
発泡樹脂成形体

【課題】微細な発泡セルを多数有する発泡樹脂成形体を提供する。
【解決手段】本発明の発泡樹脂成形体は、表面がスキン層で形成され、内部が発泡層で形成されている。発泡層には、複数の第1の発泡セル4と、第1の発泡セル4同士の間に形成され、第1の発泡セル4より小さい複数の第2の発泡セル5と、が形成されている。
【解決手段】本発明の発泡樹脂成形体は、表面がスキン層で形成され、内部が発泡層で形成されている。発泡層には、複数の第1の発泡セル4と、第1の発泡セル4同士の間に形成され、第1の発泡セル4より小さい複数の第2の発泡セル5と、が形成されている。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、射出発泡成形法などにより成形される発泡樹脂成形体に関する。
【背景技術】
【0002】
現在、環境問題等の意識の高まりから、自動車を軽量化し、自動車の燃費を向上させることが求められている。そのため、ドアトリムなどの自動車部品の基材に軽量な発泡樹脂成形体を用いることにより、自動車の軽量化を図る場合がある。発泡樹脂成形体は、軽量化のほかに、材料コストの低減という観点からも好ましく、自動車部品での採用が増加傾向にある。
【0003】
発泡樹脂成形体は、気泡(発泡セル)の割合が多い、即ち、発泡倍率が高いほど軽量であり、発泡樹脂成形体の発泡セルの径が小さいほど耐衝撃性などの物性に優れているため、発泡樹脂成形体中に形成される発泡セルは、径が小さく、数が多いことが好ましい。
【0004】
発泡樹脂成形体の成形方法は、大きく分けて物理発泡と化学発泡の2つに分類される。
【0005】
物理発泡とは、射出成形機のシリンダー内に加圧され空気、炭酸ガス、窒素、揮発性溶媒を溶解する方法である。
【0006】
化学発泡とは、射出成形機のホッパー口から母材とともに化学発泡剤を投入し熱分解もしくは化学反応により炭酸ガス、窒素、水、アンモニア等の気体を樹脂に混入させる方法である。
【0007】
物理発泡においては、溶解する圧力、温度が容易に調整できるため、超臨界状態にした炭酸ガス、窒素を直接注入できる。超臨界流体は液体のような圧縮性と気体のような拡散性があるため、樹脂内に気体の高拡散性と高溶解性を付与できる。このことにより、発泡樹脂成形体中に形成される発泡セルは、径が小さく、数が多い発泡成形体を得ることができる。
【0008】
しかしながら、物理発泡は、気体の準備に加えて、気体を樹脂に溶解させる機構や、成形前に気体が樹脂から抜けないように圧力を維持する機構などが必要であり、ユーザは新たな射出成形機を導入したり、既存の射出成形機を改造したりすることが必要となる。そのため、初期投資および維持費用等のコストが高いことが大きな課題となっている。
【0009】
化学発泡においては化学反応によって気体を生成させるため、ガスを溶解させる圧力が低く、樹脂内にガスを多量に溶融させることは難しい。したがって発泡セルが大きくなってしまう。
【0010】
発泡射出成形においては発泡樹脂成形体中の発泡セルの孔径を小さく数を多くすることを目的として、樹脂に造核剤を添加することが行われている。これは、気泡が物体の表面を起点として発生する性質を有することに基づいており、造核剤によって気泡の発生起点を増加させることができるため、化学発泡においては特に有効である。
【0011】
造核剤としては、従来からクエン酸系等の有機物が用いられている。しかし、これらの有機物は熱により分解が起こり、タール状の物質が生成される。化学発泡において、発泡剤が分解して気体を生成するためには、ある程度の高い温度が必要であり、その温度は、発泡剤の種類によっては、上記の有機物からなる造核剤が分解または劣化する温度以上となる場合もある。この場合、分解または劣化した造核剤が発泡樹脂成形体の表面の外観を損なったり、発泡セルを粗くしたり、さらには、臭いを生じる原因となったりして、発泡樹脂成形体の商品性が著しく低下してしまう。
【0012】
また、発泡剤が分解して気体を生成する温度が、造核剤が分解または劣化する温度を越えない場合であっても、長期にわたって連続して成形を続けたときに、特に射出成形では、有機物からなる造核剤が劣化し、射出成形機のスクリュなどに徐々に付着していく。そのため、付着物によってスクリュの回転が妨げられたり、射出成形機の故障が引き起こされたりするため、このような有機物からなる造核剤を多量に使用することは好ましくない。
【0013】
そこで、造核剤を熱に強くするために、造核剤として有機物ではなく無機物を用いたり、あるいは有機物と無機物とを併用したものを用いたりしている。
【0014】
化学発泡の一例を示している特許文献1には、溶融した樹脂に、発泡剤、および粒径2〜50μm、特に好ましくは5〜20μmの、造核剤となる炭酸カルシウム、タルク、あるいはマイカ等の無機化合物粉末を混入させることが示されている。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】特開2008−13780号公報
【発明の概要】
【発明が解決しようとする課題】
【0016】
上記の特許文献1では、造核剤として、粒径が1〜100μm程度の無機物のタルクなどが用いられている。この造核剤には、気泡数を増やす効果はあるものの、成形される発泡樹脂成形体に微細な発泡セルを多数有するセル構造を形成するには気泡数が十分ではない。
【0017】
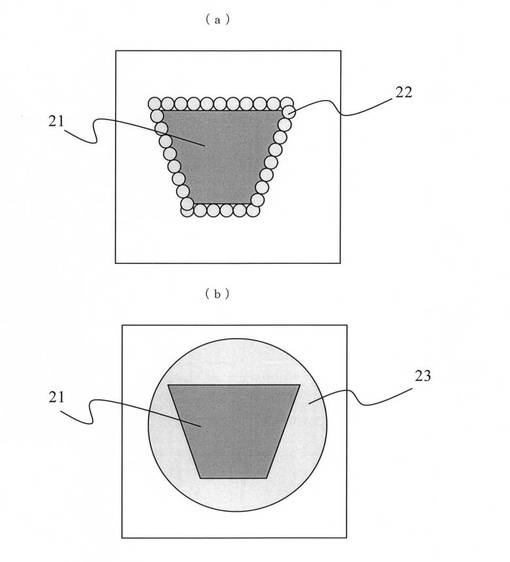
また、図9に示す気泡の成長過程からわかるように、発泡倍率を上げていくと、造核剤21の表面に多数の気泡22が生じ(図9(a)参照)、気泡の成長過程で複数の気泡22同士が結びつき、結局は大きな気泡23になってしまう(図9(b)参照)。そのため、化学発泡において、微細化した発泡セルを有する発泡樹脂成形体を具現化することはできていない。
【0018】
本発明は、上記課題を鑑みてなされたものであり、微細な発泡セルを多数有する発泡樹脂成形体を提供することを目的とする。
【課題を解決するための手段】
【0019】
本発明の発泡樹脂成形体は、表面がスキン層で形成され、内部が発泡層で形成されている。発泡層には、複数の第1の発泡セルと、第1の発泡セル同士の間に形成され、かつ、第1の発泡セルより小さい複数の第2の発泡セルと、が形成されている。
【発明の効果】
【0020】
本発明によれば、発泡樹脂成形体の発泡層に、微細な発泡セルを多数形成することができ、発泡樹脂成形体の耐衝撃性および剛性を向上させることができる。
【図面の簡単な説明】
【0021】
【図1】本発明に係る実施例1の発泡樹脂成形体の断面を、マイクロスコープを用いて撮影した写真(42倍)である。
【図2】本発明に係る実施例1の発泡樹脂成形体の断面を、走査型電子顕微鏡(SEM)を用いて撮影した写真(1000倍)である。
【図3】本発明に係る実施例1の発泡樹脂成形体の断面を、走査型電子顕微鏡(SEM)を用いて撮影した写真(3000倍)である。
【図4】比較例1の発泡樹脂成形体の断面を、マイクロスコープを用いて撮影した写真(42倍)である。
【図5】比較例1の発泡樹脂成形体の断面を、走査型電子顕微鏡(SEM)を用いて撮影した写真(1000倍)である。
【図6】比較例1の発泡樹脂成形体の断面を、走査型電子顕微鏡(SEM)を用いて撮影した写真(3000倍)である。
