
発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体

【課題】微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、機械的性能に優れ、かつ表面外観にも優れる発泡成形体を生産効率よく成形できる発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体を提供すること。
【解決手段】発泡成形用熱可塑性樹脂組成物は、ゴム強化スチレン系樹脂(A)5〜90質量%、スチレン系樹脂(B)0〜85質量%及び芳香族ポリカーボネート樹脂(C)10〜90質量%を含有し、成分(A)〜(C)の合計100質量部に対し、化学発泡剤(D)を0.1〜5質量部、タルク(E)を0.5〜18質量部、及び繊維状充填材(F)を0.5〜25質量部を配合してなる。ゴム強化スチレン系樹脂(A)の熱シクロヘキサン溶解量は、ゴム質重合体(a)を基準として1〜99質量%である。成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合は、3〜50質量%である。
【解決手段】発泡成形用熱可塑性樹脂組成物は、ゴム強化スチレン系樹脂(A)5〜90質量%、スチレン系樹脂(B)0〜85質量%及び芳香族ポリカーボネート樹脂(C)10〜90質量%を含有し、成分(A)〜(C)の合計100質量部に対し、化学発泡剤(D)を0.1〜5質量部、タルク(E)を0.5〜18質量部、及び繊維状充填材(F)を0.5〜25質量部を配合してなる。ゴム強化スチレン系樹脂(A)の熱シクロヘキサン溶解量は、ゴム質重合体(a)を基準として1〜99質量%である。成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合は、3〜50質量%である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ゴム強化スチレン系樹脂を含む発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体に関する。
【背景技術】
【0002】
熱可塑性樹脂を用いた射出成形方法において、使用する樹脂材料の低減、軽量化等を目的として発泡成形を行う射出発泡成形が従来から検討されてきた。射出発泡成形を行う方法としては、例えば、特許文献1に開示されるように、熱可塑性樹脂に、アゾジカルボン酸アミドなどの熱分解型の化学発泡剤を使用する方法が知られている。
また、化学発泡に代えて、窒素ガス、二酸化炭素などを発泡剤として用いる物理発泡剤も知られている。更に超臨界状態の物理発泡剤を用いる方法も提案されている。
【0003】
例えば、特許文献2のポリオレフィン系樹脂組成物およびそれからなる成形体においては、ポリオレフィン(A)に対して、コア−シェルグラフト共重合体(B)及びタルク等の無機充填剤(C)を混合してなり、コア−シェルグラフト共重合体(B)が、架橋ゴム状重合体(a)に、共重合可能なビニル化合物からなる単量体成分(b)をグラフト共重合してなるポリオレフィン系樹脂組成物、及びこれに発泡剤を配合して発泡成形した発泡体について開示されている。これにより、優れた加工性、耐衝撃性、剛性及び表面性を同時に呈するポリオレフィン系樹脂組成物およびそれからなる成形体を構成することができる。
【0004】
さらに、例えば、特許文献3の射出発泡成形体の製造方法においては、金型の可動側金型と固定側金型とによって形成される型内空隙に原料樹脂を充填して金型に接している部分に表皮層を形成し、原料樹脂の充填完了直後、溶融部分の発泡剤の発泡圧力が周りの表皮層を押し広げるだけの力が残っているうちに、型締めシリンダーによる締め圧を瞬時にゼロ近くまで落として、表皮層の内側に発泡層を形成している。これにより、高発泡倍率の射出成形を可能にしている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2008−133485号公報
【特許文献2】特開2002−179851号公報
【特許文献3】特開2001−302830号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、特許文献1〜3等の熱可塑性樹脂組成物によっては、高い発泡倍率で、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、機械的性能(特に剛性)にも優れた発泡成形体を得るためには十分ではない。
また、発泡成形を繰り返し行う際には、発泡に伴う揮発成分が金型の成形表面に付着し、金型が汚染されるおそれがある。そして、金型の汚染が悪化したときには、次の発泡成形前に金型の成形表面を清掃する必要があった。
【0007】
本発明は、かかる従来の問題点に鑑みてなされたもので、射出発泡成形において、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、機械的性能に優れ、かつ表面外観にも優れる発泡成形体を生産効率よく成形することができる発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体を提供しようとするものである。
【課題を解決するための手段】
【0008】
第1の発明は、ゴム質重合体(a)の存在下に芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b1)を重合してなり、かつ熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準として1〜99質量%であるゴム強化スチレン系樹脂(A)5〜90質量%と、
芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b2)を重合してなるスチレン系樹脂(B)0〜85質量%と、
芳香族ポリカーボネート樹脂(C)10〜90質量%と、
上記成分(A)〜(C)の合計100質量部に対し、化学発泡剤(D)0.1〜5質量部と、タルク(E)0.5〜18質量部と、繊維状充填材(F)0.5〜25質量部とからなり、
上記成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合が3〜50質量%であることを特徴とする発泡成形用熱可塑性樹脂組成物にある(請求項1)。
【0009】
第2の発明は、上記発泡成形用熱可塑性樹脂組成物を用い、コアバック型射出発泡成形を行って成形したことを特徴とする発泡成形体にある(請求項5)。
【発明の効果】
【0010】
第1の発明の発泡成形用熱可塑性樹脂組成物は、上記ゴム強化スチレン系樹脂(A)、スチレン系樹脂(B)、及び芳香族ポリカーボネート樹脂(C)、化学発泡剤(D)、タルク(E)及び繊維状充填材(F)を含有し、ゴム強化スチレン系樹脂(A)を組成するゴム質重合体(a)の割合を適切にしたことによって、射出発泡成形を行う際に、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、かつ機械的性能にも優れる発泡成形体を成形することができる。
【0011】
すなわち、成形する発泡成形体において、微細かつ均一な発泡セルを得るためには、本発明者の研究開発により、特に、ゴム強化スチレン系樹脂(A)の熱シクロヘキサン溶解量がゴム質重合体(a)を基準として1〜99質量%であること、及び成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合が3〜50質量%であることが必要なことがわかった。また、この組成により、機械的性能にも優れる発泡成形体が得られることがわかった。
これにより、第1の発明の発泡成形用熱可塑性樹脂組成物によれば、微細かつ均一な発泡セルを有するだけでなく、機械的性能にも優れた発泡成形体を得ることができる。
【0012】
また、第1の発明の発泡成形用熱可塑性樹脂組成物は、タルク(E)及び繊維状充填材(F)を含有することにより、発泡セルが微細かつ均一で機械的性能に一層優れ、表面外観に優れる発泡成形体を成形することができることがわかった。この理由は、次のように考えられる。
すなわち、発泡成形する際には、タルク(E)及び繊維状充填材(F)が発泡起点になり、化学発泡剤(D)による発泡を促進することができる。このとき、タルク(E)による発泡起点と繊維状充填材(F)の両端による発泡起点とが混在することにより、より効果的に微細かつ均一な発泡セルが得られると考えられる。
【0013】
また、発泡成形用熱可塑性樹脂組成物が繊維状充填材(F)を含有することにより、機械的性能として特に剛性が優れた発泡成形体を得ることができる。さらに、繊維状充填材(F)を含有することにより、発泡成形を行う金型の成形表面に汚染が発生し難くなり、表面外観に優れた発泡成形体を生産効率よく成形することができることがわかった。この理由は、次のように考えられる。
すなわち、繊維状充填材(F)が含有されていることにより、金型の成形表面に接触する溶融状態の発泡成形用熱可塑性樹脂組成物の表面が凹凸状に粗くなり、化学発泡剤(D)の発泡に伴って発生する揮発成分が、金型の成形表面と溶融状態の発泡成形用熱可塑性樹脂組成物の表面との間から外部へ抜け易くなったと考えられる。
そのため、金型の成形表面に汚染が発生し難くなり、その清掃を行う頻度を減少させることができ、生産効率よく表面外観に優れる発泡成形体を成形することができることがわかった。
【0014】
それ故、第1の発明の発泡成形用熱可塑性樹脂組成物によれば、射出発泡成形において、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、機械的性能に優れ、かつ表面外観にも優れる発泡成形体を生産効率よく成形することができる。
また、第2の発明に示すように、発泡成形体をコアバック型射出発泡成形を行って成形することにより、上記効果を容易に得ることができる。
【図面の簡単な説明】
【0015】
【図1】実施例における、第1型部及び第2型部を有する製造装置を示す断面説明図。
【図2】実施例における、充填工程において、キャビティ及び充填用隙間内に溶融樹脂を充填した状態を示す断面説明図。
【図3】実施例における、充填工程において、キャビティ及び充填用隙間内に溶融樹脂を充填した状態を、図2と直交する方向から見た状態で示す断面説明図。
【図4】実施例における、可動工程において、第1型部に対して第2型部を可動させて、キャビティ内に樹脂発泡成形体を成形した状態を示す断面説明図。
【発明を実施するための形態】
【0016】
本発明の発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体における好ましい実施の形態につき説明する。
1.発泡成形用熱可塑性樹脂組成物
上記発泡成形用熱可塑性樹脂組成物の好ましい組成について説明する。
なお、以下の説明において、「(共)重合」とは、単独重合及び共重合を意味し、「(メタ)アクリレート」とは、アクリレートとメタクリレートとの少なくとも一方を意味する。また、本発明の発泡成形用熱可塑性樹脂組成物を単に「熱可塑性樹脂組成物」と略記する場合がある。
【0017】
(1)ゴム強化スチレン系樹脂(A)
本発明の(A)成分は、ゴム質重合体(a)の存在下に芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b1)を重合してなり、かつ熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準として1〜99質量%であるゴム強化スチレン系樹脂である。
ここで使用されるゴム質重合体(a)としては、ガラス転位温度(Tg)が−10℃以下のものであり、ポリブタジエン、ポリイソプレン、ブタジエン・スチレン共重合体、ブタジエン・アクリロニトリル共重合体等の共役ジエン系ゴム、エチレン・プロピレン共重合体、エチレン・プロピレン・非共役ジエン共重合体、エチレン・1−ブテン共重合体、エチレン・1−ブテン・非共役ジエン共重合体等のオレフィン系ゴム、アクリル系ゴム、シリコーンゴム、ポリウレタン系ゴム、シリコーン・アクリル系IPNゴム、天然ゴム、共役ジエン系ブロック共重合体、水素添加共役ジエン系ブロック共重合体等が挙げられる。これらは、1種単独で、あるいは2種以上を組み合わせて用いることができる。
【0018】
上記オレフィン系ゴムは特に限定されないが、例えば、エチレンと、炭素数が3以上のα−オレフィンとを含むエチレン・α−オレフィン系ゴムが挙げられる。エチレンの含有量は、上記エチレン・α−オレフィン系ゴムを構成する単量体の全量を100質量%とした場合、好ましくは5〜95質量%、より好ましくは50〜90質量%、更に好ましくは60〜88質量%である。
上記α−オレフィンである炭素数が3以上のα−オレフィンとしては、プロピレン、1−ブテン、2−ブテン、イソブテン、1−ペンテン、2−メチル−1−ブテン、2−メチル−2−ブテン、3−メチルブテン、1−ヘキセン、4−メチル−1−ペンテン、3−メチル−1−ペンテン、1−ヘプテン、1−オクテン、1−デセン、1−ウンデセン等が挙げられる。これらのα−オレフィンは、1種単独で含まれていてもよいし、2種以上の組み合わせで含まれていてもよい。また、上記α−オレフィンのうち、プロピレン、1−ブテンが好ましい。
【0019】
上記α−オレフィンの含有量は、上記エチレン・α−オレフィン系ゴムを構成する単量体の全量を100質量%とした場合、好ましくは95〜5質量%、より好ましくは50〜10質量%、特に好ましくは40〜12質量%である。
上記エチレン・α−オレフィン系ゴムは、上記エチレン、及びα−オレフィンから構成される二元共重合体であってもよいし、これらと、更に他の化合物とから構成される重合体(三元共重合体、四元共重合体等)であってもよい。他の化合物としては、非共役ジエン化合物が挙げられる。
【0020】
上記オレフィン系ゴムに使用される非共役ジエン化合物としては、アルケニルノルボルネン類、環状ジエン類、脂肪族ジエン類などが挙げられ、好ましくは、ジシクロペンタジエン及び5−エチリデン−2−ノルボルネンである。これらの非共役ジエン化合物は単独でまたは2種以上を組み合わせて使用することができる。エチレン・α−オレフィン系ゴム中の非共役ジエン化合物単位の含有量は、通常30質量%未満、好ましくは15質量%未満である。
【0021】
上記アクリル系ゴムは特に限定されないが、アルキル基の炭素数が1〜8個の(メタ)アクリル酸アルキルエステル化合物の(共)重合体、あるいはこの(メタ)アクリル酸アルキルエステル化合物と、これと共重合可能なビニル系単量体との共重合体が好ましい。
【0022】
ここで使用されるアルキル基の炭素数が1〜8個のアクリル酸アルキルエステル化合物の具体例としては、メチルアクリレート、エチルアクリレート、プロピルアクリレート、n−ブチルアクリレート、i−ブチルアクリレート、アミルアクリレート、ヘキシルアクリレート、n−オクチルアクリレート、2−エチルヘキシルアクリレート、シクロヘキシルアクリレート等が挙げられる。メタクリル酸アルキルエステルの具体例としては、メチルメタクリレート、エチルメタクリレート、プロピルメタクリレート、n−ブチルメタクリレート、i−ブチルメタクリレート、アミルメタクリレート、ヘキシルメタクリレート、n−オクチルメタクリレート、2−エチルヘキシルメタクリレート、シクロヘキシルメタクリレート等が挙げられる。これらの化合物のうち、n−ブチルアクリレート、2−エチルヘキシルアクリレートが好ましい。また、これらは、1種単独で、あるいは2種以上を組み合わせて用いることができる。
【0023】
また、上記(メタ)アクリル酸アルキルエステル化合物と共重合可能なビニル系単量体としては、例えば、多官能性ビニル化合物、芳香族ビニル化合物、シアン化ビニル化合物等が上げられる。上記多官能性ビニル化合物とは、単量体1分子中に2個以上のビニル基を有する単量体をいい、上記(メタ)アクリル系共重合体を架橋する機能及びグラフト重合時の反応起点の役割を果たすものである。上記多官能性ビニル単量体の具体例としては、ジビニルベンゼン、ジビニルトルエン等の多官能性芳香族ビニル化合物;(ポリ)エチレングリコールジメタクリレート、トリメチロールプロパントリアクリレート等の多価アルコールの(メタ)アクリル酸エステル;ジアリルマレート、ジアリルフマレート、トリアリルシアヌレート、トリアリルシアヌレート、ジアリルフタレート、メタクリル酸アリル等が挙げられる。これらの多官能性ビニル化合物は、1種単独で、または2種以上を組み合わせて使用することができる。
【0024】
ここで使用される芳香族ビニル化合物及びシアン化ビニル化合物としては、後述するものが全て使用できる。更に、他の共重合可能な単量体として、アクリルアミド、メタクリルアミド、塩化ビニリデン、アルキル(炭素数1〜6)ビニルエーテル、アルキル基の炭素数が9個以上の(メタ)アクリル酸アルキルエステル、(メタ)アクリル酸等が挙げられ、これらは1種単独で、あるいは2種以上を組み合わせて使用される。
【0025】
上記アクリル系ゴムの好ましい単量体組成は、アルキル基の炭素数が1〜8個の(メタ)アクリル酸アルキルエステル化合物単位80〜99.99質量%、より好ましくは90〜99.95質量%、多官能性ビニル化合物単位0.01〜5質量%、より好ましくは0.05〜2.5質量%、及びこれと共重合可能な他のビニル単量体0〜20質量%、より好ましくは0〜10質量%である。ただし、単量体組成は、合計100質量%とする。
【0026】
ゴム強化スチレン系樹脂(A)の熱シクロヘキサン溶解量を、ゴム質重合体(a)を基準として1質量%以上とする目的から、上記アクリル系ゴムの製造において、多官能性ビニル化合物を使用する場合は、重合の後段階で行うことが好ましい。即ち、重合の初期段階ではアクリル酸アルキルエステル化合物、及び必要に応じて共重合可能な他のビニル単量体(b1)を重合し、重合の後期段階でアクリル酸アルキルエステル化合物及び多官能性ビニル化合物、更に必要に応じて共重合可能な他のビニル単量体(b1)を重合する方法で製造することができる。
【0027】
本発明のアクリル系ゴムの製造方法としては、(1)各種ビニル単量体を一括添加して重合する方法、(2)特定のビニル単量体を一括添加重合し、重合の後期段階で残りのビニル単量体を添加重合する方法、(3)各種ビニル単量体の一部を添加重合し、残りのビニル単量体を連続添加して重合する方法、(4)各種ビニル単量体を2段以上に分割して重合する方法等があるが、好ましくは(4)の方法であり、更に好ましくは(4)の方法で多官能性ビニル化合物を2段目以降の後期段階で使用する方法である。重合方法としては、乳化重合が特に好ましい。
上記アクリル系ゴムの体積平均粒子径は、50〜1000nmであることが好ましく、さらに好ましくは40〜700nm、特に好ましくは50〜500nmである。
【0028】
共役ジエン系ブロック共重合体としては、具体的には少なくとも1個の下記ブロックAまたは下記ブロックCと、少なくとも1個の下記ブロックBまたは下記ブロックA/Bとを含んでなる共重合体、またはブロックBもしくはA/Bによる重合体であり、アニオン重合法で公知の方法である、例えば、特公昭47−28915号公報、特公昭47−3252号公報、特公昭48−2423号公報、特公昭48−20038号公報などに開示されている方法で製造することができる。その具体的構造は、
A;芳香族ビニル化合物重合体ブロック、
B;共役ジエン重合体ブロック、
A/B;芳香族ビニル化合物/共役ジエンのランダム共重合体ブロック、
C;共役ジエンと芳香族ビニル化合物の共重合体からなり、かつ芳香族ビニル化合物が漸増するテーパーブロック、
とそれぞれ定義すると、次のような構造のものが挙げられる。
【0029】
A−B (1)
A−B−A (2)
A−B−C (3)
A−B1−B2 (4)
(ここで、B1は共役ジエン重合体ブロックまたは共役ジエンと芳香族ビニル化合物との共重合体ブロックであり、共役ジエン部分のビニル結合量は好ましくは20%以上、B2は共役ジエン重合体ブロックまたは共役ジエンと芳香族ビニル化合物の共重合体ブロックであり、共役ジエン部分のビニル結合含有量は好ましくは20%未満である。)
A−A/B (5)
A−A/B−C (6)
A−A/B−B (7)
A−A/B−A (8)
B2−B1−B2 (9)
(ここで、B1、B2は上記と同じ。)
C−B (10)
C−B−C (11)
C−A/B−C (12)
C−A−B (13)
【0030】
また、これらの基本骨格を繰り返し有する共重合体を挙げることができ、さらにそれをカップリングして得られる共役ジエン系ブロック共重合体であってもよい。上記式(4)の構造のものについては、特開平2−133406号公報、上記式(5)及び上記式(6)の構造のものについては、特開平2−305814号公報、特開平3−72512号公報に示されている。
【0031】
ここで使用される共役ジエンとしては、1,3−ブタジエン、イソプレン、2,3−ジメチル−1,3−ブタジエン、1,3−ペンタジエン、2−メチル−1,3−ペンタジエン、1,3−ヘキサジエン、4,5−ジエチル−1,3−オクタジエン、3−ブチル−1,3−オクタジエン、クロロプレンなどが挙げられるが、工業的に利用でき、また物性の優れた共役ジエン系ブロック共重合体を得るには、1,3−ブタジエン、イソプレン、1,3−ペンタジエンが好ましく、より好ましくは1,3−ブタジエンである。
【0032】
また、ここで使用される芳香族ビニル化合物としては、スチレン、t−ブチルスチレン、α−メチルスチレン、p−メチルスチレン、ヒドロキシスチレン、ビニルキシレン、モノクロルスチレン、ジクロルスチレン、モノブロムスチレン、ジブロムスチレン、フルオロスチレン、p−t−ブチルスチレン、エチルスチレン、ビニルナフタレン、ジビニルベンゼン、1,1−ジフェニルスチレン、N,N−ジエチル−p−アミノエチルスチレン、N,N−ジエチル−p−アミノエチルスチレン、ビニルピリジンなどが挙げられ、スチレン、α−メチルスチレンが好ましく、特に好ましくはスチレンである。
【0033】
上記共役ジエンブロック系共重合体中の芳香族ビニル化合物/共役ジエンの割合は、質量比で0〜70/100〜30、好ましくは0〜60/100〜40、更に好ましくは0〜50/100〜50であり、芳香族ビニル化合物を必須とする場合、好ましくは10〜70/90〜30である。ここで、芳香族ビニル化合物の含有量が70質量%を超えると樹脂状となり、ゴム成分としての効果が劣り好ましくない。
さらに、共役ジエンブロック中の共役ジエン部分のビニル結合量は、通常5〜80%の範囲である。
共役ジエン系ブロック共重合体の数平均分子量は、通常10,000〜1,000,000、好ましくは20,000〜500,000、更に好ましくは20,000〜200,000である。これらのうち、上記構造式のA部の数平均分子量は3,000から150,000、B部の数平均分子量は5,000〜200,000の範囲であることが好ましい。
【0034】
共役ジエン化合物のビニル結合量の調節は、N,N,N’,N’−テトラメチルエチレンジアミン、トリメチルアミン、トリエチルアミン、ジアゾシクロ(2,2,2)オクタアミン等のアミン類、テトラヒドロフラン、ジエチレングリコールジメチルエーテル、ジエチレングリコールジブチルエーテル等のエーテル類、チオエーテル類、ホスフィン類、ホスホアミド類、アルキルベンゼンスルホン酸塩、カリウムやナトリウムのアルコキシド等を使用して行うことができる。
【0035】
本発明で使用されるカップリング剤としては、アジピン酸ジエチル、ジビニルベンゼン、メチルジクロロシラン、四塩化珪素、ブチルトリクロロ珪素、テトラクロロ錫、ブチルトリクロロ錫、ジメチルクロロ珪素、テトラクロロゲルマニウム、1,2−ジブロモエタン、1,4−クロロメチルベンゼン、ビス(トリクロロシリル)エタン、エポキシ化アマニ油、トリレンジイソシアネート、1,2,4−ベンゼントリイソシアネート等が挙げられる。
