
短時間作用型ベンゾジアゼピンの塩およびその多型形態
本発明は、式(I):

の化合物のエシレート塩に関する。塩を調製する方法、および、特に、鎮静もしくは催眠、不安緩解、筋弛緩、または鎮痙を目的としたそれらの薬剤としての使用も説明する。本発明の一局面において、約6.2、9.2、12.3、15.0、17.2、または20.6度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す式(I)の化合物のエシレート塩(本明細書ではエシレート形態1と示す)の結晶多型も提供される。エシレート形態1結晶多型は、約6.2、9.2、12.3、15.0、17.2、および20.6度2θに特性ピークを含むXRPDパターンを示すことが好ましい。
の化合物のエシレート塩に関する。塩を調製する方法、および、特に、鎮静もしくは催眠、不安緩解、筋弛緩、または鎮痙を目的としたそれらの薬剤としての使用も説明する。本発明の一局面において、約6.2、9.2、12.3、15.0、17.2、または20.6度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す式(I)の化合物のエシレート塩(本明細書ではエシレート形態1と示す)の結晶多型も提供される。エシレート形態1結晶多型は、約6.2、9.2、12.3、15.0、17.2、および20.6度2θに特性ピークを含むXRPDパターンを示すことが好ましい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、短時間作用型ベンゾジアゼピンの塩、および、特に、鎮静もしくは催眠、不安緩解、筋弛緩、または鎮痙を目的としたこの塩の薬剤としての使用に関する。
【背景技術】
【0002】
特許文献1には、カルボン酸エステル部分を含み、非特異的組織エステラーゼにより不活性化される短時間作用型ベンゾジアゼピンが記載されている。このベンゾジアゼピンの特徴は、除去機構が器官に依存しないことであり、薬力学的プロフィールの予測および再現が容易である。この化合物は、鎮静−催眠、不安緩解、筋弛緩、および鎮痙の目的を含む治療目的に適している。この化合物は、以下の臨床設定での静脈内投与に有用な短時間作用型CNS抑制剤である:周術期の出来事に対する術前鎮静、不安緩解、および健忘のための使用;短時間の診断、手術、または内視鏡的処置時の意識下鎮静;他の麻酔薬または鎮痛薬の投与前および/または投与時に全身麻酔の誘導および維持のための成分として;ICUにおける鎮静。
【0003】
特許文献1が開示する化合物の1つ(実施例Ic−8、36ページ)は、下記式(I)に示すメチル3−[(4S)−8−ブロモ−1−メチル−6−(2−ピリジニル)−4H−イミダゾール[1,2−a][1,4]ベンゾジアゼピン−4−イル]プロパノアートである。
【0004】
【化1】
式(I)の遊離塩基は5℃で保存すると安定するが、40℃/75%相対湿度(開放)で保存した試料は潮解し、色が黄色からオレンジ色になり、最初と比べて含有量の減少が顕著になることが観察される(下記の実施例1参照)。
【0005】
現在では、意外なことに、式(I)の化合物は、様々な薬剤として許容可能な溶媒から容易に単離されるとともに、良好な熱安定性、低吸湿性、および高水溶解度を示す高結晶質のモノエシレート(エタンスルホン酸)塩を形成することが分かっている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】欧州特許第1,183,243号明細書
【発明の概要】
【課題を解決するための手段】
【0007】
本発明によれば、式(I)の化合物のエシレート(esylate)塩が提供される。この塩は結晶塩であることが好ましい。エシレート塩の多型形態の調製および特性化は、下記の実施例で説明する。
【0008】
本発明によれば、約6.2、9.2、12.3、15.0、17.2、または20.6度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す式(I)の化合物のエシレート塩(本明細書ではエシレート形態1と示す)の結晶多型も提供される。
【0009】
エシレート形態1結晶多型は、約6.2、9.2、12.3、15.0、17.2、および20.6度2θに特性ピークを含むXRPDパターンを示すことが好ましい。
【0010】
エシレート形態1結晶多型は、6.17(19.30)、9.21(20.50)、12.28(16.40)、14.97(23.40)、17.18(52.80)、20.63(100.00)[角度2θ°(相対強度パーセント)]に特性ピークを含むXRPDパターンを示すことがより好ましい。
【0011】
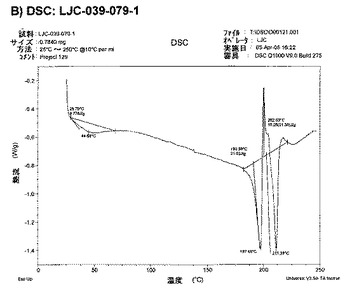
エシレート形態1結晶多型は、195〜205℃、好ましくは、約201〜202℃の範囲の示差走査熱量測定(DSC)開始溶解温度を有することが好ましい。
【0012】
本発明によれば、約3.6、6.4、7.1、12.3、14.1、または17.1度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す式(I)の化合物のエシレート塩(本明細書ではエシレート形態2として示す)の結晶多型がさらに提供される。
【0013】
エシレート形態2結晶多型は、約3.6、6.4、7.1、12.3、14.1、および17.1度2θに特性ピークを含むXRPDパターンを示すことが好ましい。
【0014】
上記結晶多型は、3.57(15.60)、6.42(21.10)、7.13(58.30)、12.29(51.50)、14.10(58.90)、17.13(68.00)[角度2θ°(相対強度パーセント)]に特性ピークを含むXRPDパターンを示すことがより好ましい。
【0015】
エシレート形態2結晶多型は、185〜195℃、好ましくは、約190〜191℃の範囲の示差走査熱量測定(DSC)開始溶解温度を有することが好ましい。
【0016】
形成、収率、純度、ならびに化学的および固体安定のロバスト性に基づくと、好ましい塩はエシレート形態1である。
【0017】
本発明によれば、式(I)の化合物のエシレート塩を作製する方法であって、式(I)の化合物の遊離塩基をエタンスルホン酸と反応させる工程を含む方法も提供される。
【0018】
また、本発明によれば、上記発明の塩を作製する方法であって、溶液において式(I)の化合物の遊離塩基をエタンスルホン酸に接触させ、それぞれのエシレート塩の沈殿物を形成する工程を含む方法が提供される。この方法は、沈殿物を単離する工程をさらに含むことが好ましい。
【0019】
遊離塩基は、トルエン、エタノール、酢酸エチル、MtBE、ジクロロメタン(DCM)、酢酸イソプロピル、ギ酸エチル、メタノール、またはアセトンに溶解することが好ましい。遊離塩基は、トルエンまたは酢酸エチルに溶解することがより好ましい。エタンスルホン酸は、エタノールに溶解することが好ましい。
【0020】
エシレート形態1は、式(I)の化合物の遊離塩基のトルエン、エタノール、酢酸エチル、MtBE、DCM、アセトン、酢酸イソプロピル、ギ酸エチル、またはメタノール溶液をエタノール中のエタンスルホン酸の溶液に接触させて上記塩の沈殿物を形成することによって調製してもよい。
【0021】
本発明によれば、上記方法で得ることが可能な式(I)の化合物のエシレート塩も提供される。
【0022】
エシレート形態2は、エシレート形態1をDCMまたはDCM水溶液(好ましくは、2.5%DCM水溶液)において室温より高い温度(好ましくは、60℃)でスラリーにして溶液を生成し、その溶液を蒸発乾固することによって調製してもよい。
【0023】
本発明によれば、上記方法で得ることができる式(I)の化合物のエシレート塩も提供される。
【0024】
本発明の塩はまた、適切な溶媒から、または適切な溶媒/貧溶媒(anti−solvent)もしくは溶媒/共溶媒混合物から式(I)の化合物のエシレートを結晶化することによって調製してもよい。適切であれば、この溶液または混合物を冷却および/または蒸発させて結晶化を行ってもよい。
【0025】
エシレート形態1は、エタノールから、またはトルエン/エタノール、酢酸エチル/エタノール、MtBE/エタノール、DCM/エタノール、アセトン/エタノール、酢酸イソプロピル/エタノール、ギ酸エチル/エタノール、メタノール/エタノール、もしくはエタノール/水から結晶化してもよい。
【0026】
エシレート形態2は、DCMまたはDCM水溶液中のエシレート形態1の溶液から結晶化してもよい(エシレート形態1は、熱溶媒、適切には、約60℃に溶解することが好ましい)。
【0027】
本発明によれば、上記方法のいずれかで得ることが可能な式(I)の化合物のエシレート塩も提供される。
【0028】
本発明の塩を調製する方法は、下記の実施例でより詳細に説明する。
【0029】
本発明の塩は、特に、鎮静もしくは催眠、不安緩解、筋弛緩、または鎮痙を目的とした薬剤として使用することができる。
【0030】
本発明の塩をバルク活性薬品として投与することは可能であるが、医薬組成物の形態で薬剤として許容可能な担体、賦形剤、または希釈剤とともに提供することが好ましい。担体、賦形剤、または希釈剤は、当然ながら、当該組成物の他の成分と適合するという意味において許容可能である必要があり、服用者に有害であってはならない。
【0031】
それゆえ、本発明は、上記発明の塩、および薬剤として許容可能な担体、賦形剤、または希釈剤を含む医薬組成物を提供する。
【0032】
本発明の医薬組成物は、経口、直腸、局所、口腔(例えば、舌下)、および非経口(例えば、皮下、筋内、皮内、または静脈内)投与に適したものを含む。
【0033】
本発明の塩は、例えば、溶液の静脈内または筋内注射による非経口投与用の医薬組成物の形態で提供されることが好ましい。医薬組成物が非経口投与用である場合、この組成物は、水溶液もしくは非水溶液、または液体混合物(菌静剤、抗酸化剤、緩衝剤、または他の薬剤として許容可能な添加剤を含み得る)であってもよい。
【0034】
本発明の塩の好ましい剤形は、pH2〜4の酸性水媒体、またはシクロデキストリン(CD)の水溶液である。これらの剤形に使用可能なシクロデキストリンは、CyDex,Inc.よりCaptisolの商品名で販売されるβ−CD、詳細には、SBE7−β−CDの陰イオン的に帯電したスルホブチルエーテル(SBE)誘導体(Critical Reviews in Therapeutic Drug Carrier Systems,14(1),1−104(1997))、またはヒドロキシプロピルCDのいずれかである。
