または90%ほどまで異なる場合があり、そして選択される特定の投与の様式に従って、液体容量、粘度等により一次的に選択される。経口、局所および非経口投与可能な組成物を調製するための実際の方法は公知であるか、または当業者には明白であり、そして詳細は例えばRemington’s Pharmaceutical Science、第19版、Mack出版社、ペンシルバニア州、イーストン(1995)(これを出典明示により本明細書の一部とする)に記載されている。
【0154】
本発明の組成物の有効量の決定を当分野において周知である標準的な経験的な方法により達成することができる。例えばM−CSFアンタゴニストの所定の投薬量で処置された対象からの血清のインビボ中和活性を、Cenciら、J Clin.Invest.1055:1279−87(2000)に記載されるように、インビトロでM−CSF誘起のネズミ単球(M−CSFに対する受容体を高レベルに発現するCD11b+細胞、CD11細胞のサブセット)の増殖および生存を遮断する血清の能力を決定するアッセイを用いて評価することができる。
【0155】
本発明の組成物を、癌転移および/または癌転移に関連する骨量減少を含む骨溶解性障害を既に患っているか、またはその傾向がある哺乳動物に、かかる疾患の進行を防御または少なくとも部分的に停止させるのに十分な量で投与する。M−CSFアンタゴニストの有効量は異なり、そして疾患の重篤度ならびに処置される患者の体重および一般状態に依存するが、一般的に約1.0mg/kgから約100mg/kg体重または約10mg/kgから90mg/kgの範囲であり、適用あたり約20mg/kgから約80mg/kgまたは約30mg/kgから約70mg/kgまたは約40mg/kgから約60mg/kgの投薬量である。例えば一つまたはそれより多い別個の投与によっても、または連続注入によっても、例えば約10mg/kgから50mg/kgまたは約20mg/kgから60mg/kgの抗MCSF抗体が患者への投与のための初期候補投薬量である。投与は毎日、隔日、毎週またはより少ない頻度であり、必要により疾患に対する応答性および患者の治療耐性に依存する。望ましい疾患病徴の抑制が生じるまで4、5、6、7、8、10もしくは12週またはそれより長いような長期間にわたる維持投薬量を必要とする場合もあり、そして必要により投薬量を調整することができる。この治療の進展は従来の技術およびアッセイにより容易にモニタリングされる。
【0156】
組成物の単回または複数回投与を実施することができ、用量レベルおよびパターンは処置する医師により選択される。疾患の防御または処置のために、抗M−CSF抗体を含むM−CSFアンタゴニストの適切な投薬量は前記されたように処置される疾患の型、疾患の重篤度および経過、アンタゴニストが防御目的で投与されても、治療目的で投与されても、以前の治療、患者の病歴およびアンタゴニストに対する応答性、ならびに担当医の判断に依存する。アンタゴニストは一度で、または一連の処置にわたって投与されるのが適当である。
【0157】
適正な医療行為に合致する仕方でアンタゴニスト組成物が処方され、投薬され(dosed)、そして投与される。この局面で考慮される因子には、処置される特定の障害、処置される特定の哺乳動物、個々の患者の臨床症状、障害の原因、薬剤の分配の部位、投与の方法、投与のスケジュール、および医療従事者に公知のその他の因子が含まれる。投与されるアンタゴニストの治療上有効な量はかかる考慮により支配され、そしてM−CSF媒介疾患、症状または障害を防御、改善または処置するために、特に癌細胞を処置するために、そしてこの上なく特に腫瘍細胞転移を処置するために必要な最少量である。かかる量は宿主に対して毒性であるかまたは宿主を有意により感染し易くする量を下回るのが好ましい。
【0158】
本発明の別の実施態様では、前記された疾患、障害または症状の処置に有用な材料を含有する製品が提供される。製品は容器およびラベルを含む。適当な容器には、例えばビン、バイアル、シリンジおよび試験管が含まれる。ガラスまたはプラスチックのような種々の材料から容器を形成することができる。容器には症状を処置するのに有効である組成物が入っており、そして容器は無菌のアクセスポートを有し得る(例えば容器は静脈内注射用溶液バッグまたは皮下注射針により突き刺すことができるストッパーを有するバイアルでよい)。組成物中の活性薬剤は本発明のM−CSFアンタゴニストまたは抗体である。容器上の、または容器に随伴されるラベルは、組成物が最適な症状を処置するために用いられることを示す。製品はさらにリン酸塩緩衝食塩水、リンガー溶液およびデキストロース溶液のような薬学的に許容されるバッファーを含む第二の容器を含む。それはその他のバッファー、希釈剤、フィルター、針、シリンジおよび使用のための指示を伴う添付文書を含む、商業的に、および使用者の立場から望ましいさらにその他の材料でよい。
【0159】
本発明を以下の実施例により説明するが、それはいかなるようにも限定されることを意図するものではない。
【実施例】
【0160】
実施例1
本実施例は動物モデルにおけるZometa(ゾレドロネート)の用量依存性、抗吸収効果を確立する(図14)。Zometa≧0.03mg/kgでの処置は骨部位で腫瘍成長により引き起こされる骨溶解性損傷を阻止した。加えてマウスを漸増濃度のZometaで処置した場合に用量応答性効果が観察された。抗マウスおよび抗ヒトM−CSF mAb 5A1および5H4の組み合わせもまたもまた骨損傷に対して保護した。重篤な骨溶解性損傷を処置したときに、M−CSF抗体単独はZometa0.03mg/kgよりも有効であった。骨溶解スコアは試験の最後の日のX線画像に基づいた(図14):0=正常;
1=正常な皮質構造を有する不確定なまたは微細な病変;
2=微細な皮質/構造破壊を伴う限局的な溶解性病変;
3=皮質/構造破壊を伴う大きな(複数の)病変;
4=保存された構造がない全体的な破壊。
【0161】
この最初の試験から、有効用量以下のZometa0.03mg/kgを用いて組み合わせ試験を実施した。6×105 MDA−MB−231Luc細胞の脛骨内接種の2週間後、Nu/Nuマウスを5A1/5H4(10mg/kg 週1回)、Zometa(0.03mg/kg 週2回)または抗体およびビスホスホネートの双方で処置した。骨病変を毎週Faxitron分析(X線テクノロジー)によりモニタリングし、そして試験の最後に全動物を最終X線に供し、そして画像を収集し、そして病変の重篤度のスコアリングのために分類した。結果の分析により、抗MCSF mAbおよびZometaの双方が骨溶解の処置に有効であったが、ZometaおよびM−CSF mAbの組み合わせ処置は骨病変の発生(図15)および程度(図16)をいずれかの単独処置よりも大きな程度まで阻止した。
【0162】
図16はZometa0.03mg/kgまたは抗MCSF抗体(5A1+5H4)10mg/kgでの処置が骨部位での腫瘍成長により引き起こされる骨溶解性損傷を阻止したことを示す。Zometa+抗MCSF抗体の組み合わせレジメンはさらに骨の溶解を阻止した。平均骨溶解スコアを1)3人の別個の志願者からの平均スコアおよび2)群平均(元来動物10匹/群)から計算した。
【0163】
骨溶解スコアは試験の最後の日のX線画像に基づいた:
0=正常;
1=正常な皮質構造を有する不確定なまたは微細な病変;
2=微細な皮質/構造破壊を伴う限局的な溶解性病変;
3=皮質/構造破壊を伴う大きな(複数の)病変;
4=保存された構造がない全体的な破壊。
【0164】
代表的なFaxitron画像の試験によりさらにZometaおよび抗MCSF抗体処置動物における比較的に微細な病変に比較して、未処置動物で見出された重篤な病変が実証された(図17)。組み合わせ処置群では有害な相互作用は観察されなかった。
【0165】
結論としては、抗MCSF抗体およびZometaの双方が骨溶解を有効に阻止し、そして二つの処置を組み合わせることにより、いずれかの単独処置と比較して抗吸収効果の上昇を招く。これは、組み合わせがビスホスホネート不耐性であるか、または既にビスホスホネート系で処置されているかのいずれかである骨疾患を有する患者にとって安全でそして有効な選択肢であり得ることを示唆している。
【0166】
実施例2
この実施例は、M−CSf活性が分化型破骨細胞活性に効果を及ぼさないことを示している(図18)。M−CSF中和抗体およびスホスホネートの分化型破骨細胞活性に及ぼす効果をヒト化Chir−RXIおよびZometaで試験した。
【0167】
ヒト骨髄CD34+細胞(Biowhittakerカタログ番号2M−101A、3×105セル/バイアル)を本明細書に記載される実験条件下で破骨細胞に分化するように誘導した。第1日にCD34+細胞を1本の凍結バイアルから培地(10%FCS、1×Pen Strepおよび1×ファンギゾンを含むアルファMEM)10mlに解凍した。細胞を1回洗浄し、そして培地2mlに再懸濁し、そしてOsteoLyseプレート(OsteoLyse(商標)アッセイキット(Human Collagen)、Cambrex)にウェルあたり100μlでプレーティングした。第2日に元来の培地を除去することなく、4×CSF−1から最終濃度30ng/ml 50μlおよび4×RANKL(sRANKL、Chemiconカタログ番号GF091、10μg/包装)から最終濃度100ng/ml 50μlを各ウェルに加えた。第7日に5×RANKLから最終濃度100ng/ml 50μlを各ウェルに加えた。
【0168】
第15日に抗体(Chir−RX1または対照抗体のいずれか)またはZometaを指示された濃度で加えた。第17日に細胞培養物の上澄10μlをサンプリングし、そしてブラック96ウェルアッセイプレート(OsteoLyseアッセイキットに含まれる)の各ウェルでフルオロフォア放出試薬200μlと混合した。
【0169】
実施例3
この実施例はZometaが分化型破骨細胞活性を用量依存的な様式で阻止することを示している(図19)。M−CSF中和抗体およびスホスホネートの分化型破骨細胞活性に及ぼす効果をヒト化Chir−RXIおよびZometaで試験した。
【0170】
ヒト骨髄CD34+細胞(Biowhittakerカタログ番号2M−101A、3×105セル/バイアル)を本明細書に記載される実験条件下で破骨細胞に分化するように誘導した。第1日にCD34+細胞を1本の凍結バイアルから培地(10%FCS、1×Pen Strepおよび1×ファンギゾンを含むアルファMEM)10mlに解凍した。細胞を1回洗浄し、そして培地2mlに再懸濁し、そしてOsteoLyseプレート(OsteoLyse(商標)アッセイキット(Human Collagen)、Cambrex)にウェルあたり100μlでプレーティングした。第2日に元来の培地を除去することなく、4×CSF−1から最終濃度30ng/ml 50μlおよび4×RANKL(sRANKL、Chemiconカタログ番号GF091、10μg/包装)から最終濃度100ng/ml 50μlを各ウェルに加えた。第7日に5×RANKLから最終濃度100ng/ml 50μlを各ウェルに加えた。
【0171】
第15日に抗体(Chir−RX1または対照抗体のいずれか)またはZometaを指示された濃度で加えた。第17日に細胞培養物の上澄10μlをサンプリングし、そしてブラック96ウェルアッセイプレート(OsteoLyseアッセイキットに含まれる)の各ウェルでフルオロフォア放出試薬200μlと混合した。
【0172】
実施例4
この実施例は霊長類においてRX1を用いた薬物動態および薬力学試験の結果を示す(図20および図21)。
【0173】
この試験の目的は、カニクイザルに単回緩徐ボーラス静脈内注射(第1日に第2および第3群)、続いて13週の観察期間か、または反復投薬(第1、43、50および57日に第1群)、続いて10週の観察期間のいずれかとして投与した場合のheRX1−10.G1、ヒト化抗ヒトM CSF抗体の薬力学および薬物動態を調査することであった。ヒト化抗M−CSF IgG1モノクローナル抗体を上腕または伏在静脈を介して緩徐ボーラス静脈内(IV)注射により投与した。
【0174】
新薬の安全性評価のために動物の使用が全世界の監督官庁により要求されている。抗体はげっ歯類で交差反応性でないが、カニクイザルで反応性であることが示されている。したがってカニクイザルは生物製剤での静脈内注射試験において使用するための許容される非げっ歯類であるので、それが選択された。
【0175】
動物を以下の群に無作為化した:
【0176】
【表5】
a 第1日に第1群は0.2mg/kg/用量を投与され、そして第43、第50および第57日に同一動物は10mg/kg/用量を投与される。
【0177】
第1日に、第2および第3群に緩徐ボーラス静脈内注射により被験処方(各々2および20mg/kg/用量)をおよそ10分間にわたって投与した。伏在静脈を介してカテーテルおよびAbbocathを用いて処方を投与した。投薬容量は4ml/kgであり、そして実際の用量は各動物の最新の実際の体重に基づいた。第1群は第1日に0.2mg/kgの用量、そして続いて第43、50および57日に10mg/kg/用量の用量が投与された。伏在静脈を介してカテーテルおよびAbbocathを用いて緩徐ボーラス静脈内注射により(およそ10分間にわたって)処方を投与した。投薬容量は4ml/kgであり、そして実際の用量は各動物の最新の実際の体重に基づいた。
【0178】
以下のように血液学および/または臨床生化学のために全動物から血液を収集した:
【0179】
【表6】
以下のパラメーターを試験した:
血液学:血液細胞形態学;赤血球恒数(MCV、MCH、MCHCおよびRDW);ヘマトクリット;ヘモグロビン;平均血小板容積;血小板数;赤血球数;網状赤血球(絶対数およびパーセント);および白血球数(総数、絶対数および種別パーセント)。
【0180】
臨床生化学::A/G比(計算値);アラニンアミノトランスフェラーゼ;アルブミン;アルカリ性ホスファターゼ;アスパラギン酸アミノトランスフェラーゼ;血中尿素窒素;カルシウム;塩化物;コレステロール;クレアチニン;グロブリン(計算値);グルコース;無機リン;カリウム;ナトリウム;総、直接型および間接型ビリルビン;総タンパク質;トリグリセリド;ならびにC反応性タンパク質。
【0181】
骨代謝回転の生化学的マーカーを以下のように分析した(骨バイオマーカーの決定のために血液およそ2mlを全動物から収集した):
【0182】
【表7】
薬物動態評価および血清M−CSF活性:
第2および第3群に関して、投薬前、第1日(注入終了の直後および注入終了の4時間後)、ならびに第3、第8、第15、第22、第29、第43、第57、第71および第85日に静脈穿刺によりSST管に血液(各1.5ml)を収集した。第1群の動物に関して、投薬前、第1日(注入終了の直後および注入終了の4時間後)、ならびに第3、第8、第15、第22、第29、第43(投薬前および注入終了の4時間後)、第50(投薬前および注入終了の4時間後)、第57(投薬前および注入終了の4時間後)、第59、第64、第71、第78、第92、第106および第120日に静脈穿刺によりSST管に血液(各1.5ml)を収集した。薬物動態評価に関して、ならびに血清M−CSF活性および残留heRX1−10.G1活性に関して試料を分析した。
【0183】
遠心する前に血液試料を室温でおよそ30分間凝血させた。およそ4℃で2700rpmで10分間の遠心により血清が得られ、そして得られた血清を四つのアリコートに分けた。
【0184】
本明細書にて言及される、および/または出願データシートに列挙される前記の全ての米国特許、米国公開特許公報、米国特許出願、外国特許、外国特許出願および非特許文献はその全てを出典明示により本明細書の一部とする。
【0185】
前記から、本発明の具体的な実施態様は本明細書では説明の目的のために記載されているが、本発明の精神および範囲から逸脱することなく種々の修飾を行うことができることは理解されよう。
【数1】
【数2】
【数3】
【数4】
【数5】
【数6】
【数7】
【数8】
【数9】
【数10】
【数11】
【数12】
【数13】
【数14】
【数15】
【数16】
【数17】
【数18】
【数19】
【数20】
【数21】
【数22】
【数23】
【数24】
【数25】
【数26】
【数27】
【数28】
【数29】
【数30】
【数31】
【数32】
【数33】
【数34】
【数35】
【数36】
【数37】
【数38】
【数39】
【数40】
【数41】
【数42】
【数43】
【数44】
【数45】
【数46】
【数47】
【数48】
【数49】
【数50】
【数51】
【数52】
【数53】
【数54】
【数55】
【数56】
【数57】
【数58】
【数59】
【数60】
【数61】
【数62】
【技術分野】
【0001】
本発明は別の治療薬と組み合わされたM−CSFアンタゴニストを対象に投与することにより、癌転移および癌転移に関連する骨量減少(bone loss)を含む骨溶解性疾患を防御および処置するための方法に関する。
【背景技術】
【0002】
(発明の背景)
骨吸収を媒介する破骨細胞は骨溶解性疾患を含む正常および異常な骨リモデリング過程に関与する。破骨細胞は造血細胞から分化する多核細胞である。破骨細胞が不完全な細胞分裂よりもむしろ、骨髄の造血幹細胞に由来する単核前駆体の融合により形成されることは一般的に知られている(Chambers、Bone and Mineral Research 6:1−25(1989);Gothlingら、Clin Orthop Relat R.120:201−228(1976);Kahnら、Nature
258:325−327(1975)、Sudaら、Endocr Rev 13:66−80、(1992);Walker、Science 180:875(1973);Walker、Science 190:785−787(1975);Walker、Science 190:784−785(1975))。これらは単球−マクロファージ細胞系と共通の幹細胞を共有する(Ashら、Nature 283:669−670(1980)、Kerbyら、J.Bone Miner Res 7:353−62(1992))。破骨細胞前駆体の成熟多核破骨細胞への分化はホルモン性および局部刺激を含む様々な因子を必要とし(Athanasouら、Bone Miner 3:317−333(1988);Feldmanら、Endocrinology 107:1137−1143(1980);Walker、Science 190:784−785(1975);Zhengら、Histochem J 23:180−188(1991))、そして生体骨および骨細胞は破骨細胞発達において非常に重要な役割を果たすことが示されている(Hagenaarsら、Bone Miner 6:179−189(1989))。骨芽または骨髄間質細胞もまた破骨細胞分化に必要とされる。破骨細胞形成を支持するこれらの細胞により生成される因子の一つはマクロファージコロニー刺激因子、M−CSFである(Wiktor−Jedrzejczakら、Proc Natl Acad Sci USA 87:4828−4832(1990);Yoshidaら、Nature 345:442−444(1990))。NF−κBリガンドに関する受容体アクチベーター(RANKL、TRANCE、ODFおよびOPGLとしても公知)は別のシグナルであり(Sudaら、Endocr Rev 13:66−80(1992))、それにより骨芽/間質細胞が破骨細胞および破骨細胞前駆体に位置する受容体、RANK(TRANCER)を介する破骨細胞形成および吸収を刺激する(Laceyら、Cell 93:165−176(1998);Tsudaら、Biochem Biophys Res Co 234:137−142(1997);Wongら、J Exp Med 186:2075−2080(1997);Wongら、J Biol.Chem 272:25190−25194(1997);Yasudaら、Endocrinology 139:1329−1337(1998);Yasudaら、Proc Natl Acad Sci US 95:3597−3602(1998))。骨芽細胞はまたオステオプロテジェリン(OPG、OCIFとしても公知)と称される、破骨細胞形成を強く阻止するタンパク質を分泌し、それはRANKLに関するデコイ受容体として作用し、したがってRANKおよびRANKLを介する破骨細胞と骨芽細胞との間の陽性シグナルを阻止する。
【0003】
破骨細胞はミネラルおよび有機骨マトリックスの双方の溶解に寄与する(Blairら、J Cell Biol 102:1164−1172(1986))。破骨細胞は特殊な膜部分ならびに、酒石酸抵抗性酸ホスファターゼ(TRAP)(Andersonら、1979)、炭酸脱水酵素II(Vaananenら、Histochemistry
78:481−485(1983))、カルシトニン受容体(Warshafskyら、Bone 6:179−185(1985))およびビトロネクチン受容体(Daviesら、J Cell Biol 109:1817−1826(1989))のようないくつかの膜および細胞質マーカーを伴う独特な極性形態学を表現する高分化型細胞を表す。多核破骨細胞は通常10個未満の核を含有するが、それらは直径10μmと100μmの間である100個までの核を含有し得る(Gothlingら、Clin Orthop Relat R 120:201−228(1976))。これによりそれらは光学顕微鏡により比較的容易に同定されることになる。それらは活性化状態にあるときに高度に空胞化され、そしてまた高い代謝速度の指標である多くのミトコンドリアを含有する(Mundy、Primer on the metabolic bone diseases and disorders of mineral metabolism、18−22頁(1990))。破骨細胞は破骨性骨転移において主要な役割を果たすので、当分野では破骨細胞刺激および機能を防御するための新しい薬剤および方法が必要とされる。
