
神経因性疼痛の治療のための、組み合わされたセロトニンおよびノルエピネフリン再取り込み阻害を有する4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンの結晶形
4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンおよびその塩の結晶形を例えば、神経因性疼痛の処置のために提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、神経因性疼痛の治療のための、組み合わされたセロトニンおよびノルエピネフリン再取り込み阻害を有する4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンの結晶形に関する。
【背景技術】
【0002】
疼痛の知覚は、身体の傷害部分から脳内の特異的受容体にシグナルが直接伝達されて、そこで知覚される疼痛がその傷害に比例するというような単純なものではない。むしろ、末梢組織への損傷および神経への傷害は、疼痛知覚に関与する中枢神経構造に、それ以降の疼痛感受性に影響を及ぼす変化を引き起こし得る。この神経可塑性は、長時間持続する侵害刺激に応答して中枢性感作を引き起こす場合があり、それ自体が、例えば慢性疼痛(すなわち、侵害刺激が停止した後でさえ疼痛の知覚が残るもの)として、または痛覚過敏(すなわち、通常、痛みを伴う刺激に対する、増大した応答)として顕在化し得る。これのさらに不思議で劇的な例の一つは「幻肢症候群」、すなわち切断前の肢に存在した疼痛の持続である。中枢神経可塑性および疼痛の最近の総説として、非特許文献1を参照されたい。
【0003】
神経因性疼痛などの慢性疼痛は、他のタイプの疼痛、例えば体性痛または内臓痛とは顕在化の仕方が異なる。疼痛は、しばしば、電撃痛(shooting)、灼熱痛(burning)、しびれ(pins and needles)、麻痺(numb)または刺痛(stabbing)と説明される。神経因性疼痛の一般的な原因には、アルコール依存症、切断、背部、脚部および腰部の問題、化学療法、糖尿病、HIV、多発性硬化症、脊椎手術、および帯状ヘルペスウイルス感染が含まれる。
【0004】
慢性疼痛の中心的コンポーネントは、例えば神経因性疼痛などの慢性疼痛が非ステロイド抗炎症薬(NSAID)およびオピオイド鎮痛薬などの古典的鎮痛薬にはあまり応答しないことが多いことの説明になるかもしれない。神経因性疼痛の処置には、アミトリプチリン(amitryline)に代表される三環系抗うつ薬(TCA)が標準になり、その作用は、セロトニントランスポータおよびノルエピネフリントランスポータに対する複合的阻害作用によって媒介されると考えられている(Clin Ther.,26,951-979,2004)。最近では、セロトニン再取り込みに対してもノルエピネフリン再取り込みに対しても阻害作用を有する、いわゆる二重作用(dual action)抗うつ薬が、神経因性疼痛の処置に臨床使用されている(非特許文献2)。二重作用性抗うつ薬の例はベンラファキシンおよびデュロキセチンであり、この種類の抗うつ薬はしばしばSNRIと呼ばれる。
【0005】
神経因性疼痛に対する選択的セロトニン再取り込み阻害剤(SSRI)の使用に関するデータは不足しているものの、一般的に限定的作用を示唆している(非特許文献3)。実際、SSRIはそれ自体が弱い抗侵害受容性しか有さないが、セロトニントランスポータの阻害が、ノルエピネフリン再取り込み阻害の抗侵害受容作用を増強するという仮説が立てられている。この考えは、SSRIよりも優れているノルエピネフリン再取り込み阻害剤と比較して、SNRIの方がさらに優れた抗侵害受容作用を有することを示す、22例の動物試験および5例のヒト試験の再検討によって裏付けられる(非特許文献4)。
【0006】
5−HT3アンタゴニストであるオダンセトロン(odansetron)に関する最近のデータは、5−HT3アンタゴニストが鎮痛作用を有し得ること、従って、神経因性疼痛の処置に役立ち得ることを暗示している(非特許文献5)。
【0007】
しかし、三環系抗うつ薬の使用は、例えば嗜眠状態、不安、不穏状態、ならびに認知および記憶障害などのような抗コリン性副作用を伴うことが公知である。従って、当分野では、神経因性疼痛を処置する代替的方法を見つけることが必要とされている。
【0008】
特許文献1として公開された国際特許出願には、例えば化合物4−[2−(4−メチルフェニルスルファニル)フェニル]ピペリジンが、遊離塩基および対応するHCl塩として開示されている。この化合物は、セロトニントランスポータおよびセロトニン受容体2C(5−HT2C)の阻害剤であると報告され、うつ病および不安などの情動障害の処置に役立つとされている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開第2003/029232号パンフレット
【非特許文献】
【0010】
【非特許文献1】Melzackら、Ann. N.Y. Acad. Sci.,933,157-174,2001
【非特許文献2】Human Psychopharm.,19,S21-S25,2004
【非特許文献3】Bas. Clin. Pharmacol.,96,399-409,2005
【非特許文献4】Pain Med. 4,310-316,2000
【非特許文献5】Anesth. Analg.,97,1474-1478,2003
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明者らは、驚いたことに、4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンが、既知の薬理学的プロファイルに加えて、セロトニン再取り込みおよびノルエピネフリン再取り込みの強力な阻害剤、セロトニン受容体3(5−HT3)のアンタゴニスト、セロトニン受容体2A(5−HT2A)のアンタゴニスト、およびα1アドレナリン作動性受容体の阻害剤であること、この化合物が、そのようなものとして、慢性疼痛などの処置に役立ち得ることを見出した。
【課題を解決するための手段】
【0012】
従って本発明は、結晶質形態にある4−[2−(4−メチルフェニル−スルファニル)フェニル]ピペリジンおよび薬学的に許容できるその塩である化合物Iに関する(ただし前記化合物は4−[2−(4−メチルフェニル−スルファニル)フェニル]−ピペリジン塩酸付加塩ではないものとする)。
【0013】
ある実施形態において、本発明は、治療に使用するための化合物Iに関する。
【0014】
ある実施形態において、本発明は、治療有効量の化合物Iをその必要がある患者に投与することを含む処置方法に関する。
【0015】
ある実施形態において、本発明は、化合物Iを含む医薬組成物に関する。
【0016】
ある実施形態において、本発明は、医薬を製造するための化合物Iの使用に関する。
【図面の簡単な説明】
【0017】
【図1】化合物IのHBr付加塩のX線回折パターンである。
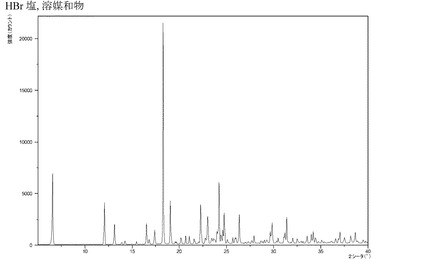
【図2】化合物IのHBr付加塩溶媒和物のX線回折パターンである。
【図3】化合物Iのパルミチン酸付加塩のX線回折パターンである。
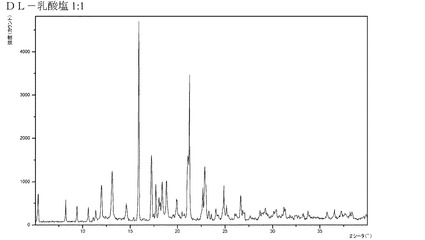
【図4】化合物IのDL−乳酸付加塩のX線回折パターンである。
【図5】化合物Iのアジピン酸付加塩(1:1)のX線回折パターン(α+β型)。
【図6】化合物Iのアジピン酸付加塩(2:1)のX線回折パターンである。
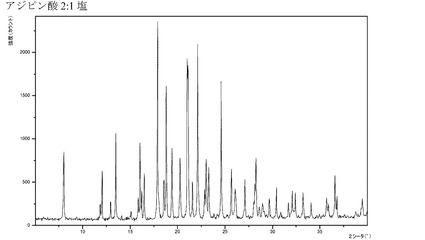
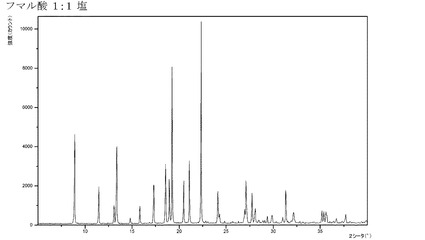
【図7】化合物Iのフマル酸付加塩(1:1)のX線回折パターンである。
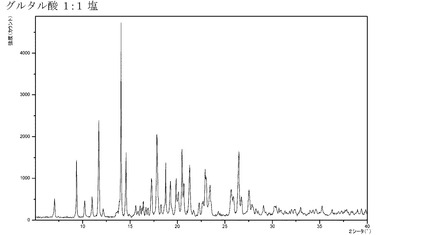
【図8】化合物Iのグルタル酸付加塩(1:1)のX線回折パターンである。
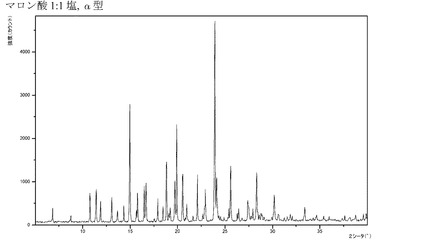
【図9】化合物Iのマロン酸付加塩(1:1)α型のX線回折パターンである。
【図10】化合物Iのマロン酸付加塩β型のX線回折パターンである。
【図11】化合物Iのシュウ酸付加塩(1:1)のX線回折パターンである。
【図12】化合物Iのセバコイン酸(sebacoinic acid)付加塩(2:1)のX線回折パターンである。
【図13】化合物Iのコハク酸付加塩(2:1)のX線回折パターンである。
【図14】化合物IのL−リンゴ酸付加塩(1:1)α型のX線回折パターンである。
【図15】化合物IのL−リンゴ酸付加塩(1:1)β型のX線回折パターンである。
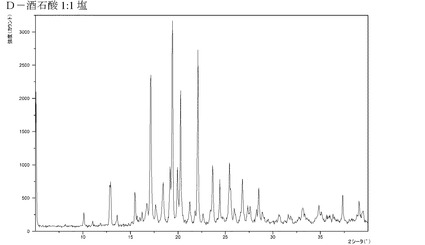
【図16】化合物IのD−酒石酸付加塩(1:1)のX線回折パターンである。
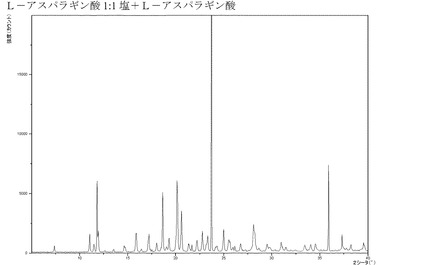
【図17】L−アスパラギン酸と混合された化合物IのL−アスパラギン酸付加塩(1:1)のX線回折パターンである。
【図18】L−アスパラギン酸と混合された化合物IのL−アスパラギン酸付加塩水和物(1:1)のX線回折パターンである。
【図19】グルタミン酸一水和物と混合された化合物Iのグルタミン酸付加塩(1:1)のX線回折パターンである。
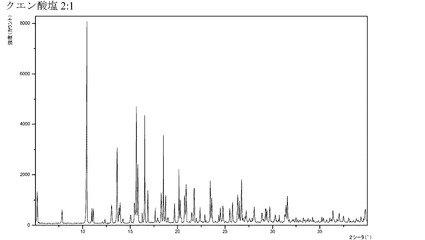
【図20】化合物Iのクエン酸付加塩(2:1)のX線回折パターンである。
【図21】化合物IのHCl酸付加塩のX線回折パターンである。
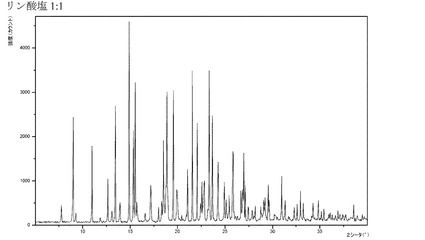
【図22】化合物Iのリン酸付加塩(1:1)のX線回折パターンである。
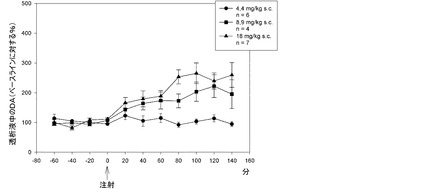
【図23】本発明の化合物を投与した後の前前頭皮質におけるドーパミンレベルである。
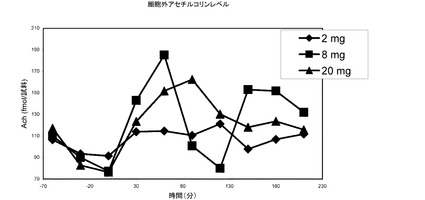
【図24】本発明の化合物を投与した後の前前頭皮質におけるアセチルコリンレベルである。
【図25a】本発明の化合物を投与した後の前前頭皮質および腹側海馬におけるアセチルコリンレベルである。
【図25b】本発明の化合物を投与した後の前前頭皮質および腹側海馬におけるアセチルコリンレベルである。
【発明を実施するための形態】
【0018】
本発明は、結晶質形態にある4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンおよび薬学的に許容できるその塩である化合物Iに関する(ただし前記化合物は塩酸付加塩ではないものとする)。4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンの構造は
【0019】
【化1】
【0020】
である。
【0021】
本発明の化合物の薬理学的プロファイルは実施例で叙述するが、次のように要約することができる。化合物は、セロトニンおよびノルエピネフリン再取り込みの阻害剤であり、セロトニン受容体2A、2Cおよび3を阻害し、そしてα−1アドレナリン作動性受容体を阻害する。
【0022】
ある実施形態において、前記薬学的に許容できる塩は、無毒性である酸の酸付加塩である。前記塩には、マレイン酸、フマル酸、安息香酸、アスコルビン酸、コハク酸、シュウ酸、ビス-メチレンサリチル酸、メタンスルホン酸、エタンジスルホン酸、酢酸、プロピオン酸、酒石酸、サリチル酸、クエン酸、グルコン酸、乳酸、リンゴ酸、マロン酸、マンデル酸、ケイ皮酸、シトラコン酸、アスパラギン酸、ステアリン酸、パルミチン酸、イタコン酸、グリコール酸、p-アミノ安息香酸、グルタミン酸、ベンゼンスルホン酸、テオフィリン酢酸、ならびに8−ハロテオフィリン、例えば8−ブロモテオフィリンなどの有機酸から製造される塩が含まれる。前記塩は、臭化水素酸、硫酸、スルファミン酸、リン酸および硝酸などの無機塩から製造することもできる。他の有用な塩を実施例1dの表(表1)に列挙する。
【0023】
ある実施形態では、本発明の化合物、すなわち式Iの化合物が、HBr付加塩である。
【0024】
ある実施形態では、本発明の化合物がDL−乳酸付加塩、特に1:1塩である。
【0025】
ある実施形態では、本発明の化合物がL−アスパラギン酸付加塩、特に1:1塩である。
【0026】
ある実施形態では、本発明の化合物がグルタミン酸付加塩、特に1:1塩である。
【0027】
ある実施形態では、本発明の化合物がグルタル酸付加塩、特に1:1塩である。
【0028】
ある実施形態では、本発明の化合物がマロン酸付加塩、特に1:1塩であり、これは二つの同質異像αおよびβで存在することが見出され、そのうちβ型は、より低い溶解度に基づいて、最も安定であると考えられる。
【0029】
ある実施形態では、本発明の化合物が精製された形態にある。「精製された形態」という用語は、その化合物が本質的に他の化合物を含まないこと、または場合によっては、他の形態、すなわち前記化合物の多形を含まないことを示すものとする。
【0030】
経口剤形、特に錠剤およびカプセル剤は、投与が容易であり、結果的にコンプライアンスが良好になるため、しばしば患者および医師に好まれる。錠剤およびカプセル剤の場合、活性成分は結晶質であることが好ましい。
【0031】
本発明の結晶は溶媒和物、すなわち溶媒分子が結晶構造の一部を形成している結晶として存在し得る。溶媒和物は水から形成させることができ、この場合、その溶媒和物はしばしば水和物と呼ばれる。代替的には、他の溶媒、例えばエタノール、アセトン、または酢酸エチルなどから溶媒和物を形成させることもできる。溶媒和物の厳密な量は、しばしば、条件に依存する。例えば水和物は、温度を増加させるにつれて、または相対湿度を低下させるにつれて、典型的には水を失う。例えば湿度などの条件が変化しても変化しないかわずかしか変化しない化合物は、医薬製剤により良く適していると一般に見なされる。水から析出させた場合にHBr付加塩が水和物を形成しないのに対して、コハク酸塩、リンゴ酸塩および酒石酸付加塩などの化合物は水和物を形成することが特筆される。
【0032】
いくつかの化合物は吸湿性である(すなわち、それらは湿気に曝露された時に水を吸収する)。吸湿性は、医薬製剤(特に錠剤またはカプセル剤などの乾燥製剤)中に存在させようとする化合物にとっては、望ましくない性質であると一般に見なされる。ある実施形態において、本発明は、吸湿性の低い結晶を提供する。
【0033】
結晶質活性成分を使用する経口剤形の場合は、前記結晶が明確に定義されていることも有益である。この文脈において「明確に定義されている(well-defined)」という用語は、特に、化学量論が明確に定義されていること、すなわち塩を形成しているイオン間の比が小さな整数の比、例えば1:1、1:2、2:1、1:1:1などであることを意味する。ある実施形態では、本発明の化合物が、明確に定義された結晶である。
【0034】
活性成分の溶解度も、バイオアベイラビリティに直接的な影響を有し得るので、剤形の選択にとって重要である。経口剤形の場合、活性成分の溶解度は高い方が、バイオアベイラビリティが増加するので有益であると一般に考えられている。一部の患者、例えば高齢の患者には、錠剤を嚥下することが困難な場合があり、経口点滴液(oral drop solution)が、錠剤を嚥下する必要を避ける適切な代替手段になり得る。経口点滴液の体積を制限するには、液中の活性成分濃度を高くする必要があり、ここでも、化合物の高い溶解度が要求される。表3に示すように、DL−乳酸、L−アスパラギン酸、グルタミン酸、グルタル酸およびマロン酸付加塩は、例外的に高い溶解度を有する。
【0035】
結晶形は化合物の濾過特性および加工特性に影響を及ぼす。針状結晶は、濾過がより困難になり、時間がかかるので、製造環境における取り扱いが、より困難になりがちである。所与の塩の厳密な結晶形は、例えば塩を析出させた条件などに依存し得る。本発明のHBr酸付加塩は、エタノール、酢酸およびプロパノールから析出させた場合に針状溶媒和結晶を成長させるが、HBr付加塩を水から析出させた場合には、針状でない非水和型の結晶を成長させて、優れた濾過特性をもたらす。
【0036】
表3には、溶液pH(Resulting pH)、すなわち塩の飽和溶液のpHも記載する。この性質は重要である。なぜなら、貯蔵中に湿気を完全に避けることはできず、湿気の蓄積が低溶液pH塩を含む錠剤中または錠剤上でのpH低下を引き起こすことになり、それが貯蔵寿命を減少させ得るからである。その上、低い溶液pHを有する塩は、錠剤を湿式造粒法で製造する場合には、加工装置の腐蝕を引き起こし得る。表3のデータは、HBr、HClおよびアジピン酸付加塩が、この点において優秀であり得ることを示唆している。
【0037】
ある実施形態では、本発明の化合物が、結晶質形態にあるHBr付加塩(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記HBr塩が、X線粉末ディフラクトグラム(XRPD)において、約6.08°、14.81°、19.26°および25.38°2θにピークを有し、特に前記HBr塩が図1に図示するXRPDを有する。
【0038】
ある実施形態では、本発明の化合物が、結晶質形態にあるDL−乳酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記DL−乳酸付加塩が、XRPDにおいて、約5.30°、8.81°、9.44°および17.24°2θにピークを有し、特に前記DL乳酸付加塩が図4に図示するXRPDを有する。
【0039】
ある実施形態では、本発明の化合物が、結晶質形態にあるL−アスパラギン酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記L−アスパラギン酸付加塩が非溶媒和物であり、XRPDにおいて約11.05°、20.16°、20.60°、25.00°2θにピークを有し、特に前記L−アスパラギン酸塩は、L−アスパラギン酸と混合した場合に、図17に図示するXRPDを有する。ある実施形態では、前記L−アスパラギン酸付加塩が、水和物(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記L−アスパラギン酸付加塩水和物が、XRPDにおいて約7.80°、13.80°、14.10°、19.63°2θにピークを有し、特に前記L−アスパラギン酸付加塩水和物が、L−アスパラギン酸と混合した場合に、図18に図示するXRPDを有する。
【0040】
ある実施形態では、本発明の化合物が、結晶質形態にあるグルタミン酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記グルタミン酸付加塩が、XRPDにおいて約7.71°、14.01°、19.26°、22.57°2θにピークを有し、特に前記グルタミン酸塩が、グルタミン酸一水和物と混合した場合に、図19に図示するXRPDを有する。
【0041】
ある実施形態では、本発明の化合物が、結晶質形態にあるマロン酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記マロン酸付加塩がα型であって、XRPDにおいて約10.77°、16.70°、19.93°、24.01°2θにピークを有するか、または前記マロン酸付加塩がβ型であって、XRPDにおいて約6.08°、10.11°、18.25°、20.26°2θにピークを有し、特に前記マロン酸付加塩は図9または図10に図示するXRPDを有する。
【0042】
ある実施形態では、本発明の化合物が、結晶質形態にあるグルタル酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記グルタル酸付加塩が、XRPDにおいて約9.39°、11.70°、14.05°、および14.58°2θにピークを有し、特に前記グルタル酸付加塩が、図8に図示するXRPDを有する。
【0043】
上述のように、本発明の化合物は、慢性疼痛の処置に特によく適している。慢性疼痛には、幻肢痛、神経因性疼痛、糖尿病性神経障害、帯状疱疹後神経痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群(CPRS)、三叉神経痛/三叉神経神経痛/疼痛性チック、外科的介入(例えば術後鎮痛薬)、糖尿病性血管障害、膵島炎に関連する毛細血管抵抗もしくは糖尿病症状、アンギナに関連する疼痛、月経に関連する疼痛、がんに関連する疼痛、歯痛、頭痛、片頭痛、緊張型頭痛、三叉神経痛、顎関節症候群、筋筋膜性疼痛筋傷害、線維筋痛症候群、骨関節痛(変形性関節症)、慢性関節リウマチ、熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫、変形性関節炎、骨粗鬆症、骨転移もしくは未知の理由による捻挫もしくは骨折骨痛、痛風、結合組織炎、筋筋膜性疼痛、胸郭出口症候群、上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄傷害(SCI)関連疼痛、中枢性卒中後痛、がん性ニューロパシー、AIDS疼痛、鎌状赤血球疼痛または老人性疼痛などの適応症が包含される。
