
神経学的障害を処置するための抗うつ剤の投薬量の段階的な漸増ならびに毎日の分割した投薬
【課題】神経学的障害の症状を処置するためにより有効な量の抗うつ剤を投与する方法を提供すること。
【解決手段】処置を必要とする患者における慢性的な神経学的障害または該神経学的障害と関連する疼痛の症状を処置する方法であって、維持投薬量に到達するまでの期間に渡って、段階的に増加する投薬量において抗うつ剤を投与する工程、を包含する、方法が提供される。別個の投薬量、ならびに維持投薬量に達するまでの期間にわたって、漸増量における別個の投薬量を摂取するための指示書を含む、抗うつ剤の投薬パックもまた提供される。
【解決手段】処置を必要とする患者における慢性的な神経学的障害または該神経学的障害と関連する疼痛の症状を処置する方法であって、維持投薬量に到達するまでの期間に渡って、段階的に増加する投薬量において抗うつ剤を投与する工程、を包含する、方法が提供される。別個の投薬量、ならびに維持投薬量に達するまでの期間にわたって、漸増量における別個の投薬量を摂取するための指示書を含む、抗うつ剤の投薬パックもまた提供される。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、NSRI化合物であるミルナシプランといった抗うつ剤の有効量を使用して神経学的障害を処置する分野であって、その抗うつ剤は、望ましくない副作用を最小化するために段階的に漸増する投薬量において投与される。
【0002】
本出願は、2002年10月3日に出願されたU.S.S.N.第60/415、739号;2002年12月6日に出願されたU.S.S.N.第60/431、550号;2003年1月28日に出願されたU.S.S.N.第60/443、203;および2003年1月28日に出願されたU.S.S.N.第60/443、081号に対する優先権を主張する
【背景技術】
【0003】
(発明の背景)
慢性疲労症候群、線維筋痛症候群、慢性的な疼痛、疼痛に対する二次的なうつ病および機能的肉体障害といった神経学的障害は、アメリカ人の寿命のある時点でその人口の大部分に影響を与える。多数のこれらの状態を処置するために抗うつ剤がしばしば使用されるが、それらの効力は、用量が制限される副作用に起因してしばしば不十分である。それゆえに、多くの抗うつ剤が、別の方法で神経学的障害の症状の処置にあたって有効であるとき、抗うつ剤が、悪影響のためにその使用において制限される。
【0004】
抗うつ剤の一つのクラスは、モノアミンノルエピネフリンおよびセロトニンの再取り込みを阻害し、セロトニン−ノルエピネフリン再取り込み阻害剤(SNRI)と呼ばれる。これらの化合物のサブクラスは、優先的に、セロトニン再取り込み阻害と同等かそれ以上の親和性で、ノルエピネフリンの再取り込みを阻害し、ノルエピネフリン−セロトニン再取り込み阻害剤(NSRI)と呼ばれる。ミルナシプラン(Z−2−アミノメチル−1−フェニル−N,N−ジエチルシクロプロパン−カルボキサミドヒドロクロリド)は、セロトニン再取り込みをこえてノルエピネフリン再取り込みを優先的に阻害する、そのようなNSRI化合物の一つである。ミルナシプランは、うつ病の処置に関してヨーロッパで認可され、販売される薬物である。うつ病の処置においてミルナシプランの臨床的有効性を実証する当局への申請資料は、ヨーロッパ、米国および日本で実行された、5732人の患者(4006人をミルナシプランで処置、394人をプラシーボで処置、940人をTCAで処置および344人をSSRIで処置した)を包含する研究に基づく。30人以上の二重盲検試験が実行され、この試験は、うつ病による入院患者および外来患者の両方が関与するDSM IIIまたはRDC診断基準(さらに最近の研究においてはDSM III−RまたはDSM IV)によって評価されるような、ミルナシプランと、プラシーボ、三環系抗うつ剤(TCA)、または選択的セロトニン再取り込み阻害剤(SSRI)のいずれかとを比較することを包含する。
【0005】
これらの研究は、ミルナシプランが主なうつ病性エピソード(成人および高齢者)において、一日二回(BID)の50mgの代表的な用量で(食事と一緒に摂取される)有効であることを示した。この用量で、ミルナシプランは、下記:
(a)プラシーボより優れた有効性:ミルナシプラン(50mg BID)とプラシーボを比較する二重盲検研究のメタアナリシス(Hamilton(HDRSまたはHAMD)と、Montgomery−Asberg(MADRS)のうつ病の評点尺度の総スコアの群の間で有意差を示す;
(b)患者の「応答者」のパーセンテージ(すなわち、HAMDおよびMADSの総スコアが50%以上減少する患者)は統計学的に、プラシーボより優れており、プラシーボに対する応答者が40%であったのに対し、全ての患者の55%、および入院患者の64%がミルナシプランに応答した;
(c)TCAに比敵した有効性:三環系(イミプラミン、アミトリプチリンおよびクロミプラリン)で67%の応答比率に対し、ミルナシプランで64%の応答比率が見られた;および、
(d)SSRIに比敵した有効性:SSRI(フルボキサミン、フルオキセチンおよびパロキセチン)で50%〜65%の応答比率に対し、ミルナシプランで64%の応答比率が見られた;
を示す。
【0006】
残念なことに、50mgを超えるミルナシプランの毎日の投薬量が、負の患者耐性(すなわち、有害反応)を付随することもまた、公知である。副作用なしに、50mgを超えるミルナシプランの毎日の投薬量を投与するために有利である。
【0007】
ミルナシプランのような抗うつ剤は、大うつ病エピソードならびに他の神経学的障害の処置において有効だが、より適切な方法が、これらの神経学的障害を処置するより有効な量の投与を行うために必要とされる。
【0008】
それゆえに、本発明の目的は、神経学的障害の症状を処置するためにより有効な量の抗うつ剤を投与する方法を提供することである。
【0009】
本発明のさらなる目的は、化合物の増加量が投与され得、ならびに正の患者の安全性プロフィールを維持し得るように、化合物の用量が制限される毒性を減少する方法を提供することである。
本発明のさらなる目的は、一日二回(BID)とは対照的に、一日一回(QD)の投薬で投与することによる抗うつ剤の適切なピーク血漿濃度(Cmax)を得る方法を提供することである。
【発明の概要】
【課題を解決するための手段】
【0010】
(発明の要旨)
坑うつ剤のより高い毎日の投薬量を投与することで、神経学的障害を処置する方法が開示され、この方法において副作用が、投薬量を段階的に増加させることにより、最小化されると説明される。より高い毎日の投薬量は、その薬物の改善された有効性、正の患者の耐性の維持、正の患者の安全性プロフィールの維持(例えば、用量が制限される毒性)、薬物の適切なピーク血漿濃度(Cmax)、および/または一日二回(BID)の投与とは対照的に一日一回(QD)の投与を結果として生じる。循環する薬物のより高いレベルが、一日一回よりもむしろ一日の経過に渡って分割された投薬において化合物を投与することによっても得られる。
本発明の好ましい実施形態では、例えば以下の方法などが提供される:
(項目1)
処置を必要とする患者における慢性的な神経学的障害または該神経学的障害と関連する疼痛の症状を処置する方法であって、以下:
維持投薬量に到達するまでの期間に渡って、段階的に増加する投薬量において抗うつ剤を投与する工程、
を包含する、方法。
(項目2)
請求項1に記載の方法であって、前記慢性的な神経学的障害が、CFS、FMS、DSP、FSD、うつ病および疼痛からなる群より選択される、方法。
(項目3)
請求項1に記載の方法であって、前記慢性的な神経学的障害が線維筋痛症候群である、方法。
(項目4)
請求項1に記載の方法であって、前記神経学的障害が、慢性疲労症候群である、方法。
(項目5)
請求項1に記載の方法であって、前記神経学的障害が、疼痛である、方法。
(項目6)
請求項1に記載の方法であって、前記抗うつ剤が、ノルアドレナリン−セロトニン再取り込み阻害剤(NSRI)、選択的セロトニン再取り込み阻害剤(SSRI)、モノアミンオキシダーゼ阻害剤;抗痙攣剤および非特異的抗うつ剤からなる群より選択される、方法。
(項目7)
請求項1に記載の方法であって、前記抗うつ剤が、鎮静特性を有する、方法。
(項目8)
請求項1に記載の方法であって、前記抗うつ剤が、NMDA−アンタゴニスト特性を有する、方法。
(項目9)
請求項1に記載の方法であって、前記抗うつ剤が、ミルナシプランである、方法。
(項目10)
請求項1に記載の方法であって、前記抗うつ剤が、漸増投薬量において、活性化化合物の循環する投薬量を増加して、副作用を回避または最小化する段階的様式において投与される、方法。
(項目11)
請求項1に記載の方法であって、前記抗うつ剤が、第一の期間において、最初の投薬量が、一日あたり100mgまでで投与され、該第一の投薬量の約1.5倍〜2.5倍である第二の投薬量が、第二の期間において投与され、前記慢性的な神経学的障害の症状を処置する、方法。
(項目12)
請求項11に記載の方法であって、第三の期間において、前記第二の投薬量の約1.5倍〜2.5倍である第三の投薬量を投与して、前記慢性的な神経学的障害の症状を処置する工程をさらに包含する、方法。
(項目13)
請求項12に記載の方法であって、第四の期間において、前記第三の投薬量の約1.5倍〜2.5倍である第四の投薬量を投与して、前記慢性的な神経学的障害の症状を処置する工程をさらに包含する、方法。
(項目14)
請求項10から請求項13のいずれかに記載の方法であって、前記各投与期間が、3日間よりも長い、方法。
(項目15)
請求項14に記載の方法であって、前記各投与期間が、2週間と12週間の間である、方法。
(項目16)
請求項1に記載の方法であって、前記抗うつ剤が、投薬量を段階的に増加する仕組みにおいて投与され、正の患者の安全性プロフィール(例えば、用量が制限される毒性)および薬物の適切なピーク血漿濃度(Cmax)を提供する、方法。
(項目17)
請求項1に記載の方法であって、別個の投薬量を含む抗うつ剤の投薬パックの提供、ならびに維持投薬量に達するまでの期間にわたって、漸増量内で別個の投薬量を摂取するための手順を含む、方法。
(項目18)
請求項17に記載の方法であって、前記投薬量パックが、ミルナシプランを含み、維持投薬量に到達するまでの日数の期間にわたって、前記投薬量が段階的に漸増される、方法。
(項目19)
請求項1に記載の方法であって、維持投薬量に達するまでの日数にわたって、抗うつ剤の漸増量を放出するために処方された抗うつ剤を提供する、方法。
(項目20)
請求項19に記載の方法であって、処方物が、持続性放出および/またはパルス状放出処方物である、方法。
(項目21)
請求項1に記載の方法であって、以下:
(a)約3日間よりも長い日数にわたって約50mgまでのミルナシプランの毎日の投薬量を必要とする患者に投与する工程;
(b)約3日間よりも長い日数にわたって約25mg〜約75mgまでのミルナシプランの毎日の投薬量を、該患者に投与する工程;および
(c)前記症状を有効に処置するための十分な期間にわたって約100mgよりも多いミルナシプランの毎日の投薬量を該患者に投与する工程;
を包含する、方法。
(項目22)
別個の投薬量、ならびに維持投薬量に達するまでの期間にわたって、漸増量における別個の投薬量を摂取するための指示書を含む、抗うつ剤の投薬パック。
(項目23)
請求項22に記載の投薬パックであって、前記投薬量が、同量であり、前記指示書が、経時的に増大した投薬量の摂取を提供する、投薬パック。
(項目24)
請求項22に記載の投薬パックであって、前記投薬量が、異なる量の抗うつ剤を含み、前記指示書が、経時的に増大した投薬量の摂取を提供する、投薬パック。
(項目25)
請求項22に記載の投薬パックであって、前記投薬量パックが、ミルナシプランを含み、前記投薬量が、維持投薬量に達する日数にわたって、段階的に増加する、投薬パック。
(項目26)
請求項22に記載の投薬パックであって、維持投薬量に達するための日数にわたって、抗うつ剤の漸増量を放出するために処方された抗うつ剤を提供する、投薬パック。
(項目27)
請求項26に記載の投薬パックであって、前記処方物が、持続性処方物および/またはパルス状放出処方物を維持される、投薬パック。
(項目28)
請求項22に記載の投薬パックであって、以下:
(a)約3日間よりも長い期間にわたる約50mgまでのミルナシプランの毎日の投薬量;
(b)約3日間よりも長い期間にわたる約25mg〜約75mgのミルナシプランの毎日の投薬量;および、
(c)症状を効率的に処置するための十分な期間にわたる約100mgよりも多いミルナシプランの毎日の投薬量;
を含む、投薬パック。
【図面の簡単な説明】
【0011】
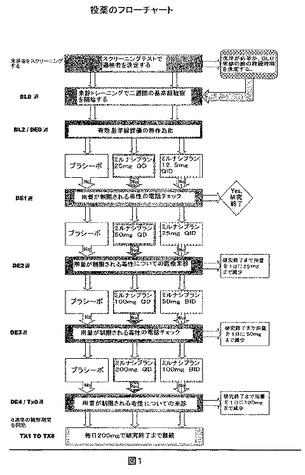
【図1】図1は、ミルナシプランの漸増する毎週の用量を患者に投薬する方法を示すフローチャートである。用量が制限される毒性は、研究を通して投薬量の段階的増加毎に評価される。
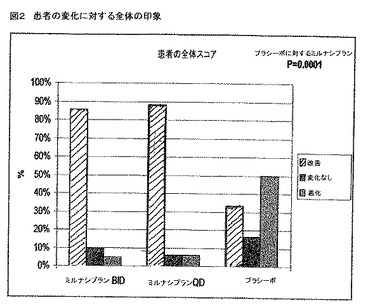
【図2】図2は、一日二回(BID)または一日一回(QD)で投与されるミルナシプランまたはプラシーボでの12週間の処置後に、改善された、不変のもしくは悪化した全体の疼痛のスコアを用いる繊維筋痛症候群(FMS)の患者のパーセントのプロットである。
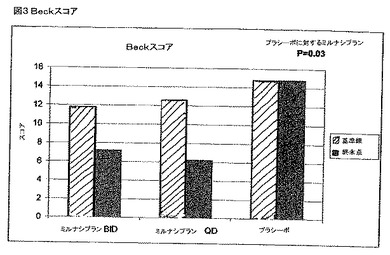
【図3】図3は、FMS患者のBeckうつ病のスコアのプロットであり、その患者は、基準ライン(治療前)およびミルナシプランのBIDまたはQDまたはプラシーボを使用した治療の終点での大うつ病(MDE)を診断される。
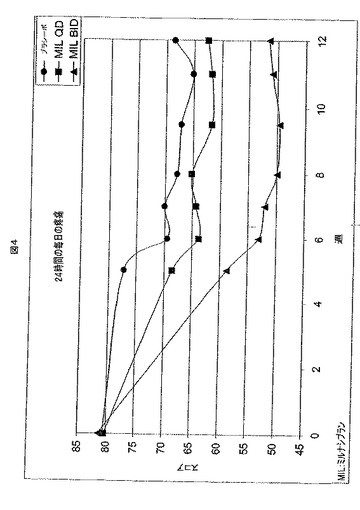
【図4】図4は、12週間の処置期間にわたって、3つのFMS処置グループの24時間の毎日の疼痛調査のスコアをプロットする。
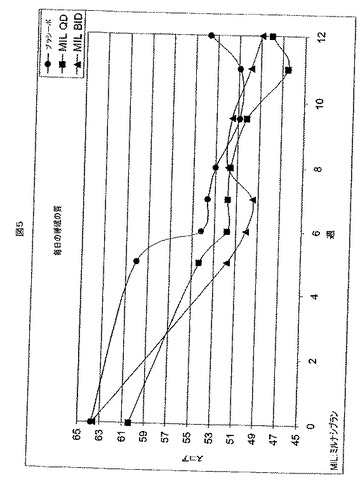
【図5】図5は、12週間の処置期間にわたって、3つの処置グループにおける患者の自己調査した毎日の睡眠の質のスコアをプロットする。
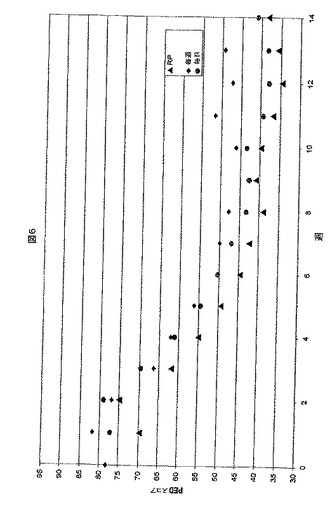
【図6】図6は、2週間の基準ライン期間および12週間の処置にわたって応答者として分類される患者の(一週間にわたって各患者について平均された)患者電子手帳の疼痛のスコアのプロットである。RP=無作為な速攻型疼痛スコア;Weekly=毎週の想起疼痛のスコア;Daily=毎日の想起疼痛スコア。
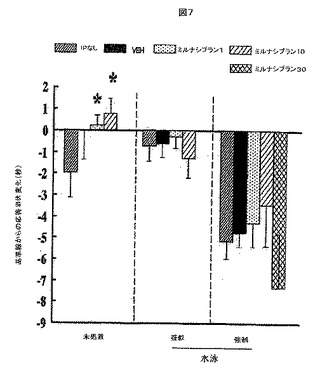
【図7】図7は、水泳ストレス試験におけるミルナシプランの注射、ビヒクルの注射または注射無しで予備処置されたラットのホットプレート潜伏期間における変化をプロットする。プロットされる変化は、水泳ストレス試験、疑似水泳試験、もしくは水泳なしの試験(未処置)を受けた後に計測された潜伏期間から、試験を受ける前に計測された潜伏を差し引いたものである。
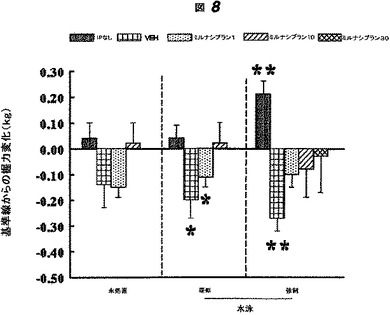
【図8】図8は、水泳ストレス試験におけるミルナシプランの注射、ビヒクルの注射または注射無しで予備処置されたラットの握力における変化をプロットする。プロットされる変化は、水泳ストレス試験、疑似水泳試験、もしくは水泳なしの試験(未処置)を受けた後に計測された潜伏期間から、試験を受ける前に計測された潜伏握力を差し引いたものである。
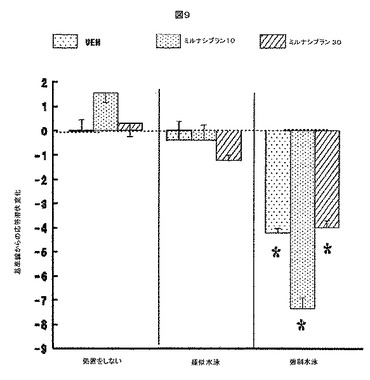
【図9】図9は、3つの水泳ストレス試験を受けた後、次いで、ストレス試験の後に、ミルナシプランの注射、ビヒクルの注射、もしくは注射無しで処置されるラットにおいて計測されたホットプレート潜伏期間をプロットする。プロットされる変化は、(1)水泳ストレス試験、疑似水泳試験、もしくは水泳なしの試験(未処置)を受け、かつ(2)試験の後にミルナシプランの注射、ビヒクルの注射、もしくは注射無しで処置された後の両方を計測した握力から、ストレス試験の前1日目に計測された潜伏期間を差し引いたものである。
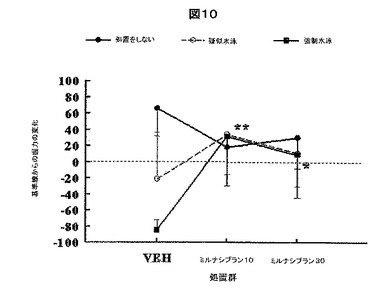
【図10】図10は、その3つの水泳ストレス試験を受けた後、次いで、そのストレス試験の後、ミルナシプランの注射、ビヒクルの注射、もしくは注射無しで処置されるラットにおいて計測された握力をプロットする。プロットされる変化は、(1)水泳ストレス試験、疑似水泳試験、もしくは水泳なしの試験(未処置)を受け、かつ(2)試験の後にミルナシプランの注射、ビヒクルの注射、もしくは注射無しで処置された後の両方計測されたその握力から、そのストレス試験の前1日目に計測された握力を差し引いたものである。
【発明を実施するための形態】
【0012】
(発明の詳細な説明)
(略語)
DSP 疼痛に対する二次的なうつ病。
CFS 慢性疲労症候群。
FMS 線維筋痛症候群。
FSD 機能的な肉体障害。
5−HT セロトニン。
NE ノルエピネフリン(ノルアドレナリン)。
NMDA N−メチル D−アスパラギン酸。
SNRI セロトニンの再取り込みが、ノルエピネフリンの再取り込みを超える、二重セロトニンノルエピネフリン再取り込み阻害剤。
NSRI ノルエピネフリンの再取り込みが、セロトニンの再取り込みを超える、二重ノルエピネフリン再取り込み阻害剤。
【0013】
(I.処置される患者)
段階的増加および/または分割された投薬量処方物で処置され得る神経学的障害は、慢性的な疼痛、神経障害性の疼痛、線維筋痛症候群、慢性疲労症候群、情動障害/気分障害、うつ病、異型のうつ病、ならびに機能的な肉体障害が挙げられる。神経学的障害の症状としては、筋骨格の疼痛、疲労、睡眠障害(sleep disorder)、睡眠障害(sleep disturbance)またはそれらの組み合わせが挙げられるが、これらに制限されない。
【0014】
神経学的障害は、その患者が、その障害に12週間より長く罹患しているが、6週間または2週間程度早期に持続的な症状に罹患しているとき、慢性的な障害と考慮される。
【0015】
慢性的な疼痛は、長期間帯にわたって(すなわち、3ヶ月より長い)持続または再発し、慢性関節リウマチといった種々の疾患または異常な状態によって生じる疼痛をいう。慢性的な疼痛は、その急性疼痛より弱いのかもしれない。疼痛に対する自動的反応が長期間持続され得ないことから、慢性的な疼痛を有する個人は、通常、脈拍の増加および急速な発汗を示さない。慢性的な痛みを有する他の者は、その環境から引きこもり得、彼らの苦痛、彼らの家族、彼らの友人および外部刺激を全く無視することにのみ、集中し得る。Mosby’s Medical,Nursing & Allied Health Dictionary,5th Editionを参照のこと。
【0016】
その慢性的な疼痛は、背部の疼痛、異形の胸痛、頭痛、骨盤痛、顔の筋膜の疼痛、腹痛および頚部痛であり得る。あるいは、その慢性的な疼痛は、関節炎、側頭部下顎関節機能障害症候群、外傷の脊髄障害、多発性硬化症、過敏性腸症候群、慢性疲労症候群、月経前症候群、多発性化学物質過敏、過換気、閉鎖性頭部外傷、線維筋痛慢性関節リウマチ、糖尿病、癌、HIVおよび間質性膀胱炎によって生じ得る。
