
神経障害の治療のための新規カルバメート誘導体
本発明は、式Iの新規カルバメート誘導体、その調製方法、並びにたとえば、神経障害性の痛み及び不安症のような神経障害を治療するための、それらを含有する医薬組成物に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規カルバメート誘導体、その調製方法、及び神経障害性の痛み及び不安症のような神経障害の治療のための、それらを含有する医薬組成物に関する。
【背景技術】
【0002】
アナンダミド及びそのほかの脂肪酸アミドは、多数の生理的な過程を調節する化学的メッセンジャーであることが知られている(Hanus L.O., Chem. Biodivers., 2007, 4, 1828)。アナンダミドは、中枢型(CB1)及び末梢型(CB2)のカンナビノイド受容体を結合することによって活性化する(Devane W. A., et al., Science, 1992, 258, 1946-1949)。アナンダミドは、痛覚、摂食、嘔吐、不安、細胞増殖、炎症及び記憶の調節に関係するとみなされると報告されている(Labar G., et al., Chem. Biodivers., 2007, 4, 1882)。
【0003】
アナンダミドの薬理作用は、その作用部位で脂肪酸アミドを分解する、中枢神経系に分布する酵素である脂肪酸アミド加水分解酵素(FAAH)によって終了する(Cravatt B.F., et al., Nature, 1996, 384, 83; Patricelli M.P., et al., Biochemistry, 1999, 38, 9804; WO 98/20119 and U.S. Pat. No. 6,271,015))。リガンドを伴ったFAAHの複合体の結晶構造が解像され、それが、三連構造、Ser−Ser−Lysを介した触媒作用を発揮することを裏付けている(Bracey M.H., et al., Science, 2002, 298, 29, 1793)。
【0004】
FAAHはまた、多数のそのほかの脂質シグナル伝達性の脂肪酸アミド(すなわち、オレアミド、N−オレオイルエタノールアミン、アラキドニルグリセロール及びパルミトイルエタノールアミド)の異化作用にも関与する。内因性のシグナル伝達性脂質のレベルを回復することによってエンドカンナビノイド系の活性を調節することは、たとえば、最近親切に概説されたようなエネルギー代謝の疾患(悪液質及び拒食症)、疼痛及び炎症、中枢神経系の疾病(卒中、多発性硬化症、パーキンソン病、ハンチントン病、アルツハイマー病、癲癇、統合失調症、不安症、うつ病及び不眠症)、循環器及び呼吸器の疾病(高血圧症、循環性ショック、心筋再潅傷害、アテローム性硬化症及び喘息)、網膜症、癌、消化器及び肝臓の疾病(炎症性大腸疾患及び肝炎)、筋骨格疾病(関節炎及び骨粗鬆症)(Pasher P., et al., Pharmacol. Rev., 2006, 58, 389及びその中の文献)のような多種多様な異なった疾患及び病態において治療上の見込みを保持することが分かった。
【0005】
FAAH−/−のKOマウスはアナンダミドを代謝することができず、繁殖力があり、一般的には正常であるが、カンナビノイド受容体にてアナンダミド及び関連する脂肪酸アミドの上昇の徴候、たとえば、疼痛鎮静の低下を示す(Cravatt B.F., et al., Proc. Natl. Acad. Sci., 2001, 98, 9371)。これは、FAAHを標的とする薬剤がアナンダミドの緊張性作用を高める可能性がある一方で、Δ9−THC及びそのほかの直接作用するカンナビノイド作動薬によって生じる複数の望ましくないことが多い効果を多分回避することを示唆している(Hall W., et al., Lancet, 1998, 352, 1611; Chaperon, F., et al., Crit. Rev. Neurobiol., 1999, 13, 243)。
【0006】
特に、不可逆的なカルバメート系阻害剤であるURB−597は、不安症のゼロプラス迷路動物モデルで有効であると共に、ラットのホットプレート及びホルマリン試験で鎮痛効果を有することが報告された(Kathuria S., et al, Nat. Med., 2003, 9, 1, 76)。この化合物は、そのほかの誘導体の中で出願WO04033422の目的である。この出願が、構造的に異なる化合物の多数を広く主張する一般式を提示するとしても、それは、本発明の化合物のどれも開示していないし、示唆もしていない。実際、支持は主としてビフェニル誘導体に関して見つけることができる。URB−597は実際、活性を付与するのに決定的である、3D−QSARモデルを介して認識されるビフェニルの足場を置換することによって親油性ビフェニル誘導体URB−524の最適化を介して特定された(Tarzia G., et al., J. Med. Chem., 2003, 46, 12, 2352)。
【0007】
WO08013963は、一般式RXYの脂肪酸アミド加水分解酵素阻害剤を記載し、式中にはカルバメート誘導体が含まれている。しかしながら、最も強力な化合物は、ケト−オキサジアゾール誘導体であると思われ;最も強力なカルバメート付加体について報告された4μmの活性に関して最も強力なものは15nmの活性を示した。本発明の化合物のどれも上記出願では記載されていないし、示唆されていない。
【0008】
出願WO03051842は、式1
【化1】
の化合物を含有するホルモン感受性のリパーゼの活性を低下させる組成物に関するものであり、式中、R1はH、置換又は非置換のアルキル、アルケニル又はシクロアルキルであることができ;XはO又はSであることができ;R2はR1の相当するものを含む多種多様な意味を有し、Lは加水分解性基である。しかしながら、本発明の化合物のどれも上記出願では記載されていないし、示唆されていない。
【0009】
WO08129129は、一般式2を有する、新規のFAAH阻害剤として複素環カルバメートを開示したが、好ましい化合物は、以下のラジカル:メトキシカルボニル、オキサゾリル、テトラゾリル、チアジアゾリル、ベンゾキサゾールカルボニル及びベンゾチアゾールカルボニルから成る基の中に好ましくは含まれるR置換基を有する。しかしながら、主張されている生体内の活性及び/又は選択性特性はどの生物実験によっても支持されていないという事実に加えて、そのような化合物は本発明の化合物とは構造的に関連しない。
【化2】
【発明の概要】
【発明が解決しようとする課題】
【0010】
FAAHを阻害する可能性のある治療上の妥当性によって、選択的で且つ強力な阻害剤を開発することへの関心がモデル化されている。そのような戦略は、様々な効果を与えることが判明している外因性のカンナビノイドの使用に対してさらに安全な代替を代表する可能性がある。FAAHを阻害することは、CB1受容体を活性化する内因性のアミド化脂質のレベルを高める理想的な方法であると思われる。従って、強力で且つ選択的なFAAH阻害剤の要望が、興味深い及び有望な目標のままである。
【課題を解決するための手段】
【0011】
本発明は、脂肪酸アミド加水分解酵素(FAAH)を阻害するための新規化合物、そのような化合物を含む組成物、並びにFAAH阻害剤を患者に投与することによって、エネルギー代謝の疾病、疼痛及び炎症、中枢神経系の疾病、循環器及び呼吸器の疾病、網膜症、癌、消化器及び肝臓の疾病、筋骨格の疾病を治療する方法を提供する。
【0012】
本発明は、一般式I
【化3】
の化合物を含み、式中、
R1は、H、(C1〜C4)−アルキル、(C3〜C6)−シクロアルキル、アリールで置換される(C1〜C6)−アルキル又は(C2〜C5)−アルキニルであり、
Xは、C又はNであり、
Yは、CH、O又はSであり、
R2は、H又は(C1〜C4)−アルキルであり、
Eは、NR3R4又はOR5であり、
R3及びR4は、同一であり、又は異なっており、H、又は任意でアリールによって置換される(C2〜C6)−アルキルであり、
R5は、任意でアリール又は(C2〜C5)−アルキニルによって置換される(C2〜C6)−アルキルであり、
X及びYを含有する環は、ヘテロ芳香族環であり、
XがNである場合、YがCHであるという条件で
薬学上許容可能なその塩と同様に、その光学活性のある形態、たとえば、エナンチオマー、ジアステレオマー、及びそのラセミ形態を含む。
【0013】
本発明の実施形態は、薬物として使用するための式Iの化合物のものである。
【0014】
さらなる実施形態では、前記薬物は、神経障害、エネルギー代謝の疾病、循環器及び呼吸器の疾病、消化器及び肝臓の疾病、筋骨格の疾病を治療するために使用される。
【0015】
好ましい実施形態では、前記薬物は神経障害を治療するために使用される。
【0016】
さらに好ましい実施形態では、前記薬物は不安症及び疼痛を治療するために使用される。
【0017】
用語「アルキル」は、好ましくは1〜約12の炭素原子を有する直鎖又は分枝鎖のアルキル基を指す。低級アルキル基は、たとえば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、tert−ブチル、ペンチル、イソペンチル、ネオペンチル、n−ヘキシルなどによって例示される。
【0018】
用語「アルコキシ」は、−OR基を指し、Rには、低級アルキル、「C3〜C10シクロアルキル」及び「ヘテロシクロアルキル」が含まれる。
【0019】
用語「ヘテロシクロアルキル」及び/又は複素環は、1又は2の窒素原子、酸素原子又はイオウ原子を含有する飽和の5又は6員環を指す。好ましいヘテロシクロアルキルには、ピロリジン、ピペリジン、ピペラジン、モルフォリン、チオモルフォリンなどが挙げられる。
【0020】
用語「アリール」は、単一の環(たとえば、フェニル)を有する、又はペンダント方式で連結されてもよく若しくは縮合されてもよい複数の環を有する6〜14の炭素原子の芳香族炭素環式の基を指す。好ましいアリールには、フェニル、ナフチル、ビフェニル、インダンなどが挙げられる。
【0021】
用語「ヘテロアリール」は、単環式のヘテロ芳香族基、又は二環式の縮合環ヘテロ芳香族基を指す。ヘテロ芳香族基の特定の例には、任意で置換されるピリジル、ピロリル、フリル又はチエニルが挙げられる。
【0022】
用語「アミノカルボニル」は、−C(O)NRR’基を指し、R、R’には独立してH、「アルキル」、「アリール」又は「アリールアミノカルボニル」が挙げられる。
【0023】
「薬学上許容可能な塩」は、所望の生物活性を保持する、式(I)の以下で特定される化合物の塩を指す。そのような塩の例には、無機酸(たとえば、塩酸、臭化水素酸、硫酸、リン酸、硝酸など)で形成される酸付加塩、並びに、たとえば、酢酸、シュウ酸、酒石酸、コハク酸、リンゴ酸、フマル酸、マレイン酸、アスコルビン酸、安息香酸、タンニン酸、パモン酸、アルギン酸、ポリグルタミン酸、ナフタレンスルホン酸、トルエンスルホン酸、ナフタレンジスルホン酸、メタンスルホン酸及びポリガラクトウロン酸のような有機酸で形成される塩が挙げられるが、これらに限定されない。塩が一酸のものである(たとえば、塩酸塩、臭化水素酸塩、p−トルエンスルホン酸塩、又は酢酸塩)、二酸の水素形態である(たとえば、水素硫酸塩又はコハク酸塩)、又は三酸の二水素形態である(たとえば、二水素リン酸塩又はクエン酸塩)場合、少なくとも1モル当量及び普通、過剰モルの酸が用いられる。しかしながら、硫酸塩、ヘミコハク酸塩、水素リン酸塩、又はリン酸塩のような塩が所望である場合、適当で正確な化学当量の酸が一般に使用される。本発明の化合物に好適な薬学上許容される塩基付加塩には、アルミニウム、カルシウム、リチウム、マグネシウム、カリウム、ナトリウム及び亜鉛から作製される金属塩、又はリジン、N,N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、メグルミン(N−メチルグルカミン)及びプロカインから作製される有機塩が挙げられる。ナトリウム塩が特に好ましい。
【0024】
本発明の実施形態は、前述の式(I)の化合物のものであり、式中、Eは、NR3R4であり、R3及びR4はHである。
【0025】
本発明の別の実施形態は、前述の式(I)の化合物のものであり、式中、R1はアリールで置換された(C1〜C6)−アルキル、又はC2〜C5)−アルキニルである。
【0026】
本発明の化合物は、従来の合成法によって調製することができ、以下で記載される。典型的な又は好ましい実験条件(すなわち、反応温度、時間、試薬のモル、溶媒など)が与えられる場合、特に言及されない限り、ほかの実験条件も使用することができることが十分に理解されるであろう。
【0027】
本発明はさらに、NEt3のような塩基の存在下で極性溶媒において、式II
【化4】
の化合物(式中、X、Y、E及びR2は上記と同義である)を式III:
R1−N=C=O(式III)の化合物と反応させることによって得ることができる式Iの化合物の調製方法を提供する。
【0028】
前記変換のすべてでは、任意の干渉反応性基を保護することができ、次いで有機化学で記載され(たとえば、Greene T.W., Wuts P.G.M., “Protective Groups in Organic Synthesis”, J. Wiley & Sons, Inc., 3rd Ed., 1999を参照)、当業者に周知の揺るぎない手順に従って脱保護することができる。
【0029】
前記変換のすべては、有機化学で記載され(たとえば、March J., “Advanced Organic Chemistry”, J. Wiley & Sons, Inc., 4th Ed., 1992を参照)、当業者に周知の揺るぎない手順の例にすぎない。
【0030】
我々は、本発明に従って調製される誘導体(I)及び薬学上許容可能なその塩が、脂肪酸アミド加水分解酵素が介在する疾患状態、疾病及び病態の治療、特に不安症及び疼痛を治療するのに有用な剤であることを見い出した。
【0031】
従って、本発明の別の目的は、治療上有効な量の上述のような式(I)の化合物を投与することを含む、脂肪酸アミド加水分解酵素が介在する疾患状態、疾病及び病態、特に不安症及び疼痛に罹っている哺乳類を治療する方法である。
【0032】
用語「治療上有効な量」は、本明細書で使用されるとき、標的とされる疾患若しくは症状を治療する、改善する、又は検出可能な治療効果を示すのに必要とされる治療剤の量を指す。
【0033】
任意の化合物について、治療上有効な用量は、最初に、細胞培養又は、普通、マウス、ラット、モルモット、ウサギ、イヌ若しくはブタである動物モデルのいずれかにて推定することができる。動物モデルを用いて、適当な濃度範囲及び投与経路を決定してもよい。次いでそのような情報を用いて、ヒトにおける投与の有用な用量及び経路を決定してもよい。ヒト相当用量(HED)を算出することにおいて、産業と評論家の文書への指針(2002, U.S. Food and Drug Administration, Rockville, Maryland, USA)で提供されている変換表を使用することが推奨される。
【0034】
医薬組成物は、十分な治療効果を生じるような量にて有効成分として少なくとも1つの式(I)の化合物を含有する。本発明に含められる組成物は、全体として従来のものであり、たとえば、RemingtonのPharmaceutical Science Handbook,Mack Pub.N.Y.の最新版で説明されるもののような製薬業界で常識である方法によって得られる。選択される投与経路に従って、組成物は、経口、非経口又は局所の投与に好適な固体又は液体の形態である。本発明に係る組成物は、有効成分と共に、少なくとも1つの薬学上許容可能なビヒクル又は賦形剤を含有する。これらは、実践上有用な製剤化の共補助剤、たとえば、可溶化剤、分散剤、懸濁剤及び乳化剤であってもよい。
【0035】
実際に投与される化合物の量は、治療される症状、選択される投与経路、投与される実際の化合物、薬剤の併用、個々の患者の年齢、体重及び応答、患者の症状の重症度などを含む関連する状況の観点から通常、医師によって決定される。一般に、有効な用量は、0.01mg/kg〜100mg/kg、好ましくは0.05mg/kg〜50mg/kgである。任意の化合物について、治療上有効な用量は、最初に、細胞培養又は、普通、マウス、ラット、モルモット、ウサギ、イヌ若しくはブタである動物モデルのいずれかにて推定することができる。ヒト対象についての正確な有効用量は、疾患の状態の重症度、対象の全身状態、対象の年齢、体重及び性別、食事、投与の時間と回数、薬剤の併用、反応の感受性、及び治療への認容性/応答に左右される。この量は、日常の実験で決定することができ、臨床医の判断の範囲内である。
【0036】
組成物は、個別に患者に投与されてもよく、又はほかの剤、薬剤又はホルモンとの併用で投与されてもよい。
【0037】
薬物は、治療剤の投与のために薬学上許容可能なキャリアも含有してもよい。キャリアがそれ自体、組成物を受け取る個体に有害な抗体の産生を誘導しないという条件で、そのようなキャリアには、抗体及びそのほかのポリペプチド、遺伝子及びたとえば、リポソームのようなそのほかの治療剤が挙げられ、それは過度の毒性なしで投与されてもよい。
【0038】
好適なキャリアは、たとえば、タンパク質、多糖類、ポリ乳酸、ポリグリコール酸、高分子アミノ酸、アミノ酸コポリマー及び不活性ウイルス粒子のような大きな、ゆっくり代謝される高分子であってもよい。
