
神経疾患治療用プロテインキナーゼ阻害剤
【課題】新規なビスN−置換されたスタウロスポリンの誘導体であり、腫瘍の阻害、炎症の抑制、免疫調整、及び心臓血管や中枢神経系の疾患の治療に用いられる神経疾患治療用プロテインキナーゼ阻害剤の提供。
【解決手段】スタウロスポリンのビス−N−置換された新規な誘導体。病的な神経細胞を治療するための方法。新規なスタウロスポリン誘導体または特定されたK−252aの機能性誘導体のいずれかを投与し、コリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、及び感覚ニューロンの機能を高める方法。
【解決手段】スタウロスポリンのビス−N−置換された新規な誘導体。病的な神経細胞を治療するための方法。新規なスタウロスポリン誘導体または特定されたK−252aの機能性誘導体のいずれかを投与し、コリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、及び感覚ニューロンの機能を高める方法。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、現在は放棄されている1992年7月24日に提出された米国特許出願第07/920,102号の一部継続出願である、1993年7月22日に提出された米国特許出願第08/096,561号のさらなる一部継続出願である、1994年10月26に提出された米国特許出願第08/329,540号のさらなる一部継続出願である。
【背景技術】
【0002】
発明の背景
プロテインキナーゼは、アミノ酸のリン酸化を行うことによって多くの細胞内タンパク質を化学的に修飾する作用のある広範な酵素のクラスである。
【0003】
プロテインキナーゼの阻害剤は構造的にはいろいろあり、神経系や他の組織に対してさまざまな(時には反対の)影響を与える。ある特定のプロテインキナーゼ阻害剤は、一種以上のプロテインキナーゼに影響するであろう。例えば、ノカルジオプシスsp.(Nocardiopsis sp.)及びアクチノマヅラsp.(Actinomadula sp.)の培養液から単離されたアルカロイド様の物質であるK−252aは、当初プロテインキナーゼCの阻害剤であると報告されたが、その後プロテインキナーゼA及びG、ミオシン軽鎖キナーゼ、及びtrk(末梢ニューロン、感覚ニューロン、それに交感ニューロンの生存性を促進する神経栄養性タンパク質である神経生長因子[NGF]によって活性化されるチロシンキナーゼ)も阻害することが判明した。
【0004】
この後者の効果と一致して、K−252aはPC−12細胞(ラットの副腎髄質腫瘍、褐色細胞腫から得られるクロム親和性細胞)に対するNGFの神経栄養性作用を遮断し、背根の神経節ニューロン及び海馬ニューロンの生存性を向上させる。しかしながらそれは、広範囲の濃度で細胞毒性を示すことがわかっており、そのため何人かの研究者は、インビボでの有用性が制限されているという結論に至っている。
【0005】
K−252aに関係のある微生物のアルカロイドであるスタウロスポリン(staurosporine)も、異なるプロテインキナーゼ及び細胞型に対してさまざまな影響を持つ。スタウロスポリンはPC−12細胞に対してNGF−様の効果を持っていて、アレチネズミの海馬が虚血後障害(post−ischemic injury)にならないように保護できることがわかっている。それは、ラットの基底前脳においてコリン作動性ニューロンに起こる損傷を回復することができる。
【0006】
K−252a及びスタロウスポリンは腫瘍の阻害剤として推奨されてきた。スタウロスポリンは殺虫剤としても提供されている。スタウロスポリンの誘導体はメチルアミン窒素で置換されたハイドロカルビルラジカルまたはアシルラジカルを持っており、以下の使用のために合成されかつ推奨されている。即ち、腫瘍の阻害、炎症の抑制、免疫調整、及び心臓血管や中枢神経系の疾患の治療である。
【発明の開示】
【0007】
発明の概要
本発明のある観点での特徴は、下式によって示される新規なビスN−置換されたスタウロスポリンの誘導体である。
[Stau]−N(CH3)−W−N(CH3)−[Stau] (I)
ここで[Stau]は下式の残基を表し、

そしてWは下式のビス(カルバミル)またはビス(チオカルバミル)ラジカルを表す。
−C(=Y)−NH−W’−NH−C(=Y)−
ここでW’は2〜20個の炭素原子からなるハイドロカルビレンラジカルであって、YはOまたはSである。
【0008】
好ましい観点では本発明は、例えば化合物1,6−ヘキサメチレン−ビス−(カルバミルスタウロスポリン)(HBCS)、p−フェニレン−ビス−(カルバミルスタウロスポリン)(PBCS)を特徴とする。
【0009】
本発明はまた、下式(II−4)によって表される新規なK−252aの誘導体を特徴とする。
ここでR1、R2、Z1、及びZ2はそれぞれ独立にHであり、Xはハイドロキシメチル基(CH2OH)であり、そしてRはOCH3 である。
【0010】
本発明はまた、下式によって表される新規なK−252aの誘導体であることを特徴とする。
ここでR1、R2、Z1、及びZ2はそれぞれ独立にHであり、XはCH2−NH−Serであり、そしてRはOH である。
【0011】
また本発明には、下式(II−49)によって表される化合物が含まれる。
ここでR1、Z1、及びZ2はそれぞれHであり、RはOHであり、R1はCH2SO2C2H5であり、そしてXはCO2CH3である。
【0012】
また本発明には下式(II−38)によって示される化合物が含まれる。
ここでR1、R2、Z1、及びZ2はそれぞれHであり、RはOHであり、そしてXはCH2NHCO2C6H5である。
【0013】
また本発明には、下式(II−45)によって示される化合物が含まれる。
ここでR1、R2はそれぞれBrであり、RはOHであり、Z1及びZ2はそれぞれHであり、そしてXはCONHC6H5 である。
【0014】
また本発明には下式(II−57)によって示される化合物が含まれる。
ここでR1、R2、Z1、及びZ2はそれぞれHであり、RはOHであり、そしてXはCH2NHCO2CH3である。
【0015】
また本発明には下式(II−72)によって示される化合物が含まれる。
ここでR1はCH2S(CH2)2NH2であり、XはCO2CH3であり、RはOHであり、そしてR2、Z1、及びZ2はそれぞれHである。
【0016】
また本発明には下式(II−75)によって示される化合物が含まれる。
ここでR1は
であり、XはCO2CH3であり、RはOHであり、そしてR2、Z1、及びZ2はそれぞれHである。
【0017】
また本発明には下式(II−79)で示される化合物が含まれる。
ここでR1はCH2S(CH2)2NH n−C4H9であり、XはCO2CH3であり、RはOHであり、そしてR2、Z1、及びZ2はそれぞれHである。
【0018】
また本発明には下式(II−80)によって示される化合物が含まれる。
ここでR1はCH2S(CH2)2N(CH3)2であり、R2はCH2S(CH2)2N(CH3)2であり、XはCO2CH3であり、RはOHであり、そしてZ1及びZ2はそれぞれHである。
【0019】
また本発明には下式(V)によって示される化合物が含まれる。
ここで、XはCO2R5(ここでR5は低級アルキル基を表す)またはCH2NHCO2R6 (ここでR6は低級アルキル基またはアリール基を表す)であり、R1は水素またはCH2SO2R7(ここでR7は低級アルキル基を表す)であるが、ただしX= CO2R5、R1= 水素は除かれる。
【0020】
化学式(V)における置換基の定義において低級アルキル基とは、直鎖状または分枝状のアルキル基であって、1〜6個の炭素原子、好ましくは1〜3個の炭素原子を有する基を意味し、例えばメチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、ネオペンチル基、及びヘキシル基である。アリール基とは、6〜10個の炭素原子を持つアリール基を意味し、例えばフェニル基及びナフチル基である。
【0021】
また本発明には、下式(VI)によって示される化合物(VI−1)が含まれる。
ここでXはCO2CH3であり、RはOHであり、R1、R2、Z1、及びZ2はそれぞれHであり、そしてR8はNHCONHC2H5である。
【0022】
また本発明には、下式(VI)によって示される化合物(VI−2)が含まれる。
ここでXはCO2CH3、それぞれのR2及びR8はNH2、RはOH、そしてR1、Z1、及びZ2はそれぞれHである。
【0023】
本発明の化合物は、薬学的に許容される酸が付加した塩、金属塩、アンモニウム塩、有機アミンが付加した塩、及びアミノ酸が付加した塩などを含む薬学的に許容される塩の形態であってもよい。
【0024】
薬学的に許容される酸が付加した塩の例には、例えば塩酸塩、硫酸塩、及び燐酸塩のような無機酸が付加した塩、また酢酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、及びクエン酸塩のような有機酸が付加した塩が挙げられる。薬学的に許容される金属塩の例には、ナトリウム塩やカリウム塩のようなアルカリ金属の塩、マグネシウム塩やカルシウム塩のようなアルカリ土類金属の塩、アルミニウム塩、及び亜鉛塩がある。薬学的に許容されるアンモニウム塩の例は、アンモニウム塩及びテトラエチルアンモニウム塩である。薬学的に許容される有機アミン付加塩は、モルホリン、ピペリジンとの塩である。薬学的に許容される有機アミノ酸が付加した塩の例は、リジン、グリシン、及びフェニルアラニンとの塩である。
【0025】
他の観点では本発明は、コリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、及び感覚ニューロン、例えば背根神経節ニューロンの機能を、治療に有効な量の新規なビス−置換されたスタウロスポリンの誘導体を例えばヒトなどの哺乳動物に投与することによって高める方法を特徴とする。この治療は、栄養に関する因子(trophic factor)とともに、好ましくはニュートロフィンファミリーの一つとともに、そして最も好ましくは神経生長因子(NGF)とともに投与することができる。本明細書において用いられるように、「栄養に関する因子」とは、栄養に関する因子に応答性の細胞の生存性または機能に直接的にまたは間接的に影響を与える分子である。ニュートロフィンファミリーとは、NGFと有意に相同なタンパク質の群であり、それにはNGFの他に、脳由来神経栄養因子(BDNF; Leibrockら、Nature 341: 149-152, 1989参照)、ニューロトロフィン−3(NT−3; Hohnら、Nature 344: 339-341, 1990参照)、及びニューロトロフィン−5(NT−4/5; Berkemeierら、Neuron 7: 857-866, 1991参照)が含まれる。
【0026】
他の観点では本発明は、治療に有効な量の新規なビス−置換されたスタウロスポリン誘導体の一つを例えばヒトのような哺乳動物に投与することによって、その哺乳動物の神経細胞を、興奮性のアミノ酸によって誘導される変性が起こらないように保護する方法を特徴とする。このような変性が起こる可能性のある症状には、アルツハイマー病、たとえば筋萎縮性側索硬化症のような運動ニューロン疾患、パーキンソン病、例えば虚血性状態のような脳血管疾患、AIDS脳症、てんかん、ハンチントン病、及び脳や脊髄に起こる震盪性または穿通性の損傷が関連する。この治療は、神経栄養性因子、好ましくはニューロトロフィンファミリーの一つ、最も好ましくは神経生長因子(NGF)とともに投与するとよい。
【0027】
他の観点では本発明は、哺乳動物、例えばヒトにおけるコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、および/または例えば背根神経節ニューロンのような感覚ニューロンの機能を、治療に有効な量のK−252aの機能性誘導体、即ち下式によって示された誘導体を前記哺乳動物に投与することによって高めるための方法を特徴とする。
【0028】
ここでは下記の表1で示された置換のいずれかが行われる。好ましくは、哺乳動物におけるコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、または例えば背根神経節ニューロンのような感覚ニューロンの機能および/または生存性を高めるための方法には、哺乳動物に、治療に有効な量の、表1に示された化合物II−3、II−20、II−30、II−33、II−38、II−49、II−51、II−65、II−69、II−72、II−73、II−79、II−80、VI−1、またはVI−2を投与することが含まれる。更に好ましくは、哺乳動物におけるコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、または感覚ニューロンの機能および/または生存性を高めるための方法には、治療に有効な量の化合物II−51を投与することが含まれる。
【0029】
表1
【0030】
(1)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
(2)NH−アミノ酸結合は、そのアミノ酸のカルボキシル基を介したアミド結合である。
(3)XおよびRは、ともに組み合わされ結合基を形成する。
(4)R3は、CH2CH=CH2であり、R4はHである。
(5)R3及びR4はそれぞれHである。
(6)R3及びR4はそれぞれCH2CH=CH2である。
(7)化合物は塩酸塩の形態を取る。
(8)R3はHであり、そしてR4はCH2CH=CH2である。
(9)IV−1及びIV−4は二つの成分の1.5〜1.0混合物である。
(10)R3= R4= CH2CH2CH2OHである。
(11)R3 =
、R4= Hである。
(12)R8= NHCONHC2H5である。
(13)R8= NH2である。
【0031】
この治療は、栄養に関する因子、好ましくはニューロトロフィンファミリー、最も好ましくは神経生長因子(NGF)とともに投与することができる。
【0032】
好ましい観点では本発明は、治療に有効な量の、下記の式(II)または(III)によって表されるK−252aの機能性誘導体を、例えばヒトのような哺乳動物に投与することによって、背根神経節の神経細胞の機能を高めるための方法を特徴とする。
【0033】
ここでは次の置換が行われる。
表2
【0034】
(1)R2は水素である。ただし、R2= Brである化合物II−20及びII−32は除く。
(2)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
(3)X及びRは、ともに組み合わされ結合基を形成する。
【0035】
この治療は、神経栄養性因子、好ましくはニューロトロフィンファミリーのうちのひとつ、最も好ましくは神経生長因子(NGF)とともに投与することができる。
【0036】
好ましい観点では本発明は、治療に有効な量の、下記の式(II)によって表されるK−252aを、例えばヒトのような哺乳動物に投与することによって、その哺乳動物のコリン作動性ニューロンの機能を高めるための方法を特徴とする。
ここでR1及びR2はそれぞれHであり、XはCO2CH3であり、RはOHであり、そしてZ1及びZ2はそれぞれHである。この治療では、栄養に関する因子、好ましくはニューロトロフィンファミリーのうちのひとつ、最も好ましくは神経生長因子(NGF)とともに投与することができる。
【0037】
好ましい観点では本発明は、治療に有効な量の、下記の式(II)、(III)、または(IV)によって表されるK−252aまたはK−252aの機能性誘導体を例えばヒトのような哺乳動物に投与することによって、線条体神経細胞の生存性や機能を高めるための方法を特徴とする。
【0038】
ここで次の置換がなされる。
表3
【0039】
(1)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
(2)R3はCH2−CH=CH2であり、R4はHである。
【0040】
好ましい観点では本発明は、治療に有効な量の、下記の式(II)によって表されるK−252aまたはK−252aの機能性誘導体を、例えばヒトのような哺乳動物に投与することによって、基底前脳神経細胞の生存性および/または機能を高めるための方法を特徴とする。
【0041】
ここで次の置換がなされる。
表4
【0042】
(1)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
【0043】
この治療では、栄養に関する因子、好ましくはニューロトロフィンファミリーのうちのひとつ、最も好ましくは神経生長因子とともに投与することができる。
【0044】
本発明の他の特徴及び利点は、以下の好ましい態様の説明、及び請求の範囲から明らかとなろう。
【0045】
好ましい態様の説明
スタウロスポリン誘導体
本発明は、新規なスタウロスポリンのビス−N−置換された誘導体、及び神経疾患のための治療薬としてのそれらの使用に関する。ここで神経疾患とは特に、損傷を受けたり、機能が不全であったり、軸索の変性が起こったり、もしくは死に至る危険性が増加したりしたニューロン細胞、または障害を受けたコリン作動性の活性のいずれかにより特徴づけられるような疾患である。これらの疾患には興奮性のアミノ酸によって誘導される疾患が含まれる。これらの新規な誘導体の治療のための利用には、この誘導体を単独で用いたり、また神経栄養性因子(好ましくはニューロトロフィンファミリーのひとつ、最も好ましくはNGF)の外因性の投与と組み合わせて用いたりできる。本発明の請求範囲に含まれる化合物は、下式によって表すことができる。
【0046】
[Stau]−N(CH3)−W−N(CH3)−[Stau] (I)
ここで[Stau]は下式の残基を表し、
そしてWはビス(カルバミル)またはビス(チオカルバミル)ラジカルを表す。
【0047】
−C(=Y)−NH−W’−NH−C(=Y)−
ここでW’は2〜20個の炭素原子からなるハイドロカルビレンラジカルであって、YはOまたはSである。W’は好ましくは、2〜10個の炭素からなり、置換されていないか、もしくは1〜3個の炭素からなる1〜3個のアルキル基で置換されたアルキレンラジカル、6〜12個の炭素からなり、置換されていないか、または1〜3個の、1〜3個の炭素からなるアルキル基、塩素、もしくは臭素で置換されたアリレンラジカルである。W’がヘキサメチレン及び1,4−フェニレンであると特に好ましい。Yは好ましくはOである。
【0048】
化学式(I)の化合物はカルバメート及びチオカルバメートの調製のための当技術分野において既知の手法を用いて合成することができる。好ましくはこの化合物は、ビス−ジイソシアネートまたはビス−ジイソチオシアネートをスタウロスポリンと反応させ、化学式(I)示される化合物(ここではそれぞれY= OまたはY= S)を得ることにより調製した。
【0049】
使用に適した中間体、ビス−ジイソシアネート及びビス−ジイソチオシアネートには、
1,6−ジイソシアナトヘキサン
トルエン−2,6−ジイソシアネート
ベンゼン−1,2−ジイソシアネート
2−メチル−1,5−ジイソシアナトペンタン
ナフタレン−2,6−ジイソシアネート
1,6−ジイソチオシアナトヘキサン
1,4−ジイソチオシアナトブタン
トルエン−2,4−ジイソシアネート
ベンゼン−1,4−ジイソシアネート
1,2−ジイソシアナトエタン
ナフタレン−1,5−ジイソシアネート
1,5−ジイソシアナトペンタン
ベンゼン−1,4−ジイソチオシアネート
2−メチル−1,5−ジイソチオシアナトペンタン
が含まれる。
【0050】
イソシアネート及びイソチオシアネートの調製のレビューとして、「シアネートおよびそのチオ誘導体の化学(The Chemistry of Cyanates and Their Thio Derivatives)、第2部、(Patai編) 、ウィリー(Wiley)、ニューヨーク、1977年におけるリッチャー(Richter)及びウルリッヒ(Ulrich)、619〜818頁」を参照のこと。この化合物は、好ましくは、ホスゲン(Y= O)またはチオホスゲン(Y= S)を対応するジアミンと反応させることによって、調製される。他の調製方法も用いることができる。例えば、1,2−ジイソシアナトエタンは、エチレン尿素をホスゲンと反応させ、続いて加熱することによって調製することができる。
【0051】
K−252aの誘導体
本発明はまた、損傷されたり、機能が不全であったり、軸索の変性が起きていたり、または死に至る危険のあるニューロンによって特徴づられるようなある種の神経疾患または障害における治療薬として、K−252aの特定の機能性誘導体を使用することに関する。この機能性誘導体は、単独で投与してもよいし、または神経栄養性因子(好ましくはニューロトロフィンファミリー、最も好ましくは神経生長因子、NGF)と組み合わせて投与してもよい。
【0052】
K−252aの「機能性誘導体」とは、望ましい生物活性、ここでは神経保護活性として定義されている活性であって、例えば、神経細胞の生存性を促進する能力、または神経繊維(例えば軸索)の生長を促進する能力、またはコリン作動性神経細胞の機能を高める能力、または例えば背根神経節の神経細胞のような感覚細胞の機能を高める能力、または線条体ニューロンの機能および/または生存性を高める能力、または基底前脳ニューロンの機能および/または生存性を高める能力を有する分子の修飾形と定義される。このような分子の修飾により、(例えば血液脳関門および細胞膜を介した)分子の溶解、吸収、輸送、生物学的半減期などを改善することができる。また他の場合では、または更に、ある部分はこの分子の毒性を減少させることができるし、またこの分子の望ましくない副作用をなくしたり減弱したりすることができる。
【0053】
本発明の範囲に含まれる化合物は、下記に示した化学式(II)[ここでは以下、化合物(II)という]、化学式(III)[ここでは以下、化合物(III)という]、化学式(IV)[ここでは以下、化合物(IV)という]、化学式(V)[ここでは以下、化合物(V)という]、化学式(VI)[ここでは以下、化合物(VI)という]で表わすことができ、
そして下記の表5で示した置換が行われる。本発明のK−252aの機能性誘導体は、当業者に知られた方法を用いる化学合成によって、デノボ(de novo)で合成することができる。例えば化合物IIの調製に用いられる手法は、ムラカタ(Murakata)ら(米国特許第4,923,986号)に記載されており、この文献は参照としてこの明細書に組み入れられる。化合物IIIを調製するために用いられる手法は、ムーディー(Moody)ら、J. Org. Chem. 57: 2105-2114 (1992)」、「ステグリッチ(Steglich)ら、Angew. Chem. Int. Ed. Engl. 19: 459-460 (1980)」、「ナカニシ(Nakanishi)ら、J. Antibiotics 39: 1066-1071 (1986)」、及び日本国特許出願第60-295172号(1985)に開示されている。他の方法は、日本国特許出願第60-295173号(1985)において化合物II−1、9、12及び15について、日本国特許出願第62-327858号(1987)において化合物II−2、3、4、24、25及び26について、日本国特許出願第62-327859号(1987)において化合物II−20について、及び日本国特許出願第60-257652号(1985)において化合物II−10について、明治製菓株式会社(Meiji Seika Kaisha Ltd.)によって開示されている。
【0054】
表5: K−252a(12)の機能性誘導体
【0055】
(1)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
(2)NH−アミノ酸結合は、そのアミノ酸のカルボキシル基を介したアミド結合である。
(3)XおよびRは、ともに組み合わされ結合基を形成する。
(4)R3は、CH2CH=CH2であり、R4はHである。
(5)R3及びR4はそれぞれHである。
(6)R3及びR4はそれぞれCH2CH=CH2である。
(7)化合物は塩酸塩の形態を取る。
(8)R3はHであり、そしてR4はCH2CH=CH2である。
(9)IV−1及びIV−4は二つの成分の1.5〜1.0混合物である。
(10)R3= R4= CH2CH2CH2OHである。
(11)R3=
、R4= Hである。
(12)K−252a自体の場合、R1= R2= H、X= CO2CH3、R= OHであり、そしてZ1及びZ2はそれぞれ水素である。
(13)R8= NHCONHC2H5である。
(14)R8= NH2である。
【0056】
本発明はまた、治療に有効な量の、上記に示された化学式(II)及び表5(記12)に示された置換によって表されるK−252aを投与することによって、コリン作動性ニューロンの機能を高めるための方法に関する。この化合物は当技術分野において記載されている手法によって調製される(マツダ(Matsuda)ら、米国特許第4,554,402号、カセ(Kase)ら、J. Antibiotics 37: 1059-1065, 1986参照)。
【0057】
「コリン作動性ニューロンの機能を高めること」とは、コリン作動性の神経細胞の生存性および/または神経繊維(例えば軸索)の生長の促進および/または神経細胞のコリン作動性機能の増強を意味する。K−252aは、栄養に関する因子、好ましくはニューロトロフィンファミリーの一つ、最も好ましくは神経生長因子(NGF)と共に投与することができるし、または同因子と組み合わせずに投与することもできる。
【0058】
化合物の使用
下記に充分に説明したように本発明は、K−252aの機能性誘導体、又は化学式Iの化合物を単独で、またはNGFのような神経栄養性因子と組み合わせて神経疾患、特に、損傷を受けたり、機能が不全であったり、軸索の変性が起こったり、もしくは死に至る危険性が増加したりしたニューロン細胞、または障害を受けたコリン作動性の活性のいずれかにより特徴づけられるような神経疾患に対する治療薬として使用する新規な方法を提供する。これらの疾患には興奮性のアミノ酸によって誘導される疾患が含まれる。神経栄養性因子との組み合わせが含まれる本発明の化合物の生物活性は、培養されたPC−12細胞におけるオルニチンデカルボキシラーゼのアッセイ、培養された脊髄または基底前脳のコリンアセチルトランスフェラーゼのアッセイ、培養された背根神経節ニューロンの生存性のアッセイ、培養された線条体ニューロンの生存性のアッセイ、培養された基底前脳ニューロンの生存性のアッセイ、成長過程でプログラムされた運動ニューロンの死についてのインオボ(in ovo)でのモデル、インビボでの大人の舌下の軸索モデル、または例えば基底核の刺激毒性障害のようなインビボでの刺激毒性の神経保護アッセイによって簡便に分析することができる。これらのアッセイについてはすべて下記に詳細に記載されている。このように本発明の化合物は、ニューロン細胞死や機能不全の危険が増加することによって特徴づけられる神経の疾患または障害を患っているヒトまたは他の哺乳動物に投与するために用いられる。これらの神経の疾患及び障害には、アルツハイマー病、たとえば筋萎縮性側索硬化症のような運動ニューロン疾患、パーキンソン病、発作や他の虚血性の障害、ハンチントン病、AIDS脳症、てんかん、脳や脊髄に起こる震盪性または穿通性の損傷、及び末梢の神経障害が含まれるが、これらに限られるわけではない。
【0059】
本明細書において提供された化合物は、薬学的に受容できる非毒性の賦形剤及び担体と混合することによって薬学的な組成物へと処方することができる。上記に記載したように、このような組成物は非経口投与、特定すると液状の溶液または懸濁剤の形態で用いるように調製することもできるし、経口投与、特定すると錠剤またはカプセルの形態で用いるように調製することもできるし、または経鼻的に、特定すると粉剤、点鼻剤、もしくは噴霧剤の形態で用いるように合成することもできる。
【0060】
この組成物は、ユニットの用量形態で便利に投与してもよいし、また、例えば「レミントンの薬剤学(Remington's Pharmaceutical Sciences)」(マック出版社(Mack Pub. Co)、イーストン、ペンシルバニア州、1980)に記載されたような薬学分野で周知の方法のいずれかによって調製してもよい。非経口投与のための配合剤は、無菌の水や食塩水、例えばポリエチレングリコールのようなポリアルキレングリコール、植物起源のオイル、水素化したナフタレンなどを通常の賦形剤として含むことができる。特定すると、生物学的適合性で生分解性のラクチド重合体、ラクチド/グリコライド共重合体、またはポリキシレン−ポリオキシプロピレン共重合体が、活性な化合物の放出を調節するための有用な賦形剤であろう。これらの活性な化合物に用いられる他の可能性のある有用な非経口輸送系としては、エチレン−ビニルアセテート共重合体の粒子、浸透性のポンプ、植え込み可能な注入系、及びリポソームがある。吸入投与のための配合剤は、賦形剤として例えばラクトースを含んでいるか、または例えばポリオキシレン−9−ラウリルエーテル、グリココレート及びデオキシコレートを含む水性溶液であってもよく、または点鼻剤の形態で投与するための油性溶液、または鼻腔内に投与できるゲルであってもよい。非経口投与のための配合剤はまた、口腔内投与のためのグリココレート、直腸投与のためのサリチレート、または膣内投与のためのクエン酸を含むことができる。経皮用の貼着剤の製剤は好ましくは、親油性の乳濁剤である。
【0061】
本発明の材料は、薬剤中の唯一の活性物質であっても、他の活性成分、神経の疾患または障害、例えば末梢神経障害におけるニューロンの生存性または軸索生長を促進する他の成長因子などと組み合わせて用いてもよい。
【0062】
本明細書に記載された化合物の治療用組成物中の濃度は、投与すべき薬物の一回用量、用いた化合物の化学的特徴(例えば疎水性)、及び投与経路などの多くの因子に依存して変化する。一般的に、本発明の化合物は、非経口投与を行うために、約0.1〜10%w/vの化合物を含む水性の生理学的緩衝溶液で用いることができる。通常の用量範囲は、1日あたり体重1kgに対して約1μg〜約1gである。好ましい用量範囲は、1日あたり体重1kgに対して約0.01mg〜約100mgである。投与すべき好ましい薬物の用量は、神経疾患の型及び進行度、特定の患者の全身的な健康状態、選択された化合物の相対的な生物効果、賦経剤の化合物の配合、及び投与経路のような変化量により決定される可能性が高い。
【0063】
本発明は次の実施例によってさらに例示されるであろう。これらの実施例は本発明の範囲を限定するものと解釈されるべきではなく、本発明の範囲は添付した請求の範囲によってのみ決定されるべきである。
【0064】
実施例1
1,6−ヘキサメチレン−ビス−(カルバミルスタウロスポリン)(6-Hexamethylene-bis-(carbamylstaurosporine))(HBCS)
酢酸エチル(無水硫酸マグネシウム上で乾燥させたもの)1.00 ml中にスタウロスポリン(カリフォルニア州サウザンドオークス、カミヤ・バイオメディカル社)1.0 mg(2.15 μmol)を溶かした溶液を、無水酢酸エチル1.00 ml中に10.75 mgのヘキサメチレン−ビス−イソシアネートを溶かした溶液17マイクロリットル(1.08 μmol)で処理した。この反応混合液をアンバーガラス反応用バイアルに入れて、室温に2日間置いた。600 μgの結晶化した沈殿物が分離した。この組成物は高速原子衝撃質量分析計(FAB-MS)によってHBCSであることが明らかになった。
M+H+ 計算値=1102 M+Na+ 計算値=1124
