
発現プロセス

ターゲットポリペプチドの産生のための発現系を提供する。発現系は、ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターを含む発現カセット、およびDNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターを含む、DNA結合性転写制御因子タンパク質を過剰発現させるための発現カセットを含む。発現カセットは直交性プロモーターの調節下にある。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はポリペプチドの発現のためのプロセス、および発現系に関する。
【背景技術】
【0002】
学術的または商業的に関心対象である多くのポリペプチド、特に療法ポリペプチドは、組換え宿主細胞における発現によって好適に産生される。特に製造環境における、こうした異種ポリペプチドの効率的な産生のため、ポリペプチドが製造における適切な段階でのみ産生されるように、ポリペプチドを調節することが好ましい。したがって、望ましい場合に適切な条件の適用によって発現が誘発可能であるように、ポリペプチドの発現は誘導因子の調節下に置かれる。例えば、いくつかの異種ポリペプチドは、宿主細胞増殖およびプラスミド安定性に有害な影響を有し、全体の生産性を減少させるため、誘導前の異種ポリペプチドの発現(いわゆる「漏出性(leaky)」発現)、すなわち基底発現の劣った調節は、一般的に望ましくない。いくつかの場合、異種ポリペプチドの漏出性発現は、クローニング中のベクター安定性の問題を生じる可能性もあり、所望のポリペプチドを発現する安定な発現ベクター構築の失敗につながりうる。
【0003】
異なる調節様式および誘導様式を持つ多くの異種ポリペプチド発現系があり、関心対象のポリペプチドを産生するための発現系/発酵プロセスの選択および最適化は大部分、経験的なプロセスとなっている。これには時間がかかり、そして望ましくない。したがって、発現の調節の改善、特に基底発現の調節の改善を提供する系が必要である。
【0004】
誘導性遺伝子発現は、遺伝子機能の分析に重要なツールである。これらの系には、誘導されない細胞において、遺伝子発現の潜在的に有害な影響を減少させ、そしてかつ誘導された遺伝子の発現による生物学的に関連する影響の明らかな区別を可能にするために、基底遺伝子発現の厳重な調節が、決定的に必要である。
【0005】
漏出性基底発現の問題を減少させるかまたは排除することを試みる、多様な改善法には、成功と失敗が入り交じってきている。例えば、大腸菌(E. coli)における基底発現を減少させるためのアプローチには、リプレッサータンパク質対オペレーター比の増加、突然変異体ファージの感染による誘導、プロモーター強度の減弱、アンチターミネーターと組み合わせた転写ターミネーターの使用が含まれる(Makrides, Microbiological Reviews, 1996, 512−538によって概説される)。タンパク質を高レベルで発現するために、トランスで供給されるT7 RNAポリメラーゼを用いるpETベクター(Novagen)に伴う基底発現の問題は、T7 RNAポリメラーゼを阻害するT7リゾチームの同時過剰発現によって減少させうる。しかし、基底発現を減少させるかまたは排除するために使用する最適な発現構成要素の使用(Studier, Journal of Molecular Biology, 1991, 219(1):37−44)は経験的であり、基底発現および誘導性に対する多数のベクター(pLysS、pLysL、pLysEおよびpLysH)の影響を決定するスクリーニングを必要とする。哺乳動物細胞において用いられるテトラサイクリンが制御する発現系の基底発現は問題である(Meyer−Ficcaら, Anal. Biochem, 2004, 334:9−19)。Laiら, Am. J. Physiol Cell Physiol, 2003, 285:711−719は、テトラサイクリンまたはエクジソン応答配列を用いて発現されるレポーター遺伝子(緑色蛍光タンパク質)を用いて、高い基底発現を持つ哺乳動物細胞がどのように生成されたかを記載する。これらの細胞は、レポーター遺伝子系と組み合わせて蛍光活性化細胞分取を用いることによって除去されうる。Beckerら, Nucleic Acids
Research, 2008, 1−13は、大腸菌における天然HU抑制タンパク質に対する置換としての真核HMGBタンパク質の使用を開示する。天然大腸菌LacZ/ベータ−ガラクトシダーゼ遺伝子の産生抑制が研究されている。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Makrides, Microbiological Reviews, 1996, 512−538
【非特許文献2】Studier, Journal of Molecular Biology, 1991, 219(1):37−44
【非特許文献3】Meyer−Ficcaら, Anal. Biochem, 2004, 334:9−19
【非特許文献4】Laiら, Am. J. Physiol Cell Physiol, 2003, 285:711−719
【非特許文献5】Beckerら, Nucleic Acids Research, 2008, 1−13
【発明の概要】
【0007】
本発明の1つの側面にしたがって、ターゲットポリペプチド産生のための発現系であって:
a)ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターを含む発現カセット;および
b)DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターを含む、DNA結合性転写制御因子タンパク質を過剰発現させるための発現カセット;
ここで該発現カセットは直交性(orthogonal)プロモーターの調節下にある
を含む、前記発現系を提供する。
【図面の簡単な説明】
【0008】
【図1】図1は、HupA遺伝子配列である。
【図2】図2は、大腸菌コドン最適化ラットHMGB−1遺伝子配列である。
【図3】図3は、IgG Fc遺伝子配列である。
【図4】図4は、実施例2の結果である。
【図5】図5は、実施例3の結果である。
【図6】図6は、実施例4の結果である。
【発明を実施するための形態】
【0009】
プロモーターは、異なる条件によって活性化される際、直交性であると認識される。これらの条件は、互いに排他的であってもよいし、または適合可能であってもよい。互いに排他的なプロモーターは、1つのプロモーターの調節下にあるDNAの発現に必要な条件が、別のプロモーターの調節下にあるDNAの発現を防止するものであり、そしてこれには、例えば、テトラサイクリン反応系が含まれ、これは、テトラサイクリンの存在下で「オフ」である(すなわち発現が防止される)が、テトラサイクリンの非存在下で「オン」である(すなわち発現が促進される)(「テトラサイクリン・オフ」)ように設計可能であるし、またはテトラサイクリンの存在下で「オン」である(「テトラサイクリン・オン」)が、その非存在下で「オフ」であるようにも設計可能である。DNA結合性転写制御因子タンパク質をコードするDNAが、テトラサイクリン・オフであるプロモーターに機能可能であるように連結され、そしてターゲットポリペプチドをコードするDNAが、テトラサイクリン・オンであるプロモーターに機能可能であるように連結される場合、テトラサイクリンの添加は、ターゲットポリペプチドの発現を誘導するが、DNA結合性転写
制御因子タンパク質のさらなる発現を防止するであろう。
【0010】
多くの態様において、機能可能であるように連結されるDNA配列は隣接する。
本発明の発現系で使用可能なDNA結合性転写制御因子タンパク質は、原核および真核生物由来のタンパク質を含む。原核タンパク質には、大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−β、組込み宿主因子(IHF)−α、IHF−βおよび相同体が含まれる。真核タンパク質は、好ましくは、酵母、例えばNHP6A、NHP6B、または哺乳動物タンパク質であり、そしてこれには、豊富配列非特異的高移動度群(HMG)タンパク質が含まれる。HMGタンパク質は、3つのファミリー: HMGBファミリー、例えばHMGB−1、HMGB−2、HMGB−3; HMGNファミリー、例えばHMGN−1、HMGN−2、HMGN−3、ならびにHMGAファミリー、例えばHMGA−1a、HMGA−1b、HMGA−1c、HMGA−2、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)などの酵母におけるミトコンドリア・ヒストン(HM)タンパク質、ならびにヒトおよび両生類ミトコンドリアで見られる相同体、ならびに細菌ヒストン様タンパク質HUに似たタンパク質をコードする植物(クリプト藻(Guillardia theta))葉緑体hlpAを含む、原核DNA結合性転写制御因子タンパク質の真核相同体に分けられる。好ましいDNA結合性転写制御因子タンパク質は、大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−βおよびHMGB−1、特にラットHMGB−1である。
【0011】
本発明の発現カセットは、宿主細胞ゲノム内に組み込まれるか、またはプラスミドなどの染色体外要素内に含まれてもよい。染色体外要素内に含まれる場合、発現カセットは、同じ染色体外要素、例えばプラスミドベクター上に含まれてもよいし、または宿主細胞に同時形質転換される、適合する別個の染色体外要素上に含まれてもよい。あるいは、発現系は、ファージまたはウイルスベクター内に取り込まれてもよく、そしてこれらを用いて、宿主細胞系内に発現系が送達されてもよい。当該技術分野に知られる方法によって、プラスミドまたは他の発現ベクターを組み立ててもよい。本発明の発現カセットは、好ましくは、どちらも別個のプラスミドの形で使用される。プラスミドは自律複製プラスミドまたは組込み性プラスミドであってもよい。プラスミドは典型的には1以上の以下も含む:選択可能マーカー、例えば抗生物質耐性を与える配列、およびcer安定性配列。
【0012】
発現カセットが別個の染色体外要素上に含まれる態様において、両方の要素を含む形質転換宿主細胞を同定するため、異なる選択可能マーカーを使用することが好ましい。
DNA結合性転写制御因子タンパク質を過剰発現するための発現カセットで使用するプロモーターは、構成的または誘導性プロモーターであってもよい。本発明の側面で使用可能な構成的プロモーターの例には、T7A1、T7A2、T7A3、spcリボソームタンパク質オペロンプロモーター、β−ラクタマーゼ遺伝子プロモーター、ファージλのPLプロモーター、複製調節プロモーターPRNAIおよびPRNAII、rrnBリボソームRNAオペロンのP1およびP2プロモーター、Lacリプレッサータンパク質プロモーターpLacI、グリセルアルデヒドリン酸デヒドロゲナーゼ(GAPDH)および形質膜H(+)−ATPアーゼ(PMA1)プロモーター、接合因子(MF)−αプロモーター、KEX2、TEF−1、サルウイルス40(SV40)初期プロモーター、ラウス肉腫ウイルス(RSV)プロモーター、サイトメガロウイルス(CMV)プロモーターおよびヒトβアクチンプロモーターが含まれる。構成的プロモーターのさらなる例には、調節領域を除去するよう修飾されている誘導性プロモーター、例えばlacまたはtacオペレーターを除去するよう修飾されているlacまたはtacプロモーターが含まれ、そしてこうしたプロモーターは、本発明の特定の態様において、特にDNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されている場合に好適である。
【0013】
本発明の側面において使用可能な誘導性プロモーターの例には、ファージRNAポリメラーゼ依存性プロモーター、特にT7 RNAポリメラーゼ依存性プロモーター系、好ましくは単一のT7プロモーターが含まれ、本明細書に援用されるStudierおよびMoffat, J. Mol. Biol. 189:113−130(1986)によって開示されるもの、特にT7遺伝子10プロモーターが含まれる。T7 RNAポリメラーゼ依存性プロモーター系を使用する場合、T7 RNAポリメラーゼの供給源が必要とされることが認識され、これは当該技術分野に知られる方法によって、そして一般的には大腸菌宿主株内に必要なファージポリメラーゼを発現するλDE3プロファージを挿入して、溶原性宿主株を生成することによって提供される。T7 RNAポリメラーゼはまた、T7 RNAポリメラーゼの遺伝子を所持する特殊化λ形質導入ファージでの感染によって細胞に送達されることも可能である。使用可能な誘導性プロモーターのさらなる例には、lac、lacUV5、trp、tac、trc、phoA、アラビノース誘導性プロモーター、温度誘導性プロモーター(高温および低温両方)、銅誘導性プロモーター、uspA、uspB、malK、浸透圧誘導性プロモーター、ガラクトース誘導性プロモーター、フェロモン誘導性プロモーター、グルコアミラーゼプロモーター、テトラサイクリン応答性プロモーター、ヒトc−fosプロモーター、エクジソン誘導性プロモーター、およびグルココルチコイド誘導性プロモーターが含まれる。プロモーターが一般的に、宿主細胞で有効であることが知られるプロモーターから選択されることが認識されるであろう。例えば、大腸菌プロモーターは、大腸菌宿主細胞において一般的に使用され、哺乳動物プロモーターは哺乳動物細胞において一般的に使用され、酵母プロモーターは、酵母細胞において一般的に使用される。原核宿主由来の多くのプロモーターは、真核宿主において使用可能であり、そして逆も可能であることが認識されるであろう。少なくとも1つの誘導性プロモーターが使用されることが好ましく、特に好ましくは、ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターが使用される。いくつかの態様において、DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結された構成的プロモーターと組み合わされた、ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターが特に好ましい。
【0014】
カセットの発現が適切な条件の改変によってスイッチ可能であるように、発現カセットにおいて、直交性プロモーターを使用する。ターゲットタンパク質の発現を誘導するように条件を調整するまで、DNA結合性転写制御因子タンパク質の過剰発現は、ターゲットポリペプチドの基底発現を減少させるかまたは防止する効果を有する。
【0015】
本発明のプロセスにおいて使用可能な誘導因子は、使用する誘導性プロモーターに対応するように選択され、そしてこれには、イソプロピル−β−D−1−チオガラクトピラノシド(IPTG)、IPTGの類似体、例えばイソブチル−C−ガラクトシド(IBCG)、ラクトースまたはメリビオース、ガラクトース、アラビノース、ラムノース、温度、pH、溶解酸素レベル、銅などの金属イオン、インドールアクリル酸、テトラサイクリン、ホモセリンラクトン、エクジソン、サリチル酸塩、リン酸塩が含まれる。他の誘導因子を用いてもよく、そしてこれは、別の箇所により詳細に記載される(例えば、The Operon, MillerおよびRenznikoff監修(1978)を参照されたい)。誘導因子を個々にまたは組み合わせて用いてもよい。
【0016】
ターゲットポリペプチドの発現を誘導する場合、DNA結合性転写制御因子タンパク質の発現を減少させるかまたは防止するように条件を選択することが好ましい。ターゲットタンパク質発現中に、DNA結合性転写制御因子タンパク質の発現を減少させるかまたは防止するために使用する方法には、1以上の以下が含まれる:
a)効能を維持するために補充を必要とする、DNA結合性転写制御因子タンパク質発現を調節するプロモーターおよび誘導因子を使用する。例えば誘導因子が代謝されるか、
無効であるように修飾されるか、または有効であるより低く濃度が低下するように細胞によって吸収される場合;
b)DNA結合性転写制御因子タンパク質の発現を調節する誘導因子を含有する培地から細胞を物理的に分離し、その後、ターゲットポリペプチドの発現を調節するのに必要な誘導因子を含有する培地内に細胞を再導入する;
c)基底発現を調節するのに十分なDNA結合性転写制御因子タンパク質が産生される一方、発現が誘導された際にターゲットポリペプチドの発現を防止しないかまたは有意に損なわないように、ターゲットポリペプチドの発現を調節するプロモーターよりも、DNA結合性転写制御因子タンパク質発現を調節するプロモーターの強度がより弱いように選択する;
