
発現微量タンパク質/ペプチドの検出・分離・同定法
本発明は、発現微量タンパク質/ペプチドの検出・分離・同定法及びそのシステムを提供するものであり、本発明は、微量の発現タンパク質及び/又はペプチドの検出・分離・同定方法であって、蛍光試薬で標識した被験試料中のタンパク質及び/又はペプチドの蛍光誘導体を、HPLCに付し、その蛍光分画を捕集した後、酵素水解に付し、その蛍光標識フラグメント及び非蛍光標識フラグメントを質量分析して得られた各フラグメントのイオン分子量情報をタンパク質及び/又はペプチドフラグメントデータベースと照合し、構造解析することを特徴とする上記タンパク質及び/又はペプチドの検出・分離・同定方法、及びその同定システム、に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、微量の発現タンパク質及び/又はペプチドの検出・分離・同定方法に関するものであり、更に詳しくは、生体において遺伝子の発現により産生される微量の発現タンパク質及び/又はペプチドを簡便な方法で、高感度に検出し、同定することを可能とする新規な発現タンパク質及び/又はペプチドの検出・分離・同定方法、及びその同定システムに関するものである。
本発明は、ポストゲノム時代において重要な役割を果たすことが期待される、発現タンパク質及び/又はペプチドを網羅的に解析するプロテオーム技術における新しい検出・分離・同定手法を提供するものとして有用である。
【背景技術】
【0002】
ポストゲノム時代において重要な課題は、遺伝子を介して発現する発現タンパク質/ペプチドの微量検出とその分離・同定である。従来、この課題達成のために、2次元電氣泳動後のペプチドフィンガープリント法が汎用されてきた(非特許文献1参照)。しかし、この方法は、煩雑な操作のために該方法の再現性に難点があった。この難点を克服する手法として、最近、多次元高速液体クロマトグラフィー(多次元HPLC)による分離・同定法、或いはICATによる手法が提案されている(非特許文献2参照)。
【0003】
これらのうち、タンパク質/ペプチドを、直接、多次元HPLCで分離・同定する方法は、全てのタンパク質/ペプチドを同時に処理するために、多大な労力と時間を要するという欠点がある。また、ICATによる手法は、チオール含有タンパク質/ペプチドのチオール基をisotope−coded affinity tags (ICAT) reagentで標識した後、それをビオチン結合カラムにて捕集し、これら全てを酵素水解し、得られたペプチドフラグメント混合物をHPLCで分離、質量分析計(MS)にて質量分析し、タンパク質/ペプチドを網羅的に解析しようとするものである。しかし、この方法は、チオール含有タンパク質/ペプチドの全てを酵素水解するため、大量に存在する目的以外のタンパク質/ペプチドのフラグメントが、目的とする微量タンパク質/ペプチドのフラグメントの検出及びその同定を妨害する、と言う欠点があり、当技術分野においては、更なる方法のブレークスルーが必要とされていた。
【0004】
【非特許文献1】Dunn MJ.Two−dimensional gel electrophoresis of proteins, J Chromatogr 1987;418:145−185
【非特許文献2】Gygi S.P, Rist B, Gerber S.A, Turecek F, Gelb M.H, Aebersold R、 Quantitative analysis of complex protein mixtures using isotope−coded affinitytags,Nature Biotechnology 1999;17:994−999
【発明の開示】
【発明が解決しようとする課題】
【0005】
このような状況の中で、本発明者らは、上記従来技術に鑑みて、上記従来技術における諸問題を抜本的に解決することを目標として鋭意研究を積み重ねた結果、従来法と異なり、被験試料中の蛍光標識可能なタンパク質及び/又はペプチドのみを蛍光選択的に分離した後、これを酵素水解に付し、分画した蛍光画分を質量分析、データベース照合、構造解析に供することにより、従来法では検出不可能であった微量の発現タンパク質及び/又はペプチドを高感度に検出し、同定することができることを見出し、本発明を完成するに至った。
【0006】
本発明は、遺伝子を介して発現する微量の発現タンパク質及び/又はペプチドを、簡便な測定手法で、高感度に検出・分離・同定することを可能とする上記発現タンパク質及び/又はペプチドの微量検出・分離・同定方法を提供することを目的とするものである。
また、本発明は、上記微量検出・分離・同定方法に使用する微量の発現タンパク質及び/又はペプチドを、高感度で検出・分離・同定するための発現タンパク質及び/又はペプチド同定システムを提供することを目的とするものである。
更に、本発明は、従来法では検出することができなかった、遺伝子を介して発現する微量の発現タンパク質及び/又はペプチドを超高感度で検出・分離・同定することを可能とする新しい分析方法及び手段を提供することを目的とする。
【課題を解決するための手段】
【0007】
上記課題を解決するための本発明は、以下の技術的手段から構成される。
(1)被験試料中の発現微量タンパク質及び/又はペプチドを高感度に検出・分離・同定する方法であって、被験試料中のタンパク質及び/又はペプチドを蛍光誘導体とした後、これを蛍光検出により分離し、その蛍光画分を質量分析に付するか、又はその蛍光画分を酵素水解に付し、そのペプチド断片を分離し、その画分を質量分析に付し、データベース照合、構造解析に供して発現タンパク質及び/又はペプチドの同定を行うことを特徴とする上記発現タンパク質及び/又はペプチドの検出・分離・同定方法。
(2)被験試料中のタンパク質及び/又はペプチドを蛍光誘導体とした後、HPLCに付し、その蛍光分画を捕集した後、酵素水解に付し、その蛍光標識フラグメント及び非蛍光標識フラグメントを質量分析又はMS/MS分析して得られた各フラグメントのイオン分子量情報をタンパク質及び/又はペプチドフラグメントデータベースと照合し、構造解析する、前記(1)に記載の方法。
(3)(a)被験試料中のタンパク質及び/又はペプチドを蛍光試薬で標識する、(b)それを1次元又は2次元のHPLC/蛍光検出により、その蛍光分画を捕集する、(c)上記蛍光分画を酵素水解に付する、(d)それを第二段階のHPLC/蛍光検出により、その蛍光クロマトグラムを得ると共に、その全ピークを質量分析に付し、データベース照合、構造解析に供する、前記(1)に記載の方法。
(4)タンパク質及び/又はペプチド試料の水溶液に、官能基特異的蛍光試薬を加え、場合により、界面活性剤及び/又はタンパク変性剤を加え、タンパク質及び/又はペプチドを蛍光標識する、前記(1)から(3)のいずれかに記載の方法。
(5)蛍光標識したタンパク質及び/又はペプチド試料を蛍光検出器付きイオン交換カラムHPLC、逆相分配HPLC、ゲル濾過HPLC、又は電気泳動に代表される分離手段に付し、蛍光をモニターしながらそのピーク分画を捕集する、前記(1)から(3)のいずれかに記載の方法。
(6)蛍光分画を、各種ペプチダーゼ、トリプシン、キモトリプシンに代表されるタンパク質分解酵素を用いて酵素水解する、前記(1)から(3)のいずれかに記載の方法。
(7)酵素水解物を蛍光検出器付き逆相HPLCに付し、蛍光ピークを検出すると共に、蛍光標識フラグメント及び蛍光非標識フラグメントの質量分析又はMS/MS分析を行う、前記(1)から(3)のいずれかに記載の方法。
(8)質量分析又はMS/MS分析に付して得られた各フラグメントのイオン分子量情報を、コンピューターによるタンパク質及び/又はペブチドフラグメントデータベースと照合し、構造解析して、酵素水解以前のタンパク質及び/又はペプチドの同定を行う、請求項1から3のいずれかに記載の方法。
(9)被験試料が、生体試料から採取したタンパク質及び/又はペプチド試料である、前記(1)から(3)のいずれかに記載の方法。
(10)タンパク質及び/又はペプチドフラグメント情報、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを用いてデータベース照合する、前記(1)から(3)のいずれかに記載の方法。
(11)前記(1)から(10)のいずれかに記載の方法に使用する発現微量タンパク質及び/又はペプチド検出・分離・同定システムであって、被験試料のタンパク質及び/又はペプチドを蛍光試薬で標識するための第一反応器、蛍光試薬で標識した蛍光誘導体を蛍光分画するための1次元又は2次元の蛍光検出器付きHPLC、蛍光分画を酵素水解するための第二反応器、酵素水解物の蛍光標識フラグメントを蛍光検出するための第二段階の蛍光検出器付きHPLC、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを搭載した構造解析装置の1種又は2種以上を構成要素として含むことを特徴とする上記検出・分離・同定システム。
(12)上記第一反応器、1次元又は2次元の蛍光検出器付きHPLC、第二反応器、第二段階の蛍光検出器付きHPLCを直列に配置してなる、前記(11)に記載のシステム。
(13)被験試料中のタンパク質及び/又はペプチドを、蛍光誘導体化試薬として、下記の化5の一般式(1)
【0008】
【化5】

【0009】
〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、−NHR′(但、R′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキル)、又は−NR′′R′′′(但し、R′′はアルキル、R′′′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキルを示す。〕で表わされる化合物、又はその同位体化合物又は下記の化2の一般式(2)
【0010】
【化6】
【0011】
(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物又はその同位体化合物、を用いて、蛍誘導体とする、請求項1に記載の方法。
(14)前記(1)に記載の方法でタンパク質及び/又はペプチドを蛍光誘導体化するために使用する蛍光誘導体化試薬であって、下記の化7の一般式(1)
【0012】
【化7】
【0013】
〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、−NHR′(但、R′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキル)、又は−NR′′R′′′(但し、R′′はアルキル、R′′′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキルを示す。〕で表わされる化合物、又はその同位体化合物又は下記の化2の一般式(2)
【0014】
【化8】
【0015】
(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物又はその同位体化合物、を用いて、蛍誘導体とする、請求項1に記載の方法。
(15)被検試料のタンパク質及び/又はペプチドを蛍光誘導体化後、HPLCで分離・検出し、分画後、酵素水解し、この水解物を直接質量分析で配列分析とタンパク質の同定を行なうことを特徴とするタンパク質及び/又はペプチドの検出・分離・同定方法。
(16)被検試料として、異なる試料A中及び試料B中のタンパク質及び/又はペプチドを、それぞれ蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬でそれぞれ誘導体化後、蛍光検出器付きHPLCで分離・検出し、分画後、各蛍光ピークをそのまま又は合体して定量に供する、及び/又は各蛍光ピークを合体して酵素水解に供し、この水解物を定量に供する、又はこの水解物をHPLC−質量分析に供し、同定を行なう、ことを特徴とするタンパク質及び/又はペプチドの検出・分離・同定方法。
(17)各蛍光ピークをそのまま又は合体してHPLCによる定量に供し、試料A中及び試料B中のタンパク質及び/又はペプチドの各誘導体の比率を算出する前記(16)に記載の方法。
(18)水解物をHPLCによる定量に供し、試料A中及び試料B中のタンパク質及び/又はペプチドの各誘導体の比率を算出する前記(16)に記載の方法。
(19)試料A中及び試料B中のタンパク質及び/又はペプチドの第1の蛍光誘導体化試薬との反応物及び第2の蛍光誘導体化試薬との反応物を合体し、2つの励起・蛍光検出の可能なHPLCに供し、分画後、各蛍光ピークを合体して酵素水解に供し、この水解物をHPLC−質量分析に供し、同定を行う前記(16)に記載の方法。
(20)試料A、Bが、2種類の細胞又は組織又は体液試料である前記(16)に記載の方法。
(21)蛍光誘導体化試薬として、DAABD−X、DAASeBD−X、及びDAAThBD−X(但し、XはCl又はF)のうちの励起・蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬でタンパク質及び/又はペプチドを誘導体化する前記(16)に記載の方法。
(22)蛍光波長の異なる蛍光誘導体化試薬として、DAABD−X、DAASeBD−X、又はDAAThBD−X(但し、XはCl又はF)と、それらの各同位体を組み合わせて使用する前記(21)に記載の方法。
(23)酵素水解した試料を直接質量分析に付しペプチドマップを得ると同時に、蛍光試薬の有する骨格並びに電荷を活用して、蛍光標識化ペプチド断片を質量分析測定部で抽出してシステイン含有ペプチド部分の構造を取得し、これらを基にタンパク質及び/又はペプチドの同定を行なう前記(16)に記載の方法。
(24)蛍光誘導体化試薬で誘導体化したタンパク質及び/又はペプチドを分解することなく分画することが可能な、少なくともミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、及びミクロ自動注入装置を具備したことを特徴とする自動分画装置。
(25)少なくとも、ミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、酵素反応装置、及びミクロ自動注入装置を備え、任意に、質量分析(MS)システムを具備したことを特徴とする微量タンパク質の高性能・簡易定量・同定解析装置。
【0016】
次に、本発明について更に詳細に説明する。
本発明は、上記従来法の難点を克服するためになされたものであり、1)微量の発現タンパク質/ペプチドを蛍光試薬で標識し、2)それをHPLC/蛍光検出にて第一段の分離・蛍光検出を行い、3)その蛍光分画(コントロール試料と比べて、被験試料に特異的に増減する蛍光画分)のみを捕集後、酵素水解し、それを第二段のHPLC/蛍光検出にて分離し、蛍光ピークの確認を行った後、HPLC/MSに付し、蛍光標識タンパク質/ペプチドフラグメントの同定を行い、当該微量タンパク質/ペプチドの特定を行う方法に関するものである。尚、タンパク質/ペプチド試料が純度の高い場合には、第一段のHPLC/蛍光検出による分離を省くことができる。本発明の方法は、従来法と異なり、蛍光標識可能なタンパク質/ペプチドのみを特異的に抽出し、検出・同定することができるという特徴を有し、微量の発現タンパク質/ペプチドを特定するために尤も相応しい方法である。
【0017】
本発明では、被験試料として、生体から採取したあらゆる種類のタンパク質及び/又はペプチドを含む試料が対象とされる。本発明の方法では、被験試料中の微量の発現タンパク質/ペプチドを蛍光試薬で標識し、蛍光誘導体化するが、この場合、タンパク質/ペプチド水溶液に、官能基特異的蛍光試薬を加え、場合により、界面活性剤及び/又はタンパク変性剤を加え、発現タンパク質/ペプチドを定量的に誘導体化することが重要である。即ち、本発明では、タンパク質/ペプチド試料の水溶液に、界面活性剤と場合によっては還元剤を添加し、これに官能基特異的蛍光試薬を加え、必要により、加温することにより、タンパク質及び/又はペプチドを蛍光標識する。本発明では、上記界面活性剤として、非イオン性、陰イオン性、陽イオン性及び両イオン性界面活性剤が用いられる。また、本発明では、上記還元剤として、好適には、Tris(2−carboxyethyl)phosphine、tributylphosphineが用いられるが、これらに制限されるものではなく、同効のものであれば同様に使用することができる。
【0018】
本発明において、上記官能基特異的蛍光試薬として、(例えば、4−Fluoro−7−nitro−2,1,3−benzoxadiazole(NBD−F)、5−(N,N−Dimethylamino)naphthalene−1−sulfonyl chloride (DNS−CL)、Orthophthaldehyde(OPA)、Fluorescamine、9−Fluorenylmethyl chloroformate (FMOC)等のアミノ基特異的蛍光試薬、Ammonium 7−fluoro−2,1,3−benzoxadiazole−4−sulfonate(SBD−F)、4−(Aminosulfonyl)−7−fluoro−2,1,3−benzoxadiazole(ABD−F)、4−(Acetylaminosulfonyl)−7−fluoro−2,1,3−benzoxadiazole(AcABD−F)、4−Fluoro−7−trichloroacetylaminosulfonyl−2,1,3−benzoxadiazole(TcAcABD−F)、monobromobimane等のチオール基特異的蛍光試薬、4−Nitro−7−N−piperazino−2,1,3−benzoxadiazole(NBD−PZ)、4−N,N−Dimethylaminosulfonyl−7−N−piperazino−2,1,3−benzoxadiazole(DBD−PZ)と縮合剤との組み合わせによるカルボキシル基特異的蛍光試薬、又は、4−(N−Chloroformylmethyl−N−methyl)amino−7−nitro−2,1,3−benzoxadiazole(NBD−COCL)等の水酸基用蛍光試薬)が例示されるが、これらに制限されない。
【0019】
本発明においては、必要により、加温する(例えば、30−100℃、望ましくは40−70℃で10−300分間、望ましくは60−180分間)ことにより、タンパク質/ペプチドを蛍光標識する。その後、反応液のほぼ全量を蛍光検出器付きイオン交換カラムHPLC、又は逆相分配HPLC、又はゲル濾過HPLCに付し、蛍光をモニターしながらピーク分画を分取する。この場合、蛍光検出は、標識蛍光体の励起・蛍光波長に相当する波長に設定して行う。例えば、NBD−F、又はSBD−Fで標識した場合には、励起波長480nm或いは380nm、励起波長520nm又は505nmに設定する。イオン交換HPLCの場合には、塩、例えば、食塩、硫酸ナトリウム、過塩素酸カリウム、酢酸アンモニウムなど、望ましくは酢酸アンモニウムのような揮発性の塩を段階的に増量し、それぞれの画分を得る。この画分そのもの又はこの画分を濃縮・乾固した試料を、酵素水解に付す。酵素としては、各種ペプチダーゼ、トリプシン、キモトリプシンなど、適宜のタンパク質分解酵素が用いられる。この際、酵素カラムを接続してオンラインで酵素水解を行うこともできる。
【0020】
この溶液の一部を蛍光検出器付き逆相分配HPLCに付し、蛍光標識体の溶出位置を確認する。次いで、この逆相HPLCカラムの出口を質量分析計(どの様な質量分析計でも対応可能であるが、望ましくはエレクトロスプレー型質量分析計を用いる)に接続し、酵素水解物の蛍光標識フラグメント及び蛍光非標識フラグメントの質量分析(蛍光標識フラグメントは一回の質量分析、蛍光非標識フラグメントは親イオンを更に質量分析する)又は質量分析/質量分析(MS/MS)を行う。この際、蛍光検出器と質量分析計を直列に接続することも可能である。このようにして得られた各フラグメントのイオン分子量情報を、コンピューターに接続したタンパク質/ペプチドフラグメントデータベースと照合し、構造解析することにより、酵素水解以前のタンパク質/ペプチドの同定を行う。この場合、本発明では、タンパク質及び/又はペプチドフラグメント情報、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを用いてデータベース照合を行う。
【0021】