実測値=1102 実測値=1124
本生成物および以下で説明されるスタウロスポリン誘導体はすべて、遮光ガラスバイアル中で保存した。
【0065】
実施例2
p−フェニレン−ビス−(カルバミルスタウロスポリン)(p-Phenylene-bis-(carbamylstraurosporine))(PBCS)
無水酢酸エチル1.00 ml中にスタウロスポリン1.0 mg(2.15 μmol)を溶かした溶液を、無水酢酸エチル1.00 ml中に3.83 mgのp−フェニレンジイソシアネート(p-phenylene diisocyanate)(トランスワールドケミカル社 P1586-1)を溶かした溶液45マイクロリットル(1.08 μmol)で処理した。反応混合液を一晩放置した。白い沈殿物が沈積した。そして、石油エーテルを0.5 ml加えた。混合液を、真空乾燥した焼結ガラスロートに濾過して入れた。全部で0.9 mgの結晶産物を採取して、高速原子衝撃質量分析計によってp−フェニレン−ビス−(カルバミルスタウロスポリン)であることを同定した。
M+H+ 計算値=1093
実測値=1093
【0066】
調製物A
N−フェニルカルバミルスタウロスポリン(N-Phenylcarbamylstraurosporine)(PCS)
参照文献:米国特許第5,093,330号
無水酢酸エチル1.50 ml中に2.0 mg(4.30 μmol)のスタウロスポリンを溶かした溶液を、無水酢酸エチル0.990 ml中に10 μlのフェニルイソシアネートを溶かした溶液468μl(4.30μmol)で処理した。反応混合液を一晩放置して、3 mlのヘキサンを少しずつ加えた。2.39 mgの無色の結晶が得られた。この産物を再結晶させた後、酢酸エチル1 mlと石油エーテル2 mlを加え、1.75 mgの結晶産物を分離した。同様の調製物からFAB-MSによってこの産物の組成がN−フェニルカルバミルスタウロスポリンであることが明らかになった。
M+H+ 計算値=586
実測値=586
【0067】
調製物B
N−フェニルチオカルバミルスタウロスポリン(N-Phenylthiocarbamylstraurosporine)(PTCS)
酢酸エチル1.00 ml中に1.0 mg(2.15 μmol)のスタウロスポリンを溶かした溶液を、酢酸エチル1.00 ml中に10 μlのフェニルイソチオシアネートを溶かした貯蔵溶液26μlで処理した。これの等量液には、290μg(2.15 μmol)のフェニルイソチオシアネートが含まれている。反応混合液を25℃に一晩置き、2 mlのヘキサンを加えた。この結果できた結晶産物を濾過して、ヘキサンで洗滌し、アルゴンガスの気流で乾燥させた。
FAB-MS 計算値: M+H+ =602
実測値 =602
【0068】
調製物C
N−エチルカルバミルスタウロスポリン(N-Ethylcarbamylstraurosporine)(ECS)
酢酸エチル900マイクロリットル中に0.9 mg(1.93 μmol)のスタウロスポリンを溶かした溶液を、1.93マイクロモルのエチルイソシアネート(無水酢酸エチル2.00 ml中に9.05 mgのエチルイソシアネートを溶かした貯蔵溶液30.2マイクロリットル)で処理した。反応混合液を25℃で一晩放置して、2.0 mlのヘキサンを加えた。結晶産物を分離し、乾燥した。
FAB-MS 計算値: M+H+=538 M+Na+=560
実測値 =538 =560
【0069】
実施例3
化合物II-4
化合物A(962 mg, 2 mmol)を、テトラヒドロフラン30 mlとメタノール10 mlの混合液に溶かし、これに760 mgの水素化ホウ素ナトリウム(20 mmol)を氷冷下で加え、その後、同じ温度で4時間、さらに室温で12時間撹拌した。これに3Nの塩酸を加えた後、溶液を食塩水溶液で洗滌し、硫酸マグネシウムで脱水して、その後、溶媒を蒸発させた。シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)で残留物を精製し、882 mg(収率97%)の化合物II-4を得た。
融点:130〜140℃
【0070】
実施例4
化合物II-14
化合物B(393 mg, 0.9 mmol)を、テトラヒドロフラン25 mlに溶解し、カルボベンゾキシ−L−セリン309 mg(1.35 mmol)、N−ヒドロキシスクシンイミド156 mg(1.35mmol)、4−メチルモルホリン0.1 ml(0.9 mmol)およびジシクロヘキシルカルボジイミド279 mg(1.35 mmol)を含むテトラヒドロフラン3 mlを氷冷下で加え、12時間撹拌した。この反応混合液を濾過し、溶媒を蒸発させた。シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)で残留物を精製し、429 mg(収率72%)の化合物Cを得た。
融点:188〜193℃
SIMS(m/z):660(M+1)+
化合物C(399 mg)をジメチルホルムアミド10 mlに溶解し、次に、炭素上10%パラジウム 300 mgを加え、水素気流中、50℃で7時間撹拌した。この反応混合液をセライトを通して濾過し、溶媒を蒸発させた。シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール/28%水酸化アンモニウム=90/10/1)で残留物を精製して得た産物を5 mlのテトラヒドロフランで溶解し、1.7 Nの塩化水素/酢酸エチル5 mlとジエチルエーテル10 mlを加えた。濾過によって沈殿物を溶液から分離し、234 mg(収率69%)の化合物II-14を得た。
融点:>300℃
【0071】
実施例5
PC-12細胞は、ラットの副腎随質の腫瘍に由来するクローン集団で、NGFの作用を研究するための、非常に有用で、広く研究されているモデルであることが分かっている(グロフ(Guroff)、神経科学における細胞培養(Cell Culture in the Neurosciences)、プレナム出版株式会社(Plenum Publishing Corporation)、245〜272頁、1985年)。これらの細胞に対するNGFの特に確実な効果の一つは、オルニチン・デカルボキシラーゼ(ODC)の活性を速やかに刺激することで、この効果は、200 nMのK-252aによって阻害されることが報告されている(Koizumiら、1988)。本実施例の実験においては、PC-12細胞(メリーランド州ベセスダの国立衛生研究所のG.グロフ博士からいただいた)を6×104細胞数/cm2の密度で48穴プレートで培養し、薬剤媒体(0.5% DMSO)、K-252a、スタウロスポリン、またはHBCSとともにインキュベートした。K-252aとスタウロスポリンは、カミヤバイオメディカル社から入手することが可能である。薬剤を添加してから4時間後に、ハフ(Huff)らによって説明されているところに従って(J. Cell Biol. 88: 189-198, 1981)、ODCアッセイのために細胞を回収した。
【0072】
これら3つの化合物はすべて、ODC活性の誘導(すなわち上昇)を示したが、効能と効力には、かなりの差違があった(図1)。K-252aは、その効果が2 nMで検出可能になり、200 nMで最大(36.3倍の誘導)になるまで増加するという、ODC活性の用量依存的な誘導を示した。同様に、スタウロスポリンの効果も、2 nMで検出可能になるが、20 nMでピーク(34.7倍の誘導)に達し、200 nMでは、かなり減少する。HBCS(実施例1)も、同様に2 nMで誘導を示したが、濃度を上昇させても効果の上昇をもたらすことはできなかった。このため、他の2つの化合物に較べて、その最大効果はずっと低かった(6.5倍の誘導)。別の実験において、PC-12細胞のODC活性に対するPTCS、PCS、およびECS(実施例2)の効果と、K-252aの効果とを比較した。200 nMの濃度でK-252aの活性を100%と表すと、PTCSはK-252aの活性の71.4%を示し、また、PCSとECSはそれぞれ、K-252aの活性の88.9%と61.9%を示した。しかし、プロテインキナーゼCインヒビターH-7は、プロテインキナーゼCの活性を阻害することが分かっている濃度である30 μMでは、ODC活性を誘導しなかった(Nakadateら、Biochem. Pharmacol. 37: 1541-1545, 1988)。
【0073】
K-252a、スタウロスポリンおよびHBCSが、NGFの生物活性を増強および/または阻害する能力を、細胞培養液 1 ml当たりNGFを10 ngを加えて、上述の濃度の上記化合物の非存在下または存在下で、上述したように細胞のODCアッセイを行なって測定した(図2)。NGFのこの濃度は、化合物の上昇効果または阻害効果を検出できるよう、中度の誘導を示す濃度を選択した。200nMのK-252aは、コイズミ(Koizumi)ら(1988)が報告しているように、ODCのNGF誘導を阻害したが、驚くべきことに、より低い濃度(2 nMと20 nM)では誘導が増強された。スタウロスポリンも、2 nMでは、NGFによる誘導を増強したが、濃度が高くなる(20 nMと200 nM)と、この効果は失われた。これに対し、HBCSは、調べたすべての濃度で、NGFの効果を増強した。この際だった効果を、HBCSのみのときの緩やかなODC誘導効果と比較して図3に示す。
【0074】
実施例6
コリンアセチルトランスフェラーゼ(ChAT)活性に対するK-252aの効果を、標準的な方法によってラット胎児から調製した解離脊髄培養物(後述参照)の中で測定した。ChATは、神経伝達物質のアセチルコリンの合成を触媒する酵素であり、コリン作用性ニューロンの特異的な生化学的マーカーである。脊髄において、コリン作用性ニューロンの大多数は運動ニューロンである。したがって、この酵素の測定は、コリン作用性ニューロンの生存および/またはこの酵素の制御に対する、一つまたは複数の因子の効果を示すために用いることができる。
【0075】
細胞を基質に接着させるために、培養開始後2〜3時間インキュベートしてから、K-252aを示された濃度で培養液に加えた。48時間後に培養液中でChAT活性を測定した。脊髄培養液中のK-252aは、ChAT活性を用量依存的に上昇させ、200〜300 nMで最大効果(2〜3倍の増加)に達した(図4)。これより濃度が高くなると、ChAT活性は低下した(図4)。インキュベートする時間を長くすると、7日目までは、ChAT活性の基底レベルが下がるために、ChAT活性は4〜5倍上昇した(図5)。この培養系において、基本的な(対照となる)条件の下では、変性したり死滅する運動ニューロンの数は増加した(McManamanら、Developmental Biol. 125: 311-320, 1988)。図4および図5に示されている結果は、培養を開始した日に1回だけK-252aを添加した結果であり、脊髄のコリン作用性ニューロンの生存および/または酵素自体の制御に対する効果が長く持続することを示している。
【0076】
ラット胎児の脊髄細胞の解離培養液を用いた実験は、ほとんど、既述されているところ(Smithら、J. Cell Biol. 101: 1608-1621, 1985)に従って行なわれた。解離細胞はトリプシンによる組織解離法を用い、当業者に既知の標準的技術によって、14日目のラット胎児から調製された(Smithら、前記)。細胞は、6 X 105細胞/cm2の濃度で、ポリ−L−オルニチンでコートしたプラスチック組織培養皿の中、無血清N2培地に撒き(培養開始し)、5%CO2/95%空気の湿潤な環境下37℃で(Bottensteinら、Proc. Natl. Acad. Sci. USA 76: 514-517, 1979)、48時間インキュベートした。イシダ(Ishida)およびデグチ(Deguchi)(J. Neurosci. 3: 1818-1823,1983)およびマクマナマンら(McManamanら、前記)(1988)に従ってフォナム(Fonnum)の方法を修正した方法を用いてChAT活性を測定した。ビシンコニン酸/Cu++反応(BCA蛋白質測定試薬、イリノイ州ロックランド、ピアス社)によって測定した全蛋白質量に対して、活性を標準化した。
【0077】
実施例7
脊髄ChATアッセイ法で、K-252aの機能性誘導体を100種以上調べて、それらの相対的な有効性を判定した。図8のデータは、300および30 nMで調べたところ、最初の機能性誘導体のうち28種が300 nMでChAT活性を有意に上昇させたことを示している。機能性誘導体の一つである化合物II-21は30 nMでも活性があった(基底レベルに対してChAT活性は30%上昇した)。K-252aや他の相同化合物では、30 nMでのChAT活性が活発には上昇しなかったことから、この化合物はこれらの化合物よりも強力であった。
【0078】
図13aは、ラットの脊髄培養物中でChAT活性を有意に上昇させることが分かった28種の最初のK-252a誘導体と、さらに30種の誘導体(化合物II-29からII-34、II-36からII-56、およびIV-1からIV-3)の能力を示している。図13bは、K-252aの誘導体であるII-66-80、IV-5、IV-6、VI-1、およびVI-2が、ラットの脊髄培養物中でChAT活性を有意に上昇させることができることを示している。図13Cは、さらに12種のK-252a誘導体がラットの脊髄培養物中でChAT活性を有意に上昇させることができることを示している。
【0079】
実施例8
K-252aおよび50種以上の機能性誘導体について、背根神経節のニューロン細胞の生存を促進する能力を評価した。細胞が生きていることは、生細胞染料であるフルオロセイン二酢酸(fluorescein diacetate)の類似合成物であるカルセイン(calcein)AMの取り込みによって測定した。カルセインは生細胞によって取り込まれ、細胞内で切断されて、生細胞の無傷の細胞膜によって保持される蛍光性の塩になる。生きたニューロンを顕微鏡下で数えた細胞数は、蛍光定量的生細胞測定法を用いて得られる相対的蛍光値と直接に正比例する。したがって、この方法は、一定の培養液中の全細胞集団における細胞の生存率に関する信頼性のおける、定量的な測定結果を提供する(Bozyczko-Coyneら、J. Neur. Meth. 50: 205-216, 1993)。
【0080】
胚齢8日目のニワトリの胚から背根神経節を切除した後、ディスパーゼ(Dispase)(中性プロテアーゼ、コラボレイティブリサーチ社)解離によって解離細胞を調製した。ニューロンを低密度(1.8 X 104細胞/cm2)で、ポリ−L−オルニチンとラミニンでコートした96穴プラスチック培養皿に接種した。細胞は、無血清N2培地(BottensteinおよびSato, 1979)、5%CO2/95%空気の湿潤な環境下37℃で48時間培養した。細胞の生存は、48時間後に上記の蛍光生細胞測定法を用いて評価した。
【0081】
背根神経節ニューロンの生存は、K-252aによって濃度依存的に強化された。約100 nMで最大活性が観察された(図6)。調査した50の類似化合物のうち24種が、DRGニューロンの生存を強化する活性があり、そのうち22種が図7に示されている。これらの類似化合物はすべて、脊髄のChAT活性を上昇させる活性もあった(実施例5参照、図8)。最初の22種と、さらに2種の活性類似化合物(II-30、II-32)が図14に示されている。24種の活性機能性誘導体によって刺激された背根神経節ニューロンを顕微鏡下で調べると、神経線維の成長が増強されていることも示された。
【0082】
実施例9
齧歯動物の脳の空洞に興奮性のアミノ酸カイニン酸(カイニン酸塩)を直接注入すると、海馬の錐体細胞のニューロン変性がもたらされる。このニューロン死は、細胞骨格蛋白質のスペクトリンの蛋白質分解が著しく促進されることで特徴づけられる。スペクトリンの分解産物は、カイニン酸投与後24時間以内に、海馬の磨砕物中で測定することができる。スペクトリンの蛋白質分解度は、海馬の錐体細胞のニューロン死の程度と高度に相関する(Simanら、J. Neurosci. 9: 1579-1590, 1989)ため、スペクトリンの蛋白質分解は、興奮性のアミノ酸によって誘導されるニューロン変性の優れた生化学マーカーになる。内因性興奮性アミノ酸の過剰放出が、発作および他の虚血性傷害、アルツハイマー病、筋萎縮性側索硬化症を含む運動ニューロン病、パーキンソン病、ハンチントン病、AIDS脳症、てんかん、脳または脊髄の振とう性ないし穿通性傷害を含む数多くの神経疾患および障害の病因であると示唆されてきた。
図9〜12に示されている結果は、以下の方法にしたがって作出されたものである。
【0083】
カイニン酸注入法: カイニン酸によって誘導されるニューロン損傷に対する、K-252aあるいはその誘導体の効果を評価した。スプラーグ−ダウレー系統ラットのメスおよびオスの成獣(175〜250 g)を、ネンブタール(Nembutal)(50 mg/kg、腹腔内)で麻酔した。カイニン酸処理(5μl)の前後にicv注入して、各ラットに試験用化合物を(全量で5μl)投与した。これは、上記の各ケースについて示された投与および注入スケジュールを用いて行われた。対照用動物には、カイニン酸および薬剤の注入の代わりに溶媒を投与した。解剖実験のために、icv注入物を、薬剤注入の約一週間前に埋め込まれたカニューレ(バージニア州ロアノーク、プラスティックワン社)を通して輸送した。このカニューレは、ブレグマの前後部で、ブレグマの1.5 mm側方、頭頂部から4.4 mm下方表面側に定位的に配置された。この処理法による結果は、後述するところの解剖学的解析を用いて、2週間後に評価した。
【0084】
カイニン酸に誘導されるスペクトリンの蛋白質分解に対するK-252a、あるいはその誘導体の効果を測るための実験では、上述の定位的配置に置いた10μlハミルトン注射器によって、5μlの薬剤または溶媒のicv注入物を、カイニン酸と同時に、麻酔したラットに投与した。これらのラットは、24時間後に屠殺して、以下に述べるような生化学的分析を行なった。
【0085】
解剖学的および生化学的分析:解剖学的分析は、以下に述べるようにして行なった。処理後2週間目にラットを断頭して屠殺し、速やかに脳を取り出してドライアイス上で凍結した。各脳から頭頂部のスライドに載せた連続切片をチオニンで染色し、顕微鏡で調べた。海馬の損傷は、脳の左右両側について、錐体細胞の損失を被った、解剖学的に定義された4つの海馬領域(CA1-4、本明細書に参照として包含される、[シェパード(Shepard)著、1979年、「脳のシナプシス構成」、オクスフォード、310頁]によって説明されているロレント・デ・ノウ(Lorente de No)の分類に従う)の総数を合計して定量した。
【0086】
生化学的解析は、以下のようにして行われた。脳スペクトリン(ホドリン)の、カルパインI感受性の蛋白質分解を、サイマン(Siman)ら(1988年、 Neuron, 1: 279-287、本明細書に参照として包含される)によって説明されているイムノブロット解析を用いて、海馬のホモジネートで測定した。要約すると、処理後24時間でラットを断頭して屠殺し、脳から速やかに背部側海馬を切除して、0.1 mMフェニルメチルスルホニルフルオリドを含む20 mMトリス塩酸(pH 7.4)の中で磨砕した。各ホモジネートの分注物から採った蛋白質をSDS-PAGEで分離し、各サンプルにおける、カイニン酸に誘導されるスペクトリン分解の量を定量するためにイムノブロット解析を用いた。
【0087】
図9は、海馬において、カイニン酸に誘導されるニューロン分解に対する、K-252aの効果を示している。カニューレを挿入したオスとメスのスプラーグ−ダウレイ(Sprague-Dawley)ラットに、カイニン酸(0.6μg)を脳の大脳外側の脳室(icv)に直接注入する30分前、または、約3時間後から24時間後に、0.4μgのK-252aまたは溶媒を投与した。2週間後に、後に説明するようにして、脳を切り出して凍結し、切片にしてから組織学的な解析をするために染色した。示されているデータは、各群についての、損傷を受けた海馬の小領域の平均数±平均値の標準誤差(S.E.M.)である。K-252aは、海馬内の損傷領域の数を、3.86±0.78(K-252aを投与しないとき)から1.18±0.4(K-252aを投与したとき)へと有意に減少させた。
【0088】
図10は、カイニン酸に誘導される、海馬におけるスペクトリンの分解に対するK-252aの効果を示したものである。メスのスプラーグ−ダウレイラットに、icv注入によって、神経毒効量(0.6μg)のカイニン酸とともに、0.4μgのK-252aまたは溶媒を投与した。擬似対照用の動物には、溶媒を注入し、カイニン酸もK-252aも投与しなかった。24時間後に、後述するようにして、背部海馬のホモジネートを解析して、スペクトリン分解産物を調べた。スペクトリンの蛋白質分解の程度を、各群のスペクトリン分解産物の増加量を擬似対照の値を上回るパーセントとして表している。示されているデータは、各群についての、スペクトリン分解産物の増加の平均パーセント(擬似対照=100%)±S.E.M.である。K-252aのicv注入は、スペクトリンの蛋白質分解の程度を、擬似対照値の140±15%(K-252aを投与しないとき)から約102±10%(K-252aを投与したとき)へと有意に減少させた。
【0089】
図11は、海馬においてカイニン酸に誘導されるニューロン分解に対する、HBCSの効果を示している。カニューレを挿入したメスのスプラーグ−ダウレイラットに、カイニン酸(0.6μg)をicv注入する40分前、または、約4時間後に、0.8μgのHBCSまたは溶媒を投与した。2週間後に、後に説明するようにして脳を切り出して凍結し、切片化してから、組織学的な解析をするために染色した。示されているデータは、各群についての、損傷を受けた海馬の小領域の平均数±S.E.M.である。HBCSは、海馬内の損傷領域の数を、約2.5±0.6(HBCS処理なし)から1.3±0.5(HBCS処理したもの)へと有意に減少させた。
【0090】
図12は、海馬におけるカイニン酸に誘導されるスペクトリン分解に対する、K-252aの機能的誘導体3種の効果を比較したものである。メスのスプラーグ−ダウレイラットに、icv注入によって、神経毒効量(0.6μg)のカイニン酸とともに、0.4μgのK-252aまたは化合物III-1かII-21、または溶媒を投与した。擬似対照用の動物には溶媒を注入し、カイニン酸もK-252aの誘導体も投与しなかった。24時間後に、後述するようにして、背部海馬のホモジネートを解析して、スペクトリン分解産物を調べた。スペクトリンの蛋白質分解の程度を、各群のスペクトリン分解産物の増加量を擬似対照の値を上回るパーセントとして表している。示されているデータは、各群についての、スペクトリン分解産物の増加の平均パーセント(擬似対照=100%)±S.E.M.である。K-252aのicv注入は、スペクトリンの蛋白質分解の程度を、擬似対照値の約128±9%(溶媒処理)から約104±4%(K-252a存在下)へと減少させた。K-252aの誘導体であるIII-1とII-21は、カイニン酸に誘導されるスペクトリン分解を阻害できなかった。
【0091】
実施例10
線条体培養液における生存を促進するK-252aの能力を測定した。線条体は、胚令17日目のラット胚から切り出し、ディスパーゼ(Dispase)(中性プロテアーゼ、コラボレイティブリサーチ社)によって細胞を分離した。ニューロンを5X104細胞/ウェル(1.5 X 105/cm2)の密度で、ポリ-l-オルニチンとラミニンで予めコートした96穴プレートに接種した。細胞は、0.05%のウシ血清アルブミンを含む無血清N2培地(BottensteinおよびSato、1979)中で、5%CO2/95%空気の湿潤な環境下において37℃で培養した。実施例8に述べたカルセイン生存細胞蛍光測定法を用いて、接種後5日目に細胞の生残数を測定した。
【0092】
線条体ニューロンの生存は、K-252aによって濃度依存的な方法で強化された。75 nMのK-252aで最大活性が観察されたが、これは対照に対して3−4倍の効果を生んだことになる(図15)。対照用の培養液では、0日目にニューロンの培養を開始したとして、5日以内に90%のニューロンが死滅したが、K-252aで処理した培養では50%のニューロンが生存していた(図16)。線条体ニューロンにおける生存効果は、培養3日後に起こり、培養7日目以上まで維持された。これらの効果は、培養開始日に1回だけK-252aを施用してもたらされたもので、ニューロンの生存に対する効果が維持されることを示している。
【0093】
図17は、対照用培養液と75 nM K-252aで処理した培養液とから撮った顕微鏡写真の組である。75 nM K-252a存在下の培養液中では細胞の生存数が増加し、神経突起の成長が見られた。
【0094】
実施例11
実施例10の線条体細胞生存測定法で、K-252aの31種の機能的誘導体の有効性と効力とを調べた。図18は、線条体ニューロンの生存を強化した18種のK-252a誘導体に関するデータを示している。
【0095】
実施例12
本発明に係る化合物が前脳基部培養物の生存を強化する能力とChAT活性の上昇について化合物の評価を行なった。これらの培養物におけるChAT活性は、海馬形成や嗅覚核、脚間核、皮質、扁桃、視床の主要なコリン作用性の供給物の代表であるコリン作用性ニューロン(培養物中の細胞の5%以下)の生化学的なマーカーである。本発明の代表的な化合物は、ChAT活性を上昇させたばかりではなく、さらに、前脳基部培養物におけるニューロンの一般的な生存数を増加させた。前脳基部は、胚令17日か18日目のラット胚から切り出し、ディスパーゼ(Dispase)(登録商標)(中性プロテアーゼ、コラボレイティブリサーチ社)によって細胞を分離した。ニューロンを5 X 104細胞/ウェル(1.5 X 105細胞/cm2)の密度で、ポリ-l-オルニチンとラミニンで予めコートした96穴プレートのウェルに接種した。細胞は、0.05%のウシ血清アルブミン(BSA)を含む無血清N2培地(Bottensteinら、前記)中で、5%CO2/95%空気の湿潤な環境下において37℃で培養した。マクマナマン(McManaman)ら(前記)、およびグリクスマン(Glicksman)ら(J. Neurochem. 61: 210-221, 1993)による前出のフォナム法(Fonnum procedure)を修正して用いて、6日目にインビトロでChAT活性を測定した。ボジツィコ−コイネ(Bozyczko-Coyne)ら(前記)によって述べられているカルセインAM蛍光定量測定法を用いて、培養開始後5日目に細胞の生存を評価した。測定する時に培地の一部を吸引して1ウェルにつき50μlにした。150μlのダルベッコの(Dulbecco's)リン酸緩衝食塩水(DPBS;Gibco BRL)中の8μM カルセインAM保存液を、96穴プレートに各ウエル当たりに200μlに、最終濃度が6μMになるように加えた。プレートを37℃で30分間インキュベートした後、200μlのDPBSで4段階希釈した。プレート読み取り用の蛍光測定計(サイトフルオル(Cytofluor)2350、ミリポア(Millipore)社)を、励起波長485 nm、蛍光波長538 nm、および感度設定#3で用いて、各ウエルの相対的蛍光量を測定した。細胞を入れてないウエル6個にカルセインAMを投与して算出した蛍光バックグランドの平均値を、すべての測定値から引いた。これらの実験で使う細胞密度の範囲について、30分間基質をインキュベートして、蛍光シグナルが直線的に増えることを確認した。K-252aと少なくとも12個のK-252a誘導体(II-3、II-5、II-10、II-20、II-21、II-22、II-30、II-32、II-51、II-62、II-63、II-64、II-65)が、前脳基部ニューロンの生存を強化した(図19)。
【0096】
実施例13
以下の試験は、ラットの基底核を傷害したときの、皮質性コリン作用機能に対する化合物II-51の効果を評価するために行なわれた。
【0097】
前脳基部から出たコリン作用性ニューロンで、海馬中隔経路を通って海馬に突き出したものか、基底-皮質経路を通って皮質に突き出したものは、アルツハイマー病の進行過程において深刻な退行変性を被る。これらのニューロンの消失と認識ないし記憶機能の減退との間には、この障害に悩む各人において、ある程度の相関関係がある(Fibiger, H. Trends in Neurosci. 14: 220-223, 1991)。コリン作用性機能不全のいくつかのモデルが、生化学マーカーを消失するだけでなく行動障害も示すようになると言われてきた。これらのモデルは、アルツハイマー病の進行に近似する(Olton, D.ら、「麻痺性痴呆:前脳基部のコリン作用系の変性によって起きる認識障害の動物モデル(Dementia: animal models of the cognitive impairments produced by degeneration of the basal forebrain cholinergic system)」、Meltzer, H.編、神経病理学:発展の第三世代(Psychopharmacology: The Third Generation of Progress)、レイブン出版(Raven Press)、ニューヨーク州、1987年、941〜953頁; Smith, S., Brain Res. Rev. 13: 103-118, 1988)。例えば、コリン作用変性モデルのひとつは、基底核の毒性刺激傷害である(Wenk, G.ら、Exp. Brain Res. 56: 335-340, 1984)。前脳基部内でのコリン作用性ニューロンに傷害が起きると、この領域でのニューロン細胞体の消失がもたらされ、続いて、前頭皮質および頭頂皮質においてコリン作用性末端マーカーの消失がもたらされる(Dunnett,S.ら、Trends Neurosci. 14: 494-501, 1991)。以下の方法を用いて、基底核に傷害を受けたラットにおいて、化合物II-51が皮質のコリン作用機能を増強することが示された。
【0098】
すべての実験について、オスのスプラーグ−ダウレイラット(225〜275グラム)を用いた。当業者に周知の方法(例えば、Wenkら、前記、参照)に、後述の修正を加えた方法を用いて、基底核大型細胞(magnocellularis)に片側性のイボテン(ibotenic)傷害を起こさせた。ラットを50 mg/kgのペントバルビタールで麻酔して、基底核大型細胞(magnocellularis)に5μgのイボテン酸(ibotenic acid)(1μlのPBS中)を片側的に注射した。座標は、パキノス(Paxinos)およびワトソン(Watson)の脳地図から用いた(1.5 mm背部側、2.8 mm側部、7.2 mm背腹方向)。注射は、6分間にわたって行われた。染色剤を注射したところ、基底核に直接注入されたことが分かった。
【0099】
化合物II-51は、30%ソルトール(Solutol)(登録商標)に0.01から0.3 mg/mlの濃度で溶解した。化合物(あるいはソルトール液)を、基底核に傷害を誘導した1日後または6時間前に皮下投与し、その後48時間毎に投与した。用量は、0.01、0.03、0.10および0.30 mg/kgであった。実験は、傷害誘発後4日から21日で終了した。フォナム(Fonnum)の方法(前記)によって、前頭壁皮質の組織ホモジネートでChAT活性を測定した。傷害した側と同側の前頭皮質のChAT活性を、対側(傷害しない側)でのChAT活性と比較して平準化した。ChAT活性は、同側の対側に対する比率で表される。
【0100】
