
皮膚粘膜毒性又は眼毒性を治療又は改善するための方法
細胞増殖及び生存シグナル変換経路を遮断することにより誘発される皮膚粘膜毒性、眼毒性、又は、副作用を治療又は改善する方法を提供する。本方法は、有効量のヒストン脱アセチル化酵素阻害剤、及び薬学的に許容可能な担体、その塩もしくは溶媒和物を、それを必要とする対象に投与するステップを含み、細胞増殖及び生存シグナル変換経路は、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムにより遮断される。

【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、皮膚疾患の治療に関するものであって、特に、HDAC 阻害剤を用いて、皮膚粘膜毒性又は眼毒性を治療することに関するものである。
【背景技術】
【0002】
関連技術の説明
多くの疾病や障害、例えば、癌、炎症性疾患、免疫学的疾患、及び、変性疾患等における、異常なシグナル伝達経路についての理解の進展と共に、細胞行動、応答、成長、生存及びアポトーシスの多くの中心的制御因子が、分子標的治療のための候補として、例えば、EGFR、VEGF、チロシンキナーゼ、セリン/トレオニンキナーゼ、及び、MAPK/ERKとして、明らかになっている(Lacouture ME. Nat Rev Cancer 6:803-812, 2006(非特許文献1))。上述による実験は、モノクローナル抗体、小化学分子、ペプチド模倣薬、及びアンチセンス・オリゴヌクレオチドを含む、革新的な分子標的技術の発展を導いている。しかし、正常な機能のためのシグナル伝達に依存している、組織中のEGFR 受容体、チロシンキナーゼ活性、又は、MAPK/ERK経路を同時に抑制することは、望まない結果をもたらす(Perez-Soler R, et al. J Clin Oncol 23:5235-5246, 2005(非特許文献2))。皮膚、粘膜、及び、角膜におけるEGFR、チロシンキナーゼ、及び、MAPK/ERK シグナル伝達は重要な機能を担うため、皮膚粘膜及び目の領域に、望まない副作用が一般に生じる。EGFR、チロシンキナーゼ活性、又は、MAPK/ERK 経路の抑制は、DNA 合成を抑制し、細胞増殖を阻み、ケラチノサイトの早発分化を促進する (Miettinen PJ, et al. Nature 376:337-341, 1995(非特許文献3))。EGFRの薬理学的な遮断は、下流経路、例えば、MAPK 経路、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt 経路、プロテインキナーゼ Cに関連するストレス活性化プロテインキナーゼ経路、及び、ヤーヌスキナーゼ (Jak)-STAT (シグナル伝達物質と転写活性化剤)経路の抑制により、EGFRに生存を依存している細胞の成長停止及びアポトーシスを招く。未分化で増殖中の表皮の基底層及び基底層直上層ならびに毛嚢外層のケラチノサイトにおいて主に現れるEGFRの抑制の結果、表皮層が薄くなり、バリア機能を損ない、更に、UV 光及び照射に対して上皮細胞を過敏にする。一方、EGFR活性の促進は、CXCL8発現の増加、ならびにCCL2、CCL5、及び、CXCL10の発現の減少に関連する (Mascia F, et al. Am J Pathol 163:303-312, 2003(非特許文献4))。反対に、EGFR 活性が抑制を受ける時、反対のパターンが発生する。マウスを用いた実験では、選択的なEGFR チロシンキナーゼ阻害剤の皮膚塗布により、接触性過敏性反応が更に重篤になり、CCL2、CCL5、及び、CXCL10の表皮レベルが上昇し、かつ、皮膚中の単球/マクロファージとT リンパ球の数が増加する。この結果から分かるのは、EGFRは、ケラチノサイトにおいて、ケモカイン発現に影響を及ぼすことにより、皮膚反応を変調することである。
【0003】
EGFRは、上皮組織の恒常的な維持、修復、及び、反応を支配する。よって、EGFR、又は、MAPK/ERK シグナル伝達経路、又は、チロシン活性に干渉する多くの標的治療薬は、発疹、皮膚乾燥、乾皮症、そう痒、角質の赤いただれ、爪周囲炎、手足反応、又は、手足症候群、毛細血管拡張症、及び、発毛又は皮膚色の変化を含む皮膚の変化を生じ得る(Galimont-Collen AFS, et al. Eur J Cancer 43:845-851, 2007(非特許文献5))。この変化は、標的治療薬に対する正常な体の副作用で、薬剤アレルギーの徴候ではない。副作用は、慢性湿疹を起こし、二次感染のリスクを増加させることがある。初期症状は、手と足の痛みを伴う過敏症である。その後、赤みと腫れが、手のひらと足の裏に発症する。最も一般的な皮膚の変化は、丘疹膿疱性の発疹である。これはしばしば座瘡に類似し、頭皮、顔、胸、上背に発症する。重症の場合、身体の他の部分を侵し、疱疹が開き、ひりひりした痛みが生じる。発疹は、通常、分子標的治療の最初の数週間が最もひどい。分子標的治療のおよそ四週目には、皮膚は、通常、かさぶたになり、非常に乾燥し、赤くなる。その数週間後、円形、平坦、又は、突起した赤色斑、及び、中心に膿を含んだ「稗粒腫」がしばしば現れる。発疹は痒みを伴うこともある。手足反応、又は、手足症候群の原因は確実には分かっていない。それは、手足の小血管の損傷、又は、血管から漏れ出して組織の損傷を起こす薬物自体と関連があるかもしれない。標的治療により生じるその他のより一般的でない副作用も、粘膜及び目に発生することがある。副作用は、下痢、吐き気、嘔吐、口のびらん、せき、及び、特に、まつげの長睫毛症を含み、長睫毛症は、目を傷つけ、著しい不快感と視力障害を引き起こし得る結膜炎及び他の眼疾患を伴うことがある。
【0004】
分子標的治療により生じる皮膚粘膜毒性もしくは眼毒性及び副作用は、痛みを伴い、身体及び精神的−社会的な不便を生じ、患者の歩行、飲食、正常な活動にも影響することがある。多くの患者は、皮膚粘膜毒性及び眼毒性の発生により、標的治療を中止したり、遅らせたりする。よって、当該毒性を治療又は改善する新規の方法が必要である。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Lacouture ME. Nat Rev Cancer 6:803-812, 2006
【非特許文献2】Perez-Soler R, et al. J Clin Oncol 23:5235-5246, 2005
【非特許文献3】Miettinen PJ, et al. Nature 376:337-341, 1995
【非特許文献4】Mascia F, et al. Am J Pathol 163:303-312, 2003
【非特許文献5】Galimont-Collen AFS, et al. Eur J Cancer 43:845-851, 2007
【発明の概要】
【0006】
一局面において、本発明は、細胞増殖及び生存シグナル変換経路の遮断から生じる皮膚粘膜毒性、眼毒性、又は、副作用を治療又は改善する方法を提供し、前記方法は、治療的有効量のヒストン脱アセチル化酵素 (HDAC) 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及び/又は、薬学的に許容可能な担体を含む薬学的組成物を、それを必要とする対象に投与するステップを含む。
【0007】
別の局面において、本発明は、それを必要とする対象において、化学療法及び/又は放射線治療を伴う又は伴わない上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF) 受容体、又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を標的とする分子治療により生じる、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を軽減する方法も提供し、前記方法は、対象の罹患皮膚又は粘膜領域に、有効量のHDAC 阻害剤、又は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸(SAHA)、トリコスタチン A、スクリプタイド、MS-27-275、及びそれらの混合物からなる群より選択される高アセチル化剤、及び、適切な塩、及び/又は、適切な担体を含む組成物を、局所的に適用するステップを含み、HDAC 阻害剤、又は、高アセチル化剤は、分子標的治療の前、分子標的治療中、及び/又は、分子標的治療の後に、患者に投与される。
【0008】
別の局面において、本発明はまた、細胞増殖及び生存シグナル変換経路の遮断により生じる皮膚粘膜毒性、眼毒性、又は、副作用を治療又は改善するための医薬の製造における、治療的有効量のヒストン脱アセチル化酵素 (HDAC) 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及び/又は、薬学的に許容可能な担体を含む薬学的組成物の使用を提供する。
【0009】
上記使用の一態様において、HDAC 阻害剤は、ヒドロキサム酸誘導体、脂肪酸誘導体、環状テトラペプチド、ベンズアミド誘導体、又は、求電子ケトン誘導体である。
【0010】
上記使用の一態様において、HDAC 阻害剤は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸(SAHA)、トリコスタチン A、スクリプタイド、又は、MS-27-275である。
【0011】
上記使用の一態様において、薬学的組成物は、非経口投与される。
【0012】
上記使用の一態様において、HDAC 阻害剤は、薬学的組成物の重量の0.00001%〜100%を占める。
【0013】
上記使用の一態様において、薬学的組成物は、クリーム剤、軟膏、ゲル剤、ペースト、粉末、水溶液、スプレー剤、懸濁液、分散剤、スラリー、ローション、パッチ、坐薬、リポソーム形態、口内洗浄剤、浣腸剤、注射液、点眼薬、点耳薬、点滴、マイクロカプセル、ナノカプセル、密封式皮膚改良剤(occlusive skin conditioning agent)、生体適合性ポリマー、又は、組織中で前記HDAC 阻害剤の保持を延長し作用を持続する物質である。
【0014】
上記使用の一態様において、薬学的組成物は、浸透促進剤、又は、pHを約3.0〜13.0の範囲で提供するためのpH 調整剤を更に含む。
【0015】
上記使用の一態様において、使用は、サイトカイン、サイトカイン阻害剤、インターロイキン、インターロイキン阻害剤、成長因子、血管形成剤、血管形成阻害剤、抗悪性腫瘍薬、抗炎症薬、ステロイド、免疫抑制剤、非ステロイド抗炎症薬、鎮痛薬、抗ヒスタミン薬、抗コリン作用薬、かゆみ止め薬、抗菌剤、抗ウイルス剤、抗真菌剤、駆虫剤、抗酸化剤、レチノイン酸、抗線維形成剤、血管作用薬、アデノシン受容体作動薬、ビタミン、ロイコトリエンモディファイヤー、インターロイキン拮抗薬、ケモカイン拮抗薬、マスト細胞阻害剤、抗IgE 抗体、選択的セロトニン再取り込み阻害剤 (SSRI)、5-ヒドロキシトリプタミン (5-HT) 受容体拮抗薬、ペルオキシソーム増殖剤活性化受容体 (PPAR) 作動薬、カルシニューリン阻害剤、ガストリン放出ペプチド受容体拮抗薬、p38 MAP キナーゼ阻害剤、ケラチノサイト成長因子 (KGF)、選択的エストロゲン受容体モジュレータ、タモキシフェン、HDAC 阻害剤、レチノイド、NFκB モジュレータ、レスベラトロール、ガバペンチン、スクラルファート、及びナロキソンからなる群より選択される少なくとも一つの第二の物質を更に投与する工程を含む。
【0016】
上記使用の一態様において、薬学的組成物及び第二の物質は、同じ処方及び同じ経路で、対象に同時に投与されるか、又は、異なる処方及び/もしくは異なる経路で連続して投与される。
【0017】
上記使用の一態様において、細胞増殖及び生存シグナル変換経路は、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムにより遮断される。
【0018】
上記使用の一態様において、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムは、上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF)、及び/又は、PI3K (ホスファチジルイノシトール3-キナーゼ)-Akt-mTOR 経路を抑制する。
【0019】
上記使用の一態様において、皮膚粘膜毒性もしくは眼毒性又は副作用は、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、ならびに/又は、爪の変形及び爪周囲の膨張を含む、皮膚又は上皮の副作用を含む。
【0020】
別の局面において、本発明はまた、化学療法及び/又は放射線治療を伴う又は伴わない上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF) 受容体、又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を標的とする分子治療により生じる、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を軽減するための医薬の製造における、有効量のHDAC 阻害剤、又は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸 (SAHA)、トリコスタチンA、スクリプタイド、MS-27-275、及び、それらの混合物からなる群より選択される高アセチル化剤、及び、適切なその塩、及び/又は、適切な担体を含む組成物の使用を提供する。
【0021】