【0039】
薬学上許容可能なキャリアの十分な議論は、RemingtonのPharmaceutical Sciences(Mack Pub.Co.,N.J.1991)で利用できる。
【0040】
治療用組成物における薬学上許容可能なキャリアは、さらに、水、生理食塩水、グリセロール及びエタノールのような液体を含有してもよい。
【0041】
さらに、たとえば、湿潤剤又は乳化剤、pH緩衝物質などのような補助物質がそのような組成物に存在してもよい。そのようなキャリアは、医薬組成物が、患者による摂取のために錠剤、丸薬、糖衣錠、カプセル、液剤、ジェル、シロップ、スラリー、懸濁液、などとして製剤化されることを可能にする。
【0042】
いったん製剤化されると、本発明の組成物は対象に直接投与することができる。治療される対象は動物であることができ、特にヒト対象を治療することができる。
【0043】
本発明の薬物は、経口の、静脈内の、筋肉内の、動脈内の、髄内の、クモ膜下の、脳室内の、経皮又は経皮の適用、皮下の、腹腔内の、鼻内の、腸内の、局所の、舌下の、膣内の、又は直腸の手段を含むが、これらに限定されないかなり多くの投与経路によって投与されてもよい。
【0044】
経口投与用の組成物は、バルク液体の溶液若しくは懸濁液、又はバルク粉末の形態を取ってもよい。しかしながら、さらに一般的には、組成物は、正確な投与を円滑にするように単位投与形態で提示される。用語「単位投与形態」は、ヒト対象及びそのほかの哺乳類にとって単位としての投与量として好適な物理的に別々の単位を指し、各単位は、好適な医薬賦形剤を伴って、所望の治療効果を生じるように算出された所定の量の活性物質を含有する。典型的な単位投与形態には、液体組成物の補充され、再測定されたアンプル若しくはシリンジ、又は固体組成物の場合における丸薬、錠剤、カプセルなどが挙げられる。そのような組成物では、本発明の化合物は普通、量的に少ない成分(約0.1〜約50重量%、又は好ましくは約1〜約40重量%)であり、残りは、種々のビヒクル又はキャリア及び所望の投与形態を形成するのに役立つ加工助剤である。調剤治療は、単回投与予定であってもよく又は複数回投与予定であってもよい。
【0045】
本発明の目的は、賦形剤及び/又は薬学上許容可能な希釈剤と組み合わせて、前述の式(I)の化合物を1以上含有する医薬組成物である。
【0046】
当該組成物は、式(I)の化合物と一緒に、既知の有効成分を含有してもよい。
【0047】
本発明のさらなる目的は、1以上の式(I)の化合物を好適な賦形剤、安定剤及び/又は薬学上許容可能な希釈剤と混合することを特徴とする医薬組成物の調製方法である。
【図面の簡単な説明】
【0048】
【図1】50mg/kgの用量での参照化合物と共に、30mg/kgの用量での選択性FAAH阻害剤、ST4068、ST3911及びST3913の鎮痛効果を示す図である。
【図2】10、30、及び100mg/kgの用量での選択性FAAH阻害剤、ST4068の鎮痛効果の用量反応を示す図である。
【図3】10、30、及び100mg/kgの用量での選択性FAAH阻害剤、ST3911の鎮痛効果の用量反応を示す図である。
【図4】10、30、及び100mg/kgの用量での選択性FAAH阻害剤、ST3913の鎮痛効果の用量反応を示す図である。
【発明を実施するための形態】
【0049】
略記
AA:アラキドン酸
AcOEt:酢酸エチル
AnNH:アラキドノイルエタノールアミド(アナンダミド)
bd:広い二重項
bs:広い一重項
CH2Cl2:ジクロロメタン
DMSO:ジメチルスルホキシド
Et2O:ジエチルエーテル
NaH:水素化ナトリウム
NaOH:水酸化ナトリウム
NEt3:トリエチルアミン
NH4OH:水酸化アンモニウム
RP−HPLC:逆相高速液体クロマトグラフィ
RT:室温
SOCl2:塩化チオニル
THF:テトラヒドロフラン
【0050】
一般的備考:1Hのスペクトルは、Bruker機器によって300MHzにて示したようなCDCl3溶液で記録した。化学シフト値はppmで、カップリング定数はHzで表す。フラッシュカラムクロマトグラフィは、シリカゲル(Merck230〜400メッシュ)を用いて実施した。
実施例1:ブチル−カルバミン酸3(3−カルバモイル−ピロール−1−イル)フェニルエステル、ST3910
【0051】
工程1:1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボアルデヒド
m−アミノフェノール(588.0mg、5.39ミリモル)の酢酸(10.0mL)と水(2.0mL)の溶液に酢酸(1.0mL)中のジメトキシテトラヒドロフランカルボキシアルデヒド(950.0mg、5.93ミリモル)を一滴ずつ加えた。溶液を100℃に15分間加熱した。溶媒を除去した後、暗茶色の反応混合物をAcOEtで希釈し、炭酸ナトリウムの飽和水溶液で中和した。後者を酢酸エチル(100mLで3回)抽出し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(1:1のn−ヘキサン/AcOEt)によって黄色固形物として純粋な生成物を得た(収率42%)。
1H−NMR(アセトン−d6)δ:6.70(s,1H),6.87(d,1H,J=7.8Hz),7.08−7.12(m,2H),7.35−7.37(m,2H),8.00(s,1H),8.84(s,1H),9.84(s,1H)
ESI−MSm/z[M−H]+186。
【0052】
工程2:1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸
3.0mLのメタノールと3.0mLの6NのNaOHにおける1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボアルデヒド(255.0mg、1.36ミリモル)の溶液に硝酸銀(370.0mg、2.18ミリモル)を加えた。次いで反応混合物を還流にて1時間撹拌した。冷却した後、溶媒を取り除いた。残留物をAcOEtによって抽出した。濃HClを用いて水性層をpH1に酸性化した。次いで後者をAcOEt(20mLで3回)で抽出した。次いで有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮して純粋な白色固形物として生成物を得た(収率73%)。
1H−NMR(アセトン−d6)δ:6.67(dd,1H,J=1.8,3.0Hz),6.84(ddd,1H,J=7.8,2.1,0.9Hz),7.04−7.11(m,2H),7.25(dd,1H,J=2.1,3.0Hz),7.34(t,1H,J=8.4Hz),7.78(dd,1H,J=1.8,2.4Hz,),8.78(s,1H),10.49(brs,1H)
ESI−MSm/z[M−H]+202
【0053】
工程3:1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸アミド
室温にて1.5mLのSOCl2に1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸(180.0mg、0.87ミリモル)を少しずつ加えた。反応混合物を還流で30分間撹拌した。冷却した後、乾燥するまで蒸発させた。得られた残留物を3.0mLのTHFに溶解し、5.0mLの濃NH4OHを室温にて慎重に加えた。混合物を7時間撹拌し、次いでAcOEtによって抽出し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(95:5のCHCl3/MeOH)によって茶色の粘性油として100mgの純粋な生成物を得た(収率57%)。
1H−NMR(アセトン−d6)δ:6.47(brs,1H),6.66(brs,1H),6.76−6.84(m,2H),6.99−7.04(m,2H),7.20−7.31(m,2H),7.85(s,1H),9.40(brs,1H)。
【0054】
工程4:ブチル−カルバミン酸3(3−カルバモイル−ピロール−1−イル)フェニルエステル、ST3910
1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸アミド(40.0mg、0.20ミリモル)の2.0mLの無水THF溶液に、n−ブチルイソシアネート(90μL、0.79ミリモル)とNEt3(110μL、0.79ミリモル)を加えた。反応混合物を室温で16時間撹拌した。溶媒を取り除き、粗精製の反応混合物をフラッシュクロマトグラフィ(98/2のCHCl3/MeOH)によって精製して、38mgの所望の付加体を導いた(収率63%)。
1H−NMR(アセトン−d6)δ:0.94(t,3H,J=7.2Hz),1.34−1.47(m,2H),1.52−1.62(m,2H),3.22(q,2H,J=6.6Hz),6.35(brs,1H),6.74(s,1H),6.91(brs,2H),7.08(d,1H,J=7.8Hz),7.28−7.50(m,4H),7.85(s,1H)
13C−NMR(アセトン−d6)δ:13.4,19.9,32.0,40.9,110.6,114.0,116.6,119.7,120.2,121.8,122.7,130.5,140.9,152.9,154.3,165.6
【0055】
実施例2〜4は、適切なイソシアネート試薬を用いて、実施例1の工程4で記載された実験条件に従って得られた。
【0056】
実施例2:ウンデカ−10−イニル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ST3911
収率:78%
1H−NMR(CD3OD)δ:1.35−1.56(m,14H),2.13(s,1H),2.16(s,2H),3.18(t,2H,J=6.9Hz),6.72(s,1H),7.08(d,1H,J=7.2Hz),7.22−7.39(m,3H),7.48(t,1H,J=8.1Hz),7.80(s,1H)
13C−NMR(CD3OD)δ:17.8,26.7,28.5,28.6,29.0,29.2,29.37,29.5,40.9,68.2,83.9,110.2,114.1,117.0,119.8,120.5,121.0,122.3,130.4,140.9,152.6,155.5,168.5
【0057】
実施例3:シクロヘキシル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ST3912
収率:62%
1H−NMR(CD3OD)δ:1.19−1.39(m,5H),1.65(d,1H,J=12.6Hz),1.78(d,2H,J=11.7Hz),1.95(d,2H,J=10.8Hz),3.39−3.48(m,1H),6.72(s,1H),7.07(d,1H,J=7.8Hz),7.21−7.38(m,3H),7.47(t,1H,J=7.8Hz),7.80(s,1H)
13C−NMR(CD3OD)δ:25.0(2),25.4,32.8(2),50.5,110.2,114.2,116.9,119.8,120.5,121.0,122.3,130.4,140.8,152.5,154.7,168.5
【0058】
実施例4:(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ST3913
収率:60%
1H−NMR(CD3OD)δ:1.38−1.40(m,4H),1.53−1.66(m,4H),2.60(t,2H,J=15.0Hz),3.17(t,2H,J=13.8Hz),6.72(s,1H),7.05−7.37(m,9H),7.46(t,1H,J=8.4Hz),7.79(s,1H)
13C−NMR(CD3OD)δ:26.5,28.8,29.5,31.5,35.6,40.9,110.2,114.1,117.0,119.8,120.5,121.0,122.3,125.5,128.1(2),128.2(2),130.4,140.8,142.7,152.5,155.5,168.5
ESI−MSm/z[M+H]+406,[M+Na]+428,[M+K]+444,[2M+H]+811,[2M+Na]+833
実施例5:ブチル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル、ST4067
【0059】
工程1:2−(3−メトキシ−ベンゾイル)−4−オキソ−ペンタン酸エチルエステル
0℃にてNaH(36.0mg、1.49ミリモル)の8.0mL無水THF懸濁液にエチル3−(3−メトキシフェニル)−3−オキソプロパノエート(262mL、1.35ミリモル)を一滴ずつ加えた。混合した反応物をこの温度で30分間撹拌した。次いでクロロアセトン(119mL、1.49ミリモル)を0℃にて一滴ずつ加え、反応混合物を室温にて48時間撹拌した。1NのHCl(3.0mL)で反応を止めた。THFを蒸発させ、残留物をAcOEt(25mLで3回)で抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(3:1のn−ヘキサン/AcOEt)によってラセミ混合物として純粋な生成物を得た(収率:67%)
1H−NMR(CDCl3)δ:1.14(t,3H,J=6.9Hz),2.21(s,3H),3.06(m,2H),3.83(s,3H),4.10(q,2H,J=6.9Hz),4.85(t,1H,J=7.0Hz),7.09−7.13(m,1H),7.33−7.39(m,1H),7.51(s,1H),7.59(d,1H,J=7.8Hz)
【0060】
工程2:2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル
2−(3−メトキシ−ベンゾイル)−4−オキソ−ペンタン酸エチルエステル(735mg、2.64ミリモル)を8mLの純粋なEtOHに溶解した。濃HCl(0.73mL)を加え、反応混合物をMW照射(150W)(CEM Discovery Microwave System)のもとで維持し、10分間還流した(最高内部温度100℃)。反応混合物を冷却し、AcOEtで希釈し、重炭酸ナトリウムの飽和水溶液で中和し、AcOEt(50mLで3回)で抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(97:3のn−ヘキサン/AcOEt)によって無色の粘性油として純粋な生成物を得た(収率90%)。
1H−NMR(CDCl3)δ:1.32(t,3H,J=6.9Hz),2.34(d,3H,J=0.9Hz),3.84(s,3H),4.28(q,2H,J=7.2Hz),6.44(d,1H,J=1.2Hz),6.92(ddd,1H,J=0.6,2.1,8.1Hz),7.32(t,1H,J=7.8Hz),7.56−7.59(m,1H),7.62−7.64(m,1H)
13C−NMR(CDCl3)δ:13.5,14.5,55.5,60.6,109.2,113.5,115.0,115.2,120.7,129.2,131.4,151.21,155.8,159.5,163.9
ESI−MSm/z[M−OEt]+215,[M+Na]+283
【0061】
工程3:2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸
EtOH(3.0mL)と水(1.0mL)における2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル(42.0mg、0.16ミリモル)の溶液にNaOH(161.0mg、4.03ミリモル)を加え、室温にて反応混合物を7時間撹拌した。EtOHを取り除き、6NのHClで残留物をpH1に酸性化した。混合物をAcOEt(20mLで3回)で抽出した。有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。さらに精製することなく、得られた化合物(37.0mg、収率100%)を次の工程に用いた。
1H−NMR(CDCl3)δ:2.37(s,3H),3.86(s,3H),6.48(s,1H),6.95(dd,1H,J=7.8,3.9Hz,),7.34(t,1H,J=7.9Hz),7.54−7.59(m,2H)
ESI−MSm/z[M−H]−231
【0062】
工程4:2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸アミド
室温にて300μLのSOCl2に2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸(68.0mg、0.29ミリモル)を少しずつ加えた。反応混合物を還流で30分間撹拌し、冷却した後、乾燥するまで蒸発させた。得られた残留物を3.0mLのTHFに溶解し、室温にて1.0mLの濃NH4OHを慎重に加えた。混合物を7時間撹拌し、AcOEtで抽出し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(95:5のCHCl3/MeOH)を介して粗精製の反応混合物を精製して茶色の粘性油として純粋な生成物を得た(65.0mg、収率97%)。
1H−NMR(CDCl3)δ:2.34(d,3H,J=0.6Hz),3.83(s,3H),5.79(brs,1H),5.90(brs,1H),6.33(d,1H,J=0.9Hz),6.92(dt,1H,J=3.0,7.5Hz),7.26−7.38(m,3H)