データはANOVA(分散分析)で解析し、タキー(Tukey's)事後検定によって処理間の差異を判定した。p<0.05であれば平均値に有意な差があるとみなした。
【0101】
基底核を傷害した動物において、皮質におけるChAT活性の時間依存的な減少があったが、傷害後3日から7日の間に最大の消失が起きた(表6)。投与経路、用量および投薬スケジュールは、成獣ラットの前脳基部でのChAT活性レベルに対して、化合物II-51の効果が示された予備的データを基礎にした。傷害をもつ動物における、化合物II-51のChAT活性レベルに対する効果(すなわち、コリン作用性機能)を見積もるために、傷害誘発の1日後に薬剤を投与し、14日から21日間続けるか、あるいは、手術の6時間前に投与して4日間続けた。
【0102】
表6
基底核大型細胞で片側傷害を誘導した後における
皮質のChAT活性減少の時間的変化a
傷害期間 注射 ChAT活性
同側/対側比
非傷害対照 − 96±8
8時間 イボテン酸(5μg ) 97±14
24時間 イボテン酸(5μg ) 105±11
72時間 イボテン酸(5μg ) 74±11b
168時間(7日間) イボテン酸(5μg ) 70±7b
a片側傷害は、ラットのNBMで誘発させた。傷害後の指示された時間に、前頭皮質のChAT活性を測定した。
b傷害なしの対照とは有意に(p<0.05)異なることを示す。
【0103】
化合物II-51による用量応答実験を用量0.01、0.03、0.10 mg/kgで行なった(表7)。イボテン酸(ibotenic acid)による傷害誘発後1日目から始めて、1日おきに21日間、化合物II-51の皮下注射を行なった。その結果、化合物II-51は0.03 mg/kgという低い用量で、皮質のChAT活性の消失を食い止める効果があることが示された(表7)。
【0104】
表7
傷害を受けたNBMラットにおける、計画的に投与された化合物II-51の
皮質ChAT活性に対する効果:用量反応実験a
傷害処理 化合物II-51の用量 ChAT活性
同側/対側比
傷害なし − 98.4±4.5
傷害あり 溶媒 67.4±7.2b
傷害あり 0.01 mg/kg QOD 70.1±11.2
傷害あり 0.03 mg/kg QOD 93.8±14.9c
傷害あり 0.10 mg/kg QOD 87.9±11.6c
a 片側傷害は、ラットのNBMで誘導した。傷害誘導後24時間目に、化合物II-51の皮下注射を開始した。傷害後21日目に動物を殺して前頭皮質のChAT活性を測定した。
b 対照とは有意に(p<0.05)異なることを示す。
c 傷害のみとは有意に異なることを示す。
【0105】
傷害誘導後4日目、14日目、21日目に測定すると、化合物II-51の全身投与により、前頭皮質におけるコリン作用性機能の減少が抑えられた(表8)。片側傷害を受けたラットにおいて、対側側のChAT活性は変わらなかった。このことは、化合物II-51が、傷害をもつニューロンにのみ影響することを示唆している。
【0106】
表8
傷害を受けたNBMラットにおける、計画的に投与された化合物II-51の
皮質ChAT活性に対する効果:時間反応実験a
傷害時間(日間) 化合物II-51の用量 ChAT活性
同側/対側比
非傷害対照 − 99±6
4日間 溶媒 77±6b
0.1 mg/kg QOD 96±12c
14日間 溶媒 72±8b
0.1 mg/kg QOD 94±6c
21日間 溶媒 66±8b
0.1 mg/kg QOD 87±7c
a 片側傷害は、ラットのNBMで誘導した。傷害誘導6時間前または1日後に、化合物II-51の皮下注射を開始した。傷害後の表示時間における前頭皮質のChAT活性を測定した。
b 対照とは有意に(p<0.05)異なることを示す。
c 同時点における傷害+溶媒とは有意に異なることを示す。
【0107】
実施例14
インオボ(in ovo)モデルは、成長過程でプログラムされた運動ニューロンの死に影響を与える目的の化合物を評価する目的で用いることができる。
【0108】
ヒナでは、体性運動ニューロンは胎児の6日目から10日目(E6からE10)の間に自然に起こる死に至る(Chu-Wangら、J. Comp. Neurol. 177:33-58, 1978、Hamburger, J. Comp. Neurol. 160: 535-546, 1975、McManamanら、Neuron 4: 891-898, 1990参照)。この期間中に、成長過程にあるヒナの胎児における腰部の脊髄の両側に存在する運動ニューロンの数は、約46,000個から23,000個に減少する。
【0109】
ヒナの胎児(E6〜E9)を、溶媒(5%のソルトール(登録商標)HS 15、BASF Aktienogesellschaft)または上述したような化合物II-51の濃度のいずれかで処理した。この処理液(50μl)を、オッペンハイム(Oppenheim)ら(Science 240: 919-921, 1988)の方法によってシェルに設けた窓を通して血管形成がなされている漿尿膜に通した。胎児をE10の時に屠殺して脊髄を取り出し、カルノイ溶液(Carnoy's solution)(10%の酢酸、60%のエタノール、30%のクロロホルム)で固定化してからパラフィンで包理し、8μmの切片に分割して前述したように(前記のOppenheimらの文献参照)チオニンで染色した。運動ニューロン(形態と位置により同定した)は、以前に確立された基準(Oppenheimら、J. Comp. Neurol. 246: 281-286, 1986参照)にしたがって10箇所の切片ごとに計器により数えた。
【0110】
化合物II-51をインオボでE6〜E9のヒナの漿尿膜に毎日通過させると、生存している腰部の運動ニューロンの数が用量−依存性で増加する結果になる(図20参照)。テストした最小有効用量(1.17μg/一個の卵)では、運動ニューロンの生存率が16%上昇していた。最大の効果は一個の卵あたり2.3μgの用量で得られ、それは対照に比べて、即ち賦形剤で処理した胎児に比べて処理をした運動ニューロンの生存率が40%増加するという結果になった。7μg/卵では運動ニューロンの生存率はそれ以上に増加せず、このことは、この状況下では最大の反応が2.3μg/卵で達成されたことを示している。
【0111】
実施例15
舌下の核に存在する運動ニューロンは、舌下神経を介して舌を支配する。成体のラットでは舌下神経を横に切開すると、舌下の核にある運動ニューロンにおいて細胞の数に影響を与えることなくChAT活性の劇的な消失が引き起こされる。ChAT活性の消失は、未熟な表現型への逆戻りのモデルとして用いられる。
【0112】
左側の舌下神経を、ネンブタール麻酔を行いつつ成体の雌のスプラーグ−ダウレイラット(120〜180g)それぞれの首にある二腹筋の下で切断した。化合物II-51の10%ソルトール(登録商標)(HS15、BASF Aktiengesellschaft)溶液、50マイクロリットル、または溶媒単独を、ゲルフォーム(gelfoam)の断片にかけ、続いてその切開された神経の近い方の端部をゲルフォームとともにパラフィンで覆った。7日目の終わりにラットを深く麻酔薬で眠らせ、ソレンソン緩衝液(Sorenson's buffer)(0.1MのNaPO4、pH7)中に入れた4%のパラホルムアルデヒドを用いて灌流した。脳幹を取り出して定着液に24時間浸漬し、続いてよくすすいでから切断する前に凍結保護のため30%のショ糖の中に入れた。脳の頭頂部分を40ミクロン切り出し、4℃のソレンソンの緩衝液中で染色されるまで保存した。舌下核は40〜48個の部分に広がっており、5個毎の部分を、前述した(Chiuら、J. Comp. Neurol. 328: 351-363, 1993参照)ような抗ChATマウスモノクローナル抗体である1E6を用いて免疫組織化学法を行うべく処理した。ChAT免疫活性ニューロンは、ベクタステイン エライトABC(Vecstastain Elite ABC)(登録商標)キット(Vector Laboratories, Burlingame, CA)を用いてアビジン−ビオチン増幅法を行うことにより部分を可視化した。
【0113】
舌下の核から得られたどの5個ごとの切片も処理をして、それぞれの動物の対照(傷つけていない)の側と軸索切断を行った側の免疫応答性を有する細胞を数えた。結果は、傷つけていない側のChAT免疫活性ニューロンの数に対する軸索切開した側のChAT免疫活性ニューロンの数のパーセントとして表されている。100μgの化合物II-51を舌下神経の切断端部に適用した場合には、有意の数のChAT-免疫応答性を有するニューロンが得られた(33.7%±9.9(平均±標準偏差))(図21参照)。対照的に、溶媒のみで処理した対照の動物では、ChAT-免疫応答性を有するニューロンはほとんどなかった(8.07%±2.9(平均±標準偏差))。
【0114】
調製方法
実施例16:化合物(V)
化合物(V)を調製する工程を下記に示す。
【0115】
工程1
化合物(V-1)(化合物Vの例であって、R1がCH2SO2R7で、XがCO2R5の化合物)は次の反応工程で調製できる。
(R5は低級アルキル基またはCH2NHCO2R6を表し、ここでR6は低級アルキル基またはアリール基を表し、そしてR7は低級アルキル基を表す。)
【0116】
出発物質(A)は日本で発行された未審査の特許出願第295588/88号(参照として本明細書に組み入れられる)に開示されている。
【0117】
化合物(V-1)は化合物(A)を1〜1.5等量の酸化剤で処理することによって得ることができる。酸化剤の例としては、m−クロロ過安息香酸が挙げられる。反応溶液としては、メチレンクロライド、クロロホルム、またはエチレンジクロライドのような水素化した炭化水素などが用いられる。反応は、−20〜30℃で0.5〜1時間で終了する。
【0118】
工程2
化学式(V-2)の化合物(化合物(V)の例であって、R1が水素、XがCH2NHCO2R6の化合物)は次の反応工程で調製することができる。
R6は低級アルキル基またはアリール基を表す。
【0119】
この出発化合物(B)は、日本で発行された未審査の特許出願第155285/87号(参照として本明細書に組み入れられる)に開示されている。
【0120】
化合物(V-2)は化合物(B)を、1〜3等量の塩基の存在下で1〜3等量のClCO2R6と反応させることによって得ることができる。塩基の例としてはトリエチルアミンが挙げられる。反応溶液としては、メチレンクロライド、クロロホルム、またはエチレンジクロライドのような水素化した炭化水素などが用いられる。反応は、−10〜30℃で0.5〜3時間で終了する。
【0121】
実施例17
化合物II-49
化合物(A;R5= CH3、R7= C2H5)(27mg、0.05mmol)を1mlのクロロホルムに溶解し、続いて10mg(0.06mmol)のm−クロロ過安息香酸をそこに氷冷しつつ添加した後、同じ温度で45分間攪拌した。クロロホルムを用いて希釈した後その混合物を、チオ硫酸ナトリウムの8%の水溶液、炭酸水素ナトリウムの飽和水溶液、水、及び生理食塩水を用いて順次洗浄し、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって、17.7mg(収率62%)の化合物II-49を得た。
【0122】
実施例18
化合物II-57
化合物(B)(43.8mg、0.1mmol)を1mlのテトラハイドロフランに溶解し、続いて9.3μl(0.12mmol)のメチルクロロホルメートと28μl(0.2mmol)のトリエチルアミンとを添加した後、氷冷しながら50分間攪拌した。テトラハイドロフランを用いて希釈した後その混合物を生理食塩水を用いて洗浄し、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって、32.6mgの化合物II-57を得た。
【0123】
実施例19
化合物II-38
実施例18と実質的に同じ方法を、43.8mg(0.1mmol)の化合物(B)と15μlのフェニルクロロホルメートを用いて繰り返すことによって27.8mg(収率50%)の化合物II-38が得られた。
【0124】
実施例20
(化合物Cから化合物Hへの合成は図22に示されている。)
化合物II-39
化合物(C)(日本で公開された未審査の特許出願第295588/88号;参照として本明細書に組み入れられる)(20mg、0.035mmol)を1mlのクロロホルムに溶解し、続いて14.6μl(0.105mmol)のトリエチルアミンと13.9μl(0.175mmol)のエチルイソシアネートとをそこに添加した後、室温で2時間攪拌した。その溶液に1mlのメタノールを添加し、続いてクロロホルムを用いて希釈した。その混合物を水と生理食塩水を用いて順次洗浄し、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)にかけることによって、21mg(収率84%)の化合物(D)を得た。
【0125】
化合物(D)(9mg、0.012mmol)を0.2mlのテトラハイドロフランと0.2mlのメタノールの混合液に溶解し、続いて2μlの28%メトキシド/メタノールをそこに添加してから室温で10分間攪拌した。その溶液にクエン酸の5%水性溶液0.1mlを添加した後、クロロホルムを用いて希釈した。この混合物を水と生理食塩水を用いて順次洗浄してから硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=9/1)にかけることによって、8mgの化合物II-39を得た。
【0126】
実施例21
化合物II-51及びII-56
化合物(E)(前記の日本で公開された未審査の特許出願第295588/88号)(60.7mg、0.1mmol)を5mlのクロロホルムと1mlのメタノールの混合溶媒に溶解し、続いて11mg(0.3mmol)の水素化ホウ素ナトリウムを氷冷しつつそこに添加した後、同じ温度で15分間攪拌した。クロロホルムを用いて希釈してからその混合物を、水と生理食塩水を用いて順次洗浄し、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール/トリエチルアミン=98/2/0.5)にかけることによって、36mg(収率59%)の化合物(F)を得た。
【0127】
化合物(F)(159mg、0.26mmol)を15mlのクロロホルムに溶解し、続いて0.8ml(10.4mmol)のエタンチオールと24mg(0.104mmol)のカンファースルホニックアシッドをそこに添加した後、室温で12時間攪拌した。この溶液を炭酸水素ナトリウムの飽和水溶液、水、及び生理食塩水を用いて順次洗浄してから、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/9−クロロホルム/メタノール=99/1)にかけることによって、43mgの化合物(G)と75mgの化合物(H)を得た。
【0128】
化合物(G)
化合物(H)
【0129】
実施例20と実質的に同じ方法を34mgの化合物(G)を用いて繰り返すことで18.7mgの化合物II-51が得られた。
【0130】
実質的に実施例20と同じ方法を30mgの化合物(H)を用いて繰り返すことで20.4mgの化合物II-56が得られた。
【0131】
実施例22
化合物IV-2
化合物II(Z1、Z2= H、R1= Br、R2= H、R= OH、X= CO2CH3)(日本で公開された未審査の特許出願第120388/87号;参照としてここに組み入れられる)(50mg、0.09mmol)を0.5mlのトリフルオロ酢酸と50μlの3N HClの混合溶媒に溶解し、続いてその溶液を室温で2日間攪拌した。その沈澱物を濾過して集め、高速液相クロマトグラフィー(ユニシル 5C18;メタノール/水=8/2)にかけることによって、8.4mgの化合物(IV-2)を得た。
【0132】
実施例23
化合物II-45は、図23に示された反応工程によって調製することができる。化合物(J)は、日本で公開された未審査の特許出願第120388/87号(参照としてここに組み入れられる)において開示されている。
【0133】
化合物II-45
化合物(J)(200mg)を1mlのジメチルホルムアミドに溶解し、続いて23.5mgの水酸化ナトリウムの水溶液0.25mlをそこに添加した後室温で4時間攪拌した。1Nの塩酸を加えて溶液のpHを1〜2に調節し、沈澱物を濾過して集めることにより178mg(収率91%)の化合物(K)が得られた。
【0134】
化合物(K)(168mg)を3mlのピリジンに溶解し、続いて0.44ml(4.7mmol)の無水酢酸をそこに添加した後室温で4日間攪拌した。その溶媒を蒸発させた後1Nの塩酸を4mlをその残渣に加え、沈澱物を濾過して集めることにより182mg(収率は定量的)の化合物(L)が得られた。
【0135】
化合物(L)(172mg)をチオニルクロライド中に懸濁してから90℃で4.5時間攪拌した。溶媒を蒸発させた後ジエチルエーテルをその残渣に添加し、沈澱物を濾過して集めることによって180mgの化合物(M)が得られた。
【0136】
化合物(M)(67mg、0.1mmol)を2mlのエチレンジクロライドに溶解し、続いてテトラハイドロフランに入れたアニリン、180μlを氷冷しながらそこに添加してから同じ温度で1時間攪拌した。溶媒を蒸発させた後でその残渣を2mlのテトラハイドロフランと0.5mlのメタノールの混合溶媒に溶解し、続いて1mlの1N NaOHをそこに添加してから室温で3時間攪拌した。この溶液に1Nの塩酸(1.2ml)を添加して中和し、続いてテトラハイドロフランを用いて希釈した。その混合物を生理食塩水を用いて洗浄してから硫酸ナトリウム上で乾燥した。この溶媒を蒸発させた後、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)にかけることによって化合物II-45(単離された化合物N、56mgから13mg)が得られた。
【0137】
実施例24
化合物II-65
【0138】
出発化合物(Q)は、日本で未審査の特許出願第295588/88号に開示されている。
【0139】
化合物(Q)(50mg、0.0861mmol)を3mlのクロロホルムに溶解し、続いて200mg(1.41mmol)の2−ジメチルアミノエタンチオール塩酸塩と49mg(0.21mmol)の(±)−10−カンファースルホニックアシッドをそこに添加した後、室温で12時間攪拌した。この反応混合物を炭酸水素ナトリウムの飽和水溶液、水、及び生理食塩水を用いて順次洗浄してから硫酸ナトリウム上で乾燥した。溶媒を蒸発させてからその残渣を、プレパラティブ薄層クロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって56.3mg(収率98%)のN,O−ジアセチル化された化合物II-65が得られた。
FAB-MS(m/z): 668(M+1)+
【0140】
N,O−ジアセチル化された化合物II-65(36.6mg、0.0548mmol)を6mlのクロロホルムと3mlのメタノールの混合溶媒に溶解し、続いて18μl(0.09mmol)の5.1Nのナトリウムメトキシドをそこに添加した後、室温で20分間攪拌した。アンバーリスト(Amberlyst)(登録商標)15イオン交換樹脂(100mg)をその反応混合物に添加してから1時間攪拌し、不溶物を濾過して分離した。溶媒を蒸発させた後その残渣をプレパラティブ薄層クロマトグラフィー(クロロホルム/メタノール=97/3)にかけることによって28.4mg(収率89%)の化合物II-65が得られた。
【0141】
実施例25
化合物II-66
化合物II-66は、例えば日本で公開された未審査の特許出願第155284/87号(参照としてここに組み入れられる)に記載された方法にしたがって調製される。
【0142】
実施例26
化合物II-75
化合物(P)(日本で公開された未審査の特許出願第295588/88号)(100mg、0.173mmol)と4−アミノ−1,2,4−トリアゾール(17.4mg、0.207mmol)を、4mlのクロロホルムと1.5mlのテトラハイドロフランの混合溶媒に溶解し、続いて0.05mlの3Nの塩酸をそこに添加した後、室温で3.5時間攪拌した。酢酸エチルをそこに加えた後、不溶性の物質を濾過して集め、シリカゲルクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって71.9mg(収率64%)のN,O−ジアセチル化された化合物II-75が得られた。
FABS−MS(m/z): 646(M+1)+
【0143】
N,O−ジアセチル化された化合物II-75(37.5mg、0.058mmol)を2mlの1,2−ジクロロエタンと0.6mlのメタノールの混合溶媒に溶解し、続いてメタノール中に入れた5.1Nのナトリウムメトキシド、11μl(0.058mmol)をそこに添加した後、室温で20分間攪拌した。アンバーリスト(Amberlyst)(登録商標)15(50mg)をその反応混合物に添加してから30分間攪拌し、不溶性物質を濾過して分離した。その不溶性物質をジクロロメタン/メタノール/水酸化アンモニウム(8/2/0.5)を用いてよく洗浄し、濾液を合わせて減圧下で濃縮した。その残渣をシリカゲルカラムクロマトグラフィーにかけることによって26.8mg(収率82%)の化合物II-75が得られた。
【0144】
実施例27
化合物II-79
化合物(Q)(日本で公開された未審査の特許出願第295588/88号)(50mg、0.0861mmol)と2−(ブチルアミノ)エタンチオール(0.127ml、0.861mmol)をクロロホルムに溶解し、続いて300mg(1.29mmol)のカンファースルホニックアシッドをそこに添加した後、室温で4日間攪拌した。炭酸水素ナトリウムの飽和水溶液をこの反応混合物に添加して、有機層を塩化ナトリウムの水溶液で洗浄してから硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させ、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって34.6mg(収率58%)のN,O−ジアセチル化された化合物II-79が得られた。
FAB-MS(m/z): 697(M+1)+
【0145】
実施例26と実質的に同じ方法を31.1mg(0.0447mmol)のN,O−ジアセチル化された化合物II-79を用いて繰り返すことによって化合物II-79が得られた(収率52%)。
【0146】
実施例28
化合物II-80
化合物F(国際公開第94/02448号(WO94/02488);参照として本明細書に組み入れられる)(6.19g、10.1mmol)を300mlの1,2−ジクロロエタンと100mlのメタノールの混合溶媒に溶解し、続いてメタノール中に入れた5.1Nのナトリウムメトキシド、0.5ml(2.55mmol)をそこに添加した後、室温で35分間攪拌した。この反応混合物を氷水に注ぎいれ、不溶性物質を濾過して集めることによって化合物II-80のビス(ジメチルアミノエチルチオメチル)の代わりにビス(ハイドロキシメチル)を持つ化合物が、4.95g(収率93%)得られた。
FAB-MS(m/z): 528(M+1)+
【0147】
実施例27と実質的に同じ方法を、化合物II-80のビス(ジメチルアミノエチルチオメチル)の代わりにビス(ハイドロキシメチル)を持つ化合物、22.1mg(0.0419mmol)と2−(ジメチルアミノ)エタンチオールハイドロクロライド、59.4mg(0.419mmol)とを用いて繰り返すことによって、13.1mg(収率45%)の化合物II-80が得られた。
【0148】
実施例29
化合物II-72
実施例27と実質的に同じ方法を、50mg(0.0861mmol)の化合物(Q)と97.8mg(0.861mmol)の2−アミノエタンチオールハイドロクロライドとを用いて繰り返すことによって、49.6mg(収率90%)のN,O−ジアセチル化された化合物II-72が得られた。
FAB-MS(m/z): 641(M+1)+
【0149】
実施例26と実質的に同じ方法を、39.5mg(0.0617mmol)のN,O−ジアセチル化された化合物II-72を用いて繰り返すことによって、30.2mg(収率88%)の化合物II-72が得られた。
【0150】
実施例30
化合物VI−1
化合物(R)(J. Antibiotics, 38: 1437, 1985、図24参照)(1g、1.81mmol)を50mlの1,2−ジクロロエタンに溶解し、続いて0.17ml(3.80mmol)の発煙硝酸を0℃でそこに滴下した後、室温で20分間攪拌した。その反応混合物をクロロホルムで希釈し、そこに炭酸水素ナトリウムの飽和水溶液を添加してから有機層を塩化ナトリウムの水溶液で洗浄し、硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させた後、40mlのジメチルホルムアミドと600mgの10%Pd/Cをその残渣に添加し、続いて水素雰囲気中で1時間、60℃で攪拌した。不溶物を濾過して除き、その濾液を減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=20/80)にかけることによって130.8mg(収率13%)のアミン誘導体が得られた。
FAB-MS(m/z): 567(M+1)+
【0151】
このアミン誘導体(23.9mg、0.0422mmol)を2mlのクロロホルムに溶解し、続いて9.2μl(0.0660mmol)のトリエチルアミンと87μl(1.10mmol)のエチルイソシアネートをそこに滴下した後、室温で2日間攪拌した。水、メタノール、及びクロロホルムをその反応混合物に加えることによってその反応を終了させ、得られた反応混合物をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液で洗浄し、硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させた後、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)にかけることによって31.4mg(収率80%)のN,O−ジアセチル化された化合物VI−1が得られた。
【0152】
実施例26と実質的に同じ方法を、21.4mg(0.0336mmol)のN,O−ジアセチル化された化合物VI−1を用いて繰り返すことで17.0mg(収率91%)の化合物VI−1が得られた。
【0153】
実施例31
化合物VI−2
実施例30と実質的に同じ方法を、5g(9.07mmol)の化合物(R;図24参照)を用いて繰り返すことで259mg(収率5%)のジアミン誘導体が得られた。
FAB-MS(m/z): 582(M+1)+
【0154】
実施例26と実質的に同じ方法を、24.5mg(0.0422mmol)のジアミン誘導体を用いて繰り返すことによって3.8mg(収率18%)の化合物VI−2が得られた。
【0155】
実施例32
化合物IV-6
化合物(S;図25参照)[J. Chem. Soc. Perkin. Trans. 1, 2475 (1990)](5.15g、13.0mmol)を30mlのジメチルホルムアミドと60mlのトルエンの混合溶媒に溶解し、続いて1.45g(12.9mmol)のtert−ブトキシカリウムをアルゴン雰囲気下、−20℃でそこに添加した後、室温で30分間攪拌した。その反応混合物を−20℃まで冷却した後、1.12ml(12.9mmol)の臭化アリールをそこに添加し、その混合物を0℃で2時間攪拌した。溶媒を減圧下で蒸発させた後、水をこの残渣に加え、続いてテトラハイドロフランを用いて抽出した。有機層を塩化ナトリウムの水溶液で洗浄し、硫酸マグネシウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/15)にかけ、ジクロロメタンを用いて粉砕することによって898.4mg(収率16%)の化合物(T-1)が唯一の位置異性体として得られた。
【0156】
化合物(T-1)(1.44g、3.30mmol)を50mlのテトラハイドロフランに溶解し、続いて4.05g(33.2mmol)の9−ボラビシクロ[3,3,1]ノナン(9−BBN)(ダイマー)をそこに添加した後、アルゴン雰囲気下で3時間、室温にて攪拌した。その反応混合物を0℃まで冷却した後、6mlの1N水酸化ナトリウムと6mlの過酸化水素の35%水溶液をそこに添加し、続いて45分間攪拌した。この反応混合物を水を用いて希釈した後、反応混合物を酢酸エチルを用いて抽出した。有機層を水と塩化ナトリウムの水溶液を用いて順次で洗浄し、硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させ、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/1)にかけることによって875.5mg(収率58%)の化合物(J−1)が得られた。
【0157】
化合物(U-1)(178.5mg、0.394mmol)を10mlのジメチルホルムアミドに溶解し、続いて309.5mg(1.18mmol)のトリフェニルホスフィンと0.060ml(1.2mmol)の臭素とをアルゴン雰囲気下で0℃にてそこに添加した後、室温で3時間攪拌した。水をその反応混合物に添加してその反応を終了させた後、その混合物を酢酸エチルを用いて抽出し、有機層を水と塩化ナトリウム水溶液を用いて順次で洗浄してから硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させ、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/8)にかけることによって134.6mg(収率66%)の化合物(W)が得られた。
【0158】
化合物(W)を5mlのジメチルホルムアミドに溶解し、続いて0.045ml(0.52mmol)のモルホリンをそこに添加した後、80℃で3時間、アルゴン雰囲気下で攪拌した。その反応混合物を室温にまで冷却した後、氷水をそこに添加し、濾過することによって生じた沈澱物を集めた。この沈澱物を減圧下で乾燥し、薄層クロマトグラフィー(クロロホルム/メタノール=25/1)にかけた。得られた生成物を10mlのテトラハイドロフランに溶解し、続いて3mlの4Nの硫酸をそこに加えてから60℃で12時間攪拌した。反応混合物を室温にまで冷却した後で氷をそこに添加し、混合物を酢酸エチルを用いて抽出した。有機層を水と塩化ナトリウムの水溶液を用いて順次洗浄してから硫酸マグネシウム上で乾燥した。その溶媒を減圧下で蒸発させ、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/2)にかけた。得られた生成物をクロロホルムと酢酸エチルの混合溶媒に溶解し、続いて酢酸エチルに入れた0.88Nの塩酸を添加した後で室温にて1時間攪拌した。生じた沈澱物を濾過して集め、酢酸エチルで洗浄してから減圧下で乾燥させることによって35.0mg(収率19%)の化合物(IV-6)が得られた。
【0159】
実施例33
化合物IV-5
【0160】
化合物(S)(J. Chem. Soc. Perkin. Trans. 1, 2475, 1990)(823.7mg、2.083mmol)を20mlのジメチルホルムアミドに溶解し、166.4mg(4.16mmol)の水素化ナトリウム(60%)を氷冷下でそこに添加した後、同じ温度で10分間攪拌した。臭化アリル(0.45ml、5.2mmol)をそこに添加してからその溶液を氷冷下で2時間攪拌した。クロロホルムを用いて希釈した後で水をそこに加えて有機層を分離し、生理食塩水を用いて洗浄してから硫酸マグネシウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/15)にかけることによって735.0mg(収率74%)の化合物(T-2)が得られた。