d)基底発現を調節するのに十分なDNA結合性転写制御因子タンパク質が産生される一方、発現が誘導された際にターゲットポリペプチドの発現を防止しないかまたは有意に損なわないように、ターゲットポリペプチドをコードするベクターよりもより少ないコピー数のDNA結合性転写制御因子タンパク質をコードするベクターを選択する;
e)DNA結合性転写制御因子タンパク質の発現を損なうための、アンチセンスまたはRNA干渉(例えばマイクロRNA(miRNA)または低分子干渉RNA(siRNA))を使用する;
f)例えば、ターゲットポリペプチドの発現が誘導される際に、阻害剤が発現されるように、選択されるDNA結合性転写制御因子タンパク質の阻害剤を発現する。多くのこうした態様において、阻害剤は、ターゲットポリペプチドと同じベクター上で発現されてもよい;
g)例えば、ターゲットポリペプチドの発現が誘導される際に、プロテアーゼが発現されるように、選択されるDNA結合性転写制御因子タンパク質をターゲットとするプロテアーゼを発現する。多くのこうした態様において、プロテアーゼは、ターゲットポリペプチドと同じベクター上で発現されてもよい;
h)例えば、ターゲットポリペプチドの発現が誘導される際に、阻害剤が発現されるように、選択されるDNA結合性転写制御因子タンパク質の発現の阻害剤を発現する。例には、DNA結合性転写制御因子タンパク質の同族(cognate)リプレッサー(例えばcIリプレッサータンパク質)の構成的発現を伴わず、通常、誘導性であるプロモーター、例えばλpLを用いることが含まれる。誘導性プロモーターを用いてターゲットポリペプチドが発現され、そして同じ誘導性プロモーターを用いて阻害剤が発現される場合、誘導因子が存在しなければ、阻害はなく、そしてDNA結合性転写制御因子タンパク質発現は構成的である。ターゲットポリペプチドの発現を誘導する誘導因子の添加はまた、阻害剤の発現も誘導し、そして阻害剤は、DNA結合性転写制御因子タンパク質の発現を防止する。多くのこうした態様において、阻害剤は、ターゲットポリペプチドと同じベクター上で発現されてもよい;
i)ある程度のリードスルーを可能にするターゲットポリペプチドの転写ターミネーターを伴い、DNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現要素を異なる方向で含むようにプラスミドベクターを設計する。ここで、DNA結合性転写制御因子タンパク質発現要素は構成的プロモーターを含み、そしてターゲットポリペプチドの発現カセットに続く。誘導因子が添加されたら、ターゲットポリペプチドからDNA結合性転写制御因子タンパク質の発現カセットに進む転写リードスルーは、DNA結合性転写制御因子タンパク質の転写を減少させるかまたは停止させるであろう;そして
j)誘導因子が添加された際、DNA結合性転写制御因子タンパク質発現要素がベクターまたは宿主ゲノムから切除され、そしてしたがってもはや発現不能となるが、ターゲットポリペプチド発現要素を損なわれず(intact)に残すように、発現系を設計する。
【0017】
DNA結合性転写制御因子タンパク質の過剰発現が、宿主細胞で通常見られるより高いレベルでのこうしたタンパク質の発現を指すことが認識されるであろう。DNA結合性転
写制御因子タンパク質が、宿主細胞に対して天然である場合、過剰発現は、こうしたタンパク質の発現を正常レベルより高く増加させることを含む。非天然DNA結合性転写制御因子タンパク質を使用する場合、宿主細胞で通常見られるレベルはゼロである。DNA結合性転写制御因子タンパク質は、ターゲットポリペプチドの基底発現が減少するかまたは防止されるレベルまで過剰発現される。
【0018】
本発明記載の発現系で使用可能なオペレーター配列には、lac、gal、deoおよびglnが含まれる。1以上、好ましくは2つのオペレーター配列を使用してもよい。特定の態様において、2以上の完全なパリンドローム・オペレーター配列を使用してもよい。多くの好ましい態様において、2つの完全なパリンドローム・オペレーター配列を使用し、最も好適には、1つのオペレーター配列はプロモーター下流に位置し、そして1つのオペレーター配列はプロモーター上流に位置する。2つのオペレーター系を使用する場合、オペレーター配列は、好ましくは、プロモーターの調節を最大にするように間隔を開けられる。2つのオペレーターは、最も一般的には、誘導性プロモーターと組み合わせて使用されることが認識されるであろう。多くの態様において、間隔は85〜150塩基対離れており、好ましくは90〜126塩基対離れており、そして最も好ましくは91または92塩基対離れている。特定の態様において、オペレーター配列は、転写開始点と重なる。
【0019】
オペレーター系は、一般的に、適切なリプレッサー配列とともに使用されることが認識されるであろう。リプレッサー配列は、リプレッサータンパク質を産生し、例えばlacオペレーターを用いる場合、lacI遺伝子配列である。他のlacリプレッサー配列もまた使用可能であり、例えばlacIQ配列を用いて、lacリプレッサータンパク質レベルを増加させてもよい。また、宿主細胞ゲノムによって、またはさらなる適合プラスミドを用いることによって、リプレッサー配列を提供してもよい。
【0020】
本発明の発現系は、ターゲットポリペプチドの基底発現の調節を提供し、そしてしたがって、より有効でそして調節可能なターゲットポリペプチド製造を提供する。さらに、本発明の発現系は、毒性の影響を有するか、あるいはベクターまたは宿主株の安定性に不都合に影響を及ぼす可能性がある、ターゲットポリペプチドの基底発現を抑制することによって、より有効なベクター組み立ておよび/またはクローニングを可能にする。
【0021】
本発明の発現系をベクター組み立てまたはクローニングで使用する場合、特定の態様において、しばしば、所望のベクター組み立てまたはクローニング後、ターゲットポリペプチドに機能可能であるように連結されたプロモーターに有利に働く(favour)条件を選択すると、前記ポリペプチドの発現が可能になるように、発現系を選択する。こうした態様は、例えばベクター組み立てで使用するものと同じ宿主細胞、最も一般的には大腸菌において、ターゲットポリペプチドを発現させようとする状況に特に適している。他の態様において、DNA結合性転写制御因子タンパク質によるターゲットポリペプチド発現の調節が非常に大きく、単にターゲットポリペプチド発現のための条件を選択するだけで、ターゲットポリペプチドがほとんどまたはまったく産生されないように発現系を選択する。こうした態様において、ターゲットポリペプチドの発現は、例えば以下によって達成可能である:
a)DNA結合性転写制御因子タンパク質を発現する細胞がもはや選択不能であるが、ターゲットポリペプチドのみを発現する細胞が選択される発現条件を使用する。こうしたアプローチは、別個のベクター上にあるDNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現カセットの使用に適している;
b)ターゲットポリペプチドを発現するが、DNA結合性転写制御因子タンパク質を発現しない宿主細胞にベクターをトランスファーする。例えば、真核生物、例えば哺乳動物または酵母細胞におけるターゲットポリペプチドの発現のためのベクターを、原核生物、
特に大腸菌においてベクター組み立てまたはクローニングする。DNA結合性転写制御因子タンパク質のプロモーターは、ターゲットポリペプチド発現宿主によって認識されない。このアプローチは、DNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現カセットが、単一ベクター上に位置する発現系に特に適しており、そしてこれはまた、DNA結合性転写制御因子タンパク質およびターゲットポリペプチドが別個のベクター上に位置する場合にもまた使用可能である;
c)DNA結合性転写制御因子タンパク質をコードする発現カセットを切除して、ベクターの構造を修飾する。こうしたアプローチは、DNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現カセットが単一ベクター上に位置する発現系、ならびにDNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現カセットが別個のベクター上に位置する発現系に適している。
【0022】
多くの好ましい態様において、対応する発現系と比較した際、ターゲットポリペプチドの発現を助け、そしてDNA結合性転写制御因子タンパク質の発現を嫌う発現条件を選択した際に、ターゲットポリペプチドの発現が防止されるかまたは損なわれないが、DNA結合性転写制御因子タンパク質発現要素を欠くように、発現系を選択する。
【0023】
特定の特に好ましい発現系は、大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−βの発現カセットを含み、最も好ましくは、これは、構成的lacプロモーターに機能可能であるように連結されている。こうした発現系は、好適には、大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−βの、そしてターゲットポリペプチドの、別個のベクターを含み、ターゲットポリペプチドのベクターは、tacプロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含み、こうしたベクターはまた、好ましくは単一のオペレーター、特に天然lacオペレーターまたは完全にパリンドロームであるlacオペレーターも含む。
【0024】
さらに特に好ましい発現系は、ラットHMGB−1の発現カセットを含み、最も好ましくは、これは、構成的lacプロモーターに機能可能であるように連結されている。こうした発現系は、好適には、ラットHMGB−1の、そしてターゲットポリペプチドの、別個のベクターを含み、ターゲットポリペプチドのベクターは、tacプロモーターまたはT7、特にT7遺伝子10プロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含む。多くのこうした態様において、ターゲットポリペプチドのベクターがtacプロモーターを含む場合、ベクターはまた、単一のオペレーター、特に天然lacオペレーターまたは完全にパリンドロームであるlacオペレーターも含む。さらなるこうした態様において、ターゲットポリペプチドのベクターが、T7、特にT7遺伝子10プロモーターを含む場合、ベクターはまた、2つの完全にパリンドロームであるオペレーター、特にlacオペレーターも含み、最も好ましくは、1つのオペレーター配列はプロモーター上流に位置し、そして1つのオペレーター配列はプロモーター下流に位置する。
【0025】
本発明の発現系を使用して、宿主細胞において、そして特に微生物において、ポリペプチドを発現させてもよい。宿主細胞は原核または真核であってもよい。原核細胞の例には、細菌細胞、例えば大腸菌、ネズミチフス菌(Salmonella typhimurium)、セラチア・マルセセンス(Serratia marsescens)および緑膿菌(Pseudomonas aeruginosa)を含むグラム陰性細菌細胞、ならびに枯草菌(Bacillus subtilis)を含むグラム陽性細菌細胞が含まれる。真核細胞の例には、酵母、例えばピキア・パストリス(Pichia pastoris)、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、ハンセヌラ・ポリモルファ(Hansenula polymorpha)、クロイベロミセス・ラクティス(Kluyveromyces lactis)、シゾ
サッカロミセス・ポンベ(Schizosaccharomyces pombe)が含まれる。使用可能な哺乳動物宿主細胞には、ヒト細胞株、例えばヒト胎児腎臓およびPERC.6細胞;ネズミ細胞株、例えばNS0細胞;ならびに特に、ハムスター細胞株、例えばベビーハムスター腎臓細胞および特にチャイニーズハムスター卵巣細胞が含まれる。他の真核宿主細胞、例えば糸状菌、植物、昆虫、両生類細胞または卵巣(ovarian)種もまた使用可能である。好ましい宿主細胞は細菌、特に腸内細菌科(enterobacteriacae)、好ましくは大腸菌、そして特にそのBまたはK12株である。
【0026】
本発明の発現系は、組換え細胞を培養することによる、ポリペプチド、特に組換えポリペプチドの製造のために使用される。
本発明のプロセスによって発現可能なポリペプチドには、療法タンパク質およびペプチドが含まれ、サイトカイン、増殖因子、抗体、抗体断片、免疫グロブリン様ポリペプチド、酵素、ワクチン、ペプチドホルモン、ケモカイン、受容体、受容体断片、キナーゼ、ホスファターゼ、イソメラーゼ、ヒドロリアーゼ、転写因子および融合ポリペプチドが含まれる。
【0027】
発現可能な抗体には、モノクローナル抗体、ポリクローナル抗体および生物学的活性を有する抗体断片が含まれ、前述のいずれかの多価および/または多重特異性型が含まれる。
【0028】
天然存在抗体は、典型的には、4つのポリペプチド鎖、ジスルフィド結合によって相互連結された2つの同一の重(H)鎖および2つの同一の軽(L)鎖を含む。各重鎖は、可変領域(VH)および定常領域(CH)を含み、CH領域は、天然型で、3つのドメインCH1、CH2およびCH3を含む。各軽鎖は、可変領域(VL)および1つのドメインCLを含む定常領域を含む。
【0029】
VHおよびVL領域をさらに、より保存されたフレームワーク領域(FR)と称される領域が介在する、相補性決定領域(CDR)と称される超可変性領域にさらに分割してもよい。各VHおよびVLは、3つのCDRおよび4つのFRで構成され、これらはアミノ末端からカルボキシ末端に、以下の順で配置される:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4。
【0030】
発現させてもよい抗体断片は、損なわれていない抗体の一部を含み、これは所望の生物学的活性を有する。抗体断片には、一般的に、少なくとも1つの抗原結合部位が含まれる。抗体断片の例には:(i)VL、CL、VHおよびCH1ドメインを有するFab断片;(ii)Fab誘導体、例えば2つのFab誘導体間のジスルフィド架橋によって二価断片を形成可能である、CH1ドメインのC末端に1以上のシステイン残基を有するFab’断片;(iii)VHおよびCH1ドメインを有するFd断片;(iv)Fd誘導体、例えばCH1ドメインのC末端に1以上のシステイン残基を有するFd誘導体;(v)抗体の単一のアームのVLおよびVHドメインを有するFv断片;(vi)一本鎖抗体分子、例えばVLおよびVHドメインが共有結合している一本鎖Fv(scFv)抗体;(vii)定常領域ドメインを含むかまたは含まない別の可変ドメイン(VHまたはVLドメインポリペプチド)に連結された、定常領域ドメインを含まないVHまたはVLドメインポリペプチド(例えばVH−VH、VH−VL、またはVL−VL)(viii)ドメイン抗体断片、例えば、VHドメイン、またはVLドメイン、およびVHまたはVLドメインいずれかの抗原結合断片、例えば単離CDR領域からなる断片;(ix)2つの抗原結合部位を含む、いわゆる「ディアボディ」、例えば同じポリペプチド鎖中で、軽鎖可変ドメイン(VL)に連結された重鎖可変ドメイン(VH);ならびに(x)相補性軽鎖ポリペプチドとともに、抗原結合領域対を形成する、タンデムFdセグメント対を含む、いわゆる直鎖抗体が含まれる。
【0031】
調製可能な好ましい抗体断片は、哺乳動物単一可変ドメイン抗体であり、免疫グロブリン可変ドメインに特徴的な配列を含み、そして抗原に特異的に結合し(すなわち500nM以下、例えば400nM以下、好ましくは250nM以下、そして最も好ましくは100nM以下の解離定数)、そして単一可変ドメインとして;すなわちいかなる相補性可変ドメインも含まずに、抗原に結合する、折りたたまれたポリペプチドドメインを含む抗体断片である。単一可変ドメイン抗体には、完全抗体可変ドメインならびに修飾可変ドメイン、例えば1以上のループが、抗体可変ドメインに特徴的でない配列で置換されているもの、あるいは一部切除(truncated)されているかまたはNもしくはC末端伸長を含む抗体可変ドメイン、ならびに可変ドメインの折りたたまれた断片が含まれる。調製可能な好ましい単一可変ドメインは、VHおよびVLの群より選択され、VカッパおよびVラムダが含まれる。最も好ましくは、単一可変ドメインは、ヒトまたはラクダ科(camelid)ドメインであり、ヒト化ラクダ科ドメインが含まれる。
【0032】
したがって、本発明はまた、宿主細胞において、本発明の第一の側面にしたがった発現系を発現する工程を含む、ターゲットポリペプチドの産生のための方法も提供する。