別の局面において、本発明は、HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及びEGFR 阻害剤を含む組み合わせを提供する。好ましくは、組み合わせは、HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及びEGFR 阻害剤を含む薬学的組成物の形態である。好ましくは、HDAC 阻害剤及びEGFR 阻害剤は、同じ処方及び同じ経路で対象に同時に投与されるか、又は、異なる処方及び/もしくは異なる経路で連続して投与される。
【0022】
別の局面において、本発明は、EGFR、チロシンキナーゼ活性、もしくは、MAPK/ERK 経路、又は、EGFRもしくはチロシンキナーゼ活性の任意の下流の生物学的効果を遮断することにより、皮膚粘膜毒性、眼毒性、又は、副作用を生じることなく、疾病又は障害を治療する方法であって、HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及びEGFR 阻害剤を含む組み合わせを投与する工程を含む、方法を提供する。関連する疾病又は障害は、癌、炎症性疾患、免疫学的疾患及び変性疾患から選択される。皮膚粘膜毒性、眼毒性、又は、副作用は、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を含む。
【0023】
別の局面において、本発明は、EGFR、チロシンキナーゼ活性、もしくは、MAPK/ERK 経路、又は、EGFRもしくはチロシンキナーゼ活性の任意の下流の生物学的効果を遮断することにより、皮膚粘膜毒性、眼毒性、又は、副作用を生じることなく、疾病または障害を治療するための医薬の製造における、HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及びEGFR 阻害剤を含む薬学的組成物の使用を提供する。関連する疾病又は障害は、癌、炎症性疾患、免疫学的疾患及び変性疾患から選択される。皮膚粘膜毒性、眼毒性、又は、副作用は、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を含む。
【0024】
詳細な説明は、添付の図面を参照して、以下の態様において提供される。
【図面の簡単な説明】
【0025】
本発明は、添付の図面を参照して、以下の詳細な説明及び実施例を読むことでより完全に理解することができる。
【0026】
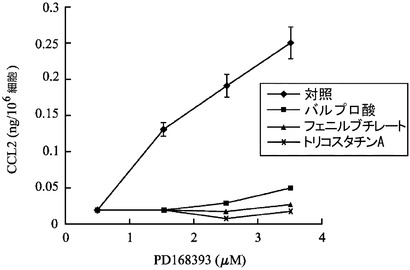
【図1】様々な HDAC 阻害剤が、皮膚の副作用に関連するケモカインの発現におけるEGFR 阻害剤の作用に影響を及ぼすことを示す。
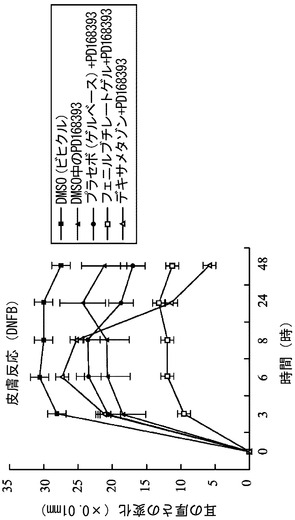
【図2】HDAC 阻害剤による局所処理により、EGFRの抑制により増加するマウス皮膚腫脹が改善されることを示す。スチューデントt検定を利用して、プラセボゲルと2.5% のフェニルブチレートゲル間の統計学的差異 (*P<0.05)を分析した。一方向 ANOVA 後、Dunnett検定を利用して、値をEGFR 抑制と比較した。†P< 0.05 を、PD168393 (すなわち、4-[(3-ブロモフェニル)アミノ]-6-アクリルアミドキナゾリン;EGFR 阻害剤)に対し顕著な差異があると見なした。
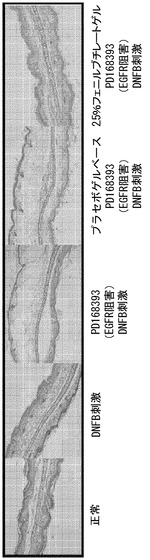
【図3】プラセボゲル、又は、2.5% フェニルブチレートゲルで、又は、それらなしで処理されたマウスの耳における、48 時間後の、EGFR 阻害剤 (PD168393)により増加したDNFB刺激に対する皮膚反応の代表例の組織学(H&E)の比較を示す。
【発明を実施するための形態】
【0027】
発明の詳細な説明
以下の説明は、本発明を実施するための最良の形態である。この説明は、本発明の一般的原理を例示するためのものであり、限定の意味で捉えるべきではない。本発明の範囲は、添付の特許請求の範囲によって最も良く定義される。
【0028】
皮膚粘膜や眼組織におけるEGFR、チロシンキナーゼ、又は、MAPK/ERK 経路の抑制は、標的細胞及び上皮細胞の異常増殖、転移、分化、及びアポトーシスを生じ、その結果、皮膚、粘膜、及び、角膜の完全性を破壊し、続いて、炎症細胞を動員する。よって、皮膚粘膜又は目の部位での副作用の局在化及び高頻度の反応は、表皮、毛嚢、爪周囲、粘膜、及び眼組織におけるEGFR、チロシンキナーゼ、及びMAPK/ERK 経路の機能を反映すると考えられる。
【0029】
本発明は、適切な担体中に調製されたHDAC 阻害剤の投与が、EGFR 阻害剤により異常に増加した皮膚反応の改善に効果的であることを証明する。よって、 HDAC 阻害剤が有利に用いられて、EGFR、チロシンキナーゼ、又は、MAPK/ERK シグナル伝達阻害剤の使用により生じる皮膚毒性を治療することができる。従って、本発明は、細胞増殖及び生存シグナル変換経路の遮断により生じる皮膚粘膜毒性、眼毒性、又は、副作用を治療、又は、改善するための、HDAC 阻害剤を薬学的に許容可能な担体中に含む薬学的組成物を提供する。
【0030】
本明細書中で用いられる「EGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤」とは、当技術分野で現在周知の、又は、将来同定される任意のEGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤のことを指し、且つ、対象へ投与されると、対象のEGFR、チロシンキナーゼ、又は、MAPK/ERK 経路に関連する生理活性(天然リガンドへのEGFRの結合により生じる任意の下流の生物学的効果を含む)を抑制する任意の化学成分を含む。このような EGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤は、EGFR、チロシンキナーゼ活性、又は、MAPK/ERK 経路、又は、EGFRの任意の下流の生物学的効果、又は、対象における癌、炎症性疾患、免疫疾患、又は、変性疾患の治療に関するチロシンキナーゼ活性を遮断するあらゆる物質を含む。このような阻害剤は、成長因子受容体の細胞外ドメイン又は細胞内ドメインと直接結合し、その関連するチロシンキナーゼ活性を抑制するか、又は、セリン/トレオニン活性により、関連する下流シグナル伝達変換カスケードに干渉することにより、作用することができる。あるいは、このようなチロシンキナーゼ阻害剤は、チロシンキナーゼ活性を有する下流のシグナルトランスデューサを抑制することにより、作用することができる。
【0031】
「チロシンキナーゼ 阻害剤」とは、チロシンキナーゼ、受容体チロシンキナーゼ、又は、当該キナーゼのATP 結合部位を抑制、又は、遮断することができる、天然、又は、合成の物質を指す。いわゆるEGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤は、これらに限定されないが、低分子量阻害剤、抗体又は抗体フラグメント、アンチセンス (DNA又はRNA)コンストラクト、ペプチド模倣薬、及びリボザイムを含む。
【0032】
HDAC 阻害剤は、ヒストンのアセチル化の変化状態により、染色質構造をリモデリングすることにより、遺伝子のサブセットを活性化、及び、抑制する(Marks et al, J. Natl. Cancer Inst., 92: 1210-1216, 2000; Kramer et al, Trends Endocrinol. Metab., 12: 294-300, 2001)。ヒストンの高アセチル化は、細胞周期阻害剤 (p21Cip1、p27Kip1、及び、p16INK4)のアップレギュレーション、癌遺伝子(Myc、及びBcl-2)のダウンレギュレーション、炎症性サイトカイン(インターロイキン (IL)-1、IL-8、TNF-α、及び、TGF-β)の抑制を生じるか、又は、変化を生じない(GAPDH、及びγ-アクチン)(Lagger et al, EMBO J., 21: 2672-2681, 2002; Richon et al, Clin. Cancer Res., 8: 662-667, 2002; Richon et al, Proc. Natl. Acad. Sci. USA., 97: 10014-10019, 2000; Van Lint et al, Gene Expr., 5: 245-243, 1996; Huang et al, Cytokine, 9: 27-36, 1997; Mishra et al, Proc. Natl. Acad. Sci. USA., 98: 2628-2633, 2001; Stockhammer et al, J. Neurosurg., 83: 672-681, 1995; Segain et al, Gut, 47: 397-403, 2000; Leoni et al, Proc. Natl. Acad. Sci. USA, 99: 2995-3000, 2002)。ヒストンの高アセチル化の誘発に加え、HDAC 阻害剤は、非ヒストンタンパク質、例えば、リボソームのS3、p53、又は、NF-κBのRel-A サブユニット、調節プロテインキナーゼ C (PKC)活性の高アセチル化を誘発し、プロテインイソプレニル化を抑制し、DNAメチル化を減少させ、核内受容体に結合される(Webb et al, J. Biol. Chem., 274: 14280-14287, 1999; Chen et al, Science, 293: 1653-1657, 2001)。HDAC 阻害剤は、細胞周期休止、細胞分化、及び、腫瘍細胞のアポトーシスを起こした細胞死を誘発し、炎症性疾患の炎症及び線維症を減少させる特性を示す(Warrell et al, J. Natl. Cancer Inst., 90: 1621-1625, 1998; Vigushin et al, Clin. Cancer Res., 7: 971-976, 2001; Saunders et al, Cancer Res., 59: 399-404, 1999; Gottlicher et al, EMBO J., 20: 6969-6978, 2001; Rombouts et al, Acta Gastroenterol. Belg., 64: 239-246, 2001)。HDAC 阻害剤の効果は、大量のヒストンのアセチル化を誘発し、アポトーシスによる細胞死、最終分化、及び腫瘍細胞の成長停止を生じるが、正常細胞には毒性がない (Garber et al, J. Natl. Cancer Inst., 94: 793-795, 2002)。加えて、HDAC阻害剤による染色質構造の変調は、更に、元々、放射線抵抗性である腫瘍細胞を放射線に増感するようにし、又、腫瘍細胞の化学療法に対する感受性を高める (Ferrandina et al, Oncol. Res., 12: 429-440, 2001; Miller et al, Int. J. Radiat. Biol., 72: 211-218, 1997; Biade et al, Int. J. Radiat. Biol., 77: 1033-1042, 2001)。
【0033】
本発明の実施に用いられる活性化合物は、通常、ヒストン高アセチル化剤、例えば、HDAC 阻害剤である。多くのこのような化合物は周知である。例えば、P. Dulski, Histone Deacetylase as Target for Antiprotozoal Agents, PCT出願 WO 97/11366 (Mar. 27, 1997)を参照のこと。このような化合物の例は、これに限定されないが、以下を含む。
【0034】
A. トリコスタチンAとその類似体、例えば: トリコスタチン A (TSA);トリコスタチン C (Koghe et al. 1998. Biochem. Pharmacol. 56:1359-1364) (トリコスタチン B は単離されているが、HDAC 阻害剤であるとは示されていない)。
【0035】
B. ペプチド、例えば: オキサムフラチン (Kim et al., Oncogene, 18:2461-2470 (1999)); トラポキシン A (TPX) (Kijima et al., J. Biol. Chem. 268, 22429-22435 (1993)); FR901228, デプシペプチド (Nakajima et al., Ex. Cell Res. 241, 126-133 (1998)); FR225497, 環状テトラペプチド (H. Mori et al., PCT出願 WO 00/08048 (Feb. 17, 2000)); アピシジン, (Darkin-Rattray et al., Proc. Natl. Acad. Sci. USA 93, 13143-13147 (1996)); アピシジン1a、アピシジンIb、アピシジンIc、アピシジンIIa、及び、アピシジンIIb (P. Dulski et al., PCT出願 WO 97/11366); HC-Toxin (Bosch et al., Plant Cell 7, 1941-1950 (1995)); WF27082 (PCT出願 WO 98/48825); 及び、クラミドシン (Bosch et al., 上記)。
【0036】
