
眼疾患の治療
本発明は、加齢黄斑変性(AMD)、脈絡膜血管新生、ヒッペル・リンダウ病、虹彩血管新生、虚血性増殖網膜症、角膜血管新生、および増殖性鎌状赤血球性網膜症からなる群から選択された新血管新生に関連した眼疾患の治療および/または予防用の医薬品を製造するための少なくとも1種のプロテアーゼの使用であって、少なくとも1種のプロテアーゼは、植物、非哺乳類動物、および微生物プロテアーゼ類からなる群から選択される使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新血管新生に関連した疾患の治療用医薬品に関する。
【背景技術】
【0002】
血管新生は、既存の血管からの内皮細胞の増殖による新しい血管の形成と定義される。この過程で、内皮細胞は、その下にある基底膜を退化させ、増殖し、隣接組織に遊走し、集合して管を構築する。最後に、管と管の連結が行われ、血流が確立する。成熟組織の変化する要求に適応する能力は、低酸素誘導因子(HIF)および血管内皮因子(VEGF)のような可溶性因子と、細胞−細胞および細胞−マトリックス相互作用とを必要とする。
【0003】
VEGFは、本来実質的な血管漏出を引き起こす因子と記述され、血管透過性因子(VPF)と呼ばれた。同じタンパク質は、内皮細胞におけるその分裂促進作用によって、後に改めて血管内皮増殖因子(VEGF)と命名された。
【0004】
VEGFは、微小血管床の透過性を増大させ、したがって血管からの流体およびタンパク質の漏出を促進する。これによって、浮腫、創傷液および漿液腫(例えば、手術後)、浸出液(例えば、慢性炎症性疾患における)、ならびに腹水(例えば、癌における)の発生が起こる。VEGFは、血管透過性の誘導においてヒスタミンより10,000倍強力である。
【0005】
さらに、VEGFは、内皮細胞増殖の最も強力な刺激剤の1つである。最後に、これは、内皮細胞からの毛細管の形成を刺激し、したがって血管新生にとって必要である事象のカスケードを促進する。新たに形成された組織における既存の血管からの新しい毛細管の増殖である新血管新生、またはさらには沈着物(歯垢などのような)は、種々の病理学的状態の発生および進行に寄与する。生理的条件下では、血管新生は、厳密に調節された過程である。この過程は、癌、関節リウマチ、子宮内膜症、乾癬、または眼血管新生のような病理学的状態においては、かなり強化され、かつ機能不全である。
【0006】
抗血管新生薬は、癌、関節リウマチ、乾癬、および眼血管新生などのような疾患の将来の治療を改良するということを示唆する証拠がますます増えている。生体内実験によって、低酸素(例えば、腫瘍壊死に近い領域における低酸素)は、様々なタイプの細胞においてVEGFとVEGF受容体(VEGFR−1)との発現を誘導できることが実証された。低酸素は、低酸素誘導因子1(HIF−1)の発現を引き起こす。引き続いて、HIF−1複合体は、細胞核内に蓄積し、DNAのHIF−1結合部位に結合し、隣接した血管を低酸素組織に発芽させることができる血管新生スイッチを始動させるVEGF−mRNAの転写を開始または上方制御する。さらに、VEGF発現は、乾癬または関節リウマチのような慢性炎症の様々なモデルで実証されているように、様々な炎症誘発性サイトカインによって誘導/上方制御することができる。
【0007】
VEGFは、プロテアーゼ活性化α2Mによってα2マクログロブリン(α2M)経路を介して循環から除去することができる。α2M−プロテアーゼ複合体は、表面の円蓋においてVEGFを結合させることができる。得られたα2M−酵素−VEGF複合体は、マクロファージおよび内皮細胞のような細胞の表面上で発現したLRP受容体に結合し(低密度リポタンパク質受容体関連タンパク質受容体)、貪食され、破壊される。タンパク質分解酵素を用いた経口療法は、活性化したα2M分子の数を増加し、したがって有機体のサイトカイン/増殖因子破壊能を高める。(デザー(Desser L)ら、「キャンサー・ケモセラピー・アンド・ファーマコロジー増補(Cancer Chemother Pharmacol Suppl)」、(2001年)、47巻:S10−S15;ラウア(Lauer D)ら、「キャンサー・ケモセラピー・アンド・ファーマコロジー増補(Cancer Chemother Pharmacol Suppl)」、(2001年)、47巻:S4−S9)。
【0008】
最近、血管新生の増大を伴う疾患、主に癌の治療のためであるが、黄斑変性症など眼における血管新生を伴う疾患のためにも、VEGF受容体遮断薬またはVEGFに対する抗体を使用した複数の治療的手法が提案されている。
【0009】
眼血管新生または新血管新生は、盲目の最も多い原因として示唆され、約20種類の異なる眼疾患の病態の基礎をなす。例えば、糖尿病において、網膜において形成された新しい毛細管は、ガラス様液に侵入し、出血および盲目を引き起こす。
【0010】
国際公開第2005/110453号は、VEGFおよびVEGF受容体を切断するためのヒト野生型および変異型タンパク質MT−SP1プロテアーゼ類の使用に関する。このような切断は、血管新生の低減を生じ、したがって血管新生を伴う病態を治療するために使用することができる。
【0011】
特開昭60−112720号は、血管新生を伴わない疾患(例えば、緑内障)を治療するパパインおよびクエン酸の使用に関する。
【0012】
国際公開第2004/046199号では、眼疾患を治療するコンドロイチン硫酸の使用が開示されている。
【0013】
国際公開第2005/056784号は、糖尿病を治療するナットウキナーゼの使用に関する。
【0014】
旧ソ連特許第1342500号では、緑内障のような眼疾患を治療するパパインの使用が記載されている。
【0015】
米国特許第6103756号は、眼障害を治療するために使用することができる抗酸化剤およびフラボノイド類を含む組成物に関する。
【発明の概要】
【発明が解決しようとする課題】
【0016】
本発明の目的は、新血管新生に関連した眼疾患を治療または予防するための医薬品を提供することである。
【課題を解決するための手段】
【0017】
したがって、本発明は、加齢黄斑変性(AMD)、脈絡膜血管新生、ヒッペル・リンダウ病、虹彩血管新生、虚血性増殖網膜症、角膜血管新生、および増殖性鎌状赤血球性網膜症からなる群から選択された新血管新生に関連した眼疾患の治療および/または予防用の医薬品を製造するための少なくとも1種のプロテアーゼの使用であって、少なくとも1種のプロテアーゼは、植物プロテアーゼ類、非哺乳類動物プロテアーゼ類、および微生物プロテアーゼ類からなる群から選択される使用に関する。
【0018】
驚くべきことに、好ましくは植物、非哺乳類動物、および微生物プロテアーゼ類からなる群から選択された少なくとも1種のプロテアーゼの組合せを含む医薬品は特に、個体に投与されたときVEGFのレベルを大幅に(本発明に従う医薬品の投与前の前記個体のVEGFレベルに比べて、少なくとも40%、好ましくは少なくとも50%、より好ましくは少なくとも60%、さらにより好ましくは少なくとも70%、最も好ましくは少なくとも80%、特に少なくとも90%)低減させることができ、したがって血管新生を低減させることが明らかになった。したがって、少なくとも1種のプロテアーゼ、好ましくは少なくとも2種類(少なくとも3種類、少なくとも4種類、少なくとも5種類、少なくとも6種類)のプロテアーゼ類の組合せを、個体における新血管新生に関連した眼疾患を予防および/または治療するために使用することができる。微生物、非哺乳類動物、および植物プロテアーゼ類は、ヒトまたは動物の細胞、特に内皮細胞と接触させたとき顕著な毒性を示さないので、前記プロテアーゼ類の投与が特に適している。実に本明細書に開示するプロテアーゼ類の組合せは、動物またはヒトへの毒性がなく、しかも血管新生に働く。
【0019】
興味深いことに、トリプシンまたはキモトリプシンのような哺乳類(すなわち、ヒトおよび動物)由来のプロテアーゼ類は血管新生を抑制または予防できないことを示し得る。したがって、このようなプロテアーゼ類の個体への単独投与は、新血管新生に関連した疾患を予防または治療するために行うことはできない。
【0020】
本明細書に定義する「医薬品」という用語は、薬剤製品だけでなく、食事補助食品も包含する。
【0021】
本明細書では、「植物プロテアーゼ類」および「動物プロテアーゼ類」および「非哺乳類動物プロテアーゼ」は、植物または非哺乳類動物において自然発生し、それらから抽出または獲得されるプロテアーゼ類とするものである。「植物プロテアーゼ類」および「動物プロテアーゼ類」および「非哺乳類動物プロテアーゼ」は、コードするDNA(例えば、cDNAとして)がそれぞれ、植物および動物(そのゲノム中に前記DNAを天然に含む)に由来しまたはそれらから獲得され、適切なベクターにクローン化され、原核(例えば、細菌)または真核細胞(例えば、昆虫細胞、哺乳類細胞)培養物で発現する組換えプロテアーゼ類でもある。
【0022】
本明細書では、「微生物プロテアーゼ類」は、細菌類および真菌類(例えば、酵母、カビ類)などの微生物において自然発生するプロテアーゼ類である。しかし、前記プロテアーゼ類は、他の細胞または有機体から単離することもできる。ただし、前記細胞および有機体は、微生物プロテアーゼのDNAを含み、組換えによって前記プロテアーゼを生成できることを条件とする。
【0023】
治療および/または予防すべき血管新生に関連した眼疾患が上記の群から選択される場合、本発明に従う医薬品の使用は特に適している。これらの疾患はすべて、大部分は体内の血管内皮増殖因子(VEGF)のレベル上昇のために血管新生の増大を示す。しかし、本発明の医薬品を加齢黄斑変性を治療するために使用することが特に好ましい。
【0024】
複数の疾患は、本発明の医薬品を使用することもできる新血管新生に関連する。
【0025】
例えば、眼血管新生は、盲目の最も多い原因である(加齢黄斑変性;ヒッペル・リンダウ病;ベーチェット症候群;特発性眼血管新生)。
【0026】
本発明の医薬品を、高いVEGFレベルによって引き起こされ、血管新生に随伴した疾患を患っている個体を予防および/または治療するために使用し、それによってこれらの疾患が大部分または完全に、増殖活性の増大を伴うわけではないことが特に好ましい。
【0027】
少なくとも1種の植物プロテアーゼは、好ましくはブロメライン、パパイン、フィシン、およびククミシンからなる群から選択される。
【0028】
好ましくは本発明に従って使用することができる植物プロテアーゼ類は上記に示す。
【0029】
これらのプロテアーゼ類は、受容者における組換え発現、または前記プロテアーゼ類を自然に産生する植物からの抽出によって得ることができ、それによってその抽出物自体を、本発明に従う医薬品を製造するために直接使用することができる。プロテアーゼ類の抽出方法は当技術分野でよく知られている。
【0030】
例えば、ブロメラインは、果実を収穫した後の植物パイナップルの切株または根部から調製される。この切株または根部を畑から回収し、皮を剥がし、圧搾して、可溶性ブロメライン酵素を含有する汁を抽出する。その後の加工としては、酵素をさらに精製するための酵素の沈殿が挙げられる。
【0031】
パパインは、パパイアの果実の乳液を回収することによって、乾燥した粗材料として生成することができる。果実の頸部に刻み目をつけた後、乳液は回収され、その後果実上で乾燥してもよく、または容器に滴下してもよい。次いで、この乳液をさらに乾燥する。この時点で、これは乾燥した粗材料に分類される。汚染物質を除去する精製工程が必要である。この精製は、活性パパイン酵素の可溶化および抽出からなる。
【0032】
本発明の好ましい実施形態によれば、微生物プロテアーゼは、ナットウキナーゼ、ブリナーゼ、プロナーゼ、セアプローゼ、セラペプターゼ、およびサブチリシンからなる群から選択される。
【0033】
微生物プロテアーゼ類は、組換え技法によって得ることもでき、または前記プロテアーゼ類を産生する微生物を含む微生物培養物から直接単離してもよい。
【0034】
例えば、ナットウキナーゼは、発酵ダイズから作られた伝統的な日本食品である納豆から、または前記プロテアーゼを産生することができる特定のバチルス・スブチリス(Bacillus subtilis)亜種(バチルス・スブチリス・バラエティー・ナットウ(Bacillus subtilis var. natto))の有機体を含む培養物によって得られる。バチルス・スブチリス・バラエティー・ナットウ(Bacillus subtilis var. natto)は、自然土壌および日本の市販納豆から単離してもよい。この菌株は、フィブリンを分解するナットウキナーゼ産物の高活性をもたらす能力を有する。炭素源、有機窒素源または無機窒素源、鉱塩、初期pH、および温度は、バチルス・スブチリス・バラエティー・ナットウ(B. subtilis var. natto)によるナットウキナーゼ産生のために最適化されなければならない。例えば、バチルス・スブチリス・バラエティー・ナットウ(B. subtilis var. natto)の最適接種サイズは約5%(体積/体積)であることがわかった。最適培地は、2.8%ダイズタンパク質、1%酵母エキス、および0.8%マルトースを含有することができる。さらに、最適pHおよび温度はそれぞれ、約6.5±0.5、および約30℃〜40℃とすることができる。最適インキュベーション時間は18〜48時間である。発酵培地におけるナットウキナーゼ活性は、40FU/ml超まで上昇することがある。
【0035】
例えば、セラペプターゼは、カイコ類の腸内に存在するセラチア(Serratia)E15細菌類から単離されたタンパク質分解酵素である。この酵素は、疼痛および炎症を自然治療するために補助食品として使用することができ、アジアおよび欧州の一部分において、臨床で使用されている。セラペプターゼは、関節炎および炎症を治療するためによく使用される非ステロイド系抗炎症薬類(NSAID類)の代替物として使用される。
【0036】
非哺乳類動物プロテアーゼは、好ましくはレプチラーゼ、オキアミ酵素、バトロキソビン、およびルンブロキナーゼからなる群から選択される。
【0037】
これらのプロテアーゼ類は、当技術分野で知られている方法で組換えによって生成することができ、またはそれぞれの動物から直接に得ることができる。
