
相同組換えにより遺伝子改変された多能性幹細胞の簡便な検出法
【課題】相同組換えにより遺伝子改変された多能性幹細胞を製造する方法および相同組換えにより遺伝子改変された多能性幹細胞の検出方法を提供する。
【解決手段】遺伝子改変された染色体断片を有する人工染色体をターゲティングベクターとして導入する工程およびSNPアレイを用いて、導入された人工染色体の一部または全体のコピー数を測定することによって相同組換えされている多能性幹細胞を選別する工程を含む多能性幹細胞を製造する方法。
【解決手段】遺伝子改変された染色体断片を有する人工染色体をターゲティングベクターとして導入する工程およびSNPアレイを用いて、導入された人工染色体の一部または全体のコピー数を測定することによって相同組換えされている多能性幹細胞を選別する工程を含む多能性幹細胞を製造する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、相同組換えにより遺伝子改変された多能性幹細胞の検出方法、ならびに、該検出方法を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞を製造する方法に関する。
【背景技術】
【0002】
内在性ゲノムDNAの一部を改変(欠失、挿入または置換)し、生物に生来備わっていない生物学的性状を付与したり、生物が生来備えている生物学的性状の発現を抑制したり、さらには該生物のある遺伝子が担う機能を解析するための方法として、該生物の内在性ゲノムDNAの所望の部位を人為的に改変する遺伝子ターゲティングが行われている(特許文献1および特許文献2)。
【0003】
遺伝子ターゲティングは、内因性遺伝子をノックアウトする(非特許文献1および非特許文献2)か、または染色体に外因性配列をノックインするために用いられている。しかし、この方法は、非常に効率が低いため(トランスフェクションされた細胞の10-6〜10-9)、多大な労力が必要である。
【0004】
そこで、遺伝子ターゲティングの効率が1,000倍以上に増大することより(非特許文献3、非特許文献4、非特許文献5、非特許文献6、非特許文献7および非特許文献8)、メガヌクレアーゼにより所望の部位を切断することで効率を上げる工夫がされている。
【0005】
しかし、いずれのクローンにおいて相同組換えが行われたか否かについては確認する必要があるため、多数の候補の中から所望の改変がなされたクローンを効率よく検出する方法が求められている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】WO1990/11354
【特許文献2】WO1991/09955
【非特許文献】
【0007】
【非特許文献1】Capecchi, M.R., Science, (1989) 244: 1288-1292
【非特許文献2】Smithies, O., Nature Medicine, (2001) 7: 1083-1086
【非特許文献3】Puchta H, et al., Nucleic Acids Res., (1993) 21: 5034-5040
【非特許文献4】Choulika A, et al., Mol. Cell. Biol., (1995) 15: 1968-1973
【非特許文献5】Puchta H, et al., Proc. Natl. Acad. Sci. U.S.A., (1996) 93: 5055-5060
【非特許文献6】Sargent RG, et al., Mol. Cell. Biol., (1997) 17: 267-277
【非特許文献7】Cohen-Tannoudji M, et al., Mol. Cell. Biol., (1998) 18: 1444-1448
【非特許文献8】Donoho G, ste al.,, Mol. Cell. Biol., (1998) 18: 4070-4078
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は、相同組換えにより遺伝子改変された多能性幹細胞の作製において、ゲノム上の所望の部位もしくは領域で相同組換えが起きたか否かを検出することを含む方法を提供することを目的とする。
【0009】
具体的には、本発明の目的は、遺伝子ターゲティングにより所望の遺伝子座を改変させた多能性幹細胞を検出することである。すなわち、本発明の目的は、ゲノム上の所望の部位で相同組換えが起きている多能性幹細胞を、ランダムインテグレーションが起きている多能性幹細胞から区別する方法を提供することである。
【課題を解決するための手段】
【0010】
本発明者らは、上記の課題を解決すべく、SNP(一塩基多型)アレイ法を用いたコピー数多型の検出方法に着目し、ターゲティングベクターとしての遺伝子改変された染色体断片を有する人工染色体ベクターが導入された多能性幹細胞中の該染色体断片に相当する領域のコピー数をSNPアレイ法により測定することにより、相同組換えが起こっているか否かを測定することができることを見出した。さらに、相同組換えが起きているか否かの確証を補助的に得るためにさらに、組込まれた選択マーカーを指標とする方法、および/または、改変部位での野生型の配列を有するアレル数を測定する方法を組み合わせることで、本発明を完成するに至った。
【0011】
すなわち、本発明は以下の通りである。
[1]次の工程(1)〜(3):
(1)多能性幹細胞に、遺伝子改変された染色体断片を有する人工染色体を導入し、遺伝子改変されたと推定される多能性幹細胞クローンからなる集団を作製する工程、
(2)前記集団の多能性幹細胞クローンについて、SNPアレイを用いて、前記導入された人工染色体の一部または全体のコピー数を測定する工程、
(3)前記コピー数が、人工染色体が導入されていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞クローンを、相同組換えにより遺伝子改変された多能性幹細胞として選択する工程、
を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の製造方法。
[2]前記工程(1)の遺伝子改変が内在性配列を保持したまま細胞ゲノムに外因性DNA断片を挿入することからなる組込みを含む、[1]に記載の方法。
[3]外因性DNAが選択マーカーをコードするDNAであって、該選択マーカーが陽性であるクローンを選択する工程をさらに含む、[2]に記載の方法。
[4]多能性幹細胞クローンの該遺伝子改変対象であるアレルの数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下しているクローンを選択する工程をさらに含む、[1]〜[3]のいずれかに記載の方法。
[5]前記多能性幹細胞がヒト多能性幹細胞である、[1]〜[4]のいずれかに記載の方法。
[6]前記人工染色体がBACクローンである、[1]〜[5]のいずれかに記載の方法。
[7]前記選択マーカーが薬剤耐性マーカーである、[3]に記載の方法。
[8]相同組換えにより遺伝子改変された多能性幹細胞の製造において、SNPアレイを用いて組換え領域のコピー数を測定し、該コピー数が、相同組換えをしていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞クローンを、相同組換えにより遺伝子改変された多能性幹細胞として検出する工程を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の検出方法。
[9]前記遺伝子改変が、内在性配列を保持したまま細胞ゲノムの外因性DNA断片を挿入することからなる組込みである、[8]に記載の方法。
[10]前記外因性DNAが選択マーカーをコードするDNAあって、該選択マーカーが陽性であるクローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する工程をさらに含む、[9]に記載の方法。
[11]多能性幹細胞クローンの該遺伝子改変対象であるアレルの数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下しているクローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する工程をさらに含む、[8]〜[10]のいずれかに記載の方法。
[12]前記多能性幹細胞がヒト多能性幹細胞である、[8]〜[11]のいずれかに記載の方法。
[13]前記選択マーカーが薬剤耐性マーカーである、[10]に記載の方法。
【発明の効果】
【0012】
本発明を用いることによって、相同組換えによる多能性幹細胞の遺伝子改変を、簡便に効率的な手順で検出することができる。本発明の方法を遺伝子ターゲティングの高効率の手法と組み合わせることができるため、本発明の方法は、多能性幹細胞の遺伝子改変の全体的な効率化に寄与する。
【図面の簡単な説明】
【0013】
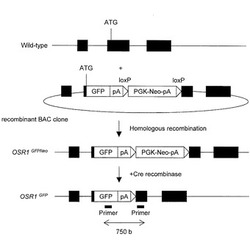
【図1】ヒトOSR1(odd-skipped related 1; M. Katoh, Int. J. Mol. Med. 2002; 10(2):221-225)遺伝子座にCre-loxPシステムを用いてGFP-PGK-Neoカセットを挿入するスキームを示す。
【図2】(A)親株または改変BACクローン導入後の薬剤耐性クローンの染色体におけるOSR1の開始コドンを挟んだ領域の野生型の存在比を定量化したグラフを示す。(B)SNPアレイを用いてiPS細胞株(3D36, 3D45, 3F3, 3l49, 3D12)の各々の第2染色体のOSR1遺伝子座付近の各プローブのコピー数を解析した結果を示す。
【発明を実施するための形態】
【0014】
本発明を以下に詳細に説明する。
本発明は、次の工程(1)〜(3):
(1)多能性幹細胞に、遺伝子改変された染色体断片を有する人工染色体を導入し、遺伝子改変されたと推定される多能性幹細胞クローンからなる集団を作製する工程、
(2)前記集団の多能性幹細胞クローンについて、SNPアレイを用いて、前記導入された人工染色体の一部または全体のコピー数を測定する工程、
(3)前記コピー数が、人工染色体が導入されていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞クローンを、相同組換えにより遺伝子改変された多能性幹細胞として選択する工程、
を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の製造方法を提供する。
【0015】
本明細書で使用する用語「相同組換え」とは、人工的に染色体もしくはゲノム上の特定遺伝子を改変する遺伝子ターゲッテイング手段であり、染色体上の目的の配列と相同的な部分をもつゲノム断片を細胞中に導入した場合、導入したゲノム断片と染色体上の対応する遺伝子座の間で塩基配列の相同性に基づいて起こる組換えである。
【0016】
また、「遺伝子改変」とは、染色体上の所望の遺伝子の遺伝子座において、外因性DNAを挿入するか、外因性DNAで遺伝子の一部もしくは全部を置換するか、あるいは遺伝子を欠損(欠失)させることを意味する。より具体的にいえば、遺伝子改変は、内在性DNA配列を保持したまま特定の遺伝子座の遺伝子の発現と連動するようにまたは恒常的に発現するように外因性DNA断片を挿入(言い換えれば、ノックイン)するか、あるいは、内在性DNA配列を改変するように該遺伝子の配列の全部もしくは部分を置換、欠損または破壊(言い換えれば、ノックアウト)することである。また、本発明において、対象となる多能性幹細胞が特定の遺伝子に変異を有する場合において、該遺伝子を正常の遺伝子配列と置換する組換えを行うための遺伝子改変は、正常の遺伝子へ改変することを意味する。
【0017】
また、改変の対象となる遺伝子は、目的によって適宜選択されてよく、例えば、疾患の原因遺伝子、細胞タイプの指標となるマーカー遺伝子、発現が減少しないハウスキーピング遺伝子などが挙げられるが、これらに限定されない。ここで、細胞タイプとは、例えば、内胚葉細胞、外胚葉細胞、中胚葉細胞、脊索中胚葉、沿軸中胚葉、中間中胚葉細胞、側板中胚葉、神経細胞、グリア細胞、造血細胞、肝細胞、膵β細胞、腎前駆細胞、内皮細胞、周皮細胞、上皮細胞、骨芽細胞、筋芽細胞、軟骨細胞など細胞が挙げられる。これらの細胞タイプの指標となるマーカー遺伝子として、GATA4、GATA5、GATA6、AFP、HNF-3β、SOX17、FOXA2、PDGFRα、FLK1、Brahcyury、Gremlin, MYH2、Nestin, SOX13, SOX21, CryM、Otx2、TP63、SOX2、PSA-NCAM、TuJ1、Thy1.2、GFAP、PAX6、A2B5、CD11b、c-kit、CD34、CD90、CD117、Albumin、CK18、CK19、PDX1、OSR1、SIX2、GATA2、VEGFR2、NG2、desmin、MUC1、BGLAP、SPP1、MyoD、MYF5、Myogenin、Aggrecan、Collagen IIおよびSox9が例示されるが、これらに限定されない。遺伝子の配列は、NCBI GenBank(hhtp://www.ncbi.nlm.nih.gov)、EMBL、DDBJなどの公知のDNAデータベースから入手できる。
【0018】
<人工染色体の導入方法>
本発明において、人工染色体としては、複製起点、セントロメアおよびテロメアなどの宿主細胞において複製に必要な機能を有する人工的に作製された染色体であり、大腸菌由来のBACベクター(Shizuya et al.(1992)Proc.Natl.Acad.Sci.U.S.A. 89:8794-8797)、P1ファージ由来のPACベクター(Ioannou et al.(1994)Nature Genetics 6:84-89;Pierce et al.(1992)Meth.Enzymol.216:549-574;Pierce et al.(1992)Proc.Natl.Acad.Sci.U.S.A.89:2056-2060;米国特許5300431号および国際PCT出願WO92/14819号)、酵母由来のYACベクター(Burke et al.(1987)Science 236:806-812)、およびヒト由来のHACベクター(WO1998/008964)などが例示される。これらの人工染色体は、外因性DNAを含む巨大なサイズのゲノム断片を保有したまま宿主細胞内で増殖することが可能である。人工染色体は、好ましくは、染色体断片を有する人工染色体、より好ましくは、ヒト染色体の断片を有する人工染色体である。DNA断片の大きさは、人工染色体が安定的に宿主内で複製が可能な大きさであり、BACまたはPACの場合は、通常、約300kb以内であり、YACの場合は1Mb以内であり、HACの場合は1Mb以上でも可能である。
【0019】
本発明の方法では、人工染色体として、導入すべきゲノム断片のサイズに応じて上記例示の人工染色体いずれかを使用することが可能であり、好ましい実施形態では人工染色体はBACベクターであり、さらに具体的には、ヒト染色体断片を保有するBACベクターとして、例えば、ローズウェル癌研究所(Roswell Park Cancer Institute、米国)が作製した平均175kbのヒトゲノム断片を有する437,000クローンのライブラリーであるRP-11が例示される。このような、ライブラリーに含まれる1つのゲノム断片を有するBACベクターをBACクローンと称する。