【0161】
水素化ホウ素ナトリウム(77.7mg、2.05mmol)を20mlのテトラハイドロフランに懸濁し、231.0mg(1.82mmol)のヨウ素をそこに0℃でアルゴン雰囲気下で添加した後、同じ温度で15分間攪拌した。化合物(T-2)(136.7mg、0.287mmol)を同じ温度でそこに加えてからその反応混合物を室温で4.5時間攪拌した。その反応混合物を0℃まで冷却した後、3.7mlの1N水酸化ナトリウムと3.7mlの過酸化水素の35%水溶液をそこに添加し、続いてさらに30分間攪拌した。この反応混合物を水を用いて希釈してから酢酸エチルを用いて抽出した。この酢酸エチル層を水と食塩水溶液を用いて順次で洗浄し、硫酸マグネシウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=15/1)にかけることによって88.9mg(収率61%)の化合物(U-2)が得られた。
【0162】
化合物(U-2)(88.9mg、0.174mmol)を10mlのテトラハイドロフランに溶解し、そこに8mlの4N硫酸を添加した後、60℃で24時間攪拌した。その反応混合物を室温にまで冷却した後で氷をそこに加え、続いて酢酸エチルによって抽出した。酢酸エチル層を水と食塩水溶液を用いて順次洗浄してから硫酸マグネシウム上で乾燥した。溶媒を蒸発させ、残渣を薄層クロマトグラフィー(クロロホルム/メタノール=15/1)にかけることによって37.6mg(収率51%)の化合物IV-5が得られた。
【0163】
実施例34
化合物II-68
化合物(Q)(50.1mg、0.0862mmol)を3mlのクロロホルムに溶解し、続いて129.5mg(0.862mmol)の2−メルカプトベンズイミダゾールと49mg(0.21mmol)の(±)−10−カンファースルホニックアシッドとをそこに添加した後、室温で12時間攪拌した。その反応混合物を炭酸水素ナトリウムの飽和水溶液、水、及び生理食塩水を用いて順次洗浄した後で硫酸ナトリウム上で乾燥した。溶媒を蒸発させてから残渣をプレパラティブ薄層クロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって、46mg(収率75%)のN,O−ジアセチル化された化合物II-68が得られた。
FAB-MS(m/z): 714(M+1)+
【0164】
実施例25と実質的に同じ方法を、33.4mg(0.0468mmol)のN,O−ジアセチル化された化合物II-68を用いて繰り返すことによって、17.5mg(収率59%)の化合物II-68が得られた。
【0165】
実施例35
化合物II-69
実施例25と実質的に同じ方法を、50mg(0.0861mmol)の化合物Q及び0.0868ml(0.861mmol)のフルフリルメルカプタンを用いて続いて行うことによって、36.0mg(収率62%)のN,O−ジアセチル化された化合物II-69が得られた。
FAB-MS(m/z): 678(M+1)+
【0166】
実施例25と実質的に同じ方法を22.7mg(0.0335mmol)のN,O−ジアセチル化された化合物II-69を用いて繰り返すことによって、17.7mg(収率89%)の化合物II-69が得られた。
【0167】
実施例36
化合物II-70
化合物(P)(100mg、0.173mmol)を4mlのクロロホルムに溶解し、続いて34.0mg(0.277mmol)の1−アミノピロリジン塩酸塩をそこに添加した後、室温で4時間攪拌した。減圧下で溶媒を蒸発させた後、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって、100.5mg(収率90%)のN,O−ジアセチル化された化合物II-70が得られた。
FAB-MS(m/z): 648(M+1)+
【0168】
実施例25と実質的に同じ方法を40mg(0.0618mmol)のN,O−ジアセチル化された化合物II-70を用いて繰り返すことによって、30mg(収率86%)の化合物II-70が得られた。
【0169】
実施例37
化合物II-71
化合物(P)(49mg、0.0846mmol)を3mlのクロロホルムに溶解し、続いて15.8mg(0.145mmol)の2−ヒドラジノピリジンのクロロホルム溶液と49mg(0.21mmol)の(±)−10−カンファースルホニックアシッドをそこに添加した後、室温で12時間攪拌した。その反応混合物を炭酸水素ナトリウムの飽和水溶液、水、及び塩溶液を用いて順次洗浄してから硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、残渣をプレパラティブ薄層クロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって、35.8mg(収率64%)のN,O−ジアセチル化された化合物II-71が得られた。
FAB-MS(m/z): 671(M+1)+
【0170】
実施例25と実質的に同じ方法を24.6mg(0.0367mmol)のN,O−ジアセチル化された化合物II-71を用いて繰り返すことによって、11.8mg(収率55%)の化合物II-71が得られた。
【0171】
実施例38
化合物II-73
実施例25と実質的に同じ方法を30mg(0.0516mmol)の化合物(Q)と52.2mg(0.516mmol)の1H−1,2,4−トリアゾール−3−チオールとを用いて続いて行うことによって、31.4mg(収率92%)のN,O−ジアセチル化された化合物II-73が得られた。
FAB-MS(m/z): 665(M+1)+
【0172】
実施例25と実質的に同じ方法を15mg(0.0226mmol)のN,O−ジアセチル化された化合物II-73を用いて繰り返すことによって、粗精製の化合物II-73が得られた。そこにクロロホルム/メタノール(90/10)を添加した後で攪拌することによって10.9mg(収率83%)の化合物II-73が沈澱物として得られた。
【0173】
実施例39
化合物II-74
化合物(P)(97.5mg、0.168mmol)を4mlのテトラハイドロフランに溶解し、続いて25.1mg(0.0950mmol)のアミノグアニジン硫酸塩の水溶液をそこに添加した後、室温で3時間攪拌した。酢酸エチルをそこに添加してから攪拌し、不溶性の物質を濾過して集めた後、シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=85/15)にかけることによって、87.1mg(収率82%)のN,O−ジアセチル化された化合物II-74が得られた。
FAB-MS(m/z): 636(M+1)+
【0174】
実施例25と実質的に同じ方法を69.6mg(0.110mmol)のN,O−ジアセチル化された化合物II-74を用いて繰り返すことによって、37.2mg(収率62%)の化合物II-77が得られた。
【0175】
実施例40
化合物II-76
化合物(P)(103.8mg、0.179mmol)を6mlのクロロホルムと3mlのメタノールの混合溶媒に溶解し、続いて0.020ml(0.207mmol)の4−アミノモルホリンの水溶液、0.5mlと3Nの塩酸、0.05mlをそこに添加した後、室温で3時間攪拌した。その反応混合物を炭酸水素ナトリウムの飽和水溶液、及び塩溶液を用いて順次洗浄してから硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=90/100)にかけることによって、82.8mg(収率70%)のN,O−ジアセチル化された化合物II-76が得られた。
FAB-MS(m/z): 663(M+1)+
【0176】
実施例25と実質的に同じ方法を50.6mg(0.0763mmol)のN,O−ジアセチル化された化合物II-76を用いて繰り返すことによって、36.4mg(収率82%)の化合物II-76が得られた。
【0177】
実施例41
化合物II-77
実施例40と実質的に同じ方法を100mg(0.173mmol)の化合物Pと16.7mg(0.173mmol)の1,1−ジメチルヒドラジン塩酸塩とを用いて続いて行うことによって、52.3mg(収率49%)のN,O−ジアセチル化された化合物II-77が得られた。
FAB-MS(m/z): 622(M+1)+
【0178】
実施例25と実質的に同じ方法を38.4mg(0.0618mmol)のN,O−ジアセチル化された化合物I−75を用いて繰り返すことによって、10.9mg(収率33%)の化合物II-77が得られた。
【0179】
実施例42
化合物II-78
実施例40と実質的に同じ方法を99.5mg(0.172mmol)の化合物(P)と42.4mgの1−アミノ−4−メチルピペラジンとを用いて続いて行うことによって、N,O−ジアセチル化された化合物II-78が得られた。
【0180】
続いて実施例25と実質的に同じ方法を、上記のN,O−ジアセチル化化合物II-78を用いて繰り返すことによって、19.4mg[化合物(P)からの収率で19%]の化合物II-78が得られた。
【0181】
実施例43
化合物II-81
化合物(AA)、即ち化合物II-80(実施例28に記載されている)のビス(ジメチルアミノエチルチオメチル)に代えて、ビス(ハイドロキシメチル)を持つ化合物(53.9mg、0.102mmol)を、2.5mlのジクロロメタンに溶解した。続いて0.18ml(2.0mmol)の2−プロパンチオールと0.03ml(0.2mmol)のトリフルオロ酢酸無水物を順次そこに添加し、アルゴン気流下で3時間、室温にて攪拌した。炭酸水素ナトリウムの飽和水溶液をその反応混合液に添加してからその混合液をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてから残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって52.6mg(収率80%)の化合物II-81が得られた。
【0182】
実施例44
化合物II-82
実施例43と実質的に同じ方法を51.9mg(0.0958mmol)の化合物(AA)と0.17ml(1.9mmol)の1−プロパンチオールと0.03ml(0.2mmol)のトリフルオロ酢酸無水物とを用いて繰り返すことによって、52.3mg(収率83%)の化合物II-82が得られた。
【0183】
実施例45
化合物II-83
実施例43と実質的に同じ方法を49.4mg(0.0937mmol)の化合物(AA)と0.20ml(1.9mmol)の1−ブタンチオールと0.03ml(0.2mmol)のトリフルオロ酢酸無水物とを用いて繰り返すことによって、51.7mg(収率82%)の化合物II-83が得られた。
【0184】
実施例46
化合物II-84
化合物(AA)(45.3mg、0.0860mmol)を0.2mlのメタノールと2mlのクロロホルムの混合溶媒に溶解し、続いて20mg(0.086mmol)のカンファースルホニックアシッドをそこに添加してから室温にて17時間攪拌した。炭酸水素ナトリウムの飽和水溶液をその反応混合液に添加して、続いてその混合液をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてから残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって23.1mg(収率48%)の化合物II-84が得られた。
【0185】
実施例47
化合物II-85
実施例46と実質的に同じ方法を、0.2mlのエタノールと2mlのクロロホルムの混合溶媒に入れた51.3mg(0.0973mmol)の化合物(AA)を用いて繰り返すことによって、24.1mg(収率42%)の化合物II-85が得られた。
【0186】
実施例48
化合物II-86
化合物(BB)(日本で公開された未審査の特許出願第295588/88号)(978mg、1.69mmol)を70mlの1,2−ジクロロエタンに溶解し、続いて0.17ml(3.8mmol)の発煙硝酸を氷冷しながらそこに滴下してから室温にて30分間攪拌した。その反応混合液をクロロホルムを用いて希釈してから炭酸水素ナトリウムの飽和水溶液をそこに添加した。不溶物を濾過して集め、乾燥した。その濾液を塩化ナトリウムの水溶液を用いて洗浄し、硫酸ナトリウム上で乾燥してから溶媒を減圧下で蒸発させた。残渣と不溶物とをあわせて946mg(収率90%)の化合(CC)が粗生成物として得られた。
FAB-MS(m/z): 625(M+1)+
【0187】
化合物(CC)(640mg、1.03mmol)を30mlの1,2−ジクロロエタンに溶解し、続いて0.3ml(3.58mmol)の1,2−エタンジチオールと0.2ml(2.0mmol)のボロントリフロライドエーテル錯体を0℃でそこに滴下してから30分間攪拌した。炭酸水素ナトリウムの飽和水溶液をその反応混合液に添加し、続いてその混合液をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム)にかけることによって579mg(収率81%)の化合物(DD)が得られた。
FAB-MS(m/z): 701(M+1)+
【0188】
化合物(DD)(579mg、0.827mmol)を56mlのN,N−ジメチルホルムアミドに溶解し、続いて400mgのパラジウム/炭素をそこに添加してから60℃で2時間、水素雰囲気下で攪拌した。不溶物を濾過して分離し、その濾液から溶媒を減圧下で蒸発させた。その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)にかけることによって193mg(収率35%)の化合物(EE)が得られた。
FAB-MS(m/z): 671(M+1)+
【0189】
化合物(EE)(193mg、0.288mmol)を10mlのクロロホルムに溶解し、続いて0.1ml(0.7mmol)のトリエチルアミンと0.2ml(2.5mmol)のエチルイソシアネートをそこに添加してから室温で20時間攪拌した。水を加えてからその混合液をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液で洗浄してから硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=96/4)にかけることによって211mg(収率99%)の化合物(FF)が得られた。
FAB-MS(m/z): 742(M+1)+
【0190】
化合物(FF)(211mg、0.285mmol)を6mlのエタノールと6mlのクロロホルムの混合溶媒に溶解し、続いて171mg(1.01mmol)の硝酸銀をそこに50℃で添加してから20分間攪拌した。反応を終了させてから不溶物を濾過して除去した。この濾液を炭酸水素ナトリウムの飽和水溶液と塩化ナトリウムの水溶液を用いて洗浄し、そして硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=97/3)にかけることによって118mg(収率62%)の化合物(GG)が得られた。
FAB-MS(m/z): 666(M+1)+
【0191】
化合物(GG)(100mg、0.150mmol)を4.5mlのクロロホルムと0.72mlのメタノールの混合溶媒に溶解し、続いて8.7mg(0.23mmol)の水素化ホウ素ナトリウムをそこに0℃で添加してから45分間攪拌した。その反応混合液を水に注入してからその混合溶液をクロロホルムで抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、続いて硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって101mg(収率100%)の化合物(HH)が得られた。
FAB-MS(m/z): 668(M+1)+
【0192】
化合物(HH)(21.7mg、0.0325mmol)を1mlの1,2−ジクロロエタンと0.3mlのメタノールの混合溶媒に溶解し、続いて6μl(0.03mmol)の5.1Nのナトリウムメトキシドのメタノール溶液をそこに添加してから1時間攪拌した。その反応混合液を水に注入してからその混合溶液をクロロホルムとメタノール(9/1)の混合溶媒で抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、続いて硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=90/10)にかけることによって14.9mg(収率79%)の化合物II-86が得られた。
【0193】
実施例49
化合物II-87
実施例43と実質的に同じ方法を29.8mg(0.0511mmol)の化合物II-86と0.14ml(1.6mmol)のエタンチオールとを用いて繰り返すことによって、24.2mg(収率76%)の化合物II-87が得られた。
【0194】
実施例50
化合物II-88
化合物(AA)(50.4mg、0.0956mmol)を0.7mlのジクロロメタンに溶解し、続いて0.09mg(0.56mmol)のトリエチルシランと0.73ml(9.5mmol)のトリフルオロ酢酸をそこに氷冷しつつ添加してから室温で10分間攪拌した。その反応混合液を1Nの水酸化ナトリウム水溶液を用いて中和してからその混合溶液をクロロホルムで抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、続いて硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=90/10)にかけることによって20.7mg(収率44%)の化合物II-88が得られた。
【0195】
実施例51
化合物II-89
化合物(AA)(4.3g、8.16mmol)を215mlのジクロロメタンに溶解し、続いて12.1mg(163mmol)のエタンチオールと2.5ml(17.7mmol)のトリフルオロ酢酸無水物をそこに連続的に添加してから室温で12時間攪拌した。炭酸水素ナトリウムの飽和水溶液をその反応混合液に添加し、続いてその混合溶液をクロロホルムで抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、続いて硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=3/7)にかけることによって4mg(収率0.08%)の化合物II-89が得られた。
【0196】
実施例52
化合物II-90
化合物(JJ)(日本で公開された未審査の特許出願第295588/88号)(18.5g、30.5mmol)を900mlのクロロホルムと145mlのメタノールの混合溶媒に溶解し、続いて3.42g(90.4mmol)の水素化ホウ素ナトリウムを氷冷しつつそこに添加してから25分間攪拌した。その反応混合液を氷水に注入し、不溶物を濾過して集めてから水を用いて洗浄を行い、減圧下で乾燥した。不溶物を555mlの1,2−ジクロロエタンと185mlのメタノールの混合溶媒に溶解し、続いて0.925ml(4.72mmol)のナトリウムメトキシドの5.1Nメタノール溶液をそれに添加してから1時間半攪拌した。この反応混合液を水に注入し、不溶物を濾過して集め、続いて減圧下で乾燥してからシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=8/2)にかけることによって0.350g(収率2.3%)の化合物II-90が得られた。
【0197】
実施例53
化合物II-91
化合物(DD)(18.6g、0.0266mmol)を1.5mlの1,2−ジクロロエタンと0.5mlのメタノールの混合溶媒に溶解し、続いて5μl(0.026mmol)のナトリウムメトキシドの5.1Nメタノール溶液をそれに添加してから1時間攪拌した。この反応混合液を水に注入し、そして得られた混合溶液をクロロホルムとメタノール(9/1)の混合溶媒を用いて抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄した後、硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって7.0mg(収率43%)の化合物II-91が得られた。
【0198】
実施例54
化合物II-92
実施例53と実質的に同じ方法を23.3mg(0.0314mmol)の化合物(FF)を用いて繰り返すことによって、14.7mg(収率71%)の化合物II-92が得られた。
【0199】
本発明はかなり詳細に列記したが、本明細書に開示された発明は実際の記載に限定されるべきではなく、添付した請求の範囲及びその等価物のすべてからなる完全な範囲が提供されるべきである。他の態様は、次の請求の範囲内に含まれる。
【図面の簡単な説明】
【0200】
【図1】図1は、PC−12細胞における基底オルニチンデカルボキシラーゼ(ODC)活性に対するK−252a誘導体である1,6−ヘキサメチレン−ビス−(カルバミルスタウロスポリン)(HBCS)およびスタウロスポリンの効果を示すグラフである。
【図2】図2は、PC−12細胞におけるNGFにより刺激されるODC活性に対するスタウロスポリン、HBCS、及びK−252aの効果を示すグラフである。
【図3】図3は、PC−12細胞におけるODC活性に対するHBCSのNGF−効力増強効果を示すグラフである。
【図4】図4は、ラット胎児の脊髄培養物におけるコリンアセチルトランスフェラーゼ(ChAT)特異的活性に対するK−252aの効果を示すグラフである。
【図5】図5は、ラット胎児の脊髄培養物におけるChAT活性に対するK−252aの効果の時間経過を示すグラフである。
【図6】図6は、ヒナ胎児の背根神経節ニューロンの生存性に対するK−252aの効果を示すグラフである。
【図7】図7は、ヒナ胎児の背根神経節ニューロンの生存性に対するK−252aの機能性誘導体の効果を示すグラフである。
【図8】図8は、ラット胎児の脊髄培養物におけるChAT活性に対するK−252aの機能性誘導体の効果を示すグラフである。
【図9】図9は、ラットの海馬へのカイニン酸塩誘導性の損傷に対するK−252aの効果を示すグラフである。
【図10】図10は、ラットの海馬におけるカイニン酸塩誘導性のスペクトリンタンパク質分解に対するK−252aの効果を示すグラフである。
【図11】図11は、海馬へのカイニン酸塩誘導性の損傷に対するHBCSの効果を示すグラフである。
【図12】図12は、ラットの海馬におけるカイニン酸塩誘導性のスペクトリンタンパク質分解に対するK−252aの機能性誘導体の効果を示すグラフである。
【図13】図13a、13b及び13cは、ラットの脊髄培養物におけるChAT活性に対するK−252a誘導体の相対的な活性を示す表である。
【図14】図14は、ヒナの背根神経節培養物におけるニューロンの生存性に対するK−252a誘導体の相対的な活性を示す表である。
【図15】図15は、K−252aの存在下での脊髄ニューロンの生存性を示すグラフである。
【図16】図16は、K−252aの存在下での脊髄細胞の生存性の時間経過を示すグラフである。
【図17】図17は、K−252aの存在下または非存在下で培養された線条体ニューロンの一対の顕微鏡写真である。
【図18】図18は、ラットの線条体培養物におけるニューロンの生存性に対するK−252a誘導体の相対的な活性を示す表である。
【図19】図19は、低密度の基底前脳ニューロンの生存性に対するK−252a誘導体の相対的な活性を示す表である。
【図20】図20は、化合物II−51が、成長過程でプログラムされた運動ニューロンの死をインオボ(in ovo)で阻止することを示す棒グラフである。
【図21】図21は、化合物II−51が、成体の舌下核におけるChAT免疫活性が軸索切断によって喪失されるのを阻止することを示す写真である。
【図22】図22は、出発化合物Cから化合物Hへの合成を示す図式である。
【図23】図23は、出発化合物Jから化合物II−45への合成を示す図式である。
【図24】図24は、化合物P、化合物Q、及び化合物Rの構造を示す図である。
【図25】図25は、出発化合物Sから化合物IV−6への合成を示す図式である。
【図26】図26は、化合物(AA)、(BB)、(CC)、(DD)及び(EE)の化学構造を示す図である。
【図27】図27は、化合物(FF)、(GG)、(HH)及び(JJ)の化学構造を示す図である。
【技術分野】
【0001】
本出願は、現在は放棄されている1992年7月24日に提出された米国特許出願第07/920,102号の一部継続出願である、1993年7月22日に提出された米国特許出願第08/096,561号のさらなる一部継続出願である、1994年10月26に提出された米国特許出願第08/329,540号のさらなる一部継続出願である。
【背景技術】
【0002】
発明の背景
プロテインキナーゼは、アミノ酸のリン酸化を行うことによって多くの細胞内タンパク質を化学的に修飾する作用のある広範な酵素のクラスである。
【0003】
プロテインキナーゼの阻害剤は構造的にはいろいろあり、神経系や他の組織に対してさまざまな(時には反対の)影響を与える。ある特定のプロテインキナーゼ阻害剤は、一種以上のプロテインキナーゼに影響するであろう。例えば、ノカルジオプシスsp.(Nocardiopsis sp.)及びアクチノマヅラsp.(Actinomadula sp.)の培養液から単離されたアルカロイド様の物質であるK−252aは、当初プロテインキナーゼCの阻害剤であると報告されたが、その後プロテインキナーゼA及びG、ミオシン軽鎖キナーゼ、及びtrk(末梢ニューロン、感覚ニューロン、それに交感ニューロンの生存性を促進する神経栄養性タンパク質である神経生長因子[NGF]によって活性化されるチロシンキナーゼ)も阻害することが判明した。
【0004】
この後者の効果と一致して、K−252aはPC−12細胞(ラットの副腎髄質腫瘍、褐色細胞腫から得られるクロム親和性細胞)に対するNGFの神経栄養性作用を遮断し、背根の神経節ニューロン及び海馬ニューロンの生存性を向上させる。しかしながらそれは、広範囲の濃度で細胞毒性を示すことがわかっており、そのため何人かの研究者は、インビボでの有用性が制限されているという結論に至っている。
【0005】
K−252aに関係のある微生物のアルカロイドであるスタウロスポリン(staurosporine)も、異なるプロテインキナーゼ及び細胞型に対してさまざまな影響を持つ。スタウロスポリンはPC−12細胞に対してNGF−様の効果を持っていて、アレチネズミの海馬が虚血後障害(post−ischemic injury)にならないように保護できることがわかっている。それは、ラットの基底前脳においてコリン作動性ニューロンに起こる損傷を回復することができる。
【0006】
K−252a及びスタロウスポリンは腫瘍の阻害剤として推奨されてきた。スタウロスポリンは殺虫剤としても提供されている。スタウロスポリンの誘導体はメチルアミン窒素で置換されたハイドロカルビルラジカルまたはアシルラジカルを持っており、以下の使用のために合成されかつ推奨されている。即ち、腫瘍の阻害、炎症の抑制、免疫調整、及び心臓血管や中枢神経系の疾患の治療である。
【発明の開示】
【0007】
発明の概要
本発明のある観点での特徴は、下式によって示される新規なビスN−置換されたスタウロスポリンの誘導体である。
[Stau]−N(CH3)−W−N(CH3)−[Stau] (I)
ここで[Stau]は下式の残基を表し、
そしてWは下式のビス(カルバミル)またはビス(チオカルバミル)ラジカルを表す。
−C(=Y)−NH−W’−NH−C(=Y)−
ここでW’は2〜20個の炭素原子からなるハイドロカルビレンラジカルであって、YはOまたはSである。
【0008】
好ましい観点では本発明は、例えば化合物1,6−ヘキサメチレン−ビス−(カルバミルスタウロスポリン)(HBCS)、p−フェニレン−ビス−(カルバミルスタウロスポリン)(PBCS)を特徴とする。
【0009】
本発明はまた、下式(II−4)によって表される新規なK−252aの誘導体を特徴とする。
ここでR1、R2、Z1、及びZ2はそれぞれ独立にHであり、Xはハイドロキシメチル基(CH2OH)であり、そしてRはOCH3 である。
【0010】
本発明はまた、下式によって表される新規なK−252aの誘導体であることを特徴とする。
ここでR1、R2、Z1、及びZ2はそれぞれ独立にHであり、XはCH2−NH−Serであり、そしてRはOH である。
【0011】
また本発明には、下式(II−49)によって表される化合物が含まれる。
ここでR1、Z1、及びZ2はそれぞれHであり、RはOHであり、R1はCH2SO2C2H5であり、そしてXはCO2CH3である。
【0012】
また本発明には下式(II−38)によって示される化合物が含まれる。
ここでR1、R2、Z1、及びZ2はそれぞれHであり、RはOHであり、そしてXはCH2NHCO2C6H5である。
【0013】
また本発明には、下式(II−45)によって示される化合物が含まれる。
ここでR1、R2はそれぞれBrであり、RはOHであり、Z1及びZ2はそれぞれHであり、そしてXはCONHC6H5 である。
【0014】
また本発明には下式(II−57)によって示される化合物が含まれる。
ここでR1、R2、Z1、及びZ2はそれぞれHであり、RはOHであり、そしてXはCH2NHCO2CH3である。
【0015】
また本発明には下式(II−72)によって示される化合物が含まれる。
ここでR1はCH2S(CH2)2NH2であり、XはCO2CH3であり、RはOHであり、そしてR2、Z1、及びZ2はそれぞれHである。
【0016】
また本発明には下式(II−75)によって示される化合物が含まれる。
ここでR1は
であり、XはCO2CH3であり、RはOHであり、そしてR2、Z1、及びZ2はそれぞれHである。
【0017】
また本発明には下式(II−79)で示される化合物が含まれる。
ここでR1はCH2S(CH2)2NH n−C4H9であり、XはCO2CH3であり、RはOHであり、そしてR2、Z1、及びZ2はそれぞれHである。
【0018】
また本発明には下式(II−80)によって示される化合物が含まれる。
ここでR1はCH2S(CH2)2N(CH3)2であり、R2はCH2S(CH2)2N(CH3)2であり、XはCO2CH3であり、RはOHであり、そしてZ1及びZ2はそれぞれHである。
【0019】
また本発明には下式(V)によって示される化合物が含まれる。
ここで、XはCO2R5(ここでR5は低級アルキル基を表す)またはCH2NHCO2R6 (ここでR6は低級アルキル基またはアリール基を表す)であり、R1は水素またはCH2SO2R7(ここでR7は低級アルキル基を表す)であるが、ただしX= CO2R5、R1= 水素は除かれる。
【0020】
化学式(V)における置換基の定義において低級アルキル基とは、直鎖状または分枝状のアルキル基であって、1〜6個の炭素原子、好ましくは1〜3個の炭素原子を有する基を意味し、例えばメチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、ペンチル基、ネオペンチル基、及びヘキシル基である。アリール基とは、6〜10個の炭素原子を持つアリール基を意味し、例えばフェニル基及びナフチル基である。
【0021】
また本発明には、下式(VI)によって示される化合物(VI−1)が含まれる。
ここでXはCO2CH3であり、RはOHであり、R1、R2、Z1、及びZ2はそれぞれHであり、そしてR8はNHCONHC2H5である。