【0017】
同様に、神経障害性の疼痛とは、末梢神経、脳神経、脊髄神経またはそれの組み合せの炎症または変性に関する疼痛をいう。その疼痛は、一般に鋭い痛み、刺すような痛みもしくは突き刺すような痛みである。根底にある障害は、末梢神経組織の破壊を生じ得、皮膚の色、温度および水腫における変化を伴い得る。Mosby’s Medical,Nursing & Allied Health Dictionary,5th Edition(1998);およびStedman’s Medical Dictionary,25th Editionを参照のこと。
【0018】
線維筋痛症候群(FMS)は、人口の2%から4%が罹患していると概算される総全身性リウマチ学的障害(common systematic rheumatologic disorder)であり、変形関節症に対してのみの有病率において第二位である。線維筋痛は、一般に圧刺激に関する疼痛について減少した許容限界に関し、そして、疲労、睡眠障害および朝のこわばりをしばしば伴う。他の通常の症候群としては、頭痛、片頭痛、非心臓性の胸痛、胸焼け、動悸、過敏性腸症候群、種々の腸嗜癖、散在性の腹痛ならびに尿頻度が挙げられる。線維筋痛に関する診断基準には、広範な疼痛の病歴だけでなく、付与される圧力に2次的な身体計側時に物理学実験における圧痛を発見することも必要とする。American College of Rheumatology(ACR)によって1990年に設定された線維筋痛についての診断基準を満たすために、個人は、軸骨格だけでなく、身体の四半分の4つ全てを含む慢性的な広範な疼痛、ならびに試験における18の圧痛点のうち11が存在することの両方を有する必要がある。
【0019】
FMSは、感覚刺激の全身化した高い感知を反映する医学的問題である。その異常性は、末梢でよりもむしろ中枢神経系(CNS)内で起こると考えられ、提案された病態生理学的欠陥は、「中枢鋭敏化」(central sensitization)と名づけられる。FMSの患者は、一般に、異痛症(電灯に触れるような非疼痛的刺激からさえも疼痛をうける)および痛覚過敏(疼痛プロセシングの漸増、(ここで疼痛刺激が拡大され、正常なボランティアの強度よりも高い強度で受ける))の両方を被る。この観点において、臨床的症状についての多くの対応があり、糖尿病ニューロパシーおよび三叉神経痛のような神経障害の疼痛の根底にあるメカニズムが提案される。
【0020】
情動障害/気分障害は、主な特徴として気分の障害により特徴付けられた種々の状態である。軽症および好都合の場合、その感情は、正常である。より重症な場合、それらは、大うつ障害または気分変調性の反応の徴候であり得るか、もしくは双極性障害の徴候を示し得る。他の気分障害は、一般の医学的状態で引き起こされ得る。Mosby’s Medical,Nursing & Allied Health Dictionary,5th Edition(1998)を参照のこと。
【0021】
うつ病は、悲しみ、絶望および落胆の感情によって特徴付けられる異常な気分障害である。うつ病は、悲しみ、憂うつ、落胆、無価値、空虚および絶望といった誇張された感情により特徴付けられた異常な感情状態をいい、その感情は、不適切で、現実に対して釣り合いが取れていない。Mosby’s Medical,Nursing & Allied Health Dictionary,5th Edition(1998)を参照のこと。
【0022】
そのうつ病は、大うつ障害のうちの少なくとも一つ(一つのエピソード、反復性のもの、軽度のもの、中程度のもの、精神病的特徴のない重傷、精神病の特徴のある重度のもの、慢性的なもの、緊張病的特徴があるもの、憂うつの特徴があるもの、異型的な特徴のあるもの、分娩後に発症するもの、一部の賓解したもの、全て賓解したもの)、気分変調性障害、うつ病の気分を有する適応障害、不安感およびうつ病の気分が混合した障害、月経前の不快性障害、小うつ病障害、反復性の短期うつ病障害、精神分裂病である精神病後のうつ病障害、パーキンソン病に関する大うつ病障害および痴呆に関するうつ病障害であり得る。
【0023】
疼痛に対して2次的なうつ病(DSP)は、疼痛の共存率および異型うつ病により特徴付けられたうつ病障害である。特に、その疼痛は、慢性的な疼痛、神経障害性の疼痛、またはそれらの組み合わせであり得る。そのDSPは、異型なうつ病および慢性的な疼痛を含み、ここで、上記慢性的な疼痛は、上記異型うつ病に先行するか、もしくは、上記異型うつ病は、上記慢性的な疼痛に先行する。あるいは、上記DSPは、異型うつ病および神経障害の疼痛を包含する。
【0024】
異型うつ病は、気分反応性、ならびに約2週間を超える期間にわたって存在する2つ以上の自律神経症状(例えば、過剰睡眠、食欲の増進または体重増加、鉛麻痺、および認められた対人拒絶に対する過敏の長期間のパターン)を含みうる。
【0025】
異型うつ病は、正の生活上の効果(気分反応性)に対する応答において、一時的に気分がよくなる能力が付随するうつ病の情動であり、加えて、過眠症、食欲の増進または体重増加、鉛麻痺およびみとめられた対人拒絶に対する過敏長期パターンの群から選択される二週間以上をこえて存在する二つ以上の自立神経症である。当業者は、自律神経症状が、他のうつ病障害において検出される症状(例えば、憂うつなうつ病)と比較して逆であり得る;それゆえ、語句「異常な」である。
【0026】
機能的肉体障害(FSD)は、組織構造または機能の疾患特異的な異常よりもむしろ症状、苦痛および能力不全によって代表的に特徴付けられるいくつかの関連する症候群を言う。機能的肉体障害を伴う患者は、肉体的には健康であるが、不具に遭遇し、医学上説明できない症状である。症状は、疲労、頭痛、関節痛、弱気、記憶障害、不安および動悸といった病状が挙げられる。一般的な機能肉体症候群は、複数の化学物質過敏性、シックハウス候群、反復ストレス傷害、慢性的な鞭打ち傷害、慢性的なライム病、シリコン胸部移植の副作用、カンジダ症感受性、湾岸戦争症候群、僧帽弁脱出および低血糖症が挙げられる。
【0027】
機能肉体障害を伴った患者は、代表的に、彼ら自身その症状についての自己診断を提供し、特定の疾患に対する彼らの症状の属性に矛盾する情報に抵抗する。これらの患者は、精神医学的な障害、特に不安、うつ病および身体型障害のより高い発生率を有する。患者の症状を増幅する心理社会的要因としては、その患者が重篤な疾患を有するという考え;状態が悪化しそうであるという患者の予測;訴訟および補償の影響を包含する「病人の役割」;およびその状態が破局的であり不能である患者による警告説明が挙げられる。それにもかかわらず、機能的肉体障害を有すると診断される患者は、考慮すべき障害ならびに不能によって特徴付けられる。
【0028】
機能的肉体障害は、疼痛にも関連し得る。その疼痛は、機能的肉体障害の発生の前に起こるかまたは後に起こり得る。その疼痛は、慢性的な疼痛または神経障害性の疼痛もしくはその二つの組み合わせであり得る。
【0029】
(II. 組成物)
(A. 活性化合物)
この方法で使用される活性化合物は、抗疼痛活性または鎮痛活性、抗うつ剤活性を有し、それゆえに疼痛、うつ病ならびに関連する疾患および症状の処置のための薬物として有用である。
【0030】
この方法で使用される化合物は、神経学的障害に関連する状態または症状を処置、防止またはより軽減するための薬物の能力を決定するための実験またはアッセイにおいて、例えば薬学研究プログラムにおいて使用するための標準化合物または参照化合物としても有用である。このようにして、本明細書で開示される化合物は、コントロール化合物または参照化合物としてそのようなアッセイで使用され得、制御標準物質として使用され得る。本発明の化合物は、市販のキットまたはそのような標準化合物もしくは参照化合物としての使用のための容器で提供され得る。
【0031】
ノルエピネフリン(NE)−セロトニン(5−HT)再取り込み阻害剤(NSRI)は、ノルエピネフリン(NE)およびセロトニン(5−HT)の両方の再取り込みを阻害する化合物クラスを言うが、5−HTの再取り込みよりもNEの再取り込みを優先的にブロックする。選択的NSRIは、少なくとも約1のNE:5−HT再取り込み阻害比を有する。具体的には、選択的NSRIは、約50までのNE:5−HTの再取り込み阻害比を有し得る。より具体的には、その選択的NSRIは、約1:1から約20:1のNE:5−HTの再取り込み阻害比を有し得る。より具体的には、選択的NSRIは、約1:1から約5:1のNE:5−HTの再取り込み阻害比を有し得る。より具体的には、その選択的NSRIは、約1:1から約3:1のNE:5−HT再取り込み阻害比を有し得る。
【0032】
NSRIは200nM以下の5−HT再取り込みのIC50および200nM以下のNE再取り込みのIC50、ならびに少なくとも1000nMドーパミン再取り込みのIC50を有する。NRSIは、少なくとも約0.5:1のNE:5−HT再取り込みの阻害比を有する。そのNE:5−HTの再取り込み阻害比は、5−HT再取り込みのIC50をNE再取り込みのIC50で除算することで計算される。例えば、化合物が、10nMのNE再取り込みのIC50および20nMの5−HTの再取り込みのIC50を有する場合、その化合物は、2:1のNE:5−HTの再取り込み阻害比を有する。特定の実施形態において、NSRIは、NE:5−HT再取り込みの約0.5:1から約50:1、約1:1から約20:1、約0.5:1から約5:1、約1:1から約5:1、約0.5:1から約3:1、もしくは約1:1から約3:1、の比の阻害を有する。
【0033】
そのNSRI化合物は、NMDAレセプターでアンタゴニスト性質を示し得る。NMDAレセプターアンタゴニストは結合し、NMDAレセプターの活性を減少する。これは、非競合的および競合的な両方の、NMDAレセプターアンタゴニスト、グリシン−部位アンタゴニスト、グルタミン酸アンタゴニストおよびアロステリックアンタゴニストが挙げられる。化合物は、当業者で公知のアッセイによってNMDAレセプターアンタゴニストであると決定され得る。
【0034】
「ミルナシプラン」(±)−シス−2−(アミノメチル)−N、N−ジメチル−1−フェニルシクロプロパンカルボキサミドヒドロクロリド(CAS 登録番号が92623−85−3)が、以下の式のNSRI化合物である:
【0035】
【化1】

ミルナシプランの調製法は、例えば、米国特許第4,478,836号およびそこで引用される参考文献で開示される。他に指摘の無い場合、ミルナシプランは、全ての立体異性体、立体異性体の混合物ならびにその薬学的に受容可能な塩を包含し得る。
【0036】
ミルナシプランの右旋性異性体は、ノルエピネフリンおよびセロトニンの再取り込みの阻害活性として、ラセミ混合体よりも約2倍の活性であり、左旋性異性体は、はるかに強力でないと考えられる。例えば、Viazzo et al.,1996,Tetrahedron Lett. 37(26):4519−4522;Deprez et al.,1998,Eur.J.Drug Metab.Pharmacokinet.23(2):166−171).を参照のこと。従って、ミルナシプランは、鏡像異性体的に純粋な形態(例えば、純粋な右旋性異性体)もしくはラセミ混合物といった右旋性異性体および左旋性異性体の混合物として投与され得る。
【0037】
必要とされる場合、ミルナシプランのラセミ混合物の分離は、キラルカラムを使用するHPLC、もしくは Thomas J.Tucker,et al.,J.Med.Chem.1994 37,2437−2444と同様のショウノウ塩化物といった分離薬物(resolving agent)を使用する分離によって達成され得る。ミルナシプランはまた、キラル触媒またはキラルリガンドを使用して直接的に合成されうる(例えば、Mark A.Huffman,et al.,J.Org.Chem.1995,60,1590−1594)。
【0038】
ミルナシプランの経口投与に対する公知の有害反応としては、吐き気、嘔吐、頭痛、ふるえ、不安、不安発作、動悸、尿貯留、起立性低血圧、発汗、胸痛、発疹、体重増加、背痛、便秘、下痢、目眩、発汗増加、心的動揺、のぼせ、震え、疲労、眠気、消化不良、嗅覚不全、緊張、口内乾燥、腹痛、不眠症または、それらの組み合わせが挙げられ得る。
【0039】
投薬量の段階的増加および/または分割された投薬量を使用して投与され得る他のSNRIとしては、以下:
ベンラファキシンヒドロクロリド(R/S)−1−[2−(デメチルアミノ)−1−(4−メトキシフェニル)エチル]シクロヘキサノールヒドロクロリド)または((±)−1−[α[(ジメチルアミノ)メチル]−p−メトキシベンジル]シクロヘキサノールヒドロクロリド);
ミルタザピン(1,2,3,4,10,14b−ヘキサヒドロ−2−メチルピラジノ [2,1−a]ピリド[2,3−c]ベンズアゼピン);
ネファゾドンヒドロクロリド(2−[3−[4−(3−クロロフェニル)−1−ピペラジニル]プロピル]−5−エチル−2,4−ジヒドロ−4−(2−フェノキシエチル)−3H−1,2,4−トリアゾール−3−オン モノヒドロクロリド);
チオリダジンヒドロクロリド(10H−フェノチアジン,10−[2−(1−メチル−2−ピペリジニル)エチル]−2−(メチルチオ)−,モノヒドロクロリド);ブプロピオン ヒドロクロリド((±)−1−(3−クロロフェニル)−2−[1,1−ジメチルエチル)アミノ]−1−プロパノン ヒドロクロリド);
モノアミンオキシダーゼ阻害剤であって、例えば、以下
トラニルシプロミンスフェート((±)−トランス−2−フェニル−シクロプロピルアミン スフェート(2:1));フェネルジンサルフェート(硫化水素フェネチルヒドラジン);
モノクロベミド(p−クロロ−N−(2−モルホリノエチル)ベンズアミド);
ピルリンドル(2,3,3a,4,5,6−ヘキサヒドロ−8−メチル−1H−ピラジノ[3,2,1−j,k]カルバゾール;
選択的セロトニン再取り込み阻害剤であって、例えば、以下:シタロプラムヒドロブロシド((±)−1−(3−ジメチルアミノプロピル)−1−(4−フルオロフェニル)−1,3−ジヒドロイソベンゾフラン−5−カルボニトリル,HBr);
パロキセチンヒドロクロリド((−)−トランス−4R−(4’−フルオロフェニル)−3S−[(3’,4’−メチレンジオコシフェノキシ)メチル]ピペリジンヒドロクロリド半水化物);
フルオキセチンヒドロクロリド((±)−N−メチル−3−フェニル−3−[(α,α,α−トリフルオロ−p−トリル)オキシ]プロピルアミンヒドロクロリド);
セルトラリンヒドロクロリド((1S−シス)−4−(3,4−ジクロロフェニル)−1,2,3,4−テトラヒドロ−N−メチル−1−ナフタレンアミンヒドロクロリド);
三環形の抗うつ剤であって例えば、以下:アミトリプチリンHCl(3−(10,11−ジヒドロ−5H−デベンゾ[a,d]シクロヘプテン−5−イリデン)−N,N−ジメチル−1−プロパンアミンヒドロクロリド)。
【0040】
デシプラミンヒドロクロリド(5H−ジベンズ[bf]アゼピン−5−プロパンアミン、10,11−ジヒドロ−N−メチル−,モノヒドロクロリド);
ドキセピンヒドロクロリド(1−プロパンアミン,3−ジベンゾ[b,e]オキセピン−11(6H)イリデン−N,N−ジメチル−,ヒドロクロリド);
マレイン酸トリミプラミン(5−(3−ジメチルアミノ−2−メチルプロピル)−10,11−ジヒドロ−5H−ジベンズ(b,f)アゼピン酸およびマレイン酸(ラセミ形態));
プロトリプチリンHCl(N−メチル−5H−ジベンゾ[a,d]−シクロヘプテン−5−プロパンアミンヒドロクロリド);
抗けいれん薬(anti−covalsant)であって、以下:
ジバルプロエックスナトリウム(水素ビス(2−プロピルペンタン酸)ナトリウム);
クロナゼパム(5−(2−クロロフェニル)−1,3−ジヒドロ−7−ニトロ−2H−1,4−ベンゾジアゼピン−2−オン);
およびアルプラゾラム(8−クロロ−1−メチル−6−フェニル−4H−s−トリアゾロ[4,3−α][1,4]ベンゾシアゼピン);
が挙げられる。
【0041】
上記化合物は、体の流体中で実質的に遊離状態であり得る。さらに、これらの化合物は、少なくとも90重量%の純度、少なくとも95重量%の純度、少なくとも98重量%の純度、少なくとも99重量%の純度であり得る。
【0042】
(B.塩類および誘導体)
その化合物の薬学的に受容可能な塩類は、その親化合物から合成され得、上記化合物は、従来の化学的方法によって塩基性部分または酸性部分を含む。一般的に、そのような塩類は、これらの化合物の遊離酸形態または遊離塩基形態と水中、有機溶媒中もしくはこの2つの混合物中で、化学量論的量の適切な酸または塩基とを反応させることで調整され得る;一般的に、エーテル、エチルアセテート、エタノール、イソプロパノールまたはアセトニトリルのような非水性媒体が好ましい。適切な塩類のリストは、Remington’s Pharmaceutical Sciences,17th ed.,Mack Publishing Company,Easton,PA,1985,p.1418に見出される。
【0043】
薬学的に受容可能な塩類の例としては、アミンおよびアルカリといった塩基性残基無機酸塩または有機酸塩が挙げられるが、これらに限定されない。その薬学的に受容可能な塩は、例えば、非毒性の無機酸または有機酸から形成されるその親化合物の従来の非毒性塩類を含む。例えば、そのような従来の非毒性塩類は、無機酸(例えば、塩酸、臭化水素酸、硫酸、砂糖、スルファミン酸、リン酸、および硝酸由来の塩類);および、有機酸(例えば、酢酸、プロピオン酸、コハク酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、パモン酸(pamoic acid)、マレイン酸、ヒドロキシマレイン酸、フェニル酢酸、グルタミン酸、安息香酸、サリチル酸、スルファニル酸、2−アセトキシ安息香酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸およびイセチオン酸)から調整される塩が挙げられる。特に、薬学的に受容可能な塩類は、塩化水素塩または塩酸(HCl)塩であり得る。
【0044】
これらの化合物は、過剰な毒性も、刺激も、アレルギー応答も、他の問題もしくは合理的な利益/リスクの比が相応につりあった合併症もなく、人および動物の組織との接触における使用に適切である。
【0045】
適切な化合物としては、プロドラッグおよび活性成分の代謝物もまた挙げられる。プロドラッグは、共有結合で結合するなんらかの物質であって、そのようなプロドラッグが、被験体に投与される場合、その物質はその活性な親薬物または他の処方物もしくは本発明の化合物をインビボにおいて放出する。
【0046】
代謝物は、生化学的プロセスから生じた任意の物質であって、このプロセスによって生細胞が、その活性化合物と相互作用し、その生細胞は、任意の代謝経路由来の産物または中間体を包含する。
【0047】
(C.活性成分の組み合わせ)
