
神経系を標的する薬理学的物質の性質を改善することに関連する方法および組成物
【課題】神経系を標的する生物活性化合物の薬理学的性質を改善することに関連する組成物および方法の提供。
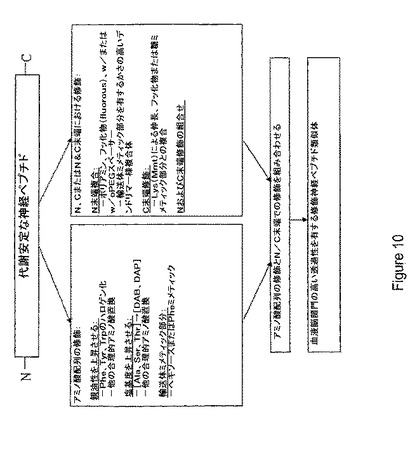
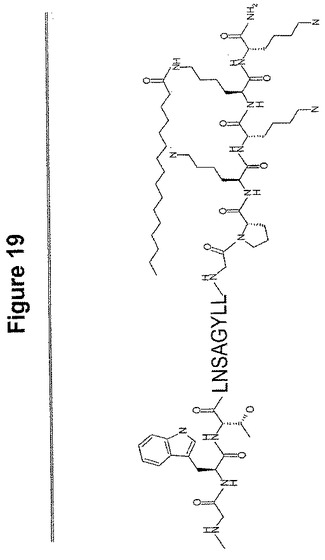
【解決手段】Sar−Trp−Thr−Leu−Asn−Ser−Ala−Gly−Tyr−Leu−Leu−Gly−Pro−Lys−Lys−(Lys−palm)−Lys−NH2の配列を有するポリペプチド及び少なくとも約90%同一のアミノ酸配列、あるいは1以上の保存的アミノ酸置換を有する前記アミノ酸配列を含む単離ポリペプチド。さらに、血液脳関門の高い透過性を有する組成物が開示され、これは、変化していない形態のペプチドと比較したとき高い親油特性と高い塩基度を有するペプチドを含む。
【解決手段】Sar−Trp−Thr−Leu−Asn−Ser−Ala−Gly−Tyr−Leu−Leu−Gly−Pro−Lys−Lys−(Lys−palm)−Lys−NH2の配列を有するポリペプチド及び少なくとも約90%同一のアミノ酸配列、あるいは1以上の保存的アミノ酸置換を有する前記アミノ酸配列を含む単離ポリペプチド。