【図7】耐寒落球衝撃試験の試験方法を示した模式図である。
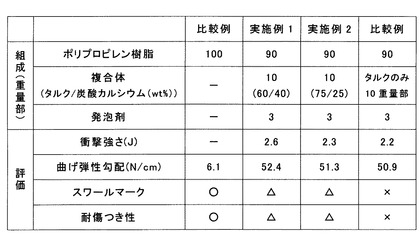
【図8】耐寒落球衝撃試験と曲げ特性の評価試験の試験結果を示す表である。
【図9】気泡の成長過程の様子を示す図であり、(a)は造核剤の表面に気泡が生じた状態、(b)は気泡同士が結びつき、1つの大きな気泡になった状態である。
【発明を実施するための形態】
【0022】
以下に、添付の図面に基づき、本発明の実施の形態を説明する。なお、同一の機能を有する構成には添付図面中、同一の番号を付与し、その説明を省略することがある。
【0023】
本発明によって成形される発泡樹脂成形体は、母材樹脂となる熱可塑性樹脂に発泡剤を混合し、さらに、造核剤として、第1の造核剤の表面に第1の造核剤よりも平均粒径の小さい第2の造核剤を付着させて形成される複合体を混合したものを射出発泡成形することによって得られる。
【0024】
[母材樹脂]
本発明に用いられる母材樹脂としては、ポリプロピレン、ポリエチレン等のポリオレフィン系樹脂が好適に用いられるが、これに限られるものではなく、例えば、ポリスチレン、ABS(アクリロニトリル、ブタジエン、スチレン共重合合成)樹脂、AS(アクリロニトリル、スチレン共重合合成)樹脂等のポリスチレン系樹脂;ナイロン6、ナイロン66、ナイロン12等のポリアミド系樹脂;ポリエチレンテレフタレート(PET)、ポリブチレンテレフタレート(PBT)、ポリトリメチレンテレフタレート(PTT)、ポリエチレンナフタレート(PEN)、ポリ乳酸等のポリエステル系樹脂;ポリ塩化ビニル;ポリカーボネート(PC);ポリアセタール(POM);ポリイミド;ポリエーテルエーテルケトン(PEEK)、などを使用することができる。これらの母材樹脂は変性されていてもよい。また、2種以上の樹脂を併用しても良い。
【0025】
[発泡剤]
本発明に用いられる発泡剤としては、化学発泡剤による熱分解型あるいは反応型の発泡剤を使用する。具体的には、アゾジカルボンアミド等のアゾ化合物、N,N−ジニトロソペンタメチレンテトラミン等のニトロソ化合物、4,4’−オキシビス(ベンゼンスルホニルヒドラジド)、ヒドラゾジカルボンアミド等のヒドラジン誘導体、炭酸水素ナトリウム等の重炭酸塩、炭酸ナトリウム、炭酸アンモニウム等の炭酸塩、亜硝酸アンモニウム等の亜硝酸塩、セミカルバジド化合物、アジド化合物、テトラゾール化合物、イソシアネート化合物、水酸化物などが好適に用いられ、また、尿素等の発泡助剤、クエン酸ナトリウム、タルク、炭酸カルシウム等の有機、無機物系造核剤を併用添加してもよい。これらの発泡剤は2種類以上の発泡剤を併用しても良い。特に、炭酸水素ナトリウムと造核剤のクエン酸ナトリウムとをマスターバッチ化したものが好適に用いられる。
【0026】
[第1の造核剤]
上述したように、本発明の造核剤は、第1の造核剤と第1の造核剤よりも平均粒径の小さい第2の造核剤との複合体からなる。第1の造核剤と後述する第2の造核剤は、発泡樹脂成形体の発泡セルとなる気泡が発生する起点となる機能を有している。
【0027】
第1の造核剤としては、タルク、マイカ、シリカ、クレー、モンモリロナイト、カオリン等のケイ酸塩;炭酸カルシウム、炭酸リチウム、炭酸マグネシウム等の炭酸塩;アルミナ、酸化チタン、酸化亜鉛等の金属酸化物;アルミニウム、鉄、銀、銅等の金属;水酸化アルミニウム、水酸化マグネシウム等の水酸化物;硫酸バリウム等の硫化物;木炭、竹炭等の炭化物;チタン酸カリウム、チタン酸バリウム等のチタン化物、などが挙げられる。これらの中では、特に、タルクがより好ましい。
【0028】
上述した無機物を用いた第1の造核剤の粒径は、0.5〜1000μmであることが好ましく、1〜10μmであることがより好ましい。粒径が0.5μm以上であれば、容易に第1の造核剤の微粒子を作製することができる。さらに、第1の造核剤の表面に多数のナノサイズ(1μm未満)の第2の造核剤を付着させることで、第1の造核剤を、母材樹脂内に第2の造核剤を分散させるためのキャリヤとして機能させることが可能となる。また、粒径が1000μm以下であれば、成形後の発泡樹脂成形体において、物性の低下や見栄えの悪化を防止することができる。
【0029】
また、第1の造核剤としては、タルク等の無機物に替えて、植物繊維、セルロース繊維、酢酸セルロース繊維、ポリエチレンテレフタレート繊維、ナイロン繊維、ポリエチレンナフタレート繊維、アラミド繊維、ビニロン繊維、ポリアリレート繊維などの繊維粉末を用いることができる。
【0030】
この繊維粉末は、母材樹脂への分散性および接着性を向上させるために、芯鞘型、あるいはサイドバイサイド型等の複合繊維であっても良い。また、軽量化および耐熱性向上のために中空型の繊維であってもよい。
【0031】
これらの繊維粉末は、平均繊維径が0.5〜250μm、平均繊維長が1〜3000μmの微細な繊維であることが望ましい。さらに、平均繊維径が1〜100μm、平均繊維長が10〜500μmであることが好ましく、平均繊維径が1〜40μm、平均繊維長が20〜300μmであることがさらに好ましい。
【0032】
[第2の造核剤]
第2の造核剤としては、例えば、タルク、シリカ、クレー、モンモリロナイト、カオリン等のケイ酸塩;炭酸カルシウム、炭酸リチウム、炭酸マグネシウム等の炭酸塩;アルミナ、酸化チタン、酸化亜鉛等の金属酸化物;アルミニウム、鉄、銀、銅等の金属;水酸化アルミニウム、水酸化マグネシウム等の水酸化物;硫酸バリウム等の硫化物;木炭、竹炭などの炭化物;チタン酸カリウム、チタン酸バリウム等のチタン化物;セルロースミクロフィブリル、酢酸セルロース等のセルロース類;フラーレン、カーボンナノチューブ等のカーボン類、などが挙げられる。これらの中の1種を単独で用いても良いし、2種以上を併用しても良い。これらの中では、炭酸カルシウム、マイカ、モンモリロナイト、酸化チタンが好ましい。特に、比較的容易にそして安価にナノサイズの粒子を作製または入手できる点から、炭酸カルシウムが好ましい。
【0033】
第2の造核剤の形状は、球状、板状、繊維状、中空状など、何れの形状でも良い。また、特定形状の微粒子を単独で用いても良いし、2種以上の異なる形状の微粒子を併用しても良い。なお、本発明の第2の造核剤には、一次粒子だけでなく、後述する第2の造核剤のサイズ(粒径)の範囲に入るものであれば、二次以上の粒子も含まれる。
【0034】
第2の造核剤のサイズは、平均粒径が1μm未満のナノサイズであることが必要である。具体的には、その平均粒径は10〜500nmである。平均粒径が10nm以上では、第2の造核剤の分散性を高める点で有利となり、平均粒径が500nm以下では、比表面積を増大させて気泡が発生する起点を増大させて発泡セルを微細化させる点で有利となる。第2の造核剤のサイズは、より好ましくは20〜200nmであり、特に好ましくは50〜100nmである。
【0035】
[第1の造核剤と第2の造核剤の複合体]
第1の造核剤および第2の造核剤は、上述したように、第1の造核剤の表面に第2の造核剤が付着して形成される複合体で母材樹脂に混合される。
【0036】