本発明では、発現タンパク質及び/又はペプチドを含む被験試料中のタンパク質及び/又はペプチドを蛍光誘導体化し、この蛍光誘導体をHPLC/蛍光検出で分離し、蛍光ピークの強さを比較して標的発現タンパク質及び/又はペプチドの蛍光誘導体を分離し、得られた標的発現タンパク質及び/又はペプチドのピーク画分を酵素分解し、次いで、質量分析又はMS/MS分析、データベース照合、構造解析により、上記タンパク質及び/又はペプチドを同定する。タンパク質及び/又はペプチドのアミノ基、チオール基、水酸基及びカルボキシル基などの機能性部分を誘導化するための多くの蛍光試薬が存在するので、本発明では、その目的に応じて、適当な試薬を任意に選択して使用することができる。後記する実施例に示されるように、例えば、Cys−含有タンパク質を誘導化するためには、チオール基に特異的な試薬である、Ammonium 7−fluoro−2,1,3−benzoxadizole−4−sulfonate(SBD−F)を使用することができる。図1に、本発明の方法のプロセスの一例を模式的に示す。後記する実施例に示されるように、実際、このようにして、ラットに10mgのデキサメタゾンを投与して2日後のランゲルハンス島におけるPancreatic polypeptide、プロインシュリン 2、78KD Glucose−regulated protein、プロテイン結合フォスファチジルアミン及びチオレドキシンが強く誘導されたことが示された。
【0022】
本発明の方法で重要な点は、各組織におけるタンパク質の量は、HPLC/蛍光検出で分離する前に、例えば、正常組織と非正常組織との組織間で定量、比較されるべきであることから、標的発現タンパク質を定量的に誘導体化することである。そのために、本発明では、適宜の界面活性剤が用いられるが、例えば、いくつかの界面活性剤についてBSAにより検討したところ、CHAPSがn−Dodecyl−β−D−maltopyranosideと比べて高い強度を示した(図2参照)。本発明では、pH、温度、反応時間及び誘導体化反応の添加剤等について、標的発現タンパク質及び/又はペプチドに応じて、最適条件を設定することで定量的な蛍光誘導体化が可能である。本発明では、これらの条件は、発現タンパク質/ペプチドの種類、分析目的等に応じて、適宜設定することができる。本発明の方法により、試験タンパク質/ペプチドのクロマトグラムは単一の蛍光ピークを示した(図3参照)。本発明の方法において、タンパク質及び/又はペプチドの検出限界は、0.2−6.0fmolであり、最適条件下での10−1000fmolの範囲で良好な直線(γ>0.9994)の測定曲線が得られ(表1参照)、検出性能は、従来法に比べて、顕著であることが分かる。表1に、蛍光検出/HPLCによる各種タンパク質及び/又はペプチドの検出限界を示した。
【0023】
【表1】
【0024】
更に、本発明では、上記方法に使用する微量の発現タンパク質及び/又はペプチド検出・分離・同定システムとして、被験試料のタンパク質及び/又はペプチドを蛍光試薬で標識するための第一反応器、蛍光試薬で標識した蛍光誘導体を蛍光分画するための1次元又は2次元の蛍光検出器付きHPLC、蛍光分画を酵素水解するための第二反応器、酵素水解物の蛍光標識フラグメントを蛍光検出するための第二段階の蛍光検出器付きHPLC、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを搭載した構造解析装置の1種又は2種以上を構成要素として含む上記検出・分離・同定システムが用いられる。この場合、上記第一反応器、2次元の蛍光検出器付きHPLC、第二反応器、第二段階の蛍光検出器付きHPLCを直列に配置することができる。これらの装置は、その使用目的に応じて、適宜の容量、形態に任意に設計することができる。
【0025】
本発明は、被験試料中のタンパク質及び/又はペプチドを、蛍光誘導体化試薬として、前記一般式(1)〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、又は−NHR′(但し、R′はN置換アルキル)を示す。〕で表わされる化合物、又は前記一般式(2)(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物、を用いて、蛍光誘導体とすることができる。更に、本発明は、これらの化合物のいずれか1種を有効成分とする新規蛍光誘導体化試薬を提供することができる。
【0026】
これらの化合物の具体例としては、好適には、例えば、以下の例があげられるが、これらに制限されるものではなく、これらと同等ないし類似の化合物であれば同様に使用することができる。本発明の化合物は、後記する実施例に具体的に記載した方法と同様にして容易に合成することができる。
(1)DAABD−Cl[4−(dimethylaminoethyl aminosulfonyl)−7−chloro−2,1,3−benzoxadiazole]
(2)TAABD−Cl(7−chloro−2,1,3−benzoxadiazole−4−sulfonylaminoethyl rimethylammonium chloride)
(3)DAABD−F[4−(dimethylaminoethyl aminosulfonyl)−7−fluoro−2,1,3−benzoxadiazole]
(4)TAABD−F(7−fluoro−2,1,3−benzoxadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
(5)DAABSeD−Cl[4−(dimethylaminoethyl aminosulfonyl)−7−chloro−2,1,3−benzoselenadiazole]
(6)TAABSeD−Cl(7−chloro−2,1,3−benzoselenadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
(7)DAABSeD−F[4−(dimethylaminoethyl aminosulfonyl)−7−fluoro−2,1,3−benzoselenadiazole]
(8)TAABSeD−F(7−fluoro−2,1,3−benzoselenadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
(9)DAABThD−Cl[4−(dimethylaminoethyl aminosulfonyl)−7−chloro−2,1,3−benzothiadiazole]
(10)TAABThD−Cl(7−chloro−2,1,3−benzothiadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
(11)DAABThD−F[4−(dimethylaminoethyl aminosulfonyl)−7−fluoro−2,1,3−benzothiadiazole]
(12)TAABThD−F(7−fluoro−2,1,3−benzothiadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
【0027】
本発明では、蛍光誘導体化試薬として、例えば、SBD−X、SBSeD−X、SBThD−X、DAABD−X、TAABD−X、DAABSeD−X、TAABSeD−X、DAABThD−X、TAABThD−X(但し、XはCl、又はF)、及びそれらの各同位体が提供される。これらの化合物は、いずれも、後記する実施例に示した化合物の場合と同様にして合成することができる。また、本発明では、前記一般式(1)で表わされる化合物の側鎖のアルキル基の鎖長を任意に変更することが可能である。本発明では、これらの化合物の、蛍光波長とHPLC分離における保持時間の相違を利用して、これらの化合物を2つ以上組み合わせて使用することができる。HPLCからの溶出は、例えば、DAABSeD−F<DAABD−Cl又はDAPABSeD−F(側鎖のアルキル:プロピル)<DAPABD−Clの順に遅くなる。また、上記化合物の各同位体を使用することにより、更なる微量検出が可能となる。
【0028】
本発明では、蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬を利用することにより、例えば、同一起源の異なる歴史を有する二つのタンパク質試料の比較を簡便、かつ同時に行なうことができる。例えば、一方が病態患者試料で一方が健常者からの試料、あるいは一つの細胞又は組織試料で、一方がある薬剤で処理された試料で、他方が未処理の試料の比較を同一のクロマトグラムに基づいて同時に行なうこと、それにより、各蛍光誘導体化試薬で誘導体化されたタンパク質及び/又はペプチドの各誘導体を簡便、かつ正確に定量すること、が可能であり、2つの試料中のタンパク質及び/又はペプチドのプロファイルを同時的に測定し、比較することが実現できる。
【0029】
本発明では、蛍光誘導体化試薬で誘導体化したタンパク質及び/又はペプチドを分解することなく分画することが可能な、少なくともミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、及びミクロ自動注入装置を具備した自動分画装置が提供される。また、本発明では、少なくとも、ミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、酵素反応装置、及びミクロ自動注入装置を備え、任意に、質量分析(MS)システムを具備した微量タンパク質及び/又はペプチドの高性能・簡易定量・同定装置が提供される。
【0030】
本発明は、例えば、微量リン酸化タンパク質や糖タンパク質を蛍光誘導体化試薬で蛍光標識後、ミクロカラム−HPLC−蛍光検出器により修飾リン酸化部分並びに糖結合部分を保持したままのタンパク質として高性能分離・検出し、ミクロフラクションコレクターにて分取する。これに酵素を添加し、標識化タンパク質を酵素水解し、次いで、この水解試料を直接質量分析に付す。得られたペプチドマップと蛍光標識化ペプチドマップ情報を基に、データベース検索ソフトを利用して、例えば、翻訳後修飾微量タンパク質の修飾部分を含む同定を行なう。本発明の手法は、蛍光検出法とミクロHPLCとを組み合わせた方法であるため、例えば、超微量タンパク質を同定できること、翻訳後修飾タンパク質をそのまま分解することなく抽出して解析できること、そのため、リン酸化部位や糖結合部位の情報を確実に得られるばかりでなく、当該タンパク質を確実に同定できること、という従来の解析手法にはない利点を有している。本発明の解析手法は、生命科学、病態化学分野で広く使われることが期待され、疾患診断、治療、人類の健康維持へ貢献することができる。
【発明の効果】
【0031】
本発明により、1)遺伝子を介して発現する発現タンパク質及び/又はペプチドを簡便な方法及び手段で、高感度に検出・分離・同定することができる、2)本発明の方法により、従来法では検出できなかった微量の発現タンパク質及び/又はペプチドを短時間で、感度良く検出・分離・同定することができる、3)また、上記検出・分離・同定方法に使用する微量の発現タンパク質及び/又はペプチドの微量検出・分離・同定システムを提供することができる、4)本発明は、プロテオームのプラットフォーム技術を提供するものとして有用である、という効果が奏される。
【発明を実施するための最良の形態】
【0032】
次に、実施例に基づいて本発明を具体的に説明するが、本発明は、以下の実施例によって何ら限定されるものではない。
【実施例1】
【0033】
ラット膵ランゲルハンス島(ラ島)中チオール含有タンパク質/ペプチドの分離・同定(1)ラ島中チオール含有タンパク質/ペプチドの蛍光誘導体化
ラ島に0.1Mホウ酸緩衝液(pH9.0)に溶解した6M塩酸グアニジン溶液50μlを加えて可溶化した。これに6M塩酸グアニジン溶液に溶解した17.5mMTCEP、17.5mMSBD−F、10mMEDTA及び50mMCHAPSをそれぞれ50μlずつ加え、混合した。この溶液を40℃にて3時間反応させることにより蛍光誘導体化を行った。
(2)イオン交換HPLCによる1次分離
上記の反応溶液をイオン交換カラムに付し、NaClのグラジエント(0、0.04、0.08、0.12及び0.3M)により蛍光タンパク質/ペプチドを溶出させ、5つのフラクションに分離した。なお、蛍光タンパク質/ペプチドの検出はSBD骨格の蛍光により行った。HPLC条件を以下に示す。
【0034】
(HPLC条件)
カラム:TSKgel DEAE−5PW 7.5×75mm(東ソー(株))
ガードカラム:C8−300−S 54.0×10mm(YMC(株))
移動相:段階溶離〔0−5分:C100%、5−15分:A100%、15−25分:A87%B13%、25−35分A73%B27%、35−45分:A60%B40%、45−55分:B100%〕
A:5mMトリス塩酸緩衝液(pH8.0)/アセトニトリル(50:50)
B:5mMトリス塩酸緩衝液(pH8.0)/アセトニトリル(50:50)
(0.3MNaCl含有)
C:5mMトリス塩酸緩衝液(pH8.0)
カラム温度:室温(約25℃))
流速:0.5ml/min
検出:Ex380nm、Em505nm
注入量:200μl
【0035】
(3)逆相HPLCによる2次分離
上記の各画分を濃縮し、アセトニトリルを蒸発させた後、逆相カラムに付し、アセトニトリルの勾配溶離によりタンパク質/ペプチドをカラムから溶出させた。なお、タンパク質/ペプチドの検出はSBD骨格の蛍光によりモニターした。HPLC条件を以下に示す。
【0036】
(HPLC条件)
カラム:カプセルパックC8 SG300 2.0×100mm((株)資生堂)
移動相:勾配溶離(0→60分:B40%→100%)
A:0.05%トリフルオロ酢酸
B:0.05%トリフルオロ酢酸/アセトニトリル(40:60)
カラム温度:室温(約25℃)
流速:0.2ml/min
検出:Ex380nm、Em505nm
注入量:50μl
【0037】
(4)酵素処理
採取したHPLCの各ピーク画分をそれぞれのチューブに0.5M炭酸水素アンモニウム溶液5μlを加えてトリフルオロ酢酸を中和後、濃縮してアセトニトリルを蒸発させた。残渣(約80μl)に20μg/mlトリプシン(プロメガ)及び10mM塩化カルシウムをそれぞれ10μlずつ添加した。これを37℃で2時間インキュベートし、HPLC−MS/MS測定用の試料とした。
【0038】
(5)MS/MS測定によるタンパク質/ペプチドの同定
上記の試料を逆相カラムHPLCに付し、エレクトロスプレー法によるMS/MS測定を行った。HPLC条件を以下に示す。なお、タンパク質/ペプチドの同定はデータベースにNCBI、サーチエンジンにMASCOTを使用した。
【0039】
(HPLC条件)
カラム:Cadenza TC−C18 2.0 ×100mm(Imtact(株))
移動相:勾配溶離(0→30分:B20%→100%)
A:0.1%ギ酸
B:0.1%ギ酸/アセトニトリル(50:50)
カラム温度:室温(約25℃)
流速:0.2ml/min
測定モード:positive
測定範囲:500−3000m/z
注入量:50μl
【0040】
上記の方法により、約130のタンパク質/ペプチドのピークが分離できた。
そのうち、約50のタンパク質/ペプチドが同定できた(表2、図6参照)。
【0041】
【表2】
【実施例2】
【0042】
SBD−Fで誘導体化したBSAを、図1に示されるプロセスにより、トリプシンで酵素水解し、得られたペプチド混合物を逆相液体クロマトグラフィー(RPLC)で分離し、蛍光検出器で検出した。次いで、各ペプチドをイオンスプレーイオントラップ質量分析計によるMS/MS分析に供した。原理的には、トリプシン分解により、BSAは、4個のアミノ酸残基以上の25のシステイン含有ペプチド及び35のシステイン非含有ペプチドが生成されるが、本実施例では、27以上の蛍光ペプチドが蛍光検出され、定量的に誘導体化が行われた(図4、A)。
【0043】
また、11のシステイン含有ペプチド及び17の非システイン含有ペプチドがマスクロマトグラフィーで検出された(図4、B)。 図5に、(M+2H)2+プレカーサー、m/z=873.4(図4で矢印で示した)から得たMS/MSスペクトルを示す。
全てのペプチドフラグメントのCIDスペクトルにより、確率的プロテイン同定法に基づくMASCOTによるデータベース照合を行い、予測通り、完全にBSAとしてタンパク質を同定した(スコア:39)。
【実施例3】
【0044】
デキサメタゾン(Dex)投与及び非投与のラット膵臓について試験した。
Dexは、肝臓のグルコース産生の増加とインシュリン耐性の誘導により主なヒト糖尿病であるタイプ2の糖尿病を誘発する。実際に、Dex処理の24時間後に、血中グルコースレベルは209.8mg/dLに達し、処理前の値の118.3mg/dLより著しく高い(p<0.05)。本実施例では、2日間Dexで処理又は未処理のラット膵臓からランゲルハンス島(60島)を採取し、SBD−Fで誘導体化した。本発明の方法を生物試料に適用するための重要な態様は、タンパク質混合物からHPLCで発現タンパク質を分離することである。本実施例では、蛍光タンパク質は、まず、SBD−Fにより生成した多くのマイナス電荷と酸性アミノ酸部分に基づいてイオン交換クロマトグラフィー(IEC)で分離された。
【0045】
IECは、塩化ナトリウムの段階溶離(0、0.04、0.08、0.12、及び0.3M NaCl)により行い、蛍光タンパク質混合物を5つの画分として得た。次いで、各画分は、更に、それらの疎水性に基づいて、逆相液体クロマトグラフィー(RPLC)により分離された。この実験でのピークの容量(n=L/(4σ)、但し、Lは分析のトータル時間及び4σはピーク幅、としてのHPLCの性能の理論値)は、各RPLCフラクションにつき40と計算され、IEC−RPLC法の5つのステップのピーク容量は、約200であった。本実施例では、各RPLCサイクルで約3−50ピーク、合計で129ピークであった(図6)。
【0046】
微量タンパク質を検出するために、IECの段階溶離ステップを増やしてピーク容量を増加させた。未処理及びDex処理ラットから得られたRPLCクロマトグラムの全ての蛍光ピークを比較した。その結果、5本の蛍光ピークが1.8以上増加し、3本の蛍光ピークがDex処理で約1.5倍減少したことが見出された(表2)。表2に、Dex投与2日後の発現タンパク質の変化を示した。これらのタンパク質(即ち、標的発現タンパク質)は、広い小孔を有するRPLC(30nm小孔径)で分離され、トリプシンで分解し、各ペプチド混合物とした。各ペプチド混合物は、通常の小孔を有するRPLC(10nm小孔径)に供し、MS/MS分析した。データベース照合により、Dex処理後2日で増加したピークは、各々、膵臓ポリペプチド、プロインシュリン2、78KDグルコース−調節タンパク質、ホスファチジルエタノールアミン結合タンパク質、及びチオレドキシンであり、減少したピークは、各々、プロテイン31、dnak−タイプ分子シャペロンhsp72−psl、及びインシュリンであった。
【実施例4】
【0047】
本実施例では、以下の化9の(1)及び(2)の反応式により、新規蛍光誘導体化試薬の合成を行った。
【0048】
【化9】
【0049】
(1)DAAB−Clの合成
4−chlorosulfonyl−7−chloro−2,1,3−benzoxadiazole(CBD−Cl)(126.53 mg)をCH3CNに溶解し、N,N−dimethylethylenediamineを滴下し、triethylamineを加えた。室温で約10分間攪拌後、反応液を減圧乾固した後、シリカゲルカラム(CH2Cl2)で精製し、4−(dimethylaminoethy laminosulfonyl)−7−chloro−2,1,3−benzoxadiazole(DAABD−Cl)(20.2mg,87.4%)を得た。
得られた化合物の確認データを以下に示す。
1H−NMR(CD3OD):7.94(1H,d,J=7.5),7.65(1H,d, J=7.5),3.06(2H,t,J=6.7),2.30(2H,t,J=6.7), 2.02(6H,s)。ESI−MS:m/z305(M+H)+
【0050】
(2)TAABD−Clの合成
4−chlorosulfonyl−7−chloro−2,1,3−benzoxadiazole(CBD−Cl)(126.53 mg)をCH3CNに溶解し、H2Oに溶かしたaminoethyl trimethylammonium chlorideを滴下し、triethylamineを加えた。室温で約20分間攪拌後、反応液を減圧乾固した後、0.1%トリフルオロ酢酸(TFA)に溶かし、ODSカラムを用いて分取し、画分には、SBD−Cl(化10)が不純物として入っていたため、陰イオン交換カラムを用いて分取し、減圧乾固して、7−chloro−2,1,3−benzoxadiazole−4−sulfoneaminoethyl trimethylammonium chloride(TAABD−Cl)(127.2mg,58.8 %)を得た。
得られた化合物の確認データを以下に示す。
1H−NMR(CD3OD):8.01(1H,d,J=7.3),7.69(1H,d, J=7.3),3.46−3.48(4H,m),3.12(9H,s)。ESI−MS :m/z319(M)+
【0051】
【化10】
【実施例5】
【0052】
本実施例では、新規蛍光誘導体化試薬の反応性について検討した。
DAABD−Cl、TAABDD−ClのSBD−Fとの比較