【0020】
染色体断片を有する人工染色体を作製するためには、当業者に周知の方法を用いて行う事が可能であり、例えば、制限酵素を用いて染色体断片中の所望の位置において切断し、適宜機能的配列を有するDNA断片を連結させる方法、ファージ由来のRed遺伝子を用いて大腸菌内で相同組換えを行う方法などが挙げられる。Red遺伝子を発現するプラスミドを有するキットは、Gene Bridges 社から購入可能であり、添付のプロトコールに従って人工染色体の改変が可能である。
【0021】
遺伝子の改変を目的として人工染色体へ挿入する外因性DNAとして、特に限定されないが、有用な(ヒトもしくは非ヒト動物由来)遺伝子類、選択マーカー遺伝子、または、これらの組み合わせ、等が例示される。外因性DNAを組み込むためのカセットにはさらに、プロモーター、IRES、部位特異的リコンビナーゼの認識配列、ターミネーターなどを含有させることができる。
【0022】
外因性DNAは、例えば、生物学的に、医学的に、あるいはクローンを選別するために、有用な機能をもつDNAであることが好ましい。
【0023】
本明細書中で使用する「有用な(ヒトもしくは非ヒト動物由来)遺伝子類」としては、例えば、機能不明の遺伝子類、疾患の原因遺伝子類、治療に有用な遺伝子類などの、医学的な研究または実用レベルで有用な遺伝子類、細胞タイプの指標となるマーカー遺伝子類、発現が減少しないハウスキーピング遺伝子類などが挙げられるが、これらに限定されない。
【0024】
本明細書で使用する用語「選択マーカー遺伝子」とは、宿主細胞を選択する指標として機能する遺伝子をいう。選択マーカーとしては、公知のポジティブマーカーまたはネガティブマーカーのいずれも使用できる。ポジティブマーカー選択マーカーとしては、蛍光マーカー、発光マーカー、および薬剤耐性マーカーが挙げられるが、これらに限定されない。「蛍光マーカー」としては、緑色蛍光プロテイン(GFP)、青色蛍光プロテイン(CFP)、黄色蛍光プロテイン(YFP)および赤色蛍光プロテイン(dsRed)のような蛍光タンパク質をコードする遺伝子が挙げられるが、これらに限定されない。「発光マーカー」としては、ルシフェラーゼのような発光タンパク質をコードする遺伝子が挙げられるが、これらに限定されない。「薬剤耐性マーカー」としてはネオマイシン(G418)耐性遺伝子との融合遺伝子(β-geo遺伝子)、CAT遺伝子、GFP遺伝子、SV40ラージT遺伝子、ネオマイシン耐性遺伝子、ピューロマイシン耐性遺伝子、ハイグロマイシン耐性遺伝子、ブラストサイジンS耐性遺伝子等をコードする遺伝子が挙げられるが、これらに限定されない。
【0025】
また、「部位特異的リコンビナーゼ」として、Creリコンビナーゼ(Gorman C, Bullock C. Curr Opin Biotechnol. (2000), 11: 455-60)、FLPリコンビナーゼ(Buchholz F, et al., Nat Biotechnol. (1998), 16: 657-662)などが例示され、これらのリコンビナーゼの標的配列としてloxP、FRTなどが例示される。2つの認識配列で挟まれた領域を削除する目的から、該認識配列は所望の領域を挟んで2ヶ所へ挿入することが望ましい。
【0026】
ターミネーターは、転写終結配列としてのポリアデニル化シグナルである。特に限定されないが、ポリアデニル化シグナル配列として、ヒトBGHポリA、SV40ポリA、ヒトβアクチンポリA、ウサギβグロブリンポリAおよび免疫グロブリンκポリAが例示される。
【0027】
上記の外因性DNAは、挿入、置換または欠損(欠失)等の目的に合わせて適宜、機能しうるように該人工染色体へ配置されてもよく、例えば、改変対象の遺伝子の置換および欠損を目的として、選択マーカーおよびターミネーターを対象遺伝子の開始コドンの後方の遺伝子配列内に連結し配置させてもよい。あるいは、選択マーカーは、改変対象の遺伝子の発現と連動して発現させるために、内在性のプロモーターと連結して配置されていてもよい。このため、選択マーカーは、口蹄疫ウイルスの2A self-cleaving peptide(Science, 322, 949-953, 2008など参照)、IRES配列などと共に連結して配置されていてもよい。さらに、挿入される選択マーカーは、人工染色体が細胞内に導入されたことを確認とするため、恒常的に発現させることを目的として外因性のプロモーターに予め連結されていてもよい。プロモーターの例としては、PGK プロモーター、EF-αプロモーター、CAGプロモーター、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、MoMuLV(モロニーマウス白血病ウイルス)LTR、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーターなどが用いられる。
【0028】
人工染色体を細胞へ導入する方法として、例えば、リン酸カルシウム沈殿法(Graham et al.(1978)Virology 52:456-457、Wigler et al.(1979)Proc.Natl.Acad.Sci.U.S.A.76 1373-1376およびCurrent Protocols in Molecular Biology Vol.1,Wiley Inter-Science,Supplement 14,Unit 9.1.1-9.1.9(1990))、ポリエチレングリコールを用いた融合方法(米国特許4684611号)、リポフェクションなどの脂質キャリアを用いる方法(Teifel et al.(1995)Biotechniques 19:79-80、Albrecht et al.(1996)Ann.Hematol.72:73-79;Holmen et al.(1995)In Vitro Cell Dev.Biol.Anim.31:347-351、Remy et al.(1994)Bioconjug.Chem. 5:647-654、Le Bolc'het al.(1995)Tetrahedron Lett.36:6681-6684、Loeffler et al.(1993)Meth.Enzymol.217:599-618およびStrauss(1996)Meth.Mol.Biol.54: 307-327)、エレクトロポレーションおよび微小核体との融合法(米国特許5240840号、4806476号、5298429号、5396767号、Fournier(1981)Proc.Natl.Acad.Sci.U.S.A. 78:6349-6353およびLambert et al(1991)Proc. Natl. Acad. Sci. U.S.A. 88:5907-59)などがある。
【0029】
上記の手法によって、遺伝子改変されたと推定される多能性幹細胞クローンからなる集団が作製される。ここで、「遺伝子改変されたと推定される」とは、多能性幹細胞クローンのほとんどが遺伝子改変されているが、一部には遺伝子改変されていない(すなわち野生型)クローンが混入しうることを意味する。該クローンには、遺伝子改変がゲノム上の所望の部位もしくは領域で起こった(すなわち相同組換えされた)クローンだけでなく、遺伝子改変がゲノム上でランダムに起こった(すなわちランダムインテグレーションされた)クローンなどが含まれる。本発明では、遺伝子改変されたと推定される多能性幹細胞クローンからなるこのような集団から、以下で説明するSNPアレイを利用することによって、相同組換えにより遺伝子改変されたクローンが選別される。これまで相同組換えされた多能性幹細胞の検出のために、SNPアレイを使用したコピー数測定法を利用することが全く行われていなかったが、本発明では、このような手法を利用することで相同組換えされた目的細胞クローンを容易に識別できる。
【0030】
<SNPアレイ>
本発明において、SNP(一塩基多型)アレイとは、アレル特異的オリゴヌクレオチドプローブを用いるSNPタイピング用のアレイであり、好ましくは、アレイシグナルの定量的特性を利用して、ゲノムコピー数を検出できるものである。好ましくは、コピー数多型を測定するためのオリゴヌクレオチドプローブを有し、2.5kb〜7kb の平均プローブ間隔であるアレイである。例えば、アフィメトリクス(Affymetrix)SNPアレイ 6.0、イルミナ CNV370-DuoおよびBead Chipsなどが例示される。
【0031】
本発明の方法において、SNPアレイを用いてコピー数を測定するために、サンプルとして抽出した染色体DNAを例えばHindIII、XbaI、NspIまたはStyI等の制限酵素で処理した後、アダプターを5’末端および3’末端に付加し、アダプター特異的なプライマーを用いてPCRにより増幅させてもよい。該増幅は、0.5kb〜1.5kb の短い制限酵素断片のみが選択的に増幅されることが望ましい。さらに、増幅されたDNA断片をDNaseIによりさらに細かく断片化したDNA断片群を、オリゴヌクレオチドプローブを有するアレイとハイブリダイゼーションさせ、当該断片の量をシグナル強度によって検出することができる。得られたシグナル強度のデータは、CNAG(copy number analyserfor gene chip)ソフトウェア(http://www.genome.umin.jp/)を用いてコピー数として算出することができる。
【0032】
より正確なコピー数を検出するため、アレル特異的にシグナルを検出することが望ましい。そのため、対照として相同組換え前の多能性幹細胞または人工染色体を導入して得られた他のクローンを用いて同様にSNPアレイを用いてシグナル強度を測定し、AsCNAR(allele-specific copy-number analysis using anonymous references)(http://www.genome.umin.jp/)を用いて解析することで、アレル別のシグナル強度を得ることができる。アレルによって同一プローブに対する親和性が異なる場合において正確なコピー数が算出できないことを、アレル特異的にシグナルを測定することで改善することができる。
【0033】
本発明で使用可能なアレイは、プローブとなるオリゴヌクレオチドが、固相担体(基板)に固定化されたマイクロアレイの形態で提供されることが好ましい。マイクロアレイの固相担体としては、例えば、ガラス基板、シリコン基板、メンブレン、ビーズなどが挙げられ、その材質、大きさ、形状は特に限定されない。マイクロアレイの形成方法は特に限定されず、当業者が利用可能ないかなる方法を用いてもよく、例えば、固相担体表面で直接プローブを合成する方法(オン・チップ法)、または予め調製したプローブを固相担体表面に結合する方法などがある。固相担体表面で直接プローブを合成する場合には、光照射で選択的に除去される保護基を用い、半導体製造に利用されるフォトリソグラフィー技術および固相合成技術を組み合わせて所定の微少なマトリックス領域でのオリゴヌクレオチドの選択的な合成を行う方法が一般的である。一方、予めプローブを調製して固相担体表面に結合する方法では、プローブ核酸の種類や固相担体の種類に応じて、スポッタ装置によりポリ陽イオン化合物やアミノ基、アルデヒド基、エポキシ基等を有するシランカップリング剤などで表面処理した固相担体の表面に点着する方法、反応活性基を導入したプローブを合成し、予め反応性基を形成させるように表面処理した固相担体表面に該プローブを点着して該プローブを固相担体表面に共有結合により結合固定させる方法などが利用できる。
【0034】
遺伝子改変された染色体断片を有する人工染色体が導入された多能性幹細胞から得られた染色体(ゲノム)中の該染色体断片のコピー数を測定することによって、該細胞ゲノムにおける該人工染色体に搭載された染色体断片のコピー数を確認することができる。この時、相同組換えにより多能性幹細胞の染色体に組換えられた場合、該多能性幹細胞中の染色体断片のコピー数に関して、該人工染色体の導入前と同等(言い換えれば、同数)のコピー数であるか、または該人工染色体の導入前より減少したコピー数である。内在性DNA配列を保持したまま細胞ゲノム中に外因性DNA断片が挿入(すなわち、ノックイン)されたときには、前記導入前と同等のコピー数となり、また、内在性DNA配列を改変するように該遺伝子の配列の全部もしくは部分が置換、欠損または破壊(すなわち、ノックアウト)されたときには、前記導入前より減少したコピー数となる。一方、ランダムインテグレーションにより、染色体断片がランダムに(言い換えれば、無作為に)細胞ゲノムに組み込まれた場合、前記導入前より増加したコピー数、例えば3コピーまたはそれ以上のコピー数を呈する。したがって、該方法でコピー数を測定することで相同組換え、具体的にはノックイン型またはノックアウト型の相同組換え、の有無の判断が可能である。
【0035】
<選択マーカーによる補助的選別>
本発明では、上記のとおりSNPアレイを用いてコピー数を測定することで相同組換えの有無を判定可能であるが、補助的に選択マーカーを利用することも可能である。
【0036】
すなわち、本発明において、選択マーカーを有する人工染色体を細胞へ導入後、選択マーカーを利用することによって、人工染色体が染色体(もしくは、ゲノム)へ組み込まれた細胞を選別することができる。ここで、薬剤選択マーカーを用いる場合、細胞培養液へ対応する薬剤を添加することで、相同組換えまたはランダムインテグレーションにより人工染色体が染色体(もしくは、ゲノム)へ組み込まれた細胞を選択マーカーが陽性である細胞として選択的に取り出すことができる。
【0037】
<アレル数測定による補助的選別>
さらに本発明では、アレル数測定による補助的選別法を行うことができる。この方法では、遺伝子改変された染色体断片を有する人工染色体を多能性幹細胞に導入後、該細胞の染色体(もしくは、ゲノム)において、遺伝子改変されていない野生型の配列を有するアレルが1つ確認できる場合、または野生型の配列を有するアレルを確認できない場合、該細胞を相同組換えにより遺伝子改変されたクローンとして選別することができる。好適には、多能性幹細胞クローンの遺伝子改変対象であるアレル数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下している、好ましくは1/2である、クローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する。常染色体上の野生型の遺伝子は通常2アレルであるが、一方の遺伝子に対して改変を行うことで野生型の遺伝子は1アレルになる。従って、野生型細胞の半数になっている場合、目的の遺伝子座において改変が行われていることが確認できる。ここで、野生型の配列を有するアレル数を測定する方法として、組換えを施した多能性幹細胞から抽出した染色体DNAを基に遺伝子を改変させた部位を含む領域をPCR法により増幅し、野生型の配列を有する場合、該配列に特徴的な大きさのPCR産物の量を測定し、野生型細胞としての遺伝子改変されていない多能性幹細胞と比較する方法が例示される。他の実施形態として、遺伝子改変された多能性幹細胞から抽出した染色体を任意の制限酵素により切断し、遺伝子が改変されることで生じる特異的な大きさのDNA断片をサザンブロット法により測定する方法が例示される。
【0038】
本発明の方法で遺伝子改変されうる多能性幹細胞について、以下で具体的に説明する。