複合体の製造方法を説明すると、予め第1の造核剤と第2の造核剤をそれぞれ上述したサイズにまで粉砕、または析出法にて調整しておく。そして、第1の造核剤と第2の造核剤と後述する表面処理のためにステアリン酸とを一緒にヘンシェルミキサなどで高速撹拌することによって複合体が得られる。撹拌条件は、乾式で回転羽根の周速が20m/s以上であることが好ましい。これにより、ナノサイズの第2の造核剤が第1の造核剤の表面を覆うように付着され複合体が形成される。
【0037】
[発泡樹脂成形体の成形方法]
次に、発泡樹脂成形体の成形方法の一例について説明する。この成形方法は一例にすぎず、この方法に限定されることはない。上述の母材樹脂と上述の複合体とを母材樹脂重量に対して複合体1〜80質量%(より好ましくは3〜50質量%、特に好ましくは5〜20質量%)の割合で2軸混練押出機に投入して混練して混ぜ合わせる。このとき、複合体が母材樹脂内で、第1の造核剤と第2の造核剤とに分離して分散するため、第2の造核剤同士は独立して存在するようになり、第2の造核剤の再凝集が防止される。また、第2の造核剤が疎水性となるように、第2の造核剤にステアリン酸にて表面処理を行っているため、第2の造核剤と母材樹脂との親和性が高くなり、より再凝集しにくくなる。
【0038】
混練の際、必要に応じて、さらに顔料、ゴム含有ポリマー、相溶化材、可塑剤、滑剤、難燃剤、抗菌剤、結晶化促進剤、酸化防止剤、紫外線吸収剤、熱安定剤、界面活性剤、帯電防止剤等の各種添加材を配合してもよい。
【0039】
複合体を母材樹脂に配合する方法としては、複合体を高濃度に樹脂に配合したマスターバッチを作製し、このマスターバッチをさらに樹脂に配合しても良い。
【0040】
そして、複合体が混ぜ合わされた母材樹脂および発泡剤、さらに、必要があれば、発泡樹脂成形体の所望のカラーバリエーションに対応した顔料入りのマスターバッチをドライブレンドする。
【0041】
ドライブレンドされた混合物は射出成形機へと供給され、一定の圧力条件下で発泡剤の発泡を抑制した状態で、2つの金型によって形成される空間、つまりキャビティへ射出される。そして、スキン層が形成された後に一方の金型を他方の金型に対して後退させることで、混合物の密度が低下し、また圧力が開放される。いわゆるキャビティ拡張法である。これより、発泡剤が分解して、第1の造核剤と第2の造核剤の表面を起点に、炭酸ガスや窒素ガスなどの気泡が発生する。この気泡が発泡セルとなり、発泡層が形成され、発泡樹脂成形体が成形される。
【0042】
なお、発泡樹脂成形体の成形方法としては、ショートショット法、エグレッション法等を用いても良い。
【0043】
上述したように、母材樹脂内に第1の造核剤と第2の造核剤とが均一に分散されている。そのため、本発明による発泡樹脂成形体における、第1の造核剤の表面で発生した気泡による第1の発泡セル、および第2の造核剤の表面で発生した気泡による第2の発泡セルは、発泡樹脂成形体内で偏りなくほぼ均一に形成される。
【0044】
次に、本発明の成形方法で成形された発泡樹脂成形体(以下の実施例1および実施例2)と、従来の成形方法で成形された発泡樹脂成形体(以下の比較例1および比較例2)とを比較した。ただし、比較例1では、発泡させていないので、発泡樹脂成形体ではなく、樹脂成形体が成形される。
【0045】
[実施例1]
母材樹脂であるポリプロピレン樹脂90質量部に対して、第1の造核剤(タルク)と第2の造核剤(炭酸カルシウム)との複合体を10質量部、発泡剤を3質量部の割合で混合し、発泡倍率が2倍となるように射出発泡成形を行い、発泡樹脂成形体を成形した。なお、複合体は、タルクが60質量%、炭酸カルシウム40質量%の割合となるように予めヘンシェルミキサで高速撹拌して得られたものである。第1の造核剤のタルクは、平均粒径が3.2μmであり、第2の造核剤の炭酸カルシウムの平均粒径は80nmである。
【0046】
[実施例2]
複合体は、タルク75%、炭酸カルシウム25%の割合とした。それ以外は実施例1と同様にして発泡樹脂成形体を成形した。
【0047】
[比較例1]
比較例1では、実施例1および実施例2において母材樹脂として用いたポリプロピレン樹脂を用いて、従来の射出成形によって樹脂成形体を成形した。第1の造核剤、第2の造核剤、および発泡剤は混合しておらず、発泡射出成形は行っていない。
【0048】
[比較例2]
比較例2では、実施例1および実施例2と同様にポリプロピレン樹脂を母材樹脂として用い、この母材樹脂90質量部に対して、発泡剤を3質量部、第1の造核剤を10質量部の割合で混合し、発泡倍率が2倍となるように射出発泡成形を行い、発泡樹脂成形体を成形した。なお、実施例1および実施例2と同様に第1の造核剤としてタルクを用いたが、第2の造核剤は使用していない。つまり、比較例2は、従来の発泡射出成形である。
【0049】
以上のようにして作成された実施例1の発泡樹脂成形体と、従来の成形方法で成形した比較例2の発泡樹脂成形体とを比較するために、マイクロスコープと走査型電子顕微鏡(SEM)による、発泡樹脂成形体の断面の観察を行った。
【0050】
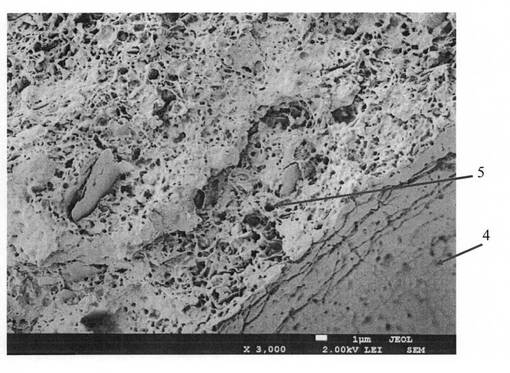
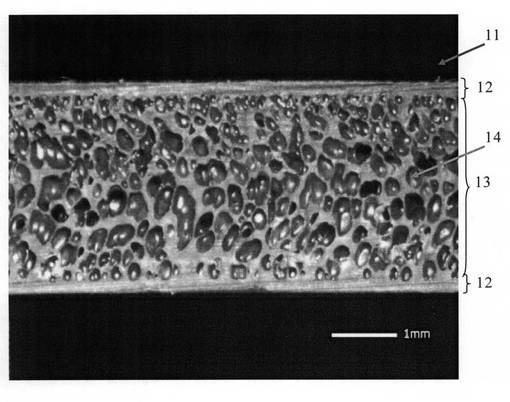
図1〜図3は、実施例1の発泡樹脂成形体の断面の拡大写真であり、図1は倍率が17倍、図2は倍率が1000倍、図3は倍率が3000倍である。また、図4〜図6は、比較例2の発泡樹脂成形体の断面の拡大写真であり、図4は倍率が17倍、図5は倍率が1000倍、図6は倍率が3000倍である。
【0051】
実施例1では、厚み約3mmの発泡樹脂成形体1が成形されており、発泡樹脂成形体1の表面には、厚みがおよそ0.2〜0.6mmのスキン層2が形成されている。スキン層2に挟まれた発泡層3には、第1の造核剤に起因して、孔径が10〜500μmの第1の発泡セル4(図1の発泡層3に確認できる発泡セル)が多数形成されていることがわかる。
【0052】
また、図1〜図3より、実施例1には、第1の発泡セル4同士の間(以降「セル壁」と称する)には、第2の造核剤に起因して、孔径がおよそ10〜1000nmの微細な第2の発泡セル5が多数形成されていることがわかる。なお、図2で確認できるセル壁の小さな穴や、図3で確認できるセル壁の小さな穴は、いずれも第2の発泡セルである。
【0053】
一方、比較例2では、厚み約3mmの発泡樹脂成形体11が成形されており、発泡樹脂成形体11の表面には、厚みがおよそ0.2〜0.6mmのスキン層12が形成されている。スキン層12に挟まれた発泡層13には、第1の造核剤に起因して、孔径10〜1000μmの第1の発泡セル14(図4の発泡層13に確認できる発泡セル)が多数形成されていることがわかる。
【0054】
また、図4〜図6の比較例2の発泡樹脂成形体11のセル壁には、実施例1で確認できる微細な発泡セルが確認できない。
【0055】
以上のことより、本発明の発泡樹脂成形体1は、第2の造核剤を使用していない従来の発泡樹脂成形体11に比べて、第1の発泡セル4を小さくすることができ、また、セル壁には、極微細な第2の発泡セル5を多数形成することができる。つまり、従来の発泡樹脂成形体11に比べて、本発明の発泡樹脂成形体1では、発泡セルの孔径を小さくし、かつ数を増やすことができる。
【0056】