発現系は、使用する細胞に関して、当該技術分野に周知の方法によって発現される。好ましい発現法には、増殖培地中で、特に発酵によって、組換え細胞を培養し、そして次いで発現されたタンパク質を回収する工程が含まれる。用語「増殖培地」は、組換え細胞を増殖させるために用いられる栄養培地を指す。多くの態様において、栄養溶液を使用する。所定の組換え細胞に適した増殖培地が当該技術分野に周知である。
【0033】
基底発現は、DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターに適した条件を選択することによって調節される。適切な場合、例えば細胞が、例えば細胞密度を監視することによって決定される所望の増殖状態に到達するまで増殖している場合、ターゲットポリペプチドをコードするDNAに機能可能であるように連結されたプロモーターに有利に働くように、条件を調整する。多くの態様において、この調整は、対数期増殖中に行われる。
【0034】
本発明を以下の実施例によって限定なしに例示する。
【実施例】
【0035】
プラスミドpAB013の構築
pAB013を生成するための出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。pZT7#2.0は、pAT153ベクター骨格、cer安定性配列、tet A/R、関心対象の遺伝子上流の単一天然lacオペレーター配列、および上流T4転写ターミネーターを有する。NcoI、EcoRIおよびXbaI制限酵素部位によって、合成オリゴヌクレオチドリンカーを用いて、T7A3プロモーターおよび二重完全パリンドロームlacオペレーターをこのプラスミド内にクローニングした。
【0036】
オリゴヌクレオチド1および2をアニーリングさせることによって、リンカー12を調製した:
【0037】
【化1】

【0038】
次いで、リンカーをプラスミドpZT7#2.0に連結させ、そしてNcoI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。形質転換体の最初のスクリーニングは、NcoIを用いた制限消化によった。配列決定によって配列を確認した。次いで、オリゴヌクレオチド3および4をアニーリングさせることによって、T7A3プロモーターカセットをこのベクター内にクローニングした:
【0039】
【化2】
【0040】
アニーリングしたオリゴヌクレオチドをベクターに連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。最初のスクリーニングは、プラスミドDNAの制限消化によった。次いで、配列決定によって配列を確認した。
【0041】
ヒト腫瘍壊死因子(hTNF)−α遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB013を生成した。このプラスミドは、T7A3プロモーター/二重完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNF−αを発現する。
【0042】
プラスミドpAB044の構築
出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。pZT7#2.0は、pAT153ベクター骨格、cer安定性配列、tet A/R、関心対象の遺伝子上流の単一天然lacオペレーター配列、および上流T4転写ターミネーターを有する。NcoI、EcoRIおよびXbaI制限酵素部位によって、合成オリゴヌクレオチドリンカーを用いて、T7A3プロモーターおよび二重完全パリンドロームlacオペレーターをこのプラスミド内にクローニングした。
【0043】
上述のように、オリゴヌクレオチド1および2をアニーリングさせることによって、リンカー12を調製した。
次いで、リンカーをプラスミドpZT7#2.0に連結させ、そしてNcoI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。形質転換体の最初のスクリーニングは、NcoIを用いた制限消化によった。配列決定によって配列を確認した。オリゴヌクレオチド17および18をアニー
リングさせることによって、tacプロモーターカセットをこのベクター内にクローニングした:
【0044】
【化3】
【0045】
アニーリングしたオリゴヌクレオチドを連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。最初のスクリーニングは、プラスミドDNAの制限消化によった。次いで、配列決定によって配列を確認した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB044を生成した。このプラスミドは、tacプロモーター/二重完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNF−αを発現する。
【0046】
プラスミドpAB235の構築
pAB235の出発ベクターはpACYC−Duet(Novagenカタログ番号71147−3)であった。DNA結合タンパク質HU(熱不安定性)−βをコードするHupB配列を大腸菌株W3110(ATCC番号ATCC27325より得られる)のゲノムDNAからPCRによって増幅した。用いたプライマーは、プライマー1: GCCATATGCAGGAAGAAGGAGAATGAATAAATCTCAATTG(配列番号7)およびプライマー2: GCCTCGAGTTAGTTTACCGCGTCTTTCAG(配列番号8)であった。PCR産物を、ベクターpCR2.1 TOPO Blunt(Invitrogenカタログ番号K280020より得られる)内にクローニングした。挿入物を、NcoI−XhoI消化(New England Biolabs)によって取り除き、そしてpACYC−Duet内にクローニングして、ベクターNBJ0585−6−1を作製した。
【0047】
HupA遺伝子(DNA結合タンパク質HU−α)を、構成的lacプロモーター(lacオペレーター配列を除く)およびHupA遺伝子除去のためのNdeI部位を含有するMluI−NcoI断片として設計した。これをベクターNBJ0585−6−1にクローニングして、プラスミドpAB235を作製した。HupA遺伝子配列を図1に提供する(配列番号9)。
【0048】
プラスミドpAB249の構築
pAB249の出発ベクターはpACYC−Duetであった。構成的lacプロモーター(lacオペレーター配列を除く)およびHupA遺伝子除去のためのNdeI部位を含有するMluI−NcoI断片として設計されたHupA遺伝子(DNA結合タンパク質HU−α)を、MluI−NcoI断片として、pACYC−Duetにクローニングして、ベクターNBJ0585−18−1を作製した。これを次いで、NdeI−XhoIで消化して、HupA遺伝子を取り除いた。次いで、大腸菌コドン最適化ラットHMGB−1(高移動度群B)遺伝子をクローニングして、pAB249を作製した。大腸菌
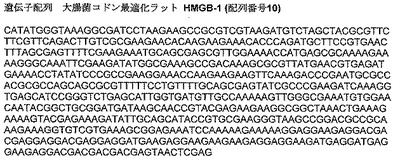
コドン最適化ラットHMGB−1遺伝子配列を図2に提供する(配列番号10)。
【0049】
pAB246の構築
pAB246の出発ベクターはpCI−Neo(Promegaカタログ番号E1841)であった。ラットHMGB−1タンパク質(チャイニーズハムスター卵巣(CHO)細胞における発現のために最適化された遺伝子配列コドン)を、クローニングのため、NheI−NotI断片として合成した。これをpCI−NeoにクローニングしてpAB246を作製した。
【0050】
pAB193の構築
pAB193の出発ベクターはpOPRSV1/MCS(Stratagene)であった。IgG Fc遺伝子配列(図3−配列番号11)をNheI/NotI断片として、SpeI/NotI消化したpOPRSVI/MCSにクローニングして、pAB193を生成した。
【0051】
実施例1
エレクトロポレーションを用いて、プラスミドpAB044を大腸菌宿主株W3110に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD047と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0052】
エレクトロポレーションを用いて、プラスミドpAB235およびpAB044を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD283と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0053】
エレクトロポレーションを用いて、プラスミドpAB235およびpAB013を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD271と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0054】
エレクトロポレーションを用いて、プラスミドpAB249およびpAB013を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD272と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0055】
エレクトロポレーションを用いて、プラスミドpAB013を大腸菌宿主株W3110に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD018と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0056】
組換え大腸菌株の要約を表1に提示する。
表1:組換え大腸菌株
【0057】
【表1】
*DNA結合タンパク質HU(熱不安定性)−α/DNA結合タンパク質HU(熱不安定性)−β
【0058】
実施例2
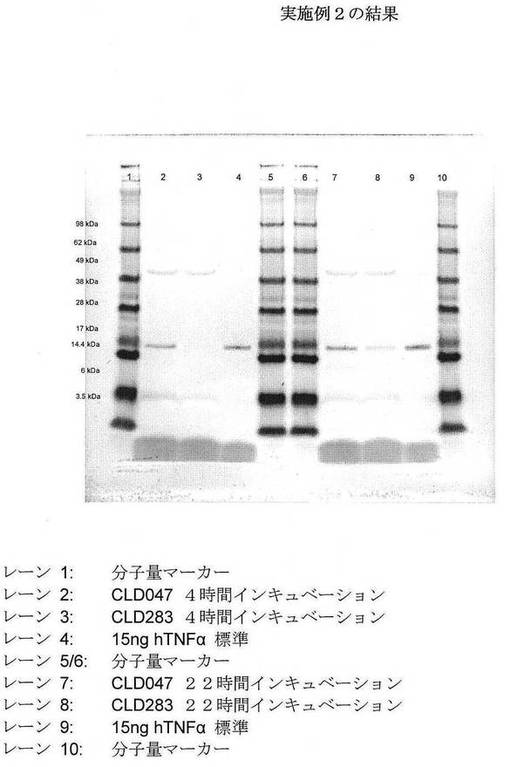
CLD047およびCLD283のバイアルを−80℃フリーザーから取り出し、そして融解させた。融解したグリセロールストック各々の10μlを、別個に、テトラサイクリン(10μg/ml、CLD047)またはCLD283用にクロラムフェニコール(34μg/ml)およびテトラサイクリン(10μg/ml)を補充した5mlルリアブロス(LB、5g/L酵母エキス(Oxoid)、10g/Lトリプトン(Oxoid)、および5g/L塩化ナトリウム)に接種した。CLD047およびCLD283の培養を37℃で、軌道振盪装置中、200rpmで16時間、インキュベーションした。次いで、500μlのこれらの培養各々を用いて、50mlのルリアブロス(上記のような組成)を含有する250mlエルレンマイヤーフラスコに別個に接種した。フラスコを37℃で、軌道振盪装置中、200rpmでインキュベーションした。上記条件下でインキュベーションを続け、その間に、増殖、細菌細胞内のhTNFαの集積を測定するために、試料を採取した(4時間および22時間)。CLD047およびCLD283の非誘導培養におけるhTNFαの基底集積レベルを、試料細菌のSDS−PAGE後のウェスタンブロット分析(抗hTNFα抗体を用いる)によって比較した。誘導の非存在下での長期のインキュベーションを用いて、観察されうるいかなる基底発現も増幅するであろう最悪条件を提供した。
【0059】
データを図4に示す。驚くべきことに、DNA結合タンパク質HU(熱不安定性)−α/DNA結合タンパク質HU(熱不安定性)−β(CLD283)の存在下でのhTNFαの基底発現は、22時間に渡る長期インキュベーション後であってさえ、有意に減少した。
【0060】
実施例3
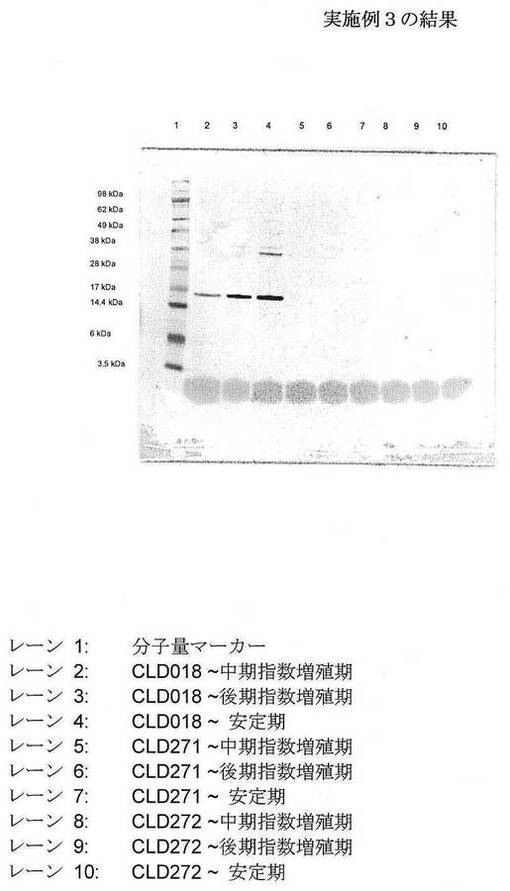
CLD018、CLD271およびCLD272のバイアルを−80℃フリーザーから取り出し、そして融解させた。融解したグリセロールストック各々の10μlを、別個に、テトラサイクリン(10μg/ml、CLD018)またはCLD271およびCLD272用にクロラムフェニコール(34μg/ml)およびテトラサイクリン(10μg/ml)を補充した5mlルリアブロス(LB、5g/L酵母エキス(Oxoid)、10g/Lトリプトン(Oxoid)、および5g/L塩化ナトリウム)に接種した。培養を37℃で、軌道振盪装置中、200rpmで16時間、インキュベーションした。次いで、500μlのこれらの培養各々を用いて、50mlのルリアブロス(上記のような組成)を含有する250mlエルレンマイヤーフラスコに別個に接種した。フラスコを37℃で、軌道振盪装置中、200rpmでインキュベーションした。OD600=0.5〜0.7(〜中期指数増殖期)まで増殖を監視し、そして試料を採取した。さらに22時間、上記条件下でインキュベーションを続け、その間に、増殖、細菌細胞内のhTNFαの
集積を測定するために、試料を採取した(3時間(後期指数増殖期)および22時間(安定期))。長期インキュベーション後のCLD018、CLD271およびCLD272の非誘導培養におけるhTNFαの基底集積レベルを、試料細菌のSDS−PAGE後のウェスタンブロット分析(抗hTNFα抗体を用いる)によって比較した。
【0061】
データを図5に示す。データによって、DNA結合タンパク質HU(熱不安定性)−α/DNA結合タンパク質HU(熱不安定性)−β(CLD271)のまたは真核ラットHMGB−1(CLD272)の存在下での、実施例2で用いたものとは異なるプロモーター/発現系を用いたhTNFαの基底発現は、長期インキュベーション後であってさえ、有意に減少することが確認される。
【0062】
実施例4
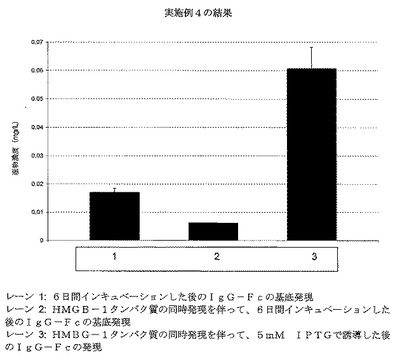
チャイニーズハムスター卵巣細胞、細胞タイプCHO−PF(ECACC、カタログ番号00102307)のアンプルを凍結ストックから生き返らせ、そして10%ウシ胎児血清(FCS)および4mM L−グルタミンを補充したイスコフの修飾ダルベッコ培地(IMDM)細胞培養増殖培地50ml中、T175静置培養内に回収した。トランスフェクション日まで、フラスコを1:5の分割比でルーチンに継代した。培地を除去し、そしてフラスコ内容物を20mlのリン酸緩衝生理食塩水溶液(PBS)で洗浄した後、4mlのトリプシンを添加し、そして37℃で3分間インキュベーションすることによって、細胞をフラスコから解離させた。細胞を解離させた際、新鮮な培地をフラスコに添加して、トリプシン活性を中和した。トランスフェクション日、細胞をフラスコから解離させ、そして試料を採取して、VicellTM自動化細胞計数装置を用いて、生存細胞濃度および生存度を決定した。細胞濃度を7.5x105細胞.ml−1に調整し、そして1mlの細胞懸濁物を6ウェル細胞培養マイクロタイタープレートの各ウェルにトランスファーした。加湿5%CO2インキュベーター(Sanyo)中、37℃で2時間プレートをインキュベーションし、そしてトランスフェクション複合体を以下のように調製した:
13.3μg pAB193および3.3μgのpAB246(等量のpCMVlacI(Stratagene、Lacリプレッサーを発現する))に加えて、80μL Fugene(登録商標)6トランスフェクション試薬(Roche、カタログ番号1814443001)および667μL IMDM。DNA/Fugene(登録商標)6複合体を室温で2時間インキュベーションした後、複合体由来の200μLを細胞培養プレート上の4ウェル各々に添加した。pAB246プラスミドDNAを除いて、上述の半量を用いて第二の複合体を調製した。これをまた2時間インキュベーションした後、6ウェル細胞培養プレートの2ウェルに添加した。細胞培養プレートを、加湿5%CO2インキュベーター中、37℃で24時間インキュベーションした。最終濃度5mMまでIPTG(イソプロピル−β−D−1−チオガラクトピラノシド)を添加することによって、pAB246を含有する細胞培養プレート上のいくつかのウェルを誘導して、所望のタンパク質IgG−Fcの誘導を確認した。細胞培養プレートを、加湿5%CO2インキュベーター中、37℃でさらに5日間インキュベーションした。酵素連結免疫吸着アッセイ(ELISA)によって、細胞培養増殖培地内へのIgG−Fcの発現/分泌を測定した。
【0063】
データを図6に示す。該データは、HMGB−1同時発現を用いて、哺乳動物発現系において、タンパク質基底発現を減少させうることを立証する。さらに、誘導因子IPTGを培養に添加すると、IgG−Fcタンパク質の誘導がなお可能であり、そして誘導後、タンパク質レベル増加が生じる。
【0064】
実施例5
プラスミドpAB007およびpAB031の構築
pAB031の生成のための出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。EcoRIおよびXbaI制限酵素部位に
よって、合成オリゴヌクレオチドリンカーを用いて、T7A3プロモーターおよび単一完全パリンドロームlacオペレーターをこのプラスミド内にクローニングした。