<多能性幹細胞>
多能性幹細胞を分化させることによってドーパミン産生神経前駆細胞を含有する細胞集団を用意する場合、使用可能な多能性幹細胞は、生体に存在するすべての細胞に分化可能である多能性を有し、かつ、増殖能をも併せもつ幹細胞であり、それには、特に限定されないが、例えば胚性幹(ES)細胞、核移植により得られるクローン胚由来の胚性幹(ntES)細胞、精子幹細胞(「GS細胞」)、胚性生殖細胞(「EG細胞」)、人工多能性幹(iPS)細胞、培養線維芽細胞や骨髄幹細胞由来の多能性細胞(Muse細胞)などが含まれる。好ましい多能性幹細胞は、ES細胞、ntES細胞、およびiPS細胞である。
【0039】
(A) 胚性幹細胞
ES細胞は、ヒトやマウスなどの哺乳動物の初期胚(例えば胚盤胞)の内部細胞塊から樹立された、多能性と自己複製による増殖能を有する幹細胞である。
【0040】
ES細胞は、受精卵の8細胞期、桑実胚後の胚である胚盤胞の内部細胞塊に由来する胚由来の幹細胞であり、成体を構成するあらゆる細胞に分化する能力、いわゆる分化多能性と、自己複製による増殖能とを有している。ES細胞は、マウスで1981年に発見され(M.J. Evans and M.H. Kaufman (1981), Nature 292:154-156)、その後、ヒト、サルなどの霊長類でもES細胞株が樹立された (J.A. Thomson et al. (1998), Science 282:1145-1147; J.A. Thomson et al. (1995), Proc. Natl. Acad. Sci. USA, 92:7844-7848;J.A. Thomson et al. (1996), Biol. Reprod., 55:254-259; J.A. Thomson and V.S. Marshall (1998), Curr. Top. Dev. Biol., 38:133-165)。
【0041】
ES細胞は、対象動物の受精卵の胚盤胞から内部細胞塊を取出し、内部細胞塊を線維芽細胞のフィーダー上で培養することによって樹立することができる。また、継代培養による細胞の維持は、白血病抑制因子(leukemia inhibitory factor (LIF))、塩基性線維芽細胞成長因子(basic fibroblast growth factor (bFGF))などの物質を添加した培養液を用いて行うことができる。ヒトおよびサルのES細胞の樹立と維持の方法については、例えばUSP5,843,780; Thomson JA, et al. (1995), Proc Natl. Acad. Sci. U S A. 92:7844-7848; Thomson JA, et al. (1998), Science. 282:1145-1147; H. Suemori et al. (2006), Biochem. Biophys. Res. Commun., 345:926-932; M. Ueno et al. (2006), Proc. Natl. Acad. Sci. USA, 103:9554-9559;H. Suemori et al. (2001), Dev. Dyn., 222:273-279;H. Kawasaki et al. (2002), Proc. Natl. Acad. Sci. USA, 99:1580-1585;Klimanskaya I, et al. (2006), Nature. 444:481-485などに記載されている。
【0042】
ES細胞作製のための培養液として、例えば0.1mM 2-メルカプトエタノール、0.1mM 非必須アミノ酸、2mM L-グルタミン酸、20% KSRおよび4ng/ml bFGFを補充したDMEM/F-12培養液を使用し、37℃、2% CO2/98% 空気の湿潤雰囲気下でヒトES細胞を維持することができる(O. Fumitaka et al. (2008), Nat. Biotechnol., 26:215-224)。また、ES細胞は、3〜4日おきに継代する必要があり、このとき、継代は、例えば1mM CaCl2および20% KSRを含有するPBS中の0.25% トリプシンおよび0.1mg/mlコラゲナーゼIVを用いて行うことができる。
【0043】
ES細胞の選択は、一般に、アルカリホスファターゼ、Oct-3/4、Nanogなどの遺伝子マーカーの発現を指標にしてReal-Time PCR法で行うことができる。特に、ヒトES細胞の選択では、OCT-3/4、NANOG、ECADなどの遺伝子マーカーの発現を指標とすることができる(E. Kroon et al. (2008), Nat. Biotechnol., 26:443-452)。
【0044】
ヒトES細胞株は、例えばWA01(H1)およびWA09(H9)は、WiCell Reserch Instituteから、KhES-1、KhES-2およびKhES-3は、京都大学再生医科学研究所(京都、日本)から入手可能である。
【0045】
(B) 精子幹細胞
精子幹細胞は、精巣由来の多能性幹細胞であり、精子形成のための起源となる細胞である。この細胞は、ES細胞と同様に、種々の系列の細胞に分化誘導可能であり、例えばマウス胚盤胞に移植するとキメラマウスを作出できるなどの性質をもつ(M. Kanatsu-Shinohara et al. (2003) Biol. Reprod., 69:612-616; K. Shinohara et al. (2004), Cell, 119:1001-1012)。神経膠細胞系由来神経栄養因子(glialcell line-derived neurotrophic factor (GDNF))を含む培養液で自己複製可能であるし、またES細胞と同様の培養条件下で継代を繰り返すことによって、精子幹細胞を得ることができる(竹林正則ら(2008),実験医学,26巻,5号(増刊),41〜46頁,羊土社(東京、日本))。
【0046】
(C) 胚性生殖細胞
胚性生殖細胞は、胎生期の始原生殖細胞から樹立される、ES細胞と同様な多能性をもつ細胞であり、LIF、bFGF、幹細胞因子(stem cell factor)などの物質の存在下で始原生殖細胞を培養することによって樹立しうる(Y. Matsui et al. (1992), Cell, 70:841-847; J.L. Resnick et al. (1992), Nature, 359:550-551)。
【0047】
(D) 人工多能性幹細胞
人工多能性幹(iPS)細胞は、特定の初期化因子を、DNAまたはタンパク質の形態で体細胞に導入することによって作製することができる、ES細胞とほぼ同等の特性、例えば分化多能性と自己複製による増殖能、を有する体細胞由来の人工の幹細胞である(K. Takahashi and S. Yamanaka (2006) Cell, 126:663-676; K. Takahashi et al. (2007), Cell, 131:861-872; J. Yu et al. (2007), Science, 318:1917-1920; Nakagawa, M.ら,Nat. Biotechnol. 26:101-106 (2008);国際公開WO 2007/069666)。初期化因子は、ES細胞に特異的に発現している遺伝子、その遺伝子産物もしくはnon-cording RNAまたはES細胞の未分化維持に重要な役割を果たす遺伝子、その遺伝子産物もしくはnon-cording RNA、あるいは低分子化合物によって構成されてもよい。初期化因子に含まれる遺伝子として、例えば、Oct3/4、Sox2、Sox1、Sox3、Sox15、Sox17、Klf4、Klf2、c-Myc、N-Myc、L-Myc、Nanog、Lin28、Fbx15、ERas、ECAT15-2、Tcl1、beta-catenin、Lin28b、Sall1、Sall4、Esrrb、Nr5a2、Tbx3またはGlis1等が例示され、これらの初期化因子は、単独で用いても良く、組み合わせて用いても良い。初期化因子の組み合わせとしては、WO2007/069666、WO2008/118820、WO2009/007852、WO2009/032194、WO2009/058413、WO2009/057831、WO2009/075119、WO2009/079007、WO2009/091659、WO2009/101084、WO2009/101407、WO2009/102983、WO2009/114949、WO2009/117439、WO2009/126250、WO2009/126251、WO2009/126655、WO2009/157593、WO2010/009015、WO2010/033906、WO2010/033920、WO2010/042800、WO2010/050626、WO 2010/056831、WO2010/068955、WO2010/098419、WO2010/102267、WO 2010/111409、WO 2010/111422、WO2010/115050、WO2010/124290、WO2010/147395、WO2010/147612、Huangfu D, et al. (2008), Nat. Biotechnol., 26: 795-797、Shi Y, et al. (2008), Cell Stem Cell, 2: 525-528、Eminli S, et al. (2008), Stem Cells. 26:2467-2474、Huangfu D, et al. (2008), Nat Biotechnol. 26:1269-1275、Shi Y, et al. (2008), Cell Stem Cell, 3, 568-574、Zhao Y, et al. (2008), Cell Stem Cell, 3:475-479、Marson A, (2008), Cell Stem Cell, 3, 132-135、Feng B, et al. (2009), Nat Cell Biol. 11:197-203、R.L. Judson et al., (2009), Nat. Biotech., 27:459-461、Lyssiotis CA, et al. (2009), Proc Natl Acad Sci U S A. 106:8912-8917、Kim JB, et al. (2009), Nature. 461:649-643、Ichida JK, et al. (2009), Cell Stem Cell. 5:491-503、Heng JC, et al. (2010), Cell Stem Cell. 6:167-74、Han J, et al. (2010), Nature. 463:1096-100、Mali P, et al. (2010), Stem Cells. 28:713-720、Maekawa M, et al. (2011), Nature. 474:225-9.に記載の組み合わせが例示される。
【0048】
上記初期化因子には、ヒストンデアセチラーゼ(HDAC)阻害剤[例えば、バルプロ酸 (VPA)、トリコスタチンA、酪酸ナトリウム、MC 1293、M344等の低分子阻害剤、HDACに対するsiRNAおよびshRNA(例、HDAC1 siRNA SmartpoolTM (Millipore)、HuSH 29mer shRNA Constructs against HDAC1 (OriGene)等)等の核酸性発現阻害剤など]、MEK阻害剤(例えば、PD184352、PD98059、U0126、SL327およびPD0325901)、Glycogen synthase kinase-3阻害剤(例えば、BioおよびCHIR99021)、DNAメチルトランスフェラーゼ阻害剤(例えば、5-azacytidine)、ヒストンメチルトランスフェラーゼ阻害剤(例えば、BIX-01294 等の低分子阻害剤、Suv39hl、Suv39h2、SetDBlおよびG9aに対するsiRNAおよびshRNA等の核酸性発現阻害剤など)、L-channel calcium agonist (例えばBayk8644)、酪酸、TGFβ阻害剤またはALK5阻害剤(例えば、LY364947、SB431542、616453およびA-83-01)、p53阻害剤(例えばp53に対するsiRNAおよびshRNA)、ARID3A阻害剤(例えば、ARID3Aに対するsiRNAおよびshRNA)、miR-291-3p、miR-294、miR-295およびmir-302などのmiRNA、Wnt Signaling(例えばsoluble Wnt3a)、神経ペプチドY、プロスタグランジン類(例えば、プロスタグランジンE2およびプロスタグランジンJ2)、hTERT、SV40LT、UTF1、IRX6、GLISl、PITX2、DMRTBl等の樹立効率を高めることを目的として用いられる因子も含まれており、本明細書においては、これらの樹立効率の改善目的にて用いられた因子についても初期化因子と別段の区別をしないものとする。
【0049】
初期化因子は、タンパク質の形態の場合、例えばリポフェクション、細胞膜透過性ペプチド(例えば、HIV由来のTATおよびポリアルギニン)との融合、マイクロインジェクションなどの手法によって体細胞内に導入してもよい。
【0050】
一方、DNAの形態の場合、例えば、ウイルス、プラスミド、人工染色体などのベクター、リポフェクション、リポソーム、マイクロインジェクションなどの手法によって体細胞内に導入することができる。ウイルスベクターとしては、レトロウイルスベクター、レンチウイルスベクター(以上、Cell, 126, pp.663-676, 2006; Cell, 131, pp.861-872, 2007; Science, 318, pp.1917-1920, 2007)、アデノウイルスベクター(Science, 322, 945-949, 2008)、アデノ随伴ウイルスベクター、センダイウイルスベクター(WO 2010/008054)などが例示される。また、人工染色体ベクターとしては、例えばヒト人工染色体(HAC)、酵母人工染色体(YAC)、細菌もしくはファージ人工染色体(BACもしくはPAC)などが含まれる。プラスミドとしては、哺乳動物細胞用プラスミドを使用しうる(Science, 322:949-953, 2008)。ベクターには、核初期化物質が発現可能なように、プロモーター、エンハンサー、リボゾーム結合配列、ターミネーター、ポリアデニル化サイトなどの制御配列を含むことができるし、さらに、必要に応じて、薬剤耐性遺伝子(例えばカナマイシン耐性遺伝子、アンピシリン耐性遺伝子、ピューロマイシン耐性遺伝子など)、チミジンキナーゼ遺伝子、ジフテリアトキシン遺伝子などの選択マーカー配列、緑色蛍光タンパク質(GFP)、βグルクロニダーゼ(GUS)、FLAGなどのレポーター遺伝子配列などを含むことができる。また、上記ベクターには、体細胞への導入後、初期化因子をコードする遺伝子もしくはプロモーターとそれに結合する初期化因子をコードする遺伝子を共に切除するために、それらの前後にLoxP配列を有してもよい。
【0051】
また、RNAの形態の場合、例えばリポフェクション、マイクロインジェクションなどの手法によって体細胞内に導入しても良く、分解を抑制するため、5-メチルシチジンおよびpseudouridine (TriLink Biotechnologies)を取り込ませたRNAを用いても良い(Warren L, (2010) Cell Stem Cell. 7:618-630)。