【0036】
本発明で使用される水素添加共役ジエン系ブロック共重合体は、上記共役ジエン系ブロック共重合体の共役ジエン部分の炭素−炭素二重結合の少なくとも30%以上、好ましくは50%以上が水素添加された部分水素添加物または完全水素添加物であり、更に好ましくは90%以上が水素添加された水素添加物である。
【0037】
上記共役ジエン系ブロック共重合体の水素添加反応は、公知の方法で行うことができるし、また、公知の方法で水素添加率を調節することにより、目的の重合体を得ることができる。具体的な方法としては、特公昭42−8704号公報、特公昭43−6636号公報、特公昭63−4841号公報、特公昭63−5401号公報、特開平2−133406号公報、特開平1−297413号公報等に開示されている方法がある。
【0038】
本発明で使用される上記ゴム質重合体(a)は、ゲル含率は70質量%以下であることが、本発明の発泡性から好ましく、更に好ましくは50質量%以下、特に好ましくは10質量%以下である。
ゲル含率は、以下に示す方法により求めることができる。
ゴム質重合体(a)1gをトルエン100mlに投入し、室温で48時間静置する。その後、100メッシュの金網(質量をW1グラムとする)で濾過したトルエン不溶分と金網を、温度80℃で6時間真空乾燥して秤量(質量W2グラムとする)する。W1及びW2を、下記式(14)に代入して、ゲル含率を得る。なお、エチレン−プロピレン系ゴム質重合体においては、エチレン結晶を有するものがあり、このようなゴム質重合体を用いる場合は、80℃の温度で溶解しゲル含率をもとめる。
ゲル含率=〔〔W2(g)−W1(g)〕/1(g)〕×100 (14)
【0039】
上記ゲル含率は、ゴム質重合体(a)の製造時に、架橋性単量体の種類及びその使用量、分子量調節剤の種類及びその使用量、重合時間、重合温度、重合転化率等を適宜設定することにより調整できる。
【0040】
本発明で使用されるゴム質重合体(a)として好ましいものは、ポリブタジエン、ブタジエン・スチレン共重合体、オレフィン系ゴム、アクリル系ゴム、シリコーンゴム、共役ジエン系ブロック共重合体、水素添加共役ジエン系ブロック共重合体であり、更に好ましくは、オレフィン系ゴム、アクリル系ゴム、水素添加共役ジエン系ブロック共重合体であり、特に好ましいものは、アクリル系ゴム、エチレン・プロピレン共重合体、エチレン・プロピレン・非共役ジエン共重合体、及び水素添加共役ジエン系ブロック共重合体であり、最も好ましいものは、アクリル系ゴムのゲル含率が10質量%以下のもので、かつ体積平均粒子径が50〜500nmのものである。
【0041】
本発明のゴム質重合体(a)は、公知の方法である乳化重合、溶液重合、塊状重合、懸濁重合等の方法で得ることができる。これらの中で、アクリル系ゴムは乳化重合により製造されたものが好ましく、エチレン・プロピレン共重合体、エチレン・プロピレン・非共役ジエン共重合体、共役ジエン系ブロック共重合体及び水素添加共役ジエン系ブロック共重合体は溶液重合、ポリブタジエン及びブタジエン・スチレン共重合体は溶液重合で製造されたものが好ましい。
【0042】
本発明の(A)成分であるゴム強化スチレン系樹脂は、上記ゴム質重合体(a)の存在下に、芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合体可能な他のビニル単量体(b1)を重合して得られる。すなわち、(b1)成分は、芳香族ビニル化合物単独でもよいし、芳香族ビニル化合物及び芳香族ビニル化合物と共重合体可能な他のビニル単量体との混合物でもよい。
ここで使用される芳香族ビニル化合物としては、上記ゴム質重合体(a)で記載したものが全て使用できる。特に好ましくはスチレン、α−メチルスチレンであり、これらは1種単独でまたは2種以上を組み合わせて使用することができる。
芳香族ビニル化合物と共重合可能な他のビニル単量体としては、ビニルシアン化合物、(メタ)アクリル酸エステル化合物、マレイミド化合物、その他の各種官能基含有不飽和化合物などが挙げられる。その他の各種官能基含有不飽和化合物としては、不飽和酸化合物、エポキシ基含有不飽和化合物、水酸基含有不飽和化合物、酸無水物基含有不飽和化合物、オキサゾリン基含有不飽和化合物、置換または非置換のアミノ基含有不飽和化合物などが挙げられる。これらの他のビニル単量体は1種単独で、または2種以上を組み合わせて使用することができる。
【0043】
上記シアン化ビニル化合物としては、アクリロニトリル、メタクリロニトリル等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。シアン化ビニル化合物を使用することにより耐薬品性が付与される。シアン化ビニル化合物の使用量は、ビニル単量体全体量(b1)中の割合として、通常0〜60質量%、好ましくは5〜50質量%である。
【0044】
(メタ)アクリル酸エステル化合物としては、アクリル酸メチル、アクリル酸エチル、アクリル酸ブチル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。(メタ)アクリル酸エステル化合物を使用することにより表面硬度が向上する。(メタ)アクリル酸エステル化合物の使用量は、ビニル単量体全体量(b1)成分中の割合として、通常0〜80質量%である。
【0045】
上記のマレイミド化合物としては、マレイミド、N−フェニルマレイミド、N−シクロヘキシルマレイミド、N−シクロヘキシルマレイミド等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。また、マレイミド単位を導入するために、無水マレイン酸を共重合させた後にイミド化してもよい。マレイミド化合物を使用することにより耐熱性が付与される。マレイミド化合物の使用量は、ビニル単量体全体量(b1)成分中の割合として、通常1〜60質量%である。
【0046】
不飽和酸化合物としては、アクリル酸、メタクリル酸、エタクリル酸、マレイン酸、フマル酸、イタコン酸、クロトン酸、桂皮酸などが挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。
【0047】
エポキシ基含有不飽和化合物としては、グリシジルアクリレート、グリシジルメタクリレート、アリルグリシジルエーテル等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。
【0048】
水酸基含有不飽和化合物としては、3−ヒドロキシ−1−プロペン、4−ヒドロキシ−1−ブテン、シス−4−ヒドロキシ−2−ブテン、トランス−4−ヒドロキシ−2−ブテン、3−ヒドロキシ−3−メチル−1−プロペン、2−ヒドロキシエチルメタクリレート、2−ヒドロキシエチルアクリレート、N−(4−ヒドロキシフェニル)マレイミド等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。
【0049】
オキサゾリン基含有不飽和化合物としては、ビニルオキサゾリン等が挙げられ、これらは、1種単独で、あるいは2種以上を組み合わせて使用することができる。
【0050】
酸無水物基含有不飽和化合物としては、無水マレイン酸、無水イタコン酸、無水シトラコン酸などが挙げられ、これらは1種単独で、あるいは2種以上を組み合わせて使用することができる。
【0051】
置換または非置換のアミノ基含有不飽和化合物としては、アクリル酸アミノエチル、アクリル酸プロピルアミノエチル、メタクリル酸ジメチルアミノエチル、メタクリル酸フェニルアミノエチル、N−ビニルジエチルアミン、N−アセチルビニルアミン、アクリルアミン、N−メチルアクリルアミン、アクリルアミド、N−メチルアクリルアミド、p−アミノスチレン等があり、これらは1種単独で、あるいは2種以上を組み合わせて使用することができる。
【0052】
上記その他の各種官能基含有不飽和化合物を使用した場合、ゴム強化スチレン系樹脂(A)、スチレン系樹脂(B)と芳香族ポリカーボネート樹脂(C)とをブレンドした際、両者の相溶性が向上する場合がある。上記その他の各種官能基含有不飽和化合物の使用量は、(A)成分と(B)成分の合計中に対して、当該官能基含有不飽和化合物の合計量として、通常0.1〜20質量%、好ましくは0.1〜10質量%である。
【0053】
ビニル単量体全体量(b1)中の芳香族ビニル化合物以外の単量体の使用量は、(b1)成分の合計を100質量%とした場合、通常80質量%以下、好ましくは60質量%以下、更に好ましくは50質量%以下である。
【0054】
ビニル単量体(b1)を構成する単量体のより好ましい組み合わせは、スチレン単独、スチレン/アクリロニトリル、スチレン/メタクリル酸メチル、スチレン/アクリロニトリル/メタクリル酸メチル、スチレン/アクリロニトリル/グリシジルメタクリレート、スチレン/アクリロニトリル/2−ヒドロキシエチルメタクリレート、スチレン/アクリロニトリル/(メタ)アクリル酸、スチレン/N−フェニルマレイミド、スチレン/アクリロニトリル/N−フェニルマレイミド、スチレン/メタクリル酸メチル/シクロヘキシルマレミド等であり、更に好ましくは、スチレン単独、スチレン/アクリロニトリル=65/45〜90/10(質量比)、スチレン/メタクリル酸メチル=80/20〜20/80(質量比)、スチレン/アクリロニトリル/メタクリル酸メチルの組み合わせで、スチレン量が20〜80質量%、アクリロニトリル及びメタクリル酸メチルの合計が20〜80質量%の範囲で任意のものである。
【0055】
本発明で使用されるゴム強化スチレン系樹脂(A)は、公知の重合法、例えば乳化重合、塊状重合、溶液重合、懸濁重合およびこれらを組み合わせた重合法で製造することができる。上記重合法は、ゴム質重合体(a)が乳化重合で得られたものは(A)成分の製造においては同じく乳化重合で製造することが、更にゴム質重合体(a)が溶液重合で得られたものである場合は、(A)成分は塊状重合、溶液重合及び懸濁重合で製造することが一般的で好ましい。ただし、溶液重合で製造されたゴム質重合体(a)であっても、本ゴム質重合体(a)を公知の方法で乳化させれば、乳化重合で(A)成分を製造することができるし、また、乳化重合で製造したゴム質重合体(a)であっても、凝固し単離した後、塊状重合、溶液重合及び懸濁重合で本発明の(A)成分を製造することができる。
【0056】
乳化重合で製造する場合、重合開始剤、連鎖移動剤、乳化剤などが使用されるが、これらは公知のものが全て使用できる。
重合開始剤としては、クメンハイドロパーオキサイド、p−メンタンハイドロパーオキサイド、ジイソプロピルベンゼンハイドロパーオキサイド、テトラメチルブチルハイドロパーオキサイド、tert−ブチルハイドロパーオキサイド、過硫酸カリウム、アゾビスイソブチロニトリル等が挙げられる。また、重合開始助剤として、各種還元剤、含糖ピロリン酸鉄処方、スルホキシレート処方等のレドックス系を使用することが好ましい。
【0057】
連鎖移動剤としては、オクチルメルカプタン、n−ドデシルメルカプタン、t−ドデシルメルカプタン、n−ヘキシルメルカプタン、ターピノーレン類などが挙げられる。
乳化剤としては、ドデシルベンゼンスルホン酸ナトリウム等のアルキルベンゼンスルホン酸塩、ラウリル硫酸ナトリウム等の脂肪族スルホン酸塩、ラウリル酸カリウム、ステアリン酸カリウム、オレイン酸カリウム、パルミチン酸カリウム等の高級脂肪酸塩、ロジン酸カリウム等のロジン酸塩などを使用することができる。
【0058】
なお、乳化重合において、ゴム質重合体(a)及びビニル系単量体の使用方法は、ゴム質重合体(a)全量の存在下にビニル系単量体を一括添加して重合してもよく、分割もしくは連続添加して重合してもよい。また、ゴム質重合体(a)の一部を重合途中で添加してもよい。
【0059】
乳化重合後、得られたラテックスは、通常、凝固剤により凝固させられる。その後、水洗、乾燥することにより、(A)成分の粉末を得る。この際、乳化重合で得た2種以上の(A)成分のラテックスを適宜ブレンドした後、凝固してもよく、また、更に(B)成分のラテックスを適宜ブレンドした後、凝固してもよい。凝固剤としては、塩化カルシウム、硫酸マグネシウム、塩化マグネシウム等の無機塩、硫酸、酢酸、クエン酸、リンゴ酸などの酸を使用することができる。また、ラテックスを噴霧乾燥することにより(A)成分の粉末を得ることもできる。
【0060】
溶液重合により本発明の(A)成分を製造する場合に使用することのできる溶剤は、通常のラジカル重合で使用される不活性重合溶媒であり、例えば、エチルベンゼン、トルエン等の芳香族炭化水素、メチルエチルケトン、アセトン等のケトン類、アセトニトリル、ジメチルホルムアミド、N−メチルピロリドン等が挙げられる。
【0061】
重合温度は、通常80〜140℃、好ましくは85〜120℃の範囲である。重合に際し、重合開始剤を使用してもよいし、重合開始剤を使用せずに、熱重合で重合してもよい。
重合開始剤としては、ケトンパーオキサイド、ジアルキルパーオキサイド、ジアシルパーオキサイド、パーオキシエステル、ハイドロパーオキサイド、アゾビスイソブチロニトリル、ベンゾイルパーオキサイド等の有機過酸化物などが好適に使用される。また、連鎖移動剤を使用する場合、例えば、メルカプタン類、ターピンーレン類、α−メチルスチレンダイマー等を使用することができる。
【0062】
また、塊状重合、懸濁重合で製造する場合、溶液重合において説明した重合開始剤、連鎖移動剤などを使用することができる。上記各重合法によって得た本発明の(A)成分中の残存する単量体量は、通常10,000ppm以下、好ましくは5,000ppm以下である。
【0063】
また、ゴム質重合体(a)の存在下にビニル系単量体を重合して得られる重合体成分には、上記のビニル系単量体がゴム質重合体(a)にグラフト共重合した共重合体とゴム質重合体(a)にグラフトしていない未グラフト成分〔上記ビニル系単量体の(共)重合体〕が含まれる。
【0064】
上記のゴム強化スチレン系樹脂(A)のグラフト率は、通常5〜100質量%、好ましくは10〜90質量%、更に好ましくは15〜85質量%、特に好ましくは20〜80質量%にコントロールすることが好ましい。グラフト率は、重合開始剤の種類、使用量、連鎖移動剤の種類、使用量、重合方法、重合時の単量体とゴム質重合体(a)の接触時間、ゴム質重合体種、重合温度等の各種要因で変わるが、一般的にはグラフト率を上げる方向で(A)成分から熱シクロヘキサンに溶解する成分が少なくなるが、当該溶解成分がなくなることにより本発明の組成物の発泡性が悪くなる。
なお、グラフト率は以下の式(15)により求めることができる。
グラフト率(質量%)={(T−S)/S}×100 (15)
【0065】
上記式(15)中、Tはゴム強化スチレン系樹脂(A)1gをアセトン20ml(アクリル系ゴムの場合はアセトニトリル)に投入し、振とう機により2時間振とうした後、遠心分離機(回転数;23,000rpm)で60分間遠心分離し、不溶分と可溶分とを分離して得られる不溶分の質量(g)であり、Sはゴム強化スチレン系樹脂(A)1gに含まれるゴム質重合体(a)の質量(g)である。
なお、ビニル単量体として芳香族ビニル化合物のみを用いた場合は、アセトンの代わりにメチルエチルケトンを用いて測定する。
【0066】
また、本発明で使用されるゴム強化スチレン系樹脂(A)のアセトン可溶分(アクリル系ゴムの場合はアセトニトリル)の極限粘度〔η〕(溶媒としてメチルエチルケトンを使用し、30℃で測定)は、通常0.15〜1.2dl/g、好ましくは0.2〜1.0dl/g、更に好ましくは0.2〜0.8dl/gである。本発明で使用するゴム強化スチレン系樹脂(A)中に分散するグラフト化ゴム質重合体粒子の平均粒子径は、通常50〜3,000nm、好ましくは40〜2,5000nm、特に好ましくは50〜2,000nmである。ゴム粒子径が50nm未満では耐衝撃性が劣る傾向にあり、3,000nmを超えると成形品表面外観が劣る傾向にある。また、使用するゴム質重合体(a)とビニル系単量体の共重合体の屈折率を実質的に合わせること及び/または分散するゴム質重合体(a)の粒子径を実質的に可視光の波長以下(通常1,500nm以下)にすることで透明性を有する(A)成分を得ることができるが、これらの透明性樹脂も本発明の(A)成分として用いることができる。
【0067】
本発明の組成物に用いられる(A)成分は、発泡性の発現のためには、下記条件で示した熱シクロヘキサン溶解量が、使用したゴム質重合体(a)を基準として、1〜99質量%であることが必要であり、好ましくは2質量%以上、更に好ましくは4質量%以上、特に好ましくは5〜80質量%である。なお、ここで溶解してくる成分の主成分は、ゴム質重合体(a)である。
また、特にゴム質重合体(a)がアクリル系ゴムの場合、熱シクロヘキサン溶解量は、好ましくは1〜40質量%、さらに好ましくは2〜30質量%、特に好ましくは3〜20質量%である。ゴム質重合体(a)がオレフィン系ゴムの場合、好ましくは5〜60質量%、さらに好ましくは10〜50質量%、特に好ましくは20〜40質量%である。
【0068】
熱シクロヘキサン溶解量は、以下に示す方法により求めることができる。すなわち、ゴム強化スチレン系樹脂(A)〔ここで(A)成分中のゴム量は、W1グラム〕を、ソックスレー抽出器を用いて、常圧下で、シクロヘキサンを8時間還流させる。シクロヘキサン溶液を乾固し、抽出物の重量を測定し(W2グラム)、下記式(16)で、熱シクロヘキサン溶解量を算出する。
【0069】
熱シクロヘキサン溶解量(%)=W2/W1×100 (16)
【0070】
本発明は、本発明で用いるゴム変性スチレン系樹脂(A)の熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準として1質量%以上である場合、具体的には成分(A)中のゴム質重合体(a)が熱シクロヘキサンに溶解すること、即ちゴム質重合体(a)が未架橋、またはルーズな架橋状態にある場合に、樹脂の発泡性が向上するというものである。
【0071】
ゴム質重合体(a)の含有量は、(A)〜(C)成分の合計量を100質量%として、3〜50質量%、好ましくは3〜40質量%、さらに好ましくは5〜35質量%である。(a)成分が、この範囲にあると、発泡性、発泡成形体の外観、機械的特性に優れる。
【0072】
(2)スチレン系樹脂(B)
本発明の(B)成分は、芳香族ビニル化合物、又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b2)を重合してなるスチレン系樹脂である。すなわち、(b2)成分は、芳香族ビニル化合物単独でもよいし、芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体との混合物でもよい。ここで使用される芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体としては、上記(A)成分で記載したものが全て使用できる。また、ビニル単量体(b2)は、上記ビニル単量体(b1)と同一であってもよいし、異なっていてもよい。
【0073】
好ましい(B)成分としては、スチレンの単独重合体、スチレン・アクリロニトリル共重合体、スチレン・メタクリル酸メチル共重合体、スチレン・アクリロニトリル・メタクリル酸メチル共重合体、スチレン・マレイミド化合物共重合体、スチレン・アクリロニトリル・マレイミド化合物共重合体及びこれらと上記官能基含有不飽和化合物との共重合体である。
【0074】
本発明の(B)成分は、上記した(A)成分の製造法で記載した公知の重合法である乳化重合、塊状重合、溶液重合、懸濁重合及びこれらを組み合わせた方法で製造することができる。
【0075】
(3)芳香族ポリカーボネート樹脂(C)
本発明で使用される芳香族ポリカーボネート樹脂(C)は、ジヒドロキシアリール化合物とホスゲンとの界面重縮合法、ジヒドロキシアリール化合物とジフェニルカーボネート等のカーボネート化合物とのエステル交換反応(溶融重縮合)によって得られるもの等、公知の重合法によって得られるものが全て使用できる。
【0076】
上記ジヒドロキシアリール化合物としては、ビス(4−ヒドロキシフェニル)メタン、1,1−ビス(4−ヒドロキシフェニル)エタン、2,2−ビス(4−ヒドロキシフェニル)ブタン、2,2−ビス(4−ヒドロキシフェニル)オクタン、ビス(4−ヒドロキシフェニル)プロパン、2,2−ビス(4−ヒドロキシ−3−t−ブチルフェニル)プロパン、2,2−ビス(4−ヒドロキシ−3−t−ブチルフェニル)プロパン、1,1−ビス(4−ヒドロキシフェニル)シクロペンタン、1,1−ビス(4−ヒドロキシフェニル)シクロヘキサン、4,4’−ジヒドロキシフェニルエーテル、4、4’−ジヒドロキシフェニルスルフィド、4,4’−ジヒドロキシフェニルスルホン、4,4’−ジヒドロキシ−3,3’−ジメチルジフェニルスルホン、ヒドロキノン、レゾルシン等が挙げられる。更に、ヒドロキシアリールオキシ末端かされたポリオルガノシロキサン(例えば、米国特許第3,419,634号明細書参照)等がある。これらは1種単独で、または2種以上を組み合わせて使用することができる。これらの中では、2,2−ビス(4−ヒドロキシフェニルプロパン(ビスフェノールA)が好ましい。
【0077】
ポリカーボネート樹脂(C)の粘度平均分子量は、好ましくは12,000〜40,000、さらに好ましくは15,000〜35,000、特に好ましくは18,000〜30,000である。分子量が高い方が得られる発泡成形体の機械的強度が高くなるが、流動性が低下し、均一なセルが得られず、発泡成形体の外観が低下する傾向となる。また、分子量の異なる2種以上のポリカーボネートを用いることもできる。
【0078】
上記の芳香族ポリカーボネートの粘度平均分子量は、通常、塩化メチレンを溶媒として、20℃、濃度〔0.7g/100ml(塩化メチレン)〕で測定した比粘度(ηsp)を以下の式(17)に挿入して算出できる。
【0079】
粘度平均分子量=(〔η〕×8130)1.205 (17)
ここで、〔η〕=〔(ηsp×1.12+1)1/2−1〕/0.56Cである。なお、Cは濃度を示す。
【0080】
界面重縮合で得られるポリカーボネート系樹脂は、各種の塩素化合物を含む場合があるが、この塩素化合物は、本発明の組成物の耐久性に悪影響する場合がある。このことから、塩素化合物含有量は、塩素原子として、通常300ppm以下、好ましくは100ppm以下とされる。
【0081】
(4)成分(A)〜(C)の配合割合(上記樹脂成分の配合割合)
本発明のゴム強化スチレン系樹脂(A)の配合量は、(A)〜(C)成分の合計を100質量%としたとき、5〜90質量%、好ましくは6〜60質量%、更に好ましくは7〜40質量%である。ゴム強化スチレン系樹脂(A)が5質量%未満及び90質量%超過では発泡性が劣る。
【0082】
本発明のスチレン系樹脂(B)の配合量は、(A)〜(C)成分の合計を100質量%としたとき、0〜85質量%、好ましくは5〜50質量%、更に好ましくは10〜30質量%である。スチレン系樹脂(B)が85質量%超過では発泡性が劣り、優れた外観を有する発泡成形体を得ることが困難となる。(B)成分は、(A)〜(C)成分中のゴム質重合体(a)の量を3〜50質量%に調整すること、(共)重合体種を変えて本発明の組成物に機能を付与すること、及び、他の熱可塑性重合体との相溶性を向上させることを目的として配合される。
【0083】
また、本発明に用いられる熱可塑性樹脂組成物中の芳香族ポリカーボネート樹脂(C)の含有量は、10〜90質量%、好ましくは30〜75質量%、さらに好ましくは50〜70質量%である。芳香族ポリカーボネート樹脂(C)の含有量が10質量%未満では、均一なセル径を有する発泡成形体を得ることが困難となり、一方、芳香族ポリカーボネート樹脂(C)の含有量が95質量%を超えると、優れた外観を有する発泡成形体を得ることが困難となる。