10μM還元型グルタチオン、システイン、ホモシステイン混合液100μLとDAABD−Cl又はTAABD−Cl 100μLを混合し、pH9、40℃、10〜120分間反応させた。尚、各試薬は5mM EDTAを含む0.10M ホウ酸緩衝液(pH 9)に溶解した。0.1%ぎ酸で反応停止後、生成物をHPLCを用いて測定した。
【0053】
図7に、蛍光誘導体化反応時間と蛍光強度との関係(左図:DAABD−Cl、右図:TAABD−Cl)を示す。
SBD−F(化11)の場合、40℃で誘導体化を行うと120分間の反応時間が必要であったが、DAABD−Clは10〜20分、TAABD−Clは20〜30分で反応が終了することがわかった。
したがって、反応時間はDAABD−Clの場合は20分、TAABD−Clの場合は30分が好適である。
【0054】
【化11】
【0055】
(2)新規蛍光誘導体化試薬のMSでの感度
上記(1)で作成したサンプルをLC−MSにより検出し、蛍光誘導体化試薬でラベル化していないもの及びSBD−Fで誘導体化したものと、相対強度を比較した。
【0056】
蛍光誘導体化試薬でラベル化していないcysteine、homocysteine、GSHの高さをそれぞれ1としたときの相対強度は表3の通りであった。これより、DAABD−ClがMSでの感度が最も高いことがわかった。また、移動相が酸性であるので、DAABD化誘導体はプラスに荷電され水溶性であると考えられる。
【0057】
【表3】
【実施例6】
【0058】
(1)TAABD−Clのペプチドへの応用
10μMの以下に示した4種類のペプチド標品、17.5mM TAABD−Cl、10 mM EDTA、50mM CHAPS(界面活性剤)、2.5mM TCEP(還元剤)それぞれ50μLを混合し、pH9.0、40℃で、30,60,90,120分間反応させた。尚、各試薬は6.0M 塩酸グアニジン(タンパク変性剤)を含む0.10 M ホウ酸緩衝液(pH9.0)に溶解した。生成したTAABD化ペプチドをHPLCを用いて測定した。
1. vasopressin
2. oxytocin
3. somatostatin
4. amylin(rat)
図8に TAABD−Clとの反応時間と蛍光強度との関係を示す。
図8より、反応時間が60分までは生成量が増加した。反応停止後は氷冷下で保存し、−20℃にて保存すると、48時間はほとんど分解しなかった。
【0059】
(2)DAABD化ペプチド・タンパクの検出限界
表4に示した10種類のペプチド・タンパク質標品10μM混合液、2.5mM TCEP、17.5mM DAABD−Cl、10mM EDTA、50mM CHAPSそれぞれ50μLを混合し、pH9.0、40℃で、30分間反応させた。尚、各試薬は6.0 M 塩酸グアニジンを含む0.10M ホウ酸緩衝液(pH9.0)に溶解した。生成したDAABD化ペプチド・タンパク質はHPLCを用いて測定し、蛍光検出の検出限界をSBD−Fと比較した。
【0060】
【表4】
【0061】
(3)DAABD化ペプチド・タンパク質の同定
上記(2)で誘導体化した物質のうち、vasopressin、oxytocin、somatostatin、calcitonin、amylinはLC−MSにより同定できた。それらの分子量を以下に示す。
m/z 541.8(M+3H)3+[DAABD−vasopressin]
516.0(M+3H)3+[DAABD−oxytocin]
726.6(M+3H)3+[DAABD−somatostatin]
989.9(M+4H)4+[DAABD−calcitonin]
892.8(M+5H)5+[DAABD−amylin]
【0062】
これら全ての分子量は、それぞれのペプチドの2つのシステイン残基にDAABDが付加したとしたときの分子量であり、多価イオンピークの検出結果よりDAABD−Clによる誘導体化において、これらのペプチドのシステイン残基間のS−S結合が還元され、二つのチオール基両方に試薬が反応したことがわかった。
また、タンパク質の場合は酵素によってペプチドに分解する必要があるため、酵素トリプシンで消化し、LC−MS/MS検出及びMASCOTによるデータベース検索を行って同定を試みた結果、システインを含まないペプチドのアミノ酸配列も併せて決定し、タンパク質を同定することができた。
【実施例7】
【0063】
(SBSeD−Fの合成)
2−fluoroacetanilideを硝酸で処理して、1−acetylamino−2−nitro−6−fluorobenzeneとし、これを脱アセチル化して、2−fluoro−6−nitroanillineとし、次いで、パラジウム担持炭素触媒を用いて水素化して、3−fluoro−o−phenylenediamineを得た。
Selenium dioxideエタノール加熱溶液を、3−fluoro−o−phenylenediamine(60mg,0.48mmol)のエタノール加熱溶液に加え、混合物を30分加熱した。これを、シリカゲルカラムによるクロマトグラフィーに供し、溶離液のジクロロメタンで溶出し、4−fluoro−2,1,3−benzoselenadiazoleを白色粉末(88mg)として得た。得られた化合物の確認データを以下に示す。
mp.129℃、NMR(methanol−d4):δH7.55(1H,d,J=9.2)、7.41(1H,m)、7.06(1H,m)、ESI−MS:m/z202.8[(M+H)]。
【0064】
このようにして得た4−fluoro−2,1,3−benzoselenadiazoleをfuming sulfuric acid(60%)に溶かし、130℃で3時間還流した。この溶液を冷却し、冷水(30ml)に注ぎ、28%ammonium hydroxideで中和した。この中性溶液にエタノール100mlを加え、濾過物を減圧乾固させた。残渣を水(1.0ml)に溶解し、更に、以下のHPLCで精製した。即ち、残渣の100μlをHPLC分離に供した。HPLCカラム:TSK−gel ODS−120T、150×4.6mm i.d.、東ソー、溶離液:蒸留水、流速0.5ml/min、検出:280nm。SBSeD−Fに相当するフラクションを集め、減圧して白色粉末(50mg)を得た。得られた化合物の確認データを以下に示す。
m.P.>300℃、NMR(methanol−d4):δH7.97(1H,d d,J=7.6,J=5.4)、7.11(1H,dd,J=7.6,J=10.1)、ESI−MS:m/z280.8[(M−H)]。
【実施例8】
【0065】
(SBThD−Fの合成)
N−thionylaniline(0.49g,3.5mmol)を3−fluoro−o−phenylenediamine(200mg,1.6mmol)トルエン(2ml)溶液に加えた。反応混合物を100−120℃で4時間加熱し、溶媒を濾別した後、残渣をジクロロメタンに溶かし、溶液を10%HCl溶液及び水で各々洗浄した。有機相を乾燥し、減圧乾固させた。これをシリカゲルによるクロマトグラフィーに供し、溶離液のクロロホルムで溶出し、4−fluoro−2,1,3−benzothiadiazoleを淡黄色油として得た。得られた化合物の確認データを以下に示す。
NMR(methanol−d4):δH7.69(1H,d,J=8.9)、7.50(1H,m)、7.20(1H,m)、ESI−MS:m/z154.9[(M+H)]。
【0066】
このようにして得た4−fluoro−2,1,3−benzothiadiazole(30ml)をfuming sulfuric acid(60%)に溶かし、130℃で3時間還流した。次いで、この溶液を冷却し、ゆっくり冷水(30ml)に注ぎ、28%ammonium hydroxideで中和した。中性溶液にエタノール100mlを加え、得られた濾過物を減圧乾固した。残渣を水(1.0ml)に溶かし、更に、以下の条件でHPLCで精製した。SBThD−Fに相当するフラクションを集め、減圧し、白色粉末(25mg)を得た。得られた化合物の確認データを以下に示す。
decomp.265℃、NMR(methanol−d4):δH8.06(1H,dd,J=7.9,J=4.9)、7.11(1H,dd,J=7.9,J=9.8)、ESI−MS:m/z232.8[(M−H)]。
上記方法により合成したSBSeD−F及びSBThD−Fを下記の化12に示す。
【0067】
【化12】
【実施例9】
【0068】
(1)システイン誘導体の蛍光スペクトル
1mMEDTAを含む0.1Mホウ酸緩衝液(pH9.0)による上記SBSeD−F、SBThD−F又はSBD−Fの各蛍光試薬溶液(4mM)の500μl部分を、0.1Mホウ酸緩衝液(pH9.0)によるシステイン(0.4mM)溶液の同量と混合した。この混合物を60℃で8時間放置した。反応の後、反応混合物をHPLC分離し、各システイン誘導体に相当するフラクションを集め、それらの蛍光スペクトルを測定した。
【0069】
SBSeD−FとSBThD−Fのシステインに対する反応性
4mMの各試薬、SBSeD−F、SBThD−F又はSBD−F及び1mM EDTAを含む0.1Mホウ酸緩衝液(pH9.0又はpH10)の500μl部分を、0.1Mホウ酸緩衝液(pH9.0又はpH10)によるシステイン溶液(0.4mM)の同量と混合した。反応混合物をHPLC分離し、60℃での反応をモニターした。
【0070】
(2)結果
SBD−Fのような水溶性試薬は、そのsulfonic acid残渣により水溶体中での誘導体の可溶性を増加させ、結果として、誘導体の吸収又は沈殿が減少する。したがって、インシュリンのような比較的疎水性ペプチドのSBD−Fによる誘導体は、逆相カラムで溶出され、高感度に検出されたが、本実施例では、更に、benzoselenadiazole又はbenzothiadiazole骨格をもつ蛍光試薬としてSBSeD−F及びSBThD−Fを合成し、それらのシステインに対する反応性及びその誘導体の蛍光特性を調べた。
【0071】
最大励起(λex)及び発光波長(λex)、及び誘導体の保持時間を表5に示す。各システイン誘導体の質量数([M+H])は、SBSeD−F(m/z381.9)、SBThD−F(m/z334.0)及びSBD−F(m/z318.0)について、理論値(各々382.0、334.0及び318.0)と一致した。誘導体の最大励起波長は、SBSeD−F(340nm)及びSBThD−F(315nm)はSBD−F(365nm)より短く、誘導体の最大発光波長は、SBSeD−F(542nm)は、SBSeD−F(517nm)及びSBD−F(514nm)よりも長かった。SBSeD−F自体は蛍光が少ないが、SBThD−Fは多少の蛍光を与えた(λex;350nm,λem:424nm)。SBSeD−F、SBThD−F及びSBD−Fのシステイン誘導体の逆相カラム(C18)に対するpH2.0の移動相による保持時間(tR)は、各々4.5、5.3及び4.8分であった。これから、SBSeD−Fは、これらの中で最も高い親水性の蛍光試薬であった。
【0072】
SBD−Fの場合、至適反応条件は60℃でpH9.5で1時間であるが、SBSeD−F及びSBThD−Fの反応性は、SBD−Fと比べて低く、蛍光強度は8−24時間で徐々に増加し(図9、10)、24時間後でも、反応は最大に達しなかった(図9)。pH10.0及び60℃では、SBSeD−F又はSBThD−Fとシステインの量的反応時間は、8時間以上であり、SBD−Fでは、1時間以内で完全に反応した(図10)。
このように、水溶性の蛍光試薬SBSeD−F及びSBThD−Fは、SBD−Fと比べて蛍光特性及び疎水性の点で異なっており、プロテオーム解析のための新規蛍光誘導体化試薬として有用である。
【実施例10】
【0073】
DAABD−Clによる線虫(C.elegans)タンパク質の誘導体化及び同定
(1)方法
線虫(Bristol N2株)を、大腸菌(E.coli)のOP50株を栄養源として20℃で、NGM寒天上に培養し、M9バッファーにより浮遊させてバクテリアから分離した。上記線虫をM9バッファーで2回洗った後、−80℃で保管して用いた。この線虫を等量の10mM CHAPSに懸濁し、超音波で溶解した。可溶性のフラクションを4℃で10,000rpm、5分の遠心分離により集めた。上澄を可溶性フラクションとして−20℃で保管した。このフラクションのタンパク質濃度をBSAを標準に用いるBradford methodで決定した。上澄の約20μL(100μgタンパク質)を同容量の2.5mM TCEP,17.5mM DAABD−Cl、10mM Na2 EDTA及び50mM CHAPSを6.0Mグアニジンを含む100mMホウ酸塩緩衝液(pH9.0)中で混合した。反応混合物を40℃で30分インキュベートした後、反応を200μLの0.1%ギ酸で停止し、次いで、反応混合物(10μgタンパク質)の30μLをHPLCシステムに注入した。
【0074】
RPカラムがPROTEIN(30nm孔径、250×4.6mm i.d.)(Imtakt)、移動相が溶離液(A)0.1%トリフルオロ酢酸及び溶離液(B)水/CH3CN/トリフルオロ酢酸(70/30/0.1)、グラジエントシステムが流速0.25mL/minで100分かけて30から70%Bの条件でHPLCを行った。蛍光検出は508nm、励起波長は387nmで行った。同定のために、蛍光タンパク質誘導体のいくつかのピークフラクションを分離し、減圧下に10μLに濃縮した。各フラクションは、2μg/mLトリプシン及び1.0mM塩化カルシウムを含む90μLの5.0mM重炭酸アンモニウム溶液(pH7.8)で希釈し、37℃で2時間インキュベートした。各タンパク質加水分解ペプチド混合物を直接エレクトロスプレーイオントラップ質量分析装置を用いたLC−MS/MSに供した。クロマトグラフィーは、HP1090シリーズIIシステム及びCadenza TC−18 column(12nmポーラスシリカ、100×2.0mm i.d.)のカラムを用いて実施した。移動相は溶離液(A)1.0mMギ酸アンモニウム及び溶離液(B)1.0mMギ酸アンモニウム/CH3CN(50/50)とした。グラジエント溶出は、流速0.2mL/minで60分かけて0から100%で行った。タンパク質の同定は、システインのチオール残基に結合したDAABDを記憶するMASCOT(Matrix Science Ltd.,U.K.)データベースサーチアルゴリズムを用いてNCBInrデータベースより行った。
【0075】
(2)結果
図11は、DAABD−Clで誘導体化した、線虫の可溶性フラクションから得られたタンパク質(約10μg)のクロマトグラムを示す。本実施例では、タンパク質の分離、トリプシンによる加水分解及び任意に選択されたピークフラクションのLC−MS/MS同定により、10種類のタンパク質が同定された。
図中、1はリボゾームタンパク質S3a(MW=28942)、2はカルレティキュリン(calreticulin)前駆体(MW=45588)、3はリボゾームタンパク質L1(MW=38635)、4は伸長因子(elongation factor)1−アルファ(MW=50636)、5はリンゴ酸デヒドロゲナーゼ(MW=35098)、6は40Sリボゾームタンパク質(MW=22044)、7はビテロゲニン(vitellogenin)(MW=193098)、8はアルギニンキナーゼ(MW=41969)、9はHSP−1熱ショック(heat shock)70kdタンパク質A(MW=69680)及び10はリボゾームタンパク質L7Ae(MW=13992)を示す。本実施例では、任意に選択された10種類のタンパク質が同定されたが、本発明では、同様にして、他のタンパク質の同定をすることが可能である。
【実施例11】
【0076】
7−クロロ−N−(2−ジメチルアミノプロピル)−2,1,3−ベンゾキサジアゾール−4−スルフォンアミド
4−クロロ−7−クロロスルフォニル−2,1,3−ベンゾキサジアゾール(0.25g、0.99mmol)をアセトニトリル(8ml)に溶解し、室温で撹拌した。次いで、これにN,N−ジエチルエチレンジアミン(0.21ml、1.49mmol)及びトリエチルアミン(0.21ml、1.49ml)を加えた。反応混合物を30分間撹拌し、次いで、減圧で濃縮した。残渣をクロロホルムに溶解し、飽和アンモニウムクロライド水溶液及び塩水で洗浄し、有機相をNa2SO4で乾燥し、減圧で濃縮した。残渣をシリカゲルクロマトグラフィー(10%メタノール/クロロホルム)で精製し、生成物を得た。得られた固体をメチレンクロライド及びジイソプロピルエステルから再結晶し、明るい黄色の小板状の生成物(0.22g、0.66mmol、67%)を得た。1H−NMR(CDCl3,500MHz)δ7.97(d,J=7.4Hz,1H),7.53(d,J=7.4Hz,1H),3.05(t,J=5.7Hz,2H),2.48(t,J=5.7Hz,2H),2.33(q,J=7.5Hz,4H),0.87(t,J=7.5Hz,6H);13C−NMR(CDCl3,125MHz)δ148.77,145.00,133.40,129.15,127.88,127.47,51.14,46.09,40.45,11.36;IR(KBr,cm−1)3446,3209,3101,2976,2817,1525,1347,1164.
【実施例12】
【0077】
4−クロロ−7−クロロスルフォニル−2,1,3−ベンゾキサジアゾール(0.21g、0.88mmol)をアセトニトリル(8ml)に溶解し、0℃で撹拌した。次いで、これにN,N−ジエチル−1,3−プロパンジアミン(0.20ml、1.25mmol)及びトリエチルアミン(0.17ml、1.25ml)を加えた。反応混合物を30分間撹拌し、次いで、減圧で濃縮した。残渣をクロロホルムに溶解し、飽和アンモニウムクロライド水溶液及び塩水で洗浄し、有機相をNa2SO4で乾燥し、減圧で濃縮した。残渣をシルカゲルクロマトグラフィー(10%メタノール/クロロホルム)で精製し、明るい黄色固体の生成物(0.10g、0.29mmol、35%)を得た。1H−NMR(CD3OD,500MHz)δ8.06(d,J=7.5Hz,1H),7.77(d,J=7.5Hz,1H),3.19(m,8H),1.93(m,2H),1.32(t,J=7.5Hz,6H);13C−NMR(CD3OD,125MHz)δ150.39,146.66,135.58,131.33,129.33,128.10,50.41,41.21,25.69,9.26;IR(KBr,cm−1)3501,3428,3209,2973,2733,2675,1524,1338,1160.
【実施例13】
【0078】
N,N−ジメチルエチレンジアミン−d6の合成
ジメチルアンモニウムクロライド−d6(2.46g)及びブロモアセトニトリル(3.37g)をジエチルエーテル(20ml)に溶解した。10℃でこれに50%NaOH(4.5g)を添加した後、混合物を同じ温度で2時間撹拌した。エーテル層を分離し、水層をエーテル(10ml×3)で抽出した。合体したエーテル層をMgSO4で乾燥し、次いで、減圧で濃縮してN,N−ジメチルアミノアセトニトリル−d6溶液(10g)を得て、次いで、それを10℃でテトラハイドロフラン(40ml)中のLiAlH4(1.28g)及びスルフォン酸(1.69g)の混合物に加えた。反応混合物を室温で13時間よく撹拌、混合した。これにエーテル(30ml)を加えた後、混合物をNaOH(水6mlに4g溶解)で処理した。エーテル層を分離し、水層をエーテル(10ml×2)で抽出した。合体したエーテル層をMgSO4で乾燥し、次いで、(5gになるまで)減圧濃縮した。残渣を希釈し、フラクション(70−80℃)を合わせて、1.79g(収率52.5%)のN,N−ジメチルエチレンジアミン−d6(77.4% THF溶液)を得た。1H−NMR(CDCl3):δ1.82−1.88(1.54H,m for THF,4H),2.33(2H,t,J=6Hz),2.77(2H,t,J=6.4Hz),3.72−3.76(1.48H,m for THF,4H)
【実施例14】
【0079】
7−クロロ−4−(ジメチルアミノエチルアミノスルフォニル)−2,1,3−ベンゾキサジアゾール−d6(DAABD−Cl−d6)の合成
4−クロロ−7−クロロスルフォニル−2,1,3−ベンゾキサジアゾール(3.28g)をCH3CN(60ml)に溶解した。この溶液に、N,N−ジメチルエチレンジアミン−d6(1.0g)及びトリエチルアミン(1.92ml)を各々加えて、氷で冷却して、同じ温度に1時間保持し、室温で1.5時間撹拌、混合した。次いで、反応混合を減圧濃縮し、残渣をAcOEtに溶解した。エチルアセテート溶液を飽和NaHCO3、蒸留水、飽和NaCl溶液で各々洗浄し、MgSO4で乾燥した。ろ過した溶液を減圧濃縮し、残渣をCH2Cl2:MeOH(50:1)を溶出液としてシリカゲルカラムで精製した。溶出した溶液を減圧乾燥し、残渣をEtOH−AcOEtで再結晶して1.36g(収率41.3%)のDAABD−Cl−d6:mp、110℃を得た。1H−NMR(CDCl3):δ7.99(1H,d,J=7.3Hz),7.54(1H,d,J=7.3Hz),3.13(2H,m),2.33(2H,m),ESI−MS:m/z311(M+H)+,IR(KBr)cm−1;1342and1166.