【0052】
iPS細胞誘導のための培養液としては、例えば、10〜15%FBSを含有するDMEM、DMEM/F12またはDME培養液(これらの培養液にはさらに、LIF、penicillin/streptomycin、puromycin、L-グルタミン、非必須アミノ酸類、β-メルカプトエタノールなどを適宜含むことができる。)または市販の培養液[例えば、マウスES細胞培養用培養液(TX-WES培養液、トロンボX社)、霊長類ES細胞培養用培養液(霊長類ES/iPS細胞用培養液、リプロセル社)、無血清培地(mTeSR、Stemcell Technology社)]などが含まれる。
【0053】
培養法の例としては、たとえば、37℃、5%CO2存在下にて、10%FBS含有DMEMまたはDMEM/F12培養液上で体細胞と初期化因子とを接触させ約4〜7日間培養し、その後、細胞をフィーダー細胞(たとえば、マイトマイシンC処理STO細胞、SNL細胞等)上にまきなおし、体細胞と初期化因子の接触から約10日後からbFGF含有霊長類ES細胞培養用培養液で培養し、該接触から約30〜約45日またはそれ以上ののちにiPS様コロニーを生じさせることができる。
【0054】
あるいは、37℃、5% CO2存在下にて、フィーダー細胞(たとえば、マイトマイシンC処理STO細胞、SNL細胞等)上で10%FBS含有DMEM培養液(これにはさらに、LIF、ペニシリン/ストレプトマイシン、ピューロマイシン、L-グルタミン、非必須アミノ酸類、β-メルカプトエタノールなどを適宜含むことができる。)で培養し、約25〜約30日またはそれ以上ののちにES様コロニーを生じさせることができる。望ましくは、フィーダー細胞の代わりに、初期化される体細胞そのものを用いる(Takahashi K, et al. (2009), PLoS One. 4:e8067またはWO2010/137746)、もしくは細胞外基質(例えば、Laminin-5(WO2009/123349)およびマトリゲル(BD社))を用いる方法が例示される。
この他にも、血清を含有しない培地を用いて培養する方法も例示される(Sun N, et al. (2009), Proc Natl Acad Sci U S A. 106:15720-15725)。さらに、樹立効率を上げるため、低酸素条件(0.1%以上、15%以下の酸素濃度)によりiPS細胞を樹立しても良い(Yoshida Y, et al. (2009), Cell Stem Cell. 5:237-241またはWO2010/013845)。
【0055】
上記培養の間には、培養開始2日目以降から毎日1回新鮮な培養液と培養液交換を行う。また、核初期化に使用する体細胞の細胞数は、限定されないが、培養ディッシュ100cm2あたり約5×103〜約5×106細胞の範囲である。
【0056】
iPS細胞は、形成したコロニーの形状により選択することが可能である。一方、体細胞が初期化された場合に発現する遺伝子(例えば、Oct3/4、Nanog)と連動して発現する薬剤耐性遺伝子をマーカー遺伝子として導入した場合は、対応する薬剤を含む培養液(選択培養液)で培養を行うことにより樹立したiPS細胞を選択することができる。また、マーカー遺伝子が蛍光タンパク質遺伝子の場合は蛍光顕微鏡で観察することによって、発光酵素遺伝子の場合は発光基質を加えることによって、また発色酵素遺伝子の場合は発色基質を加えることによって、iPS細胞を選択することができる。
【0057】
本明細書中で使用する「体細胞」なる用語は、卵子、卵母細胞などの生殖系列細胞または分化全能性細胞、あるいはES細胞、を除くあらゆる動物細胞(好ましくは、ヒトを含む哺乳動物細胞)をいう。体細胞には、非限定的に、胎児(仔)の体細胞、新生児(仔)の体細胞、および成熟した健全なもしくは疾患性の体細胞のいずれも包含されるし、また、初代培養細胞、継代細胞、および株化細胞のいずれも包含される。具体的には、体細胞は、例えば(1)神経幹細胞、造血幹細胞、間葉系幹細胞、歯髄幹細胞等の組織幹細胞(体性幹細胞)、(2)組織前駆細胞、(3)リンパ球、上皮細胞、内皮細胞、筋肉細胞、線維芽細胞(皮膚細胞等)、毛細胞、肝細胞、胃粘膜細胞、腸細胞、脾細胞、膵細胞(膵外分泌細胞等)、脳細胞、肺細胞、腎細胞および脂肪細胞等の分化した細胞などが例示される。
【0058】
また、iPS細胞を移植用細胞の材料として用いる場合、拒絶反応が起こらないという観点から、移植先の個体のHLA遺伝子型が同一もしくは実質的に同一である体細胞を用いることが望ましい。ここで、「実質的に同一」とは、移植した細胞に対して免疫抑制剤により免疫反応が抑制できる程度にHLA遺伝子型が一致していることであり、例えば、HLA-A、HLA-BおよびHLA-DRの3遺伝子座あるいはHLA-Cを加えた4遺伝子座が一致するHLA型を有する体細胞である。
【0059】
(E) 核移植により得られたクローン胚由来のES細胞
nt ES細胞は、核移植技術によって作製されたクローン胚由来のES細胞であり、受精卵由来のES細胞とほぼ同じ特性を有している(T. Wakayama et al. (2001), Science, 292:740-743; S. Wakayama et al. (2005), Biol. Reprod., 72:932-936; J. Byrne et al. (2007), Nature, 450:497-502)。すなわち、未受精卵の核を体細胞の核と置換することによって得られたクローン胚由来の胚盤胞の内部細胞塊から樹立されたES細胞がnt ES(nuclear transfer ES)細胞である。nt ES細胞の作製のためには、核移植技術(J.B. Cibelli et al. (1998), Nature Biotechnol., 16:642-646)とES細胞作製技術(上記)との組み合わせが利用される(若山清香ら(2008),実験医学,26巻,5号(増刊), 47〜52頁)。核移植においては、哺乳動物の除核した未受精卵に、体細胞の核を注入し、数時間培養することで初期化することができる。
【0060】
(F) Multilineage-differentiating Stress Enduring cells(Muse細胞)
Muse細胞は、WO2011/007900に記載された方法にて製造された多能性幹細胞であり、詳細には、線維芽細胞または骨髄間質細胞を長時間トリプシン処理、好ましくは8時間または16時間トリプシン処理した後、浮遊培養することで得られる多能性を有した細胞であり、SSEA-3およびCD105が陽性である。
【実施例】
【0061】
以下に、本発明の実施例と比較例について説明するが、本実施例は本発明の再現を補助する目的でその一実施態様を示すものであって、本実施例は本発明の範囲を制限するものではない。
【0062】
<組換えヒトBACクローンの作製>
OSR1遺伝子座から86.3kb上流および89.8kb下流を含むヒトBACクローン(RP11-458J18)は、BACPAC Resources Center (Oakland, CA)より購入した。Lee, E.C. et al. (2001) Genomics 73, 56-65に記載の方法で組換えを行った。簡潔には、hOSR1-EGFP-S: TCTTCTTTTCTTTGCAGATCCGGATTGAGAAGCCACTGCAACTACCGAACACCATGGTGAGCAAGGGCGAGGA (配列番号1)およびhOSR1-PNL-AS: GTTCACTGCCTGAAGGAAGGAGTAGTTGGTGAGCTGCAGGGAAGGGTGGAGTCGACGGCGAGCTCAGACG(配列番号2) のプライマーを用いてPCRにより5’および3’側にホモロジーアームを有するEGFP-polyA-LoxP-PGK-Neo-LoxP(EGFP-pA-PNL)カセットを作製した。このカセットおよびヒトBACクローンを大腸菌DH10Bへ導入し、相同組換えのためにリコンビナーゼを活性化させ、ヒトBACクローン中のOSR1の開始コドン直後の配列をEGFP-pA-PNLカセットを挿入した(図1中のrecomninant BAC clone)。
【0063】
<相同組換えiPS細胞の作製>
上述した方法で作製した組換えヒトBACクローンを制限酵素で切断し、一本鎖DNAとした。30μg一本鎖DNAをY27632(Wako)およびトリプシンで処理したヒトiPS細胞(201B7)にエレクトロポレーション(250 V, 500 mF単回パルス)により導入した。導入2時間後、iPS細胞を薬剤選択用培地中で培養した。
【0064】
<PCR分析>
得られた130個の薬剤耐性クローンiPS細胞から染色体DNAを抽出し、OSR1F(5’-GGATTGAGAAGCCACTGCAACT-3’(配列番号3))およびOSR1R(5’-CCGTTCACTGCCTGAAGGA-3’(配列番号4))のプライマーを用いて定量PCRを行った(図2A)。この結果、3D36、3D45、3F3および3I49の4つクローンにおいてOSR1の開始コドン付近が欠損していることが確認された。
【0065】
<SNPアレイ分析>
染色体DNAを抽出し、GeneChipTM Mapping 250K NSP arrays (Affymetrix)を用いて得られたデータをソフトウェア(CNAG/AsCNAR)により対立遺伝子特異的コピー数解析を3D36、3D45、3F3、3I49および3D12に対して行った。GeneChipに含有された各プローブの検出値からCNAG/AsCNAR 解析により得られたプローブ領域のコピー数を縦軸に、該プローブの遺伝子上での位置を横軸にとりプロットした(図2B)。その結果、3D36、3D45、3F3および3I49は、OSR1の遺伝子座の領域が2コピー含有することが示された。このことは、相同組換えにより、EGFP-pA-PNLカセットが挿入されたことを示している。一方、3D12 は、OSR1領域が3コピー含有することが示された。このことは、組換えヒトBACクローンが相同組換えではなく、染色体にランダムに組み込まれたことを示唆している。以上より、相同組換えによりEGFP-pA-PNLカセットが所望の位置にノックインされたヒトiPS細胞(3D36、3D45、3F3および3I49)が得られた。また、解析領域を拡大して調べたところ、3I49には第9番染色体にコピー数多型を有している事が確認された。これより、培養中または遺伝子改変の際に、3I49は第9染色体において異常な遺伝子の重複が起きたと考えられる。
【0066】
<核型解析>
Gバンド解析により核型解析を行ったところ、3D45は正常の核型を有していることが確認された。
【0067】
<Creリコンビナーゼ処理>
OSR1がタゲーゲティングされたヒトiPS細胞(3D36、3D45、3F3および3I49)を10μM Y27,632添加培地で1日培養した後、トリプシン処理により分離させ、エレクトロポレーションによりCre発現ベクターを導入した。導入後、loxP配列を挟むようにGFP配列部位およびOSR1遺伝子の開始コドン以降に設計された図1に記載のプライマーで染色体DNAをPCRしたところ、全てのクローンにおいてPGK-Neo-pAが除外されたことが確認された。
【産業上の利用可能性】
【0068】
本発明によれば、人工染色体をターゲティングベクターとして用いることにより高効率で相同組換えが起こった多能性幹細胞を選択的に製造することが可能となる。また、SNPアレイを用いることにより簡便に相同組換えされた多能性幹細胞を検出することが可能となる。したがって、本発明を用いて相同組換えされた多能性幹細胞を製造することで、簡便に治療剤または分化誘導剤のスクリーニングすることもしくは治療用の細胞の確保が可能となる。
【技術分野】
【0001】
本発明は、相同組換えにより遺伝子改変された多能性幹細胞の検出方法、ならびに、該検出方法を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞を製造する方法に関する。
【背景技術】
【0002】
内在性ゲノムDNAの一部を改変(欠失、挿入または置換)し、生物に生来備わっていない生物学的性状を付与したり、生物が生来備えている生物学的性状の発現を抑制したり、さらには該生物のある遺伝子が担う機能を解析するための方法として、該生物の内在性ゲノムDNAの所望の部位を人為的に改変する遺伝子ターゲティングが行われている(特許文献1および特許文献2)。
【0003】
遺伝子ターゲティングは、内因性遺伝子をノックアウトする(非特許文献1および非特許文献2)か、または染色体に外因性配列をノックインするために用いられている。しかし、この方法は、非常に効率が低いため(トランスフェクションされた細胞の10-6〜10-9)、多大な労力が必要である。
【0004】
そこで、遺伝子ターゲティングの効率が1,000倍以上に増大することより(非特許文献3、非特許文献4、非特許文献5、非特許文献6、非特許文献7および非特許文献8)、メガヌクレアーゼにより所望の部位を切断することで効率を上げる工夫がされている。
【0005】
しかし、いずれのクローンにおいて相同組換えが行われたか否かについては確認する必要があるため、多数の候補の中から所望の改変がなされたクローンを効率よく検出する方法が求められている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】WO1990/11354
【特許文献2】WO1991/09955
【非特許文献】
【0007】
【非特許文献1】Capecchi, M.R., Science, (1989) 244: 1288-1292
【非特許文献2】Smithies, O., Nature Medicine, (2001) 7: 1083-1086
【非特許文献3】Puchta H, et al., Nucleic Acids Res., (1993) 21: 5034-5040
【非特許文献4】Choulika A, et al., Mol. Cell. Biol., (1995) 15: 1968-1973
【非特許文献5】Puchta H, et al., Proc. Natl. Acad. Sci. U.S.A., (1996) 93: 5055-5060
【非特許文献6】Sargent RG, et al., Mol. Cell. Biol., (1997) 17: 267-277
【非特許文献7】Cohen-Tannoudji M, et al., Mol. Cell. Biol., (1998) 18: 1444-1448
【非特許文献8】Donoho G, ste al.,, Mol. Cell. Biol., (1998) 18: 4070-4078
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は、相同組換えにより遺伝子改変された多能性幹細胞の作製において、ゲノム上の所望の部位もしくは領域で相同組換えが起きたか否かを検出することを含む方法を提供することを目的とする。
【0009】
具体的には、本発明の目的は、遺伝子ターゲティングにより所望の遺伝子座を改変させた多能性幹細胞を検出することである。すなわち、本発明の目的は、ゲノム上の所望の部位で相同組換えが起きている多能性幹細胞を、ランダムインテグレーションが起きている多能性幹細胞から区別する方法を提供することである。
