
神経変性障害の治療のためのホスホリパーゼDの修飾
本発明は、神経変性疾患を治療する方法に関し、該方法は、そのような治療を必要とする被験体に、ホスホリパーゼD1および/またはホスホリパーゼD2などのホスホリパーゼDファミリーの酵素の、触媒活性をはじめとする作用を阻害するかまたは減少させる1種以上の薬剤を投与するステップを含む。本発明はまた、ホスホリパーゼDファミリーの酵素の活性を阻害するかまたは減少させる薬剤を同定するために用いることができ、かつ神経変性疾患の治療で用いることができる細胞ベースのアッセイにも関する。
【発明の詳細な説明】
【技術分野】
【0001】
助成金情報
本発明は、米国国立衛生研究所により授与された政府補助金第NIH RO1 NS056049号を用いてなされた。政府は、本発明における一部の権利を有する。
【0002】
優先権主張
本出願は、米国仮出願第61/230,447号(2009年7月31日出願)および米国仮出願第61/182,609号(2009年5月29日)に対する優先権を主張する(これら両方の内容はその全体が参照により本明細書中に組み入れられる)。
【0003】
1. 導入
本発明は、神経変性疾患を治療する方法に関し、該方法は、そのような治療を必要とする被験体に、ホスホリパーゼD1および/またはホスホリパーゼD2などのホスホリパーゼDファミリーの酵素の、触媒活性をはじめとする作用を阻害するかまたは減少させる1種以上の薬剤を投与するステップを含む。本発明はまた、ホスホリパーゼDファミリーの酵素の活性を阻害するかまたは減少させ、かつ神経変性疾患の治療方法で用いることができる薬剤を同定するために用いることができる細胞ベースのアッセイにも関する。
【背景技術】
【0004】
2. 本発明の背景
神経変性疾患は、シナプス機能不全を特徴とし、認知能力および機能的能力の進行性の低下を伴い、多くの場合に死に至る、様々な障害を包含する。アルツハイマー病(AD)は、最も一般的な加齢に関連する衰弱性神経変性障害であり、約400万人の米国人および世界中で約2000〜3000万人が罹患している。ADの古典的な神経病理学的特徴としては、海馬、扁桃体、ならびに側頭葉、前頭葉および頭頂葉の連合皮質での老人(β-アミロイド含有)斑および神経原線維変化の存在が挙げられる。より微妙な変化としては、反応性の星状膠細胞の変化、ならびに内嗅皮質および前脳基底部でのニューロンおよびシナプスの減少が挙げられる。
【0005】
アルツハイマー病の病因は完全には明らかになっていないが、この疾患と、膜タンパク質であるアミロイド前駆体タンパク質(APP)の切断産物との間に関連があることが知られている。γ-セクレターゼは、APPのアミロイド-β(Aβ)ドメインのC末端切断を媒介し、これにより、α-セクレターゼ(ADAM10およびTACE)またはβ-セクレターゼ(BACE1)による細胞外ドメイン切断を通じて生成される膜結合型APP C末端断片からAβ/p3が放出される。γ-セクレターゼ切断は、2種類の主要なAβアイソフォームであるAβ40およびAβ42を生成する。プレセニリン遺伝子であるPS1およびPS2のすべての突然変異が、γ-セクレターゼ活性の変化をもたらし、これが、場合によってはより無害なAβ40ペプチドを減らして、アミロイド生成性および神経毒性が非常に高いAβ42分子種の生成の上昇につながることが、十分に証明されている。
【0006】
ホスホイノシチド(「PI」)は、様々な細胞内経路でシグナル伝達分子として機能し(Williams, 1999, Biochim. Biophys. Acta 1441: 255-267;Rhee and Bai, 1997, J. Biol. Chem. 272(24): 15045-15048;Katan, 1998. Biochim. Biophys. Acta 1436: 5-17)、一部の細胞タイプでのPIの異常の調節は、種々のヒト疾患状態を促進することが示されている(Pendaries et al., 2003, FEBS Lett. 546(1):25-31)。PIシグナル伝達は、多数のキナーゼ、ホスファターゼ、およびホスホリパーゼにより厳重に調節されている。ホスホリパーゼC(PLC)によるホスファチジルイノシトール4,5-ビスリン酸(phosphotidylinositol 4,5-biphosphate)(PIP2)の加水分解は、様々な細胞機能の調節での初期の重要な事象である。Aβ42が、PIP2レベルの低下を引き起こすことが明らかになっている(国際特許出願第PCT/US2007/085274号、WO 2008/064244、参照により本明細書中に組み入れられる)。
【0007】
ホスホリパーゼD(PLD)は、ホスファチジルコリンの加水分解を触媒して、ホスファチジン酸を形成させる(国際特許出願第PCT/US2007/085274号、WO 2008/064244、参照により本明細書中に組み入れられる;Sweeney et al., 2002, J. Biol. Chem. 277:3030-3039; Exton et al., 2002, FEBS Lett 531:58-61;Schields and Arvan, 1999, Curr. Opin. Cell Biol. 11:489-494)。PLDは、種々の膜輸送ステップ(例えば、分泌小胞の放出、エンドサイトーシスおよびエキソサイトーシス)を調節することが報告されている(Chen et al., 1997, J Cell Biol. 138:495-504;Shen et al., 2001, Mol. Cell. Biol. 21:595-602;Humeau et al., 2001, Proc. Natl. Acad. Sci. 98:15300-5305;Cockcroft, 2001, Cell. Nol. Life Sci. 58:1674-1687)。PLD1およびPLD2は、この酵素の2種類の異なるアイソフォームであり(Hammond et al., 1995, J. Biol. Chem. 270:29640;Colley et al., 1997, Curr. Biol. 7:191;Steed et al., 1998, FASEB J. 12:1309;図1A〜Bを参照されたい)、異なる細胞内機能を有することが報告されている(Choi et al., 2002, J. Immunol. 168:5682-5689)。Caiら(2006, Proc. Natl. Acad. Sci. U.S.A.)には、PLD1がβAPPの細胞内輸送を調節することが報告されており、随伴する論文(Cai et al., 2006, Proc. Natl .Acad. Sci. U.S.A. 103:1941-1946)には、独立したメカニズムにより、PLD1がγ-セクレターゼ複合体の完全性を損なわせ、β-アミロイド形成を阻害することが報告されている。Caiらは、PLD代謝の欠陥が、アルツハイマー病の病因に関与している可能性があることを示唆している。
【発明の概要】
【0008】
3. 発明の概要
本発明は、神経変性疾患の治療方法に関し、該方法は、そのような治療を必要とする被験体に、ホスホリパーゼD1および/またはホスホリパーゼD2などのホスホリパーゼDファミリーの酵素の、触媒活性をはじめとする作用を阻害するかまたは減少させる1種以上の薬剤を投与するステップを含む。本発明はまた、ホスホリパーゼDファミリーの酵素の活性を阻害するかまたは減少させる薬剤を同定するために用いることができる細胞ベースのアッセイにも関する。
【0009】
さらなる実施形態では、本発明は、PLD阻害剤、好ましくはPLD2阻害剤がガングリオシドレベルを低下させるという知見に基づいて、上昇したガングリオシドレベルに関連する障害の治療を提供する。
【0010】
本明細書中で用いる場合、「治療する」との用語は、疾患の症状または徴候の進行速度を緩和または低下または減少させること、あるいは、限定するものではないが記憶障害(短期もしくは長期)および/または認知症をはじめとする疾患または障害の発症リスクを減少させることを意味する。本発明に従って治療することができる神経変性疾患の非限定的な例としては、アルツハイマー病、軽度認知障害、パーキンソン病、ハンチントン舞踏病、老年性認知症およびクロイツフェルト・ヤコブ病が挙げられる。本発明はまた、進行性記憶障害を抑制するためにも用いることができる。本発明に従って治療することができる上昇したガングリオシドレベルを有する障害の非限定的な例としては、GM1ガングリオシドーシス、モルキオ病B型、テイ・サックス病、サンドホフ病AB型、およびニーマン・ピック病C型が挙げられる。
【図面の簡単な説明】
【0011】
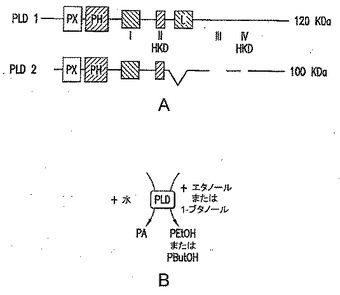
【図1】(A)PLD1/PLD2のドメイン構造を示す図である。(B)PLD酵素により媒介されるトランスホスファチジル化反応を示す図である。この反応は、それぞれ、エタノールおよび1-ブタノールの存在下で、ホスファチジルエタノール(PEtOH)またはホスファチジルブタノール(PButOH)の合成をもたらす。
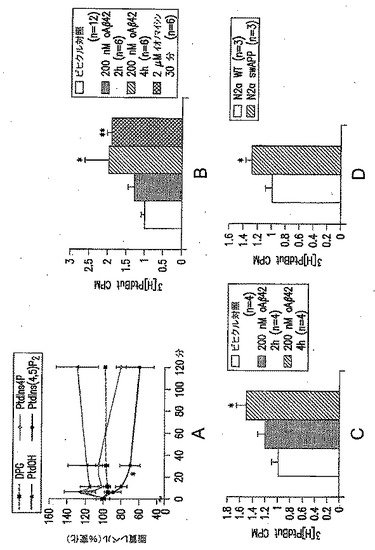
【図2−1】(A) 200nM合成oAβ42で処理した初代培養皮質ニューロンでの、ジホスホグリセリン酸(「DPG」)、PEtOH、ホスファチジルイノシトール4リン酸(「PtdIns4P」)およびPIP2(「PtdIns(4,5)P2」とも表記される)のレベルを表す図である(n=9)。(B) oAβ42または2μM Ca++イオノフォアであるイオノマイシンの存在下または非存在下における、初代培養皮質ニューロンでの、1-ブタノールの存在下でのトランスホスファチジル化反応を介した[3H]-ホスファチジルブタノールの合成により測定したPLD活性を表す図である。(C) oAβ42の存在下または非存在下における、培養N2a細胞での[3H]ホスファチジルブタノールの合成により測定したPLD活性を表す図である。(D)培養N2a細胞またはswAPP突然変異体を発現する培養N2a細胞のいずれかでの、[3H]ホスファチジルブタノールの合成により測定したPLD活性を表す図である。値は、平均±SEMを表す。ns=有意差なし。*p<0.05;**p<0.01;***p<0.001。
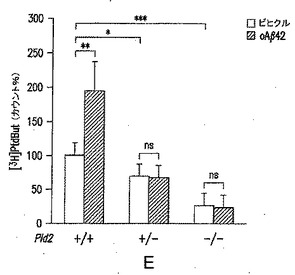
【図2−2】(E) PLD2の野生型(+/+)またはヘテロ接合(+/-)もしくはホモ接合(-/-)の突然変異体であるマウスから調製した初代培養皮質ニューロン培養物での、oAβ42に応答した[3H]ホスファチジルブタノール産生により測定したPLD2活性を表す図である。値は、平均±SEMを表す。ns=有意差なし。*p<0.05;**p<0.01;***p<0.001。
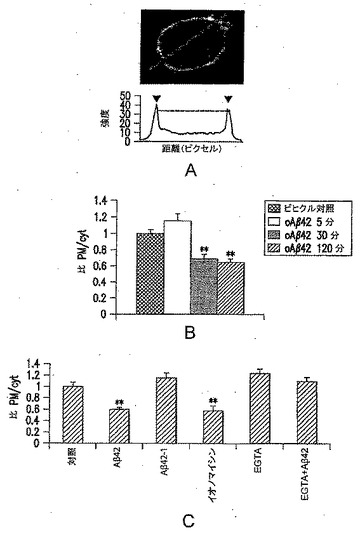
【図3−1】PC12細胞でのAβ誘導性GFP-PLD2局在変化を表す図である。(A)形質膜/細胞質比の計算を表す図である。(B)200nM合成オリゴマーAβ42の作用の定量化を表す図である。対照(n=31細胞);5分(n=29);30分(n=32)および120分(n=30)。(C) GFPPLD2の局在変化はCa2+依存的であることを表す図である。対照(n=12);200nM Aβ42(n=13);200nM逆方向ペプチドAβ42-1(n=12);2μMイオノマイシン(n=13);2mM EGTA(n=14);2mM EGTAおよび200nM oAβ42(n=13)。すべての処置は、30分間であった。
【図3−2】PC12細胞でのAβ誘導性GFP-PLD2局在変化を表す図である。(D)PLD2-GFP構築物を表す図である。
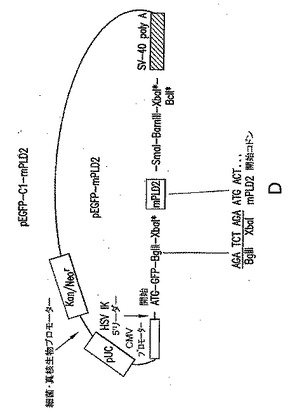
【図4】(A) PLD2免疫反応性は、Pld2 KOマウスから調製した成体脳抽出物(脱核上清(postnuclear supernatant)には存在しないことを表す図である。ウエスタンブロット分析は、示したタンパク質に対する抗体を用いたECLを用いて行なった。(B)マウスへのエタノールのIP注入に続くホスファチジルエタノール(PEtOH)生成により測定した、Pld2 KO脳での総PLD活性の減少を表す図である。PEtOHは、LC-MSにより測定した。対照として、ホスファチジルセリンのレベルを示す。N=4。
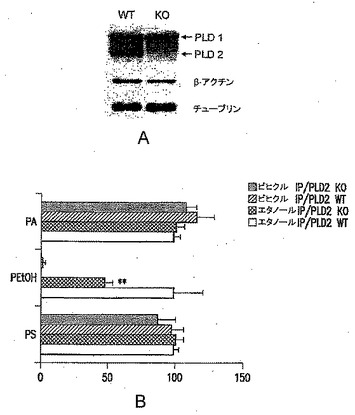
【図5】Pld2 KO皮質ニューロンでの分泌Aβレベルの低下を表す図である。培養物を、第14日にswAPPレンチウイルスに感染させ、Aβ40およびAβ42のELISA測定のために、第16日に培地を回収した。(A)総タンパク質に対して標準化し、非感染Pld2 WT培養物に対する%として表したAβレベルである。(B)標準化したAβ40およびAβ42レベル(%WT)である。N=3。
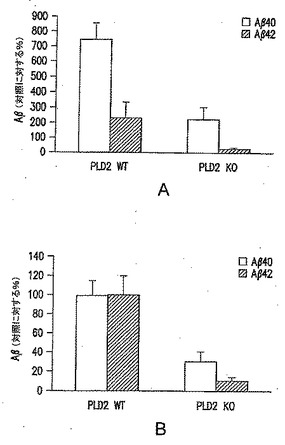
【図6】PLD2KOマウス由来の海馬は、oAβ42の存在下で正常なLTPを示すことを表す図である。ビヒクルの存在下では、PLD2WTスライス(n=10)とPLD2KOスライス(n=9)との間でLTPに差がなかった(F1,17=0.00、p=0.947)。PLD2WTスライスは、200nM oAβ42の浴浸漬(bath application)に続いてLTPの減少を示したが(n=8)(F1,16=5.19、p=0.038、ビヒクルとの比較)、PLD2KOスライスは、ペプチドの存在下でLTPの差異を示さなかった(n=8)(F1,14=0.01、p=0.919、ビヒクルとの比較)。fEPSP、CA1領域興奮性シナプス後電位。バーは、oAβ42の浴浸漬の時間を表す。3本の矢印は、増強を誘発するために用いたバースト刺激を表す。動物は、約3ヵ月齢であった。
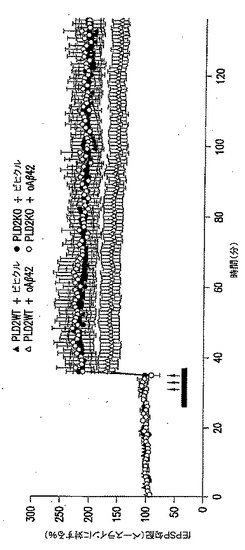
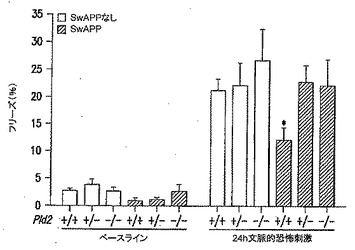