【0044】
特に、本発明の化合物は、上に列挙した慢性疼痛適応症に関連するうつ病などの気分障害の処置に役立つ。
【0045】
疼痛は、国際疼痛学会(IASP)により、「現実のもしくは潜在的な組織損傷に関連付けられる、またはそのような組織損傷との関連で説明される、不快な感覚的および情動的体験」と定義されている(IASP Classification of chronic pain、第2版、IASP Press(2002)、210)。疼痛は常に主観的であるが、その原因または症候群は分類することができる。「神経因性疼痛」は、サブタイプとして、IASPにより、「神経系における原発病変または機能障害によって開始されるまたは引き起こされる疼痛」と定義されている。
【0046】
神経因性疼痛の異なるサブタイプがIASPによって認識され、その例を以下に挙げる。
【0047】
「通常は疼痛を惹起しない刺激による疼痛」と定義される異痛である。
【0048】
「しばしば血管運動および発汗促進機能障害ならびにその後の栄養変化を併発する、外傷性神経病変後の持続的灼熱痛、異痛およびヒペルパシーの症候群」と定義されるカウザルギーである。
【0049】
「知覚を除く、刺激に対する増大した感受性」と定義される知覚過敏である。
【0050】
「1または複数の神経の分布範囲における疼痛」と定義される神経痛である。
【0051】
「1または複数の神経の炎症」と定義される神経炎である。
【0052】
「ある神経における機能の障害または病理学的変化、単一の神経では、単ニューロパシー、いくつかの神経では、多発性単ニューロパシー、散在性および両側性の場合は、多発ニューロパシー」と定義されるニューロパシーである。ニューロパシーは、例えば糖尿病に関連付けられる場合があり、その場合は糖尿病性神経障害と呼ばれる。
【0053】
「通常、痛みを伴う刺激に対する増大した応答」と定義される痛覚過敏である。
【0054】
「刺激、特に反復刺激に対する異常に痛い反応、ならびに上昇した閾値とを特徴とする、有痛症候群」と定義されるヒペルパシーである。
神経因性疼痛を惹起する刺激は、機械的刺激または熱刺激であり得る。
【0055】
本発明の化合物はユニークな薬理学的プロファイルを有するため、慢性疼痛に直接関係しない他の疾患の処置にも適している。5−HT2C受容体は例えばドーパミン作動性ニューロン上に位置していて、そこでは活性化がドーパミン放出に対して緊張性抑制効果を発揮し、5−HT2Cアンタゴニストはドーパミンレベルの増加をもたらす。実施例2Eに提示するデータは、本発明の化合物が、実際に、脳内で細胞外ドーパミンレベルの用量依存的増加を引き起こすことを示している。これに基づいて、5−HT2Cアンタゴニストは、選択的セロトニン再取り込み阻害剤による処置に不応性であるうつ病の処置に、特に適しているという仮説を立てることができる(Psychopharmacol. Bull.,39,147-166,2006)。この仮説の裏付けは、臨床応答が不充分な抑うつ患者(治療抵抗性うつ病、TRD、または不応性うつ病)の処置にはSSRI単独よりもミルタジピン(mirtazipine)とSSRIとの組合せの方が優れていることを示すいくつかの臨床研究に見出される(Psychother. Psychosom.,75,139-153,2006)。ミルタザピンは5−HT2および5−HT3アンタゴニストでもあり、セロトニン再取り込み阻害作用を5−HT2および5−HT3拮抗作用と合わせて発揮する化合物、例えば本発明の化合物が、TRDの処置に役立つこと、すなわち治療抵抗性うつ病を患っている患者の寛解率を増加させることを示している。
【0056】
実施例2Fおよび2Gに提示するデータは、本発明の化合物が、前前頭皮質および腹側海馬におけるアセチルコリンの細胞外レベルの増加を引き起こすことを示している。脳内のアセチルコリンレベルを増加させることはアルツハイマー病および認知障害全般を処置するための方法になるという長年にわたる臨床的証拠がある(アルツハイマー病の処置におけるアセチルコリンエステラーゼ阻害剤の使用を参照されたい)。これに基づいて、本発明の化合物は、アルツハイマー病および認知障害の処置に役立ち、アルツハイマー病および認知障害に関連するうつ病などの気分障害の処置にも役立つと考えられる。
【0057】
一部の抑うつ患者は、彼らがMADRDやHAMDなどの臨床上有意義なうつ病尺度において改善を示すという意味では、例えばSSRIによる処置に応答するが、他の症状、例えば睡眠障害および認知障害は残ることになる。これに関連して、これらの患者を部分応答者(partial responder)と呼ぶ。アセチルコリンレベルに対する上述の作用のための、本発明の化合物は、うつ病だけでなく認知障害の処置にも役立つと予想される。α−1アドレナリン作動性受容体アンタゴニストである化合物プラゾシンが睡眠障害を減少させることは、臨床研究によって示されている(Biol. Psychiatry, 61,928-934,2007)。さらにまた、本発明の化合物の5−HT2Aおよび5−HT2C拮抗作用は、鎮静、睡眠改善作用を有するとも考えられ(Neuropharmacol, 33,467-471,1994)、従って、本発明の化合物は部分応答者の処置に役立ち、言い換えると、本発明の化合物で抑うつ患者を処置することにより、部分応答者の割合が減る。
【0058】
注意欠陥多動障害(ADHD)は、最も一般的な神経行動学的障害の一つである。ADHDは、限定的、反復的または定型的行動を伴う社会性およびコミュニケーション障害の三主徴を特徴とする。ADHDは通常、小児期または青年期に始まるが、症状は成人まで継続し得る。アトモキセチンは、ADHDの処置にFDAによって承認された、現在唯一の非中枢刺激薬である(Drugs,64,205-222,2004)。アトモキセチンはノルエピネフリン再取り込み阻害剤であり、ADHDの処置に本発明の化合物を使用し得ることを示唆している。また、本発明の化合物は、上述のα−1アドレナリン作動性受容体および5−HT2拮抗作用のための、ADHDの処置に有益な鎮静作用を有し得る。
【0059】
メランコリーは、しばしば重症うつ病に結びつけられるうつ病の特別なサブタイプであり、このタイプのうつ病はメランコリー型うつ病とも呼ばれる。メランコリーは不安、未来恐怖、不眠、および食欲不振に関連付けられる。セロトニン再取り込みとノルエピネフリン再取り込みの両方を阻害する化合物、例えばベンラファキシンは、重症うつ病およびメランコリーの患者の処置に、特に有効であることが示されている(Depres. Anxiety,12,50-54,2000)。上述のように、5−HT2C拮抗作用を発揮する化合物はドーパミンレベルを増加させ、従って、そのような化合物はメランコリーの処置に有効であると予想される(Psychpharm. Bull.,39,147-166,2006)。また、本発明の化合物のα−1アドレナリン作動性受容体および5−HT2拮抗作用は、睡眠を正常化するのに役立つと予想され、従って、前記化合物はメランコリーの処置に役立つ。
【0060】
FDAは最近、心的外傷後ストレス障害(PTSD)の処置に、二つのSSRI、セルトラリンおよびパロキセチンを承認した。さらにまた、5−HT2Aアンタゴニスト活性を有する化合物は、PTSD患者の動揺、不眠および爆発性を抑制することができると予想されるので、有用である(Curr opinion Invest. Drug,4,37-41,2003)。従って、本発明の化合物はPTSDの処置に役立つと予想される。
【0061】
顔面紅潮は閉経移行期に関連する症状である。一部の女性は、睡眠または活動全般が妨げられ、処置が必要になるほど、これに悩まされ得る。何十年も前からエストロゲンによるホルモン補充療法が確立された治療になっているが、最近は、乳がんや心イベントなどの副作用に関する懸念が表明されている。SSRIおよびSNRIを使った臨床試験では、これらの化合物は、顔面紅潮に対して効果を有するが、その効果はエストロゲンほどではないことが示されている(J.Am.Med.Ass.,295,2057-2071,2006)。しかし、セロトニンおよび/またはノルエピネフリン再取り込みを阻害する化合物、例えば本発明の化合物による顔面紅潮の処置は、エストロゲンを許容できない女性またはエストロゲンを許容しないであろう女性にとって、代替処置になり得る。
【0062】
睡眠時無呼吸症候群もしくは閉塞性睡眠時無呼吸低呼吸症候群または閉塞性睡眠呼吸障害は、有効な薬物療法がまだ同定されていない障害である。しかし、動物で行われたいくつかの研究は、5−HT3アンタゴニスト、例えば本発明の化合物が治療的介入に有効であり得ることを示唆している(Sleep,21,131-136,1998;Sleep,8,871,878,2001)。
【0063】
5−HT3アンタゴニストであるオダンセトロンは、最近、渇望ならびにアルコールおよび薬物乱用の処置に有効であることが示されている(Drug Alc. Depend.,84,256-263,2006;Pharmacol Therapeut.,111,855-876,2006)。5−HT3アンタゴニスト、例えば本発明の化合物が、渇望、例えばアルコール、ニコチンまたは炭水化物渇望、ならびにアルコールおよび薬物乱用の処置に役立ち得るという考えを裏付けるものと思われる。
【0064】
5−HT3アンタゴニストの用途として提案されるものには、他にも、嘔吐、特に化学療法誘発性嘔吐、摂食障害、例えば過食症、および過敏性腸症候群(IBS)などがある(Exp. Opin. Ther. Targets,11,527-540,2007)。
【0065】
ユニークな薬理学的プロファイルを有する本発明の化合物は、さらに、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病またはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症および腹圧性尿失禁の処置にも役立つと予想される。
【0066】
ある実施形態において、本発明は、慢性疼痛;部分応答者のうつ病;治療抵抗性うつ病;アルツハイマー病;認知障害;ADHD;メランコリー;PTSD;顔面紅潮;睡眠時無呼吸;アルコール、ニコチンもしくは炭水化物渇望;物質乱用;アルコールもしくは薬物乱用;嘔吐;摂食障害;IBS;情動障害;うつ病;大うつ病性障害;産後うつ病;双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病;不安;全般性不安障害;社会不安障害;強迫性障害;パニック障害;パニック発作;恐怖症;社会恐怖症;広場恐怖症または腹圧性尿失禁を処置する方法であって、その必要がある患者に治療有効量の化合物Iを投与することを含む方法に関する。ある実施形態において、上に列挙した疾患のいずれかについて処置される前記患者は、始めに前記疾患の診断を下されている。
【0067】
ある実施形態において、本発明は、慢性疼痛を処置する方法であって、その必要がある患者に治療有効量の化合物Iを投与することを含む方法に関する。ある実施形態では、前記慢性疼痛が、幻肢痛;神経因性疼痛;糖尿病性神経障害;帯状疱疹後神経痛(PHN);手根管症候群(CTS);HIVニューロパシー;複合性局所疼痛症候群(CPRS);三叉神経痛/三叉神経神経痛/疼痛性チック;外科的介入(例えば術後鎮痛薬);糖尿病性血管障害;膵島炎に関連する毛細血管抵抗もしくは糖尿病症状;アンギナに関連する疼痛;月経に関連する疼痛;がんに関連する疼痛;歯痛;頭痛;片頭痛;緊張型頭痛;三叉神経痛;顎関節症候群;筋筋膜性疼痛筋傷害;線維筋痛症候群;骨関節痛(変形性関節症);慢性関節リウマチ;熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫;変形性関節炎、骨粗鬆症、骨転移もしくは未知の理由による捻挫もしくは骨折骨痛;痛風;結合組織炎;筋筋膜性疼痛;胸郭出口症候群;上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの);骨盤痛;心臓性胸痛;非心臓性胸痛;脊髄傷害(SCI)関連疼痛;中枢性卒中後痛;がん性ニューロパシー;AIDS疼痛;鎌状赤血球疼痛または老人性疼痛から選択される。
【0068】
ある実施形態では、前記慢性疼痛が神経因性疼痛である。
【0069】
ある実施形態では、前記神経因性疼痛が、ヒペルパシー、痛覚過敏、ニューロパシー、糖尿病性神経障害、神経炎、神経痛、知覚過敏、カウザルギー、および異痛から選択される。
【0070】
ある実施形態では、本発明の化合物が、1日あたり約0.001〜約100mg/kg体重の量で投与される。
【0071】
典型的経口投薬量は、1日あたり約0.001〜約100mg/kg体重、好ましくは1日あたり約0.01〜約50mg/kg体重の範囲にあり、それが1回以上の投薬、例えば1〜3回の投薬で投与される。厳密な投薬量は、投与頻度および投与様式、処置される対象の性別、年齢、体重および全身状態、処置される状態の性質および重症度、ならびに処置されるべき併発疾患および当業者には明白な他の因子に依存する。
【0072】
成人の場合、典型的経口投薬量は、1〜100mg/日の本発明の化合物、例えば1〜30mg/日、または5〜25mg/日の範囲にある。これは典型的には、0.1〜50mg、例えば1〜25mg、例えば1、5、10、15、20または25mgの本発明の化合物を1日に1回または2回投与することによって達成され得る。
【0073】
本明細書において使用する化合物の「治療有効量」とは、前記化合物の投与を含む治療的介入において、所与の疾患およびその合併症の臨床症状を治癒、軽減または部分的に抑止するのに充分な量を意味する。これを達成するのに充分な量は「治療有効量」と定義される。この用語は、前記化合物の投与を含む処置において、所与の疾患およびその合併症の臨床症状を治癒、軽減または部分的に抑止するのに充分な量も包含する。各目的のための有効量は、その疾患または傷害の重症度ならびに対象の体重および全身状態に依存する。適当な投薬量の決定は、値のマトリクスを作成し、そのマトリクス中の異なる点を試験することにより、日常的な実験を使って達成することができ、それが全て、熟練した医師の通常の技量に含まれることは、理解される。
【0074】
本明細書において使用する用語「処置」および「処置する」は、疾患または障害などの状態と闘うためになされる患者の管理および医療を意味する。この用語は、患者が患っている所与の状態に関する処置の全範囲、例えば症状もしくは合併症を軽減するため、その疾患、障害もしくは状態の進行を遅延させるため、症状および合併症を軽減し、もしくは緩和するため、および/またはその疾患、障害もしくは状態を治癒させ、もしくは排除するため、ならびにその状態を防止するためになされる活性化合物の投与を包含するものとし、この場合、防止は、その疾患、状態、または障害と闘うためになされる患者の管理および医療と解釈され、症状または合併症の発生を防止するための活性化合物の投与を包含する。それでもなお、予防的(防止的)処置と治療的(治癒的)処置は、本発明の二つの別個の態様である。処置される患者は、好ましくは哺乳動物、特にヒトである。
【0075】
ある実施形態において、本発明は、慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁を処置するための医薬の製造における本発明の使用に関する。
【0076】
ある実施形態において、本発明は、神経因性疼痛などの慢性疼痛を処置するための医薬の製造における本発明の使用に関する。
【0077】
ある実施形態において、本発明は、慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁を処置するための医薬として使用するための本発明の化合物に関する。
【0078】
ある実施形態において、本発明は、神経因性疼痛などの慢性疼痛を処置するための医薬として使用するための本発明の化合物に関する。
【0079】
本発明の化合物は、純粋な化合物として単独で、または薬学的に許容できる担体もしくは賦形剤と組み合わせて、単回投与または複数回投与で投与することができる。本発明の医薬組成物は、「Remington: The Science and Practice of Pharmacy」(第19版、Gennaro編、Mack Publishing Co.、ペンシルバニア州イーストン、1995)に開示されているような従来の技法に従って、薬学的に許容できる担体または希釈剤、ならびに他の任意の公知佐剤および賦形剤を使って製剤することができる。
【0080】
医薬組成物は、経口、直腸、鼻、肺、局所外用(口腔内および舌下を含む)、経皮、槽内、腹腔内、膣および非経口(皮下、筋肉内、髄腔内、静脈内および皮内を含む)経路など、任意の適切な経路による投与に合わせて、具体的に製剤化することができ、経口経路は好ましい。好ましい経路が、処置される対象の全身状態および年齢、処置される状態の性質および選択した活性成分に依存することは理解される。
【0081】
経口投与用の医薬組成物には、カプセル剤、錠剤、糖衣丸、丸剤、口中錠、粉末剤および顆粒剤などの固形剤形が含まれる。それらは適宜、コーティングを施して調製することができる。
【0082】
経口投与用の液状剤形には、溶液剤、乳剤、懸濁剤、シロップ剤およびエリキシル剤が含まれる。
【0083】
非経口投与用の医薬組成物には、滅菌された水性および非水性の注射可能な溶液剤、分散剤、懸濁剤または乳剤、ならびに使用に先だって滅菌注射可能溶液または分散液に再構成される滅菌粉末剤が含まれる。
【0084】
他の適切な投与形態には、坐剤、噴霧剤、軟膏、クリーム剤、ゲル剤、吸入剤、皮膚パッチ、インプラントなどがある。
【0085】
本発明の化合物は、約0.1〜50mgの量の前記化合物(例えば1mg、5mg、10mg、15mg、20mgまたは25mgの本発明の化合物)を含有する単位剤形(unit dosage form)で投与すると好都合である。
【0086】
静脈内投与、髄腔内投与、筋肉内投与及び同様の投与などの非経口経路の場合、典型的に、用量は経口投与に用いられる用量の約半分程度である。
【0087】
非経口投与には、滅菌水性溶液、水性プロピレングリコール、水性ビタミンEまたはゴマ油もしくはラッカセイ油中の本発明の化合物の溶液を使用することができる。そのような水性溶液は、必要であれば適切に緩衝化されるべきであり、最初に希釈液を充分な食塩水またはグルコースで等張性にする。水性溶液は静脈内、筋肉内、皮下および腹腔内投与には特に適している。使用される滅菌水性媒質は全て、当業者に公知である標準的技法により、容易に入手することができる。
【0088】
適切な医薬担体には、不活性固形希釈剤または増量剤、滅菌水性溶液および種々の有機溶剤が含まれる。固形担体の例は、ラクトース、白土、スクロース、シクロデキストリン、タルク、ゼラチン、寒天、ペクチン、アラビアゴム、ステアリン酸マグネシウム、ステアリン酸およびセルロースの低級アルキルエーテルである。液状担体の例は、シロップ、ラッカセイ油、オリーブ油、リン脂質、脂肪酸、脂肪酸アミン、ポリオキシエチレンおよび水である。本発明の化合物と薬学的に許容できる担体とを組み合わせることによって形成された医薬組成物は、次に、開示した投与経路に適した様々な剤形で、容易に投与することができる。
【0089】
経口投与に適した本発明の製剤は、それぞれが予め決定された量の活性成分を含有し、適切な賦形剤を含んでもよい、カプセル剤または錠剤などの不連続な単位として提示することができる。さらにまた、経口使用できる製剤は、粉末状もしくは顆粒状であるか、水性もしくは非水性液体中の溶液もしくは懸濁液であるか、水中油型もしくは油中水型の液状乳剤であることができる。
【0090】
経口投与に固形担体を使用する場合、その調製物は錠剤であるか、例えば粉末もしくはペレット状にして硬ゼラチンカプセルに入れるか、またはトローチ剤もしくは口中剤の形態を取り得る。固形担体の量は様々であり得るが、通常は約25mg〜約1gである。
【0091】
液状担体を使用する場合、その調製物はシロップ剤、乳剤、軟ゼラチンカプセル剤または滅菌注射用液剤、例えば水性もしくは非水性の液状懸濁剤もしくは溶液剤の形態を取り得る。
【0092】
錠剤は、活性成分を通常の佐剤および/または希釈剤と混合した後、その混合物を従来の打錠機で圧縮することによって調製することができる。佐剤または希釈剤の例には、トウモロコシデンプン、ジャガイモデンプン、タルク、ステアリン酸マグネシウム、ゼラチン、ラクトース、ゴムなどが含まれる。そのような目的に通常使用される他の佐剤または添加剤、例えば着色剤、着香剤、保存剤などはいずれも、それらが活性成分と適合するのであれば、使用することができる。
【0093】
