
皮下使用の高濃度免疫グロブリン製剤を生成する方法
本発明は、貯蔵された血漿から、皮下注射用の高濃縮免疫グロブリン組成物を調製する新規の改良された方法に関する。皮下注射用途に適した20%以上の免疫グロブリンを含む組成物も記載される。別の態様では、本発明は、血漿から濃縮されたIgG組成物を調製する方法を提供し、(1)限外濾過により血漿製剤中の蛋白質を5%(重量/体積)又はほぼその濃度に濃縮する工程と、(2)透析濾過により製剤中の蛋白質を20%(重量/体積)又はほぼその濃度に更に濃縮する工程とを含む改善を有する。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2009年5月27日に出願され、全ての目的のために本明細書に参照としてそのまま明確に組み込まれている、米国仮特許出願第61/181,606号の利益を請求する。
【背景技術】
【0002】
ヒト血漿からの免疫グロブリン製品は、1952年、免疫不全を治療するために最初に利用された。初期は、IgGの筋肉内投与又は皮下投与が、選択方法であった。しかしながら、様々な疾病に対する有効な治療に必要なIgGを多量に注入するために、IgG濃度の低い(50mg/mL)、静脈注射で投与可能な製品が開発された。通常、静脈注射用免疫グロブリン(IVIG)は、千人を超える血液提供者の血漿から貯留された免疫グロブリンG(IgG)(単数)、免疫グロブリン(複数)を含む。通常、Fc依存性作動因子機能が無傷である95%超過の非修飾IgGと微量の免疫グロブリンA(IgA)又は免疫グロブリンM(IgM)とを含むIVIGは、3つの主分類の病状:1.X連鎖無ガンマグロブリン血症、低ガンマグロブリン血症(原発性免疫不全)、後天性免疫不全疾患(続発性免疫不全)等の低抗体レベルを特徴とする免疫不全、2.炎症性及び自己免疫疾患、及び、3.急性感染症を治療するのに主に利用される、無菌の浄化されたIgG製品である。
【0003】
数多くのIVIGの市場供給者は、様々なIVIG製品を提供している。再溶出後の溶液中に50mg/mL蛋白質のみを含む、旧型の凍結乾燥されたIVIG製品と比較して、最新の開発品は、高度に浄化、濃縮されたヒトIgG抗体の100mg/mLの使用準備済みの無菌の液体製剤である。IVIG等のIgG製品は、貯蔵されたヒト血漿から製造されるので、提供者の血液からの病因汚染物(特に、ヒトに様々な疾病を引き起こすことで知られているウイルス)は、生産プロセスでの重大な懸念事項である。IgG製品についての別の重大な検討事項は、特に、使用準備済みの製剤としての、それら製品の貯蔵中の安定性である。IVIGと比べると、皮下投与可能な免疫グロブリン製剤は、家庭での治療処置が可能にし、副作用があまりないという利点がある。部位当たりの注入体積が少ないという不利な点をなくすために、IgGの濃度をより高くする(例えば、100mg/mLの代わりに200mg/mLを含む)ことは、明らかに有利になり得る。
【0004】
血清及び血漿蛋白質の調製及び特性に関して一連の影響力をもつ論文のうちの第4の連載記事において、コーンら(アメリカ化学学会誌、1946年、第68(3)号、459〜475頁)は、ヒト血漿からIgG濃縮成分を単離するのを可能にする、血漿蛋白質アルコール分別方法(方法6)を最初に記載している。数年後、オンクレイら(アメリカ化学学会誌、1949年、第71(2)号、541〜550頁)は、コーンの方法を拡張し、純粋なIgG製剤を単離することになる方法(方法9)を発表した。
【0005】
これらの方法は、血漿由来血液因子の全産業の基礎を築く一方で、川崎病、免疫血小板減少性紫斑病、及び原発性免疫不全を含む、幾つかの免疫関連疾患を治療するのに十分な高濃度のIgG製剤を与えることができなかった。そのように、高純度で高濃度のIgG製剤を提供するために、イオン交換クロマトグラフィー等の様々な技術を用いる追加の方法論が開発された。ホップら(ミュンヘン医事週報誌、1967年、(34)、1749〜1752頁)、ファルクスヴェーデン(スウェーデン特許、第348942号)、ファルクスヴェーデン及びルンドブラッド(血漿蛋白質分別方法、1980年)は、この目的のためにイオン交換クロマトグラフィーを先駆けて用いた。
【0006】
様々な最新の方法は、カプリル酸塩沈殿(レービングら、国際輸血学会誌、2003年、(84)、193〜201頁)、カラムクロマトグラフィーと組み合わせたコーン分画(I+)II+IIIエタノール沈殿(タナカら、ブラジル医療生体研究誌、2000年、(33)、37〜30頁)等の沈殿工程を用いる。最近では、テシュナーら(国際輸血学会誌、2007年、(92)、42〜55頁)は、貯蔵された血漿から寒冷沈殿物を除去し、次に、変更されたコーン・オンクレイ冷却エタノール分別、続いて、中間イオン交換クロマトグラフィーのS/D処理、ナノ濾過、及び任意選択で限外濾過/透析濾過を実行する、10%IVIG製品生成方法を記載している。
【発明の概要】
【発明が解決しようとする課題】
【0007】
しかしながら、これらのIgG製造方法によりもたらされる純度、安全性、及び産出量が向上しているにもかかわらず、皮下投与及び/又は筋肉内投与に適した高濃度のIgG製剤は、尚も必要とされている。本発明は、これらの他の必要性を充たし、安定で高度に浄化され、ウイルスに不活性で使用準備済みの、IgG濃度の高い製品を製造する方法を記載する。
【課題を解決するための手段】
【0008】
一態様では、本発明は、組成物1リットル当たり約180グラム超過の蛋白質を含み、少なくとも95%の蛋白質が、ヒトIgG等のIgGである、水性組成物を提供する。幾つかの実施形態では、組成物は、皮下投与及び筋肉内投与に適した製品を与えるプロセスにより生成され、10〜22%の蛋白質濃度範囲でどの濃度に調節されるのかに関係なく、ウイルスを不活性にするために最終容器内で高温で処理され得る。幾つかの事例では、組成物中の蛋白質濃度は、20%(重量/体積)であるか、又は、ほぼその濃度である。他の事例では、組成物は、約0.1〜0.3Mグリシンを更に含み得る。本発明の組成物は、pHが約3〜6又は約4〜6のように変化し得る。
【0009】
別の態様では、本発明は、血漿から濃縮されたIgG組成物を調製する方法を提供し、(1)限外濾過により血漿製剤中の蛋白質を5%(重量/体積)又はほぼその濃度に濃縮する工程と、(2)透析濾過により製剤中の蛋白質を20%(重量/体積)又はほぼその濃度に更に濃縮する工程とを含む改善を有する。組成物中の少なくとも95%の上記蛋白質は、ヒトIgG等のIgGである。幾つかの実施形態では、工程(1)は、公称分画分子量(NMWCO)が100kDa以下である限外濾過薄膜を利用して実行される。他の実施形態では、工程(2)は、グリシンの透析濾過緩衝液を用いて4.2±0.1のpHで実行される。幾つかの事例では、透析濾過緩衝体は、0.25Mグリシンを有し、pHが4.0である。幾つかの特定の実施形態では、工程(2)後の蛋白質濃度は、20%(重量/体積)よりも高く、続いて、透析濾過緩衝体で、20%(重量/体積)に又はほぼその濃度に調節される。
【0010】
別の態様では、本発明は、以下の工程を含む、血漿から濃縮IgG組成物を調製する方法を提供する:
(1)遠心分離により血漿から液体と沈殿物とを分離すること、
(2)事前に冷却されたエタノールを(1)からの液体と混合して、エタノール濃度が8%(体積/体積)又はほぼその値である混合物を作ること、
(3)遠心分離により、(2)の混合物から液体と沈殿物とを分離すること、
(4)(3)からの液体のpH及びエタノール濃度を、それぞれ、7.0及び20〜25%(体積/体積)又はそれらの値に調節することにより、混合物を作ること、
(5)遠心分離により、(4)の混合物から液体と沈殿物とを分離すること、
(6)(5)の沈殿物を緩衝液で、1〜15又はほぼそれらの値の重量比に再び懸濁して、懸濁液を作ること、
(7)二酸化シリコン(SiO2)を(6)からの懸濁液と混合し、濾過により濾液を得ること、
(8)洗浄剤と冷却アルコールを、(7)の濾液と混合し、遠心分離により沈殿物を得ること、
(9)溶媒又は洗浄剤を含む水溶液中に前記沈殿物を溶解し、その溶液を少なくとも60分にわたり維持すること、
(10)(9)の後の溶液を陽イオン交換クロマトグラフィーカラムに通し、カラムに吸収された蛋白質を溶出液中に溶出させること、
(11)(10)からの溶出液を陰イオン交換クロマトグラフィーカラムに通して、流出液を生成すること、
(12)流出液をナノ濾過器に通して、ナノ濾液を生成すること、
(13)ナノ濾液を限外濾過薄膜に通して、限外濾液を生成すること、
(14)限外濾液を透析濾過緩衝体液で透析濾過して、蛋白質濃度が20%(重量/体積)又はほぼその値の溶液を生成すること、並びに、
(15)前記溶液を0.2μm以下のフィルタに通して濾過することで(14)からの溶液を減菌することにより、濃縮されたIgG組成物を得ること。
【0011】
幾つかの実施形態では、工程(2)は、約−2〜0℃の温度で実行される、又は、工程(2)の混合物は、少なくとも15分にわたり混合され、次に、少なくとも2時間にわたり約−2〜0℃の温度に保たれる。幾つかの実施形態では、工程(4)は、少なくとも15分にわたり混合され、次に、少なくとも8時間にわたり、−7℃又はほぼその温度に保たれる。幾つかの実施形態では、工程(6)の懸濁液は、約40〜160分にわたり、約2℃〜8℃の温度で、5.0又はほぼその値のpHで攪拌される、又は、混合物は、約2〜8℃の温度で、少なくとも50分にわたり実行される。幾つかの実施形態では、工程(8)は、約−5〜−10℃の温度で実行される。幾つかの実施形態では、工程(9)の溶液は、1.0%(体積/体積)のトリトンX−100と、0.3%(体積/体積)のトゥイーン80と、0.3%(体積/体積)燐酸トリ−(n−ブチル)(TNBP)とを含む。幾つかの実施形態では、工程(9)の溶液は、約18〜25℃の温度で保持される。幾つかの実施形態では、工程(10)の陽イオン交換クロマトグラフィーカラムは、pHが5.5±0.1の10mMの酢酸塩緩衝液で洗浄され、pHが8.5±0.1、伝導率が5.0±0.2mS/cmの、35mM一塩基性燐酸ナトリウム、10mMトリス緩衝液で溶出される。幾つかの実施形態では、工程(10)の溶出液は、工程(11)の前に、6.4±0.2のpH、約1.5〜2.5mS/cmの伝導率に調節される。幾つかの実施形態では、工程(11)の流出液は、工程(12)の前に、0.2μm以下の孔径のフィルタを通る。幾つかの実施形態では、工程(13)の限外濾液は、蛋白質濃度が、5±1%(重量/体積)又はほぼその値である。他の実施形態では、工程(13)の限外濾過薄膜は、公称分画分子量(NMWCO)が、50kDa以下である。幾つかの実施形態では、工程(14)の血液濾過緩衝液は、pHが4.2±0.1の0.25Mグリシン溶液である。他の実施形態では、工程(14)からの溶液は、蛋白質濃度が20%(重量/体積)よりも大きく、続いて、血液透析緩衝液で、約20.4±0.4(重量/体積)又はほぼその濃度に調節される。幾つかの実施形態では、工程(13)及び(14)は、約2〜8℃の温度で実行される。他の実施形態では、調製方法は、容器を密封する前に、工程(15)の減菌された溶液を無菌状態下で容器内に分注する工程を更に含む。
【0012】
他の実施形態では、上記の方法は、密封された容器を約30〜32℃で約21〜22日にわたり保存する工程を更に含んでもよく、その方法は、20%IgG製品を10%の従来技術の静注製剤と同程度に安定させる調製工程を更に含んでもよい。
【0013】
更に別の態様では、本発明は、上記の調製方法により生成され、少なくとも18%(重量/体積)の免疫グロブリン、例えば、少なくとも20%(重量/体積)の免疫グロブリンを含む、水性組成物を提供する。
【0014】
別の態様では、本発明は、有効量の上記の組成物を患者に投与することを含む、免疫不全、自己免疫不全、又は急性感染症に罹患した患者を治療する方法を提供する。
【図面の簡単な説明】
【0015】
【図1A】新規の限外濾過/透析濾過システムの図面表示である。
【図1B】新規の限外濾過/透析濾過システムの図面表示である。
【図1C】新規の限外濾過/透析濾過システムの図面表示である。
【図1D】新規の限外濾過/透析濾過システムの図面表示である。
【図1E】新規の限外濾過/透析濾過システムの図面表示である。
【図2】図2である。
【図3】図3である。
【発明を実施するための形態】
【0016】
「抗体」は、分析物(抗原)と結合してその分析物を認識する免疫グロブリン遺伝子(単数)又は免疫グロブリン遺伝子(複数)により実質的に符号化されたポリペプチド、若しくは、それらの断片を指す。認識される免疫グロブリン遺伝子として、カッパ、ラムダ、アルファ、ガンマ、デルタ、イプシロン及びミュー一定領域遺伝子、並びに、ミリアド免疫グロブリン可変領域遺伝子が挙げられる。軽鎖は、カッパ又はラムダのいずれかに分類される。重鎖は、ガンマ、ミュー、アルファ、デルタ、又はイプシロンに分類され、それぞれ、IgG、IgM、IgA、IgD、及びIgEの順で、免疫グロブリン型を定義する。
【0017】
典型的な免疫グロブリン(抗体)構造ユニットは、2対のポリペプチド鎖で構成され、各々の対は、1つの「軽」鎖(約25kD)と1つの「重」鎖(約50〜70kD)とを有する。各々の鎖のN末端は、抗原認識に寄与する約100〜110以上のアミノ酸の可変領域を定める。用語、可変軽鎖(VL)及び可変重鎖(VH)は、それぞれ、これらの軽鎖及び重鎖を指す。
【0018】
用語「限外濾過(UF)」は、水圧が液体を半浸透性膜に押し付ける、様々な薄膜濾過方法を包含する。懸濁された高分子量の固体及び溶質は、保持されるが、水及び低分子量の溶質は、薄膜を通過する。この分離プロセスは、マクロ分子(103〜106Da)溶液、特に、蛋白質溶液を浄化し、濃縮するのに利用されることが多い。数多くの限外薄膜は、それらの膜が保持する分子の大きさに応じて、入手することができる。限外濾過は、通常、1〜1000kDaの薄膜孔径及び0.01〜10barの操作圧力を特徴とし、特に、糖類及び塩類等の小さな分子から、蛋白質等のコロイドを分離するのに有用である。
【0019】
用語「透析濾過」は、限外濾過と同じ薄膜を用いて行われ、流れが接線方向の濾過である。透析濾過の間、緩衝液を再利用タンク内に導入するのに対し、濾液をユニット操作から除去する。製品が残余物(例えば、IgG)内にあるプロセスでは、透析濾過は、成分を製品貯蔵槽から濾液内へ洗い出し、それにより、緩衝液を交換し、望ましくない種の濃度を低減させる。
【0020】
用語「約」は、本明細書で用いられる場合、特定値から正負10%の近似範囲を表す。例えば、語句「約20%」は、18〜22%の範囲を含む。
【0021】
用語「混合」は、いずれかの形式の攪拌により溶液又は懸濁液中に2つ以上の化合物又は物質を等しく配分する行為を記述する。この用語が本出願に利用される場合、「混合」の結果として、溶液又は懸濁液中で全ての成分を完全に均等に配分する必要はない。
【0022】
本出願では、用語「溶媒」は、1つ以上の他の物質を溶解又は懸濁することを可能にする、いずれかの液体物質を含む。溶媒は、本質的に、水等の無機質であってもよく、エタノール、アセトン、酢酸メチル、酢酸エチル、ヘキサン、石油エーテル等のような有機液体であってもよい。溶媒は、「溶媒洗浄剤処理」で用いられる場合、溶液中の脂質包膜ウイルスを不活性にするのに利用される溶媒洗浄剤混合物の一部である、有機溶媒(例えば、燐酸トリ−N−ブチル)を表す。
【0023】
用語「洗浄剤」は、本出願で、用語「界面活性剤」又は「表面活性剤」と交換可能に利用される。界面活性剤は、通常、両媒性の、即ち、疎水基(「尾部」)と親水基(「頭部」)の両方を含む有機化合物であり、それらの官能基により、界面活性剤は、有機溶媒と水の両方に溶ける。界面活性剤は、その頭部が形式上帯電された塩基の存在により分類され得る。非イオン性界面活性剤は、塩基の頭部が帯電されていないのに対し、イオン性界面活性剤は、その頭部が電荷を帯びている。双性イオン界面活性剤は、2つの反対に帯電された塩基を有する頭部を含む。通常の界面活性剤の幾つかの実例として、陰イオン性(硫酸塩陰イオン、スルホン酸塩陰イオン又はカルボン酸塩陰イオン)では、ペルフルオロオクタン酸塩(PFOA又はPFO)、ペルフルオロオクタンスルホン酸塩(PFOS)、ドデシル硫酸ナトリウム(SDS)、ラウリル硫酸アンモニウム、及び他のアルキル硫酸塩、ラウリル硫酸ナトリウム(ラウリルエーテル硫酸ナトリウム、又はSLESとしても知られている)、アルキルベンゼンスルホン酸塩と、陽イオン性(第四級アンモニウム陽イオンに基づく)では、セチルトリメチルアンモニウム臭化物(CTAB)、別名、ヘキサデシルトリメチルアンモニウムブロミド、及び他のアルキルトリメチルアンモニウム塩類、セチルピリジニウム塩化物(CPC)、ポリエトキシル化獣脂アミン(POEA)、塩化ベンザルコニウム(BAC)、塩化ベンゼトニウム(BZT)と、カプリル酸塩、カプリル酸、ヘプタン酸塩、ヘキサン酸、ヘプタン酸、ナノ酸、デカン酸等を含む長鎖脂肪酸及びそれらの塩類と、双性イオン性(両性)では、ドデシルベタイン、ヤシ酸アミドプロピルベタイン、ココ両性グリシネートと、非イオン性では、アルキルポリ(エチレンオキシド)、アルキルフェノールポリ(エチレンオキシド)、ポリ(エチレンオキシド)とポリ(プロピレンオキシド)との共重合体(ポロキサマ類又はポロキサミン類として市場で知られている)、オクチルグルコシド、デシルマルトシド、脂肪アルコール(例えば、セチルアルコール及びオレイルアルコール)、コカミドMEA、コカミドDEA、ポリソルベート類(ツイン20、ツイン80等)、トリトン洗浄剤、及びドデシルジメチルアミンオキシドを含むアルキルポリグルコシド類が挙げられる。
【0024】
用語「静脈注射IgG」又は「IVIG」処置は、本明細書で用いられる場合、一般に、IgG免疫グロブリンの組成物を患者に静脈注射で、皮下に又は筋肉内に投与し、免疫不全、炎症性疾患、及び自己免疫疾患等の数多くの病状に対処する治療方法を指す。IgG免疫グロブリンは、通常、貯蔵されており、血漿から調製される。全ての抗体又は断片を利用することができる。IgG免疫グロブリンは、皮下投与又は筋肉内投与のために高濃度(10%超過)に調合され得る。これは、特に、特定の抗原(例えば、Rh(D)因子、百日咳毒素、破傷風毒素、ボツリヌス毒素、狂犬病等)の平均滴定量よりも高い滴定量で調製される、特化されたIgG製剤に共通のことである。考察し易くするために、そのような皮下投与又は筋肉内投与用に調合されるIgG組成物も、本出願では、用語「IVIG」に含まれる。
【0025】
「治療上有効な量又は投与量」又は「十分な/有効な量又は投与量」は、投与されて効果を作り出す投与量を意味する。正確な投与量は、治療目的に依存することになり、当業者により、周知の技術を利用して確認され得る(例えば、リバーマン、医薬適用法(第1〜3巻、1992年)、ロイド、医薬調合の技術、科学技術(1999年)、ピッカー、投与量計算法(1999年)、及び、レミングトン、調剤科学及び実践、第20版(2003年、ジェンナロ、リピンコット、ウィリアムズ&ウィルキンス出版)を参照すること。これらの開示内容は、全ての目的のために、参照として本明細書にそのまま組み込まれている)。
【0026】
最新医療で日常実践されているように、濃縮免疫グロブリン(特に、IgG)の殺菌製剤は、免疫不全、炎症性及び自己免疫疾患、及び急性感染症の3つの分類内にある病状に対処するのに利用される。通常利用される1つのIgG製品、静脈注射用免疫グロブリン又はIVIGは、静脈内投与用に、例えば、10%濃度に調合される。濃縮された免疫グロブリンは、皮下投与又は筋肉内投与用に、20%の濃度又はほぼその濃度にも調合され得る。
【0027】
一態様では、本発明は、貯蔵された血漿から、高純度で高濃度の免疫グロブリン組成物を生成する新規の改善された方法に関する。以前利用されたIgG浄化及び濃縮方法と比較して、本発明者は、IgGを著しく損なわずにIgG濃度がより高くなり、最終製剤でのpHが低く保たれる、限外濾過及び調合工程を組み込んでいる。通常、製品は、蛋白質濃度が、少なくとも18%(重量/体積)であり、そのうちの大半(通常、95%以上)は、IgGであり、pHは、pH3〜6の範囲であり、それは、血漿中に存在し得るウイルス等の病原を不活性にし易くする。それらのIgG濃度が高く、それにより、投与体積を少なくなるので、本発明の製品は、皮下及び/又は筋肉内投与に適している。幾つかの実施形態では、IgG製品は、粘性が18mパスカル−秒未満であり、従って、静脈内投与にも適用し得る。静脈注射用製品の品質属性を、皮下及び筋肉内注射用製品に必要とされる高濃度と組み合わせることができるので、簡単な希釈により、静脈内投与することもできる。本発明のIgG組成物の別の利点は、貯蔵中の安定性が優れていることである。
【0028】
ある態様では、本発明は、最終蛋白質濃度が約17%よりも大きく、IgG純度が少なくとも約95%である、高濃度IgG製剤を調製する方法を提供する。ある態様では、製剤は、安定性が長期にわたり、静脈、皮下、及び/又は筋肉内投与用に調合される。
【0029】
別の態様では、本発明は、本明細書中で与えられる改良された製造方法に従って調製されたIgG組成物の医薬組成物及び製剤を提供する。ある実施形態では、これらの組成物及び製剤は、現在市場にある他のIVIG組成物と比べて、向上した特性を与える。例えば、ある実施形態では、本明細書中で与えられる組成物及び製剤は、長期にわたり安定している。別の実施形態では、本明細書中で与えられる組成物及び製剤は、現在市場にある他のIVIG組成物と比べて、IgG濃度が高い。更に他の実施形態で、本明細書中で与えられる組成物及び製剤は、IgG濃度が高く、長期にわたり安定している。
【0030】
更に別の態様では、本発明は、本明細書中で与えられる改良された方法を利用して調製されるIgG組成物を投与することを含む、免疫不全、炎症性及び自己免疫疾患、及び急性感染症を治療する方法を提供する。
【0031】
I.濃縮され、浄化されたIgG製剤の生成
幾つかの自己免疫容態に対処するための、全ての抗体を含むIVIG組成物が記述されている(例えば、米国特許出願第US2002/0114802号、第US2003/0099635号、及び第US2002/0098182号を参照)。これらの参照文献に開示されるIVIG組成物は、ポリクローナル抗体を含む。
【0032】
一般に、本発明による免疫グロブリン製剤は、適切な開始材料、例えば、回収血漿又は原血漿から調製され得る。一般例では、血液又は血漿は、健康な提供者から収集される。通常、血液は、免疫グロブリン製剤(一般に、「同種の」免疫グロブリンと呼ばれる)が投与されることになる被験体としての同じ動物種から収集される。免疫グロブリンは、例えば、沈殿(アルコール分別又はポリエチレングリコール分別)、クロマトグラフィー法(イオン交換クロマトグラフィー、親和性クロマトグラフィー、免疫親和性クロマトグラフィー)の超遠視分離、及び電気泳動による調製等のような適切な手順により、血液から単離される(例えば、コーンら、アメリカ化学学会誌、第68巻、459〜75頁(1946年)、オンクレイら、アメリカ化学学会誌、第71巻、541〜50頁(1949年)、バルンデルンら、国際輸血学会誌、第7巻、157〜74頁(1962年)、コベルトら、国際輸血学会誌、第13巻、93〜102頁(1967年)、米国特許第5,122,373号及び第5,177,194号を参照すること。それらの開示内容は、全ての目的のために、参照として本明細書にそのまま組み込まれている)。
【0033】
上記の方法とは異なり、一態様では、本発明は、寒冷貯蔵された開始材料を利用する、濃縮IgG組成物を調製する方法を提供する。一般に、本明細書中で与えられる方法は、変更されたコーン・オンクレイ・アルコール分別工程とイオン交換クロマトグラフィーの両方を利用して、優れたIgG産出量を与え、一方では、向上しない場合は、現在利用可能な市販のIVIG製剤で見出されるものと同じ品質を保つ。
【0034】
多くの事例では、免疫グロブリンは、アルコール分別及び/又はイオン交換、並びに、当業者に周知のクロマトグラフィー方法により生成されるガンマグロブリン含有製品から調製される。通常、浄化されたコーン分画IIが利用される。開始コーン分画IIペーストは、通常、95%又はほぼその率のIgGであり、4つのIgG亜類型から構成される。異なる亜類型は、それらの亜類型が得られる貯蔵されたヒト血漿内に見られるのとほぼ同じ比率で分画II内に存在する。分画IIは、投与可能な製品に調合される前に、更に浄化される。例えば、分画IIペーストは、冷い浄化水性アルコール溶液中に溶解され、沈殿及び濾過を介して除去され得る。最終濾過の後に、免疫グロブリン懸濁液は、透析され、又は、(公称分子量限界が100,000ダルトン以下である限外薄膜を利用して)透析濾過され、アルコールを除去し得る。溶液は、所望の蛋白質濃度を得るために、濃縮又は希釈されてもよく、更に、当業者に十分に知られている技術により浄化され得る。
【0035】
分取工程を利用して、特定の同類型又は亜類型の免疫グロブリンを濃縮することができる。例えば、蛋白質A、蛋白質G又は蛋白質セファロースクロマトグラフィーを利用して、IgGの又は特定のIgG亜類型の免疫グロブリンの混合物を濃縮することができる。(一般に、ハーロー及びレイン、抗体を用いて(コールドスプリングハーバー研究所出版、1999年)、ハーロー及びレイン、抗体、実験マニュアル(コールドスプリングハーバー研究所出版、1988年)、米国特許第5,180,810号を参照すること。)
【0036】
以下に詳細に記載されるように、本発明の高濃度IgG製品は、IVIG生成プロセスと同じ又は類似の多くの工程を有するプロセスにより生成される。生産プロセスの終了付近での、開チャンネル薄膜を利用した、特別設定の後洗浄製剤を用いての限界濾過/透析濾過の追加工程により、その結果得られるIgG組成物は、従来技術のIVIG(例えば、ガンマガード(登録商標)リキッド)と比べて、産出量と保管安全性に悪影響を与えずに、蛋白質濃度(200mg/mL)の高さがほぼ2倍になる。大抵の市販の限外濾過薄膜では、主要蛋白質が損なわれずに、200mg/mLのIgG濃度に達することができない。これらの薄膜は早期に詰まることになり、従って、適切な後洗浄液を得るのは難しい。従って、開チャンネル薄膜構成を用いなければならない。開チャンネル薄膜でも、特別設定の後洗手段を利用して、蛋白質を著しく損なわずに(損失が2%未満)、要求される濃度を得なければならない。更に意外なこととして、200mg/mLの高い蛋白質濃度により、低pH保存工程のウイルス不活性能力は左右されない。高濃度IgG組成物を生成する一般的なプロセスは、以下の工程を含む:
【0037】
A.寒冷沈殿物の分離
浄化プロセスは、通常、安全性及び品質の検討のために既に確認された、事前に凍結された貯蔵血漿を解凍することで開始する。解凍は、通常、6℃未満の温度で実行される。次に、遠心分離又は濾過を冷気中で実行して、血漿が解凍された後、通常は、解凍と同じ温度で、固体と液体とを分離する。次に、液体部分(遠心分離又は濾過により、新鮮な解凍された血漿から寒冷不溶性蛋白質が除去された後は、「寒冷貯蔵血漿」とも呼ばれる)は、次の工程で処理される。この際、実施例1で詳細に記載される、第8阻害因子迂回活性複合体(FEIBA)、第4因子複合体、第7因子濃縮物、又は抗凝固第3因子複合体を単離するために、様々な追加工程を取り上げることができる。
【0038】
B.分画Iの上澄みの取得
この工程では、寒冷貯蔵血漿は、通常、0±1℃又はほぼその温度に冷却され、そのpHは、7.0又はほぼ7.0に調節される。ある実施形態では、pHは、約7.0〜約7.5に、好ましくは、7.1又はほぼ7.1と7.3又はほぼ7.3との間に、さらに好ましくは、7.2又はほぼ7.2に調節される。ある実施形態では、pHは、7.0に又はほぼ7.0に調節される。別の実施形態では、pHは、7.1又はほぼ7.1に調節される、別の実施形態では、pHは、7.2又はほぼ7.2に調節される。別の実施形態では、pHは、7.3又はほぼ7.3に調節される。別の実施形態では、pHは、7.4又はほぼ7.4に調節される。別の実施形態では、pHは、7.5又はほぼ7.5に調節される。次に、事前に冷却されたエタノールを加え、血漿を攪拌しながら、8%体積/体積の標的エタノール濃度にする。同時に、温度を更に、約−4〜約0℃に、好ましくは、約−2℃に下げ、α2−マクログロブリン、β1A及びβ1Cグロブリン、フィブリノゲン、及び第8因子等の汚染物を沈殿させる。通常、沈殿事象は、少なくとも1時間の保持時間を含むことになるが、より短い又はより長い保持時間を用いてもよい。続いて、理想的には、寒冷貯蔵血漿中に存在するIgG含有量の全体を含む上澄み(上澄みI)は、遠心分離、濾過、又は別の適切な方法により収集される。
【0039】
C.分画II+IIIの沈殿
IgG含有量を更に濃縮し、その分画の純度を更に上げるために、上澄みIは、第2沈殿工程に曝される。一般に、溶液のpHは、約6.8〜約7.2のpHに、好ましくは、7.0又は約7.0のpHに調節される。次に、その溶液に、攪拌しながら、アルコール、好ましくは、エタノールを加え、約20%〜約25%(体積/体積)の最終濃度にする。一実施形態では、最終アルコール濃度は、20%又はほぼその値である。別の実施形態では、最終アルコール濃度は、21%又はほぼその値である。別の実施形態では、最終アルコール濃度は、22%又はほぼその値である。別の実施形態では、最終アルコール濃度は、23%又はほぼその値である。別の実施形態では、最終アルコール濃度は、24%又はほぼその値である。別の実施形態では、最終アルコール濃度は、25%又はほぼその値である。分画II+III上澄みとも呼ばれる液体部分を更に処理して、第5因子を抽出する。この工程からの沈殿物を、次の工程で更に処理する。一実施形態では、工程B及びCを共に実行することもできる。
【0040】
D.分画II及びIII沈殿物からの抽出
冷却抽出緩衝液を利用して、分画II+III沈殿物を、沈殿物1部に対して抽出緩衝液15部の通常比に再び懸濁する。模範的抽出緩衝液は、5mMの一塩基性燐酸ナトリウムと5mMの酢酸とを含み、pHが、4.5±0.2又はほぼその値であり、伝導率が、0.7〜0.9mS/cm又はほぼその値である。一実施形態では、抽出緩衝液の伝導率は、0.7mS/cm又はほぼその値である。別の実施形態では、抽出緩衝液の伝導率は、0.8mS/cm又はほぼその値である。更に別の実施形態では、抽出緩衝液の伝導率は、0.9mS/cm又はほぼその値である。抽出プロセスは、2〜8℃又はほぼその値の温度に実行される。
【0041】
他の適切な再懸濁比を利用してもよく、例えば、約1:8〜約1:30、又は、約1:10〜約1:20、又は、約1:12〜約1:18、又は、約1:13〜約1:17、又は、約1:14〜約1:16である。ある実施形態では、再懸濁比は、1:8、1:9、1:10、1:11、1:12、1:13、1:14、1:15、1:16、1:17、1:18、1:19、1:20、1:21、1:21、1:22、1:23、1:24、1:25、1:26、1:27、1:28、1:29、1:30、又はそれ以上、若しくは、ほぼそれらの値であり得る。
【0042】