本発明の発泡樹脂成形体1における第1の発泡セル4は、従来の発泡樹脂成形体11における第1の発泡セル14よりも小さい理由を考察してみる。発泡剤によって母材樹脂内に発生するガスの量は同じであると仮定する。従来の発泡樹脂成形体11では、発生したガスは第1の造核剤に起因して気泡を生じる。一方、本発明では、第1の造核剤のほかに、微細な第2の造核剤を使用しているため、発生したガスの一部は第1の造核剤に起因して気泡を生じ、発生したガスの他の一部は第2の造核剤に起因して気泡を生じる。つまり、発生したガスは、第1の造核剤と第2の造核剤とに分散して気泡を生じることになるため、第1の発泡セル4が小さくなったと考えられる。また、第2の造核剤の粒子は非常に小さく、母材樹脂内に均一に分散しているため、第2の造核剤に起因して生じる気泡は小さく、気泡同士が結びついて大きな気泡になってしまうことが少なく、図2や図3からわかるように、第2の発泡セル5は小さい。このことから、第2の発泡セル5は大きくならないと考えられる。
【0057】
次に、実施例1、2および比較例1、2の物性の違いを調べた。はじめに試験方法について説明する。
【0058】
(耐寒落球衝撃強さ)
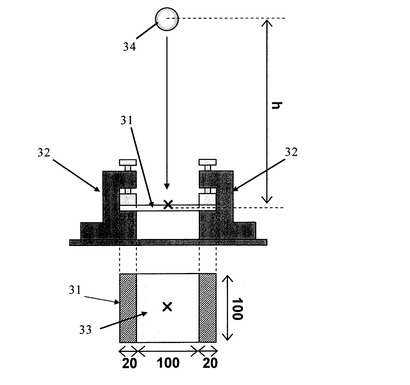
試験片31は厚さ3mmの発泡樹脂成形体から長さ×幅=140mm×100mmに切り出したものを用いた。試験方法は、図7に示すように、試験片31の長さ方向の両端をそれぞれ20mm金属製の治具32に挟んで保持し、治具32から露出している部分である、長さ×幅=100mm×100mmの領域を打撃面33とした。なお、治具32には5Nのトルクをかけることで、試験片31を保持した。雰囲気は−30℃であり、半径25mmの球状頭部を持つ質量500gの錘34を打撃面33の中心の上方から落下させた。この試験は、錘34を落とす高さhを変えて行い、各高さhあたり5回試験を実施した。錘34を落下させた後、試験片31の状態を観察し、試験片31に変化が無いまたは白化したものを「○」、亀裂又は破壊したものを「×」とした。この結果から、○の数が50%になる高さhを算出し、この高さhより衝撃強さを算出した。その結果を図8に示している。
【0059】
(曲げ弾性勾配)
「JIS K 7171 プラスチック−曲げ特性の求め方」を試験方法として採用し、曲げ弾性勾配を測定した。試験片は、厚さ3mmの発泡樹脂成形体から、樹脂流れ方向(MD)と樹脂流れ方向に直交する直交方向(TD)とからそれぞれ長さ×幅=150mm×50mmに切り出したものを用いた。試験機には半径2mmの支持台および圧子を取り付け、支点間距離100mm、試験速度50mm/sで試験を実施した。試験結果は、MD、TDの平均値とし、図8に示している。
【0060】
(スワールマーク)
成形品の外観を目視にて観察し、スワールマークの有無について、以下の基準で評価した。なお、スワールマークは成形時にフローフロントで発泡が起こり、その気泡を引きずった跡が成形体の表面に外観不良として現れるものであり、発泡射出成形ではこの発生が避けられないが、発泡セル径が小さいほど目立ちにくくなる。以下の基準で評価した。「○」:問題無し、「△」:目立たない程度に(一部に)スワールマークがみられる、「×」:目立つ程度にスワールマークがみられる、である。試験結果を図8に示す。
【0061】
(耐傷つき性)
成形品に、先端の直径が1mmの鋼鉄針を用い、500gの荷重をかけ、1000mm/minの引っかき速度で、2mmの間隔で碁盤目状に傷をつける。その後、成形品の外観を目視にて観察し傷つきの有無について、以下の基準で評価した。「○」:問題無し、「△」:目立たない程度に(一部に)傷がみられる、「×」:目立つ程度に傷がみられる、である。試験結果を図8に示す。
【0062】
単なる射出成形を行った樹脂成形体である比較例1と、発泡樹脂成形体である実施例1、2および比較例2を比較すると、曲げ弾性勾配は、単なる射出成形体である比較例1に比べて発泡樹脂成形体である実施例1、2および比較例2のほうが著しく大きい。つまり、発泡樹脂成形体である実施例1、2および比較例2は、単なる射出成形体である比較例1に比べて剛性が著しく向上することがわかる。
【0063】
比較例2と実施例1および実施例2とを比較すると、比較例2に比べて、実施例1および実施例2のほうが、衝撃に強いことがわかる。
【0064】
実施例1および実施例2の優れた耐衝撃性は、ナノサイズの第2の造核剤に起因する第2の発泡セルによるものと考えられる。
【0065】
実施例1および実施例2のスワールマークおよび傷つき性は、比較例2に比べて改善されることがわかる。つまり、比較例2に比べて意匠性が向上する。これにより、本発明の発泡樹脂成形体は、従来の発泡樹脂成形体に比べて塗装量を削減可能となり、コストを削減することができる。さらに、発泡樹脂成形体を適応できる製品や用途を拡大させることができる。
【0066】
以上の結果から、複合体における第1の造核剤と第2の造核剤との重量比は、75:25〜60:40の間であることが好ましい。また、複合体は、母材樹脂100質量部に対して10質量部以上、混合物に混入されていることが好ましい。
【符号の説明】
【0067】
1、11 発泡樹脂成形体
2、12 スキン層
3、13 発泡層
4、14 第1の発泡セル
5 第2の発泡セル
【技術分野】
【0001】
本発明は、射出発泡成形法などにより成形される発泡樹脂成形体に関する。
【背景技術】
【0002】
現在、環境問題等の意識の高まりから、自動車を軽量化し、自動車の燃費を向上させることが求められている。そのため、ドアトリムなどの自動車部品の基材に軽量な発泡樹脂成形体を用いることにより、自動車の軽量化を図る場合がある。発泡樹脂成形体は、軽量化のほかに、材料コストの低減という観点からも好ましく、自動車部品での採用が増加傾向にある。
【0003】
発泡樹脂成形体は、気泡(発泡セル)の割合が多い、即ち、発泡倍率が高いほど軽量であり、発泡樹脂成形体の発泡セルの径が小さいほど耐衝撃性などの物性に優れているため、発泡樹脂成形体中に形成される発泡セルは、径が小さく、数が多いことが好ましい。
【0004】
発泡樹脂成形体の成形方法は、大きく分けて物理発泡と化学発泡の2つに分類される。
【0005】
物理発泡とは、射出成形機のシリンダー内に加圧され空気、炭酸ガス、窒素、揮発性溶媒を溶解する方法である。
【0006】
化学発泡とは、射出成形機のホッパー口から母材とともに化学発泡剤を投入し熱分解もしくは化学反応により炭酸ガス、窒素、水、アンモニア等の気体を樹脂に混入させる方法である。
【0007】
物理発泡においては、溶解する圧力、温度が容易に調整できるため、超臨界状態にした炭酸ガス、窒素を直接注入できる。超臨界流体は液体のような圧縮性と気体のような拡散性があるため、樹脂内に気体の高拡散性と高溶解性を付与できる。このことにより、発泡樹脂成形体中に形成される発泡セルは、径が小さく、数が多い発泡成形体を得ることができる。
【0008】
しかしながら、物理発泡は、気体の準備に加えて、気体を樹脂に溶解させる機構や、成形前に気体が樹脂から抜けないように圧力を維持する機構などが必要であり、ユーザは新たな射出成形機を導入したり、既存の射出成形機を改造したりすることが必要となる。そのため、初期投資および維持費用等のコストが高いことが大きな課題となっている。
【0009】
化学発泡においては化学反応によって気体を生成させるため、ガスを溶解させる圧力が低く、樹脂内にガスを多量に溶融させることは難しい。したがって発泡セルが大きくなってしまう。