【図7】PLD2除去がSwAPPマウスでの学習および記憶を改善することを表す図である。SwAPPマウス(Tg2576)をPld2ノックアウトマウスと交配させて、得られた子孫[Pld2+/+/tgなし(n=14);Pld2+/-/tgなし(n=14);Pld2-/-/tgなし(n=11);Pld2+/+/SwAPP(n=10);Pld2+/-/SwAPP(n=12);Pld2-/-/SwAPP(n=11)]を文脈的恐怖記憶についてのトレーニングに供し、足ショックの24時間後に評価した。5〜6ヵ月齢の動物を用いた。*、p<0.05(スチューデントの片側(one tail)t検定)。
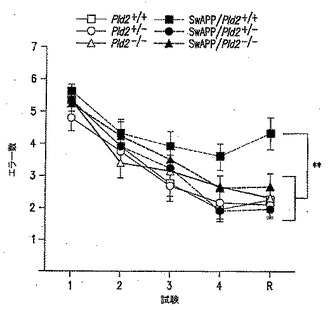
【図8】PLD2除去がSwAPPマウスでの学習および記憶を改善することを表す図である。12ヵ月齢マウスを、放射状水迷路(RAWM)試験に供した。試験の最後の3日間で、エラーをスコア付けした。Pld2-/-/SwAPP(n=7)およびPld2+/+/SwAPP(n=6)を除くすべての遺伝子型について、n値は8であった。**、p<0.01。値は平均±SEMを表す。
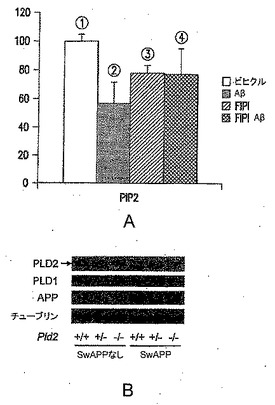
【図9−1】(A) FIPIがoAβ42誘導性のPIP2レベルの低下を部分的に回復させることを表す図である。2週齢の初代皮質神経培養物を、ビヒクル(「1」)、200nM oAβ42で2時間(「2」)、750nM FIPIで3時間(「3」)、または750nM FIPIで1時間の前処理とそれに続く200nM oAβ42で2時間の処理(「4」)のいずれかで急性的に処理した。n=3。(B)野生型またはPLD2のホモ接合突然変異体(±SwAPP)であるマウスでのPLD1およびPLD2レベルを示す図である。
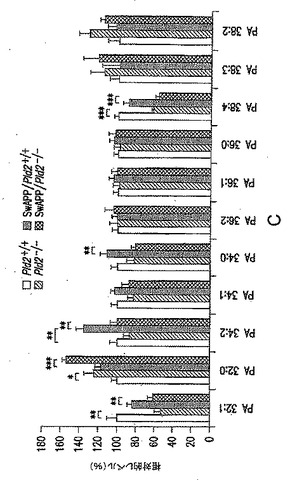
【図9−2】(C)対照と比較した、示した突然変異体マウスでのPA分子種の相対量を示す図である。値は平均±SEMを表す(n=6〜8)。*p<0.05;**p<0.01;***p<0.001。
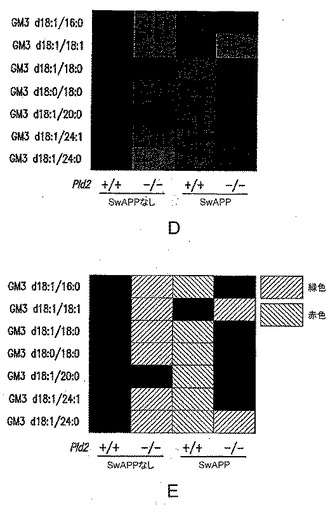
【図9−3】(D)野生型またはPLD2のホモ接合突然変異体(±SwAPP)であるマウスでのGM3レベルを示す図である(カラーバージョン)。(E)野生型またはPLD2のホモ接合突然変異体(±SwAPP)であるマウスでのGM3レベルを示す図である(図9Dの白黒の簡素化;色を表すための平行線模様:右上がり=緑色、右下がり=赤色、色のグラデーションは示していない)。
【図10】FIPI誘導性のAβの産生低下を示す図である。コンフルエンスに達したswAPP発現N2a細胞を、新しい培地に置き換え、ビヒクル(黒色)またはFIPI(灰色)で6時間処理した。培地を回収し、Aβ40(A)およびAβ42(B)の両方をELISAにより測定した。n=6。
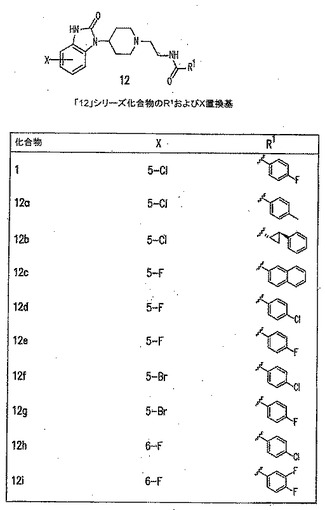
【図11A】PLD阻害剤を示す図である。
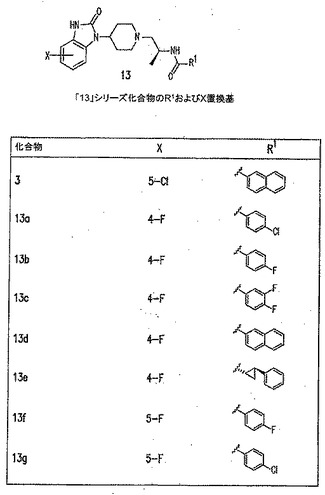
【図11B】PLD阻害剤を示す図である。
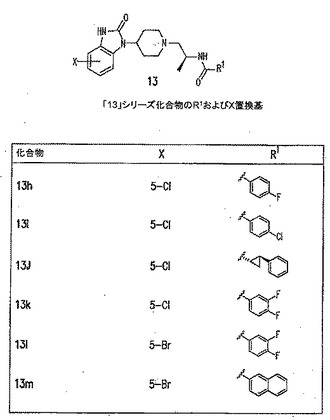
【図11C】PLD阻害剤を示す図である。
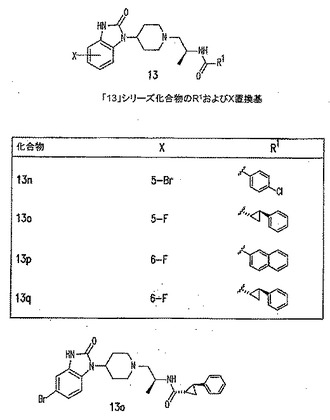
【図11D】PLD阻害剤を示す図である。
【図11E】PLD阻害剤を示す図である。
【図11F】PLD阻害剤を示す図である。
【図11G】PLD阻害剤を示す図である。
【図11H】PLD阻害剤を示す図である。
【図11I】PLD阻害剤を示す図である。
【図11J】PLD阻害剤を示す図である。
【図11K】PLD阻害剤を示す図である。
【図11L】PLD阻害剤を示す図である。
【図11M】PLD阻害剤を示す図である。
【発明を実施するための形態】
【0012】
5. 発明の詳細な説明
本発明は、ホスホリパーゼD1(PLD1)および/またはホスホリパーゼD2(PLD2)などのホスホリパーゼDファミリーの酵素の、触媒活性をはじめとする作用を阻害するかまたは減少させることにより、アミロイドβの生成を減少させ、かつ/またはアミロイドβの毒性作用を阻害する薬剤の使用に関し、神経変性疾患の治療のためのそのような薬剤の使用を含む。本発明はまた、ホスホリパーゼDファミリーの酵素の作用および/もしくは活性を阻害するかまたは減少させ、かつ本明細書中に記載した治療方法で用いることができる薬剤を同定するために用いることができるアッセイシステムにも関する。
【0013】
明確にするために、限定ではなく、本発明の詳細な説明を、以下の小節に分ける:
(i) PLD阻害剤;
(ii)アッセイシステム;および
(iii)治療方法。
【0014】
5.1 PLD阻害剤
PLD1阻害剤およびPLD2阻害剤を含むPLD活性の阻害剤を、本発明に従って用いることができる。PLD阻害剤は、それを投与される被験体体内に存在するPLD活性の量を減少させ、酵素活性の直接的阻害ならびにPLDの量または利用可能性の減少をはじめとするいずれのメカニズムによってそれを行なってもよい。
【0015】
一部の非限定的実施形態では、本発明は、PLD(PLD1および/またはPLD2であり得るが、PLD2の阻害が好ましい)の酵素活性を阻害する薬剤の使用を提供する。
【0016】
1つの非限定的実施形態では、PLD2をはじめとするPLDを阻害する薬剤は、5-フルオロ-2-インドリルデス-クロロハロペミド(「FIPI」)である。
【0017】
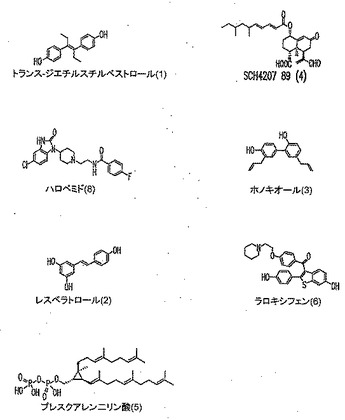
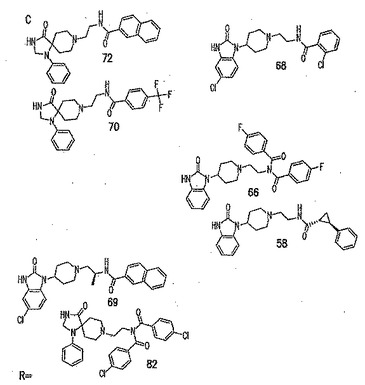
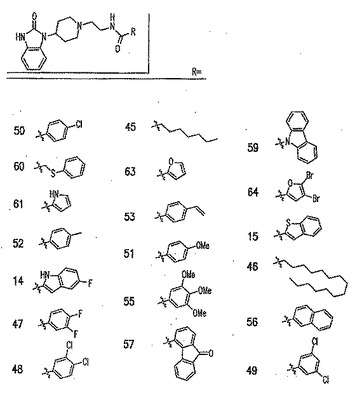
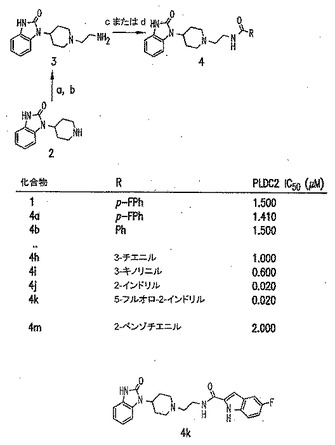
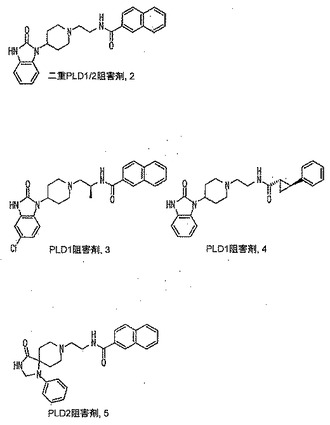
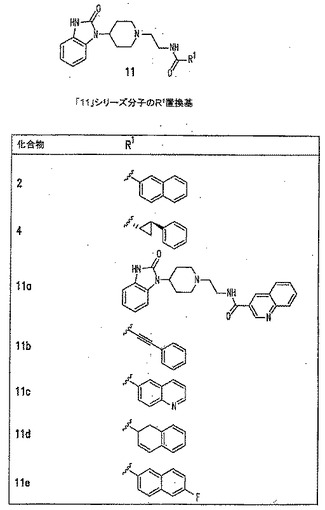
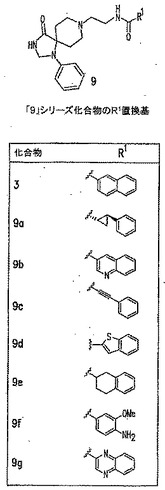
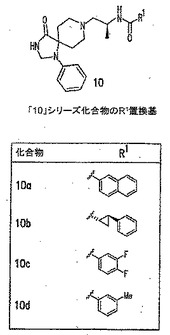
本発明に従って用いることができるさらなるPLD阻害剤としては、限定するものではないが、以下のものが挙げられる:ジエチルスチベストロール、レスベラトロール、ホンキオール、SCH420789、プレスクアレン二リン酸、ラロキシフェン、ハロペミド、4-ヒドロキシタモキシフェン、図11A〜Cに示された化合物(Scott et al., 2009, Nat Chem Biol 5(2):108-117およびNature Chemical Biology 10.1038/nchembio.140でオンラインで入手可能な補足情報);ハロペミド誘導体、特に2-インドリル部分を含むハロペミド誘導体、図11Dに示された化合物を含む(Monovich et al., 2007, Bioorg. Med. Chem. Lett. 17:2310-2311);図11E〜Jに示された化合物、限定するものではないが、ハロゲン化ピペリジニルベンゾイミダゾロン部分およびS-メチル部分を含むハロペミド誘導体を含む(Lewis et al., 2009, Bioorg. Med. Chem. Letts. 19:1916-1920);図11Eの化合物5の誘導体、1,3,8-トリアザスピロ[4,5]デカン-4-オン構造を含む化合物を含む、図11K〜Lに示された化合物を含む(Lavieri et al., 2009, Bioorg. Med. Chem. Lett. 19:2240-2243);および図11Mに示された化合物(Lavieri et al., 2009, Bioorg. Med. Chem. Lett. 19:2240-2243)。
【0018】
特に好ましい非限定的実施形態では、PLD阻害剤は、限定するものではないが、以下のものなどのPLD2選択的阻害剤である:4-OHタモキシフェン;図11Bの化合物72および82(Scott et al., 2009, Nat Chem Biol 5(2):108-117);図11Dの化合物4jおよび4k(Monovich et al., 2007, Bioorg. Med. Chem. Lett. 17:2310-2311);図11Eの化合物5(Lewis et al., 2009, Bioorg. Med. Chem. Letts. 19:1916-1920);および図11Eの化合物5の誘導体、1,3,8-トリアザスピロ[4,5]デカン-4-オン構造を含む化合物を含む、図11K〜Lに示された化合物を含む(Lavieri et al., 2009, Bioorg. Med. Chem. Lett. 19:2240-2243)。上記の参考文献のそれぞれおよびそれらに関連付けられたいかなる公衆に供給された補足情報、ならびにその中に示されたいかなる化合物および/または合成スキームも、その全体が参照により本明細書中に組み入れられる。
【0019】
本発明の別の好ましい非限定的実施形態では、PLD阻害剤は、図11Cの化合物56(図11Eの二重PLD1/2阻害剤2および図11Fの化合物2でもある)であり、これは、N-(2-(4-(2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)エチル)-2-ナフタミドとしても知られ、81nMのPLD1 IC50、240nMのPLD2 IC50、380nMの293-PLD2 IC50、および21nMのCalu-1 IC50を有する(Scott et al., 2009, Nature Chemical Biology 5:108-117)。
【0020】
本発明の別の好ましい非限定的実施形態では、PLD阻害剤は、図11Jの化合物13rであり、これは、(1R,2R)-N-((S)-1-(4-(5-ブロモ-2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)プロパン-2-イル)-2-フェニルシクロプロパンカルボキサミドとしても知られ、15nMのPLD1 IC50、1100nMのPLD2 IC50、6400nMの293-PLD2 IC50、および3.7nMのCalu-1 IC50を有し(Lewis et al., 2009, Bioorganic and Medicinal Chemistry Letters 19:1916-1920);この化合物は、PLD1選択的である。
【0021】
本発明の別の好ましい非限定的実施形態では、PLD阻害剤は、図11Kの化合物9bであり、これは、N-(2-(4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4,5]デカン-8-イル)エチル)キノリン-3-カルボキサミドとしても知られ、20,000nMのPLD1 IC50、500nMのPLD2 IC50、90nMの293-PLD2 IC50、および1900nMのCalu-1 IC50を有し(Lavieri et al., 2009, Bioorganic and Medicinal Chemistry Letters 19:2240-2243);この化合物は、PLD2選択的である。
【0022】
さらなるPLD阻害剤は、当技術分野で公知の方法により同定することができ、そのような方法としては、限定するものではないが、Scott et al., 2009, Nat Chem Biol. Feb;5(2):108-17;Monovich et al., 2007, Bioorg. Med. Chem. Lett. 17:2310-2311;Lewis et al., 2009, Bioorg. Med. Chem. Letts. 19:1916-1920;またはLavieri et al., 2009, Bioorg. Med. Chem. Lett. 19:2240-2243に記載されたアッセイが挙げられる。
【0023】
あるいは、PLD阻害剤は、PLD、特にPLD2の発現を減少させる分子であり得、例えば、PLD1および/またはPLD2遺伝子に相補的な部分を含む小分子干渉RNAまたはアンチセンスRNAである。
【0024】
5.2 アッセイシステム
一部の非限定的実施形態では、本発明は、PLD(例えば、PLD2)を阻害する薬剤を同定するためのアッセイシステムを提供し、このアッセイシステムは、図3A〜Cに示されている特徴を有する。例えば、被験薬剤が細胞内(例えば、PC12細胞;Ca++イオンの存在下)でのPLD2(例えば、検出可能にタグ付加されたPLD2(例えば、緑色蛍光タンパク質でタグ付加されたもの))の移行を変化させる能力を試験することができ、ここで、被験薬剤がAβ42誘導性の細胞膜から細胞質へのPLD2の移行を阻害する能力は、その薬剤が潜在的な本発明の治療剤であること、および場合によっては、その治療活性を、例えば限定するものではないが、Tg2576マウスでのin vivo試験で確認することができる(図8を参照されたい)ことを示す。
【0025】
非限定的実施形態では、本発明はさらに、PLD(好ましくはPLD2)または膜から移行することができるその一部分の蛍光型を発現するように遺伝子操作された細胞(好ましくは神経細胞)または細胞株(好ましくは神経細胞株)を利用するアッセイシステムを提供する。蛍光タンパク質は、例えば、限定するものではないが、緑色蛍光タンパク質、増強型緑色蛍光タンパク質、黄色蛍光タンパク質、赤色蛍光タンパク質、または当技術分野で公知の他の蛍光タンパク質であり得る。PLDは、ヒト、ラットもしくはマウス由来のPLDタンパク質、またはその移行可能な断片であり得る(例えば、限定するものではないが、GenBank登録番号AAH15033、AAH56871、NP002654、EHW90405、EHW90406、AA021120、AAD04197、AAB96656、AAB96655、NP002653、NP001123553、AAH68976、CAB76564、NP150641、AAM48521、BAA24078、U87557、NP032902、NP032901、AAH68144、またはNC000077.5を参照されたい)。例えば、そのようなアッセイシステムでは、限定するものではないが、PC12細胞またはN2a細胞などの神経細胞株を用いることができる。あるいは、限定するものではないが、CHO、NIH 3t3、HEK293、またはHeLa(72)などの非神経細胞株を用いることができる。