【実施例15】
【0080】
7−フルオロ−N−[2−(ジメチルアミノ)エチル]−2,1,3−ベンゾセレナジアゾール−4−スルフォンアミド(DAABSeD−F)の合成
7−フルオロ−2,1,3−ベンゾセレナジアゾール−4−スルフォニルクロライド(75mg)を3mlのアセトニトリルに溶解した。これに、N,N−ジメチルエチレンジアミン−(22mg)及びトリエチルアミン(35μl)を加えた後、混合物を氷の上で30分間撹拌した。反応混合を減圧で関そうし、残渣をCH2Cl2に溶解し,CH2Cl2−CH3OH(93:7)を用いてシリカゲルクロマトグラフィーに供し、7−フルオロ−N−[2−(ジメチルアミノ)エチル]−2,1,3−ベンゾセレナジアゾール−4−スルフォンアミド(49mg、56%)を赤い粉末として生成した。mp:117−120℃。1H−NMRδH8.09(1H,d,J=7.5Hz),7.24(1H,d,J=7.5Hz),2.93(2H,t,J=6.7Hz),2.27(2H,t,J=6.7Hz),1.98(6H,s)in CD3OD;ESI−MSm/z353[(M+H)+]
【実施例16】
【0081】
(1)DAABSeD−Fによる低分子量チオールの誘導体化反応及び誘導体の蛍光特性
1mM(システイン、ホモシステイン、アラニン、セリン、又はグルタチオン)溶液の20μLを、同容量の2.5mMTCEP、17.5mMDAANSeD−F、10mMNa2EDTA及び50mMCHAPSと混合した。各試薬は、6.0Mグアニジンを含む100mMホウ酸バッファーに溶解した。反応混合物を40℃で10−120分間加熱し、反応を0.1%トリフルオロ酢酸の200μLで停止した。混合溶液を300μLのメタノールで希釈し、蛍光スペクトルを蛍光スペクトロメーターで測定した。その結果、最大励起波長は約420nm、最大蛍光波長は約540nmであった。
【0082】
(2)低分子量チオールのDAABSeD−誘導体の分離及び同定
0.1%トリフルオロ酢酸で反応を停止した上記反応混合物の10μLをHPLCシステムに供した。HPLCシステムは、ポンプ(L−7100、日立社製)、分離カラムTSKgel120−TQA(内径250×4.6mm)(東ソー社製)及び蛍光検出器(FL−2025、ジャスコ社製)で構成した。蛍光検出は540nmで、励起波長は420nmで行った。移動相は、150mMリン酸バッファー/CH3CN(94/6)であり、流速は0.75ml/分とした。
同定のために、反応混合物をHP1090シリーズIIシステム(Hewlett−Packard(GmbH)のカラムに直接注入して、エレクトロスプレー法によるMS及びMS/MSスペクトルを得た。クロマトグラフィーは、Cadenza TC−18カラム(シリカの12nm孔、内径100×2.0mm)(Imtact(株))で行った。移動相は、0.1%ギ酸の溶出液(A)及び水/CH3CN/ギ酸(50/50/0.1)の溶出液(B)で構成した。0から100%Bで流速0.2ml/分で15分間グラジエント溶出を行った。このとき得られたクロマトグラムは、システイン、ホモシステイン、グルタチオンの3つのピークを与えた。
【0083】
(3)DAABSeD−Fによるペプチド及びタンパク質の誘導体化反応
100μMのペプチド及びタンパク質(インシュリン、トリプシン阻害剤、α−酸グリコプロテイン、カルボニックアンヒドラーゼ、α−ラクトアルブミン)溶液又は10μM BSA溶液の20μLを同容量の2.5mM TCEP、17.5mM DAABD−Cl(比較として)、10mM Na2EDTA及び50mM CHAPSと混合した。各反応混合物を6.0Mグアニジンを含む100mMホウ酸バッファー(pH9.0)に溶解した。各反応混合物を40℃で60分間(DAABSeD−F)又は20分間(DAABD−Cl)加熱し、反応を0.1%トリフルオロ酢酸の200μLで停止した。混合物をメタノール300μLで希釈し、蛍光スペクトルを蛍光スペクトロメーターで測定した。その結果、最大励起波長は約425nm、最大蛍光波長は約535nmであった。
【0084】
(4)ペプチド及びタンパク質のDAABSeD誘導体の分離
メタノールを除いた上記反応混合物の55μLをHPLCカラムに供した。HPLCシステムは、ポンプ(L−7100、日立社製)、タンパク質用のRPカラム(30nm孔サイズ、内径250×4.6mm)(Imtact社製)及び蛍光検出器(FL−2025、ジャスコ社製)で構成した。蛍光検出は、550nm(励起波長450nm)(DAABSeD−F)又は490nm(励起波長370nm)(DAABD−Cl)で行った。溶出液(A)は、水/CH3CN/トリフルオロ酢酸(90/10/0.1)からなり、溶出液(B)は、水/CH3CN/トリフルオロ酢酸(30/70/0.1)で構成した。0から100%Bで流速0.5mL/分で200分間のグラジエント溶出を行った。
【0085】
(5)異なる波長でのタンパク質誘導体のHPLC分離・定量及びタンパク質の同定
α−ラクトアルブミンを含み、10mMEDTA、50mMCHAPS、2.5mMTCEP、6.0Mグアニジンを含む混合溶液を2つの部分に分け、試料Aには1μMのα−ラクトアルブミン及び試料Bには2μMのα−ラクトアルブミンを含むように調整した。試料AはDAABD−Clと40℃で20分間反応させ、試料BはDAABSeD−Fと40℃で30分間反応させ、両者の反応溶液を再度混合した。混合物を2つの蛍光検出器に直列に連結したHPLCに供した。一つは、370nmで励起して蛍光波長490nmでDAABD誘導体をモニターし、もう一つは、450nmで励起して蛍光波長550nmでDAABSeD誘導体をモニターした。HPLCシステムは上記(4)と同じである。
【0086】
タンパク質の同定のために、試料A中のα−ラクトアルブミンのDAABD誘導体及び試料B中のα−ラクトアルブミンのDAABSeD誘導体に相当するピークフラクションを合体し、pH7.8に調整し、トリプシンで加水分解した。次いで、得られたペプチド混合物を窒素ガスで濃縮し、タンパク質同定アルゴリズムを備えた、シスラインのチオール基に結合したDAABD(MW、3900)又はDAABSeD(MW、4539)を記憶しているMASCOTを有するHPLC−MS/MSに供した。クロマトグラフィーは、Agilent1100シリーズシステム(Agilent Technologies社製)及び分離カラム、Cadenza TC−18カラム(シリカの12nm孔、内径100×2.0mm)(Imtact社製)、0.1%ギ酸の溶出液(A)及び水/CH3CN/キ酸(50/50/0.1)の溶出液(B)で構成した。0から100%Bで流速0.2mL/分で60分間のグラジエント溶出を行った。その結果、実施例10と同様にα−ラクトアルブミン(MW16228)が同定できた。
【実施例17】
【0087】
(1)DAABSeD−Fの合成
前述のように、ベンゾキサジアゾール試薬の、7−クロロ−N−[2−(ジメチルアミノ)エチル]−2,1,3−ベンゾキサジアゾール−4−スルフォンアミド(DAABD−Cl)は、感度のよい、選択的なチオール特異的試薬であり、プロテオミクス研究に有用である。DAABD−Clは、ペプチド及びタンパク質のチオールと反応して誘導体化し、390nmの励起で502nmで蛍光を発する。本実施例では、図12の合成経路により、7−フルオロ−N−[2−(ジメチルアミノ)エチル]−2,1,3−ベンゾセレナジアゾール−4−スルフォンアミド(DAABSeD−F)をDDABD−Clのカウンターパートとして合成した。
【0088】
本発明者は、7−フルオロ−2,1,3−ベンゾセレナジアゾール−4−スルフォネート(SBSeD−F)及び7−フルオロ−2,1,3−ベンゾチアジアゾール−4−スルフォネート(SBThD−F)をチオール特異的蛍光試薬として合成した。これらのシステインとの誘導体の蛍光波長は、各340nm及び315nmの励起波長で、それぞれ542nm及び517nmであった。対応するベンゾキサジアゾール試薬の、7−フルオロ−2,1,3−ベンゾキサジアゾール−4−スルフォネート(SBD−F)は、365nmの励起で514nmで蛍光を発する誘導体を与える。SBSeD−FとSBD−Fでの誘導体の最大波長の大きな相違にヒントを得て、本発明者は、ベンゾセレナジアゾール試薬、DAABSeD−FをDAABD−Clのカウンターパートとして合成した(図12)。
【0089】
(2)チオールに対するDAABSeD−Fの誘導体化反応の反応条件及び反応性
DAABSeD−Fによる低分子量チオール(システイン、ホモシステイン、グルタチオン、アラニン、セリン及びトリプシン)の誘導体化反応を40℃(pH9.0)で行った。ペプチド及びタンパク質(インシュリン、α1−酸グリコタンパク質、トリプシン阻害剤、カルボニックアンヒドラーゼ、α−ラクトアルブミン、BSA等)の場合、反応を促進するために、CHAPS及びグアニジンの存在下で行った。予測したように、DAABSeD−Fは、アラリン(アミノ及びカルボキシル基を含む)セリン(アミノ、カルボキシル及び水酸基を含む)及びチロシン(アミノ、カルボキシル、フェノール性水酸基を含む)のようなチオールを含まない化合物との反応では蛍光を示さなかった。これに対して、チオール化合物の場合には、蛍光を発し、その強度は30分で最大に達したが、これは、DAABSeD−Fはペプチド及びタンパク質と同様に低分子量のチオールと特異的に反応し、蛍光を生じ、反応速度は、DAABD−Clと同様であることを示している。
【0090】
(3)DAABD−Fと比較したDAABSeD−F誘導体の蛍光特性
DAABSeD誘導体は、最大励起及び蛍光波長が、それぞれ423−429nm及び524−542nmの範囲にあることを示した(表5)。
【0091】
【表5】
【0092】
(4)LC−MSによるチオールのDAABSeD誘導体の同定及び検出
DAABSeD−Fで誘導体化したチオール混合物をエレクトロスプレーイオントラップ質量分析計を備えたHPLCに供した。各チオールについて単一ピークが得られた。マススペクトルから、各低分子量チオールのDAABSeD誘導体が、システイン(MW=121)、ホモシステイン(MW=135)及びグルタチオン誘導体(MW=307)について、ベースイオンピークがm/z=454(M+H)+、468(M+H)+及び640(M+H)+として検出された。本実験では、用いたエレクトロスプレー法による分子イオン検出の限界(MWが3000以下)のため、タンパク質のDAASeD誘導体の同定は困難であった。
【0093】
(5)DAABSeDで誘導体化したペプチド及びタンパク質の検出限界
DAABSeD−Fで誘導体化したタンパク質から得られるクロマトグラムは、各誘導体の単一ピークを示し、重なるピークは観察されなかった。試薬自体は蛍光を発しないので、DAABSeD−Fピークは現れなかった。上述の誘導体のピーク高さは、注入量に比例した。バソプレシン、α−ラクトアルブミン及びBSAの検出限界は、それぞれ、7.5、7.2、及び7.2fmolであった。これらの検出限界はDAABD誘導体のものと同様であった。DAABD誘導体に比較して、DAABSeD誘導体は早く溶出したので、DAABSeD誘導体は、DAABD誘導体よりも、HPLC固定相に対する低い親和性を有していた。同様の傾向は、SBD誘導体と比べて、SBSeD誘導体についても観察された。
【0094】
(6)DAABSeD及びDAABD誘導体の同時検出
DAABD及びDAABSeD誘導体を併用した、二つの誘導体の同時蛍光検出は、同一起源の異なる歴史を有する二つのタンパク質試料の比較を簡便にする。例えば、一方が病態患者試料で一方が健常者からの試料、あるいは、二つの細胞又は組織試料で、一方がある薬剤で処理され、他方が未処理のものが例示される。本実験では、試料A中のα−ラクトアルブミンのDAABD誘導体の反応溶液と、試料B中のα−ラクトアルブミンのDAASeBD誘導体の反応溶液を合体し、二つの蛍光検出器に連結したHPLCに供した。それぞれの検出波長は、DAABD誘導体では370nmの励起で490nmで行い、DAASeBD誘導体では、450nmの励起で550nmで行なった。二つの誘導体は、クロマトグラムで各単一ピークを与えた。α−ラクトアルブミンのDAABD誘導体及びα−ラクトアルブミンのDAASeBD誘導体の保持時間は、53.3分、及び50.0分であった。
【0095】
試料A中のα−ラクトアルブミンのDAABD誘導体は、観察されたが、試料B中のα−ラクトアルブミンのDAABSeD誘導体は観察されなかった。試料B中のα−ラクトアルブミンのDAABSeD誘導体は観察され、試料A中のα−ラクトアルブミンのDAABD誘導体は観察されなかった。したがって、この方法で、試料A中に存在するα−ラクトアルブミンと試料B中に存在するα−ラクトアルブミンを区別することができ、同じ試料中のそれらの量を計測し、比較することができる。
【0096】
分離された相当するピークフラクションを合体した混合物をトリプシンのような酵素で処理することで、タンパク質の定量及び同定が可能であった。例えば、試料A中のDAABD誘導体及び試料B中のα−ラクトアルブミンのDAABSeDに相当する各ピークを合体し、トリプシンで分解し、得られたペプチド混合物を、2つの蛍光検出器に直結したHPLCにて分離・定量するとそのタンパク質(α−ラクトアルブミン)の定量が可能であり、更に得られたペプチド混合物をシステインのチオール残基に結合したDAABD又はDAABSeDを記憶している、タンパク質の同定アルゴリズム、MASCOTを備えた、HPLC−MS/MSに供した。その結果、実施例10と同様にα−ラクトアルブミン(MW16228)が同定された。
【0097】
本実験では、異なった試料A及びB中のタンパク質α−ラクトアルブミンを取り上げ、比較した。これに対して、一つの試料中に多くの化合物が存在する実際のバイオ試料の、誘導化したタンパク質溶液を合体した混合物は、複雑なHPLCクロマトグラムのパターンを与えるので、クロマトグラムでタンパク質ピークを区別することは非常に困難になる。しかし、標的タンパク質の標準品があり、異なったクロマトフラフィー条件で二次元クロマトグラフィーを行なえば、二つのクロマトグラフィー上のタンパク質の二つの誘導体の保持時間を知ることができるので、クロマトグラム上でDAABD−Cl及びDAABSeD−Fの各誘導体のピーク分画を分離した後、これらを合体したピークフラクションを第2のクロマトグラフィーで再クロマトすることにより、精製されたタンパク質の各誘導体が定量できる。更に、ピーク分画を上記の酵素処理、蛍光検出HPLC又はLC−質量分析計に付し、異なった試料中のタンパク質の定量又は同定ができる。
【産業上の利用可能性】
【0098】
以上詳述したように、本発明は、微量の発現タンパク質及び/又はペプチドの検出・分離・同定方法及びそのシステムに係るものであり、本発明により、遺伝子を介して発現する発現タンパク質及び/又はペプチドを簡便な方法及び手段で、高感度に検出・分離・同定することができる。本発明の方法により、従来法では検出できなかった微量の発現タンパク質及び/又はペプチドを短時間で、感度良く検出・分離・同定することができる。また、上記検出・分離・同定方法に使用する微量の発現タンパク質及び/又はペプチドの微量検出・分離・同定システムを提供することができる。本発明は、プロテオームのプラットフォーム技術を提供するものとして有用である。
【図面の簡単な説明】
【0099】
【図1】本発明の方法の操作工程の一例を示す。
【図2】界面活性剤の種類と蛍光誘導体の生成の度合いとの関係を示す。
【図3】本発明の方法により試験した蛍光誘導体タンパク質/ペプチドのそれぞれの蛍光ピークを示す。
【図4】酵素水解物の蛍光クロマトグラム(A)、及びマスクロマトグラム(B)を示す。
【図5】MS/MSによるマススペクトルを示す。
【図6】実施例3における逆相クロマトグラフィー(RPLC)によるクロマトグラムを示す。
【図7】蛍光誘導体化の反応時間と蛍光強度との関係を示す(左図:DAABD−Cl,右図:TAABD−Cl)。
【図8】TAABD−Clとの反応時間と蛍光強度との関係を示す。
【図9】新規蛍光試薬によるシステインの蛍光誘導体化(pH9.0)の反応時間とピーク領域との関係を示す。
【図10】新規蛍光試薬によるシステインの蛍光誘導体化(pH10.0)の反応時間とピーク領域との関係を示す。
【図11】DAABD−Clで誘導体化した、線虫(C.elegans)の可溶性フラクションが得られたタンパク質(約10μg)のクロマトグラムを示す。
【図12】DAABSeD−Xの合成経路を示す。
【技術分野】
【0001】
本発明は、微量の発現タンパク質及び/又はペプチドの検出・分離・同定方法に関するものであり、更に詳しくは、生体において遺伝子の発現により産生される微量の発現タンパク質及び/又はペプチドを簡便な方法で、高感度に検出し、同定することを可能とする新規な発現タンパク質及び/又はペプチドの検出・分離・同定方法、及びその同定システムに関するものである。
本発明は、ポストゲノム時代において重要な役割を果たすことが期待される、発現タンパク質及び/又はペプチドを網羅的に解析するプロテオーム技術における新しい検出・分離・同定手法を提供するものとして有用である。
【背景技術】
【0002】
ポストゲノム時代において重要な課題は、遺伝子を介して発現する発現タンパク質/ペプチドの微量検出とその分離・同定である。従来、この課題達成のために、2次元電氣泳動後のペプチドフィンガープリント法が汎用されてきた(非特許文献1参照)。しかし、この方法は、煩雑な操作のために該方法の再現性に難点があった。この難点を克服する手法として、最近、多次元高速液体クロマトグラフィー(多次元HPLC)による分離・同定法、或いはICATによる手法が提案されている(非特許文献2参照)。
【0003】
これらのうち、タンパク質/ペプチドを、直接、多次元HPLCで分離・同定する方法は、全てのタンパク質/ペプチドを同時に処理するために、多大な労力と時間を要するという欠点がある。また、ICATによる手法は、チオール含有タンパク質/ペプチドのチオール基をisotope−coded affinity tags (ICAT) reagentで標識した後、それをビオチン結合カラムにて捕集し、これら全てを酵素水解し、得られたペプチドフラグメント混合物をHPLCで分離、質量分析計(MS)にて質量分析し、タンパク質/ペプチドを網羅的に解析しようとするものである。しかし、この方法は、チオール含有タンパク質/ペプチドの全てを酵素水解するため、大量に存在する目的以外のタンパク質/ペプチドのフラグメントが、目的とする微量タンパク質/ペプチドのフラグメントの検出及びその同定を妨害する、と言う欠点があり、当技術分野においては、更なる方法のブレークスルーが必要とされていた。
【0004】
【非特許文献1】Dunn MJ.Two−dimensional gel electrophoresis of proteins, J Chromatogr 1987;418:145−185
【非特許文献2】Gygi S.P, Rist B, Gerber S.A, Turecek F, Gelb M.H, Aebersold R、 Quantitative analysis of complex protein mixtures using isotope−coded affinitytags,Nature Biotechnology 1999;17:994−999
【発明の開示】
【発明が解決しようとする課題】
【0005】
このような状況の中で、本発明者らは、上記従来技術に鑑みて、上記従来技術における諸問題を抜本的に解決することを目標として鋭意研究を積み重ねた結果、従来法と異なり、被験試料中の蛍光標識可能なタンパク質及び/又はペプチドのみを蛍光選択的に分離した後、これを酵素水解に付し、分画した蛍光画分を質量分析、データベース照合、構造解析に供することにより、従来法では検出不可能であった微量の発現タンパク質及び/又はペプチドを高感度に検出し、同定することができることを見出し、本発明を完成するに至った。
【0006】
本発明は、遺伝子を介して発現する微量の発現タンパク質及び/又はペプチドを、簡便な測定手法で、高感度に検出・分離・同定することを可能とする上記発現タンパク質及び/又はペプチドの微量検出・分離・同定方法を提供することを目的とするものである。
また、本発明は、上記微量検出・分離・同定方法に使用する微量の発現タンパク質及び/又はペプチドを、高感度で検出・分離・同定するための発現タンパク質及び/又はペプチド同定システムを提供することを目的とするものである。
更に、本発明は、従来法では検出することができなかった、遺伝子を介して発現する微量の発現タンパク質及び/又はペプチドを超高感度で検出・分離・同定することを可能とする新しい分析方法及び手段を提供することを目的とする。
【課題を解決するための手段】
【0007】
上記課題を解決するための本発明は、以下の技術的手段から構成される。
(1)被験試料中の発現微量タンパク質及び/又はペプチドを高感度に検出・分離・同定する方法であって、被験試料中のタンパク質及び/又はペプチドを蛍光誘導体とした後、これを蛍光検出により分離し、その蛍光画分を質量分析に付するか、又はその蛍光画分を酵素水解に付し、そのペプチド断片を分離し、その画分を質量分析に付し、データベース照合、構造解析に供して発現タンパク質及び/又はペプチドの同定を行うことを特徴とする上記発現タンパク質及び/又はペプチドの検出・分離・同定方法。
(2)被験試料中のタンパク質及び/又はペプチドを蛍光誘導体とした後、HPLCに付し、その蛍光分画を捕集した後、酵素水解に付し、その蛍光標識フラグメント及び非蛍光標識フラグメントを質量分析又はMS/MS分析して得られた各フラグメントのイオン分子量情報をタンパク質及び/又はペプチドフラグメントデータベースと照合し、構造解析する、前記(1)に記載の方法。
(3)(a)被験試料中のタンパク質及び/又はペプチドを蛍光試薬で標識する、(b)それを1次元又は2次元のHPLC/蛍光検出により、その蛍光分画を捕集する、(c)上記蛍光分画を酵素水解に付する、(d)それを第二段階のHPLC/蛍光検出により、その蛍光クロマトグラムを得ると共に、その全ピークを質量分析に付し、データベース照合、構造解析に供する、前記(1)に記載の方法。
(4)タンパク質及び/又はペプチド試料の水溶液に、官能基特異的蛍光試薬を加え、場合により、界面活性剤及び/又はタンパク変性剤を加え、タンパク質及び/又はペプチドを蛍光標識する、前記(1)から(3)のいずれかに記載の方法。
(5)蛍光標識したタンパク質及び/又はペプチド試料を蛍光検出器付きイオン交換カラムHPLC、逆相分配HPLC、ゲル濾過HPLC、又は電気泳動に代表される分離手段に付し、蛍光をモニターしながらそのピーク分画を捕集する、前記(1)から(3)のいずれかに記載の方法。
(6)蛍光分画を、各種ペプチダーゼ、トリプシン、キモトリプシンに代表されるタンパク質分解酵素を用いて酵素水解する、前記(1)から(3)のいずれかに記載の方法。
(7)酵素水解物を蛍光検出器付き逆相HPLCに付し、蛍光ピークを検出すると共に、蛍光標識フラグメント及び蛍光非標識フラグメントの質量分析又はMS/MS分析を行う、前記(1)から(3)のいずれかに記載の方法。
(8)質量分析又はMS/MS分析に付して得られた各フラグメントのイオン分子量情報を、コンピューターによるタンパク質及び/又はペブチドフラグメントデータベースと照合し、構造解析して、酵素水解以前のタンパク質及び/又はペプチドの同定を行う、請求項1から3のいずれかに記載の方法。
(9)被験試料が、生体試料から採取したタンパク質及び/又はペプチド試料である、前記(1)から(3)のいずれかに記載の方法。
(10)タンパク質及び/又はペプチドフラグメント情報、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを用いてデータベース照合する、前記(1)から(3)のいずれかに記載の方法。
(11)前記(1)から(10)のいずれかに記載の方法に使用する発現微量タンパク質及び/又はペプチド検出・分離・同定システムであって、被験試料のタンパク質及び/又はペプチドを蛍光試薬で標識するための第一反応器、蛍光試薬で標識した蛍光誘導体を蛍光分画するための1次元又は2次元の蛍光検出器付きHPLC、蛍光分画を酵素水解するための第二反応器、酵素水解物の蛍光標識フラグメントを蛍光検出するための第二段階の蛍光検出器付きHPLC、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを搭載した構造解析装置の1種又は2種以上を構成要素として含むことを特徴とする上記検出・分離・同定システム。
(12)上記第一反応器、1次元又は2次元の蛍光検出器付きHPLC、第二反応器、第二段階の蛍光検出器付きHPLCを直列に配置してなる、前記(11)に記載のシステム。
(13)被験試料中のタンパク質及び/又はペプチドを、蛍光誘導体化試薬として、下記の化5の一般式(1)
