
神経刺激性ステロイドの医薬組成物及びその使用
本発明は、気分障害及び同様の障害を処置する際に使用するために所望の性質を有する、神経刺激性ステロイドの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン又はその医薬的に許容される塩もしくは溶媒和物の医薬組成物に関する。本発明の医薬組成物は3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルをもたらす。本発明はまた、医薬組成物を投与することによってこれらの障害を処置する方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬品化学の分野、並びに気分障害などを処置するために有用な医薬組成物に関する。より具体的には、本発明は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルをもたらす、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン又はその医薬的に許容される塩もしくは溶媒和物の医薬組成物に関する。本発明はまた、本発明の医薬組成物を投与することによってこれらの障害を処置する方法に関する。
【背景技術】
【0002】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンは神経刺激性の合成ステロイドである。その主な分子的標的はγ−アミノ酪酸タイプA(GABAA)受容体であり、この場合、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンはチャ
ネル機能の正のアロステリック調節因子として作用する。他のクラスのGABAA調節因子(例えば、ベンゾジアゼピン系化合物及び他のベンゾジアゼピン部位リガンドなど)と同様に、様々な神経刺激性ステロイドが、例えば、下記の障害を処置するために、可能性のある数多くの適応を有する:睡眠障害(例えば、Edgar, D.M.他、J. Pharmacol. Exp. Ther.、282:420-29(1997); Friess, E.他、Am. J. Physiol.、272(Endocrin. Metab. 35):E885-91(1997)を参照のこと)、不安(例えば、Purdy, R.H.他、Proc. Natl. Acad. Sci.、88:4553-57(1991); Vanover, K.E.他、J. Pharmacol. Exp. Ther. 295:337-45(2000); Strohle, A.他、Arch. Gen. Psychiatry、60:161-68(2003)を参照のこと)、うつ病(例えば、Dong, E.他、Proc. Natl. Acad. Sci.、98:2849-54(2001); Rupprecht, R.及びHolsboer, F.、Trends Neurosci.、22:410-16(1999); Uzunova, V.他、Proc. Natl. Acad. Sci.、95:3239-44(1998)を参照のこと)、てんかん(例えば、Carter, R.B.他、J. Pharmacol. Exp. Ther.、280:1284-95(1997); Laxer, K.他、Epilepsia、41:1187-94(2000); Kerrigan, J.F.他、Epilepsy Res.、42:133-39(2000)を参照のこと)、並びに月経前症候群(PMS)及び月経前不快障害(PMDD)(例えば、Rapkin, A.J.他、Obs. Gyn.、90:709-14(1997); Monteleone, P.他、Eu. J. Endocrinol.、142:269-73(2000); Smith, M.J.他、Biol. Psychiatry、54:757-62(2003)を参照のこと)。
【0003】
米国特許第5,939,545号及び同第6,277,838号には、下記式の化合物:
【化1】

が、ストレス又は不安、気分障害(うつ病、月経前症候群又は出産後うつ病を含む)の処置又は予防において有用であるとして議論される。例えば、‘838号特許の第60欄60行〜第61欄65行を参照のこと。1つのそのような化合物が下記式の3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンである:
【化2】
Vanover, K.E.他、Psychopharmarmacology、155:285-91(2001)を参照のこと。
【発明の開示】
【0004】
本発明の第1の態様は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン又はその医薬的に許容される塩もしくは溶媒和物と、1つ又はそれ以上の医薬的に許容される賦形剤とを含み、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態血漿中レベルをもたらす医薬組成物に関する。ヒトにおける気分障害及び同様の障害などを処置するための目標血漿中レベルは約5ng/mL〜約500ng/mLの範囲であり、特に約50ng/mL〜約250ng/mLの範囲である。CNS副作用及び他の副作用が、約500ng/mLを越える血漿中レベルで生じることが予想される。
【0005】
本発明の第2の態様は、その処置が3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルから利益を受ける状態又は障害を処置する方法に関する。1つの実施形態において、そのような状態は気分障害である。
【発明を実施するための最良の形態】
【0006】
本発明は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルをもたらす、ヒトにおける1つ又はそれ以上の状態又は障害を処置するために有用な医薬組成物を提供する。1つの実施形態において、医薬組成物は固体の経口用製剤である。本発明の医薬組成物により処置可能な状態又は障害は、とりわけ、不安、うつ病、アルコール嗜癖、アルコール依存、月経前緊張、月経前症候群及び月経前不快障害から選択することができる。不安には、例えば、全般性不安障害、恐慌性障害、強迫性障害、心的外傷後ストレス障害及び社会不安障害などが含まれる。
【0007】
臨床研究では、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンが下記の薬物動態学的性質を経口服用後にヒトにおいて有することが示唆される:(1)Tmaxが約1時間〜約3時間の範囲である迅速な吸収;(2)被験者間での可変なCmaxレベル;(3)用量比例を上回るCmax値;及び(4)5つの異なる服用群の間で平均して約12時間であったT1/2値。下記の表1を参照のこと。本発明の関連では、薬物動態学的パラメータ(例えば、AUC、Cmax及びtmaxなど)は平均値を示す。括弧において報告される値は標準偏差に対応する。
表I
【表1】
【0008】
これらの薬物動態学的性質を考えると、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの血漿中レベルを規定の治療的範囲においてもたらすことができる組成物は、この薬物の持続した治療的血漿中レベルを必要とする障害(例えば、気分障害など)を処置することにおいて有益であると考えられる。このような持続した血漿中レベルは、適切な技術を使用して、例えば、制御放出配合物を使用して達成することができる。そのような配合物は、即時放出配合物を上回る利点(例えば、持続した効力、低下した副作用、及び服用の改善された容易さなど)をもたらすと考えられる。
【0009】
濃度勾配又は血漿中曲線を、Cmax、tmax及びAUCなどのパラメータによって記述することができる。これらのパラメータは、特定の薬物配合物の薬物動態学的性質を記述することにおいて重要である。
【0010】
血漿中曲線を記述するパラメータは、例えば、活性薬剤(例えば、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンなど)を多数の好適な試験被験者に投与することによって臨床試験で得ることができる。その後、個々の試験者の血漿中での値が平均化される(例えば、AUC、Cmax及びtmaxの平均値が得られる)。本発明の関連では、薬物動態学的パラメータ(例えば、AUC、Cmax及びtmaxなど)は平均値を示す。
【0011】
薬物動態学的パラメータ(例えば、平均tmax、平均Cmax及び平均AUCなど)が健康なヒト被験者について測定されるならば、そのようなパラメータは、典型的には、血漿中での値の発達を健康なヒト被験者の適切な試験集団において経時的に測定することによって得られる。
【0012】
被験者の血液サンプルを、任意の好適な時間間隔で、好ましくは、下記時点のいずれか1つ又は組合せにおいて採取することができる:服用前、服用後0.25時間、0.5時間、1.0時間、1.5時間、2時間、3時間、4時間、5時間、6時間、8時間、10時間、12時間、24時間、30時間、36時間、48時間、60時間、72時間、84時間及び研究終了時(これは、例えば、服用後96時間である場合がある)。
【0013】
この関連での用語「健康な」ヒト被験者は、身長、体重及び生理学的パラメータ(例えば、血圧など)に関して平均値を有する、通常の場合には白人、黒人、アジア人又は他の指定された起源の典型的な男性又は女性を示す。本発明の目的のための健康なヒト被験者は、臨床試験協調化のための国際会議(ICH)の勧告に基づき、また、勧告に従う参加基準及び除外基準に従って選択される。本発明の目的のために、健康な被験者は、実施例において概略されるような参加基準及び除外基準に従って特定することができる。
【0014】
従って、参加基準には、21歳超〜45歳未満の年齢;男性について40kg〜100kgの範囲の体重及び30kg/m2未満のボディマス指数(BMI);病歴、身体検査、生命徴候、研究室評価、12誘導線心電図(ECG)、及びホルターモニターを24時間にわたって使用する携帯型ECGでの著しく異常な知見がないことによって明確に示される全般的に良好な健康状態が含まれる。さらに、被験者は、好ましくは、ランダム化前30日間において、好ましくは、所与の時間枠内で夜間に6.5時間〜8.5時間の睡眠を取る。
【0015】
典型的な除外パラメータには、過去6ヶ月の期間中における臨床的に著しい睡眠異常の何らかの前歴;夜間労働又は交代勤務を含む、ランダム化前30日間における何らかの著しい睡眠不規則;ランダム化前30日間において3つ以上の時間帯を越える移動;パルスオキシメトリーによって測定されたときの94%以下の酸素飽和度(SpO2);ランダム化前30日間における(15分以上の)日課的な昼寝;向精神性薬物又は催眠性薬物に対する過敏性の何らかの前歴;精神医学的障害(例えば、精神病、強迫性障害、大うつ病又は不安障害/恐慌性障害など)の何らかの病歴;薬物の吸収、分布、代謝又は排出を妨害する恐れがある病歴又は何らかの現在の状態;ランダム化前30日以内における何らかの催眠剤又は睡眠補助薬(メラトニンを含む)の使用;薬物乱用又はアルコール中毒の前歴;過去1年間における発作又は閉鎖性頭部外傷の前歴;過去3ヶ月以内における喫煙の前歴又はニコチン含有製品の使用;ランダム化前48時間以内におけるアルコール飲料物の消費;ランダム化前30日間における1日に5杯以上の茶、コーヒー又はソーダの日課的消費;服用前3日間の期間中におけるカフェイン含有の食物又は飲料物の消費;ランダム化前30日間の期間中における何らかの臨床的に著しい病気;ランダム化前7日間の期間中における何らかの薬物(処方薬及び大衆薬を含む)、1日摂取値の300%を超える何らかのビタミン剤及び/又はミネラル補助物、グレープフルーツジュース及びセイヨウオトギリソウ;服用前10時間及び研究用薬物投与後4時間の絶食を拒否すること、並びにアルコール、カフェイン又はキサンチンを含有する食物又は飲料物を絶つことを全研究期間にわたって拒否すること;ランダム化前30日間における臨床研究への参加;ランダム化前30日間における献血又は血液製剤提供;尿中薬物スクリーニング、尿中ニコチン、血中アルコール、HBsAg、HBsAb(被験者が免疫化されていない限り)、抗HCV又は抗HIVの陽性結果;7日前から1日前までの睡眠記録によって、又は1日前の夜(登録後最初の夜)でのアクチグラフィー(actigraphy)によって示されるような30分を超える睡眠潜時、あるいは、85%以下又は95%超の睡眠効率が含まれる。
【0016】
薬物動態学的パラメータ(例えば、平均tmax、平均Cmax及び平均AUCなど)が被験者において得られるならば、被験者群は好適な数の被験者を含む。妥当な数の被験者は、例えば、4名、8名、10名、20名、30名、40名、50名、60名、72名又はより一層多数の患者である。被験者は、処置される状態の症状に従って選択される。被験者は、例えば、用量増加を調べるために、好適なサイズのコホートにグループ分けすることができる。本発明の目的のために、被験者は、実施例において提供される参加基準及び除外基準に従って選択することができる。
【0017】
上記及び下記において示されるような薬物動態学的パラメータの値は、実施例7で得られたデータ(それらのすべてが健康なヒト被験者における単用量研究に関連する)に基づいて推定されていることを理解しなければならない。しかしながら、比較可能な結果が、健康なヒト被験者における定常状態での投与、又はヒト患者における単用量及び定常状態での投与の時に得られることが推定される。
【0018】
本発明による医薬組成物は、約1.5時間〜約5時間の範囲で、好ましくは約1.3時間〜約3.5時間の範囲で、より好ましくは約1.6時間〜約3.2時間の範囲でTmax値を示す。臨床研究では、本発明による組成物が、好ましくは、活性薬剤(すなわち、本発明による医薬組成物に存在する3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)の量に依存することなく、迅速に吸収されることが示唆される。
【0019】
本発明の医薬組成物のCmax値は、1mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約1.0ng/mL〜約1.8ng/mLの範囲であり、好ましくは約1.2ng/mL〜約1.6ng/mLの範囲である。
【0020】
本発明の医薬組成物のCmax値は、3mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約4.0ng/mL〜約8.0ng/mLの範囲であり、好ましくは約4.0ng/mL〜約6.5ng/mLの範囲であり、より好ましくは約5.0ng/mL〜約6.0ng/mLの範囲である。
【0021】
