
硫黄含有化合物及びその製造方法
【課題】高い屈折率を有し、レンズ等の光学部材用の樹脂を形成しうる単量体として用いることができる化合物を提供する。
【解決手段】下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を有する硫黄含有化合物。その好ましい例は、下記の一般式(1)で示される。

(式中、Rは水素原子又はメチル基であり、k、l、m、nは各々独立に0〜2の整数であり、k+l=1又は2、m+n=1又は2であり、p、qは各々独立に0〜2の整数である。)
【解決手段】下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を有する硫黄含有化合物。その好ましい例は、下記の一般式(1)で示される。
(式中、Rは水素原子又はメチル基であり、k、l、m、nは各々独立に0〜2の整数であり、k+l=1又は2、m+n=1又は2であり、p、qは各々独立に0〜2の整数である。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、レンズ等の光学部品の原料である単量体として用いるための硫黄含有化合物及びその製造方法に関する。
【背景技術】
【0002】
プラスチックレンズは、ガラス等の無機材料からなるレンズに比べて、軽量で割れ難いなどの特長を有するため、眼鏡レンズ、カメラレンズ等の用途に広く用いられている。
近年、眼鏡レンズの中心厚が小さくなる傾向があるなどの事情の下、プラスチックレンズの材料として、より高い屈折率を有する光学用樹脂が望まれている。
このような高い屈折率を有する光学用樹脂(重合体)を得るための単量体として、硫黄原子を含有する化合物(特許文献1〜2)や、芳香環を有するウレタン(メタ)アクリレート(特許文献3)が知られている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2007−211021号公報
【特許文献2】特開平8−325337号公報
【特許文献3】特開平5−255464号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
従来、高い屈折率を有する放射線硬化性化合物を得るためには、複雑な工程が必要であった。
また、前記の特許文献3に記載されているような、芳香環を有する単量体を用いる場合、芳香環が嵩高いため、高屈折率化が困難であった。
本発明は、高い屈折率を有し、レンズ等の光学部材を形成するための単量体として用いうる化合物であって、少ない工程数で製造することのできる化合物、及び、その製造方法を提供することを目的とする。
【課題を解決するための手段】
【0005】
本発明者は、上記課題を解決するために鋭意検討した結果、特定の化学構造を有する硫黄含有化合物によれば、前記の課題を解決しうることを見出し、本発明を完成した。
すなわち、本発明は、以下の[1]〜[6]を提供するものである。
[1] 下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を有する硫黄含有化合物。
【化1】
[2] 下記の一般式(1)で表される前記[1]に記載の硫黄含有化合物。
【化2】
(式中、Rは水素原子又はメチル基であり、k、l、m、nは各々独立に0〜2の整数であり、k+l=1又は2、m+n=1又は2であり、p、qは各々独立に0〜2の整数である。)
[3] 屈折率nDが1.60以上である前記[1]又は[2]に記載の硫黄含有化合物。
[4] 両端にチオールを有し、かつ分岐鎖として(メタ)アクリロイル基を導入可能な官能基を有する化合物と、テトラメチルチオメタンを反応させて、上記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を導入可能な上記官能基を有する化合物を得た後、該官能基に対して(メタ)アクリロイル基を導入する、前記[1]〜[3]のいずれかに記載の硫黄含有化合物の製造方法。
[5] 前記[1]〜[3]のいずれかに記載の硫黄含有化合物を含む組成物を重合させてなる硬化体。
[6] 前記[5]に記載の硬化体からなる光学部材。
【発明の効果】
【0006】
本発明の化合物は、下記式(2)で示される構造を有するスピロ骨格を備えているため、例えば屈折率nDが1.60以上の高い屈折率を有する。
【化3】
本発明の化合物は、光学部材の材料である単量体として用いることができる。具体的には、レンズ(例えば、眼鏡レンズ、カメラレンズ等)の材料、ナノインプリント用材料、光学接着剤等の用途に用いることができる。
本発明の化合物の製造方法によれば、目的とする化合物を少ない工程数で短時間に容易に得ることができる。
【図面の簡単な説明】
【0007】
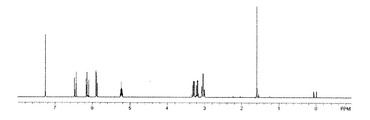
【図1】実施例1で得られた化合物の1H NMRスペクトルを示す図である。
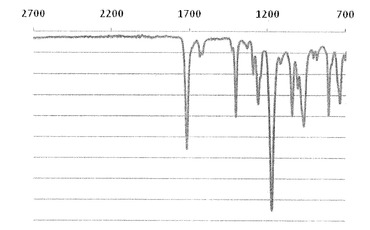
【図2】実施例1で得られた化合物のIRスペクトルを示す図である。
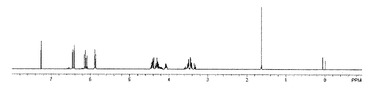
【図3】実施例2で得られた化合物の1H NMRスペクトルを示す図である。
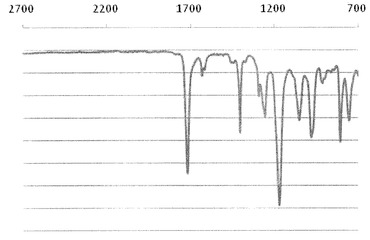
【図4】実施例2で得られた化合物のIRスペクトルを示す図である。
【発明を実施するための形態】
【0008】
本発明の化合物は、下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を有する硫黄含有化合物である。
【化4】
本発明の化合物の好ましい一例として、下記の一般式(1)で表されるものが挙げられる。
【化5】
(式中、Rは水素原子又はメチル基であり、k、l、m、nは各々独立に0〜2の整数であり、k+l=1又は2、m+n=1又は2であり、p、qは各々独立に0〜2の整数である。)
【0009】
本発明の化合物の製造方法の一例としては、両端にチオールを有し、かつ分岐鎖として(メタ)アクリロイル基を導入可能な官能基を有する化合物と、テトラメチルチオメタンを反応させて、下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を導入可能な上記官能基を有する化合物を得た後、該官能基に対して(メタ)アクリロイル基を導入する方法が挙げられる。
【化6】
この方法の一例を下記の合成フロー1、他の例を下記の合成フロー2に示す。なお、下記の合成フロー1〜2中、符号1で示す化合物は、以下、「化合物1」と称する。同様に、符号2〜12で示す化合物は、各々「化合物2」〜「化合物12」と称する。
【0010】
[合成フロー1]
【化7】
【0011】
前記の合成フロー1中、まず、1,3−ジブロモ−2−プロパノールを、溶媒(例えば、塩化メチレン)に溶解させた状態で、触媒(例えばトリエチルアミン)の存在下に、ベンゾイルクロライドと反応させて、化合物1を生成させる。
ベンゾイルクロライドの量は、1,3−ジブロモ−2−プロパノールに対して当量比で好ましくは1.0以上、より好ましくは1.0〜1.2である。トリエチルアミンの量は、1,3−ジブロモ−2−プロパノールに対して当量比で、好ましくは1.0〜1.2である。反応条件は、1,3−ジブロモ−2−プロパノールの濃度が好ましくは0.1〜2M、温度が好ましくは0〜60℃、より好ましくは10〜40℃、反応時間が好ましくは0.5時間以上、より好ましくは3時間以上である。