本発明の化合物を含むカプセル剤は、前記化合物を含む粉末を微結晶セルロースおよびステアリン酸マグネシウムと混合し、前記粉末を硬ゼラチンカプセルに入れることによって調製することができる。場合により、適切な色素を使って前記カプセル剤を着色してもよい。典型的には、カプセル剤は、0.25〜20%の本発明の化合物、例えば0.5〜1.0%、3.0〜4.0%、14.0〜16.0%の本発明の化合物を含む。これらの力価は、単位剤形中の1、5、10、15、20および25mgの本発明の化合物を便利に送達するために使用することができる。
【0094】
注射用溶液剤は、活性成分と場合によっては添加剤とを注射用溶剤(好ましくは滅菌水)の一部に溶解し、その溶液を所望の体積に調節し、その溶液を滅菌し、それを適切なアンプルまたはバイアルに充填することによって調製することができる。浸透圧調節剤、保存剤、酸化防止剤など、当分野で従来から使用されている任意の適切な添加剤を加えることができる。
【0095】
化合物Iは、国際公開第2003/029232号パンフレットに概説されているように調製することができる。化合物Iの塩は、適当な酸を添加した後、析出させることによって製造することができる。析出は、例えば冷却、溶剤の除去、他の溶剤の添加、またはそれらの併用によって引き起こすことができる。
【0096】
本明細書で言及する参考文献は、刊行物、特許出願、および特許を含めて全て、その文書の組み込みが本明細書のどこか他の項で別途なされているかどうかにかかわらず、各参考文献が参照により、組み込まれることが個別にかつ明示的に示され、本明細書にその全体が(法が許す範囲で最大限に)記載されているかのように、参照により本明細書に組み込まれる。
【0097】
本発明の説明における用語「a」および「an」ならびに「the」および類似する指示物(referent)の使用は、本明細書に別段の表示がない限り、または文脈上、明らかに矛盾しない限り、単数および複数の両方を包含するとみなすべきである。例えば「化合物(the compound)」という表現は、別段の表示がない限り、本発明の様々な「化合物(compounds)」または記載された特定の態様を指すと理解すべきである。
【0098】
別段の表示がない限り、本明細書に記載する厳密な値は全て、対応する近似値の代表である(例えば、特定の因子または測定値に関して記載される厳密な例示的値は全て、対応する近似測定値(適宜「約」によって修飾されるもの)をも記載しているとみなすことができる)。
【0099】
1または複数の要素に関して「を含む(comprising)」「を有する(having)」「を包含する(including)」または「含有する(containing)」などの用語を使ってなされる本発明の任意の1または複数の態様の、本明細書における説明は、別段の明記がない限り、または文脈上、明らかに矛盾しない限り、その特定の1または複数の要素「からなる(consists of)」「から本質的になる(consists essentially of)」または「を実質的に含む(substantially comprise)」本発明の類似する1または複数の態様の裏付けを提供するものである(例えば、特定の要素を含むと本明細書に記載されている組成物は、別段の明記がない限り、または文脈上、明らかに矛盾しない限り、その要素からなる組成物も記載していると理解すべきである)。
【実施例】
【0100】
分析方法
X線粉末ディフラクトグラム(XRPD)は、CuKα1放射線を使ってPANalytical X’Pert PRO X線回折計で測定した。試料は、X’celerator検出器を使用し、反射モードにより、2θ範囲5〜40°で測定した。元素組成(CHN)はElementarのElementar Vario EL装置で測定した。約4mgの試料を各測定に使用し、結果を2回の測定の平均値として記載する。
【0101】
実施例1a 化合物IのHBr塩
撹拌して、わずかに(約45℃)加熱した油状の4−(2−p−トリルスルファニル−フェニル)−ピペリジン−1−カルボン酸エチルエステル442グラムにAcOH中の33wt%HBr(5.7M、2.5等量)545mlを加えた。この混合によって10℃の発熱が生じる。最終添加後に、反応混合物を80℃まで加熱し、18時間放置する。試料を取り出してHPLCで分析し、もし完了していなければ、AcOH中の33wt%HBrを追加しなければならない。そうでない場合は、その混合物を25℃まで冷却して、生成物4−(2−p−トリルスルファニル−フェニル)−ピペリジン臭化水素酸塩を析出させる。25℃で1時間後、その濃厚懸濁液にジエチルエーテル800mlを加える。撹拌をさらに1時間続けてから、生成物を濾過によって単離し、ジエチルエーテル400mlで洗浄し、真空中、40℃で一晩乾燥する。化合物Iの臭化水素酸塩は白色固体として単離された。
【0102】
実施例1b 化合物IのHBr塩
【0103】
2−(4−トリルスルファニル)−フェニルブロミド
窒素で覆った撹拌反応器中で、N−メチル−ピロリドン、NMP(4.5L)に、窒素を20分間吹き込んだ。4−メチルベンゼンチオール(900g、7.25mol)を加え、次に1,2−ジブロモベンゼン(1709g、7.25mol)を加えた。最終的にカリウムtert−ブトキシド(813g、7.25mol)を最後の反応物として加えた。反応は発熱的であり、反応混合物の温度を70℃まで上昇させた。次に反応混合物を120℃に2〜3時間加熱した。反応混合物を室温まで冷却した。酢酸エチル(4L)および塩化ナトリウム水溶液(15%、2.5L)を加えた。その混合物を20分間撹拌した。水相を分離し、新たな酢酸エチル(2L)の一部分で抽出した。水相を分離し、有機相を合わせ、塩化ナトリウム溶液(15%、2.5L)で洗浄した。有機相を分離し、硫酸ナトリウムで乾燥し、減圧下で蒸発させることにより、20〜30%のNMPを含有する赤色油状物を得た。その油状物をメタノールで2倍の体積に希釈し、その混合物を還流した。透明な赤色溶液が得られるまで、メタノールをさらに追加した。その溶液に種晶を入れて室温までゆっくり冷却した。生成物はオフホワイトの結晶として結晶化し、それらを濾過によって単離し、メタノールで洗浄し、真空オーブン中、40℃で恒量まで乾燥した。
【0104】
エチル4−ヒドロキシ−4−(2−(4−トリルスルファニル)フェニル)−ピペリジン−1−カルボキシレート
窒素で覆われた撹拌反応器中で、2−(4−トリルスルファニル)−フェニルブロミド(600g、2.15mol)をヘプタン(4.5L)に懸濁した。室温でヘキサン中の10M BuLi(235mL、2.36mol)を10分かけて加えた。わずかな発熱しか認められなかった。その懸濁液を周囲温度で1時間撹拌した後、−40℃まで冷却した。THF(1.5L)に溶解した1−カルベトキシ−4−ピペリドン(368g、2.15mol)を反応温度が−40℃未満に保たれる速さを上回らない速さで加えた。反応が完了したら、それを0℃まで温め、温度を10℃未満に保ちながら、1M HCl(1L)を加えた。酸性水相を分離し、酢酸エチル(1L)で抽出した。有機相を合わせ、塩化ナトリウム溶液(15%、1L)で抽出した。有機相を硫酸ナトリウムで乾燥し、蒸発させることにより、半結晶質塊を得た。それをエチルエーテル(250mL)でスラリー化し、濾別した。真空オーブン中、40℃で恒量まで乾燥した。
【0105】
エチル4−(2−(4−トリルスルファニル)フェニル)−ピペリジン−1−カルボキシレート
トリフルオロ酢酸(2.8kg、24.9mol)およびトリエチルシラン(362g、3.1mol)を効率の良い撹拌機を有する反応器に投入した。エチル4−ヒドロキシ−4−(2−(4−トリルスルファニル)フェニル)−ピペリジン−1−カルボキシレート(462g、1.24mol)を粉末ロートから少しずつ加えた。反応はわずかに発熱的だった。温度は50℃まで上昇した。添加を終わらせた後、反応混合物を60℃に18時間温めた。反応混合物を室温まで冷却した。トルエン(750mL)および水(750mL)を加えた。有機相を単離し、水相を新たなトルエン(750mL)の一部分で抽出した。有機相を合わせ、塩化ナトリウム溶液(15%、500mL)で洗浄し、硫酸ナトリウムで乾燥した。硫酸ナトリウムを濾去し、濾液を減圧下で蒸発させることによって赤色油状物とし、それをさらに次のステップで加工した。
【0106】
4−(2−(4−トリルスルファニル)フェニル)−ピペリジン臭化水素酸塩
実施例3で得た赤色油状物である粗エチル4−(2−(4−トリルスルファニル)フェニル)−ピペリジン−1−カルボキシレートを撹拌反応器において、酢酸中の臭化水素酸(40%、545mL、3.11mol)と混合した。その混合物を80℃で18時間加熱した。その反応混合物を室温まで冷却した。冷却中に生成物が晶出した。室温で1時間後、エチルエーテル(800mL)を反応混合物に加え、その混合物をさらに1時間撹拌した。生成物を濾別し、エチルエーテルで洗浄し、真空オーブン中、50℃で恒量まで乾燥した。
【0107】
実施例1c 化合物IのHBr塩の再結晶
化合物IのHBr塩(例えば上記のように調製したもの)10.0グラムの混合物をH2O 100ml中で加熱還流した。混合物は80〜90℃で透明になり、完全に溶解した。その透明な溶液にチャコール1グラムを加え、還流を15分間続けてから、濾過し、室温まで自然放冷した。冷却中に白色固体の析出が起こり、その懸濁液を室温で1時間撹拌した。濾過し、真空下40℃で一晩乾燥することにより、化合物IのHBr酸付加塩6.9グラム(69%)を得た。XRPDについては図1を参照されたい。元素分析:3.92%N、59.36%C、6.16%H(理論値:3.85%N、59.34%C、6.09%H)。
【0108】
実施例1d 遊離塩基の原液の調製
酢酸エチル500mlおよびH2O 200mlの混合物に化合物IのHBr塩50グラムを加えて、二相スラリーを生成させた。このスラリーに約25mlの濃NaOHを加えたところ、透明な二相溶液の形成が起こった(pHは13〜14と測定された)。その溶液を激しく15分間撹拌し、有機相を分離した。有機相をH2O 200mlで洗浄し、Na2SO4で乾燥し、濾過し、真空下、60℃で蒸発させることにより、遊離塩基を38グラムの収量(99%)で、ほぼ無色の油状物として得た。
【0109】
酢酸エチルを使って10グラムの油状物を溶解し、体積を150mlに調節することによって、酢酸エチル中の0.235M原液を生成し、そこから一定分量1.5ml(遊離塩基100mg)を使用した。
【0110】
96vol%EtOHを使って10グラムの油状物を溶解し、体積を100mlに調節することによって、EtOH中の0.353M原液を生成し、そこから一定分量1.0ml(遊離塩基100mg)を使用した。
【0111】
実施例1e 遊離塩基の原液を使った塩の形成
所与の一定分量を試験管に入れ、撹拌しながら、表1に示すように適当な量の酸を加えた。酸が液体である場合はニートで加え、そうでない場合は、所与の溶剤に溶解してから加えた。混合および析出の後、撹拌を一晩続け、析出物を濾過によって集めた。真空下、30℃で乾燥する前に、少量の参照試料を取り出し、真空せずに室温で乾燥した。この手順は溶媒和物について調べるために含まれた。いくつかの結果を表1に提示する。XRPDディフラクトグラムを図1〜22に示し、選択したピークの位置を表2に要約する。表3に、水における本発明の化合物の溶解度を結果として得られる飽和溶液のpHと共に示す。「析出物」という欄は、溶解度決定後に単離された析出物が溶解した化合物と同一であるかどうかを表し、これは水和物の形成を示す。
【0112】
【表1】
【0113】
【表2】
【0114】
【表3】
【0115】
実施例2A セロトニン(5−HT)およびノルエピネフリン(NE)再取り込み阻害
試験化合物およびラット皮質シナプトソーム調製物の一定分量を37℃で10分間プレインキュベートした後、[3H]NEまたは[3H]5−HT(最終濃度10nM)を加えた。10μMタルスプラムまたはシタロプラムの存在下で非特異的取り込み量を決定し、緩衝液の存在下で総取り込み量を決定した。一定分量を37℃で15分間インキュベートした。インキュベーション後に、シナプトソームによって取り込まれた[3H]NEまたは[3H]5−HTを0.1%PEIに30分間予浸したUnifilter GF/Cでの濾過により、Tomtec Cell Harvesterプログラムを使って分離した。フィルターを洗浄し、Wallac MicroBetaカウンターでカウントした。
【0116】
NETにおいて、本発明の化合物は23nMのIC50値を示す。SERTにおいて、本発明の化合物は8nMのIC50値を示す。
【0117】
実施例2B 5−HT2A拮抗作用
本発明の化合物をセロトニン受容体に対する親和性について試験したところ、これらは5−HT2A受容体に親和性を有するアンタゴニストプロファイル(Ki 54nM)を示すことが分かった。親和性は、Y=100/(1+10(X−logIC50))から算出される(式中、Yは結合%を表し、Xは化合物の濃度を表す)。5濃度の化合物(1、10、30、100、1000nM)を使って、IC50を算出した。Kiは、Cheng Prusoff式、Ki=(IC50/(1+([L]/Kd))から算出した。親和性はMDL Pharmaservicesカタログ番号271650で決定した。
【0118】
ヒト5−HT2A受容体を発現させる哺乳動物細胞において、本発明の化合物は競合的アンタゴニスト特性を示す。化合物は5−HT2A受容体に<100nMのKiで結合し、機能アッセイでは、5−HTによって惹起される細胞内貯蔵からのCa2+の放出を化合物が67nMのKbで拮抗する。シルド解析により、100mMのKbを有する競合的拮抗作用が明らかになった。
【0119】
実験は以下のように行った。実験の2日または3日前に、250fmol/mgのヒト5−HT2A受容体を発現させるCHO細胞を実験日にコンフルエント単層を得るのに充分な密度でプレーティングする。その細胞に、湿度95%の5%CO2インキュベータ中、37℃で60分間、色素負荷する(Molecular DevicesのCa2+キット)。蛍光イメージングプレートリーダまたはMolecular Devices(カリフォルニア州サニーベール)のFLIPR384を使用し、488nmの励起波長および500〜560nmの蛍光範囲で、基礎蛍光をモニターした。レーザー強度は、約8000〜10000蛍光ユニットの基礎値を得るのに適したレベルに設定した。基礎蛍光の変動は10%未満にするべきである。少なくとも3桁をカバーし、増加する一連の試験化合物濃度を使ってEC50値を評価する。pA2値の評価は、4つの異なる化合物濃度(150、400、1500および4000nM)で5−HTの全用量応答曲線を検証して行った。Kb値の評価も、EC85の5−HTで、2桁にわたる試験物質濃度を検証して行った。試験物質は5−HTの5分前に細胞に加える。Ki値はCheng−Prusoff式を使って算出される。
【0120】
実施例2C 5−HT3A受容体拮抗作用
ヒトホモマー5−HT3A受容体を発現させる卵母細胞において、5−HTは2600nMのEC50で電流を作動させる。この電流は、オンダンセトロンなどの古典的5−HT3アンタゴニストで拮抗することができる。オンダンセトロンはこの系において1nM未満のKi値を示す。本発明の化合物は、低濃度(0.1nM〜100nM)では強力な拮抗作用を示し(IC50約10nM/Kb約2nM)、より高濃度(100〜100000nM)で適用した場合にはアゴニスト特性を示して(EC50約2600nM)、5−HTそのものによって誘起される最大電流の約70〜80%の最大電流に達する。ラットホモマー5−HT3A受容体を発現させる卵母細胞において、5−HTは3.3μMのEC50で電流を作動させる。実験は次のように行った。0.4%MS−222中で10〜15分間麻酔した成熟雌アフリカツメガエル(Xenepus laevis)から、卵母細胞を外科的に摘出した。次にその卵母細胞をOR2緩衝液(82.5mN NaCl、2.0mM KCl、1.0mM MgCl2および5.0mM HEPES、pH7.6))中の0.5mg/mlコラゲナーゼ(タイプIA Sigma−Aldrich)により、室温で2〜3時間消化した。卵胞層が取り除かれた卵母細胞を選択し、2mMピルビン酸ナトリウム、0.1U/lペニシリンおよび0.1μg/lストレプトマイシンを補足した変法バース塩類緩衝液(Modified Barth's Saline buffer)[88mM NaCl、1mM KCl、15mM HEPES、2.4mM NaHCO3、0.41mM CaCl2、0.82mM MgSO4、0.3mM Ca(NO3)2]中で24時間インキュベートした。ステージIV−IV卵母細胞を同定し、ヒト5−HT3A受容体をコードするcRNA 14〜50pgを含有するヌクレアーゼフリー水12〜48nlを注入し、それを電気生理学的記録に使用するまで18℃でインキュベートしておいた(注入後1〜7日間)。ヒト5−HT3受容体を発現させている卵母細胞を1mlバスに入れ、リンゲル緩衝液(115mM NaCl、2.5mM KCl、10mM HEPES、1.8mM CaCl2、0.1mM MgCl2、pH7.5)で潅流した。寒天で栓をした3M KClを含有する0.5〜1MΩ電極で細胞を固定し、GeneClamp 500B増幅器により、−90mVで電圧固定した。卵母細胞をリンゲル緩衝液で潅流し続け、薬物をその潅流液に適用した。5−HTアゴニスト溶液を10〜30秒間適用した。10μM 5−HT刺激に対する濃度応答を測定することによって5−HT3受容体アンタゴニストの効力を調べた。
【0121】
実施例2D α1A受容体拮抗作用
本発明の化合物をα1A受容体に対する親和性について試験したところ、これらはα1A受容体に対して中等度の親和性を有するアンタゴニストプロファイルを示すことが分かった(Ki=34nM)。
【0122】
実験日に膜(膜調製の説明については下記参照)を融解し、ウルトラタラックスを使って緩衝液中でホモジナイズし、所望の濃度に希釈した(5μg/ウェル〜5μg/900μl、使用時まで氷上の保存)。
【0123】
試験化合物50μl、[3H]−プラゾシン50μlおよび膜900μlを混合することによって実験を開始し、その混合物を25℃で20分間インキュベートする。10μM WB−4101の存在下で非特異的結合量を決定し、緩衝液の存在下で総結合量を決定する。インキュベーション後に、0.1%PEIに30分間予浸したUnifilter GF/Bでの濾過により、Tomtec Cell Harvesterプログラム(D4.2..4)を使って、結合リガンドを未結合リガンドから分離する。96ウェル。フィルターを氷冷緩衝液1mlで3回洗浄し、50℃で乾燥し、35μl/ウェルのシンチレーション液をフィルターに加える。結合している放射能をWallac OY 1450 MicroBetaでカウントする。親和性はY=100/(1+10(X−logIC50))から算出される(式中、Yは結合%を表し、Xは化合物の濃度を表す)。2桁をカバーする化合物濃度を使ってIC50値を算出した。KiはCheng Prusoff式、Ki=(IC50/(1+([L]/Kd))から算出した。
【0124】
機能アッセイにおいて、本発明の化合物は、アドレナリンが惹起する細胞内貯蔵からのCa2+の放出を拮抗し、機能アッセイにより、化合物はアンタゴニストであることが明らかになった。
【0125】
これらの実験は基本的に以下に述べるように行った。
【0126】
細胞は全て、10%BCS、4mM L−グルタミン(またはCOS−7の場合は2mM)、および100単位/mlペニシリン+100μg/mlストレプトマイシンを補足したDMEM培地において、5%CO2中、37℃で培養した。
【0127】
アッセイの24時間前に、ヒトアルファ1A−7受容体を発現させるCHO細胞をポリ−D−リジンで被覆された384ウェルブラックウォールマイクロタイタープレートに播種した。培養培地を吸引し、細胞への色素負荷をハンクス平衡塩類溶液(138mM NaCl、5mM KCl、1.3mM CaCl2、0.5mM MgCl2、0.4mM MgSO4、0.3mM KH2PO4、0.3mM Na2HPO4、5.6mMグルコース)+20mM HEPES pH 7.4、0.05%BSAおよび2.5mMプロベニシド(probenicid)(50μl/ウェル)から構成されるアッセイ緩衝液中の1.5μM Fluo−4を使って、5%CO2中、37℃で1時間行った。過剰な色素を捨てた後、細胞をアッセイ緩衝液中で洗浄し、45μl/ウェル(またはアンタゴニストアッセイの場合は30μl/ウェル)に匹敵する最終液量を重層した。アンタゴニスト評価の場合は、この時点で、アンタゴニストまたはビヒクルを4×最終濃度で4%DMSO含有緩衝液中の15μl一定分量(最終DMSO=1%)として加えた後、20分間インキュベートした。蛍光イメージングプレートリーダまたはMolecular Devices(カリフォルニア州サニーベール)のFLIPR(商標)を使用し、488nmの励起波長および500〜560nmの蛍光範囲で、基礎蛍光をモニターした。レーザー励起エネルギーは、基礎蛍光測定値が約8000相対蛍光ユニット(RFU)になるように調節した。次に、アッセイ緩衝液(15μl)に希釈したアゴニストを使って細胞を室温で刺激し、RFUを2.5分間にわたって1.5秒間隔で測定した。蛍光の最大変化を各ウェルについて算出した。蛍光の最大変化から導いた濃度応答曲線を非線形回帰(Hill式)によって解析した。アンタゴニスト決定の場合、20分間の化合物インキュベーション(上記)後に、固定濃度の標準アゴニスト、セロトニンを加えた。