本発明の医薬組成物のCmax値は、10mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約16.9ng/mL〜約46.3ng/mLの範囲であり、好ましくは約21.0ng/mL〜約32.0ng/mLの範囲であり、より好ましくは約25.0ng/mL〜約28.0ng/mLの範囲である。
【0022】
本発明の医薬組成物のCmax値は、30mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約95.6ng/mL〜約154.0ng/mLの範囲であり、好ましくは約108.0ng/mL〜約134.0ng/mLの範囲であり、より好ましくは約115.0ng/mL〜約125.0ng/mLの範囲である。
【0023】
本発明の医薬組成物のCmax値は、60mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約218.0ng/mL〜約479.0ng/mLの範囲であり、好ましくは約275.0ng/mL〜約385.0ng/mLの範囲であり、より好ましくは約300ng/mL〜約350ng/mLの範囲である。
【0024】
さらに、本発明による医薬組成物を投与することによって、用量比例よりも大きいCmax値を得ることができる。
【0025】
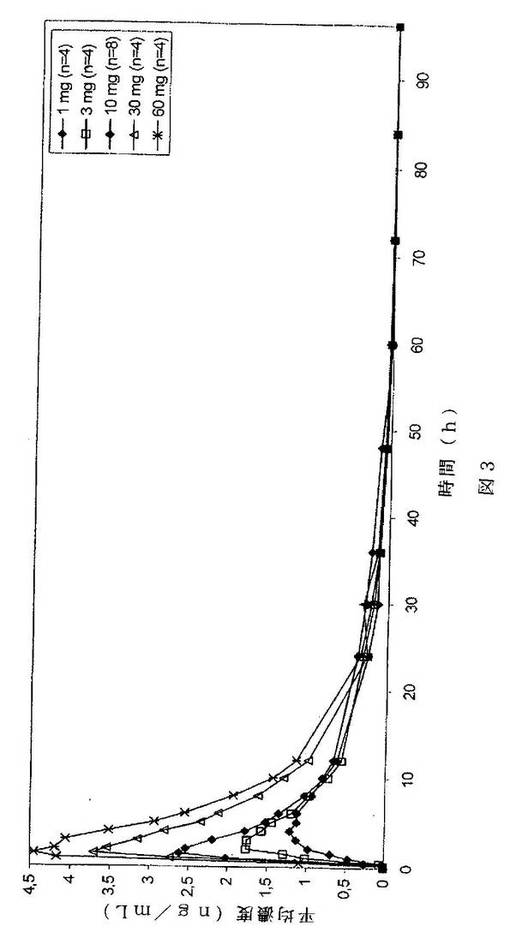
用量比例よりも大きいCmax値は、本発明の関連では、活性薬剤の正規化されたCmax値が一定でないことを意味する。別の言い方をすれば、量1(例えば、10mg)の活性薬剤についての正規化されたCmax値が、量2(例えば、1mg)の活性薬剤についての正規化されたCmax値により大きい。活性薬剤(3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)の、用量比例よりも大きいCmax挙動もまた図3に示される。図3は、用量により正規化された血漿中濃度の時間に対する図を示す。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む本発明による3mg製剤、10mg製剤、30mg製剤及び60mg製剤についての正規化されたCmax値が、本発明による1mg製剤について得られたCmaxよりも大きい。1mgを超える活性薬剤を含む製剤について得られる正規化されたCmax値は、本発明による1mg製剤から得られたCmax値よりも少なくとも約2倍、少なくとも約2.5倍、少なくとも約3倍、少なくとも約4倍、少なくとも約5倍大きい正規化されたCmax値を有することができる。60mgを超える3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤は、少なくとも約5を超える用量比例したCmax値を示すことが推定され得る。何らかの理論によって限定されることはないが、本発明による1mg製剤のCmax値と比較して、少なくとも約7、少なくとも約10、少なくとも約15又は少なくとも約20又は少なくとも約25を超える正規化されたCmax値が本発明の製剤により達成され得ることが推定され得る。
【0026】
本発明による医薬組成物の、用量比例よりも大きいCmax値は、活性薬剤の量をほんのわずかに変更するか、又はほぼ同じレベルで維持しながら、Cmaxの著しい増大を可能にすることができる。
【0027】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルは約5ng/mL〜約500ng/mLの範囲が可能である。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの他の治療的範囲には、約50ng/mL〜約500ng/mL、約50ng/mL〜約400ng/mL、約50ng/mL〜約325ng/mL、約50ng/mL〜約250ng/mL、約50ng/mL〜約100ng/mL、及び約100ng/mL〜約250ng/mLが含まれる。
【0028】
これらの定常状態血漿中レベルは、適切な技術を使用して達成することができ、例えば、適切な放出プロフィールを有する制御放出配合物を使用して達成することができる。適切な放出プロフィールを、例えば、単一送達システム又は多粒子送達システムを使用して達成することができる。単一送達システムの例には、ワックスマトリックス錠剤、親水性マトリックス錠剤、及び制御放出被覆を伴う錠剤が含まれるが、これらに限定されない。多粒子システムの例には、制御放出被覆に基づく溶融押出し多粒子剤又は溶融押出し多粒子システムなどのマトリックスシステム(例えば、被覆ビーズなど)が含まれるが、これらに限定されない。
【0029】
本発明によるこれらの制御放出型医薬組成物は、活性薬剤と一緒にマトリックスに配合される制御放出物質、又は活性薬剤を含む基質を覆う制御放出被覆剤として適用される制御放出物質を含むことができる。用語「基質」は、ビーズ、ペレット、球状物、錠剤、錠剤コアなどを含むことができる。制御放出物質は、所望により疎水性又は親水性であり得る。本発明による製剤は、好ましくは経口用製剤であり、例えば顆粒、球状物、ペレットなどとして提供することができる。他方で、本発明の製剤は、制御放出被覆剤により被覆された錠剤として、又は活性薬剤のマトリックスと、制御放出物質と、場合により、他の医薬的に許容される成分(例えば、結合剤、希釈剤、着色剤、滑剤など)とを含む錠剤として調製することができる。
【0030】
制御放出配合物についての例には、WO01/32148、米国特許第4,861,598号、米国特許第4,990,341号、米国特許第4,884,909号、それらにおいて引用される参考文献、及び当分野において開示される他の参考文献に開示される配合物が含まれ得る。
【0031】
1つの実施形態において、本発明の医薬組成物は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的な定常状態血漿中レベルを投与後の約12時間〜約24時間の持続期間にわたってもたらす。別の実施形態において、本発明の医薬組成物は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的な定常状態血漿中レベルを投与後の約6時間〜約12時間の持続期間にわたってもたらす。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンは光学異性体(エナンチオマー)として存在する場合があり、本発明は、そのような光学異性体のラセミ混合物及びエナンチオマー濃縮された混合物の両方、並びに当業者には周知である方法に従って分離することができる個々のエナンチオマーを含む。
【0032】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンは、米国特許出願第60/604,447号(これは本明細書によりその全体が参考として組み込まれる)に記載されるように同形の結晶性晶癖として存在する場合がある。
【0033】
本発明の範囲にはまた、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの溶媒和形態(特に、水和形態)を含む、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態治療的血漿中レベルをもたらす組成物が含まれる。水和は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、又は3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む組成物を製造しているときに生じ得るか、あるいは、水和は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの吸湿性のために時間とともに生じ得る。本発明の範囲にはまた、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの医薬的に許容される塩を含む、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態治療的血漿中レベルをもたらす組成物が含まれる。医薬的に許容される酸付加塩が、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの溶液を医薬的に許容される非毒性の酸(例えば、塩酸、フマル酸、マレイン酸、コハク酸、酢酸、クエン酸、酒石酸、炭酸、リン酸、シュウ酸及びジクロロ酢酸など)の溶液と混合することによって形成される。医薬的に許容される塩基性塩が、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの溶液を医薬的に許容される非毒性の塩基(例えば、水酸化ナトリウム、水酸化カリウム、水酸化コリン及び炭酸ナトリウムなど)の溶液と混合することによって形成される。
【0034】
本発明はまた、その処置が3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルから利益を受ける状態又は障害を処置する方法に関する。この処置方法は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態治療的レベルをもたらす医薬組成物を、そのような処置を必要としている被験者に投与することを含む。そのような状態及び障害には、気分障害、例えば、不安、うつ病、アルコール嗜癖及び/又はアルコール依存、月経前緊張、月経前症候群及び月経前不快障害などが含まれる。不安には、例えば、全般性不安障害、恐慌性障害、強迫性障害、心的外傷後ストレス障害及び社会不安障害などが含まれる。
【0035】
定義
状態又は障害を「処置する」、あるいは、状態又は障害の「処置」の用語は、本明細書中で用いられる場合、(i)状態又は障害を阻害すること、すなわち、状態又は障害又はその臨床的症状の発達を停止させること、及び/又は(ii)状態又は障害を軽減すること、すなわち、状態又は障害又はその臨床的症状の一時的又は永続的な退行を生じさせることを示す。
【0036】
用語「キラル中心」は、4つの異なる基が結合する炭素原子、又は3つ異なる基が結合するイオウ原子(この場合、イオウ原子及びその結合した基は、スルホキシド、スルフィン酸エステル、スルホニウム塩又はスルフィトを形成する)を示す。
【0037】
用語「エナンチオマー」又は用語「エナンチオマー状」は、その鏡像と重ねることができず、従って、光学活性である分子を示し、この場合、エナンチオマーは、偏光した光の面を一方の方向に回転させ、その鏡像体は、偏光した光の面を反対方向に回転させる。
【0038】
用語「ラセミ(状)」は、光学的に不活性である、等しい量のエナンチオマーの混合物を示す。
【0039】
用語「分割」は、分子の2つのエナンチオマー形態の一方を分離又は濃縮又は減損することを示す。表現「エナンチオマー濃縮された」は、一方のエナンチオマーが、その鏡像分子よりも高い濃度で存在する混合物を示す。
【0040】
Cmax値は、活性薬剤(すなわち、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)の最大血漿中濃度を示す。
【0041】
tmax値は、Cmax値に達する時点を示す。別の言い方をすれば、tmaxは最大の観測された血漿中濃度の時点である。
【0042】
AUC(曲線下面積)値は濃度曲線の面積に対応する。AUC値は、全体では血液循環に吸収された活性薬剤(すなわち、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)の量に比例しており、従って、生物学的利用能についての尺度である。
【0043】
AUC(0〜24)値は、投与時から投与後24時間までの血漿中濃度−時間曲線下の面積についての値である。
【0044】
AUC(0〜最後)値は、投与時から最後の測定可能な濃度までの血漿中濃度−時間曲線下の面積についての値である。
【0045】
用語「正規化されたCmax」は、活性薬剤の特定の投薬量のCmax値と、活性薬剤の量との比率を示す。
【0046】
用語「生物学的利用能」は、本発明の目的のためには、活性薬剤(例えば、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)が製剤から吸収される程度として定義される。
【0047】
本発明の関連での用語「平均」は、少なくとも2名の被験者のデータの平均を示す。
【0048】
用語「持続放出」は、本発明の目的のためには、血中レベルが、約6時間又は約12時間又は約24時間又はより一層長い時間の期間にわたって治療的範囲内で、しかし、毒性レベル未満で維持されるような速度での3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの放出として定義される。用語「持続放出」により、本発明による調製物は「即時放出」調製物から区別される。用語「持続放出」及び用語「制御放出」は交換可能に使用される。
【0049】
用語t1/2は、本発明の目的のためには、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの吸収可能な用量の半分が血漿に移されるために必要な時間の量として定義される。この値は、「見かけ」の吸収半減期ではなく、むしろ、「真の」値(これは排除プロセスの影響が考慮に入れられる)として計算することができる。
【0050】
用語「定常状態」は、所与の薬物についての血漿中レベルが達成され、そのようなレベルが、薬物のその後の服用により、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンについての最小有効治療的レベルであるか、又は最小有効治療的レベルを上回り、かつ、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンについての最小毒性血漿中レベルよりも低いレベルで維持されることを意味する。
【0051】
最小有効治療的レベルは、部分的には、所与の患者において達成された所望される効果の程度によって決定される。この測定は非常に主観的であり、また、大きい個体変動が被験者間に存在し得ることが当業者によって十分に理解される。それぞれの用量を投与した後、濃度が最大値を通過し、その後、最小値に再び低下することは明らかである。
【0052】
定常状態は下記のように記載することができる:時間t=0(最初の用量が投与される時間)で、濃度Cもまた0である。その後、濃度は第1の最大値を通過し、その後、第1の最小値に低下する。濃度が0に低下する前に、別の用量が投与され、その結果、濃度における第2の増大が0において始まらない。この第1の濃度最小値の上に加わって、曲線は、第2の用量が投与された後の第2の最大値(これは第1の最大値を上回る)を通過し、その後、第1の最小値を上回る第2の最小値に低下する。従って、血漿中曲線は、吸収及び排出が均衡状態にある点に落ち着くまで、活性薬剤の繰り返された服用及び付随する段階毎の蓄積のために上昇する。この状態は、吸収及び排出が平衡状態にあり、濃度が、規定された最小値と、規定された最大値との間で絶えず振動する状態であり、この状態が定常状態と呼ばれる。
