
短時間作用型ベンゾジアゼピンの塩およびそれらの多形形態
【課題】鎮静または催眠、不安緩解、筋弛緩、または抗痙攣の目的のための薬剤としての短時間作用型ベンゾジアゼピンの塩の提供。
【解決手段】下記式で表されるベンゾジアゼピンのベシル酸塩。

【解決手段】下記式で表されるベンゾジアゼピンのベシル酸塩。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、短時間作用型ベンゾジアゼピンの塩、および特に鎮静または催眠、不安緩解、筋弛緩、または抗痙攣の目的のための薬剤としての該塩の使用に関する。
【背景技術】
【0002】
特許文献1は、カルボン酸エステル部分を含み、非特異的組織エステラーゼによって不活性化される短時間作用型ベンゾジアゼピンを記載している。器官に依存しない排除機構が、これらのベンゾジアゼピンの特徴であることが予想され、より予想可能かつ再現性のある薬力学的プロフィールを提供する。これらの化合物は、鎮静−催眠、不安緩解、筋弛緩および抗痙攣の目的を含めた治療用途に適する。これらの化合物は、次の臨床状態(clinical setting):術前の鎮静、不安緩解、および手術前後の記憶消失的使用;短期診断、手術または内視鏡的処置中の意識下鎮静;他の麻酔薬または鎮痛薬の投与の前および/または同時の全身麻酔の誘導および維持のための成分として;ICU鎮静において、静脈内投与されるのに有用な短時間作用型CNS抑制剤である。
【0003】
特許文献1(実施例Ic−8、36頁)に開示された化合物の1つは、以下の式(I)に示されたメチル3−[(4S)−8−ブロモ−1−メチル−6−(2−ピリジニル)−4H−イミダゾール[1,2−a][1,4]ベンゾジアゼピン−4−イル]プロパノエートである。
【0004】
【化1】
【先行技術文献】
【特許文献】
【0005】
【特許文献1】欧州特許第1,183,243号明細書
【発明の概要】
【発明が解決しようとする課題】
【0006】
式(I)の遊離塩基は、5℃で保存される場合に安定であるが、40℃/相対湿度75%(開放)で保存されたサンプルは、潮解し、色が黄色から橙色になり、初期に比べて含有量の著しい減少を示すことが観測される(以下の実施例1を参照されたい)。
【課題を解決するための手段】
【0007】
驚くべきことに、式(I)の化合物は、高結晶質のモノ(ベンゼンスルホン酸)ベシル酸塩を形成し、これは一連の薬学的に許容される溶媒から容易に単離され、良好な熱安定性、低い吸湿性および高い水溶性を示すことが見出された。
【0008】
本発明によれば、式(I)の化合物のベシル酸塩が提供される。好ましくは、この塩は、結晶塩である。好ましくは、この結晶塩は、式(I)の化合物:ベシル酸塩の1:1の化学量論比を有する。ベシル酸の多形相の調製および特徴づけは、以下の実施例に記載されている。
【0009】
本発明によれば、式(I)の化合物のベシル酸塩の結晶多形(本明細書でベシル酸塩形態1と呼ぶ)が提供され、これは、約7.3、7.8、9.4、12.1、14.1、14.4、14.7,または15.6度2θにおける特性ピークを含む粉末X線回折(XRPD)パターンを示す。
【0010】
好ましくは、ベシル酸塩形態1の結晶多形は、約7.3、7.8、9.4、12.1、14.1、14.4、14.7、および15.6度2θにおける特性ピークを含むXRPDパターンを示す。
【0011】
より好ましくは、ベシル酸塩形態1の結晶多形は、7.25(10.60)、7.84(72.60)、9.36(12.10)、12.13(32.50)、14.06(48.50)、14.41(74.30)、14.70(50.70)、15.60(26.90)[回折角2θ度(%相対強度)]における特性ピークを含むXRPDパターンを示す。
【0012】
好ましくは、ベシル酸塩形態1の結晶多形は、187〜204℃、好ましくは、約191〜192℃の範囲の示差走査熱量測定(DSC)での開始融解温度を有する。
【0013】
形態1の結晶構造は、190K(6.3のRファクター)で分解された。形態1は、化合物:ベシル酸塩の1:1の化学量論比を有する。その結晶非対称単位は、2つの独立した化合物分子および2つのベシル酸塩分子を含有する。2つの独立した化合物分子は、イミダゾール環上で1価プロトン化されている。この結晶構造は、a=7.6868Å、b=29.2607Å、c=12.3756Å、α=90°、β=97.7880°、γ=90°の単位格子寸法およびP21の空間群を有する。この結晶構造は、実施例9により詳細に記載されており、結晶学的座標は、表17に提供されている。形態1の結合の長さおよび角度は、それぞれ表19および20に提供されている。
【0014】
a=7.6868Å、b=29.2607Å、c=12.3756Å、α=90°、β=97.7880°、γ=90°の単位格子寸法を有する結晶を含む結晶多形である式(I)の化合物のベシル酸塩が、本発明によって提供される。
【0015】
表17に示される構造座標によって定義される結晶構造を有する結晶多形である式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0016】
それぞれ表19および20に示される結合の長さおよび角度を有する式(I)の化合物のベシル酸塩が、本発明によってさらに提供される。
【0017】
約8.6、10.5、12.0、13.1、14.4、または15.9度2θにおける特性ピークを含むXRPDパターンを示す式(I)の化合物のベシル酸塩の結晶多形(以下、ベシル酸塩形態2と呼ぶ)が、本発明によってさらに提供される。
【0018】
好ましくは、ベシル酸塩形態2の結晶多形は、約8.6、10.5、12.0、13.1、14.4、および15.9度2θにおける特性ピークを含むXRPDパターンを示す。
【0019】
より好ましくは、8.64(17.60)、10.46(21.00)、12.03(22.80)、13.14(27.70)、14.42(11.20)、15.91(100.00)[回折角2θ度(%相対強度)]における特性ピークを含むXRPDパターンを示すベシル酸塩形態2の結晶多形を示す。
【0020】
好ましくは、ベシル酸塩形態2の結晶多形は、170〜200℃の範囲、好ましくは、約180℃の示差走査熱量測定(DSC)での開始融解温度を有する。
【0021】
形態2の結晶構造は、190K(3.8のRファクター)で分解された。形態2は、化合物:ベシル酸塩の1:1の化学量論比を有する。その結晶学的非対称単位は、1つの化合物分子および1つのベシル酸塩分子を含有する。この化合物分子は、イミダゾール環上で1価プロトン化されている。この結晶構造は、a=8.92130Å、b=11.1536Å、c=25.8345Å、α=90°、β=90°、γ=90°の単位格子寸法、およびP212121の空間群を有する。この結晶構造は、実施例10により詳細に記載されており、結晶学的座標は、表18に提供されている。形態2の結合の長さおよび角度は、それぞれ表21および22に提供されている。
【0022】
本発明によれば、a=8.92130Å、b=11.1536Å、c=25.8345Å、α=90°、β=90°、γ=90°の単位格子寸法を有する結晶を含む結晶多形である式(I)の化合物のベシル酸塩が提供される。
【0023】
表18に示される構造座標によって定義される結晶構造を有する結晶多形である式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0024】
それぞれ表21および22に示される結合の長さおよび角度を有する式(I)の化合物のベシル酸塩が、本発明によってさらに提供される。
【0025】
約7.6、11.2、12.4、14.6、15.2、16.4、または17.7度2θにおける特性ピークを含む粉末X線回折(XRPD)パターンを示す式(I)の化合物のベシル酸塩の結晶多形(以下、ベシル酸塩形態3と呼ぶ)が、本発明によってさらに提供される。
【0026】
好ましくは、ベシル酸塩形態3の結晶多形は、約7.6、11.2、12.4、14.6、15.2、16.4、および17.7度2θにおける特性ピークを含むXRPDパターンを示す。
【0027】
より好ましくは、ベシル酸塩形態3の結晶多形は、7.61(65.70)、11.19(33.20)、12.38(48.70)、14.63(30.60)、15.18(33.20)、16.40(29.60)、17.68(51.30)[回折角2θ°(%相対強度)]における特性ピークを含むXRPDパターンを示す。
【0028】
好ましくは、ベシル酸塩形態3の結晶多形は、195−205℃、好ましくは、約200−201℃の範囲の示差走査熱量測定(DSC)での開始融解温度を有する。
【0029】
約7.6、10.8、15.2、15.9、または22.0度2θにおける特性ピークを含むXRPDパターンを示す式(I)の化合物のベシル酸塩の結晶多形(以下、ベシル酸塩形態4と呼ぶ)が、本発明によってさらに提供される。
【0030】
好ましくは、ベシル酸塩形態4の結晶多形は、約7.6、10.8、15.2、15.9、および22.0度2θにおける特性ピークを含むXRPDパターンを示す。
【0031】
好ましくは、ベシル酸塩形態4の結晶多形は、7.62(83.50)、10.75(14.70)、15.17(37.80)、15.85(28.70)、22.03(100)[回折角2θ°(%相対強度)]における特性ピークを含むXRPDパターンを示す。
【0032】
好ましくは、ベシル酸塩形態4の結晶多形は、180−185℃、好ましくは、約182℃の範囲の示差走査熱量測定(DSC)での開始融解温度を有する。
【0033】
好ましい塩は、形成、収量、純度および化学的および固体安定性の頑健性に基づいて、ベシル酸塩形態1である。
【0034】
式(I)の化合物の遊離塩基をベンゼンスルホン酸と反応させることを含む式(I)の化合物のベシル酸塩の製造方法も、本発明によって提供される。
【0035】
同様に、本発明によれば、溶液中で式(I)の化合物の遊離塩基をベンゼンスルホン酸と接触させベシル酸塩の沈殿の形成を生じさせることを含む本発明の塩の製造の方法が提供される。好ましくは、この方法は、沈殿を単離することをさらに含む。
【0036】
好ましくは、上記遊離塩基は、トルエン、エタノール、酢酸エチル、MtBE、ジクロロメタン(DCM)、酢酸イソプロピル、ギ酸エチル、メタノール、またはアセトンに溶解される。より好ましくは、上記遊離塩基は、トルエンまたは酢酸エチルに溶解される。好ましくは、ベンゼンスルホン酸は、エタノールに溶解される。
【0037】
ベシル酸塩形態1は、式(I)の化合物の遊離塩基のトルエン、酢酸エチル、アセトン、酢酸イソプロピル、またはギ酸エチル中の溶液とベンゼンスルホン酸のエタノール中の溶液とを接触させて塩の沈殿の形成を生じさせることによって調製され得る。
【0038】
上記方法によって得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0039】
ベシル酸塩形態2は、メタノール中の式(I)の化合物の遊離塩基の溶液とエタノール中のベンゼンスルホン酸の溶液とを接触させて塩の沈殿の形成を生じさせることによって調製され得る。好ましくは、この混合物は周囲温度未満に冷却される(例えば4℃)。
【0040】
上記方法で得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0041】
ベシル酸塩形態3は、形態1を用いて、酢酸エチル/エタノールからの形態1の結晶化から得られる液体に播種することによって調製され得る。好ましくは、この液体は、周囲温度未満に冷却される(例えば4℃)。
【0042】
一実施形態によれば、ベシル酸塩形態3は、式(I)の化合物の酢酸エチル中の溶液とベンゼンスルホン酸のエタノール中の溶液とを接触させて形成される沈殿から分離される濾過溶液に、式(I)の化合物のベシル酸塩形態1の結晶塩で播種してベシル酸塩形態3結晶多形を生じさせることによって調製され得る。
【0043】
上記方法のいずれかによって得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0044】
ベシル酸塩形態4は、酢酸イソプロピル/エタノール、好ましくは、40%酢酸イソプロピル/エタノールからベシル酸塩形態1を再結晶化させることによって調製され得る。
【0045】
上記方法によって得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0046】
本発明の塩は、適切な溶媒から、または適切な溶媒/貧溶媒(anti−solvent)もしくは溶媒/共溶媒混合物から式(I)の化合物のベシル酸塩を結晶化させることによっても調製され得る。この溶液または混合物は、適切な場合、冷却および/または蒸発して結晶化を達成し得る。
【0047】
本発明者らは、形態2の結晶化は、極性(例えば、アセトニトリル:水)もしくは親油性(n−ノナン)のいずれかの、または両方(ジメチルスルホキシド:1,2−ジクロロベンゼン)の極端な状態が存在する条件において観測されることを見出した。
【0048】
形態2の結晶化のための溶媒の例は、ノナン;メタノールである。
【0049】
形態1の結晶化のための溶媒/貧溶媒混合物の例は、ジメチルアセトアミド/メチルイソブチルケトン;ジメチルアセトアミド/テトラクロロエチレン;アセトニトリル/3−メチルブタン−1−オール;アセトニトリル/1,2−ジクロロベンゼン;アセトニトリル/酢酸ペンチル;メタノール/3−メチルブタン−1−オール;メタノール/メチルイソブチルケトン;2,2,2−トリフルオロエタノール/1,4−ジメチルベンゼン;エタノール/メチルイソブチルケトン;エタノール/1,4−ジメチルベンゼン;プロパン−1−オール/1,2−ジクロロベンゼン;プロパン−1−オール/テトラクロロエチレン;プロパン−2−オール/1,2−ジクロロベンゼン;プロパン−2−オール/n−ノナン;2−メトキシエタノール/水;2−メトキシエタノール/酢酸ペンチル;2−メトキシエタノール/1,4−ジメチルベンゼン;テトラヒドロフラン/水;テトラヒドロフラン/3−メチルブタン−1−オール;テトラヒドロフラン/1,2−ジクロロベンゼン;テトラヒドロフラン/酢酸エチル;テトラヒドロフラン/1,3−ジメチルベンゼンである。
【0050】
形態2の結晶化のための溶媒/貧溶媒混合物の例は、エタノール/酢酸エチル;エタノール/メチルイソブチルケトン;エタノール/p−シメン;ジメチルスルホキシド/1,2−ジクロロベンゼン;アセトニトリル/水;エタノール/1,2−ジクロロベンゼン;エタノール/テトラクロロエチレン;テトラヒドロフラン/1,2−ジクロロベンゼン;テトラヒドロフラン/酢酸エチルである。
【0051】
好ましい実施形態によれば、形態1は、2−メトキシエタノール/酢酸ペンチルから結晶化される。
【0052】
好ましい実施形態によれば、形態2は、エタノール/酢酸エチルから結晶化される。
【0053】
好ましい実施形態によれば、形態2は、メタノール/エタノールから結晶化される(周囲温度未満、例えば4℃でメタノール/エタノール中の式(I)の化合物のベシル酸塩の溶液を冷却することによるのが好ましい)。
【0054】
好ましい実施形態によれば、形態3は、エタノール/酢酸エチルから結晶化される(周囲温度未満、例えば4℃に混合物を冷却することによるのが適切である)。
【0055】
好ましい実施形態によれば、形態4は、酢酸イソプロピル/エタノールから結晶化される(周囲温度に酢酸イソプロピル/エタノール中の式(I)の化合物の溶液を冷却することによるのが好ましい)。
【0056】
上記方法のいずれかによって得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0057】
本発明の塩の調製方法は、以下の実施例に詳細に記載される。
【0058】
本発明の塩は、特に鎮静または催眠、不安緩解、筋弛緩、または抗痙攣の目的のための薬剤として使用され得る。
【0059】
本発明の塩は、バルクの活性薬品として投与されることが可能であるが、この塩は、好ましくは、薬剤組成物の形態で、薬学的に許容される担体、賦形剤、または希釈剤と一緒に提供される。担体、賦形剤、または希釈剤は、当然、組成物の他の成分と相溶性があるという意味において許容されなければならず、レシピエントに有害であってはならない。
【0060】
したがって、本発明は、本発明の塩および薬学的に許容される担体、賦形剤、または希釈剤を含む薬剤組成物を提供する。
【0061】
本発明の薬剤組成物には、経口、直腸、局所、口腔(例えば、舌下)および非経口(例えば、皮下、筋肉内または静脈内)投与に適するものが含まれる。
【0062】
好ましくは、本発明の塩は、例えば、溶液の静脈内または筋肉内注射による非経口投与のための薬剤組成物の形態で提供される。薬剤組成物が、非経口投与用である場合、この組成物は、静菌剤、抗酸化剤、緩衝剤または他の薬学的に許容される添加剤を含み得る水性もしくは非水性溶液または液体の混合物であり得る。
【0063】
本発明の塩の好ましい製剤は、pH2〜4の水性酸性媒体またはサイクロデキストリン(CD)の水溶液中である。これらの製剤に使用され得るサイクロデキストリンは、β−CDの陰イオン性に帯電されたスルホブチルエーテル(SBE)誘導体、具体的にはCyDex,Inc.によってCaptisolの商標で市販されているSBE7−β−CD(CriticalReviews in Therapeutic Drug Carrier Systems、14巻(1号)、1〜104頁(1997年))、またはヒドロキシプロピルCDのいずれかである。
【0064】
本発明の塩のさらに好ましい製剤は、この塩に加えて、次の試剤:アスコルビン酸、クエン酸、マレイン酸、リン酸、グリシン、塩酸グリシン、コハク酸または酒水酸の少なくとも1つを含む凍結乾燥製剤である。これらの試剤は、緩衝剤、粘結剤または可視化剤(visualization agent)として有用であると考えられる。ある場合には、製剤に塩化ナトリウム、マンニトール、ポリビニルピロリドン、または他の成分を含むことが有益であり得る。
【0065】
製剤化の好ましい方法(すなわち、酸緩衝剤またはCDベース)は、特定の塩の物理化学的性質(例えば、水溶解度、pKaなど)によって決まり得る。別法として、上記塩は、水(注射用)またはブドウ糖または食塩水での再構成用の凍結乾燥固体として提供され得る。このような製剤は、アンプルまたは使い捨て注射器具などの単位剤形で通常提供される。それらは、それから適切な用量が抜き取られるボトルなどの複数用量形態でも提示され得る。全てのこのような製剤は無菌でなければならない。
【0066】
本発明によれば、対象に有効な鎮静量または催眠量の本発明の塩を投与することを含む、対象において鎮静または催眠を生じるための方法が提供される。
【0067】
対象に有効な不安緩解量の本発明の塩を投与することを含む、対象に不安緩解(anxiolysis)を誘導するための方法も、本発明によって提供される。
【0068】
対象に有効な筋弛緩量の本発明の塩を投与することを含む、対象において筋弛緩を誘導するための方法が、本発明によってさらに提供される。
【0069】
対象に有効な抗痙攣量の本発明の塩を投与することを含む、対象において痙攣を治療するための方法が、本発明によってさらに提供される。
【0070】
本発明によれば、対象に鎮静または催眠を生ずるための薬剤の製造における鎮静量または催眠量の本発明の塩の使用も提供される。
【0071】
本発明によれば、対象に鎮静または催眠を生ずるための本発明の塩も提供される。
【0072】
対象における不安緩解を生ずるための薬剤の製造における不安緩解量の本発明の塩の使用も、本発明によって提供される。
【0073】
対象における不安緩解を生ずるための本発明の塩も、本発明によって提供される。
【0074】
対象における筋弛緩を生じるための薬剤の製造における、筋弛緩量の本発明の塩の使用が本発明によってさらに提供される。
【0075】
対象における筋弛緩を生じるための本発明の塩が本発明によってさらに提供される。
【0076】
対象における痙攣を治療するための、薬剤の製造における抗痙攣量の本発明の塩の使用が本発明によってさらに提供される。
【0077】
対象における痙攣を治療するための本発明の塩が本発明によってさらに提供される。
【0078】
上記対象は、適切には哺乳類、好ましくは、ヒトである。
【0079】
ヒトへの投与に適する非経口製剤は、好ましくは、0.1から20mg/mlの溶液中の本発明の塩または複数用量バイアル用にはその倍数を含有する。
【0080】
静脈内投与は、ボーラス注入法または、より適切には、連続注入の形態を取り得る。それぞれの対象のための用量は変わり得るが、哺乳類において鎮静または催眠を得るのに適する本発明の塩の静脈内投与量または用量は、0.01から5.0mg/体重1kg、より詳しくは、0.02から0.5mg/体重1kgである(上記は、活性成分である上記塩の重量に基づく)。哺乳類において不安緩解を得るのに適する本発明の塩の静脈内投与量または用量は、0.01から5.0mg/体重1kg、より詳しくは、0.02から0.5mg/体重1kgである(上記は、活性成分である上記塩の重量に基づく)。哺乳類において筋弛緩を得るのに適する本発明の塩の静脈内投与量または用量は、0.01から5.0mg/体重1kg、より詳しくは、0.02から0.5mg/体重1kgである(上記は、活性成分である上記塩の重量に基づく)。哺乳類において痙攣を治療するのに適する本発明の塩の静脈内投与量または用量は、0.01から5.0mg/体重1kg、より詳しくは、0.02から0.5mg/体重1kgである(上記は、活性成分である上記塩の重量に基づく)。
【0081】
本発明の塩は、次の臨床状態:術前の鎮静、不安緩解、および手術前後の記憶消失使用;短期診断、手術または内視鏡的処置中の意識下鎮静;他の麻酔薬または鎮痛薬の投与の前および/または同時の全身麻酔の誘導および維持のための成分として;ICU鎮静において、静脈内投与されるのに有用な短時間作用型CNS抑制剤である。
【0082】
本発明の好ましい実施形態は、添付図を参照して以下の実施例に記載される。
例えば、本発明は、以下の項目を提供する。
(項目1)
式(I):
【化4】
の化合物のベシル酸塩。
(項目2)
結晶塩である、項目1に記載の塩。
(項目3)
約7.3、7.8、9.4、12.1、14.1、14.4、14.7、および15.6度2θにおける特性ピークを含む粉末X線回折(XRPD)パターンを示す結晶多形である、項目2に記載のベシル酸塩。
(項目4)