【0035】
本発明の塩のさらに好ましい剤形は、当該塩の他に以下の薬剤の少なくとも1つを含む凍結乾燥製剤である:アスコルビン酸、クエン酸、マレイン酸、リン酸、グリシン、塩酸グリシン、コハク酸、または酒石酸。これらの薬剤は、緩衝剤、ケーキング剤、または可視化剤に有用であると考えられる。上記製剤に塩化ナトリウム、マンニトール、ポリビニルピロリドン、または他の成分を含むことが有利な場合もある。
【0036】
好ましい調合法(すなわち、酸緩衝剤またはCD系)は、特定の塩の物理化学的特性(例えば、水溶解度、pKa等)に依存し得る。代替的には、上記塩は、(注射のために)水で再構成する凍結乾燥固体、またはデキストロースもしくは食塩水として用意してもよい。そのような製剤は、通常、アンプルまたは使い捨て注射器具等の単位投与形態として用意される。これらはまた、適切な用量を取り出すことが可能なボトル等の複数回投与形態として用意してもよい。そのような製剤の全ては無菌である必要がある。
【0037】
本発明によれば、被験者を鎮静化または催眠する方法であって、上記発明の塩を鎮静性の有効量または催眠性の有効量で被験者に投与する工程を含む方法が提供される。
【0038】
本発明によれば、被験者の不安を緩解する方法であって、上記発明の塩を不安緩解性の有効量で被験者に投与する工程を含む方法も提供される。
【0039】
本発明によれば、被験者の筋肉を弛緩する方法であって、上記発明の塩を筋弛緩に効果的な量で被験者に投与する工程を含む方法がさらに提供される。
【0040】
本発明によれば、被験者のけいれんを処置する方法であって、上記発明の塩を鎮痙に効果的な量で被験者に投与する工程を含む方法がさらに提供される。
【0041】
本発明によれば、被験者を鎮静化または催眠する薬剤の製造における上記発明の塩の鎮静性の量または催眠性の量での使用も提供される。
【0042】
本発明によれば、被験者を鎮静化または催眠する上記発明の塩も提供される。
【0043】
本発明によれば、被験者の不安を緩解する薬剤の製造における上記発明の塩の不安緩解性の量での使用も提供される。
【0044】
本発明によれば、被験者の不安を緩解する上記発明の塩も提供される。
【0045】
本発明によれば、被験者の筋肉を弛緩する薬剤の製造における上記発明の塩の筋弛緩性の量での使用がさらに提供される。
【0046】
本発明によれば、被験者の筋肉を弛緩する上記発明の塩がさらに提供される。
【0047】
本発明によれば、被験者のけいれんを処置する薬剤の製造における上記発明の塩の鎮痙性の量での使用がさらに提供される。
【0048】
本発明によれば、被験者のけいれんを治療する上記発明の塩がさらに提供される。
【0049】
被験者としては哺乳動物が適しており、ヒトであることが好ましい。
【0050】
ヒトに対する投与に適した非経口医薬品は、本発明の塩の溶液を0.1〜20mg/ml含有するか、または複数回投与バイアル用にその数倍含有することが好ましい。
【0051】
静脈内投与は、ボーラス注入、または、より適切には、持続注入の形態を取るようにすることができる。各被験者に対する投与量は異なり得るが、哺乳動物の鎮静化または催眠に適した本発明の塩の静脈内量または投与量は、体重1kgにつき0.01〜5.0mg、より詳細には、体重1kgにつき0.02〜0.5mgであり、これは、活性成分である塩の質量に基づく。哺乳動物の不安を緩解するために適した本発明の塩の静脈内量または投与量は、体重1kgにつき0.01〜5.0mg、より詳細には、体重1kgにつき0.02〜0.5mgであり、これは、活性成分である塩の質量に基づく。哺乳動物の筋肉を弛緩するために適した本発明の塩の静脈内量または投与量は、体重1kgにつき0.01〜5.0mg、より詳細には、体重1kgにつき0.02〜0.5mgであり、これは、活性成分である塩の質量に基づく。哺乳動物のけいれんの治療に適した本発明の塩の静脈内量または投与量は、体重1kgにつき0.01〜5.0mg、より詳細には、体重1kgにつき0.02〜0.5mgであり、これは、活性成分である塩の質量に基づく。
【0052】
本発明の塩は、以下の臨床設定での静脈内投与に有用な短時間作用型CNS抑制剤である:周術期の出来事に対する術前鎮静、不安緩解、および健忘のための使用;短時間の診断、手術、または内視鏡的処置時の意識下鎮静;他の麻酔薬または鎮痛薬の投与前および/または投与時に全身麻酔の誘導および維持のための成分として;ICUにおける鎮静。
【0053】
下記の実施例において添付の図面を参照しながら本発明の好適な実施態様を説明する。
【図面の簡単な説明】
【0054】
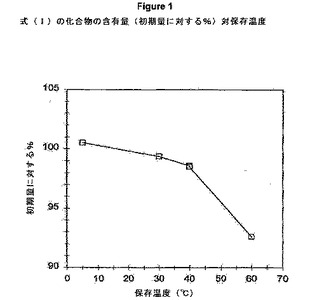
【図1】式(I)の化合物の含有量(初期量に対する%)対保存温度のグラフを示す。
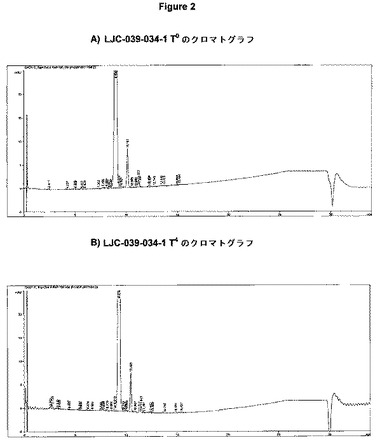
【図2】T0およびT4でのLJC−039−034−1(エシレート塩)のクロマトグラフを示す(表10の結果に対応する)。
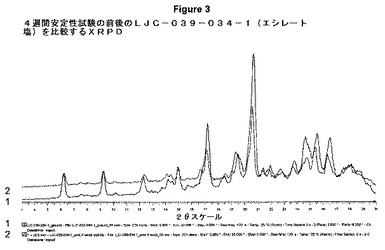
【図3】4週間安定性試験の前後のLJC−039−034−1(エシレート塩)を比較するXRPDを示す。
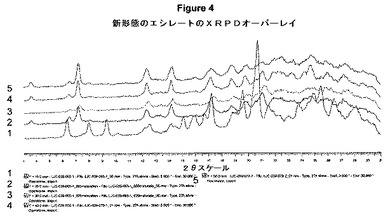
【図4】新形態のエシレートのXRPDオーバーレイを示す。
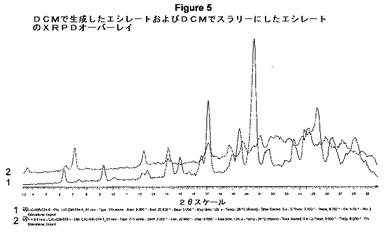
【図5】DCMで生成したエシレートおよびDCMでスラリーにしたエシレートのXRPDオーバーレイを示す。
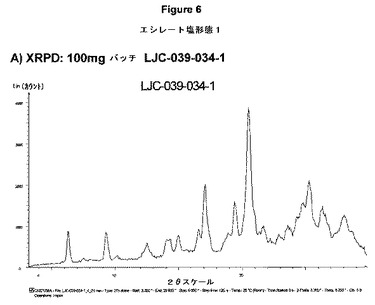
【図6A】エシレート形態1の結果を示す:A)100mgバッチLJC−039−034−1のXRPD。
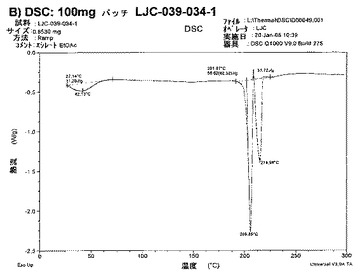
【図6B】エシレート形態1の結果を示す;B)100mgバッチLJC−039−034−1のDSC。
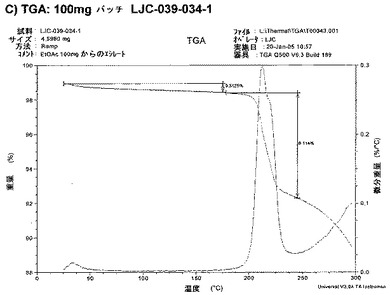
【図6C】エシレート形態1の結果を示す;C)100mgバッチLJC−039−034−1のTGA。
【図6D】エシレート形態1の結果を示す;D)100mgスケールバッチLJC−039−034−1の1H NMR。
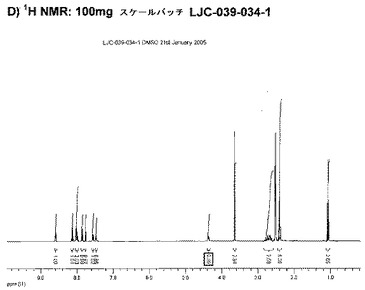
【図6E】エシレート形態1の結果を示す;E)100mgバッチLJC−039−034−1のGVS。
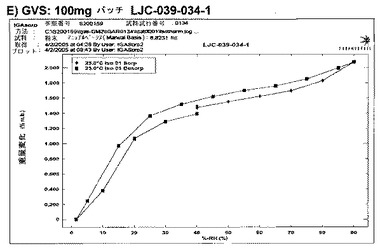
【図6F】エシレート形態1の結果を示す;F)100mgバッチLJC−039−034−1のGVS後のXRPD。
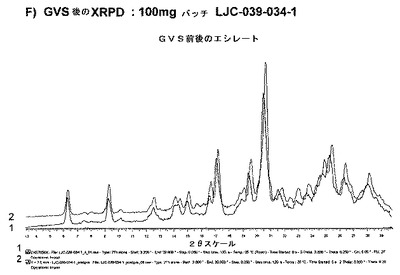
【図6G】エシレート形態1の結果を示す;G)LJC−039−034−1の40℃/75%RHでの安定性試験後のXRPD。
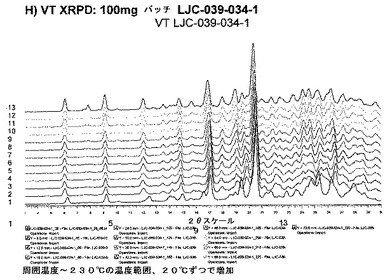
【図6H】エシレート形態1の結果を示す;H)100mgバッチLJC−039−034−1のVT XRPD。
【図6I】エシレート形態1の結果を示す;I)100mgバッチLJC−039−034−1の偏光顕微鏡検査。
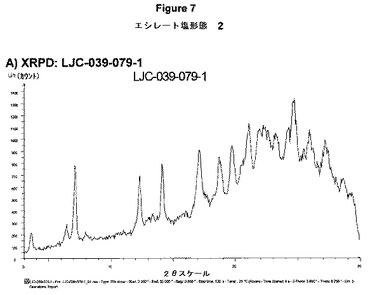
【図7A】エシレート形態2の結果を示す:A)LJC−039−079−1のXRPD。
【図7B】エシレート形態2の結果を示す:B)LJC−039−079−1のDSC。
【発明を実施するための形態】
【0055】
(実施例1)
式(I)の化合物の固体安定性試験
方法/手法 正確に計量した式(I)の化合物の2mgの試料を4mL透明ガラスねじ口バイアルに入れた。試料を最初にテストしてから5℃/周囲相対湿度(AMRH)(密閉)、30℃/60%RH(密閉)、40℃/75%RH(開放)、および60℃/AMRH(密閉)で保存し、34日後にテストした。
【0056】
試料の外観を目視検査した。式(I)の化合物の含有量の値を表1のHPLC法で判定した。式(I)の化合物の標準的な試料(Batch U12438/79/1)に対する重量%/重量(%w/w)の値を測定した。式(I)の化合物のピーク面積を総ピーク面積で割って面積%の値を求めた。
【0057】
【表1】
結果
外観 表2は、外観についての結果を示す。
【0058】
【表2】
式(I)の化合物の含有量(%w/w) %w/w含有量の値(表3参照)は、ばらつきが非常に大きく、初期値と5℃/AMRH(密閉)、30℃/60%RH(密閉)、または40℃/75%RH(開放)での34日後に測定した値の誤差を検出できないことを示す。60℃/AMRH(密閉)で保存した試料の34日後に測定した平均%w/wは、初期値から10%w/wの減少を示す。
【0059】
式(I)の化合物の含有量(%面積) 式(I)の化合物の面積%含有量(表3および図1参照)は、5℃/AMRH(密閉)で保存した34日後には有意な変化を示していないが、30℃/60%RH(密閉)、40℃/75%RH(開放)、または60℃/AMRH(密閉)の試料では保存温度の上昇とともに徐々に減少している。主要な分解ピークは、RRT0.68、0.87、およびRRT0.90で観察されるが、最初の時点(23個のピーク)でも相対的に複雑なクロマトグラムは、多数の新たな小分解ピーク(small degradent peaks)(例えば、30℃/60%RH(密閉)では7個のピーク;60℃/AMRH(密閉)では13〜20個のピーク)も示している。これらの観察結果は、複数の分解経路を示唆している。RRT0.68の分解生成物(degradant)は、暫定的にエステル加水分解生成物(式(I)の化合物の遊離酸)と同定する。これは、加水分解生成物について予想されるように、40℃/75%RH(開放)試料で最も優勢である。
【0060】
【表3】
注釈
1. オートサンプラーシーケンサーのエラーにより1つの試料しかテストしなかった。
【0061】
結論
式(I)の化合物は、5℃/AMRH(密閉)で保存してから少なくとも34日間は外観および含有量に関して安定している。30℃/60%RH(密閉)では外観に変化は見られなかったが、初期面積%に対する式(I)の化合物の含有量にはおよそ0.6%の低下が観察された。40℃/75%RH(開放)または60℃/AMRH(密閉)で保存した試料は潮解し、色が黄色からオレンジ色になり、式(I)の化合物の含有量は初期値から顕著な減少(1.5〜8%)を示した。RRT0.68、0.87、およびRRT0.90の主要な分解ピークが、多数のより小さなピークとともに観察されており、複数の分解経路を示唆している。RRT0.68の分解生成物は、暫定的にエステル加水分解生成物として同定する。これらの結果は、式(I)の化合物は長期間保存する場合には冷蔵保存する必要があることを示す。