【0038】
特に好ましい医薬品は、植物プロテアーゼ類であるブロメラインおよび/またはパパイン、ならびに任意選択で微生物プロテアーゼであるナットウキナーゼを含む。本発明に従う医薬品中のこれらのプロテアーゼ類の好ましい比は、下記の表で見られ、これによればパパイン、ナットウキナーゼ、および/またはブロメラインの量は互いに独立して、医薬品中に存在するプロテアーゼの全量の0%〜80%、好ましくは10%〜75%、より好ましくは15(または16,67)%〜75%で変わり得る。
【0039】
【表1】

【0040】
本発明の別の好ましい実施形態によれば、少なくとも1種の植物、非哺乳類動物、および/または微生物プロテアーゼは、医薬品中に10重量/重量%〜90重量/重量%、好ましくは20重量/重量%〜80重量/重量%、より好ましくは30重量/重量%〜70重量/重量%の量で含まれる。
【0041】
好ましくは、少なくとも1種の植物、非哺乳類動物、および/または微生物プロテアーゼは、1mg/体重1kg〜100mg/体重1kg、好ましくは2mg/体重1kg〜50mg/体重1kg、より好ましくは5mg/体重1kg〜20mg/体重1kgの所定量で個体に投与される。
【0042】
好ましくは、医薬品は、さらに少なくとも1種の医薬として許容される担体、希釈剤、および/または賦形剤、好ましくは結合剤、充填剤、崩壊剤、滑沢剤、保存剤、および/またはコーティング剤を含むことができる。
【0043】
本発明に従う医薬品の薬剤処方に応じて、賦形剤、コーティング剤などのような様々な他の物質を使用してもよい。
【0044】
さらに、本発明の医薬品は、好ましくは経口、局所、経腸、または非経口投与に適合させることができる。
【0045】
本発明の好ましい実施形態によれば、医薬品は、点眼剤、点耳剤、点鼻剤、スプレー式点鼻剤、錠剤、好ましくは可溶性錠剤、発泡錠、胃耐性錠剤、および舌下錠、カプセル剤、好ましくは胃耐性カプセル剤、散剤、顆粒剤、経口用液剤、経口用ドロップ剤、軟膏剤、ローション剤、乳剤、ヒドロゲル剤、坐剤、腟坐剤、輸注剤、および注射剤からなる群から選択された剤形で用いられる。
【0046】
特定の本発明の好ましい実施形態では、医薬品を経口投与に適合させる。この投与モードは、非侵襲性であり、したがって医薬品の反復投与を(患者を害することなく)可能にする。
【0047】
本発明の医薬品は、(1)経口投与用、例えば水薬(水性もしくは非水性液剤または懸濁剤)、錠剤、ボーラス、散剤、顆粒剤、舌に適用するためのペースト剤;(2)例えば滅菌液剤もしくは懸濁剤としての例えば皮下、筋肉内、または静脈注射による非経口投与用;(3)例えば皮膚に適用されるクリーム剤、軟膏剤、またはスプレーとしての局所塗布用;あるいは(4)例えば腟坐剤、クリーム剤、またはフォーム剤としての腟内または直腸内投与用に適合されたものを含めて、固体または液体の形で投与するために特別に製剤することができる。
【0048】
本明細書で「医薬として許容される」という語句は、正しい医学的判断の範囲内で、過剰の毒性、刺激、アレルギー応答、または他の問題もしくは合併症なしにヒトおよび動物の組織と接触させて使用するのに適し、妥当な利益/危険比に見合った化合物、材料、組成物、および/または剤形を意味するように使用される。
【0049】
医薬として許容される担体として機能することができる材料の例としては、下記が挙げられる。(1)ラクトース、グルコース、およびスクロースなどの糖類;(2)トウモロコシデンプンおよび馬鈴薯デンプンなどのデンプン類;(3)カルボキシメチルセルロースナトリウム、エチルセルロース、および酢酸セルロースなどのセルロースおよびその誘導体;(4)トラガカント末;(5)麦芽;(6)ゼラチン;(7)タルク;(8)カカオバターおよび座剤ワックス類などの賦形剤;(9)落花生油、綿実油、紅花油、ゴマ油、オリーブ油、トウモロコシ油、およびダイズ油などの油類;(10)プロピレングリコールなどのグリコール類;(11)グリセリン、ソルビトール、マンニトール、およびポリエチレングリコールなどのポリオール類;(12)オレイン酸エチルおよびラウリン酸エチルなどのエステル類;(13)寒天;(14)水酸化マグネシウムおよび水酸化アルミニウムなどの緩衝剤;(15)アルギン酸;(16)発熱物質を含まない水;(17)等張性生理食塩水;(18)リンゲル液;(19)エチルアルコール;(20)リン酸緩衝溶液類;ならびに(21)薬剤処方で使用される他の無毒性相溶物質類。
【0050】
湿潤化剤、ラウリル硫酸ナトリウムやステアリン酸マグネシウムなどの乳化剤および滑沢剤、ならびに着色剤、遊離剤、コーティング剤、甘味剤、着香剤、および芳香剤、保存剤、ならびに抗酸化剤も、本発明に従う医薬品中に存在することができる。医薬として許容される抗酸化剤としては、例えば(1)アスコルビン酸、システイン塩酸塩、重硫酸ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウムなどの水溶性抗酸化剤;(2)パルミチン酸アスコルビル、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、α−トコフェロールなどの油溶性抗酸化剤;ならびに(3)クエン酸、エチレンジアミン四酢酸(EDTA)、ソルビトール、酒石酸、リン酸などの金属キレート剤が挙げられる。
【0051】
本発明の製剤としては、経口、経鼻、局所(頬側および舌下を含む)、直腸、腟内、および/または非経口投与に適した製剤が挙げられる。製剤は、好都合なことに単位剤形で提供することができ、薬学技術分野でよく知られている任意の方法で調製することができる。担体材料と組み合わせて、単一剤形を生成することができる活性成分の量は、治療されている受容者および特定の投与モードに応じて異なる。担体材料と組み合わせて、単一剤形を生成することができる活性成分の量は、一般に治療効果をもたらす化合物のその量である。
【0052】
本発明に従う医薬品および製剤を調製する方法としては、本発明の化合物を担体lgおよび任意選択で1つまたは複数の副成分と結合させる工程が挙げられる。一般に、製剤は、本発明の化合物を液体担体もしくは微細化固体担体または両方と均一かつ緊密に結合させ、次いで必要なら、生成物を造形することによって調製される。経口投与に適した本発明の製剤は、カプセル剤、カシェ剤、丸剤、錠剤、ロゼンジ剤(着香基材、通常はスクロースおよびアカシアまたはトラガカントを使用する)、散剤、顆粒剤の形、または水性もしくは非水性液体中の液剤もしくは懸濁剤、または水中油型もしくは油中水型液体乳剤、またはシロップ剤、または香錠剤(ゼラチンおよびグリセリンなどの不活性基材、またはスクロースおよびアカシアを使用する)とすることができ、それぞれ、活性成分として所定量の本発明のプロテアーゼの組合せを含有する。
【0053】
本発明のプロテアーゼは、ボーラス、舐剤、またはペースト剤として投与することもできる。経口投与用の本発明の固体剤形(カプセル剤、錠剤、丸剤、糖衣錠、散剤、顆粒剤など)では、活性成分をクエン酸ナトリウムもしくは第二リン酸カルシウムなど1種もしくは複数の医薬として許容される担体、および/または次のいずれかと混合する:(1)デンプン類、ラクトース、スクロース、グルコース、マンニトール、および/またはケイ酸などの充填剤または増量剤;(2)例えばカルボキシメチルセルロース、アルギン酸塩/エステル/アルギナート類、ゼラチン、ポリビニルピロリドン、スクロース、および/またはアカシアなどの結合剤;(3)グリセロールなどの保湿剤;(4)寒天、炭酸カルシウム、馬鈴薯またはタピオカデンプン、アルギン酸、ある種のシリケート類、および炭酸ナトリウムなどの崩壊剤;(5)パラフィンなどの溶解遅延剤;(6)第四級アンモニウム化合物などの吸収促進剤;(7)例えばセチルアルコールおよびグリセロールモノステアレートなどの湿潤化剤;(8)カオリンおよびベントナイト粘土などの吸収剤;(9)タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール類、ラウリル硫酸ナトリウム、およびそれらの混合物などの滑沢剤;ならびに(10)着色剤。
【0054】
カプセル剤、錠剤、および丸剤の場合は、医薬組成物は緩衝剤も含むことができる。同様のタイプの固体組成物も、ラクトースまたは乳糖類のような賦形剤および高分子量ポリエチレングリコール類などを使用する軟質および硬質充填ゼラチンカプセル剤中の充填剤として使用することができる。
【0055】
錠剤は、任意選択で1種または複数の副成分と共に圧縮または成形によって作製することができる。圧縮錠は、結合剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース)、滑沢剤、不活性希釈剤、保存剤、崩壊剤(例えば、デンプングリコール酸ナトリウムまたは架橋カルボキシメチルセルロースナトリウム)、表面活性または分散剤を使用して調製することができる。成形錠剤は、不活性液体希釈剤で湿潤させた粉末化合物の混合物を適当な機械で成形することによって作製することができる。本発明の医薬組成物の錠剤、ならびに他の固体剤形、具体的には糖衣錠、カプセル剤、丸剤、および顆粒剤は、任意選択で製薬技術分野で周知の腸溶性コーティングおよび他のコーティングなどのコーティングおよびシェルを用いて刻み目をつけ、または調製することができる。これらは、その中の活性成分をスローまたは制御放出するように、例えば所望の放出プロファイルをもたらすための様々な比率のヒドロキシプロピルメチルセルロース、他のポリマーマトリックス類、リポソーム類、および/またはマイクロスフェア類を使用して製剤することもできる。これらは、例えば細菌保持フィルターによる濾過によって、あるいは使用直前に滅菌水または何らかの他の無菌注射用媒体に溶解することができる無菌固体組成物の形で滅菌剤を組み込むことによって、無菌化することができる。これらの組成物は、場合によっては不透明化剤を含有してもよく、活性成分のみをまたはそれを優先的に、消化管のある部分で、場合によっては遅延方式で放出する組成物の特徴を示すことがある。使用することができる包埋組成物としては、例えば高分子物質類およびワックス類が挙げられる。プロテアーゼ類は、適切な場合には上記の賦形剤の1種または複数と共にマイクロカプセルの形とすることもできる。本発明の化合物の経口投与用の液体剤形としては、医薬として許容される乳剤、マイクロエマルジョン、液剤、懸濁剤、シロップ剤、およびエリキシル剤が挙げられる。液体剤形は、活性成分に加えて、例えば水または他の溶媒、可溶化剤および乳化剤、具体的にはエチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油類(特に、綿実油、落花生油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油、およびゴマ油)、グリセロール、ポリエチレングリコール類、およびソルビタンの脂肪酸エステル、ならびにそれらの混合物など、当技術分野でよく使用される不活性希釈剤を含有することができる。不活性希釈剤の他に、経口組成物は、湿潤化剤、乳化および懸濁化剤、甘味剤、着香剤、着色剤、芳香剤、ならびに保存剤などのアジュバントも含むことができる。懸濁剤は、活性化合物に加えて、例えばエトキシ化イソステアリルアルコール類、ポリオキシエチレンソルビトールおよびソルビタンエステル類、微結晶セルロース、メタ水酸化アルミニウム、ベントナイト、寒天、およびトラガカント、ならびにそれらの混合物として懸濁化剤を含有することができる。
【0056】
直腸または腟内投与用の本発明の医薬組成物の製剤は、本発明のプロテアーゼ類を例えばカカオバター、ポリエチレングリコール、座剤ワックスまたはサリチラートを含む1種または複数の適当な非刺激性賦形剤または担体と混合することによって調製することができ、室温では固体であるが体温では液体となり、したがって直腸または膣腔で溶融し、活性化合物を放出する坐剤として提供することもできる。腟内投与に適した本発明の製剤としては、当技術分野で適切であると知られているような担体を含有する腟坐剤、タンポン剤、クリーム剤、ゲル剤、ペースト剤、フォーム剤、またはスプレー製剤も挙げられる。本発明の化合物の局所または経皮投与用の剤形としては、散剤、スプレー剤、軟膏剤、ペースト剤、クリーム剤、ローション剤、ゲル剤、液剤、貼付剤、および吸入剤が挙げられる。プロテアーゼ類は、医薬として許容される担体、および必要とされる場合がある任意の保存剤、緩衝剤、または噴射剤と無菌条件下で混合してもよい。軟膏剤、ペースト剤、クリーム剤、およびゲル剤は、本発明の活性化合物に加えて、動物および植物性脂肪類、油類、ワックス類、パラフィン類、デンプン、トラガカント、セルロース誘導体類、ポリエチレングリコール類、シリコーン類、ベントナイト類、ケイ酸、タルク、ならびに酸化亜鉛、またはそれらの混合物などの賦形剤を含有することができる。
【0057】
散剤およびスプレー剤は、本発明のプロテアーゼ類に加えて、ラクトース、タルク、ケイ酸、水酸化アルミニウム、ケイ酸カルシウム類、およびポリアミド粉末、またはこれらの物質の混合物などの賦形剤を含有することができる。スプレー剤は、さらにクロロフルオロ炭化水素類、ならびにブタンおよびプロパンなどの揮発性非置換炭化水素類など慣例の噴射剤を含有することができる。
【0058】
経皮貼付剤には、本発明のプロテアーゼ類の体内への送達を制御する利点が付加されている。このような剤形は、プロテアーゼ類を適切な媒体に溶解または分散することによって作製することができる。吸収促進剤を使用して、皮膚を通過するプロテアーゼ類の流入を増大させることもできる。このような流入の速度は、速度制御膜を備えることによって、または化合物をポリマーマトリックスもしくはゲルに分散することによって制御することができる。
【0059】
眼科用製剤、眼軟膏剤、散剤、液剤なども本発明の範囲内であると考えられる。
【0060】
