
神経変性疾患治療薬のスクリーニング方法
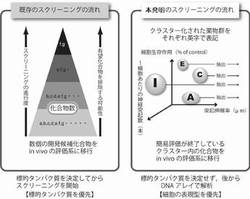
【課題】複雑な作用メカニズムが関与すると考えられる神経変性疾患治療薬の開発には、より膨大なスクリーニング試験が必要と考えられるが、本発明の方法は、この創薬過程における膨大なスクリーニング工程を効率化するものである。
【解決手段】神経モデル細胞(例えばPC12細胞)の生存維持作用、並びに当該細胞に対する神経突起伸展作用及び/又は神経突起数増加作用を指標としてヒストンの修飾又はDNAの修飾に関与する酵素の阻害剤をクラスター化した後、クラスター毎に評価および/またはクラスター毎に解析する工程を含む神経変性疾患治療薬のスクリーニング方法を提供する。ターゲット分子を特定する必要がない表現型による本発明のスクリーニング方法は、実験動物を用いたin vivoのスクリーニング系への移行を飛躍的に早期化し、その結果、創薬化の効率を高めるものである。
【解決手段】神経モデル細胞(例えばPC12細胞)の生存維持作用、並びに当該細胞に対する神経突起伸展作用及び/又は神経突起数増加作用を指標としてヒストンの修飾又はDNAの修飾に関与する酵素の阻害剤をクラスター化した後、クラスター毎に評価および/またはクラスター毎に解析する工程を含む神経変性疾患治療薬のスクリーニング方法を提供する。ターゲット分子を特定する必要がない表現型による本発明のスクリーニング方法は、実験動物を用いたin vivoのスクリーニング系への移行を飛躍的に早期化し、その結果、創薬化の効率を高めるものである。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、神経変性疾患治療薬のスクリーニング方法に関する。
【背景技術】
【0002】
現在、製薬企業を主体とする医薬品の開発では、病気の原因となる分子を先ず見出し、何十万検体から分子、細胞、実験動物を用いたスクリーニング(企業によっては、大規模なハイスループットスクリーニング(HTS))を経て薬効を有する化合物(リード化合物)を抽出した後、化合物の更なる化学修飾・解析を行うことによってヒトでの有効性を高めた有望化合物を得て、その後、臨床試験(治験)を行い、ヒトへの有効性を確認している。
【0003】
脳精神神経疾患治療薬(アルツハイマー病、パーキンソン病、ハンチントン病などを適応症とする)においては、神経細胞を死滅させない役割を担う分子や逆に神経細胞を死滅させる役割を担う分子をターゲット分子として前臨床試験や臨床試験が行われてきた。
【0004】
アルツハイマー病に対しては、コリン作動性神経細胞の活動に必須であるアセチルコリンのシナプス内での濃度維持によるコリン作動性神経細胞の活動維持と生存維持を目的として、アセチルコリン分解酵素をターゲット分子とした阻害剤が、既に上市されている。しかし、生存維持が可能な期間が限られており、完治を可能とする医薬品ではない。
【0005】
また、パーキンソン病では、黒質ドーパミン作動性神経細胞の活動維持と生存維持を目的として、ドーパミン受容体をターゲット分子としたプロドラッグ(ドーパミン前駆分子)であるL-DOPAが上市されている。
また、ドーパミン受容体作動薬、ドーパミン発現上昇薬などのドーパミン作動性神経細胞をターゲットとした医薬品も上市されている。
【0006】
ハンチントン病は、この疾患が環境要因から引き起こされる孤発性疾患ではなく、完全にヒトが持つゲノム中の特定遺伝子(huntintin)のポリグルタミン鎖の伸長度に依存する遺伝性疾患であり、huntintinのポリグルタミン伸長鎖を変える様な薬物療法は、現在のところ存在しない。
【0007】
癲癇に関しては、神経系の興奮伝播につながるナトリウムチャネルの不活性状態を維持する薬剤、カルシウムチャネルを不活性化する薬剤、抑制性神経細胞の神経伝達物質であるGABAの作用を増強する薬剤が上市されている。バルプロ酸(VPA)は、ヒストン脱アセチル化酵素(HDAC)阻害作用を有しているが、ヒストン脱アセチル化酵素(HDAC)阻害剤として開発された医薬品ではなく、脳神経細胞の抑制性の神経伝達物質であるGABAの産生量を減少させないために、GABAトランスアミノブチラーゼ阻害剤として上市された。
【0008】
ヒストン (histone) は、真核生物のクロマチン(染色体)を構成するタンパク質の一群で、非常に長い分子であるDNA を核内に収納する役割を担っている。
ヒストンは強い塩基性のタンパク質であり、酸性のDNAとの高い親和性を示す。ヒストンが塩基性を持つのは、正の電荷を持つアミノ酸の含量が高いからである。各ヒストンを構成するアミノ酸のうち、20%以上がリシンまたはアルギニンである。ヌクレオソームヒストンの構造は球形のカルボキシル末端と、直鎖状のアミノ末端(ヒストンテイル)からなっている。ヒストンテイルのリシンやアスパラギン残基はアセチル化、メチル化、リン酸化、ユビキチン化といった化学修飾を受けることが知られている。これらの化学修飾は、遺伝子発現等、数々のクロマチン機能の制御に関わっていることが証明されつつある。
【0009】
ヒストン脱アセチル化酵素(以下、HDACという。)は、ヌクレオソーム中のヒストンタンパク質の、所謂、ヒストンテイルの特定リシン残基のアセチル基を脱アセチル化させることを触媒するタンパク質である。このタンパク質の作用により、八量体から成るヒストンとそれに巻き付いているDNA鎖に存在している僅かな空間が閉鎖され、転写を抑制することが知られている。HDAC阻害剤は、このHDACの作用を阻害することから、転写活性が上昇し、各種の遺伝子群を発現誘導する。その結果として、細胞が様々な形質変化を起こし、特定の表現型を獲得する。HDAC阻害剤は、癌治療薬としての開発が進んでいる。本発明者等も、多数のHDAC阻害剤を合成し、抗癌効果について検討した(特許文献1、非特許文献1)。
【0010】
脳精神疾患では、多くの先行医薬品が上市されているが、未だヒストンの修飾に関与する酵素阻害剤は上市されておらず、また、HDAC阻害剤としての治療薬は上市されていない。HDAC阻害剤の脳精神神経系疾患治療薬としての開発は進行している。しかし、本発明に見られるターゲット分子を特定する必要がない新規スクリーニング系を用いた創薬方法は存在しない。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特開2007−1885号公報
【非特許文献】
【0012】
【非特許文献1】Bioorganic and Medical Chemistry 18, 3925-3933,2010
【発明の概要】
【発明が解決しようとする課題】
【0013】
神経変性疾患には数多くの蛋白質が関わっていると考えられ、ターゲット分子に対する作用を評価するスクリーニング方法では、膨大なスクリーニング試験が必要となる。また、ヒストンの修飾に関与する酵素阻害剤およびDNAの修飾に関与する酵素阻害剤は細胞に様々な形質変化を起こし、特定の表現型を獲得し、この表現型獲得には複数の遺伝子が関わっていると考えられる。
このような、複雑な作用メカニズムが関与すると考えられる神経変性疾患治療剤の開発には、より膨大なスクリーニング試験が必要と考えられ、本発明の方法は、この創薬過程における膨大なスクリーニング工程を効率化するものである。
【課題を解決するための手段】
【0014】
すなわち、本発明は以下からなる。
1.神経モデル細胞の生存維持作用、並びに神経モデル細胞に対する神経突起伸展作用及び/又は神経突起数増加作用を指標としてヒストンの修飾又はDNAの修飾に関与する酵素の阻害剤をクラスター化した後、クラスター毎に評価および/またはクラスター毎に解析する工程を含む神経変性疾患治療薬のスクリーニング方法。
2.修飾がアセチル化、メチル化、リン酸化、又はユビキチン化である前項1に記載のスクリーニング方法。
3.神経モデル細胞がPC12細胞である前項1または2に記載のスクリーニング方法。
4.クラスター毎に解析する工程がDNAマイクロアレイを用いた遺伝子発現の解析工程である前項1〜3のいずれか1に記載のスクリーニング方法。
【発明の効果】
【0015】
本発明の、ターゲット分子を特定する必要がない表現型による新規スクリーニング方法は、複数のタンパク質(遺伝子産物群)による発現変動が関与する神経変性疾患の治療薬のスクリーニングにおいて大幅に効率を上昇させるものである。さらに本発明は、ヒストンの修飾又はDNAの修飾に関与する酵素阻害剤を、細胞が有する機能を指標としてクラスター化することによるスクリーニング方法であり、同一の表現型を発現する同一のクラスターに属する物質は、その表現型を獲得するための発現変動が類似している遺伝子群に関与すると考えられ、クラスター毎に治療効果を評価することができ、同一のクラスターに属する物質を、それ以降のスクリーニングから排除することが出来る。
【0016】
このように本発明は、上記操作により、スクリーニング効率を飛躍的に上昇させるものである。すなわち、治療効果の評価系において低い評価しか得られない場合には、同じクラスターからではなく、恐らく異なる遺伝子発現群に関与するクラスターからの化合物を選定することができる。また、その別のクラスターから1個、または、複数個を選定する事により、創薬化の過程で必要な同一の基本骨格を有する複数の類縁化合物の新規合成(化合物の最適化)に見られる様な無駄な化学修飾処理も排することができることから効率よく創薬化することができる。それは、本発明の基本となる複数遺伝子の発現変動を有する化合物におけるスクリーニングにおいて、クラスター化された化合物群は、治療を可能とする指標である細胞生存率上昇作用、神経突起伸展作用、神経突起数増加作用の評価を既に終えた化合物群から構成されていることやリード化合物から化学基を結合させた僅かな構造変化を伴う類縁化合物でも遺伝子の発現群が異なってくることから、表現型の評価段階で化合物の有用性が想定し得るからである。この事実は、本発明の実施例にも記載のあるバルプロ酸が、他の化合物が有する表現型の評価の値を凌駕し、既に上市されていることからも明らかである。