II+III沈殿物を抽出するのに適切な溶液は、一般に、pHが、約4.0〜約5.5である。ある実施形態では、溶液は、pHが、約4.0〜約5.0である。別の実施形態では、溶液は、pHが、約4.5〜約5.0である。他の実施形態では、抽出液は、pHが、約4.0、4.1、4.2、4.3、4.4、4.5、4.6、4.7、4.8、4.9、5.0、5.1、5.2、5.3、5.4、又は5.5である。一実施形態では、抽出液のpHは、4.5又はほぼその値である。別の実施形態では、抽出液のpHは、4.6又はほぼその値である。抽出液のpHは、4.7又はほぼその値である。抽出液のpHは、4.8又はほぼその値である。抽出液のpHは、4.9又はほぼその値である。抽出液のpHは、5.0又はほぼその値である。
【0043】
抽出緩衝液は、好ましくは、伝導率が、約0.5mS/cm〜約2.0mS/cmである。例えば、ある実施形態では、抽出液の伝導率は、0.5mS/cm又はほぼその値であり、若しくは、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9又はほぼそれらの値であり、若しくは、約2.0mS/cmである。当業者は、適切な伝導率を有する抽出緩衝液を生成する方法を知っている。
【0044】
E.煙霧シリカ処理及び濾過
ある実施形態では、先の工程からの懸濁液に煙霧シリカ(例えば、アエロジル380又はその等価物)を加え、40g/kg又はほぼその値の懸濁液濃度にする、又は、寒冷貯蔵血漿の1.8g/リットルと等価にする。混合は、少なくとも50〜70分にわたり約2〜8℃で行われる。幾つかの事例では、濾過補助剤(例えば、ワールドミネラルズからの、0.5kg/kg又はほぼその値の懸濁液濃度で利用されるハイフロスーパーセル)を加えて、濾過により液体/固体に分離する後続の工程を容易にする。抽出緩衝液を利用して、圧濾器を後洗浄する。濾液プロセスは、約2〜8℃の温度に保たれる。
【0045】
F.沈殿物Gの分別
次に、先の工程からの濾液をポリソルベート−80と混合して、0.2%重量/体積又はその値の濃度にし、少なくとも30分にわたり約2℃〜8℃の温度で攪拌する。次に、クエン酸ナトリウム二無水物を溶液内に8g/リットル又はその値で混合し、更に30分にわたり約2℃〜8℃の温度で攪拌する。次に、溶液のpHを7.0±0.1又はその値に調節する。ある実施形態では、pHを水酸化ナトリウム又は酢酸のどちらかで調節する。次に、冷却アルコールを溶液に加えて、25%体積/体積の濃度にし、コーン分画IIと同じ沈殿工程を行う。
【0046】
G.沈殿物Gの懸濁液
先の工程からの沈殿物を溶解し、0.2μmの公称孔径の深層フィルタ(例えば、キュノVR06フィルタ又はその等価物)で濾過して、透明な濾液を得る。別の実施形態では、沈殿物を溶解し、次に、遠心分離して、浄化された上澄みを回収する。
【0047】
H.溶媒及び洗浄剤処理
先の工程からの濾液を溶媒/洗浄剤処理に利用する。通常の溶媒/洗浄剤処理混合物は、1.0%(体積/体積)トリトンX−100、0.3%(体積/体積)ツイン−80、及び0.3%(体積/体積)TNBPを含み、混合物は、通常、少なくとも60分にわたり約18℃〜25℃の温度に保たれる。血漿由来成分を洗浄剤処理する方法は、当該技術分野で十分に知られている。一般に、本明細書中で与えられる方法と共に、いずれかの標準的な非イオン性洗浄剤処理を利用してもよい。
【0048】
I.陽イオン交換クロマトグラフィー
S/D処理されたPptG濾液からのIgGを更に浄化し、濃縮するために、陽イオン交換及び/又は陰イオン交換クロマトグラフィーを用いることができる。イオン交換クロマトグラフィーを利用してIgGを浄化し、濃縮する方法は、当該技術分野で十分に知られている。例えば、米国特許第5,886,154号は、分画II+III沈殿物を低いpH(約3.8〜4.5)で抽出した後に、カプリル酸を利用してIgGを沈殿させ、最終的に、2つの陰イオン交換クロマトグラフィー工程を実施する方法を記載している。米国特許第6,069,236号は、アルコール沈殿に全く依存しないクロマトグラフィーによるIgG浄化方式を記載している。PCT出願第WO2005/073252号は、分画II+III沈殿物の抽出、カプリル酸処理、PEG処理、及び単一の陰イオン交換クロマトグラフィー工程を含むIgG浄化方法を記載している。米国特許第7,186,410号は、分画I+II+III沈殿物又は分画II沈殿物のいずれかの抽出と、続く、アルカリ性pHで実行される単一の陰イオン交換工程とを含むIgG浄化方法を記載している。米国特許第7,553,938号は、分画I+II+III沈殿物又は分画II+III沈殿物のいずれかの抽出と、カプリル酸処理と、一回又は二回の陰イオン交換クロマトグラフィー工程とを含む方法を記載している。米国特許第6,093,324号は、約6.0〜約6.6のpHで操作されるマクロ多孔性陰イオン交換樹脂の利用と、を含む浄化方法を記載している。米国特許第6,835,379号は、アルコール分別を用いずに陽イオン交換クロマトグラフィーに依存する浄化方法を記載している。
【0049】
一実施形態では、先の工程からの蛋白質溶液を含む溶剤/洗浄剤は、次に、陽イオン交換カラムに通され、溶媒と洗浄剤とを除去する。SD剤を洗い落とした後、次に、吸着された蛋白質を高pHの溶出緩衝液で溶出する。一実施形態では、溶出緩衝液は、pHが、約7.5〜約9.5である。別の実施形態では、溶出緩衝液は、pHが、約8.0〜約9.0である。好ましい実施形態では、溶出緩衝液は、pHが、8.5±0.1又はほぼその値である。
【0050】
J.陰イオン交換クロマトグラフィー
先の工程からの溶出物は、pH6に調節され、次の平衡陰イオン交換カラムに適した伝導率になるように希釈される。充填及び洗浄中のカラム通過画分は、別の処理のために収集される。
【0051】
K.ナノ濾過
本明細書中で与えられるIgG組成物のウイルス負荷を更に削減するために、適切なナノ濾過装置を利用して、陰イオン交換カラム流出物をナノ濾過する。ある実施形態では、ナノ濾過装置は、平均孔径が、約15nm〜約200nmである。この用途に適したナノフィルタの実例として、DVD、DV50、DV20(パール社)、バイレゾルブNFP、バイレゾルブNFP(ミリポア社)、プラノバ15N、20N、35N、及び75N(プラノバ社)が挙げられるが、これらに限定されない。特定の実施形態では、ナノフィルタは、平均孔径が、約15nm〜約72nm、又は、約19nm〜約35nm、又は、15nm、19nm、35nm、又は72nm若しくはほぼそれらの値であり得る。好ましい実施形態では、アサヒ・プラノバ35Nフィルタ又はその等価物等のナノフィルタは、平均孔径が、35nm又はほぼその値である。
【0052】
L.限外濾過及び透析濾過
ナノ濾過の後、限外濾過により、濾液を更に、5±1%の蛋白質濃度に濃縮する。幾つかの実施例では、除外濾過は、開経路篩を有するカセット内で実行され、除外濾過薄膜は、公称分画分子量(NMWCO)が、50kDa以下である。
【0053】
一実施形態では、限外濾過により、ナノ濾液を約2%〜約10%(重量/体積)の蛋白質濃度に濃縮してもよい。ある実施形態では、除外濾過は、開経路篩を有するカセット内で実行され、除外濾過薄膜は、公称分画分子量(NMWCO)が、約100kDa未満である、若しくは、約90、80、70、60、50、40、30kDa未満である、若しくは、それらよりも少ない。好ましい実施形態では、限外濾過薄膜は、NMWCOが、50kDa以下である。
【0054】
限外濾過工程が完了した後、静脈又は筋肉内投与に適した溶液で透析濾過することにより、濃縮液を更に濃縮してもよい。ある実施形態では、透析濾過溶液は、安定剤及び/又は緩衝剤を含んでもよい。好ましい実施形態では、安定剤及び緩衝剤は、適切な濃度の、例えば、0.20M〜0.30Mの、又は約0.22M〜約0.28Mの、又は約0.24M〜約0.26mMの、若しくは、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9、又は3.0或いはほぼそれらの値の濃度の濃度のグリシンである。好ましい実施形態では、透析緩衝液は、0.25M又はほぼその値のグリシンを含む。
【0055】
好ましい実施形態では、除外濾過工程が完了した後、濃縮液をpHの低い0.25Mグリシン溶液で透析濾過する。通常、最小限の交換体積は、当初の濃縮液の体積の6倍であり、溶液は、20%重量/体積よりも大きな蛋白質濃度に濃縮される。透析濾過及び濃縮工程の終わりでは、溶液のpHは、通常、4.4〜4.9である。
【0056】
通常、最小限の交換体積は、当初の濃縮液の体積の少なくとも約3倍である、又は、当初の濃縮液の体積の少なくとも約4,5、6、7、8、9倍又はそれ以上である。IgG溶液は、約5%〜約22%(重量/体積)の、又は約10%〜約22%(重量/体積)の、又は約15%〜約22%(重量/体積)の、又は約18%〜約22%(重量/体積)の、約20%〜約22%(重量/体積)の最終蛋白質濃度に、若しくは、約5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21%、22%又はほぼそれらの値、若しくは、それらよりも高い最終濃度に濃縮され得る。好ましい実施形態では、IgG溶液は、20%、約20%〜約22%、22%の最終蛋白質濃度に濃縮される。通常、濃縮工程の終わりでは、溶液のpHは、約4.6〜5.1である。
【0057】
M.調合
透析工程が完了した後、溶液の蛋白質濃度は、透析緩衝液で、約5%〜約20%(重量/体積)、又は約10%〜約20%(重量/体積)、又は約15%〜約20%(重量/体積)、又は約18%〜約20%(重量/体積)の最終濃度に、若しくは、約5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、又は20%の最終濃度に調節される。好ましい実施形態では、溶液の最終蛋白質濃度は、19%、約19%〜約21%、21%である。好ましい実施形態では、透析濾過が完了した後、溶液の蛋白質濃度は、透析濾過緩衝液で、20%重量/体積よりも僅かに大きく、例えば、20.4±04%重量/体積又はほぼその値に調節される。
【0058】
N.更なる殺菌
調合されたバルク溶液は、絶対孔径が0.2ミクロン以下の薄膜フィルタを通して最初に濾過することにより、更に殺菌される。次に、溶液は、適切に密封するための最終容器内に無菌で分注され、検査用の試料が採取される。最終工程として、密封された容器を長期間、例えば、21〜22日にわたり30〜32℃で保存する。
【0059】
II.濃縮IgG組成物
一態様では、本発明は、本明細書中で与えられる方法により調製された水性IgG組成物に関する。一般に、本明細書に記載される新規の方法により調製されたIgG組成物は、IgG含有量及び純度が高い。例えば、本明細書中で与えられるIgG組成物は、蛋白質濃度が少なくとも15%(重量/体積)であり、IgG含有量が、90%純度よりも大きい。これらの高純度のIgG組成物は、治療投与に、例えば、皮下及び/又は筋肉内投与に適している。一実施形態では、本明細書中で与えられるIgG組成物は、静脈内投与に適しており、例えば、投与前に希釈される。一実施形態では、IgGの濃度は、20%又はほぼその値であり、皮下投与又は筋肉内投与に利用される。
【0060】
一実施形態では、本発明は、以下の工程を含む方法により調製された水性IgG組成物を提供する:
(1)遠心分離により血漿から液体と沈殿物とを分離すること、
(2)事前に冷却されたエタノールを(1)からの液体と混合して、エタノール濃度が8%(体積/体積)又はほぼその値である混合物を作ること、
(3)遠心分離により、(2)の混合物から液体と沈殿物とを分離すること、
(4)(3)からの液体のpH及びエタノール濃度を、それぞれ、7.0及び20〜25%(体積/体積)又はそれらの値に調節することにより、混合物を作ること、
(5)遠心分離により、(4)の混合物から液体と沈殿物とを分離すること、
(6)(5)の沈殿物を緩衝液で約1〜15の重量比に再び懸濁して、懸濁液を作ること、
(7)二酸化シリコン(SiO2)を(6)からの懸濁液と混合し、濾過により濾液を得ること、
(8)洗浄剤と冷却アルコールを、(7)の濾液と混合し、遠心分離により沈殿物を得ること、
(9)溶媒又は洗浄剤を含む水溶液中に前記沈殿物を溶解し、その溶液を少なくとも60分にわたり維持すること、
(10)(9)の後の溶液を陽イオン交換クロマトグラフィーカラムに通し、カラムに吸収された蛋白質を溶出液中に溶出させること、
(11)(10)からの溶出液を陰イオン交換クロマトグラフィーカラムに通して、流出液を生成すること、
(12)流出液をナノ濾過器に通して、ナノ濾液を生成すること、
(13)ナノ濾液を限外濾過薄膜に通して、限外濾液を生成すること、
(14)限外濾液を透析濾過緩衝液で透析濾過して、蛋白質濃度が20%(重量/体積)又はほぼその値の溶液を生成すること、並びに、
(15)前記溶液を0.2μm以下のフィルタに通して濾過することで(14)からの溶液を減菌することにより、濃縮されたIgGの組成物を得ること。
【0061】
一実施形態では、本発明は、蛋白質濃度が約150g/L〜約250g/Lの水性IgG組成物を提供する。ある実施形態では、IgG組成物の蛋白質濃度は、約175g/L〜約225g/L、又は約200g/L〜約225g/L、又はこれらの範囲内のいずれかの適切な濃度、例えば、150g/L、155g/L、160g/L、165g/L、170g/L、175g/L、180g/L、185g/L、190g/L、195g/L、200g/L、205g/L、210g/L、215g/L、220g/L、225g/L、230g/L、235g/L、240g/L、245g/L、250g/L又はほぼそれらの値、若しくは、それらよりも高い値の濃度である。好ましい実施形態では、水性IgG組成物は、蛋白質濃度が200g/L又はほぼその値である。特に好ましい実施形態では、水性IgG組成物は、蛋白質濃度が204g/L又はほぼその値である。
【0062】
本明細書中で与えられる方法により、純度レベルが極めて高いIgG組成物を調製することができる。例えば、一実施形態では、本明細書中で与えられる組成物中の総蛋白質の少なくとも約95%がIgGである。他の実施形態では、蛋白質の少なくとも約96%がIgGである、又は、組成物の総蛋白質の少なくとも約97%、98%、99%、99.5%又はそれ以上がIgGである。
【0063】
同様に、本明細書中で与えられる方法により、汚染液が極めて低レベルであるIgG組成物を調製することができる。例えば、ある実施形態では、約100mg/L未満のIgAを含むIgG組成物が提供される。他の実施形態では、IgG組成物は、約50mg/L未満のIgA、好ましくは、約35mg/L未満のIgA、最も好ましくは、20mg/L未満のIgAを含む。
III.医薬組成物
【0064】
別の態様では、本発明は、本明細書中で与えられる方法により調製された純粋なIgGを含む医薬組成物及び調合物を提供する。一般に、本明細書中で与えられる新規の方法により調製されたIgG医薬組成物及び調合物は、IgG含有量及び純度が高い。例えば、本明細書中で与えられるIgG医薬組成物及び調合物は、蛋白質濃度が、少なくとも約15%(重量/体積)、IgG含有量が、90%純度よりも大きくてもよい。これらの高純度IgG組成物は、治療投与に、例えば、皮下及び/又は筋肉内投与に適している。一実施形態では、本明細書中で与えられるIgG組成物は、静脈内投与に適しており、例えば、投与前に希釈される。一実施形態では、IgGの濃度は、20%又はほぼその値であり、皮下投与又は筋肉内投与に利用される。
【0065】
一実施形態では、本明細書中で与えられる医薬組成物は、本明細書中で与えられる方法を利用して単離された水性IgG組成物を調合することにより調製される。一般に、調合された組成物は、少なくとも1つの、好ましくは、少なくとも2つの、最も好ましくは、少なくとも3つのウイルス不活性化又は除去工程に曝されている。本明細書中で与えられる方法と共に利用され得るウイルス不活性化又は除去工程の限定されない実施例として、溶剤洗浄剤処理(ホロヴィッツら、血液凝固線溶、1994年(第5号付録3)、21頁〜28頁、クレイルら、輸血、2003年、第4号、1023〜1028頁、それらの両方とも、全ての目的のために、本明細書に参照としてそのまま、明確に組み込まれている)、ナノ濾過(ハマモトら、国際輸血学会誌、1989年、第56号、230〜236頁、及びユアサら、一般ウイルス学会誌、1991年(第72号(部編8))、2021〜2024頁、それらの両方とも、全ての目的のために、本明細書に参照としてそのまま、明確に組み込まれている)、高温での低pH恒温放置(ケンプフら、輸血、1991年、第31号、423〜427頁、及び、ルイら、生物学、1994年、第22号、13〜19号)が挙げられる。
【0066】
ある実施形態では、IgG含有量が約175g/L IgG〜約225g/L IgGの医薬製剤が提供される。一般に、これらのIVIG製剤は、本明細書に記載される方法を利用して血漿からのIgG組成物を単離し、その組成物を濃縮し、濃縮された組成物を、静脈内投与に適した溶液中に調合することにより調製される。IgG組成物は、当業者に周知のいずれかの適切な方法を利用して濃縮され得る。一実施形態では、組成物は、限外濾過/透析濾過により濃縮される。幾つかの実施形態では、組成物を濃縮するのに利用される限外濾過装置は、公称分画分子量(NMWCO)が約100kDa未満、又は、約90、80、70、60、50、40、30kDa未満、又は、それらの値よりも小さい限外濾過薄膜を用いる。好ましい実施形態では、限外濾過薄膜は、NMWCOが、50kDa未満である。緩衝液交換は、当業者に周知のいずれかの適切な技術を利用して行われてもよい。特定の実施形態では、緩衝液交換は、透析濾過により行われる。
【0067】
特定の実施形態では、IgG医薬組成物が提供され、そのIgG組成物は、以下の工程を含む方法を利用して、血漿から浄化される:
(1)遠心分離により、血漿から液体と沈殿物とを分離すること、
(2)事前に冷却されたエタノールを(1)からの液体と混合して、エタノール濃度が8%(体積/体積)又はほぼその値である混合物を作ること、
(3)遠心分離により、(2)の混合物から液体と沈殿物とを分離すること、
(4)(3)からの液体のpH及びエタノール濃度を、それぞれ、7.0及び20〜25%(体積/体積)又はそれらの値に調節することにより、混合物を作ること、
(5)遠心分離により、(4)の混合物から液体と沈殿物とを分離すること、
(6)(5)の沈殿物を緩衝液で約1〜15の重量比で再び懸濁して、懸濁液を作ること、
(7)二酸化シリコン(SiO2)を(6)からの懸濁液と混合し、濾過により濾液を得ること、
(8)洗浄剤と冷却アルコールを、(7)の濾液と混合し、遠心分離により沈殿物を得ること、
(9)溶媒又は洗浄剤を含む水溶液中に前記沈殿物を溶解し、その溶液を少なくとも60分にわたり維持すること、
(10)(9)の後の溶液を陽イオン交換クロマトグラフィーカラムに通し、カラムに吸収された蛋白質を溶出液中に溶出させること、
(11)(10)からの溶出液を陰イオン交換クロマトグラフィーカラムに通して、流出液を生成すること、
(12)流出液をナノ濾過器に通して、ナノ濾液を生成すること、
(13)ナノ濾液を限外濾過薄膜に通して、限外濾液を生成すること、
(14)限外濾液を透析濾過緩衝液で透析濾過して、蛋白質濃度が20%(重量/体積)又はほぼその値の溶液を生成すること、並びに、
(15)前記溶液を0.2μm以下のフィルタに通して濾過することで(14)からの溶液を減菌することにより、濃縮されたIgGの組成物を得ること。
【0068】
一実施形態では、本発明は、蛋白質濃度が約150g/L〜約250g/Lの医薬IgG組成物を提供する。ある実施形態では、IgG組成物の蛋白質濃度は、約175g/L〜約225g/L、又は約200g/L〜約225g/L、又はこれらの範囲内のいずれかの適切な濃度、例えば、150g/L、155g/L、160g/L、165g/L、170g/L、175g/L、180g/L、185g/L、190g/L、195g/L、200g/L、205g/L、210g/L、215g/L、220g/L、225g/L、230g/L、235g/L、240g/L、245g/L、250g/L又はほぼそれらの値、若しくは、それらよりも高い値の濃度である。好ましい実施形態では、水性IgG組成物は、蛋白質濃度が200g/L又はほぼその値である。特に好ましい実施形態では、水性IgG組成物は、蛋白質濃度が204g/L又はほぼその値である。
【0069】
本明細書で与えられる方法により、純度レベルが極めて高いIgG医薬組成物を調製することができる。例えば、一実施形態では、本明細書中で与えられる組成物中の総蛋白質の少なくとも約95%がIgGである。他の実施形態では、蛋白質の少なくとも約96%がIgGである、又は、組成物の総蛋白質の少なくとも約97%、98%、99%、99.5%又はそれ以上がIgGである。
【0070】
同様に、本明細書で与えられる方法により、汚染液が極めて低レベルであるIgG医薬組成物を調製することができる。例えば、ある実施形態では、約100mg/L未満のIgAを含むIgG組成物が提供される。他の実施形態では、IgG組成物は、約50mg/L未満のIgA、好ましくは、約35mg/L未満のIgA、最も好ましくは、20mg/L未満のIgAを含む。
【0071】
本明細書で与えられる医薬組成物は、通常、静脈内投与に適した1つ以上の緩衝剤又はpH安定剤を含む。本明細書中で与えられるIgG組成物を調合するのに適した緩衝剤の限定されない実例として、グリシン、クエン酸塩、燐酸塩、酢酸塩、グルタミン酸塩、酒石酸塩、安息香酸塩、乳酸塩、ヒスチジン又は他のアミノ酸類、グルコン酸塩、リンゴ酸塩、蟻酸塩、プロピオン酸塩、炭酸塩、又は、適切なpHに調節されたそれらのいずれかの組み合わせが挙げられる。一般に、緩衝剤は、調合物中で適切なpHを長期間にわたり維持するのに十分なものである。好ましい実施形態では、緩衝剤は、グリシンである。
【0072】
幾つかの実施形態では、調合物中の緩衝剤の濃度は、約100mM〜約400mMであり、好ましくは、約150mM〜約350mM、より好ましくは、約150mM〜約300mM、最も好ましくは、約175mM〜約225mMである。特に好ましい実施形態では、濃縮されたIgG組成物は、約150mM〜約250mMのグリシン、最も好ましくは、約200mMのグリシンを含む。別の好ましい実施形態では、濃縮されたIgG組成物は、250mM又はほぼその値のグリシンを含む。
【0073】
ある実施形態では、調合物のpHは、約4.1〜約5.6であり、好ましくは、約4.4〜約5.3、最も好ましくは、約4.6〜約5.1である。特定の実施形態では、調合物のpHは、約4.1、4.2、4.3、4.4、4.5、4.6、4.7、4.8、4.9、5.0、5.1、5.2、5.3、5.4、5.5、又は5.6であってもよい。
【0074】
幾つかの実施形態では、本明細書中で与えられる医薬組成物は、任意選択で、組成物のモル浸透圧濃度を調節する薬剤を更に含んでもよい。モル浸透圧濃度の限定されない実施例として、マンニトール、ソルビトール、グリセロール、蔗糖、グルコース、ブドウ糖、左旋糖、果糖、乳糖、ポリエチレングリコール類、燐酸類、塩化ナトリウム、塩化カリウム、塩化カルシウム、グルコノグルコヘプトン酸カルシウム、ジメチルスルホン等が挙げられる。
【0075】
通常、本明細書中で与えられる調合物は、モル浸透圧濃度が、約285mOsmol/kg(レイシーら、薬物情報ハンドブック、レキシ−コンプ、1999年、1254頁)の生理上のモル浸透圧濃度に匹敵する。ある実施形態では、調合物のモル浸透圧濃度は、約200mOsmol/kg〜約350mOsmol/kg、好ましくは、約240mOsmol/kg〜約300mOsmol/kgである。特定の実施形態では、調合物のモル浸透圧濃度は、約200mOsmol/kg、又は210mOsmol/kg、220mOsmol/kg、230mOsmol/kg、240mOsmol/kg、245mOsmol/kg、250mOsmol/kg、255mOsmol/kg、260mOsmol/kg、265mOsmol/kg、270mOsmol/kg、275mOsmol/kg、280mOsmol/kg、285mOsmol/kg、290mOsmol/kg、295mOsmol/kg、300mOsmol/kg、310mOsmol/kg、320mOsmol/kg、330mOsmol/kg、340mOsmol/kg、340mOsmol/kg、又は350mOsmol/kgである。
【0076】
本明細書中で与えられるIgG調合物は、一般に、液体形態で、長期間にわたり安定である。ある実施形態では、調合物は、室温で少なくとも約3ヶ月にわたり、又は、室温で少なくとも約4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、又は24ヶ月にわたり安定である。調合物は、その上、一般に、冷凍条件下(通常、約2℃〜約8℃)で少なくとも約18ヶ月にわたり、或いは、冷凍条件下で少なくとも約21、24、27、30、33、36、39、42、又は45ヶ月にわたり安定である。
IV.IgG製剤の投与
【0077】
最新医療において日常実施されるように、濃縮免疫グロブリン(特に、IgG)の無菌製剤を利用して、免疫不全、炎症性及び自己免疫疾患、及び急性感染症の3つの主要な分類内にある病状に対処する。これらのIgG製剤は、多発性硬化症(特に、再発寛解型多発性硬化症又はRRMS)、アルツハイマー病、及びパーキンソン病を治療するにも有用であり得る。本発明の浄化されたIgG製剤は、これらの目的に、並びに、IgG製剤の他の臨床上許容される用途に適している。
【0078】
FDAは、同種骨髄移植、慢性リンパ性白血病、特発性血小板減少性紫斑病(ITP)、小児HIV、原発性免疫不全、川崎病、慢性炎症性脱髄性多発神経炎(CIDP)、及び、抗体の高い移植患者又はABO不適合提供者の腎臓移植を含む、様々な適応症に対処するのにIVIGを利用することを承認している。ある実施形態では、本明細書中で与えられるIVIG組成物は、これらの疾患及び容態の治療又は対処に有用である。
【0079】
更に、通常、IVIGの適応外使用を患者に施して、様々な適応症、例えば、慢性疲労症候群、大腸炎、皮膚筋炎及び多発性筋炎、グレーブス眼症、ギラン・バレー症候群、筋萎縮症、封入体筋炎、ランバート・イートン症候群、紅斑性狼瘡、多巣性運動ニューロパチー、多発性硬化症(MS)、重症筋無力症、新生児同種免疫性血小板減少症、パルボウイルスB19感染症、尋常性天疱瘡、輸血後紫斑病、腎臓移植拒絶、自然流産/流産、スティッフパーソン症候群、眼球クローヌス・ミオクローヌス、重篤疾患成人の重症敗血症及び敗血症性ショック、中毒性表皮壊死症、慢性リンパ性白血病、多発性骨髄腫、X連鎖無ガンマグロブリン血症、及び低ガンマグロブリン血症を治療又は対処する。ある実施形態では、本明細書中で与えられるIVIG組成物は、これらの疾病及び容態の治療又は対処に有用である。
【0080】
最終的に、原発性免疫不全、RRMS、アルツハイマー病、及びパーキンソン病を含む疾病の治療又は処置のためのIVIGの実験的使用が提案されている(米国特許出願公報第US2009/0148463号。これは、全ての目的のために参照としてそのまま本明細書に組み込まれている)。ある実施形態では、本明細書中で与えられるIVIG組成物は、原発性免疫不全、RRMS、アルツハイマー病、又はパーキンソン病の治療又は処置に有用である。日常の投与を含むある実施形態では、被験体に投与される有効量は、医者により、年齢、体重、疾病の重症度、投与経路(例えば、静脈注射又は皮下注射)及び治療に対する応答の個別の違いを考慮して定められ得る。ある実施形態では、本発明の免疫グロブリン製剤は、毎日約5mg/キログラム〜約2000mg/キログラムで被験体に投与され得る。追加の実施形態では、免疫グロブリン製剤は、少なくとも約10mg/キログラム、少なくとも15mg/キログラム、少なくとも20mg/キログラム、少なくとも25mg/キログラム、少なくとも30mg/キログラム、又は少なくとも50mg/キログラムの量で投与され得る。追加の実施形態では、免疫グロブリン製剤は、被験体に、約100mg/キログラム、約150mg/キログラム、約200mg/キログラム、約250mg/キログラム、約300mg/キログラム、約400mg/キログラムまでの投与量で投与され得る。他の実施形態では、免疫グロブリン製剤の投与量は、より多く又はより少ない場合がある。更に、免疫グロブリン製剤は、毎日1以上の投与回数で投与され得る。IgG製剤により治療される疾病に精通している臨床医は、当該分野で知られている基準に従って、患者に適した投与量を定めることができる。
【0081】
1つの通常利用されるIgG製品、静脈注射用免疫グロブリン又はIVIGは、静脈注射投与用に調合される。本発明のIgG製剤では、極めて高い免疫グロブリン濃度(20%重量/体積又はそれよりも高い)が得られ、その濃度により、治療効果のあるのための体積が著しく削減され、本発明の組成物は、患者の皮下及び/又は筋肉内に投与するのに特に有利であるが、静脈内投与が、投与のための選択肢として残る。
【0082】
用語「有効量」は、被験体中の処置される病状(例えば、原発性免疫不全、RRMS、アルツハイマー病、パーキンソン病等)を改善する又は治療することになる、免疫グロブリン特にIgG製剤の量を指す。被験体に投与される有効量は、医者により、年齢、体重、疾病の重症度、投与経路(例えば、静脈注射又は皮下注射)及び治療に対する応答の個別の違いを考慮して定められ得る。ある実施形態では、本発明の免疫グロブリン製剤は、毎日約5mg/キログラム〜約2000mg/キログラムで被験体に投与され得る。追加の実施形態では、免疫グロブリン製剤は、少なくとも約10mg/キログラム、少なくとも15mg/キログラム、少なくとも20mg/キログラム、少なくとも25mg/キログラム、少なくとも30mg/キログラム、又は少なくとも50mg/キログラムの量で投与され得る。追加の実施形態では、免疫グロブリン製剤は、被験体に、約100mg/キログラム、約150mg/キログラム、約200mg/キログラム、約250mg/キログラム、約300mg/キログラム、約400mg/キログラムまでの投与量で投与され得る。他の実施形態では、免疫グロブリン製剤の投与量は、より多い又はより少ない場合がある。更に、免疫グロブリン製剤は、毎日1以上の投与回数で投与され得る。IgG製剤により治療される疾病に精通している臨床医は、当該分野で知られている基準に従って、患者に適した投与量を定めることができる。
【0083】
ある実施形態では、濃縮されたIgG製剤は、投与につき約5mg/キログラム〜約2000mg/キログラム被験体に投与され得る。ある実施形態では、投与量は、少なくとも約5mg/kg、又は少なくとも約10mg/kg、又は少なくとも約20mg/kg、30mg/kg、40mg/kg、50mg/kg、60mg/kg、70mg/kg、80mg/kg、90mg/kg、100mg/kg、125mg/kg、150mg/kg、175mg/kg、200mg/kg、250mg/kg、300mg/kg、350mg/kg、400mg/kg、450mg/kg、500mg/kg、550mg/kg、600mg/kg、650mg/kg、700mg/kg、750mg/kg、800mg/kg、850mg/kg、900mg/kg、950mg/kg、1000mg/kg、1100mg/kg、1200mg/kg、1300mg/kg、1400mg/kg、1500mg/kg、1600mg/kg、1700mg/kg、1800mg/kg、1900mg/kg、又は少なくとも約2000mg/kgであり得る。
【0084】