【0084】
(5)化学発泡剤(D)
使用する化学発泡剤(D)として、特に限定はないが好ましいものとしては、例えば分解されて炭酸ガスを発生する熱分解型無機発泡剤(炭酸水素ナトリウム、炭酸アンモニウム、炭酸水素アンモニウムなど)、分解されて窒素ガスを発生する熱分解型発泡剤(アゾジカルボンアミド(ADCA)、N,N’−ジニトロソペンタメチレンテトラミン(DPT)、4,4’−オキシビス(ベンゼンスルホニルヒドラジド)(OBSH)、アゾビスイソブチロニトリル、p−トルエンスルホニルヒドラジド、5−フェニルテトラゾールなど公知の熱分解型発泡性化合物が挙げられる。
【0085】
化学発泡剤(D)の含有量は、所望の発泡倍率が得られるように、用いる化学発泡剤や樹脂の種類に応じて適宜選択されるものであるが、上記成分(A)〜(C)の合計100質量部に対して化学発泡剤0.1〜5質量部であり、好ましくは0.2〜4質量部、更に好ましくは0.3〜3質量部である。化学発泡剤の含有量が0.1質量部未満である場合には、化学発泡剤の含有量が少なくて、発泡の各セル径を均一にすることが困難になる。一方、化学発泡剤の含有量が5質量部を超える場合には、化学発泡剤の含有量が多くて、化学発泡剤の残渣による金型汚染が生じ、外観に優れた発泡成形体を得ることが困難になる。
【0086】
溶融状態可塑性樹脂への発泡剤の配合方法としては、熱可塑性樹脂組成物のペレットと発泡剤マスターバッチペレットをドライブレンドした後、成形機に供給し、成形機内で樹脂を可塑化させ、金型内で発泡させる方法が好ましく用いられる。また、物理発泡剤を併用してもよい。物理発泡剤としては、具体的には、プロパン、ブタン、水、炭酸ガス等が挙げられる。
【0087】
(6)タルク(E)
本発明で用いられるタルク(E)は特に制限はないが、一般的には含水珪酸マグネシウム塩の粘土鉱物の一種で、その組成は(MgO)x(SiO2)y・zH2Oである(x、y、zは正値)。また、タルク中のMgの一部がCa2+等の2価の金属イオンに置換されてもよい。タルクの粒径は、特に制限されないが、レーザー散乱法による平均粒子径として、通常0.1〜50μm、好ましくは0.3〜25μm、更に好ましくは0.5〜20μmである。タルクの平均粒子径が0.1μm未満では、熱可塑性樹脂組成物中でのタルクの分散性が不十分となり、発泡性が劣り、優れた外観を有する発泡成形体を得ることが困難となる。一方、タルクの平均粒径が50μmを超えると、発泡性が劣り、優れた外観を有する発泡成形体を得ることが困難となる。
【0088】
本発明におけるタルク(E)の含有量は、上記成分(A)〜(C)の合計100質量部に対して、0.5〜18質量部、好ましくは1〜15質量部、更に好ましくは2〜12質量部である。タルクの含有量が0.5質量部未満の場合及びタルクの含有量が18質量部を超える場合には、発泡性が劣り、優れた外観を有する発泡成形体を得ることが困難となる。
【0089】
(7)繊維状充填材(F)
本発明における繊維状充填材(F)は、繊維状であれば特に限定はないが、例えばガラス繊維、炭素繊維、アラミド繊維等の有機繊維、セラミック系ウィスカー等の無機繊維、金属繊維等が挙げられる。このうち、発泡成形体の剛性向上の観点から、ガラス繊維を用いることが好ましい。
【0090】
本発明に用いられるガラス繊維の組成は、珪酸塩ガラス、ほう酸珪酸ガラス、燐酸塩ガラス等が挙げられる。またガラスの種類としては、Eガラス、Cガラス、Aガラス、Sガラス、Mガラス、ARガラス、Lガラス等が挙げられるが、Eガラス、Cガラスが好ましい。本発明に用いられるガラス繊維には、適当なサイジング剤を用いても構わない。サイジング剤としては、表面処理剤、フィルム形成剤、潤滑剤、界面活性剤、帯電防止剤等が挙げられる。表面処理剤としては、アミン系、シラン系、エポキシ系等のカップリング剤が挙げられる。本発明に用いられるガラス繊維は、ロービングを用いた長繊維タイプでもよく、チョップドストランドであってもよい。
【0091】
本発明に用いられるガラス繊維の直径は特に指定はないが、通常φ1〜500μm、好ましくはφ5〜200μm、更に好ましくはφ5〜100μmである。ガラス繊維の直径がφ1μm未満では、機械的強度が不十分となるおそれがある。一方、ガラス繊維の直径がφ500μmを超えると、発泡性、発泡成形体の外観が悪化するおそれがある。
【0092】
また、ガラス繊維の長さは特に指定はないが、通常0.01〜10mm、好ましくは0.05〜5mmである。ガラス繊維の長さが0.01mm未満では、発泡性が劣り、機械的強度が不十分となるおそれがある。一方、ガラス繊維の長さが10mmを超えると、発泡性が不十分となるおそれがある。
ガラス繊維の長さ(μm)/直径(μm)は、通常5〜1000、好ましくは8〜500である。ガラス繊維の長さ/直径が5未満では、ガラス繊維の両端が発泡起点とならず、発泡性が不十分となるおそれがある。一方、ガラス繊維の長さ/直径が1000を超えると、ガラス繊維の数の減少により、発泡起点の数が減少し、発泡性が不十分となるおそれがある。
また、成形した発泡成形体中に分散しているガラス繊維の残存平均繊維長さは、0.01〜1mmが好ましく、0.02〜0.8mmがより好ましく、0.03〜0.7mmが更に好ましい。ガラス繊維の残存平均繊維長さが上記範囲にあると、発泡性、発泡成形体の外観、機械的特性が十分になる。
【0093】
炭素繊維としては、PAN系、ピッチ系等が用いられる。また上記炭素繊維は、カーボンナノチューブなどの炭素繊維構造体であってもよい。
本発明に用いられる炭素繊維の直径は特に指定はないが、0.5μm〜200μmが好ましく、1μm〜50μmがより好ましく、5μm〜50μmがさらに好ましい。繊維の長さは特に指定はないが、発泡成形体中で20μm以上であることが好ましい。
【0094】
本発明における繊維状充填材(F)の配合量は、上記成分(A)〜(C)の合計100質量部に対して、0.5〜25質量部、好ましくは0.5〜18質量部、更に好ましくは2〜18質量部、特に好ましくは4〜18質量部である。繊維状充填材の含有量が0.5質量部未満の場合及び25質量部を超える場合には、発泡性、発泡成形体の外観、金型汚染性、機械的強度が悪化するおそれがある。
【0095】
本発明に用いられる熱可塑性樹脂組成物には、熱老化防止剤を配合することができる。熱老化防止剤としては、フェノール系、リン系、硫黄系などが挙げられ、好ましくはフェノール系、リン系および硫黄系の3種混合系である。熱老化防止剤として、この3種混合系を用いると、長時間、高温下に曝された時の、引張り伸び率を保持するという効果が得られる。
【0096】
熱老化防止剤のうち、フェノール系としては、2,6−ジ−t−ブチルフェノール誘導体、2−メチル−6−t−ブチルフェノール誘導体、オクタデシル3(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート、4,4’−ブチリデン−ビス(6−t−ブチル−m−クレゾール)、ペンタエリスリチル・テトラキス〔3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート〕、2〔1−(2−ヒドロキシ−3,5−ジ−t−ペンチルフェニル)−エチル〕−4,6−ジ−t−ペンチルフェニルアクリレート、2−t−ブチル−6(3−t−ブチル−2−ヒドロキシ−5−メチルベンジル)−4−メチルフェニルアクリレートなどが挙げられる。
【0097】
リン系としては、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト、サイクリックネオペンタンテトライルビス(2,4−ジ−t−ブチルフェニルホスファイト)、ジステアリルペンタエリスリトールジホスファイト、リン酸2水素ナトリウム、リン酸1水素2ナトリウムなどが挙げられる。
硫黄系としては、3,3’−チオビスプロピオン酸ジドデシルエステル、3,3’−チオビスプロピオン酸ジオクタデシルエステル、ペンタエリスリトール−テトラキス(3−ラウリルプロピオネート)、ジラウリル3,3’−チオジプロピオネートなどが挙げられる。
【0098】
本発明に用いられる熱可塑性樹脂組成物中の熱老化防止剤の割合は、0〜5質量%、好ましくは0〜3質量%である。本発明の熱可塑性樹脂組成物において、ポリカーボネート樹脂(A)以外のゴム強化ビニル系樹脂(B)成分は、熱老化防止剤を添加することで、熱老化特性が改良されるが、芳香族ポリカーボネート樹脂(C)は、熱老化防止剤が加水分解を促進する触媒として働くことがあり、熱老化防止剤を入れない方が劣化を抑制する傾向もある。これらの相反する効果を鑑みて、5質量%を上限として上記熱老化防止剤を添加すれば、最適な熱老化防止効果が得られる。
【0099】
また、本発明に用いられる発泡成形用熱可塑性樹脂組成物には、公知の耐候剤、滑剤、着色剤、帯電防止剤、シリコーンオイル、タルク(E)及び繊維状充填材(F)を除く無機フィラーなどの添加剤を配合することができる。このうち、耐候剤としては、ベンゾトリアゾール系、トリアジン系、ベンゾフェノン系などが好ましい。滑剤としては、エチレンビスステアリルアミド、硬化ヒマシ油などが好ましい。着色剤としては、カーボンブラック、ベンガラなどが挙げられる。帯電防止剤としては、ポリエーテル、アルキル基を有するスルホン酸塩などが挙げられる。
【0100】
本発明の発泡成形用熱可塑性樹脂組成物には、本発明の目的とする性能を損なわない範囲で、更に他の熱可塑性樹脂を配合することができる。熱可塑性樹脂としては、ゴム強化スチレン系樹脂〔ただし、(A)成分は除く〕、ポリオレフィン系樹脂、塩化ビニル系樹脂、アクリル系樹脂、ポリエステル系樹脂、ポリアミド系樹脂、ポリアセタール系樹脂、ポリフェニレンエーテル系樹脂、ポリアリーレンスルフィド系樹脂等があり、これらの熱可塑性樹脂は1種単独で、または2種以上を組み合わせて使用することができる。
【0101】
また、第1の発明において、上記ゴム質重合体(a)は、オレフィン系ゴム、アクリル系ゴム及び水素添加共役ジエン系ブロック共重合体からなる群から選ばれた少なくとも1種であることが好ましい(請求項2)。
この場合には、ゴム質重合体が適切であり、微細で均一な発泡セルを有する発泡成形体を安定して得ることができる。
【0102】
また、上記アクリル系ゴムは、ゲル含率が70質量%以下であり、体積平均粒子径が50〜300nmであることが好ましい(請求項3)。
この場合には、ゴム質重合体が適切であり、微細で均一な発泡セルを有する発泡成形体を安定して得ることができる。
【0103】
また、上記発泡成形用熱可塑性樹脂組成物は、コアバック型射出発泡成形に用いることが好ましい(請求項4)。
この場合には、微細で均一な発泡セルを有する発泡成形体を安定して得ることができる。
【0104】
2.熱可塑性樹脂組成物の製造
本発明に用いられる熱可塑性樹脂組成物は、各種押出機、バンバリーミキサー、ニーダ
ー、ロールなどを用いて混練することができる。例えば、(A)、(B)、(C)、(D)、(E)及び(F)成分、並びに必要に応じてその他の添加剤を混練することにより熱可塑性樹脂組成物のペレットを得ることができる。具体的には、2軸押出機によって(A)〜(F)成分を溶融させる方法などが挙げられる。混練温度は、熱可塑性樹脂組成物の配合によって適宜選択されるが、本発明においては、通常220〜260℃である。
【0105】
3.発泡成形体(発泡成形品)の成形方法
本発明の発泡成形用熱可塑性樹脂組成物を用いて発泡成形体を成型する方法としては、射出発泡成形、押出発泡成形等公知の方法を用いることができる。
射出発泡成形方法では、上記射出発泡成形用熱可塑性樹脂組成物を、射出成形機の金型内に形成されたキャビティ空間に射出し、直ちに、あるいは所定時間が経過した後、可動型、あるいは可動型に内設された可動コアを所定の速度で所定位置まで後退させ、キャビティ空間を拡大することにより発泡させる、所謂、コアバック方式の射出成形法によって発泡成形体を得ることができる。金型の温度は、通常、射出される際の熱可塑性樹脂組成物の温度より相当に低いため、キャビティの表面に接して形成される発泡成形体の表面には、ほとんど発泡していない緻密なスキン層が形成される。
【0106】
発泡成形体は、樹脂製等の基材の表面に接するように一体に形成することもできる。このような積層品は、キャビティ空間に予め基材を配置しておき、その表面に熱可塑性樹脂組成物を射出することにより形成することができる。また、2本の射出ユニットが搭載された射出成形機を使用し、先ず、基材となる樹脂等を射出して基材を形成し、その後、可動型に内設された可動コアを後退させて熱可塑性樹脂組成物を射出するためのキャビティ空間を形成し、次いで、熱可塑性樹脂組成物を射出し、その後、可動コアを更に後退させてキャビティ空間を拡大し、発泡させて、基材の表面に発泡成形体が積層された積層品とすることもできる。
【0107】
本発明の射出発泡成形方法では、可動型の後退速度、あるいは可動型に内設して設けられた可動コアの後退速度、即ち、上記「型開速度」は0.05〜20mm/秒である。この型開速度は、好ましくは0.1〜10mm/秒である。このような型開速度とすることにより、平均セル径が50〜500μmと適度に微細である均質な発泡成形体とすることができる。
【0108】
型開速度が0.05mm/秒未満であると、冷却が進んで発泡不足が発生し、発泡成形体の表面に凹凸が生じる。一方、型開速度が20mm/秒を越えると、セル径が大きく、また不均一な発泡成形体となる。
更に、射出される熱可塑性樹脂組成物の温度は、好ましくは200〜280℃、特に好ましくは220〜270℃である。この温度が200℃未満であると、熱可塑性樹脂組成物の流動性が不十分となり、特に、末端部では充填不良が発生することがある。一方、280℃を越えると、熱可塑性樹脂組成物の組成によっては熱劣化等が懸念される。
【0109】
また、金型温度は、好ましくは20〜80℃、特に好ましくは30〜70℃である。この温度が20℃未満であると、金型内表面と接触した熱可塑性樹脂組成物が急激に冷却され、均質な発泡成形体とすることができず、末端部で充填不良が発生することもある。一方、80℃を越えると、発泡成形体のキャビティの表面に接して形成された部分に均質なスキン層が形成されないことがあり、好ましくない。
【0110】
また、熱可塑性樹脂組成物を射出してから可動型、あるいは可動型に内設された可動コアの後退を開始するまでの時間(金型後退遅延時間)は、型開速度にもよるが、3秒以下とすることが好ましく、射出完了後、直ちに後退を開始してもよい。この金型後退遅延時間は、好ましくは0.1〜2.5秒、特に好ましくは0.1〜1.5秒である。金型後退遅延時間が3秒を越えると、冷却が進んで均質な発泡成形体とすることができない場合がある。
【0111】
金型の後退量は所定の発泡倍率により設定すればよく、限定されないが、特に、機器の機枠等では、金型内キャビティ空間に充填された素材の初期肉厚に対して発泡成形体の最終肉厚が1.1〜3.0倍となるように金型を後退させる、即ち、型開きすることが好ましい。この肉厚の比を発泡倍率とすれば、発泡倍率は、好ましくは1.1〜3倍、更に好ましくは1.5〜2倍であり、発泡成形体の肉厚が5〜30mm、特に5〜25mmである製品が多いことを考慮すれば、金型の後退量は、通常、2.5〜30mmである。
【0112】
なお、冷却時間は発泡成形体の寸法、あるいは冷却方法にもよるが、脱型時の発泡成形体の温度が40〜80℃程度にまで低下しておればよく、一般に30秒以上であればよく、大型の製品であっても100秒で十分である。
本発明の発泡成形体の成形方法において、射出充填時に、金型内にファブリックやフィルムをインサートしてもよい。
【0113】
4.発泡成形体
本発明の成形方法により得られた発泡成形体は、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり機械的性能に優れる発泡成形体である。具体的には、発泡セル径の平均径が好ましくは50〜500μm、より好ましくは70〜450μm、さらに好ましくは100〜400μmであり、発泡セル径が均一であり、粒径分布の狭いことが好ましい。特に発泡成形体発泡セルの大半のセル径が400μm以下の均一発泡成形体であることが好ましい。
本発明の成形方法により得られた発泡成形体は、さらに発泡倍率が1.01〜3.0倍、好ましくは1.1〜2.7倍、より好ましくは1.5〜2.5倍の所望の倍率にすることができる。
【0114】
本発明の成形方法により得られた発泡成形体の形状は、目的、用途などにより選択され、板状(シート状)、筒状、半筒状、棒状、線状、塊状等とすることができる。
本発明の発泡成形体は、表示板、コンクリートパネル、屋上断熱材、畳芯材、襖、システムキッチンの木材代替材、風呂蓋、テーブル板などの土木・建築関連資材、サイドモール、吸音材、バンパー、ドアハンドル、コンソールボックス、天井材、ピラー、センターロークラスタフィニッシュパネル、カウルサイドトリム、センターアウトレット、ドアライニング、アッシュトレイ、フットレスト、ステアリングコラムカバー、ロアインサート、ロアハンドルパネル、ホイルキャップ、スポイラーなどの車両用内外装関連資材、容器、トレー、通い箱等の日用雑貨用品、テレビ、ビデオ、エアコンのハウジング、パラボナアンテナ、エアコン室外機等の電気・電子部品、ビート板、プロテクターなどのスポーツ用品、壁、床、機枠、家具、化粧シート、間仕切り、ラティス、フェンス、雨樋、サイジングボード、カーポート等の住宅・事務所用内外装材、玩具・遊技機等の機枠、緩衝材、補強材、断熱材、芯材、代替合板等として用いることができる。
さらに、本発明の発泡成形体は、用途によっては、他の成形品、部材等と一体化させ、複合化させてなる物品として用いることができる。
【実施例】
【0115】
以下に、本発明の発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体にかかる実施例につき、図面を参照して説明する。
本例の発泡成形用熱可塑性樹脂組成物は、ゴム強化スチレン系樹脂(A)5〜90質量%、スチレン系樹脂(B)0〜85質量%、及び芳香族ポリカーボネート樹脂(C)10〜90質量%を含有し、成分(A)〜(C)の合計100質量部に対し、化学発泡剤(D)を0.1〜5質量部、タルク(E)を0.5〜18質量部、及び繊維状充填材(F)を0.5〜25質量部配合してなる。
【0116】
ゴム強化スチレン系樹脂(A)は、ゴム質重合体(a)の存在下に、芳香族ビニル化合物、又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b1)を重合してなり、かつ熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準(100質量%)として1〜99質量%である。また、成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合は、3〜50質量%である。
スチレン系樹脂(B)は、芳香族ビニル化合物、又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b2)を重合してなる。
【0117】
以下に、本例の発泡成形用熱可塑性樹脂組成物、これを用いた発泡成形体6及び発泡成形体6の成形方法につき、図1〜図4を参照して詳説する。
本例においては、上記組成の発泡成形用熱可塑性樹脂組成物を用いることによって、射出発泡成形を行う際に、微細な発泡セル構造を発現し、発泡成形体6の部位によらず発泡セルの大きさが均一であり、機械的性能にも優れ、かつ表面外観にも優れた発泡成形体6を生産効率よく成形することができる。
【0118】
本例においては、上記発泡成形用熱可塑性樹脂組成物を用いて発泡成形体6を成形するに当たり、次の製造装置1を用いる。
製造装置1は、図1〜図4に示すごとく、金型として、第1型部2と、第1型部2に対して相対的に可動する第2型部3とを備えている。そして、製造装置1は、第2型部3と第1型部2との間に形成したキャビティ41A内に、熱可塑性樹脂組成物を溶融させた溶融樹脂60を充填し、第1型部2と第2型部3とをキャビティ41Aの容積が拡大する離隔方向Rに相対的に可動させるよう構成してある。
【0119】
図1に示すごとく、第2型部3は、第1型部2に設けたキャビティ形成凹部21内に配置するキャビティ形成凸部31を設けてなる。第1型部2及び第2型部3においては、キャビティ形成凹部21において第1型部2と第2型部3との可動方向Dに平行に形成した内側面211と、キャビティ形成凸部31において可動方向Dに平行に形成した外側面311との間には、溶融樹脂60を充填するための充填用隙間42がキャビティ41Aと連通して形成されている。
本例の製造装置1は、溶融樹脂60をキャビティ41A内に充填すると共に充填用隙間42に充填した後、第1型部2と第2型部3とを離隔方向Rに相対的に可動させるよう構成してある。
【0120】
図1、図4に示すごとく、本例の第1型部2は、キャビティ41内に溶融樹脂60を充填するための樹脂注入口22を設けた固定型部である。本例の第2型部3は、第1型部2に対して離隔方向Rに可動する可動型部である。
また、本例の第2型部3は、第1型部2との間に形成するキャビティ41の容積を1.1〜2.0倍に拡大させるように、第1型部2に対して離隔方向Rへ後退するよう構成してある。そして、本例において発泡成形する発泡成形体6の発泡倍率は、1.1〜2.0倍である。
図2、図3に示すごとく、本例の製造装置1は、第1型部2に設けた樹脂注入口22に接続して、上記キャビティ41A内に溶融樹脂60を注入するための注入ノズル25を有している。本例の第2型部3は、油圧、空気圧、電力等によって動作する駆動源によって、第1型部2に対して進退する(可動方向Dに移動する)よう構成してある。
【0121】
本例においては、第1型部2及び第2型部3を用いて、略直方体形状であって、四角形状断面を有し、一方向Lに長い板形状の発泡成形体6を発泡成形する。
図4に示すごとく、本例の第2型部3は、キャビティ形成凸部31の先端面312が充填用隙間42の可動方向Dの端部まで移動するまで第1型部2に対して離隔方向Rに可動するよう構成してある。
第2型部3が第1型部2に対する原位置301にあるときには、キャビティ41に連通して、溶融樹脂60を充填するための充填用隙間42が形成される。この充填用隙間42は、溶融樹脂60を充填した際に、充填用隙間42の全体にスキン層61を形成することができる幅(隙間)に形成する。そして、図1、図2に示すごとく、第2型部3が離隔方向Rに可動する前の原位置301にあるときには、第2型部3と第1型部2との間には、容積が縮小したキャビティ41A及び充填用隙間42が形成されると共に、図4に示すごとく、第2型部3が離隔方向Rに可動した可動位置302にあるときには、第2型部3と第1型部2との間には、容積が拡大したキャビティ41Bが形成される。
【0122】
図1に示すごとく、本例の第1型部2は、キャビティ41における樹脂注入側の表面212(発泡成形体6の固定側表面を成形する底面)、及び樹脂注入側の表面に直交する全周の側面211(内側面211、発泡成形体6の全側面を成形する面)を形成するためのキャビティ形成凹部21を有している。本例の第2型部3は、キャビティ41における樹脂受け側の表面312(発泡成形体6の可動側表面を成形する先端面)、及び樹脂注入側の表面に直交する全周の側面311(外側面311)を形成するためのキャビティ形成凸部31を有している。
第1型部2のキャビティ形成凹部21における内側面211、及び第2型部3のキャビティ形成凸部31における外側面311は、可動方向Dに平行に形成してある。