さらに、血液脳関門の高い透過性を有する組成物が開示され、これは、変化していない形態のペプチドと比較したとき高い親油特性と高い塩基度を有するペプチドを含む。
Notice: Undefined index: DEJ in /mnt/www/gzt_disp.php on line 298
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
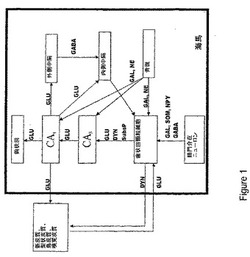
【図1】


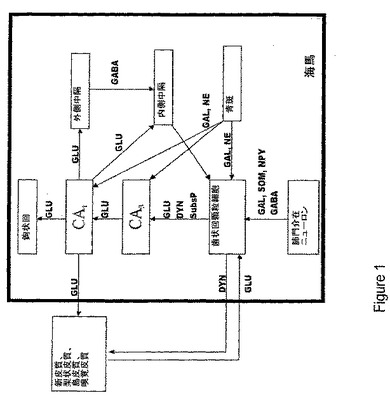
【図2】
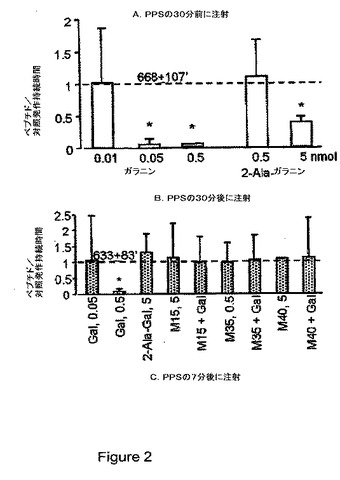
【図3】
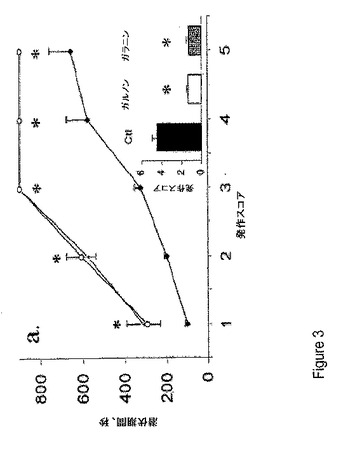
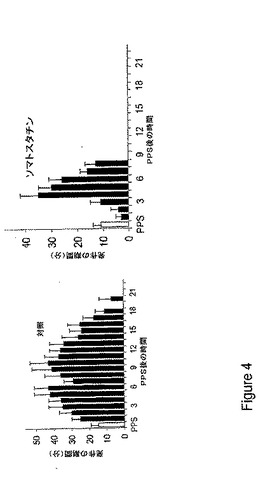
【図4】
