
神経炎症のイメージング
本発明は、インビボイメージング、特に末梢ベンゾジアゼピンレセプター(PBR)のインビボイメージングに関する。高い親和性をもってPBRに結合し、投与後には脳内への良好な取込みを示し、高いレベルのPBRを発現する組織と優先的に結合する四環式インドール型インビボイメージング剤が提供される。本発明はまた、本発明のインビボイメージング剤の合成において有用な前駆体化合物、並びに前記前駆体化合物の使用を含む前記インビボイメージング剤の合成方法及び前記方法を実施するためのキットも提供する。インビボイメージング剤の自動化合成のためのカセットも提供される。加えて本発明は、本発明のインビボイメージング剤を含む放射性医薬組成物並びに前記インビボイメージング剤の使用方法も提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、インビボイメージング、特に末梢ベンゾジアゼピンレセプター(PBR)のインビボイメージングに関する。高い親和性をもってPBRに結合し、投与後には脳内への良好な取込みを示し、高いレベルのPBRを発現する組織と優先的に結合する四環式インドール型インビボイメージング剤が提供される。本発明はまた、本発明のインビボイメージング剤の合成において有用な前駆体化合物、並びに前記前駆体化合物の使用を含む前記インビボイメージング剤の合成方法及び前記方法を実施するためのキットも提供する。インビボイメージング剤の自動化合成のためのカセットも提供される。加えて本発明は、本発明のインビボイメージング剤を含む放射性医薬組成物並びに前記インビボイメージング剤の使用方法も提供する。
【背景技術】
【0002】
トランスロケータータンパク質(TSPO)としても知られる末梢ベンゾジアゼピンレセプター(PBR)は、主として末梢組織及びグリア細胞に局在することが知られているが、それの生理学的機能はまだ明確に解明されていない。細胞レベル以下では、PBRはミトコンドリア外膜上に局在することが知られていて、これはミトコンドリア機能の調節及び及び免疫系において役割を果たす可能性を表している。さらに、PBRが細胞増殖、ステロイド生成、カルシウム流れ及び細胞呼吸に関係することも仮定されてきた。PBRは、急性及び慢性ストレス、不安、うつ病、パーキンソン病、アルツハイマー病、脳損傷、癌(Gavish et al,Pharm.Rev.1999;51:629)、ハンチントン病(Messmer and Reynolds,Neurosci.Lett.1998;241:53−6)、喘息(Pelaia et al,Gen.Pharmacol.1997;28(4):495−8)、慢性関節リウマチ(Bribes et al,Eur.J.Pharmacol.2002;452(1):111−22)、アテローム性動脈硬化症(Davies et al,J.Nucl.Med.2004;45:1898−1907)及び多発性硬化症(Banati et al,2000 Brain;123:2321)を始めとする各種の状態と関連していた。PBRはニューロパシー性疼痛にも関連する可能性があり、Tsuda et alはニューロパシー性疼痛をもった被験者で小グリア細胞の活性化を観察している(2005 TINS 28(2)pp101−7)。
【0003】
PBRに対して親和性を有するリガンドは当技術分野で公知である。米国特許第6,451,795号には、PBRに対して親和性(最も活性の高い化合物に関して0.2〜0.5nMのIC50値)を有する1群のインドール化合物が開示されている。この特許には、かかる化合物が末梢ニューロパシーの予防又は治療及び中枢神経変性疾患の治療のために有用であると述べられている。Okubo et al(Bioorganic & Medicinal Chemistry,2004;12:3569−80)は、PBRに対して親和性(約0.4nMという低いIC50値)を有する1群の四環式インドール化合物の設計、合成及び構造を記載している。Okubo et alによるこの文献には、かかる化合物の特別な用途は論議されていない。
【0004】
PBRのインビボイメージングも当技術分野で公知である。PBR選択性リガンドを用いる陽電子放出断層撮影(PET)イメージングでは、(R)−[11C]PK11195が中枢神経系(CNS)炎症の包括的指示薬を提供する。(R)−[11C]PK11195の使用の成功にもかかわらず、それには制約がある。それは、高いタンパク質結合性及び低特異性乃至非特異性結合を有することが知られている(Lockhart et al.Nucl Med Biol.30(2):199−206)。それの放射性標識代謝産物の役割は知られておらず、結合の定量化には複雑なモデル化が要求される。PBRに対して高い親和性及び選択性を有することでCNSにおけるPBR測定の向上を可能にする化合物を得るための努力が行われてきた。[11C]DAA1106及び[18F]FEDAA1106はアリールオキシアニリン化合物に基づくPET放射性リガンドであり、ヒトにおいて研究されてきた(Ikomo et al,J.Cereb.Blood Flow Metab.2007;27:173−84及びFujimura et al,J.Nuc.Med.2006;47:43−50)。しかし、これらの化合物の動力学的性質は理想的でなく、その用途は定量的研究に制限されることがある。これらの放射性リガンドにさらなる改良を加えようという努力の中で、Briard et al(J.Med.Chem.2008;51:17−30)は別のアリールオキシアニリン誘導体PBR28を報告した。PBR28の11C標識バージョンをサルに注射し、PETを用いてその脳内動力学を評価した。[11C]PBR28は、高い脳内取込み、PBR発現組織に対する良好な特異的結合、及びインビボイメージングのために一層適した動力学的性質を示した。PBR結合ピラゾロピリミジン化合物もまた、PBRを標的化するためのPET放射性リガンドとして評価された。James et al(J.Nuc.Med.2008;49(5):814−22)は、PET放射性リガンド[18F]−DPA−714が静脈内投与後のヒヒ脳においてPBRに対する高い親和性及びPBRによる選択的取込みを有することを報告している。[18F]−DPA−714の脳内取込みの動力学は[11C]DAA1106及び[18F]FEDAA1106より遅いが、事実上はそれに類似していることが報告された。国際公開第2007/057705号には、イメージング成分で標識された四環式インドール化合物であって、インビボイメージングのために適したものが開示されている。国際公開第2007/057705号中に例示されたインビボイメージング剤は、PBRに対して良好な親和性を有し、[3H]−PK−11195に対する競合アッセイにおけるKi値が1.0nM乃至0.1nMであることが示された。しかし、このたび本発明者らは、PBR発現組織に対するこれらの化合物の選択性が中枢神経系におけるPBS発現のインビボイメージングのためには理想的でないことを見出した。
【0005】
中枢神経系におけるPBS発現の評価のための代替インビボイメージング剤を得るためには、既知の四環式インドール化合物に改良を加える余地が存在している。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開第2007/057705号パンフレット
【発明の概要】
【課題を解決するための手段】
【0007】
本発明は、四環式インドール化合物に基づくインビボイメージング剤を提供する。四環式インドール化合物に基づく既知のインビボイメージング剤に比べて、本発明のインビボイメージング剤はインビボイメージングのための良好な性質を有している。本発明のインビボイメージング剤は、末梢ベンゾジアゼピンレセプターに対する良好な結合性を有すると共に、被験体への投与後における良好な脳内取込み及びインビボ動力学を有している。
【図面の簡単な説明】
【0008】
【図1】図1は、FNAラットの損傷(右側の)顔面神経核におけるインビボイメージング剤1の結合を示す代表的なオートラジオグラフである。
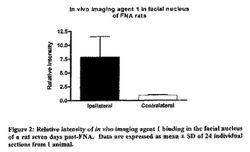
【図2】図2は、FNAから7日後のラットの顔面神経核に結合したインビボイメージング剤1の相対強度を示している。データは1頭の動物からの24の個別切片の平均±SDとして表されている。
【発明を実施するための形態】
【0009】
インビボイメージング剤
一態様では、本発明は、次の式Iのインビボイメージング剤或いはそれの塩又は溶媒和物であって、前記式Iのインビボイメージング剤の少なくとも1つの原子がインビボイメージングのために適した放射性同位体であり、前記放射性同位体が炭素の放射性同位体である場合にそれはカルボニル炭素であるインビボイメージング剤を提供する。
【0010】
【化1】

式中、
Qは水素又はフッ素であり、
Xは水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−6アルキル、C1−6ハロアルキル、C1−6アルコキシ又はC1−6アルキルアミドであり、
YはS、SO又はSO2であり、
Rは水素、C1−6アルキル又はC1−6フルオロアルキルであり、
ただしYがSである場合にはQ及びXが共に水素ではないことを条件とする。
【0011】
本明細書中で使用する「インビボイメージング」という用語は、本発明の被験体の内部構造の全部又は一部の画像を非侵襲的に生成する技法をいう。本発明で使用するための好ましいインビボイメージング方法は、単光子放出コンピューター断層撮影(SPECT)及び陽電子放出断層撮影(PET)であり、PETが特に好ましい。本発明の方法においてPETが好ましいのは、それが優れた感度及び分解能を有する結果、病変部における比較的小さい変化でも経時的に観察できるからである。PETスキャナーは、日常的にピコモル範囲内の放射能濃度を測定している。現在、マイクロPETスキャナーは約1mmの空間分解能に接近しているが、臨床スキャナーは約4〜5mmである。
【0012】
式Iの「インビボイメージング剤」は、インビボイメージングのために適した放射性同位体を含んでいる。この「インビボイメージングのために適した放射性同位体」は、式Iのインビボイメージング剤に関して上記に定義した原子の1つの放射性同位体形態である。本明細書で定義されるインビボイメージングのために適するには、放射性同位体はγ線放射体又は陽電子放射体であり、それによって投与後にインビボイメージング剤を被験体の外部で検出できることが好ましい。
【0013】
本発明に係る好適な塩には、(i)鉱酸(例えば、塩酸、臭化水素酸、リン酸、メタリン酸、硝酸及び硫酸)から導かれるもの並びに有機酸(例えば、酒石酸、トリフルオロ酢酸、クエン酸、リンゴ酸、乳酸、フマル酸、安息香酸、グリコール酸、グルコン酸、コハク酸、メタンスルホン酸及びp−トルエンスルホン酸)から導かれるもののような生理学的に許容される酸付加塩、並びに(ii)アンモニウム塩、アルカリ金属塩(例えば、ナトリウム塩及びカリウム塩)、アルカリ土類金属塩(例えば、カルシウム塩及びマグネシウム塩)、有機塩基(例えば、トリエタノールアミン、N−メチル−D−グルカミン、ピペリジン、ピリジン、ピペラジン及びモルホリン)との塩、及びアミノ酸(例えば、アルギニン及びリシン)との塩のような生理学的に許容される塩基塩がある。
【0014】
本発明に係る好適な溶媒和物には、エタノール、水、食塩水、生理的緩衝液及びグリコールと共に生成されるものがある。
【0015】
特記しない限り、単独で又は組み合わせて使用される「アルキル」という用語は、好ましくは1〜6の炭素原子、最も好ましくは1〜4の炭素原子を含む直鎖又は枝分れしたアルキル基を意味する。かかる基の例には、特に限定されないが、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソアミル及びヘキシルがある。
【0016】
特記しない限り、単独で又は組み合わせて使用される「アルコキシ」という用語はアルキルエーテル基を意味し、ここでアルキルという用語は上記に定義した通りである。好適なアルキルエーテル基の例には、特に限定されないが、メトキシ、エトキシ、n−プロポキシ、イソプロポキシ、n−ブトキシ、イソブトキシ、sec−ブトキシ及びtert−ブトキシがある。
【0017】
「アルキルアミド」は上記に定義したアルキル基がアミドに結合したものであり、ここでアミドは−C(=O)−NR’R”基(式中、R’及びR”は独立に水素又は炭化水素基である。)である。
【0018】
「ハロゲン」又は「ハロ」という用語は、フッ素、塩素、臭素及びヨウ素から選択される置換基を意味する。「ハロアルキル」は、上記に定義したアルキル基が1以上のハロゲンで置換されたものである。
【0019】
「ヒドロキシ」という用語は−OH基をいう。
【0020】
好ましい実施形態では、Qは水素である。
【0021】
Xは好ましくは水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ又はC1−4アルキルアミドである。Xは最も好ましくは水素又はC1−4アルコキシである。
【0022】
Yは好ましくはS又はSO2である。Yは最も好ましくはSである。
【0023】
Rは好ましくは水素、C1−4アルキル又はC1−4フルオロアルキルである。Rは好ましくは水素、C1−4アルキル又はC1−4フルオロアルキルである。Rは好ましくはC1−4フルオロアルキルである。
【0024】
式Iの好ましい実施形態では、
Xは水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ又はC1−4アルキルアミドであり、
YはS又はSO2であり、
Rは水素、C1−4アルキル又はC1−4フルオロアルキルである。
【0025】
式Iの最も好ましい実施形態では、
Qは水素であり、
XはC1−4アルコキシであり、
YはSであり、
RはC1−4フルオロアルキルである。
【0026】
式Iの別の好ましい実施形態では、
Qはフッ素であり、
Xは水素であり、
YはSであり、
RはC1−4フルオロアルキルである。
【0027】
本発明のインビボイメージングのために適した好ましい放射性同位体は、γ線放出型放射性ハロゲン及び陽電子放出型放射性非金属である。
【0028】
本発明で使用するのに適したγ線放出型放射性ハロゲンの例は、123I、131I及び77Brである。好ましいγ線放出型放射性ハロゲンは123Iである。
【0029】
本発明で使用するのに適した陽電子放出型放射性非金属の例は、11C、13N、18F及び124Iである。好ましい陽電子放出型放射性非金属は18Fである。18Fは本発明のインビボイメージングのために適する最も好ましい放射性同位体である。
【0030】
式Iのインビボイメージング剤に関する好ましい実施形態では、Xは123I、124I、131I、18F又はC1−4[18F]−フルオロアルキルである。
【0031】
式Iのインビボイメージング剤に関する別の好ましい実施形態では、RはC1−4[18F]−フルオロアルキルである。
【0032】
式Iのインビボイメージング剤に関するさらに別の好ましい実施形態では、カルボニル炭素は11Cである。
【0033】
本発明の若干の好ましいインビボイメージング剤の非限定的な例は以下の通りである。
【0034】
【化2】
上記のインビボイメージング剤のうちでは、イメージング剤2が最も好ましい。
【0035】
これらのインビボイメージング剤を得るために使用される合成方法は、以下の実験セクションに記載されている。本発明のインビボイメージング剤のこれらの非放射性バージョンの効力は、実施例10に記載されるように、インビトロアッセイで測定した。
【0036】
実施例7乃至実施例9には、放射性フッ素化インビボイメージング剤1〜7を得るための方法が記載されている。当業者には知られている通り、18Fを取り扱う場合、使用するスケール及び条件は安全上及び実際上の考慮事項に応じて異なる。18F PETトレーサーの製造に関する総説は、“Principles and Practice of Positron Emission Tomography”(2002 Lippincott Williams & Wilkins;Wahl and Buchanan,Eds.)の第1章及び第2章を参照されたい。かかるインビボイメージング剤を動物体内分布モデルで試験し(実施例11)、その体内分布を(国際公開第2007/057705号の実施例14に従って製造した)先行技術の化合物[18F]FE−PBRの体内分布と比較した。
【0037】
【化3】
以下の表1に、インビトロ親和性アッセイ並びにインビボ体内分布試験で得られたデータを示す。非放射性類似体をインビトロ親和性アッセイで試験し、放射性標識バージョンを体内分布アッセイで評価した。
【0038】
【表1】
かかるデータは、インビボイメージング剤1〜7の非放射性バージョンの効力が先行技術の化合物[18F]FE−PBRの効力に比べて勝ることを示している。加えて、かかるデータは、本発明のインビボイメージング剤1〜7が[18F]FE−PBRに比べて注射から30分後に線条体よりOB中に顕著に多く保持されることを示している。PBRはラット脳の他の領域に比べてOBにおいて高度に発現されることが知られているので(“Handbook of Substance Abuse”by Tarter,Ammerman and Ott;Springer 1998:398−99を参照されたい)、これらのデータは意外にも、インビボイメージング剤1〜7が先に例示したインビボイメージング剤[18F]FE−PBRよりPBRに対して向上した選択性を有することを実証している。
【0039】
以下の実施例12に記載されるように、インビボイメージング剤1をさらにオートラジオグラフィーモデルで分析した。顔面神経核の損傷側において顕著に高いレベルの放射能が検出された(図1及び図2参照)。損傷側の平均強度は、非損傷側が3.73±0.36であるのに比べて7.75±0.95であった。両側間の比は8.23±2.36であった。損傷部は正常部に比べて高いPBR発現を示すので、これらのデータは、インビボイメージング剤1がPBRに対して良好な選択性を有するという生体分布データからの結論を支持している。
【0040】
したがって、本発明のインビボイメージング剤は、既知の四環式インドール型PBR結合インビボイメージング剤に比べ、PBRのインビボイメージングのための意外に優れた性質を有している。
【0041】
前駆体化合物
別の態様では、本発明は次の式IIを有する前駆体化合物を提供する。
【0042】
【化4】
式中、R1、X1及びZ1の1つが本明細書で好適なもの及び好ましいものとして定義した放射性同位体の適当な供給源と反応する化学基を含む結果、当該前駆体化合物を前記放射性同位体の前記適当な供給源と反応せれば、本明細書で好適なもの及び好ましいものとして定義したインビボイメージング剤が生成され、
R1が前記化学基を含まない場合、それは式IのRに関して本明細書で好適なもの及び好ましいものとして定義した通りであり、任意にはさらに保護基を含み、
X1が前記化学基を含まない場合、それは式IのXに関して本明細書で好適なもの及び好ましいものとして定義した通りであり、任意にはさらに保護基を含み、
Z1が前記化学基を含まない場合、それは−C(=O)−N−(CH2−CH3)2であり、任意にはさらに保護基を含み、
Q1は式IのQに関して本明細書で好適なもの及び好ましいものとして定義した通りであり、
Y1は式IのYに関して本明細書で好適なもの及び好ましいものとして定義した通りであり、任意にはさらに保護基を含む。
【0043】
「前駆体化合物」は、好都合な化学形態の検出可能な標識との化学反応が部位特異的に起こり、最小数の段階(理想的にはただ1つの段階)で反応を実施でき、かつ格別の精製の必要なしに(理想的にはいかなる追加の精製も必要なしに)所望のインビボイメージング剤が得られるように設計された、放射性標識化合物の誘導体からなる。かかる前駆体化合物は合成品であり、良好な化学純度で簡便に得ることができる。前駆体化合物は、任意には前駆体化合物のある種の官能基に関して保護基を含むことができる。
【0044】
「保護基」という用語は、望ましくない化学反応を阻止又は抑制するが、分子の残部を変質させない十分に温和な条件下で問題の官能基から脱離させ得るのに十分な反応性を有するように設計された基を意味する。脱保護後には所望の生成物が得られる。保護基は当業者にとって公知であり、アミン基に関してはBoc(ここでBocはtert−ブチルオキシカルボニルである。)、Fmoc(ここでFmocはフルオレニルメトキシカルボニルである。)、トリフルオロアセチル、アリルオキシカルボニル、Dde[即ち、1−(4,4−ジメチル−2,6−ジオキソシクロヘキシリデン)エチル]及びNpys(即ち、3−ニトロ−2−ピリジンスルフェニル)から適宜に選択され、カルボキシル基に関してはメチルエステル、tert−ブチルエステル及びベンジルエステルから適宜に選択される。ヒドロキシル基に関しては、好適な保護基は、メチル、エチル又はtert−ブチル、アルコキシメチル又はアルコキシエチル、ベンジル、アセチル、ベンゾイル、トリチル(Trt)、又はテトラブチルジメチルシリルのようなトリアルキルシリルである。さらに他の保護基の使用は、‘Protective Groups in Organic Synthesis’,Theorodora W.Greene and Peter G.M.Wuts(Third Edition,John Wiley & Sons,1999)に記載されている。
【0045】
「放射性同位体の適当な供給源」という用語は、放射性同位体が前駆体化合物に共有結合するようにして前駆体化合物の置換基と反応し得る化学形態の放射性同位体を意味する。
【0046】
以下のセクションに示される特定の放射性同位体の各々に関しては、放射性同位体の1種以上の適当な供給源が論議される。インビボイメージング剤の分野の当業者は、本発明における用途に適した放射性同位体のこれらの供給源及びその他の供給源に精通しているであろう。
【0047】
インビボイメージング剤の放射性同位体が18Fある場合、放射性フッ素原子はフルオロアルキル基又はフルオロアルコキシ基の一部をなすことができる。これは、アルキルフルオリドがインビボでの代謝に耐えるからである。別法として、放射性フッ素原子は直接共有結合によってベンゼン環のような芳香環に結合できる。
【0048】
放射性フッ素化は、18F−フッ化物と良好な脱離基を有する前駆体化合物(例えば、アルキルブロミド、アルキルメシレート又はアルキルトシレート)中の適当な化学基との反応を用いる直接標識によって実施できる。
【0049】
18Fはまた、18F(CH2)3OMs又は18F(CH2)3OBrでヒドロキシル基をO−アルキル化することによっても導入できる。
【0050】
アリール系に関しては、アリールジアゾニウム塩、アリールニトロ化合物又はアリール第四級アンモニウム塩からの18F−フッ化物求核置換が、アリール−18F誘導体への好適な経路である。かかる方策は、例えば、式Iの1〜4位又は7〜10位に18Fを導入するために適している。
【0051】
別法として、18Fによる標識は、式Iの誘導体からの脱離基の求核置換によって達成できる。好適な脱離基には、Cl、Br、I、トシレート(OTs)、メシレート(OMs)及びトリフレート(OTf)がある。かかる誘導体は、本発明のインビボイメージング化合物を製造するための前駆体化合物である。
【0052】
別の方策は、上記に定義したような好適な脱離基を、前駆体化合物上に存在するアルキルアミド基上に設けることである。このようにすれば、通常は核反応18O(p,n)18Fからの水溶液として得られ、次いで陽性対イオンの添加及びそれに続く水の除去によって反応性にされる[18F]−フッ化物イオン(18F−)の適当な供給源との反応によって前駆体化合物を一段階で標識できる。この方法のためには、前駆体化合物は、放射性フッ素化が化合物上の特定の部位で起こるように選択的に化学保護されるのが普通である。好適な保護基は既に前述したものである。
【0053】
放射性同位体が18Fである場合、X1又はR1は以下のいずれかを含むことが好ましい。
(i)求核置換のためのアルキルハライド又はアルキルスルホネート(例えば、アルキルブロミド、アルキルメシレート又はアルキルトシレート)、或いは