【0012】
次に、化合物1を、溶媒(例えば、N,N−ジメチルホルムアミド;DMF)に溶解させた状態で、チオ酢酸カリウム(KSAc)と反応させて、化合物2を生成させる。
チオ酢酸カリウムの量は、化合物1に対して当量比で、好ましくは2.0以上、好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物1の濃度が好ましくは0.5M以下、温度が好ましくは10〜90℃、より好ましくは50〜70℃(溶媒がDMFである場合)、反応時間が好ましくは0.5時間以上、より好ましくは3時間以上である。なお、溶媒としては、N,N−ジメチルホルムアミド(DMF)に代えて、テトラヒドロフラン(THF)、ジメチルスルホキシド(DMSO)等を用いてもよい。
【0013】
次に、化合物2を、溶媒(例えば、クロロホルム及びメタノール)に溶解させた状態で、濃塩酸と反応させて、化合物3を生成させる。
塩酸の量は、化合物2に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物2の濃度が好ましくは0.1〜1.0M、温度が好ましくは10〜90℃、より好ましくは50〜70℃、反応時間が好ましくは10時間以上、より好ましくは20時間以上である。
【0014】
次に、化合物3を、溶媒(例えば、トルエン)に溶解させた状態で、触媒であるH+(例えば、p−トルエンスルホン酸)の存在下に、テトラメチルチオメタンと反応させて、化合物4を生成させる。
テトラメチルチオメタンの量は、化合物3に対して当量比で、好ましくは0.50以上、より好ましくは0.50〜0.55である。
p−トルエンスルホン酸の量は、化合物3に対して当量比で、好ましくは0.01〜0.10である。反応条件は、反応時の反応液中の化合物3の濃度が好ましくは0.3〜0.5M、温度が好ましくは60〜120℃、より好ましくは80〜120℃、反応時間が好ましくは10時間以上、より好ましくは15時間以上、液性が酸性(好ましくはpH4以下)である。
【0015】
次に、化合物4を、溶媒(例えば、クロロホルム及びメタノール)に溶解させた状態で、炭酸カリウムと反応させて、化合物5を生成させる。
炭酸カリウムの量は、化合物4に対して当量比で、好ましくは3以上、より好ましくは6以上である。反応条件は、反応時の反応液中の化合物4の濃度が好ましくは0.1〜1.0M、温度が好ましくは0〜60℃、より好ましくは5〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
【0016】
次に、化合物5を、溶媒(例えば、塩化メチレン)に溶解させた状態で、触媒(例えば、トリエチルアミン)の存在下に、アクリル酸クロライドと反応させて、化合物6を生成させる。
アクリル酸クロライドの量は、化合物5に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。トリエチルアミンの量は、化合物5に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物5の濃度が好ましくは0.1〜1.0M、温度が好ましくは0〜60℃、より好ましくは5〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
【0017】
[合成フロー2]
【化8】
【0018】
前記の合成フロー2中、まず、1,2−ジブロモ−3−プロパノールを、溶媒(例えば、塩化メチレン)に溶解させた状態で、触媒(例えば、トリエチルアミン)の存在下に、ベンゾイルクロライドと反応させて、化合物7を生成させる。
ベンゾイルクロライドの量は、1,2−ジブロモ−3−プロパノールに対して当量比で、好ましくは1.0以上、より好ましくは1.0〜1.2である。トリエチルアミンの量は、1,2−ジブロモ−3−プロパノールに対して当量比で、好ましくは1.0〜1.2である。反応条件は、1,2−ジブロモ−3−プロパノールの濃度が好ましくは0.1〜2M、温度が好ましくは0〜60℃、より好ましくは15〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
なお、前記の合成フロー2中、室温を「r.t.」、出発物質(化合物7)を「S.M.」、反応時間が6時間であることを「6h」と記載している。
【0019】
次に、化合物7を、溶媒(例えば、N,N−ジメチルホルムアミド;DMF)に溶解させた状態で、触媒(例えば水素化ナトリウム)の存在下にチオ酢酸と反応させて、化合物8を生成させる。
チオ酢酸の量は、化合物7に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。水素化ナトリウムの量は、化合物7に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物7の濃度が好ましくは0.2〜1.0M、温度が好ましくは10〜90℃、より好ましくは50〜70℃(溶媒がDMFである場合)、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。なお、溶媒としては、N,N−ジメチルホルムアミド(DMF)に代えて、テトラヒドロフラン(THF)、ジメチルスルホキシド(DMSO)等を用いてもよい。
【0020】
次に、化合物8を、溶媒(例えば、クロロホルム及びメタノール)に溶解させた状態で、濃塩酸と反応させて、化合物9を生成させる。
塩酸の量は、化合物8に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物8の濃度が好ましくは0.1〜1.0M、温度が好ましくは10〜90℃、より好ましくは50〜70℃、反応時間が好ましくは10時間以上、より好ましくは20時間以上である。
【0021】
次に、化合物9を、溶媒(例えば、トルエン)に溶解させた状態で、触媒であるH+(例えば、p−トルエンスルホン酸)の存在下に、テトラメチルチオメタンと反応させて、化合物10を生成させる。
テトラメチルチオメタンの量は、化合物9に対して当量比で、好ましくは0.50以上、より好ましくは0.50〜0.55である。p−トルエンスルホン酸の量は、化合物9に対して当量比で、好ましくは0.01〜0.10である。反応条件は、反応時の反応液中の化合物9の濃度が好ましくは0.3〜0.5M、温度が好ましくは60〜120℃、より好ましくは80〜120℃、反応時間が好ましくは10時間以上、より好ましくは15時間以上である。
【0022】
次に、化合物10を、溶媒(例えば、クロロホルム及びメタノール)に溶解させた状態で、炭酸カリウムと反応させて、化合物11を生成させる。
炭酸カリウムの量は、化合物10に対して当量比で、好ましくは3以上、より好ましくは4以上である。反応条件は、反応時の反応液中の化合物10の濃度が好ましくは0.1〜1.0M、温度が好ましくは0〜60℃、より好ましくは5〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
【0023】