a=7.6868Å、b=29.2607Å、c=12.3756Å、α=90°、β=97.7880°、γ=90°の単位格子寸法を有する結晶を含む結晶多形である、項目2または3に記載のベシル酸塩。
(項目5)
表17に示された構造座標によって定義される結晶構造を有する結晶多形である、項目2から4のいずれかに記載のベシル酸塩。
(項目6)
表19および20に示された結合の長さおよび角度を有する結晶構造を有する結晶多形である、項目2から5のいずれかに記載のベシル酸塩。
(項目7)
約8.6、10.5、12.0、13.1、14.4、および15.9度2θにおける特性ピークを含むXRPDパターンを示す結晶多形である、項目2に記載のベシル酸塩。
(項目8)
a=8.92130Å、b=11.1536Å、c=25.8345Å、α=90°、β=90°、γ=90°の単位格子寸法を有する結晶を含む結晶多形である、項目2または7に記載のベシル酸塩。
(項目9)
表18に示された構造座標によって定義される結晶構造を有する結晶多形である、項目2、7または8のいずれかに記載のベシル酸塩。
(項目10)
表21および22に示された結合の長さおよび角度を有する結晶構造を有する結晶多形である、項目2、または7から9のいずれかに記載のベシル酸塩。
(項目11)
約7.6、11.2、12.4、14.6、15.2、16.4、および17.7度2θにおける特性ピークを含む粉末X線回折(XRPD)パターンを示す式(I)の化合物のベシル酸塩の結晶多形である、項目2に記載のベシル酸塩。
(項目12)
約7.6、10.8、15.2、15.9および22.0度2θにおける特性ピークを含むXRPDパターンを示す式(I)の化合物のベシル酸塩の結晶多形である、項目2に記載のベシル酸塩。
(項目13)
項目1から12のいずれかに記載の塩、および薬学的に許容される担体、賦形剤、または希釈剤を含む薬剤組成物。
(項目14)
薬剤としての使用のための項目1から12のいずれかに記載の塩。
(項目15)
対象において鎮静または催眠を生じるための薬剤の製造における、鎮静量または催眠量の項目1から12のいずれかに記載の塩の使用。
(項目16)
対象において不安緩解を生じるための薬剤の製造における、不安緩解量の項目1から12のいずれかに記載の塩の使用。
(項目17)
対象において筋弛緩を生じるための薬剤の製造における、筋弛緩量の項目1から12のいずれかに記載の塩の使用。
(項目18)
対象において痙攣を治療するための薬剤の製造における、抗痙攣量の項目1から12のいずれかに記載の塩の使用。
(項目19)
式(I)の化合物の遊離塩基をベンゼンスルホン酸と反応させる工程を含む、項目1に記載の塩の製造方法。
(項目20)
溶液中で前記遊離塩基をベンゼンスルホン酸と接触させて、前記ベシル酸塩の沈殿の形成を引き起こす工程を含む、項目19に記載の方法。
(項目21)
前記沈殿を単離する工程をさらに含む、項目20に記載の方法。
(項目22)
前記遊離塩基が、トルエンまたは酢酸エチルに溶解される、項目20または21に記載の方法。
(項目23)
前記ベンゼンスルホン酸が、エタノールに溶解される、項目20から22のいずれかに記載の方法。
(項目24)
項目3から6のいずれかに記載の塩を調製する項目20に記載の方法であって、トルエン、酢酸エチル、アセトン、酢酸イソプロピル、またはギ酸エチル中の式(I)の化合物の遊離塩基の溶液を、エタノール中のベンゼンスルホン酸の溶液と接触させて該塩の沈殿の形成を引き起こす工程を含む方法。
(項目25)
項目7から10のいずれかに記載の塩を調製する項目20に記載の方法であって、メタノール中の式(I)の化合物の遊離塩基の溶液を、エタノール中のベンゼンスルホン酸の溶液と接触させて該塩の沈殿の形成を引き起こす工程を含む方法。
(項目26)
項目11に記載の塩を調製する方法であって、酢酸エチル中の式(I)の化合物の溶液を、エタノール中のベンゼンスルホン酸の溶液と接触させることによって形成される沈殿から分離される濾液溶液に、式(I)の化合物のベシル酸塩形態1結晶塩を用いて播種して、結晶多形を生じる工程を含む方法。
(項目27)
酢酸イソプロピル/エタノールから式(I)の化合物のベシル酸塩形態1結晶塩を再結晶化する工程を含む、項目12に記載の塩を調製する方法。
(項目28)
溶媒から、または溶媒/貧溶媒もしくは溶媒/共溶媒混合物から式(I)の化合物のベシル酸塩を結晶化させる工程を含む、項目2から12のいずれかに記載の塩を調製する方法。
(項目29)
対象において鎮静または催眠を生じる方法であって、該対象に有効な鎮静量または催眠量の項目1から12のいずれかに記載の塩を投与する工程を含む方法。
(項目30)
対象において不安緩解を誘発する方法であって、該対象に有効な不安緩解量の項目1から12のいずれかに記載の塩を投与する工程を含む方法。
(項目31)
対象において筋弛緩を誘発する方法であって、該対象に有効な筋弛緩量の項目1から12のいずれかに記載の塩を投与する工程を含む方法。
(項目32)
対象において痙攣を治療する方法であって、該対象に有効な抗痙攣量の項目1から12のいずれかに記載の塩を投与する工程を含む方法。
【図面の簡単な説明】
【0083】
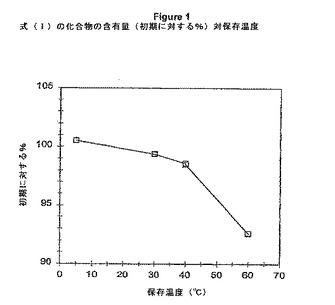
【図1】式(I)の化合物含有量(初期に対する%)対保存温度のグラフを示す図である。
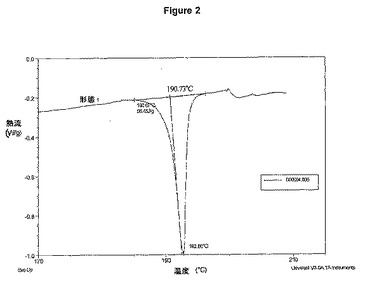
【図2】LJC−039−081−1の示差走査熱量測定(DSC)を示す図である。
【図3】LJC−039−081−2(点線)と重ね合わせたLJC−039−081−1(実線)のDSCを示す図である。
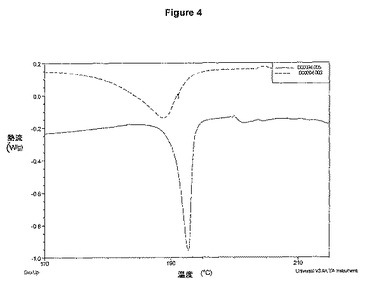
【図4】ベシル酸塩形態のDSCを示す図である(形態1実線、形態2一点鎖線)。
【図5】ベシル酸塩形態のDSCを示す図である(形態1実線、形態3点線および一点鎖線)。
【図6】T0およびT4におけるLJC−039−037−1のクロマトグラフを示す図である(表10の結果に関連)。
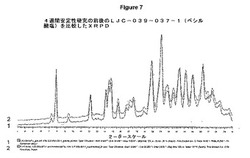
【図7】4週間安定性研究の前後でLJC−039−037−1(ベシル酸塩)を比較したXRPDを示す図である。
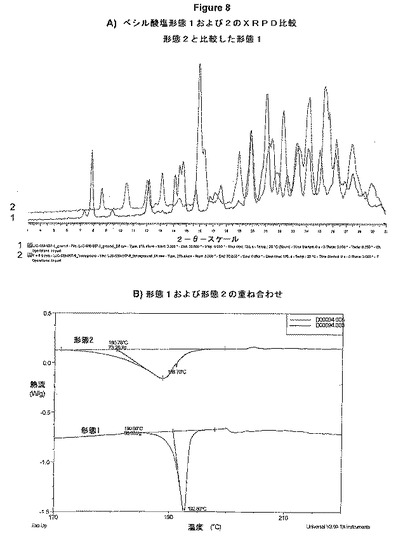
【図8】図8Aは、ベシル酸塩形態1および2のXRPD比較を示す図である。図8Bは、形態1および2の重ね合わせの示差走査熱量測定(DSC)を示す図である。
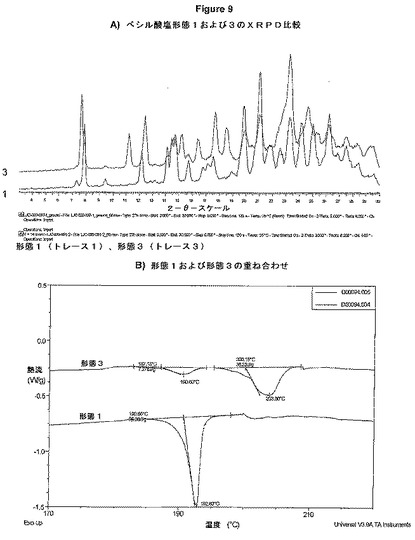
【図9】図9Aは、ベシル酸塩形態1および3のXRPD比較を示す図である。図9Bは、形態1および3の重ね合わせを示す図である。
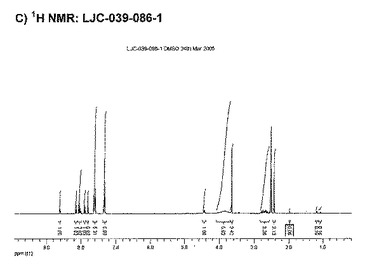
【図10】LJC−039−086−1のDSCを示す図である(ベシル酸塩形態4)。
【図11A】ベシル酸塩形態1についての結果を示す図である:A)100mgバッチLJC−039−037−1についてのXRPD。
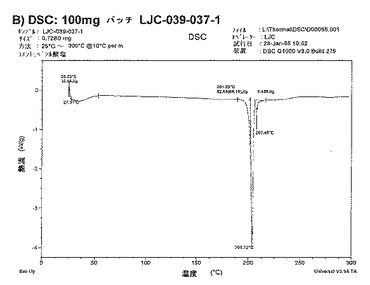
【図11B】ベシル酸塩形態1についての結果を示す図である:B)100mgバッチLJC−039−037−1についてのDSC。
【図11C】ベシル酸塩形態1についての結果を示す図である:C)100mgバッチLJC−039−037−1についてのTGA。
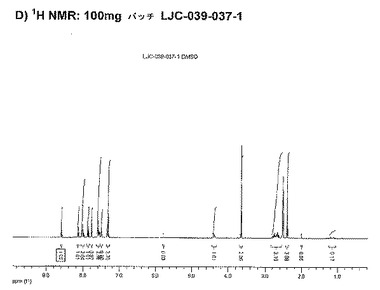
【図11D】ベシル酸塩形態1についての結果を示す図である:D)100mgバッチLJC−039−037−1についての1H NMR。
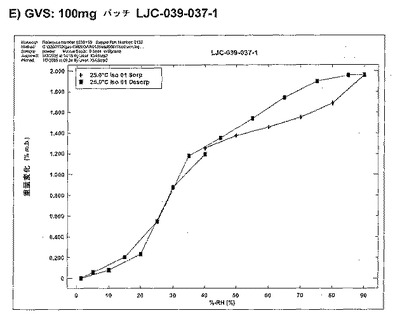
【図11E】ベシル酸塩形態1についての結果を示す図である:E)100mgバッチLJC−039−037−1についてのGVS。
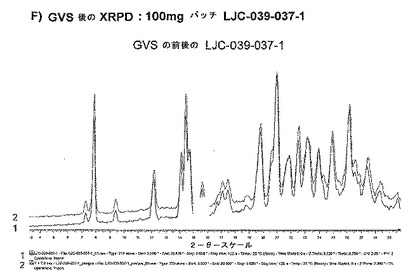
【図11F】ベシル酸塩形態1についての結果を示す図である:F)100mgバッチLJC−039−037−1についてのGVS後のXRPD。
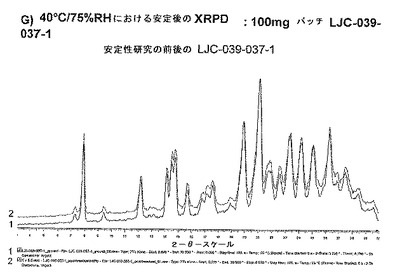
【図11G】ベシル酸塩形態1についての結果を示す図である:G)100mgバッチLJC−039−037−1についての40℃/75%RHにおける安定後のXRPD。
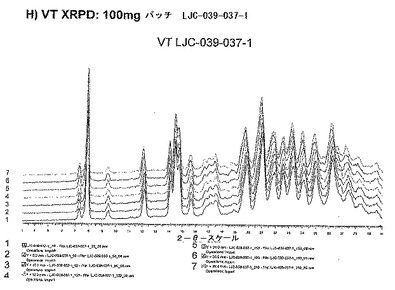
【図11H】ベシル酸塩形態1についての結果を示す図である:H)100mgバッチLJC−039−037−1についてのVT XRPD。
【図11I】ベシル酸塩形態1についての結果を示す図である:I)100mgバッチLJC−039−037−1についての偏光顕微鏡検査。
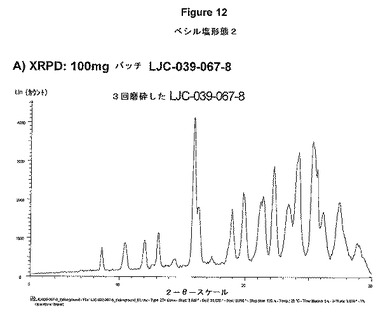
【図12A】ベシル酸塩形態2についての結果を示す図である:A)100mgバッチLJC−039−067−8についてのXRPD。
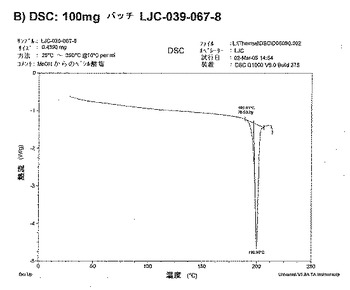
【図12B】ベシル酸塩形態2についての結果を示す図である:B)100mgバッチLJC−039−067−8についてのDSC。
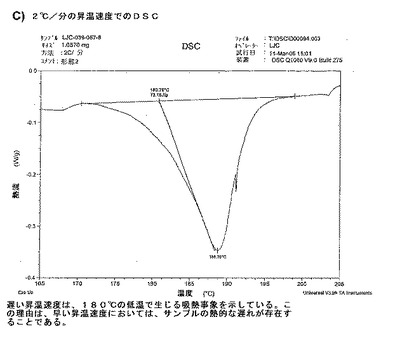
【図12C】ベシル酸塩形態2についての結果を示す図である:C)2℃/分の昇温速度でのDSC。
【図12D】ベシル酸塩形態2についての結果を示す図である:D)LJC−039−067−8についての1H NMR。
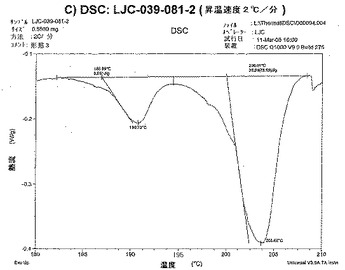
【図13A】ベシル酸塩形態3についての結果を示す図である:A)LJC−039−081−2についてのXRPD(LJC−039−081−1の液体からの2回目の採集)。
【図13B】ベシル酸塩形態3についての結果を示す図である:B)LJC−039−081−2についてのDSC。
【図13C】ベシル酸塩形態3についての結果を示す図である:C)LJC−039−081−2についてのDSC(2℃/分の昇温速度)。
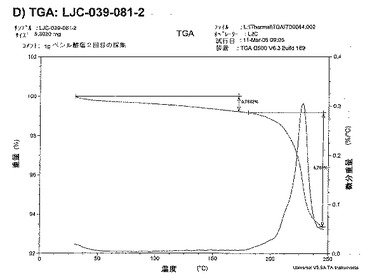
【図13D】ベシル酸塩形態3についての結果を示す図である:D)LJC−039−081−2についてのTGA。
【図13E】ベシル酸塩形態3についての結果を示す図である:E)LJC−039−081−2についての1H NMR。
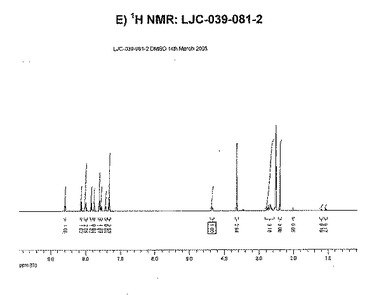
【図13F】ベシル酸塩形態3についての結果を示す図である:F)LJC−039−081−2についてのGVS。
【図13G】ベシル酸塩形態3についての結果を示す図である:G)LJC−039−081−2についてのGVS後のXRPD。
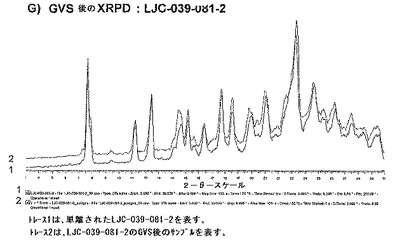
【図14A】ベシル酸塩形態4についての結果を示す図である:A)LJC−039−086−1についてのXRPD。
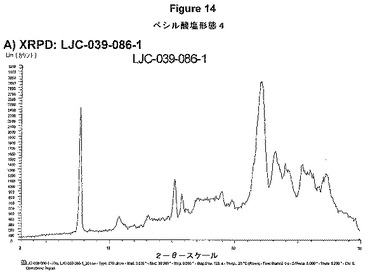
【図14B】ベシル酸塩形態4についての結果を示す図である:B)LJC−039−086−1についてのDSC。
【図14C】ベシル酸塩形態4についての結果を示す図である:C)LJC−039−086−1についての1H NMR。
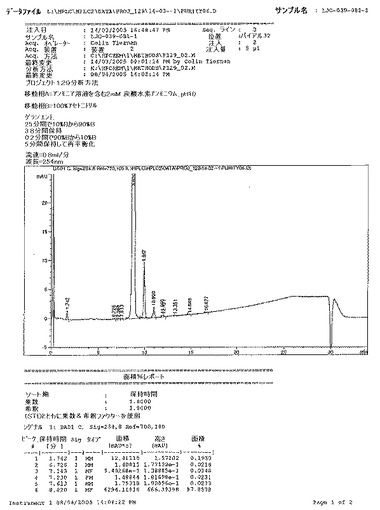
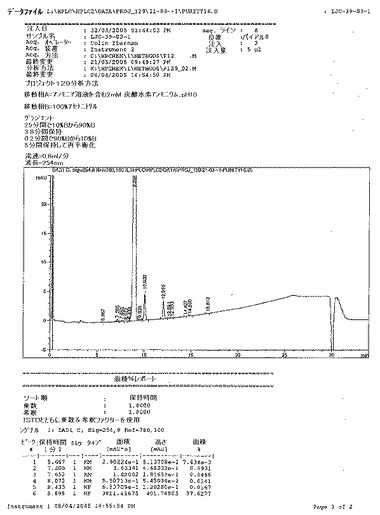
【図15A】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15B】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15C】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15D】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15E】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15F】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
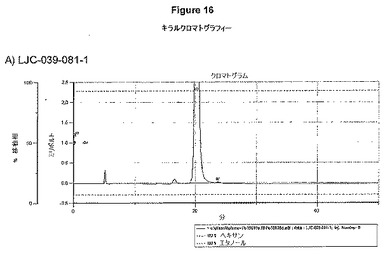
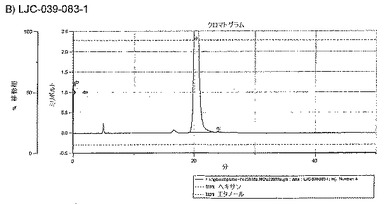
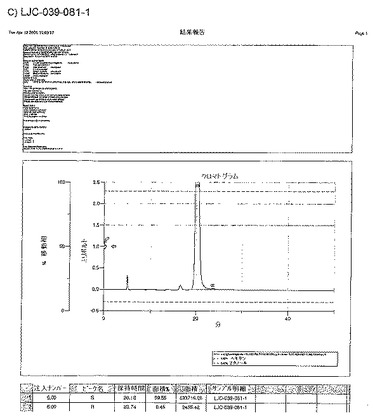
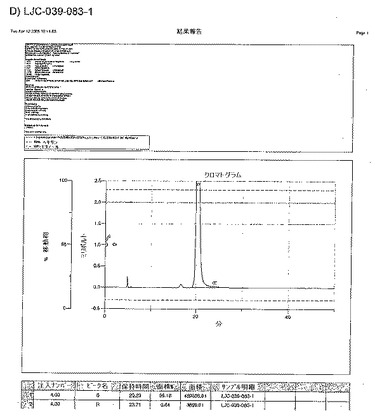
【図16A】LJC−039−081−1、およびLJC−039−083−1についてのキラルクロマトグラフィーを示す図である。
【図16B】LJC−039−081−1、およびLJC−039−083−1についてのキラルクロマトグラフィーを示す図である。
【図16C】LJC−039−081−1、およびLJC−039−083−1についてのキラルクロマトグラフィーを示す図である。
【図16D】LJC−039−081−1、およびLJC−039−083−1についてのキラルクロマトグラフィーを示す図である。
【図17】(約4−8mmの視野径)式(I)の化合物のベシル酸塩の結晶化において観測された固体の典型画像を示す図である。
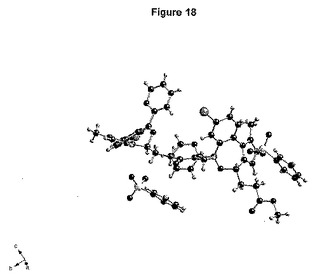
【図18】形態1における非対称単位の内容を示す図である。
【図19】2−メトキシエタノール:酢酸ペンチル溶液から成長させた式(I)の化合物のベシル酸塩、形態1の結晶の単結晶X線回折によって求められた分子構造を示す図である(原子は熱振動楕円体によって表されている)。結晶構造中に特に位置する水素のみが示されている。
【図20】形態1の2つの独立した分子によってとられる立体配座を示す図である。
【図21】形態1の1つの独立した分子によってとられる立体配座(上部)および形態2の立体配座(下部)の比較を示す図である。
【図22】2つの異なる方向に沿って見た、2つの独立した形態1のベシル酸塩によってとられる立体配座の比較を示す図である。
【図23】1つの独立した形態1のベシル酸塩によってとられる立体配座(上部)および形態2の立体配座(下部)の比較を示す図である。
【図24−1】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、2−メトキシエタノール:酢酸ペンチル溶液から成長させた式(I)の化合物のベシル酸塩の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
【図24−2】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、2−メトキシエタノール:酢酸ペンチル溶液から成長させた式(I)の化合物のベシル酸塩の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
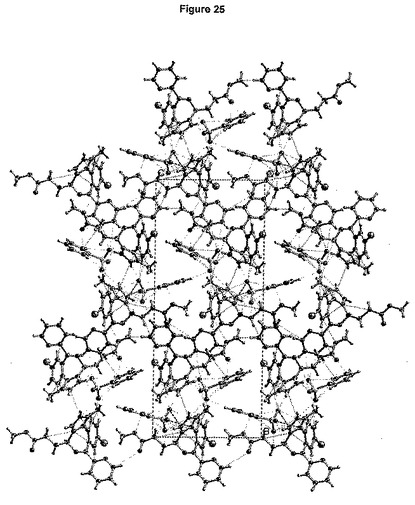
【図25】形態1についてのショートコンタクトC−O<3.6Å、C−C<3.6Å、およびN−O<3.5Åを示す図である。
【図26】形態1についての単結晶X線回折データから計算された粉末パターン回折を示す図である。
【図27】式(I)の化合物のベシル酸塩の形態2について観測された板状の形態の結晶を示す図である。
【図28】形態2の非対称単位の内容を示す図である。
【図29】式(I)の化合物のベシル酸塩、形態2の結晶の単結晶X線回折によって求められた分子構造を示す図である(原子は熱振動楕円体によって表されている)。結晶構造中に特に位置する水素のみが示されている。
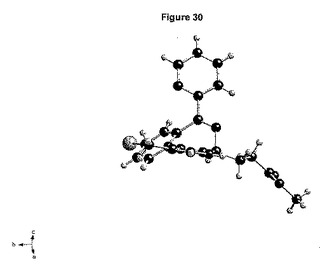
【図30】形態2の独立した分子によってとられる立体配座を示す図である。
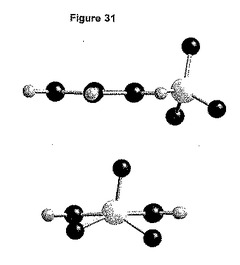
【図31】2つの異なる方向に沿って見た、形態2の独立したベシル酸塩によってとられる立体配座を示す図である。
【図32−1】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、式(I)の化合物のベシル酸塩の形態2の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
【図32−2】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、式(I)の化合物のベシル酸塩の形態2の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
【図32−3】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、式(I)の化合物のベシル酸塩の形態2の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
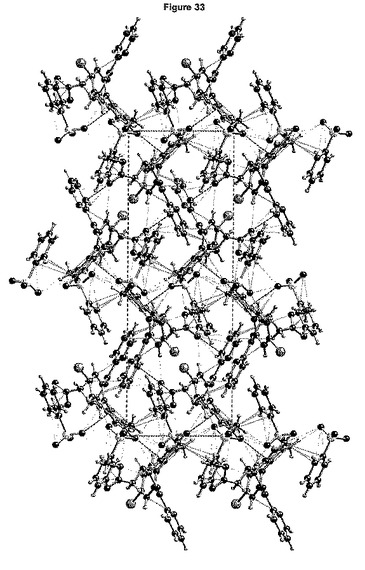
【図33】形態2についてのショートコンタクトC−O<3.6Å、C−C<3.6ÅおよびN−O<3.5Åを示す図である。
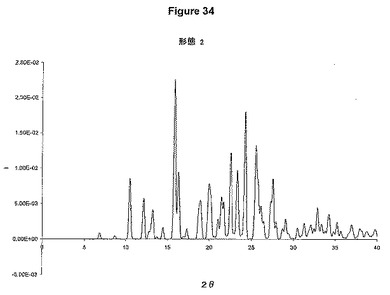
【図34】形態2についての単結晶X線回折データから計算された粉末パターン回折を示す図である。
【図35】式(I)の化合物のベシル酸塩形態1についての原子中心の標識付けを示す図である。
【図36】式(I)の化合物のベシル酸塩形態2についての原子中心の標識付けを示す図である。
【発明を実施するための形態】
【0084】
(実施例1)
式(I)の化合物の固体状態安定性の研究
方法/技術 正確に秤量した2mgの式(I)の化合物のサンプルを4mL透明ガラスねじ口バイアル中に置いた。最初に、および5℃/周囲相対湿度(AMRH)密栓、30℃/60%RH密栓、40℃/75%RH開放および60℃/AMRH密栓で保存された34日後に、サンプルを試験した。
【0085】
サンプルを、外観について目視で検査した。式(I)の化合物の含有量を、表1においてHPLC法によって求めた。%重量/重量(%w/w)値を、式(I)の化合物の標準サンプル、バッチU12438/79/1に対して測定した。%面積値を、式(I)の化合物のピーク面積を総ピーク面積で割ることによって得た。
【0086】
【表1】
結果
外観 表2は外観の結果を示す。
【0087】
【表2】
式(I)の化合物の含有量(%w/w) %w/w含有量(表3を参照されたい)は、初期値および5℃/AMRH密栓、30℃/60%RH密栓または40℃/75%RH開放での34日後に測定された値の間の差異を検出するには多すぎるばらつきを示している。60℃/AMRH密栓で34日保存されたサンプルについて測定された平均%w/wは、初期値から10%w/w減少を示している。
【0088】
式(I)の化合物の含有量(%面積) 式(I)の化合物の%面積含有量(表3および図1を参照されたい)は、5℃/AMRH密栓で保存された34日後に有意な変化を示さないが、30℃/60%RH密栓、40℃/75%RH開放または60℃/AMRH密栓におけるサンプルについては、保存温度増加とともに着実に減少する。主要な分解ピークは、RRT0.68、0.87およびRRT0.90において観測されるが、クロマトグラムも(初期においてさえも比較的複雑である(23個のピーク))多くの新しい小さな分解のピークを示している(例えば、30℃/60%RH密栓において7個のピーク;60℃/AMRH密栓において13〜20個のピーク)。これらの観測は、複数の分解経路を示唆している。RRT0.68での分解物(degradent)は、エステル加水分解生成物(式(I)の化合物の遊離酸)として暫定的に同定されている。それは、加水分解生成物について予想されるように、40℃/75%RH開放サンプルにおいて最も優勢である。
【0089】
【表3】
結論
式(I)の化合物は、5℃/AMRH密栓で保存された少なくとも34日間は外観および含有量について安定している。30℃/60%RH密栓で外観における変化は指摘されなかったが、初期の%面積に対して式(I)の化合物の含有量における約0.6%低下が観測された。40℃/75%RH開放または60℃/AMRH密栓で保存されたサンプルは、潮解し、色が黄色から橙色になり、初期に比べて式(I)の化合物の含有量における著しい低下(1.5から8%)を示した。RRT0.68、0.87およびRRT0.90における主要な分解ピークが、多数のより小さなピークと一緒に観測され、複数の分解経路が示唆された。RRT0.68における分解物は、エステル加水分解生成物として暫定的に同定されている。これらの結果は、長期保存のためには冷蔵されるべきであることを示している。
【0090】
(実施例2)
式(I)の化合物の溶解度が、広範囲の有機溶媒で測定された。溶解度データが、以下の表4に示されている。
【0091】
【表4】
データは、式(I)の化合物は、通常の有機溶媒において高い溶解度を有することを明らかに示している。好ましい溶媒は、エタノールおよびトルエンである。
【0092】
化合物の遊離塩基の2つの塩基性中心が、pKaについて測定された。しかし、ピリジン環の塩基性中心は、1.99のpKaを有した。イミダゾール環の塩基性中心のpKaは、4.53と測定された。
【0093】
ベンゼンスルホン酸を、使用して式(I)の化合物のベシル酸塩を生成した。実験を、6体積の溶媒を使用する20mgスケールで実施した。全ての反応を、溶解性に応じてエタノール中の原液(1M)としてまたは固体として酸を装入して周囲温度で実施した。
【0094】
単離された固体は、塩の形成を立証する1H NMRにおける有意なピークシフトを示した。粉末X線回折(XRPD)は、塩が結晶性の徴候を有することを示した。表5は、単離された塩形態を要約している。
【0095】
【表5】
塩を、その後40℃/75%RHで2週間保存し、次いで材料の安定性を評価するために化学的純度についてXRPDおよびHPLCで再分析した。塩は、湿度条件への曝露後に同じ粉末パターンを保持し、安定性改善を支持する高い化学的純度も保持した。
【0096】
トルエンからのベシル酸塩は、安定性研究の前後で高い純度を示したことが、単離塩のT1純度結果からわかる(以下の表6)。
【0097】
【表6】