【0063】
工程5:2−(3−ヒドロキシ−フェニル)−5−メチル−フラン−3−カルボン酸アミド
2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸アミド(188.0mg、0.81ミリモル)の無水CH2Cl2(6.0mL)懸濁液にBBr3(ジクロロメタン中の1M溶液、2.4mL、2.44ミリモル)を−78℃にて加えた。次いで反応混合物を室温に温め、8時間撹拌した。炭酸ナトリウムの飽和水溶液を加えて反応を止めた。残留物をAcOEt(20mLで3回)で抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮して、茶色っぽい固形物として純粋な生成物を得た(175.0mg、収率98%)。
1H−NMR(アセトン−d6)δ:2.33(d,3H,J=0.9Hz),6.42(d,1H,J=0.9Hz),6.56(brs,1H),6.81(ddd,1H,J=0.9,2.7,4.8Hz),6.94(brs,1H),7.22(t,1H,J=7.8Hz),7.45(dt,1H,J=7.8,1.5Hz),7.53(t,1H,J=1.8Hz),8.48(s,1H)
【0064】
工程6:ブチル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル
2−(3−ヒドロキシ−フェニル)−5−メチル−フラン−3−カルボン酸アミド(36.0mg、0.17ミリモル)の3.0mL無水THF溶液にn−ブチルイソシアネート(80μL、0.75ミリモル)とNEt3(100μL、0.75ミリモル)を加えた。反応混合物を室温にて16時間撹拌した。溶媒を取り除き、カラムクロマトグラフィ(99:1のCHCl3/MeOH)による精製によって無色の油として純粋な生成物を得た(40.0mg、収率77%)。
1H−NMR(CDCl3)δ:0.96(t,3H,J=7.5Hz),1.36−1.43(m,2H),1.50−1.57(m,2H),2.35(d,3H,J=0.9Hz),3.15−3.21(m,2H),6.38(d,1H,J=0.9Hz),7.07(ddd,1H,J=0.9,2.4,4.8Hz),7.38(t,1H,J=7.8Hz),7.63(t,1H,J=1.8Hz),7.70(dt,1H,J=1.5,5.4Hz)
13C−NMR(CDCl3)δ:12.0,12.9,19.8,31.7,40.5,107.9,118.2,120.2,121.6,123.7,129.0,131.6,151.4,151.5,151.9,155.9,168.0
【0065】
実施例6は、n−ブチルイソシアネートの代わりにフェニルヘキシルイソシアネートを用いて実施例5の工程6に記載された実験条件に従って合成された。
【0066】
実施例6:(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、ST4068
収率:71%
1H−NMR(CDCl3)δ:1.38(brs,4H),1.55−1.66(m,4H),2.33(s,3H),2.61(t,2H,J=7.5Hz),3.24(q,2H,J=6.6Hz),5.10(brs,1H),5.61(brs,1H),5.82(brs,1H),6.32(s,1H),7.11−7.19(m,4H),7.25−7.30(m,2H),7.39(d,1H,J=7.8Hz),7.58(s,1H),7.64(d,1H,J=7.8Hz)
13C−NMR(CDCl3)δ:13.6,26.8,29.1,29.9,31.6,36.1,41.5,108.5,118.1,121.2,122.3,124.7,125.9,128.5(2),128.6(2),129.8,131.3,142.8,151.3,151.5,152.1,154.6,165.9
実施例7:(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステル、ST4069
【0067】
工程1:2−(3−メトキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸エチルエステル
2−(3−メトキシ−ベンゾイル)−4−オキソペンタン酸エチルエステル(500.0mg、1.8ミリモル)の5.0mLトルエン溶液にLawessonの試薬(2.00g)を加えた。還流のもと(最高内部温度100℃)でマイクロ波照射下(150W)(CEM Discovery Microwave System)で10分間反応を続けた。冷却した後、セライトを介して混合物を濾過し、減圧下で溶媒を除去した。フラッシュカラムクロマトグラフィ(99:1のn−ヘキサン/酢酸エチル)によって無色の油として純粋な生成物を得た(240.mg、収率48%)。
1H−NMR(CDCl3)δ:1.18(t,3H,J=7.2Hz),2.47(d,3H,J=0.9Hz),3.82(s,3H),4.17(q,2H,J=6.9Hz),6.90(ddd,1H,J=0.9,2.7,8.4Hz),7.00−7.05(m,2H),7.15(d,1H,J=0.9Hz),7.28(t,1H,J=7.8Hz)
ESI−MSm/z[M−OEt]+231,[M+Na]+299
【0068】
工程2:2−(3−メトキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸
実施例5の工程3で記載された実験条件に従って化合物を得た。
収率:97%。精製することなくその化合物を次の工程に用いた。
1H−NMR(CDCl3)δ:2.46(d,3H,J=0.6Hz),3.81(s,3H),6.92(dd,1H,J=2.4,8.4Hz),7.03−7.07(m,2H),7.19(d,1H,J=0.9Hz),7.28(t,1H,J=8.1Hz)
【0069】
工程3:2−(3−メトキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸アミド
実施例5の工程4で記載された実験条件に従って化合物を得た。
収率:78%
1H−NMR(アセトン−d6)δ:2.46(d,3H,J=0.6Hz),3.82(s,3H),6.50(brs,2H),6.93(dd,1H,J=1.8,8.4Hz),6.99(d,1H,J=1.2Hz),7.06−7.11(m,2H),7.32(t,1H,J=8.1Hz)
【0070】
工程4:2−(3−ヒドロキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸アミド
実施例5の工程5で記載された実験条件に従って化合物を得た。
収率:95%
1H−NMR(アセトン−d6)δ:2.45(d,3H,J=0.9Hz),6.49(brs,2H),6.84(ddd,1H,J=0.9,2.4,8.1Hz),6.96−7.00(m,3H),7.22(t,1H,J=7.5Hz),8.55(s,1H)
【0071】
工程5:(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステル
2−(3−ヒドロキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸アミドをフェニルヘキシルイソシアネートと反応させて、実施例5の工程6で記載された実験条件に従って化合物を得た。
収率:80%
1H−NMR(CDCl3)δ:1.36−1.40(m,4H),1.54−1.69(m,4H),2.45(d,3H,J=1.2Hz),2.61(t,2H,J=7.5Hz),3.24(q,2H,J=6.9Hz),5.13(t,1H,J=5.4Hz),5.53(brs,2H),7.12−7.19(m,5H),7.25−7.32(m,4H),7.39(t,1H,J=7.8Hz)
13C−NMR(CDCl3)δ:15.3,26.8,29.1,29.9,31.6,36.1,41.5,122.3,123.1,125.9,126.6,127.9,128.5(2),128.6(2),129.9,132.6,134.3,139.5,141.7,142.8,151.4,154.5,166.0
実施例8:シクロヘキシル−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル、ST4104
【0072】
工程1:2−[2−(3−メトキシ−フェニル)−2−オキソ−エチル]−3−オキソ−酪酸エチルエステル
エチル3−(3−メトキシフェニル)−3−オキソプロパノエートとクロロアセトンの代わりにエチルアセトアセテートと2−ブロモ−1−(3−メトキシフェニル)エタノンを用いて、実施例5の工程1で記載された実験条件に従って化合物を得た。
収率:79%
1H−NMR(CDCl3)δ:1.29(t,3H,J=7.2Hz),2.45(s,3H),3.51(dd,1H,J=18.6,5.7Hz),3.71(dd,1H,J=18.6,8.3Hz),3.85(s,3H),4.23(m,3H),7.12(m,1H),7.37(m,1H),7.47(m,1H),7.58(m,1H);ESI−MSm/z[M+H]+279,[M+Na]+301(100)
【0073】
工程2:5−(3−メトキシ−フェニル)−2−メチル−フラン−3−カルボン酸エチルエステル
実施例5の工程2で記載された実験条件に従って化合物を得た。
収率:72%
1H−NMR(CDCl3)δ:1.35(t,3H,J=7.2Hz),2.62(s,3H),3.81(s,3H),4.29(q,2H,J=7.2Hz),6.80(m,1H),6.86(s,1H),7.15−7.28(m,3H)
13C−NMR(CDCl3)δ:14.1,14.6,55.4,60.4,106.0,109.1,113.6,115.6,116.4,129.9,131.5,151.74,158.8,160.1,164.1
【0074】
工程3:5−(3−メトキシ−フェニル)−2−メチル−フラン−3−カルボン酸
実施例5の工程3で記載された実験条件に従って化合物を得た。
収率:98%。精製することなくその化合物を次の工程に用いた。
1H−NMR(CDCl3)δ:2.69(s,3H),3.86(s,3H),6.84(m,1H),6.92(s,1H),7.18−7.33(m,3H),10.83(brs,1H)
【0075】
工程4:5−(3−メトキシ−フェニル)−2−メチル−フラン−3−カルボン酸アミド
実施例1の工程3で記載された実験条件に従って化合物を得た。
収率:74%
1H−NMR(CDCl3)δ:2.67(s,3H),3.85(s,3H),5.68(brs,2H),6.67(s,1H),6.83(m,1H),7.16(m,1H),7.21(m,1H),7.26−7.32(m,1H)
ESI−MSm/z[M+Na]+254(100),[M+K]+270,[2M+Na]+485
【0076】
工程5:5−(3−ヒドロキシ−フェニル)−2−メチル−フラン−3−カルボン酸アミド
実施例5の工程5で記載された実験条件に従って化合物を得た。
収率:74%
1H−NMR(CD3OD)δ:2.58(s,3H),6.70(m,1H),6.92(s,1H),7.09(m,2H),7.17(m,1H);ESI−MSm/z[M+Na]+240(100)
【0077】
工程6:シクロヘキシル−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル、ST4104
5−(3−メトキシ−フェニル)−2−メチル−フラン−3−カルボン酸アミド(45.0mg、0.21ミリモル)の2.0mL無水THF溶液にシクロヘキシルイソシアネート(106μL、0.83ミリモル)とNEt3(116μL、0.83ミリモル)を加えた。室温にて反応混合物を16時間撹拌した。溶媒を除去し、カラムクロマトグラフィ(9:1のCHCl3/MeOH)によって無色の固形物として純粋な生成物を得た(収率:34%)。
1H−NMR(CD3OD)δ:1.19−1.39(m,5H),1.65(m,1H),1.79(m,2H),1.96(m,2H),2.61(s,3H),3.38(m,1H),7.02(m,2H),7.39(m,2H),7.50(m,1H)
13C−NMR(CD3OD)δ:12.5,24.9,25.4,32.8,50.5,104.9,116.7,117.3,120.1,120.8,129.6,131.5,150.9,152.0,155.0,157.2,167.3
ESI−MSm/z[M+Na]+365,[2M+Na]+707
【0078】
実施例9:(6−フェニル−ヘキシル)−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル、ST4105
シクロヘキシルイソシアネートの代わりにフェニルヘキシルイソシアネートを用いて(Mor M., et al., J. Med. Chem., 2008, 51, 12, 3487)、実施例8の工程5で記載された実験条件に従って化合物を得た。
収率:40%
1H−NMR(CD3OD)δ:1.37(m,4H),1.52−1.63(m,4H),2.58(m,5H),3.16(t,2H,J=6.9Hz),6.99(m,2H),7.12(m,3H),7.22(m,2H),7.35(m,2H),7.46(m,1H).
13C−NMR(CD3OD)δ:12.6,26.5,28.8,29.5,31.5,35.7,40.9,104.9,116.7,117.3,120.1,120.8,125.5,128.1,128.2,129.7,131.6,142.7,150.9,152.0,155.8,157.2,167.3.
ESI−MSm/z[M+Na]+443,[M+K]+459,[2M+Na]+863
実施例10:2−(3−ブチルカルバモイルオキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル、ST4110
【0079】
工程1:2−(3−ヒドロキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル
実施例5の工程5で記載された実験条件に従って化合物を得た。
収率:60%
1H−NMR(CD3OD)δ:1.30(t,3H,J=7.5Hz),2.33(s,3H),4.24(q,2H,J=7.2Hz),6.43(s,1H),6.80(dd,1H,J=1.8,6.9Hz),7.21(t,1H,J=8.1Hz),7.36(t,2H,J=7.2Hz)
【0080】
工程2:2−(3−ブチルカルバモイルオキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル
実施例5の工程6で記載された実験条件に従って化合物を得た。
収率:81%
1H−NMR(CDCl3)δ:0.94(t,3H,J=6.9Hz),1.29−1.40(m,5H),1.49−1.59(m,2H),2.32(s,3H),3.25(q,2H,J=6.6Hz),4.28(q,2H,J=6.6Hz),5.12(brs,1H),6.42(s,1H),7.14(d,1H,J=7.8Hz),7.37(t,1H,J=8.4Hz),7.77(s,1H),7.84(d,1H,J=7.5Hz)
13C−NMR(CDCl3)δ:13.5,13.9,14.4,20.1,32.1,41.2,60.7,109.2,115.2,121.4,122.4,125.1,129.0,131.4,151.1,151.5,154.7,154.9,163.9
ESI−MSm/z[M+Na]+368,[2M+Na]+713
実施例11:1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸−ウンデカ−10−イニルエステル、ST4112
【0081】
工程1:1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸−ウンデカ−10−イニルエステル
1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸(100.0mg、0.49ミリモル)と10−ウンデシン−1−オール(90mL、0.49ミリモル)の2.0mL無水THFの撹拌された溶液に、0℃にてトリフェニルホスフィン(129mg、0.49ミリモル)とDIAD(90mL、0.49ミリモル)を一滴ずつ加えた。室温にて反応混合物を48時間撹拌した。減圧下で溶媒を取り除いた。残留物に重炭酸ナトリウムの飽和水溶液を加え、得られた混合物をAcOEt(25mLで3回)で抽出し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。フラッシュクロマトグラフィ(1:9のAcOEt/n−ヘキサン)によって精製を行い、無色の固形物として120mgの所望の生成物(収率69%)を得た。
1H−NMR(CDCl3)δ:1.32−1.55(m,12H),1.70−1.75(m,2H),1.93(t,1H,J=2.7Hz),2.17(dd,2H,J=2.7,7.2Hz),4.25(t,2H,J=6.9Hz),5.52(brs,1H),6.74(q,1H,J=1.8Hz),6.79(dd,1HJ=2.1,8.1Hz),6.92(t,1H,J=2.1Hz),6.95−7.00(m,2H),7.30(t,1H,J=8.1Hz),7.66(dd,1H,J=1.5,2.1Hz).