次に、化合物11を、溶媒(例えば、塩化メチレン)に溶解させた状態で、触媒(例えば、トリエチルアミン)の存在下に、アクリル酸クロライドと反応させて、化合物12を生成させる。
アクリル酸クロライドの量は、化合物11に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜4.0である。トリエチルアミンの量は、化合物11に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜3.0である。反応条件は、反応時の反応液中の化合物11の濃度が好ましくは0.1〜1.0M、温度が好ましくは0〜60℃、より好ましくは5〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
【0024】
前記合成フロー1、2において、化合物1、7の合成時に用いるジブロモプロパノールに代えて、他のジハロゲン化アルコール化合物を用いることができる。
他のジハロゲン化アルコール化合物の例としては、1,3−ジヨウ化−2−プロパノール等が挙げられる。
【0025】
本発明の化合物の屈折率nD(液屈折率)は、好ましくは1.60以上である。
本発明の化合物を含む組成物は、例えば、本発明の化合物と、他の化合物(例えば、本発明の化合物に該当しない(メタ)アクリレート化合物)と、光重合開始剤とを含む。
他の化合物の例として、ペンタエリスリトールテトラアクリレート、ビスフェノールA−EO変性ジアクリレート等が挙げられる。
光重合開始剤の例として、1−ヒドロキシシクロヘキシルフェニルケトン、2−メチル−1−[4−(メチルチオ)フェニル]−2−モルホリノ−プロパン−1−オン等が挙げられる。
各成分の配合割合は、例えば、本発明の化合物が50〜99質量%、他の化合物が0〜45質量%、光重合開始剤が1〜10質量%とすることができる。
【0026】
本発明の化合物を含む組成物を重合させてなる硬化体の屈折率nDは、好ましくは
1.62以上である。
【実施例】
【0027】
[テトラメチルチオメタンの合成方法]
本発明の化合物の製造に用いられる、テトラメチルチオメタンの合成方法の一例を下記の合成フロー3に示す。なお、下記合成フロー3中、符号13で示す化合物は、以下、「化合物13」と称する。
[合成フロー3]
【化9】
【0028】
500mLの2口フラスコに、チオウレア(50g、657mmol)、蒸留水23mLを加えた。次いで、氷浴にて0℃に冷却し、滴下漏斗を用いて硫酸ジメチル(45g、373mmol)を30分以上かけて滴下した。その後、130℃にて1時間攪拌し、生成した析出物をエタノールで洗浄後、濾別、乾燥することで化合物13(59g、657mmol)を得た。
【0029】
500mLの2口フラスコに、化合物13(35g、380mmol)、1N塩酸水14mL、及び蒸留水90mLを加えた。次いで、氷浴にて0℃に冷却し、ジエチルエーテル21mL、亜硝酸ナトリウム(35g、507mmol)を加えて、二酸化窒素の発生が止まるまで攪拌した。さらに、蒸留水100mLと、化合物13(28g、311mmol)を加えて、室温にて30分攪拌した。次いで、25%アンモニア水50mLを加えて、室温にて30分攪拌した。その後、窒素の発生が治まるのを確認してから、50℃にて1時間攪拌した。反応終了後、反応混合物に酢酸エチルを加えて有機層を抽出した。得られた有機層に硫酸マグネシウムを加えて乾燥、濾過し、溶剤を除去した後、残留物を減圧蒸留することでテトラメチルチオメタンを得た(5g、25mmol)。
【0030】
[実施例1]
前記の合成フロー1に従って、1,3−ジブロモ−2−プロパノールを出発物質にして、目的とする硫黄含有化合物(合成フロー1中の化合物6)を得た。
[化合物1の合成方法]
200mLの2口フラスコに90mLの塩化メチレン、1,3−ジブロモ−2−プロパノール(5g、22.9mmol)、及びトリエチルアミン(2.58g、25.5mmol)を加えた。次いで、氷浴にて0℃に冷却し、ベンゾイルクロライド(3.58g、25.5mmol)を滴下した。混合物を徐々に室温に戻した後、6時間室温(15〜25℃である。以下、同じ。)にて攪拌した。反応終了後、飽和重層水を加えて30分攪拌し、溶媒を真空下で除去した。次いで、反応混合物に蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層を飽和食塩水で洗浄した後、硫酸マグネシウムを加えて、乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=1/1)することにより化合物1を得た(無色透明液体、収率96%、7.08g、22.0mmol)。
【0031】
[化合物2の合成方法]
200mLの2口フラスコに50mLのDMF、化合物1(3.65g、11.3mmol)を加えた。次いで、チオ酢酸カリウム(2.84g、24.9mmol)を加えて、60℃にて6時間攪拌した。反応終了後、反応混合物に蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層を1N塩酸水、飽和食塩水で洗浄した後、硫酸マグネシウムを加えて、乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/酢酸エチルの体積比=100/1)することにより化合物2を得た(黄色液体、収率95%、3.35g、10.7mmol)。
【0032】
[化合物3の合成方法]
200mLのフラスコに20mLのクロロホルム、化合物2(3.17g、10.2mmol)、4mLの濃塩酸、及び40mLのメタノールを加えて、60℃にて24時間攪拌した。反応終了後、反応混合物に蒸留水及びクロロホルムを加えて有機層を抽出した。得られた有機層を飽和食塩水にて洗浄した後、硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/酢酸エチルの体積比=100/1)することにより化合物3を得た(黄色液体、収率85%、1.96g、8.6mmol)。
【0033】
[化合物4の合成方法]
100mLの2口フラスコにテトラメチルチオメタン(0.37g、1.86mmol)、9.3mLのトルエン、化合物3(0.85g、3.71mmol)、及びp−トルエンスルホン酸(0.032g、0.19mmol)を加えて、100℃にて16時間攪拌した。反応終了後、真空下で反応混合物から溶剤を除去したのち、残留物をカラムクロマトグラフィーにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=1/1)することにより化合物4を得た(黄色液体、収率65%、0.28g、0.60mmol)。
【0034】
[化合物5の合成]
200mLの2口フラスコに化合物4(0.48g、1.0mmol)、7mLのクロロホルム、7mLのメタノール、及び炭酸カリウム(0.85g、6.1mmol)を加えて、室温にて6時間攪拌した。反応終了後、反応混合物に蒸留水及びクロロホルムを加えて有機層を抽出した。さらに、水層に1N塩酸を加えて酸性にした後、クロロホルムで有機層の再抽出を行った。得られた有機層に硫酸マグネシウムを加えて、乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;酢酸エチル/クロロホルムの体積比=1/1)することにより化合物5を得た(無色透明粘調性液体、収率75%、0.2g、0.75mmol)。