【0062】
(実施例2)
様々な有機溶媒における式(I)の化合物の溶解度を判定した。溶解度データを下記の表4に示す。
【0063】
【表4】
上記データは、式(I)の化合物が一般的な有機溶媒において高い溶解度を有することを明確に示す。好ましい溶媒はエタノールおよびトルエンである。
【0064】
上記化合物の遊離塩基の2つの塩基中心のpKaを測定した。しかしながら、ピリジン環の塩基中心のpKaは1.99であった。イミダゾール環の塩基中心のpKaは、4.53と測定された。
【0065】
エタンスルホン酸を用いて式(I)の化合物のエシレート塩を生成した。6種類の容積の溶媒を用いて20mgスケールで実験を行った。全ての反応は、溶解度に応じて、酸をエタノール(1M)原液または固体として充填し周囲温度で行った。
【0066】
単離した全ての固体は、1H NMRにおいて有意なピークシフトを示し、塩形成が確認された。X線粉末回折(XRPD)は、全ての塩が結晶化の徴候を有することを示した。表5は、単離した塩形態の一覧を示す。
【0067】
【表5】
これらの塩を引き続き40℃/75%RHで2週間保存した後、化学的純度についてXRPDおよびHPLCで再分析し、上記原料の安定性を評価した。上記塩は、上記湿度条件に曝した後も同じ粉末パターンを保持するとともに、高い化学的純度を保持しており、安定性が改善したことを裏付けている。
【0068】
単離した塩のT1の純度結果(下記の表6)から、特に、トルエンのエシレート塩が安定性試験の前後に高い純度値を示したことが分かる。
【0069】
【表6】
上記の結果は、エシレート塩形態が高い純度および好ましい安定性結果を示したことを示す。
【0070】
(実施例3)
実施例2のデータに基づいてエシレート塩を100mgに増量した。トルエンがエシレート塩の単離に好ましい溶媒であることが分かった。
【0071】
式(I)の化合物のエシレート塩
上記プロセスの規模が大きくなるかどうか、および単離した原料がより小規模の実験で見られた結晶形態(形態1)と同じであることを確認するために、投入原料を50mgに増量した。分析により上記塩が形態1であること、およびその特性が予想したものと一致していることを確認すると、完全な特性化、および試料の40℃/75%RHでの4週間安定性試験を可能にするために100mgの投入原料で他の規模の拡大化を行った。規模を拡大した反応はともに、エタンスルホン酸をエタノール溶液(1M)として加えたトルエンで行った。この段階で、トルエンは相対的に高い収率での高結晶質原料の生成に関して最良の結果を示し、選択溶媒に関しても同様であった。
【0072】
エシレート実験手順
式(I)の化合物の遊離塩基(100mg、バッチ704−17)をバイアルに充填し、酢酸エチル(600μl)を周囲温度で加えた。この溶液にエタンスルホン酸(250μl、1Mのエタノール溶液)を加え、反応混合物を一晩撹拌した。一晩撹拌した後固体が沈殿し、溶液をろ過し、酢酸エチルで洗浄し、真空下において40℃でオーブン乾燥した。XRPDによる分析は、この固体が生成した他のエシレートと同一の粉末パターンであることを示し、1H−NMRにおいて、有意なピークシフトおよびエタンスルホン酸対イオンに対応するピークにより塩形成を確認した。
【0073】
上記エシレート塩は、5つの異なる溶媒(トルエン、エタノール、酢酸エチル、MtBE、およびDCM)から単離した際に同じ粉末パターンを示した。上記酢酸エチルから単離した塩を、完全な特性化を行う塩として選択した(表7)。
【0074】
【表7】
プロセス最適化
エシレート塩(形態1)の収率をさらに改善するために、4種類の溶媒(酢酸イソプロピル、ギ酸エチル、メタノール、およびアセトン)をスクリーニングした。関連する酸をエタノール原液として加えたこれらの溶媒において100mgスケール反応を合計8回行い、先の実験の全てと比較した。式(I)の化合物(バッチ704−38、100mg)を溶媒(600μl)に周囲温度に溶解した。酸(250μl、1Mエタノール原液)を加え、全ての反応混合物を周囲温度で48時間静置した。結果を表8に示す。
【0075】
【表8】
全ての反応が形態1を示した。
【0076】
上記研究から酢酸イソプロピル等の溶媒が塩の純度を増したが、回収率を低減させたと結論づけた。先に選択した溶媒(酢酸エチル)により、純度値が高い塩が高収率で得られたため、最終的なスケールアップ実験に酢酸エチルを用いることを決定した。
【0077】
エシレート水溶媒実験
実施例2では、エタノールからのエシレート塩の形成により、純度が低減しただけでなく、エステル加水分解の結果として酸と思われる不純物が生じたことが観察された。これに該当するかどうかを判定するために、水の量を変更しながらエタノールを溶媒として用いて実験を行った。基本手順は以下のとおりであった。
【0078】
式(I)の化合物(4×20mg)をエタノール(4×120μl、加熱、2、5、または10%H2O)に溶解した。エタンスルホン酸(50μl、1Mエタノール溶液)を上記溶液に加えた。反応混合物を周囲温度で16時間静置した後、全てが溶液のままであった。これらの溶液を16時間蒸発濃縮した。5%および10%H2Oを入れたバイアルに沈殿を促進するために貧溶媒処理を必要としたが、ニートエタノール反応混合物が油状になったため、ジエチルエーテルで粉末にした。この2%H2Oを含有する反応混合物は、油が結晶化したような固体を含んでいた。XRPD分析では4つの固体全てが結晶質であり、それら全てが先に単離したエシレート塩と同じ粉末パターンを示した。これらの試料の化学的純度を調べた。
【0079】
【表9】
上記実験は、ニートまたは含水エタノール混合物から得た試料の純度に有意な変化がないことを示した。HPLCにより微量の酸不純物が存在することも示されており、系内の少量の水は加水分解を触媒するには不十分であったことを暗示している。
【0080】
次の段階では、全ての塩を4週間の間40℃/75%RHの条件にし、それらの純度をHPLCで監視することでそれらの安定性を判定した(表10)。
【0081】
(実施例4)
塩安定性試験
【0082】
【表10】
エシレートの結晶質試料を合計4週間の間40℃/75%RHで保存し、7日ごとにHPLCのために試料を取り出した。試験中、これらのエシレート塩には純度の変化はなかった。
【0083】
0週および4週目の時点のエシレート塩形態のクロマトグラフを図2に示す。
【0084】
図2に示すクロマトグラフから、エシレート塩の不純物プロフィールに変化がほとんどないことが分かる。親ピーク上に小さなショルダー部ができている。
【0085】
湿度実験前後の塩の粉末パターンから、形態に変化がないことが分かる。
【0086】
図3は、4週間安定性試験の前(トレース1)および後(トレース2)のLJC−039−034−1(エシレート塩)を比較するXRPDを示す。
【0087】
(実施例5)
多型性検査
多型性を示すエシレート塩の傾向を判定するために、30種類の溶媒(15種類のニートおよびそれらの2.5%含水対応物)を用いて熟成実験を試みた。各種の溶媒中で1週間周囲温度〜60℃の加熱/冷却周期で固体をスラリーにした(表11参照)。1週間後、それらのスラリーを蒸発させ、その固体をXRPDおよびHPLCで分析した。
【0088】
表11 エシレートについての多型性検査の結果
・開始純度98.6%
【0089】
【表11】
エシレートの熟成試験では、DCMおよびDCM水溶液から新たな形態(形態2)が示された。この熟成後の純度結果は、メタノール、MEK水溶液、およびジオキサン水溶液でスラリー化したものだけが分解したことを示しており、エシレートの高温での溶液安定性は良好であることを示唆している。
【0090】
新形態のエシレートの検査
上記の新たに同定した形態についてさらなる情報を得るために、より大きなLJC−039−058−1の試料をDCMおよび2.5%DCM水溶液中において60℃でスラリーにした。両方の試料を溶解し、周囲温度で蒸発乾固させて分析した。両方の試料について粉末パターンは同じであり、熟成試験で観察したものと一致した。
【0091】
図4は、上記新形態のエシレートのXRPDオーバーレイを示す。トレース1は、熟成試験の投入原料として用いられたエシレート塩(LJC−039−065−1)を示す。トレース2および3は、それぞれ、DCMおよびDCM水溶液の熟成結果を示す。トレース4および5は、異なるバッチのエシレート(LJC−039−058−1)を用いたDCMおよびDCM水溶液の反復熟成試験を示す。
【0092】
興味深いことに、エシレートが先にDCMから単離し、他の溶媒から単離したエシレート塩と同じ形態(すなわち、形態1)を示した。形態1をDCMにおいてより高い温度でスラリーにした場合のみ形態2が顕著になった。
【0093】
図5は、DCMで生成したエシレートおよびDCMでスラリーにしたエシレートのXRPDオーバーレイを示す。トレース1は、DCM(LJC−039−034−5)から単離した形態1のエシレートを表し、トレース2は、DCM(LJC−039−079−1)でスラリーにした後の形態1のエシレートの結果を表す。
【0094】
塩のスクリーニング検査では、式(I)の化合物がpKaの適切な範囲内で多数の塩を形成すること、およびそれらが様々な溶媒から容易に単離されることが示された。これらの塩の完全な特性化から、それらエシレート塩が湿度について良好な安定性を有すると判定した。エシレートには2つの多型形態があると結論づけた。
【0095】
完全な分析データを添付の図6〜図7に示す。
【0096】
実施例2〜5の実験方法
実施例2
式(I)の化合物(5mg/well)をHPLCバイアル内の溶媒1(30μl)に溶解した。これらの溶液に、エタンスルホン酸(11.4μl、1Mエタノール溶液)を加え、その反応混合物を周囲温度で一晩静置した。固体を入れたこれらのバイアルを真空下において40℃で乾燥させ、溶液のままであったものを蒸発濃縮した後、ヘプタンで処理した。沈殿したものを上述したように乾燥させ、油状になったものを4℃で保存した。
【0097】
1エタノール、トルエン、およびアセトントリル(acetontrile)
エシレート形態1の増量
式(I)の化合物(100mg)を酢酸エチル(600μl)に溶解し、エタンスルホン酸(250μl、1Mエタノール溶液)を加えた。およそ5分後に沈殿が生じ、その反応混合物を80分間周囲温度で撹拌した。この固体をろ過し、酢酸エチルで洗浄し、真空下において40℃で16時間オーブン乾燥した。