この処置方法で使用される各治療薬剤は、独立的に任意の投薬形態であり得、また、組み合せで投与され得る。その薬剤は、単体の投薬ユニット(すなわち、一つのカプセル、錠剤、粉末、または液体等の中で一緒に併用される)において組み合せの産物として共に処方され得、単体の化合物として同時に、もしくは単体の投薬ユニットとして共に処方されない場合に任意の順序で投与され得る。好ましくは、その薬剤は、分割して投与される場合、お互いに一時間以内で投与される。
【0048】
同時投与薬剤の適切な投薬量は、当業者において、本開示に基づいて当該分野の開業医によって容易に確認できる。一般的な指導を介して、代表的には、毎日の投薬量は、各成分の約100ミリグラムから約1.5グラムであり得る。一つ以上の化合物が投与される場合、典型的投薬量は、各薬剤の約100ミリグラムから約1.5グラムであり得るであり得る。一つより多い化合物が投与される場合、次いで代表的には、毎日の投薬量は、各薬剤の100ミリグラムから1.5グラムであり得る。一般的な指導を介して、複数の薬剤が、組み合せされて投与されるとき、各薬剤の投薬量は、組み合せの相乗作用効果の視点から、個別に投与される場合の通常の投薬量と比較して約70%〜80%まで減少し得る。
【0049】
(D.処方物および賦形剤)
(処方物)
活性成分は、固形の投薬形態(例えばカプセル、錠剤および散剤)または液状の投薬形態(例えば、エリキシル剤、シロップおよび懸濁液)で経口的に投与され得る。活性成分はまた、無菌の液状の投薬形態において、非経口的に投与され得る。添加剤はまた、物理学的な外形を向上し、安定性を改善し、そして投薬後の崩壊を補助するための処方物において包含されうる。例えば、経口投与のための液状の投薬形態は、患者の受容を増加するために着色剤および矯味矯臭剤を含み得る。
【0050】
活性成分の毎日の投薬量は、一日一回(QD)の投薬量または代替の分割された投薬量、例えば一日二回(BID)の投薬量で投与され得る。
【0051】
ゼラチンのカプセルは、活性成分ならびに、ラクトース、デンプン、セルロース誘導体、ステアリン酸マグネシウム、ステアリン酸などといった粉末の担体を含む。類似する希釈剤が、圧縮錠剤を作製するのに使用され得る。錠剤およびカプセルの両方が、徐放性生成物として製造され得、数時間または数日間にわたって薬物の持続的な放出を提供し、ならびに移植、または経皮/経粘膜配達のために処方される。そのような処方物は、代表的には、生物分解もしくは生物侵食されて、ミルナシプランの一部を放出するポリマーを包含する。処方物は、マイクロカプセル、リポソーム、固体のモノリシック(monolithic)移植物、ゲル、粘性流体、椎間板、もしくは附着フィルムの形式を有し得る。
【0052】
圧縮錠剤は、好ましくない味覚を覆い隠し、その錠剤を大気から保護するために糖被覆またはフィルム被覆され得るか、または胃腸管において選択的崩壊のために腸溶性の被覆され得る。
【0053】
(キット)
活性成分は、キットとして処方され得、上記キットは、分割された毎日の投薬当量または投薬量ユニットを維持する容器と組み合わされる漸増濃度の用量ならびにその用量を投与するための手順を伴ったプリントされた説明書(printed indicia)が挙げられる。
【0054】
好ましくは、四つの工程の投薬量の段階的増加において使用されるキットは、第一投薬量ユニットである100mgまでの活性成分を含む。第二の投薬量は、第一の投薬量よりも1.5倍〜2.5倍多い投薬量が含まれる。第三の投薬量は、第二の投薬量よりも1.5倍〜2.5倍多い投薬量が含まれる。第四の投薬量は、第三の投薬量よりも1.5倍〜2.5倍多い投薬量が含まれる。このキットは、四つの投薬量ユニットを維持するための容器ならびにその用量を投与するための指示を有するプリントされた説明書を含む。第一のユニット投薬量形態は、一日一回(QD)の投薬量の形態であり得る。このキットの形式は、より多い、または少ない漸増投薬量の投薬ユニットを包含するように改変され得る。
【0055】
本キットの好ましい実施形態は、100mgまでのミルナシプランの第一の投薬量および100mgより多いミルナシプランの第二の投薬量ユニットを含む。このキットはまた、二つの投薬量ユニット形態を維持するための容器ならびにその用量を投与するための指示を有する、プリントされた説明書を含む。このキットは、投薬量ユニット形態を維持するための容器およびその投薬量を投与するための指示とともに、(1)20mg〜30mg、(2)40mg〜60mg、(3)75mg〜125mg、(4)175mg〜225mgのミルナシプランの四つの段階的増加投薬量を含むように改変され得る。
【0056】
(賦形剤)
活性成分または治療成分に加えて、錠剤は多くの不活性物質を含む。後者は添加物または「付加物」として公知である。それらは、最終錠剤において作用する部分に従って分類され得る。第一の群は、処方物に対して十分な圧縮特性を与えるように補助するものを含む。これらは、(1)希釈剤、(2)結合剤および(3)滑沢剤が挙げられる。添加物質の第二の群は、最終錠剤にさらなる望ましい物理学的特性をあたえるように補助する。この群に包含される添加物質は、(1)崩壊剤、(2)着色剤、ならびにチュアブル錠剤の場合においては、(3)矯味矯臭剤および(4)甘味剤がある。
【0057】
しばしば、活性成分の単体の投薬は、わずかであり、不活性物質は、錠剤を圧縮によって実用的なサイズにするためにバルクを増加するために追加される。この目的について使用される希釈剤は、希釈剤と活性成分の間で適合性に準じて、燐酸二カルシウム、硫酸カルシウム、乳酸、カオリン、マンニトール、塩化ナトリウム、乾燥デンプン、ならびに粉末糖が挙げられる。
【0058】
粉末物質に対して粘着特性を付与するのに使用される薬剤は、結合剤または顆粒剤といわれる。それらは、錠剤処方物に粘着性を付与し、圧縮後その錠剤がインタクトなままであること、ならびに望ましい硬度およびサイズの顆粒剤の処方物による自由流動特性の改善を確実にする。一般的に結合剤として使用される物質としては、デンプン、ゼラチン、ならびにスクロース、グルコース、デキストロース、モラセスおよびラクトースといった糖類が挙げられる。使用される天然および人工のガムは、アラビアゴム、アルギン酸ナトリウム、トチャカ抽出物、パンワーゴム、ガティゴム、イサポール(isapol)さやの粘液、カルボキシメチルセルロース、メチルセルロース、ポリビニルピロリドン、ハチゴムならびにカラマツアラボガラクタンが挙げられる。ある状況下では結合剤と考えられ得る他の薬剤には、ポリエチレングリコール、エチルセルロース、ワックス、水およびアルコールがある。
【0059】
滑沢剤は、錠剤顆粒の流速の改善、ダイやせん孔の表面に対する錠剤物質の付着の防護、粒子間摩擦の減少、ならびにダイキャビティからの錠剤排出の促進のための錠剤製品において使用される。一般的に、使用される滑沢剤としては、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸および硬化植物油が挙げられる。
【0060】
崩壊剤は、物質もしくは物質の混合物であり、投与後の崩壊または分解を促進するために錠剤に対して添加される。崩壊剤として働く物質は、化学的にはデンプン、粘土、セルロース、アルギン(align)もしくはゴムとして分類される。
【0061】
デンプンに加え、多種の物質が使用され、崩壊剤として有効であると報告されている。この群は、ビーガム(Veegum HV)、メチルセルロース、寒天、ベントナイト、セルロースおよび、木製品、天然のスポンジ、陽イオン交換樹脂、アルギン酸、グアールガム、柑橘類のパルプならびにカルボキシメチラーゼが挙げられる。デンプンとの組み合せにおいてラウリル硫酸ナトリウムは、有効な崩壊剤であることが実証されている。
【0062】
非水溶性の担体もしくは賦形剤は、生体適合性、および望ましい速度で血流に活性成分を放出するのに十分な哺乳動物の体温で液状もしくは柔軟性の任意の物質である。その担体は、通常疎水性、かつ一般的に、例えば、植物、動物、無機質および合成物の起源もしくは誘導の油もしくは脂肪といった有機物である。好ましくは、その担体は、水において非混和性であり、および/または一般的に脂肪溶媒として公知の物質中において溶解性である。
【0063】
代表的に、水、適切な油、生理食塩水、水性デキストロース(グルコース)および関連糖質溶液ならびにプロピレングリコールおよびポリエチレングリコールといったグリコールは、非経口の溶液について適切な担体である。非経口投与のための溶液は、好ましくはミルナシプランの水溶性塩、適切な安定薬剤、ならびに必要とする場合緩衝物質を包含する。重硫酸塩ナトリウム、硫酸ナトリウム、もしくはアスコルビン酸、これらの単体もしくは組み合せといった抗酸化薬剤は、適切な安定薬剤である。また、使用されるのは、クエン酸およびその塩ならびにEDTAナトリウムである。さらに、非経口の溶液は、塩化ベンザアルコニウム塩化物、メチルパラベンまたはプロピルパラベンならびにクロロブタノールといった保存剤を含み得る。適切な薬学的担体は、当該分野で公知である。
【0064】
(III.使用法)
(A.処置プロトコル)
活性化合物の投与は、患者に対する有害反応(薬物(例えばミルナシプラン)に対する、異常な、有害な、もしくは予測外の反応の減少もしくは欠如を意味する)が実質的にないと同時に、神経学的障害の症状を処置するのに有効な量であるべきである。語句、「有害反応が実質的にない」は、活性成分の特定の投薬量での有害反応の回数、性質および程度に関係する。活性化合物は、神経学的障害の症状を回復もしくは排除するための治療的に有効な期間にわたって投与されるべきである。
【0065】
有害反応は、下記:皮膚、中枢神経系および末梢神経系、視覚、精神医学、胃腸系、肝臓および胆汁系、内分泌および代謝系、心血管系、呼吸系、赤血球、白血球、血小板、血液、泌尿器系、生殖系ならびに新生物のうちの少なくとも一つに関連し得る。その有害反応は、下記:悪心、嘔吐、頭痛、臆病、不安、不安発作、動悸、尿貯留、起立性低血圧、発汗、胸痛、発疹、体重増加、背痛、便秘、目眩、発汗増加、動揺、のぼせ、震え、疲労、眠気、消化不良、嗅覚異常、緊張感、口内乾燥、腹痛および不眠のうちの少なくとも一つと関連し得る。
【0066】
その化合物は単体で投与されうるが、好ましくは、選択された投与経路ならびに医薬品の実行基準に基づいて選択された薬学的担体とともに投与される。それらは、個々の治療薬剤もしくはその治療薬剤の組み合せのいずれかとして、医薬品とともに使用するために利用可能である任意の従来の手段によって投与され得る。
【0067】
好ましくは、活性成分の毎日の投薬量は、患者において循環する活性化合物の治療的量に達するように一定期間にわたって段階的に増加される(例えば、第一の期間および/または第二の期間)。
【0068】
毎日の投薬量の漸進的段階的増加は、投与される活性成分に対する患者の耐性の改善を意図する。活性成分で処置された患者は、(1)有害反応を経験しない;(2)わずかな有害反応を経験する;(3)重傷度の少ない程度の有害反応を経験する;もしくはその組み合わせである。この耐性は、投薬の段階的増加手順なしに活性成分の同投薬量に対する哺乳動物の耐性と比較して、特定の投薬量での活性成分に対する哺乳動物の耐性に関連する。
【0069】
一般的に、活性化合物は段階的形式における漸増投薬量において投与され得、有害反応を回避もしくは最小化するための患者の耐性の増加を伴って活性化合物の循環投薬量を増加する。活性化合物の毎日の投薬量は、各期間(すなわち各工程)の間で異なり得るが、ただし各投薬量は、その工程の間に割り当てられた投薬量の範囲内である。
【0070】
例えば、4段階の段階的増加において、活性化合物は、第一の期間に100mgまでの毎日の投薬量で投与され、そして第二の期間に最初の投薬量の1.5倍〜2.5倍量まで段階的に増加される。第二の投薬期間は、第一の期間と同様の持続期間であり得、第一の期間の後、活性成分の投薬量は、第三の投薬期間に第二の投薬量の1.5倍〜2.5倍量まで再び段階的に増加される。第三の投薬期間は、第一および第二の期間と同様の持続期間であり得、その第一の期間または第二の期間の後、活性成分の投薬量は、神経学的障害の症状を処置するのに治療的に有効な期間に第三の投薬量の1.5倍〜2.5倍量まで再び段階的に増加する。あるいは、2段階の段階的増加において、その投薬量は、第二の投薬量に対して一度だけ段階的に増加され得、第二の期間は、神経学的障害の症状を処置するのに治療的に有効な期間であり得る。
【0071】
投薬量の段階的増加での各工程の期間は、3日間、3日間をこえた時間、もしくは2週間、4週間、6週間、8週間、10週間、12週間もしくは20週間よりも長い期間であり得る。毎日の投薬量は、一回投与され得るか、もしくは一日に二回以上(例えば、2回、3回、4回、または5回)までに分割され得る。
【0072】
ある実施形態は、3日間より長い期間にわたって100mgまでのミルナシプランの毎日の投薬量を投与し、そして神経学的障害の症状を処置するために治療的に有効な期間にわたって100mgより多い毎日の投薬量の段階的増加により、2段階の段階的増加を要する。
【0073】
別の実施形態は、3日間より長い期間にわたって、最初の毎日の約10mgと約50mgの間の投薬量でのミルナシプランの投与、そして3日間より長い期間にわたる約25mg〜約75mgへの毎日の投薬量の段階的増加ならびに神経学的障害の症状を処置するために治療的に有効な期間にわたる100mgより多い毎日の投薬量の段階的増加によって、3段階の段階的増加を要する。
【0074】
別の実施形態は、最初の毎日の約20mgと約30mgの間の投薬量で7日間にわたるミルナシプランの投与、7日間にわたる約40mg〜約60mgへの毎日の投薬量の段階的増加、および7日間にわたる約75mg〜約125mgへの毎日の投薬量の段階的増加、ならびに神経学的障害の症状を処置するために治療的に有効な期間にわたる175mg〜225mgへの毎日の投薬量の段階的増加によって4段階の段階的増加を要する。
【0075】
これらの投与のプロトコルは、上述の任意の抗うつ剤化合物にも適用され得る。
【0076】
(B.効果的な投薬量の範囲)
投与される投薬量は、特定の薬剤ならびにその投与形式および投与経路の薬力学特性;レシピエントの年齢、健康および体重;その症状の性質および程度;同時発生的な処置の種類;処置の頻度;ならびに望ましい効果といった既知の要因に依存して変化する。活性成分の毎日の投薬量は、体重1キログラムあたり約0.001mg〜約1000mgまであり、好ましい投薬は約0.1mg/kg〜100mg/kgであり、好ましくは一日に数回投与されることが期待される。
【0077】
本明細書において開示される活性成分(例えば、ミルナシプラン)の投薬量は、神経学的傷害の症状を処置することにおいて適切な活性を有し、そのような活性は例えば以下である:(1)活性成分の改善された効力、(2)正の患者耐性の維持、(3)正の患者の安全性プロフィールの維持(例えば、用量が制限される毒性)、(4)活性成分の適切なピーク血漿濃度(Cmax)、および/または(5)一日二回(BID)の投与とは対照的に一日一回(QD)。
【0078】
NSRI抗うつ剤は、うつ病を処置するための抗うつ剤の推奨される毎日の投薬量よりも多い(例えば、約1.5倍〜約4.0倍多い)毎日の投薬量において投与され得る。本明細書において開示される抗うつ剤においての推奨される毎日の投薬量は、うつ病を処置するために、例えば、(Physisician’s Desk Referance)(PDR),55th Edition(2001);およびインターネット薬物索引ウェブサイト(www.RxList.com);もしくはSLS 精神医学ならびに薬物関連ウェブサイト(http://sl.schofield3.home.att.net/medicine/psychiatric_drugs_chart.html)で見
られ得る。
【0079】
NSRI抗うつ剤は、他の神経学的障害の症状を処置する場合、うつ病を処置するのに推奨されるよりもより分割された投薬量において投与され得る。抗うつ剤について推奨される毎日の投薬量は、抗うつ剤を処置するために、例えば、Physisician’s
Desk Referance)(PDR),55th Edition(2001);およびインターネット薬物索引ウェブサイト(www.RxList.com);もしくはSLS 精神医学ならびに薬物関連ウェブサイト(http://sl.schof
ield3.home.att.net/medicine/psychiatric_drugs_chart.html)で見られ得る。
【0080】
一般的に、2段階の投薬量の段階的増加において、第一の投薬量は、一日あたり約100mgまでであり、第二の投薬量は一日あたり約100mgよりも多い。好ましくは、次の段階的増加工程の投薬量は、一般的に前の投薬量の1.5倍〜2.5倍である。そのようにして、4段階の段階的増加のプロトコルにおいて、第二の毎日の投薬量は、第一の毎日の投薬量の約1.5倍〜約2.5倍である;第三の毎日の投薬量は、第二の毎日の投薬量の約1.5倍〜約2.5倍である;および第四の毎日の投薬量は、第三の毎日の投薬量の約1.5倍〜約2.5倍であって、神経学的障害の症状を効果的に処置する十分な期間にわたって投与される。
【0081】
例えば、3段階のミルナシプランの投薬量の段階的増加において好ましい投薬量の様式は、約3日間の間1日50mgであり、続いて約3日間の間で25mg〜75mgであり、次いで神経学的障害の症状を効率的に処置するための十分な期間にわたって100mgより多い投薬量である。
【0082】
別の例として、4段階のミルナシプランの投薬量の段階的増加の好ましい様式は、約7日間の間に約20mg〜約30mg、続いて約7日間の間約40mg〜約60mgの毎日の投薬量が投与され、約7日間の間約75mg〜約125mgの毎日の投薬量が続き、最終的に、神経学的障害の症状を効果的に処置する十分な期間にわたって約175mg〜約225mgである。
【0083】
好ましくは、ミルナシプランは100mg/日よりも多く投与され、より好ましくは、200mg/日よりも多く投与される。1つの実施形態において、ミルナシプランは、体量の約100mg/70kgよりも多く投与される。
【0084】
(増加した毎日の投与においての分割された毎日の投薬量)
より高い毎日の投薬量は、一回の毎日の投薬量を分割すること、ならびに一日二回以上(例えば、2回,3回,4回または5回)投与することで達成され得る。任意の一つ以上の抗うつ剤は、本明細書で記載される毎日の投薬量において、分割された投薬量で投与され得、長期間にわたって有効な血清中の薬物濃度を得る。
【0085】
例えば、約200mgのミルナシプランの毎日の投薬量の投与は、約100mgで一日二回(BID)投与され得る。同様に、ミルナシプランの約400mgの毎日の投薬量の投与は、約200mgを一日二回(BID)投与され得る。この技術は、抗うつ剤のより高い投薬量を投与するために拡大され得る:
ミルナシプランは、一日あたり約50mgと約800mgの間で;好ましくは、一日あたり約100mgと約400mgの間で、最も好ましくは、一日あたり約200mgと約300mgの間で投与され得る。
【0086】