【0065】
上述のように、オリゴヌクレオチド3および4によって、T7A3プロモーターを含有するリンカーを作製した。
オリゴヌクレオチド3および4をアニーリングさせ、次いで、形成されたリンカーをプラスミドpZT7#2.0に連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。最初のスクリーニングは、プラスミドDNAの制限消化によった。次いで、配列決定によって配列を確認した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB031を生成した。このプラスミドは、T7A3プロモーター/単一完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNF−αを発現する。
【0066】
プラスミドpAB040の構築
pAB040の生成のための出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。EcoRIおよびXbaI制限酵素部位によって、合成オリゴヌクレオチドリンカーを用いて、tacプロモーターおよび単一完全パリンドロームlacオペレーターをこのプラスミド内にクローニングした。
【0067】
オリゴヌクレオチド13および14をアニーリングさせることによって、リンカー1314を調製した。
【0068】
【化4】
【0069】
次いで、リンカーをプラスミドpZT7#2.0に連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。形質転換体の最初のスクリーニングは、NcoIを用いた制限消化によった。配列決定によって配列を確認した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB040を生成した。このプラスミドは、Tacプロモーター/単一完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNFαを発現する。
【0070】
プラスミドpAB041の構築
pAB041の生成のための出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。EcoRIおよびXbaI制限酵素部位によって、合成オリゴヌクレオチドリンカーを用いて、tacプロモーターおよび単一天然lacオペレーターをこのプラスミド内にクローニングした。
【0071】
オリゴヌクレオチド11および12をアニーリングさせることによって、リンカー11
12を調製した。
【0072】
【化5】
【0073】
次いで、リンカーをプラスミドpZT7#2.0に連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。形質転換体の最初のスクリーニングは、NcoIを用いた制限消化によった。配列決定によって配列を確認した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB041を生成した。このプラスミドは、Tacプロモーター/単一天然lacオペレーター配列/lacリプレッサー系の調節下でhTNFαを発現する。
【0074】
プラスミドpAB350の構築
pZT7#3.3発現プラスミドをUS 6,537,779に記載されるように調製した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB350を生成した。このプラスミドは、T7遺伝子10プロモーター/二重完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNFαを発現する。
【0075】
株構築
CLD032
エレクトロポレーションを用いて、プラスミドpAB031を大腸菌宿主株W3110に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD032と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0076】
CLD408
エレクトロポレーションを用いて、プラスミドpAB235およびpAB031を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD408と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0077】
CLD409
エレクトロポレーションを用いて、プラスミドpAB249およびpAB031を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD409と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0078】
CLD042
エレクトロポレーションを用いて、プラスミドpAB041を大腸菌宿主株W3110に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD042と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0079】
CLD410
エレクトロポレーションを用いて、プラスミドpAB235およびpAB041を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD410と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0080】
CLD411
エレクトロポレーションを用いて、プラスミドpAB249およびpAB041を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD411と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0081】
CLD412
エレクトロポレーションを用いて、プラスミドpAB235およびpAB040を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD412と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0082】
CLD413
エレクトロポレーションを用いて、プラスミドpAB249およびpAB040を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD413と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0083】
CLD023
エレクトロポレーションを用いて、プラスミドpAB350を大腸菌宿主株BL21(λDE3)に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD023と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0084】
CLD414
エレクトロポレーションを用いて、プラスミドpAB235およびpAB350を大腸菌宿主株BL21(λDE3)に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD414と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0085】
CLD415
エレクトロポレーションを用いて、プラスミドpAB249およびpAB350を大腸菌宿主株BL21(λDE3)に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、
コロニーを選択した。CLD415と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0086】
実施例5で使用する組換え大腸菌株の要約を表2に示す。
表2
【0087】
【表2】
*DNA結合タンパク質HU(熱不安定性)−α/DNA結合タンパク質HU(熱不安定性)−β
【0088】
CLD032、CLD408、CLD409、CLD042、CLD410、CLD411、CLD0412、CLD0413、CLD023、CLD414およびCLD415のバイアルを−80℃フリーザーから取り出し、そして融解させた。融解したグリセロールストック各々の10μlを、別個に、テトラサイクリン(10μg/ml、CLD0032、CLD042、CLD043およびCLD023)またはCLD408、CLD409、CLD410、CLD411、CLD412、CLD413およびCLD414用にクロラムフェニコール(34μg/ml)およびテトラサイクリン(10μg/ml)を補充した5mlルリアブロス(LB、5g/L酵母エキス(Oxoid)、10g/Lトリプトン(Oxoid)、および5g/L塩化ナトリウム)に接種した。培養を37℃で、軌道振盪装置中、200rpmで16時間、インキュベーションした。次いで、500μlのこれらの培養各々を用いて、50mlのルリアブロス(上記のような組成)を含有する250mlエルレンマイヤーフラスコに別個に接種した。フラスコを37℃で、軌道振盪装置中、200rpmでインキュベーションした。OD600=0.5〜0.7(〜中期指数増殖期)まで増殖を監視した。試料を採取し、そしてフラスコ各対の一方を0.5mM IPTGで誘導した。さらに22時間、上記条件下でインキュベーションを続け、その間に、増殖、細菌細胞内のhTNFαの集積を測定するために、試料を採取した(3時間(後期指数増殖期)および22時間(安定期))。長期インキュベーション後の培養中のhTNFα集積レベルを、試料細菌のSDS−PAGEによって比較した。SimplyBlue染色したSDS−PAGEゲルのレーザー濃度計によって、hTNF
α集積レベル(%総細胞微生物タンパク質(%TCP)として表わす)を決定し、そして株CLD032、CLD408、CLD409、CLD042、CLD410、CLD411、CLD023、CLD414およびCLD415に関して検出されたレベルを、以下の表3に示す。株CLD0412およびCLD0413に関して検出されたレベルを以下の表4に示す。
【0089】
表3のデータは、本発明記載の発現系を含む例示的な株が、DNA結合性転写制御因子タンパク質の発現カセットを欠く対応する系と比較して、hTNFαの基底発現の有意な減少を示すことを示す。しかし、hTNFα発現がIPTG添加によって誘導された際、本発明記載の株によって産生されるhTNFαのレベルは、DNA結合性転写制御因子タンパク質の発現カセットを欠く対応する系によって産生されるレベルに匹敵する。
【0090】
表4のデータは、本発明記載の発現系を含むこれらの株に関するhTNFαの基底発現の優れた調節および優れた誘導産生を示す。
表3. hTNFα発現レベル
【0091】
【表3】
*=本発明記載
【0092】
表4. hTNFα発現レベル
【0093】
【表4】
【技術分野】
【0001】
本発明はポリペプチドの発現のためのプロセス、および発現系に関する。
【背景技術】
【0002】
学術的または商業的に関心対象である多くのポリペプチド、特に療法ポリペプチドは、組換え宿主細胞における発現によって好適に産生される。特に製造環境における、こうした異種ポリペプチドの効率的な産生のため、ポリペプチドが製造における適切な段階でのみ産生されるように、ポリペプチドを調節することが好ましい。したがって、望ましい場合に適切な条件の適用によって発現が誘発可能であるように、ポリペプチドの発現は誘導因子の調節下に置かれる。例えば、いくつかの異種ポリペプチドは、宿主細胞増殖およびプラスミド安定性に有害な影響を有し、全体の生産性を減少させるため、誘導前の異種ポリペプチドの発現(いわゆる「漏出性(leaky)」発現)、すなわち基底発現の劣った調節は、一般的に望ましくない。いくつかの場合、異種ポリペプチドの漏出性発現は、クローニング中のベクター安定性の問題を生じる可能性もあり、所望のポリペプチドを発現する安定な発現ベクター構築の失敗につながりうる。
【0003】
異なる調節様式および誘導様式を持つ多くの異種ポリペプチド発現系があり、関心対象のポリペプチドを産生するための発現系/発酵プロセスの選択および最適化は大部分、経験的なプロセスとなっている。これには時間がかかり、そして望ましくない。したがって、発現の調節の改善、特に基底発現の調節の改善を提供する系が必要である。
【0004】
誘導性遺伝子発現は、遺伝子機能の分析に重要なツールである。これらの系には、誘導されない細胞において、遺伝子発現の潜在的に有害な影響を減少させ、そしてかつ誘導された遺伝子の発現による生物学的に関連する影響の明らかな区別を可能にするために、基底遺伝子発現の厳重な調節が、決定的に必要である。
【0005】
漏出性基底発現の問題を減少させるかまたは排除することを試みる、多様な改善法には、成功と失敗が入り交じってきている。例えば、大腸菌(E. coli)における基底発現を減少させるためのアプローチには、リプレッサータンパク質対オペレーター比の増加、突然変異体ファージの感染による誘導、プロモーター強度の減弱、アンチターミネーターと組み合わせた転写ターミネーターの使用が含まれる(Makrides, Microbiological Reviews, 1996, 512−538によって概説される)。タンパク質を高レベルで発現するために、トランスで供給されるT7 RNAポリメラーゼを用いるpETベクター(Novagen)に伴う基底発現の問題は、T7 RNAポリメラーゼを阻害するT7リゾチームの同時過剰発現によって減少させうる。しかし、基底発現を減少させるかまたは排除するために使用する最適な発現構成要素の使用(Studier, Journal of Molecular Biology, 1991, 219(1):37−44)は経験的であり、基底発現および誘導性に対する多数のベクター(pLysS、pLysL、pLysEおよびpLysH)の影響を決定するスクリーニングを必要とする。哺乳動物細胞において用いられるテトラサイクリンが制御する発現系の基底発現は問題である(Meyer−Ficcaら, Anal. Biochem, 2004, 334:9−19)。Laiら, Am. J. Physiol Cell Physiol, 2003, 285:711−719は、テトラサイクリンまたはエクジソン応答配列を用いて発現されるレポーター遺伝子(緑色蛍光タンパク質)を用いて、高い基底発現を持つ哺乳動物細胞がどのように生成されたかを記載する。これらの細胞は、レポーター遺伝子系と組み合わせて蛍光活性化細胞分取を用いることによって除去されうる。Beckerら, Nucleic Acids
Research, 2008, 1−13は、大腸菌における天然HU抑制タンパク質に対する置換としての真核HMGBタンパク質の使用を開示する。天然大腸菌LacZ/ベータ−ガラクトシダーゼ遺伝子の産生抑制が研究されている。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Makrides, Microbiological Reviews, 1996, 512−538
【非特許文献2】Studier, Journal of Molecular Biology, 1991, 219(1):37−44
【非特許文献3】Meyer−Ficcaら, Anal. Biochem, 2004, 334:9−19
【非特許文献4】Laiら, Am. J. Physiol Cell Physiol, 2003, 285:711−719
【非特許文献5】Beckerら, Nucleic Acids Research, 2008, 1−13
【発明の概要】
【0007】
本発明の1つの側面にしたがって、ターゲットポリペプチド産生のための発現系であって:
a)ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターを含む発現カセット;および
b)DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターを含む、DNA結合性転写制御因子タンパク質を過剰発現させるための発現カセット;
ここで該発現カセットは直交性(orthogonal)プロモーターの調節下にある
を含む、前記発現系を提供する。
【図面の簡単な説明】
【0008】
【図1】図1は、HupA遺伝子配列である。
【図2】図2は、大腸菌コドン最適化ラットHMGB−1遺伝子配列である。
【図3】図3は、IgG Fc遺伝子配列である。
【図4】図4は、実施例2の結果である。
【図5】図5は、実施例3の結果である。
【図6】図6は、実施例4の結果である。
【発明を実施するための形態】
【0009】