【0098】
分析方法
示差走査熱量測定(DSC)
50ポジションオートサンプラーを備えたTA instrument社製Q1000でDSCデータを収集した。エネルギーおよび温度較正基準はインジウムとした。25〜350℃の間で10℃/分の割合で試料を加熱した。試料に対して30ml/分の窒素パージを維持した。特に明示しない限り、0.5〜3mgの試料を用いて、全ての試料をピンホールアルミパンに入れた。
【0099】
熱重量分析(TGA)
アルメルで較正し、10℃/分の走査速度で動作するTA Instrument社製Q500 TGA1でTGAデータを収集した。試料に対して60ml/分の窒素パージを維持した。典型的には、特に明示しない限り、5〜10mgの試料を予めタラ秤量した(pre−tared)白金るつぼ上に載せた。
【0100】
NMR
オートサンプラーを備えたBruker社製400MHz機で全てのスペクトルを収集した。特に明示しない限り、試料はd6−DMSOで調製した。
【0101】
XRPD(X線粉末回折)
Bruker社製AXS C2 GADDS回折装置
Cu−Kα線(40kV、40mA)、自動XYZステージ、自動的な試料の位置決めのためのレーザービデオ顕微鏡、およびHiStar2次元面積検出器を用いてBruker社製AXS C2 GADDS回折装置で試料のX線粉末回折パターンを得た。X線光学は、0.3mmのピンホールコリメータに接続した単一のゲーベル多層膜ミラーで構成される。
【0102】
ビーム広がり、すなわち、試料上において効果的なX線ビームのサイズはおよそ4mmであった。試料と検出器との距離を3.2〜29.8°の効果的な2θ範囲が得られる20cmにしてθ−θ連続走査モードを用いた。試料の典型的な露光時間は120秒であった。
【0103】
周囲条件下でテストした試料を、未粉砕の粉末を用いて平板試験片として調製した。およそ1〜2mgの試料をスライドガラス上に軽く押えつけて表面を平にした。非周囲条件下でテストした試料を、熱伝導性化合物を塗布したシリコンウエハ上に載せた。次いで、この試料を約20℃/分で適温に加熱し、引き続き約1分間等温に維持した後データ収集を開始した。
【0104】
純度分析:
化学的方法
Agilent社のHP1100で純度分析を行った。
【0105】
方法:勾配、逆相
方法実施時間(分):34
カラム:Phenomenex社製Gemini C18 5μm(2.0×50mm)(ガードカートリッジ:Phenomenex社製Gemini C18ガードカートリッジ2×4mm)
カラム温度(℃):40
注入(μl):5
流量(ml/分):0.8
検出:UV
波長(nm):255(90nmの帯域幅)、240(80nmの帯域幅)、254(8nmの帯域幅)
相A:2mmol NH4HCO3(NH3溶液でpH10に調節)
相B:アセトニトリル
タイムテーブル:
【0106】
【表12】
キラル法
Gilson社製HPLCシステムで純度分析を行った。
【0107】
方法:無勾配、順相
方法実施時間(分):50
カラム:Diacel Chrialcel OJ−H(5μm)4.6×250mm(ガードカートリッジ:Diacel Chrialcel OJ−H分析ガードカートリッジ5μm 4.0×10mm)
カラム温度(℃):40
注入(μl):10
流量(ml/分):1.0
検出:UV
波長(nm):225(単一波長検出器)
相A:ヘキサン
相B:エタノール
タイムテーブル:
【0108】
【表13】
重量蒸気収着(Gravimetric Vapour Sorption)(GVS)試験
CFRSorpソフトウェアを実行するHiden社製IGASorp水分収着分析装置で全ての試料をテストした。試料サイズは、典型的には、10mgであった。水分吸脱着等温線法を下記に示すように行った(2回の走査で1周期を完了する)。典型的な室内湿度および温度(40%RH、25℃)で全ての試料を脱着した。GVS後のXRPD分析で全ての試料を分析した。特に明示しない限り、0〜90%RH範囲に対して10%RH間隔で25℃で基準等温線法を行った。
【0109】
【表14】
溶解度
この測定は、0.25mlの溶媒(水)中に十分な化合物を懸濁し、10mg/mlの親遊離形態の化合物の最大最終濃度を得ることにより行った。25℃で24時間懸濁液を平衡化した後、pHチェックを行い、グラスファイバーC96ウェルプレートでろ過した。次いで、ろ液を101倍に希釈する。HPLCにより、およそ0.1mg/mlのDMSOに溶解した基準溶液を参照して定量を行った。異なる容積の基準希釈試験液および不希釈試験液を注入した。基準液注入におけるピーク最大値と同じ保持時間で見られるピーク面積を積分して溶解度を算出した。ろ板に十分な固体が存在する場合、XRPDは、通常、相変化、水和物形成、非晶質化、結晶化等についてチェックされる。
【0110】
【表15】
pKa判定
D−PASアタッチメントを装着したSirius社製GIpKa器具でpKa判定を行った。25℃のMeOH:H2O混合物において電位差滴定で測定を行った。滴定媒体は、0.15M KCIでイオン強度を調節した。MeOH:H2O混合物で見られた値をYasuda−Shedlovsky外挿法により0%共溶媒に当てはめて推定した。
【0111】
3種類の比のOctonol:ISA水を用いてSirius社製GIpKa器具で行った電位差滴定によりLog P、Log Pion、およびLog D値を得た。
【0112】
高温顕微鏡検査(Hot Stage Microscopy)
25〜350℃の温度範囲において10〜20℃/分の範囲の通常の加熱速度でMettler−Toledo社製MTFP82HTホットステージと組み合わせたLeica社製LM/DM偏光顕微鏡を用いて高温顕微鏡検査(Hot Stage Microscopy)を行った。少量の試料を個々の粒子ができる限り分離するようにスライドガラス上に分散した。20倍対物レンズを備えた(λ擬似カラーフィルタに接続した)ライトの普通光または交差偏光の下で試料を観察した。
【0113】
キラル純度法
システム構成
ポンプ:Gilson社製322バイナリポンプ
検出器:Gilson社製152UV/Vis
オートサンプラー:Gilson社製233XLラック+Gilson社製402デュアルシリンジポンプ
カラムオーブン:Phenomenex社製Thermasphere TS−130
ソフトウェア:Gilson社製Unipoint LCソフトウェア
カラム:Daicel社製Chiralcel OJ−H、5μm、4.6×250mm
ガードカラム:Daicel社製Chiralcel OJ−H分析ガードカートリッジ、5μm、4.6×10mm。
【0114】
HPLC条件
チャネルA:ヘキサン(93%)
チャネルB:エタノール(7%)
流量:1.0ml/分
検出器波長 225nm
カラム温度:40℃
実行時間:50.0分。
【0115】
試料条件
およそ0.2mgの試料を適切な体積のヘキサン:エタノール(1:1(v/v))に溶解し、0.2mg/ml溶液を得た。これに蓋をして、約15秒の間高速でボルテックスミキサーにかけた。この時点で固体が残っている場合、試料バイアルをおよそ10秒間超音波分解した後、さらに10〜15秒間ボルテックスミキサーにかけた。10μlをHPLCシステムに注入した。繰り返し注入の最初にヘキサン:エタノール(1:1(v/v))をブランクとして注入した後、試料を繰り返し注入した。
【技術分野】
【0001】
本発明は、短時間作用型ベンゾジアゼピンの塩、および、特に、鎮静もしくは催眠、不安緩解、筋弛緩、または鎮痙を目的としたこの塩の薬剤としての使用に関する。
【背景技術】
【0002】
特許文献1には、カルボン酸エステル部分を含み、非特異的組織エステラーゼにより不活性化される短時間作用型ベンゾジアゼピンが記載されている。このベンゾジアゼピンの特徴は、除去機構が器官に依存しないことであり、薬力学的プロフィールの予測および再現が容易である。この化合物は、鎮静−催眠、不安緩解、筋弛緩、および鎮痙の目的を含む治療目的に適している。この化合物は、以下の臨床設定での静脈内投与に有用な短時間作用型CNS抑制剤である:周術期の出来事に対する術前鎮静、不安緩解、および健忘のための使用;短時間の診断、手術、または内視鏡的処置時の意識下鎮静;他の麻酔薬または鎮痛薬の投与前および/または投与時に全身麻酔の誘導および維持のための成分として;ICUにおける鎮静。
【0003】
特許文献1が開示する化合物の1つ(実施例Ic−8、36ページ)は、下記式(I)に示すメチル3−[(4S)−8−ブロモ−1−メチル−6−(2−ピリジニル)−4H−イミダゾール[1,2−a][1,4]ベンゾジアゼピン−4−イル]プロパノアートである。
【0004】
【化1】
式(I)の遊離塩基は5℃で保存すると安定するが、40℃/75%相対湿度(開放)で保存した試料は潮解し、色が黄色からオレンジ色になり、最初と比べて含有量の減少が顕著になることが観察される(下記の実施例1参照)。
【0005】
現在では、意外なことに、式(I)の化合物は、様々な薬剤として許容可能な溶媒から容易に単離されるとともに、良好な熱安定性、低吸湿性、および高水溶解度を示す高結晶質のモノエシレート(エタンスルホン酸)塩を形成することが分かっている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】欧州特許第1,183,243号明細書
【発明の概要】
【課題を解決するための手段】
【0007】
本発明によれば、式(I)の化合物のエシレート(esylate)塩が提供される。この塩は結晶塩であることが好ましい。エシレート塩の多型形態の調製および特性化は、下記の実施例で説明する。
【0008】
本発明によれば、約6.2、9.2、12.3、15.0、17.2、または20.6度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す式(I)の化合物のエシレート塩(本明細書ではエシレート形態1と示す)の結晶多型も提供される。
【0009】
エシレート形態1結晶多型は、約6.2、9.2、12.3、15.0、17.2、および20.6度2θに特性ピークを含むXRPDパターンを示すことが好ましい。
【0010】
エシレート形態1結晶多型は、6.17(19.30)、9.21(20.50)、12.28(16.40)、14.97(23.40)、17.18(52.80)、20.63(100.00)[角度2θ°(相対強度パーセント)]に特性ピークを含むXRPDパターンを示すことがより好ましい。