上記の結果は、ベシル酸形態は、高純度および好ましい安定性結果を示したことを示している。
【0098】
(実施例3)
実施例2のデータに基づいて、ベシル酸塩の100mgへのスケールアップを実施した。トルエンは、ベシル酸塩の単離用の好ましい溶媒であることが見出された。
【0099】
式(I)の化合物のベシル酸塩
プロセスがスケールアップするか否かを確かめるため、および単離される材料が、先のより小さなスケールの実験で見られた同じ結晶形態(形態1)であることを確かめるために、50mgの投入材料へのスケールアップを実施した。塩が形態1であったことおよび性質が期待されるものと一致していたことが分析で確認され次第、完全な特徴づけを行うためおよび40℃/75%RHでの4週間の安定性研究用のサンプルを提出するために、100mgの投入材料を用いてもう1つのスケールアップを実施した。両方のスケールアップした反応を、エタノール中の溶液(1M)としてベンゼンスルホン酸を添加して、トルエン中で実施した。
【0100】
ベシル酸塩の実験手順
式(I)の化合物の遊離塩基(100mg、バッチ704−17)を、バイアルに装入し、トルエン(600μl)を周囲温度で添加した。この溶液に、ベンゼンスルホン酸(250μl、エタノール中1M)を添加し、反応混合物を15分間撹拌し、その後、固体が溶液から沈殿し、この沈殿を濾過し、トルエンで洗浄し、真空下40℃でオーブン乾燥した。XRPDによる分析は、固体が、生成された他のベシル酸塩と同じ粉末パターンであることを示し、1H NMRは、有意なピークシフトにより塩形成を確認した。
【0101】
【表7】
LJC−039−037−1の鏡像体過剰率は、わずかに94.4であり、したがって、この結果を、同じ条件で単離された別のバッチのベシル酸塩(LJC−039−081−1)と比較した。このバッチの鏡像体過剰率は99.1%であった。
プロセス最適化
ベシル酸塩(形態1)の収量をさらに改良するために、4種の溶媒をスクリーニングした(酢酸イソプロピル、ギ酸エチル、メタノールおよびアセトン)。合計8つの100mgスケールの反応を、先の実験との比較のために関連のある酸をエタノール中の原液として添加して、これらの溶媒中で実施した。
【0102】
式(I)の化合物(バッチ704−38、100mg)を、周囲温度で溶媒(600μl)中に溶解した。酸(250μl、エタノール中の1M原液)を添加し、全ての反応混合物を、周囲温度で48時間静置した。結果は表8に要約されている。
【0103】
【表8】
メタノール中のベシル酸塩の形成の反応以外の全ての反応は、形態1を示した。メタノール反応物を4℃で保存した。得られたデータは、無水ベシル酸塩1:1を立証し、材料の粉末パターンは、新たな形態(形態2)の存在を立証した。
【0104】
酢酸イソプロピルなどの溶媒は、塩の純度を増すが、回収を減少させることが、この研究から結論付けられた。溶媒の先の選択(酢酸エチル)が、高純度の高い収量の塩をもたらしたので、最終スケールアップ実験用に酢酸エチルを使用することが決定された。
ベシル酸塩(形態1)1gスケールアップ
ベシル酸塩の1gの形成を実施した。これにより、首尾よく950mg(70%収率)の形態1が生成された。この液体は、非常に色が濃く(黄色)、したがって、少量の形態1で播種して回収を助けた。この液体を4℃で16時間保存した。得られた固体は、新しい粉末パターン(形態3)を示した。この固体を、熱分析および様々な温度でのXRPDで分析して、真正の多形であるか溶媒和物であるか否かを確かめた。1H NMR証拠からの分析の解釈によって、この固体が溶媒和物ではないことが結論付けられ、DSCが、高温顕微鏡により確認された2つの吸熱事象を示した(図3)。形態1の種は187℃で融解し、形態3は200℃で融解すると解釈された。形態1がXRPDによって同定されなかった理由は、これが顕微鏡検査に比べて感度の低い技術であることである。
【0105】
形態3は、形態1より低い温度で沈殿する。
【0106】
多形の間の関係を提案するために、多形の特徴づけを実施した。
【0107】
【表9】
LJC−039−081−2に存在する少量の形態1のより低い融点は、潜在的に低い純度が原因であり得る(LJC−039−081−1の97.9%と比較して97.2%)。
【0108】
図4は、ベシル酸塩の形態1(実線)および2(一点鎖線)のDSCを示している。
【0109】
図5は、ベシル酸塩の形態1(実線)および3(点線および一点鎖線)のDSCを示している。
【0110】
(実施例4)
塩の安定性研究
【0111】
【表10】
ベシル酸塩の結晶サンプルを、40℃/75%RHで合計4週間保存し、サンプルを7日毎にHPLC用に取り出した。ベシル酸塩hplc純度は、96.7%に達したT3まで一定であった。しかし、この値はT4まで一定であった。
【0112】
ベシル酸塩形態のhplcクロマトグラフは、0週から4週の時点の間、図6に示されている。
【0113】
λmaxが親ピークのλmaxと一致しないので、親のピークの前の主要なピークは、汚染からであると考えられる。それは、T1、T2、T3およびT4の不純物プロフィールにも存在しない。
【0114】
形態における変化がないことは、湿度研究の前後の塩の粉末パターンからわかる。
【0115】
図7は、4週間安定性研究の前後のLJC−039−037−1(ベシル酸塩)の比較のXRPDを示している。
【0116】
(実施例5)
多形研究
・多形を示すベシル酸塩の傾向を求めるために、30種の溶媒(15種のニートに加えてそれらの2.5%水性対応物)を使用して成熟実験を設定した。周囲温度から60℃の加熱/冷却サイクルで1週間、固体を様々な溶媒(表11を参照されたい)中でスラリーにした。1週間後、スラリーを蒸発し、固体をXRPDおよびHPLCで分析した。
【0117】
表11 ベシル酸塩(LJC−039−058−2)についての多形研究の結果
・初期hplc純度97.7%
【0118】
【表11】
ベシル酸塩を使用した成熟研究は、新しい形態を示さなかった。成熟後の純度の結果は、アセトニトリル、水性THF、水性IPA、水性MEK、水性ジオキサンおよび水性アセトニトリル中にスラリーにしたものは分解したことを示している。これは、ベシル酸塩(形態1)は、高温でのニート有機溶媒中で良好な溶液安定性を有することを示唆している。
【0119】
ベシル酸塩の新しい形態の研究
ベシル酸塩の新しい形態は、成熟研究からは見られなかったが、メタノール中で結晶が成長した場合に、新しい形態が見られた。メタノールから得られる単結晶を、粉末パターンを得るために磨砕した。このパターンは、形態1と異なることがわかった。形態2のさらなる供給を得るために、反復実験を実施した。溶媒を蒸発させること(これは形態1をもたらす)の反対に、液体からの16時間かけた沈殿から形態2を単離することが可能であった。興味深いことに、2つの晶癖(針および塊)が存在した。両方とも単結晶構造の測定に使用された針晶癖と同じ粉末パターンを示した。
【0120】
形態2の完全な分析を実施した。単結晶データが無水ベシル酸塩1:1を立証したので、形態2は真正の多形であることが結論付けられた。
【0121】
図8Aは、ベシル酸塩形態1および2のXRPD比較を示している。形態1(トレース1)および形態2(トレース2)の間には明らかな差異がある。2つの粉末パターンからわかるように、両方の形態は非常に異なる。2つの形態の融点を比較するために熱解析を実施し、熱力学的溶解度の測定値も記録した。
【0122】
図8Bは、形態1および2の重ね合わせを示している。形態1および2は、1つの吸熱事象(融解)を示している。
【0123】
形態3が、2回目の採集がLJC−039−081−1(1gスケールアップ反応)の液体から単離されたときに同定された。それが溶媒和物であるか否かおよびどのように形態が相互変換するのかを決定するために分析を実施した。
【0124】
図9Aは、ベシル酸塩形態1および3のXRPD比較を示している。図9Bは、形態1、および3の重ね合わせを示している。
【0125】
形態1は、1つの吸熱事象(融解)を示しているが、形態3は、2つの事象を示している。形態3の高温顕微鏡は、互いの20℃以内の2つの融解を明らかに示している。より低い感度の技術である様々な温度のXRPDにおいて検出されないために、少量のより低融点の多形が存在することが仮定される。形態3が単離される液体を播種するのに形態1が使用されたので、最初の吸熱事象は形態1を表すことはあり得る。
【0126】
溶解度データは、全ての3つの形態は、pH3において7.8から8.3mg/mlの非常に似た水溶解度を有することを示している。
【0127】
ベシル酸塩形態4
ベシル酸塩形態1(LJC−039−083−1)の公表バッチ(release batch)は、高純度(97.6%)であったが、遊離塩基から運ばれた少量の不純物を含有した(0.78%、11.9分RT)。この不純物は、吸熱変換(130℃で開始)を示すことがDSC実験で観測された。このピークは、親ピークのものと無関係のλmaxを有することが確認された。
【0128】
100mgサンプルを、40%酢酸イソプロピル/エタノールからの再結晶化の試みのために取った。再結晶化を、最小量の熱した溶媒中に塩を溶解し、次いで周囲温度にゆっくりと冷却して沈殿を得ることによって慣習的に実施した。乾燥した固体を、XRPDで分析し、新しい形態が示され、熱分析および1H NMRによって、多形であって溶媒和物ではないことが確認された。図10は、LJC−039−086−1のDSCを示している。
【0129】
塩の選別研究は、式(I)の化合物は、適切なpKa範囲内で多くの塩を形成すること、および、それらは、一連の溶媒から容易に単離されることを示した。塩の完全な特性決定から、ベシル酸塩は、湿度に関して良好な安定性を有することが決定された。ベシル酸塩の2つの多形があることが結論付けられた。形態3は、形態1で播種後のLJC−039−081−1の液体の2回目の採集から生じた。形態4は、40%酢酸イソプロピル/エタノールから形態1の再結晶化が実施された後に観測された。
【0130】
完全な分析データが以下の図11−14に示されている。
【0131】
実施例2〜5のための実験方法
(実施例2)
式(I)の化合物(5mg/ウェル)を、HPLCバイアル中で溶媒1(30μl)に溶解した。この溶液に、ベンゼンスルホン酸(11.4μl、エタノール中の1M)を添加し、この反応混合物を常温で終夜放置した。固体を含有したバイアルは、真空下40℃で乾燥し、溶液のままであったものは、蒸発により濃縮し、次いでヘプタンで処理した。沈殿したものは、前述のように乾燥し、溶けたものは、4℃で保存した。
【0132】
ベシル酸塩形態1のスケールアップ
式(I)の化合物(100mg)を酢酸エチル(600μl)に溶解し、ベンゼンスルホン酸(250μl、エタノール中の1M)を添加した。沈殿が即座に生じ、この反応混合物を常温で24時間撹拌した。固体を濾過し、酢酸エチルで洗浄し、真空下で40℃において16時間オーブン乾燥した。
【0133】
分析方法
示差走査熱量測定(DSC)
DSCデータを、50位置のオートサンプラーを備えたTA Instrument Q1000で集めた。エネルギーおよび温度較正基準はインジウムであった。サンプルを、25から350℃の間、10℃/分の速度で加熱した。30ml/分の窒素パージを、サンプル上で維持した。
【0134】
特に指定のない限り、0.5から3mgの間のサンプルを使用し、全てのサンプルはピンホールアルミ板状(pin holed aluminium pan)中で試験された。
【0135】
熱重量分析(TGA)
TGAデータは、Alumelで較正され、10℃/分の走査速度で測定するTA Instrument Q500 TGAで集めた。60ml/分の窒素パージを、サンプル上で維持した。
【0136】
1エタノール、トルエンおよびアセトニトリル。
【0137】
特に指定のない限り、一般的に5〜10mgのサンプルが、予め風袋計量した(pre−tared)白金るつぼに装入された。
【0138】
NMR
全てのスペクトルは、オートサンプラーを備えたBruker 400MHzで集められた。特に指定のない限り、サンプルは、d6−DMSO中で調製された。
【0139】
XRPD(粉末X線回折)
Bruker AXS C2 GADDS回折計
サンプルの粉末X線回折パターンは、Cu Kα放射(40kV、40mA)、自動化XYZステージ、自動サンプル位置決めのためのレーザービデオ顕微鏡およびHiStar2次元面積検出器を使用するBruker AXS C2 GADDS回折計で取得した。X線光学機器は、0.3mmのピンホールコリメータと一体となった単一Gobel多層鏡から成る。
【0140】
ビーム広がり、すなわち、サンプル上のX線ビームの有効なサイズは、約4mmであった。3.2〜29.8°の有効2θ範囲をもたらす20cmのサンプルと検出器の距離を有するθ−θ連続走査モードが使用された。サンプルの一般的な曝露時間は120秒であろう。
【0141】
周囲条件下で測定されるサンプルを、磨砕せずに得られた粉末を用いてフラットプレート試料として調製した。約1〜2mgのサンプルを、ガラススライド上で軽く圧縮して平面を得た。非周囲条件下で測定されるサンプルを、熱伝導化合物とともにシリコンウエハー上に載せた。サンプルを、次いで、約20℃/分で適切な温度に加熱し、続いて約1分間等温的に保持した後、データ収集を開始した。
【0142】
純度分析:
化学的方法
純度分析をHP1100 Agilentで実施した。
方法:グラジエント、逆相
方法持続時間/分:34
カラム:Phenomenex Gemini C18 5μm(2.0×50mm)(保護カートリッジ Phenomenex Gemini C18 保護カートリッジ2×4mm)
カラム温度/℃:40
注入/μl:5
流速ml/分:0.8
検出:UV
波長/nm:255(90nmのバンド幅)、240(80nmのバンド幅)、254(8nmのバンド幅)
相A:2mmol NH4HCO3(NH3溶液でpH10に調節された)
相B:アセトニトリル
タイムテーブル:
【0143】
【表11−1】
キラル法
純度分析を、Gilson HPLCシステムで行った。
方法:アイソクラチック、順相
方法持続時間/分:50
カラム:Diacel Chrialcel OJ−H(5μm)4.6×250mm(保護カートリッジDiacel Chrialcel OJ−H分析保護カートリッジ5μm 4.0×10mm)
カラム温度/℃:40
注入/μl:10
流速ml/分:1.0
検出:UV
波長/nm:225(単一波長検出器)
相A:ヘキサン
相B:エタノール
タイムテーブル:
【0144】
【表11−2】
重量蒸気収着(gravimetric vapour sorption)(GVS)研究
全てのサンプルを、CFRSorpソフトウエアを使用するHiden IGASorp水分収着分析機で測定した。サンプルサイズは一般的に10mgであった。水分吸着脱着等温線を、以下に記載のとおり実施した(2つの走査で1つの完全なサイクルが得られる)。全てのサンプルを、常湿常温(40%RH、25℃)で装入/取り出しした。全てのサンプルを、GVS分析後XRPDによって分析した。特に指定のない限り、標準の等温線を、0〜90%RH範囲にわたる10%RH間隔で25℃において実施した。
【0145】
【表11−3】
溶解度
溶解度を、0.25mlの溶媒(水)中に十分な化合物を懸濁させて、この化合物の親遊離形態の10mg/mlの最大最終濃度を得ることによって測定した。懸濁液を、25℃で24時間平衡化し、続いてpH確認し、ガラス繊維C96ウェルプレートを通して濾過した。次いで、この濾液を101×に希釈した。定量は、約0.1mg/mlでDMSO中に溶解した標準を参照するHPLCによった。様々な量の標準、希釈および非希釈試薬を注入した。標準の注入における最大ピークと同じ保持時間において見出されたピーク面積の積分によって、溶解度を算出した。フィルタープレート中に十分な固体がある場合、相変化、水和物形成、アモルファス化、結晶化等について、XRPDを標準的に確認する。
【0146】
【表11−4】
pKa測定
pKa測定を、D−PASアタッチメントを有するSirius GlpKa装置で行った。25℃におけるMeOH:H2O混合物中の電位差滴定法によって、測定を行った。滴定媒体は、0.15M KClでイオン強度調節された。MeOH:H2O混合物中で見出された値を、Yasuda−Shedlovsky外挿法により0%共溶媒に外挿した。
【0147】
高温顕微鏡検査
高温顕微鏡検査を、10〜20℃/分の範囲の一般的な加熱速度での25〜350℃の温度範囲のMettler−Toledo MTFP82HTホットステージを組み合わせたLeica LM/DM偏光顕微鏡を使用して実施した。少量のサンプルを、個々の粒子ができるだけ隔たるようにガラススライド上に分散した。サンプルを、×20対物レンズを用いて正または交差偏光下(λ着色フィルターと組み合わせた)で見た。
【0148】
キラル純度法
システム設定
ポンプ:Gilson 322バイナリーポンプ
検出器:Gilson 152UV/Vis
オートサンプラー:Gilson 233XLラック+Gilson 402デュアルシリンジポンプ
カラムオーブン:Phenomenex Thermasphere TS−130
ソフトウエア:Gilson Unipoint LCソフトウエア
カラム:Daicel Chiralcel OJ−H、5μm、4.6×250mm
保護カラム:Daicel Chiralcel OJ−H分析保護カートリッジ、5μm、4.6×10mm。
【0149】
HPLC条件
経路A:ヘキサン(93%)
経路B:エタノール(7%)
流速:1.0ml/分
検出器波長:225nm
カラム温度:40℃
ランタイム:50.0分。
【0150】
サンプル条件
約0.2mgのサンプルを、適切な量のヘキサン:エタノールの1:1v/vに溶解して0.2mg/ml溶液を得た。これにフタをして約15秒間、高速度のボルテックスミキサーにかけた。この時点で固体が残った場合、サンプルバイアルを、約10秒間、超音波処理し、続いてさらに10から15秒間ボルテックスミキサーにかけた。HPLCシステムに、10μlを注入した。ブランクとしてのヘキサン:エタノールの1:1v/vの最初の二重の注入の後に、サンプルを二重に注入した。
【0151】
(実施例5)
薬理試験例
本発明のベシル酸塩形態1の麻酔および鎮静効果を評価した。ベシル酸(ベンゼンスルホン酸)塩を、動物への試験組成物の投与のために生理的食塩水に溶解した。試験組成物を、マウスに投与し、個々のPlexiglasケージ(20×10×10cm)中に置いた。マウスに静脈経路でビヒクルまたは試験物質のいずれかを注入した。睡眠への待ち時間および麻酔状態の期間(最大:試験物質投与後90分)を記録した。麻酔状態は、立直り反射喪失(LRR)によって示される。立直り反射試験は、動物が落ち着いたと見え次第、約20〜30秒毎に実施した。立直り反射がなくなり次第、その後20〜30秒毎に立直り反射の復帰について試験することによって立直り反射喪失の期間を測定した。1グループ当たり8匹のマウスを研究し、試験をブラインドで実施した。この研究の結果は、下表に記載されている。
【0152】
【表11−5】
Mann−Whitney Uテスト:NS=有意ではない;*=p<0.05;**=p<0.01
Fisherの厳密テスト(LRRのあったマウスの数):兆候なし=有意ではない;+=p<0.05;++=p<0.01
(#):n<3の場合、計算されず
(##):最大=注入後90分。
【0153】
上記の表の結果は、ベシル酸塩形態1は、立直り反射喪失への短い待ち時間を有し、したがって動物における麻酔状態への短い誘導時間を有することを示している。さらに、マウスは、短い立直り反射喪失の期間によって示されるようにすばやく麻酔状態から回復する。したがって、この化合物は、迅速な麻酔状態導入および麻酔状態からの回復を提供し得る。
【0154】
(実施例6)
形態2、3、および4の結晶化のためのさらなる条件
先に報告された形態2、3、および4の結晶化を再現する試みにおいて、さらなる条件が試験された。しかし、以下に記載のように、報告されたスケールを実質的に減らし、方法をそれにより改変した。
【0155】
形態2
5mgの固体を25ulのメタノールに溶解し、10ulのエタノールを添加し、次いで、この溶液を4℃に3日間冷蔵した。
【0156】
形態3
3つの変形を試みた。
1.5mgの固体を50ulのエタノールに溶解し、120ulの酢酸エチルを添加し、次いで、この溶液を4℃に3日間冷蔵した。
2.10.1mgの固体を300ulのエタノールに溶解し、120ulの酢酸エチルを添加し、次いで、この溶液を4℃に3日間冷蔵した。
3.2.5mgの固体を、シラン処理したバイアル中で50ulのエタノールに溶解し、100ulの酢酸エチルを添加し、次いで、この溶液を4℃に3日間冷蔵した。
【0157】
形態4
3つの変形を試みた。
1.酢酸イソプロピル:エタノール(40%:60%v/v)の温めた(70℃)混合物を、固体が溶解するまで20ulアリコートで5mgの温めた固体に添加し(合計で60ulの溶媒混合物)、次いで、この溶液を、数時間かけて、最初70℃のサーモスタットで温度調節した水浴中で、ゆっくりと常温に冷却した。
2.5mgの固体を、180ulの温めた(50℃)酢酸イソプロピル:エタノール(40%:60%v/v)の溶媒に溶解し、この溶液を、数時間かけて、サーモスタットで温度調節した水浴(最初50℃)中で、ゆっくりと常温に冷却した。
3.5mgポーションの固体を、シラン処理したバイアル中で100ulの温めた(50℃)酢酸イソプロピル:エタノール(40%:60%v/v)の溶媒に溶解し、この溶液を、数時間かけて、サーモスタットで温度調節した水浴(最初50℃)中で、ゆっくりと常温に冷却した。
【0158】
それぞれの結晶化は、羽根状および板状のような晶癖を有する固体材料を生じ、形態4の結晶化も針のような材料を生じた。
【0159】
(実施例7)
式(I)の化合物のベシル酸塩の特性決定
式(I)の化合物のベシル酸塩は、キラルであり、以下の単一の鏡像体、すなわち、S鏡像体(後に求められた結晶構図と一致する)であると考えられる。
【0160】
【化2】
上記複素環構造は、イミダゾール環中に塩基性窒素(約5のpKa)、およびピリジル環中に弱塩基性窒素(約2のpKa)を含有する。イミダゾール−窒素は、一般的に、水溶液中で強酸性ベシル酸塩(約−0.6のpKa)の存在下でプロトン化され、ピリジニル−窒素も、潜在的に、過剰なベシル酸塩の条件下でプロトン化される。