【0010】
発泡射出成形においては発泡樹脂成形体中の発泡セルの孔径を小さく数を多くすることを目的として、樹脂に造核剤を添加することが行われている。これは、気泡が物体の表面を起点として発生する性質を有することに基づいており、造核剤によって気泡の発生起点を増加させることができるため、化学発泡においては特に有効である。
【0011】
造核剤としては、従来からクエン酸系等の有機物が用いられている。しかし、これらの有機物は熱により分解が起こり、タール状の物質が生成される。化学発泡において、発泡剤が分解して気体を生成するためには、ある程度の高い温度が必要であり、その温度は、発泡剤の種類によっては、上記の有機物からなる造核剤が分解または劣化する温度以上となる場合もある。この場合、分解または劣化した造核剤が発泡樹脂成形体の表面の外観を損なったり、発泡セルを粗くしたり、さらには、臭いを生じる原因となったりして、発泡樹脂成形体の商品性が著しく低下してしまう。
【0012】
また、発泡剤が分解して気体を生成する温度が、造核剤が分解または劣化する温度を越えない場合であっても、長期にわたって連続して成形を続けたときに、特に射出成形では、有機物からなる造核剤が劣化し、射出成形機のスクリュなどに徐々に付着していく。そのため、付着物によってスクリュの回転が妨げられたり、射出成形機の故障が引き起こされたりするため、このような有機物からなる造核剤を多量に使用することは好ましくない。
【0013】
そこで、造核剤を熱に強くするために、造核剤として有機物ではなく無機物を用いたり、あるいは有機物と無機物とを併用したものを用いたりしている。
【0014】
化学発泡の一例を示している特許文献1には、溶融した樹脂に、発泡剤、および粒径2〜50μm、特に好ましくは5〜20μmの、造核剤となる炭酸カルシウム、タルク、あるいはマイカ等の無機化合物粉末を混入させることが示されている。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】特開2008−13780号公報
【発明の概要】
【発明が解決しようとする課題】
【0016】
上記の特許文献1では、造核剤として、粒径が1〜100μm程度の無機物のタルクなどが用いられている。この造核剤には、気泡数を増やす効果はあるものの、成形される発泡樹脂成形体に微細な発泡セルを多数有するセル構造を形成するには気泡数が十分ではない。
【0017】
また、図9に示す気泡の成長過程からわかるように、発泡倍率を上げていくと、造核剤21の表面に多数の気泡22が生じ(図9(a)参照)、気泡の成長過程で複数の気泡22同士が結びつき、結局は大きな気泡23になってしまう(図9(b)参照)。そのため、化学発泡において、微細化した発泡セルを有する発泡樹脂成形体を具現化することはできていない。
【0018】
本発明は、上記課題を鑑みてなされたものであり、微細な発泡セルを多数有する発泡樹脂成形体を提供することを目的とする。
【課題を解決するための手段】
【0019】
本発明の発泡樹脂成形体は、表面がスキン層で形成され、内部が発泡層で形成されている。発泡層には、複数の第1の発泡セルと、第1の発泡セル同士の間に形成され、かつ、第1の発泡セルより小さい複数の第2の発泡セルと、が形成されている。
【発明の効果】
【0020】
本発明によれば、発泡樹脂成形体の発泡層に、微細な発泡セルを多数形成することができ、発泡樹脂成形体の耐衝撃性および剛性を向上させることができる。
【図面の簡単な説明】
【0021】
【図1】本発明に係る実施例1の発泡樹脂成形体の断面を、マイクロスコープを用いて撮影した写真(42倍)である。
【図2】本発明に係る実施例1の発泡樹脂成形体の断面を、走査型電子顕微鏡(SEM)を用いて撮影した写真(1000倍)である。
【図3】本発明に係る実施例1の発泡樹脂成形体の断面を、走査型電子顕微鏡(SEM)を用いて撮影した写真(3000倍)である。
【図4】比較例1の発泡樹脂成形体の断面を、マイクロスコープを用いて撮影した写真(42倍)である。
【図5】比較例1の発泡樹脂成形体の断面を、走査型電子顕微鏡(SEM)を用いて撮影した写真(1000倍)である。
【図6】比較例1の発泡樹脂成形体の断面を、走査型電子顕微鏡(SEM)を用いて撮影した写真(3000倍)である。
【図7】耐寒落球衝撃試験の試験方法を示した模式図である。
【図8】耐寒落球衝撃試験と曲げ特性の評価試験の試験結果を示す表である。
【図9】気泡の成長過程の様子を示す図であり、(a)は造核剤の表面に気泡が生じた状態、(b)は気泡同士が結びつき、1つの大きな気泡になった状態である。
【発明を実施するための形態】
【0022】
以下に、添付の図面に基づき、本発明の実施の形態を説明する。なお、同一の機能を有する構成には添付図面中、同一の番号を付与し、その説明を省略することがある。
【0023】
本発明によって成形される発泡樹脂成形体は、母材樹脂となる熱可塑性樹脂に発泡剤を混合し、さらに、造核剤として、第1の造核剤の表面に第1の造核剤よりも平均粒径の小さい第2の造核剤を付着させて形成される複合体を混合したものを射出発泡成形することによって得られる。
【0024】
[母材樹脂]
本発明に用いられる母材樹脂としては、ポリプロピレン、ポリエチレン等のポリオレフィン系樹脂が好適に用いられるが、これに限られるものではなく、例えば、ポリスチレン、ABS(アクリロニトリル、ブタジエン、スチレン共重合合成)樹脂、AS(アクリロニトリル、スチレン共重合合成)樹脂等のポリスチレン系樹脂;ナイロン6、ナイロン66、ナイロン12等のポリアミド系樹脂;ポリエチレンテレフタレート(PET)、ポリブチレンテレフタレート(PBT)、ポリトリメチレンテレフタレート(PTT)、ポリエチレンナフタレート(PEN)、ポリ乳酸等のポリエステル系樹脂;ポリ塩化ビニル;ポリカーボネート(PC);ポリアセタール(POM);ポリイミド;ポリエーテルエーテルケトン(PEEK)、などを使用することができる。これらの母材樹脂は変性されていてもよい。また、2種以上の樹脂を併用しても良い。
【0025】
[発泡剤]
本発明に用いられる発泡剤としては、化学発泡剤による熱分解型あるいは反応型の発泡剤を使用する。具体的には、アゾジカルボンアミド等のアゾ化合物、N,N−ジニトロソペンタメチレンテトラミン等のニトロソ化合物、4,4’−オキシビス(ベンゼンスルホニルヒドラジド)、ヒドラゾジカルボンアミド等のヒドラジン誘導体、炭酸水素ナトリウム等の重炭酸塩、炭酸ナトリウム、炭酸アンモニウム等の炭酸塩、亜硝酸アンモニウム等の亜硝酸塩、セミカルバジド化合物、アジド化合物、テトラゾール化合物、イソシアネート化合物、水酸化物などが好適に用いられ、また、尿素等の発泡助剤、クエン酸ナトリウム、タルク、炭酸カルシウム等の有機、無機物系造核剤を併用添加してもよい。これらの発泡剤は2種類以上の発泡剤を併用しても良い。特に、炭酸水素ナトリウムと造核剤のクエン酸ナトリウムとをマスターバッチ化したものが好適に用いられる。
【0026】
[第1の造核剤]
上述したように、本発明の造核剤は、第1の造核剤と第1の造核剤よりも平均粒径の小さい第2の造核剤との複合体からなる。第1の造核剤と後述する第2の造核剤は、発泡樹脂成形体の発泡セルとなる気泡が発生する起点となる機能を有している。
【0027】