本発明に従って、治療工程を完了するのに必要な時間は、医者が定めることができ、1日程の短いものから1〜6ヶ月までの範囲であり得る。
【0085】
濃縮IgG治療の投与量及び頻度は、他の因子の中でも、患者の治療される疾病又は容態並びに疾病又は容態の重症度に依存することになる。一般に、原発性免疫不全では、体重当たり約100mg/kg〜約400mg/kgの投与量が、3〜4週毎に投与されることになる。神経性及び自己免疫疾患では、体重当たり2g/kgが、3〜6ヶ月間、月に1度、連続5日間にわたり施される。これは、一般に、体重当たり約100mg/kg〜約400mg/kgを、ほぼ3〜4週毎に1度、投与することを含む維持治療で補われる。一般に、患者は、ほぼ14〜35日毎に1度、又は、ほぼ21〜28日毎に1度、投与又は処方を受ける。治療の頻度は、他の因子の中でも、患者の治療される疾病又は容態並びに疾病又は容態の重症度に依存することになる。
【0086】
好ましい実施形態では、ヒトの免疫不全、自己免疫疾患、又は急性感染症を治療する方法は、その必要に応じて与えられ、その方法は、本発明の医薬IgG組成物を投与することを含む。
【実施例】
【0087】
以下の実施例は、限定するものとしてではなく、単に説明のために与えられる。当業者は、様々な危急ではないパラメータを変化又は変更して、本質的に同じ又は類似の結果を出し得ることを容易に認識することになる。
【0088】
実施例1:様々なIgG濃度及び製剤の安定性検査
1.目的
この検査の目的は、高い蛋白質濃度での低pH(濃縮溶液中で計測される場合、0.25Mグリシン4.4〜4.9)の保存安定性を、筋肉内及び皮下注入可能な免疫グロブリンに現在利用されるような中性pH製剤(22.5g/lグリシン、3g/lNaClpH7)と比較することである。
【0089】
2.実験計画
全行程は、蛋白質が5%になるようにナノ濾過を濃縮することで開始する。0.15Mのグリシン(調査される最低グリシン濃度)で10回の緩衝剤交換が行われ、続いて、蛋白質が20%を超える、標的値への最終濃縮が行われる。実験の第1工程では、分画分子量が30Kである0.5m2のミリポア社ポリエーテルスルホン薄膜(通常の篩)が利用されるが、実験の第2工程は、開孔篩を有する0.5m2のミリポア社ポリエーテルスルホン薄膜で行われ、その薄膜は、粘性の高い溶液により適しており、洗浄後の成分は、薄膜表面が更に小さな第2の限外/透析濾過装置(0.1m2、開孔篩)により濃縮されて、産出量の損失が低減される。
【0090】
最終容器を調合し、低pHで保存する、又は、低pH保存をバルク溶液で行い、その後に、保存前に、最終容器を中性pHで調合する。
【表1】

【0091】
pH:メトラー・トレド製Lot405−DPA−SC−S8/120電極とPT1000温度探触子とを備えたクニックポータメス型911XPHで、pHを検査した。
【0092】
蛋白質:ビウレットで蛋白質値を確定した。
【0093】
高能率分子篩クロマトグラフィーを利用して、分子径分布を検査した。
【0094】
欧州薬局方に記載されるように、ACA滴定量を検査した。
【0095】
以下の許容基準が定義される:
・高速液体クロマトグラフィーによる分子径分布:
i.単量体及びオリゴマー/二量体:>90%
ii.凝集体:<10%(<%、静脈内投与に対して)
・ACA滴定量:
静脈内投与に対して、50%未満のCH50単位、消費された/mg蛋白質
【0096】
5.結果及び議論
5.1.低pH4.7及びpH7.0に調整された製剤中の凝集体及び断片含有量とACA滴定量との比較
以下の表2で示されるのは、異なる蛋白質濃度での標準的な製剤(pH4.7、0.25Mグリシン、又はpH7.0、22.5g/Lグリシン、3g/L NaCl)に対する、28〜30℃で3ヶ月間保存した後の凝集体及び断片含有量並びにACA的定量である。
【表2】
【0097】
データは、低pHの製剤では、28〜30℃で3ヶ月間保存した後、凝集体が低く、ACA滴定量が低いことを明らかに示す。pH7製剤の全てのACA滴定量は、この検査で定められる許容基準を超える。表2には、2〜8℃で3ヶ月後の値が与えられる。
【表3】
【0098】
2〜8℃での結果では、28〜30℃で見られる傾向が確認される。ACA滴定量は、全て、許容基準として定められた限定値よりも低いが、pH7.0製剤は、値がより高いようである。蛋白質値は、検査されるパラメータの結果に影響を与えない。
【0099】
5.2 製剤IGSC60(Ph4.7、A篩)及びIGSC62(Ph4.7、V篩、より小さな除外濾過システムでの洗浄後濃縮)中の、凝集体及び断片含有量並びにACA滴定量への異なる濃縮手段の影響
[0099]以下の表4で示されるのは、異なる濃縮手段を用いた、異なる蛋白質濃度での低Ph製剤に対する、28〜30℃で3ヶ月間保存した後の凝集体及び断片含有量並びにACA滴定量である。
【表4】
【0100】
両方の濃縮様式について、3ヶ月間保存した後のデータは、類似の結果を示した。表5では、2〜8℃での対応する値が示される。
【表5】
【0101】
2〜8℃で得られる値では、28〜30℃で得られた結果が確認された。濃縮方法は、製品の安定性に影響を与えない。
【0102】
主要濃縮工程の一方のシステムと後洗浄液を濃縮する他方のシステムの、2つのシステムを用いての取り組みにより産出量が高くなるので、この方法は、大規模な製造に更に適している。
【0103】
以下の結論が、この検査にある結果から引き出される:
・低pH製剤により、2〜8℃及び28〜30℃での3ヶ月間の保存の後、ACA値は更に低く、凝集体含有量が更に低く、断片含有量が更に低くなる。
・18〜30℃での3ヶ月間の保存の後、中性pH製剤のACA値は、許容基準を超える。
・蛋白質値は、検査されるパラメータの結果に影響を与えない。
・濃縮方法は、製品の安定性に影響を与えない。
・開孔篩薄膜を用いて適切な後洗浄液を得ることができる。
【0104】
これらの結果に基づいて、低pHでの調合、後洗浄液を濃縮する第2小型装置と開孔薄膜付き除外/透析濾過装置とを利用する濃縮方法を用いて、皮下の投与ための、20%±2%の蛋白質濃度の新規のIgG製品を生成すると結論を下された。
【0105】
実施例2:サブキュヴィアNGの除外濾過及び調合
高濃度前臨床用20%ロットを生産する間に、情報を収集した。ナノ濾過工程までの、20%ロットを製造するのに利用されるプロセスが上に記載された。限外/透析濾過を向上させて、溶液を20%(限界:18〜22%)まで濃縮した。産出量損失を最小限に低減するために、透析濾過に利用される限外濾過装置の後洗浄液は、同じ薄膜を備えた第2小型装置により濃縮され、その後、バルク溶液に加えられる。
【0106】
意外にも、低いpHで保存する間のウイルスの不活性は、溶液の蛋白質含有量に左右されないことを示すことができた。10%溶液(ガンマガード(登録商標)リキッド)及び20%溶液において、類似のウイルスの低減を得た。従って、20%製品に対して、低pHでの保存をウイルス低減工程として維持した。
【0107】
2.プロセス注釈
限外濾過
ナノ濾液のグリシン濃度を0.25Mの標的に調節する。限外濾過(UF)により、溶液を6±2%重量/体積%の蛋白質濃度に濃縮する。通常、蛋白質濃度は、AU280−320の計測により定められる。14.1の消衰係数を利用する。pHを5.2±0.2に調節する。利用されるUF薄膜は、公称分画分子量(NMWCO)が50,000ダルトン以下であり、特に、高粘度製品用に設計されている(ミリポア社からのV篩)。例えば、ミリポア社ペリコンバイオマックスは、NMWCOが50Kダルトン以下である。薄膜材料は、ポリエーテルサルフォンである。
【0108】
pH4.2±0.2の0.25Mグリシン溶液で濃縮液を透析濾過する。最小交換体積は、当初の濃縮液体積の10倍である。限外濾過/透析濾過操作を通じて、溶液を4℃〜20℃に保つ。
【0109】
透析濾過後、溶液を、最小限の22%重量/体積の蛋白質濃度に濃縮する。溶液温度を2℃〜8℃に調節する。UV読み出しにより、14.1の消衰係数値を利用して、蛋白質濃度を確定してもよい。
【0110】
システム中の完全な残留蛋白質を回収するために、第1大型限外濾過システムの後洗浄は、再循環様式で、死容積の少なくとも2倍で行われ、全ての蛋白質が洗い出されたことを確証する。次に、第1システムの10分の1以下の寸法の、同じ型式の薄膜を備えた第2限外/透析濾過システムを用いて、第1限外濾過システムの後洗浄液を少なくとも22%重量/体積の蛋白質濃度に濃縮する。後洗浄濃縮液をバルク溶液に加える。次に、第2限外濾過システムを後洗浄する。工程14の最終バルクの蛋白質濃度を調節するのにこの後洗浄液を利用する。溶液温度は、2℃〜8℃に調節される。
【0111】
調合
更に、第2小型限外濾過システムの後洗浄液で及び/又は透析濾過緩衝剤で、蛋白質濃縮液を20.4±0.4%に調節する。
必要であれば、pHを4.4〜4.0に調節する。
実施例3:20%ロットの製造
【0112】
1.序文
本報告は、前臨床ロット生産を記載し、皮下用途の20%(重量/体積)液体多価ヒト免疫グロブリン製剤である、バクスター社の新規の検査対象の免疫グロブリン製剤「サブキュヴィアNG、20%」の結果を要約する。
【0113】
「本発明の詳細な技術」で記載されたように上記のような濃縮方法を用いて、製造が行われた。分別は、寒冷沈殿物の分離で開始する。次に、後続の冷却アルコール分別の前に様々な粗製凝固因子及び阻害因子を単離するために、寒冷保存血漿が利用され得る。サブキュヴィアNG、20%を浄化するまえに寒冷貯蔵血漿から粗製凝固因子及び阻害因子をバッチ処理で吸着するために、幾つかの経路を選択し、それらの経路は、以下の表で経路1〜7と呼ばれる。
【表6】
【0114】
前臨床サブキュヴィアNG、20%の生成では、多岐にわたる異なる吸着選択肢を包含するように、経路1、3及び6から誘導されるコーン開始材料を選択した。様々な吸着プロセスは、テシュナーら、国際輸血学会誌(2007年、第92号、42〜55頁)、ポルスラーら、国際輸血学会誌(2008年、第94号、184〜192頁)、米国特許第6,395,880号及び第5,409,990号、及び、プロトロンビン複合体:ブルンメルヒュイス、血漿蛋白質分別方法(J.M.カーリング編者、アカデミック出版、1980年)に記載される。
【0115】
変更されたコーンアルコール分別は、沈殿物Gと呼ばれる、主要な中間IgG成分をもたらし、その成分は、クロマトグラフィー浄化を利用して、最終製品に更に処理される。下流製造工程は、陽イオン交換クロマトグラフィー(CMセファロース高速流)と陰イオン交換クロマトグラフィー(ANXセファロース高速流)とを含み、つまり、3つの独立したウイルス不活性又は除去工程を含む。
・溶媒/洗浄剤処理(1%トリトンX−100、0.3%燐酸トリ−N−ブチル及び0.3%ポリソルベート−80の混合物)
・ナノ濾過(アサヒ・プラノバ35nm)及び
・3週間にわたる高温での低pH保存
【0116】
皮下用途のための高蛋白質濃度に到達するために、限外/透析濾過工程で開経路篩を利用しなければならない。好ましくは、第2限外/透析濾過装置を利用して、後洗浄成分を濃縮し、第1装置から全蛋白質を回収する。
【0117】
サブキュヴィアNG、20%は、免疫グロブリンG(IgG)の液体製剤であり、そのうちの少なくとも95%の蛋白質は、ガンマグロブリンである。製品は、等浸透圧性のものであり、低pHに調合される。最終濃縮工程の間、10%蛋白質の濃度で、pHは、4.4〜4.9である。長期間の安定性検査の結果が有効になった後、20%溶液の最終pHが定められる。最終溶液は、180〜220gの蛋白質と、僅かの賦形剤として、リットル当たり0.1〜0.3モルのグリシンとを含む。液体製剤は、僅かに薄い黄色の透明であり、実質的に目に見えない粒子である。
2.前臨床ロットのデータ及び質量均衡
1.前臨床ロット:SC00107NG
吸着選択肢1:選択肢6:F IX、F VII、AT−III
開始材料のロット番号:沈殿物G VNELG171(米国源)
最終容器のロット番号:SC00107NG
2.前臨床ロット:SC00207NG
吸着選択肢3:FEIBA、AT−III
開始材料のロット番号:沈殿物G VNELG173(米国源)
最終容器のロット番号:SC00207NG
3.前臨床ロット:SC00307NG
吸着選択肢1:吸着工程なし
開始材料のロット番号:沈殿物G LB079301(米国源)
最終容器のロット番号:SC00307NG
【表7】
【0118】
最終バルクレベルでは、製剤の純度が、低レベルの主要不純物により示され、それらの不純物は、全IgGの0.1%を十分に下回る。本プロセスのこの最終工程での20%製品の分子径分布は、ガンマガード(登録商標)リキッド/キオヴィグ最終容器の分子径分布と類似している。これは、20%蛋白質までの濃縮が、IgG分子の保全性に負の影響を与えないことを示す。
【0119】
3.前臨床バッチの評価からの追加の結果
最終容器暫定放出基準は、皮下ヒト免疫グロブリンに対する許可局からの要求、皮下投与用の現行製品の最終容器仕様、及びガンマガード(登録商標)リキッド/キオヴィグ仕様に基づいて定められた。製品及び/又はプロセス関連不純物の程度を評価するために、追加の品質管理検査が実行された。
【0120】
更に、最終容器の関連する抗体スペクトルの評価が行われ、前臨床IVIG、10%TVRロットからの結果と比較された。
【0121】
以下の表(表8)に結果が与えられる。
【表8】
【0122】
3つの前臨床サブキュヴィアNG20%最終容器と前臨床ガンマガード(登録商標)リキッド/キオヴィグロットに対する抗体滴定量及び濃縮率は、同程度の大きさである。
【表9】
【0123】
製品及びプロセス関連不純物の除去は、全ての3つのロットに対して十分である。
【0124】
4.結論
サブキュヴィアNG20%の3つの最終容器ロットは、200リットルの大きさで成功裏に製造された。アルコール分別の前に多岐にわたる吸着工程を包含ために、3つの吸着経路を選択した。つまり、
−選択肢1、吸着工程を用いない米国源血漿
−選択肢3、FEIBA、AT−III吸着後の米国源血漿
−選択肢6、F−IX、F−VII、AT−III吸着後の米国源血漿
【0125】
前臨床生産及び中間体の評価の間に監視される処理パラメータについて、最終製品では、3つのロットの間で検出可能な明らかな違いが見られなかった。
【0126】
全ての最終容器は、どの種類の開始材料が選択されたのかに関係なく、製品に関する暫定仕様に適合する。
【0127】
実施例4:20%製剤の保存検査
1.序文
サブキュヴィアNG、20%は、皮下用途の20%(重量/体積)液体多価ヒト免疫グロブリン製剤である。サブキュヴィアNG、20%は、上記のように生産された。
【0128】
この検査は、3つの前臨床ロット及び1つの実行可能性ロットの2〜8℃及び28〜30℃(実行可能性ロットのみ)での6ヶ月までにわたる保存データを要約する。
【0129】
2.目的
本検査の目的として、サブキュヴィアNG20%最終容器の2〜8℃及び28〜30℃での保存安定性を評価する。
【0130】
3.安定性指標パラメータ
主要な劣化様式は、変性、凝集及び壊裂であり、試料の分子径分布が変化することになる。従って、高能率分子篩クロマトグラフィーによる分子径分布の分析は、主要な指標パラメータである。
【0131】
4.バッチ検査及び主要梱包
本報告中にある安定性データは、前臨床ロットSC00107、SC00207NG及びSC00307NG並びに実行可能性ロットIgGSC62/1で作成される。
【0132】
5.結果
以下の表11に、12ヶ月までの保管後の前臨床最終容器の結果を示す。
【表10】
【表11】
【0133】
6.結論
データでは、製品が、18ヶ月までの2〜8℃及び28〜30℃の保管について調査されるパラメータについての定義済みの仕様に準拠することが確認された。
【0134】
実施例5:ウイルス不活性のための低pH処理
目的及び合理性
序文
以下に、サブキュヴィアNG溶液と呼ばれる、免疫グロブリン皮下(ヒト)14〜20%3回ウイルス削減(TVR)溶液は、貯蔵されたヒト血漿から製造される。凝固因子/阻害因子を除去する塊捕獲工程の後に、サブキュヴィアNGの浄化が、変更されたコーンアルコール分別で開始され、主要な中間沈殿物Gを得て、クロマトグラフィー浄化並びに3つの異なるウイルス不活性/除去工程を含む下流プロセスに続く。本検査では、最終容器の低pH保存のウイルス削減能力が、詳細に調査された。
【0135】
下流浄化プロセスは、以下の工程を含む:再懸濁された沈殿物Gが、溶媒/洗浄剤(S/D)処理、カルボキシメチル(CM)セファロース高速流を利用した陽イオン交換クロマトグラフィー及びANXセファロース(登録商標)高速流を利用した陰イオン交換クロマトグラフィー、ナノ濾過、除外濾過、pH調整、並びに減菌濾過及び充填に曝される。無菌充填後、サブキュヴィアNG溶液は、最終容器内で低pH保存に曝される(製造プロセスを概略的に示すために、以下のプロセス方式を参照すること)。
プロセス方式
【0136】
図2に示されるプロセス体系Aは、工程10から始まる、サブキュヴィアNG溶液の製造方式の下流部分の概略を与える。本検査に利用される特別仕様の中間体は、プロセス工程「最終容器、前保存」に等しい。
【0137】
図2に示されるプロセス方式Bは、ウイルス滴定の試料採取を含む本検査に利用される、システム規模縮小型プロセスの概略を与える。
【0138】
潜在的なウイルス伝搬に対する高い安全性の余地を与えるために、作用様式が相補する3つの専用のウイルス不活性/除去工程は、サブキュヴィアNG溶液の製造プロセスに一体化される:
・溶媒/洗浄剤処理(工程11)
・ナノ濾過(工程14)
・最中容器中の高温での低pH保存(後無菌充填)
【0139】
ウイルス不活性に関する低pH及び高温保存の能力及び堅牢性は、以前の検査行程中に既に調査されており、その際、サブキュヴィアNGに等価であるが静脈注射用の10%蛋白質に調整された製品であるIGIV10%VRTが利用された。この検査から得られた結果では、調査される全ての脂質包膜ウイルスが、有効に且つ堅牢に不活性にされたことが実証された。ウシウイルス性下痢ウイルス(BVDV)では、不活性動態が最も低かったことが示された。その上、このプロセス行程が、小さな非脂質包膜DNAウイルスに対する製造プロセスのウイルス安全性に更に寄与することが実証することができた。サブキュヴィアNG及びIGIV10%TVRは、免疫グロブリン製品であり、サブキュヴィアNGは、皮下投与用の変異形態であり、IGIV10%TVRは、静脈内投与用の製品変異形態である。両方が、限外濾過/透析濾過及び調合行程までの同じ製造プロセスを共有し、サブキュヴィアNGの蛋白質濃度が、10%の代わりに、14%〜20%の範囲に調節されることのみ異なる。
【0140】
従って、BVDV及びMMV不活性に関する、サブキュヴィアNG製造中の低pH及び高温保存行程の能力及び堅牢性(欧州医薬品評価機構、ヒト用薬剤評価(2001):CPMPバイオ技術作業部会を参照すること。血漿誘導性医薬品についての手引き、CPMP/BWP/269/95(第3改訂版)に注目すること)を評価するために、選択された基準プロセスパラメータに、製造プロセス用に定められた上限及び下限(即ち、時間、温度には下限、pHには、上限)を設定した。加えて、このウイルス不活性工程の堅牢性を更に調査するために、システム規模縮小行程の間、温度を所定の時間期間にわたって低くした。
【0141】
上に考察されたように、以下の包膜及び非包膜ウイルスを利用した。
・C型肝炎ウイルス(HCV)及び他の小さな脂質包膜RVAウイルスのモデルとして、ウシウイルス性下痢ウイルス(BVDV)。
・ヒトパルボウイルスB19(B19V)及び他の小さな非包膜DNAウイルスのモデルとして、マウス微小ウイルス(MMV)。
【0142】
CPMP指針268/95(欧州医薬品評価機構、ヒト用薬剤評価ユニット(1996):CPMPバイオ技術作業部会。ウイルス検証検査についての手引き:ウイルスの不活性及び除去を検証する検査の設計、寄与及び解釈、CPMP/BWP/268/95(改訂版)に注目すること)に従って、各々の製造プロセスのシステム規模縮小型モデルで検査を行った。サブキュヴィアNG溶液の製造中のウイルス不活性に関して、システム規模縮小型製造工程「低pH及び高温での保存」で得られた結果の有効性を実証するために、大規模製造プロセス中のこの製造工程用に定められたパラメータと、システム規模縮小型モデルのプロセスパラメータとを比較した。加えて、選択された生体化学パラメータを監視し、大規模プロセスの各々のパラメータと比較した。
【0143】
材料、方法及び設備
ウイルス及び細胞
アメリカ培養細胞系統保存機関(ATCC、ロックビル、メリーランド州)から得られるBVDV、NADL株(生体的にクローン化された、ATCC VR−1422)を利用する。標準操作手続に従ってMDBK細胞(ATCC CCL−22)上でウイルスを増殖し、BT細胞(ATCC CRL−1390)上で滴定する。
【0144】
メリーランド州、ロックビルのアメリカ培養細胞系統保存機関から、MMV、プロトタイプ株(ATCC VR−1346)を得た。標準操作手続に従ってウイルスを増殖し、A9細胞(ATCC CCL−1.4)上で滴定した。
【0145】
試験項目
オーストリア国、ウイーン、産業通り131のバクスターのPPD製品対応部門から、「低pH及び高温での保存」前の、即ち、製造工程無菌充填の後の特別仕様のサブキュヴィアNG中間体の以下のロットを得た。
・蛋白質濃度が13.5%のロット番号IGSC64
・蛋白質濃度が20.9%のロット番号IGSC64
【0146】
材料を+2℃〜+8℃の最終容器内に移送し、6ヶ月以内に利用した。
【0147】
緩衝液/溶液
pH調節用溶液
0.5MNaOH又は0.5MHClを利用して、pHを調節した。(室温で両方の溶液を保存し、12ヶ月以内に利用した。)
【表12】
【0148】
薬剤、即ち、蒸留水とHClを室温で組み合わせた。
【表13】
【0149】
それぞれの量のNaOHを、室温で蒸留水中に溶解した。
【0150】
pH計測用溶液
pHを、直接的に及び欧州薬局方(EP)に従う希釈溶液中で計測した。EP方法に従ってpHを計測するために、0.9NaCl溶液を利用して、蛋白質濃度を1%に希釈した。
【0151】
0.9%NaCl溶液を以下のように調製した:9.0±0.9gNaClを1000mlの蒸留水中に溶解した。溶液を0.2μmのフィルタで濾過し、室温で保存し、12ヶ月以内に利用した。
【0152】
細胞培養液
BVDV及びMMVウイルス滴定のための標準操作手続に従って、細胞培養又はウイルス滴定用の培地を調製した。
【0153】
ウイルス滴定アッセイ
TCID50アッセイ
標準操作手続に従うTCID50アッセイにより、ウイルスを含む試料を滴定した。端的に、連続1/2対数希釈液をそれぞれの組織培養液中で調製し、それぞれの指標細胞株を播種されたマイクロ滴定プレートの8ウェルの各々に100μlの各希釈液を加えた。36℃±2℃の温度の加湿されたCO2調節保存ユニット中でマイクロ滴定プレートを保存する。細胞変性効果の評価は、7日保存後に、顕微鏡での細胞の視覚検査により得られる。
【0154】
ポワソン分布に従って50%組織培養感染量(TCID50)を算出し、log10[TCID50/ml]で表した。
【0155】
BVDVバルク滴定
MMVが添加された試料についてMMV検査を行った。低pH保存の20〜27日後にサブキュヴィアNG中間体から取り出されたBVDV添加試料の検出限界を低くするために、これらの試料を以下のように滴定した。標準TCID50アッセイに加えて、1:3.16希釈液(0.5対数)及び以下の2つの1/2対数希釈液(それぞれの細胞培養液を希釈に利用する)の試料を、それぞれの指標細胞株を播種された96ウェルマイクロ滴定プレートの全てのウェル(ウェル当たり100μl)に加える。細胞の保存及びそれぞれのウイルスの細胞変性効果の評価は、以下のように行われた(TCOD50アッセイ)。結果を以下のように算出した。バルク滴定で滴定された体積と通常のTCID50アッセイ滴定された体積の比Rvolを算出した。TCID50アッセイで確定されたウイルス滴定量からRvolのlog10を引き、バルク滴定により確定されたウイルス滴定量としての結果を得る。
【0156】
実施例:
−連続0.5対数希釈(全て12列)のマイクロ滴定プレート上で試料を検定し、その後、希釈0(即ち、希釈されていない)からウェルが負であることを見出す。ポワソン分布に従う計算は、0.11log10[TCID50/ml]よりも小さいウイルス滴定量を与える。検定される試料の対応する全体積は、1.17mlである。
−同じ試料(希釈0)をマイクロ滴定プレートの全てのウェル上に付与する(バルク滴定)。従って、付与される総試料体積は、9.6mlである。
−体積比は、RVol=9.6:1.17=8.21、log10(RVol)=0.91である。
−次に、計算されたウイルス滴定量は、0.11−0.91よりも小さい、即ち、−0.80よりも小さい。ウイルス滴定量の信頼区間の上限を同様に計算する。
連続運動試料中の感染していない場合のウイルス滴定量の計算が、記録される。
【0157】
pH保存の完了後、連続運動試料中で最終試料までウイルス感染性が検出されなかった場合、検定検出限界を算出するために、明らかに負の結果であるTCID50アッセイ中のウェルに利用される全ての連続負性試料の体積を考慮に入れる。このために、以下の式を利用した(付録1も参照すること):
【数1】
式中、
Xi(i=1、2,3、...n)は、n連続負性試料の個々のウイルス滴定量[log10(TCID50/ml)]である。
【0158】
全ての試料が同じ滴定量、即ち、xを有する場合、式は、次式に簡略化される:
【数2】
【0159】
細胞毒性
細胞毒性判定用の各々の細胞株上で、擬似添加された、即ち、ウイルス原液の代わりに、感染細胞用BT培地(5%ウマ血清)*を添加されたサブキュヴィアNG中間体から、並びに、擬似添加され、pHが調節され(pH4.9±0.1)、0.45μmのフィルタで濾過された開始材料から、利用されるウイルス指標細胞株に対するプロセス中間体の細胞毒性を判定した。試料は、ウイルス含有試料と同様に検定された、即ち、希釈され、続いて、指標細胞株と共に保存された。保存後、最も高い非細胞毒性濃度を確定した。これらの生体系の変化に関して、ウイルス添加試料と擬似添加利用の細胞毒性と比較した場合、±0.5対数工程の逸脱は、大きくないと見なされた。
* 感染細胞用BT培地の組成は、以下である:DMEM(4.5g/lのDグルコースを含む)+1%(体積/体積)Lグルタミン(200mM)+1%(体積/体積)硫酸ゲンタマイシン(10mg/ml)+1%(体積/体積)ピルビン酸ナトリウム+2%(体積/体積)重炭酸ナトリウム(7.5%)+1%(体積/体積)非必須アミノ酸+5%(体積/体積)ウマ血清
【0160】
干渉
低ウイルス滴定量の試料に対して、感染性検定の性能への試料基質の影響を評価する必要がある。高いウイルス滴定量を含む試料に対して、希釈系列の重要部分、即ち、ウイルス正性及びウイルス負性ウェルの部分を記録することができ、高い試料希釈で生じる。その結果、ウイルス感染性の検出に対する、希釈された幾つかのlog10工程でもある試料基質の影響を無視することができる。サブキュヴィアNG中間体を含む試料に対する低ウイルス滴定量の確定との干渉は、以下のように計測された。擬似添加され、pH調節され(pH4.9±0.1)、0.45μmのフィルタで濾過された開始材料(サブキュヴィアNG中間体)から試料を採取し、適切な組織培養液で1:3.16に希釈し、希釈済みの(適切な組織培養液、即ち、BT細胞/BVDV用BT培地及びA9細胞/MMV用A9培地中で)ウイルス原液の懸濁液を用いて1:100で、log10(TCID50/ml)の2又は3名目滴定量に添加し、上記のように滴定した。添加プロセス中間体に対して得られた滴定量は、添加細胞培養液の対照の滴定量と比較された。
【0161】
ウイルス低減率の計算
CPMP指針268/95[3]に従って、以下の式を用いして、プロセスのウイルス不活性能力の分析を行った。
【数3】
式中
R=ウイルス低減率
V1=開始材料の体積
T1=開始材料中のウイルス濃度[TCID50/ml]
V2=工程後の材料の体積
T2=工程後のウイルスの濃度[TCID50/ml]
【0162】
処理前及び処理後の各添加試料の体積及び滴定量を用いて、Rを計算した。ウイルスが検出不可能である場合でも、計算のために、ウイルス滴定量として検出限界を取り上げた。標準的な操作手続に従って、小数点以下2桁まで与えられるウイルス滴定量log10(TCID50/ml)を用いて計算を実行した。低減率は、小数点以下1桁まで丸められた。
【0163】
検証パラメータの確定
時間、温度[℃]
タイマで保存時間を計測した。Pt−100センサで温度を計測し、連続的に記録した。
【0164】
pH値
標準的な操作手続に従って、標準的な実験用pHメータを用いて直接的に計測するばかりでなく、欧州薬局方(EP)により定められた方法を実施するにより、即ち、0.9%NaCl溶液を用いて1%蛋白質に希釈した後、pH値を確定した。以下では、添え字(EP)を有するpHは、EP方法に従うpH計測を意味するのに対し、添え字なしの「pH」は、直接計測を意味する。
他の羽他メータの確定
【0165】
分子径分布(MSD)
分子径分布のHPLC−SEC(高性能液体クロマトグラフィー−分子篩クロマトグラフィー)分析は、オーストリア国、ウイーンのバクスター社の分析生体化学部門により、標準的な操作手順に従って行われた。「システム規模拡大」ロット(オーストリア国、ウイーンのバクスター社のPPD製品対応部門で生産される)の全ての試料のHPLC−SEC分析は、この検査に従って実行された。従って、(この検査行程中の検査システム規模縮小プロセス内で蓄積された)擬似添加行程の試料の検査結果と、「システム規模拡大」ロットの検査結果との間の比較可能性を保証するために、擬似添加行程の試料のMSDの分析が、同じ部内で同じ検査に従って行われた。
【0166】
酢酸セルロース電気泳動
オーストリア国、ウイーンのバクスター社のACV化学部により標準的な操作手続に従って酢酸セルロース電気泳動(CAE)を実行した。
【0167】
丸め
標準的な手順に従って丸めを実行した。
・製造プロセス中のそれぞれのパラメータ用に指定されるような、又は、検定の精度により確定されるような小数点以下の同数桁まで、検定結果を丸めた。
・検定結果を丸めずに計算を行い、最終結果のみ丸めらたれた。
設備
・標準的な実験用pHメータを利用した。
・メトラー社AT−100分析天秤及びザルトリウス社BP3100P実験用天秤を利用した。
・PVDF薄膜フィルタ(ミレックスHV、又は均等物)を用いて0.45μm濾過を実行した。
・実験用タイマを用いてプロセス時間を計測した。
・生体安全性分類IIの小室を利用した。
・湿度が調節されたヘラウス社CO2恒温装置内で細胞を保存した。
・混合用に磁気攪拌機/攪拌棒を利用した。
・ハッケ社又はユラボ社低温維持装置及び/又は攪拌加熱ブロックにより温度を制御した。
・横河電機社μR1000レコーダに接続されたPt−100センサを利用して、温度を計測した。
【0168】
検査計画
ウイルス不活性に関する、サブキュヴィアNGの製造工程「低pH及び高温での保存」の堅牢性を調査するために、ウイルス不活性の効率を向上させるための主要パラメータを以下のように定めた。低pH保存によるウイルス不活性の堅牢性を調査するのに適した条件下で行程を実行した。大規模プロセスの条件とシステム規模縮小行程の条件の比較は、表Aに与えられる。