【0053】
本発明の医薬組成物は、その意図された目的を達成する任意の手段によって投与することができる。例えば、投与は、皮下経路、静脈内経路、筋肉内経路、腹腔内経路、口内経路又は眼経路、直腸的、非経口的、全身的(intrasystemically)、膣内、局所的(粉末、軟膏、滴剤又は経皮パッチによるような局所的)、あるいは、口腔スプレー又は鼻スプレーが可能である。代わりとして、又は同時に、投与は経口経路によることが可能である。経口経路による本発明の医薬組成物の投与が現時点では好ましい。
【0054】
投与頻度は、投与された組成物について予期される作用持続期間にわたって適切でなければならない。例えば、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルを投与後の約12時間〜約24時間の持続期間にわたってもたらす組成物については、組成物を1日に1回与えることができる。同様に、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルを投与後の約6時間〜約12時間の持続期間にわたってもたらす組成物については、組成物を1日に2回与えることができる。
【0055】
下記の実施例は本発明の組成物及び方法を例示するが、本発明の組成物及び方法を限定しない。当業者には自明である、臨床治療において通常の場合には遭遇する多様な条件及びパラメータの他の好適な改変及び適合化は本発明の精神及び範囲に含まれる。
(実施例)
【0056】
3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オンを、Hogenkamp他、「Synthesis and in Vitro Activity of 3β-Suibstituted-3α-hydroxypregnan-20-ones: Allosteric Modulators of the GABAA Receptor」、J. Med. Chem.、40:61-72(1997)に記載されるように、(3R)−スピロ[オキシラン−2α,5α−プレグナン]−20−オン及びナトリウムメトキシドから調製することができる。
【0057】
服用が即時放出懸濁物を用いて行われた3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの単用量試験に基づいて、1日に1回与えられる約30mgの用量(遊離化合物に基づく)が、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルを投与後の約12時間〜約24時間の持続期間にわたってもたらす組成物については適切であることが予想される。この量が下記の実施例では使用され、しかし、必要な用量は約20mg/日〜約40mg/日の範囲が可能であることが理解される。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンが塩の形態であるならば、そのような量に対する適切な調節を行うことができる。そのような修正は十分に当業者の知識及び能力の範囲内である。
【0058】
投薬量は、所望される作用持続期間を達成するために調節することができる。例えば、1日に2回与えられる約15mgの用量(遊離化合物に基づく)が、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルを投与後の約6時間〜約12時間の持続期間にわたってもたらす組成物については適切であることが予想される。
【実施例1】
【0059】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの調製
【0060】
表題化合物及びその塩酸塩を下記にように調製することができる。
a)21−ブロモ−3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オン。3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オン(30.0g、82.9mmol)を約900mLのメタノールに溶解した溶液に、この溶液を室温で撹拌しながら、3滴の48% HBr水溶液を加える。その後、臭素(13.9g、87.1mmol)を約200mLのメタノールにおける溶液として約2時間かけて滴下して加える。その期間中、反応液を遮光する。TLC(1%アセトン/CH2Cl2)を使用して、出発物質の不在、及び極性がより低い生成物の形成を(さらに約30分後に)示すことができる。反応液を約300mLに濃縮する。CH2Cl2(約400mL)を加え、反応液を約200mLの水を含有する分液漏斗に注ぐ。相を分離し、水相をCH2Cl2で抽出する(約100mL、3回)。有機相を一緒にし、約200mLの飽和NaHCO3水溶液により洗浄し、Na2SO4で乾燥し、減圧下で濃縮して、ブロミドを淡黄色の泡状物として得る。この生成物は、さらに精製することなく次の工程で使用することができる。
b)3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン。上記のように調製されたブロミド(36.7g、82.9mmol)を約800mLのCH3CNに懸濁した懸濁物に、イミダゾール(28.2g、415mmol)を加え、反応物をAr下で加熱還流する。TLC(95:4.5:0.5のCH2Cl2:MeOH:トリエチルアミン)を使用して、反応の完了を(約1時間の還流の後で)示すことができる。反応物を室温に冷却し、真空下で濃縮する。得られたオイルを約600mLのCH2Cl2に溶解し、希NaHCO3溶液により洗浄し(約200mL、4回)、Na2SO4で乾燥し、真空下で濃縮する。95:4.5:0.5のCH2Cl2:MeOH:トリエチルアミンにより溶出するシリカゲルでのフラッシュクロマトグラフィーによる精製により、約18gの表題化合物を白色の固体として得る:m.p.185〜187℃(およそ)。元素分析、計算値(C26H40N2O3):C、72.86;H、9.41;N、6.54。1H NMR(300MHz、CDCl3)δ(およそ)7.40(s、1H)、7.08(s、1H)、6.84(s、1H)、4.72(d、1H、J=17.7Hz)、4.64(d、1H、J=18Hz)、3.39(s、3H)、3.18(s、2H)、2.57(t、1H、J=8.7Hz)、0.76(s、3H)、0.66(s、3H)。
c)3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、塩酸塩。塩化水素ガスを3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン(1.00g、2.33mmol、約35mLのCH2Cl2に溶解する)の溶液に約7分間吹き込む。白色の析出物が形成する。溶媒を真空下で除いて、約1.10gの塩酸塩を白色の固体として得る:m.p.230〜233℃(およそ)。1H NMR(300MHz、CDCl3)δ(およそ)9.66(s、1H)、7.31(s、1H)、7.05(s、1H)、5.45(d、1H、J=18Hz)、5.26(d、1H、J=18Hz)、3.39(s、3H)、3.19(s、2H)、2.72(t、1H、J=8.7Hz)、0.76(s、3H)、0.70(s、3H)。
【実施例2】
【0061】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む親水性マトリックス錠剤
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン 30mg
ヒドロキシプロピルメチルセルロース 30mg
噴霧乾燥ラクトース 88.9mg
コロイド状二酸化ケイ素 0.15mg
ステアリン酸マグネシウム 1mg
合計 150mg
【0062】
コロイド状二酸化ケイ素と、ラクトースの一部とを混合する。混合物を篩分けする。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、ヒドロキシプロピルメチルセルロース及び残りのラクトースを混合物に加え、混合する。ステアリン酸マグネシウムを加え、混合する。最終混合物を150mgの目標重量に圧縮成形する。
【実施例3】
【0063】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含むカプセル化溶融押出し多粒子剤(MEM)
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン 30mg
Eudragit RLPO 50mg
Eudragit RSPO 132mg
ステアリルアルコール 28mg
グリセリルベヘナート 10mg
合計 250mg
サイズ#1のハードゼラチンカプセル外皮
【0064】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、Eudragit RSPO、Eudragit RLPO、ステアリルアルコール及びグリセリルベヘナートを混合する。ホットメルト押出し機を使用して、約1mmの直径のストランドに押し出し、ストランドを約1mmの長さに切断して、MEMを作製する。MEMを250mgの目標を満たす重量(target fill weight)でハードゼラチンカプセルに入れる。
【実施例4】
【0065】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む疎水性マトリックス錠剤
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン 30mg
噴霧乾燥ラクトース 70mg
ヒドロキシエチルセルロース 10mg
セトステアリルアルコール 25mg
タルク 3mg
ステアリン酸マグネシウム 2mg
合計 140mg
【0066】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、ラクトース及びセトステアリルアルコールを、ヒドロキシエチルセルロースを結合剤として使用して湿式造粒する。乾燥し、篩分けした顆粒をタルクと混合する。ステアリン酸マグネシウムを加え、混合する。140mgの目標重量に圧縮成形する。
【実施例5】
【0067】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む溶融押出し顆粒(MEG)錠剤
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン 30mg
ステアリルアルコール 20mg
グリセリルベヘナート 10mg
二塩基性リン酸カルシウム 29mg
微結晶性セルロース 30mg
ステアリン酸マグネシウム 1mg
合計 120mg
【0068】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、ステアリルアルコール、グリセリルベヘナート及び約半分の二塩基性リン酸カルシウムを混合する。混合物を、ステアリルアルコール及びグリセリルベヘナートを融解するために十分な温度で、直径が数mmのストランドに押し出す。ストランドを数mmの長さに切断する。得られた押出し物片を、高速度ミルを使用して粉砕する。粉砕された押出し物を残りの二塩基性リン酸カルシウム及び微結晶性セルロースと混合する。ステアリン酸マグネシウムを加え、混合する。120mgの目標重量に圧縮成形する。
【実施例6】
【0069】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む制御放出ビーズ
【表2】
【0070】
工程1.3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを、ミキサーを使用して容器において563mLの水に溶解する。その後、Opadry Clearをこの溶液に溶解する。1kgの流動層において、上記溶液を約40℃の入口温度において約10mL/分でNuParielビーズに噴霧する。
【0071】
工程2.トリエチルシトラート及びタルクを水に分散し、次いで、Eudragit L30D分散物を、ミキサーを使用して加える。分散させたとき、この分散物を、約40℃の入口温度において約10mL/分で、1kgの流動層において工程1からの薬物負荷ビーズに噴霧する。
【実施例7】
【0072】
本発明による医薬組成物の全体的影響及び薬物動態学的パラメータを明らかにするために、健康な男性被験者における増量単用量ランダム化二重盲検プラセボ対照試験が行われている。本発明による医薬組成物は経口用懸濁物として投与されている。
【0073】
予想された最初の用量増加系列は、1mg、3mg、10mg、30mg、100mg、300mg、600mg及び1000mgである。さらに、活性薬剤の60mg用量が試験されている。
【0074】
試験集団、参加基準及び除外基準
6名の被験者からなる群での合計で72名までの被験者が米国での第I相臨床試験に登録された。
【0075】
研究参加者が、下記に列記される参加基準及び除外基準に従って選択された。一般には、年齢が21歳〜45歳の範囲にあり、正常な睡眠履歴が明らかにされた健康な男性被験者が、本研究のための好適な被験者である。
【0076】
具体的には、被験者を下記の参加基準に従って選択した:
年齢が21歳〜45歳の男性被験者。
・40kg〜100kgの範囲での体重及び30kg/m2未満のボディマス指数(BMI)を有する被験者。
・注目するほどではない病歴、身体検査、生命徴候、研究室評価、12誘導線心電図(ECG)、及びホルターモニターを24時間にわたって使用する携帯型ECGによって明らかにされるように健康である被験者。
・ランダム化前30日間において夜間に6.5時間〜8.5時間の睡眠を取る被験者。夜の就寝日課は試験参加前の少なくとも1週間については午後9:30〜午前12:00の間である。
・プロトコル特異的な手順を開始する前にインフォームドコンセントに署名する被験者。
【0077】
本研究から除外された被験者は下記の被験者であった:
・過去6ヶ月の期間中において臨床的に著しい睡眠異常の何らかの前歴を有する被験者。
・夜間労働又は交代勤務を含む、ランダム化前30日間において何らかの著しい睡眠不規則を有する被験者。
・ランダム化前30日間において3つ以上の時間帯を越えて移動する被験者。
・パルスオキシメトリーによって測定されたときに94%より少ない酸素飽和度(SpO2)を有する被験者。
・ランダム化前30日間において(15分以上の)日課的な昼寝を取る被験者。
・向精神性薬物又は催眠性薬物に対する過敏性の何らかの前歴を有する被験者。