【0022】
また本発明には、下式(VI)によって示される化合物(VI−2)が含まれる。
ここでXはCO2CH3、それぞれのR2及びR8はNH2、RはOH、そしてR1、Z1、及びZ2はそれぞれHである。
【0023】
本発明の化合物は、薬学的に許容される酸が付加した塩、金属塩、アンモニウム塩、有機アミンが付加した塩、及びアミノ酸が付加した塩などを含む薬学的に許容される塩の形態であってもよい。
【0024】
薬学的に許容される酸が付加した塩の例には、例えば塩酸塩、硫酸塩、及び燐酸塩のような無機酸が付加した塩、また酢酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、及びクエン酸塩のような有機酸が付加した塩が挙げられる。薬学的に許容される金属塩の例には、ナトリウム塩やカリウム塩のようなアルカリ金属の塩、マグネシウム塩やカルシウム塩のようなアルカリ土類金属の塩、アルミニウム塩、及び亜鉛塩がある。薬学的に許容されるアンモニウム塩の例は、アンモニウム塩及びテトラエチルアンモニウム塩である。薬学的に許容される有機アミン付加塩は、モルホリン、ピペリジンとの塩である。薬学的に許容される有機アミノ酸が付加した塩の例は、リジン、グリシン、及びフェニルアラニンとの塩である。
【0025】
他の観点では本発明は、コリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、及び感覚ニューロン、例えば背根神経節ニューロンの機能を、治療に有効な量の新規なビス−置換されたスタウロスポリンの誘導体を例えばヒトなどの哺乳動物に投与することによって高める方法を特徴とする。この治療は、栄養に関する因子(trophic factor)とともに、好ましくはニュートロフィンファミリーの一つとともに、そして最も好ましくは神経生長因子(NGF)とともに投与することができる。本明細書において用いられるように、「栄養に関する因子」とは、栄養に関する因子に応答性の細胞の生存性または機能に直接的にまたは間接的に影響を与える分子である。ニュートロフィンファミリーとは、NGFと有意に相同なタンパク質の群であり、それにはNGFの他に、脳由来神経栄養因子(BDNF; Leibrockら、Nature 341: 149-152, 1989参照)、ニューロトロフィン−3(NT−3; Hohnら、Nature 344: 339-341, 1990参照)、及びニューロトロフィン−5(NT−4/5; Berkemeierら、Neuron 7: 857-866, 1991参照)が含まれる。
【0026】
他の観点では本発明は、治療に有効な量の新規なビス−置換されたスタウロスポリン誘導体の一つを例えばヒトのような哺乳動物に投与することによって、その哺乳動物の神経細胞を、興奮性のアミノ酸によって誘導される変性が起こらないように保護する方法を特徴とする。このような変性が起こる可能性のある症状には、アルツハイマー病、たとえば筋萎縮性側索硬化症のような運動ニューロン疾患、パーキンソン病、例えば虚血性状態のような脳血管疾患、AIDS脳症、てんかん、ハンチントン病、及び脳や脊髄に起こる震盪性または穿通性の損傷が関連する。この治療は、神経栄養性因子、好ましくはニューロトロフィンファミリーの一つ、最も好ましくは神経生長因子(NGF)とともに投与するとよい。
【0027】
他の観点では本発明は、哺乳動物、例えばヒトにおけるコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、および/または例えば背根神経節ニューロンのような感覚ニューロンの機能を、治療に有効な量のK−252aの機能性誘導体、即ち下式によって示された誘導体を前記哺乳動物に投与することによって高めるための方法を特徴とする。
【0028】
ここでは下記の表1で示された置換のいずれかが行われる。好ましくは、哺乳動物におけるコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、または例えば背根神経節ニューロンのような感覚ニューロンの機能および/または生存性を高めるための方法には、哺乳動物に、治療に有効な量の、表1に示された化合物II−3、II−20、II−30、II−33、II−38、II−49、II−51、II−65、II−69、II−72、II−73、II−79、II−80、VI−1、またはVI−2を投与することが含まれる。更に好ましくは、哺乳動物におけるコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、または感覚ニューロンの機能および/または生存性を高めるための方法には、治療に有効な量の化合物II−51を投与することが含まれる。
【0029】
表1
【0030】
(1)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
(2)NH−アミノ酸結合は、そのアミノ酸のカルボキシル基を介したアミド結合である。
(3)XおよびRは、ともに組み合わされ結合基を形成する。
(4)R3は、CH2CH=CH2であり、R4はHである。
(5)R3及びR4はそれぞれHである。
(6)R3及びR4はそれぞれCH2CH=CH2である。
(7)化合物は塩酸塩の形態を取る。
(8)R3はHであり、そしてR4はCH2CH=CH2である。
(9)IV−1及びIV−4は二つの成分の1.5〜1.0混合物である。
(10)R3= R4= CH2CH2CH2OHである。
(11)R3 =
、R4= Hである。
(12)R8= NHCONHC2H5である。
(13)R8= NH2である。
【0031】
この治療は、栄養に関する因子、好ましくはニューロトロフィンファミリー、最も好ましくは神経生長因子(NGF)とともに投与することができる。
【0032】
好ましい観点では本発明は、治療に有効な量の、下記の式(II)または(III)によって表されるK−252aの機能性誘導体を、例えばヒトのような哺乳動物に投与することによって、背根神経節の神経細胞の機能を高めるための方法を特徴とする。
【0033】
ここでは次の置換が行われる。
表2
【0034】
(1)R2は水素である。ただし、R2= Brである化合物II−20及びII−32は除く。
(2)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
(3)X及びRは、ともに組み合わされ結合基を形成する。
【0035】
この治療は、神経栄養性因子、好ましくはニューロトロフィンファミリーのうちのひとつ、最も好ましくは神経生長因子(NGF)とともに投与することができる。
【0036】
好ましい観点では本発明は、治療に有効な量の、下記の式(II)によって表されるK−252aを、例えばヒトのような哺乳動物に投与することによって、その哺乳動物のコリン作動性ニューロンの機能を高めるための方法を特徴とする。
ここでR1及びR2はそれぞれHであり、XはCO2CH3であり、RはOHであり、そしてZ1及びZ2はそれぞれHである。この治療では、栄養に関する因子、好ましくはニューロトロフィンファミリーのうちのひとつ、最も好ましくは神経生長因子(NGF)とともに投与することができる。
【0037】
好ましい観点では本発明は、治療に有効な量の、下記の式(II)、(III)、または(IV)によって表されるK−252aまたはK−252aの機能性誘導体を例えばヒトのような哺乳動物に投与することによって、線条体神経細胞の生存性や機能を高めるための方法を特徴とする。
【0038】
ここで次の置換がなされる。
表3
【0039】
(1)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
(2)R3はCH2−CH=CH2であり、R4はHである。
【0040】
好ましい観点では本発明は、治療に有効な量の、下記の式(II)によって表されるK−252aまたはK−252aの機能性誘導体を、例えばヒトのような哺乳動物に投与することによって、基底前脳神経細胞の生存性および/または機能を高めるための方法を特徴とする。
【0041】
ここで次の置換がなされる。
表4
【0042】
(1)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
【0043】
この治療では、栄養に関する因子、好ましくはニューロトロフィンファミリーのうちのひとつ、最も好ましくは神経生長因子とともに投与することができる。
【0044】
本発明の他の特徴及び利点は、以下の好ましい態様の説明、及び請求の範囲から明らかとなろう。
【0045】
好ましい態様の説明
スタウロスポリン誘導体
本発明は、新規なスタウロスポリンのビス−N−置換された誘導体、及び神経疾患のための治療薬としてのそれらの使用に関する。ここで神経疾患とは特に、損傷を受けたり、機能が不全であったり、軸索の変性が起こったり、もしくは死に至る危険性が増加したりしたニューロン細胞、または障害を受けたコリン作動性の活性のいずれかにより特徴づけられるような疾患である。これらの疾患には興奮性のアミノ酸によって誘導される疾患が含まれる。これらの新規な誘導体の治療のための利用には、この誘導体を単独で用いたり、また神経栄養性因子(好ましくはニューロトロフィンファミリーのひとつ、最も好ましくはNGF)の外因性の投与と組み合わせて用いたりできる。本発明の請求範囲に含まれる化合物は、下式によって表すことができる。
【0046】
[Stau]−N(CH3)−W−N(CH3)−[Stau] (I)
ここで[Stau]は下式の残基を表し、
そしてWはビス(カルバミル)またはビス(チオカルバミル)ラジカルを表す。
【0047】
−C(=Y)−NH−W’−NH−C(=Y)−
ここでW’は2〜20個の炭素原子からなるハイドロカルビレンラジカルであって、YはOまたはSである。W’は好ましくは、2〜10個の炭素からなり、置換されていないか、もしくは1〜3個の炭素からなる1〜3個のアルキル基で置換されたアルキレンラジカル、6〜12個の炭素からなり、置換されていないか、または1〜3個の、1〜3個の炭素からなるアルキル基、塩素、もしくは臭素で置換されたアリレンラジカルである。W’がヘキサメチレン及び1,4−フェニレンであると特に好ましい。Yは好ましくはOである。
【0048】
化学式(I)の化合物はカルバメート及びチオカルバメートの調製のための当技術分野において既知の手法を用いて合成することができる。好ましくはこの化合物は、ビス−ジイソシアネートまたはビス−ジイソチオシアネートをスタウロスポリンと反応させ、化学式(I)示される化合物(ここではそれぞれY= OまたはY= S)を得ることにより調製した。
【0049】
使用に適した中間体、ビス−ジイソシアネート及びビス−ジイソチオシアネートには、
1,6−ジイソシアナトヘキサン
トルエン−2,6−ジイソシアネート
ベンゼン−1,2−ジイソシアネート
2−メチル−1,5−ジイソシアナトペンタン
ナフタレン−2,6−ジイソシアネート
1,6−ジイソチオシアナトヘキサン
1,4−ジイソチオシアナトブタン
トルエン−2,4−ジイソシアネート
ベンゼン−1,4−ジイソシアネート
1,2−ジイソシアナトエタン
ナフタレン−1,5−ジイソシアネート
1,5−ジイソシアナトペンタン
ベンゼン−1,4−ジイソチオシアネート
2−メチル−1,5−ジイソチオシアナトペンタン
が含まれる。
【0050】
イソシアネート及びイソチオシアネートの調製のレビューとして、「シアネートおよびそのチオ誘導体の化学(The Chemistry of Cyanates and Their Thio Derivatives)、第2部、(Patai編) 、ウィリー(Wiley)、ニューヨーク、1977年におけるリッチャー(Richter)及びウルリッヒ(Ulrich)、619〜818頁」を参照のこと。この化合物は、好ましくは、ホスゲン(Y= O)またはチオホスゲン(Y= S)を対応するジアミンと反応させることによって、調製される。他の調製方法も用いることができる。例えば、1,2−ジイソシアナトエタンは、エチレン尿素をホスゲンと反応させ、続いて加熱することによって調製することができる。
【0051】
K−252aの誘導体
本発明はまた、損傷されたり、機能が不全であったり、軸索の変性が起きていたり、または死に至る危険のあるニューロンによって特徴づられるようなある種の神経疾患または障害における治療薬として、K−252aの特定の機能性誘導体を使用することに関する。この機能性誘導体は、単独で投与してもよいし、または神経栄養性因子(好ましくはニューロトロフィンファミリー、最も好ましくは神経生長因子、NGF)と組み合わせて投与してもよい。
【0052】
K−252aの「機能性誘導体」とは、望ましい生物活性、ここでは神経保護活性として定義されている活性であって、例えば、神経細胞の生存性を促進する能力、または神経繊維(例えば軸索)の生長を促進する能力、またはコリン作動性神経細胞の機能を高める能力、または例えば背根神経節の神経細胞のような感覚細胞の機能を高める能力、または線条体ニューロンの機能および/または生存性を高める能力、または基底前脳ニューロンの機能および/または生存性を高める能力を有する分子の修飾形と定義される。このような分子の修飾により、(例えば血液脳関門および細胞膜を介した)分子の溶解、吸収、輸送、生物学的半減期などを改善することができる。また他の場合では、または更に、ある部分はこの分子の毒性を減少させることができるし、またこの分子の望ましくない副作用をなくしたり減弱したりすることができる。
【0053】
本発明の範囲に含まれる化合物は、下記に示した化学式(II)[ここでは以下、化合物(II)という]、化学式(III)[ここでは以下、化合物(III)という]、化学式(IV)[ここでは以下、化合物(IV)という]、化学式(V)[ここでは以下、化合物(V)という]、化学式(VI)[ここでは以下、化合物(VI)という]で表わすことができ、
そして下記の表5で示した置換が行われる。本発明のK−252aの機能性誘導体は、当業者に知られた方法を用いる化学合成によって、デノボ(de novo)で合成することができる。例えば化合物IIの調製に用いられる手法は、ムラカタ(Murakata)ら(米国特許第4,923,986号)に記載されており、この文献は参照としてこの明細書に組み入れられる。化合物IIIを調製するために用いられる手法は、ムーディー(Moody)ら、J. Org. Chem. 57: 2105-2114 (1992)」、「ステグリッチ(Steglich)ら、Angew. Chem. Int. Ed. Engl. 19: 459-460 (1980)」、「ナカニシ(Nakanishi)ら、J. Antibiotics 39: 1066-1071 (1986)」、及び日本国特許出願第60-295172号(1985)に開示されている。他の方法は、日本国特許出願第60-295173号(1985)において化合物II−1、9、12及び15について、日本国特許出願第62-327858号(1987)において化合物II−2、3、4、24、25及び26について、日本国特許出願第62-327859号(1987)において化合物II−20について、及び日本国特許出願第60-257652号(1985)において化合物II−10について、明治製菓株式会社(Meiji Seika Kaisha Ltd.)によって開示されている。
【0054】
表5: K−252a(12)の機能性誘導体
【0055】
(1)Z1及びZ2は両方とも水素であるか、または示した部位でともに組み合わされ酸素を意味する。
(2)NH−アミノ酸結合は、そのアミノ酸のカルボキシル基を介したアミド結合である。
(3)XおよびRは、ともに組み合わされ結合基を形成する。
(4)R3は、CH2CH=CH2であり、R4はHである。
(5)R3及びR4はそれぞれHである。
(6)R3及びR4はそれぞれCH2CH=CH2である。
(7)化合物は塩酸塩の形態を取る。
(8)R3はHであり、そしてR4はCH2CH=CH2である。
(9)IV−1及びIV−4は二つの成分の1.5〜1.0混合物である。
(10)R3= R4= CH2CH2CH2OHである。
(11)R3=
、R4= Hである。
(12)K−252a自体の場合、R1= R2= H、X= CO2CH3、R= OHであり、そしてZ1及びZ2はそれぞれ水素である。
(13)R8= NHCONHC2H5である。
(14)R8= NH2である。
【0056】
本発明はまた、治療に有効な量の、上記に示された化学式(II)及び表5(記12)に示された置換によって表されるK−252aを投与することによって、コリン作動性ニューロンの機能を高めるための方法に関する。この化合物は当技術分野において記載されている手法によって調製される(マツダ(Matsuda)ら、米国特許第4,554,402号、カセ(Kase)ら、J. Antibiotics 37: 1059-1065, 1986参照)。
【0057】
「コリン作動性ニューロンの機能を高めること」とは、コリン作動性の神経細胞の生存性および/または神経繊維(例えば軸索)の生長の促進および/または神経細胞のコリン作動性機能の増強を意味する。K−252aは、栄養に関する因子、好ましくはニューロトロフィンファミリーの一つ、最も好ましくは神経生長因子(NGF)と共に投与することができるし、または同因子と組み合わせずに投与することもできる。
【0058】
化合物の使用
下記に充分に説明したように本発明は、K−252aの機能性誘導体、又は化学式Iの化合物を単独で、またはNGFのような神経栄養性因子と組み合わせて神経疾患、特に、損傷を受けたり、機能が不全であったり、軸索の変性が起こったり、もしくは死に至る危険性が増加したりしたニューロン細胞、または障害を受けたコリン作動性の活性のいずれかにより特徴づけられるような神経疾患に対する治療薬として使用する新規な方法を提供する。これらの疾患には興奮性のアミノ酸によって誘導される疾患が含まれる。神経栄養性因子との組み合わせが含まれる本発明の化合物の生物活性は、培養されたPC−12細胞におけるオルニチンデカルボキシラーゼのアッセイ、培養された脊髄または基底前脳のコリンアセチルトランスフェラーゼのアッセイ、培養された背根神経節ニューロンの生存性のアッセイ、培養された線条体ニューロンの生存性のアッセイ、培養された基底前脳ニューロンの生存性のアッセイ、成長過程でプログラムされた運動ニューロンの死についてのインオボ(in ovo)でのモデル、インビボでの大人の舌下の軸索モデル、または例えば基底核の刺激毒性障害のようなインビボでの刺激毒性の神経保護アッセイによって簡便に分析することができる。これらのアッセイについてはすべて下記に詳細に記載されている。このように本発明の化合物は、ニューロン細胞死や機能不全の危険が増加することによって特徴づけられる神経の疾患または障害を患っているヒトまたは他の哺乳動物に投与するために用いられる。これらの神経の疾患及び障害には、アルツハイマー病、たとえば筋萎縮性側索硬化症のような運動ニューロン疾患、パーキンソン病、発作や他の虚血性の障害、ハンチントン病、AIDS脳症、てんかん、脳や脊髄に起こる震盪性または穿通性の損傷、及び末梢の神経障害が含まれるが、これらに限られるわけではない。
【0059】
本明細書において提供された化合物は、薬学的に受容できる非毒性の賦形剤及び担体と混合することによって薬学的な組成物へと処方することができる。上記に記載したように、このような組成物は非経口投与、特定すると液状の溶液または懸濁剤の形態で用いるように調製することもできるし、経口投与、特定すると錠剤またはカプセルの形態で用いるように調製することもできるし、または経鼻的に、特定すると粉剤、点鼻剤、もしくは噴霧剤の形態で用いるように合成することもできる。
【0060】
この組成物は、ユニットの用量形態で便利に投与してもよいし、また、例えば「レミントンの薬剤学(Remington's Pharmaceutical Sciences)」(マック出版社(Mack Pub. Co)、イーストン、ペンシルバニア州、1980)に記載されたような薬学分野で周知の方法のいずれかによって調製してもよい。非経口投与のための配合剤は、無菌の水や食塩水、例えばポリエチレングリコールのようなポリアルキレングリコール、植物起源のオイル、水素化したナフタレンなどを通常の賦形剤として含むことができる。特定すると、生物学的適合性で生分解性のラクチド重合体、ラクチド/グリコライド共重合体、またはポリキシレン−ポリオキシプロピレン共重合体が、活性な化合物の放出を調節するための有用な賦形剤であろう。これらの活性な化合物に用いられる他の可能性のある有用な非経口輸送系としては、エチレン−ビニルアセテート共重合体の粒子、浸透性のポンプ、植え込み可能な注入系、及びリポソームがある。吸入投与のための配合剤は、賦形剤として例えばラクトースを含んでいるか、または例えばポリオキシレン−9−ラウリルエーテル、グリココレート及びデオキシコレートを含む水性溶液であってもよく、または点鼻剤の形態で投与するための油性溶液、または鼻腔内に投与できるゲルであってもよい。非経口投与のための配合剤はまた、口腔内投与のためのグリココレート、直腸投与のためのサリチレート、または膣内投与のためのクエン酸を含むことができる。経皮用の貼着剤の製剤は好ましくは、親油性の乳濁剤である。
【0061】
本発明の材料は、薬剤中の唯一の活性物質であっても、他の活性成分、神経の疾患または障害、例えば末梢神経障害におけるニューロンの生存性または軸索生長を促進する他の成長因子などと組み合わせて用いてもよい。
【0062】
本明細書に記載された化合物の治療用組成物中の濃度は、投与すべき薬物の一回用量、用いた化合物の化学的特徴(例えば疎水性)、及び投与経路などの多くの因子に依存して変化する。一般的に、本発明の化合物は、非経口投与を行うために、約0.1〜10%w/vの化合物を含む水性の生理学的緩衝溶液で用いることができる。通常の用量範囲は、1日あたり体重1kgに対して約1μg〜約1gである。好ましい用量範囲は、1日あたり体重1kgに対して約0.01mg〜約100mgである。投与すべき好ましい薬物の用量は、神経疾患の型及び進行度、特定の患者の全身的な健康状態、選択された化合物の相対的な生物効果、賦経剤の化合物の配合、及び投与経路のような変化量により決定される可能性が高い。
【0063】
本発明は次の実施例によってさらに例示されるであろう。これらの実施例は本発明の範囲を限定するものと解釈されるべきではなく、本発明の範囲は添付した請求の範囲によってのみ決定されるべきである。
【0064】
実施例1
1,6−ヘキサメチレン−ビス−(カルバミルスタウロスポリン)(6-Hexamethylene-bis-(carbamylstaurosporine))(HBCS)
酢酸エチル(無水硫酸マグネシウム上で乾燥させたもの)1.00 ml中にスタウロスポリン(カリフォルニア州サウザンドオークス、カミヤ・バイオメディカル社)1.0 mg(2.15 μmol)を溶かした溶液を、無水酢酸エチル1.00 ml中に10.75 mgのヘキサメチレン−ビス−イソシアネートを溶かした溶液17マイクロリットル(1.08 μmol)で処理した。この反応混合液をアンバーガラス反応用バイアルに入れて、室温に2日間置いた。600 μgの結晶化した沈殿物が分離した。この組成物は高速原子衝撃質量分析計(FAB-MS)によってHBCSであることが明らかになった。
M+H+ 計算値=1102 M+Na+ 計算値=1124
実測値=1102 実測値=1124
本生成物および以下で説明されるスタウロスポリン誘導体はすべて、遮光ガラスバイアル中で保存した。
【0065】
実施例2
p−フェニレン−ビス−(カルバミルスタウロスポリン)(p-Phenylene-bis-(carbamylstraurosporine))(PBCS)
無水酢酸エチル1.00 ml中にスタウロスポリン1.0 mg(2.15 μmol)を溶かした溶液を、無水酢酸エチル1.00 ml中に3.83 mgのp−フェニレンジイソシアネート(p-phenylene diisocyanate)(トランスワールドケミカル社 P1586-1)を溶かした溶液45マイクロリットル(1.08 μmol)で処理した。反応混合液を一晩放置した。白い沈殿物が沈積した。そして、石油エーテルを0.5 ml加えた。混合液を、真空乾燥した焼結ガラスロートに濾過して入れた。全部で0.9 mgの結晶産物を採取して、高速原子衝撃質量分析計によってp−フェニレン−ビス−(カルバミルスタウロスポリン)であることを同定した。
M+H+ 計算値=1093
実測値=1093
【0066】
調製物A
N−フェニルカルバミルスタウロスポリン(N-Phenylcarbamylstraurosporine)(PCS)
参照文献:米国特許第5,093,330号
無水酢酸エチル1.50 ml中に2.0 mg(4.30 μmol)のスタウロスポリンを溶かした溶液を、無水酢酸エチル0.990 ml中に10 μlのフェニルイソシアネートを溶かした溶液468μl(4.30μmol)で処理した。反応混合液を一晩放置して、3 mlのヘキサンを少しずつ加えた。2.39 mgの無色の結晶が得られた。この産物を再結晶させた後、酢酸エチル1 mlと石油エーテル2 mlを加え、1.75 mgの結晶産物を分離した。同様の調製物からFAB-MSによってこの産物の組成がN−フェニルカルバミルスタウロスポリンであることが明らかになった。
M+H+ 計算値=586
実測値=586
【0067】
調製物B
N−フェニルチオカルバミルスタウロスポリン(N-Phenylthiocarbamylstraurosporine)(PTCS)
酢酸エチル1.00 ml中に1.0 mg(2.15 μmol)のスタウロスポリンを溶かした溶液を、酢酸エチル1.00 ml中に10 μlのフェニルイソチオシアネートを溶かした貯蔵溶液26μlで処理した。これの等量液には、290μg(2.15 μmol)のフェニルイソチオシアネートが含まれている。反応混合液を25℃に一晩置き、2 mlのヘキサンを加えた。この結果できた結晶産物を濾過して、ヘキサンで洗滌し、アルゴンガスの気流で乾燥させた。
FAB-MS 計算値: M+H+ =602
実測値 =602
【0068】
調製物C
N−エチルカルバミルスタウロスポリン(N-Ethylcarbamylstraurosporine)(ECS)
酢酸エチル900マイクロリットル中に0.9 mg(1.93 μmol)のスタウロスポリンを溶かした溶液を、1.93マイクロモルのエチルイソシアネート(無水酢酸エチル2.00 ml中に9.05 mgのエチルイソシアネートを溶かした貯蔵溶液30.2マイクロリットル)で処理した。反応混合液を25℃で一晩放置して、2.0 mlのヘキサンを加えた。結晶産物を分離し、乾燥した。
FAB-MS 計算値: M+H+=538 M+Na+=560
実測値 =538 =560
【0069】
実施例3
化合物II-4
化合物A(962 mg, 2 mmol)を、テトラヒドロフラン30 mlとメタノール10 mlの混合液に溶かし、これに760 mgの水素化ホウ素ナトリウム(20 mmol)を氷冷下で加え、その後、同じ温度で4時間、さらに室温で12時間撹拌した。これに3Nの塩酸を加えた後、溶液を食塩水溶液で洗滌し、硫酸マグネシウムで脱水して、その後、溶媒を蒸発させた。シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)で残留物を精製し、882 mg(収率97%)の化合物II-4を得た。
融点:130〜140℃
【0070】
実施例4
化合物II-14
化合物B(393 mg, 0.9 mmol)を、テトラヒドロフラン25 mlに溶解し、カルボベンゾキシ−L−セリン309 mg(1.35 mmol)、N−ヒドロキシスクシンイミド156 mg(1.35mmol)、4−メチルモルホリン0.1 ml(0.9 mmol)およびジシクロヘキシルカルボジイミド279 mg(1.35 mmol)を含むテトラヒドロフラン3 mlを氷冷下で加え、12時間撹拌した。この反応混合液を濾過し、溶媒を蒸発させた。シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)で残留物を精製し、429 mg(収率72%)の化合物Cを得た。
融点:188〜193℃
SIMS(m/z):660(M+1)+
化合物C(399 mg)をジメチルホルムアミド10 mlに溶解し、次に、炭素上10%パラジウム 300 mgを加え、水素気流中、50℃で7時間撹拌した。この反応混合液をセライトを通して濾過し、溶媒を蒸発させた。シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール/28%水酸化アンモニウム=90/10/1)で残留物を精製して得た産物を5 mlのテトラヒドロフランで溶解し、1.7 Nの塩化水素/酢酸エチル5 mlとジエチルエーテル10 mlを加えた。濾過によって沈殿物を溶液から分離し、234 mg(収率69%)の化合物II-14を得た。
融点:>300℃
【0071】
実施例5
PC-12細胞は、ラットの副腎随質の腫瘍に由来するクローン集団で、NGFの作用を研究するための、非常に有用で、広く研究されているモデルであることが分かっている(グロフ(Guroff)、神経科学における細胞培養(Cell Culture in the Neurosciences)、プレナム出版株式会社(Plenum Publishing Corporation)、245〜272頁、1985年)。これらの細胞に対するNGFの特に確実な効果の一つは、オルニチン・デカルボキシラーゼ(ODC)の活性を速やかに刺激することで、この効果は、200 nMのK-252aによって阻害されることが報告されている(Koizumiら、1988)。本実施例の実験においては、PC-12細胞(メリーランド州ベセスダの国立衛生研究所のG.グロフ博士からいただいた)を6×104細胞数/cm2の密度で48穴プレートで培養し、薬剤媒体(0.5% DMSO)、K-252a、スタウロスポリン、またはHBCSとともにインキュベートした。K-252aとスタウロスポリンは、カミヤバイオメディカル社から入手することが可能である。薬剤を添加してから4時間後に、ハフ(Huff)らによって説明されているところに従って(J. Cell Biol. 88: 189-198, 1981)、ODCアッセイのために細胞を回収した。
【0072】
これら3つの化合物はすべて、ODC活性の誘導(すなわち上昇)を示したが、効能と効力には、かなりの差違があった(図1)。K-252aは、その効果が2 nMで検出可能になり、200 nMで最大(36.3倍の誘導)になるまで増加するという、ODC活性の用量依存的な誘導を示した。同様に、スタウロスポリンの効果も、2 nMで検出可能になるが、20 nMでピーク(34.7倍の誘導)に達し、200 nMでは、かなり減少する。HBCS(実施例1)も、同様に2 nMで誘導を示したが、濃度を上昇させても効果の上昇をもたらすことはできなかった。このため、他の2つの化合物に較べて、その最大効果はずっと低かった(6.5倍の誘導)。別の実験において、PC-12細胞のODC活性に対するPTCS、PCS、およびECS(実施例2)の効果と、K-252aの効果とを比較した。200 nMの濃度でK-252aの活性を100%と表すと、PTCSはK-252aの活性の71.4%を示し、また、PCSとECSはそれぞれ、K-252aの活性の88.9%と61.9%を示した。しかし、プロテインキナーゼCインヒビターH-7は、プロテインキナーゼCの活性を阻害することが分かっている濃度である30 μMでは、ODC活性を誘導しなかった(Nakadateら、Biochem. Pharmacol. 37: 1541-1545, 1988)。