非経口投与に適した本発明の医薬組成物は、1種または複数の医薬として許容される無菌の水性もしくは非水性等張液剤、分散剤、懸濁剤、もしくは乳剤、または使用直前に無菌注射用液剤もしくは分散剤に再構成することができる無菌粉末と組み合わせて本発明のプロテアーゼ類を含み、抗酸化剤、緩衝剤、静菌剤、製剤を所期のレシピエントの血液と等張にする溶質、または懸濁化もしくは粘稠化剤を含有することができる。本発明の医薬組成物中で使用される適当な水性および非水性担体としては、例えば水、エタノール、ポリオール類(グリセロール、プロピレングリコール、ポリエチレングリコールなど)、およびそれらの適当な混合物、オリーブ油などの植物油類、ならびにオレイン酸エチルなどの注射用有機エステル類が挙げられる。適切な流動性は、例えばレシチンなどのコーティング材料の使用によって、分散剤の場合は必要とされる粒径の維持によって、および界面活性剤の使用によって維持することができる。これらの組成物は、保存剤、湿潤化剤、乳化剤および分散化剤などのアジュバントも含有することができる。微生物の本組成物に対する作用の予防は、様々な抗菌剤および抗真菌剤、例えばパラベン、クロロブタノール、フェノール ソルビン酸などの包含によって確実にすることができる。糖類、塩化ナトリウムなどの等張剤を組成物に含めることも望ましい場合もある。さらに、注射用剤形の吸収は、吸収を遅延させる作用剤、具体的にはモノステアリン酸アルミニウムおよびゼラチンを含めることによって延長することができる。場合によっては、薬物の効果を延長させるために、皮下または筋肉内注射による薬物の吸収を遅くすることが望ましい。これは、水溶性が不十分な結晶質または非晶質材料の液体懸濁剤を使用することによって実現することができる。あるいは、非経口投与された薬物剤形の遅延吸収は、薬物を油ビヒクルに溶解または懸濁することによって実現される。注射用デポー剤形は、ポリラクチド−ポリグリコリドなどの生分解性ポリマー類中に本化合物のマイクロカプセル化マトリックスを形成することによって作製される。薬物とポリマーの比、および使用する特定のポリマーの性質に応じて、薬物放出速度を制御することができる。他の生分解性ポリマー類としては、例えばポリ(オルトエステル)類)およびポリ(無水物)類が挙げられる。
【0061】
デポー注射用製剤は、体組織と相溶性があるリポソームまたはマイクロエマルジョン中に薬物を封入することによっても調製される。本発明のプロテアーゼ類は、医薬としてヒトおよび動物に投与するとき、それ自体、または例えば0.1%〜99.5%(より好ましくは0.5%〜90%)のプロテアーゼ類を医薬として許容される担体と組み合わせて含有する医薬組成物として投与することができる。本発明の調製物は、経口、非経口、局所、または直腸投与することができる。これらは、当然、各投与経路に適した剤形によって投与される。例えば、これらは、錠剤またはカプセル剤形で、注射、吸入、点眼剤、軟膏剤、坐剤などによって投与され;注射、輸注、または吸入によって投与され;ローション剤または軟膏剤によって局所投与され;あるいは坐剤によって直腸投与される。経口および局所投与が好ましい。
【0062】
本明細書では「非経口投与」および「非経口投与された」という語句は、経腸および局所投与以外の投与モード、通常は注射による投与モードを意味し、静脈内、筋肉内、動脈内、髄腔内、嚢内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下、関節内、嚢下、クモ膜下、脊髄内、および胸骨内注射および輸注が挙げられるが、これらに限定されるものではない。
【0063】
ヒトおよび他の動物に、治療のために本発明の医薬品を経口、例えばスプレー剤によるような経鼻、直腸、腟内、非経口、大槽内、ならびに散剤、軟膏剤、またはドロップ剤によるような頬側および舌下を含めて局所投与を含めて、任意の適当な投与経路によって投与することができる。選択された投与経路にかかわらず、適当な水和した形で使用することができる本発明のプロテアーゼ類および/または本発明の医薬組成物を、当業者に周知の通常の方法で医薬として許容される剤形に製剤する。
【0064】
本発明の医薬組成物中の活性成分の実際の用量レベルは、患者への毒性がなく、特定の患者、組成物、および投与モードで所望の治療効果を実現するのに有効な一定量の活性成分が得られるように変更することができる。用量レベルの選択は、使用する本発明の特定のプロテアーゼの活性、投与経路、投与時間、使用する特定の化合物の排泄速度、治療期間、使用するプロテアーゼ類と組み合わせて使用する他の薬物、化合物、および/または材料、治療対象の患者の年齢、性別、体重、病態、全身的健康状態、および前病歴、ならびに医療技術分野で周知の同様な要因を含めて、様々な要因に依存する。当技術分野の医師または獣医は、必要とされる医薬組成物の有効量を容易に確定し、処方することができる。例えば、医師または獣医であれば、医薬組成物中で使用される本発明のプロテアーゼ類の用量を、所望の治療効果を実現するために必要とされるレベルより低いレベルで開始し、所望の効果が実現されるまで投与量を徐々に増加させることができるであろう。
【0065】
本発明のプロテアーゼ類は単独投与することができるが、プロテアーゼ類を医薬製剤(組成物)として投与することが好ましい。
【0066】
本発明の別の好ましい実施形態によれば、医薬品は、さらに少なくとも1種の別の活性成分を含む。
【0067】
前記活性成分は、本発明に従うプロテアーゼ類を用いた血管新生疾患の予防および治療を支援することができる任意の活性成分であり得る。しかし、前記プロテアーゼ類以外の効果を示す活性成分を添加することも当然可能である。
【0068】
少なくとも1種の別の活性成分は、好ましくはフラボノイド類、特にバイオフラボノイド類、抗酸化剤、またはシロヤナギ樹皮抽出物のような他の物質からなる群から選択される。
【0069】
本発明の別の好ましい実施形態によれば、医薬品は、ブロメライン、パパイン、フィシン、ナットウキナーゼ、ブリナーゼ、プロナーゼ、セラペプターゼ、レプチラーゼ、オキアミ酵素、バトロキソビン、ルンブロキナーゼ、ククミシン、サブチリシン、セアプローゼからなる群から選択された少なくとも1種(2種、3種、またはさらには4種)のプロテアーゼ類、および任意選択で別の活性成分を含み、前記別の活性成分は、好ましくはフラボノイド、特にルチンである。
【0070】
本発明の好ましい実施形態によれば、フラボノイドは、ルチンまたはその誘導体からなる群から選択される。
【0071】
例えば、ブロメラインおよびパパインは、チオールプロテアーゼ類と呼ばれ、活性部位にシステイン残基を含む。酸化条件下で、このシステインのチオール基は水素原子を失い、別のチオール基と架橋し、ジスルフィド橋を形成し、あるいは同じ酸化過程で別の残基と架橋することができる。この酸化状態では、ブロメラインおよびパパインは活性を失う。抗酸化剤であるビタミンC、ルチンおよびプロアントシアニジン類のようなバイオフラボノイド類を包含することによって、チオールプロテアーゼ類の活性スルフヒドリル基の酸化を防止することができる。
【0072】
前記別の活性成分は、好ましくは本発明の医薬品中に5重量/重量%〜35重量/重量%、好ましくは10重量/重量%〜30重量/重量%、より好ましくは15重量/重量%〜25重量/重量%の量で含まれる。
【0073】
次の図面および実施例によって、本発明をさらに説明する。
【図面の簡単な説明】
【0074】
【図1】酵素処理したHUVECの上清へのLDH放出を示す(毒性試験)。
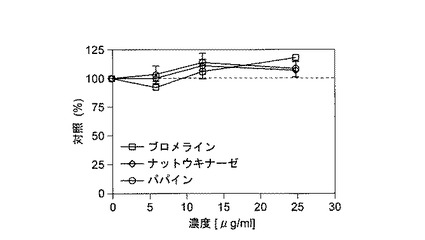
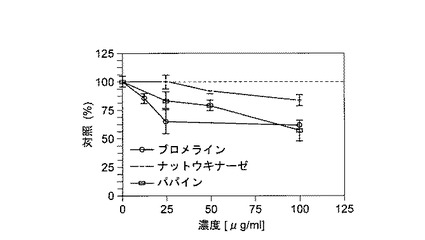
【図2】酵素処理したHUVECを用いたMTTアッセイを示す(抗増殖作用)。
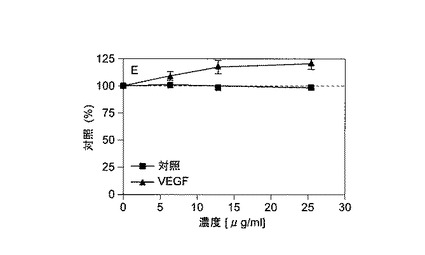
【図3】Aは、VEGF誘導管腔構造形成の25%ブロメライン、50%ナットウキナーゼ、および25%パパインの組合せによる抑制を示し、Bは、VEGFでのみ処理した対照を示す。VEGF対照では、形成された管腔構造の狭いパターンが見られるが、酵素処理試料は、酵素カクテルの抗血管新生活性を示唆する非管腔構造形成の広域を示す。
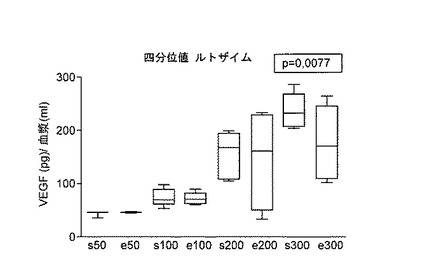
【図4】ルトザイム(Rutozym)で治療した患者のVEGF血中濃度を示す。
【図5】VEGF刺激HUVECおよびルトシド(Rutosid)を用いたMTTアッセイを示す。増殖の抑制は見られない。
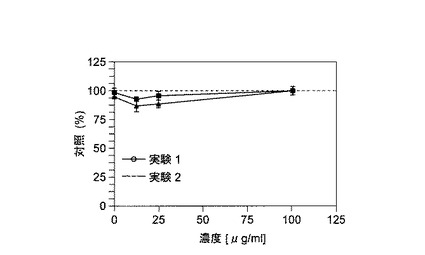
【図6】ルトシドのHUVECに及ぼす毒性効果を示す。静止期のHUVECにおいては、毒性効果が見られないが、VEGFによって活性化されたHUVECはわずかな効果を示す。
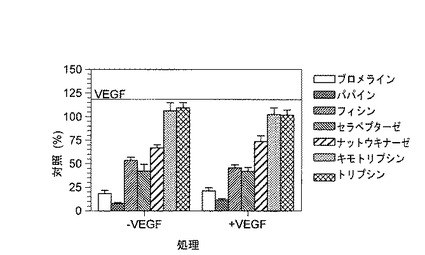
【図7】左側は、HUVECにおける自発的管腔構造形成の抑制を示す。ブロメライン、フィシン、ナットウキナーゼ、パパイン、およびセラペプターゼは管腔構造形成を抑制したが、キモトリプシンまたはトリプシンは抑制しなかった。右側は、HUVECにおけるVEGF誘導管腔構造形成の抑制を示す。ブロメライン、フィシン、ナットウキナーゼ、パパイン、およびセラペプターゼは、無処理HUVECの場合とほぼ同じ程度に管腔構造形成を抑制したが、キモトリプシンまたはトリプシンは抑制しなかった。
【実施例】
【0075】
材料:
シグマ・アルドリッチ(Sigma Aldrich)(オーストリア)から、活性3.51U/mgのパイナップル茎由来ブロメラインを入手した。
株式会社日本生物科学研究所(Japan Bio Science Laboratory Co, Ltd.)から、活性10.000U/mlのナットウキナーゼを購入した。
シグマ・アルドリッチ(Sigma Aldrich)(オーストリア)から、活性>3U/mgのカリカ・パパイア(Carica Papaya)由来パパインを入手した。
【実施例1】
【0076】
毒性試験
ブロメライン、ナットウキナーゼ、およびパパインの抗増殖作用を、乳酸デヒドロゲナーゼ(LDH)アッセイで評価した。セミコンフルエントな状態まで培養したヒト臍帯静脈内皮細胞(HUVEC)をトリプシンで処理することによって回収し、予めヒトフィブロネクチンでコーティングした96ウェルマイクロプレートに、2500個の細胞/ウェルの密度で播種した。適切な付着を可能にするために、10%ウシ胎仔血清、60μg/mlの内皮細胞増殖サプリメントhrEGF、hrFGF2、hrIGF、hrVEGF、アスコルビン酸、およびヘパリンを含有する内皮細胞基本培地2MV(ケンブレックス・バイオケミカルズ(Cambrex Biochemicals))中で、細胞を24時間インキュベートした。付着した後、増殖因子を含まない培地199+10%ウシ胎仔血清(FCS)中37℃/湿度95%でインキュベーションすることによって、細胞を飢餓状態にした。24時間後、10%FCS、VEGFおよび様々な濃度の酵素を含有する培地199で、上清を置換した。48時間インキュベートした後、上清を回収し、製造業者(プロメガ(Promega)、ドイツ)の指示書に従ってLDHアッセイを行った。すべてのウェルから50μlずつ、新しい96ウェル平底(酵素アッセイ)プレートに移した。Assay BufferをSubstrate Mixに添加し、穏やかに混合した。再構成したSubstrate Mixを各ウェルに50μlずつ添加した。プレートを室温で30分間インキュベートした。Stop Solutionを各ウェルに50μlずつ添加した。1時間以内に、490nmの光学密度を参照波長620nmで測定した。結果を無処理対照(%)で表す。
【0077】
結果を図1に示す。これらの結果から、2日間インキュベートした後、ブロメライン、ナットウキナーゼ、およびパパインは25μg/mlを含むレベルまで、HUVECに毒性効果を示さなかったことが明らかである。
【実施例2】
【0078】
抗増殖作用
ブロメライン、ナットウキナーゼ、およびパパイン、ならびにそれらの混合物の抗増殖作用を、3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(MTT)アッセイで評価した。セミコンフルエントな状態まで培養したヒト臍帯静脈内皮細胞(HUVEC)をトリプシンで処理することによって回収し、予めヒトフィブロネクチンでコーティングした96ウェルマイクロプレートに、1000個の細胞/ウェルの密度で播種した。適切な付着を可能にするために、10%ウシ胎仔血清、60μg/mlの内皮細胞増殖サプリメントhrEGF、hrFGF2、hrIGF、hrVEGF、アスコルビン酸、およびヘパリンを含有する内皮細胞基本培地2MV(ケンブレックス・バイオケミカルズ(Cambrex Biochemicals))中で細胞を24時間インキュベートした。付着した後、増殖因子を含まない培地199+10%ウシ胎仔血清(FCS)中37℃/湿度95%でインキュベーションすることによって、細胞を飢餓状態にした。24時間後、10%FCS、様々な濃度のブロメライン、ナットウキナーゼ、およびパパインを含有する培地199で、上清を置換した。細胞を、37℃/湿度95%でさらに48時間インキュベートした。EZ4U MTTキット(バイオメディカ(Biomedica)、オーストリア;製造業者の指示書に従う)を使用して、MTTアッセイを実施した。450nmの光学密度を参照波長620nmで測定した。結果を増殖(%)で表す。100%はVEGF処理対照の増殖である。
【0079】
濃度反応実験の結果を図2に示す。これらの結果は、ブロメライン、ナットウキナーゼ、およびパパインの明らかな抗増殖効果を示し、ブロメラインおよびパパインは、25μg/mlと低い濃度で75%の増殖に到達した。LDH放出試験の結果とまとめると、これらのデータは、明らかに抗増殖性ではあるが細胞毒性のない作用を示唆する。
【0080】
これらの混合物の結果を表1に示す。明らかな抗増殖効果が見られる。
【0081】
【表2】
【実施例3】
【0082】
抗血管新生活性
方法:
抗血管新生活性を管腔構造形成アッセイで評価した。成長因子低減Matrigel(ベクトン・ディッキンソン(Becton Dickinson)、ウィーン)を4℃の温度で解凍した。96ウェルマイクロタイタープレートのウェルに、50μl/ウェルをピペットで加えた。プレートを4℃で24時間放置した。実験する前に、プレートを37℃で30〜60分間インキュベートして、ゲルを凝固させた。HUVECをトリプシンで処理することによって回収し、製造業者の指示書に従ってアスコルビン酸およびヒドロコルチゾンを補充した内皮増殖培地2(MV)(ロンザ(Lonza)、ブリュッセル)、ならびに5000U/mlのヘパリンおよび1%ウシ胎仔血清中のMatrigelでコーティングした96ウェルマイクロプレートに、20.000個の細胞/ウェルの密度で播種した。薬物およびVEGFを所望の濃度に添加した。さらに16〜18時間後、各ウェルの写真を撮影した。管腔構造の全長は、Neuron Length Determination Pluginで測定し、ImageJ Softwareを使用して確定した。
【0083】
結果
酵素および酵素混合物の管腔構造形成アッセイの結果を図3、図7、および表1に示す。
【0084】
表1は、ブロメライン、ナットウキナーゼ、およびパパインの様々な混合物による管腔構造形成の抑制を示す。結果は、次のことを明らかに示す。
1.管腔構造形成は、酵素であるブロメライン、フィシン、ナットウキナーゼ、パパイン、およびセラペプターゼによって抑制されている。
2.ブロメライン、ナットウキナーゼ、およびパパインの組合せは、これら薬物の単独より効果が高い。
【0085】
【表3】
【0086】
表1:VEGFの存在下HUVECにおける管腔構造形成(対照(%))。結果を管腔構造形成(%)で示す。100%は、VEGFでのみ処理した試料における管腔構造形成に等しい。示していない組合せは管腔構造形成を抑制しなかった。
【0087】
結論
VEGFによって媒介された管腔構造のような微小血管の形成は、ブロメライン、フィシン、ナットウキナーゼ、パパイン、またはセラペプターゼ単独で処理した後、ならびに酵素であるブロメライン、ナットウキナーゼ、およびパパインの混合物で処理した後、大幅に低減したことが認められた。
【実施例4】
【0088】
この実施例では、酵素療法(酵素混合物:ナットウキナーゼ、ブロメライン、パパイン+ルチンバイオフラボノイド、シロヤナギ樹皮抽出物)のVEGF血中濃度の量に及ぼす効果を検討した。酵素療法はヒト血中のVEGF濃度の上昇を大幅に低減するということを明らかにする可能性がある(下記の結果を参照のこと)。
【0089】
試験は、2型糖尿病患者男女111名を対象に、並行比較2群として多施設共同無作為化非盲検パイロット研究として行った。54名の患者は、酵素混合物(ナットウキナーゼ(20000FU/gm)25mg、ブロメライン(2450GDU/gm)90mg、パパインN.F.(2.400USP単位/mg)100mg、ルチンバイオフラボノイド複合体(ルトシド(rutoside)類およびルチノシド類)120mg、シロヤナギ樹皮抽出物(15%サリシン/7%ポリフェノール類)100mg、マーリン・ニュートラシューティカルズ(Marlyn Nutraceuticals)、米国)を4週間摂取した。患者の血漿中のVEGF濃度を、補給前および補給4週間直後に試験した。患者は、自分自身が初期値で自己対照として機能する。
【0090】
VEGF血中濃度を異なる4群に分けた(四分位値;図4を参照のこと):患者における治療前VEGF血中濃度<50ng/ml;(s50=開始<50ng/ml;e50=終わり)s100:治療前VEGF濃度<100ng/ml;s200:治療前<200ng/ml、およびs300:治療前VEGF>200ng。植物および/または微生物を供給源とするプロテアーゼ類を含む本発明に従う医薬品の使用は、VEGF血中レベルを低減するために使用することができ、したがって新血管新生に関連した疾患の治療に使用できることを示し得る。
【実施例5】
【0091】
ルトシドのHUVECに及ぼす抗増殖性効果
ルトシドの抗増殖作用の可能性を評価するために、3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(MTT)アッセイを利用した。セミコンフルエントな状態まで培養したヒト臍帯静脈内皮細胞(HUVEC)をトリプシンで処理することによって回収し、予めヒトフィブロネクチンでコーティングした96ウェルマイクロプレートに、1000個の細胞/ウェルの密度で播種した。適切な付着を可能にするために、10%ウシ胎仔血清、60μg/mlの内皮細胞増殖サプリメントhrEGF、hrFGF−2、hrIGF、hrVEGF、アスコルビン酸、およびヘパリンを含有する内皮細胞基本培地2MV(ケンブレックス・バイオケミカルズ(Cambrex Biochemicals))中で、細胞を24時間インキュベートした。付着した後、増殖因子を含まない培地199+10%ウシ胎仔血清(FCS)中37℃/湿度95%でインキュベーションすることによって、細胞を飢餓状態にした。24時間後、10%FCS、VEGFおよび様々な濃度のルトシドおよびVEGFを含有する培地199で、上清を置換した。細胞を、37℃/湿度95%でさらに48時間インキュベートした。EZ4U MTTキット(バイオメディカ(Biomedica)、オーストリア)を製造業者の指示書に従って使用して、MTTアッセイを実施した。450nmの光学密度を参照波長620nmで測定した。結果を増殖(%)で表す。100%はVEGF処理対照の増殖である。
【0092】
結果は、ルトシドがVEGF刺激HUVEC増殖を抑制しないことを明らかに示す(図5を参照のこと)。
【実施例6】
【0093】
ルトシドのHUVECに及ぼす毒性効果
ルトシドの細胞毒性の可能性を試験するために、乳酸デヒドロゲナーゼ(LDH)アッセイを利用した。セミコンフルエントな状態まで培養したヒト臍帯静脈内皮細胞(HUVEC)をトリプシンで処理することによって回収し、予めヒトフィブロネクチンでコーティングした96ウェルマイクロプレートに、2500個の細胞/ウェルの密度で播種した。適切な付着を可能にするために、10%ウシ胎仔血清、60μg/mlの内皮細胞増殖サプリメントhrEGF、hrFGF−2、hrIGF、hrVEGF、アスコルビン酸、およびヘパリンを含有する内皮細胞基本培地2MV(ケンブレックス・バイオケミカルズ(Cambrex Biochemicals))中で、細胞を24時間インキュベートした。付着した後、増殖因子を含まない培地199+10%ウシ胎仔血清(FCS)中37℃/湿度95%でインキュベーションすることによって、細胞を飢餓状態にした。24時間後、10%FCS、VEGFおよび様々な濃度の酵素を含有する培地199で、上清を置換した。48時間インキュベートした後、上清を回収し、製造業者(プロメガ(Promega)、ドイツ)の指示書に従ってLDHアッセイを行った。すべてのウェルから50μlずつ、新しい96ウェル平底(酵素アッセイ)プレートに移した。Assay BufferをSubstrate Mixに添加し、穏やかに混合した。再構成したSubstrate Mixを各ウェルに50μlずつ添加した。プレートを室温で30分間インキュベートした。Stop Solutionを各ウェルに50μlずつ添加した。1時間以内に、490nmの光学密度を参照波長620nmで測定した。結果を無処理対照(%)で表す。
【0094】
結果は、ルトシドの毒性がHUVECにおいて無視できる程度であることを明らかに示す(図6を参照のこと)。
【実施例7】
【0095】
酸素誘発網膜症およびレーザー誘発脈絡膜血管新生のマウスモデルにおける植物由来のプロテアーゼ類によるVEGFの抑制
血管増殖性網膜症は、工業先進国の重度視力喪失の主な原因であり、基礎疾患は糖尿病、網膜静脈閉塞症、未熟児網膜症、または後期の加齢黄斑変性(AMD)である。虚血性網膜疾患の標準的治療は、VEGFのような血管新生因子の産生を最小限に抑えるための末梢網膜組織の破壊に基づいている。神経組織の破壊は不可逆であるので、血管新生因子阻害薬を用いた局所療法は、網膜または脈絡膜血管新生の発症リスクが上昇している患者を保護するために望ましい。一方、AMDでは、血管形成阻害薬を用いた局所療法が、標準的ケアになった。
【0096】
方法
A.酸素誘発網膜症(OIR)
マウス酸素誘発網膜症(OIR)モデルを、スミス(Smith)および同僚が記述している(Invest. Ophthalmol. Vis. Sci.(1994年)、35巻:p.101−111)ように樹立した。7日(P7)齢C57/B16Jマウスを、75%酸素中にP12まで入れた。正常な酸素に戻した後、マウスは相対的に低酸素のため網膜血管新生を発生した。この効果は、一方の眼に硝子体内注射された被検物質の影響を受けた。他方の眼には、対照液剤を注射した。P17に、フルオレセインデキストランで灌流した後、フラットマウントによって網膜増殖を評価した。これらのホールマウントによって、網膜脈管構造の血管病変をコード化した形で評価することが可能になる。各フラットマウントについて、評価システムに従ったスコアを決定し、ウィルコクソン順位和検定法で比較し、処理と対照の間で有意差を得た。合計30匹のマウス/群を使用した。
【0097】
第2の試験では、P12に、被検物質を腹腔内注射した。この場合、2つの眼の間での個人内比較は可能でなく、したがって対照群がさらに必要であった。合計25匹のマウス/群を使用した。
【0098】
B.レーザー誘発脈絡膜血管新生(レーザー誘発CNV)
マウスレーザー誘発脈絡膜血管新生モデルを、カンポキアーロ(Campochiaro)および同僚が記述しているように樹立した(トベ(Tobe)ら、Am. J. Pathol.(1998年)、153巻:p.1641−1646。0日(d0)に、12週齢より若くないC57/B16Jマウスに麻酔をかけ、視覚制御されたレーザーによる3つの熱傷を網膜に与えて、新生血管形成を誘発させた。マウスは、創傷後2週間以内にレーザー部位で脈絡膜血管新生を発生した。d7すなわち7日に、被検物質を一方の眼に、かつ対照液剤を他方の眼に硝子体内注射して、網膜または脈絡膜血管新生に影響があるかどうかを確認した。13日より後のd14に、マウスをデキストラン−フルオレセインで灌流し、脈絡膜ホールマウントを調製した。ホールマウントによって、クロリドの血管病変およびCNV膜のサイズを評価することが可能になる。各レーザー部位の値をウィルコクソン順位和検定法で比較し、処理と対照の間で有意差を得た。合計30匹のマウス/群を使用した。
【0099】
別の実験では、P7すなわち7日に、被検物質を腹腔内注射した。この場合、2つの眼の間での個人内比較は可能でなく、したがって対照群がさらに必要であった。合計25匹のマウス/群を使用した。
【技術分野】
【0001】
本発明は、新血管新生に関連した疾患の治療用医薬品に関する。
【背景技術】
【0002】
血管新生は、既存の血管からの内皮細胞の増殖による新しい血管の形成と定義される。この過程で、内皮細胞は、その下にある基底膜を退化させ、増殖し、隣接組織に遊走し、集合して管を構築する。最後に、管と管の連結が行われ、血流が確立する。成熟組織の変化する要求に適応する能力は、低酸素誘導因子(HIF)および血管内皮因子(VEGF)のような可溶性因子と、細胞−細胞および細胞−マトリックス相互作用とを必要とする。
【0003】
VEGFは、本来実質的な血管漏出を引き起こす因子と記述され、血管透過性因子(VPF)と呼ばれた。同じタンパク質は、内皮細胞におけるその分裂促進作用によって、後に改めて血管内皮増殖因子(VEGF)と命名された。
【0004】
VEGFは、微小血管床の透過性を増大させ、したがって血管からの流体およびタンパク質の漏出を促進する。これによって、浮腫、創傷液および漿液腫(例えば、手術後)、浸出液(例えば、慢性炎症性疾患における)、ならびに腹水(例えば、癌における)の発生が起こる。VEGFは、血管透過性の誘導においてヒスタミンより10,000倍強力である。
【0005】
さらに、VEGFは、内皮細胞増殖の最も強力な刺激剤の1つである。最後に、これは、内皮細胞からの毛細管の形成を刺激し、したがって血管新生にとって必要である事象のカスケードを促進する。新たに形成された組織における既存の血管からの新しい毛細管の増殖である新血管新生、またはさらには沈着物(歯垢などのような)は、種々の病理学的状態の発生および進行に寄与する。生理的条件下では、血管新生は、厳密に調節された過程である。この過程は、癌、関節リウマチ、子宮内膜症、乾癬、または眼血管新生のような病理学的状態においては、かなり強化され、かつ機能不全である。
【0006】
抗血管新生薬は、癌、関節リウマチ、乾癬、および眼血管新生などのような疾患の将来の治療を改良するということを示唆する証拠がますます増えている。生体内実験によって、低酸素(例えば、腫瘍壊死に近い領域における低酸素)は、様々なタイプの細胞においてVEGFとVEGF受容体(VEGFR−1)との発現を誘導できることが実証された。低酸素は、低酸素誘導因子1(HIF−1)の発現を引き起こす。引き続いて、HIF−1複合体は、細胞核内に蓄積し、DNAのHIF−1結合部位に結合し、隣接した血管を低酸素組織に発芽させることができる血管新生スイッチを始動させるVEGF−mRNAの転写を開始または上方制御する。さらに、VEGF発現は、乾癬または関節リウマチのような慢性炎症の様々なモデルで実証されているように、様々な炎症誘発性サイトカインによって誘導/上方制御することができる。
【0007】
VEGFは、プロテアーゼ活性化α2Mによってα2マクログロブリン(α2M)経路を介して循環から除去することができる。α2M−プロテアーゼ複合体は、表面の円蓋においてVEGFを結合させることができる。得られたα2M−酵素−VEGF複合体は、マクロファージおよび内皮細胞のような細胞の表面上で発現したLRP受容体に結合し(低密度リポタンパク質受容体関連タンパク質受容体)、貪食され、破壊される。タンパク質分解酵素を用いた経口療法は、活性化したα2M分子の数を増加し、したがって有機体のサイトカイン/増殖因子破壊能を高める。(デザー(Desser L)ら、「キャンサー・ケモセラピー・アンド・ファーマコロジー増補(Cancer Chemother Pharmacol Suppl)」、(2001年)、47巻:S10−S15;ラウア(Lauer D)ら、「キャンサー・ケモセラピー・アンド・ファーマコロジー増補(Cancer Chemother Pharmacol Suppl)」、(2001年)、47巻:S4−S9)。
【0008】