C. ヒドロキサム酸ベースのハイブリッド極性化合物 (HPCs)、例えば: サリチルヒドロキサム酸(SBHA) (Andrews et al., International J. Parasitology 30, 761-768 (2000)); スベロイルアニリドヒドロキサム酸 (SAHA) (Richon et al., Proc. Natl. Acad. Sci. USA 95, 3003-3007 (1998)); アゼライン酸ビスヒドロキサム酸(ABHA) (Andrews et al., 上記); アゼライン-1-ヒドロオキサメート-9-アニリド(AAHA) (Qiu et al., Mol. Biol. Cell 11, 2069-2083 (2000)); M-カルボキシ桂皮酸ビスヒドロキサミド (CBHA) (Ricon et al., 上記); 6-(3-クロロフェニルウレイド)カーポイックヒドロキサム酸 (3-Cl-UCHA) (Richon et al., 上記); MW2796 (Andrews et al., 上記); 及び、MW2996 (Andrews et al., 上記)である。HDAC 阻害剤としての効果がない類似体は: ヘキサメチレンビスアセタミド (HBMA) (Richon et al. 1998, PNAS, 95:3003-3007); 及び、ジエチルビス(ペンタメチレン-N,N-ジメチルカルボキサミド) マロネート (EMBA) (Richon et al. 1998, PNAS, 95:3003-3007)である。
【0037】
D. 短鎖脂肪酸 (SCFA) 化合物、例えば: 酪酸ナトリウム (Cousens et al., J. Biol. Chem. 254, 1716-1723 (1979)); イソ吉草酸 (McBain et al., Biochem. Pharm. 53:1357-1368 (1997)); バルプロ酸; 吉草酸 (McBain et al., 上記); 4-フェニルブチレート (4-PBA) (Lea and Tulsyan, Anticancer Research, 15, 879-873 (1995)); フェニルブチレート (PB) (Wang et al., Cancer Research, 59, 2766-2799 (1999)); プロピオネート (McBain et al., 上記); ブチルアミド (Lea and Tulsyan, 上記); イソブチルアミド (Lea and Tulsyan, 上記);酢酸フェニル (Lea and Tulsyan, 上記); 3-ブロモプロピオネート (Lea and Tulsyan, 上記); 及び、トリブチリン (Guan et al., Cancer Research, 60, 749-755 (2000))。
【0038】
E. ベンズアミド誘導体、例えば: MS-27-275 [N-(2-アミノフェニル)-4-[N-(ピリジン-3-イル-メトキシカルボニル) アミノメチル]ベンズアミド] (Saito et al., Proc. Natl. Acad. Sci. USA 96, 4592-4597 (1999));及び、MS-27-275の3'-アミノ誘導体 (Saito et al., 上記)。
【0039】
F. その他の阻害剤、例えば:デプデシン [その類似体(モノ-MTM-デプデシン及びデプデシン-ビスエーテル)はHDACを抑制しない] (Kwon et al. 1998. PNAS 95:3356-3361);及び、スクリプタイド (Su et al. 2000 Cancer Research, 60:3137-3142)。
【0040】
HDAC阻害剤と組み合わせる第二の化合物は、これに限定されないが、サイトカイン、サイトカイン阻害剤、インターロイキン、インターロイキン阻害剤、成長因子、血管形成剤、血管形成阻害剤、抗悪性腫瘍薬、抗炎症薬、ステロイド、免疫抑制剤、非ステロイド抗炎症薬、鎮痛剤、抗ヒスタミン薬、抗コリン作用薬、かゆみ止め薬、抗菌剤、抗ウイルス剤、抗真菌剤、駆虫剤、抗酸化剤、レチノイン酸、抗線維形成剤、血管作用薬、アデノシン受容体作動薬、ビタミン、ロイコトリエンモディファイヤー、インターロイキン拮抗薬、ケモカイン拮抗薬、マスト細胞阻害剤、抗IgE 抗体、選択的セロトニン再取り込み阻害剤 (SSRI)、5-ヒドロキシトリプタミン (5-HT) 受容体拮抗薬、ペルオキシソーム増殖剤活性化受容体(PPAR) 作動薬、カルシニューリン阻害剤、ガストリン放出ペプチド 受容体拮抗薬、p38 MAP キナーゼ阻害剤、ケラチノサイト成長因子 (KGF)、HDAC 阻害剤、レチノイド、NFκBモジュレータ、ガバペンチン、スクラルファート、及び、ナロキソンを含む。
【0041】
本発明の化合物は、薬学的組成物として調製することができる。このような組成物は、必要に応じて、従来の毒性のない薬学的に許容可能な担体、アジュバント、及び、賦形剤を含む投与単位製剤で、経口、非経口、吸入スプレー、直腸、経膣、皮内、又は、局所で投与することができる。局所投与は、経皮投与、例えば、経皮パッチ又はイオントフォレーゼ装置の使用も含む。本明細書中で用いられる「非経口」という用語は、皮下、静脈、筋肉内、又は、胸骨内への注射又は注入の技術を含む。薬剤の調製は、例えば、Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa. (1975)、及び、Liberman, H. A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y. (1980)に記述されている。
【0042】
一態様において、EGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤の使用により生じる皮膚の副作用を治療する薬剤は、一般に、皮膚の管理容易性が増加した状態を提供することを目的とする。スキンケア組成物の製剤には、クリーム、軟膏、ゲル、スプレー、ローション、スキントニック、シャンプー、又は、ムースを含むカテゴリーがある。一般に、皮膚用スプレーは、エアロゾル化したコポリマー、例えば、ポリビニルピロリドン、酢酸ビニル等から構成され、セットローションの機能も有する。スキンジェル製剤は、組成は、スプレーと類似するが、ゲル状で、且つ、ノンアルコール形式で、皮膚表面を覆うことができる。スキンムースは、加圧するとエアロゾルの缶から放出される泡である。本発明によるHDAC 阻害剤の、局所用スキンケア組成物中の含量は、好ましくは、濃度が組成物の総重量の0.00001〜100.00%、又は、容量が1 〜1000 mgである。本発明による、皮膚病変を治療するためのスキンケア組成物は、疎水性又は親水性クリーム、軟膏、ゲル、皮膚軟化剤、スプレー、ローション、スキントニック、シャンプー、又は、ムースとして調製され、スキンケア組成物での使用に適した当技術分野で周知のタイプの追加成分を有し、このような更なる成分は、ワセリン、ワックス、ラノリン、シリコーン、リポソーム、植物、鉱物油、可塑剤、香料、保存剤、浸透促進剤、pH調整剤、又は、局所用皮膚組成物のためのその他の適切な成分を含む。このような成分は、肌に潤いを与え、活性化合物を安定化させ、薬剤と皮膚との接触及び局所濃度を増加させ、薬剤の持続放出を制御し、及び/又は、皮膚の破損の減少を助け、皮膚萎縮、線維症、及び、感染症を防止し、皮膚の創傷治癒を促進する。pH 調整剤は、製剤の pHを約 3.0〜13.0の幅に調整するよう提供される。
【0043】
本発明は、更に、本明細書に記載されているようなEGFR、チロシンキナーゼ、VEGF、セリン/トレオニンキナーゼ、又は、MAPK/ERK 阻害剤の使用により生じる皮膚の副作用を治療する方法を提供し、少なくともHDAC 阻害剤を、サイトカイン、サイトカイン阻害剤、インターロイキン、インターロイキン阻害剤、成長因子、血管形成剤、血管形成阻害剤、抗悪性腫瘍薬、抗炎症薬、ステロイド、免疫抑制剤、非ステロイド抗炎症薬、鎮痛剤、抗ヒスタミン薬、抗コリン作用薬、かゆみ止め薬、抗菌剤、抗ウイルス剤、抗真菌剤、駆虫剤、抗酸化剤、レチノイン酸、抗線維形成剤、血管作用薬、アデノシン受容体作動薬、ビタミン、ロイコトリエンモディファイヤー、インターロイキン拮抗薬、ケモカイン拮抗薬、マスト細胞阻害剤、抗IgE 抗体、選択的セロトニン再取り込み阻害剤 (SSRI)、5-ヒドロキシトリプタミン (5-HT) 受容体拮抗薬、ペルオキシソーム増殖剤活性化受容体 (PPAR) 作動薬、カルシニューリン阻害剤、ガストリン放出ペプチド受容体拮抗薬、p38 MAP キナーゼ阻害剤、ケラチノサイト成長因子 (KGF)、HDAC 阻害剤、レチノイド、NFκB モジュレータ、ガバペンチン、スクラルファート、及び、ナロキソンを含む少なくとも一つ又はそれ以上の他の薬剤と一緒に提供する組成物を含み、この活性成分は、遊離型、又は、薬学的に許容可能な塩もしくは溶媒和物の形態で存在し、任意で、少なくとも一つの薬学的に許容可能な担体を有し、この組成物とその他の薬剤は、全身又は局所に、同時又は個別もしくは連続的に使用される。
【0044】
皮膚粘膜毒性、眼毒性、又は、副作用は、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長増加、ならびに/又は、脆化した変形爪及び爪周囲の膨張等の皮膚又は上皮の副作用を含む。EGFR、チロシンキナーゼ、VEGF、セリン/トレオニンキナーゼ、又は、MAPK/ERK 阻害剤は、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムであってよく、上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF)、及び/又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を抑制する。
【0045】
本発明において用いられる成分の適切な塩はまた、無機陽イオンを含むもの、例えば、アルカリ金属塩、特に、ナトリウム塩、カリウム塩、又は、アンモニウム塩、アルカリ土類金属塩、例えば、マグネシウム塩又はカルシウム塩、二価、又は、四価陽イオンを含む塩、例えば、亜鉛塩、アルミニウム塩、又は、ジルコニウム塩である。また、有機塩基を含む塩、例えば、ジシクロヘキシルアミン塩;メチル-D-グルカミン; アルギニン、リジン、ヒスチジン、グルタミン等のアミノ酸を含む塩も企図される。また、塩基性の窒素含有基は、低級ハロゲン化アルキル、例えば、塩化、臭化、及びヨウ化メチル、エチル、プロピル、及び、ブチル; 硫酸ジアルキル、例えば、硫酸ジメチル、硫酸ジエチル、硫酸ジブチル、及び、硫酸ジアミル;長鎖ハロゲン化物、例えば、塩化、臭化、及びヨウ化デシル、ラウリル、ミリスチル、及び、ステアリル; 芳香族ハロゲン化物、例えば、臭化ベンジル、及び、臭化フェネチル; ならびに、その他の物質により四級化される。塩形成物質、例えば、低分子量アルキルアミン、例えば、メチルアミン、エチルアミン、又は、トリエチルアミンも用いることができる。これにより、水溶性もしくは油溶性、又は、分散性製品が得られる。
【0046】
担体材料と組み合わされて、単一投与量を形成する。活性成分の含量は、対象及び特定の投与様式によって異なる。必要な投与量は、当業者によく知られているいくつかの要因に従って変化し、当該要因は、これらに限定されないが、使用される化合物、対象の種、対象のサイズ、及び、関連する病状の重症度を含む。化合物は、単回投与されてもよく、24時間内に複数回投与されても、又は、持続注入されてもよい。持続注入により投与される場合、化合物は、当技術分野で周知の方法、例えば、これらに限定されないが、静脈重力点滴、静脈注入ポンプ、埋め込み型注入ポンプ、又は、任意の局所経路により供給されてよい。処置時間の長さは、多くの要因、例えば、皮膚又は粘膜の病変の期間及び重症度によって変化する。HDAC 阻害剤、又は、本発明のその他の薬剤と組み合わせた対象の治療が、皮膚粘膜又は眼の障害が消失するまで、又は、対象が生存する限り、継続される。
【0047】
粘膜病変を治療するための一態様において、生体適合性ポリマーが選択されて、それにより、生体適合性ポリマーが治療用組成物に取り入れられると、治療用組成物の粘性が、典型的には37℃である生理的温度の増加と共に増加する。このように、治療用組成物は、低温下で、低粘性の流動性流体媒質として投与され、治療用組成物の粘性は、治療用組成物が生理的温度へと温められるにつれて増加する。好ましい一態様において、1℃からホストの生理的温度(例えば、ヒトでは37℃)である少なくとも特定の範囲に対し、治療用組成物は逆熱(reverse-thermal)粘性特性を示す。本発明の治療用組成物を形成するのに用いられ得る生体適合性ポリマーは、一般に、生体接着性ポリマー、カチオン性ポリマー、粘性ポリマーゲル、ヒドロゲル、天然ポリマー、ポリオキシアルキレンブロック共重合体、及び、逆熱ゲル化ポリマーを含む。治療用組成物は、生体適合性ポリマーに加え、治療用組成物の生体接着性を増加する別個の生体接着剤を含んでもよい。生体接着剤は、しばしば、第二ポリマーとなり、高い生体接着性を有する。
【0048】
治療用組成物は、更に、スクラルファートと共に、増粘ゲル形態に調製することもできる。これは、スプーンで口に運んで嚥下して、消化管全体を覆うことができる。本発明の薬学的物質は、送達領域によって異なる製品形態に生成され得る。治療用組成物を投与するための可能な製品形態の例には、経口液剤、膀胱洗浄液、ゲル剤、スラリー、口内洗浄剤、トローチ、タブレット、フィルム、パッチ、棒つきキャンディ、スプレー、点滴、又は、坐薬が含まれる。例えば、座薬に調製されたゲルは、直腸又は膣の粘膜表面の治療用に投与するための好ましい製品の一つである。スラリー又は経口液剤は、口腔、食道、及び/又は、消化管の粘膜表面の治療に用いることができる。
【実施例】
【0049】
実施例 1: HADC 阻害剤によるCCL2の抑制
EGFR経路の抑制は、CCL2の表皮レベルの増加により、皮膚反応を生じ得るので、EGFR 阻害剤 PD168393、及び/又は、異なる HDAC 阻害剤を用いて、又は、用いないで処理されたケラチノサイト培養物の上清中のCCL2レベルを、ELISAにより検出した。EGFR 阻害剤 (PD168393)による24時間の処理によるケモカイン CCL2 レベルのアップレギュレーションは、それぞれ、異なる HDAC 阻害剤 (バルプロ酸 (5 mM)、フェニルブチレート (5 mM)、及び、トリコスタチンA (5 nM))で、2 時間、前処理されたケラチノサイトからの上清において抑制された(図1)。その結果は、3回の独立した実験±SDの平均値として示される。