【0123】
また、同図に示すごとく、本例の第1型部2においてキャビティ形成凹部21を形成する型壁部の開口先端部23の全周には、開口先端部23と第2型部3における外側面311との間を閉塞する閉塞型部5が設けてある。
そして、製造装置1は、第1型部2に対して第2型部3を離隔方向Rに可動させるときには、第2型部3における外側面311が閉塞型部5における内周面51と摺動することにより、充填用隙間42を形成した状態を維持して、キャビティ41Aの容積を拡大させるよう構成してある。
【0124】
また、図4に示すごとく、製造装置1は、第1型部2に対して第2型部3を離隔方向Rに相対的に可動させたときには、第2型部3における先端面312と閉塞型部5における内側端面52とが一致するよう構成してある。そして、上記可動工程において、第1型部2に対して第2型部3を離隔方向Rに可動させたときには、発泡成形体6の全周の表面にスキン層61を形成すると共に、スキン層61の内側部分に発泡層62を形成することができる。
【0125】
なお、成形する発泡成形体6の形状によっては、第2型部3における先端面312と閉塞型部5における内側端面52とが一致するまでは第2型部3を離隔方向Rに可動させずに、発泡成形体6からスキン層61による突出部が突出した形状を形成することもできる。
また、本例の充填用隙間42の幅(隙間)は適宜変更することができ、充填用隙間42に形成するスキン層61の厚みを適宜変更することができる。
【0126】
次に、上記製造装置1を用いて、発泡成形体6を成形する方法を詳説する。
本例の成形方法においては、充填工程として、図2、図3に示すごとく、第2型部3と第1型部2との間に形成したキャビティ41A内に、溶融樹脂60を充填してキャビティ41Aの表面に接触する溶融樹脂60の部分にスキン層61を形成し、可動工程として、図4に示すごとく、第1型部2と第2型部3とをキャビティ41Aの容積が拡大する離隔方向Rに相対的に可動させて、スキン層61に対する内側部分に溶融樹脂60を発泡させた発泡層62を形成する。
本例の発泡成形体6の成形方法は、溶融樹脂60を充填したキャビティ41Aの容積を拡大させて、溶融樹脂60を発泡させる方法であり、発泡成形する発泡成形体6の全周の表面に、溶融樹脂60がほとんど発泡せずに硬化したスキン層61を効果的に形成することができるものである。
【0127】
本例においては、まず、充填工程として、図2、図3に示すごとく、注入ノズル25に保持する溶融樹脂60を、第1型部2における樹脂注入口22から縮小した状態のキャビティ41A内に注入する。このとき、溶融樹脂60は、キャビティ41Aから充填用隙間42へと流入し、縮小した状態のキャビティ41及び充填用隙間42の全体に溶融樹脂60が充填される。そして、第1型部2のキャビティ形成凹部21の底面212及び内側面211と、第2型部3のキャビティ形成凸部31の先端面312及び外側面311に接触する溶融樹脂60の部分は、他の溶融樹脂60の部分(キャビティ41の内部(内側部分)における溶融樹脂60の部分)よりも早く冷却硬化して半硬化状態のスキン層61が形成される。そのため、キャビティ41における接触表面だけでなく、充填用隙間42における接触表面にも、溶融樹脂60が半硬化して未発泡のスキン層61が形成される。本例では、充填用隙間42の全体にスキン層61が形成される。
【0128】
次いで、可動工程として、図4に示すごとく、第1型部2に対して第2型部3を離隔方向Rに可動させる。このとき、充填用隙間42内に充填されて硬化した溶融樹脂60によるスキン層61に対して、第2型部3のキャビティ形成凸部31における外側面311が摺動し、このスキン層61の内側部分に溶融樹脂60が流入して発泡する。また、第1型部2のキャビティ形成凹部21における樹脂注入側の表面212、及び第2型部3のキャビティ形成凸部31における樹脂受け側の表面312に形成されたスキン層61の内側部分にも溶融樹脂60が流入して発泡する。
こうして、発泡成形体6の内部に溶融樹脂60が発泡した発泡層62を形成すると共に、発泡成形体6の全周の表面にスキン層61を形成することができる。そのため、全周の表面に安定してスキン層61を形成することができ、発泡成形した発泡成形体6の機械的強度を効果的に向上させることができる。
【0129】
それ故、本例の発泡成形体6の成形方法によれば、発泡成形体6の全周の表面にスキン層61を形成することができ、発泡成形体6の機械的強度を向上させることができる。
【0130】
また、本例においては、樹脂成形を行う所定の温度において、粘度が高い芳香族ポリカーボネート樹脂(C)の存在により、発泡によるセル径を均一にすることができ、発泡成形体6の外観を向上させることができる。また、樹脂成形を行う所定の温度において、芳香族ポリカーボネート樹脂(C)よりも粘度が低いゴム強化スチレン系樹脂(A)の存在により、発泡成形体6の表面に安定してスキン層61を形成することができる。
【0131】
本例の発泡成形用熱可塑性樹脂組成物は、上記ゴム強化スチレン系樹脂(A)、スチレン系樹脂(B)、及び芳香族ポリカーボネート樹脂(C)、化学発泡剤(D)、タルク(E)及び繊維状充填材(F)を含有し、ゴム強化スチレン系樹脂(A)を組成するゴム質重合体(a)の割合を適切にしたことによって、射出発泡成形を行う際に、微細な発泡セル構造を発現し、発泡成形体6の部位によらず発泡セルの大きさが均一であり、かつ機械的性能にも優れる発泡成形体6を成形することができる。
【0132】
また、本例の発泡成形用熱可塑性樹脂組成物は、タルク(E)及び繊維状充填材(F)を含有することにより、発泡性、機械的性能に一層優れ、表面外観に優れる発泡成形体6を成形することができる。この理由は、次のように考えられる。
すなわち、発泡成形する際には、タルク(E)及び繊維状充填材(F)が発泡起点になり、化学発泡剤(D)による発泡を促進することができる。このとき、タルク(E)による発泡起点と繊維状充填材(F)の両端による発泡起点とが混在することにより、より効果的に微細かつ均一な発泡セルが得られると考えられる。
【0133】
また、発泡成形用熱可塑性樹脂組成物が繊維状充填材(F)を含有することにより、機械的性能として特に剛性が優れた発泡成形体6を得ることができる。さらに、繊維状充填材(F)を含有することにより、発泡成形を行う金型の成形表面(第1型部2及び第2型部3におけるキャビティ41及び充填用隙間42の表面)に汚染が発生し難くなり、表面外観に優れた発泡成形体6を生産効率よく成形することができることがわかった。この理由は、次のように考えられる。
すなわち、繊維状充填材(F)が含有されていることにより、金型の成形表面に接触する溶融状態の発泡成形用熱可塑性樹脂組成物の表面が凹凸状に粗くなり、化学発泡剤(D)の発泡に伴って発生する揮発成分が、金型の成形表面と溶融状態の発泡成形用熱可塑性樹脂組成物の表面との間から外部へ抜け易くなったためであると考えられる。
そのため、金型の成形表面に汚染が発生し難くなり、その清掃を行う頻度を減少させることができ、生産効率よく発泡成形体6を成形することができる。
【0134】
それ故、本例の発泡成形用熱可塑性樹脂組成物によれば、射出発泡成形において、微細な発泡セル構造を発現し、発泡成形体6の部位によらず発泡セルの大きさが均一であり、機械的性能に優れ、かつ表面外観にも優れる発泡成形体6を生産効率よく成形することができる。
【0135】
(確認試験)
本確認試験においては、上記発泡成形用熱可塑性樹脂組成物(発明品1〜13)及び比較のための熱可塑性樹脂組成物(比較品1〜9)を用いて発泡成形体を成形し、この発泡成形体の外観及び内部の観察と、機械的性能としての曲げモジュラスの測定と、金型の汚染の観察とを行った。
以下に具体的な組成の熱可塑性樹脂組成物を挙げ、本発明を更に詳細に説明するが、本発明の主旨を超えない限り、本発明は以下の例に限定されるものではない。なお、以下の記載において「部」及び「%」は、特に断らない限り質量基準である。
【0136】
本確認試験においては、次の各成分を含有する発泡成形用熱可塑性樹脂組成物を用いた。
<(A)成分;ゴム強化スチレン系樹脂>
(重合体A1;アクリル系ゴム/スチレン/アクリロニトリル共重合体)
攪拌機を備えたガラス製フラスコに、窒素気流中で、イオン交換水160部、ドデシルベンゼンスルホン酸ナトリウム1部、クメンハイドロパーオキサイド0.002部、エチレンジアミン四酢酸四ナトリウム塩0.004部、硫酸第1鉄7水和物0.001部とn−ブチルアクリレート10部、アリルメタクリレート0.02部を投入し、撹拌しながら昇温した。60℃に達したところで、ナトリウムホルムアルデヒドスルホキシレート0.75部(20%水溶液)を添加した。内温を60℃に保持し80分経過後、n−ブチルアクリレート40部、アリルメタクリレート0.65部、及びクメンハイドロパーオキサイド0.01部、ドデシルベンゼンスルホン酸ナトリウム0.3部、イオン交換水7部、を180分間かけて連続に添加し、添加終了後更に60分重合を継続した。60分経過後、アクリル系ゴムラテックスの一部をサンプリングし評価したところ、体積平均粒子径0.1μm、ゲル含率6%であった。
【0137】
その後、ドデシルベンゼンスルホン酸ナトリウム5.2部(4%水溶液)、ナトリウムホルムアルデヒドスルホキシレート3部(20%水溶液)を添加し、スチレン38部、アクリロニトリル12部、及びt−ブチルハイドロパーオキサイド0.2部、ドデシルベンゼンスルホン酸ナトリウム0.3部、イオン交換水20部を5時間かけて連続に添加した。添加終了後更に45分間重合を継続後、2,2’−メチレンビス(4−エチル−6−t−ブチルフェノール)0.2部を添加して重合を終了した。重合転化率は99%であった。反応生成物のラテックスを硫酸マグネシウム水溶液で凝固、水洗した後、乾燥してASA樹脂(A1)を得た。
この重合体A1のグラフト率は65%、アセトン可溶分の極限粘度は〔η〕は0.38dl/gであった。また、熱シクロヘキサン溶解量は5%であった。
【0138】
(重合体A2;エチレン・プロピレン系ゴム質重合体/スチレン/アクリロニトリ共重合体)
リボン型撹拌翼を備えたステンレス製オートクレーブに窒素気流中で、エチレン・プロピレン系ゴム質重合体〔エチレン含量63%、非共役ジエン成分はジシクロペンタジエン、ヨウ素価10、ムーニー粘度(ML1+4、100℃)33、ゲル含率0%〕を30部、スチレン45部、アクリロニトリル25部、トルエン140部を仕込み、内温を75℃に昇温して、オートクレーブ内容物を1時間撹拌して均一溶液とした。その後、t−ブチルパーオキシイソプロピルモノカーボネート0.45部を添加し、内温を更に昇温し、100℃に達した後、この温度を保持しながら、撹拌回転数100rpmとして重合反応を行った。
【0139】
重合反応開始後、4時間経過後から内温を120℃に昇温し、この温度を保持しながら更に2時間反応を行って終了した。内温を100℃まで冷却したあと、オクタデシル−3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェノール)−プロピオネート0.2部を添加した。重合転化率は95%であった。反応混合物をオートクレーブより抜き出し、水蒸気蒸留により未反応物と溶媒とを留去し、40mmφの真空ベント付き押出機でシリンダー温度を220℃、真空度760mmHgに調節して揮発分を実質的に脱揮させ、ペレット化し重合体A2を得た。
この重合体A2のグラフト率は60%、アセトン可溶分の極限粘度〔η〕は0.45dl/gであり、熱シクロヘキサン溶解量は35%であった。
【0140】
(重合体A3;ブタジエン系ゴム質重合体/スチレン/アクリロニトリ共重合体)
重合体A2を得るために用いたエチレン・プロピレン系ゴム質重合体の代わりに、ポリブタジエン〔JSR社製、「BR51」、ハイシスタイプ、ムーニー粘度(ML1+4、100℃)33、ゲル含率0%〕15部、スチレン64部、アクリロニトリル21部に変えた以外は、重合体A2の場合と同様の方法で製造し、重合体A3を得た。
この重合体A3の重合転化率91%であり、グラフト率68%、アセトン可溶分の極限粘度〔η〕は0.39dl/gであり、熱シクロヘキサン溶解量は2%であった。
【0141】
(重合体A4;水素添加共役ジエン系ブロック共重合体/スチレン/アクリロニトリル/メタクリル酸メチル共重合体)
リボン型撹拌翼を備えたステンレス製オートクレーブに窒素気流中で、水素添加ブタジエン系ブロック共重合体〔JSR社製ダイナロン4600P(商品名)ゲル含率0%〕30部、メタクリル酸メチル50部、スチレン10部、アクリロニトリル10部、トルエン100部仕込み、内温を75℃に昇温させながら、撹拌により溶解させ均一溶液を得た後、t−ブチルパーオキシイソプロピルカーボネート0.5部、t−ドデシルメルカプタン0.1部を添加し、内温を更に昇温し、100℃に達した後は温度一定に制御しながら、撹拌回転数100rpmにて6時間重合反応を行った。その後、オクタデシル−3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェノール)−プロピオネート0.2部添加した後、反応混合物をオートクレーブより抜き出した。なお、重合転化率は91%であった。水蒸気蒸留により未反応物と溶媒を留去し、40mmφの真空ベント付き押出機(220℃、760mmHg真空)にて、実質的に揮発分を脱揮させ、重合体A4のペレットを得た。
この重合体A4のグラフト率は35%、アセトン可溶分の極限粘度〔η〕は0.30であり、熱シクロヘキサン溶解量は65%であった。
【0142】
(重合体A5;ポリブタジエン/スチレン/アクリロニトリル共重合体)
重合体A1を得るために用いたアクリル系ゴムラテックスの代わりに、ポリブタジエンラテックス(平均ゴム粒径0.3μm、ゲル含率85%)を用いた以外は、重合体A1の場合と同様の方法で製造し、重合体A5を得た。
この重合体A5の重合転化率は99%、得られた重合体D1のグラフト率は78%、アセトン可溶分の極限粘度〔η〕は、0.38dl/gであった。また熱シクロヘキサン溶解量は0%であった。
【0143】
<(B)成分;スチレン系樹脂>
(重合体B1;スチレン/アクリロニトリル共重合体)
リボン翼を備えたステンレス製オートクレーブを2基連結し、窒素置換した後、1基目の反応容器にスチレン75部、アクリロニトリル25部、トルエン20部を連続的に添加した。分子量調節剤としてtert−ドデシルメルカプタン0.14部及びトルエン5部の溶液、及び重合開始剤として、1,1’−アゾビス(シクロヘキサン−1−カーボニトリル)0.1部、及びトルエン5部の溶液を連続的に供給した。1基目の重合温度は、110℃にコントロールし、平均滞留時間2.0時間、重合転化率57%であった。得られた重合体溶液は、1基目の反応容器の外部に設けたポンプにより、スチレン、アクリロニトリル、トルエン、分子量調節剤、及び重合開始剤の供給量と同量を連続的に取り出し2基目の反応容器に供給した。2基目の反応容器の重合温度は、130℃で行い、重合転化率は75%であった。2基目の反応容器で得られた共重合溶液は、2軸3段ベント付き押出機を使用して、直接未反応単量体と溶剤を脱揮し、極限粘度〔η〕0.45dl/gの重合体B1を得た。
【0144】
<(C)成分;芳香族ポリカーボネート樹脂>
芳香族ポリカーボネート樹脂として、三菱エンジニアリングプラスチックス社製「ノバレックス 7022PJ」を用いた。
【0145】
<(D)成分;化学発泡剤>
化学発泡剤としては、永和化成工業社製「ポリスレンEB106」、マスターバッチ(ADCA(発泡剤)/ABS(樹脂)=10/90(質量比))を用いた。化学発泡剤の配合量は、樹脂成分((A)〜(C)成分)の合計100質量部に対し、0.35質量部とした。
【0146】
<(E)成分;タルク>
日本タルク社製の微粉タルク「MICRO ACE SG−200」(商品名)を用いた。レーザー回折法によるD50(重量平均)は1μmである。
【0147】
<(F)成分;繊維状充填材>
ガラス繊維として、エヌエスジー・ヴェトロテックス社製のマイクログラス・チョップドストランド「RES 03−TP89Z」(商品名)を用いた。ガラス繊維の直径は13μm、長さは、3mmである。
【0148】
<(G)成分;酸化防止剤>
酸化防止剤としては、ADEKA社製アデカスタブAO−412S(AO1)と、ADEKA社製アデカスタブ2112(AO2)と、住友化学工業社製スミライザーGS(AO3)とを用いた。酸化防止剤の配合量は、樹脂成分((A)〜(C)成分)の合計100質量部に対し、それぞれ0.1質量部とした。
【0149】
<その他の成分;ガラスビーズ>
ポッターズ・バロティーニ社製のフィラー用ガラスビーズ「GB731」(商品名)を用いた。材質はソーダ石灰ガラスであり、重量平均粒子径は32μmであった。
【0150】
上記A1〜A5のいずれかによる(A)成分、(B)〜(G)成分、その他の成分を用いて製造した熱可塑性樹脂組成物の組成を表1に発明品1〜13として示し、比較のために製造した熱可塑性樹脂組成物の組成を表2に比較品1〜9として示す。表1、表2において、溶解量は、(A)成分中における熱シクロヘキサン溶解量(%)を示す。
【0151】
【表1】

【0152】
【表2】
【0153】
表1における発明品1〜13及び表2における比較品1〜9は、次のようにして得た。
表1に記載した配合割合で、(A)〜(C)、(E)、(F)、(G)、その他の成分をヘンシエルミキサーにてブレンドした後、日本製鋼所製の二軸押出機TEX44を用いて、250℃にて押し出し、発泡成形前の熱可塑性樹脂ペレットを得た。
発泡成形機としては、日本製鋼所製110(t)電動成形機(J110AD)を用いた。得られた熱可塑性樹脂ペレットと発泡剤マスターバッチ((D)成分)をドライブレンドして発泡成形機に供給して射出発泡成形を行い、評価用試験片としての発泡成形体を得た。
また、射出発泡成形における充填時間は1秒、型開速度は0.5mm/秒とした。
【0154】
(A)成分(ゴム強化スチレン系樹脂)における熱シクロヘキサン溶解量は、上述した測定方法によって測定した。
なお、ゴム質重合体(a)のゲル含率、ゴム強化スチレン系樹脂(A)のグラフト率、ゴム強化スチレン系樹脂(A)及びスチレン系樹脂(B)のアセトン可溶分の極限粘度〔η〕についても、上述した測定方法によって測定した。
【0155】
発明品1〜13及び比較品1〜9について、最高発泡倍率の測定結果、外観観察及び内部観察の評価結果、曲げモジュラス(曲げ弾性率)、金型の汚染の評価結果を表1、表2に示す。
最高発泡倍率は、発泡倍率を1.1倍から0.1倍ずつ上げていき、外観観察の結果が○から△に変わる倍率(成形外観が悪化する倍率)として求めた。また、発泡倍率は、容積拡張前のキャビティの隙間を3mmとし、キャビティの隙間を拡張する量を0.3mmずつ拡大して求めた。
【0156】
発泡成形体の外観観察は、発泡成形体の表面を目視観察し、良好な平面が得られた場合
を○、一部に波打ちしている面があった場合を△、歪んで波打ちしている場合を×として
評価した。
発泡成形体の内部観察は、発泡倍率を1.5倍として得られた発泡成形体の断面を目視観察し、均一で微細な発泡セルが形成されていた場合を○、発泡セルの外径に大小の分布があった場合を△、発泡セルのほとんどが大きな外径になっていた場合を×として評価した。
【0157】
曲げモジュラス(曲げ弾性率)(MPa)は、発泡倍率を1.5倍として得られた発泡成形体を用い、ISO178に準拠して測定した。
なお、発泡成形体の内部観察及び曲げモジュラスは、発泡成形体が波打っていたり、歪んでいた場合には、測定を実施しなかった。この場合は、表中に「−」と記載した。
金型の汚染は、発泡倍率を1.5倍として発泡成形体を成形し、発泡成形後に金型の成形表面に汚染が見られなかった場合を○、金型の成形表面の一部に汚染が見られた場合を△、金型の成形表面の全体に渡って汚染が見られた場合を×として評価した。
なお、最高発泡倍率が1.5倍以上に達しないものは、金型の汚染の評価を行わなかった。
【0158】
発明品1〜13については、発泡成形用熱可塑性樹脂組成物を用いて成形した発泡成形体であり、発泡性に優れ(最高発泡倍率が高く)、成形外観が良好であり、また、内部の観察において、ほぼ微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさがほぼ均一であった。また、機械的性能としての曲げモジュラスも高い値が得られた。さらに、金型の汚染も成形表面の一部に見られる程度であった。
【0159】
これに対し、比較品1、2については、繊維状充填材(ガラス繊維)(F)の代わりにガラスビーズを用いて発泡成形したことにより、発泡セルが大きくなり、金型の成形表面の全体に渡って汚染が見られた。比較品3については、ガラス繊維(F)の配合量が多いために、成形外観が劣り、発泡セルが大きくなった。比較品4については、タルクが含まれないために、発泡セルが大きくなった。比較品5については、タルク(E)の配合量が多いために、成形外観が劣り、発泡セルが大きくなった。
【0160】
また、比較品6については、ゴム強化スチレン系樹脂(A)及びゴム質重合体(a)が少ないことにより、比較品7については、ゴム強化スチレン系樹脂(A)が多いことにより、比較品8については、ゴム強化スチレン系樹脂(A)中の熱シクロヘキサン溶解量が0(%)であることにより、最高発泡倍率が低く、成形外観が劣り、発泡セルが大きくなり、金型の成形表面の全体に渡って汚染が見られた。また、比較品9については、繊維状充填材を含有していないことにより、金型の成形表面の全体に渡って汚染が見られた。
なお、比較品1〜8については、内部観察結果に劣ったため、曲げモジュラスは評価しなかった。
【0161】
そして、特に、成形する発泡成形体において、微細かつ均一な発泡セルを得るためには、ゴム強化スチレン系樹脂(A)の熱シクロヘキサン溶解量がゴム質重合体(a)を基準として1〜99質量%であること、及び樹脂成分100質量%におけるゴム質重合体(a)の割合が3〜50質量%であることがわかった。また、この組成により、機械的強度及び成形外観に優れた発泡成形体が得られ、金型の汚染度合いも少ないことがわかった。
【符号の説明】
【0162】
1 製造装置
2 第1型部
21 キャビティ形成凹部
22 樹脂注入口
3 第2型部
31 キャビティ形成凸部
41A、B キャビティ
42 充填用隙間
6 発泡成形体
60 溶融樹脂(熱可塑性樹脂組成物)
【技術分野】
【0001】
本発明は、ゴム強化スチレン系樹脂を含む発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体に関する。
【背景技術】
【0002】
熱可塑性樹脂を用いた射出成形方法において、使用する樹脂材料の低減、軽量化等を目的として発泡成形を行う射出発泡成形が従来から検討されてきた。射出発泡成形を行う方法としては、例えば、特許文献1に開示されるように、熱可塑性樹脂に、アゾジカルボン酸アミドなどの熱分解型の化学発泡剤を使用する方法が知られている。
また、化学発泡に代えて、窒素ガス、二酸化炭素などを発泡剤として用いる物理発泡剤も知られている。更に超臨界状態の物理発泡剤を用いる方法も提案されている。
【0003】
例えば、特許文献2のポリオレフィン系樹脂組成物およびそれからなる成形体においては、ポリオレフィン(A)に対して、コア−シェルグラフト共重合体(B)及びタルク等の無機充填剤(C)を混合してなり、コア−シェルグラフト共重合体(B)が、架橋ゴム状重合体(a)に、共重合可能なビニル化合物からなる単量体成分(b)をグラフト共重合してなるポリオレフィン系樹脂組成物、及びこれに発泡剤を配合して発泡成形した発泡体について開示されている。これにより、優れた加工性、耐衝撃性、剛性及び表面性を同時に呈するポリオレフィン系樹脂組成物およびそれからなる成形体を構成することができる。