第1の造核剤としては、タルク、マイカ、シリカ、クレー、モンモリロナイト、カオリン等のケイ酸塩;炭酸カルシウム、炭酸リチウム、炭酸マグネシウム等の炭酸塩;アルミナ、酸化チタン、酸化亜鉛等の金属酸化物;アルミニウム、鉄、銀、銅等の金属;水酸化アルミニウム、水酸化マグネシウム等の水酸化物;硫酸バリウム等の硫化物;木炭、竹炭等の炭化物;チタン酸カリウム、チタン酸バリウム等のチタン化物、などが挙げられる。これらの中では、特に、タルクがより好ましい。
【0028】
上述した無機物を用いた第1の造核剤の粒径は、0.5〜1000μmであることが好ましく、1〜10μmであることがより好ましい。粒径が0.5μm以上であれば、容易に第1の造核剤の微粒子を作製することができる。さらに、第1の造核剤の表面に多数のナノサイズ(1μm未満)の第2の造核剤を付着させることで、第1の造核剤を、母材樹脂内に第2の造核剤を分散させるためのキャリヤとして機能させることが可能となる。また、粒径が1000μm以下であれば、成形後の発泡樹脂成形体において、物性の低下や見栄えの悪化を防止することができる。
【0029】
また、第1の造核剤としては、タルク等の無機物に替えて、植物繊維、セルロース繊維、酢酸セルロース繊維、ポリエチレンテレフタレート繊維、ナイロン繊維、ポリエチレンナフタレート繊維、アラミド繊維、ビニロン繊維、ポリアリレート繊維などの繊維粉末を用いることができる。
【0030】
この繊維粉末は、母材樹脂への分散性および接着性を向上させるために、芯鞘型、あるいはサイドバイサイド型等の複合繊維であっても良い。また、軽量化および耐熱性向上のために中空型の繊維であってもよい。
【0031】
これらの繊維粉末は、平均繊維径が0.5〜250μm、平均繊維長が1〜3000μmの微細な繊維であることが望ましい。さらに、平均繊維径が1〜100μm、平均繊維長が10〜500μmであることが好ましく、平均繊維径が1〜40μm、平均繊維長が20〜300μmであることがさらに好ましい。
【0032】
[第2の造核剤]
第2の造核剤としては、例えば、タルク、シリカ、クレー、モンモリロナイト、カオリン等のケイ酸塩;炭酸カルシウム、炭酸リチウム、炭酸マグネシウム等の炭酸塩;アルミナ、酸化チタン、酸化亜鉛等の金属酸化物;アルミニウム、鉄、銀、銅等の金属;水酸化アルミニウム、水酸化マグネシウム等の水酸化物;硫酸バリウム等の硫化物;木炭、竹炭などの炭化物;チタン酸カリウム、チタン酸バリウム等のチタン化物;セルロースミクロフィブリル、酢酸セルロース等のセルロース類;フラーレン、カーボンナノチューブ等のカーボン類、などが挙げられる。これらの中の1種を単独で用いても良いし、2種以上を併用しても良い。これらの中では、炭酸カルシウム、マイカ、モンモリロナイト、酸化チタンが好ましい。特に、比較的容易にそして安価にナノサイズの粒子を作製または入手できる点から、炭酸カルシウムが好ましい。
【0033】
第2の造核剤の形状は、球状、板状、繊維状、中空状など、何れの形状でも良い。また、特定形状の微粒子を単独で用いても良いし、2種以上の異なる形状の微粒子を併用しても良い。なお、本発明の第2の造核剤には、一次粒子だけでなく、後述する第2の造核剤のサイズ(粒径)の範囲に入るものであれば、二次以上の粒子も含まれる。
【0034】
第2の造核剤のサイズは、平均粒径が1μm未満のナノサイズであることが必要である。具体的には、その平均粒径は10〜500nmである。平均粒径が10nm以上では、第2の造核剤の分散性を高める点で有利となり、平均粒径が500nm以下では、比表面積を増大させて気泡が発生する起点を増大させて発泡セルを微細化させる点で有利となる。第2の造核剤のサイズは、より好ましくは20〜200nmであり、特に好ましくは50〜100nmである。
【0035】
[第1の造核剤と第2の造核剤の複合体]
第1の造核剤および第2の造核剤は、上述したように、第1の造核剤の表面に第2の造核剤が付着して形成される複合体で母材樹脂に混合される。
【0036】
複合体の製造方法を説明すると、予め第1の造核剤と第2の造核剤をそれぞれ上述したサイズにまで粉砕、または析出法にて調整しておく。そして、第1の造核剤と第2の造核剤と後述する表面処理のためにステアリン酸とを一緒にヘンシェルミキサなどで高速撹拌することによって複合体が得られる。撹拌条件は、乾式で回転羽根の周速が20m/s以上であることが好ましい。これにより、ナノサイズの第2の造核剤が第1の造核剤の表面を覆うように付着され複合体が形成される。
【0037】
[発泡樹脂成形体の成形方法]
次に、発泡樹脂成形体の成形方法の一例について説明する。この成形方法は一例にすぎず、この方法に限定されることはない。上述の母材樹脂と上述の複合体とを母材樹脂重量に対して複合体1〜80質量%(より好ましくは3〜50質量%、特に好ましくは5〜20質量%)の割合で2軸混練押出機に投入して混練して混ぜ合わせる。このとき、複合体が母材樹脂内で、第1の造核剤と第2の造核剤とに分離して分散するため、第2の造核剤同士は独立して存在するようになり、第2の造核剤の再凝集が防止される。また、第2の造核剤が疎水性となるように、第2の造核剤にステアリン酸にて表面処理を行っているため、第2の造核剤と母材樹脂との親和性が高くなり、より再凝集しにくくなる。
【0038】
混練の際、必要に応じて、さらに顔料、ゴム含有ポリマー、相溶化材、可塑剤、滑剤、難燃剤、抗菌剤、結晶化促進剤、酸化防止剤、紫外線吸収剤、熱安定剤、界面活性剤、帯電防止剤等の各種添加材を配合してもよい。
【0039】
複合体を母材樹脂に配合する方法としては、複合体を高濃度に樹脂に配合したマスターバッチを作製し、このマスターバッチをさらに樹脂に配合しても良い。
【0040】
そして、複合体が混ぜ合わされた母材樹脂および発泡剤、さらに、必要があれば、発泡樹脂成形体の所望のカラーバリエーションに対応した顔料入りのマスターバッチをドライブレンドする。
【0041】
ドライブレンドされた混合物は射出成形機へと供給され、一定の圧力条件下で発泡剤の発泡を抑制した状態で、2つの金型によって形成される空間、つまりキャビティへ射出される。そして、スキン層が形成された後に一方の金型を他方の金型に対して後退させることで、混合物の密度が低下し、また圧力が開放される。いわゆるキャビティ拡張法である。これより、発泡剤が分解して、第1の造核剤と第2の造核剤の表面を起点に、炭酸ガスや窒素ガスなどの気泡が発生する。この気泡が発泡セルとなり、発泡層が形成され、発泡樹脂成形体が成形される。
【0042】
なお、発泡樹脂成形体の成形方法としては、ショートショット法、エグレッション法等を用いても良い。
【0043】
上述したように、母材樹脂内に第1の造核剤と第2の造核剤とが均一に分散されている。そのため、本発明による発泡樹脂成形体における、第1の造核剤の表面で発生した気泡による第1の発泡セル、および第2の造核剤の表面で発生した気泡による第2の発泡セルは、発泡樹脂成形体内で偏りなくほぼ均一に形成される。
【0044】
次に、本発明の成形方法で成形された発泡樹脂成形体(以下の実施例1および実施例2)と、従来の成形方法で成形された発泡樹脂成形体(以下の比較例1および比較例2)とを比較した。ただし、比較例1では、発泡させていないので、発泡樹脂成形体ではなく、樹脂成形体が成形される。
【0045】
[実施例1]