(ii)(例えば、18F(CH2)3OMs又は18F(CH2)3OBrでヒドロキシル基をO−アルキル化することによる18Fの導入のための)ヒドロキシル。
【0054】
本発明の若干の18Fインビボイメージング剤を得るための一般反応スキームを以下に示す。
【0055】
【化5】
式中、Xは式Iに関して定義した通りであり、nは0〜5である。RTは室温を表し、OMsはメシレートを表す。
【0056】
本発明の若干の18Fインビボイメージング剤を得るための別の一般反応スキームを以下に示す。
【0057】
【化6】
式中、Xは式Iに関して定義した通りであり、nは0〜5であり、OTsはトシレートを表す。
【0058】
11C標識PETトレーサー化合物は、前駆体化合物を11C−ヨウ化メチルと反応させることで合成できる。11Cの半減期は20.4分にすぎないので、11C−ヨウ化メチル中間体は高い比放射能を有すること、したがってできるだけ速い反応プロセスを用いてそれを製造することが重要である。かかる11C標識技法に関する徹底的な総説を、Antoni et al,“Aspects on the Synthesis of 11C−Labelled Compounds”in Handbook of Radiopharmaceuticals,Ed.M.J.Welch and C.S.Redvanly(2003,John Wiley and Sons)に見出すことができる。
【0059】
本発明のインビボイメージング剤が11Cで標識される場合、11Cはカルボニル炭素である。したがってこれは、11Cが式Iのカルボニル炭素の位置に存在し得るか、或いは別法としてXがC1−6アルキルアミドである場合にXの位置に存在し得ることを意味する。
【0060】
式Iの11C標識インビボイメージング剤は、以下の反応スキームを用いて得ることができる。
【0061】
【化7】
式中、式IIb及び式IdのR3、X3及びY3は、式IのR、X及びYに関して記載した通りであり、
Z3は、遷移金属触媒に適した基質(例えば、水素、ハロゲン化物、ボロン酸、OTf又は有機スズ)である。
【0062】
13N標識化合物の合成方法は、Clark and Aigbirhio(“Chemistry of Nitrogen−13 and Oxygen−15”in“Handbook of Radiopharmaceuticals”;2003 Wiley:Welch and Redvanly,Eds.)によって記載されている。例えば、式Iのインビボイメージング剤は、適当な前駆体化合物中のハロゲンを13N標識ジエチルアミンで求核置換して所望のアミドを生成することで得ることができる。
【0063】
イメージング成分が放射性ヨウ素である場合、好ましい前駆体化合物は、求電子又は求核ヨウ素化を受ける誘導体或いは標識アルデヒド又はケトンとの縮合を受ける誘導体を含むものである。第1のカテゴリーの例は以下の(a)及び(b)である。
(a)トリアルキルスタンナン(例えば、トリメチルスタンニル又はトリブチルスタンニル)、トリアルキルシラン(例えば、トリメチルシリル)或いは有機ホウ素化合物(例えば、ボロネートエステル又はオルガノトリフルオロボレート)のような有機金属誘導体。
(b)求電子ヨウ素化に向けて活性化された芳香環(例えば、フェノール類)及び求核ヨウ素化に向けて活性化された芳香環(例えば、アリールヨードニウム塩、アリールジアゾニウム塩、アリールトリアルキルアンモニウム塩又はニトロアリール誘導体)。
【0064】
放射性ヨウ素化のために好ましくは、前駆体化合物は、(放射性ヨウ素交換を可能にするための)アリールヨージド又はブロミド、活性化前駆体化合物アリール環(例えば、フェノール基)、有機金属前駆体化合物(例えば、トリアルキルスズ、トリアルキルシリル又は有機ホウ素化合物)、或いはトリアゼンのような有機前駆体化合物又は求核置換のための良好な脱離基(例えば、ヨードニウム塩)からなる。放射性ヨウ素を有機分子中に導入するための前駆体化合物及び方法は、Bolton(J.Lab.Comp.Radiopharm.2002;45:485−528)によって記載されている。放射性ヨウ素をタンパク質中に導入するための前駆体化合物及び方法は、Wilbur(Bioconj.Chem.1992;3(6):433−470)によって記載されている。好適なボロネートエステル有機ホウ素化合物及びその製法は、Kabalka et al(Nucl.Med.Biol.2002;29:841−843及び2003;30:369−373)によって記載されている。好適なオルガノトリフルオロボレート及びその製法は、Kabalka et al(Nucl.Med.Biol.2004;31:935−938)によって記載されている。放射性ヨウ素化用の好ましい前駆体化合物は有機金属前駆体化合物からなり、最も好ましくはトリアルキルスズからなる。
【0065】
放射性ヨウ素を結合できるアリール基の例を以下に示す。
【0066】
【化8】
これらの基はいずれも、芳香環上への容易な放射性ヨウ素置換を可能にする置換基を含んでいる。放射性ヨウ素を含む別の置換基は、例えば次式のように、放射性ハロゲン交換による直接ヨウ素化で合成することができる。
【0067】
【化9】
放射性同位体が放射性ヨウ素である場合、式IIのX1は、そけが結合した芳香族基と共に、
(i)有機金属誘導体又は有機ホウ素化合物で置換された芳香環を形成するか、
(ii)求電子ヨウ素化に向けて活性化された芳香環(例えば、フェノール類)を形成するか、或いは
(iii)求核放射性ヨウ素化に向けて活性化された芳香環(例えば、アリールヨードニウム塩、アリールジアゾニウム塩、アリールトリアルキルアンモニウム塩又はニトロアリール誘導体)を形成する。
【0068】
これらの前駆体化合物は、放射性ヨウ素置換により、本発明の放射性ヨウ素化インビボイメージング剤に容易に転化される。
【0069】
放射性臭素化は、放射性ヨウ素化に関して上記に記載したものと同様な方法によって達成できる。Kabalka and Varmaは、放射性臭素化化合物を含む放射性ハロゲン化化合物を合成するための様々な方法を総説している(Tetrahedron 1989;45(21):6601−21)。
【0070】
本発明の前駆体化合物は、理想的には無菌で非発熱性の形態で供給される。したがって前駆体化合物は、インビボイメージング剤を哺乳動物への投与に適した生体適合性キャリヤーと共に含んでなる医薬組成物の製造のために使用できる。前駆体化合物はまた、かかる医薬組成物を製造するためのキット中に一成分として含めるためにも適している。
【0071】
好ましい実施形態では、前駆体化合物はキット又は自動化合成装置で使用するように設計されたカセットの一部として溶液状態で供給される。これらの態様は、本発明の追加の態様に関連して以下に一層詳しく論議される。
【0072】
別の好ましい実施形態では、前駆体化合物は固相に結合されている。前駆体化合物は、好ましくは固体担体マトリックスに共有結合した状態で供給される。このようにすれば、所望の生成物は溶液状態で生成される一方、出発原料及び不純物は固相に結合したままに保たれる。かかる系の例としては、18F−フッ化物による固相求電子フッ素化用の前駆体化合物が国際公開第03/002489号に記載されており、18F−フッ化物による固相求核フッ素化用の前駆体化合物が国際公開第03/002157号に記載されている。
【0073】
インビボイメージング剤の製造方法
さらに別の態様では、本発明は、本発明のインビボイメージング剤の製造方法であって、
(i)上記に定義した式IIの前駆体化合物を用意する段階、
(ii)上記に定義した前記放射性同位体の適当な供給源を用意する段階、並びに
(iii)段階(i)の前駆体化合物を段階(ii)の放射性同位体と反応させて本発明のインビボイメージング剤を得る段階
を含んでなる方法を提供する。
【0074】
段階(i)では、前駆体化合物は、前駆体化合物の説明において上記に記載したように、キット又は自動化合成装置で使用するのに適したカセット中に溶液状態で供給するか、或いは別法として固体担体に結合した状態で供給することができる。かかるキット及びカセットは本発明の追加の態様をなしており、以下に一層詳しく論議される。
【0075】
放射性同位体の適当な供給源は、本発明の前駆体化合物に関連して上記に記載した通りである。
【0076】
前駆体化合物を放射性同位体と「反応させる」段階は、できるだけ高い放射化学収率(RCY)で所望のインビボイメージング剤を生成するのに適した反応条件下で2種の反応体を合わせることを含んでいる。本発明のインビボイメージング剤を得るための特定の合成経路は、以下の実験セクションに示される。
【0077】
インビボイメージング剤の製造用キット
さらに別の態様では、本発明は、本発明のインビボイメージング剤を製造するためのキットを提供する。前記キットは上記に記載された式IIの前駆体化合物を含む結果、放射性同位体の無菌供給源との反応により最小数の操作で所望のインビボイメージング剤が得られる。かかる考慮事項は、放射性同位体が比較的短い半減期を有する場合において、取扱いを容易にし、したがって放射線薬剤師に対する放射線量を低減させるため特に重要である。前駆体化合物は好ましくは凍結乾燥形態でキット中に存在しており、かかるキットの再構成用反応媒質は好ましくは生体適合性キャリヤーである。
【0078】
「生体適合性キャリヤー」は、組成物が生理学的に認容され得るようにして(即ち、毒性又は過度の不快感なしに哺乳動物体に投与できるようにして)インビボイメージング剤を懸濁又は溶解するための流体(特に液体)である。生体適合性キャリヤーは、好適には、無菌のパイロジェンフリー注射用水、(有利には注射用の最終生成物が等張性又は非低張性になるように平衡させ得る)食塩水のような水溶液、或いは1種以上の張度調整物質(例えば、血漿陽イオンと生体適合性対イオンとの塩)、糖(例えば、グルコース又はスクロース)、糖アルコール(例えば、ソルビトール又はマンニトール)、グリコール(例えば、グリセロール)又は他の非イオン性ポリオール物質(例えば、ポリエチレングリコール、プロピレングリコールなど)の水溶液のような注射可能なキャリヤー液体である。生体適合性キャリヤーはまた、エタノールのような生体適合性有機溶媒を含んでいてもよい。かかる有機溶媒は、親油性の高い化合物又は配合物を可溶化するために有用である。好ましくは、生体適合性キャリヤーはパイロジェンフリー注射用水、等張食塩水又はエタノール水溶液である。静脈内注射用生体適合性キャリヤーのpHは好適には4.0〜10.5の範囲内にある。
【0079】
本発明のキットでは、前駆体化合物は好ましくは、注射器による溶液の追加及び抜取りを許しながら、無菌保全性及び/又は放射能安全性の維持、さらに任意には不活性ヘッドスペースガス(例えば、窒素又はアルゴン)の維持を可能にする密封容器に入れて供給される。好ましい密封容器は、気密クロージャーを(通例はアルミニウムからなる)オーバーシールと共にクリンプ加工した隔壁密封バイアルである。かかる密封容器は、例えばヘッドスペースガスの変更又は溶液のガス抜きのために所望される場合、クロージャーが真空に耐え得るという追加の利点を有している。
【0080】
キット中に使用する場合、前駆体化合物の好ましい実施形態は、本明細書で記載した通りである。
【0081】
キット中に使用するための前駆体化合物を無菌製造条件下で使用すれば、所望の無菌で非発熱性の材料を得ることができる。別法として、前駆体化合物を非無菌条件下で使用し、次いで例えばγ線照射、オートクレーブ処理、乾熱又は(例えば、エチレンオキシドによる)化学処理を用いる終末滅菌を施すこともできる。好ましくは、前駆体化合物は無菌で非発熱性の形態で供給される。最も好ましくは、無菌で非発熱性の前駆体化合物は上述したような密封容器に入れて供給される。
【0082】
好ましくは、試験間での汚染の可能性を最小限に抑えると共に無菌性及び品質保証を確実にするため、キットのすべての構成要素は使い捨てである。
【0083】
好ましい実施形態では、キットは、以下に一層詳しく記載されるように、適宜に改造された自動化合成装置に挿入し得るカセットを含むことができる。かかるキットは、通例はフッ化物イオンによるフッ素化のための手段を含み、また不要のフッ化物イオンを除去するためのカラムを含むことができる。合成のために必要な試薬、溶媒及び他の消耗品もまた、濃度、体積、送出時間などに関する最終ユーザーの要求条件を満たすように自動化合成装置を運転させるソフトウェアを保持したコンパクトディスクのようなデータ媒体と共に含めることができる。
【0084】
現在、PETイメージング用の[18F]放射性トレーサーはしばしば自動化放射合成装置で簡便に製造されている。かかる装置には、Tracerlab及びFastlab(GE Healthcare社)を始めとするいくつかの市販例が存在している。かかる装置は、通常、放射化学を実施するために(しばしば使い捨ての)「カセット」を含んでおり、これを装置に取り付けることで放射合成が実施される。普通、カセットは流体通路、反応器、及び試薬バイアルを受け入れるためのポート並びに放射合成後の清掃段階で使用される固相抽出カートリッジを含んでいる。
【0085】
したがって本発明は、別の態様では、本明細書で前述した密封容器に入れた前駆体化合物を含んでなる自動化合成装置用カセットを提供する。本発明はまた、本明細書で定義したインビボイメージング剤の自動化合成のためのカセットであって、
(i)本明細書で定義した前駆体化合物を含む容器、及び
(ii)本明細書で定義した放射性同位体の適当な供給源を用いて容器を溶出するための手段
を含んでなるカセットを提供する。
【0086】
かかるカセットはさらに、
(iii)過剰の放射性標識を除去するためのイオン交換カートリッジ、及び任意には
(iv)得られた放射性標識生成物を脱保護して本明細書で定義したインビボイメージング剤を生成するためのカートリッジ
を含むことができる。
【0087】
放射性医薬組成物
さらに別の態様では、本発明は「放射性医薬組成物」を提供するが、これは本発明のインビボイメージング剤を、哺乳動物への投与に適した形態の生体適合性キャリヤーと共に含んでいる。生体適合性キャリヤーは、本発明のキットに関連して上記に定義した通りである。
【0088】
かかる放射性医薬組成物は非経口的に(即ち、注射によって)投与でき、最も好ましくは水溶液である。かかる組成物は、緩衝剤、薬学的に許容される可溶化剤(例えば、シクロデキストリン或いはPluronic、Tween又はリン脂質のような界面活性剤)、薬学的に許容される安定剤又は酸化防止剤(例えば、アスコルビン酸、ゲンチジン酸又はパラアミノ安息香酸)のような追加成分を任意に含み得る。本発明のインビボイメージング剤が放射性医薬組成物として提供される場合、前記インビボイメージング剤の製造方法はさらに、放射性医薬組成物を得るために必要な段階(例えば、有機溶媒の除去、生体適合性緩衝剤及び任意の追加成分の添加)を含むことができる。非経口的投与のためには、放射性医薬組成物が無菌性かつ無発熱原性であることを保証するための手段を講じることも必要である。
【0089】
インビボイメージング方法
さらに別の態様では、本発明は、被験体におけるPBR発現の体内分布及び/又は程度を決定するためのインビボイメージング方法であって、
(i)本明細書で定義したインビボイメージング剤を前記被験体に投与する段階、
(ii)前記イメージング剤を前記被験体中のPBRに結合させる段階、
(iii)前記イメージング剤の放射性同位体から放出される信号をインビボイメージング技法によって検出する段階、
(iv)前記信号の位置及び/又は量を表す画像を生成する段階、並びに
(v)前記被験体におけるPBR発現の分布及び程度を決定する段階であって、前記発現は前記インビボイメージング剤から放出される前記信号と直接に相関している段階
を含んでなるインビボイメージング方法を提供する。
【0090】
本発明のインビボイメージング方法に関しては、インビボイメージング剤は本明細書でおいて前記に定義した通りである。
【0091】
インビボイメージング剤を「投与する」段階は、好ましくは非経口的に実施され、最も好ましくは静脈内に実施される。静脈内経路は、インビボイメージング剤を被験体の身体全域に送達するため、したがって血液脳関門(BBB)を横切って前記被験体で発現されたPBRに接触させるための最も効率的な方法である。本発明のインビボイメージング剤は、好ましくは本明細書で定義した本発明の医薬組成物として投与される。
【0092】
投与段階後かつ検出段階前に、インビボイメージング剤をPBRに結合させる。例えば、被験体がインタクトな哺乳動物である場合、インビボイメージング剤は哺乳動物の身体を通って動的に移動し、体内の様々な組織に接触する。ひとたびインビボイメージング剤がPBRに接触すれば、特異的な相互作用が起こる結果、PBRをもった組織からのインビボイメージング剤のクリアランスは、PBRをもたない組織又はPBRの少ない組織よりも長い時間がかかる。一定の時点に達すれば、PBRをもった組織に結合したインビボイメージング剤とPBRをもたない組織又はPBRの少ない組織に結合したインビボイメージング剤との比の結果として、PBRに特異的に結合したインビボイメージング剤の検出が可能となる。理想的なかかる比は約2:1である。
【0093】
本発明の方法の「検出」段階は、放射性同位体から放出される信号を、前記信号に対して感受性を有する検出器によって検出することを含んでいる。この検出段階はまた、信号データの取得として理解することもできる。単光子放出コンピューター断層撮影(SPECT)及び陽電子放出断層撮影(PET)が、本発明の方法で使用するための最も好適なインビボイメージング技法である。PETが、本発明の方法で使用するための好ましいインビボイメージング技法である。
【0094】
本発明の方法の「生成」段階は、取得された信号データに再構築アルゴリズムを適用してデータセットを得るコンピューターによって実施される。次いで、このデータセットを操作することで、前記放射性同位体から放出される信号の位置及び/又は量を示す画像が生成される。放出される信号はPBRの発現と直接に相関する結果、生成された画像を評価することで「決定」段階を行うことができる。
【0095】
本発明の「被験体」は、任意のヒト又は動物被験体であり得る。好ましくは、本発明の被験体は哺乳動物である。最も好ましくは、前記被験体はインタクトな哺乳動物生体である。特に好ましい実施形態では、本発明の被験体はヒトである。かかるイメージング方法は、健常被験体或いはPBRの異常発現に関連する病的状態(「PBR状態」)を有することが知られ又は疑われる被験体においてPBRを検査するために使用できる。好ましくは、前記方法はPBR状態を有することが疑われる被験体のインビボイメージングに関し、したがって前記状態の診断方法において有用である。インビボイメージングが役に立つかかる状態の例には、神経炎症が存在するパーキンソン病、多発性硬化症、アルツハイマー病及びハンチントン病のような神経疾患がある。本発明の化合物によるイメージングが有用であり得る他のPBR状態には、ニューロパシー性疼痛、関節炎、喘息、アテローム性動脈硬化症、並びに結腸直腸癌及び乳癌のような悪性疾患がある。本発明のインビボイメージング剤は、その良好な脳内取込みのため、中枢神経系(CNS)のインビボイメージングに特に適している。
【0096】
別の実施形態では、本発明のインビボイメージング方法は前記被験体に関する治療計画の進行中に繰り返して実施することができ、前記計画はPBR状態と戦うための薬物の投与を含んでいる。例えば、本発明のインビボイメージング方法は、PBR状態と戦うための薬物による治療前、治療中及び治療後に実施できる。このようによすれば、前記治療の効果を経時的なモニターすることができる。この実施形態のために好ましくは、インビボイメージング技法はPETである。PETは優れた感度及び分解能を有する結果、病変部における比較的小さい変化でも経時的に観察でき、これは治療モニタリングのために有利である。PETスキャナーは、日常的にピコモル範囲内の放射能濃度を測定している。現在、マイクロPETスキャナーは約1mmの空間分解能に接近しているが、臨床スキャナーは約4〜5mmである。
【0097】
さらに別の態様では、本発明はPBR状態の診断方法を提供する。本発明の診断方法は、上記に定義したインビボイメージング方法を、PBR発現の分布及び程度を特定の臨床像に帰因させる追加段階(vi)(即ち、演繹的な医学的決断段階)と共に含んでいる。
【0098】
別の態様では、本発明は、本明細書中で定義した診断方法で使用するための、本明細書中で定義したインビボイメージング剤を提供する。
【0099】
さらに別の態様では、本発明は、本明細書中で定義した診断方法で使用するための本明細書中で定義した放射性医薬組成物の製造で使用するための、本明細書中で定義したインビボイメージング剤を提供する。
【実施例】
【0100】
実施例の簡単な説明
すべての試薬はSigma Aldrich社から入手した。
【0101】
実施例1乃至実施例6は、本発明の様々なインビボイメージング剤の非放射性バージョンの合成法を記載している。
【0102】
実施例7乃至実施例9は、本発明の18F標識インビボイメージング剤を得るための方法を記載している。
【0103】
実施例10は、本発明のイメージング剤のPBR親和性を測定するために使用したインビトロ効力アッセイを記載している。
【0104】
実施例11は、動物体内分布試験の実施方法を記載している。
【0105】
実施例12は、顔面神経軸索切断動物モデル及びオートラジオグラフィー試験におけるそれの使用を記載している。
【0106】
実施例中で使用される略語のリスト
DCM ジクロロメタン
DMF ジメチルホルムアミド
DMSO ジメチルスルホキシド
EtOAc 酢酸エチル
FNA 顔面神経軸索切断
g グラム
h 時間
HRMS 高分解能質量分析法
K222 クリプトフィックス2.2.2
M モル濃度=溶質のモル数/溶液1リットル
MHz メガヘルツ
ml ミリリットル
mmol ミリモル
N 規定度=当量数/溶液1L
NMR 核磁気共鳴
PBR 末梢ベンゾジアゼピンレセプター
RT 室温
実施例1:(±)−11−(2−フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤1)の製造
実施例1(i):(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド
【0107】
【化10】
T.Okubo et al(Bioorg.Med.Chem.2004;12:3569−3580)に記載されているようにして製造した(±)−4−オキソ−チオクロマン−2−カルボン酸(10.4g、50mmol)の乾燥DCM(100ml)溶液を、塩化オキサリル(12.6g、100mmol)及び1滴のDMFと共に窒素雰囲気下において室温で18時間撹拌した。次いで、反応物を真空中で蒸発させてガム状にし、次いでDCM(100ml)に再溶解し、氷浴上で0℃に冷却し、撹拌しながらDCM(20ml)中のジエチルアミン(8.03g、110mmol)を1時間かけて滴下することで処理した。反応物を1時間かけて室温まで放温し、10%炭酸カリウム水溶液(100ml)を添加し、反応混合物を激しく撹拌した。DCM溶液を分離した。水溶液をDCMの追加バッチ(100ml)で2回抽出し、合わせた抽出液を硫酸マグネシウム上で乾燥した。DCM溶液を真空中で濃縮して暗緑色の油状物を得たが、これは静置することで結晶化した。結晶質固体をジエチルエーテル(50ml)でトリチュレートして濾過することで、標記化合物(8.57g、65%)を淡緑色の固体として得た。
【0108】
1H NMR(300MHz,CDCl3)δ 1.06(t,J=7.1Hz,3H)、1.23(t,J=7.1Hz,3H)、3.0−3.5(m,6H)、4.25(m,1H)、7.15−7.21(m,2H)、7.32−7.39(m,1H)、8.10−8.14(m,1H)。
【0109】
13C NMR(75MHz,CDCl3)δ 12.9、14.8、40.1、40.7、42.3、42.5、125.8、127.2、128.7、130.8、133.4、137.9、167.9、193.1。
【0110】
実施例1(ii):(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0111】
【化11】