【0004】
癌転移は癌患者における術後または治療後の再発の主な原因である。処置を開発するための集中的な努力にもかかわらず、癌転移は治療に対して依然実質的に不応性である。骨は種々の型のヒト癌(乳腺、肺、前立腺および甲状腺癌)の転移の最も一般的な部位の一つである。破骨性骨転移の発症は難治性の疼痛、高度な骨折し易さ、神経圧迫および高カルシウム血症による深刻な病的状態を引き起こす。これらの臨床上の問題にもかかわらず、癌転移に関連する骨量減少に利用可能な処置はわずかしかない。
【0005】
骨溶解性疾患を標的とするいくつかの治療計画が現在用いられているか、または開発中であり、ここでは主に破骨細胞の形成または活性を阻止することにより骨吸収を遮断する薬物の開発に尽力している。ビスホスホネート系(BPs)、骨に集中するピロリン酸類似体は今日までの最も有効な骨吸収の阻害剤である。BPsは破骨細胞に取り込まれ、その活性を阻止し、そして細胞にアポトーシスを被らせ、それにより骨吸収を阻止する。アレンドロネートは脊椎/股関節骨折の有意な減少を示す最初の骨吸収のBP阻害剤であり、そして骨粗鬆症の処置のために承認されている。最新世代のBPであるZometaは高カルシウム血症ならびに固形腫瘍および多発性骨髄腫における骨疾患の処置のために承認されており、そしてパジェット病ならびに固形腫瘍および多発性骨髄腫の結果である骨転移の処置の可能性に関して調査中である。Zometaは非常に低用量で作用し、そして月1回の15分間静脈内注入として与えられるが、骨芽細胞にも影響し、そして腎毒性および顎の骨壊死のような副作用を引き起こし得る(非特許文献1;非特許文献2;非特許文献3;非特許文献4;非特許文献5;非特許文献6;非特許文献7;非特許文献8)。したがって骨溶解性疾患および/または骨溶解性骨転移を含む癌転移を防御または処置するための新しい薬剤および方法を同定することが当分野では依然必要とされている。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】FromigueおよびBrody、J.Endocrinol.Invest.(2002)25:39−46
【非特許文献2】Ibrahim,A.ら、Clin.Canc.Res.(2003)9:2394−99
【非特許文献3】Body,J.J.、The Breast.(2003)S2:S37−44
【非特許文献4】Yaccoby,S.ら、Brit.J.Hemat.(2002)116:278−80
【非特許文献5】Corey,E.ら、Clin.Canc.Res.(2003)9:295−306
【非特許文献6】Coleman,R.E.、Sem.Oncol.,(2002)29(6):43−49
【非特許文献7】Coleman,R.E.、Eur.Soc.Med.Oncol.(2005)16:687−95
【非特許文献8】Bamiasら、J Clin Oncol(2005)13:8580−8587
【発明の概要】
【課題を解決するための手段】
【0007】
(発明の要旨)
本発明の組成物および方法は当分野において前記された、およびその他の関連する必要性を満足する。本発明の一つの実施態様では、骨溶解性障害を患うかまたはその危険性を有する対象に単剤治療の量のM−CSFアンタゴニストおよび単剤治療上有効な量の第二の抗破骨細胞薬を約1日から1年の移行期間(transition period)の間投与し、その間M−CSFアンタゴニストは活性破骨細胞の数を治療上望ましいレベルまで低下させることを含む、その対象を処置するための方法が提供される。M−CSFアンタゴニストの実例には、M−CSF抗体が挙げられ、そして第二の抗破骨細胞薬の実例には、ビスホスホネート系および抗RANKL抗体を含むRANKL阻害剤が挙げられる。抗M−CSF抗体および破骨細胞阻害剤を伴う方法および/または使用は本明細書では場合によっては国際公開番号第WO2005/068503号に開示されるRX1、5H4、MCIおよびMC3由来の抗体の使用を排除する。移行期間の長さは、例えば少なくとも1日から1年まででよく、そして例えば破骨細胞成長または活性の関連マーカーによりモニタリングすることができる。これに代えて、それらを同時に与えることができる。
【0008】
実例として骨形成のマーカーには、限定するものではないがカルシウム、ならびに全および骨特異的アルカリ性ホスファターゼ(BAP)、オステオカルシン(OC、骨glaタンパク質)、IC型プロコラーゲンプロペプチド(PICP)、IN型プロコラーゲンプロペプチド(PINP)が挙げられ、そして骨吸収のマーカーには、限定するものではないがNTX(骨コラーゲンのN末端架橋テロペプチド)およびCTX(骨コラーゲンのC末端架橋テロペプチド)、ピリジニウム架橋(ピリジノリンおよびデオキシピリジノリン(DPD))および関連ペプチド、骨のI型コラーゲン分解産物、ヒドロキシプロリンおよびヒドロキシリジングリコシド、酒石酸抵抗性酸ホスファターゼ(TRACP)、ならびに骨シアロタンパク質(BSP)が挙げられる。Fohrら、J.Clin.Endocrinol.Metab.,88(11):5059−5075(2003年11月)を参照のこと。
【0009】
関連する実施態様では、第二の抗破骨細胞薬が移行期間の後に中断される前記された方法が提供される。その他の関連する実施態様では、第二の抗破骨細胞薬を移行期間の後に減少させる前記された方法が提供される。さらなる関連する実施態様では、M−CSFアンタゴニストの量を移行期間の後に減少させる前記された方法が提供される。
【0010】
本発明の方法がM−CSFとその受容体(M−CSFR)との間の相互作用を阻止することによりその治療可能性を達成すると企図される。M−CSF/M−CSFR相互作用の阻止が破骨細胞増殖および/または分化を阻止することがさらに企図される。本発明のいずれかの方法または組成物では、M−CSFアンタゴニストは抗M−CSF抗体を含むポリペプチド;抗M−CSFR抗体を含むポリペプチド;M−CSFムテインもしくはその誘導体を含む可溶性ポリペプチド;またはM−CSFRムテインもしくはその誘導体を含む可溶性ポリペプチド;またはM−CSFもしくはM−CSFRの発現を阻止する核酸分子でよい。種々のM−CSFアンタゴニストの同定、生成および修飾は国際公開番号第WO2005/068503号に記載され、その全てを出典明示により本明細書の一部とする。
【0011】
M−CSF抗体はポリクローナル抗体;モノクローナル抗体;ヒト化抗体;ヒト抗体;ヒト改変抗体;キメラ抗体;Fab、F(ab’)2もしくはFv抗体フラグメント;または前記された抗体のいずれか一つの抗体のムテインでよい。
【0012】
骨溶解を阻止する本発明のM−CSF抗体は国際公開番号第WO2005/068503号(M−CSF抗体に関するその教示に関してその全てを出典明示により本明細書の一部とする)に記載されている。
【0013】
本発明の一つの実施態様では、図1、3および4の各々に示されるアミノ酸配列を有するネズミモノクローナル抗体RX1、MC1またはMC3のいずれか一つと同一のM−CSFのエピトープに特異的に結合する機能的フラグメントを含む非ネズミモノクローナル抗体が提供される。関連する実施態様では抗体がポリクローナル抗体;Human Engineered(商標)抗体を含むモノクローナル抗体;ヒト化抗体;ヒト抗体;;キメラ抗体;Fab、F(ab’)2;Fv;Sc FvまたはSCA抗体フラグメント;ダイアボディー;直鎖状抗体;または好ましくは少なくとも10−7、10−8もしくは10−9またはそれより高い結合親和性を保持するこれらの抗体のいずれか一つのムテイン;からなる群から選択される前記された抗体が提供される。M−CSFへの結合に関して、図1に示されるアミノ酸配列を有するモノクローナル抗体RX1、MC1および/またはMC3と75%を超えて競合する機能的フラグメントを含む非ネズミモノクローナル抗体もまた企図される。
【0014】
別の実施態様では、非ネズミモノクローナル抗体またはその機能的フラグメントが図7のアミノ酸98−105の少なくとも4、5、6、7または8個の近接する残基を含むM−CSFのエピトープに結合する機能的フラグメントを含む該非ネズミモノクローナル抗体が提供される。
【0015】
別の実施態様では、本発明は非ネズミモノクローナル抗体またはその機能的フラグメントが図7のアミノ酸65−73または138−144の少なくとも4、5、6、7または8個の近接する残基を含むM−CSFのエピトープ(5H4またはMC3により認識されるM−CSFエピトープに相当する)に結合する機能的フラグメントを含む該非ネズミモノクローナル抗体を提供する。
【0016】
さらに別の実施態様では、図7のアミノ酸98−105を含むM−CSFのエピトープに結合する前記された抗体またはフラグメントが提供される。関連する実施態様では、図1AのCDR3を含む前記された抗体が提供される。別の実施態様では、図1Aで示されるネズミ抗体RX1の少なくとも1、2、3、4、5または6個のCDRを含む抗体が提供される。ネズミ抗体RX1の少なくとも1、2、3、4または5個のCDRを含むかかる抗体はまた図8A−Bで示される抗体5H4の6個のCDRのいずれかの少なくとも1、2、3、4または5個のCDRを含んでなってよい。これに代えてネズミ抗体RX1の少なくとも1、2、3、4または5個のCDRを含む抗体はまた図8A−Bで示される抗体MC1の6個のCDRのいずれかの少なくとも1、2、3、4または5個のCDRを含んでなってよい。さらに別の代替えでは、前記された抗体はまた図8A−Bで示される抗体MC3の6個のCDRのいずれかの少なくとも1、2、3、4または5個のCDRを含んでなってよい。関連する実施態様では、ネズミ抗体RX1の少なくとも1、2、3、4または5個のCDRを含む抗体は提供される図8A−Bで示されるコンセンサスCDRの少なくとも1、2、3、4または5個のCDRを含んでなってよい。さらに別の関連する実施態様では、前記された抗体では(複数の)コンセンサスCDRの一つまたはそれより多い残基はネズミ抗体RX1、5H4、MC1またはMC3のCDRのいずれかの対応する残基により置換されている。抗体の一つまたはそれより多いアミノ酸が例えばCDRの保存置換、および/または低および中リスク残基における保存もしくは非保存変化により変異していたとしても、望ましい結合親和性を保持することができる。
【0017】
本発明の別の実施態様では、図1A、2、3または4で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変重鎖アミノ酸配列を含む前記された抗体のバリアントが提供される。関連する実施態様では、抗体は図1A、2、3または4で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変軽鎖アミノ酸配列を含む。
【0018】
さらに別の実施態様では、抗体はヒト抗体配列の定常領域ならびに一つまたはそれより多い重および軽鎖可変フレームワーク領域を含む。関連する実施態様では、抗体はヒトIgG1、IgG2、IgG3またはIgG4の修飾または未修飾定常領域を含む。好ましい実施態様では、定常領域はヒトIgG1またはIgG4であり、それは場合によっては特定の特性を強化または低下させるために修飾され得る。IgG1の場合、定常領域、特にヒンジまたはCH2領域に対する修飾は、ADCCおよび/またはCDC活性を含むエフェクター機能を上昇または低下させ得る。その他の実施態様では、IgG2定常領域を修飾して抗体−抗原凝集形成を低下させる。IgG4の場合、定常領域、特にヒンジ領域に対する修飾は、半抗体の形成を低下させ得る。
【0019】
本発明の一つの実施態様では、国際公開番号第WO2005/068503号に記載されるようなネズミ抗体RX1、5H4、MC1もしくはMC3のいずれか一つと同一のM−CSFのエピトープに特異的に結合するか、またはM−CSFに対する結合に関して10%を超えて、さらに好ましくは25%を超えて、なおさらに好ましくは50%を超えて、もっとさらに好ましくは75%を超えて、そして最も好ましくは90%を超えて前記されたネズミ抗体のいずれか一つと拮抗する非ネズミモノクローナル抗体が提供される。キメラ、ヒト、ヒト化、ヒト改変抗体またはそれのフラグメントもしくはムテインもしくは化学的に誘導体化されたものを含むかかるネズミ抗体の配列由来の抗体が第WO2005/068503号に記載されている。
【0020】
M−CSFに結合する能力を保持する「RX1由来の抗体」には以下のいずれか一つが含まれる:
1)相同性決定に関する類似アミノ酸を考慮して、図1で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変重鎖アミノ酸配列を含む、および/または図1で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変軽鎖アミノ酸配列を含むバリアントを含む、図1に示されるアミノ酸配列を有するネズミ抗体RX1のアミノ酸バリアント;
2)図1で示されるアミノ酸配列を有するネズミ抗体RX1の一つまたはそれより多い相補性決定領域(CDR)を含んでなり、好ましくはRX1重鎖の少なくともCDR3を含んでなり、そして好ましくは二つもしくはそれより多い、または三つもしくはそれより多い、または四つもしくはそれより多い、または五つもしくはそれより多い、または六つ全部のCDRを含むM−CSF結合ポリペプチド(ネズミ抗体RX1を除く);
3)図9Bから12Bで示される重および軽鎖アミノ酸配列を有するHuman Engineered(商標)抗体または図9Bから12Bの元来のHuman Engineered(商標)重もしくは軽鎖と少なくとも60%のアミノ酸配列同一性、さらに好ましくは少なくとも80%、さらに好ましくは少なくとも85%、さらに好ましくは少なくとも90%、そして最も好ましくは例えば65%、70%、75%、80%、81%、82%、83%、84%、85%。86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%および100%を含む少なくとも95%の同一性を有する重もしくは軽鎖を含むそのバリアント;
4)図9Bから12BのHuman Engineered(商標)抗体の一つまたはそれより多いCDRの高リスク残基を含んでなり、そして好ましくは二つもしくはそれより多い、または三つもしくはそれより多い、または四つもしくはそれより多い、または五つもしくはそれより多い、または六つ全部のCDRの高リスク残基を含むM−CSF結合ポリペプチド(ネズミ抗体RX1を除く);
5)図1Bで示される高リスクアミノ酸残基を保持し、そして図1Bで示される低または中リスク残基の一つまたはそれより多い変化を含む;
例えば図1Bで示される低リスク残基での一つまたはそれより多い変化および中リスク残基での保存置換を含むか;または
例えば図1Bで示される中および高リスクアミノ酸残基を保持し、そして低リスク残基で一つまたはそれより多い変化を含んでなり;
ここで変化は挿入、欠失または置換を含み、そして保存置換でよいか、または改変抗体の配列をヒト軽鎖もしくは重鎖配列、ヒト生殖系軽鎖もしくは重鎖配列、コンセンサスヒト軽鎖もしくは重鎖配列、またはコンセンサスヒト生殖系軽鎖もしくは重鎖配列に近づけることができる;
Human Engineered(商標)抗体またはバリアント。かかる抗体は好ましくは少なくとも10−7、10−8もしくは10−9またはそれより高い親和性を伴ってM−CSFに結合し、そして好ましくはM−CSFの活性を誘起する破骨細胞形成を中和する。
【0021】
同様にM−CSFに結合する能力を保持する「MC3由来の抗体」には以下のいずれか一つが含まれる:
1)相同性決定に関する類似アミノ酸を考慮して、図4で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変重鎖アミノ酸配列を含む、および/または図4で示されるアミノ酸配列に対して少なくとも60、65、70、75、80、85、90、91、92、93、94、95、96、97、98または99%相同である可変軽鎖アミノ酸配列を含むバリアントを含む、図4に示されるアミノ酸配列を有するネズミ抗体MC3のアミノ酸バリアント;
2)図4で示されるアミノ酸配列を有するネズミ抗体MC3の一つまたはそれより多い相補性決定領域(CDR)を含んでなり、好ましくはMC3重鎖の少なくともCDR3を含んでなり、そして好ましくは二つもしくはそれより多い、または三つもしくはそれより多い、または四つもしくはそれより多い、または五つもしくはそれより多い、または六つ全部のCDRを含むM−CSF結合ポリペプチド(場合によってはネズミ抗体MC3を含むかまたは除く);
3)Studnickaら、米国特許第5,766,886号および本明細書の実施例4Aに示される方法に従って、図13C−13Eに示されるKabat番号付けを用いて、低、中および高リスク残基を同定するためにネズミ配列を変化させることにより作成されたHuman Engineered(商標)抗体;かかる抗体は少なくとも一つの以下の重鎖および少なくとも一つの以下の軽鎖を含む:(a)低リスク残基の全てが修飾されており、必要によりヒト参照免疫グロブリン配列と同一の残基になる重鎖、または(b)低および中リスク残基の全てが修飾されており、必要によりヒト参照免疫グロブリン配列と同一の残基になる重鎖、(c)低リスク残基の全てが修飾されており、必要によりヒト参照免疫グロブリン配列と同一の残基になる軽鎖、または(d)低および中リスク残基の全てが修飾されており、必要によりヒト参照免疫グロブリン配列と同一の残基になる軽鎖;
4)元来のHuman Engineered(商標)重または軽鎖と少なくとも60%のアミノ酸配列同一性、さらに好ましくは少なくとも80%、さらに好ましくは少なくとも85%、さらに好ましくは少なくとも90%、そして最も好ましくは例えば65%、70%、75%、80%、81%、82%、83%、84%、85%。86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%および100%を含む少なくとも95%の同一性を有する重または軽鎖を含む前記のパラグラフ(3)において前記された抗体のバリアント;
5)図4のネズミMC3抗体の一つまたはそれより多いCDRの高リスク残基を含んでなり、そして好ましくは二つもしくはそれより多い、または三つもしくはそれより多い、または四つもしくはそれより多い、または五つもしくはそれより多い、または六つ全部のCDRの高リスク残基を含むM−CSF結合ポリペプチド(場合によってはネズミ抗体MC3を含むかまたは除く);
6)ネズミMC3抗体の高リスクアミノ酸残基を保持し、そして低または中リスク残基で一つまたはそれより多い変化を含む;
例えば低リスク残基での一つまたはそれより多い変化および中リスク残基での保存置換を含むか;または
例えば中および高リスクアミノ酸残基を保持し、そして低リスク残基で一つまたはそれより多い変化を含んでなり;
ここで変化は挿入、欠失または置換を含み、そして保存置換でよいか、または改変抗体の配列をヒト軽鎖もしくは重鎖配列、ヒト生殖系軽鎖もしくは重鎖配列、コンセンサスヒト軽鎖もしくは重鎖配列、またはコンセンサスヒト生殖系軽鎖もしくは重鎖配列に近づけることができる;
Human Engineered(商標)抗体またはバリアント。かかる抗体は好ましくは少なくとも10−7、10−8もしくは10−9またはそれより高い親和性を伴ってM−CSFに結合し、そして好ましくはM−CSFの活性を誘起する破骨細胞形成を中和する。
【0022】
「5H4由来の抗体」または「MC1由来の抗体」なる用語は前記の記載に従って同様に定義される。
【0023】
本明細書にて詳細に記載されるように、Human Engineered(商標)抗体またはバリアントを含むRX1、5H4、MC1またはMC3由来の抗体は、IgG、IgA、IgMまたはIgEのような異なるアイソタイプのものでよい。IgGクラスの抗体は異なる定常領域を含むことができ、例えばIgG2抗体をIgG1またはIgG4定常領域を表示するように修飾することができる。好ましい実施態様では、本発明は修飾または未修飾IgG1またはIgG4定常領域を含むHuman Engineered(商標)抗体またはバリアントを提供する。IgG1の場合、定常領域、特にヒンジまたはCH2領域に対する修飾は、ADCCおよび/またはCDC活性を含むエフェクター機能を上昇または低下させることができる。その他の実施態様では、IgG2定常領域を修飾して抗体−抗原凝集形成を低下させる。IgG4の場合、定常領域、特にヒンジ領域に対する修飾は半抗体の形成を低下させることができる。具体的な例示的な実施態様では、IgG4ヒンジ配列Cys−Pro−Ser−CysをIgG1ヒンジ配列Cys−Pro−Pro−Cysに変異させることが提供される。
【0024】
前記されたM−CSFアンタゴニストまたはM−CSF抗体および薬学的に許容される担体、賦形剤または希釈剤を含む医薬組成物を本発明に従って投与することができる。
【0025】
二つまたはそれより多いM−CSFアンタゴニストを一緒に混合するか、またはM−CSFアンタゴニストおよび第二の抗破骨細胞薬を同時投与して、癌転移および/または癌転移に関連する骨量減少を含む本発明の骨溶解性疾患に対して改善された効果を提供することはさらに有利であり得る。