【0035】
[化合物6の合成]
100mLの2口フラスコに塩化メチレン10mL、化合物5(0.54g、2.1mmol)、及びトリエチルアミン(0.46g、4.6mmol)を加えた。次いで、氷浴にて冷却し、アクリル酸クロライド(0.38g、4.6mmol)を滴下しながら徐々に室温に戻し、6時間攪拌した。反応終了後、反応混合物に飽和重層水を加えて30分攪拌した後、真空下で溶剤を除去した。その後、蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層に硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=6/1)することにより化合物6を得た(淡白色液体、収率52%、0.39g、1.09mmol)。
【0036】
[化合物6の物性評価]
(1)液屈折率の測定
シリコンウエハー上に化合物6を塗布したのち、n&k Technology社製のアッベ屈折計「n&k Analyzer 1510」を用いて、液屈折率の測定を行った。その結果、化合物の波長589nmにおける液屈折率は、1.621、アッベ数は25だった。
【0037】
[実施例2]
前記の合成フロー2に従って、1,2−ジブロモ−3−プロパノールを出発物質にして、目的とする硫黄含有化合物(合成フロー2中の化合物12)を得た。
[化合物7の合成方法]
200mLの2口フラスコに90mLの塩化メチレン、1,2−ジブロモ−3−プロパノール(5g、22.9mmol)、及びトリエチルアミン(2.58g、22.5mmol)を加えた。次いで、氷浴にて0℃に冷却し、ベンゾイルクロライド(3.58g、25.5mmol)を滴下した。混合物を徐々に室温に戻した後、6時間室温にて攪拌した。反応終了後、飽和重層水を加えて30分攪拌し、溶媒を真空下で除去した。次いで、反応混合物に蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層を飽和食塩水で洗浄した後、硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=1/1)することにより化合物7を得た(無色透明液体、収率87%)。
【0038】
[化合物8の合成方法]
窒素雰囲気下にて200mLの2口フラスコに、水素化ナトリウム(1.03g、43mmol)、脱水DMF(100mL)、を加えた。次いで、氷浴にて0℃に冷却し、チオ酢酸(3.4g、45mmol)を滴下した。その後、氷浴を外して、室温にて30分攪拌した。その後、10mLのDMFに溶解させた化合物7(6.28g、20mmol)を反応混合物中に滴下し、60℃にて6時間攪拌した。反応終了後、反応混合物に蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層を1N塩酸水、及び飽和食塩水で洗浄した後、硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;ヘキサン/酢酸エチルの体積比=6/1)することにより化合物8を得た(黄色液体、収率77%、4.67g、15mmol)。
【0039】
[化合物9の合成方法]
200mLのフラスコに、15mLのクロロホルム、化合物8(2.03g、6.47mmol)、3mLの濃塩酸、及び30mLのメタノールを加えて、60℃にて24時間攪拌した。反応終了後、反応混合物に蒸留水及びクロロホルムを加えて有機層を抽出した。得られた有機層を飽和食塩水にて洗浄した後、硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/酢酸エチルの体積比=100/1)することにより化合物9を得た(黄色液体、収率87%、1.29g、5.66mmol)。
【0040】
[化合物10の合成方法]
100mLの2口フラスコに、テトラメチルチオメタン(0.57g、2.83mmol)、14mLのトルエン、化合物9(1.29g、5.66mmol)、及びp−トルエンスルホン酸(0.049g、0.28mmol)を加えて、100℃で16時間攪拌した。反応終了後、真空下で反応混合物から溶剤を除去したのち、残留物をカラムクロマトグラフィーにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=1/1)することにより化合物10を得た(黄色液体、収率72%、0.94g、2.03mmol)。
【0041】
[化合物11の合成方法]
200mLの2口フラスコに、化合物10(2.32g、5mmol)、30mLのクロロホルム、30mLのメタノール、及び炭酸カリウム(2.77g、20mmol)を加えて、室温にて6時間攪拌した。反応終了後、反応混合物に蒸留水及びクロロホルムを加えて有機層を抽出した。さらに水層に1N塩酸を加えて酸性にした後、クロロホルムで有機層の再抽出を行った。得られた有機層に硫酸マグネシウムを加えて、乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;酢酸エチル/クロロホルムの体積比=1/1)することにより化合物11を得た(薄黄色液体、収率61%、0.79g、3.07mmol)。
【0042】
[化合物12の合成方法]
100mLの2口フラスコに、塩化メチレン15mL、化合物11(0.79g、3.07mmol)、及びトリエチルアミン(0.78g、7.68mmol)を加えた。次いで、氷浴にて0℃に冷却し、アクリル酸クロライド(0.83g、9.21mmol)を
滴下しながら徐々に室温に戻し、6時間室温にて攪拌した。反応終了後、反応混合物に飽和重層水を加えて30分攪拌後、真空下で溶剤を除去した。その後、蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層に硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=6/1)することにより化合物12を得た(黄色液体、収率67%、0.75g、2.05mmol)。
【0043】
[化合物12の物性評価]
(1)液屈折率の測定
実施例1と同様にして、液屈折率の測定を行った。その結果、化合物の波長589nmにおける液屈折率は、1.610、アッベ数は36だった。
【技術分野】
【0001】
本発明は、レンズ等の光学部品の原料である単量体として用いるための硫黄含有化合物及びその製造方法に関する。
【背景技術】
【0002】
プラスチックレンズは、ガラス等の無機材料からなるレンズに比べて、軽量で割れ難いなどの特長を有するため、眼鏡レンズ、カメラレンズ等の用途に広く用いられている。
近年、眼鏡レンズの中心厚が小さくなる傾向があるなどの事情の下、プラスチックレンズの材料として、より高い屈折率を有する光学用樹脂が望まれている。
このような高い屈折率を有する光学用樹脂(重合体)を得るための単量体として、硫黄原子を含有する化合物(特許文献1〜2)や、芳香環を有するウレタン(メタ)アクリレート(特許文献3)が知られている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開2007−211021号公報