(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(1.32g、5.0mmol、実施例1(i))及び4−メトキシフェニルヒドラジン塩酸塩(0.87g、5.0mmol)のエタノール(10ml)溶液に、濃硫酸(0.73ml、1.35g、13.8mmol)を窒素下で添加した。反応混合物を24時間加熱還流した。冷却後、反応混合物を濾過し、固体をエタノールで洗浄し、(45℃で)真空乾燥することで、標記化合物(1.05g、57%)を淡黄色の固体として得た。
【0112】
1H NMR(300MHz,DMSO−d6)δ 0.97(t,J=6.8Hz,3H)、1.28(t,J=6.8Hz,3H)、3.25(m,2H)、3.60(m,2H)、3.74(s,3H)、5.59(s,1H)、6.80(m,2H)、7.10−7.35(m,4H)、7.75(d,J=7.3Hz,1H)、11.52(s,1H,NH)。
【0113】
13C NMR(75MHz,DMSO−d6)δ 10.5、12.7、32.7、37.9、39.5、53.0、97.6、103.3、109.87、109.92、120.3、123.5、123.8、124.3、124.7、124.9、127.8、129.4、131.8、151.3、166.2。
【0114】
m/z(ES+)367.1(M+H)。
【0115】
実施例1(iii):(±)−11−(2−フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0116】
【化12】
(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(150mg、0.41mmol、実施例1(ii))の無水DMF(4ml)溶液に、L.Cronin et al(J.Org.Chem.2004;69:5934−5946)に記載されているようにして製造した2−フルオロエチルトシレート(166mg、0.82mmol)を添加し、次いで鉱油中の水素化ナトリウム60%分散液(34mg、0.82mmol)を窒素下で添加した。反応混合物を80℃で1時間加熱した。冷却後、溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。5〜10%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製した。粗生成物をエーテル/石油スピリットで奪活し、濾過し、(45℃で)真空乾燥することで、標記化合物(77mg、46%)を淡褐色の固体として得た。
【0117】
1H NMR(300MHz,CDCl3)δ 1.12(t,J=7.0Hz,3H)、1.36(t,J=7.0Hz,3H)、3.25−3.70(m,4H)、3.83(s,3H)、4.45−4.70(m,2H)、4.80(t,J=5.2Hz,1H)、4.96(t,J=5.2Hz,1H)、5.09(s,1H)、6.84−6.93(m,2H)、7.13−7.32(m,3H)、7.46(m,1H)、7.58(d,J=8.0Hz,1H)。
【0118】
13C NMR(75MHz,CDCl3)δ 12.9、14.9、37.3、41.1、42.5、45.5、45.8、55.9、81.2、83.5、100.4、110.1、111.09、111.12、112.8、124.31、124.35、125.2、126.5、127.1、127.6、128.8、132.2、134.4、137.0、154.8、168.0。
【0119】
19F NMR(282MHz,CDCl3)δ −219.4、−219.5、−219.6、−219.65、−219.73、−219.9、−219.9。
【0120】
m/z(ES+)413.1(M+H)。
【0121】
実施例2:(±)−11−(2−フルオロエチル)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤3)の製造
実施例2(i):(±)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0122】
【化13】
この化合物は、4−メトキシフェニルヒドラジン塩酸塩の代わりに2−メトキシフェニルヒドラジン塩酸塩を使用した点を除き、実施例1(ii)に関して記載したようにして製造した。化合物は40%の収率で得られた。
【0123】
m/z(ES+)367.0(M+H)。
【0124】
実施例2(ii):(±)−11−(2−フルオロエチル)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0125】
【化14】
この化合物は、(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの代わりに(±)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例2(i))を使用した点を除き、実施例1(iii)に関して記載したようにして製造した。再結晶(エーテル)後、化合物は白色の固体として10%の収率で得られた。
【0126】
1H NMR(300MHz,CDCl3)δ 1.09(t,J=7.0Hz,3H)、1.35(t,J=7.0Hz,3H)、3.25−3.67(m,4H)、3.95(s,3H)、4.70−4.96(m,4H)、5.04(s,1H)、6.67(m,1H)、7.04(m,2H)、7.16(m,1H)、7.29(m,1H)、7.45(m,1H)、7.77(m,1H)。
【0127】
m/z(ES+)413.1(M+H)。
【0128】
実施例3:(±)−4−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤4)の製造
実施例3(i):(±)−8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸
【0129】
【化15】
丸底フラスコ内において、トルエン(12mL)中の2−フルオロチオフェノール(5.0g、39.0mmol、4.16mL)及びフラン−2,5−ジオン(3.82g、39.0mmol)を50℃で40分間撹拌した。次いで、反応温度が60℃を超えないようにしながら、トルエン(5mL)中のトリエチルアミン(100μl)を10分かけて添加した。次いで、反応物を70℃で20分間加熱した。次いで、反応物を高真空下で濃縮することで、粗生成物を油状物として得た。この物質をDCM(75mL)に溶解し、氷浴上で冷却し、温度を10℃未満に保つようにしながら三塩化アルミニウム(7.78g、58.5mmol)を少量ずつ加えて処理した。反応物をRTまで温めたところ、塩化水素ガスの激しい発生が起こり、反応物は非常に粘稠になると共に赤色に変わった。RTで1.5時間撹拌した後、反応混合物をDCM(50mL)で希釈してその粘度を低下させ、2L三角フラスコ内の激しく撹拌した濃塩酸(30mL)及び氷(30g)中にゆっくりと注ぎ込んだ。反応物を激しく撹拌し、追加のDCM(500mL)及びイソプロピルアルコール(50mL)で希釈して晶出した固体を溶解した。DCM層を分離し、硫酸マグネシウム上で乾燥し、真空中で濃縮して褐色の固体を得た。粗生成物を精製し、ジエチルエーテルでトリチュレートし、クリーム色の固体を濾過により集めて2.5g(28%)の8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸を得た。1H NMR(300MHz;DMSO−d3):δ 3.04−3.20(2H,m,3−H)、4.51(1H,dd,J=4及び6Hz,2−H)、7.26−7.34(1H,m,6−H)、7.45−7.52(1H,m,7−H)、7.82(1H,dd,J=1及び8Hz,5H)。13C NMR(75MHz;DMSO−d3):δ 40.5、40.7、119.8、120.1、123.88、123.92、126.0、126.1、131.9、156.1、159.2、171.5、191.2、191.3。
【0130】
実施例3(ii):(±)−8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド
【0131】
【化16】
乾燥DCM(50ml)中の8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸(2.5g、11.1mmol)を、反応を触媒するための塩化オキサリル(2.81g、22.1mmol、1.93mL)及び1滴のDMFと共に窒素雰囲気下において室温で18時間撹拌した。酸は最初は不溶性であったが、反応するに従って溶解し、2時間後にはオレンジ色の透明溶液となり、次いで24時間後には黒色に変わった。次いで、反応物を真空中で蒸発させてガム状にして過剰の塩化オキサリルを除去し、CDCl3中で1H NMR及び13C NMRを実施して反応の完了を確認した。次いで、反応物をDCM(50ml)に再溶解し、氷浴上で0℃に冷却し、撹拌しながらDCM(20ml)中のジエチルアミン(1.66g、22.7mmol、2.05mL)を1時間かけて滴下することで処理した。反応物を1時間かけて室温まで放温した。次いで、反応物を5%炭酸カリウム溶液(100ml)の添加によって奪活し、反応混合物を激しく撹拌した。DCM溶液を分離し、硫酸マグネシウム上で乾燥した。DCMの追加バッチ(100ml)を水溶液と共に振盪し、次いで分離して硫酸マグネシウム上で乾燥することを2回繰り返した。合わせたDCM溶液を真空中で濃縮して褐色の固体を得た。粗固体を酢酸エチル及びガソリンからの高温再結晶によって精製することで、1.76g(56%)の8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミドを黄色の結晶として得た。1H NMR(300MHz;CDCl3):δ 1.07(3H,t,J=7Hz,N(CH2CH3)2)、1.26(3H,t,J=7Hz,N(CH2CH3)2)、3.02−3.55(6H,m,2−H及びN(CH2CH3)2)、4.24−4.27(1H,m,2−H)、7.15−7.19(2H,m,6−H及び7−H)、7.93−7.97(1H,m,5−H)。
【0132】
LC−MS:C14H16FNO2Sに関するm/z計算値、281.1;実測値、282.0(M+H)+。
【0133】
実施例3(iii):(±)−4−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0134】
【化17】
エタノール(10mL)及び硫酸(濃、0.8mL)中の8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(1.7g、6.0mmol)及びフェニルヒドラジン(0.65g、6.0mmol、0.6mL)を、還流させながら一晩撹拌した。冷却後、反応物を濾過し、白色の固体を集めて1.4g(80%)の粗物質(純度90%)を得た。粗固体(500mg)をエタノールからの高温再結晶によって精製することで、277mg(13%)の4−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の結晶として得た。構造は1H NMRによって確認した。1H NMR(300MHz;DMSO−d6):δ 0.96(3H,t,J=7Hz,N(CH2CH3)2)、1.30(3H,t,J=9Hz,N(CH2CH3)2)、3.19−3.25(2H,m,N(CH2CH3)2)、3.56−3.66(2H,m,N(CH2CH3)2)、5.76(1H,s,6−H)、7.02−7.45(6H,m,ArH)、7.65(1H,dd,J=1及び6Hz,ArH)、11.8(1H,s,NH)。
【0135】
LC−MS:C20H19FN2OSに関するm/zの計算値、354.2;実測値、355.0(M+H)+。
【0136】
実施例3(iv):(±)−4−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0137】
【化18】
(±)−4−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(0.10g、0.28mmol)を窒素下において室温で乾燥DMF(6mL)に溶解した。フルオロエチルトシレート(0.12g、0.12mmol)を添加し、次いでNaH(0.02g、0.56mmol、油中60%)を添加した。反応物を80℃で1時間加熱した。溶媒を減圧下で除去し、残留物をDCMに溶解し、水で洗浄した。有機物をMgSO4上で乾燥し、濾過し、蒸発乾固させた。粗物質をメタノールから晶出させることで、34.4mg(30%)の4−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得た。1H NMR(300MHz,DMSO−d6)δ 0.94(3H,t,J=7Hz,N(CH2CH3)2)、1.29(3H,t,J=7Hz,N(CH2CH3)2)、3.14−3.26(2H,m,N(CH2CH3)2)、3.55−3.65(2H,m,N(CH2CH3)2)、4.65−4.95(4H,m,NCH2CH2F)、5.62(1H,s,6−H)、7.12−7.37(4H,m,ArH)、7.48(1H,d,J=9Hz,ArH)、7.61−7.68(2H,m,ArH)。
【0138】
LC−MS:C22H22F2N2OSに関するm/zの計算値、401.1;実測値、401.1(M+H)+。
【0139】
実施例4:(±)−3−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤5)の製造
実施例4(i):(±)−7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸
【0140】
【化19】
丸底フラスコ内において、トルエン(12mL)中の3−フルオロチオフェノール(10.0g、71.3mmol、8.85mL)及びフラン−2,5−ジオン(7.0g、71.3mmol)を50℃で40分間撹拌した。次いで、反応温度が60℃を超えないようにしながら、トルエン(1mL)中のトリエチルアミン(26μl)を10分かけて添加した。次いで、反応物を70℃で20分間加熱した。次いで、反応物を高真空下で濃縮することで、粗生成物を油状物として得た。この物質をDCM(75mL)に溶解し、氷浴上で冷却し、温度を10℃未満に保つようにしながら三塩化アルミニウム(7.78g、58.5mmol)を少量ずつ加えて処理した。反応物をRTまで温めたところ、塩化水素ガスの激しい発生が起こり、反応物は非常に粘稠になると共に赤色に変わった。室温で1.5時間撹拌した後、反応混合物をDCM(50mL)で希釈してその粘度を低下させ、2L三角フラスコ内の激しく撹拌した濃塩酸(30mL)及び氷(30g)中にゆっくりと注ぎ込んだ。反応物を激しく撹拌し、追加のDCM(500mL)及びイソプロピルアルコール(50mL)で希釈して晶出した固体を溶解した。DCM層を分離し、硫酸マグネシウム上で乾燥し、真空中で濃縮して褐色の固体を得た。固体をジエチルエーテルでトリチュレートし、次いで濾過することで、4.2g(48%)の7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸をクリーム色の固体として得た。1H NMR(300MHz;DMSO−d3):δ 3.00−3.16(2H,m,3−H)、4.44(1H,dd,J=5及び10Hz,2−H)、7.08(1H,td,J1=3及び9Hz,6−H)、7.30(1H,dd,J=5及び10Hz,ArH)、8.01(1H,dd,J1=5及び10Hz,ArH)。13C NMR(75MHz;DMSO−d3):δ 38.0、39.6、111.1、111.3、111.5、111.8、125.0、125.1、129.0、129.2、139.6、139.7、160.9、164.3、169.5、188.9。
【0141】
実施例4(ii):(±)−7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド
【0142】
【化20】
乾燥DCM(50ml)中の7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸(4g、17.7mmol)を、反応を触媒するための塩化オキサリル(4.49g、35.4mmol、3.1mL)及び1滴のDMFと共に窒素雰囲気下において室温で18時間撹拌した。酸は最初は不溶性であったが、反応するに従って溶解し、2時間後にはオレンジ色の透明溶液となり、次いで24時間後には黒色に変わった。次いで、反応物を真空中で蒸発させてガム状にして過剰の塩化オキサリルを除去し、CDCl3中で1H NMR及び13C NMRを実施して反応の完了を確認した。次いで、反応物をDCM(50ml)に再溶解し、氷浴上で0℃に冷却し、撹拌しながらDCM(10ml)中のジエチルアミンを1時間かけて滴下することで処理した。反応物を1時間かけて室温まで放温した。次いで、反応物を5%炭酸カリウム溶液(50ml)の添加によって奪活し、反応混合物を激しく撹拌した。DCM溶液を分離し、硫酸マグネシウム上で乾燥した。DCMの追加バッチ(50ml)を水溶液と共に振盪し、次いで分離して硫酸マグネシウム上で乾燥することを2回繰り返した。合わせたDCM溶液を真空中で濃縮して褐色の固体を得、これを静置して結晶化させることで、5.03g(クアント(quant))の7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミドを得た。構造は1H NMRによって確認した。1H NMR(300MHz;CDCl3):δ 1.07(3H,t,J=7Hz,N(CH2CH3)2)、1.24(3H,t,J=7Hz,N(CH2CH3)2)、2.99−3.50(6H,m,2−H及びN(CH2CH3)2)、4.24−4.27(1H,m,2−H)、6.83−6.94(2H,m,6−H及び8−H)、8.15(1H,dd,J=6及び9Hz,5−H)。
【0143】
LC−MS:C14H16FNO2Sに関するm/zの計算値、281.1;実測値、282.0(M+H)+。
【0144】
実施例4(iii):(±)−3−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0145】
【化21】
エタノール(10mL)及び硫酸(濃、1.2mL)中の7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(2.5g、8.9mmol)及びフェニルヒドラジン(0.96g、8.9mmol、0.9mL)を、還流させながら一晩撹拌した。粗固体をエタノールからの高温再結晶によって精製することで、1.49g(47%)の3−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の結晶として得た。1H NMR(300MHz;DMSO−d6):δ 0.96(3H,t,J=6Hz,N(CH2CH3)2)、1.29(3H,t,J=6Hz,N(CH2CH3)2)、3.19−3.25(2H,m,N(CH2CH3)2)、3.55−3.61(2H,m,N(CH2CH3)2)、5.66(1H,s,6−H)、7.03(1H,td,J=1及び8Hz,ArH)、7.09−7.18(2H,m,ArH)、7.25(1H,dd,J=3及び9Hz,ArH)、7.35(1H,d,J=8Hz,ArH)、7.41(1H,d,J=8Hz,ArH)、7.81(1H,dd,J=6及び9Hz,ArH)、11.68(1H,s,NH)。
【0146】
LC−MS:C20H19FN2OSに関するm/zの計算値、352.1;実測値、353.2(M+H)+。
【0147】
実施例4(iv):(±)−3−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0148】
【化22】
(±)−3−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(0.20g、0.56mmol)を窒素下において室温で乾燥DMF(6mL)に溶解した。フルオロエチルトシレート(0.25g、1.13mmol)を添加し、次いでNaH(0.05g、1.13mmol、油中60%)を添加した。反応物を80℃で1時間加熱した。溶媒を減圧下で除去し、残留物をDCMに溶解し、水で洗浄した。有機物をMgSO4上で乾燥し、濾過し、蒸発乾固させた。水(A)及びアセトニトリル(B)を溶離剤とする半分取HPLC(Gemini 5μ、C18、110A、150×21mm、20分で5〜95%B、21mL/分)によって粗物質を精製することで、79.9mg(35%)の3−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得た。1H NMR(300MHz,DMSO−d6)δ 0.95(3H,t,J=9Hz,N(CH2CH3)2)、1.88(3H,t,J=9Hz,N(CH2CH3)2)、3.14−3.26(2H,m,N(CH2CH3)2)、3.51−3.67(2H,m,N(CH2CH3)2)、4.58−4.97(4H,m,NCH2CH2F)、5.53(1H,s,6−H)、7.12−7.27(3H,m,ArH)、7.38−4.47(2H,m,ArH)、7.61(1H,d,J=9Hz,ArH)、7.80−7.86(1H,m,ArH)。
【0149】
LC−MS:C22H22F2N2OSに関するm/zの計算値、401.1;実測値、401.1(M+H)+。
【0150】
実施例5:(±)−8−エトキシ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤6)の製造
実施例5(i):(±)−11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0151】
【化23】
実施例1(ii)の(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(2.0g、5.40mmol)の無水DMF(20ml)溶液に、鉱油中の水素化ナトリウム60%分散液(240mg、6.0mmol)を添加し、混合物を窒素下において室温で5分間撹拌した。2−(ブロモエトキシ)−tert−ブチル−ジメチルシラン(2.6g、10.8mmol)を添加し、混合物を4時間撹拌した。溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。3%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、標記化合物(2.0g、70%)を黄色の固体として得た。
【0152】
実施例5(ii):(±)−11−[2−ヒドロキシエチル]−8−ヒドロキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0153】
【化24】
(±)−11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(1.0g、1.91mmol)の−78℃の乾燥DCM(60ml)溶液に、三臭化ホウ素(11.5ml、DCM中1M、11.5mmol)を添加した。溶液をRTまで昇温させ、24時間撹拌した。溶媒を真空中で除去し、メタノール(40ml)で奪活し、1M HCl(10ml)を添加し、1時間還流した。溶媒を真空中で除去し、混合物をメタノール(5ml)に溶解し、水(100ml)で奪活し、濾過し、(45℃で)真空乾燥することで、標記化合物(0.77g、100%)を淡褐色の粉末として得た。
【0154】
実施例5(iii):(±)−11−[2−ヒドロキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0155】
【化25】
(±)−11−[2−ヒドロキシエチル]−8−ヒドロキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(400mg、1.01mmol)の0℃の無水DMF(4ml)溶液に、鉱油中の水素化ナトリウム60%分散液(40mg、1.01mmol)を添加した。混合物を窒素下において0℃で10分間撹拌した。臭化エチル(218mg、2.0mmol、150μl)を添加し、混合物を24時間撹拌した。溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。40〜60%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、標記化合物(340mg、79%)を白色の固体として得た。