【0082】
工程2:1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸−ウンデカ−10−イニルエステル
1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸−ウンデカ−10−イニルエステルから出発して実施例5の工程6で記載された実験条件に従って表題の化合物を得た。
1H−NMR(CDCl3)δ:0.94(t,3H,J=7.2Hz),1.30−1.74(m,18H),1.93(s,1H),2.16(dd,2H,J=4.5,6.3Hz),3.26(q,2H,J=6.3Hz),4.22(t,2H,J=6.3Hz),5.22(brs,1H),6.73(s,1H),6.99(s,1H),7.07(d,1H,J=8.1Hz),7.19−7.26(m,2H),7.39(t,1H,J=8.1Hz),7.65(s,1H)
13C−NMR(CDCl3)δ:14.0,18.6,20.1,26.2,28.7,28.9,29.1,29.2,29.5,29.6,32.1,41.3,64.3,68.3,85.0,111.9,114.8,117.7,118.6,120.2,120.7,124.5,130.5,140.8,152.2,154.3,165.0
ESI−MSm/z[M+H]+453,M+Na]+475,[M+K]+491
実施例12:1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸6−フェニルヘキシルエステル、ST4114
【0083】
工程1:1−(3−ヒドロキシ−フェニル)−H−ピロール−カルボン酸6−フェニル−ヘキシルエステル
10−ウンデシン−1−オールの代わりに6−フェニル−1−ヘキサノールを用いて、実施例11の工程1で記載された実験条件に従って表題の化合物を得た。
収率:63%
1H−NMR(CDCl3)δ:1.38−1.48(m,4H),1.49−1.76(m,4H),2.62(t,2H,J=6.9Hz),4.25(t,2H,J=6.9Hz),6.73−6.99(m,6H),7.16−7.30(m,6H),7.67−7.68(m,1H).
【0084】
工程2:1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸6−フェニルヘキシルエステル
1−(3−ヒドロキシ−フェニル)−H−ピロール−カルボン酸6−フェニル−ヘキシルエステルから出発して、実施例11の工程2で記載された実験条件に従って表題の化合物を得た。
収率:95%
1H−NMR(CDCl3)δ:0.94(t,3H,J=7.2Hz),1.34−1.81(m,11H),2.62(t,3H,J=7.2Hz),3.27(q,2H,J=6.6Hz),4.24(t,2H,J=6.6Hz),5.21(t,1H,J=5.4Hz),6.75(s,1H),6.99−7.43(m,10H),7.67(s,1H)
13C−NMR(CDCl3)δ:14.0,20.1,26.2,29.0,29.2,31.6,32.1,36.1,41.3,64.3,112.0,114.9,117.7,118.6,120.2,120.8,124.5,125.8,128.5(2),128.6(2),130.6,140.8,142.9,152.2,154.3,165.0
ESI−MSm/z[M+H]+461,[M+Na]+485,[M+K]+501
【0085】
実施例13:シクロヘキシル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、ST4049
n−ブチルイソシアネートの代わりにシクロヘキシルイソシアネートを用いて、実施例5の工程6で記載された実験条件に従って実施例6を合成した。
収率:79%
1H−NMR(DMSO−d6)δ:1.13−1.28(m,5H),1.52−1.59(m,1H),1.67−1.70(m,2H),1.79−1.83(m,2H),2.31(s,3H),3.28−3.30(m,1H),6.48(s,1H),7.04(dd,1H,J1=0.9Hz,J2=7.5Hz),7.27(bs,1H),7.37(t,1H,J=8.1Hz),7.63−7.74(m,4H)
13C−NMR(DMSO−d6)δ:13.79,25.24,25.81,33.22,50.44,109.28,119.29,120.57,122.26,123.75,129.73,131.82,150.74,151.46,151.59,154.05,165.69
【0086】
実施例14:シクロヘキシル−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステル、ST4050
2−(3−ヒドロキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸アミドをシクロヘキシルイソシアネートと反応させて、実施例5の工程6で記載された実験条件に従って化合物を得た。
収率:72%
1H−NMR(CD3OD)δ:1.22−1.39(m,5H),1.64(bd,1H,J=12.6Hz),1.78(bd,2H,J=12.0Hz),1.94(bd,2H,J=10.5Hz),2.47(s,3H),3.38−3.48(m,1H),6.97(m,1H),7.09(d,1H,J=8.1Hz),7.21(s,1H),7.30−7.41(m,2H)
13C−NMR(CD3OD)δ:13.71,24.98,25.42,32.80,50.48,121.46,122.06,125.57,126.79,129.30,132.89,134.67,139.75,141.39,151.5,155.03,167.13
ESI−MSm/z[M+Na]+381,[2M+Na]+740
【0087】
調製1:11−イソシアナト−ウンデカ−1−イン
ウンデカ−10−イニルアミン(Crisp G.T., et al., Tetrahedron, 1997, 53, 4, 1505, 514 mg)を33mLのCH2Cl2に溶解した。溶液を0℃に冷却し、重炭酸ナトリウムの飽和水溶液(30mL)を加えた。二相混合物を0℃で10分間撹拌し、層を分離し、シリンジを介して、ホスゲンの溶液(トルエン中20%、3.23mL)を有機層に直接加えた。15分後、水性層をCH2Cl2で抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。さらに精製することなく化合物を使用した。
収率:94%
1H−NMR(CDCl3)δ:1.27(m,4H),1.51(m,4H),2.51(t,2H,J=7.7Hz),3.14(t,2H,J=6.6Hz),7.07(m,3H),7.17(m,2H)
【0088】
生物学的結果
FAAHアッセイ
本発明の化合物は、親和性を示し、脂肪酸アミド加水分解酵素の活性を阻害する。
【0089】
RP−HPLCを用いて、[1−14C]AnNH(52mCi/ミリモル)からの[14C]AAの放出を測定することによってFAAH(EC3.5.1.4)のアッセイを行った。また、[1−14C]AnNHについて以下で記載される同様の実験条件下で[3H]AnNH(205Ci/ミリモル)を基質として用いて[3H]AAの放出を測定することができた。種々の濃度での本発明の化合物を、2mLのエッペンドルフ管における200μLの加水分解酵素アッセイ緩衝液(50mMトリス−HCl、pH9.0)に加え、20分後、10μMの濃度までの[1−14C]AnNH2を加えた。マウスの脳ホモジネート(40μg)を添加することによって反応を開始し、37℃にて15分間インキュベートした後、撹拌しながら、800μLの氷冷メタノール/クロロホルム(2:1、v/v)の添加によって反応を止めた。この混合物を室温で30分間放置し、次いで、撹拌しながら、240μLのクロロホルムと240μLの水を加えた。室温にて10分後、混合物を3000gにて5分間遠心し、上部の水性層を吸引によって取り除き、DNA MINIスピードバック(Heto−Holten、Denmark)にて試料を30℃、100ミリバールで30分間遠心することによって下部の有機層を乾燥させた。残留物を50μLのメタノールに溶解し、以下に詳述するように、AAの定量用のRP−HPLCに供した。FAAHに特異的な活性は、放出されたピコモルAA/分/タンパク質のmgとして表した。0〜12μMの濃度範囲にて、[1−14C]AnNH、[1−14C]ODNHEtOH又は[1−14C]ODNH2を用いて、Lineweaver−Burkの解析によって動態試験を行った。線形回帰プログラム(Kaleidagraph 3.0)による実験点のフィッティングによってr値>0.97の直線が得られた。
【0090】
【表1】
【0091】
選択性特性
Maccarrone,M.ら、J.Biol.Chem.,2000,275,13484,Fezza、F.ら、Anal Biochem.,2005,339,113,Dinh、T.Pら、2002,Proc.Natl.Acad.Sci.99,10819,Bisogno、T.ら、2003,J.Cell Biol.,163,463,Maccarrone、M.ら、J.Biol.Chem.,2000,275,31938,Ross、R.A.ら、Br.J.Pharmacol.,2001,132,631に記載された手順に従って、以下の標的:AMT、NAPE−PLD、MAGL、DAGL、CB1/CB2及びTRPV1に対する選択性特性に関して本発明の化合物の評価も行った。結果を表2に示す。
【0092】
【表2】
【0093】
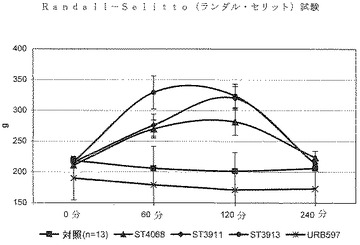
生体内鎮痛活性
足引っ込め試験を用いて機械的な痛覚過敏を評価した。Randall−Selitto(ランダル・セリット)の痛覚計(イタリア、VareseのUgo Basile)を用いて、左右の後肢に高い圧力を適用することによってグラムで表現する傷害受容の閾値を測定した。傷害受容の閾値を定量するのに使用したパラメータは、ラットがその足を引っ込める圧力(グラム)として定義した。試験手順と実験に先立つ週での研究者による取り扱いにラットを慣らせた。30mg/kgの用量でのST4068、ST3911及びST3913による急性の経口処理は、十分な鎮痛活性を示した一方で、50mg/kg用量での周知の参照化合物URB597は活性を示さなかった(図1)。
【0094】
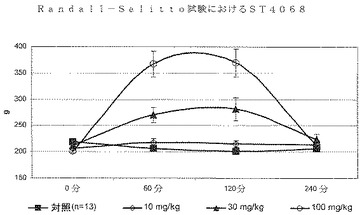
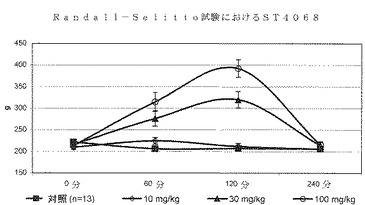
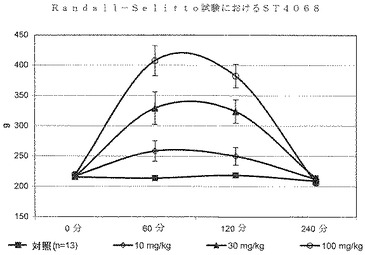
ST4068、ST3911及びST3913によって、10、30及び100mg/kgでの用量反応実験も行った。化合物はすべて30及び100mg/kgの用量計画で鎮痛活性を発揮し、後者の投与量はさらに高い効果を付与した(図2、3及び4)。
【技術分野】
【0001】
本発明は、新規カルバメート誘導体、その調製方法、及び神経障害性の痛み及び不安症のような神経障害の治療のための、それらを含有する医薬組成物に関する。
【背景技術】
【0002】
アナンダミド及びそのほかの脂肪酸アミドは、多数の生理的な過程を調節する化学的メッセンジャーであることが知られている(Hanus L.O., Chem. Biodivers., 2007, 4, 1828)。アナンダミドは、中枢型(CB1)及び末梢型(CB2)のカンナビノイド受容体を結合することによって活性化する(Devane W. A., et al., Science, 1992, 258, 1946-1949)。アナンダミドは、痛覚、摂食、嘔吐、不安、細胞増殖、炎症及び記憶の調節に関係するとみなされると報告されている(Labar G., et al., Chem. Biodivers., 2007, 4, 1882)。
【0003】
アナンダミドの薬理作用は、その作用部位で脂肪酸アミドを分解する、中枢神経系に分布する酵素である脂肪酸アミド加水分解酵素(FAAH)によって終了する(Cravatt B.F., et al., Nature, 1996, 384, 83; Patricelli M.P., et al., Biochemistry, 1999, 38, 9804; WO 98/20119 and U.S. Pat. No. 6,271,015))。リガンドを伴ったFAAHの複合体の結晶構造が解像され、それが、三連構造、Ser−Ser−Lysを介した触媒作用を発揮することを裏付けている(Bracey M.H., et al., Science, 2002, 298, 29, 1793)。
【0004】
FAAHはまた、多数のそのほかの脂質シグナル伝達性の脂肪酸アミド(すなわち、オレアミド、N−オレオイルエタノールアミン、アラキドニルグリセロール及びパルミトイルエタノールアミド)の異化作用にも関与する。内因性のシグナル伝達性脂質のレベルを回復することによってエンドカンナビノイド系の活性を調節することは、たとえば、最近親切に概説されたようなエネルギー代謝の疾患(悪液質及び拒食症)、疼痛及び炎症、中枢神経系の疾病(卒中、多発性硬化症、パーキンソン病、ハンチントン病、アルツハイマー病、癲癇、統合失調症、不安症、うつ病及び不眠症)、循環器及び呼吸器の疾病(高血圧症、循環性ショック、心筋再潅傷害、アテローム性硬化症及び喘息)、網膜症、癌、消化器及び肝臓の疾病(炎症性大腸疾患及び肝炎)、筋骨格疾病(関節炎及び骨粗鬆症)(Pasher P., et al., Pharmacol. Rev., 2006, 58, 389及びその中の文献)のような多種多様な異なった疾患及び病態において治療上の見込みを保持することが分かった。
【0005】
FAAH−/−のKOマウスはアナンダミドを代謝することができず、繁殖力があり、一般的には正常であるが、カンナビノイド受容体にてアナンダミド及び関連する脂肪酸アミドの上昇の徴候、たとえば、疼痛鎮静の低下を示す(Cravatt B.F., et al., Proc. Natl. Acad. Sci., 2001, 98, 9371)。これは、FAAHを標的とする薬剤がアナンダミドの緊張性作用を高める可能性がある一方で、Δ9−THC及びそのほかの直接作用するカンナビノイド作動薬によって生じる複数の望ましくないことが多い効果を多分回避することを示唆している(Hall W., et al., Lancet, 1998, 352, 1611; Chaperon, F., et al., Crit. Rev. Neurobiol., 1999, 13, 243)。
【0006】
特に、不可逆的なカルバメート系阻害剤であるURB−597は、不安症のゼロプラス迷路動物モデルで有効であると共に、ラットのホットプレート及びホルマリン試験で鎮痛効果を有することが報告された(Kathuria S., et al, Nat. Med., 2003, 9, 1, 76)。この化合物は、そのほかの誘導体の中で出願WO04033422の目的である。この出願が、構造的に異なる化合物の多数を広く主張する一般式を提示するとしても、それは、本発明の化合物のどれも開示していないし、示唆もしていない。実際、支持は主としてビフェニル誘導体に関して見つけることができる。URB−597は実際、活性を付与するのに決定的である、3D−QSARモデルを介して認識されるビフェニルの足場を置換することによって親油性ビフェニル誘導体URB−524の最適化を介して特定された(Tarzia G., et al., J. Med. Chem., 2003, 46, 12, 2352)。
【0007】
WO08013963は、一般式RXYの脂肪酸アミド加水分解酵素阻害剤を記載し、式中にはカルバメート誘導体が含まれている。しかしながら、最も強力な化合物は、ケト−オキサジアゾール誘導体であると思われ;最も強力なカルバメート付加体について報告された4μmの活性に関して最も強力なものは15nmの活性を示した。本発明の化合物のどれも上記出願では記載されていないし、示唆されていない。
【0008】
出願WO03051842は、式1
【化1】
