
神経変性疾患の予防及び/又は治療
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物を含み、神経変性疾患の予防及び/又は治療に用いられる薬剤組成物。本薬剤組成物は、特に好ましくは、アルツハイマー病の予防及び/又は治療に用いることができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、神経変性疾患の予防及び/又は治療に関する。
【背景技術】
【0002】
加齢に関連する病気の発生率は、平均余命が延びるとともに増加している。アルツハイマー病(AD)は、命にかかわり、進行性かつ不可逆性の神経変性疾患であって、記憶喪失、意識障害、判断力低下、人格変化、見当識失調、言語能力失調の症状を発症する神経変性疾患である。
【0003】
ADは、神経原線維変化(NFT)及び神経毒性を有するβ−アミロイド(Aβ)の脳沈着により特徴付けられる。ADにおけるβ−セクレターゼ(BACE−1:アミロイド前駆体タンパク質をβ切断するエンザイム−1、β−セクレターゼ−1)によるアミロイド前駆体タンパク質(APP)の分裂(切断)は、アミロイド斑の主成分であるアミロイドβペプチドを生成する初期の律速段階である。α、β及びγセクレターゼによるAPPの切断により様々なペプチドを生成し、当該ペプチドのうちβ及びγセクレターゼによる逐次反応により生成されるAβ1-40及びAβ1-42は、アミロイド生成性であり、神経毒性を有する。APPの幾つかの病理学的変異体においてはBACE−1による切断に対するAPPの感受性が大きくなることが見出されており、従って、BACE−1活性を制御することが、製薬における重要なターゲットとなっている。
【0004】
硫酸化ヘパラン(HS)はアミロイド斑の構成成分として確認され、これによるアミロイドタンパク質、ペプチド及び原線維に対する反応性、アグリゲーションの促進化及び線維の安定化は、これまでの研究により裏付けられている。可溶性のヘパリンやヘパリン様物質(ヘプリン類縁体)もまた、インビトロ及びインビボのいずれにおいても当該プロセスを抑制することが示されている。
【0005】
最近、HSがBACE−1によるAPPの切断を直接的に制御し得ることが発見されたことにより、HSの新たな機能が見出された。牛肺ヘパリン(BLH)、豚粘膜のHS及びそれらの誘導体は、恐らくは酵素活性領域へのアクセスをブロックする作用から、α又はγ−セクレターゼによるAPP切断プロセスを妨害することなくBACE−1活性を抑制することが判明した(非特許文献1参照)。
【0006】
HSやHSの高度に硫酸化された構造を有するヘパリン様物質は、N−アセチル又はN−スルホ−α−D−グルコサミンのいずれかと結合したα−L−イズロン酸又はβ−D−グルクロン酸による1,4結合されたジサッカライド(二糖)繰返し単位から成るグリコサミノグリカンである。O−硫酸化の主位置は、イズロン酸のC−2位やグルコサミンのC−6位であり、さらに、まれにグルコサミンのC−3位である。生合成における様々な置換が、かなりの多様性をもたらすことになる。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Scholefield,Z.et al、Journal of Cell Biology 2003,163,97−107
【発明の概要】
【発明が解決しようとする課題】
【0008】
ヘパリンは、HSよりも高度な硫酸化や均質性を示し、凝固因子XaとIIaを示すアンチトロンビンIIIとセリンプロテアーゼとの間の不可逆な複合体の生成速度を大きくすることにより抗凝固作用を発揮し得るものとして広く使用される薬剤である。ヘパリンは、HS様物質として広範囲に利用され、しばしばHSが関与する多くの生物学的プロセスにおける優れた活性剤とされてきた。しかし、ヘパリンの誘導体が新薬として使用されるように開発された場合に、ヘパリンの抗凝固作用が弱まることは重大な問題である。
【0009】
HSや他のグリコサミノグリカンとアミロイド生成経路との相互作用に関するこれまでの研究は、それらを生成する酵素よりも、ヘパリンや高度に硫酸化された他の化合物とアミロイドタンパク質やペプチドとの相互作用にこれまで向けられてきた。しかし、この点においてHS活性の構造的要件に関する研究は、比較的少ない
BACE−1活性は、Aβペプチドの生成やADの進行に対する基礎的な重要性をもつが、現在のところ、BACE−1をターゲットとする効果的な療法はない。BACE−1に対する治療試薬としての非修飾ヘパリンの使用は、多くの副作用を疑いなく引き起こし、特に内出血の発症や血液凝固能作用の低下のリスクを増大させ得る。このことは、効果的な服用量に重大な制限を与えるため、標準的なヘパリン糖類の臨床使用の障害となる。
【0010】
平均余命が延びるとともに、加齢に関連する神経変性疾患の疾患、例えばAD、に対する効果的で新規な治療法の必要性が益々高まってきている。
本発明の目的は、神経変性疾患の予防及び/又は治療に用いられる化合物を提供することである。
【課題を解決するための手段】
【0011】
本発明の第1の観点によれば、1以上(1個以上)のジサッカライド(二糖)単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物であって、神経変性疾患の予防及び/又は治療に用いられる化合物が提供される。
【0012】
また、本発明は、BACE−1に対する強い抑制活性を維持しつつ、HSと比較して明確に低減された抗凝固活性を示すHS様物質(HS類縁体)とみなされる化合物を提供する。
【0013】
当該化合物におけるウロン酸部位の2−O原子の少なくとも約60%が水素原子に置換されていることが望ましい。より好ましくは、当該化合物におけるウロン酸部位の2−O原子の75%以上が水素原子に置換されていることが望ましい。さらに好ましくは、当該化合物におけるウロン酸部位の2−O原子のさらに大部分(85%から95%)が水素原子に置換されていることが望ましい。最も好ましくは、当該化合物におけるウロン酸部位の2−O原子の全て、すなわち、100%、が水素原子に置換されていることが望ましい。
【0014】
本発明の化合物に存在する各々のグルコサミン部位の6−O原子に関しては、単一又は複数の6−O原子が実質的に硫酸化される限りは、その望ましい硫酸化レベルは特に限定されないが、実質的に少なくとも約60%の硫酸化レベル(硫酸化率)で硫酸化されていることが望ましく、さらに好ましくは、少なくとも約75%の硫酸化レベルで硫酸化されていることが望ましい。さらに好ましくは、グルコサミンの6−O原子の少なくとも約85%から約95%が硫酸化されていることが望ましく、最も好ましくは、グルコサミン部位の6−O原子の全てが硫酸化され、グルコサミン部位の6−O原子の硫酸化レベルが100%に達していることが望ましい。
【0015】
ウロン酸の2−O原子に対する水素置換レベル(水素置換率)及び/又はグルコサミンの6−O原子の硫酸化レベルが100%より小さい場合には、複数のジサッカライドから成る当該化合物が、ウロン酸の2−O原子及び/又はグルコサミンの6−O原子の置換に関して異なるパターンを有する隣接するジサッカライドから成ることになる。
【0016】
例えば、本発明による化合物は、4つのジサッカライドを含み、75%のウロン酸の2−O原子の水素置換レベルを有することができ、すなわち、4つのジサッカライドのうち3つは、2−O原子が水素原子を有するウロン酸部位を含有することができる。この場合において、水素置換された2−Oウロン酸を含む3つのジサッカライドは、水素置換されたヘキササッカライド(6糖)の一端又は他端にて水素置換されない2−Oウロン酸原子を含むヘキササッカライドにより相互に共有結合される。別の態様として、水素置換されない2−Oウロン酸原子を含むジサッカライドは、水素置換された2−Oウロン酸原子を含む3つのジサッカライドのうちいずれか2つの間に提供されることもできる。
【0017】
さらなる一例としては、6−Oグルコサミン原子の60%が硫酸化されたイコササッカライド(20糖)から成る本発明の化合物は、グルコサミンの各々のO−6位が硫酸化された6つのジサッカライドを含む。当該6つの6−O硫酸化されたジサッカライドは、望ましい直鎖配列にて残りの4つの6−O硫酸化されないジサッカライド(すなわち、グルコサミン部位のO−6位にて硫酸化されないジサッカライド)と結合されることができ、例えば、6−O硫酸化されたジサッカライドが、6−O硫酸化されないオクタサッカライド(8糖)と相互結合して6−O硫酸化されたドデカサッカライド(12糖)を形成したり、また、6−O硫酸化されたジサッカライドの3つが、相互結合して、最初に一端を6−O硫酸化されないジサッカライドで結合されて、以降6−O硫酸化されたジサッカライドと6−O硫酸化されないジサッカライドとの繰返し配列により6−O硫酸化されたヘキササッカライド(6糖)を提供する。
【0018】
上述したScholefieldの研究では、最も活性なBACE−1インヒビターは、O−2位及びO−6位にて高度に硫酸化された牛肺から生成されるN−アセチル化されたヘパリンであると結論している。(上記Scholefield,Z.らに関して)O−2位又はO−6位における硫酸基の脱離は、化合物のBACE−1活性をかなりの程度減少させ、1又は複数の硫酸基(N−硫酸基のみ以外)の脱離は、当該化合物のBACE−1に対する抑制力に有害な影響をもたらすことを示唆している。
【0019】
さらに、当該研究では、BLHがHSよりもBACE−1活性のインヒビターとして優れていると結論付けている。HSがジサッカライドあたり約1.5の硫酸基を含むのに対して、ヘパリンがジサッカライドあたり約2.6〜約2.9の硫酸基を含むことから、ヘパリンに比べてHSの活性が低いことは、少なくとも部分的には、タンパク結合を還元するものと考えられる選択的脱硫酸化に関与する電荷の減少が原因として挙げられる。硫酸化の度合いとHSに対するヘパリンの活性度レベルとの関連は、高度に硫酸化されたHSプロテオグリカンが低度に硫酸化されたHSプロテオグリカンより線維芽細胞増殖因子と強固に結合していることを見出した他の研究により示されている(Kreuger,J.,P.Jemthら、J Biochem2005,389(Pt1),145-150)。Scholefieldの研究で述べられた結論に寄与するさらなる要因としては、2−O硫酸基の脱硫酸化により、ヘパリンが異なるコンホメーションのイズロン酸環そして恐らくはグリコシド結合を呈することが挙げられる。
【0020】
このように、Scholefield,Z.らやKreuger,J.らの研究は、一般的に脱硫酸化が活性を低下させることを示している。本発明の基礎を形成する化合物は、ウロン酸部位の2−O位が実質的に脱硫酸化され、そして、グルコサミン部位のN−2位が完全に脱硫酸化されているものであり、従って、2−O硫酸基やN−硫酸基を含有する対応化合物に比べて当該活性の低下を示すことが想定される。しかし、驚くべきことに、本発明の化合物について実施された実験の結果(詳細は後述)は、実質的には全体的に硫酸化レベルを低下させているにも関わらず、2−O硫酸基及びN−硫酸基の両方を除去することにより、予想外に高いBACE−1抑制活性を有する化合物が得られることを示した。
【0021】
また、本発明の第2の観点によれば、1以上(1個以上)のジサッカライド(二糖)単位から成り、当該ジサッカライド単位は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が硫酸基と置換され、グルコサミン部位の2−N原子が硫酸基以外の原子又は置換基と置換された化合物の使用であって、神経変性疾患の予防及び/又は治療に用いられる医薬の調製(製造)における使用が提供される。
【0022】
また、本発明の第3の観点によれば、1以上(1個以上)のジサッカライド単位から成り、当該ジサッカライド単位は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物を患者に有効量投与することを含む神経変性疾患を予防及び/又は治療する方法が提供される。
また、本発明の第4の観点によれば、本願にて定義され、本発明の様々な側面にて利用される化合物は、合成原料又は天然原料から生成される。
【0023】
当該化合物は、全体又は一部分が合成される。当該化合物は、天然の糖類、例えば、HS様物質である豚腸粘膜ヘパリン(PIMH)や牛肺ヘパリン(BLH)から化学修飾により生成可能である。PIMHは、後述する実施例にて例示する。
【0024】
また、本発明の上記第4の観点による化合物の生成には、解重合(脱重合)工程を含むことが好ましい。当該解重合工程は、亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、フリーラジカル(遊離基)処理及びこれらの組み合わせにより成る群の中から選択されることが好ましい。
【0025】
また、本発明の第5の観点によれば、本願にて定義された本発明の様々な側面の一部を形成する化合物の生産方法であって、1又は複数の合成原料又は1又は複数の天然原料からの生産方法が提供される。
【0026】
当該生産方法は、解重合工程を含むことが好ましい。当該解重合工程は、亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、フリーラジカル処理及びこれらの組み合わせから成る群の中から選択されることが好ましい。
当該解重合工程を選択することにより、詳細は後述するが、少なくとも部分的には、本化合物の1以上(1つ以上)の末端基構造に影響を与えることができる。
【0027】
本発明による化合物の基礎部分であるジサッカライド繰返し単位において、ウロン酸部位(好ましくは、ウロン酸残基がグルコサミン残基に結合することにより得られたもの)は、所望のエピマー形態をとることができる。当該ウロン酸部位は、α−L−イズロン酸部位(以下の式(I)、(II)及び(III)においては、便宜上、これのみを例示している)、β−D−グルクロン酸部位及びα−L−ガラクツロン酸部位から成る群の中から選択されることができる。
【0028】
6−Oウロン酸原子、3−Oウロン酸原子及び3−Oグルコサミン原子に結合した置換基の各々は、それぞれが独立して、水素原子、O−エーテル糖環置換基を形成する置換又は非置換のアルキル基、置換又は非置換のアルコキシ基、置換又は非置換のアリール基、O−エステル糖環置換基を形成する置換又は非置換のアシル基、置換又は非置換のアミド基(例えば、フタルアミド基)、硫酸基及びリン酸基から成る群の中から選択される。さらに、6−Cウロン酸のカルボキシル基を化学修飾して、6−Cウロン酸のアルコール基やエステル基を生成するようにしてもよい。
【0029】
6−Oウロン酸原子、3−Oウロン酸原子、3−Oグルコサミン原子、環状炭素原子のうちいずれか1つに結合したアルキル基は、直鎖構造又は分岐構造であり、好ましくは、C1-6アルキル基(炭素数1〜6のアルキル基)であり、1以上(1個以上)の原子や置換基、例えば、ハロゲン原子(例えばフッ素、塩素又は臭素)やアリール基、アシル基、アミド基(例えばフタルアミド基)又はリン酸基により置換されていてもよい。
【0030】
6−Oウロン酸原子、3−Oウロン酸原子、3−Oグルコサミン原子、環状炭素原子のうちいずれか1つに結合した置換又は非置換のアシル基は、直鎖構造(例えばペンタノニル基)又は分岐構造(例えばピバロイル基)であり、C1-6の置換又は非置換のアシル基であることが好ましい。当該アシル基は、アリールアシル基、例えばベンゾイル基とすることができる。また、当該アシル基は、1以上(1個以上)のハロゲン原子、特にフッ素原子、塩素原子又は臭素原子で置換されることができる。従って、好ましいアシル基は、モノ−、ジ−及びトリ−フルオロアセチル基である。当該アシル基は、さらに好ましくは、フタロイル基である。当該アシル基は、置換又は非置換のアセチル基、置換又は非置換のプロプリオニル基及び置換又は非置換のブタノイル基から成る群から選択されることが好ましい。当該アシル基は、最も好ましくは、非置換のアセチル基である。
【0031】
ウロン酸部位やグルコサミン部位の環状炭素原子における置換パターンは、望ましい特性、例えば本発明のジサッカライド化合物における疎水性の度合い、を与えるように選択される。ウロン酸部位及び/又はグルコサミン部位の2−、3−又は6−炭素原子の各々は、水素原子、置換又は非置換のアルキル基(好ましくはメチル基やエチル基)、エーテル基を形成する置換又は非置換のアルコキシ基、置換又は非置換のアリール基(例えばベンジル基)、置換又は非置換のアシル基(例えばアセチル基)、エステル基を形成する置換又は非置換のカルボキシル基、置換又は非置換のアミド基(例えばフタルアミド基)、硫酸基又はリン酸基により成る群の中から選択された置換基により置換されることができる。
【0032】
本発明の化合物は、多くの解重合工程によりポリサッカライド(多糖類)から得ることができる。本発明の化合物の末端基は、利用した製造法の特性に応じて広い多様な形態を備えることができる。さらに、当該化合物は、任意の数のモノサッカライド(単糖)を含むことができ、当該化合物におけるモノサッカライドの総数は、偶数個でもよいし、奇数個でもよい。
【0033】
解重合方法としては、以下に限定されないが一例として、亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、又はフリーラジカル処理のうちのいずれか又は組み合わせを含むことができる。
【0034】
非還元末端モノサッカライドは、グルコサミン残基、又はその誘導体、又はウロン酸部位(例えば、α−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位)又は誘導体、若しくは断片(フラグメント)、例えば、Δ(デルタ)4−5不飽和環(すなわち、環内の炭素原子4と炭素原子5の間のC−C二重結合)を含有するα−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位とすることができる。そのような不飽和化は、例えば、細菌リアーゼ酵素処理又は化学的ベータ脱離(ヘパリン/HSを断片化するのに一般的に用いられる)による分解により、本化合物を形成するポリサッカライドの断片ができるときに生じ得る。
【0035】
還元末端モノサッカライドは、ウロン酸部位(例えば、α−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位)、又はその誘導体、グルコサミン部位、又はその誘導体、若しくはその断片(フラグメント)、2,5-アンヒドロマンノース、2,5-アンヒドロマンニトール、1,6アンヒドロ(二環)環構造、又はマンノサミン残基とすることができる。当該化合物の生成には、亜硝酸分解を含むことができ、この場合、還元末端モノサッカライドは、2,5-アンヒドロマンニトールであることが多く、これは、通常、化学還元されて2,5-アンヒドロマンニトールになる。化学的ベータ脱離法を用いて本化合物を製造する場合、還元末端の幾つかは、1,6アンヒドロ(二環)環構造(一般的に6−O硫酸化されたグルコサミン残基から誘導される)としても存在する。さらに、化学的ベータ脱離法は、グルコサミン残基のエピマー化を引き起こしてマンノサミン残基を形成することもできる。
【0036】
上記のように定義された本発明の好ましい具体例としては、以下の式(I)に示される化合物が挙げられる。なお、式(I)は、便宜上の理由のみにより、ウロン酸部位がα−L−イズロン酸部位により示されているが、式(I)は、当該ウロン酸部位がα−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位である化合物にも及ぶと解すべきである。