ベンラファキシンヒドロクロリドは、一日あたり約75mgと約1,500mgの間で;好ましくは、一日あたり約112.5mgと約900mgの間で、最も好ましくは、一日あたり約225mgと約600mgの間で投与され得る。
【0087】
ミルタザピンは、一日あたり約15mgと約180mgの間で;好ましくは、一日あたり約30mgと約120mgの間で、最も好ましくは、一日あたり約45mgと約60mgの間で投与され得る。
【0088】
ネファゾドンは、一日あたり約200mgと約2,400mgの間で;好ましくは、一日あたり約300mgと約1200mgの間で、最も好ましくは、一日あたり約600mgと約800mgの間で投与され得る。
【0089】
チオリダジンヒドロクロリドは、一日あたり約50mgと約800mgの間で;好ましくは、一日あたり約100mgと約400mgの間で、最も好ましくは、一日あたり約200mgと約300mgの間で投与され得る。
【0090】
ブプロピオンヒドロクロリドは、一日あたり約150mgと約1,600mgの間で;好ましくは、一日あたり約300mgと約1200mgの間で、最も好ましくは、一日あたり約400mgと約600mgの間で投与され得る。
【0091】
フェネルジンスルフェートは、一日あたり約15mgと約360mgの間で;好ましくは、一日あたり約60mgと約240mgの間で、最も好ましくは、一日あたり約90mgと約135mgの間で投与され得る。
【0092】
硫酸トラニルシプロミンは、一日あたり約30mgと約160mgの間で;好ましくは、一日あたり約45mgと約120mgの間で、最も好ましくは、一日あたり約60mgと約80mgの間で投与され得る。
【0093】
モクロベミンドは、一日あたり約400mgと約3,600mgの間で;好ましくは、一日あたり約500mgと約2400mgの間で、最も好ましくは、一日あたり約900mgと約1200mgの間で投与され得る。
【0094】
ピルリンドルは、一日あたり約200mgと約1,600mgの間で;好ましくは、一日あたり約300mgと約1,200mgの間で、最も好ましくは、一日あたり約400mgと約800mgの間で投与され得る。
【0095】
シタロプラムブロミドは、一日あたり約20mgと約240mgの間で;好ましくは、一日あたり約40mgと約160mgの間で、最も好ましくは、一日あたり約60mgと約80mgの間で投与され得る。
【0096】
パロキセチンヒドロクロリドは、一日あたり約20mgと約250mgの間で;好ましくは、一日あたり約50mgと約200mgの間で、最も好ましくは、一日あたり約62.5mgと約80mgの間で投与され得る。
【0097】
フルオキセチンヒドロクロリドは、一日あたり約20mgと約320mgの間で;好ましくは、一日あたり約60mgと約240mgの間で、最も好ましくは、一日あたり約80mgと約120mgの間で投与され得る。
【0098】
セルトラリンヒドロクロリドは、一日あたり約25mgと約800mgの間で;好ましくは、一日あたり約50mgと約200mgの間で、最も好ましくは、一日あたり約75mgと約100mgの間で投与され得る。
【0099】
アミトリプチリンヒドロクロリドは、一日あたり約50mgと約600mgの間で;好ましくは、一日あたり約100mgと約400mgの間で、最も好ましくは、一日あたり約150mgと約200mgの間で投与され得る。
【0100】
パーフェナジンおよびアミトリプチンヒドロクロリドは、それぞれ、約6mg〜約22mg、約75mg〜約350mgの間で;好ましくは、一日あたり、それぞれ約9mg〜約15mg、約100mg〜約200mgの間で投与され得る。
【0101】
デシプラミンヒドロクロリドは、一日あたり約100mgと約1200mgの間で;好ましくは、一日あたり約200mgと約800mgの間で、最も好ましくは、一日あたり約300mgと約400mgの間で投与され得る。
【0102】
ドキセピンヒドロクロリドは、一日あたり約75mgと約1200mgの間で;好ましくは、一日あたり約150mgと約600mgの間で、最も好ましくは、一日あたり約300mgと約450mgの間で投与され得る。
【0103】
マレイン酸トリミプラミンは、一日あたり約75mgと約800mgの間で;好ましくは、一日あたり約150mgと約600mgの間で、最も好ましくは、一日あたり約200mgと約300mgの間で投与され得る。
【0104】
抗うつ剤は、必要な回数および間隔において一定期間にわたって、維持投薬量に達するように、摂取する指示とともにパッケージされた使用について現在認可された投薬処方物においてもしくは投薬パックにおいて提供され得る。例えば、抗うつ剤の投薬パックは、別個の投薬量、ならびに維持投薬量に達するまでの期間にわたって漸増量における別個の投薬量を服用するための手順を含み得る。ある実施形態において、投薬量は同量であり、手順は経時的に投薬量の増加した回数の服用を提供する。他の実施形態において、投薬パックは、異なる量の抗うつ剤を含む投薬量を含み、その指示は経時的に漸増量においての投薬量の服用を提供する。あるいは、その投薬パックは、維持投薬量に達するための期間にわたって、抗うつ剤の漸増量を放出するために処方され得るか、もしくは、その処方物は徐放性処方物および/またはパルス状放出処方物であり得る。
【0105】
代表的な投薬パックは、以下:
(a)約3日間よりも長い期間にわたる。約50mgまでのミルナシプランの毎日の投薬量;
(b)3日間よりも長い期間にわたる。約25mg〜約75mgまでのミルナシプランの毎日の投薬量;および、
(c)その症状を効果的に処置するのに十分な期間にわたる約100mgを超えるミルナシプランの毎日の投薬量;
を含む。
【0106】
本明細書で開示される任意の特許、もしくは特許文書は、本発明に参考として援用され、本発明の一部を形成する。
【実施例】
【0107】
(実施例1:漸進的または投薬量の段階的増加ならびに線維筋痛の処置におけるミルナシプランの有効性)
この研究を、線維筋痛症候群(FMS)の処置においてミルナシプランの効力を特徴付けるのに行った。第二の目的は、投与される毎日の用量、投薬の頻度ならびにFMSの処置の有効性の間の関連性を評価することであった。より高い投薬量が、より有効であることが理解されているので、第三の目的は、投薬量の漸進的な段階的増加がミルナシプランの投薬耐性を増加し得るかどうかを決定することであった。以前の研究では、患者に、50mg、100mgまたは200mgのミルナシプランの最初の毎日の投薬量を与えると、200mgとの有害な事象プロフィールは、100mgまたは50mgよりも実質的に悪かった。この研究において、毎日の投薬量は、漸進的に段階的に増加した。
【0108】
(方法)
線維筋痛症候群についての1990年のACR基準を満たす被検体は、登録についての適格者であった。患者は、抗うつ剤、睡眠薬および潜在的に効力の計測に干渉し得る他の薬物の排除後、最初の二週間にわたって基準の症状を記録した。患者を、一日一回のミルナシプランの投薬処置群、一日二回のミルナシプランの投薬処置群、もしくはプラシーボコントロールを1.5:1.5:1の比で無作為化した。活性処置の患者に、最初に25mgのミルナシプランを一日一回(25mg QD)もしくは一日二回(12.5mg BID)の投薬量を、最初の一週間にわたって与えた。患者がこの投薬で耐容性を示す場合、その患者は、二週目に50mgまでの毎日の投薬量、三週目に100mgまでの毎日の投薬量、四週目に200mgまでの毎日の投薬量へと段階的に増やし、もしくはプラシーボに対応するように増加した。用量が制限される毒性にいずれかの所定の週において遭遇した場合、患者を、以前の週の投薬量に減少し、その研究の余剰期間にわたって、その投薬量で維持した。被検体は、彼らの最大耐性用量(200mgまで)をさらなる8週間にわたって服用し続けた。患者は、計12週の投与を受け得る。用量が制限される毒性を、薬物関連の等級3〜4の有害な事象が発生すると定義する。
【0109】
(効力の計測)
効力を、最初の投薬を行う前ならびに一ヶ月ごとの通院の間、FMS状態の評価を使用して計測した。これらは、線維筋痛影響質問表(Impact Question naire)、マッギール疼痛(Mcgill Pain)、患者の臨床的な全体の印象、患者の全体の疼痛状態、生命測定のSF−36特性および、誘発した疼痛の測定を含んだ。
【0110】
(結果)
以前の研究において、患者は、彼らの最後の毎日の投薬量へと、短い期間(通常一週間以下)で段階的に増加した。200mgの投薬量では、50mgまたは100mgの投薬量よりも実質的に悪化した副作用の発生頻度を与えた。これらの結果に基づき、本研究では、患者の約50%が、200mgの毎日の投薬に耐性を示すことができず、および100mg/日で処置されることが期待された。意外にも、本研究において大部分の患者が、200mg/日に達し、この投薬量においてよく耐性を示した。ミルナシプランで処置された約70人の内、わずか9人のみが、200mg/日で耐性を示すのに失敗した。
【0111】
非段階的な最初の100mgのミルナシプランの投薬量を与える患者についての副作用は、処置の最初の週において悪化し、2週〜4週までにより低いレベルに沈静化した。これは、よりゆっくりとした投薬の段階的増加は、200mgのミルナシプランの毎日の投薬量により多くの患者が耐性を示したことの原因であることを示唆している。
【0112】
(実施例2:線維筋痛症候群を処置するためのミルナシプランの使用)
(方法)
12週間の無作為化、二重盲検プラシーボコントロール投薬の段階的増加単独療法治験を、FMSと診断された患者においてミルナシプランを評価するために行った。最初の期間の後、患者を疼痛投薬、中枢神経作用性刺激薬、抗うつ剤および鎮静睡眠薬を排除した場合、二週間の基準期間を開始した。基準期間が首尾よく完了した後、患者を、プラシーボ、QDミルナシプラン、またはBIDミルナシプランを1:1.5:1.5の比で無作為化した。全ての患者で、4週間の期間にわたって、一週間に一度の工程を毎日25mgから毎日50mg、毎日100mg、および最終的に毎日200mgまで、もしくは用量が制限される毒性(DLT)が明らかになるまで、段階的に増加した。DLTの存在を、患者を次のより高い投薬量に進めるように認定する前に、本研究機関により各週に評価した。DLTが明らかである事象において、患者は、以前の良く耐性を示す投薬量において安定し、安定した用量治療で八週間の間、この用量を維持した。
【0113】
任意の所定の投薬レベルにおいて、ミルナシプランQDの患者では、朝にミルナシプランの全量を摂取し、夜にプラシーボを摂取した。ミルナシプランBIDの患者では、朝および夜に分割した投薬量が与えられて同じ総量で摂取した。本研究の間、患者に、電子手帳を携帯し疼痛、疲労、睡眠および身体的情報を記録するように要請した。特注デザインの手帳は、複数の方法で自発的な疼痛を捕捉し、その方法としては以下:
(1)無作為な毎日のプロンプト(一日あたり4回〜5回彼らの現在の疼痛レベルを患者に対して記録するように知らせる装置);
(2)以前の24時間の疼痛について質問する朝の毎日のプロンプト;
(3)過去一週間の間の患者の平均的疼痛について提出を求める金曜日の夜の毎週のレポート;
が、挙げられる。
【0114】
電気的な評価を、クリニック来診の間で伝統的な疼痛、気分、および生活の質の調査表と共に追加した。対数Gracely 固定化(Gracely auchored)疼痛スケールの電子バージョンをその手帳において実行した。このスケールは、患者に、その疼痛を説明する感覚を「極めて激しい」、「非常に激しい」、「激しい」、「強力」、「わずかながら激しい」、「わずかに強力な」、「中程度」、「軽度」、「非常に軽度」、「弱い」、「非常に弱い」、「かすかな」、「痛みの無い」の中で選択することを求める。
【0115】
処置の第4週、第8週、第12週の間で、患者は、クリニックに来診し、ここで、多くの基準結果の測定を、SF−36、マッギ−ル疼痛(Mcgill Pain)アンケート、BECK、STPI、線維筋痛影響アンケートおよびいくつかの疼痛測定を含めて、実行した。安全性情報は、これらのクリニック来診時に収集した。
【0116】
主要な終点は、患者の電子手帳(PED)で収集された疼痛スコアに基づく基準線から終点までの疼痛スコアの変化として定義された。終点は、単一の値(クリニック測定のような)で評価のための12週、もしくは手帳基準の結果においての11週および12週の平均スコアとして定義した。「応答者」は、少なくとも30%の疼痛スコアを改善する患者として定義した。
【0117】
表1は、治験に関する登録基準をおよび除外基準を示す。
【0118】
【表1】
(結果)
表2は、用量の段階的増加の失敗の割合を示す。ミルナシプランの一日二回の投薬は、一日一回の投薬よりも低い割合の投薬耐性を生じた。この違いは、小サンプルサイズ(p=0.07)であったにも拘らず、ほぼ統計学的有意性に達した(表における「盲検」は、この日付における患者の処置群が、研究者にも伏せられる患者をいう)。
【0119】
【表2】
図2は、おそらくミルナシプランの効果の最も著しい証拠をあらわす。この図は、全て患者においての全体疼痛スコアをプロットし、上記患者は、この分析時間で終点に達した(38 MILおよび12プラシーボ)。1〜7スケールにおいて、1は非常によく改善し、4は変化がなく、および7は非常に悪化し、プラシーボ患者においての平均値が4.3であった一方、MIL患者においての平均値は2.3であった。図2において、1〜3のスコアは改善と示され、4は変化なし、ならびに5〜7は悪化である。ミルナシプラン群とプラシーボとの間の違いは、統計学的にp=0.0001で有意であった。
【0120】
図3は、治験の処置段階の間のBeckスコアの変化を示した。この治験は、うつ病患者においては選択しなかったが、FMS集団においてかなり高い割合の大うつ病およびうつ病の気分が存在した。このサンプルにおいて、14人の患者がMDEの定義を満たした。プールしたミルナシプランの処置群とプラシーボ群との間の終点でのスコアにおける違いは、p=0.03で有意であって、MILの公知の抗うつ効果がこの集団の処置において重要な助けとなることを示す。
【0121】
図4は、この研究の経過にわたって三つの処置群において報告された24時間の毎日の疼痛スコアを示す。BID投薬 は、処置期間全体を通して、QD投薬より有効であった。
【0122】
図5は、研究機関にわたって3つの処置群の患者によって報告された。毎日の睡眠の質のスコアを示す。睡眠の質では、プラシーボと比較したミルナシプランでの有意な改善が見られず、QD投薬とBID投薬の違いが検出されなかった。反対に、これは夜において用量を投与することを包含する。BID投薬においてさえも、睡眠の質の悪化は見られなかった。このことは、抗うつ剤の一般的副作用が睡眠の質に干渉するので顕著である。
【0123】
図6は、ミルナシプランをBIDで摂取した「応答者」として分類される患者の患者電子手帳(PED)の疼痛スコアを示す。スコアが各データポイントについて平均された患者の数が、1週目で34であり、13週目および14週目で24に減少した。RPは、一週間にわたって平均化した無作為な刺激疼痛スコアである。毎週(weekly)は、毎週の想起スコアである。毎日(Daily)は、毎日の想起スコアである。最初の二週間は基準ライン期間であり、3〜6週間は投薬の段階的増加期間、ならびに6週間から終了は、安定した投薬処置期間であることを留意する必要がある。投薬の段階的増加をともなう2〜6週間にわたる疼痛スコアにおける改善は、ミルナシプランのより高い投薬量が、FMSに関係する疼痛の処置においてより有効であることを示唆する。
【0124】
(結論)
ミルナシプランは、線維筋痛症候群に関係する疼痛を効果的に処置した。FMSを伴ったうつ病患者における気分もまた改善した。一日あたり200mgの相対的に高い用量を使用した。用量が、200mgに段階的に増加する場合に疼痛スコアにおける改善は、この用量が疼痛の緩和において重要であることを示す。一日二回の投薬は、疼痛の軽減において一日一回の投薬よりも有効であった。一日二回の投薬によって、一日一回の投薬をおこなうよりも少ない用量関連の有害な事象ならびに使用される漸増した投薬量に対して、より少ない不耐性もまた得た。
【0125】
(実施例3:水泳ストレス誘導の筋痛覚過敏に対するミルナシプランを使用した処置)
頻回の不可避の水泳ストレスが、ラットにおいて、熱性および長期の化学的に有害な両方の刺激に対する遅延性および長期間の皮膚の痛覚過敏を産生することが示された。この水泳ストレス誘導痛覚過敏(SSIH)モデルは、FMSと複数の特性を共有する。これらの類似性としては、(1)皮膚の痛覚過敏の存在、(2)ストレスの有意な役割、(3)痛覚過敏応答におけるNMDAレセプター機構の関与、および(4)抗うつ剤の有効性、が挙げられる。特に、クロルミプラミンおよびフルオキセチンによる前処置は、頻回にストレスを与えられたラットにおける皮膚の痛覚過敏の発症を防護し、ある抗うつ剤が、FMSにおける疼痛および睡眠異常を改善する。
【0126】
(方法)
ミルナシプラン(MIL)を、通常の生理食塩水で混合し、Sprague−Dawleyラット(200g〜300g)に対し腹腔内注射(IP)を介して投与した。
【0127】
ラットの処置群は、以下:(1)ストレス−水20cmにおいて水泳を強制;(2)疑似−水2cm〜3cmにおける水泳;(3)未処置−ケージにおいて平穏なまま、である。ラットに、注射無し、食塩水のIP QDもしくはミルナシプランの10mg/kg/日または30mg/kg/日 IP QDを与えた。
【0128】
ラットに、ホットプレート応答潜伏(秒)による熱性の痛覚の閾値について試験をおこなった。ラットに、痛覚計によって握力(kg)を計測することによる筋痛覚過敏について試験をおこなった。
【0129】
ミルナシプランによる前処置の有効性を試験する場合、ラットに、1日目〜11日目に薬物を与え、8日目〜10日目に水泳ストレスを与えた。ホットプレート応答の潜伏および握力に関するデータを、7日目および11日目に得た。ミルナシプランの処置後の有効性の試験をする場合、ホットプレートの応答潜伏および握力を、1日目、5日目および10日目に計測した;水泳ストレスを、2日目〜4日目に施行し、薬物処置を、5日目〜10日目に施行した。
【0130】
(結果)
図7は、ホットプレートの潜伏における水泳ストレス誘導の減少を示し、これらの減少は、ミルナシプランの前処置によって防護されなかった。
【0131】
図8は、頻回のストレス(IPの注射および強制/疑似水泳)が握力の減少の原因となったことを示す。ミルナシプランによる前処理は、握力におけるこのストレス誘導減少を防護した。このようにして、ミルナシプランによる前処置はFMSのこのモデルにおける筋痛覚過敏を防護した。
【0132】
図9は、水泳ストレス後のミルナシプランの処置が、ホットプレート応答の潜伏における水泳ストレス誘導の減少を改善しないことを示す。
【0133】
しかし図10は、ミルナシプランが、頻回の強制水泳ストレスによる握力の減少を改善することを示す。
【0134】
(結論)
水泳ストレスによって誘起される熱性の皮膚の感覚過敏は持続性であり、条件付け数日後の状態において、基本的に不変であった。
【0135】
水泳ストレス後の頻回のIPの注射は、筋肉の異痛に改善するようである握力の減少を誘導する。