の化合物を含有するホルモン感受性のリパーゼの活性を低下させる組成物に関するものであり、式中、R1はH、置換又は非置換のアルキル、アルケニル又はシクロアルキルであることができ;XはO又はSであることができ;R2はR1の相当するものを含む多種多様な意味を有し、Lは加水分解性基である。しかしながら、本発明の化合物のどれも上記出願では記載されていないし、示唆されていない。
【0009】
WO08129129は、一般式2を有する、新規のFAAH阻害剤として複素環カルバメートを開示したが、好ましい化合物は、以下のラジカル:メトキシカルボニル、オキサゾリル、テトラゾリル、チアジアゾリル、ベンゾキサゾールカルボニル及びベンゾチアゾールカルボニルから成る基の中に好ましくは含まれるR置換基を有する。しかしながら、主張されている生体内の活性及び/又は選択性特性はどの生物実験によっても支持されていないという事実に加えて、そのような化合物は本発明の化合物とは構造的に関連しない。
【化2】
【発明の概要】
【発明が解決しようとする課題】
【0010】
FAAHを阻害する可能性のある治療上の妥当性によって、選択的で且つ強力な阻害剤を開発することへの関心がモデル化されている。そのような戦略は、様々な効果を与えることが判明している外因性のカンナビノイドの使用に対してさらに安全な代替を代表する可能性がある。FAAHを阻害することは、CB1受容体を活性化する内因性のアミド化脂質のレベルを高める理想的な方法であると思われる。従って、強力で且つ選択的なFAAH阻害剤の要望が、興味深い及び有望な目標のままである。
【課題を解決するための手段】
【0011】
本発明は、脂肪酸アミド加水分解酵素(FAAH)を阻害するための新規化合物、そのような化合物を含む組成物、並びにFAAH阻害剤を患者に投与することによって、エネルギー代謝の疾病、疼痛及び炎症、中枢神経系の疾病、循環器及び呼吸器の疾病、網膜症、癌、消化器及び肝臓の疾病、筋骨格の疾病を治療する方法を提供する。
【0012】
本発明は、一般式I
【化3】
の化合物を含み、式中、
R1は、H、(C1〜C4)−アルキル、(C3〜C6)−シクロアルキル、アリールで置換される(C1〜C6)−アルキル又は(C2〜C5)−アルキニルであり、
Xは、C又はNであり、
Yは、CH、O又はSであり、
R2は、H又は(C1〜C4)−アルキルであり、
Eは、NR3R4又はOR5であり、
R3及びR4は、同一であり、又は異なっており、H、又は任意でアリールによって置換される(C2〜C6)−アルキルであり、
R5は、任意でアリール又は(C2〜C5)−アルキニルによって置換される(C2〜C6)−アルキルであり、
X及びYを含有する環は、ヘテロ芳香族環であり、
XがNである場合、YがCHであるという条件で
薬学上許容可能なその塩と同様に、その光学活性のある形態、たとえば、エナンチオマー、ジアステレオマー、及びそのラセミ形態を含む。
【0013】
本発明の実施形態は、薬物として使用するための式Iの化合物のものである。
【0014】
さらなる実施形態では、前記薬物は、神経障害、エネルギー代謝の疾病、循環器及び呼吸器の疾病、消化器及び肝臓の疾病、筋骨格の疾病を治療するために使用される。
【0015】
好ましい実施形態では、前記薬物は神経障害を治療するために使用される。
【0016】
さらに好ましい実施形態では、前記薬物は不安症及び疼痛を治療するために使用される。
【0017】
用語「アルキル」は、好ましくは1〜約12の炭素原子を有する直鎖又は分枝鎖のアルキル基を指す。低級アルキル基は、たとえば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、tert−ブチル、ペンチル、イソペンチル、ネオペンチル、n−ヘキシルなどによって例示される。
【0018】
用語「アルコキシ」は、−OR基を指し、Rには、低級アルキル、「C3〜C10シクロアルキル」及び「ヘテロシクロアルキル」が含まれる。
【0019】
用語「ヘテロシクロアルキル」及び/又は複素環は、1又は2の窒素原子、酸素原子又はイオウ原子を含有する飽和の5又は6員環を指す。好ましいヘテロシクロアルキルには、ピロリジン、ピペリジン、ピペラジン、モルフォリン、チオモルフォリンなどが挙げられる。
【0020】
用語「アリール」は、単一の環(たとえば、フェニル)を有する、又はペンダント方式で連結されてもよく若しくは縮合されてもよい複数の環を有する6〜14の炭素原子の芳香族炭素環式の基を指す。好ましいアリールには、フェニル、ナフチル、ビフェニル、インダンなどが挙げられる。
【0021】
用語「ヘテロアリール」は、単環式のヘテロ芳香族基、又は二環式の縮合環ヘテロ芳香族基を指す。ヘテロ芳香族基の特定の例には、任意で置換されるピリジル、ピロリル、フリル又はチエニルが挙げられる。
【0022】
用語「アミノカルボニル」は、−C(O)NRR’基を指し、R、R’には独立してH、「アルキル」、「アリール」又は「アリールアミノカルボニル」が挙げられる。
【0023】
「薬学上許容可能な塩」は、所望の生物活性を保持する、式(I)の以下で特定される化合物の塩を指す。そのような塩の例には、無機酸(たとえば、塩酸、臭化水素酸、硫酸、リン酸、硝酸など)で形成される酸付加塩、並びに、たとえば、酢酸、シュウ酸、酒石酸、コハク酸、リンゴ酸、フマル酸、マレイン酸、アスコルビン酸、安息香酸、タンニン酸、パモン酸、アルギン酸、ポリグルタミン酸、ナフタレンスルホン酸、トルエンスルホン酸、ナフタレンジスルホン酸、メタンスルホン酸及びポリガラクトウロン酸のような有機酸で形成される塩が挙げられるが、これらに限定されない。塩が一酸のものである(たとえば、塩酸塩、臭化水素酸塩、p−トルエンスルホン酸塩、又は酢酸塩)、二酸の水素形態である(たとえば、水素硫酸塩又はコハク酸塩)、又は三酸の二水素形態である(たとえば、二水素リン酸塩又はクエン酸塩)場合、少なくとも1モル当量及び普通、過剰モルの酸が用いられる。しかしながら、硫酸塩、ヘミコハク酸塩、水素リン酸塩、又はリン酸塩のような塩が所望である場合、適当で正確な化学当量の酸が一般に使用される。本発明の化合物に好適な薬学上許容される塩基付加塩には、アルミニウム、カルシウム、リチウム、マグネシウム、カリウム、ナトリウム及び亜鉛から作製される金属塩、又はリジン、N,N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、メグルミン(N−メチルグルカミン)及びプロカインから作製される有機塩が挙げられる。ナトリウム塩が特に好ましい。
【0024】
本発明の実施形態は、前述の式(I)の化合物のものであり、式中、Eは、NR3R4であり、R3及びR4はHである。
【0025】
本発明の別の実施形態は、前述の式(I)の化合物のものであり、式中、R1はアリールで置換された(C1〜C6)−アルキル、又はC2〜C5)−アルキニルである。
【0026】
本発明の化合物は、従来の合成法によって調製することができ、以下で記載される。典型的な又は好ましい実験条件(すなわち、反応温度、時間、試薬のモル、溶媒など)が与えられる場合、特に言及されない限り、ほかの実験条件も使用することができることが十分に理解されるであろう。
【0027】
本発明はさらに、NEt3のような塩基の存在下で極性溶媒において、式II
【化4】
の化合物(式中、X、Y、E及びR2は上記と同義である)を式III:
R1−N=C=O(式III)の化合物と反応させることによって得ることができる式Iの化合物の調製方法を提供する。
【0028】
前記変換のすべてでは、任意の干渉反応性基を保護することができ、次いで有機化学で記載され(たとえば、Greene T.W., Wuts P.G.M., “Protective Groups in Organic Synthesis”, J. Wiley & Sons, Inc., 3rd Ed., 1999を参照)、当業者に周知の揺るぎない手順に従って脱保護することができる。
【0029】
前記変換のすべては、有機化学で記載され(たとえば、March J., “Advanced Organic Chemistry”, J. Wiley & Sons, Inc., 4th Ed., 1992を参照)、当業者に周知の揺るぎない手順の例にすぎない。
【0030】
我々は、本発明に従って調製される誘導体(I)及び薬学上許容可能なその塩が、脂肪酸アミド加水分解酵素が介在する疾患状態、疾病及び病態の治療、特に不安症及び疼痛を治療するのに有用な剤であることを見い出した。
【0031】
従って、本発明の別の目的は、治療上有効な量の上述のような式(I)の化合物を投与することを含む、脂肪酸アミド加水分解酵素が介在する疾患状態、疾病及び病態、特に不安症及び疼痛に罹っている哺乳類を治療する方法である。
【0032】
用語「治療上有効な量」は、本明細書で使用されるとき、標的とされる疾患若しくは症状を治療する、改善する、又は検出可能な治療効果を示すのに必要とされる治療剤の量を指す。
【0033】
任意の化合物について、治療上有効な用量は、最初に、細胞培養又は、普通、マウス、ラット、モルモット、ウサギ、イヌ若しくはブタである動物モデルのいずれかにて推定することができる。動物モデルを用いて、適当な濃度範囲及び投与経路を決定してもよい。次いでそのような情報を用いて、ヒトにおける投与の有用な用量及び経路を決定してもよい。ヒト相当用量(HED)を算出することにおいて、産業と評論家の文書への指針(2002, U.S. Food and Drug Administration, Rockville, Maryland, USA)で提供されている変換表を使用することが推奨される。
【0034】
医薬組成物は、十分な治療効果を生じるような量にて有効成分として少なくとも1つの式(I)の化合物を含有する。本発明に含められる組成物は、全体として従来のものであり、たとえば、RemingtonのPharmaceutical Science Handbook,Mack Pub.N.Y.の最新版で説明されるもののような製薬業界で常識である方法によって得られる。選択される投与経路に従って、組成物は、経口、非経口又は局所の投与に好適な固体又は液体の形態である。本発明に係る組成物は、有効成分と共に、少なくとも1つの薬学上許容可能なビヒクル又は賦形剤を含有する。これらは、実践上有用な製剤化の共補助剤、たとえば、可溶化剤、分散剤、懸濁剤及び乳化剤であってもよい。
【0035】
実際に投与される化合物の量は、治療される症状、選択される投与経路、投与される実際の化合物、薬剤の併用、個々の患者の年齢、体重及び応答、患者の症状の重症度などを含む関連する状況の観点から通常、医師によって決定される。一般に、有効な用量は、0.01mg/kg〜100mg/kg、好ましくは0.05mg/kg〜50mg/kgである。任意の化合物について、治療上有効な用量は、最初に、細胞培養又は、普通、マウス、ラット、モルモット、ウサギ、イヌ若しくはブタである動物モデルのいずれかにて推定することができる。ヒト対象についての正確な有効用量は、疾患の状態の重症度、対象の全身状態、対象の年齢、体重及び性別、食事、投与の時間と回数、薬剤の併用、反応の感受性、及び治療への認容性/応答に左右される。この量は、日常の実験で決定することができ、臨床医の判断の範囲内である。
【0036】
組成物は、個別に患者に投与されてもよく、又はほかの剤、薬剤又はホルモンとの併用で投与されてもよい。
【0037】
薬物は、治療剤の投与のために薬学上許容可能なキャリアも含有してもよい。キャリアがそれ自体、組成物を受け取る個体に有害な抗体の産生を誘導しないという条件で、そのようなキャリアには、抗体及びそのほかのポリペプチド、遺伝子及びたとえば、リポソームのようなそのほかの治療剤が挙げられ、それは過度の毒性なしで投与されてもよい。
【0038】
好適なキャリアは、たとえば、タンパク質、多糖類、ポリ乳酸、ポリグリコール酸、高分子アミノ酸、アミノ酸コポリマー及び不活性ウイルス粒子のような大きな、ゆっくり代謝される高分子であってもよい。
【0039】
薬学上許容可能なキャリアの十分な議論は、RemingtonのPharmaceutical Sciences(Mack Pub.Co.,N.J.1991)で利用できる。
【0040】
治療用組成物における薬学上許容可能なキャリアは、さらに、水、生理食塩水、グリセロール及びエタノールのような液体を含有してもよい。
【0041】
さらに、たとえば、湿潤剤又は乳化剤、pH緩衝物質などのような補助物質がそのような組成物に存在してもよい。そのようなキャリアは、医薬組成物が、患者による摂取のために錠剤、丸薬、糖衣錠、カプセル、液剤、ジェル、シロップ、スラリー、懸濁液、などとして製剤化されることを可能にする。
【0042】
いったん製剤化されると、本発明の組成物は対象に直接投与することができる。治療される対象は動物であることができ、特にヒト対象を治療することができる。
【0043】
本発明の薬物は、経口の、静脈内の、筋肉内の、動脈内の、髄内の、クモ膜下の、脳室内の、経皮又は経皮の適用、皮下の、腹腔内の、鼻内の、腸内の、局所の、舌下の、膣内の、又は直腸の手段を含むが、これらに限定されないかなり多くの投与経路によって投与されてもよい。
【0044】
経口投与用の組成物は、バルク液体の溶液若しくは懸濁液、又はバルク粉末の形態を取ってもよい。しかしながら、さらに一般的には、組成物は、正確な投与を円滑にするように単位投与形態で提示される。用語「単位投与形態」は、ヒト対象及びそのほかの哺乳類にとって単位としての投与量として好適な物理的に別々の単位を指し、各単位は、好適な医薬賦形剤を伴って、所望の治療効果を生じるように算出された所定の量の活性物質を含有する。典型的な単位投与形態には、液体組成物の補充され、再測定されたアンプル若しくはシリンジ、又は固体組成物の場合における丸薬、錠剤、カプセルなどが挙げられる。そのような組成物では、本発明の化合物は普通、量的に少ない成分(約0.1〜約50重量%、又は好ましくは約1〜約40重量%)であり、残りは、種々のビヒクル又はキャリア及び所望の投与形態を形成するのに役立つ加工助剤である。調剤治療は、単回投与予定であってもよく又は複数回投与予定であってもよい。
【0045】
本発明の目的は、賦形剤及び/又は薬学上許容可能な希釈剤と組み合わせて、前述の式(I)の化合物を1以上含有する医薬組成物である。
【0046】
当該組成物は、式(I)の化合物と一緒に、既知の有効成分を含有してもよい。
【0047】
本発明のさらなる目的は、1以上の式(I)の化合物を好適な賦形剤、安定剤及び/又は薬学上許容可能な希釈剤と混合することを特徴とする医薬組成物の調製方法である。
【図面の簡単な説明】
【0048】
【図1】50mg/kgの用量での参照化合物と共に、30mg/kgの用量での選択性FAAH阻害剤、ST4068、ST3911及びST3913の鎮痛効果を示す図である。
【図2】10、30、及び100mg/kgの用量での選択性FAAH阻害剤、ST4068の鎮痛効果の用量反応を示す図である。
【図3】10、30、及び100mg/kgの用量での選択性FAAH阻害剤、ST3911の鎮痛効果の用量反応を示す図である。
【図4】10、30、及び100mg/kgの用量での選択性FAAH阻害剤、ST3913の鎮痛効果の用量反応を示す図である。
【発明を実施するための形態】
【0049】
略記
AA:アラキドン酸
AcOEt:酢酸エチル
AnNH:アラキドノイルエタノールアミド(アナンダミド)
bd:広い二重項
bs:広い一重項
CH2Cl2:ジクロロメタン
DMSO:ジメチルスルホキシド
Et2O:ジエチルエーテル
NaH:水素化ナトリウム
NaOH:水酸化ナトリウム
NEt3:トリエチルアミン
NH4OH:水酸化アンモニウム
RP−HPLC:逆相高速液体クロマトグラフィ
RT:室温
SOCl2:塩化チオニル
THF:テトラヒドロフラン
【0050】
一般的備考:1Hのスペクトルは、Bruker機器によって300MHzにて示したようなCDCl3溶液で記録した。化学シフト値はppmで、カップリング定数はHzで表す。フラッシュカラムクロマトグラフィは、シリカゲル(Merck230〜400メッシュ)を用いて実施した。
実施例1:ブチル−カルバミン酸3(3−カルバモイル−ピロール−1−イル)フェニルエステル、ST3910
【0051】