さらに、分子生物学的な作用機序の解析を早期に終了させることができる。
また、既存のスクリーニングに見られる様なターゲット分子を選定しないため、これまでに低い評価で候補医薬品として対象外になっていた化合物からの再スクリーニングにより有効な医薬品を開発できる可能性が広がると考えられる。
【図面の簡単な説明】
【0017】
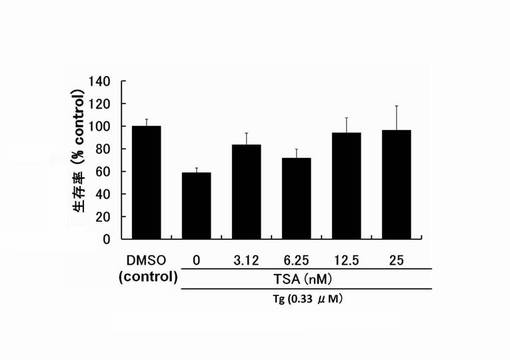
【図1】サプシガルギン(Tg)により細胞死誘導されたPC12細胞へのトリコスタチンA(以下TSAという。)の生存維持作用を示したグラフである。
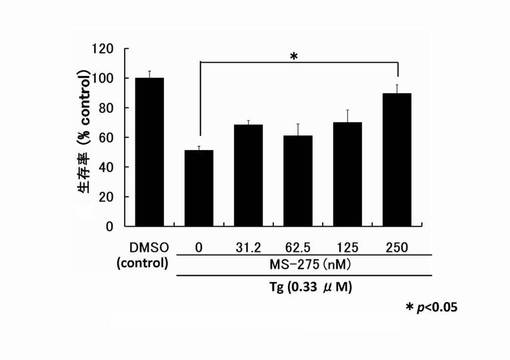
【図2】サプシガルギン(Tg)により細胞死誘導されたPC12細胞へのMS-275の生存維持作用を示したグラフである。
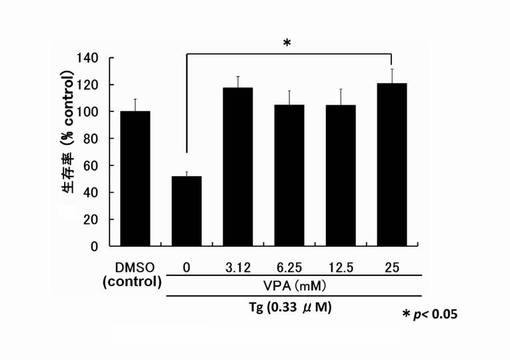
【図3】サプシガルギン(Tg)により細胞死誘導されたPC12細胞へのバルプロ酸(以下VPAという。)の生存維持作用を示したグラフである。
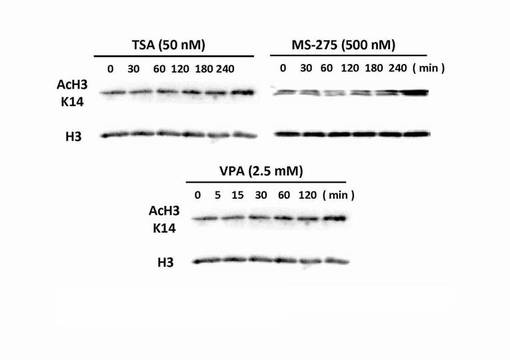
【図4】TSA、MS-275およびVPAで処理されたPC12細胞のヒストンH3(K14)のアセチル化を示した電気泳動(SDS-PAGE)後のウエスタンブロットによるヒストンH3のアセチル化の亢進を確認した写真である。
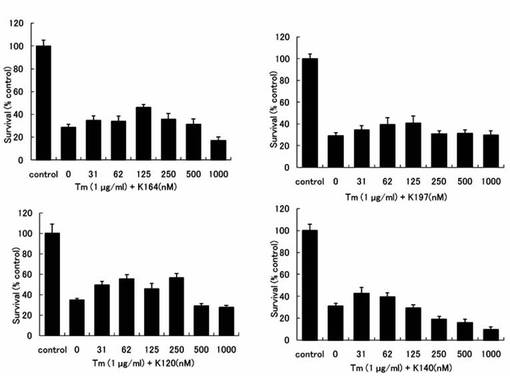
【図5】ツニカマイシン(Tm)により細胞死誘導されたPC12細胞へのK164、K197、K120およびK140の生存維持作用を示したグラフである。
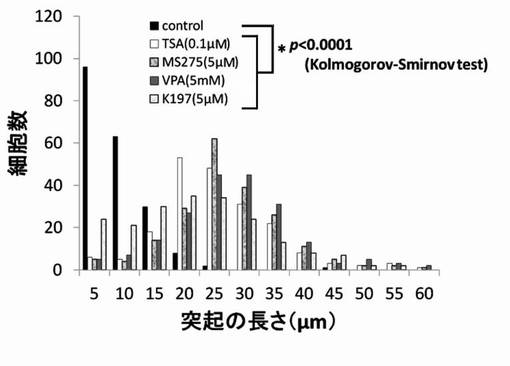
【図6】TSA、MS-275、VPAおよびK197のPC12細胞への神経突起伸展作用を示したグラフである。
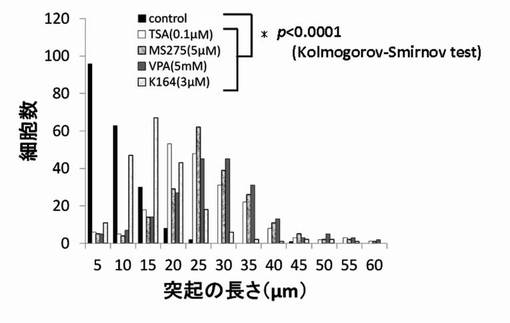
【図7】TSA、MS-275、VPAおよびK164のPC12細胞への神経突起伸展作用を示したグラフである。
【図8】TSA、MS-275、VPAおよびK120のPC12細胞への神経突起伸展作用を示したグラフである。
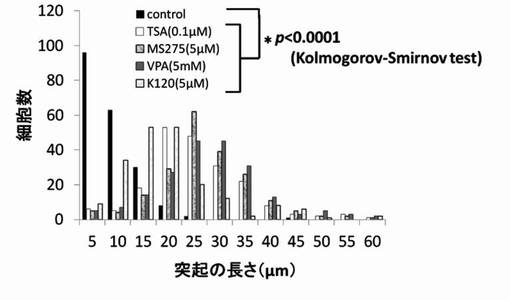
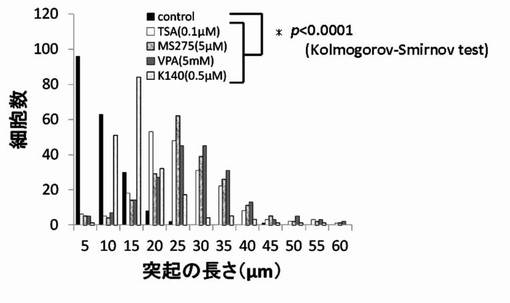
【図9】TSA、MS-275、VPAおよびK140のPC12細胞への神経突起伸展作用を示したグラフである。
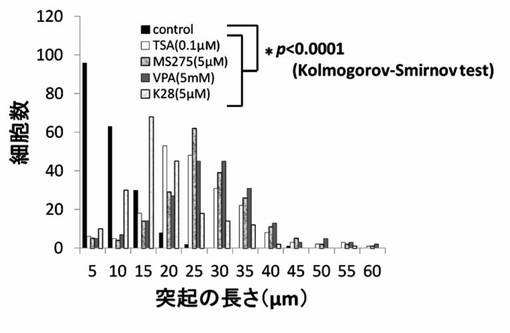
【図10】TSA、MS-275、VPAおよびK28のPC12細胞への神経突起展作用を示したグラフである。
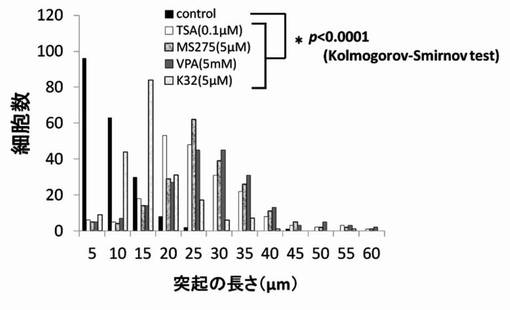
【図11】TSA、MS-275、VPAおよびK32のPC12細胞への神経突起伸展作用を示したグラフである。
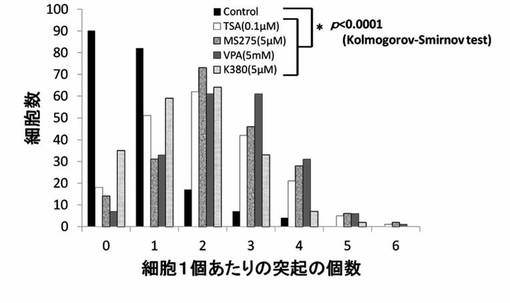
【図12】TSA、MS-275、VPAおよびK380のPC12細胞への神経突起伸展作用を示したグラフである。
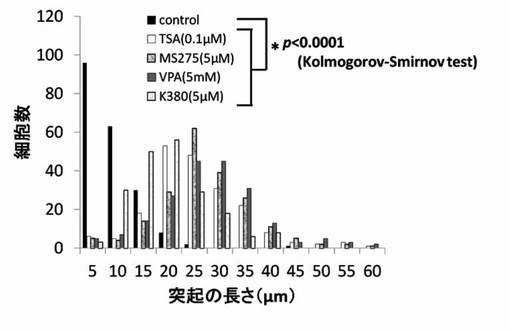
【図13】TSA、MS-275、VPAおよびK197のPC12細胞への神経突起数増加作用を示したグラフである。
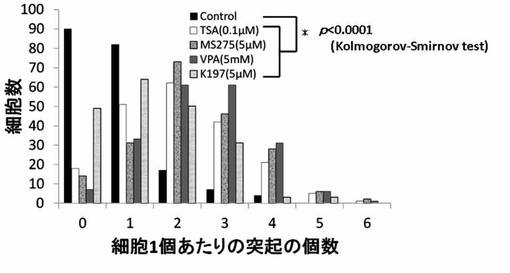
【図14】TSA、MS-275、VPAおよびK164のPC12細胞への神経突起数増加作用を示したグラフである。
【図15】TSA、MS-275、VPAおよびK120のPC12細胞への神経突起数増加作用を示したグラフである。
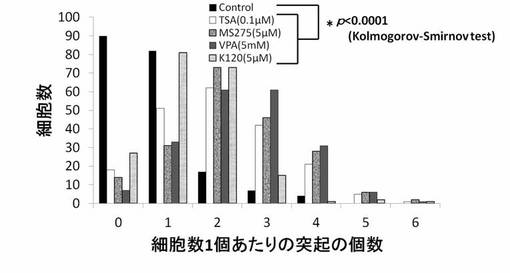
【図16】TSA、MS-275、VPAおよびK140のPC12細胞への神経突起数増加作用を示したグラフである。
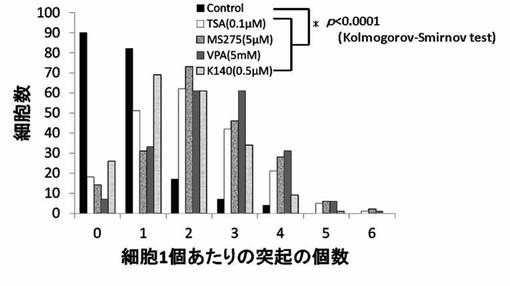
【図17】TSA、MS-275、VPAおよびK28のPC12細胞への神経突起数増加作用を示したグラフである。
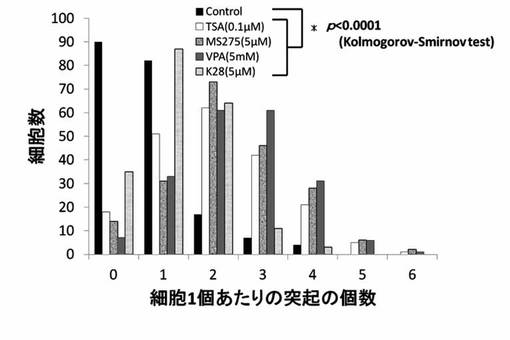
【図18】TSA、MS-275、VPAおよびK32のPC12細胞への神経突起数増加作用を示したグラフである。
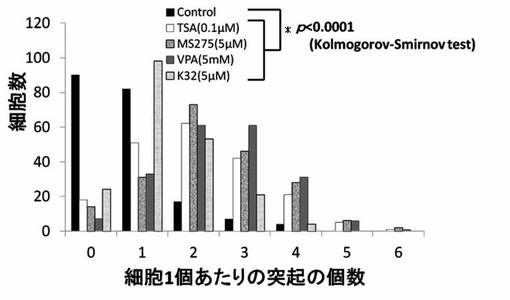
【図19】TSA、MS-275、VPAおよびK380のPC12細胞への神経突起数増加作用を示したグラフである。
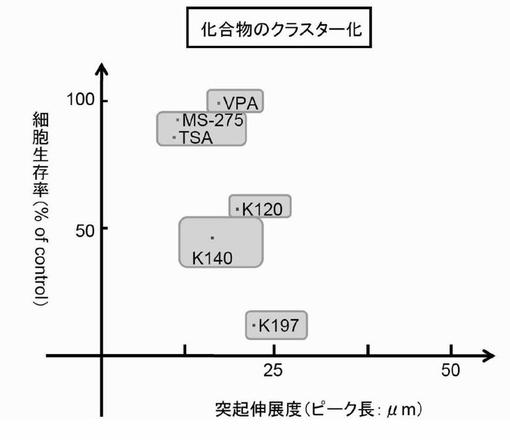
【図20】HDAC阻害剤のクラスター化を示したグラフである。
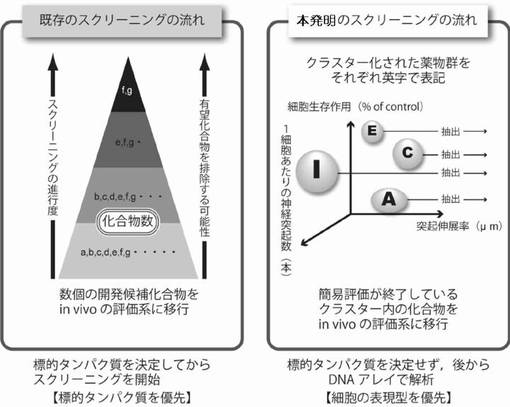
【図21】本発明の3次元のクラスター化によるスクリーニング方法の概念図を示した説明図である。
【発明を実施するための形態】
【0018】
本発明における神経変性疾患とは、中枢神経系の神経細胞のプログラム細胞死が原因とされている疾患であり、例えば、アルツハイマー病、パーキンソン病、ハンチントン病等が挙げられる。
【0019】
本発明のヒストンの修飾又はDNAの修飾に関与する酵素阻害剤(以下、修飾酵素阻害剤という。)とは、ヒストン又はDNAの化学修飾を触媒する酵素の阻害剤および脱化学修飾を触媒する酵素の阻害剤を含む。
【0020】
ヒストンは、アセチル化、メチル化、リン酸化、ユビキチン化といった化学修飾を受けるタンパク質であり、それに関与する酵素とはこれらの化学修飾を触媒する酵素または脱化学修飾を触媒する酵素である。また、ここにおける酵素は、ヒストン以外の修飾や脱修飾により、遺伝子の発現を変動させる場合がある。これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
【0021】
ヒストンのアルギニン残基、リシン残基のメチル化はゲノムの活性化、不活性化の主たる決定要素となっている。メチル化にはメチラーゼやメチルトランスフェラーゼ、脱メチル化にはデメチラーゼ等の酵素が関与している。これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
【0022】
アセチル化には、ヒストンアセチルトランスフェラーゼ、脱アセチル化にはヒストン脱アセチル化酵素(HDAC)が関与している。これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
ヒストン脱アセチル化酵素(以下、HDACという。)には複数のタイプが存在し、多くのタンパク質との複合体の状態で働く。現在、HDACは18種類のタンパク質が知られており、4つのグループに分類されている。
クラスI HDAC1, HDAC2, HDAC3, HDAC8
クラスII HDAC4, HDAC5, HDAC6, HDAC7 HDAC9, HDAC10
クラスIII SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, SIRT7
クラスIV HDAC11
【0023】
本発明で用いるHDAC阻害剤とは、HDACの活性を阻害する物質であれば何ら限定されない。低分子化合物、ポリマー、ペプチドなどを含む。
現在すでに多くのHDAC阻害剤が知られている。例えば次の4種類が挙げられる。
a)脂肪酸(Butyrate,phenyl butyrate,valproic acid(VPA)など)
b)ヒドロキサム酸(Trichostatin A (TSA),oxamflatin,suberolyanilide hydroxamic acid(SAHA)など)
c)環状ペプチド(Trapoxin (TPX),apicidin,FK228など)
d)ベンズアミド(MS-275,MGCD0103,CI-994など)
【0024】
また、リン酸化による遺伝子発現制御機構も知られており、これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
修飾酵素としてリン酸化に関与する酵素としては、mitogen- and stress-activated kinases (MSK)などがあり、ヒストンH3内のアミノ酸27残基目のリシンをトリメチル化する事と同時に、ヒストンH3内のアミノ酸28残基目のセリンをリン酸化する事により、神経細胞様に細胞が分化することが知られている。この現象は、ポリコームグループタンパク質やそれを含む複合体により、ヒストンH3内のアミノ酸27残基目のリシンをトリメチル化する事によって、遺伝子の発現を抑制することが知られている。上述のMSKは、ポリコームグループタンパク質との協調作用によって作用する。また、ヒストンH3内のアミノ酸14残基目のリシンをアセチル化する事と同時に、ヒストンH3内のアミノ酸10残基目のセリンをリン酸化する事により、ストレスに関する記憶を生じさせることが知られている。このリン酸化には、extracellular signal-regulated kinase 1/2 (ERK1/2)を介したMSKの活性化が重要であり、c−Junやc−Fosの発現を上昇することが知られている。