【0026】
本発明の例示的な実施態様では、第二の抗破骨細胞薬がビスホスホネートである前記された方法が提供される。さらなる実施態様では、ビスホスホネートはゾレドロネート(zoledronate)、パミドロネート(pamidronate)、クロドロネート(clodronate)、エチドロネート(etidronate)、チルドロネート(tiludronate)、アレンドロネート(alendronate)、イバンドロナート(ibandronate)またはリセドロネート(risedronate)である。その他の抗破骨細胞薬の実例にはビスホスホネート系、PTHrP中和剤(例えば抗体、アンチセンス、siRNA)、カテプシンK阻害剤、MIP−1−αアンタゴニスト、RANK/RANKL中和剤(例えば抗RANK抗体、抗RANKL抗体またはアンチセンス、可溶性RANKL受容体またはそのムテイン)、RANKLワクチン、オステオプロテグリン(OPG)、血小板由来成長因子(PDGF)、srcキナーゼ阻害剤、マロン酸ガリウムおよびマトリックスメタロプロテイナーゼ(MMP)阻害剤が挙げられる。
【0027】
本発明の治療方法をさらに癌化学療法剤のような第三の治療薬と、または放射線治療もしくは外科的手術と組み合わせることができる。癌化学療法剤には、限定するものではないがカルボプラチンおよびシスプラチンのようなアルキル化剤;ナイトロジェンマスタードアルキル化剤;カルムスチン(BCNU)のようなニトロソウレアアルキル化剤;メトトレキサートのような代謝拮抗剤;プリン類似体代謝拮抗剤、メルカプトプリン;フルオロウラシル(5−FU)およびゲムシタビンのようなピリミジン類似体代謝拮抗剤;ゴセレリン、リュープロリドおよびタモキシフェンのような抗腫瘍性ホルモン薬;アルデスロイキン、インターロイキン−2、ドセタキセル、エトポシド(VP−16)、インターフェロンアルファ、パクリタキセルおよびトレチノイン(ATRA)のような天然抗悪性腫瘍薬;ブレオマイシン、ダクチノマイシン、ダウノルビシン、ドキソルビシンおよびマイトマイシンのような抗菌性天然抗悪性腫瘍薬;ならびにビンブラスチン、ビンクリスチン、ビンデシンのようなビンカアルカロイド天然抗悪性腫瘍薬;ヒドロキシウレア;アセグラトン、アドリアマイシン、イフォスファミド、エノシタビン、エピチオスタノール、アクラルビシン、アンシタビン、ニムスチン、塩酸プロカルバジン、カルボコン、カルボプラチン、カルモフール、クロモマイシンA3、抗腫瘍性多糖類、抗腫瘍性血小板因子、シクロホスファミド、シゾフィラン、シタラビン、ダカルバジン、チオイノシン、チオテパ、テガフル、ネオカルチノスタチン、OK−432、ブレオマイシン、フルツロン、ブロクスウリジン、ブスルファン、ホンバン、ペプロマイシン、ベスタチン(ウベニメクス)、インターフェロン−β、メピチオスタン、ミトブロニトール、メルファラン、ラミニンペプチド、レンチナン,カワラタケ(Coriolus versicolor)抽出物、テガフル/ウラシル、エストラムスチン(エストロゲン/メクロレタミン)が含まれる。
【0028】
さらに、癌患者の補助的療法として用いられるさらなる薬剤には、EPO、G−CSF、ガンシクロビル;抗生物質、ロイプロリド;メペリジン;ジドブジン(AZT);変異体および類似体を含むインターロイキン1から18;インターフェロンα、βおよびγのようなインターフェロン系またはサイトカイン系;黄体形成ホルモン放出ホルモン(LHRH)および類似体、および生殖腺刺激ホルモン放出ホルモン(GnRH)のようなホルモン系;形質転換成長因子β(TGF−β)、線維芽細胞成長因子(FGF)、神経成長因子(NGF)、成長ホルモン放出因子(GHRF)、上皮細胞成長因子(EGF)、線維芽細胞成長因子相同因子(FGFHF)、肝細胞成長因子(HGF)およびインスリン成長因子(IGF)のような成長因子;腫瘍壊死因子α&β(TNF−α&β);浸潤阻止因子(invasion inhibiting factor)2(IIF−2);骨形成タンパク質1−7(BMP1−7);ソマトスタチン;チモシンα−1;γグロブリン;スーパーオキシドジスムターゼ(SOD);補体因子;抗血管形成因子;抗原性物質;プロドラッグ;成長因子受容体キナーゼ阻害剤;抗Her2抗体;ならびにVEGF中和抗体が含まれる。
【0029】
移行期間に続いて、治療効果を達成するために必要とされるM−CSFアンタゴニストの量または第二の抗破骨細胞薬の量を減少させることができる。したがってかかる期間の後、M−CSFアンタゴニストが第二の抗破骨細胞薬の効果を改善するか、または第二の抗破骨細胞薬の投与に随伴される副作用を低減するか、または第二の抗破骨細胞薬の安全性を改善することができる。M−CSFアンタゴニストはまた効果を改善するか、癌化学療法剤、その他の補助的療法、外科的手術または放射線治療のような第三の治療法の副作用を低減するか、または安全性を改善することができる。本発明の別の実施態様では、M−CSFアンタゴニストを含む医薬品、ならびにその医薬品が第二および/もしくは第三の治療薬との、および/または外科的手術もしくは放射線治療との組み合わせで用いられるべきであるとする説明書を含む包装、バイアルまたは容器が提供される。
【0030】
多くの骨溶解性障害は本発明による処置に適していると企図される。本明細書にて使用される際には、「骨溶解性障害」は破骨細胞活性の上昇の結果である任意の症状である。骨溶解性障害の危険性を有する対象は、骨溶解性障害を発達させる傾向がある群の対象、または骨破壊活性の上昇を引き起こすかもしくはそれに寄与する疾患を患う対象でよい。本発明の例示的な実施態様では骨溶解性障害は、内分泌疾患(副腎皮質ホルモン過剰症、性腺機能低下症、原発性または二次性副甲状腺機能亢進症、甲状腺機能亢進症)、高カルシウム血症、欠乏状態(クル病/骨軟化症、壊血病、栄養障害)、慢性疾患(吸収不良症候群、慢性腎不全(腎性骨異栄養症)、慢性肝疾患(肝性骨異栄養症))、薬物(糖質コルチコイド(糖質コルチコイド誘起骨粗鬆症)、ヘパリン、アルコール)または遺伝性疾患(骨形成不全症、ホモシスチン尿症)、癌、骨粗鬆症、大理石骨病、関節炎および関節リウマチに関連する骨の炎症、歯周病、線維性骨異形成症および/またはパジェット病を含む破骨細胞活性の相対的な上昇に関連する代謝性骨疾患でよい。
【0031】
その他の例示的な実施態様では、骨溶解性障害は骨への転移性癌でよく、ここで転移性癌は乳腺、肺、腎臓、多発性骨髄腫、甲状腺、前立腺、腺癌、白血病およびリンパ腫を含む血液細胞悪性腫瘍;頭頸部癌;食道癌、胃癌、結腸癌、腸癌、結腸直腸癌、直腸癌、膵臓癌、肝臓癌、胆管もしくは胆嚢の癌を含む胃腸管系癌;卵巣癌腫、子宮内膜癌、膣癌もしくは子宮頸癌を含む女性生殖器の悪性腫瘍;膀胱癌;神経芽細胞腫を含む脳癌;肉腫、骨肉腫;または悪性メラノーマもしくは扁平上皮癌を含む皮膚癌である。
【0032】
本発明の例示的な実施態様では、前記の方法のいずれかは骨量減少を防御するかまたは低減させることができるか、または疾患に関連する骨転移もしくは骨量減少の重篤度を防御もしくは低減させる。
【0033】
本発明に従って投与されるM−CSF抗体を約2μg/kgから30mg/kg、0.1mg/kgから30mg/kg、または0.1mg/kgから10mg/kg体重の間の用量で与えることができる。
例えば、本願発明は以下の項目を提供する。
(項目1)
単剤治療上有効な用量の抗M−CSF抗体および単剤治療上有効な用量の破骨細胞阻害剤を骨溶解性障害を患う患者に投与する工程を含む、該患者を処置する方法。
(項目2)
単剤治療上有効な用量の抗M−CSF抗体および単剤治療上有効な用量のビスホスホネートを骨溶解性障害を患う患者に投与する工程を含む該患者を処置する方法。
(項目3)
前記破骨細胞阻害剤がRANKL阻害剤である項目1に記載の方法。
(項目4)
前記抗MCSF抗体がRX1由来の抗体、MC1由来の抗体、MC3由来の抗体または5H4由来の抗体である項目2に記載の方法。
(項目5)
前記抗MCSF抗体がheRX1である項目2に記載の方法。
(項目6)
前記骨溶解性障害が骨粗鬆症、癌転移に関連する骨量減少、パジェット病またはプロテーゼ周囲骨量減少である項目1から5のいずれかに記載の方法。
(項目7)
前記破骨細胞阻害剤および前記MCSF抗体が同時に投与される項目1から6のいずれかに記載の方法。
(項目8)
前記RANKL阻害剤が抗RANKL抗体、可溶性RANKL受容体およびRANKLワクチンからなる群から選択される項目に記載の方法。
(項目9)
前記抗RANKL抗体がAMG−162である項目3、4、6および7に記載の方法。
(項目10)
前記ビスホスホネートがゾレドロネート、パミドロネート、クロドロネート、エチドロネート、チルドロネート、アレンドロネート、イバンドロナートおよびリセドロネートからなる群から選択される項目2、4、6および7に記載の方法。
(項目11)
前記ビスホスホネートがゾレドロネートである項目10に記載の方法。
(項目12)
移行期間に単剤治療上有効な用量の抗M−CSF抗体および単剤治療上有効な用量のビスホスホネートを骨溶解性障害を患う患者に投与する工程を含む、該患者を処置する方法。
(項目13)
前記破骨細胞阻害剤がRANKL阻害剤である項目12に記載の方法。
(項目14)
前記移行期間がおよそ1−7日である項目13に記載の方法。
(項目15)
前記移行期間が1週間から1か月である項目13に記載の方法。
(項目16)
前記移行期間が1か月から3か月である項目13に記載の方法。
(項目17)
前記移行期間が3から6か月である項目13に記載の方法。
(項目18)
前記移行期間が6から12か月である項目13に記載の方法。
(項目19)
前記移行期間後に前記ビスホスホネート治療を中断する工程をさらに含む項目13に記載の方法。
(項目20)
前記移行期間後に前記ビスホスホネートの用量を減少させる工程をさらに含む項目13に記載の方法。
(項目21)
前記移行期間の直後にビスホスホネートの用量を減少させる項目13に記載の方法。
(項目22)
前記移行期間後に抗MCSF抗体の用量を減少させる工程をさらに含む項目13に記載の方法。
(項目24)
前記移行期間の直後に抗MCSF抗体の用量を減少させる項目13に記載の方法。
(項目25)
前記移行期間後に前記ビスホスホネートが少なくとも1回投与される項目13に記載の方法。
(項目26)
前記抗MCSF抗体が配列番号5および配列番号6のネズミRX1抗体からの少なくとも二つのCDRを含む項目1から25のいずれかに記載の方法。
(項目27)
前記抗MCSF抗体が配列番号5および配列番号6のネズミRX1抗体からの少なくとも三つのCDRを含む項目1から26のいずれかに記載の方法。
(項目28)
前記抗MCSF抗体が配列番号5および配列番号6のM−CSFに対する結合に関して少なくとも75%、ネズミRX1抗体と競合する項目1から27のいずれかに記載の方法。
(項目29)
前記破骨細胞阻害剤がゾレドロネートであり、そして前記MCSF抗体がRX1由来の抗体である項目6、13および20に記載の方法。
(項目30)
前記ゾレドロネートが0.5mgと4mgの間の用量で投与される項目29に記載の方法。
(項目31)
前記抗MCSF抗体がRX1、MC1、MC3または5H4抗体と同一のエピトープに結合する項目4、6、8、10、11または12に記載の方法。
【図面の簡単な説明】
【0034】
【図1A】M−CSF特異的ネズミ抗体RX1のアミノ酸配列(配列番号2および4)(アメリカン・タイプ・カルチャー・コレクション(米国、バージニア州、マナサス)でATCC寄託番号PTA−6113の下に寄託されたプラスミドのcDNAインサートによりコードされる)および対応する核酸配列(配列番号1および3)を示す。CDR領域は番号付けされ、そして太字で示される。
【図1B】各々M−CSF特異的ネズミ抗体RX1軽鎖(配列番号5)および重鎖(配列番号6)のアミノ酸配列を示し、Studnickaら、第WO93/11794号に従って同定された高リスク(太字)、中リスク(下線)および低リスク残基を伴う。
【図1C】各々M−CSF特異的ネズミ抗体RX1軽鎖(配列番号5)および重鎖(配列番号6)のアミノ酸配列を示し、Studnickaら、第WO93/11794号に従って同定された高リスク(太字)、中リスク(下線)および低リスク残基を伴う。
【図2】各々M−CSF特異的ネズミ抗体5H4(配列番号10および11)、MC1(配列番号12および13)(ATCC寄託番号PTA−6263の下に寄託されたハイブリドーマにより生成される)およびMC3(配列番号14および15)(ATCC寄託番号PTA−6264の下に寄託されたハイブリドーマにより生成される)のアミノ酸配列を示す。
【図3】各々M−CSF特異的ネズミ抗体5H4(配列番号10および11)、MC1(配列番号12および13)(ATCC寄託番号PTA−6263の下に寄託されたハイブリドーマにより生成される)およびMC3(配列番号14および15)(ATCC寄託番号PTA−6264の下に寄託されたハイブリドーマにより生成される)のアミノ酸配列を示す。
【図4】各々M−CSF特異的ネズミ抗体5H4(配列番号10および11)、MC1(配列番号12および13)(ATCC寄託番号PTA−6263の下に寄託されたハイブリドーマにより生成される)およびMC3(配列番号14および15)(ATCC寄託番号PTA−6264の下に寄託されたハイブリドーマにより生成される)のアミノ酸配列を示す。
【図5】M−CSFαのアミノ酸配列(配列番号7)である。
【図6】M−CSFβのアミノ酸配列(配列番号8)である。
【図7】M−CSFγのアミノ酸配列(配列番号9)である。DNA配列における多くの多型はアミノ酸の差異を招き得る。例えば共通の多型は104位でProよりむしろAlaを提供する。
【図8A】ヒトM−CSF特異的ネズミ抗体RX1;5H4;MC1;およびMC3の重および軽鎖アミノ酸配列(配列番号16−38)のCDR領域のアラインメントである。
【図8B】ヒトM−CSF特異的ネズミ抗体RX1;5H4;MC1;およびMC3の重および軽鎖アミノ酸配列(配列番号16−38)のCDR領域のアラインメントである。
【図9A】(a)ネズミRX1重鎖のリスクライン(H=高リスク、M=中リスク、L=低リスク)、(b)RX1重鎖アミノ酸配列(配列番号6)、(c)RX1とアラインメントされた最も近いヒトコンセンサス配列、KabatVh2コンセンサスのアミノ酸配列(配列番号39)および(d)二つのHuman Engineered(商標)配列(配列番号41および43)の実例を生成させた変化を示す。
【図9B】「低リスク」および「低+中リスク」と称される二つの重鎖Human Engineered(商標)配列の実例のアミノ酸配列(配列番号41および43)ならびに対応する核酸配列(配列番号40および42)を示す。
【図10A】(a)ネズミRX1軽鎖のリスクライン(H=高リスク、M=中リスク、L=低リスク)、(b)RX1軽鎖アミノ酸配列(配列番号5)、(c)RX1とアラインメントされた最も近いヒトコンセンサス配列、KabatVk3コンセンサスのアミノ酸配列(配列番号49)および(d)二つのHuman Engineered(商標)配列(配列番号45および47)の実例を生成させた変化を示す。
【図10B】「低リスク」および「低+中リスク」と称される二つの軽鎖Human Engineered(商標)配列の実例のアミノ酸配列(配列番号45および47)ならびに対応する核酸配列(配列番号44および46)を示す。
【図11A】(a)ネズミRX1軽鎖のリスクライン(H=高リスク、M=中リスク、L=低リスク)、(b)RX1軽鎖アミノ酸配列(配列番号5)、(c)RX1とアラインメントされた最も近いヒトコンセンサス配列、KabatVk3コンセンサスのアミノ酸配列(配列番号49)および(d)54−56位が変化しなかった(すなわちネズミ配列のままである)代替えのアミノ酸配列の実例(配列番号48)を示す。
【図11B】二つの代替えの軽鎖Human Engineered(商標)配列の実例のアミノ酸配列(配列番号48、87)ならびに対応する核酸配列(配列番号88および86)を示す。
【図12A】(a)ネズミRX1軽鎖のリスクライン(H=高リスク、M=中リスク、L=低リスク)、(b)RX1軽鎖アミノ酸配列(配列番号5)、(c)RX1とアラインメントされた最も近いヒトコンセンサス生殖系配列、Vk6サブグループ2−1−(1)A14のアミノ酸配列(配列番号50)および(d)二つのHuman Engineered(商標)配列(配列番号51および53)の実例を生成させた変化を示す。
【図12B】「低リスク」および「低+中リスク」と称される二つの軽鎖Human Engineered(商標)配列の実例のアミノ酸配列(配列番号51および53)ならびに対応する核酸配列(配列番号52)を示す。
【図13A】Kabat番号付けシステム(「POS」と称される一直線で示されるアミノ酸番号付け)を用いて種々のヒトコンセンサスおよびヒト生殖系コンセンサス配列を伴うネズミRX1重鎖アミノ酸配列(配列番号54)のアラインメント(配列番号55−83)を示す。
【図13B】Kabat番号付けシステム(「POS」と称される一直線で示されるアミノ酸番号付け)を用いて種々のヒトコンセンサスおよびヒト生殖系コンセンサス配列を伴うネズミRX1重鎖アミノ酸配列(配列番号54)のアラインメント(配列番号55−83)を示す。
【図13C】Kabat番号付けシステムに対応する抗体5H4、MC1およびMC3のアミノ酸残基(各々配列番号10および11;配列番号12および13;配列番号14および15)がどのようになっているかを示す。
【図13D】Kabat番号付けシステムに対応する抗体5H4、MC1およびMC3のアミノ酸残基(各々配列番号10および11;配列番号12および13;配列番号14および15)がどのようになっているかを示す。
【図13E】Kabat番号付けシステムに対応する抗体5H4、MC1およびMC3のアミノ酸残基(各々配列番号10および11;配列番号12および13;配列番号14および15)がどのようになっているかを示す。
【図14】動物モデルにおけるZometaの抗吸収効果を示す。
【図15】検出可能な骨溶解を伴う各群の動物のパーセントを示す。
【図16】試験の最後の日のX線画像分析に基づく平均骨溶解スコアを示す。
【図17】試験の最終日の脛骨(腫瘍接種部位)の代表的なFaxitron X線画像を示す。
【図18】破骨細胞活性に及ぼすRX1の効果を示す。
【図19】Zometaによる破骨細胞活性の阻止を示す。
【図20】霊長類におけるRX1での薬物動態試験の結果を示す。
【図21】霊長類におけるRX1での薬物動態試験の結果を示す。
【発明を実施するための形態】
【0035】
(詳細な説明)
マクロファージコロニー刺激因子(M−CSF)としても公知のコロニー刺激因子(CSF−1)は破骨細胞形成に重要であることが分かっている。加えて、M−CSFは成熟破骨細胞の骨破壊機能、その他の可溶性因子と協働してその遊走およびその生存性、ならびに骨芽細胞および線維芽細胞により提供される細胞間相互作用を変調することが示されている(FixeおよびPraloran、Cytokine 10:3−7(1998);Martinら、Critical Rev.in Eukaryotic Gene Expression 8:107−23(1998))。
【0036】
全長ヒトM−CSF mRNAは554個のアミノ酸の前駆体タンパク質をコードする。選択的mRNAスプライシングおよび差次的翻訳後タンパク質プロセシングにより、M−CSFは糖タンパク質もしくは硫酸コンドロイチン含有プロテオグリカンとして循環中に分泌されるか、またはM−CSF産生細胞の表面の膜を貫通する糖タンパク質として発現されるかのいずれかになり得る。完全なインビトロ生物学的活性に必要とされる最小配列であるヒトM−CSFの細菌発現されたアミノ末端の150個のアミノ酸の三次元構造は、このタンパク質は各単量体が四つのアルファヘリックス束および逆平行ベータシートからなる、ジスルフィド連結された二量体であることを示している(Panditら、Science 258:1358−62(1992))。三つの異なるM−CSF種は選択的mRNAスプライシングにより生成される。三つのポリペプチド前駆体は256個のアミノ酸のM−CFSα、554個のアミノ酸のM−CSFβ、および438個のアミノ酸のM−CSFγである。M−CSFβは膜結合形態では生じない分泌タンパク質である。M−CSFαはタンパク質切断により緩徐に放出される膜内在性タンパク質として発現される。M−CSFαは図5で示される配列のアミノ酸191−197で切断されている。M−CSFの膜結合形態は近くの細胞の受容体と相互作用することができ、そしてしたがって特異的細胞間接触を媒介する。「M−CSF」なる用語はまた図7のアミノ酸36−438をも含み得る。
【0037】
種々の形態のM−CSFは標的細胞でその受容体M−CSFRに結合することにより機能する。M−CSFRは五つの細胞外免疫グロブリン様ドメイン、膜貫通ドメインおよび細胞内分断Src関連チロシンキナーゼドメインを有する膜を貫通する分子である。M−CSFRはc−fms癌原遺伝子によりコードされる。M−CSFRの細胞外ドメインへのM−CSFの結合により、受容体の二量体化に至り、それは細胞質キナーゼドメインを活性化し、自己リン酸化およびその他の細胞タンパク質のリン酸化に至る(Hamilton J.A.、J Leukoc Biol.62(2):145−55(1997);Hamilton J.A.、Immuno Today.,18(7):313−7(1997))。
【0038】
リン酸化された細胞タンパク質は細胞応答に至る生化学的事象のカスケード:有糸分裂、サイトカインの分泌、膜ラフリングおよびその独自の受容体の転写の調節を誘起する(FixeおよびPraloran、Cytokine 10:32−37(1998))。