・精神医学的障害(例えば、精神病、強迫性障害、大うつ病又は不安障害/恐慌性障害など)の何らかの病歴を有する被験者。
・薬物の吸収、分布、代謝又は排出を妨害する恐れがある病歴又は何らかの現在の状態を有する被験者。
・ランダム化前30日以内において何らかの催眠剤又は睡眠補助薬(メラトニンを含む)を使用する被験者。
・薬物乱用又はアルコール中毒の前歴を有する被験者。
・過去1年間において発作又は閉鎖性頭部外傷の前歴を有する被験者。
・過去3ヶ月以内において喫煙の前歴又はニコチン含有製品の使用を有する被験者。
・ランダム化前48時間以内においてアルコール飲料物を消費する被験者。
・ランダム化前30日間において1日に5杯以上の茶、コーヒー又はソーダを日課的に消費する被験者。
・服用前3日間の期間中においてカフェイン含有の食物又は飲料物を消費する被験者。
・ランダム化前30日間の期間中において何らかの臨床的に著しい病気を有する被験者。
・ランダム化前7日間の期間中において、何らかの薬物(処方薬及び大衆薬を含む)、1日摂取値の300%を超える何らかのビタミン剤及び/又はミネラル補助物、グレープフルーツジュース及びセイヨウオトギリソウを使用する被験者。
・服用前10時間及び研究用薬物投与後4時間の絶食を拒否し、また、アルコール、カフェイン又はキサンチンを含有する食物又は飲料物を絶つことを全研究期間にわたって拒否する被験者。
・ランダム化前30日間において臨床研究に参加した被験者。
・ランダム化前30日間において献血したか、又は血液製剤を提供した被験者。
・尿中薬物スクリーニング、尿中ニコチン、血中アルコール、HBsAg、HBsAb(被験者が免疫化されていない限り)、抗HCV又は抗HIVの陽性結果を有する被験者。
・7日前から1日前までの睡眠記録によって、又は1日前の夜(登録後最初の夜)でのアクチグラフィーによって示されるような30分を超える睡眠潜時、あるいは、85%より少ない又は95%超の睡眠効率を有する被験者。
【0078】
参加基準又は除外基準からの小さいずれは、その被験者が研究に参加する前の、治験依頼者の医学的モニターからの特別な許可によってのみ許される。
【0079】
研究設計:
本研究は、健康な成人男性被験者における増量単用量ランダム化二重盲検プラセボ対照試験として設計される。
【0080】
本研究は6名の被験者のコホートで行われ、そのうちの4名が、活性な薬物を受けるためにランダム化され、また、そのうちの2名がプラセボを受ける。12までの群を研究することができる。
【0081】
予想された最初の用量増加系列は、1mg、3mg、10mg、30mg、100mg、300mg、600mg及び1000mgである。この系列は、下記の原則に基づいて、指示されたならば、変更することができる:1)前回の用量レベルでのさらなる被験者群を、臨床での観察結果を高めるために研究することができる;2)先行する最大用量よりも低い用量の研究を含み、その後のコホートのために増量比を小さくすること;及び3)生物的利用能に対する食物の影響を明らかにするために、所定の用量レベルを食物(例えば、高脂肪食)とともに投与することができる。食物とともに投与されるとき、用量は、絶食条件のもとでの前回に投与された十分に許容された用量よりも1/10を超えない。用量増加のための判断基準は、研究薬物をそれぞれのコホートに投与した後での安全性、許容性、及び利用可能な薬物動態学的データである。
【0082】
用量増加は、用量を制限する毒性に遭遇するまで、又は最大用量に達するまで続けられる。
【0083】
基本的には、本研究は下記の段階において設計される:
・スクリーニング
・ベースライン期間
・処置期間
【0084】
スクリーニング期間はランダム化前の28日まで続けることができる。この期間中に、被験者は研究適格性について評価され、また、研究ユニットへの許可がなされる前に、連続した6夜について睡眠記録を保存する。
【0085】
下記の評価が、被験者がインフォームドコンセントに署名した後のスクリーニング期間において7日前又は7日前以前に行われる。
・病歴
・身体検査
・生命徴候
・指パルスオキシメトリーによって測定される酸素飽和度
・研究室評価
・従来的な12誘導線ECG
・ホルターモニター又はH−12デジタルレコーダーを用いた24時間ECG
【0086】
参加基準及び除外基準
被験者がホルターレコーダー又はH−12デジタルレコーダーを着用している24時間の期間中、被験者は、激しい身体活動又は運動をしないように制限される。
【0087】
上記の研究室評価には、血液学、血清化学、肝機能検査、HBsAg、HBsAb、抗HCV及び抗HIVについての血清学、尿検査、血中アルコール、並びに尿中薬物/ニコチンスクリーニングが含まれる。
【0088】
被験者の適格性についての予備的評価が、上記で具体的に述べられた参加基準及び除外基準、並びに上記試験の結果に基づいて行われる。
【0089】
ベースライン期間は、2日前の夕方での登録から、1日目での最初の服用前測定の直前までである。この期間中に、被験者は研究施設に滞在し、更新された病歴、身体検査、生命徴候、パルスオキシメトリー、研究室検査、薬物スクリーニング、24時間ECG、24時間EEG、終夜アクチグラフィー、並びにCCT(コンピューター認知検査)及びSSS(スタンフォード眠気スケール)訓練によってその適格性についてさらに評価される。1日前からのその睡眠記録が完了し、1日目の午前中に検討される。登録基準を満たす被験者がベースライン期間終了時にランダム化される。
【0090】
ベースライン期間は、研究ユニットへの参加が2日前の夕方に許可されたときに始まり、1日目での最初の服用前測定の直前で終わる。
【0091】
被験者は、研究ユニットでの参加が2日前に許可され、研究3日目まで施設に収容される(4泊5日)。食事及び間食が研究センターによって提供される。食物又は飲み物は、被験者による研究施設内への持ち込みが禁止される。グレープフルーツ、グレープフルーツジュース、アルコール、カフェイン又はキサンチンを含有する食物又は飲料物(チョコレートを含む)が、滞在期間中を通して禁止される。喫煙、又は研究薬物以外の薬物(大衆薬及び生薬(例えば、セイヨウオトギリソウなど)を含む)が禁止される。たとえ、有害事象を処置するためであっても、何らかの薬物がベースライン期間中に使用されるならば、その被験者は研究から外される。
【0092】
ベースラインの生命徴候及びSpO2が、服用前のおよそ24時間前、23.75時間前、23.5時間前、23時間前、22.5時間前、22時間前、21時間前、20時間前、19時間前、18時間前、16時間前、14時間前及び12時間前に、被験者が仰臥位で3分間休息した後で測定される。
【0093】
加えて、ベースラインでの、SpO2を伴う従来の12誘導線ECG及びEEGが行われる。
【0094】
血中アルコール、尿中薬物/ニコチンスクリーニング、及び研究室検査(血液学、血清化学、肝機能検査及び尿検査など)が行われる。過去6日間にわたる被験者の睡眠記録が適格性について検討される。
【0095】
処置期間は継続して5日までである。服用日の午前中に、被験者は、活性な研究薬物又はプラセボのいずれかの経口用量物を受けるためにランダム化される。被験者は、この期間中、安全性について綿密にモニターされ、また、様々な薬力学的測定を使用して試験される。例えば、血液サンプル及び尿サンプルが、活性薬剤及びその代謝産物の濃度を求めるために集められる。
【0096】
この二重盲検期は1日目での最初の服用前測定の収集から始まり、5日目での研究終了まで続く。
【0097】
服用前の生命徴候測定、パルスオキシメトリー及びECGが、服用前の30分以内に上記順序で行われる。さらに、服用前の血液サンプル及び研究室検査(血液学、血清化学、肝機能検査及び尿検査)が、生命徴候、SpO2及びECGが測定された後に集められる。
【0098】
続いて、服用前のSSS(スタンフォード眠気スケール)及びCCT(コンピューター化認知検査)が、服用前の血液サンプルを集めた後に行われる。SSSが最初に行われ、次いで、CCTが下記の順で行われる:単純反応時間(Simple Reaction Time)、選択反応時間(Choice Reaction Time)、次いで、指ビジランス(Digit Vigilance)。
【0099】
投与後、生命徴候及びSpO2が、服用後0.25時間、0.5時間、1.0時間、1.5時間、2時間、3時間、4時間、5時間、6時間、8時間、10時間、12時間、24時間、30時間、36時間、48時間、60時間、72時間、84時間及び研究終了時(これは服用後96時間である)において測定される。
【0100】
薬物動態学用サンプルが、薬物暴露の程度を調べるためにそれぞれのコホートについて分析される。血液サンプルが、活性薬剤、その代謝産物、及び関連物質を下記の時点で測定するために集められる:服用後0.25時間、0.5時間、1.0時間、1.5時間、2時間、3時間、4時間、5時間、6時間、8時間、10時間、12時間、24時間、30時間、36時間、48時間、60時間、72時間、84時間及び研究終了時。5回までのさらなる服用後の血液サンプル採取を行うことができ、かつ/又は、薬物動態学的パラメータを得る時期を、以前のコホートからの血液サンプルを分析した後で、指示されるならば変更することができる。生命徴候、パルスオキシメトリー及びECGが得られた後、サンプルが集められる。
【0101】
薬物動態学用サンプルが、1mg、3mg、10mg、30mg及び60mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む本発明による医薬組成物から得られている。分析された被験者の平均血漿中濃度が表IIに示される。
表II
【表3】
【0102】
加えて、研究室検査(血液学、血清化学、肝機能検査及び尿検査)が、服用後24時間及び48時間並びに研究終了時に行われる。
【0103】
SSS及びCCTを使用する薬力学的評価が、服用後0.5時間、1時間、1.5時間、2時間、3時間、4時間、5時間、6時間、8時間、10時間及び12時間、並びに生命徴候、パルスオキシメトリー評価及び薬物動態学用サンプル採取の後で(服用前と同じ順序により)行われる。SSSがCCTの前に行われる。
【0104】
研究期間中、食事が所定の時間で与えられ、また、必要ならば、研究者は、被験者のための特定の食事を、そのような食事のときにおける鎮静の程度に依存して抜くように決定することができる。
【0105】
被験者は、48時間の手順が完了した後で施設から出ることができ、また、指定された時間でのその後の手順(例えば、生命徴候測定及び採血)のために施設に戻る。被験者は、何らかの有害事象又は安全性に関連した他の理由のために、研究者の判断で研究施設に留まり続けるように依頼される場合がある。
【0106】
研究終了時の評価が、服用後96時間で、又は早期中断時に行われる。これには、生命徴候(血圧、脈拍数、呼吸数及び体温)、パルスオキシメトリー、身体検査、ECG、研究室検査(血液学、血清化学、肝機能検査及び尿検査)及び薬物動態学用血液採取が含まれる。
【0107】
ここまで本発明が詳しく記載されているが、本発明は、本発明の範囲又はその任意の実施形態に影響を及ぼすことなく、広範囲かつ同等な様々な条件、配合物及び他のパラメータの範囲内で行われ得ることが当業者によって理解される。本明細書中に引用されたすべての特許及び刊行物は、それらの全体において、参考として本明細書中に全体が組み込まれる。
【図面の簡単な説明】
【0108】
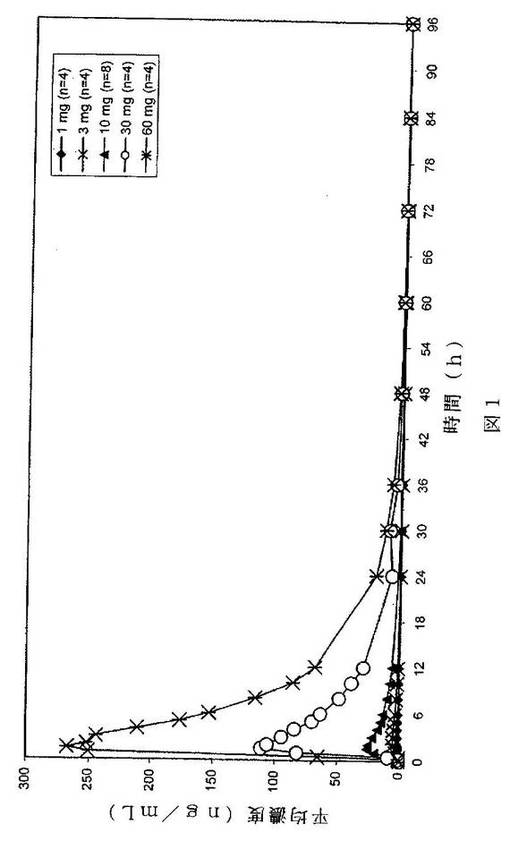
【図1】経時的な血漿中濃度レベルを示すグラフである。
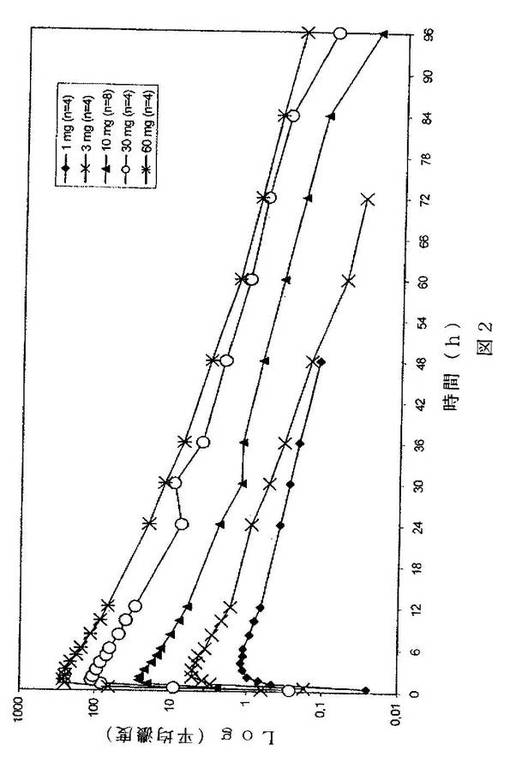
【図2】経時的な血漿中濃度レベルを半対数により示すグラフである。
【図3】経時的な血漿中濃度の用量正規化プロットを示すグラフである。
【技術分野】
【0001】
本発明は、医薬品化学の分野、並びに気分障害などを処置するために有用な医薬組成物に関する。より具体的には、本発明は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルをもたらす、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン又はその医薬的に許容される塩もしくは溶媒和物の医薬組成物に関する。本発明はまた、本発明の医薬組成物を投与することによってこれらの障害を処置する方法に関する。
【背景技術】
【0002】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンは神経刺激性の合成ステロイドである。その主な分子的標的はγ−アミノ酪酸タイプA(GABAA)受容体であり、この場合、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンはチャ
ネル機能の正のアロステリック調節因子として作用する。他のクラスのGABAA調節因子(例えば、ベンゾジアゼピン系化合物及び他のベンゾジアゼピン部位リガンドなど)と同様に、様々な神経刺激性ステロイドが、例えば、下記の障害を処置するために、可能性のある数多くの適応を有する:睡眠障害(例えば、Edgar, D.M.他、J. Pharmacol. Exp. Ther.、282:420-29(1997); Friess, E.他、Am. J. Physiol.、272(Endocrin. Metab. 35):E885-91(1997)を参照のこと)、不安(例えば、Purdy, R.H.他、Proc. Natl. Acad. Sci.、88:4553-57(1991); Vanover, K.E.他、J. Pharmacol. Exp. Ther. 295:337-45(2000); Strohle, A.他、Arch. Gen. Psychiatry、60:161-68(2003)を参照のこと)、うつ病(例えば、Dong, E.他、Proc. Natl. Acad. Sci.、98:2849-54(2001); Rupprecht, R.及びHolsboer, F.、Trends Neurosci.、22:410-16(1999); Uzunova, V.他、Proc. Natl. Acad. Sci.、95:3239-44(1998)を参照のこと)、てんかん(例えば、Carter, R.B.他、J. Pharmacol. Exp. Ther.、280:1284-95(1997); Laxer, K.他、Epilepsia、41:1187-94(2000); Kerrigan, J.F.他、Epilepsy Res.、42:133-39(2000)を参照のこと)、並びに月経前症候群(PMS)及び月経前不快障害(PMDD)(例えば、Rapkin, A.J.他、Obs. Gyn.、90:709-14(1997); Monteleone, P.他、Eu. J. Endocrinol.、142:269-73(2000); Smith, M.J.他、Biol. Psychiatry、54:757-62(2003)を参照のこと)。