最近、血管新生の増大を伴う疾患、主に癌の治療のためであるが、黄斑変性症など眼における血管新生を伴う疾患のためにも、VEGF受容体遮断薬またはVEGFに対する抗体を使用した複数の治療的手法が提案されている。
【0009】
眼血管新生または新血管新生は、盲目の最も多い原因として示唆され、約20種類の異なる眼疾患の病態の基礎をなす。例えば、糖尿病において、網膜において形成された新しい毛細管は、ガラス様液に侵入し、出血および盲目を引き起こす。
【0010】
国際公開第2005/110453号は、VEGFおよびVEGF受容体を切断するためのヒト野生型および変異型タンパク質MT−SP1プロテアーゼ類の使用に関する。このような切断は、血管新生の低減を生じ、したがって血管新生を伴う病態を治療するために使用することができる。
【0011】
特開昭60−112720号は、血管新生を伴わない疾患(例えば、緑内障)を治療するパパインおよびクエン酸の使用に関する。
【0012】
国際公開第2004/046199号では、眼疾患を治療するコンドロイチン硫酸の使用が開示されている。
【0013】
国際公開第2005/056784号は、糖尿病を治療するナットウキナーゼの使用に関する。
【0014】
旧ソ連特許第1342500号では、緑内障のような眼疾患を治療するパパインの使用が記載されている。
【0015】
米国特許第6103756号は、眼障害を治療するために使用することができる抗酸化剤およびフラボノイド類を含む組成物に関する。
【発明の概要】
【発明が解決しようとする課題】
【0016】
本発明の目的は、新血管新生に関連した眼疾患を治療または予防するための医薬品を提供することである。
【課題を解決するための手段】
【0017】
したがって、本発明は、加齢黄斑変性(AMD)、脈絡膜血管新生、ヒッペル・リンダウ病、虹彩血管新生、虚血性増殖網膜症、角膜血管新生、および増殖性鎌状赤血球性網膜症からなる群から選択された新血管新生に関連した眼疾患の治療および/または予防用の医薬品を製造するための少なくとも1種のプロテアーゼの使用であって、少なくとも1種のプロテアーゼは、植物プロテアーゼ類、非哺乳類動物プロテアーゼ類、および微生物プロテアーゼ類からなる群から選択される使用に関する。
【0018】
驚くべきことに、好ましくは植物、非哺乳類動物、および微生物プロテアーゼ類からなる群から選択された少なくとも1種のプロテアーゼの組合せを含む医薬品は特に、個体に投与されたときVEGFのレベルを大幅に(本発明に従う医薬品の投与前の前記個体のVEGFレベルに比べて、少なくとも40%、好ましくは少なくとも50%、より好ましくは少なくとも60%、さらにより好ましくは少なくとも70%、最も好ましくは少なくとも80%、特に少なくとも90%)低減させることができ、したがって血管新生を低減させることが明らかになった。したがって、少なくとも1種のプロテアーゼ、好ましくは少なくとも2種類(少なくとも3種類、少なくとも4種類、少なくとも5種類、少なくとも6種類)のプロテアーゼ類の組合せを、個体における新血管新生に関連した眼疾患を予防および/または治療するために使用することができる。微生物、非哺乳類動物、および植物プロテアーゼ類は、ヒトまたは動物の細胞、特に内皮細胞と接触させたとき顕著な毒性を示さないので、前記プロテアーゼ類の投与が特に適している。実に本明細書に開示するプロテアーゼ類の組合せは、動物またはヒトへの毒性がなく、しかも血管新生に働く。
【0019】
興味深いことに、トリプシンまたはキモトリプシンのような哺乳類(すなわち、ヒトおよび動物)由来のプロテアーゼ類は血管新生を抑制または予防できないことを示し得る。したがって、このようなプロテアーゼ類の個体への単独投与は、新血管新生に関連した疾患を予防または治療するために行うことはできない。
【0020】
本明細書に定義する「医薬品」という用語は、薬剤製品だけでなく、食事補助食品も包含する。
【0021】
本明細書では、「植物プロテアーゼ類」および「動物プロテアーゼ類」および「非哺乳類動物プロテアーゼ」は、植物または非哺乳類動物において自然発生し、それらから抽出または獲得されるプロテアーゼ類とするものである。「植物プロテアーゼ類」および「動物プロテアーゼ類」および「非哺乳類動物プロテアーゼ」は、コードするDNA(例えば、cDNAとして)がそれぞれ、植物および動物(そのゲノム中に前記DNAを天然に含む)に由来しまたはそれらから獲得され、適切なベクターにクローン化され、原核(例えば、細菌)または真核細胞(例えば、昆虫細胞、哺乳類細胞)培養物で発現する組換えプロテアーゼ類でもある。
【0022】
本明細書では、「微生物プロテアーゼ類」は、細菌類および真菌類(例えば、酵母、カビ類)などの微生物において自然発生するプロテアーゼ類である。しかし、前記プロテアーゼ類は、他の細胞または有機体から単離することもできる。ただし、前記細胞および有機体は、微生物プロテアーゼのDNAを含み、組換えによって前記プロテアーゼを生成できることを条件とする。
【0023】
治療および/または予防すべき血管新生に関連した眼疾患が上記の群から選択される場合、本発明に従う医薬品の使用は特に適している。これらの疾患はすべて、大部分は体内の血管内皮増殖因子(VEGF)のレベル上昇のために血管新生の増大を示す。しかし、本発明の医薬品を加齢黄斑変性を治療するために使用することが特に好ましい。
【0024】
複数の疾患は、本発明の医薬品を使用することもできる新血管新生に関連する。
【0025】
例えば、眼血管新生は、盲目の最も多い原因である(加齢黄斑変性;ヒッペル・リンダウ病;ベーチェット症候群;特発性眼血管新生)。
【0026】
本発明の医薬品を、高いVEGFレベルによって引き起こされ、血管新生に随伴した疾患を患っている個体を予防および/または治療するために使用し、それによってこれらの疾患が大部分または完全に、増殖活性の増大を伴うわけではないことが特に好ましい。
【0027】
少なくとも1種の植物プロテアーゼは、好ましくはブロメライン、パパイン、フィシン、およびククミシンからなる群から選択される。
【0028】
好ましくは本発明に従って使用することができる植物プロテアーゼ類は上記に示す。
【0029】
これらのプロテアーゼ類は、受容者における組換え発現、または前記プロテアーゼ類を自然に産生する植物からの抽出によって得ることができ、それによってその抽出物自体を、本発明に従う医薬品を製造するために直接使用することができる。プロテアーゼ類の抽出方法は当技術分野でよく知られている。
【0030】
例えば、ブロメラインは、果実を収穫した後の植物パイナップルの切株または根部から調製される。この切株または根部を畑から回収し、皮を剥がし、圧搾して、可溶性ブロメライン酵素を含有する汁を抽出する。その後の加工としては、酵素をさらに精製するための酵素の沈殿が挙げられる。
【0031】
パパインは、パパイアの果実の乳液を回収することによって、乾燥した粗材料として生成することができる。果実の頸部に刻み目をつけた後、乳液は回収され、その後果実上で乾燥してもよく、または容器に滴下してもよい。次いで、この乳液をさらに乾燥する。この時点で、これは乾燥した粗材料に分類される。汚染物質を除去する精製工程が必要である。この精製は、活性パパイン酵素の可溶化および抽出からなる。
【0032】
本発明の好ましい実施形態によれば、微生物プロテアーゼは、ナットウキナーゼ、ブリナーゼ、プロナーゼ、セアプローゼ、セラペプターゼ、およびサブチリシンからなる群から選択される。
【0033】
微生物プロテアーゼ類は、組換え技法によって得ることもでき、または前記プロテアーゼ類を産生する微生物を含む微生物培養物から直接単離してもよい。
【0034】
例えば、ナットウキナーゼは、発酵ダイズから作られた伝統的な日本食品である納豆から、または前記プロテアーゼを産生することができる特定のバチルス・スブチリス(Bacillus subtilis)亜種(バチルス・スブチリス・バラエティー・ナットウ(Bacillus subtilis var. natto))の有機体を含む培養物によって得られる。バチルス・スブチリス・バラエティー・ナットウ(Bacillus subtilis var. natto)は、自然土壌および日本の市販納豆から単離してもよい。この菌株は、フィブリンを分解するナットウキナーゼ産物の高活性をもたらす能力を有する。炭素源、有機窒素源または無機窒素源、鉱塩、初期pH、および温度は、バチルス・スブチリス・バラエティー・ナットウ(B. subtilis var. natto)によるナットウキナーゼ産生のために最適化されなければならない。例えば、バチルス・スブチリス・バラエティー・ナットウ(B. subtilis var. natto)の最適接種サイズは約5%(体積/体積)であることがわかった。最適培地は、2.8%ダイズタンパク質、1%酵母エキス、および0.8%マルトースを含有することができる。さらに、最適pHおよび温度はそれぞれ、約6.5±0.5、および約30℃〜40℃とすることができる。最適インキュベーション時間は18〜48時間である。発酵培地におけるナットウキナーゼ活性は、40FU/ml超まで上昇することがある。
【0035】
例えば、セラペプターゼは、カイコ類の腸内に存在するセラチア(Serratia)E15細菌類から単離されたタンパク質分解酵素である。この酵素は、疼痛および炎症を自然治療するために補助食品として使用することができ、アジアおよび欧州の一部分において、臨床で使用されている。セラペプターゼは、関節炎および炎症を治療するためによく使用される非ステロイド系抗炎症薬類(NSAID類)の代替物として使用される。
【0036】
非哺乳類動物プロテアーゼは、好ましくはレプチラーゼ、オキアミ酵素、バトロキソビン、およびルンブロキナーゼからなる群から選択される。
【0037】
これらのプロテアーゼ類は、当技術分野で知られている方法で組換えによって生成することができ、またはそれぞれの動物から直接に得ることができる。
【0038】
特に好ましい医薬品は、植物プロテアーゼ類であるブロメラインおよび/またはパパイン、ならびに任意選択で微生物プロテアーゼであるナットウキナーゼを含む。本発明に従う医薬品中のこれらのプロテアーゼ類の好ましい比は、下記の表で見られ、これによればパパイン、ナットウキナーゼ、および/またはブロメラインの量は互いに独立して、医薬品中に存在するプロテアーゼの全量の0%〜80%、好ましくは10%〜75%、より好ましくは15(または16,67)%〜75%で変わり得る。
【0039】
【表1】
【0040】
本発明の別の好ましい実施形態によれば、少なくとも1種の植物、非哺乳類動物、および/または微生物プロテアーゼは、医薬品中に10重量/重量%〜90重量/重量%、好ましくは20重量/重量%〜80重量/重量%、より好ましくは30重量/重量%〜70重量/重量%の量で含まれる。
【0041】
好ましくは、少なくとも1種の植物、非哺乳類動物、および/または微生物プロテアーゼは、1mg/体重1kg〜100mg/体重1kg、好ましくは2mg/体重1kg〜50mg/体重1kg、より好ましくは5mg/体重1kg〜20mg/体重1kgの所定量で個体に投与される。
【0042】
好ましくは、医薬品は、さらに少なくとも1種の医薬として許容される担体、希釈剤、および/または賦形剤、好ましくは結合剤、充填剤、崩壊剤、滑沢剤、保存剤、および/またはコーティング剤を含むことができる。
【0043】
本発明に従う医薬品の薬剤処方に応じて、賦形剤、コーティング剤などのような様々な他の物質を使用してもよい。
【0044】
さらに、本発明の医薬品は、好ましくは経口、局所、経腸、または非経口投与に適合させることができる。
【0045】
本発明の好ましい実施形態によれば、医薬品は、点眼剤、点耳剤、点鼻剤、スプレー式点鼻剤、錠剤、好ましくは可溶性錠剤、発泡錠、胃耐性錠剤、および舌下錠、カプセル剤、好ましくは胃耐性カプセル剤、散剤、顆粒剤、経口用液剤、経口用ドロップ剤、軟膏剤、ローション剤、乳剤、ヒドロゲル剤、坐剤、腟坐剤、輸注剤、および注射剤からなる群から選択された剤形で用いられる。
【0046】
特定の本発明の好ましい実施形態では、医薬品を経口投与に適合させる。この投与モードは、非侵襲性であり、したがって医薬品の反復投与を(患者を害することなく)可能にする。
【0047】
本発明の医薬品は、(1)経口投与用、例えば水薬(水性もしくは非水性液剤または懸濁剤)、錠剤、ボーラス、散剤、顆粒剤、舌に適用するためのペースト剤;(2)例えば滅菌液剤もしくは懸濁剤としての例えば皮下、筋肉内、または静脈注射による非経口投与用;(3)例えば皮膚に適用されるクリーム剤、軟膏剤、またはスプレーとしての局所塗布用;あるいは(4)例えば腟坐剤、クリーム剤、またはフォーム剤としての腟内または直腸内投与用に適合されたものを含めて、固体または液体の形で投与するために特別に製剤することができる。
【0048】
本明細書で「医薬として許容される」という語句は、正しい医学的判断の範囲内で、過剰の毒性、刺激、アレルギー応答、または他の問題もしくは合併症なしにヒトおよび動物の組織と接触させて使用するのに適し、妥当な利益/危険比に見合った化合物、材料、組成物、および/または剤形を意味するように使用される。
【0049】
医薬として許容される担体として機能することができる材料の例としては、下記が挙げられる。(1)ラクトース、グルコース、およびスクロースなどの糖類;(2)トウモロコシデンプンおよび馬鈴薯デンプンなどのデンプン類;(3)カルボキシメチルセルロースナトリウム、エチルセルロース、および酢酸セルロースなどのセルロースおよびその誘導体;(4)トラガカント末;(5)麦芽;(6)ゼラチン;(7)タルク;(8)カカオバターおよび座剤ワックス類などの賦形剤;(9)落花生油、綿実油、紅花油、ゴマ油、オリーブ油、トウモロコシ油、およびダイズ油などの油類;(10)プロピレングリコールなどのグリコール類;(11)グリセリン、ソルビトール、マンニトール、およびポリエチレングリコールなどのポリオール類;(12)オレイン酸エチルおよびラウリン酸エチルなどのエステル類;(13)寒天;(14)水酸化マグネシウムおよび水酸化アルミニウムなどの緩衝剤;(15)アルギン酸;(16)発熱物質を含まない水;(17)等張性生理食塩水;(18)リンゲル液;(19)エチルアルコール;(20)リン酸緩衝溶液類;ならびに(21)薬剤処方で使用される他の無毒性相溶物質類。