【0156】
実施例5(iv):(±)−11−[2−メタンスルホキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0157】
【化26】
(±)−11−[2−ヒドロキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(0.34g、0.80mmol)の無水DCM(15ml)懸濁液に、ピリジン(0.63g、8.0mmol、0.65ml)を添加した。反応物を0℃に冷却し、メタンスルホニルクロリド(0.37g、3.2mmol、0.25ml)を添加した。反応混合物をRTで3時間撹拌した。混合物を05M HCl(2×20ml)で洗浄し、次いで水(2×20ml)で洗浄し、(MgSO4で)乾燥し、溶媒を減圧下で除去した。20%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製した。残留物をエーテル/石油スピリットで奪活し、濾過し、(45℃で)真空乾燥することで、標記化合物(0.38g、95%)を淡黄色の固体として得た。
【0158】
実施例5(v):(±)−11−[2−フルオロエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0159】
【化27】
窒素下にある(±)−11−[2−メタンスルホキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(100mg、0.20mmol)の無水アセトニトリル(5ml)溶液に、THF中の1.0M TBAF(0.4ml、0.4mmol)を添加した。混合物を80℃で2時間加熱した。溶媒を真空中で除去し、5〜10%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、標記化合物(26mg、31%)を黄色の固体として得た。
【0160】
実施例6:(±)−7−メトキシ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤2)及び(±)−9−メトキシ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤7)の製造
実施例6(i):7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0161】
【化28】
(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(3.33g、12.6mmol、実施例1(i))及び3−メトキシフェニルヒドラジン塩酸塩(2.2g、12.6mmol)のエタノール(30ml)溶液に、濃硫酸(1.83ml、3.40g、11.5mmol)を窒素下で添加した。反応混合物を24時間加熱還流した。冷却後、反応混合物を濾過し、固体をエタノールで洗浄し、(45℃で)真空乾燥することで、7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの混合物(3.2g、69%)を淡白色の固体として得た。
【0162】
実施例6(ii):11−(2−フルオロエチル)−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−(2−フルオロエチル)−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0163】
【化29】
(実施例6(i)に従って製造した)7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの異性体混合物(1.0g、2.73mmol)の無水DMF(10mL)溶液に、2−フルオロエチルトシレート(1.2g、5.46mmol)を添加し、次いで鉱油中の水素化ナトリウム60%分散液(131mg、5.46mmol)を窒素下で添加した。反応混合物を80℃で1時間加熱した。冷却後、溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。5〜10%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、異性体混合物(1.0g、89%)を得た。次いで、水(A)及びメタノール(B)を溶離剤とするHPLC(Gemini 5μ、C18、110A、150×21mm、20分で70〜95%B、21mL/分)によってこの物質(400mg)を精製することで、240mgの9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを黄色の固体として得ると共に、100mgの7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得た。
【0164】
実施例7:18F標識イメージング剤2及び7の製造
実施例7(i):11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0165】
【化30】
実施例6(i)に従って製造した異性体混合物(2.0g、5.46mmol)の無水DMF(20ml)溶液に、鉱油中の水素化ナトリウム60%分散液(240mg、6.0mmol)を添加し、混合物を窒素下において室温で5分間撹拌した。2−(ブロモエトキシ)−tert−ブチル−ジメチルシラン(2.6g、10.9mmol)を添加し、混合物を4時間撹拌した。溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。5%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの異性体混合物(2.53g、88%)を黄色の固体として得た。
【0166】
実施例7(ii):11−[2−ヒドロキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−[2−(2−ヒドロキシオキシ)エチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0167】
【化31】
実施例7(i)に従って製造した異性体混合物(2.5g、4.76mmol)の無水THF(40ml)溶液に、THF中の1.0M TBAF(9.5ml、9.5mmol)を窒素下で添加した。混合物を室温で4時間撹拌した。溶媒を真空中で除去し、40%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、11−[2−ヒドロキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−[2−(2−ヒドロキシオキシ)エチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの異性体混合物(1.90g、97%)を淡黄色の固体として得た。
【0168】
実施例7(iii):(±)−11−[2−メタンスルホキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び(±)−11−[2−メタンスルホキシエチル]−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0169】
【化32】
実施例7(ii)に従って製造した異性体混合物(1.0g、2.4mmol)の無水DCM(30ml)溶液に、ピリジン(1.9g、24.0mmol、1.9ml)を添加した。反応物を0℃に冷却し、メタンスルホニルクロリド(1.1g、9.6mmol、0.74ml)を添加した。反応混合物をRTで4時間撹拌した。混合物を05M HCl(2×20ml)で洗浄し、次いで水(2×20ml)で洗浄し、(MgSO4で)乾燥し、溶媒を減圧下で除去した。20%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、異性体混合物(1.0g、85%)を得た。次いで、
水(A)及びメタノール(B)を溶離剤とするHPLC(Gemini 5μ、C18、110A、150×21mm、30分で5〜95%B、21mL/分)によってこの物質(400mg)を精製することで、170mgの11−[2−メタンスルホキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得ると共に、60mgの11−2−(メタンスルホキシエチル)−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得た。
【0170】
実施例7(iv):直接標識方法
実施例7(iii)に従って製造した前駆体化合物11−[2−メタンスルホキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−2−(メタンスルホキシエチル)−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを直接標識方法によって放射性標識することで、それぞれイメー
ジング剤2及び7を得た。18F−水を反応器に添加し、次いでアセトニトリル(500μl)中のK222(2mg)及びKHCO3(0.1mol dm−3、50μl)を添加し、100℃で20〜30分間乾燥した。アセトニトリル(1000μl)中の前駆体(0.5〜1mg)を添加した。反応器を密封し、100℃で10分間加熱した。反応混合物を冷却し、水(1.5ml)で反応器から洗い出し、半分取HPLCによって精製した。主な放射性生成物を含む画分を集め、H2Oで10mlの体積に希釈した。これをコンディショニングされたライトC18 sep pak上に装填し、H2O(1×2ml)でフラッシュし、生成物をEtOH(0.5ml)でP6バイアル中に溶出し、PBS(5ml)を添加した。
【0171】
実施例8:18F標識イメージング剤1、3、4及び5の製造
実施例8(i):前駆体化合物の製造
以下の前駆体化合物を製造した。
(a)(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例1(ii)に従って製造した)。
(b)(±)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例2(i)に従って製造した)。
(c)(±)−4−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例3(iii)に従って製造した)。
(d)(±)−3−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例4(iv)に従って製造した)。
以下に記載する間接標識方法を用いてこれらの前駆体化合物を放射性標識することで、それぞれ18F標識イメージング剤1、3、4及び5を得た。
【0172】
実施例8(ii):間接標識方法
反応器内のK222(4mg)、K2CO3水溶液(0.1モル溶液の50μl)及びアセトニトリル(500μl)に18F−/水を添加し、窒素流下において100℃で20〜30分間乾燥した。アセトニトリル(1000μl)中のエチル−1,2−ジトシレート(4mg)を添加し、100℃で10分間加熱した。反応混合物を冷却し、半分取HPLCによって精製し、18F−フルオロエチルトシレートを含む画分を集めた。この画分をH2Oで約20mlの体積に希釈し、コンディショニングされたライトt−C18 sep pak上に装填し、H2O(1×2ml)でフラッシュした。このsep pakを、高流量のN2ライン上で20分間乾燥した。次いで、18F−フルオロエチルトシレートをDMF(500μl)で溶出した。
【0173】
別途に、DMF(250μl)中の前駆体(13mg)を第2の反応器に添加し、N2で5分間パージした。次いで、DMF(2×250μl)中のNaH(13mg)を窒素下で添加し、反応器を45℃で0.5〜1時間加熱した。次いで、これに上記のようにして調製したDMF中の18F−フルオロエチルトシレートを添加し、N2でパージした反応器内において100℃で10分間加熱した。反応物を冷却し、水(1ml)で反応器から洗い出した。溶液をシリンジフィルターで濾過し、分取HPLCで精製した。主な放射性ピークを含む画分を集めた。これをH2Oで約10mlの体積に希釈し、コンディショニングされたライトC18 sep pak上に装填し、H2O(1×2ml)でフラッシュし、EtOH(0.5ml)でP6バイアル中に溶出し、リン酸緩衝食塩水(5ml)を添加した。
【0174】
実施例9:18F標識イメージング剤6の製造
(実施例5(iv)に従って製造した)(±)−11−[2−メタンスルホキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを、上記実施例7(iv)に記載した直接標識方法を用いて放射性標識した。
【0175】
実施例10:インビトロ効力アッセイ
Le Fur et al(Life Sci.1983;USA 33:449−57)の方法を改変した方法を用いて、PBRに対する親和性に関する化合物のスクリーニングを行った。
【0176】
(1%DMSOを含む50mMトリス−HCl、pH7.4、10mM MgCl2に溶解した)試験すべき化合物を、ウィスター系ラットの心臓PBRとの結合に関して0.3nM[3H]−PK−11195と競合させた。反応は50mMトリス−HCl、pH7.4、10mM MgCl2中において25℃で15分間実施した。
【0177】
化合物のスクリーニングは、推定Kiの周辺の300倍の濃度範囲にわたる6種の濃度で行った。結果は上記表1に示されており、本発明の化合物の効力が先行技術の化合物の効力に比べて勝ることを実証している。
【0178】
実施例11:インビボ体内分布方法
上記実施例7、実施例8及び実施例9で製造したインビボイメージング剤1〜7をインビボ体内分布モデルで試験し、その体内分布を(国際公開第2007/057705号の実施例14に従って製造した)先行技術の化合物[18F]FE−PBRの体内分布と比較した。
【0179】
雄ウィスター系ラット成体(200〜300g)に、側尾静脈を通して1〜3MBqの各インビボイメージング剤を注射した。注射から2分後、10分後、30分後又は60分後(n=3)に、ラットを安楽死させ、液体シンチレーションカウンティングによる放射能測定のために組織又は体液の試料を採取した。
【0180】
[18F]FE−PBRに比べて、本発明の化合物は高い嗅球:線条体結合比を示した(上記表1参照)。
【0181】
実施例12:顔面神経軸索切断(FNA)モデルを用いたオートラジオグラフィー
インビボ試験のためには、雄ウィスター系ラット(180〜200g)を使用した。イソフルラン麻酔下で、右側の耳領域から毛を除去した。耳内切開を行い、顔面神経の主幹を確認した。耳の後方において、茎乳突孔からの出口で顔面神経を切断した。切創を縫合し、動物を回復させた。手術から7日後の動物に、側尾静脈を通して約5〜10MBqのインビボイメージング剤1を注射した。30分後に脳幹を取り出し、イソペンタン中で凍結した。両顔面神経核を含む脳幹のクリオスタット切片(12μm)をスライドガラス上にマウントし、蛍光スクリーンに一晩露光させた。
【0182】
次いで、Storm(GE Healthcare社)蛍光イメージャー上でスクリーンを走査し、ImageQuant TL(GE Healthcare社)を用いて得られたスキャンの分析及び定量化を行った。図1及び図2は、この実験で得られたデータを示している。
【技術分野】
【0001】
本発明は、インビボイメージング、特に末梢ベンゾジアゼピンレセプター(PBR)のインビボイメージングに関する。高い親和性をもってPBRに結合し、投与後には脳内への良好な取込みを示し、高いレベルのPBRを発現する組織と優先的に結合する四環式インドール型インビボイメージング剤が提供される。本発明はまた、本発明のインビボイメージング剤の合成において有用な前駆体化合物、並びに前記前駆体化合物の使用を含む前記インビボイメージング剤の合成方法及び前記方法を実施するためのキットも提供する。インビボイメージング剤の自動化合成のためのカセットも提供される。加えて本発明は、本発明のインビボイメージング剤を含む放射性医薬組成物並びに前記インビボイメージング剤の使用方法も提供する。
【背景技術】
【0002】
トランスロケータータンパク質(TSPO)としても知られる末梢ベンゾジアゼピンレセプター(PBR)は、主として末梢組織及びグリア細胞に局在することが知られているが、それの生理学的機能はまだ明確に解明されていない。細胞レベル以下では、PBRはミトコンドリア外膜上に局在することが知られていて、これはミトコンドリア機能の調節及び及び免疫系において役割を果たす可能性を表している。さらに、PBRが細胞増殖、ステロイド生成、カルシウム流れ及び細胞呼吸に関係することも仮定されてきた。PBRは、急性及び慢性ストレス、不安、うつ病、パーキンソン病、アルツハイマー病、脳損傷、癌(Gavish et al,Pharm.Rev.1999;51:629)、ハンチントン病(Messmer and Reynolds,Neurosci.Lett.1998;241:53−6)、喘息(Pelaia et al,Gen.Pharmacol.1997;28(4):495−8)、慢性関節リウマチ(Bribes et al,Eur.J.Pharmacol.2002;452(1):111−22)、アテローム性動脈硬化症(Davies et al,J.Nucl.Med.2004;45:1898−1907)及び多発性硬化症(Banati et al,2000 Brain;123:2321)を始めとする各種の状態と関連していた。PBRはニューロパシー性疼痛にも関連する可能性があり、Tsuda et alはニューロパシー性疼痛をもった被験者で小グリア細胞の活性化を観察している(2005 TINS 28(2)pp101−7)。
【0003】
PBRに対して親和性を有するリガンドは当技術分野で公知である。米国特許第6,451,795号には、PBRに対して親和性(最も活性の高い化合物に関して0.2〜0.5nMのIC50値)を有する1群のインドール化合物が開示されている。この特許には、かかる化合物が末梢ニューロパシーの予防又は治療及び中枢神経変性疾患の治療のために有用であると述べられている。Okubo et al(Bioorganic & Medicinal Chemistry,2004;12:3569−80)は、PBRに対して親和性(約0.4nMという低いIC50値)を有する1群の四環式インドール化合物の設計、合成及び構造を記載している。Okubo et alによるこの文献には、かかる化合物の特別な用途は論議されていない。
【0004】
PBRのインビボイメージングも当技術分野で公知である。PBR選択性リガンドを用いる陽電子放出断層撮影(PET)イメージングでは、(R)−[11C]PK11195が中枢神経系(CNS)炎症の包括的指示薬を提供する。(R)−[11C]PK11195の使用の成功にもかかわらず、それには制約がある。それは、高いタンパク質結合性及び低特異性乃至非特異性結合を有することが知られている(Lockhart et al.Nucl Med Biol.30(2):199−206)。それの放射性標識代謝産物の役割は知られておらず、結合の定量化には複雑なモデル化が要求される。PBRに対して高い親和性及び選択性を有することでCNSにおけるPBR測定の向上を可能にする化合物を得るための努力が行われてきた。[11C]DAA1106及び[18F]FEDAA1106はアリールオキシアニリン化合物に基づくPET放射性リガンドであり、ヒトにおいて研究されてきた(Ikomo et al,J.Cereb.Blood Flow Metab.2007;27:173−84及びFujimura et al,J.Nuc.Med.2006;47:43−50)。しかし、これらの化合物の動力学的性質は理想的でなく、その用途は定量的研究に制限されることがある。これらの放射性リガンドにさらなる改良を加えようという努力の中で、Briard et al(J.Med.Chem.2008;51:17−30)は別のアリールオキシアニリン誘導体PBR28を報告した。PBR28の11C標識バージョンをサルに注射し、PETを用いてその脳内動力学を評価した。[11C]PBR28は、高い脳内取込み、PBR発現組織に対する良好な特異的結合、及びインビボイメージングのために一層適した動力学的性質を示した。PBR結合ピラゾロピリミジン化合物もまた、PBRを標的化するためのPET放射性リガンドとして評価された。James et al(J.Nuc.Med.2008;49(5):814−22)は、PET放射性リガンド[18F]−DPA−714が静脈内投与後のヒヒ脳においてPBRに対する高い親和性及びPBRによる選択的取込みを有することを報告している。[18F]−DPA−714の脳内取込みの動力学は[11C]DAA1106及び[18F]FEDAA1106より遅いが、事実上はそれに類似していることが報告された。国際公開第2007/057705号には、イメージング成分で標識された四環式インドール化合物であって、インビボイメージングのために適したものが開示されている。国際公開第2007/057705号中に例示されたインビボイメージング剤は、PBRに対して良好な親和性を有し、[3H]−PK−11195に対する競合アッセイにおけるKi値が1.0nM乃至0.1nMであることが示された。しかし、このたび本発明者らは、PBR発現組織に対するこれらの化合物の選択性が中枢神経系におけるPBS発現のインビボイメージングのためには理想的でないことを見出した。
【0005】
中枢神経系におけるPBS発現の評価のための代替インビボイメージング剤を得るためには、既知の四環式インドール化合物に改良を加える余地が存在している。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開第2007/057705号パンフレット
【発明の概要】
【課題を解決するための手段】
【0007】
本発明は、四環式インドール化合物に基づくインビボイメージング剤を提供する。四環式インドール化合物に基づく既知のインビボイメージング剤に比べて、本発明のインビボイメージング剤はインビボイメージングのための良好な性質を有している。本発明のインビボイメージング剤は、末梢ベンゾジアゼピンレセプターに対する良好な結合性を有すると共に、被験体への投与後における良好な脳内取込み及びインビボ動力学を有している。
【図面の簡単な説明】
【0008】
【図1】図1は、FNAラットの損傷(右側の)顔面神経核におけるインビボイメージング剤1の結合を示す代表的なオートラジオグラフである。
【図2】図2は、FNAから7日後のラットの顔面神経核に結合したインビボイメージング剤1の相対強度を示している。データは1頭の動物からの24の個別切片の平均±SDとして表されている。
【発明を実施するための形態】
【0009】
インビボイメージング剤
一態様では、本発明は、次の式Iのインビボイメージング剤或いはそれの塩又は溶媒和物であって、前記式Iのインビボイメージング剤の少なくとも1つの原子がインビボイメージングのために適した放射性同位体であり、前記放射性同位体が炭素の放射性同位体である場合にそれはカルボニル炭素であるインビボイメージング剤を提供する。
【0010】
【化1】
式中、
Qは水素又はフッ素であり、
Xは水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−6アルキル、C1−6ハロアルキル、C1−6アルコキシ又はC1−6アルキルアミドであり、
YはS、SO又はSO2であり、
Rは水素、C1−6アルキル又はC1−6フルオロアルキルであり、
ただしYがSである場合にはQ及びXが共に水素ではないことを条件とする。
【0011】