・蛋白質濃度:サブキュヴィアNGは、開発中の製品であり、調合されたバルクの蛋白質濃度範囲は、現時点では、ある程度広く定められる、つまり、約14%〜20%であり、後により狭くされ得る。従って、低pH処理の堅牢性を調査するために、2つの行程(行程計画)を、2つの極端な可能な蛋白質濃度で実行した。行程計画1を13.5%蛋白質濃度で実行した。行程計画2を20.9%蛋白質濃度で実行した。
特別仕様の材料の蛋白質濃度は、PPD製品対応部門により与えられた。特別仕様の材料の蛋白質濃度は、分析からデータが既に利用可能であったので再度定められず、特別仕様のサブキュヴィアNG中間体を生成した後に実行された。システム規模縮小プロセスに利用された材料は、減菌濾過され、システム規模縮小プロセスを開始するまで、2℃〜8℃で保存された(即ち、いずれの凍結/解凍手段にも曝されない)。最終的に調合されて減菌濾過された中間体に対する蛋白質濃度の変化は、予期されない。
・pH値:生産では、4.4〜4.9のpH範囲(直接計測)が、低pH保存を通過したサブキュヴィアNG最終容器で示される。ウイルス不活性にあまり好ましくはない条件下の低pH処理の有効性及び堅牢性を調査するために、2つの行程が、定められた上限で、即ち、4.9±0.1のpHで実行された。pHは、添加後に計測され、必要な場合、上限に調節された。低pHで27日±5時間にわたり保存した後、pHが、直接pH計測により、及び、欧州薬局方により推奨される方法により、即ち、0.9%NaCl溶液で1%蛋白質に希釈して再び計測された。
・温度:生産プロセスでは、低pH保存の間の温度は、30〜32℃に設定される。ウイルス不活性にあまり好ましない条件下での低pH保存の堅牢性を調査するために、全てのシステム規模縮小行程が、温度範囲の下限で、即ち、30±1℃で実行された。温度揺らぎの潜在的な影響を調査するために、温度は、毎週一回、6時間以上にわたり25±1℃に低くされた(即ち、温度低下は、保存第0日、第7日、第14日、第20日及び第27日に開始された。日数は、暦の日を指す。即ち、第0日は、保存が開始された日である)。誤って、6時間にわたる25℃1±1℃への温度低下が、第1週及び第2週の保存中に、週毎に2回、即ち、第1週の保存第0日及び第4日、低pH処理の第2週の保存第7日及び第10日に実行された。
・時間:保存時間は、通常、21日(BVDVの有効な不活性に必要とされる)〜28日(技術面/物流面の理由による上限)の範囲である。ウイルス不活性にあまり好ましくはない条件下の低pH処理の有効性及び堅牢性を調査するために、全てのシステム規模縮小行程が、上述の保存時間範囲の限界よりも下で、即ち、短い選択肢に対して20日±4日にわたり、長い選択肢に対して27日±5日にわたり実行された。添加された中間体を、低pH及び高温で27日±5日にわたり保存したが、ウイルス滴定用の試料を、20日±4日保存した後に採取し、3週保存した後にウイルス排除を調査した(表Aも参照すること)。
【0169】
総合的に、総計、4つのウイルス添加行程(BVDVで2つ、MMVで2つ)を実行して、製造工程「低pH及び高温での保存」のウイルス不活性能の堅牢性を調査した。更に、2つの添加されていない対照行程を実行して、生体化学パラメータを選定するための試料を生成した。加えて、2つの擬似添加対照行程を実行して、これらの行程から取り出された試料を利用して、ウイルス滴定検定へのプロセス中間体の潜在的な影響(即ち、細胞毒性及び干渉)を調査した。
【0170】
システム規模縮小プロセスと大規模プロセスの均等性更に示すために、最終製品について、低pH保存前及び保存後に以下の生体化学分析:分子径分布及び酢酸セルロース電気泳動を実行した。
【表14】
1 サブキュヴィアNGは、開発中の製品であり、蛋白質濃度は、尚、狭く定められていない。従って、2つのシステム規模縮小行程を、2つの極端な可能な蛋白質濃度で実行した。
2 保存期間は、尚、狭く定められておらず、ウイルス排除検査の結果に依存する。
3 両方の行程を、27日±5時間にわたり実行し、ウイルス滴定用の試料を保存第20日で取り出したので、ウイルス不活性データは、21日〜22日保存の大規模プロセス選択肢に利用することができた。
4 温度を、保存の週毎に1回、6時間にわたり25±1℃に低くした(即ち、温度低下は、第0日、第7日、第14日、第20日及び第27日に開始された。日数は、暦の日を指す。即ち、第0日は、保存が開始された日である)。第4.1.1章(「温度分布」)で考察されるように、6時間にわたる25±1℃への温度低下を、誤って、保存第1週及び第2週に、週毎に2回実行した(DR1_0407も参照すること)。
5 欧州薬局方による方法
【0171】
実験手順
その複雑性により、ウイルス添加工程、pH調節工程b及び0.45mm濾過工程の序列が、図3に見られる流れ図に示される(矢印の厚さは、相対的な体積を示す)。
【0172】
開始材料
行程1では、蛋白質濃度が13.5%の特別試料の材料(ロット番号IGSC64)を取り入れた。この材料を利用して、可能な蛋白質濃度の下限で、pH範囲の上限(即ち、BVDV:4.97及びMMV4.93)で、及び温度範囲の下限で(即ち、29.4℃で)行程を実行した。
【0173】
行程2では、蛋白質濃度が20.9%の特別仕様の材料(ロット番号IGSC64)を取り入れた。この材料を利用して、可能な蛋白質濃度の上限で、pH範囲の上限(即ち、BVDV:4.92及びMMV4.86)で、及び温度範囲の下限で(即ち、29.4℃で)行程を実行した。
【0174】
45.5mlの各々の開始材料に4.5mlのウイルス原液懸濁液を添加し、1〜2分の攪拌後に1mlの試料を採取した。即座に、試料をそれぞれの細胞培養液で1:3.16に希釈し(即ち、1体積の試料に2.16体積の細胞培養液を加え)、滴定した。続いて、別の3mlの試料を採取し、0.45μmPVDF薄膜を通して濾過した。即座に、1mlの濾過された材料をそれぞれの細胞培養液で1:3.16に希釈し、滴定した。
【0175】
pHの調節
一定分量の35mlのウイルス添加開始材料を0.5M HCl溶液を用いて攪拌下で(両方の行程で)4.9±0.1のpHに希釈した。次に、材料を2つの一定分量に分割した。一方の30mlの一定分量を「低pH保存」に利用し、5mlの一定分量を「温度保持対照」に利用した(「保持対照」の章を参照すること)。
【0176】
別の一定分量の10mlのウイルス添加開始材料を、0.5M NaOH溶液を用いて7.0±0.1のpHに調節した。5mlのpH調節された材料を「pH保持対照」に利用した。
【0177】
残りの5mlのpH調節された材料を「pH温度組み合わせ型保持対照」に利用した(「保持対照」の章を参照すること)。
【0178】
保持対照
ウイルス不活性の機能を、即ち、pH又は温度によるその機能を調査するために、3つの保持対照を異なる条件下で保存した。
【0179】
各々の5ml一定分量のウイルス添加pH調節(pH7.0±0.1)開始材料を、0.45μmPVDF薄膜フィルタを通して寒冷薬瓶内に濾過した。一定分量を、添加プロセス材料と共に30±1℃で保存し、次に、プロセスの終わりまでこの温度で保存した(pH保持対照、「pHHC」)。即座に、他の一定分量を+2℃〜+8℃で、27日±5時間にわたり保存した(pH温度組み合わせ型保持対照)。
【0180】
5mlの一定分量のウイルス添加pH調節(pH4.9±0.1)0.45μ濾過開始材料(上の「pH調節」を参照すること)を即座に+2℃〜+8℃に、プロセスの終わりまで保存した(温度保持対照、「tHC」)。
【0181】
濾過後の全ての保持対照の最小体積は、常に3mlを上回り、従って、保存期間後のウイルス滴定用に適した体積を利用することができた。
【0182】
低pH保存
30ml一定分量のウイルス添加pH調節(pH4.9±0.1)サブキュヴィアNG中間体を、0.45μmPVDF薄膜フィルタを通して、50ml無菌ガラス瓶内に濾過した。濾過後のサブキュヴィアNG中間体の最小体積は、常に、24mlを上回った。したがって、保存期間後のウイルス滴定に十分な体積を利用することができた。続いて、水槽を利用し、前後の緩やかな動きの攪拌下で、温度を30±1℃に保ち、外部温度センサを介して温度調節低温維持装置により、その水槽を調節した。外部センサ及び追加のPt100電極を、低pH保存に利用されたガラス瓶と等価な50mlガラス瓶内に置いた。各々のガラス瓶を、濾過後の添加プロセス材料の最大可能量である30mlの水で充たした。温度を連続的に記録した。材料が29℃の温度に達すると直ぐに、試料取り出しを開始した。低pHによる更なるウイルスの不活性を妨げるために、即座に、各々の試料を、それぞれの冷却細胞培養液(+2℃〜+8℃で保存)で1:3.16に希釈し(即ち、1体積の試料に2.16体積の細胞培養液を加え)、滴定した。全ての行程では、27日±5時間にわたる全プロセスを通して、サブキュヴィアNG中間体を(前後の緩やかな動きの)攪拌下で30±1℃に保存し、保存の毎週1回、少なくとも6時間(6時間以上)にわたり温度を25±1℃に下げた。誤って、6時間にわたる25±1℃への温度低下を、保存第1週及び第2週の週毎に2回、即ち、第1週の保存第0日と第4日、及び、低pH処理の第2週の保存第7日と第10日に行った。考察のために、第4.1.1章(「温度分布」)を参照すること。他の試料を抽出計画(第3.2章を参照すること)に従って採取し、即座に上記のように希釈し、滴定した。30℃±1℃での低PH保存が完了した後、直接計測により、及び、欧州薬局方により推奨される方法(0.9%NaCl溶液で1%蛋白質に希釈すること)によりpHを確定した。
【0183】
ウイルスを用いない対照行程
2つの対照行程を、ウイルス添加行程で記載されたように実行した。添加されずに0.45μm濾過されたサブキュヴィアNG中間体を処理した。材料をpH調節しなかったことを除いて、ウイルス添加行程1及び2でそれぞれ示されるのと同じプロセスパラメータを用いた。生体化学パラメータを確定するための試料を、抽出計画(第3.2章を参照すること)に従って取り出した。
【0184】
擬似添加開始材料(感染細胞用BT培地で0.9:10に添加された*;滴定前に上記のように試料を濾過する)、及び、0.45μm濾過後の擬似添加pH調節サブキュヴィアNG中間体の細胞毒性を判定した。細胞毒性を判定するための試料を、抽出計画(第3.2章を参照すること)に従って取り出した。
* 感染細胞用BT培地の組成は、以下である:DMEM(4.5g/lのDグルコースを含む)+1%(体積/体積)Lグルタミン(200mM)+1%(体積/体積)硫酸ゲンタマイシン(10mg/ml)+1%(体積/体積)ピルビン酸ナトリウム+2%(体積/体積)重炭酸ナトリウム(7.5%)+1%(体積/体積)非必須アミノ酸+5%(体積/体積)ウマ血清
【0185】
潜在的干渉
ウイルス検出と、pH(pH4.9±0.1)調節後のサブキュヴィアNG中間体を含む試料に対する試料基質の干渉を、以下のように各々の指標細胞株に対して2通りで調査した。擬似添加され、pH調節され、0.45μm濾過されたサブキュヴィアNG中間体から採取された1mlの試料を、それぞれの冷却細胞培養液(+2℃〜+8℃で保存)で1:3.16(体積/体積)に希釈した。続いて、1.8mlの希釈された材料に、0.2mlの希釈済みの*ウイルス原液懸濁液を1:10で添加し、2.0及び3.0log10[TCID50/ml]の計算された滴定量にした。混合後、試料を採取し、即座に滴定した。対照として、1.8mlのそれぞれの冷却細胞培養液(+2℃〜+8℃で保存)に、まさに同じ方法で、滴定前に、希釈済みのウイルス原液懸濁液を添加した。
* ウイルス原液懸濁液を事前に希釈するのに、適切な細胞培養液[BT細胞用BT培地(BVDV)、A9細胞用A9培地(MMV)]を利用した。
【0186】
抽出計画
ウイルス滴定
試料採取には、以下の許容範囲が適用される:1日(±1時間)保存後、2〜6日(±2時間)保存後、7〜13日(±3時間)保存後、14〜20日(±4時間)保存後、及び21〜27日(±5時間)保存後。試料符号についでは、第1.1章を参照すること(プロセス方式)
【0187】
即座に、試料を更に保存せずに滴定した。
【表15】
【0188】
細胞毒性の判定
即座に、試料を更に保存せず滴定した。
【表16】
【0189】
干渉の判定
即座に、試料を更に保存することなく滴定した。
【表17】
【0190】
生体化学パラメータの判定
各々の無添加対照行程からのみ試料を採取し、分析するまで、+2〜+8℃で保存した。
【表18】
1 システム規模縮小プロセスを評価するために、2つの「システム規模拡大」ロット並びに特別仕様材料(ロット番号IGSC64)の生産中に生成されたデータも考慮した。
2 システム規模縮小プロセスを評価するために、2つの「システム規模拡大」ロットの生産中に生成されたデータも考慮した。
3 製品の開発段階に関して、28日の保存期間の後の生体化学パラメータのデータを利用することができるとは考えられない。
【0191】
結果及び考察
この工程のシステム規模縮小型実験用モデルを利用して、脂質包膜ウイルスと脂質非包膜ウイルスの両方に専用の不活性工程としてのサブキュヴィアNGの低pH及び高温での保存の有効性及び堅牢性をBVDVとMMVで調査した。検証パラメータ及び生体化学パラメータを定義し、システム規模縮小プロセスで計測し、製造プロセス条件でのそれらの結果を比較することにより、2つのプロセスの均等性を実証した。
検証パラメータ及び生体化学パラメータについての結果
【0192】
検証パラメータ
ウイルス不活性に重要なパラメータ、即ち、プロセス材料のpH、低pH保存中の温度及び保存時間を判定することにより、システム規模縮小手段の有効性を確認した。
【0193】
低pH及び高温での保存前のpH値
添加プロセス材料のpHは、計測され、全ての行程において必要に応じて、4.9±0.1に調節され、従って、検査計画で示された限界内であった。保存後に確定されたpH値を第4.1.2章(他のプロセスパラメータ及び生体化学パラメータ)で考察する。
【0194】
温度分布
温度変化に対する、低pH及び高温での保存工程の堅牢性を調査するために、保存の毎週1回、温度を30℃±1℃から25±1℃に6時間にわたり下げた。温度は、常に、検査計画で示された範囲内であった。しかしながら、保存第1週及び第2週では、6時間にわたる25±1℃への温度低下を、週に2回、即ち、第1週の第0日及び第4日、第2週の第7日及び第10日に、誤って実行した。この事象が、行程概要を、ウイルス不活性に関してより悪い状況に移したので、ウイルス不活性の堅牢性を、当初意図したものよりも更に広範囲にわたり調査した。
【0195】
保存時間
低pH及び高温での保存の継続時間は、サブキュヴィアNGの製造プロセスで示されていない。現時点での検査結果に応じて、幾つかの選択肢がある:保存時間の継続時間の第1選択肢は、21〜22日である。第2選択肢は、約28日であってもよい。ウイルス不活性にあまり好ましくない条件下での低pH処理の有効性及び堅牢性を調査するために、全てのシステム規模縮小行程を、可能な保存時間の下限よりも下で、即ち、第1選択肢に対して20日±4時間にわたり、第2選択肢に対して27日±5時間にわたり実行した。添加された中間体を、低pH及び高温で、26日+22時間(MMV、両方の行程)にわたり、27日+1時間(BVDV、両方の行程)にわたり保存した。3週間保存した後のウイルス排除を調査するために、MMVに対して20日+1時間保存した後に、BVDVに対して20日+2時間後に、ウイルス滴定用試料を採取した。
【0196】
保存時間は、全ての行程において、システム規模縮小で示された限界、即ち、20日±4時間、27日±5時間以内であった。その限界は、現時点で考えられる製造中の保存時間の下限を下回る。
他のプロセスパラメータ及び生体化学パラメータ
【0197】
低pH保存後のpH
低pH保存に続いて、pH値を再び、直接的に、並びに、欧州薬局方(EP)により推奨される方法に従って、即ち、0.9%NaCl溶液で1%蛋白質に希釈して計測した。処理後に直接計測により計測されるpHは、低pH保存の前では、pH値に近かった。即ち、差は、−0.07〜+0.04(保存前のpHを引いた後のpH)の範囲であった。EP方法に従って計測されたpHは、常に、直接計測方法と比べて0.2〜0.3だけ高かった。EP方法を利用する際のこの僅かなpH値の増加は、pHを定めるこれらの2つの異なる方法の典型である。この事実は、製造規模で定められた限界内にあるとも考えられる。低pH処理後のpHの限界は、直接計測により定められる場合、4.4〜4.9であり、EP方法により定められる場合、4.6〜5.1である。
【0198】
分子径分布(MSD)
2つの無添加対照行程での保存の前後に、分子径分布(HPLC分析による)を調査した。サブキュヴィアNGの開発工程中に生産された3つの「システム規模拡大」ロットについて、及び、特別仕様の材料IGSC64(14%並びに20%蛋白質濃度)について検査結果を比較した。「システム規模拡大」ロットとも表記されるロットIGSC64は、この行程では十分な材料を入手することができなかったので、製造中に低pH及び高温での保存に曝されなかった。全ての結果は、互いに十分に比較された。分子径分布の値、特に、IgG単量体の百分率は、「システム規模拡大」行程で観察されたものに近かった。保存後のIgG単量体の濃度が数パーセント低かった、第2対照行程で留意することは、既に、保存前の材料のIgG単量体の濃度が比較的低く、その低い濃度は、「システム規模拡大」プロセス内で検査される材料にも適用されることに留意すること。従って、分子径分布データは、大規模プロセスとシステム規模縮小の等価性に対応する。
【0199】
ガンマグロブリンの純度
2つの無添加対照行程からの試料のガンマグロブリンの純度(CA電気泳動による)を確定し、2つの「システム規模拡大」ロットから、及び、特別仕様の材料IGSC64(14%並びに20%蛋白質濃度、低pH処理直前の値、説明は、上を参照すること)から得られた結果と比較した。システム規模縮小プロセス中のガンマグロブリンの純度について定められる全ての値は、「システム規模拡大」ロットで得られる結果に十分に匹敵し、検定精度以内であった(相対的標準偏差は、1.5%である)。これらの結果は、製造プロセスとシステム規模縮小プロセスの同等性を実証する。
ウイルス滴定の結果
【0200】
細胞毒性
低pH及び高温での保存前に得られた中間体の細胞毒性を、ウイルス添加行程で記載されたようにそれぞれのpHに調節された擬似添加材料を利用した対照行程で調査した。pH調節前後の擬似添加プロセス中間体では、BT細胞上での行程2を除いた全ての2つの行程で利用される細胞への細胞毒性の極めて弱い影響のみが見られた。pH調節された中間体では、1.0log10希釈及びそれ以降から、細胞毒性の影響が見られなかった。ウイルス添加行程について細胞毒性の著しい差は見られなかった。
【0201】
干渉検査
干渉検査からの結果では、低滴定量のBVDV及びMMVの検出と試料基質の著しい干渉は見られなかった。添加細胞培養液対照とサブキュヴィアNG中間体との間の添加ウイルス滴定量の差は、−1.0log10〜0.1log10であって、ウイルス滴定検定の精度内にある。従って、ウイルス不活性行程中に得られた滴定量は、干渉効果により歪まず、試料の実際の滴定量を示した。
【0202】
ウイルス不活性
ウイルスBVDV及びMMVの各々を用いて2つの行程を実行した。即ち、行程1は、13.5%蛋白質濃度であり、行程2は、20.9%蛋白質濃度であった。両方の行程を、4.9±0.1のpH、30±1℃で実行した。温度は、週毎に1回、最小6時間にわたり25±1℃に下げられた。2つの行程の結果の比較では、サブキュヴィアNGの可能な蛋白質濃度の上限及び下限で実行された2つの行程の間で、ウイルス不活性動態の著しい違いが現れなかった。
【0203】
ウイルス不活性にあまり好ましくない条件、即ち、保存時間及び温度の下限並びにpHの上限を用いて、BVDVについて、20日±4時間保存した後のBVDVの十分な不活性と、27日±5時間保存した後にそれぞれのサブキュヴィアNG中間体内に添加され得る全てのウイルスの完全な不活性が、調査される条件にかわわりなく実証された。両方の行程では、BVDVからの残留干渉性を、第20日に、尚も検出することができた。しかしながら、全てのBVDVは、27日保存の終わりに、検出限界以下に不活性化された。MMVの低減率では、脂質膜のないウイルスに関しても、この工程が、製品のウイルス安全性に大きく寄与することが実証された。利用されたウイルスに対する個別のウイルス滴定量及び低減率を以下でより詳細に考察する。加えて、ウイルス不活性動態の図表示が、各々のウイルスに対して与えられ、それぞれの結果表に追従している。
【0204】
BVDV
BVDVは、保存20日によるほぼ検出限界で(行程1)又は検出限界で(行程2)不活性化され、行程1及び2では、それぞれ、5.3log10及び5.5log10の低減率が与えられた。27日保存した後に、サブキュヴィアNG内に添加された全てのBVDVは、両方の行程で検出限界以下に不活性化され、低減率は、行程1及び2で、それぞれ、6.6log10及び6.5log10よりも大きかった。2つの行程の不活性動態の大きな差を観察することができなかった。行程1の中間体(蛋白質濃度が13.5%)と行程2の中間体(蛋白質濃度が20.9%)の両方で、不活性動態が拮抗していることが示された。
【0205】
高温での低pH処理では、20日±4時間保存した後のBVDVの不活性が有効であることが実証された。計算された平均低減率は、5.4log10であった。27日間低pH及び高温で保存した後、BVDVの検出限界以下への完全な不活性が得られ、6.6log10を上回る平均低減率が算出された。
【0206】
MMV
MMVでは、20日間低pH及び高温で保存した後に、行程1及び2に対して、それぞれ、2.9log10及び3.1log10の低減率が算出された。算出された平均低減率は、3.0log10である。27日間低pH処理した後、3.4log10及び3.7log10の低減率が得られた。算出された平均低減率は、3.6log10である。これらの低減率では、生理化学不活性に極めて耐性のあるパルボウイルスに関して、このプロセス行程が、製造プロセスのウイルス安全性に大きく寄与することが実証された。両方の行程でのウイルス不活性動態は二相性であり、不活性は、最初の7日間はより速く、続く3週間は多少より緩やかであった。
【0207】
保持対照
ウイルス不活性の機能、即ち、ウイルス不活性がpHにより、又は、温度により、又は、上記2つの組み合わせにより仲介されるのかを調査するために、3つの保持対照が、それぞれの条件下に保たれた。
【0208】
27日±5時間にわたり2℃〜8℃に保たれた「組み合わせ型保持対照」(pH7.0±0.1)を滴定した結果、調査された両方のウイルスに対して、ウイルス滴定量は、行程1のMMVを除いたウイルス添加開始材料に匹敵することになった。滴定量の少量の損失(1.6log10)が観察された。
【0209】
30±1℃及びpH7.0±0.1での保存(「pH保持対照」)では、脂質包膜ウイルスBVDVが、高温での保存に極めて鋭敏であることが示された。BVDVは、両方の行程で、4.6log10で不活性化された。その上、脂質膜のないウイルスMMVは、行程1及び行程2で、それぞれ、3.3log10及び3.8log10で不活性化された。
【0210】
27日±5時間にわたり低pHで2℃〜8℃に保たれた「温度保持対照」(tHC)を滴定した結果、調査された両方のウイルスに対して、ウイルス滴定量は、ウイルス添加開始材料に匹敵することになった。これらの結果で、BVDV並びにMMVは、4.4〜4.9の範囲内の低pH及び低温での保存に耐性があることが実証された。
【0211】
総じて、3つの保持対照の結果は、以下を示唆する:
・ 温度は、BVDVの不活性に最も重要な因子であり、低pHは、ある程度寄与し得る(30±1℃及び中性pHでの対照が、27日後に、完全ではないが実質的に不活性化されたことに基づく)。
・ MMVの不活性では、27日後の低pH動態試料のウイルス滴定量が、中性pH及び30℃での対照と実質的に同一であるので、温度は、唯一の重要な因子であると思われる。
【0212】
本検査行程では、サブキュヴィアNGの製造での低pH及び高温での保存のシステム規模縮小モデルを実証し、幾つかのプロセスパラメータを判定することにより製造手段のその等価物を実証した。加えて、システム規模縮小モデルと製造プロセスからの中間体の生体化学パラメータの比較は、システム規模縮小プロセスで得られる低減率が大規模プロセスに対しても有効であるという、両方のプロセスの等価性を裏付けた。ウイルス不活性に関する堅牢性を調査するために、サブキュヴィアNGの異なる蛋白質濃度の影響を調査した。添加開始材料のpHを上限に調節して、製造プロセスと比較し、温度の周期的低下の影響を調査した。本検査で得られるウイルス低減率及び不活性動態は、脂質包膜ウイルスBVDVが、27日内の低pH及び高温での保存により有効に且つ堅牢に不活性化されることを実証した。その上、20日にわたり低pH及び高温で保存した後、BVDVについて、著しいBVDVの不活性が得られ、平均低減率は、5.4log10であった。パルボウイルスモデルMMVに対して算出された平均低減率、即ち、20日保存後の3.0log10、20日保存後の3.6log10は、このプロセス行程が、生理化学耐性の高い小さな脂質膜のないDNAウイルスに関して、20日並びに27日の保存時間にわたる製造工程のウイルス安全性に更に寄与することを示す。
【0213】
調査された両方の蛋白質濃度では、MMV及びBVDVに対して同じ不活性動態が示され、濃縮免疫グロブリン溶液並びにウイルスBVDV及びMMVに対して、ウイルス不活性能への蛋白質濃度の影響が大きくないことを示唆する。
【0214】
本明細書に記載される実施例及び実施形態は、単に説明目的のためのものであり、それらに照らし合わせて様々な修正又は変更が、当業者に暗示されることになり、本出願の趣旨及び権限並びに付属の請求項の範囲内に含まれるべきであると理解される。本明細書で引用される全ての公報、特許、及び特許出願は、全ての目的のために、参照としてそのまま本明細書に組み込まれている。
【技術分野】
【0001】
本出願は、2009年5月27日に出願され、全ての目的のために本明細書に参照としてそのまま明確に組み込まれている、米国仮特許出願第61/181,606号の利益を請求する。
【背景技術】
【0002】
ヒト血漿からの免疫グロブリン製品は、1952年、免疫不全を治療するために最初に利用された。初期は、IgGの筋肉内投与又は皮下投与が、選択方法であった。しかしながら、様々な疾病に対する有効な治療に必要なIgGを多量に注入するために、IgG濃度の低い(50mg/mL)、静脈注射で投与可能な製品が開発された。通常、静脈注射用免疫グロブリン(IVIG)は、千人を超える血液提供者の血漿から貯留された免疫グロブリンG(IgG)(単数)、免疫グロブリン(複数)を含む。通常、Fc依存性作動因子機能が無傷である95%超過の非修飾IgGと微量の免疫グロブリンA(IgA)又は免疫グロブリンM(IgM)とを含むIVIGは、3つの主分類の病状:1.X連鎖無ガンマグロブリン血症、低ガンマグロブリン血症(原発性免疫不全)、後天性免疫不全疾患(続発性免疫不全)等の低抗体レベルを特徴とする免疫不全、2.炎症性及び自己免疫疾患、及び、3.急性感染症を治療するのに主に利用される、無菌の浄化されたIgG製品である。
【0003】
数多くのIVIGの市場供給者は、様々なIVIG製品を提供している。再溶出後の溶液中に50mg/mL蛋白質のみを含む、旧型の凍結乾燥されたIVIG製品と比較して、最新の開発品は、高度に浄化、濃縮されたヒトIgG抗体の100mg/mLの使用準備済みの無菌の液体製剤である。IVIG等のIgG製品は、貯蔵されたヒト血漿から製造されるので、提供者の血液からの病因汚染物(特に、ヒトに様々な疾病を引き起こすことで知られているウイルス)は、生産プロセスでの重大な懸念事項である。IgG製品についての別の重大な検討事項は、特に、使用準備済みの製剤としての、それら製品の貯蔵中の安定性である。IVIGと比べると、皮下投与可能な免疫グロブリン製剤は、家庭での治療処置が可能にし、副作用があまりないという利点がある。部位当たりの注入体積が少ないという不利な点をなくすために、IgGの濃度をより高くする(例えば、100mg/mLの代わりに200mg/mLを含む)ことは、明らかに有利になり得る。
【0004】
血清及び血漿蛋白質の調製及び特性に関して一連の影響力をもつ論文のうちの第4の連載記事において、コーンら(アメリカ化学学会誌、1946年、第68(3)号、459〜475頁)は、ヒト血漿からIgG濃縮成分を単離するのを可能にする、血漿蛋白質アルコール分別方法(方法6)を最初に記載している。数年後、オンクレイら(アメリカ化学学会誌、1949年、第71(2)号、541〜550頁)は、コーンの方法を拡張し、純粋なIgG製剤を単離することになる方法(方法9)を発表した。
【0005】
これらの方法は、血漿由来血液因子の全産業の基礎を築く一方で、川崎病、免疫血小板減少性紫斑病、及び原発性免疫不全を含む、幾つかの免疫関連疾患を治療するのに十分な高濃度のIgG製剤を与えることができなかった。そのように、高純度で高濃度のIgG製剤を提供するために、イオン交換クロマトグラフィー等の様々な技術を用いる追加の方法論が開発された。ホップら(ミュンヘン医事週報誌、1967年、(34)、1749〜1752頁)、ファルクスヴェーデン(スウェーデン特許、第348942号)、ファルクスヴェーデン及びルンドブラッド(血漿蛋白質分別方法、1980年)は、この目的のためにイオン交換クロマトグラフィーを先駆けて用いた。
【0006】
様々な最新の方法は、カプリル酸塩沈殿(レービングら、国際輸血学会誌、2003年、(84)、193〜201頁)、カラムクロマトグラフィーと組み合わせたコーン分画(I+)II+IIIエタノール沈殿(タナカら、ブラジル医療生体研究誌、2000年、(33)、37〜30頁)等の沈殿工程を用いる。最近では、テシュナーら(国際輸血学会誌、2007年、(92)、42〜55頁)は、貯蔵された血漿から寒冷沈殿物を除去し、次に、変更されたコーン・オンクレイ冷却エタノール分別、続いて、中間イオン交換クロマトグラフィーのS/D処理、ナノ濾過、及び任意選択で限外濾過/透析濾過を実行する、10%IVIG製品生成方法を記載している。
【発明の概要】
【発明が解決しようとする課題】
【0007】
しかしながら、これらのIgG製造方法によりもたらされる純度、安全性、及び産出量が向上しているにもかかわらず、皮下投与及び/又は筋肉内投与に適した高濃度のIgG製剤は、尚も必要とされている。本発明は、これらの他の必要性を充たし、安定で高度に浄化され、ウイルスに不活性で使用準備済みの、IgG濃度の高い製品を製造する方法を記載する。
【課題を解決するための手段】
【0008】
一態様では、本発明は、組成物1リットル当たり約180グラム超過の蛋白質を含み、少なくとも95%の蛋白質が、ヒトIgG等のIgGである、水性組成物を提供する。幾つかの実施形態では、組成物は、皮下投与及び筋肉内投与に適した製品を与えるプロセスにより生成され、10〜22%の蛋白質濃度範囲でどの濃度に調節されるのかに関係なく、ウイルスを不活性にするために最終容器内で高温で処理され得る。幾つかの事例では、組成物中の蛋白質濃度は、20%(重量/体積)であるか、又は、ほぼその濃度である。他の事例では、組成物は、約0.1〜0.3Mグリシンを更に含み得る。本発明の組成物は、pHが約3〜6又は約4〜6のように変化し得る。