【0161】
化合物の中性遊離塩基形態(すなわち、プロトン化されていない)は、いくらか親油性(logPオクタノール:水約4.0)であると予想され、したがって、水性環境よりいくらか親油性環境を好むであろう。さらに、それは、モノプロトン化(pH3においてlogDオクタノール:水約2)された場合でさえ、ある程度の親油性を保持しそうであるが、ベシル酸塩対イオンの影響は、その固有の親水性によって、この傾向を改善しそうである。親油性の程度は、二プロトン化形態(pH0においてlogDオクタノール:水約0.6)ではさらに減少する。
【0162】
化合物は、過剰な水素結合受容体も有し、したがって、水素結合供与溶媒と適切にパートナーになる。したがって、化合物は、特に、部分的に親油性の、水素結合供与環境を提供するアルコールなどの一連の極性有機溶媒における可溶化を好むことが予想される。これは、実験的証拠によって支持された(使用される溶媒の詳細は、実施例8で示される)。
【0163】
【表11−6】
可溶性(>5mg/ml)、部分的に可溶性(2.5〜5mg/ml)、部分的に不溶性(0.5〜2.5mg/ml)、不溶性(<0.5mg/ml)
引用された値は近似であるが、実験的に確認されている。
【0164】
これらの結果は、多様な極性有機溶媒における化合物の良好な溶解性を強調している。特に、2,2,2−トリフルオロエタノールおよびヘキサフルオロプロパン−2−オールは、両方とも、この化合物の極めて良好な溶媒であることが確認されている。このことは、両方の溶媒が強い水素結合供与体であるという上記に論じた考察と一致する。同様に、より実質的に親油性の溶媒は、不十分な溶媒であり、したがって結晶化の潜在的な貧溶媒であることが確認されている。
【0165】
(実施例8)
式(I)の化合物のベシル酸塩の結晶化
式(I)の化合物のベシル酸塩の形態1および2の結晶材料を得ることの助けとなる様々な条件が記載される。成分としてアルコールまたはアセトニトリル溶媒を、それらのそれぞれに相溶性の貧溶媒または共溶媒とともに含む結晶化条件は、有用な結晶材料を得るのに最も有望な条件を提供すると考えられる。溶媒/貧溶媒二成分混合物を使用する結晶化を主に使用した。結晶化を、常温および低温(4℃)において、溶媒/貧溶媒混合物中の化合物の半飽和溶液からの遅延蒸発によって実施した。結晶化は、一般的に、調製の3〜5日以内に観測された。
【0166】
サンプル量が許される場合、全ての結晶化条件を、ガラス96ウェルプレートフォーマット中で二重に実施し、それぞれのウェルプレートの半分は、ウェルプレートのもう半分における条件を複製するのに使用した。ウェル間の交差汚染を設計によって最小化する。試験した条件の全てが、少なくとも二重に再現可能な挙動を示し、大部分が、さらなる分析に適する固体材料を生じた。
【0167】
全ての場合で、サンプルおよび結晶化媒体と接触する器具は、様々な溶媒および試薬で綿密に洗浄した後、エタノール中に浸し、豊富な蒸発窒素を用いて通気乾燥した。
【0168】
表12に記載されたとおりの、市販供給者からの高品質の溶媒が使用された。
【0169】
【表12】
得られた結晶形態の目視分析を、デジタルカメラを装着し、必要に応じて透過および反射照明の両方を用いる双眼顕微鏡(約10×〜40×倍率)を使用して達成した。
【0170】
固体材料の目視による特性決定を、以下の表14に要約している。独特の結晶または球晶のいずれかとしての、羽根状または平面状/板状の形態の優勢を観察した。全体的に、溶媒としてエタノールを用いた結晶化(この結晶化では、球晶およびインターフェース型成長への傾向が温度低下に伴って減少した)を例外として、周囲温度で実施された結晶化と4℃での結晶化との間で形態的な差異は少ししかなかった。貧溶媒の使用は、実質的に結晶材料の質を改善し得ることは注目される。
【0171】
観測された結晶材料の画像の例が図17に示されている。この図に示されたように、アセトニトリルは、一般的に不十分な核生成、したがって不十分な質の結晶表面からの成長の結果として見られる球晶成長を生じる傾向を有する。対照的に、2−メトキシエタノールは、羽根状/針のような形態の独特の結晶を生じる傾向を有する。
【0172】
形態1が条件の多くから結晶化する一般的な傾向があるようである。しかし、形態2も、形態3および4を得るためのスケールダウンした類似物(実施例6に記載された)を含めた、いくつかの結晶化条件から観測されたことは注目される。形態2は、極性(アセトニトリル:水)または親油性(n−ノナン)または両方(ジメチルスルホキシド:1,2−ジクロロベンゼン)のいずれかの極端な状態が存在する条件において観測される。一般的に、形態2の結晶は、その卓越した品質および特徴的な十分に形成された板状/平面状の晶癖において顕著であった。
【0173】
単結晶X線回折格子の測定
生成された結晶形態の補強証拠を提供するために、適切な品質のいくつかの結晶の格子パラメーターを、単結晶X線回折を使用して求めた。結晶単位格子パラメーターを、Mo放射を備え、結晶が油を含むガラス繊維上に載せられ、260Kに保たれるKappa CCD回折計を使用して求めた。表13に要約されたとおりの形態1および形態2のパラメーターを求めた。
【0174】
【表13】
単結晶X線回折単位格子の結果を含む、式(I)の化合物のベシル酸塩についての溶媒/共溶媒および溶媒/貧溶媒条件からの結晶化の結果を表14に記載する。
【0175】
【表14−1】
【0176】
【表14−2】
完全な単結晶X線回折結晶構造測定に適する品質の様々な結晶を達成し、形態1および2のための完全な構造を得た。これらの結晶構造が実施例9および10で報告される。
【0177】
(実施例9)
形態1の結晶構造
針晶癖を有する、2−メトキシエタノール:酢酸ペンチル溶液から成長させた式(I)の化合物のベシル酸塩の結晶の画像を図17に示した。
【0178】
単一の針晶癖結晶(サイズが約0.8×0.04×0.02mm)を選択し、その格子パラメーターを260Kで、次いで190Kで測定した。260〜190Kの温度に低下させることで変換は観測されなかった。本発明で分析された構造は190Kにおけるデータについてであり、結晶のパラメーターおよびX線回折の精密化を表15に示す。
【0179】
【表15−1】
【0180】
【表15−2】
非対称単位の内容が図18に示されている。それは、2種の独立した化合物の分子および2種の独立したベシル酸塩対イオンから成る。それぞれの化合物は、プロトン化されたイミダゾール−窒素を有する。
【0181】
フラック「エナンチオポール」パラメーター(Flack“Enantiopole”parameter)は、0.03(1)と測定され、したがって、本明細書に記載された構造の立体化学は、十分に確立されており、上記化合物のために意図された立体化学と一致する。
【0182】
【化3】
結晶学的座標および他の関連データを、表17にSHELXファイルの形態で記載している。
【0183】
配座障害は、図19に示された原子位置の「熱振動楕円体(thermal ellipsoid)」によって表し得る(最初の近似において)。障害の主要な領域は、メチル基およびベシル酸塩にあることがわかる。
【0184】
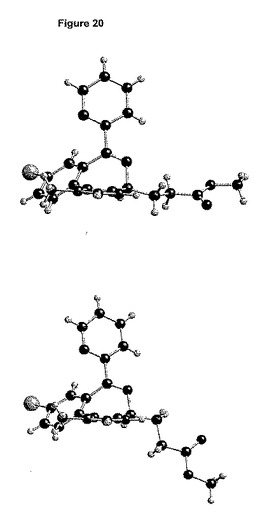
2つの独立した分子の間の差異は、主として図20に見られるエステル鎖に由来する。1つの分子は、イミダゾール環と同一平面上にあるエステル鎖を有するが、もう1つの分子は、直交するエステル鎖を有する。
【0185】
これらのエステル鎖の立体配座は、形態2の立体配座と異なる(図21)。形態1で観測される直交する立体配座は、形態2に見出される立体配座と最大の類似性を有する。
【0186】
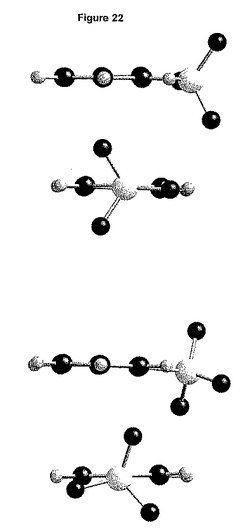
2種の独立したベシル酸塩は、ねじれ形立体配座を有する(図22)。結合の長さの実質的な差異は明らかではない。
【0187】
あるベシル酸塩は、形態2のベシル酸塩について観測される立体配座をとる(図23)。
【0188】
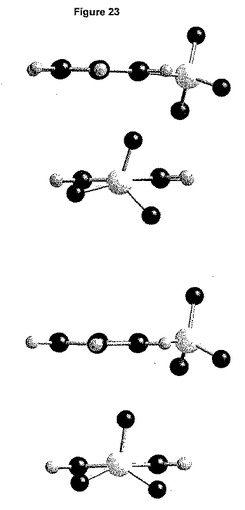
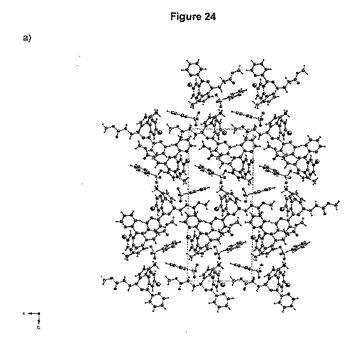
結晶軸a、bおよびcに沿って見た分割された結晶構造を、それぞれ図24a、bおよびcに示す。図25は、結晶充てんにおいて観測される最短のコンタクト(contact)を要約している。
【0189】
それぞれの化合物は、2つの独立したベシル酸塩と相互に作用する。特に、短い距離(水素結合タイプ)は、1種のベシル酸塩の1個の酸素原子および化合物のイミダゾール環のプロトン化窒素の間で確立される。第二の独立した化合物は、同様に、但し第二の独立したベシル酸塩と相互作用する。
【0190】
他の近接したコンタクト(C−O、H−O)は、主としてイミダゾールおよびピリジル環の近くで化合物およびベシル酸塩の間で観測される。いくつかの近接したコンタクトは、2つの化合物同士の間(Br−N、C−C、O−H)および2つのベシル酸塩同士の間(O−Hコンタクト)でも観測されるが、後者の程度は低い。
【0191】
実験で求めた結晶構造を使用して、形態1についての粉末回折パターンを、CrystalDiffract(登録商標)(CrystalDiffractはCrystalMaker Ltdの登録商標である)を使用して算出し、図26に示している。この粉末パターンは、形態1について報告された実験的粉末パターンに一致する。
【0192】
(実施例10)
形態2の結晶構造
板状状晶癖を有する式(I)の化合物のベシル酸塩形態2の画像を図27に示している。
【0193】
単一板状状晶癖結晶(サイズ約0.7×0.30×0.25mm)を選択し、その格子パラメーターを260Kで、次いで190Kで求めた。260〜190Kの間の温度への低下で変換は観測されなかった。本発明で分析された構造は、190Kにおけるデータについてであり、結晶のパラメーターおよびX線回折の精密化は表16に示されている。
【0194】
【表16−1】
【0195】
【表16−2】
非対称単位の内容が図28に示されている。これは、1種の独立した化合物の分子および1種の独立したベシル酸塩から成る。化合物は、プロトン化されたイミダゾール−窒素を有する。
【0196】
フラック「エナンチオポール」パラメーターは、0.011(9)と測定され、したがって、本明細書に記載された構造の立体化学は、十分に確立されており、上記化合物のために意図された立体化学と一致する。結晶学的座標および他の関連データを、表18にSHELXファイルの形態で記載している。
【0197】
配座障害は、図29に示された原子位置の「熱振動楕円体」によって表し得る(最初の近似において)。障害の主要な領域は、ベシル酸塩にあることがわかる。
【0198】
上記に記載のとおり、図30に示された形態2のエステル鎖の立体配座は、形態1でとられる立体配座と異なる。
【0199】
しかし、ベシル酸塩の立体配座は、形態1のベシル酸塩の1つについて観測された立体配座と類似している(図31)。
【0200】
結晶軸a、bおよびcに沿って見た分割された結晶構造を、それぞれ図32a、bおよびcに示し、図33は、結晶充てんにおいて観測される最短のコンタクトを要約している。化合物は、イミダゾール環のそのプロトン化された窒素を介してベシル酸塩の1個の酸素原子とショートコンタクト(short contact)(水素結合タイプ)を確立する。他のショートコンタクト(C−C、C−O、H−O)は、イミダゾール環を介して化合物およびベシル酸塩の間で観測される。
【0201】
いくつかの近接したコンタクトは、2つの化合物同士の間(Br−C、C−C、O−C、O−H)でも観測され、この多くは、エステル鎖を介する。ベシル酸塩同士の間には近接したコンタクトはない。
【0202】
実験で求めた結晶構造を使用して、形態2についての粉末回折パターンを、CrystalDiffract(登録商標)を使用して算出した(図34)。この粉末パターンは、形態2について報告された実験的粉末パターンに一致する。
【0203】
表17.式(I)の化合物のベシル酸塩形態1についてのSHELXファイルの形態にまとめた結晶学的座標および他の関連データ。
【0204】
【表17−1】
【0205】
【表17−2】
【0206】
【表17−3】
【0207】
【表17−4】
【0208】
【表17−5】
【0209】
【表17−6】
【0210】
【表17−7】
表18.式(I)の化合物のベシル酸塩形態2についてのSHELXファイルの形態にまとめた結晶学的座標および他の関連データ。
【0211】
【表18−1】
【0212】
【表18−2】
【0213】
【表18−3】
【0214】
【表18−4】
表19.式(I)の化合物のベシル酸塩形態1の結合の長さ。
【0215】
【表19−1】
【0216】
【表19−2】
表20.式(I)の化合物のベシル酸塩形態1の角度。
【0217】
【表20−1】
【0218】
【表20−2】
【0219】
【表20−3】
表21.式(I)の化合物のベシル酸塩形態2の結合の長さ。
【0220】
【表21】
表22.式(I)の化合物のベシル酸塩形態2の角度。
【0221】
【表22−1】
【0222】
【表22−2】
【技術分野】
【0001】
本発明は、短時間作用型ベンゾジアゼピンの塩、および特に鎮静または催眠、不安緩解、筋弛緩、または抗痙攣の目的のための薬剤としての該塩の使用に関する。
【背景技術】
【0002】
特許文献1は、カルボン酸エステル部分を含み、非特異的組織エステラーゼによって不活性化される短時間作用型ベンゾジアゼピンを記載している。器官に依存しない排除機構が、これらのベンゾジアゼピンの特徴であることが予想され、より予想可能かつ再現性のある薬力学的プロフィールを提供する。これらの化合物は、鎮静−催眠、不安緩解、筋弛緩および抗痙攣の目的を含めた治療用途に適する。これらの化合物は、次の臨床状態(clinical setting):術前の鎮静、不安緩解、および手術前後の記憶消失的使用;短期診断、手術または内視鏡的処置中の意識下鎮静;他の麻酔薬または鎮痛薬の投与の前および/または同時の全身麻酔の誘導および維持のための成分として;ICU鎮静において、静脈内投与されるのに有用な短時間作用型CNS抑制剤である。
【0003】
特許文献1(実施例Ic−8、36頁)に開示された化合物の1つは、以下の式(I)に示されたメチル3−[(4S)−8−ブロモ−1−メチル−6−(2−ピリジニル)−4H−イミダゾール[1,2−a][1,4]ベンゾジアゼピン−4−イル]プロパノエートである。
【0004】
【化1】
【先行技術文献】
【特許文献】
【0005】
【特許文献1】欧州特許第1,183,243号明細書
【発明の概要】
【発明が解決しようとする課題】
【0006】
式(I)の遊離塩基は、5℃で保存される場合に安定であるが、40℃/相対湿度75%(開放)で保存されたサンプルは、潮解し、色が黄色から橙色になり、初期に比べて含有量の著しい減少を示すことが観測される(以下の実施例1を参照されたい)。
【課題を解決するための手段】
【0007】
驚くべきことに、式(I)の化合物は、高結晶質のモノ(ベンゼンスルホン酸)ベシル酸塩を形成し、これは一連の薬学的に許容される溶媒から容易に単離され、良好な熱安定性、低い吸湿性および高い水溶性を示すことが見出された。
【0008】
本発明によれば、式(I)の化合物のベシル酸塩が提供される。好ましくは、この塩は、結晶塩である。好ましくは、この結晶塩は、式(I)の化合物:ベシル酸塩の1:1の化学量論比を有する。ベシル酸の多形相の調製および特徴づけは、以下の実施例に記載されている。
【0009】
本発明によれば、式(I)の化合物のベシル酸塩の結晶多形(本明細書でベシル酸塩形態1と呼ぶ)が提供され、これは、約7.3、7.8、9.4、12.1、14.1、14.4、14.7,または15.6度2θにおける特性ピークを含む粉末X線回折(XRPD)パターンを示す。
【0010】
好ましくは、ベシル酸塩形態1の結晶多形は、約7.3、7.8、9.4、12.1、14.1、14.4、14.7、および15.6度2θにおける特性ピークを含むXRPDパターンを示す。
【0011】
より好ましくは、ベシル酸塩形態1の結晶多形は、7.25(10.60)、7.84(72.60)、9.36(12.10)、12.13(32.50)、14.06(48.50)、14.41(74.30)、14.70(50.70)、15.60(26.90)[回折角2θ度(%相対強度)]における特性ピークを含むXRPDパターンを示す。
【0012】
好ましくは、ベシル酸塩形態1の結晶多形は、187〜204℃、好ましくは、約191〜192℃の範囲の示差走査熱量測定(DSC)での開始融解温度を有する。
【0013】
形態1の結晶構造は、190K(6.3のRファクター)で分解された。形態1は、化合物:ベシル酸塩の1:1の化学量論比を有する。その結晶非対称単位は、2つの独立した化合物分子および2つのベシル酸塩分子を含有する。2つの独立した化合物分子は、イミダゾール環上で1価プロトン化されている。この結晶構造は、a=7.6868Å、b=29.2607Å、c=12.3756Å、α=90°、β=97.7880°、γ=90°の単位格子寸法およびP21の空間群を有する。この結晶構造は、実施例9により詳細に記載されており、結晶学的座標は、表17に提供されている。形態1の結合の長さおよび角度は、それぞれ表19および20に提供されている。
【0014】
a=7.6868Å、b=29.2607Å、c=12.3756Å、α=90°、β=97.7880°、γ=90°の単位格子寸法を有する結晶を含む結晶多形である式(I)の化合物のベシル酸塩が、本発明によって提供される。
【0015】
表17に示される構造座標によって定義される結晶構造を有する結晶多形である式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0016】
それぞれ表19および20に示される結合の長さおよび角度を有する式(I)の化合物のベシル酸塩が、本発明によってさらに提供される。
【0017】
約8.6、10.5、12.0、13.1、14.4、または15.9度2θにおける特性ピークを含むXRPDパターンを示す式(I)の化合物のベシル酸塩の結晶多形(以下、ベシル酸塩形態2と呼ぶ)が、本発明によってさらに提供される。
【0018】
好ましくは、ベシル酸塩形態2の結晶多形は、約8.6、10.5、12.0、13.1、14.4、および15.9度2θにおける特性ピークを含むXRPDパターンを示す。
【0019】
より好ましくは、8.64(17.60)、10.46(21.00)、12.03(22.80)、13.14(27.70)、14.42(11.20)、15.91(100.00)[回折角2θ度(%相対強度)]における特性ピークを含むXRPDパターンを示すベシル酸塩形態2の結晶多形を示す。
【0020】
好ましくは、ベシル酸塩形態2の結晶多形は、170〜200℃の範囲、好ましくは、約180℃の示差走査熱量測定(DSC)での開始融解温度を有する。
【0021】
形態2の結晶構造は、190K(3.8のRファクター)で分解された。形態2は、化合物:ベシル酸塩の1:1の化学量論比を有する。その結晶学的非対称単位は、1つの化合物分子および1つのベシル酸塩分子を含有する。この化合物分子は、イミダゾール環上で1価プロトン化されている。この結晶構造は、a=8.92130Å、b=11.1536Å、c=25.8345Å、α=90°、β=90°、γ=90°の単位格子寸法、およびP212121の空間群を有する。この結晶構造は、実施例10により詳細に記載されており、結晶学的座標は、表18に提供されている。形態2の結合の長さおよび角度は、それぞれ表21および22に提供されている。
【0022】
本発明によれば、a=8.92130Å、b=11.1536Å、c=25.8345Å、α=90°、β=90°、γ=90°の単位格子寸法を有する結晶を含む結晶多形である式(I)の化合物のベシル酸塩が提供される。
【0023】
表18に示される構造座標によって定義される結晶構造を有する結晶多形である式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0024】
それぞれ表21および22に示される結合の長さおよび角度を有する式(I)の化合物のベシル酸塩が、本発明によってさらに提供される。
【0025】
約7.6、11.2、12.4、14.6、15.2、16.4、または17.7度2θにおける特性ピークを含む粉末X線回折(XRPD)パターンを示す式(I)の化合物のベシル酸塩の結晶多形(以下、ベシル酸塩形態3と呼ぶ)が、本発明によってさらに提供される。
【0026】
好ましくは、ベシル酸塩形態3の結晶多形は、約7.6、11.2、12.4、14.6、15.2、16.4、および17.7度2θにおける特性ピークを含むXRPDパターンを示す。
【0027】
より好ましくは、ベシル酸塩形態3の結晶多形は、7.61(65.70)、11.19(33.20)、12.38(48.70)、14.63(30.60)、15.18(33.20)、16.40(29.60)、17.68(51.30)[回折角2θ°(%相対強度)]における特性ピークを含むXRPDパターンを示す。
【0028】
好ましくは、ベシル酸塩形態3の結晶多形は、195−205℃、好ましくは、約200−201℃の範囲の示差走査熱量測定(DSC)での開始融解温度を有する。
【0029】
約7.6、10.8、15.2、15.9、または22.0度2θにおける特性ピークを含むXRPDパターンを示す式(I)の化合物のベシル酸塩の結晶多形(以下、ベシル酸塩形態4と呼ぶ)が、本発明によってさらに提供される。
【0030】
好ましくは、ベシル酸塩形態4の結晶多形は、約7.6、10.8、15.2、15.9、および22.0度2θにおける特性ピークを含むXRPDパターンを示す。
【0031】
好ましくは、ベシル酸塩形態4の結晶多形は、7.62(83.50)、10.75(14.70)、15.17(37.80)、15.85(28.70)、22.03(100)[回折角2θ°(%相対強度)]における特性ピークを含むXRPDパターンを示す。
【0032】
好ましくは、ベシル酸塩形態4の結晶多形は、180−185℃、好ましくは、約182℃の範囲の示差走査熱量測定(DSC)での開始融解温度を有する。
【0033】
好ましい塩は、形成、収量、純度および化学的および固体安定性の頑健性に基づいて、ベシル酸塩形態1である。
【0034】