【0128】
実施例2E ドーパミンの増加
本発明の化合物の単回注射は、ラット前頭皮質における細胞外DAレベルを用量依存的に増加させた。図23に図示するように、本発明の化合物は、8.9mg/kgおよび18mg/kg s.c.で、DAレベルをベースラインレベルよりそれぞれ約100%および150%上昇させた。量は遊離塩基として算出する。
【0129】
方法
初期体重275〜300gの雄スプレーグ−ドーリーラットを使用した。動物を室内温度(21±2℃)および湿度(55±5%)が一定になるように制御された条件下、12時間明/暗周期で飼育し、食物および水道水を自由に摂取させた。三日間の処置実験に、浸透圧ミニポンプ(Alzet、2ML1)を使用した。ポンプへの充填を無菌条件下で行い、セボフルランス(sevoflurance)麻酔下で皮下に植え込んだ。実験は内蔵のミニポンプを使って行った。3日間の処置後の試験化合物の血漿レベルを測定するための血液試料を実験の最後に収集した。
【0130】
外科手術および微小透析実験
動物をヒプノルム(hypnorm)/ドルミカム(2ml/kg)で麻酔し、透析プローブチップが腹側海馬内(座標:ブレグマの前方5.6mm、側方−5.0mm、硬膜の腹側7.0mm)または前頭皮質内(座標:ブレグマの前方3.2mm;側方3.0mm;硬膜の腹側4.0mm)に配置されるように、脳内ガイドカニューレ(CMA/12)を海馬中に定位的に植え込んだ。ガイドカニューレを固定するために固定ネジおよびアクリルセメントを使用した。動物の体温を直腸プローブでモニターし、37℃に維持した。ラットを一匹ずつケージに入れて、外科手術から2日間回復させた。実験日に、ガイドカニューレを通して微小透析プローブ(CMA/12、直径0.5mm、長さ3mm)を挿入した。2チャンネルスイベルを通してプローブを微量注入ポンプに接続した。濾過したリンゲル液(145mm NaCl、3mM KCl、1mM MgCl2、1.2mM CaCl2)による微小透析プローブの潅流を脳内にプローブを挿入する直前に開始し、実験の継続中は1(1.3)μL/分の一定流量で続けた。180分間の安定化後に、実験を開始した。透析液を20(30)分ごとに収集した。
【0131】
実験後に断頭によってラットを屠殺し、その脳を摘出し、凍結し、プローブの配置を検証するために薄切した。
【0132】
透析液の分析
透析液中のドーパミンの濃度は電気化学的検出によるHPLCを利用して分析した。モノアミン類を逆相液体クロマトグラフィー(ODS 150×3mm、3μM)で分離した。ドーパミン:流速0.5ml/分の90mM NaH2PO4、50mMクエン酸ナトリウム、367mg/l 1−オクタンスルホン酸ナトリウム、50μM EDTAおよび8%アセトニトリル(pH4.0)からなる移動相である。電量検出器を使って電気化学的検出を行った;電位は250mVに設定(ガードセルは350mV)(Coulochem II、ESA)。
【0133】
実施例2F アセチルコリンの増加
本発明の化合物が自由行動ラットの前前頭皮質におけるアセチルコリンの細胞外レベルに及ぼす作用を評価するために、実験を計画した。
【0134】
実験には雄ウィスターラット(280〜350g;Harlan、オランダ・ザイスト)を使用した。ラットをプラスチック製ケージ(30×30×40cm)で個別に飼育し、食物および水を自由に摂取させておいた。
【0135】
イソフルラン(2%、400mL/分 N2O、400ml/分 O2)を使ってラットを麻酔した。局所麻酔にはリドカイン(10%m/v)を使用した。各動物を定位固定装置(Kopf instrument、USA)に入れ、PaxinosおよびWatsonのラット脳アトラス(1982)を使って、自作のI字状プローブ(Hospal AN 69メンブレン、露出表面4mm)を内側前前頭皮質(mPFC)に挿入した。プローブの先端の座標はmPFC[AP=3.4mm、L=−0.8mm、V=5.0mm]だった。次にプローブを歯科用セメントとネジで頭骨に固定した。フルニキシン(1mg/kg s.c.)を術後鎮痛薬として投与した。
【0136】
実験は外科手術の24〜48時間後に行った。実験日に、可撓性PEEKチューブでラットを微小潅流ポンプ(CMA102)に接続し、147mM NaCl、3.0mM KCl、1.2mM CaCl2、および1.2mM MgCl2を含有する流速1.5μL/分のリンゲル緩衝液で、透析プローブを潅流した。アセチルコリン決定用に、55μLの0.02Mギ酸が含有されるミニバイアルに、微小透析試料を30分間隔で収集した。自動フラクションコレクター(CMA142)を使って試料を収集し、分析するまで−80℃で保存した。実験の完了後にラットを屠殺した。脳を摘出し、パラホルムアルデヒド溶液(4%m/v)中で硬化させた。脳の冠状切片を作製することにより、PaxinosおよびWatson(1982)に従って、各プローブの位置を組織学的に検証した。
【0137】
試験化合物を10%2−OH−プロピル−ベータ−シクロデキストリンに溶解し、液量5mL/kgの皮下注射により、異なる用量で投与を行った。
【0138】
アセチルコリンの濃度は、HPLCとタンデム質量分析(MS/MS)検出で決定した。
【0139】
自動試料インジェクター(PerkinElmer Instruments、シリーズ200)を使って一定分量(25μL)をHPLCカラムに注入した。クロマトグラフィー分離は、4×2.0mmガードカラム(Phenomenex Synergy MAX−RP AJO−6073、Bester)で保護された逆相150×2.00mm(4μm)分析用カラム(Phenomenex Synergy MAX−RP、Bester)で行った(どちらのカラムも温度を30℃に維持)。移動相(定組成)は超精製水(UP)、アセトニトリル(ACN)、およびトリフルオロ酢酸(TFA)からなった(UP:ACN:TFA=95.0:0.5:0.1v/v/v%)。移動相をHPLCポンプ(PerkinElmer Instruments、シリーズ200マイクロポンプ)により、0.300mL/分の流量で、システムに流した。
【0140】
LC/MS分析は、API 4000 MS/MS検出器およびTurbo Ion Sprayインターフェース(どちらもApplied Biosystem(オランダ)製)からなるAPI4000 MS/MSシステムを使って行った。取得は正イオン化モードで行い、イオンスプレー電圧は5.5kV、ネブライザーガス圧は50psig(SCIEXスケール0〜90で)、プローブ温度は600℃に設定した。アセチルコリン(前駆体146.1Da、生成物86.8Da)を検出するために、マルチプルリアクションモニタリング(MRM)モードで装置を作動させた。衝突エネルギーを21.0eVとし、衝突ガス(窒素)圧を7(0〜12のSCIEXスケールで)に保った。Analyst(商標)データシステム(Applied Biosystem、バージョン1.2)を使って、データを較正し、定量した。
【0141】
変化量が50%未満である二つの連続微小透析試料をベースラインレベルとし、それを100%に設定した。アセチルコリン濃度の変化を同じ対象内でのベースラインのパーセントとして表す。
【0142】
データを図24に示す。
【0143】
実施例2G アセチルコリンの増加
本発明の化合物が自由行動ラットの前前頭皮質および腹側海馬におけるアセチルコリンの細胞外レベルに及ぼす作用を評価するために、実験を計画した。
【0144】
初期体重275〜300gの雄スプレーグ−ドーリーラットを使用した。動物を室内温度(21±2℃)および湿度(55±5%)が一定になるように制御された条件下、12時間明/暗周期で飼育し、食物および水道水を自由に摂取させた。
【0145】
外科手術および微小透析実験
ラットをヒプノルム/ドルミカム(2ml/kg)で麻酔し、透析プローブチップを腹側海馬内(座標:ブレグマの前方5.6mm、側方−5.0mm、硬膜の腹側7.0mm)または前頭皮質内(座標:ブレグマの前方3.2mm;側方0.8mm;硬膜の腹側4.0mm)に配置することを目標として、脳内ガイドカニューレ(CMA/12)を海馬中に定位的に植え込んだ。ガイドカニューレを固定するために固定ネジおよびアクリルセメントを使用した。動物の体温を直腸プローブでモニターし、37℃に維持した。ラットを一匹ずつケージに入れて、外科手術から2日間回復させた。実験日に、ガイドカニューレを通して微小透析プローブ(CMA/12、直径0.5mm、長さ3mm)を挿入した。
【0146】
2チャンネルスイベルを通してプローブを微量注入ポンプに接続した。濾過したリンゲル液(0.5μMネオスチグミンを含有する145mm NaCl、3mM KCl、1mM MgCl2、1.2mM CaCl2)による微小透析プローブの潅流を脳内にプローブを挿入する直前に開始し、実験の継続中は1μL/分の一定流量で続けた。180分間の安定化後に、実験を開始した。透析液を20分ごとに収集した。実験後に動物を屠殺し、その脳を摘出し、凍結し、プローブの配置を検証するために薄切した。
【0147】
透析液アセチルコリンの分析
透析液中のアセチルコリン(ACh)濃度は、100mMリン酸水素二ナトリウム、2.0mMオクタンスルホン酸、0.5mMテトラメチルアンモニウムクロリドおよび0.005%MB(ESA)、pH8.0からなる移動相を使用し、電気化学的検出によるHPLCを利用して分析した。分析用カラム(ESA ACH−250)、流量0.35ml/分、温度:35℃でのAChの分離に先だって、固定化コリンオキシダーゼを含有するプレカラム酵素リアクタ(ESA)により、注入された試料(10μl)からコリンが除去された。分析用カラム後に、試料は、固定化アセチルコリンエステラーゼおよびコリンオキシダーゼを含有するポストカラム固相リアクタ(ESA)を通過した。後者のリアクタはAChをコリンに変換した後、コリンをベタインおよびH2O2に変換した。後者を白金電極(分析用セル:ESA、モデル5040)を使って電気化学的に検出した。
【0148】
データ提示
単回注射実験では、化合物投与直前の三つの連続するACh試料の平均値を各実験の基礎レベルとし、データを基礎値に対するパーセンテージに変換した(平均基礎注射前値を100%に標準化)。データを図25aおよび25bに提示する。
【0149】
図24に提示するデータは、アセチルコリンレベルの予想外の低下(例えば8mg/kgを参照)を示しているが、これを説明することは困難であり、実験的不確定性によるものであると考えられる。実施例2Fおよび2Gから得られるデータセットはどちらも、全体として、脳内の細胞外アセチルコリンレベルの同じ(すなわち用量依存的な)増加を示している。この前臨床的知見は、例えばアルツハイマー患者、部分応答者、認知障害などのような認知障害を特徴とする疾患の処置などに有用な、臨床状況における認知の改善につながると予想される。
【0150】
実施例3 神経因性疼痛に対する効果
神経因性疼痛に対する効力を実証するために、本発明の化合物を神経因性疼痛のホルマリンモデル(Neuropharm.,48,252-263,2005;Pain,51,5-17,1992)で試験した。このモデルでは、マウスの左後肢の足底表面にホルマリン(4.5%、20μl)を注射してから、そのマウスを観察のために個別にガラス製ビーカー(容量2l)に入れる。ホルマリン注射が引き起こす刺激は、傷害を受けた肢をなめるのに費やす時間の量として定量化される特徴的な二相性行動応答を誘起する。第1相(約0〜10分)は直接的な化学刺激および侵害受容を表すのに対して、第2相(約20〜30分)は神経因性の疼痛を表すと考えられる。これら二つの相は休止時間によって隔てられ、その間は行動が正常に戻る。傷害を受けた肢をなめるのに費やした時間の量を二つの相で測定することにより、疼痛刺激の低減に関する試験化合物の有効性が評価される。
【0151】
各群8匹のC57/B6マウス(約25g)を試験した。次の表4は、二つの相、すなわちホルマリン注射の0〜5分後および20〜30分後において、傷害を受けた肢をなめるのに費やされた時間の量を示している。投与した化合物の量は遊離塩基として算出されている。
【0152】
【表4】
【0153】
表4のデータは、直接的な化学刺激および侵害受容を表す第1相では、本発明の化合物がほとんど効果を有さないことを明らかにしている。より注目すべきことに、このデータは、第2相において肢をなめるのに費やされた時間の明確かつ用量依存的な短縮も明らかにし、神経因性疼痛の処置における本発明の化合物の効果を示している。
実施例4 カプセル剤
第1ステップでは、4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジン臭化水素酸塩を微結晶セルロースと混合した。第2ステップではステアリン酸マグネシウムを混ぜ合わせた。四つの力価を有するカプセル剤を調製した。活性成分は遊離塩基として記載する。
【0154】
【表5】
【0155】
各バッチから10,000個のカプセル剤が調製された。
【技術分野】
【0001】
本発明は、神経因性疼痛の治療のための、組み合わされたセロトニンおよびノルエピネフリン再取り込み阻害を有する4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンの結晶形に関する。
【背景技術】
【0002】
疼痛の知覚は、身体の傷害部分から脳内の特異的受容体にシグナルが直接伝達されて、そこで知覚される疼痛がその傷害に比例するというような単純なものではない。むしろ、末梢組織への損傷および神経への傷害は、疼痛知覚に関与する中枢神経構造に、それ以降の疼痛感受性に影響を及ぼす変化を引き起こし得る。この神経可塑性は、長時間持続する侵害刺激に応答して中枢性感作を引き起こす場合があり、それ自体が、例えば慢性疼痛(すなわち、侵害刺激が停止した後でさえ疼痛の知覚が残るもの)として、または痛覚過敏(すなわち、通常、痛みを伴う刺激に対する、増大した応答)として顕在化し得る。これのさらに不思議で劇的な例の一つは「幻肢症候群」、すなわち切断前の肢に存在した疼痛の持続である。中枢神経可塑性および疼痛の最近の総説として、非特許文献1を参照されたい。
【0003】
神経因性疼痛などの慢性疼痛は、他のタイプの疼痛、例えば体性痛または内臓痛とは顕在化の仕方が異なる。疼痛は、しばしば、電撃痛(shooting)、灼熱痛(burning)、しびれ(pins and needles)、麻痺(numb)または刺痛(stabbing)と説明される。神経因性疼痛の一般的な原因には、アルコール依存症、切断、背部、脚部および腰部の問題、化学療法、糖尿病、HIV、多発性硬化症、脊椎手術、および帯状ヘルペスウイルス感染が含まれる。
【0004】
慢性疼痛の中心的コンポーネントは、例えば神経因性疼痛などの慢性疼痛が非ステロイド抗炎症薬(NSAID)およびオピオイド鎮痛薬などの古典的鎮痛薬にはあまり応答しないことが多いことの説明になるかもしれない。神経因性疼痛の処置には、アミトリプチリン(amitryline)に代表される三環系抗うつ薬(TCA)が標準になり、その作用は、セロトニントランスポータおよびノルエピネフリントランスポータに対する複合的阻害作用によって媒介されると考えられている(Clin Ther.,26,951-979,2004)。最近では、セロトニン再取り込みに対してもノルエピネフリン再取り込みに対しても阻害作用を有する、いわゆる二重作用(dual action)抗うつ薬が、神経因性疼痛の処置に臨床使用されている(非特許文献2)。二重作用性抗うつ薬の例はベンラファキシンおよびデュロキセチンであり、この種類の抗うつ薬はしばしばSNRIと呼ばれる。
【0005】
神経因性疼痛に対する選択的セロトニン再取り込み阻害剤(SSRI)の使用に関するデータは不足しているものの、一般的に限定的作用を示唆している(非特許文献3)。実際、SSRIはそれ自体が弱い抗侵害受容性しか有さないが、セロトニントランスポータの阻害が、ノルエピネフリン再取り込み阻害の抗侵害受容作用を増強するという仮説が立てられている。この考えは、SSRIよりも優れているノルエピネフリン再取り込み阻害剤と比較して、SNRIの方がさらに優れた抗侵害受容作用を有することを示す、22例の動物試験および5例のヒト試験の再検討によって裏付けられる(非特許文献4)。
【0006】
5−HT3アンタゴニストであるオダンセトロン(odansetron)に関する最近のデータは、5−HT3アンタゴニストが鎮痛作用を有し得ること、従って、神経因性疼痛の処置に役立ち得ることを暗示している(非特許文献5)。
【0007】
しかし、三環系抗うつ薬の使用は、例えば嗜眠状態、不安、不穏状態、ならびに認知および記憶障害などのような抗コリン性副作用を伴うことが公知である。従って、当分野では、神経因性疼痛を処置する代替的方法を見つけることが必要とされている。
【0008】
特許文献1として公開された国際特許出願には、例えば化合物4−[2−(4−メチルフェニルスルファニル)フェニル]ピペリジンが、遊離塩基および対応するHCl塩として開示されている。この化合物は、セロトニントランスポータおよびセロトニン受容体2C(5−HT2C)の阻害剤であると報告され、うつ病および不安などの情動障害の処置に役立つとされている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開第2003/029232号パンフレット
【非特許文献】
【0010】
【非特許文献1】Melzackら、Ann. N.Y. Acad. Sci.,933,157-174,2001
【非特許文献2】Human Psychopharm.,19,S21-S25,2004
【非特許文献3】Bas. Clin. Pharmacol.,96,399-409,2005
【非特許文献4】Pain Med. 4,310-316,2000
【非特許文献5】Anesth. Analg.,97,1474-1478,2003
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明者らは、驚いたことに、4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンが、既知の薬理学的プロファイルに加えて、セロトニン再取り込みおよびノルエピネフリン再取り込みの強力な阻害剤、セロトニン受容体3(5−HT3)のアンタゴニスト、セロトニン受容体2A(5−HT2A)のアンタゴニスト、およびα1アドレナリン作動性受容体の阻害剤であること、この化合物が、そのようなものとして、慢性疼痛などの処置に役立ち得ることを見出した。
【課題を解決するための手段】
【0012】
従って本発明は、結晶質形態にある4−[2−(4−メチルフェニル−スルファニル)フェニル]ピペリジンおよび薬学的に許容できるその塩である化合物Iに関する(ただし前記化合物は4−[2−(4−メチルフェニル−スルファニル)フェニル]−ピペリジン塩酸付加塩ではないものとする)。
【0013】
ある実施形態において、本発明は、治療に使用するための化合物Iに関する。
【0014】
ある実施形態において、本発明は、治療有効量の化合物Iをその必要がある患者に投与することを含む処置方法に関する。
【0015】
ある実施形態において、本発明は、化合物Iを含む医薬組成物に関する。
【0016】
ある実施形態において、本発明は、医薬を製造するための化合物Iの使用に関する。
【図面の簡単な説明】
【0017】
【図1】化合物IのHBr付加塩のX線回折パターンである。
【図2】化合物IのHBr付加塩溶媒和物のX線回折パターンである。
【図3】化合物Iのパルミチン酸付加塩のX線回折パターンである。
【図4】化合物IのDL−乳酸付加塩のX線回折パターンである。
【図5】化合物Iのアジピン酸付加塩(1:1)のX線回折パターン(α+β型)。
【図6】化合物Iのアジピン酸付加塩(2:1)のX線回折パターンである。
【図7】化合物Iのフマル酸付加塩(1:1)のX線回折パターンである。
【図8】化合物Iのグルタル酸付加塩(1:1)のX線回折パターンである。
【図9】化合物Iのマロン酸付加塩(1:1)α型のX線回折パターンである。
【図10】化合物Iのマロン酸付加塩β型のX線回折パターンである。
【図11】化合物Iのシュウ酸付加塩(1:1)のX線回折パターンである。
【図12】化合物Iのセバコイン酸(sebacoinic acid)付加塩(2:1)のX線回折パターンである。
【図13】化合物Iのコハク酸付加塩(2:1)のX線回折パターンである。
【図14】化合物IのL−リンゴ酸付加塩(1:1)α型のX線回折パターンである。
【図15】化合物IのL−リンゴ酸付加塩(1:1)β型のX線回折パターンである。
【図16】化合物IのD−酒石酸付加塩(1:1)のX線回折パターンである。
【図17】L−アスパラギン酸と混合された化合物IのL−アスパラギン酸付加塩(1:1)のX線回折パターンである。