【0009】
別の態様では、本発明は、血漿から濃縮されたIgG組成物を調製する方法を提供し、(1)限外濾過により血漿製剤中の蛋白質を5%(重量/体積)又はほぼその濃度に濃縮する工程と、(2)透析濾過により製剤中の蛋白質を20%(重量/体積)又はほぼその濃度に更に濃縮する工程とを含む改善を有する。組成物中の少なくとも95%の上記蛋白質は、ヒトIgG等のIgGである。幾つかの実施形態では、工程(1)は、公称分画分子量(NMWCO)が100kDa以下である限外濾過薄膜を利用して実行される。他の実施形態では、工程(2)は、グリシンの透析濾過緩衝液を用いて4.2±0.1のpHで実行される。幾つかの事例では、透析濾過緩衝体は、0.25Mグリシンを有し、pHが4.0である。幾つかの特定の実施形態では、工程(2)後の蛋白質濃度は、20%(重量/体積)よりも高く、続いて、透析濾過緩衝体で、20%(重量/体積)に又はほぼその濃度に調節される。
【0010】
別の態様では、本発明は、以下の工程を含む、血漿から濃縮IgG組成物を調製する方法を提供する:
(1)遠心分離により血漿から液体と沈殿物とを分離すること、
(2)事前に冷却されたエタノールを(1)からの液体と混合して、エタノール濃度が8%(体積/体積)又はほぼその値である混合物を作ること、
(3)遠心分離により、(2)の混合物から液体と沈殿物とを分離すること、
(4)(3)からの液体のpH及びエタノール濃度を、それぞれ、7.0及び20〜25%(体積/体積)又はそれらの値に調節することにより、混合物を作ること、
(5)遠心分離により、(4)の混合物から液体と沈殿物とを分離すること、
(6)(5)の沈殿物を緩衝液で、1〜15又はほぼそれらの値の重量比に再び懸濁して、懸濁液を作ること、
(7)二酸化シリコン(SiO2)を(6)からの懸濁液と混合し、濾過により濾液を得ること、
(8)洗浄剤と冷却アルコールを、(7)の濾液と混合し、遠心分離により沈殿物を得ること、
(9)溶媒又は洗浄剤を含む水溶液中に前記沈殿物を溶解し、その溶液を少なくとも60分にわたり維持すること、
(10)(9)の後の溶液を陽イオン交換クロマトグラフィーカラムに通し、カラムに吸収された蛋白質を溶出液中に溶出させること、
(11)(10)からの溶出液を陰イオン交換クロマトグラフィーカラムに通して、流出液を生成すること、
(12)流出液をナノ濾過器に通して、ナノ濾液を生成すること、
(13)ナノ濾液を限外濾過薄膜に通して、限外濾液を生成すること、
(14)限外濾液を透析濾過緩衝体液で透析濾過して、蛋白質濃度が20%(重量/体積)又はほぼその値の溶液を生成すること、並びに、
(15)前記溶液を0.2μm以下のフィルタに通して濾過することで(14)からの溶液を減菌することにより、濃縮されたIgG組成物を得ること。
【0011】
幾つかの実施形態では、工程(2)は、約−2〜0℃の温度で実行される、又は、工程(2)の混合物は、少なくとも15分にわたり混合され、次に、少なくとも2時間にわたり約−2〜0℃の温度に保たれる。幾つかの実施形態では、工程(4)は、少なくとも15分にわたり混合され、次に、少なくとも8時間にわたり、−7℃又はほぼその温度に保たれる。幾つかの実施形態では、工程(6)の懸濁液は、約40〜160分にわたり、約2℃〜8℃の温度で、5.0又はほぼその値のpHで攪拌される、又は、混合物は、約2〜8℃の温度で、少なくとも50分にわたり実行される。幾つかの実施形態では、工程(8)は、約−5〜−10℃の温度で実行される。幾つかの実施形態では、工程(9)の溶液は、1.0%(体積/体積)のトリトンX−100と、0.3%(体積/体積)のトゥイーン80と、0.3%(体積/体積)燐酸トリ−(n−ブチル)(TNBP)とを含む。幾つかの実施形態では、工程(9)の溶液は、約18〜25℃の温度で保持される。幾つかの実施形態では、工程(10)の陽イオン交換クロマトグラフィーカラムは、pHが5.5±0.1の10mMの酢酸塩緩衝液で洗浄され、pHが8.5±0.1、伝導率が5.0±0.2mS/cmの、35mM一塩基性燐酸ナトリウム、10mMトリス緩衝液で溶出される。幾つかの実施形態では、工程(10)の溶出液は、工程(11)の前に、6.4±0.2のpH、約1.5〜2.5mS/cmの伝導率に調節される。幾つかの実施形態では、工程(11)の流出液は、工程(12)の前に、0.2μm以下の孔径のフィルタを通る。幾つかの実施形態では、工程(13)の限外濾液は、蛋白質濃度が、5±1%(重量/体積)又はほぼその値である。他の実施形態では、工程(13)の限外濾過薄膜は、公称分画分子量(NMWCO)が、50kDa以下である。幾つかの実施形態では、工程(14)の血液濾過緩衝液は、pHが4.2±0.1の0.25Mグリシン溶液である。他の実施形態では、工程(14)からの溶液は、蛋白質濃度が20%(重量/体積)よりも大きく、続いて、血液透析緩衝液で、約20.4±0.4(重量/体積)又はほぼその濃度に調節される。幾つかの実施形態では、工程(13)及び(14)は、約2〜8℃の温度で実行される。他の実施形態では、調製方法は、容器を密封する前に、工程(15)の減菌された溶液を無菌状態下で容器内に分注する工程を更に含む。
【0012】
他の実施形態では、上記の方法は、密封された容器を約30〜32℃で約21〜22日にわたり保存する工程を更に含んでもよく、その方法は、20%IgG製品を10%の従来技術の静注製剤と同程度に安定させる調製工程を更に含んでもよい。
【0013】
更に別の態様では、本発明は、上記の調製方法により生成され、少なくとも18%(重量/体積)の免疫グロブリン、例えば、少なくとも20%(重量/体積)の免疫グロブリンを含む、水性組成物を提供する。
【0014】
別の態様では、本発明は、有効量の上記の組成物を患者に投与することを含む、免疫不全、自己免疫不全、又は急性感染症に罹患した患者を治療する方法を提供する。
【図面の簡単な説明】
【0015】
【図1A】新規の限外濾過/透析濾過システムの図面表示である。
【図1B】新規の限外濾過/透析濾過システムの図面表示である。
【図1C】新規の限外濾過/透析濾過システムの図面表示である。
【図1D】新規の限外濾過/透析濾過システムの図面表示である。
【図1E】新規の限外濾過/透析濾過システムの図面表示である。
【図2】図2である。
【図3】図3である。
【発明を実施するための形態】
【0016】
「抗体」は、分析物(抗原)と結合してその分析物を認識する免疫グロブリン遺伝子(単数)又は免疫グロブリン遺伝子(複数)により実質的に符号化されたポリペプチド、若しくは、それらの断片を指す。認識される免疫グロブリン遺伝子として、カッパ、ラムダ、アルファ、ガンマ、デルタ、イプシロン及びミュー一定領域遺伝子、並びに、ミリアド免疫グロブリン可変領域遺伝子が挙げられる。軽鎖は、カッパ又はラムダのいずれかに分類される。重鎖は、ガンマ、ミュー、アルファ、デルタ、又はイプシロンに分類され、それぞれ、IgG、IgM、IgA、IgD、及びIgEの順で、免疫グロブリン型を定義する。
【0017】
典型的な免疫グロブリン(抗体)構造ユニットは、2対のポリペプチド鎖で構成され、各々の対は、1つの「軽」鎖(約25kD)と1つの「重」鎖(約50〜70kD)とを有する。各々の鎖のN末端は、抗原認識に寄与する約100〜110以上のアミノ酸の可変領域を定める。用語、可変軽鎖(VL)及び可変重鎖(VH)は、それぞれ、これらの軽鎖及び重鎖を指す。
【0018】
用語「限外濾過(UF)」は、水圧が液体を半浸透性膜に押し付ける、様々な薄膜濾過方法を包含する。懸濁された高分子量の固体及び溶質は、保持されるが、水及び低分子量の溶質は、薄膜を通過する。この分離プロセスは、マクロ分子(103〜106Da)溶液、特に、蛋白質溶液を浄化し、濃縮するのに利用されることが多い。数多くの限外薄膜は、それらの膜が保持する分子の大きさに応じて、入手することができる。限外濾過は、通常、1〜1000kDaの薄膜孔径及び0.01〜10barの操作圧力を特徴とし、特に、糖類及び塩類等の小さな分子から、蛋白質等のコロイドを分離するのに有用である。
【0019】
用語「透析濾過」は、限外濾過と同じ薄膜を用いて行われ、流れが接線方向の濾過である。透析濾過の間、緩衝液を再利用タンク内に導入するのに対し、濾液をユニット操作から除去する。製品が残余物(例えば、IgG)内にあるプロセスでは、透析濾過は、成分を製品貯蔵槽から濾液内へ洗い出し、それにより、緩衝液を交換し、望ましくない種の濃度を低減させる。
【0020】
用語「約」は、本明細書で用いられる場合、特定値から正負10%の近似範囲を表す。例えば、語句「約20%」は、18〜22%の範囲を含む。
【0021】
用語「混合」は、いずれかの形式の攪拌により溶液又は懸濁液中に2つ以上の化合物又は物質を等しく配分する行為を記述する。この用語が本出願に利用される場合、「混合」の結果として、溶液又は懸濁液中で全ての成分を完全に均等に配分する必要はない。
【0022】
本出願では、用語「溶媒」は、1つ以上の他の物質を溶解又は懸濁することを可能にする、いずれかの液体物質を含む。溶媒は、本質的に、水等の無機質であってもよく、エタノール、アセトン、酢酸メチル、酢酸エチル、ヘキサン、石油エーテル等のような有機液体であってもよい。溶媒は、「溶媒洗浄剤処理」で用いられる場合、溶液中の脂質包膜ウイルスを不活性にするのに利用される溶媒洗浄剤混合物の一部である、有機溶媒(例えば、燐酸トリ−N−ブチル)を表す。
【0023】
用語「洗浄剤」は、本出願で、用語「界面活性剤」又は「表面活性剤」と交換可能に利用される。界面活性剤は、通常、両媒性の、即ち、疎水基(「尾部」)と親水基(「頭部」)の両方を含む有機化合物であり、それらの官能基により、界面活性剤は、有機溶媒と水の両方に溶ける。界面活性剤は、その頭部が形式上帯電された塩基の存在により分類され得る。非イオン性界面活性剤は、塩基の頭部が帯電されていないのに対し、イオン性界面活性剤は、その頭部が電荷を帯びている。双性イオン界面活性剤は、2つの反対に帯電された塩基を有する頭部を含む。通常の界面活性剤の幾つかの実例として、陰イオン性(硫酸塩陰イオン、スルホン酸塩陰イオン又はカルボン酸塩陰イオン)では、ペルフルオロオクタン酸塩(PFOA又はPFO)、ペルフルオロオクタンスルホン酸塩(PFOS)、ドデシル硫酸ナトリウム(SDS)、ラウリル硫酸アンモニウム、及び他のアルキル硫酸塩、ラウリル硫酸ナトリウム(ラウリルエーテル硫酸ナトリウム、又はSLESとしても知られている)、アルキルベンゼンスルホン酸塩と、陽イオン性(第四級アンモニウム陽イオンに基づく)では、セチルトリメチルアンモニウム臭化物(CTAB)、別名、ヘキサデシルトリメチルアンモニウムブロミド、及び他のアルキルトリメチルアンモニウム塩類、セチルピリジニウム塩化物(CPC)、ポリエトキシル化獣脂アミン(POEA)、塩化ベンザルコニウム(BAC)、塩化ベンゼトニウム(BZT)と、カプリル酸塩、カプリル酸、ヘプタン酸塩、ヘキサン酸、ヘプタン酸、ナノ酸、デカン酸等を含む長鎖脂肪酸及びそれらの塩類と、双性イオン性(両性)では、ドデシルベタイン、ヤシ酸アミドプロピルベタイン、ココ両性グリシネートと、非イオン性では、アルキルポリ(エチレンオキシド)、アルキルフェノールポリ(エチレンオキシド)、ポリ(エチレンオキシド)とポリ(プロピレンオキシド)との共重合体(ポロキサマ類又はポロキサミン類として市場で知られている)、オクチルグルコシド、デシルマルトシド、脂肪アルコール(例えば、セチルアルコール及びオレイルアルコール)、コカミドMEA、コカミドDEA、ポリソルベート類(ツイン20、ツイン80等)、トリトン洗浄剤、及びドデシルジメチルアミンオキシドを含むアルキルポリグルコシド類が挙げられる。
【0024】
用語「静脈注射IgG」又は「IVIG」処置は、本明細書で用いられる場合、一般に、IgG免疫グロブリンの組成物を患者に静脈注射で、皮下に又は筋肉内に投与し、免疫不全、炎症性疾患、及び自己免疫疾患等の数多くの病状に対処する治療方法を指す。IgG免疫グロブリンは、通常、貯蔵されており、血漿から調製される。全ての抗体又は断片を利用することができる。IgG免疫グロブリンは、皮下投与又は筋肉内投与のために高濃度(10%超過)に調合され得る。これは、特に、特定の抗原(例えば、Rh(D)因子、百日咳毒素、破傷風毒素、ボツリヌス毒素、狂犬病等)の平均滴定量よりも高い滴定量で調製される、特化されたIgG製剤に共通のことである。考察し易くするために、そのような皮下投与又は筋肉内投与用に調合されるIgG組成物も、本出願では、用語「IVIG」に含まれる。
【0025】
「治療上有効な量又は投与量」又は「十分な/有効な量又は投与量」は、投与されて効果を作り出す投与量を意味する。正確な投与量は、治療目的に依存することになり、当業者により、周知の技術を利用して確認され得る(例えば、リバーマン、医薬適用法(第1〜3巻、1992年)、ロイド、医薬調合の技術、科学技術(1999年)、ピッカー、投与量計算法(1999年)、及び、レミングトン、調剤科学及び実践、第20版(2003年、ジェンナロ、リピンコット、ウィリアムズ&ウィルキンス出版)を参照すること。これらの開示内容は、全ての目的のために、参照として本明細書にそのまま組み込まれている)。
【0026】
最新医療で日常実践されているように、濃縮免疫グロブリン(特に、IgG)の殺菌製剤は、免疫不全、炎症性及び自己免疫疾患、及び急性感染症の3つの分類内にある病状に対処するのに利用される。通常利用される1つのIgG製品、静脈注射用免疫グロブリン又はIVIGは、静脈内投与用に、例えば、10%濃度に調合される。濃縮された免疫グロブリンは、皮下投与又は筋肉内投与用に、20%の濃度又はほぼその濃度にも調合され得る。
【0027】
一態様では、本発明は、貯蔵された血漿から、高純度で高濃度の免疫グロブリン組成物を生成する新規の改善された方法に関する。以前利用されたIgG浄化及び濃縮方法と比較して、本発明者は、IgGを著しく損なわずにIgG濃度がより高くなり、最終製剤でのpHが低く保たれる、限外濾過及び調合工程を組み込んでいる。通常、製品は、蛋白質濃度が、少なくとも18%(重量/体積)であり、そのうちの大半(通常、95%以上)は、IgGであり、pHは、pH3〜6の範囲であり、それは、血漿中に存在し得るウイルス等の病原を不活性にし易くする。それらのIgG濃度が高く、それにより、投与体積を少なくなるので、本発明の製品は、皮下及び/又は筋肉内投与に適している。幾つかの実施形態では、IgG製品は、粘性が18mパスカル−秒未満であり、従って、静脈内投与にも適用し得る。静脈注射用製品の品質属性を、皮下及び筋肉内注射用製品に必要とされる高濃度と組み合わせることができるので、簡単な希釈により、静脈内投与することもできる。本発明のIgG組成物の別の利点は、貯蔵中の安定性が優れていることである。
【0028】
ある態様では、本発明は、最終蛋白質濃度が約17%よりも大きく、IgG純度が少なくとも約95%である、高濃度IgG製剤を調製する方法を提供する。ある態様では、製剤は、安定性が長期にわたり、静脈、皮下、及び/又は筋肉内投与用に調合される。
【0029】
別の態様では、本発明は、本明細書中で与えられる改良された製造方法に従って調製されたIgG組成物の医薬組成物及び製剤を提供する。ある実施形態では、これらの組成物及び製剤は、現在市場にある他のIVIG組成物と比べて、向上した特性を与える。例えば、ある実施形態では、本明細書中で与えられる組成物及び製剤は、長期にわたり安定している。別の実施形態では、本明細書中で与えられる組成物及び製剤は、現在市場にある他のIVIG組成物と比べて、IgG濃度が高い。更に他の実施形態で、本明細書中で与えられる組成物及び製剤は、IgG濃度が高く、長期にわたり安定している。
【0030】
更に別の態様では、本発明は、本明細書中で与えられる改良された方法を利用して調製されるIgG組成物を投与することを含む、免疫不全、炎症性及び自己免疫疾患、及び急性感染症を治療する方法を提供する。
【0031】
I.濃縮され、浄化されたIgG製剤の生成
幾つかの自己免疫容態に対処するための、全ての抗体を含むIVIG組成物が記述されている(例えば、米国特許出願第US2002/0114802号、第US2003/0099635号、及び第US2002/0098182号を参照)。これらの参照文献に開示されるIVIG組成物は、ポリクローナル抗体を含む。
【0032】
一般に、本発明による免疫グロブリン製剤は、適切な開始材料、例えば、回収血漿又は原血漿から調製され得る。一般例では、血液又は血漿は、健康な提供者から収集される。通常、血液は、免疫グロブリン製剤(一般に、「同種の」免疫グロブリンと呼ばれる)が投与されることになる被験体としての同じ動物種から収集される。免疫グロブリンは、例えば、沈殿(アルコール分別又はポリエチレングリコール分別)、クロマトグラフィー法(イオン交換クロマトグラフィー、親和性クロマトグラフィー、免疫親和性クロマトグラフィー)の超遠視分離、及び電気泳動による調製等のような適切な手順により、血液から単離される(例えば、コーンら、アメリカ化学学会誌、第68巻、459〜75頁(1946年)、オンクレイら、アメリカ化学学会誌、第71巻、541〜50頁(1949年)、バルンデルンら、国際輸血学会誌、第7巻、157〜74頁(1962年)、コベルトら、国際輸血学会誌、第13巻、93〜102頁(1967年)、米国特許第5,122,373号及び第5,177,194号を参照すること。それらの開示内容は、全ての目的のために、参照として本明細書にそのまま組み込まれている)。
【0033】
上記の方法とは異なり、一態様では、本発明は、寒冷貯蔵された開始材料を利用する、濃縮IgG組成物を調製する方法を提供する。一般に、本明細書中で与えられる方法は、変更されたコーン・オンクレイ・アルコール分別工程とイオン交換クロマトグラフィーの両方を利用して、優れたIgG産出量を与え、一方では、向上しない場合は、現在利用可能な市販のIVIG製剤で見出されるものと同じ品質を保つ。
【0034】
多くの事例では、免疫グロブリンは、アルコール分別及び/又はイオン交換、並びに、当業者に周知のクロマトグラフィー方法により生成されるガンマグロブリン含有製品から調製される。通常、浄化されたコーン分画IIが利用される。開始コーン分画IIペーストは、通常、95%又はほぼその率のIgGであり、4つのIgG亜類型から構成される。異なる亜類型は、それらの亜類型が得られる貯蔵されたヒト血漿内に見られるのとほぼ同じ比率で分画II内に存在する。分画IIは、投与可能な製品に調合される前に、更に浄化される。例えば、分画IIペーストは、冷い浄化水性アルコール溶液中に溶解され、沈殿及び濾過を介して除去され得る。最終濾過の後に、免疫グロブリン懸濁液は、透析され、又は、(公称分子量限界が100,000ダルトン以下である限外薄膜を利用して)透析濾過され、アルコールを除去し得る。溶液は、所望の蛋白質濃度を得るために、濃縮又は希釈されてもよく、更に、当業者に十分に知られている技術により浄化され得る。
【0035】
分取工程を利用して、特定の同類型又は亜類型の免疫グロブリンを濃縮することができる。例えば、蛋白質A、蛋白質G又は蛋白質セファロースクロマトグラフィーを利用して、IgGの又は特定のIgG亜類型の免疫グロブリンの混合物を濃縮することができる。(一般に、ハーロー及びレイン、抗体を用いて(コールドスプリングハーバー研究所出版、1999年)、ハーロー及びレイン、抗体、実験マニュアル(コールドスプリングハーバー研究所出版、1988年)、米国特許第5,180,810号を参照すること。)
【0036】
以下に詳細に記載されるように、本発明の高濃度IgG製品は、IVIG生成プロセスと同じ又は類似の多くの工程を有するプロセスにより生成される。生産プロセスの終了付近での、開チャンネル薄膜を利用した、特別設定の後洗浄製剤を用いての限界濾過/透析濾過の追加工程により、その結果得られるIgG組成物は、従来技術のIVIG(例えば、ガンマガード(登録商標)リキッド)と比べて、産出量と保管安全性に悪影響を与えずに、蛋白質濃度(200mg/mL)の高さがほぼ2倍になる。大抵の市販の限外濾過薄膜では、主要蛋白質が損なわれずに、200mg/mLのIgG濃度に達することができない。これらの薄膜は早期に詰まることになり、従って、適切な後洗浄液を得るのは難しい。従って、開チャンネル薄膜構成を用いなければならない。開チャンネル薄膜でも、特別設定の後洗手段を利用して、蛋白質を著しく損なわずに(損失が2%未満)、要求される濃度を得なければならない。更に意外なこととして、200mg/mLの高い蛋白質濃度により、低pH保存工程のウイルス不活性能力は左右されない。高濃度IgG組成物を生成する一般的なプロセスは、以下の工程を含む:
【0037】
A.寒冷沈殿物の分離
浄化プロセスは、通常、安全性及び品質の検討のために既に確認された、事前に凍結された貯蔵血漿を解凍することで開始する。解凍は、通常、6℃未満の温度で実行される。次に、遠心分離又は濾過を冷気中で実行して、血漿が解凍された後、通常は、解凍と同じ温度で、固体と液体とを分離する。次に、液体部分(遠心分離又は濾過により、新鮮な解凍された血漿から寒冷不溶性蛋白質が除去された後は、「寒冷貯蔵血漿」とも呼ばれる)は、次の工程で処理される。この際、実施例1で詳細に記載される、第8阻害因子迂回活性複合体(FEIBA)、第4因子複合体、第7因子濃縮物、又は抗凝固第3因子複合体を単離するために、様々な追加工程を取り上げることができる。
【0038】
B.分画Iの上澄みの取得
この工程では、寒冷貯蔵血漿は、通常、0±1℃又はほぼその温度に冷却され、そのpHは、7.0又はほぼ7.0に調節される。ある実施形態では、pHは、約7.0〜約7.5に、好ましくは、7.1又はほぼ7.1と7.3又はほぼ7.3との間に、さらに好ましくは、7.2又はほぼ7.2に調節される。ある実施形態では、pHは、7.0に又はほぼ7.0に調節される。別の実施形態では、pHは、7.1又はほぼ7.1に調節される、別の実施形態では、pHは、7.2又はほぼ7.2に調節される。別の実施形態では、pHは、7.3又はほぼ7.3に調節される。別の実施形態では、pHは、7.4又はほぼ7.4に調節される。別の実施形態では、pHは、7.5又はほぼ7.5に調節される。次に、事前に冷却されたエタノールを加え、血漿を攪拌しながら、8%体積/体積の標的エタノール濃度にする。同時に、温度を更に、約−4〜約0℃に、好ましくは、約−2℃に下げ、α2−マクログロブリン、β1A及びβ1Cグロブリン、フィブリノゲン、及び第8因子等の汚染物を沈殿させる。通常、沈殿事象は、少なくとも1時間の保持時間を含むことになるが、より短い又はより長い保持時間を用いてもよい。続いて、理想的には、寒冷貯蔵血漿中に存在するIgG含有量の全体を含む上澄み(上澄みI)は、遠心分離、濾過、又は別の適切な方法により収集される。
【0039】
C.分画II+IIIの沈殿
IgG含有量を更に濃縮し、その分画の純度を更に上げるために、上澄みIは、第2沈殿工程に曝される。一般に、溶液のpHは、約6.8〜約7.2のpHに、好ましくは、7.0又は約7.0のpHに調節される。次に、その溶液に、攪拌しながら、アルコール、好ましくは、エタノールを加え、約20%〜約25%(体積/体積)の最終濃度にする。一実施形態では、最終アルコール濃度は、20%又はほぼその値である。別の実施形態では、最終アルコール濃度は、21%又はほぼその値である。別の実施形態では、最終アルコール濃度は、22%又はほぼその値である。別の実施形態では、最終アルコール濃度は、23%又はほぼその値である。別の実施形態では、最終アルコール濃度は、24%又はほぼその値である。別の実施形態では、最終アルコール濃度は、25%又はほぼその値である。分画II+III上澄みとも呼ばれる液体部分を更に処理して、第5因子を抽出する。この工程からの沈殿物を、次の工程で更に処理する。一実施形態では、工程B及びCを共に実行することもできる。
【0040】
D.分画II及びIII沈殿物からの抽出
冷却抽出緩衝液を利用して、分画II+III沈殿物を、沈殿物1部に対して抽出緩衝液15部の通常比に再び懸濁する。模範的抽出緩衝液は、5mMの一塩基性燐酸ナトリウムと5mMの酢酸とを含み、pHが、4.5±0.2又はほぼその値であり、伝導率が、0.7〜0.9mS/cm又はほぼその値である。一実施形態では、抽出緩衝液の伝導率は、0.7mS/cm又はほぼその値である。別の実施形態では、抽出緩衝液の伝導率は、0.8mS/cm又はほぼその値である。更に別の実施形態では、抽出緩衝液の伝導率は、0.9mS/cm又はほぼその値である。抽出プロセスは、2〜8℃又はほぼその値の温度に実行される。
【0041】
他の適切な再懸濁比を利用してもよく、例えば、約1:8〜約1:30、又は、約1:10〜約1:20、又は、約1:12〜約1:18、又は、約1:13〜約1:17、又は、約1:14〜約1:16である。ある実施形態では、再懸濁比は、1:8、1:9、1:10、1:11、1:12、1:13、1:14、1:15、1:16、1:17、1:18、1:19、1:20、1:21、1:21、1:22、1:23、1:24、1:25、1:26、1:27、1:28、1:29、1:30、又はそれ以上、若しくは、ほぼそれらの値であり得る。
【0042】
II+III沈殿物を抽出するのに適切な溶液は、一般に、pHが、約4.0〜約5.5である。ある実施形態では、溶液は、pHが、約4.0〜約5.0である。別の実施形態では、溶液は、pHが、約4.5〜約5.0である。他の実施形態では、抽出液は、pHが、約4.0、4.1、4.2、4.3、4.4、4.5、4.6、4.7、4.8、4.9、5.0、5.1、5.2、5.3、5.4、又は5.5である。一実施形態では、抽出液のpHは、4.5又はほぼその値である。別の実施形態では、抽出液のpHは、4.6又はほぼその値である。抽出液のpHは、4.7又はほぼその値である。抽出液のpHは、4.8又はほぼその値である。抽出液のpHは、4.9又はほぼその値である。抽出液のpHは、5.0又はほぼその値である。
【0043】
抽出緩衝液は、好ましくは、伝導率が、約0.5mS/cm〜約2.0mS/cmである。例えば、ある実施形態では、抽出液の伝導率は、0.5mS/cm又はほぼその値であり、若しくは、0.6、0.7、0.8、0.9、1.0、1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9又はほぼそれらの値であり、若しくは、約2.0mS/cmである。当業者は、適切な伝導率を有する抽出緩衝液を生成する方法を知っている。
【0044】
E.煙霧シリカ処理及び濾過
ある実施形態では、先の工程からの懸濁液に煙霧シリカ(例えば、アエロジル380又はその等価物)を加え、40g/kg又はほぼその値の懸濁液濃度にする、又は、寒冷貯蔵血漿の1.8g/リットルと等価にする。混合は、少なくとも50〜70分にわたり約2〜8℃で行われる。幾つかの事例では、濾過補助剤(例えば、ワールドミネラルズからの、0.5kg/kg又はほぼその値の懸濁液濃度で利用されるハイフロスーパーセル)を加えて、濾過により液体/固体に分離する後続の工程を容易にする。抽出緩衝液を利用して、圧濾器を後洗浄する。濾液プロセスは、約2〜8℃の温度に保たれる。
【0045】
F.沈殿物Gの分別
次に、先の工程からの濾液をポリソルベート−80と混合して、0.2%重量/体積又はその値の濃度にし、少なくとも30分にわたり約2℃〜8℃の温度で攪拌する。次に、クエン酸ナトリウム二無水物を溶液内に8g/リットル又はその値で混合し、更に30分にわたり約2℃〜8℃の温度で攪拌する。次に、溶液のpHを7.0±0.1又はその値に調節する。ある実施形態では、pHを水酸化ナトリウム又は酢酸のどちらかで調節する。次に、冷却アルコールを溶液に加えて、25%体積/体積の濃度にし、コーン分画IIと同じ沈殿工程を行う。
【0046】
G.沈殿物Gの懸濁液
先の工程からの沈殿物を溶解し、0.2μmの公称孔径の深層フィルタ(例えば、キュノVR06フィルタ又はその等価物)で濾過して、透明な濾液を得る。別の実施形態では、沈殿物を溶解し、次に、遠心分離して、浄化された上澄みを回収する。
【0047】
H.溶媒及び洗浄剤処理
先の工程からの濾液を溶媒/洗浄剤処理に利用する。通常の溶媒/洗浄剤処理混合物は、1.0%(体積/体積)トリトンX−100、0.3%(体積/体積)ツイン−80、及び0.3%(体積/体積)TNBPを含み、混合物は、通常、少なくとも60分にわたり約18℃〜25℃の温度に保たれる。血漿由来成分を洗浄剤処理する方法は、当該技術分野で十分に知られている。一般に、本明細書中で与えられる方法と共に、いずれかの標準的な非イオン性洗浄剤処理を利用してもよい。
【0048】
I.陽イオン交換クロマトグラフィー
S/D処理されたPptG濾液からのIgGを更に浄化し、濃縮するために、陽イオン交換及び/又は陰イオン交換クロマトグラフィーを用いることができる。イオン交換クロマトグラフィーを利用してIgGを浄化し、濃縮する方法は、当該技術分野で十分に知られている。例えば、米国特許第5,886,154号は、分画II+III沈殿物を低いpH(約3.8〜4.5)で抽出した後に、カプリル酸を利用してIgGを沈殿させ、最終的に、2つの陰イオン交換クロマトグラフィー工程を実施する方法を記載している。米国特許第6,069,236号は、アルコール沈殿に全く依存しないクロマトグラフィーによるIgG浄化方式を記載している。PCT出願第WO2005/073252号は、分画II+III沈殿物の抽出、カプリル酸処理、PEG処理、及び単一の陰イオン交換クロマトグラフィー工程を含むIgG浄化方法を記載している。米国特許第7,186,410号は、分画I+II+III沈殿物又は分画II沈殿物のいずれかの抽出と、続く、アルカリ性pHで実行される単一の陰イオン交換工程とを含むIgG浄化方法を記載している。米国特許第7,553,938号は、分画I+II+III沈殿物又は分画II+III沈殿物のいずれかの抽出と、カプリル酸処理と、一回又は二回の陰イオン交換クロマトグラフィー工程とを含む方法を記載している。米国特許第6,093,324号は、約6.0〜約6.6のpHで操作されるマクロ多孔性陰イオン交換樹脂の利用と、を含む浄化方法を記載している。米国特許第6,835,379号は、アルコール分別を用いずに陽イオン交換クロマトグラフィーに依存する浄化方法を記載している。
【0049】
一実施形態では、先の工程からの蛋白質溶液を含む溶剤/洗浄剤は、次に、陽イオン交換カラムに通され、溶媒と洗浄剤とを除去する。SD剤を洗い落とした後、次に、吸着された蛋白質を高pHの溶出緩衝液で溶出する。一実施形態では、溶出緩衝液は、pHが、約7.5〜約9.5である。別の実施形態では、溶出緩衝液は、pHが、約8.0〜約9.0である。好ましい実施形態では、溶出緩衝液は、pHが、8.5±0.1又はほぼその値である。
【0050】
J.陰イオン交換クロマトグラフィー
先の工程からの溶出物は、pH6に調節され、次の平衡陰イオン交換カラムに適した伝導率になるように希釈される。充填及び洗浄中のカラム通過画分は、別の処理のために収集される。
【0051】
K.ナノ濾過