【0039】
「腫瘍」とは本明細書にて使用される際には、悪性であろうと良性であろうと、全ての新生物の細胞成長および増殖、ならびに全ての前癌性および癌性細胞および組織を指す。
【0040】
「癌」および「癌性」なる用語は、典型的には無秩序な細胞成長を特徴とする哺乳動物における生理学的症状を指すかまたは記載する。癌の実例には、限定するものではないが癌腫;リンパ腫、芽細胞腫、肉腫;および白血病が挙げられる。かかる癌のさらに特定の実例には、乳癌、前立腺癌、結腸癌、扁平上皮癌、小細胞肺癌、非小細胞肺癌、胃腸管系の癌、膵臓癌、神経膠芽腫、子宮頸癌、卵巣癌、肝臓癌、膀胱癌、肝細胞腫、結腸直腸癌、子宮内膜癌腫、唾液腺癌腫;腎臓癌、肝臓癌、外陰癌、甲状腺癌、肝細胞癌腫および種々の型の頭頸部癌が挙げられる。
【0041】
「処置」は障害の発達を防御するか、または病理を変化させる意図を伴って実施される介入である。したがって「処置」は治療的処置および予防的または防御的手段の双方を指す。処置を必要とするものには既に障害を有するもの、および障害が防御されるべきものが含まれる。腫瘍(例えば癌)処置において、治療薬は腫瘍細胞の病理を直接低減させるか、またはその他の治療薬、例えば放射線および/もしくは化学療法による処置に対する腫瘍細胞の感受性をさらに高めることができる。癌の「病理」には患者の健康を危険にさらす全ての現象が含まれる。これには限定するものではないが、異常なまたは制御できない細胞成長、転移、近隣細胞の正常な機能の妨害、サイトカインまたはその他の分泌性生成物の異常レベルでの放出、炎症性または免疫学的応答の抑制または激化等が含まれる。
【0042】
処置の目的のための「哺乳動物」とはヒト、家庭用および農場用の動物、ならびに動物園、スポーツまたはペット用動物、例えばイヌ、ウマ、ネコ、ウシ、等を含む哺乳動物として分類される任意の動物を指す。好ましくは、哺乳動物はヒトである。
【0043】
本明細書にて使用される際には、「転移性癌」なる語句は身体のその他の部分、特に骨にまで広がる可能性を有する癌として定義される。種々の癌が骨に転移することができるが、最も一般的な転移する癌は乳腺、肺、腎臓、多発性骨髄腫、甲状腺および前立腺である。実例としては、骨に転移する可能性を有するその他の癌には、限定するものではないが腺癌、白血病およびリンパ腫を含む血液細胞悪性腫瘍;頭頸部癌;食道癌、胃癌、結腸癌、腸癌、結腸直腸癌、直腸癌、膵臓癌、肝臓癌、胆管もしくは胆嚢の癌を含む胃腸管系癌;卵巣癌腫、子宮内膜癌、膣癌および子宮頸癌を含む女性生殖器の悪性腫瘍;膀胱癌;神経芽細胞腫を含む脳癌;肉腫、骨肉腫;ならびに悪性メラノーマおよび扁平上皮癌を含む皮膚癌が挙げられる。本発明は特に骨における腫瘍誘起の骨溶解性病変の防御および処置を企図する。
【0044】
本明細書にて使用される際には、「治療上有効な量」なる語句は、望ましい処置レジメンに従って投与された場合、望ましい治療的または予防的効果または応答を引き出すM−CSF抗体のような本発明の実施態様に適切であろう、治療用または予防用のM−CSFアンタゴニストの量を指すと意図されることを指す。
【0045】
ヒト「M−CSF」とは、本明細書にて使用される際には、Kawasakiら、Science 230:291(1985)、Cerrettiら、Molecular Immunology,25:761(1988)、またはLadnerら、EMBO Journal 6:2693(1987)(その各々を出典明示により本明細書の一部とする)に記載される成熟ヒトM−CSFα、M−CSFβまたはM−CSFγポリペプチドと実質的に同一のアミノ酸配列を有するヒトポリペプチドを指す。かかる用語法は、三つの成熟M−CSFが前記で記載されたように異なるアミノ酸配列を有し、そしてM−CSFの活性形態はジスルフィド結合した二量体であることの理解を反映しており;したがって「M−CSF」なる用語が生物学的に活性な形態を指す場合、二量体形態が意図される。「M−CSF二量体」とは二量体化している二つのM−CSFポリペプチド単量体を指し、そして双方のホモ二量体(同一の型のM−CSF単量体の二つからなる)およびヘテロ二量体(二つの異なる単量体からなる)を含む。米国特許第4,929,700号(出典明示により本明細書の一部とする)に記載されるように、M−CSF単量体をインビトロでM−CSF二量体に変換することができる。
【0046】
1.アンタゴニスト
本明細書にて使用される際には、「アンタゴニスト」なる用語は一般的に、例えば一つの分子のと別の分子との結合を妨害するための分子、化合物もしくはその他の薬剤の特性、または立体障害、立体構造の変化もしくはその他の生化学的メカニズムのいずれかによる一つ細胞の別の細胞による刺激を指す。一つの点については、アンタゴニストなる用語は、受容体のそのリガンドに対する結合、例えばそれによりM−CSFが誘因となるシグナル伝達経路を阻止するM−CSFのM−CSFRとの結合を防御する薬剤の特性を指す。アンタゴニストなる用語は任意の具体的な作用メカニズムにより限定されないが、むしろ一般的に現在定義されている機能的特性を指す。本発明のアンタゴニストには、限定するものではないが:M−CSF抗体およびそのフラグメントおよびムテインおよび修飾体、可溶性M−CSFおよびそのフラグメントおよびムテインおよび修飾体、M−CSFR抗体およびそのフラグメントおよびムテインおよび修飾体、可溶性M−CSFRおよびそのフラグメントおよびムテインおよび修飾体、ならびにM−CSFまたはM−CSFRに結合するペプチドおよびその他の化学的化合物および分子、ならびにM−CSFおよびM−CSFRの発現を阻止するアンチセンスまたはRNAiのような核酸分子が含まれる。本発明のアンタゴニストのいずれかを当分野において公知の任意の様式で投与することができる。例えばM−CSFムテイン、M−CSFRムテインまたはM−CSFもしくはM−CSFRに結合する抗体フラグメントを、遺伝子治療を介して投与することができる。
【0047】
本発明のM−CSFアンタゴニストには、適用可能な場合、機能的均等物が含まれる。例えば分子は長さ、構造、構成成分等で異なり得るが、依然として一つまたはそれより多い、定義された機能を保持し得る。さらに特に、本発明の抗体、抗体フラグメントまたはペプチドの機能的均等物には、擬似化合物、すなわち抗原結合に関して適切な立体構造および/または配向を擬似するように設計された構築物が含まれ得る。
【0048】
好ましいM−CSFアンタゴニストを、場合によっては側鎖の付加等により、例えばアミノ末端アシル化、カルボキシ末端アミド化またはアミノ酸側鎖へのさらなる基の結合により修飾することができる。アンタゴニストはまた一つまたはそれより多い保存アミノ酸置換を含むことができる。「保存アミノ酸置換」とは全体的な電荷、疎水性/親水性および/または置換されたアミノ酸の立体容積を保存する、アミノ酸配列におけるこれらの変化を意味する。例えば以下の基の間の置換は保存的である:Gly/Ala、Val/Ile/Leu、Asp/Glu、Lys/Arg、Asn/Gln、Ser/Cys/ThrおよびPhe/Trp/Tyr。かかる修飾は実質的にはM−CSFアンタゴニストの効果を減じることはなく、そして例えばインビボで半減期の増加または毒性の低下のような望ましい特性を付与し得る。
【0049】
本発明はまたアミノ酸残基の挿入、欠失または置換以外の修飾を担持するポリペプチドを含むことも意図される。実例としては、修飾は天然の共有結合性でよく、そして例えば重合体、脂質、その他の有機および無機部分との化学的結合を含み得る。かかる誘導体を調製して、ポリペプチドの循環半減期を増すことができるか、またはポリペプチドが望ましい細胞、組織もしくは器官を標的とする能力を改善するように設計することができる。同様に、本発明はさらにポリエチレングリコール、ポリオキシエチレングリコールまたはポリプロピレングリコールのような一つまたはそれより多い水溶性重合体連結を含むように共有結合的に修飾されているM−CSFまたはM−CSFRポリペプチドを包含する。
【0050】
A.M−CSF抗体
「抗体」なる用語は最も広義で用いられ、そして完全に組み立てられた抗体、モノクローナル抗体、ポリクローナル抗体、多重特異性抗体(例えば二重特異性抗体)、抗原に結合できる抗体フラグメント(例えばFab’、F’(ab)2、Fv、一本鎖抗体、ダイアボディー)および望ましい生物学的活性を呈する限り前記のものを含む組換えペプチドを含む。
【0051】
「モノクローナル抗体」なる用語は本明細書にて使用される際には、実質的に均一な抗体の集団から得られる抗体を指し、すなわち集団を含む個々の抗体は少量で存在し得る可能な天然発生変異を除いて同一である。モノクローナル抗体は高度に特異的であり、単一の抗原部位に対して指向する。さらに典型的には異なる決定基(エピトープ)に対して指向する異なる抗体を含む従来の(ポリクローナル)抗体調製物とは対照的に、各モノクローナル抗体は抗原上の単一の決定基に対して指向する。その特異性に加えて、モノクローナル抗体は均一な培養により合成され、異なる特異性および特徴を有するその他の免疫グロブリンにより汚染されていないという点で有利である。
【0052】
「モノクローナル」なる修飾語句は抗体の実質的に均一な集団から得られるような抗体の性状を示し、そして任意の特定の方法による抗体の産生を必要とすると解釈されるべきではない。例えば本発明に従って用いられるモノクローナル抗体を、Kohlerら、Nature,256:495(1975)により最初に記載されたハイブリドーマ法により作成することができるか、または組換えDNA法(例えば米国特許第4,816,567号参照)により作成することができる。「モノクローナル抗体」を例えばClacksonら、Nature,352:624628(1991)およびMarksら、J.Mol.Biol.,222:581−597(1991)に記載される技術を用いてファージ抗体ライブラリーから単離することもできる。
【0053】
その重鎖の定常ドメインのアミノ酸配列に依存して、免疫グロブリンを異なるクラスに割り当てることができる。IgA、IgD、IgE、IgGおよびIgMの五つの主要なクラスがあり、そしてこれらのうちのいくつかをさらにサブクラスまたはアイソタイプ、例えばIgG1、IgG2、IgG3、IgG4、IgA1およびIgA2に分けることができる。免疫グロブリンの異なるクラスに相当する重鎖定常ドメインを各々アルファ、デルタ、イプシロン、ガンマおよびミューと称する。免疫グロブリンの異なるクラスのサブユニット構造および三次元立体構造は周知である。異なるアイソタイプは異なるエフェクター機能を有し、例えばIgG1およびIgG3アイソタイプはADCC活性を有する。
【0054】
「抗体フラグメント」はインタクトな全長抗体の一部、好ましくはインタクトな抗体の抗原結合または可変領域を含む。抗体フラグメントの実例には、Fab、Fab’、F(ab’)2およびFvフラグメント;ダイアボディー;直鎖状抗体(Zapataら、Protein Eng.,8(10):1057−1062(1995));一本鎖抗体分子;および抗体フラグメントから形成された多重特異性抗体が挙げられる。抗体のパパイン消化により「Fab」フラグメントと称される二つの同一の抗原結合フラグメントが生成され、各々単一の抗原結合部位および、残りの「Fc」フラグメントを有し、「Fc」フラグメントの名前は35を容易に結晶化するその能力を反映する。ペプシン処理により、抗体のVHおよびVLドメイン(これらのドメインは単一のポリペプチド鎖に存在する)を含む二つの「一本鎖Fv」すなわち「sFv」抗体フラグメントを有するF(ab’)2フラグメントを生じる。好ましくは、FvポリペプチドはさらにVHおよびVLドメイン間でポリペプチドリンカーを含んでなり、それはFvが抗原結合に関して望ましい構造を形成するのを可能にする。sFvの概説に関しては、Pluckthun、The
Pharmacology of Monoclonal Antibodies、113巻、RosenburgおよびMoore編、Springer−Verlag(ニューヨーク)、269−315頁(1994)を参照のこと。
【0055】
「超可変」領域なる用語は抗原結合に寄与する抗体のアミノ酸残基を指す。超可変領域は相補性決定領域、すなわちCDRからのアミノ酸残基(すなわちKabatら、Sequences of Proteins of Immunological Interest、第5版、公衆衛生局、国立衛生研究所、メリーランド州、ベセスダ(1991)に記載されるように、軽鎖可変ドメインの残基24−34(L1)、50−56(L2)および89−97(L3)ならびに重鎖可変ドメインの31−35(H1)、50−65(H2)および95−102(H3))および/または超可変ループからのこれらの残基(すなわちChothiaら、J.Mol.Biol.196:901−917(1987)に記載されるように、軽鎖可変ドメインの残基26−32(L1)、50−52(L2)および91−96(L3)ならびに重鎖可変ドメインの26−32(H1)、53−55(H2)および96−101(H3)を含む。
【0056】
「フレームワーク」またはFR残基は超可変領域残基以外のこれらの可変ドメイン残基である。
【0057】
「ダイアボディー」なる用語は二つの抗原結合部位を有する小型の抗体フラグメントを指し、そのフラグメントは同一のポリペプチド鎖(VH VL)で軽鎖可変ドメイン(VL)に連結された重鎖可変ドメイン(VH)を含む。非常に短くて同一鎖上の二つのドメイン間で対形成することができないリンカーを用いることにより、ドメインをその他の鎖の相補的ドメインと対形成させ、そして二つの抗原結合部位を創成する。ダイアボディーは例えば欧州特許第404,097号;第WO93/11161号;および30Hollingerら、Proc.Natl.Acad.Sci.USA,90:6444−6448(1993)にてさらに十分に記載されている。
【0058】
いくつかの実施態様では、少なくとも二つの異なるエピトープに関して結合特異性を有するモノクローナル、ヒト、ヒト化、Human Engineered(商標)またはバリアント抗M−CSF抗体を含む多重特異性(例えば二重特異性)モノクローナル抗体を作成するのが望ましい場合もある。二重特異性抗体の実例はM−CSFの二つの異なるエピトープに結合し得る。これに代えて、抗M−CSFアームをT細胞受容体分子(例えばCD2またはCD3)のような白血球上のトリガー分子、またはFcγRI(CD64)、FcγRII(CD32)およびFcγRIII(CD16)のようなIgGに関するFc受容体(FcγR)に結合するアームと組み合わせて、M−CSF発現細胞に対する細胞防衛メカニズムに集中させることができる。二重特異性抗体を用いてM−CSFを発現する細胞に細胞毒性薬を局在化させることもできる。これらの抗体はM−CSF結合アームおよび細胞毒性薬(例えばサポリン、抗インターフェロン60、ビンカアルカロイド、リシンA鎖、メトトレキサートまたは放射性同位元素ハプテン)に結合するアームを有する。二重特異性抗体を全長抗体または抗体フラグメント(例えばF(ab’).sub.2二重特異性抗体)として調製することができる。
【0059】
二重特異性抗体を作成するための別の研究法に従って、抗体分子の対の間のインターフェースを操作して組換え細胞培養から回収されるヘテロ二量体のパーセンテージを最大にすることができる。好ましいインターフェースは抗体定常ドメインのCH3ドメインの少なくとも一部を含む。この方法では、最初の抗体分子のインターフェースからの一つまたはそれより多い小型アミノ酸側鎖をより大型の側鎖(例えばチロシンまたはトリプトファン)と置き換える。大型のアミノ酸側鎖を小型のもの(例えばアラニンまたはスレオニン)と置き換えることにより、(複数の)大型側鎖と同一かまたは類似した大きさの代償性「空洞」が第二の抗体分子のインターフェースに創成される。これにより、ホモ二量体のようなその他の望ましくない最終生成物よりもヘテロ二量体の収量を増加させるためのメカニズムが提供される。1996年9月6日公開の第WO96/27011号を参照のこと。
【0060】
二重特異性抗体には架橋または「ヘテロ抱合」抗体が含まれる。例えばヘテロ抱合の抗体の一方をアビジンに結合させ、他方をビオチンに結合させることができる。任意の便宜的な架橋方法を用いてヘテロ抱合抗体を作成することができる。適当な架橋剤は当分野において周知であり、そして米国特許第4,676,980号にて、多くの架橋技術と一緒に開示されている。
【0061】
抗体フラグメントから二重特異性抗体を作成するための技術もまた文献に記載されている。例えば二重特異性抗体を、化学的連結を用いて調製することができる。Brennanら、Science 229:81(1985)は、インタクトな抗体がタンパク質分解により切断されてF(ab’)2フラグメントを作成する手順を記載している。これらのフラグメントはジチオール錯化剤、亜ヒ酸ナトリウムの存在下で還元されて隣接ジチオールを安定化し、そして分子間ジスルフィド形成を防御する。作成されたFab’フラグメントは次いでチオニトロ安息香酸(TNB)誘導体に変換される。Fab’−TNB誘導体の一つは次いでメルカプトエチルアミンでの還元によりFab’−チオールに再変換され、そして等モル濃度の量のその他のFab’−TNB誘導体と混合されて二重特異性抗体を形成する。生成された二重特異性抗体を酵素の選択的固定のための薬剤として用いることができる。なおさらなる実施態様では、大腸菌から直接回収されたFab’−SHフラグメントをインビトロで化学的に結合させて二重特異性抗体を形成することができる。(Shalabyら、J.Exp.Med.175:217−225(1992))。
【0062】
Shalabyら、J.Exp.Med.175:217−225(1992)は完全なヒト化二重特異性抗体F(ab’)2分子の生成を記載している。各Fab’フラグメントは大腸菌から別個に分泌され、そしてインビトロで指示された化学的結合に供されて二重特異性抗体を形成した。このように形成された二重特異性抗体はHER2受容体を過剰発現する細胞および正常なヒトT細胞に結合でき、そしてヒト乳腺腫瘍標的に対するヒト細胞毒性リンパ球の溶解活性の誘因となることができた。
【0063】
組換え細胞培養から直接二重特異性抗体フラグメントを作成および単離するための種々の技術もまた記載されている。例えばロイシンジッパーを用いて二重特異性抗体が生成されている(Kostelnyら、J.Immunol.148(5):1547−1553(1992))。FosおよびJunタンパク質からのロイシンジッパーペプチドは遺伝子融合により二つの異なる抗体のFab’部分に連結された。抗体ホモ二量体はヒンジ領域で還元されて単量体を形成し、そして次に再酸化されて抗体ヘテロ二量体を形成する。この方法を抗体ホモ二量体の生成に利用することもできる。Hollingerら、Proc.Natl.Acad.Sci.USA 90:6444−6448(1993)に記載される「ダイアボディー」テクノロジーは二重特異性抗体フラグメントを作成するための代替えメカニズムを提供している。
【0064】
フラグメントは、短すぎて同一鎖上の二つのドメイン間で対形成できないリンカーにより、軽鎖可変領域(VL)に連結されている重鎖可変領域(VH)を含む。したがって、一つのフラグメントのVHおよびVLドメインは別のフラグメントの相補的VLおよびVHドメインと対形成を強いられ、それにより二つの抗原部位を形成する。一本鎖Fv(sFv)二量体の使用による二重特異性抗体フラグメントを作成するための別の計画もまた報告されている。Gruberら、J.Immunol.152:5368(1994)を参照のこと。
【0065】
これに代えて、二重特異性抗体はZapataら、Protein Eng.8(10):1057−1062(1995)に記載されるように生成された「直鎖状抗体」でよい。簡単には、これらの抗体は一対の抗原結合領域を形成する一対のタンデムFdセグメント(VH−CH1−VH−CH1)を含む。直鎖状抗体は二重特異性または単一特異性でよい。
【0066】
2価を超える抗体もまた企図される。例えば三重特異性抗体を調製することができる。(Tuffら、J.lmmunol.147:60(1991))
特定の実施態様では、モノクローナル、ヒト、ヒト化、Human Engineered(商標)またはバリアント抗M−CSF抗体はRX1、5H4、MC1またはMC3抗体フラグメントのような抗体フラグメントである。抗体フラグメントを生成するための種々の技術が開発されている。伝統的にはこれらのフラグメントはインタクトな抗体のタンパク質消化を介して誘導される(例えばMorimotoら、Journal of Biochemical and Biophysical Methods 24:107−117(1992)およびBrennanら、Science 229:81(1985)を参照のこと)。しかしながら、今やこれらのフラグメントを組換え宿主細胞により直接生成することができる。Betterら、Science 240:1041−1043(1988)は細菌からの機能的抗体フラグメントの分泌を開示している(例えばBetterら、Skerraら、Science 240:1038−1041(1988)を参照のこと)。例えばFab’−SHフラグメントを大腸菌から直接回収し、そして化学的に連結させてF(ab’)2フラグメントを形成することができる(Carterら、Bio/Technology 10:163−167(1992))。別の実施態様では、ロイシンジッパーGCN4を用いてF(ab’)2分子の組み立てを促進してF(ab’)2を形成する。別の研究法に従って、Fv、FabまたはF(ab’)2フラグメントを宿主細胞培養から直接単離することができる。抗体フラグメントの生成のためのその他の技術は当業者には明白であろう。