【図18】L−アスパラギン酸と混合された化合物IのL−アスパラギン酸付加塩水和物(1:1)のX線回折パターンである。
【図19】グルタミン酸一水和物と混合された化合物Iのグルタミン酸付加塩(1:1)のX線回折パターンである。
【図20】化合物Iのクエン酸付加塩(2:1)のX線回折パターンである。
【図21】化合物IのHCl酸付加塩のX線回折パターンである。
【図22】化合物Iのリン酸付加塩(1:1)のX線回折パターンである。
【図23】本発明の化合物を投与した後の前前頭皮質におけるドーパミンレベルである。
【図24】本発明の化合物を投与した後の前前頭皮質におけるアセチルコリンレベルである。
【図25a】本発明の化合物を投与した後の前前頭皮質および腹側海馬におけるアセチルコリンレベルである。
【図25b】本発明の化合物を投与した後の前前頭皮質および腹側海馬におけるアセチルコリンレベルである。
【発明を実施するための形態】
【0018】
本発明は、結晶質形態にある4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンおよび薬学的に許容できるその塩である化合物Iに関する(ただし前記化合物は塩酸付加塩ではないものとする)。4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジンの構造は
【0019】
【化1】
【0020】
である。
【0021】
本発明の化合物の薬理学的プロファイルは実施例で叙述するが、次のように要約することができる。化合物は、セロトニンおよびノルエピネフリン再取り込みの阻害剤であり、セロトニン受容体2A、2Cおよび3を阻害し、そしてα−1アドレナリン作動性受容体を阻害する。
【0022】
ある実施形態において、前記薬学的に許容できる塩は、無毒性である酸の酸付加塩である。前記塩には、マレイン酸、フマル酸、安息香酸、アスコルビン酸、コハク酸、シュウ酸、ビス-メチレンサリチル酸、メタンスルホン酸、エタンジスルホン酸、酢酸、プロピオン酸、酒石酸、サリチル酸、クエン酸、グルコン酸、乳酸、リンゴ酸、マロン酸、マンデル酸、ケイ皮酸、シトラコン酸、アスパラギン酸、ステアリン酸、パルミチン酸、イタコン酸、グリコール酸、p-アミノ安息香酸、グルタミン酸、ベンゼンスルホン酸、テオフィリン酢酸、ならびに8−ハロテオフィリン、例えば8−ブロモテオフィリンなどの有機酸から製造される塩が含まれる。前記塩は、臭化水素酸、硫酸、スルファミン酸、リン酸および硝酸などの無機塩から製造することもできる。他の有用な塩を実施例1dの表(表1)に列挙する。
【0023】
ある実施形態では、本発明の化合物、すなわち式Iの化合物が、HBr付加塩である。
【0024】
ある実施形態では、本発明の化合物がDL−乳酸付加塩、特に1:1塩である。
【0025】
ある実施形態では、本発明の化合物がL−アスパラギン酸付加塩、特に1:1塩である。
【0026】
ある実施形態では、本発明の化合物がグルタミン酸付加塩、特に1:1塩である。
【0027】
ある実施形態では、本発明の化合物がグルタル酸付加塩、特に1:1塩である。
【0028】
ある実施形態では、本発明の化合物がマロン酸付加塩、特に1:1塩であり、これは二つの同質異像αおよびβで存在することが見出され、そのうちβ型は、より低い溶解度に基づいて、最も安定であると考えられる。
【0029】
ある実施形態では、本発明の化合物が精製された形態にある。「精製された形態」という用語は、その化合物が本質的に他の化合物を含まないこと、または場合によっては、他の形態、すなわち前記化合物の多形を含まないことを示すものとする。
【0030】
経口剤形、特に錠剤およびカプセル剤は、投与が容易であり、結果的にコンプライアンスが良好になるため、しばしば患者および医師に好まれる。錠剤およびカプセル剤の場合、活性成分は結晶質であることが好ましい。
【0031】
本発明の結晶は溶媒和物、すなわち溶媒分子が結晶構造の一部を形成している結晶として存在し得る。溶媒和物は水から形成させることができ、この場合、その溶媒和物はしばしば水和物と呼ばれる。代替的には、他の溶媒、例えばエタノール、アセトン、または酢酸エチルなどから溶媒和物を形成させることもできる。溶媒和物の厳密な量は、しばしば、条件に依存する。例えば水和物は、温度を増加させるにつれて、または相対湿度を低下させるにつれて、典型的には水を失う。例えば湿度などの条件が変化しても変化しないかわずかしか変化しない化合物は、医薬製剤により良く適していると一般に見なされる。水から析出させた場合にHBr付加塩が水和物を形成しないのに対して、コハク酸塩、リンゴ酸塩および酒石酸付加塩などの化合物は水和物を形成することが特筆される。
【0032】
いくつかの化合物は吸湿性である(すなわち、それらは湿気に曝露された時に水を吸収する)。吸湿性は、医薬製剤(特に錠剤またはカプセル剤などの乾燥製剤)中に存在させようとする化合物にとっては、望ましくない性質であると一般に見なされる。ある実施形態において、本発明は、吸湿性の低い結晶を提供する。
【0033】
結晶質活性成分を使用する経口剤形の場合は、前記結晶が明確に定義されていることも有益である。この文脈において「明確に定義されている(well-defined)」という用語は、特に、化学量論が明確に定義されていること、すなわち塩を形成しているイオン間の比が小さな整数の比、例えば1:1、1:2、2:1、1:1:1などであることを意味する。ある実施形態では、本発明の化合物が、明確に定義された結晶である。
【0034】
活性成分の溶解度も、バイオアベイラビリティに直接的な影響を有し得るので、剤形の選択にとって重要である。経口剤形の場合、活性成分の溶解度は高い方が、バイオアベイラビリティが増加するので有益であると一般に考えられている。一部の患者、例えば高齢の患者には、錠剤を嚥下することが困難な場合があり、経口点滴液(oral drop solution)が、錠剤を嚥下する必要を避ける適切な代替手段になり得る。経口点滴液の体積を制限するには、液中の活性成分濃度を高くする必要があり、ここでも、化合物の高い溶解度が要求される。表3に示すように、DL−乳酸、L−アスパラギン酸、グルタミン酸、グルタル酸およびマロン酸付加塩は、例外的に高い溶解度を有する。
【0035】
結晶形は化合物の濾過特性および加工特性に影響を及ぼす。針状結晶は、濾過がより困難になり、時間がかかるので、製造環境における取り扱いが、より困難になりがちである。所与の塩の厳密な結晶形は、例えば塩を析出させた条件などに依存し得る。本発明のHBr酸付加塩は、エタノール、酢酸およびプロパノールから析出させた場合に針状溶媒和結晶を成長させるが、HBr付加塩を水から析出させた場合には、針状でない非水和型の結晶を成長させて、優れた濾過特性をもたらす。
【0036】
表3には、溶液pH(Resulting pH)、すなわち塩の飽和溶液のpHも記載する。この性質は重要である。なぜなら、貯蔵中に湿気を完全に避けることはできず、湿気の蓄積が低溶液pH塩を含む錠剤中または錠剤上でのpH低下を引き起こすことになり、それが貯蔵寿命を減少させ得るからである。その上、低い溶液pHを有する塩は、錠剤を湿式造粒法で製造する場合には、加工装置の腐蝕を引き起こし得る。表3のデータは、HBr、HClおよびアジピン酸付加塩が、この点において優秀であり得ることを示唆している。
【0037】
ある実施形態では、本発明の化合物が、結晶質形態にあるHBr付加塩(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記HBr塩が、X線粉末ディフラクトグラム(XRPD)において、約6.08°、14.81°、19.26°および25.38°2θにピークを有し、特に前記HBr塩が図1に図示するXRPDを有する。
【0038】
ある実施形態では、本発明の化合物が、結晶質形態にあるDL−乳酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記DL−乳酸付加塩が、XRPDにおいて、約5.30°、8.81°、9.44°および17.24°2θにピークを有し、特に前記DL乳酸付加塩が図4に図示するXRPDを有する。
【0039】
ある実施形態では、本発明の化合物が、結晶質形態にあるL−アスパラギン酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記L−アスパラギン酸付加塩が非溶媒和物であり、XRPDにおいて約11.05°、20.16°、20.60°、25.00°2θにピークを有し、特に前記L−アスパラギン酸塩は、L−アスパラギン酸と混合した場合に、図17に図示するXRPDを有する。ある実施形態では、前記L−アスパラギン酸付加塩が、水和物(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記L−アスパラギン酸付加塩水和物が、XRPDにおいて約7.80°、13.80°、14.10°、19.63°2θにピークを有し、特に前記L−アスパラギン酸付加塩水和物が、L−アスパラギン酸と混合した場合に、図18に図示するXRPDを有する。
【0040】
ある実施形態では、本発明の化合物が、結晶質形態にあるグルタミン酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記グルタミン酸付加塩が、XRPDにおいて約7.71°、14.01°、19.26°、22.57°2θにピークを有し、特に前記グルタミン酸塩が、グルタミン酸一水和物と混合した場合に、図19に図示するXRPDを有する。
【0041】
ある実施形態では、本発明の化合物が、結晶質形態にあるマロン酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記マロン酸付加塩がα型であって、XRPDにおいて約10.77°、16.70°、19.93°、24.01°2θにピークを有するか、または前記マロン酸付加塩がβ型であって、XRPDにおいて約6.08°、10.11°、18.25°、20.26°2θにピークを有し、特に前記マロン酸付加塩は図9または図10に図示するXRPDを有する。
【0042】
ある実施形態では、本発明の化合物が、結晶質形態にあるグルタル酸付加塩(1:1)(特に、精製された形態にあるもの)である。さらにもう一つの実施形態では、前記グルタル酸付加塩が、XRPDにおいて約9.39°、11.70°、14.05°、および14.58°2θにピークを有し、特に前記グルタル酸付加塩が、図8に図示するXRPDを有する。
【0043】
上述のように、本発明の化合物は、慢性疼痛の処置に特によく適している。慢性疼痛には、幻肢痛、神経因性疼痛、糖尿病性神経障害、帯状疱疹後神経痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群(CPRS)、三叉神経痛/三叉神経神経痛/疼痛性チック、外科的介入(例えば術後鎮痛薬)、糖尿病性血管障害、膵島炎に関連する毛細血管抵抗もしくは糖尿病症状、アンギナに関連する疼痛、月経に関連する疼痛、がんに関連する疼痛、歯痛、頭痛、片頭痛、緊張型頭痛、三叉神経痛、顎関節症候群、筋筋膜性疼痛筋傷害、線維筋痛症候群、骨関節痛(変形性関節症)、慢性関節リウマチ、熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫、変形性関節炎、骨粗鬆症、骨転移もしくは未知の理由による捻挫もしくは骨折骨痛、痛風、結合組織炎、筋筋膜性疼痛、胸郭出口症候群、上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄傷害(SCI)関連疼痛、中枢性卒中後痛、がん性ニューロパシー、AIDS疼痛、鎌状赤血球疼痛または老人性疼痛などの適応症が包含される。
【0044】
特に、本発明の化合物は、上に列挙した慢性疼痛適応症に関連するうつ病などの気分障害の処置に役立つ。
【0045】
疼痛は、国際疼痛学会(IASP)により、「現実のもしくは潜在的な組織損傷に関連付けられる、またはそのような組織損傷との関連で説明される、不快な感覚的および情動的体験」と定義されている(IASP Classification of chronic pain、第2版、IASP Press(2002)、210)。疼痛は常に主観的であるが、その原因または症候群は分類することができる。「神経因性疼痛」は、サブタイプとして、IASPにより、「神経系における原発病変または機能障害によって開始されるまたは引き起こされる疼痛」と定義されている。
【0046】
神経因性疼痛の異なるサブタイプがIASPによって認識され、その例を以下に挙げる。
【0047】
「通常は疼痛を惹起しない刺激による疼痛」と定義される異痛である。
【0048】
「しばしば血管運動および発汗促進機能障害ならびにその後の栄養変化を併発する、外傷性神経病変後の持続的灼熱痛、異痛およびヒペルパシーの症候群」と定義されるカウザルギーである。
【0049】
「知覚を除く、刺激に対する増大した感受性」と定義される知覚過敏である。
【0050】
「1または複数の神経の分布範囲における疼痛」と定義される神経痛である。
【0051】
「1または複数の神経の炎症」と定義される神経炎である。
【0052】
「ある神経における機能の障害または病理学的変化、単一の神経では、単ニューロパシー、いくつかの神経では、多発性単ニューロパシー、散在性および両側性の場合は、多発ニューロパシー」と定義されるニューロパシーである。ニューロパシーは、例えば糖尿病に関連付けられる場合があり、その場合は糖尿病性神経障害と呼ばれる。
【0053】
「通常、痛みを伴う刺激に対する増大した応答」と定義される痛覚過敏である。
【0054】
「刺激、特に反復刺激に対する異常に痛い反応、ならびに上昇した閾値とを特徴とする、有痛症候群」と定義されるヒペルパシーである。
神経因性疼痛を惹起する刺激は、機械的刺激または熱刺激であり得る。
【0055】
本発明の化合物はユニークな薬理学的プロファイルを有するため、慢性疼痛に直接関係しない他の疾患の処置にも適している。5−HT2C受容体は例えばドーパミン作動性ニューロン上に位置していて、そこでは活性化がドーパミン放出に対して緊張性抑制効果を発揮し、5−HT2Cアンタゴニストはドーパミンレベルの増加をもたらす。実施例2Eに提示するデータは、本発明の化合物が、実際に、脳内で細胞外ドーパミンレベルの用量依存的増加を引き起こすことを示している。これに基づいて、5−HT2Cアンタゴニストは、選択的セロトニン再取り込み阻害剤による処置に不応性であるうつ病の処置に、特に適しているという仮説を立てることができる(Psychopharmacol. Bull.,39,147-166,2006)。この仮説の裏付けは、臨床応答が不充分な抑うつ患者(治療抵抗性うつ病、TRD、または不応性うつ病)の処置にはSSRI単独よりもミルタジピン(mirtazipine)とSSRIとの組合せの方が優れていることを示すいくつかの臨床研究に見出される(Psychother. Psychosom.,75,139-153,2006)。ミルタザピンは5−HT2および5−HT3アンタゴニストでもあり、セロトニン再取り込み阻害作用を5−HT2および5−HT3拮抗作用と合わせて発揮する化合物、例えば本発明の化合物が、TRDの処置に役立つこと、すなわち治療抵抗性うつ病を患っている患者の寛解率を増加させることを示している。
【0056】
実施例2Fおよび2Gに提示するデータは、本発明の化合物が、前前頭皮質および腹側海馬におけるアセチルコリンの細胞外レベルの増加を引き起こすことを示している。脳内のアセチルコリンレベルを増加させることはアルツハイマー病および認知障害全般を処置するための方法になるという長年にわたる臨床的証拠がある(アルツハイマー病の処置におけるアセチルコリンエステラーゼ阻害剤の使用を参照されたい)。これに基づいて、本発明の化合物は、アルツハイマー病および認知障害の処置に役立ち、アルツハイマー病および認知障害に関連するうつ病などの気分障害の処置にも役立つと考えられる。
【0057】
一部の抑うつ患者は、彼らがMADRDやHAMDなどの臨床上有意義なうつ病尺度において改善を示すという意味では、例えばSSRIによる処置に応答するが、他の症状、例えば睡眠障害および認知障害は残ることになる。これに関連して、これらの患者を部分応答者(partial responder)と呼ぶ。アセチルコリンレベルに対する上述の作用のための、本発明の化合物は、うつ病だけでなく認知障害の処置にも役立つと予想される。α−1アドレナリン作動性受容体アンタゴニストである化合物プラゾシンが睡眠障害を減少させることは、臨床研究によって示されている(Biol. Psychiatry, 61,928-934,2007)。さらにまた、本発明の化合物の5−HT2Aおよび5−HT2C拮抗作用は、鎮静、睡眠改善作用を有するとも考えられ(Neuropharmacol, 33,467-471,1994)、従って、本発明の化合物は部分応答者の処置に役立ち、言い換えると、本発明の化合物で抑うつ患者を処置することにより、部分応答者の割合が減る。
【0058】
注意欠陥多動障害(ADHD)は、最も一般的な神経行動学的障害の一つである。ADHDは、限定的、反復的または定型的行動を伴う社会性およびコミュニケーション障害の三主徴を特徴とする。ADHDは通常、小児期または青年期に始まるが、症状は成人まで継続し得る。アトモキセチンは、ADHDの処置にFDAによって承認された、現在唯一の非中枢刺激薬である(Drugs,64,205-222,2004)。アトモキセチンはノルエピネフリン再取り込み阻害剤であり、ADHDの処置に本発明の化合物を使用し得ることを示唆している。また、本発明の化合物は、上述のα−1アドレナリン作動性受容体および5−HT2拮抗作用のための、ADHDの処置に有益な鎮静作用を有し得る。
【0059】
メランコリーは、しばしば重症うつ病に結びつけられるうつ病の特別なサブタイプであり、このタイプのうつ病はメランコリー型うつ病とも呼ばれる。メランコリーは不安、未来恐怖、不眠、および食欲不振に関連付けられる。セロトニン再取り込みとノルエピネフリン再取り込みの両方を阻害する化合物、例えばベンラファキシンは、重症うつ病およびメランコリーの患者の処置に、特に有効であることが示されている(Depres. Anxiety,12,50-54,2000)。上述のように、5−HT2C拮抗作用を発揮する化合物はドーパミンレベルを増加させ、従って、そのような化合物はメランコリーの処置に有効であると予想される(Psychpharm. Bull.,39,147-166,2006)。また、本発明の化合物のα−1アドレナリン作動性受容体および5−HT2拮抗作用は、睡眠を正常化するのに役立つと予想され、従って、前記化合物はメランコリーの処置に役立つ。
【0060】
FDAは最近、心的外傷後ストレス障害(PTSD)の処置に、二つのSSRI、セルトラリンおよびパロキセチンを承認した。さらにまた、5−HT2Aアンタゴニスト活性を有する化合物は、PTSD患者の動揺、不眠および爆発性を抑制することができると予想されるので、有用である(Curr opinion Invest. Drug,4,37-41,2003)。従って、本発明の化合物はPTSDの処置に役立つと予想される。
【0061】
顔面紅潮は閉経移行期に関連する症状である。一部の女性は、睡眠または活動全般が妨げられ、処置が必要になるほど、これに悩まされ得る。何十年も前からエストロゲンによるホルモン補充療法が確立された治療になっているが、最近は、乳がんや心イベントなどの副作用に関する懸念が表明されている。SSRIおよびSNRIを使った臨床試験では、これらの化合物は、顔面紅潮に対して効果を有するが、その効果はエストロゲンほどではないことが示されている(J.Am.Med.Ass.,295,2057-2071,2006)。しかし、セロトニンおよび/またはノルエピネフリン再取り込みを阻害する化合物、例えば本発明の化合物による顔面紅潮の処置は、エストロゲンを許容できない女性またはエストロゲンを許容しないであろう女性にとって、代替処置になり得る。
【0062】
睡眠時無呼吸症候群もしくは閉塞性睡眠時無呼吸低呼吸症候群または閉塞性睡眠呼吸障害は、有効な薬物療法がまだ同定されていない障害である。しかし、動物で行われたいくつかの研究は、5−HT3アンタゴニスト、例えば本発明の化合物が治療的介入に有効であり得ることを示唆している(Sleep,21,131-136,1998;Sleep,8,871,878,2001)。
【0063】
5−HT3アンタゴニストであるオダンセトロンは、最近、渇望ならびにアルコールおよび薬物乱用の処置に有効であることが示されている(Drug Alc. Depend.,84,256-263,2006;Pharmacol Therapeut.,111,855-876,2006)。5−HT3アンタゴニスト、例えば本発明の化合物が、渇望、例えばアルコール、ニコチンまたは炭水化物渇望、ならびにアルコールおよび薬物乱用の処置に役立ち得るという考えを裏付けるものと思われる。
【0064】