【0136】
ミルナシプランは、水泳ストレスによって生じる筋肉の異痛の改善および防護において有効である。しかし、ミルナシプランは、皮膚の熱性の感覚過敏を無効もしくは防護しない。
【0137】
皮膚および筋肉の侵害受容の調節は、存在し得、薬理学的に独立して影響を受け得ることから、皮膚および筋肉の痛覚の侵害受容は、この動物モデルにおいては分し得る
【技術分野】
【0001】
(発明の分野)
本発明は、NSRI化合物であるミルナシプランといった抗うつ剤の有効量を使用して神経学的障害を処置する分野であって、その抗うつ剤は、望ましくない副作用を最小化するために段階的に漸増する投薬量において投与される。
【0002】
本出願は、2002年10月3日に出願されたU.S.S.N.第60/415、739号;2002年12月6日に出願されたU.S.S.N.第60/431、550号;2003年1月28日に出願されたU.S.S.N.第60/443、203;および2003年1月28日に出願されたU.S.S.N.第60/443、081号に対する優先権を主張する
【背景技術】
【0003】
(発明の背景)
慢性疲労症候群、線維筋痛症候群、慢性的な疼痛、疼痛に対する二次的なうつ病および機能的肉体障害といった神経学的障害は、アメリカ人の寿命のある時点でその人口の大部分に影響を与える。多数のこれらの状態を処置するために抗うつ剤がしばしば使用されるが、それらの効力は、用量が制限される副作用に起因してしばしば不十分である。それゆえに、多くの抗うつ剤が、別の方法で神経学的障害の症状の処置にあたって有効であるとき、抗うつ剤が、悪影響のためにその使用において制限される。
【0004】
抗うつ剤の一つのクラスは、モノアミンノルエピネフリンおよびセロトニンの再取り込みを阻害し、セロトニン−ノルエピネフリン再取り込み阻害剤(SNRI)と呼ばれる。これらの化合物のサブクラスは、優先的に、セロトニン再取り込み阻害と同等かそれ以上の親和性で、ノルエピネフリンの再取り込みを阻害し、ノルエピネフリン−セロトニン再取り込み阻害剤(NSRI)と呼ばれる。ミルナシプラン(Z−2−アミノメチル−1−フェニル−N,N−ジエチルシクロプロパン−カルボキサミドヒドロクロリド)は、セロトニン再取り込みをこえてノルエピネフリン再取り込みを優先的に阻害する、そのようなNSRI化合物の一つである。ミルナシプランは、うつ病の処置に関してヨーロッパで認可され、販売される薬物である。うつ病の処置においてミルナシプランの臨床的有効性を実証する当局への申請資料は、ヨーロッパ、米国および日本で実行された、5732人の患者(4006人をミルナシプランで処置、394人をプラシーボで処置、940人をTCAで処置および344人をSSRIで処置した)を包含する研究に基づく。30人以上の二重盲検試験が実行され、この試験は、うつ病による入院患者および外来患者の両方が関与するDSM IIIまたはRDC診断基準(さらに最近の研究においてはDSM III−RまたはDSM IV)によって評価されるような、ミルナシプランと、プラシーボ、三環系抗うつ剤(TCA)、または選択的セロトニン再取り込み阻害剤(SSRI)のいずれかとを比較することを包含する。
【0005】
これらの研究は、ミルナシプランが主なうつ病性エピソード(成人および高齢者)において、一日二回(BID)の50mgの代表的な用量で(食事と一緒に摂取される)有効であることを示した。この用量で、ミルナシプランは、下記:
(a)プラシーボより優れた有効性:ミルナシプラン(50mg BID)とプラシーボを比較する二重盲検研究のメタアナリシス(Hamilton(HDRSまたはHAMD)と、Montgomery−Asberg(MADRS)のうつ病の評点尺度の総スコアの群の間で有意差を示す;
(b)患者の「応答者」のパーセンテージ(すなわち、HAMDおよびMADSの総スコアが50%以上減少する患者)は統計学的に、プラシーボより優れており、プラシーボに対する応答者が40%であったのに対し、全ての患者の55%、および入院患者の64%がミルナシプランに応答した;
(c)TCAに比敵した有効性:三環系(イミプラミン、アミトリプチリンおよびクロミプラリン)で67%の応答比率に対し、ミルナシプランで64%の応答比率が見られた;および、
(d)SSRIに比敵した有効性:SSRI(フルボキサミン、フルオキセチンおよびパロキセチン)で50%〜65%の応答比率に対し、ミルナシプランで64%の応答比率が見られた;
を示す。
【0006】
残念なことに、50mgを超えるミルナシプランの毎日の投薬量が、負の患者耐性(すなわち、有害反応)を付随することもまた、公知である。副作用なしに、50mgを超えるミルナシプランの毎日の投薬量を投与するために有利である。
【0007】
ミルナシプランのような抗うつ剤は、大うつ病エピソードならびに他の神経学的障害の処置において有効だが、より適切な方法が、これらの神経学的障害を処置するより有効な量の投与を行うために必要とされる。
【0008】
それゆえに、本発明の目的は、神経学的障害の症状を処置するためにより有効な量の抗うつ剤を投与する方法を提供することである。
【0009】
本発明のさらなる目的は、化合物の増加量が投与され得、ならびに正の患者の安全性プロフィールを維持し得るように、化合物の用量が制限される毒性を減少する方法を提供することである。
本発明のさらなる目的は、一日二回(BID)とは対照的に、一日一回(QD)の投薬で投与することによる抗うつ剤の適切なピーク血漿濃度(Cmax)を得る方法を提供することである。
【発明の概要】
【課題を解決するための手段】
【0010】
(発明の要旨)
坑うつ剤のより高い毎日の投薬量を投与することで、神経学的障害を処置する方法が開示され、この方法において副作用が、投薬量を段階的に増加させることにより、最小化されると説明される。より高い毎日の投薬量は、その薬物の改善された有効性、正の患者の耐性の維持、正の患者の安全性プロフィールの維持(例えば、用量が制限される毒性)、薬物の適切なピーク血漿濃度(Cmax)、および/または一日二回(BID)の投与とは対照的に一日一回(QD)の投与を結果として生じる。循環する薬物のより高いレベルが、一日一回よりもむしろ一日の経過に渡って分割された投薬において化合物を投与することによっても得られる。
本発明の好ましい実施形態では、例えば以下の方法などが提供される:
(項目1)
処置を必要とする患者における慢性的な神経学的障害または該神経学的障害と関連する疼痛の症状を処置する方法であって、以下:
維持投薬量に到達するまでの期間に渡って、段階的に増加する投薬量において抗うつ剤を投与する工程、
を包含する、方法。
(項目2)
請求項1に記載の方法であって、前記慢性的な神経学的障害が、CFS、FMS、DSP、FSD、うつ病および疼痛からなる群より選択される、方法。
(項目3)
請求項1に記載の方法であって、前記慢性的な神経学的障害が線維筋痛症候群である、方法。
(項目4)
請求項1に記載の方法であって、前記神経学的障害が、慢性疲労症候群である、方法。
(項目5)
請求項1に記載の方法であって、前記神経学的障害が、疼痛である、方法。
(項目6)
請求項1に記載の方法であって、前記抗うつ剤が、ノルアドレナリン−セロトニン再取り込み阻害剤(NSRI)、選択的セロトニン再取り込み阻害剤(SSRI)、モノアミンオキシダーゼ阻害剤;抗痙攣剤および非特異的抗うつ剤からなる群より選択される、方法。
(項目7)
請求項1に記載の方法であって、前記抗うつ剤が、鎮静特性を有する、方法。
(項目8)
請求項1に記載の方法であって、前記抗うつ剤が、NMDA−アンタゴニスト特性を有する、方法。
(項目9)
請求項1に記載の方法であって、前記抗うつ剤が、ミルナシプランである、方法。
(項目10)
請求項1に記載の方法であって、前記抗うつ剤が、漸増投薬量において、活性化化合物の循環する投薬量を増加して、副作用を回避または最小化する段階的様式において投与される、方法。
(項目11)
請求項1に記載の方法であって、前記抗うつ剤が、第一の期間において、最初の投薬量が、一日あたり100mgまでで投与され、該第一の投薬量の約1.5倍〜2.5倍である第二の投薬量が、第二の期間において投与され、前記慢性的な神経学的障害の症状を処置する、方法。
(項目12)
請求項11に記載の方法であって、第三の期間において、前記第二の投薬量の約1.5倍〜2.5倍である第三の投薬量を投与して、前記慢性的な神経学的障害の症状を処置する工程をさらに包含する、方法。
(項目13)
請求項12に記載の方法であって、第四の期間において、前記第三の投薬量の約1.5倍〜2.5倍である第四の投薬量を投与して、前記慢性的な神経学的障害の症状を処置する工程をさらに包含する、方法。
(項目14)
請求項10から請求項13のいずれかに記載の方法であって、前記各投与期間が、3日間よりも長い、方法。
(項目15)
請求項14に記載の方法であって、前記各投与期間が、2週間と12週間の間である、方法。
(項目16)
請求項1に記載の方法であって、前記抗うつ剤が、投薬量を段階的に増加する仕組みにおいて投与され、正の患者の安全性プロフィール(例えば、用量が制限される毒性)および薬物の適切なピーク血漿濃度(Cmax)を提供する、方法。
(項目17)
請求項1に記載の方法であって、別個の投薬量を含む抗うつ剤の投薬パックの提供、ならびに維持投薬量に達するまでの期間にわたって、漸増量内で別個の投薬量を摂取するための手順を含む、方法。
(項目18)
請求項17に記載の方法であって、前記投薬量パックが、ミルナシプランを含み、維持投薬量に到達するまでの日数の期間にわたって、前記投薬量が段階的に漸増される、方法。
(項目19)
請求項1に記載の方法であって、維持投薬量に達するまでの日数にわたって、抗うつ剤の漸増量を放出するために処方された抗うつ剤を提供する、方法。
(項目20)
請求項19に記載の方法であって、処方物が、持続性放出および/またはパルス状放出処方物である、方法。
(項目21)
請求項1に記載の方法であって、以下:
(a)約3日間よりも長い日数にわたって約50mgまでのミルナシプランの毎日の投薬量を必要とする患者に投与する工程;
(b)約3日間よりも長い日数にわたって約25mg〜約75mgまでのミルナシプランの毎日の投薬量を、該患者に投与する工程;および
(c)前記症状を有効に処置するための十分な期間にわたって約100mgよりも多いミルナシプランの毎日の投薬量を該患者に投与する工程;
を包含する、方法。
(項目22)
別個の投薬量、ならびに維持投薬量に達するまでの期間にわたって、漸増量における別個の投薬量を摂取するための指示書を含む、抗うつ剤の投薬パック。
(項目23)
請求項22に記載の投薬パックであって、前記投薬量が、同量であり、前記指示書が、経時的に増大した投薬量の摂取を提供する、投薬パック。
(項目24)
請求項22に記載の投薬パックであって、前記投薬量が、異なる量の抗うつ剤を含み、前記指示書が、経時的に増大した投薬量の摂取を提供する、投薬パック。
(項目25)
請求項22に記載の投薬パックであって、前記投薬量パックが、ミルナシプランを含み、前記投薬量が、維持投薬量に達する日数にわたって、段階的に増加する、投薬パック。
(項目26)
請求項22に記載の投薬パックであって、維持投薬量に達するための日数にわたって、抗うつ剤の漸増量を放出するために処方された抗うつ剤を提供する、投薬パック。
(項目27)
請求項26に記載の投薬パックであって、前記処方物が、持続性処方物および/またはパルス状放出処方物を維持される、投薬パック。
(項目28)
請求項22に記載の投薬パックであって、以下:
(a)約3日間よりも長い期間にわたる約50mgまでのミルナシプランの毎日の投薬量;
(b)約3日間よりも長い期間にわたる約25mg〜約75mgのミルナシプランの毎日の投薬量;および、
(c)症状を効率的に処置するための十分な期間にわたる約100mgよりも多いミルナシプランの毎日の投薬量;
を含む、投薬パック。
【図面の簡単な説明】
【0011】
【図1】図1は、ミルナシプランの漸増する毎週の用量を患者に投薬する方法を示すフローチャートである。用量が制限される毒性は、研究を通して投薬量の段階的増加毎に評価される。
【図2】図2は、一日二回(BID)または一日一回(QD)で投与されるミルナシプランまたはプラシーボでの12週間の処置後に、改善された、不変のもしくは悪化した全体の疼痛のスコアを用いる繊維筋痛症候群(FMS)の患者のパーセントのプロットである。
【図3】図3は、FMS患者のBeckうつ病のスコアのプロットであり、その患者は、基準ライン(治療前)およびミルナシプランのBIDまたはQDまたはプラシーボを使用した治療の終点での大うつ病(MDE)を診断される。
【図4】図4は、12週間の処置期間にわたって、3つのFMS処置グループの24時間の毎日の疼痛調査のスコアをプロットする。
【図5】図5は、12週間の処置期間にわたって、3つの処置グループにおける患者の自己調査した毎日の睡眠の質のスコアをプロットする。
【図6】図6は、2週間の基準ライン期間および12週間の処置にわたって応答者として分類される患者の(一週間にわたって各患者について平均された)患者電子手帳の疼痛のスコアのプロットである。RP=無作為な速攻型疼痛スコア;Weekly=毎週の想起疼痛のスコア;Daily=毎日の想起疼痛スコア。
【図7】図7は、水泳ストレス試験におけるミルナシプランの注射、ビヒクルの注射または注射無しで予備処置されたラットのホットプレート潜伏期間における変化をプロットする。プロットされる変化は、水泳ストレス試験、疑似水泳試験、もしくは水泳なしの試験(未処置)を受けた後に計測された潜伏期間から、試験を受ける前に計測された潜伏を差し引いたものである。
【図8】図8は、水泳ストレス試験におけるミルナシプランの注射、ビヒクルの注射または注射無しで予備処置されたラットの握力における変化をプロットする。プロットされる変化は、水泳ストレス試験、疑似水泳試験、もしくは水泳なしの試験(未処置)を受けた後に計測された潜伏期間から、試験を受ける前に計測された潜伏握力を差し引いたものである。
【図9】図9は、3つの水泳ストレス試験を受けた後、次いで、ストレス試験の後に、ミルナシプランの注射、ビヒクルの注射、もしくは注射無しで処置されるラットにおいて計測されたホットプレート潜伏期間をプロットする。プロットされる変化は、(1)水泳ストレス試験、疑似水泳試験、もしくは水泳なしの試験(未処置)を受け、かつ(2)試験の後にミルナシプランの注射、ビヒクルの注射、もしくは注射無しで処置された後の両方を計測した握力から、ストレス試験の前1日目に計測された潜伏期間を差し引いたものである。
【図10】図10は、その3つの水泳ストレス試験を受けた後、次いで、そのストレス試験の後、ミルナシプランの注射、ビヒクルの注射、もしくは注射無しで処置されるラットにおいて計測された握力をプロットする。プロットされる変化は、(1)水泳ストレス試験、疑似水泳試験、もしくは水泳なしの試験(未処置)を受け、かつ(2)試験の後にミルナシプランの注射、ビヒクルの注射、もしくは注射無しで処置された後の両方計測されたその握力から、そのストレス試験の前1日目に計測された握力を差し引いたものである。
【発明を実施するための形態】
【0012】
(発明の詳細な説明)
(略語)
DSP 疼痛に対する二次的なうつ病。
CFS 慢性疲労症候群。
FMS 線維筋痛症候群。
FSD 機能的な肉体障害。
5−HT セロトニン。
NE ノルエピネフリン(ノルアドレナリン)。
NMDA N−メチル D−アスパラギン酸。
SNRI セロトニンの再取り込みが、ノルエピネフリンの再取り込みを超える、二重セロトニンノルエピネフリン再取り込み阻害剤。
NSRI ノルエピネフリンの再取り込みが、セロトニンの再取り込みを超える、二重ノルエピネフリン再取り込み阻害剤。
【0013】
(I.処置される患者)
段階的増加および/または分割された投薬量処方物で処置され得る神経学的障害は、慢性的な疼痛、神経障害性の疼痛、線維筋痛症候群、慢性疲労症候群、情動障害/気分障害、うつ病、異型のうつ病、ならびに機能的な肉体障害が挙げられる。神経学的障害の症状としては、筋骨格の疼痛、疲労、睡眠障害(sleep disorder)、睡眠障害(sleep disturbance)またはそれらの組み合わせが挙げられるが、これらに制限されない。
【0014】
神経学的障害は、その患者が、その障害に12週間より長く罹患しているが、6週間または2週間程度早期に持続的な症状に罹患しているとき、慢性的な障害と考慮される。
【0015】
慢性的な疼痛は、長期間帯にわたって(すなわち、3ヶ月より長い)持続または再発し、慢性関節リウマチといった種々の疾患または異常な状態によって生じる疼痛をいう。慢性的な疼痛は、その急性疼痛より弱いのかもしれない。疼痛に対する自動的反応が長期間持続され得ないことから、慢性的な疼痛を有する個人は、通常、脈拍の増加および急速な発汗を示さない。慢性的な痛みを有する他の者は、その環境から引きこもり得、彼らの苦痛、彼らの家族、彼らの友人および外部刺激を全く無視することにのみ、集中し得る。Mosby’s Medical,Nursing & Allied Health Dictionary,5th Editionを参照のこと。
【0016】
その慢性的な疼痛は、背部の疼痛、異形の胸痛、頭痛、骨盤痛、顔の筋膜の疼痛、腹痛および頚部痛であり得る。あるいは、その慢性的な疼痛は、関節炎、側頭部下顎関節機能障害症候群、外傷の脊髄障害、多発性硬化症、過敏性腸症候群、慢性疲労症候群、月経前症候群、多発性化学物質過敏、過換気、閉鎖性頭部外傷、線維筋痛慢性関節リウマチ、糖尿病、癌、HIVおよび間質性膀胱炎によって生じ得る。
【0017】
同様に、神経障害性の疼痛とは、末梢神経、脳神経、脊髄神経またはそれの組み合せの炎症または変性に関する疼痛をいう。その疼痛は、一般に鋭い痛み、刺すような痛みもしくは突き刺すような痛みである。根底にある障害は、末梢神経組織の破壊を生じ得、皮膚の色、温度および水腫における変化を伴い得る。Mosby’s Medical,Nursing & Allied Health Dictionary,5th Edition(1998);およびStedman’s Medical Dictionary,25th Editionを参照のこと。
【0018】
線維筋痛症候群(FMS)は、人口の2%から4%が罹患していると概算される総全身性リウマチ学的障害(common systematic rheumatologic disorder)であり、変形関節症に対してのみの有病率において第二位である。線維筋痛は、一般に圧刺激に関する疼痛について減少した許容限界に関し、そして、疲労、睡眠障害および朝のこわばりをしばしば伴う。他の通常の症候群としては、頭痛、片頭痛、非心臓性の胸痛、胸焼け、動悸、過敏性腸症候群、種々の腸嗜癖、散在性の腹痛ならびに尿頻度が挙げられる。線維筋痛に関する診断基準には、広範な疼痛の病歴だけでなく、付与される圧力に2次的な身体計側時に物理学実験における圧痛を発見することも必要とする。American College of Rheumatology(ACR)によって1990年に設定された線維筋痛についての診断基準を満たすために、個人は、軸骨格だけでなく、身体の四半分の4つ全てを含む慢性的な広範な疼痛、ならびに試験における18の圧痛点のうち11が存在することの両方を有する必要がある。