【特許文献2】特開平8−325337号公報
【特許文献3】特開平5−255464号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
従来、高い屈折率を有する放射線硬化性化合物を得るためには、複雑な工程が必要であった。
また、前記の特許文献3に記載されているような、芳香環を有する単量体を用いる場合、芳香環が嵩高いため、高屈折率化が困難であった。
本発明は、高い屈折率を有し、レンズ等の光学部材を形成するための単量体として用いうる化合物であって、少ない工程数で製造することのできる化合物、及び、その製造方法を提供することを目的とする。
【課題を解決するための手段】
【0005】
本発明者は、上記課題を解決するために鋭意検討した結果、特定の化学構造を有する硫黄含有化合物によれば、前記の課題を解決しうることを見出し、本発明を完成した。
すなわち、本発明は、以下の[1]〜[6]を提供するものである。
[1] 下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を有する硫黄含有化合物。
【化1】
[2] 下記の一般式(1)で表される前記[1]に記載の硫黄含有化合物。
【化2】
(式中、Rは水素原子又はメチル基であり、k、l、m、nは各々独立に0〜2の整数であり、k+l=1又は2、m+n=1又は2であり、p、qは各々独立に0〜2の整数である。)
[3] 屈折率nDが1.60以上である前記[1]又は[2]に記載の硫黄含有化合物。
[4] 両端にチオールを有し、かつ分岐鎖として(メタ)アクリロイル基を導入可能な官能基を有する化合物と、テトラメチルチオメタンを反応させて、上記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を導入可能な上記官能基を有する化合物を得た後、該官能基に対して(メタ)アクリロイル基を導入する、前記[1]〜[3]のいずれかに記載の硫黄含有化合物の製造方法。
[5] 前記[1]〜[3]のいずれかに記載の硫黄含有化合物を含む組成物を重合させてなる硬化体。
[6] 前記[5]に記載の硬化体からなる光学部材。
【発明の効果】
【0006】
本発明の化合物は、下記式(2)で示される構造を有するスピロ骨格を備えているため、例えば屈折率nDが1.60以上の高い屈折率を有する。
【化3】
本発明の化合物は、光学部材の材料である単量体として用いることができる。具体的には、レンズ(例えば、眼鏡レンズ、カメラレンズ等)の材料、ナノインプリント用材料、光学接着剤等の用途に用いることができる。
本発明の化合物の製造方法によれば、目的とする化合物を少ない工程数で短時間に容易に得ることができる。
【図面の簡単な説明】
【0007】
【図1】実施例1で得られた化合物の1H NMRスペクトルを示す図である。
【図2】実施例1で得られた化合物のIRスペクトルを示す図である。
【図3】実施例2で得られた化合物の1H NMRスペクトルを示す図である。
【図4】実施例2で得られた化合物のIRスペクトルを示す図である。
【発明を実施するための形態】
【0008】
本発明の化合物は、下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を有する硫黄含有化合物である。
【化4】
本発明の化合物の好ましい一例として、下記の一般式(1)で表されるものが挙げられる。
【化5】
(式中、Rは水素原子又はメチル基であり、k、l、m、nは各々独立に0〜2の整数であり、k+l=1又は2、m+n=1又は2であり、p、qは各々独立に0〜2の整数である。)
【0009】
本発明の化合物の製造方法の一例としては、両端にチオールを有し、かつ分岐鎖として(メタ)アクリロイル基を導入可能な官能基を有する化合物と、テトラメチルチオメタンを反応させて、下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を導入可能な上記官能基を有する化合物を得た後、該官能基に対して(メタ)アクリロイル基を導入する方法が挙げられる。
【化6】
この方法の一例を下記の合成フロー1、他の例を下記の合成フロー2に示す。なお、下記の合成フロー1〜2中、符号1で示す化合物は、以下、「化合物1」と称する。同様に、符号2〜12で示す化合物は、各々「化合物2」〜「化合物12」と称する。
【0010】
[合成フロー1]
【化7】
【0011】
前記の合成フロー1中、まず、1,3−ジブロモ−2−プロパノールを、溶媒(例えば、塩化メチレン)に溶解させた状態で、触媒(例えばトリエチルアミン)の存在下に、ベンゾイルクロライドと反応させて、化合物1を生成させる。
ベンゾイルクロライドの量は、1,3−ジブロモ−2−プロパノールに対して当量比で好ましくは1.0以上、より好ましくは1.0〜1.2である。トリエチルアミンの量は、1,3−ジブロモ−2−プロパノールに対して当量比で、好ましくは1.0〜1.2である。反応条件は、1,3−ジブロモ−2−プロパノールの濃度が好ましくは0.1〜2M、温度が好ましくは0〜60℃、より好ましくは10〜40℃、反応時間が好ましくは0.5時間以上、より好ましくは3時間以上である。
【0012】
次に、化合物1を、溶媒(例えば、N,N−ジメチルホルムアミド;DMF)に溶解させた状態で、チオ酢酸カリウム(KSAc)と反応させて、化合物2を生成させる。
チオ酢酸カリウムの量は、化合物1に対して当量比で、好ましくは2.0以上、好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物1の濃度が好ましくは0.5M以下、温度が好ましくは10〜90℃、より好ましくは50〜70℃(溶媒がDMFである場合)、反応時間が好ましくは0.5時間以上、より好ましくは3時間以上である。なお、溶媒としては、N,N−ジメチルホルムアミド(DMF)に代えて、テトラヒドロフラン(THF)、ジメチルスルホキシド(DMSO)等を用いてもよい。
【0013】
次に、化合物2を、溶媒(例えば、クロロホルム及びメタノール)に溶解させた状態で、濃塩酸と反応させて、化合物3を生成させる。
塩酸の量は、化合物2に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物2の濃度が好ましくは0.1〜1.0M、温度が好ましくは10〜90℃、より好ましくは50〜70℃、反応時間が好ましくは10時間以上、より好ましくは20時間以上である。
【0014】
次に、化合物3を、溶媒(例えば、トルエン)に溶解させた状態で、触媒であるH+(例えば、p−トルエンスルホン酸)の存在下に、テトラメチルチオメタンと反応させて、化合物4を生成させる。
テトラメチルチオメタンの量は、化合物3に対して当量比で、好ましくは0.50以上、より好ましくは0.50〜0.55である。
p−トルエンスルホン酸の量は、化合物3に対して当量比で、好ましくは0.01〜0.10である。反応条件は、反応時の反応液中の化合物3の濃度が好ましくは0.3〜0.5M、温度が好ましくは60〜120℃、より好ましくは80〜120℃、反応時間が好ましくは10時間以上、より好ましくは15時間以上、液性が酸性(好ましくはpH4以下)である。
【0015】
次に、化合物4を、溶媒(例えば、クロロホルム及びメタノール)に溶解させた状態で、炭酸カリウムと反応させて、化合物5を生成させる。