母材樹脂であるポリプロピレン樹脂90質量部に対して、第1の造核剤(タルク)と第2の造核剤(炭酸カルシウム)との複合体を10質量部、発泡剤を3質量部の割合で混合し、発泡倍率が2倍となるように射出発泡成形を行い、発泡樹脂成形体を成形した。なお、複合体は、タルクが60質量%、炭酸カルシウム40質量%の割合となるように予めヘンシェルミキサで高速撹拌して得られたものである。第1の造核剤のタルクは、平均粒径が3.2μmであり、第2の造核剤の炭酸カルシウムの平均粒径は80nmである。
【0046】
[実施例2]
複合体は、タルク75%、炭酸カルシウム25%の割合とした。それ以外は実施例1と同様にして発泡樹脂成形体を成形した。
【0047】
[比較例1]
比較例1では、実施例1および実施例2において母材樹脂として用いたポリプロピレン樹脂を用いて、従来の射出成形によって樹脂成形体を成形した。第1の造核剤、第2の造核剤、および発泡剤は混合しておらず、発泡射出成形は行っていない。
【0048】
[比較例2]
比較例2では、実施例1および実施例2と同様にポリプロピレン樹脂を母材樹脂として用い、この母材樹脂90質量部に対して、発泡剤を3質量部、第1の造核剤を10質量部の割合で混合し、発泡倍率が2倍となるように射出発泡成形を行い、発泡樹脂成形体を成形した。なお、実施例1および実施例2と同様に第1の造核剤としてタルクを用いたが、第2の造核剤は使用していない。つまり、比較例2は、従来の発泡射出成形である。
【0049】
以上のようにして作成された実施例1の発泡樹脂成形体と、従来の成形方法で成形した比較例2の発泡樹脂成形体とを比較するために、マイクロスコープと走査型電子顕微鏡(SEM)による、発泡樹脂成形体の断面の観察を行った。
【0050】
図1〜図3は、実施例1の発泡樹脂成形体の断面の拡大写真であり、図1は倍率が17倍、図2は倍率が1000倍、図3は倍率が3000倍である。また、図4〜図6は、比較例2の発泡樹脂成形体の断面の拡大写真であり、図4は倍率が17倍、図5は倍率が1000倍、図6は倍率が3000倍である。
【0051】
実施例1では、厚み約3mmの発泡樹脂成形体1が成形されており、発泡樹脂成形体1の表面には、厚みがおよそ0.2〜0.6mmのスキン層2が形成されている。スキン層2に挟まれた発泡層3には、第1の造核剤に起因して、孔径が10〜500μmの第1の発泡セル4(図1の発泡層3に確認できる発泡セル)が多数形成されていることがわかる。
【0052】
また、図1〜図3より、実施例1には、第1の発泡セル4同士の間(以降「セル壁」と称する)には、第2の造核剤に起因して、孔径がおよそ10〜1000nmの微細な第2の発泡セル5が多数形成されていることがわかる。なお、図2で確認できるセル壁の小さな穴や、図3で確認できるセル壁の小さな穴は、いずれも第2の発泡セルである。
【0053】
一方、比較例2では、厚み約3mmの発泡樹脂成形体11が成形されており、発泡樹脂成形体11の表面には、厚みがおよそ0.2〜0.6mmのスキン層12が形成されている。スキン層12に挟まれた発泡層13には、第1の造核剤に起因して、孔径10〜1000μmの第1の発泡セル14(図4の発泡層13に確認できる発泡セル)が多数形成されていることがわかる。
【0054】
また、図4〜図6の比較例2の発泡樹脂成形体11のセル壁には、実施例1で確認できる微細な発泡セルが確認できない。
【0055】
以上のことより、本発明の発泡樹脂成形体1は、第2の造核剤を使用していない従来の発泡樹脂成形体11に比べて、第1の発泡セル4を小さくすることができ、また、セル壁には、極微細な第2の発泡セル5を多数形成することができる。つまり、従来の発泡樹脂成形体11に比べて、本発明の発泡樹脂成形体1では、発泡セルの孔径を小さくし、かつ数を増やすことができる。
【0056】
本発明の発泡樹脂成形体1における第1の発泡セル4は、従来の発泡樹脂成形体11における第1の発泡セル14よりも小さい理由を考察してみる。発泡剤によって母材樹脂内に発生するガスの量は同じであると仮定する。従来の発泡樹脂成形体11では、発生したガスは第1の造核剤に起因して気泡を生じる。一方、本発明では、第1の造核剤のほかに、微細な第2の造核剤を使用しているため、発生したガスの一部は第1の造核剤に起因して気泡を生じ、発生したガスの他の一部は第2の造核剤に起因して気泡を生じる。つまり、発生したガスは、第1の造核剤と第2の造核剤とに分散して気泡を生じることになるため、第1の発泡セル4が小さくなったと考えられる。また、第2の造核剤の粒子は非常に小さく、母材樹脂内に均一に分散しているため、第2の造核剤に起因して生じる気泡は小さく、気泡同士が結びついて大きな気泡になってしまうことが少なく、図2や図3からわかるように、第2の発泡セル5は小さい。このことから、第2の発泡セル5は大きくならないと考えられる。
【0057】
次に、実施例1、2および比較例1、2の物性の違いを調べた。はじめに試験方法について説明する。
【0058】
(耐寒落球衝撃強さ)
試験片31は厚さ3mmの発泡樹脂成形体から長さ×幅=140mm×100mmに切り出したものを用いた。試験方法は、図7に示すように、試験片31の長さ方向の両端をそれぞれ20mm金属製の治具32に挟んで保持し、治具32から露出している部分である、長さ×幅=100mm×100mmの領域を打撃面33とした。なお、治具32には5Nのトルクをかけることで、試験片31を保持した。雰囲気は−30℃であり、半径25mmの球状頭部を持つ質量500gの錘34を打撃面33の中心の上方から落下させた。この試験は、錘34を落とす高さhを変えて行い、各高さhあたり5回試験を実施した。錘34を落下させた後、試験片31の状態を観察し、試験片31に変化が無いまたは白化したものを「○」、亀裂又は破壊したものを「×」とした。この結果から、○の数が50%になる高さhを算出し、この高さhより衝撃強さを算出した。その結果を図8に示している。
【0059】
(曲げ弾性勾配)
「JIS K 7171 プラスチック−曲げ特性の求め方」を試験方法として採用し、曲げ弾性勾配を測定した。試験片は、厚さ3mmの発泡樹脂成形体から、樹脂流れ方向(MD)と樹脂流れ方向に直交する直交方向(TD)とからそれぞれ長さ×幅=150mm×50mmに切り出したものを用いた。試験機には半径2mmの支持台および圧子を取り付け、支点間距離100mm、試験速度50mm/sで試験を実施した。試験結果は、MD、TDの平均値とし、図8に示している。
【0060】
(スワールマーク)
成形品の外観を目視にて観察し、スワールマークの有無について、以下の基準で評価した。なお、スワールマークは成形時にフローフロントで発泡が起こり、その気泡を引きずった跡が成形体の表面に外観不良として現れるものであり、発泡射出成形ではこの発生が避けられないが、発泡セル径が小さいほど目立ちにくくなる。以下の基準で評価した。「○」:問題無し、「△」:目立たない程度に(一部に)スワールマークがみられる、「×」:目立つ程度にスワールマークがみられる、である。試験結果を図8に示す。
【0061】
(耐傷つき性)
成形品に、先端の直径が1mmの鋼鉄針を用い、500gの荷重をかけ、1000mm/minの引っかき速度で、2mmの間隔で碁盤目状に傷をつける。その後、成形品の外観を目視にて観察し傷つきの有無について、以下の基準で評価した。「○」:問題無し、「△」:目立たない程度に(一部に)傷がみられる、「×」:目立つ程度に傷がみられる、である。試験結果を図8に示す。