【0025】
また、ユビキチン化による遺伝子発現制御機構も知られており、これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
修飾酵素としてユビキチン化に関与するタンパク質としては、E3ユビキチンリガーゼなどがポリユビチン化酵素として知られており、MED19やMED26などを代表とした神経細胞に分化する多くの遺伝子の発現を抑制しているrepressor element 1-silencing transcription factor (REST)をポリユビキチン化する。そして、タンパク質の分解の場として知られるプロテアソームによってRESTが分解され、その結果、神経細胞へと分化が促される一連の細部内分子機構が知られている。
【0026】
DNAの修飾に関与する酵素としては、DNAのメチル化酵素が例示される。例えば、DNAメチルトランスフェラーゼなどである。
特定遺伝子のプロモーター領域近傍などには、CpGアイランドと呼ばれるシトシン、リン酸、グアニンというDNA中の塩基に特殊な繰り返し配列が存在する。このCpGに対し、DNAメチルトランスフェラーゼが作用すると、シトシンにメチル基が付加され、シトシンは、5−メチルシトシンに変わる。その結果、遺伝子の転写が抑制され、その遺伝子の翻訳産物であるタンパク質の発現が減少することが知られている。
【0027】
本発明のスクリーニング方法で用いられる神経モデル細胞は、神経細胞様に分化し得る細胞であればよい。
神経モデル細胞としては、例えばPC12細胞、ES細胞、Neuro2a細胞、ヒト神経線維芽細胞 (NH-12細胞)、NG108-15細胞などが例示される。これら細胞はPC12細胞と、神経突起を進展させる細胞内分子機構が大よそ同一であり、神経突起を構成するタンパク質群がおおよそ同一であることからPC12と同様に本発明のスクリーニングに用いることができる。
【0028】
本発明のスクリーニング方法で用いられる好ましい細胞であるPC12細胞は、副腎褐色細胞腫由来の細胞株であり、ニューロン前駆体細胞からニューロン様の細胞への分化の実験モデル細胞として周知である。
【0029】
神経モデル細胞の生存維持作用とは、通常の方法で培養されている神経モデル細胞に、細胞死誘導剤を接触させて細胞死誘導させた細胞に対する、修飾酵素阻害剤の生存維持作用である。
【0030】
アルツハイマー病やパーキンソン病に見られる特異的な現象として、神経細胞の小胞体内に構造異常タンパク質が蓄積し、その蓄積による刺激(小胞体ストレスと呼ぶ。)が細胞死を引き起こしていることが明らかになっている。従って、上述の現象を模倣するように小胞体内に構造異常タンパク質が蓄積し、細胞死を引き起こすことが知られる物質を細胞死誘導剤として用いることが望ましい。特に好ましい細胞死誘導剤としてはサプシガルギン(以下Tgという。)やツニカマイシン(以下Tmという。)が挙げられる。
【0031】
好ましい実施態様としては、生育用培地に懸濁されたPC12細胞を、通常の培養用プレート1cm2あたり1.0×105〜2.0×105個で播種し、通常の方法で12〜30時間好ましくは16〜24時間培養後、細胞死誘導剤とHDAC阻害剤をPC12細胞に接触せしめ、10〜40時間後好ましくは20〜28時間後の細胞生存率を測定する。
【0032】
HDAC阻害剤により様々な遺伝子が発現される。HDAC阻害剤を添加してから細胞生存率を測定するまでの培養時間はこの発現に要する時間であることが好ましい。この時間は遺伝子に依存するが、10〜40時間、好ましくは20〜28時間である。
【0033】
好ましい生育用培地としては、5/5-DMEM(非動化した5%ウマ血清と5%ウシ胎児血清含有Dulbecco's Modified Eagle's Medium)が挙げられる。
培養は、5%の炭酸ガス気相下、37℃、100%湿度の条件に設定したCO2インキュベーター内で培養することが好ましい。
【0034】
培養用プレートは、コラーゲンによってコーティング処理を行った細胞培養専用のプレートが好ましく、96穴プレート、24穴プレート等が挙げられる。好ましくは96穴プレートである。
【0035】
細胞死誘導剤とHDAC阻害剤をPC12細胞に接触させる方法は、好ましくは、細胞がプレートの底辺に安定して付着した状態になったところで、細胞死誘導剤とHDAC阻害剤を添加する。新たに添加する培地である無血清Dulbecco's Modified Eagle's Medium(以下、SF-DMEMという。)に予めTg又はTm並びにHDAC阻害剤を混合し、その混合容液を添加すると簡便でより好ましい。
【0036】
HDAC阻害剤は、培地中1nM〜50mM、好ましくは1nM〜5μMの濃度になるように添加する。
HDAC阻害剤は、DMSO(ジメチルスルフォキシド)を溶媒とした溶液中で保存している。従って、細胞に対してHDAC阻害剤を添加する際には、DMSOも同時に添加される。添加されるDMSOを最小限に抑えなければ、細胞にダメージを与えてしまう。よって、HDAC阻害剤の添加量は、培地の液量に対して1000分の1の液量となる様に調整しなければならない。
細胞生存率の測定用の好ましい培地としては、SF-DMEMが挙げられる。
【0037】
神経モデル細胞に対する神経突起伸展作用とは、培養されている神経モデル細胞へ、修飾酵素阻害剤を添加した場合の神経突起の伸展作用である。
【0038】
好ましい実施態様は、PC12細胞の神経突起伸展作用の定量化であり、PC12細胞はSF-DMEMによる約24時間程度の培養では神経突起はほぼ伸展していない状態のため、神経突起伸展作用は、PC12細胞の曲面を起点とした神経突起の長さとして示される。
生育用培地に懸濁されたPC12細胞を、通常の培養用プレート1cm2あたり0.01×105〜2.0×105個、好ましくは0.1×105〜0.5×105で播種し、通常の方法で12〜30時間好ましくは16〜24時間培養を行う。その後、HDAC阻害剤をPC12細胞に接触させて10〜40時間後、好ましくは20〜28時間経過後の細胞突起の長さを測定する。
【0039】
HDAC阻害剤により様々な遺伝子が発現される。HDAC阻害剤を添加してから神経突起の長さを測定するまでの培養時間はこの発現に要する時間であることが好ましい。この時間は遺伝子に依存するが、18〜40時間、好ましくは20〜28時間である。
【0040】
好ましい生育用培地としては、5/5-DMEMが挙げられる。
培養は、5%の炭酸ガス気相下、37℃、100%湿度の条件に設定したCO2インキュベーター内で培養することが好ましい。
【0041】
培養用プレートは、コラーゲンによってコーティング処理を行った細胞培養専用のプレートが好ましく、96穴プレート、24穴プレート等が挙げられる。好ましくは24穴プレートである。
【0042】
HDAC阻害剤をPC12細胞に接触させる方法は、好ましくは、細胞がプレートの底辺に安定して付着した状態になったところで、HDAC阻害剤を添加する。新たに添加する培地SF-DMEMに予めHDAC阻害剤を混合し、その混合容液を添加すると簡便でより好ましい。
【0043】
HDAC阻害剤は、培地中1nM〜50mM、好ましくは1nM〜5μMの濃度になるように添加する。
HDAC阻害剤は、DMSO(ジメチルスルフォキシド)を溶媒とした溶液中で保存している。従って、細胞に対してHDAC阻害剤を添加する際には、DMSOも同時に添加される。添加されるDMSOを最小限に抑えなければ、細胞にダメージを与えてしまう。よって、HDAC阻害剤の添加量は、培地の液量に対して1000分の1の液量となる様に調整しなければならない。
細胞突起の長さの測定用の好ましい培地としては、SF-DMEMが挙げられる。
【0044】
神経モデル細胞に対する神経突起数増加作用とは、培養されている神経モデル細胞へ修飾酵素阻害剤を添加した場合の神経突起数の増加作用である。
【0045】
好ましい実施態様は、PC12細胞の神経突起数増加作用であり、PC12細胞は、SF-DMEMによる約24時間程度の培養では神経突起はほぼ伸展していない状態のため、神経突起数増加作用は、1細胞当りの神経突起の本数で示すことができる。細胞の培養方法とHDAC阻害剤の添加方法は上記神経突起の長さの測定と場合と同様である。
【0046】
修飾酵素阻害剤をクラスター化するとは、細胞に同様の発現型を生じさせる修飾酵素阻害剤をグループ化するという意味である。より具体的には以下のようにグループ化する。
神経モデル細胞の生存維持作用と神経モデル細胞に対する神経突起伸展作用を指標として2次元のグラフを作成して、同様の表現型を有する修飾酵素阻害剤をグループ化する。
または、神経モデル細胞の生存維持作用と神経モデル細胞に対する神経突起数増加作用を指標として2次元のグラフを作成して、同様の表現型を有する修飾酵素阻害剤をグループ化する。
または、神経モデル細胞の生存維持作用並びに神経モデル細胞に対する神経突起伸展作用及び神経突起数増加作用を指標として3次元のグラフを作成して、同様の表現型を有する修飾酵素阻害剤をグループ化する。