工程1:1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボアルデヒド
m−アミノフェノール(588.0mg、5.39ミリモル)の酢酸(10.0mL)と水(2.0mL)の溶液に酢酸(1.0mL)中のジメトキシテトラヒドロフランカルボキシアルデヒド(950.0mg、5.93ミリモル)を一滴ずつ加えた。溶液を100℃に15分間加熱した。溶媒を除去した後、暗茶色の反応混合物をAcOEtで希釈し、炭酸ナトリウムの飽和水溶液で中和した。後者を酢酸エチル(100mLで3回)抽出し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(1:1のn−ヘキサン/AcOEt)によって黄色固形物として純粋な生成物を得た(収率42%)。
1H−NMR(アセトン−d6)δ:6.70(s,1H),6.87(d,1H,J=7.8Hz),7.08−7.12(m,2H),7.35−7.37(m,2H),8.00(s,1H),8.84(s,1H),9.84(s,1H)
ESI−MSm/z[M−H]+186。
【0052】
工程2:1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸
3.0mLのメタノールと3.0mLの6NのNaOHにおける1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボアルデヒド(255.0mg、1.36ミリモル)の溶液に硝酸銀(370.0mg、2.18ミリモル)を加えた。次いで反応混合物を還流にて1時間撹拌した。冷却した後、溶媒を取り除いた。残留物をAcOEtによって抽出した。濃HClを用いて水性層をpH1に酸性化した。次いで後者をAcOEt(20mLで3回)で抽出した。次いで有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮して純粋な白色固形物として生成物を得た(収率73%)。
1H−NMR(アセトン−d6)δ:6.67(dd,1H,J=1.8,3.0Hz),6.84(ddd,1H,J=7.8,2.1,0.9Hz),7.04−7.11(m,2H),7.25(dd,1H,J=2.1,3.0Hz),7.34(t,1H,J=8.4Hz),7.78(dd,1H,J=1.8,2.4Hz,),8.78(s,1H),10.49(brs,1H)
ESI−MSm/z[M−H]+202
【0053】
工程3:1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸アミド
室温にて1.5mLのSOCl2に1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸(180.0mg、0.87ミリモル)を少しずつ加えた。反応混合物を還流で30分間撹拌した。冷却した後、乾燥するまで蒸発させた。得られた残留物を3.0mLのTHFに溶解し、5.0mLの濃NH4OHを室温にて慎重に加えた。混合物を7時間撹拌し、次いでAcOEtによって抽出し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(95:5のCHCl3/MeOH)によって茶色の粘性油として100mgの純粋な生成物を得た(収率57%)。
1H−NMR(アセトン−d6)δ:6.47(brs,1H),6.66(brs,1H),6.76−6.84(m,2H),6.99−7.04(m,2H),7.20−7.31(m,2H),7.85(s,1H),9.40(brs,1H)。
【0054】
工程4:ブチル−カルバミン酸3(3−カルバモイル−ピロール−1−イル)フェニルエステル、ST3910
1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸アミド(40.0mg、0.20ミリモル)の2.0mLの無水THF溶液に、n−ブチルイソシアネート(90μL、0.79ミリモル)とNEt3(110μL、0.79ミリモル)を加えた。反応混合物を室温で16時間撹拌した。溶媒を取り除き、粗精製の反応混合物をフラッシュクロマトグラフィ(98/2のCHCl3/MeOH)によって精製して、38mgの所望の付加体を導いた(収率63%)。
1H−NMR(アセトン−d6)δ:0.94(t,3H,J=7.2Hz),1.34−1.47(m,2H),1.52−1.62(m,2H),3.22(q,2H,J=6.6Hz),6.35(brs,1H),6.74(s,1H),6.91(brs,2H),7.08(d,1H,J=7.8Hz),7.28−7.50(m,4H),7.85(s,1H)
13C−NMR(アセトン−d6)δ:13.4,19.9,32.0,40.9,110.6,114.0,116.6,119.7,120.2,121.8,122.7,130.5,140.9,152.9,154.3,165.6
【0055】
実施例2〜4は、適切なイソシアネート試薬を用いて、実施例1の工程4で記載された実験条件に従って得られた。
【0056】
実施例2:ウンデカ−10−イニル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ST3911
収率:78%
1H−NMR(CD3OD)δ:1.35−1.56(m,14H),2.13(s,1H),2.16(s,2H),3.18(t,2H,J=6.9Hz),6.72(s,1H),7.08(d,1H,J=7.2Hz),7.22−7.39(m,3H),7.48(t,1H,J=8.1Hz),7.80(s,1H)
13C−NMR(CD3OD)δ:17.8,26.7,28.5,28.6,29.0,29.2,29.37,29.5,40.9,68.2,83.9,110.2,114.1,117.0,119.8,120.5,121.0,122.3,130.4,140.9,152.6,155.5,168.5
【0057】
実施例3:シクロヘキシル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ST3912
収率:62%
1H−NMR(CD3OD)δ:1.19−1.39(m,5H),1.65(d,1H,J=12.6Hz),1.78(d,2H,J=11.7Hz),1.95(d,2H,J=10.8Hz),3.39−3.48(m,1H),6.72(s,1H),7.07(d,1H,J=7.8Hz),7.21−7.38(m,3H),7.47(t,1H,J=7.8Hz),7.80(s,1H)
13C−NMR(CD3OD)δ:25.0(2),25.4,32.8(2),50.5,110.2,114.2,116.9,119.8,120.5,121.0,122.3,130.4,140.8,152.5,154.7,168.5
【0058】
実施例4:(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ST3913
収率:60%
1H−NMR(CD3OD)δ:1.38−1.40(m,4H),1.53−1.66(m,4H),2.60(t,2H,J=15.0Hz),3.17(t,2H,J=13.8Hz),6.72(s,1H),7.05−7.37(m,9H),7.46(t,1H,J=8.4Hz),7.79(s,1H)
13C−NMR(CD3OD)δ:26.5,28.8,29.5,31.5,35.6,40.9,110.2,114.1,117.0,119.8,120.5,121.0,122.3,125.5,128.1(2),128.2(2),130.4,140.8,142.7,152.5,155.5,168.5
ESI−MSm/z[M+H]+406,[M+Na]+428,[M+K]+444,[2M+H]+811,[2M+Na]+833
実施例5:ブチル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル、ST4067
【0059】
工程1:2−(3−メトキシ−ベンゾイル)−4−オキソ−ペンタン酸エチルエステル
0℃にてNaH(36.0mg、1.49ミリモル)の8.0mL無水THF懸濁液にエチル3−(3−メトキシフェニル)−3−オキソプロパノエート(262mL、1.35ミリモル)を一滴ずつ加えた。混合した反応物をこの温度で30分間撹拌した。次いでクロロアセトン(119mL、1.49ミリモル)を0℃にて一滴ずつ加え、反応混合物を室温にて48時間撹拌した。1NのHCl(3.0mL)で反応を止めた。THFを蒸発させ、残留物をAcOEt(25mLで3回)で抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(3:1のn−ヘキサン/AcOEt)によってラセミ混合物として純粋な生成物を得た(収率:67%)
1H−NMR(CDCl3)δ:1.14(t,3H,J=6.9Hz),2.21(s,3H),3.06(m,2H),3.83(s,3H),4.10(q,2H,J=6.9Hz),4.85(t,1H,J=7.0Hz),7.09−7.13(m,1H),7.33−7.39(m,1H),7.51(s,1H),7.59(d,1H,J=7.8Hz)
【0060】
工程2:2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル
2−(3−メトキシ−ベンゾイル)−4−オキソ−ペンタン酸エチルエステル(735mg、2.64ミリモル)を8mLの純粋なEtOHに溶解した。濃HCl(0.73mL)を加え、反応混合物をMW照射(150W)(CEM Discovery Microwave System)のもとで維持し、10分間還流した(最高内部温度100℃)。反応混合物を冷却し、AcOEtで希釈し、重炭酸ナトリウムの飽和水溶液で中和し、AcOEt(50mLで3回)で抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(97:3のn−ヘキサン/AcOEt)によって無色の粘性油として純粋な生成物を得た(収率90%)。
1H−NMR(CDCl3)δ:1.32(t,3H,J=6.9Hz),2.34(d,3H,J=0.9Hz),3.84(s,3H),4.28(q,2H,J=7.2Hz),6.44(d,1H,J=1.2Hz),6.92(ddd,1H,J=0.6,2.1,8.1Hz),7.32(t,1H,J=7.8Hz),7.56−7.59(m,1H),7.62−7.64(m,1H)
13C−NMR(CDCl3)δ:13.5,14.5,55.5,60.6,109.2,113.5,115.0,115.2,120.7,129.2,131.4,151.21,155.8,159.5,163.9
ESI−MSm/z[M−OEt]+215,[M+Na]+283
【0061】
工程3:2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸
EtOH(3.0mL)と水(1.0mL)における2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル(42.0mg、0.16ミリモル)の溶液にNaOH(161.0mg、4.03ミリモル)を加え、室温にて反応混合物を7時間撹拌した。EtOHを取り除き、6NのHClで残留物をpH1に酸性化した。混合物をAcOEt(20mLで3回)で抽出した。有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。さらに精製することなく、得られた化合物(37.0mg、収率100%)を次の工程に用いた。
1H−NMR(CDCl3)δ:2.37(s,3H),3.86(s,3H),6.48(s,1H),6.95(dd,1H,J=7.8,3.9Hz,),7.34(t,1H,J=7.9Hz),7.54−7.59(m,2H)
ESI−MSm/z[M−H]−231
【0062】
工程4:2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸アミド
室温にて300μLのSOCl2に2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸(68.0mg、0.29ミリモル)を少しずつ加えた。反応混合物を還流で30分間撹拌し、冷却した後、乾燥するまで蒸発させた。得られた残留物を3.0mLのTHFに溶解し、室温にて1.0mLの濃NH4OHを慎重に加えた。混合物を7時間撹拌し、AcOEtで抽出し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。カラムクロマトグラフィ(95:5のCHCl3/MeOH)を介して粗精製の反応混合物を精製して茶色の粘性油として純粋な生成物を得た(65.0mg、収率97%)。
1H−NMR(CDCl3)δ:2.34(d,3H,J=0.6Hz),3.83(s,3H),5.79(brs,1H),5.90(brs,1H),6.33(d,1H,J=0.9Hz),6.92(dt,1H,J=3.0,7.5Hz),7.26−7.38(m,3H)
【0063】
工程5:2−(3−ヒドロキシ−フェニル)−5−メチル−フラン−3−カルボン酸アミド
2−(3−メトキシ−フェニル)−5−メチル−フラン−3−カルボン酸アミド(188.0mg、0.81ミリモル)の無水CH2Cl2(6.0mL)懸濁液にBBr3(ジクロロメタン中の1M溶液、2.4mL、2.44ミリモル)を−78℃にて加えた。次いで反応混合物を室温に温め、8時間撹拌した。炭酸ナトリウムの飽和水溶液を加えて反応を止めた。残留物をAcOEt(20mLで3回)で抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮して、茶色っぽい固形物として純粋な生成物を得た(175.0mg、収率98%)。
1H−NMR(アセトン−d6)δ:2.33(d,3H,J=0.9Hz),6.42(d,1H,J=0.9Hz),6.56(brs,1H),6.81(ddd,1H,J=0.9,2.7,4.8Hz),6.94(brs,1H),7.22(t,1H,J=7.8Hz),7.45(dt,1H,J=7.8,1.5Hz),7.53(t,1H,J=1.8Hz),8.48(s,1H)
【0064】
工程6:ブチル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル
2−(3−ヒドロキシ−フェニル)−5−メチル−フラン−3−カルボン酸アミド(36.0mg、0.17ミリモル)の3.0mL無水THF溶液にn−ブチルイソシアネート(80μL、0.75ミリモル)とNEt3(100μL、0.75ミリモル)を加えた。反応混合物を室温にて16時間撹拌した。溶媒を取り除き、カラムクロマトグラフィ(99:1のCHCl3/MeOH)による精製によって無色の油として純粋な生成物を得た(40.0mg、収率77%)。
1H−NMR(CDCl3)δ:0.96(t,3H,J=7.5Hz),1.36−1.43(m,2H),1.50−1.57(m,2H),2.35(d,3H,J=0.9Hz),3.15−3.21(m,2H),6.38(d,1H,J=0.9Hz),7.07(ddd,1H,J=0.9,2.4,4.8Hz),7.38(t,1H,J=7.8Hz),7.63(t,1H,J=1.8Hz),7.70(dt,1H,J=1.5,5.4Hz)
13C−NMR(CDCl3)δ:12.0,12.9,19.8,31.7,40.5,107.9,118.2,120.2,121.6,123.7,129.0,131.6,151.4,151.5,151.9,155.9,168.0
【0065】
実施例6は、n−ブチルイソシアネートの代わりにフェニルヘキシルイソシアネートを用いて実施例5の工程6に記載された実験条件に従って合成された。
【0066】
実施例6:(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、ST4068
収率:71%
1H−NMR(CDCl3)δ:1.38(brs,4H),1.55−1.66(m,4H),2.33(s,3H),2.61(t,2H,J=7.5Hz),3.24(q,2H,J=6.6Hz),5.10(brs,1H),5.61(brs,1H),5.82(brs,1H),6.32(s,1H),7.11−7.19(m,4H),7.25−7.30(m,2H),7.39(d,1H,J=7.8Hz),7.58(s,1H),7.64(d,1H,J=7.8Hz)