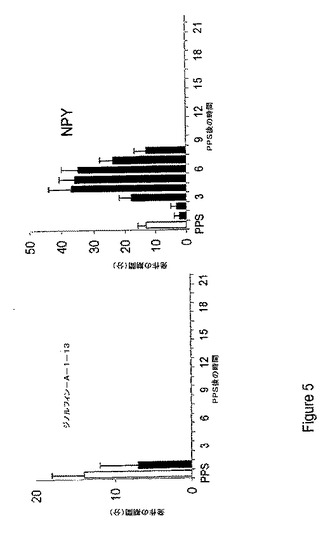
【図5】
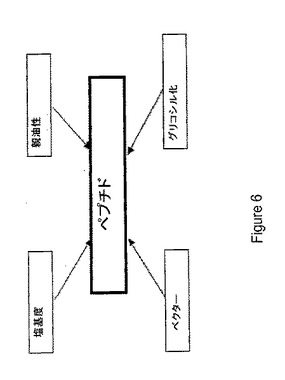
【図6】
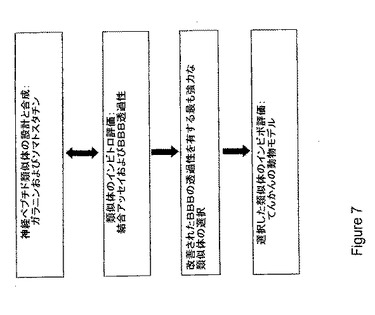
【図7】
【図8】
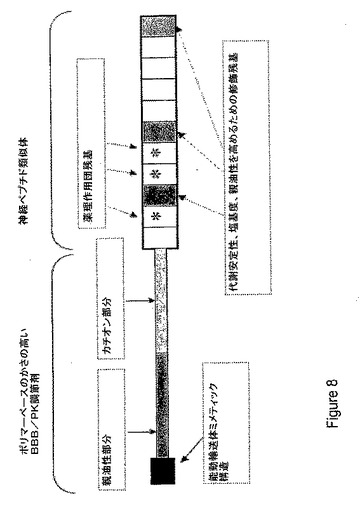
【図9】
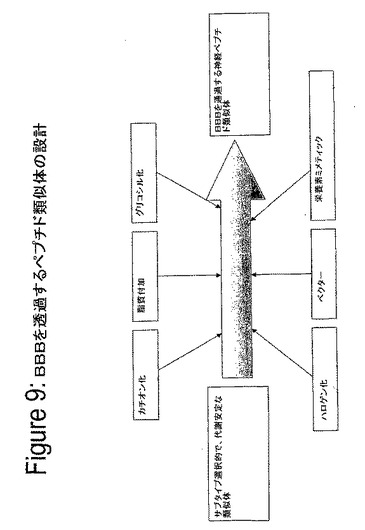
【図10】
【図11】
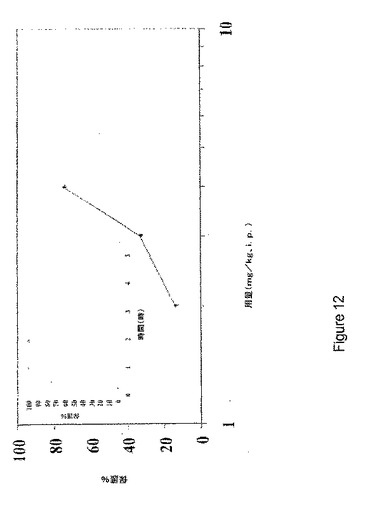
【図12】
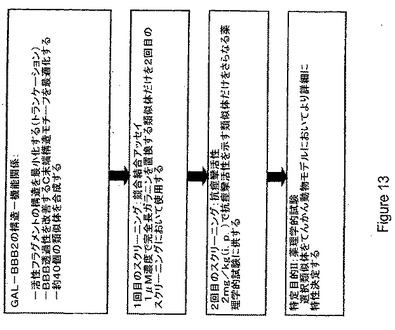
【図13】
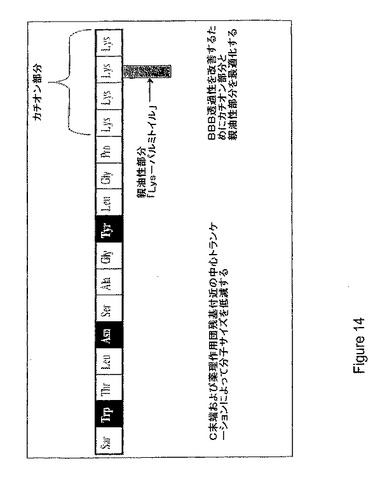
【図14】
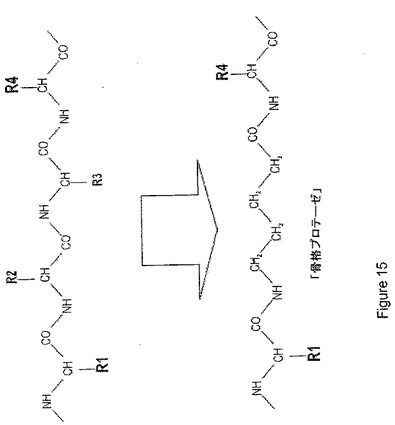
【図15】
【図16】
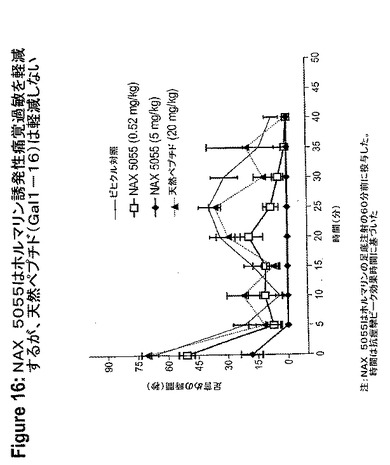
【図17】