グループ化は化合物の多少を問わずに決定する。同様の表現型を呈する1つのクラスター中のHDAC阻害剤は、ほぼ同一の遺伝子群の発現を活性化すると考えられる。
【0047】
クラスター毎に評価する工程とは、グラフの中から最適と判断されたクラスター中から化合物を選択し、その後の実験動物を用いたin vivoのスクリーニング系で評価をすることである。1つのクラスターから化合物の基本骨格が異なる1〜6、好ましくは、1〜4、さらに好ましくは1〜3の化合物を選択する。その後のin vivoのスクリーニング系で低い評価であった場合は、おそらく異なる遺伝子発現群からなる他のクラスターから化合物を選択することにより、これまでのスクリーニングで行われてきた改めて再スクリーニングを行うことや発現遺伝子群の評価を行っていない基本骨格が異なるバックアップ化合物を候補化合物として選択しなおすなどの非効率な過程を排する効率的なスクリーニングができる。
【0048】
クラスター毎に解析する工程とは、クラスターから化合物を選択し、その化合物により発現が促進される遺伝子群を解析することである。1つのクラスターに属する修飾酵素阻害剤は、その主作用とそこから得られる表現型から推察される通り、おそらくほぼ同一の遺伝子群を発現することより、その作用機序の解明はクラスター毎に解析すれば足りる。よって、素早く目的の修飾酵素阻害剤の作用機序の解析を終了することができる。
【0049】
クラスターの遺伝子発現を簡易に解析するために、既知遺伝子の機能を基に作製された遺伝子ライブラリーを乗せたDNAアレイを用いることができる。例えば、細胞生存率の上昇作用を評価するためには、Akt、GSK-3、phospatidylinositol 3-kinase、Bcl-2、Bcl-XL、Mcl-1などの遺伝子群を予めアレイ化しておくか、あるいは、通常のDNAマイクロアレイにより、遺伝子発現の上昇を評価する。
【実施例】
【0050】
<HDAC阻害剤>
HDAC阻害剤としてよく知られている、トリコスタチンA(TSA)、MS-275、バソプロ酸(VPA)および本発明者らが合成したK28、K32、K120、K140、K164、K197およびK380(Bioorganic and Medical Chemistry 18, 3925-3933,2010)を使用した。
K28、K32、K120、K140、K164、K197又はK380の構造を以下に示す。
【0051】
1.K28
【化1】

【0052】
2.K32
【化2】
【0053】
3.K120
【化3】
【0054】
4.K140
【化4】
【0055】
5.K164
【化5】
【0056】
6.K197
【化6】
【0057】
7.K380
【化71】
【0058】
<PC12細胞の培養方法>
PC12細胞は、ダルベッコ改変イーグル培地(DMEM)に対し、最終濃度5%のウシ胎児血清、最終濃度5%のウマ血清、0.1%ペニシリン-ストレプトマイシン混合用液(Gibco BRL社製)を添加した培地(5/5-DMEM)を用いて生育(継代)を行った。また、PC12細胞は、100%の湿度、5%の二酸化炭素存在下、37℃にて培養を行った。
尚、2日に1回の割合で細胞の培地交換を行った。その際に、凝集した細胞群が見られれば、先にチップを付けた10 mlのプラスチックピペットを用い、広く満遍なく散乱するように細胞をピペッティングによって引き剥がし、再付着させる操作を行った。
【0059】
<PC12細胞生存維持作用>
生育(継代)された5/5-DMEM 中のPC12細胞を、コラーゲンコートを行った96穴プレート(Falcon社製)に、1cm2あたり1.0〜2.0×105個で100μlの液量になる様に播種した。コラーゲンコートは、各穴に100μlのコラーゲン溶液(ラット尾部より抽出したコラーゲンの0.15M 酢酸溶液)を添加後、デシケーター内に33%アンモニア水溶液を、2ml加えた小瓶を上記の96穴プレートと共に密閉し、約2時間後にデシケーターから取り出すことにより行った。コラーゲンコートした96穴プレートは、血清無添加のダルベッコ改変イーグル培地(SF-DMEM)を用い、それぞれの穴に200μlずつ、2回添加することにより洗浄した。
【0060】
バルプロ酸、トリコスタチンAおよびMS-275のPC12細胞生存率の測定:
96穴プレートに対し、PC12細胞を播種し、24時間培養した。その後、予めSF-DMEM中にTgを0.33μMになるように添加して溶液を作製した。さらに、HDAC阻害剤をその溶液に添加し攪拌した。溶液中のHDAC阻害剤の濃度は、トリコスタチンA(TSA)が3.12〜25nM、MS-275が31.2〜250nM、バルプロ酸(VPA)が3.12〜25mM として溶液を添加した。そして、それぞれの穴中でさらに24時間培養を行った。
その後、培地をそれぞれの穴から抜き取り、100μlのMTT溶液(1mg/ml 臭化3-(4,5-ジメチル-2-チアゾリル)-2,5-ジフェニル-2H-テトラゾリウム(生理食塩水中で5mg/mlで保存しているものを希釈して使用)とSF-DMEMの1:4混合溶液)を10分間反応させた。反応後は、MTT溶液を抜き取り、pHが4.7のLysis buffer(20% ドデシル硫酸ナトリウム、50% N,N-ジメチルホルムアミド、30% ミリQ水)を100μlずつ添加し、プレートを60分攪拌する。それから、プレートリーダー(大日本住友製薬製)を用い、570 nmの吸光度を測定し、その吸光度から得られる値を以下の式に代入し、細胞生存率とした。
式:{(テストした細胞群のサンプルの値)-(無細胞ブランクの値)/(薬剤無添加細胞群をコントロールとしたサンプルの値)-(無細胞ブランクの値)} × 100
結果を図1〜図3に示す。
【0061】
HDAC阻害剤(K28、K32、K120、K140、K164、K197およびK380)のPC12細胞生存率の測定:
96穴プレートに対し、PC12細胞を播種し、24時間培養した。その後、予めSF-DMEM中にTmを1μg/mlになるように添加して溶液を作製した。さらに、HDAC阻害剤(K28、K32、K120、K140、K164、K197およびK380)を溶液中の濃度が31nM〜1mMになるように添加した。それ以外は、上記バルプロ酸等と同一条件にてPC12細胞生存維持作用を測定した。K120、K140、K164、K197の結果を図5に示す。
【0062】
<PC12細胞に対する神経突起伸展作用>
神経突起伸長度の測定:
上記と同様に5/5-DMEMによって生育(継代)させたPC12細胞を、コラーゲンコートした24穴プレートに1cm2あたり0.1〜0.5×105個で播種した。
播種から16〜24時間経過後、SF-DMEMにHDAC阻害剤であるバルプロ酸、トリコスタチンA、MS-275、K28、K32、K120、K140、K164、K197またはK380を予め溶解させ、24穴プレートに500μlとなる様にそれぞれを添加し、約24時間培養した。HDAC阻害剤のSF-DMEM中の濃度は、バルプロ酸5mM、トリコスタチンAは0.1μM、それ以外のHDAC阻害剤の濃度は0.5μM〜5μMとした。HDAC阻害剤の添加後24時間経過したPC12細胞を4%パラホルムアルデヒド水溶液によって30分間固定を行った。
40倍の対物レンズを用い、位相差顕微鏡の画像を得た。引き続き、位相差顕微鏡の画像計測・解析ソフトであるBZ-2解析アプリケーション(KEYENCE社製)を用い解析した。すなわち、PC12細胞を位相差画像としてデジタル写真化した後、それらの画像を保存し、1細胞中の最長突起の長さを測定した。よって、神経突起伸展度は、1細胞当たりの最長突起の長さとして数値化した(単位は、μm)。また、画像毎に、20個の細胞を無作為抽出し、画像を10枚撮影後、PC12細胞の突起伸長度を測定した(合計200個)。同時に、生存している細胞の神経突起を測定している事を確かめるために、ヘキスト33258水溶液を用い、核を染色した。そして、それぞれの位相差画像は、BZ-2解析アプリケーションによって重ね合わした。次に、重ね合わせた画像は、細胞を円形もしくは楕円形とし、その曲面上になく突出しているものを神経突起とした。
尚、測定値は、集計後、分布をヒストグラムとし、さらに、ヒストグラムから最大値(ピーク長)を抽出し、各薬剤が示す神経突起伸展度とした。そして、陰性コントロールと各HDAC阻害剤の添加細胞群の神経突起伸展度をKolmogorov-Smirnov検定により有意差がある事を確認した。結果を図6〜図12に示す。
【0063】
<PC12細胞に対する神経突起数増加作用>
1細胞当たりの神経突起数の測定:
神経突起伸展度の測定と同様にPC12細胞を培養し、HDAC阻害剤を添加後、細胞の神経突起数が増加したPC12細胞を得て、1細胞あたりの神経突起数を測定した。位相差顕微鏡の画像計測・解析ソフトであるBZ-2解析アプリケーションを用い、PC12細胞の1細胞あたりの神経突起の数を測定した。神経突起は、神経突起伸展度の測定の場合と同様の定義とした。また、神経突起数の測定でも画像毎に、20個の細胞を無作為抽出し測定した。位相差画像は10枚撮影し、合計200個の細胞が有する神経突起の個数を測定した。測定後、集計し平均値を求め、1細胞当たりの神経突起数とした。結果を図13〜図19に示す。
【0064】
<HDAC阻害剤のクラスター化>
細胞生存率と突起伸展度の二次元のグラフに各HDAC阻害剤の値をプロットしてクラスター化を行った(図20)。細胞生存率は、上述の細胞生存率の測定に基づき測定した最高の値とし、突起伸展度は、1つの細胞が有する神経突起の最長突起を、細胞200個分の分布図として作製した後のピーク値を神経突起伸展度の値とした。