【0019】
FMSは、感覚刺激の全身化した高い感知を反映する医学的問題である。その異常性は、末梢でよりもむしろ中枢神経系(CNS)内で起こると考えられ、提案された病態生理学的欠陥は、「中枢鋭敏化」(central sensitization)と名づけられる。FMSの患者は、一般に、異痛症(電灯に触れるような非疼痛的刺激からさえも疼痛をうける)および痛覚過敏(疼痛プロセシングの漸増、(ここで疼痛刺激が拡大され、正常なボランティアの強度よりも高い強度で受ける))の両方を被る。この観点において、臨床的症状についての多くの対応があり、糖尿病ニューロパシーおよび三叉神経痛のような神経障害の疼痛の根底にあるメカニズムが提案される。
【0020】
情動障害/気分障害は、主な特徴として気分の障害により特徴付けられた種々の状態である。軽症および好都合の場合、その感情は、正常である。より重症な場合、それらは、大うつ障害または気分変調性の反応の徴候であり得るか、もしくは双極性障害の徴候を示し得る。他の気分障害は、一般の医学的状態で引き起こされ得る。Mosby’s Medical,Nursing & Allied Health Dictionary,5th Edition(1998)を参照のこと。
【0021】
うつ病は、悲しみ、絶望および落胆の感情によって特徴付けられる異常な気分障害である。うつ病は、悲しみ、憂うつ、落胆、無価値、空虚および絶望といった誇張された感情により特徴付けられた異常な感情状態をいい、その感情は、不適切で、現実に対して釣り合いが取れていない。Mosby’s Medical,Nursing & Allied Health Dictionary,5th Edition(1998)を参照のこと。
【0022】
そのうつ病は、大うつ障害のうちの少なくとも一つ(一つのエピソード、反復性のもの、軽度のもの、中程度のもの、精神病的特徴のない重傷、精神病の特徴のある重度のもの、慢性的なもの、緊張病的特徴があるもの、憂うつの特徴があるもの、異型的な特徴のあるもの、分娩後に発症するもの、一部の賓解したもの、全て賓解したもの)、気分変調性障害、うつ病の気分を有する適応障害、不安感およびうつ病の気分が混合した障害、月経前の不快性障害、小うつ病障害、反復性の短期うつ病障害、精神分裂病である精神病後のうつ病障害、パーキンソン病に関する大うつ病障害および痴呆に関するうつ病障害であり得る。
【0023】
疼痛に対して2次的なうつ病(DSP)は、疼痛の共存率および異型うつ病により特徴付けられたうつ病障害である。特に、その疼痛は、慢性的な疼痛、神経障害性の疼痛、またはそれらの組み合わせであり得る。そのDSPは、異型なうつ病および慢性的な疼痛を含み、ここで、上記慢性的な疼痛は、上記異型うつ病に先行するか、もしくは、上記異型うつ病は、上記慢性的な疼痛に先行する。あるいは、上記DSPは、異型うつ病および神経障害の疼痛を包含する。
【0024】
異型うつ病は、気分反応性、ならびに約2週間を超える期間にわたって存在する2つ以上の自律神経症状(例えば、過剰睡眠、食欲の増進または体重増加、鉛麻痺、および認められた対人拒絶に対する過敏の長期間のパターン)を含みうる。
【0025】
異型うつ病は、正の生活上の効果(気分反応性)に対する応答において、一時的に気分がよくなる能力が付随するうつ病の情動であり、加えて、過眠症、食欲の増進または体重増加、鉛麻痺およびみとめられた対人拒絶に対する過敏長期パターンの群から選択される二週間以上をこえて存在する二つ以上の自立神経症である。当業者は、自律神経症状が、他のうつ病障害において検出される症状(例えば、憂うつなうつ病)と比較して逆であり得る;それゆえ、語句「異常な」である。
【0026】
機能的肉体障害(FSD)は、組織構造または機能の疾患特異的な異常よりもむしろ症状、苦痛および能力不全によって代表的に特徴付けられるいくつかの関連する症候群を言う。機能的肉体障害を伴う患者は、肉体的には健康であるが、不具に遭遇し、医学上説明できない症状である。症状は、疲労、頭痛、関節痛、弱気、記憶障害、不安および動悸といった病状が挙げられる。一般的な機能肉体症候群は、複数の化学物質過敏性、シックハウス候群、反復ストレス傷害、慢性的な鞭打ち傷害、慢性的なライム病、シリコン胸部移植の副作用、カンジダ症感受性、湾岸戦争症候群、僧帽弁脱出および低血糖症が挙げられる。
【0027】
機能肉体障害を伴った患者は、代表的に、彼ら自身その症状についての自己診断を提供し、特定の疾患に対する彼らの症状の属性に矛盾する情報に抵抗する。これらの患者は、精神医学的な障害、特に不安、うつ病および身体型障害のより高い発生率を有する。患者の症状を増幅する心理社会的要因としては、その患者が重篤な疾患を有するという考え;状態が悪化しそうであるという患者の予測;訴訟および補償の影響を包含する「病人の役割」;およびその状態が破局的であり不能である患者による警告説明が挙げられる。それにもかかわらず、機能的肉体障害を有すると診断される患者は、考慮すべき障害ならびに不能によって特徴付けられる。
【0028】
機能的肉体障害は、疼痛にも関連し得る。その疼痛は、機能的肉体障害の発生の前に起こるかまたは後に起こり得る。その疼痛は、慢性的な疼痛または神経障害性の疼痛もしくはその二つの組み合わせであり得る。
【0029】
(II. 組成物)
(A. 活性化合物)
この方法で使用される活性化合物は、抗疼痛活性または鎮痛活性、抗うつ剤活性を有し、それゆえに疼痛、うつ病ならびに関連する疾患および症状の処置のための薬物として有用である。
【0030】
この方法で使用される化合物は、神経学的障害に関連する状態または症状を処置、防止またはより軽減するための薬物の能力を決定するための実験またはアッセイにおいて、例えば薬学研究プログラムにおいて使用するための標準化合物または参照化合物としても有用である。このようにして、本明細書で開示される化合物は、コントロール化合物または参照化合物としてそのようなアッセイで使用され得、制御標準物質として使用され得る。本発明の化合物は、市販のキットまたはそのような標準化合物もしくは参照化合物としての使用のための容器で提供され得る。
【0031】
ノルエピネフリン(NE)−セロトニン(5−HT)再取り込み阻害剤(NSRI)は、ノルエピネフリン(NE)およびセロトニン(5−HT)の両方の再取り込みを阻害する化合物クラスを言うが、5−HTの再取り込みよりもNEの再取り込みを優先的にブロックする。選択的NSRIは、少なくとも約1のNE:5−HT再取り込み阻害比を有する。具体的には、選択的NSRIは、約50までのNE:5−HTの再取り込み阻害比を有し得る。より具体的には、その選択的NSRIは、約1:1から約20:1のNE:5−HTの再取り込み阻害比を有し得る。より具体的には、選択的NSRIは、約1:1から約5:1のNE:5−HTの再取り込み阻害比を有し得る。より具体的には、その選択的NSRIは、約1:1から約3:1のNE:5−HT再取り込み阻害比を有し得る。
【0032】
NSRIは200nM以下の5−HT再取り込みのIC50および200nM以下のNE再取り込みのIC50、ならびに少なくとも1000nMドーパミン再取り込みのIC50を有する。NRSIは、少なくとも約0.5:1のNE:5−HT再取り込みの阻害比を有する。そのNE:5−HTの再取り込み阻害比は、5−HT再取り込みのIC50をNE再取り込みのIC50で除算することで計算される。例えば、化合物が、10nMのNE再取り込みのIC50および20nMの5−HTの再取り込みのIC50を有する場合、その化合物は、2:1のNE:5−HTの再取り込み阻害比を有する。特定の実施形態において、NSRIは、NE:5−HT再取り込みの約0.5:1から約50:1、約1:1から約20:1、約0.5:1から約5:1、約1:1から約5:1、約0.5:1から約3:1、もしくは約1:1から約3:1、の比の阻害を有する。
【0033】
そのNSRI化合物は、NMDAレセプターでアンタゴニスト性質を示し得る。NMDAレセプターアンタゴニストは結合し、NMDAレセプターの活性を減少する。これは、非競合的および競合的な両方の、NMDAレセプターアンタゴニスト、グリシン−部位アンタゴニスト、グルタミン酸アンタゴニストおよびアロステリックアンタゴニストが挙げられる。化合物は、当業者で公知のアッセイによってNMDAレセプターアンタゴニストであると決定され得る。
【0034】
「ミルナシプラン」(±)−シス−2−(アミノメチル)−N、N−ジメチル−1−フェニルシクロプロパンカルボキサミドヒドロクロリド(CAS 登録番号が92623−85−3)が、以下の式のNSRI化合物である:
【0035】
【化1】
ミルナシプランの調製法は、例えば、米国特許第4,478,836号およびそこで引用される参考文献で開示される。他に指摘の無い場合、ミルナシプランは、全ての立体異性体、立体異性体の混合物ならびにその薬学的に受容可能な塩を包含し得る。
【0036】
ミルナシプランの右旋性異性体は、ノルエピネフリンおよびセロトニンの再取り込みの阻害活性として、ラセミ混合体よりも約2倍の活性であり、左旋性異性体は、はるかに強力でないと考えられる。例えば、Viazzo et al.,1996,Tetrahedron Lett. 37(26):4519−4522;Deprez et al.,1998,Eur.J.Drug Metab.Pharmacokinet.23(2):166−171).を参照のこと。従って、ミルナシプランは、鏡像異性体的に純粋な形態(例えば、純粋な右旋性異性体)もしくはラセミ混合物といった右旋性異性体および左旋性異性体の混合物として投与され得る。
【0037】
必要とされる場合、ミルナシプランのラセミ混合物の分離は、キラルカラムを使用するHPLC、もしくは Thomas J.Tucker,et al.,J.Med.Chem.1994 37,2437−2444と同様のショウノウ塩化物といった分離薬物(resolving agent)を使用する分離によって達成され得る。ミルナシプランはまた、キラル触媒またはキラルリガンドを使用して直接的に合成されうる(例えば、Mark A.Huffman,et al.,J.Org.Chem.1995,60,1590−1594)。
【0038】
ミルナシプランの経口投与に対する公知の有害反応としては、吐き気、嘔吐、頭痛、ふるえ、不安、不安発作、動悸、尿貯留、起立性低血圧、発汗、胸痛、発疹、体重増加、背痛、便秘、下痢、目眩、発汗増加、心的動揺、のぼせ、震え、疲労、眠気、消化不良、嗅覚不全、緊張、口内乾燥、腹痛、不眠症または、それらの組み合わせが挙げられ得る。
【0039】
投薬量の段階的増加および/または分割された投薬量を使用して投与され得る他のSNRIとしては、以下:
ベンラファキシンヒドロクロリド(R/S)−1−[2−(デメチルアミノ)−1−(4−メトキシフェニル)エチル]シクロヘキサノールヒドロクロリド)または((±)−1−[α[(ジメチルアミノ)メチル]−p−メトキシベンジル]シクロヘキサノールヒドロクロリド);
ミルタザピン(1,2,3,4,10,14b−ヘキサヒドロ−2−メチルピラジノ [2,1−a]ピリド[2,3−c]ベンズアゼピン);
ネファゾドンヒドロクロリド(2−[3−[4−(3−クロロフェニル)−1−ピペラジニル]プロピル]−5−エチル−2,4−ジヒドロ−4−(2−フェノキシエチル)−3H−1,2,4−トリアゾール−3−オン モノヒドロクロリド);
チオリダジンヒドロクロリド(10H−フェノチアジン,10−[2−(1−メチル−2−ピペリジニル)エチル]−2−(メチルチオ)−,モノヒドロクロリド);ブプロピオン ヒドロクロリド((±)−1−(3−クロロフェニル)−2−[1,1−ジメチルエチル)アミノ]−1−プロパノン ヒドロクロリド);
モノアミンオキシダーゼ阻害剤であって、例えば、以下
トラニルシプロミンスフェート((±)−トランス−2−フェニル−シクロプロピルアミン スフェート(2:1));フェネルジンサルフェート(硫化水素フェネチルヒドラジン);
モノクロベミド(p−クロロ−N−(2−モルホリノエチル)ベンズアミド);
ピルリンドル(2,3,3a,4,5,6−ヘキサヒドロ−8−メチル−1H−ピラジノ[3,2,1−j,k]カルバゾール;
選択的セロトニン再取り込み阻害剤であって、例えば、以下:シタロプラムヒドロブロシド((±)−1−(3−ジメチルアミノプロピル)−1−(4−フルオロフェニル)−1,3−ジヒドロイソベンゾフラン−5−カルボニトリル,HBr);
パロキセチンヒドロクロリド((−)−トランス−4R−(4’−フルオロフェニル)−3S−[(3’,4’−メチレンジオコシフェノキシ)メチル]ピペリジンヒドロクロリド半水化物);
フルオキセチンヒドロクロリド((±)−N−メチル−3−フェニル−3−[(α,α,α−トリフルオロ−p−トリル)オキシ]プロピルアミンヒドロクロリド);
セルトラリンヒドロクロリド((1S−シス)−4−(3,4−ジクロロフェニル)−1,2,3,4−テトラヒドロ−N−メチル−1−ナフタレンアミンヒドロクロリド);
三環形の抗うつ剤であって例えば、以下:アミトリプチリンHCl(3−(10,11−ジヒドロ−5H−デベンゾ[a,d]シクロヘプテン−5−イリデン)−N,N−ジメチル−1−プロパンアミンヒドロクロリド)。
【0040】
デシプラミンヒドロクロリド(5H−ジベンズ[bf]アゼピン−5−プロパンアミン、10,11−ジヒドロ−N−メチル−,モノヒドロクロリド);
ドキセピンヒドロクロリド(1−プロパンアミン,3−ジベンゾ[b,e]オキセピン−11(6H)イリデン−N,N−ジメチル−,ヒドロクロリド);
マレイン酸トリミプラミン(5−(3−ジメチルアミノ−2−メチルプロピル)−10,11−ジヒドロ−5H−ジベンズ(b,f)アゼピン酸およびマレイン酸(ラセミ形態));
プロトリプチリンHCl(N−メチル−5H−ジベンゾ[a,d]−シクロヘプテン−5−プロパンアミンヒドロクロリド);
抗けいれん薬(anti−covalsant)であって、以下:
ジバルプロエックスナトリウム(水素ビス(2−プロピルペンタン酸)ナトリウム);
クロナゼパム(5−(2−クロロフェニル)−1,3−ジヒドロ−7−ニトロ−2H−1,4−ベンゾジアゼピン−2−オン);
およびアルプラゾラム(8−クロロ−1−メチル−6−フェニル−4H−s−トリアゾロ[4,3−α][1,4]ベンゾシアゼピン);
が挙げられる。
【0041】
上記化合物は、体の流体中で実質的に遊離状態であり得る。さらに、これらの化合物は、少なくとも90重量%の純度、少なくとも95重量%の純度、少なくとも98重量%の純度、少なくとも99重量%の純度であり得る。
【0042】
(B.塩類および誘導体)
その化合物の薬学的に受容可能な塩類は、その親化合物から合成され得、上記化合物は、従来の化学的方法によって塩基性部分または酸性部分を含む。一般的に、そのような塩類は、これらの化合物の遊離酸形態または遊離塩基形態と水中、有機溶媒中もしくはこの2つの混合物中で、化学量論的量の適切な酸または塩基とを反応させることで調整され得る;一般的に、エーテル、エチルアセテート、エタノール、イソプロパノールまたはアセトニトリルのような非水性媒体が好ましい。適切な塩類のリストは、Remington’s Pharmaceutical Sciences,17th ed.,Mack Publishing Company,Easton,PA,1985,p.1418に見出される。
【0043】
薬学的に受容可能な塩類の例としては、アミンおよびアルカリといった塩基性残基無機酸塩または有機酸塩が挙げられるが、これらに限定されない。その薬学的に受容可能な塩は、例えば、非毒性の無機酸または有機酸から形成されるその親化合物の従来の非毒性塩類を含む。例えば、そのような従来の非毒性塩類は、無機酸(例えば、塩酸、臭化水素酸、硫酸、砂糖、スルファミン酸、リン酸、および硝酸由来の塩類);および、有機酸(例えば、酢酸、プロピオン酸、コハク酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、アスコルビン酸、パモン酸(pamoic acid)、マレイン酸、ヒドロキシマレイン酸、フェニル酢酸、グルタミン酸、安息香酸、サリチル酸、スルファニル酸、2−アセトキシ安息香酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸およびイセチオン酸)から調整される塩が挙げられる。特に、薬学的に受容可能な塩類は、塩化水素塩または塩酸(HCl)塩であり得る。
【0044】
これらの化合物は、過剰な毒性も、刺激も、アレルギー応答も、他の問題もしくは合理的な利益/リスクの比が相応につりあった合併症もなく、人および動物の組織との接触における使用に適切である。
【0045】
適切な化合物としては、プロドラッグおよび活性成分の代謝物もまた挙げられる。プロドラッグは、共有結合で結合するなんらかの物質であって、そのようなプロドラッグが、被験体に投与される場合、その物質はその活性な親薬物または他の処方物もしくは本発明の化合物をインビボにおいて放出する。
【0046】
代謝物は、生化学的プロセスから生じた任意の物質であって、このプロセスによって生細胞が、その活性化合物と相互作用し、その生細胞は、任意の代謝経路由来の産物または中間体を包含する。
【0047】
(C.活性成分の組み合わせ)
この処置方法で使用される各治療薬剤は、独立的に任意の投薬形態であり得、また、組み合せで投与され得る。その薬剤は、単体の投薬ユニット(すなわち、一つのカプセル、錠剤、粉末、または液体等の中で一緒に併用される)において組み合せの産物として共に処方され得、単体の化合物として同時に、もしくは単体の投薬ユニットとして共に処方されない場合に任意の順序で投与され得る。好ましくは、その薬剤は、分割して投与される場合、お互いに一時間以内で投与される。
【0048】
同時投与薬剤の適切な投薬量は、当業者において、本開示に基づいて当該分野の開業医によって容易に確認できる。一般的な指導を介して、代表的には、毎日の投薬量は、各成分の約100ミリグラムから約1.5グラムであり得る。一つ以上の化合物が投与される場合、典型的投薬量は、各薬剤の約100ミリグラムから約1.5グラムであり得るであり得る。一つより多い化合物が投与される場合、次いで代表的には、毎日の投薬量は、各薬剤の100ミリグラムから1.5グラムであり得る。一般的な指導を介して、複数の薬剤が、組み合せされて投与されるとき、各薬剤の投薬量は、組み合せの相乗作用効果の視点から、個別に投与される場合の通常の投薬量と比較して約70%〜80%まで減少し得る。
【0049】
(D.処方物および賦形剤)
(処方物)
活性成分は、固形の投薬形態(例えばカプセル、錠剤および散剤)または液状の投薬形態(例えば、エリキシル剤、シロップおよび懸濁液)で経口的に投与され得る。活性成分はまた、無菌の液状の投薬形態において、非経口的に投与され得る。添加剤はまた、物理学的な外形を向上し、安定性を改善し、そして投薬後の崩壊を補助するための処方物において包含されうる。例えば、経口投与のための液状の投薬形態は、患者の受容を増加するために着色剤および矯味矯臭剤を含み得る。