【0008】
【化5】
【0009】
〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、−NHR′(但、R′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキル)、又は−NR′′R′′′(但し、R′′はアルキル、R′′′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキルを示す。〕で表わされる化合物、又はその同位体化合物又は下記の化2の一般式(2)
【0010】
【化6】
【0011】
(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物又はその同位体化合物、を用いて、蛍誘導体とする、請求項1に記載の方法。
(14)前記(1)に記載の方法でタンパク質及び/又はペプチドを蛍光誘導体化するために使用する蛍光誘導体化試薬であって、下記の化7の一般式(1)
【0012】
【化7】
【0013】
〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、−NHR′(但、R′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキル)、又は−NR′′R′′′(但し、R′′はアルキル、R′′′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキルを示す。〕で表わされる化合物、又はその同位体化合物又は下記の化2の一般式(2)
【0014】
【化8】
【0015】
(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物又はその同位体化合物、を用いて、蛍誘導体とする、請求項1に記載の方法。
(15)被検試料のタンパク質及び/又はペプチドを蛍光誘導体化後、HPLCで分離・検出し、分画後、酵素水解し、この水解物を直接質量分析で配列分析とタンパク質の同定を行なうことを特徴とするタンパク質及び/又はペプチドの検出・分離・同定方法。
(16)被検試料として、異なる試料A中及び試料B中のタンパク質及び/又はペプチドを、それぞれ蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬でそれぞれ誘導体化後、蛍光検出器付きHPLCで分離・検出し、分画後、各蛍光ピークをそのまま又は合体して定量に供する、及び/又は各蛍光ピークを合体して酵素水解に供し、この水解物を定量に供する、又はこの水解物をHPLC−質量分析に供し、同定を行なう、ことを特徴とするタンパク質及び/又はペプチドの検出・分離・同定方法。
(17)各蛍光ピークをそのまま又は合体してHPLCによる定量に供し、試料A中及び試料B中のタンパク質及び/又はペプチドの各誘導体の比率を算出する前記(16)に記載の方法。
(18)水解物をHPLCによる定量に供し、試料A中及び試料B中のタンパク質及び/又はペプチドの各誘導体の比率を算出する前記(16)に記載の方法。
(19)試料A中及び試料B中のタンパク質及び/又はペプチドの第1の蛍光誘導体化試薬との反応物及び第2の蛍光誘導体化試薬との反応物を合体し、2つの励起・蛍光検出の可能なHPLCに供し、分画後、各蛍光ピークを合体して酵素水解に供し、この水解物をHPLC−質量分析に供し、同定を行う前記(16)に記載の方法。
(20)試料A、Bが、2種類の細胞又は組織又は体液試料である前記(16)に記載の方法。
(21)蛍光誘導体化試薬として、DAABD−X、DAASeBD−X、及びDAAThBD−X(但し、XはCl又はF)のうちの励起・蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬でタンパク質及び/又はペプチドを誘導体化する前記(16)に記載の方法。
(22)蛍光波長の異なる蛍光誘導体化試薬として、DAABD−X、DAASeBD−X、又はDAAThBD−X(但し、XはCl又はF)と、それらの各同位体を組み合わせて使用する前記(21)に記載の方法。
(23)酵素水解した試料を直接質量分析に付しペプチドマップを得ると同時に、蛍光試薬の有する骨格並びに電荷を活用して、蛍光標識化ペプチド断片を質量分析測定部で抽出してシステイン含有ペプチド部分の構造を取得し、これらを基にタンパク質及び/又はペプチドの同定を行なう前記(16)に記載の方法。
(24)蛍光誘導体化試薬で誘導体化したタンパク質及び/又はペプチドを分解することなく分画することが可能な、少なくともミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、及びミクロ自動注入装置を具備したことを特徴とする自動分画装置。
(25)少なくとも、ミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、酵素反応装置、及びミクロ自動注入装置を備え、任意に、質量分析(MS)システムを具備したことを特徴とする微量タンパク質の高性能・簡易定量・同定解析装置。
【0016】
次に、本発明について更に詳細に説明する。
本発明は、上記従来法の難点を克服するためになされたものであり、1)微量の発現タンパク質/ペプチドを蛍光試薬で標識し、2)それをHPLC/蛍光検出にて第一段の分離・蛍光検出を行い、3)その蛍光分画(コントロール試料と比べて、被験試料に特異的に増減する蛍光画分)のみを捕集後、酵素水解し、それを第二段のHPLC/蛍光検出にて分離し、蛍光ピークの確認を行った後、HPLC/MSに付し、蛍光標識タンパク質/ペプチドフラグメントの同定を行い、当該微量タンパク質/ペプチドの特定を行う方法に関するものである。尚、タンパク質/ペプチド試料が純度の高い場合には、第一段のHPLC/蛍光検出による分離を省くことができる。本発明の方法は、従来法と異なり、蛍光標識可能なタンパク質/ペプチドのみを特異的に抽出し、検出・同定することができるという特徴を有し、微量の発現タンパク質/ペプチドを特定するために尤も相応しい方法である。
【0017】
本発明では、被験試料として、生体から採取したあらゆる種類のタンパク質及び/又はペプチドを含む試料が対象とされる。本発明の方法では、被験試料中の微量の発現タンパク質/ペプチドを蛍光試薬で標識し、蛍光誘導体化するが、この場合、タンパク質/ペプチド水溶液に、官能基特異的蛍光試薬を加え、場合により、界面活性剤及び/又はタンパク変性剤を加え、発現タンパク質/ペプチドを定量的に誘導体化することが重要である。即ち、本発明では、タンパク質/ペプチド試料の水溶液に、界面活性剤と場合によっては還元剤を添加し、これに官能基特異的蛍光試薬を加え、必要により、加温することにより、タンパク質及び/又はペプチドを蛍光標識する。本発明では、上記界面活性剤として、非イオン性、陰イオン性、陽イオン性及び両イオン性界面活性剤が用いられる。また、本発明では、上記還元剤として、好適には、Tris(2−carboxyethyl)phosphine、tributylphosphineが用いられるが、これらに制限されるものではなく、同効のものであれば同様に使用することができる。
【0018】
本発明において、上記官能基特異的蛍光試薬として、(例えば、4−Fluoro−7−nitro−2,1,3−benzoxadiazole(NBD−F)、5−(N,N−Dimethylamino)naphthalene−1−sulfonyl chloride (DNS−CL)、Orthophthaldehyde(OPA)、Fluorescamine、9−Fluorenylmethyl chloroformate (FMOC)等のアミノ基特異的蛍光試薬、Ammonium 7−fluoro−2,1,3−benzoxadiazole−4−sulfonate(SBD−F)、4−(Aminosulfonyl)−7−fluoro−2,1,3−benzoxadiazole(ABD−F)、4−(Acetylaminosulfonyl)−7−fluoro−2,1,3−benzoxadiazole(AcABD−F)、4−Fluoro−7−trichloroacetylaminosulfonyl−2,1,3−benzoxadiazole(TcAcABD−F)、monobromobimane等のチオール基特異的蛍光試薬、4−Nitro−7−N−piperazino−2,1,3−benzoxadiazole(NBD−PZ)、4−N,N−Dimethylaminosulfonyl−7−N−piperazino−2,1,3−benzoxadiazole(DBD−PZ)と縮合剤との組み合わせによるカルボキシル基特異的蛍光試薬、又は、4−(N−Chloroformylmethyl−N−methyl)amino−7−nitro−2,1,3−benzoxadiazole(NBD−COCL)等の水酸基用蛍光試薬)が例示されるが、これらに制限されない。
【0019】
本発明においては、必要により、加温する(例えば、30−100℃、望ましくは40−70℃で10−300分間、望ましくは60−180分間)ことにより、タンパク質/ペプチドを蛍光標識する。その後、反応液のほぼ全量を蛍光検出器付きイオン交換カラムHPLC、又は逆相分配HPLC、又はゲル濾過HPLCに付し、蛍光をモニターしながらピーク分画を分取する。この場合、蛍光検出は、標識蛍光体の励起・蛍光波長に相当する波長に設定して行う。例えば、NBD−F、又はSBD−Fで標識した場合には、励起波長480nm或いは380nm、励起波長520nm又は505nmに設定する。イオン交換HPLCの場合には、塩、例えば、食塩、硫酸ナトリウム、過塩素酸カリウム、酢酸アンモニウムなど、望ましくは酢酸アンモニウムのような揮発性の塩を段階的に増量し、それぞれの画分を得る。この画分そのもの又はこの画分を濃縮・乾固した試料を、酵素水解に付す。酵素としては、各種ペプチダーゼ、トリプシン、キモトリプシンなど、適宜のタンパク質分解酵素が用いられる。この際、酵素カラムを接続してオンラインで酵素水解を行うこともできる。
【0020】
この溶液の一部を蛍光検出器付き逆相分配HPLCに付し、蛍光標識体の溶出位置を確認する。次いで、この逆相HPLCカラムの出口を質量分析計(どの様な質量分析計でも対応可能であるが、望ましくはエレクトロスプレー型質量分析計を用いる)に接続し、酵素水解物の蛍光標識フラグメント及び蛍光非標識フラグメントの質量分析(蛍光標識フラグメントは一回の質量分析、蛍光非標識フラグメントは親イオンを更に質量分析する)又は質量分析/質量分析(MS/MS)を行う。この際、蛍光検出器と質量分析計を直列に接続することも可能である。このようにして得られた各フラグメントのイオン分子量情報を、コンピューターに接続したタンパク質/ペプチドフラグメントデータベースと照合し、構造解析することにより、酵素水解以前のタンパク質/ペプチドの同定を行う。この場合、本発明では、タンパク質及び/又はペプチドフラグメント情報、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを用いてデータベース照合を行う。
【0021】
本発明では、発現タンパク質及び/又はペプチドを含む被験試料中のタンパク質及び/又はペプチドを蛍光誘導体化し、この蛍光誘導体をHPLC/蛍光検出で分離し、蛍光ピークの強さを比較して標的発現タンパク質及び/又はペプチドの蛍光誘導体を分離し、得られた標的発現タンパク質及び/又はペプチドのピーク画分を酵素分解し、次いで、質量分析又はMS/MS分析、データベース照合、構造解析により、上記タンパク質及び/又はペプチドを同定する。タンパク質及び/又はペプチドのアミノ基、チオール基、水酸基及びカルボキシル基などの機能性部分を誘導化するための多くの蛍光試薬が存在するので、本発明では、その目的に応じて、適当な試薬を任意に選択して使用することができる。後記する実施例に示されるように、例えば、Cys−含有タンパク質を誘導化するためには、チオール基に特異的な試薬である、Ammonium 7−fluoro−2,1,3−benzoxadizole−4−sulfonate(SBD−F)を使用することができる。図1に、本発明の方法のプロセスの一例を模式的に示す。後記する実施例に示されるように、実際、このようにして、ラットに10mgのデキサメタゾンを投与して2日後のランゲルハンス島におけるPancreatic polypeptide、プロインシュリン 2、78KD Glucose−regulated protein、プロテイン結合フォスファチジルアミン及びチオレドキシンが強く誘導されたことが示された。
【0022】
本発明の方法で重要な点は、各組織におけるタンパク質の量は、HPLC/蛍光検出で分離する前に、例えば、正常組織と非正常組織との組織間で定量、比較されるべきであることから、標的発現タンパク質を定量的に誘導体化することである。そのために、本発明では、適宜の界面活性剤が用いられるが、例えば、いくつかの界面活性剤についてBSAにより検討したところ、CHAPSがn−Dodecyl−β−D−maltopyranosideと比べて高い強度を示した(図2参照)。本発明では、pH、温度、反応時間及び誘導体化反応の添加剤等について、標的発現タンパク質及び/又はペプチドに応じて、最適条件を設定することで定量的な蛍光誘導体化が可能である。本発明では、これらの条件は、発現タンパク質/ペプチドの種類、分析目的等に応じて、適宜設定することができる。本発明の方法により、試験タンパク質/ペプチドのクロマトグラムは単一の蛍光ピークを示した(図3参照)。本発明の方法において、タンパク質及び/又はペプチドの検出限界は、0.2−6.0fmolであり、最適条件下での10−1000fmolの範囲で良好な直線(γ>0.9994)の測定曲線が得られ(表1参照)、検出性能は、従来法に比べて、顕著であることが分かる。表1に、蛍光検出/HPLCによる各種タンパク質及び/又はペプチドの検出限界を示した。
【0023】
【表1】
【0024】
更に、本発明では、上記方法に使用する微量の発現タンパク質及び/又はペプチド検出・分離・同定システムとして、被験試料のタンパク質及び/又はペプチドを蛍光試薬で標識するための第一反応器、蛍光試薬で標識した蛍光誘導体を蛍光分画するための1次元又は2次元の蛍光検出器付きHPLC、蛍光分画を酵素水解するための第二反応器、酵素水解物の蛍光標識フラグメントを蛍光検出するための第二段階の蛍光検出器付きHPLC、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを搭載した構造解析装置の1種又は2種以上を構成要素として含む上記検出・分離・同定システムが用いられる。この場合、上記第一反応器、2次元の蛍光検出器付きHPLC、第二反応器、第二段階の蛍光検出器付きHPLCを直列に配置することができる。これらの装置は、その使用目的に応じて、適宜の容量、形態に任意に設計することができる。
【0025】
本発明は、被験試料中のタンパク質及び/又はペプチドを、蛍光誘導体化試薬として、前記一般式(1)〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、又は−NHR′(但し、R′はN置換アルキル)を示す。〕で表わされる化合物、又は前記一般式(2)(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物、を用いて、蛍光誘導体とすることができる。更に、本発明は、これらの化合物のいずれか1種を有効成分とする新規蛍光誘導体化試薬を提供することができる。
【0026】
これらの化合物の具体例としては、好適には、例えば、以下の例があげられるが、これらに制限されるものではなく、これらと同等ないし類似の化合物であれば同様に使用することができる。本発明の化合物は、後記する実施例に具体的に記載した方法と同様にして容易に合成することができる。
(1)DAABD−Cl[4−(dimethylaminoethyl aminosulfonyl)−7−chloro−2,1,3−benzoxadiazole]
(2)TAABD−Cl(7−chloro−2,1,3−benzoxadiazole−4−sulfonylaminoethyl rimethylammonium chloride)
(3)DAABD−F[4−(dimethylaminoethyl aminosulfonyl)−7−fluoro−2,1,3−benzoxadiazole]
(4)TAABD−F(7−fluoro−2,1,3−benzoxadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
(5)DAABSeD−Cl[4−(dimethylaminoethyl aminosulfonyl)−7−chloro−2,1,3−benzoselenadiazole]
(6)TAABSeD−Cl(7−chloro−2,1,3−benzoselenadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
(7)DAABSeD−F[4−(dimethylaminoethyl aminosulfonyl)−7−fluoro−2,1,3−benzoselenadiazole]
(8)TAABSeD−F(7−fluoro−2,1,3−benzoselenadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
(9)DAABThD−Cl[4−(dimethylaminoethyl aminosulfonyl)−7−chloro−2,1,3−benzothiadiazole]
(10)TAABThD−Cl(7−chloro−2,1,3−benzothiadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
(11)DAABThD−F[4−(dimethylaminoethyl aminosulfonyl)−7−fluoro−2,1,3−benzothiadiazole]
(12)TAABThD−F(7−fluoro−2,1,3−benzothiadiazole−4−sulfonylaminoethyl trimethylammonium chloride)
【0027】
本発明では、蛍光誘導体化試薬として、例えば、SBD−X、SBSeD−X、SBThD−X、DAABD−X、TAABD−X、DAABSeD−X、TAABSeD−X、DAABThD−X、TAABThD−X(但し、XはCl、又はF)、及びそれらの各同位体が提供される。これらの化合物は、いずれも、後記する実施例に示した化合物の場合と同様にして合成することができる。また、本発明では、前記一般式(1)で表わされる化合物の側鎖のアルキル基の鎖長を任意に変更することが可能である。本発明では、これらの化合物の、蛍光波長とHPLC分離における保持時間の相違を利用して、これらの化合物を2つ以上組み合わせて使用することができる。HPLCからの溶出は、例えば、DAABSeD−F<DAABD−Cl又はDAPABSeD−F(側鎖のアルキル:プロピル)<DAPABD−Clの順に遅くなる。また、上記化合物の各同位体を使用することにより、更なる微量検出が可能となる。
【0028】
本発明では、蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬を利用することにより、例えば、同一起源の異なる歴史を有する二つのタンパク質試料の比較を簡便、かつ同時に行なうことができる。例えば、一方が病態患者試料で一方が健常者からの試料、あるいは一つの細胞又は組織試料で、一方がある薬剤で処理された試料で、他方が未処理の試料の比較を同一のクロマトグラムに基づいて同時に行なうこと、それにより、各蛍光誘導体化試薬で誘導体化されたタンパク質及び/又はペプチドの各誘導体を簡便、かつ正確に定量すること、が可能であり、2つの試料中のタンパク質及び/又はペプチドのプロファイルを同時的に測定し、比較することが実現できる。
【0029】
本発明では、蛍光誘導体化試薬で誘導体化したタンパク質及び/又はペプチドを分解することなく分画することが可能な、少なくともミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、及びミクロ自動注入装置を具備した自動分画装置が提供される。また、本発明では、少なくとも、ミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、酵素反応装置、及びミクロ自動注入装置を備え、任意に、質量分析(MS)システムを具備した微量タンパク質及び/又はペプチドの高性能・簡易定量・同定装置が提供される。
【0030】
本発明は、例えば、微量リン酸化タンパク質や糖タンパク質を蛍光誘導体化試薬で蛍光標識後、ミクロカラム−HPLC−蛍光検出器により修飾リン酸化部分並びに糖結合部分を保持したままのタンパク質として高性能分離・検出し、ミクロフラクションコレクターにて分取する。これに酵素を添加し、標識化タンパク質を酵素水解し、次いで、この水解試料を直接質量分析に付す。得られたペプチドマップと蛍光標識化ペプチドマップ情報を基に、データベース検索ソフトを利用して、例えば、翻訳後修飾微量タンパク質の修飾部分を含む同定を行なう。本発明の手法は、蛍光検出法とミクロHPLCとを組み合わせた方法であるため、例えば、超微量タンパク質を同定できること、翻訳後修飾タンパク質をそのまま分解することなく抽出して解析できること、そのため、リン酸化部位や糖結合部位の情報を確実に得られるばかりでなく、当該タンパク質を確実に同定できること、という従来の解析手法にはない利点を有している。本発明の解析手法は、生命科学、病態化学分野で広く使われることが期待され、疾患診断、治療、人類の健康維持へ貢献することができる。
【発明の効果】
【0031】
本発明により、1)遺伝子を介して発現する発現タンパク質及び/又はペプチドを簡便な方法及び手段で、高感度に検出・分離・同定することができる、2)本発明の方法により、従来法では検出できなかった微量の発現タンパク質及び/又はペプチドを短時間で、感度良く検出・分離・同定することができる、3)また、上記検出・分離・同定方法に使用する微量の発現タンパク質及び/又はペプチドの微量検出・分離・同定システムを提供することができる、4)本発明は、プロテオームのプラットフォーム技術を提供するものとして有用である、という効果が奏される。
【発明を実施するための最良の形態】
【0032】
次に、実施例に基づいて本発明を具体的に説明するが、本発明は、以下の実施例によって何ら限定されるものではない。
【実施例1】
【0033】
ラット膵ランゲルハンス島(ラ島)中チオール含有タンパク質/ペプチドの分離・同定(1)ラ島中チオール含有タンパク質/ペプチドの蛍光誘導体化
ラ島に0.1Mホウ酸緩衝液(pH9.0)に溶解した6M塩酸グアニジン溶液50μlを加えて可溶化した。これに6M塩酸グアニジン溶液に溶解した17.5mMTCEP、17.5mMSBD−F、10mMEDTA及び50mMCHAPSをそれぞれ50μlずつ加え、混合した。この溶液を40℃にて3時間反応させることにより蛍光誘導体化を行った。
(2)イオン交換HPLCによる1次分離
上記の反応溶液をイオン交換カラムに付し、NaClのグラジエント(0、0.04、0.08、0.12及び0.3M)により蛍光タンパク質/ペプチドを溶出させ、5つのフラクションに分離した。なお、蛍光タンパク質/ペプチドの検出はSBD骨格の蛍光により行った。HPLC条件を以下に示す。
【0034】
(HPLC条件)
カラム:TSKgel DEAE−5PW 7.5×75mm(東ソー(株))
ガードカラム:C8−300−S 54.0×10mm(YMC(株))
移動相:段階溶離〔0−5分:C100%、5−15分:A100%、15−25分:A87%B13%、25−35分A73%B27%、35−45分:A60%B40%、45−55分:B100%〕
A:5mMトリス塩酸緩衝液(pH8.0)/アセトニトリル(50:50)
B:5mMトリス塩酸緩衝液(pH8.0)/アセトニトリル(50:50)
(0.3MNaCl含有)
C:5mMトリス塩酸緩衝液(pH8.0)
カラム温度:室温(約25℃))
流速:0.5ml/min
検出:Ex380nm、Em505nm
注入量:200μl
【0035】
(3)逆相HPLCによる2次分離
上記の各画分を濃縮し、アセトニトリルを蒸発させた後、逆相カラムに付し、アセトニトリルの勾配溶離によりタンパク質/ペプチドをカラムから溶出させた。なお、タンパク質/ペプチドの検出はSBD骨格の蛍光によりモニターした。HPLC条件を以下に示す。
【0036】
(HPLC条件)
カラム:カプセルパックC8 SG300 2.0×100mm((株)資生堂)
移動相:勾配溶離(0→60分:B40%→100%)
A:0.05%トリフルオロ酢酸