【0037】
【化1】

【0038】
式(I)において、実質的に全てのR1は水素原子であり、実質的に全てのR2が硫酸基であり、実質的に全てのR3が硫酸基以外の原子又は置換基であり、nは1以上の整数であり、R4、R5及びR6は水素原子、置換又は非置換のアルキル基、置換又は非置換のアルコキシ基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、硫酸基又はリン酸基により成る群の中からそれぞれ独立して個々に選択され、XとYは水素、末端モノサッカライド(単糖)基、末端ジサッカライド(二糖)基及び/又はそれらのフラグメント(断片)若しくはそれらの誘導体より成る群の中からそれぞれ独立して選択される。
【0039】
単なる例示であるが、本発明の化合物が式(I)のサッカライド単位のみから成る場合、XとYの一方が水素であり、XとYの他方がモノサッカライドであるときには、当該化合物は全体として奇数個のモノサッカライド単位から構成され、これに対し、XとYが同じとき(例えばXとYが共に水素、モノサッカライド、又はジサッカライド)には偶数個のモノサッカライド単位から成ると解される。さらに、当該化合物は、XとYの一方がモノサッカライドであり、XとYの他方がジサッカライドの場合には、当該化合物は奇数個のモノサッカライド単位から成るものである。従って、式(I)、並びに後記の式(II)及び式(III)のいずれも、奇数個のモノサッカライド単位を含む当該化合物と偶数個のモノサッカライド単位を含む当該化合物の両方に及ぶよう意図されている。
【0040】
Xが、末端モノサッカライド基である場合には、Xはグルコサミン部位、又はその誘導体、若しくは断片(フラグメント)であることが好ましい。Xは、式(I)のグルコサミン部位と同じ構造を呈してもよい(この場合、R2、R3及びR4は上記で定義された通りである)。
【0041】
Xが末端ジサッカライド基である場合には、Xは、カッコで示されたジサッカライド繰返し構造を有して、非還元末端モノサッカライドが、式(I)のウロン酸部位、すなわち、α−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位、と同じ一般的構造(ここで、R1、R4及びR5は上記で定義した通りである)を有することが好ましい。ジサッカライド単位Xは、カッコで示すジサッカライド単位の一部を形成するモノサッカライドの一方又は両方の誘導体を含んでいてもよい。
【0042】
非還元末端モノサッカライトは、Δ4−5不飽和環(すなわち、環内の炭素原子4と炭素原子5の間のC−C二重結合)を有するα−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位とすることができる。そのような不飽和化は、例えば、本化合物を形成するポリサッカライドフラグメントが細菌リアーゼ酵素処理又は化学的ベータ脱離(ヘパリン/HSを断片化するのに一般的に使用される)による分解により生じるときに起こる。
【0043】
Yが末端モノサッカライドである場合には、Yは、ウロン酸部位、又はその誘導体、若しくはその断片(フラグメント)であることが好ましい。Yは、式(I)のウロン酸部位、すなわち、α−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位、と同じ一般的構造(ここで、R1、R4及びR5は上記で定義された通りである)を有することが好ましい。
【0044】
Yが末端ジサッカライド基である場合には、Yは、式(I)のカッコで示されたジサッカライド繰返し構造を有して、還元末端モノサッカライドが、式(I)のグルコサミン部位と同じ一般的構造(ここで、R2、R3及びR4は上記で定義した通りである)を有することが好ましい。ジサッカライド単位であるYは、カッコで示されたジサッカライド繰返し単位の一部を形成する1又は2つのモノサッカライドからの誘導体を含むこともできる。還元末端モノサッカライドは、2,5-アンヒドロマンノース、2,5-アンヒドロマンニトール、1,6アンヒドロ(二環)環構造、又はマンノサミン残基とすることができる。本化合物の生成には、亜硝酸分解を含むことができ、この場合、還元末端モノサッカライドは、2,5-アンヒドロマンニトールであることが多く、これは、通常、化学還元されて2,5-アンヒドロマンニトールになる。化学的ベータ脱離法を用いて本化合物を製造する場合、還元末端の幾つかは、1,6アンヒドロ(二環)環構造(一般的に6−O硫酸化されたグルコサミン残基から誘導される)としても存在する。さらに、化学的ベータ脱離法は、グルコサミン残基のエピマー化を引き起こしてマンノサミン残基を形成することもできる。
【0045】
式(I)の化合物の好ましい実施形態においては、R5及びR6がともに水素原子であり、以下の式(II)にて示された構造を有することができ、この構造においては、上記の式(I)にて定義されたR1、R2、R3、n、X及びYが用いられ、また、ウロン酸部位は、便宜上、α−L−イズロン酸部位のみにより示されているが、α−L−イズロン酸部位、β−D−グルクロン酸部位、又はα−L−ガラクツロン酸部位である場合も含む。
【0046】
【化2】
【0047】
グルコサミン部位の2−N原子(上記の式(I)及び式(II)におけるR3)は、2−N置換基が硫酸基(SO3-)以外であるという条件のもとで、望ましい有機系又は無機系の置換基により置換できる。2−Nグルコサミン原子は、水素原子、置換又は非置換のアルキル基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、及びリン酸基により成る群の中から選択された置換基により置換されることができる。
【0048】
2−Nアルキル基は、直鎖構造、分岐構造、又は環構造とすることができ、好ましくは、C1-6アルキル基であり、1以上の原子や置換基、例えば、ハロゲン原子(例えばフッ素、塩素又は臭素)やアリール基、アシル基、アミド基(例えばフタルアミド基)又はリン酸基により置換されていてもよい。
【0049】
2−Nグルコサミン原子に直接結合したアミド基及び/又は2−Nグルコサミン原子に結合したアルキル基と結合したアミド基は、任意の都合のよい形態をとることができ、例えば、メチルアミド基、エチルアミド基又はフタルアミド基となる。
【0050】
2−Nグルコサミン原子に直接結合した置換又は非置換のアシル基及び/又は2−Nグルコサミン原子に結合したアルキル基と結合したアシル基は、直鎖構造(例えばペンタノニル基)又は分岐構造(例えばピバロイル基)であり、C1-6の置換又は非置換のアシル基であることが好ましい。当該アシル基は、アリールアシル基、例えばベンゾイル基とすることができる。また、当該アシル基は、1以上(1個以上)のハロゲン原子、特にフッ素原子、塩素原子又は臭素原子で置換されることができる。従って、好ましいアシル基は、モノ−、ジ−及びトリ−フルオロアセチル基である。当該アシル基は、さらに好ましくは、フタロイル基である。2−Nグルコサミン原子は、置換又は非置換のアセチル基、置換又は非置換のプロプリオニル基及び置換又は非置換のブタノイル基から成る群から選択されるアシル基により置換されることが好ましい。当該アシル基は、最も好ましくは、以下の式(III)に示すような非置換のアセチル基である。
【0051】
R基及び上記式(I)にて定義されたR基群及びnに関して、本発明の特に好ましい化合物の実施態様においては、全てのR1が水素原子であり、全てのR2が硫酸基であり、R3がアセチル基であり、R4、R5及びR6が全て水素原子である。この好適な実施態様は、以下の式(III)にて示され、当該式においてX及びYは上記式(I)で定義された通りであり、ウロン酸部位はα−L−イズロン酸部位(式中、便宜上これのみが示されているが)、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位とすることができる。
【0052】
【化3】
【0053】
本発明の化合物は、任意の適切な長さを有することができ、そして、異なる構造(すなわち、異なる置換パターンを有する構造)のジサッカライドが望ましい直鎖配列にて結合されているようにすることができる。さらに、本発明の化合物は、天然のオリゴ糖やポリサッカライド(多糖類)やその断片(フラグメント)による適切な化学修飾により生産されることができる。
【0054】
本発明の化合物は、奇数個又は偶数個のモノサッカライド単位から成ることができる。本化合物がジサッカライド繰返し単位のみから成る場合には、モノサッカライド単位の総数は偶数個となるが、本化合物がジサッカライド繰返し単位を含み、且つ当該分子のいずれか一端に単一の末端サッカライドを含むような場合には、奇数個のモノサッカライドから成る化合物となる。
【0055】
例として、上記式(I)に関して、本化合物は、後述の3つの好ましい構造(式(IV)、式(V)及び式(VI))のうちの1つにより示され、ここで、全てのR基やnは上記式(I)にて定義されたとおりであり、ウロン酸部位は、便宜上α−L−イズロン酸部位のみが示されているが、α−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位のいずれでもよい。
式(IV)において、Xは、カッコで示されたジサッカライド繰返し構造に含まれると同じ一般的構造を有する末端グルコサミン部位である。
【0056】
【化4】
式(V)において、Yは、カッコで示されたジサッカライド繰返し構造に含まれると同じ一般的構造を有する末端ウロン酸部位である。
【0057】
【化5】
式(VI)において、Xは、カッコで示されたジサッカライド繰返し構造に含まれると同じ一般的構造を有する末端グルコサミン部位であり、Yは、カッコで示されたジサッカライド繰返し構造に含まれると同じ一般的構造を有する末端ウロン酸部位である。
【0058】
【化6】
【0059】
Scholefieldの研究では、BLHの10以上のモノサッカライドから成るオリゴ糖は、BACE−1活性を抑制するが、当サイズ未満のオリゴ糖では、有意の活性は見出されなかったと結論付けている。(上記Scholefield,Z.らに関して)しかし、予想外なことに、10未満のモノサッカライドから成るオリゴ糖を含む本発明の化合物は、BACE−1活性を抑制することが示された。本発明の化合物は、4から24までの(4〜24個の)モノサッカライド単位(例えば、上記式(I)から式(III)においてn=2から12、X=Y=H)から成ることが好ましい。本化合物は、より少ない個数のモノサッカライドから成ることが好ましい。なぜなら、より小さい化合物のほうが容易に血液脳関門を通過することができるためである。本化合物は、さらに好ましくは、6から20までの(6〜20個の)モノサッカライド単位(好ましくは、n=3から10、X=Y=H、さらに好ましくは、n=2から9、X=ジサッカライド、Y=H)、さらに好ましくは、6から18までの(6〜18個の)モノサッカライド単位(好ましくは、n=3から9、X=Y=H、さらに好ましくは、n=2から7、X=モノサッカライド、Y=モノサッカライド)から成るものである。本発明の化合物のサイズ範囲としては、6から16までの(6〜16個の)モノサッカライド単位であることが好ましく、これは約1500ダルトン(Dalton)から約5000ダルトンまでの化合物の分子質量範囲に相当する。
【発明の効果】
【0060】
神経変性疾患、例えば、老人性痴呆症、初老期痴呆症、多発梗塞性痴呆及び他の神経変性の疾患や障害の予防及び/又は治療のために、医学及び獣医学のともに役立つ薬品として使用される化合物が待ち望まれている。本発明による化合物は、アルツハイマー病の予防及び/又は治療のための利用に大いに適する。
【0061】
本発明の第1の観点による化合物が、本発明の第2の観点と合わせて、神経変性疾患、特にアルツハイマー病の予防及び/又は治療のための医薬の調剤としての利用に大いに適することは、当業者にとって明らかである。
【0062】
本発明の第3の観点の神経変性疾患の予防法及び/又は治療法は、本発明の第1の観点による化合物を利用することが望ましい。
【0063】
また、本発明の第6の観点によれば、1以上(1つ以上)のジサッカライド単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、実質的に、ウロン酸部位の2−O原子が水素原子と置換され、グルコサミン部位の6−O原子が硫酸基と置換され、グルコサミン部位の2−N原子が硫酸基以外の原子又は置換基と置換された化合物から成る薬剤組成物であって、神経変性疾患の予防及び/又は治療に用いられる薬剤組成物が提供される。
【0064】
本化合物は、本発明の第6の観点から成る薬剤組成物として、薬学的に許容される基材、希釈剤、又は添加剤と組み合せて利用されることが好ましい。
【0065】
また、本発明の第7の観点によれば、1以上(1個以上)のジサッカライド単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、実質的に、ウロン酸部位の2−O原子が水素原子と置換され、グルコサミン部位の6−O原子が硫酸基と置換される化合物であって、グルコサミン部位の2−N原子が硫酸基以外の原子又は置換基に置換された化合物が提供される。
【0066】
下記の実施例の分析結果から、本発明の化合物は、BACE−1インヒビターとしてのIC50が約100μg/mlより小さく、さらに好ましくは、約10μg/mlより小さく、さらに好ましくは、約1μg/mlより小さく、さらにより好ましくは、約0.1μg/mlより小さく、最も好ましくは、約0.05μg/mlより小さい値を示す。
【0067】
抗凝固作用に関して、本発明の化合物は、非修飾の豚腸粘膜ヘパリン(PIMH)における抗凝固活性の約50%より小さいことが好ましい。本発明の化合物は、好ましくは、非修飾PIMHの抗凝固活性の約20%より小さく、さらに好ましくは、当該抗凝固活性の約5%より小さいことが望ましい。本発明による化合物は、特に好ましくは、当該抗凝固活性の約1%より小さく、さらに好ましくは、当該抗凝固活性の約0.5%より小さいことが望ましい。本発明による化合物は、さらに好ましくは、当該抗凝固活性の約0.1%より小さく、最も好ましくは、当該抗凝固活性の約0.03%より小さいことが望ましい。
【0068】
本発明の化合物の治療可能比率(抗BACE−1活性(以下の実施例にて記載された分析結果により決定される)と抗セリンプロテアーゼ凝固因子Xaとの比率)は、好ましくは、約100より大きく、さらに好ましくは、約500より大きく、さらに好ましくは、約1000より大きく、最も好ましくは、1000より大きい。本発明の化合物の治療可能比率は、特に好ましくは、100から2000までの範囲であり、さらに好ましくは、200から1500までの範囲であり、最も好ましくは、500から1500までの範囲である。本発明による化合物は、本発明の好適な実施例において、当該治療可能比率が約1092であることを示す。
【0069】
本発明の化合物は、特に化合物の利用方法に応じて多種の形態をとることが可能である。すなわち、本化合物は、例えば、粉薬、錠剤、カプセル、液体、軟膏、乳液、ジェル、ヒドロゲル、エアゾル、スプレー、ミセル、経皮貼布、リポソーム、又は人や動物への投与に適切な他の形態として提供されることができる。本化合物の媒体(賦形剤)は、投与された患者に対して優れた耐性があり、本化合物を脳まで運搬できるものである。
【0070】
また、本化合物は、点眼薬又は眼軟膏剤の形態により経眼投与することができ、液体形状又は固体形状により経口投与することもできる。経口投与に適する化合物としては、丸薬、カプセル、顆粒剤、錠剤及び粉薬のような固体形状や、水薬、シロップ剤、エリキシル剤及び懸濁液のような液体形状が含まれる。非経口投与として有効な形態としては、殺菌済み水薬、乳剤及び懸濁液が含まれる。
【0071】
本発明による化合物は、多くの方法により利用されることができる。例えば、当該化合物が、全身投与することが要求されることもあるが、錠剤、カプセル、又は液状の形態で経口投与された場合に要求される。あるいはまた、本化合物は、注射により血流に注入されることができる。当該注射は、静脈注射(ボーラスや浸剤)や皮下注射(ボーラスや浸剤)で行うことができる。また、本化合物は、吸入投与(例えば経鼻投与)されることもできる。
本化合物は、また、脳内、脳室内、クモ膜下腔内の搬送作用により、中枢に投与されることができる。
【0072】
本化合物は、また、緩速又は遅延型の送出装置と併用されることが出来る。当該送出装置は、例えば、患者の上皮又は皮下に挿入され、本化合物は、数週間又はさらに数ヶ月にわたり送出されることができる。当該装置は、本化合物の頻繁な通常投与を必要とする場合(例えば、患者に対して少なくとも毎日の錠剤摂取や毎日の注射による投与の場合)に、特に有用である。
【0073】
本化合物の必要摂取量は、生物学的活性やバイオアベイラビリティ(生体利用性)により決定され、そして、それらの特性は、本化合物の投与形態、本化合物の利用時の物理化学的特性及び本化合物の利用形態が単独療法であるか複合療法であるかに依存する。本化合物の投与頻度もまた、上記に述べた要因及び特に患者に投与された化合物の半減期に影響される。
【0074】
本化合物の最適な投与量は、当業者により決定されることができ、使用する化合物の種類、薬剤の効力、投与形態及び病状の進行に応じて変化する。治療を受けている特定の患者に応じた他の要因、例えば、患者の年齢、体重、性別、食事及び投与時間も考慮して、投与量を調整することが必要となる。
【0075】
製薬業にて常套的に利用される公知の方法(例えば、インビボでの実験や臨床試験など)では、調剤された混合物や構成物や管理体制の定式化を用いて、化合物や成分を特定割合に調合し正確な治療計画(例えば、本化合物の1日の投与量や投与頻度)を確立することができる。使用する化合物の種類にも依存するが、一般的には、体重の0.01μg/kgから1.0g/kgまでの範囲の日用量(1日の投与量)でADの治療に利用可能である。さらに好ましくは、本化合物の日用量は、体重の0.01mg/kgから100mg/kgまでの範囲が望ましい。
【0076】
毎日の投与は、単一投与(例えば、経口投与のための1日用の錠剤や1日1回の注射)として与えられることができる。その代わりとして、本化合物は、1日2回以上の投与を必要とされることもある。例えば、ADの患者は、錠剤にて25mgから5000mgまでの2回以上の服用により投与される。治療中の患者は、起床時に第1の服用を行い、夜に第2の服用を行う(1日2回の服用の場合)か、以降3時間又は4時間ごとに服用を行うことができる。別のやり方として、遅延化送出装置を用いて、繰返し投与の必要なく患者への本化合物の適用量を提供することもできる。
【0077】
本発明は、治療に有効な量の本発明による化合物及び好ましくは薬学的に許容される賦形剤(ベヒクル)を含む薬剤組成物を提供する。本発明における”治療に効果的な量”とは、本化合物が効果を発揮する疾患の患者に投与される場合に、当該疾患を減少、緩和、又は軽減する本化合物又はその組成物の量である。”患者”としては、脊椎動物、哺乳動物、ペット、又は人間が該当する。本発明の実施において、”薬学的に許容される賦形剤”は、薬剤組成物の形成に有用な当該技術分野における当業者に公知である生理的な賦形剤であれば種類は問わない。
【0078】