【0003】
米国特許第5,939,545号及び同第6,277,838号には、下記式の化合物:
【化1】
が、ストレス又は不安、気分障害(うつ病、月経前症候群又は出産後うつ病を含む)の処置又は予防において有用であるとして議論される。例えば、‘838号特許の第60欄60行〜第61欄65行を参照のこと。1つのそのような化合物が下記式の3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンである:
【化2】
Vanover, K.E.他、Psychopharmarmacology、155:285-91(2001)を参照のこと。
【発明の開示】
【0004】
本発明の第1の態様は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン又はその医薬的に許容される塩もしくは溶媒和物と、1つ又はそれ以上の医薬的に許容される賦形剤とを含み、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態血漿中レベルをもたらす医薬組成物に関する。ヒトにおける気分障害及び同様の障害などを処置するための目標血漿中レベルは約5ng/mL〜約500ng/mLの範囲であり、特に約50ng/mL〜約250ng/mLの範囲である。CNS副作用及び他の副作用が、約500ng/mLを越える血漿中レベルで生じることが予想される。
【0005】
本発明の第2の態様は、その処置が3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルから利益を受ける状態又は障害を処置する方法に関する。1つの実施形態において、そのような状態は気分障害である。
【発明を実施するための最良の形態】
【0006】
本発明は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルをもたらす、ヒトにおける1つ又はそれ以上の状態又は障害を処置するために有用な医薬組成物を提供する。1つの実施形態において、医薬組成物は固体の経口用製剤である。本発明の医薬組成物により処置可能な状態又は障害は、とりわけ、不安、うつ病、アルコール嗜癖、アルコール依存、月経前緊張、月経前症候群及び月経前不快障害から選択することができる。不安には、例えば、全般性不安障害、恐慌性障害、強迫性障害、心的外傷後ストレス障害及び社会不安障害などが含まれる。
【0007】
臨床研究では、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンが下記の薬物動態学的性質を経口服用後にヒトにおいて有することが示唆される:(1)Tmaxが約1時間〜約3時間の範囲である迅速な吸収;(2)被験者間での可変なCmaxレベル;(3)用量比例を上回るCmax値;及び(4)5つの異なる服用群の間で平均して約12時間であったT1/2値。下記の表1を参照のこと。本発明の関連では、薬物動態学的パラメータ(例えば、AUC、Cmax及びtmaxなど)は平均値を示す。括弧において報告される値は標準偏差に対応する。
表I
【表1】
【0008】
これらの薬物動態学的性質を考えると、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの血漿中レベルを規定の治療的範囲においてもたらすことができる組成物は、この薬物の持続した治療的血漿中レベルを必要とする障害(例えば、気分障害など)を処置することにおいて有益であると考えられる。このような持続した血漿中レベルは、適切な技術を使用して、例えば、制御放出配合物を使用して達成することができる。そのような配合物は、即時放出配合物を上回る利点(例えば、持続した効力、低下した副作用、及び服用の改善された容易さなど)をもたらすと考えられる。
【0009】
濃度勾配又は血漿中曲線を、Cmax、tmax及びAUCなどのパラメータによって記述することができる。これらのパラメータは、特定の薬物配合物の薬物動態学的性質を記述することにおいて重要である。
【0010】
血漿中曲線を記述するパラメータは、例えば、活性薬剤(例えば、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンなど)を多数の好適な試験被験者に投与することによって臨床試験で得ることができる。その後、個々の試験者の血漿中での値が平均化される(例えば、AUC、Cmax及びtmaxの平均値が得られる)。本発明の関連では、薬物動態学的パラメータ(例えば、AUC、Cmax及びtmaxなど)は平均値を示す。
【0011】
薬物動態学的パラメータ(例えば、平均tmax、平均Cmax及び平均AUCなど)が健康なヒト被験者について測定されるならば、そのようなパラメータは、典型的には、血漿中での値の発達を健康なヒト被験者の適切な試験集団において経時的に測定することによって得られる。
【0012】
被験者の血液サンプルを、任意の好適な時間間隔で、好ましくは、下記時点のいずれか1つ又は組合せにおいて採取することができる:服用前、服用後0.25時間、0.5時間、1.0時間、1.5時間、2時間、3時間、4時間、5時間、6時間、8時間、10時間、12時間、24時間、30時間、36時間、48時間、60時間、72時間、84時間及び研究終了時(これは、例えば、服用後96時間である場合がある)。
【0013】
この関連での用語「健康な」ヒト被験者は、身長、体重及び生理学的パラメータ(例えば、血圧など)に関して平均値を有する、通常の場合には白人、黒人、アジア人又は他の指定された起源の典型的な男性又は女性を示す。本発明の目的のための健康なヒト被験者は、臨床試験協調化のための国際会議(ICH)の勧告に基づき、また、勧告に従う参加基準及び除外基準に従って選択される。本発明の目的のために、健康な被験者は、実施例において概略されるような参加基準及び除外基準に従って特定することができる。
【0014】
従って、参加基準には、21歳超〜45歳未満の年齢;男性について40kg〜100kgの範囲の体重及び30kg/m2未満のボディマス指数(BMI);病歴、身体検査、生命徴候、研究室評価、12誘導線心電図(ECG)、及びホルターモニターを24時間にわたって使用する携帯型ECGでの著しく異常な知見がないことによって明確に示される全般的に良好な健康状態が含まれる。さらに、被験者は、好ましくは、ランダム化前30日間において、好ましくは、所与の時間枠内で夜間に6.5時間〜8.5時間の睡眠を取る。
【0015】
典型的な除外パラメータには、過去6ヶ月の期間中における臨床的に著しい睡眠異常の何らかの前歴;夜間労働又は交代勤務を含む、ランダム化前30日間における何らかの著しい睡眠不規則;ランダム化前30日間において3つ以上の時間帯を越える移動;パルスオキシメトリーによって測定されたときの94%以下の酸素飽和度(SpO2);ランダム化前30日間における(15分以上の)日課的な昼寝;向精神性薬物又は催眠性薬物に対する過敏性の何らかの前歴;精神医学的障害(例えば、精神病、強迫性障害、大うつ病又は不安障害/恐慌性障害など)の何らかの病歴;薬物の吸収、分布、代謝又は排出を妨害する恐れがある病歴又は何らかの現在の状態;ランダム化前30日以内における何らかの催眠剤又は睡眠補助薬(メラトニンを含む)の使用;薬物乱用又はアルコール中毒の前歴;過去1年間における発作又は閉鎖性頭部外傷の前歴;過去3ヶ月以内における喫煙の前歴又はニコチン含有製品の使用;ランダム化前48時間以内におけるアルコール飲料物の消費;ランダム化前30日間における1日に5杯以上の茶、コーヒー又はソーダの日課的消費;服用前3日間の期間中におけるカフェイン含有の食物又は飲料物の消費;ランダム化前30日間の期間中における何らかの臨床的に著しい病気;ランダム化前7日間の期間中における何らかの薬物(処方薬及び大衆薬を含む)、1日摂取値の300%を超える何らかのビタミン剤及び/又はミネラル補助物、グレープフルーツジュース及びセイヨウオトギリソウ;服用前10時間及び研究用薬物投与後4時間の絶食を拒否すること、並びにアルコール、カフェイン又はキサンチンを含有する食物又は飲料物を絶つことを全研究期間にわたって拒否すること;ランダム化前30日間における臨床研究への参加;ランダム化前30日間における献血又は血液製剤提供;尿中薬物スクリーニング、尿中ニコチン、血中アルコール、HBsAg、HBsAb(被験者が免疫化されていない限り)、抗HCV又は抗HIVの陽性結果;7日前から1日前までの睡眠記録によって、又は1日前の夜(登録後最初の夜)でのアクチグラフィー(actigraphy)によって示されるような30分を超える睡眠潜時、あるいは、85%以下又は95%超の睡眠効率が含まれる。
【0016】
薬物動態学的パラメータ(例えば、平均tmax、平均Cmax及び平均AUCなど)が被験者において得られるならば、被験者群は好適な数の被験者を含む。妥当な数の被験者は、例えば、4名、8名、10名、20名、30名、40名、50名、60名、72名又はより一層多数の患者である。被験者は、処置される状態の症状に従って選択される。被験者は、例えば、用量増加を調べるために、好適なサイズのコホートにグループ分けすることができる。本発明の目的のために、被験者は、実施例において提供される参加基準及び除外基準に従って選択することができる。
【0017】
上記及び下記において示されるような薬物動態学的パラメータの値は、実施例7で得られたデータ(それらのすべてが健康なヒト被験者における単用量研究に関連する)に基づいて推定されていることを理解しなければならない。しかしながら、比較可能な結果が、健康なヒト被験者における定常状態での投与、又はヒト患者における単用量及び定常状態での投与の時に得られることが推定される。
【0018】
本発明による医薬組成物は、約1.5時間〜約5時間の範囲で、好ましくは約1.3時間〜約3.5時間の範囲で、より好ましくは約1.6時間〜約3.2時間の範囲でTmax値を示す。臨床研究では、本発明による組成物が、好ましくは、活性薬剤(すなわち、本発明による医薬組成物に存在する3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)の量に依存することなく、迅速に吸収されることが示唆される。
【0019】
本発明の医薬組成物のCmax値は、1mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約1.0ng/mL〜約1.8ng/mLの範囲であり、好ましくは約1.2ng/mL〜約1.6ng/mLの範囲である。
【0020】
本発明の医薬組成物のCmax値は、3mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約4.0ng/mL〜約8.0ng/mLの範囲であり、好ましくは約4.0ng/mL〜約6.5ng/mLの範囲であり、より好ましくは約5.0ng/mL〜約6.0ng/mLの範囲である。
【0021】
本発明の医薬組成物のCmax値は、10mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約16.9ng/mL〜約46.3ng/mLの範囲であり、好ましくは約21.0ng/mL〜約32.0ng/mLの範囲であり、より好ましくは約25.0ng/mL〜約28.0ng/mLの範囲である。
【0022】
本発明の医薬組成物のCmax値は、30mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約95.6ng/mL〜約154.0ng/mLの範囲であり、好ましくは約108.0ng/mL〜約134.0ng/mLの範囲であり、より好ましくは約115.0ng/mL〜約125.0ng/mLの範囲である。
【0023】
本発明の医薬組成物のCmax値は、60mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤については約218.0ng/mL〜約479.0ng/mLの範囲であり、好ましくは約275.0ng/mL〜約385.0ng/mLの範囲であり、より好ましくは約300ng/mL〜約350ng/mLの範囲である。
【0024】
さらに、本発明による医薬組成物を投与することによって、用量比例よりも大きいCmax値を得ることができる。
【0025】
用量比例よりも大きいCmax値は、本発明の関連では、活性薬剤の正規化されたCmax値が一定でないことを意味する。別の言い方をすれば、量1(例えば、10mg)の活性薬剤についての正規化されたCmax値が、量2(例えば、1mg)の活性薬剤についての正規化されたCmax値により大きい。活性薬剤(3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)の、用量比例よりも大きいCmax挙動もまた図3に示される。図3は、用量により正規化された血漿中濃度の時間に対する図を示す。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む本発明による3mg製剤、10mg製剤、30mg製剤及び60mg製剤についての正規化されたCmax値が、本発明による1mg製剤について得られたCmaxよりも大きい。1mgを超える活性薬剤を含む製剤について得られる正規化されたCmax値は、本発明による1mg製剤から得られたCmax値よりも少なくとも約2倍、少なくとも約2.5倍、少なくとも約3倍、少なくとも約4倍、少なくとも約5倍大きい正規化されたCmax値を有することができる。60mgを超える3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む製剤は、少なくとも約5を超える用量比例したCmax値を示すことが推定され得る。何らかの理論によって限定されることはないが、本発明による1mg製剤のCmax値と比較して、少なくとも約7、少なくとも約10、少なくとも約15又は少なくとも約20又は少なくとも約25を超える正規化されたCmax値が本発明の製剤により達成され得ることが推定され得る。
【0026】
本発明による医薬組成物の、用量比例よりも大きいCmax値は、活性薬剤の量をほんのわずかに変更するか、又はほぼ同じレベルで維持しながら、Cmaxの著しい増大を可能にすることができる。
【0027】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルは約5ng/mL〜約500ng/mLの範囲が可能である。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの他の治療的範囲には、約50ng/mL〜約500ng/mL、約50ng/mL〜約400ng/mL、約50ng/mL〜約325ng/mL、約50ng/mL〜約250ng/mL、約50ng/mL〜約100ng/mL、及び約100ng/mL〜約250ng/mLが含まれる。