B:0.05%トリフルオロ酢酸/アセトニトリル(40:60)
カラム温度:室温(約25℃)
流速:0.2ml/min
検出:Ex380nm、Em505nm
注入量:50μl
【0037】
(4)酵素処理
採取したHPLCの各ピーク画分をそれぞれのチューブに0.5M炭酸水素アンモニウム溶液5μlを加えてトリフルオロ酢酸を中和後、濃縮してアセトニトリルを蒸発させた。残渣(約80μl)に20μg/mlトリプシン(プロメガ)及び10mM塩化カルシウムをそれぞれ10μlずつ添加した。これを37℃で2時間インキュベートし、HPLC−MS/MS測定用の試料とした。
【0038】
(5)MS/MS測定によるタンパク質/ペプチドの同定
上記の試料を逆相カラムHPLCに付し、エレクトロスプレー法によるMS/MS測定を行った。HPLC条件を以下に示す。なお、タンパク質/ペプチドの同定はデータベースにNCBI、サーチエンジンにMASCOTを使用した。
【0039】
(HPLC条件)
カラム:Cadenza TC−C18 2.0 ×100mm(Imtact(株))
移動相:勾配溶離(0→30分:B20%→100%)
A:0.1%ギ酸
B:0.1%ギ酸/アセトニトリル(50:50)
カラム温度:室温(約25℃)
流速:0.2ml/min
測定モード:positive
測定範囲:500−3000m/z
注入量:50μl
【0040】
上記の方法により、約130のタンパク質/ペプチドのピークが分離できた。
そのうち、約50のタンパク質/ペプチドが同定できた(表2、図6参照)。
【0041】
【表2】
【実施例2】
【0042】
SBD−Fで誘導体化したBSAを、図1に示されるプロセスにより、トリプシンで酵素水解し、得られたペプチド混合物を逆相液体クロマトグラフィー(RPLC)で分離し、蛍光検出器で検出した。次いで、各ペプチドをイオンスプレーイオントラップ質量分析計によるMS/MS分析に供した。原理的には、トリプシン分解により、BSAは、4個のアミノ酸残基以上の25のシステイン含有ペプチド及び35のシステイン非含有ペプチドが生成されるが、本実施例では、27以上の蛍光ペプチドが蛍光検出され、定量的に誘導体化が行われた(図4、A)。
【0043】
また、11のシステイン含有ペプチド及び17の非システイン含有ペプチドがマスクロマトグラフィーで検出された(図4、B)。 図5に、(M+2H)2+プレカーサー、m/z=873.4(図4で矢印で示した)から得たMS/MSスペクトルを示す。
全てのペプチドフラグメントのCIDスペクトルにより、確率的プロテイン同定法に基づくMASCOTによるデータベース照合を行い、予測通り、完全にBSAとしてタンパク質を同定した(スコア:39)。
【実施例3】
【0044】
デキサメタゾン(Dex)投与及び非投与のラット膵臓について試験した。
Dexは、肝臓のグルコース産生の増加とインシュリン耐性の誘導により主なヒト糖尿病であるタイプ2の糖尿病を誘発する。実際に、Dex処理の24時間後に、血中グルコースレベルは209.8mg/dLに達し、処理前の値の118.3mg/dLより著しく高い(p<0.05)。本実施例では、2日間Dexで処理又は未処理のラット膵臓からランゲルハンス島(60島)を採取し、SBD−Fで誘導体化した。本発明の方法を生物試料に適用するための重要な態様は、タンパク質混合物からHPLCで発現タンパク質を分離することである。本実施例では、蛍光タンパク質は、まず、SBD−Fにより生成した多くのマイナス電荷と酸性アミノ酸部分に基づいてイオン交換クロマトグラフィー(IEC)で分離された。
【0045】
IECは、塩化ナトリウムの段階溶離(0、0.04、0.08、0.12、及び0.3M NaCl)により行い、蛍光タンパク質混合物を5つの画分として得た。次いで、各画分は、更に、それらの疎水性に基づいて、逆相液体クロマトグラフィー(RPLC)により分離された。この実験でのピークの容量(n=L/(4σ)、但し、Lは分析のトータル時間及び4σはピーク幅、としてのHPLCの性能の理論値)は、各RPLCフラクションにつき40と計算され、IEC−RPLC法の5つのステップのピーク容量は、約200であった。本実施例では、各RPLCサイクルで約3−50ピーク、合計で129ピークであった(図6)。
【0046】
微量タンパク質を検出するために、IECの段階溶離ステップを増やしてピーク容量を増加させた。未処理及びDex処理ラットから得られたRPLCクロマトグラムの全ての蛍光ピークを比較した。その結果、5本の蛍光ピークが1.8以上増加し、3本の蛍光ピークがDex処理で約1.5倍減少したことが見出された(表2)。表2に、Dex投与2日後の発現タンパク質の変化を示した。これらのタンパク質(即ち、標的発現タンパク質)は、広い小孔を有するRPLC(30nm小孔径)で分離され、トリプシンで分解し、各ペプチド混合物とした。各ペプチド混合物は、通常の小孔を有するRPLC(10nm小孔径)に供し、MS/MS分析した。データベース照合により、Dex処理後2日で増加したピークは、各々、膵臓ポリペプチド、プロインシュリン2、78KDグルコース−調節タンパク質、ホスファチジルエタノールアミン結合タンパク質、及びチオレドキシンであり、減少したピークは、各々、プロテイン31、dnak−タイプ分子シャペロンhsp72−psl、及びインシュリンであった。
【実施例4】
【0047】
本実施例では、以下の化9の(1)及び(2)の反応式により、新規蛍光誘導体化試薬の合成を行った。
【0048】
【化9】
【0049】
(1)DAAB−Clの合成
4−chlorosulfonyl−7−chloro−2,1,3−benzoxadiazole(CBD−Cl)(126.53 mg)をCH3CNに溶解し、N,N−dimethylethylenediamineを滴下し、triethylamineを加えた。室温で約10分間攪拌後、反応液を減圧乾固した後、シリカゲルカラム(CH2Cl2)で精製し、4−(dimethylaminoethy laminosulfonyl)−7−chloro−2,1,3−benzoxadiazole(DAABD−Cl)(20.2mg,87.4%)を得た。
得られた化合物の確認データを以下に示す。
1H−NMR(CD3OD):7.94(1H,d,J=7.5),7.65(1H,d, J=7.5),3.06(2H,t,J=6.7),2.30(2H,t,J=6.7), 2.02(6H,s)。ESI−MS:m/z305(M+H)+
【0050】
(2)TAABD−Clの合成
4−chlorosulfonyl−7−chloro−2,1,3−benzoxadiazole(CBD−Cl)(126.53 mg)をCH3CNに溶解し、H2Oに溶かしたaminoethyl trimethylammonium chlorideを滴下し、triethylamineを加えた。室温で約20分間攪拌後、反応液を減圧乾固した後、0.1%トリフルオロ酢酸(TFA)に溶かし、ODSカラムを用いて分取し、画分には、SBD−Cl(化10)が不純物として入っていたため、陰イオン交換カラムを用いて分取し、減圧乾固して、7−chloro−2,1,3−benzoxadiazole−4−sulfoneaminoethyl trimethylammonium chloride(TAABD−Cl)(127.2mg,58.8 %)を得た。
得られた化合物の確認データを以下に示す。
1H−NMR(CD3OD):8.01(1H,d,J=7.3),7.69(1H,d, J=7.3),3.46−3.48(4H,m),3.12(9H,s)。ESI−MS :m/z319(M)+
【0051】
【化10】
【実施例5】
【0052】
本実施例では、新規蛍光誘導体化試薬の反応性について検討した。
DAABD−Cl、TAABDD−ClのSBD−Fとの比較
10μM還元型グルタチオン、システイン、ホモシステイン混合液100μLとDAABD−Cl又はTAABD−Cl 100μLを混合し、pH9、40℃、10〜120分間反応させた。尚、各試薬は5mM EDTAを含む0.10M ホウ酸緩衝液(pH 9)に溶解した。0.1%ぎ酸で反応停止後、生成物をHPLCを用いて測定した。
【0053】
図7に、蛍光誘導体化反応時間と蛍光強度との関係(左図:DAABD−Cl、右図:TAABD−Cl)を示す。
SBD−F(化11)の場合、40℃で誘導体化を行うと120分間の反応時間が必要であったが、DAABD−Clは10〜20分、TAABD−Clは20〜30分で反応が終了することがわかった。
したがって、反応時間はDAABD−Clの場合は20分、TAABD−Clの場合は30分が好適である。
【0054】
【化11】
【0055】
(2)新規蛍光誘導体化試薬のMSでの感度
上記(1)で作成したサンプルをLC−MSにより検出し、蛍光誘導体化試薬でラベル化していないもの及びSBD−Fで誘導体化したものと、相対強度を比較した。
【0056】
蛍光誘導体化試薬でラベル化していないcysteine、homocysteine、GSHの高さをそれぞれ1としたときの相対強度は表3の通りであった。これより、DAABD−ClがMSでの感度が最も高いことがわかった。また、移動相が酸性であるので、DAABD化誘導体はプラスに荷電され水溶性であると考えられる。
【0057】
【表3】
【実施例6】
【0058】
(1)TAABD−Clのペプチドへの応用
10μMの以下に示した4種類のペプチド標品、17.5mM TAABD−Cl、10 mM EDTA、50mM CHAPS(界面活性剤)、2.5mM TCEP(還元剤)それぞれ50μLを混合し、pH9.0、40℃で、30,60,90,120分間反応させた。尚、各試薬は6.0M 塩酸グアニジン(タンパク変性剤)を含む0.10 M ホウ酸緩衝液(pH9.0)に溶解した。生成したTAABD化ペプチドをHPLCを用いて測定した。
1. vasopressin
2. oxytocin
3. somatostatin
4. amylin(rat)
図8に TAABD−Clとの反応時間と蛍光強度との関係を示す。
図8より、反応時間が60分までは生成量が増加した。反応停止後は氷冷下で保存し、−20℃にて保存すると、48時間はほとんど分解しなかった。
【0059】
(2)DAABD化ペプチド・タンパクの検出限界
表4に示した10種類のペプチド・タンパク質標品10μM混合液、2.5mM TCEP、17.5mM DAABD−Cl、10mM EDTA、50mM CHAPSそれぞれ50μLを混合し、pH9.0、40℃で、30分間反応させた。尚、各試薬は6.0 M 塩酸グアニジンを含む0.10M ホウ酸緩衝液(pH9.0)に溶解した。生成したDAABD化ペプチド・タンパク質はHPLCを用いて測定し、蛍光検出の検出限界をSBD−Fと比較した。
【0060】
【表4】
【0061】
(3)DAABD化ペプチド・タンパク質の同定
上記(2)で誘導体化した物質のうち、vasopressin、oxytocin、somatostatin、calcitonin、amylinはLC−MSにより同定できた。それらの分子量を以下に示す。
m/z 541.8(M+3H)3+[DAABD−vasopressin]
516.0(M+3H)3+[DAABD−oxytocin]
726.6(M+3H)3+[DAABD−somatostatin]
989.9(M+4H)4+[DAABD−calcitonin]
892.8(M+5H)5+[DAABD−amylin]
【0062】
これら全ての分子量は、それぞれのペプチドの2つのシステイン残基にDAABDが付加したとしたときの分子量であり、多価イオンピークの検出結果よりDAABD−Clによる誘導体化において、これらのペプチドのシステイン残基間のS−S結合が還元され、二つのチオール基両方に試薬が反応したことがわかった。
また、タンパク質の場合は酵素によってペプチドに分解する必要があるため、酵素トリプシンで消化し、LC−MS/MS検出及びMASCOTによるデータベース検索を行って同定を試みた結果、システインを含まないペプチドのアミノ酸配列も併せて決定し、タンパク質を同定することができた。
【実施例7】
【0063】
(SBSeD−Fの合成)
2−fluoroacetanilideを硝酸で処理して、1−acetylamino−2−nitro−6−fluorobenzeneとし、これを脱アセチル化して、2−fluoro−6−nitroanillineとし、次いで、パラジウム担持炭素触媒を用いて水素化して、3−fluoro−o−phenylenediamineを得た。
Selenium dioxideエタノール加熱溶液を、3−fluoro−o−phenylenediamine(60mg,0.48mmol)のエタノール加熱溶液に加え、混合物を30分加熱した。これを、シリカゲルカラムによるクロマトグラフィーに供し、溶離液のジクロロメタンで溶出し、4−fluoro−2,1,3−benzoselenadiazoleを白色粉末(88mg)として得た。得られた化合物の確認データを以下に示す。
mp.129℃、NMR(methanol−d4):δH7.55(1H,d,J=9.2)、7.41(1H,m)、7.06(1H,m)、ESI−MS:m/z202.8[(M+H)]。
【0064】
このようにして得た4−fluoro−2,1,3−benzoselenadiazoleをfuming sulfuric acid(60%)に溶かし、130℃で3時間還流した。この溶液を冷却し、冷水(30ml)に注ぎ、28%ammonium hydroxideで中和した。この中性溶液にエタノール100mlを加え、濾過物を減圧乾固させた。残渣を水(1.0ml)に溶解し、更に、以下のHPLCで精製した。即ち、残渣の100μlをHPLC分離に供した。HPLCカラム:TSK−gel ODS−120T、150×4.6mm i.d.、東ソー、溶離液:蒸留水、流速0.5ml/min、検出:280nm。SBSeD−Fに相当するフラクションを集め、減圧して白色粉末(50mg)を得た。得られた化合物の確認データを以下に示す。
m.P.>300℃、NMR(methanol−d4):δH7.97(1H,d d,J=7.6,J=5.4)、7.11(1H,dd,J=7.6,J=10.1)、ESI−MS:m/z280.8[(M−H)]。
【実施例8】
【0065】
(SBThD−Fの合成)
N−thionylaniline(0.49g,3.5mmol)を3−fluoro−o−phenylenediamine(200mg,1.6mmol)トルエン(2ml)溶液に加えた。反応混合物を100−120℃で4時間加熱し、溶媒を濾別した後、残渣をジクロロメタンに溶かし、溶液を10%HCl溶液及び水で各々洗浄した。有機相を乾燥し、減圧乾固させた。これをシリカゲルによるクロマトグラフィーに供し、溶離液のクロロホルムで溶出し、4−fluoro−2,1,3−benzothiadiazoleを淡黄色油として得た。得られた化合物の確認データを以下に示す。
NMR(methanol−d4):δH7.69(1H,d,J=8.9)、7.50(1H,m)、7.20(1H,m)、ESI−MS:m/z154.9[(M+H)]。
【0066】
このようにして得た4−fluoro−2,1,3−benzothiadiazole(30ml)をfuming sulfuric acid(60%)に溶かし、130℃で3時間還流した。次いで、この溶液を冷却し、ゆっくり冷水(30ml)に注ぎ、28%ammonium hydroxideで中和した。中性溶液にエタノール100mlを加え、得られた濾過物を減圧乾固した。残渣を水(1.0ml)に溶かし、更に、以下の条件でHPLCで精製した。SBThD−Fに相当するフラクションを集め、減圧し、白色粉末(25mg)を得た。得られた化合物の確認データを以下に示す。
decomp.265℃、NMR(methanol−d4):δH8.06(1H,dd,J=7.9,J=4.9)、7.11(1H,dd,J=7.9,J=9.8)、ESI−MS:m/z232.8[(M−H)]。
上記方法により合成したSBSeD−F及びSBThD−Fを下記の化12に示す。
【0067】
【化12】
【実施例9】
【0068】
(1)システイン誘導体の蛍光スペクトル
1mMEDTAを含む0.1Mホウ酸緩衝液(pH9.0)による上記SBSeD−F、SBThD−F又はSBD−Fの各蛍光試薬溶液(4mM)の500μl部分を、0.1Mホウ酸緩衝液(pH9.0)によるシステイン(0.4mM)溶液の同量と混合した。この混合物を60℃で8時間放置した。反応の後、反応混合物をHPLC分離し、各システイン誘導体に相当するフラクションを集め、それらの蛍光スペクトルを測定した。
【0069】
SBSeD−FとSBThD−Fのシステインに対する反応性
4mMの各試薬、SBSeD−F、SBThD−F又はSBD−F及び1mM EDTAを含む0.1Mホウ酸緩衝液(pH9.0又はpH10)の500μl部分を、0.1Mホウ酸緩衝液(pH9.0又はpH10)によるシステイン溶液(0.4mM)の同量と混合した。反応混合物をHPLC分離し、60℃での反応をモニターした。
【0070】
(2)結果
SBD−Fのような水溶性試薬は、そのsulfonic acid残渣により水溶体中での誘導体の可溶性を増加させ、結果として、誘導体の吸収又は沈殿が減少する。したがって、インシュリンのような比較的疎水性ペプチドのSBD−Fによる誘導体は、逆相カラムで溶出され、高感度に検出されたが、本実施例では、更に、benzoselenadiazole又はbenzothiadiazole骨格をもつ蛍光試薬としてSBSeD−F及びSBThD−Fを合成し、それらのシステインに対する反応性及びその誘導体の蛍光特性を調べた。
【0071】
最大励起(λex)及び発光波長(λex)、及び誘導体の保持時間を表5に示す。各システイン誘導体の質量数([M+H])は、SBSeD−F(m/z381.9)、SBThD−F(m/z334.0)及びSBD−F(m/z318.0)について、理論値(各々382.0、334.0及び318.0)と一致した。誘導体の最大励起波長は、SBSeD−F(340nm)及びSBThD−F(315nm)はSBD−F(365nm)より短く、誘導体の最大発光波長は、SBSeD−F(542nm)は、SBSeD−F(517nm)及びSBD−F(514nm)よりも長かった。SBSeD−F自体は蛍光が少ないが、SBThD−Fは多少の蛍光を与えた(λex;350nm,λem:424nm)。SBSeD−F、SBThD−F及びSBD−Fのシステイン誘導体の逆相カラム(C18)に対するpH2.0の移動相による保持時間(tR)は、各々4.5、5.3及び4.8分であった。これから、SBSeD−Fは、これらの中で最も高い親水性の蛍光試薬であった。
【0072】
SBD−Fの場合、至適反応条件は60℃でpH9.5で1時間であるが、SBSeD−F及びSBThD−Fの反応性は、SBD−Fと比べて低く、蛍光強度は8−24時間で徐々に増加し(図9、10)、24時間後でも、反応は最大に達しなかった(図9)。pH10.0及び60℃では、SBSeD−F又はSBThD−Fとシステインの量的反応時間は、8時間以上であり、SBD−Fでは、1時間以内で完全に反応した(図10)。
このように、水溶性の蛍光試薬SBSeD−F及びSBThD−Fは、SBD−Fと比べて蛍光特性及び疎水性の点で異なっており、プロテオーム解析のための新規蛍光誘導体化試薬として有用である。
【実施例10】
【0073】
DAABD−Clによる線虫(C.elegans)タンパク質の誘導体化及び同定
(1)方法
線虫(Bristol N2株)を、大腸菌(E.coli)のOP50株を栄養源として20℃で、NGM寒天上に培養し、M9バッファーにより浮遊させてバクテリアから分離した。上記線虫をM9バッファーで2回洗った後、−80℃で保管して用いた。この線虫を等量の10mM CHAPSに懸濁し、超音波で溶解した。可溶性のフラクションを4℃で10,000rpm、5分の遠心分離により集めた。上澄を可溶性フラクションとして−20℃で保管した。このフラクションのタンパク質濃度をBSAを標準に用いるBradford methodで決定した。上澄の約20μL(100μgタンパク質)を同容量の2.5mM TCEP,17.5mM DAABD−Cl、10mM Na2 EDTA及び50mM CHAPSを6.0Mグアニジンを含む100mMホウ酸塩緩衝液(pH9.0)中で混合した。反応混合物を40℃で30分インキュベートした後、反応を200μLの0.1%ギ酸で停止し、次いで、反応混合物(10μgタンパク質)の30μLをHPLCシステムに注入した。
【0074】
RPカラムがPROTEIN(30nm孔径、250×4.6mm i.d.)(Imtakt)、移動相が溶離液(A)0.1%トリフルオロ酢酸及び溶離液(B)水/CH3CN/トリフルオロ酢酸(70/30/0.1)、グラジエントシステムが流速0.25mL/minで100分かけて30から70%Bの条件でHPLCを行った。蛍光検出は508nm、励起波長は387nmで行った。同定のために、蛍光タンパク質誘導体のいくつかのピークフラクションを分離し、減圧下に10μLに濃縮した。各フラクションは、2μg/mLトリプシン及び1.0mM塩化カルシウムを含む90μLの5.0mM重炭酸アンモニウム溶液(pH7.8)で希釈し、37℃で2時間インキュベートした。各タンパク質加水分解ペプチド混合物を直接エレクトロスプレーイオントラップ質量分析装置を用いたLC−MS/MSに供した。クロマトグラフィーは、HP1090シリーズIIシステム及びCadenza TC−18 column(12nmポーラスシリカ、100×2.0mm i.d.)のカラムを用いて実施した。移動相は溶離液(A)1.0mMギ酸アンモニウム及び溶離液(B)1.0mMギ酸アンモニウム/CH3CN(50/50)とした。グラジエント溶出は、流速0.2mL/minで60分かけて0から100%で行った。タンパク質の同定は、システインのチオール残基に結合したDAABDを記憶するMASCOT(Matrix Science Ltd.,U.K.)データベースサーチアルゴリズムを用いてNCBInrデータベースより行った。
【0075】
(2)結果
図11は、DAABD−Clで誘導体化した、線虫の可溶性フラクションから得られたタンパク質(約10μg)のクロマトグラムを示す。本実施例では、タンパク質の分離、トリプシンによる加水分解及び任意に選択されたピークフラクションのLC−MS/MS同定により、10種類のタンパク質が同定された。
図中、1はリボゾームタンパク質S3a(MW=28942)、2はカルレティキュリン(calreticulin)前駆体(MW=45588)、3はリボゾームタンパク質L1(MW=38635)、4は伸長因子(elongation factor)1−アルファ(MW=50636)、5はリンゴ酸デヒドロゲナーゼ(MW=35098)、6は40Sリボゾームタンパク質(MW=22044)、7はビテロゲニン(vitellogenin)(MW=193098)、8はアルギニンキナーゼ(MW=41969)、9はHSP−1熱ショック(heat shock)70kdタンパク質A(MW=69680)及び10はリボゾームタンパク質L7Ae(MW=13992)を示す。本実施例では、任意に選択された10種類のタンパク質が同定されたが、本発明では、同様にして、他のタンパク質の同定をすることが可能である。
【実施例11】
【0076】
7−クロロ−N−(2−ジメチルアミノプロピル)−2,1,3−ベンゾキサジアゾール−4−スルフォンアミド
4−クロロ−7−クロロスルフォニル−2,1,3−ベンゾキサジアゾール(0.25g、0.99mmol)をアセトニトリル(8ml)に溶解し、室温で撹拌した。次いで、これにN,N−ジエチルエチレンジアミン(0.21ml、1.49mmol)及びトリエチルアミン(0.21ml、1.49ml)を加えた。反応混合物を30分間撹拌し、次いで、減圧で濃縮した。残渣をクロロホルムに溶解し、飽和アンモニウムクロライド水溶液及び塩水で洗浄し、有機相をNa2SO4で乾燥し、減圧で濃縮した。残渣をシリカゲルクロマトグラフィー(10%メタノール/クロロホルム)で精製し、生成物を得た。得られた固体をメチレンクロライド及びジイソプロピルエステルから再結晶し、明るい黄色の小板状の生成物(0.22g、0.66mmol、67%)を得た。1H−NMR(CDCl3,500MHz)δ7.97(d,J=7.4Hz,1H),7.53(d,J=7.4Hz,1H),3.05(t,J=5.7Hz,2H),2.48(t,J=5.7Hz,2H),2.33(q,J=7.5Hz,4H),0.87(t,J=7.5Hz,6H);13C−NMR(CDCl3,125MHz)δ148.77,145.00,133.40,129.15,127.88,127.47,51.14,46.09,40.45,11.36;IR(KBr,cm−1)3446,3209,3101,2976,2817,1525,1347,1164.