1つの実施態様としては、本発明の第6の観点に従う組成物における当該化合物の量を、約0.01mgから約800mgまでとすることができる。また、他の実施態様としては、当該化合物の量を、約0.01mgから約500mgまでとすることができる。また、他の実施態様としては、当該化合物の量を、約0.01mgから約250mgまでとすることができる。また、他の実施態様としては、当該化合物の量を、約0.1mgから約60mgまでとすることができる。さらに別の実施態様としては、当該化合物の量を、約1mgから約20mgまでとすることができる。
【0079】
1つの実施態様においては、本発明の第4の観点から成る組成物において用いられる薬学的な賦形剤は、液体状とすることができ、当該薬剤組成物は、水薬の形態とすることができる。また、他の実施例においては、当該薬学的に許容される賦形剤は、固体状とすることができ、当該薬剤組成物は、粉薬又は錠剤の形態とすることができる。さらに、他の実施例においては、当該薬学的に許容される賦形剤は、ジェル状とすることができ、当該薬剤組成物は、座薬又は乳液の形態とすることができる。さらに、他の実施態様においては、当該化合物又はその組成物は、薬学的に許容される経皮貼布の一部として形成されることができる。
【0080】
本発明の第6の観点から成る組成物において用いられる固体状の賦形剤は、香味添加剤、潤滑剤、溶解剤、懸濁剤、賦形剤、滑剤、圧縮剤、結合剤、又は口腔内崩壊錠としても作用する1以上の構成要素を、カプセル剤として含めることも可能である。粉薬においては、賦形剤は、細分化された活性成分を混合された細分化された固体状である。錠剤においては、活性成分は、適切な形状にて必要な圧縮特性を備え、望ましい形状やサイズで圧縮される賦形剤とともに混合される。粉薬及び錠剤は、最大で99%以下の活性成分を含むことが好ましい。適切な固体状の賦形剤としては、例えば、リン酸カルシウム、ステアリン酸マグネシウム、タルク、砂糖、デキストリン、でんぷん、ゼラチン、セルロース、ポリビニルピロリジン、低溶性ワックス及びイオン交換樹脂を挙げることができる。
【0081】
液体状の賦形剤は、水薬、懸濁液、乳剤、シロップ剤、エリキシル剤及び本発明の第6の観点により形成される加圧された組成物を調製するのに利用することができる。本発明の第1の観点により形成される化合物は、水、有機溶媒、若しくはその混合剤、又は薬学的に許容された油脂や脂肪のような薬学的に許容される液体状の賦形剤にて溶解又は懸濁されることができる。当該液体状の賦形剤は、溶解剤、乳化剤、緩衝液、防腐剤、甘味料、香料、懸濁化剤、増粘剤、着色料、粘性調節剤、安定剤、又は浸透圧調節のような他の適切な薬学的な添加物を含めることができる。本発明の第1の観点から成る化合物の経口投与や非経口投与に用いられる液体状の賦形剤の好適な一例としては、水(部分的に上記添加物、例えば、セルロース、誘導体、好ましくは、カルボキシメチルセルロースナトリウム溶液、を含む)、アルコール類(一価アルコールや多価アルコール、例えば、グリコール、を含む)、その誘導体及び油脂(例えば、分留されたココナッツオイルやピーナッツオイル)を挙げることができる。当該賦形剤は、また、非経口投与の場合には、オレイン酸エチル及びミリスチン酸イソプロピルのような油性エステルとすることができる。無菌の液体状の賦形剤は、非経口投与のための無菌の液体状の当該組成物において有用である。加圧された組成物に対する液体状の賦形剤には、ハロゲン化炭化水素、又は他の薬学的に許容された推進剤を用いることができる。
【0082】
本発明の第1の観点から成る化合物は、他の溶質や、懸濁化剤(例えば、溶液を等浸透圧化させる含塩物やグルコース)、胆汁酸塩、アラビアゴム、ゼラチン、モノオレイン酸ソルビタン、ポリソルベート80(ソルビトール及びその無水物がエチレンオキサイドと共重合されたオレイン酸エステル)等を含有する無菌の水薬又は懸濁液として経口投与されることができる。
【0083】
無菌の水薬又は懸濁液のような液体状で薬学的に許容される組成物は、例えば、筋肉内、クモ膜下腔内、硬膜外、腹腔内、又は皮下の注射により利用されることができる。無菌の水薬は、また静脈内に投与することもできる。本発明の化合物は、本発明の第6の観点に従い無菌の固体状の化合物として調製することもでき、当該組成物は、無菌の水、生理的食塩水、又は他の適切な無菌の注射用媒体を用いた投与時に溶解又は懸濁されることができる。賦形剤には、必要且つ不活性な結合剤、懸濁化剤、潤滑剤、香味剤、甘味剤、防腐剤、着色料、又はコーティング剤を含めることができる。
【0084】
本発明の化合物は、予防療法に利用されることに大いに適する。”予防療法”とは、ADのような神経変性疾患の影響を防止又は軽減する療法であれば特に限定されずに含まれるものとする。予防療法は、例えば、規定年齢に到達した人や神経変性疾患への遺伝的な素因を有する人に定期的に与えることができる。別のやり方として、予防療法は、神経変性疾患の発症を引き起こしやすい状況にさらされている人にケースバイケースで与えられることもできる。
【0085】
本発明は、後述する実施例を用いてさらに容易に理解されることができる。しかし、当該実施例における特定の手段や結論は、後述する請求項にてより広範に記載される本発明の一部を説明するものに過ぎないということは当業者には明らかなことであろう。
本発明の実施の態様を実施例に沿って説明するが、当該実施例は例示のものにすぎず本発明を限定するものではない。
【図面の簡単な説明】
【0086】
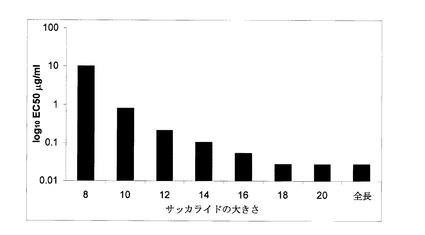
【図1】実施例1に記載されているように非修飾PIMHのフラグメントのBACE−1抑制活性をサッカライドの大きさを関数として示すグラフである。
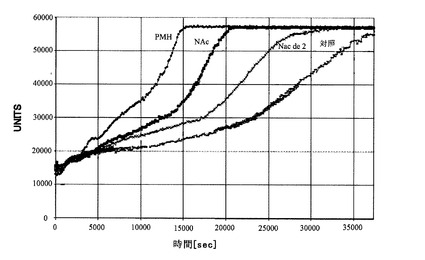
【図2】実施例2に記載されているようにA−βペプチドの凝集速度(アグリゲーション率)に対する修飾ヘパリン誘導体の効果を示すグラフである。また、化合物を添加しない場合の凝集速度を「control(対照)」として示している。
【実施例1】
【0087】
伝達性海綿状脳症(TSEs)に対する懸念が表明された後は、BLHの利用度や薬剤的使用は最近少なくなっているが、このことは本発明者に豚腸粘膜ヘパリン(PIMH)を用いてBACE−1抑制活性を調べることを促した。PIMHは、BLHよりもβ−D−グルクロン酸のレベル(量)が高いが、いずれもこれまでに広く利用されておりTSEの危険性については知られていないからである。
【0088】
一連の化学修飾されたPIMH誘導体(式(VII)、表1に示す)を、後述の付録(アペンディックス)に記載の手法で調製した。所定のジサッカライド繰返し構造に応じて、O−、N−硫酸化及びN−アセチル化のパターンを変化させて、活性の体系的検討ができるようにした。各誘導体について、BACE−1のAPP切断抑制能、抗凝固剤としての効能(抗凝固因子Xa)及び他のアスパルチルプロテアーゼ系分子に対する抑制能を評価した。
【0089】
PIMH誘導体の1つとして、化合物4(式(III)のウロン酸部位がα−L−イズロン酸部位である下記の式(VIII))は、本発明による化合物の好ましい例の1つである。
【0090】
【化7】
実施された実験結果は、表1に示され、詳細は以下に述べる。
【0091】
【化8】
【0092】
【表1】
【0093】
表1において、抗凝固活性は、PIMH(100%として)に対する割合で示される。置換パターンは、それぞれ、イズロン酸の2位、グルコサミンの6位及びグルコサミンの2位に相当するR1、R2及びR3により定義される。治療可能比率は、BACE−1に対するIC50/抗凝固活性により算出した。UAは、ウロン酸であり、α−L−イズロン酸又はβ−D−グルクロン酸のいずれかである。また、GlcNは、α−D−グルコサミンである。#化合物(9)は、イズロン酸とグルコサミン残基のともに3位にて硫酸化されている。当該IC50値は、当然のことながら、使用された分析手段の性質に依存する。
【0094】
(BACE−1抑制活性)
(1)、(2)、(3)及び(5)に該当する置換パターンのBLHにおけるBACE−1抑制特性は、BLHを出発原料としてScholefield,Z.らにより既に調べられた。(上記Scholefield,Z.らに関して)
FRETペプチド分裂分析(FRET peptide cleavage assay)を使用して、BACE−1によるインビトロのAPP分断を測定した。非修飾PIMH(1)の次に、最も効果的なBACE−1インヒビターは、N−脱硫酸化され、N−再アセチル化されたPIMH(2)である。同様のIC50値であり、活性で3番目と4番目にランクされた化合物は、2−O脱硫酸化された(3)と、これまで報告されたことのない置換パターンを備える2−O脱硫酸化/N−アセチル化されたPIMHとしての本発明の化合物(4)である。これは、N−硫酸も2−O−硫酸も、6−O硫酸化を伴う場合には、高活性を得るための絶対要件ではないことを示している。このようにして、本発明の好ましい化合物(4)は、最も活性の高いモノ−硫酸化された化合物であった。
【0095】
6−O硫酸基の除去は、最も活性の低いジ硫酸化化合物(化合物(5))を生成した。化合物(4)は、6−O脱硫酸化された(5)又は6−O脱硫酸化/N−アセチル化されたヘパリン(6)のいずれよりも活性が高い。これは、化合物(4)が化合物(5)よりもジサッカライドあたりの硫酸基が少なく、化合物(6)とジサッカライドあたりの硫酸基が同数であるという事実からみて想定外の結果である。
【0096】
最も活性が低いモノ−硫酸化化合物は、2−O硫酸基と6−O硫酸基とが共に脱硫酸化されてN−硫酸基のみが残存する化合物(7)であった。全硫酸基を脱硫酸化した化合物(8)は、PIMHの抗BACE−1活性がほぼ無くなっていた。
【0097】
BLHに対して既に報告された置換パターン(上記Scholefield,Z.ら)を備える本発明の化合物(4)及び化合物(5)の活性から、BACE−1活性の抑制において6−O硫酸化が重要な役割を果たすことが示された。
【0098】
本発明者らは、また、イズロン酸及びグルコサミン残基のいずれにおいても3位が更に硫酸付加された過硫酸化ヘパリン(化合物(9))の効果も分析した。本化合物は、予期しないことに、非修飾PIMHと同等のIC50を示した。硫酸化レベルとBACE−1抑制活性との直接的な相関関係が存在しないことから、当該活性が電荷密度と単純に相関しないことが明確に立証された。
【0099】
本発明者らは、特定の理論に束縛されることを望んでいないが、当該活性と置換パターンとの関係は、複雑であり、立体配置(コンホメーション)効果に依存しているようである。イズロン酸環とグリコシド結合の形状の両方が、置換パターンに依存しているからである。
【0100】
本発明者らは、3−アミノ−1プロパンスルホン酸(3APS,Alzehemed(商標名))(化合物10)の効果も調べた。この化合物は、ADの治療法として臨床試験中であり、Aβアグリゲーションを抑制する作用が提唱され、「ヘパリンミメチック(擬似ヘパリン)」と言われる。結果としては、3APSは、高濃度(1000μg/ml)であってもBACE−1を抑制しなかった。この結果は、「ヘパリンミメチック(擬似ヘパリン)」の活性が、BACE−1抑制までは拡張することはできないということを示している。
【0101】
(抗凝固活性)
N−硫酸基の除去及びN−アセチル基による置換は、化学修飾されたPIMHによるアンチトロンビンIII/凝固因子Xa複合体の妨害能を大幅に低減した。N−アセチル基を有する化合物(本発明の好ましい化合物(4)を含む)の抗第Xa因子活性は、PIMHよりも少なくとも3000倍低い(表1)。
他方、2−O硫酸基又は6−O硫酸基のいずれかの脱硫酸化は、アンチトロンビンIII/凝固因子Xaの活性を約200倍低減させた。
【0102】
ヘパリン/HSの抗第Xa因子活性が特定のペンタサッカライド(5糖類)配列-4)GlcNAc(6S)α(1-4)GlcAβ(1-4)GlcAβ(1-4)GlcNS(3,6S)α(1-4)IdoA(2S)α(1-4)GlcNS(6S)α(1-によることは、確かである。
【0103】
中央のグルコサミン残基内の3−O硫酸基の存在は、抗第Xa因子活性に重要であり、それを除去すると活性が消滅してしまう。これに対して、中央のグルコサミン残基からN−硫酸基又はイズロン酸残基から2−O硫酸基のいずれかを除去することは、それほど劇的ではないが有害な作用をもたらす。
【0104】
ヘパリン誘導体においてN−硫酸基を除去してN−アセチル基で置換すること(化合物(1)から化合物(2)への化学修飾)は、化合物(2)にて示される抗凝固活性の損失として説明される。しかし、高アルカリ条件下で発生するイズロン酸のO−脱硫酸化も、また、抗第Xa因子活性のかなりの低下を生じたが、これには2つの理由が考えられる。
【0105】
先ず、2−O硫酸基が除去されること(本発明に従う好ましい化合物(4)におけるように)であるが、これに加えて、稀ではあるが3−O硫酸基とN−硫酸基の両方を有するグルコサミン残基(例えば、ペンタサッカライド配列AGA*IA)において第2の化学修飾が起こり、N−硫酸化アジリジン基が形成し3−O硫酸基が消滅することである。
【0106】
化合物(5)又は化合物(8)のように(これらの化合物は異なる条件下で調製された)O−脱硫酸化された他のヘパリン誘導体は、当該化学修飾を含まず、当該活性の低下は、当該ペンタサッカライド(5糖)配列に含まれる関連基の離脱に起因すると考えられる。特に、3−O硫酸基は、温和な6−O脱硫酸下にて安定であることは注目に値する。
【0107】
(BACE−1と構造的に関連する他のプロテアーゼの抑制)
BACE−1と構造的に最も近い類縁体は、アスパルチルプロテアーゼペプシン、カテプシンD及びレニンである。これらの酵素は、各々、消化機能、血圧調節及びリソソームのタンパク質の分解機能を有する。効果的なBACE−1インヒビターは、また、これらの酵素とも反応し、薬学的に投与された場合には、望まない副作用を引き起こす可能性がある。
【0108】
Scholefieldの研究により既に調査された置換パターンを有する化合物(1)、(2)及び(3)や、本発明による好ましい化合物(4)のアスパルチルプロテアーゼへ抵抗する活性は、FRETペプチド分裂分析により測定された。当該化合物は、いずれも、最大1000μg/mlの濃度であってもレニンに対して抑制作用を及ぼさなかった。
【0109】
興味深いことに、非修飾PIMH(化合物(1))は、ペプシン及びカテプシンDの両方に対する抑制活性を示し、IC50値は各々0.23μg/ml及び0.1μg/mlであった。N−アセチル基(化合物2)や好ましくは化合物(4)であるN−アセチル化/Ido−2−OH化されたPIMHは、PIMH(化合物1)と比較してペプシンやレニンの両方に対する抑制活性が著しく減少することを示した。
【0110】
N−アセチル化されたPIMH(化合物2)のペプシンに対するIC50は、PIMH(化合物1)よりも14倍弱く、3.27μg/mlを示した。これに対して、N−アセチル化/Ido−2−OH化されたPIMH(好ましい化合物である化合物4)は1000μg/mlより高い濃度にてペプシンを抑制できなかった。
【0111】
化合物(2)及び化合物(3)のカテプシンDに対するIC50値は、各々、0.27μg/ml及び0.77μg/mlを示した。このように、高レベルの抗BACE−1活性を示すPIMHの化学修飾された化合物(好ましい化合物(4)を含む)は、レニン、ペプシン及びカテプシンDを有意に抑制せず、非修飾PIMHよりも高いIC50値を示す。
【0112】
(オリゴ糖によるBACE−1抑制)
全長PIMHを、酵素分解し、ゲル濾過クロマトグラフィーにより分別し、得られた断片(フラグメント)を用いてFRETペプチド分裂分析によりBACE−1抑制の有効サイズを決定した。
【0113】
図1は、糖類のサイズを関数として非修飾PIMH断片のBACE−1抑制活性を示すグラフである。図1に記載された第1の特徴は、当該活性が、オクタサッカライド(8糖)に見出されたことであり、高レベルの抗BACE−1活性の最低要件は、約8個のサッカライド(8糖)であることを示す。図1に示された結果が、活性に関して既知の研究(上記Scholefield,Z.ら)により提案されたデカサッカライド(10糖)の値よりも低い最小長を示すという事実から、当該活性はジサッカライド(二糖)のような低いレベルのサッカライドでも期待されるものと想定される。Scholefieldの研究結果と比較して活性レベルの低いことは予想外である。ポリサッカライド(多糖類)とオリゴサッカライド(オリゴ糖)とは活性のレベル(大きさ)が同じではないという証拠があるので、ポリサッカライドをより小さく断片化したものが全長分子と同等に挙動するとは予想できないからである。オクタサッカライドに比べてデカサッカライドに抑制活性のシフト(10倍の増加)が見られ、このことは10個のサッカライドの方がより好ましい最小長であることを示唆している。
【0114】
18個のサッカライド単位(糖単位)においては、当該活性は、最長PIMHと同等である。3000ダルトン(糖単位としては12糖までと同等)と同等なサイズのヘパリン糖類が血液脳関門(BBB)を通過するため、当該データは、インビボ投与に際して有望である。
【0115】
標準的なヘパリンの投与に関して他に起こり得る副作用としては、ヘパリン−血小板第4因子複合体に対する抗体の産生により引き起こされるヘパリン起因性血小板減少症(HIT)があるが、一方で、分子量及び硫酸化レベルを減少させることにより、当該症状を改善させることが示されている。
【0116】
硫酸化レベルを低下されたオリゴ糖(例えば、本発明に従う好ましい化合物(4))、においてBACE−1抵抗活性が維持されることは、脳への潜在的な取込みや抗凝固作用の低下や他の望まれない副作用を改善して薬学的に利用できる大いなる可能性を示している。N−アセチル化されたPIMH(化合物2)やN−アセチル化/Ido−2−OH化されたPIMH(本発明の好ましい化合物(4))は、抗凝固活性がほとんど無い状態で高いBACE−1抑制活性を示した。
従って、本発明に従う好ましい化合物(4)は、ADのような神経変性疾患の予防及び/又は治療にて利用される優れた候補の代表例である。
【0117】
(実施例1の付録(アペンディックス))
(化学修飾されたヘパリンの調製)
化合物(1)から化合物(9)までの化学修飾されたヘパリンは、下記反応(a)から(g)までの以下組み合わせにより調製された。
(1).PIMH出発原料(Celsus Labs,Cincinnati,OH)
(2).N−アセチル化ヘパリン(d)(f)
(3).Ido 2−O脱硫酸化ヘパリン(a)
(4).Ido 2−O脱硫酸化、N−アセチル化ヘパリン(a)(d)(f)
(5).6−O脱硫酸化ヘパリン(b)(e)
(6).6−O脱硫酸化、N−アセチル化ヘパリン(b)(f)
(7).6−O脱硫酸化、2−O脱硫酸化ヘパリン(c)(e)
(8).6−O脱硫酸化、2−O脱硫酸化、N−アセチル化ヘパリン(c)(f)
(9).過硫酸化ヘパリン(g)(e)
化合物は、既知のように1Hや13CのNMRにより特性が決定された。(Yates,E.A.;Santini,F.;Guerrini,M.;Naggi,A.;Torri,G.ら、Carbohydrate Research 1996,294,15-27.)化合物は、分析前に、適切な緩衝剤にて脱塩され、凍結乾燥され、再懸濁された。
【0118】
(化学反応)