本明細書中で与えられるIgG組成物のウイルス負荷を更に削減するために、適切なナノ濾過装置を利用して、陰イオン交換カラム流出物をナノ濾過する。ある実施形態では、ナノ濾過装置は、平均孔径が、約15nm〜約200nmである。この用途に適したナノフィルタの実例として、DVD、DV50、DV20(パール社)、バイレゾルブNFP、バイレゾルブNFP(ミリポア社)、プラノバ15N、20N、35N、及び75N(プラノバ社)が挙げられるが、これらに限定されない。特定の実施形態では、ナノフィルタは、平均孔径が、約15nm〜約72nm、又は、約19nm〜約35nm、又は、15nm、19nm、35nm、又は72nm若しくはほぼそれらの値であり得る。好ましい実施形態では、アサヒ・プラノバ35Nフィルタ又はその等価物等のナノフィルタは、平均孔径が、35nm又はほぼその値である。
【0052】
L.限外濾過及び透析濾過
ナノ濾過の後、限外濾過により、濾液を更に、5±1%の蛋白質濃度に濃縮する。幾つかの実施例では、除外濾過は、開経路篩を有するカセット内で実行され、除外濾過薄膜は、公称分画分子量(NMWCO)が、50kDa以下である。
【0053】
一実施形態では、限外濾過により、ナノ濾液を約2%〜約10%(重量/体積)の蛋白質濃度に濃縮してもよい。ある実施形態では、除外濾過は、開経路篩を有するカセット内で実行され、除外濾過薄膜は、公称分画分子量(NMWCO)が、約100kDa未満である、若しくは、約90、80、70、60、50、40、30kDa未満である、若しくは、それらよりも少ない。好ましい実施形態では、限外濾過薄膜は、NMWCOが、50kDa以下である。
【0054】
限外濾過工程が完了した後、静脈又は筋肉内投与に適した溶液で透析濾過することにより、濃縮液を更に濃縮してもよい。ある実施形態では、透析濾過溶液は、安定剤及び/又は緩衝剤を含んでもよい。好ましい実施形態では、安定剤及び緩衝剤は、適切な濃度の、例えば、0.20M〜0.30Mの、又は約0.22M〜約0.28Mの、又は約0.24M〜約0.26mMの、若しくは、2.0、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9、又は3.0或いはほぼそれらの値の濃度の濃度のグリシンである。好ましい実施形態では、透析緩衝液は、0.25M又はほぼその値のグリシンを含む。
【0055】
好ましい実施形態では、除外濾過工程が完了した後、濃縮液をpHの低い0.25Mグリシン溶液で透析濾過する。通常、最小限の交換体積は、当初の濃縮液の体積の6倍であり、溶液は、20%重量/体積よりも大きな蛋白質濃度に濃縮される。透析濾過及び濃縮工程の終わりでは、溶液のpHは、通常、4.4〜4.9である。
【0056】
通常、最小限の交換体積は、当初の濃縮液の体積の少なくとも約3倍である、又は、当初の濃縮液の体積の少なくとも約4,5、6、7、8、9倍又はそれ以上である。IgG溶液は、約5%〜約22%(重量/体積)の、又は約10%〜約22%(重量/体積)の、又は約15%〜約22%(重量/体積)の、又は約18%〜約22%(重量/体積)の、約20%〜約22%(重量/体積)の最終蛋白質濃度に、若しくは、約5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、21%、22%又はほぼそれらの値、若しくは、それらよりも高い最終濃度に濃縮され得る。好ましい実施形態では、IgG溶液は、20%、約20%〜約22%、22%の最終蛋白質濃度に濃縮される。通常、濃縮工程の終わりでは、溶液のpHは、約4.6〜5.1である。
【0057】
M.調合
透析工程が完了した後、溶液の蛋白質濃度は、透析緩衝液で、約5%〜約20%(重量/体積)、又は約10%〜約20%(重量/体積)、又は約15%〜約20%(重量/体積)、又は約18%〜約20%(重量/体積)の最終濃度に、若しくは、約5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、又は20%の最終濃度に調節される。好ましい実施形態では、溶液の最終蛋白質濃度は、19%、約19%〜約21%、21%である。好ましい実施形態では、透析濾過が完了した後、溶液の蛋白質濃度は、透析濾過緩衝液で、20%重量/体積よりも僅かに大きく、例えば、20.4±04%重量/体積又はほぼその値に調節される。
【0058】
N.更なる殺菌
調合されたバルク溶液は、絶対孔径が0.2ミクロン以下の薄膜フィルタを通して最初に濾過することにより、更に殺菌される。次に、溶液は、適切に密封するための最終容器内に無菌で分注され、検査用の試料が採取される。最終工程として、密封された容器を長期間、例えば、21〜22日にわたり30〜32℃で保存する。
【0059】
II.濃縮IgG組成物
一態様では、本発明は、本明細書中で与えられる方法により調製された水性IgG組成物に関する。一般に、本明細書に記載される新規の方法により調製されたIgG組成物は、IgG含有量及び純度が高い。例えば、本明細書中で与えられるIgG組成物は、蛋白質濃度が少なくとも15%(重量/体積)であり、IgG含有量が、90%純度よりも大きい。これらの高純度のIgG組成物は、治療投与に、例えば、皮下及び/又は筋肉内投与に適している。一実施形態では、本明細書中で与えられるIgG組成物は、静脈内投与に適しており、例えば、投与前に希釈される。一実施形態では、IgGの濃度は、20%又はほぼその値であり、皮下投与又は筋肉内投与に利用される。
【0060】
一実施形態では、本発明は、以下の工程を含む方法により調製された水性IgG組成物を提供する:
(1)遠心分離により血漿から液体と沈殿物とを分離すること、
(2)事前に冷却されたエタノールを(1)からの液体と混合して、エタノール濃度が8%(体積/体積)又はほぼその値である混合物を作ること、
(3)遠心分離により、(2)の混合物から液体と沈殿物とを分離すること、
(4)(3)からの液体のpH及びエタノール濃度を、それぞれ、7.0及び20〜25%(体積/体積)又はそれらの値に調節することにより、混合物を作ること、
(5)遠心分離により、(4)の混合物から液体と沈殿物とを分離すること、
(6)(5)の沈殿物を緩衝液で約1〜15の重量比に再び懸濁して、懸濁液を作ること、
(7)二酸化シリコン(SiO2)を(6)からの懸濁液と混合し、濾過により濾液を得ること、
(8)洗浄剤と冷却アルコールを、(7)の濾液と混合し、遠心分離により沈殿物を得ること、
(9)溶媒又は洗浄剤を含む水溶液中に前記沈殿物を溶解し、その溶液を少なくとも60分にわたり維持すること、
(10)(9)の後の溶液を陽イオン交換クロマトグラフィーカラムに通し、カラムに吸収された蛋白質を溶出液中に溶出させること、
(11)(10)からの溶出液を陰イオン交換クロマトグラフィーカラムに通して、流出液を生成すること、
(12)流出液をナノ濾過器に通して、ナノ濾液を生成すること、
(13)ナノ濾液を限外濾過薄膜に通して、限外濾液を生成すること、
(14)限外濾液を透析濾過緩衝液で透析濾過して、蛋白質濃度が20%(重量/体積)又はほぼその値の溶液を生成すること、並びに、
(15)前記溶液を0.2μm以下のフィルタに通して濾過することで(14)からの溶液を減菌することにより、濃縮されたIgGの組成物を得ること。
【0061】
一実施形態では、本発明は、蛋白質濃度が約150g/L〜約250g/Lの水性IgG組成物を提供する。ある実施形態では、IgG組成物の蛋白質濃度は、約175g/L〜約225g/L、又は約200g/L〜約225g/L、又はこれらの範囲内のいずれかの適切な濃度、例えば、150g/L、155g/L、160g/L、165g/L、170g/L、175g/L、180g/L、185g/L、190g/L、195g/L、200g/L、205g/L、210g/L、215g/L、220g/L、225g/L、230g/L、235g/L、240g/L、245g/L、250g/L又はほぼそれらの値、若しくは、それらよりも高い値の濃度である。好ましい実施形態では、水性IgG組成物は、蛋白質濃度が200g/L又はほぼその値である。特に好ましい実施形態では、水性IgG組成物は、蛋白質濃度が204g/L又はほぼその値である。
【0062】
本明細書中で与えられる方法により、純度レベルが極めて高いIgG組成物を調製することができる。例えば、一実施形態では、本明細書中で与えられる組成物中の総蛋白質の少なくとも約95%がIgGである。他の実施形態では、蛋白質の少なくとも約96%がIgGである、又は、組成物の総蛋白質の少なくとも約97%、98%、99%、99.5%又はそれ以上がIgGである。
【0063】
同様に、本明細書中で与えられる方法により、汚染液が極めて低レベルであるIgG組成物を調製することができる。例えば、ある実施形態では、約100mg/L未満のIgAを含むIgG組成物が提供される。他の実施形態では、IgG組成物は、約50mg/L未満のIgA、好ましくは、約35mg/L未満のIgA、最も好ましくは、20mg/L未満のIgAを含む。
III.医薬組成物
【0064】
別の態様では、本発明は、本明細書中で与えられる方法により調製された純粋なIgGを含む医薬組成物及び調合物を提供する。一般に、本明細書中で与えられる新規の方法により調製されたIgG医薬組成物及び調合物は、IgG含有量及び純度が高い。例えば、本明細書中で与えられるIgG医薬組成物及び調合物は、蛋白質濃度が、少なくとも約15%(重量/体積)、IgG含有量が、90%純度よりも大きくてもよい。これらの高純度IgG組成物は、治療投与に、例えば、皮下及び/又は筋肉内投与に適している。一実施形態では、本明細書中で与えられるIgG組成物は、静脈内投与に適しており、例えば、投与前に希釈される。一実施形態では、IgGの濃度は、20%又はほぼその値であり、皮下投与又は筋肉内投与に利用される。
【0065】
一実施形態では、本明細書中で与えられる医薬組成物は、本明細書中で与えられる方法を利用して単離された水性IgG組成物を調合することにより調製される。一般に、調合された組成物は、少なくとも1つの、好ましくは、少なくとも2つの、最も好ましくは、少なくとも3つのウイルス不活性化又は除去工程に曝されている。本明細書中で与えられる方法と共に利用され得るウイルス不活性化又は除去工程の限定されない実施例として、溶剤洗浄剤処理(ホロヴィッツら、血液凝固線溶、1994年(第5号付録3)、21頁〜28頁、クレイルら、輸血、2003年、第4号、1023〜1028頁、それらの両方とも、全ての目的のために、本明細書に参照としてそのまま、明確に組み込まれている)、ナノ濾過(ハマモトら、国際輸血学会誌、1989年、第56号、230〜236頁、及びユアサら、一般ウイルス学会誌、1991年(第72号(部編8))、2021〜2024頁、それらの両方とも、全ての目的のために、本明細書に参照としてそのまま、明確に組み込まれている)、高温での低pH恒温放置(ケンプフら、輸血、1991年、第31号、423〜427頁、及び、ルイら、生物学、1994年、第22号、13〜19号)が挙げられる。
【0066】
ある実施形態では、IgG含有量が約175g/L IgG〜約225g/L IgGの医薬製剤が提供される。一般に、これらのIVIG製剤は、本明細書に記載される方法を利用して血漿からのIgG組成物を単離し、その組成物を濃縮し、濃縮された組成物を、静脈内投与に適した溶液中に調合することにより調製される。IgG組成物は、当業者に周知のいずれかの適切な方法を利用して濃縮され得る。一実施形態では、組成物は、限外濾過/透析濾過により濃縮される。幾つかの実施形態では、組成物を濃縮するのに利用される限外濾過装置は、公称分画分子量(NMWCO)が約100kDa未満、又は、約90、80、70、60、50、40、30kDa未満、又は、それらの値よりも小さい限外濾過薄膜を用いる。好ましい実施形態では、限外濾過薄膜は、NMWCOが、50kDa未満である。緩衝液交換は、当業者に周知のいずれかの適切な技術を利用して行われてもよい。特定の実施形態では、緩衝液交換は、透析濾過により行われる。
【0067】
特定の実施形態では、IgG医薬組成物が提供され、そのIgG組成物は、以下の工程を含む方法を利用して、血漿から浄化される:
(1)遠心分離により、血漿から液体と沈殿物とを分離すること、
(2)事前に冷却されたエタノールを(1)からの液体と混合して、エタノール濃度が8%(体積/体積)又はほぼその値である混合物を作ること、
(3)遠心分離により、(2)の混合物から液体と沈殿物とを分離すること、
(4)(3)からの液体のpH及びエタノール濃度を、それぞれ、7.0及び20〜25%(体積/体積)又はそれらの値に調節することにより、混合物を作ること、
(5)遠心分離により、(4)の混合物から液体と沈殿物とを分離すること、
(6)(5)の沈殿物を緩衝液で約1〜15の重量比で再び懸濁して、懸濁液を作ること、
(7)二酸化シリコン(SiO2)を(6)からの懸濁液と混合し、濾過により濾液を得ること、
(8)洗浄剤と冷却アルコールを、(7)の濾液と混合し、遠心分離により沈殿物を得ること、
(9)溶媒又は洗浄剤を含む水溶液中に前記沈殿物を溶解し、その溶液を少なくとも60分にわたり維持すること、
(10)(9)の後の溶液を陽イオン交換クロマトグラフィーカラムに通し、カラムに吸収された蛋白質を溶出液中に溶出させること、
(11)(10)からの溶出液を陰イオン交換クロマトグラフィーカラムに通して、流出液を生成すること、
(12)流出液をナノ濾過器に通して、ナノ濾液を生成すること、
(13)ナノ濾液を限外濾過薄膜に通して、限外濾液を生成すること、
(14)限外濾液を透析濾過緩衝液で透析濾過して、蛋白質濃度が20%(重量/体積)又はほぼその値の溶液を生成すること、並びに、
(15)前記溶液を0.2μm以下のフィルタに通して濾過することで(14)からの溶液を減菌することにより、濃縮されたIgGの組成物を得ること。
【0068】
一実施形態では、本発明は、蛋白質濃度が約150g/L〜約250g/Lの医薬IgG組成物を提供する。ある実施形態では、IgG組成物の蛋白質濃度は、約175g/L〜約225g/L、又は約200g/L〜約225g/L、又はこれらの範囲内のいずれかの適切な濃度、例えば、150g/L、155g/L、160g/L、165g/L、170g/L、175g/L、180g/L、185g/L、190g/L、195g/L、200g/L、205g/L、210g/L、215g/L、220g/L、225g/L、230g/L、235g/L、240g/L、245g/L、250g/L又はほぼそれらの値、若しくは、それらよりも高い値の濃度である。好ましい実施形態では、水性IgG組成物は、蛋白質濃度が200g/L又はほぼその値である。特に好ましい実施形態では、水性IgG組成物は、蛋白質濃度が204g/L又はほぼその値である。
【0069】
本明細書で与えられる方法により、純度レベルが極めて高いIgG医薬組成物を調製することができる。例えば、一実施形態では、本明細書中で与えられる組成物中の総蛋白質の少なくとも約95%がIgGである。他の実施形態では、蛋白質の少なくとも約96%がIgGである、又は、組成物の総蛋白質の少なくとも約97%、98%、99%、99.5%又はそれ以上がIgGである。
【0070】
同様に、本明細書で与えられる方法により、汚染液が極めて低レベルであるIgG医薬組成物を調製することができる。例えば、ある実施形態では、約100mg/L未満のIgAを含むIgG組成物が提供される。他の実施形態では、IgG組成物は、約50mg/L未満のIgA、好ましくは、約35mg/L未満のIgA、最も好ましくは、20mg/L未満のIgAを含む。
【0071】
本明細書で与えられる医薬組成物は、通常、静脈内投与に適した1つ以上の緩衝剤又はpH安定剤を含む。本明細書中で与えられるIgG組成物を調合するのに適した緩衝剤の限定されない実例として、グリシン、クエン酸塩、燐酸塩、酢酸塩、グルタミン酸塩、酒石酸塩、安息香酸塩、乳酸塩、ヒスチジン又は他のアミノ酸類、グルコン酸塩、リンゴ酸塩、蟻酸塩、プロピオン酸塩、炭酸塩、又は、適切なpHに調節されたそれらのいずれかの組み合わせが挙げられる。一般に、緩衝剤は、調合物中で適切なpHを長期間にわたり維持するのに十分なものである。好ましい実施形態では、緩衝剤は、グリシンである。
【0072】
幾つかの実施形態では、調合物中の緩衝剤の濃度は、約100mM〜約400mMであり、好ましくは、約150mM〜約350mM、より好ましくは、約150mM〜約300mM、最も好ましくは、約175mM〜約225mMである。特に好ましい実施形態では、濃縮されたIgG組成物は、約150mM〜約250mMのグリシン、最も好ましくは、約200mMのグリシンを含む。別の好ましい実施形態では、濃縮されたIgG組成物は、250mM又はほぼその値のグリシンを含む。
【0073】
ある実施形態では、調合物のpHは、約4.1〜約5.6であり、好ましくは、約4.4〜約5.3、最も好ましくは、約4.6〜約5.1である。特定の実施形態では、調合物のpHは、約4.1、4.2、4.3、4.4、4.5、4.6、4.7、4.8、4.9、5.0、5.1、5.2、5.3、5.4、5.5、又は5.6であってもよい。
【0074】
幾つかの実施形態では、本明細書中で与えられる医薬組成物は、任意選択で、組成物のモル浸透圧濃度を調節する薬剤を更に含んでもよい。モル浸透圧濃度の限定されない実施例として、マンニトール、ソルビトール、グリセロール、蔗糖、グルコース、ブドウ糖、左旋糖、果糖、乳糖、ポリエチレングリコール類、燐酸類、塩化ナトリウム、塩化カリウム、塩化カルシウム、グルコノグルコヘプトン酸カルシウム、ジメチルスルホン等が挙げられる。
【0075】
通常、本明細書中で与えられる調合物は、モル浸透圧濃度が、約285mOsmol/kg(レイシーら、薬物情報ハンドブック、レキシ−コンプ、1999年、1254頁)の生理上のモル浸透圧濃度に匹敵する。ある実施形態では、調合物のモル浸透圧濃度は、約200mOsmol/kg〜約350mOsmol/kg、好ましくは、約240mOsmol/kg〜約300mOsmol/kgである。特定の実施形態では、調合物のモル浸透圧濃度は、約200mOsmol/kg、又は210mOsmol/kg、220mOsmol/kg、230mOsmol/kg、240mOsmol/kg、245mOsmol/kg、250mOsmol/kg、255mOsmol/kg、260mOsmol/kg、265mOsmol/kg、270mOsmol/kg、275mOsmol/kg、280mOsmol/kg、285mOsmol/kg、290mOsmol/kg、295mOsmol/kg、300mOsmol/kg、310mOsmol/kg、320mOsmol/kg、330mOsmol/kg、340mOsmol/kg、340mOsmol/kg、又は350mOsmol/kgである。
【0076】
本明細書中で与えられるIgG調合物は、一般に、液体形態で、長期間にわたり安定である。ある実施形態では、調合物は、室温で少なくとも約3ヶ月にわたり、又は、室温で少なくとも約4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、又は24ヶ月にわたり安定である。調合物は、その上、一般に、冷凍条件下(通常、約2℃〜約8℃)で少なくとも約18ヶ月にわたり、或いは、冷凍条件下で少なくとも約21、24、27、30、33、36、39、42、又は45ヶ月にわたり安定である。
IV.IgG製剤の投与
【0077】
最新医療において日常実施されるように、濃縮免疫グロブリン(特に、IgG)の無菌製剤を利用して、免疫不全、炎症性及び自己免疫疾患、及び急性感染症の3つの主要な分類内にある病状に対処する。これらのIgG製剤は、多発性硬化症(特に、再発寛解型多発性硬化症又はRRMS)、アルツハイマー病、及びパーキンソン病を治療するにも有用であり得る。本発明の浄化されたIgG製剤は、これらの目的に、並びに、IgG製剤の他の臨床上許容される用途に適している。
【0078】
FDAは、同種骨髄移植、慢性リンパ性白血病、特発性血小板減少性紫斑病(ITP)、小児HIV、原発性免疫不全、川崎病、慢性炎症性脱髄性多発神経炎(CIDP)、及び、抗体の高い移植患者又はABO不適合提供者の腎臓移植を含む、様々な適応症に対処するのにIVIGを利用することを承認している。ある実施形態では、本明細書中で与えられるIVIG組成物は、これらの疾患及び容態の治療又は対処に有用である。
【0079】
更に、通常、IVIGの適応外使用を患者に施して、様々な適応症、例えば、慢性疲労症候群、大腸炎、皮膚筋炎及び多発性筋炎、グレーブス眼症、ギラン・バレー症候群、筋萎縮症、封入体筋炎、ランバート・イートン症候群、紅斑性狼瘡、多巣性運動ニューロパチー、多発性硬化症(MS)、重症筋無力症、新生児同種免疫性血小板減少症、パルボウイルスB19感染症、尋常性天疱瘡、輸血後紫斑病、腎臓移植拒絶、自然流産/流産、スティッフパーソン症候群、眼球クローヌス・ミオクローヌス、重篤疾患成人の重症敗血症及び敗血症性ショック、中毒性表皮壊死症、慢性リンパ性白血病、多発性骨髄腫、X連鎖無ガンマグロブリン血症、及び低ガンマグロブリン血症を治療又は対処する。ある実施形態では、本明細書中で与えられるIVIG組成物は、これらの疾病及び容態の治療又は対処に有用である。
【0080】
最終的に、原発性免疫不全、RRMS、アルツハイマー病、及びパーキンソン病を含む疾病の治療又は処置のためのIVIGの実験的使用が提案されている(米国特許出願公報第US2009/0148463号。これは、全ての目的のために参照としてそのまま本明細書に組み込まれている)。ある実施形態では、本明細書中で与えられるIVIG組成物は、原発性免疫不全、RRMS、アルツハイマー病、又はパーキンソン病の治療又は処置に有用である。日常の投与を含むある実施形態では、被験体に投与される有効量は、医者により、年齢、体重、疾病の重症度、投与経路(例えば、静脈注射又は皮下注射)及び治療に対する応答の個別の違いを考慮して定められ得る。ある実施形態では、本発明の免疫グロブリン製剤は、毎日約5mg/キログラム〜約2000mg/キログラムで被験体に投与され得る。追加の実施形態では、免疫グロブリン製剤は、少なくとも約10mg/キログラム、少なくとも15mg/キログラム、少なくとも20mg/キログラム、少なくとも25mg/キログラム、少なくとも30mg/キログラム、又は少なくとも50mg/キログラムの量で投与され得る。追加の実施形態では、免疫グロブリン製剤は、被験体に、約100mg/キログラム、約150mg/キログラム、約200mg/キログラム、約250mg/キログラム、約300mg/キログラム、約400mg/キログラムまでの投与量で投与され得る。他の実施形態では、免疫グロブリン製剤の投与量は、より多く又はより少ない場合がある。更に、免疫グロブリン製剤は、毎日1以上の投与回数で投与され得る。IgG製剤により治療される疾病に精通している臨床医は、当該分野で知られている基準に従って、患者に適した投与量を定めることができる。
【0081】
1つの通常利用されるIgG製品、静脈注射用免疫グロブリン又はIVIGは、静脈注射投与用に調合される。本発明のIgG製剤では、極めて高い免疫グロブリン濃度(20%重量/体積又はそれよりも高い)が得られ、その濃度により、治療効果のあるのための体積が著しく削減され、本発明の組成物は、患者の皮下及び/又は筋肉内に投与するのに特に有利であるが、静脈内投与が、投与のための選択肢として残る。
【0082】
用語「有効量」は、被験体中の処置される病状(例えば、原発性免疫不全、RRMS、アルツハイマー病、パーキンソン病等)を改善する又は治療することになる、免疫グロブリン特にIgG製剤の量を指す。被験体に投与される有効量は、医者により、年齢、体重、疾病の重症度、投与経路(例えば、静脈注射又は皮下注射)及び治療に対する応答の個別の違いを考慮して定められ得る。ある実施形態では、本発明の免疫グロブリン製剤は、毎日約5mg/キログラム〜約2000mg/キログラムで被験体に投与され得る。追加の実施形態では、免疫グロブリン製剤は、少なくとも約10mg/キログラム、少なくとも15mg/キログラム、少なくとも20mg/キログラム、少なくとも25mg/キログラム、少なくとも30mg/キログラム、又は少なくとも50mg/キログラムの量で投与され得る。追加の実施形態では、免疫グロブリン製剤は、被験体に、約100mg/キログラム、約150mg/キログラム、約200mg/キログラム、約250mg/キログラム、約300mg/キログラム、約400mg/キログラムまでの投与量で投与され得る。他の実施形態では、免疫グロブリン製剤の投与量は、より多い又はより少ない場合がある。更に、免疫グロブリン製剤は、毎日1以上の投与回数で投与され得る。IgG製剤により治療される疾病に精通している臨床医は、当該分野で知られている基準に従って、患者に適した投与量を定めることができる。
【0083】
ある実施形態では、濃縮されたIgG製剤は、投与につき約5mg/キログラム〜約2000mg/キログラム被験体に投与され得る。ある実施形態では、投与量は、少なくとも約5mg/kg、又は少なくとも約10mg/kg、又は少なくとも約20mg/kg、30mg/kg、40mg/kg、50mg/kg、60mg/kg、70mg/kg、80mg/kg、90mg/kg、100mg/kg、125mg/kg、150mg/kg、175mg/kg、200mg/kg、250mg/kg、300mg/kg、350mg/kg、400mg/kg、450mg/kg、500mg/kg、550mg/kg、600mg/kg、650mg/kg、700mg/kg、750mg/kg、800mg/kg、850mg/kg、900mg/kg、950mg/kg、1000mg/kg、1100mg/kg、1200mg/kg、1300mg/kg、1400mg/kg、1500mg/kg、1600mg/kg、1700mg/kg、1800mg/kg、1900mg/kg、又は少なくとも約2000mg/kgであり得る。
【0084】
本発明に従って、治療工程を完了するのに必要な時間は、医者が定めることができ、1日程の短いものから1〜6ヶ月までの範囲であり得る。
【0085】
濃縮IgG治療の投与量及び頻度は、他の因子の中でも、患者の治療される疾病又は容態並びに疾病又は容態の重症度に依存することになる。一般に、原発性免疫不全では、体重当たり約100mg/kg〜約400mg/kgの投与量が、3〜4週毎に投与されることになる。神経性及び自己免疫疾患では、体重当たり2g/kgが、3〜6ヶ月間、月に1度、連続5日間にわたり施される。これは、一般に、体重当たり約100mg/kg〜約400mg/kgを、ほぼ3〜4週毎に1度、投与することを含む維持治療で補われる。一般に、患者は、ほぼ14〜35日毎に1度、又は、ほぼ21〜28日毎に1度、投与又は処方を受ける。治療の頻度は、他の因子の中でも、患者の治療される疾病又は容態並びに疾病又は容態の重症度に依存することになる。
【0086】
好ましい実施形態では、ヒトの免疫不全、自己免疫疾患、又は急性感染症を治療する方法は、その必要に応じて与えられ、その方法は、本発明の医薬IgG組成物を投与することを含む。
【実施例】
【0087】
以下の実施例は、限定するものとしてではなく、単に説明のために与えられる。当業者は、様々な危急ではないパラメータを変化又は変更して、本質的に同じ又は類似の結果を出し得ることを容易に認識することになる。
【0088】
実施例1:様々なIgG濃度及び製剤の安定性検査
1.目的
この検査の目的は、高い蛋白質濃度での低pH(濃縮溶液中で計測される場合、0.25Mグリシン4.4〜4.9)の保存安定性を、筋肉内及び皮下注入可能な免疫グロブリンに現在利用されるような中性pH製剤(22.5g/lグリシン、3g/lNaClpH7)と比較することである。
【0089】
2.実験計画
全行程は、蛋白質が5%になるようにナノ濾過を濃縮することで開始する。0.15Mのグリシン(調査される最低グリシン濃度)で10回の緩衝剤交換が行われ、続いて、蛋白質が20%を超える、標的値への最終濃縮が行われる。実験の第1工程では、分画分子量が30Kである0.5m2のミリポア社ポリエーテルスルホン薄膜(通常の篩)が利用されるが、実験の第2工程は、開孔篩を有する0.5m2のミリポア社ポリエーテルスルホン薄膜で行われ、その薄膜は、粘性の高い溶液により適しており、洗浄後の成分は、薄膜表面が更に小さな第2の限外/透析濾過装置(0.1m2、開孔篩)により濃縮されて、産出量の損失が低減される。
【0090】
最終容器を調合し、低pHで保存する、又は、低pH保存をバルク溶液で行い、その後に、保存前に、最終容器を中性pHで調合する。
【表1】
【0091】
pH:メトラー・トレド製Lot405−DPA−SC−S8/120電極とPT1000温度探触子とを備えたクニックポータメス型911XPHで、pHを検査した。
【0092】
蛋白質:ビウレットで蛋白質値を確定した。
【0093】
高能率分子篩クロマトグラフィーを利用して、分子径分布を検査した。
【0094】
欧州薬局方に記載されるように、ACA滴定量を検査した。
【0095】
以下の許容基準が定義される:
・高速液体クロマトグラフィーによる分子径分布:
i.単量体及びオリゴマー/二量体:>90%
ii.凝集体:<10%(<%、静脈内投与に対して)
・ACA滴定量:
静脈内投与に対して、50%未満のCH50単位、消費された/mg蛋白質
【0096】
5.結果及び議論
5.1.低pH4.7及びpH7.0に調整された製剤中の凝集体及び断片含有量とACA滴定量との比較
以下の表2で示されるのは、異なる蛋白質濃度での標準的な製剤(pH4.7、0.25Mグリシン、又はpH7.0、22.5g/Lグリシン、3g/L NaCl)に対する、28〜30℃で3ヶ月間保存した後の凝集体及び断片含有量並びにACA的定量である。
【表2】
【0097】
データは、低pHの製剤では、28〜30℃で3ヶ月間保存した後、凝集体が低く、ACA滴定量が低いことを明らかに示す。pH7製剤の全てのACA滴定量は、この検査で定められる許容基準を超える。表2には、2〜8℃で3ヶ月後の値が与えられる。
【表3】
【0098】
2〜8℃での結果では、28〜30℃で見られる傾向が確認される。ACA滴定量は、全て、許容基準として定められた限定値よりも低いが、pH7.0製剤は、値がより高いようである。蛋白質値は、検査されるパラメータの結果に影響を与えない。
【0099】
5.2 製剤IGSC60(Ph4.7、A篩)及びIGSC62(Ph4.7、V篩、より小さな除外濾過システムでの洗浄後濃縮)中の、凝集体及び断片含有量並びにACA滴定量への異なる濃縮手段の影響
[0099]以下の表4で示されるのは、異なる濃縮手段を用いた、異なる蛋白質濃度での低Ph製剤に対する、28〜30℃で3ヶ月間保存した後の凝集体及び断片含有量並びにACA滴定量である。
【表4】
【0100】
両方の濃縮様式について、3ヶ月間保存した後のデータは、類似の結果を示した。表5では、2〜8℃での対応する値が示される。
【表5】
【0101】
2〜8℃で得られる値では、28〜30℃で得られた結果が確認された。濃縮方法は、製品の安定性に影響を与えない。
【0102】
主要濃縮工程の一方のシステムと後洗浄液を濃縮する他方のシステムの、2つのシステムを用いての取り組みにより産出量が高くなるので、この方法は、大規模な製造に更に適している。
【0103】
以下の結論が、この検査にある結果から引き出される:
・低pH製剤により、2〜8℃及び28〜30℃での3ヶ月間の保存の後、ACA値は更に低く、凝集体含有量が更に低く、断片含有量が更に低くなる。
・18〜30℃での3ヶ月間の保存の後、中性pH製剤のACA値は、許容基準を超える。
・蛋白質値は、検査されるパラメータの結果に影響を与えない。
・濃縮方法は、製品の安定性に影響を与えない。
・開孔篩薄膜を用いて適切な後洗浄液を得ることができる。