【0067】
「単離された」抗体は同定され、そしてその天然の環境の構成成分から分離されて、そして回収されているものである。その天然の環境の夾雑する構成成分は抗体の診断的または治療的使用を妨害する材料であり、そして酵素、ホルモンおよびその他のタンパク質性または非タンパク質性溶質を含み得る。好ましい実施態様では、抗体は(1)ローリー法により決定されるように、抗体の95重量%、そして最も好ましくは99重量%を超えるまで、(2)スピニングカップシークエネーターの使用により、N末端、もしくは内部のアミノ酸配列の少なくとも15残基を得るのに十分な程度まで、または(3)クーマシーブルーもしくは好ましくは銀染色を用いて還元もしくは非還元条件下でSDS−PAGEにより均一になるまで精製される。組換え細胞内で原位置の抗体は、抗体の天然の環境の少なくとも一つの構成成分が存在しないので、単離された抗体に含まれる。しかしながら通常単離された抗体は少なくとも一つの精製工程により調製される。
【0068】
抗体の構造および作成の詳細な記載に関してはRoth,D.B.およびCraig,N.L.、Cell,94:411−414(1998)および米国特許第6,255,458号(その全てを出典明示により本明細書の一部とする)を参照のこと。簡単には重および軽鎖免疫グロブリン遺伝子をコードするDNAを作成するための過程は一次的にはB細胞の発達の際に生じる。種々の免疫グロブリン遺伝子セグメントの再配列および結合の前に、V、D、Jおよび定常(C)遺伝子セグメントは一般的に単一の染色体上の比較的近接近して見出される。B細胞分化の間に、V、D、J(または軽鎖遺伝子の場合VおよびJのみ)遺伝子セグメントの適切なファミリーメンバーの各々の一つは組換えられて機能的に再配列された重および軽鎖免疫グロブリン遺伝子を形成する。この遺伝子セグメント再配列過程は逐次的であると思われる。最初に重鎖D−J結合、続いて重鎖V−DJ結合および軽鎖V−J結合が作られる。
【0069】
機能的重および軽鎖可変領域を形成する可変領域遺伝子セグメントの組換えは、組換え能力のあるV、DおよびJセグメントをフランキングする組換えシグナル配列(RSS’s)により媒介される。直接組換えに必要および十分なRSS’sは二分子対称七量体、ATリッチ九量体および12または23塩基対のいずれかの介在スペーサー領域を含む。これらのシグナルは異なる遺伝子座およびD−J(またはV−J)組換えを担持する種間で保存され、そして機能的に互換性である。Oettingerら、Science 248:1517−1523(1990)およびそこに引用される参照文献を参照のこと。七量体は配列CACAGTGまたはその類似体、続いて非保存配列のスペーサー、そして次に配列ACAAAAACCまたはその類似体を有する九量体を含む。これらの配列は各VおよびD遺伝子セグメントのJまたは下流側に見出される。生殖系列DおよびJセグメントの直ぐ先は再度二つの組換えシグナル配列、再度非保存配列により分けられた、最初に九量体、そして次に七量体がある。VL、VHまたはDセグメントに続く七量体および九量体配列は、それらが組換えるJL、DまたはJHセグメントに先行するものに相補的である。七量体配列と九量体配列の間のスペーサーは12塩基対の長さか、または22塩基対と24塩基対の間の長さのいずれかである。
【0070】
V、DおよびJセグメントの再配列に加えて、軽鎖のVおよびJセグメントが結合し、そして重鎖のDおよびJセグメントが結合する位置の可変組換えにより、免疫グロブリン重および軽鎖の一次的なレパートリーでさらなる多様性を生じる。軽鎖におけるかかる変化は典型的にはV遺伝子セグメントの最後のコドンおよびJセグメントの最初のコドン内で生じる。結合における類似の不正確さはDセグメントとJHセグメントの間の重鎖染色体上で生じ、そして10ヌクレオチドほどまで伸長し得る。さらに、DとJH遺伝子セグメントの間およびVHとD遺伝子セグメントの間で、ゲノムDNAによりコードされないいくつかのヌクレオチドが挿入され得る。これらのヌクレオチドの付加はN領域多様性として公知である。
【0071】
可変領域遺伝子セグメントにおけるかかる再配列およびかかる結合の間に生じ得る可変組換えの正味の効果は一次抗体レパートリーの生成である。
【0072】
「Fv」は完全抗原認識および結合部位を含有する最小抗体フラグメントである。この領域は緊密な、非共有結合性会合の一つの重鎖および一つの軽鎖可変ドメインの二量体からなる。各可変ドメインの三つのCDRが相互作用してVH V1二量体の表面で抗原結合部位を定義するのはこの立体構造においてである。六つのCDRが集団で抗体に対する抗原結合特異性に寄与する。しかしながら単一の可変ドメイン(または抗原に特異的な三つのCDRのみを含むFvの半分)でさえ抗原を認識し、そして結合する能力を有しているが、親和性は全結合部位よりも低い。
【0073】
Fabフラグメントもまた軽鎖の定常ドメインおよび重鎖の第一定常ドメイン(CH1)を含有する。Fabフラグメントは抗体ヒンジ領域からの一つまたはそれより多いシステインを含む重鎖CH1ドメインのカルボキシ末端における数個の残基の付加によりFab’フラグメントと異なる。本明細書ではFab’−SHは定常ドメインの(複数の)システイン残基が遊離チオール基を担持するFab’に関する名称である。F(ab’)2抗体フラグメントは元来それらの間にヒンジシステインを有するFab’フラグメントの対として生成された。
【0074】
「中和抗体」とは、それが結合する標的抗原のエフェクター機能を排除するかまたは有意に低減させることができる抗体分子を意味する。したがって抗標的「中和」抗体は酵素活性、リガンド結合または細胞内シグナリングのようなエフェクター機能を排除するかまたは有意に低減させることができる。
【0075】
本明細書にて提供されるような癌転移または癌転移に関連する骨量減少の処置のための組成物および方法は、望ましい効果を達成するために単独でまたはその他の治療と組み合わされて使用される一つまたはそれより多い抗体を利用できる。環境抗原との直接接触または抗原での免疫のいずれかの結果として、本発明による抗体を、抗体を産生する動物から単離することができる。これに代えて、当分野において周知の抗体発現系の一つを用いて組換えDNA方法論により抗体を産生することができる(例えばHarlowおよびLane、Antibodies:A Laboratory Manual、Cold Spring Harbor Laboratory(1988)を参照のこと)。かかる抗体は組換えIgG、免疫グロブリンから誘導される配列または「Human Engineered(商標)」抗体を有するキメラ融合タンパク質を含むことができ、それを全て本発明による癌転移および/または癌転移に関連する骨量減少の処置のために用いることができる。インタクトな全長分子に加えて、「抗体」なる用語はまたそのフラグメント(例えばscFv、Fv、Fd、Fab、Fab’およびF(ab)’2フラグメントのような)またはM−CSF(またはM−CSFR)に結合するインタクトな分子および/もしくはフラグメントの多量体もしくは凝集体をも指す。これらの抗体フラグメントは抗原に結合し、そして例えばガラクトース残基の組み込みによるクリアランスおよび取り込みを促進する構造的様相を呈するように誘導体化され得る。
【0076】
本発明の一つの実施態様では、本質的にHalenbeckら、米国特許第5,491,065号(1997)(出典明示により本明細書の一部とする)に記載されるようにM−CSFモノクローナル抗体を調製することができる。M−CSFモノクローナル抗体の実例としては、付随する生物学的活性の中和を伴う組換えまたは未変性二量体M−CSFに随伴される見かけの立体構造エピトープに結合するものが挙げられる。これらの抗体は単量体および化学的に誘導体化された二量体M−CSFを含む生物学的に不活性な形態のM−CSFとは実質的には反応しない。
【0077】
本発明のその他の実施態様では、Human Engineered(商標)抗M−CSFモノクローナル抗体が提供される。「Human Engineered(商標)抗体」なる語句は非ヒト抗体、典型的にはマウスモノクローナル抗体から誘導された抗体を指す。これに代えて、親、非ヒト抗体の抗原結合特性を保持するかまたは実質的に保持するが、ヒトに投与された場合、親抗体と比較して免疫原性の低下を呈するキメラ抗体からHuman Engineered(商標)抗体を誘導することができる。本明細書にて使用される際には、「キメラ抗体」なる語句は典型的には異なる種を起源とする二つの異なる抗体から誘導される配列を含有する抗体(例えば米国特許第4,816,567号を参照のこと)を指す。最も典型的には、キメラ抗体はヒトおよびネズミ抗体フラグメント、一般的にはヒト定常およびマウス可変領域を含む。
【0078】
「相補性決定領域」なる語句または「CDR」なる用語は未変性免疫グロブリン結合部位の天然Fv領域の結合親和性および特異性を一緒に定義するアミノ酸配列を指す(例えばChothiaら、J.Mol.Biol.196:901−917(1987);Kabatら、米国保健社会福祉省NIH出版第91 3242号(1991)を参照のこと)。「定常領域」なる語句はエフェクター機能を付与する抗体分子の部分を指す。本発明では、マウス定常領域はヒト定常領域により置換されるのが好ましい。対象抗体の定常領域はヒト免疫グロブリンから誘導される。重鎖定常領域を五つのアイソタイプ:アルファ、デルタ、イプシロン、ガンマまたはミューのいずれかから選択することができる。
【0079】
本発明の抗体は、約106M−1以上、好ましくは約107M−1以上、さらに好ましくは約108M−1以上、そして最も好ましくは約109M−1、1010M−1、1011M−1または1012M−1以上のKaで抗原に結合する場合、免疫特異的または特異的に結合するとされる。抗M−CSF抗体は、宿主/対象組織により発現されるもの、および腫瘍により発現されるものを含むM−CSFの様々な天然発生形態に結合し得る。RX1、5H4、MC1またはMC3抗体のような本明細書にて開示されるモノクローナル抗体はM−CSFに関して親和性を有し、そして少なくとも10−4M、好ましくは少なくとも約10−7Mから約10−8M、さらに好ましくは少なくとも約l0−8M、10−10M、l0−11Mまたは10−12Mの解離平衡定数(Kd)を特徴とする。平衡透析による;製造者により概説される一般的な手順を用いてBlAcore2000装置を使用することによる;125I標識M−CSFを用いるラジオイムノアッセイによる;または当業者に公知の別の方法によるような従来の技術を用いてかかる親和性を容易に決定することができる。例えばScatchardら、Ann N.Y.Acad.Sci.,51:660(1949)の方法により親和性データを分析することができる。このように好ましいM−CSF抗体はM−CSFに関して高度な特性を呈し、そしてその他の分子とは実質的に低い親和性で結合することは明白である。好ましい抗体は、図4のネズミRX1がM−CSFと結合するのと類似の親和性でM−CSFに結合し、低い免疫原性を呈し、そして転移性疾患動物モデルにおいて試験した場合、癌の転移を阻止する。その他の抗体の実例は図2、3または4のネズミ5H4、MC1またはMC3が各々M−CSFと結合するのと類似の親和性でM−CSFに結合する。
【0080】
抗体の生成に使用される抗原は例えば、場合によってはエピトープがその未変性立体構造を表示することを可能にする別のポリペプチドに融合された、望ましいエピトープを保持するインタクトなM−CSFまたはM−CSFのフラグメントでよい。これに代えて、その細胞表面でM−CSFを発現する細胞を用いて抗体を作成することができる。かかる細胞は形質転換されてM−CSFを発現できるか、またはM−CSFを発現するその他の天然発生細胞でよい。抗体の作成に有用なM−CSFのその他の形態は当業者には明白であろう。
【0081】
i.ポリクローナル抗体
ポリクローナル抗体を好ましくは動物において関連抗原およびアジュバントの複数回皮下(sc)または腹腔内(ip)注射により上昇させる。二機能性または誘導体化剤、例えばマレイミドベンゾイルスルホスクシンイミドエステル(システイン残基により抱合)、N−ヒドロキシスクシンイミド(リジン残基による)、グルタルアルデヒド、無水コハク酸または当分野において公知のその他の薬剤を用いて、関連抗原を免疫される種で免疫原性であるタンパク質、例えばキーホールリンペットヘモシアニン、血清アルブミン、ウシチログロブリン、大豆トリプシン阻害剤に抱合させることにより改善された抗体応答を得ることができる。
【0082】
例えば100μgまたは5μgのタンパク質または抱合体(各々ウサギまたはマウスに関して)をフロイント完全アジュバント3容量と組み合わせ、そして溶液を複数の部位に皮内注射することにより、動物を抗原、免疫原性抱合体または誘導体に対して免疫する。1か月後、フロイント完全アジュバント中1/5(分画(1/10))から元来の量のペプチドまたは包合体で複数の部位に皮下注射することにより動物をブーストする。ブースター注射後7−14日に動物を出血させ、そして血清を抗体力価に関して検定する。力価がプラトーになるまで動物をブーストする。好ましくは同一抗原であるが、異なるタンパク質に、および/または異なる架橋剤により抱合させた抱合体で動物をブーストする。組換え細胞培養でタンパク質融合体として抱合体を作成することもできる。またミョウバンのような凝集剤を用いて免疫応答を強化するのが適当である。
【0083】
ii.モノクローナル抗体
モノクローナル抗体を、Kohlerら、Nature,256:495(1975)により最初に記載されたハイブリドーマ法を用いて作成できるか、または組換えDNA法により作成できる。
【0084】
ハイブリドーマ法では、マウスまたはハムスターもしくはマカクザルのようなその他の適切な宿主動物を本明細書に記載されるように免疫して、免疫に用いられるタンパク質に特異的に結合する抗体を産生するか、または産生できるリンパ球を導く。これに代えてリンパ球をインビトロで免疫できる。次いでポリエチレングリコールのような適当な融合剤を用いてリンパ球を骨髄腫細胞と融合してハイブリドーマ細胞を形成する(Goding、Monoclonal Antibodies:Principles and Practice、59−103頁(Academic Press、1986))。
【0085】
このように調製されたハイブリドーマ細胞を、好ましくは融合されていない親骨髄腫細胞の成長または生存を阻止する一つまたはそれより多い物質を含有する適当な培養培地に播種し、そして成長させる。例えば親骨髄腫細胞が酵素ヒポキサンチングアニンホスホリボシルトランスフェラーゼ(HGPRTまたはHPRT)を欠く場合、ハイブリドーマのための培養培地は典型的にはヒポキサンチン、アミノプテリンおよびチミジンを含み(HAT培地)、その物質はHGPRT欠損細胞の成長を防御する。
【0086】
好ましい骨髄腫細胞は効率的に融合し、選択された抗体産生細胞による安定した高レベルの抗体の産生を支持し、そして培地に対して感受性があるものである。ヒト骨髄腫およびマウス−ヒトヘテロ骨髄腫細胞系はまたヒトモノクローナル抗体の産生に関して記載されている(Kozbor、J.Immunol.,133:3001(1984);Brodeurら、Monoclonal Antibody Production Techniques and Applications、51−63頁(Marcel Dekker社、ニューヨーク、1987))。ネズミ骨髄腫系の実例にはソーク研究所セル・ディストリビューション・センター(米国、カリフォルニア州、サンディエゴ)から入手可能なMOP−21およびM.C.−11マウス腫瘍、ならびにアメリカン・タイプ・カルチャー・コレクション(米国、メリーランド州、ロックビル)から入手可能なSP−2またはX63−Ag8−653細胞から誘導されるものが挙げられる。
【0087】
ハイブリドーマ細胞が成長している培養培地を、抗原に対して指向するモノクローナル抗体の産生に関して検定する。好ましくはハイブリドーマ細胞により産生されたモノクローナル抗体の結合特異性を免疫沈殿により、またはラジオイムノアッセイ(RIA)もしくは酵素結合免疫吸着アッセイ(ELISA)のようなインビトロ結合アッセイにより決定する。モノクローナル抗体の結合親和性を例えばスカッチャード分析により決定することができる(Munsonら、Anal.Biochem.,107:220(1980))。
【0088】
望ましい特異性、親和性および/または活性の抗体を産生するハイブリドーマ細胞を同定した後、限界希釈手順によりクローンをサブクローニングし、そして標準的な方法により成長させることができる(Goding、Monoclonal Antibodies:Principles and Practice、59−103頁(Academic Press、1986))。この目的のための適当な培養培地には、例えばD−MEMまたはRPMI−1640培地が含まれる。加えて、ハイブリドーマ細胞をインビボで動物の腹水腫瘍として成長させることができる。サブクローンにより分泌されるモノクローナル抗体を例えばプロテインAセファロース、ヒドロキシルアパタイトクロマトグラフィー、ゲル電気泳動、透析または親和性クロマトグラフィーのような従来の免疫グロブリン精製手順により培養培地、腹水または血清から分離するのが適当である。
【0089】
本発明の抗体は、約106M−1以上、好ましくは約107M−1以上、さらに好ましくは約108M−1以上、そして最も好ましくは約109M−1、1010M−1、1011M−1または1012M−1以上のKaで抗原に結合する場合、免疫特異的または特異的に結合するとされる。抗M−CSF抗体は、宿主/対象組織により発現されるもの、および腫瘍により発現されるものを含むM−CSFの様々な天然発生形態に結合し得る。RX1、5H4、MC1またはMC3抗体のような本明細書にて開示されるモノクローナル抗体はM−CSFに関して親和性を有し、そして少なくとも10−4M、好ましくは少なくとも約10−7Mから約10−8M、さらに好ましくは少なくとも約l0−8M、10−10M、l0−11Mまたは10−12Mの解離平衡定数(Kd)を特徴とする。平衡透析による;製造者により概説される一般的な手順を用いてBlAcore2000装置を使用することによる;125I標識M−CSFを用いるラジオイムノアッセイによる;または当業者に公知の別の方法によるような従来の技術を用いてかかる親和性を容易に決定することができる。例えばScatchardら、Ann N.Y.Acad.Sci.,51:660(1949)の方法により親和性データを分析することができる。このように好ましいM−CSF抗体はM−CSFに関して高度な特異性を呈し、そしてその他の分子とは実質的に低い親和性で結合する。好ましい抗体は、図1のネズミRX1がM−CSFと結合するのと類似の親和性でM−CSFに結合し、低い免疫原性を呈し、そして転移性疾患動物モデルにおいて試験した場合、癌の転移を阻止する。その他の抗体の実例は図2、3または4のネズミ5H4、MC1またはMC3が各々M−CSFと結合するのと類似の親和性でM−CSFに結合する。
【0090】
保存置換を表1に「好ましい置換」の見出しの下で示す。かかる置換が生物学的活性の変化を招く場合、次いで表1の「置換の実例」と表示される、またはアミノ酸クラスに関して以下にさらに記載されるようなさらに実質的な変化を導入し、そして生成物をスクリーニングすることができる。
【0091】
【表1】
抗体の生物学的特性における実質的な修飾は、(a)置換の部分のポリペプチドバックボーンの構造、例えばシートもしくはヘリックス構造として;(b)標的部位での分子の電荷もしくは疎水性;または(c)側鎖の容積;の維持に及ぼすその効果において有意に異なる置換基を選択することにより達成される。天然発生残基は共通の側鎖特性に基づいて群に分けられる:
(1)疎水性:ノルロイシン、met、ala、val、leu、ile;
(2)中性親水性:cys、ser、thr;
(3)酸性:asp、glu;
(4)塩基性:asn、gln、his、lys、arg;
(5)鎖配向に影響する残基:gly、pro;および
(6)芳香族:trp、tyr、phe。
【0092】
非保存置換はこれらのクラスの一つのメンバーを別のクラスのメンバーと置き換えることを伴う。
【0093】
ヒト化またはバリアント抗体の適切な立体構造の維持に関与しない任意のシステイン残基はまた、分子の酸化安定性の改善および異常架橋の防御のために一般的にセリンで置換され得る。逆にその安定性の改善のために(複数の)システイン結合を抗体に付加できる(特に抗体がFvフラグメントのような抗体フラグメントである場合)。
【0094】
B.M−CSFムテイン
本発明はさらに本発明の方法によるMCSFアンタゴニストとして用いることができるM−CSFムテインを提供する。
【0095】
「フラグメント」は本明細書にて使用される際には、インタクトな未変性分子の一部を意味し;例えばフラグメントポリペプチドは、N末端またはC末端のいずれかからの一つまたはそれより多いアミノ酸が欠失している未変性ポリペプチドのフラグメントである。
【0096】
「ムテイン」はポリペプチドに関して本明細書にて使用される際には、インタクトな未変性分子のバリアントまたは一つもしくはそれより多いアミノ酸が置換、挿入もしくは欠失している未変性分子のフラグメントのバリアントを意味する。かかる置換、挿入または欠失はN末端、C末端または分子の内部でよい。したがって「ムテイン」なる用語にはその範囲内に未変性分子のフラグメントが含まれる。挿入ムテインにはN末端またはC末端での融合、例えば半減期を増すための免疫グロブリンのFc部分への融合が含まれる。