【0026】
具体的な非限定的実施形態では、褐色細胞腫細胞株であるPC12を、PLD(好ましくはPLD2)のコード配列全体またはAβオリゴマーでの処理に際して細胞質に移行するそのような配列の一部分に連結された蛍光タンパク質を含む融合タンパク質をコードする構築物でトランスフェクションすることができる。16〜24時間後、落射蛍光顕微鏡法を用いて、細胞質と比較した形質膜での蛍光PLD2の分布を可視化するために用いることができる。対照細胞では、蛍光は、細胞同士を隔てる辺縁部として見えるはずであり、したがって、形質膜に濃縮されている。oAβ42(oAβ42は、オリゴマーAβまたはAβ42もしくはAβ40をはじめとする他のAβ分子種の他の誘導体を意味する)を用いた細胞の処理は、数分以内に、形質膜からのプローブの著明な消失および細胞質での蛍光レベルの対応する増加を誘導するはずであり、より拡散して見えるはずである。この作用は、イオノマイシンを用いた処理により模倣することができる。oAβ42の非存在下でのPLD2トランスフェクション細胞では、被験薬剤が細胞表面でのPLD2の局在を増加させる能力は、細胞質の平均蛍光強度に対する形質膜の蛍光強度の比の増大として検出することができる。
【0027】
したがって、本発明は、細胞表面結合型PLD(好ましくはPLD2)を増加させる薬剤を同定する方法を提供し、該方法は、(i)蛍光PLD(好ましくはPLD2)センサーを含む宿主細胞を準備するステップ;(ii)被験薬剤を該宿主細胞に投与するステップ;および(iii)細胞質の平均蛍光に対する形質膜の蛍光の比を測定するステップを含み、ここで、この比の増大が、宿主細胞表面でのPLD(好ましくはPLD2)レベルの増大を示す。
【0028】
別の実施形態では、本発明は、oAβ42の毒性作用を阻害する薬剤を同定する方法を提供し、該方法は、以下のステップ:(i)蛍光PLD(好ましくはPLD2)センサーを含む宿主細胞を準備するステップ;(ii)該宿主細胞を毒性濃度のoAβ42に曝露するステップ;(iii)該宿主細胞に被験薬剤を投与するステップ;および(iv)形質膜および細胞質での蛍光を測定するステップを含み、ここで、細胞質に対する形質膜での蛍光の比の増大が、該被験薬剤がoAβ42の毒性作用を阻害することを示す。
【0029】
また別の非限定的実施形態では、本発明は、PLD(好ましくはPLD2)の阻害剤および/またはADに対する治療剤である薬剤を同定する方法を提供し、該方法は、以下のステップ:(i)宿主細胞を準備するステップ;(ii)該宿主細胞に被験薬剤を投与するステップ;(iii)該被験薬剤の投与が、該宿主細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルを低下させるか否かを決定するステップを含み、ここで、PA 34:2、PA 34:0、および/またはGM3のレベルの低下が、該被験薬剤がPLD(例えば、PLD2)の阻害剤であり、ADを治療するために用いることができることを示す。例えば、図9C〜Dを参照されたい。例えば、被験薬剤に曝露された宿主細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルを、適切な対照細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルと比較することができる。リン脂質脂肪酸(PA)組成物に対する命名法は、合計鎖長:不飽和結合の数として表す。これらの実施形態では、宿主細胞は、限定するものではないが、神経細胞もしくは神経細胞株または組織移植片(例えば、皮質組織)であり得、あるいは、宿主細胞は、マウス、例えば限定するものではないが、SwAPP突然変異を保持するマウスなどの非ヒト試験動物体内のものであり得る。1つの具体的な非限定的実施形態では、宿主細胞は、褐色細胞腫細胞、例えば、PC12細胞であり得る。
【0030】
5.3 治療方法
一部の非限定的実施形態では、本発明は、そのような治療を必要とする被験体に、有効量のPLDの阻害剤、例えば、PLD1および/またはPLD2阻害剤を投与することによる、アミロイド生成性ペプチドに関連するシナプス機能不全、記憶障害および/または神経変性を抑制する方法を提供する。
【0031】
そのような治療を必要とする被験体は、PLD酵素を有するヒトまたは非ヒト被験体であり得る。前記被験体は、シナプス機能不全、記憶障害および/もしくは神経変性に罹患している場合があるか、または、例えば限定するものではないが、年齢、家族歴、もしくは毒性物質への曝露により、これらの状態の1以上を発症するリスクがある場合がある。
【0032】
一部の非限定的実施形態では、本発明は、ホスホリパーゼD1および/またはホスホリパーゼD2などのホスホリパーゼD酵素の作用をブロックするかまたは部分的にブロックすることにより、アミロイド生成(すなわち、Aβ40およびAβ42などの毒性Aβ分子種の生成)を減少させる方法を提供する。一部の関連する非限定的実施形態では、本発明は、神経細胞に対するAβ42ペプチドの毒性作用から保護する方法を提供し、該方法は、該細胞を有効量のホスホリパーゼD阻害剤に曝露するステップを含む。
【0033】
一部の非限定的実施形態では、本発明は、神経変性疾患または障害を治療する(例えば、その症状を減少させ、かつ/またはその進行を遅らせ、かつ/または発症リスクを減少させる)方法を提供し、そのような疾患または障害とは、限定するものではないが、アルツハイマー病、軽度認知障害、パーキンソン病、ハンチントン舞踏病、老年性認知症、および/またはクロイツフェルト・ヤコブ病などのプリオン関連疾患(prior-related disease)などである。
【0034】
一部の非限定的実施形態では、本発明は、被験体での記憶障害の進行を抑制する方法を提供し、該方法は、該被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む。一部の非限定的実施形態では、本発明は、記憶障害および/または認知症の発症リスクを減少させる方法を提供し、該方法は、被験体(例えば、少なくとも約40歳または少なくとも約50歳または少なくとも約60歳のヒト被験体)に、有効量のPLD阻害剤を投与するステップを含む。
【0035】
さらなる非限定的実施形態では、本発明は、ガングリオシドレベルの上昇に関連する障害を治療する方法を提供し、該方法は、そのような治療を必要とする被験体に、有効量のPLD阻害剤、好ましくはPLD2阻害剤を投与し、それにより該被験体の臨床状態を緩和し、かつ/またはガングリオシドレベルを低下させるステップを含む。ガングリオシドレベルの上昇に関連する障害の非限定的な例は、GM1ガングリオシドーシス、モルキオ病B型、テイ・サックス病、サンドホフ病AB型、およびニーマン・ピック病C型である。
【0036】
本節で論じた方法で用いることができるPLD阻害剤は、上記の5.1節に示されている。
【0037】
PLD阻害剤は、限定するものではないが、経口投与、皮下投与、筋肉内投与、静脈内投与、髄腔内投与、吸入投与、または直腸投与によるものをはじめとする、当技術分野で公知のいずれかの好適な経路により投与することができる。
【0038】
特定の非限定的実施形態では、PLD阻害剤はFIPIであり、約50〜2500nM、または約250〜2000nM、または約250〜1000nMの脳脊髄液中濃度を達成するために投与される。PLD阻害剤がFIPIでない場合、(FIPIでない)PLD阻害剤についての用量範囲は、FIPIのEC50に対する該PLD阻害剤のEC50の比を、FIPIについての上記の用量範囲に乗算することにより、決定することができる。EC50は、例えば限定するものではないが、本明細書中に記載されたアッセイにより測定され、該薬剤が、oAβ42誘導性の形質膜から細胞質へのPLDの移行を阻害する、または初代培養皮質ニューロンでのoAβ42誘導性のPIP2の減少を阻害する、またはswAPPを発現する培養ニューロンでのAβ42生成を減少させる、またはTg2576マウスなどのADの動物モデルでの行動遂行を改善する能力などである。
【0039】
PLD阻害剤が図11Cの化合物56(これは、図11Eの二重PLD1/2阻害剤2および図11Fの化合物2でもある)(N-(2-(4-(2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)エチル)-2-ナフタミドとしても知られ、81nMのPLD1 IC50、240nMのPLD2 IC50、380nMの293-PLD2 IC50、および21nMのCalu-1 IC50を有する)である1つの特定の非限定的実施形態では、該PLD阻害剤は、約10〜2000nM、または約10〜1000nM、または約10〜500nM、または約200〜1000nM、または約200〜500nMの脳脊髄液中濃度を達成するために投与することができる。
【0040】
PLD阻害剤が図11Jの化合物13r((1R,2R)-N-((S)-1-(4-(5-ブロモ-2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)プロパン-2-イル)-2-フェニルシクロプロパンカルボキサミドとしても知られ、15nMのPLD1 IC50、1100nMのPLD2 IC50、6400nMの293-PLD2 IC50、および3.7nMのCalu-1 IC50を有する)である別の特定の非限定的実施形態では、該PLD阻害剤は、約2〜10,000nM、または約2〜200nM、または約2〜100nM、または約2〜50nM、または約500〜8000nM、または約500〜2000nMの脳脊髄液中濃度を達成するために投与することができる。
【0041】
PLD阻害剤が図11KのPLD2選択的阻害剤化合物9b(N-(2-(4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4,5]デカン-8-イル)エチル)キノリン-3-カルボキサミドとしても知られ、20,000nMのPLD1 IC50、500nMのPLD2 IC50、90nMの293-PLD2 IC50、および1900nMのCalu-1 IC50を有する)である別の特定の非限定的実施形態では、該PLD阻害剤は、約30〜1500nM、または約50〜1000nM、または50〜800nM、または50〜600nMの脳脊髄液中濃度を達成するために投与することができる。
【0042】
特定の非限定的実施形態では、PLD阻害剤は、1日1回以上、週1回以上、または月1回以上、投与することができる。治療期間は、連続的でも不連続的であってもよい。
【実施例】
【0043】
6. 実施例1
PLD1/PLD2のドメイン構造を、図1Aに示している。領域I〜IVは、触媒ドメインの主要な決定要素であり、HKD特徴モチーフ(IIおよびIV)は、コンセンサス配列HxK(x)4D(x)6GSxNを示す。PLD1は、触媒活性の調節に関与する追加の領域(活性化ループ、L)を含む。PLD酵素により媒介されるトランスホスファチジル化反応は、図1Bに示され、この反応は、それぞれ、エタノールおよび1-ブタノールの存在下で、ホスファチジルエタノール(PEtOH)またはホスファチジルブタノール(PButOH)の合成をもたらす。
【0044】
初代培養皮質ニューロンを200nM合成オリゴマー化Aβ42(「oAβ42」)で処理した場合、ホスファチジン酸(PtdOH)のレベルは、上昇することが観察された(図2A)。さらに、oAβ42の急性的な細胞外添加(200nM)は、新生マウス由来の初代培養皮質ニューロン(図2B)および神経芽細胞腫細胞株Neuro2A(N2A)(図2C)において、ホスホリパーゼD(PLD)の酵素活性を刺激することが見出された。神経芽細胞腫細胞株N2Aでのアミロイド前駆体タンパク質のスウェーデン型突然変異体(swAPP)の過剰発現は、Aβの生成および分泌の増加をもたらすことが示されており、PLDの酵素活性を刺激することも見出された(図2D)。
【0045】
図2Eに示されているように、PLD2を欠損した培養ニューロンは、Aβ42オリゴマーを用いた処理に応答しない。初代培養皮質ニューロン培養物を第12日に[3H]ミリスチン酸で標識し、第15日に処理を行ない、続いて脂質を抽出し、[3H]ホスファチジルブタノールカウント/合計カウントの比をPLD活性の指標として用いた。ビヒクルまたは200μM oAβ42を用いた4時間の処理を行なった後に、Pld2+/+(ビヒクルおよびoAβ42処理のそれぞれに対して、n=19および12)、Pld2+/-(n=7)、およびPld2-/-(それぞれ、n=5および7)でのPLD活性測定を行なった。
【0046】
PLD2の細胞内局在をモニタリングするために、GFPタグ付加PLD2構築物を作製した(図3D)。構築物の作製では、標準的な細菌株ではメチル化がXbaI部位およびBclI部位での切断を妨害するが、mPLD2はXbaIおよびSmaI部位を用いて切断することができた(内部にBamHI部位およびBglII部位がある)。pEGFPは約4.7kb長であり、mPLD2 cDNAは約3kb長であった。哺乳動物細胞に最適化されたGFPの赤側シフト変異体をコードするので、pEGFP(Clontech)を用いた。PLD2(マウス)のGenBank登録番号は、U87557である。GFP-PLD2コード構築物を、Lipofectamine 2000によりPC12細胞に導入した(Hammond et al., 1995, J. Biol. Chem. 270:29640-29643;Colley et al., 1997, Current Biology 7:191-201;Sung et al., 1997, EMBO J. 16:4519-4530;Sung et al., 1999, J. Biol. Chem. 274:3659-3666;およびSung et al., 1999, J. Biol. Chem. 274:494-502を参照されたい)。oAβ42の急性的な細胞外添加(200nM)は、Ca2+依存的に(すなわち、EGTAなどの細胞外Ca2+を捕捉する薬剤によりブロックされる作用)、褐色細胞腫細胞株PC12で、細胞表面から細胞質へのGFPタグ付加ホスホリパーゼD2(PLD2)の移行をもたらす(図3A〜C)。
【0047】