本明細書中で使用する「インビボイメージング」という用語は、本発明の被験体の内部構造の全部又は一部の画像を非侵襲的に生成する技法をいう。本発明で使用するための好ましいインビボイメージング方法は、単光子放出コンピューター断層撮影(SPECT)及び陽電子放出断層撮影(PET)であり、PETが特に好ましい。本発明の方法においてPETが好ましいのは、それが優れた感度及び分解能を有する結果、病変部における比較的小さい変化でも経時的に観察できるからである。PETスキャナーは、日常的にピコモル範囲内の放射能濃度を測定している。現在、マイクロPETスキャナーは約1mmの空間分解能に接近しているが、臨床スキャナーは約4〜5mmである。
【0012】
式Iの「インビボイメージング剤」は、インビボイメージングのために適した放射性同位体を含んでいる。この「インビボイメージングのために適した放射性同位体」は、式Iのインビボイメージング剤に関して上記に定義した原子の1つの放射性同位体形態である。本明細書で定義されるインビボイメージングのために適するには、放射性同位体はγ線放射体又は陽電子放射体であり、それによって投与後にインビボイメージング剤を被験体の外部で検出できることが好ましい。
【0013】
本発明に係る好適な塩には、(i)鉱酸(例えば、塩酸、臭化水素酸、リン酸、メタリン酸、硝酸及び硫酸)から導かれるもの並びに有機酸(例えば、酒石酸、トリフルオロ酢酸、クエン酸、リンゴ酸、乳酸、フマル酸、安息香酸、グリコール酸、グルコン酸、コハク酸、メタンスルホン酸及びp−トルエンスルホン酸)から導かれるもののような生理学的に許容される酸付加塩、並びに(ii)アンモニウム塩、アルカリ金属塩(例えば、ナトリウム塩及びカリウム塩)、アルカリ土類金属塩(例えば、カルシウム塩及びマグネシウム塩)、有機塩基(例えば、トリエタノールアミン、N−メチル−D−グルカミン、ピペリジン、ピリジン、ピペラジン及びモルホリン)との塩、及びアミノ酸(例えば、アルギニン及びリシン)との塩のような生理学的に許容される塩基塩がある。
【0014】
本発明に係る好適な溶媒和物には、エタノール、水、食塩水、生理的緩衝液及びグリコールと共に生成されるものがある。
【0015】
特記しない限り、単独で又は組み合わせて使用される「アルキル」という用語は、好ましくは1〜6の炭素原子、最も好ましくは1〜4の炭素原子を含む直鎖又は枝分れしたアルキル基を意味する。かかる基の例には、特に限定されないが、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、ペンチル、イソアミル及びヘキシルがある。
【0016】
特記しない限り、単独で又は組み合わせて使用される「アルコキシ」という用語はアルキルエーテル基を意味し、ここでアルキルという用語は上記に定義した通りである。好適なアルキルエーテル基の例には、特に限定されないが、メトキシ、エトキシ、n−プロポキシ、イソプロポキシ、n−ブトキシ、イソブトキシ、sec−ブトキシ及びtert−ブトキシがある。
【0017】
「アルキルアミド」は上記に定義したアルキル基がアミドに結合したものであり、ここでアミドは−C(=O)−NR’R”基(式中、R’及びR”は独立に水素又は炭化水素基である。)である。
【0018】
「ハロゲン」又は「ハロ」という用語は、フッ素、塩素、臭素及びヨウ素から選択される置換基を意味する。「ハロアルキル」は、上記に定義したアルキル基が1以上のハロゲンで置換されたものである。
【0019】
「ヒドロキシ」という用語は−OH基をいう。
【0020】
好ましい実施形態では、Qは水素である。
【0021】
Xは好ましくは水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ又はC1−4アルキルアミドである。Xは最も好ましくは水素又はC1−4アルコキシである。
【0022】
Yは好ましくはS又はSO2である。Yは最も好ましくはSである。
【0023】
Rは好ましくは水素、C1−4アルキル又はC1−4フルオロアルキルである。Rは好ましくは水素、C1−4アルキル又はC1−4フルオロアルキルである。Rは好ましくはC1−4フルオロアルキルである。
【0024】
式Iの好ましい実施形態では、
Xは水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ又はC1−4アルキルアミドであり、
YはS又はSO2であり、
Rは水素、C1−4アルキル又はC1−4フルオロアルキルである。
【0025】
式Iの最も好ましい実施形態では、
Qは水素であり、
XはC1−4アルコキシであり、
YはSであり、
RはC1−4フルオロアルキルである。
【0026】
式Iの別の好ましい実施形態では、
Qはフッ素であり、
Xは水素であり、
YはSであり、
RはC1−4フルオロアルキルである。
【0027】
本発明のインビボイメージングのために適した好ましい放射性同位体は、γ線放出型放射性ハロゲン及び陽電子放出型放射性非金属である。
【0028】
本発明で使用するのに適したγ線放出型放射性ハロゲンの例は、123I、131I及び77Brである。好ましいγ線放出型放射性ハロゲンは123Iである。
【0029】
本発明で使用するのに適した陽電子放出型放射性非金属の例は、11C、13N、18F及び124Iである。好ましい陽電子放出型放射性非金属は18Fである。18Fは本発明のインビボイメージングのために適する最も好ましい放射性同位体である。
【0030】
式Iのインビボイメージング剤に関する好ましい実施形態では、Xは123I、124I、131I、18F又はC1−4[18F]−フルオロアルキルである。
【0031】
式Iのインビボイメージング剤に関する別の好ましい実施形態では、RはC1−4[18F]−フルオロアルキルである。
【0032】
式Iのインビボイメージング剤に関するさらに別の好ましい実施形態では、カルボニル炭素は11Cである。
【0033】
本発明の若干の好ましいインビボイメージング剤の非限定的な例は以下の通りである。
【0034】
【化2】
上記のインビボイメージング剤のうちでは、イメージング剤2が最も好ましい。
【0035】
これらのインビボイメージング剤を得るために使用される合成方法は、以下の実験セクションに記載されている。本発明のインビボイメージング剤のこれらの非放射性バージョンの効力は、実施例10に記載されるように、インビトロアッセイで測定した。
【0036】
実施例7乃至実施例9には、放射性フッ素化インビボイメージング剤1〜7を得るための方法が記載されている。当業者には知られている通り、18Fを取り扱う場合、使用するスケール及び条件は安全上及び実際上の考慮事項に応じて異なる。18F PETトレーサーの製造に関する総説は、“Principles and Practice of Positron Emission Tomography”(2002 Lippincott Williams & Wilkins;Wahl and Buchanan,Eds.)の第1章及び第2章を参照されたい。かかるインビボイメージング剤を動物体内分布モデルで試験し(実施例11)、その体内分布を(国際公開第2007/057705号の実施例14に従って製造した)先行技術の化合物[18F]FE−PBRの体内分布と比較した。
【0037】
【化3】
以下の表1に、インビトロ親和性アッセイ並びにインビボ体内分布試験で得られたデータを示す。非放射性類似体をインビトロ親和性アッセイで試験し、放射性標識バージョンを体内分布アッセイで評価した。
【0038】
【表1】
かかるデータは、インビボイメージング剤1〜7の非放射性バージョンの効力が先行技術の化合物[18F]FE−PBRの効力に比べて勝ることを示している。加えて、かかるデータは、本発明のインビボイメージング剤1〜7が[18F]FE−PBRに比べて注射から30分後に線条体よりOB中に顕著に多く保持されることを示している。PBRはラット脳の他の領域に比べてOBにおいて高度に発現されることが知られているので(“Handbook of Substance Abuse”by Tarter,Ammerman and Ott;Springer 1998:398−99を参照されたい)、これらのデータは意外にも、インビボイメージング剤1〜7が先に例示したインビボイメージング剤[18F]FE−PBRよりPBRに対して向上した選択性を有することを実証している。
【0039】
以下の実施例12に記載されるように、インビボイメージング剤1をさらにオートラジオグラフィーモデルで分析した。顔面神経核の損傷側において顕著に高いレベルの放射能が検出された(図1及び図2参照)。損傷側の平均強度は、非損傷側が3.73±0.36であるのに比べて7.75±0.95であった。両側間の比は8.23±2.36であった。損傷部は正常部に比べて高いPBR発現を示すので、これらのデータは、インビボイメージング剤1がPBRに対して良好な選択性を有するという生体分布データからの結論を支持している。
【0040】
したがって、本発明のインビボイメージング剤は、既知の四環式インドール型PBR結合インビボイメージング剤に比べ、PBRのインビボイメージングのための意外に優れた性質を有している。
【0041】
前駆体化合物
別の態様では、本発明は次の式IIを有する前駆体化合物を提供する。
【0042】
【化4】
式中、R1、X1及びZ1の1つが本明細書で好適なもの及び好ましいものとして定義した放射性同位体の適当な供給源と反応する化学基を含む結果、当該前駆体化合物を前記放射性同位体の前記適当な供給源と反応せれば、本明細書で好適なもの及び好ましいものとして定義したインビボイメージング剤が生成され、
R1が前記化学基を含まない場合、それは式IのRに関して本明細書で好適なもの及び好ましいものとして定義した通りであり、任意にはさらに保護基を含み、
X1が前記化学基を含まない場合、それは式IのXに関して本明細書で好適なもの及び好ましいものとして定義した通りであり、任意にはさらに保護基を含み、
Z1が前記化学基を含まない場合、それは−C(=O)−N−(CH2−CH3)2であり、任意にはさらに保護基を含み、
Q1は式IのQに関して本明細書で好適なもの及び好ましいものとして定義した通りであり、
Y1は式IのYに関して本明細書で好適なもの及び好ましいものとして定義した通りであり、任意にはさらに保護基を含む。
【0043】
「前駆体化合物」は、好都合な化学形態の検出可能な標識との化学反応が部位特異的に起こり、最小数の段階(理想的にはただ1つの段階)で反応を実施でき、かつ格別の精製の必要なしに(理想的にはいかなる追加の精製も必要なしに)所望のインビボイメージング剤が得られるように設計された、放射性標識化合物の誘導体からなる。かかる前駆体化合物は合成品であり、良好な化学純度で簡便に得ることができる。前駆体化合物は、任意には前駆体化合物のある種の官能基に関して保護基を含むことができる。
【0044】
「保護基」という用語は、望ましくない化学反応を阻止又は抑制するが、分子の残部を変質させない十分に温和な条件下で問題の官能基から脱離させ得るのに十分な反応性を有するように設計された基を意味する。脱保護後には所望の生成物が得られる。保護基は当業者にとって公知であり、アミン基に関してはBoc(ここでBocはtert−ブチルオキシカルボニルである。)、Fmoc(ここでFmocはフルオレニルメトキシカルボニルである。)、トリフルオロアセチル、アリルオキシカルボニル、Dde[即ち、1−(4,4−ジメチル−2,6−ジオキソシクロヘキシリデン)エチル]及びNpys(即ち、3−ニトロ−2−ピリジンスルフェニル)から適宜に選択され、カルボキシル基に関してはメチルエステル、tert−ブチルエステル及びベンジルエステルから適宜に選択される。ヒドロキシル基に関しては、好適な保護基は、メチル、エチル又はtert−ブチル、アルコキシメチル又はアルコキシエチル、ベンジル、アセチル、ベンゾイル、トリチル(Trt)、又はテトラブチルジメチルシリルのようなトリアルキルシリルである。さらに他の保護基の使用は、‘Protective Groups in Organic Synthesis’,Theorodora W.Greene and Peter G.M.Wuts(Third Edition,John Wiley & Sons,1999)に記載されている。
【0045】
「放射性同位体の適当な供給源」という用語は、放射性同位体が前駆体化合物に共有結合するようにして前駆体化合物の置換基と反応し得る化学形態の放射性同位体を意味する。
【0046】
以下のセクションに示される特定の放射性同位体の各々に関しては、放射性同位体の1種以上の適当な供給源が論議される。インビボイメージング剤の分野の当業者は、本発明における用途に適した放射性同位体のこれらの供給源及びその他の供給源に精通しているであろう。
【0047】
インビボイメージング剤の放射性同位体が18Fある場合、放射性フッ素原子はフルオロアルキル基又はフルオロアルコキシ基の一部をなすことができる。これは、アルキルフルオリドがインビボでの代謝に耐えるからである。別法として、放射性フッ素原子は直接共有結合によってベンゼン環のような芳香環に結合できる。
【0048】
放射性フッ素化は、18F−フッ化物と良好な脱離基を有する前駆体化合物(例えば、アルキルブロミド、アルキルメシレート又はアルキルトシレート)中の適当な化学基との反応を用いる直接標識によって実施できる。
【0049】
18Fはまた、18F(CH2)3OMs又は18F(CH2)3OBrでヒドロキシル基をO−アルキル化することによっても導入できる。
【0050】
アリール系に関しては、アリールジアゾニウム塩、アリールニトロ化合物又はアリール第四級アンモニウム塩からの18F−フッ化物求核置換が、アリール−18F誘導体への好適な経路である。かかる方策は、例えば、式Iの1〜4位又は7〜10位に18Fを導入するために適している。
【0051】
別法として、18Fによる標識は、式Iの誘導体からの脱離基の求核置換によって達成できる。好適な脱離基には、Cl、Br、I、トシレート(OTs)、メシレート(OMs)及びトリフレート(OTf)がある。かかる誘導体は、本発明のインビボイメージング化合物を製造するための前駆体化合物である。
【0052】
別の方策は、上記に定義したような好適な脱離基を、前駆体化合物上に存在するアルキルアミド基上に設けることである。このようにすれば、通常は核反応18O(p,n)18Fからの水溶液として得られ、次いで陽性対イオンの添加及びそれに続く水の除去によって反応性にされる[18F]−フッ化物イオン(18F−)の適当な供給源との反応によって前駆体化合物を一段階で標識できる。この方法のためには、前駆体化合物は、放射性フッ素化が化合物上の特定の部位で起こるように選択的に化学保護されるのが普通である。好適な保護基は既に前述したものである。
【0053】
放射性同位体が18Fである場合、X1又はR1は以下のいずれかを含むことが好ましい。
(i)求核置換のためのアルキルハライド又はアルキルスルホネート(例えば、アルキルブロミド、アルキルメシレート又はアルキルトシレート)、或いは
(ii)(例えば、18F(CH2)3OMs又は18F(CH2)3OBrでヒドロキシル基をO−アルキル化することによる18Fの導入のための)ヒドロキシル。
【0054】
本発明の若干の18Fインビボイメージング剤を得るための一般反応スキームを以下に示す。
【0055】
【化5】
式中、Xは式Iに関して定義した通りであり、nは0〜5である。RTは室温を表し、OMsはメシレートを表す。
【0056】
本発明の若干の18Fインビボイメージング剤を得るための別の一般反応スキームを以下に示す。
【0057】
【化6】
式中、Xは式Iに関して定義した通りであり、nは0〜5であり、OTsはトシレートを表す。
【0058】
11C標識PETトレーサー化合物は、前駆体化合物を11C−ヨウ化メチルと反応させることで合成できる。11Cの半減期は20.4分にすぎないので、11C−ヨウ化メチル中間体は高い比放射能を有すること、したがってできるだけ速い反応プロセスを用いてそれを製造することが重要である。かかる11C標識技法に関する徹底的な総説を、Antoni et al,“Aspects on the Synthesis of 11C−Labelled Compounds”in Handbook of Radiopharmaceuticals,Ed.M.J.Welch and C.S.Redvanly(2003,John Wiley and Sons)に見出すことができる。
【0059】
本発明のインビボイメージング剤が11Cで標識される場合、11Cはカルボニル炭素である。したがってこれは、11Cが式Iのカルボニル炭素の位置に存在し得るか、或いは別法としてXがC1−6アルキルアミドである場合にXの位置に存在し得ることを意味する。
【0060】
式Iの11C標識インビボイメージング剤は、以下の反応スキームを用いて得ることができる。
【0061】
【化7】
式中、式IIb及び式IdのR3、X3及びY3は、式IのR、X及びYに関して記載した通りであり、
Z3は、遷移金属触媒に適した基質(例えば、水素、ハロゲン化物、ボロン酸、OTf又は有機スズ)である。
【0062】
13N標識化合物の合成方法は、Clark and Aigbirhio(“Chemistry of Nitrogen−13 and Oxygen−15”in“Handbook of Radiopharmaceuticals”;2003 Wiley:Welch and Redvanly,Eds.)によって記載されている。例えば、式Iのインビボイメージング剤は、適当な前駆体化合物中のハロゲンを13N標識ジエチルアミンで求核置換して所望のアミドを生成することで得ることができる。
【0063】
イメージング成分が放射性ヨウ素である場合、好ましい前駆体化合物は、求電子又は求核ヨウ素化を受ける誘導体或いは標識アルデヒド又はケトンとの縮合を受ける誘導体を含むものである。第1のカテゴリーの例は以下の(a)及び(b)である。
(a)トリアルキルスタンナン(例えば、トリメチルスタンニル又はトリブチルスタンニル)、トリアルキルシラン(例えば、トリメチルシリル)或いは有機ホウ素化合物(例えば、ボロネートエステル又はオルガノトリフルオロボレート)のような有機金属誘導体。
(b)求電子ヨウ素化に向けて活性化された芳香環(例えば、フェノール類)及び求核ヨウ素化に向けて活性化された芳香環(例えば、アリールヨードニウム塩、アリールジアゾニウム塩、アリールトリアルキルアンモニウム塩又はニトロアリール誘導体)。
【0064】
放射性ヨウ素化のために好ましくは、前駆体化合物は、(放射性ヨウ素交換を可能にするための)アリールヨージド又はブロミド、活性化前駆体化合物アリール環(例えば、フェノール基)、有機金属前駆体化合物(例えば、トリアルキルスズ、トリアルキルシリル又は有機ホウ素化合物)、或いはトリアゼンのような有機前駆体化合物又は求核置換のための良好な脱離基(例えば、ヨードニウム塩)からなる。放射性ヨウ素を有機分子中に導入するための前駆体化合物及び方法は、Bolton(J.Lab.Comp.Radiopharm.2002;45:485−528)によって記載されている。放射性ヨウ素をタンパク質中に導入するための前駆体化合物及び方法は、Wilbur(Bioconj.Chem.1992;3(6):433−470)によって記載されている。好適なボロネートエステル有機ホウ素化合物及びその製法は、Kabalka et al(Nucl.Med.Biol.2002;29:841−843及び2003;30:369−373)によって記載されている。好適なオルガノトリフルオロボレート及びその製法は、Kabalka et al(Nucl.Med.Biol.2004;31:935−938)によって記載されている。放射性ヨウ素化用の好ましい前駆体化合物は有機金属前駆体化合物からなり、最も好ましくはトリアルキルスズからなる。
【0065】
放射性ヨウ素を結合できるアリール基の例を以下に示す。
【0066】
【化8】
これらの基はいずれも、芳香環上への容易な放射性ヨウ素置換を可能にする置換基を含んでいる。放射性ヨウ素を含む別の置換基は、例えば次式のように、放射性ハロゲン交換による直接ヨウ素化で合成することができる。
【0067】
【化9】
放射性同位体が放射性ヨウ素である場合、式IIのX1は、そけが結合した芳香族基と共に、
(i)有機金属誘導体又は有機ホウ素化合物で置換された芳香環を形成するか、
(ii)求電子ヨウ素化に向けて活性化された芳香環(例えば、フェノール類)を形成するか、或いは
(iii)求核放射性ヨウ素化に向けて活性化された芳香環(例えば、アリールヨードニウム塩、アリールジアゾニウム塩、アリールトリアルキルアンモニウム塩又はニトロアリール誘導体)を形成する。
【0068】
これらの前駆体化合物は、放射性ヨウ素置換により、本発明の放射性ヨウ素化インビボイメージング剤に容易に転化される。
【0069】
放射性臭素化は、放射性ヨウ素化に関して上記に記載したものと同様な方法によって達成できる。Kabalka and Varmaは、放射性臭素化化合物を含む放射性ハロゲン化化合物を合成するための様々な方法を総説している(Tetrahedron 1989;45(21):6601−21)。
【0070】
本発明の前駆体化合物は、理想的には無菌で非発熱性の形態で供給される。したがって前駆体化合物は、インビボイメージング剤を哺乳動物への投与に適した生体適合性キャリヤーと共に含んでなる医薬組成物の製造のために使用できる。前駆体化合物はまた、かかる医薬組成物を製造するためのキット中に一成分として含めるためにも適している。
【0071】
好ましい実施形態では、前駆体化合物はキット又は自動化合成装置で使用するように設計されたカセットの一部として溶液状態で供給される。これらの態様は、本発明の追加の態様に関連して以下に一層詳しく論議される。
【0072】
別の好ましい実施形態では、前駆体化合物は固相に結合されている。前駆体化合物は、好ましくは固体担体マトリックスに共有結合した状態で供給される。このようにすれば、所望の生成物は溶液状態で生成される一方、出発原料及び不純物は固相に結合したままに保たれる。かかる系の例としては、18F−フッ化物による固相求電子フッ素化用の前駆体化合物が国際公開第03/002489号に記載されており、18F−フッ化物による固相求核フッ素化用の前駆体化合物が国際公開第03/002157号に記載されている。
【0073】
インビボイメージング剤の製造方法
さらに別の態様では、本発明は、本発明のインビボイメージング剤の製造方法であって、
(i)上記に定義した式IIの前駆体化合物を用意する段階、
(ii)上記に定義した前記放射性同位体の適当な供給源を用意する段階、並びに
(iii)段階(i)の前駆体化合物を段階(ii)の放射性同位体と反応させて本発明のインビボイメージング剤を得る段階
を含んでなる方法を提供する。
【0074】
段階(i)では、前駆体化合物は、前駆体化合物の説明において上記に記載したように、キット又は自動化合成装置で使用するのに適したカセット中に溶液状態で供給するか、或いは別法として固体担体に結合した状態で供給することができる。かかるキット及びカセットは本発明の追加の態様をなしており、以下に一層詳しく論議される。
【0075】
放射性同位体の適当な供給源は、本発明の前駆体化合物に関連して上記に記載した通りである。
【0076】
前駆体化合物を放射性同位体と「反応させる」段階は、できるだけ高い放射化学収率(RCY)で所望のインビボイメージング剤を生成するのに適した反応条件下で2種の反応体を合わせることを含んでいる。本発明のインビボイメージング剤を得るための特定の合成経路は、以下の実験セクションに示される。
【0077】
インビボイメージング剤の製造用キット
さらに別の態様では、本発明は、本発明のインビボイメージング剤を製造するためのキットを提供する。前記キットは上記に記載された式IIの前駆体化合物を含む結果、放射性同位体の無菌供給源との反応により最小数の操作で所望のインビボイメージング剤が得られる。かかる考慮事項は、放射性同位体が比較的短い半減期を有する場合において、取扱いを容易にし、したがって放射線薬剤師に対する放射線量を低減させるため特に重要である。前駆体化合物は好ましくは凍結乾燥形態でキット中に存在しており、かかるキットの再構成用反応媒質は好ましくは生体適合性キャリヤーである。
【0078】