プロモーターは、異なる条件によって活性化される際、直交性であると認識される。これらの条件は、互いに排他的であってもよいし、または適合可能であってもよい。互いに排他的なプロモーターは、1つのプロモーターの調節下にあるDNAの発現に必要な条件が、別のプロモーターの調節下にあるDNAの発現を防止するものであり、そしてこれには、例えば、テトラサイクリン反応系が含まれ、これは、テトラサイクリンの存在下で「オフ」である(すなわち発現が防止される)が、テトラサイクリンの非存在下で「オン」である(すなわち発現が促進される)(「テトラサイクリン・オフ」)ように設計可能であるし、またはテトラサイクリンの存在下で「オン」である(「テトラサイクリン・オン」)が、その非存在下で「オフ」であるようにも設計可能である。DNA結合性転写制御因子タンパク質をコードするDNAが、テトラサイクリン・オフであるプロモーターに機能可能であるように連結され、そしてターゲットポリペプチドをコードするDNAが、テトラサイクリン・オンであるプロモーターに機能可能であるように連結される場合、テトラサイクリンの添加は、ターゲットポリペプチドの発現を誘導するが、DNA結合性転写
制御因子タンパク質のさらなる発現を防止するであろう。
【0010】
多くの態様において、機能可能であるように連結されるDNA配列は隣接する。
本発明の発現系で使用可能なDNA結合性転写制御因子タンパク質は、原核および真核生物由来のタンパク質を含む。原核タンパク質には、大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−β、組込み宿主因子(IHF)−α、IHF−βおよび相同体が含まれる。真核タンパク質は、好ましくは、酵母、例えばNHP6A、NHP6B、または哺乳動物タンパク質であり、そしてこれには、豊富配列非特異的高移動度群(HMG)タンパク質が含まれる。HMGタンパク質は、3つのファミリー: HMGBファミリー、例えばHMGB−1、HMGB−2、HMGB−3; HMGNファミリー、例えばHMGN−1、HMGN−2、HMGN−3、ならびにHMGAファミリー、例えばHMGA−1a、HMGA−1b、HMGA−1c、HMGA−2、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)などの酵母におけるミトコンドリア・ヒストン(HM)タンパク質、ならびにヒトおよび両生類ミトコンドリアで見られる相同体、ならびに細菌ヒストン様タンパク質HUに似たタンパク質をコードする植物(クリプト藻(Guillardia theta))葉緑体hlpAを含む、原核DNA結合性転写制御因子タンパク質の真核相同体に分けられる。好ましいDNA結合性転写制御因子タンパク質は、大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−βおよびHMGB−1、特にラットHMGB−1である。
【0011】
本発明の発現カセットは、宿主細胞ゲノム内に組み込まれるか、またはプラスミドなどの染色体外要素内に含まれてもよい。染色体外要素内に含まれる場合、発現カセットは、同じ染色体外要素、例えばプラスミドベクター上に含まれてもよいし、または宿主細胞に同時形質転換される、適合する別個の染色体外要素上に含まれてもよい。あるいは、発現系は、ファージまたはウイルスベクター内に取り込まれてもよく、そしてこれらを用いて、宿主細胞系内に発現系が送達されてもよい。当該技術分野に知られる方法によって、プラスミドまたは他の発現ベクターを組み立ててもよい。本発明の発現カセットは、好ましくは、どちらも別個のプラスミドの形で使用される。プラスミドは自律複製プラスミドまたは組込み性プラスミドであってもよい。プラスミドは典型的には1以上の以下も含む:選択可能マーカー、例えば抗生物質耐性を与える配列、およびcer安定性配列。
【0012】
発現カセットが別個の染色体外要素上に含まれる態様において、両方の要素を含む形質転換宿主細胞を同定するため、異なる選択可能マーカーを使用することが好ましい。
DNA結合性転写制御因子タンパク質を過剰発現するための発現カセットで使用するプロモーターは、構成的または誘導性プロモーターであってもよい。本発明の側面で使用可能な構成的プロモーターの例には、T7A1、T7A2、T7A3、spcリボソームタンパク質オペロンプロモーター、β−ラクタマーゼ遺伝子プロモーター、ファージλのPLプロモーター、複製調節プロモーターPRNAIおよびPRNAII、rrnBリボソームRNAオペロンのP1およびP2プロモーター、Lacリプレッサータンパク質プロモーターpLacI、グリセルアルデヒドリン酸デヒドロゲナーゼ(GAPDH)および形質膜H(+)−ATPアーゼ(PMA1)プロモーター、接合因子(MF)−αプロモーター、KEX2、TEF−1、サルウイルス40(SV40)初期プロモーター、ラウス肉腫ウイルス(RSV)プロモーター、サイトメガロウイルス(CMV)プロモーターおよびヒトβアクチンプロモーターが含まれる。構成的プロモーターのさらなる例には、調節領域を除去するよう修飾されている誘導性プロモーター、例えばlacまたはtacオペレーターを除去するよう修飾されているlacまたはtacプロモーターが含まれ、そしてこうしたプロモーターは、本発明の特定の態様において、特にDNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されている場合に好適である。
【0013】
本発明の側面において使用可能な誘導性プロモーターの例には、ファージRNAポリメラーゼ依存性プロモーター、特にT7 RNAポリメラーゼ依存性プロモーター系、好ましくは単一のT7プロモーターが含まれ、本明細書に援用されるStudierおよびMoffat, J. Mol. Biol. 189:113−130(1986)によって開示されるもの、特にT7遺伝子10プロモーターが含まれる。T7 RNAポリメラーゼ依存性プロモーター系を使用する場合、T7 RNAポリメラーゼの供給源が必要とされることが認識され、これは当該技術分野に知られる方法によって、そして一般的には大腸菌宿主株内に必要なファージポリメラーゼを発現するλDE3プロファージを挿入して、溶原性宿主株を生成することによって提供される。T7 RNAポリメラーゼはまた、T7 RNAポリメラーゼの遺伝子を所持する特殊化λ形質導入ファージでの感染によって細胞に送達されることも可能である。使用可能な誘導性プロモーターのさらなる例には、lac、lacUV5、trp、tac、trc、phoA、アラビノース誘導性プロモーター、温度誘導性プロモーター(高温および低温両方)、銅誘導性プロモーター、uspA、uspB、malK、浸透圧誘導性プロモーター、ガラクトース誘導性プロモーター、フェロモン誘導性プロモーター、グルコアミラーゼプロモーター、テトラサイクリン応答性プロモーター、ヒトc−fosプロモーター、エクジソン誘導性プロモーター、およびグルココルチコイド誘導性プロモーターが含まれる。プロモーターが一般的に、宿主細胞で有効であることが知られるプロモーターから選択されることが認識されるであろう。例えば、大腸菌プロモーターは、大腸菌宿主細胞において一般的に使用され、哺乳動物プロモーターは哺乳動物細胞において一般的に使用され、酵母プロモーターは、酵母細胞において一般的に使用される。原核宿主由来の多くのプロモーターは、真核宿主において使用可能であり、そして逆も可能であることが認識されるであろう。少なくとも1つの誘導性プロモーターが使用されることが好ましく、特に好ましくは、ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターが使用される。いくつかの態様において、DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結された構成的プロモーターと組み合わされた、ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターが特に好ましい。
【0014】
カセットの発現が適切な条件の改変によってスイッチ可能であるように、発現カセットにおいて、直交性プロモーターを使用する。ターゲットタンパク質の発現を誘導するように条件を調整するまで、DNA結合性転写制御因子タンパク質の過剰発現は、ターゲットポリペプチドの基底発現を減少させるかまたは防止する効果を有する。
【0015】
本発明のプロセスにおいて使用可能な誘導因子は、使用する誘導性プロモーターに対応するように選択され、そしてこれには、イソプロピル−β−D−1−チオガラクトピラノシド(IPTG)、IPTGの類似体、例えばイソブチル−C−ガラクトシド(IBCG)、ラクトースまたはメリビオース、ガラクトース、アラビノース、ラムノース、温度、pH、溶解酸素レベル、銅などの金属イオン、インドールアクリル酸、テトラサイクリン、ホモセリンラクトン、エクジソン、サリチル酸塩、リン酸塩が含まれる。他の誘導因子を用いてもよく、そしてこれは、別の箇所により詳細に記載される(例えば、The Operon, MillerおよびRenznikoff監修(1978)を参照されたい)。誘導因子を個々にまたは組み合わせて用いてもよい。
【0016】
ターゲットポリペプチドの発現を誘導する場合、DNA結合性転写制御因子タンパク質の発現を減少させるかまたは防止するように条件を選択することが好ましい。ターゲットタンパク質発現中に、DNA結合性転写制御因子タンパク質の発現を減少させるかまたは防止するために使用する方法には、1以上の以下が含まれる:
a)効能を維持するために補充を必要とする、DNA結合性転写制御因子タンパク質発現を調節するプロモーターおよび誘導因子を使用する。例えば誘導因子が代謝されるか、
無効であるように修飾されるか、または有効であるより低く濃度が低下するように細胞によって吸収される場合;
b)DNA結合性転写制御因子タンパク質の発現を調節する誘導因子を含有する培地から細胞を物理的に分離し、その後、ターゲットポリペプチドの発現を調節するのに必要な誘導因子を含有する培地内に細胞を再導入する;
c)基底発現を調節するのに十分なDNA結合性転写制御因子タンパク質が産生される一方、発現が誘導された際にターゲットポリペプチドの発現を防止しないかまたは有意に損なわないように、ターゲットポリペプチドの発現を調節するプロモーターよりも、DNA結合性転写制御因子タンパク質発現を調節するプロモーターの強度がより弱いように選択する;
d)基底発現を調節するのに十分なDNA結合性転写制御因子タンパク質が産生される一方、発現が誘導された際にターゲットポリペプチドの発現を防止しないかまたは有意に損なわないように、ターゲットポリペプチドをコードするベクターよりもより少ないコピー数のDNA結合性転写制御因子タンパク質をコードするベクターを選択する;
e)DNA結合性転写制御因子タンパク質の発現を損なうための、アンチセンスまたはRNA干渉(例えばマイクロRNA(miRNA)または低分子干渉RNA(siRNA))を使用する;
f)例えば、ターゲットポリペプチドの発現が誘導される際に、阻害剤が発現されるように、選択されるDNA結合性転写制御因子タンパク質の阻害剤を発現する。多くのこうした態様において、阻害剤は、ターゲットポリペプチドと同じベクター上で発現されてもよい;
g)例えば、ターゲットポリペプチドの発現が誘導される際に、プロテアーゼが発現されるように、選択されるDNA結合性転写制御因子タンパク質をターゲットとするプロテアーゼを発現する。多くのこうした態様において、プロテアーゼは、ターゲットポリペプチドと同じベクター上で発現されてもよい;
h)例えば、ターゲットポリペプチドの発現が誘導される際に、阻害剤が発現されるように、選択されるDNA結合性転写制御因子タンパク質の発現の阻害剤を発現する。例には、DNA結合性転写制御因子タンパク質の同族(cognate)リプレッサー(例えばcIリプレッサータンパク質)の構成的発現を伴わず、通常、誘導性であるプロモーター、例えばλpLを用いることが含まれる。誘導性プロモーターを用いてターゲットポリペプチドが発現され、そして同じ誘導性プロモーターを用いて阻害剤が発現される場合、誘導因子が存在しなければ、阻害はなく、そしてDNA結合性転写制御因子タンパク質発現は構成的である。ターゲットポリペプチドの発現を誘導する誘導因子の添加はまた、阻害剤の発現も誘導し、そして阻害剤は、DNA結合性転写制御因子タンパク質の発現を防止する。多くのこうした態様において、阻害剤は、ターゲットポリペプチドと同じベクター上で発現されてもよい;
i)ある程度のリードスルーを可能にするターゲットポリペプチドの転写ターミネーターを伴い、DNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現要素を異なる方向で含むようにプラスミドベクターを設計する。ここで、DNA結合性転写制御因子タンパク質発現要素は構成的プロモーターを含み、そしてターゲットポリペプチドの発現カセットに続く。誘導因子が添加されたら、ターゲットポリペプチドからDNA結合性転写制御因子タンパク質の発現カセットに進む転写リードスルーは、DNA結合性転写制御因子タンパク質の転写を減少させるかまたは停止させるであろう;そして
j)誘導因子が添加された際、DNA結合性転写制御因子タンパク質発現要素がベクターまたは宿主ゲノムから切除され、そしてしたがってもはや発現不能となるが、ターゲットポリペプチド発現要素を損なわれず(intact)に残すように、発現系を設計する。
【0017】
DNA結合性転写制御因子タンパク質の過剰発現が、宿主細胞で通常見られるより高いレベルでのこうしたタンパク質の発現を指すことが認識されるであろう。DNA結合性転
写制御因子タンパク質が、宿主細胞に対して天然である場合、過剰発現は、こうしたタンパク質の発現を正常レベルより高く増加させることを含む。非天然DNA結合性転写制御因子タンパク質を使用する場合、宿主細胞で通常見られるレベルはゼロである。DNA結合性転写制御因子タンパク質は、ターゲットポリペプチドの基底発現が減少するかまたは防止されるレベルまで過剰発現される。
【0018】
本発明記載の発現系で使用可能なオペレーター配列には、lac、gal、deoおよびglnが含まれる。1以上、好ましくは2つのオペレーター配列を使用してもよい。特定の態様において、2以上の完全なパリンドローム・オペレーター配列を使用してもよい。多くの好ましい態様において、2つの完全なパリンドローム・オペレーター配列を使用し、最も好適には、1つのオペレーター配列はプロモーター下流に位置し、そして1つのオペレーター配列はプロモーター上流に位置する。2つのオペレーター系を使用する場合、オペレーター配列は、好ましくは、プロモーターの調節を最大にするように間隔を開けられる。2つのオペレーターは、最も一般的には、誘導性プロモーターと組み合わせて使用されることが認識されるであろう。多くの態様において、間隔は85〜150塩基対離れており、好ましくは90〜126塩基対離れており、そして最も好ましくは91または92塩基対離れている。特定の態様において、オペレーター配列は、転写開始点と重なる。
【0019】
オペレーター系は、一般的に、適切なリプレッサー配列とともに使用されることが認識されるであろう。リプレッサー配列は、リプレッサータンパク質を産生し、例えばlacオペレーターを用いる場合、lacI遺伝子配列である。他のlacリプレッサー配列もまた使用可能であり、例えばlacIQ配列を用いて、lacリプレッサータンパク質レベルを増加させてもよい。また、宿主細胞ゲノムによって、またはさらなる適合プラスミドを用いることによって、リプレッサー配列を提供してもよい。
【0020】
本発明の発現系は、ターゲットポリペプチドの基底発現の調節を提供し、そしてしたがって、より有効でそして調節可能なターゲットポリペプチド製造を提供する。さらに、本発明の発現系は、毒性の影響を有するか、あるいはベクターまたは宿主株の安定性に不都合に影響を及ぼす可能性がある、ターゲットポリペプチドの基底発現を抑制することによって、より有効なベクター組み立ておよび/またはクローニングを可能にする。
【0021】
本発明の発現系をベクター組み立てまたはクローニングで使用する場合、特定の態様において、しばしば、所望のベクター組み立てまたはクローニング後、ターゲットポリペプチドに機能可能であるように連結されたプロモーターに有利に働く(favour)条件を選択すると、前記ポリペプチドの発現が可能になるように、発現系を選択する。こうした態様は、例えばベクター組み立てで使用するものと同じ宿主細胞、最も一般的には大腸菌において、ターゲットポリペプチドを発現させようとする状況に特に適している。他の態様において、DNA結合性転写制御因子タンパク質によるターゲットポリペプチド発現の調節が非常に大きく、単にターゲットポリペプチド発現のための条件を選択するだけで、ターゲットポリペプチドがほとんどまたはまったく産生されないように発現系を選択する。こうした態様において、ターゲットポリペプチドの発現は、例えば以下によって達成可能である:
a)DNA結合性転写制御因子タンパク質を発現する細胞がもはや選択不能であるが、ターゲットポリペプチドのみを発現する細胞が選択される発現条件を使用する。こうしたアプローチは、別個のベクター上にあるDNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現カセットの使用に適している;
b)ターゲットポリペプチドを発現するが、DNA結合性転写制御因子タンパク質を発現しない宿主細胞にベクターをトランスファーする。例えば、真核生物、例えば哺乳動物または酵母細胞におけるターゲットポリペプチドの発現のためのベクターを、原核生物、
特に大腸菌においてベクター組み立てまたはクローニングする。DNA結合性転写制御因子タンパク質のプロモーターは、ターゲットポリペプチド発現宿主によって認識されない。このアプローチは、DNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現カセットが、単一ベクター上に位置する発現系に特に適しており、そしてこれはまた、DNA結合性転写制御因子タンパク質およびターゲットポリペプチドが別個のベクター上に位置する場合にもまた使用可能である;
c)DNA結合性転写制御因子タンパク質をコードする発現カセットを切除して、ベクターの構造を修飾する。こうしたアプローチは、DNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現カセットが単一ベクター上に位置する発現系、ならびにDNA結合性転写制御因子タンパク質およびターゲットポリペプチドの発現カセットが別個のベクター上に位置する発現系に適している。
【0022】
多くの好ましい態様において、対応する発現系と比較した際、ターゲットポリペプチドの発現を助け、そしてDNA結合性転写制御因子タンパク質の発現を嫌う発現条件を選択した際に、ターゲットポリペプチドの発現が防止されるかまたは損なわれないが、DNA結合性転写制御因子タンパク質発現要素を欠くように、発現系を選択する。
【0023】
特定の特に好ましい発現系は、大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−βの発現カセットを含み、最も好ましくは、これは、構成的lacプロモーターに機能可能であるように連結されている。こうした発現系は、好適には、大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−βの、そしてターゲットポリペプチドの、別個のベクターを含み、ターゲットポリペプチドのベクターは、tacプロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含み、こうしたベクターはまた、好ましくは単一のオペレーター、特に天然lacオペレーターまたは完全にパリンドロームであるlacオペレーターも含む。
【0024】
さらに特に好ましい発現系は、ラットHMGB−1の発現カセットを含み、最も好ましくは、これは、構成的lacプロモーターに機能可能であるように連結されている。こうした発現系は、好適には、ラットHMGB−1の、そしてターゲットポリペプチドの、別個のベクターを含み、ターゲットポリペプチドのベクターは、tacプロモーターまたはT7、特にT7遺伝子10プロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含む。多くのこうした態様において、ターゲットポリペプチドのベクターがtacプロモーターを含む場合、ベクターはまた、単一のオペレーター、特に天然lacオペレーターまたは完全にパリンドロームであるlacオペレーターも含む。さらなるこうした態様において、ターゲットポリペプチドのベクターが、T7、特にT7遺伝子10プロモーターを含む場合、ベクターはまた、2つの完全にパリンドロームであるオペレーター、特にlacオペレーターも含み、最も好ましくは、1つのオペレーター配列はプロモーター上流に位置し、そして1つのオペレーター配列はプロモーター下流に位置する。
【0025】
本発明の発現系を使用して、宿主細胞において、そして特に微生物において、ポリペプチドを発現させてもよい。宿主細胞は原核または真核であってもよい。原核細胞の例には、細菌細胞、例えば大腸菌、ネズミチフス菌(Salmonella typhimurium)、セラチア・マルセセンス(Serratia marsescens)および緑膿菌(Pseudomonas aeruginosa)を含むグラム陰性細菌細胞、ならびに枯草菌(Bacillus subtilis)を含むグラム陽性細菌細胞が含まれる。真核細胞の例には、酵母、例えばピキア・パストリス(Pichia pastoris)、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、ハンセヌラ・ポリモルファ(Hansenula polymorpha)、クロイベロミセス・ラクティス(Kluyveromyces lactis)、シゾ
サッカロミセス・ポンベ(Schizosaccharomyces pombe)が含まれる。使用可能な哺乳動物宿主細胞には、ヒト細胞株、例えばヒト胎児腎臓およびPERC.6細胞;ネズミ細胞株、例えばNS0細胞;ならびに特に、ハムスター細胞株、例えばベビーハムスター腎臓細胞および特にチャイニーズハムスター卵巣細胞が含まれる。他の真核宿主細胞、例えば糸状菌、植物、昆虫、両生類細胞または卵巣(ovarian)種もまた使用可能である。好ましい宿主細胞は細菌、特に腸内細菌科(enterobacteriacae)、好ましくは大腸菌、そして特にそのBまたはK12株である。
【0026】
本発明の発現系は、組換え細胞を培養することによる、ポリペプチド、特に組換えポリペプチドの製造のために使用される。
本発明のプロセスによって発現可能なポリペプチドには、療法タンパク質およびペプチドが含まれ、サイトカイン、増殖因子、抗体、抗体断片、免疫グロブリン様ポリペプチド、酵素、ワクチン、ペプチドホルモン、ケモカイン、受容体、受容体断片、キナーゼ、ホスファターゼ、イソメラーゼ、ヒドロリアーゼ、転写因子および融合ポリペプチドが含まれる。
【0027】
発現可能な抗体には、モノクローナル抗体、ポリクローナル抗体および生物学的活性を有する抗体断片が含まれ、前述のいずれかの多価および/または多重特異性型が含まれる。
【0028】
天然存在抗体は、典型的には、4つのポリペプチド鎖、ジスルフィド結合によって相互連結された2つの同一の重(H)鎖および2つの同一の軽(L)鎖を含む。各重鎖は、可変領域(VH)および定常領域(CH)を含み、CH領域は、天然型で、3つのドメインCH1、CH2およびCH3を含む。各軽鎖は、可変領域(VL)および1つのドメインCLを含む定常領域を含む。
【0029】
VHおよびVL領域をさらに、より保存されたフレームワーク領域(FR)と称される領域が介在する、相補性決定領域(CDR)と称される超可変性領域にさらに分割してもよい。各VHおよびVLは、3つのCDRおよび4つのFRで構成され、これらはアミノ末端からカルボキシ末端に、以下の順で配置される:FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4。
【0030】
発現させてもよい抗体断片は、損なわれていない抗体の一部を含み、これは所望の生物学的活性を有する。抗体断片には、一般的に、少なくとも1つの抗原結合部位が含まれる。抗体断片の例には:(i)VL、CL、VHおよびCH1ドメインを有するFab断片;(ii)Fab誘導体、例えば2つのFab誘導体間のジスルフィド架橋によって二価断片を形成可能である、CH1ドメインのC末端に1以上のシステイン残基を有するFab’断片;(iii)VHおよびCH1ドメインを有するFd断片;(iv)Fd誘導体、例えばCH1ドメインのC末端に1以上のシステイン残基を有するFd誘導体;(v)抗体の単一のアームのVLおよびVHドメインを有するFv断片;(vi)一本鎖抗体分子、例えばVLおよびVHドメインが共有結合している一本鎖Fv(scFv)抗体;(vii)定常領域ドメインを含むかまたは含まない別の可変ドメイン(VHまたはVLドメインポリペプチド)に連結された、定常領域ドメインを含まないVHまたはVLドメインポリペプチド(例えばVH−VH、VH−VL、またはVL−VL)(viii)ドメイン抗体断片、例えば、VHドメイン、またはVLドメイン、およびVHまたはVLドメインいずれかの抗原結合断片、例えば単離CDR領域からなる断片;(ix)2つの抗原結合部位を含む、いわゆる「ディアボディ」、例えば同じポリペプチド鎖中で、軽鎖可変ドメイン(VL)に連結された重鎖可変ドメイン(VH);ならびに(x)相補性軽鎖ポリペプチドとともに、抗原結合領域対を形成する、タンデムFdセグメント対を含む、いわゆる直鎖抗体が含まれる。
【0031】
調製可能な好ましい抗体断片は、哺乳動物単一可変ドメイン抗体であり、免疫グロブリン可変ドメインに特徴的な配列を含み、そして抗原に特異的に結合し(すなわち500nM以下、例えば400nM以下、好ましくは250nM以下、そして最も好ましくは100nM以下の解離定数)、そして単一可変ドメインとして;すなわちいかなる相補性可変ドメインも含まずに、抗原に結合する、折りたたまれたポリペプチドドメインを含む抗体断片である。単一可変ドメイン抗体には、完全抗体可変ドメインならびに修飾可変ドメイン、例えば1以上のループが、抗体可変ドメインに特徴的でない配列で置換されているもの、あるいは一部切除(truncated)されているかまたはNもしくはC末端伸長を含む抗体可変ドメイン、ならびに可変ドメインの折りたたまれた断片が含まれる。調製可能な好ましい単一可変ドメインは、VHおよびVLの群より選択され、VカッパおよびVラムダが含まれる。最も好ましくは、単一可変ドメインは、ヒトまたはラクダ科(camelid)ドメインであり、ヒト化ラクダ科ドメインが含まれる。
【0032】
したがって、本発明はまた、宿主細胞において、本発明の第一の側面にしたがった発現系を発現する工程を含む、ターゲットポリペプチドの産生のための方法も提供する。
発現系は、使用する細胞に関して、当該技術分野に周知の方法によって発現される。好ましい発現法には、増殖培地中で、特に発酵によって、組換え細胞を培養し、そして次いで発現されたタンパク質を回収する工程が含まれる。用語「増殖培地」は、組換え細胞を増殖させるために用いられる栄養培地を指す。多くの態様において、栄養溶液を使用する。所定の組換え細胞に適した増殖培地が当該技術分野に周知である。
【0033】
基底発現は、DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターに適した条件を選択することによって調節される。適切な場合、例えば細胞が、例えば細胞密度を監視することによって決定される所望の増殖状態に到達するまで増殖している場合、ターゲットポリペプチドをコードするDNAに機能可能であるように連結されたプロモーターに有利に働くように、条件を調整する。多くの態様において、この調整は、対数期増殖中に行われる。
【0034】
本発明を以下の実施例によって限定なしに例示する。
【実施例】
【0035】
プラスミドpAB013の構築
pAB013を生成するための出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。pZT7#2.0は、pAT153ベクター骨格、cer安定性配列、tet A/R、関心対象の遺伝子上流の単一天然lacオペレーター配列、および上流T4転写ターミネーターを有する。NcoI、EcoRIおよびXbaI制限酵素部位によって、合成オリゴヌクレオチドリンカーを用いて、T7A3プロモーターおよび二重完全パリンドロームlacオペレーターをこのプラスミド内にクローニングした。
【0036】
オリゴヌクレオチド1および2をアニーリングさせることによって、リンカー12を調製した:
【0037】
【化1】
【0038】
次いで、リンカーをプラスミドpZT7#2.0に連結させ、そしてNcoI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。形質転換体の最初のスクリーニングは、NcoIを用いた制限消化によった。配列決定によって配列を確認した。次いで、オリゴヌクレオチド3および4をアニーリングさせることによって、T7A3プロモーターカセットをこのベクター内にクローニングした:
【0039】
【化2】
【0040】
アニーリングしたオリゴヌクレオチドをベクターに連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。最初のスクリーニングは、プラスミドDNAの制限消化によった。次いで、配列決定によって配列を確認した。
【0041】
ヒト腫瘍壊死因子(hTNF)−α遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB013を生成した。このプラスミドは、T7A3プロモーター/二重完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNF−αを発現する。
【0042】
プラスミドpAB044の構築
出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。pZT7#2.0は、pAT153ベクター骨格、cer安定性配列、tet A/R、関心対象の遺伝子上流の単一天然lacオペレーター配列、および上流T4転写ターミネーターを有する。NcoI、EcoRIおよびXbaI制限酵素部位によって、合成オリゴヌクレオチドリンカーを用いて、T7A3プロモーターおよび二重完全パリンドロームlacオペレーターをこのプラスミド内にクローニングした。
【0043】
上述のように、オリゴヌクレオチド1および2をアニーリングさせることによって、リンカー12を調製した。
次いで、リンカーをプラスミドpZT7#2.0に連結させ、そしてNcoI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。形質転換体の最初のスクリーニングは、NcoIを用いた制限消化によった。配列決定によって配列を確認した。オリゴヌクレオチド17および18をアニー
リングさせることによって、tacプロモーターカセットをこのベクター内にクローニングした:
【0044】
【化3】
【0045】
アニーリングしたオリゴヌクレオチドを連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。最初のスクリーニングは、プラスミドDNAの制限消化によった。次いで、配列決定によって配列を確認した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB044を生成した。このプラスミドは、tacプロモーター/二重完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNF−αを発現する。
【0046】
プラスミドpAB235の構築
pAB235の出発ベクターはpACYC−Duet(Novagenカタログ番号71147−3)であった。DNA結合タンパク質HU(熱不安定性)−βをコードするHupB配列を大腸菌株W3110(ATCC番号ATCC27325より得られる)のゲノムDNAからPCRによって増幅した。用いたプライマーは、プライマー1: GCCATATGCAGGAAGAAGGAGAATGAATAAATCTCAATTG(配列番号7)およびプライマー2: GCCTCGAGTTAGTTTACCGCGTCTTTCAG(配列番号8)であった。PCR産物を、ベクターpCR2.1 TOPO Blunt(Invitrogenカタログ番号K280020より得られる)内にクローニングした。挿入物を、NcoI−XhoI消化(New England Biolabs)によって取り除き、そしてpACYC−Duet内にクローニングして、ベクターNBJ0585−6−1を作製した。
【0047】
HupA遺伝子(DNA結合タンパク質HU−α)を、構成的lacプロモーター(lacオペレーター配列を除く)およびHupA遺伝子除去のためのNdeI部位を含有するMluI−NcoI断片として設計した。これをベクターNBJ0585−6−1にクローニングして、プラスミドpAB235を作製した。HupA遺伝子配列を図1に提供する(配列番号9)。
【0048】
プラスミドpAB249の構築
pAB249の出発ベクターはpACYC−Duetであった。構成的lacプロモーター(lacオペレーター配列を除く)およびHupA遺伝子除去のためのNdeI部位を含有するMluI−NcoI断片として設計されたHupA遺伝子(DNA結合タンパク質HU−α)を、MluI−NcoI断片として、pACYC−Duetにクローニングして、ベクターNBJ0585−18−1を作製した。これを次いで、NdeI−XhoIで消化して、HupA遺伝子を取り除いた。次いで、大腸菌コドン最適化ラットHMGB−1(高移動度群B)遺伝子をクローニングして、pAB249を作製した。大腸菌