【0011】
エシレート形態1結晶多型は、195〜205℃、好ましくは、約201〜202℃の範囲の示差走査熱量測定(DSC)開始溶解温度を有することが好ましい。
【0012】
本発明によれば、約3.6、6.4、7.1、12.3、14.1、または17.1度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す式(I)の化合物のエシレート塩(本明細書ではエシレート形態2として示す)の結晶多型がさらに提供される。
【0013】
エシレート形態2結晶多型は、約3.6、6.4、7.1、12.3、14.1、および17.1度2θに特性ピークを含むXRPDパターンを示すことが好ましい。
【0014】
上記結晶多型は、3.57(15.60)、6.42(21.10)、7.13(58.30)、12.29(51.50)、14.10(58.90)、17.13(68.00)[角度2θ°(相対強度パーセント)]に特性ピークを含むXRPDパターンを示すことがより好ましい。
【0015】
エシレート形態2結晶多型は、185〜195℃、好ましくは、約190〜191℃の範囲の示差走査熱量測定(DSC)開始溶解温度を有することが好ましい。
【0016】
形成、収率、純度、ならびに化学的および固体安定のロバスト性に基づくと、好ましい塩はエシレート形態1である。
【0017】
本発明によれば、式(I)の化合物のエシレート塩を作製する方法であって、式(I)の化合物の遊離塩基をエタンスルホン酸と反応させる工程を含む方法も提供される。
【0018】
また、本発明によれば、上記発明の塩を作製する方法であって、溶液において式(I)の化合物の遊離塩基をエタンスルホン酸に接触させ、それぞれのエシレート塩の沈殿物を形成する工程を含む方法が提供される。この方法は、沈殿物を単離する工程をさらに含むことが好ましい。
【0019】
遊離塩基は、トルエン、エタノール、酢酸エチル、MtBE、ジクロロメタン(DCM)、酢酸イソプロピル、ギ酸エチル、メタノール、またはアセトンに溶解することが好ましい。遊離塩基は、トルエンまたは酢酸エチルに溶解することがより好ましい。エタンスルホン酸は、エタノールに溶解することが好ましい。
【0020】
エシレート形態1は、式(I)の化合物の遊離塩基のトルエン、エタノール、酢酸エチル、MtBE、DCM、アセトン、酢酸イソプロピル、ギ酸エチル、またはメタノール溶液をエタノール中のエタンスルホン酸の溶液に接触させて上記塩の沈殿物を形成することによって調製してもよい。
【0021】
本発明によれば、上記方法で得ることが可能な式(I)の化合物のエシレート塩も提供される。
【0022】
エシレート形態2は、エシレート形態1をDCMまたはDCM水溶液(好ましくは、2.5%DCM水溶液)において室温より高い温度(好ましくは、60℃)でスラリーにして溶液を生成し、その溶液を蒸発乾固することによって調製してもよい。
【0023】
本発明によれば、上記方法で得ることができる式(I)の化合物のエシレート塩も提供される。
【0024】
本発明の塩はまた、適切な溶媒から、または適切な溶媒/貧溶媒(anti−solvent)もしくは溶媒/共溶媒混合物から式(I)の化合物のエシレートを結晶化することによって調製してもよい。適切であれば、この溶液または混合物を冷却および/または蒸発させて結晶化を行ってもよい。
【0025】
エシレート形態1は、エタノールから、またはトルエン/エタノール、酢酸エチル/エタノール、MtBE/エタノール、DCM/エタノール、アセトン/エタノール、酢酸イソプロピル/エタノール、ギ酸エチル/エタノール、メタノール/エタノール、もしくはエタノール/水から結晶化してもよい。
【0026】
エシレート形態2は、DCMまたはDCM水溶液中のエシレート形態1の溶液から結晶化してもよい(エシレート形態1は、熱溶媒、適切には、約60℃に溶解することが好ましい)。
【0027】
本発明によれば、上記方法のいずれかで得ることが可能な式(I)の化合物のエシレート塩も提供される。
【0028】
本発明の塩を調製する方法は、下記の実施例でより詳細に説明する。
【0029】
本発明の塩は、特に、鎮静もしくは催眠、不安緩解、筋弛緩、または鎮痙を目的とした薬剤として使用することができる。
【0030】
本発明の塩をバルク活性薬品として投与することは可能であるが、医薬組成物の形態で薬剤として許容可能な担体、賦形剤、または希釈剤とともに提供することが好ましい。担体、賦形剤、または希釈剤は、当然ながら、当該組成物の他の成分と適合するという意味において許容可能である必要があり、服用者に有害であってはならない。
【0031】
それゆえ、本発明は、上記発明の塩、および薬剤として許容可能な担体、賦形剤、または希釈剤を含む医薬組成物を提供する。
【0032】
本発明の医薬組成物は、経口、直腸、局所、口腔(例えば、舌下)、および非経口(例えば、皮下、筋内、皮内、または静脈内)投与に適したものを含む。
【0033】
本発明の塩は、例えば、溶液の静脈内または筋内注射による非経口投与用の医薬組成物の形態で提供されることが好ましい。医薬組成物が非経口投与用である場合、この組成物は、水溶液もしくは非水溶液、または液体混合物(菌静剤、抗酸化剤、緩衝剤、または他の薬剤として許容可能な添加剤を含み得る)であってもよい。
【0034】
本発明の塩の好ましい剤形は、pH2〜4の酸性水媒体、またはシクロデキストリン(CD)の水溶液である。これらの剤形に使用可能なシクロデキストリンは、CyDex,Inc.よりCaptisolの商品名で販売されるβ−CD、詳細には、SBE7−β−CDの陰イオン的に帯電したスルホブチルエーテル(SBE)誘導体(Critical Reviews in Therapeutic Drug Carrier Systems,14(1),1−104(1997))、またはヒドロキシプロピルCDのいずれかである。
【0035】
本発明の塩のさらに好ましい剤形は、当該塩の他に以下の薬剤の少なくとも1つを含む凍結乾燥製剤である:アスコルビン酸、クエン酸、マレイン酸、リン酸、グリシン、塩酸グリシン、コハク酸、または酒石酸。これらの薬剤は、緩衝剤、ケーキング剤、または可視化剤に有用であると考えられる。上記製剤に塩化ナトリウム、マンニトール、ポリビニルピロリドン、または他の成分を含むことが有利な場合もある。
【0036】
好ましい調合法(すなわち、酸緩衝剤またはCD系)は、特定の塩の物理化学的特性(例えば、水溶解度、pKa等)に依存し得る。代替的には、上記塩は、(注射のために)水で再構成する凍結乾燥固体、またはデキストロースもしくは食塩水として用意してもよい。そのような製剤は、通常、アンプルまたは使い捨て注射器具等の単位投与形態として用意される。これらはまた、適切な用量を取り出すことが可能なボトル等の複数回投与形態として用意してもよい。そのような製剤の全ては無菌である必要がある。
【0037】
本発明によれば、被験者を鎮静化または催眠する方法であって、上記発明の塩を鎮静性の有効量または催眠性の有効量で被験者に投与する工程を含む方法が提供される。
【0038】
本発明によれば、被験者の不安を緩解する方法であって、上記発明の塩を不安緩解性の有効量で被験者に投与する工程を含む方法も提供される。
【0039】
本発明によれば、被験者の筋肉を弛緩する方法であって、上記発明の塩を筋弛緩に効果的な量で被験者に投与する工程を含む方法がさらに提供される。
【0040】
本発明によれば、被験者のけいれんを処置する方法であって、上記発明の塩を鎮痙に効果的な量で被験者に投与する工程を含む方法がさらに提供される。
【0041】
本発明によれば、被験者を鎮静化または催眠する薬剤の製造における上記発明の塩の鎮静性の量または催眠性の量での使用も提供される。
【0042】
本発明によれば、被験者を鎮静化または催眠する上記発明の塩も提供される。
【0043】
本発明によれば、被験者の不安を緩解する薬剤の製造における上記発明の塩の不安緩解性の量での使用も提供される。
【0044】
本発明によれば、被験者の不安を緩解する上記発明の塩も提供される。
【0045】
本発明によれば、被験者の筋肉を弛緩する薬剤の製造における上記発明の塩の筋弛緩性の量での使用がさらに提供される。
【0046】
本発明によれば、被験者の筋肉を弛緩する上記発明の塩がさらに提供される。
【0047】
本発明によれば、被験者のけいれんを処置する薬剤の製造における上記発明の塩の鎮痙性の量での使用がさらに提供される。
【0048】
本発明によれば、被験者のけいれんを治療する上記発明の塩がさらに提供される。
【0049】
被験者としては哺乳動物が適しており、ヒトであることが好ましい。
【0050】
ヒトに対する投与に適した非経口医薬品は、本発明の塩の溶液を0.1〜20mg/ml含有するか、または複数回投与バイアル用にその数倍含有することが好ましい。
【0051】
静脈内投与は、ボーラス注入、または、より適切には、持続注入の形態を取るようにすることができる。各被験者に対する投与量は異なり得るが、哺乳動物の鎮静化または催眠に適した本発明の塩の静脈内量または投与量は、体重1kgにつき0.01〜5.0mg、より詳細には、体重1kgにつき0.02〜0.5mgであり、これは、活性成分である塩の質量に基づく。哺乳動物の不安を緩解するために適した本発明の塩の静脈内量または投与量は、体重1kgにつき0.01〜5.0mg、より詳細には、体重1kgにつき0.02〜0.5mgであり、これは、活性成分である塩の質量に基づく。哺乳動物の筋肉を弛緩するために適した本発明の塩の静脈内量または投与量は、体重1kgにつき0.01〜5.0mg、より詳細には、体重1kgにつき0.02〜0.5mgであり、これは、活性成分である塩の質量に基づく。哺乳動物のけいれんの治療に適した本発明の塩の静脈内量または投与量は、体重1kgにつき0.01〜5.0mg、より詳細には、体重1kgにつき0.02〜0.5mgであり、これは、活性成分である塩の質量に基づく。
【0052】
本発明の塩は、以下の臨床設定での静脈内投与に有用な短時間作用型CNS抑制剤である:周術期の出来事に対する術前鎮静、不安緩解、および健忘のための使用;短時間の診断、手術、または内視鏡的処置時の意識下鎮静;他の麻酔薬または鎮痛薬の投与前および/または投与時に全身麻酔の誘導および維持のための成分として;ICUにおける鎮静。
【0053】
下記の実施例において添付の図面を参照しながら本発明の好適な実施態様を説明する。
【図面の簡単な説明】
【0054】
【図1】式(I)の化合物の含有量(初期量に対する%)対保存温度のグラフを示す。