「生体適合性キャリヤー」は、組成物が生理学的に認容され得るようにして(即ち、毒性又は過度の不快感なしに哺乳動物体に投与できるようにして)インビボイメージング剤を懸濁又は溶解するための流体(特に液体)である。生体適合性キャリヤーは、好適には、無菌のパイロジェンフリー注射用水、(有利には注射用の最終生成物が等張性又は非低張性になるように平衡させ得る)食塩水のような水溶液、或いは1種以上の張度調整物質(例えば、血漿陽イオンと生体適合性対イオンとの塩)、糖(例えば、グルコース又はスクロース)、糖アルコール(例えば、ソルビトール又はマンニトール)、グリコール(例えば、グリセロール)又は他の非イオン性ポリオール物質(例えば、ポリエチレングリコール、プロピレングリコールなど)の水溶液のような注射可能なキャリヤー液体である。生体適合性キャリヤーはまた、エタノールのような生体適合性有機溶媒を含んでいてもよい。かかる有機溶媒は、親油性の高い化合物又は配合物を可溶化するために有用である。好ましくは、生体適合性キャリヤーはパイロジェンフリー注射用水、等張食塩水又はエタノール水溶液である。静脈内注射用生体適合性キャリヤーのpHは好適には4.0〜10.5の範囲内にある。
【0079】
本発明のキットでは、前駆体化合物は好ましくは、注射器による溶液の追加及び抜取りを許しながら、無菌保全性及び/又は放射能安全性の維持、さらに任意には不活性ヘッドスペースガス(例えば、窒素又はアルゴン)の維持を可能にする密封容器に入れて供給される。好ましい密封容器は、気密クロージャーを(通例はアルミニウムからなる)オーバーシールと共にクリンプ加工した隔壁密封バイアルである。かかる密封容器は、例えばヘッドスペースガスの変更又は溶液のガス抜きのために所望される場合、クロージャーが真空に耐え得るという追加の利点を有している。
【0080】
キット中に使用する場合、前駆体化合物の好ましい実施形態は、本明細書で記載した通りである。
【0081】
キット中に使用するための前駆体化合物を無菌製造条件下で使用すれば、所望の無菌で非発熱性の材料を得ることができる。別法として、前駆体化合物を非無菌条件下で使用し、次いで例えばγ線照射、オートクレーブ処理、乾熱又は(例えば、エチレンオキシドによる)化学処理を用いる終末滅菌を施すこともできる。好ましくは、前駆体化合物は無菌で非発熱性の形態で供給される。最も好ましくは、無菌で非発熱性の前駆体化合物は上述したような密封容器に入れて供給される。
【0082】
好ましくは、試験間での汚染の可能性を最小限に抑えると共に無菌性及び品質保証を確実にするため、キットのすべての構成要素は使い捨てである。
【0083】
好ましい実施形態では、キットは、以下に一層詳しく記載されるように、適宜に改造された自動化合成装置に挿入し得るカセットを含むことができる。かかるキットは、通例はフッ化物イオンによるフッ素化のための手段を含み、また不要のフッ化物イオンを除去するためのカラムを含むことができる。合成のために必要な試薬、溶媒及び他の消耗品もまた、濃度、体積、送出時間などに関する最終ユーザーの要求条件を満たすように自動化合成装置を運転させるソフトウェアを保持したコンパクトディスクのようなデータ媒体と共に含めることができる。
【0084】
現在、PETイメージング用の[18F]放射性トレーサーはしばしば自動化放射合成装置で簡便に製造されている。かかる装置には、Tracerlab及びFastlab(GE Healthcare社)を始めとするいくつかの市販例が存在している。かかる装置は、通常、放射化学を実施するために(しばしば使い捨ての)「カセット」を含んでおり、これを装置に取り付けることで放射合成が実施される。普通、カセットは流体通路、反応器、及び試薬バイアルを受け入れるためのポート並びに放射合成後の清掃段階で使用される固相抽出カートリッジを含んでいる。
【0085】
したがって本発明は、別の態様では、本明細書で前述した密封容器に入れた前駆体化合物を含んでなる自動化合成装置用カセットを提供する。本発明はまた、本明細書で定義したインビボイメージング剤の自動化合成のためのカセットであって、
(i)本明細書で定義した前駆体化合物を含む容器、及び
(ii)本明細書で定義した放射性同位体の適当な供給源を用いて容器を溶出するための手段
を含んでなるカセットを提供する。
【0086】
かかるカセットはさらに、
(iii)過剰の放射性標識を除去するためのイオン交換カートリッジ、及び任意には
(iv)得られた放射性標識生成物を脱保護して本明細書で定義したインビボイメージング剤を生成するためのカートリッジ
を含むことができる。
【0087】
放射性医薬組成物
さらに別の態様では、本発明は「放射性医薬組成物」を提供するが、これは本発明のインビボイメージング剤を、哺乳動物への投与に適した形態の生体適合性キャリヤーと共に含んでいる。生体適合性キャリヤーは、本発明のキットに関連して上記に定義した通りである。
【0088】
かかる放射性医薬組成物は非経口的に(即ち、注射によって)投与でき、最も好ましくは水溶液である。かかる組成物は、緩衝剤、薬学的に許容される可溶化剤(例えば、シクロデキストリン或いはPluronic、Tween又はリン脂質のような界面活性剤)、薬学的に許容される安定剤又は酸化防止剤(例えば、アスコルビン酸、ゲンチジン酸又はパラアミノ安息香酸)のような追加成分を任意に含み得る。本発明のインビボイメージング剤が放射性医薬組成物として提供される場合、前記インビボイメージング剤の製造方法はさらに、放射性医薬組成物を得るために必要な段階(例えば、有機溶媒の除去、生体適合性緩衝剤及び任意の追加成分の添加)を含むことができる。非経口的投与のためには、放射性医薬組成物が無菌性かつ無発熱原性であることを保証するための手段を講じることも必要である。
【0089】
インビボイメージング方法
さらに別の態様では、本発明は、被験体におけるPBR発現の体内分布及び/又は程度を決定するためのインビボイメージング方法であって、
(i)本明細書で定義したインビボイメージング剤を前記被験体に投与する段階、
(ii)前記イメージング剤を前記被験体中のPBRに結合させる段階、
(iii)前記イメージング剤の放射性同位体から放出される信号をインビボイメージング技法によって検出する段階、
(iv)前記信号の位置及び/又は量を表す画像を生成する段階、並びに
(v)前記被験体におけるPBR発現の分布及び程度を決定する段階であって、前記発現は前記インビボイメージング剤から放出される前記信号と直接に相関している段階
を含んでなるインビボイメージング方法を提供する。
【0090】
本発明のインビボイメージング方法に関しては、インビボイメージング剤は本明細書でおいて前記に定義した通りである。
【0091】
インビボイメージング剤を「投与する」段階は、好ましくは非経口的に実施され、最も好ましくは静脈内に実施される。静脈内経路は、インビボイメージング剤を被験体の身体全域に送達するため、したがって血液脳関門(BBB)を横切って前記被験体で発現されたPBRに接触させるための最も効率的な方法である。本発明のインビボイメージング剤は、好ましくは本明細書で定義した本発明の医薬組成物として投与される。
【0092】
投与段階後かつ検出段階前に、インビボイメージング剤をPBRに結合させる。例えば、被験体がインタクトな哺乳動物である場合、インビボイメージング剤は哺乳動物の身体を通って動的に移動し、体内の様々な組織に接触する。ひとたびインビボイメージング剤がPBRに接触すれば、特異的な相互作用が起こる結果、PBRをもった組織からのインビボイメージング剤のクリアランスは、PBRをもたない組織又はPBRの少ない組織よりも長い時間がかかる。一定の時点に達すれば、PBRをもった組織に結合したインビボイメージング剤とPBRをもたない組織又はPBRの少ない組織に結合したインビボイメージング剤との比の結果として、PBRに特異的に結合したインビボイメージング剤の検出が可能となる。理想的なかかる比は約2:1である。
【0093】
本発明の方法の「検出」段階は、放射性同位体から放出される信号を、前記信号に対して感受性を有する検出器によって検出することを含んでいる。この検出段階はまた、信号データの取得として理解することもできる。単光子放出コンピューター断層撮影(SPECT)及び陽電子放出断層撮影(PET)が、本発明の方法で使用するための最も好適なインビボイメージング技法である。PETが、本発明の方法で使用するための好ましいインビボイメージング技法である。
【0094】
本発明の方法の「生成」段階は、取得された信号データに再構築アルゴリズムを適用してデータセットを得るコンピューターによって実施される。次いで、このデータセットを操作することで、前記放射性同位体から放出される信号の位置及び/又は量を示す画像が生成される。放出される信号はPBRの発現と直接に相関する結果、生成された画像を評価することで「決定」段階を行うことができる。
【0095】
本発明の「被験体」は、任意のヒト又は動物被験体であり得る。好ましくは、本発明の被験体は哺乳動物である。最も好ましくは、前記被験体はインタクトな哺乳動物生体である。特に好ましい実施形態では、本発明の被験体はヒトである。かかるイメージング方法は、健常被験体或いはPBRの異常発現に関連する病的状態(「PBR状態」)を有することが知られ又は疑われる被験体においてPBRを検査するために使用できる。好ましくは、前記方法はPBR状態を有することが疑われる被験体のインビボイメージングに関し、したがって前記状態の診断方法において有用である。インビボイメージングが役に立つかかる状態の例には、神経炎症が存在するパーキンソン病、多発性硬化症、アルツハイマー病及びハンチントン病のような神経疾患がある。本発明の化合物によるイメージングが有用であり得る他のPBR状態には、ニューロパシー性疼痛、関節炎、喘息、アテローム性動脈硬化症、並びに結腸直腸癌及び乳癌のような悪性疾患がある。本発明のインビボイメージング剤は、その良好な脳内取込みのため、中枢神経系(CNS)のインビボイメージングに特に適している。
【0096】
別の実施形態では、本発明のインビボイメージング方法は前記被験体に関する治療計画の進行中に繰り返して実施することができ、前記計画はPBR状態と戦うための薬物の投与を含んでいる。例えば、本発明のインビボイメージング方法は、PBR状態と戦うための薬物による治療前、治療中及び治療後に実施できる。このようによすれば、前記治療の効果を経時的なモニターすることができる。この実施形態のために好ましくは、インビボイメージング技法はPETである。PETは優れた感度及び分解能を有する結果、病変部における比較的小さい変化でも経時的に観察でき、これは治療モニタリングのために有利である。PETスキャナーは、日常的にピコモル範囲内の放射能濃度を測定している。現在、マイクロPETスキャナーは約1mmの空間分解能に接近しているが、臨床スキャナーは約4〜5mmである。
【0097】
さらに別の態様では、本発明はPBR状態の診断方法を提供する。本発明の診断方法は、上記に定義したインビボイメージング方法を、PBR発現の分布及び程度を特定の臨床像に帰因させる追加段階(vi)(即ち、演繹的な医学的決断段階)と共に含んでいる。
【0098】
別の態様では、本発明は、本明細書中で定義した診断方法で使用するための、本明細書中で定義したインビボイメージング剤を提供する。
【0099】
さらに別の態様では、本発明は、本明細書中で定義した診断方法で使用するための本明細書中で定義した放射性医薬組成物の製造で使用するための、本明細書中で定義したインビボイメージング剤を提供する。
【実施例】
【0100】
実施例の簡単な説明
すべての試薬はSigma Aldrich社から入手した。
【0101】
実施例1乃至実施例6は、本発明の様々なインビボイメージング剤の非放射性バージョンの合成法を記載している。
【0102】
実施例7乃至実施例9は、本発明の18F標識インビボイメージング剤を得るための方法を記載している。
【0103】
実施例10は、本発明のイメージング剤のPBR親和性を測定するために使用したインビトロ効力アッセイを記載している。
【0104】
実施例11は、動物体内分布試験の実施方法を記載している。
【0105】
実施例12は、顔面神経軸索切断動物モデル及びオートラジオグラフィー試験におけるそれの使用を記載している。
【0106】
実施例中で使用される略語のリスト
DCM ジクロロメタン
DMF ジメチルホルムアミド
DMSO ジメチルスルホキシド
EtOAc 酢酸エチル
FNA 顔面神経軸索切断
g グラム
h 時間
HRMS 高分解能質量分析法
K222 クリプトフィックス2.2.2
M モル濃度=溶質のモル数/溶液1リットル
MHz メガヘルツ
ml ミリリットル
mmol ミリモル
N 規定度=当量数/溶液1L
NMR 核磁気共鳴
PBR 末梢ベンゾジアゼピンレセプター
RT 室温
実施例1:(±)−11−(2−フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤1)の製造
実施例1(i):(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド
【0107】
【化10】
T.Okubo et al(Bioorg.Med.Chem.2004;12:3569−3580)に記載されているようにして製造した(±)−4−オキソ−チオクロマン−2−カルボン酸(10.4g、50mmol)の乾燥DCM(100ml)溶液を、塩化オキサリル(12.6g、100mmol)及び1滴のDMFと共に窒素雰囲気下において室温で18時間撹拌した。次いで、反応物を真空中で蒸発させてガム状にし、次いでDCM(100ml)に再溶解し、氷浴上で0℃に冷却し、撹拌しながらDCM(20ml)中のジエチルアミン(8.03g、110mmol)を1時間かけて滴下することで処理した。反応物を1時間かけて室温まで放温し、10%炭酸カリウム水溶液(100ml)を添加し、反応混合物を激しく撹拌した。DCM溶液を分離した。水溶液をDCMの追加バッチ(100ml)で2回抽出し、合わせた抽出液を硫酸マグネシウム上で乾燥した。DCM溶液を真空中で濃縮して暗緑色の油状物を得たが、これは静置することで結晶化した。結晶質固体をジエチルエーテル(50ml)でトリチュレートして濾過することで、標記化合物(8.57g、65%)を淡緑色の固体として得た。
【0108】
1H NMR(300MHz,CDCl3)δ 1.06(t,J=7.1Hz,3H)、1.23(t,J=7.1Hz,3H)、3.0−3.5(m,6H)、4.25(m,1H)、7.15−7.21(m,2H)、7.32−7.39(m,1H)、8.10−8.14(m,1H)。
【0109】
13C NMR(75MHz,CDCl3)δ 12.9、14.8、40.1、40.7、42.3、42.5、125.8、127.2、128.7、130.8、133.4、137.9、167.9、193.1。
【0110】
実施例1(ii):(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0111】
【化11】
(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(1.32g、5.0mmol、実施例1(i))及び4−メトキシフェニルヒドラジン塩酸塩(0.87g、5.0mmol)のエタノール(10ml)溶液に、濃硫酸(0.73ml、1.35g、13.8mmol)を窒素下で添加した。反応混合物を24時間加熱還流した。冷却後、反応混合物を濾過し、固体をエタノールで洗浄し、(45℃で)真空乾燥することで、標記化合物(1.05g、57%)を淡黄色の固体として得た。
【0112】
1H NMR(300MHz,DMSO−d6)δ 0.97(t,J=6.8Hz,3H)、1.28(t,J=6.8Hz,3H)、3.25(m,2H)、3.60(m,2H)、3.74(s,3H)、5.59(s,1H)、6.80(m,2H)、7.10−7.35(m,4H)、7.75(d,J=7.3Hz,1H)、11.52(s,1H,NH)。
【0113】
13C NMR(75MHz,DMSO−d6)δ 10.5、12.7、32.7、37.9、39.5、53.0、97.6、103.3、109.87、109.92、120.3、123.5、123.8、124.3、124.7、124.9、127.8、129.4、131.8、151.3、166.2。
【0114】
m/z(ES+)367.1(M+H)。
【0115】
実施例1(iii):(±)−11−(2−フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0116】
【化12】
(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(150mg、0.41mmol、実施例1(ii))の無水DMF(4ml)溶液に、L.Cronin et al(J.Org.Chem.2004;69:5934−5946)に記載されているようにして製造した2−フルオロエチルトシレート(166mg、0.82mmol)を添加し、次いで鉱油中の水素化ナトリウム60%分散液(34mg、0.82mmol)を窒素下で添加した。反応混合物を80℃で1時間加熱した。冷却後、溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。5〜10%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製した。粗生成物をエーテル/石油スピリットで奪活し、濾過し、(45℃で)真空乾燥することで、標記化合物(77mg、46%)を淡褐色の固体として得た。
【0117】
1H NMR(300MHz,CDCl3)δ 1.12(t,J=7.0Hz,3H)、1.36(t,J=7.0Hz,3H)、3.25−3.70(m,4H)、3.83(s,3H)、4.45−4.70(m,2H)、4.80(t,J=5.2Hz,1H)、4.96(t,J=5.2Hz,1H)、5.09(s,1H)、6.84−6.93(m,2H)、7.13−7.32(m,3H)、7.46(m,1H)、7.58(d,J=8.0Hz,1H)。
【0118】
13C NMR(75MHz,CDCl3)δ 12.9、14.9、37.3、41.1、42.5、45.5、45.8、55.9、81.2、83.5、100.4、110.1、111.09、111.12、112.8、124.31、124.35、125.2、126.5、127.1、127.6、128.8、132.2、134.4、137.0、154.8、168.0。
【0119】
19F NMR(282MHz,CDCl3)δ −219.4、−219.5、−219.6、−219.65、−219.73、−219.9、−219.9。
【0120】
m/z(ES+)413.1(M+H)。
【0121】
実施例2:(±)−11−(2−フルオロエチル)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤3)の製造
実施例2(i):(±)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0122】
【化13】
この化合物は、4−メトキシフェニルヒドラジン塩酸塩の代わりに2−メトキシフェニルヒドラジン塩酸塩を使用した点を除き、実施例1(ii)に関して記載したようにして製造した。化合物は40%の収率で得られた。
【0123】
m/z(ES+)367.0(M+H)。
【0124】
実施例2(ii):(±)−11−(2−フルオロエチル)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0125】
【化14】
この化合物は、(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの代わりに(±)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例2(i))を使用した点を除き、実施例1(iii)に関して記載したようにして製造した。再結晶(エーテル)後、化合物は白色の固体として10%の収率で得られた。
【0126】
1H NMR(300MHz,CDCl3)δ 1.09(t,J=7.0Hz,3H)、1.35(t,J=7.0Hz,3H)、3.25−3.67(m,4H)、3.95(s,3H)、4.70−4.96(m,4H)、5.04(s,1H)、6.67(m,1H)、7.04(m,2H)、7.16(m,1H)、7.29(m,1H)、7.45(m,1H)、7.77(m,1H)。
【0127】
m/z(ES+)413.1(M+H)。
【0128】
実施例3:(±)−4−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤4)の製造
実施例3(i):(±)−8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸
【0129】
【化15】
丸底フラスコ内において、トルエン(12mL)中の2−フルオロチオフェノール(5.0g、39.0mmol、4.16mL)及びフラン−2,5−ジオン(3.82g、39.0mmol)を50℃で40分間撹拌した。次いで、反応温度が60℃を超えないようにしながら、トルエン(5mL)中のトリエチルアミン(100μl)を10分かけて添加した。次いで、反応物を70℃で20分間加熱した。次いで、反応物を高真空下で濃縮することで、粗生成物を油状物として得た。この物質をDCM(75mL)に溶解し、氷浴上で冷却し、温度を10℃未満に保つようにしながら三塩化アルミニウム(7.78g、58.5mmol)を少量ずつ加えて処理した。反応物をRTまで温めたところ、塩化水素ガスの激しい発生が起こり、反応物は非常に粘稠になると共に赤色に変わった。RTで1.5時間撹拌した後、反応混合物をDCM(50mL)で希釈してその粘度を低下させ、2L三角フラスコ内の激しく撹拌した濃塩酸(30mL)及び氷(30g)中にゆっくりと注ぎ込んだ。反応物を激しく撹拌し、追加のDCM(500mL)及びイソプロピルアルコール(50mL)で希釈して晶出した固体を溶解した。DCM層を分離し、硫酸マグネシウム上で乾燥し、真空中で濃縮して褐色の固体を得た。粗生成物を精製し、ジエチルエーテルでトリチュレートし、クリーム色の固体を濾過により集めて2.5g(28%)の8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸を得た。1H NMR(300MHz;DMSO−d3):δ 3.04−3.20(2H,m,3−H)、4.51(1H,dd,J=4及び6Hz,2−H)、7.26−7.34(1H,m,6−H)、7.45−7.52(1H,m,7−H)、7.82(1H,dd,J=1及び8Hz,5H)。13C NMR(75MHz;DMSO−d3):δ 40.5、40.7、119.8、120.1、123.88、123.92、126.0、126.1、131.9、156.1、159.2、171.5、191.2、191.3。
【0130】
実施例3(ii):(±)−8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド
【0131】
【化16】
乾燥DCM(50ml)中の8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸(2.5g、11.1mmol)を、反応を触媒するための塩化オキサリル(2.81g、22.1mmol、1.93mL)及び1滴のDMFと共に窒素雰囲気下において室温で18時間撹拌した。酸は最初は不溶性であったが、反応するに従って溶解し、2時間後にはオレンジ色の透明溶液となり、次いで24時間後には黒色に変わった。次いで、反応物を真空中で蒸発させてガム状にして過剰の塩化オキサリルを除去し、CDCl3中で1H NMR及び13C NMRを実施して反応の完了を確認した。次いで、反応物をDCM(50ml)に再溶解し、氷浴上で0℃に冷却し、撹拌しながらDCM(20ml)中のジエチルアミン(1.66g、22.7mmol、2.05mL)を1時間かけて滴下することで処理した。反応物を1時間かけて室温まで放温した。次いで、反応物を5%炭酸カリウム溶液(100ml)の添加によって奪活し、反応混合物を激しく撹拌した。DCM溶液を分離し、硫酸マグネシウム上で乾燥した。DCMの追加バッチ(100ml)を水溶液と共に振盪し、次いで分離して硫酸マグネシウム上で乾燥することを2回繰り返した。合わせたDCM溶液を真空中で濃縮して褐色の固体を得た。粗固体を酢酸エチル及びガソリンからの高温再結晶によって精製することで、1.76g(56%)の8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミドを黄色の結晶として得た。1H NMR(300MHz;CDCl3):δ 1.07(3H,t,J=7Hz,N(CH2CH3)2)、1.26(3H,t,J=7Hz,N(CH2CH3)2)、3.02−3.55(6H,m,2−H及びN(CH2CH3)2)、4.24−4.27(1H,m,2−H)、7.15−7.19(2H,m,6−H及び7−H)、7.93−7.97(1H,m,5−H)。