神経系でのPLD2の役割を調べるためのin vivoモデル系を提供するために、Cre-LoxP技術を用いて、PLD2「ノックアウト」マウス(「PLD2KOマウス」)を作製した。PLD2に対する免疫反応性は、これらのマウスの脳抽出物には存在しないことが示され、このことは、ノックアウト動物ではPLD2が発現されていないことを示す(図4A)。さらに、Pldノックアウトマウスの脳では、合計のPLD活性が約半分まで減少していることが示された(図4B)。PLD2ノックアウトマウスは生存可能であり、これまでに、明らかな異常を示すことは観察されていない。
【0048】
PLD2 KO動物でAβ42の作用を調べるための実験の第1シリーズでは、WTおよびKOマウスの初代皮質ニューロンを回収し、培養で確立し、swAPPレンチウイルスを感染させた。ニューロンでの全長swAPPの発現は、典型的には、多量の分泌型Aβ40およびAβ42をもたらした。得られたPld2 KOニューロンは、野生型培養物と比較して、少量のAβ40およびAβ42を分泌することが観察された(図5A〜B)。
【0049】
次に、長期増強(「LTP」)に対するPLD2KOの影響を、KO動物から作製した海馬脳スライスで試験した。LTPは、電気生理学的技術を用いて測定され、多くの事例で学習および記憶と関連する現象である。Aβ(および特にAβオリゴマー)がLTPを妨害することが、いくつかのグループにより以前に示されており、軽度認知障害およびアルツハイマー病に伴う認知障害に対する考えられる基礎を与えている。図6に示されているように、oAβ42に曝露されたPLD2野生型マウス由来の海馬はLTPの減弱を示した一方で、PLD2KOマウス由来の海馬はoAβ42の存在下で正常なLTPを示した。
【0050】
Aβ42過剰発現の状況でのPLD2KOのin vivoでの影響を試験するために、PLD2KOマウスをswAPPマウスと交配させた。PLD2の除去に関してホモ接合およびヘテロ接合の子孫を作製し、続いて2種類の行動試験の遂行(パフォーマンス)を対照動物(野生型PLD2を有するswAPP突然変異体動物を含む)と比較した。文脈的恐怖条件付け(FC)パラダイムでの試験結果を図7に示し、放射状水迷路(RAWM)パラダイムでの試験結果を図8に示す。これらの試験パラダイムの両方で、swAPPを過剰発現するマウス(Tg2576系統)でのPld2の1コピーまたは2コピーの遺伝的不活性化が、Tg2576(swAPP発現)マウスの特徴である学習障害を緩和した。
【0051】
PLD2の遺伝的除去のAβ42保護効果が上記の実験で実証されたため、この酵素の化学的阻害の影響を評価するために実験を行なった。図9Aに示されているように、PLD(PLD2を含む)の薬理学的阻害剤である5-フルオロ-2-インドリルデス-クロロハロペミド(「FIPI」)が、oAβ42の急性的な細胞外添加(200nM)に続く、初代培養皮質ニューロンでのPIP2欠損を部分的に回復させることが見出された。FIPIは、ハロペミドの類似体であり、元々はMonovichらによって特性決定され(Monovich, L. et al. Optimization of halopemide for phospholipase D2 inhibition. Bioorg Med Chem Lett 17, 2310-2311 (2007))、Michael Frohmanのグループによりさらに特性決定された(Su, W. et al. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol 75, 437-446 (2009))。
【0052】
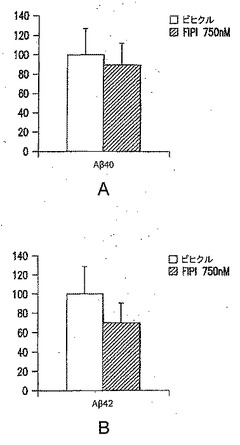
次に、PAおよびガングリオシドGM3レベルに対するSwAPP過剰発現およびPld2遺伝子型の影響を調べるために、実験を行なった。図9Bに示されているように、PLD2、PLD1、APPおよびチューブリンのウエスタンブロット分析により、タンパク質レベルを評価した(代表的なブロットを示す)。SwAPPトランスジーンを有するか有しないPld2+/+およびPld2-/-マウスから前脳脂質を抽出し、LC-MS分析に供し、PA分子種の相対量を突然変異体マウスで測定し、対照マウス(Pld2+/+、SwAPPなし)と比較した。結果を図9Cに示す。PA分子種である34:2および34:0(矢印)は、アルツハイマー病のバイオマーカー候補であり、Pld2ノックアウト(KO)でのその減少は、PLD2阻害剤の作用をモニタリングするためにこれらのバイオマーカーを用いることができることを示す。リン脂質脂肪酸組成物に対する命名法は、合計鎖長:不飽和結合の数として表す。図9Dに示されているように、糖スフィンゴ脂質(ガングリオシド)であるGM3は、アルツハイマー病マウス突然変異体(赤色)で上昇し、この疾患のバイオマーカーとして用いることができる。GM3のレベルは、Pld2 KO(緑色)では低下する。SwAPP突然変異体では、PLD2の除去が、GM3の正常レベルを回復させる(黒色)。
【0053】
さらに、FIPIで処理した場合、swAPPを発現するN2A細胞が、より少量のAβ42を分泌することが見出された(図10)。FIPIはPLD1およびPLD2の両方をブロックするので、この薬物の保護効果が、PLD1、PLD2またはその両方のどれを含むかは明らかでない。
【0054】
まとめると、PLD2の遺伝的除去(部分的除去および完全除去の両方)それ自体は、ノックアウトマウスに対して生体レベルでは実質的な影響を有しないようであり、PLD2の遺伝的除去は種々のアルツハイマー病実験モデルの設定では保護を付与するので、PLD2が、薬理学的阻害の合理的な標的として浮上してくる。同様に、PLD1の除去は、いかなる明らかな異常をもたらすことも観察されておらず、このことは、PLD1阻害に依存する治療薬も、治療的効果を有し得ることを示唆している。
【0055】
7. 実施例2
徐々に多くなる証拠は、アルツハイマー病(AD)が、脂質代謝での深刻な変化に関連すること、そしてこれらの変化は、シナプス機能不全および認知低下の基盤となる分子経路の揺らぎの原因である可能性があることを示している。本研究では、本発明者らは、アミロイドβ(Aβ)と、複数の細胞内プロセスを制御する重要なシグナル伝達リン脂質であるホスファチジン酸(PtdOH)との関連を調べた。本発明者らは、可溶性Aβ1-42オリゴマーでの培養ニューロンの処理が、PtdOHレベルを上昇させることを以前に報告していた。ホスホリパーゼD(PLD)経路はPtdOHの生体活性プールの主要な供給源であるため、本発明者らは、このファミリーの脂質酵素に着目した。その結果、Aβ1-42オリゴマーでの培養ニューロンおよび神経芽細胞腫細胞の処理ならびにAPPのスウェーデン型突然変異体(swAPP)の発現は、PLD活性の著明な増大をもたらす。本発明者らはまた、Aβ1-42オリゴマー処理が、形質膜から細胞質へのPLD2の移行を促進することも示し、このことは、PLD2がAβシグナル伝達経路に存在することを示唆する。ADにおけるPLD経路の重要性を遺伝学的に調べるために、本発明者らは、Pld2遺伝子の条件付き欠失を有するマウスを作製した。本発明者らの結果は、いかなる明白な表現型も引き起こさないPld2除去が、海馬スライスでの長期増強(LTP)に対するAβのシナプス障害作用を抑制することを示し、このことは、Pld2除去が細胞毒性ペプチドに対する保護を与えることを示唆する。際立って、本発明者らによる行動解析は、Pld2の1コピー(swAPP/Pld2+/-)または2コピー(swAPP/Pld2-/-)のいずれかを欠損しているADのトランスジェニックマウスモデル(swAPP)で、文脈的学習が改善していることを示す。つまり、これらの知見は、PLD2経路が、Aβオリゴマーの細胞毒性作用の一部を仲介していること、およびこの経路をブロックすることにより、ADに関連するシナプス機能不全および認知低下を軽減できることを示唆する。
【0056】
種々の刊行物を本明細書中で引用したが、これによりそれらの内容はその全体が本明細書中に組み入れられる。
【技術分野】
【0001】
助成金情報
本発明は、米国国立衛生研究所により授与された政府補助金第NIH RO1 NS056049号を用いてなされた。政府は、本発明における一部の権利を有する。
【0002】
優先権主張
本出願は、米国仮出願第61/230,447号(2009年7月31日出願)および米国仮出願第61/182,609号(2009年5月29日)に対する優先権を主張する(これら両方の内容はその全体が参照により本明細書中に組み入れられる)。
【0003】
1. 導入
本発明は、神経変性疾患を治療する方法に関し、該方法は、そのような治療を必要とする被験体に、ホスホリパーゼD1および/またはホスホリパーゼD2などのホスホリパーゼDファミリーの酵素の、触媒活性をはじめとする作用を阻害するかまたは減少させる1種以上の薬剤を投与するステップを含む。本発明はまた、ホスホリパーゼDファミリーの酵素の活性を阻害するかまたは減少させ、かつ神経変性疾患の治療方法で用いることができる薬剤を同定するために用いることができる細胞ベースのアッセイにも関する。
【背景技術】
【0004】
2. 本発明の背景
神経変性疾患は、シナプス機能不全を特徴とし、認知能力および機能的能力の進行性の低下を伴い、多くの場合に死に至る、様々な障害を包含する。アルツハイマー病(AD)は、最も一般的な加齢に関連する衰弱性神経変性障害であり、約400万人の米国人および世界中で約2000〜3000万人が罹患している。ADの古典的な神経病理学的特徴としては、海馬、扁桃体、ならびに側頭葉、前頭葉および頭頂葉の連合皮質での老人(β-アミロイド含有)斑および神経原線維変化の存在が挙げられる。より微妙な変化としては、反応性の星状膠細胞の変化、ならびに内嗅皮質および前脳基底部でのニューロンおよびシナプスの減少が挙げられる。
【0005】
アルツハイマー病の病因は完全には明らかになっていないが、この疾患と、膜タンパク質であるアミロイド前駆体タンパク質(APP)の切断産物との間に関連があることが知られている。γ-セクレターゼは、APPのアミロイド-β(Aβ)ドメインのC末端切断を媒介し、これにより、α-セクレターゼ(ADAM10およびTACE)またはβ-セクレターゼ(BACE1)による細胞外ドメイン切断を通じて生成される膜結合型APP C末端断片からAβ/p3が放出される。γ-セクレターゼ切断は、2種類の主要なAβアイソフォームであるAβ40およびAβ42を生成する。プレセニリン遺伝子であるPS1およびPS2のすべての突然変異が、γ-セクレターゼ活性の変化をもたらし、これが、場合によってはより無害なAβ40ペプチドを減らして、アミロイド生成性および神経毒性が非常に高いAβ42分子種の生成の上昇につながることが、十分に証明されている。
【0006】
ホスホイノシチド(「PI」)は、様々な細胞内経路でシグナル伝達分子として機能し(Williams, 1999, Biochim. Biophys. Acta 1441: 255-267;Rhee and Bai, 1997, J. Biol. Chem. 272(24): 15045-15048;Katan, 1998. Biochim. Biophys. Acta 1436: 5-17)、一部の細胞タイプでのPIの異常の調節は、種々のヒト疾患状態を促進することが示されている(Pendaries et al., 2003, FEBS Lett. 546(1):25-31)。PIシグナル伝達は、多数のキナーゼ、ホスファターゼ、およびホスホリパーゼにより厳重に調節されている。ホスホリパーゼC(PLC)によるホスファチジルイノシトール4,5-ビスリン酸(phosphotidylinositol 4,5-biphosphate)(PIP2)の加水分解は、様々な細胞機能の調節での初期の重要な事象である。Aβ42が、PIP2レベルの低下を引き起こすことが明らかになっている(国際特許出願第PCT/US2007/085274号、WO 2008/064244、参照により本明細書中に組み入れられる)。
【0007】
ホスホリパーゼD(PLD)は、ホスファチジルコリンの加水分解を触媒して、ホスファチジン酸を形成させる(国際特許出願第PCT/US2007/085274号、WO 2008/064244、参照により本明細書中に組み入れられる;Sweeney et al., 2002, J. Biol. Chem. 277:3030-3039; Exton et al., 2002, FEBS Lett 531:58-61;Schields and Arvan, 1999, Curr. Opin. Cell Biol. 11:489-494)。PLDは、種々の膜輸送ステップ(例えば、分泌小胞の放出、エンドサイトーシスおよびエキソサイトーシス)を調節することが報告されている(Chen et al., 1997, J Cell Biol. 138:495-504;Shen et al., 2001, Mol. Cell. Biol. 21:595-602;Humeau et al., 2001, Proc. Natl. Acad. Sci. 98:15300-5305;Cockcroft, 2001, Cell. Nol. Life Sci. 58:1674-1687)。PLD1およびPLD2は、この酵素の2種類の異なるアイソフォームであり(Hammond et al., 1995, J. Biol. Chem. 270:29640;Colley et al., 1997, Curr. Biol. 7:191;Steed et al., 1998, FASEB J. 12:1309;図1A〜Bを参照されたい)、異なる細胞内機能を有することが報告されている(Choi et al., 2002, J. Immunol. 168:5682-5689)。Caiら(2006, Proc. Natl. Acad. Sci. U.S.A.)には、PLD1がβAPPの細胞内輸送を調節することが報告されており、随伴する論文(Cai et al., 2006, Proc. Natl .Acad. Sci. U.S.A. 103:1941-1946)には、独立したメカニズムにより、PLD1がγ-セクレターゼ複合体の完全性を損なわせ、β-アミロイド形成を阻害することが報告されている。Caiらは、PLD代謝の欠陥が、アルツハイマー病の病因に関与している可能性があることを示唆している。