式(I)の化合物の遊離塩基をベンゼンスルホン酸と反応させることを含む式(I)の化合物のベシル酸塩の製造方法も、本発明によって提供される。
【0035】
同様に、本発明によれば、溶液中で式(I)の化合物の遊離塩基をベンゼンスルホン酸と接触させベシル酸塩の沈殿の形成を生じさせることを含む本発明の塩の製造の方法が提供される。好ましくは、この方法は、沈殿を単離することをさらに含む。
【0036】
好ましくは、上記遊離塩基は、トルエン、エタノール、酢酸エチル、MtBE、ジクロロメタン(DCM)、酢酸イソプロピル、ギ酸エチル、メタノール、またはアセトンに溶解される。より好ましくは、上記遊離塩基は、トルエンまたは酢酸エチルに溶解される。好ましくは、ベンゼンスルホン酸は、エタノールに溶解される。
【0037】
ベシル酸塩形態1は、式(I)の化合物の遊離塩基のトルエン、酢酸エチル、アセトン、酢酸イソプロピル、またはギ酸エチル中の溶液とベンゼンスルホン酸のエタノール中の溶液とを接触させて塩の沈殿の形成を生じさせることによって調製され得る。
【0038】
上記方法によって得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0039】
ベシル酸塩形態2は、メタノール中の式(I)の化合物の遊離塩基の溶液とエタノール中のベンゼンスルホン酸の溶液とを接触させて塩の沈殿の形成を生じさせることによって調製され得る。好ましくは、この混合物は周囲温度未満に冷却される(例えば4℃)。
【0040】
上記方法で得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0041】
ベシル酸塩形態3は、形態1を用いて、酢酸エチル/エタノールからの形態1の結晶化から得られる液体に播種することによって調製され得る。好ましくは、この液体は、周囲温度未満に冷却される(例えば4℃)。
【0042】
一実施形態によれば、ベシル酸塩形態3は、式(I)の化合物の酢酸エチル中の溶液とベンゼンスルホン酸のエタノール中の溶液とを接触させて形成される沈殿から分離される濾過溶液に、式(I)の化合物のベシル酸塩形態1の結晶塩で播種してベシル酸塩形態3結晶多形を生じさせることによって調製され得る。
【0043】
上記方法のいずれかによって得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0044】
ベシル酸塩形態4は、酢酸イソプロピル/エタノール、好ましくは、40%酢酸イソプロピル/エタノールからベシル酸塩形態1を再結晶化させることによって調製され得る。
【0045】
上記方法によって得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0046】
本発明の塩は、適切な溶媒から、または適切な溶媒/貧溶媒(anti−solvent)もしくは溶媒/共溶媒混合物から式(I)の化合物のベシル酸塩を結晶化させることによっても調製され得る。この溶液または混合物は、適切な場合、冷却および/または蒸発して結晶化を達成し得る。
【0047】
本発明者らは、形態2の結晶化は、極性(例えば、アセトニトリル:水)もしくは親油性(n−ノナン)のいずれかの、または両方(ジメチルスルホキシド:1,2−ジクロロベンゼン)の極端な状態が存在する条件において観測されることを見出した。
【0048】
形態2の結晶化のための溶媒の例は、ノナン;メタノールである。
【0049】
形態1の結晶化のための溶媒/貧溶媒混合物の例は、ジメチルアセトアミド/メチルイソブチルケトン;ジメチルアセトアミド/テトラクロロエチレン;アセトニトリル/3−メチルブタン−1−オール;アセトニトリル/1,2−ジクロロベンゼン;アセトニトリル/酢酸ペンチル;メタノール/3−メチルブタン−1−オール;メタノール/メチルイソブチルケトン;2,2,2−トリフルオロエタノール/1,4−ジメチルベンゼン;エタノール/メチルイソブチルケトン;エタノール/1,4−ジメチルベンゼン;プロパン−1−オール/1,2−ジクロロベンゼン;プロパン−1−オール/テトラクロロエチレン;プロパン−2−オール/1,2−ジクロロベンゼン;プロパン−2−オール/n−ノナン;2−メトキシエタノール/水;2−メトキシエタノール/酢酸ペンチル;2−メトキシエタノール/1,4−ジメチルベンゼン;テトラヒドロフラン/水;テトラヒドロフラン/3−メチルブタン−1−オール;テトラヒドロフラン/1,2−ジクロロベンゼン;テトラヒドロフラン/酢酸エチル;テトラヒドロフラン/1,3−ジメチルベンゼンである。
【0050】
形態2の結晶化のための溶媒/貧溶媒混合物の例は、エタノール/酢酸エチル;エタノール/メチルイソブチルケトン;エタノール/p−シメン;ジメチルスルホキシド/1,2−ジクロロベンゼン;アセトニトリル/水;エタノール/1,2−ジクロロベンゼン;エタノール/テトラクロロエチレン;テトラヒドロフラン/1,2−ジクロロベンゼン;テトラヒドロフラン/酢酸エチルである。
【0051】
好ましい実施形態によれば、形態1は、2−メトキシエタノール/酢酸ペンチルから結晶化される。
【0052】
好ましい実施形態によれば、形態2は、エタノール/酢酸エチルから結晶化される。
【0053】
好ましい実施形態によれば、形態2は、メタノール/エタノールから結晶化される(周囲温度未満、例えば4℃でメタノール/エタノール中の式(I)の化合物のベシル酸塩の溶液を冷却することによるのが好ましい)。
【0054】
好ましい実施形態によれば、形態3は、エタノール/酢酸エチルから結晶化される(周囲温度未満、例えば4℃に混合物を冷却することによるのが適切である)。
【0055】
好ましい実施形態によれば、形態4は、酢酸イソプロピル/エタノールから結晶化される(周囲温度に酢酸イソプロピル/エタノール中の式(I)の化合物の溶液を冷却することによるのが好ましい)。
【0056】
上記方法のいずれかによって得られる式(I)の化合物のベシル酸塩も、本発明によって提供される。
【0057】
本発明の塩の調製方法は、以下の実施例に詳細に記載される。
【0058】
本発明の塩は、特に鎮静または催眠、不安緩解、筋弛緩、または抗痙攣の目的のための薬剤として使用され得る。
【0059】
本発明の塩は、バルクの活性薬品として投与されることが可能であるが、この塩は、好ましくは、薬剤組成物の形態で、薬学的に許容される担体、賦形剤、または希釈剤と一緒に提供される。担体、賦形剤、または希釈剤は、当然、組成物の他の成分と相溶性があるという意味において許容されなければならず、レシピエントに有害であってはならない。
【0060】
したがって、本発明は、本発明の塩および薬学的に許容される担体、賦形剤、または希釈剤を含む薬剤組成物を提供する。
【0061】
本発明の薬剤組成物には、経口、直腸、局所、口腔(例えば、舌下)および非経口(例えば、皮下、筋肉内または静脈内)投与に適するものが含まれる。
【0062】
好ましくは、本発明の塩は、例えば、溶液の静脈内または筋肉内注射による非経口投与のための薬剤組成物の形態で提供される。薬剤組成物が、非経口投与用である場合、この組成物は、静菌剤、抗酸化剤、緩衝剤または他の薬学的に許容される添加剤を含み得る水性もしくは非水性溶液または液体の混合物であり得る。
【0063】
本発明の塩の好ましい製剤は、pH2〜4の水性酸性媒体またはサイクロデキストリン(CD)の水溶液中である。これらの製剤に使用され得るサイクロデキストリンは、β−CDの陰イオン性に帯電されたスルホブチルエーテル(SBE)誘導体、具体的にはCyDex,Inc.によってCaptisolの商標で市販されているSBE7−β−CD(CriticalReviews in Therapeutic Drug Carrier Systems、14巻(1号)、1〜104頁(1997年))、またはヒドロキシプロピルCDのいずれかである。
【0064】
本発明の塩のさらに好ましい製剤は、この塩に加えて、次の試剤:アスコルビン酸、クエン酸、マレイン酸、リン酸、グリシン、塩酸グリシン、コハク酸または酒水酸の少なくとも1つを含む凍結乾燥製剤である。これらの試剤は、緩衝剤、粘結剤または可視化剤(visualization agent)として有用であると考えられる。ある場合には、製剤に塩化ナトリウム、マンニトール、ポリビニルピロリドン、または他の成分を含むことが有益であり得る。
【0065】
製剤化の好ましい方法(すなわち、酸緩衝剤またはCDベース)は、特定の塩の物理化学的性質(例えば、水溶解度、pKaなど)によって決まり得る。別法として、上記塩は、水(注射用)またはブドウ糖または食塩水での再構成用の凍結乾燥固体として提供され得る。このような製剤は、アンプルまたは使い捨て注射器具などの単位剤形で通常提供される。それらは、それから適切な用量が抜き取られるボトルなどの複数用量形態でも提示され得る。全てのこのような製剤は無菌でなければならない。
【0066】
本発明によれば、対象に有効な鎮静量または催眠量の本発明の塩を投与することを含む、対象において鎮静または催眠を生じるための方法が提供される。
【0067】
対象に有効な不安緩解量の本発明の塩を投与することを含む、対象に不安緩解(anxiolysis)を誘導するための方法も、本発明によって提供される。
【0068】
対象に有効な筋弛緩量の本発明の塩を投与することを含む、対象において筋弛緩を誘導するための方法が、本発明によってさらに提供される。
【0069】
対象に有効な抗痙攣量の本発明の塩を投与することを含む、対象において痙攣を治療するための方法が、本発明によってさらに提供される。
【0070】
本発明によれば、対象に鎮静または催眠を生ずるための薬剤の製造における鎮静量または催眠量の本発明の塩の使用も提供される。
【0071】
本発明によれば、対象に鎮静または催眠を生ずるための本発明の塩も提供される。
【0072】
対象における不安緩解を生ずるための薬剤の製造における不安緩解量の本発明の塩の使用も、本発明によって提供される。
【0073】
対象における不安緩解を生ずるための本発明の塩も、本発明によって提供される。
【0074】
対象における筋弛緩を生じるための薬剤の製造における、筋弛緩量の本発明の塩の使用が本発明によってさらに提供される。
【0075】
対象における筋弛緩を生じるための本発明の塩が本発明によってさらに提供される。
【0076】
対象における痙攣を治療するための、薬剤の製造における抗痙攣量の本発明の塩の使用が本発明によってさらに提供される。
【0077】
対象における痙攣を治療するための本発明の塩が本発明によってさらに提供される。
【0078】
上記対象は、適切には哺乳類、好ましくは、ヒトである。
【0079】
ヒトへの投与に適する非経口製剤は、好ましくは、0.1から20mg/mlの溶液中の本発明の塩または複数用量バイアル用にはその倍数を含有する。
【0080】
静脈内投与は、ボーラス注入法または、より適切には、連続注入の形態を取り得る。それぞれの対象のための用量は変わり得るが、哺乳類において鎮静または催眠を得るのに適する本発明の塩の静脈内投与量または用量は、0.01から5.0mg/体重1kg、より詳しくは、0.02から0.5mg/体重1kgである(上記は、活性成分である上記塩の重量に基づく)。哺乳類において不安緩解を得るのに適する本発明の塩の静脈内投与量または用量は、0.01から5.0mg/体重1kg、より詳しくは、0.02から0.5mg/体重1kgである(上記は、活性成分である上記塩の重量に基づく)。哺乳類において筋弛緩を得るのに適する本発明の塩の静脈内投与量または用量は、0.01から5.0mg/体重1kg、より詳しくは、0.02から0.5mg/体重1kgである(上記は、活性成分である上記塩の重量に基づく)。哺乳類において痙攣を治療するのに適する本発明の塩の静脈内投与量または用量は、0.01から5.0mg/体重1kg、より詳しくは、0.02から0.5mg/体重1kgである(上記は、活性成分である上記塩の重量に基づく)。
【0081】
本発明の塩は、次の臨床状態:術前の鎮静、不安緩解、および手術前後の記憶消失使用;短期診断、手術または内視鏡的処置中の意識下鎮静;他の麻酔薬または鎮痛薬の投与の前および/または同時の全身麻酔の誘導および維持のための成分として;ICU鎮静において、静脈内投与されるのに有用な短時間作用型CNS抑制剤である。
【0082】
本発明の好ましい実施形態は、添付図を参照して以下の実施例に記載される。
例えば、本発明は、以下の項目を提供する。
(項目1)
式(I):
【化4】
の化合物のベシル酸塩。
(項目2)
結晶塩である、項目1に記載の塩。
(項目3)
約7.3、7.8、9.4、12.1、14.1、14.4、14.7、および15.6度2θにおける特性ピークを含む粉末X線回折(XRPD)パターンを示す結晶多形である、項目2に記載のベシル酸塩。
(項目4)
a=7.6868Å、b=29.2607Å、c=12.3756Å、α=90°、β=97.7880°、γ=90°の単位格子寸法を有する結晶を含む結晶多形である、項目2または3に記載のベシル酸塩。
(項目5)
表17に示された構造座標によって定義される結晶構造を有する結晶多形である、項目2から4のいずれかに記載のベシル酸塩。
(項目6)
表19および20に示された結合の長さおよび角度を有する結晶構造を有する結晶多形である、項目2から5のいずれかに記載のベシル酸塩。
(項目7)
約8.6、10.5、12.0、13.1、14.4、および15.9度2θにおける特性ピークを含むXRPDパターンを示す結晶多形である、項目2に記載のベシル酸塩。
(項目8)
a=8.92130Å、b=11.1536Å、c=25.8345Å、α=90°、β=90°、γ=90°の単位格子寸法を有する結晶を含む結晶多形である、項目2または7に記載のベシル酸塩。
(項目9)
表18に示された構造座標によって定義される結晶構造を有する結晶多形である、項目2、7または8のいずれかに記載のベシル酸塩。
(項目10)
表21および22に示された結合の長さおよび角度を有する結晶構造を有する結晶多形である、項目2、または7から9のいずれかに記載のベシル酸塩。
(項目11)
約7.6、11.2、12.4、14.6、15.2、16.4、および17.7度2θにおける特性ピークを含む粉末X線回折(XRPD)パターンを示す式(I)の化合物のベシル酸塩の結晶多形である、項目2に記載のベシル酸塩。
(項目12)
約7.6、10.8、15.2、15.9および22.0度2θにおける特性ピークを含むXRPDパターンを示す式(I)の化合物のベシル酸塩の結晶多形である、項目2に記載のベシル酸塩。
(項目13)
項目1から12のいずれかに記載の塩、および薬学的に許容される担体、賦形剤、または希釈剤を含む薬剤組成物。
(項目14)
薬剤としての使用のための項目1から12のいずれかに記載の塩。
(項目15)
対象において鎮静または催眠を生じるための薬剤の製造における、鎮静量または催眠量の項目1から12のいずれかに記載の塩の使用。
(項目16)
対象において不安緩解を生じるための薬剤の製造における、不安緩解量の項目1から12のいずれかに記載の塩の使用。
(項目17)
対象において筋弛緩を生じるための薬剤の製造における、筋弛緩量の項目1から12のいずれかに記載の塩の使用。
(項目18)
対象において痙攣を治療するための薬剤の製造における、抗痙攣量の項目1から12のいずれかに記載の塩の使用。
(項目19)
式(I)の化合物の遊離塩基をベンゼンスルホン酸と反応させる工程を含む、項目1に記載の塩の製造方法。
(項目20)
溶液中で前記遊離塩基をベンゼンスルホン酸と接触させて、前記ベシル酸塩の沈殿の形成を引き起こす工程を含む、項目19に記載の方法。
(項目21)
前記沈殿を単離する工程をさらに含む、項目20に記載の方法。
(項目22)
前記遊離塩基が、トルエンまたは酢酸エチルに溶解される、項目20または21に記載の方法。
(項目23)
前記ベンゼンスルホン酸が、エタノールに溶解される、項目20から22のいずれかに記載の方法。
(項目24)
項目3から6のいずれかに記載の塩を調製する項目20に記載の方法であって、トルエン、酢酸エチル、アセトン、酢酸イソプロピル、またはギ酸エチル中の式(I)の化合物の遊離塩基の溶液を、エタノール中のベンゼンスルホン酸の溶液と接触させて該塩の沈殿の形成を引き起こす工程を含む方法。
(項目25)
項目7から10のいずれかに記載の塩を調製する項目20に記載の方法であって、メタノール中の式(I)の化合物の遊離塩基の溶液を、エタノール中のベンゼンスルホン酸の溶液と接触させて該塩の沈殿の形成を引き起こす工程を含む方法。
(項目26)
項目11に記載の塩を調製する方法であって、酢酸エチル中の式(I)の化合物の溶液を、エタノール中のベンゼンスルホン酸の溶液と接触させることによって形成される沈殿から分離される濾液溶液に、式(I)の化合物のベシル酸塩形態1結晶塩を用いて播種して、結晶多形を生じる工程を含む方法。
(項目27)
酢酸イソプロピル/エタノールから式(I)の化合物のベシル酸塩形態1結晶塩を再結晶化する工程を含む、項目12に記載の塩を調製する方法。
(項目28)
溶媒から、または溶媒/貧溶媒もしくは溶媒/共溶媒混合物から式(I)の化合物のベシル酸塩を結晶化させる工程を含む、項目2から12のいずれかに記載の塩を調製する方法。
(項目29)
対象において鎮静または催眠を生じる方法であって、該対象に有効な鎮静量または催眠量の項目1から12のいずれかに記載の塩を投与する工程を含む方法。
(項目30)
対象において不安緩解を誘発する方法であって、該対象に有効な不安緩解量の項目1から12のいずれかに記載の塩を投与する工程を含む方法。
(項目31)
対象において筋弛緩を誘発する方法であって、該対象に有効な筋弛緩量の項目1から12のいずれかに記載の塩を投与する工程を含む方法。
(項目32)
対象において痙攣を治療する方法であって、該対象に有効な抗痙攣量の項目1から12のいずれかに記載の塩を投与する工程を含む方法。
【図面の簡単な説明】
【0083】
【図1】式(I)の化合物含有量(初期に対する%)対保存温度のグラフを示す図である。
【図2】LJC−039−081−1の示差走査熱量測定(DSC)を示す図である。
【図3】LJC−039−081−2(点線)と重ね合わせたLJC−039−081−1(実線)のDSCを示す図である。
【図4】ベシル酸塩形態のDSCを示す図である(形態1実線、形態2一点鎖線)。
【図5】ベシル酸塩形態のDSCを示す図である(形態1実線、形態3点線および一点鎖線)。
【図6】T0およびT4におけるLJC−039−037−1のクロマトグラフを示す図である(表10の結果に関連)。
【図7】4週間安定性研究の前後でLJC−039−037−1(ベシル酸塩)を比較したXRPDを示す図である。
【図8】図8Aは、ベシル酸塩形態1および2のXRPD比較を示す図である。図8Bは、形態1および2の重ね合わせの示差走査熱量測定(DSC)を示す図である。
【図9】図9Aは、ベシル酸塩形態1および3のXRPD比較を示す図である。図9Bは、形態1および3の重ね合わせを示す図である。
【図10】LJC−039−086−1のDSCを示す図である(ベシル酸塩形態4)。
【図11A】ベシル酸塩形態1についての結果を示す図である:A)100mgバッチLJC−039−037−1についてのXRPD。
【図11B】ベシル酸塩形態1についての結果を示す図である:B)100mgバッチLJC−039−037−1についてのDSC。
【図11C】ベシル酸塩形態1についての結果を示す図である:C)100mgバッチLJC−039−037−1についてのTGA。
【図11D】ベシル酸塩形態1についての結果を示す図である:D)100mgバッチLJC−039−037−1についての1H NMR。
【図11E】ベシル酸塩形態1についての結果を示す図である:E)100mgバッチLJC−039−037−1についてのGVS。
【図11F】ベシル酸塩形態1についての結果を示す図である:F)100mgバッチLJC−039−037−1についてのGVS後のXRPD。
【図11G】ベシル酸塩形態1についての結果を示す図である:G)100mgバッチLJC−039−037−1についての40℃/75%RHにおける安定後のXRPD。
【図11H】ベシル酸塩形態1についての結果を示す図である:H)100mgバッチLJC−039−037−1についてのVT XRPD。
【図11I】ベシル酸塩形態1についての結果を示す図である:I)100mgバッチLJC−039−037−1についての偏光顕微鏡検査。
【図12A】ベシル酸塩形態2についての結果を示す図である:A)100mgバッチLJC−039−067−8についてのXRPD。
【図12B】ベシル酸塩形態2についての結果を示す図である:B)100mgバッチLJC−039−067−8についてのDSC。
【図12C】ベシル酸塩形態2についての結果を示す図である:C)2℃/分の昇温速度でのDSC。
【図12D】ベシル酸塩形態2についての結果を示す図である:D)LJC−039−067−8についての1H NMR。
【図13A】ベシル酸塩形態3についての結果を示す図である:A)LJC−039−081−2についてのXRPD(LJC−039−081−1の液体からの2回目の採集)。
【図13B】ベシル酸塩形態3についての結果を示す図である:B)LJC−039−081−2についてのDSC。
【図13C】ベシル酸塩形態3についての結果を示す図である:C)LJC−039−081−2についてのDSC(2℃/分の昇温速度)。
【図13D】ベシル酸塩形態3についての結果を示す図である:D)LJC−039−081−2についてのTGA。
【図13E】ベシル酸塩形態3についての結果を示す図である:E)LJC−039−081−2についての1H NMR。
【図13F】ベシル酸塩形態3についての結果を示す図である:F)LJC−039−081−2についてのGVS。
【図13G】ベシル酸塩形態3についての結果を示す図である:G)LJC−039−081−2についてのGVS後のXRPD。
【図14A】ベシル酸塩形態4についての結果を示す図である:A)LJC−039−086−1についてのXRPD。
【図14B】ベシル酸塩形態4についての結果を示す図である:B)LJC−039−086−1についてのDSC。
【図14C】ベシル酸塩形態4についての結果を示す図である:C)LJC−039−086−1についての1H NMR。