【0104】
これらの結果に基づいて、低pHでの調合、後洗浄液を濃縮する第2小型装置と開孔薄膜付き除外/透析濾過装置とを利用する濃縮方法を用いて、皮下の投与ための、20%±2%の蛋白質濃度の新規のIgG製品を生成すると結論を下された。
【0105】
実施例2:サブキュヴィアNGの除外濾過及び調合
高濃度前臨床用20%ロットを生産する間に、情報を収集した。ナノ濾過工程までの、20%ロットを製造するのに利用されるプロセスが上に記載された。限外/透析濾過を向上させて、溶液を20%(限界:18〜22%)まで濃縮した。産出量損失を最小限に低減するために、透析濾過に利用される限外濾過装置の後洗浄液は、同じ薄膜を備えた第2小型装置により濃縮され、その後、バルク溶液に加えられる。
【0106】
意外にも、低いpHで保存する間のウイルスの不活性は、溶液の蛋白質含有量に左右されないことを示すことができた。10%溶液(ガンマガード(登録商標)リキッド)及び20%溶液において、類似のウイルスの低減を得た。従って、20%製品に対して、低pHでの保存をウイルス低減工程として維持した。
【0107】
2.プロセス注釈
限外濾過
ナノ濾液のグリシン濃度を0.25Mの標的に調節する。限外濾過(UF)により、溶液を6±2%重量/体積%の蛋白質濃度に濃縮する。通常、蛋白質濃度は、AU280−320の計測により定められる。14.1の消衰係数を利用する。pHを5.2±0.2に調節する。利用されるUF薄膜は、公称分画分子量(NMWCO)が50,000ダルトン以下であり、特に、高粘度製品用に設計されている(ミリポア社からのV篩)。例えば、ミリポア社ペリコンバイオマックスは、NMWCOが50Kダルトン以下である。薄膜材料は、ポリエーテルサルフォンである。
【0108】
pH4.2±0.2の0.25Mグリシン溶液で濃縮液を透析濾過する。最小交換体積は、当初の濃縮液体積の10倍である。限外濾過/透析濾過操作を通じて、溶液を4℃〜20℃に保つ。
【0109】
透析濾過後、溶液を、最小限の22%重量/体積の蛋白質濃度に濃縮する。溶液温度を2℃〜8℃に調節する。UV読み出しにより、14.1の消衰係数値を利用して、蛋白質濃度を確定してもよい。
【0110】
システム中の完全な残留蛋白質を回収するために、第1大型限外濾過システムの後洗浄は、再循環様式で、死容積の少なくとも2倍で行われ、全ての蛋白質が洗い出されたことを確証する。次に、第1システムの10分の1以下の寸法の、同じ型式の薄膜を備えた第2限外/透析濾過システムを用いて、第1限外濾過システムの後洗浄液を少なくとも22%重量/体積の蛋白質濃度に濃縮する。後洗浄濃縮液をバルク溶液に加える。次に、第2限外濾過システムを後洗浄する。工程14の最終バルクの蛋白質濃度を調節するのにこの後洗浄液を利用する。溶液温度は、2℃〜8℃に調節される。
【0111】
調合
更に、第2小型限外濾過システムの後洗浄液で及び/又は透析濾過緩衝剤で、蛋白質濃縮液を20.4±0.4%に調節する。
必要であれば、pHを4.4〜4.0に調節する。
実施例3:20%ロットの製造
【0112】
1.序文
本報告は、前臨床ロット生産を記載し、皮下用途の20%(重量/体積)液体多価ヒト免疫グロブリン製剤である、バクスター社の新規の検査対象の免疫グロブリン製剤「サブキュヴィアNG、20%」の結果を要約する。
【0113】
「本発明の詳細な技術」で記載されたように上記のような濃縮方法を用いて、製造が行われた。分別は、寒冷沈殿物の分離で開始する。次に、後続の冷却アルコール分別の前に様々な粗製凝固因子及び阻害因子を単離するために、寒冷保存血漿が利用され得る。サブキュヴィアNG、20%を浄化するまえに寒冷貯蔵血漿から粗製凝固因子及び阻害因子をバッチ処理で吸着するために、幾つかの経路を選択し、それらの経路は、以下の表で経路1〜7と呼ばれる。
【表6】
【0114】
前臨床サブキュヴィアNG、20%の生成では、多岐にわたる異なる吸着選択肢を包含するように、経路1、3及び6から誘導されるコーン開始材料を選択した。様々な吸着プロセスは、テシュナーら、国際輸血学会誌(2007年、第92号、42〜55頁)、ポルスラーら、国際輸血学会誌(2008年、第94号、184〜192頁)、米国特許第6,395,880号及び第5,409,990号、及び、プロトロンビン複合体:ブルンメルヒュイス、血漿蛋白質分別方法(J.M.カーリング編者、アカデミック出版、1980年)に記載される。
【0115】
変更されたコーンアルコール分別は、沈殿物Gと呼ばれる、主要な中間IgG成分をもたらし、その成分は、クロマトグラフィー浄化を利用して、最終製品に更に処理される。下流製造工程は、陽イオン交換クロマトグラフィー(CMセファロース高速流)と陰イオン交換クロマトグラフィー(ANXセファロース高速流)とを含み、つまり、3つの独立したウイルス不活性又は除去工程を含む。
・溶媒/洗浄剤処理(1%トリトンX−100、0.3%燐酸トリ−N−ブチル及び0.3%ポリソルベート−80の混合物)
・ナノ濾過(アサヒ・プラノバ35nm)及び
・3週間にわたる高温での低pH保存
【0116】
皮下用途のための高蛋白質濃度に到達するために、限外/透析濾過工程で開経路篩を利用しなければならない。好ましくは、第2限外/透析濾過装置を利用して、後洗浄成分を濃縮し、第1装置から全蛋白質を回収する。
【0117】
サブキュヴィアNG、20%は、免疫グロブリンG(IgG)の液体製剤であり、そのうちの少なくとも95%の蛋白質は、ガンマグロブリンである。製品は、等浸透圧性のものであり、低pHに調合される。最終濃縮工程の間、10%蛋白質の濃度で、pHは、4.4〜4.9である。長期間の安定性検査の結果が有効になった後、20%溶液の最終pHが定められる。最終溶液は、180〜220gの蛋白質と、僅かの賦形剤として、リットル当たり0.1〜0.3モルのグリシンとを含む。液体製剤は、僅かに薄い黄色の透明であり、実質的に目に見えない粒子である。
2.前臨床ロットのデータ及び質量均衡
1.前臨床ロット:SC00107NG
吸着選択肢1:選択肢6:F IX、F VII、AT−III
開始材料のロット番号:沈殿物G VNELG171(米国源)
最終容器のロット番号:SC00107NG
2.前臨床ロット:SC00207NG
吸着選択肢3:FEIBA、AT−III
開始材料のロット番号:沈殿物G VNELG173(米国源)
最終容器のロット番号:SC00207NG
3.前臨床ロット:SC00307NG
吸着選択肢1:吸着工程なし
開始材料のロット番号:沈殿物G LB079301(米国源)
最終容器のロット番号:SC00307NG
【表7】
【0118】
最終バルクレベルでは、製剤の純度が、低レベルの主要不純物により示され、それらの不純物は、全IgGの0.1%を十分に下回る。本プロセスのこの最終工程での20%製品の分子径分布は、ガンマガード(登録商標)リキッド/キオヴィグ最終容器の分子径分布と類似している。これは、20%蛋白質までの濃縮が、IgG分子の保全性に負の影響を与えないことを示す。
【0119】
3.前臨床バッチの評価からの追加の結果
最終容器暫定放出基準は、皮下ヒト免疫グロブリンに対する許可局からの要求、皮下投与用の現行製品の最終容器仕様、及びガンマガード(登録商標)リキッド/キオヴィグ仕様に基づいて定められた。製品及び/又はプロセス関連不純物の程度を評価するために、追加の品質管理検査が実行された。
【0120】
更に、最終容器の関連する抗体スペクトルの評価が行われ、前臨床IVIG、10%TVRロットからの結果と比較された。
【0121】
以下の表(表8)に結果が与えられる。
【表8】
【0122】
3つの前臨床サブキュヴィアNG20%最終容器と前臨床ガンマガード(登録商標)リキッド/キオヴィグロットに対する抗体滴定量及び濃縮率は、同程度の大きさである。
【表9】
【0123】
製品及びプロセス関連不純物の除去は、全ての3つのロットに対して十分である。
【0124】
4.結論
サブキュヴィアNG20%の3つの最終容器ロットは、200リットルの大きさで成功裏に製造された。アルコール分別の前に多岐にわたる吸着工程を包含ために、3つの吸着経路を選択した。つまり、
−選択肢1、吸着工程を用いない米国源血漿
−選択肢3、FEIBA、AT−III吸着後の米国源血漿
−選択肢6、F−IX、F−VII、AT−III吸着後の米国源血漿
【0125】
前臨床生産及び中間体の評価の間に監視される処理パラメータについて、最終製品では、3つのロットの間で検出可能な明らかな違いが見られなかった。
【0126】
全ての最終容器は、どの種類の開始材料が選択されたのかに関係なく、製品に関する暫定仕様に適合する。
【0127】
実施例4:20%製剤の保存検査
1.序文
サブキュヴィアNG、20%は、皮下用途の20%(重量/体積)液体多価ヒト免疫グロブリン製剤である。サブキュヴィアNG、20%は、上記のように生産された。
【0128】
この検査は、3つの前臨床ロット及び1つの実行可能性ロットの2〜8℃及び28〜30℃(実行可能性ロットのみ)での6ヶ月までにわたる保存データを要約する。
【0129】
2.目的
本検査の目的として、サブキュヴィアNG20%最終容器の2〜8℃及び28〜30℃での保存安定性を評価する。
【0130】
3.安定性指標パラメータ
主要な劣化様式は、変性、凝集及び壊裂であり、試料の分子径分布が変化することになる。従って、高能率分子篩クロマトグラフィーによる分子径分布の分析は、主要な指標パラメータである。
【0131】
4.バッチ検査及び主要梱包
本報告中にある安定性データは、前臨床ロットSC00107、SC00207NG及びSC00307NG並びに実行可能性ロットIgGSC62/1で作成される。
【0132】
5.結果
以下の表11に、12ヶ月までの保管後の前臨床最終容器の結果を示す。
【表10】
【表11】
【0133】
6.結論
データでは、製品が、18ヶ月までの2〜8℃及び28〜30℃の保管について調査されるパラメータについての定義済みの仕様に準拠することが確認された。
【0134】
実施例5:ウイルス不活性のための低pH処理
目的及び合理性
序文
以下に、サブキュヴィアNG溶液と呼ばれる、免疫グロブリン皮下(ヒト)14〜20%3回ウイルス削減(TVR)溶液は、貯蔵されたヒト血漿から製造される。凝固因子/阻害因子を除去する塊捕獲工程の後に、サブキュヴィアNGの浄化が、変更されたコーンアルコール分別で開始され、主要な中間沈殿物Gを得て、クロマトグラフィー浄化並びに3つの異なるウイルス不活性/除去工程を含む下流プロセスに続く。本検査では、最終容器の低pH保存のウイルス削減能力が、詳細に調査された。
【0135】
下流浄化プロセスは、以下の工程を含む:再懸濁された沈殿物Gが、溶媒/洗浄剤(S/D)処理、カルボキシメチル(CM)セファロース高速流を利用した陽イオン交換クロマトグラフィー及びANXセファロース(登録商標)高速流を利用した陰イオン交換クロマトグラフィー、ナノ濾過、除外濾過、pH調整、並びに減菌濾過及び充填に曝される。無菌充填後、サブキュヴィアNG溶液は、最終容器内で低pH保存に曝される(製造プロセスを概略的に示すために、以下のプロセス方式を参照すること)。
プロセス方式
【0136】
図2に示されるプロセス体系Aは、工程10から始まる、サブキュヴィアNG溶液の製造方式の下流部分の概略を与える。本検査に利用される特別仕様の中間体は、プロセス工程「最終容器、前保存」に等しい。
【0137】
図2に示されるプロセス方式Bは、ウイルス滴定の試料採取を含む本検査に利用される、システム規模縮小型プロセスの概略を与える。
【0138】
潜在的なウイルス伝搬に対する高い安全性の余地を与えるために、作用様式が相補する3つの専用のウイルス不活性/除去工程は、サブキュヴィアNG溶液の製造プロセスに一体化される:
・溶媒/洗浄剤処理(工程11)
・ナノ濾過(工程14)
・最中容器中の高温での低pH保存(後無菌充填)
【0139】
ウイルス不活性に関する低pH及び高温保存の能力及び堅牢性は、以前の検査行程中に既に調査されており、その際、サブキュヴィアNGに等価であるが静脈注射用の10%蛋白質に調整された製品であるIGIV10%VRTが利用された。この検査から得られた結果では、調査される全ての脂質包膜ウイルスが、有効に且つ堅牢に不活性にされたことが実証された。ウシウイルス性下痢ウイルス(BVDV)では、不活性動態が最も低かったことが示された。その上、このプロセス行程が、小さな非脂質包膜DNAウイルスに対する製造プロセスのウイルス安全性に更に寄与することが実証することができた。サブキュヴィアNG及びIGIV10%TVRは、免疫グロブリン製品であり、サブキュヴィアNGは、皮下投与用の変異形態であり、IGIV10%TVRは、静脈内投与用の製品変異形態である。両方が、限外濾過/透析濾過及び調合行程までの同じ製造プロセスを共有し、サブキュヴィアNGの蛋白質濃度が、10%の代わりに、14%〜20%の範囲に調節されることのみ異なる。
【0140】
従って、BVDV及びMMV不活性に関する、サブキュヴィアNG製造中の低pH及び高温保存行程の能力及び堅牢性(欧州医薬品評価機構、ヒト用薬剤評価(2001):CPMPバイオ技術作業部会を参照すること。血漿誘導性医薬品についての手引き、CPMP/BWP/269/95(第3改訂版)に注目すること)を評価するために、選択された基準プロセスパラメータに、製造プロセス用に定められた上限及び下限(即ち、時間、温度には下限、pHには、上限)を設定した。加えて、このウイルス不活性工程の堅牢性を更に調査するために、システム規模縮小行程の間、温度を所定の時間期間にわたって低くした。
【0141】
上に考察されたように、以下の包膜及び非包膜ウイルスを利用した。
・C型肝炎ウイルス(HCV)及び他の小さな脂質包膜RVAウイルスのモデルとして、ウシウイルス性下痢ウイルス(BVDV)。
・ヒトパルボウイルスB19(B19V)及び他の小さな非包膜DNAウイルスのモデルとして、マウス微小ウイルス(MMV)。
【0142】
CPMP指針268/95(欧州医薬品評価機構、ヒト用薬剤評価ユニット(1996):CPMPバイオ技術作業部会。ウイルス検証検査についての手引き:ウイルスの不活性及び除去を検証する検査の設計、寄与及び解釈、CPMP/BWP/268/95(改訂版)に注目すること)に従って、各々の製造プロセスのシステム規模縮小型モデルで検査を行った。サブキュヴィアNG溶液の製造中のウイルス不活性に関して、システム規模縮小型製造工程「低pH及び高温での保存」で得られた結果の有効性を実証するために、大規模製造プロセス中のこの製造工程用に定められたパラメータと、システム規模縮小型モデルのプロセスパラメータとを比較した。加えて、選択された生体化学パラメータを監視し、大規模プロセスの各々のパラメータと比較した。
【0143】
材料、方法及び設備
ウイルス及び細胞
アメリカ培養細胞系統保存機関(ATCC、ロックビル、メリーランド州)から得られるBVDV、NADL株(生体的にクローン化された、ATCC VR−1422)を利用する。標準操作手続に従ってMDBK細胞(ATCC CCL−22)上でウイルスを増殖し、BT細胞(ATCC CRL−1390)上で滴定する。
【0144】
メリーランド州、ロックビルのアメリカ培養細胞系統保存機関から、MMV、プロトタイプ株(ATCC VR−1346)を得た。標準操作手続に従ってウイルスを増殖し、A9細胞(ATCC CCL−1.4)上で滴定した。
【0145】
試験項目
オーストリア国、ウイーン、産業通り131のバクスターのPPD製品対応部門から、「低pH及び高温での保存」前の、即ち、製造工程無菌充填の後の特別仕様のサブキュヴィアNG中間体の以下のロットを得た。
・蛋白質濃度が13.5%のロット番号IGSC64
・蛋白質濃度が20.9%のロット番号IGSC64
【0146】
材料を+2℃〜+8℃の最終容器内に移送し、6ヶ月以内に利用した。
【0147】
緩衝液/溶液
pH調節用溶液
0.5MNaOH又は0.5MHClを利用して、pHを調節した。(室温で両方の溶液を保存し、12ヶ月以内に利用した。)
【表12】
【0148】
薬剤、即ち、蒸留水とHClを室温で組み合わせた。
【表13】
【0149】
それぞれの量のNaOHを、室温で蒸留水中に溶解した。
【0150】
pH計測用溶液
pHを、直接的に及び欧州薬局方(EP)に従う希釈溶液中で計測した。EP方法に従ってpHを計測するために、0.9NaCl溶液を利用して、蛋白質濃度を1%に希釈した。
【0151】
0.9%NaCl溶液を以下のように調製した:9.0±0.9gNaClを1000mlの蒸留水中に溶解した。溶液を0.2μmのフィルタで濾過し、室温で保存し、12ヶ月以内に利用した。
【0152】
細胞培養液
BVDV及びMMVウイルス滴定のための標準操作手続に従って、細胞培養又はウイルス滴定用の培地を調製した。
【0153】
ウイルス滴定アッセイ
TCID50アッセイ
標準操作手続に従うTCID50アッセイにより、ウイルスを含む試料を滴定した。端的に、連続1/2対数希釈液をそれぞれの組織培養液中で調製し、それぞれの指標細胞株を播種されたマイクロ滴定プレートの8ウェルの各々に100μlの各希釈液を加えた。36℃±2℃の温度の加湿されたCO2調節保存ユニット中でマイクロ滴定プレートを保存する。細胞変性効果の評価は、7日保存後に、顕微鏡での細胞の視覚検査により得られる。
【0154】
ポワソン分布に従って50%組織培養感染量(TCID50)を算出し、log10[TCID50/ml]で表した。
【0155】
BVDVバルク滴定
MMVが添加された試料についてMMV検査を行った。低pH保存の20〜27日後にサブキュヴィアNG中間体から取り出されたBVDV添加試料の検出限界を低くするために、これらの試料を以下のように滴定した。標準TCID50アッセイに加えて、1:3.16希釈液(0.5対数)及び以下の2つの1/2対数希釈液(それぞれの細胞培養液を希釈に利用する)の試料を、それぞれの指標細胞株を播種された96ウェルマイクロ滴定プレートの全てのウェル(ウェル当たり100μl)に加える。細胞の保存及びそれぞれのウイルスの細胞変性効果の評価は、以下のように行われた(TCOD50アッセイ)。結果を以下のように算出した。バルク滴定で滴定された体積と通常のTCID50アッセイ滴定された体積の比Rvolを算出した。TCID50アッセイで確定されたウイルス滴定量からRvolのlog10を引き、バルク滴定により確定されたウイルス滴定量としての結果を得る。
【0156】
実施例:
−連続0.5対数希釈(全て12列)のマイクロ滴定プレート上で試料を検定し、その後、希釈0(即ち、希釈されていない)からウェルが負であることを見出す。ポワソン分布に従う計算は、0.11log10[TCID50/ml]よりも小さいウイルス滴定量を与える。検定される試料の対応する全体積は、1.17mlである。
−同じ試料(希釈0)をマイクロ滴定プレートの全てのウェル上に付与する(バルク滴定)。従って、付与される総試料体積は、9.6mlである。
−体積比は、RVol=9.6:1.17=8.21、log10(RVol)=0.91である。
−次に、計算されたウイルス滴定量は、0.11−0.91よりも小さい、即ち、−0.80よりも小さい。ウイルス滴定量の信頼区間の上限を同様に計算する。
連続運動試料中の感染していない場合のウイルス滴定量の計算が、記録される。
【0157】
pH保存の完了後、連続運動試料中で最終試料までウイルス感染性が検出されなかった場合、検定検出限界を算出するために、明らかに負の結果であるTCID50アッセイ中のウェルに利用される全ての連続負性試料の体積を考慮に入れる。このために、以下の式を利用した(付録1も参照すること):
【数1】
式中、
Xi(i=1、2,3、...n)は、n連続負性試料の個々のウイルス滴定量[log10(TCID50/ml)]である。
【0158】
全ての試料が同じ滴定量、即ち、xを有する場合、式は、次式に簡略化される:
【数2】
【0159】
細胞毒性
細胞毒性判定用の各々の細胞株上で、擬似添加された、即ち、ウイルス原液の代わりに、感染細胞用BT培地(5%ウマ血清)*を添加されたサブキュヴィアNG中間体から、並びに、擬似添加され、pHが調節され(pH4.9±0.1)、0.45μmのフィルタで濾過された開始材料から、利用されるウイルス指標細胞株に対するプロセス中間体の細胞毒性を判定した。試料は、ウイルス含有試料と同様に検定された、即ち、希釈され、続いて、指標細胞株と共に保存された。保存後、最も高い非細胞毒性濃度を確定した。これらの生体系の変化に関して、ウイルス添加試料と擬似添加利用の細胞毒性と比較した場合、±0.5対数工程の逸脱は、大きくないと見なされた。
* 感染細胞用BT培地の組成は、以下である:DMEM(4.5g/lのDグルコースを含む)+1%(体積/体積)Lグルタミン(200mM)+1%(体積/体積)硫酸ゲンタマイシン(10mg/ml)+1%(体積/体積)ピルビン酸ナトリウム+2%(体積/体積)重炭酸ナトリウム(7.5%)+1%(体積/体積)非必須アミノ酸+5%(体積/体積)ウマ血清
【0160】
干渉
低ウイルス滴定量の試料に対して、感染性検定の性能への試料基質の影響を評価する必要がある。高いウイルス滴定量を含む試料に対して、希釈系列の重要部分、即ち、ウイルス正性及びウイルス負性ウェルの部分を記録することができ、高い試料希釈で生じる。その結果、ウイルス感染性の検出に対する、希釈された幾つかのlog10工程でもある試料基質の影響を無視することができる。サブキュヴィアNG中間体を含む試料に対する低ウイルス滴定量の確定との干渉は、以下のように計測された。擬似添加され、pH調節され(pH4.9±0.1)、0.45μmのフィルタで濾過された開始材料(サブキュヴィアNG中間体)から試料を採取し、適切な組織培養液で1:3.16に希釈し、希釈済みの(適切な組織培養液、即ち、BT細胞/BVDV用BT培地及びA9細胞/MMV用A9培地中で)ウイルス原液の懸濁液を用いて1:100で、log10(TCID50/ml)の2又は3名目滴定量に添加し、上記のように滴定した。添加プロセス中間体に対して得られた滴定量は、添加細胞培養液の対照の滴定量と比較された。
【0161】
ウイルス低減率の計算
CPMP指針268/95[3]に従って、以下の式を用いして、プロセスのウイルス不活性能力の分析を行った。
【数3】
式中
R=ウイルス低減率
V1=開始材料の体積
T1=開始材料中のウイルス濃度[TCID50/ml]
V2=工程後の材料の体積
T2=工程後のウイルスの濃度[TCID50/ml]
【0162】
処理前及び処理後の各添加試料の体積及び滴定量を用いて、Rを計算した。ウイルスが検出不可能である場合でも、計算のために、ウイルス滴定量として検出限界を取り上げた。標準的な操作手続に従って、小数点以下2桁まで与えられるウイルス滴定量log10(TCID50/ml)を用いて計算を実行した。低減率は、小数点以下1桁まで丸められた。
【0163】
検証パラメータの確定
時間、温度[℃]
タイマで保存時間を計測した。Pt−100センサで温度を計測し、連続的に記録した。
【0164】
pH値
標準的な操作手続に従って、標準的な実験用pHメータを用いて直接的に計測するばかりでなく、欧州薬局方(EP)により定められた方法を実施するにより、即ち、0.9%NaCl溶液を用いて1%蛋白質に希釈した後、pH値を確定した。以下では、添え字(EP)を有するpHは、EP方法に従うpH計測を意味するのに対し、添え字なしの「pH」は、直接計測を意味する。
他の羽他メータの確定
【0165】
分子径分布(MSD)
分子径分布のHPLC−SEC(高性能液体クロマトグラフィー−分子篩クロマトグラフィー)分析は、オーストリア国、ウイーンのバクスター社の分析生体化学部門により、標準的な操作手順に従って行われた。「システム規模拡大」ロット(オーストリア国、ウイーンのバクスター社のPPD製品対応部門で生産される)の全ての試料のHPLC−SEC分析は、この検査に従って実行された。従って、(この検査行程中の検査システム規模縮小プロセス内で蓄積された)擬似添加行程の試料の検査結果と、「システム規模拡大」ロットの検査結果との間の比較可能性を保証するために、擬似添加行程の試料のMSDの分析が、同じ部内で同じ検査に従って行われた。
【0166】
酢酸セルロース電気泳動
オーストリア国、ウイーンのバクスター社のACV化学部により標準的な操作手続に従って酢酸セルロース電気泳動(CAE)を実行した。
【0167】
丸め
標準的な手順に従って丸めを実行した。
・製造プロセス中のそれぞれのパラメータ用に指定されるような、又は、検定の精度により確定されるような小数点以下の同数桁まで、検定結果を丸めた。
・検定結果を丸めずに計算を行い、最終結果のみ丸めらたれた。
設備
・標準的な実験用pHメータを利用した。
・メトラー社AT−100分析天秤及びザルトリウス社BP3100P実験用天秤を利用した。
・PVDF薄膜フィルタ(ミレックスHV、又は均等物)を用いて0.45μm濾過を実行した。
・実験用タイマを用いてプロセス時間を計測した。
・生体安全性分類IIの小室を利用した。
・湿度が調節されたヘラウス社CO2恒温装置内で細胞を保存した。
・混合用に磁気攪拌機/攪拌棒を利用した。
・ハッケ社又はユラボ社低温維持装置及び/又は攪拌加熱ブロックにより温度を制御した。
・横河電機社μR1000レコーダに接続されたPt−100センサを利用して、温度を計測した。
【0168】
検査計画
ウイルス不活性に関する、サブキュヴィアNGの製造工程「低pH及び高温での保存」の堅牢性を調査するために、ウイルス不活性の効率を向上させるための主要パラメータを以下のように定めた。低pH保存によるウイルス不活性の堅牢性を調査するのに適した条件下で行程を実行した。大規模プロセスの条件とシステム規模縮小行程の条件の比較は、表Aに与えられる。
・蛋白質濃度:サブキュヴィアNGは、開発中の製品であり、調合されたバルクの蛋白質濃度範囲は、現時点では、ある程度広く定められる、つまり、約14%〜20%であり、後により狭くされ得る。従って、低pH処理の堅牢性を調査するために、2つの行程(行程計画)を、2つの極端な可能な蛋白質濃度で実行した。行程計画1を13.5%蛋白質濃度で実行した。行程計画2を20.9%蛋白質濃度で実行した。
特別仕様の材料の蛋白質濃度は、PPD製品対応部門により与えられた。特別仕様の材料の蛋白質濃度は、分析からデータが既に利用可能であったので再度定められず、特別仕様のサブキュヴィアNG中間体を生成した後に実行された。システム規模縮小プロセスに利用された材料は、減菌濾過され、システム規模縮小プロセスを開始するまで、2℃〜8℃で保存された(即ち、いずれの凍結/解凍手段にも曝されない)。最終的に調合されて減菌濾過された中間体に対する蛋白質濃度の変化は、予期されない。
・pH値:生産では、4.4〜4.9のpH範囲(直接計測)が、低pH保存を通過したサブキュヴィアNG最終容器で示される。ウイルス不活性にあまり好ましくはない条件下の低pH処理の有効性及び堅牢性を調査するために、2つの行程が、定められた上限で、即ち、4.9±0.1のpHで実行された。pHは、添加後に計測され、必要な場合、上限に調節された。低pHで27日±5時間にわたり保存した後、pHが、直接pH計測により、及び、欧州薬局方により推奨される方法により、即ち、0.9%NaCl溶液で1%蛋白質に希釈して再び計測された。
・温度:生産プロセスでは、低pH保存の間の温度は、30〜32℃に設定される。ウイルス不活性にあまり好ましない条件下での低pH保存の堅牢性を調査するために、全てのシステム規模縮小行程が、温度範囲の下限で、即ち、30±1℃で実行された。温度揺らぎの潜在的な影響を調査するために、温度は、毎週一回、6時間以上にわたり25±1℃に低くされた(即ち、温度低下は、保存第0日、第7日、第14日、第20日及び第27日に開始された。日数は、暦の日を指す。即ち、第0日は、保存が開始された日である)。誤って、6時間にわたる25℃1±1℃への温度低下が、第1週及び第2週の保存中に、週毎に2回、即ち、第1週の保存第0日及び第4日、低pH処理の第2週の保存第7日及び第10日に実行された。
・時間:保存時間は、通常、21日(BVDVの有効な不活性に必要とされる)〜28日(技術面/物流面の理由による上限)の範囲である。ウイルス不活性にあまり好ましくはない条件下の低pH処理の有効性及び堅牢性を調査するために、全てのシステム規模縮小行程が、上述の保存時間範囲の限界よりも下で、即ち、短い選択肢に対して20日±4日にわたり、長い選択肢に対して27日±5日にわたり実行された。添加された中間体を、低pH及び高温で27日±5日にわたり保存したが、ウイルス滴定用の試料を、20日±4日保存した後に採取し、3週保存した後にウイルス排除を調査した(表Aも参照すること)。
【0169】
総合的に、総計、4つのウイルス添加行程(BVDVで2つ、MMVで2つ)を実行して、製造工程「低pH及び高温での保存」のウイルス不活性能の堅牢性を調査した。更に、2つの添加されていない対照行程を実行して、生体化学パラメータを選定するための試料を生成した。加えて、2つの擬似添加対照行程を実行して、これらの行程から取り出された試料を利用して、ウイルス滴定検定へのプロセス中間体の潜在的な影響(即ち、細胞毒性及び干渉)を調査した。
【0170】
システム規模縮小プロセスと大規模プロセスの均等性更に示すために、最終製品について、低pH保存前及び保存後に以下の生体化学分析:分子径分布及び酢酸セルロース電気泳動を実行した。
【表14】
1 サブキュヴィアNGは、開発中の製品であり、蛋白質濃度は、尚、狭く定められていない。従って、2つのシステム規模縮小行程を、2つの極端な可能な蛋白質濃度で実行した。
2 保存期間は、尚、狭く定められておらず、ウイルス排除検査の結果に依存する。
3 両方の行程を、27日±5時間にわたり実行し、ウイルス滴定用の試料を保存第20日で取り出したので、ウイルス不活性データは、21日〜22日保存の大規模プロセス選択肢に利用することができた。
4 温度を、保存の週毎に1回、6時間にわたり25±1℃に低くした(即ち、温度低下は、第0日、第7日、第14日、第20日及び第27日に開始された。日数は、暦の日を指す。即ち、第0日は、保存が開始された日である)。第4.1.1章(「温度分布」)で考察されるように、6時間にわたる25±1℃への温度低下を、誤って、保存第1週及び第2週に、週毎に2回実行した(DR1_0407も参照すること)。
5 欧州薬局方による方法
【0171】
実験手順
その複雑性により、ウイルス添加工程、pH調節工程b及び0.45mm濾過工程の序列が、図3に見られる流れ図に示される(矢印の厚さは、相対的な体積を示す)。
【0172】
開始材料
行程1では、蛋白質濃度が13.5%の特別試料の材料(ロット番号IGSC64)を取り入れた。この材料を利用して、可能な蛋白質濃度の下限で、pH範囲の上限(即ち、BVDV:4.97及びMMV4.93)で、及び温度範囲の下限で(即ち、29.4℃で)行程を実行した。
【0173】
行程2では、蛋白質濃度が20.9%の特別仕様の材料(ロット番号IGSC64)を取り入れた。この材料を利用して、可能な蛋白質濃度の上限で、pH範囲の上限(即ち、BVDV:4.92及びMMV4.86)で、及び温度範囲の下限で(即ち、29.4℃で)行程を実行した。
【0174】
45.5mlの各々の開始材料に4.5mlのウイルス原液懸濁液を添加し、1〜2分の攪拌後に1mlの試料を採取した。即座に、試料をそれぞれの細胞培養液で1:3.16に希釈し(即ち、1体積の試料に2.16体積の細胞培養液を加え)、滴定した。続いて、別の3mlの試料を採取し、0.45μmPVDF薄膜を通して濾過した。即座に、1mlの濾過された材料をそれぞれの細胞培養液で1:3.16に希釈し、滴定した。