【産業上の利用可能性】
【0065】
HDAC阻害剤を表現型でスクリーニングすることにより神経変性疾患治療薬のスクリーニング効率が上がる。また、略同一の表現型でクラスター化することにより、スクリーニング効率が飛躍的に上昇し、新薬の創出が容易になる。また、本発明の新規のスクリーニング系によってこれまで見落としていた有用性のある薬剤を再びスクリーニングすることにより、既存の化合物ライブラリーから新薬を見出すことができる。
【技術分野】
【0001】
本発明は、神経変性疾患治療薬のスクリーニング方法に関する。
【背景技術】
【0002】
現在、製薬企業を主体とする医薬品の開発では、病気の原因となる分子を先ず見出し、何十万検体から分子、細胞、実験動物を用いたスクリーニング(企業によっては、大規模なハイスループットスクリーニング(HTS))を経て薬効を有する化合物(リード化合物)を抽出した後、化合物の更なる化学修飾・解析を行うことによってヒトでの有効性を高めた有望化合物を得て、その後、臨床試験(治験)を行い、ヒトへの有効性を確認している。
【0003】
脳精神神経疾患治療薬(アルツハイマー病、パーキンソン病、ハンチントン病などを適応症とする)においては、神経細胞を死滅させない役割を担う分子や逆に神経細胞を死滅させる役割を担う分子をターゲット分子として前臨床試験や臨床試験が行われてきた。
【0004】
アルツハイマー病に対しては、コリン作動性神経細胞の活動に必須であるアセチルコリンのシナプス内での濃度維持によるコリン作動性神経細胞の活動維持と生存維持を目的として、アセチルコリン分解酵素をターゲット分子とした阻害剤が、既に上市されている。しかし、生存維持が可能な期間が限られており、完治を可能とする医薬品ではない。
【0005】
また、パーキンソン病では、黒質ドーパミン作動性神経細胞の活動維持と生存維持を目的として、ドーパミン受容体をターゲット分子としたプロドラッグ(ドーパミン前駆分子)であるL-DOPAが上市されている。
また、ドーパミン受容体作動薬、ドーパミン発現上昇薬などのドーパミン作動性神経細胞をターゲットとした医薬品も上市されている。
【0006】
ハンチントン病は、この疾患が環境要因から引き起こされる孤発性疾患ではなく、完全にヒトが持つゲノム中の特定遺伝子(huntintin)のポリグルタミン鎖の伸長度に依存する遺伝性疾患であり、huntintinのポリグルタミン伸長鎖を変える様な薬物療法は、現在のところ存在しない。
【0007】
癲癇に関しては、神経系の興奮伝播につながるナトリウムチャネルの不活性状態を維持する薬剤、カルシウムチャネルを不活性化する薬剤、抑制性神経細胞の神経伝達物質であるGABAの作用を増強する薬剤が上市されている。バルプロ酸(VPA)は、ヒストン脱アセチル化酵素(HDAC)阻害作用を有しているが、ヒストン脱アセチル化酵素(HDAC)阻害剤として開発された医薬品ではなく、脳神経細胞の抑制性の神経伝達物質であるGABAの産生量を減少させないために、GABAトランスアミノブチラーゼ阻害剤として上市された。
【0008】
ヒストン (histone) は、真核生物のクロマチン(染色体)を構成するタンパク質の一群で、非常に長い分子であるDNA を核内に収納する役割を担っている。
ヒストンは強い塩基性のタンパク質であり、酸性のDNAとの高い親和性を示す。ヒストンが塩基性を持つのは、正の電荷を持つアミノ酸の含量が高いからである。各ヒストンを構成するアミノ酸のうち、20%以上がリシンまたはアルギニンである。ヌクレオソームヒストンの構造は球形のカルボキシル末端と、直鎖状のアミノ末端(ヒストンテイル)からなっている。ヒストンテイルのリシンやアスパラギン残基はアセチル化、メチル化、リン酸化、ユビキチン化といった化学修飾を受けることが知られている。これらの化学修飾は、遺伝子発現等、数々のクロマチン機能の制御に関わっていることが証明されつつある。
【0009】
ヒストン脱アセチル化酵素(以下、HDACという。)は、ヌクレオソーム中のヒストンタンパク質の、所謂、ヒストンテイルの特定リシン残基のアセチル基を脱アセチル化させることを触媒するタンパク質である。このタンパク質の作用により、八量体から成るヒストンとそれに巻き付いているDNA鎖に存在している僅かな空間が閉鎖され、転写を抑制することが知られている。HDAC阻害剤は、このHDACの作用を阻害することから、転写活性が上昇し、各種の遺伝子群を発現誘導する。その結果として、細胞が様々な形質変化を起こし、特定の表現型を獲得する。HDAC阻害剤は、癌治療薬としての開発が進んでいる。本発明者等も、多数のHDAC阻害剤を合成し、抗癌効果について検討した(特許文献1、非特許文献1)。
【0010】
脳精神疾患では、多くの先行医薬品が上市されているが、未だヒストンの修飾に関与する酵素阻害剤は上市されておらず、また、HDAC阻害剤としての治療薬は上市されていない。HDAC阻害剤の脳精神神経系疾患治療薬としての開発は進行している。しかし、本発明に見られるターゲット分子を特定する必要がない新規スクリーニング系を用いた創薬方法は存在しない。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特開2007−1885号公報
【非特許文献】
【0012】
【非特許文献1】Bioorganic and Medical Chemistry 18, 3925-3933,2010
【発明の概要】
【発明が解決しようとする課題】
【0013】
神経変性疾患には数多くの蛋白質が関わっていると考えられ、ターゲット分子に対する作用を評価するスクリーニング方法では、膨大なスクリーニング試験が必要となる。また、ヒストンの修飾に関与する酵素阻害剤およびDNAの修飾に関与する酵素阻害剤は細胞に様々な形質変化を起こし、特定の表現型を獲得し、この表現型獲得には複数の遺伝子が関わっていると考えられる。
このような、複雑な作用メカニズムが関与すると考えられる神経変性疾患治療剤の開発には、より膨大なスクリーニング試験が必要と考えられ、本発明の方法は、この創薬過程における膨大なスクリーニング工程を効率化するものである。
【課題を解決するための手段】
【0014】
すなわち、本発明は以下からなる。
1.神経モデル細胞の生存維持作用、並びに神経モデル細胞に対する神経突起伸展作用及び/又は神経突起数増加作用を指標としてヒストンの修飾又はDNAの修飾に関与する酵素の阻害剤をクラスター化した後、クラスター毎に評価および/またはクラスター毎に解析する工程を含む神経変性疾患治療薬のスクリーニング方法。
2.修飾がアセチル化、メチル化、リン酸化、又はユビキチン化である前項1に記載のスクリーニング方法。
3.神経モデル細胞がPC12細胞である前項1または2に記載のスクリーニング方法。
4.クラスター毎に解析する工程がDNAマイクロアレイを用いた遺伝子発現の解析工程である前項1〜3のいずれか1に記載のスクリーニング方法。
【発明の効果】
【0015】
本発明の、ターゲット分子を特定する必要がない表現型による新規スクリーニング方法は、複数のタンパク質(遺伝子産物群)による発現変動が関与する神経変性疾患の治療薬のスクリーニングにおいて大幅に効率を上昇させるものである。さらに本発明は、ヒストンの修飾又はDNAの修飾に関与する酵素阻害剤を、細胞が有する機能を指標としてクラスター化することによるスクリーニング方法であり、同一の表現型を発現する同一のクラスターに属する物質は、その表現型を獲得するための発現変動が類似している遺伝子群に関与すると考えられ、クラスター毎に治療効果を評価することができ、同一のクラスターに属する物質を、それ以降のスクリーニングから排除することが出来る。
【0016】
このように本発明は、上記操作により、スクリーニング効率を飛躍的に上昇させるものである。すなわち、治療効果の評価系において低い評価しか得られない場合には、同じクラスターからではなく、恐らく異なる遺伝子発現群に関与するクラスターからの化合物を選定することができる。また、その別のクラスターから1個、または、複数個を選定する事により、創薬化の過程で必要な同一の基本骨格を有する複数の類縁化合物の新規合成(化合物の最適化)に見られる様な無駄な化学修飾処理も排することができることから効率よく創薬化することができる。それは、本発明の基本となる複数遺伝子の発現変動を有する化合物におけるスクリーニングにおいて、クラスター化された化合物群は、治療を可能とする指標である細胞生存率上昇作用、神経突起伸展作用、神経突起数増加作用の評価を既に終えた化合物群から構成されていることやリード化合物から化学基を結合させた僅かな構造変化を伴う類縁化合物でも遺伝子の発現群が異なってくることから、表現型の評価段階で化合物の有用性が想定し得るからである。この事実は、本発明の実施例にも記載のあるバルプロ酸が、他の化合物が有する表現型の評価の値を凌駕し、既に上市されていることからも明らかである。
さらに、分子生物学的な作用機序の解析を早期に終了させることができる。
また、既存のスクリーニングに見られる様なターゲット分子を選定しないため、これまでに低い評価で候補医薬品として対象外になっていた化合物からの再スクリーニングにより有効な医薬品を開発できる可能性が広がると考えられる。
【図面の簡単な説明】
【0017】
【図1】サプシガルギン(Tg)により細胞死誘導されたPC12細胞へのトリコスタチンA(以下TSAという。)の生存維持作用を示したグラフである。