【0050】
湿潤化剤、ラウリル硫酸ナトリウムやステアリン酸マグネシウムなどの乳化剤および滑沢剤、ならびに着色剤、遊離剤、コーティング剤、甘味剤、着香剤、および芳香剤、保存剤、ならびに抗酸化剤も、本発明に従う医薬品中に存在することができる。医薬として許容される抗酸化剤としては、例えば(1)アスコルビン酸、システイン塩酸塩、重硫酸ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウムなどの水溶性抗酸化剤;(2)パルミチン酸アスコルビル、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、α−トコフェロールなどの油溶性抗酸化剤;ならびに(3)クエン酸、エチレンジアミン四酢酸(EDTA)、ソルビトール、酒石酸、リン酸などの金属キレート剤が挙げられる。
【0051】
本発明の製剤としては、経口、経鼻、局所(頬側および舌下を含む)、直腸、腟内、および/または非経口投与に適した製剤が挙げられる。製剤は、好都合なことに単位剤形で提供することができ、薬学技術分野でよく知られている任意の方法で調製することができる。担体材料と組み合わせて、単一剤形を生成することができる活性成分の量は、治療されている受容者および特定の投与モードに応じて異なる。担体材料と組み合わせて、単一剤形を生成することができる活性成分の量は、一般に治療効果をもたらす化合物のその量である。
【0052】
本発明に従う医薬品および製剤を調製する方法としては、本発明の化合物を担体lgおよび任意選択で1つまたは複数の副成分と結合させる工程が挙げられる。一般に、製剤は、本発明の化合物を液体担体もしくは微細化固体担体または両方と均一かつ緊密に結合させ、次いで必要なら、生成物を造形することによって調製される。経口投与に適した本発明の製剤は、カプセル剤、カシェ剤、丸剤、錠剤、ロゼンジ剤(着香基材、通常はスクロースおよびアカシアまたはトラガカントを使用する)、散剤、顆粒剤の形、または水性もしくは非水性液体中の液剤もしくは懸濁剤、または水中油型もしくは油中水型液体乳剤、またはシロップ剤、または香錠剤(ゼラチンおよびグリセリンなどの不活性基材、またはスクロースおよびアカシアを使用する)とすることができ、それぞれ、活性成分として所定量の本発明のプロテアーゼの組合せを含有する。
【0053】
本発明のプロテアーゼは、ボーラス、舐剤、またはペースト剤として投与することもできる。経口投与用の本発明の固体剤形(カプセル剤、錠剤、丸剤、糖衣錠、散剤、顆粒剤など)では、活性成分をクエン酸ナトリウムもしくは第二リン酸カルシウムなど1種もしくは複数の医薬として許容される担体、および/または次のいずれかと混合する:(1)デンプン類、ラクトース、スクロース、グルコース、マンニトール、および/またはケイ酸などの充填剤または増量剤;(2)例えばカルボキシメチルセルロース、アルギン酸塩/エステル/アルギナート類、ゼラチン、ポリビニルピロリドン、スクロース、および/またはアカシアなどの結合剤;(3)グリセロールなどの保湿剤;(4)寒天、炭酸カルシウム、馬鈴薯またはタピオカデンプン、アルギン酸、ある種のシリケート類、および炭酸ナトリウムなどの崩壊剤;(5)パラフィンなどの溶解遅延剤;(6)第四級アンモニウム化合物などの吸収促進剤;(7)例えばセチルアルコールおよびグリセロールモノステアレートなどの湿潤化剤;(8)カオリンおよびベントナイト粘土などの吸収剤;(9)タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール類、ラウリル硫酸ナトリウム、およびそれらの混合物などの滑沢剤;ならびに(10)着色剤。
【0054】
カプセル剤、錠剤、および丸剤の場合は、医薬組成物は緩衝剤も含むことができる。同様のタイプの固体組成物も、ラクトースまたは乳糖類のような賦形剤および高分子量ポリエチレングリコール類などを使用する軟質および硬質充填ゼラチンカプセル剤中の充填剤として使用することができる。
【0055】
錠剤は、任意選択で1種または複数の副成分と共に圧縮または成形によって作製することができる。圧縮錠は、結合剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース)、滑沢剤、不活性希釈剤、保存剤、崩壊剤(例えば、デンプングリコール酸ナトリウムまたは架橋カルボキシメチルセルロースナトリウム)、表面活性または分散剤を使用して調製することができる。成形錠剤は、不活性液体希釈剤で湿潤させた粉末化合物の混合物を適当な機械で成形することによって作製することができる。本発明の医薬組成物の錠剤、ならびに他の固体剤形、具体的には糖衣錠、カプセル剤、丸剤、および顆粒剤は、任意選択で製薬技術分野で周知の腸溶性コーティングおよび他のコーティングなどのコーティングおよびシェルを用いて刻み目をつけ、または調製することができる。これらは、その中の活性成分をスローまたは制御放出するように、例えば所望の放出プロファイルをもたらすための様々な比率のヒドロキシプロピルメチルセルロース、他のポリマーマトリックス類、リポソーム類、および/またはマイクロスフェア類を使用して製剤することもできる。これらは、例えば細菌保持フィルターによる濾過によって、あるいは使用直前に滅菌水または何らかの他の無菌注射用媒体に溶解することができる無菌固体組成物の形で滅菌剤を組み込むことによって、無菌化することができる。これらの組成物は、場合によっては不透明化剤を含有してもよく、活性成分のみをまたはそれを優先的に、消化管のある部分で、場合によっては遅延方式で放出する組成物の特徴を示すことがある。使用することができる包埋組成物としては、例えば高分子物質類およびワックス類が挙げられる。プロテアーゼ類は、適切な場合には上記の賦形剤の1種または複数と共にマイクロカプセルの形とすることもできる。本発明の化合物の経口投与用の液体剤形としては、医薬として許容される乳剤、マイクロエマルジョン、液剤、懸濁剤、シロップ剤、およびエリキシル剤が挙げられる。液体剤形は、活性成分に加えて、例えば水または他の溶媒、可溶化剤および乳化剤、具体的にはエチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油類(特に、綿実油、落花生油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油、およびゴマ油)、グリセロール、ポリエチレングリコール類、およびソルビタンの脂肪酸エステル、ならびにそれらの混合物など、当技術分野でよく使用される不活性希釈剤を含有することができる。不活性希釈剤の他に、経口組成物は、湿潤化剤、乳化および懸濁化剤、甘味剤、着香剤、着色剤、芳香剤、ならびに保存剤などのアジュバントも含むことができる。懸濁剤は、活性化合物に加えて、例えばエトキシ化イソステアリルアルコール類、ポリオキシエチレンソルビトールおよびソルビタンエステル類、微結晶セルロース、メタ水酸化アルミニウム、ベントナイト、寒天、およびトラガカント、ならびにそれらの混合物として懸濁化剤を含有することができる。
【0056】
直腸または腟内投与用の本発明の医薬組成物の製剤は、本発明のプロテアーゼ類を例えばカカオバター、ポリエチレングリコール、座剤ワックスまたはサリチラートを含む1種または複数の適当な非刺激性賦形剤または担体と混合することによって調製することができ、室温では固体であるが体温では液体となり、したがって直腸または膣腔で溶融し、活性化合物を放出する坐剤として提供することもできる。腟内投与に適した本発明の製剤としては、当技術分野で適切であると知られているような担体を含有する腟坐剤、タンポン剤、クリーム剤、ゲル剤、ペースト剤、フォーム剤、またはスプレー製剤も挙げられる。本発明の化合物の局所または経皮投与用の剤形としては、散剤、スプレー剤、軟膏剤、ペースト剤、クリーム剤、ローション剤、ゲル剤、液剤、貼付剤、および吸入剤が挙げられる。プロテアーゼ類は、医薬として許容される担体、および必要とされる場合がある任意の保存剤、緩衝剤、または噴射剤と無菌条件下で混合してもよい。軟膏剤、ペースト剤、クリーム剤、およびゲル剤は、本発明の活性化合物に加えて、動物および植物性脂肪類、油類、ワックス類、パラフィン類、デンプン、トラガカント、セルロース誘導体類、ポリエチレングリコール類、シリコーン類、ベントナイト類、ケイ酸、タルク、ならびに酸化亜鉛、またはそれらの混合物などの賦形剤を含有することができる。
【0057】
散剤およびスプレー剤は、本発明のプロテアーゼ類に加えて、ラクトース、タルク、ケイ酸、水酸化アルミニウム、ケイ酸カルシウム類、およびポリアミド粉末、またはこれらの物質の混合物などの賦形剤を含有することができる。スプレー剤は、さらにクロロフルオロ炭化水素類、ならびにブタンおよびプロパンなどの揮発性非置換炭化水素類など慣例の噴射剤を含有することができる。
【0058】
経皮貼付剤には、本発明のプロテアーゼ類の体内への送達を制御する利点が付加されている。このような剤形は、プロテアーゼ類を適切な媒体に溶解または分散することによって作製することができる。吸収促進剤を使用して、皮膚を通過するプロテアーゼ類の流入を増大させることもできる。このような流入の速度は、速度制御膜を備えることによって、または化合物をポリマーマトリックスもしくはゲルに分散することによって制御することができる。
【0059】
眼科用製剤、眼軟膏剤、散剤、液剤なども本発明の範囲内であると考えられる。
【0060】
非経口投与に適した本発明の医薬組成物は、1種または複数の医薬として許容される無菌の水性もしくは非水性等張液剤、分散剤、懸濁剤、もしくは乳剤、または使用直前に無菌注射用液剤もしくは分散剤に再構成することができる無菌粉末と組み合わせて本発明のプロテアーゼ類を含み、抗酸化剤、緩衝剤、静菌剤、製剤を所期のレシピエントの血液と等張にする溶質、または懸濁化もしくは粘稠化剤を含有することができる。本発明の医薬組成物中で使用される適当な水性および非水性担体としては、例えば水、エタノール、ポリオール類(グリセロール、プロピレングリコール、ポリエチレングリコールなど)、およびそれらの適当な混合物、オリーブ油などの植物油類、ならびにオレイン酸エチルなどの注射用有機エステル類が挙げられる。適切な流動性は、例えばレシチンなどのコーティング材料の使用によって、分散剤の場合は必要とされる粒径の維持によって、および界面活性剤の使用によって維持することができる。これらの組成物は、保存剤、湿潤化剤、乳化剤および分散化剤などのアジュバントも含有することができる。微生物の本組成物に対する作用の予防は、様々な抗菌剤および抗真菌剤、例えばパラベン、クロロブタノール、フェノール ソルビン酸などの包含によって確実にすることができる。糖類、塩化ナトリウムなどの等張剤を組成物に含めることも望ましい場合もある。さらに、注射用剤形の吸収は、吸収を遅延させる作用剤、具体的にはモノステアリン酸アルミニウムおよびゼラチンを含めることによって延長することができる。場合によっては、薬物の効果を延長させるために、皮下または筋肉内注射による薬物の吸収を遅くすることが望ましい。これは、水溶性が不十分な結晶質または非晶質材料の液体懸濁剤を使用することによって実現することができる。あるいは、非経口投与された薬物剤形の遅延吸収は、薬物を油ビヒクルに溶解または懸濁することによって実現される。注射用デポー剤形は、ポリラクチド−ポリグリコリドなどの生分解性ポリマー類中に本化合物のマイクロカプセル化マトリックスを形成することによって作製される。薬物とポリマーの比、および使用する特定のポリマーの性質に応じて、薬物放出速度を制御することができる。他の生分解性ポリマー類としては、例えばポリ(オルトエステル)類)およびポリ(無水物)類が挙げられる。
【0061】
デポー注射用製剤は、体組織と相溶性があるリポソームまたはマイクロエマルジョン中に薬物を封入することによっても調製される。本発明のプロテアーゼ類は、医薬としてヒトおよび動物に投与するとき、それ自体、または例えば0.1%〜99.5%(より好ましくは0.5%〜90%)のプロテアーゼ類を医薬として許容される担体と組み合わせて含有する医薬組成物として投与することができる。本発明の調製物は、経口、非経口、局所、または直腸投与することができる。これらは、当然、各投与経路に適した剤形によって投与される。例えば、これらは、錠剤またはカプセル剤形で、注射、吸入、点眼剤、軟膏剤、坐剤などによって投与され;注射、輸注、または吸入によって投与され;ローション剤または軟膏剤によって局所投与され;あるいは坐剤によって直腸投与される。経口および局所投与が好ましい。
【0062】
本明細書では「非経口投与」および「非経口投与された」という語句は、経腸および局所投与以外の投与モード、通常は注射による投与モードを意味し、静脈内、筋肉内、動脈内、髄腔内、嚢内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、表皮下、関節内、嚢下、クモ膜下、脊髄内、および胸骨内注射および輸注が挙げられるが、これらに限定されるものではない。
【0063】
ヒトおよび他の動物に、治療のために本発明の医薬品を経口、例えばスプレー剤によるような経鼻、直腸、腟内、非経口、大槽内、ならびに散剤、軟膏剤、またはドロップ剤によるような頬側および舌下を含めて局所投与を含めて、任意の適当な投与経路によって投与することができる。選択された投与経路にかかわらず、適当な水和した形で使用することができる本発明のプロテアーゼ類および/または本発明の医薬組成物を、当業者に周知の通常の方法で医薬として許容される剤形に製剤する。
【0064】
本発明の医薬組成物中の活性成分の実際の用量レベルは、患者への毒性がなく、特定の患者、組成物、および投与モードで所望の治療効果を実現するのに有効な一定量の活性成分が得られるように変更することができる。用量レベルの選択は、使用する本発明の特定のプロテアーゼの活性、投与経路、投与時間、使用する特定の化合物の排泄速度、治療期間、使用するプロテアーゼ類と組み合わせて使用する他の薬物、化合物、および/または材料、治療対象の患者の年齢、性別、体重、病態、全身的健康状態、および前病歴、ならびに医療技術分野で周知の同様な要因を含めて、様々な要因に依存する。当技術分野の医師または獣医は、必要とされる医薬組成物の有効量を容易に確定し、処方することができる。例えば、医師または獣医であれば、医薬組成物中で使用される本発明のプロテアーゼ類の用量を、所望の治療効果を実現するために必要とされるレベルより低いレベルで開始し、所望の効果が実現されるまで投与量を徐々に増加させることができるであろう。