【課題を解決するための手段】
【0010】
本発明者らは、上記の課題を解決すべく、SNP(一塩基多型)アレイ法を用いたコピー数多型の検出方法に着目し、ターゲティングベクターとしての遺伝子改変された染色体断片を有する人工染色体ベクターが導入された多能性幹細胞中の該染色体断片に相当する領域のコピー数をSNPアレイ法により測定することにより、相同組換えが起こっているか否かを測定することができることを見出した。さらに、相同組換えが起きているか否かの確証を補助的に得るためにさらに、組込まれた選択マーカーを指標とする方法、および/または、改変部位での野生型の配列を有するアレル数を測定する方法を組み合わせることで、本発明を完成するに至った。
【0011】
すなわち、本発明は以下の通りである。
[1]次の工程(1)〜(3):
(1)多能性幹細胞に、遺伝子改変された染色体断片を有する人工染色体を導入し、遺伝子改変されたと推定される多能性幹細胞クローンからなる集団を作製する工程、
(2)前記集団の多能性幹細胞クローンについて、SNPアレイを用いて、前記導入された人工染色体の一部または全体のコピー数を測定する工程、
(3)前記コピー数が、人工染色体が導入されていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞クローンを、相同組換えにより遺伝子改変された多能性幹細胞として選択する工程、
を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の製造方法。
[2]前記工程(1)の遺伝子改変が内在性配列を保持したまま細胞ゲノムに外因性DNA断片を挿入することからなる組込みを含む、[1]に記載の方法。
[3]外因性DNAが選択マーカーをコードするDNAであって、該選択マーカーが陽性であるクローンを選択する工程をさらに含む、[2]に記載の方法。
[4]多能性幹細胞クローンの該遺伝子改変対象であるアレルの数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下しているクローンを選択する工程をさらに含む、[1]〜[3]のいずれかに記載の方法。
[5]前記多能性幹細胞がヒト多能性幹細胞である、[1]〜[4]のいずれかに記載の方法。
[6]前記人工染色体がBACクローンである、[1]〜[5]のいずれかに記載の方法。
[7]前記選択マーカーが薬剤耐性マーカーである、[3]に記載の方法。
[8]相同組換えにより遺伝子改変された多能性幹細胞の製造において、SNPアレイを用いて組換え領域のコピー数を測定し、該コピー数が、相同組換えをしていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞クローンを、相同組換えにより遺伝子改変された多能性幹細胞として検出する工程を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の検出方法。
[9]前記遺伝子改変が、内在性配列を保持したまま細胞ゲノムの外因性DNA断片を挿入することからなる組込みである、[8]に記載の方法。
[10]前記外因性DNAが選択マーカーをコードするDNAあって、該選択マーカーが陽性であるクローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する工程をさらに含む、[9]に記載の方法。
[11]多能性幹細胞クローンの該遺伝子改変対象であるアレルの数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下しているクローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する工程をさらに含む、[8]〜[10]のいずれかに記載の方法。
[12]前記多能性幹細胞がヒト多能性幹細胞である、[8]〜[11]のいずれかに記載の方法。
[13]前記選択マーカーが薬剤耐性マーカーである、[10]に記載の方法。
【発明の効果】
【0012】
本発明を用いることによって、相同組換えによる多能性幹細胞の遺伝子改変を、簡便に効率的な手順で検出することができる。本発明の方法を遺伝子ターゲティングの高効率の手法と組み合わせることができるため、本発明の方法は、多能性幹細胞の遺伝子改変の全体的な効率化に寄与する。
【図面の簡単な説明】
【0013】
【図1】ヒトOSR1(odd-skipped related 1; M. Katoh, Int. J. Mol. Med. 2002; 10(2):221-225)遺伝子座にCre-loxPシステムを用いてGFP-PGK-Neoカセットを挿入するスキームを示す。
【図2】(A)親株または改変BACクローン導入後の薬剤耐性クローンの染色体におけるOSR1の開始コドンを挟んだ領域の野生型の存在比を定量化したグラフを示す。(B)SNPアレイを用いてiPS細胞株(3D36, 3D45, 3F3, 3l49, 3D12)の各々の第2染色体のOSR1遺伝子座付近の各プローブのコピー数を解析した結果を示す。
【発明を実施するための形態】
【0014】
本発明を以下に詳細に説明する。
本発明は、次の工程(1)〜(3):
(1)多能性幹細胞に、遺伝子改変された染色体断片を有する人工染色体を導入し、遺伝子改変されたと推定される多能性幹細胞クローンからなる集団を作製する工程、
(2)前記集団の多能性幹細胞クローンについて、SNPアレイを用いて、前記導入された人工染色体の一部または全体のコピー数を測定する工程、
(3)前記コピー数が、人工染色体が導入されていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞クローンを、相同組換えにより遺伝子改変された多能性幹細胞として選択する工程、
を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の製造方法を提供する。
【0015】
本明細書で使用する用語「相同組換え」とは、人工的に染色体もしくはゲノム上の特定遺伝子を改変する遺伝子ターゲッテイング手段であり、染色体上の目的の配列と相同的な部分をもつゲノム断片を細胞中に導入した場合、導入したゲノム断片と染色体上の対応する遺伝子座の間で塩基配列の相同性に基づいて起こる組換えである。
【0016】
また、「遺伝子改変」とは、染色体上の所望の遺伝子の遺伝子座において、外因性DNAを挿入するか、外因性DNAで遺伝子の一部もしくは全部を置換するか、あるいは遺伝子を欠損(欠失)させることを意味する。より具体的にいえば、遺伝子改変は、内在性DNA配列を保持したまま特定の遺伝子座の遺伝子の発現と連動するようにまたは恒常的に発現するように外因性DNA断片を挿入(言い換えれば、ノックイン)するか、あるいは、内在性DNA配列を改変するように該遺伝子の配列の全部もしくは部分を置換、欠損または破壊(言い換えれば、ノックアウト)することである。また、本発明において、対象となる多能性幹細胞が特定の遺伝子に変異を有する場合において、該遺伝子を正常の遺伝子配列と置換する組換えを行うための遺伝子改変は、正常の遺伝子へ改変することを意味する。
【0017】
また、改変の対象となる遺伝子は、目的によって適宜選択されてよく、例えば、疾患の原因遺伝子、細胞タイプの指標となるマーカー遺伝子、発現が減少しないハウスキーピング遺伝子などが挙げられるが、これらに限定されない。ここで、細胞タイプとは、例えば、内胚葉細胞、外胚葉細胞、中胚葉細胞、脊索中胚葉、沿軸中胚葉、中間中胚葉細胞、側板中胚葉、神経細胞、グリア細胞、造血細胞、肝細胞、膵β細胞、腎前駆細胞、内皮細胞、周皮細胞、上皮細胞、骨芽細胞、筋芽細胞、軟骨細胞など細胞が挙げられる。これらの細胞タイプの指標となるマーカー遺伝子として、GATA4、GATA5、GATA6、AFP、HNF-3β、SOX17、FOXA2、PDGFRα、FLK1、Brahcyury、Gremlin, MYH2、Nestin, SOX13, SOX21, CryM、Otx2、TP63、SOX2、PSA-NCAM、TuJ1、Thy1.2、GFAP、PAX6、A2B5、CD11b、c-kit、CD34、CD90、CD117、Albumin、CK18、CK19、PDX1、OSR1、SIX2、GATA2、VEGFR2、NG2、desmin、MUC1、BGLAP、SPP1、MyoD、MYF5、Myogenin、Aggrecan、Collagen IIおよびSox9が例示されるが、これらに限定されない。遺伝子の配列は、NCBI GenBank(hhtp://www.ncbi.nlm.nih.gov)、EMBL、DDBJなどの公知のDNAデータベースから入手できる。
【0018】
<人工染色体の導入方法>
本発明において、人工染色体としては、複製起点、セントロメアおよびテロメアなどの宿主細胞において複製に必要な機能を有する人工的に作製された染色体であり、大腸菌由来のBACベクター(Shizuya et al.(1992)Proc.Natl.Acad.Sci.U.S.A. 89:8794-8797)、P1ファージ由来のPACベクター(Ioannou et al.(1994)Nature Genetics 6:84-89;Pierce et al.(1992)Meth.Enzymol.216:549-574;Pierce et al.(1992)Proc.Natl.Acad.Sci.U.S.A.89:2056-2060;米国特許5300431号および国際PCT出願WO92/14819号)、酵母由来のYACベクター(Burke et al.(1987)Science 236:806-812)、およびヒト由来のHACベクター(WO1998/008964)などが例示される。これらの人工染色体は、外因性DNAを含む巨大なサイズのゲノム断片を保有したまま宿主細胞内で増殖することが可能である。人工染色体は、好ましくは、染色体断片を有する人工染色体、より好ましくは、ヒト染色体の断片を有する人工染色体である。DNA断片の大きさは、人工染色体が安定的に宿主内で複製が可能な大きさであり、BACまたはPACの場合は、通常、約300kb以内であり、YACの場合は1Mb以内であり、HACの場合は1Mb以上でも可能である。
【0019】
本発明の方法では、人工染色体として、導入すべきゲノム断片のサイズに応じて上記例示の人工染色体いずれかを使用することが可能であり、好ましい実施形態では人工染色体はBACベクターであり、さらに具体的には、ヒト染色体断片を保有するBACベクターとして、例えば、ローズウェル癌研究所(Roswell Park Cancer Institute、米国)が作製した平均175kbのヒトゲノム断片を有する437,000クローンのライブラリーであるRP-11が例示される。このような、ライブラリーに含まれる1つのゲノム断片を有するBACベクターをBACクローンと称する。
【0020】
染色体断片を有する人工染色体を作製するためには、当業者に周知の方法を用いて行う事が可能であり、例えば、制限酵素を用いて染色体断片中の所望の位置において切断し、適宜機能的配列を有するDNA断片を連結させる方法、ファージ由来のRed遺伝子を用いて大腸菌内で相同組換えを行う方法などが挙げられる。Red遺伝子を発現するプラスミドを有するキットは、Gene Bridges 社から購入可能であり、添付のプロトコールに従って人工染色体の改変が可能である。
【0021】
遺伝子の改変を目的として人工染色体へ挿入する外因性DNAとして、特に限定されないが、有用な(ヒトもしくは非ヒト動物由来)遺伝子類、選択マーカー遺伝子、または、これらの組み合わせ、等が例示される。外因性DNAを組み込むためのカセットにはさらに、プロモーター、IRES、部位特異的リコンビナーゼの認識配列、ターミネーターなどを含有させることができる。
【0022】
外因性DNAは、例えば、生物学的に、医学的に、あるいはクローンを選別するために、有用な機能をもつDNAであることが好ましい。
【0023】
本明細書中で使用する「有用な(ヒトもしくは非ヒト動物由来)遺伝子類」としては、例えば、機能不明の遺伝子類、疾患の原因遺伝子類、治療に有用な遺伝子類などの、医学的な研究または実用レベルで有用な遺伝子類、細胞タイプの指標となるマーカー遺伝子類、発現が減少しないハウスキーピング遺伝子類などが挙げられるが、これらに限定されない。
【0024】
本明細書で使用する用語「選択マーカー遺伝子」とは、宿主細胞を選択する指標として機能する遺伝子をいう。選択マーカーとしては、公知のポジティブマーカーまたはネガティブマーカーのいずれも使用できる。ポジティブマーカー選択マーカーとしては、蛍光マーカー、発光マーカー、および薬剤耐性マーカーが挙げられるが、これらに限定されない。「蛍光マーカー」としては、緑色蛍光プロテイン(GFP)、青色蛍光プロテイン(CFP)、黄色蛍光プロテイン(YFP)および赤色蛍光プロテイン(dsRed)のような蛍光タンパク質をコードする遺伝子が挙げられるが、これらに限定されない。「発光マーカー」としては、ルシフェラーゼのような発光タンパク質をコードする遺伝子が挙げられるが、これらに限定されない。「薬剤耐性マーカー」としてはネオマイシン(G418)耐性遺伝子との融合遺伝子(β-geo遺伝子)、CAT遺伝子、GFP遺伝子、SV40ラージT遺伝子、ネオマイシン耐性遺伝子、ピューロマイシン耐性遺伝子、ハイグロマイシン耐性遺伝子、ブラストサイジンS耐性遺伝子等をコードする遺伝子が挙げられるが、これらに限定されない。
【0025】
また、「部位特異的リコンビナーゼ」として、Creリコンビナーゼ(Gorman C, Bullock C. Curr Opin Biotechnol. (2000), 11: 455-60)、FLPリコンビナーゼ(Buchholz F, et al., Nat Biotechnol. (1998), 16: 657-662)などが例示され、これらのリコンビナーゼの標的配列としてloxP、FRTなどが例示される。2つの認識配列で挟まれた領域を削除する目的から、該認識配列は所望の領域を挟んで2ヶ所へ挿入することが望ましい。
【0026】
ターミネーターは、転写終結配列としてのポリアデニル化シグナルである。特に限定されないが、ポリアデニル化シグナル配列として、ヒトBGHポリA、SV40ポリA、ヒトβアクチンポリA、ウサギβグロブリンポリAおよび免疫グロブリンκポリAが例示される。
【0027】
上記の外因性DNAは、挿入、置換または欠損(欠失)等の目的に合わせて適宜、機能しうるように該人工染色体へ配置されてもよく、例えば、改変対象の遺伝子の置換および欠損を目的として、選択マーカーおよびターミネーターを対象遺伝子の開始コドンの後方の遺伝子配列内に連結し配置させてもよい。あるいは、選択マーカーは、改変対象の遺伝子の発現と連動して発現させるために、内在性のプロモーターと連結して配置されていてもよい。このため、選択マーカーは、口蹄疫ウイルスの2A self-cleaving peptide(Science, 322, 949-953, 2008など参照)、IRES配列などと共に連結して配置されていてもよい。さらに、挿入される選択マーカーは、人工染色体が細胞内に導入されたことを確認とするため、恒常的に発現させることを目的として外因性のプロモーターに予め連結されていてもよい。プロモーターの例としては、PGK プロモーター、EF-αプロモーター、CAGプロモーター、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMV(サイトメガロウイルス)プロモーター、RSV(ラウス肉腫ウイルス)プロモーター、MoMuLV(モロニーマウス白血病ウイルス)LTR、HSV-TK(単純ヘルペスウイルスチミジンキナーゼ)プロモーターなどが用いられる。