【実施例12】
【0077】
4−クロロ−7−クロロスルフォニル−2,1,3−ベンゾキサジアゾール(0.21g、0.88mmol)をアセトニトリル(8ml)に溶解し、0℃で撹拌した。次いで、これにN,N−ジエチル−1,3−プロパンジアミン(0.20ml、1.25mmol)及びトリエチルアミン(0.17ml、1.25ml)を加えた。反応混合物を30分間撹拌し、次いで、減圧で濃縮した。残渣をクロロホルムに溶解し、飽和アンモニウムクロライド水溶液及び塩水で洗浄し、有機相をNa2SO4で乾燥し、減圧で濃縮した。残渣をシルカゲルクロマトグラフィー(10%メタノール/クロロホルム)で精製し、明るい黄色固体の生成物(0.10g、0.29mmol、35%)を得た。1H−NMR(CD3OD,500MHz)δ8.06(d,J=7.5Hz,1H),7.77(d,J=7.5Hz,1H),3.19(m,8H),1.93(m,2H),1.32(t,J=7.5Hz,6H);13C−NMR(CD3OD,125MHz)δ150.39,146.66,135.58,131.33,129.33,128.10,50.41,41.21,25.69,9.26;IR(KBr,cm−1)3501,3428,3209,2973,2733,2675,1524,1338,1160.
【実施例13】
【0078】
N,N−ジメチルエチレンジアミン−d6の合成
ジメチルアンモニウムクロライド−d6(2.46g)及びブロモアセトニトリル(3.37g)をジエチルエーテル(20ml)に溶解した。10℃でこれに50%NaOH(4.5g)を添加した後、混合物を同じ温度で2時間撹拌した。エーテル層を分離し、水層をエーテル(10ml×3)で抽出した。合体したエーテル層をMgSO4で乾燥し、次いで、減圧で濃縮してN,N−ジメチルアミノアセトニトリル−d6溶液(10g)を得て、次いで、それを10℃でテトラハイドロフラン(40ml)中のLiAlH4(1.28g)及びスルフォン酸(1.69g)の混合物に加えた。反応混合物を室温で13時間よく撹拌、混合した。これにエーテル(30ml)を加えた後、混合物をNaOH(水6mlに4g溶解)で処理した。エーテル層を分離し、水層をエーテル(10ml×2)で抽出した。合体したエーテル層をMgSO4で乾燥し、次いで、(5gになるまで)減圧濃縮した。残渣を希釈し、フラクション(70−80℃)を合わせて、1.79g(収率52.5%)のN,N−ジメチルエチレンジアミン−d6(77.4% THF溶液)を得た。1H−NMR(CDCl3):δ1.82−1.88(1.54H,m for THF,4H),2.33(2H,t,J=6Hz),2.77(2H,t,J=6.4Hz),3.72−3.76(1.48H,m for THF,4H)
【実施例14】
【0079】
7−クロロ−4−(ジメチルアミノエチルアミノスルフォニル)−2,1,3−ベンゾキサジアゾール−d6(DAABD−Cl−d6)の合成
4−クロロ−7−クロロスルフォニル−2,1,3−ベンゾキサジアゾール(3.28g)をCH3CN(60ml)に溶解した。この溶液に、N,N−ジメチルエチレンジアミン−d6(1.0g)及びトリエチルアミン(1.92ml)を各々加えて、氷で冷却して、同じ温度に1時間保持し、室温で1.5時間撹拌、混合した。次いで、反応混合を減圧濃縮し、残渣をAcOEtに溶解した。エチルアセテート溶液を飽和NaHCO3、蒸留水、飽和NaCl溶液で各々洗浄し、MgSO4で乾燥した。ろ過した溶液を減圧濃縮し、残渣をCH2Cl2:MeOH(50:1)を溶出液としてシリカゲルカラムで精製した。溶出した溶液を減圧乾燥し、残渣をEtOH−AcOEtで再結晶して1.36g(収率41.3%)のDAABD−Cl−d6:mp、110℃を得た。1H−NMR(CDCl3):δ7.99(1H,d,J=7.3Hz),7.54(1H,d,J=7.3Hz),3.13(2H,m),2.33(2H,m),ESI−MS:m/z311(M+H)+,IR(KBr)cm−1;1342and1166.
【実施例15】
【0080】
7−フルオロ−N−[2−(ジメチルアミノ)エチル]−2,1,3−ベンゾセレナジアゾール−4−スルフォンアミド(DAABSeD−F)の合成
7−フルオロ−2,1,3−ベンゾセレナジアゾール−4−スルフォニルクロライド(75mg)を3mlのアセトニトリルに溶解した。これに、N,N−ジメチルエチレンジアミン−(22mg)及びトリエチルアミン(35μl)を加えた後、混合物を氷の上で30分間撹拌した。反応混合を減圧で関そうし、残渣をCH2Cl2に溶解し,CH2Cl2−CH3OH(93:7)を用いてシリカゲルクロマトグラフィーに供し、7−フルオロ−N−[2−(ジメチルアミノ)エチル]−2,1,3−ベンゾセレナジアゾール−4−スルフォンアミド(49mg、56%)を赤い粉末として生成した。mp:117−120℃。1H−NMRδH8.09(1H,d,J=7.5Hz),7.24(1H,d,J=7.5Hz),2.93(2H,t,J=6.7Hz),2.27(2H,t,J=6.7Hz),1.98(6H,s)in CD3OD;ESI−MSm/z353[(M+H)+]
【実施例16】
【0081】
(1)DAABSeD−Fによる低分子量チオールの誘導体化反応及び誘導体の蛍光特性
1mM(システイン、ホモシステイン、アラニン、セリン、又はグルタチオン)溶液の20μLを、同容量の2.5mMTCEP、17.5mMDAANSeD−F、10mMNa2EDTA及び50mMCHAPSと混合した。各試薬は、6.0Mグアニジンを含む100mMホウ酸バッファーに溶解した。反応混合物を40℃で10−120分間加熱し、反応を0.1%トリフルオロ酢酸の200μLで停止した。混合溶液を300μLのメタノールで希釈し、蛍光スペクトルを蛍光スペクトロメーターで測定した。その結果、最大励起波長は約420nm、最大蛍光波長は約540nmであった。
【0082】
(2)低分子量チオールのDAABSeD−誘導体の分離及び同定
0.1%トリフルオロ酢酸で反応を停止した上記反応混合物の10μLをHPLCシステムに供した。HPLCシステムは、ポンプ(L−7100、日立社製)、分離カラムTSKgel120−TQA(内径250×4.6mm)(東ソー社製)及び蛍光検出器(FL−2025、ジャスコ社製)で構成した。蛍光検出は540nmで、励起波長は420nmで行った。移動相は、150mMリン酸バッファー/CH3CN(94/6)であり、流速は0.75ml/分とした。
同定のために、反応混合物をHP1090シリーズIIシステム(Hewlett−Packard(GmbH)のカラムに直接注入して、エレクトロスプレー法によるMS及びMS/MSスペクトルを得た。クロマトグラフィーは、Cadenza TC−18カラム(シリカの12nm孔、内径100×2.0mm)(Imtact(株))で行った。移動相は、0.1%ギ酸の溶出液(A)及び水/CH3CN/ギ酸(50/50/0.1)の溶出液(B)で構成した。0から100%Bで流速0.2ml/分で15分間グラジエント溶出を行った。このとき得られたクロマトグラムは、システイン、ホモシステイン、グルタチオンの3つのピークを与えた。
【0083】
(3)DAABSeD−Fによるペプチド及びタンパク質の誘導体化反応
100μMのペプチド及びタンパク質(インシュリン、トリプシン阻害剤、α−酸グリコプロテイン、カルボニックアンヒドラーゼ、α−ラクトアルブミン)溶液又は10μM BSA溶液の20μLを同容量の2.5mM TCEP、17.5mM DAABD−Cl(比較として)、10mM Na2EDTA及び50mM CHAPSと混合した。各反応混合物を6.0Mグアニジンを含む100mMホウ酸バッファー(pH9.0)に溶解した。各反応混合物を40℃で60分間(DAABSeD−F)又は20分間(DAABD−Cl)加熱し、反応を0.1%トリフルオロ酢酸の200μLで停止した。混合物をメタノール300μLで希釈し、蛍光スペクトルを蛍光スペクトロメーターで測定した。その結果、最大励起波長は約425nm、最大蛍光波長は約535nmであった。
【0084】
(4)ペプチド及びタンパク質のDAABSeD誘導体の分離
メタノールを除いた上記反応混合物の55μLをHPLCカラムに供した。HPLCシステムは、ポンプ(L−7100、日立社製)、タンパク質用のRPカラム(30nm孔サイズ、内径250×4.6mm)(Imtact社製)及び蛍光検出器(FL−2025、ジャスコ社製)で構成した。蛍光検出は、550nm(励起波長450nm)(DAABSeD−F)又は490nm(励起波長370nm)(DAABD−Cl)で行った。溶出液(A)は、水/CH3CN/トリフルオロ酢酸(90/10/0.1)からなり、溶出液(B)は、水/CH3CN/トリフルオロ酢酸(30/70/0.1)で構成した。0から100%Bで流速0.5mL/分で200分間のグラジエント溶出を行った。
【0085】
(5)異なる波長でのタンパク質誘導体のHPLC分離・定量及びタンパク質の同定
α−ラクトアルブミンを含み、10mMEDTA、50mMCHAPS、2.5mMTCEP、6.0Mグアニジンを含む混合溶液を2つの部分に分け、試料Aには1μMのα−ラクトアルブミン及び試料Bには2μMのα−ラクトアルブミンを含むように調整した。試料AはDAABD−Clと40℃で20分間反応させ、試料BはDAABSeD−Fと40℃で30分間反応させ、両者の反応溶液を再度混合した。混合物を2つの蛍光検出器に直列に連結したHPLCに供した。一つは、370nmで励起して蛍光波長490nmでDAABD誘導体をモニターし、もう一つは、450nmで励起して蛍光波長550nmでDAABSeD誘導体をモニターした。HPLCシステムは上記(4)と同じである。
【0086】
タンパク質の同定のために、試料A中のα−ラクトアルブミンのDAABD誘導体及び試料B中のα−ラクトアルブミンのDAABSeD誘導体に相当するピークフラクションを合体し、pH7.8に調整し、トリプシンで加水分解した。次いで、得られたペプチド混合物を窒素ガスで濃縮し、タンパク質同定アルゴリズムを備えた、シスラインのチオール基に結合したDAABD(MW、3900)又はDAABSeD(MW、4539)を記憶しているMASCOTを有するHPLC−MS/MSに供した。クロマトグラフィーは、Agilent1100シリーズシステム(Agilent Technologies社製)及び分離カラム、Cadenza TC−18カラム(シリカの12nm孔、内径100×2.0mm)(Imtact社製)、0.1%ギ酸の溶出液(A)及び水/CH3CN/キ酸(50/50/0.1)の溶出液(B)で構成した。0から100%Bで流速0.2mL/分で60分間のグラジエント溶出を行った。その結果、実施例10と同様にα−ラクトアルブミン(MW16228)が同定できた。
【実施例17】
【0087】
(1)DAABSeD−Fの合成
前述のように、ベンゾキサジアゾール試薬の、7−クロロ−N−[2−(ジメチルアミノ)エチル]−2,1,3−ベンゾキサジアゾール−4−スルフォンアミド(DAABD−Cl)は、感度のよい、選択的なチオール特異的試薬であり、プロテオミクス研究に有用である。DAABD−Clは、ペプチド及びタンパク質のチオールと反応して誘導体化し、390nmの励起で502nmで蛍光を発する。本実施例では、図12の合成経路により、7−フルオロ−N−[2−(ジメチルアミノ)エチル]−2,1,3−ベンゾセレナジアゾール−4−スルフォンアミド(DAABSeD−F)をDDABD−Clのカウンターパートとして合成した。
【0088】
本発明者は、7−フルオロ−2,1,3−ベンゾセレナジアゾール−4−スルフォネート(SBSeD−F)及び7−フルオロ−2,1,3−ベンゾチアジアゾール−4−スルフォネート(SBThD−F)をチオール特異的蛍光試薬として合成した。これらのシステインとの誘導体の蛍光波長は、各340nm及び315nmの励起波長で、それぞれ542nm及び517nmであった。対応するベンゾキサジアゾール試薬の、7−フルオロ−2,1,3−ベンゾキサジアゾール−4−スルフォネート(SBD−F)は、365nmの励起で514nmで蛍光を発する誘導体を与える。SBSeD−FとSBD−Fでの誘導体の最大波長の大きな相違にヒントを得て、本発明者は、ベンゾセレナジアゾール試薬、DAABSeD−FをDAABD−Clのカウンターパートとして合成した(図12)。
【0089】
(2)チオールに対するDAABSeD−Fの誘導体化反応の反応条件及び反応性
DAABSeD−Fによる低分子量チオール(システイン、ホモシステイン、グルタチオン、アラニン、セリン及びトリプシン)の誘導体化反応を40℃(pH9.0)で行った。ペプチド及びタンパク質(インシュリン、α1−酸グリコタンパク質、トリプシン阻害剤、カルボニックアンヒドラーゼ、α−ラクトアルブミン、BSA等)の場合、反応を促進するために、CHAPS及びグアニジンの存在下で行った。予測したように、DAABSeD−Fは、アラリン(アミノ及びカルボキシル基を含む)セリン(アミノ、カルボキシル及び水酸基を含む)及びチロシン(アミノ、カルボキシル、フェノール性水酸基を含む)のようなチオールを含まない化合物との反応では蛍光を示さなかった。これに対して、チオール化合物の場合には、蛍光を発し、その強度は30分で最大に達したが、これは、DAABSeD−Fはペプチド及びタンパク質と同様に低分子量のチオールと特異的に反応し、蛍光を生じ、反応速度は、DAABD−Clと同様であることを示している。
【0090】
(3)DAABD−Fと比較したDAABSeD−F誘導体の蛍光特性
DAABSeD誘導体は、最大励起及び蛍光波長が、それぞれ423−429nm及び524−542nmの範囲にあることを示した(表5)。
【0091】
【表5】
【0092】
(4)LC−MSによるチオールのDAABSeD誘導体の同定及び検出
DAABSeD−Fで誘導体化したチオール混合物をエレクトロスプレーイオントラップ質量分析計を備えたHPLCに供した。各チオールについて単一ピークが得られた。マススペクトルから、各低分子量チオールのDAABSeD誘導体が、システイン(MW=121)、ホモシステイン(MW=135)及びグルタチオン誘導体(MW=307)について、ベースイオンピークがm/z=454(M+H)+、468(M+H)+及び640(M+H)+として検出された。本実験では、用いたエレクトロスプレー法による分子イオン検出の限界(MWが3000以下)のため、タンパク質のDAASeD誘導体の同定は困難であった。
【0093】
(5)DAABSeDで誘導体化したペプチド及びタンパク質の検出限界
DAABSeD−Fで誘導体化したタンパク質から得られるクロマトグラムは、各誘導体の単一ピークを示し、重なるピークは観察されなかった。試薬自体は蛍光を発しないので、DAABSeD−Fピークは現れなかった。上述の誘導体のピーク高さは、注入量に比例した。バソプレシン、α−ラクトアルブミン及びBSAの検出限界は、それぞれ、7.5、7.2、及び7.2fmolであった。これらの検出限界はDAABD誘導体のものと同様であった。DAABD誘導体に比較して、DAABSeD誘導体は早く溶出したので、DAABSeD誘導体は、DAABD誘導体よりも、HPLC固定相に対する低い親和性を有していた。同様の傾向は、SBD誘導体と比べて、SBSeD誘導体についても観察された。
【0094】
(6)DAABSeD及びDAABD誘導体の同時検出
DAABD及びDAABSeD誘導体を併用した、二つの誘導体の同時蛍光検出は、同一起源の異なる歴史を有する二つのタンパク質試料の比較を簡便にする。例えば、一方が病態患者試料で一方が健常者からの試料、あるいは、二つの細胞又は組織試料で、一方がある薬剤で処理され、他方が未処理のものが例示される。本実験では、試料A中のα−ラクトアルブミンのDAABD誘導体の反応溶液と、試料B中のα−ラクトアルブミンのDAASeBD誘導体の反応溶液を合体し、二つの蛍光検出器に連結したHPLCに供した。それぞれの検出波長は、DAABD誘導体では370nmの励起で490nmで行い、DAASeBD誘導体では、450nmの励起で550nmで行なった。二つの誘導体は、クロマトグラムで各単一ピークを与えた。α−ラクトアルブミンのDAABD誘導体及びα−ラクトアルブミンのDAASeBD誘導体の保持時間は、53.3分、及び50.0分であった。
【0095】
試料A中のα−ラクトアルブミンのDAABD誘導体は、観察されたが、試料B中のα−ラクトアルブミンのDAABSeD誘導体は観察されなかった。試料B中のα−ラクトアルブミンのDAABSeD誘導体は観察され、試料A中のα−ラクトアルブミンのDAABD誘導体は観察されなかった。したがって、この方法で、試料A中に存在するα−ラクトアルブミンと試料B中に存在するα−ラクトアルブミンを区別することができ、同じ試料中のそれらの量を計測し、比較することができる。
【0096】
分離された相当するピークフラクションを合体した混合物をトリプシンのような酵素で処理することで、タンパク質の定量及び同定が可能であった。例えば、試料A中のDAABD誘導体及び試料B中のα−ラクトアルブミンのDAABSeDに相当する各ピークを合体し、トリプシンで分解し、得られたペプチド混合物を、2つの蛍光検出器に直結したHPLCにて分離・定量するとそのタンパク質(α−ラクトアルブミン)の定量が可能であり、更に得られたペプチド混合物をシステインのチオール残基に結合したDAABD又はDAABSeDを記憶している、タンパク質の同定アルゴリズム、MASCOTを備えた、HPLC−MS/MSに供した。その結果、実施例10と同様にα−ラクトアルブミン(MW16228)が同定された。
【0097】
本実験では、異なった試料A及びB中のタンパク質α−ラクトアルブミンを取り上げ、比較した。これに対して、一つの試料中に多くの化合物が存在する実際のバイオ試料の、誘導化したタンパク質溶液を合体した混合物は、複雑なHPLCクロマトグラムのパターンを与えるので、クロマトグラムでタンパク質ピークを区別することは非常に困難になる。しかし、標的タンパク質の標準品があり、異なったクロマトフラフィー条件で二次元クロマトグラフィーを行なえば、二つのクロマトグラフィー上のタンパク質の二つの誘導体の保持時間を知ることができるので、クロマトグラム上でDAABD−Cl及びDAABSeD−Fの各誘導体のピーク分画を分離した後、これらを合体したピークフラクションを第2のクロマトグラフィーで再クロマトすることにより、精製されたタンパク質の各誘導体が定量できる。更に、ピーク分画を上記の酵素処理、蛍光検出HPLC又はLC−質量分析計に付し、異なった試料中のタンパク質の定量又は同定ができる。
【産業上の利用可能性】
【0098】
以上詳述したように、本発明は、微量の発現タンパク質及び/又はペプチドの検出・分離・同定方法及びそのシステムに係るものであり、本発明により、遺伝子を介して発現する発現タンパク質及び/又はペプチドを簡便な方法及び手段で、高感度に検出・分離・同定することができる。本発明の方法により、従来法では検出できなかった微量の発現タンパク質及び/又はペプチドを短時間で、感度良く検出・分離・同定することができる。また、上記検出・分離・同定方法に使用する微量の発現タンパク質及び/又はペプチドの微量検出・分離・同定システムを提供することができる。本発明は、プロテオームのプラットフォーム技術を提供するものとして有用である。
【図面の簡単な説明】
【0099】
【図1】本発明の方法の操作工程の一例を示す。
【図2】界面活性剤の種類と蛍光誘導体の生成の度合いとの関係を示す。
【図3】本発明の方法により試験した蛍光誘導体タンパク質/ペプチドのそれぞれの蛍光ピークを示す。
【図4】酵素水解物の蛍光クロマトグラム(A)、及びマスクロマトグラム(B)を示す。
【図5】MS/MSによるマススペクトルを示す。
【図6】実施例3における逆相クロマトグラフィー(RPLC)によるクロマトグラムを示す。
【図7】蛍光誘導体化の反応時間と蛍光強度との関係を示す(左図:DAABD−Cl,右図:TAABD−Cl)。
【図8】TAABD−Clとの反応時間と蛍光強度との関係を示す。
【図9】新規蛍光試薬によるシステインの蛍光誘導体化(pH9.0)の反応時間とピーク領域との関係を示す。
【図10】新規蛍光試薬によるシステインの蛍光誘導体化(pH10.0)の反応時間とピーク領域との関係を示す。