コドン最適化ラットHMGB−1遺伝子配列を図2に提供する(配列番号10)。
【0049】
pAB246の構築
pAB246の出発ベクターはpCI−Neo(Promegaカタログ番号E1841)であった。ラットHMGB−1タンパク質(チャイニーズハムスター卵巣(CHO)細胞における発現のために最適化された遺伝子配列コドン)を、クローニングのため、NheI−NotI断片として合成した。これをpCI−NeoにクローニングしてpAB246を作製した。
【0050】
pAB193の構築
pAB193の出発ベクターはpOPRSV1/MCS(Stratagene)であった。IgG Fc遺伝子配列(図3−配列番号11)をNheI/NotI断片として、SpeI/NotI消化したpOPRSVI/MCSにクローニングして、pAB193を生成した。
【0051】
実施例1
エレクトロポレーションを用いて、プラスミドpAB044を大腸菌宿主株W3110に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD047と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0052】
エレクトロポレーションを用いて、プラスミドpAB235およびpAB044を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD283と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0053】
エレクトロポレーションを用いて、プラスミドpAB235およびpAB013を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD271と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0054】
エレクトロポレーションを用いて、プラスミドpAB249およびpAB013を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD272と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0055】
エレクトロポレーションを用いて、プラスミドpAB013を大腸菌宿主株W3110に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD018と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0056】
組換え大腸菌株の要約を表1に提示する。
表1:組換え大腸菌株
【0057】
【表1】
*DNA結合タンパク質HU(熱不安定性)−α/DNA結合タンパク質HU(熱不安定性)−β
【0058】
実施例2
CLD047およびCLD283のバイアルを−80℃フリーザーから取り出し、そして融解させた。融解したグリセロールストック各々の10μlを、別個に、テトラサイクリン(10μg/ml、CLD047)またはCLD283用にクロラムフェニコール(34μg/ml)およびテトラサイクリン(10μg/ml)を補充した5mlルリアブロス(LB、5g/L酵母エキス(Oxoid)、10g/Lトリプトン(Oxoid)、および5g/L塩化ナトリウム)に接種した。CLD047およびCLD283の培養を37℃で、軌道振盪装置中、200rpmで16時間、インキュベーションした。次いで、500μlのこれらの培養各々を用いて、50mlのルリアブロス(上記のような組成)を含有する250mlエルレンマイヤーフラスコに別個に接種した。フラスコを37℃で、軌道振盪装置中、200rpmでインキュベーションした。上記条件下でインキュベーションを続け、その間に、増殖、細菌細胞内のhTNFαの集積を測定するために、試料を採取した(4時間および22時間)。CLD047およびCLD283の非誘導培養におけるhTNFαの基底集積レベルを、試料細菌のSDS−PAGE後のウェスタンブロット分析(抗hTNFα抗体を用いる)によって比較した。誘導の非存在下での長期のインキュベーションを用いて、観察されうるいかなる基底発現も増幅するであろう最悪条件を提供した。
【0059】
データを図4に示す。驚くべきことに、DNA結合タンパク質HU(熱不安定性)−α/DNA結合タンパク質HU(熱不安定性)−β(CLD283)の存在下でのhTNFαの基底発現は、22時間に渡る長期インキュベーション後であってさえ、有意に減少した。
【0060】
実施例3
CLD018、CLD271およびCLD272のバイアルを−80℃フリーザーから取り出し、そして融解させた。融解したグリセロールストック各々の10μlを、別個に、テトラサイクリン(10μg/ml、CLD018)またはCLD271およびCLD272用にクロラムフェニコール(34μg/ml)およびテトラサイクリン(10μg/ml)を補充した5mlルリアブロス(LB、5g/L酵母エキス(Oxoid)、10g/Lトリプトン(Oxoid)、および5g/L塩化ナトリウム)に接種した。培養を37℃で、軌道振盪装置中、200rpmで16時間、インキュベーションした。次いで、500μlのこれらの培養各々を用いて、50mlのルリアブロス(上記のような組成)を含有する250mlエルレンマイヤーフラスコに別個に接種した。フラスコを37℃で、軌道振盪装置中、200rpmでインキュベーションした。OD600=0.5〜0.7(〜中期指数増殖期)まで増殖を監視し、そして試料を採取した。さらに22時間、上記条件下でインキュベーションを続け、その間に、増殖、細菌細胞内のhTNFαの
集積を測定するために、試料を採取した(3時間(後期指数増殖期)および22時間(安定期))。長期インキュベーション後のCLD018、CLD271およびCLD272の非誘導培養におけるhTNFαの基底集積レベルを、試料細菌のSDS−PAGE後のウェスタンブロット分析(抗hTNFα抗体を用いる)によって比較した。
【0061】
データを図5に示す。データによって、DNA結合タンパク質HU(熱不安定性)−α/DNA結合タンパク質HU(熱不安定性)−β(CLD271)のまたは真核ラットHMGB−1(CLD272)の存在下での、実施例2で用いたものとは異なるプロモーター/発現系を用いたhTNFαの基底発現は、長期インキュベーション後であってさえ、有意に減少することが確認される。
【0062】
実施例4
チャイニーズハムスター卵巣細胞、細胞タイプCHO−PF(ECACC、カタログ番号00102307)のアンプルを凍結ストックから生き返らせ、そして10%ウシ胎児血清(FCS)および4mM L−グルタミンを補充したイスコフの修飾ダルベッコ培地(IMDM)細胞培養増殖培地50ml中、T175静置培養内に回収した。トランスフェクション日まで、フラスコを1:5の分割比でルーチンに継代した。培地を除去し、そしてフラスコ内容物を20mlのリン酸緩衝生理食塩水溶液(PBS)で洗浄した後、4mlのトリプシンを添加し、そして37℃で3分間インキュベーションすることによって、細胞をフラスコから解離させた。細胞を解離させた際、新鮮な培地をフラスコに添加して、トリプシン活性を中和した。トランスフェクション日、細胞をフラスコから解離させ、そして試料を採取して、VicellTM自動化細胞計数装置を用いて、生存細胞濃度および生存度を決定した。細胞濃度を7.5x105細胞.ml−1に調整し、そして1mlの細胞懸濁物を6ウェル細胞培養マイクロタイタープレートの各ウェルにトランスファーした。加湿5%CO2インキュベーター(Sanyo)中、37℃で2時間プレートをインキュベーションし、そしてトランスフェクション複合体を以下のように調製した:
13.3μg pAB193および3.3μgのpAB246(等量のpCMVlacI(Stratagene、Lacリプレッサーを発現する))に加えて、80μL Fugene(登録商標)6トランスフェクション試薬(Roche、カタログ番号1814443001)および667μL IMDM。DNA/Fugene(登録商標)6複合体を室温で2時間インキュベーションした後、複合体由来の200μLを細胞培養プレート上の4ウェル各々に添加した。pAB246プラスミドDNAを除いて、上述の半量を用いて第二の複合体を調製した。これをまた2時間インキュベーションした後、6ウェル細胞培養プレートの2ウェルに添加した。細胞培養プレートを、加湿5%CO2インキュベーター中、37℃で24時間インキュベーションした。最終濃度5mMまでIPTG(イソプロピル−β−D−1−チオガラクトピラノシド)を添加することによって、pAB246を含有する細胞培養プレート上のいくつかのウェルを誘導して、所望のタンパク質IgG−Fcの誘導を確認した。細胞培養プレートを、加湿5%CO2インキュベーター中、37℃でさらに5日間インキュベーションした。酵素連結免疫吸着アッセイ(ELISA)によって、細胞培養増殖培地内へのIgG−Fcの発現/分泌を測定した。
【0063】
データを図6に示す。該データは、HMGB−1同時発現を用いて、哺乳動物発現系において、タンパク質基底発現を減少させうることを立証する。さらに、誘導因子IPTGを培養に添加すると、IgG−Fcタンパク質の誘導がなお可能であり、そして誘導後、タンパク質レベル増加が生じる。
【0064】
実施例5
プラスミドpAB007およびpAB031の構築
pAB031の生成のための出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。EcoRIおよびXbaI制限酵素部位に
よって、合成オリゴヌクレオチドリンカーを用いて、T7A3プロモーターおよび単一完全パリンドロームlacオペレーターをこのプラスミド内にクローニングした。
【0065】
上述のように、オリゴヌクレオチド3および4によって、T7A3プロモーターを含有するリンカーを作製した。
オリゴヌクレオチド3および4をアニーリングさせ、次いで、形成されたリンカーをプラスミドpZT7#2.0に連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。最初のスクリーニングは、プラスミドDNAの制限消化によった。次いで、配列決定によって配列を確認した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB031を生成した。このプラスミドは、T7A3プロモーター/単一完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNF−αを発現する。
【0066】
プラスミドpAB040の構築
pAB040の生成のための出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。EcoRIおよびXbaI制限酵素部位によって、合成オリゴヌクレオチドリンカーを用いて、tacプロモーターおよび単一完全パリンドロームlacオペレーターをこのプラスミド内にクローニングした。
【0067】
オリゴヌクレオチド13および14をアニーリングさせることによって、リンカー1314を調製した。
【0068】
【化4】
【0069】
次いで、リンカーをプラスミドpZT7#2.0に連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。形質転換体の最初のスクリーニングは、NcoIを用いた制限消化によった。配列決定によって配列を確認した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB040を生成した。このプラスミドは、Tacプロモーター/単一完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNFαを発現する。
【0070】
プラスミドpAB041の構築
pAB041の生成のための出発ベクターは、US 6,537,779に記載されるように調製されたpZT7#2.0であった。EcoRIおよびXbaI制限酵素部位によって、合成オリゴヌクレオチドリンカーを用いて、tacプロモーターおよび単一天然lacオペレーターをこのプラスミド内にクローニングした。
【0071】
オリゴヌクレオチド11および12をアニーリングさせることによって、リンカー11
12を調製した。
【0072】
【化5】
【0073】
次いで、リンカーをプラスミドpZT7#2.0に連結させ、そしてXbaI/EcoRI断片として、クローニング宿主株XL−1 Blue MR(Stratagene)に形質転換した。形質転換体の最初のスクリーニングは、NcoIを用いた制限消化によった。配列決定によって配列を確認した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB041を生成した。このプラスミドは、Tacプロモーター/単一天然lacオペレーター配列/lacリプレッサー系の調節下でhTNFαを発現する。
【0074】
プラスミドpAB350の構築
pZT7#3.3発現プラスミドをUS 6,537,779に記載されるように調製した。ヒトTNFα遺伝子をNdeI/XhoI断片として、このプラスミド内にクローニングして、pAB350を生成した。このプラスミドは、T7遺伝子10プロモーター/二重完全パリンドロームlacオペレーター配列/lacリプレッサー系の調節下でhTNFαを発現する。
【0075】
株構築
CLD032
エレクトロポレーションを用いて、プラスミドpAB031を大腸菌宿主株W3110に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD032と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0076】
CLD408
エレクトロポレーションを用いて、プラスミドpAB235およびpAB031を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD408と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0077】
CLD409
エレクトロポレーションを用いて、プラスミドpAB249およびpAB031を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD409と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0078】
CLD042
エレクトロポレーションを用いて、プラスミドpAB041を大腸菌宿主株W3110に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD042と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0079】
CLD410
エレクトロポレーションを用いて、プラスミドpAB235およびpAB041を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD410と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0080】
CLD411
エレクトロポレーションを用いて、プラスミドpAB249およびpAB041を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD411と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0081】
CLD412
エレクトロポレーションを用いて、プラスミドpAB235およびpAB040を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD412と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0082】
CLD413
エレクトロポレーションを用いて、プラスミドpAB249およびpAB040を大腸菌宿主株W3110に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD413と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0083】
CLD023
エレクトロポレーションを用いて、プラスミドpAB350を大腸菌宿主株BL21(λDE3)に形質転換した。テトラサイクリンを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD023と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0084】