【図2】T0およびT4でのLJC−039−034−1(エシレート塩)のクロマトグラフを示す(表10の結果に対応する)。
【図3】4週間安定性試験の前後のLJC−039−034−1(エシレート塩)を比較するXRPDを示す。
【図4】新形態のエシレートのXRPDオーバーレイを示す。
【図5】DCMで生成したエシレートおよびDCMでスラリーにしたエシレートのXRPDオーバーレイを示す。
【図6A】エシレート形態1の結果を示す:A)100mgバッチLJC−039−034−1のXRPD。
【図6B】エシレート形態1の結果を示す;B)100mgバッチLJC−039−034−1のDSC。
【図6C】エシレート形態1の結果を示す;C)100mgバッチLJC−039−034−1のTGA。
【図6D】エシレート形態1の結果を示す;D)100mgスケールバッチLJC−039−034−1の1H NMR。
【図6E】エシレート形態1の結果を示す;E)100mgバッチLJC−039−034−1のGVS。
【図6F】エシレート形態1の結果を示す;F)100mgバッチLJC−039−034−1のGVS後のXRPD。
【図6G】エシレート形態1の結果を示す;G)LJC−039−034−1の40℃/75%RHでの安定性試験後のXRPD。
【図6H】エシレート形態1の結果を示す;H)100mgバッチLJC−039−034−1のVT XRPD。
【図6I】エシレート形態1の結果を示す;I)100mgバッチLJC−039−034−1の偏光顕微鏡検査。
【図7A】エシレート形態2の結果を示す:A)LJC−039−079−1のXRPD。
【図7B】エシレート形態2の結果を示す:B)LJC−039−079−1のDSC。
【発明を実施するための形態】
【0055】
(実施例1)
式(I)の化合物の固体安定性試験
方法/手法 正確に計量した式(I)の化合物の2mgの試料を4mL透明ガラスねじ口バイアルに入れた。試料を最初にテストしてから5℃/周囲相対湿度(AMRH)(密閉)、30℃/60%RH(密閉)、40℃/75%RH(開放)、および60℃/AMRH(密閉)で保存し、34日後にテストした。
【0056】
試料の外観を目視検査した。式(I)の化合物の含有量の値を表1のHPLC法で判定した。式(I)の化合物の標準的な試料(Batch U12438/79/1)に対する重量%/重量(%w/w)の値を測定した。式(I)の化合物のピーク面積を総ピーク面積で割って面積%の値を求めた。
【0057】
【表1】
結果
外観 表2は、外観についての結果を示す。
【0058】
【表2】
式(I)の化合物の含有量(%w/w) %w/w含有量の値(表3参照)は、ばらつきが非常に大きく、初期値と5℃/AMRH(密閉)、30℃/60%RH(密閉)、または40℃/75%RH(開放)での34日後に測定した値の誤差を検出できないことを示す。60℃/AMRH(密閉)で保存した試料の34日後に測定した平均%w/wは、初期値から10%w/wの減少を示す。
【0059】
式(I)の化合物の含有量(%面積) 式(I)の化合物の面積%含有量(表3および図1参照)は、5℃/AMRH(密閉)で保存した34日後には有意な変化を示していないが、30℃/60%RH(密閉)、40℃/75%RH(開放)、または60℃/AMRH(密閉)の試料では保存温度の上昇とともに徐々に減少している。主要な分解ピークは、RRT0.68、0.87、およびRRT0.90で観察されるが、最初の時点(23個のピーク)でも相対的に複雑なクロマトグラムは、多数の新たな小分解ピーク(small degradent peaks)(例えば、30℃/60%RH(密閉)では7個のピーク;60℃/AMRH(密閉)では13〜20個のピーク)も示している。これらの観察結果は、複数の分解経路を示唆している。RRT0.68の分解生成物(degradant)は、暫定的にエステル加水分解生成物(式(I)の化合物の遊離酸)と同定する。これは、加水分解生成物について予想されるように、40℃/75%RH(開放)試料で最も優勢である。
【0060】
【表3】
注釈
1. オートサンプラーシーケンサーのエラーにより1つの試料しかテストしなかった。
【0061】
結論
式(I)の化合物は、5℃/AMRH(密閉)で保存してから少なくとも34日間は外観および含有量に関して安定している。30℃/60%RH(密閉)では外観に変化は見られなかったが、初期面積%に対する式(I)の化合物の含有量にはおよそ0.6%の低下が観察された。40℃/75%RH(開放)または60℃/AMRH(密閉)で保存した試料は潮解し、色が黄色からオレンジ色になり、式(I)の化合物の含有量は初期値から顕著な減少(1.5〜8%)を示した。RRT0.68、0.87、およびRRT0.90の主要な分解ピークが、多数のより小さなピークとともに観察されており、複数の分解経路を示唆している。RRT0.68の分解生成物は、暫定的にエステル加水分解生成物として同定する。これらの結果は、式(I)の化合物は長期間保存する場合には冷蔵保存する必要があることを示す。
【0062】
(実施例2)
様々な有機溶媒における式(I)の化合物の溶解度を判定した。溶解度データを下記の表4に示す。
【0063】
【表4】
上記データは、式(I)の化合物が一般的な有機溶媒において高い溶解度を有することを明確に示す。好ましい溶媒はエタノールおよびトルエンである。
【0064】
上記化合物の遊離塩基の2つの塩基中心のpKaを測定した。しかしながら、ピリジン環の塩基中心のpKaは1.99であった。イミダゾール環の塩基中心のpKaは、4.53と測定された。
【0065】
エタンスルホン酸を用いて式(I)の化合物のエシレート塩を生成した。6種類の容積の溶媒を用いて20mgスケールで実験を行った。全ての反応は、溶解度に応じて、酸をエタノール(1M)原液または固体として充填し周囲温度で行った。
【0066】
単離した全ての固体は、1H NMRにおいて有意なピークシフトを示し、塩形成が確認された。X線粉末回折(XRPD)は、全ての塩が結晶化の徴候を有することを示した。表5は、単離した塩形態の一覧を示す。
【0067】
【表5】
これらの塩を引き続き40℃/75%RHで2週間保存した後、化学的純度についてXRPDおよびHPLCで再分析し、上記原料の安定性を評価した。上記塩は、上記湿度条件に曝した後も同じ粉末パターンを保持するとともに、高い化学的純度を保持しており、安定性が改善したことを裏付けている。
【0068】
単離した塩のT1の純度結果(下記の表6)から、特に、トルエンのエシレート塩が安定性試験の前後に高い純度値を示したことが分かる。
【0069】
【表6】
上記の結果は、エシレート塩形態が高い純度および好ましい安定性結果を示したことを示す。
【0070】
(実施例3)
実施例2のデータに基づいてエシレート塩を100mgに増量した。トルエンがエシレート塩の単離に好ましい溶媒であることが分かった。
【0071】
式(I)の化合物のエシレート塩
上記プロセスの規模が大きくなるかどうか、および単離した原料がより小規模の実験で見られた結晶形態(形態1)と同じであることを確認するために、投入原料を50mgに増量した。分析により上記塩が形態1であること、およびその特性が予想したものと一致していることを確認すると、完全な特性化、および試料の40℃/75%RHでの4週間安定性試験を可能にするために100mgの投入原料で他の規模の拡大化を行った。規模を拡大した反応はともに、エタンスルホン酸をエタノール溶液(1M)として加えたトルエンで行った。この段階で、トルエンは相対的に高い収率での高結晶質原料の生成に関して最良の結果を示し、選択溶媒に関しても同様であった。
【0072】
エシレート実験手順
式(I)の化合物の遊離塩基(100mg、バッチ704−17)をバイアルに充填し、酢酸エチル(600μl)を周囲温度で加えた。この溶液にエタンスルホン酸(250μl、1Mのエタノール溶液)を加え、反応混合物を一晩撹拌した。一晩撹拌した後固体が沈殿し、溶液をろ過し、酢酸エチルで洗浄し、真空下において40℃でオーブン乾燥した。XRPDによる分析は、この固体が生成した他のエシレートと同一の粉末パターンであることを示し、1H−NMRにおいて、有意なピークシフトおよびエタンスルホン酸対イオンに対応するピークにより塩形成を確認した。
【0073】
上記エシレート塩は、5つの異なる溶媒(トルエン、エタノール、酢酸エチル、MtBE、およびDCM)から単離した際に同じ粉末パターンを示した。上記酢酸エチルから単離した塩を、完全な特性化を行う塩として選択した(表7)。
【0074】
【表7】
プロセス最適化
エシレート塩(形態1)の収率をさらに改善するために、4種類の溶媒(酢酸イソプロピル、ギ酸エチル、メタノール、およびアセトン)をスクリーニングした。関連する酸をエタノール原液として加えたこれらの溶媒において100mgスケール反応を合計8回行い、先の実験の全てと比較した。式(I)の化合物(バッチ704−38、100mg)を溶媒(600μl)に周囲温度に溶解した。酸(250μl、1Mエタノール原液)を加え、全ての反応混合物を周囲温度で48時間静置した。結果を表8に示す。
【0075】
【表8】
全ての反応が形態1を示した。
【0076】
上記研究から酢酸イソプロピル等の溶媒が塩の純度を増したが、回収率を低減させたと結論づけた。先に選択した溶媒(酢酸エチル)により、純度値が高い塩が高収率で得られたため、最終的なスケールアップ実験に酢酸エチルを用いることを決定した。
【0077】
エシレート水溶媒実験
実施例2では、エタノールからのエシレート塩の形成により、純度が低減しただけでなく、エステル加水分解の結果として酸と思われる不純物が生じたことが観察された。これに該当するかどうかを判定するために、水の量を変更しながらエタノールを溶媒として用いて実験を行った。基本手順は以下のとおりであった。
【0078】
式(I)の化合物(4×20mg)をエタノール(4×120μl、加熱、2、5、または10%H2O)に溶解した。エタンスルホン酸(50μl、1Mエタノール溶液)を上記溶液に加えた。反応混合物を周囲温度で16時間静置した後、全てが溶液のままであった。これらの溶液を16時間蒸発濃縮した。5%および10%H2Oを入れたバイアルに沈殿を促進するために貧溶媒処理を必要としたが、ニートエタノール反応混合物が油状になったため、ジエチルエーテルで粉末にした。この2%H2Oを含有する反応混合物は、油が結晶化したような固体を含んでいた。XRPD分析では4つの固体全てが結晶質であり、それら全てが先に単離したエシレート塩と同じ粉末パターンを示した。これらの試料の化学的純度を調べた。
【0079】