【発明の概要】
【0008】
3. 発明の概要
本発明は、神経変性疾患の治療方法に関し、該方法は、そのような治療を必要とする被験体に、ホスホリパーゼD1および/またはホスホリパーゼD2などのホスホリパーゼDファミリーの酵素の、触媒活性をはじめとする作用を阻害するかまたは減少させる1種以上の薬剤を投与するステップを含む。本発明はまた、ホスホリパーゼDファミリーの酵素の活性を阻害するかまたは減少させる薬剤を同定するために用いることができる細胞ベースのアッセイにも関する。
【0009】
さらなる実施形態では、本発明は、PLD阻害剤、好ましくはPLD2阻害剤がガングリオシドレベルを低下させるという知見に基づいて、上昇したガングリオシドレベルに関連する障害の治療を提供する。
【0010】
本明細書中で用いる場合、「治療する」との用語は、疾患の症状または徴候の進行速度を緩和または低下または減少させること、あるいは、限定するものではないが記憶障害(短期もしくは長期)および/または認知症をはじめとする疾患または障害の発症リスクを減少させることを意味する。本発明に従って治療することができる神経変性疾患の非限定的な例としては、アルツハイマー病、軽度認知障害、パーキンソン病、ハンチントン舞踏病、老年性認知症およびクロイツフェルト・ヤコブ病が挙げられる。本発明はまた、進行性記憶障害を抑制するためにも用いることができる。本発明に従って治療することができる上昇したガングリオシドレベルを有する障害の非限定的な例としては、GM1ガングリオシドーシス、モルキオ病B型、テイ・サックス病、サンドホフ病AB型、およびニーマン・ピック病C型が挙げられる。
【図面の簡単な説明】
【0011】
【図1】(A)PLD1/PLD2のドメイン構造を示す図である。(B)PLD酵素により媒介されるトランスホスファチジル化反応を示す図である。この反応は、それぞれ、エタノールおよび1-ブタノールの存在下で、ホスファチジルエタノール(PEtOH)またはホスファチジルブタノール(PButOH)の合成をもたらす。
【図2−1】(A) 200nM合成oAβ42で処理した初代培養皮質ニューロンでの、ジホスホグリセリン酸(「DPG」)、PEtOH、ホスファチジルイノシトール4リン酸(「PtdIns4P」)およびPIP2(「PtdIns(4,5)P2」とも表記される)のレベルを表す図である(n=9)。(B) oAβ42または2μM Ca++イオノフォアであるイオノマイシンの存在下または非存在下における、初代培養皮質ニューロンでの、1-ブタノールの存在下でのトランスホスファチジル化反応を介した[3H]-ホスファチジルブタノールの合成により測定したPLD活性を表す図である。(C) oAβ42の存在下または非存在下における、培養N2a細胞での[3H]ホスファチジルブタノールの合成により測定したPLD活性を表す図である。(D)培養N2a細胞またはswAPP突然変異体を発現する培養N2a細胞のいずれかでの、[3H]ホスファチジルブタノールの合成により測定したPLD活性を表す図である。値は、平均±SEMを表す。ns=有意差なし。*p<0.05;**p<0.01;***p<0.001。
【図2−2】(E) PLD2の野生型(+/+)またはヘテロ接合(+/-)もしくはホモ接合(-/-)の突然変異体であるマウスから調製した初代培養皮質ニューロン培養物での、oAβ42に応答した[3H]ホスファチジルブタノール産生により測定したPLD2活性を表す図である。値は、平均±SEMを表す。ns=有意差なし。*p<0.05;**p<0.01;***p<0.001。
【図3−1】PC12細胞でのAβ誘導性GFP-PLD2局在変化を表す図である。(A)形質膜/細胞質比の計算を表す図である。(B)200nM合成オリゴマーAβ42の作用の定量化を表す図である。対照(n=31細胞);5分(n=29);30分(n=32)および120分(n=30)。(C) GFPPLD2の局在変化はCa2+依存的であることを表す図である。対照(n=12);200nM Aβ42(n=13);200nM逆方向ペプチドAβ42-1(n=12);2μMイオノマイシン(n=13);2mM EGTA(n=14);2mM EGTAおよび200nM oAβ42(n=13)。すべての処置は、30分間であった。
【図3−2】PC12細胞でのAβ誘導性GFP-PLD2局在変化を表す図である。(D)PLD2-GFP構築物を表す図である。
【図4】(A) PLD2免疫反応性は、Pld2 KOマウスから調製した成体脳抽出物(脱核上清(postnuclear supernatant)には存在しないことを表す図である。ウエスタンブロット分析は、示したタンパク質に対する抗体を用いたECLを用いて行なった。(B)マウスへのエタノールのIP注入に続くホスファチジルエタノール(PEtOH)生成により測定した、Pld2 KO脳での総PLD活性の減少を表す図である。PEtOHは、LC-MSにより測定した。対照として、ホスファチジルセリンのレベルを示す。N=4。
【図5】Pld2 KO皮質ニューロンでの分泌Aβレベルの低下を表す図である。培養物を、第14日にswAPPレンチウイルスに感染させ、Aβ40およびAβ42のELISA測定のために、第16日に培地を回収した。(A)総タンパク質に対して標準化し、非感染Pld2 WT培養物に対する%として表したAβレベルである。(B)標準化したAβ40およびAβ42レベル(%WT)である。N=3。
【図6】PLD2KOマウス由来の海馬は、oAβ42の存在下で正常なLTPを示すことを表す図である。ビヒクルの存在下では、PLD2WTスライス(n=10)とPLD2KOスライス(n=9)との間でLTPに差がなかった(F1,17=0.00、p=0.947)。PLD2WTスライスは、200nM oAβ42の浴浸漬(bath application)に続いてLTPの減少を示したが(n=8)(F1,16=5.19、p=0.038、ビヒクルとの比較)、PLD2KOスライスは、ペプチドの存在下でLTPの差異を示さなかった(n=8)(F1,14=0.01、p=0.919、ビヒクルとの比較)。fEPSP、CA1領域興奮性シナプス後電位。バーは、oAβ42の浴浸漬の時間を表す。3本の矢印は、増強を誘発するために用いたバースト刺激を表す。動物は、約3ヵ月齢であった。
【図7】PLD2除去がSwAPPマウスでの学習および記憶を改善することを表す図である。SwAPPマウス(Tg2576)をPld2ノックアウトマウスと交配させて、得られた子孫[Pld2+/+/tgなし(n=14);Pld2+/-/tgなし(n=14);Pld2-/-/tgなし(n=11);Pld2+/+/SwAPP(n=10);Pld2+/-/SwAPP(n=12);Pld2-/-/SwAPP(n=11)]を文脈的恐怖記憶についてのトレーニングに供し、足ショックの24時間後に評価した。5〜6ヵ月齢の動物を用いた。*、p<0.05(スチューデントの片側(one tail)t検定)。
【図8】PLD2除去がSwAPPマウスでの学習および記憶を改善することを表す図である。12ヵ月齢マウスを、放射状水迷路(RAWM)試験に供した。試験の最後の3日間で、エラーをスコア付けした。Pld2-/-/SwAPP(n=7)およびPld2+/+/SwAPP(n=6)を除くすべての遺伝子型について、n値は8であった。**、p<0.01。値は平均±SEMを表す。
【図9−1】(A) FIPIがoAβ42誘導性のPIP2レベルの低下を部分的に回復させることを表す図である。2週齢の初代皮質神経培養物を、ビヒクル(「1」)、200nM oAβ42で2時間(「2」)、750nM FIPIで3時間(「3」)、または750nM FIPIで1時間の前処理とそれに続く200nM oAβ42で2時間の処理(「4」)のいずれかで急性的に処理した。n=3。(B)野生型またはPLD2のホモ接合突然変異体(±SwAPP)であるマウスでのPLD1およびPLD2レベルを示す図である。
【図9−2】(C)対照と比較した、示した突然変異体マウスでのPA分子種の相対量を示す図である。値は平均±SEMを表す(n=6〜8)。*p<0.05;**p<0.01;***p<0.001。
【図9−3】(D)野生型またはPLD2のホモ接合突然変異体(±SwAPP)であるマウスでのGM3レベルを示す図である(カラーバージョン)。(E)野生型またはPLD2のホモ接合突然変異体(±SwAPP)であるマウスでのGM3レベルを示す図である(図9Dの白黒の簡素化;色を表すための平行線模様:右上がり=緑色、右下がり=赤色、色のグラデーションは示していない)。
【図10】FIPI誘導性のAβの産生低下を示す図である。コンフルエンスに達したswAPP発現N2a細胞を、新しい培地に置き換え、ビヒクル(黒色)またはFIPI(灰色)で6時間処理した。培地を回収し、Aβ40(A)およびAβ42(B)の両方をELISAにより測定した。n=6。
【図11A】PLD阻害剤を示す図である。
【図11B】PLD阻害剤を示す図である。
【図11C】PLD阻害剤を示す図である。
【図11D】PLD阻害剤を示す図である。
【図11E】PLD阻害剤を示す図である。
【図11F】PLD阻害剤を示す図である。
【図11G】PLD阻害剤を示す図である。
【図11H】PLD阻害剤を示す図である。
【図11I】PLD阻害剤を示す図である。
【図11J】PLD阻害剤を示す図である。
【図11K】PLD阻害剤を示す図である。
【図11L】PLD阻害剤を示す図である。
【図11M】PLD阻害剤を示す図である。
【発明を実施するための形態】
【0012】
5. 発明の詳細な説明
本発明は、ホスホリパーゼD1(PLD1)および/またはホスホリパーゼD2(PLD2)などのホスホリパーゼDファミリーの酵素の、触媒活性をはじめとする作用を阻害するかまたは減少させることにより、アミロイドβの生成を減少させ、かつ/またはアミロイドβの毒性作用を阻害する薬剤の使用に関し、神経変性疾患の治療のためのそのような薬剤の使用を含む。本発明はまた、ホスホリパーゼDファミリーの酵素の作用および/もしくは活性を阻害するかまたは減少させ、かつ本明細書中に記載した治療方法で用いることができる薬剤を同定するために用いることができるアッセイシステムにも関する。
【0013】
明確にするために、限定ではなく、本発明の詳細な説明を、以下の小節に分ける:
(i) PLD阻害剤;
(ii)アッセイシステム;および
(iii)治療方法。
【0014】
5.1 PLD阻害剤
PLD1阻害剤およびPLD2阻害剤を含むPLD活性の阻害剤を、本発明に従って用いることができる。PLD阻害剤は、それを投与される被験体体内に存在するPLD活性の量を減少させ、酵素活性の直接的阻害ならびにPLDの量または利用可能性の減少をはじめとするいずれのメカニズムによってそれを行なってもよい。
【0015】
一部の非限定的実施形態では、本発明は、PLD(PLD1および/またはPLD2であり得るが、PLD2の阻害が好ましい)の酵素活性を阻害する薬剤の使用を提供する。
【0016】
1つの非限定的実施形態では、PLD2をはじめとするPLDを阻害する薬剤は、5-フルオロ-2-インドリルデス-クロロハロペミド(「FIPI」)である。
【0017】
本発明に従って用いることができるさらなるPLD阻害剤としては、限定するものではないが、以下のものが挙げられる:ジエチルスチベストロール、レスベラトロール、ホンキオール、SCH420789、プレスクアレン二リン酸、ラロキシフェン、ハロペミド、4-ヒドロキシタモキシフェン、図11A〜Cに示された化合物(Scott et al., 2009, Nat Chem Biol 5(2):108-117およびNature Chemical Biology 10.1038/nchembio.140でオンラインで入手可能な補足情報);ハロペミド誘導体、特に2-インドリル部分を含むハロペミド誘導体、図11Dに示された化合物を含む(Monovich et al., 2007, Bioorg. Med. Chem. Lett. 17:2310-2311);図11E〜Jに示された化合物、限定するものではないが、ハロゲン化ピペリジニルベンゾイミダゾロン部分およびS-メチル部分を含むハロペミド誘導体を含む(Lewis et al., 2009, Bioorg. Med. Chem. Letts. 19:1916-1920);図11Eの化合物5の誘導体、1,3,8-トリアザスピロ[4,5]デカン-4-オン構造を含む化合物を含む、図11K〜Lに示された化合物を含む(Lavieri et al., 2009, Bioorg. Med. Chem. Lett. 19:2240-2243);および図11Mに示された化合物(Lavieri et al., 2009, Bioorg. Med. Chem. Lett. 19:2240-2243)。
【0018】
特に好ましい非限定的実施形態では、PLD阻害剤は、限定するものではないが、以下のものなどのPLD2選択的阻害剤である:4-OHタモキシフェン;図11Bの化合物72および82(Scott et al., 2009, Nat Chem Biol 5(2):108-117);図11Dの化合物4jおよび4k(Monovich et al., 2007, Bioorg. Med. Chem. Lett. 17:2310-2311);図11Eの化合物5(Lewis et al., 2009, Bioorg. Med. Chem. Letts. 19:1916-1920);および図11Eの化合物5の誘導体、1,3,8-トリアザスピロ[4,5]デカン-4-オン構造を含む化合物を含む、図11K〜Lに示された化合物を含む(Lavieri et al., 2009, Bioorg. Med. Chem. Lett. 19:2240-2243)。上記の参考文献のそれぞれおよびそれらに関連付けられたいかなる公衆に供給された補足情報、ならびにその中に示されたいかなる化合物および/または合成スキームも、その全体が参照により本明細書中に組み入れられる。
【0019】