【0028】
人工染色体を細胞へ導入する方法として、例えば、リン酸カルシウム沈殿法(Graham et al.(1978)Virology 52:456-457、Wigler et al.(1979)Proc.Natl.Acad.Sci.U.S.A.76 1373-1376およびCurrent Protocols in Molecular Biology Vol.1,Wiley Inter-Science,Supplement 14,Unit 9.1.1-9.1.9(1990))、ポリエチレングリコールを用いた融合方法(米国特許4684611号)、リポフェクションなどの脂質キャリアを用いる方法(Teifel et al.(1995)Biotechniques 19:79-80、Albrecht et al.(1996)Ann.Hematol.72:73-79;Holmen et al.(1995)In Vitro Cell Dev.Biol.Anim.31:347-351、Remy et al.(1994)Bioconjug.Chem. 5:647-654、Le Bolc'het al.(1995)Tetrahedron Lett.36:6681-6684、Loeffler et al.(1993)Meth.Enzymol.217:599-618およびStrauss(1996)Meth.Mol.Biol.54: 307-327)、エレクトロポレーションおよび微小核体との融合法(米国特許5240840号、4806476号、5298429号、5396767号、Fournier(1981)Proc.Natl.Acad.Sci.U.S.A. 78:6349-6353およびLambert et al(1991)Proc. Natl. Acad. Sci. U.S.A. 88:5907-59)などがある。
【0029】
上記の手法によって、遺伝子改変されたと推定される多能性幹細胞クローンからなる集団が作製される。ここで、「遺伝子改変されたと推定される」とは、多能性幹細胞クローンのほとんどが遺伝子改変されているが、一部には遺伝子改変されていない(すなわち野生型)クローンが混入しうることを意味する。該クローンには、遺伝子改変がゲノム上の所望の部位もしくは領域で起こった(すなわち相同組換えされた)クローンだけでなく、遺伝子改変がゲノム上でランダムに起こった(すなわちランダムインテグレーションされた)クローンなどが含まれる。本発明では、遺伝子改変されたと推定される多能性幹細胞クローンからなるこのような集団から、以下で説明するSNPアレイを利用することによって、相同組換えにより遺伝子改変されたクローンが選別される。これまで相同組換えされた多能性幹細胞の検出のために、SNPアレイを使用したコピー数測定法を利用することが全く行われていなかったが、本発明では、このような手法を利用することで相同組換えされた目的細胞クローンを容易に識別できる。
【0030】
<SNPアレイ>
本発明において、SNP(一塩基多型)アレイとは、アレル特異的オリゴヌクレオチドプローブを用いるSNPタイピング用のアレイであり、好ましくは、アレイシグナルの定量的特性を利用して、ゲノムコピー数を検出できるものである。好ましくは、コピー数多型を測定するためのオリゴヌクレオチドプローブを有し、2.5kb〜7kb の平均プローブ間隔であるアレイである。例えば、アフィメトリクス(Affymetrix)SNPアレイ 6.0、イルミナ CNV370-DuoおよびBead Chipsなどが例示される。
【0031】
本発明の方法において、SNPアレイを用いてコピー数を測定するために、サンプルとして抽出した染色体DNAを例えばHindIII、XbaI、NspIまたはStyI等の制限酵素で処理した後、アダプターを5’末端および3’末端に付加し、アダプター特異的なプライマーを用いてPCRにより増幅させてもよい。該増幅は、0.5kb〜1.5kb の短い制限酵素断片のみが選択的に増幅されることが望ましい。さらに、増幅されたDNA断片をDNaseIによりさらに細かく断片化したDNA断片群を、オリゴヌクレオチドプローブを有するアレイとハイブリダイゼーションさせ、当該断片の量をシグナル強度によって検出することができる。得られたシグナル強度のデータは、CNAG(copy number analyserfor gene chip)ソフトウェア(http://www.genome.umin.jp/)を用いてコピー数として算出することができる。
【0032】
より正確なコピー数を検出するため、アレル特異的にシグナルを検出することが望ましい。そのため、対照として相同組換え前の多能性幹細胞または人工染色体を導入して得られた他のクローンを用いて同様にSNPアレイを用いてシグナル強度を測定し、AsCNAR(allele-specific copy-number analysis using anonymous references)(http://www.genome.umin.jp/)を用いて解析することで、アレル別のシグナル強度を得ることができる。アレルによって同一プローブに対する親和性が異なる場合において正確なコピー数が算出できないことを、アレル特異的にシグナルを測定することで改善することができる。
【0033】
本発明で使用可能なアレイは、プローブとなるオリゴヌクレオチドが、固相担体(基板)に固定化されたマイクロアレイの形態で提供されることが好ましい。マイクロアレイの固相担体としては、例えば、ガラス基板、シリコン基板、メンブレン、ビーズなどが挙げられ、その材質、大きさ、形状は特に限定されない。マイクロアレイの形成方法は特に限定されず、当業者が利用可能ないかなる方法を用いてもよく、例えば、固相担体表面で直接プローブを合成する方法(オン・チップ法)、または予め調製したプローブを固相担体表面に結合する方法などがある。固相担体表面で直接プローブを合成する場合には、光照射で選択的に除去される保護基を用い、半導体製造に利用されるフォトリソグラフィー技術および固相合成技術を組み合わせて所定の微少なマトリックス領域でのオリゴヌクレオチドの選択的な合成を行う方法が一般的である。一方、予めプローブを調製して固相担体表面に結合する方法では、プローブ核酸の種類や固相担体の種類に応じて、スポッタ装置によりポリ陽イオン化合物やアミノ基、アルデヒド基、エポキシ基等を有するシランカップリング剤などで表面処理した固相担体の表面に点着する方法、反応活性基を導入したプローブを合成し、予め反応性基を形成させるように表面処理した固相担体表面に該プローブを点着して該プローブを固相担体表面に共有結合により結合固定させる方法などが利用できる。
【0034】
遺伝子改変された染色体断片を有する人工染色体が導入された多能性幹細胞から得られた染色体(ゲノム)中の該染色体断片のコピー数を測定することによって、該細胞ゲノムにおける該人工染色体に搭載された染色体断片のコピー数を確認することができる。この時、相同組換えにより多能性幹細胞の染色体に組換えられた場合、該多能性幹細胞中の染色体断片のコピー数に関して、該人工染色体の導入前と同等(言い換えれば、同数)のコピー数であるか、または該人工染色体の導入前より減少したコピー数である。内在性DNA配列を保持したまま細胞ゲノム中に外因性DNA断片が挿入(すなわち、ノックイン)されたときには、前記導入前と同等のコピー数となり、また、内在性DNA配列を改変するように該遺伝子の配列の全部もしくは部分が置換、欠損または破壊(すなわち、ノックアウト)されたときには、前記導入前より減少したコピー数となる。一方、ランダムインテグレーションにより、染色体断片がランダムに(言い換えれば、無作為に)細胞ゲノムに組み込まれた場合、前記導入前より増加したコピー数、例えば3コピーまたはそれ以上のコピー数を呈する。したがって、該方法でコピー数を測定することで相同組換え、具体的にはノックイン型またはノックアウト型の相同組換え、の有無の判断が可能である。
【0035】
<選択マーカーによる補助的選別>
本発明では、上記のとおりSNPアレイを用いてコピー数を測定することで相同組換えの有無を判定可能であるが、補助的に選択マーカーを利用することも可能である。
【0036】
すなわち、本発明において、選択マーカーを有する人工染色体を細胞へ導入後、選択マーカーを利用することによって、人工染色体が染色体(もしくは、ゲノム)へ組み込まれた細胞を選別することができる。ここで、薬剤選択マーカーを用いる場合、細胞培養液へ対応する薬剤を添加することで、相同組換えまたはランダムインテグレーションにより人工染色体が染色体(もしくは、ゲノム)へ組み込まれた細胞を選択マーカーが陽性である細胞として選択的に取り出すことができる。
【0037】
<アレル数測定による補助的選別>
さらに本発明では、アレル数測定による補助的選別法を行うことができる。この方法では、遺伝子改変された染色体断片を有する人工染色体を多能性幹細胞に導入後、該細胞の染色体(もしくは、ゲノム)において、遺伝子改変されていない野生型の配列を有するアレルが1つ確認できる場合、または野生型の配列を有するアレルを確認できない場合、該細胞を相同組換えにより遺伝子改変されたクローンとして選別することができる。好適には、多能性幹細胞クローンの遺伝子改変対象であるアレル数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下している、好ましくは1/2である、クローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する。常染色体上の野生型の遺伝子は通常2アレルであるが、一方の遺伝子に対して改変を行うことで野生型の遺伝子は1アレルになる。従って、野生型細胞の半数になっている場合、目的の遺伝子座において改変が行われていることが確認できる。ここで、野生型の配列を有するアレル数を測定する方法として、組換えを施した多能性幹細胞から抽出した染色体DNAを基に遺伝子を改変させた部位を含む領域をPCR法により増幅し、野生型の配列を有する場合、該配列に特徴的な大きさのPCR産物の量を測定し、野生型細胞としての遺伝子改変されていない多能性幹細胞と比較する方法が例示される。他の実施形態として、遺伝子改変された多能性幹細胞から抽出した染色体を任意の制限酵素により切断し、遺伝子が改変されることで生じる特異的な大きさのDNA断片をサザンブロット法により測定する方法が例示される。
【0038】
本発明の方法で遺伝子改変されうる多能性幹細胞について、以下で具体的に説明する。
<多能性幹細胞>
多能性幹細胞を分化させることによってドーパミン産生神経前駆細胞を含有する細胞集団を用意する場合、使用可能な多能性幹細胞は、生体に存在するすべての細胞に分化可能である多能性を有し、かつ、増殖能をも併せもつ幹細胞であり、それには、特に限定されないが、例えば胚性幹(ES)細胞、核移植により得られるクローン胚由来の胚性幹(ntES)細胞、精子幹細胞(「GS細胞」)、胚性生殖細胞(「EG細胞」)、人工多能性幹(iPS)細胞、培養線維芽細胞や骨髄幹細胞由来の多能性細胞(Muse細胞)などが含まれる。好ましい多能性幹細胞は、ES細胞、ntES細胞、およびiPS細胞である。
【0039】
(A) 胚性幹細胞
ES細胞は、ヒトやマウスなどの哺乳動物の初期胚(例えば胚盤胞)の内部細胞塊から樹立された、多能性と自己複製による増殖能を有する幹細胞である。
【0040】
ES細胞は、受精卵の8細胞期、桑実胚後の胚である胚盤胞の内部細胞塊に由来する胚由来の幹細胞であり、成体を構成するあらゆる細胞に分化する能力、いわゆる分化多能性と、自己複製による増殖能とを有している。ES細胞は、マウスで1981年に発見され(M.J. Evans and M.H. Kaufman (1981), Nature 292:154-156)、その後、ヒト、サルなどの霊長類でもES細胞株が樹立された (J.A. Thomson et al. (1998), Science 282:1145-1147; J.A. Thomson et al. (1995), Proc. Natl. Acad. Sci. USA, 92:7844-7848;J.A. Thomson et al. (1996), Biol. Reprod., 55:254-259; J.A. Thomson and V.S. Marshall (1998), Curr. Top. Dev. Biol., 38:133-165)。
【0041】
ES細胞は、対象動物の受精卵の胚盤胞から内部細胞塊を取出し、内部細胞塊を線維芽細胞のフィーダー上で培養することによって樹立することができる。また、継代培養による細胞の維持は、白血病抑制因子(leukemia inhibitory factor (LIF))、塩基性線維芽細胞成長因子(basic fibroblast growth factor (bFGF))などの物質を添加した培養液を用いて行うことができる。ヒトおよびサルのES細胞の樹立と維持の方法については、例えばUSP5,843,780; Thomson JA, et al. (1995), Proc Natl. Acad. Sci. U S A. 92:7844-7848; Thomson JA, et al. (1998), Science. 282:1145-1147; H. Suemori et al. (2006), Biochem. Biophys. Res. Commun., 345:926-932; M. Ueno et al. (2006), Proc. Natl. Acad. Sci. USA, 103:9554-9559;H. Suemori et al. (2001), Dev. Dyn., 222:273-279;H. Kawasaki et al. (2002), Proc. Natl. Acad. Sci. USA, 99:1580-1585;Klimanskaya I, et al. (2006), Nature. 444:481-485などに記載されている。
【0042】
ES細胞作製のための培養液として、例えば0.1mM 2-メルカプトエタノール、0.1mM 非必須アミノ酸、2mM L-グルタミン酸、20% KSRおよび4ng/ml bFGFを補充したDMEM/F-12培養液を使用し、37℃、2% CO2/98% 空気の湿潤雰囲気下でヒトES細胞を維持することができる(O. Fumitaka et al. (2008), Nat. Biotechnol., 26:215-224)。また、ES細胞は、3〜4日おきに継代する必要があり、このとき、継代は、例えば1mM CaCl2および20% KSRを含有するPBS中の0.25% トリプシンおよび0.1mg/mlコラゲナーゼIVを用いて行うことができる。
【0043】
ES細胞の選択は、一般に、アルカリホスファターゼ、Oct-3/4、Nanogなどの遺伝子マーカーの発現を指標にしてReal-Time PCR法で行うことができる。特に、ヒトES細胞の選択では、OCT-3/4、NANOG、ECADなどの遺伝子マーカーの発現を指標とすることができる(E. Kroon et al. (2008), Nat. Biotechnol., 26:443-452)。