【表9】
上記実験は、ニートまたは含水エタノール混合物から得た試料の純度に有意な変化がないことを示した。HPLCにより微量の酸不純物が存在することも示されており、系内の少量の水は加水分解を触媒するには不十分であったことを暗示している。
【0080】
次の段階では、全ての塩を4週間の間40℃/75%RHの条件にし、それらの純度をHPLCで監視することでそれらの安定性を判定した(表10)。
【0081】
(実施例4)
塩安定性試験
【0082】
【表10】
エシレートの結晶質試料を合計4週間の間40℃/75%RHで保存し、7日ごとにHPLCのために試料を取り出した。試験中、これらのエシレート塩には純度の変化はなかった。
【0083】
0週および4週目の時点のエシレート塩形態のクロマトグラフを図2に示す。
【0084】
図2に示すクロマトグラフから、エシレート塩の不純物プロフィールに変化がほとんどないことが分かる。親ピーク上に小さなショルダー部ができている。
【0085】
湿度実験前後の塩の粉末パターンから、形態に変化がないことが分かる。
【0086】
図3は、4週間安定性試験の前(トレース1)および後(トレース2)のLJC−039−034−1(エシレート塩)を比較するXRPDを示す。
【0087】
(実施例5)
多型性検査
多型性を示すエシレート塩の傾向を判定するために、30種類の溶媒(15種類のニートおよびそれらの2.5%含水対応物)を用いて熟成実験を試みた。各種の溶媒中で1週間周囲温度〜60℃の加熱/冷却周期で固体をスラリーにした(表11参照)。1週間後、それらのスラリーを蒸発させ、その固体をXRPDおよびHPLCで分析した。
【0088】
表11 エシレートについての多型性検査の結果
・開始純度98.6%
【0089】
【表11】
エシレートの熟成試験では、DCMおよびDCM水溶液から新たな形態(形態2)が示された。この熟成後の純度結果は、メタノール、MEK水溶液、およびジオキサン水溶液でスラリー化したものだけが分解したことを示しており、エシレートの高温での溶液安定性は良好であることを示唆している。
【0090】
新形態のエシレートの検査
上記の新たに同定した形態についてさらなる情報を得るために、より大きなLJC−039−058−1の試料をDCMおよび2.5%DCM水溶液中において60℃でスラリーにした。両方の試料を溶解し、周囲温度で蒸発乾固させて分析した。両方の試料について粉末パターンは同じであり、熟成試験で観察したものと一致した。
【0091】
図4は、上記新形態のエシレートのXRPDオーバーレイを示す。トレース1は、熟成試験の投入原料として用いられたエシレート塩(LJC−039−065−1)を示す。トレース2および3は、それぞれ、DCMおよびDCM水溶液の熟成結果を示す。トレース4および5は、異なるバッチのエシレート(LJC−039−058−1)を用いたDCMおよびDCM水溶液の反復熟成試験を示す。
【0092】
興味深いことに、エシレートが先にDCMから単離し、他の溶媒から単離したエシレート塩と同じ形態(すなわち、形態1)を示した。形態1をDCMにおいてより高い温度でスラリーにした場合のみ形態2が顕著になった。
【0093】
図5は、DCMで生成したエシレートおよびDCMでスラリーにしたエシレートのXRPDオーバーレイを示す。トレース1は、DCM(LJC−039−034−5)から単離した形態1のエシレートを表し、トレース2は、DCM(LJC−039−079−1)でスラリーにした後の形態1のエシレートの結果を表す。
【0094】
塩のスクリーニング検査では、式(I)の化合物がpKaの適切な範囲内で多数の塩を形成すること、およびそれらが様々な溶媒から容易に単離されることが示された。これらの塩の完全な特性化から、それらエシレート塩が湿度について良好な安定性を有すると判定した。エシレートには2つの多型形態があると結論づけた。
【0095】
完全な分析データを添付の図6〜図7に示す。
【0096】
実施例2〜5の実験方法
実施例2
式(I)の化合物(5mg/well)をHPLCバイアル内の溶媒1(30μl)に溶解した。これらの溶液に、エタンスルホン酸(11.4μl、1Mエタノール溶液)を加え、その反応混合物を周囲温度で一晩静置した。固体を入れたこれらのバイアルを真空下において40℃で乾燥させ、溶液のままであったものを蒸発濃縮した後、ヘプタンで処理した。沈殿したものを上述したように乾燥させ、油状になったものを4℃で保存した。
【0097】
1エタノール、トルエン、およびアセトントリル(acetontrile)
エシレート形態1の増量
式(I)の化合物(100mg)を酢酸エチル(600μl)に溶解し、エタンスルホン酸(250μl、1Mエタノール溶液)を加えた。およそ5分後に沈殿が生じ、その反応混合物を80分間周囲温度で撹拌した。この固体をろ過し、酢酸エチルで洗浄し、真空下において40℃で16時間オーブン乾燥した。
【0098】
分析方法
示差走査熱量測定(DSC)
50ポジションオートサンプラーを備えたTA instrument社製Q1000でDSCデータを収集した。エネルギーおよび温度較正基準はインジウムとした。25〜350℃の間で10℃/分の割合で試料を加熱した。試料に対して30ml/分の窒素パージを維持した。特に明示しない限り、0.5〜3mgの試料を用いて、全ての試料をピンホールアルミパンに入れた。
【0099】
熱重量分析(TGA)
アルメルで較正し、10℃/分の走査速度で動作するTA Instrument社製Q500 TGA1でTGAデータを収集した。試料に対して60ml/分の窒素パージを維持した。典型的には、特に明示しない限り、5〜10mgの試料を予めタラ秤量した(pre−tared)白金るつぼ上に載せた。
【0100】
NMR
オートサンプラーを備えたBruker社製400MHz機で全てのスペクトルを収集した。特に明示しない限り、試料はd6−DMSOで調製した。
【0101】
XRPD(X線粉末回折)
Bruker社製AXS C2 GADDS回折装置
Cu−Kα線(40kV、40mA)、自動XYZステージ、自動的な試料の位置決めのためのレーザービデオ顕微鏡、およびHiStar2次元面積検出器を用いてBruker社製AXS C2 GADDS回折装置で試料のX線粉末回折パターンを得た。X線光学は、0.3mmのピンホールコリメータに接続した単一のゲーベル多層膜ミラーで構成される。
【0102】
ビーム広がり、すなわち、試料上において効果的なX線ビームのサイズはおよそ4mmであった。試料と検出器との距離を3.2〜29.8°の効果的な2θ範囲が得られる20cmにしてθ−θ連続走査モードを用いた。試料の典型的な露光時間は120秒であった。
【0103】
周囲条件下でテストした試料を、未粉砕の粉末を用いて平板試験片として調製した。およそ1〜2mgの試料をスライドガラス上に軽く押えつけて表面を平にした。非周囲条件下でテストした試料を、熱伝導性化合物を塗布したシリコンウエハ上に載せた。次いで、この試料を約20℃/分で適温に加熱し、引き続き約1分間等温に維持した後データ収集を開始した。
【0104】
純度分析:
化学的方法
Agilent社のHP1100で純度分析を行った。
【0105】
方法:勾配、逆相
方法実施時間(分):34
カラム:Phenomenex社製Gemini C18 5μm(2.0×50mm)(ガードカートリッジ:Phenomenex社製Gemini C18ガードカートリッジ2×4mm)
カラム温度(℃):40
注入(μl):5
流量(ml/分):0.8
検出:UV
波長(nm):255(90nmの帯域幅)、240(80nmの帯域幅)、254(8nmの帯域幅)
相A:2mmol NH4HCO3(NH3溶液でpH10に調節)
相B:アセトニトリル
タイムテーブル:
【0106】
【表12】
キラル法
Gilson社製HPLCシステムで純度分析を行った。
【0107】
方法:無勾配、順相
方法実施時間(分):50
カラム:Diacel Chrialcel OJ−H(5μm)4.6×250mm(ガードカートリッジ:Diacel Chrialcel OJ−H分析ガードカートリッジ5μm 4.0×10mm)
カラム温度(℃):40
注入(μl):10
流量(ml/分):1.0
検出:UV
波長(nm):225(単一波長検出器)
相A:ヘキサン
相B:エタノール
タイムテーブル:
【0108】
【表13】
重量蒸気収着(Gravimetric Vapour Sorption)(GVS)試験
CFRSorpソフトウェアを実行するHiden社製IGASorp水分収着分析装置で全ての試料をテストした。試料サイズは、典型的には、10mgであった。水分吸脱着等温線法を下記に示すように行った(2回の走査で1周期を完了する)。典型的な室内湿度および温度(40%RH、25℃)で全ての試料を脱着した。GVS後のXRPD分析で全ての試料を分析した。特に明示しない限り、0〜90%RH範囲に対して10%RH間隔で25℃で基準等温線法を行った。
【0109】
【表14】
溶解度
この測定は、0.25mlの溶媒(水)中に十分な化合物を懸濁し、10mg/mlの親遊離形態の化合物の最大最終濃度を得ることにより行った。25℃で24時間懸濁液を平衡化した後、pHチェックを行い、グラスファイバーC96ウェルプレートでろ過した。次いで、ろ液を101倍に希釈する。HPLCにより、およそ0.1mg/mlのDMSOに溶解した基準溶液を参照して定量を行った。異なる容積の基準希釈試験液および不希釈試験液を注入した。基準液注入におけるピーク最大値と同じ保持時間で見られるピーク面積を積分して溶解度を算出した。ろ板に十分な固体が存在する場合、XRPDは、通常、相変化、水和物形成、非晶質化、結晶化等についてチェックされる。
【0110】
【表15】
pKa判定
D−PASアタッチメントを装着したSirius社製GIpKa器具でpKa判定を行った。25℃のMeOH:H2O混合物において電位差滴定で測定を行った。滴定媒体は、0.15M KCIでイオン強度を調節した。MeOH:H2O混合物で見られた値をYasuda−Shedlovsky外挿法により0%共溶媒に当てはめて推定した。
【0111】
3種類の比のOctonol:ISA水を用いてSirius社製GIpKa器具で行った電位差滴定によりLog P、Log Pion、およびLog D値を得た。
【0112】
高温顕微鏡検査(Hot Stage Microscopy)