【0097】
スミス・ウォーターマン相同性検索アルゴリズム(Meth.Mol.Biol.70:173−187(1997))により決定され、以下の検索パラメーター:ギャップオープンペナルティー12およびギャップ伸長ペナルティー1でアフィンギャップ検索を用いてMSPRCHプログラム(Oxford Molecular)で実行されるように、本発明による好ましいムテインは未変性ポリペプチドに対して少なくとも約65%、70%、75%、80%、85%、90%、95%、97%またはそれより高い配列同一性(相同性)を呈する。その他の周知の、および日常的に用いられる相同性/同一性走査アルゴリズムプログラムには、PearsonおよびLipman、PNAS USA,85:2444−2448(1988);LipmanおよびPearson、Science,222:1435(1985);Devereauxら、Nuc.Acids Res.,12:387−395(1984);またはAltschulら、Mol.Biol.,215:403−410(1990)のBLASTP、BLASTNまたはBLASTXアルゴリズムが含まれる。これらのアルゴリズムを用いるコンピュータープログラムもまた利用可能であり、そして限定するものではないが:GAP、BESTFIT、BLAST、FASTAおよびTFASTA(Genetics Computing Group(GCG)パッケージ、バージョン8(米国、ウィスコンシン州、マジソン)から市販により入手可能である);ならびにPC/GeneプログラムのCLUSTAL(Intellegenetics(カリフォルニア州マウンテンビュー)による)が含まれる。好ましくはプログラムにより決定されるデフォルトパラメーターを用いて配列同一性のパーセンテージを決定する。
【0098】
「修飾」は本明細書にて使用される際には、望ましい活性(アゴニストまたはアンタゴニスト)が保持されている限り、グリコシル化、リン酸化、重合体抱合(例えばポリエチレングリコールとの)またはその他の外来分子の付加のような未変性ポリペプチド、フラグメントまたはムテインの任意の修飾を意味する。
【0099】
米国特許第6,025,146号およびKoths、Mol.Reprod.Dev.46(1):31−38(1997年1月)(双方共にその全てを出典明示により本明細書の一部とする)はM−CSF単独およびMCSF−Rと複合化されたM−CSFの結晶化を記載し、そしてM−CSF三次元構造および受容体結合に関与する残基を特徴付けしている。米国特許第6,025,146号はまた、構造情報に基づいてM−CSFにおけるアミノ酸置換候補を選択するための方法を記載している。この形態のM−CSFの全体的なトポロジーは逆平行の四つのアルファヘリックス束のものであり、そこでヘリックスは、たいてい四つのヘリックス束の、より一般的に観察される上下上下連結とは異なり、上上下下と走る。長い交差接続がヘリックスAをヘリックスBに連結し、そして類似の接続がヘリックスCとDの間で見出される。ジスルフィド連結された二量体形態では、束は終端間で連結され、極めて平らな細長い構造を形成する(直径およそ85×35×25)。各単量体で三つの分子内ジスルフィド結合(Cys7−Cys90、Cys48−Cys139、Cys102−Cys146)があり、その全てが分子の遠位末端にある。一つの鎖間ジスルフィド結合(Cys31−−Cys31)は図2で示されるようにそれを通過する非結晶学的な2回対称軸を有する二量体インターフェースに位置する。変異実験によりこの形態のM−CSFでは全てのシステイン残基が完全な生物学的活性に必要であり得ることが示される。本明細書にて記載される構造は、その役割が受容体認識に関連しているというよりむしろ一次構造的であることを示唆している。米国特許第6,025,146号は、配列のアミノ酸残基のアルファ炭素位置により同定されるような、トランケートされた組換えM−CSFα二量体の三次元的構造を提供する。
【0100】
ヘリックスA、CおよびDの具体的な残基は受容体結合相互作用の特異性に関与するようである。M−CSFβがシステイン157および/または159を伴う鎖内ジスルフィド結合を有するので、M−CSFのC末端領域は構造の「後方」から伸びてM−CSFの膜結合形態に関する可変長「テザー」を提供する。したがってM−CSFの「正面」または受容体結合領域は分子の反対側にあり、未変性M−CSFの各々約6から26、71から90、および110から130の残基を含むヘリックスA、CおよびDの、またはそれの近くの溶媒接触可能残基からなる。部位特異的変異誘発によりこれらの領域における溶媒接触可能残基を変化させて受容体との側鎖相互作用を上昇または低下させることにより、M−CSFアゴニストまたはアンタゴニストを作成することができる。トリペプチドgly−x−glyの場合、接触可能なアミノ酸の表面積の正規化に基づいて、約0.25を超える、および好ましくは約0.4を超える溶媒接触可能表面積を有する残基が好ましい(Kabsch,W.ら、Biopolymers 22:2577(1983))。好ましくは、単量体の相対的配向を維持し、そしてタンパク質フォールディングの過程を乱すのを回避するために、二量体インターフェースのようなタンパク質のその他の部分と相互作用しない残基が選択される。場合によってさらに考えられることは、ヒトM−CSFとマウスM−CSF間で保存されない残基を選択することであり、それはヒトM−CSF受容体を認識しない。候補アミノ酸は好ましくは、MCSF−R残基との水素結合および/または疎水性相互作用を破壊するために、非保存アミノ酸での置換のために選択される。例えば一つまたはそれより多いヒスチジンを類似の大きさの非水素供与性アミノ酸に変えることにより、変化した受容体結合能力を有するM−CSFを創成できる。置換に好ましいアミノ酸には、限定するものではないが:H15;Q79;R86;E115;E41;K93;D99;L55;S18;Q20;175;V78;L85;D69;N70;H9;N63;およびT34が含まれる。受容体シグナリングに重要なM−CSF残基は、M−CSFの不連続領域から構成されると考えられる。投与された可能性のあるM−CSF基盤のタンパク質性薬物に対する抗体形成の可能性を最小限にするために、可能な限り溶媒接触可能な親M−CSF残基を保持するのが望ましい。
【0101】
N末端/Aヘリックス領域のアミノ酸H15およびH9の変異誘発は有意に低い生物学的活性および有意に低いMCSF−R結合能力を有するムテインに至った。この結果により、生物学的活性の低下は受容体結合親和性の低下によるものであったことが示され
;したがってこれらのヒスチジンアミノ酸は、M−CSF受容体結合親和性に重要である接触を表し、そして完全な受容体結合能力が望ましい場合、未変化にしておくべきである。Y6およびS13のような直ぐ近くの溶媒接触可能残基ならびにその他のものもまたM−CSF受容体コンタクト残基を表し得る。ヘリックスAおよびCの中央部分の溶媒接触可能残基の重要性を試験するために、M−CSFの二重変異体(Q20A、V78K)を構築した。この二重ムテインの生物学的活性はわずかに低く(8−10倍)、そしてしたがって受容体結合活性が低かった。残基Q17、R21、E115およびE119の変異誘発は目的の部分の溶媒接触可能アミノ酸の側鎖特性を変化させたが、生物学的特異活性には影響しなかったが、これはアンタゴニスト活性を有するように設計されたムテインでこれらの残基を変化させる必要がないことを示唆している。
【0102】
一つの実施態様では、本発明は受容体結合に関与するヘリックスAおよび/またはCおよび/またはDの残基(例えばアミノ酸6から26、71から90および/または110から130)が非保存的に変異しているM−CSFムテインの使用を企図する。かかるムテインは好ましくはヘリックスA、CまたはD内の未変性配列に対して少なくとも65%、70%、75%、80%、85%または90%の類似性を保持する(すなわち同一であるかまたは類似の特性を有するアミノ酸)が、残りのペプチドの未変性配列に対して高い類似性、例えば少なくとも95%、98%または99%の類似性を有する。加えて、受容体結合部位の三次元立体構造を支持する残基は非保存的に変異され得る。
【0103】
別の実施態様では、M−CSFムテインはM−CSFの単量体形態である。M−CSFの二量体形態は生物学的に活性な形態であり、そしてM−CSFの単量体形態は一般的に不活性である。単量体のジスルフィド結合はCys31−Cys31鎖間連結により生じるようである。したがってM−CSFの単量体形態がアンタゴニストとしての使用に適当であり得ることが企図される。かかる形態にはCys31および/もしくはその他のシステインのシステイン欠失および/もしくはシステイン置き換え(例えばシステインのアラニンへの置換)を含むムテイン、または(複数の)システイン、特にCys31が化学的に修飾されてジスルフィド結合に利用できなくなっているムテインが含まれる。
【0104】
さらに別の実施態様では、M−CSFムテインは一つもしくはそれより多いヘリックスA、CもしくはD、または受容体結合に関与するその部分を単独で、または適切な三次元立体構造でフラグメントの表示を可能にするその他のポリペプチドに融合されて含む。
【0105】
組換え生成または化学的合成を含む当分野において周知の技術を用いて、任意の望ましい保存および/または非保存ムテインを含有するムテインが容易に調製される。
【0106】
保存置換、特にリガンド−受容体結合に直接関与する領域の外側の置換はM−CSFムテイン(またはM−CSFRムテイン)の結合特性を有意に変化しないと予測される。物理学的特性ならびに二次および三次タンパク質構造に対する寄与に従ってアミノ酸を分類することができる。保存置換は一つのアミノ酸の、類似の特性を有する別のアミノ酸との置換として当分野において認識されている。保存置換の実例を直ぐ下の表2(1997年3月13日公開の第WO97/09433号、10頁より(PCT/GB96/02197、9/6/96出願)に示す。
【0107】
【表2】
これに代えて保存アミノ酸をLehninger(Biochemistry、第2版;Worth Publishers社、ニューヨーク州、ニューヨーク(1975)、71−77頁)に記載されるように群分けすることができ、直ぐ下の表3に示す。
【0108】
【表3】
さらに別の代替えとして、保存置換の実例を直ぐ下の表4に示す。
【0109】
【表4】
M−CSFをコードするDNA配列の利用性により望ましいポリペプチドを生成するための種々の発現系の使用が可能になる。発現ベクターの構築および適切なDNA配列からの組換え体の生成は当分野において周知の方法により実施される。これらの技術および種々のその他の技術は一般的にSambrookら、Molecular Cloning−A Laboratory Manual、Cold Spring Harbor Laboratory、ニューヨーク州、コールドスプリングハーバー(1989)、およびKriegler,M.、Gene Transfer and Expression,A Laboratory Manual、Stockton Press、ニューヨーク(1990)に従って実施される(双方共に出典明示により本明細書の一部とする)。
【0110】
周知の組換えDNA技術に従って望ましい構造(例えばM−CSFの受容体結合能力)を破壊せずにDNA配列によりコードされるアミノ酸の欠失、付加または変更によりM−CSFの一次配列に対して特定の修飾を作ることができる。さらに、個々のアミノ酸を置換するかまたは酸化、還元もしくはその他の修飾により修飾することができ、そして活性結合部位および構造情報を保持するフラグメントを得るためにポリペプチドを切断することができることは当業者には理解されよう。かかる置換および変更により、「成熟M−CSFα(配列番号7)、M−CSFβ(配列番号8)およびM−CSFγ(配列番号9)ポリペプチドと実質的に同一のアミノ酸配列を有している」ポリペプチドの定義に入るアミノ酸配列を有するポリペプチドが導かれる。
【0111】
当分野において公知の化学的合成または組換え生成技術によりポリペプチドを生成することができる。
【0112】
タンパク質の関連性を、そのコード化核酸の関連性により特徴づけることもできる。ポリヌクレオチド配列の同一性および/または類似性を決定する方法は前記で記載される。加えて、穏やかにまたは高度にストリンジェントな条件下でハイブリダイズするその能力を試験することにより、ポリヌクレオチド配列の類似性を決定する方法を以下のように決定することができる。穏やかにストリンジェントな条件の実例は以下のとおりである:50%ホルムアミド、1%SDS、1M NaCl、10%硫酸デキストランを含むハイブリダイゼーション溶液中42℃でハイブリダイゼーション、そして0.1×SSCおよび1%SDSを含む洗浄溶液中60℃で30分間2回洗浄。高度にストリンジェントな条件には0.1×SSCおよび1%SDSを含む洗浄溶液中68℃での洗浄が含まれる。当分野において記載されるような(Ausubelら(編)、Protocols in Molecular Biology、John Wiley & Sons(1994)、6.0.3から6.4.10頁)温度およびバッファーまたは塩濃度の変化により均等なストリンジェンシーの条件を達成できることは当分野において理解されている。ハイブリダイゼーション条件の修飾を経験的に決定できるか、またはプローブのグアノシン/シトシン(GC)塩基対の長さおよびパーセンテージに基づいて正確に計算することができる。Sambrookら(編)、Molecular Cloning:A Laboratory Manual、Cold Spring Harbor Laboratory Press:ニューヨーク州、コールドスプリングハーバー(1989)、9.47から9.51頁に記載されるように、ハイブリダイゼーション条件を計算することができる。
【0113】
C.可溶性M−CSFR
本発明によるM−CSFRフラグメントの実例は一つもしくはそれより多い、または二つもしくはそれより多いM−CSF/受容体結合に関与するドメイン(ドメイン1、2および3と考えられる)を含んでなり得る。好ましいM−CSFRフラグメントはM−CSFRのドメイン1、2および3の三つ全てを含む。かかるフラグメントに対する、またはM−CSFRの細胞外ドメイン全体に対するさらなる変異および/または修飾が企図され、そして前記でM−CSFムテインに関するセクションで記載されるように生成され得る。
【0114】
M−CSFR(配列番号84および85)は五つの細胞外免疫グロブリン様ドメイン(そのドメイン1−3はリガンド−受容体結合に関与すると考えられる)、膜貫通ドメインおよび細胞内分断Src関連チロシンキナーゼドメインを有する、膜を貫通する分子である。配列番号85に関して、前記されたドメインは以下のように位置する:Igドメイン1:アミノ酸27−102;Igドメイン2:アミノ酸112−196;Igドメイン3:アミノ酸215−285;Igドメイン4:アミノ酸308−399;Igドメイン5:アミノ酸410−492;膜貫通ドメイン:アミノ酸515−537;およびキナーゼドメイン:アミノ酸582−910。「典型的な」免疫グロブリン様ドメインは通常各ループの先端で二つのシステインの間のジスルフィド結合により繋留されるループ構造を含有する。M−CSF−Rでは、Ig様ループを形成するこれらのシステインは以下のアミノ酸位置にある:ドメイン1:42、84;ドメイン2:127、177;ドメイン3:224、278;ドメイン4:関与するシステインなし;ドメイン5:419、485。
【0115】
M−CSFRのインタクトな細胞外部分または抗原性を保持するその任意のフラグメント、例えば一つまたはそれより多いIg様ループを用いて未変性受容体に結合する抗体を上昇させることができる。ポリクローナル、モノクローナル、キメラ、CDRグラフト、ヒト化、完全ヒト抗体およびその抗原結合フラグメントをM−CSFに対する抗体に関して前記されたように調製することができる。本明細書にて「スクリーニング方法」の表題のセクションに記載されるようなアッセイを用いて、または当分野において公知の任意の適当なアッセイを用いてMCSFアンタゴニストとしての活性に関して、および本発明の処置方法における適性に関して抗体産生物をスクリーニングすることができる。
【0116】
受容体の細胞外ドメイン内の一つまたはそれより多い前記されたIg様ループは、M−CSFとM−CSFRとの間の相互作用を阻止するのに十分であり得る。したがってM−CSFRおよびそのムテインの細胞外ドメインのフラグメントを当分野において周知の組換えまたは化学的合成手段を用いて容易に調製することができる。本明細書にて「スクリーニング方法」の表題のセクションに記載されるようなアッセイを用いて、または当分野において公知の任意の適当なアッセイを用いてMCSFアンタゴニストとしての活性に関して、および本発明の処置方法における適性に関して産生物をスクリーニングすることができる。
【0117】
D.遺伝子治療
治療用タンパク質の適切な細胞への分配を、物理的DNA移入法(例えばリポソームまたは化学的処理)の使用を含む当分野において公知の任意の適当な研究法の使用による、またはウイルスベクター(例えばアデノウイルス、アデノ随伴ウイルスまたはレトロウイルス)使用によるエキソビボ、インサイチュまたはインビボ遺伝子治療により行うことができる。アンチセンス化合物およびそれを用いる方法もまた本発明により提供される。遺伝子発現レベルを低下させるための周知のアンチセンス、遺伝子「ノックアウト」、リボザイム、三重ヘリックス、二重ヘリックスまたはRNAi法の使用によりM−CSFまたはM−CSFR活性のレベルを低下させることができる。かかる分子の生成および使用のための技術は当業者に周知である。
【0118】
本明細書にて使用される際には、「ペプチド擬似物質」なる用語は、ファルマコフォアの空間的配向が天然のペプチドの生理活性立体構造を実質的に擬似するように足場に支持された、アミノ酸側鎖の集合体またはファルマコフォア、またはその適当な誘導体を含む非ペプチド化合物である。例えばペプチド擬似物質はアミノ酸またはペプチド結合を欠くが、結合活性に必要である親ペプチドからのペプチド鎖群の特定の三次元配置を保持する。足場は二環式、三環式またはさらに高次の多環式炭素もしくはヘテロ原子骨格を含んでなり得るか、または一つもしくはそれより多い環構造(例えばピリジン、インダゾール等)もしくはアミド結合に基づき得る。この足場をコアの一方の末端でスペーサーにより酸性基(例えばカルボン酸官能基)に、および他方の末端で塩基性基(例えばアミジンまたはグアニジンのようなN含有部分)に連結させることができる。ペプチド擬似物質を合成するための技術の実例は2003年10月23日公開の米国特許出願番号第20030199531号、2003年7月24日公開の米国特許出願番号第20030139348号に記載されている。
【0119】
抗体およびその他のタンパク質に加えて、本発明はまた、限定するものではないがM−CSFとM−CSFRの間の相互作用またはM−CSFRの活性化を阻止するのにも有効であるペプチドまたは小型有機分子を含む代替えのM−CSFアンタゴニストをも企図する。
【0120】
II.組み合わせ治療
M−CSFアンタゴニストおよび第二の抗破骨細胞薬のような本発明による二つの治療薬の併用投与は、薬剤がその治療効果を奏する期間に重複が存在する限り、同時にまたは同一経路により薬剤が投与される必要はない。異なる日または週の投与のような同時または逐次的投与が企図される。
【0121】
M−CSF抗体(M−CSFアンタゴニストの実例)での処置の開始後、治療効果が観察される有意なタイムラグが見出されることから、移行期間の間の作用がより迅速に発現する第二の抗破骨細胞薬の同時投与が望ましくなる。移行期間の間、二つの薬剤は単剤治療で有効な量で投与されなければならない。移行期間に続いて、第二の抗破骨細胞薬を中止するか、または投薬量を減少させることができる。M−CSFアンタゴニストおよび第二の抗破骨細胞薬が相乗効果を奏する場合、移行期間後に一つまたは双方の用量を減らすことができる。
【0122】
本発明の組成物を、癌転移および/または癌転移に関連する骨量減少、または骨粗鬆症のようなその他の骨量減少に関連する疾患を含む骨溶解性障害を既に患っているか、またはその傾向がある哺乳動物に、かかる疾患の進行を防御または少なくとも部分的に停止させるのに十分な量で投与する。治療薬が単独で与えられる(第二の治療薬と組み合わされない)場合にこれを達成するのに十分な治療薬の量は「単剤治療上有効な用量」として定義される。
【0123】
本発明の組み合わせ治療では、M−CSF抗体のようなM−CSFアンタゴニストおよび第二の抗破骨細胞薬を同時にまたは異なる時間に投与することができる。二つの薬剤を例えば互いに8時間、1日、14日、30日、3か月、6か月、9か月または1年以内に投与することができる。
【0124】
第二の抗破骨細胞薬の実例には、限定するものではないがゾレドロネート、パミドロネート、クロドロネート、エチドロネート、チルドロネート、アレンドロネート、イバンドロナートまたはリセドロネートを含むビスホスホネート系が挙げられる。その他の抗破骨細胞薬の実例には、ビスホスホネート系、PTHrP中和剤(例えば抗体、アンチセンス、siRNA)、カテプシンK阻害剤、MIP−1−αアンタゴニスト、RANK/RANKL中和剤(例えばAMG−162のような抗RANK抗体、またはアンチセンス、可溶性RANKL受容体もしくはそのムテイン)、RANKLワクチン、オステオプロテグリン(OPG)、血小板由来成長因子(PDGF)、srcキナーゼ阻害剤、マロン酸ガリウムおよびマトリックスメタロプロテイナーゼ(MMP)阻害剤が挙げられる。