【0028】
これらの定常状態血漿中レベルは、適切な技術を使用して達成することができ、例えば、適切な放出プロフィールを有する制御放出配合物を使用して達成することができる。適切な放出プロフィールを、例えば、単一送達システム又は多粒子送達システムを使用して達成することができる。単一送達システムの例には、ワックスマトリックス錠剤、親水性マトリックス錠剤、及び制御放出被覆を伴う錠剤が含まれるが、これらに限定されない。多粒子システムの例には、制御放出被覆に基づく溶融押出し多粒子剤又は溶融押出し多粒子システムなどのマトリックスシステム(例えば、被覆ビーズなど)が含まれるが、これらに限定されない。
【0029】
本発明によるこれらの制御放出型医薬組成物は、活性薬剤と一緒にマトリックスに配合される制御放出物質、又は活性薬剤を含む基質を覆う制御放出被覆剤として適用される制御放出物質を含むことができる。用語「基質」は、ビーズ、ペレット、球状物、錠剤、錠剤コアなどを含むことができる。制御放出物質は、所望により疎水性又は親水性であり得る。本発明による製剤は、好ましくは経口用製剤であり、例えば顆粒、球状物、ペレットなどとして提供することができる。他方で、本発明の製剤は、制御放出被覆剤により被覆された錠剤として、又は活性薬剤のマトリックスと、制御放出物質と、場合により、他の医薬的に許容される成分(例えば、結合剤、希釈剤、着色剤、滑剤など)とを含む錠剤として調製することができる。
【0030】
制御放出配合物についての例には、WO01/32148、米国特許第4,861,598号、米国特許第4,990,341号、米国特許第4,884,909号、それらにおいて引用される参考文献、及び当分野において開示される他の参考文献に開示される配合物が含まれ得る。
【0031】
1つの実施形態において、本発明の医薬組成物は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的な定常状態血漿中レベルを投与後の約12時間〜約24時間の持続期間にわたってもたらす。別の実施形態において、本発明の医薬組成物は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的な定常状態血漿中レベルを投与後の約6時間〜約12時間の持続期間にわたってもたらす。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンは光学異性体(エナンチオマー)として存在する場合があり、本発明は、そのような光学異性体のラセミ混合物及びエナンチオマー濃縮された混合物の両方、並びに当業者には周知である方法に従って分離することができる個々のエナンチオマーを含む。
【0032】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンは、米国特許出願第60/604,447号(これは本明細書によりその全体が参考として組み込まれる)に記載されるように同形の結晶性晶癖として存在する場合がある。
【0033】
本発明の範囲にはまた、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの溶媒和形態(特に、水和形態)を含む、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態治療的血漿中レベルをもたらす組成物が含まれる。水和は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、又は3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む組成物を製造しているときに生じ得るか、あるいは、水和は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの吸湿性のために時間とともに生じ得る。本発明の範囲にはまた、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの医薬的に許容される塩を含む、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態治療的血漿中レベルをもたらす組成物が含まれる。医薬的に許容される酸付加塩が、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの溶液を医薬的に許容される非毒性の酸(例えば、塩酸、フマル酸、マレイン酸、コハク酸、酢酸、クエン酸、酒石酸、炭酸、リン酸、シュウ酸及びジクロロ酢酸など)の溶液と混合することによって形成される。医薬的に許容される塩基性塩が、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの溶液を医薬的に許容される非毒性の塩基(例えば、水酸化ナトリウム、水酸化カリウム、水酸化コリン及び炭酸ナトリウムなど)の溶液と混合することによって形成される。
【0034】
本発明はまた、その処置が3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの持続した治療的血漿中レベルから利益を受ける状態又は障害を処置する方法に関する。この処置方法は、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態治療的レベルをもたらす医薬組成物を、そのような処置を必要としている被験者に投与することを含む。そのような状態及び障害には、気分障害、例えば、不安、うつ病、アルコール嗜癖及び/又はアルコール依存、月経前緊張、月経前症候群及び月経前不快障害などが含まれる。不安には、例えば、全般性不安障害、恐慌性障害、強迫性障害、心的外傷後ストレス障害及び社会不安障害などが含まれる。
【0035】
定義
状態又は障害を「処置する」、あるいは、状態又は障害の「処置」の用語は、本明細書中で用いられる場合、(i)状態又は障害を阻害すること、すなわち、状態又は障害又はその臨床的症状の発達を停止させること、及び/又は(ii)状態又は障害を軽減すること、すなわち、状態又は障害又はその臨床的症状の一時的又は永続的な退行を生じさせることを示す。
【0036】
用語「キラル中心」は、4つの異なる基が結合する炭素原子、又は3つ異なる基が結合するイオウ原子(この場合、イオウ原子及びその結合した基は、スルホキシド、スルフィン酸エステル、スルホニウム塩又はスルフィトを形成する)を示す。
【0037】
用語「エナンチオマー」又は用語「エナンチオマー状」は、その鏡像と重ねることができず、従って、光学活性である分子を示し、この場合、エナンチオマーは、偏光した光の面を一方の方向に回転させ、その鏡像体は、偏光した光の面を反対方向に回転させる。
【0038】
用語「ラセミ(状)」は、光学的に不活性である、等しい量のエナンチオマーの混合物を示す。
【0039】
用語「分割」は、分子の2つのエナンチオマー形態の一方を分離又は濃縮又は減損することを示す。表現「エナンチオマー濃縮された」は、一方のエナンチオマーが、その鏡像分子よりも高い濃度で存在する混合物を示す。
【0040】
Cmax値は、活性薬剤(すなわち、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)の最大血漿中濃度を示す。
【0041】
tmax値は、Cmax値に達する時点を示す。別の言い方をすれば、tmaxは最大の観測された血漿中濃度の時点である。
【0042】
AUC(曲線下面積)値は濃度曲線の面積に対応する。AUC値は、全体では血液循環に吸収された活性薬剤(すなわち、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)の量に比例しており、従って、生物学的利用能についての尺度である。
【0043】
AUC(0〜24)値は、投与時から投与後24時間までの血漿中濃度−時間曲線下の面積についての値である。
【0044】
AUC(0〜最後)値は、投与時から最後の測定可能な濃度までの血漿中濃度−時間曲線下の面積についての値である。
【0045】
用語「正規化されたCmax」は、活性薬剤の特定の投薬量のCmax値と、活性薬剤の量との比率を示す。
【0046】
用語「生物学的利用能」は、本発明の目的のためには、活性薬剤(例えば、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン)が製剤から吸収される程度として定義される。
【0047】
本発明の関連での用語「平均」は、少なくとも2名の被験者のデータの平均を示す。
【0048】
用語「持続放出」は、本発明の目的のためには、血中レベルが、約6時間又は約12時間又は約24時間又はより一層長い時間の期間にわたって治療的範囲内で、しかし、毒性レベル未満で維持されるような速度での3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの放出として定義される。用語「持続放出」により、本発明による調製物は「即時放出」調製物から区別される。用語「持続放出」及び用語「制御放出」は交換可能に使用される。
【0049】
用語t1/2は、本発明の目的のためには、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの吸収可能な用量の半分が血漿に移されるために必要な時間の量として定義される。この値は、「見かけ」の吸収半減期ではなく、むしろ、「真の」値(これは排除プロセスの影響が考慮に入れられる)として計算することができる。
【0050】
用語「定常状態」は、所与の薬物についての血漿中レベルが達成され、そのようなレベルが、薬物のその後の服用により、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンについての最小有効治療的レベルであるか、又は最小有効治療的レベルを上回り、かつ、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンについての最小毒性血漿中レベルよりも低いレベルで維持されることを意味する。
【0051】
最小有効治療的レベルは、部分的には、所与の患者において達成された所望される効果の程度によって決定される。この測定は非常に主観的であり、また、大きい個体変動が被験者間に存在し得ることが当業者によって十分に理解される。それぞれの用量を投与した後、濃度が最大値を通過し、その後、最小値に再び低下することは明らかである。
【0052】
定常状態は下記のように記載することができる:時間t=0(最初の用量が投与される時間)で、濃度Cもまた0である。その後、濃度は第1の最大値を通過し、その後、第1の最小値に低下する。濃度が0に低下する前に、別の用量が投与され、その結果、濃度における第2の増大が0において始まらない。この第1の濃度最小値の上に加わって、曲線は、第2の用量が投与された後の第2の最大値(これは第1の最大値を上回る)を通過し、その後、第1の最小値を上回る第2の最小値に低下する。従って、血漿中曲線は、吸収及び排出が均衡状態にある点に落ち着くまで、活性薬剤の繰り返された服用及び付随する段階毎の蓄積のために上昇する。この状態は、吸収及び排出が平衡状態にあり、濃度が、規定された最小値と、規定された最大値との間で絶えず振動する状態であり、この状態が定常状態と呼ばれる。
【0053】
本発明の医薬組成物は、その意図された目的を達成する任意の手段によって投与することができる。例えば、投与は、皮下経路、静脈内経路、筋肉内経路、腹腔内経路、口内経路又は眼経路、直腸的、非経口的、全身的(intrasystemically)、膣内、局所的(粉末、軟膏、滴剤又は経皮パッチによるような局所的)、あるいは、口腔スプレー又は鼻スプレーが可能である。代わりとして、又は同時に、投与は経口経路によることが可能である。経口経路による本発明の医薬組成物の投与が現時点では好ましい。
【0054】
投与頻度は、投与された組成物について予期される作用持続期間にわたって適切でなければならない。例えば、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルを投与後の約12時間〜約24時間の持続期間にわたってもたらす組成物については、組成物を1日に1回与えることができる。同様に、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルを投与後の約6時間〜約12時間の持続期間にわたってもたらす組成物については、組成物を1日に2回与えることができる。
【0055】
下記の実施例は本発明の組成物及び方法を例示するが、本発明の組成物及び方法を限定しない。当業者には自明である、臨床治療において通常の場合には遭遇する多様な条件及びパラメータの他の好適な改変及び適合化は本発明の精神及び範囲に含まれる。
(実施例)
【0056】
3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オンを、Hogenkamp他、「Synthesis and in Vitro Activity of 3β-Suibstituted-3α-hydroxypregnan-20-ones: Allosteric Modulators of the GABAA Receptor」、J. Med. Chem.、40:61-72(1997)に記載されるように、(3R)−スピロ[オキシラン−2α,5α−プレグナン]−20−オン及びナトリウムメトキシドから調製することができる。
【0057】
服用が即時放出懸濁物を用いて行われた3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの単用量試験に基づいて、1日に1回与えられる約30mgの用量(遊離化合物に基づく)が、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルを投与後の約12時間〜約24時間の持続期間にわたってもたらす組成物については適切であることが予想される。この量が下記の実施例では使用され、しかし、必要な用量は約20mg/日〜約40mg/日の範囲が可能であることが理解される。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンが塩の形態であるならば、そのような量に対する適切な調節を行うことができる。そのような修正は十分に当業者の知識及び能力の範囲内である。
【0058】
投薬量は、所望される作用持続期間を達成するために調節することができる。例えば、1日に2回与えられる約15mgの用量(遊離化合物に基づく)が、3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの治療的血漿中レベルを投与後の約6時間〜約12時間の持続期間にわたってもたらす組成物については適切であることが予想される。
【実施例1】