【0050】
実施例 2:皮膚治療用のHDAC 阻害剤の異なる製剤の調製
A:フェニルブチレートのゲルの調製
第一の部分: 10 g のStabileze QM.RTM.、380.561 g の脱イオン水をビーカー中で混合し、70℃で加熱した。
【0051】
第二の部分: 5.739 g のナトリウム4-フェニルブチレート (Merck)、0.125 g のメチルパラベン (Merck)、0.075 g のプロピルパラベン (Merck)、83.5 g の1,2-プロパンジオール、及び、20 g の10% NaOH をビーカー中で混合し、70℃で加熱した。
【0052】
第二の部分をゆっくりと第一の部分に加え、400 rpmで20 分間連続攪拌して、混合物を形成した。混合物を室温で冷却した。
【0053】
B:フェニルブチレートのリポソーム製剤の調製
このリポソーム製剤では、卵ホスファチジルコリン (EPC)及びコレステロールが、第一脂質成分として等モル濃度又は異なるモル濃度で用いられた。ナトリウム4-フェニルブチレートを有する様々なリポソームを、脂質とフェニルブチレートの割合を変化させることにより得た。リポソームは、薄膜水和法により調製され、膜押し出し法により大きさを調整され、且つ、物理的に評価された。
【0054】
C:トリコスタチンAの軟膏の調製
472.5 gの白色ワセリン (Riedel-de Haen)、27 gのパラフィンろう 50/52 (ローカルサプライヤー)、及び、0.5 gのトリコスタチン A (sigma)をビーカー中で混合し、70℃で加熱して、ペーストを形成した。このペーストを、400rpmで、1時間攪拌し、その後、室温で冷却した。
【0055】
D:トリコスタチン Aの油性軟膏の調製
67.5 gの白色ワセリン (Riedel-de Haen)、16 gのセチルアルコール (Riedel-de Haen)、260.5 gの軟パラフィン (Merck)、155.5 gの流動パラフィン (Merck)、及び、0.5 gのトリコスタチンA (sigma)をビーカー中で混合して、70℃で加熱し、ペーストを形成した。このペーストを、400rpmで、1時間攪拌し、その後、室温で冷却した。
【0056】
E:バルプロ酸のクリームの調製
第一の部分: 70 g のTefose 63.RTM.、20 gのSuperpolystate.RTM.、10 g の Coster 5000.RTM.、15 gのMyriyol 318.RTM.、15 gのCoster 5088.RTM.、及び15 gのGMS SE.RTM. (すべてローカルサプライヤーにより市販されている)をビーカー中で混合し、70℃で加熱した。
【0057】
第二の部分: 5.739 g のバルプロ酸 (sigma)、0.125 g のメチルパラベン (Merck)、0.075 g のプロピルパラベン (Merck)、及び、149.061 g の脱イオン水をビーカー中で混合し、70℃で加熱した。
【0058】
第二の部分をゆっくりと第一の部分に加え、400 rpm で、5 分間連続攪拌し、混合物を形成した。2% のStabileze QM.RTM.D (2 g のStabileze QM.RTM.を98 g の脱イオン水に溶解し、70℃で加熱及び攪拌して、ペーストを形成し、室温に冷却することにより調製した) を混合物に加えて、5 分間攪拌した。混合物のpHを、0.85% のリン酸 (Merck) により5.34 に調整し、 600 rpm で20 分間攪拌した。混合物を室温で冷却した。
【0059】
実施例 3: インビボでのHDAC 阻害剤によるEGFR阻害剤の抑制
EGFR 阻害剤により増大した皮膚反応の試験において、それぞれ、体重22 ± 2 g のBALB/c 雄性マウスの群(各n=5)に、実験動物の右耳上を、10μLの0.5% の2,4-ジニトロフルオロベンゼン (DNFB)により刺激する30分前に、それぞれ、各耳の各側に、10μLの賦形剤 (DMSO/無水エタノール 1/10)、又は、PD168393の溶液 (4 mmol/L) を局所適用した (Pastore S, et al. J Immunol 174:5047-5056, 2005)。
【0060】
EGFR 阻害剤誘発性の皮膚反応を処置するために、第0日と第1日の前、2.5% の4-フェニルブチレートゲル、又は、プラセボ (ゲルベース)を、3時間間隔で3回、右耳に局所適用した。第1日目に、フェニルブチレート、又は、プラセボの第一の処理の60分後、右耳を0.5% DNFBで刺激する30分前に、PD168393を局所適用した。第1日目の、フェニルブチレート及びプラセボによる第二回及び第三回の処理は、DNFB 刺激の1 時間後と3 時間後に適用された。DNFB刺激の0、3、6、8、24 、及び、48 時間後の耳の腫脹を、Dyerモデルマイクロメータ計測器で測定した。比較のための実験対照として、DNFB刺激の60分前と15分後に、強力なステロイドデキサメタゾン (0.3 mg) を局所投与した。各マウスの左右の耳の厚さを、Dyerモデルマイクロメータ計測器で測定した。耳の浮腫は、右耳 (処理された耳)から左耳(正常対照)の厚さを引くことにより計算された。
【0061】
図2及び3を参照すると、4 mMのPD168393のみ、DMSOのみ、プラセボゲルベースのみ、又は、2.5% のフェニルブチレートゲルの局所投与は、耳の厚さに影響せず、正常な皮膚組織像の変化を誘発することもなかった。一方、DNFB刺激の30分前に適用された4 mM のPD168393 により、DNFB誘発性の皮膚反応の悪化が起こった。しかし、プラセボゲルベースにより前処理された皮膚と比較した、2.5% のフェニルブチレートゲルにより前処理された皮膚は、DNFB刺激の3、6、8、24、及び、48 時間後にDNFB刺激により誘発された、EGFR 阻害剤により増大した耳の腫脹の有意な減少をもたらした(図 2と3)。デキサメタゾンは、DNFB刺激の3、6、及び、8 時間後のピークとなるEGFR 阻害剤により増大した皮膚反応を抑制しないが、DNFB刺激の24 時間後に、皮膚腫脹の有意な抑制効果を示す。DNFB刺激の24時間後の皮膚腫脹に対するデキサメタゾンによる抑制の程度は、フェニルブチレートによるものより大きい。よって、この結果は、デキサメタゾンが EGFR 阻害剤により増大する皮膚反応の抑制に対して効果がないこと、及び、2.5% のフェニルブチレートゲルが、EGFRの抑制により増大する皮膚副作用に対する治療的有用性を有することを示唆する。
【0062】
実施例及び好ましい態様により本発明を説明してきたが、本発明は、開示された態様により限定されるものではないことを理解されたい。むしろ、各種の改変及び同様のアレンジを包含することが意図されている。従って、添付の特許請求の範囲の保護範囲は、このような改変及び同様のアレンジをすべて包含するべく、最も広い解釈に基づく。
【技術分野】
【0001】
発明の分野
本発明は、皮膚疾患の治療に関するものであって、特に、HDAC 阻害剤を用いて、皮膚粘膜毒性又は眼毒性を治療することに関するものである。
【背景技術】
【0002】
関連技術の説明
多くの疾病や障害、例えば、癌、炎症性疾患、免疫学的疾患、及び、変性疾患等における、異常なシグナル伝達経路についての理解の進展と共に、細胞行動、応答、成長、生存及びアポトーシスの多くの中心的制御因子が、分子標的治療のための候補として、例えば、EGFR、VEGF、チロシンキナーゼ、セリン/トレオニンキナーゼ、及び、MAPK/ERKとして、明らかになっている(Lacouture ME. Nat Rev Cancer 6:803-812, 2006(非特許文献1))。上述による実験は、モノクローナル抗体、小化学分子、ペプチド模倣薬、及びアンチセンス・オリゴヌクレオチドを含む、革新的な分子標的技術の発展を導いている。しかし、正常な機能のためのシグナル伝達に依存している、組織中のEGFR 受容体、チロシンキナーゼ活性、又は、MAPK/ERK経路を同時に抑制することは、望まない結果をもたらす(Perez-Soler R, et al. J Clin Oncol 23:5235-5246, 2005(非特許文献2))。皮膚、粘膜、及び、角膜におけるEGFR、チロシンキナーゼ、及び、MAPK/ERK シグナル伝達は重要な機能を担うため、皮膚粘膜及び目の領域に、望まない副作用が一般に生じる。EGFR、チロシンキナーゼ活性、又は、MAPK/ERK 経路の抑制は、DNA 合成を抑制し、細胞増殖を阻み、ケラチノサイトの早発分化を促進する (Miettinen PJ, et al. Nature 376:337-341, 1995(非特許文献3))。EGFRの薬理学的な遮断は、下流経路、例えば、MAPK 経路、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt 経路、プロテインキナーゼ Cに関連するストレス活性化プロテインキナーゼ経路、及び、ヤーヌスキナーゼ (Jak)-STAT (シグナル伝達物質と転写活性化剤)経路の抑制により、EGFRに生存を依存している細胞の成長停止及びアポトーシスを招く。未分化で増殖中の表皮の基底層及び基底層直上層ならびに毛嚢外層のケラチノサイトにおいて主に現れるEGFRの抑制の結果、表皮層が薄くなり、バリア機能を損ない、更に、UV 光及び照射に対して上皮細胞を過敏にする。一方、EGFR活性の促進は、CXCL8発現の増加、ならびにCCL2、CCL5、及び、CXCL10の発現の減少に関連する (Mascia F, et al. Am J Pathol 163:303-312, 2003(非特許文献4))。反対に、EGFR 活性が抑制を受ける時、反対のパターンが発生する。マウスを用いた実験では、選択的なEGFR チロシンキナーゼ阻害剤の皮膚塗布により、接触性過敏性反応が更に重篤になり、CCL2、CCL5、及び、CXCL10の表皮レベルが上昇し、かつ、皮膚中の単球/マクロファージとT リンパ球の数が増加する。この結果から分かるのは、EGFRは、ケラチノサイトにおいて、ケモカイン発現に影響を及ぼすことにより、皮膚反応を変調することである。
【0003】
EGFRは、上皮組織の恒常的な維持、修復、及び、反応を支配する。よって、EGFR、又は、MAPK/ERK シグナル伝達経路、又は、チロシン活性に干渉する多くの標的治療薬は、発疹、皮膚乾燥、乾皮症、そう痒、角質の赤いただれ、爪周囲炎、手足反応、又は、手足症候群、毛細血管拡張症、及び、発毛又は皮膚色の変化を含む皮膚の変化を生じ得る(Galimont-Collen AFS, et al. Eur J Cancer 43:845-851, 2007(非特許文献5))。この変化は、標的治療薬に対する正常な体の副作用で、薬剤アレルギーの徴候ではない。副作用は、慢性湿疹を起こし、二次感染のリスクを増加させることがある。初期症状は、手と足の痛みを伴う過敏症である。その後、赤みと腫れが、手のひらと足の裏に発症する。最も一般的な皮膚の変化は、丘疹膿疱性の発疹である。これはしばしば座瘡に類似し、頭皮、顔、胸、上背に発症する。重症の場合、身体の他の部分を侵し、疱疹が開き、ひりひりした痛みが生じる。発疹は、通常、分子標的治療の最初の数週間が最もひどい。分子標的治療のおよそ四週目には、皮膚は、通常、かさぶたになり、非常に乾燥し、赤くなる。その数週間後、円形、平坦、又は、突起した赤色斑、及び、中心に膿を含んだ「稗粒腫」がしばしば現れる。発疹は痒みを伴うこともある。手足反応、又は、手足症候群の原因は確実には分かっていない。それは、手足の小血管の損傷、又は、血管から漏れ出して組織の損傷を起こす薬物自体と関連があるかもしれない。標的治療により生じるその他のより一般的でない副作用も、粘膜及び目に発生することがある。副作用は、下痢、吐き気、嘔吐、口のびらん、せき、及び、特に、まつげの長睫毛症を含み、長睫毛症は、目を傷つけ、著しい不快感と視力障害を引き起こし得る結膜炎及び他の眼疾患を伴うことがある。
【0004】
分子標的治療により生じる皮膚粘膜毒性もしくは眼毒性及び副作用は、痛みを伴い、身体及び精神的−社会的な不便を生じ、患者の歩行、飲食、正常な活動にも影響することがある。多くの患者は、皮膚粘膜毒性及び眼毒性の発生により、標的治療を中止したり、遅らせたりする。よって、当該毒性を治療又は改善する新規の方法が必要である。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Lacouture ME. Nat Rev Cancer 6:803-812, 2006
【非特許文献2】Perez-Soler R, et al. J Clin Oncol 23:5235-5246, 2005
【非特許文献3】Miettinen PJ, et al. Nature 376:337-341, 1995
【非特許文献4】Mascia F, et al. Am J Pathol 163:303-312, 2003
【非特許文献5】Galimont-Collen AFS, et al. Eur J Cancer 43:845-851, 2007
【発明の概要】
【0006】
一局面において、本発明は、細胞増殖及び生存シグナル変換経路の遮断から生じる皮膚粘膜毒性、眼毒性、又は、副作用を治療又は改善する方法を提供し、前記方法は、治療的有効量のヒストン脱アセチル化酵素 (HDAC) 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及び/又は、薬学的に許容可能な担体を含む薬学的組成物を、それを必要とする対象に投与するステップを含む。
【0007】