【0125】
ビスホスホネート系の用量の実例には、静脈内投与4mgが挙げられる。3.5mg、3.3mgまたは3.0mgを含むより少ない投薬量を投与することもできる。皮下および第WO02/087555号に記載されるようなものを含むその他の投与経路が可能である。M−CSF抗体の有効量は異なり、そして疾患の重篤度ならびに処置される患者の体重および一般状態に依存するが、一般的に約1.0mg/kgから約100mg/kg体重または約10mg/kgから約30mg/kgの範囲であり、適用あたり約0.1mg/kgから約10mg/kgまたは約1mg/kgから約10mg/kgの投薬量がさらに一般的に用いられる。例えば一つまたはそれより多い別個の投与によっても、または連続注入によっても、例えば約1011g/kgから5mg/kgまたは約30mg/kgから1mg/kgの抗体が患者への投与のための初期候補投薬量である。投与は毎日、隔日、毎週またはより少ない頻度であり、必要により疾患に対する応答性および患者の治療耐性に依存する。望ましい疾患病徴の抑制が生じるまで4、5、6、7、8、10もしくは12週またはそれより長いような長期間にわたる維持投薬量を必要とする場合もあり、そして必要により投薬量を調整することができる。この治療の進展は従来の技術およびアッセイにより容易にモニタリングされる。
【0126】
本発明の方法は癌の全段階で有用であり得るが、進行性または転移性癌において特に適切であり得る。化学療法剤処置を受けたことのない患者では、治療方法を化学療法または放射線レジメンと組み合わせるのが好ましいであろうが、一つまたはそれより多い化学療法剤を投与されている患者には本発明の治療方法での処置が指示され得る。加えて本発明の治療方法により、特に化学療法剤の毒性に対して十分な耐性のない患者では、併用化学療法の投薬量を減少させて使用することも可能になる。
【0127】
本発明の方法は単一の抗M−CSF抗体および異なる抗体の組み合わせまたは「カクテル」の投与を企図する。かかる抗体カクテルは異なるエフェクターメカニズムを引き出す抗体を含有するか、または細胞毒性抗体を免疫エフェクター機能性に依存する抗体と直接組み合わせるので、特定の利点を有し得る。組み合わされたかかる抗体は相乗的治療効果を奏し得る。
【0128】
本発明の方法を癌治療のようなさらにその他の治療方法と組み合わせて使用することができる。癌治療薬および/または手順の実例には、限定するものではないが種々の化学療法剤、アンドロゲン遮断薬および免疫モジュレーター(例えばIL−2、GM−CSF、SLC)、ビスホスホネート(系)、例えばアレディア(すなわちパミドロネート、パミドロン酸、パミドロン酸二ナトリウム、パミドロン酸二ナトリウム五水和物);Zometa(すなわちアクラスタ、ゾレドロン酸、ゾレドロネート);クロンドロネート(すなわちボネフォス、ロロン、クロドロン酸二ナトリウム、クロンドロン酸ナトリウム);フォサマックス(すなわちアレンドロネート、アレンドロン酸ナトリウム塩三水和物、アレンドロン酸);フォサバンス(すなわちビタミンDと共に処方されたフォサマックス);ボンドロナト(Bondronat)またはボンビバまたはボニバ(すなわちイバンドロナート、イバンドロン酸、イバンドロン酸ナトリウム);アクトネル(すなわちリセドロネート、リセドロン酸ナトリウム、リセドロン酸);ジドロネルまたはジドロカル(すなわちエチドロネート、エチドロン酸、エチドロン酸二ナトリウム);ネリキシア(Nerixia)(すなわちネリドロネート、ネリドロン酸);スケリッド(すなわちチルドロネート、チルドロン酸);ジメチル−APD(すなわちオルパドロネート、オルパドロン酸);およびメドロン酸またはメドロナート)、外科的手術、放射線、細胞毒性化学療法、ホルモン療法(例えばタモキシフェン;抗アンドロゲン療法)、抗体療法(例えばRANKL/RANK中和抗体;PTHrP中和、抗Her2、抗CD20、抗CD40、CD22、VEGF、IGFR−1、EphA2、HAAH、TMEFF2、CAIX抗体)、治療用タンパク質療法(例えば可溶性RANKL受容体;OPG、ならびにPDGFおよびMMP阻害剤)、小型分子薬物治療(例えばSrcキナーゼ阻害剤)、成長因子受容体のキナーゼ阻害剤、またはRANKL阻害剤、オリゴヌクレオチド療法(例えばRANKLまたはRANKまたはPTHrPアンチセンス)、遺伝子治療(例えば抗RANKL抗体のようなRANKLまたはRANK阻害剤)、ペプチド療法(例えばRANKLのムテイン)ならびに本明細書に記載されるこれれのタンパク質、ペプチド、化合物および小型分子が挙げられる。
【0129】
癌化学療法剤には、限定するものではないがカルボプラチンおよびシスプラチンのようなアルキル化剤;ナイトロジェンマスタードアルキル化剤;カルムスチン(BCNU)のようなニトロソウレアアルキル化剤;メトトレキサートのような代謝拮抗剤;葉酸;プリン類似体代謝拮抗剤、メルカプトプリン;フルオロウラシル(5−FU)およびゲムシタビン(Gemzar(登録商標))のようなピリミジン類似体代謝拮抗剤;ゴセレリン、リュープロリドおよびタモキシフェンのような抗腫瘍性ホルモン薬;アルデスロイキン、インターロイキン−2、ドセタキセル、エトポシド(VP−16)、インターフェロンアルファ、パクリタキセル(Taxol(登録商標))およびトレチノイン(ATRA)のような天然抗悪性腫瘍薬;ブレオマイシン、ダクチノマイシン、ダウノルビシン、ドキソルビシン、ダウノマイシンおよびマイトマイシンCを含むマイトマイシンのような抗菌性天然抗悪性腫瘍薬;ならびにビンブラスチン、ビンクリスチン、ビンデシンのようなビンカアルカロイド天然抗悪性腫瘍薬;ヒドロキシウレア;アセグラトン、アドリアマイシン、イフォスファミド、エノシタビン、エピチオスタノール、アクラルビシン、アンシタビン、ニムスチン、塩酸プロカルバジン、カルボコン、カルボプラチン、カルモフール、クロモマイシンA3、抗腫瘍性多糖類、抗腫瘍性血小板因子、シクロホスファミド(Cytoxin(登録商標))、シゾフィラン、シタラビン(シトシンアラビノシド)、ダカルバジン、チオイノシン、チオテパ、テガフル、ドラスタチン系、アウリスタチンのようなドラスタチン類似体、CPT−11(イリノテカン)、ミトザントロン、ビノレルビン、テニポシド、アミノプテリン、カルミノマイシン、エスペラミシン系(例えば米国特許第4,675,187号参照)、ネオカルチノスタチン、OK−432、ブレオマイシン、フルツロン、ブロクスウリジン、ブスルファン、ホンバン、ペプロマイシン、ベスタチン(Ubenimex(登録商標))、インターフェロン−β、メピチオスタン、ミトブロニトール、メルファラン、ラミニンペプチド、レンチナン,カワラタケ(Coriolus versicolor)抽出物、テガフル/ウラシル、エストラムスチン(エストロゲン/メクロレタミン)が含まれる。
【0130】
さらに、癌患者のための治療として使用されるさらなる薬剤には、EPO、G−CSF、ガンシクロビル;抗生物質、ロイプロリド;メペリジン;ジドブジン(AZT);変異体および類似体を含むインターロイキン1から18;インターフェロンα、βおよびγのようなインターフェロン系またはサイトカイン系;黄体形成ホルモン放出ホルモン(LHRH)および類似体、および生殖腺刺激ホルモン放出ホルモン(GnRH)のようなホルモン系;形質転換成長因子β(TGF−β)、線維芽細胞成長因子(FGF)、神経成長因子(NGF)、成長ホルモン放出因子(GHRF)、上皮細胞成長因子(EGF)、線維芽細胞成長因子相同因子(FGFHF)、肝細胞成長因子(HGF)およびインスリン成長因子(IGF)のような成長因子;腫瘍壊死因子α&β(TNF−α&β);浸潤阻止因子(invasion inhibiting factor)2(IIF−2);骨形成タンパク質1−7(BMP1−7);ソマトスタチン;チモシンα−1;γグロブリン;スーパーオキシドジスムターゼ(SOD);補体因子;抗血管形成因子;抗原性物質;ならびにプロドラッグが含まれる。
【0131】
プロドラッグとは、親薬物と比較して腫瘍細胞に対して細胞毒性が弱いかまたは細胞無毒性であり、そして酵素により活性化されるかまたは活性なもしくはさらに活性な親形態に変換されることが可能である、医薬的に活性な物質の前駆体または誘導体形態を指す。例えばWilman、「Prodrugs in Cancer Chemotherapy」、Biochemical Society Transactions、14、375−382頁、第615回ベルファスト会議(1986)およびStellaら、「Prodrugs:A Chemical Approach to Targeted
Drug Deliver」、Directed Drug Delivery、Borchardtら(編)、247−267頁、Humana Press(1985)参照。プロドラッグには限定するものではないが、さらに活性な細胞毒性遊離薬物に変換することができるリン酸含有プロドラッグ、チオリン酸含有プロドラッグ、硫酸含有プロドラッグ、ペプチド含有プロドラッグ、Dアミノ酸修飾プロドラッグ、グリコシル化プロドラッグ、βラクタム含有プロドラッグ、場合によっては置換されたフェノキシアセトアミド含有プロドラッグまたは場合によっては置換されたフェニルアセトアミド含有プロドラッグ、5−フルオロシトシンおよびその他の5−フルオロウリジンプロドラッグが含まれる。本明細書における使用のためのプロドラッグ形態に誘導体化できる細胞毒性薬物の実例には、限定するものではないが前記されたこれらの化学療法剤が挙げられる。
【0132】
III.投与および調製
M−CSFアンタゴニストの有効量は異なり、そして疾患の重篤度ならびに処置される患者の体重および一般状態に依存するが、一般的に約1.0μg/kgから約100mg/kg体重の範囲であり、適用あたり約10μg/kgから約10mg/kgの投薬量がさらに一般的に用いられる。本発明の組成物の有効量の決定を当分野において周知である標準的な経験的な方法により達成することができる。例えば所定の投薬量のM−CSFアンタゴニストで処置された対象からの血清のインビボ中和活性を、Cenciら、J Clin.Invest.1055:1279−87(2000)に記載されるように、インビトロでM−CSF誘起のネズミ単球(M−CSFに対する受容体を高レベルに発現するCD11b+細胞、CD11細胞のサブセット)の増殖および生存を遮断する血清の能力を決定するアッセイを用いて評価することができる。
【0133】
投与は毎日、2日毎、3日毎、週2回、毎週またはより少ない頻度であり、必要により疾患に対する応答性および患者の治療耐性に依存する。長期間にわたる維持投薬量を必要とする場合もあり、そして必要により投薬量を調整することができる。
【0134】
組成物の単回または複数回投与を実施することができ、用量レベルおよびパターンは処置する医師により選択される。
【0135】
本発明の方法の実施において使用される抗M−CSF抗体を含むM−CSFアンタゴニストを望ましい分配方法に適当な担体を含む医薬組成物に処方することができる。適当な担体には、M−CSFアンタゴニストと組み合わされた場合にアンタゴニストの抗腫瘍機能を保持し、そして対象の免疫系とは非反応性である任意の材料が含まれる。実例には、限定するものではないが滅菌リン酸塩緩衝生理食塩水、静菌水等のようないずれかの多くの標準的な医薬用担体が挙げられる。種々の水性担体、例えば水、緩衝用水、0.4%生理食塩水、0.3%グリシン等が用いられ、そして穏やかな化学的修飾等に供された、アルブミン、リポタンパク質、グロブリン等のような安定性を強化するためのその他のタンパク質を含むことができる。
【0136】
望ましい純度を有するアンタゴニストを場合によっては生理学的に許容される担体、賦形剤または安定化剤(Remington’s Pharmaceutical Sciences、第16版、Osol,A.編(1980))と混合することにより、凍結乾燥処方または水溶液の形態の保存用のアンタゴニストの治療用処方を調製する。許容される担体、賦形剤または安定化剤は用いられると投薬量および濃度でレシピエントに対して無毒性であり、そしてリン酸塩、クエン酸塩およびその他の有機酸のようなバッファー;アスコルビン酸およびメチオニンを含む抗酸化剤;保存剤(例えば塩化オクタデシルジメチルベンジルアンモニウム;塩化ヘキサメトニウム;塩化ベンザルコニウム、塩化ベンゼトニウム;フェノール、ブチルまたはベンジルアルコール;メチルまたはプロピルパラベンのようなアルキルパラベン;カテコール;レソルシノール;シクロヘキサノール;3−ペンタノール;およびm−クレゾール);低分子量(約10残基未満)ポリペプチド;血清アルブミン、ゼラチンもしくは免疫グロブリンのようなタンパク質;ポリビニルピロリドンのような親水性重合体;グリシン、グルタミン、アスパラギン、ヒスチジン、アルギニンもしくはリジンのようなアミノ酸;グルコース、マンノースもしくはデキストリンを含む単糖類、二糖類およびその他の炭水化物;EDTAのようなキレート剤;スクロース、マンニトール、トレハロースもしくはソルビトールのような糖;ナトリウムのような塩形成対イオン;金属複合体(例えばZnタンパク質複合体);ならびに/またはTWEEN(商標)、PLURONICS(商標)もしくはポリエチレングリコール(PEG)のような非イオン性界面活性剤が含まれる。
【0137】
本明細書では処方はまた、処置される特定の適応症に関して必要により一つより多い活性化合物、好ましくは互いに悪影響を及ぼさない相補的活性を有するものを含有し得る。例えばさらに免疫抑制剤を提供するのが望ましい場合もある。かかる分子は意図される目的のために有効である量で組み合わされて存在するのが適当である。
【0138】
例えばコアセルベーション技術により、または界面重合により調製されたマイクロカプセル、例えばヒドロキシメチルセルロースまたはゼラチンマイクロカプセルおよびポリ−(メチルメタクリラート)マイクロカプセルに、各々コロイド薬物分配系(例えばリポソーム、アルブミンマイクロスフェア、マイクロエマルジョン、ナノ粒子およびナノカプセル)で、またはマクロエマルジョンで活性成分を封入することができる。かかる技術はRemington’s Pharmaceutical Sciences、第16版、Osol,A.編(1980)に開示されている。
【0139】
インビボ投与に用いられる処方は滅菌されていなければならない。これは滅菌ろ過膜を通してろ過することにより容易に達成される。
【0140】
非経口、皮下、腹腔内、肺内および鼻内を含む任意の適当な手段により、ならびに所望により局部処置、病巣内投与用にアンタゴニストが投与される。非経口注入には静脈内、動脈内、腹腔内、筋肉内、皮内または皮下投与が含まれる。加えてアンタゴニストをパルス注入により、特にアンタゴニストの漸減用量で投与するのが適当である。好ましくは投与が短期か長期かにある程度依存して、注射により、最も好ましくは静脈内または皮下注射により用量を与える。例えば望ましい部位に接近して配置されたカテーテルにより、局所、特に経皮、経粘膜、直腸、経口または局部投与を含むその他の投与方法が企図される。
【0141】
本発明の組成物を例えば顆粒、粉末、錠剤、カプセル、シロップ、坐剤、注射、エマルジョン、エリキシル、懸濁液または溶液の形態にできる。本組成物を例えば経口投与による、鼻内投与による、直腸投与、皮下注射、静脈内注射、筋肉内注射または腹腔内注射による種々の投与経路用に処方できる。
【0142】
注射用投薬形態には一般的に、適当な分散剤または湿潤剤および懸濁化剤を用いて調製できる水性懸濁液または油性懸濁液が含まれる。注射用形態は溶媒または希釈剤で調製される溶液相または懸濁液の形態でよい。許容される溶媒またはベヒクルには、滅菌水、リンガー溶液または等張水性生理食塩水が含まれる。これに代えて、滅菌油を溶媒または懸濁化剤として用いることができる。好ましくは油または脂肪酸は非揮発性であり、天然または合成油、脂肪酸、モノ、ジまたはトリグリセリドを含む。
【0143】
注射用の医薬用処方および/または医薬品は前記されたような適切な溶液での再構築に適当な粉末でよい。これの実例には限定するものではないが、凍結−乾燥、回転乾燥またはスプレー乾燥粉末、非晶質粉末、顆粒、沈殿物または微粒子が挙げられる。注射用の処方は場合によっては安定化剤、pH調整剤、界面活性剤、バイオアベイラビリティー調整剤およびこれらの組み合わせを含有し得る。
【0144】
徐放性調製物を調製することができる。徐放性調製物の適当な実例にはアンタゴニストを含有する固体疎水性重合体の半透性マトリックスが挙げられ、そのマトリックスは造形物、例えばフィルムまたはマイクロカプセルの形態である。徐放性マトリックスの実例には、ポリエステル、ヒドロゲル(例えばポリ(2−ヒドロキシエチル−メタクリラート)、またはポリ(ビニルアルコール))、ポリラクチド系(米国特許第3,773,919号)、L−グルタミン酸およびγエチル−L−グルタマートの共重合体、非分解性エチレン−ビニルアセタート、Lupron Depot(商標)(乳酸−グリコール酸共重合体および酢酸ロイプロリドから構成される注射用マイクロスフェア)のような分解性乳酸−グリコール酸共重合体およびポリ−D−(−)−3−ヒドロキシ酪酸が挙げられる。エチレン−ビニルアセタートおよび乳酸−グリコール酸のような重合体は100日間にわたる分子の放出を可能にするが、特定のヒドロゲルはタンパク質をより短い期間で放出する。カプセル化アンタゴニストは体内に長時間留まる場合、37℃で水分に暴露された結果、変性するかまたは凝集して生物学的活性の喪失および免疫原性の変化の可能性を招き得る。関与するメカニズムに依存して安定化のために合理的な計画を考案することができる。例えば凝集メカニズムがチオ−ジスルフィド交換による分子間S−S結合形成であることが見出された場合、スルフヒドリル残基を修飾し、酸性溶液から凍結乾燥し、水分含量を制御し、適切な添加剤を使用し、そして具体的な重合体マトリックス組成物を開発することにより安定化を達成することができる。
【0145】
本発明の処方を本明細書にて前記されたような短時間作用型、即時放出型、長時間作用型または徐放型になるように設計することができる。したがって医薬用処方を放出制御用または放出遅延用に処方することもできる。
【0146】
本組成物は例えばミセルもしくはリポソーム、またはその他の何らかのカプセル化形態を含むか、または放出延長形態で投与されて長時間保存および/または分配効果を提供することができる。したがって医薬用処方および医薬品をペレットまたはシリンダーに圧縮し、そしてデポー注射として、またはステントのようなインプラントとして筋肉内または皮下に埋め込むことができる。かかるインプラントはシリコンおよび生分解性重合体のような公知の不活性材料を用いることができる。
【0147】
前記したようなこれらの代表的な投薬形態に加えて、薬学的に許容される賦形剤および担体が一般的に当業者に公知であり、そしてしたがって本発明に含まれる。かかる賦形剤および担体は例えば「Remingtons Pharmaceutical Sciences」、Mack出版社、ニュージャージー州(1991)に記載されており、それを出典明示により本明細書の一部とする。
【0148】
具体的な投薬量を疾患の症状、対象の年齢、体重、一般健康状態、性別および食事、投薬間隔、投与経路、排泄速度、ならびに薬物の組み合わせに依存して調整することができる。有効量を含有する前記のいずれかの投薬形態は十分に通常の実験の範疇であり、そしてしたがって十分に本発明の範囲内である。
【0149】
本発明による治療薬として有用なM−CSFアンタゴニストまたは抗体はしばしばその他の天然発生免疫グロブリンまたはその他の生物学的分子を実質的に含まずに調製される。好ましいM−CSFアンタゴニストはまた癌転移および/または癌転移に関連する骨量減少を含む骨溶解性障害に苦しむか、またはそれを患う傾向がある哺乳動物に投与される場合、最小限の毒性しか呈さない。
【0150】
本発明の組成物を従来の周知の滅菌技術により滅菌することができる。得られた溶液を使用のために包装するかまたは無菌条件下でろ過し、そして凍結乾燥することができ、凍結乾燥された調製物を投与前に滅菌溶液と組み合わせる。組成物はpH調整剤および緩衝剤、等張化剤等、例えば酢酸ナトリウム、酪酸ナトリウム、塩化ナトリウム、塩化カリウム、塩化カルシウムのような生理学的状態に近づけるのに必要とされるような薬学的に許容される補助物質ならびに安定化剤(例えば120%マルトース等)を含有し得る。
【0151】
また本発明のM−CSFアンタゴニストを、薬物(例えば本明細書に開示されるアンタゴニストおよび場合によっては化学療法剤)の分配に有用である種々の型の脂質および/またはリン脂質および/または界面活性剤から構成される小型ベシクルであるリポソームを介して投与することもできる。リポソームはエマルジョン、フォーム、ミセル、不溶性単層、リン脂質分散物、ラメラ層等を含み、そしてM−CSFアンタゴニストを特定の組織に標的化し、そして組成物の半減期を増すベヒクルとして提供され得る。リポソームを調製するために、例えば米国特許第4,837,028号および第5,019,369号(これらの特許を出典明示により本明細書の一部とする)に記載されるような種々の方法が利用可能である。
【0152】