本発明の別の好ましい非限定的実施形態では、PLD阻害剤は、図11Cの化合物56(図11Eの二重PLD1/2阻害剤2および図11Fの化合物2でもある)であり、これは、N-(2-(4-(2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)エチル)-2-ナフタミドとしても知られ、81nMのPLD1 IC50、240nMのPLD2 IC50、380nMの293-PLD2 IC50、および21nMのCalu-1 IC50を有する(Scott et al., 2009, Nature Chemical Biology 5:108-117)。
【0020】
本発明の別の好ましい非限定的実施形態では、PLD阻害剤は、図11Jの化合物13rであり、これは、(1R,2R)-N-((S)-1-(4-(5-ブロモ-2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)プロパン-2-イル)-2-フェニルシクロプロパンカルボキサミドとしても知られ、15nMのPLD1 IC50、1100nMのPLD2 IC50、6400nMの293-PLD2 IC50、および3.7nMのCalu-1 IC50を有し(Lewis et al., 2009, Bioorganic and Medicinal Chemistry Letters 19:1916-1920);この化合物は、PLD1選択的である。
【0021】
本発明の別の好ましい非限定的実施形態では、PLD阻害剤は、図11Kの化合物9bであり、これは、N-(2-(4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4,5]デカン-8-イル)エチル)キノリン-3-カルボキサミドとしても知られ、20,000nMのPLD1 IC50、500nMのPLD2 IC50、90nMの293-PLD2 IC50、および1900nMのCalu-1 IC50を有し(Lavieri et al., 2009, Bioorganic and Medicinal Chemistry Letters 19:2240-2243);この化合物は、PLD2選択的である。
【0022】
さらなるPLD阻害剤は、当技術分野で公知の方法により同定することができ、そのような方法としては、限定するものではないが、Scott et al., 2009, Nat Chem Biol. Feb;5(2):108-17;Monovich et al., 2007, Bioorg. Med. Chem. Lett. 17:2310-2311;Lewis et al., 2009, Bioorg. Med. Chem. Letts. 19:1916-1920;またはLavieri et al., 2009, Bioorg. Med. Chem. Lett. 19:2240-2243に記載されたアッセイが挙げられる。
【0023】
あるいは、PLD阻害剤は、PLD、特にPLD2の発現を減少させる分子であり得、例えば、PLD1および/またはPLD2遺伝子に相補的な部分を含む小分子干渉RNAまたはアンチセンスRNAである。
【0024】
5.2 アッセイシステム
一部の非限定的実施形態では、本発明は、PLD(例えば、PLD2)を阻害する薬剤を同定するためのアッセイシステムを提供し、このアッセイシステムは、図3A〜Cに示されている特徴を有する。例えば、被験薬剤が細胞内(例えば、PC12細胞;Ca++イオンの存在下)でのPLD2(例えば、検出可能にタグ付加されたPLD2(例えば、緑色蛍光タンパク質でタグ付加されたもの))の移行を変化させる能力を試験することができ、ここで、被験薬剤がAβ42誘導性の細胞膜から細胞質へのPLD2の移行を阻害する能力は、その薬剤が潜在的な本発明の治療剤であること、および場合によっては、その治療活性を、例えば限定するものではないが、Tg2576マウスでのin vivo試験で確認することができる(図8を参照されたい)ことを示す。
【0025】
非限定的実施形態では、本発明はさらに、PLD(好ましくはPLD2)または膜から移行することができるその一部分の蛍光型を発現するように遺伝子操作された細胞(好ましくは神経細胞)または細胞株(好ましくは神経細胞株)を利用するアッセイシステムを提供する。蛍光タンパク質は、例えば、限定するものではないが、緑色蛍光タンパク質、増強型緑色蛍光タンパク質、黄色蛍光タンパク質、赤色蛍光タンパク質、または当技術分野で公知の他の蛍光タンパク質であり得る。PLDは、ヒト、ラットもしくはマウス由来のPLDタンパク質、またはその移行可能な断片であり得る(例えば、限定するものではないが、GenBank登録番号AAH15033、AAH56871、NP002654、EHW90405、EHW90406、AA021120、AAD04197、AAB96656、AAB96655、NP002653、NP001123553、AAH68976、CAB76564、NP150641、AAM48521、BAA24078、U87557、NP032902、NP032901、AAH68144、またはNC000077.5を参照されたい)。例えば、そのようなアッセイシステムでは、限定するものではないが、PC12細胞またはN2a細胞などの神経細胞株を用いることができる。あるいは、限定するものではないが、CHO、NIH 3t3、HEK293、またはHeLa(72)などの非神経細胞株を用いることができる。
【0026】
具体的な非限定的実施形態では、褐色細胞腫細胞株であるPC12を、PLD(好ましくはPLD2)のコード配列全体またはAβオリゴマーでの処理に際して細胞質に移行するそのような配列の一部分に連結された蛍光タンパク質を含む融合タンパク質をコードする構築物でトランスフェクションすることができる。16〜24時間後、落射蛍光顕微鏡法を用いて、細胞質と比較した形質膜での蛍光PLD2の分布を可視化するために用いることができる。対照細胞では、蛍光は、細胞同士を隔てる辺縁部として見えるはずであり、したがって、形質膜に濃縮されている。oAβ42(oAβ42は、オリゴマーAβまたはAβ42もしくはAβ40をはじめとする他のAβ分子種の他の誘導体を意味する)を用いた細胞の処理は、数分以内に、形質膜からのプローブの著明な消失および細胞質での蛍光レベルの対応する増加を誘導するはずであり、より拡散して見えるはずである。この作用は、イオノマイシンを用いた処理により模倣することができる。oAβ42の非存在下でのPLD2トランスフェクション細胞では、被験薬剤が細胞表面でのPLD2の局在を増加させる能力は、細胞質の平均蛍光強度に対する形質膜の蛍光強度の比の増大として検出することができる。
【0027】
したがって、本発明は、細胞表面結合型PLD(好ましくはPLD2)を増加させる薬剤を同定する方法を提供し、該方法は、(i)蛍光PLD(好ましくはPLD2)センサーを含む宿主細胞を準備するステップ;(ii)被験薬剤を該宿主細胞に投与するステップ;および(iii)細胞質の平均蛍光に対する形質膜の蛍光の比を測定するステップを含み、ここで、この比の増大が、宿主細胞表面でのPLD(好ましくはPLD2)レベルの増大を示す。
【0028】
別の実施形態では、本発明は、oAβ42の毒性作用を阻害する薬剤を同定する方法を提供し、該方法は、以下のステップ:(i)蛍光PLD(好ましくはPLD2)センサーを含む宿主細胞を準備するステップ;(ii)該宿主細胞を毒性濃度のoAβ42に曝露するステップ;(iii)該宿主細胞に被験薬剤を投与するステップ;および(iv)形質膜および細胞質での蛍光を測定するステップを含み、ここで、細胞質に対する形質膜での蛍光の比の増大が、該被験薬剤がoAβ42の毒性作用を阻害することを示す。
【0029】
また別の非限定的実施形態では、本発明は、PLD(好ましくはPLD2)の阻害剤および/またはADに対する治療剤である薬剤を同定する方法を提供し、該方法は、以下のステップ:(i)宿主細胞を準備するステップ;(ii)該宿主細胞に被験薬剤を投与するステップ;(iii)該被験薬剤の投与が、該宿主細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルを低下させるか否かを決定するステップを含み、ここで、PA 34:2、PA 34:0、および/またはGM3のレベルの低下が、該被験薬剤がPLD(例えば、PLD2)の阻害剤であり、ADを治療するために用いることができることを示す。例えば、図9C〜Dを参照されたい。例えば、被験薬剤に曝露された宿主細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルを、適切な対照細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルと比較することができる。リン脂質脂肪酸(PA)組成物に対する命名法は、合計鎖長:不飽和結合の数として表す。これらの実施形態では、宿主細胞は、限定するものではないが、神経細胞もしくは神経細胞株または組織移植片(例えば、皮質組織)であり得、あるいは、宿主細胞は、マウス、例えば限定するものではないが、SwAPP突然変異を保持するマウスなどの非ヒト試験動物体内のものであり得る。1つの具体的な非限定的実施形態では、宿主細胞は、褐色細胞腫細胞、例えば、PC12細胞であり得る。
【0030】
5.3 治療方法
一部の非限定的実施形態では、本発明は、そのような治療を必要とする被験体に、有効量のPLDの阻害剤、例えば、PLD1および/またはPLD2阻害剤を投与することによる、アミロイド生成性ペプチドに関連するシナプス機能不全、記憶障害および/または神経変性を抑制する方法を提供する。
【0031】
そのような治療を必要とする被験体は、PLD酵素を有するヒトまたは非ヒト被験体であり得る。前記被験体は、シナプス機能不全、記憶障害および/もしくは神経変性に罹患している場合があるか、または、例えば限定するものではないが、年齢、家族歴、もしくは毒性物質への曝露により、これらの状態の1以上を発症するリスクがある場合がある。
【0032】
一部の非限定的実施形態では、本発明は、ホスホリパーゼD1および/またはホスホリパーゼD2などのホスホリパーゼD酵素の作用をブロックするかまたは部分的にブロックすることにより、アミロイド生成(すなわち、Aβ40およびAβ42などの毒性Aβ分子種の生成)を減少させる方法を提供する。一部の関連する非限定的実施形態では、本発明は、神経細胞に対するAβ42ペプチドの毒性作用から保護する方法を提供し、該方法は、該細胞を有効量のホスホリパーゼD阻害剤に曝露するステップを含む。
【0033】
一部の非限定的実施形態では、本発明は、神経変性疾患または障害を治療する(例えば、その症状を減少させ、かつ/またはその進行を遅らせ、かつ/または発症リスクを減少させる)方法を提供し、そのような疾患または障害とは、限定するものではないが、アルツハイマー病、軽度認知障害、パーキンソン病、ハンチントン舞踏病、老年性認知症、および/またはクロイツフェルト・ヤコブ病などのプリオン関連疾患(prior-related disease)などである。
【0034】
一部の非限定的実施形態では、本発明は、被験体での記憶障害の進行を抑制する方法を提供し、該方法は、該被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む。一部の非限定的実施形態では、本発明は、記憶障害および/または認知症の発症リスクを減少させる方法を提供し、該方法は、被験体(例えば、少なくとも約40歳または少なくとも約50歳または少なくとも約60歳のヒト被験体)に、有効量のPLD阻害剤を投与するステップを含む。
【0035】
さらなる非限定的実施形態では、本発明は、ガングリオシドレベルの上昇に関連する障害を治療する方法を提供し、該方法は、そのような治療を必要とする被験体に、有効量のPLD阻害剤、好ましくはPLD2阻害剤を投与し、それにより該被験体の臨床状態を緩和し、かつ/またはガングリオシドレベルを低下させるステップを含む。ガングリオシドレベルの上昇に関連する障害の非限定的な例は、GM1ガングリオシドーシス、モルキオ病B型、テイ・サックス病、サンドホフ病AB型、およびニーマン・ピック病C型である。
【0036】
本節で論じた方法で用いることができるPLD阻害剤は、上記の5.1節に示されている。
【0037】
PLD阻害剤は、限定するものではないが、経口投与、皮下投与、筋肉内投与、静脈内投与、髄腔内投与、吸入投与、または直腸投与によるものをはじめとする、当技術分野で公知のいずれかの好適な経路により投与することができる。
【0038】
特定の非限定的実施形態では、PLD阻害剤はFIPIであり、約50〜2500nM、または約250〜2000nM、または約250〜1000nMの脳脊髄液中濃度を達成するために投与される。PLD阻害剤がFIPIでない場合、(FIPIでない)PLD阻害剤についての用量範囲は、FIPIのEC50に対する該PLD阻害剤のEC50の比を、FIPIについての上記の用量範囲に乗算することにより、決定することができる。EC50は、例えば限定するものではないが、本明細書中に記載されたアッセイにより測定され、該薬剤が、oAβ42誘導性の形質膜から細胞質へのPLDの移行を阻害する、または初代培養皮質ニューロンでのoAβ42誘導性のPIP2の減少を阻害する、またはswAPPを発現する培養ニューロンでのAβ42生成を減少させる、またはTg2576マウスなどのADの動物モデルでの行動遂行を改善する能力などである。
【0039】
PLD阻害剤が図11Cの化合物56(これは、図11Eの二重PLD1/2阻害剤2および図11Fの化合物2でもある)(N-(2-(4-(2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)エチル)-2-ナフタミドとしても知られ、81nMのPLD1 IC50、240nMのPLD2 IC50、380nMの293-PLD2 IC50、および21nMのCalu-1 IC50を有する)である1つの特定の非限定的実施形態では、該PLD阻害剤は、約10〜2000nM、または約10〜1000nM、または約10〜500nM、または約200〜1000nM、または約200〜500nMの脳脊髄液中濃度を達成するために投与することができる。
【0040】