13C−NMR(CDCl3)δ:13.6,26.8,29.1,29.9,31.6,36.1,41.5,108.5,118.1,121.2,122.3,124.7,125.9,128.5(2),128.6(2),129.8,131.3,142.8,151.3,151.5,152.1,154.6,165.9
実施例7:(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステル、ST4069
【0067】
工程1:2−(3−メトキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸エチルエステル
2−(3−メトキシ−ベンゾイル)−4−オキソペンタン酸エチルエステル(500.0mg、1.8ミリモル)の5.0mLトルエン溶液にLawessonの試薬(2.00g)を加えた。還流のもと(最高内部温度100℃)でマイクロ波照射下(150W)(CEM Discovery Microwave System)で10分間反応を続けた。冷却した後、セライトを介して混合物を濾過し、減圧下で溶媒を除去した。フラッシュカラムクロマトグラフィ(99:1のn−ヘキサン/酢酸エチル)によって無色の油として純粋な生成物を得た(240.mg、収率48%)。
1H−NMR(CDCl3)δ:1.18(t,3H,J=7.2Hz),2.47(d,3H,J=0.9Hz),3.82(s,3H),4.17(q,2H,J=6.9Hz),6.90(ddd,1H,J=0.9,2.7,8.4Hz),7.00−7.05(m,2H),7.15(d,1H,J=0.9Hz),7.28(t,1H,J=7.8Hz)
ESI−MSm/z[M−OEt]+231,[M+Na]+299
【0068】
工程2:2−(3−メトキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸
実施例5の工程3で記載された実験条件に従って化合物を得た。
収率:97%。精製することなくその化合物を次の工程に用いた。
1H−NMR(CDCl3)δ:2.46(d,3H,J=0.6Hz),3.81(s,3H),6.92(dd,1H,J=2.4,8.4Hz),7.03−7.07(m,2H),7.19(d,1H,J=0.9Hz),7.28(t,1H,J=8.1Hz)
【0069】
工程3:2−(3−メトキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸アミド
実施例5の工程4で記載された実験条件に従って化合物を得た。
収率:78%
1H−NMR(アセトン−d6)δ:2.46(d,3H,J=0.6Hz),3.82(s,3H),6.50(brs,2H),6.93(dd,1H,J=1.8,8.4Hz),6.99(d,1H,J=1.2Hz),7.06−7.11(m,2H),7.32(t,1H,J=8.1Hz)
【0070】
工程4:2−(3−ヒドロキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸アミド
実施例5の工程5で記載された実験条件に従って化合物を得た。
収率:95%
1H−NMR(アセトン−d6)δ:2.45(d,3H,J=0.9Hz),6.49(brs,2H),6.84(ddd,1H,J=0.9,2.4,8.1Hz),6.96−7.00(m,3H),7.22(t,1H,J=7.5Hz),8.55(s,1H)
【0071】
工程5:(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステル
2−(3−ヒドロキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸アミドをフェニルヘキシルイソシアネートと反応させて、実施例5の工程6で記載された実験条件に従って化合物を得た。
収率:80%
1H−NMR(CDCl3)δ:1.36−1.40(m,4H),1.54−1.69(m,4H),2.45(d,3H,J=1.2Hz),2.61(t,2H,J=7.5Hz),3.24(q,2H,J=6.9Hz),5.13(t,1H,J=5.4Hz),5.53(brs,2H),7.12−7.19(m,5H),7.25−7.32(m,4H),7.39(t,1H,J=7.8Hz)
13C−NMR(CDCl3)δ:15.3,26.8,29.1,29.9,31.6,36.1,41.5,122.3,123.1,125.9,126.6,127.9,128.5(2),128.6(2),129.9,132.6,134.3,139.5,141.7,142.8,151.4,154.5,166.0
実施例8:シクロヘキシル−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル、ST4104
【0072】
工程1:2−[2−(3−メトキシ−フェニル)−2−オキソ−エチル]−3−オキソ−酪酸エチルエステル
エチル3−(3−メトキシフェニル)−3−オキソプロパノエートとクロロアセトンの代わりにエチルアセトアセテートと2−ブロモ−1−(3−メトキシフェニル)エタノンを用いて、実施例5の工程1で記載された実験条件に従って化合物を得た。
収率:79%
1H−NMR(CDCl3)δ:1.29(t,3H,J=7.2Hz),2.45(s,3H),3.51(dd,1H,J=18.6,5.7Hz),3.71(dd,1H,J=18.6,8.3Hz),3.85(s,3H),4.23(m,3H),7.12(m,1H),7.37(m,1H),7.47(m,1H),7.58(m,1H);ESI−MSm/z[M+H]+279,[M+Na]+301(100)
【0073】
工程2:5−(3−メトキシ−フェニル)−2−メチル−フラン−3−カルボン酸エチルエステル
実施例5の工程2で記載された実験条件に従って化合物を得た。
収率:72%
1H−NMR(CDCl3)δ:1.35(t,3H,J=7.2Hz),2.62(s,3H),3.81(s,3H),4.29(q,2H,J=7.2Hz),6.80(m,1H),6.86(s,1H),7.15−7.28(m,3H)
13C−NMR(CDCl3)δ:14.1,14.6,55.4,60.4,106.0,109.1,113.6,115.6,116.4,129.9,131.5,151.74,158.8,160.1,164.1
【0074】
工程3:5−(3−メトキシ−フェニル)−2−メチル−フラン−3−カルボン酸
実施例5の工程3で記載された実験条件に従って化合物を得た。
収率:98%。精製することなくその化合物を次の工程に用いた。
1H−NMR(CDCl3)δ:2.69(s,3H),3.86(s,3H),6.84(m,1H),6.92(s,1H),7.18−7.33(m,3H),10.83(brs,1H)
【0075】
工程4:5−(3−メトキシ−フェニル)−2−メチル−フラン−3−カルボン酸アミド
実施例1の工程3で記載された実験条件に従って化合物を得た。
収率:74%
1H−NMR(CDCl3)δ:2.67(s,3H),3.85(s,3H),5.68(brs,2H),6.67(s,1H),6.83(m,1H),7.16(m,1H),7.21(m,1H),7.26−7.32(m,1H)
ESI−MSm/z[M+Na]+254(100),[M+K]+270,[2M+Na]+485
【0076】
工程5:5−(3−ヒドロキシ−フェニル)−2−メチル−フラン−3−カルボン酸アミド
実施例5の工程5で記載された実験条件に従って化合物を得た。
収率:74%
1H−NMR(CD3OD)δ:2.58(s,3H),6.70(m,1H),6.92(s,1H),7.09(m,2H),7.17(m,1H);ESI−MSm/z[M+Na]+240(100)
【0077】
工程6:シクロヘキシル−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル、ST4104
5−(3−メトキシ−フェニル)−2−メチル−フラン−3−カルボン酸アミド(45.0mg、0.21ミリモル)の2.0mL無水THF溶液にシクロヘキシルイソシアネート(106μL、0.83ミリモル)とNEt3(116μL、0.83ミリモル)を加えた。室温にて反応混合物を16時間撹拌した。溶媒を除去し、カラムクロマトグラフィ(9:1のCHCl3/MeOH)によって無色の固形物として純粋な生成物を得た(収率:34%)。
1H−NMR(CD3OD)δ:1.19−1.39(m,5H),1.65(m,1H),1.79(m,2H),1.96(m,2H),2.61(s,3H),3.38(m,1H),7.02(m,2H),7.39(m,2H),7.50(m,1H)
13C−NMR(CD3OD)δ:12.5,24.9,25.4,32.8,50.5,104.9,116.7,117.3,120.1,120.8,129.6,131.5,150.9,152.0,155.0,157.2,167.3
ESI−MSm/z[M+Na]+365,[2M+Na]+707
【0078】
実施例9:(6−フェニル−ヘキシル)−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル、ST4105
シクロヘキシルイソシアネートの代わりにフェニルヘキシルイソシアネートを用いて(Mor M., et al., J. Med. Chem., 2008, 51, 12, 3487)、実施例8の工程5で記載された実験条件に従って化合物を得た。
収率:40%
1H−NMR(CD3OD)δ:1.37(m,4H),1.52−1.63(m,4H),2.58(m,5H),3.16(t,2H,J=6.9Hz),6.99(m,2H),7.12(m,3H),7.22(m,2H),7.35(m,2H),7.46(m,1H).
13C−NMR(CD3OD)δ:12.6,26.5,28.8,29.5,31.5,35.7,40.9,104.9,116.7,117.3,120.1,120.8,125.5,128.1,128.2,129.7,131.6,142.7,150.9,152.0,155.8,157.2,167.3.
ESI−MSm/z[M+Na]+443,[M+K]+459,[2M+Na]+863
実施例10:2−(3−ブチルカルバモイルオキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル、ST4110
【0079】
工程1:2−(3−ヒドロキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル
実施例5の工程5で記載された実験条件に従って化合物を得た。
収率:60%
1H−NMR(CD3OD)δ:1.30(t,3H,J=7.5Hz),2.33(s,3H),4.24(q,2H,J=7.2Hz),6.43(s,1H),6.80(dd,1H,J=1.8,6.9Hz),7.21(t,1H,J=8.1Hz),7.36(t,2H,J=7.2Hz)
【0080】
工程2:2−(3−ブチルカルバモイルオキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル
実施例5の工程6で記載された実験条件に従って化合物を得た。
収率:81%
1H−NMR(CDCl3)δ:0.94(t,3H,J=6.9Hz),1.29−1.40(m,5H),1.49−1.59(m,2H),2.32(s,3H),3.25(q,2H,J=6.6Hz),4.28(q,2H,J=6.6Hz),5.12(brs,1H),6.42(s,1H),7.14(d,1H,J=7.8Hz),7.37(t,1H,J=8.4Hz),7.77(s,1H),7.84(d,1H,J=7.5Hz)
13C−NMR(CDCl3)δ:13.5,13.9,14.4,20.1,32.1,41.2,60.7,109.2,115.2,121.4,122.4,125.1,129.0,131.4,151.1,151.5,154.7,154.9,163.9
ESI−MSm/z[M+Na]+368,[2M+Na]+713
実施例11:1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸−ウンデカ−10−イニルエステル、ST4112
【0081】
工程1:1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸−ウンデカ−10−イニルエステル
1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸(100.0mg、0.49ミリモル)と10−ウンデシン−1−オール(90mL、0.49ミリモル)の2.0mL無水THFの撹拌された溶液に、0℃にてトリフェニルホスフィン(129mg、0.49ミリモル)とDIAD(90mL、0.49ミリモル)を一滴ずつ加えた。室温にて反応混合物を48時間撹拌した。減圧下で溶媒を取り除いた。残留物に重炭酸ナトリウムの飽和水溶液を加え、得られた混合物をAcOEt(25mLで3回)で抽出し、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。フラッシュクロマトグラフィ(1:9のAcOEt/n−ヘキサン)によって精製を行い、無色の固形物として120mgの所望の生成物(収率69%)を得た。
1H−NMR(CDCl3)δ:1.32−1.55(m,12H),1.70−1.75(m,2H),1.93(t,1H,J=2.7Hz),2.17(dd,2H,J=2.7,7.2Hz),4.25(t,2H,J=6.9Hz),5.52(brs,1H),6.74(q,1H,J=1.8Hz),6.79(dd,1HJ=2.1,8.1Hz),6.92(t,1H,J=2.1Hz),6.95−7.00(m,2H),7.30(t,1H,J=8.1Hz),7.66(dd,1H,J=1.5,2.1Hz).
【0082】
工程2:1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸−ウンデカ−10−イニルエステル
1−(3−ヒドロキシ−フェニル)−1H−ピロール−3−カルボン酸−ウンデカ−10−イニルエステルから出発して実施例5の工程6で記載された実験条件に従って表題の化合物を得た。
1H−NMR(CDCl3)δ:0.94(t,3H,J=7.2Hz),1.30−1.74(m,18H),1.93(s,1H),2.16(dd,2H,J=4.5,6.3Hz),3.26(q,2H,J=6.3Hz),4.22(t,2H,J=6.3Hz),5.22(brs,1H),6.73(s,1H),6.99(s,1H),7.07(d,1H,J=8.1Hz),7.19−7.26(m,2H),7.39(t,1H,J=8.1Hz),7.65(s,1H)
13C−NMR(CDCl3)δ:14.0,18.6,20.1,26.2,28.7,28.9,29.1,29.2,29.5,29.6,32.1,41.3,64.3,68.3,85.0,111.9,114.8,117.7,118.6,120.2,120.7,124.5,130.5,140.8,152.2,154.3,165.0
ESI−MSm/z[M+H]+453,M+Na]+475,[M+K]+491
実施例12:1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸6−フェニルヘキシルエステル、ST4114
【0083】
工程1:1−(3−ヒドロキシ−フェニル)−H−ピロール−カルボン酸6−フェニル−ヘキシルエステル
10−ウンデシン−1−オールの代わりに6−フェニル−1−ヘキサノールを用いて、実施例11の工程1で記載された実験条件に従って表題の化合物を得た。
収率:63%
1H−NMR(CDCl3)δ:1.38−1.48(m,4H),1.49−1.76(m,4H),2.62(t,2H,J=6.9Hz),4.25(t,2H,J=6.9Hz),6.73−6.99(m,6H),7.16−7.30(m,6H),7.67−7.68(m,1H).