別の局面において、本発明は、それを必要とする対象において、化学療法及び/又は放射線治療を伴う又は伴わない上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF) 受容体、又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を標的とする分子治療により生じる、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を軽減する方法も提供し、前記方法は、対象の罹患皮膚又は粘膜領域に、有効量のHDAC 阻害剤、又は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸(SAHA)、トリコスタチン A、スクリプタイド、MS-27-275、及びそれらの混合物からなる群より選択される高アセチル化剤、及び、適切な塩、及び/又は、適切な担体を含む組成物を、局所的に適用するステップを含み、HDAC 阻害剤、又は、高アセチル化剤は、分子標的治療の前、分子標的治療中、及び/又は、分子標的治療の後に、患者に投与される。
【0008】
別の局面において、本発明はまた、細胞増殖及び生存シグナル変換経路の遮断により生じる皮膚粘膜毒性、眼毒性、又は、副作用を治療又は改善するための医薬の製造における、治療的有効量のヒストン脱アセチル化酵素 (HDAC) 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及び/又は、薬学的に許容可能な担体を含む薬学的組成物の使用を提供する。
【0009】
上記使用の一態様において、HDAC 阻害剤は、ヒドロキサム酸誘導体、脂肪酸誘導体、環状テトラペプチド、ベンズアミド誘導体、又は、求電子ケトン誘導体である。
【0010】
上記使用の一態様において、HDAC 阻害剤は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸(SAHA)、トリコスタチン A、スクリプタイド、又は、MS-27-275である。
【0011】
上記使用の一態様において、薬学的組成物は、非経口投与される。
【0012】
上記使用の一態様において、HDAC 阻害剤は、薬学的組成物の重量の0.00001%〜100%を占める。
【0013】
上記使用の一態様において、薬学的組成物は、クリーム剤、軟膏、ゲル剤、ペースト、粉末、水溶液、スプレー剤、懸濁液、分散剤、スラリー、ローション、パッチ、坐薬、リポソーム形態、口内洗浄剤、浣腸剤、注射液、点眼薬、点耳薬、点滴、マイクロカプセル、ナノカプセル、密封式皮膚改良剤(occlusive skin conditioning agent)、生体適合性ポリマー、又は、組織中で前記HDAC 阻害剤の保持を延長し作用を持続する物質である。
【0014】
上記使用の一態様において、薬学的組成物は、浸透促進剤、又は、pHを約3.0〜13.0の範囲で提供するためのpH 調整剤を更に含む。
【0015】
上記使用の一態様において、使用は、サイトカイン、サイトカイン阻害剤、インターロイキン、インターロイキン阻害剤、成長因子、血管形成剤、血管形成阻害剤、抗悪性腫瘍薬、抗炎症薬、ステロイド、免疫抑制剤、非ステロイド抗炎症薬、鎮痛薬、抗ヒスタミン薬、抗コリン作用薬、かゆみ止め薬、抗菌剤、抗ウイルス剤、抗真菌剤、駆虫剤、抗酸化剤、レチノイン酸、抗線維形成剤、血管作用薬、アデノシン受容体作動薬、ビタミン、ロイコトリエンモディファイヤー、インターロイキン拮抗薬、ケモカイン拮抗薬、マスト細胞阻害剤、抗IgE 抗体、選択的セロトニン再取り込み阻害剤 (SSRI)、5-ヒドロキシトリプタミン (5-HT) 受容体拮抗薬、ペルオキシソーム増殖剤活性化受容体 (PPAR) 作動薬、カルシニューリン阻害剤、ガストリン放出ペプチド受容体拮抗薬、p38 MAP キナーゼ阻害剤、ケラチノサイト成長因子 (KGF)、選択的エストロゲン受容体モジュレータ、タモキシフェン、HDAC 阻害剤、レチノイド、NFκB モジュレータ、レスベラトロール、ガバペンチン、スクラルファート、及びナロキソンからなる群より選択される少なくとも一つの第二の物質を更に投与する工程を含む。
【0016】
上記使用の一態様において、薬学的組成物及び第二の物質は、同じ処方及び同じ経路で、対象に同時に投与されるか、又は、異なる処方及び/もしくは異なる経路で連続して投与される。
【0017】
上記使用の一態様において、細胞増殖及び生存シグナル変換経路は、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムにより遮断される。
【0018】
上記使用の一態様において、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムは、上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF)、及び/又は、PI3K (ホスファチジルイノシトール3-キナーゼ)-Akt-mTOR 経路を抑制する。
【0019】
上記使用の一態様において、皮膚粘膜毒性もしくは眼毒性又は副作用は、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、ならびに/又は、爪の変形及び爪周囲の膨張を含む、皮膚又は上皮の副作用を含む。
【0020】
別の局面において、本発明はまた、化学療法及び/又は放射線治療を伴う又は伴わない上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF) 受容体、又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を標的とする分子治療により生じる、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を軽減するための医薬の製造における、有効量のHDAC 阻害剤、又は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸 (SAHA)、トリコスタチンA、スクリプタイド、MS-27-275、及び、それらの混合物からなる群より選択される高アセチル化剤、及び、適切なその塩、及び/又は、適切な担体を含む組成物の使用を提供する。
【0021】
別の局面において、本発明は、HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及びEGFR 阻害剤を含む組み合わせを提供する。好ましくは、組み合わせは、HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及びEGFR 阻害剤を含む薬学的組成物の形態である。好ましくは、HDAC 阻害剤及びEGFR 阻害剤は、同じ処方及び同じ経路で対象に同時に投与されるか、又は、異なる処方及び/もしくは異なる経路で連続して投与される。
【0022】
別の局面において、本発明は、EGFR、チロシンキナーゼ活性、もしくは、MAPK/ERK 経路、又は、EGFRもしくはチロシンキナーゼ活性の任意の下流の生物学的効果を遮断することにより、皮膚粘膜毒性、眼毒性、又は、副作用を生じることなく、疾病又は障害を治療する方法であって、HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及びEGFR 阻害剤を含む組み合わせを投与する工程を含む、方法を提供する。関連する疾病又は障害は、癌、炎症性疾患、免疫学的疾患及び変性疾患から選択される。皮膚粘膜毒性、眼毒性、又は、副作用は、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を含む。
【0023】
別の局面において、本発明は、EGFR、チロシンキナーゼ活性、もしくは、MAPK/ERK 経路、又は、EGFRもしくはチロシンキナーゼ活性の任意の下流の生物学的効果を遮断することにより、皮膚粘膜毒性、眼毒性、又は、副作用を生じることなく、疾病または障害を治療するための医薬の製造における、HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及びEGFR 阻害剤を含む薬学的組成物の使用を提供する。関連する疾病又は障害は、癌、炎症性疾患、免疫学的疾患及び変性疾患から選択される。皮膚粘膜毒性、眼毒性、又は、副作用は、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を含む。
【0024】
詳細な説明は、添付の図面を参照して、以下の態様において提供される。
【図面の簡単な説明】
【0025】
本発明は、添付の図面を参照して、以下の詳細な説明及び実施例を読むことでより完全に理解することができる。
【0026】
【図1】様々な HDAC 阻害剤が、皮膚の副作用に関連するケモカインの発現におけるEGFR 阻害剤の作用に影響を及ぼすことを示す。
【図2】HDAC 阻害剤による局所処理により、EGFRの抑制により増加するマウス皮膚腫脹が改善されることを示す。スチューデントt検定を利用して、プラセボゲルと2.5% のフェニルブチレートゲル間の統計学的差異 (*P<0.05)を分析した。一方向 ANOVA 後、Dunnett検定を利用して、値をEGFR 抑制と比較した。†P< 0.05 を、PD168393 (すなわち、4-[(3-ブロモフェニル)アミノ]-6-アクリルアミドキナゾリン;EGFR 阻害剤)に対し顕著な差異があると見なした。
【図3】プラセボゲル、又は、2.5% フェニルブチレートゲルで、又は、それらなしで処理されたマウスの耳における、48 時間後の、EGFR 阻害剤 (PD168393)により増加したDNFB刺激に対する皮膚反応の代表例の組織学(H&E)の比較を示す。
【発明を実施するための形態】
【0027】
発明の詳細な説明
以下の説明は、本発明を実施するための最良の形態である。この説明は、本発明の一般的原理を例示するためのものであり、限定の意味で捉えるべきではない。本発明の範囲は、添付の特許請求の範囲によって最も良く定義される。
【0028】
皮膚粘膜や眼組織におけるEGFR、チロシンキナーゼ、又は、MAPK/ERK 経路の抑制は、標的細胞及び上皮細胞の異常増殖、転移、分化、及びアポトーシスを生じ、その結果、皮膚、粘膜、及び、角膜の完全性を破壊し、続いて、炎症細胞を動員する。よって、皮膚粘膜又は目の部位での副作用の局在化及び高頻度の反応は、表皮、毛嚢、爪周囲、粘膜、及び眼組織におけるEGFR、チロシンキナーゼ、及びMAPK/ERK 経路の機能を反映すると考えられる。
【0029】
本発明は、適切な担体中に調製されたHDAC 阻害剤の投与が、EGFR 阻害剤により異常に増加した皮膚反応の改善に効果的であることを証明する。よって、 HDAC 阻害剤が有利に用いられて、EGFR、チロシンキナーゼ、又は、MAPK/ERK シグナル伝達阻害剤の使用により生じる皮膚毒性を治療することができる。従って、本発明は、細胞増殖及び生存シグナル変換経路の遮断により生じる皮膚粘膜毒性、眼毒性、又は、副作用を治療、又は、改善するための、HDAC 阻害剤を薬学的に許容可能な担体中に含む薬学的組成物を提供する。
【0030】
本明細書中で用いられる「EGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤」とは、当技術分野で現在周知の、又は、将来同定される任意のEGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤のことを指し、且つ、対象へ投与されると、対象のEGFR、チロシンキナーゼ、又は、MAPK/ERK 経路に関連する生理活性(天然リガンドへのEGFRの結合により生じる任意の下流の生物学的効果を含む)を抑制する任意の化学成分を含む。このような EGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤は、EGFR、チロシンキナーゼ活性、又は、MAPK/ERK 経路、又は、EGFRの任意の下流の生物学的効果、又は、対象における癌、炎症性疾患、免疫疾患、又は、変性疾患の治療に関するチロシンキナーゼ活性を遮断するあらゆる物質を含む。このような阻害剤は、成長因子受容体の細胞外ドメイン又は細胞内ドメインと直接結合し、その関連するチロシンキナーゼ活性を抑制するか、又は、セリン/トレオニン活性により、関連する下流シグナル伝達変換カスケードに干渉することにより、作用することができる。あるいは、このようなチロシンキナーゼ阻害剤は、チロシンキナーゼ活性を有する下流のシグナルトランスデューサを抑制することにより、作用することができる。
【0031】
「チロシンキナーゼ 阻害剤」とは、チロシンキナーゼ、受容体チロシンキナーゼ、又は、当該キナーゼのATP 結合部位を抑制、又は、遮断することができる、天然、又は、合成の物質を指す。いわゆるEGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤は、これらに限定されないが、低分子量阻害剤、抗体又は抗体フラグメント、アンチセンス (DNA又はRNA)コンストラクト、ペプチド模倣薬、及びリボザイムを含む。
【0032】