【0044】
ヒトES細胞株は、例えばWA01(H1)およびWA09(H9)は、WiCell Reserch Instituteから、KhES-1、KhES-2およびKhES-3は、京都大学再生医科学研究所(京都、日本)から入手可能である。
【0045】
(B) 精子幹細胞
精子幹細胞は、精巣由来の多能性幹細胞であり、精子形成のための起源となる細胞である。この細胞は、ES細胞と同様に、種々の系列の細胞に分化誘導可能であり、例えばマウス胚盤胞に移植するとキメラマウスを作出できるなどの性質をもつ(M. Kanatsu-Shinohara et al. (2003) Biol. Reprod., 69:612-616; K. Shinohara et al. (2004), Cell, 119:1001-1012)。神経膠細胞系由来神経栄養因子(glialcell line-derived neurotrophic factor (GDNF))を含む培養液で自己複製可能であるし、またES細胞と同様の培養条件下で継代を繰り返すことによって、精子幹細胞を得ることができる(竹林正則ら(2008),実験医学,26巻,5号(増刊),41〜46頁,羊土社(東京、日本))。
【0046】
(C) 胚性生殖細胞
胚性生殖細胞は、胎生期の始原生殖細胞から樹立される、ES細胞と同様な多能性をもつ細胞であり、LIF、bFGF、幹細胞因子(stem cell factor)などの物質の存在下で始原生殖細胞を培養することによって樹立しうる(Y. Matsui et al. (1992), Cell, 70:841-847; J.L. Resnick et al. (1992), Nature, 359:550-551)。
【0047】
(D) 人工多能性幹細胞
人工多能性幹(iPS)細胞は、特定の初期化因子を、DNAまたはタンパク質の形態で体細胞に導入することによって作製することができる、ES細胞とほぼ同等の特性、例えば分化多能性と自己複製による増殖能、を有する体細胞由来の人工の幹細胞である(K. Takahashi and S. Yamanaka (2006) Cell, 126:663-676; K. Takahashi et al. (2007), Cell, 131:861-872; J. Yu et al. (2007), Science, 318:1917-1920; Nakagawa, M.ら,Nat. Biotechnol. 26:101-106 (2008);国際公開WO 2007/069666)。初期化因子は、ES細胞に特異的に発現している遺伝子、その遺伝子産物もしくはnon-cording RNAまたはES細胞の未分化維持に重要な役割を果たす遺伝子、その遺伝子産物もしくはnon-cording RNA、あるいは低分子化合物によって構成されてもよい。初期化因子に含まれる遺伝子として、例えば、Oct3/4、Sox2、Sox1、Sox3、Sox15、Sox17、Klf4、Klf2、c-Myc、N-Myc、L-Myc、Nanog、Lin28、Fbx15、ERas、ECAT15-2、Tcl1、beta-catenin、Lin28b、Sall1、Sall4、Esrrb、Nr5a2、Tbx3またはGlis1等が例示され、これらの初期化因子は、単独で用いても良く、組み合わせて用いても良い。初期化因子の組み合わせとしては、WO2007/069666、WO2008/118820、WO2009/007852、WO2009/032194、WO2009/058413、WO2009/057831、WO2009/075119、WO2009/079007、WO2009/091659、WO2009/101084、WO2009/101407、WO2009/102983、WO2009/114949、WO2009/117439、WO2009/126250、WO2009/126251、WO2009/126655、WO2009/157593、WO2010/009015、WO2010/033906、WO2010/033920、WO2010/042800、WO2010/050626、WO 2010/056831、WO2010/068955、WO2010/098419、WO2010/102267、WO 2010/111409、WO 2010/111422、WO2010/115050、WO2010/124290、WO2010/147395、WO2010/147612、Huangfu D, et al. (2008), Nat. Biotechnol., 26: 795-797、Shi Y, et al. (2008), Cell Stem Cell, 2: 525-528、Eminli S, et al. (2008), Stem Cells. 26:2467-2474、Huangfu D, et al. (2008), Nat Biotechnol. 26:1269-1275、Shi Y, et al. (2008), Cell Stem Cell, 3, 568-574、Zhao Y, et al. (2008), Cell Stem Cell, 3:475-479、Marson A, (2008), Cell Stem Cell, 3, 132-135、Feng B, et al. (2009), Nat Cell Biol. 11:197-203、R.L. Judson et al., (2009), Nat. Biotech., 27:459-461、Lyssiotis CA, et al. (2009), Proc Natl Acad Sci U S A. 106:8912-8917、Kim JB, et al. (2009), Nature. 461:649-643、Ichida JK, et al. (2009), Cell Stem Cell. 5:491-503、Heng JC, et al. (2010), Cell Stem Cell. 6:167-74、Han J, et al. (2010), Nature. 463:1096-100、Mali P, et al. (2010), Stem Cells. 28:713-720、Maekawa M, et al. (2011), Nature. 474:225-9.に記載の組み合わせが例示される。
【0048】
上記初期化因子には、ヒストンデアセチラーゼ(HDAC)阻害剤[例えば、バルプロ酸 (VPA)、トリコスタチンA、酪酸ナトリウム、MC 1293、M344等の低分子阻害剤、HDACに対するsiRNAおよびshRNA(例、HDAC1 siRNA SmartpoolTM (Millipore)、HuSH 29mer shRNA Constructs against HDAC1 (OriGene)等)等の核酸性発現阻害剤など]、MEK阻害剤(例えば、PD184352、PD98059、U0126、SL327およびPD0325901)、Glycogen synthase kinase-3阻害剤(例えば、BioおよびCHIR99021)、DNAメチルトランスフェラーゼ阻害剤(例えば、5-azacytidine)、ヒストンメチルトランスフェラーゼ阻害剤(例えば、BIX-01294 等の低分子阻害剤、Suv39hl、Suv39h2、SetDBlおよびG9aに対するsiRNAおよびshRNA等の核酸性発現阻害剤など)、L-channel calcium agonist (例えばBayk8644)、酪酸、TGFβ阻害剤またはALK5阻害剤(例えば、LY364947、SB431542、616453およびA-83-01)、p53阻害剤(例えばp53に対するsiRNAおよびshRNA)、ARID3A阻害剤(例えば、ARID3Aに対するsiRNAおよびshRNA)、miR-291-3p、miR-294、miR-295およびmir-302などのmiRNA、Wnt Signaling(例えばsoluble Wnt3a)、神経ペプチドY、プロスタグランジン類(例えば、プロスタグランジンE2およびプロスタグランジンJ2)、hTERT、SV40LT、UTF1、IRX6、GLISl、PITX2、DMRTBl等の樹立効率を高めることを目的として用いられる因子も含まれており、本明細書においては、これらの樹立効率の改善目的にて用いられた因子についても初期化因子と別段の区別をしないものとする。
【0049】
初期化因子は、タンパク質の形態の場合、例えばリポフェクション、細胞膜透過性ペプチド(例えば、HIV由来のTATおよびポリアルギニン)との融合、マイクロインジェクションなどの手法によって体細胞内に導入してもよい。
【0050】
一方、DNAの形態の場合、例えば、ウイルス、プラスミド、人工染色体などのベクター、リポフェクション、リポソーム、マイクロインジェクションなどの手法によって体細胞内に導入することができる。ウイルスベクターとしては、レトロウイルスベクター、レンチウイルスベクター(以上、Cell, 126, pp.663-676, 2006; Cell, 131, pp.861-872, 2007; Science, 318, pp.1917-1920, 2007)、アデノウイルスベクター(Science, 322, 945-949, 2008)、アデノ随伴ウイルスベクター、センダイウイルスベクター(WO 2010/008054)などが例示される。また、人工染色体ベクターとしては、例えばヒト人工染色体(HAC)、酵母人工染色体(YAC)、細菌もしくはファージ人工染色体(BACもしくはPAC)などが含まれる。プラスミドとしては、哺乳動物細胞用プラスミドを使用しうる(Science, 322:949-953, 2008)。ベクターには、核初期化物質が発現可能なように、プロモーター、エンハンサー、リボゾーム結合配列、ターミネーター、ポリアデニル化サイトなどの制御配列を含むことができるし、さらに、必要に応じて、薬剤耐性遺伝子(例えばカナマイシン耐性遺伝子、アンピシリン耐性遺伝子、ピューロマイシン耐性遺伝子など)、チミジンキナーゼ遺伝子、ジフテリアトキシン遺伝子などの選択マーカー配列、緑色蛍光タンパク質(GFP)、βグルクロニダーゼ(GUS)、FLAGなどのレポーター遺伝子配列などを含むことができる。また、上記ベクターには、体細胞への導入後、初期化因子をコードする遺伝子もしくはプロモーターとそれに結合する初期化因子をコードする遺伝子を共に切除するために、それらの前後にLoxP配列を有してもよい。
【0051】
また、RNAの形態の場合、例えばリポフェクション、マイクロインジェクションなどの手法によって体細胞内に導入しても良く、分解を抑制するため、5-メチルシチジンおよびpseudouridine (TriLink Biotechnologies)を取り込ませたRNAを用いても良い(Warren L, (2010) Cell Stem Cell. 7:618-630)。
【0052】
iPS細胞誘導のための培養液としては、例えば、10〜15%FBSを含有するDMEM、DMEM/F12またはDME培養液(これらの培養液にはさらに、LIF、penicillin/streptomycin、puromycin、L-グルタミン、非必須アミノ酸類、β-メルカプトエタノールなどを適宜含むことができる。)または市販の培養液[例えば、マウスES細胞培養用培養液(TX-WES培養液、トロンボX社)、霊長類ES細胞培養用培養液(霊長類ES/iPS細胞用培養液、リプロセル社)、無血清培地(mTeSR、Stemcell Technology社)]などが含まれる。
【0053】
培養法の例としては、たとえば、37℃、5%CO2存在下にて、10%FBS含有DMEMまたはDMEM/F12培養液上で体細胞と初期化因子とを接触させ約4〜7日間培養し、その後、細胞をフィーダー細胞(たとえば、マイトマイシンC処理STO細胞、SNL細胞等)上にまきなおし、体細胞と初期化因子の接触から約10日後からbFGF含有霊長類ES細胞培養用培養液で培養し、該接触から約30〜約45日またはそれ以上ののちにiPS様コロニーを生じさせることができる。
【0054】
あるいは、37℃、5% CO2存在下にて、フィーダー細胞(たとえば、マイトマイシンC処理STO細胞、SNL細胞等)上で10%FBS含有DMEM培養液(これにはさらに、LIF、ペニシリン/ストレプトマイシン、ピューロマイシン、L-グルタミン、非必須アミノ酸類、β-メルカプトエタノールなどを適宜含むことができる。)で培養し、約25〜約30日またはそれ以上ののちにES様コロニーを生じさせることができる。望ましくは、フィーダー細胞の代わりに、初期化される体細胞そのものを用いる(Takahashi K, et al. (2009), PLoS One. 4:e8067またはWO2010/137746)、もしくは細胞外基質(例えば、Laminin-5(WO2009/123349)およびマトリゲル(BD社))を用いる方法が例示される。
この他にも、血清を含有しない培地を用いて培養する方法も例示される(Sun N, et al. (2009), Proc Natl Acad Sci U S A. 106:15720-15725)。さらに、樹立効率を上げるため、低酸素条件(0.1%以上、15%以下の酸素濃度)によりiPS細胞を樹立しても良い(Yoshida Y, et al. (2009), Cell Stem Cell. 5:237-241またはWO2010/013845)。
【0055】
上記培養の間には、培養開始2日目以降から毎日1回新鮮な培養液と培養液交換を行う。また、核初期化に使用する体細胞の細胞数は、限定されないが、培養ディッシュ100cm2あたり約5×103〜約5×106細胞の範囲である。
【0056】
iPS細胞は、形成したコロニーの形状により選択することが可能である。一方、体細胞が初期化された場合に発現する遺伝子(例えば、Oct3/4、Nanog)と連動して発現する薬剤耐性遺伝子をマーカー遺伝子として導入した場合は、対応する薬剤を含む培養液(選択培養液)で培養を行うことにより樹立したiPS細胞を選択することができる。また、マーカー遺伝子が蛍光タンパク質遺伝子の場合は蛍光顕微鏡で観察することによって、発光酵素遺伝子の場合は発光基質を加えることによって、また発色酵素遺伝子の場合は発色基質を加えることによって、iPS細胞を選択することができる。
【0057】
本明細書中で使用する「体細胞」なる用語は、卵子、卵母細胞などの生殖系列細胞または分化全能性細胞、あるいはES細胞、を除くあらゆる動物細胞(好ましくは、ヒトを含む哺乳動物細胞)をいう。体細胞には、非限定的に、胎児(仔)の体細胞、新生児(仔)の体細胞、および成熟した健全なもしくは疾患性の体細胞のいずれも包含されるし、また、初代培養細胞、継代細胞、および株化細胞のいずれも包含される。具体的には、体細胞は、例えば(1)神経幹細胞、造血幹細胞、間葉系幹細胞、歯髄幹細胞等の組織幹細胞(体性幹細胞)、(2)組織前駆細胞、(3)リンパ球、上皮細胞、内皮細胞、筋肉細胞、線維芽細胞(皮膚細胞等)、毛細胞、肝細胞、胃粘膜細胞、腸細胞、脾細胞、膵細胞(膵外分泌細胞等)、脳細胞、肺細胞、腎細胞および脂肪細胞等の分化した細胞などが例示される。
【0058】