【0073】
K-252a、スタウロスポリンおよびHBCSが、NGFの生物活性を増強および/または阻害する能力を、細胞培養液 1 ml当たりNGFを10 ngを加えて、上述の濃度の上記化合物の非存在下または存在下で、上述したように細胞のODCアッセイを行なって測定した(図2)。NGFのこの濃度は、化合物の上昇効果または阻害効果を検出できるよう、中度の誘導を示す濃度を選択した。200nMのK-252aは、コイズミ(Koizumi)ら(1988)が報告しているように、ODCのNGF誘導を阻害したが、驚くべきことに、より低い濃度(2 nMと20 nM)では誘導が増強された。スタウロスポリンも、2 nMでは、NGFによる誘導を増強したが、濃度が高くなる(20 nMと200 nM)と、この効果は失われた。これに対し、HBCSは、調べたすべての濃度で、NGFの効果を増強した。この際だった効果を、HBCSのみのときの緩やかなODC誘導効果と比較して図3に示す。
【0074】
実施例6
コリンアセチルトランスフェラーゼ(ChAT)活性に対するK-252aの効果を、標準的な方法によってラット胎児から調製した解離脊髄培養物(後述参照)の中で測定した。ChATは、神経伝達物質のアセチルコリンの合成を触媒する酵素であり、コリン作用性ニューロンの特異的な生化学的マーカーである。脊髄において、コリン作用性ニューロンの大多数は運動ニューロンである。したがって、この酵素の測定は、コリン作用性ニューロンの生存および/またはこの酵素の制御に対する、一つまたは複数の因子の効果を示すために用いることができる。
【0075】
細胞を基質に接着させるために、培養開始後2〜3時間インキュベートしてから、K-252aを示された濃度で培養液に加えた。48時間後に培養液中でChAT活性を測定した。脊髄培養液中のK-252aは、ChAT活性を用量依存的に上昇させ、200〜300 nMで最大効果(2〜3倍の増加)に達した(図4)。これより濃度が高くなると、ChAT活性は低下した(図4)。インキュベートする時間を長くすると、7日目までは、ChAT活性の基底レベルが下がるために、ChAT活性は4〜5倍上昇した(図5)。この培養系において、基本的な(対照となる)条件の下では、変性したり死滅する運動ニューロンの数は増加した(McManamanら、Developmental Biol. 125: 311-320, 1988)。図4および図5に示されている結果は、培養を開始した日に1回だけK-252aを添加した結果であり、脊髄のコリン作用性ニューロンの生存および/または酵素自体の制御に対する効果が長く持続することを示している。
【0076】
ラット胎児の脊髄細胞の解離培養液を用いた実験は、ほとんど、既述されているところ(Smithら、J. Cell Biol. 101: 1608-1621, 1985)に従って行なわれた。解離細胞はトリプシンによる組織解離法を用い、当業者に既知の標準的技術によって、14日目のラット胎児から調製された(Smithら、前記)。細胞は、6 X 105細胞/cm2の濃度で、ポリ−L−オルニチンでコートしたプラスチック組織培養皿の中、無血清N2培地に撒き(培養開始し)、5%CO2/95%空気の湿潤な環境下37℃で(Bottensteinら、Proc. Natl. Acad. Sci. USA 76: 514-517, 1979)、48時間インキュベートした。イシダ(Ishida)およびデグチ(Deguchi)(J. Neurosci. 3: 1818-1823,1983)およびマクマナマンら(McManamanら、前記)(1988)に従ってフォナム(Fonnum)の方法を修正した方法を用いてChAT活性を測定した。ビシンコニン酸/Cu++反応(BCA蛋白質測定試薬、イリノイ州ロックランド、ピアス社)によって測定した全蛋白質量に対して、活性を標準化した。
【0077】
実施例7
脊髄ChATアッセイ法で、K-252aの機能性誘導体を100種以上調べて、それらの相対的な有効性を判定した。図8のデータは、300および30 nMで調べたところ、最初の機能性誘導体のうち28種が300 nMでChAT活性を有意に上昇させたことを示している。機能性誘導体の一つである化合物II-21は30 nMでも活性があった(基底レベルに対してChAT活性は30%上昇した)。K-252aや他の相同化合物では、30 nMでのChAT活性が活発には上昇しなかったことから、この化合物はこれらの化合物よりも強力であった。
【0078】
図13aは、ラットの脊髄培養物中でChAT活性を有意に上昇させることが分かった28種の最初のK-252a誘導体と、さらに30種の誘導体(化合物II-29からII-34、II-36からII-56、およびIV-1からIV-3)の能力を示している。図13bは、K-252aの誘導体であるII-66-80、IV-5、IV-6、VI-1、およびVI-2が、ラットの脊髄培養物中でChAT活性を有意に上昇させることができることを示している。図13Cは、さらに12種のK-252a誘導体がラットの脊髄培養物中でChAT活性を有意に上昇させることができることを示している。
【0079】
実施例8
K-252aおよび50種以上の機能性誘導体について、背根神経節のニューロン細胞の生存を促進する能力を評価した。細胞が生きていることは、生細胞染料であるフルオロセイン二酢酸(fluorescein diacetate)の類似合成物であるカルセイン(calcein)AMの取り込みによって測定した。カルセインは生細胞によって取り込まれ、細胞内で切断されて、生細胞の無傷の細胞膜によって保持される蛍光性の塩になる。生きたニューロンを顕微鏡下で数えた細胞数は、蛍光定量的生細胞測定法を用いて得られる相対的蛍光値と直接に正比例する。したがって、この方法は、一定の培養液中の全細胞集団における細胞の生存率に関する信頼性のおける、定量的な測定結果を提供する(Bozyczko-Coyneら、J. Neur. Meth. 50: 205-216, 1993)。
【0080】
胚齢8日目のニワトリの胚から背根神経節を切除した後、ディスパーゼ(Dispase)(中性プロテアーゼ、コラボレイティブリサーチ社)解離によって解離細胞を調製した。ニューロンを低密度(1.8 X 104細胞/cm2)で、ポリ−L−オルニチンとラミニンでコートした96穴プラスチック培養皿に接種した。細胞は、無血清N2培地(BottensteinおよびSato, 1979)、5%CO2/95%空気の湿潤な環境下37℃で48時間培養した。細胞の生存は、48時間後に上記の蛍光生細胞測定法を用いて評価した。
【0081】
背根神経節ニューロンの生存は、K-252aによって濃度依存的に強化された。約100 nMで最大活性が観察された(図6)。調査した50の類似化合物のうち24種が、DRGニューロンの生存を強化する活性があり、そのうち22種が図7に示されている。これらの類似化合物はすべて、脊髄のChAT活性を上昇させる活性もあった(実施例5参照、図8)。最初の22種と、さらに2種の活性類似化合物(II-30、II-32)が図14に示されている。24種の活性機能性誘導体によって刺激された背根神経節ニューロンを顕微鏡下で調べると、神経線維の成長が増強されていることも示された。
【0082】
実施例9
齧歯動物の脳の空洞に興奮性のアミノ酸カイニン酸(カイニン酸塩)を直接注入すると、海馬の錐体細胞のニューロン変性がもたらされる。このニューロン死は、細胞骨格蛋白質のスペクトリンの蛋白質分解が著しく促進されることで特徴づけられる。スペクトリンの分解産物は、カイニン酸投与後24時間以内に、海馬の磨砕物中で測定することができる。スペクトリンの蛋白質分解度は、海馬の錐体細胞のニューロン死の程度と高度に相関する(Simanら、J. Neurosci. 9: 1579-1590, 1989)ため、スペクトリンの蛋白質分解は、興奮性のアミノ酸によって誘導されるニューロン変性の優れた生化学マーカーになる。内因性興奮性アミノ酸の過剰放出が、発作および他の虚血性傷害、アルツハイマー病、筋萎縮性側索硬化症を含む運動ニューロン病、パーキンソン病、ハンチントン病、AIDS脳症、てんかん、脳または脊髄の振とう性ないし穿通性傷害を含む数多くの神経疾患および障害の病因であると示唆されてきた。
図9〜12に示されている結果は、以下の方法にしたがって作出されたものである。
【0083】
カイニン酸注入法: カイニン酸によって誘導されるニューロン損傷に対する、K-252aあるいはその誘導体の効果を評価した。スプラーグ−ダウレー系統ラットのメスおよびオスの成獣(175〜250 g)を、ネンブタール(Nembutal)(50 mg/kg、腹腔内)で麻酔した。カイニン酸処理(5μl)の前後にicv注入して、各ラットに試験用化合物を(全量で5μl)投与した。これは、上記の各ケースについて示された投与および注入スケジュールを用いて行われた。対照用動物には、カイニン酸および薬剤の注入の代わりに溶媒を投与した。解剖実験のために、icv注入物を、薬剤注入の約一週間前に埋め込まれたカニューレ(バージニア州ロアノーク、プラスティックワン社)を通して輸送した。このカニューレは、ブレグマの前後部で、ブレグマの1.5 mm側方、頭頂部から4.4 mm下方表面側に定位的に配置された。この処理法による結果は、後述するところの解剖学的解析を用いて、2週間後に評価した。
【0084】
カイニン酸に誘導されるスペクトリンの蛋白質分解に対するK-252a、あるいはその誘導体の効果を測るための実験では、上述の定位的配置に置いた10μlハミルトン注射器によって、5μlの薬剤または溶媒のicv注入物を、カイニン酸と同時に、麻酔したラットに投与した。これらのラットは、24時間後に屠殺して、以下に述べるような生化学的分析を行なった。
【0085】
解剖学的および生化学的分析:解剖学的分析は、以下に述べるようにして行なった。処理後2週間目にラットを断頭して屠殺し、速やかに脳を取り出してドライアイス上で凍結した。各脳から頭頂部のスライドに載せた連続切片をチオニンで染色し、顕微鏡で調べた。海馬の損傷は、脳の左右両側について、錐体細胞の損失を被った、解剖学的に定義された4つの海馬領域(CA1-4、本明細書に参照として包含される、[シェパード(Shepard)著、1979年、「脳のシナプシス構成」、オクスフォード、310頁]によって説明されているロレント・デ・ノウ(Lorente de No)の分類に従う)の総数を合計して定量した。
【0086】
生化学的解析は、以下のようにして行われた。脳スペクトリン(ホドリン)の、カルパインI感受性の蛋白質分解を、サイマン(Siman)ら(1988年、 Neuron, 1: 279-287、本明細書に参照として包含される)によって説明されているイムノブロット解析を用いて、海馬のホモジネートで測定した。要約すると、処理後24時間でラットを断頭して屠殺し、脳から速やかに背部側海馬を切除して、0.1 mMフェニルメチルスルホニルフルオリドを含む20 mMトリス塩酸(pH 7.4)の中で磨砕した。各ホモジネートの分注物から採った蛋白質をSDS-PAGEで分離し、各サンプルにおける、カイニン酸に誘導されるスペクトリン分解の量を定量するためにイムノブロット解析を用いた。
【0087】
図9は、海馬において、カイニン酸に誘導されるニューロン分解に対する、K-252aの効果を示している。カニューレを挿入したオスとメスのスプラーグ−ダウレイ(Sprague-Dawley)ラットに、カイニン酸(0.6μg)を脳の大脳外側の脳室(icv)に直接注入する30分前、または、約3時間後から24時間後に、0.4μgのK-252aまたは溶媒を投与した。2週間後に、後に説明するようにして、脳を切り出して凍結し、切片にしてから組織学的な解析をするために染色した。示されているデータは、各群についての、損傷を受けた海馬の小領域の平均数±平均値の標準誤差(S.E.M.)である。K-252aは、海馬内の損傷領域の数を、3.86±0.78(K-252aを投与しないとき)から1.18±0.4(K-252aを投与したとき)へと有意に減少させた。
【0088】
図10は、カイニン酸に誘導される、海馬におけるスペクトリンの分解に対するK-252aの効果を示したものである。メスのスプラーグ−ダウレイラットに、icv注入によって、神経毒効量(0.6μg)のカイニン酸とともに、0.4μgのK-252aまたは溶媒を投与した。擬似対照用の動物には、溶媒を注入し、カイニン酸もK-252aも投与しなかった。24時間後に、後述するようにして、背部海馬のホモジネートを解析して、スペクトリン分解産物を調べた。スペクトリンの蛋白質分解の程度を、各群のスペクトリン分解産物の増加量を擬似対照の値を上回るパーセントとして表している。示されているデータは、各群についての、スペクトリン分解産物の増加の平均パーセント(擬似対照=100%)±S.E.M.である。K-252aのicv注入は、スペクトリンの蛋白質分解の程度を、擬似対照値の140±15%(K-252aを投与しないとき)から約102±10%(K-252aを投与したとき)へと有意に減少させた。
【0089】
図11は、海馬においてカイニン酸に誘導されるニューロン分解に対する、HBCSの効果を示している。カニューレを挿入したメスのスプラーグ−ダウレイラットに、カイニン酸(0.6μg)をicv注入する40分前、または、約4時間後に、0.8μgのHBCSまたは溶媒を投与した。2週間後に、後に説明するようにして脳を切り出して凍結し、切片化してから、組織学的な解析をするために染色した。示されているデータは、各群についての、損傷を受けた海馬の小領域の平均数±S.E.M.である。HBCSは、海馬内の損傷領域の数を、約2.5±0.6(HBCS処理なし)から1.3±0.5(HBCS処理したもの)へと有意に減少させた。
【0090】
図12は、海馬におけるカイニン酸に誘導されるスペクトリン分解に対する、K-252aの機能的誘導体3種の効果を比較したものである。メスのスプラーグ−ダウレイラットに、icv注入によって、神経毒効量(0.6μg)のカイニン酸とともに、0.4μgのK-252aまたは化合物III-1かII-21、または溶媒を投与した。擬似対照用の動物には溶媒を注入し、カイニン酸もK-252aの誘導体も投与しなかった。24時間後に、後述するようにして、背部海馬のホモジネートを解析して、スペクトリン分解産物を調べた。スペクトリンの蛋白質分解の程度を、各群のスペクトリン分解産物の増加量を擬似対照の値を上回るパーセントとして表している。示されているデータは、各群についての、スペクトリン分解産物の増加の平均パーセント(擬似対照=100%)±S.E.M.である。K-252aのicv注入は、スペクトリンの蛋白質分解の程度を、擬似対照値の約128±9%(溶媒処理)から約104±4%(K-252a存在下)へと減少させた。K-252aの誘導体であるIII-1とII-21は、カイニン酸に誘導されるスペクトリン分解を阻害できなかった。
【0091】
実施例10
線条体培養液における生存を促進するK-252aの能力を測定した。線条体は、胚令17日目のラット胚から切り出し、ディスパーゼ(Dispase)(中性プロテアーゼ、コラボレイティブリサーチ社)によって細胞を分離した。ニューロンを5X104細胞/ウェル(1.5 X 105/cm2)の密度で、ポリ-l-オルニチンとラミニンで予めコートした96穴プレートに接種した。細胞は、0.05%のウシ血清アルブミンを含む無血清N2培地(BottensteinおよびSato、1979)中で、5%CO2/95%空気の湿潤な環境下において37℃で培養した。実施例8に述べたカルセイン生存細胞蛍光測定法を用いて、接種後5日目に細胞の生残数を測定した。
【0092】
線条体ニューロンの生存は、K-252aによって濃度依存的な方法で強化された。75 nMのK-252aで最大活性が観察されたが、これは対照に対して3−4倍の効果を生んだことになる(図15)。対照用の培養液では、0日目にニューロンの培養を開始したとして、5日以内に90%のニューロンが死滅したが、K-252aで処理した培養では50%のニューロンが生存していた(図16)。線条体ニューロンにおける生存効果は、培養3日後に起こり、培養7日目以上まで維持された。これらの効果は、培養開始日に1回だけK-252aを施用してもたらされたもので、ニューロンの生存に対する効果が維持されることを示している。
【0093】
図17は、対照用培養液と75 nM K-252aで処理した培養液とから撮った顕微鏡写真の組である。75 nM K-252a存在下の培養液中では細胞の生存数が増加し、神経突起の成長が見られた。
【0094】
実施例11
実施例10の線条体細胞生存測定法で、K-252aの31種の機能的誘導体の有効性と効力とを調べた。図18は、線条体ニューロンの生存を強化した18種のK-252a誘導体に関するデータを示している。
【0095】
実施例12
本発明に係る化合物が前脳基部培養物の生存を強化する能力とChAT活性の上昇について化合物の評価を行なった。これらの培養物におけるChAT活性は、海馬形成や嗅覚核、脚間核、皮質、扁桃、視床の主要なコリン作用性の供給物の代表であるコリン作用性ニューロン(培養物中の細胞の5%以下)の生化学的なマーカーである。本発明の代表的な化合物は、ChAT活性を上昇させたばかりではなく、さらに、前脳基部培養物におけるニューロンの一般的な生存数を増加させた。前脳基部は、胚令17日か18日目のラット胚から切り出し、ディスパーゼ(Dispase)(登録商標)(中性プロテアーゼ、コラボレイティブリサーチ社)によって細胞を分離した。ニューロンを5 X 104細胞/ウェル(1.5 X 105細胞/cm2)の密度で、ポリ-l-オルニチンとラミニンで予めコートした96穴プレートのウェルに接種した。細胞は、0.05%のウシ血清アルブミン(BSA)を含む無血清N2培地(Bottensteinら、前記)中で、5%CO2/95%空気の湿潤な環境下において37℃で培養した。マクマナマン(McManaman)ら(前記)、およびグリクスマン(Glicksman)ら(J. Neurochem. 61: 210-221, 1993)による前出のフォナム法(Fonnum procedure)を修正して用いて、6日目にインビトロでChAT活性を測定した。ボジツィコ−コイネ(Bozyczko-Coyne)ら(前記)によって述べられているカルセインAM蛍光定量測定法を用いて、培養開始後5日目に細胞の生存を評価した。測定する時に培地の一部を吸引して1ウェルにつき50μlにした。150μlのダルベッコの(Dulbecco's)リン酸緩衝食塩水(DPBS;Gibco BRL)中の8μM カルセインAM保存液を、96穴プレートに各ウエル当たりに200μlに、最終濃度が6μMになるように加えた。プレートを37℃で30分間インキュベートした後、200μlのDPBSで4段階希釈した。プレート読み取り用の蛍光測定計(サイトフルオル(Cytofluor)2350、ミリポア(Millipore)社)を、励起波長485 nm、蛍光波長538 nm、および感度設定#3で用いて、各ウエルの相対的蛍光量を測定した。細胞を入れてないウエル6個にカルセインAMを投与して算出した蛍光バックグランドの平均値を、すべての測定値から引いた。これらの実験で使う細胞密度の範囲について、30分間基質をインキュベートして、蛍光シグナルが直線的に増えることを確認した。K-252aと少なくとも12個のK-252a誘導体(II-3、II-5、II-10、II-20、II-21、II-22、II-30、II-32、II-51、II-62、II-63、II-64、II-65)が、前脳基部ニューロンの生存を強化した(図19)。
【0096】
実施例13
以下の試験は、ラットの基底核を傷害したときの、皮質性コリン作用機能に対する化合物II-51の効果を評価するために行なわれた。
【0097】
前脳基部から出たコリン作用性ニューロンで、海馬中隔経路を通って海馬に突き出したものか、基底-皮質経路を通って皮質に突き出したものは、アルツハイマー病の進行過程において深刻な退行変性を被る。これらのニューロンの消失と認識ないし記憶機能の減退との間には、この障害に悩む各人において、ある程度の相関関係がある(Fibiger, H. Trends in Neurosci. 14: 220-223, 1991)。コリン作用性機能不全のいくつかのモデルが、生化学マーカーを消失するだけでなく行動障害も示すようになると言われてきた。これらのモデルは、アルツハイマー病の進行に近似する(Olton, D.ら、「麻痺性痴呆:前脳基部のコリン作用系の変性によって起きる認識障害の動物モデル(Dementia: animal models of the cognitive impairments produced by degeneration of the basal forebrain cholinergic system)」、Meltzer, H.編、神経病理学:発展の第三世代(Psychopharmacology: The Third Generation of Progress)、レイブン出版(Raven Press)、ニューヨーク州、1987年、941〜953頁; Smith, S., Brain Res. Rev. 13: 103-118, 1988)。例えば、コリン作用変性モデルのひとつは、基底核の毒性刺激傷害である(Wenk, G.ら、Exp. Brain Res. 56: 335-340, 1984)。前脳基部内でのコリン作用性ニューロンに傷害が起きると、この領域でのニューロン細胞体の消失がもたらされ、続いて、前頭皮質および頭頂皮質においてコリン作用性末端マーカーの消失がもたらされる(Dunnett,S.ら、Trends Neurosci. 14: 494-501, 1991)。以下の方法を用いて、基底核に傷害を受けたラットにおいて、化合物II-51が皮質のコリン作用機能を増強することが示された。
【0098】
すべての実験について、オスのスプラーグ−ダウレイラット(225〜275グラム)を用いた。当業者に周知の方法(例えば、Wenkら、前記、参照)に、後述の修正を加えた方法を用いて、基底核大型細胞(magnocellularis)に片側性のイボテン(ibotenic)傷害を起こさせた。ラットを50 mg/kgのペントバルビタールで麻酔して、基底核大型細胞(magnocellularis)に5μgのイボテン酸(ibotenic acid)(1μlのPBS中)を片側的に注射した。座標は、パキノス(Paxinos)およびワトソン(Watson)の脳地図から用いた(1.5 mm背部側、2.8 mm側部、7.2 mm背腹方向)。注射は、6分間にわたって行われた。染色剤を注射したところ、基底核に直接注入されたことが分かった。
【0099】
化合物II-51は、30%ソルトール(Solutol)(登録商標)に0.01から0.3 mg/mlの濃度で溶解した。化合物(あるいはソルトール液)を、基底核に傷害を誘導した1日後または6時間前に皮下投与し、その後48時間毎に投与した。用量は、0.01、0.03、0.10および0.30 mg/kgであった。実験は、傷害誘発後4日から21日で終了した。フォナム(Fonnum)の方法(前記)によって、前頭壁皮質の組織ホモジネートでChAT活性を測定した。傷害した側と同側の前頭皮質のChAT活性を、対側(傷害しない側)でのChAT活性と比較して平準化した。ChAT活性は、同側の対側に対する比率で表される。
【0100】
データはANOVA(分散分析)で解析し、タキー(Tukey's)事後検定によって処理間の差異を判定した。p<0.05であれば平均値に有意な差があるとみなした。
【0101】
基底核を傷害した動物において、皮質におけるChAT活性の時間依存的な減少があったが、傷害後3日から7日の間に最大の消失が起きた(表6)。投与経路、用量および投薬スケジュールは、成獣ラットの前脳基部でのChAT活性レベルに対して、化合物II-51の効果が示された予備的データを基礎にした。傷害をもつ動物における、化合物II-51のChAT活性レベルに対する効果(すなわち、コリン作用性機能)を見積もるために、傷害誘発の1日後に薬剤を投与し、14日から21日間続けるか、あるいは、手術の6時間前に投与して4日間続けた。
【0102】
表6
基底核大型細胞で片側傷害を誘導した後における
皮質のChAT活性減少の時間的変化a
傷害期間 注射 ChAT活性
同側/対側比
非傷害対照 − 96±8
8時間 イボテン酸(5μg ) 97±14
24時間 イボテン酸(5μg ) 105±11
72時間 イボテン酸(5μg ) 74±11b
168時間(7日間) イボテン酸(5μg ) 70±7b
a片側傷害は、ラットのNBMで誘発させた。傷害後の指示された時間に、前頭皮質のChAT活性を測定した。
b傷害なしの対照とは有意に(p<0.05)異なることを示す。
【0103】
化合物II-51による用量応答実験を用量0.01、0.03、0.10 mg/kgで行なった(表7)。イボテン酸(ibotenic acid)による傷害誘発後1日目から始めて、1日おきに21日間、化合物II-51の皮下注射を行なった。その結果、化合物II-51は0.03 mg/kgという低い用量で、皮質のChAT活性の消失を食い止める効果があることが示された(表7)。
【0104】
表7
傷害を受けたNBMラットにおける、計画的に投与された化合物II-51の
皮質ChAT活性に対する効果:用量反応実験a
傷害処理 化合物II-51の用量 ChAT活性
同側/対側比
傷害なし − 98.4±4.5
傷害あり 溶媒 67.4±7.2b
傷害あり 0.01 mg/kg QOD 70.1±11.2
傷害あり 0.03 mg/kg QOD 93.8±14.9c
傷害あり 0.10 mg/kg QOD 87.9±11.6c
a 片側傷害は、ラットのNBMで誘導した。傷害誘導後24時間目に、化合物II-51の皮下注射を開始した。傷害後21日目に動物を殺して前頭皮質のChAT活性を測定した。
b 対照とは有意に(p<0.05)異なることを示す。
c 傷害のみとは有意に異なることを示す。
【0105】
傷害誘導後4日目、14日目、21日目に測定すると、化合物II-51の全身投与により、前頭皮質におけるコリン作用性機能の減少が抑えられた(表8)。片側傷害を受けたラットにおいて、対側側のChAT活性は変わらなかった。このことは、化合物II-51が、傷害をもつニューロンにのみ影響することを示唆している。
【0106】
表8
傷害を受けたNBMラットにおける、計画的に投与された化合物II-51の
皮質ChAT活性に対する効果:時間反応実験a
傷害時間(日間) 化合物II-51の用量 ChAT活性
同側/対側比
非傷害対照 − 99±6
4日間 溶媒 77±6b
0.1 mg/kg QOD 96±12c
14日間 溶媒 72±8b
0.1 mg/kg QOD 94±6c
21日間 溶媒 66±8b
0.1 mg/kg QOD 87±7c
a 片側傷害は、ラットのNBMで誘導した。傷害誘導6時間前または1日後に、化合物II-51の皮下注射を開始した。傷害後の表示時間における前頭皮質のChAT活性を測定した。
b 対照とは有意に(p<0.05)異なることを示す。
c 同時点における傷害+溶媒とは有意に異なることを示す。
【0107】
実施例14
インオボ(in ovo)モデルは、成長過程でプログラムされた運動ニューロンの死に影響を与える目的の化合物を評価する目的で用いることができる。
【0108】
ヒナでは、体性運動ニューロンは胎児の6日目から10日目(E6からE10)の間に自然に起こる死に至る(Chu-Wangら、J. Comp. Neurol. 177:33-58, 1978、Hamburger, J. Comp. Neurol. 160: 535-546, 1975、McManamanら、Neuron 4: 891-898, 1990参照)。この期間中に、成長過程にあるヒナの胎児における腰部の脊髄の両側に存在する運動ニューロンの数は、約46,000個から23,000個に減少する。
【0109】
ヒナの胎児(E6〜E9)を、溶媒(5%のソルトール(登録商標)HS 15、BASF Aktienogesellschaft)または上述したような化合物II-51の濃度のいずれかで処理した。この処理液(50μl)を、オッペンハイム(Oppenheim)ら(Science 240: 919-921, 1988)の方法によってシェルに設けた窓を通して血管形成がなされている漿尿膜に通した。胎児をE10の時に屠殺して脊髄を取り出し、カルノイ溶液(Carnoy's solution)(10%の酢酸、60%のエタノール、30%のクロロホルム)で固定化してからパラフィンで包理し、8μmの切片に分割して前述したように(前記のOppenheimらの文献参照)チオニンで染色した。運動ニューロン(形態と位置により同定した)は、以前に確立された基準(Oppenheimら、J. Comp. Neurol. 246: 281-286, 1986参照)にしたがって10箇所の切片ごとに計器により数えた。
【0110】
化合物II-51をインオボでE6〜E9のヒナの漿尿膜に毎日通過させると、生存している腰部の運動ニューロンの数が用量−依存性で増加する結果になる(図20参照)。テストした最小有効用量(1.17μg/一個の卵)では、運動ニューロンの生存率が16%上昇していた。最大の効果は一個の卵あたり2.3μgの用量で得られ、それは対照に比べて、即ち賦形剤で処理した胎児に比べて処理をした運動ニューロンの生存率が40%増加するという結果になった。7μg/卵では運動ニューロンの生存率はそれ以上に増加せず、このことは、この状況下では最大の反応が2.3μg/卵で達成されたことを示している。
【0111】
実施例15
舌下の核に存在する運動ニューロンは、舌下神経を介して舌を支配する。成体のラットでは舌下神経を横に切開すると、舌下の核にある運動ニューロンにおいて細胞の数に影響を与えることなくChAT活性の劇的な消失が引き起こされる。ChAT活性の消失は、未熟な表現型への逆戻りのモデルとして用いられる。
【0112】
左側の舌下神経を、ネンブタール麻酔を行いつつ成体の雌のスプラーグ−ダウレイラット(120〜180g)それぞれの首にある二腹筋の下で切断した。化合物II-51の10%ソルトール(登録商標)(HS15、BASF Aktiengesellschaft)溶液、50マイクロリットル、または溶媒単独を、ゲルフォーム(gelfoam)の断片にかけ、続いてその切開された神経の近い方の端部をゲルフォームとともにパラフィンで覆った。7日目の終わりにラットを深く麻酔薬で眠らせ、ソレンソン緩衝液(Sorenson's buffer)(0.1MのNaPO4、pH7)中に入れた4%のパラホルムアルデヒドを用いて灌流した。脳幹を取り出して定着液に24時間浸漬し、続いてよくすすいでから切断する前に凍結保護のため30%のショ糖の中に入れた。脳の頭頂部分を40ミクロン切り出し、4℃のソレンソンの緩衝液中で染色されるまで保存した。舌下核は40〜48個の部分に広がっており、5個毎の部分を、前述した(Chiuら、J. Comp. Neurol. 328: 351-363, 1993参照)ような抗ChATマウスモノクローナル抗体である1E6を用いて免疫組織化学法を行うべく処理した。ChAT免疫活性ニューロンは、ベクタステイン エライトABC(Vecstastain Elite ABC)(登録商標)キット(Vector Laboratories, Burlingame, CA)を用いてアビジン−ビオチン増幅法を行うことにより部分を可視化した。
【0113】