【0132】
LC−MS:C14H16FNO2Sに関するm/z計算値、281.1;実測値、282.0(M+H)+。
【0133】
実施例3(iii):(±)−4−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0134】
【化17】
エタノール(10mL)及び硫酸(濃、0.8mL)中の8−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(1.7g、6.0mmol)及びフェニルヒドラジン(0.65g、6.0mmol、0.6mL)を、還流させながら一晩撹拌した。冷却後、反応物を濾過し、白色の固体を集めて1.4g(80%)の粗物質(純度90%)を得た。粗固体(500mg)をエタノールからの高温再結晶によって精製することで、277mg(13%)の4−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の結晶として得た。構造は1H NMRによって確認した。1H NMR(300MHz;DMSO−d6):δ 0.96(3H,t,J=7Hz,N(CH2CH3)2)、1.30(3H,t,J=9Hz,N(CH2CH3)2)、3.19−3.25(2H,m,N(CH2CH3)2)、3.56−3.66(2H,m,N(CH2CH3)2)、5.76(1H,s,6−H)、7.02−7.45(6H,m,ArH)、7.65(1H,dd,J=1及び6Hz,ArH)、11.8(1H,s,NH)。
【0135】
LC−MS:C20H19FN2OSに関するm/zの計算値、354.2;実測値、355.0(M+H)+。
【0136】
実施例3(iv):(±)−4−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0137】
【化18】
(±)−4−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(0.10g、0.28mmol)を窒素下において室温で乾燥DMF(6mL)に溶解した。フルオロエチルトシレート(0.12g、0.12mmol)を添加し、次いでNaH(0.02g、0.56mmol、油中60%)を添加した。反応物を80℃で1時間加熱した。溶媒を減圧下で除去し、残留物をDCMに溶解し、水で洗浄した。有機物をMgSO4上で乾燥し、濾過し、蒸発乾固させた。粗物質をメタノールから晶出させることで、34.4mg(30%)の4−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得た。1H NMR(300MHz,DMSO−d6)δ 0.94(3H,t,J=7Hz,N(CH2CH3)2)、1.29(3H,t,J=7Hz,N(CH2CH3)2)、3.14−3.26(2H,m,N(CH2CH3)2)、3.55−3.65(2H,m,N(CH2CH3)2)、4.65−4.95(4H,m,NCH2CH2F)、5.62(1H,s,6−H)、7.12−7.37(4H,m,ArH)、7.48(1H,d,J=9Hz,ArH)、7.61−7.68(2H,m,ArH)。
【0138】
LC−MS:C22H22F2N2OSに関するm/zの計算値、401.1;実測値、401.1(M+H)+。
【0139】
実施例4:(±)−3−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤5)の製造
実施例4(i):(±)−7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸
【0140】
【化19】
丸底フラスコ内において、トルエン(12mL)中の3−フルオロチオフェノール(10.0g、71.3mmol、8.85mL)及びフラン−2,5−ジオン(7.0g、71.3mmol)を50℃で40分間撹拌した。次いで、反応温度が60℃を超えないようにしながら、トルエン(1mL)中のトリエチルアミン(26μl)を10分かけて添加した。次いで、反応物を70℃で20分間加熱した。次いで、反応物を高真空下で濃縮することで、粗生成物を油状物として得た。この物質をDCM(75mL)に溶解し、氷浴上で冷却し、温度を10℃未満に保つようにしながら三塩化アルミニウム(7.78g、58.5mmol)を少量ずつ加えて処理した。反応物をRTまで温めたところ、塩化水素ガスの激しい発生が起こり、反応物は非常に粘稠になると共に赤色に変わった。室温で1.5時間撹拌した後、反応混合物をDCM(50mL)で希釈してその粘度を低下させ、2L三角フラスコ内の激しく撹拌した濃塩酸(30mL)及び氷(30g)中にゆっくりと注ぎ込んだ。反応物を激しく撹拌し、追加のDCM(500mL)及びイソプロピルアルコール(50mL)で希釈して晶出した固体を溶解した。DCM層を分離し、硫酸マグネシウム上で乾燥し、真空中で濃縮して褐色の固体を得た。固体をジエチルエーテルでトリチュレートし、次いで濾過することで、4.2g(48%)の7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸をクリーム色の固体として得た。1H NMR(300MHz;DMSO−d3):δ 3.00−3.16(2H,m,3−H)、4.44(1H,dd,J=5及び10Hz,2−H)、7.08(1H,td,J1=3及び9Hz,6−H)、7.30(1H,dd,J=5及び10Hz,ArH)、8.01(1H,dd,J1=5及び10Hz,ArH)。13C NMR(75MHz;DMSO−d3):δ 38.0、39.6、111.1、111.3、111.5、111.8、125.0、125.1、129.0、129.2、139.6、139.7、160.9、164.3、169.5、188.9。
【0141】
実施例4(ii):(±)−7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド
【0142】
【化20】
乾燥DCM(50ml)中の7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸(4g、17.7mmol)を、反応を触媒するための塩化オキサリル(4.49g、35.4mmol、3.1mL)及び1滴のDMFと共に窒素雰囲気下において室温で18時間撹拌した。酸は最初は不溶性であったが、反応するに従って溶解し、2時間後にはオレンジ色の透明溶液となり、次いで24時間後には黒色に変わった。次いで、反応物を真空中で蒸発させてガム状にして過剰の塩化オキサリルを除去し、CDCl3中で1H NMR及び13C NMRを実施して反応の完了を確認した。次いで、反応物をDCM(50ml)に再溶解し、氷浴上で0℃に冷却し、撹拌しながらDCM(10ml)中のジエチルアミンを1時間かけて滴下することで処理した。反応物を1時間かけて室温まで放温した。次いで、反応物を5%炭酸カリウム溶液(50ml)の添加によって奪活し、反応混合物を激しく撹拌した。DCM溶液を分離し、硫酸マグネシウム上で乾燥した。DCMの追加バッチ(50ml)を水溶液と共に振盪し、次いで分離して硫酸マグネシウム上で乾燥することを2回繰り返した。合わせたDCM溶液を真空中で濃縮して褐色の固体を得、これを静置して結晶化させることで、5.03g(クアント(quant))の7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミドを得た。構造は1H NMRによって確認した。1H NMR(300MHz;CDCl3):δ 1.07(3H,t,J=7Hz,N(CH2CH3)2)、1.24(3H,t,J=7Hz,N(CH2CH3)2)、2.99−3.50(6H,m,2−H及びN(CH2CH3)2)、4.24−4.27(1H,m,2−H)、6.83−6.94(2H,m,6−H及び8−H)、8.15(1H,dd,J=6及び9Hz,5−H)。
【0143】
LC−MS:C14H16FNO2Sに関するm/zの計算値、281.1;実測値、282.0(M+H)+。
【0144】
実施例4(iii):(±)−3−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0145】
【化21】
エタノール(10mL)及び硫酸(濃、1.2mL)中の7−フルオロ−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(2.5g、8.9mmol)及びフェニルヒドラジン(0.96g、8.9mmol、0.9mL)を、還流させながら一晩撹拌した。粗固体をエタノールからの高温再結晶によって精製することで、1.49g(47%)の3−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の結晶として得た。1H NMR(300MHz;DMSO−d6):δ 0.96(3H,t,J=6Hz,N(CH2CH3)2)、1.29(3H,t,J=6Hz,N(CH2CH3)2)、3.19−3.25(2H,m,N(CH2CH3)2)、3.55−3.61(2H,m,N(CH2CH3)2)、5.66(1H,s,6−H)、7.03(1H,td,J=1及び8Hz,ArH)、7.09−7.18(2H,m,ArH)、7.25(1H,dd,J=3及び9Hz,ArH)、7.35(1H,d,J=8Hz,ArH)、7.41(1H,d,J=8Hz,ArH)、7.81(1H,dd,J=6及び9Hz,ArH)、11.68(1H,s,NH)。
【0146】
LC−MS:C20H19FN2OSに関するm/zの計算値、352.1;実測値、353.2(M+H)+。
【0147】
実施例4(iv):(±)−3−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0148】
【化22】
(±)−3−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(0.20g、0.56mmol)を窒素下において室温で乾燥DMF(6mL)に溶解した。フルオロエチルトシレート(0.25g、1.13mmol)を添加し、次いでNaH(0.05g、1.13mmol、油中60%)を添加した。反応物を80℃で1時間加熱した。溶媒を減圧下で除去し、残留物をDCMに溶解し、水で洗浄した。有機物をMgSO4上で乾燥し、濾過し、蒸発乾固させた。水(A)及びアセトニトリル(B)を溶離剤とする半分取HPLC(Gemini 5μ、C18、110A、150×21mm、20分で5〜95%B、21mL/分)によって粗物質を精製することで、79.9mg(35%)の3−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得た。1H NMR(300MHz,DMSO−d6)δ 0.95(3H,t,J=9Hz,N(CH2CH3)2)、1.88(3H,t,J=9Hz,N(CH2CH3)2)、3.14−3.26(2H,m,N(CH2CH3)2)、3.51−3.67(2H,m,N(CH2CH3)2)、4.58−4.97(4H,m,NCH2CH2F)、5.53(1H,s,6−H)、7.12−7.27(3H,m,ArH)、7.38−4.47(2H,m,ArH)、7.61(1H,d,J=9Hz,ArH)、7.80−7.86(1H,m,ArH)。
【0149】
LC−MS:C22H22F2N2OSに関するm/zの計算値、401.1;実測値、401.1(M+H)+。
【0150】
実施例5:(±)−8−エトキシ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤6)の製造
実施例5(i):(±)−11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0151】
【化23】
実施例1(ii)の(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(2.0g、5.40mmol)の無水DMF(20ml)溶液に、鉱油中の水素化ナトリウム60%分散液(240mg、6.0mmol)を添加し、混合物を窒素下において室温で5分間撹拌した。2−(ブロモエトキシ)−tert−ブチル−ジメチルシラン(2.6g、10.8mmol)を添加し、混合物を4時間撹拌した。溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。3%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、標記化合物(2.0g、70%)を黄色の固体として得た。
【0152】
実施例5(ii):(±)−11−[2−ヒドロキシエチル]−8−ヒドロキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0153】
【化24】
(±)−11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(1.0g、1.91mmol)の−78℃の乾燥DCM(60ml)溶液に、三臭化ホウ素(11.5ml、DCM中1M、11.5mmol)を添加した。溶液をRTまで昇温させ、24時間撹拌した。溶媒を真空中で除去し、メタノール(40ml)で奪活し、1M HCl(10ml)を添加し、1時間還流した。溶媒を真空中で除去し、混合物をメタノール(5ml)に溶解し、水(100ml)で奪活し、濾過し、(45℃で)真空乾燥することで、標記化合物(0.77g、100%)を淡褐色の粉末として得た。
【0154】
実施例5(iii):(±)−11−[2−ヒドロキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0155】
【化25】
(±)−11−[2−ヒドロキシエチル]−8−ヒドロキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(400mg、1.01mmol)の0℃の無水DMF(4ml)溶液に、鉱油中の水素化ナトリウム60%分散液(40mg、1.01mmol)を添加した。混合物を窒素下において0℃で10分間撹拌した。臭化エチル(218mg、2.0mmol、150μl)を添加し、混合物を24時間撹拌した。溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。40〜60%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、標記化合物(340mg、79%)を白色の固体として得た。
【0156】
実施例5(iv):(±)−11−[2−メタンスルホキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0157】
【化26】
(±)−11−[2−ヒドロキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(0.34g、0.80mmol)の無水DCM(15ml)懸濁液に、ピリジン(0.63g、8.0mmol、0.65ml)を添加した。反応物を0℃に冷却し、メタンスルホニルクロリド(0.37g、3.2mmol、0.25ml)を添加した。反応混合物をRTで3時間撹拌した。混合物を05M HCl(2×20ml)で洗浄し、次いで水(2×20ml)で洗浄し、(MgSO4で)乾燥し、溶媒を減圧下で除去した。20%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製した。残留物をエーテル/石油スピリットで奪活し、濾過し、(45℃で)真空乾燥することで、標記化合物(0.38g、95%)を淡黄色の固体として得た。
【0158】
実施例5(v):(±)−11−[2−フルオロエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0159】
【化27】
窒素下にある(±)−11−[2−メタンスルホキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(100mg、0.20mmol)の無水アセトニトリル(5ml)溶液に、THF中の1.0M TBAF(0.4ml、0.4mmol)を添加した。混合物を80℃で2時間加熱した。溶媒を真空中で除去し、5〜10%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、標記化合物(26mg、31%)を黄色の固体として得た。
【0160】
実施例6:(±)−7−メトキシ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤2)及び(±)−9−メトキシ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性イメージング剤7)の製造
実施例6(i):7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0161】
【化28】
(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(3.33g、12.6mmol、実施例1(i))及び3−メトキシフェニルヒドラジン塩酸塩(2.2g、12.6mmol)のエタノール(30ml)溶液に、濃硫酸(1.83ml、3.40g、11.5mmol)を窒素下で添加した。反応混合物を24時間加熱還流した。冷却後、反応混合物を濾過し、固体をエタノールで洗浄し、(45℃で)真空乾燥することで、7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの混合物(3.2g、69%)を淡白色の固体として得た。
【0162】
実施例6(ii):11−(2−フルオロエチル)−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−(2−フルオロエチル)−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0163】
【化29】
(実施例6(i)に従って製造した)7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの異性体混合物(1.0g、2.73mmol)の無水DMF(10mL)溶液に、2−フルオロエチルトシレート(1.2g、5.46mmol)を添加し、次いで鉱油中の水素化ナトリウム60%分散液(131mg、5.46mmol)を窒素下で添加した。反応混合物を80℃で1時間加熱した。冷却後、溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。5〜10%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、異性体混合物(1.0g、89%)を得た。次いで、水(A)及びメタノール(B)を溶離剤とするHPLC(Gemini 5μ、C18、110A、150×21mm、20分で70〜95%B、21mL/分)によってこの物質(400mg)を精製することで、240mgの9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを黄色の固体として得ると共に、100mgの7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得た。
【0164】
実施例7:18F標識イメージング剤2及び7の製造
実施例7(i):11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0165】
【化30】
実施例6(i)に従って製造した異性体混合物(2.0g、5.46mmol)の無水DMF(20ml)溶液に、鉱油中の水素化ナトリウム60%分散液(240mg、6.0mmol)を添加し、混合物を窒素下において室温で5分間撹拌した。2−(ブロモエトキシ)−tert−ブチル−ジメチルシラン(2.6g、10.9mmol)を添加し、混合物を4時間撹拌した。溶媒を真空中で除去し、残留物を水(30ml)で奪活し、DCM(2×30ml)で抽出し、(MgSO4で)乾燥し、溶媒を真空中で除去した。5%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−[2−(tert−ブチル−ジメチル−シラニルオキシ)エチル]−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの異性体混合物(2.53g、88%)を黄色の固体として得た。
【0166】
実施例7(ii):11−[2−ヒドロキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−[2−(2−ヒドロキシオキシ)エチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0167】
【化31】
実施例7(i)に従って製造した異性体混合物(2.5g、4.76mmol)の無水THF(40ml)溶液に、THF中の1.0M TBAF(9.5ml、9.5mmol)を窒素下で添加した。混合物を室温で4時間撹拌した。溶媒を真空中で除去し、40%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、11−[2−ヒドロキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−[2−(2−ヒドロキシオキシ)エチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドの異性体混合物(1.90g、97%)を淡黄色の固体として得た。
【0168】
実施例7(iii):(±)−11−[2−メタンスルホキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び(±)−11−[2−メタンスルホキシエチル]−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド
【0169】
【化32】
実施例7(ii)に従って製造した異性体混合物(1.0g、2.4mmol)の無水DCM(30ml)溶液に、ピリジン(1.9g、24.0mmol、1.9ml)を添加した。反応物を0℃に冷却し、メタンスルホニルクロリド(1.1g、9.6mmol、0.74ml)を添加した。反応混合物をRTで4時間撹拌した。混合物を05M HCl(2×20ml)で洗浄し、次いで水(2×20ml)で洗浄し、(MgSO4で)乾燥し、溶媒を減圧下で除去した。20%EtOAc/CH2Cl2を溶離剤とするシリカ上でのカラムクロマトグラフィーによって残留物を精製することで、異性体混合物(1.0g、85%)を得た。次いで、
水(A)及びメタノール(B)を溶離剤とするHPLC(Gemini 5μ、C18、110A、150×21mm、30分で5〜95%B、21mL/分)によってこの物質(400mg)を精製することで、170mgの11−[2−メタンスルホキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得ると共に、60mgの11−2−(メタンスルホキシエチル)−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを白色の固体として得た。
【0170】
実施例7(iv):直接標識方法
実施例7(iii)に従って製造した前駆体化合物11−[2−メタンスルホキシエチル]−7−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド及び11−2−(メタンスルホキシエチル)−9−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを直接標識方法によって放射性標識することで、それぞれイメー