また、iPS細胞を移植用細胞の材料として用いる場合、拒絶反応が起こらないという観点から、移植先の個体のHLA遺伝子型が同一もしくは実質的に同一である体細胞を用いることが望ましい。ここで、「実質的に同一」とは、移植した細胞に対して免疫抑制剤により免疫反応が抑制できる程度にHLA遺伝子型が一致していることであり、例えば、HLA-A、HLA-BおよびHLA-DRの3遺伝子座あるいはHLA-Cを加えた4遺伝子座が一致するHLA型を有する体細胞である。
【0059】
(E) 核移植により得られたクローン胚由来のES細胞
nt ES細胞は、核移植技術によって作製されたクローン胚由来のES細胞であり、受精卵由来のES細胞とほぼ同じ特性を有している(T. Wakayama et al. (2001), Science, 292:740-743; S. Wakayama et al. (2005), Biol. Reprod., 72:932-936; J. Byrne et al. (2007), Nature, 450:497-502)。すなわち、未受精卵の核を体細胞の核と置換することによって得られたクローン胚由来の胚盤胞の内部細胞塊から樹立されたES細胞がnt ES(nuclear transfer ES)細胞である。nt ES細胞の作製のためには、核移植技術(J.B. Cibelli et al. (1998), Nature Biotechnol., 16:642-646)とES細胞作製技術(上記)との組み合わせが利用される(若山清香ら(2008),実験医学,26巻,5号(増刊), 47〜52頁)。核移植においては、哺乳動物の除核した未受精卵に、体細胞の核を注入し、数時間培養することで初期化することができる。
【0060】
(F) Multilineage-differentiating Stress Enduring cells(Muse細胞)
Muse細胞は、WO2011/007900に記載された方法にて製造された多能性幹細胞であり、詳細には、線維芽細胞または骨髄間質細胞を長時間トリプシン処理、好ましくは8時間または16時間トリプシン処理した後、浮遊培養することで得られる多能性を有した細胞であり、SSEA-3およびCD105が陽性である。
【実施例】
【0061】
以下に、本発明の実施例と比較例について説明するが、本実施例は本発明の再現を補助する目的でその一実施態様を示すものであって、本実施例は本発明の範囲を制限するものではない。
【0062】
<組換えヒトBACクローンの作製>
OSR1遺伝子座から86.3kb上流および89.8kb下流を含むヒトBACクローン(RP11-458J18)は、BACPAC Resources Center (Oakland, CA)より購入した。Lee, E.C. et al. (2001) Genomics 73, 56-65に記載の方法で組換えを行った。簡潔には、hOSR1-EGFP-S: TCTTCTTTTCTTTGCAGATCCGGATTGAGAAGCCACTGCAACTACCGAACACCATGGTGAGCAAGGGCGAGGA (配列番号1)およびhOSR1-PNL-AS: GTTCACTGCCTGAAGGAAGGAGTAGTTGGTGAGCTGCAGGGAAGGGTGGAGTCGACGGCGAGCTCAGACG(配列番号2) のプライマーを用いてPCRにより5’および3’側にホモロジーアームを有するEGFP-polyA-LoxP-PGK-Neo-LoxP(EGFP-pA-PNL)カセットを作製した。このカセットおよびヒトBACクローンを大腸菌DH10Bへ導入し、相同組換えのためにリコンビナーゼを活性化させ、ヒトBACクローン中のOSR1の開始コドン直後の配列をEGFP-pA-PNLカセットを挿入した(図1中のrecomninant BAC clone)。
【0063】
<相同組換えiPS細胞の作製>
上述した方法で作製した組換えヒトBACクローンを制限酵素で切断し、一本鎖DNAとした。30μg一本鎖DNAをY27632(Wako)およびトリプシンで処理したヒトiPS細胞(201B7)にエレクトロポレーション(250 V, 500 mF単回パルス)により導入した。導入2時間後、iPS細胞を薬剤選択用培地中で培養した。
【0064】
<PCR分析>
得られた130個の薬剤耐性クローンiPS細胞から染色体DNAを抽出し、OSR1F(5’-GGATTGAGAAGCCACTGCAACT-3’(配列番号3))およびOSR1R(5’-CCGTTCACTGCCTGAAGGA-3’(配列番号4))のプライマーを用いて定量PCRを行った(図2A)。この結果、3D36、3D45、3F3および3I49の4つクローンにおいてOSR1の開始コドン付近が欠損していることが確認された。
【0065】
<SNPアレイ分析>
染色体DNAを抽出し、GeneChipTM Mapping 250K NSP arrays (Affymetrix)を用いて得られたデータをソフトウェア(CNAG/AsCNAR)により対立遺伝子特異的コピー数解析を3D36、3D45、3F3、3I49および3D12に対して行った。GeneChipに含有された各プローブの検出値からCNAG/AsCNAR 解析により得られたプローブ領域のコピー数を縦軸に、該プローブの遺伝子上での位置を横軸にとりプロットした(図2B)。その結果、3D36、3D45、3F3および3I49は、OSR1の遺伝子座の領域が2コピー含有することが示された。このことは、相同組換えにより、EGFP-pA-PNLカセットが挿入されたことを示している。一方、3D12 は、OSR1領域が3コピー含有することが示された。このことは、組換えヒトBACクローンが相同組換えではなく、染色体にランダムに組み込まれたことを示唆している。以上より、相同組換えによりEGFP-pA-PNLカセットが所望の位置にノックインされたヒトiPS細胞(3D36、3D45、3F3および3I49)が得られた。また、解析領域を拡大して調べたところ、3I49には第9番染色体にコピー数多型を有している事が確認された。これより、培養中または遺伝子改変の際に、3I49は第9染色体において異常な遺伝子の重複が起きたと考えられる。
【0066】
<核型解析>
Gバンド解析により核型解析を行ったところ、3D45は正常の核型を有していることが確認された。
【0067】
<Creリコンビナーゼ処理>
OSR1がタゲーゲティングされたヒトiPS細胞(3D36、3D45、3F3および3I49)を10μM Y27,632添加培地で1日培養した後、トリプシン処理により分離させ、エレクトロポレーションによりCre発現ベクターを導入した。導入後、loxP配列を挟むようにGFP配列部位およびOSR1遺伝子の開始コドン以降に設計された図1に記載のプライマーで染色体DNAをPCRしたところ、全てのクローンにおいてPGK-Neo-pAが除外されたことが確認された。
【産業上の利用可能性】
【0068】
本発明によれば、人工染色体をターゲティングベクターとして用いることにより高効率で相同組換えが起こった多能性幹細胞を選択的に製造することが可能となる。また、SNPアレイを用いることにより簡便に相同組換えされた多能性幹細胞を検出することが可能となる。したがって、本発明を用いて相同組換えされた多能性幹細胞を製造することで、簡便に治療剤または分化誘導剤のスクリーニングすることもしくは治療用の細胞の確保が可能となる。
【特許請求の範囲】
【請求項1】
次の工程(1)〜(3):
(1)多能性幹細胞に、遺伝子改変された染色体断片を有する人工染色体を導入し、遺伝子改変されたと推定される多能性幹細胞クローンからなる集団を作製する工程、
(2)前記集団の多能性幹細胞クローンについて、SNPアレイを用いて、前記導入された人工染色体の一部または全体のコピー数を測定する工程、
(3)前記コピー数が、人工染色体が導入されていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞クローンを、相同組換えにより遺伝子改変された多能性幹細胞として選択する工程、
を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の製造方法。
【請求項2】
前記工程(1)の遺伝子改変が内在性配列を保持したまま細胞ゲノムに外因性DNA断片を挿入することからなる組込みを含む、請求項1に記載の方法。
【請求項3】
外因性DNAが選択マーカーをコードするDNAであって、該選択マーカーが陽性であるクローンを選択する工程をさらに含む、請求項2に記載の方法。
【請求項4】
多能性幹細胞クローンの該遺伝子改変対象であるアレルの数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下しているクローンを選択する工程をさらに含む、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記多能性幹細胞がヒト多能性幹細胞である、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記人工染色体がBACクローンである、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記選択マーカーが薬剤耐性マーカーである、請求項3に記載の方法。
【請求項8】
相同組換えにより遺伝子改変された多能性幹細胞の製造において、SNPアレイを用いて組換え領域のコピー数を測定し、該コピー数が、相同組換えをしていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞ローンを、相同組換えにより遺伝子改変された多能性幹細胞として検出する工程を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の検出方法。
【請求項9】
前記遺伝子改変が、内在性配列を保持したまま細胞ゲノムに外因性DNA断片を挿入することからなる組込みである、請求項8に記載の方法。
【請求項10】
前記外因性DNAが選択マーカーをコードするDNAあって、該選択マーカーが陽性であるクローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する工程をさらに含む、請求項9に記載の方法。
【請求項11】
多能性幹細胞クローンの該遺伝子改変対象であるアレルの数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下しているクローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する工程をさらに含む、請求項8〜10のいずれか1項に記載の方法。
【請求項12】
前記多能性幹細胞がヒト多能性幹細胞である、請求項8〜11のいずれか1項に記載の方法。
【請求項13】
前記選択マーカーが薬剤耐性マーカーである、請求項10に記載の方法。
【請求項1】
次の工程(1)〜(3):
(1)多能性幹細胞に、遺伝子改変された染色体断片を有する人工染色体を導入し、遺伝子改変されたと推定される多能性幹細胞クローンからなる集団を作製する工程、
(2)前記集団の多能性幹細胞クローンについて、SNPアレイを用いて、前記導入された人工染色体の一部または全体のコピー数を測定する工程、
(3)前記コピー数が、人工染色体が導入されていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞クローンを、相同組換えにより遺伝子改変された多能性幹細胞として選択する工程、
を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の製造方法。
【請求項2】
前記工程(1)の遺伝子改変が内在性配列を保持したまま細胞ゲノムに外因性DNA断片を挿入することからなる組込みを含む、請求項1に記載の方法。
【請求項3】
外因性DNAが選択マーカーをコードするDNAであって、該選択マーカーが陽性であるクローンを選択する工程をさらに含む、請求項2に記載の方法。
【請求項4】
多能性幹細胞クローンの該遺伝子改変対象であるアレルの数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下しているクローンを選択する工程をさらに含む、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記多能性幹細胞がヒト多能性幹細胞である、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記人工染色体がBACクローンである、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記選択マーカーが薬剤耐性マーカーである、請求項3に記載の方法。
【請求項8】
相同組換えにより遺伝子改変された多能性幹細胞の製造において、SNPアレイを用いて組換え領域のコピー数を測定し、該コピー数が、相同組換えをしていない野生型細胞のコピー数と同等であるまたは減少している多能性幹細胞ローンを、相同組換えにより遺伝子改変された多能性幹細胞として検出する工程を含むことを特徴とする、相同組換えにより遺伝子改変された多能性幹細胞の検出方法。
【請求項9】
前記遺伝子改変が、内在性配列を保持したまま細胞ゲノムに外因性DNA断片を挿入することからなる組込みである、請求項8に記載の方法。
【請求項10】
前記外因性DNAが選択マーカーをコードするDNAあって、該選択マーカーが陽性であるクローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する工程をさらに含む、請求項9に記載の方法。
【請求項11】
多能性幹細胞クローンの該遺伝子改変対象であるアレルの数が、相同組換えをしていない野生型細胞の該アレル数と比較して低下しているクローンを相同組換えにより遺伝子改変された多能性幹細胞として検出する工程をさらに含む、請求項8〜10のいずれか1項に記載の方法。
【請求項12】
前記多能性幹細胞がヒト多能性幹細胞である、請求項8〜11のいずれか1項に記載の方法。
【請求項13】
前記選択マーカーが薬剤耐性マーカーである、請求項10に記載の方法。
【図1】


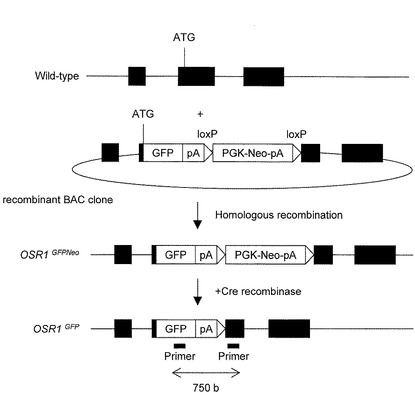
【図2】
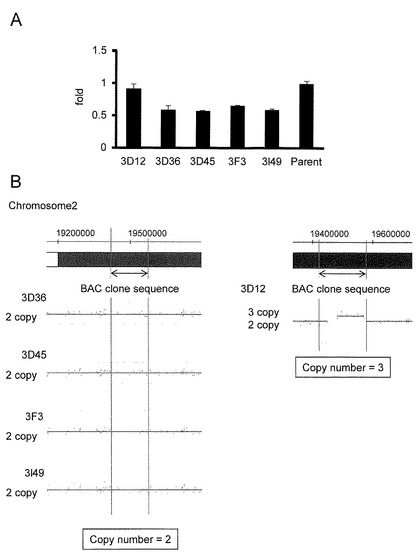
【図2】
【公開番号】特開2013−66386(P2013−66386A)
【公開日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願番号】特願2011−204950(P2011−204950)
【出願日】平成23年9月20日(2011.9.20)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成23年度文部科学省、「科学技術試験研究委託事業」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
【公開日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願日】平成23年9月20日(2011.9.20)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成23年度文部科学省、「科学技術試験研究委託事業」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
[ Back to top ]