HDAC 阻害剤は、ヒストンのアセチル化の変化状態により、染色質構造をリモデリングすることにより、遺伝子のサブセットを活性化、及び、抑制する(Marks et al, J. Natl. Cancer Inst., 92: 1210-1216, 2000; Kramer et al, Trends Endocrinol. Metab., 12: 294-300, 2001)。ヒストンの高アセチル化は、細胞周期阻害剤 (p21Cip1、p27Kip1、及び、p16INK4)のアップレギュレーション、癌遺伝子(Myc、及びBcl-2)のダウンレギュレーション、炎症性サイトカイン(インターロイキン (IL)-1、IL-8、TNF-α、及び、TGF-β)の抑制を生じるか、又は、変化を生じない(GAPDH、及びγ-アクチン)(Lagger et al, EMBO J., 21: 2672-2681, 2002; Richon et al, Clin. Cancer Res., 8: 662-667, 2002; Richon et al, Proc. Natl. Acad. Sci. USA., 97: 10014-10019, 2000; Van Lint et al, Gene Expr., 5: 245-243, 1996; Huang et al, Cytokine, 9: 27-36, 1997; Mishra et al, Proc. Natl. Acad. Sci. USA., 98: 2628-2633, 2001; Stockhammer et al, J. Neurosurg., 83: 672-681, 1995; Segain et al, Gut, 47: 397-403, 2000; Leoni et al, Proc. Natl. Acad. Sci. USA, 99: 2995-3000, 2002)。ヒストンの高アセチル化の誘発に加え、HDAC 阻害剤は、非ヒストンタンパク質、例えば、リボソームのS3、p53、又は、NF-κBのRel-A サブユニット、調節プロテインキナーゼ C (PKC)活性の高アセチル化を誘発し、プロテインイソプレニル化を抑制し、DNAメチル化を減少させ、核内受容体に結合される(Webb et al, J. Biol. Chem., 274: 14280-14287, 1999; Chen et al, Science, 293: 1653-1657, 2001)。HDAC 阻害剤は、細胞周期休止、細胞分化、及び、腫瘍細胞のアポトーシスを起こした細胞死を誘発し、炎症性疾患の炎症及び線維症を減少させる特性を示す(Warrell et al, J. Natl. Cancer Inst., 90: 1621-1625, 1998; Vigushin et al, Clin. Cancer Res., 7: 971-976, 2001; Saunders et al, Cancer Res., 59: 399-404, 1999; Gottlicher et al, EMBO J., 20: 6969-6978, 2001; Rombouts et al, Acta Gastroenterol. Belg., 64: 239-246, 2001)。HDAC 阻害剤の効果は、大量のヒストンのアセチル化を誘発し、アポトーシスによる細胞死、最終分化、及び腫瘍細胞の成長停止を生じるが、正常細胞には毒性がない (Garber et al, J. Natl. Cancer Inst., 94: 793-795, 2002)。加えて、HDAC阻害剤による染色質構造の変調は、更に、元々、放射線抵抗性である腫瘍細胞を放射線に増感するようにし、又、腫瘍細胞の化学療法に対する感受性を高める (Ferrandina et al, Oncol. Res., 12: 429-440, 2001; Miller et al, Int. J. Radiat. Biol., 72: 211-218, 1997; Biade et al, Int. J. Radiat. Biol., 77: 1033-1042, 2001)。
【0033】
本発明の実施に用いられる活性化合物は、通常、ヒストン高アセチル化剤、例えば、HDAC 阻害剤である。多くのこのような化合物は周知である。例えば、P. Dulski, Histone Deacetylase as Target for Antiprotozoal Agents, PCT出願 WO 97/11366 (Mar. 27, 1997)を参照のこと。このような化合物の例は、これに限定されないが、以下を含む。
【0034】
A. トリコスタチンAとその類似体、例えば: トリコスタチン A (TSA);トリコスタチン C (Koghe et al. 1998. Biochem. Pharmacol. 56:1359-1364) (トリコスタチン B は単離されているが、HDAC 阻害剤であるとは示されていない)。
【0035】
B. ペプチド、例えば: オキサムフラチン (Kim et al., Oncogene, 18:2461-2470 (1999)); トラポキシン A (TPX) (Kijima et al., J. Biol. Chem. 268, 22429-22435 (1993)); FR901228, デプシペプチド (Nakajima et al., Ex. Cell Res. 241, 126-133 (1998)); FR225497, 環状テトラペプチド (H. Mori et al., PCT出願 WO 00/08048 (Feb. 17, 2000)); アピシジン, (Darkin-Rattray et al., Proc. Natl. Acad. Sci. USA 93, 13143-13147 (1996)); アピシジン1a、アピシジンIb、アピシジンIc、アピシジンIIa、及び、アピシジンIIb (P. Dulski et al., PCT出願 WO 97/11366); HC-Toxin (Bosch et al., Plant Cell 7, 1941-1950 (1995)); WF27082 (PCT出願 WO 98/48825); 及び、クラミドシン (Bosch et al., 上記)。
【0036】
C. ヒドロキサム酸ベースのハイブリッド極性化合物 (HPCs)、例えば: サリチルヒドロキサム酸(SBHA) (Andrews et al., International J. Parasitology 30, 761-768 (2000)); スベロイルアニリドヒドロキサム酸 (SAHA) (Richon et al., Proc. Natl. Acad. Sci. USA 95, 3003-3007 (1998)); アゼライン酸ビスヒドロキサム酸(ABHA) (Andrews et al., 上記); アゼライン-1-ヒドロオキサメート-9-アニリド(AAHA) (Qiu et al., Mol. Biol. Cell 11, 2069-2083 (2000)); M-カルボキシ桂皮酸ビスヒドロキサミド (CBHA) (Ricon et al., 上記); 6-(3-クロロフェニルウレイド)カーポイックヒドロキサム酸 (3-Cl-UCHA) (Richon et al., 上記); MW2796 (Andrews et al., 上記); 及び、MW2996 (Andrews et al., 上記)である。HDAC 阻害剤としての効果がない類似体は: ヘキサメチレンビスアセタミド (HBMA) (Richon et al. 1998, PNAS, 95:3003-3007); 及び、ジエチルビス(ペンタメチレン-N,N-ジメチルカルボキサミド) マロネート (EMBA) (Richon et al. 1998, PNAS, 95:3003-3007)である。
【0037】
D. 短鎖脂肪酸 (SCFA) 化合物、例えば: 酪酸ナトリウム (Cousens et al., J. Biol. Chem. 254, 1716-1723 (1979)); イソ吉草酸 (McBain et al., Biochem. Pharm. 53:1357-1368 (1997)); バルプロ酸; 吉草酸 (McBain et al., 上記); 4-フェニルブチレート (4-PBA) (Lea and Tulsyan, Anticancer Research, 15, 879-873 (1995)); フェニルブチレート (PB) (Wang et al., Cancer Research, 59, 2766-2799 (1999)); プロピオネート (McBain et al., 上記); ブチルアミド (Lea and Tulsyan, 上記); イソブチルアミド (Lea and Tulsyan, 上記);酢酸フェニル (Lea and Tulsyan, 上記); 3-ブロモプロピオネート (Lea and Tulsyan, 上記); 及び、トリブチリン (Guan et al., Cancer Research, 60, 749-755 (2000))。
【0038】
E. ベンズアミド誘導体、例えば: MS-27-275 [N-(2-アミノフェニル)-4-[N-(ピリジン-3-イル-メトキシカルボニル) アミノメチル]ベンズアミド] (Saito et al., Proc. Natl. Acad. Sci. USA 96, 4592-4597 (1999));及び、MS-27-275の3'-アミノ誘導体 (Saito et al., 上記)。
【0039】
F. その他の阻害剤、例えば:デプデシン [その類似体(モノ-MTM-デプデシン及びデプデシン-ビスエーテル)はHDACを抑制しない] (Kwon et al. 1998. PNAS 95:3356-3361);及び、スクリプタイド (Su et al. 2000 Cancer Research, 60:3137-3142)。
【0040】
HDAC阻害剤と組み合わせる第二の化合物は、これに限定されないが、サイトカイン、サイトカイン阻害剤、インターロイキン、インターロイキン阻害剤、成長因子、血管形成剤、血管形成阻害剤、抗悪性腫瘍薬、抗炎症薬、ステロイド、免疫抑制剤、非ステロイド抗炎症薬、鎮痛剤、抗ヒスタミン薬、抗コリン作用薬、かゆみ止め薬、抗菌剤、抗ウイルス剤、抗真菌剤、駆虫剤、抗酸化剤、レチノイン酸、抗線維形成剤、血管作用薬、アデノシン受容体作動薬、ビタミン、ロイコトリエンモディファイヤー、インターロイキン拮抗薬、ケモカイン拮抗薬、マスト細胞阻害剤、抗IgE 抗体、選択的セロトニン再取り込み阻害剤 (SSRI)、5-ヒドロキシトリプタミン (5-HT) 受容体拮抗薬、ペルオキシソーム増殖剤活性化受容体(PPAR) 作動薬、カルシニューリン阻害剤、ガストリン放出ペプチド 受容体拮抗薬、p38 MAP キナーゼ阻害剤、ケラチノサイト成長因子 (KGF)、HDAC 阻害剤、レチノイド、NFκBモジュレータ、ガバペンチン、スクラルファート、及び、ナロキソンを含む。
【0041】
本発明の化合物は、薬学的組成物として調製することができる。このような組成物は、必要に応じて、従来の毒性のない薬学的に許容可能な担体、アジュバント、及び、賦形剤を含む投与単位製剤で、経口、非経口、吸入スプレー、直腸、経膣、皮内、又は、局所で投与することができる。局所投与は、経皮投与、例えば、経皮パッチ又はイオントフォレーゼ装置の使用も含む。本明細書中で用いられる「非経口」という用語は、皮下、静脈、筋肉内、又は、胸骨内への注射又は注入の技術を含む。薬剤の調製は、例えば、Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa. (1975)、及び、Liberman, H. A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y. (1980)に記述されている。
【0042】
一態様において、EGFR、チロシンキナーゼ、又は、MAPK/ERK 阻害剤の使用により生じる皮膚の副作用を治療する薬剤は、一般に、皮膚の管理容易性が増加した状態を提供することを目的とする。スキンケア組成物の製剤には、クリーム、軟膏、ゲル、スプレー、ローション、スキントニック、シャンプー、又は、ムースを含むカテゴリーがある。一般に、皮膚用スプレーは、エアロゾル化したコポリマー、例えば、ポリビニルピロリドン、酢酸ビニル等から構成され、セットローションの機能も有する。スキンジェル製剤は、組成は、スプレーと類似するが、ゲル状で、且つ、ノンアルコール形式で、皮膚表面を覆うことができる。スキンムースは、加圧するとエアロゾルの缶から放出される泡である。本発明によるHDAC 阻害剤の、局所用スキンケア組成物中の含量は、好ましくは、濃度が組成物の総重量の0.00001〜100.00%、又は、容量が1 〜1000 mgである。本発明による、皮膚病変を治療するためのスキンケア組成物は、疎水性又は親水性クリーム、軟膏、ゲル、皮膚軟化剤、スプレー、ローション、スキントニック、シャンプー、又は、ムースとして調製され、スキンケア組成物での使用に適した当技術分野で周知のタイプの追加成分を有し、このような更なる成分は、ワセリン、ワックス、ラノリン、シリコーン、リポソーム、植物、鉱物油、可塑剤、香料、保存剤、浸透促進剤、pH調整剤、又は、局所用皮膚組成物のためのその他の適切な成分を含む。このような成分は、肌に潤いを与え、活性化合物を安定化させ、薬剤と皮膚との接触及び局所濃度を増加させ、薬剤の持続放出を制御し、及び/又は、皮膚の破損の減少を助け、皮膚萎縮、線維症、及び、感染症を防止し、皮膚の創傷治癒を促進する。pH 調整剤は、製剤の pHを約 3.0〜13.0の幅に調整するよう提供される。
【0043】
本発明は、更に、本明細書に記載されているようなEGFR、チロシンキナーゼ、VEGF、セリン/トレオニンキナーゼ、又は、MAPK/ERK 阻害剤の使用により生じる皮膚の副作用を治療する方法を提供し、少なくともHDAC 阻害剤を、サイトカイン、サイトカイン阻害剤、インターロイキン、インターロイキン阻害剤、成長因子、血管形成剤、血管形成阻害剤、抗悪性腫瘍薬、抗炎症薬、ステロイド、免疫抑制剤、非ステロイド抗炎症薬、鎮痛剤、抗ヒスタミン薬、抗コリン作用薬、かゆみ止め薬、抗菌剤、抗ウイルス剤、抗真菌剤、駆虫剤、抗酸化剤、レチノイン酸、抗線維形成剤、血管作用薬、アデノシン受容体作動薬、ビタミン、ロイコトリエンモディファイヤー、インターロイキン拮抗薬、ケモカイン拮抗薬、マスト細胞阻害剤、抗IgE 抗体、選択的セロトニン再取り込み阻害剤 (SSRI)、5-ヒドロキシトリプタミン (5-HT) 受容体拮抗薬、ペルオキシソーム増殖剤活性化受容体 (PPAR) 作動薬、カルシニューリン阻害剤、ガストリン放出ペプチド受容体拮抗薬、p38 MAP キナーゼ阻害剤、ケラチノサイト成長因子 (KGF)、HDAC 阻害剤、レチノイド、NFκB モジュレータ、ガバペンチン、スクラルファート、及び、ナロキソンを含む少なくとも一つ又はそれ以上の他の薬剤と一緒に提供する組成物を含み、この活性成分は、遊離型、又は、薬学的に許容可能な塩もしくは溶媒和物の形態で存在し、任意で、少なくとも一つの薬学的に許容可能な担体を有し、この組成物とその他の薬剤は、全身又は局所に、同時又は個別もしくは連続的に使用される。