【図15A】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15B】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15C】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15D】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15E】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図15F】ベシル酸塩の公表バッチについてのHPLCクロマトグラフおよび次いでの結果を詳述するAgilent ChemStationレポートを示す図である。
【図16A】LJC−039−081−1、およびLJC−039−083−1についてのキラルクロマトグラフィーを示す図である。
【図16B】LJC−039−081−1、およびLJC−039−083−1についてのキラルクロマトグラフィーを示す図である。
【図16C】LJC−039−081−1、およびLJC−039−083−1についてのキラルクロマトグラフィーを示す図である。
【図16D】LJC−039−081−1、およびLJC−039−083−1についてのキラルクロマトグラフィーを示す図である。
【図17】(約4−8mmの視野径)式(I)の化合物のベシル酸塩の結晶化において観測された固体の典型画像を示す図である。
【図18】形態1における非対称単位の内容を示す図である。
【図19】2−メトキシエタノール:酢酸ペンチル溶液から成長させた式(I)の化合物のベシル酸塩、形態1の結晶の単結晶X線回折によって求められた分子構造を示す図である(原子は熱振動楕円体によって表されている)。結晶構造中に特に位置する水素のみが示されている。
【図20】形態1の2つの独立した分子によってとられる立体配座を示す図である。
【図21】形態1の1つの独立した分子によってとられる立体配座(上部)および形態2の立体配座(下部)の比較を示す図である。
【図22】2つの異なる方向に沿って見た、2つの独立した形態1のベシル酸塩によってとられる立体配座の比較を示す図である。
【図23】1つの独立した形態1のベシル酸塩によってとられる立体配座(上部)および形態2の立体配座(下部)の比較を示す図である。
【図24−1】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、2−メトキシエタノール:酢酸ペンチル溶液から成長させた式(I)の化合物のベシル酸塩の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
【図24−2】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、2−メトキシエタノール:酢酸ペンチル溶液から成長させた式(I)の化合物のベシル酸塩の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
【図25】形態1についてのショートコンタクトC−O<3.6Å、C−C<3.6Å、およびN−O<3.5Åを示す図である。
【図26】形態1についての単結晶X線回折データから計算された粉末パターン回折を示す図である。
【図27】式(I)の化合物のベシル酸塩の形態2について観測された板状の形態の結晶を示す図である。
【図28】形態2の非対称単位の内容を示す図である。
【図29】式(I)の化合物のベシル酸塩、形態2の結晶の単結晶X線回折によって求められた分子構造を示す図である(原子は熱振動楕円体によって表されている)。結晶構造中に特に位置する水素のみが示されている。
【図30】形態2の独立した分子によってとられる立体配座を示す図である。
【図31】2つの異なる方向に沿って見た、形態2の独立したベシル酸塩によってとられる立体配座を示す図である。
【図32−1】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、式(I)の化合物のベシル酸塩の形態2の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
【図32−2】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、式(I)の化合物のベシル酸塩の形態2の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
【図32−3】結晶のa軸(a)、b軸(b)、およびc軸(c)に沿って見た、式(I)の化合物のベシル酸塩の形態2の結晶の単結晶X線回折によって求められた結晶構造を示す図である。
【図33】形態2についてのショートコンタクトC−O<3.6Å、C−C<3.6ÅおよびN−O<3.5Åを示す図である。
【図34】形態2についての単結晶X線回折データから計算された粉末パターン回折を示す図である。
【図35】式(I)の化合物のベシル酸塩形態1についての原子中心の標識付けを示す図である。
【図36】式(I)の化合物のベシル酸塩形態2についての原子中心の標識付けを示す図である。
【発明を実施するための形態】
【0084】
(実施例1)
式(I)の化合物の固体状態安定性の研究
方法/技術 正確に秤量した2mgの式(I)の化合物のサンプルを4mL透明ガラスねじ口バイアル中に置いた。最初に、および5℃/周囲相対湿度(AMRH)密栓、30℃/60%RH密栓、40℃/75%RH開放および60℃/AMRH密栓で保存された34日後に、サンプルを試験した。
【0085】
サンプルを、外観について目視で検査した。式(I)の化合物の含有量を、表1においてHPLC法によって求めた。%重量/重量(%w/w)値を、式(I)の化合物の標準サンプル、バッチU12438/79/1に対して測定した。%面積値を、式(I)の化合物のピーク面積を総ピーク面積で割ることによって得た。
【0086】
【表1】
結果
外観 表2は外観の結果を示す。
【0087】
【表2】
式(I)の化合物の含有量(%w/w) %w/w含有量(表3を参照されたい)は、初期値および5℃/AMRH密栓、30℃/60%RH密栓または40℃/75%RH開放での34日後に測定された値の間の差異を検出するには多すぎるばらつきを示している。60℃/AMRH密栓で34日保存されたサンプルについて測定された平均%w/wは、初期値から10%w/w減少を示している。
【0088】
式(I)の化合物の含有量(%面積) 式(I)の化合物の%面積含有量(表3および図1を参照されたい)は、5℃/AMRH密栓で保存された34日後に有意な変化を示さないが、30℃/60%RH密栓、40℃/75%RH開放または60℃/AMRH密栓におけるサンプルについては、保存温度増加とともに着実に減少する。主要な分解ピークは、RRT0.68、0.87およびRRT0.90において観測されるが、クロマトグラムも(初期においてさえも比較的複雑である(23個のピーク))多くの新しい小さな分解のピークを示している(例えば、30℃/60%RH密栓において7個のピーク;60℃/AMRH密栓において13〜20個のピーク)。これらの観測は、複数の分解経路を示唆している。RRT0.68での分解物(degradent)は、エステル加水分解生成物(式(I)の化合物の遊離酸)として暫定的に同定されている。それは、加水分解生成物について予想されるように、40℃/75%RH開放サンプルにおいて最も優勢である。
【0089】
【表3】
結論
式(I)の化合物は、5℃/AMRH密栓で保存された少なくとも34日間は外観および含有量について安定している。30℃/60%RH密栓で外観における変化は指摘されなかったが、初期の%面積に対して式(I)の化合物の含有量における約0.6%低下が観測された。40℃/75%RH開放または60℃/AMRH密栓で保存されたサンプルは、潮解し、色が黄色から橙色になり、初期に比べて式(I)の化合物の含有量における著しい低下(1.5から8%)を示した。RRT0.68、0.87およびRRT0.90における主要な分解ピークが、多数のより小さなピークと一緒に観測され、複数の分解経路が示唆された。RRT0.68における分解物は、エステル加水分解生成物として暫定的に同定されている。これらの結果は、長期保存のためには冷蔵されるべきであることを示している。
【0090】
(実施例2)
式(I)の化合物の溶解度が、広範囲の有機溶媒で測定された。溶解度データが、以下の表4に示されている。
【0091】
【表4】
データは、式(I)の化合物は、通常の有機溶媒において高い溶解度を有することを明らかに示している。好ましい溶媒は、エタノールおよびトルエンである。
【0092】
化合物の遊離塩基の2つの塩基性中心が、pKaについて測定された。しかし、ピリジン環の塩基性中心は、1.99のpKaを有した。イミダゾール環の塩基性中心のpKaは、4.53と測定された。
【0093】
ベンゼンスルホン酸を、使用して式(I)の化合物のベシル酸塩を生成した。実験を、6体積の溶媒を使用する20mgスケールで実施した。全ての反応を、溶解性に応じてエタノール中の原液(1M)としてまたは固体として酸を装入して周囲温度で実施した。
【0094】
単離された固体は、塩の形成を立証する1H NMRにおける有意なピークシフトを示した。粉末X線回折(XRPD)は、塩が結晶性の徴候を有することを示した。表5は、単離された塩形態を要約している。
【0095】
【表5】
塩を、その後40℃/75%RHで2週間保存し、次いで材料の安定性を評価するために化学的純度についてXRPDおよびHPLCで再分析した。塩は、湿度条件への曝露後に同じ粉末パターンを保持し、安定性改善を支持する高い化学的純度も保持した。
【0096】
トルエンからのベシル酸塩は、安定性研究の前後で高い純度を示したことが、単離塩のT1純度結果からわかる(以下の表6)。
【0097】
【表6】
上記の結果は、ベシル酸形態は、高純度および好ましい安定性結果を示したことを示している。
【0098】
(実施例3)
実施例2のデータに基づいて、ベシル酸塩の100mgへのスケールアップを実施した。トルエンは、ベシル酸塩の単離用の好ましい溶媒であることが見出された。
【0099】
式(I)の化合物のベシル酸塩
プロセスがスケールアップするか否かを確かめるため、および単離される材料が、先のより小さなスケールの実験で見られた同じ結晶形態(形態1)であることを確かめるために、50mgの投入材料へのスケールアップを実施した。塩が形態1であったことおよび性質が期待されるものと一致していたことが分析で確認され次第、完全な特徴づけを行うためおよび40℃/75%RHでの4週間の安定性研究用のサンプルを提出するために、100mgの投入材料を用いてもう1つのスケールアップを実施した。両方のスケールアップした反応を、エタノール中の溶液(1M)としてベンゼンスルホン酸を添加して、トルエン中で実施した。
【0100】
ベシル酸塩の実験手順
式(I)の化合物の遊離塩基(100mg、バッチ704−17)を、バイアルに装入し、トルエン(600μl)を周囲温度で添加した。この溶液に、ベンゼンスルホン酸(250μl、エタノール中1M)を添加し、反応混合物を15分間撹拌し、その後、固体が溶液から沈殿し、この沈殿を濾過し、トルエンで洗浄し、真空下40℃でオーブン乾燥した。XRPDによる分析は、固体が、生成された他のベシル酸塩と同じ粉末パターンであることを示し、1H NMRは、有意なピークシフトにより塩形成を確認した。
【0101】
【表7】
LJC−039−037−1の鏡像体過剰率は、わずかに94.4であり、したがって、この結果を、同じ条件で単離された別のバッチのベシル酸塩(LJC−039−081−1)と比較した。このバッチの鏡像体過剰率は99.1%であった。
プロセス最適化
ベシル酸塩(形態1)の収量をさらに改良するために、4種の溶媒をスクリーニングした(酢酸イソプロピル、ギ酸エチル、メタノールおよびアセトン)。合計8つの100mgスケールの反応を、先の実験との比較のために関連のある酸をエタノール中の原液として添加して、これらの溶媒中で実施した。
【0102】
式(I)の化合物(バッチ704−38、100mg)を、周囲温度で溶媒(600μl)中に溶解した。酸(250μl、エタノール中の1M原液)を添加し、全ての反応混合物を、周囲温度で48時間静置した。結果は表8に要約されている。
【0103】
【表8】
メタノール中のベシル酸塩の形成の反応以外の全ての反応は、形態1を示した。メタノール反応物を4℃で保存した。得られたデータは、無水ベシル酸塩1:1を立証し、材料の粉末パターンは、新たな形態(形態2)の存在を立証した。
【0104】
酢酸イソプロピルなどの溶媒は、塩の純度を増すが、回収を減少させることが、この研究から結論付けられた。溶媒の先の選択(酢酸エチル)が、高純度の高い収量の塩をもたらしたので、最終スケールアップ実験用に酢酸エチルを使用することが決定された。
ベシル酸塩(形態1)1gスケールアップ
ベシル酸塩の1gの形成を実施した。これにより、首尾よく950mg(70%収率)の形態1が生成された。この液体は、非常に色が濃く(黄色)、したがって、少量の形態1で播種して回収を助けた。この液体を4℃で16時間保存した。得られた固体は、新しい粉末パターン(形態3)を示した。この固体を、熱分析および様々な温度でのXRPDで分析して、真正の多形であるか溶媒和物であるか否かを確かめた。1H NMR証拠からの分析の解釈によって、この固体が溶媒和物ではないことが結論付けられ、DSCが、高温顕微鏡により確認された2つの吸熱事象を示した(図3)。形態1の種は187℃で融解し、形態3は200℃で融解すると解釈された。形態1がXRPDによって同定されなかった理由は、これが顕微鏡検査に比べて感度の低い技術であることである。
【0105】
形態3は、形態1より低い温度で沈殿する。
【0106】
多形の間の関係を提案するために、多形の特徴づけを実施した。
【0107】
【表9】
LJC−039−081−2に存在する少量の形態1のより低い融点は、潜在的に低い純度が原因であり得る(LJC−039−081−1の97.9%と比較して97.2%)。
【0108】
図4は、ベシル酸塩の形態1(実線)および2(一点鎖線)のDSCを示している。
【0109】
図5は、ベシル酸塩の形態1(実線)および3(点線および一点鎖線)のDSCを示している。
【0110】
(実施例4)
塩の安定性研究
【0111】
【表10】
ベシル酸塩の結晶サンプルを、40℃/75%RHで合計4週間保存し、サンプルを7日毎にHPLC用に取り出した。ベシル酸塩hplc純度は、96.7%に達したT3まで一定であった。しかし、この値はT4まで一定であった。
【0112】
ベシル酸塩形態のhplcクロマトグラフは、0週から4週の時点の間、図6に示されている。
【0113】
λmaxが親ピークのλmaxと一致しないので、親のピークの前の主要なピークは、汚染からであると考えられる。それは、T1、T2、T3およびT4の不純物プロフィールにも存在しない。
【0114】
形態における変化がないことは、湿度研究の前後の塩の粉末パターンからわかる。
【0115】
図7は、4週間安定性研究の前後のLJC−039−037−1(ベシル酸塩)の比較のXRPDを示している。
【0116】
(実施例5)
多形研究
・多形を示すベシル酸塩の傾向を求めるために、30種の溶媒(15種のニートに加えてそれらの2.5%水性対応物)を使用して成熟実験を設定した。周囲温度から60℃の加熱/冷却サイクルで1週間、固体を様々な溶媒(表11を参照されたい)中でスラリーにした。1週間後、スラリーを蒸発し、固体をXRPDおよびHPLCで分析した。
【0117】
表11 ベシル酸塩(LJC−039−058−2)についての多形研究の結果
・初期hplc純度97.7%
【0118】
【表11】
ベシル酸塩を使用した成熟研究は、新しい形態を示さなかった。成熟後の純度の結果は、アセトニトリル、水性THF、水性IPA、水性MEK、水性ジオキサンおよび水性アセトニトリル中にスラリーにしたものは分解したことを示している。これは、ベシル酸塩(形態1)は、高温でのニート有機溶媒中で良好な溶液安定性を有することを示唆している。
【0119】
ベシル酸塩の新しい形態の研究
ベシル酸塩の新しい形態は、成熟研究からは見られなかったが、メタノール中で結晶が成長した場合に、新しい形態が見られた。メタノールから得られる単結晶を、粉末パターンを得るために磨砕した。このパターンは、形態1と異なることがわかった。形態2のさらなる供給を得るために、反復実験を実施した。溶媒を蒸発させること(これは形態1をもたらす)の反対に、液体からの16時間かけた沈殿から形態2を単離することが可能であった。興味深いことに、2つの晶癖(針および塊)が存在した。両方とも単結晶構造の測定に使用された針晶癖と同じ粉末パターンを示した。
【0120】
形態2の完全な分析を実施した。単結晶データが無水ベシル酸塩1:1を立証したので、形態2は真正の多形であることが結論付けられた。
【0121】
図8Aは、ベシル酸塩形態1および2のXRPD比較を示している。形態1(トレース1)および形態2(トレース2)の間には明らかな差異がある。2つの粉末パターンからわかるように、両方の形態は非常に異なる。2つの形態の融点を比較するために熱解析を実施し、熱力学的溶解度の測定値も記録した。
【0122】
図8Bは、形態1および2の重ね合わせを示している。形態1および2は、1つの吸熱事象(融解)を示している。
【0123】
形態3が、2回目の採集がLJC−039−081−1(1gスケールアップ反応)の液体から単離されたときに同定された。それが溶媒和物であるか否かおよびどのように形態が相互変換するのかを決定するために分析を実施した。
【0124】
図9Aは、ベシル酸塩形態1および3のXRPD比較を示している。図9Bは、形態1、および3の重ね合わせを示している。
【0125】
形態1は、1つの吸熱事象(融解)を示しているが、形態3は、2つの事象を示している。形態3の高温顕微鏡は、互いの20℃以内の2つの融解を明らかに示している。より低い感度の技術である様々な温度のXRPDにおいて検出されないために、少量のより低融点の多形が存在することが仮定される。形態3が単離される液体を播種するのに形態1が使用されたので、最初の吸熱事象は形態1を表すことはあり得る。
【0126】
溶解度データは、全ての3つの形態は、pH3において7.8から8.3mg/mlの非常に似た水溶解度を有することを示している。
【0127】
ベシル酸塩形態4
ベシル酸塩形態1(LJC−039−083−1)の公表バッチ(release batch)は、高純度(97.6%)であったが、遊離塩基から運ばれた少量の不純物を含有した(0.78%、11.9分RT)。この不純物は、吸熱変換(130℃で開始)を示すことがDSC実験で観測された。このピークは、親ピークのものと無関係のλmaxを有することが確認された。
【0128】
100mgサンプルを、40%酢酸イソプロピル/エタノールからの再結晶化の試みのために取った。再結晶化を、最小量の熱した溶媒中に塩を溶解し、次いで周囲温度にゆっくりと冷却して沈殿を得ることによって慣習的に実施した。乾燥した固体を、XRPDで分析し、新しい形態が示され、熱分析および1H NMRによって、多形であって溶媒和物ではないことが確認された。図10は、LJC−039−086−1のDSCを示している。
【0129】
塩の選別研究は、式(I)の化合物は、適切なpKa範囲内で多くの塩を形成すること、および、それらは、一連の溶媒から容易に単離されることを示した。塩の完全な特性決定から、ベシル酸塩は、湿度に関して良好な安定性を有することが決定された。ベシル酸塩の2つの多形があることが結論付けられた。形態3は、形態1で播種後のLJC−039−081−1の液体の2回目の採集から生じた。形態4は、40%酢酸イソプロピル/エタノールから形態1の再結晶化が実施された後に観測された。
【0130】
完全な分析データが以下の図11−14に示されている。
【0131】
実施例2〜5のための実験方法
(実施例2)
式(I)の化合物(5mg/ウェル)を、HPLCバイアル中で溶媒1(30μl)に溶解した。この溶液に、ベンゼンスルホン酸(11.4μl、エタノール中の1M)を添加し、この反応混合物を常温で終夜放置した。固体を含有したバイアルは、真空下40℃で乾燥し、溶液のままであったものは、蒸発により濃縮し、次いでヘプタンで処理した。沈殿したものは、前述のように乾燥し、溶けたものは、4℃で保存した。
【0132】
ベシル酸塩形態1のスケールアップ
式(I)の化合物(100mg)を酢酸エチル(600μl)に溶解し、ベンゼンスルホン酸(250μl、エタノール中の1M)を添加した。沈殿が即座に生じ、この反応混合物を常温で24時間撹拌した。固体を濾過し、酢酸エチルで洗浄し、真空下で40℃において16時間オーブン乾燥した。
【0133】
分析方法
示差走査熱量測定(DSC)
DSCデータを、50位置のオートサンプラーを備えたTA Instrument Q1000で集めた。エネルギーおよび温度較正基準はインジウムであった。サンプルを、25から350℃の間、10℃/分の速度で加熱した。30ml/分の窒素パージを、サンプル上で維持した。
【0134】
特に指定のない限り、0.5から3mgの間のサンプルを使用し、全てのサンプルはピンホールアルミ板状(pin holed aluminium pan)中で試験された。
【0135】
熱重量分析(TGA)
TGAデータは、Alumelで較正され、10℃/分の走査速度で測定するTA Instrument Q500 TGAで集めた。60ml/分の窒素パージを、サンプル上で維持した。
【0136】
1エタノール、トルエンおよびアセトニトリル。
【0137】
特に指定のない限り、一般的に5〜10mgのサンプルが、予め風袋計量した(pre−tared)白金るつぼに装入された。
【0138】
NMR
全てのスペクトルは、オートサンプラーを備えたBruker 400MHzで集められた。特に指定のない限り、サンプルは、d6−DMSO中で調製された。
【0139】
XRPD(粉末X線回折)
Bruker AXS C2 GADDS回折計
サンプルの粉末X線回折パターンは、Cu Kα放射(40kV、40mA)、自動化XYZステージ、自動サンプル位置決めのためのレーザービデオ顕微鏡およびHiStar2次元面積検出器を使用するBruker AXS C2 GADDS回折計で取得した。X線光学機器は、0.3mmのピンホールコリメータと一体となった単一Gobel多層鏡から成る。
【0140】
ビーム広がり、すなわち、サンプル上のX線ビームの有効なサイズは、約4mmであった。3.2〜29.8°の有効2θ範囲をもたらす20cmのサンプルと検出器の距離を有するθ−θ連続走査モードが使用された。サンプルの一般的な曝露時間は120秒であろう。
【0141】