【0175】
pHの調節
一定分量の35mlのウイルス添加開始材料を0.5M HCl溶液を用いて攪拌下で(両方の行程で)4.9±0.1のpHに希釈した。次に、材料を2つの一定分量に分割した。一方の30mlの一定分量を「低pH保存」に利用し、5mlの一定分量を「温度保持対照」に利用した(「保持対照」の章を参照すること)。
【0176】
別の一定分量の10mlのウイルス添加開始材料を、0.5M NaOH溶液を用いて7.0±0.1のpHに調節した。5mlのpH調節された材料を「pH保持対照」に利用した。
【0177】
残りの5mlのpH調節された材料を「pH温度組み合わせ型保持対照」に利用した(「保持対照」の章を参照すること)。
【0178】
保持対照
ウイルス不活性の機能を、即ち、pH又は温度によるその機能を調査するために、3つの保持対照を異なる条件下で保存した。
【0179】
各々の5ml一定分量のウイルス添加pH調節(pH7.0±0.1)開始材料を、0.45μmPVDF薄膜フィルタを通して寒冷薬瓶内に濾過した。一定分量を、添加プロセス材料と共に30±1℃で保存し、次に、プロセスの終わりまでこの温度で保存した(pH保持対照、「pHHC」)。即座に、他の一定分量を+2℃〜+8℃で、27日±5時間にわたり保存した(pH温度組み合わせ型保持対照)。
【0180】
5mlの一定分量のウイルス添加pH調節(pH4.9±0.1)0.45μ濾過開始材料(上の「pH調節」を参照すること)を即座に+2℃〜+8℃に、プロセスの終わりまで保存した(温度保持対照、「tHC」)。
【0181】
濾過後の全ての保持対照の最小体積は、常に3mlを上回り、従って、保存期間後のウイルス滴定用に適した体積を利用することができた。
【0182】
低pH保存
30ml一定分量のウイルス添加pH調節(pH4.9±0.1)サブキュヴィアNG中間体を、0.45μmPVDF薄膜フィルタを通して、50ml無菌ガラス瓶内に濾過した。濾過後のサブキュヴィアNG中間体の最小体積は、常に、24mlを上回った。したがって、保存期間後のウイルス滴定に十分な体積を利用することができた。続いて、水槽を利用し、前後の緩やかな動きの攪拌下で、温度を30±1℃に保ち、外部温度センサを介して温度調節低温維持装置により、その水槽を調節した。外部センサ及び追加のPt100電極を、低pH保存に利用されたガラス瓶と等価な50mlガラス瓶内に置いた。各々のガラス瓶を、濾過後の添加プロセス材料の最大可能量である30mlの水で充たした。温度を連続的に記録した。材料が29℃の温度に達すると直ぐに、試料取り出しを開始した。低pHによる更なるウイルスの不活性を妨げるために、即座に、各々の試料を、それぞれの冷却細胞培養液(+2℃〜+8℃で保存)で1:3.16に希釈し(即ち、1体積の試料に2.16体積の細胞培養液を加え)、滴定した。全ての行程では、27日±5時間にわたる全プロセスを通して、サブキュヴィアNG中間体を(前後の緩やかな動きの)攪拌下で30±1℃に保存し、保存の毎週1回、少なくとも6時間(6時間以上)にわたり温度を25±1℃に下げた。誤って、6時間にわたる25±1℃への温度低下を、保存第1週及び第2週の週毎に2回、即ち、第1週の保存第0日と第4日、及び、低pH処理の第2週の保存第7日と第10日に行った。考察のために、第4.1.1章(「温度分布」)を参照すること。他の試料を抽出計画(第3.2章を参照すること)に従って採取し、即座に上記のように希釈し、滴定した。30℃±1℃での低PH保存が完了した後、直接計測により、及び、欧州薬局方により推奨される方法(0.9%NaCl溶液で1%蛋白質に希釈すること)によりpHを確定した。
【0183】
ウイルスを用いない対照行程
2つの対照行程を、ウイルス添加行程で記載されたように実行した。添加されずに0.45μm濾過されたサブキュヴィアNG中間体を処理した。材料をpH調節しなかったことを除いて、ウイルス添加行程1及び2でそれぞれ示されるのと同じプロセスパラメータを用いた。生体化学パラメータを確定するための試料を、抽出計画(第3.2章を参照すること)に従って取り出した。
【0184】
擬似添加開始材料(感染細胞用BT培地で0.9:10に添加された*;滴定前に上記のように試料を濾過する)、及び、0.45μm濾過後の擬似添加pH調節サブキュヴィアNG中間体の細胞毒性を判定した。細胞毒性を判定するための試料を、抽出計画(第3.2章を参照すること)に従って取り出した。
* 感染細胞用BT培地の組成は、以下である:DMEM(4.5g/lのDグルコースを含む)+1%(体積/体積)Lグルタミン(200mM)+1%(体積/体積)硫酸ゲンタマイシン(10mg/ml)+1%(体積/体積)ピルビン酸ナトリウム+2%(体積/体積)重炭酸ナトリウム(7.5%)+1%(体積/体積)非必須アミノ酸+5%(体積/体積)ウマ血清
【0185】
潜在的干渉
ウイルス検出と、pH(pH4.9±0.1)調節後のサブキュヴィアNG中間体を含む試料に対する試料基質の干渉を、以下のように各々の指標細胞株に対して2通りで調査した。擬似添加され、pH調節され、0.45μm濾過されたサブキュヴィアNG中間体から採取された1mlの試料を、それぞれの冷却細胞培養液(+2℃〜+8℃で保存)で1:3.16(体積/体積)に希釈した。続いて、1.8mlの希釈された材料に、0.2mlの希釈済みの*ウイルス原液懸濁液を1:10で添加し、2.0及び3.0log10[TCID50/ml]の計算された滴定量にした。混合後、試料を採取し、即座に滴定した。対照として、1.8mlのそれぞれの冷却細胞培養液(+2℃〜+8℃で保存)に、まさに同じ方法で、滴定前に、希釈済みのウイルス原液懸濁液を添加した。
* ウイルス原液懸濁液を事前に希釈するのに、適切な細胞培養液[BT細胞用BT培地(BVDV)、A9細胞用A9培地(MMV)]を利用した。
【0186】
抽出計画
ウイルス滴定
試料採取には、以下の許容範囲が適用される:1日(±1時間)保存後、2〜6日(±2時間)保存後、7〜13日(±3時間)保存後、14〜20日(±4時間)保存後、及び21〜27日(±5時間)保存後。試料符号についでは、第1.1章を参照すること(プロセス方式)
【0187】
即座に、試料を更に保存せずに滴定した。
【表15】
【0188】
細胞毒性の判定
即座に、試料を更に保存せず滴定した。
【表16】
【0189】
干渉の判定
即座に、試料を更に保存することなく滴定した。
【表17】
【0190】
生体化学パラメータの判定
各々の無添加対照行程からのみ試料を採取し、分析するまで、+2〜+8℃で保存した。
【表18】
1 システム規模縮小プロセスを評価するために、2つの「システム規模拡大」ロット並びに特別仕様材料(ロット番号IGSC64)の生産中に生成されたデータも考慮した。
2 システム規模縮小プロセスを評価するために、2つの「システム規模拡大」ロットの生産中に生成されたデータも考慮した。
3 製品の開発段階に関して、28日の保存期間の後の生体化学パラメータのデータを利用することができるとは考えられない。
【0191】
結果及び考察
この工程のシステム規模縮小型実験用モデルを利用して、脂質包膜ウイルスと脂質非包膜ウイルスの両方に専用の不活性工程としてのサブキュヴィアNGの低pH及び高温での保存の有効性及び堅牢性をBVDVとMMVで調査した。検証パラメータ及び生体化学パラメータを定義し、システム規模縮小プロセスで計測し、製造プロセス条件でのそれらの結果を比較することにより、2つのプロセスの均等性を実証した。
検証パラメータ及び生体化学パラメータについての結果
【0192】
検証パラメータ
ウイルス不活性に重要なパラメータ、即ち、プロセス材料のpH、低pH保存中の温度及び保存時間を判定することにより、システム規模縮小手段の有効性を確認した。
【0193】
低pH及び高温での保存前のpH値
添加プロセス材料のpHは、計測され、全ての行程において必要に応じて、4.9±0.1に調節され、従って、検査計画で示された限界内であった。保存後に確定されたpH値を第4.1.2章(他のプロセスパラメータ及び生体化学パラメータ)で考察する。
【0194】
温度分布
温度変化に対する、低pH及び高温での保存工程の堅牢性を調査するために、保存の毎週1回、温度を30℃±1℃から25±1℃に6時間にわたり下げた。温度は、常に、検査計画で示された範囲内であった。しかしながら、保存第1週及び第2週では、6時間にわたる25±1℃への温度低下を、週に2回、即ち、第1週の第0日及び第4日、第2週の第7日及び第10日に、誤って実行した。この事象が、行程概要を、ウイルス不活性に関してより悪い状況に移したので、ウイルス不活性の堅牢性を、当初意図したものよりも更に広範囲にわたり調査した。
【0195】
保存時間
低pH及び高温での保存の継続時間は、サブキュヴィアNGの製造プロセスで示されていない。現時点での検査結果に応じて、幾つかの選択肢がある:保存時間の継続時間の第1選択肢は、21〜22日である。第2選択肢は、約28日であってもよい。ウイルス不活性にあまり好ましくない条件下での低pH処理の有効性及び堅牢性を調査するために、全てのシステム規模縮小行程を、可能な保存時間の下限よりも下で、即ち、第1選択肢に対して20日±4時間にわたり、第2選択肢に対して27日±5時間にわたり実行した。添加された中間体を、低pH及び高温で、26日+22時間(MMV、両方の行程)にわたり、27日+1時間(BVDV、両方の行程)にわたり保存した。3週間保存した後のウイルス排除を調査するために、MMVに対して20日+1時間保存した後に、BVDVに対して20日+2時間後に、ウイルス滴定用試料を採取した。
【0196】
保存時間は、全ての行程において、システム規模縮小で示された限界、即ち、20日±4時間、27日±5時間以内であった。その限界は、現時点で考えられる製造中の保存時間の下限を下回る。
他のプロセスパラメータ及び生体化学パラメータ
【0197】
低pH保存後のpH
低pH保存に続いて、pH値を再び、直接的に、並びに、欧州薬局方(EP)により推奨される方法に従って、即ち、0.9%NaCl溶液で1%蛋白質に希釈して計測した。処理後に直接計測により計測されるpHは、低pH保存の前では、pH値に近かった。即ち、差は、−0.07〜+0.04(保存前のpHを引いた後のpH)の範囲であった。EP方法に従って計測されたpHは、常に、直接計測方法と比べて0.2〜0.3だけ高かった。EP方法を利用する際のこの僅かなpH値の増加は、pHを定めるこれらの2つの異なる方法の典型である。この事実は、製造規模で定められた限界内にあるとも考えられる。低pH処理後のpHの限界は、直接計測により定められる場合、4.4〜4.9であり、EP方法により定められる場合、4.6〜5.1である。
【0198】
分子径分布(MSD)
2つの無添加対照行程での保存の前後に、分子径分布(HPLC分析による)を調査した。サブキュヴィアNGの開発工程中に生産された3つの「システム規模拡大」ロットについて、及び、特別仕様の材料IGSC64(14%並びに20%蛋白質濃度)について検査結果を比較した。「システム規模拡大」ロットとも表記されるロットIGSC64は、この行程では十分な材料を入手することができなかったので、製造中に低pH及び高温での保存に曝されなかった。全ての結果は、互いに十分に比較された。分子径分布の値、特に、IgG単量体の百分率は、「システム規模拡大」行程で観察されたものに近かった。保存後のIgG単量体の濃度が数パーセント低かった、第2対照行程で留意することは、既に、保存前の材料のIgG単量体の濃度が比較的低く、その低い濃度は、「システム規模拡大」プロセス内で検査される材料にも適用されることに留意すること。従って、分子径分布データは、大規模プロセスとシステム規模縮小の等価性に対応する。
【0199】
ガンマグロブリンの純度
2つの無添加対照行程からの試料のガンマグロブリンの純度(CA電気泳動による)を確定し、2つの「システム規模拡大」ロットから、及び、特別仕様の材料IGSC64(14%並びに20%蛋白質濃度、低pH処理直前の値、説明は、上を参照すること)から得られた結果と比較した。システム規模縮小プロセス中のガンマグロブリンの純度について定められる全ての値は、「システム規模拡大」ロットで得られる結果に十分に匹敵し、検定精度以内であった(相対的標準偏差は、1.5%である)。これらの結果は、製造プロセスとシステム規模縮小プロセスの同等性を実証する。
ウイルス滴定の結果
【0200】
細胞毒性
低pH及び高温での保存前に得られた中間体の細胞毒性を、ウイルス添加行程で記載されたようにそれぞれのpHに調節された擬似添加材料を利用した対照行程で調査した。pH調節前後の擬似添加プロセス中間体では、BT細胞上での行程2を除いた全ての2つの行程で利用される細胞への細胞毒性の極めて弱い影響のみが見られた。pH調節された中間体では、1.0log10希釈及びそれ以降から、細胞毒性の影響が見られなかった。ウイルス添加行程について細胞毒性の著しい差は見られなかった。
【0201】
干渉検査
干渉検査からの結果では、低滴定量のBVDV及びMMVの検出と試料基質の著しい干渉は見られなかった。添加細胞培養液対照とサブキュヴィアNG中間体との間の添加ウイルス滴定量の差は、−1.0log10〜0.1log10であって、ウイルス滴定検定の精度内にある。従って、ウイルス不活性行程中に得られた滴定量は、干渉効果により歪まず、試料の実際の滴定量を示した。
【0202】
ウイルス不活性
ウイルスBVDV及びMMVの各々を用いて2つの行程を実行した。即ち、行程1は、13.5%蛋白質濃度であり、行程2は、20.9%蛋白質濃度であった。両方の行程を、4.9±0.1のpH、30±1℃で実行した。温度は、週毎に1回、最小6時間にわたり25±1℃に下げられた。2つの行程の結果の比較では、サブキュヴィアNGの可能な蛋白質濃度の上限及び下限で実行された2つの行程の間で、ウイルス不活性動態の著しい違いが現れなかった。
【0203】
ウイルス不活性にあまり好ましくない条件、即ち、保存時間及び温度の下限並びにpHの上限を用いて、BVDVについて、20日±4時間保存した後のBVDVの十分な不活性と、27日±5時間保存した後にそれぞれのサブキュヴィアNG中間体内に添加され得る全てのウイルスの完全な不活性が、調査される条件にかわわりなく実証された。両方の行程では、BVDVからの残留干渉性を、第20日に、尚も検出することができた。しかしながら、全てのBVDVは、27日保存の終わりに、検出限界以下に不活性化された。MMVの低減率では、脂質膜のないウイルスに関しても、この工程が、製品のウイルス安全性に大きく寄与することが実証された。利用されたウイルスに対する個別のウイルス滴定量及び低減率を以下でより詳細に考察する。加えて、ウイルス不活性動態の図表示が、各々のウイルスに対して与えられ、それぞれの結果表に追従している。
【0204】
BVDV
BVDVは、保存20日によるほぼ検出限界で(行程1)又は検出限界で(行程2)不活性化され、行程1及び2では、それぞれ、5.3log10及び5.5log10の低減率が与えられた。27日保存した後に、サブキュヴィアNG内に添加された全てのBVDVは、両方の行程で検出限界以下に不活性化され、低減率は、行程1及び2で、それぞれ、6.6log10及び6.5log10よりも大きかった。2つの行程の不活性動態の大きな差を観察することができなかった。行程1の中間体(蛋白質濃度が13.5%)と行程2の中間体(蛋白質濃度が20.9%)の両方で、不活性動態が拮抗していることが示された。
【0205】
高温での低pH処理では、20日±4時間保存した後のBVDVの不活性が有効であることが実証された。計算された平均低減率は、5.4log10であった。27日間低pH及び高温で保存した後、BVDVの検出限界以下への完全な不活性が得られ、6.6log10を上回る平均低減率が算出された。
【0206】
MMV
MMVでは、20日間低pH及び高温で保存した後に、行程1及び2に対して、それぞれ、2.9log10及び3.1log10の低減率が算出された。算出された平均低減率は、3.0log10である。27日間低pH処理した後、3.4log10及び3.7log10の低減率が得られた。算出された平均低減率は、3.6log10である。これらの低減率では、生理化学不活性に極めて耐性のあるパルボウイルスに関して、このプロセス行程が、製造プロセスのウイルス安全性に大きく寄与することが実証された。両方の行程でのウイルス不活性動態は二相性であり、不活性は、最初の7日間はより速く、続く3週間は多少より緩やかであった。
【0207】
保持対照
ウイルス不活性の機能、即ち、ウイルス不活性がpHにより、又は、温度により、又は、上記2つの組み合わせにより仲介されるのかを調査するために、3つの保持対照が、それぞれの条件下に保たれた。
【0208】
27日±5時間にわたり2℃〜8℃に保たれた「組み合わせ型保持対照」(pH7.0±0.1)を滴定した結果、調査された両方のウイルスに対して、ウイルス滴定量は、行程1のMMVを除いたウイルス添加開始材料に匹敵することになった。滴定量の少量の損失(1.6log10)が観察された。
【0209】
30±1℃及びpH7.0±0.1での保存(「pH保持対照」)では、脂質包膜ウイルスBVDVが、高温での保存に極めて鋭敏であることが示された。BVDVは、両方の行程で、4.6log10で不活性化された。その上、脂質膜のないウイルスMMVは、行程1及び行程2で、それぞれ、3.3log10及び3.8log10で不活性化された。
【0210】
27日±5時間にわたり低pHで2℃〜8℃に保たれた「温度保持対照」(tHC)を滴定した結果、調査された両方のウイルスに対して、ウイルス滴定量は、ウイルス添加開始材料に匹敵することになった。これらの結果で、BVDV並びにMMVは、4.4〜4.9の範囲内の低pH及び低温での保存に耐性があることが実証された。
【0211】
総じて、3つの保持対照の結果は、以下を示唆する:
・ 温度は、BVDVの不活性に最も重要な因子であり、低pHは、ある程度寄与し得る(30±1℃及び中性pHでの対照が、27日後に、完全ではないが実質的に不活性化されたことに基づく)。
・ MMVの不活性では、27日後の低pH動態試料のウイルス滴定量が、中性pH及び30℃での対照と実質的に同一であるので、温度は、唯一の重要な因子であると思われる。
【0212】
本検査行程では、サブキュヴィアNGの製造での低pH及び高温での保存のシステム規模縮小モデルを実証し、幾つかのプロセスパラメータを判定することにより製造手段のその等価物を実証した。加えて、システム規模縮小モデルと製造プロセスからの中間体の生体化学パラメータの比較は、システム規模縮小プロセスで得られる低減率が大規模プロセスに対しても有効であるという、両方のプロセスの等価性を裏付けた。ウイルス不活性に関する堅牢性を調査するために、サブキュヴィアNGの異なる蛋白質濃度の影響を調査した。添加開始材料のpHを上限に調節して、製造プロセスと比較し、温度の周期的低下の影響を調査した。本検査で得られるウイルス低減率及び不活性動態は、脂質包膜ウイルスBVDVが、27日内の低pH及び高温での保存により有効に且つ堅牢に不活性化されることを実証した。その上、20日にわたり低pH及び高温で保存した後、BVDVについて、著しいBVDVの不活性が得られ、平均低減率は、5.4log10であった。パルボウイルスモデルMMVに対して算出された平均低減率、即ち、20日保存後の3.0log10、20日保存後の3.6log10は、このプロセス行程が、生理化学耐性の高い小さな脂質膜のないDNAウイルスに関して、20日並びに27日の保存時間にわたる製造工程のウイルス安全性に更に寄与することを示す。
【0213】
調査された両方の蛋白質濃度では、MMV及びBVDVに対して同じ不活性動態が示され、濃縮免疫グロブリン溶液並びにウイルスBVDV及びMMVに対して、ウイルス不活性能への蛋白質濃度の影響が大きくないことを示唆する。
【0214】
本明細書に記載される実施例及び実施形態は、単に説明目的のためのものであり、それらに照らし合わせて様々な修正又は変更が、当業者に暗示されることになり、本出願の趣旨及び権限並びに付属の請求項の範囲内に含まれるべきであると理解される。本明細書で引用される全ての公報、特許、及び特許出願は、全ての目的のために、参照としてそのまま本明細書に組み込まれている。
【特許請求の範囲】
【請求項1】
水性組成物1リットル当たり約180グラム超過の蛋白質を含み、前記蛋白質の少なくとも95%がIgGである、該組成物。
【請求項2】
皮下投与及び静脈内投与に適した製品をもたらすプロセスにより生成される組成物であり、10〜22%の蛋白質濃度範囲でどの濃度に調節されるのかに関係なく、前記組成物が、ウイルスを不活性にするために最終容器内で高温で処理され得る、請求項1の組成物。
【請求項3】
前記IgGがヒトIgGである、請求項1に記載の組成物。
【請求項4】
前記蛋白質濃度が、約20%(重量/体積)である、請求項1に記載の組成物。
【請求項5】
約0.1〜0.3Mグリシンを更に含む、請求項1に記載の組成物。
【請求項6】
pHが約3〜6である、請求項1に記載の組成物。
【請求項7】
pHが約4〜6である、請求項6に記載の組成物。
【請求項8】
血漿から濃縮IgGの組成物を調製する方法において、
(1)限外濾過により血漿製剤中の蛋白質を約5%(重量/体積)に濃縮する工程と、
(2)透析濾過により前記製剤中の前記蛋白質を約20%(重量/体積)に更に濃縮する工程
から成る改善を含み、前記蛋白質の少なくとも95%がIgGである、前記方法。
【請求項9】
工程(1)が、限外濾過薄膜を利用して実行され、公称分画分子量(NMWCO)が、100kDa以下である、請求項8に記載の方法。
【請求項10】
工程(2)が、pHが4.2±0.1のグリシン透析濾過緩衝液で実行される、請求項8に記載の方法。
【請求項11】
前記透析濾過緩衝液が、0.25Mグリシンを有し、pHが4.0である、請求項10に記載の方法。
【請求項12】
工程(2)後の前記蛋白質濃度が、20%(重量/体積)よりも高く、透析濾過緩衝液で約20%(重量/体積)に実質的に調節される、請求項8に記載の方法。
【請求項13】
前記IgGがヒトIgGである、請求項8に記載の方法。
【請求項14】
血漿から濃縮IgGの組成物を調製する方法であって、
(1)遠心分離により、血漿から液体と沈殿物とを分離する工程と、
(2)冷却済みのエタノールを(1)からの前記液体と混合して、エタノール濃度が約8%(体積/体積)の混合物を作る工程と、
(3)遠心分離により、(2)の前記混合物から液体と沈殿物とを分離する工程と、
(4)(3)からの前記液体のpH及びエタノール濃度を、それぞれ、7.0及び20〜25%(体積/体積)に調節することにより、混合物を作る工程と、
(5)遠心分離により、(4)の前記混合物から液体と沈殿物とを分離する工程と、
(6)(5)の前記沈殿物を緩衝液で約1〜15の重量比で再び懸濁して、懸濁液を作る工程と、
(7)二酸化シリコン(SiO2)を(6)からの前記懸濁液と混合し、濾過により濾液を得る工程と、
(8)洗浄剤と冷却アルコールを、(7)の前記濾液と混合し、遠心分離により沈殿物を得る工程と、
(9)溶媒又は洗浄剤を含む水溶液中に前記沈殿物を溶解し、前記溶液を少なくとも60分にわたり維持する工程と、
(10)(9)の後の前記溶液を陽イオン交換クロマトグラフィーカラムに通し、前記カラムに吸収された蛋白質を溶出液中に溶出させる工程と、
(11)(10)からの前記溶出液を陰イオン交換クロマトグラフィーカラムに通して、流出液を生成する工程と、
(12)前記流出液をナノ濾過器に通して、ナノ濾液を生成する工程と、
(13)前記ナノ濾液を限外濾過薄膜に通して、限外濾液を生成する工程と、
(14)前記限外濾液を透析濾過緩衝液で透析濾過して、蛋白質濃度が約20%(重量/体積)の溶液を生成する工程と、
(15)前記溶液を0.2μm以下のフィルタに通して濾過することで(14)からの前記溶液を減菌することにより、濃縮されたIgGの組成物を得る工程と
を含む、前記方法。
【請求項15】
前記密封された容器を約21〜22日にわたり約30〜32℃で保存する工程を更に含む、請求項14に記載の方法。
【請求項16】
前記20%IgG製品を10%の従来技術の静注製剤と同程度に安定させる調合工程を更に含む、請求項14に記載の方法。
【請求項17】
請求項14〜15のいずれか一項の方法により生成され、少なくとも18%(重量/体積)の免疫グロブリンを含む、水性組成物。
【請求項18】
少なくとも20%(重量/体積)の免疫グロブリンを含む、請求項17に記載の水性組成物。
【請求項19】
請求項1〜16及び請求項17〜18のいずれか一項の有効量の前記組成物を患者に投与することを含む、免疫不全、自己免疫不全、又は急性感染症に罹患した患者を治療する方法。
【請求項1】
水性組成物1リットル当たり約180グラム超過の蛋白質を含み、前記蛋白質の少なくとも95%がIgGである、該組成物。
【請求項2】
皮下投与及び静脈内投与に適した製品をもたらすプロセスにより生成される組成物であり、10〜22%の蛋白質濃度範囲でどの濃度に調節されるのかに関係なく、前記組成物が、ウイルスを不活性にするために最終容器内で高温で処理され得る、請求項1の組成物。
【請求項3】
前記IgGがヒトIgGである、請求項1に記載の組成物。
【請求項4】
前記蛋白質濃度が、約20%(重量/体積)である、請求項1に記載の組成物。
【請求項5】
約0.1〜0.3Mグリシンを更に含む、請求項1に記載の組成物。
【請求項6】
pHが約3〜6である、請求項1に記載の組成物。
【請求項7】
pHが約4〜6である、請求項6に記載の組成物。
【請求項8】
血漿から濃縮IgGの組成物を調製する方法において、
(1)限外濾過により血漿製剤中の蛋白質を約5%(重量/体積)に濃縮する工程と、
(2)透析濾過により前記製剤中の前記蛋白質を約20%(重量/体積)に更に濃縮する工程
から成る改善を含み、前記蛋白質の少なくとも95%がIgGである、前記方法。
【請求項9】
工程(1)が、限外濾過薄膜を利用して実行され、公称分画分子量(NMWCO)が、100kDa以下である、請求項8に記載の方法。
【請求項10】
工程(2)が、pHが4.2±0.1のグリシン透析濾過緩衝液で実行される、請求項8に記載の方法。
【請求項11】
前記透析濾過緩衝液が、0.25Mグリシンを有し、pHが4.0である、請求項10に記載の方法。
【請求項12】
工程(2)後の前記蛋白質濃度が、20%(重量/体積)よりも高く、透析濾過緩衝液で約20%(重量/体積)に実質的に調節される、請求項8に記載の方法。
【請求項13】
前記IgGがヒトIgGである、請求項8に記載の方法。
【請求項14】
血漿から濃縮IgGの組成物を調製する方法であって、
(1)遠心分離により、血漿から液体と沈殿物とを分離する工程と、
(2)冷却済みのエタノールを(1)からの前記液体と混合して、エタノール濃度が約8%(体積/体積)の混合物を作る工程と、
(3)遠心分離により、(2)の前記混合物から液体と沈殿物とを分離する工程と、
(4)(3)からの前記液体のpH及びエタノール濃度を、それぞれ、7.0及び20〜25%(体積/体積)に調節することにより、混合物を作る工程と、
(5)遠心分離により、(4)の前記混合物から液体と沈殿物とを分離する工程と、
(6)(5)の前記沈殿物を緩衝液で約1〜15の重量比で再び懸濁して、懸濁液を作る工程と、
(7)二酸化シリコン(SiO2)を(6)からの前記懸濁液と混合し、濾過により濾液を得る工程と、
(8)洗浄剤と冷却アルコールを、(7)の前記濾液と混合し、遠心分離により沈殿物を得る工程と、
(9)溶媒又は洗浄剤を含む水溶液中に前記沈殿物を溶解し、前記溶液を少なくとも60分にわたり維持する工程と、
(10)(9)の後の前記溶液を陽イオン交換クロマトグラフィーカラムに通し、前記カラムに吸収された蛋白質を溶出液中に溶出させる工程と、
(11)(10)からの前記溶出液を陰イオン交換クロマトグラフィーカラムに通して、流出液を生成する工程と、
(12)前記流出液をナノ濾過器に通して、ナノ濾液を生成する工程と、
(13)前記ナノ濾液を限外濾過薄膜に通して、限外濾液を生成する工程と、
(14)前記限外濾液を透析濾過緩衝液で透析濾過して、蛋白質濃度が約20%(重量/体積)の溶液を生成する工程と、
(15)前記溶液を0.2μm以下のフィルタに通して濾過することで(14)からの前記溶液を減菌することにより、濃縮されたIgGの組成物を得る工程と
を含む、前記方法。
【請求項15】
前記密封された容器を約21〜22日にわたり約30〜32℃で保存する工程を更に含む、請求項14に記載の方法。
【請求項16】
前記20%IgG製品を10%の従来技術の静注製剤と同程度に安定させる調合工程を更に含む、請求項14に記載の方法。
【請求項17】
請求項14〜15のいずれか一項の方法により生成され、少なくとも18%(重量/体積)の免疫グロブリンを含む、水性組成物。
【請求項18】
少なくとも20%(重量/体積)の免疫グロブリンを含む、請求項17に記載の水性組成物。
【請求項19】
請求項1〜16及び請求項17〜18のいずれか一項の有効量の前記組成物を患者に投与することを含む、免疫不全、自己免疫不全、又は急性感染症に罹患した患者を治療する方法。
【図1A】

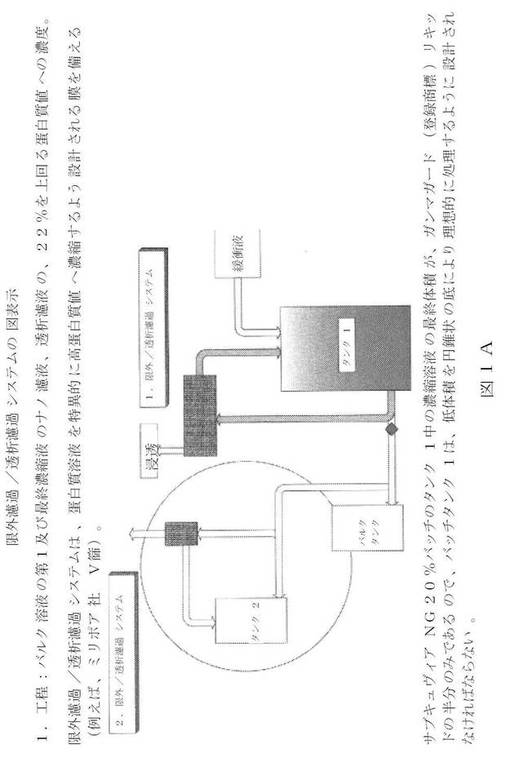
【図1B】
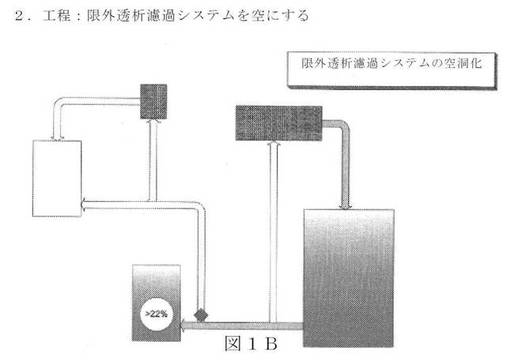
【図1C】
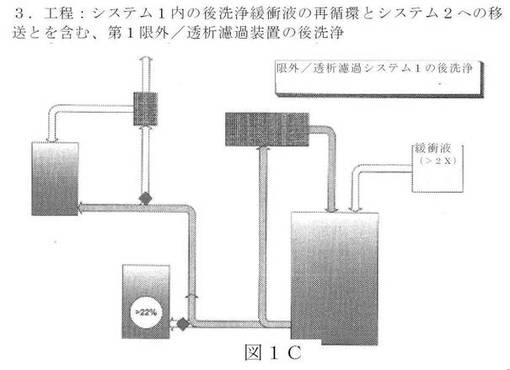
【図1D】
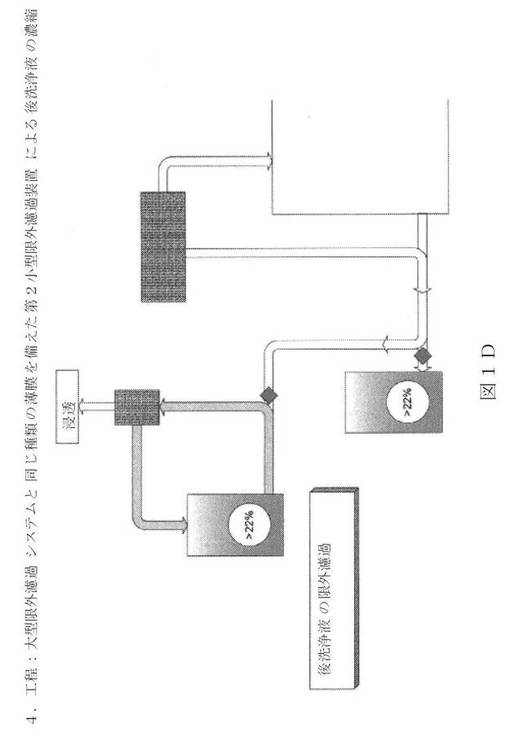
【図1E】
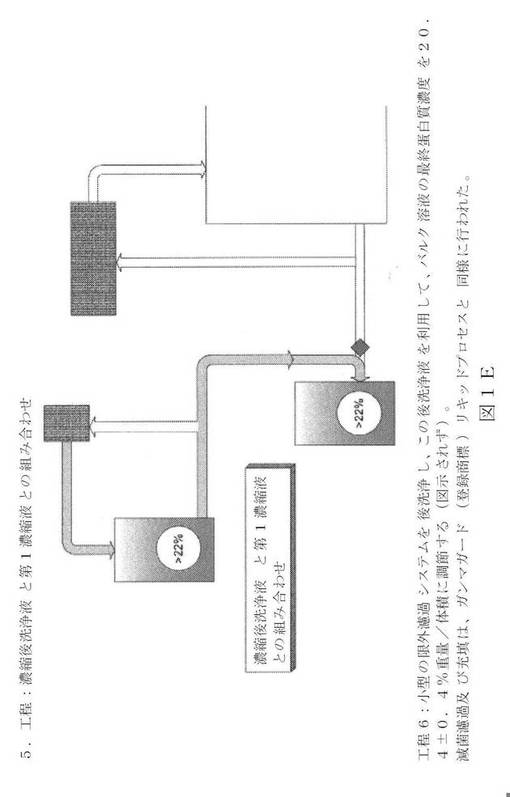
【図2】

【図3】
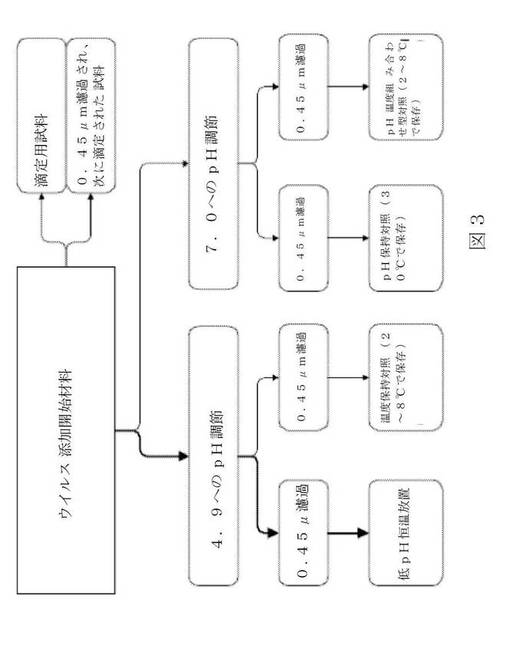
【図1B】
【図1C】
【図1D】
【図1E】
【図2】
【図3】
【公表番号】特表2012−528190(P2012−528190A)
【公表日】平成24年11月12日(2012.11.12)
【国際特許分類】
【出願番号】特願2012−513260(P2012−513260)
【出願日】平成22年5月27日(2010.5.27)
【国際出願番号】PCT/US2010/036430
【国際公開番号】WO2010/138736
【国際公開日】平成22年12月2日(2010.12.2)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【出願人】(501453189)バクスター・ヘルスケヤー・ソシエテ・アノニム (289)
【氏名又は名称原語表記】BAXTER HEALTHCARE S.A.
【Fターム(参考)】
【公表日】平成24年11月12日(2012.11.12)
【国際特許分類】
【出願日】平成22年5月27日(2010.5.27)
【国際出願番号】PCT/US2010/036430
【国際公開番号】WO2010/138736
【国際公開日】平成22年12月2日(2010.12.2)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【出願人】(501453189)バクスター・ヘルスケヤー・ソシエテ・アノニム (289)
【氏名又は名称原語表記】BAXTER HEALTHCARE S.A.
【Fターム(参考)】
[ Back to top ]