5−HT3アンタゴニストの用途として提案されるものには、他にも、嘔吐、特に化学療法誘発性嘔吐、摂食障害、例えば過食症、および過敏性腸症候群(IBS)などがある(Exp. Opin. Ther. Targets,11,527-540,2007)。
【0065】
ユニークな薬理学的プロファイルを有する本発明の化合物は、さらに、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病またはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症および腹圧性尿失禁の処置にも役立つと予想される。
【0066】
ある実施形態において、本発明は、慢性疼痛;部分応答者のうつ病;治療抵抗性うつ病;アルツハイマー病;認知障害;ADHD;メランコリー;PTSD;顔面紅潮;睡眠時無呼吸;アルコール、ニコチンもしくは炭水化物渇望;物質乱用;アルコールもしくは薬物乱用;嘔吐;摂食障害;IBS;情動障害;うつ病;大うつ病性障害;産後うつ病;双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病;不安;全般性不安障害;社会不安障害;強迫性障害;パニック障害;パニック発作;恐怖症;社会恐怖症;広場恐怖症または腹圧性尿失禁を処置する方法であって、その必要がある患者に治療有効量の化合物Iを投与することを含む方法に関する。ある実施形態において、上に列挙した疾患のいずれかについて処置される前記患者は、始めに前記疾患の診断を下されている。
【0067】
ある実施形態において、本発明は、慢性疼痛を処置する方法であって、その必要がある患者に治療有効量の化合物Iを投与することを含む方法に関する。ある実施形態では、前記慢性疼痛が、幻肢痛;神経因性疼痛;糖尿病性神経障害;帯状疱疹後神経痛(PHN);手根管症候群(CTS);HIVニューロパシー;複合性局所疼痛症候群(CPRS);三叉神経痛/三叉神経神経痛/疼痛性チック;外科的介入(例えば術後鎮痛薬);糖尿病性血管障害;膵島炎に関連する毛細血管抵抗もしくは糖尿病症状;アンギナに関連する疼痛;月経に関連する疼痛;がんに関連する疼痛;歯痛;頭痛;片頭痛;緊張型頭痛;三叉神経痛;顎関節症候群;筋筋膜性疼痛筋傷害;線維筋痛症候群;骨関節痛(変形性関節症);慢性関節リウマチ;熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫;変形性関節炎、骨粗鬆症、骨転移もしくは未知の理由による捻挫もしくは骨折骨痛;痛風;結合組織炎;筋筋膜性疼痛;胸郭出口症候群;上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの);骨盤痛;心臓性胸痛;非心臓性胸痛;脊髄傷害(SCI)関連疼痛;中枢性卒中後痛;がん性ニューロパシー;AIDS疼痛;鎌状赤血球疼痛または老人性疼痛から選択される。
【0068】
ある実施形態では、前記慢性疼痛が神経因性疼痛である。
【0069】
ある実施形態では、前記神経因性疼痛が、ヒペルパシー、痛覚過敏、ニューロパシー、糖尿病性神経障害、神経炎、神経痛、知覚過敏、カウザルギー、および異痛から選択される。
【0070】
ある実施形態では、本発明の化合物が、1日あたり約0.001〜約100mg/kg体重の量で投与される。
【0071】
典型的経口投薬量は、1日あたり約0.001〜約100mg/kg体重、好ましくは1日あたり約0.01〜約50mg/kg体重の範囲にあり、それが1回以上の投薬、例えば1〜3回の投薬で投与される。厳密な投薬量は、投与頻度および投与様式、処置される対象の性別、年齢、体重および全身状態、処置される状態の性質および重症度、ならびに処置されるべき併発疾患および当業者には明白な他の因子に依存する。
【0072】
成人の場合、典型的経口投薬量は、1〜100mg/日の本発明の化合物、例えば1〜30mg/日、または5〜25mg/日の範囲にある。これは典型的には、0.1〜50mg、例えば1〜25mg、例えば1、5、10、15、20または25mgの本発明の化合物を1日に1回または2回投与することによって達成され得る。
【0073】
本明細書において使用する化合物の「治療有効量」とは、前記化合物の投与を含む治療的介入において、所与の疾患およびその合併症の臨床症状を治癒、軽減または部分的に抑止するのに充分な量を意味する。これを達成するのに充分な量は「治療有効量」と定義される。この用語は、前記化合物の投与を含む処置において、所与の疾患およびその合併症の臨床症状を治癒、軽減または部分的に抑止するのに充分な量も包含する。各目的のための有効量は、その疾患または傷害の重症度ならびに対象の体重および全身状態に依存する。適当な投薬量の決定は、値のマトリクスを作成し、そのマトリクス中の異なる点を試験することにより、日常的な実験を使って達成することができ、それが全て、熟練した医師の通常の技量に含まれることは、理解される。
【0074】
本明細書において使用する用語「処置」および「処置する」は、疾患または障害などの状態と闘うためになされる患者の管理および医療を意味する。この用語は、患者が患っている所与の状態に関する処置の全範囲、例えば症状もしくは合併症を軽減するため、その疾患、障害もしくは状態の進行を遅延させるため、症状および合併症を軽減し、もしくは緩和するため、および/またはその疾患、障害もしくは状態を治癒させ、もしくは排除するため、ならびにその状態を防止するためになされる活性化合物の投与を包含するものとし、この場合、防止は、その疾患、状態、または障害と闘うためになされる患者の管理および医療と解釈され、症状または合併症の発生を防止するための活性化合物の投与を包含する。それでもなお、予防的(防止的)処置と治療的(治癒的)処置は、本発明の二つの別個の態様である。処置される患者は、好ましくは哺乳動物、特にヒトである。
【0075】
ある実施形態において、本発明は、慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁を処置するための医薬の製造における本発明の使用に関する。
【0076】
ある実施形態において、本発明は、神経因性疼痛などの慢性疼痛を処置するための医薬の製造における本発明の使用に関する。
【0077】
ある実施形態において、本発明は、慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁を処置するための医薬として使用するための本発明の化合物に関する。
【0078】
ある実施形態において、本発明は、神経因性疼痛などの慢性疼痛を処置するための医薬として使用するための本発明の化合物に関する。
【0079】
本発明の化合物は、純粋な化合物として単独で、または薬学的に許容できる担体もしくは賦形剤と組み合わせて、単回投与または複数回投与で投与することができる。本発明の医薬組成物は、「Remington: The Science and Practice of Pharmacy」(第19版、Gennaro編、Mack Publishing Co.、ペンシルバニア州イーストン、1995)に開示されているような従来の技法に従って、薬学的に許容できる担体または希釈剤、ならびに他の任意の公知佐剤および賦形剤を使って製剤することができる。
【0080】
医薬組成物は、経口、直腸、鼻、肺、局所外用(口腔内および舌下を含む)、経皮、槽内、腹腔内、膣および非経口(皮下、筋肉内、髄腔内、静脈内および皮内を含む)経路など、任意の適切な経路による投与に合わせて、具体的に製剤化することができ、経口経路は好ましい。好ましい経路が、処置される対象の全身状態および年齢、処置される状態の性質および選択した活性成分に依存することは理解される。
【0081】
経口投与用の医薬組成物には、カプセル剤、錠剤、糖衣丸、丸剤、口中錠、粉末剤および顆粒剤などの固形剤形が含まれる。それらは適宜、コーティングを施して調製することができる。
【0082】
経口投与用の液状剤形には、溶液剤、乳剤、懸濁剤、シロップ剤およびエリキシル剤が含まれる。
【0083】
非経口投与用の医薬組成物には、滅菌された水性および非水性の注射可能な溶液剤、分散剤、懸濁剤または乳剤、ならびに使用に先だって滅菌注射可能溶液または分散液に再構成される滅菌粉末剤が含まれる。
【0084】
他の適切な投与形態には、坐剤、噴霧剤、軟膏、クリーム剤、ゲル剤、吸入剤、皮膚パッチ、インプラントなどがある。
【0085】
本発明の化合物は、約0.1〜50mgの量の前記化合物(例えば1mg、5mg、10mg、15mg、20mgまたは25mgの本発明の化合物)を含有する単位剤形(unit dosage form)で投与すると好都合である。
【0086】
静脈内投与、髄腔内投与、筋肉内投与及び同様の投与などの非経口経路の場合、典型的に、用量は経口投与に用いられる用量の約半分程度である。
【0087】
非経口投与には、滅菌水性溶液、水性プロピレングリコール、水性ビタミンEまたはゴマ油もしくはラッカセイ油中の本発明の化合物の溶液を使用することができる。そのような水性溶液は、必要であれば適切に緩衝化されるべきであり、最初に希釈液を充分な食塩水またはグルコースで等張性にする。水性溶液は静脈内、筋肉内、皮下および腹腔内投与には特に適している。使用される滅菌水性媒質は全て、当業者に公知である標準的技法により、容易に入手することができる。
【0088】
適切な医薬担体には、不活性固形希釈剤または増量剤、滅菌水性溶液および種々の有機溶剤が含まれる。固形担体の例は、ラクトース、白土、スクロース、シクロデキストリン、タルク、ゼラチン、寒天、ペクチン、アラビアゴム、ステアリン酸マグネシウム、ステアリン酸およびセルロースの低級アルキルエーテルである。液状担体の例は、シロップ、ラッカセイ油、オリーブ油、リン脂質、脂肪酸、脂肪酸アミン、ポリオキシエチレンおよび水である。本発明の化合物と薬学的に許容できる担体とを組み合わせることによって形成された医薬組成物は、次に、開示した投与経路に適した様々な剤形で、容易に投与することができる。
【0089】
経口投与に適した本発明の製剤は、それぞれが予め決定された量の活性成分を含有し、適切な賦形剤を含んでもよい、カプセル剤または錠剤などの不連続な単位として提示することができる。さらにまた、経口使用できる製剤は、粉末状もしくは顆粒状であるか、水性もしくは非水性液体中の溶液もしくは懸濁液であるか、水中油型もしくは油中水型の液状乳剤であることができる。
【0090】
経口投与に固形担体を使用する場合、その調製物は錠剤であるか、例えば粉末もしくはペレット状にして硬ゼラチンカプセルに入れるか、またはトローチ剤もしくは口中剤の形態を取り得る。固形担体の量は様々であり得るが、通常は約25mg〜約1gである。
【0091】
液状担体を使用する場合、その調製物はシロップ剤、乳剤、軟ゼラチンカプセル剤または滅菌注射用液剤、例えば水性もしくは非水性の液状懸濁剤もしくは溶液剤の形態を取り得る。
【0092】
錠剤は、活性成分を通常の佐剤および/または希釈剤と混合した後、その混合物を従来の打錠機で圧縮することによって調製することができる。佐剤または希釈剤の例には、トウモロコシデンプン、ジャガイモデンプン、タルク、ステアリン酸マグネシウム、ゼラチン、ラクトース、ゴムなどが含まれる。そのような目的に通常使用される他の佐剤または添加剤、例えば着色剤、着香剤、保存剤などはいずれも、それらが活性成分と適合するのであれば、使用することができる。
【0093】
本発明の化合物を含むカプセル剤は、前記化合物を含む粉末を微結晶セルロースおよびステアリン酸マグネシウムと混合し、前記粉末を硬ゼラチンカプセルに入れることによって調製することができる。場合により、適切な色素を使って前記カプセル剤を着色してもよい。典型的には、カプセル剤は、0.25〜20%の本発明の化合物、例えば0.5〜1.0%、3.0〜4.0%、14.0〜16.0%の本発明の化合物を含む。これらの力価は、単位剤形中の1、5、10、15、20および25mgの本発明の化合物を便利に送達するために使用することができる。
【0094】
注射用溶液剤は、活性成分と場合によっては添加剤とを注射用溶剤(好ましくは滅菌水)の一部に溶解し、その溶液を所望の体積に調節し、その溶液を滅菌し、それを適切なアンプルまたはバイアルに充填することによって調製することができる。浸透圧調節剤、保存剤、酸化防止剤など、当分野で従来から使用されている任意の適切な添加剤を加えることができる。
【0095】
化合物Iは、国際公開第2003/029232号パンフレットに概説されているように調製することができる。化合物Iの塩は、適当な酸を添加した後、析出させることによって製造することができる。析出は、例えば冷却、溶剤の除去、他の溶剤の添加、またはそれらの併用によって引き起こすことができる。
【0096】
本明細書で言及する参考文献は、刊行物、特許出願、および特許を含めて全て、その文書の組み込みが本明細書のどこか他の項で別途なされているかどうかにかかわらず、各参考文献が参照により、組み込まれることが個別にかつ明示的に示され、本明細書にその全体が(法が許す範囲で最大限に)記載されているかのように、参照により本明細書に組み込まれる。
【0097】
本発明の説明における用語「a」および「an」ならびに「the」および類似する指示物(referent)の使用は、本明細書に別段の表示がない限り、または文脈上、明らかに矛盾しない限り、単数および複数の両方を包含するとみなすべきである。例えば「化合物(the compound)」という表現は、別段の表示がない限り、本発明の様々な「化合物(compounds)」または記載された特定の態様を指すと理解すべきである。
【0098】
別段の表示がない限り、本明細書に記載する厳密な値は全て、対応する近似値の代表である(例えば、特定の因子または測定値に関して記載される厳密な例示的値は全て、対応する近似測定値(適宜「約」によって修飾されるもの)をも記載しているとみなすことができる)。
【0099】
1または複数の要素に関して「を含む(comprising)」「を有する(having)」「を包含する(including)」または「含有する(containing)」などの用語を使ってなされる本発明の任意の1または複数の態様の、本明細書における説明は、別段の明記がない限り、または文脈上、明らかに矛盾しない限り、その特定の1または複数の要素「からなる(consists of)」「から本質的になる(consists essentially of)」または「を実質的に含む(substantially comprise)」本発明の類似する1または複数の態様の裏付けを提供するものである(例えば、特定の要素を含むと本明細書に記載されている組成物は、別段の明記がない限り、または文脈上、明らかに矛盾しない限り、その要素からなる組成物も記載していると理解すべきである)。
【実施例】
【0100】
分析方法
X線粉末ディフラクトグラム(XRPD)は、CuKα1放射線を使ってPANalytical X’Pert PRO X線回折計で測定した。試料は、X’celerator検出器を使用し、反射モードにより、2θ範囲5〜40°で測定した。元素組成(CHN)はElementarのElementar Vario EL装置で測定した。約4mgの試料を各測定に使用し、結果を2回の測定の平均値として記載する。
【0101】
実施例1a 化合物IのHBr塩
撹拌して、わずかに(約45℃)加熱した油状の4−(2−p−トリルスルファニル−フェニル)−ピペリジン−1−カルボン酸エチルエステル442グラムにAcOH中の33wt%HBr(5.7M、2.5等量)545mlを加えた。この混合によって10℃の発熱が生じる。最終添加後に、反応混合物を80℃まで加熱し、18時間放置する。試料を取り出してHPLCで分析し、もし完了していなければ、AcOH中の33wt%HBrを追加しなければならない。そうでない場合は、その混合物を25℃まで冷却して、生成物4−(2−p−トリルスルファニル−フェニル)−ピペリジン臭化水素酸塩を析出させる。25℃で1時間後、その濃厚懸濁液にジエチルエーテル800mlを加える。撹拌をさらに1時間続けてから、生成物を濾過によって単離し、ジエチルエーテル400mlで洗浄し、真空中、40℃で一晩乾燥する。化合物Iの臭化水素酸塩は白色固体として単離された。
【0102】
実施例1b 化合物IのHBr塩
【0103】
2−(4−トリルスルファニル)−フェニルブロミド
窒素で覆った撹拌反応器中で、N−メチル−ピロリドン、NMP(4.5L)に、窒素を20分間吹き込んだ。4−メチルベンゼンチオール(900g、7.25mol)を加え、次に1,2−ジブロモベンゼン(1709g、7.25mol)を加えた。最終的にカリウムtert−ブトキシド(813g、7.25mol)を最後の反応物として加えた。反応は発熱的であり、反応混合物の温度を70℃まで上昇させた。次に反応混合物を120℃に2〜3時間加熱した。反応混合物を室温まで冷却した。酢酸エチル(4L)および塩化ナトリウム水溶液(15%、2.5L)を加えた。その混合物を20分間撹拌した。水相を分離し、新たな酢酸エチル(2L)の一部分で抽出した。水相を分離し、有機相を合わせ、塩化ナトリウム溶液(15%、2.5L)で洗浄した。有機相を分離し、硫酸ナトリウムで乾燥し、減圧下で蒸発させることにより、20〜30%のNMPを含有する赤色油状物を得た。その油状物をメタノールで2倍の体積に希釈し、その混合物を還流した。透明な赤色溶液が得られるまで、メタノールをさらに追加した。その溶液に種晶を入れて室温までゆっくり冷却した。生成物はオフホワイトの結晶として結晶化し、それらを濾過によって単離し、メタノールで洗浄し、真空オーブン中、40℃で恒量まで乾燥した。
【0104】
エチル4−ヒドロキシ−4−(2−(4−トリルスルファニル)フェニル)−ピペリジン−1−カルボキシレート
窒素で覆われた撹拌反応器中で、2−(4−トリルスルファニル)−フェニルブロミド(600g、2.15mol)をヘプタン(4.5L)に懸濁した。室温でヘキサン中の10M BuLi(235mL、2.36mol)を10分かけて加えた。わずかな発熱しか認められなかった。その懸濁液を周囲温度で1時間撹拌した後、−40℃まで冷却した。THF(1.5L)に溶解した1−カルベトキシ−4−ピペリドン(368g、2.15mol)を反応温度が−40℃未満に保たれる速さを上回らない速さで加えた。反応が完了したら、それを0℃まで温め、温度を10℃未満に保ちながら、1M HCl(1L)を加えた。酸性水相を分離し、酢酸エチル(1L)で抽出した。有機相を合わせ、塩化ナトリウム溶液(15%、1L)で抽出した。有機相を硫酸ナトリウムで乾燥し、蒸発させることにより、半結晶質塊を得た。それをエチルエーテル(250mL)でスラリー化し、濾別した。真空オーブン中、40℃で恒量まで乾燥した。
【0105】
エチル4−(2−(4−トリルスルファニル)フェニル)−ピペリジン−1−カルボキシレート
トリフルオロ酢酸(2.8kg、24.9mol)およびトリエチルシラン(362g、3.1mol)を効率の良い撹拌機を有する反応器に投入した。エチル4−ヒドロキシ−4−(2−(4−トリルスルファニル)フェニル)−ピペリジン−1−カルボキシレート(462g、1.24mol)を粉末ロートから少しずつ加えた。反応はわずかに発熱的だった。温度は50℃まで上昇した。添加を終わらせた後、反応混合物を60℃に18時間温めた。反応混合物を室温まで冷却した。トルエン(750mL)および水(750mL)を加えた。有機相を単離し、水相を新たなトルエン(750mL)の一部分で抽出した。有機相を合わせ、塩化ナトリウム溶液(15%、500mL)で洗浄し、硫酸ナトリウムで乾燥した。硫酸ナトリウムを濾去し、濾液を減圧下で蒸発させることによって赤色油状物とし、それをさらに次のステップで加工した。
【0106】
4−(2−(4−トリルスルファニル)フェニル)−ピペリジン臭化水素酸塩
実施例3で得た赤色油状物である粗エチル4−(2−(4−トリルスルファニル)フェニル)−ピペリジン−1−カルボキシレートを撹拌反応器において、酢酸中の臭化水素酸(40%、545mL、3.11mol)と混合した。その混合物を80℃で18時間加熱した。その反応混合物を室温まで冷却した。冷却中に生成物が晶出した。室温で1時間後、エチルエーテル(800mL)を反応混合物に加え、その混合物をさらに1時間撹拌した。生成物を濾別し、エチルエーテルで洗浄し、真空オーブン中、50℃で恒量まで乾燥した。
【0107】
実施例1c 化合物IのHBr塩の再結晶
化合物IのHBr塩(例えば上記のように調製したもの)10.0グラムの混合物をH2O 100ml中で加熱還流した。混合物は80〜90℃で透明になり、完全に溶解した。その透明な溶液にチャコール1グラムを加え、還流を15分間続けてから、濾過し、室温まで自然放冷した。冷却中に白色固体の析出が起こり、その懸濁液を室温で1時間撹拌した。濾過し、真空下40℃で一晩乾燥することにより、化合物IのHBr酸付加塩6.9グラム(69%)を得た。XRPDについては図1を参照されたい。元素分析:3.92%N、59.36%C、6.16%H(理論値:3.85%N、59.34%C、6.09%H)。
【0108】
実施例1d 遊離塩基の原液の調製