周囲条件下で測定されるサンプルを、磨砕せずに得られた粉末を用いてフラットプレート試料として調製した。約1〜2mgのサンプルを、ガラススライド上で軽く圧縮して平面を得た。非周囲条件下で測定されるサンプルを、熱伝導化合物とともにシリコンウエハー上に載せた。サンプルを、次いで、約20℃/分で適切な温度に加熱し、続いて約1分間等温的に保持した後、データ収集を開始した。
【0142】
純度分析:
化学的方法
純度分析をHP1100 Agilentで実施した。
方法:グラジエント、逆相
方法持続時間/分:34
カラム:Phenomenex Gemini C18 5μm(2.0×50mm)(保護カートリッジ Phenomenex Gemini C18 保護カートリッジ2×4mm)
カラム温度/℃:40
注入/μl:5
流速ml/分:0.8
検出:UV
波長/nm:255(90nmのバンド幅)、240(80nmのバンド幅)、254(8nmのバンド幅)
相A:2mmol NH4HCO3(NH3溶液でpH10に調節された)
相B:アセトニトリル
タイムテーブル:
【0143】
【表11−1】
キラル法
純度分析を、Gilson HPLCシステムで行った。
方法:アイソクラチック、順相
方法持続時間/分:50
カラム:Diacel Chrialcel OJ−H(5μm)4.6×250mm(保護カートリッジDiacel Chrialcel OJ−H分析保護カートリッジ5μm 4.0×10mm)
カラム温度/℃:40
注入/μl:10
流速ml/分:1.0
検出:UV
波長/nm:225(単一波長検出器)
相A:ヘキサン
相B:エタノール
タイムテーブル:
【0144】
【表11−2】
重量蒸気収着(gravimetric vapour sorption)(GVS)研究
全てのサンプルを、CFRSorpソフトウエアを使用するHiden IGASorp水分収着分析機で測定した。サンプルサイズは一般的に10mgであった。水分吸着脱着等温線を、以下に記載のとおり実施した(2つの走査で1つの完全なサイクルが得られる)。全てのサンプルを、常湿常温(40%RH、25℃)で装入/取り出しした。全てのサンプルを、GVS分析後XRPDによって分析した。特に指定のない限り、標準の等温線を、0〜90%RH範囲にわたる10%RH間隔で25℃において実施した。
【0145】
【表11−3】
溶解度
溶解度を、0.25mlの溶媒(水)中に十分な化合物を懸濁させて、この化合物の親遊離形態の10mg/mlの最大最終濃度を得ることによって測定した。懸濁液を、25℃で24時間平衡化し、続いてpH確認し、ガラス繊維C96ウェルプレートを通して濾過した。次いで、この濾液を101×に希釈した。定量は、約0.1mg/mlでDMSO中に溶解した標準を参照するHPLCによった。様々な量の標準、希釈および非希釈試薬を注入した。標準の注入における最大ピークと同じ保持時間において見出されたピーク面積の積分によって、溶解度を算出した。フィルタープレート中に十分な固体がある場合、相変化、水和物形成、アモルファス化、結晶化等について、XRPDを標準的に確認する。
【0146】
【表11−4】
pKa測定
pKa測定を、D−PASアタッチメントを有するSirius GlpKa装置で行った。25℃におけるMeOH:H2O混合物中の電位差滴定法によって、測定を行った。滴定媒体は、0.15M KClでイオン強度調節された。MeOH:H2O混合物中で見出された値を、Yasuda−Shedlovsky外挿法により0%共溶媒に外挿した。
【0147】
高温顕微鏡検査
高温顕微鏡検査を、10〜20℃/分の範囲の一般的な加熱速度での25〜350℃の温度範囲のMettler−Toledo MTFP82HTホットステージを組み合わせたLeica LM/DM偏光顕微鏡を使用して実施した。少量のサンプルを、個々の粒子ができるだけ隔たるようにガラススライド上に分散した。サンプルを、×20対物レンズを用いて正または交差偏光下(λ着色フィルターと組み合わせた)で見た。
【0148】
キラル純度法
システム設定
ポンプ:Gilson 322バイナリーポンプ
検出器:Gilson 152UV/Vis
オートサンプラー:Gilson 233XLラック+Gilson 402デュアルシリンジポンプ
カラムオーブン:Phenomenex Thermasphere TS−130
ソフトウエア:Gilson Unipoint LCソフトウエア
カラム:Daicel Chiralcel OJ−H、5μm、4.6×250mm
保護カラム:Daicel Chiralcel OJ−H分析保護カートリッジ、5μm、4.6×10mm。
【0149】
HPLC条件
経路A:ヘキサン(93%)
経路B:エタノール(7%)
流速:1.0ml/分
検出器波長:225nm
カラム温度:40℃
ランタイム:50.0分。
【0150】
サンプル条件
約0.2mgのサンプルを、適切な量のヘキサン:エタノールの1:1v/vに溶解して0.2mg/ml溶液を得た。これにフタをして約15秒間、高速度のボルテックスミキサーにかけた。この時点で固体が残った場合、サンプルバイアルを、約10秒間、超音波処理し、続いてさらに10から15秒間ボルテックスミキサーにかけた。HPLCシステムに、10μlを注入した。ブランクとしてのヘキサン:エタノールの1:1v/vの最初の二重の注入の後に、サンプルを二重に注入した。
【0151】
(実施例5)
薬理試験例
本発明のベシル酸塩形態1の麻酔および鎮静効果を評価した。ベシル酸(ベンゼンスルホン酸)塩を、動物への試験組成物の投与のために生理的食塩水に溶解した。試験組成物を、マウスに投与し、個々のPlexiglasケージ(20×10×10cm)中に置いた。マウスに静脈経路でビヒクルまたは試験物質のいずれかを注入した。睡眠への待ち時間および麻酔状態の期間(最大:試験物質投与後90分)を記録した。麻酔状態は、立直り反射喪失(LRR)によって示される。立直り反射試験は、動物が落ち着いたと見え次第、約20〜30秒毎に実施した。立直り反射がなくなり次第、その後20〜30秒毎に立直り反射の復帰について試験することによって立直り反射喪失の期間を測定した。1グループ当たり8匹のマウスを研究し、試験をブラインドで実施した。この研究の結果は、下表に記載されている。
【0152】
【表11−5】
Mann−Whitney Uテスト:NS=有意ではない;*=p<0.05;**=p<0.01
Fisherの厳密テスト(LRRのあったマウスの数):兆候なし=有意ではない;+=p<0.05;++=p<0.01
(#):n<3の場合、計算されず
(##):最大=注入後90分。
【0153】
上記の表の結果は、ベシル酸塩形態1は、立直り反射喪失への短い待ち時間を有し、したがって動物における麻酔状態への短い誘導時間を有することを示している。さらに、マウスは、短い立直り反射喪失の期間によって示されるようにすばやく麻酔状態から回復する。したがって、この化合物は、迅速な麻酔状態導入および麻酔状態からの回復を提供し得る。
【0154】
(実施例6)
形態2、3、および4の結晶化のためのさらなる条件
先に報告された形態2、3、および4の結晶化を再現する試みにおいて、さらなる条件が試験された。しかし、以下に記載のように、報告されたスケールを実質的に減らし、方法をそれにより改変した。
【0155】
形態2
5mgの固体を25ulのメタノールに溶解し、10ulのエタノールを添加し、次いで、この溶液を4℃に3日間冷蔵した。
【0156】
形態3
3つの変形を試みた。
1.5mgの固体を50ulのエタノールに溶解し、120ulの酢酸エチルを添加し、次いで、この溶液を4℃に3日間冷蔵した。
2.10.1mgの固体を300ulのエタノールに溶解し、120ulの酢酸エチルを添加し、次いで、この溶液を4℃に3日間冷蔵した。
3.2.5mgの固体を、シラン処理したバイアル中で50ulのエタノールに溶解し、100ulの酢酸エチルを添加し、次いで、この溶液を4℃に3日間冷蔵した。
【0157】
形態4
3つの変形を試みた。
1.酢酸イソプロピル:エタノール(40%:60%v/v)の温めた(70℃)混合物を、固体が溶解するまで20ulアリコートで5mgの温めた固体に添加し(合計で60ulの溶媒混合物)、次いで、この溶液を、数時間かけて、最初70℃のサーモスタットで温度調節した水浴中で、ゆっくりと常温に冷却した。
2.5mgの固体を、180ulの温めた(50℃)酢酸イソプロピル:エタノール(40%:60%v/v)の溶媒に溶解し、この溶液を、数時間かけて、サーモスタットで温度調節した水浴(最初50℃)中で、ゆっくりと常温に冷却した。
3.5mgポーションの固体を、シラン処理したバイアル中で100ulの温めた(50℃)酢酸イソプロピル:エタノール(40%:60%v/v)の溶媒に溶解し、この溶液を、数時間かけて、サーモスタットで温度調節した水浴(最初50℃)中で、ゆっくりと常温に冷却した。
【0158】
それぞれの結晶化は、羽根状および板状のような晶癖を有する固体材料を生じ、形態4の結晶化も針のような材料を生じた。
【0159】
(実施例7)
式(I)の化合物のベシル酸塩の特性決定
式(I)の化合物のベシル酸塩は、キラルであり、以下の単一の鏡像体、すなわち、S鏡像体(後に求められた結晶構図と一致する)であると考えられる。
【0160】
【化2】
上記複素環構造は、イミダゾール環中に塩基性窒素(約5のpKa)、およびピリジル環中に弱塩基性窒素(約2のpKa)を含有する。イミダゾール−窒素は、一般的に、水溶液中で強酸性ベシル酸塩(約−0.6のpKa)の存在下でプロトン化され、ピリジニル−窒素も、潜在的に、過剰なベシル酸塩の条件下でプロトン化される。
【0161】
化合物の中性遊離塩基形態(すなわち、プロトン化されていない)は、いくらか親油性(logPオクタノール:水約4.0)であると予想され、したがって、水性環境よりいくらか親油性環境を好むであろう。さらに、それは、モノプロトン化(pH3においてlogDオクタノール:水約2)された場合でさえ、ある程度の親油性を保持しそうであるが、ベシル酸塩対イオンの影響は、その固有の親水性によって、この傾向を改善しそうである。親油性の程度は、二プロトン化形態(pH0においてlogDオクタノール:水約0.6)ではさらに減少する。
【0162】
化合物は、過剰な水素結合受容体も有し、したがって、水素結合供与溶媒と適切にパートナーになる。したがって、化合物は、特に、部分的に親油性の、水素結合供与環境を提供するアルコールなどの一連の極性有機溶媒における可溶化を好むことが予想される。これは、実験的証拠によって支持された(使用される溶媒の詳細は、実施例8で示される)。
【0163】
【表11−6】
可溶性(>5mg/ml)、部分的に可溶性(2.5〜5mg/ml)、部分的に不溶性(0.5〜2.5mg/ml)、不溶性(<0.5mg/ml)
引用された値は近似であるが、実験的に確認されている。
【0164】
これらの結果は、多様な極性有機溶媒における化合物の良好な溶解性を強調している。特に、2,2,2−トリフルオロエタノールおよびヘキサフルオロプロパン−2−オールは、両方とも、この化合物の極めて良好な溶媒であることが確認されている。このことは、両方の溶媒が強い水素結合供与体であるという上記に論じた考察と一致する。同様に、より実質的に親油性の溶媒は、不十分な溶媒であり、したがって結晶化の潜在的な貧溶媒であることが確認されている。
【0165】
(実施例8)
式(I)の化合物のベシル酸塩の結晶化
式(I)の化合物のベシル酸塩の形態1および2の結晶材料を得ることの助けとなる様々な条件が記載される。成分としてアルコールまたはアセトニトリル溶媒を、それらのそれぞれに相溶性の貧溶媒または共溶媒とともに含む結晶化条件は、有用な結晶材料を得るのに最も有望な条件を提供すると考えられる。溶媒/貧溶媒二成分混合物を使用する結晶化を主に使用した。結晶化を、常温および低温(4℃)において、溶媒/貧溶媒混合物中の化合物の半飽和溶液からの遅延蒸発によって実施した。結晶化は、一般的に、調製の3〜5日以内に観測された。
【0166】
サンプル量が許される場合、全ての結晶化条件を、ガラス96ウェルプレートフォーマット中で二重に実施し、それぞれのウェルプレートの半分は、ウェルプレートのもう半分における条件を複製するのに使用した。ウェル間の交差汚染を設計によって最小化する。試験した条件の全てが、少なくとも二重に再現可能な挙動を示し、大部分が、さらなる分析に適する固体材料を生じた。
【0167】
全ての場合で、サンプルおよび結晶化媒体と接触する器具は、様々な溶媒および試薬で綿密に洗浄した後、エタノール中に浸し、豊富な蒸発窒素を用いて通気乾燥した。
【0168】
表12に記載されたとおりの、市販供給者からの高品質の溶媒が使用された。
【0169】
【表12】
得られた結晶形態の目視分析を、デジタルカメラを装着し、必要に応じて透過および反射照明の両方を用いる双眼顕微鏡(約10×〜40×倍率)を使用して達成した。
【0170】
固体材料の目視による特性決定を、以下の表14に要約している。独特の結晶または球晶のいずれかとしての、羽根状または平面状/板状の形態の優勢を観察した。全体的に、溶媒としてエタノールを用いた結晶化(この結晶化では、球晶およびインターフェース型成長への傾向が温度低下に伴って減少した)を例外として、周囲温度で実施された結晶化と4℃での結晶化との間で形態的な差異は少ししかなかった。貧溶媒の使用は、実質的に結晶材料の質を改善し得ることは注目される。
【0171】
観測された結晶材料の画像の例が図17に示されている。この図に示されたように、アセトニトリルは、一般的に不十分な核生成、したがって不十分な質の結晶表面からの成長の結果として見られる球晶成長を生じる傾向を有する。対照的に、2−メトキシエタノールは、羽根状/針のような形態の独特の結晶を生じる傾向を有する。
【0172】
形態1が条件の多くから結晶化する一般的な傾向があるようである。しかし、形態2も、形態3および4を得るためのスケールダウンした類似物(実施例6に記載された)を含めた、いくつかの結晶化条件から観測されたことは注目される。形態2は、極性(アセトニトリル:水)または親油性(n−ノナン)または両方(ジメチルスルホキシド:1,2−ジクロロベンゼン)のいずれかの極端な状態が存在する条件において観測される。一般的に、形態2の結晶は、その卓越した品質および特徴的な十分に形成された板状/平面状の晶癖において顕著であった。
【0173】
単結晶X線回折格子の測定
生成された結晶形態の補強証拠を提供するために、適切な品質のいくつかの結晶の格子パラメーターを、単結晶X線回折を使用して求めた。結晶単位格子パラメーターを、Mo放射を備え、結晶が油を含むガラス繊維上に載せられ、260Kに保たれるKappa CCD回折計を使用して求めた。表13に要約されたとおりの形態1および形態2のパラメーターを求めた。
【0174】
【表13】
単結晶X線回折単位格子の結果を含む、式(I)の化合物のベシル酸塩についての溶媒/共溶媒および溶媒/貧溶媒条件からの結晶化の結果を表14に記載する。
【0175】
【表14−1】
【0176】
【表14−2】
完全な単結晶X線回折結晶構造測定に適する品質の様々な結晶を達成し、形態1および2のための完全な構造を得た。これらの結晶構造が実施例9および10で報告される。
【0177】
(実施例9)
形態1の結晶構造
針晶癖を有する、2−メトキシエタノール:酢酸ペンチル溶液から成長させた式(I)の化合物のベシル酸塩の結晶の画像を図17に示した。
【0178】
単一の針晶癖結晶(サイズが約0.8×0.04×0.02mm)を選択し、その格子パラメーターを260Kで、次いで190Kで測定した。260〜190Kの温度に低下させることで変換は観測されなかった。本発明で分析された構造は190Kにおけるデータについてであり、結晶のパラメーターおよびX線回折の精密化を表15に示す。
【0179】
【表15−1】
【0180】
【表15−2】
非対称単位の内容が図18に示されている。それは、2種の独立した化合物の分子および2種の独立したベシル酸塩対イオンから成る。それぞれの化合物は、プロトン化されたイミダゾール−窒素を有する。
【0181】
フラック「エナンチオポール」パラメーター(Flack“Enantiopole”parameter)は、0.03(1)と測定され、したがって、本明細書に記載された構造の立体化学は、十分に確立されており、上記化合物のために意図された立体化学と一致する。
【0182】
【化3】
結晶学的座標および他の関連データを、表17にSHELXファイルの形態で記載している。
【0183】
配座障害は、図19に示された原子位置の「熱振動楕円体(thermal ellipsoid)」によって表し得る(最初の近似において)。障害の主要な領域は、メチル基およびベシル酸塩にあることがわかる。
【0184】
2つの独立した分子の間の差異は、主として図20に見られるエステル鎖に由来する。1つの分子は、イミダゾール環と同一平面上にあるエステル鎖を有するが、もう1つの分子は、直交するエステル鎖を有する。
【0185】
これらのエステル鎖の立体配座は、形態2の立体配座と異なる(図21)。形態1で観測される直交する立体配座は、形態2に見出される立体配座と最大の類似性を有する。
【0186】
2種の独立したベシル酸塩は、ねじれ形立体配座を有する(図22)。結合の長さの実質的な差異は明らかではない。
【0187】
あるベシル酸塩は、形態2のベシル酸塩について観測される立体配座をとる(図23)。
【0188】
結晶軸a、bおよびcに沿って見た分割された結晶構造を、それぞれ図24a、bおよびcに示す。図25は、結晶充てんにおいて観測される最短のコンタクト(contact)を要約している。
【0189】
それぞれの化合物は、2つの独立したベシル酸塩と相互に作用する。特に、短い距離(水素結合タイプ)は、1種のベシル酸塩の1個の酸素原子および化合物のイミダゾール環のプロトン化窒素の間で確立される。第二の独立した化合物は、同様に、但し第二の独立したベシル酸塩と相互作用する。
【0190】
他の近接したコンタクト(C−O、H−O)は、主としてイミダゾールおよびピリジル環の近くで化合物およびベシル酸塩の間で観測される。いくつかの近接したコンタクトは、2つの化合物同士の間(Br−N、C−C、O−H)および2つのベシル酸塩同士の間(O−Hコンタクト)でも観測されるが、後者の程度は低い。
【0191】
実験で求めた結晶構造を使用して、形態1についての粉末回折パターンを、CrystalDiffract(登録商標)(CrystalDiffractはCrystalMaker Ltdの登録商標である)を使用して算出し、図26に示している。この粉末パターンは、形態1について報告された実験的粉末パターンに一致する。
【0192】
(実施例10)
形態2の結晶構造
板状状晶癖を有する式(I)の化合物のベシル酸塩形態2の画像を図27に示している。
【0193】
単一板状状晶癖結晶(サイズ約0.7×0.30×0.25mm)を選択し、その格子パラメーターを260Kで、次いで190Kで求めた。260〜190Kの間の温度への低下で変換は観測されなかった。本発明で分析された構造は、190Kにおけるデータについてであり、結晶のパラメーターおよびX線回折の精密化は表16に示されている。
【0194】
【表16−1】
【0195】
【表16−2】
非対称単位の内容が図28に示されている。これは、1種の独立した化合物の分子および1種の独立したベシル酸塩から成る。化合物は、プロトン化されたイミダゾール−窒素を有する。
【0196】
フラック「エナンチオポール」パラメーターは、0.011(9)と測定され、したがって、本明細書に記載された構造の立体化学は、十分に確立されており、上記化合物のために意図された立体化学と一致する。結晶学的座標および他の関連データを、表18にSHELXファイルの形態で記載している。
【0197】
配座障害は、図29に示された原子位置の「熱振動楕円体」によって表し得る(最初の近似において)。障害の主要な領域は、ベシル酸塩にあることがわかる。
【0198】
上記に記載のとおり、図30に示された形態2のエステル鎖の立体配座は、形態1でとられる立体配座と異なる。
【0199】
しかし、ベシル酸塩の立体配座は、形態1のベシル酸塩の1つについて観測された立体配座と類似している(図31)。
【0200】
結晶軸a、bおよびcに沿って見た分割された結晶構造を、それぞれ図32a、bおよびcに示し、図33は、結晶充てんにおいて観測される最短のコンタクトを要約している。化合物は、イミダゾール環のそのプロトン化された窒素を介してベシル酸塩の1個の酸素原子とショートコンタクト(short contact)(水素結合タイプ)を確立する。他のショートコンタクト(C−C、C−O、H−O)は、イミダゾール環を介して化合物およびベシル酸塩の間で観測される。
【0201】
いくつかの近接したコンタクトは、2つの化合物同士の間(Br−C、C−C、O−C、O−H)でも観測され、この多くは、エステル鎖を介する。ベシル酸塩同士の間には近接したコンタクトはない。
【0202】
実験で求めた結晶構造を使用して、形態2についての粉末回折パターンを、CrystalDiffract(登録商標)を使用して算出した(図34)。この粉末パターンは、形態2について報告された実験的粉末パターンに一致する。
【0203】
表17.式(I)の化合物のベシル酸塩形態1についてのSHELXファイルの形態にまとめた結晶学的座標および他の関連データ。
【0204】
【表17−1】
【0205】
【表17−2】
【0206】
【表17−3】
【0207】
【表17−4】
【0208】
【表17−5】
【0209】
【表17−6】
【0210】
【表17−7】
表18.式(I)の化合物のベシル酸塩形態2についてのSHELXファイルの形態にまとめた結晶学的座標および他の関連データ。
【0211】
【表18−1】
【0212】
【表18−2】
【0213】
【表18−3】
【0214】
【表18−4】
表19.式(I)の化合物のベシル酸塩形態1の結合の長さ。
【0215】
【表19−1】
【0216】
【表19−2】
表20.式(I)の化合物のベシル酸塩形態1の角度。
【0217】
【表20−1】
【0218】
【表20−2】
【0219】
【表20−3】
表21.式(I)の化合物のベシル酸塩形態2の結合の長さ。
【0220】
【表21】
表22.式(I)の化合物のベシル酸塩形態2の角度。
【0221】
【表22−1】
【0222】
【表22−2】
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
【図1】