PLD阻害剤が図11Jの化合物13r((1R,2R)-N-((S)-1-(4-(5-ブロモ-2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)プロパン-2-イル)-2-フェニルシクロプロパンカルボキサミドとしても知られ、15nMのPLD1 IC50、1100nMのPLD2 IC50、6400nMの293-PLD2 IC50、および3.7nMのCalu-1 IC50を有する)である別の特定の非限定的実施形態では、該PLD阻害剤は、約2〜10,000nM、または約2〜200nM、または約2〜100nM、または約2〜50nM、または約500〜8000nM、または約500〜2000nMの脳脊髄液中濃度を達成するために投与することができる。
【0041】
PLD阻害剤が図11KのPLD2選択的阻害剤化合物9b(N-(2-(4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4,5]デカン-8-イル)エチル)キノリン-3-カルボキサミドとしても知られ、20,000nMのPLD1 IC50、500nMのPLD2 IC50、90nMの293-PLD2 IC50、および1900nMのCalu-1 IC50を有する)である別の特定の非限定的実施形態では、該PLD阻害剤は、約30〜1500nM、または約50〜1000nM、または50〜800nM、または50〜600nMの脳脊髄液中濃度を達成するために投与することができる。
【0042】
特定の非限定的実施形態では、PLD阻害剤は、1日1回以上、週1回以上、または月1回以上、投与することができる。治療期間は、連続的でも不連続的であってもよい。
【実施例】
【0043】
6. 実施例1
PLD1/PLD2のドメイン構造を、図1Aに示している。領域I〜IVは、触媒ドメインの主要な決定要素であり、HKD特徴モチーフ(IIおよびIV)は、コンセンサス配列HxK(x)4D(x)6GSxNを示す。PLD1は、触媒活性の調節に関与する追加の領域(活性化ループ、L)を含む。PLD酵素により媒介されるトランスホスファチジル化反応は、図1Bに示され、この反応は、それぞれ、エタノールおよび1-ブタノールの存在下で、ホスファチジルエタノール(PEtOH)またはホスファチジルブタノール(PButOH)の合成をもたらす。
【0044】
初代培養皮質ニューロンを200nM合成オリゴマー化Aβ42(「oAβ42」)で処理した場合、ホスファチジン酸(PtdOH)のレベルは、上昇することが観察された(図2A)。さらに、oAβ42の急性的な細胞外添加(200nM)は、新生マウス由来の初代培養皮質ニューロン(図2B)および神経芽細胞腫細胞株Neuro2A(N2A)(図2C)において、ホスホリパーゼD(PLD)の酵素活性を刺激することが見出された。神経芽細胞腫細胞株N2Aでのアミロイド前駆体タンパク質のスウェーデン型突然変異体(swAPP)の過剰発現は、Aβの生成および分泌の増加をもたらすことが示されており、PLDの酵素活性を刺激することも見出された(図2D)。
【0045】
図2Eに示されているように、PLD2を欠損した培養ニューロンは、Aβ42オリゴマーを用いた処理に応答しない。初代培養皮質ニューロン培養物を第12日に[3H]ミリスチン酸で標識し、第15日に処理を行ない、続いて脂質を抽出し、[3H]ホスファチジルブタノールカウント/合計カウントの比をPLD活性の指標として用いた。ビヒクルまたは200μM oAβ42を用いた4時間の処理を行なった後に、Pld2+/+(ビヒクルおよびoAβ42処理のそれぞれに対して、n=19および12)、Pld2+/-(n=7)、およびPld2-/-(それぞれ、n=5および7)でのPLD活性測定を行なった。
【0046】
PLD2の細胞内局在をモニタリングするために、GFPタグ付加PLD2構築物を作製した(図3D)。構築物の作製では、標準的な細菌株ではメチル化がXbaI部位およびBclI部位での切断を妨害するが、mPLD2はXbaIおよびSmaI部位を用いて切断することができた(内部にBamHI部位およびBglII部位がある)。pEGFPは約4.7kb長であり、mPLD2 cDNAは約3kb長であった。哺乳動物細胞に最適化されたGFPの赤側シフト変異体をコードするので、pEGFP(Clontech)を用いた。PLD2(マウス)のGenBank登録番号は、U87557である。GFP-PLD2コード構築物を、Lipofectamine 2000によりPC12細胞に導入した(Hammond et al., 1995, J. Biol. Chem. 270:29640-29643;Colley et al., 1997, Current Biology 7:191-201;Sung et al., 1997, EMBO J. 16:4519-4530;Sung et al., 1999, J. Biol. Chem. 274:3659-3666;およびSung et al., 1999, J. Biol. Chem. 274:494-502を参照されたい)。oAβ42の急性的な細胞外添加(200nM)は、Ca2+依存的に(すなわち、EGTAなどの細胞外Ca2+を捕捉する薬剤によりブロックされる作用)、褐色細胞腫細胞株PC12で、細胞表面から細胞質へのGFPタグ付加ホスホリパーゼD2(PLD2)の移行をもたらす(図3A〜C)。
【0047】
神経系でのPLD2の役割を調べるためのin vivoモデル系を提供するために、Cre-LoxP技術を用いて、PLD2「ノックアウト」マウス(「PLD2KOマウス」)を作製した。PLD2に対する免疫反応性は、これらのマウスの脳抽出物には存在しないことが示され、このことは、ノックアウト動物ではPLD2が発現されていないことを示す(図4A)。さらに、Pldノックアウトマウスの脳では、合計のPLD活性が約半分まで減少していることが示された(図4B)。PLD2ノックアウトマウスは生存可能であり、これまでに、明らかな異常を示すことは観察されていない。
【0048】
PLD2 KO動物でAβ42の作用を調べるための実験の第1シリーズでは、WTおよびKOマウスの初代皮質ニューロンを回収し、培養で確立し、swAPPレンチウイルスを感染させた。ニューロンでの全長swAPPの発現は、典型的には、多量の分泌型Aβ40およびAβ42をもたらした。得られたPld2 KOニューロンは、野生型培養物と比較して、少量のAβ40およびAβ42を分泌することが観察された(図5A〜B)。
【0049】
次に、長期増強(「LTP」)に対するPLD2KOの影響を、KO動物から作製した海馬脳スライスで試験した。LTPは、電気生理学的技術を用いて測定され、多くの事例で学習および記憶と関連する現象である。Aβ(および特にAβオリゴマー)がLTPを妨害することが、いくつかのグループにより以前に示されており、軽度認知障害およびアルツハイマー病に伴う認知障害に対する考えられる基礎を与えている。図6に示されているように、oAβ42に曝露されたPLD2野生型マウス由来の海馬はLTPの減弱を示した一方で、PLD2KOマウス由来の海馬はoAβ42の存在下で正常なLTPを示した。
【0050】
Aβ42過剰発現の状況でのPLD2KOのin vivoでの影響を試験するために、PLD2KOマウスをswAPPマウスと交配させた。PLD2の除去に関してホモ接合およびヘテロ接合の子孫を作製し、続いて2種類の行動試験の遂行(パフォーマンス)を対照動物(野生型PLD2を有するswAPP突然変異体動物を含む)と比較した。文脈的恐怖条件付け(FC)パラダイムでの試験結果を図7に示し、放射状水迷路(RAWM)パラダイムでの試験結果を図8に示す。これらの試験パラダイムの両方で、swAPPを過剰発現するマウス(Tg2576系統)でのPld2の1コピーまたは2コピーの遺伝的不活性化が、Tg2576(swAPP発現)マウスの特徴である学習障害を緩和した。
【0051】
PLD2の遺伝的除去のAβ42保護効果が上記の実験で実証されたため、この酵素の化学的阻害の影響を評価するために実験を行なった。図9Aに示されているように、PLD(PLD2を含む)の薬理学的阻害剤である5-フルオロ-2-インドリルデス-クロロハロペミド(「FIPI」)が、oAβ42の急性的な細胞外添加(200nM)に続く、初代培養皮質ニューロンでのPIP2欠損を部分的に回復させることが見出された。FIPIは、ハロペミドの類似体であり、元々はMonovichらによって特性決定され(Monovich, L. et al. Optimization of halopemide for phospholipase D2 inhibition. Bioorg Med Chem Lett 17, 2310-2311 (2007))、Michael Frohmanのグループによりさらに特性決定された(Su, W. et al. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol Pharmacol 75, 437-446 (2009))。
【0052】
次に、PAおよびガングリオシドGM3レベルに対するSwAPP過剰発現およびPld2遺伝子型の影響を調べるために、実験を行なった。図9Bに示されているように、PLD2、PLD1、APPおよびチューブリンのウエスタンブロット分析により、タンパク質レベルを評価した(代表的なブロットを示す)。SwAPPトランスジーンを有するか有しないPld2+/+およびPld2-/-マウスから前脳脂質を抽出し、LC-MS分析に供し、PA分子種の相対量を突然変異体マウスで測定し、対照マウス(Pld2+/+、SwAPPなし)と比較した。結果を図9Cに示す。PA分子種である34:2および34:0(矢印)は、アルツハイマー病のバイオマーカー候補であり、Pld2ノックアウト(KO)でのその減少は、PLD2阻害剤の作用をモニタリングするためにこれらのバイオマーカーを用いることができることを示す。リン脂質脂肪酸組成物に対する命名法は、合計鎖長:不飽和結合の数として表す。図9Dに示されているように、糖スフィンゴ脂質(ガングリオシド)であるGM3は、アルツハイマー病マウス突然変異体(赤色)で上昇し、この疾患のバイオマーカーとして用いることができる。GM3のレベルは、Pld2 KO(緑色)では低下する。SwAPP突然変異体では、PLD2の除去が、GM3の正常レベルを回復させる(黒色)。
【0053】
さらに、FIPIで処理した場合、swAPPを発現するN2A細胞が、より少量のAβ42を分泌することが見出された(図10)。FIPIはPLD1およびPLD2の両方をブロックするので、この薬物の保護効果が、PLD1、PLD2またはその両方のどれを含むかは明らかでない。
【0054】
まとめると、PLD2の遺伝的除去(部分的除去および完全除去の両方)それ自体は、ノックアウトマウスに対して生体レベルでは実質的な影響を有しないようであり、PLD2の遺伝的除去は種々のアルツハイマー病実験モデルの設定では保護を付与するので、PLD2が、薬理学的阻害の合理的な標的として浮上してくる。同様に、PLD1の除去は、いかなる明らかな異常をもたらすことも観察されておらず、このことは、PLD1阻害に依存する治療薬も、治療的効果を有し得ることを示唆している。
【0055】
7. 実施例2
徐々に多くなる証拠は、アルツハイマー病(AD)が、脂質代謝での深刻な変化に関連すること、そしてこれらの変化は、シナプス機能不全および認知低下の基盤となる分子経路の揺らぎの原因である可能性があることを示している。本研究では、本発明者らは、アミロイドβ(Aβ)と、複数の細胞内プロセスを制御する重要なシグナル伝達リン脂質であるホスファチジン酸(PtdOH)との関連を調べた。本発明者らは、可溶性Aβ1-42オリゴマーでの培養ニューロンの処理が、PtdOHレベルを上昇させることを以前に報告していた。ホスホリパーゼD(PLD)経路はPtdOHの生体活性プールの主要な供給源であるため、本発明者らは、このファミリーの脂質酵素に着目した。その結果、Aβ1-42オリゴマーでの培養ニューロンおよび神経芽細胞腫細胞の処理ならびにAPPのスウェーデン型突然変異体(swAPP)の発現は、PLD活性の著明な増大をもたらす。本発明者らはまた、Aβ1-42オリゴマー処理が、形質膜から細胞質へのPLD2の移行を促進することも示し、このことは、PLD2がAβシグナル伝達経路に存在することを示唆する。ADにおけるPLD経路の重要性を遺伝学的に調べるために、本発明者らは、Pld2遺伝子の条件付き欠失を有するマウスを作製した。本発明者らの結果は、いかなる明白な表現型も引き起こさないPld2除去が、海馬スライスでの長期増強(LTP)に対するAβのシナプス障害作用を抑制することを示し、このことは、Pld2除去が細胞毒性ペプチドに対する保護を与えることを示唆する。際立って、本発明者らによる行動解析は、Pld2の1コピー(swAPP/Pld2+/-)または2コピー(swAPP/Pld2-/-)のいずれかを欠損しているADのトランスジェニックマウスモデル(swAPP)で、文脈的学習が改善していることを示す。つまり、これらの知見は、PLD2経路が、Aβオリゴマーの細胞毒性作用の一部を仲介していること、およびこの経路をブロックすることにより、ADに関連するシナプス機能不全および認知低下を軽減できることを示唆する。
【0056】
種々の刊行物を本明細書中で引用したが、これによりそれらの内容はその全体が本明細書中に組み入れられる。
【特許請求の範囲】
【請求項1】
神経変性疾患を治療する方法であって、そのような治療を必要とする被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む、上記方法。
【請求項2】
神経変性疾患の発症リスクを減少させる方法であって、そのような治療を必要とする被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む、上記方法。
【請求項3】
被験体での記憶障害の進行を抑制する方法であって、該被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む、上記方法。
【請求項4】
神経細胞に対するAβ42ペプチドの毒性作用から保護する方法であって、該細胞を、有効量のホスホリパーゼD阻害剤に曝露するステップを含む、上記方法。
【請求項5】
ホスホリパーゼD阻害剤が、ホスホリパーゼD2阻害剤である、請求項1、2、3または4に記載の方法。
【請求項6】
ホスホリパーゼD阻害剤が、ハロペミド誘導体である、請求項1、2、3または4に記載の方法。
【請求項7】