(a)イズロン酸の2−O−硫酸基の選択的脱硫酸は、Jaseja及びPerlinに記載のように行った(Jaseja,M.;Rej,R.N.;Sauriol,F;Perlin,A.S.Can.J.Chem.1989,67,1449-1456.)。なお、N−や3−O−硫酸化されたグルコサミン単位の少数にて随伴する化学修飾があることに留意する(Santini,F.;Bisio,A.;Guerrini,M.;Yates,E.A.Carbohydrate Research 1997,302,103-108)。
【0119】
(b)グルコサミンの6−O−硫酸基の選択的除去(脱硫酸)は、既知方法に従い化学修飾することにより(Yates, E. A.他、上記)実施した(Inoue,S.;Miyawaki,M.Analytical Biochemistry 1975,65,164-174.)。
【0120】
(c)O−やN−硫酸基の完全脱硫酸は、既述の方法(Inoue,S.;Miyawaki,M.上記)により、可溶媒分解性の脱硫酸を用いることにより行った。
【0121】
(d)N−硫酸基の選択的脱硫は、既知のように動力学制御下による制御された可溶媒分解性の脱硫酸を用いて実施した(Inoue,Y.;Nagasawa,K.Carbohydrate Research 1976,46,87-95)。
【0122】
(e)N−再硫酸化は、既報に従いトリメチルアミン硫黄三酸化物複合体を用いることにより行った(Lloyd,A.G.;Embery,G.;Fowler,L.j.Biochemical Pharmacology 1971,20,637-648.)。
【0123】
(f)N−再硫酸化は、飽和炭酸水素ナトリウムにおける酸無水物を用いることにより行われる(上記Yates,E.A.ら)。
【0124】
(g)有効なヒドロキシル基(水酸基)の完全なO−硫酸化は、既報に従い、ピリジン中のヘパリンのテトラブチルアンモニウム塩に過剰の三酸化イオウ−ピリジン複合体を用いて(Yates,E.A.;Santini,F.;De Cristofano,B.;Payre,N.;Cosentino,C.ら、Carbohydrate Research 2000,329,239-247.)、次いで、N−再硫酸化する(Lloyd,A.G.他、上記)ことにより行い、この際、異常なN−スルホアジリジンの形成が起こらないよう注意した(Yates,E.A.;Santini,F.;Bisio,A.;Cosentino,C. Carbohydrate Research 1997,298,335-340.)。
【0125】
(NMR分光分析)
化学療法の有効性は、それぞれ500MHz及び125MHzにおける1H及び13CのNMRにより観察した(D2O、27℃)。化学シフト;δ/ppm(外部標準)は、確定したモデル化合物と完全に一致した。(Yates,E.A.他、上記)
【0126】
(オリゴ糖の調製)
豚粘膜ヘパリン及び化学修飾N−アセチル化ヘパリンは、pH7.0にて、100mUヘパラティナーゼII(Ibex Technologies Inc., Montreal, Canada)、100mM当たり100mgの酢酸ナトリウム、0.1mMの酢酸カルシウムを用いて消化分解した。消化分解で得られた断片(フラグメント)は、ゲル濾過クロマトグラフィー(Superdex-30,Amersham Pharmacia,UK,2000mm x 30mm, 100mM 炭酸水素アンモニウム)により分別し、サイズに応じて定義された標準に基づいて同定した。
【0127】
(インビトロでのペプチド分裂分析によるBACE−1抑制の測定)
APPのBACE−1分裂を抑制する化合物の能力は、スウェーデン型アミノ酸変異体を含有するFRETペプチド5-FAM-Glu-Val-Asn-Leu-Asp-Ala-Phe-Lys(QXL520)-OH(Anaspec,Inc.,CA,USA)を用いて蛍光共鳴エネルギー移動(FRET)ペプチド分裂分析を用いて測定された。無調整の場合、アミノ酸末端のフルオロフェア(蛍光体)は、消光するが、酵素分解が発生する場合には、当該フルオロフェアは、消光から解放され、蛍光(520nm)を発する。分析は、96ウェルの黒プレートを用いて3回行った(20mM酢酸ナトリウム、0.1%トリトン−X−100、pH4.5、ウェルごとに2.5μMのペプチド、遺伝子組換えヒトBACE−1(Sigma)の4.0×10-3ユニット/ウェル)。酵素活性及びバックグラウンド蛍光のコントロール(対照)を用いて、プレートを温置した(1h,25℃、2.5M酢酸ナトリウムを用いて停止する)。インヒビターは、1000μg/mlから0.0001μg/mlの範囲の濃度で添加した。
480ex/520nmの蛍光は、Polarstarプレートリーダー(BMG LabTechnologies,UK)にて測定され、データは抑制パーセント比率に対して抑制濃度のlog10対数をプロットし、BioDataFit 1.02(Chang Bioscience,USA)を用いて4パラメータのS字カーブに適合させて分析された。
【0128】
(抗凝固活性)
抗第Xa因子活性は、既知の活性を有するによる豚腸粘膜ヘパリン(PIMH)を標準として(Sigma,UK)測定した。用いたのは、診断グレードのコーティングされたヘパリン試験用キット(Chromogenix,MA,USA)であり、96ウェルの黒プレートを利用し、A405の読み込みを行って測定された(Polarstarプレートリーダー(BMG LabTechnologies,UK))。
【0129】
(他のプロテアーゼに対する活性)
構造的に関連するプロテアーゼペプシン及びカテプシンD(Sigma,UK)を抑制する化合物の能力は、FRET分裂分析(メーカーによる使用法に従い、5 pmol 酵素/ウェル、EnzChekプロテアーゼ分析キット(Molecular Probes,UK)にて)を用いて測定された。ヒトの組み換えレニン(Cayman Chemical,MI,USA)に対する活性は、FRETペプチド分裂分析(メーカーによる使用法に従い、0.08 pmol 酵素/ウェル、Renin Substrate 1(Molecular Probes,UK)にて)を用いて測定された。IC50値は、上記と同様に算出された。
【実施例2】
【0130】
本発明者らは、神経変性症状を治療するための本発明の化合物の効果を示すために、さらなる実験を行った。
なお、特に言及しない限り、実施例1(及びその付録)に使用された方法は、本実施例にても適用されるものとする。
【0131】
(BACEインヒビターとして2−O脱硫酸化されたN−修飾ヘパリン)
2−O脱硫酸化されていると共にN−修飾されたヘパリンの多くの例について、BACEインヒビターとして試験し、親(非修飾)ヘパリンと比較した。これらのポリサッカライド(多糖類)は、N−脱硫酸化によりグルコサミン残基を生成し、その後、遊離アミノ酸残基に様々な化学置換基を置換し、2−O位において適切な脱硫酸化を行うことにより、化学修飾される。
【0132】
2−O脱硫酸化によりN−修飾されたヘパリンは、目的以外の活性の低い有効なBACEインヒビターである。一例として、N−アセチル2−O脱硫酸化PMH(約82%の6−O硫酸基含有率、約0.1%未満の2−O硫酸基含有率)は、非修飾PMHよりもEC50が約3倍低い(表2)。さらなる例としては、N−プロピオン酸2−O脱硫酸化PMH(約82%の6−O硫酸基含有率、約0.1%未満の2−O硫酸基含有率)があり、当該PMHは、非修飾PMHよりもEC50が約8倍低いにも関わらず、無処理のヘパリンに比べて、約1/100の抗第Xa因子活性を有しており、これらの化合物が構造変化(この場合、N−置換)に対して予期し得ない程敏感であることが特徴である。さらに、上記N−修飾されたヘパリンは、いずれも、非常に弱い抗凝固活性を示した(非修飾PMHと比較して1/100から1/3000)。
【0133】
【表2】
【0134】
(アミロイド−βペプチドアグリゲーションに対する化学修飾ヘパリンの効果)
抗凝固活性に加えて、神経変性疾患の治療に直接関連するヘパリンの生じ得る副作用としては、広く知られているように、アミロイドβ(Aβ)ペプチドのアグリゲーション(凝集)がある。(Watson,D.J.,Lander,A.D. and Selkoe,D.J.(1997)Heparin-binding properties of the amyloidogenic peptides Aβ and amylin. Dependence on aggregation state and inhibition by Congo red. J Biol Chem,272,31617-24)当該アグリゲーションを低減することは望ましい特性である。従って、本発明者らは、Aβアグリゲーションを分析して非修飾の豚粘膜ヘパリンとの比較を行い、化学修飾されたヘパリンの活性を調べた。当該試験の結果は、図2に記載している。
【0135】
当該アグリゲーションの最大化は、非修飾ヘパリン“PMH”において観察される。ヘパリンを脱硫酸化することにより当該アグリゲーションが驚くほど低減することが観察された。幾つかの化合物(例えば、N−アセチル化ヘパリン“NAc”やido-2-脱硫酸化されたN−アセチル化ヘパリン“NAc de 2”)は、望ましいBACE−1抑制活性を有しており、好ましい抗凝固活性(抗第Xa因子活性)(Patey et al(2006)49 6129-6132)はまた、Aβアグリゲーションを改善する(それぞれ50%及び100%遅くする)。すなわち、100μg/mlの濃度において、N−アセチル化または0−脱硫酸化PMHのいずれかは、ペプチドアグリゲーションが最大となる半分の時間に50%の増加となり、N−アセチル化や2-de−O硫酸化されたPMHは、100%の増加となり、ほぼ対照(コントロール)レベル(すなわち、ヘパリンを添加しない場合のアグリゲーション)に達する。このように、本発明の請求項1に定義された好ましい化学修飾は、ヘパリンのAβアグリゲーションの促進力を著しく低減させ、結果的に、非修飾ヘパリンと比較して改善された治療可能比率をもたらす。
【0136】
(Aβアグリゲーション分析手順)
Aβペプチドは、1mlのヘキサフルオロ−2−プロパノールを1mlのAβペプチド(アミロイドβプロテイン断片1−42;Sigma A9810)に室温で1時間添加して調製した。生成物は、50μl(50μg)のアリコートに分割され、約2時間Speed Vacにて乾燥され、20℃で保管された。Thioflavin T(Sigma T-3516)溶液は、50mM Glycine-NaOH (pH8.5)中の5mM溶液として調製された。20μlのThioflavin T溶液に加えて50μlの1MのDDTと430μlの分析用緩衝液(10mM HEPES/100mM NaCl Ph7.4)とを含むThioflavin T分析緩衝液を氷上保持した。20mM NaOHの50μlをAβペプチドの50μgに付加した。その後、Thioflavin T分析緩衝液の2.5mlを当該Aβペプチド溶液に付加し、“Aβ緩衝液”を生成した。Thioflavin T分析緩衝液の残りの2.5mlは、ブランク緩衝液であり、共に氷上維持した。6つのサンプル希釈剤(分析緩衝液にて1:1)を96ウェルの黒プレートにピペット注入(Greiner 655076;100μl/ウェル)し、当該プレートを氷上維持した。96ウェルの黒プレートの読み取りを行い(450nmに励起波長があり、490nmに発光波長がある)、37℃にて150秒ごとの読み取りを250サイクル行った。
【産業上の利用可能性】
【0137】
本薬剤は、アルツハイマー病(AD)の予防及び/又は治療に用いることができる。
【技術分野】
【0001】
本発明は、神経変性疾患の予防及び/又は治療に関する。
【背景技術】
【0002】
加齢に関連する病気の発生率は、平均余命が延びるとともに増加している。アルツハイマー病(AD)は、命にかかわり、進行性かつ不可逆性の神経変性疾患であって、記憶喪失、意識障害、判断力低下、人格変化、見当識失調、言語能力失調の症状を発症する神経変性疾患である。
【0003】
ADは、神経原線維変化(NFT)及び神経毒性を有するβ−アミロイド(Aβ)の脳沈着により特徴付けられる。ADにおけるβ−セクレターゼ(BACE−1:アミロイド前駆体タンパク質をβ切断するエンザイム−1、β−セクレターゼ−1)によるアミロイド前駆体タンパク質(APP)の分裂(切断)は、アミロイド斑の主成分であるアミロイドβペプチドを生成する初期の律速段階である。α、β及びγセクレターゼによるAPPの切断により様々なペプチドを生成し、当該ペプチドのうちβ及びγセクレターゼによる逐次反応により生成されるAβ1-40及びAβ1-42は、アミロイド生成性であり、神経毒性を有する。APPの幾つかの病理学的変異体においてはBACE−1による切断に対するAPPの感受性が大きくなることが見出されており、従って、BACE−1活性を制御することが、製薬における重要なターゲットとなっている。
【0004】
硫酸化ヘパラン(HS)はアミロイド斑の構成成分として確認され、これによるアミロイドタンパク質、ペプチド及び原線維に対する反応性、アグリゲーションの促進化及び線維の安定化は、これまでの研究により裏付けられている。可溶性のヘパリンやヘパリン様物質(ヘプリン類縁体)もまた、インビトロ及びインビボのいずれにおいても当該プロセスを抑制することが示されている。
【0005】
最近、HSがBACE−1によるAPPの切断を直接的に制御し得ることが発見されたことにより、HSの新たな機能が見出された。牛肺ヘパリン(BLH)、豚粘膜のHS及びそれらの誘導体は、恐らくは酵素活性領域へのアクセスをブロックする作用から、α又はγ−セクレターゼによるAPP切断プロセスを妨害することなくBACE−1活性を抑制することが判明した(非特許文献1参照)。
【0006】
HSやHSの高度に硫酸化された構造を有するヘパリン様物質は、N−アセチル又はN−スルホ−α−D−グルコサミンのいずれかと結合したα−L−イズロン酸又はβ−D−グルクロン酸による1,4結合されたジサッカライド(二糖)繰返し単位から成るグリコサミノグリカンである。O−硫酸化の主位置は、イズロン酸のC−2位やグルコサミンのC−6位であり、さらに、まれにグルコサミンのC−3位である。生合成における様々な置換が、かなりの多様性をもたらすことになる。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Scholefield,Z.et al、Journal of Cell Biology 2003,163,97−107
【発明の概要】
【発明が解決しようとする課題】
【0008】
ヘパリンは、HSよりも高度な硫酸化や均質性を示し、凝固因子XaとIIaを示すアンチトロンビンIIIとセリンプロテアーゼとの間の不可逆な複合体の生成速度を大きくすることにより抗凝固作用を発揮し得るものとして広く使用される薬剤である。ヘパリンは、HS様物質として広範囲に利用され、しばしばHSが関与する多くの生物学的プロセスにおける優れた活性剤とされてきた。しかし、ヘパリンの誘導体が新薬として使用されるように開発された場合に、ヘパリンの抗凝固作用が弱まることは重大な問題である。
【0009】
HSや他のグリコサミノグリカンとアミロイド生成経路との相互作用に関するこれまでの研究は、それらを生成する酵素よりも、ヘパリンや高度に硫酸化された他の化合物とアミロイドタンパク質やペプチドとの相互作用にこれまで向けられてきた。しかし、この点においてHS活性の構造的要件に関する研究は、比較的少ない
BACE−1活性は、Aβペプチドの生成やADの進行に対する基礎的な重要性をもつが、現在のところ、BACE−1をターゲットとする効果的な療法はない。BACE−1に対する治療試薬としての非修飾ヘパリンの使用は、多くの副作用を疑いなく引き起こし、特に内出血の発症や血液凝固能作用の低下のリスクを増大させ得る。このことは、効果的な服用量に重大な制限を与えるため、標準的なヘパリン糖類の臨床使用の障害となる。
【0010】
平均余命が延びるとともに、加齢に関連する神経変性疾患の疾患、例えばAD、に対する効果的で新規な治療法の必要性が益々高まってきている。
本発明の目的は、神経変性疾患の予防及び/又は治療に用いられる化合物を提供することである。
【課題を解決するための手段】
【0011】
本発明の第1の観点によれば、1以上(1個以上)のジサッカライド(二糖)単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物であって、神経変性疾患の予防及び/又は治療に用いられる化合物が提供される。
【0012】
また、本発明は、BACE−1に対する強い抑制活性を維持しつつ、HSと比較して明確に低減された抗凝固活性を示すHS様物質(HS類縁体)とみなされる化合物を提供する。
【0013】
当該化合物におけるウロン酸部位の2−O原子の少なくとも約60%が水素原子に置換されていることが望ましい。より好ましくは、当該化合物におけるウロン酸部位の2−O原子の75%以上が水素原子に置換されていることが望ましい。さらに好ましくは、当該化合物におけるウロン酸部位の2−O原子のさらに大部分(85%から95%)が水素原子に置換されていることが望ましい。最も好ましくは、当該化合物におけるウロン酸部位の2−O原子の全て、すなわち、100%、が水素原子に置換されていることが望ましい。
【0014】
本発明の化合物に存在する各々のグルコサミン部位の6−O原子に関しては、単一又は複数の6−O原子が実質的に硫酸化される限りは、その望ましい硫酸化レベルは特に限定されないが、実質的に少なくとも約60%の硫酸化レベル(硫酸化率)で硫酸化されていることが望ましく、さらに好ましくは、少なくとも約75%の硫酸化レベルで硫酸化されていることが望ましい。さらに好ましくは、グルコサミンの6−O原子の少なくとも約85%から約95%が硫酸化されていることが望ましく、最も好ましくは、グルコサミン部位の6−O原子の全てが硫酸化され、グルコサミン部位の6−O原子の硫酸化レベルが100%に達していることが望ましい。
【0015】
ウロン酸の2−O原子に対する水素置換レベル(水素置換率)及び/又はグルコサミンの6−O原子の硫酸化レベルが100%より小さい場合には、複数のジサッカライドから成る当該化合物が、ウロン酸の2−O原子及び/又はグルコサミンの6−O原子の置換に関して異なるパターンを有する隣接するジサッカライドから成ることになる。
【0016】
例えば、本発明による化合物は、4つのジサッカライドを含み、75%のウロン酸の2−O原子の水素置換レベルを有することができ、すなわち、4つのジサッカライドのうち3つは、2−O原子が水素原子を有するウロン酸部位を含有することができる。この場合において、水素置換された2−Oウロン酸を含む3つのジサッカライドは、水素置換されたヘキササッカライド(6糖)の一端又は他端にて水素置換されない2−Oウロン酸原子を含むヘキササッカライドにより相互に共有結合される。別の態様として、水素置換されない2−Oウロン酸原子を含むジサッカライドは、水素置換された2−Oウロン酸原子を含む3つのジサッカライドのうちいずれか2つの間に提供されることもできる。
【0017】
さらなる一例としては、6−Oグルコサミン原子の60%が硫酸化されたイコササッカライド(20糖)から成る本発明の化合物は、グルコサミンの各々のO−6位が硫酸化された6つのジサッカライドを含む。当該6つの6−O硫酸化されたジサッカライドは、望ましい直鎖配列にて残りの4つの6−O硫酸化されないジサッカライド(すなわち、グルコサミン部位のO−6位にて硫酸化されないジサッカライド)と結合されることができ、例えば、6−O硫酸化されたジサッカライドが、6−O硫酸化されないオクタサッカライド(8糖)と相互結合して6−O硫酸化されたドデカサッカライド(12糖)を形成したり、また、6−O硫酸化されたジサッカライドの3つが、相互結合して、最初に一端を6−O硫酸化されないジサッカライドで結合されて、以降6−O硫酸化されたジサッカライドと6−O硫酸化されないジサッカライドとの繰返し配列により6−O硫酸化されたヘキササッカライド(6糖)を提供する。