【0004】
さらに、例えば、特許文献3の射出発泡成形体の製造方法においては、金型の可動側金型と固定側金型とによって形成される型内空隙に原料樹脂を充填して金型に接している部分に表皮層を形成し、原料樹脂の充填完了直後、溶融部分の発泡剤の発泡圧力が周りの表皮層を押し広げるだけの力が残っているうちに、型締めシリンダーによる締め圧を瞬時にゼロ近くまで落として、表皮層の内側に発泡層を形成している。これにより、高発泡倍率の射出成形を可能にしている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2008−133485号公報
【特許文献2】特開2002−179851号公報
【特許文献3】特開2001−302830号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかしながら、特許文献1〜3等の熱可塑性樹脂組成物によっては、高い発泡倍率で、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、機械的性能(特に剛性)にも優れた発泡成形体を得るためには十分ではない。
また、発泡成形を繰り返し行う際には、発泡に伴う揮発成分が金型の成形表面に付着し、金型が汚染されるおそれがある。そして、金型の汚染が悪化したときには、次の発泡成形前に金型の成形表面を清掃する必要があった。
【0007】
本発明は、かかる従来の問題点に鑑みてなされたもので、射出発泡成形において、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、機械的性能に優れ、かつ表面外観にも優れる発泡成形体を生産効率よく成形することができる発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体を提供しようとするものである。
【課題を解決するための手段】
【0008】
第1の発明は、ゴム質重合体(a)の存在下に芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b1)を重合してなり、かつ熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準として1〜99質量%であるゴム強化スチレン系樹脂(A)5〜90質量%と、
芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b2)を重合してなるスチレン系樹脂(B)0〜85質量%と、
芳香族ポリカーボネート樹脂(C)10〜90質量%と、
上記成分(A)〜(C)の合計100質量部に対し、化学発泡剤(D)0.1〜5質量部と、タルク(E)0.5〜18質量部と、繊維状充填材(F)0.5〜25質量部とからなり、
上記成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合が3〜50質量%であることを特徴とする発泡成形用熱可塑性樹脂組成物にある(請求項1)。
【0009】
第2の発明は、上記発泡成形用熱可塑性樹脂組成物を用い、コアバック型射出発泡成形を行って成形したことを特徴とする発泡成形体にある(請求項5)。
【発明の効果】
【0010】
第1の発明の発泡成形用熱可塑性樹脂組成物は、上記ゴム強化スチレン系樹脂(A)、スチレン系樹脂(B)、及び芳香族ポリカーボネート樹脂(C)、化学発泡剤(D)、タルク(E)及び繊維状充填材(F)を含有し、ゴム強化スチレン系樹脂(A)を組成するゴム質重合体(a)の割合を適切にしたことによって、射出発泡成形を行う際に、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、かつ機械的性能にも優れる発泡成形体を成形することができる。
【0011】
すなわち、成形する発泡成形体において、微細かつ均一な発泡セルを得るためには、本発明者の研究開発により、特に、ゴム強化スチレン系樹脂(A)の熱シクロヘキサン溶解量がゴム質重合体(a)を基準として1〜99質量%であること、及び成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合が3〜50質量%であることが必要なことがわかった。また、この組成により、機械的性能にも優れる発泡成形体が得られることがわかった。
これにより、第1の発明の発泡成形用熱可塑性樹脂組成物によれば、微細かつ均一な発泡セルを有するだけでなく、機械的性能にも優れた発泡成形体を得ることができる。
【0012】
また、第1の発明の発泡成形用熱可塑性樹脂組成物は、タルク(E)及び繊維状充填材(F)を含有することにより、発泡セルが微細かつ均一で機械的性能に一層優れ、表面外観に優れる発泡成形体を成形することができることがわかった。この理由は、次のように考えられる。
すなわち、発泡成形する際には、タルク(E)及び繊維状充填材(F)が発泡起点になり、化学発泡剤(D)による発泡を促進することができる。このとき、タルク(E)による発泡起点と繊維状充填材(F)の両端による発泡起点とが混在することにより、より効果的に微細かつ均一な発泡セルが得られると考えられる。
【0013】
また、発泡成形用熱可塑性樹脂組成物が繊維状充填材(F)を含有することにより、機械的性能として特に剛性が優れた発泡成形体を得ることができる。さらに、繊維状充填材(F)を含有することにより、発泡成形を行う金型の成形表面に汚染が発生し難くなり、表面外観に優れた発泡成形体を生産効率よく成形することができることがわかった。この理由は、次のように考えられる。
すなわち、繊維状充填材(F)が含有されていることにより、金型の成形表面に接触する溶融状態の発泡成形用熱可塑性樹脂組成物の表面が凹凸状に粗くなり、化学発泡剤(D)の発泡に伴って発生する揮発成分が、金型の成形表面と溶融状態の発泡成形用熱可塑性樹脂組成物の表面との間から外部へ抜け易くなったと考えられる。
そのため、金型の成形表面に汚染が発生し難くなり、その清掃を行う頻度を減少させることができ、生産効率よく表面外観に優れる発泡成形体を成形することができることがわかった。
【0014】
それ故、第1の発明の発泡成形用熱可塑性樹脂組成物によれば、射出発泡成形において、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり、機械的性能に優れ、かつ表面外観にも優れる発泡成形体を生産効率よく成形することができる。
また、第2の発明に示すように、発泡成形体をコアバック型射出発泡成形を行って成形することにより、上記効果を容易に得ることができる。
【図面の簡単な説明】
【0015】
【図1】実施例における、第1型部及び第2型部を有する製造装置を示す断面説明図。
【図2】実施例における、充填工程において、キャビティ及び充填用隙間内に溶融樹脂を充填した状態を示す断面説明図。
【図3】実施例における、充填工程において、キャビティ及び充填用隙間内に溶融樹脂を充填した状態を、図2と直交する方向から見た状態で示す断面説明図。
【図4】実施例における、可動工程において、第1型部に対して第2型部を可動させて、キャビティ内に樹脂発泡成形体を成形した状態を示す断面説明図。
【発明を実施するための形態】
【0016】
本発明の発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体における好ましい実施の形態につき説明する。
1.発泡成形用熱可塑性樹脂組成物
上記発泡成形用熱可塑性樹脂組成物の好ましい組成について説明する。
なお、以下の説明において、「(共)重合」とは、単独重合及び共重合を意味し、「(メタ)アクリレート」とは、アクリレートとメタクリレートとの少なくとも一方を意味する。また、本発明の発泡成形用熱可塑性樹脂組成物を単に「熱可塑性樹脂組成物」と略記する場合がある。
【0017】
(1)ゴム強化スチレン系樹脂(A)
本発明の(A)成分は、ゴム質重合体(a)の存在下に芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b1)を重合してなり、かつ熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準として1〜99質量%であるゴム強化スチレン系樹脂である。
ここで使用されるゴム質重合体(a)としては、ガラス転位温度(Tg)が−10℃以下のものであり、ポリブタジエン、ポリイソプレン、ブタジエン・スチレン共重合体、ブタジエン・アクリロニトリル共重合体等の共役ジエン系ゴム、エチレン・プロピレン共重合体、エチレン・プロピレン・非共役ジエン共重合体、エチレン・1−ブテン共重合体、エチレン・1−ブテン・非共役ジエン共重合体等のオレフィン系ゴム、アクリル系ゴム、シリコーンゴム、ポリウレタン系ゴム、シリコーン・アクリル系IPNゴム、天然ゴム、共役ジエン系ブロック共重合体、水素添加共役ジエン系ブロック共重合体等が挙げられる。これらは、1種単独で、あるいは2種以上を組み合わせて用いることができる。
【0018】
上記オレフィン系ゴムは特に限定されないが、例えば、エチレンと、炭素数が3以上のα−オレフィンとを含むエチレン・α−オレフィン系ゴムが挙げられる。エチレンの含有量は、上記エチレン・α−オレフィン系ゴムを構成する単量体の全量を100質量%とした場合、好ましくは5〜95質量%、より好ましくは50〜90質量%、更に好ましくは60〜88質量%である。
上記α−オレフィンである炭素数が3以上のα−オレフィンとしては、プロピレン、1−ブテン、2−ブテン、イソブテン、1−ペンテン、2−メチル−1−ブテン、2−メチル−2−ブテン、3−メチルブテン、1−ヘキセン、4−メチル−1−ペンテン、3−メチル−1−ペンテン、1−ヘプテン、1−オクテン、1−デセン、1−ウンデセン等が挙げられる。これらのα−オレフィンは、1種単独で含まれていてもよいし、2種以上の組み合わせで含まれていてもよい。また、上記α−オレフィンのうち、プロピレン、1−ブテンが好ましい。
【0019】
上記α−オレフィンの含有量は、上記エチレン・α−オレフィン系ゴムを構成する単量体の全量を100質量%とした場合、好ましくは95〜5質量%、より好ましくは50〜10質量%、特に好ましくは40〜12質量%である。
上記エチレン・α−オレフィン系ゴムは、上記エチレン、及びα−オレフィンから構成される二元共重合体であってもよいし、これらと、更に他の化合物とから構成される重合体(三元共重合体、四元共重合体等)であってもよい。他の化合物としては、非共役ジエン化合物が挙げられる。
【0020】
上記オレフィン系ゴムに使用される非共役ジエン化合物としては、アルケニルノルボルネン類、環状ジエン類、脂肪族ジエン類などが挙げられ、好ましくは、ジシクロペンタジエン及び5−エチリデン−2−ノルボルネンである。これらの非共役ジエン化合物は単独でまたは2種以上を組み合わせて使用することができる。エチレン・α−オレフィン系ゴム中の非共役ジエン化合物単位の含有量は、通常30質量%未満、好ましくは15質量%未満である。
【0021】
上記アクリル系ゴムは特に限定されないが、アルキル基の炭素数が1〜8個の(メタ)アクリル酸アルキルエステル化合物の(共)重合体、あるいはこの(メタ)アクリル酸アルキルエステル化合物と、これと共重合可能なビニル系単量体との共重合体が好ましい。
【0022】
ここで使用されるアルキル基の炭素数が1〜8個のアクリル酸アルキルエステル化合物の具体例としては、メチルアクリレート、エチルアクリレート、プロピルアクリレート、n−ブチルアクリレート、i−ブチルアクリレート、アミルアクリレート、ヘキシルアクリレート、n−オクチルアクリレート、2−エチルヘキシルアクリレート、シクロヘキシルアクリレート等が挙げられる。メタクリル酸アルキルエステルの具体例としては、メチルメタクリレート、エチルメタクリレート、プロピルメタクリレート、n−ブチルメタクリレート、i−ブチルメタクリレート、アミルメタクリレート、ヘキシルメタクリレート、n−オクチルメタクリレート、2−エチルヘキシルメタクリレート、シクロヘキシルメタクリレート等が挙げられる。これらの化合物のうち、n−ブチルアクリレート、2−エチルヘキシルアクリレートが好ましい。また、これらは、1種単独で、あるいは2種以上を組み合わせて用いることができる。
【0023】
また、上記(メタ)アクリル酸アルキルエステル化合物と共重合可能なビニル系単量体としては、例えば、多官能性ビニル化合物、芳香族ビニル化合物、シアン化ビニル化合物等が上げられる。上記多官能性ビニル化合物とは、単量体1分子中に2個以上のビニル基を有する単量体をいい、上記(メタ)アクリル系共重合体を架橋する機能及びグラフト重合時の反応起点の役割を果たすものである。上記多官能性ビニル単量体の具体例としては、ジビニルベンゼン、ジビニルトルエン等の多官能性芳香族ビニル化合物;(ポリ)エチレングリコールジメタクリレート、トリメチロールプロパントリアクリレート等の多価アルコールの(メタ)アクリル酸エステル;ジアリルマレート、ジアリルフマレート、トリアリルシアヌレート、トリアリルシアヌレート、ジアリルフタレート、メタクリル酸アリル等が挙げられる。これらの多官能性ビニル化合物は、1種単独で、または2種以上を組み合わせて使用することができる。
【0024】
ここで使用される芳香族ビニル化合物及びシアン化ビニル化合物としては、後述するものが全て使用できる。更に、他の共重合可能な単量体として、アクリルアミド、メタクリルアミド、塩化ビニリデン、アルキル(炭素数1〜6)ビニルエーテル、アルキル基の炭素数が9個以上の(メタ)アクリル酸アルキルエステル、(メタ)アクリル酸等が挙げられ、これらは1種単独で、あるいは2種以上を組み合わせて使用される。
【0025】
上記アクリル系ゴムの好ましい単量体組成は、アルキル基の炭素数が1〜8個の(メタ)アクリル酸アルキルエステル化合物単位80〜99.99質量%、より好ましくは90〜99.95質量%、多官能性ビニル化合物単位0.01〜5質量%、より好ましくは0.05〜2.5質量%、及びこれと共重合可能な他のビニル単量体0〜20質量%、より好ましくは0〜10質量%である。ただし、単量体組成は、合計100質量%とする。
【0026】
ゴム強化スチレン系樹脂(A)の熱シクロヘキサン溶解量を、ゴム質重合体(a)を基準として1質量%以上とする目的から、上記アクリル系ゴムの製造において、多官能性ビニル化合物を使用する場合は、重合の後段階で行うことが好ましい。即ち、重合の初期段階ではアクリル酸アルキルエステル化合物、及び必要に応じて共重合可能な他のビニル単量体(b1)を重合し、重合の後期段階でアクリル酸アルキルエステル化合物及び多官能性ビニル化合物、更に必要に応じて共重合可能な他のビニル単量体(b1)を重合する方法で製造することができる。
【0027】
本発明のアクリル系ゴムの製造方法としては、(1)各種ビニル単量体を一括添加して重合する方法、(2)特定のビニル単量体を一括添加重合し、重合の後期段階で残りのビニル単量体を添加重合する方法、(3)各種ビニル単量体の一部を添加重合し、残りのビニル単量体を連続添加して重合する方法、(4)各種ビニル単量体を2段以上に分割して重合する方法等があるが、好ましくは(4)の方法であり、更に好ましくは(4)の方法で多官能性ビニル化合物を2段目以降の後期段階で使用する方法である。重合方法としては、乳化重合が特に好ましい。
上記アクリル系ゴムの体積平均粒子径は、50〜1000nmであることが好ましく、さらに好ましくは40〜700nm、特に好ましくは50〜500nmである。
【0028】
共役ジエン系ブロック共重合体としては、具体的には少なくとも1個の下記ブロックAまたは下記ブロックCと、少なくとも1個の下記ブロックBまたは下記ブロックA/Bとを含んでなる共重合体、またはブロックBもしくはA/Bによる重合体であり、アニオン重合法で公知の方法である、例えば、特公昭47−28915号公報、特公昭47−3252号公報、特公昭48−2423号公報、特公昭48−20038号公報などに開示されている方法で製造することができる。その具体的構造は、
A;芳香族ビニル化合物重合体ブロック、
B;共役ジエン重合体ブロック、
A/B;芳香族ビニル化合物/共役ジエンのランダム共重合体ブロック、
C;共役ジエンと芳香族ビニル化合物の共重合体からなり、かつ芳香族ビニル化合物が漸増するテーパーブロック、
とそれぞれ定義すると、次のような構造のものが挙げられる。
【0029】
A−B (1)
A−B−A (2)
A−B−C (3)
A−B1−B2 (4)
(ここで、B1は共役ジエン重合体ブロックまたは共役ジエンと芳香族ビニル化合物との共重合体ブロックであり、共役ジエン部分のビニル結合量は好ましくは20%以上、B2は共役ジエン重合体ブロックまたは共役ジエンと芳香族ビニル化合物の共重合体ブロックであり、共役ジエン部分のビニル結合含有量は好ましくは20%未満である。)
A−A/B (5)
A−A/B−C (6)
A−A/B−B (7)
A−A/B−A (8)
B2−B1−B2 (9)
(ここで、B1、B2は上記と同じ。)
C−B (10)
C−B−C (11)
C−A/B−C (12)
C−A−B (13)
【0030】
また、これらの基本骨格を繰り返し有する共重合体を挙げることができ、さらにそれをカップリングして得られる共役ジエン系ブロック共重合体であってもよい。上記式(4)の構造のものについては、特開平2−133406号公報、上記式(5)及び上記式(6)の構造のものについては、特開平2−305814号公報、特開平3−72512号公報に示されている。
【0031】
ここで使用される共役ジエンとしては、1,3−ブタジエン、イソプレン、2,3−ジメチル−1,3−ブタジエン、1,3−ペンタジエン、2−メチル−1,3−ペンタジエン、1,3−ヘキサジエン、4,5−ジエチル−1,3−オクタジエン、3−ブチル−1,3−オクタジエン、クロロプレンなどが挙げられるが、工業的に利用でき、また物性の優れた共役ジエン系ブロック共重合体を得るには、1,3−ブタジエン、イソプレン、1,3−ペンタジエンが好ましく、より好ましくは1,3−ブタジエンである。
【0032】
また、ここで使用される芳香族ビニル化合物としては、スチレン、t−ブチルスチレン、α−メチルスチレン、p−メチルスチレン、ヒドロキシスチレン、ビニルキシレン、モノクロルスチレン、ジクロルスチレン、モノブロムスチレン、ジブロムスチレン、フルオロスチレン、p−t−ブチルスチレン、エチルスチレン、ビニルナフタレン、ジビニルベンゼン、1,1−ジフェニルスチレン、N,N−ジエチル−p−アミノエチルスチレン、N,N−ジエチル−p−アミノエチルスチレン、ビニルピリジンなどが挙げられ、スチレン、α−メチルスチレンが好ましく、特に好ましくはスチレンである。
【0033】
上記共役ジエンブロック系共重合体中の芳香族ビニル化合物/共役ジエンの割合は、質量比で0〜70/100〜30、好ましくは0〜60/100〜40、更に好ましくは0〜50/100〜50であり、芳香族ビニル化合物を必須とする場合、好ましくは10〜70/90〜30である。ここで、芳香族ビニル化合物の含有量が70質量%を超えると樹脂状となり、ゴム成分としての効果が劣り好ましくない。
さらに、共役ジエンブロック中の共役ジエン部分のビニル結合量は、通常5〜80%の範囲である。
共役ジエン系ブロック共重合体の数平均分子量は、通常10,000〜1,000,000、好ましくは20,000〜500,000、更に好ましくは20,000〜200,000である。これらのうち、上記構造式のA部の数平均分子量は3,000から150,000、B部の数平均分子量は5,000〜200,000の範囲であることが好ましい。
【0034】
共役ジエン化合物のビニル結合量の調節は、N,N,N’,N’−テトラメチルエチレンジアミン、トリメチルアミン、トリエチルアミン、ジアゾシクロ(2,2,2)オクタアミン等のアミン類、テトラヒドロフラン、ジエチレングリコールジメチルエーテル、ジエチレングリコールジブチルエーテル等のエーテル類、チオエーテル類、ホスフィン類、ホスホアミド類、アルキルベンゼンスルホン酸塩、カリウムやナトリウムのアルコキシド等を使用して行うことができる。
【0035】
本発明で使用されるカップリング剤としては、アジピン酸ジエチル、ジビニルベンゼン、メチルジクロロシラン、四塩化珪素、ブチルトリクロロ珪素、テトラクロロ錫、ブチルトリクロロ錫、ジメチルクロロ珪素、テトラクロロゲルマニウム、1,2−ジブロモエタン、1,4−クロロメチルベンゼン、ビス(トリクロロシリル)エタン、エポキシ化アマニ油、トリレンジイソシアネート、1,2,4−ベンゼントリイソシアネート等が挙げられる。
【0036】
本発明で使用される水素添加共役ジエン系ブロック共重合体は、上記共役ジエン系ブロック共重合体の共役ジエン部分の炭素−炭素二重結合の少なくとも30%以上、好ましくは50%以上が水素添加された部分水素添加物または完全水素添加物であり、更に好ましくは90%以上が水素添加された水素添加物である。
【0037】
上記共役ジエン系ブロック共重合体の水素添加反応は、公知の方法で行うことができるし、また、公知の方法で水素添加率を調節することにより、目的の重合体を得ることができる。具体的な方法としては、特公昭42−8704号公報、特公昭43−6636号公報、特公昭63−4841号公報、特公昭63−5401号公報、特開平2−133406号公報、特開平1−297413号公報等に開示されている方法がある。
【0038】
本発明で使用される上記ゴム質重合体(a)は、ゲル含率は70質量%以下であることが、本発明の発泡性から好ましく、更に好ましくは50質量%以下、特に好ましくは10質量%以下である。
ゲル含率は、以下に示す方法により求めることができる。
ゴム質重合体(a)1gをトルエン100mlに投入し、室温で48時間静置する。その後、100メッシュの金網(質量をW1グラムとする)で濾過したトルエン不溶分と金網を、温度80℃で6時間真空乾燥して秤量(質量W2グラムとする)する。W1及びW2を、下記式(14)に代入して、ゲル含率を得る。なお、エチレン−プロピレン系ゴム質重合体においては、エチレン結晶を有するものがあり、このようなゴム質重合体を用いる場合は、80℃の温度で溶解しゲル含率をもとめる。
ゲル含率=〔〔W2(g)−W1(g)〕/1(g)〕×100 (14)
【0039】
上記ゲル含率は、ゴム質重合体(a)の製造時に、架橋性単量体の種類及びその使用量、分子量調節剤の種類及びその使用量、重合時間、重合温度、重合転化率等を適宜設定することにより調整できる。
【0040】
本発明で使用されるゴム質重合体(a)として好ましいものは、ポリブタジエン、ブタジエン・スチレン共重合体、オレフィン系ゴム、アクリル系ゴム、シリコーンゴム、共役ジエン系ブロック共重合体、水素添加共役ジエン系ブロック共重合体であり、更に好ましくは、オレフィン系ゴム、アクリル系ゴム、水素添加共役ジエン系ブロック共重合体であり、特に好ましいものは、アクリル系ゴム、エチレン・プロピレン共重合体、エチレン・プロピレン・非共役ジエン共重合体、及び水素添加共役ジエン系ブロック共重合体であり、最も好ましいものは、アクリル系ゴムのゲル含率が10質量%以下のもので、かつ体積平均粒子径が50〜500nmのものである。
【0041】
本発明のゴム質重合体(a)は、公知の方法である乳化重合、溶液重合、塊状重合、懸濁重合等の方法で得ることができる。これらの中で、アクリル系ゴムは乳化重合により製造されたものが好ましく、エチレン・プロピレン共重合体、エチレン・プロピレン・非共役ジエン共重合体、共役ジエン系ブロック共重合体及び水素添加共役ジエン系ブロック共重合体は溶液重合、ポリブタジエン及びブタジエン・スチレン共重合体は溶液重合で製造されたものが好ましい。