【0059】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの調製
【0060】
表題化合物及びその塩酸塩を下記にように調製することができる。
a)21−ブロモ−3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オン。3α−ヒドロキシ−3β−メトキシメチル−5α−プレグナン−20−オン(30.0g、82.9mmol)を約900mLのメタノールに溶解した溶液に、この溶液を室温で撹拌しながら、3滴の48% HBr水溶液を加える。その後、臭素(13.9g、87.1mmol)を約200mLのメタノールにおける溶液として約2時間かけて滴下して加える。その期間中、反応液を遮光する。TLC(1%アセトン/CH2Cl2)を使用して、出発物質の不在、及び極性がより低い生成物の形成を(さらに約30分後に)示すことができる。反応液を約300mLに濃縮する。CH2Cl2(約400mL)を加え、反応液を約200mLの水を含有する分液漏斗に注ぐ。相を分離し、水相をCH2Cl2で抽出する(約100mL、3回)。有機相を一緒にし、約200mLの飽和NaHCO3水溶液により洗浄し、Na2SO4で乾燥し、減圧下で濃縮して、ブロミドを淡黄色の泡状物として得る。この生成物は、さらに精製することなく次の工程で使用することができる。
b)3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン。上記のように調製されたブロミド(36.7g、82.9mmol)を約800mLのCH3CNに懸濁した懸濁物に、イミダゾール(28.2g、415mmol)を加え、反応物をAr下で加熱還流する。TLC(95:4.5:0.5のCH2Cl2:MeOH:トリエチルアミン)を使用して、反応の完了を(約1時間の還流の後で)示すことができる。反応物を室温に冷却し、真空下で濃縮する。得られたオイルを約600mLのCH2Cl2に溶解し、希NaHCO3溶液により洗浄し(約200mL、4回)、Na2SO4で乾燥し、真空下で濃縮する。95:4.5:0.5のCH2Cl2:MeOH:トリエチルアミンにより溶出するシリカゲルでのフラッシュクロマトグラフィーによる精製により、約18gの表題化合物を白色の固体として得る:m.p.185〜187℃(およそ)。元素分析、計算値(C26H40N2O3):C、72.86;H、9.41;N、6.54。1H NMR(300MHz、CDCl3)δ(およそ)7.40(s、1H)、7.08(s、1H)、6.84(s、1H)、4.72(d、1H、J=17.7Hz)、4.64(d、1H、J=18Hz)、3.39(s、3H)、3.18(s、2H)、2.57(t、1H、J=8.7Hz)、0.76(s、3H)、0.66(s、3H)。
c)3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、塩酸塩。塩化水素ガスを3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン(1.00g、2.33mmol、約35mLのCH2Cl2に溶解する)の溶液に約7分間吹き込む。白色の析出物が形成する。溶媒を真空下で除いて、約1.10gの塩酸塩を白色の固体として得る:m.p.230〜233℃(およそ)。1H NMR(300MHz、CDCl3)δ(およそ)9.66(s、1H)、7.31(s、1H)、7.05(s、1H)、5.45(d、1H、J=18Hz)、5.26(d、1H、J=18Hz)、3.39(s、3H)、3.19(s、2H)、2.72(t、1H、J=8.7Hz)、0.76(s、3H)、0.70(s、3H)。
【実施例2】
【0061】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む親水性マトリックス錠剤
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン 30mg
ヒドロキシプロピルメチルセルロース 30mg
噴霧乾燥ラクトース 88.9mg
コロイド状二酸化ケイ素 0.15mg
ステアリン酸マグネシウム 1mg
合計 150mg
【0062】
コロイド状二酸化ケイ素と、ラクトースの一部とを混合する。混合物を篩分けする。3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、ヒドロキシプロピルメチルセルロース及び残りのラクトースを混合物に加え、混合する。ステアリン酸マグネシウムを加え、混合する。最終混合物を150mgの目標重量に圧縮成形する。
【実施例3】
【0063】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含むカプセル化溶融押出し多粒子剤(MEM)
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン 30mg
Eudragit RLPO 50mg
Eudragit RSPO 132mg
ステアリルアルコール 28mg
グリセリルベヘナート 10mg
合計 250mg
サイズ#1のハードゼラチンカプセル外皮
【0064】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、Eudragit RSPO、Eudragit RLPO、ステアリルアルコール及びグリセリルベヘナートを混合する。ホットメルト押出し機を使用して、約1mmの直径のストランドに押し出し、ストランドを約1mmの長さに切断して、MEMを作製する。MEMを250mgの目標を満たす重量(target fill weight)でハードゼラチンカプセルに入れる。
【実施例4】
【0065】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む疎水性マトリックス錠剤
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン 30mg
噴霧乾燥ラクトース 70mg
ヒドロキシエチルセルロース 10mg
セトステアリルアルコール 25mg
タルク 3mg
ステアリン酸マグネシウム 2mg
合計 140mg
【0066】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、ラクトース及びセトステアリルアルコールを、ヒドロキシエチルセルロースを結合剤として使用して湿式造粒する。乾燥し、篩分けした顆粒をタルクと混合する。ステアリン酸マグネシウムを加え、混合する。140mgの目標重量に圧縮成形する。
【実施例5】
【0067】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む溶融押出し顆粒(MEG)錠剤
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン 30mg
ステアリルアルコール 20mg
グリセリルベヘナート 10mg
二塩基性リン酸カルシウム 29mg
微結晶性セルロース 30mg
ステアリン酸マグネシウム 1mg
合計 120mg
【0068】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン、ステアリルアルコール、グリセリルベヘナート及び約半分の二塩基性リン酸カルシウムを混合する。混合物を、ステアリルアルコール及びグリセリルベヘナートを融解するために十分な温度で、直径が数mmのストランドに押し出す。ストランドを数mmの長さに切断する。得られた押出し物片を、高速度ミルを使用して粉砕する。粉砕された押出し物を残りの二塩基性リン酸カルシウム及び微結晶性セルロースと混合する。ステアリン酸マグネシウムを加え、混合する。120mgの目標重量に圧縮成形する。
【実施例6】
【0069】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む制御放出ビーズ
【表2】
【0070】
工程1.3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを、ミキサーを使用して容器において563mLの水に溶解する。その後、Opadry Clearをこの溶液に溶解する。1kgの流動層において、上記溶液を約40℃の入口温度において約10mL/分でNuParielビーズに噴霧する。
【0071】
工程2.トリエチルシトラート及びタルクを水に分散し、次いで、Eudragit L30D分散物を、ミキサーを使用して加える。分散させたとき、この分散物を、約40℃の入口温度において約10mL/分で、1kgの流動層において工程1からの薬物負荷ビーズに噴霧する。
【実施例7】
【0072】
本発明による医薬組成物の全体的影響及び薬物動態学的パラメータを明らかにするために、健康な男性被験者における増量単用量ランダム化二重盲検プラセボ対照試験が行われている。本発明による医薬組成物は経口用懸濁物として投与されている。
【0073】
予想された最初の用量増加系列は、1mg、3mg、10mg、30mg、100mg、300mg、600mg及び1000mgである。さらに、活性薬剤の60mg用量が試験されている。
【0074】
試験集団、参加基準及び除外基準
6名の被験者からなる群での合計で72名までの被験者が米国での第I相臨床試験に登録された。
【0075】
研究参加者が、下記に列記される参加基準及び除外基準に従って選択された。一般には、年齢が21歳〜45歳の範囲にあり、正常な睡眠履歴が明らかにされた健康な男性被験者が、本研究のための好適な被験者である。
【0076】
具体的には、被験者を下記の参加基準に従って選択した:
年齢が21歳〜45歳の男性被験者。
・40kg〜100kgの範囲での体重及び30kg/m2未満のボディマス指数(BMI)を有する被験者。
・注目するほどではない病歴、身体検査、生命徴候、研究室評価、12誘導線心電図(ECG)、及びホルターモニターを24時間にわたって使用する携帯型ECGによって明らかにされるように健康である被験者。
・ランダム化前30日間において夜間に6.5時間〜8.5時間の睡眠を取る被験者。夜の就寝日課は試験参加前の少なくとも1週間については午後9:30〜午前12:00の間である。
・プロトコル特異的な手順を開始する前にインフォームドコンセントに署名する被験者。
【0077】
本研究から除外された被験者は下記の被験者であった:
・過去6ヶ月の期間中において臨床的に著しい睡眠異常の何らかの前歴を有する被験者。
・夜間労働又は交代勤務を含む、ランダム化前30日間において何らかの著しい睡眠不規則を有する被験者。
・ランダム化前30日間において3つ以上の時間帯を越えて移動する被験者。
・パルスオキシメトリーによって測定されたときに94%より少ない酸素飽和度(SpO2)を有する被験者。
・ランダム化前30日間において(15分以上の)日課的な昼寝を取る被験者。
・向精神性薬物又は催眠性薬物に対する過敏性の何らかの前歴を有する被験者。
・精神医学的障害(例えば、精神病、強迫性障害、大うつ病又は不安障害/恐慌性障害など)の何らかの病歴を有する被験者。
・薬物の吸収、分布、代謝又は排出を妨害する恐れがある病歴又は何らかの現在の状態を有する被験者。
・ランダム化前30日以内において何らかの催眠剤又は睡眠補助薬(メラトニンを含む)を使用する被験者。
・薬物乱用又はアルコール中毒の前歴を有する被験者。
・過去1年間において発作又は閉鎖性頭部外傷の前歴を有する被験者。
・過去3ヶ月以内において喫煙の前歴又はニコチン含有製品の使用を有する被験者。
・ランダム化前48時間以内においてアルコール飲料物を消費する被験者。
・ランダム化前30日間において1日に5杯以上の茶、コーヒー又はソーダを日課的に消費する被験者。
・服用前3日間の期間中においてカフェイン含有の食物又は飲料物を消費する被験者。
・ランダム化前30日間の期間中において何らかの臨床的に著しい病気を有する被験者。
・ランダム化前7日間の期間中において、何らかの薬物(処方薬及び大衆薬を含む)、1日摂取値の300%を超える何らかのビタミン剤及び/又はミネラル補助物、グレープフルーツジュース及びセイヨウオトギリソウを使用する被験者。
・服用前10時間及び研究用薬物投与後4時間の絶食を拒否し、また、アルコール、カフェイン又はキサンチンを含有する食物又は飲料物を絶つことを全研究期間にわたって拒否する被験者。
・ランダム化前30日間において臨床研究に参加した被験者。
・ランダム化前30日間において献血したか、又は血液製剤を提供した被験者。
・尿中薬物スクリーニング、尿中ニコチン、血中アルコール、HBsAg、HBsAb(被験者が免疫化されていない限り)、抗HCV又は抗HIVの陽性結果を有する被験者。
・7日前から1日前までの睡眠記録によって、又は1日前の夜(登録後最初の夜)でのアクチグラフィーによって示されるような30分を超える睡眠潜時、あるいは、85%より少ない又は95%超の睡眠効率を有する被験者。
【0078】
参加基準又は除外基準からの小さいずれは、その被験者が研究に参加する前の、治験依頼者の医学的モニターからの特別な許可によってのみ許される。
【0079】
研究設計:
本研究は、健康な成人男性被験者における増量単用量ランダム化二重盲検プラセボ対照試験として設計される。
【0080】
本研究は6名の被験者のコホートで行われ、そのうちの4名が、活性な薬物を受けるためにランダム化され、また、そのうちの2名がプラセボを受ける。12までの群を研究することができる。
【0081】
予想された最初の用量増加系列は、1mg、3mg、10mg、30mg、100mg、300mg、600mg及び1000mgである。この系列は、下記の原則に基づいて、指示されたならば、変更することができる:1)前回の用量レベルでのさらなる被験者群を、臨床での観察結果を高めるために研究することができる;2)先行する最大用量よりも低い用量の研究を含み、その後のコホートのために増量比を小さくすること;及び3)生物的利用能に対する食物の影響を明らかにするために、所定の用量レベルを食物(例えば、高脂肪食)とともに投与することができる。食物とともに投与されるとき、用量は、絶食条件のもとでの前回に投与された十分に許容された用量よりも1/10を超えない。用量増加のための判断基準は、研究薬物をそれぞれのコホートに投与した後での安全性、許容性、及び利用可能な薬物動態学的データである。
【0082】
用量増加は、用量を制限する毒性に遭遇するまで、又は最大用量に達するまで続けられる。
【0083】
基本的には、本研究は下記の段階において設計される:
・スクリーニング
・ベースライン期間
・処置期間
【0084】
スクリーニング期間はランダム化前の28日まで続けることができる。この期間中に、被験者は研究適格性について評価され、また、研究ユニットへの許可がなされる前に、連続した6夜について睡眠記録を保存する。
【0085】