【図11】DAABD−Clで誘導体化した、線虫(C.elegans)の可溶性フラクションが得られたタンパク質(約10μg)のクロマトグラムを示す。
【図12】DAABSeD−Xの合成経路を示す。
【特許請求の範囲】
【請求項1】
被験試料中の発現微量タンパク質及び/又はペプチドを高感度に検出・分離・同定する方法であって、被験試料中のタンパク質及び/又はペプチドを蛍光誘導体とした後、これを蛍光検出により分離し、その蛍光画分を質量分析に付するか、又はその蛍光画分を酵素水解に付し、そのペプチド断片を分離し、その画分を質量分析に付し、データベース照合、構造解析に供して発現タンパク質及び/又はペプチドの同定を行うことを特徴とする上記発現タンパク質及び/又はペプチドの検出・分離・同定方法。
【請求項2】
被験試料中のタンパク質及び/又はペプチドを蛍光誘導体とした後、HPLCに付し、その蛍光分画を捕集した後、酵素水解に付し、その蛍光標識フラグメント及び非蛍光標識フラグメントを質量分析又はMS/MS分析して得られた各フラグメントのイオン分子量情報をタンパク質及び/又はペプチドフラグメントデータベースと照合し、構造解析する、請求項1に記載の方法。
【請求項3】
(1)被験試料中のタンパク質及び/又はペプチドを蛍光試薬で標識する、(2)それを1次元又は2次元のHPLC/蛍光検出により、その蛍光分画を捕集する、(3)上記蛍光分画を酵素水解に付する、(4)それを第二段階のHPLC/蛍光検出により、その蛍光クロマトグラムを得ると共に、その全ピークを質量分析に付し、データベース照合、構造解析に供する、請求項1に記載の方法。
【請求項4】
タンパク質及び/又はペプチド試料の水溶液に、官能基特異的蛍光試薬を加え、場合により、界面活性剤及び/又はタンパク変性剤を加え、タンパク質及び/又はペプチドを蛍光標識する、請求項1から3のいずれかに記載の方法。
【請求項5】
蛍光標識したタンパク質及び/又はペプチド試料を蛍光検出器付きイオン交換カラムHPLC、逆相分配HPLC、ゲル濾過HPLC、又は電気泳動に代表される分離手段に付し、蛍光をモニターしながらそのピーク分画を捕集する、請求項1から3のいずれかに記載の方法。
【請求項6】
蛍光分画を、各種ペプチダーゼ、トリプシン、キモトリプシンに代表されるタンパク質分解酵素を用いて酵素水解する、請求項1から3のいずれかに記載の方法。
【請求項7】
酵素水解物を蛍光検出器付き逆相HPLCに付し、蛍光ピークを検出すると共に、蛍光標識フラグメント及び蛍光非標識フラグメントの質量分析又はMS/MS分析を行う、請求項1から3のいずれかに記載の方法。
【請求項8】
質量分析又はMS/MS分析に付して得られた各フラグメントのイオン分子量情報を、コンピューターによるタンパク質及び/又はペブチドフラグメントデータベースと照合し、構造解析して、酵素水解以前のタンパク質及び/又はペプチドの同定を行う、請求項1から3のいずれかに記載の方法。
【請求項9】
被験試料が、生体試料から採取したタンパク質及び/又はペプチド試料である、請求項1から3のいずれかに記載の方法。
【請求項10】
タンパク質及び/又はペプチドフラグメント情報、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを用いてデータベース照合する、請求項1から3のいずれかに記載の方法。
【請求項11】
請求項1から10のいずれかに記載の方法に使用する発現微量タンパク質及び/又はペプチド検出・分離・同定システムであって、被験試料のタンパク質及び/又はペプチドを蛍光試薬で標識するための第一反応器、蛍光試薬で標識した蛍光誘導体を蛍光分画するための1次元又は2次元の蛍光検出器付きHPLC、蛍光分画を酵素水解するための第二反応器、酵素水解物の蛍光標識フラグメントを蛍光検出するための第二段階の蛍光検出器付きHPLC、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを搭載した構造解析装置の1種又は2種以上を構成要素として含むことを特徴とする上記検出・分離・同定システム。
【請求項12】
上記第一反応器、1次元又は2次元の蛍光検出器付きHPLC、第二反応器、第二段階の蛍光検出器付きHPLCを直列に配置してなる、請求項11に記載のシステム。
【請求項13】
被験試料中のタンパク質及び/又はペプチドを、蛍光誘導体化試薬として、下記の化1の一般式(1)
【化1】
〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、−NHR′(但、R′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキル)、又は−NR′′R′′′(但し、R′′はアルキル、R′′′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキルを示す。〕で表わされる化合物、又はその同位体化合物又は下記の化2の一般式(2)
【化2】
(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物又はその同位体化合物、を用いて、蛍誘導体とする、請求項1に記載の方法。
【請求項14】
請求項1に記載の方法でタンパク質及び/又はペプチドを蛍光誘導化するために使用する蛍光誘導体化試薬であって、下記の化3の一般式(1)
【化3】
〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、−NHR′(但、R′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキル)、又は−NR′′R′′′(但し、R′′はアルキル、R′′′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキルを示す。〕で表わされる化合物、又はその同位体化合物又は下記の化2の一般式(2)
【化4】
(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物又はその同位体化合物、を用いて、蛍誘導体とする、請求項1に記載の方法。
【請求項15】
被検試料のタンパク質及び/又はペプチドを蛍光誘導体化後、HPLCで分離・検出し、分画後、酵素水解し、この水解物を直接質量分析で配列分析とタンパク質の同定を行なうことを特徴とするタンパク質及び/又はペプチドの検出・分離・同定方法。
【請求項16】
被検試料として、異なる試料A中及び試料B中のタンパク質及び/又はペプチドを、それぞれ蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬でそれぞれ誘導体化後、蛍光検出器付きHPLCで分離・検出し、分画後、各蛍光ピークをそのまま又は合体して定量に供する、及び/又は各蛍光ピークを合体して酵素水解に供し、この水解物を定量に供する、又はこの水解物をHPLC−質量分析に供し、同定を行なう、ことを特徴とするタンパク質及び/又はペプチドの検出・分離・同定方法。
【請求項17】
各蛍光ピークをそのまま又は合体してHPLCによる定量に供し、試料A中及び試料B中のタンパク質及び/又はペプチドの各誘導体の比率を算出する請求項16に記載の方法。
【請求項18】
水解物をHPLCによる定量に供し、試料A中及び試料B中のタンパク質及び/又はペプチドの各誘導体の比率を算出する請求項16に記載の方法。
【請求項19】
試料A中及び試料B中のタンパク質及び/又はペプチドの第1の蛍光誘導体化試薬との反応物及び第2の蛍光誘導体化試薬との反応物を合体し、2つの励起・蛍光検出の可能なHPLCに供し、分画後、各蛍光ピークを合体して酵素水解に供し、この水解物をHPLC−質量分析に供し、同定を行う請求項16に記載の方法。
【請求項20】
試料A、Bが、2種類の細胞又は組織又は体液試料である請求項16に記載の方法。
【請求項21】
蛍光誘導体化試薬として、DAABD−X、DAASeBD−X、及びDAAThBD−X(但し、XはCl又はF)のうちの励起・蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬でタンパク質及び/又はペプチドを誘導体化する請求項16に記載の方法。
【請求項22】
蛍光波長の異なる蛍光誘導体化試薬として、DAABD−X、DAASeBD−X、又はDAAThBD−X(但し、XはCl又はF)と、それらの各同位体を組み合わせて使用する請求項21に記載の方法。
【請求項23】
酵素水解した試料を直接質量分析に付しペプチドマップを得ると同時に、蛍光試薬の有する骨格並びに電荷を活用して、蛍光標識化ペプチド断片を質量分析測定部で抽出してシステイン含有ペプチド部分の構造を取得し、これらを基にタンパク質及び/又はペプチドの同定を行なう請求項16に記載の方法。
【請求項24】
蛍光誘導体化試薬で誘導体化したタンパク質及び/又はペプチドを分解することなく分画することが可能な、少なくともミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、及びミクロ自動注入装置を具備したことを特徴とする自動分画装置。
【請求項25】
少なくとも、ミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、酵素反応装置、及びミクロ自動注入装置を備え、任意に、質量分析(MS)システムを具備したことを特徴とする微量タンパク質の高性能・簡易定量・同定解析装置。
【請求項1】
被験試料中の発現微量タンパク質及び/又はペプチドを高感度に検出・分離・同定する方法であって、被験試料中のタンパク質及び/又はペプチドを蛍光誘導体とした後、これを蛍光検出により分離し、その蛍光画分を質量分析に付するか、又はその蛍光画分を酵素水解に付し、そのペプチド断片を分離し、その画分を質量分析に付し、データベース照合、構造解析に供して発現タンパク質及び/又はペプチドの同定を行うことを特徴とする上記発現タンパク質及び/又はペプチドの検出・分離・同定方法。
【請求項2】
被験試料中のタンパク質及び/又はペプチドを蛍光誘導体とした後、HPLCに付し、その蛍光分画を捕集した後、酵素水解に付し、その蛍光標識フラグメント及び非蛍光標識フラグメントを質量分析又はMS/MS分析して得られた各フラグメントのイオン分子量情報をタンパク質及び/又はペプチドフラグメントデータベースと照合し、構造解析する、請求項1に記載の方法。
【請求項3】
(1)被験試料中のタンパク質及び/又はペプチドを蛍光試薬で標識する、(2)それを1次元又は2次元のHPLC/蛍光検出により、その蛍光分画を捕集する、(3)上記蛍光分画を酵素水解に付する、(4)それを第二段階のHPLC/蛍光検出により、その蛍光クロマトグラムを得ると共に、その全ピークを質量分析に付し、データベース照合、構造解析に供する、請求項1に記載の方法。
【請求項4】
タンパク質及び/又はペプチド試料の水溶液に、官能基特異的蛍光試薬を加え、場合により、界面活性剤及び/又はタンパク変性剤を加え、タンパク質及び/又はペプチドを蛍光標識する、請求項1から3のいずれかに記載の方法。
【請求項5】
蛍光標識したタンパク質及び/又はペプチド試料を蛍光検出器付きイオン交換カラムHPLC、逆相分配HPLC、ゲル濾過HPLC、又は電気泳動に代表される分離手段に付し、蛍光をモニターしながらそのピーク分画を捕集する、請求項1から3のいずれかに記載の方法。
【請求項6】
蛍光分画を、各種ペプチダーゼ、トリプシン、キモトリプシンに代表されるタンパク質分解酵素を用いて酵素水解する、請求項1から3のいずれかに記載の方法。
【請求項7】
酵素水解物を蛍光検出器付き逆相HPLCに付し、蛍光ピークを検出すると共に、蛍光標識フラグメント及び蛍光非標識フラグメントの質量分析又はMS/MS分析を行う、請求項1から3のいずれかに記載の方法。
【請求項8】
質量分析又はMS/MS分析に付して得られた各フラグメントのイオン分子量情報を、コンピューターによるタンパク質及び/又はペブチドフラグメントデータベースと照合し、構造解析して、酵素水解以前のタンパク質及び/又はペプチドの同定を行う、請求項1から3のいずれかに記載の方法。
【請求項9】
被験試料が、生体試料から採取したタンパク質及び/又はペプチド試料である、請求項1から3のいずれかに記載の方法。
【請求項10】
タンパク質及び/又はペプチドフラグメント情報、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを用いてデータベース照合する、請求項1から3のいずれかに記載の方法。
【請求項11】
請求項1から10のいずれかに記載の方法に使用する発現微量タンパク質及び/又はペプチド検出・分離・同定システムであって、被験試料のタンパク質及び/又はペプチドを蛍光試薬で標識するための第一反応器、蛍光試薬で標識した蛍光誘導体を蛍光分画するための1次元又は2次元の蛍光検出器付きHPLC、蛍光分画を酵素水解するための第二反応器、酵素水解物の蛍光標識フラグメントを蛍光検出するための第二段階の蛍光検出器付きHPLC、及び蛍光試薬で標識したアミノ酸の情報を含んだデータベースを搭載した構造解析装置の1種又は2種以上を構成要素として含むことを特徴とする上記検出・分離・同定システム。
【請求項12】
上記第一反応器、1次元又は2次元の蛍光検出器付きHPLC、第二反応器、第二段階の蛍光検出器付きHPLCを直列に配置してなる、請求項11に記載のシステム。
【請求項13】
被験試料中のタンパク質及び/又はペプチドを、蛍光誘導体化試薬として、下記の化1の一般式(1)
【化1】
〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、−NHR′(但、R′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキル)、又は−NR′′R′′′(但し、R′′はアルキル、R′′′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキルを示す。〕で表わされる化合物、又はその同位体化合物又は下記の化2の一般式(2)
【化2】
(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物又はその同位体化合物、を用いて、蛍誘導体とする、請求項1に記載の方法。
【請求項14】
請求項1に記載の方法でタンパク質及び/又はペプチドを蛍光誘導化するために使用する蛍光誘導体化試薬であって、下記の化3の一般式(1)
【化3】
〔式中、Xは、ハロゲン、Yは、O、Se又はS、Rは、−NH2、−NHR′(但、R′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキル)、又は−NR′′R′′′(但し、R′′はアルキル、R′′′はアルキル置換Nアルキル、ジアルキル置換Nアルキル又はトリアルキル置換Nアルキルを示す。〕で表わされる化合物、又はその同位体化合物又は下記の化2の一般式(2)
【化4】
(式中、Xは、ハロゲン、Yは、Se又はSを示す。)で表わされる化合物又はその同位体化合物、を用いて、蛍誘導体とする、請求項1に記載の方法。
【請求項15】
被検試料のタンパク質及び/又はペプチドを蛍光誘導体化後、HPLCで分離・検出し、分画後、酵素水解し、この水解物を直接質量分析で配列分析とタンパク質の同定を行なうことを特徴とするタンパク質及び/又はペプチドの検出・分離・同定方法。
【請求項16】
被検試料として、異なる試料A中及び試料B中のタンパク質及び/又はペプチドを、それぞれ蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬でそれぞれ誘導体化後、蛍光検出器付きHPLCで分離・検出し、分画後、各蛍光ピークをそのまま又は合体して定量に供する、及び/又は各蛍光ピークを合体して酵素水解に供し、この水解物を定量に供する、又はこの水解物をHPLC−質量分析に供し、同定を行なう、ことを特徴とするタンパク質及び/又はペプチドの検出・分離・同定方法。
【請求項17】
各蛍光ピークをそのまま又は合体してHPLCによる定量に供し、試料A中及び試料B中のタンパク質及び/又はペプチドの各誘導体の比率を算出する請求項16に記載の方法。
【請求項18】
水解物をHPLCによる定量に供し、試料A中及び試料B中のタンパク質及び/又はペプチドの各誘導体の比率を算出する請求項16に記載の方法。
【請求項19】
試料A中及び試料B中のタンパク質及び/又はペプチドの第1の蛍光誘導体化試薬との反応物及び第2の蛍光誘導体化試薬との反応物を合体し、2つの励起・蛍光検出の可能なHPLCに供し、分画後、各蛍光ピークを合体して酵素水解に供し、この水解物をHPLC−質量分析に供し、同定を行う請求項16に記載の方法。
【請求項20】
試料A、Bが、2種類の細胞又は組織又は体液試料である請求項16に記載の方法。
【請求項21】
蛍光誘導体化試薬として、DAABD−X、DAASeBD−X、及びDAAThBD−X(但し、XはCl又はF)のうちの励起・蛍光波長の異なる少なくとも2つの蛍光誘導体化試薬でタンパク質及び/又はペプチドを誘導体化する請求項16に記載の方法。
【請求項22】
蛍光波長の異なる蛍光誘導体化試薬として、DAABD−X、DAASeBD−X、又はDAAThBD−X(但し、XはCl又はF)と、それらの各同位体を組み合わせて使用する請求項21に記載の方法。
【請求項23】
酵素水解した試料を直接質量分析に付しペプチドマップを得ると同時に、蛍光試薬の有する骨格並びに電荷を活用して、蛍光標識化ペプチド断片を質量分析測定部で抽出してシステイン含有ペプチド部分の構造を取得し、これらを基にタンパク質及び/又はペプチドの同定を行なう請求項16に記載の方法。
【請求項24】
蛍光誘導体化試薬で誘導体化したタンパク質及び/又はペプチドを分解することなく分画することが可能な、少なくともミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、及びミクロ自動注入装置を具備したことを特徴とする自動分画装置。
【請求項25】
少なくとも、ミクロカラム−HPLC、ミクロ蛍光検出器、ミクロフラクションコレクター、酵素反応装置、及びミクロ自動注入装置を備え、任意に、質量分析(MS)システムを具備したことを特徴とする微量タンパク質の高性能・簡易定量・同定解析装置。
【図1】


【図2】
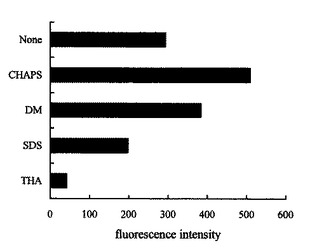
【図3】

【図4】
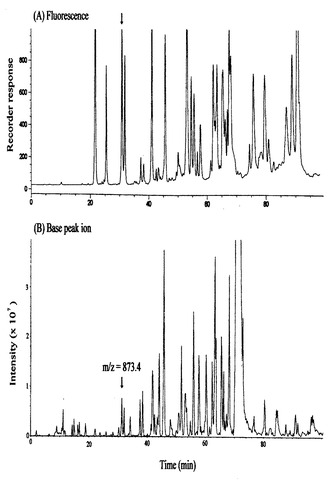
【図5】
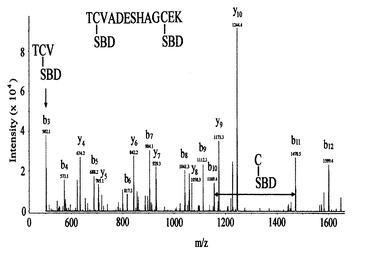
【図6】
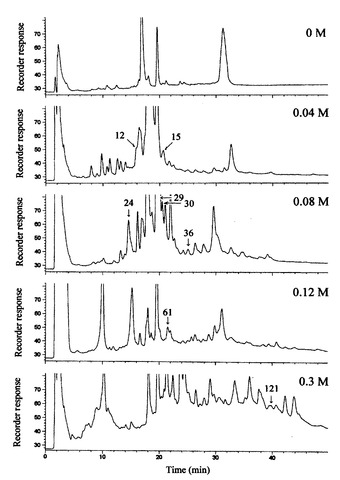
【図7】
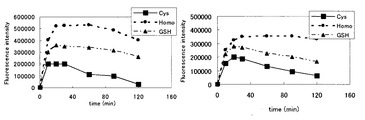
【図8】

【図9】
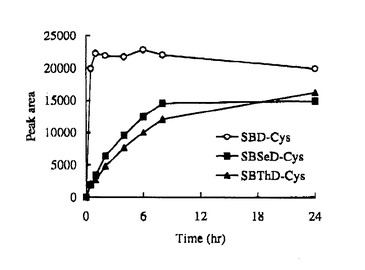
【図10】
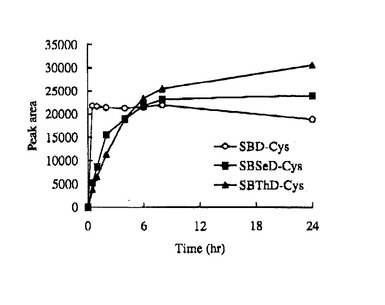
【図11】
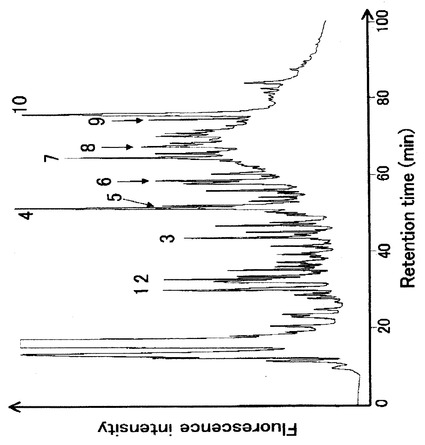
【図12】
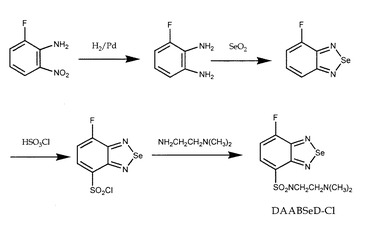
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【国際公開番号】WO2005/056146
【国際公開日】平成17年6月23日(2005.6.23)
【発行日】平成19年12月6日(2007.12.6)
【国際特許分類】
【出願番号】特願2005−516223(P2005−516223)
【国際出願番号】PCT/JP2004/018592
【国際出願日】平成16年12月13日(2004.12.13)
【出願人】(592191793)
【Fターム(参考)】
【国際公開日】平成17年6月23日(2005.6.23)
【発行日】平成19年12月6日(2007.12.6)
【国際特許分類】
【国際出願番号】PCT/JP2004/018592
【国際出願日】平成16年12月13日(2004.12.13)
【出願人】(592191793)
【Fターム(参考)】
[ Back to top ]