ジング剤2及び7を得た。18F−水を反応器に添加し、次いでアセトニトリル(500μl)中のK222(2mg)及びKHCO3(0.1mol dm−3、50μl)を添加し、100℃で20〜30分間乾燥した。アセトニトリル(1000μl)中の前駆体(0.5〜1mg)を添加した。反応器を密封し、100℃で10分間加熱した。反応混合物を冷却し、水(1.5ml)で反応器から洗い出し、半分取HPLCによって精製した。主な放射性生成物を含む画分を集め、H2Oで10mlの体積に希釈した。これをコンディショニングされたライトC18 sep pak上に装填し、H2O(1×2ml)でフラッシュし、生成物をEtOH(0.5ml)でP6バイアル中に溶出し、PBS(5ml)を添加した。
【0171】
実施例8:18F標識イメージング剤1、3、4及び5の製造
実施例8(i):前駆体化合物の製造
以下の前駆体化合物を製造した。
(a)(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例1(ii)に従って製造した)。
(b)(±)−10−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例2(i)に従って製造した)。
(c)(±)−4−フルオロ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例3(iii)に従って製造した)。
(d)(±)−3−フルオロ−11−(2−フルオロエチル)−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(実施例4(iv)に従って製造した)。
以下に記載する間接標識方法を用いてこれらの前駆体化合物を放射性標識することで、それぞれ18F標識イメージング剤1、3、4及び5を得た。
【0172】
実施例8(ii):間接標識方法
反応器内のK222(4mg)、K2CO3水溶液(0.1モル溶液の50μl)及びアセトニトリル(500μl)に18F−/水を添加し、窒素流下において100℃で20〜30分間乾燥した。アセトニトリル(1000μl)中のエチル−1,2−ジトシレート(4mg)を添加し、100℃で10分間加熱した。反応混合物を冷却し、半分取HPLCによって精製し、18F−フルオロエチルトシレートを含む画分を集めた。この画分をH2Oで約20mlの体積に希釈し、コンディショニングされたライトt−C18 sep pak上に装填し、H2O(1×2ml)でフラッシュした。このsep pakを、高流量のN2ライン上で20分間乾燥した。次いで、18F−フルオロエチルトシレートをDMF(500μl)で溶出した。
【0173】
別途に、DMF(250μl)中の前駆体(13mg)を第2の反応器に添加し、N2で5分間パージした。次いで、DMF(2×250μl)中のNaH(13mg)を窒素下で添加し、反応器を45℃で0.5〜1時間加熱した。次いで、これに上記のようにして調製したDMF中の18F−フルオロエチルトシレートを添加し、N2でパージした反応器内において100℃で10分間加熱した。反応物を冷却し、水(1ml)で反応器から洗い出した。溶液をシリンジフィルターで濾過し、分取HPLCで精製した。主な放射性ピークを含む画分を集めた。これをH2Oで約10mlの体積に希釈し、コンディショニングされたライトC18 sep pak上に装填し、H2O(1×2ml)でフラッシュし、EtOH(0.5ml)でP6バイアル中に溶出し、リン酸緩衝食塩水(5ml)を添加した。
【0174】
実施例9:18F標識イメージング剤6の製造
(実施例5(iv)に従って製造した)(±)−11−[2−メタンスルホキシエチル]−8−エトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミドを、上記実施例7(iv)に記載した直接標識方法を用いて放射性標識した。
【0175】
実施例10:インビトロ効力アッセイ
Le Fur et al(Life Sci.1983;USA 33:449−57)の方法を改変した方法を用いて、PBRに対する親和性に関する化合物のスクリーニングを行った。
【0176】
(1%DMSOを含む50mMトリス−HCl、pH7.4、10mM MgCl2に溶解した)試験すべき化合物を、ウィスター系ラットの心臓PBRとの結合に関して0.3nM[3H]−PK−11195と競合させた。反応は50mMトリス−HCl、pH7.4、10mM MgCl2中において25℃で15分間実施した。
【0177】
化合物のスクリーニングは、推定Kiの周辺の300倍の濃度範囲にわたる6種の濃度で行った。結果は上記表1に示されており、本発明の化合物の効力が先行技術の化合物の効力に比べて勝ることを実証している。
【0178】
実施例11:インビボ体内分布方法
上記実施例7、実施例8及び実施例9で製造したインビボイメージング剤1〜7をインビボ体内分布モデルで試験し、その体内分布を(国際公開第2007/057705号の実施例14に従って製造した)先行技術の化合物[18F]FE−PBRの体内分布と比較した。
【0179】
雄ウィスター系ラット成体(200〜300g)に、側尾静脈を通して1〜3MBqの各インビボイメージング剤を注射した。注射から2分後、10分後、30分後又は60分後(n=3)に、ラットを安楽死させ、液体シンチレーションカウンティングによる放射能測定のために組織又は体液の試料を採取した。
【0180】
[18F]FE−PBRに比べて、本発明の化合物は高い嗅球:線条体結合比を示した(上記表1参照)。
【0181】
実施例12:顔面神経軸索切断(FNA)モデルを用いたオートラジオグラフィー
インビボ試験のためには、雄ウィスター系ラット(180〜200g)を使用した。イソフルラン麻酔下で、右側の耳領域から毛を除去した。耳内切開を行い、顔面神経の主幹を確認した。耳の後方において、茎乳突孔からの出口で顔面神経を切断した。切創を縫合し、動物を回復させた。手術から7日後の動物に、側尾静脈を通して約5〜10MBqのインビボイメージング剤1を注射した。30分後に脳幹を取り出し、イソペンタン中で凍結した。両顔面神経核を含む脳幹のクリオスタット切片(12μm)をスライドガラス上にマウントし、蛍光スクリーンに一晩露光させた。
【0182】
次いで、Storm(GE Healthcare社)蛍光イメージャー上でスクリーンを走査し、ImageQuant TL(GE Healthcare社)を用いて得られたスキャンの分析及び定量化を行った。図1及び図2は、この実験で得られたデータを示している。
【特許請求の範囲】
【請求項1】
次の式Iのインビボイメージング剤或いはそれの塩又は溶媒和物であって、前記式Iのインビボイメージング剤の少なくとも1つの原子がインビボイメージングのために適した放射性同位体であり、前記放射性同位体が炭素の放射性同位体である場合にそれはカルボニル炭素である、インビボイメージング剤。
【化1】
(式中、
Qは水素又はフッ素であり、
Xは水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−6アルキル、C1−6ハロアルキル、C1−6アルコキシ又はC1−6アルキルアミドであり、
YはS、SO又はSO2であり、
Rは水素、C1−6アルキル又はC1−6フルオロアルキルであり、
ただしYがSである場合にはQ及びXが共に水素ではないことを条件とする。)
【請求項2】
Qが水素である、請求項1記載のインビボイメージング剤。
【請求項3】
Xが水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ又はC1−4アルキルアミドである、請求項1又は請求項2記載のインビボイメージング剤。
【請求項4】
Xが水素又はC1−4アルコキシである、請求項3記載のインビボイメージング剤。
【請求項5】
YがS又はSO2である、請求項1乃至請求項4のいずれか1項記載のインビボイメージング剤。
【請求項6】
YがSである、請求項5記載のインビボイメージング剤。
【請求項7】
Rが水素、C1−4アルキル又はC1−4フルオロアルキルである、請求項1乃至請求項6のいずれか1項記載のインビボイメージング剤。
【請求項8】
RがC1−4フルオロアルキルである、請求項7記載のインビボイメージング剤。
【請求項9】
Xが水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ又はC1−4アルキルアミドであり、
YがS又はSO2であり、
Rが水素、C1−4アルキル又はC1−4フルオロアルキルである、請求項1記載のインビボイメージング剤。
【請求項10】
Qが水素であり、
XがC1−4アルコキシであり、
YがSであり、
RがC1−4フルオロアルキルである、請求項8記載のインビボイメージング剤。
【請求項11】
Qがフッ素であり、
Xが水素であり、
YがSであり、
RがC1−4フルオロアルキルである、請求項8記載のインビボイメージング剤。
【請求項12】
前記放射性同位体がγ線放出型放射性ハロゲン又は陽電子放出型放射性非金属である、請求項1乃至請求項11のいずれか1項記載のインビボイメージング剤。
【請求項13】
前記γ線放出型放射性ハロゲンが123I、131I及び77Brから選択される、請求項12記載のインビボイメージング剤。
【請求項14】
前記陽電子放出型放射性非金属が11C、13N、18F及び124Iから選択される、請求項12記載のインビボイメージング剤。
【請求項15】
RがC1−4[18F]−フルオロアルキルである、請求項14記載のインビボイメージング剤。
【請求項16】
以下のものから選択される、請求項1記載のインビボイメージング剤。
【化2】
【請求項17】
請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤の製造において有用な前駆体化合物であって、次の式IIを有する前駆体化合物。
【化3】
(式中、R1、X1及びZ1の1つが請求項1及び請求項12乃至請求項14のいずれか1項記載の放射性同位体の適当な供給源と反応する化学基を含む結果、当該前駆体化合物を前記放射性同位体の前記適当な供給源と反応せれば前記インビボイメージング剤が生成され、
R1が前記化学基を含まない場合、それは請求項1及び請求項7乃至請求項11のいずれか1項でRに関して定義した通りであり、任意にはさらに保護基を含み、
X1が前記化学基を含まない場合、それは請求項1、請求項3、請求項4及び請求項9乃至請求項11のいずれか1項でXに関して定義した通りであり、任意にはさらに保護基を含み、
Z1が前記化学基を含まない場合、それは−C(=O)−N−(CH2−CH3)2であり、任意にはさらに保護基を含み、
Q1は請求項1、請求項2及び請求項9乃至請求項11のいずれか1項でQに関して定義した通りであり、
Y1は請求項1、請求項5、請求項6及び請求項9乃至請求項11のいずれか1項でYに関して定義した通りであり、任意にはさらに保護基を含む。)
【請求項18】
前記放射性同位体の前記適当な供給源が18Fの供給源であり、前記化学基が
(i)求核置換用のアルキルハライド又はアルキルスルホネート、及び
(ii)ヒドロキシル
から選択される、請求項17記載の前駆体化合物。
【請求項19】
式IIのR1又はX1が請求項17に定義した通りであり、前記化学基を含む、請求項18記載の前駆体化合物。
【請求項20】
請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤の製造方法であって、
(i)請求項17乃至請求項19のいずれか1項記載の式IIの前駆体化合物を用意する段階、
(ii)請求項1及び請求項12乃至請求項14のいずれか1項記載の前記放射性同位体の適当な供給源を用意する段階、並びに
(iii)段階(i)の前駆体化合物を段階(ii)の放射性同位体の供給源と反応させて前記インビボイメージング剤を得る段階
を含んでなる方法。
【請求項21】
請求項1記載の方法を実施するためのキットであって、請求項17乃至請求項19のいずれか1項記載の前駆体化合物を含んでなるキット。
【請求項22】
請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤の自動化合成のためのカセットであって、
(i)請求項17乃至請求項19のいずれか1項記載の前駆体化合物を含む容器、並びに
(ii)請求項1及び請求項12乃至請求項14のいずれか1項記載の放射性同位体の適当な供給源を用いて容器を溶出するための手段
を含んでなるカセット。
【請求項23】
さらに、
(iii)過剰の放射性標識を除去するためのイオン交換カートリッジ、及び任意には
(iv)得られた放射性標識生成物を脱保護して請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤を生成するためのカートリッジ
を含む、請求項22記載のカセット。
【請求項24】
請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤を、哺乳動物への投与に適した形態の生体適合性キャリヤーと共に含んでなる放射性医薬組成物。
【請求項25】
被験体におけるPBR発現の体内分布及び/又は程度を決定するためのインビボイメージング方法であって、
(i)請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤を前記被験体に投与する段階、
(ii)前記イメージング剤を前記被験体中のPBRに結合させる段階、
(iii)前記イメージング剤の放射性同位体から放出される信号をインビボイメージング技法によって検出する段階、
(iv)前記信号の位置及び/又は量を表す画像を生成する段階、並びに
(v)前記被験体におけるPBR発現の分布及び程度を決定する段階であって、前記発現は前記インビボイメージング剤から放出される前記信号と直接に相関している段階
を含んでなるインビボイメージング方法。
【請求項26】
前記被験体に関する治療計画の進行中に繰り返して実施される請求項25記載のインビボイメージング方法であって、前記計画がPBR状態と戦うための薬物の投与を含むインビボイメージング方法。
【請求項27】
PBRがアップレギュレートされた状態の診断方法であって、請求項25又は請求項26のいずれか記載のインビボイメージング方法を、PBR発現の分布及び程度を特定の臨床像に帰因させる追加段階(vi)と共に含んでなる診断方法。
【請求項28】
請求項27記載の診断方法で使用するための、請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤。
【請求項29】
請求項27記載の診断方法で使用するための請求項24記載の放射性医薬組成物の製造で使用するための、請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤。
【請求項1】
次の式Iのインビボイメージング剤或いはそれの塩又は溶媒和物であって、前記式Iのインビボイメージング剤の少なくとも1つの原子がインビボイメージングのために適した放射性同位体であり、前記放射性同位体が炭素の放射性同位体である場合にそれはカルボニル炭素である、インビボイメージング剤。
【化1】
(式中、
Qは水素又はフッ素であり、
Xは水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−6アルキル、C1−6ハロアルキル、C1−6アルコキシ又はC1−6アルキルアミドであり、
YはS、SO又はSO2であり、
Rは水素、C1−6アルキル又はC1−6フルオロアルキルであり、
ただしYがSである場合にはQ及びXが共に水素ではないことを条件とする。)
【請求項2】
Qが水素である、請求項1記載のインビボイメージング剤。
【請求項3】
Xが水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ又はC1−4アルキルアミドである、請求項1又は請求項2記載のインビボイメージング剤。
【請求項4】
Xが水素又はC1−4アルコキシである、請求項3記載のインビボイメージング剤。
【請求項5】
YがS又はSO2である、請求項1乃至請求項4のいずれか1項記載のインビボイメージング剤。
【請求項6】
YがSである、請求項5記載のインビボイメージング剤。
【請求項7】
Rが水素、C1−4アルキル又はC1−4フルオロアルキルである、請求項1乃至請求項6のいずれか1項記載のインビボイメージング剤。
【請求項8】
RがC1−4フルオロアルキルである、請求項7記載のインビボイメージング剤。
【請求項9】
Xが水素、フルオロ、ブロモ、ヨード、ヒドロキシ、C1−4アルキル、C1−4ハロアルキル、C1−4アルコキシ又はC1−4アルキルアミドであり、
YがS又はSO2であり、
Rが水素、C1−4アルキル又はC1−4フルオロアルキルである、請求項1記載のインビボイメージング剤。
【請求項10】
Qが水素であり、
XがC1−4アルコキシであり、
YがSであり、
RがC1−4フルオロアルキルである、請求項8記載のインビボイメージング剤。
【請求項11】
Qがフッ素であり、
Xが水素であり、
YがSであり、
RがC1−4フルオロアルキルである、請求項8記載のインビボイメージング剤。
【請求項12】
前記放射性同位体がγ線放出型放射性ハロゲン又は陽電子放出型放射性非金属である、請求項1乃至請求項11のいずれか1項記載のインビボイメージング剤。
【請求項13】
前記γ線放出型放射性ハロゲンが123I、131I及び77Brから選択される、請求項12記載のインビボイメージング剤。
【請求項14】
前記陽電子放出型放射性非金属が11C、13N、18F及び124Iから選択される、請求項12記載のインビボイメージング剤。
【請求項15】
RがC1−4[18F]−フルオロアルキルである、請求項14記載のインビボイメージング剤。
【請求項16】
以下のものから選択される、請求項1記載のインビボイメージング剤。
【化2】
【請求項17】
請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤の製造において有用な前駆体化合物であって、次の式IIを有する前駆体化合物。
【化3】
(式中、R1、X1及びZ1の1つが請求項1及び請求項12乃至請求項14のいずれか1項記載の放射性同位体の適当な供給源と反応する化学基を含む結果、当該前駆体化合物を前記放射性同位体の前記適当な供給源と反応せれば前記インビボイメージング剤が生成され、
R1が前記化学基を含まない場合、それは請求項1及び請求項7乃至請求項11のいずれか1項でRに関して定義した通りであり、任意にはさらに保護基を含み、
X1が前記化学基を含まない場合、それは請求項1、請求項3、請求項4及び請求項9乃至請求項11のいずれか1項でXに関して定義した通りであり、任意にはさらに保護基を含み、
Z1が前記化学基を含まない場合、それは−C(=O)−N−(CH2−CH3)2であり、任意にはさらに保護基を含み、
Q1は請求項1、請求項2及び請求項9乃至請求項11のいずれか1項でQに関して定義した通りであり、
Y1は請求項1、請求項5、請求項6及び請求項9乃至請求項11のいずれか1項でYに関して定義した通りであり、任意にはさらに保護基を含む。)
【請求項18】
前記放射性同位体の前記適当な供給源が18Fの供給源であり、前記化学基が
(i)求核置換用のアルキルハライド又はアルキルスルホネート、及び
(ii)ヒドロキシル
から選択される、請求項17記載の前駆体化合物。
【請求項19】
式IIのR1又はX1が請求項17に定義した通りであり、前記化学基を含む、請求項18記載の前駆体化合物。
【請求項20】
請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤の製造方法であって、
(i)請求項17乃至請求項19のいずれか1項記載の式IIの前駆体化合物を用意する段階、
(ii)請求項1及び請求項12乃至請求項14のいずれか1項記載の前記放射性同位体の適当な供給源を用意する段階、並びに
(iii)段階(i)の前駆体化合物を段階(ii)の放射性同位体の供給源と反応させて前記インビボイメージング剤を得る段階
を含んでなる方法。
【請求項21】
請求項1記載の方法を実施するためのキットであって、請求項17乃至請求項19のいずれか1項記載の前駆体化合物を含んでなるキット。
【請求項22】
請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤の自動化合成のためのカセットであって、
(i)請求項17乃至請求項19のいずれか1項記載の前駆体化合物を含む容器、並びに
(ii)請求項1及び請求項12乃至請求項14のいずれか1項記載の放射性同位体の適当な供給源を用いて容器を溶出するための手段
を含んでなるカセット。
【請求項23】
さらに、
(iii)過剰の放射性標識を除去するためのイオン交換カートリッジ、及び任意には
(iv)得られた放射性標識生成物を脱保護して請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤を生成するためのカートリッジ
を含む、請求項22記載のカセット。
【請求項24】
請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤を、哺乳動物への投与に適した形態の生体適合性キャリヤーと共に含んでなる放射性医薬組成物。
【請求項25】
被験体におけるPBR発現の体内分布及び/又は程度を決定するためのインビボイメージング方法であって、
(i)請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤を前記被験体に投与する段階、
(ii)前記イメージング剤を前記被験体中のPBRに結合させる段階、
(iii)前記イメージング剤の放射性同位体から放出される信号をインビボイメージング技法によって検出する段階、
(iv)前記信号の位置及び/又は量を表す画像を生成する段階、並びに
(v)前記被験体におけるPBR発現の分布及び程度を決定する段階であって、前記発現は前記インビボイメージング剤から放出される前記信号と直接に相関している段階
を含んでなるインビボイメージング方法。
【請求項26】
前記被験体に関する治療計画の進行中に繰り返して実施される請求項25記載のインビボイメージング方法であって、前記計画がPBR状態と戦うための薬物の投与を含むインビボイメージング方法。
【請求項27】
PBRがアップレギュレートされた状態の診断方法であって、請求項25又は請求項26のいずれか記載のインビボイメージング方法を、PBR発現の分布及び程度を特定の臨床像に帰因させる追加段階(vi)と共に含んでなる診断方法。
【請求項28】
請求項27記載の診断方法で使用するための、請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤。
【請求項29】
請求項27記載の診断方法で使用するための請求項24記載の放射性医薬組成物の製造で使用するための、請求項1乃至請求項16のいずれか1項記載のインビボイメージング剤。
【図1】


【図2】

【図2】
【公表番号】特表2012−504581(P2012−504581A)
【公表日】平成24年2月23日(2012.2.23)
【国際特許分類】
【出願番号】特願2011−529567(P2011−529567)
【出願日】平成21年10月2日(2009.10.2)
【国際出願番号】PCT/EP2009/062827
【国際公開番号】WO2010/037851
【国際公開日】平成22年4月8日(2010.4.8)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
【公表日】平成24年2月23日(2012.2.23)
【国際特許分類】
【出願日】平成21年10月2日(2009.10.2)
【国際出願番号】PCT/EP2009/062827
【国際公開番号】WO2010/037851
【国際公開日】平成22年4月8日(2010.4.8)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
[ Back to top ]