【0018】
上述したScholefieldの研究では、最も活性なBACE−1インヒビターは、O−2位及びO−6位にて高度に硫酸化された牛肺から生成されるN−アセチル化されたヘパリンであると結論している。(上記Scholefield,Z.らに関して)O−2位又はO−6位における硫酸基の脱離は、化合物のBACE−1活性をかなりの程度減少させ、1又は複数の硫酸基(N−硫酸基のみ以外)の脱離は、当該化合物のBACE−1に対する抑制力に有害な影響をもたらすことを示唆している。
【0019】
さらに、当該研究では、BLHがHSよりもBACE−1活性のインヒビターとして優れていると結論付けている。HSがジサッカライドあたり約1.5の硫酸基を含むのに対して、ヘパリンがジサッカライドあたり約2.6〜約2.9の硫酸基を含むことから、ヘパリンに比べてHSの活性が低いことは、少なくとも部分的には、タンパク結合を還元するものと考えられる選択的脱硫酸化に関与する電荷の減少が原因として挙げられる。硫酸化の度合いとHSに対するヘパリンの活性度レベルとの関連は、高度に硫酸化されたHSプロテオグリカンが低度に硫酸化されたHSプロテオグリカンより線維芽細胞増殖因子と強固に結合していることを見出した他の研究により示されている(Kreuger,J.,P.Jemthら、J Biochem2005,389(Pt1),145-150)。Scholefieldの研究で述べられた結論に寄与するさらなる要因としては、2−O硫酸基の脱硫酸化により、ヘパリンが異なるコンホメーションのイズロン酸環そして恐らくはグリコシド結合を呈することが挙げられる。
【0020】
このように、Scholefield,Z.らやKreuger,J.らの研究は、一般的に脱硫酸化が活性を低下させることを示している。本発明の基礎を形成する化合物は、ウロン酸部位の2−O位が実質的に脱硫酸化され、そして、グルコサミン部位のN−2位が完全に脱硫酸化されているものであり、従って、2−O硫酸基やN−硫酸基を含有する対応化合物に比べて当該活性の低下を示すことが想定される。しかし、驚くべきことに、本発明の化合物について実施された実験の結果(詳細は後述)は、実質的には全体的に硫酸化レベルを低下させているにも関わらず、2−O硫酸基及びN−硫酸基の両方を除去することにより、予想外に高いBACE−1抑制活性を有する化合物が得られることを示した。
【0021】
また、本発明の第2の観点によれば、1以上(1個以上)のジサッカライド(二糖)単位から成り、当該ジサッカライド単位は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が硫酸基と置換され、グルコサミン部位の2−N原子が硫酸基以外の原子又は置換基と置換された化合物の使用であって、神経変性疾患の予防及び/又は治療に用いられる医薬の調製(製造)における使用が提供される。
【0022】
また、本発明の第3の観点によれば、1以上(1個以上)のジサッカライド単位から成り、当該ジサッカライド単位は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物を患者に有効量投与することを含む神経変性疾患を予防及び/又は治療する方法が提供される。
また、本発明の第4の観点によれば、本願にて定義され、本発明の様々な側面にて利用される化合物は、合成原料又は天然原料から生成される。
【0023】
当該化合物は、全体又は一部分が合成される。当該化合物は、天然の糖類、例えば、HS様物質である豚腸粘膜ヘパリン(PIMH)や牛肺ヘパリン(BLH)から化学修飾により生成可能である。PIMHは、後述する実施例にて例示する。
【0024】
また、本発明の上記第4の観点による化合物の生成には、解重合(脱重合)工程を含むことが好ましい。当該解重合工程は、亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、フリーラジカル(遊離基)処理及びこれらの組み合わせにより成る群の中から選択されることが好ましい。
【0025】
また、本発明の第5の観点によれば、本願にて定義された本発明の様々な側面の一部を形成する化合物の生産方法であって、1又は複数の合成原料又は1又は複数の天然原料からの生産方法が提供される。
【0026】
当該生産方法は、解重合工程を含むことが好ましい。当該解重合工程は、亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、フリーラジカル処理及びこれらの組み合わせから成る群の中から選択されることが好ましい。
当該解重合工程を選択することにより、詳細は後述するが、少なくとも部分的には、本化合物の1以上(1つ以上)の末端基構造に影響を与えることができる。
【0027】
本発明による化合物の基礎部分であるジサッカライド繰返し単位において、ウロン酸部位(好ましくは、ウロン酸残基がグルコサミン残基に結合することにより得られたもの)は、所望のエピマー形態をとることができる。当該ウロン酸部位は、α−L−イズロン酸部位(以下の式(I)、(II)及び(III)においては、便宜上、これのみを例示している)、β−D−グルクロン酸部位及びα−L−ガラクツロン酸部位から成る群の中から選択されることができる。
【0028】
6−Oウロン酸原子、3−Oウロン酸原子及び3−Oグルコサミン原子に結合した置換基の各々は、それぞれが独立して、水素原子、O−エーテル糖環置換基を形成する置換又は非置換のアルキル基、置換又は非置換のアルコキシ基、置換又は非置換のアリール基、O−エステル糖環置換基を形成する置換又は非置換のアシル基、置換又は非置換のアミド基(例えば、フタルアミド基)、硫酸基及びリン酸基から成る群の中から選択される。さらに、6−Cウロン酸のカルボキシル基を化学修飾して、6−Cウロン酸のアルコール基やエステル基を生成するようにしてもよい。
【0029】
6−Oウロン酸原子、3−Oウロン酸原子、3−Oグルコサミン原子、環状炭素原子のうちいずれか1つに結合したアルキル基は、直鎖構造又は分岐構造であり、好ましくは、C1-6アルキル基(炭素数1〜6のアルキル基)であり、1以上(1個以上)の原子や置換基、例えば、ハロゲン原子(例えばフッ素、塩素又は臭素)やアリール基、アシル基、アミド基(例えばフタルアミド基)又はリン酸基により置換されていてもよい。
【0030】
6−Oウロン酸原子、3−Oウロン酸原子、3−Oグルコサミン原子、環状炭素原子のうちいずれか1つに結合した置換又は非置換のアシル基は、直鎖構造(例えばペンタノニル基)又は分岐構造(例えばピバロイル基)であり、C1-6の置換又は非置換のアシル基であることが好ましい。当該アシル基は、アリールアシル基、例えばベンゾイル基とすることができる。また、当該アシル基は、1以上(1個以上)のハロゲン原子、特にフッ素原子、塩素原子又は臭素原子で置換されることができる。従って、好ましいアシル基は、モノ−、ジ−及びトリ−フルオロアセチル基である。当該アシル基は、さらに好ましくは、フタロイル基である。当該アシル基は、置換又は非置換のアセチル基、置換又は非置換のプロプリオニル基及び置換又は非置換のブタノイル基から成る群から選択されることが好ましい。当該アシル基は、最も好ましくは、非置換のアセチル基である。
【0031】
ウロン酸部位やグルコサミン部位の環状炭素原子における置換パターンは、望ましい特性、例えば本発明のジサッカライド化合物における疎水性の度合い、を与えるように選択される。ウロン酸部位及び/又はグルコサミン部位の2−、3−又は6−炭素原子の各々は、水素原子、置換又は非置換のアルキル基(好ましくはメチル基やエチル基)、エーテル基を形成する置換又は非置換のアルコキシ基、置換又は非置換のアリール基(例えばベンジル基)、置換又は非置換のアシル基(例えばアセチル基)、エステル基を形成する置換又は非置換のカルボキシル基、置換又は非置換のアミド基(例えばフタルアミド基)、硫酸基又はリン酸基により成る群の中から選択された置換基により置換されることができる。
【0032】
本発明の化合物は、多くの解重合工程によりポリサッカライド(多糖類)から得ることができる。本発明の化合物の末端基は、利用した製造法の特性に応じて広い多様な形態を備えることができる。さらに、当該化合物は、任意の数のモノサッカライド(単糖)を含むことができ、当該化合物におけるモノサッカライドの総数は、偶数個でもよいし、奇数個でもよい。
【0033】
解重合方法としては、以下に限定されないが一例として、亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、又はフリーラジカル処理のうちのいずれか又は組み合わせを含むことができる。
【0034】
非還元末端モノサッカライドは、グルコサミン残基、又はその誘導体、又はウロン酸部位(例えば、α−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位)又は誘導体、若しくは断片(フラグメント)、例えば、Δ(デルタ)4−5不飽和環(すなわち、環内の炭素原子4と炭素原子5の間のC−C二重結合)を含有するα−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位とすることができる。そのような不飽和化は、例えば、細菌リアーゼ酵素処理又は化学的ベータ脱離(ヘパリン/HSを断片化するのに一般的に用いられる)による分解により、本化合物を形成するポリサッカライドの断片ができるときに生じ得る。
【0035】
還元末端モノサッカライドは、ウロン酸部位(例えば、α−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位)、又はその誘導体、グルコサミン部位、又はその誘導体、若しくはその断片(フラグメント)、2,5-アンヒドロマンノース、2,5-アンヒドロマンニトール、1,6アンヒドロ(二環)環構造、又はマンノサミン残基とすることができる。当該化合物の生成には、亜硝酸分解を含むことができ、この場合、還元末端モノサッカライドは、2,5-アンヒドロマンニトールであることが多く、これは、通常、化学還元されて2,5-アンヒドロマンニトールになる。化学的ベータ脱離法を用いて本化合物を製造する場合、還元末端の幾つかは、1,6アンヒドロ(二環)環構造(一般的に6−O硫酸化されたグルコサミン残基から誘導される)としても存在する。さらに、化学的ベータ脱離法は、グルコサミン残基のエピマー化を引き起こしてマンノサミン残基を形成することもできる。
【0036】
上記のように定義された本発明の好ましい具体例としては、以下の式(I)に示される化合物が挙げられる。なお、式(I)は、便宜上の理由のみにより、ウロン酸部位がα−L−イズロン酸部位により示されているが、式(I)は、当該ウロン酸部位がα−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位である化合物にも及ぶと解すべきである。
【0037】
【化1】
【0038】
式(I)において、実質的に全てのR1は水素原子であり、実質的に全てのR2が硫酸基であり、実質的に全てのR3が硫酸基以外の原子又は置換基であり、nは1以上の整数であり、R4、R5及びR6は水素原子、置換又は非置換のアルキル基、置換又は非置換のアルコキシ基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、硫酸基又はリン酸基により成る群の中からそれぞれ独立して個々に選択され、XとYは水素、末端モノサッカライド(単糖)基、末端ジサッカライド(二糖)基及び/又はそれらのフラグメント(断片)若しくはそれらの誘導体より成る群の中からそれぞれ独立して選択される。
【0039】
単なる例示であるが、本発明の化合物が式(I)のサッカライド単位のみから成る場合、XとYの一方が水素であり、XとYの他方がモノサッカライドであるときには、当該化合物は全体として奇数個のモノサッカライド単位から構成され、これに対し、XとYが同じとき(例えばXとYが共に水素、モノサッカライド、又はジサッカライド)には偶数個のモノサッカライド単位から成ると解される。さらに、当該化合物は、XとYの一方がモノサッカライドであり、XとYの他方がジサッカライドの場合には、当該化合物は奇数個のモノサッカライド単位から成るものである。従って、式(I)、並びに後記の式(II)及び式(III)のいずれも、奇数個のモノサッカライド単位を含む当該化合物と偶数個のモノサッカライド単位を含む当該化合物の両方に及ぶよう意図されている。
【0040】
Xが、末端モノサッカライド基である場合には、Xはグルコサミン部位、又はその誘導体、若しくは断片(フラグメント)であることが好ましい。Xは、式(I)のグルコサミン部位と同じ構造を呈してもよい(この場合、R2、R3及びR4は上記で定義された通りである)。
【0041】
Xが末端ジサッカライド基である場合には、Xは、カッコで示されたジサッカライド繰返し構造を有して、非還元末端モノサッカライドが、式(I)のウロン酸部位、すなわち、α−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位、と同じ一般的構造(ここで、R1、R4及びR5は上記で定義した通りである)を有することが好ましい。ジサッカライド単位Xは、カッコで示すジサッカライド単位の一部を形成するモノサッカライドの一方又は両方の誘導体を含んでいてもよい。
【0042】
非還元末端モノサッカライトは、Δ4−5不飽和環(すなわち、環内の炭素原子4と炭素原子5の間のC−C二重結合)を有するα−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位とすることができる。そのような不飽和化は、例えば、本化合物を形成するポリサッカライドフラグメントが細菌リアーゼ酵素処理又は化学的ベータ脱離(ヘパリン/HSを断片化するのに一般的に使用される)による分解により生じるときに起こる。
【0043】
Yが末端モノサッカライドである場合には、Yは、ウロン酸部位、又はその誘導体、若しくはその断片(フラグメント)であることが好ましい。Yは、式(I)のウロン酸部位、すなわち、α−L−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位、と同じ一般的構造(ここで、R1、R4及びR5は上記で定義された通りである)を有することが好ましい。
【0044】
Yが末端ジサッカライド基である場合には、Yは、式(I)のカッコで示されたジサッカライド繰返し構造を有して、還元末端モノサッカライドが、式(I)のグルコサミン部位と同じ一般的構造(ここで、R2、R3及びR4は上記で定義した通りである)を有することが好ましい。ジサッカライド単位であるYは、カッコで示されたジサッカライド繰返し単位の一部を形成する1又は2つのモノサッカライドからの誘導体を含むこともできる。還元末端モノサッカライドは、2,5-アンヒドロマンノース、2,5-アンヒドロマンニトール、1,6アンヒドロ(二環)環構造、又はマンノサミン残基とすることができる。本化合物の生成には、亜硝酸分解を含むことができ、この場合、還元末端モノサッカライドは、2,5-アンヒドロマンニトールであることが多く、これは、通常、化学還元されて2,5-アンヒドロマンニトールになる。化学的ベータ脱離法を用いて本化合物を製造する場合、還元末端の幾つかは、1,6アンヒドロ(二環)環構造(一般的に6−O硫酸化されたグルコサミン残基から誘導される)としても存在する。さらに、化学的ベータ脱離法は、グルコサミン残基のエピマー化を引き起こしてマンノサミン残基を形成することもできる。
【0045】
式(I)の化合物の好ましい実施形態においては、R5及びR6がともに水素原子であり、以下の式(II)にて示された構造を有することができ、この構造においては、上記の式(I)にて定義されたR1、R2、R3、n、X及びYが用いられ、また、ウロン酸部位は、便宜上、α−L−イズロン酸部位のみにより示されているが、α−L−イズロン酸部位、β−D−グルクロン酸部位、又はα−L−ガラクツロン酸部位である場合も含む。
【0046】
【化2】
【0047】
グルコサミン部位の2−N原子(上記の式(I)及び式(II)におけるR3)は、2−N置換基が硫酸基(SO3-)以外であるという条件のもとで、望ましい有機系又は無機系の置換基により置換できる。2−Nグルコサミン原子は、水素原子、置換又は非置換のアルキル基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、及びリン酸基により成る群の中から選択された置換基により置換されることができる。
【0048】
2−Nアルキル基は、直鎖構造、分岐構造、又は環構造とすることができ、好ましくは、C1-6アルキル基であり、1以上の原子や置換基、例えば、ハロゲン原子(例えばフッ素、塩素又は臭素)やアリール基、アシル基、アミド基(例えばフタルアミド基)又はリン酸基により置換されていてもよい。
【0049】
2−Nグルコサミン原子に直接結合したアミド基及び/又は2−Nグルコサミン原子に結合したアルキル基と結合したアミド基は、任意の都合のよい形態をとることができ、例えば、メチルアミド基、エチルアミド基又はフタルアミド基となる。
【0050】
2−Nグルコサミン原子に直接結合した置換又は非置換のアシル基及び/又は2−Nグルコサミン原子に結合したアルキル基と結合したアシル基は、直鎖構造(例えばペンタノニル基)又は分岐構造(例えばピバロイル基)であり、C1-6の置換又は非置換のアシル基であることが好ましい。当該アシル基は、アリールアシル基、例えばベンゾイル基とすることができる。また、当該アシル基は、1以上(1個以上)のハロゲン原子、特にフッ素原子、塩素原子又は臭素原子で置換されることができる。従って、好ましいアシル基は、モノ−、ジ−及びトリ−フルオロアセチル基である。当該アシル基は、さらに好ましくは、フタロイル基である。2−Nグルコサミン原子は、置換又は非置換のアセチル基、置換又は非置換のプロプリオニル基及び置換又は非置換のブタノイル基から成る群から選択されるアシル基により置換されることが好ましい。当該アシル基は、最も好ましくは、以下の式(III)に示すような非置換のアセチル基である。
【0051】
R基及び上記式(I)にて定義されたR基群及びnに関して、本発明の特に好ましい化合物の実施態様においては、全てのR1が水素原子であり、全てのR2が硫酸基であり、R3がアセチル基であり、R4、R5及びR6が全て水素原子である。この好適な実施態様は、以下の式(III)にて示され、当該式においてX及びYは上記式(I)で定義された通りであり、ウロン酸部位はα−L−イズロン酸部位(式中、便宜上これのみが示されているが)、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位とすることができる。
【0052】
【化3】
【0053】
本発明の化合物は、任意の適切な長さを有することができ、そして、異なる構造(すなわち、異なる置換パターンを有する構造)のジサッカライドが望ましい直鎖配列にて結合されているようにすることができる。さらに、本発明の化合物は、天然のオリゴ糖やポリサッカライド(多糖類)やその断片(フラグメント)による適切な化学修飾により生産されることができる。
【0054】