【0042】
本発明の(A)成分であるゴム強化スチレン系樹脂は、上記ゴム質重合体(a)の存在下に、芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合体可能な他のビニル単量体(b1)を重合して得られる。すなわち、(b1)成分は、芳香族ビニル化合物単独でもよいし、芳香族ビニル化合物及び芳香族ビニル化合物と共重合体可能な他のビニル単量体との混合物でもよい。
ここで使用される芳香族ビニル化合物としては、上記ゴム質重合体(a)で記載したものが全て使用できる。特に好ましくはスチレン、α−メチルスチレンであり、これらは1種単独でまたは2種以上を組み合わせて使用することができる。
芳香族ビニル化合物と共重合可能な他のビニル単量体としては、ビニルシアン化合物、(メタ)アクリル酸エステル化合物、マレイミド化合物、その他の各種官能基含有不飽和化合物などが挙げられる。その他の各種官能基含有不飽和化合物としては、不飽和酸化合物、エポキシ基含有不飽和化合物、水酸基含有不飽和化合物、酸無水物基含有不飽和化合物、オキサゾリン基含有不飽和化合物、置換または非置換のアミノ基含有不飽和化合物などが挙げられる。これらの他のビニル単量体は1種単独で、または2種以上を組み合わせて使用することができる。
【0043】
上記シアン化ビニル化合物としては、アクリロニトリル、メタクリロニトリル等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。シアン化ビニル化合物を使用することにより耐薬品性が付与される。シアン化ビニル化合物の使用量は、ビニル単量体全体量(b1)中の割合として、通常0〜60質量%、好ましくは5〜50質量%である。
【0044】
(メタ)アクリル酸エステル化合物としては、アクリル酸メチル、アクリル酸エチル、アクリル酸ブチル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。(メタ)アクリル酸エステル化合物を使用することにより表面硬度が向上する。(メタ)アクリル酸エステル化合物の使用量は、ビニル単量体全体量(b1)成分中の割合として、通常0〜80質量%である。
【0045】
上記のマレイミド化合物としては、マレイミド、N−フェニルマレイミド、N−シクロヘキシルマレイミド、N−シクロヘキシルマレイミド等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。また、マレイミド単位を導入するために、無水マレイン酸を共重合させた後にイミド化してもよい。マレイミド化合物を使用することにより耐熱性が付与される。マレイミド化合物の使用量は、ビニル単量体全体量(b1)成分中の割合として、通常1〜60質量%である。
【0046】
不飽和酸化合物としては、アクリル酸、メタクリル酸、エタクリル酸、マレイン酸、フマル酸、イタコン酸、クロトン酸、桂皮酸などが挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。
【0047】
エポキシ基含有不飽和化合物としては、グリシジルアクリレート、グリシジルメタクリレート、アリルグリシジルエーテル等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。
【0048】
水酸基含有不飽和化合物としては、3−ヒドロキシ−1−プロペン、4−ヒドロキシ−1−ブテン、シス−4−ヒドロキシ−2−ブテン、トランス−4−ヒドロキシ−2−ブテン、3−ヒドロキシ−3−メチル−1−プロペン、2−ヒドロキシエチルメタクリレート、2−ヒドロキシエチルアクリレート、N−(4−ヒドロキシフェニル)マレイミド等が挙げられ、これらは1種単独で、または2種以上を組み合わせて使用することができる。
【0049】
オキサゾリン基含有不飽和化合物としては、ビニルオキサゾリン等が挙げられ、これらは、1種単独で、あるいは2種以上を組み合わせて使用することができる。
【0050】
酸無水物基含有不飽和化合物としては、無水マレイン酸、無水イタコン酸、無水シトラコン酸などが挙げられ、これらは1種単独で、あるいは2種以上を組み合わせて使用することができる。
【0051】
置換または非置換のアミノ基含有不飽和化合物としては、アクリル酸アミノエチル、アクリル酸プロピルアミノエチル、メタクリル酸ジメチルアミノエチル、メタクリル酸フェニルアミノエチル、N−ビニルジエチルアミン、N−アセチルビニルアミン、アクリルアミン、N−メチルアクリルアミン、アクリルアミド、N−メチルアクリルアミド、p−アミノスチレン等があり、これらは1種単独で、あるいは2種以上を組み合わせて使用することができる。
【0052】
上記その他の各種官能基含有不飽和化合物を使用した場合、ゴム強化スチレン系樹脂(A)、スチレン系樹脂(B)と芳香族ポリカーボネート樹脂(C)とをブレンドした際、両者の相溶性が向上する場合がある。上記その他の各種官能基含有不飽和化合物の使用量は、(A)成分と(B)成分の合計中に対して、当該官能基含有不飽和化合物の合計量として、通常0.1〜20質量%、好ましくは0.1〜10質量%である。
【0053】
ビニル単量体全体量(b1)中の芳香族ビニル化合物以外の単量体の使用量は、(b1)成分の合計を100質量%とした場合、通常80質量%以下、好ましくは60質量%以下、更に好ましくは50質量%以下である。
【0054】
ビニル単量体(b1)を構成する単量体のより好ましい組み合わせは、スチレン単独、スチレン/アクリロニトリル、スチレン/メタクリル酸メチル、スチレン/アクリロニトリル/メタクリル酸メチル、スチレン/アクリロニトリル/グリシジルメタクリレート、スチレン/アクリロニトリル/2−ヒドロキシエチルメタクリレート、スチレン/アクリロニトリル/(メタ)アクリル酸、スチレン/N−フェニルマレイミド、スチレン/アクリロニトリル/N−フェニルマレイミド、スチレン/メタクリル酸メチル/シクロヘキシルマレミド等であり、更に好ましくは、スチレン単独、スチレン/アクリロニトリル=65/45〜90/10(質量比)、スチレン/メタクリル酸メチル=80/20〜20/80(質量比)、スチレン/アクリロニトリル/メタクリル酸メチルの組み合わせで、スチレン量が20〜80質量%、アクリロニトリル及びメタクリル酸メチルの合計が20〜80質量%の範囲で任意のものである。
【0055】
本発明で使用されるゴム強化スチレン系樹脂(A)は、公知の重合法、例えば乳化重合、塊状重合、溶液重合、懸濁重合およびこれらを組み合わせた重合法で製造することができる。上記重合法は、ゴム質重合体(a)が乳化重合で得られたものは(A)成分の製造においては同じく乳化重合で製造することが、更にゴム質重合体(a)が溶液重合で得られたものである場合は、(A)成分は塊状重合、溶液重合及び懸濁重合で製造することが一般的で好ましい。ただし、溶液重合で製造されたゴム質重合体(a)であっても、本ゴム質重合体(a)を公知の方法で乳化させれば、乳化重合で(A)成分を製造することができるし、また、乳化重合で製造したゴム質重合体(a)であっても、凝固し単離した後、塊状重合、溶液重合及び懸濁重合で本発明の(A)成分を製造することができる。
【0056】
乳化重合で製造する場合、重合開始剤、連鎖移動剤、乳化剤などが使用されるが、これらは公知のものが全て使用できる。
重合開始剤としては、クメンハイドロパーオキサイド、p−メンタンハイドロパーオキサイド、ジイソプロピルベンゼンハイドロパーオキサイド、テトラメチルブチルハイドロパーオキサイド、tert−ブチルハイドロパーオキサイド、過硫酸カリウム、アゾビスイソブチロニトリル等が挙げられる。また、重合開始助剤として、各種還元剤、含糖ピロリン酸鉄処方、スルホキシレート処方等のレドックス系を使用することが好ましい。
【0057】
連鎖移動剤としては、オクチルメルカプタン、n−ドデシルメルカプタン、t−ドデシルメルカプタン、n−ヘキシルメルカプタン、ターピノーレン類などが挙げられる。
乳化剤としては、ドデシルベンゼンスルホン酸ナトリウム等のアルキルベンゼンスルホン酸塩、ラウリル硫酸ナトリウム等の脂肪族スルホン酸塩、ラウリル酸カリウム、ステアリン酸カリウム、オレイン酸カリウム、パルミチン酸カリウム等の高級脂肪酸塩、ロジン酸カリウム等のロジン酸塩などを使用することができる。
【0058】
なお、乳化重合において、ゴム質重合体(a)及びビニル系単量体の使用方法は、ゴム質重合体(a)全量の存在下にビニル系単量体を一括添加して重合してもよく、分割もしくは連続添加して重合してもよい。また、ゴム質重合体(a)の一部を重合途中で添加してもよい。
【0059】
乳化重合後、得られたラテックスは、通常、凝固剤により凝固させられる。その後、水洗、乾燥することにより、(A)成分の粉末を得る。この際、乳化重合で得た2種以上の(A)成分のラテックスを適宜ブレンドした後、凝固してもよく、また、更に(B)成分のラテックスを適宜ブレンドした後、凝固してもよい。凝固剤としては、塩化カルシウム、硫酸マグネシウム、塩化マグネシウム等の無機塩、硫酸、酢酸、クエン酸、リンゴ酸などの酸を使用することができる。また、ラテックスを噴霧乾燥することにより(A)成分の粉末を得ることもできる。
【0060】
溶液重合により本発明の(A)成分を製造する場合に使用することのできる溶剤は、通常のラジカル重合で使用される不活性重合溶媒であり、例えば、エチルベンゼン、トルエン等の芳香族炭化水素、メチルエチルケトン、アセトン等のケトン類、アセトニトリル、ジメチルホルムアミド、N−メチルピロリドン等が挙げられる。
【0061】
重合温度は、通常80〜140℃、好ましくは85〜120℃の範囲である。重合に際し、重合開始剤を使用してもよいし、重合開始剤を使用せずに、熱重合で重合してもよい。
重合開始剤としては、ケトンパーオキサイド、ジアルキルパーオキサイド、ジアシルパーオキサイド、パーオキシエステル、ハイドロパーオキサイド、アゾビスイソブチロニトリル、ベンゾイルパーオキサイド等の有機過酸化物などが好適に使用される。また、連鎖移動剤を使用する場合、例えば、メルカプタン類、ターピンーレン類、α−メチルスチレンダイマー等を使用することができる。
【0062】
また、塊状重合、懸濁重合で製造する場合、溶液重合において説明した重合開始剤、連鎖移動剤などを使用することができる。上記各重合法によって得た本発明の(A)成分中の残存する単量体量は、通常10,000ppm以下、好ましくは5,000ppm以下である。
【0063】
また、ゴム質重合体(a)の存在下にビニル系単量体を重合して得られる重合体成分には、上記のビニル系単量体がゴム質重合体(a)にグラフト共重合した共重合体とゴム質重合体(a)にグラフトしていない未グラフト成分〔上記ビニル系単量体の(共)重合体〕が含まれる。
【0064】
上記のゴム強化スチレン系樹脂(A)のグラフト率は、通常5〜100質量%、好ましくは10〜90質量%、更に好ましくは15〜85質量%、特に好ましくは20〜80質量%にコントロールすることが好ましい。グラフト率は、重合開始剤の種類、使用量、連鎖移動剤の種類、使用量、重合方法、重合時の単量体とゴム質重合体(a)の接触時間、ゴム質重合体種、重合温度等の各種要因で変わるが、一般的にはグラフト率を上げる方向で(A)成分から熱シクロヘキサンに溶解する成分が少なくなるが、当該溶解成分がなくなることにより本発明の組成物の発泡性が悪くなる。
なお、グラフト率は以下の式(15)により求めることができる。
グラフト率(質量%)={(T−S)/S}×100 (15)
【0065】
上記式(15)中、Tはゴム強化スチレン系樹脂(A)1gをアセトン20ml(アクリル系ゴムの場合はアセトニトリル)に投入し、振とう機により2時間振とうした後、遠心分離機(回転数;23,000rpm)で60分間遠心分離し、不溶分と可溶分とを分離して得られる不溶分の質量(g)であり、Sはゴム強化スチレン系樹脂(A)1gに含まれるゴム質重合体(a)の質量(g)である。
なお、ビニル単量体として芳香族ビニル化合物のみを用いた場合は、アセトンの代わりにメチルエチルケトンを用いて測定する。
【0066】
また、本発明で使用されるゴム強化スチレン系樹脂(A)のアセトン可溶分(アクリル系ゴムの場合はアセトニトリル)の極限粘度〔η〕(溶媒としてメチルエチルケトンを使用し、30℃で測定)は、通常0.15〜1.2dl/g、好ましくは0.2〜1.0dl/g、更に好ましくは0.2〜0.8dl/gである。本発明で使用するゴム強化スチレン系樹脂(A)中に分散するグラフト化ゴム質重合体粒子の平均粒子径は、通常50〜3,000nm、好ましくは40〜2,5000nm、特に好ましくは50〜2,000nmである。ゴム粒子径が50nm未満では耐衝撃性が劣る傾向にあり、3,000nmを超えると成形品表面外観が劣る傾向にある。また、使用するゴム質重合体(a)とビニル系単量体の共重合体の屈折率を実質的に合わせること及び/または分散するゴム質重合体(a)の粒子径を実質的に可視光の波長以下(通常1,500nm以下)にすることで透明性を有する(A)成分を得ることができるが、これらの透明性樹脂も本発明の(A)成分として用いることができる。
【0067】
本発明の組成物に用いられる(A)成分は、発泡性の発現のためには、下記条件で示した熱シクロヘキサン溶解量が、使用したゴム質重合体(a)を基準として、1〜99質量%であることが必要であり、好ましくは2質量%以上、更に好ましくは4質量%以上、特に好ましくは5〜80質量%である。なお、ここで溶解してくる成分の主成分は、ゴム質重合体(a)である。
また、特にゴム質重合体(a)がアクリル系ゴムの場合、熱シクロヘキサン溶解量は、好ましくは1〜40質量%、さらに好ましくは2〜30質量%、特に好ましくは3〜20質量%である。ゴム質重合体(a)がオレフィン系ゴムの場合、好ましくは5〜60質量%、さらに好ましくは10〜50質量%、特に好ましくは20〜40質量%である。
【0068】
熱シクロヘキサン溶解量は、以下に示す方法により求めることができる。すなわち、ゴム強化スチレン系樹脂(A)〔ここで(A)成分中のゴム量は、W1グラム〕を、ソックスレー抽出器を用いて、常圧下で、シクロヘキサンを8時間還流させる。シクロヘキサン溶液を乾固し、抽出物の重量を測定し(W2グラム)、下記式(16)で、熱シクロヘキサン溶解量を算出する。
【0069】
熱シクロヘキサン溶解量(%)=W2/W1×100 (16)
【0070】
本発明は、本発明で用いるゴム変性スチレン系樹脂(A)の熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準として1質量%以上である場合、具体的には成分(A)中のゴム質重合体(a)が熱シクロヘキサンに溶解すること、即ちゴム質重合体(a)が未架橋、またはルーズな架橋状態にある場合に、樹脂の発泡性が向上するというものである。
【0071】
ゴム質重合体(a)の含有量は、(A)〜(C)成分の合計量を100質量%として、3〜50質量%、好ましくは3〜40質量%、さらに好ましくは5〜35質量%である。(a)成分が、この範囲にあると、発泡性、発泡成形体の外観、機械的特性に優れる。
【0072】
(2)スチレン系樹脂(B)
本発明の(B)成分は、芳香族ビニル化合物、又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b2)を重合してなるスチレン系樹脂である。すなわち、(b2)成分は、芳香族ビニル化合物単独でもよいし、芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体との混合物でもよい。ここで使用される芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体としては、上記(A)成分で記載したものが全て使用できる。また、ビニル単量体(b2)は、上記ビニル単量体(b1)と同一であってもよいし、異なっていてもよい。
【0073】
好ましい(B)成分としては、スチレンの単独重合体、スチレン・アクリロニトリル共重合体、スチレン・メタクリル酸メチル共重合体、スチレン・アクリロニトリル・メタクリル酸メチル共重合体、スチレン・マレイミド化合物共重合体、スチレン・アクリロニトリル・マレイミド化合物共重合体及びこれらと上記官能基含有不飽和化合物との共重合体である。
【0074】
本発明の(B)成分は、上記した(A)成分の製造法で記載した公知の重合法である乳化重合、塊状重合、溶液重合、懸濁重合及びこれらを組み合わせた方法で製造することができる。
【0075】
(3)芳香族ポリカーボネート樹脂(C)
本発明で使用される芳香族ポリカーボネート樹脂(C)は、ジヒドロキシアリール化合物とホスゲンとの界面重縮合法、ジヒドロキシアリール化合物とジフェニルカーボネート等のカーボネート化合物とのエステル交換反応(溶融重縮合)によって得られるもの等、公知の重合法によって得られるものが全て使用できる。
【0076】
上記ジヒドロキシアリール化合物としては、ビス(4−ヒドロキシフェニル)メタン、1,1−ビス(4−ヒドロキシフェニル)エタン、2,2−ビス(4−ヒドロキシフェニル)ブタン、2,2−ビス(4−ヒドロキシフェニル)オクタン、ビス(4−ヒドロキシフェニル)プロパン、2,2−ビス(4−ヒドロキシ−3−t−ブチルフェニル)プロパン、2,2−ビス(4−ヒドロキシ−3−t−ブチルフェニル)プロパン、1,1−ビス(4−ヒドロキシフェニル)シクロペンタン、1,1−ビス(4−ヒドロキシフェニル)シクロヘキサン、4,4’−ジヒドロキシフェニルエーテル、4、4’−ジヒドロキシフェニルスルフィド、4,4’−ジヒドロキシフェニルスルホン、4,4’−ジヒドロキシ−3,3’−ジメチルジフェニルスルホン、ヒドロキノン、レゾルシン等が挙げられる。更に、ヒドロキシアリールオキシ末端かされたポリオルガノシロキサン(例えば、米国特許第3,419,634号明細書参照)等がある。これらは1種単独で、または2種以上を組み合わせて使用することができる。これらの中では、2,2−ビス(4−ヒドロキシフェニルプロパン(ビスフェノールA)が好ましい。
【0077】
ポリカーボネート樹脂(C)の粘度平均分子量は、好ましくは12,000〜40,000、さらに好ましくは15,000〜35,000、特に好ましくは18,000〜30,000である。分子量が高い方が得られる発泡成形体の機械的強度が高くなるが、流動性が低下し、均一なセルが得られず、発泡成形体の外観が低下する傾向となる。また、分子量の異なる2種以上のポリカーボネートを用いることもできる。
【0078】
上記の芳香族ポリカーボネートの粘度平均分子量は、通常、塩化メチレンを溶媒として、20℃、濃度〔0.7g/100ml(塩化メチレン)〕で測定した比粘度(ηsp)を以下の式(17)に挿入して算出できる。
【0079】
粘度平均分子量=(〔η〕×8130)1.205 (17)
ここで、〔η〕=〔(ηsp×1.12+1)1/2−1〕/0.56Cである。なお、Cは濃度を示す。
【0080】
界面重縮合で得られるポリカーボネート系樹脂は、各種の塩素化合物を含む場合があるが、この塩素化合物は、本発明の組成物の耐久性に悪影響する場合がある。このことから、塩素化合物含有量は、塩素原子として、通常300ppm以下、好ましくは100ppm以下とされる。
【0081】
(4)成分(A)〜(C)の配合割合(上記樹脂成分の配合割合)
本発明のゴム強化スチレン系樹脂(A)の配合量は、(A)〜(C)成分の合計を100質量%としたとき、5〜90質量%、好ましくは6〜60質量%、更に好ましくは7〜40質量%である。ゴム強化スチレン系樹脂(A)が5質量%未満及び90質量%超過では発泡性が劣る。
【0082】
本発明のスチレン系樹脂(B)の配合量は、(A)〜(C)成分の合計を100質量%としたとき、0〜85質量%、好ましくは5〜50質量%、更に好ましくは10〜30質量%である。スチレン系樹脂(B)が85質量%超過では発泡性が劣り、優れた外観を有する発泡成形体を得ることが困難となる。(B)成分は、(A)〜(C)成分中のゴム質重合体(a)の量を3〜50質量%に調整すること、(共)重合体種を変えて本発明の組成物に機能を付与すること、及び、他の熱可塑性重合体との相溶性を向上させることを目的として配合される。
【0083】
また、本発明に用いられる熱可塑性樹脂組成物中の芳香族ポリカーボネート樹脂(C)の含有量は、10〜90質量%、好ましくは30〜75質量%、さらに好ましくは50〜70質量%である。芳香族ポリカーボネート樹脂(C)の含有量が10質量%未満では、均一なセル径を有する発泡成形体を得ることが困難となり、一方、芳香族ポリカーボネート樹脂(C)の含有量が95質量%を超えると、優れた外観を有する発泡成形体を得ることが困難となる。
【0084】
(5)化学発泡剤(D)
使用する化学発泡剤(D)として、特に限定はないが好ましいものとしては、例えば分解されて炭酸ガスを発生する熱分解型無機発泡剤(炭酸水素ナトリウム、炭酸アンモニウム、炭酸水素アンモニウムなど)、分解されて窒素ガスを発生する熱分解型発泡剤(アゾジカルボンアミド(ADCA)、N,N’−ジニトロソペンタメチレンテトラミン(DPT)、4,4’−オキシビス(ベンゼンスルホニルヒドラジド)(OBSH)、アゾビスイソブチロニトリル、p−トルエンスルホニルヒドラジド、5−フェニルテトラゾールなど公知の熱分解型発泡性化合物が挙げられる。
【0085】
化学発泡剤(D)の含有量は、所望の発泡倍率が得られるように、用いる化学発泡剤や樹脂の種類に応じて適宜選択されるものであるが、上記成分(A)〜(C)の合計100質量部に対して化学発泡剤0.1〜5質量部であり、好ましくは0.2〜4質量部、更に好ましくは0.3〜3質量部である。化学発泡剤の含有量が0.1質量部未満である場合には、化学発泡剤の含有量が少なくて、発泡の各セル径を均一にすることが困難になる。一方、化学発泡剤の含有量が5質量部を超える場合には、化学発泡剤の含有量が多くて、化学発泡剤の残渣による金型汚染が生じ、外観に優れた発泡成形体を得ることが困難になる。
【0086】
溶融状態可塑性樹脂への発泡剤の配合方法としては、熱可塑性樹脂組成物のペレットと発泡剤マスターバッチペレットをドライブレンドした後、成形機に供給し、成形機内で樹脂を可塑化させ、金型内で発泡させる方法が好ましく用いられる。また、物理発泡剤を併用してもよい。物理発泡剤としては、具体的には、プロパン、ブタン、水、炭酸ガス等が挙げられる。
【0087】
(6)タルク(E)
本発明で用いられるタルク(E)は特に制限はないが、一般的には含水珪酸マグネシウム塩の粘土鉱物の一種で、その組成は(MgO)x(SiO2)y・zH2Oである(x、y、zは正値)。また、タルク中のMgの一部がCa2+等の2価の金属イオンに置換されてもよい。タルクの粒径は、特に制限されないが、レーザー散乱法による平均粒子径として、通常0.1〜50μm、好ましくは0.3〜25μm、更に好ましくは0.5〜20μmである。タルクの平均粒子径が0.1μm未満では、熱可塑性樹脂組成物中でのタルクの分散性が不十分となり、発泡性が劣り、優れた外観を有する発泡成形体を得ることが困難となる。一方、タルクの平均粒径が50μmを超えると、発泡性が劣り、優れた外観を有する発泡成形体を得ることが困難となる。
【0088】
本発明におけるタルク(E)の含有量は、上記成分(A)〜(C)の合計100質量部に対して、0.5〜18質量部、好ましくは1〜15質量部、更に好ましくは2〜12質量部である。タルクの含有量が0.5質量部未満の場合及びタルクの含有量が18質量部を超える場合には、発泡性が劣り、優れた外観を有する発泡成形体を得ることが困難となる。
【0089】
(7)繊維状充填材(F)