ホスホリパーゼD阻害剤が、図11A〜Mに示されたホスホリパーゼD阻害剤からなる群より選択される、請求項1、2、3または4に記載の方法。
【請求項8】
ホスホリパーゼD阻害剤が、5-フルオロ-2-インドリルデス-クロロハロペミド(「FIPI」)である、請求項1、2、3または4に記載の方法。
【請求項9】
ホスホリパーゼD阻害剤が、N-(2-(4-(2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)エチル)-2-ナフタミドである、請求項1、2、3または4に記載の方法。
【請求項10】
ホスホリパーゼD阻害剤が、(1R,2R)-N-((S)-1-(4-(5-ブロモ-2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)プロパン-2-イル)-2-フェニルシクロプロパンカルボキサミドである、請求項1、2、3または4に記載の方法。
【請求項11】
ホスホリパーゼD阻害剤が、N-(2-(4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4,5]デカン-8-イル)エチル)キノリン-3-カルボキサミドである、請求項1、2、3または4に記載の方法。
【請求項12】
神経変性障害が、アルツハイマー病、軽度認知障害、パーキンソン病、ハンチントン舞踏病、老年性認知症、およびプリオン関連疾患からなる群より選択される、請求項1または2に記載の方法。
【請求項13】
神経変性障害の治療のための、ホスホリパーゼD阻害剤。
【請求項14】
神経変性疾患のリスクを減少させるための、ホスホリパーゼD阻害剤。
【請求項15】
記憶障害の進行を抑制するための、ホスホリパーゼD阻害剤。
【請求項16】
神経細胞に対するAβ42ペプチドの毒性作用から保護するための、ホスホリパーゼD阻害剤。
【請求項17】
ホスホリパーゼD2阻害剤である、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項18】
ハロペミド誘導体である、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項19】
図11A〜Mに示されたホスホリパーゼD阻害剤からなる群より選択される、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項20】
5-フルオロ-2-インドリルデス-クロロハロペミド(「FIPI」)である、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項21】
N-(2-(4-(2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)エチル)-2-ナフタミドである、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項22】
(1R,2R)-N-((S)-1-(4-(5-ブロモ-2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)プロパン-2-イル)-2-フェニルシクロプロパンカルボキサミドである、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項23】
N-(2-(4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4,5]デカン-8-イル)エチル)キノリン-3-カルボキサミドである、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項24】
神経変性障害が、アルツハイマー病、軽度認知障害、パーキンソン病、ハンチントン舞踏病、老年性認知症、およびプリオン関連疾患からなる群より選択される、請求項13または14に記載のホスホリパーゼD阻害剤。
【請求項25】
PLD2の阻害剤である薬剤を同定する方法であって、以下のステップ:(i)宿主細胞を準備するステップ;(ii)被験薬剤を該宿主細胞に投与するステップ;(iii)該被験薬剤の投与が、該宿主細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルを低下させるか否かを決定するステップを含み、ここで、PA 34:2、PA 34:0、および/またはGM3のレベルの低下が、該被験薬剤がPLD2の阻害剤であることを示す、上記方法。
【請求項26】
アルツハイマー病の治療剤である薬剤を同定する方法であって、以下のステップ:(i)宿主細胞を準備するステップ;(ii)被験薬剤を該宿主細胞に投与するステップ;(iii)該被験薬剤の投与が、該宿主細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルを低下させるか否かを決定するステップを含み、ここで、PA 34:2、PA 34:0、および/またはGM3のレベルの低下が、該被験薬剤を用いてADを治療することができることを示す、上記方法。
【請求項27】
ガングリオシドレベルの上昇に関連する障害を治療する方法であって、そのような治療を必要とする被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む、上記方法。
【請求項28】
ホスホリパーゼD阻害剤が、ホスホリパーゼD2阻害剤である、請求項27に記載の方法。
【請求項29】
前記障害が、GM1ガングリオシドーシス、モルキオ病B型、テイ・サックス病、サンドホフ病AB型、およびニーマン・ピック病C型からなる群より選択される、請求項27または28に記載の方法。
【請求項1】
神経変性疾患を治療する方法であって、そのような治療を必要とする被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む、上記方法。
【請求項2】
神経変性疾患の発症リスクを減少させる方法であって、そのような治療を必要とする被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む、上記方法。
【請求項3】
被験体での記憶障害の進行を抑制する方法であって、該被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む、上記方法。
【請求項4】
神経細胞に対するAβ42ペプチドの毒性作用から保護する方法であって、該細胞を、有効量のホスホリパーゼD阻害剤に曝露するステップを含む、上記方法。
【請求項5】
ホスホリパーゼD阻害剤が、ホスホリパーゼD2阻害剤である、請求項1、2、3または4に記載の方法。
【請求項6】
ホスホリパーゼD阻害剤が、ハロペミド誘導体である、請求項1、2、3または4に記載の方法。
【請求項7】
ホスホリパーゼD阻害剤が、図11A〜Mに示されたホスホリパーゼD阻害剤からなる群より選択される、請求項1、2、3または4に記載の方法。
【請求項8】
ホスホリパーゼD阻害剤が、5-フルオロ-2-インドリルデス-クロロハロペミド(「FIPI」)である、請求項1、2、3または4に記載の方法。
【請求項9】
ホスホリパーゼD阻害剤が、N-(2-(4-(2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)エチル)-2-ナフタミドである、請求項1、2、3または4に記載の方法。
【請求項10】
ホスホリパーゼD阻害剤が、(1R,2R)-N-((S)-1-(4-(5-ブロモ-2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)プロパン-2-イル)-2-フェニルシクロプロパンカルボキサミドである、請求項1、2、3または4に記載の方法。
【請求項11】
ホスホリパーゼD阻害剤が、N-(2-(4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4,5]デカン-8-イル)エチル)キノリン-3-カルボキサミドである、請求項1、2、3または4に記載の方法。
【請求項12】
神経変性障害が、アルツハイマー病、軽度認知障害、パーキンソン病、ハンチントン舞踏病、老年性認知症、およびプリオン関連疾患からなる群より選択される、請求項1または2に記載の方法。
【請求項13】
神経変性障害の治療のための、ホスホリパーゼD阻害剤。
【請求項14】
神経変性疾患のリスクを減少させるための、ホスホリパーゼD阻害剤。
【請求項15】
記憶障害の進行を抑制するための、ホスホリパーゼD阻害剤。
【請求項16】
神経細胞に対するAβ42ペプチドの毒性作用から保護するための、ホスホリパーゼD阻害剤。
【請求項17】
ホスホリパーゼD2阻害剤である、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項18】
ハロペミド誘導体である、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項19】
図11A〜Mに示されたホスホリパーゼD阻害剤からなる群より選択される、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項20】
5-フルオロ-2-インドリルデス-クロロハロペミド(「FIPI」)である、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項21】
N-(2-(4-(2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)エチル)-2-ナフタミドである、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項22】
(1R,2R)-N-((S)-1-(4-(5-ブロモ-2-オキソ-2,3-ジヒドロ-1H-ベンゾ[d]イミダゾール-1-イル)ピペリジン-1-イル)プロパン-2-イル)-2-フェニルシクロプロパンカルボキサミドである、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項23】
N-(2-(4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4,5]デカン-8-イル)エチル)キノリン-3-カルボキサミドである、請求項13、14、15または16に記載のホスホリパーゼD阻害剤。
【請求項24】
神経変性障害が、アルツハイマー病、軽度認知障害、パーキンソン病、ハンチントン舞踏病、老年性認知症、およびプリオン関連疾患からなる群より選択される、請求項13または14に記載のホスホリパーゼD阻害剤。
【請求項25】
PLD2の阻害剤である薬剤を同定する方法であって、以下のステップ:(i)宿主細胞を準備するステップ;(ii)被験薬剤を該宿主細胞に投与するステップ;(iii)該被験薬剤の投与が、該宿主細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルを低下させるか否かを決定するステップを含み、ここで、PA 34:2、PA 34:0、および/またはGM3のレベルの低下が、該被験薬剤がPLD2の阻害剤であることを示す、上記方法。
【請求項26】
アルツハイマー病の治療剤である薬剤を同定する方法であって、以下のステップ:(i)宿主細胞を準備するステップ;(ii)被験薬剤を該宿主細胞に投与するステップ;(iii)該被験薬剤の投与が、該宿主細胞でのPA 34:2、PA 34:0、および/またはGM3のレベルを低下させるか否かを決定するステップを含み、ここで、PA 34:2、PA 34:0、および/またはGM3のレベルの低下が、該被験薬剤を用いてADを治療することができることを示す、上記方法。
【請求項27】
ガングリオシドレベルの上昇に関連する障害を治療する方法であって、そのような治療を必要とする被験体に、有効量のホスホリパーゼD阻害剤を投与するステップを含む、上記方法。
【請求項28】
ホスホリパーゼD阻害剤が、ホスホリパーゼD2阻害剤である、請求項27に記載の方法。
【請求項29】
前記障害が、GM1ガングリオシドーシス、モルキオ病B型、テイ・サックス病、サンドホフ病AB型、およびニーマン・ピック病C型からなる群より選択される、請求項27または28に記載の方法。
【図1】


【図2−1】
【図2−2】
【図3−1】
【図3−2】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9−1】
【図9−2】
【図9−3】
【図10】
【図11A】
【図11B】
【図11C】
【図11D】
【図11E】
【図11F】
【図11G】
【図11H】
【図11I】
【図11J】
【図11K】
【図11L】
【図11M】
【図2−1】
【図2−2】
【図3−1】
【図3−2】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9−1】
【図9−2】
【図9−3】
【図10】
【図11A】
【図11B】
【図11C】
【図11D】
【図11E】
【図11F】
【図11G】
【図11H】
【図11I】
【図11J】
【図11K】
【図11L】
【図11M】
【公表番号】特表2012−528201(P2012−528201A)
【公表日】平成24年11月12日(2012.11.12)
【国際特許分類】
【出願番号】特願2012−513315(P2012−513315)
【出願日】平成22年5月28日(2010.5.28)
【国際出願番号】PCT/US2010/036660
【国際公開番号】WO2010/138869
【国際公開日】平成22年12月2日(2010.12.2)
【出願人】(501306715)ザ トラスティース オブ コロンビア ユニバーシティ イン ザ シティ オブ ニューヨーク (11)
【Fターム(参考)】
【公表日】平成24年11月12日(2012.11.12)
【国際特許分類】
【出願日】平成22年5月28日(2010.5.28)
【国際出願番号】PCT/US2010/036660
【国際公開番号】WO2010/138869
【国際公開日】平成22年12月2日(2010.12.2)
【出願人】(501306715)ザ トラスティース オブ コロンビア ユニバーシティ イン ザ シティ オブ ニューヨーク (11)
【Fターム(参考)】
[ Back to top ]