本発明の化合物は、奇数個又は偶数個のモノサッカライド単位から成ることができる。本化合物がジサッカライド繰返し単位のみから成る場合には、モノサッカライド単位の総数は偶数個となるが、本化合物がジサッカライド繰返し単位を含み、且つ当該分子のいずれか一端に単一の末端サッカライドを含むような場合には、奇数個のモノサッカライドから成る化合物となる。
【0055】
例として、上記式(I)に関して、本化合物は、後述の3つの好ましい構造(式(IV)、式(V)及び式(VI))のうちの1つにより示され、ここで、全てのR基やnは上記式(I)にて定義されたとおりであり、ウロン酸部位は、便宜上α−L−イズロン酸部位のみが示されているが、α−イズロン酸部位、β−D−グルクロン酸部位又はα−L−ガラクツロン酸部位のいずれでもよい。
式(IV)において、Xは、カッコで示されたジサッカライド繰返し構造に含まれると同じ一般的構造を有する末端グルコサミン部位である。
【0056】
【化4】
式(V)において、Yは、カッコで示されたジサッカライド繰返し構造に含まれると同じ一般的構造を有する末端ウロン酸部位である。
【0057】
【化5】
式(VI)において、Xは、カッコで示されたジサッカライド繰返し構造に含まれると同じ一般的構造を有する末端グルコサミン部位であり、Yは、カッコで示されたジサッカライド繰返し構造に含まれると同じ一般的構造を有する末端ウロン酸部位である。
【0058】
【化6】
【0059】
Scholefieldの研究では、BLHの10以上のモノサッカライドから成るオリゴ糖は、BACE−1活性を抑制するが、当サイズ未満のオリゴ糖では、有意の活性は見出されなかったと結論付けている。(上記Scholefield,Z.らに関して)しかし、予想外なことに、10未満のモノサッカライドから成るオリゴ糖を含む本発明の化合物は、BACE−1活性を抑制することが示された。本発明の化合物は、4から24までの(4〜24個の)モノサッカライド単位(例えば、上記式(I)から式(III)においてn=2から12、X=Y=H)から成ることが好ましい。本化合物は、より少ない個数のモノサッカライドから成ることが好ましい。なぜなら、より小さい化合物のほうが容易に血液脳関門を通過することができるためである。本化合物は、さらに好ましくは、6から20までの(6〜20個の)モノサッカライド単位(好ましくは、n=3から10、X=Y=H、さらに好ましくは、n=2から9、X=ジサッカライド、Y=H)、さらに好ましくは、6から18までの(6〜18個の)モノサッカライド単位(好ましくは、n=3から9、X=Y=H、さらに好ましくは、n=2から7、X=モノサッカライド、Y=モノサッカライド)から成るものである。本発明の化合物のサイズ範囲としては、6から16までの(6〜16個の)モノサッカライド単位であることが好ましく、これは約1500ダルトン(Dalton)から約5000ダルトンまでの化合物の分子質量範囲に相当する。
【発明の効果】
【0060】
神経変性疾患、例えば、老人性痴呆症、初老期痴呆症、多発梗塞性痴呆及び他の神経変性の疾患や障害の予防及び/又は治療のために、医学及び獣医学のともに役立つ薬品として使用される化合物が待ち望まれている。本発明による化合物は、アルツハイマー病の予防及び/又は治療のための利用に大いに適する。
【0061】
本発明の第1の観点による化合物が、本発明の第2の観点と合わせて、神経変性疾患、特にアルツハイマー病の予防及び/又は治療のための医薬の調剤としての利用に大いに適することは、当業者にとって明らかである。
【0062】
本発明の第3の観点の神経変性疾患の予防法及び/又は治療法は、本発明の第1の観点による化合物を利用することが望ましい。
【0063】
また、本発明の第6の観点によれば、1以上(1つ以上)のジサッカライド単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、実質的に、ウロン酸部位の2−O原子が水素原子と置換され、グルコサミン部位の6−O原子が硫酸基と置換され、グルコサミン部位の2−N原子が硫酸基以外の原子又は置換基と置換された化合物から成る薬剤組成物であって、神経変性疾患の予防及び/又は治療に用いられる薬剤組成物が提供される。
【0064】
本化合物は、本発明の第6の観点から成る薬剤組成物として、薬学的に許容される基材、希釈剤、又は添加剤と組み合せて利用されることが好ましい。
【0065】
また、本発明の第7の観点によれば、1以上(1個以上)のジサッカライド単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、実質的に、ウロン酸部位の2−O原子が水素原子と置換され、グルコサミン部位の6−O原子が硫酸基と置換される化合物であって、グルコサミン部位の2−N原子が硫酸基以外の原子又は置換基に置換された化合物が提供される。
【0066】
下記の実施例の分析結果から、本発明の化合物は、BACE−1インヒビターとしてのIC50が約100μg/mlより小さく、さらに好ましくは、約10μg/mlより小さく、さらに好ましくは、約1μg/mlより小さく、さらにより好ましくは、約0.1μg/mlより小さく、最も好ましくは、約0.05μg/mlより小さい値を示す。
【0067】
抗凝固作用に関して、本発明の化合物は、非修飾の豚腸粘膜ヘパリン(PIMH)における抗凝固活性の約50%より小さいことが好ましい。本発明の化合物は、好ましくは、非修飾PIMHの抗凝固活性の約20%より小さく、さらに好ましくは、当該抗凝固活性の約5%より小さいことが望ましい。本発明による化合物は、特に好ましくは、当該抗凝固活性の約1%より小さく、さらに好ましくは、当該抗凝固活性の約0.5%より小さいことが望ましい。本発明による化合物は、さらに好ましくは、当該抗凝固活性の約0.1%より小さく、最も好ましくは、当該抗凝固活性の約0.03%より小さいことが望ましい。
【0068】
本発明の化合物の治療可能比率(抗BACE−1活性(以下の実施例にて記載された分析結果により決定される)と抗セリンプロテアーゼ凝固因子Xaとの比率)は、好ましくは、約100より大きく、さらに好ましくは、約500より大きく、さらに好ましくは、約1000より大きく、最も好ましくは、1000より大きい。本発明の化合物の治療可能比率は、特に好ましくは、100から2000までの範囲であり、さらに好ましくは、200から1500までの範囲であり、最も好ましくは、500から1500までの範囲である。本発明による化合物は、本発明の好適な実施例において、当該治療可能比率が約1092であることを示す。
【0069】
本発明の化合物は、特に化合物の利用方法に応じて多種の形態をとることが可能である。すなわち、本化合物は、例えば、粉薬、錠剤、カプセル、液体、軟膏、乳液、ジェル、ヒドロゲル、エアゾル、スプレー、ミセル、経皮貼布、リポソーム、又は人や動物への投与に適切な他の形態として提供されることができる。本化合物の媒体(賦形剤)は、投与された患者に対して優れた耐性があり、本化合物を脳まで運搬できるものである。
【0070】
また、本化合物は、点眼薬又は眼軟膏剤の形態により経眼投与することができ、液体形状又は固体形状により経口投与することもできる。経口投与に適する化合物としては、丸薬、カプセル、顆粒剤、錠剤及び粉薬のような固体形状や、水薬、シロップ剤、エリキシル剤及び懸濁液のような液体形状が含まれる。非経口投与として有効な形態としては、殺菌済み水薬、乳剤及び懸濁液が含まれる。
【0071】
本発明による化合物は、多くの方法により利用されることができる。例えば、当該化合物が、全身投与することが要求されることもあるが、錠剤、カプセル、又は液状の形態で経口投与された場合に要求される。あるいはまた、本化合物は、注射により血流に注入されることができる。当該注射は、静脈注射(ボーラスや浸剤)や皮下注射(ボーラスや浸剤)で行うことができる。また、本化合物は、吸入投与(例えば経鼻投与)されることもできる。
本化合物は、また、脳内、脳室内、クモ膜下腔内の搬送作用により、中枢に投与されることができる。
【0072】
本化合物は、また、緩速又は遅延型の送出装置と併用されることが出来る。当該送出装置は、例えば、患者の上皮又は皮下に挿入され、本化合物は、数週間又はさらに数ヶ月にわたり送出されることができる。当該装置は、本化合物の頻繁な通常投与を必要とする場合(例えば、患者に対して少なくとも毎日の錠剤摂取や毎日の注射による投与の場合)に、特に有用である。
【0073】
本化合物の必要摂取量は、生物学的活性やバイオアベイラビリティ(生体利用性)により決定され、そして、それらの特性は、本化合物の投与形態、本化合物の利用時の物理化学的特性及び本化合物の利用形態が単独療法であるか複合療法であるかに依存する。本化合物の投与頻度もまた、上記に述べた要因及び特に患者に投与された化合物の半減期に影響される。
【0074】
本化合物の最適な投与量は、当業者により決定されることができ、使用する化合物の種類、薬剤の効力、投与形態及び病状の進行に応じて変化する。治療を受けている特定の患者に応じた他の要因、例えば、患者の年齢、体重、性別、食事及び投与時間も考慮して、投与量を調整することが必要となる。
【0075】
製薬業にて常套的に利用される公知の方法(例えば、インビボでの実験や臨床試験など)では、調剤された混合物や構成物や管理体制の定式化を用いて、化合物や成分を特定割合に調合し正確な治療計画(例えば、本化合物の1日の投与量や投与頻度)を確立することができる。使用する化合物の種類にも依存するが、一般的には、体重の0.01μg/kgから1.0g/kgまでの範囲の日用量(1日の投与量)でADの治療に利用可能である。さらに好ましくは、本化合物の日用量は、体重の0.01mg/kgから100mg/kgまでの範囲が望ましい。
【0076】
毎日の投与は、単一投与(例えば、経口投与のための1日用の錠剤や1日1回の注射)として与えられることができる。その代わりとして、本化合物は、1日2回以上の投与を必要とされることもある。例えば、ADの患者は、錠剤にて25mgから5000mgまでの2回以上の服用により投与される。治療中の患者は、起床時に第1の服用を行い、夜に第2の服用を行う(1日2回の服用の場合)か、以降3時間又は4時間ごとに服用を行うことができる。別のやり方として、遅延化送出装置を用いて、繰返し投与の必要なく患者への本化合物の適用量を提供することもできる。
【0077】
本発明は、治療に有効な量の本発明による化合物及び好ましくは薬学的に許容される賦形剤(ベヒクル)を含む薬剤組成物を提供する。本発明における”治療に効果的な量”とは、本化合物が効果を発揮する疾患の患者に投与される場合に、当該疾患を減少、緩和、又は軽減する本化合物又はその組成物の量である。”患者”としては、脊椎動物、哺乳動物、ペット、又は人間が該当する。本発明の実施において、”薬学的に許容される賦形剤”は、薬剤組成物の形成に有用な当該技術分野における当業者に公知である生理的な賦形剤であれば種類は問わない。
【0078】
1つの実施態様としては、本発明の第6の観点に従う組成物における当該化合物の量を、約0.01mgから約800mgまでとすることができる。また、他の実施態様としては、当該化合物の量を、約0.01mgから約500mgまでとすることができる。また、他の実施態様としては、当該化合物の量を、約0.01mgから約250mgまでとすることができる。また、他の実施態様としては、当該化合物の量を、約0.1mgから約60mgまでとすることができる。さらに別の実施態様としては、当該化合物の量を、約1mgから約20mgまでとすることができる。
【0079】
1つの実施態様においては、本発明の第4の観点から成る組成物において用いられる薬学的な賦形剤は、液体状とすることができ、当該薬剤組成物は、水薬の形態とすることができる。また、他の実施例においては、当該薬学的に許容される賦形剤は、固体状とすることができ、当該薬剤組成物は、粉薬又は錠剤の形態とすることができる。さらに、他の実施例においては、当該薬学的に許容される賦形剤は、ジェル状とすることができ、当該薬剤組成物は、座薬又は乳液の形態とすることができる。さらに、他の実施態様においては、当該化合物又はその組成物は、薬学的に許容される経皮貼布の一部として形成されることができる。
【0080】
本発明の第6の観点から成る組成物において用いられる固体状の賦形剤は、香味添加剤、潤滑剤、溶解剤、懸濁剤、賦形剤、滑剤、圧縮剤、結合剤、又は口腔内崩壊錠としても作用する1以上の構成要素を、カプセル剤として含めることも可能である。粉薬においては、賦形剤は、細分化された活性成分を混合された細分化された固体状である。錠剤においては、活性成分は、適切な形状にて必要な圧縮特性を備え、望ましい形状やサイズで圧縮される賦形剤とともに混合される。粉薬及び錠剤は、最大で99%以下の活性成分を含むことが好ましい。適切な固体状の賦形剤としては、例えば、リン酸カルシウム、ステアリン酸マグネシウム、タルク、砂糖、デキストリン、でんぷん、ゼラチン、セルロース、ポリビニルピロリジン、低溶性ワックス及びイオン交換樹脂を挙げることができる。
【0081】
液体状の賦形剤は、水薬、懸濁液、乳剤、シロップ剤、エリキシル剤及び本発明の第6の観点により形成される加圧された組成物を調製するのに利用することができる。本発明の第1の観点により形成される化合物は、水、有機溶媒、若しくはその混合剤、又は薬学的に許容された油脂や脂肪のような薬学的に許容される液体状の賦形剤にて溶解又は懸濁されることができる。当該液体状の賦形剤は、溶解剤、乳化剤、緩衝液、防腐剤、甘味料、香料、懸濁化剤、増粘剤、着色料、粘性調節剤、安定剤、又は浸透圧調節のような他の適切な薬学的な添加物を含めることができる。本発明の第1の観点から成る化合物の経口投与や非経口投与に用いられる液体状の賦形剤の好適な一例としては、水(部分的に上記添加物、例えば、セルロース、誘導体、好ましくは、カルボキシメチルセルロースナトリウム溶液、を含む)、アルコール類(一価アルコールや多価アルコール、例えば、グリコール、を含む)、その誘導体及び油脂(例えば、分留されたココナッツオイルやピーナッツオイル)を挙げることができる。当該賦形剤は、また、非経口投与の場合には、オレイン酸エチル及びミリスチン酸イソプロピルのような油性エステルとすることができる。無菌の液体状の賦形剤は、非経口投与のための無菌の液体状の当該組成物において有用である。加圧された組成物に対する液体状の賦形剤には、ハロゲン化炭化水素、又は他の薬学的に許容された推進剤を用いることができる。
【0082】
本発明の第1の観点から成る化合物は、他の溶質や、懸濁化剤(例えば、溶液を等浸透圧化させる含塩物やグルコース)、胆汁酸塩、アラビアゴム、ゼラチン、モノオレイン酸ソルビタン、ポリソルベート80(ソルビトール及びその無水物がエチレンオキサイドと共重合されたオレイン酸エステル)等を含有する無菌の水薬又は懸濁液として経口投与されることができる。
【0083】
無菌の水薬又は懸濁液のような液体状で薬学的に許容される組成物は、例えば、筋肉内、クモ膜下腔内、硬膜外、腹腔内、又は皮下の注射により利用されることができる。無菌の水薬は、また静脈内に投与することもできる。本発明の化合物は、本発明の第6の観点に従い無菌の固体状の化合物として調製することもでき、当該組成物は、無菌の水、生理的食塩水、又は他の適切な無菌の注射用媒体を用いた投与時に溶解又は懸濁されることができる。賦形剤には、必要且つ不活性な結合剤、懸濁化剤、潤滑剤、香味剤、甘味剤、防腐剤、着色料、又はコーティング剤を含めることができる。
【0084】
本発明の化合物は、予防療法に利用されることに大いに適する。”予防療法”とは、ADのような神経変性疾患の影響を防止又は軽減する療法であれば特に限定されずに含まれるものとする。予防療法は、例えば、規定年齢に到達した人や神経変性疾患への遺伝的な素因を有する人に定期的に与えることができる。別のやり方として、予防療法は、神経変性疾患の発症を引き起こしやすい状況にさらされている人にケースバイケースで与えられることもできる。
【0085】
本発明は、後述する実施例を用いてさらに容易に理解されることができる。しかし、当該実施例における特定の手段や結論は、後述する請求項にてより広範に記載される本発明の一部を説明するものに過ぎないということは当業者には明らかなことであろう。
本発明の実施の態様を実施例に沿って説明するが、当該実施例は例示のものにすぎず本発明を限定するものではない。
【図面の簡単な説明】
【0086】
【図1】実施例1に記載されているように非修飾PIMHのフラグメントのBACE−1抑制活性をサッカライドの大きさを関数として示すグラフである。
【図2】実施例2に記載されているようにA−βペプチドの凝集速度(アグリゲーション率)に対する修飾ヘパリン誘導体の効果を示すグラフである。また、化合物を添加しない場合の凝集速度を「control(対照)」として示している。
【実施例1】
【0087】
伝達性海綿状脳症(TSEs)に対する懸念が表明された後は、BLHの利用度や薬剤的使用は最近少なくなっているが、このことは本発明者に豚腸粘膜ヘパリン(PIMH)を用いてBACE−1抑制活性を調べることを促した。PIMHは、BLHよりもβ−D−グルクロン酸のレベル(量)が高いが、いずれもこれまでに広く利用されておりTSEの危険性については知られていないからである。
【0088】
一連の化学修飾されたPIMH誘導体(式(VII)、表1に示す)を、後述の付録(アペンディックス)に記載の手法で調製した。所定のジサッカライド繰返し構造に応じて、O−、N−硫酸化及びN−アセチル化のパターンを変化させて、活性の体系的検討ができるようにした。各誘導体について、BACE−1のAPP切断抑制能、抗凝固剤としての効能(抗凝固因子Xa)及び他のアスパルチルプロテアーゼ系分子に対する抑制能を評価した。
【0089】
PIMH誘導体の1つとして、化合物4(式(III)のウロン酸部位がα−L−イズロン酸部位である下記の式(VIII))は、本発明による化合物の好ましい例の1つである。
【0090】
【化7】
実施された実験結果は、表1に示され、詳細は以下に述べる。
【0091】
【化8】
【0092】
【表1】
【0093】
表1において、抗凝固活性は、PIMH(100%として)に対する割合で示される。置換パターンは、それぞれ、イズロン酸の2位、グルコサミンの6位及びグルコサミンの2位に相当するR1、R2及びR3により定義される。治療可能比率は、BACE−1に対するIC50/抗凝固活性により算出した。UAは、ウロン酸であり、α−L−イズロン酸又はβ−D−グルクロン酸のいずれかである。また、GlcNは、α−D−グルコサミンである。#化合物(9)は、イズロン酸とグルコサミン残基のともに3位にて硫酸化されている。当該IC50値は、当然のことながら、使用された分析手段の性質に依存する。
【0094】
(BACE−1抑制活性)
(1)、(2)、(3)及び(5)に該当する置換パターンのBLHにおけるBACE−1抑制特性は、BLHを出発原料としてScholefield,Z.らにより既に調べられた。(上記Scholefield,Z.らに関して)
FRETペプチド分裂分析(FRET peptide cleavage assay)を使用して、BACE−1によるインビトロのAPP分断を測定した。非修飾PIMH(1)の次に、最も効果的なBACE−1インヒビターは、N−脱硫酸化され、N−再アセチル化されたPIMH(2)である。同様のIC50値であり、活性で3番目と4番目にランクされた化合物は、2−O脱硫酸化された(3)と、これまで報告されたことのない置換パターンを備える2−O脱硫酸化/N−アセチル化されたPIMHとしての本発明の化合物(4)である。これは、N−硫酸も2−O−硫酸も、6−O硫酸化を伴う場合には、高活性を得るための絶対要件ではないことを示している。このようにして、本発明の好ましい化合物(4)は、最も活性の高いモノ−硫酸化された化合物であった。