【0065】
本発明のプロテアーゼ類は単独投与することができるが、プロテアーゼ類を医薬製剤(組成物)として投与することが好ましい。
【0066】
本発明の別の好ましい実施形態によれば、医薬品は、さらに少なくとも1種の別の活性成分を含む。
【0067】
前記活性成分は、本発明に従うプロテアーゼ類を用いた血管新生疾患の予防および治療を支援することができる任意の活性成分であり得る。しかし、前記プロテアーゼ類以外の効果を示す活性成分を添加することも当然可能である。
【0068】
少なくとも1種の別の活性成分は、好ましくはフラボノイド類、特にバイオフラボノイド類、抗酸化剤、またはシロヤナギ樹皮抽出物のような他の物質からなる群から選択される。
【0069】
本発明の別の好ましい実施形態によれば、医薬品は、ブロメライン、パパイン、フィシン、ナットウキナーゼ、ブリナーゼ、プロナーゼ、セラペプターゼ、レプチラーゼ、オキアミ酵素、バトロキソビン、ルンブロキナーゼ、ククミシン、サブチリシン、セアプローゼからなる群から選択された少なくとも1種(2種、3種、またはさらには4種)のプロテアーゼ類、および任意選択で別の活性成分を含み、前記別の活性成分は、好ましくはフラボノイド、特にルチンである。
【0070】
本発明の好ましい実施形態によれば、フラボノイドは、ルチンまたはその誘導体からなる群から選択される。
【0071】
例えば、ブロメラインおよびパパインは、チオールプロテアーゼ類と呼ばれ、活性部位にシステイン残基を含む。酸化条件下で、このシステインのチオール基は水素原子を失い、別のチオール基と架橋し、ジスルフィド橋を形成し、あるいは同じ酸化過程で別の残基と架橋することができる。この酸化状態では、ブロメラインおよびパパインは活性を失う。抗酸化剤であるビタミンC、ルチンおよびプロアントシアニジン類のようなバイオフラボノイド類を包含することによって、チオールプロテアーゼ類の活性スルフヒドリル基の酸化を防止することができる。
【0072】
前記別の活性成分は、好ましくは本発明の医薬品中に5重量/重量%〜35重量/重量%、好ましくは10重量/重量%〜30重量/重量%、より好ましくは15重量/重量%〜25重量/重量%の量で含まれる。
【0073】
次の図面および実施例によって、本発明をさらに説明する。
【図面の簡単な説明】
【0074】
【図1】酵素処理したHUVECの上清へのLDH放出を示す(毒性試験)。
【図2】酵素処理したHUVECを用いたMTTアッセイを示す(抗増殖作用)。
【図3】Aは、VEGF誘導管腔構造形成の25%ブロメライン、50%ナットウキナーゼ、および25%パパインの組合せによる抑制を示し、Bは、VEGFでのみ処理した対照を示す。VEGF対照では、形成された管腔構造の狭いパターンが見られるが、酵素処理試料は、酵素カクテルの抗血管新生活性を示唆する非管腔構造形成の広域を示す。
【図4】ルトザイム(Rutozym)で治療した患者のVEGF血中濃度を示す。
【図5】VEGF刺激HUVECおよびルトシド(Rutosid)を用いたMTTアッセイを示す。増殖の抑制は見られない。
【図6】ルトシドのHUVECに及ぼす毒性効果を示す。静止期のHUVECにおいては、毒性効果が見られないが、VEGFによって活性化されたHUVECはわずかな効果を示す。
【図7】左側は、HUVECにおける自発的管腔構造形成の抑制を示す。ブロメライン、フィシン、ナットウキナーゼ、パパイン、およびセラペプターゼは管腔構造形成を抑制したが、キモトリプシンまたはトリプシンは抑制しなかった。右側は、HUVECにおけるVEGF誘導管腔構造形成の抑制を示す。ブロメライン、フィシン、ナットウキナーゼ、パパイン、およびセラペプターゼは、無処理HUVECの場合とほぼ同じ程度に管腔構造形成を抑制したが、キモトリプシンまたはトリプシンは抑制しなかった。
【実施例】
【0075】
材料:
シグマ・アルドリッチ(Sigma Aldrich)(オーストリア)から、活性3.51U/mgのパイナップル茎由来ブロメラインを入手した。
株式会社日本生物科学研究所(Japan Bio Science Laboratory Co, Ltd.)から、活性10.000U/mlのナットウキナーゼを購入した。
シグマ・アルドリッチ(Sigma Aldrich)(オーストリア)から、活性>3U/mgのカリカ・パパイア(Carica Papaya)由来パパインを入手した。
【実施例1】
【0076】
毒性試験
ブロメライン、ナットウキナーゼ、およびパパインの抗増殖作用を、乳酸デヒドロゲナーゼ(LDH)アッセイで評価した。セミコンフルエントな状態まで培養したヒト臍帯静脈内皮細胞(HUVEC)をトリプシンで処理することによって回収し、予めヒトフィブロネクチンでコーティングした96ウェルマイクロプレートに、2500個の細胞/ウェルの密度で播種した。適切な付着を可能にするために、10%ウシ胎仔血清、60μg/mlの内皮細胞増殖サプリメントhrEGF、hrFGF2、hrIGF、hrVEGF、アスコルビン酸、およびヘパリンを含有する内皮細胞基本培地2MV(ケンブレックス・バイオケミカルズ(Cambrex Biochemicals))中で、細胞を24時間インキュベートした。付着した後、増殖因子を含まない培地199+10%ウシ胎仔血清(FCS)中37℃/湿度95%でインキュベーションすることによって、細胞を飢餓状態にした。24時間後、10%FCS、VEGFおよび様々な濃度の酵素を含有する培地199で、上清を置換した。48時間インキュベートした後、上清を回収し、製造業者(プロメガ(Promega)、ドイツ)の指示書に従ってLDHアッセイを行った。すべてのウェルから50μlずつ、新しい96ウェル平底(酵素アッセイ)プレートに移した。Assay BufferをSubstrate Mixに添加し、穏やかに混合した。再構成したSubstrate Mixを各ウェルに50μlずつ添加した。プレートを室温で30分間インキュベートした。Stop Solutionを各ウェルに50μlずつ添加した。1時間以内に、490nmの光学密度を参照波長620nmで測定した。結果を無処理対照(%)で表す。
【0077】
結果を図1に示す。これらの結果から、2日間インキュベートした後、ブロメライン、ナットウキナーゼ、およびパパインは25μg/mlを含むレベルまで、HUVECに毒性効果を示さなかったことが明らかである。
【実施例2】
【0078】
抗増殖作用
ブロメライン、ナットウキナーゼ、およびパパイン、ならびにそれらの混合物の抗増殖作用を、3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(MTT)アッセイで評価した。セミコンフルエントな状態まで培養したヒト臍帯静脈内皮細胞(HUVEC)をトリプシンで処理することによって回収し、予めヒトフィブロネクチンでコーティングした96ウェルマイクロプレートに、1000個の細胞/ウェルの密度で播種した。適切な付着を可能にするために、10%ウシ胎仔血清、60μg/mlの内皮細胞増殖サプリメントhrEGF、hrFGF2、hrIGF、hrVEGF、アスコルビン酸、およびヘパリンを含有する内皮細胞基本培地2MV(ケンブレックス・バイオケミカルズ(Cambrex Biochemicals))中で細胞を24時間インキュベートした。付着した後、増殖因子を含まない培地199+10%ウシ胎仔血清(FCS)中37℃/湿度95%でインキュベーションすることによって、細胞を飢餓状態にした。24時間後、10%FCS、様々な濃度のブロメライン、ナットウキナーゼ、およびパパインを含有する培地199で、上清を置換した。細胞を、37℃/湿度95%でさらに48時間インキュベートした。EZ4U MTTキット(バイオメディカ(Biomedica)、オーストリア;製造業者の指示書に従う)を使用して、MTTアッセイを実施した。450nmの光学密度を参照波長620nmで測定した。結果を増殖(%)で表す。100%はVEGF処理対照の増殖である。
【0079】
濃度反応実験の結果を図2に示す。これらの結果は、ブロメライン、ナットウキナーゼ、およびパパインの明らかな抗増殖効果を示し、ブロメラインおよびパパインは、25μg/mlと低い濃度で75%の増殖に到達した。LDH放出試験の結果とまとめると、これらのデータは、明らかに抗増殖性ではあるが細胞毒性のない作用を示唆する。
【0080】
これらの混合物の結果を表1に示す。明らかな抗増殖効果が見られる。
【0081】
【表2】
【実施例3】
【0082】
抗血管新生活性
方法:
抗血管新生活性を管腔構造形成アッセイで評価した。成長因子低減Matrigel(ベクトン・ディッキンソン(Becton Dickinson)、ウィーン)を4℃の温度で解凍した。96ウェルマイクロタイタープレートのウェルに、50μl/ウェルをピペットで加えた。プレートを4℃で24時間放置した。実験する前に、プレートを37℃で30〜60分間インキュベートして、ゲルを凝固させた。HUVECをトリプシンで処理することによって回収し、製造業者の指示書に従ってアスコルビン酸およびヒドロコルチゾンを補充した内皮増殖培地2(MV)(ロンザ(Lonza)、ブリュッセル)、ならびに5000U/mlのヘパリンおよび1%ウシ胎仔血清中のMatrigelでコーティングした96ウェルマイクロプレートに、20.000個の細胞/ウェルの密度で播種した。薬物およびVEGFを所望の濃度に添加した。さらに16〜18時間後、各ウェルの写真を撮影した。管腔構造の全長は、Neuron Length Determination Pluginで測定し、ImageJ Softwareを使用して確定した。
【0083】
結果
酵素および酵素混合物の管腔構造形成アッセイの結果を図3、図7、および表1に示す。
【0084】
表1は、ブロメライン、ナットウキナーゼ、およびパパインの様々な混合物による管腔構造形成の抑制を示す。結果は、次のことを明らかに示す。
1.管腔構造形成は、酵素であるブロメライン、フィシン、ナットウキナーゼ、パパイン、およびセラペプターゼによって抑制されている。
2.ブロメライン、ナットウキナーゼ、およびパパインの組合せは、これら薬物の単独より効果が高い。
【0085】
【表3】
【0086】
表1:VEGFの存在下HUVECにおける管腔構造形成(対照(%))。結果を管腔構造形成(%)で示す。100%は、VEGFでのみ処理した試料における管腔構造形成に等しい。示していない組合せは管腔構造形成を抑制しなかった。
【0087】
結論
VEGFによって媒介された管腔構造のような微小血管の形成は、ブロメライン、フィシン、ナットウキナーゼ、パパイン、またはセラペプターゼ単独で処理した後、ならびに酵素であるブロメライン、ナットウキナーゼ、およびパパインの混合物で処理した後、大幅に低減したことが認められた。
【実施例4】
【0088】
この実施例では、酵素療法(酵素混合物:ナットウキナーゼ、ブロメライン、パパイン+ルチンバイオフラボノイド、シロヤナギ樹皮抽出物)のVEGF血中濃度の量に及ぼす効果を検討した。酵素療法はヒト血中のVEGF濃度の上昇を大幅に低減するということを明らかにする可能性がある(下記の結果を参照のこと)。
【0089】
試験は、2型糖尿病患者男女111名を対象に、並行比較2群として多施設共同無作為化非盲検パイロット研究として行った。54名の患者は、酵素混合物(ナットウキナーゼ(20000FU/gm)25mg、ブロメライン(2450GDU/gm)90mg、パパインN.F.(2.400USP単位/mg)100mg、ルチンバイオフラボノイド複合体(ルトシド(rutoside)類およびルチノシド類)120mg、シロヤナギ樹皮抽出物(15%サリシン/7%ポリフェノール類)100mg、マーリン・ニュートラシューティカルズ(Marlyn Nutraceuticals)、米国)を4週間摂取した。患者の血漿中のVEGF濃度を、補給前および補給4週間直後に試験した。患者は、自分自身が初期値で自己対照として機能する。
【0090】
VEGF血中濃度を異なる4群に分けた(四分位値;図4を参照のこと):患者における治療前VEGF血中濃度<50ng/ml;(s50=開始<50ng/ml;e50=終わり)s100:治療前VEGF濃度<100ng/ml;s200:治療前<200ng/ml、およびs300:治療前VEGF>200ng。植物および/または微生物を供給源とするプロテアーゼ類を含む本発明に従う医薬品の使用は、VEGF血中レベルを低減するために使用することができ、したがって新血管新生に関連した疾患の治療に使用できることを示し得る。
【実施例5】
【0091】
ルトシドのHUVECに及ぼす抗増殖性効果
ルトシドの抗増殖作用の可能性を評価するために、3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(MTT)アッセイを利用した。セミコンフルエントな状態まで培養したヒト臍帯静脈内皮細胞(HUVEC)をトリプシンで処理することによって回収し、予めヒトフィブロネクチンでコーティングした96ウェルマイクロプレートに、1000個の細胞/ウェルの密度で播種した。適切な付着を可能にするために、10%ウシ胎仔血清、60μg/mlの内皮細胞増殖サプリメントhrEGF、hrFGF−2、hrIGF、hrVEGF、アスコルビン酸、およびヘパリンを含有する内皮細胞基本培地2MV(ケンブレックス・バイオケミカルズ(Cambrex Biochemicals))中で、細胞を24時間インキュベートした。付着した後、増殖因子を含まない培地199+10%ウシ胎仔血清(FCS)中37℃/湿度95%でインキュベーションすることによって、細胞を飢餓状態にした。24時間後、10%FCS、VEGFおよび様々な濃度のルトシドおよびVEGFを含有する培地199で、上清を置換した。細胞を、37℃/湿度95%でさらに48時間インキュベートした。EZ4U MTTキット(バイオメディカ(Biomedica)、オーストリア)を製造業者の指示書に従って使用して、MTTアッセイを実施した。450nmの光学密度を参照波長620nmで測定した。結果を増殖(%)で表す。100%はVEGF処理対照の増殖である。