【0050】
活性成分の毎日の投薬量は、一日一回(QD)の投薬量または代替の分割された投薬量、例えば一日二回(BID)の投薬量で投与され得る。
【0051】
ゼラチンのカプセルは、活性成分ならびに、ラクトース、デンプン、セルロース誘導体、ステアリン酸マグネシウム、ステアリン酸などといった粉末の担体を含む。類似する希釈剤が、圧縮錠剤を作製するのに使用され得る。錠剤およびカプセルの両方が、徐放性生成物として製造され得、数時間または数日間にわたって薬物の持続的な放出を提供し、ならびに移植、または経皮/経粘膜配達のために処方される。そのような処方物は、代表的には、生物分解もしくは生物侵食されて、ミルナシプランの一部を放出するポリマーを包含する。処方物は、マイクロカプセル、リポソーム、固体のモノリシック(monolithic)移植物、ゲル、粘性流体、椎間板、もしくは附着フィルムの形式を有し得る。
【0052】
圧縮錠剤は、好ましくない味覚を覆い隠し、その錠剤を大気から保護するために糖被覆またはフィルム被覆され得るか、または胃腸管において選択的崩壊のために腸溶性の被覆され得る。
【0053】
(キット)
活性成分は、キットとして処方され得、上記キットは、分割された毎日の投薬当量または投薬量ユニットを維持する容器と組み合わされる漸増濃度の用量ならびにその用量を投与するための手順を伴ったプリントされた説明書(printed indicia)が挙げられる。
【0054】
好ましくは、四つの工程の投薬量の段階的増加において使用されるキットは、第一投薬量ユニットである100mgまでの活性成分を含む。第二の投薬量は、第一の投薬量よりも1.5倍〜2.5倍多い投薬量が含まれる。第三の投薬量は、第二の投薬量よりも1.5倍〜2.5倍多い投薬量が含まれる。第四の投薬量は、第三の投薬量よりも1.5倍〜2.5倍多い投薬量が含まれる。このキットは、四つの投薬量ユニットを維持するための容器ならびにその用量を投与するための指示を有するプリントされた説明書を含む。第一のユニット投薬量形態は、一日一回(QD)の投薬量の形態であり得る。このキットの形式は、より多い、または少ない漸増投薬量の投薬ユニットを包含するように改変され得る。
【0055】
本キットの好ましい実施形態は、100mgまでのミルナシプランの第一の投薬量および100mgより多いミルナシプランの第二の投薬量ユニットを含む。このキットはまた、二つの投薬量ユニット形態を維持するための容器ならびにその用量を投与するための指示を有する、プリントされた説明書を含む。このキットは、投薬量ユニット形態を維持するための容器およびその投薬量を投与するための指示とともに、(1)20mg〜30mg、(2)40mg〜60mg、(3)75mg〜125mg、(4)175mg〜225mgのミルナシプランの四つの段階的増加投薬量を含むように改変され得る。
【0056】
(賦形剤)
活性成分または治療成分に加えて、錠剤は多くの不活性物質を含む。後者は添加物または「付加物」として公知である。それらは、最終錠剤において作用する部分に従って分類され得る。第一の群は、処方物に対して十分な圧縮特性を与えるように補助するものを含む。これらは、(1)希釈剤、(2)結合剤および(3)滑沢剤が挙げられる。添加物質の第二の群は、最終錠剤にさらなる望ましい物理学的特性をあたえるように補助する。この群に包含される添加物質は、(1)崩壊剤、(2)着色剤、ならびにチュアブル錠剤の場合においては、(3)矯味矯臭剤および(4)甘味剤がある。
【0057】
しばしば、活性成分の単体の投薬は、わずかであり、不活性物質は、錠剤を圧縮によって実用的なサイズにするためにバルクを増加するために追加される。この目的について使用される希釈剤は、希釈剤と活性成分の間で適合性に準じて、燐酸二カルシウム、硫酸カルシウム、乳酸、カオリン、マンニトール、塩化ナトリウム、乾燥デンプン、ならびに粉末糖が挙げられる。
【0058】
粉末物質に対して粘着特性を付与するのに使用される薬剤は、結合剤または顆粒剤といわれる。それらは、錠剤処方物に粘着性を付与し、圧縮後その錠剤がインタクトなままであること、ならびに望ましい硬度およびサイズの顆粒剤の処方物による自由流動特性の改善を確実にする。一般的に結合剤として使用される物質としては、デンプン、ゼラチン、ならびにスクロース、グルコース、デキストロース、モラセスおよびラクトースといった糖類が挙げられる。使用される天然および人工のガムは、アラビアゴム、アルギン酸ナトリウム、トチャカ抽出物、パンワーゴム、ガティゴム、イサポール(isapol)さやの粘液、カルボキシメチルセルロース、メチルセルロース、ポリビニルピロリドン、ハチゴムならびにカラマツアラボガラクタンが挙げられる。ある状況下では結合剤と考えられ得る他の薬剤には、ポリエチレングリコール、エチルセルロース、ワックス、水およびアルコールがある。
【0059】
滑沢剤は、錠剤顆粒の流速の改善、ダイやせん孔の表面に対する錠剤物質の付着の防護、粒子間摩擦の減少、ならびにダイキャビティからの錠剤排出の促進のための錠剤製品において使用される。一般的に、使用される滑沢剤としては、タルク、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸および硬化植物油が挙げられる。
【0060】
崩壊剤は、物質もしくは物質の混合物であり、投与後の崩壊または分解を促進するために錠剤に対して添加される。崩壊剤として働く物質は、化学的にはデンプン、粘土、セルロース、アルギン(align)もしくはゴムとして分類される。
【0061】
デンプンに加え、多種の物質が使用され、崩壊剤として有効であると報告されている。この群は、ビーガム(Veegum HV)、メチルセルロース、寒天、ベントナイト、セルロースおよび、木製品、天然のスポンジ、陽イオン交換樹脂、アルギン酸、グアールガム、柑橘類のパルプならびにカルボキシメチラーゼが挙げられる。デンプンとの組み合せにおいてラウリル硫酸ナトリウムは、有効な崩壊剤であることが実証されている。
【0062】
非水溶性の担体もしくは賦形剤は、生体適合性、および望ましい速度で血流に活性成分を放出するのに十分な哺乳動物の体温で液状もしくは柔軟性の任意の物質である。その担体は、通常疎水性、かつ一般的に、例えば、植物、動物、無機質および合成物の起源もしくは誘導の油もしくは脂肪といった有機物である。好ましくは、その担体は、水において非混和性であり、および/または一般的に脂肪溶媒として公知の物質中において溶解性である。
【0063】
代表的に、水、適切な油、生理食塩水、水性デキストロース(グルコース)および関連糖質溶液ならびにプロピレングリコールおよびポリエチレングリコールといったグリコールは、非経口の溶液について適切な担体である。非経口投与のための溶液は、好ましくはミルナシプランの水溶性塩、適切な安定薬剤、ならびに必要とする場合緩衝物質を包含する。重硫酸塩ナトリウム、硫酸ナトリウム、もしくはアスコルビン酸、これらの単体もしくは組み合せといった抗酸化薬剤は、適切な安定薬剤である。また、使用されるのは、クエン酸およびその塩ならびにEDTAナトリウムである。さらに、非経口の溶液は、塩化ベンザアルコニウム塩化物、メチルパラベンまたはプロピルパラベンならびにクロロブタノールといった保存剤を含み得る。適切な薬学的担体は、当該分野で公知である。
【0064】
(III.使用法)
(A.処置プロトコル)
活性化合物の投与は、患者に対する有害反応(薬物(例えばミルナシプラン)に対する、異常な、有害な、もしくは予測外の反応の減少もしくは欠如を意味する)が実質的にないと同時に、神経学的障害の症状を処置するのに有効な量であるべきである。語句、「有害反応が実質的にない」は、活性成分の特定の投薬量での有害反応の回数、性質および程度に関係する。活性化合物は、神経学的障害の症状を回復もしくは排除するための治療的に有効な期間にわたって投与されるべきである。
【0065】
有害反応は、下記:皮膚、中枢神経系および末梢神経系、視覚、精神医学、胃腸系、肝臓および胆汁系、内分泌および代謝系、心血管系、呼吸系、赤血球、白血球、血小板、血液、泌尿器系、生殖系ならびに新生物のうちの少なくとも一つに関連し得る。その有害反応は、下記:悪心、嘔吐、頭痛、臆病、不安、不安発作、動悸、尿貯留、起立性低血圧、発汗、胸痛、発疹、体重増加、背痛、便秘、目眩、発汗増加、動揺、のぼせ、震え、疲労、眠気、消化不良、嗅覚異常、緊張感、口内乾燥、腹痛および不眠のうちの少なくとも一つと関連し得る。
【0066】
その化合物は単体で投与されうるが、好ましくは、選択された投与経路ならびに医薬品の実行基準に基づいて選択された薬学的担体とともに投与される。それらは、個々の治療薬剤もしくはその治療薬剤の組み合せのいずれかとして、医薬品とともに使用するために利用可能である任意の従来の手段によって投与され得る。
【0067】
好ましくは、活性成分の毎日の投薬量は、患者において循環する活性化合物の治療的量に達するように一定期間にわたって段階的に増加される(例えば、第一の期間および/または第二の期間)。
【0068】
毎日の投薬量の漸進的段階的増加は、投与される活性成分に対する患者の耐性の改善を意図する。活性成分で処置された患者は、(1)有害反応を経験しない;(2)わずかな有害反応を経験する;(3)重傷度の少ない程度の有害反応を経験する;もしくはその組み合わせである。この耐性は、投薬の段階的増加手順なしに活性成分の同投薬量に対する哺乳動物の耐性と比較して、特定の投薬量での活性成分に対する哺乳動物の耐性に関連する。
【0069】
一般的に、活性化合物は段階的形式における漸増投薬量において投与され得、有害反応を回避もしくは最小化するための患者の耐性の増加を伴って活性化合物の循環投薬量を増加する。活性化合物の毎日の投薬量は、各期間(すなわち各工程)の間で異なり得るが、ただし各投薬量は、その工程の間に割り当てられた投薬量の範囲内である。
【0070】
例えば、4段階の段階的増加において、活性化合物は、第一の期間に100mgまでの毎日の投薬量で投与され、そして第二の期間に最初の投薬量の1.5倍〜2.5倍量まで段階的に増加される。第二の投薬期間は、第一の期間と同様の持続期間であり得、第一の期間の後、活性成分の投薬量は、第三の投薬期間に第二の投薬量の1.5倍〜2.5倍量まで再び段階的に増加される。第三の投薬期間は、第一および第二の期間と同様の持続期間であり得、その第一の期間または第二の期間の後、活性成分の投薬量は、神経学的障害の症状を処置するのに治療的に有効な期間に第三の投薬量の1.5倍〜2.5倍量まで再び段階的に増加する。あるいは、2段階の段階的増加において、その投薬量は、第二の投薬量に対して一度だけ段階的に増加され得、第二の期間は、神経学的障害の症状を処置するのに治療的に有効な期間であり得る。
【0071】
投薬量の段階的増加での各工程の期間は、3日間、3日間をこえた時間、もしくは2週間、4週間、6週間、8週間、10週間、12週間もしくは20週間よりも長い期間であり得る。毎日の投薬量は、一回投与され得るか、もしくは一日に二回以上(例えば、2回、3回、4回、または5回)までに分割され得る。
【0072】
ある実施形態は、3日間より長い期間にわたって100mgまでのミルナシプランの毎日の投薬量を投与し、そして神経学的障害の症状を処置するために治療的に有効な期間にわたって100mgより多い毎日の投薬量の段階的増加により、2段階の段階的増加を要する。
【0073】
別の実施形態は、3日間より長い期間にわたって、最初の毎日の約10mgと約50mgの間の投薬量でのミルナシプランの投与、そして3日間より長い期間にわたる約25mg〜約75mgへの毎日の投薬量の段階的増加ならびに神経学的障害の症状を処置するために治療的に有効な期間にわたる100mgより多い毎日の投薬量の段階的増加によって、3段階の段階的増加を要する。
【0074】
別の実施形態は、最初の毎日の約20mgと約30mgの間の投薬量で7日間にわたるミルナシプランの投与、7日間にわたる約40mg〜約60mgへの毎日の投薬量の段階的増加、および7日間にわたる約75mg〜約125mgへの毎日の投薬量の段階的増加、ならびに神経学的障害の症状を処置するために治療的に有効な期間にわたる175mg〜225mgへの毎日の投薬量の段階的増加によって4段階の段階的増加を要する。
【0075】
これらの投与のプロトコルは、上述の任意の抗うつ剤化合物にも適用され得る。
【0076】
(B.効果的な投薬量の範囲)
投与される投薬量は、特定の薬剤ならびにその投与形式および投与経路の薬力学特性;レシピエントの年齢、健康および体重;その症状の性質および程度;同時発生的な処置の種類;処置の頻度;ならびに望ましい効果といった既知の要因に依存して変化する。活性成分の毎日の投薬量は、体重1キログラムあたり約0.001mg〜約1000mgまであり、好ましい投薬は約0.1mg/kg〜100mg/kgであり、好ましくは一日に数回投与されることが期待される。
【0077】
本明細書において開示される活性成分(例えば、ミルナシプラン)の投薬量は、神経学的傷害の症状を処置することにおいて適切な活性を有し、そのような活性は例えば以下である:(1)活性成分の改善された効力、(2)正の患者耐性の維持、(3)正の患者の安全性プロフィールの維持(例えば、用量が制限される毒性)、(4)活性成分の適切なピーク血漿濃度(Cmax)、および/または(5)一日二回(BID)の投与とは対照的に一日一回(QD)。
【0078】
NSRI抗うつ剤は、うつ病を処置するための抗うつ剤の推奨される毎日の投薬量よりも多い(例えば、約1.5倍〜約4.0倍多い)毎日の投薬量において投与され得る。本明細書において開示される抗うつ剤においての推奨される毎日の投薬量は、うつ病を処置するために、例えば、(Physisician’s Desk Referance)(PDR),55th Edition(2001);およびインターネット薬物索引ウェブサイト(www.RxList.com);もしくはSLS 精神医学ならびに薬物関連ウェブサイト(http://sl.schofield3.home.att.net/medicine/psychiatric_drugs_chart.html)で見
られ得る。
【0079】
NSRI抗うつ剤は、他の神経学的障害の症状を処置する場合、うつ病を処置するのに推奨されるよりもより分割された投薬量において投与され得る。抗うつ剤について推奨される毎日の投薬量は、抗うつ剤を処置するために、例えば、Physisician’s
Desk Referance)(PDR),55th Edition(2001);およびインターネット薬物索引ウェブサイト(www.RxList.com);もしくはSLS 精神医学ならびに薬物関連ウェブサイト(http://sl.schof
ield3.home.att.net/medicine/psychiatric_drugs_chart.html)で見られ得る。
【0080】
一般的に、2段階の投薬量の段階的増加において、第一の投薬量は、一日あたり約100mgまでであり、第二の投薬量は一日あたり約100mgよりも多い。好ましくは、次の段階的増加工程の投薬量は、一般的に前の投薬量の1.5倍〜2.5倍である。そのようにして、4段階の段階的増加のプロトコルにおいて、第二の毎日の投薬量は、第一の毎日の投薬量の約1.5倍〜約2.5倍である;第三の毎日の投薬量は、第二の毎日の投薬量の約1.5倍〜約2.5倍である;および第四の毎日の投薬量は、第三の毎日の投薬量の約1.5倍〜約2.5倍であって、神経学的障害の症状を効果的に処置する十分な期間にわたって投与される。
【0081】
例えば、3段階のミルナシプランの投薬量の段階的増加において好ましい投薬量の様式は、約3日間の間1日50mgであり、続いて約3日間の間で25mg〜75mgであり、次いで神経学的障害の症状を効率的に処置するための十分な期間にわたって100mgより多い投薬量である。
【0082】
別の例として、4段階のミルナシプランの投薬量の段階的増加の好ましい様式は、約7日間の間に約20mg〜約30mg、続いて約7日間の間約40mg〜約60mgの毎日の投薬量が投与され、約7日間の間約75mg〜約125mgの毎日の投薬量が続き、最終的に、神経学的障害の症状を効果的に処置する十分な期間にわたって約175mg〜約225mgである。
【0083】
好ましくは、ミルナシプランは100mg/日よりも多く投与され、より好ましくは、200mg/日よりも多く投与される。1つの実施形態において、ミルナシプランは、体量の約100mg/70kgよりも多く投与される。
【0084】
(増加した毎日の投与においての分割された毎日の投薬量)
より高い毎日の投薬量は、一回の毎日の投薬量を分割すること、ならびに一日二回以上(例えば、2回,3回,4回または5回)投与することで達成され得る。任意の一つ以上の抗うつ剤は、本明細書で記載される毎日の投薬量において、分割された投薬量で投与され得、長期間にわたって有効な血清中の薬物濃度を得る。
【0085】
例えば、約200mgのミルナシプランの毎日の投薬量の投与は、約100mgで一日二回(BID)投与され得る。同様に、ミルナシプランの約400mgの毎日の投薬量の投与は、約200mgを一日二回(BID)投与され得る。この技術は、抗うつ剤のより高い投薬量を投与するために拡大され得る:
ミルナシプランは、一日あたり約50mgと約800mgの間で;好ましくは、一日あたり約100mgと約400mgの間で、最も好ましくは、一日あたり約200mgと約300mgの間で投与され得る。
【0086】
ベンラファキシンヒドロクロリドは、一日あたり約75mgと約1,500mgの間で;好ましくは、一日あたり約112.5mgと約900mgの間で、最も好ましくは、一日あたり約225mgと約600mgの間で投与され得る。
【0087】
ミルタザピンは、一日あたり約15mgと約180mgの間で;好ましくは、一日あたり約30mgと約120mgの間で、最も好ましくは、一日あたり約45mgと約60mgの間で投与され得る。
【0088】
ネファゾドンは、一日あたり約200mgと約2,400mgの間で;好ましくは、一日あたり約300mgと約1200mgの間で、最も好ましくは、一日あたり約600mgと約800mgの間で投与され得る。
【0089】
チオリダジンヒドロクロリドは、一日あたり約50mgと約800mgの間で;好ましくは、一日あたり約100mgと約400mgの間で、最も好ましくは、一日あたり約200mgと約300mgの間で投与され得る。
【0090】
ブプロピオンヒドロクロリドは、一日あたり約150mgと約1,600mgの間で;好ましくは、一日あたり約300mgと約1200mgの間で、最も好ましくは、一日あたり約400mgと約600mgの間で投与され得る。
【0091】
フェネルジンスルフェートは、一日あたり約15mgと約360mgの間で;好ましくは、一日あたり約60mgと約240mgの間で、最も好ましくは、一日あたり約90mgと約135mgの間で投与され得る。
【0092】
硫酸トラニルシプロミンは、一日あたり約30mgと約160mgの間で;好ましくは、一日あたり約45mgと約120mgの間で、最も好ましくは、一日あたり約60mgと約80mgの間で投与され得る。
【0093】