本発明における繊維状充填材(F)は、繊維状であれば特に限定はないが、例えばガラス繊維、炭素繊維、アラミド繊維等の有機繊維、セラミック系ウィスカー等の無機繊維、金属繊維等が挙げられる。このうち、発泡成形体の剛性向上の観点から、ガラス繊維を用いることが好ましい。
【0090】
本発明に用いられるガラス繊維の組成は、珪酸塩ガラス、ほう酸珪酸ガラス、燐酸塩ガラス等が挙げられる。またガラスの種類としては、Eガラス、Cガラス、Aガラス、Sガラス、Mガラス、ARガラス、Lガラス等が挙げられるが、Eガラス、Cガラスが好ましい。本発明に用いられるガラス繊維には、適当なサイジング剤を用いても構わない。サイジング剤としては、表面処理剤、フィルム形成剤、潤滑剤、界面活性剤、帯電防止剤等が挙げられる。表面処理剤としては、アミン系、シラン系、エポキシ系等のカップリング剤が挙げられる。本発明に用いられるガラス繊維は、ロービングを用いた長繊維タイプでもよく、チョップドストランドであってもよい。
【0091】
本発明に用いられるガラス繊維の直径は特に指定はないが、通常φ1〜500μm、好ましくはφ5〜200μm、更に好ましくはφ5〜100μmである。ガラス繊維の直径がφ1μm未満では、機械的強度が不十分となるおそれがある。一方、ガラス繊維の直径がφ500μmを超えると、発泡性、発泡成形体の外観が悪化するおそれがある。
【0092】
また、ガラス繊維の長さは特に指定はないが、通常0.01〜10mm、好ましくは0.05〜5mmである。ガラス繊維の長さが0.01mm未満では、発泡性が劣り、機械的強度が不十分となるおそれがある。一方、ガラス繊維の長さが10mmを超えると、発泡性が不十分となるおそれがある。
ガラス繊維の長さ(μm)/直径(μm)は、通常5〜1000、好ましくは8〜500である。ガラス繊維の長さ/直径が5未満では、ガラス繊維の両端が発泡起点とならず、発泡性が不十分となるおそれがある。一方、ガラス繊維の長さ/直径が1000を超えると、ガラス繊維の数の減少により、発泡起点の数が減少し、発泡性が不十分となるおそれがある。
また、成形した発泡成形体中に分散しているガラス繊維の残存平均繊維長さは、0.01〜1mmが好ましく、0.02〜0.8mmがより好ましく、0.03〜0.7mmが更に好ましい。ガラス繊維の残存平均繊維長さが上記範囲にあると、発泡性、発泡成形体の外観、機械的特性が十分になる。
【0093】
炭素繊維としては、PAN系、ピッチ系等が用いられる。また上記炭素繊維は、カーボンナノチューブなどの炭素繊維構造体であってもよい。
本発明に用いられる炭素繊維の直径は特に指定はないが、0.5μm〜200μmが好ましく、1μm〜50μmがより好ましく、5μm〜50μmがさらに好ましい。繊維の長さは特に指定はないが、発泡成形体中で20μm以上であることが好ましい。
【0094】
本発明における繊維状充填材(F)の配合量は、上記成分(A)〜(C)の合計100質量部に対して、0.5〜25質量部、好ましくは0.5〜18質量部、更に好ましくは2〜18質量部、特に好ましくは4〜18質量部である。繊維状充填材の含有量が0.5質量部未満の場合及び25質量部を超える場合には、発泡性、発泡成形体の外観、金型汚染性、機械的強度が悪化するおそれがある。
【0095】
本発明に用いられる熱可塑性樹脂組成物には、熱老化防止剤を配合することができる。熱老化防止剤としては、フェノール系、リン系、硫黄系などが挙げられ、好ましくはフェノール系、リン系および硫黄系の3種混合系である。熱老化防止剤として、この3種混合系を用いると、長時間、高温下に曝された時の、引張り伸び率を保持するという効果が得られる。
【0096】
熱老化防止剤のうち、フェノール系としては、2,6−ジ−t−ブチルフェノール誘導体、2−メチル−6−t−ブチルフェノール誘導体、オクタデシル3(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート、4,4’−ブチリデン−ビス(6−t−ブチル−m−クレゾール)、ペンタエリスリチル・テトラキス〔3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート〕、2〔1−(2−ヒドロキシ−3,5−ジ−t−ペンチルフェニル)−エチル〕−4,6−ジ−t−ペンチルフェニルアクリレート、2−t−ブチル−6(3−t−ブチル−2−ヒドロキシ−5−メチルベンジル)−4−メチルフェニルアクリレートなどが挙げられる。
【0097】
リン系としては、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト、サイクリックネオペンタンテトライルビス(2,4−ジ−t−ブチルフェニルホスファイト)、ジステアリルペンタエリスリトールジホスファイト、リン酸2水素ナトリウム、リン酸1水素2ナトリウムなどが挙げられる。
硫黄系としては、3,3’−チオビスプロピオン酸ジドデシルエステル、3,3’−チオビスプロピオン酸ジオクタデシルエステル、ペンタエリスリトール−テトラキス(3−ラウリルプロピオネート)、ジラウリル3,3’−チオジプロピオネートなどが挙げられる。
【0098】
本発明に用いられる熱可塑性樹脂組成物中の熱老化防止剤の割合は、0〜5質量%、好ましくは0〜3質量%である。本発明の熱可塑性樹脂組成物において、ポリカーボネート樹脂(A)以外のゴム強化ビニル系樹脂(B)成分は、熱老化防止剤を添加することで、熱老化特性が改良されるが、芳香族ポリカーボネート樹脂(C)は、熱老化防止剤が加水分解を促進する触媒として働くことがあり、熱老化防止剤を入れない方が劣化を抑制する傾向もある。これらの相反する効果を鑑みて、5質量%を上限として上記熱老化防止剤を添加すれば、最適な熱老化防止効果が得られる。
【0099】
また、本発明に用いられる発泡成形用熱可塑性樹脂組成物には、公知の耐候剤、滑剤、着色剤、帯電防止剤、シリコーンオイル、タルク(E)及び繊維状充填材(F)を除く無機フィラーなどの添加剤を配合することができる。このうち、耐候剤としては、ベンゾトリアゾール系、トリアジン系、ベンゾフェノン系などが好ましい。滑剤としては、エチレンビスステアリルアミド、硬化ヒマシ油などが好ましい。着色剤としては、カーボンブラック、ベンガラなどが挙げられる。帯電防止剤としては、ポリエーテル、アルキル基を有するスルホン酸塩などが挙げられる。
【0100】
本発明の発泡成形用熱可塑性樹脂組成物には、本発明の目的とする性能を損なわない範囲で、更に他の熱可塑性樹脂を配合することができる。熱可塑性樹脂としては、ゴム強化スチレン系樹脂〔ただし、(A)成分は除く〕、ポリオレフィン系樹脂、塩化ビニル系樹脂、アクリル系樹脂、ポリエステル系樹脂、ポリアミド系樹脂、ポリアセタール系樹脂、ポリフェニレンエーテル系樹脂、ポリアリーレンスルフィド系樹脂等があり、これらの熱可塑性樹脂は1種単独で、または2種以上を組み合わせて使用することができる。
【0101】
また、第1の発明において、上記ゴム質重合体(a)は、オレフィン系ゴム、アクリル系ゴム及び水素添加共役ジエン系ブロック共重合体からなる群から選ばれた少なくとも1種であることが好ましい(請求項2)。
この場合には、ゴム質重合体が適切であり、微細で均一な発泡セルを有する発泡成形体を安定して得ることができる。
【0102】
また、上記アクリル系ゴムは、ゲル含率が70質量%以下であり、体積平均粒子径が50〜300nmであることが好ましい(請求項3)。
この場合には、ゴム質重合体が適切であり、微細で均一な発泡セルを有する発泡成形体を安定して得ることができる。
【0103】
また、上記発泡成形用熱可塑性樹脂組成物は、コアバック型射出発泡成形に用いることが好ましい(請求項4)。
この場合には、微細で均一な発泡セルを有する発泡成形体を安定して得ることができる。
【0104】
2.熱可塑性樹脂組成物の製造
本発明に用いられる熱可塑性樹脂組成物は、各種押出機、バンバリーミキサー、ニーダ
ー、ロールなどを用いて混練することができる。例えば、(A)、(B)、(C)、(D)、(E)及び(F)成分、並びに必要に応じてその他の添加剤を混練することにより熱可塑性樹脂組成物のペレットを得ることができる。具体的には、2軸押出機によって(A)〜(F)成分を溶融させる方法などが挙げられる。混練温度は、熱可塑性樹脂組成物の配合によって適宜選択されるが、本発明においては、通常220〜260℃である。
【0105】
3.発泡成形体(発泡成形品)の成形方法
本発明の発泡成形用熱可塑性樹脂組成物を用いて発泡成形体を成型する方法としては、射出発泡成形、押出発泡成形等公知の方法を用いることができる。
射出発泡成形方法では、上記射出発泡成形用熱可塑性樹脂組成物を、射出成形機の金型内に形成されたキャビティ空間に射出し、直ちに、あるいは所定時間が経過した後、可動型、あるいは可動型に内設された可動コアを所定の速度で所定位置まで後退させ、キャビティ空間を拡大することにより発泡させる、所謂、コアバック方式の射出成形法によって発泡成形体を得ることができる。金型の温度は、通常、射出される際の熱可塑性樹脂組成物の温度より相当に低いため、キャビティの表面に接して形成される発泡成形体の表面には、ほとんど発泡していない緻密なスキン層が形成される。
【0106】
発泡成形体は、樹脂製等の基材の表面に接するように一体に形成することもできる。このような積層品は、キャビティ空間に予め基材を配置しておき、その表面に熱可塑性樹脂組成物を射出することにより形成することができる。また、2本の射出ユニットが搭載された射出成形機を使用し、先ず、基材となる樹脂等を射出して基材を形成し、その後、可動型に内設された可動コアを後退させて熱可塑性樹脂組成物を射出するためのキャビティ空間を形成し、次いで、熱可塑性樹脂組成物を射出し、その後、可動コアを更に後退させてキャビティ空間を拡大し、発泡させて、基材の表面に発泡成形体が積層された積層品とすることもできる。
【0107】
本発明の射出発泡成形方法では、可動型の後退速度、あるいは可動型に内設して設けられた可動コアの後退速度、即ち、上記「型開速度」は0.05〜20mm/秒である。この型開速度は、好ましくは0.1〜10mm/秒である。このような型開速度とすることにより、平均セル径が50〜500μmと適度に微細である均質な発泡成形体とすることができる。
【0108】
型開速度が0.05mm/秒未満であると、冷却が進んで発泡不足が発生し、発泡成形体の表面に凹凸が生じる。一方、型開速度が20mm/秒を越えると、セル径が大きく、また不均一な発泡成形体となる。
更に、射出される熱可塑性樹脂組成物の温度は、好ましくは200〜280℃、特に好ましくは220〜270℃である。この温度が200℃未満であると、熱可塑性樹脂組成物の流動性が不十分となり、特に、末端部では充填不良が発生することがある。一方、280℃を越えると、熱可塑性樹脂組成物の組成によっては熱劣化等が懸念される。
【0109】
また、金型温度は、好ましくは20〜80℃、特に好ましくは30〜70℃である。この温度が20℃未満であると、金型内表面と接触した熱可塑性樹脂組成物が急激に冷却され、均質な発泡成形体とすることができず、末端部で充填不良が発生することもある。一方、80℃を越えると、発泡成形体のキャビティの表面に接して形成された部分に均質なスキン層が形成されないことがあり、好ましくない。
【0110】
また、熱可塑性樹脂組成物を射出してから可動型、あるいは可動型に内設された可動コアの後退を開始するまでの時間(金型後退遅延時間)は、型開速度にもよるが、3秒以下とすることが好ましく、射出完了後、直ちに後退を開始してもよい。この金型後退遅延時間は、好ましくは0.1〜2.5秒、特に好ましくは0.1〜1.5秒である。金型後退遅延時間が3秒を越えると、冷却が進んで均質な発泡成形体とすることができない場合がある。
【0111】
金型の後退量は所定の発泡倍率により設定すればよく、限定されないが、特に、機器の機枠等では、金型内キャビティ空間に充填された素材の初期肉厚に対して発泡成形体の最終肉厚が1.1〜3.0倍となるように金型を後退させる、即ち、型開きすることが好ましい。この肉厚の比を発泡倍率とすれば、発泡倍率は、好ましくは1.1〜3倍、更に好ましくは1.5〜2倍であり、発泡成形体の肉厚が5〜30mm、特に5〜25mmである製品が多いことを考慮すれば、金型の後退量は、通常、2.5〜30mmである。
【0112】
なお、冷却時間は発泡成形体の寸法、あるいは冷却方法にもよるが、脱型時の発泡成形体の温度が40〜80℃程度にまで低下しておればよく、一般に30秒以上であればよく、大型の製品であっても100秒で十分である。
本発明の発泡成形体の成形方法において、射出充填時に、金型内にファブリックやフィルムをインサートしてもよい。
【0113】
4.発泡成形体
本発明の成形方法により得られた発泡成形体は、微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさが均一であり機械的性能に優れる発泡成形体である。具体的には、発泡セル径の平均径が好ましくは50〜500μm、より好ましくは70〜450μm、さらに好ましくは100〜400μmであり、発泡セル径が均一であり、粒径分布の狭いことが好ましい。特に発泡成形体発泡セルの大半のセル径が400μm以下の均一発泡成形体であることが好ましい。
本発明の成形方法により得られた発泡成形体は、さらに発泡倍率が1.01〜3.0倍、好ましくは1.1〜2.7倍、より好ましくは1.5〜2.5倍の所望の倍率にすることができる。
【0114】
本発明の成形方法により得られた発泡成形体の形状は、目的、用途などにより選択され、板状(シート状)、筒状、半筒状、棒状、線状、塊状等とすることができる。
本発明の発泡成形体は、表示板、コンクリートパネル、屋上断熱材、畳芯材、襖、システムキッチンの木材代替材、風呂蓋、テーブル板などの土木・建築関連資材、サイドモール、吸音材、バンパー、ドアハンドル、コンソールボックス、天井材、ピラー、センターロークラスタフィニッシュパネル、カウルサイドトリム、センターアウトレット、ドアライニング、アッシュトレイ、フットレスト、ステアリングコラムカバー、ロアインサート、ロアハンドルパネル、ホイルキャップ、スポイラーなどの車両用内外装関連資材、容器、トレー、通い箱等の日用雑貨用品、テレビ、ビデオ、エアコンのハウジング、パラボナアンテナ、エアコン室外機等の電気・電子部品、ビート板、プロテクターなどのスポーツ用品、壁、床、機枠、家具、化粧シート、間仕切り、ラティス、フェンス、雨樋、サイジングボード、カーポート等の住宅・事務所用内外装材、玩具・遊技機等の機枠、緩衝材、補強材、断熱材、芯材、代替合板等として用いることができる。
さらに、本発明の発泡成形体は、用途によっては、他の成形品、部材等と一体化させ、複合化させてなる物品として用いることができる。
【実施例】
【0115】
以下に、本発明の発泡成形用熱可塑性樹脂組成物及びこれを用いた発泡成形体にかかる実施例につき、図面を参照して説明する。
本例の発泡成形用熱可塑性樹脂組成物は、ゴム強化スチレン系樹脂(A)5〜90質量%、スチレン系樹脂(B)0〜85質量%、及び芳香族ポリカーボネート樹脂(C)10〜90質量%を含有し、成分(A)〜(C)の合計100質量部に対し、化学発泡剤(D)を0.1〜5質量部、タルク(E)を0.5〜18質量部、及び繊維状充填材(F)を0.5〜25質量部配合してなる。
【0116】
ゴム強化スチレン系樹脂(A)は、ゴム質重合体(a)の存在下に、芳香族ビニル化合物、又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b1)を重合してなり、かつ熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準(100質量%)として1〜99質量%である。また、成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合は、3〜50質量%である。
スチレン系樹脂(B)は、芳香族ビニル化合物、又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b2)を重合してなる。
【0117】
以下に、本例の発泡成形用熱可塑性樹脂組成物、これを用いた発泡成形体6及び発泡成形体6の成形方法につき、図1〜図4を参照して詳説する。
本例においては、上記組成の発泡成形用熱可塑性樹脂組成物を用いることによって、射出発泡成形を行う際に、微細な発泡セル構造を発現し、発泡成形体6の部位によらず発泡セルの大きさが均一であり、機械的性能にも優れ、かつ表面外観にも優れた発泡成形体6を生産効率よく成形することができる。
【0118】
本例においては、上記発泡成形用熱可塑性樹脂組成物を用いて発泡成形体6を成形するに当たり、次の製造装置1を用いる。
製造装置1は、図1〜図4に示すごとく、金型として、第1型部2と、第1型部2に対して相対的に可動する第2型部3とを備えている。そして、製造装置1は、第2型部3と第1型部2との間に形成したキャビティ41A内に、熱可塑性樹脂組成物を溶融させた溶融樹脂60を充填し、第1型部2と第2型部3とをキャビティ41Aの容積が拡大する離隔方向Rに相対的に可動させるよう構成してある。
【0119】
図1に示すごとく、第2型部3は、第1型部2に設けたキャビティ形成凹部21内に配置するキャビティ形成凸部31を設けてなる。第1型部2及び第2型部3においては、キャビティ形成凹部21において第1型部2と第2型部3との可動方向Dに平行に形成した内側面211と、キャビティ形成凸部31において可動方向Dに平行に形成した外側面311との間には、溶融樹脂60を充填するための充填用隙間42がキャビティ41Aと連通して形成されている。
本例の製造装置1は、溶融樹脂60をキャビティ41A内に充填すると共に充填用隙間42に充填した後、第1型部2と第2型部3とを離隔方向Rに相対的に可動させるよう構成してある。
【0120】
図1、図4に示すごとく、本例の第1型部2は、キャビティ41内に溶融樹脂60を充填するための樹脂注入口22を設けた固定型部である。本例の第2型部3は、第1型部2に対して離隔方向Rに可動する可動型部である。
また、本例の第2型部3は、第1型部2との間に形成するキャビティ41の容積を1.1〜2.0倍に拡大させるように、第1型部2に対して離隔方向Rへ後退するよう構成してある。そして、本例において発泡成形する発泡成形体6の発泡倍率は、1.1〜2.0倍である。
図2、図3に示すごとく、本例の製造装置1は、第1型部2に設けた樹脂注入口22に接続して、上記キャビティ41A内に溶融樹脂60を注入するための注入ノズル25を有している。本例の第2型部3は、油圧、空気圧、電力等によって動作する駆動源によって、第1型部2に対して進退する(可動方向Dに移動する)よう構成してある。
【0121】
本例においては、第1型部2及び第2型部3を用いて、略直方体形状であって、四角形状断面を有し、一方向Lに長い板形状の発泡成形体6を発泡成形する。
図4に示すごとく、本例の第2型部3は、キャビティ形成凸部31の先端面312が充填用隙間42の可動方向Dの端部まで移動するまで第1型部2に対して離隔方向Rに可動するよう構成してある。
第2型部3が第1型部2に対する原位置301にあるときには、キャビティ41に連通して、溶融樹脂60を充填するための充填用隙間42が形成される。この充填用隙間42は、溶融樹脂60を充填した際に、充填用隙間42の全体にスキン層61を形成することができる幅(隙間)に形成する。そして、図1、図2に示すごとく、第2型部3が離隔方向Rに可動する前の原位置301にあるときには、第2型部3と第1型部2との間には、容積が縮小したキャビティ41A及び充填用隙間42が形成されると共に、図4に示すごとく、第2型部3が離隔方向Rに可動した可動位置302にあるときには、第2型部3と第1型部2との間には、容積が拡大したキャビティ41Bが形成される。
【0122】
図1に示すごとく、本例の第1型部2は、キャビティ41における樹脂注入側の表面212(発泡成形体6の固定側表面を成形する底面)、及び樹脂注入側の表面に直交する全周の側面211(内側面211、発泡成形体6の全側面を成形する面)を形成するためのキャビティ形成凹部21を有している。本例の第2型部3は、キャビティ41における樹脂受け側の表面312(発泡成形体6の可動側表面を成形する先端面)、及び樹脂注入側の表面に直交する全周の側面311(外側面311)を形成するためのキャビティ形成凸部31を有している。
第1型部2のキャビティ形成凹部21における内側面211、及び第2型部3のキャビティ形成凸部31における外側面311は、可動方向Dに平行に形成してある。
【0123】
また、同図に示すごとく、本例の第1型部2においてキャビティ形成凹部21を形成する型壁部の開口先端部23の全周には、開口先端部23と第2型部3における外側面311との間を閉塞する閉塞型部5が設けてある。
そして、製造装置1は、第1型部2に対して第2型部3を離隔方向Rに可動させるときには、第2型部3における外側面311が閉塞型部5における内周面51と摺動することにより、充填用隙間42を形成した状態を維持して、キャビティ41Aの容積を拡大させるよう構成してある。
【0124】
また、図4に示すごとく、製造装置1は、第1型部2に対して第2型部3を離隔方向Rに相対的に可動させたときには、第2型部3における先端面312と閉塞型部5における内側端面52とが一致するよう構成してある。そして、上記可動工程において、第1型部2に対して第2型部3を離隔方向Rに可動させたときには、発泡成形体6の全周の表面にスキン層61を形成すると共に、スキン層61の内側部分に発泡層62を形成することができる。
【0125】
なお、成形する発泡成形体6の形状によっては、第2型部3における先端面312と閉塞型部5における内側端面52とが一致するまでは第2型部3を離隔方向Rに可動させずに、発泡成形体6からスキン層61による突出部が突出した形状を形成することもできる。
また、本例の充填用隙間42の幅(隙間)は適宜変更することができ、充填用隙間42に形成するスキン層61の厚みを適宜変更することができる。
【0126】
次に、上記製造装置1を用いて、発泡成形体6を成形する方法を詳説する。
本例の成形方法においては、充填工程として、図2、図3に示すごとく、第2型部3と第1型部2との間に形成したキャビティ41A内に、溶融樹脂60を充填してキャビティ41Aの表面に接触する溶融樹脂60の部分にスキン層61を形成し、可動工程として、図4に示すごとく、第1型部2と第2型部3とをキャビティ41Aの容積が拡大する離隔方向Rに相対的に可動させて、スキン層61に対する内側部分に溶融樹脂60を発泡させた発泡層62を形成する。
本例の発泡成形体6の成形方法は、溶融樹脂60を充填したキャビティ41Aの容積を拡大させて、溶融樹脂60を発泡させる方法であり、発泡成形する発泡成形体6の全周の表面に、溶融樹脂60がほとんど発泡せずに硬化したスキン層61を効果的に形成することができるものである。
【0127】
本例においては、まず、充填工程として、図2、図3に示すごとく、注入ノズル25に保持する溶融樹脂60を、第1型部2における樹脂注入口22から縮小した状態のキャビティ41A内に注入する。このとき、溶融樹脂60は、キャビティ41Aから充填用隙間42へと流入し、縮小した状態のキャビティ41及び充填用隙間42の全体に溶融樹脂60が充填される。そして、第1型部2のキャビティ形成凹部21の底面212及び内側面211と、第2型部3のキャビティ形成凸部31の先端面312及び外側面311に接触する溶融樹脂60の部分は、他の溶融樹脂60の部分(キャビティ41の内部(内側部分)における溶融樹脂60の部分)よりも早く冷却硬化して半硬化状態のスキン層61が形成される。そのため、キャビティ41における接触表面だけでなく、充填用隙間42における接触表面にも、溶融樹脂60が半硬化して未発泡のスキン層61が形成される。本例では、充填用隙間42の全体にスキン層61が形成される。