CLD414
エレクトロポレーションを用いて、プラスミドpAB235およびpAB350を大腸菌宿主株BL21(λDE3)に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、コロニーを選択した。CLD414と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0085】
CLD415
エレクトロポレーションを用いて、プラスミドpAB249およびpAB350を大腸菌宿主株BL21(λDE3)に同時形質転換した。テトラサイクリンおよびクロラムフェニコールを補充したルリアブロス(LB)寒天上にプレーティングすることによって、
コロニーを選択した。CLD415と称される、生じた組換え株を精製し、そしてグリセロールストック中、−80℃で維持した。
【0086】
実施例5で使用する組換え大腸菌株の要約を表2に示す。
表2
【0087】
【表2】
*DNA結合タンパク質HU(熱不安定性)−α/DNA結合タンパク質HU(熱不安定性)−β
【0088】
CLD032、CLD408、CLD409、CLD042、CLD410、CLD411、CLD0412、CLD0413、CLD023、CLD414およびCLD415のバイアルを−80℃フリーザーから取り出し、そして融解させた。融解したグリセロールストック各々の10μlを、別個に、テトラサイクリン(10μg/ml、CLD0032、CLD042、CLD043およびCLD023)またはCLD408、CLD409、CLD410、CLD411、CLD412、CLD413およびCLD414用にクロラムフェニコール(34μg/ml)およびテトラサイクリン(10μg/ml)を補充した5mlルリアブロス(LB、5g/L酵母エキス(Oxoid)、10g/Lトリプトン(Oxoid)、および5g/L塩化ナトリウム)に接種した。培養を37℃で、軌道振盪装置中、200rpmで16時間、インキュベーションした。次いで、500μlのこれらの培養各々を用いて、50mlのルリアブロス(上記のような組成)を含有する250mlエルレンマイヤーフラスコに別個に接種した。フラスコを37℃で、軌道振盪装置中、200rpmでインキュベーションした。OD600=0.5〜0.7(〜中期指数増殖期)まで増殖を監視した。試料を採取し、そしてフラスコ各対の一方を0.5mM IPTGで誘導した。さらに22時間、上記条件下でインキュベーションを続け、その間に、増殖、細菌細胞内のhTNFαの集積を測定するために、試料を採取した(3時間(後期指数増殖期)および22時間(安定期))。長期インキュベーション後の培養中のhTNFα集積レベルを、試料細菌のSDS−PAGEによって比較した。SimplyBlue染色したSDS−PAGEゲルのレーザー濃度計によって、hTNF
α集積レベル(%総細胞微生物タンパク質(%TCP)として表わす)を決定し、そして株CLD032、CLD408、CLD409、CLD042、CLD410、CLD411、CLD023、CLD414およびCLD415に関して検出されたレベルを、以下の表3に示す。株CLD0412およびCLD0413に関して検出されたレベルを以下の表4に示す。
【0089】
表3のデータは、本発明記載の発現系を含む例示的な株が、DNA結合性転写制御因子タンパク質の発現カセットを欠く対応する系と比較して、hTNFαの基底発現の有意な減少を示すことを示す。しかし、hTNFα発現がIPTG添加によって誘導された際、本発明記載の株によって産生されるhTNFαのレベルは、DNA結合性転写制御因子タンパク質の発現カセットを欠く対応する系によって産生されるレベルに匹敵する。
【0090】
表4のデータは、本発明記載の発現系を含むこれらの株に関するhTNFαの基底発現の優れた調節および優れた誘導産生を示す。
表3. hTNFα発現レベル
【0091】
【表3】
*=本発明記載
【0092】
表4. hTNFα発現レベル
【0093】
【表4】
【特許請求の範囲】
【請求項1】
ターゲットポリペプチド産生のための発現系であって:
a)ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターを含む発現カセット;および
b)DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターを含む、DNA結合性転写制御因子タンパク質を過剰発現させるための発現カセット;
ここで該発現カセットは直交性プロモーターの調節下にある
を含む、前記発現系。
【請求項2】
ターゲットポリペプチドをコードする発現カセットおよびDNA結合性転写制御因子タンパク質を過剰発現させるための発現カセットが異なるベクター上に位置する、請求項1記載の発現系。
【請求項3】
a)ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターを含む発現カセット;および
b)DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターを含む、DNA結合性転写制御因子タンパク質を過剰発現させるための発現カセット;
ここで該発現カセットは直交性プロモーターの調節下にある
を含む、ベクター。
【請求項4】
請求項1または2記載の発現系、あるいは請求項3記載のベクターを含む、宿主細胞。
【請求項5】
大腸菌(E coli)である、請求項4記載の宿主細胞。
【請求項6】
請求項1または2記載の発現系の発現を含む、ターゲットポリペプチドの産生のためのプロセス。
【請求項7】
DNA結合性転写制御因子タンパク質の発現に有利に働く条件下で、細胞を所望の増殖状態に増殖させ、そして細胞が所望の増殖状態に到達したら、条件をターゲットポリペプチドの発現に有利に働くものに調整する、請求項6記載のプロセス。
【請求項8】
DNA結合性転写制御因子タンパク質が、DNA結合タンパク質HU−α、DNA結合タンパク質HU−β、およびHMGBファミリーからなる群より選択される、先行する請求項いずれか記載の発現系、ベクター、宿主細胞またはプロセス。
【請求項9】
DNA結合性転写制御因子タンパク質が、構成的プロモーターに機能可能であるように連結されている、先行する請求項いずれか記載の発現系、ベクター、宿主細胞またはプロセス。
【請求項10】
プロモーターが、T7A1、T7A2、T7A3、spcリボソームタンパク質オペロンプロモーター、β−ラクタマーゼ遺伝子プロモーター、ファージλのPLプロモーター、複製調節プロモーターPRNAIおよびPRNAII、rrnBリボソームRNAオペロンのP1およびP2プロモーター、Lacリプレッサータンパク質プロモーターpLacI、グリセルアルデヒドリン酸デヒドロゲナーゼおよび形質膜H(+)−ATPアーゼプロモーター、接合因子−αプロモーター、KEX2、TEF−1、サルウイルス40初期プロモーター、ラウス肉腫ウイルスプロモーター、サイトメガロウイルスプロモーターおよびヒトβアクチンプロモーターからなる群より選択される、請求項9記載の発現系、
ベクター、宿主細胞またはプロセス。
【請求項11】
誘導性プロモーターが、T7 RNAポリメラーゼ依存性プロモーター、lac、lacUV5、trp、tac、trc、phoA、アラビノース誘導性プロモーター、温度誘導性プロモーター、銅誘導性プロモーター、uspA、uspB、malK、浸透圧誘導性プロモーター、ガラクトース誘導性プロモーター、フェロモン誘導性プロモーター、グルコアミラーゼプロモーター、テトラサイクリン応答性プロモーター、ヒトc−fosプロモーター、エクジソン誘導性プロモーターおよびグルココルチコイド誘導性プロモーターからなる群より選択される、先行する請求項いずれか記載の発現系、ベクター、宿主細胞またはプロセス。
【請求項12】
DNA結合性転写制御因子タンパク質の発現カセットが、構成的lacプロモーターに機能可能であるように連結された大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−βの発現カセットを含み、そしてターゲットポリペプチドの発現カセットがtacプロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含み、そしてまた単一のオペレーターも含む、請求項2記載の発現系。
【請求項13】
ターゲットポリペプチドの発現カセットに含まれるオペレーターが、天然lacオペレーターまたは完全にパリンドロームであるlacオペレーターである、請求項12記載の発現系。
【請求項14】
DNA結合性転写制御因子タンパク質の発現カセットが、構成的lacプロモーターに機能可能であるように連結されたラットHMGB−1の発現カセットを含み、そしてターゲットポリペプチドの発現カセットが、a)tacプロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含み、そしてまた単一のオペレーターも含むか;またはb)T7プロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含み、そしてまた2つの完全にパリンドロームであるオペレーターも含み、1つのオペレーターがプロモーターの上流に位置し、そして1つのオペレーターがプロモーターの下流に位置する、請求項2記載の発現系。
【請求項15】
請求項12、13または14記載の発現系の発現を含む、ターゲットポリペプチドの産生のためのプロセス。
【請求項16】
DNA結合性転写制御因子タンパク質の発現に有利に働く条件下で、細胞を所望の増殖状態に増殖させ、そして細胞が所望の増殖状態に到達したら、条件をターゲットポリペプチドの発現に有利に働くものに調整する、請求項15記載のプロセス。
【請求項17】
請求項12、13または14記載の発現系を含む、宿主細胞。
【請求項1】
ターゲットポリペプチド産生のための発現系であって:
a)ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターを含む発現カセット;および
b)DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターを含む、DNA結合性転写制御因子タンパク質を過剰発現させるための発現カセット;
ここで該発現カセットは直交性プロモーターの調節下にある
を含む、前記発現系。
【請求項2】
ターゲットポリペプチドをコードする発現カセットおよびDNA結合性転写制御因子タンパク質を過剰発現させるための発現カセットが異なるベクター上に位置する、請求項1記載の発現系。
【請求項3】
a)ターゲットポリペプチドをコードするDNAに機能可能であるように連結された誘導性プロモーターを含む発現カセット;および
b)DNA結合性転写制御因子タンパク質をコードするDNAに機能可能であるように連結されたプロモーターを含む、DNA結合性転写制御因子タンパク質を過剰発現させるための発現カセット;
ここで該発現カセットは直交性プロモーターの調節下にある
を含む、ベクター。
【請求項4】
請求項1または2記載の発現系、あるいは請求項3記載のベクターを含む、宿主細胞。
【請求項5】
大腸菌(E coli)である、請求項4記載の宿主細胞。
【請求項6】
請求項1または2記載の発現系の発現を含む、ターゲットポリペプチドの産生のためのプロセス。
【請求項7】
DNA結合性転写制御因子タンパク質の発現に有利に働く条件下で、細胞を所望の増殖状態に増殖させ、そして細胞が所望の増殖状態に到達したら、条件をターゲットポリペプチドの発現に有利に働くものに調整する、請求項6記載のプロセス。
【請求項8】
DNA結合性転写制御因子タンパク質が、DNA結合タンパク質HU−α、DNA結合タンパク質HU−β、およびHMGBファミリーからなる群より選択される、先行する請求項いずれか記載の発現系、ベクター、宿主細胞またはプロセス。
【請求項9】
DNA結合性転写制御因子タンパク質が、構成的プロモーターに機能可能であるように連結されている、先行する請求項いずれか記載の発現系、ベクター、宿主細胞またはプロセス。
【請求項10】
プロモーターが、T7A1、T7A2、T7A3、spcリボソームタンパク質オペロンプロモーター、β−ラクタマーゼ遺伝子プロモーター、ファージλのPLプロモーター、複製調節プロモーターPRNAIおよびPRNAII、rrnBリボソームRNAオペロンのP1およびP2プロモーター、Lacリプレッサータンパク質プロモーターpLacI、グリセルアルデヒドリン酸デヒドロゲナーゼおよび形質膜H(+)−ATPアーゼプロモーター、接合因子−αプロモーター、KEX2、TEF−1、サルウイルス40初期プロモーター、ラウス肉腫ウイルスプロモーター、サイトメガロウイルスプロモーターおよびヒトβアクチンプロモーターからなる群より選択される、請求項9記載の発現系、
ベクター、宿主細胞またはプロセス。
【請求項11】
誘導性プロモーターが、T7 RNAポリメラーゼ依存性プロモーター、lac、lacUV5、trp、tac、trc、phoA、アラビノース誘導性プロモーター、温度誘導性プロモーター、銅誘導性プロモーター、uspA、uspB、malK、浸透圧誘導性プロモーター、ガラクトース誘導性プロモーター、フェロモン誘導性プロモーター、グルコアミラーゼプロモーター、テトラサイクリン応答性プロモーター、ヒトc−fosプロモーター、エクジソン誘導性プロモーターおよびグルココルチコイド誘導性プロモーターからなる群より選択される、先行する請求項いずれか記載の発現系、ベクター、宿主細胞またはプロセス。
【請求項12】
DNA結合性転写制御因子タンパク質の発現カセットが、構成的lacプロモーターに機能可能であるように連結された大腸菌DNA結合タンパク質HU−α/DNA結合タンパク質HU−βの発現カセットを含み、そしてターゲットポリペプチドの発現カセットがtacプロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含み、そしてまた単一のオペレーターも含む、請求項2記載の発現系。
【請求項13】
ターゲットポリペプチドの発現カセットに含まれるオペレーターが、天然lacオペレーターまたは完全にパリンドロームであるlacオペレーターである、請求項12記載の発現系。
【請求項14】
DNA結合性転写制御因子タンパク質の発現カセットが、構成的lacプロモーターに機能可能であるように連結されたラットHMGB−1の発現カセットを含み、そしてターゲットポリペプチドの発現カセットが、a)tacプロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含み、そしてまた単一のオペレーターも含むか;またはb)T7プロモーターに機能可能であるように連結されたターゲットポリペプチドの発現カセットを含み、そしてまた2つの完全にパリンドロームであるオペレーターも含み、1つのオペレーターがプロモーターの上流に位置し、そして1つのオペレーターがプロモーターの下流に位置する、請求項2記載の発現系。
【請求項15】
請求項12、13または14記載の発現系の発現を含む、ターゲットポリペプチドの産生のためのプロセス。
【請求項16】
DNA結合性転写制御因子タンパク質の発現に有利に働く条件下で、細胞を所望の増殖状態に増殖させ、そして細胞が所望の増殖状態に到達したら、条件をターゲットポリペプチドの発現に有利に働くものに調整する、請求項15記載のプロセス。
【請求項17】
請求項12、13または14記載の発現系を含む、宿主細胞。
【図1】


【図2】
【図3】

【図6】
【図4】
【図5】
【図2】
【図3】
【図6】
【図4】
【図5】
【公表番号】特表2012−521752(P2012−521752A)
【公表日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願番号】特願2012−501373(P2012−501373)
【出願日】平成22年3月24日(2010.3.24)
【国際出願番号】PCT/GB2010/000547
【国際公開番号】WO2010/109187
【国際公開日】平成22年9月30日(2010.9.30)
【出願人】(508236033)フジフィルム・ダイオシンス・バイオテクノロジーズ ・ユーケイ・リミテッド (8)
【Fターム(参考)】
【公表日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願日】平成22年3月24日(2010.3.24)
【国際出願番号】PCT/GB2010/000547
【国際公開番号】WO2010/109187
【国際公開日】平成22年9月30日(2010.9.30)
【出願人】(508236033)フジフィルム・ダイオシンス・バイオテクノロジーズ ・ユーケイ・リミテッド (8)
【Fターム(参考)】
[ Back to top ]