舌下の核から得られたどの5個ごとの切片も処理をして、それぞれの動物の対照(傷つけていない)の側と軸索切断を行った側の免疫応答性を有する細胞を数えた。結果は、傷つけていない側のChAT免疫活性ニューロンの数に対する軸索切開した側のChAT免疫活性ニューロンの数のパーセントとして表されている。100μgの化合物II-51を舌下神経の切断端部に適用した場合には、有意の数のChAT-免疫応答性を有するニューロンが得られた(33.7%±9.9(平均±標準偏差))(図21参照)。対照的に、溶媒のみで処理した対照の動物では、ChAT-免疫応答性を有するニューロンはほとんどなかった(8.07%±2.9(平均±標準偏差))。
【0114】
調製方法
実施例16:化合物(V)
化合物(V)を調製する工程を下記に示す。
【0115】
工程1
化合物(V-1)(化合物Vの例であって、R1がCH2SO2R7で、XがCO2R5の化合物)は次の反応工程で調製できる。
(R5は低級アルキル基またはCH2NHCO2R6を表し、ここでR6は低級アルキル基またはアリール基を表し、そしてR7は低級アルキル基を表す。)
【0116】
出発物質(A)は日本で発行された未審査の特許出願第295588/88号(参照として本明細書に組み入れられる)に開示されている。
【0117】
化合物(V-1)は化合物(A)を1〜1.5等量の酸化剤で処理することによって得ることができる。酸化剤の例としては、m−クロロ過安息香酸が挙げられる。反応溶液としては、メチレンクロライド、クロロホルム、またはエチレンジクロライドのような水素化した炭化水素などが用いられる。反応は、−20〜30℃で0.5〜1時間で終了する。
【0118】
工程2
化学式(V-2)の化合物(化合物(V)の例であって、R1が水素、XがCH2NHCO2R6の化合物)は次の反応工程で調製することができる。
R6は低級アルキル基またはアリール基を表す。
【0119】
この出発化合物(B)は、日本で発行された未審査の特許出願第155285/87号(参照として本明細書に組み入れられる)に開示されている。
【0120】
化合物(V-2)は化合物(B)を、1〜3等量の塩基の存在下で1〜3等量のClCO2R6と反応させることによって得ることができる。塩基の例としてはトリエチルアミンが挙げられる。反応溶液としては、メチレンクロライド、クロロホルム、またはエチレンジクロライドのような水素化した炭化水素などが用いられる。反応は、−10〜30℃で0.5〜3時間で終了する。
【0121】
実施例17
化合物II-49
化合物(A;R5= CH3、R7= C2H5)(27mg、0.05mmol)を1mlのクロロホルムに溶解し、続いて10mg(0.06mmol)のm−クロロ過安息香酸をそこに氷冷しつつ添加した後、同じ温度で45分間攪拌した。クロロホルムを用いて希釈した後その混合物を、チオ硫酸ナトリウムの8%の水溶液、炭酸水素ナトリウムの飽和水溶液、水、及び生理食塩水を用いて順次洗浄し、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって、17.7mg(収率62%)の化合物II-49を得た。
【0122】
実施例18
化合物II-57
化合物(B)(43.8mg、0.1mmol)を1mlのテトラハイドロフランに溶解し、続いて9.3μl(0.12mmol)のメチルクロロホルメートと28μl(0.2mmol)のトリエチルアミンとを添加した後、氷冷しながら50分間攪拌した。テトラハイドロフランを用いて希釈した後その混合物を生理食塩水を用いて洗浄し、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって、32.6mgの化合物II-57を得た。
【0123】
実施例19
化合物II-38
実施例18と実質的に同じ方法を、43.8mg(0.1mmol)の化合物(B)と15μlのフェニルクロロホルメートを用いて繰り返すことによって27.8mg(収率50%)の化合物II-38が得られた。
【0124】
実施例20
(化合物Cから化合物Hへの合成は図22に示されている。)
化合物II-39
化合物(C)(日本で公開された未審査の特許出願第295588/88号;参照として本明細書に組み入れられる)(20mg、0.035mmol)を1mlのクロロホルムに溶解し、続いて14.6μl(0.105mmol)のトリエチルアミンと13.9μl(0.175mmol)のエチルイソシアネートとをそこに添加した後、室温で2時間攪拌した。その溶液に1mlのメタノールを添加し、続いてクロロホルムを用いて希釈した。その混合物を水と生理食塩水を用いて順次洗浄し、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)にかけることによって、21mg(収率84%)の化合物(D)を得た。
【0125】
化合物(D)(9mg、0.012mmol)を0.2mlのテトラハイドロフランと0.2mlのメタノールの混合液に溶解し、続いて2μlの28%メトキシド/メタノールをそこに添加してから室温で10分間攪拌した。その溶液にクエン酸の5%水性溶液0.1mlを添加した後、クロロホルムを用いて希釈した。この混合物を水と生理食塩水を用いて順次洗浄してから硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=9/1)にかけることによって、8mgの化合物II-39を得た。
【0126】
実施例21
化合物II-51及びII-56
化合物(E)(前記の日本で公開された未審査の特許出願第295588/88号)(60.7mg、0.1mmol)を5mlのクロロホルムと1mlのメタノールの混合溶媒に溶解し、続いて11mg(0.3mmol)の水素化ホウ素ナトリウムを氷冷しつつそこに添加した後、同じ温度で15分間攪拌した。クロロホルムを用いて希釈してからその混合物を、水と生理食塩水を用いて順次洗浄し、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール/トリエチルアミン=98/2/0.5)にかけることによって、36mg(収率59%)の化合物(F)を得た。
【0127】
化合物(F)(159mg、0.26mmol)を15mlのクロロホルムに溶解し、続いて0.8ml(10.4mmol)のエタンチオールと24mg(0.104mmol)のカンファースルホニックアシッドをそこに添加した後、室温で12時間攪拌した。この溶液を炭酸水素ナトリウムの飽和水溶液、水、及び生理食塩水を用いて順次洗浄してから、硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/9−クロロホルム/メタノール=99/1)にかけることによって、43mgの化合物(G)と75mgの化合物(H)を得た。
【0128】
化合物(G)
化合物(H)
【0129】
実施例20と実質的に同じ方法を34mgの化合物(G)を用いて繰り返すことで18.7mgの化合物II-51が得られた。
【0130】
実質的に実施例20と同じ方法を30mgの化合物(H)を用いて繰り返すことで20.4mgの化合物II-56が得られた。
【0131】
実施例22
化合物IV-2
化合物II(Z1、Z2= H、R1= Br、R2= H、R= OH、X= CO2CH3)(日本で公開された未審査の特許出願第120388/87号;参照としてここに組み入れられる)(50mg、0.09mmol)を0.5mlのトリフルオロ酢酸と50μlの3N HClの混合溶媒に溶解し、続いてその溶液を室温で2日間攪拌した。その沈澱物を濾過して集め、高速液相クロマトグラフィー(ユニシル 5C18;メタノール/水=8/2)にかけることによって、8.4mgの化合物(IV-2)を得た。
【0132】
実施例23
化合物II-45は、図23に示された反応工程によって調製することができる。化合物(J)は、日本で公開された未審査の特許出願第120388/87号(参照としてここに組み入れられる)において開示されている。
【0133】
化合物II-45
化合物(J)(200mg)を1mlのジメチルホルムアミドに溶解し、続いて23.5mgの水酸化ナトリウムの水溶液0.25mlをそこに添加した後室温で4時間攪拌した。1Nの塩酸を加えて溶液のpHを1〜2に調節し、沈澱物を濾過して集めることにより178mg(収率91%)の化合物(K)が得られた。
【0134】
化合物(K)(168mg)を3mlのピリジンに溶解し、続いて0.44ml(4.7mmol)の無水酢酸をそこに添加した後室温で4日間攪拌した。その溶媒を蒸発させた後1Nの塩酸を4mlをその残渣に加え、沈澱物を濾過して集めることにより182mg(収率は定量的)の化合物(L)が得られた。
【0135】
化合物(L)(172mg)をチオニルクロライド中に懸濁してから90℃で4.5時間攪拌した。溶媒を蒸発させた後ジエチルエーテルをその残渣に添加し、沈澱物を濾過して集めることによって180mgの化合物(M)が得られた。
【0136】
化合物(M)(67mg、0.1mmol)を2mlのエチレンジクロライドに溶解し、続いてテトラハイドロフランに入れたアニリン、180μlを氷冷しながらそこに添加してから同じ温度で1時間攪拌した。溶媒を蒸発させた後でその残渣を2mlのテトラハイドロフランと0.5mlのメタノールの混合溶媒に溶解し、続いて1mlの1N NaOHをそこに添加してから室温で3時間攪拌した。この溶液に1Nの塩酸(1.2ml)を添加して中和し、続いてテトラハイドロフランを用いて希釈した。その混合物を生理食塩水を用いて洗浄してから硫酸ナトリウム上で乾燥した。この溶媒を蒸発させた後、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)にかけることによって化合物II-45(単離された化合物N、56mgから13mg)が得られた。
【0137】
実施例24
化合物II-65
【0138】
出発化合物(Q)は、日本で未審査の特許出願第295588/88号に開示されている。
【0139】
化合物(Q)(50mg、0.0861mmol)を3mlのクロロホルムに溶解し、続いて200mg(1.41mmol)の2−ジメチルアミノエタンチオール塩酸塩と49mg(0.21mmol)の(±)−10−カンファースルホニックアシッドをそこに添加した後、室温で12時間攪拌した。この反応混合物を炭酸水素ナトリウムの飽和水溶液、水、及び生理食塩水を用いて順次洗浄してから硫酸ナトリウム上で乾燥した。溶媒を蒸発させてからその残渣を、プレパラティブ薄層クロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって56.3mg(収率98%)のN,O−ジアセチル化された化合物II-65が得られた。
FAB-MS(m/z): 668(M+1)+
【0140】
N,O−ジアセチル化された化合物II-65(36.6mg、0.0548mmol)を6mlのクロロホルムと3mlのメタノールの混合溶媒に溶解し、続いて18μl(0.09mmol)の5.1Nのナトリウムメトキシドをそこに添加した後、室温で20分間攪拌した。アンバーリスト(Amberlyst)(登録商標)15イオン交換樹脂(100mg)をその反応混合物に添加してから1時間攪拌し、不溶物を濾過して分離した。溶媒を蒸発させた後その残渣をプレパラティブ薄層クロマトグラフィー(クロロホルム/メタノール=97/3)にかけることによって28.4mg(収率89%)の化合物II-65が得られた。
【0141】
実施例25
化合物II-66
化合物II-66は、例えば日本で公開された未審査の特許出願第155284/87号(参照としてここに組み入れられる)に記載された方法にしたがって調製される。
【0142】
実施例26
化合物II-75
化合物(P)(日本で公開された未審査の特許出願第295588/88号)(100mg、0.173mmol)と4−アミノ−1,2,4−トリアゾール(17.4mg、0.207mmol)を、4mlのクロロホルムと1.5mlのテトラハイドロフランの混合溶媒に溶解し、続いて0.05mlの3Nの塩酸をそこに添加した後、室温で3.5時間攪拌した。酢酸エチルをそこに加えた後、不溶性の物質を濾過して集め、シリカゲルクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって71.9mg(収率64%)のN,O−ジアセチル化された化合物II-75が得られた。
FABS−MS(m/z): 646(M+1)+
【0143】
N,O−ジアセチル化された化合物II-75(37.5mg、0.058mmol)を2mlの1,2−ジクロロエタンと0.6mlのメタノールの混合溶媒に溶解し、続いてメタノール中に入れた5.1Nのナトリウムメトキシド、11μl(0.058mmol)をそこに添加した後、室温で20分間攪拌した。アンバーリスト(Amberlyst)(登録商標)15(50mg)をその反応混合物に添加してから30分間攪拌し、不溶性物質を濾過して分離した。その不溶性物質をジクロロメタン/メタノール/水酸化アンモニウム(8/2/0.5)を用いてよく洗浄し、濾液を合わせて減圧下で濃縮した。その残渣をシリカゲルカラムクロマトグラフィーにかけることによって26.8mg(収率82%)の化合物II-75が得られた。
【0144】
実施例27
化合物II-79
化合物(Q)(日本で公開された未審査の特許出願第295588/88号)(50mg、0.0861mmol)と2−(ブチルアミノ)エタンチオール(0.127ml、0.861mmol)をクロロホルムに溶解し、続いて300mg(1.29mmol)のカンファースルホニックアシッドをそこに添加した後、室温で4日間攪拌した。炭酸水素ナトリウムの飽和水溶液をこの反応混合物に添加して、有機層を塩化ナトリウムの水溶液で洗浄してから硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させ、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって34.6mg(収率58%)のN,O−ジアセチル化された化合物II-79が得られた。
FAB-MS(m/z): 697(M+1)+
【0145】
実施例26と実質的に同じ方法を31.1mg(0.0447mmol)のN,O−ジアセチル化された化合物II-79を用いて繰り返すことによって化合物II-79が得られた(収率52%)。
【0146】
実施例28
化合物II-80
化合物F(国際公開第94/02448号(WO94/02488);参照として本明細書に組み入れられる)(6.19g、10.1mmol)を300mlの1,2−ジクロロエタンと100mlのメタノールの混合溶媒に溶解し、続いてメタノール中に入れた5.1Nのナトリウムメトキシド、0.5ml(2.55mmol)をそこに添加した後、室温で35分間攪拌した。この反応混合物を氷水に注ぎいれ、不溶性物質を濾過して集めることによって化合物II-80のビス(ジメチルアミノエチルチオメチル)の代わりにビス(ハイドロキシメチル)を持つ化合物が、4.95g(収率93%)得られた。
FAB-MS(m/z): 528(M+1)+
【0147】
実施例27と実質的に同じ方法を、化合物II-80のビス(ジメチルアミノエチルチオメチル)の代わりにビス(ハイドロキシメチル)を持つ化合物、22.1mg(0.0419mmol)と2−(ジメチルアミノ)エタンチオールハイドロクロライド、59.4mg(0.419mmol)とを用いて繰り返すことによって、13.1mg(収率45%)の化合物II-80が得られた。
【0148】
実施例29
化合物II-72
実施例27と実質的に同じ方法を、50mg(0.0861mmol)の化合物(Q)と97.8mg(0.861mmol)の2−アミノエタンチオールハイドロクロライドとを用いて繰り返すことによって、49.6mg(収率90%)のN,O−ジアセチル化された化合物II-72が得られた。
FAB-MS(m/z): 641(M+1)+
【0149】
実施例26と実質的に同じ方法を、39.5mg(0.0617mmol)のN,O−ジアセチル化された化合物II-72を用いて繰り返すことによって、30.2mg(収率88%)の化合物II-72が得られた。
【0150】
実施例30
化合物VI−1
化合物(R)(J. Antibiotics, 38: 1437, 1985、図24参照)(1g、1.81mmol)を50mlの1,2−ジクロロエタンに溶解し、続いて0.17ml(3.80mmol)の発煙硝酸を0℃でそこに滴下した後、室温で20分間攪拌した。その反応混合物をクロロホルムで希釈し、そこに炭酸水素ナトリウムの飽和水溶液を添加してから有機層を塩化ナトリウムの水溶液で洗浄し、硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させた後、40mlのジメチルホルムアミドと600mgの10%Pd/Cをその残渣に添加し、続いて水素雰囲気中で1時間、60℃で攪拌した。不溶物を濾過して除き、その濾液を減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=20/80)にかけることによって130.8mg(収率13%)のアミン誘導体が得られた。
FAB-MS(m/z): 567(M+1)+
【0151】
このアミン誘導体(23.9mg、0.0422mmol)を2mlのクロロホルムに溶解し、続いて9.2μl(0.0660mmol)のトリエチルアミンと87μl(1.10mmol)のエチルイソシアネートをそこに滴下した後、室温で2日間攪拌した。水、メタノール、及びクロロホルムをその反応混合物に加えることによってその反応を終了させ、得られた反応混合物をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液で洗浄し、硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させた後、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)にかけることによって31.4mg(収率80%)のN,O−ジアセチル化された化合物VI−1が得られた。
【0152】
実施例26と実質的に同じ方法を、21.4mg(0.0336mmol)のN,O−ジアセチル化された化合物VI−1を用いて繰り返すことで17.0mg(収率91%)の化合物VI−1が得られた。
【0153】
実施例31
化合物VI−2
実施例30と実質的に同じ方法を、5g(9.07mmol)の化合物(R;図24参照)を用いて繰り返すことで259mg(収率5%)のジアミン誘導体が得られた。
FAB-MS(m/z): 582(M+1)+
【0154】
実施例26と実質的に同じ方法を、24.5mg(0.0422mmol)のジアミン誘導体を用いて繰り返すことによって3.8mg(収率18%)の化合物VI−2が得られた。
【0155】
実施例32
化合物IV-6
化合物(S;図25参照)[J. Chem. Soc. Perkin. Trans. 1, 2475 (1990)](5.15g、13.0mmol)を30mlのジメチルホルムアミドと60mlのトルエンの混合溶媒に溶解し、続いて1.45g(12.9mmol)のtert−ブトキシカリウムをアルゴン雰囲気下、−20℃でそこに添加した後、室温で30分間攪拌した。その反応混合物を−20℃まで冷却した後、1.12ml(12.9mmol)の臭化アリールをそこに添加し、その混合物を0℃で2時間攪拌した。溶媒を減圧下で蒸発させた後、水をこの残渣に加え、続いてテトラハイドロフランを用いて抽出した。有機層を塩化ナトリウムの水溶液で洗浄し、硫酸マグネシウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/15)にかけ、ジクロロメタンを用いて粉砕することによって898.4mg(収率16%)の化合物(T-1)が唯一の位置異性体として得られた。
【0156】
化合物(T-1)(1.44g、3.30mmol)を50mlのテトラハイドロフランに溶解し、続いて4.05g(33.2mmol)の9−ボラビシクロ[3,3,1]ノナン(9−BBN)(ダイマー)をそこに添加した後、アルゴン雰囲気下で3時間、室温にて攪拌した。その反応混合物を0℃まで冷却した後、6mlの1N水酸化ナトリウムと6mlの過酸化水素の35%水溶液をそこに添加し、続いて45分間攪拌した。この反応混合物を水を用いて希釈した後、反応混合物を酢酸エチルを用いて抽出した。有機層を水と塩化ナトリウムの水溶液を用いて順次で洗浄し、硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させ、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=100/1)にかけることによって875.5mg(収率58%)の化合物(J−1)が得られた。
【0157】
化合物(U-1)(178.5mg、0.394mmol)を10mlのジメチルホルムアミドに溶解し、続いて309.5mg(1.18mmol)のトリフェニルホスフィンと0.060ml(1.2mmol)の臭素とをアルゴン雰囲気下で0℃にてそこに添加した後、室温で3時間攪拌した。水をその反応混合物に添加してその反応を終了させた後、その混合物を酢酸エチルを用いて抽出し、有機層を水と塩化ナトリウム水溶液を用いて順次で洗浄してから硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させ、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/8)にかけることによって134.6mg(収率66%)の化合物(W)が得られた。
【0158】
化合物(W)を5mlのジメチルホルムアミドに溶解し、続いて0.045ml(0.52mmol)のモルホリンをそこに添加した後、80℃で3時間、アルゴン雰囲気下で攪拌した。その反応混合物を室温にまで冷却した後、氷水をそこに添加し、濾過することによって生じた沈澱物を集めた。この沈澱物を減圧下で乾燥し、薄層クロマトグラフィー(クロロホルム/メタノール=25/1)にかけた。得られた生成物を10mlのテトラハイドロフランに溶解し、続いて3mlの4Nの硫酸をそこに加えてから60℃で12時間攪拌した。反応混合物を室温にまで冷却した後で氷をそこに添加し、混合物を酢酸エチルを用いて抽出した。有機層を水と塩化ナトリウムの水溶液を用いて順次洗浄してから硫酸マグネシウム上で乾燥した。その溶媒を減圧下で蒸発させ、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/2)にかけた。得られた生成物をクロロホルムと酢酸エチルの混合溶媒に溶解し、続いて酢酸エチルに入れた0.88Nの塩酸を添加した後で室温にて1時間攪拌した。生じた沈澱物を濾過して集め、酢酸エチルで洗浄してから減圧下で乾燥させることによって35.0mg(収率19%)の化合物(IV-6)が得られた。
【0159】
実施例33
化合物IV-5
【0160】
化合物(S)(J. Chem. Soc. Perkin. Trans. 1, 2475, 1990)(823.7mg、2.083mmol)を20mlのジメチルホルムアミドに溶解し、166.4mg(4.16mmol)の水素化ナトリウム(60%)を氷冷下でそこに添加した後、同じ温度で10分間攪拌した。臭化アリル(0.45ml、5.2mmol)をそこに添加してからその溶液を氷冷下で2時間攪拌した。クロロホルムを用いて希釈した後で水をそこに加えて有機層を分離し、生理食塩水を用いて洗浄してから硫酸マグネシウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=1/15)にかけることによって735.0mg(収率74%)の化合物(T-2)が得られた。
【0161】
水素化ホウ素ナトリウム(77.7mg、2.05mmol)を20mlのテトラハイドロフランに懸濁し、231.0mg(1.82mmol)のヨウ素をそこに0℃でアルゴン雰囲気下で添加した後、同じ温度で15分間攪拌した。化合物(T-2)(136.7mg、0.287mmol)を同じ温度でそこに加えてからその反応混合物を室温で4.5時間攪拌した。その反応混合物を0℃まで冷却した後、3.7mlの1N水酸化ナトリウムと3.7mlの過酸化水素の35%水溶液をそこに添加し、続いてさらに30分間攪拌した。この反応混合物を水を用いて希釈してから酢酸エチルを用いて抽出した。この酢酸エチル層を水と食塩水溶液を用いて順次で洗浄し、硫酸マグネシウム上で乾燥した。溶媒を蒸発させた後、その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=15/1)にかけることによって88.9mg(収率61%)の化合物(U-2)が得られた。
【0162】
化合物(U-2)(88.9mg、0.174mmol)を10mlのテトラハイドロフランに溶解し、そこに8mlの4N硫酸を添加した後、60℃で24時間攪拌した。その反応混合物を室温にまで冷却した後で氷をそこに加え、続いて酢酸エチルによって抽出した。酢酸エチル層を水と食塩水溶液を用いて順次洗浄してから硫酸マグネシウム上で乾燥した。溶媒を蒸発させ、残渣を薄層クロマトグラフィー(クロロホルム/メタノール=15/1)にかけることによって37.6mg(収率51%)の化合物IV-5が得られた。
【0163】
実施例34
化合物II-68
化合物(Q)(50.1mg、0.0862mmol)を3mlのクロロホルムに溶解し、続いて129.5mg(0.862mmol)の2−メルカプトベンズイミダゾールと49mg(0.21mmol)の(±)−10−カンファースルホニックアシッドとをそこに添加した後、室温で12時間攪拌した。その反応混合物を炭酸水素ナトリウムの飽和水溶液、水、及び生理食塩水を用いて順次洗浄した後で硫酸ナトリウム上で乾燥した。溶媒を蒸発させてから残渣をプレパラティブ薄層クロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって、46mg(収率75%)のN,O−ジアセチル化された化合物II-68が得られた。
FAB-MS(m/z): 714(M+1)+
【0164】
実施例25と実質的に同じ方法を、33.4mg(0.0468mmol)のN,O−ジアセチル化された化合物II-68を用いて繰り返すことによって、17.5mg(収率59%)の化合物II-68が得られた。
【0165】
実施例35
化合物II-69
実施例25と実質的に同じ方法を、50mg(0.0861mmol)の化合物Q及び0.0868ml(0.861mmol)のフルフリルメルカプタンを用いて続いて行うことによって、36.0mg(収率62%)のN,O−ジアセチル化された化合物II-69が得られた。
FAB-MS(m/z): 678(M+1)+
【0166】
実施例25と実質的に同じ方法を22.7mg(0.0335mmol)のN,O−ジアセチル化された化合物II-69を用いて繰り返すことによって、17.7mg(収率89%)の化合物II-69が得られた。
【0167】
実施例36
化合物II-70
化合物(P)(100mg、0.173mmol)を4mlのクロロホルムに溶解し、続いて34.0mg(0.277mmol)の1−アミノピロリジン塩酸塩をそこに添加した後、室温で4時間攪拌した。減圧下で溶媒を蒸発させた後、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって、100.5mg(収率90%)のN,O−ジアセチル化された化合物II-70が得られた。
FAB-MS(m/z): 648(M+1)+
【0168】
実施例25と実質的に同じ方法を40mg(0.0618mmol)のN,O−ジアセチル化された化合物II-70を用いて繰り返すことによって、30mg(収率86%)の化合物II-70が得られた。
【0169】
実施例37
化合物II-71
化合物(P)(49mg、0.0846mmol)を3mlのクロロホルムに溶解し、続いて15.8mg(0.145mmol)の2−ヒドラジノピリジンのクロロホルム溶液と49mg(0.21mmol)の(±)−10−カンファースルホニックアシッドをそこに添加した後、室温で12時間攪拌した。その反応混合物を炭酸水素ナトリウムの飽和水溶液、水、及び塩溶液を用いて順次洗浄してから硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、残渣をプレパラティブ薄層クロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって、35.8mg(収率64%)のN,O−ジアセチル化された化合物II-71が得られた。
FAB-MS(m/z): 671(M+1)+
【0170】
実施例25と実質的に同じ方法を24.6mg(0.0367mmol)のN,O−ジアセチル化された化合物II-71を用いて繰り返すことによって、11.8mg(収率55%)の化合物II-71が得られた。
【0171】
実施例38
化合物II-73
実施例25と実質的に同じ方法を30mg(0.0516mmol)の化合物(Q)と52.2mg(0.516mmol)の1H−1,2,4−トリアゾール−3−チオールとを用いて続いて行うことによって、31.4mg(収率92%)のN,O−ジアセチル化された化合物II-73が得られた。
FAB-MS(m/z): 665(M+1)+
【0172】
実施例25と実質的に同じ方法を15mg(0.0226mmol)のN,O−ジアセチル化された化合物II-73を用いて繰り返すことによって、粗精製の化合物II-73が得られた。そこにクロロホルム/メタノール(90/10)を添加した後で攪拌することによって10.9mg(収率83%)の化合物II-73が沈澱物として得られた。
【0173】
実施例39
化合物II-74
化合物(P)(97.5mg、0.168mmol)を4mlのテトラハイドロフランに溶解し、続いて25.1mg(0.0950mmol)のアミノグアニジン硫酸塩の水溶液をそこに添加した後、室温で3時間攪拌した。酢酸エチルをそこに添加してから攪拌し、不溶性の物質を濾過して集めた後、シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=85/15)にかけることによって、87.1mg(収率82%)のN,O−ジアセチル化された化合物II-74が得られた。
FAB-MS(m/z): 636(M+1)+
【0174】
実施例25と実質的に同じ方法を69.6mg(0.110mmol)のN,O−ジアセチル化された化合物II-74を用いて繰り返すことによって、37.2mg(収率62%)の化合物II-77が得られた。
【0175】
実施例40
化合物II-76
化合物(P)(103.8mg、0.179mmol)を6mlのクロロホルムと3mlのメタノールの混合溶媒に溶解し、続いて0.020ml(0.207mmol)の4−アミノモルホリンの水溶液、0.5mlと3Nの塩酸、0.05mlをそこに添加した後、室温で3時間攪拌した。その反応混合物を炭酸水素ナトリウムの飽和水溶液、及び塩溶液を用いて順次洗浄してから硫酸ナトリウム上で乾燥した。溶媒を蒸発させた後、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=90/100)にかけることによって、82.8mg(収率70%)のN,O−ジアセチル化された化合物II-76が得られた。
FAB-MS(m/z): 663(M+1)+
【0176】
実施例25と実質的に同じ方法を50.6mg(0.0763mmol)のN,O−ジアセチル化された化合物II-76を用いて繰り返すことによって、36.4mg(収率82%)の化合物II-76が得られた。
【0177】
実施例41
化合物II-77