【0062】
単なる射出成形を行った樹脂成形体である比較例1と、発泡樹脂成形体である実施例1、2および比較例2を比較すると、曲げ弾性勾配は、単なる射出成形体である比較例1に比べて発泡樹脂成形体である実施例1、2および比較例2のほうが著しく大きい。つまり、発泡樹脂成形体である実施例1、2および比較例2は、単なる射出成形体である比較例1に比べて剛性が著しく向上することがわかる。
【0063】
比較例2と実施例1および実施例2とを比較すると、比較例2に比べて、実施例1および実施例2のほうが、衝撃に強いことがわかる。
【0064】
実施例1および実施例2の優れた耐衝撃性は、ナノサイズの第2の造核剤に起因する第2の発泡セルによるものと考えられる。
【0065】
実施例1および実施例2のスワールマークおよび傷つき性は、比較例2に比べて改善されることがわかる。つまり、比較例2に比べて意匠性が向上する。これにより、本発明の発泡樹脂成形体は、従来の発泡樹脂成形体に比べて塗装量を削減可能となり、コストを削減することができる。さらに、発泡樹脂成形体を適応できる製品や用途を拡大させることができる。
【0066】
以上の結果から、複合体における第1の造核剤と第2の造核剤との重量比は、75:25〜60:40の間であることが好ましい。また、複合体は、母材樹脂100質量部に対して10質量部以上、混合物に混入されていることが好ましい。
【符号の説明】
【0067】
1、11 発泡樹脂成形体
2、12 スキン層
3、13 発泡層
4、14 第1の発泡セル
5 第2の発泡セル
【特許請求の範囲】
【請求項1】
表面がスキン層で形成され、内部が発泡層で形成されている発泡樹脂成形体であって、
前記発泡層には、複数の第1の発泡セルと、前記第1の発泡セル同士の間に形成され、前記第1の発泡セルより小さい複数の第2の発泡セルと、が形成されている、発泡樹脂成形体。
【請求項2】
前記第1の発泡セルの平均孔径が10〜1000μmであり、前記第2の発泡セルの平均孔径が10〜1000nmである、請求項1に記載の発泡樹脂成形体。
【請求項3】
前記第1の発泡セルを形成させるための第1の造核剤と、前記第2の発泡セルを形成させるための第2の造核剤と、を有する請求項1または2に記載の発泡樹脂成形体。
【請求項4】
表面がスキン層で形成され、内部が発泡層で形成されている発泡樹脂成形体の成形方法であって、
第1の造核剤の表面に、前記第1の造核剤よりも小さな第2の造核剤を付着させて、前記第1の造核剤と前記第2の造核剤との複合体を形成し、
樹脂と、発泡剤と、前記複合体とを混合して混合物を形成し、
前記混合物を発泡射出成形する、発泡樹脂成形体の成形方法。
【請求項5】
前記第1の造核剤の粒径は、0.5〜1000μmであり、前記第2の造核剤の粒径は、10〜500nmである、請求項4に記載の発泡樹脂成形体の成形方法。
【請求項6】
前記第1の造核剤の粒径は、1〜10μmであり、前記第2の造核剤の粒径は、50〜100nmである、請求項5に記載の発泡樹脂成形体の成形方法。
【請求項7】
前記第1の造核剤および前記第2の造核剤は無機物である、請求項4から6のいずれか1項に記載の発泡樹脂成形体の成形方法。
【請求項8】
前記第1の造核剤はタルクであり、前記第2の造核剤は炭酸カルシウムである、請求項7に記載の発泡樹脂成形体の成形方法。
【請求項9】
前記第2の造核剤は、疎水性を有するように表面処理されている、請求項4から8のいずれか1項に記載の発泡樹脂成形体の成形方法。
【請求項10】
前記複合体における、前記第1の造核剤と前記第2の造核剤との重量比は75:25〜60:40の間である、請求項4から9のいずれか1項に記載の発泡樹脂成形体の成形方法。
【請求項11】
前記複合体は、前記樹脂の質量に対して1〜80質量%、前記混合物に混入されている、請求項4から10のいずれか1項に記載の発泡樹脂成形体の成形方法。
【請求項1】
表面がスキン層で形成され、内部が発泡層で形成されている発泡樹脂成形体であって、
前記発泡層には、複数の第1の発泡セルと、前記第1の発泡セル同士の間に形成され、前記第1の発泡セルより小さい複数の第2の発泡セルと、が形成されている、発泡樹脂成形体。
【請求項2】
前記第1の発泡セルの平均孔径が10〜1000μmであり、前記第2の発泡セルの平均孔径が10〜1000nmである、請求項1に記載の発泡樹脂成形体。
【請求項3】
前記第1の発泡セルを形成させるための第1の造核剤と、前記第2の発泡セルを形成させるための第2の造核剤と、を有する請求項1または2に記載の発泡樹脂成形体。
【請求項4】
表面がスキン層で形成され、内部が発泡層で形成されている発泡樹脂成形体の成形方法であって、
第1の造核剤の表面に、前記第1の造核剤よりも小さな第2の造核剤を付着させて、前記第1の造核剤と前記第2の造核剤との複合体を形成し、
樹脂と、発泡剤と、前記複合体とを混合して混合物を形成し、
前記混合物を発泡射出成形する、発泡樹脂成形体の成形方法。
【請求項5】
前記第1の造核剤の粒径は、0.5〜1000μmであり、前記第2の造核剤の粒径は、10〜500nmである、請求項4に記載の発泡樹脂成形体の成形方法。
【請求項6】
前記第1の造核剤の粒径は、1〜10μmであり、前記第2の造核剤の粒径は、50〜100nmである、請求項5に記載の発泡樹脂成形体の成形方法。
【請求項7】
前記第1の造核剤および前記第2の造核剤は無機物である、請求項4から6のいずれか1項に記載の発泡樹脂成形体の成形方法。
【請求項8】
前記第1の造核剤はタルクであり、前記第2の造核剤は炭酸カルシウムである、請求項7に記載の発泡樹脂成形体の成形方法。
【請求項9】
前記第2の造核剤は、疎水性を有するように表面処理されている、請求項4から8のいずれか1項に記載の発泡樹脂成形体の成形方法。
【請求項10】
前記複合体における、前記第1の造核剤と前記第2の造核剤との重量比は75:25〜60:40の間である、請求項4から9のいずれか1項に記載の発泡樹脂成形体の成形方法。
【請求項11】
前記複合体は、前記樹脂の質量に対して1〜80質量%、前記混合物に混入されている、請求項4から10のいずれか1項に記載の発泡樹脂成形体の成形方法。
【図7】


【図8】
【図1】

【図2】

【図3】
【図4】
【図5】

【図6】

【図9】
【図8】
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【図9】
【公開番号】特開2012−233055(P2012−233055A)
【公開日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願番号】特願2011−101712(P2011−101712)
【出願日】平成23年4月28日(2011.4.28)
【出願人】(390031451)株式会社林技術研究所 (83)
【Fターム(参考)】
【公開日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願日】平成23年4月28日(2011.4.28)
【出願人】(390031451)株式会社林技術研究所 (83)
【Fターム(参考)】
[ Back to top ]