モクロベミンドは、一日あたり約400mgと約3,600mgの間で;好ましくは、一日あたり約500mgと約2400mgの間で、最も好ましくは、一日あたり約900mgと約1200mgの間で投与され得る。
【0094】
ピルリンドルは、一日あたり約200mgと約1,600mgの間で;好ましくは、一日あたり約300mgと約1,200mgの間で、最も好ましくは、一日あたり約400mgと約800mgの間で投与され得る。
【0095】
シタロプラムブロミドは、一日あたり約20mgと約240mgの間で;好ましくは、一日あたり約40mgと約160mgの間で、最も好ましくは、一日あたり約60mgと約80mgの間で投与され得る。
【0096】
パロキセチンヒドロクロリドは、一日あたり約20mgと約250mgの間で;好ましくは、一日あたり約50mgと約200mgの間で、最も好ましくは、一日あたり約62.5mgと約80mgの間で投与され得る。
【0097】
フルオキセチンヒドロクロリドは、一日あたり約20mgと約320mgの間で;好ましくは、一日あたり約60mgと約240mgの間で、最も好ましくは、一日あたり約80mgと約120mgの間で投与され得る。
【0098】
セルトラリンヒドロクロリドは、一日あたり約25mgと約800mgの間で;好ましくは、一日あたり約50mgと約200mgの間で、最も好ましくは、一日あたり約75mgと約100mgの間で投与され得る。
【0099】
アミトリプチリンヒドロクロリドは、一日あたり約50mgと約600mgの間で;好ましくは、一日あたり約100mgと約400mgの間で、最も好ましくは、一日あたり約150mgと約200mgの間で投与され得る。
【0100】
パーフェナジンおよびアミトリプチンヒドロクロリドは、それぞれ、約6mg〜約22mg、約75mg〜約350mgの間で;好ましくは、一日あたり、それぞれ約9mg〜約15mg、約100mg〜約200mgの間で投与され得る。
【0101】
デシプラミンヒドロクロリドは、一日あたり約100mgと約1200mgの間で;好ましくは、一日あたり約200mgと約800mgの間で、最も好ましくは、一日あたり約300mgと約400mgの間で投与され得る。
【0102】
ドキセピンヒドロクロリドは、一日あたり約75mgと約1200mgの間で;好ましくは、一日あたり約150mgと約600mgの間で、最も好ましくは、一日あたり約300mgと約450mgの間で投与され得る。
【0103】
マレイン酸トリミプラミンは、一日あたり約75mgと約800mgの間で;好ましくは、一日あたり約150mgと約600mgの間で、最も好ましくは、一日あたり約200mgと約300mgの間で投与され得る。
【0104】
抗うつ剤は、必要な回数および間隔において一定期間にわたって、維持投薬量に達するように、摂取する指示とともにパッケージされた使用について現在認可された投薬処方物においてもしくは投薬パックにおいて提供され得る。例えば、抗うつ剤の投薬パックは、別個の投薬量、ならびに維持投薬量に達するまでの期間にわたって漸増量における別個の投薬量を服用するための手順を含み得る。ある実施形態において、投薬量は同量であり、手順は経時的に投薬量の増加した回数の服用を提供する。他の実施形態において、投薬パックは、異なる量の抗うつ剤を含む投薬量を含み、その指示は経時的に漸増量においての投薬量の服用を提供する。あるいは、その投薬パックは、維持投薬量に達するための期間にわたって、抗うつ剤の漸増量を放出するために処方され得るか、もしくは、その処方物は徐放性処方物および/またはパルス状放出処方物であり得る。
【0105】
代表的な投薬パックは、以下:
(a)約3日間よりも長い期間にわたる。約50mgまでのミルナシプランの毎日の投薬量;
(b)3日間よりも長い期間にわたる。約25mg〜約75mgまでのミルナシプランの毎日の投薬量;および、
(c)その症状を効果的に処置するのに十分な期間にわたる約100mgを超えるミルナシプランの毎日の投薬量;
を含む。
【0106】
本明細書で開示される任意の特許、もしくは特許文書は、本発明に参考として援用され、本発明の一部を形成する。
【実施例】
【0107】
(実施例1:漸進的または投薬量の段階的増加ならびに線維筋痛の処置におけるミルナシプランの有効性)
この研究を、線維筋痛症候群(FMS)の処置においてミルナシプランの効力を特徴付けるのに行った。第二の目的は、投与される毎日の用量、投薬の頻度ならびにFMSの処置の有効性の間の関連性を評価することであった。より高い投薬量が、より有効であることが理解されているので、第三の目的は、投薬量の漸進的な段階的増加がミルナシプランの投薬耐性を増加し得るかどうかを決定することであった。以前の研究では、患者に、50mg、100mgまたは200mgのミルナシプランの最初の毎日の投薬量を与えると、200mgとの有害な事象プロフィールは、100mgまたは50mgよりも実質的に悪かった。この研究において、毎日の投薬量は、漸進的に段階的に増加した。
【0108】
(方法)
線維筋痛症候群についての1990年のACR基準を満たす被検体は、登録についての適格者であった。患者は、抗うつ剤、睡眠薬および潜在的に効力の計測に干渉し得る他の薬物の排除後、最初の二週間にわたって基準の症状を記録した。患者を、一日一回のミルナシプランの投薬処置群、一日二回のミルナシプランの投薬処置群、もしくはプラシーボコントロールを1.5:1.5:1の比で無作為化した。活性処置の患者に、最初に25mgのミルナシプランを一日一回(25mg QD)もしくは一日二回(12.5mg BID)の投薬量を、最初の一週間にわたって与えた。患者がこの投薬で耐容性を示す場合、その患者は、二週目に50mgまでの毎日の投薬量、三週目に100mgまでの毎日の投薬量、四週目に200mgまでの毎日の投薬量へと段階的に増やし、もしくはプラシーボに対応するように増加した。用量が制限される毒性にいずれかの所定の週において遭遇した場合、患者を、以前の週の投薬量に減少し、その研究の余剰期間にわたって、その投薬量で維持した。被検体は、彼らの最大耐性用量(200mgまで)をさらなる8週間にわたって服用し続けた。患者は、計12週の投与を受け得る。用量が制限される毒性を、薬物関連の等級3〜4の有害な事象が発生すると定義する。
【0109】
(効力の計測)
効力を、最初の投薬を行う前ならびに一ヶ月ごとの通院の間、FMS状態の評価を使用して計測した。これらは、線維筋痛影響質問表(Impact Question naire)、マッギール疼痛(Mcgill Pain)、患者の臨床的な全体の印象、患者の全体の疼痛状態、生命測定のSF−36特性および、誘発した疼痛の測定を含んだ。
【0110】
(結果)
以前の研究において、患者は、彼らの最後の毎日の投薬量へと、短い期間(通常一週間以下)で段階的に増加した。200mgの投薬量では、50mgまたは100mgの投薬量よりも実質的に悪化した副作用の発生頻度を与えた。これらの結果に基づき、本研究では、患者の約50%が、200mgの毎日の投薬に耐性を示すことができず、および100mg/日で処置されることが期待された。意外にも、本研究において大部分の患者が、200mg/日に達し、この投薬量においてよく耐性を示した。ミルナシプランで処置された約70人の内、わずか9人のみが、200mg/日で耐性を示すのに失敗した。
【0111】
非段階的な最初の100mgのミルナシプランの投薬量を与える患者についての副作用は、処置の最初の週において悪化し、2週〜4週までにより低いレベルに沈静化した。これは、よりゆっくりとした投薬の段階的増加は、200mgのミルナシプランの毎日の投薬量により多くの患者が耐性を示したことの原因であることを示唆している。
【0112】
(実施例2:線維筋痛症候群を処置するためのミルナシプランの使用)
(方法)
12週間の無作為化、二重盲検プラシーボコントロール投薬の段階的増加単独療法治験を、FMSと診断された患者においてミルナシプランを評価するために行った。最初の期間の後、患者を疼痛投薬、中枢神経作用性刺激薬、抗うつ剤および鎮静睡眠薬を排除した場合、二週間の基準期間を開始した。基準期間が首尾よく完了した後、患者を、プラシーボ、QDミルナシプラン、またはBIDミルナシプランを1:1.5:1.5の比で無作為化した。全ての患者で、4週間の期間にわたって、一週間に一度の工程を毎日25mgから毎日50mg、毎日100mg、および最終的に毎日200mgまで、もしくは用量が制限される毒性(DLT)が明らかになるまで、段階的に増加した。DLTの存在を、患者を次のより高い投薬量に進めるように認定する前に、本研究機関により各週に評価した。DLTが明らかである事象において、患者は、以前の良く耐性を示す投薬量において安定し、安定した用量治療で八週間の間、この用量を維持した。
【0113】
任意の所定の投薬レベルにおいて、ミルナシプランQDの患者では、朝にミルナシプランの全量を摂取し、夜にプラシーボを摂取した。ミルナシプランBIDの患者では、朝および夜に分割した投薬量が与えられて同じ総量で摂取した。本研究の間、患者に、電子手帳を携帯し疼痛、疲労、睡眠および身体的情報を記録するように要請した。特注デザインの手帳は、複数の方法で自発的な疼痛を捕捉し、その方法としては以下:
(1)無作為な毎日のプロンプト(一日あたり4回〜5回彼らの現在の疼痛レベルを患者に対して記録するように知らせる装置);
(2)以前の24時間の疼痛について質問する朝の毎日のプロンプト;
(3)過去一週間の間の患者の平均的疼痛について提出を求める金曜日の夜の毎週のレポート;
が、挙げられる。
【0114】
電気的な評価を、クリニック来診の間で伝統的な疼痛、気分、および生活の質の調査表と共に追加した。対数Gracely 固定化(Gracely auchored)疼痛スケールの電子バージョンをその手帳において実行した。このスケールは、患者に、その疼痛を説明する感覚を「極めて激しい」、「非常に激しい」、「激しい」、「強力」、「わずかながら激しい」、「わずかに強力な」、「中程度」、「軽度」、「非常に軽度」、「弱い」、「非常に弱い」、「かすかな」、「痛みの無い」の中で選択することを求める。
【0115】
処置の第4週、第8週、第12週の間で、患者は、クリニックに来診し、ここで、多くの基準結果の測定を、SF−36、マッギ−ル疼痛(Mcgill Pain)アンケート、BECK、STPI、線維筋痛影響アンケートおよびいくつかの疼痛測定を含めて、実行した。安全性情報は、これらのクリニック来診時に収集した。
【0116】
主要な終点は、患者の電子手帳(PED)で収集された疼痛スコアに基づく基準線から終点までの疼痛スコアの変化として定義された。終点は、単一の値(クリニック測定のような)で評価のための12週、もしくは手帳基準の結果においての11週および12週の平均スコアとして定義した。「応答者」は、少なくとも30%の疼痛スコアを改善する患者として定義した。
【0117】
表1は、治験に関する登録基準をおよび除外基準を示す。
【0118】
【表1】
(結果)
表2は、用量の段階的増加の失敗の割合を示す。ミルナシプランの一日二回の投薬は、一日一回の投薬よりも低い割合の投薬耐性を生じた。この違いは、小サンプルサイズ(p=0.07)であったにも拘らず、ほぼ統計学的有意性に達した(表における「盲検」は、この日付における患者の処置群が、研究者にも伏せられる患者をいう)。
【0119】
【表2】
図2は、おそらくミルナシプランの効果の最も著しい証拠をあらわす。この図は、全て患者においての全体疼痛スコアをプロットし、上記患者は、この分析時間で終点に達した(38 MILおよび12プラシーボ)。1〜7スケールにおいて、1は非常によく改善し、4は変化がなく、および7は非常に悪化し、プラシーボ患者においての平均値が4.3であった一方、MIL患者においての平均値は2.3であった。図2において、1〜3のスコアは改善と示され、4は変化なし、ならびに5〜7は悪化である。ミルナシプラン群とプラシーボとの間の違いは、統計学的にp=0.0001で有意であった。
【0120】
図3は、治験の処置段階の間のBeckスコアの変化を示した。この治験は、うつ病患者においては選択しなかったが、FMS集団においてかなり高い割合の大うつ病およびうつ病の気分が存在した。このサンプルにおいて、14人の患者がMDEの定義を満たした。プールしたミルナシプランの処置群とプラシーボ群との間の終点でのスコアにおける違いは、p=0.03で有意であって、MILの公知の抗うつ効果がこの集団の処置において重要な助けとなることを示す。
【0121】
図4は、この研究の経過にわたって三つの処置群において報告された24時間の毎日の疼痛スコアを示す。BID投薬 は、処置期間全体を通して、QD投薬より有効であった。
【0122】
図5は、研究機関にわたって3つの処置群の患者によって報告された。毎日の睡眠の質のスコアを示す。睡眠の質では、プラシーボと比較したミルナシプランでの有意な改善が見られず、QD投薬とBID投薬の違いが検出されなかった。反対に、これは夜において用量を投与することを包含する。BID投薬においてさえも、睡眠の質の悪化は見られなかった。このことは、抗うつ剤の一般的副作用が睡眠の質に干渉するので顕著である。
【0123】
図6は、ミルナシプランをBIDで摂取した「応答者」として分類される患者の患者電子手帳(PED)の疼痛スコアを示す。スコアが各データポイントについて平均された患者の数が、1週目で34であり、13週目および14週目で24に減少した。RPは、一週間にわたって平均化した無作為な刺激疼痛スコアである。毎週(weekly)は、毎週の想起スコアである。毎日(Daily)は、毎日の想起スコアである。最初の二週間は基準ライン期間であり、3〜6週間は投薬の段階的増加期間、ならびに6週間から終了は、安定した投薬処置期間であることを留意する必要がある。投薬の段階的増加をともなう2〜6週間にわたる疼痛スコアにおける改善は、ミルナシプランのより高い投薬量が、FMSに関係する疼痛の処置においてより有効であることを示唆する。
【0124】
(結論)
ミルナシプランは、線維筋痛症候群に関係する疼痛を効果的に処置した。FMSを伴ったうつ病患者における気分もまた改善した。一日あたり200mgの相対的に高い用量を使用した。用量が、200mgに段階的に増加する場合に疼痛スコアにおける改善は、この用量が疼痛の緩和において重要であることを示す。一日二回の投薬は、疼痛の軽減において一日一回の投薬よりも有効であった。一日二回の投薬によって、一日一回の投薬をおこなうよりも少ない用量関連の有害な事象ならびに使用される漸増した投薬量に対して、より少ない不耐性もまた得た。
【0125】
(実施例3:水泳ストレス誘導の筋痛覚過敏に対するミルナシプランを使用した処置)
頻回の不可避の水泳ストレスが、ラットにおいて、熱性および長期の化学的に有害な両方の刺激に対する遅延性および長期間の皮膚の痛覚過敏を産生することが示された。この水泳ストレス誘導痛覚過敏(SSIH)モデルは、FMSと複数の特性を共有する。これらの類似性としては、(1)皮膚の痛覚過敏の存在、(2)ストレスの有意な役割、(3)痛覚過敏応答におけるNMDAレセプター機構の関与、および(4)抗うつ剤の有効性、が挙げられる。特に、クロルミプラミンおよびフルオキセチンによる前処置は、頻回にストレスを与えられたラットにおける皮膚の痛覚過敏の発症を防護し、ある抗うつ剤が、FMSにおける疼痛および睡眠異常を改善する。
【0126】
(方法)
ミルナシプラン(MIL)を、通常の生理食塩水で混合し、Sprague−Dawleyラット(200g〜300g)に対し腹腔内注射(IP)を介して投与した。
【0127】
ラットの処置群は、以下:(1)ストレス−水20cmにおいて水泳を強制;(2)疑似−水2cm〜3cmにおける水泳;(3)未処置−ケージにおいて平穏なまま、である。ラットに、注射無し、食塩水のIP QDもしくはミルナシプランの10mg/kg/日または30mg/kg/日 IP QDを与えた。
【0128】
ラットに、ホットプレート応答潜伏(秒)による熱性の痛覚の閾値について試験をおこなった。ラットに、痛覚計によって握力(kg)を計測することによる筋痛覚過敏について試験をおこなった。
【0129】
ミルナシプランによる前処置の有効性を試験する場合、ラットに、1日目〜11日目に薬物を与え、8日目〜10日目に水泳ストレスを与えた。ホットプレート応答の潜伏および握力に関するデータを、7日目および11日目に得た。ミルナシプランの処置後の有効性の試験をする場合、ホットプレートの応答潜伏および握力を、1日目、5日目および10日目に計測した;水泳ストレスを、2日目〜4日目に施行し、薬物処置を、5日目〜10日目に施行した。
【0130】
(結果)
図7は、ホットプレートの潜伏における水泳ストレス誘導の減少を示し、これらの減少は、ミルナシプランの前処置によって防護されなかった。
【0131】
図8は、頻回のストレス(IPの注射および強制/疑似水泳)が握力の減少の原因となったことを示す。ミルナシプランによる前処理は、握力におけるこのストレス誘導減少を防護した。このようにして、ミルナシプランによる前処置はFMSのこのモデルにおける筋痛覚過敏を防護した。
【0132】
図9は、水泳ストレス後のミルナシプランの処置が、ホットプレート応答の潜伏における水泳ストレス誘導の減少を改善しないことを示す。
【0133】
しかし図10は、ミルナシプランが、頻回の強制水泳ストレスによる握力の減少を改善することを示す。
【0134】
(結論)
水泳ストレスによって誘起される熱性の皮膚の感覚過敏は持続性であり、条件付け数日後の状態において、基本的に不変であった。
【0135】
水泳ストレス後の頻回のIPの注射は、筋肉の異痛に改善するようである握力の減少を誘導する。
【0136】
ミルナシプランは、水泳ストレスによって生じる筋肉の異痛の改善および防護において有効である。しかし、ミルナシプランは、皮膚の熱性の感覚過敏を無効もしくは防護しない。
【0137】
皮膚および筋肉の侵害受容の調節は、存在し得、薬理学的に独立して影響を受け得ることから、皮膚および筋肉の痛覚の侵害受容は、この動物モデルにおいては分し得る
【特許請求の範囲】
【請求項1】
本明細書中に記載の発明。
【請求項1】
本明細書中に記載の発明。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2010−275314(P2010−275314A)
【公開日】平成22年12月9日(2010.12.9)
【国際特許分類】
【出願番号】特願2010−173175(P2010−173175)
【出願日】平成22年7月30日(2010.7.30)
【分割の表示】特願2005−500374(P2005−500374)の分割
【原出願日】平成15年10月3日(2003.10.3)
【出願人】(504045075)サイプレス バイオサイエンス, インコーポレイテッド (5)
【Fターム(参考)】
【公開日】平成22年12月9日(2010.12.9)
【国際特許分類】
【出願日】平成22年7月30日(2010.7.30)
【分割の表示】特願2005−500374(P2005−500374)の分割
【原出願日】平成15年10月3日(2003.10.3)
【出願人】(504045075)サイプレス バイオサイエンス, インコーポレイテッド (5)
【Fターム(参考)】
[ Back to top ]