実施例40と実質的に同じ方法を100mg(0.173mmol)の化合物Pと16.7mg(0.173mmol)の1,1−ジメチルヒドラジン塩酸塩とを用いて続いて行うことによって、52.3mg(収率49%)のN,O−ジアセチル化された化合物II-77が得られた。
FAB-MS(m/z): 622(M+1)+
【0178】
実施例25と実質的に同じ方法を38.4mg(0.0618mmol)のN,O−ジアセチル化された化合物I−75を用いて繰り返すことによって、10.9mg(収率33%)の化合物II-77が得られた。
【0179】
実施例42
化合物II-78
実施例40と実質的に同じ方法を99.5mg(0.172mmol)の化合物(P)と42.4mgの1−アミノ−4−メチルピペラジンとを用いて続いて行うことによって、N,O−ジアセチル化された化合物II-78が得られた。
【0180】
続いて実施例25と実質的に同じ方法を、上記のN,O−ジアセチル化化合物II-78を用いて繰り返すことによって、19.4mg[化合物(P)からの収率で19%]の化合物II-78が得られた。
【0181】
実施例43
化合物II-81
化合物(AA)、即ち化合物II-80(実施例28に記載されている)のビス(ジメチルアミノエチルチオメチル)に代えて、ビス(ハイドロキシメチル)を持つ化合物(53.9mg、0.102mmol)を、2.5mlのジクロロメタンに溶解した。続いて0.18ml(2.0mmol)の2−プロパンチオールと0.03ml(0.2mmol)のトリフルオロ酢酸無水物を順次そこに添加し、アルゴン気流下で3時間、室温にて攪拌した。炭酸水素ナトリウムの飽和水溶液をその反応混合液に添加してからその混合液をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてから残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって52.6mg(収率80%)の化合物II-81が得られた。
【0182】
実施例44
化合物II-82
実施例43と実質的に同じ方法を51.9mg(0.0958mmol)の化合物(AA)と0.17ml(1.9mmol)の1−プロパンチオールと0.03ml(0.2mmol)のトリフルオロ酢酸無水物とを用いて繰り返すことによって、52.3mg(収率83%)の化合物II-82が得られた。
【0183】
実施例45
化合物II-83
実施例43と実質的に同じ方法を49.4mg(0.0937mmol)の化合物(AA)と0.20ml(1.9mmol)の1−ブタンチオールと0.03ml(0.2mmol)のトリフルオロ酢酸無水物とを用いて繰り返すことによって、51.7mg(収率82%)の化合物II-83が得られた。
【0184】
実施例46
化合物II-84
化合物(AA)(45.3mg、0.0860mmol)を0.2mlのメタノールと2mlのクロロホルムの混合溶媒に溶解し、続いて20mg(0.086mmol)のカンファースルホニックアシッドをそこに添加してから室温にて17時間攪拌した。炭酸水素ナトリウムの飽和水溶液をその反応混合液に添加して、続いてその混合液をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてから残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=99/1)にかけることによって23.1mg(収率48%)の化合物II-84が得られた。
【0185】
実施例47
化合物II-85
実施例46と実質的に同じ方法を、0.2mlのエタノールと2mlのクロロホルムの混合溶媒に入れた51.3mg(0.0973mmol)の化合物(AA)を用いて繰り返すことによって、24.1mg(収率42%)の化合物II-85が得られた。
【0186】
実施例48
化合物II-86
化合物(BB)(日本で公開された未審査の特許出願第295588/88号)(978mg、1.69mmol)を70mlの1,2−ジクロロエタンに溶解し、続いて0.17ml(3.8mmol)の発煙硝酸を氷冷しながらそこに滴下してから室温にて30分間攪拌した。その反応混合液をクロロホルムを用いて希釈してから炭酸水素ナトリウムの飽和水溶液をそこに添加した。不溶物を濾過して集め、乾燥した。その濾液を塩化ナトリウムの水溶液を用いて洗浄し、硫酸ナトリウム上で乾燥してから溶媒を減圧下で蒸発させた。残渣と不溶物とをあわせて946mg(収率90%)の化合(CC)が粗生成物として得られた。
FAB-MS(m/z): 625(M+1)+
【0187】
化合物(CC)(640mg、1.03mmol)を30mlの1,2−ジクロロエタンに溶解し、続いて0.3ml(3.58mmol)の1,2−エタンジチオールと0.2ml(2.0mmol)のボロントリフロライドエーテル錯体を0℃でそこに滴下してから30分間攪拌した。炭酸水素ナトリウムの飽和水溶液をその反応混合液に添加し、続いてその混合液をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム)にかけることによって579mg(収率81%)の化合物(DD)が得られた。
FAB-MS(m/z): 701(M+1)+
【0188】
化合物(DD)(579mg、0.827mmol)を56mlのN,N−ジメチルホルムアミドに溶解し、続いて400mgのパラジウム/炭素をそこに添加してから60℃で2時間、水素雰囲気下で攪拌した。不溶物を濾過して分離し、その濾液から溶媒を減圧下で蒸発させた。その残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=98/2)にかけることによって193mg(収率35%)の化合物(EE)が得られた。
FAB-MS(m/z): 671(M+1)+
【0189】
化合物(EE)(193mg、0.288mmol)を10mlのクロロホルムに溶解し、続いて0.1ml(0.7mmol)のトリエチルアミンと0.2ml(2.5mmol)のエチルイソシアネートをそこに添加してから室温で20時間攪拌した。水を加えてからその混合液をクロロホルムを用いて抽出した。有機層を塩化ナトリウムの水溶液で洗浄してから硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=96/4)にかけることによって211mg(収率99%)の化合物(FF)が得られた。
FAB-MS(m/z): 742(M+1)+
【0190】
化合物(FF)(211mg、0.285mmol)を6mlのエタノールと6mlのクロロホルムの混合溶媒に溶解し、続いて171mg(1.01mmol)の硝酸銀をそこに50℃で添加してから20分間攪拌した。反応を終了させてから不溶物を濾過して除去した。この濾液を炭酸水素ナトリウムの飽和水溶液と塩化ナトリウムの水溶液を用いて洗浄し、そして硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=97/3)にかけることによって118mg(収率62%)の化合物(GG)が得られた。
FAB-MS(m/z): 666(M+1)+
【0191】
化合物(GG)(100mg、0.150mmol)を4.5mlのクロロホルムと0.72mlのメタノールの混合溶媒に溶解し、続いて8.7mg(0.23mmol)の水素化ホウ素ナトリウムをそこに0℃で添加してから45分間攪拌した。その反応混合液を水に注入してからその混合溶液をクロロホルムで抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、続いて硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって101mg(収率100%)の化合物(HH)が得られた。
FAB-MS(m/z): 668(M+1)+
【0192】
化合物(HH)(21.7mg、0.0325mmol)を1mlの1,2−ジクロロエタンと0.3mlのメタノールの混合溶媒に溶解し、続いて6μl(0.03mmol)の5.1Nのナトリウムメトキシドのメタノール溶液をそこに添加してから1時間攪拌した。その反応混合液を水に注入してからその混合溶液をクロロホルムとメタノール(9/1)の混合溶媒で抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、続いて硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=90/10)にかけることによって14.9mg(収率79%)の化合物II-86が得られた。
【0193】
実施例49
化合物II-87
実施例43と実質的に同じ方法を29.8mg(0.0511mmol)の化合物II-86と0.14ml(1.6mmol)のエタンチオールとを用いて繰り返すことによって、24.2mg(収率76%)の化合物II-87が得られた。
【0194】
実施例50
化合物II-88
化合物(AA)(50.4mg、0.0956mmol)を0.7mlのジクロロメタンに溶解し、続いて0.09mg(0.56mmol)のトリエチルシランと0.73ml(9.5mmol)のトリフルオロ酢酸をそこに氷冷しつつ添加してから室温で10分間攪拌した。その反応混合液を1Nの水酸化ナトリウム水溶液を用いて中和してからその混合溶液をクロロホルムで抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、続いて硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=90/10)にかけることによって20.7mg(収率44%)の化合物II-88が得られた。
【0195】
実施例51
化合物II-89
化合物(AA)(4.3g、8.16mmol)を215mlのジクロロメタンに溶解し、続いて12.1mg(163mmol)のエタンチオールと2.5ml(17.7mmol)のトリフルオロ酢酸無水物をそこに連続的に添加してから室温で12時間攪拌した。炭酸水素ナトリウムの飽和水溶液をその反応混合液に添加し、続いてその混合溶液をクロロホルムで抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄し、続いて硫酸ナトリウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/トルエン=3/7)にかけることによって4mg(収率0.08%)の化合物II-89が得られた。
【0196】
実施例52
化合物II-90
化合物(JJ)(日本で公開された未審査の特許出願第295588/88号)(18.5g、30.5mmol)を900mlのクロロホルムと145mlのメタノールの混合溶媒に溶解し、続いて3.42g(90.4mmol)の水素化ホウ素ナトリウムを氷冷しつつそこに添加してから25分間攪拌した。その反応混合液を氷水に注入し、不溶物を濾過して集めてから水を用いて洗浄を行い、減圧下で乾燥した。不溶物を555mlの1,2−ジクロロエタンと185mlのメタノールの混合溶媒に溶解し、続いて0.925ml(4.72mmol)のナトリウムメトキシドの5.1Nメタノール溶液をそれに添加してから1時間半攪拌した。この反応混合液を水に注入し、不溶物を濾過して集め、続いて減圧下で乾燥してからシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=8/2)にかけることによって0.350g(収率2.3%)の化合物II-90が得られた。
【0197】
実施例53
化合物II-91
化合物(DD)(18.6g、0.0266mmol)を1.5mlの1,2−ジクロロエタンと0.5mlのメタノールの混合溶媒に溶解し、続いて5μl(0.026mmol)のナトリウムメトキシドの5.1Nメタノール溶液をそれに添加してから1時間攪拌した。この反応混合液を水に注入し、そして得られた混合溶液をクロロホルムとメタノール(9/1)の混合溶媒を用いて抽出した。有機層を塩化ナトリウムの水溶液を用いて洗浄した後、硫酸マグネシウム上で乾燥した。溶媒を減圧下で蒸発させてからその残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/メタノール=95/5)にかけることによって7.0mg(収率43%)の化合物II-91が得られた。
【0198】
実施例54
化合物II-92
実施例53と実質的に同じ方法を23.3mg(0.0314mmol)の化合物(FF)を用いて繰り返すことによって、14.7mg(収率71%)の化合物II-92が得られた。
【0199】
本発明はかなり詳細に列記したが、本明細書に開示された発明は実際の記載に限定されるべきではなく、添付した請求の範囲及びその等価物のすべてからなる完全な範囲が提供されるべきである。他の態様は、次の請求の範囲内に含まれる。
【図面の簡単な説明】
【0200】
【図1】図1は、PC−12細胞における基底オルニチンデカルボキシラーゼ(ODC)活性に対するK−252a誘導体である1,6−ヘキサメチレン−ビス−(カルバミルスタウロスポリン)(HBCS)およびスタウロスポリンの効果を示すグラフである。
【図2】図2は、PC−12細胞におけるNGFにより刺激されるODC活性に対するスタウロスポリン、HBCS、及びK−252aの効果を示すグラフである。
【図3】図3は、PC−12細胞におけるODC活性に対するHBCSのNGF−効力増強効果を示すグラフである。
【図4】図4は、ラット胎児の脊髄培養物におけるコリンアセチルトランスフェラーゼ(ChAT)特異的活性に対するK−252aの効果を示すグラフである。
【図5】図5は、ラット胎児の脊髄培養物におけるChAT活性に対するK−252aの効果の時間経過を示すグラフである。
【図6】図6は、ヒナ胎児の背根神経節ニューロンの生存性に対するK−252aの効果を示すグラフである。
【図7】図7は、ヒナ胎児の背根神経節ニューロンの生存性に対するK−252aの機能性誘導体の効果を示すグラフである。
【図8】図8は、ラット胎児の脊髄培養物におけるChAT活性に対するK−252aの機能性誘導体の効果を示すグラフである。
【図9】図9は、ラットの海馬へのカイニン酸塩誘導性の損傷に対するK−252aの効果を示すグラフである。
【図10】図10は、ラットの海馬におけるカイニン酸塩誘導性のスペクトリンタンパク質分解に対するK−252aの効果を示すグラフである。
【図11】図11は、海馬へのカイニン酸塩誘導性の損傷に対するHBCSの効果を示すグラフである。
【図12】図12は、ラットの海馬におけるカイニン酸塩誘導性のスペクトリンタンパク質分解に対するK−252aの機能性誘導体の効果を示すグラフである。
【図13】図13a、13b及び13cは、ラットの脊髄培養物におけるChAT活性に対するK−252a誘導体の相対的な活性を示す表である。
【図14】図14は、ヒナの背根神経節培養物におけるニューロンの生存性に対するK−252a誘導体の相対的な活性を示す表である。
【図15】図15は、K−252aの存在下での脊髄ニューロンの生存性を示すグラフである。
【図16】図16は、K−252aの存在下での脊髄細胞の生存性の時間経過を示すグラフである。
【図17】図17は、K−252aの存在下または非存在下で培養された線条体ニューロンの一対の顕微鏡写真である。
【図18】図18は、ラットの線条体培養物におけるニューロンの生存性に対するK−252a誘導体の相対的な活性を示す表である。
【図19】図19は、低密度の基底前脳ニューロンの生存性に対するK−252a誘導体の相対的な活性を示す表である。
【図20】図20は、化合物II−51が、成長過程でプログラムされた運動ニューロンの死をインオボ(in ovo)で阻止することを示す棒グラフである。
【図21】図21は、化合物II−51が、成体の舌下核におけるChAT免疫活性が軸索切断によって喪失されるのを阻止することを示す写真である。
【図22】図22は、出発化合物Cから化合物Hへの合成を示す図式である。
【図23】図23は、出発化合物Jから化合物II−45への合成を示す図式である。
【図24】図24は、化合物P、化合物Q、及び化合物Rの構造を示す図である。
【図25】図25は、出発化合物Sから化合物IV−6への合成を示す図式である。
【図26】図26は、化合物(AA)、(BB)、(CC)、(DD)及び(EE)の化学構造を示す図である。
【図27】図27は、化合物(FF)、(GG)、(HH)及び(JJ)の化学構造を示す図である。
【特許請求の範囲】
【請求項1】
下式(I)の化合物。
[Stau]−N(CH3)−W−N(CH3)−[Stau] (I)
ここで[Stau]は下式の残基を表し、
そしてWは下式のラジカルを表す。
−C(=Y)−NH−W’−NH−C(=Y)−
ここでW’は2〜20個の炭素原子からなるハイドロカルビレンラジカルであって、YはOまたはSである。
【請求項2】
請求項1記載の化合物を含む組成物。
【請求項3】
哺乳動物のコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、及び感覚ニューロンの機能を高める方法であって、治療に有効な量の、請求項1に記載の組成物を前記哺乳動物に投与することを含む、方法。
【請求項4】
感覚ニューロンが背根の神経節ニューロンである、請求項3に記載の方法。
【請求項5】
治療に有効な量の、請求項1に記載の組成物を哺乳動物に投与することを含む、興奮性のアミノ酸によって誘導される神経細胞の変性を治療する方法。
【請求項6】
神経細胞の変性がアルツハイマー病と関連するものである、請求項5に記載の方法。
【請求項7】
神経細胞の変性が運動ニューロン疾患と関連するものである、請求項4に記載の方法。
【請求項8】
運動ニューロン疾患が筋萎縮性側索硬化症である、請求項7に記載の方法。
【請求項9】
神経細胞の変性がパーキンソン病と関連するものである、請求項5に記載の方法。
【請求項10】
神経細胞の変性が脳血管疾患と関連するものである、請求項5に記載の方法。
【請求項11】
脳血管疾患が虚血性のものである、請求項10に記載の方法。
【請求項12】
神経細胞の変性がAIDS脳症と関連するものである、請求項5に記載の方法。
【請求項13】
神経細胞の変性がてんかんと関連するものである、請求項5に記載の方法。
【請求項14】
神経細胞の変性が脳に起こる震盪性の損傷と関連するものである、請求項5に記載の方法。
【請求項15】
神経細胞の変性が脊髄に起こる震盪性の損傷と関連するものである、請求項5に記載の方法。
【請求項16】
神経細胞の変性が脳に起こる穿通性の損傷と関連するものである、請求項5に記載の方法。
【請求項17】
神経細胞の変性が脊髄に起こる穿通性の損傷と関連するものである、請求項5に記載の方法。
【請求項18】
神経細胞の変性がハンチントン病と関連するものである、請求項5に記載の方法。
【請求項19】
組成物が栄養に関する因子とともに投与される、請求項3に記載の方法。
【請求項20】
組成物が栄養に関する因子とともに投与される、請求項5に記載の方法。
【請求項21】
栄養に関する因子がニューロトロフィンファミリーの一つである、請求項19または20に記載の方法。
【請求項22】
ニューロトロフィンファミリーの一つが、神経生長因子(NGF)である、請求項21に記載の方法。
【特許請求の範囲】
【請求項1】
下式(I)の化合物;
[Stau]−N(CH3)−W−N(CH3)−[Stau] (I)
ここで[Stau]は下式の残基を表し、
そしてWは下式のラジカルを表す。
−C(=Y)−NH−W’−NH−C(=Y)−
ここでW’は2〜20個の炭素原子からなるハイドロカルビレンラジカルであって、YはOまたはSである。
【請求項2】
請求項1記載の化合物を含む医薬組成物。
【請求項3】
請求項1に記載の化合物を有効成分として含有する、哺乳動物のコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、及び感覚ニューロンの機能を高めるための医薬組成物。
【請求項4】
感覚ニューロンが背根の神経節ニューロンである、請求項3に記載の医薬組成物。
【請求項5】
請求項1に記載の化合物を有効成分として含有する、哺乳動物の興奮性のアミノ酸によって誘導される神経細胞の変性の治療用医薬組成物。
【請求項6】
神経細胞の変性がアルツハイマー病と関連するものである、請求項5に記載の医薬組成物。
【請求項7】
神経細胞の変性が運動ニューロン疾患と関連するものである、請求項4に記載の医薬組成物。
【請求項8】
運動ニューロン疾患が筋萎縮性側索硬化症である、請求項7に記載の医薬組成物。
【請求項9】
神経細胞の変性がパーキンソン病と関連するものである、請求項5に記載の医薬組成物。
【請求項10】
神経細胞の変性が脳血管疾患と関連するものである、請求項5に記載の医薬組成物。
【請求項11】
脳血管疾患が虚血性のものである、請求項10に記載の医薬組成物。
【請求項12】
神経細胞の変性がAIDS脳症と関連するものである、請求項5に記載の医薬組成物。
【請求項13】
神経細胞の変性がてんかんと関連するものである、請求項5に記載の医薬組成物。
【請求項14】
神経細胞の変性が脳に起こる震盪性の損傷と関連するものである、請求項5に記載の医薬組成物。
【請求項15】
神経細胞の変性が脊髄に起こる震盪性の損傷と関連するものである、請求項5に記載の医薬組成物。
【請求項16】
神経細胞の変性が脳に起こる穿通性の損傷と関連するものである、請求項5に記載の医薬組成物。
【請求項17】
神経細胞の変性が脊髄に起こる穿通性の損傷と関連するものである、請求項5に記載の医薬組成物。
【請求項18】
神経細胞の変性がハンチントン病と関連するものである、請求項5に記載の医薬組成物。
【請求項19】
前記医薬組成物が、栄養に関する因子を含む、請求項3に記載の医薬組成物。
【請求項20】
前記医薬組成物が、栄養に関する因子を含む、請求項5に記載の医薬組成物。
【請求項21】
前記栄養に関する因子がニューロトロフィンファミリーの一つである、請求項19または20に記載の医薬組成物。
【請求項22】
前記ニューロトロフィンファミリーの一つが、神経生長因子(NGF)である、請求項21に記載の医薬組成物。
【請求項1】
下式(I)の化合物。
[Stau]−N(CH3)−W−N(CH3)−[Stau] (I)
ここで[Stau]は下式の残基を表し、
そしてWは下式のラジカルを表す。
−C(=Y)−NH−W’−NH−C(=Y)−
ここでW’は2〜20個の炭素原子からなるハイドロカルビレンラジカルであって、YはOまたはSである。
【請求項2】
請求項1記載の化合物を含む組成物。
【請求項3】
哺乳動物のコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、及び感覚ニューロンの機能を高める方法であって、治療に有効な量の、請求項1に記載の組成物を前記哺乳動物に投与することを含む、方法。
【請求項4】
感覚ニューロンが背根の神経節ニューロンである、請求項3に記載の方法。
【請求項5】
治療に有効な量の、請求項1に記載の組成物を哺乳動物に投与することを含む、興奮性のアミノ酸によって誘導される神経細胞の変性を治療する方法。
【請求項6】
神経細胞の変性がアルツハイマー病と関連するものである、請求項5に記載の方法。
【請求項7】
神経細胞の変性が運動ニューロン疾患と関連するものである、請求項4に記載の方法。
【請求項8】
運動ニューロン疾患が筋萎縮性側索硬化症である、請求項7に記載の方法。
【請求項9】
神経細胞の変性がパーキンソン病と関連するものである、請求項5に記載の方法。
【請求項10】
神経細胞の変性が脳血管疾患と関連するものである、請求項5に記載の方法。
【請求項11】
脳血管疾患が虚血性のものである、請求項10に記載の方法。
【請求項12】
神経細胞の変性がAIDS脳症と関連するものである、請求項5に記載の方法。
【請求項13】
神経細胞の変性がてんかんと関連するものである、請求項5に記載の方法。
【請求項14】
神経細胞の変性が脳に起こる震盪性の損傷と関連するものである、請求項5に記載の方法。
【請求項15】
神経細胞の変性が脊髄に起こる震盪性の損傷と関連するものである、請求項5に記載の方法。
【請求項16】
神経細胞の変性が脳に起こる穿通性の損傷と関連するものである、請求項5に記載の方法。
【請求項17】
神経細胞の変性が脊髄に起こる穿通性の損傷と関連するものである、請求項5に記載の方法。
【請求項18】
神経細胞の変性がハンチントン病と関連するものである、請求項5に記載の方法。
【請求項19】
組成物が栄養に関する因子とともに投与される、請求項3に記載の方法。
【請求項20】
組成物が栄養に関する因子とともに投与される、請求項5に記載の方法。
【請求項21】
栄養に関する因子がニューロトロフィンファミリーの一つである、請求項19または20に記載の方法。
【請求項22】
ニューロトロフィンファミリーの一つが、神経生長因子(NGF)である、請求項21に記載の方法。
【特許請求の範囲】
【請求項1】
下式(I)の化合物;
[Stau]−N(CH3)−W−N(CH3)−[Stau] (I)
ここで[Stau]は下式の残基を表し、
そしてWは下式のラジカルを表す。
−C(=Y)−NH−W’−NH−C(=Y)−
ここでW’は2〜20個の炭素原子からなるハイドロカルビレンラジカルであって、YはOまたはSである。
【請求項2】
請求項1記載の化合物を含む医薬組成物。
【請求項3】
請求項1に記載の化合物を有効成分として含有する、哺乳動物のコリン作動性ニューロン、線条体ニューロン、基底前脳ニューロン、及び感覚ニューロンの機能を高めるための医薬組成物。
【請求項4】
感覚ニューロンが背根の神経節ニューロンである、請求項3に記載の医薬組成物。
【請求項5】
請求項1に記載の化合物を有効成分として含有する、哺乳動物の興奮性のアミノ酸によって誘導される神経細胞の変性の治療用医薬組成物。
【請求項6】
神経細胞の変性がアルツハイマー病と関連するものである、請求項5に記載の医薬組成物。
【請求項7】
神経細胞の変性が運動ニューロン疾患と関連するものである、請求項4に記載の医薬組成物。
【請求項8】
運動ニューロン疾患が筋萎縮性側索硬化症である、請求項7に記載の医薬組成物。
【請求項9】
神経細胞の変性がパーキンソン病と関連するものである、請求項5に記載の医薬組成物。
【請求項10】
神経細胞の変性が脳血管疾患と関連するものである、請求項5に記載の医薬組成物。
【請求項11】
脳血管疾患が虚血性のものである、請求項10に記載の医薬組成物。
【請求項12】
神経細胞の変性がAIDS脳症と関連するものである、請求項5に記載の医薬組成物。
【請求項13】
神経細胞の変性がてんかんと関連するものである、請求項5に記載の医薬組成物。
【請求項14】
神経細胞の変性が脳に起こる震盪性の損傷と関連するものである、請求項5に記載の医薬組成物。
【請求項15】
神経細胞の変性が脊髄に起こる震盪性の損傷と関連するものである、請求項5に記載の医薬組成物。
【請求項16】
神経細胞の変性が脳に起こる穿通性の損傷と関連するものである、請求項5に記載の医薬組成物。
【請求項17】
神経細胞の変性が脊髄に起こる穿通性の損傷と関連するものである、請求項5に記載の医薬組成物。
【請求項18】
神経細胞の変性がハンチントン病と関連するものである、請求項5に記載の医薬組成物。
【請求項19】
前記医薬組成物が、栄養に関する因子を含む、請求項3に記載の医薬組成物。
【請求項20】
前記医薬組成物が、栄養に関する因子を含む、請求項5に記載の医薬組成物。
【請求項21】
前記栄養に関する因子がニューロトロフィンファミリーの一つである、請求項19または20に記載の医薬組成物。
【請求項22】
前記ニューロトロフィンファミリーの一つが、神経生長因子(NGF)である、請求項21に記載の医薬組成物。
【図1】

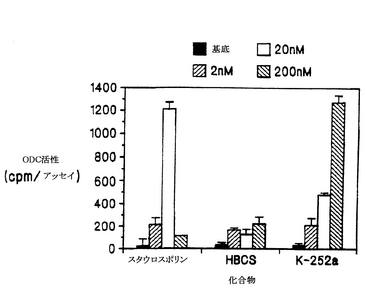
【図2】
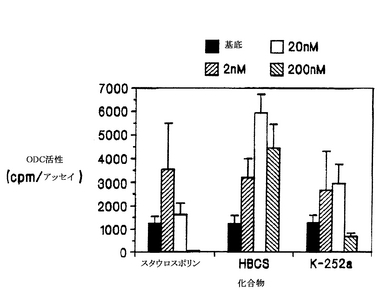
【図3】
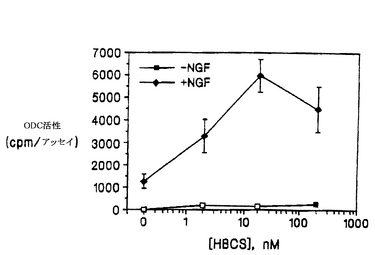
【図4】
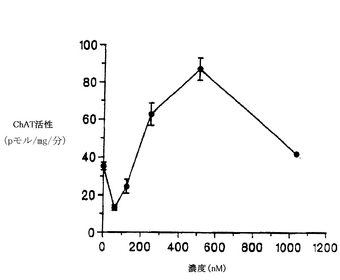
【図5】
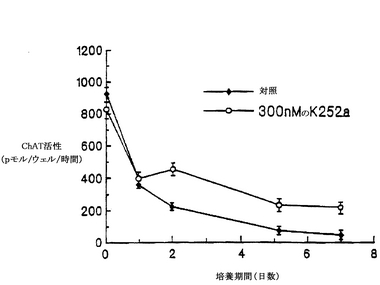
【図6】
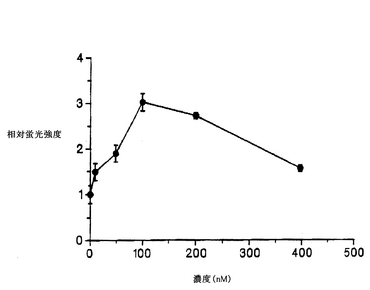
【図7】
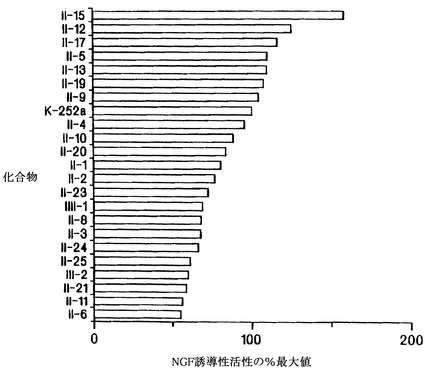
【図8】
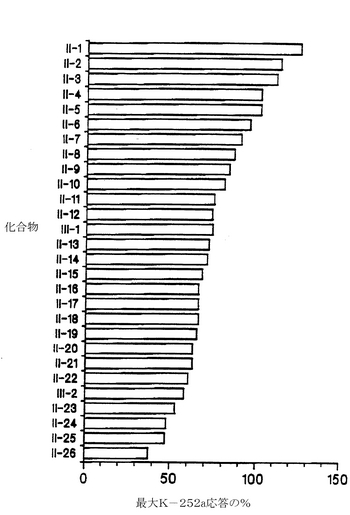
【図9】
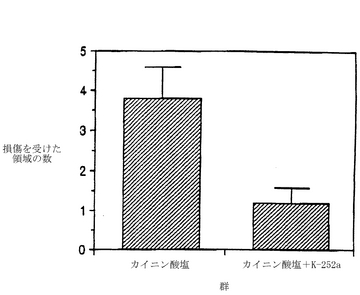
【図10】
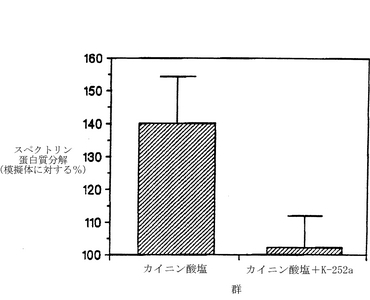
【図11】
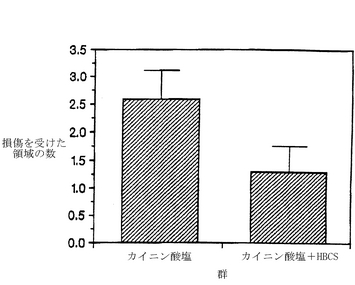
【図12】
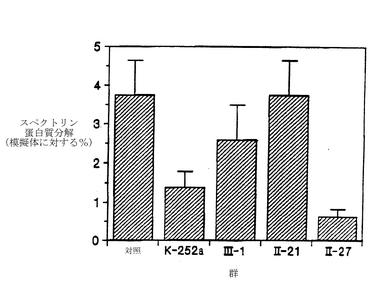
【図13a】

【図13b】
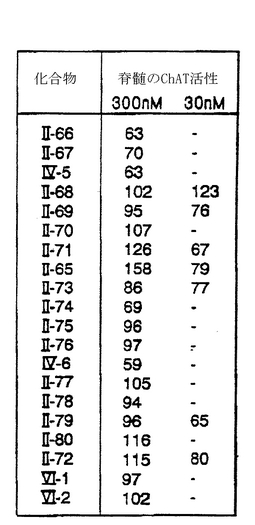
【図13c】
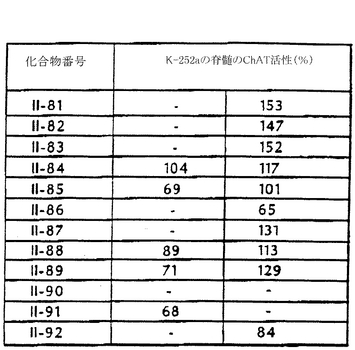
【図14】

【図15】
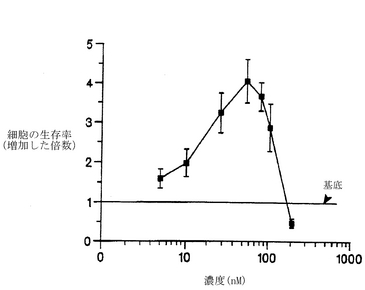
【図16】
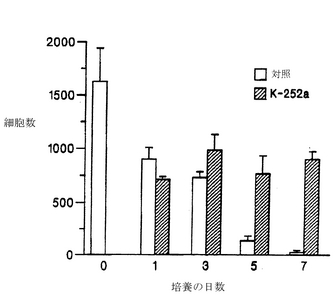
【図17】

【図18】
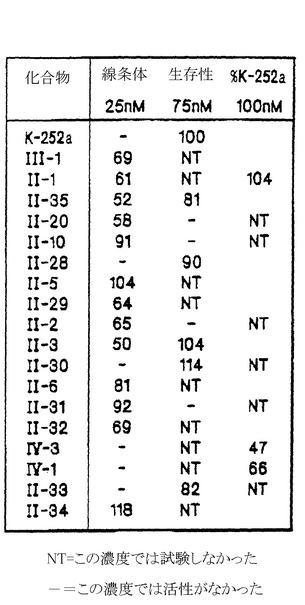
【図19】
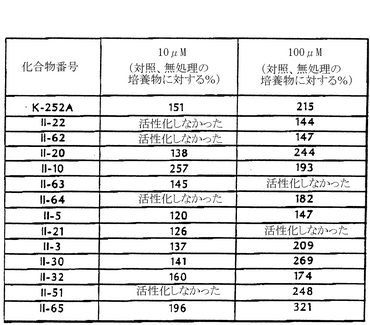
【図20】
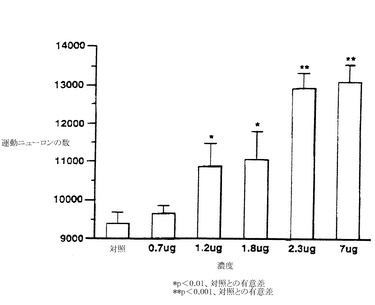
【図21】

【図22】
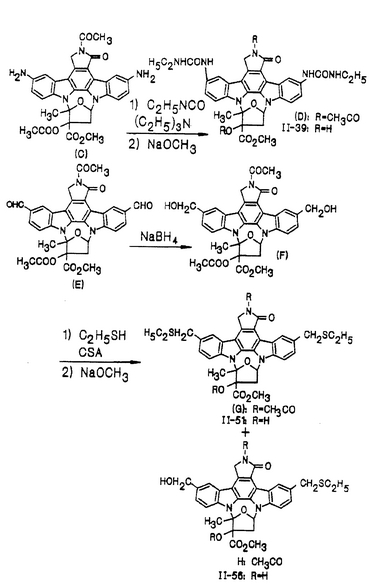
【図23】
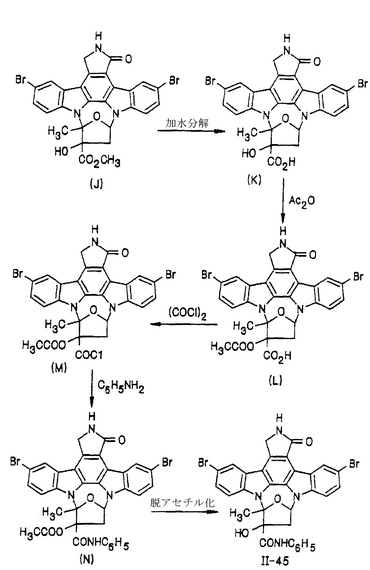
【図24】
【図25】
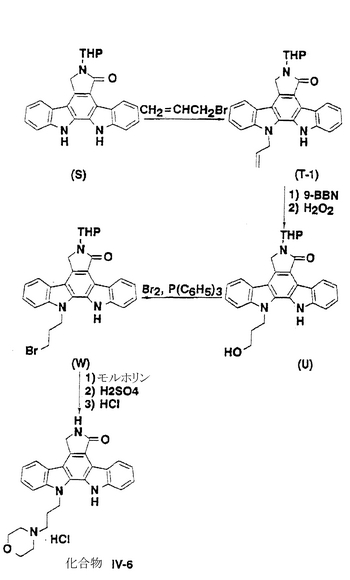
【図26】
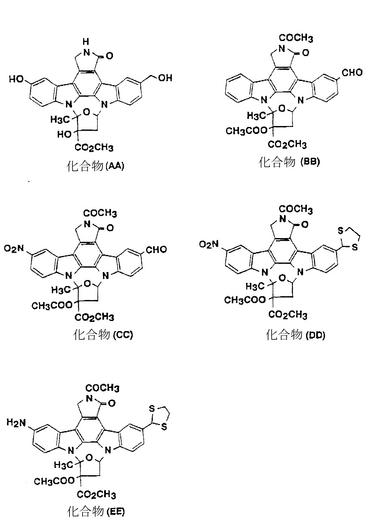
【図27】
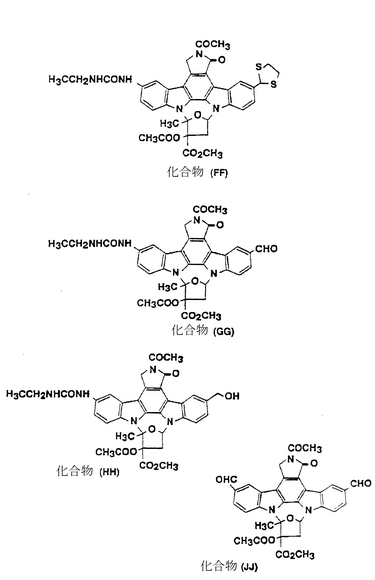
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13a】
【図13b】
【図13c】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2006−117690(P2006−117690A)
【公開日】平成18年5月11日(2006.5.11)
【国際特許分類】
【出願番号】特願2005−357071(P2005−357071)
【出願日】平成17年12月12日(2005.12.12)
【分割の表示】特願平8−514605の分割
【原出願日】平成7年10月4日(1995.10.4)
【出願人】(505191685)セファロン インク. (2)
【出願人】(000001029)協和醗酵工業株式会社 (276)
【Fターム(参考)】
【公開日】平成18年5月11日(2006.5.11)
【国際特許分類】
【出願日】平成17年12月12日(2005.12.12)
【分割の表示】特願平8−514605の分割
【原出願日】平成7年10月4日(1995.10.4)
【出願人】(505191685)セファロン インク. (2)
【出願人】(000001029)協和醗酵工業株式会社 (276)
【Fターム(参考)】
[ Back to top ]