炭酸カリウムの量は、化合物4に対して当量比で、好ましくは3以上、より好ましくは6以上である。反応条件は、反応時の反応液中の化合物4の濃度が好ましくは0.1〜1.0M、温度が好ましくは0〜60℃、より好ましくは5〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
【0016】
次に、化合物5を、溶媒(例えば、塩化メチレン)に溶解させた状態で、触媒(例えば、トリエチルアミン)の存在下に、アクリル酸クロライドと反応させて、化合物6を生成させる。
アクリル酸クロライドの量は、化合物5に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。トリエチルアミンの量は、化合物5に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物5の濃度が好ましくは0.1〜1.0M、温度が好ましくは0〜60℃、より好ましくは5〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
【0017】
[合成フロー2]
【化8】
【0018】
前記の合成フロー2中、まず、1,2−ジブロモ−3−プロパノールを、溶媒(例えば、塩化メチレン)に溶解させた状態で、触媒(例えば、トリエチルアミン)の存在下に、ベンゾイルクロライドと反応させて、化合物7を生成させる。
ベンゾイルクロライドの量は、1,2−ジブロモ−3−プロパノールに対して当量比で、好ましくは1.0以上、より好ましくは1.0〜1.2である。トリエチルアミンの量は、1,2−ジブロモ−3−プロパノールに対して当量比で、好ましくは1.0〜1.2である。反応条件は、1,2−ジブロモ−3−プロパノールの濃度が好ましくは0.1〜2M、温度が好ましくは0〜60℃、より好ましくは15〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
なお、前記の合成フロー2中、室温を「r.t.」、出発物質(化合物7)を「S.M.」、反応時間が6時間であることを「6h」と記載している。
【0019】
次に、化合物7を、溶媒(例えば、N,N−ジメチルホルムアミド;DMF)に溶解させた状態で、触媒(例えば水素化ナトリウム)の存在下にチオ酢酸と反応させて、化合物8を生成させる。
チオ酢酸の量は、化合物7に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。水素化ナトリウムの量は、化合物7に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物7の濃度が好ましくは0.2〜1.0M、温度が好ましくは10〜90℃、より好ましくは50〜70℃(溶媒がDMFである場合)、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。なお、溶媒としては、N,N−ジメチルホルムアミド(DMF)に代えて、テトラヒドロフラン(THF)、ジメチルスルホキシド(DMSO)等を用いてもよい。
【0020】
次に、化合物8を、溶媒(例えば、クロロホルム及びメタノール)に溶解させた状態で、濃塩酸と反応させて、化合物9を生成させる。
塩酸の量は、化合物8に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜2.5である。反応条件は、反応時の反応液中の化合物8の濃度が好ましくは0.1〜1.0M、温度が好ましくは10〜90℃、より好ましくは50〜70℃、反応時間が好ましくは10時間以上、より好ましくは20時間以上である。
【0021】
次に、化合物9を、溶媒(例えば、トルエン)に溶解させた状態で、触媒であるH+(例えば、p−トルエンスルホン酸)の存在下に、テトラメチルチオメタンと反応させて、化合物10を生成させる。
テトラメチルチオメタンの量は、化合物9に対して当量比で、好ましくは0.50以上、より好ましくは0.50〜0.55である。p−トルエンスルホン酸の量は、化合物9に対して当量比で、好ましくは0.01〜0.10である。反応条件は、反応時の反応液中の化合物9の濃度が好ましくは0.3〜0.5M、温度が好ましくは60〜120℃、より好ましくは80〜120℃、反応時間が好ましくは10時間以上、より好ましくは15時間以上である。
【0022】
次に、化合物10を、溶媒(例えば、クロロホルム及びメタノール)に溶解させた状態で、炭酸カリウムと反応させて、化合物11を生成させる。
炭酸カリウムの量は、化合物10に対して当量比で、好ましくは3以上、より好ましくは4以上である。反応条件は、反応時の反応液中の化合物10の濃度が好ましくは0.1〜1.0M、温度が好ましくは0〜60℃、より好ましくは5〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
【0023】
次に、化合物11を、溶媒(例えば、塩化メチレン)に溶解させた状態で、触媒(例えば、トリエチルアミン)の存在下に、アクリル酸クロライドと反応させて、化合物12を生成させる。
アクリル酸クロライドの量は、化合物11に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜4.0である。トリエチルアミンの量は、化合物11に対して当量比で、好ましくは2.0以上、より好ましくは2.0〜3.0である。反応条件は、反応時の反応液中の化合物11の濃度が好ましくは0.1〜1.0M、温度が好ましくは0〜60℃、より好ましくは5〜40℃、反応時間が好ましくは3時間以上、より好ましくは5時間以上である。
【0024】
前記合成フロー1、2において、化合物1、7の合成時に用いるジブロモプロパノールに代えて、他のジハロゲン化アルコール化合物を用いることができる。
他のジハロゲン化アルコール化合物の例としては、1,3−ジヨウ化−2−プロパノール等が挙げられる。
【0025】
本発明の化合物の屈折率nD(液屈折率)は、好ましくは1.60以上である。
本発明の化合物を含む組成物は、例えば、本発明の化合物と、他の化合物(例えば、本発明の化合物に該当しない(メタ)アクリレート化合物)と、光重合開始剤とを含む。
他の化合物の例として、ペンタエリスリトールテトラアクリレート、ビスフェノールA−EO変性ジアクリレート等が挙げられる。
光重合開始剤の例として、1−ヒドロキシシクロヘキシルフェニルケトン、2−メチル−1−[4−(メチルチオ)フェニル]−2−モルホリノ−プロパン−1−オン等が挙げられる。
各成分の配合割合は、例えば、本発明の化合物が50〜99質量%、他の化合物が0〜45質量%、光重合開始剤が1〜10質量%とすることができる。
【0026】
本発明の化合物を含む組成物を重合させてなる硬化体の屈折率nDは、好ましくは
1.62以上である。
【実施例】
【0027】
[テトラメチルチオメタンの合成方法]
本発明の化合物の製造に用いられる、テトラメチルチオメタンの合成方法の一例を下記の合成フロー3に示す。なお、下記合成フロー3中、符号13で示す化合物は、以下、「化合物13」と称する。
[合成フロー3]
【化9】
【0028】