【0044】
皮膚粘膜毒性、眼毒性、又は、副作用は、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長増加、ならびに/又は、脆化した変形爪及び爪周囲の膨張等の皮膚又は上皮の副作用を含む。EGFR、チロシンキナーゼ、VEGF、セリン/トレオニンキナーゼ、又は、MAPK/ERK 阻害剤は、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムであってよく、上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF)、及び/又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を抑制する。
【0045】
本発明において用いられる成分の適切な塩はまた、無機陽イオンを含むもの、例えば、アルカリ金属塩、特に、ナトリウム塩、カリウム塩、又は、アンモニウム塩、アルカリ土類金属塩、例えば、マグネシウム塩又はカルシウム塩、二価、又は、四価陽イオンを含む塩、例えば、亜鉛塩、アルミニウム塩、又は、ジルコニウム塩である。また、有機塩基を含む塩、例えば、ジシクロヘキシルアミン塩;メチル-D-グルカミン; アルギニン、リジン、ヒスチジン、グルタミン等のアミノ酸を含む塩も企図される。また、塩基性の窒素含有基は、低級ハロゲン化アルキル、例えば、塩化、臭化、及びヨウ化メチル、エチル、プロピル、及び、ブチル; 硫酸ジアルキル、例えば、硫酸ジメチル、硫酸ジエチル、硫酸ジブチル、及び、硫酸ジアミル;長鎖ハロゲン化物、例えば、塩化、臭化、及びヨウ化デシル、ラウリル、ミリスチル、及び、ステアリル; 芳香族ハロゲン化物、例えば、臭化ベンジル、及び、臭化フェネチル; ならびに、その他の物質により四級化される。塩形成物質、例えば、低分子量アルキルアミン、例えば、メチルアミン、エチルアミン、又は、トリエチルアミンも用いることができる。これにより、水溶性もしくは油溶性、又は、分散性製品が得られる。
【0046】
担体材料と組み合わされて、単一投与量を形成する。活性成分の含量は、対象及び特定の投与様式によって異なる。必要な投与量は、当業者によく知られているいくつかの要因に従って変化し、当該要因は、これらに限定されないが、使用される化合物、対象の種、対象のサイズ、及び、関連する病状の重症度を含む。化合物は、単回投与されてもよく、24時間内に複数回投与されても、又は、持続注入されてもよい。持続注入により投与される場合、化合物は、当技術分野で周知の方法、例えば、これらに限定されないが、静脈重力点滴、静脈注入ポンプ、埋め込み型注入ポンプ、又は、任意の局所経路により供給されてよい。処置時間の長さは、多くの要因、例えば、皮膚又は粘膜の病変の期間及び重症度によって変化する。HDAC 阻害剤、又は、本発明のその他の薬剤と組み合わせた対象の治療が、皮膚粘膜又は眼の障害が消失するまで、又は、対象が生存する限り、継続される。
【0047】
粘膜病変を治療するための一態様において、生体適合性ポリマーが選択されて、それにより、生体適合性ポリマーが治療用組成物に取り入れられると、治療用組成物の粘性が、典型的には37℃である生理的温度の増加と共に増加する。このように、治療用組成物は、低温下で、低粘性の流動性流体媒質として投与され、治療用組成物の粘性は、治療用組成物が生理的温度へと温められるにつれて増加する。好ましい一態様において、1℃からホストの生理的温度(例えば、ヒトでは37℃)である少なくとも特定の範囲に対し、治療用組成物は逆熱(reverse-thermal)粘性特性を示す。本発明の治療用組成物を形成するのに用いられ得る生体適合性ポリマーは、一般に、生体接着性ポリマー、カチオン性ポリマー、粘性ポリマーゲル、ヒドロゲル、天然ポリマー、ポリオキシアルキレンブロック共重合体、及び、逆熱ゲル化ポリマーを含む。治療用組成物は、生体適合性ポリマーに加え、治療用組成物の生体接着性を増加する別個の生体接着剤を含んでもよい。生体接着剤は、しばしば、第二ポリマーとなり、高い生体接着性を有する。
【0048】
治療用組成物は、更に、スクラルファートと共に、増粘ゲル形態に調製することもできる。これは、スプーンで口に運んで嚥下して、消化管全体を覆うことができる。本発明の薬学的物質は、送達領域によって異なる製品形態に生成され得る。治療用組成物を投与するための可能な製品形態の例には、経口液剤、膀胱洗浄液、ゲル剤、スラリー、口内洗浄剤、トローチ、タブレット、フィルム、パッチ、棒つきキャンディ、スプレー、点滴、又は、坐薬が含まれる。例えば、座薬に調製されたゲルは、直腸又は膣の粘膜表面の治療用に投与するための好ましい製品の一つである。スラリー又は経口液剤は、口腔、食道、及び/又は、消化管の粘膜表面の治療に用いることができる。
【実施例】
【0049】
実施例 1: HADC 阻害剤によるCCL2の抑制
EGFR経路の抑制は、CCL2の表皮レベルの増加により、皮膚反応を生じ得るので、EGFR 阻害剤 PD168393、及び/又は、異なる HDAC 阻害剤を用いて、又は、用いないで処理されたケラチノサイト培養物の上清中のCCL2レベルを、ELISAにより検出した。EGFR 阻害剤 (PD168393)による24時間の処理によるケモカイン CCL2 レベルのアップレギュレーションは、それぞれ、異なる HDAC 阻害剤 (バルプロ酸 (5 mM)、フェニルブチレート (5 mM)、及び、トリコスタチンA (5 nM))で、2 時間、前処理されたケラチノサイトからの上清において抑制された(図1)。その結果は、3回の独立した実験±SDの平均値として示される。
【0050】
実施例 2:皮膚治療用のHDAC 阻害剤の異なる製剤の調製
A:フェニルブチレートのゲルの調製
第一の部分: 10 g のStabileze QM.RTM.、380.561 g の脱イオン水をビーカー中で混合し、70℃で加熱した。
【0051】
第二の部分: 5.739 g のナトリウム4-フェニルブチレート (Merck)、0.125 g のメチルパラベン (Merck)、0.075 g のプロピルパラベン (Merck)、83.5 g の1,2-プロパンジオール、及び、20 g の10% NaOH をビーカー中で混合し、70℃で加熱した。
【0052】
第二の部分をゆっくりと第一の部分に加え、400 rpmで20 分間連続攪拌して、混合物を形成した。混合物を室温で冷却した。
【0053】
B:フェニルブチレートのリポソーム製剤の調製
このリポソーム製剤では、卵ホスファチジルコリン (EPC)及びコレステロールが、第一脂質成分として等モル濃度又は異なるモル濃度で用いられた。ナトリウム4-フェニルブチレートを有する様々なリポソームを、脂質とフェニルブチレートの割合を変化させることにより得た。リポソームは、薄膜水和法により調製され、膜押し出し法により大きさを調整され、且つ、物理的に評価された。
【0054】
C:トリコスタチンAの軟膏の調製
472.5 gの白色ワセリン (Riedel-de Haen)、27 gのパラフィンろう 50/52 (ローカルサプライヤー)、及び、0.5 gのトリコスタチン A (sigma)をビーカー中で混合し、70℃で加熱して、ペーストを形成した。このペーストを、400rpmで、1時間攪拌し、その後、室温で冷却した。
【0055】
D:トリコスタチン Aの油性軟膏の調製
67.5 gの白色ワセリン (Riedel-de Haen)、16 gのセチルアルコール (Riedel-de Haen)、260.5 gの軟パラフィン (Merck)、155.5 gの流動パラフィン (Merck)、及び、0.5 gのトリコスタチンA (sigma)をビーカー中で混合して、70℃で加熱し、ペーストを形成した。このペーストを、400rpmで、1時間攪拌し、その後、室温で冷却した。
【0056】
E:バルプロ酸のクリームの調製
第一の部分: 70 g のTefose 63.RTM.、20 gのSuperpolystate.RTM.、10 g の Coster 5000.RTM.、15 gのMyriyol 318.RTM.、15 gのCoster 5088.RTM.、及び15 gのGMS SE.RTM. (すべてローカルサプライヤーにより市販されている)をビーカー中で混合し、70℃で加熱した。
【0057】
第二の部分: 5.739 g のバルプロ酸 (sigma)、0.125 g のメチルパラベン (Merck)、0.075 g のプロピルパラベン (Merck)、及び、149.061 g の脱イオン水をビーカー中で混合し、70℃で加熱した。
【0058】
第二の部分をゆっくりと第一の部分に加え、400 rpm で、5 分間連続攪拌し、混合物を形成した。2% のStabileze QM.RTM.D (2 g のStabileze QM.RTM.を98 g の脱イオン水に溶解し、70℃で加熱及び攪拌して、ペーストを形成し、室温に冷却することにより調製した) を混合物に加えて、5 分間攪拌した。混合物のpHを、0.85% のリン酸 (Merck) により5.34 に調整し、 600 rpm で20 分間攪拌した。混合物を室温で冷却した。
【0059】
実施例 3: インビボでのHDAC 阻害剤によるEGFR阻害剤の抑制
EGFR 阻害剤により増大した皮膚反応の試験において、それぞれ、体重22 ± 2 g のBALB/c 雄性マウスの群(各n=5)に、実験動物の右耳上を、10μLの0.5% の2,4-ジニトロフルオロベンゼン (DNFB)により刺激する30分前に、それぞれ、各耳の各側に、10μLの賦形剤 (DMSO/無水エタノール 1/10)、又は、PD168393の溶液 (4 mmol/L) を局所適用した (Pastore S, et al. J Immunol 174:5047-5056, 2005)。
【0060】
EGFR 阻害剤誘発性の皮膚反応を処置するために、第0日と第1日の前、2.5% の4-フェニルブチレートゲル、又は、プラセボ (ゲルベース)を、3時間間隔で3回、右耳に局所適用した。第1日目に、フェニルブチレート、又は、プラセボの第一の処理の60分後、右耳を0.5% DNFBで刺激する30分前に、PD168393を局所適用した。第1日目の、フェニルブチレート及びプラセボによる第二回及び第三回の処理は、DNFB 刺激の1 時間後と3 時間後に適用された。DNFB刺激の0、3、6、8、24 、及び、48 時間後の耳の腫脹を、Dyerモデルマイクロメータ計測器で測定した。比較のための実験対照として、DNFB刺激の60分前と15分後に、強力なステロイドデキサメタゾン (0.3 mg) を局所投与した。各マウスの左右の耳の厚さを、Dyerモデルマイクロメータ計測器で測定した。耳の浮腫は、右耳 (処理された耳)から左耳(正常対照)の厚さを引くことにより計算された。
【0061】
図2及び3を参照すると、4 mMのPD168393のみ、DMSOのみ、プラセボゲルベースのみ、又は、2.5% のフェニルブチレートゲルの局所投与は、耳の厚さに影響せず、正常な皮膚組織像の変化を誘発することもなかった。一方、DNFB刺激の30分前に適用された4 mM のPD168393 により、DNFB誘発性の皮膚反応の悪化が起こった。しかし、プラセボゲルベースにより前処理された皮膚と比較した、2.5% のフェニルブチレートゲルにより前処理された皮膚は、DNFB刺激の3、6、8、24、及び、48 時間後にDNFB刺激により誘発された、EGFR 阻害剤により増大した耳の腫脹の有意な減少をもたらした(図 2と3)。デキサメタゾンは、DNFB刺激の3、6、及び、8 時間後のピークとなるEGFR 阻害剤により増大した皮膚反応を抑制しないが、DNFB刺激の24 時間後に、皮膚腫脹の有意な抑制効果を示す。DNFB刺激の24時間後の皮膚腫脹に対するデキサメタゾンによる抑制の程度は、フェニルブチレートによるものより大きい。よって、この結果は、デキサメタゾンが EGFR 阻害剤により増大する皮膚反応の抑制に対して効果がないこと、及び、2.5% のフェニルブチレートゲルが、EGFRの抑制により増大する皮膚副作用に対する治療的有用性を有することを示唆する。
【0062】
実施例及び好ましい態様により本発明を説明してきたが、本発明は、開示された態様により限定されるものではないことを理解されたい。むしろ、各種の改変及び同様のアレンジを包含することが意図されている。従って、添付の特許請求の範囲の保護範囲は、このような改変及び同様のアレンジをすべて包含するべく、最も広い解釈に基づく。
【特許請求の範囲】
【請求項1】
細胞増殖及び生存シグナル変換経路の遮断により生じる皮膚粘膜毒性もしくは眼毒性又は副作用を治療又は改善する方法であって、治療的有効量のヒストン脱アセチル化酵素 (HDAC) 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及び/又は、薬学的に許容可能な担体を含む薬学的組成物を、それを必要とする対象に投与するステップを含む、方法。
【請求項2】
前記HDAC 阻害剤が、ヒドロキサム酸誘導体、脂肪酸誘導体、環状テトラペプチド、ベンズアミド誘導体、又は、求電子ケトン誘導体である、請求項1に記載の方法。
【請求項3】