【0084】
工程2:1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸6−フェニルヘキシルエステル
1−(3−ヒドロキシ−フェニル)−H−ピロール−カルボン酸6−フェニル−ヘキシルエステルから出発して、実施例11の工程2で記載された実験条件に従って表題の化合物を得た。
収率:95%
1H−NMR(CDCl3)δ:0.94(t,3H,J=7.2Hz),1.34−1.81(m,11H),2.62(t,3H,J=7.2Hz),3.27(q,2H,J=6.6Hz),4.24(t,2H,J=6.6Hz),5.21(t,1H,J=5.4Hz),6.75(s,1H),6.99−7.43(m,10H),7.67(s,1H)
13C−NMR(CDCl3)δ:14.0,20.1,26.2,29.0,29.2,31.6,32.1,36.1,41.3,64.3,112.0,114.9,117.7,118.6,120.2,120.8,124.5,125.8,128.5(2),128.6(2),130.6,140.8,142.9,152.2,154.3,165.0
ESI−MSm/z[M+H]+461,[M+Na]+485,[M+K]+501
【0085】
実施例13:シクロヘキシル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、ST4049
n−ブチルイソシアネートの代わりにシクロヘキシルイソシアネートを用いて、実施例5の工程6で記載された実験条件に従って実施例6を合成した。
収率:79%
1H−NMR(DMSO−d6)δ:1.13−1.28(m,5H),1.52−1.59(m,1H),1.67−1.70(m,2H),1.79−1.83(m,2H),2.31(s,3H),3.28−3.30(m,1H),6.48(s,1H),7.04(dd,1H,J1=0.9Hz,J2=7.5Hz),7.27(bs,1H),7.37(t,1H,J=8.1Hz),7.63−7.74(m,4H)
13C−NMR(DMSO−d6)δ:13.79,25.24,25.81,33.22,50.44,109.28,119.29,120.57,122.26,123.75,129.73,131.82,150.74,151.46,151.59,154.05,165.69
【0086】
実施例14:シクロヘキシル−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステル、ST4050
2−(3−ヒドロキシ−フェニル)−5−メチル−チオフェン−3−カルボン酸アミドをシクロヘキシルイソシアネートと反応させて、実施例5の工程6で記載された実験条件に従って化合物を得た。
収率:72%
1H−NMR(CD3OD)δ:1.22−1.39(m,5H),1.64(bd,1H,J=12.6Hz),1.78(bd,2H,J=12.0Hz),1.94(bd,2H,J=10.5Hz),2.47(s,3H),3.38−3.48(m,1H),6.97(m,1H),7.09(d,1H,J=8.1Hz),7.21(s,1H),7.30−7.41(m,2H)
13C−NMR(CD3OD)δ:13.71,24.98,25.42,32.80,50.48,121.46,122.06,125.57,126.79,129.30,132.89,134.67,139.75,141.39,151.5,155.03,167.13
ESI−MSm/z[M+Na]+381,[2M+Na]+740
【0087】
調製1:11−イソシアナト−ウンデカ−1−イン
ウンデカ−10−イニルアミン(Crisp G.T., et al., Tetrahedron, 1997, 53, 4, 1505, 514 mg)を33mLのCH2Cl2に溶解した。溶液を0℃に冷却し、重炭酸ナトリウムの飽和水溶液(30mL)を加えた。二相混合物を0℃で10分間撹拌し、層を分離し、シリンジを介して、ホスゲンの溶液(トルエン中20%、3.23mL)を有機層に直接加えた。15分後、水性層をCH2Cl2で抽出した。合わせた有機層を硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。さらに精製することなく化合物を使用した。
収率:94%
1H−NMR(CDCl3)δ:1.27(m,4H),1.51(m,4H),2.51(t,2H,J=7.7Hz),3.14(t,2H,J=6.6Hz),7.07(m,3H),7.17(m,2H)
【0088】
生物学的結果
FAAHアッセイ
本発明の化合物は、親和性を示し、脂肪酸アミド加水分解酵素の活性を阻害する。
【0089】
RP−HPLCを用いて、[1−14C]AnNH(52mCi/ミリモル)からの[14C]AAの放出を測定することによってFAAH(EC3.5.1.4)のアッセイを行った。また、[1−14C]AnNHについて以下で記載される同様の実験条件下で[3H]AnNH(205Ci/ミリモル)を基質として用いて[3H]AAの放出を測定することができた。種々の濃度での本発明の化合物を、2mLのエッペンドルフ管における200μLの加水分解酵素アッセイ緩衝液(50mMトリス−HCl、pH9.0)に加え、20分後、10μMの濃度までの[1−14C]AnNH2を加えた。マウスの脳ホモジネート(40μg)を添加することによって反応を開始し、37℃にて15分間インキュベートした後、撹拌しながら、800μLの氷冷メタノール/クロロホルム(2:1、v/v)の添加によって反応を止めた。この混合物を室温で30分間放置し、次いで、撹拌しながら、240μLのクロロホルムと240μLの水を加えた。室温にて10分後、混合物を3000gにて5分間遠心し、上部の水性層を吸引によって取り除き、DNA MINIスピードバック(Heto−Holten、Denmark)にて試料を30℃、100ミリバールで30分間遠心することによって下部の有機層を乾燥させた。残留物を50μLのメタノールに溶解し、以下に詳述するように、AAの定量用のRP−HPLCに供した。FAAHに特異的な活性は、放出されたピコモルAA/分/タンパク質のmgとして表した。0〜12μMの濃度範囲にて、[1−14C]AnNH、[1−14C]ODNHEtOH又は[1−14C]ODNH2を用いて、Lineweaver−Burkの解析によって動態試験を行った。線形回帰プログラム(Kaleidagraph 3.0)による実験点のフィッティングによってr値>0.97の直線が得られた。
【0090】
【表1】
【0091】
選択性特性
Maccarrone,M.ら、J.Biol.Chem.,2000,275,13484,Fezza、F.ら、Anal Biochem.,2005,339,113,Dinh、T.Pら、2002,Proc.Natl.Acad.Sci.99,10819,Bisogno、T.ら、2003,J.Cell Biol.,163,463,Maccarrone、M.ら、J.Biol.Chem.,2000,275,31938,Ross、R.A.ら、Br.J.Pharmacol.,2001,132,631に記載された手順に従って、以下の標的:AMT、NAPE−PLD、MAGL、DAGL、CB1/CB2及びTRPV1に対する選択性特性に関して本発明の化合物の評価も行った。結果を表2に示す。
【0092】
【表2】
【0093】
生体内鎮痛活性
足引っ込め試験を用いて機械的な痛覚過敏を評価した。Randall−Selitto(ランダル・セリット)の痛覚計(イタリア、VareseのUgo Basile)を用いて、左右の後肢に高い圧力を適用することによってグラムで表現する傷害受容の閾値を測定した。傷害受容の閾値を定量するのに使用したパラメータは、ラットがその足を引っ込める圧力(グラム)として定義した。試験手順と実験に先立つ週での研究者による取り扱いにラットを慣らせた。30mg/kgの用量でのST4068、ST3911及びST3913による急性の経口処理は、十分な鎮痛活性を示した一方で、50mg/kg用量での周知の参照化合物URB597は活性を示さなかった(図1)。
【0094】
ST4068、ST3911及びST3913によって、10、30及び100mg/kgでの用量反応実験も行った。化合物はすべて30及び100mg/kgの用量計画で鎮痛活性を発揮し、後者の投与量はさらに高い効果を付与した(図2、3及び4)。
【特許請求の範囲】
【請求項1】
一般式I:
【化1】
[式中、
R1は、H、(C1〜C4)−アルキル、(C3〜C6)−シクロアルキル、アリールで置換される(C1〜C6)−アルキル又は(C2〜C5)−アルキニルであり、
Xは、C又はNであり、
Yは、CH、O又はSであり、
R2は、H又は(C1〜C4)−アルキルであり、
Eは、NR3R4又はOR5であり、
R3及びR4は、同一であり、又は異なっており、H、又はアリールによって置換されていてもよい(C2〜C6)−アルキルであり、
R5は、アリール又は(C2〜C5)−アルキニルによって置換されていてもよい(C2〜C6)−アルキルであり、
X及びYを含有する環は、ヘテロ芳香族環を意味する;
ただし、XがNである場合、YはCHである]
で示される化合物、エナンチオマー、ジアステレオマー、及びそのラセミ形態などのその光学活性のある形態、並びに薬学上許容可能なその塩。
【請求項2】
R1が、アリールで置換される(C1〜C6)−アルキル又は(C2〜C5)−アルキニルである請求項1に記載の化合物。
【請求項3】
R3及びR4がHである請求項1又は2に記載の化合物。
【請求項4】
ブチル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ウンデカ−10−イニル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、シクロヘキシル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ブチル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、(6−フェニル−ヘキシル)カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステル、シクロヘキシル−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、(6−フェニル−ヘキシル)−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、2−(3−ブチルカルバモイルオキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル、1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸ウンデカ−10−イニルエステル、1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸6−フェニル−ヘキシルエステル、シクロヘキシル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル及びシクロヘキシル−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステルから成る群から選択される請求項1〜3のいずれかに記載の化合物。
【請求項5】
請求項1〜4のいずれか1項に記載の化合物の薬物としての使用。
【請求項6】
FAAH活性の調節が患者の健康の改善を生じる病的状態を治療するために薬物を調製するための、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項7】
神経障害、エネルギー代謝の疾患、循環器及び呼吸器の疾病、消化器及び肝臓の疾病、網膜症、癌及び筋骨格疾病から成る群から選択される病的状態を治療するための、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項8】
障害が神経障害である請求項7に記載の使用。
【請求項9】
障害が不安症である請求項8に記載の使用。
【請求項10】
障害が神経障害性の痛みである請求項8に記載の使用。
【請求項11】
障害がパーキンソン病である請求項8に記載の使用。
【請求項12】
少なくとも1つの薬学上許容可能なビヒクル及び/又は賦形剤との混合で有効成分としての請求項1〜4に記載の化合物を少なくとも1つ含有する医薬組成物。
【請求項13】
FAAH活性の調節が患者の健康の改善を生じる病的状態に冒された哺乳類に、有効量の請求項1〜4の化合物を投与する工程を含む、FAAHを阻害する方法。
【請求項14】
NEt3などの塩基の存在下、極性溶媒にて、式II:
【化2】
(式中、X、Y、E及びR2は請求項1と同義である)の化合物を式III:
R1−N=C=O(式III)(式中、R1は請求項1と同義)と反応させることによって得ることができる請求項1の化合物を合成する方法。
【請求項15】
請求項1〜4のいずれかに記載の化合物と薬学上許容可能なビヒクル及び/又は賦形剤を密な混合剤にすることを含む、請求項12に記載の医薬組成物を調製する方法。
【請求項1】
一般式I:
【化1】
[式中、
R1は、H、(C1〜C4)−アルキル、(C3〜C6)−シクロアルキル、アリールで置換される(C1〜C6)−アルキル又は(C2〜C5)−アルキニルであり、
Xは、C又はNであり、
Yは、CH、O又はSであり、
R2は、H又は(C1〜C4)−アルキルであり、
Eは、NR3R4又はOR5であり、
R3及びR4は、同一であり、又は異なっており、H、又はアリールによって置換されていてもよい(C2〜C6)−アルキルであり、
R5は、アリール又は(C2〜C5)−アルキニルによって置換されていてもよい(C2〜C6)−アルキルであり、
X及びYを含有する環は、ヘテロ芳香族環を意味する;
ただし、XがNである場合、YはCHである]
で示される化合物、エナンチオマー、ジアステレオマー、及びそのラセミ形態などのその光学活性のある形態、並びに薬学上許容可能なその塩。
【請求項2】
R1が、アリールで置換される(C1〜C6)−アルキル又は(C2〜C5)−アルキニルである請求項1に記載の化合物。
【請求項3】
R3及びR4がHである請求項1又は2に記載の化合物。
【請求項4】
ブチル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ウンデカ−10−イニル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、シクロヘキシル−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−ピロール−1−イル)−フェニルエステル、ブチル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、(6−フェニル−ヘキシル)カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、(6−フェニル−ヘキシル)−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステル、シクロヘキシル−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、(6−フェニル−ヘキシル)−カルバミン酸3−(4−カルバモイル−5−メチル−フラン−2−イル)−フェニルエステル、2−(3−ブチルカルバモイルオキシ−フェニル)−5−メチル−フラン−3−カルボン酸エチルエステル、1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸ウンデカ−10−イニルエステル、1−(3−ブチルカルバモイルオキシ−フェニル)−1H−ピロール−3−カルボン酸6−フェニル−ヘキシルエステル、シクロヘキシル−カルバミン酸3−(3−カルバモイル−5−メチル−フラン−2−イル)フェニルエステル及びシクロヘキシル−カルバミン酸3−(3−カルバモイル−5−メチル−チオフェン−2−イル)−フェニルエステルから成る群から選択される請求項1〜3のいずれかに記載の化合物。
【請求項5】
請求項1〜4のいずれか1項に記載の化合物の薬物としての使用。
【請求項6】
FAAH活性の調節が患者の健康の改善を生じる病的状態を治療するために薬物を調製するための、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項7】
神経障害、エネルギー代謝の疾患、循環器及び呼吸器の疾病、消化器及び肝臓の疾病、網膜症、癌及び筋骨格疾病から成る群から選択される病的状態を治療するための、請求項1〜4のいずれか1項に記載の化合物の使用。
【請求項8】
障害が神経障害である請求項7に記載の使用。
【請求項9】
障害が不安症である請求項8に記載の使用。
【請求項10】
障害が神経障害性の痛みである請求項8に記載の使用。
【請求項11】
障害がパーキンソン病である請求項8に記載の使用。
【請求項12】
少なくとも1つの薬学上許容可能なビヒクル及び/又は賦形剤との混合で有効成分としての請求項1〜4に記載の化合物を少なくとも1つ含有する医薬組成物。
【請求項13】
FAAH活性の調節が患者の健康の改善を生じる病的状態に冒された哺乳類に、有効量の請求項1〜4の化合物を投与する工程を含む、FAAHを阻害する方法。
【請求項14】
NEt3などの塩基の存在下、極性溶媒にて、式II:
【化2】
(式中、X、Y、E及びR2は請求項1と同義である)の化合物を式III:
R1−N=C=O(式III)(式中、R1は請求項1と同義)と反応させることによって得ることができる請求項1の化合物を合成する方法。
【請求項15】
請求項1〜4のいずれかに記載の化合物と薬学上許容可能なビヒクル及び/又は賦形剤を密な混合剤にすることを含む、請求項12に記載の医薬組成物を調製する方法。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公表番号】特表2012−520841(P2012−520841A)
【公表日】平成24年9月10日(2012.9.10)
【国際特許分類】
【出願番号】特願2012−500180(P2012−500180)
【出願日】平成22年3月8日(2010.3.8)
【国際出願番号】PCT/EP2010/052884
【国際公開番号】WO2010/105930
【国際公開日】平成22年9月23日(2010.9.23)
【出願人】(591043248)シグマ−タウ・インドゥストリエ・ファルマチェウチケ・リウニテ・ソシエタ・ペル・アチオニ (92)
【氏名又は名称原語表記】SIGMA−TAU INDUSTRIE FARMACEUTICHE RIUNITE SOCIETA PER AZIONI
【Fターム(参考)】
【公表日】平成24年9月10日(2012.9.10)
【国際特許分類】
【出願日】平成22年3月8日(2010.3.8)
【国際出願番号】PCT/EP2010/052884
【国際公開番号】WO2010/105930
【国際公開日】平成22年9月23日(2010.9.23)
【出願人】(591043248)シグマ−タウ・インドゥストリエ・ファルマチェウチケ・リウニテ・ソシエタ・ペル・アチオニ (92)
【氏名又は名称原語表記】SIGMA−TAU INDUSTRIE FARMACEUTICHE RIUNITE SOCIETA PER AZIONI
【Fターム(参考)】
[ Back to top ]