【図2】サプシガルギン(Tg)により細胞死誘導されたPC12細胞へのMS-275の生存維持作用を示したグラフである。
【図3】サプシガルギン(Tg)により細胞死誘導されたPC12細胞へのバルプロ酸(以下VPAという。)の生存維持作用を示したグラフである。
【図4】TSA、MS-275およびVPAで処理されたPC12細胞のヒストンH3(K14)のアセチル化を示した電気泳動(SDS-PAGE)後のウエスタンブロットによるヒストンH3のアセチル化の亢進を確認した写真である。
【図5】ツニカマイシン(Tm)により細胞死誘導されたPC12細胞へのK164、K197、K120およびK140の生存維持作用を示したグラフである。
【図6】TSA、MS-275、VPAおよびK197のPC12細胞への神経突起伸展作用を示したグラフである。
【図7】TSA、MS-275、VPAおよびK164のPC12細胞への神経突起伸展作用を示したグラフである。
【図8】TSA、MS-275、VPAおよびK120のPC12細胞への神経突起伸展作用を示したグラフである。
【図9】TSA、MS-275、VPAおよびK140のPC12細胞への神経突起伸展作用を示したグラフである。
【図10】TSA、MS-275、VPAおよびK28のPC12細胞への神経突起展作用を示したグラフである。
【図11】TSA、MS-275、VPAおよびK32のPC12細胞への神経突起伸展作用を示したグラフである。
【図12】TSA、MS-275、VPAおよびK380のPC12細胞への神経突起伸展作用を示したグラフである。
【図13】TSA、MS-275、VPAおよびK197のPC12細胞への神経突起数増加作用を示したグラフである。
【図14】TSA、MS-275、VPAおよびK164のPC12細胞への神経突起数増加作用を示したグラフである。
【図15】TSA、MS-275、VPAおよびK120のPC12細胞への神経突起数増加作用を示したグラフである。
【図16】TSA、MS-275、VPAおよびK140のPC12細胞への神経突起数増加作用を示したグラフである。
【図17】TSA、MS-275、VPAおよびK28のPC12細胞への神経突起数増加作用を示したグラフである。
【図18】TSA、MS-275、VPAおよびK32のPC12細胞への神経突起数増加作用を示したグラフである。
【図19】TSA、MS-275、VPAおよびK380のPC12細胞への神経突起数増加作用を示したグラフである。
【図20】HDAC阻害剤のクラスター化を示したグラフである。
【図21】本発明の3次元のクラスター化によるスクリーニング方法の概念図を示した説明図である。
【発明を実施するための形態】
【0018】
本発明における神経変性疾患とは、中枢神経系の神経細胞のプログラム細胞死が原因とされている疾患であり、例えば、アルツハイマー病、パーキンソン病、ハンチントン病等が挙げられる。
【0019】
本発明のヒストンの修飾又はDNAの修飾に関与する酵素阻害剤(以下、修飾酵素阻害剤という。)とは、ヒストン又はDNAの化学修飾を触媒する酵素の阻害剤および脱化学修飾を触媒する酵素の阻害剤を含む。
【0020】
ヒストンは、アセチル化、メチル化、リン酸化、ユビキチン化といった化学修飾を受けるタンパク質であり、それに関与する酵素とはこれらの化学修飾を触媒する酵素または脱化学修飾を触媒する酵素である。また、ここにおける酵素は、ヒストン以外の修飾や脱修飾により、遺伝子の発現を変動させる場合がある。これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
【0021】
ヒストンのアルギニン残基、リシン残基のメチル化はゲノムの活性化、不活性化の主たる決定要素となっている。メチル化にはメチラーゼやメチルトランスフェラーゼ、脱メチル化にはデメチラーゼ等の酵素が関与している。これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
【0022】
アセチル化には、ヒストンアセチルトランスフェラーゼ、脱アセチル化にはヒストン脱アセチル化酵素(HDAC)が関与している。これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
ヒストン脱アセチル化酵素(以下、HDACという。)には複数のタイプが存在し、多くのタンパク質との複合体の状態で働く。現在、HDACは18種類のタンパク質が知られており、4つのグループに分類されている。
クラスI HDAC1, HDAC2, HDAC3, HDAC8
クラスII HDAC4, HDAC5, HDAC6, HDAC7 HDAC9, HDAC10
クラスIII SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, SIRT7
クラスIV HDAC11
【0023】
本発明で用いるHDAC阻害剤とは、HDACの活性を阻害する物質であれば何ら限定されない。低分子化合物、ポリマー、ペプチドなどを含む。
現在すでに多くのHDAC阻害剤が知られている。例えば次の4種類が挙げられる。
a)脂肪酸(Butyrate,phenyl butyrate,valproic acid(VPA)など)
b)ヒドロキサム酸(Trichostatin A (TSA),oxamflatin,suberolyanilide hydroxamic acid(SAHA)など)
c)環状ペプチド(Trapoxin (TPX),apicidin,FK228など)
d)ベンズアミド(MS-275,MGCD0103,CI-994など)
【0024】
また、リン酸化による遺伝子発現制御機構も知られており、これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
修飾酵素としてリン酸化に関与する酵素としては、mitogen- and stress-activated kinases (MSK)などがあり、ヒストンH3内のアミノ酸27残基目のリシンをトリメチル化する事と同時に、ヒストンH3内のアミノ酸28残基目のセリンをリン酸化する事により、神経細胞様に細胞が分化することが知られている。この現象は、ポリコームグループタンパク質やそれを含む複合体により、ヒストンH3内のアミノ酸27残基目のリシンをトリメチル化する事によって、遺伝子の発現を抑制することが知られている。上述のMSKは、ポリコームグループタンパク質との協調作用によって作用する。また、ヒストンH3内のアミノ酸14残基目のリシンをアセチル化する事と同時に、ヒストンH3内のアミノ酸10残基目のセリンをリン酸化する事により、ストレスに関する記憶を生じさせることが知られている。このリン酸化には、extracellular signal-regulated kinase 1/2 (ERK1/2)を介したMSKの活性化が重要であり、c−Junやc−Fosの発現を上昇することが知られている。
【0025】
また、ユビキチン化による遺伝子発現制御機構も知られており、これらの酵素を阻害することにより、遺伝子発現が上昇され、または抑制され、複数の遺伝子発現が変化する。
修飾酵素としてユビキチン化に関与するタンパク質としては、E3ユビキチンリガーゼなどがポリユビチン化酵素として知られており、MED19やMED26などを代表とした神経細胞に分化する多くの遺伝子の発現を抑制しているrepressor element 1-silencing transcription factor (REST)をポリユビキチン化する。そして、タンパク質の分解の場として知られるプロテアソームによってRESTが分解され、その結果、神経細胞へと分化が促される一連の細部内分子機構が知られている。
【0026】
DNAの修飾に関与する酵素としては、DNAのメチル化酵素が例示される。例えば、DNAメチルトランスフェラーゼなどである。
特定遺伝子のプロモーター領域近傍などには、CpGアイランドと呼ばれるシトシン、リン酸、グアニンというDNA中の塩基に特殊な繰り返し配列が存在する。このCpGに対し、DNAメチルトランスフェラーゼが作用すると、シトシンにメチル基が付加され、シトシンは、5−メチルシトシンに変わる。その結果、遺伝子の転写が抑制され、その遺伝子の翻訳産物であるタンパク質の発現が減少することが知られている。
【0027】
本発明のスクリーニング方法で用いられる神経モデル細胞は、神経細胞様に分化し得る細胞であればよい。
神経モデル細胞としては、例えばPC12細胞、ES細胞、Neuro2a細胞、ヒト神経線維芽細胞 (NH-12細胞)、NG108-15細胞などが例示される。これら細胞はPC12細胞と、神経突起を進展させる細胞内分子機構が大よそ同一であり、神経突起を構成するタンパク質群がおおよそ同一であることからPC12と同様に本発明のスクリーニングに用いることができる。
【0028】
本発明のスクリーニング方法で用いられる好ましい細胞であるPC12細胞は、副腎褐色細胞腫由来の細胞株であり、ニューロン前駆体細胞からニューロン様の細胞への分化の実験モデル細胞として周知である。
【0029】
神経モデル細胞の生存維持作用とは、通常の方法で培養されている神経モデル細胞に、細胞死誘導剤を接触させて細胞死誘導させた細胞に対する、修飾酵素阻害剤の生存維持作用である。
【0030】