【0095】
6−O硫酸基の除去は、最も活性の低いジ硫酸化化合物(化合物(5))を生成した。化合物(4)は、6−O脱硫酸化された(5)又は6−O脱硫酸化/N−アセチル化されたヘパリン(6)のいずれよりも活性が高い。これは、化合物(4)が化合物(5)よりもジサッカライドあたりの硫酸基が少なく、化合物(6)とジサッカライドあたりの硫酸基が同数であるという事実からみて想定外の結果である。
【0096】
最も活性が低いモノ−硫酸化化合物は、2−O硫酸基と6−O硫酸基とが共に脱硫酸化されてN−硫酸基のみが残存する化合物(7)であった。全硫酸基を脱硫酸化した化合物(8)は、PIMHの抗BACE−1活性がほぼ無くなっていた。
【0097】
BLHに対して既に報告された置換パターン(上記Scholefield,Z.ら)を備える本発明の化合物(4)及び化合物(5)の活性から、BACE−1活性の抑制において6−O硫酸化が重要な役割を果たすことが示された。
【0098】
本発明者らは、また、イズロン酸及びグルコサミン残基のいずれにおいても3位が更に硫酸付加された過硫酸化ヘパリン(化合物(9))の効果も分析した。本化合物は、予期しないことに、非修飾PIMHと同等のIC50を示した。硫酸化レベルとBACE−1抑制活性との直接的な相関関係が存在しないことから、当該活性が電荷密度と単純に相関しないことが明確に立証された。
【0099】
本発明者らは、特定の理論に束縛されることを望んでいないが、当該活性と置換パターンとの関係は、複雑であり、立体配置(コンホメーション)効果に依存しているようである。イズロン酸環とグリコシド結合の形状の両方が、置換パターンに依存しているからである。
【0100】
本発明者らは、3−アミノ−1プロパンスルホン酸(3APS,Alzehemed(商標名))(化合物10)の効果も調べた。この化合物は、ADの治療法として臨床試験中であり、Aβアグリゲーションを抑制する作用が提唱され、「ヘパリンミメチック(擬似ヘパリン)」と言われる。結果としては、3APSは、高濃度(1000μg/ml)であってもBACE−1を抑制しなかった。この結果は、「ヘパリンミメチック(擬似ヘパリン)」の活性が、BACE−1抑制までは拡張することはできないということを示している。
【0101】
(抗凝固活性)
N−硫酸基の除去及びN−アセチル基による置換は、化学修飾されたPIMHによるアンチトロンビンIII/凝固因子Xa複合体の妨害能を大幅に低減した。N−アセチル基を有する化合物(本発明の好ましい化合物(4)を含む)の抗第Xa因子活性は、PIMHよりも少なくとも3000倍低い(表1)。
他方、2−O硫酸基又は6−O硫酸基のいずれかの脱硫酸化は、アンチトロンビンIII/凝固因子Xaの活性を約200倍低減させた。
【0102】
ヘパリン/HSの抗第Xa因子活性が特定のペンタサッカライド(5糖類)配列-4)GlcNAc(6S)α(1-4)GlcAβ(1-4)GlcAβ(1-4)GlcNS(3,6S)α(1-4)IdoA(2S)α(1-4)GlcNS(6S)α(1-によることは、確かである。
【0103】
中央のグルコサミン残基内の3−O硫酸基の存在は、抗第Xa因子活性に重要であり、それを除去すると活性が消滅してしまう。これに対して、中央のグルコサミン残基からN−硫酸基又はイズロン酸残基から2−O硫酸基のいずれかを除去することは、それほど劇的ではないが有害な作用をもたらす。
【0104】
ヘパリン誘導体においてN−硫酸基を除去してN−アセチル基で置換すること(化合物(1)から化合物(2)への化学修飾)は、化合物(2)にて示される抗凝固活性の損失として説明される。しかし、高アルカリ条件下で発生するイズロン酸のO−脱硫酸化も、また、抗第Xa因子活性のかなりの低下を生じたが、これには2つの理由が考えられる。
【0105】
先ず、2−O硫酸基が除去されること(本発明に従う好ましい化合物(4)におけるように)であるが、これに加えて、稀ではあるが3−O硫酸基とN−硫酸基の両方を有するグルコサミン残基(例えば、ペンタサッカライド配列AGA*IA)において第2の化学修飾が起こり、N−硫酸化アジリジン基が形成し3−O硫酸基が消滅することである。
【0106】
化合物(5)又は化合物(8)のように(これらの化合物は異なる条件下で調製された)O−脱硫酸化された他のヘパリン誘導体は、当該化学修飾を含まず、当該活性の低下は、当該ペンタサッカライド(5糖)配列に含まれる関連基の離脱に起因すると考えられる。特に、3−O硫酸基は、温和な6−O脱硫酸下にて安定であることは注目に値する。
【0107】
(BACE−1と構造的に関連する他のプロテアーゼの抑制)
BACE−1と構造的に最も近い類縁体は、アスパルチルプロテアーゼペプシン、カテプシンD及びレニンである。これらの酵素は、各々、消化機能、血圧調節及びリソソームのタンパク質の分解機能を有する。効果的なBACE−1インヒビターは、また、これらの酵素とも反応し、薬学的に投与された場合には、望まない副作用を引き起こす可能性がある。
【0108】
Scholefieldの研究により既に調査された置換パターンを有する化合物(1)、(2)及び(3)や、本発明による好ましい化合物(4)のアスパルチルプロテアーゼへ抵抗する活性は、FRETペプチド分裂分析により測定された。当該化合物は、いずれも、最大1000μg/mlの濃度であってもレニンに対して抑制作用を及ぼさなかった。
【0109】
興味深いことに、非修飾PIMH(化合物(1))は、ペプシン及びカテプシンDの両方に対する抑制活性を示し、IC50値は各々0.23μg/ml及び0.1μg/mlであった。N−アセチル基(化合物2)や好ましくは化合物(4)であるN−アセチル化/Ido−2−OH化されたPIMHは、PIMH(化合物1)と比較してペプシンやレニンの両方に対する抑制活性が著しく減少することを示した。
【0110】
N−アセチル化されたPIMH(化合物2)のペプシンに対するIC50は、PIMH(化合物1)よりも14倍弱く、3.27μg/mlを示した。これに対して、N−アセチル化/Ido−2−OH化されたPIMH(好ましい化合物である化合物4)は1000μg/mlより高い濃度にてペプシンを抑制できなかった。
【0111】
化合物(2)及び化合物(3)のカテプシンDに対するIC50値は、各々、0.27μg/ml及び0.77μg/mlを示した。このように、高レベルの抗BACE−1活性を示すPIMHの化学修飾された化合物(好ましい化合物(4)を含む)は、レニン、ペプシン及びカテプシンDを有意に抑制せず、非修飾PIMHよりも高いIC50値を示す。
【0112】
(オリゴ糖によるBACE−1抑制)
全長PIMHを、酵素分解し、ゲル濾過クロマトグラフィーにより分別し、得られた断片(フラグメント)を用いてFRETペプチド分裂分析によりBACE−1抑制の有効サイズを決定した。
【0113】
図1は、糖類のサイズを関数として非修飾PIMH断片のBACE−1抑制活性を示すグラフである。図1に記載された第1の特徴は、当該活性が、オクタサッカライド(8糖)に見出されたことであり、高レベルの抗BACE−1活性の最低要件は、約8個のサッカライド(8糖)であることを示す。図1に示された結果が、活性に関して既知の研究(上記Scholefield,Z.ら)により提案されたデカサッカライド(10糖)の値よりも低い最小長を示すという事実から、当該活性はジサッカライド(二糖)のような低いレベルのサッカライドでも期待されるものと想定される。Scholefieldの研究結果と比較して活性レベルの低いことは予想外である。ポリサッカライド(多糖類)とオリゴサッカライド(オリゴ糖)とは活性のレベル(大きさ)が同じではないという証拠があるので、ポリサッカライドをより小さく断片化したものが全長分子と同等に挙動するとは予想できないからである。オクタサッカライドに比べてデカサッカライドに抑制活性のシフト(10倍の増加)が見られ、このことは10個のサッカライドの方がより好ましい最小長であることを示唆している。
【0114】
18個のサッカライド単位(糖単位)においては、当該活性は、最長PIMHと同等である。3000ダルトン(糖単位としては12糖までと同等)と同等なサイズのヘパリン糖類が血液脳関門(BBB)を通過するため、当該データは、インビボ投与に際して有望である。
【0115】
標準的なヘパリンの投与に関して他に起こり得る副作用としては、ヘパリン−血小板第4因子複合体に対する抗体の産生により引き起こされるヘパリン起因性血小板減少症(HIT)があるが、一方で、分子量及び硫酸化レベルを減少させることにより、当該症状を改善させることが示されている。
【0116】
硫酸化レベルを低下されたオリゴ糖(例えば、本発明に従う好ましい化合物(4))、においてBACE−1抵抗活性が維持されることは、脳への潜在的な取込みや抗凝固作用の低下や他の望まれない副作用を改善して薬学的に利用できる大いなる可能性を示している。N−アセチル化されたPIMH(化合物2)やN−アセチル化/Ido−2−OH化されたPIMH(本発明の好ましい化合物(4))は、抗凝固活性がほとんど無い状態で高いBACE−1抑制活性を示した。
従って、本発明に従う好ましい化合物(4)は、ADのような神経変性疾患の予防及び/又は治療にて利用される優れた候補の代表例である。
【0117】
(実施例1の付録(アペンディックス))
(化学修飾されたヘパリンの調製)
化合物(1)から化合物(9)までの化学修飾されたヘパリンは、下記反応(a)から(g)までの以下組み合わせにより調製された。
(1).PIMH出発原料(Celsus Labs,Cincinnati,OH)
(2).N−アセチル化ヘパリン(d)(f)
(3).Ido 2−O脱硫酸化ヘパリン(a)
(4).Ido 2−O脱硫酸化、N−アセチル化ヘパリン(a)(d)(f)
(5).6−O脱硫酸化ヘパリン(b)(e)
(6).6−O脱硫酸化、N−アセチル化ヘパリン(b)(f)
(7).6−O脱硫酸化、2−O脱硫酸化ヘパリン(c)(e)
(8).6−O脱硫酸化、2−O脱硫酸化、N−アセチル化ヘパリン(c)(f)
(9).過硫酸化ヘパリン(g)(e)
化合物は、既知のように1Hや13CのNMRにより特性が決定された。(Yates,E.A.;Santini,F.;Guerrini,M.;Naggi,A.;Torri,G.ら、Carbohydrate Research 1996,294,15-27.)化合物は、分析前に、適切な緩衝剤にて脱塩され、凍結乾燥され、再懸濁された。
【0118】
(化学反応)
(a)イズロン酸の2−O−硫酸基の選択的脱硫酸は、Jaseja及びPerlinに記載のように行った(Jaseja,M.;Rej,R.N.;Sauriol,F;Perlin,A.S.Can.J.Chem.1989,67,1449-1456.)。なお、N−や3−O−硫酸化されたグルコサミン単位の少数にて随伴する化学修飾があることに留意する(Santini,F.;Bisio,A.;Guerrini,M.;Yates,E.A.Carbohydrate Research 1997,302,103-108)。
【0119】
(b)グルコサミンの6−O−硫酸基の選択的除去(脱硫酸)は、既知方法に従い化学修飾することにより(Yates, E. A.他、上記)実施した(Inoue,S.;Miyawaki,M.Analytical Biochemistry 1975,65,164-174.)。
【0120】
(c)O−やN−硫酸基の完全脱硫酸は、既述の方法(Inoue,S.;Miyawaki,M.上記)により、可溶媒分解性の脱硫酸を用いることにより行った。
【0121】
(d)N−硫酸基の選択的脱硫は、既知のように動力学制御下による制御された可溶媒分解性の脱硫酸を用いて実施した(Inoue,Y.;Nagasawa,K.Carbohydrate Research 1976,46,87-95)。
【0122】
(e)N−再硫酸化は、既報に従いトリメチルアミン硫黄三酸化物複合体を用いることにより行った(Lloyd,A.G.;Embery,G.;Fowler,L.j.Biochemical Pharmacology 1971,20,637-648.)。
【0123】
(f)N−再硫酸化は、飽和炭酸水素ナトリウムにおける酸無水物を用いることにより行われる(上記Yates,E.A.ら)。
【0124】
(g)有効なヒドロキシル基(水酸基)の完全なO−硫酸化は、既報に従い、ピリジン中のヘパリンのテトラブチルアンモニウム塩に過剰の三酸化イオウ−ピリジン複合体を用いて(Yates,E.A.;Santini,F.;De Cristofano,B.;Payre,N.;Cosentino,C.ら、Carbohydrate Research 2000,329,239-247.)、次いで、N−再硫酸化する(Lloyd,A.G.他、上記)ことにより行い、この際、異常なN−スルホアジリジンの形成が起こらないよう注意した(Yates,E.A.;Santini,F.;Bisio,A.;Cosentino,C. Carbohydrate Research 1997,298,335-340.)。
【0125】
(NMR分光分析)
化学療法の有効性は、それぞれ500MHz及び125MHzにおける1H及び13CのNMRにより観察した(D2O、27℃)。化学シフト;δ/ppm(外部標準)は、確定したモデル化合物と完全に一致した。(Yates,E.A.他、上記)
【0126】
(オリゴ糖の調製)
豚粘膜ヘパリン及び化学修飾N−アセチル化ヘパリンは、pH7.0にて、100mUヘパラティナーゼII(Ibex Technologies Inc., Montreal, Canada)、100mM当たり100mgの酢酸ナトリウム、0.1mMの酢酸カルシウムを用いて消化分解した。消化分解で得られた断片(フラグメント)は、ゲル濾過クロマトグラフィー(Superdex-30,Amersham Pharmacia,UK,2000mm x 30mm, 100mM 炭酸水素アンモニウム)により分別し、サイズに応じて定義された標準に基づいて同定した。
【0127】
(インビトロでのペプチド分裂分析によるBACE−1抑制の測定)
APPのBACE−1分裂を抑制する化合物の能力は、スウェーデン型アミノ酸変異体を含有するFRETペプチド5-FAM-Glu-Val-Asn-Leu-Asp-Ala-Phe-Lys(QXL520)-OH(Anaspec,Inc.,CA,USA)を用いて蛍光共鳴エネルギー移動(FRET)ペプチド分裂分析を用いて測定された。無調整の場合、アミノ酸末端のフルオロフェア(蛍光体)は、消光するが、酵素分解が発生する場合には、当該フルオロフェアは、消光から解放され、蛍光(520nm)を発する。分析は、96ウェルの黒プレートを用いて3回行った(20mM酢酸ナトリウム、0.1%トリトン−X−100、pH4.5、ウェルごとに2.5μMのペプチド、遺伝子組換えヒトBACE−1(Sigma)の4.0×10-3ユニット/ウェル)。酵素活性及びバックグラウンド蛍光のコントロール(対照)を用いて、プレートを温置した(1h,25℃、2.5M酢酸ナトリウムを用いて停止する)。インヒビターは、1000μg/mlから0.0001μg/mlの範囲の濃度で添加した。
480ex/520nmの蛍光は、Polarstarプレートリーダー(BMG LabTechnologies,UK)にて測定され、データは抑制パーセント比率に対して抑制濃度のlog10対数をプロットし、BioDataFit 1.02(Chang Bioscience,USA)を用いて4パラメータのS字カーブに適合させて分析された。
【0128】
(抗凝固活性)
抗第Xa因子活性は、既知の活性を有するによる豚腸粘膜ヘパリン(PIMH)を標準として(Sigma,UK)測定した。用いたのは、診断グレードのコーティングされたヘパリン試験用キット(Chromogenix,MA,USA)であり、96ウェルの黒プレートを利用し、A405の読み込みを行って測定された(Polarstarプレートリーダー(BMG LabTechnologies,UK))。
【0129】
(他のプロテアーゼに対する活性)
構造的に関連するプロテアーゼペプシン及びカテプシンD(Sigma,UK)を抑制する化合物の能力は、FRET分裂分析(メーカーによる使用法に従い、5 pmol 酵素/ウェル、EnzChekプロテアーゼ分析キット(Molecular Probes,UK)にて)を用いて測定された。ヒトの組み換えレニン(Cayman Chemical,MI,USA)に対する活性は、FRETペプチド分裂分析(メーカーによる使用法に従い、0.08 pmol 酵素/ウェル、Renin Substrate 1(Molecular Probes,UK)にて)を用いて測定された。IC50値は、上記と同様に算出された。
【実施例2】
【0130】
本発明者らは、神経変性症状を治療するための本発明の化合物の効果を示すために、さらなる実験を行った。
なお、特に言及しない限り、実施例1(及びその付録)に使用された方法は、本実施例にても適用されるものとする。
【0131】
(BACEインヒビターとして2−O脱硫酸化されたN−修飾ヘパリン)
2−O脱硫酸化されていると共にN−修飾されたヘパリンの多くの例について、BACEインヒビターとして試験し、親(非修飾)ヘパリンと比較した。これらのポリサッカライド(多糖類)は、N−脱硫酸化によりグルコサミン残基を生成し、その後、遊離アミノ酸残基に様々な化学置換基を置換し、2−O位において適切な脱硫酸化を行うことにより、化学修飾される。
【0132】
2−O脱硫酸化によりN−修飾されたヘパリンは、目的以外の活性の低い有効なBACEインヒビターである。一例として、N−アセチル2−O脱硫酸化PMH(約82%の6−O硫酸基含有率、約0.1%未満の2−O硫酸基含有率)は、非修飾PMHよりもEC50が約3倍低い(表2)。さらなる例としては、N−プロピオン酸2−O脱硫酸化PMH(約82%の6−O硫酸基含有率、約0.1%未満の2−O硫酸基含有率)があり、当該PMHは、非修飾PMHよりもEC50が約8倍低いにも関わらず、無処理のヘパリンに比べて、約1/100の抗第Xa因子活性を有しており、これらの化合物が構造変化(この場合、N−置換)に対して予期し得ない程敏感であることが特徴である。さらに、上記N−修飾されたヘパリンは、いずれも、非常に弱い抗凝固活性を示した(非修飾PMHと比較して1/100から1/3000)。
【0133】
【表2】
【0134】
(アミロイド−βペプチドアグリゲーションに対する化学修飾ヘパリンの効果)
抗凝固活性に加えて、神経変性疾患の治療に直接関連するヘパリンの生じ得る副作用としては、広く知られているように、アミロイドβ(Aβ)ペプチドのアグリゲーション(凝集)がある。(Watson,D.J.,Lander,A.D. and Selkoe,D.J.(1997)Heparin-binding properties of the amyloidogenic peptides Aβ and amylin. Dependence on aggregation state and inhibition by Congo red. J Biol Chem,272,31617-24)当該アグリゲーションを低減することは望ましい特性である。従って、本発明者らは、Aβアグリゲーションを分析して非修飾の豚粘膜ヘパリンとの比較を行い、化学修飾されたヘパリンの活性を調べた。当該試験の結果は、図2に記載している。
【0135】