500mLの2口フラスコに、チオウレア(50g、657mmol)、蒸留水23mLを加えた。次いで、氷浴にて0℃に冷却し、滴下漏斗を用いて硫酸ジメチル(45g、373mmol)を30分以上かけて滴下した。その後、130℃にて1時間攪拌し、生成した析出物をエタノールで洗浄後、濾別、乾燥することで化合物13(59g、657mmol)を得た。
【0029】
500mLの2口フラスコに、化合物13(35g、380mmol)、1N塩酸水14mL、及び蒸留水90mLを加えた。次いで、氷浴にて0℃に冷却し、ジエチルエーテル21mL、亜硝酸ナトリウム(35g、507mmol)を加えて、二酸化窒素の発生が止まるまで攪拌した。さらに、蒸留水100mLと、化合物13(28g、311mmol)を加えて、室温にて30分攪拌した。次いで、25%アンモニア水50mLを加えて、室温にて30分攪拌した。その後、窒素の発生が治まるのを確認してから、50℃にて1時間攪拌した。反応終了後、反応混合物に酢酸エチルを加えて有機層を抽出した。得られた有機層に硫酸マグネシウムを加えて乾燥、濾過し、溶剤を除去した後、残留物を減圧蒸留することでテトラメチルチオメタンを得た(5g、25mmol)。
【0030】
[実施例1]
前記の合成フロー1に従って、1,3−ジブロモ−2−プロパノールを出発物質にして、目的とする硫黄含有化合物(合成フロー1中の化合物6)を得た。
[化合物1の合成方法]
200mLの2口フラスコに90mLの塩化メチレン、1,3−ジブロモ−2−プロパノール(5g、22.9mmol)、及びトリエチルアミン(2.58g、25.5mmol)を加えた。次いで、氷浴にて0℃に冷却し、ベンゾイルクロライド(3.58g、25.5mmol)を滴下した。混合物を徐々に室温に戻した後、6時間室温(15〜25℃である。以下、同じ。)にて攪拌した。反応終了後、飽和重層水を加えて30分攪拌し、溶媒を真空下で除去した。次いで、反応混合物に蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層を飽和食塩水で洗浄した後、硫酸マグネシウムを加えて、乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=1/1)することにより化合物1を得た(無色透明液体、収率96%、7.08g、22.0mmol)。
【0031】
[化合物2の合成方法]
200mLの2口フラスコに50mLのDMF、化合物1(3.65g、11.3mmol)を加えた。次いで、チオ酢酸カリウム(2.84g、24.9mmol)を加えて、60℃にて6時間攪拌した。反応終了後、反応混合物に蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層を1N塩酸水、飽和食塩水で洗浄した後、硫酸マグネシウムを加えて、乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/酢酸エチルの体積比=100/1)することにより化合物2を得た(黄色液体、収率95%、3.35g、10.7mmol)。
【0032】
[化合物3の合成方法]
200mLのフラスコに20mLのクロロホルム、化合物2(3.17g、10.2mmol)、4mLの濃塩酸、及び40mLのメタノールを加えて、60℃にて24時間攪拌した。反応終了後、反応混合物に蒸留水及びクロロホルムを加えて有機層を抽出した。得られた有機層を飽和食塩水にて洗浄した後、硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/酢酸エチルの体積比=100/1)することにより化合物3を得た(黄色液体、収率85%、1.96g、8.6mmol)。
【0033】
[化合物4の合成方法]
100mLの2口フラスコにテトラメチルチオメタン(0.37g、1.86mmol)、9.3mLのトルエン、化合物3(0.85g、3.71mmol)、及びp−トルエンスルホン酸(0.032g、0.19mmol)を加えて、100℃にて16時間攪拌した。反応終了後、真空下で反応混合物から溶剤を除去したのち、残留物をカラムクロマトグラフィーにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=1/1)することにより化合物4を得た(黄色液体、収率65%、0.28g、0.60mmol)。
【0034】
[化合物5の合成]
200mLの2口フラスコに化合物4(0.48g、1.0mmol)、7mLのクロロホルム、7mLのメタノール、及び炭酸カリウム(0.85g、6.1mmol)を加えて、室温にて6時間攪拌した。反応終了後、反応混合物に蒸留水及びクロロホルムを加えて有機層を抽出した。さらに、水層に1N塩酸を加えて酸性にした後、クロロホルムで有機層の再抽出を行った。得られた有機層に硫酸マグネシウムを加えて、乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;酢酸エチル/クロロホルムの体積比=1/1)することにより化合物5を得た(無色透明粘調性液体、収率75%、0.2g、0.75mmol)。
【0035】
[化合物6の合成]
100mLの2口フラスコに塩化メチレン10mL、化合物5(0.54g、2.1mmol)、及びトリエチルアミン(0.46g、4.6mmol)を加えた。次いで、氷浴にて冷却し、アクリル酸クロライド(0.38g、4.6mmol)を滴下しながら徐々に室温に戻し、6時間攪拌した。反応終了後、反応混合物に飽和重層水を加えて30分攪拌した後、真空下で溶剤を除去した。その後、蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層に硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=6/1)することにより化合物6を得た(淡白色液体、収率52%、0.39g、1.09mmol)。
【0036】
[化合物6の物性評価]
(1)液屈折率の測定
シリコンウエハー上に化合物6を塗布したのち、n&k Technology社製のアッベ屈折計「n&k Analyzer 1510」を用いて、液屈折率の測定を行った。その結果、化合物の波長589nmにおける液屈折率は、1.621、アッベ数は25だった。
【0037】
[実施例2]
前記の合成フロー2に従って、1,2−ジブロモ−3−プロパノールを出発物質にして、目的とする硫黄含有化合物(合成フロー2中の化合物12)を得た。
[化合物7の合成方法]
200mLの2口フラスコに90mLの塩化メチレン、1,2−ジブロモ−3−プロパノール(5g、22.9mmol)、及びトリエチルアミン(2.58g、22.5mmol)を加えた。次いで、氷浴にて0℃に冷却し、ベンゾイルクロライド(3.58g、25.5mmol)を滴下した。混合物を徐々に室温に戻した後、6時間室温にて攪拌した。反応終了後、飽和重層水を加えて30分攪拌し、溶媒を真空下で除去した。次いで、反応混合物に蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層を飽和食塩水で洗浄した後、硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=1/1)することにより化合物7を得た(無色透明液体、収率87%)。
【0038】