アルツハイマー病やパーキンソン病に見られる特異的な現象として、神経細胞の小胞体内に構造異常タンパク質が蓄積し、その蓄積による刺激(小胞体ストレスと呼ぶ。)が細胞死を引き起こしていることが明らかになっている。従って、上述の現象を模倣するように小胞体内に構造異常タンパク質が蓄積し、細胞死を引き起こすことが知られる物質を細胞死誘導剤として用いることが望ましい。特に好ましい細胞死誘導剤としてはサプシガルギン(以下Tgという。)やツニカマイシン(以下Tmという。)が挙げられる。
【0031】
好ましい実施態様としては、生育用培地に懸濁されたPC12細胞を、通常の培養用プレート1cm2あたり1.0×105〜2.0×105個で播種し、通常の方法で12〜30時間好ましくは16〜24時間培養後、細胞死誘導剤とHDAC阻害剤をPC12細胞に接触せしめ、10〜40時間後好ましくは20〜28時間後の細胞生存率を測定する。
【0032】
HDAC阻害剤により様々な遺伝子が発現される。HDAC阻害剤を添加してから細胞生存率を測定するまでの培養時間はこの発現に要する時間であることが好ましい。この時間は遺伝子に依存するが、10〜40時間、好ましくは20〜28時間である。
【0033】
好ましい生育用培地としては、5/5-DMEM(非動化した5%ウマ血清と5%ウシ胎児血清含有Dulbecco's Modified Eagle's Medium)が挙げられる。
培養は、5%の炭酸ガス気相下、37℃、100%湿度の条件に設定したCO2インキュベーター内で培養することが好ましい。
【0034】
培養用プレートは、コラーゲンによってコーティング処理を行った細胞培養専用のプレートが好ましく、96穴プレート、24穴プレート等が挙げられる。好ましくは96穴プレートである。
【0035】
細胞死誘導剤とHDAC阻害剤をPC12細胞に接触させる方法は、好ましくは、細胞がプレートの底辺に安定して付着した状態になったところで、細胞死誘導剤とHDAC阻害剤を添加する。新たに添加する培地である無血清Dulbecco's Modified Eagle's Medium(以下、SF-DMEMという。)に予めTg又はTm並びにHDAC阻害剤を混合し、その混合容液を添加すると簡便でより好ましい。
【0036】
HDAC阻害剤は、培地中1nM〜50mM、好ましくは1nM〜5μMの濃度になるように添加する。
HDAC阻害剤は、DMSO(ジメチルスルフォキシド)を溶媒とした溶液中で保存している。従って、細胞に対してHDAC阻害剤を添加する際には、DMSOも同時に添加される。添加されるDMSOを最小限に抑えなければ、細胞にダメージを与えてしまう。よって、HDAC阻害剤の添加量は、培地の液量に対して1000分の1の液量となる様に調整しなければならない。
細胞生存率の測定用の好ましい培地としては、SF-DMEMが挙げられる。
【0037】
神経モデル細胞に対する神経突起伸展作用とは、培養されている神経モデル細胞へ、修飾酵素阻害剤を添加した場合の神経突起の伸展作用である。
【0038】
好ましい実施態様は、PC12細胞の神経突起伸展作用の定量化であり、PC12細胞はSF-DMEMによる約24時間程度の培養では神経突起はほぼ伸展していない状態のため、神経突起伸展作用は、PC12細胞の曲面を起点とした神経突起の長さとして示される。
生育用培地に懸濁されたPC12細胞を、通常の培養用プレート1cm2あたり0.01×105〜2.0×105個、好ましくは0.1×105〜0.5×105で播種し、通常の方法で12〜30時間好ましくは16〜24時間培養を行う。その後、HDAC阻害剤をPC12細胞に接触させて10〜40時間後、好ましくは20〜28時間経過後の細胞突起の長さを測定する。
【0039】
HDAC阻害剤により様々な遺伝子が発現される。HDAC阻害剤を添加してから神経突起の長さを測定するまでの培養時間はこの発現に要する時間であることが好ましい。この時間は遺伝子に依存するが、18〜40時間、好ましくは20〜28時間である。
【0040】
好ましい生育用培地としては、5/5-DMEMが挙げられる。
培養は、5%の炭酸ガス気相下、37℃、100%湿度の条件に設定したCO2インキュベーター内で培養することが好ましい。
【0041】
培養用プレートは、コラーゲンによってコーティング処理を行った細胞培養専用のプレートが好ましく、96穴プレート、24穴プレート等が挙げられる。好ましくは24穴プレートである。
【0042】
HDAC阻害剤をPC12細胞に接触させる方法は、好ましくは、細胞がプレートの底辺に安定して付着した状態になったところで、HDAC阻害剤を添加する。新たに添加する培地SF-DMEMに予めHDAC阻害剤を混合し、その混合容液を添加すると簡便でより好ましい。
【0043】
HDAC阻害剤は、培地中1nM〜50mM、好ましくは1nM〜5μMの濃度になるように添加する。
HDAC阻害剤は、DMSO(ジメチルスルフォキシド)を溶媒とした溶液中で保存している。従って、細胞に対してHDAC阻害剤を添加する際には、DMSOも同時に添加される。添加されるDMSOを最小限に抑えなければ、細胞にダメージを与えてしまう。よって、HDAC阻害剤の添加量は、培地の液量に対して1000分の1の液量となる様に調整しなければならない。
細胞突起の長さの測定用の好ましい培地としては、SF-DMEMが挙げられる。
【0044】
神経モデル細胞に対する神経突起数増加作用とは、培養されている神経モデル細胞へ修飾酵素阻害剤を添加した場合の神経突起数の増加作用である。
【0045】
好ましい実施態様は、PC12細胞の神経突起数増加作用であり、PC12細胞は、SF-DMEMによる約24時間程度の培養では神経突起はほぼ伸展していない状態のため、神経突起数増加作用は、1細胞当りの神経突起の本数で示すことができる。細胞の培養方法とHDAC阻害剤の添加方法は上記神経突起の長さの測定と場合と同様である。
【0046】
修飾酵素阻害剤をクラスター化するとは、細胞に同様の発現型を生じさせる修飾酵素阻害剤をグループ化するという意味である。より具体的には以下のようにグループ化する。
神経モデル細胞の生存維持作用と神経モデル細胞に対する神経突起伸展作用を指標として2次元のグラフを作成して、同様の表現型を有する修飾酵素阻害剤をグループ化する。
または、神経モデル細胞の生存維持作用と神経モデル細胞に対する神経突起数増加作用を指標として2次元のグラフを作成して、同様の表現型を有する修飾酵素阻害剤をグループ化する。
または、神経モデル細胞の生存維持作用並びに神経モデル細胞に対する神経突起伸展作用及び神経突起数増加作用を指標として3次元のグラフを作成して、同様の表現型を有する修飾酵素阻害剤をグループ化する。
グループ化は化合物の多少を問わずに決定する。同様の表現型を呈する1つのクラスター中のHDAC阻害剤は、ほぼ同一の遺伝子群の発現を活性化すると考えられる。
【0047】
クラスター毎に評価する工程とは、グラフの中から最適と判断されたクラスター中から化合物を選択し、その後の実験動物を用いたin vivoのスクリーニング系で評価をすることである。1つのクラスターから化合物の基本骨格が異なる1〜6、好ましくは、1〜4、さらに好ましくは1〜3の化合物を選択する。その後のin vivoのスクリーニング系で低い評価であった場合は、おそらく異なる遺伝子発現群からなる他のクラスターから化合物を選択することにより、これまでのスクリーニングで行われてきた改めて再スクリーニングを行うことや発現遺伝子群の評価を行っていない基本骨格が異なるバックアップ化合物を候補化合物として選択しなおすなどの非効率な過程を排する効率的なスクリーニングができる。
【0048】
クラスター毎に解析する工程とは、クラスターから化合物を選択し、その化合物により発現が促進される遺伝子群を解析することである。1つのクラスターに属する修飾酵素阻害剤は、その主作用とそこから得られる表現型から推察される通り、おそらくほぼ同一の遺伝子群を発現することより、その作用機序の解明はクラスター毎に解析すれば足りる。よって、素早く目的の修飾酵素阻害剤の作用機序の解析を終了することができる。
【0049】
クラスターの遺伝子発現を簡易に解析するために、既知遺伝子の機能を基に作製された遺伝子ライブラリーを乗せたDNAアレイを用いることができる。例えば、細胞生存率の上昇作用を評価するためには、Akt、GSK-3、phospatidylinositol 3-kinase、Bcl-2、Bcl-XL、Mcl-1などの遺伝子群を予めアレイ化しておくか、あるいは、通常のDNAマイクロアレイにより、遺伝子発現の上昇を評価する。
【実施例】
【0050】
<HDAC阻害剤>
HDAC阻害剤としてよく知られている、トリコスタチンA(TSA)、MS-275、バソプロ酸(VPA)および本発明者らが合成したK28、K32、K120、K140、K164、K197およびK380(Bioorganic and Medical Chemistry 18, 3925-3933,2010)を使用した。
K28、K32、K120、K140、K164、K197又はK380の構造を以下に示す。
【0051】
1.K28
【化1】
【0052】
2.K32
【化2】
【0053】
3.K120
【化3】
【0054】
4.K140
【化4】
【0055】
5.K164
【化5】
【0056】
6.K197
【化6】
【0057】
7.K380
【化71】
【0058】
<PC12細胞の培養方法>
PC12細胞は、ダルベッコ改変イーグル培地(DMEM)に対し、最終濃度5%のウシ胎児血清、最終濃度5%のウマ血清、0.1%ペニシリン-ストレプトマイシン混合用液(Gibco BRL社製)を添加した培地(5/5-DMEM)を用いて生育(継代)を行った。また、PC12細胞は、100%の湿度、5%の二酸化炭素存在下、37℃にて培養を行った。