酢酸エチル500mlおよびH2O 200mlの混合物に化合物IのHBr塩50グラムを加えて、二相スラリーを生成させた。このスラリーに約25mlの濃NaOHを加えたところ、透明な二相溶液の形成が起こった(pHは13〜14と測定された)。その溶液を激しく15分間撹拌し、有機相を分離した。有機相をH2O 200mlで洗浄し、Na2SO4で乾燥し、濾過し、真空下、60℃で蒸発させることにより、遊離塩基を38グラムの収量(99%)で、ほぼ無色の油状物として得た。
【0109】
酢酸エチルを使って10グラムの油状物を溶解し、体積を150mlに調節することによって、酢酸エチル中の0.235M原液を生成し、そこから一定分量1.5ml(遊離塩基100mg)を使用した。
【0110】
96vol%EtOHを使って10グラムの油状物を溶解し、体積を100mlに調節することによって、EtOH中の0.353M原液を生成し、そこから一定分量1.0ml(遊離塩基100mg)を使用した。
【0111】
実施例1e 遊離塩基の原液を使った塩の形成
所与の一定分量を試験管に入れ、撹拌しながら、表1に示すように適当な量の酸を加えた。酸が液体である場合はニートで加え、そうでない場合は、所与の溶剤に溶解してから加えた。混合および析出の後、撹拌を一晩続け、析出物を濾過によって集めた。真空下、30℃で乾燥する前に、少量の参照試料を取り出し、真空せずに室温で乾燥した。この手順は溶媒和物について調べるために含まれた。いくつかの結果を表1に提示する。XRPDディフラクトグラムを図1〜22に示し、選択したピークの位置を表2に要約する。表3に、水における本発明の化合物の溶解度を結果として得られる飽和溶液のpHと共に示す。「析出物」という欄は、溶解度決定後に単離された析出物が溶解した化合物と同一であるかどうかを表し、これは水和物の形成を示す。
【0112】
【表1】
【0113】
【表2】
【0114】
【表3】
【0115】
実施例2A セロトニン(5−HT)およびノルエピネフリン(NE)再取り込み阻害
試験化合物およびラット皮質シナプトソーム調製物の一定分量を37℃で10分間プレインキュベートした後、[3H]NEまたは[3H]5−HT(最終濃度10nM)を加えた。10μMタルスプラムまたはシタロプラムの存在下で非特異的取り込み量を決定し、緩衝液の存在下で総取り込み量を決定した。一定分量を37℃で15分間インキュベートした。インキュベーション後に、シナプトソームによって取り込まれた[3H]NEまたは[3H]5−HTを0.1%PEIに30分間予浸したUnifilter GF/Cでの濾過により、Tomtec Cell Harvesterプログラムを使って分離した。フィルターを洗浄し、Wallac MicroBetaカウンターでカウントした。
【0116】
NETにおいて、本発明の化合物は23nMのIC50値を示す。SERTにおいて、本発明の化合物は8nMのIC50値を示す。
【0117】
実施例2B 5−HT2A拮抗作用
本発明の化合物をセロトニン受容体に対する親和性について試験したところ、これらは5−HT2A受容体に親和性を有するアンタゴニストプロファイル(Ki 54nM)を示すことが分かった。親和性は、Y=100/(1+10(X−logIC50))から算出される(式中、Yは結合%を表し、Xは化合物の濃度を表す)。5濃度の化合物(1、10、30、100、1000nM)を使って、IC50を算出した。Kiは、Cheng Prusoff式、Ki=(IC50/(1+([L]/Kd))から算出した。親和性はMDL Pharmaservicesカタログ番号271650で決定した。
【0118】
ヒト5−HT2A受容体を発現させる哺乳動物細胞において、本発明の化合物は競合的アンタゴニスト特性を示す。化合物は5−HT2A受容体に<100nMのKiで結合し、機能アッセイでは、5−HTによって惹起される細胞内貯蔵からのCa2+の放出を化合物が67nMのKbで拮抗する。シルド解析により、100mMのKbを有する競合的拮抗作用が明らかになった。
【0119】
実験は以下のように行った。実験の2日または3日前に、250fmol/mgのヒト5−HT2A受容体を発現させるCHO細胞を実験日にコンフルエント単層を得るのに充分な密度でプレーティングする。その細胞に、湿度95%の5%CO2インキュベータ中、37℃で60分間、色素負荷する(Molecular DevicesのCa2+キット)。蛍光イメージングプレートリーダまたはMolecular Devices(カリフォルニア州サニーベール)のFLIPR384を使用し、488nmの励起波長および500〜560nmの蛍光範囲で、基礎蛍光をモニターした。レーザー強度は、約8000〜10000蛍光ユニットの基礎値を得るのに適したレベルに設定した。基礎蛍光の変動は10%未満にするべきである。少なくとも3桁をカバーし、増加する一連の試験化合物濃度を使ってEC50値を評価する。pA2値の評価は、4つの異なる化合物濃度(150、400、1500および4000nM)で5−HTの全用量応答曲線を検証して行った。Kb値の評価も、EC85の5−HTで、2桁にわたる試験物質濃度を検証して行った。試験物質は5−HTの5分前に細胞に加える。Ki値はCheng−Prusoff式を使って算出される。
【0120】
実施例2C 5−HT3A受容体拮抗作用
ヒトホモマー5−HT3A受容体を発現させる卵母細胞において、5−HTは2600nMのEC50で電流を作動させる。この電流は、オンダンセトロンなどの古典的5−HT3アンタゴニストで拮抗することができる。オンダンセトロンはこの系において1nM未満のKi値を示す。本発明の化合物は、低濃度(0.1nM〜100nM)では強力な拮抗作用を示し(IC50約10nM/Kb約2nM)、より高濃度(100〜100000nM)で適用した場合にはアゴニスト特性を示して(EC50約2600nM)、5−HTそのものによって誘起される最大電流の約70〜80%の最大電流に達する。ラットホモマー5−HT3A受容体を発現させる卵母細胞において、5−HTは3.3μMのEC50で電流を作動させる。実験は次のように行った。0.4%MS−222中で10〜15分間麻酔した成熟雌アフリカツメガエル(Xenepus laevis)から、卵母細胞を外科的に摘出した。次にその卵母細胞をOR2緩衝液(82.5mN NaCl、2.0mM KCl、1.0mM MgCl2および5.0mM HEPES、pH7.6))中の0.5mg/mlコラゲナーゼ(タイプIA Sigma−Aldrich)により、室温で2〜3時間消化した。卵胞層が取り除かれた卵母細胞を選択し、2mMピルビン酸ナトリウム、0.1U/lペニシリンおよび0.1μg/lストレプトマイシンを補足した変法バース塩類緩衝液(Modified Barth's Saline buffer)[88mM NaCl、1mM KCl、15mM HEPES、2.4mM NaHCO3、0.41mM CaCl2、0.82mM MgSO4、0.3mM Ca(NO3)2]中で24時間インキュベートした。ステージIV−IV卵母細胞を同定し、ヒト5−HT3A受容体をコードするcRNA 14〜50pgを含有するヌクレアーゼフリー水12〜48nlを注入し、それを電気生理学的記録に使用するまで18℃でインキュベートしておいた(注入後1〜7日間)。ヒト5−HT3受容体を発現させている卵母細胞を1mlバスに入れ、リンゲル緩衝液(115mM NaCl、2.5mM KCl、10mM HEPES、1.8mM CaCl2、0.1mM MgCl2、pH7.5)で潅流した。寒天で栓をした3M KClを含有する0.5〜1MΩ電極で細胞を固定し、GeneClamp 500B増幅器により、−90mVで電圧固定した。卵母細胞をリンゲル緩衝液で潅流し続け、薬物をその潅流液に適用した。5−HTアゴニスト溶液を10〜30秒間適用した。10μM 5−HT刺激に対する濃度応答を測定することによって5−HT3受容体アンタゴニストの効力を調べた。
【0121】
実施例2D α1A受容体拮抗作用
本発明の化合物をα1A受容体に対する親和性について試験したところ、これらはα1A受容体に対して中等度の親和性を有するアンタゴニストプロファイルを示すことが分かった(Ki=34nM)。
【0122】
実験日に膜(膜調製の説明については下記参照)を融解し、ウルトラタラックスを使って緩衝液中でホモジナイズし、所望の濃度に希釈した(5μg/ウェル〜5μg/900μl、使用時まで氷上の保存)。
【0123】
試験化合物50μl、[3H]−プラゾシン50μlおよび膜900μlを混合することによって実験を開始し、その混合物を25℃で20分間インキュベートする。10μM WB−4101の存在下で非特異的結合量を決定し、緩衝液の存在下で総結合量を決定する。インキュベーション後に、0.1%PEIに30分間予浸したUnifilter GF/Bでの濾過により、Tomtec Cell Harvesterプログラム(D4.2..4)を使って、結合リガンドを未結合リガンドから分離する。96ウェル。フィルターを氷冷緩衝液1mlで3回洗浄し、50℃で乾燥し、35μl/ウェルのシンチレーション液をフィルターに加える。結合している放射能をWallac OY 1450 MicroBetaでカウントする。親和性はY=100/(1+10(X−logIC50))から算出される(式中、Yは結合%を表し、Xは化合物の濃度を表す)。2桁をカバーする化合物濃度を使ってIC50値を算出した。KiはCheng Prusoff式、Ki=(IC50/(1+([L]/Kd))から算出した。
【0124】
機能アッセイにおいて、本発明の化合物は、アドレナリンが惹起する細胞内貯蔵からのCa2+の放出を拮抗し、機能アッセイにより、化合物はアンタゴニストであることが明らかになった。
【0125】
これらの実験は基本的に以下に述べるように行った。
【0126】
細胞は全て、10%BCS、4mM L−グルタミン(またはCOS−7の場合は2mM)、および100単位/mlペニシリン+100μg/mlストレプトマイシンを補足したDMEM培地において、5%CO2中、37℃で培養した。
【0127】
アッセイの24時間前に、ヒトアルファ1A−7受容体を発現させるCHO細胞をポリ−D−リジンで被覆された384ウェルブラックウォールマイクロタイタープレートに播種した。培養培地を吸引し、細胞への色素負荷をハンクス平衡塩類溶液(138mM NaCl、5mM KCl、1.3mM CaCl2、0.5mM MgCl2、0.4mM MgSO4、0.3mM KH2PO4、0.3mM Na2HPO4、5.6mMグルコース)+20mM HEPES pH 7.4、0.05%BSAおよび2.5mMプロベニシド(probenicid)(50μl/ウェル)から構成されるアッセイ緩衝液中の1.5μM Fluo−4を使って、5%CO2中、37℃で1時間行った。過剰な色素を捨てた後、細胞をアッセイ緩衝液中で洗浄し、45μl/ウェル(またはアンタゴニストアッセイの場合は30μl/ウェル)に匹敵する最終液量を重層した。アンタゴニスト評価の場合は、この時点で、アンタゴニストまたはビヒクルを4×最終濃度で4%DMSO含有緩衝液中の15μl一定分量(最終DMSO=1%)として加えた後、20分間インキュベートした。蛍光イメージングプレートリーダまたはMolecular Devices(カリフォルニア州サニーベール)のFLIPR(商標)を使用し、488nmの励起波長および500〜560nmの蛍光範囲で、基礎蛍光をモニターした。レーザー励起エネルギーは、基礎蛍光測定値が約8000相対蛍光ユニット(RFU)になるように調節した。次に、アッセイ緩衝液(15μl)に希釈したアゴニストを使って細胞を室温で刺激し、RFUを2.5分間にわたって1.5秒間隔で測定した。蛍光の最大変化を各ウェルについて算出した。蛍光の最大変化から導いた濃度応答曲線を非線形回帰(Hill式)によって解析した。アンタゴニスト決定の場合、20分間の化合物インキュベーション(上記)後に、固定濃度の標準アゴニスト、セロトニンを加えた。
【0128】
実施例2E ドーパミンの増加
本発明の化合物の単回注射は、ラット前頭皮質における細胞外DAレベルを用量依存的に増加させた。図23に図示するように、本発明の化合物は、8.9mg/kgおよび18mg/kg s.c.で、DAレベルをベースラインレベルよりそれぞれ約100%および150%上昇させた。量は遊離塩基として算出する。
【0129】
方法
初期体重275〜300gの雄スプレーグ−ドーリーラットを使用した。動物を室内温度(21±2℃)および湿度(55±5%)が一定になるように制御された条件下、12時間明/暗周期で飼育し、食物および水道水を自由に摂取させた。三日間の処置実験に、浸透圧ミニポンプ(Alzet、2ML1)を使用した。ポンプへの充填を無菌条件下で行い、セボフルランス(sevoflurance)麻酔下で皮下に植え込んだ。実験は内蔵のミニポンプを使って行った。3日間の処置後の試験化合物の血漿レベルを測定するための血液試料を実験の最後に収集した。
【0130】
外科手術および微小透析実験
動物をヒプノルム(hypnorm)/ドルミカム(2ml/kg)で麻酔し、透析プローブチップが腹側海馬内(座標:ブレグマの前方5.6mm、側方−5.0mm、硬膜の腹側7.0mm)または前頭皮質内(座標:ブレグマの前方3.2mm;側方3.0mm;硬膜の腹側4.0mm)に配置されるように、脳内ガイドカニューレ(CMA/12)を海馬中に定位的に植え込んだ。ガイドカニューレを固定するために固定ネジおよびアクリルセメントを使用した。動物の体温を直腸プローブでモニターし、37℃に維持した。ラットを一匹ずつケージに入れて、外科手術から2日間回復させた。実験日に、ガイドカニューレを通して微小透析プローブ(CMA/12、直径0.5mm、長さ3mm)を挿入した。2チャンネルスイベルを通してプローブを微量注入ポンプに接続した。濾過したリンゲル液(145mm NaCl、3mM KCl、1mM MgCl2、1.2mM CaCl2)による微小透析プローブの潅流を脳内にプローブを挿入する直前に開始し、実験の継続中は1(1.3)μL/分の一定流量で続けた。180分間の安定化後に、実験を開始した。透析液を20(30)分ごとに収集した。
【0131】
実験後に断頭によってラットを屠殺し、その脳を摘出し、凍結し、プローブの配置を検証するために薄切した。
【0132】
透析液の分析
透析液中のドーパミンの濃度は電気化学的検出によるHPLCを利用して分析した。モノアミン類を逆相液体クロマトグラフィー(ODS 150×3mm、3μM)で分離した。ドーパミン:流速0.5ml/分の90mM NaH2PO4、50mMクエン酸ナトリウム、367mg/l 1−オクタンスルホン酸ナトリウム、50μM EDTAおよび8%アセトニトリル(pH4.0)からなる移動相である。電量検出器を使って電気化学的検出を行った;電位は250mVに設定(ガードセルは350mV)(Coulochem II、ESA)。
【0133】
実施例2F アセチルコリンの増加
本発明の化合物が自由行動ラットの前前頭皮質におけるアセチルコリンの細胞外レベルに及ぼす作用を評価するために、実験を計画した。
【0134】
実験には雄ウィスターラット(280〜350g;Harlan、オランダ・ザイスト)を使用した。ラットをプラスチック製ケージ(30×30×40cm)で個別に飼育し、食物および水を自由に摂取させておいた。
【0135】
イソフルラン(2%、400mL/分 N2O、400ml/分 O2)を使ってラットを麻酔した。局所麻酔にはリドカイン(10%m/v)を使用した。各動物を定位固定装置(Kopf instrument、USA)に入れ、PaxinosおよびWatsonのラット脳アトラス(1982)を使って、自作のI字状プローブ(Hospal AN 69メンブレン、露出表面4mm)を内側前前頭皮質(mPFC)に挿入した。プローブの先端の座標はmPFC[AP=3.4mm、L=−0.8mm、V=5.0mm]だった。次にプローブを歯科用セメントとネジで頭骨に固定した。フルニキシン(1mg/kg s.c.)を術後鎮痛薬として投与した。
【0136】
実験は外科手術の24〜48時間後に行った。実験日に、可撓性PEEKチューブでラットを微小潅流ポンプ(CMA102)に接続し、147mM NaCl、3.0mM KCl、1.2mM CaCl2、および1.2mM MgCl2を含有する流速1.5μL/分のリンゲル緩衝液で、透析プローブを潅流した。アセチルコリン決定用に、55μLの0.02Mギ酸が含有されるミニバイアルに、微小透析試料を30分間隔で収集した。自動フラクションコレクター(CMA142)を使って試料を収集し、分析するまで−80℃で保存した。実験の完了後にラットを屠殺した。脳を摘出し、パラホルムアルデヒド溶液(4%m/v)中で硬化させた。脳の冠状切片を作製することにより、PaxinosおよびWatson(1982)に従って、各プローブの位置を組織学的に検証した。
【0137】
試験化合物を10%2−OH−プロピル−ベータ−シクロデキストリンに溶解し、液量5mL/kgの皮下注射により、異なる用量で投与を行った。
【0138】
アセチルコリンの濃度は、HPLCとタンデム質量分析(MS/MS)検出で決定した。
【0139】
自動試料インジェクター(PerkinElmer Instruments、シリーズ200)を使って一定分量(25μL)をHPLCカラムに注入した。クロマトグラフィー分離は、4×2.0mmガードカラム(Phenomenex Synergy MAX−RP AJO−6073、Bester)で保護された逆相150×2.00mm(4μm)分析用カラム(Phenomenex Synergy MAX−RP、Bester)で行った(どちらのカラムも温度を30℃に維持)。移動相(定組成)は超精製水(UP)、アセトニトリル(ACN)、およびトリフルオロ酢酸(TFA)からなった(UP:ACN:TFA=95.0:0.5:0.1v/v/v%)。移動相をHPLCポンプ(PerkinElmer Instruments、シリーズ200マイクロポンプ)により、0.300mL/分の流量で、システムに流した。
【0140】
LC/MS分析は、API 4000 MS/MS検出器およびTurbo Ion Sprayインターフェース(どちらもApplied Biosystem(オランダ)製)からなるAPI4000 MS/MSシステムを使って行った。取得は正イオン化モードで行い、イオンスプレー電圧は5.5kV、ネブライザーガス圧は50psig(SCIEXスケール0〜90で)、プローブ温度は600℃に設定した。アセチルコリン(前駆体146.1Da、生成物86.8Da)を検出するために、マルチプルリアクションモニタリング(MRM)モードで装置を作動させた。衝突エネルギーを21.0eVとし、衝突ガス(窒素)圧を7(0〜12のSCIEXスケールで)に保った。Analyst(商標)データシステム(Applied Biosystem、バージョン1.2)を使って、データを較正し、定量した。
【0141】
変化量が50%未満である二つの連続微小透析試料をベースラインレベルとし、それを100%に設定した。アセチルコリン濃度の変化を同じ対象内でのベースラインのパーセントとして表す。
【0142】
データを図24に示す。
【0143】
実施例2G アセチルコリンの増加
本発明の化合物が自由行動ラットの前前頭皮質および腹側海馬におけるアセチルコリンの細胞外レベルに及ぼす作用を評価するために、実験を計画した。
【0144】
初期体重275〜300gの雄スプレーグ−ドーリーラットを使用した。動物を室内温度(21±2℃)および湿度(55±5%)が一定になるように制御された条件下、12時間明/暗周期で飼育し、食物および水道水を自由に摂取させた。
【0145】
外科手術および微小透析実験
ラットをヒプノルム/ドルミカム(2ml/kg)で麻酔し、透析プローブチップを腹側海馬内(座標:ブレグマの前方5.6mm、側方−5.0mm、硬膜の腹側7.0mm)または前頭皮質内(座標:ブレグマの前方3.2mm;側方0.8mm;硬膜の腹側4.0mm)に配置することを目標として、脳内ガイドカニューレ(CMA/12)を海馬中に定位的に植え込んだ。ガイドカニューレを固定するために固定ネジおよびアクリルセメントを使用した。動物の体温を直腸プローブでモニターし、37℃に維持した。ラットを一匹ずつケージに入れて、外科手術から2日間回復させた。実験日に、ガイドカニューレを通して微小透析プローブ(CMA/12、直径0.5mm、長さ3mm)を挿入した。
【0146】
2チャンネルスイベルを通してプローブを微量注入ポンプに接続した。濾過したリンゲル液(0.5μMネオスチグミンを含有する145mm NaCl、3mM KCl、1mM MgCl2、1.2mM CaCl2)による微小透析プローブの潅流を脳内にプローブを挿入する直前に開始し、実験の継続中は1μL/分の一定流量で続けた。180分間の安定化後に、実験を開始した。透析液を20分ごとに収集した。実験後に動物を屠殺し、その脳を摘出し、凍結し、プローブの配置を検証するために薄切した。
【0147】
透析液アセチルコリンの分析