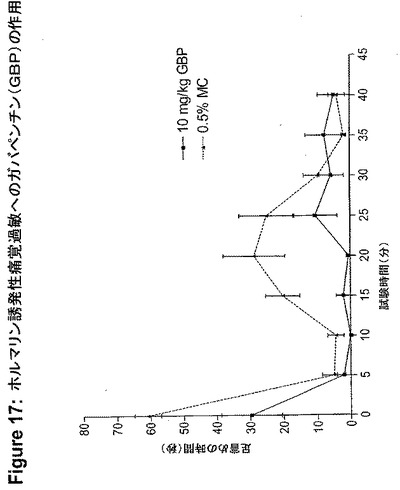
【図18】
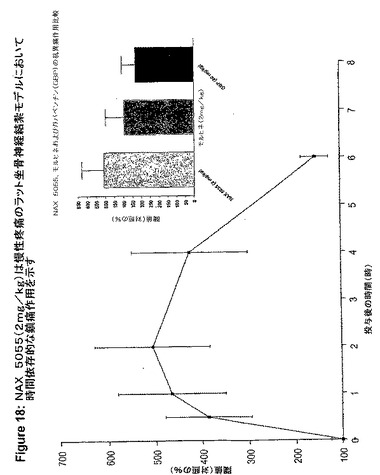
【図19】
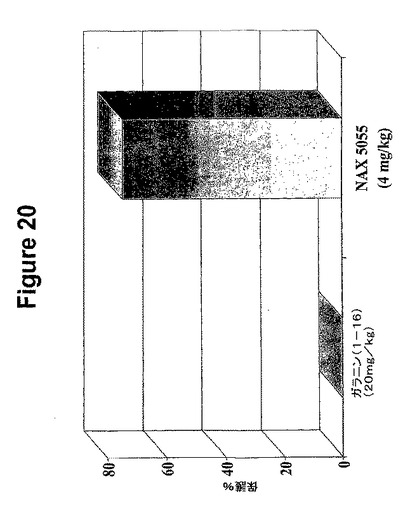
【図20】
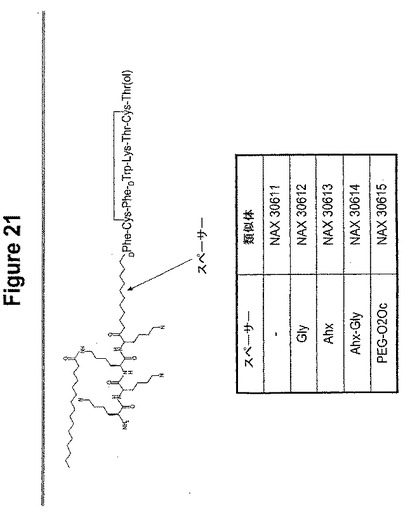
【図21】
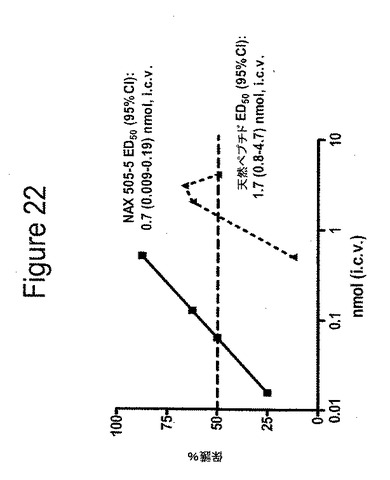
【図22】
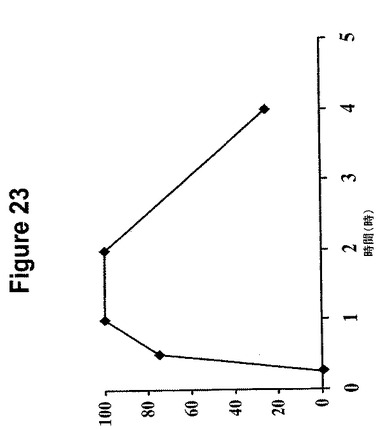
【図23】
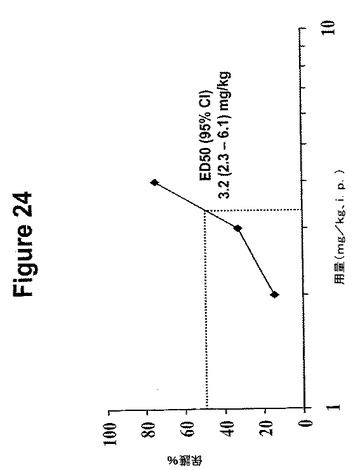
【図24】
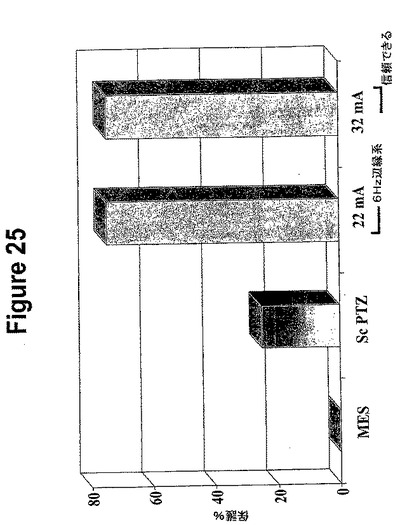
【図25】
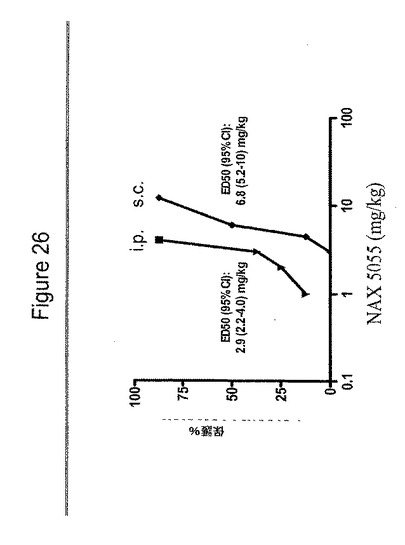
【図26】
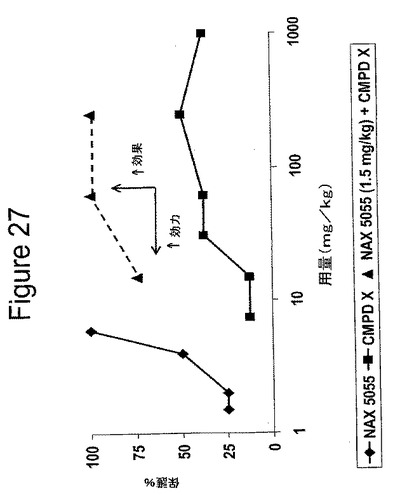
【図27】
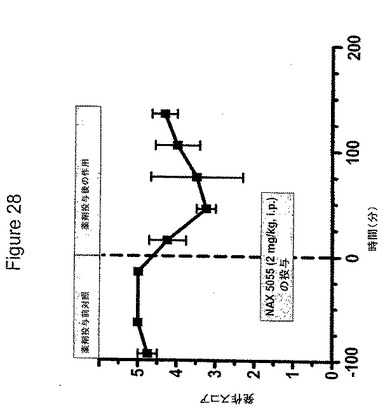