【0128】
次いで、可動工程として、図4に示すごとく、第1型部2に対して第2型部3を離隔方向Rに可動させる。このとき、充填用隙間42内に充填されて硬化した溶融樹脂60によるスキン層61に対して、第2型部3のキャビティ形成凸部31における外側面311が摺動し、このスキン層61の内側部分に溶融樹脂60が流入して発泡する。また、第1型部2のキャビティ形成凹部21における樹脂注入側の表面212、及び第2型部3のキャビティ形成凸部31における樹脂受け側の表面312に形成されたスキン層61の内側部分にも溶融樹脂60が流入して発泡する。
こうして、発泡成形体6の内部に溶融樹脂60が発泡した発泡層62を形成すると共に、発泡成形体6の全周の表面にスキン層61を形成することができる。そのため、全周の表面に安定してスキン層61を形成することができ、発泡成形した発泡成形体6の機械的強度を効果的に向上させることができる。
【0129】
それ故、本例の発泡成形体6の成形方法によれば、発泡成形体6の全周の表面にスキン層61を形成することができ、発泡成形体6の機械的強度を向上させることができる。
【0130】
また、本例においては、樹脂成形を行う所定の温度において、粘度が高い芳香族ポリカーボネート樹脂(C)の存在により、発泡によるセル径を均一にすることができ、発泡成形体6の外観を向上させることができる。また、樹脂成形を行う所定の温度において、芳香族ポリカーボネート樹脂(C)よりも粘度が低いゴム強化スチレン系樹脂(A)の存在により、発泡成形体6の表面に安定してスキン層61を形成することができる。
【0131】
本例の発泡成形用熱可塑性樹脂組成物は、上記ゴム強化スチレン系樹脂(A)、スチレン系樹脂(B)、及び芳香族ポリカーボネート樹脂(C)、化学発泡剤(D)、タルク(E)及び繊維状充填材(F)を含有し、ゴム強化スチレン系樹脂(A)を組成するゴム質重合体(a)の割合を適切にしたことによって、射出発泡成形を行う際に、微細な発泡セル構造を発現し、発泡成形体6の部位によらず発泡セルの大きさが均一であり、かつ機械的性能にも優れる発泡成形体6を成形することができる。
【0132】
また、本例の発泡成形用熱可塑性樹脂組成物は、タルク(E)及び繊維状充填材(F)を含有することにより、発泡性、機械的性能に一層優れ、表面外観に優れる発泡成形体6を成形することができる。この理由は、次のように考えられる。
すなわち、発泡成形する際には、タルク(E)及び繊維状充填材(F)が発泡起点になり、化学発泡剤(D)による発泡を促進することができる。このとき、タルク(E)による発泡起点と繊維状充填材(F)の両端による発泡起点とが混在することにより、より効果的に微細かつ均一な発泡セルが得られると考えられる。
【0133】
また、発泡成形用熱可塑性樹脂組成物が繊維状充填材(F)を含有することにより、機械的性能として特に剛性が優れた発泡成形体6を得ることができる。さらに、繊維状充填材(F)を含有することにより、発泡成形を行う金型の成形表面(第1型部2及び第2型部3におけるキャビティ41及び充填用隙間42の表面)に汚染が発生し難くなり、表面外観に優れた発泡成形体6を生産効率よく成形することができることがわかった。この理由は、次のように考えられる。
すなわち、繊維状充填材(F)が含有されていることにより、金型の成形表面に接触する溶融状態の発泡成形用熱可塑性樹脂組成物の表面が凹凸状に粗くなり、化学発泡剤(D)の発泡に伴って発生する揮発成分が、金型の成形表面と溶融状態の発泡成形用熱可塑性樹脂組成物の表面との間から外部へ抜け易くなったためであると考えられる。
そのため、金型の成形表面に汚染が発生し難くなり、その清掃を行う頻度を減少させることができ、生産効率よく発泡成形体6を成形することができる。
【0134】
それ故、本例の発泡成形用熱可塑性樹脂組成物によれば、射出発泡成形において、微細な発泡セル構造を発現し、発泡成形体6の部位によらず発泡セルの大きさが均一であり、機械的性能に優れ、かつ表面外観にも優れる発泡成形体6を生産効率よく成形することができる。
【0135】
(確認試験)
本確認試験においては、上記発泡成形用熱可塑性樹脂組成物(発明品1〜13)及び比較のための熱可塑性樹脂組成物(比較品1〜9)を用いて発泡成形体を成形し、この発泡成形体の外観及び内部の観察と、機械的性能としての曲げモジュラスの測定と、金型の汚染の観察とを行った。
以下に具体的な組成の熱可塑性樹脂組成物を挙げ、本発明を更に詳細に説明するが、本発明の主旨を超えない限り、本発明は以下の例に限定されるものではない。なお、以下の記載において「部」及び「%」は、特に断らない限り質量基準である。
【0136】
本確認試験においては、次の各成分を含有する発泡成形用熱可塑性樹脂組成物を用いた。
<(A)成分;ゴム強化スチレン系樹脂>
(重合体A1;アクリル系ゴム/スチレン/アクリロニトリル共重合体)
攪拌機を備えたガラス製フラスコに、窒素気流中で、イオン交換水160部、ドデシルベンゼンスルホン酸ナトリウム1部、クメンハイドロパーオキサイド0.002部、エチレンジアミン四酢酸四ナトリウム塩0.004部、硫酸第1鉄7水和物0.001部とn−ブチルアクリレート10部、アリルメタクリレート0.02部を投入し、撹拌しながら昇温した。60℃に達したところで、ナトリウムホルムアルデヒドスルホキシレート0.75部(20%水溶液)を添加した。内温を60℃に保持し80分経過後、n−ブチルアクリレート40部、アリルメタクリレート0.65部、及びクメンハイドロパーオキサイド0.01部、ドデシルベンゼンスルホン酸ナトリウム0.3部、イオン交換水7部、を180分間かけて連続に添加し、添加終了後更に60分重合を継続した。60分経過後、アクリル系ゴムラテックスの一部をサンプリングし評価したところ、体積平均粒子径0.1μm、ゲル含率6%であった。
【0137】
その後、ドデシルベンゼンスルホン酸ナトリウム5.2部(4%水溶液)、ナトリウムホルムアルデヒドスルホキシレート3部(20%水溶液)を添加し、スチレン38部、アクリロニトリル12部、及びt−ブチルハイドロパーオキサイド0.2部、ドデシルベンゼンスルホン酸ナトリウム0.3部、イオン交換水20部を5時間かけて連続に添加した。添加終了後更に45分間重合を継続後、2,2’−メチレンビス(4−エチル−6−t−ブチルフェノール)0.2部を添加して重合を終了した。重合転化率は99%であった。反応生成物のラテックスを硫酸マグネシウム水溶液で凝固、水洗した後、乾燥してASA樹脂(A1)を得た。
この重合体A1のグラフト率は65%、アセトン可溶分の極限粘度は〔η〕は0.38dl/gであった。また、熱シクロヘキサン溶解量は5%であった。
【0138】
(重合体A2;エチレン・プロピレン系ゴム質重合体/スチレン/アクリロニトリ共重合体)
リボン型撹拌翼を備えたステンレス製オートクレーブに窒素気流中で、エチレン・プロピレン系ゴム質重合体〔エチレン含量63%、非共役ジエン成分はジシクロペンタジエン、ヨウ素価10、ムーニー粘度(ML1+4、100℃)33、ゲル含率0%〕を30部、スチレン45部、アクリロニトリル25部、トルエン140部を仕込み、内温を75℃に昇温して、オートクレーブ内容物を1時間撹拌して均一溶液とした。その後、t−ブチルパーオキシイソプロピルモノカーボネート0.45部を添加し、内温を更に昇温し、100℃に達した後、この温度を保持しながら、撹拌回転数100rpmとして重合反応を行った。
【0139】
重合反応開始後、4時間経過後から内温を120℃に昇温し、この温度を保持しながら更に2時間反応を行って終了した。内温を100℃まで冷却したあと、オクタデシル−3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェノール)−プロピオネート0.2部を添加した。重合転化率は95%であった。反応混合物をオートクレーブより抜き出し、水蒸気蒸留により未反応物と溶媒とを留去し、40mmφの真空ベント付き押出機でシリンダー温度を220℃、真空度760mmHgに調節して揮発分を実質的に脱揮させ、ペレット化し重合体A2を得た。
この重合体A2のグラフト率は60%、アセトン可溶分の極限粘度〔η〕は0.45dl/gであり、熱シクロヘキサン溶解量は35%であった。
【0140】
(重合体A3;ブタジエン系ゴム質重合体/スチレン/アクリロニトリ共重合体)
重合体A2を得るために用いたエチレン・プロピレン系ゴム質重合体の代わりに、ポリブタジエン〔JSR社製、「BR51」、ハイシスタイプ、ムーニー粘度(ML1+4、100℃)33、ゲル含率0%〕15部、スチレン64部、アクリロニトリル21部に変えた以外は、重合体A2の場合と同様の方法で製造し、重合体A3を得た。
この重合体A3の重合転化率91%であり、グラフト率68%、アセトン可溶分の極限粘度〔η〕は0.39dl/gであり、熱シクロヘキサン溶解量は2%であった。
【0141】
(重合体A4;水素添加共役ジエン系ブロック共重合体/スチレン/アクリロニトリル/メタクリル酸メチル共重合体)
リボン型撹拌翼を備えたステンレス製オートクレーブに窒素気流中で、水素添加ブタジエン系ブロック共重合体〔JSR社製ダイナロン4600P(商品名)ゲル含率0%〕30部、メタクリル酸メチル50部、スチレン10部、アクリロニトリル10部、トルエン100部仕込み、内温を75℃に昇温させながら、撹拌により溶解させ均一溶液を得た後、t−ブチルパーオキシイソプロピルカーボネート0.5部、t−ドデシルメルカプタン0.1部を添加し、内温を更に昇温し、100℃に達した後は温度一定に制御しながら、撹拌回転数100rpmにて6時間重合反応を行った。その後、オクタデシル−3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェノール)−プロピオネート0.2部添加した後、反応混合物をオートクレーブより抜き出した。なお、重合転化率は91%であった。水蒸気蒸留により未反応物と溶媒を留去し、40mmφの真空ベント付き押出機(220℃、760mmHg真空)にて、実質的に揮発分を脱揮させ、重合体A4のペレットを得た。
この重合体A4のグラフト率は35%、アセトン可溶分の極限粘度〔η〕は0.30であり、熱シクロヘキサン溶解量は65%であった。
【0142】
(重合体A5;ポリブタジエン/スチレン/アクリロニトリル共重合体)
重合体A1を得るために用いたアクリル系ゴムラテックスの代わりに、ポリブタジエンラテックス(平均ゴム粒径0.3μm、ゲル含率85%)を用いた以外は、重合体A1の場合と同様の方法で製造し、重合体A5を得た。
この重合体A5の重合転化率は99%、得られた重合体D1のグラフト率は78%、アセトン可溶分の極限粘度〔η〕は、0.38dl/gであった。また熱シクロヘキサン溶解量は0%であった。
【0143】
<(B)成分;スチレン系樹脂>
(重合体B1;スチレン/アクリロニトリル共重合体)
リボン翼を備えたステンレス製オートクレーブを2基連結し、窒素置換した後、1基目の反応容器にスチレン75部、アクリロニトリル25部、トルエン20部を連続的に添加した。分子量調節剤としてtert−ドデシルメルカプタン0.14部及びトルエン5部の溶液、及び重合開始剤として、1,1’−アゾビス(シクロヘキサン−1−カーボニトリル)0.1部、及びトルエン5部の溶液を連続的に供給した。1基目の重合温度は、110℃にコントロールし、平均滞留時間2.0時間、重合転化率57%であった。得られた重合体溶液は、1基目の反応容器の外部に設けたポンプにより、スチレン、アクリロニトリル、トルエン、分子量調節剤、及び重合開始剤の供給量と同量を連続的に取り出し2基目の反応容器に供給した。2基目の反応容器の重合温度は、130℃で行い、重合転化率は75%であった。2基目の反応容器で得られた共重合溶液は、2軸3段ベント付き押出機を使用して、直接未反応単量体と溶剤を脱揮し、極限粘度〔η〕0.45dl/gの重合体B1を得た。
【0144】
<(C)成分;芳香族ポリカーボネート樹脂>
芳香族ポリカーボネート樹脂として、三菱エンジニアリングプラスチックス社製「ノバレックス 7022PJ」を用いた。
【0145】
<(D)成分;化学発泡剤>
化学発泡剤としては、永和化成工業社製「ポリスレンEB106」、マスターバッチ(ADCA(発泡剤)/ABS(樹脂)=10/90(質量比))を用いた。化学発泡剤の配合量は、樹脂成分((A)〜(C)成分)の合計100質量部に対し、0.35質量部とした。
【0146】
<(E)成分;タルク>
日本タルク社製の微粉タルク「MICRO ACE SG−200」(商品名)を用いた。レーザー回折法によるD50(重量平均)は1μmである。
【0147】
<(F)成分;繊維状充填材>
ガラス繊維として、エヌエスジー・ヴェトロテックス社製のマイクログラス・チョップドストランド「RES 03−TP89Z」(商品名)を用いた。ガラス繊維の直径は13μm、長さは、3mmである。
【0148】
<(G)成分;酸化防止剤>
酸化防止剤としては、ADEKA社製アデカスタブAO−412S(AO1)と、ADEKA社製アデカスタブ2112(AO2)と、住友化学工業社製スミライザーGS(AO3)とを用いた。酸化防止剤の配合量は、樹脂成分((A)〜(C)成分)の合計100質量部に対し、それぞれ0.1質量部とした。
【0149】
<その他の成分;ガラスビーズ>
ポッターズ・バロティーニ社製のフィラー用ガラスビーズ「GB731」(商品名)を用いた。材質はソーダ石灰ガラスであり、重量平均粒子径は32μmであった。
【0150】
上記A1〜A5のいずれかによる(A)成分、(B)〜(G)成分、その他の成分を用いて製造した熱可塑性樹脂組成物の組成を表1に発明品1〜13として示し、比較のために製造した熱可塑性樹脂組成物の組成を表2に比較品1〜9として示す。表1、表2において、溶解量は、(A)成分中における熱シクロヘキサン溶解量(%)を示す。
【0151】
【表1】
【0152】
【表2】
【0153】
表1における発明品1〜13及び表2における比較品1〜9は、次のようにして得た。
表1に記載した配合割合で、(A)〜(C)、(E)、(F)、(G)、その他の成分をヘンシエルミキサーにてブレンドした後、日本製鋼所製の二軸押出機TEX44を用いて、250℃にて押し出し、発泡成形前の熱可塑性樹脂ペレットを得た。
発泡成形機としては、日本製鋼所製110(t)電動成形機(J110AD)を用いた。得られた熱可塑性樹脂ペレットと発泡剤マスターバッチ((D)成分)をドライブレンドして発泡成形機に供給して射出発泡成形を行い、評価用試験片としての発泡成形体を得た。
また、射出発泡成形における充填時間は1秒、型開速度は0.5mm/秒とした。
【0154】
(A)成分(ゴム強化スチレン系樹脂)における熱シクロヘキサン溶解量は、上述した測定方法によって測定した。
なお、ゴム質重合体(a)のゲル含率、ゴム強化スチレン系樹脂(A)のグラフト率、ゴム強化スチレン系樹脂(A)及びスチレン系樹脂(B)のアセトン可溶分の極限粘度〔η〕についても、上述した測定方法によって測定した。
【0155】
発明品1〜13及び比較品1〜9について、最高発泡倍率の測定結果、外観観察及び内部観察の評価結果、曲げモジュラス(曲げ弾性率)、金型の汚染の評価結果を表1、表2に示す。
最高発泡倍率は、発泡倍率を1.1倍から0.1倍ずつ上げていき、外観観察の結果が○から△に変わる倍率(成形外観が悪化する倍率)として求めた。また、発泡倍率は、容積拡張前のキャビティの隙間を3mmとし、キャビティの隙間を拡張する量を0.3mmずつ拡大して求めた。
【0156】
発泡成形体の外観観察は、発泡成形体の表面を目視観察し、良好な平面が得られた場合
を○、一部に波打ちしている面があった場合を△、歪んで波打ちしている場合を×として
評価した。
発泡成形体の内部観察は、発泡倍率を1.5倍として得られた発泡成形体の断面を目視観察し、均一で微細な発泡セルが形成されていた場合を○、発泡セルの外径に大小の分布があった場合を△、発泡セルのほとんどが大きな外径になっていた場合を×として評価した。
【0157】
曲げモジュラス(曲げ弾性率)(MPa)は、発泡倍率を1.5倍として得られた発泡成形体を用い、ISO178に準拠して測定した。
なお、発泡成形体の内部観察及び曲げモジュラスは、発泡成形体が波打っていたり、歪んでいた場合には、測定を実施しなかった。この場合は、表中に「−」と記載した。
金型の汚染は、発泡倍率を1.5倍として発泡成形体を成形し、発泡成形後に金型の成形表面に汚染が見られなかった場合を○、金型の成形表面の一部に汚染が見られた場合を△、金型の成形表面の全体に渡って汚染が見られた場合を×として評価した。
なお、最高発泡倍率が1.5倍以上に達しないものは、金型の汚染の評価を行わなかった。
【0158】
発明品1〜13については、発泡成形用熱可塑性樹脂組成物を用いて成形した発泡成形体であり、発泡性に優れ(最高発泡倍率が高く)、成形外観が良好であり、また、内部の観察において、ほぼ微細な発泡セル構造を発現し、発泡成形体の部位によらず発泡セルの大きさがほぼ均一であった。また、機械的性能としての曲げモジュラスも高い値が得られた。さらに、金型の汚染も成形表面の一部に見られる程度であった。
【0159】
これに対し、比較品1、2については、繊維状充填材(ガラス繊維)(F)の代わりにガラスビーズを用いて発泡成形したことにより、発泡セルが大きくなり、金型の成形表面の全体に渡って汚染が見られた。比較品3については、ガラス繊維(F)の配合量が多いために、成形外観が劣り、発泡セルが大きくなった。比較品4については、タルクが含まれないために、発泡セルが大きくなった。比較品5については、タルク(E)の配合量が多いために、成形外観が劣り、発泡セルが大きくなった。
【0160】
また、比較品6については、ゴム強化スチレン系樹脂(A)及びゴム質重合体(a)が少ないことにより、比較品7については、ゴム強化スチレン系樹脂(A)が多いことにより、比較品8については、ゴム強化スチレン系樹脂(A)中の熱シクロヘキサン溶解量が0(%)であることにより、最高発泡倍率が低く、成形外観が劣り、発泡セルが大きくなり、金型の成形表面の全体に渡って汚染が見られた。また、比較品9については、繊維状充填材を含有していないことにより、金型の成形表面の全体に渡って汚染が見られた。
なお、比較品1〜8については、内部観察結果に劣ったため、曲げモジュラスは評価しなかった。
【0161】
そして、特に、成形する発泡成形体において、微細かつ均一な発泡セルを得るためには、ゴム強化スチレン系樹脂(A)の熱シクロヘキサン溶解量がゴム質重合体(a)を基準として1〜99質量%であること、及び樹脂成分100質量%におけるゴム質重合体(a)の割合が3〜50質量%であることがわかった。また、この組成により、機械的強度及び成形外観に優れた発泡成形体が得られ、金型の汚染度合いも少ないことがわかった。
【符号の説明】
【0162】
1 製造装置
2 第1型部
21 キャビティ形成凹部
22 樹脂注入口
3 第2型部
31 キャビティ形成凸部
41A、B キャビティ
42 充填用隙間
6 発泡成形体
60 溶融樹脂(熱可塑性樹脂組成物)
【特許請求の範囲】
【請求項1】
ゴム質重合体(a)の存在下に芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b1)を重合してなり、かつ熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準として1〜99質量%であるゴム強化スチレン系樹脂(A)5〜90質量%と、
芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b2)を重合してなるスチレン系樹脂(B)0〜85質量%と、
芳香族ポリカーボネート樹脂(C)10〜90質量%と、
上記成分(A)〜(C)の合計100質量部に対し、化学発泡剤(D)0.1〜5質量部と、タルク(E)0.5〜18質量部と、繊維状充填材(F)0.5〜25質量部とからなり、
上記成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合が3〜50質量%であることを特徴とする発泡成形用熱可塑性樹脂組成物。
【請求項2】
請求項1において、上記ゴム質重合体(a)は、オレフィン系ゴム、アクリル系ゴム及び水素添加共役ジエン系ブロック共重合体からなる群から選ばれた少なくとも1種であることを特徴とする発泡成形用熱可塑性樹脂組成物。
【請求項3】
請求項2において、上記アクリル系ゴムは、ゲル含率が70質量%以下であり、体積平均粒子径が50〜300nmであることを特徴とする発泡成形用熱可塑性樹脂組成物。
【請求項4】
請求項1〜3のいずれか一項において、コアバック型射出発泡成形に用いることを特徴とする発泡成形用熱可塑性樹脂組成物。
【請求項5】
請求項1〜3のいずれか一項に記載の発泡成形用熱可塑性樹脂組成物を用い、コアバック型射出発泡成形を行って成形したことを特徴とする発泡成形体。
【請求項1】
ゴム質重合体(a)の存在下に芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b1)を重合してなり、かつ熱シクロヘキサン溶解量が、ゴム質重合体(a)を基準として1〜99質量%であるゴム強化スチレン系樹脂(A)5〜90質量%と、
芳香族ビニル化合物又は芳香族ビニル化合物及び芳香族ビニル化合物と共重合可能な他のビニル単量体(b2)を重合してなるスチレン系樹脂(B)0〜85質量%と、
芳香族ポリカーボネート樹脂(C)10〜90質量%と、
上記成分(A)〜(C)の合計100質量部に対し、化学発泡剤(D)0.1〜5質量部と、タルク(E)0.5〜18質量部と、繊維状充填材(F)0.5〜25質量部とからなり、
上記成分(A)〜(C)の合計100質量%におけるゴム質重合体(a)の割合が3〜50質量%であることを特徴とする発泡成形用熱可塑性樹脂組成物。
【請求項2】
請求項1において、上記ゴム質重合体(a)は、オレフィン系ゴム、アクリル系ゴム及び水素添加共役ジエン系ブロック共重合体からなる群から選ばれた少なくとも1種であることを特徴とする発泡成形用熱可塑性樹脂組成物。
【請求項3】
請求項2において、上記アクリル系ゴムは、ゲル含率が70質量%以下であり、体積平均粒子径が50〜300nmであることを特徴とする発泡成形用熱可塑性樹脂組成物。
【請求項4】
請求項1〜3のいずれか一項において、コアバック型射出発泡成形に用いることを特徴とする発泡成形用熱可塑性樹脂組成物。
【請求項5】
請求項1〜3のいずれか一項に記載の発泡成形用熱可塑性樹脂組成物を用い、コアバック型射出発泡成形を行って成形したことを特徴とする発泡成形体。
【図1】


【図2】
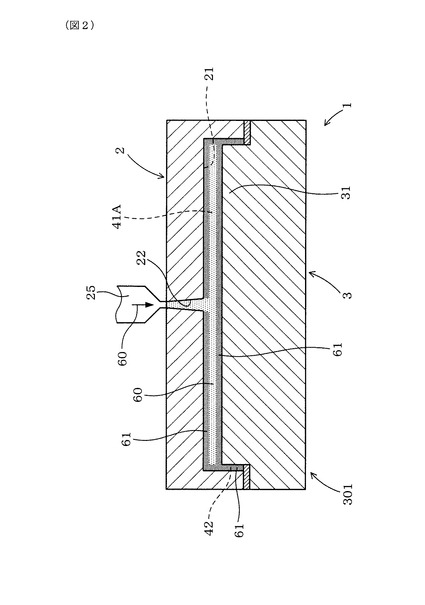
【図3】
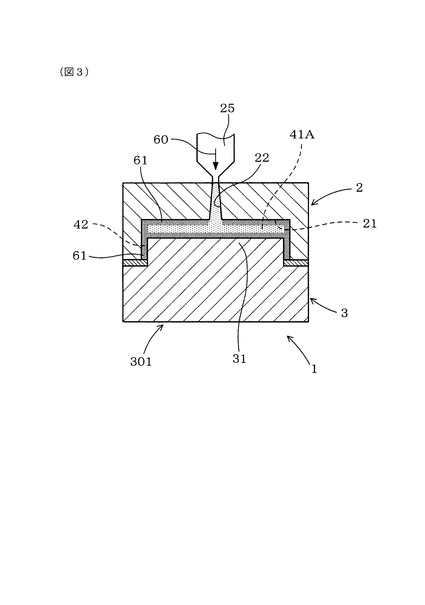
【図4】
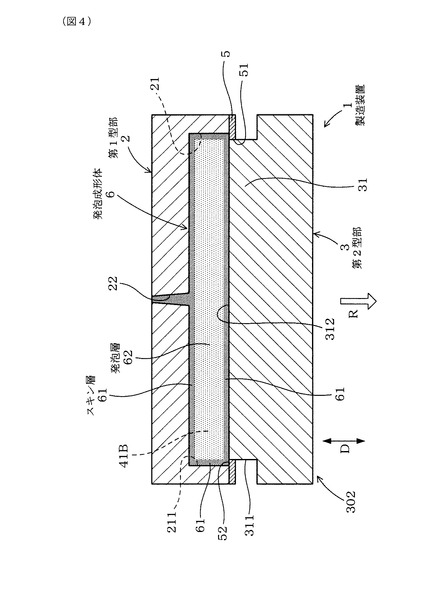
【図2】
【図3】
【図4】
【公開番号】特開2011−37925(P2011−37925A)
【公開日】平成23年2月24日(2011.2.24)
【国際特許分類】
【出願番号】特願2009−183813(P2009−183813)
【出願日】平成21年8月6日(2009.8.6)
【出願人】(396021575)テクノポリマー株式会社 (278)
【Fターム(参考)】
【公開日】平成23年2月24日(2011.2.24)
【国際特許分類】
【出願日】平成21年8月6日(2009.8.6)
【出願人】(396021575)テクノポリマー株式会社 (278)
【Fターム(参考)】
[ Back to top ]