25〜350℃の温度範囲において10〜20℃/分の範囲の通常の加熱速度でMettler−Toledo社製MTFP82HTホットステージと組み合わせたLeica社製LM/DM偏光顕微鏡を用いて高温顕微鏡検査(Hot Stage Microscopy)を行った。少量の試料を個々の粒子ができる限り分離するようにスライドガラス上に分散した。20倍対物レンズを備えた(λ擬似カラーフィルタに接続した)ライトの普通光または交差偏光の下で試料を観察した。
【0113】
キラル純度法
システム構成
ポンプ:Gilson社製322バイナリポンプ
検出器:Gilson社製152UV/Vis
オートサンプラー:Gilson社製233XLラック+Gilson社製402デュアルシリンジポンプ
カラムオーブン:Phenomenex社製Thermasphere TS−130
ソフトウェア:Gilson社製Unipoint LCソフトウェア
カラム:Daicel社製Chiralcel OJ−H、5μm、4.6×250mm
ガードカラム:Daicel社製Chiralcel OJ−H分析ガードカートリッジ、5μm、4.6×10mm。
【0114】
HPLC条件
チャネルA:ヘキサン(93%)
チャネルB:エタノール(7%)
流量:1.0ml/分
検出器波長 225nm
カラム温度:40℃
実行時間:50.0分。
【0115】
試料条件
およそ0.2mgの試料を適切な体積のヘキサン:エタノール(1:1(v/v))に溶解し、0.2mg/ml溶液を得た。これに蓋をして、約15秒の間高速でボルテックスミキサーにかけた。この時点で固体が残っている場合、試料バイアルをおよそ10秒間超音波分解した後、さらに10〜15秒間ボルテックスミキサーにかけた。10μlをHPLCシステムに注入した。繰り返し注入の最初にヘキサン:エタノール(1:1(v/v))をブランクとして注入した後、試料を繰り返し注入した。
【特許請求の範囲】
【請求項1】
式(I):
【化2】
の化合物のエシレート塩。
【請求項2】
結晶塩である、請求項1に記載の塩。
【請求項3】
約6.2、9.2、12.3、15.0、17.2、および20.6度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す結晶多型である、請求項2に記載のエシレート塩。
【請求項4】
前記塩は、約3.6、6.4、7.1、12.3、14.1、および17.1度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す結晶多型である、請求項2に記載のエシレート塩。
【請求項5】
請求項1〜4のいずれかに記載の塩、および薬剤として許容可能な担体、賦形剤、または希釈剤を含む医薬組成物。
【請求項6】
薬剤として使用するための請求項1〜4のいずれかに記載の塩。
【請求項7】
対象において鎮静または催眠を生成するための薬剤の製造における、鎮静性の量または催眠性の量の請求項1〜4のいずれかに記載の塩の使用。
【請求項8】
対象において不安緩解を生成するための薬剤の製造における、不安緩解性の量の請求項1〜4のいずれかに記載の塩の使用。
【請求項9】
対象において筋弛緩を生成するための薬剤の製造における、筋弛緩性の量の請求項1〜4のいずれかに記載の塩の使用。
【請求項10】
対象のけいれんを処置するための薬剤の製造における、鎮痙性の量の請求項1〜4のいずれかに記載の塩の使用。
【請求項11】
請求項1に記載の塩を作製する方法であって、式(I)の化合物の遊離塩基をエタンスルホン酸と反応させる工程を包含する、方法。
【請求項12】
溶液において式(I)の化合物の遊離塩基をエタンスルホン酸と接触させて前記エシレート塩の沈殿物を形成する工程を包含する、請求項11に記載の方法。
【請求項13】
前記沈殿物を単離する工程をさらに包含する、請求項12に記載の方法。
【請求項14】
前記遊離塩基は、トルエンまたは酢酸エチルに溶解される、請求項12または13に記載の方法。
【請求項15】
前記エタンスルホン酸は、エタノールに溶解される、請求項12〜14のいずれかに記載の方法。
【請求項16】
請求項3に記載の塩を調製する請求項12に記載の方法であって、トルエン、エタノール、酢酸エチル(ethyle acetate)、MtBE、DCM、アセトン、酢酸イソプロピル、ギ酸エチル、またはメタノール中の式(I)の化合物の遊離塩基の溶液をエタノール中のエタンスルホン酸の溶液と接触させて該塩の沈殿物を形成する工程を包含する、方法。
【請求項17】
請求項4に記載の塩を調製する請求項12に記載の方法であって、請求項3に記載の塩をDCMまたはDCM水溶液において室温より高い温度でスラリーにして溶液を生成する工程、および該溶液を蒸発乾固する工程を包含する、方法。
【請求項18】
請求項2〜4のいずれかに記載の塩を調製する方法であって、式(I)の化合物のエシレートを溶媒から、または溶媒/貧溶媒混合物もしくは溶媒/共溶媒混合物から結晶化する工程を包含する、方法。
【請求項19】
対象において鎮静または催眠を生成するための方法であって、請求項1〜4のいずれかに記載の塩を鎮静性の有効量または催眠性の有効量で該対象に投与する工程を包含する、方法。
【請求項20】
対象において不安緩解を生成するための方法であって、請求項1〜4のいずれかに記載の塩を不安緩解性の有効量で該対象に投与する工程を包含する、方法。
【請求項21】
対象において筋弛緩を生成するための方法であって、請求項1〜4のいずれかに記載の塩を筋弛緩性の有効量で該対象に投与する工程を包含する、方法。
【請求項22】
対象のけいれんを処置するための方法であって、請求項1〜4のいずれかに記載の塩を鎮痙性の有効量で該対象に投与する工程を包含する、方法。
【請求項1】
式(I):
【化2】
の化合物のエシレート塩。
【請求項2】
結晶塩である、請求項1に記載の塩。
【請求項3】
約6.2、9.2、12.3、15.0、17.2、および20.6度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す結晶多型である、請求項2に記載のエシレート塩。
【請求項4】
前記塩は、約3.6、6.4、7.1、12.3、14.1、および17.1度2θに特性ピークを含むX線粉末回折(XRPD)パターンを示す結晶多型である、請求項2に記載のエシレート塩。
【請求項5】
請求項1〜4のいずれかに記載の塩、および薬剤として許容可能な担体、賦形剤、または希釈剤を含む医薬組成物。
【請求項6】
薬剤として使用するための請求項1〜4のいずれかに記載の塩。
【請求項7】
対象において鎮静または催眠を生成するための薬剤の製造における、鎮静性の量または催眠性の量の請求項1〜4のいずれかに記載の塩の使用。
【請求項8】
対象において不安緩解を生成するための薬剤の製造における、不安緩解性の量の請求項1〜4のいずれかに記載の塩の使用。
【請求項9】
対象において筋弛緩を生成するための薬剤の製造における、筋弛緩性の量の請求項1〜4のいずれかに記載の塩の使用。
【請求項10】
対象のけいれんを処置するための薬剤の製造における、鎮痙性の量の請求項1〜4のいずれかに記載の塩の使用。
【請求項11】
請求項1に記載の塩を作製する方法であって、式(I)の化合物の遊離塩基をエタンスルホン酸と反応させる工程を包含する、方法。
【請求項12】
溶液において式(I)の化合物の遊離塩基をエタンスルホン酸と接触させて前記エシレート塩の沈殿物を形成する工程を包含する、請求項11に記載の方法。
【請求項13】
前記沈殿物を単離する工程をさらに包含する、請求項12に記載の方法。
【請求項14】
前記遊離塩基は、トルエンまたは酢酸エチルに溶解される、請求項12または13に記載の方法。
【請求項15】
前記エタンスルホン酸は、エタノールに溶解される、請求項12〜14のいずれかに記載の方法。
【請求項16】
請求項3に記載の塩を調製する請求項12に記載の方法であって、トルエン、エタノール、酢酸エチル(ethyle acetate)、MtBE、DCM、アセトン、酢酸イソプロピル、ギ酸エチル、またはメタノール中の式(I)の化合物の遊離塩基の溶液をエタノール中のエタンスルホン酸の溶液と接触させて該塩の沈殿物を形成する工程を包含する、方法。
【請求項17】
請求項4に記載の塩を調製する請求項12に記載の方法であって、請求項3に記載の塩をDCMまたはDCM水溶液において室温より高い温度でスラリーにして溶液を生成する工程、および該溶液を蒸発乾固する工程を包含する、方法。
【請求項18】
請求項2〜4のいずれかに記載の塩を調製する方法であって、式(I)の化合物のエシレートを溶媒から、または溶媒/貧溶媒混合物もしくは溶媒/共溶媒混合物から結晶化する工程を包含する、方法。
【請求項19】
対象において鎮静または催眠を生成するための方法であって、請求項1〜4のいずれかに記載の塩を鎮静性の有効量または催眠性の有効量で該対象に投与する工程を包含する、方法。
【請求項20】
対象において不安緩解を生成するための方法であって、請求項1〜4のいずれかに記載の塩を不安緩解性の有効量で該対象に投与する工程を包含する、方法。
【請求項21】
対象において筋弛緩を生成するための方法であって、請求項1〜4のいずれかに記載の塩を筋弛緩性の有効量で該対象に投与する工程を包含する、方法。
【請求項22】
対象のけいれんを処置するための方法であって、請求項1〜4のいずれかに記載の塩を鎮痙性の有効量で該対象に投与する工程を包含する、方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図6G】
【図6H】
【図6I】

【図7A】
【図7B】
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【図6G】
【図6H】
【図6I】
【図7A】
【図7B】
【公表番号】特表2009−542787(P2009−542787A)
【公表日】平成21年12月3日(2009.12.3)
【国際特許分類】
【出願番号】特願2009−518957(P2009−518957)
【出願日】平成19年7月10日(2007.7.10)
【国際出願番号】PCT/GB2007/002583
【国際公開番号】WO2008/007081
【国際公開日】平成20年1月17日(2008.1.17)
【出願人】(509011824)パイオン ユーケー リミテッド (5)
【Fターム(参考)】
【公表日】平成21年12月3日(2009.12.3)
【国際特許分類】
【出願日】平成19年7月10日(2007.7.10)
【国際出願番号】PCT/GB2007/002583
【国際公開番号】WO2008/007081
【国際公開日】平成20年1月17日(2008.1.17)
【出願人】(509011824)パイオン ユーケー リミテッド (5)
【Fターム(参考)】
[ Back to top ]