【図28】
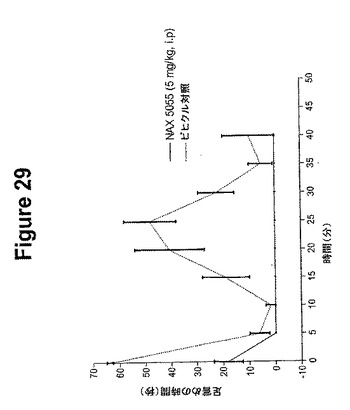
【図29】
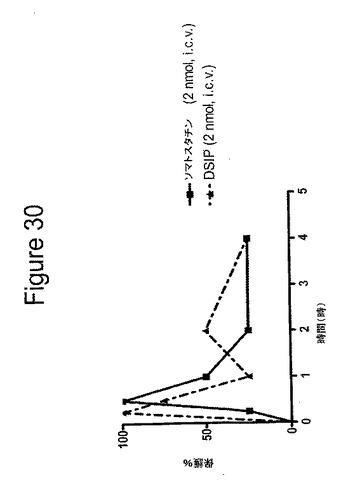
【図30】
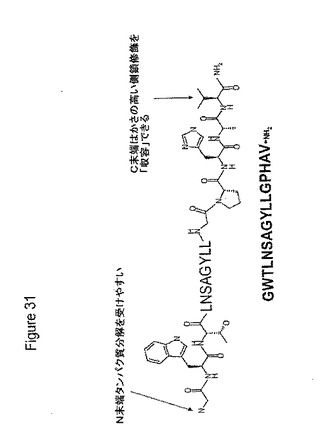
【図31】
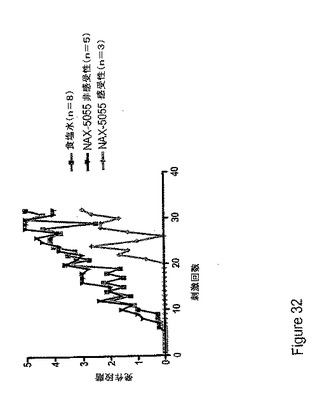
【図32】
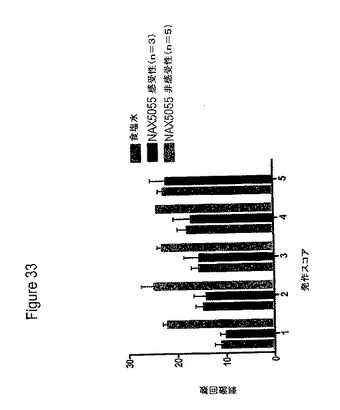
【図33】
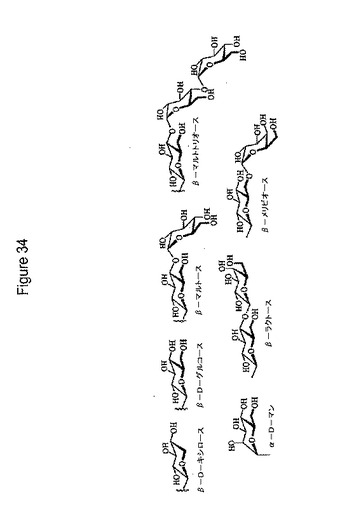
【図34】
【図35】

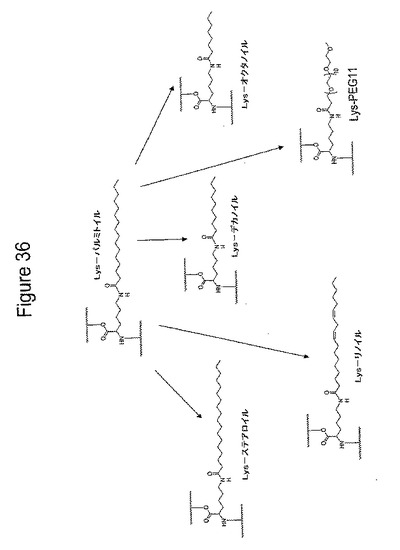
【図36】
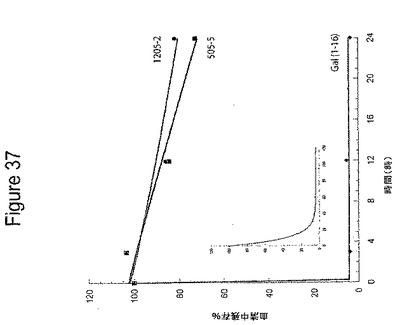
【図37】
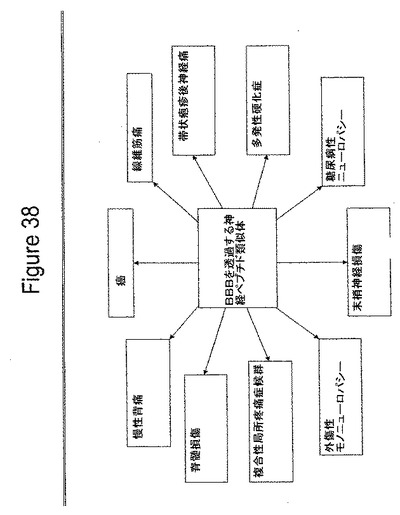
【図38】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【公開番号】特開2012−229241(P2012−229241A)
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−152861(P2012−152861)
【出願日】平成24年7月6日(2012.7.6)
【分割の表示】特願2008−549570(P2008−549570)の分割
【原出願日】平成19年1月5日(2007.1.5)
【出願人】(506051429)ユニバーシティ オブ ユタ リサーチ ファウンデーション (25)
【Fターム(参考)】
【公開日】平成24年11月22日(2012.11.22)
【国際特許分類】
【出願番号】特願2012−152861(P2012−152861)
【出願日】平成24年7月6日(2012.7.6)
【分割の表示】特願2008−549570(P2008−549570)の分割
【原出願日】平成19年1月5日(2007.1.5)
【出願人】(506051429)ユニバーシティ オブ ユタ リサーチ ファウンデーション (25)
【Fターム(参考)】
[ Back to top ]