下記の評価が、被験者がインフォームドコンセントに署名した後のスクリーニング期間において7日前又は7日前以前に行われる。
・病歴
・身体検査
・生命徴候
・指パルスオキシメトリーによって測定される酸素飽和度
・研究室評価
・従来的な12誘導線ECG
・ホルターモニター又はH−12デジタルレコーダーを用いた24時間ECG
【0086】
参加基準及び除外基準
被験者がホルターレコーダー又はH−12デジタルレコーダーを着用している24時間の期間中、被験者は、激しい身体活動又は運動をしないように制限される。
【0087】
上記の研究室評価には、血液学、血清化学、肝機能検査、HBsAg、HBsAb、抗HCV及び抗HIVについての血清学、尿検査、血中アルコール、並びに尿中薬物/ニコチンスクリーニングが含まれる。
【0088】
被験者の適格性についての予備的評価が、上記で具体的に述べられた参加基準及び除外基準、並びに上記試験の結果に基づいて行われる。
【0089】
ベースライン期間は、2日前の夕方での登録から、1日目での最初の服用前測定の直前までである。この期間中に、被験者は研究施設に滞在し、更新された病歴、身体検査、生命徴候、パルスオキシメトリー、研究室検査、薬物スクリーニング、24時間ECG、24時間EEG、終夜アクチグラフィー、並びにCCT(コンピューター認知検査)及びSSS(スタンフォード眠気スケール)訓練によってその適格性についてさらに評価される。1日前からのその睡眠記録が完了し、1日目の午前中に検討される。登録基準を満たす被験者がベースライン期間終了時にランダム化される。
【0090】
ベースライン期間は、研究ユニットへの参加が2日前の夕方に許可されたときに始まり、1日目での最初の服用前測定の直前で終わる。
【0091】
被験者は、研究ユニットでの参加が2日前に許可され、研究3日目まで施設に収容される(4泊5日)。食事及び間食が研究センターによって提供される。食物又は飲み物は、被験者による研究施設内への持ち込みが禁止される。グレープフルーツ、グレープフルーツジュース、アルコール、カフェイン又はキサンチンを含有する食物又は飲料物(チョコレートを含む)が、滞在期間中を通して禁止される。喫煙、又は研究薬物以外の薬物(大衆薬及び生薬(例えば、セイヨウオトギリソウなど)を含む)が禁止される。たとえ、有害事象を処置するためであっても、何らかの薬物がベースライン期間中に使用されるならば、その被験者は研究から外される。
【0092】
ベースラインの生命徴候及びSpO2が、服用前のおよそ24時間前、23.75時間前、23.5時間前、23時間前、22.5時間前、22時間前、21時間前、20時間前、19時間前、18時間前、16時間前、14時間前及び12時間前に、被験者が仰臥位で3分間休息した後で測定される。
【0093】
加えて、ベースラインでの、SpO2を伴う従来の12誘導線ECG及びEEGが行われる。
【0094】
血中アルコール、尿中薬物/ニコチンスクリーニング、及び研究室検査(血液学、血清化学、肝機能検査及び尿検査など)が行われる。過去6日間にわたる被験者の睡眠記録が適格性について検討される。
【0095】
処置期間は継続して5日までである。服用日の午前中に、被験者は、活性な研究薬物又はプラセボのいずれかの経口用量物を受けるためにランダム化される。被験者は、この期間中、安全性について綿密にモニターされ、また、様々な薬力学的測定を使用して試験される。例えば、血液サンプル及び尿サンプルが、活性薬剤及びその代謝産物の濃度を求めるために集められる。
【0096】
この二重盲検期は1日目での最初の服用前測定の収集から始まり、5日目での研究終了まで続く。
【0097】
服用前の生命徴候測定、パルスオキシメトリー及びECGが、服用前の30分以内に上記順序で行われる。さらに、服用前の血液サンプル及び研究室検査(血液学、血清化学、肝機能検査及び尿検査)が、生命徴候、SpO2及びECGが測定された後に集められる。
【0098】
続いて、服用前のSSS(スタンフォード眠気スケール)及びCCT(コンピューター化認知検査)が、服用前の血液サンプルを集めた後に行われる。SSSが最初に行われ、次いで、CCTが下記の順で行われる:単純反応時間(Simple Reaction Time)、選択反応時間(Choice Reaction Time)、次いで、指ビジランス(Digit Vigilance)。
【0099】
投与後、生命徴候及びSpO2が、服用後0.25時間、0.5時間、1.0時間、1.5時間、2時間、3時間、4時間、5時間、6時間、8時間、10時間、12時間、24時間、30時間、36時間、48時間、60時間、72時間、84時間及び研究終了時(これは服用後96時間である)において測定される。
【0100】
薬物動態学用サンプルが、薬物暴露の程度を調べるためにそれぞれのコホートについて分析される。血液サンプルが、活性薬剤、その代謝産物、及び関連物質を下記の時点で測定するために集められる:服用後0.25時間、0.5時間、1.0時間、1.5時間、2時間、3時間、4時間、5時間、6時間、8時間、10時間、12時間、24時間、30時間、36時間、48時間、60時間、72時間、84時間及び研究終了時。5回までのさらなる服用後の血液サンプル採取を行うことができ、かつ/又は、薬物動態学的パラメータを得る時期を、以前のコホートからの血液サンプルを分析した後で、指示されるならば変更することができる。生命徴候、パルスオキシメトリー及びECGが得られた後、サンプルが集められる。
【0101】
薬物動態学用サンプルが、1mg、3mg、10mg、30mg及び60mgの3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む本発明による医薬組成物から得られている。分析された被験者の平均血漿中濃度が表IIに示される。
表II
【表3】
【0102】
加えて、研究室検査(血液学、血清化学、肝機能検査及び尿検査)が、服用後24時間及び48時間並びに研究終了時に行われる。
【0103】
SSS及びCCTを使用する薬力学的評価が、服用後0.5時間、1時間、1.5時間、2時間、3時間、4時間、5時間、6時間、8時間、10時間及び12時間、並びに生命徴候、パルスオキシメトリー評価及び薬物動態学用サンプル採取の後で(服用前と同じ順序により)行われる。SSSがCCTの前に行われる。
【0104】
研究期間中、食事が所定の時間で与えられ、また、必要ならば、研究者は、被験者のための特定の食事を、そのような食事のときにおける鎮静の程度に依存して抜くように決定することができる。
【0105】
被験者は、48時間の手順が完了した後で施設から出ることができ、また、指定された時間でのその後の手順(例えば、生命徴候測定及び採血)のために施設に戻る。被験者は、何らかの有害事象又は安全性に関連した他の理由のために、研究者の判断で研究施設に留まり続けるように依頼される場合がある。
【0106】
研究終了時の評価が、服用後96時間で、又は早期中断時に行われる。これには、生命徴候(血圧、脈拍数、呼吸数及び体温)、パルスオキシメトリー、身体検査、ECG、研究室検査(血液学、血清化学、肝機能検査及び尿検査)及び薬物動態学用血液採取が含まれる。
【0107】
ここまで本発明が詳しく記載されているが、本発明は、本発明の範囲又はその任意の実施形態に影響を及ぼすことなく、広範囲かつ同等な様々な条件、配合物及び他のパラメータの範囲内で行われ得ることが当業者によって理解される。本明細書中に引用されたすべての特許及び刊行物は、それらの全体において、参考として本明細書中に全体が組み込まれる。
【図面の簡単な説明】
【0108】
【図1】経時的な血漿中濃度レベルを示すグラフである。
【図2】経時的な血漿中濃度レベルを半対数により示すグラフである。
【図3】経時的な血漿中濃度の用量正規化プロットを示すグラフである。
【特許請求の範囲】
【請求項1】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン又はその医薬的に許容される塩もしくは溶媒和物と、1つ又はそれ以上の医薬的に許容される賦形剤とを含み、約5ng/mL〜約500ng/mLの範囲での3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態血漿中レベルをもたらす、医薬組成物。
【請求項2】
約50ng/mL〜約500ng/mLの範囲、約50ng/mL〜約400ng/mLの範囲、約50ng/mL〜約325ng/mLの範囲、約50ng/mL〜約100ng/mLの範囲又は約100ng/mL〜約250ng/mLの範囲での3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態血漿中レベルをもたらす、請求項1に記載の医薬組成物。
【請求項3】
約50ng/mL〜約250ng/mLの範囲での3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態血漿中レベルをもたらす、請求項1に記載の医薬組成物。
【請求項4】
約20mg〜約40mg(好ましくは約30mg)の3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む、請求項3に記載の医薬組成物。
【請求項5】
経口投与のために好適である、請求項1に記載の医薬組成物。
【請求項6】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの前記定常状態血漿中レベルを投与後の約12時間〜約24時間の持続期間にわたってもたらす、請求項1に記載の医薬組成物。
【請求項7】
親水性マトリックス錠剤、カプセル化された溶融押出し多粒子剤、疎水性マトリックス錠剤、溶融押出し顆粒又は制御型ビーズの形態である、請求項3に記載の医薬組成物。
【請求項8】
全般性不安障害、恐慌性障害、強迫性障害、心的外傷後ストレス障害、社会不安障害、うつ病、アルコール嗜癖及び/又はアルコール依存、月経前緊張、月経前症候群又は月経前不快障害のための処置を必要とする患者を処置する方法であって、請求項1〜7のいずれかに記載の医薬組成物を前記患者に投与することを含む、方法。
【請求項9】
全般性不安障害、恐慌性障害、強迫性障害、心的外傷後ストレス障害、社会不安障害、うつ病、アルコール嗜癖及び/又はアルコール依存、月経前緊張、月経前症候群又は月経前不快障害を処置するための医薬品の調製における、請求項1〜7のいずれか一項に記載の医薬組成物の使用。
【請求項10】
実質的には本明細書中に記載及び例示されるような医薬品の製造における医薬組成物の使用。
【請求項11】
実質的には本明細書中に記載及び例示されるような医薬組成物。
【請求項12】
必要性のある患者を実質的には本明細書中に記載及び例示されるように処置する方法。
【請求項1】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オン又はその医薬的に許容される塩もしくは溶媒和物と、1つ又はそれ以上の医薬的に許容される賦形剤とを含み、約5ng/mL〜約500ng/mLの範囲での3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態血漿中レベルをもたらす、医薬組成物。
【請求項2】
約50ng/mL〜約500ng/mLの範囲、約50ng/mL〜約400ng/mLの範囲、約50ng/mL〜約325ng/mLの範囲、約50ng/mL〜約100ng/mLの範囲又は約100ng/mL〜約250ng/mLの範囲での3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態血漿中レベルをもたらす、請求項1に記載の医薬組成物。
【請求項3】
約50ng/mL〜約250ng/mLの範囲での3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの定常状態血漿中レベルをもたらす、請求項1に記載の医薬組成物。
【請求項4】
約20mg〜約40mg(好ましくは約30mg)の3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンを含む、請求項3に記載の医薬組成物。
【請求項5】
経口投与のために好適である、請求項1に記載の医薬組成物。
【請求項6】
3α−ヒドロキシ−3β−メトキシメチル−21−(1’−イミダゾリル)−5α−プレグナン−20−オンの前記定常状態血漿中レベルを投与後の約12時間〜約24時間の持続期間にわたってもたらす、請求項1に記載の医薬組成物。
【請求項7】
親水性マトリックス錠剤、カプセル化された溶融押出し多粒子剤、疎水性マトリックス錠剤、溶融押出し顆粒又は制御型ビーズの形態である、請求項3に記載の医薬組成物。
【請求項8】
全般性不安障害、恐慌性障害、強迫性障害、心的外傷後ストレス障害、社会不安障害、うつ病、アルコール嗜癖及び/又はアルコール依存、月経前緊張、月経前症候群又は月経前不快障害のための処置を必要とする患者を処置する方法であって、請求項1〜7のいずれかに記載の医薬組成物を前記患者に投与することを含む、方法。
【請求項9】
全般性不安障害、恐慌性障害、強迫性障害、心的外傷後ストレス障害、社会不安障害、うつ病、アルコール嗜癖及び/又はアルコール依存、月経前緊張、月経前症候群又は月経前不快障害を処置するための医薬品の調製における、請求項1〜7のいずれか一項に記載の医薬組成物の使用。
【請求項10】
実質的には本明細書中に記載及び例示されるような医薬品の製造における医薬組成物の使用。
【請求項11】
実質的には本明細書中に記載及び例示されるような医薬組成物。
【請求項12】
必要性のある患者を実質的には本明細書中に記載及び例示されるように処置する方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公表番号】特表2008−542419(P2008−542419A)
【公表日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願番号】特願2008−515150(P2008−515150)
【出願日】平成18年6月9日(2006.6.9)
【国際出願番号】PCT/EP2006/005574
【国際公開番号】WO2006/131392
【国際公開日】平成18年12月14日(2006.12.14)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
【公表日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願日】平成18年6月9日(2006.6.9)
【国際出願番号】PCT/EP2006/005574
【国際公開番号】WO2006/131392
【国際公開日】平成18年12月14日(2006.12.14)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
[ Back to top ]