[化合物8の合成方法]
窒素雰囲気下にて200mLの2口フラスコに、水素化ナトリウム(1.03g、43mmol)、脱水DMF(100mL)、を加えた。次いで、氷浴にて0℃に冷却し、チオ酢酸(3.4g、45mmol)を滴下した。その後、氷浴を外して、室温にて30分攪拌した。その後、10mLのDMFに溶解させた化合物7(6.28g、20mmol)を反応混合物中に滴下し、60℃にて6時間攪拌した。反応終了後、反応混合物に蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層を1N塩酸水、及び飽和食塩水で洗浄した後、硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;ヘキサン/酢酸エチルの体積比=6/1)することにより化合物8を得た(黄色液体、収率77%、4.67g、15mmol)。
【0039】
[化合物9の合成方法]
200mLのフラスコに、15mLのクロロホルム、化合物8(2.03g、6.47mmol)、3mLの濃塩酸、及び30mLのメタノールを加えて、60℃にて24時間攪拌した。反応終了後、反応混合物に蒸留水及びクロロホルムを加えて有機層を抽出した。得られた有機層を飽和食塩水にて洗浄した後、硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/酢酸エチルの体積比=100/1)することにより化合物9を得た(黄色液体、収率87%、1.29g、5.66mmol)。
【0040】
[化合物10の合成方法]
100mLの2口フラスコに、テトラメチルチオメタン(0.57g、2.83mmol)、14mLのトルエン、化合物9(1.29g、5.66mmol)、及びp−トルエンスルホン酸(0.049g、0.28mmol)を加えて、100℃で16時間攪拌した。反応終了後、真空下で反応混合物から溶剤を除去したのち、残留物をカラムクロマトグラフィーにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=1/1)することにより化合物10を得た(黄色液体、収率72%、0.94g、2.03mmol)。
【0041】
[化合物11の合成方法]
200mLの2口フラスコに、化合物10(2.32g、5mmol)、30mLのクロロホルム、30mLのメタノール、及び炭酸カリウム(2.77g、20mmol)を加えて、室温にて6時間攪拌した。反応終了後、反応混合物に蒸留水及びクロロホルムを加えて有機層を抽出した。さらに水層に1N塩酸を加えて酸性にした後、クロロホルムで有機層の再抽出を行った。得られた有機層に硫酸マグネシウムを加えて、乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;酢酸エチル/クロロホルムの体積比=1/1)することにより化合物11を得た(薄黄色液体、収率61%、0.79g、3.07mmol)。
【0042】
[化合物12の合成方法]
100mLの2口フラスコに、塩化メチレン15mL、化合物11(0.79g、3.07mmol)、及びトリエチルアミン(0.78g、7.68mmol)を加えた。次いで、氷浴にて0℃に冷却し、アクリル酸クロライド(0.83g、9.21mmol)を
滴下しながら徐々に室温に戻し、6時間室温にて攪拌した。反応終了後、反応混合物に飽和重層水を加えて30分攪拌後、真空下で溶剤を除去した。その後、蒸留水及び酢酸エチルを加えて有機層を抽出した。得られた有機層に硫酸マグネシウムを加えて乾燥、濾過し、さらに溶剤を除去したのち、残留物をカラムクロマトグラフィにて精製(展開溶剤;クロロホルム/ヘキサンの体積比=6/1)することにより化合物12を得た(黄色液体、収率67%、0.75g、2.05mmol)。
【0043】
[化合物12の物性評価]
(1)液屈折率の測定
実施例1と同様にして、液屈折率の測定を行った。その結果、化合物の波長589nmにおける液屈折率は、1.610、アッベ数は36だった。
【特許請求の範囲】
【請求項1】
下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を有する硫黄含有化合物。
【化1】
【請求項2】
下記の一般式(1)で表される請求項1に記載の硫黄含有化合物。
【化2】
(式中、Rは水素原子又はメチル基であり、k、l、m、nは各々独立に0〜2の整数であり、k+l=1又は2、m+n=1又は2であり、p、qは各々独立に0〜2の整数である。)
【請求項3】
屈折率nDが1.60以上である請求項1又は2に記載の硫黄含有化合物。
【請求項4】
両端にチオールを有し、かつ分岐鎖として、(メタ)アクリロイル基を導入可能な官能基を有する化合物と、テトラメチルチオメタンを反応させて、下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を導入可能な上記官能基を有する化合物を得た後、該官能基に対して(メタ)アクリロイル基を導入する、請求項1〜3のいずれか1項に記載の硫黄含有化合物の製造方法。
【化3】
【請求項5】
請求項1〜3のいずれか1項に記載の硫黄含有化合物を含む組成物を重合させてなる硬化体。
【請求項6】
請求項5に記載の硬化体からなる光学部材。
【請求項1】
下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を有する硫黄含有化合物。
【化1】
【請求項2】
下記の一般式(1)で表される請求項1に記載の硫黄含有化合物。
【化2】
(式中、Rは水素原子又はメチル基であり、k、l、m、nは各々独立に0〜2の整数であり、k+l=1又は2、m+n=1又は2であり、p、qは各々独立に0〜2の整数である。)
【請求項3】
屈折率nDが1.60以上である請求項1又は2に記載の硫黄含有化合物。
【請求項4】
両端にチオールを有し、かつ分岐鎖として、(メタ)アクリロイル基を導入可能な官能基を有する化合物と、テトラメチルチオメタンを反応させて、下記式(2)で示される構造を有するスピロ骨格、及び、(メタ)アクリロイル基を導入可能な上記官能基を有する化合物を得た後、該官能基に対して(メタ)アクリロイル基を導入する、請求項1〜3のいずれか1項に記載の硫黄含有化合物の製造方法。
【化3】
【請求項5】
請求項1〜3のいずれか1項に記載の硫黄含有化合物を含む組成物を重合させてなる硬化体。
【請求項6】
請求項5に記載の硬化体からなる光学部材。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2011−148719(P2011−148719A)
【公開日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願番号】特願2010−9876(P2010−9876)
【出願日】平成22年1月20日(2010.1.20)
【出願人】(000004178)JSR株式会社 (3,320)
【Fターム(参考)】
【公開日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願日】平成22年1月20日(2010.1.20)
【出願人】(000004178)JSR株式会社 (3,320)
【Fターム(参考)】
[ Back to top ]