【図2】
【図3】

【図4】
【図5】

【図6】

【図7】

【図8】
【図9】
【図10】

【図11A】

【図11B】
【図11C】

【図11D】
【図11E】
【図11F】
【図11G】
【図11H】
【図11I】

【図12A】
【図12B】
【図12C】
【図12D】

【図13A】

【図13B】

【図13C】
【図13D】
【図13E】
【図13F】

【図13G】
【図14A】
【図14B】

【図14C】
【図15A】

【図15B】

【図15C】
【図15D】

【図15E】
【図15F】

【図16A】
【図16B】
【図16C】
【図16D】
【図17】

【図18】
【図19】

【図20】
【図21】

【図22】
【図23】
【図24−1】
【図24−2】

【図25】
【図26】

【図27】

【図28】

【図29】

【図30】
【図31】
【図32−1】

【図32−2】

【図32−3】

【図33】
【図34】
【図35】

【図36】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11A】
【図11B】
【図11C】
【図11D】
【図11E】
【図11F】
【図11G】
【図11H】
【図11I】
【図12A】
【図12B】
【図12C】
【図12D】
【図13A】
【図13B】
【図13C】
【図13D】
【図13E】
【図13F】
【図13G】
【図14A】
【図14B】
【図14C】
【図15A】
【図15B】
【図15C】
【図15D】
【図15E】
【図15F】
【図16A】
【図16B】
【図16C】
【図16D】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24−1】
【図24−2】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32−1】
【図32−2】
【図32−3】
【図33】
【図34】
【図35】
【図36】
【公開番号】特開2013−49690(P2013−49690A)
【公開日】平成25年3月14日(2013.3.14)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−235328(P2012−235328)
【出願日】平成24年10月25日(2012.10.25)
【分割の表示】特願2009−518952(P2009−518952)の分割
【原出願日】平成19年7月10日(2007.7.10)
【出願人】(509011824)パイオン ユーケー リミテッド (5)
【Fターム(参考)】
【公開日】平成25年3月14日(2013.3.14)
【国際特許分類】
【出願番号】特願2012−235328(P2012−235328)
【出願日】平成24年10月25日(2012.10.25)
【分割の表示】特願2009−518952(P2009−518952)の分割
【原出願日】平成19年7月10日(2007.7.10)
【出願人】(509011824)パイオン ユーケー リミテッド (5)
【Fターム(参考)】
[ Back to top ]