透析液中のアセチルコリン(ACh)濃度は、100mMリン酸水素二ナトリウム、2.0mMオクタンスルホン酸、0.5mMテトラメチルアンモニウムクロリドおよび0.005%MB(ESA)、pH8.0からなる移動相を使用し、電気化学的検出によるHPLCを利用して分析した。分析用カラム(ESA ACH−250)、流量0.35ml/分、温度:35℃でのAChの分離に先だって、固定化コリンオキシダーゼを含有するプレカラム酵素リアクタ(ESA)により、注入された試料(10μl)からコリンが除去された。分析用カラム後に、試料は、固定化アセチルコリンエステラーゼおよびコリンオキシダーゼを含有するポストカラム固相リアクタ(ESA)を通過した。後者のリアクタはAChをコリンに変換した後、コリンをベタインおよびH2O2に変換した。後者を白金電極(分析用セル:ESA、モデル5040)を使って電気化学的に検出した。
【0148】
データ提示
単回注射実験では、化合物投与直前の三つの連続するACh試料の平均値を各実験の基礎レベルとし、データを基礎値に対するパーセンテージに変換した(平均基礎注射前値を100%に標準化)。データを図25aおよび25bに提示する。
【0149】
図24に提示するデータは、アセチルコリンレベルの予想外の低下(例えば8mg/kgを参照)を示しているが、これを説明することは困難であり、実験的不確定性によるものであると考えられる。実施例2Fおよび2Gから得られるデータセットはどちらも、全体として、脳内の細胞外アセチルコリンレベルの同じ(すなわち用量依存的な)増加を示している。この前臨床的知見は、例えばアルツハイマー患者、部分応答者、認知障害などのような認知障害を特徴とする疾患の処置などに有用な、臨床状況における認知の改善につながると予想される。
【0150】
実施例3 神経因性疼痛に対する効果
神経因性疼痛に対する効力を実証するために、本発明の化合物を神経因性疼痛のホルマリンモデル(Neuropharm.,48,252-263,2005;Pain,51,5-17,1992)で試験した。このモデルでは、マウスの左後肢の足底表面にホルマリン(4.5%、20μl)を注射してから、そのマウスを観察のために個別にガラス製ビーカー(容量2l)に入れる。ホルマリン注射が引き起こす刺激は、傷害を受けた肢をなめるのに費やす時間の量として定量化される特徴的な二相性行動応答を誘起する。第1相(約0〜10分)は直接的な化学刺激および侵害受容を表すのに対して、第2相(約20〜30分)は神経因性の疼痛を表すと考えられる。これら二つの相は休止時間によって隔てられ、その間は行動が正常に戻る。傷害を受けた肢をなめるのに費やした時間の量を二つの相で測定することにより、疼痛刺激の低減に関する試験化合物の有効性が評価される。
【0151】
各群8匹のC57/B6マウス(約25g)を試験した。次の表4は、二つの相、すなわちホルマリン注射の0〜5分後および20〜30分後において、傷害を受けた肢をなめるのに費やされた時間の量を示している。投与した化合物の量は遊離塩基として算出されている。
【0152】
【表4】
【0153】
表4のデータは、直接的な化学刺激および侵害受容を表す第1相では、本発明の化合物がほとんど効果を有さないことを明らかにしている。より注目すべきことに、このデータは、第2相において肢をなめるのに費やされた時間の明確かつ用量依存的な短縮も明らかにし、神経因性疼痛の処置における本発明の化合物の効果を示している。
実施例4 カプセル剤
第1ステップでは、4−[2−(4−メチルフェニルスルファニル)−フェニル]ピペリジン臭化水素酸塩を微結晶セルロースと混合した。第2ステップではステアリン酸マグネシウムを混ぜ合わせた。四つの力価を有するカプセル剤を調製した。活性成分は遊離塩基として記載する。
【0154】
【表5】
【0155】
各バッチから10,000個のカプセル剤が調製された。
【特許請求の範囲】
【請求項1】
結晶質形態にある4−[2−(4−メチルフェニル−スルファニル)フェニル]ピペリジン
【化1】
および薬学的に許容できるその塩である化合物I(ただし前記化合物は4−[2−(4−メチルフェニルスルファニル)フェニル]−ピペリジン塩酸付加塩ではないものとする)。
【請求項2】
HBr付加塩である、請求項1に記載の化合物。
【請求項3】
XPRDにおける約6.08、14.81、19.26および25.38°2θのピークによって特徴付けられる、請求項2に記載の化合物。
【請求項4】
図1に図示するXRPDによって特徴付けられる、請求項2に記載の化合物。
【請求項5】
DL−乳酸付加塩である、請求項1に記載の化合物。
【請求項6】
XRPDにおける約5.30、8.18、9.44および17.24°2θのピークによって特徴付けられる、請求項5に記載の化合物。
【請求項7】
図4に図示するXRPDによって特徴付けられる、請求項5に記載の化合物。
【請求項8】
グルタル酸付加塩(1:1)である、請求項1に記載の化合物。
【請求項9】
XRPDにおける約9.39、11.70、14.05および14.58°2θのピークによって特徴付けられる、請求項8に記載の化合物。
【請求項10】
図8に図示するXRPDによって特徴付けられる、請求項8に記載の化合物。
【請求項11】
マロン酸付加塩(1:1)である、請求項1に記載の化合物。
【請求項12】
XRPDにおける10.77°、16.70°、19.93°および24.01°2θ、または6.08°、10.11°、18.25°および20.26°2θのピークによって特徴付けられる、請求項11に記載の化合物。
【請求項13】
図9または10に図示するXRPDによって特徴付けられる、請求項11に記載の化合物。
【請求項14】
L−アスパラギン酸付加塩(1:1)またはL−アスパラギン酸付加塩水和物(1:1)である、請求項1に記載の化合物。
【請求項15】
グルタミン酸付加塩(1:1)またはグルタミン酸付加塩一水和物である、請求項1に記載の化合物。
【請求項16】
治療に使用するための請求項1〜15のいずれかに記載の化合物。
【請求項17】
請求項1〜15のいずれかに記載の化合物を薬学的に許容できる賦形剤と共に含む、医薬組成物。
【請求項18】
慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁から選択される疾患を処置する方法であって、その必要がある患者に治療有効量の請求項1〜15のいずれかに記載の化合物を投与することを含む、方法。
【請求項19】
前記疾患が慢性疼痛である、請求項18に記載の方法。
【請求項20】
前記慢性疼痛が幻肢痛、神経因性疼痛、糖尿病性神経障害、帯状疱疹後神経痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群(CPRS)、三叉神経痛/三叉神経神経痛/疼痛性チック、外科的介入(例えば術後鎮痛薬)、糖尿病性血管障害、膵島炎に関連する毛細血管抵抗または糖尿病症状、アンギナに関連する疼痛、月経に関連する疼痛、がんに関連する疼痛、歯痛、頭痛、片頭痛、緊張型頭痛、三叉神経痛、顎関節症候群、筋筋膜性疼痛筋傷害、線維筋痛症候群、骨関節痛(変形性関節症)、慢性関節リウマチ、熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫、変形性関節炎、骨粗鬆症、骨転移または未知の理由による捻挫または骨折骨痛、痛風、結合組織炎、筋筋膜性疼痛、胸郭出口症候群、上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄傷害(SCI)関連疼痛、中枢性卒中後痛、がん性ニューロパシー、AIDS疼痛、鎌状赤血球疼痛および老人性疼痛から選択される、請求項19に記載の方法。
【請求項21】
前記慢性疼痛が神経因性疼痛である、請求項20に記載の方法。
【請求項22】
前記神経因性疼痛がヒペルパシー、痛覚過敏、ニューロパシー、糖尿病性神経障害、神経炎、神経痛、知覚過敏、カウザルギー、および異痛から選択される、請求項21に記載の方法。
【請求項23】
慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁から選択される疾患を処置するための医薬の製造における、請求項1〜15のいずれかに記載の化合物の使用。
【請求項24】
前記疾患が慢性疼痛である、請求項23に記載の使用。
【請求項25】
前記慢性疼痛が幻肢痛、神経因性疼痛、糖尿病性神経障害、帯状疱疹後神経痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群(CPRS)、三叉神経痛/三叉神経神経痛/疼痛性チック、外科的介入(例えば術後鎮痛薬)、糖尿病性血管障害、膵島炎に関連する毛細血管抵抗または糖尿病症状、アンギナに関連する疼痛、月経に関連する疼痛、がんに関連する疼痛、歯痛、頭痛、片頭痛、緊張型頭痛、三叉神経痛、顎関節症候群、筋筋膜性疼痛筋傷害、線維筋痛症候群、骨関節痛(変形性関節症)、慢性関節リウマチ、熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫、変形性関節炎、骨粗鬆症、骨転移または未知の理由による捻挫または骨折骨痛、痛風、結合組織炎、筋筋膜性疼痛、胸郭出口症候群、上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄傷害(SCI)関連疼痛、中枢性卒中後痛、がん性ニューロパシー、AIDS疼痛、鎌状赤血球疼痛および老人性疼痛から選択される、請求項24に記載の使用。
【請求項26】
前記慢性疼痛が神経因性疼痛である、請求項25に記載の使用。
【請求項27】
前記神経因性疼痛がヒペルパシー、痛覚過敏、ニューロパシー、糖尿病性神経障害、神経炎、神経痛、知覚過敏、カウザルギー、および異痛から選択される、請求項26に記載の使用。
【請求項28】
慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁から選択される疾患の処置において使用するための、請求項1〜15のいずれかに記載の化合物。
【請求項29】
慢性疼痛の処置において使用するための、請求項28に記載の化合物。
【請求項30】
幻肢痛、神経因性疼痛、糖尿病性神経障害、帯状疱疹後神経痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群(CPRS)、三叉神経痛/三叉神経神経痛/疼痛性チック、外科的介入(例えば術後鎮痛薬)、糖尿病性血管障害、膵島炎に関連する毛細血管抵抗もしくは糖尿病症状、アンギナに関連する疼痛、月経に関連する疼痛、がんに関連する疼痛、歯痛、頭痛、片頭痛、緊張型頭痛、三叉神経痛、顎関節症候群、筋筋膜性疼痛筋傷害、線維筋痛症候群、骨関節痛(変形性関節症)、慢性関節リウマチ、熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫、変形性関節炎、骨粗鬆症、骨転移もしくは未知の理由による捻挫もしくは骨折骨痛、痛風、結合組織炎、筋筋膜性疼痛、胸郭出口症候群、上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄傷害(SCI)関連疼痛、中枢性卒中後痛、がん性ニューロパシー、AIDS疼痛、鎌状赤血球疼痛または老人性疼痛の処置において使用するための、請求項29に記載の化合物。
【請求項31】
神経因性疼痛の処置において使用するための、請求項30に記載の化合物。
【請求項32】
前記神経因性疼痛がヒペルパシー、痛覚過敏、ニューロパシー、糖尿病性神経障害、神経炎、神経痛、知覚過敏、カウザルギー、および異痛から選択される、請求項31に記載の化合物。
【請求項1】
結晶質形態にある4−[2−(4−メチルフェニル−スルファニル)フェニル]ピペリジン
【化1】
および薬学的に許容できるその塩である化合物I(ただし前記化合物は4−[2−(4−メチルフェニルスルファニル)フェニル]−ピペリジン塩酸付加塩ではないものとする)。
【請求項2】
HBr付加塩である、請求項1に記載の化合物。
【請求項3】
XPRDにおける約6.08、14.81、19.26および25.38°2θのピークによって特徴付けられる、請求項2に記載の化合物。
【請求項4】
図1に図示するXRPDによって特徴付けられる、請求項2に記載の化合物。
【請求項5】
DL−乳酸付加塩である、請求項1に記載の化合物。
【請求項6】
XRPDにおける約5.30、8.18、9.44および17.24°2θのピークによって特徴付けられる、請求項5に記載の化合物。
【請求項7】
図4に図示するXRPDによって特徴付けられる、請求項5に記載の化合物。
【請求項8】
グルタル酸付加塩(1:1)である、請求項1に記載の化合物。
【請求項9】
XRPDにおける約9.39、11.70、14.05および14.58°2θのピークによって特徴付けられる、請求項8に記載の化合物。
【請求項10】
図8に図示するXRPDによって特徴付けられる、請求項8に記載の化合物。
【請求項11】
マロン酸付加塩(1:1)である、請求項1に記載の化合物。
【請求項12】
XRPDにおける10.77°、16.70°、19.93°および24.01°2θ、または6.08°、10.11°、18.25°および20.26°2θのピークによって特徴付けられる、請求項11に記載の化合物。
【請求項13】
図9または10に図示するXRPDによって特徴付けられる、請求項11に記載の化合物。
【請求項14】
L−アスパラギン酸付加塩(1:1)またはL−アスパラギン酸付加塩水和物(1:1)である、請求項1に記載の化合物。
【請求項15】
グルタミン酸付加塩(1:1)またはグルタミン酸付加塩一水和物である、請求項1に記載の化合物。
【請求項16】
治療に使用するための請求項1〜15のいずれかに記載の化合物。
【請求項17】
請求項1〜15のいずれかに記載の化合物を薬学的に許容できる賦形剤と共に含む、医薬組成物。
【請求項18】
慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁から選択される疾患を処置する方法であって、その必要がある患者に治療有効量の請求項1〜15のいずれかに記載の化合物を投与することを含む、方法。
【請求項19】
前記疾患が慢性疼痛である、請求項18に記載の方法。
【請求項20】
前記慢性疼痛が幻肢痛、神経因性疼痛、糖尿病性神経障害、帯状疱疹後神経痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群(CPRS)、三叉神経痛/三叉神経神経痛/疼痛性チック、外科的介入(例えば術後鎮痛薬)、糖尿病性血管障害、膵島炎に関連する毛細血管抵抗または糖尿病症状、アンギナに関連する疼痛、月経に関連する疼痛、がんに関連する疼痛、歯痛、頭痛、片頭痛、緊張型頭痛、三叉神経痛、顎関節症候群、筋筋膜性疼痛筋傷害、線維筋痛症候群、骨関節痛(変形性関節症)、慢性関節リウマチ、熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫、変形性関節炎、骨粗鬆症、骨転移または未知の理由による捻挫または骨折骨痛、痛風、結合組織炎、筋筋膜性疼痛、胸郭出口症候群、上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄傷害(SCI)関連疼痛、中枢性卒中後痛、がん性ニューロパシー、AIDS疼痛、鎌状赤血球疼痛および老人性疼痛から選択される、請求項19に記載の方法。
【請求項21】
前記慢性疼痛が神経因性疼痛である、請求項20に記載の方法。
【請求項22】
前記神経因性疼痛がヒペルパシー、痛覚過敏、ニューロパシー、糖尿病性神経障害、神経炎、神経痛、知覚過敏、カウザルギー、および異痛から選択される、請求項21に記載の方法。
【請求項23】
慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁から選択される疾患を処置するための医薬の製造における、請求項1〜15のいずれかに記載の化合物の使用。
【請求項24】
前記疾患が慢性疼痛である、請求項23に記載の使用。
【請求項25】
前記慢性疼痛が幻肢痛、神経因性疼痛、糖尿病性神経障害、帯状疱疹後神経痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群(CPRS)、三叉神経痛/三叉神経神経痛/疼痛性チック、外科的介入(例えば術後鎮痛薬)、糖尿病性血管障害、膵島炎に関連する毛細血管抵抗または糖尿病症状、アンギナに関連する疼痛、月経に関連する疼痛、がんに関連する疼痛、歯痛、頭痛、片頭痛、緊張型頭痛、三叉神経痛、顎関節症候群、筋筋膜性疼痛筋傷害、線維筋痛症候群、骨関節痛(変形性関節症)、慢性関節リウマチ、熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫、変形性関節炎、骨粗鬆症、骨転移または未知の理由による捻挫または骨折骨痛、痛風、結合組織炎、筋筋膜性疼痛、胸郭出口症候群、上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄傷害(SCI)関連疼痛、中枢性卒中後痛、がん性ニューロパシー、AIDS疼痛、鎌状赤血球疼痛および老人性疼痛から選択される、請求項24に記載の使用。
【請求項26】
前記慢性疼痛が神経因性疼痛である、請求項25に記載の使用。
【請求項27】
前記神経因性疼痛がヒペルパシー、痛覚過敏、ニューロパシー、糖尿病性神経障害、神経炎、神経痛、知覚過敏、カウザルギー、および異痛から選択される、請求項26に記載の使用。
【請求項28】
慢性疼痛、部分応答者のうつ病、治療抵抗性うつ病、アルツハイマー病、認知障害、ADHD、メランコリー、PTSD、顔面紅潮、睡眠時無呼吸、アルコール、ニコチンもしくは炭水化物渇望、物質乱用、アルコールもしくは薬物乱用、嘔吐、摂食障害、IBS、情動障害、うつ病、大うつ病性障害、産後うつ病、双極性障害、アルツハイマー病、精神病もしくはパーキンソン病に関連するうつ病、不安、全般性不安障害、社会不安障害、強迫性障害、パニック障害、パニック発作、恐怖症、社会恐怖症、広場恐怖症または腹圧性尿失禁から選択される疾患の処置において使用するための、請求項1〜15のいずれかに記載の化合物。
【請求項29】
慢性疼痛の処置において使用するための、請求項28に記載の化合物。
【請求項30】
幻肢痛、神経因性疼痛、糖尿病性神経障害、帯状疱疹後神経痛(PHN)、手根管症候群(CTS)、HIVニューロパシー、複合性局所疼痛症候群(CPRS)、三叉神経痛/三叉神経神経痛/疼痛性チック、外科的介入(例えば術後鎮痛薬)、糖尿病性血管障害、膵島炎に関連する毛細血管抵抗もしくは糖尿病症状、アンギナに関連する疼痛、月経に関連する疼痛、がんに関連する疼痛、歯痛、頭痛、片頭痛、緊張型頭痛、三叉神経痛、顎関節症候群、筋筋膜性疼痛筋傷害、線維筋痛症候群、骨関節痛(変形性関節症)、慢性関節リウマチ、熱傷に関連する外傷に起因する慢性関節リウマチおよび浮腫、変形性関節炎、骨粗鬆症、骨転移もしくは未知の理由による捻挫もしくは骨折骨痛、痛風、結合組織炎、筋筋膜性疼痛、胸郭出口症候群、上背部痛または下背部痛(背痛が全身性、局所性、または原発性脊椎疾患(神経根障害)に起因するもの)、骨盤痛、心臓性胸痛、非心臓性胸痛、脊髄傷害(SCI)関連疼痛、中枢性卒中後痛、がん性ニューロパシー、AIDS疼痛、鎌状赤血球疼痛または老人性疼痛の処置において使用するための、請求項29に記載の化合物。
【請求項31】
神経因性疼痛の処置において使用するための、請求項30に記載の化合物。
【請求項32】
前記神経因性疼痛がヒペルパシー、痛覚過敏、ニューロパシー、糖尿病性神経障害、神経炎、神経痛、知覚過敏、カウザルギー、および異痛から選択される、請求項31に記載の化合物。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】

【図13】
【図14】
【図15】

【図16】
【図17】
【図18】

【図19】

【図20】
【図21】

【図22】
【図23】
【図24】
【図25a】

【図25b】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25a】
【図25b】
【公表番号】特表2009−539890(P2009−539890A)
【公表日】平成21年11月19日(2009.11.19)
【国際特許分類】
【出願番号】特願2009−514643(P2009−514643)
【出願日】平成19年6月15日(2007.6.15)
【国際出願番号】PCT/DK2007/050076
【国際公開番号】WO2007/144006
【国際公開日】平成19年12月21日(2007.12.21)
【出願人】(591143065)ハー・ルンドベック・アクチエゼルスカベット (129)
【Fターム(参考)】
【公表日】平成21年11月19日(2009.11.19)
【国際特許分類】
【出願日】平成19年6月15日(2007.6.15)
【国際出願番号】PCT/DK2007/050076
【国際公開番号】WO2007/144006
【国際公開日】平成19年12月21日(2007.12.21)
【出願人】(591143065)ハー・ルンドベック・アクチエゼルスカベット (129)
【Fターム(参考)】
[ Back to top ]