当該アグリゲーションの最大化は、非修飾ヘパリン“PMH”において観察される。ヘパリンを脱硫酸化することにより当該アグリゲーションが驚くほど低減することが観察された。幾つかの化合物(例えば、N−アセチル化ヘパリン“NAc”やido-2-脱硫酸化されたN−アセチル化ヘパリン“NAc de 2”)は、望ましいBACE−1抑制活性を有しており、好ましい抗凝固活性(抗第Xa因子活性)(Patey et al(2006)49 6129-6132)はまた、Aβアグリゲーションを改善する(それぞれ50%及び100%遅くする)。すなわち、100μg/mlの濃度において、N−アセチル化または0−脱硫酸化PMHのいずれかは、ペプチドアグリゲーションが最大となる半分の時間に50%の増加となり、N−アセチル化や2-de−O硫酸化されたPMHは、100%の増加となり、ほぼ対照(コントロール)レベル(すなわち、ヘパリンを添加しない場合のアグリゲーション)に達する。このように、本発明の請求項1に定義された好ましい化学修飾は、ヘパリンのAβアグリゲーションの促進力を著しく低減させ、結果的に、非修飾ヘパリンと比較して改善された治療可能比率をもたらす。
【0136】
(Aβアグリゲーション分析手順)
Aβペプチドは、1mlのヘキサフルオロ−2−プロパノールを1mlのAβペプチド(アミロイドβプロテイン断片1−42;Sigma A9810)に室温で1時間添加して調製した。生成物は、50μl(50μg)のアリコートに分割され、約2時間Speed Vacにて乾燥され、20℃で保管された。Thioflavin T(Sigma T-3516)溶液は、50mM Glycine-NaOH (pH8.5)中の5mM溶液として調製された。20μlのThioflavin T溶液に加えて50μlの1MのDDTと430μlの分析用緩衝液(10mM HEPES/100mM NaCl Ph7.4)とを含むThioflavin T分析緩衝液を氷上保持した。20mM NaOHの50μlをAβペプチドの50μgに付加した。その後、Thioflavin T分析緩衝液の2.5mlを当該Aβペプチド溶液に付加し、“Aβ緩衝液”を生成した。Thioflavin T分析緩衝液の残りの2.5mlは、ブランク緩衝液であり、共に氷上維持した。6つのサンプル希釈剤(分析緩衝液にて1:1)を96ウェルの黒プレートにピペット注入(Greiner 655076;100μl/ウェル)し、当該プレートを氷上維持した。96ウェルの黒プレートの読み取りを行い(450nmに励起波長があり、490nmに発光波長がある)、37℃にて150秒ごとの読み取りを250サイクル行った。
【産業上の利用可能性】
【0137】
本薬剤は、アルツハイマー病(AD)の予防及び/又は治療に用いることができる。
【特許請求の範囲】
【請求項1】
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々が、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物であって、神経変性疾患の予防及び/又は治療に用いられる
化合物。
【請求項2】
請求項1に記載の化合物において、
少なくとも約60%のウロン酸部位の2−O原子が、水素原子と置換されている
化合物。
【請求項3】
請求項1に記載の化合物において、
すべての2−Oウロン酸原子が、水素原子と置換されている
化合物。
【請求項4】
請求項1〜請求項3のいずれかに記載の化合物において、
少なくとも約60%のグルコサミン部位の6−O原子が、硫酸基と置換されている
化合物。
【請求項5】
請求項1〜請求項3のいずれかに記載の化合物において、
すべてのグルコサミン部位の6−O原子が、硫酸基と置換されている
化合物。
【請求項6】
請求項1〜請求項5のいずれかに記載の化合物において、
ウロン酸部位が、イズロン酸部位、グルクロン酸部位及びガラクツロン酸部位から成る群の中から選択されている
化合物。
【請求項7】
請求項1〜請求項6のいずれかに記載の化合物において、
グルコサミン部位の2−N原子が、水素原子、置換又は非置換のアルキル基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、及びリン酸基により成る群の中から選択された置換基により置換されている
化合物。
【請求項8】
請求項7に記載の化合物において、
置換又は非置換のアシル基が、置換又は非置換のC1-6アシル基である
化合物。
【請求項9】
請求項7に記載の化合物において、
置換又は非置換のアシル基が、置換又は非置換のアセチル基、置換又は非置換のプロプリオニル基及び置換又は非置換のブタノイル基から成る群から選択されている
化合物。
【請求項10】
請求項1〜請求項9のいずれかに記載の化合物において、
4から24までのモノサッカライド単位から成る
化合物。
【請求項11】
請求項1〜請求項9のいずれかに記載の化合物において、
6から20までのモノサッカライド単位から成る
化合物。
【請求項12】
請求項1〜請求項9のいずれかに記載の化合物において、
6から18までのモノサッカライド単位から成る
化合物。
【請求項13】
請求項1〜請求項9のいずれかに記載の化合物において、
6から16までのモノサッカライド単位から成る
化合物。
【請求項14】
請求項1に記載の化合物において、
下記の式(I)にて示され、式(I)中、実質的に全てのR1基は水素原子であり、実質的に全てのR2が硫酸基であり、実質的に全てのR3が硫酸基以外の原子又は置換基であり、nは1以上の整数であり、R4、R5及びR6は水素原子、置換又は非置換のアルキル基、置換又は非置換のアルコキシ基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、硫酸基及びリン酸基により成る群の中からそれぞれ独立して選択され、XとYは水素、末端モノサッカライド基、末端ジサッカライド基及び/又はそれらのフラグメント若しくはそれらの誘導体より成る群の中からそれぞれ独立して選択されている
化合物。
【化1】
【請求項15】
請求項14に記載の化合物において、
R3基が、水素原子、置換又は非置換のアルキル基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、リン酸基により成る群の中から選択されている
化合物。
【請求項16】
請求項15に記載の化合物において、
置換又は非置換のアシル基が、置換又は非置換のC1-6アシル基である
化合物。
【請求項17】
請求項15に記載の化合物において、
置換又は非置換のアシル基が、置換又は非置換のアセチル基、置換又は非置換のプロプリオニル基及び置換又は非置換のブタノイル基から成る群から選択されている
化合物。
【請求項18】
請求項14〜請求項17のいずれかに記載の化合物において、
R5基及びR6基が、水素原子である
化合物。
【請求項19】
請求項14〜請求項18のいずれかに記載の化合物において、
R4基が、水素原子である
化合物。
【請求項20】
請求項14〜請求項19のいずれかに記載の化合物において、
nが、2から12である
化合物。
【請求項21】
請求項1に記載の化合物において、
下記の式(III)にて示され、式(III)中、nは1以上の整数であり、XとYは水素、末端モノサッカライド基、末端ジサッカライド基及び/又はそれらのフラグメント若しくはそれらの誘導体より成る群の中からそれぞれ独立して選択されている
化合物。
【化2】
【請求項22】
請求項21に記載の化合物において、
nが、2から12である
化合物。
【請求項23】
請求項21に記載の化合物において、
nが、3から8である
化合物。
【請求項24】
請求項1〜請求項23のいずれかに記載の化合物において、
神経変性疾患が、アルツハイマー病である
化合物。
【請求項25】
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々が、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物の使用であって、神経変性疾患の予防及び/又は治療用の薬剤の製造における
使用。
【請求項26】
請求項25に記載の化合物の使用において、
化合物が、請求項2〜請求項23のいずれかに記載の化合物である
化合物の使用。
【請求項27】
請求項25又は請求項26に記載の化合物の使用において、
神経変性疾患が、アルツハイマー病である
化合物の使用。
【請求項28】
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々が、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物を患者に有効量投与することを含む神経変性疾患を予防及び/又は治療する
方法。
【請求項29】
請求項28に記載の方法において、
化合物が、請求項2〜請求項23のいずれかに記載の化合物である
方法。
【請求項30】
請求項28又は請求項29に記載の方法において、
神経変性疾患が、アルツハイマー病である
方法。
【請求項31】
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物を含み、神経変性疾患の予防及び/又は治療に用いられる
薬剤組成物。
【請求項32】
請求項31に記載の薬剤組成物において、
化合物が、請求項2〜請求項23のいずれかに記載の化合物である
薬剤組成物。
【請求項33】
請求項31又は請求項32に記載の薬剤組成物において、
神経変性疾患が、アルツハイマー病である
薬剤組成物。
【請求項34】
請求項1〜請求項24のいずれかに記載の化合物において、
1若しくは複数の合成原料、又は、1若しくは複数の天然原料から生成されている
化合物。
【請求項35】
請求項1〜請求項24のいずれかに記載の化合物において、
化合物の生産が、亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、フリーラジカル処理及びこれらの組み合わせにより成る群の中から選択されている解重合工程を含む
化合物。
【請求項36】
請求項1〜請求項24のいずれかに記載の化合物の生産方法において、
1若しくは複数の合成原料、又は、1若しくは複数の天然原料を用いる
生産方法。
【請求項37】
請求項1〜請求項24のいずれかに記載の化合物の生産方法において、
亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、フリーラジカル処理及びこれらの組み合わせにより成る群の中から選択される解重合工程を含む
生産方法。
【請求項1】
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々が、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物であって、神経変性疾患の予防及び/又は治療に用いられる
化合物。
【請求項2】
請求項1に記載の化合物において、
少なくとも約60%のウロン酸部位の2−O原子が、水素原子と置換されている
化合物。
【請求項3】
請求項1に記載の化合物において、
すべての2−Oウロン酸原子が、水素原子と置換されている
化合物。
【請求項4】
請求項1〜請求項3のいずれかに記載の化合物において、
少なくとも約60%のグルコサミン部位の6−O原子が、硫酸基と置換されている
化合物。
【請求項5】
請求項1〜請求項3のいずれかに記載の化合物において、
すべてのグルコサミン部位の6−O原子が、硫酸基と置換されている
化合物。
【請求項6】
請求項1〜請求項5のいずれかに記載の化合物において、
ウロン酸部位が、イズロン酸部位、グルクロン酸部位及びガラクツロン酸部位から成る群の中から選択されている
化合物。
【請求項7】
請求項1〜請求項6のいずれかに記載の化合物において、
グルコサミン部位の2−N原子が、水素原子、置換又は非置換のアルキル基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、及びリン酸基により成る群の中から選択された置換基により置換されている
化合物。
【請求項8】
請求項7に記載の化合物において、
置換又は非置換のアシル基が、置換又は非置換のC1-6アシル基である
化合物。
【請求項9】
請求項7に記載の化合物において、
置換又は非置換のアシル基が、置換又は非置換のアセチル基、置換又は非置換のプロプリオニル基及び置換又は非置換のブタノイル基から成る群から選択されている
化合物。
【請求項10】
請求項1〜請求項9のいずれかに記載の化合物において、
4から24までのモノサッカライド単位から成る
化合物。
【請求項11】
請求項1〜請求項9のいずれかに記載の化合物において、
6から20までのモノサッカライド単位から成る
化合物。
【請求項12】
請求項1〜請求項9のいずれかに記載の化合物において、
6から18までのモノサッカライド単位から成る
化合物。
【請求項13】
請求項1〜請求項9のいずれかに記載の化合物において、
6から16までのモノサッカライド単位から成る
化合物。
【請求項14】
請求項1に記載の化合物において、
下記の式(I)にて示され、式(I)中、実質的に全てのR1基は水素原子であり、実質的に全てのR2が硫酸基であり、実質的に全てのR3が硫酸基以外の原子又は置換基であり、nは1以上の整数であり、R4、R5及びR6は水素原子、置換又は非置換のアルキル基、置換又は非置換のアルコキシ基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、硫酸基及びリン酸基により成る群の中からそれぞれ独立して選択され、XとYは水素、末端モノサッカライド基、末端ジサッカライド基及び/又はそれらのフラグメント若しくはそれらの誘導体より成る群の中からそれぞれ独立して選択されている
化合物。
【化1】
【請求項15】
請求項14に記載の化合物において、
R3基が、水素原子、置換又は非置換のアルキル基、置換又は非置換のアリール基、置換又は非置換のアシル基、置換又は非置換のアミド基、リン酸基により成る群の中から選択されている
化合物。
【請求項16】
請求項15に記載の化合物において、
置換又は非置換のアシル基が、置換又は非置換のC1-6アシル基である
化合物。
【請求項17】
請求項15に記載の化合物において、
置換又は非置換のアシル基が、置換又は非置換のアセチル基、置換又は非置換のプロプリオニル基及び置換又は非置換のブタノイル基から成る群から選択されている
化合物。
【請求項18】
請求項14〜請求項17のいずれかに記載の化合物において、
R5基及びR6基が、水素原子である
化合物。
【請求項19】
請求項14〜請求項18のいずれかに記載の化合物において、
R4基が、水素原子である
化合物。
【請求項20】
請求項14〜請求項19のいずれかに記載の化合物において、
nが、2から12である
化合物。
【請求項21】
請求項1に記載の化合物において、
下記の式(III)にて示され、式(III)中、nは1以上の整数であり、XとYは水素、末端モノサッカライド基、末端ジサッカライド基及び/又はそれらのフラグメント若しくはそれらの誘導体より成る群の中からそれぞれ独立して選択されている
化合物。
【化2】
【請求項22】
請求項21に記載の化合物において、
nが、2から12である
化合物。
【請求項23】
請求項21に記載の化合物において、
nが、3から8である
化合物。
【請求項24】
請求項1〜請求項23のいずれかに記載の化合物において、
神経変性疾患が、アルツハイマー病である
化合物。
【請求項25】
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々が、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物の使用であって、神経変性疾患の予防及び/又は治療用の薬剤の製造における
使用。
【請求項26】
請求項25に記載の化合物の使用において、
化合物が、請求項2〜請求項23のいずれかに記載の化合物である
化合物の使用。
【請求項27】
請求項25又は請求項26に記載の化合物の使用において、
神経変性疾患が、アルツハイマー病である
化合物の使用。
【請求項28】
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々が、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物を患者に有効量投与することを含む神経変性疾患を予防及び/又は治療する
方法。
【請求項29】
請求項28に記載の方法において、
化合物が、請求項2〜請求項23のいずれかに記載の化合物である
方法。
【請求項30】
請求項28又は請求項29に記載の方法において、
神経変性疾患が、アルツハイマー病である
方法。
【請求項31】
1以上のジサッカライド単位から成り、当該ジサッカライド単位の各々は、グルコサミン部位と結合したウロン酸部位で構成され、ウロン酸部位の2−O原子が実質的に水素原子と置換され、グルコサミン部位の6−O原子が実質的に硫酸基と置換され、グルコサミン部位の2−N原子が実質的に硫酸基以外の原子又は置換基と置換された化合物を含み、神経変性疾患の予防及び/又は治療に用いられる
薬剤組成物。
【請求項32】
請求項31に記載の薬剤組成物において、
化合物が、請求項2〜請求項23のいずれかに記載の化合物である
薬剤組成物。
【請求項33】
請求項31又は請求項32に記載の薬剤組成物において、
神経変性疾患が、アルツハイマー病である
薬剤組成物。
【請求項34】
請求項1〜請求項24のいずれかに記載の化合物において、
1若しくは複数の合成原料、又は、1若しくは複数の天然原料から生成されている
化合物。
【請求項35】
請求項1〜請求項24のいずれかに記載の化合物において、
化合物の生産が、亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、フリーラジカル処理及びこれらの組み合わせにより成る群の中から選択されている解重合工程を含む
化合物。
【請求項36】
請求項1〜請求項24のいずれかに記載の化合物の生産方法において、
1若しくは複数の合成原料、又は、1若しくは複数の天然原料を用いる
生産方法。
【請求項37】
請求項1〜請求項24のいずれかに記載の化合物の生産方法において、
亜硝酸開裂、細菌リアーゼ酵素処理、過ヨウ素酸酸化、アルカリ条件下の化学的ベータ脱離、フリーラジカル処理及びこれらの組み合わせにより成る群の中から選択される解重合工程を含む
生産方法。
【図1】

【図2】
【図2】
【公表番号】特表2009−537622(P2009−537622A)
【公表日】平成21年10月29日(2009.10.29)
【国際特許分類】
【出願番号】特願2009−511571(P2009−511571)
【出願日】平成19年5月23日(2007.5.23)
【国際出願番号】PCT/GB2007/001895
【国際公開番号】WO2007/138263
【国際公開日】平成19年12月6日(2007.12.6)
【出願人】(508346000)ザ ユニバーシティ オブ リバプール (5)
【氏名又は名称原語表記】THE UNIVERSITY OF LIVERPOOL
【Fターム(参考)】
【公表日】平成21年10月29日(2009.10.29)
【国際特許分類】
【出願日】平成19年5月23日(2007.5.23)
【国際出願番号】PCT/GB2007/001895
【国際公開番号】WO2007/138263
【国際公開日】平成19年12月6日(2007.12.6)
【出願人】(508346000)ザ ユニバーシティ オブ リバプール (5)
【氏名又は名称原語表記】THE UNIVERSITY OF LIVERPOOL
【Fターム(参考)】
[ Back to top ]