前記HDAC 阻害剤が、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシンA、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸 (SAHA)、トリコスタチン A、スクリプタイド、又は、MS-27-275である、請求項1に記載の方法。
【請求項4】
前記薬学的組成物が、非経口投与される、請求項1に記載の方法。
【請求項5】
前記HDAC 阻害剤が、前記薬学的組成物の重量の0.00001% 〜100%を占める、請求項1に記載の方法。
【請求項6】
前記薬学的組成物が、クリーム剤、軟膏、ゲル剤、ペースト、粉末、水溶液、スプレー剤、懸濁剤、分散剤、スラリー、ローション、パッチ、坐薬、リポソーム形態、口内洗浄剤、浣腸剤、注射液、点眼薬、点耳薬、点滴、マイクロカプセル、ナノカプセル、密封式皮膚改良剤(occlusive skin conditioning agent)、生体適合性ポリマー、又は、組織中で前記HDAC 阻害剤の保持を延長し作用を持続する物質である、請求項1に記載の方法。
【請求項7】
前記薬学的組成物が、浸透促進剤、又は、pHを約3.0 〜13.0の範囲で提供するためのpH 調整剤を更に含む、請求項1に記載の方法。
【請求項8】
サイトカイン、サイトカイン阻害剤、インターロイキン、インターロイキン阻害剤、成長因子、血管形成剤、血管形成阻害剤、抗悪性腫瘍薬、抗炎症薬、ステロイド、免疫抑制剤、非ステロイド抗炎症薬、鎮痛剤、抗ヒスタミン薬、抗コリン作用薬、かゆみ止め薬、抗菌剤、抗ウイルス剤、抗真菌剤、駆虫剤、抗酸化剤、レチノイン酸、抗線維形成剤、血管作用薬、アデノシン受容体作動薬、ビタミン、ロイコトリエンモディファイヤー、インターロイキン拮抗薬、ケモカイン拮抗薬、マスト細胞阻害剤、抗IgE 抗体、選択的セロトニン再取り込み阻害剤 (SSRI)、5-ヒドロキシトリプタミン (5-HT) 受容体拮抗薬、ペルオキシソーム増殖剤活性化受容体 (PPAR) 作動薬、カルシニューリン阻害剤、ガストリン放出ペプチド受容体拮抗薬、p38 MAP キナーゼ阻害剤、ケラチノサイト成長因子 (KGF)、選択的エストロゲン受容体モジュレータ、タモキシフェン、HDAC 阻害剤、レチノイド、NFκB モジュレータ、レスベラトロール、ガバペンチン、スクラルファート、及び、ナロキソンからなる群より選択される少なくとも一つの第二の物質を更に投与する、請求項1に記載の方法。
【請求項9】
前記薬学的組成物及び前記第二の物質が、同じ処方及び同じ経路で対象に同時に投与されるか、又は、異なる処方及び/もしくは異なる経路で連続して投与される、請求項8に記載の方法。
【請求項10】
前記細胞増殖及び生存シグナル変換経路が、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムにより遮断される、請求項1に記載の方法。
【請求項11】
前記抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムが、上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF)、及び/又は、PI3K (ホスファチジルイノシトール3-キナーゼ)-Akt-mTOR 経路を抑制する、請求項10に記載の方法。
【請求項12】
前記皮膚粘膜毒性もしくは眼毒性又は副作用が、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、ならびに/又は、脆化した変形爪及び爪周囲の膨張を含む、皮膚又は上皮の副作用を含む、請求項1に記載の方法。
【請求項13】
化学療法及び/又は放射線治療を伴う又は伴わない上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF) 受容体、又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を標的とする分子治療により生じる、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を軽減する方法であって、対象の罹患皮膚又は粘膜領域に、有効量のHDAC 阻害剤、又は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸 (SAHA)、トリコスタチンA、スクリプタイド、MS-27-275、及び、それらの混合物からなる群より選択される高アセチル化剤、及び、適切な塩、及び/又は、適切な担体を含む組成物を、局所的に適用するステップを含み、HDAC 阻害剤、又は、高アセチル化剤が、分子標的治療の前、分子標的治療中、及び/又は、分子標的治療の後に、患者に投与される、方法。
【請求項14】
化学療法及び/又は放射線治療を伴う又は伴わない上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF) 受容体、又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を標的とする分子治療により生じる、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である、皮膚又は上皮の副作用を軽減するための医薬の製造のための、有効量のHDAC 阻害剤、又は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸(SAHA)、トリコスタチン A、スクリプタイド、MS-27-275、及び、それらの混合物からなる群より選択される高アセチル化剤、及び、適切な塩、及び/又は、適切な担体を含む組成物の使用。
【請求項15】
HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及び、EGFR 阻害剤を含む、組み合わせ。
【請求項1】
細胞増殖及び生存シグナル変換経路の遮断により生じる皮膚粘膜毒性もしくは眼毒性又は副作用を治療又は改善する方法であって、治療的有効量のヒストン脱アセチル化酵素 (HDAC) 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及び/又は、薬学的に許容可能な担体を含む薬学的組成物を、それを必要とする対象に投与するステップを含む、方法。
【請求項2】
前記HDAC 阻害剤が、ヒドロキサム酸誘導体、脂肪酸誘導体、環状テトラペプチド、ベンズアミド誘導体、又は、求電子ケトン誘導体である、請求項1に記載の方法。
【請求項3】
前記HDAC 阻害剤が、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシンA、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸 (SAHA)、トリコスタチン A、スクリプタイド、又は、MS-27-275である、請求項1に記載の方法。
【請求項4】
前記薬学的組成物が、非経口投与される、請求項1に記載の方法。
【請求項5】
前記HDAC 阻害剤が、前記薬学的組成物の重量の0.00001% 〜100%を占める、請求項1に記載の方法。
【請求項6】
前記薬学的組成物が、クリーム剤、軟膏、ゲル剤、ペースト、粉末、水溶液、スプレー剤、懸濁剤、分散剤、スラリー、ローション、パッチ、坐薬、リポソーム形態、口内洗浄剤、浣腸剤、注射液、点眼薬、点耳薬、点滴、マイクロカプセル、ナノカプセル、密封式皮膚改良剤(occlusive skin conditioning agent)、生体適合性ポリマー、又は、組織中で前記HDAC 阻害剤の保持を延長し作用を持続する物質である、請求項1に記載の方法。
【請求項7】
前記薬学的組成物が、浸透促進剤、又は、pHを約3.0 〜13.0の範囲で提供するためのpH 調整剤を更に含む、請求項1に記載の方法。
【請求項8】
サイトカイン、サイトカイン阻害剤、インターロイキン、インターロイキン阻害剤、成長因子、血管形成剤、血管形成阻害剤、抗悪性腫瘍薬、抗炎症薬、ステロイド、免疫抑制剤、非ステロイド抗炎症薬、鎮痛剤、抗ヒスタミン薬、抗コリン作用薬、かゆみ止め薬、抗菌剤、抗ウイルス剤、抗真菌剤、駆虫剤、抗酸化剤、レチノイン酸、抗線維形成剤、血管作用薬、アデノシン受容体作動薬、ビタミン、ロイコトリエンモディファイヤー、インターロイキン拮抗薬、ケモカイン拮抗薬、マスト細胞阻害剤、抗IgE 抗体、選択的セロトニン再取り込み阻害剤 (SSRI)、5-ヒドロキシトリプタミン (5-HT) 受容体拮抗薬、ペルオキシソーム増殖剤活性化受容体 (PPAR) 作動薬、カルシニューリン阻害剤、ガストリン放出ペプチド受容体拮抗薬、p38 MAP キナーゼ阻害剤、ケラチノサイト成長因子 (KGF)、選択的エストロゲン受容体モジュレータ、タモキシフェン、HDAC 阻害剤、レチノイド、NFκB モジュレータ、レスベラトロール、ガバペンチン、スクラルファート、及び、ナロキソンからなる群より選択される少なくとも一つの第二の物質を更に投与する、請求項1に記載の方法。
【請求項9】
前記薬学的組成物及び前記第二の物質が、同じ処方及び同じ経路で対象に同時に投与されるか、又は、異なる処方及び/もしくは異なる経路で連続して投与される、請求項8に記載の方法。
【請求項10】
前記細胞増殖及び生存シグナル変換経路が、抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムにより遮断される、請求項1に記載の方法。
【請求項11】
前記抗体標的治療剤、抗体フラグメント、標的小分子化合物、低分子量チロシンキナーゼ阻害剤、ペプチド模倣薬、アンチセンス・オリゴヌクレオチド (DNA及び/又はRNA)、又は、リボザイムが、上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF)、及び/又は、PI3K (ホスファチジルイノシトール3-キナーゼ)-Akt-mTOR 経路を抑制する、請求項10に記載の方法。
【請求項12】
前記皮膚粘膜毒性もしくは眼毒性又は副作用が、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、ならびに/又は、脆化した変形爪及び爪周囲の膨張を含む、皮膚又は上皮の副作用を含む、請求項1に記載の方法。
【請求項13】
化学療法及び/又は放射線治療を伴う又は伴わない上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF) 受容体、又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を標的とする分子治療により生じる、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である皮膚又は上皮の副作用を軽減する方法であって、対象の罹患皮膚又は粘膜領域に、有効量のHDAC 阻害剤、又は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸 (SAHA)、トリコスタチンA、スクリプタイド、MS-27-275、及び、それらの混合物からなる群より選択される高アセチル化剤、及び、適切な塩、及び/又は、適切な担体を含む組成物を、局所的に適用するステップを含み、HDAC 阻害剤、又は、高アセチル化剤が、分子標的治療の前、分子標的治療中、及び/又は、分子標的治療の後に、患者に投与される、方法。
【請求項14】
化学療法及び/又は放射線治療を伴う又は伴わない上皮細胞成長因子受容体 (EGFR)、MAPK(マイトジェン活性化プロテインキナーゼ)/ERK (細胞外調節キナーゼ)、血管内皮成長因子受容体 (VEGF) 受容体、又は、PI3K (ホスファチジルイノシトール 3-キナーゼ)-Akt-mTOR 経路を標的とする分子治療により生じる、丘疹膿疱性の発疹、皮膚乾燥、そう痒、皮膚炎、手足症候群、粘膜炎、抜け毛、睫毛と顔の毛の成長の増加、脆化した変形爪及び爪周囲の膨張である、皮膚又は上皮の副作用を軽減するための医薬の製造のための、有効量のHDAC 阻害剤、又は、バルプロ酸、フェニルブチレート、アルギニンブチレート、デプデシン、トラポキシン A、デプシペプチド、オキサムフラチン、スベロイルアニリドヒドロキサム酸(SAHA)、トリコスタチン A、スクリプタイド、MS-27-275、及び、それらの混合物からなる群より選択される高アセチル化剤、及び、適切な塩、及び/又は、適切な担体を含む組成物の使用。
【請求項15】
HDAC 阻害剤、及び/又は、薬学的に許容可能なその塩もしくは溶媒和物、及び、EGFR 阻害剤を含む、組み合わせ。
【図1】

【図2】
【図3】
【図2】
【図3】
【公表番号】特表2012−530076(P2012−530076A)
【公表日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願番号】特願2012−515308(P2012−515308)
【出願日】平成21年6月26日(2009.6.26)
【国際出願番号】PCT/CN2009/072474
【国際公開番号】WO2010/148572
【国際公開日】平成22年12月29日(2010.12.29)
【出願人】(511305483)アサン ラボラトリーズ カンパニー, リミテッド (1)
【Fターム(参考)】
【公表日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願日】平成21年6月26日(2009.6.26)
【国際出願番号】PCT/CN2009/072474
【国際公開番号】WO2010/148572
【国際公開日】平成22年12月29日(2010.12.29)
【出願人】(511305483)アサン ラボラトリーズ カンパニー, リミテッド (1)
【Fターム(参考)】
[ Back to top ]