【0092】
結果は、ルトシドがVEGF刺激HUVEC増殖を抑制しないことを明らかに示す(図5を参照のこと)。
【実施例6】
【0093】
ルトシドのHUVECに及ぼす毒性効果
ルトシドの細胞毒性の可能性を試験するために、乳酸デヒドロゲナーゼ(LDH)アッセイを利用した。セミコンフルエントな状態まで培養したヒト臍帯静脈内皮細胞(HUVEC)をトリプシンで処理することによって回収し、予めヒトフィブロネクチンでコーティングした96ウェルマイクロプレートに、2500個の細胞/ウェルの密度で播種した。適切な付着を可能にするために、10%ウシ胎仔血清、60μg/mlの内皮細胞増殖サプリメントhrEGF、hrFGF−2、hrIGF、hrVEGF、アスコルビン酸、およびヘパリンを含有する内皮細胞基本培地2MV(ケンブレックス・バイオケミカルズ(Cambrex Biochemicals))中で、細胞を24時間インキュベートした。付着した後、増殖因子を含まない培地199+10%ウシ胎仔血清(FCS)中37℃/湿度95%でインキュベーションすることによって、細胞を飢餓状態にした。24時間後、10%FCS、VEGFおよび様々な濃度の酵素を含有する培地199で、上清を置換した。48時間インキュベートした後、上清を回収し、製造業者(プロメガ(Promega)、ドイツ)の指示書に従ってLDHアッセイを行った。すべてのウェルから50μlずつ、新しい96ウェル平底(酵素アッセイ)プレートに移した。Assay BufferをSubstrate Mixに添加し、穏やかに混合した。再構成したSubstrate Mixを各ウェルに50μlずつ添加した。プレートを室温で30分間インキュベートした。Stop Solutionを各ウェルに50μlずつ添加した。1時間以内に、490nmの光学密度を参照波長620nmで測定した。結果を無処理対照(%)で表す。
【0094】
結果は、ルトシドの毒性がHUVECにおいて無視できる程度であることを明らかに示す(図6を参照のこと)。
【実施例7】
【0095】
酸素誘発網膜症およびレーザー誘発脈絡膜血管新生のマウスモデルにおける植物由来のプロテアーゼ類によるVEGFの抑制
血管増殖性網膜症は、工業先進国の重度視力喪失の主な原因であり、基礎疾患は糖尿病、網膜静脈閉塞症、未熟児網膜症、または後期の加齢黄斑変性(AMD)である。虚血性網膜疾患の標準的治療は、VEGFのような血管新生因子の産生を最小限に抑えるための末梢網膜組織の破壊に基づいている。神経組織の破壊は不可逆であるので、血管新生因子阻害薬を用いた局所療法は、網膜または脈絡膜血管新生の発症リスクが上昇している患者を保護するために望ましい。一方、AMDでは、血管形成阻害薬を用いた局所療法が、標準的ケアになった。
【0096】
方法
A.酸素誘発網膜症(OIR)
マウス酸素誘発網膜症(OIR)モデルを、スミス(Smith)および同僚が記述している(Invest. Ophthalmol. Vis. Sci.(1994年)、35巻:p.101−111)ように樹立した。7日(P7)齢C57/B16Jマウスを、75%酸素中にP12まで入れた。正常な酸素に戻した後、マウスは相対的に低酸素のため網膜血管新生を発生した。この効果は、一方の眼に硝子体内注射された被検物質の影響を受けた。他方の眼には、対照液剤を注射した。P17に、フルオレセインデキストランで灌流した後、フラットマウントによって網膜増殖を評価した。これらのホールマウントによって、網膜脈管構造の血管病変をコード化した形で評価することが可能になる。各フラットマウントについて、評価システムに従ったスコアを決定し、ウィルコクソン順位和検定法で比較し、処理と対照の間で有意差を得た。合計30匹のマウス/群を使用した。
【0097】
第2の試験では、P12に、被検物質を腹腔内注射した。この場合、2つの眼の間での個人内比較は可能でなく、したがって対照群がさらに必要であった。合計25匹のマウス/群を使用した。
【0098】
B.レーザー誘発脈絡膜血管新生(レーザー誘発CNV)
マウスレーザー誘発脈絡膜血管新生モデルを、カンポキアーロ(Campochiaro)および同僚が記述しているように樹立した(トベ(Tobe)ら、Am. J. Pathol.(1998年)、153巻:p.1641−1646。0日(d0)に、12週齢より若くないC57/B16Jマウスに麻酔をかけ、視覚制御されたレーザーによる3つの熱傷を網膜に与えて、新生血管形成を誘発させた。マウスは、創傷後2週間以内にレーザー部位で脈絡膜血管新生を発生した。d7すなわち7日に、被検物質を一方の眼に、かつ対照液剤を他方の眼に硝子体内注射して、網膜または脈絡膜血管新生に影響があるかどうかを確認した。13日より後のd14に、マウスをデキストラン−フルオレセインで灌流し、脈絡膜ホールマウントを調製した。ホールマウントによって、クロリドの血管病変およびCNV膜のサイズを評価することが可能になる。各レーザー部位の値をウィルコクソン順位和検定法で比較し、処理と対照の間で有意差を得た。合計30匹のマウス/群を使用した。
【0099】
別の実験では、P7すなわち7日に、被検物質を腹腔内注射した。この場合、2つの眼の間での個人内比較は可能でなく、したがって対照群がさらに必要であった。合計25匹のマウス/群を使用した。
【特許請求の範囲】
【請求項1】
加齢黄斑変性(AMD)、脈絡膜血管新生、ヒッペル・リンダウ病、虹彩血管新生、虚血性増殖網膜症、角膜血管新生、および増殖性鎌状赤血球性網膜症からなる群から選択された新血管新生に関連した眼疾患の治療および/または予防用の医薬品を製造するための少なくとも1種のプロテアーゼの使用であって、前記少なくとも1種のプロテアーゼは、植物、非哺乳類動物、および微生物プロテアーゼ類からなる群から選択される使用。
【請求項2】
前記少なくとも1種の植物プロテアーゼが、ブロメライン、パパイン、フィシン、およびククミシンからなる群から選択されることを特徴とする請求項1に記載の使用。
【請求項3】
前記微生物プロテアーゼが、ナットウキナーゼ、プロナーゼ、ブリナーゼ、セアプローゼ、セラペプターゼ、およびサブチリシンからなる群から選択されることを特徴とする請求項1に記載の使用。
【請求項4】
前記非哺乳類動物プロテアーゼが、レプチラーゼ、オキアミ酵素、バトロキソビン、およびルンブロキナーゼからなる群から選択されることを特徴とする請求項1に記載の使用。
【請求項5】
前記少なくとも1種の植物、非哺乳類動物、および/または微生物プロテアーゼが、医薬品中に10重量/重量%〜90重量/重量%、好ましくは20重量/重量%〜80重量/重量%、より好ましくは30重量/重量%〜70重量/重量%の量で含まれることを特徴とする請求項1から4のいずれか一項に記載の使用。
【請求項6】
前記少なくとも1種の植物、非哺乳類動物、および/または微生物プロテアーゼが、1mg/体重1kg〜100mg/kg、好ましくは2mg/体重1kg〜50mg/kg、より好ましくは5mg/体重1kg〜20mg/kgの所定量で個体に投与されることを特徴とする請求項1から5のいずれか一項に記載の使用。
【請求項7】
前記医薬品が、さらに少なくとも1種の医薬として許容される担体、希釈剤、および/または賦形剤、好ましくは結合剤、充填剤、崩壊剤、滑沢剤、保存剤、および/またはコーティングを含むことを特徴とする請求項1から6のいずれか一項に記載の使用。
【請求項8】
前記医薬品が、眼内、経口、局所、経腸、または非経口投与に適合されていることを特徴とする請求項1から7のいずれか一項に記載の使用。
【請求項9】
前記医薬品が、点眼剤、点耳剤、点鼻剤、スプレー式点鼻剤、錠剤、好ましくは可溶性錠剤、発泡錠、胃耐性錠剤、および舌下錠、カプセル剤、好ましくは胃耐性カプセル剤、散剤、顆粒剤、経口用液剤、経口用ドロップ剤、軟膏剤、ローション剤、乳剤、ヒドロゲル剤、坐剤、腟坐剤、輸注剤、ならびに注射剤からなる群から選択された剤形で用いられることを特徴とする請求項1から8のいずれか一項に記載の使用。
【請求項10】
前記医薬品が、さらに少なくとも1種の別の活性成分を含むことを特徴とする請求項1から9のいずれか一項に記載の使用。
【請求項11】
前記少なくとも1種の別の活性成分が、フラボノイドおよび/または抗酸化剤であることを特徴とする請求項10に記載の使用。
【請求項12】
前記フラボノイドがルチンであることを特徴とする請求項11に記載の使用。
【請求項13】
前記別の活性成分が、医薬品中に5重量/重量%〜35重量/重量%、好ましくは10重量/重量%〜30重量/重量%、より好ましくは15重量/重量%〜25重量/重量%の量で含まれることを特徴とする特許請求項10から12のいずれか一項に記載の使用。
【請求項14】
前記医薬品が、ブロメライン、パパイン、フィシン、ナットウキナーゼ、ブリナーゼ、プロナーゼ、セラペプターゼ、レプチラーゼ、オキアミ酵素、バトロキソビン、ルンブロキナーゼ、ククミシン、サブチリシン、セアプローゼからなる群から選択された少なくとも1種のプロテアーゼ、および任意選択で別の活性成分を含み、前記別の活性成分は、好ましくはフラボノイド、特にルチンであることを特徴とする請求項1から13のいずれか一項に記載の使用。
【請求項1】
加齢黄斑変性(AMD)、脈絡膜血管新生、ヒッペル・リンダウ病、虹彩血管新生、虚血性増殖網膜症、角膜血管新生、および増殖性鎌状赤血球性網膜症からなる群から選択された新血管新生に関連した眼疾患の治療および/または予防用の医薬品を製造するための少なくとも1種のプロテアーゼの使用であって、前記少なくとも1種のプロテアーゼは、植物、非哺乳類動物、および微生物プロテアーゼ類からなる群から選択される使用。
【請求項2】
前記少なくとも1種の植物プロテアーゼが、ブロメライン、パパイン、フィシン、およびククミシンからなる群から選択されることを特徴とする請求項1に記載の使用。
【請求項3】
前記微生物プロテアーゼが、ナットウキナーゼ、プロナーゼ、ブリナーゼ、セアプローゼ、セラペプターゼ、およびサブチリシンからなる群から選択されることを特徴とする請求項1に記載の使用。
【請求項4】
前記非哺乳類動物プロテアーゼが、レプチラーゼ、オキアミ酵素、バトロキソビン、およびルンブロキナーゼからなる群から選択されることを特徴とする請求項1に記載の使用。
【請求項5】
前記少なくとも1種の植物、非哺乳類動物、および/または微生物プロテアーゼが、医薬品中に10重量/重量%〜90重量/重量%、好ましくは20重量/重量%〜80重量/重量%、より好ましくは30重量/重量%〜70重量/重量%の量で含まれることを特徴とする請求項1から4のいずれか一項に記載の使用。
【請求項6】
前記少なくとも1種の植物、非哺乳類動物、および/または微生物プロテアーゼが、1mg/体重1kg〜100mg/kg、好ましくは2mg/体重1kg〜50mg/kg、より好ましくは5mg/体重1kg〜20mg/kgの所定量で個体に投与されることを特徴とする請求項1から5のいずれか一項に記載の使用。
【請求項7】
前記医薬品が、さらに少なくとも1種の医薬として許容される担体、希釈剤、および/または賦形剤、好ましくは結合剤、充填剤、崩壊剤、滑沢剤、保存剤、および/またはコーティングを含むことを特徴とする請求項1から6のいずれか一項に記載の使用。
【請求項8】
前記医薬品が、眼内、経口、局所、経腸、または非経口投与に適合されていることを特徴とする請求項1から7のいずれか一項に記載の使用。
【請求項9】
前記医薬品が、点眼剤、点耳剤、点鼻剤、スプレー式点鼻剤、錠剤、好ましくは可溶性錠剤、発泡錠、胃耐性錠剤、および舌下錠、カプセル剤、好ましくは胃耐性カプセル剤、散剤、顆粒剤、経口用液剤、経口用ドロップ剤、軟膏剤、ローション剤、乳剤、ヒドロゲル剤、坐剤、腟坐剤、輸注剤、ならびに注射剤からなる群から選択された剤形で用いられることを特徴とする請求項1から8のいずれか一項に記載の使用。
【請求項10】
前記医薬品が、さらに少なくとも1種の別の活性成分を含むことを特徴とする請求項1から9のいずれか一項に記載の使用。
【請求項11】
前記少なくとも1種の別の活性成分が、フラボノイドおよび/または抗酸化剤であることを特徴とする請求項10に記載の使用。
【請求項12】
前記フラボノイドがルチンであることを特徴とする請求項11に記載の使用。
【請求項13】
前記別の活性成分が、医薬品中に5重量/重量%〜35重量/重量%、好ましくは10重量/重量%〜30重量/重量%、より好ましくは15重量/重量%〜25重量/重量%の量で含まれることを特徴とする特許請求項10から12のいずれか一項に記載の使用。
【請求項14】
前記医薬品が、ブロメライン、パパイン、フィシン、ナットウキナーゼ、ブリナーゼ、プロナーゼ、セラペプターゼ、レプチラーゼ、オキアミ酵素、バトロキソビン、ルンブロキナーゼ、ククミシン、サブチリシン、セアプローゼからなる群から選択された少なくとも1種のプロテアーゼ、および任意選択で別の活性成分を含み、前記別の活性成分は、好ましくはフラボノイド、特にルチンであることを特徴とする請求項1から13のいずれか一項に記載の使用。
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2010−500382(P2010−500382A)
【公表日】平成22年1月7日(2010.1.7)
【国際特許分類】
【出願番号】特願2009−524045(P2009−524045)
【出願日】平成19年8月16日(2007.8.16)
【国際出願番号】PCT/AT2007/000393
【国際公開番号】WO2008/019417
【国際公開日】平成20年2月21日(2008.2.21)
【出願人】(509039840)マーリン ニュートラシューティカルズ, インコーポレイテッド (1)
【Fターム(参考)】
【公表日】平成22年1月7日(2010.1.7)
【国際特許分類】
【出願日】平成19年8月16日(2007.8.16)
【国際出願番号】PCT/AT2007/000393
【国際公開番号】WO2008/019417
【国際公開日】平成20年2月21日(2008.2.21)
【出願人】(509039840)マーリン ニュートラシューティカルズ, インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]