アンタゴニストを含有するリポソームは、Epsteinら、Proc.Natl.Acad.Sci.USA 82:3688(1985);Hwangら、Proc.Natl Acad.Sci.USA 77:4030(1980);ならびに米国特許第4,485,045号および第4,544,545号に記載されるように当分野において公知の方法により調製される。循環時間が延長されたリポソームが米国特許第5,013,556号に開示されている。特に有用なリポソームをホスファチジルコリン、コレステロールおよびPEG誘導体化ホスファチジルエタノールアミン(PEG−PE)を含む脂質組成物を用いる逆相蒸発法により作成することができる。望ましい直径を有するリポソームを生じるために規定の孔径のフィルターを通してリポソームを押し出す。Martinら、J.Biol.Chem.257:286−288(1982)に記載されるように、ジスルフィド交換反応により本発明のM−CSF抗体のFab’フラグメントをリポソームに抱合させることができる。化学療法剤(例えばドキソルビシン)は場合によってはリポソームに含有される(例えばGabizonら、J.National Cancer Inst.81(19):1484(1989)参照)。
【0153】
これらの組成物中のM−CSFアンタゴニストの濃度は大きく、すなわち重量で約10%未満、通常少なくとも約25%から75%または90%ほどまで異なる場合があり、そして選択される特定の投与の様式に従って、液体容量、粘度等により一次的に選択される。経口、局所および非経口投与可能な組成物を調製するための実際の方法は公知であるか、または当業者には明白であり、そして詳細は例えばRemington’s Pharmaceutical Science、第19版、Mack出版社、ペンシルバニア州、イーストン(1995)(これを出典明示により本明細書の一部とする)に記載されている。
【0154】
本発明の組成物の有効量の決定を当分野において周知である標準的な経験的な方法により達成することができる。例えばM−CSFアンタゴニストの所定の投薬量で処置された対象からの血清のインビボ中和活性を、Cenciら、J Clin.Invest.1055:1279−87(2000)に記載されるように、インビトロでM−CSF誘起のネズミ単球(M−CSFに対する受容体を高レベルに発現するCD11b+細胞、CD11細胞のサブセット)の増殖および生存を遮断する血清の能力を決定するアッセイを用いて評価することができる。
【0155】
本発明の組成物を、癌転移および/または癌転移に関連する骨量減少を含む骨溶解性障害を既に患っているか、またはその傾向がある哺乳動物に、かかる疾患の進行を防御または少なくとも部分的に停止させるのに十分な量で投与する。M−CSFアンタゴニストの有効量は異なり、そして疾患の重篤度ならびに処置される患者の体重および一般状態に依存するが、一般的に約1.0mg/kgから約100mg/kg体重または約10mg/kgから90mg/kgの範囲であり、適用あたり約20mg/kgから約80mg/kgまたは約30mg/kgから約70mg/kgまたは約40mg/kgから約60mg/kgの投薬量である。例えば一つまたはそれより多い別個の投与によっても、または連続注入によっても、例えば約10mg/kgから50mg/kgまたは約20mg/kgから60mg/kgの抗MCSF抗体が患者への投与のための初期候補投薬量である。投与は毎日、隔日、毎週またはより少ない頻度であり、必要により疾患に対する応答性および患者の治療耐性に依存する。望ましい疾患病徴の抑制が生じるまで4、5、6、7、8、10もしくは12週またはそれより長いような長期間にわたる維持投薬量を必要とする場合もあり、そして必要により投薬量を調整することができる。この治療の進展は従来の技術およびアッセイにより容易にモニタリングされる。
【0156】
組成物の単回または複数回投与を実施することができ、用量レベルおよびパターンは処置する医師により選択される。疾患の防御または処置のために、抗M−CSF抗体を含むM−CSFアンタゴニストの適切な投薬量は前記されたように処置される疾患の型、疾患の重篤度および経過、アンタゴニストが防御目的で投与されても、治療目的で投与されても、以前の治療、患者の病歴およびアンタゴニストに対する応答性、ならびに担当医の判断に依存する。アンタゴニストは一度で、または一連の処置にわたって投与されるのが適当である。
【0157】
適正な医療行為に合致する仕方でアンタゴニスト組成物が処方され、投薬され(dosed)、そして投与される。この局面で考慮される因子には、処置される特定の障害、処置される特定の哺乳動物、個々の患者の臨床症状、障害の原因、薬剤の分配の部位、投与の方法、投与のスケジュール、および医療従事者に公知のその他の因子が含まれる。投与されるアンタゴニストの治療上有効な量はかかる考慮により支配され、そしてM−CSF媒介疾患、症状または障害を防御、改善または処置するために、特に癌細胞を処置するために、そしてこの上なく特に腫瘍細胞転移を処置するために必要な最少量である。かかる量は宿主に対して毒性であるかまたは宿主を有意により感染し易くする量を下回るのが好ましい。
【0158】
本発明の別の実施態様では、前記された疾患、障害または症状の処置に有用な材料を含有する製品が提供される。製品は容器およびラベルを含む。適当な容器には、例えばビン、バイアル、シリンジおよび試験管が含まれる。ガラスまたはプラスチックのような種々の材料から容器を形成することができる。容器には症状を処置するのに有効である組成物が入っており、そして容器は無菌のアクセスポートを有し得る(例えば容器は静脈内注射用溶液バッグまたは皮下注射針により突き刺すことができるストッパーを有するバイアルでよい)。組成物中の活性薬剤は本発明のM−CSFアンタゴニストまたは抗体である。容器上の、または容器に随伴されるラベルは、組成物が最適な症状を処置するために用いられることを示す。製品はさらにリン酸塩緩衝食塩水、リンガー溶液およびデキストロース溶液のような薬学的に許容されるバッファーを含む第二の容器を含む。それはその他のバッファー、希釈剤、フィルター、針、シリンジおよび使用のための指示を伴う添付文書を含む、商業的に、および使用者の立場から望ましいさらにその他の材料でよい。
【0159】
本発明を以下の実施例により説明するが、それはいかなるようにも限定されることを意図するものではない。
【実施例】
【0160】
実施例1
本実施例は動物モデルにおけるZometa(ゾレドロネート)の用量依存性、抗吸収効果を確立する(図14)。Zometa≧0.03mg/kgでの処置は骨部位で腫瘍成長により引き起こされる骨溶解性損傷を阻止した。加えてマウスを漸増濃度のZometaで処置した場合に用量応答性効果が観察された。抗マウスおよび抗ヒトM−CSF mAb 5A1および5H4の組み合わせもまたもまた骨損傷に対して保護した。重篤な骨溶解性損傷を処置したときに、M−CSF抗体単独はZometa0.03mg/kgよりも有効であった。骨溶解スコアは試験の最後の日のX線画像に基づいた(図14):0=正常;
1=正常な皮質構造を有する不確定なまたは微細な病変;
2=微細な皮質/構造破壊を伴う限局的な溶解性病変;
3=皮質/構造破壊を伴う大きな(複数の)病変;
4=保存された構造がない全体的な破壊。
【0161】
この最初の試験から、有効用量以下のZometa0.03mg/kgを用いて組み合わせ試験を実施した。6×105 MDA−MB−231Luc細胞の脛骨内接種の2週間後、Nu/Nuマウスを5A1/5H4(10mg/kg 週1回)、Zometa(0.03mg/kg 週2回)または抗体およびビスホスホネートの双方で処置した。骨病変を毎週Faxitron分析(X線テクノロジー)によりモニタリングし、そして試験の最後に全動物を最終X線に供し、そして画像を収集し、そして病変の重篤度のスコアリングのために分類した。結果の分析により、抗MCSF mAbおよびZometaの双方が骨溶解の処置に有効であったが、ZometaおよびM−CSF mAbの組み合わせ処置は骨病変の発生(図15)および程度(図16)をいずれかの単独処置よりも大きな程度まで阻止した。
【0162】
図16はZometa0.03mg/kgまたは抗MCSF抗体(5A1+5H4)10mg/kgでの処置が骨部位での腫瘍成長により引き起こされる骨溶解性損傷を阻止したことを示す。Zometa+抗MCSF抗体の組み合わせレジメンはさらに骨の溶解を阻止した。平均骨溶解スコアを1)3人の別個の志願者からの平均スコアおよび2)群平均(元来動物10匹/群)から計算した。
【0163】
骨溶解スコアは試験の最後の日のX線画像に基づいた:
0=正常;
1=正常な皮質構造を有する不確定なまたは微細な病変;
2=微細な皮質/構造破壊を伴う限局的な溶解性病変;
3=皮質/構造破壊を伴う大きな(複数の)病変;
4=保存された構造がない全体的な破壊。
【0164】
代表的なFaxitron画像の試験によりさらにZometaおよび抗MCSF抗体処置動物における比較的に微細な病変に比較して、未処置動物で見出された重篤な病変が実証された(図17)。組み合わせ処置群では有害な相互作用は観察されなかった。
【0165】
結論としては、抗MCSF抗体およびZometaの双方が骨溶解を有効に阻止し、そして二つの処置を組み合わせることにより、いずれかの単独処置と比較して抗吸収効果の上昇を招く。これは、組み合わせがビスホスホネート不耐性であるか、または既にビスホスホネート系で処置されているかのいずれかである骨疾患を有する患者にとって安全でそして有効な選択肢であり得ることを示唆している。
【0166】
実施例2
この実施例は、M−CSf活性が分化型破骨細胞活性に効果を及ぼさないことを示している(図18)。M−CSF中和抗体およびスホスホネートの分化型破骨細胞活性に及ぼす効果をヒト化Chir−RXIおよびZometaで試験した。
【0167】
ヒト骨髄CD34+細胞(Biowhittakerカタログ番号2M−101A、3×105セル/バイアル)を本明細書に記載される実験条件下で破骨細胞に分化するように誘導した。第1日にCD34+細胞を1本の凍結バイアルから培地(10%FCS、1×Pen Strepおよび1×ファンギゾンを含むアルファMEM)10mlに解凍した。細胞を1回洗浄し、そして培地2mlに再懸濁し、そしてOsteoLyseプレート(OsteoLyse(商標)アッセイキット(Human Collagen)、Cambrex)にウェルあたり100μlでプレーティングした。第2日に元来の培地を除去することなく、4×CSF−1から最終濃度30ng/ml 50μlおよび4×RANKL(sRANKL、Chemiconカタログ番号GF091、10μg/包装)から最終濃度100ng/ml 50μlを各ウェルに加えた。第7日に5×RANKLから最終濃度100ng/ml 50μlを各ウェルに加えた。
【0168】
第15日に抗体(Chir−RX1または対照抗体のいずれか)またはZometaを指示された濃度で加えた。第17日に細胞培養物の上澄10μlをサンプリングし、そしてブラック96ウェルアッセイプレート(OsteoLyseアッセイキットに含まれる)の各ウェルでフルオロフォア放出試薬200μlと混合した。
【0169】
実施例3
この実施例はZometaが分化型破骨細胞活性を用量依存的な様式で阻止することを示している(図19)。M−CSF中和抗体およびスホスホネートの分化型破骨細胞活性に及ぼす効果をヒト化Chir−RXIおよびZometaで試験した。
【0170】
ヒト骨髄CD34+細胞(Biowhittakerカタログ番号2M−101A、3×105セル/バイアル)を本明細書に記載される実験条件下で破骨細胞に分化するように誘導した。第1日にCD34+細胞を1本の凍結バイアルから培地(10%FCS、1×Pen Strepおよび1×ファンギゾンを含むアルファMEM)10mlに解凍した。細胞を1回洗浄し、そして培地2mlに再懸濁し、そしてOsteoLyseプレート(OsteoLyse(商標)アッセイキット(Human Collagen)、Cambrex)にウェルあたり100μlでプレーティングした。第2日に元来の培地を除去することなく、4×CSF−1から最終濃度30ng/ml 50μlおよび4×RANKL(sRANKL、Chemiconカタログ番号GF091、10μg/包装)から最終濃度100ng/ml 50μlを各ウェルに加えた。第7日に5×RANKLから最終濃度100ng/ml 50μlを各ウェルに加えた。
【0171】
第15日に抗体(Chir−RX1または対照抗体のいずれか)またはZometaを指示された濃度で加えた。第17日に細胞培養物の上澄10μlをサンプリングし、そしてブラック96ウェルアッセイプレート(OsteoLyseアッセイキットに含まれる)の各ウェルでフルオロフォア放出試薬200μlと混合した。
【0172】
実施例4
この実施例は霊長類においてRX1を用いた薬物動態および薬力学試験の結果を示す(図20および図21)。
【0173】
この試験の目的は、カニクイザルに単回緩徐ボーラス静脈内注射(第1日に第2および第3群)、続いて13週の観察期間か、または反復投薬(第1、43、50および57日に第1群)、続いて10週の観察期間のいずれかとして投与した場合のheRX1−10.G1、ヒト化抗ヒトM CSF抗体の薬力学および薬物動態を調査することであった。ヒト化抗M−CSF IgG1モノクローナル抗体を上腕または伏在静脈を介して緩徐ボーラス静脈内(IV)注射により投与した。
【0174】
新薬の安全性評価のために動物の使用が全世界の監督官庁により要求されている。抗体はげっ歯類で交差反応性でないが、カニクイザルで反応性であることが示されている。したがってカニクイザルは生物製剤での静脈内注射試験において使用するための許容される非げっ歯類であるので、それが選択された。
【0175】
動物を以下の群に無作為化した:
【0176】
【表5】
a 第1日に第1群は0.2mg/kg/用量を投与され、そして第43、第50および第57日に同一動物は10mg/kg/用量を投与される。
【0177】
第1日に、第2および第3群に緩徐ボーラス静脈内注射により被験処方(各々2および20mg/kg/用量)をおよそ10分間にわたって投与した。伏在静脈を介してカテーテルおよびAbbocathを用いて処方を投与した。投薬容量は4ml/kgであり、そして実際の用量は各動物の最新の実際の体重に基づいた。第1群は第1日に0.2mg/kgの用量、そして続いて第43、50および57日に10mg/kg/用量の用量が投与された。伏在静脈を介してカテーテルおよびAbbocathを用いて緩徐ボーラス静脈内注射により(およそ10分間にわたって)処方を投与した。投薬容量は4ml/kgであり、そして実際の用量は各動物の最新の実際の体重に基づいた。
【0178】
以下のように血液学および/または臨床生化学のために全動物から血液を収集した:
【0179】
【表6】
以下のパラメーターを試験した:
血液学:血液細胞形態学;赤血球恒数(MCV、MCH、MCHCおよびRDW);ヘマトクリット;ヘモグロビン;平均血小板容積;血小板数;赤血球数;網状赤血球(絶対数およびパーセント);および白血球数(総数、絶対数および種別パーセント)。
【0180】
臨床生化学::A/G比(計算値);アラニンアミノトランスフェラーゼ;アルブミン;アルカリ性ホスファターゼ;アスパラギン酸アミノトランスフェラーゼ;血中尿素窒素;カルシウム;塩化物;コレステロール;クレアチニン;グロブリン(計算値);グルコース;無機リン;カリウム;ナトリウム;総、直接型および間接型ビリルビン;総タンパク質;トリグリセリド;ならびにC反応性タンパク質。
【0181】
骨代謝回転の生化学的マーカーを以下のように分析した(骨バイオマーカーの決定のために血液およそ2mlを全動物から収集した):
【0182】
【表7】
薬物動態評価および血清M−CSF活性:
第2および第3群に関して、投薬前、第1日(注入終了の直後および注入終了の4時間後)、ならびに第3、第8、第15、第22、第29、第43、第57、第71および第85日に静脈穿刺によりSST管に血液(各1.5ml)を収集した。第1群の動物に関して、投薬前、第1日(注入終了の直後および注入終了の4時間後)、ならびに第3、第8、第15、第22、第29、第43(投薬前および注入終了の4時間後)、第50(投薬前および注入終了の4時間後)、第57(投薬前および注入終了の4時間後)、第59、第64、第71、第78、第92、第106および第120日に静脈穿刺によりSST管に血液(各1.5ml)を収集した。薬物動態評価に関して、ならびに血清M−CSF活性および残留heRX1−10.G1活性に関して試料を分析した。
【0183】
遠心する前に血液試料を室温でおよそ30分間凝血させた。およそ4℃で2700rpmで10分間の遠心により血清が得られ、そして得られた血清を四つのアリコートに分けた。
【0184】
本明細書にて言及される、および/または出願データシートに列挙される前記の全ての米国特許、米国公開特許公報、米国特許出願、外国特許、外国特許出願および非特許文献はその全てを出典明示により本明細書の一部とする。
【0185】
前記から、本発明の具体的な実施態様は本明細書では説明の目的のために記載されているが、本発明の精神および範囲から逸脱することなく種々の修飾を行うことができることは理解されよう。
【数1】
【数2】
【数3】
【数4】
【数5】
【数6】
【数7】
【数8】
【数9】
【数10】
【数11】
【数12】
【数13】
【数14】
【数15】
【数16】
【数17】
【数18】
【数19】
【数20】
【数21】
【数22】
【数23】
【数24】
【数25】
【数26】
【数27】
【数28】
【数29】
【数30】
【数31】
【数32】
【数33】
【数34】
【数35】
【数36】
【数37】
【数38】
【数39】
【数40】
【数41】
【数42】
【数43】
【数44】
【数45】
【数46】
【数47】
【数48】
【数49】
【数50】
【数51】
【数52】
【数53】
【数54】
【数55】
【数56】
【数57】
【数58】
【数59】
【数60】
【数61】
【数62】
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1A】


【図1B】

【図1C】

【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8A】
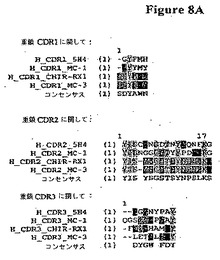
【図8B】
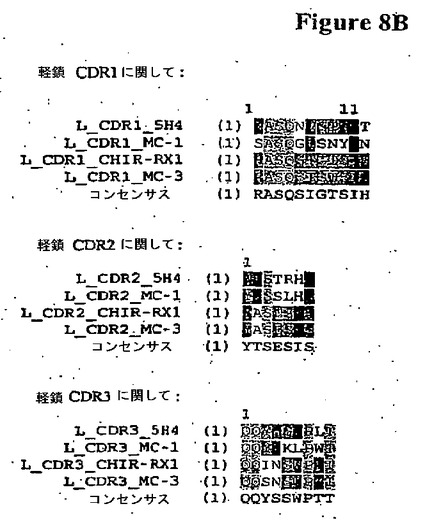
【図9A】
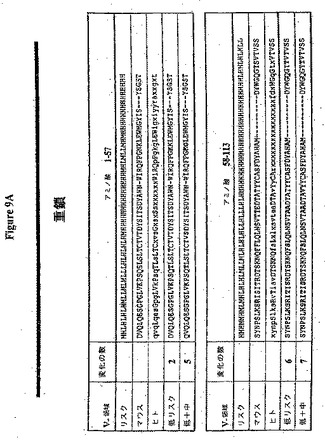
【図9B】

【図10A】
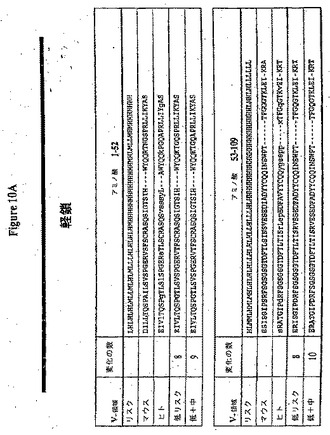
【図10B】

【図11A】
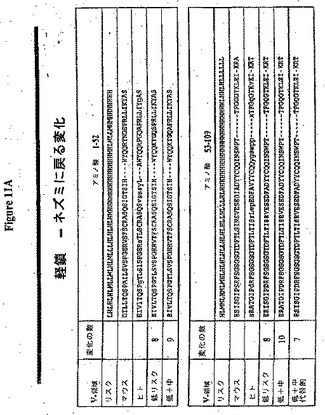
【図11B】

【図12A】
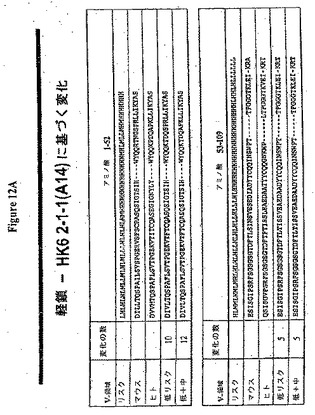
【図12B】

【図13A】

【図13B】

【図13C】

【図13D】

【図13E】

【図14】
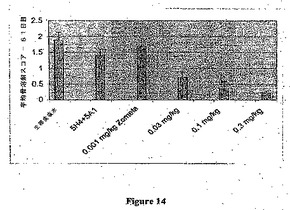
【図15】
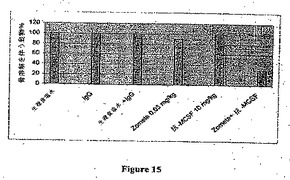
【図16】
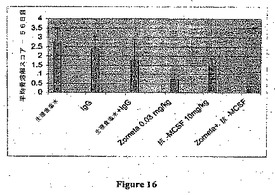
【図17】
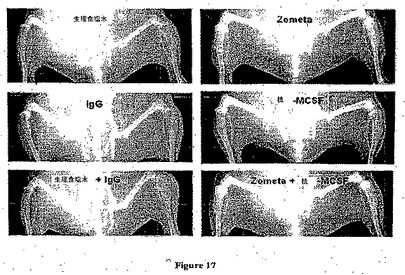
【図18】
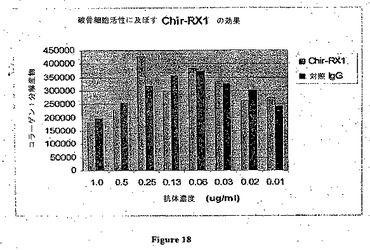
【図19】
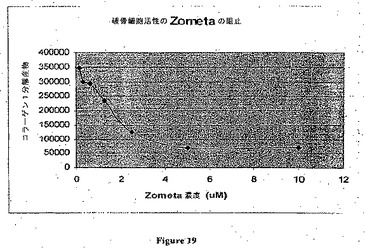
【図20】
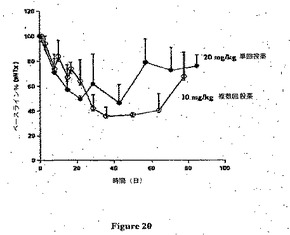
【図21】
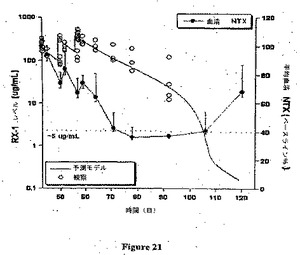
【図1B】
【図1C】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図9A】
【図9B】
【図10A】
【図10B】
【図11A】
【図11B】
【図12A】
【図12B】
【図13A】
【図13B】
【図13C】
【図13D】
【図13E】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公開番号】特開2012−229271(P2012−229271A)
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−186613(P2012−186613)
【出願日】平成24年8月27日(2012.8.27)
【分割の表示】特願2008−549592(P2008−549592)の分割
【原出願日】平成19年1月4日(2007.1.4)
【出願人】(504389991)ノバルティス アーゲー (806)
【出願人】(500570793)ゾーマ テクノロジー リミテッド (26)
【Fターム(参考)】
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【出願番号】特願2012−186613(P2012−186613)
【出願日】平成24年8月27日(2012.8.27)
【分割の表示】特願2008−549592(P2008−549592)の分割
【原出願日】平成19年1月4日(2007.1.4)
【出願人】(504389991)ノバルティス アーゲー (806)
【出願人】(500570793)ゾーマ テクノロジー リミテッド (26)
【Fターム(参考)】
[ Back to top ]