尚、2日に1回の割合で細胞の培地交換を行った。その際に、凝集した細胞群が見られれば、先にチップを付けた10 mlのプラスチックピペットを用い、広く満遍なく散乱するように細胞をピペッティングによって引き剥がし、再付着させる操作を行った。
【0059】
<PC12細胞生存維持作用>
生育(継代)された5/5-DMEM 中のPC12細胞を、コラーゲンコートを行った96穴プレート(Falcon社製)に、1cm2あたり1.0〜2.0×105個で100μlの液量になる様に播種した。コラーゲンコートは、各穴に100μlのコラーゲン溶液(ラット尾部より抽出したコラーゲンの0.15M 酢酸溶液)を添加後、デシケーター内に33%アンモニア水溶液を、2ml加えた小瓶を上記の96穴プレートと共に密閉し、約2時間後にデシケーターから取り出すことにより行った。コラーゲンコートした96穴プレートは、血清無添加のダルベッコ改変イーグル培地(SF-DMEM)を用い、それぞれの穴に200μlずつ、2回添加することにより洗浄した。
【0060】
バルプロ酸、トリコスタチンAおよびMS-275のPC12細胞生存率の測定:
96穴プレートに対し、PC12細胞を播種し、24時間培養した。その後、予めSF-DMEM中にTgを0.33μMになるように添加して溶液を作製した。さらに、HDAC阻害剤をその溶液に添加し攪拌した。溶液中のHDAC阻害剤の濃度は、トリコスタチンA(TSA)が3.12〜25nM、MS-275が31.2〜250nM、バルプロ酸(VPA)が3.12〜25mM として溶液を添加した。そして、それぞれの穴中でさらに24時間培養を行った。
その後、培地をそれぞれの穴から抜き取り、100μlのMTT溶液(1mg/ml 臭化3-(4,5-ジメチル-2-チアゾリル)-2,5-ジフェニル-2H-テトラゾリウム(生理食塩水中で5mg/mlで保存しているものを希釈して使用)とSF-DMEMの1:4混合溶液)を10分間反応させた。反応後は、MTT溶液を抜き取り、pHが4.7のLysis buffer(20% ドデシル硫酸ナトリウム、50% N,N-ジメチルホルムアミド、30% ミリQ水)を100μlずつ添加し、プレートを60分攪拌する。それから、プレートリーダー(大日本住友製薬製)を用い、570 nmの吸光度を測定し、その吸光度から得られる値を以下の式に代入し、細胞生存率とした。
式:{(テストした細胞群のサンプルの値)-(無細胞ブランクの値)/(薬剤無添加細胞群をコントロールとしたサンプルの値)-(無細胞ブランクの値)} × 100
結果を図1〜図3に示す。
【0061】
HDAC阻害剤(K28、K32、K120、K140、K164、K197およびK380)のPC12細胞生存率の測定:
96穴プレートに対し、PC12細胞を播種し、24時間培養した。その後、予めSF-DMEM中にTmを1μg/mlになるように添加して溶液を作製した。さらに、HDAC阻害剤(K28、K32、K120、K140、K164、K197およびK380)を溶液中の濃度が31nM〜1mMになるように添加した。それ以外は、上記バルプロ酸等と同一条件にてPC12細胞生存維持作用を測定した。K120、K140、K164、K197の結果を図5に示す。
【0062】
<PC12細胞に対する神経突起伸展作用>
神経突起伸長度の測定:
上記と同様に5/5-DMEMによって生育(継代)させたPC12細胞を、コラーゲンコートした24穴プレートに1cm2あたり0.1〜0.5×105個で播種した。
播種から16〜24時間経過後、SF-DMEMにHDAC阻害剤であるバルプロ酸、トリコスタチンA、MS-275、K28、K32、K120、K140、K164、K197またはK380を予め溶解させ、24穴プレートに500μlとなる様にそれぞれを添加し、約24時間培養した。HDAC阻害剤のSF-DMEM中の濃度は、バルプロ酸5mM、トリコスタチンAは0.1μM、それ以外のHDAC阻害剤の濃度は0.5μM〜5μMとした。HDAC阻害剤の添加後24時間経過したPC12細胞を4%パラホルムアルデヒド水溶液によって30分間固定を行った。
40倍の対物レンズを用い、位相差顕微鏡の画像を得た。引き続き、位相差顕微鏡の画像計測・解析ソフトであるBZ-2解析アプリケーション(KEYENCE社製)を用い解析した。すなわち、PC12細胞を位相差画像としてデジタル写真化した後、それらの画像を保存し、1細胞中の最長突起の長さを測定した。よって、神経突起伸展度は、1細胞当たりの最長突起の長さとして数値化した(単位は、μm)。また、画像毎に、20個の細胞を無作為抽出し、画像を10枚撮影後、PC12細胞の突起伸長度を測定した(合計200個)。同時に、生存している細胞の神経突起を測定している事を確かめるために、ヘキスト33258水溶液を用い、核を染色した。そして、それぞれの位相差画像は、BZ-2解析アプリケーションによって重ね合わした。次に、重ね合わせた画像は、細胞を円形もしくは楕円形とし、その曲面上になく突出しているものを神経突起とした。
尚、測定値は、集計後、分布をヒストグラムとし、さらに、ヒストグラムから最大値(ピーク長)を抽出し、各薬剤が示す神経突起伸展度とした。そして、陰性コントロールと各HDAC阻害剤の添加細胞群の神経突起伸展度をKolmogorov-Smirnov検定により有意差がある事を確認した。結果を図6〜図12に示す。
【0063】
<PC12細胞に対する神経突起数増加作用>
1細胞当たりの神経突起数の測定:
神経突起伸展度の測定と同様にPC12細胞を培養し、HDAC阻害剤を添加後、細胞の神経突起数が増加したPC12細胞を得て、1細胞あたりの神経突起数を測定した。位相差顕微鏡の画像計測・解析ソフトであるBZ-2解析アプリケーションを用い、PC12細胞の1細胞あたりの神経突起の数を測定した。神経突起は、神経突起伸展度の測定の場合と同様の定義とした。また、神経突起数の測定でも画像毎に、20個の細胞を無作為抽出し測定した。位相差画像は10枚撮影し、合計200個の細胞が有する神経突起の個数を測定した。測定後、集計し平均値を求め、1細胞当たりの神経突起数とした。結果を図13〜図19に示す。
【0064】
<HDAC阻害剤のクラスター化>
細胞生存率と突起伸展度の二次元のグラフに各HDAC阻害剤の値をプロットしてクラスター化を行った(図20)。細胞生存率は、上述の細胞生存率の測定に基づき測定した最高の値とし、突起伸展度は、1つの細胞が有する神経突起の最長突起を、細胞200個分の分布図として作製した後のピーク値を神経突起伸展度の値とした。
【産業上の利用可能性】
【0065】
HDAC阻害剤を表現型でスクリーニングすることにより神経変性疾患治療薬のスクリーニング効率が上がる。また、略同一の表現型でクラスター化することにより、スクリーニング効率が飛躍的に上昇し、新薬の創出が容易になる。また、本発明の新規のスクリーニング系によってこれまで見落としていた有用性のある薬剤を再びスクリーニングすることにより、既存の化合物ライブラリーから新薬を見出すことができる。
【特許請求の範囲】
【請求項1】
神経モデル細胞の生存維持作用、並びに神経モデル細胞に対する神経突起伸展作用及び/又は神経突起数増加作用を指標としてヒストンの修飾又はDNAの修飾に関与する酵素の阻害剤をクラスター化した後、クラスター毎に評価および/またはクラスター毎に解析する工程を含む神経変性疾患治療薬のスクリーニング方法。
【請求項2】
修飾がアセチル化、メチル化、リン酸化、又はユビキチン化である請求項1に記載のスクリーニング方法。
【請求項3】
神経モデル細胞がPC12細胞である請求項1または2に記載のスクリーニング方法。
【請求項4】
クラスター毎に解析する工程がDNAマイクロアレイを用いた遺伝子発現の解析工程である請求項1〜3のいずれか1に記載のスクリーニング方法。
【請求項1】
神経モデル細胞の生存維持作用、並びに神経モデル細胞に対する神経突起伸展作用及び/又は神経突起数増加作用を指標としてヒストンの修飾又はDNAの修飾に関与する酵素の阻害剤をクラスター化した後、クラスター毎に評価および/またはクラスター毎に解析する工程を含む神経変性疾患治療薬のスクリーニング方法。
【請求項2】
修飾がアセチル化、メチル化、リン酸化、又はユビキチン化である請求項1に記載のスクリーニング方法。
【請求項3】
神経モデル細胞がPC12細胞である請求項1または2に記載のスクリーニング方法。
【請求項4】
クラスター毎に解析する工程がDNAマイクロアレイを用いた遺伝子発現の解析工程である請求項1〜3のいずれか1に記載のスクリーニング方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】

【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公開番号】特開2012−95588(P2012−95588A)
【公開日】平成24年5月24日(2012.5.24)
【国際特許分類】
【出願番号】特願2010−245306(P2010−245306)
【出願日】平成22年11月1日(2010.11.1)
【出願人】(399030060)学校法人 関西大学 (208)
【Fターム(参考)】
【公開日】平成24年5月24日(2012.5.24)
【国際特許分類】
【出願日】平成22年11月1日(2010.11.1)
【出願人】(399030060)学校法人 関西大学 (208)
【Fターム(参考)】
[ Back to top ]