
第4級アンモニウム塩および電解質並びに電気化学デバイス
【課題】 高い電気伝導性、耐電圧を有する第4級アンモニウム塩、耐電圧、電気伝導度の高い電解質、耐電圧、電気伝導性が高い電解液、並びに高電圧、高放電容量、大電流放電性能を有する電気化学デバイスを提供する。
【解決手段】 (1)で表される第4級アンモニウム塩。
【化1】

(式中、R1〜R2は、共にメチル基を示す。X−は、BF4−を示す。)
【解決手段】 (1)で表される第4級アンモニウム塩。
【化1】
(式中、R1〜R2は、共にメチル基を示す。X−は、BF4−を示す。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、第4級アンモニウム塩および電解質、電解液並びに電気化学デバイスに係る。詳しくは、有機溶媒に対する溶解性が高く、耐電圧、電気伝導性が高い電解質として使用できる機能性材料に関する。
【背景技術】
【0002】
近年、バッテリーやキャパシタをはじめとする電気化学デバイスの出力密度、エネルギー密度向上の要求が高まっており、耐電圧性の観点から電解液は水系よりも有機系が多用されてきている。有機電解液としてはプロピレンカーボネートなどの有機溶媒にアルカリ金属塩や固体アンモニウム塩を溶解させた例が挙げられ、前者はリチウムイオン電池用の電解液として、後者は電気二重層キャパシタ用の電解液として使用されている。有機電解液は水系に比べて電気伝導性が劣っており、電気伝導性を向上するために有機溶媒や電解質に関する研究が数多くおこなわれてきた。この結果、特許文献1(特開平3−58526号公報)では電気二重層キャパシタの電解質として非対称型アンモニウム塩が示されている。テトラアルキルアンモニウム塩の種類と電気伝導性に関してはUe et al., J. Electrochem. Soc. 141(2989) 1994で詳細に検討されており、テトラエチルアンモニウムテトラフルオロボレートやトリエチルメチルアンモニウムテトラフルオロボレートが用いられているのが一般的である。
【発明の開示】
【発明が解決しようとする課題】
【0003】
こうした固体状電解質を溶媒に溶解させた非水電解液では、電解液の電気伝導性は電解質の濃度とともに変化する。濃度の上昇とともに電解液中のイオン濃度が増加することによって電気伝導度が増加するがやがて極大点に達する。電気伝導度が極大点に達し減少し始めるのは電解液中にイオンの数が増すにつれて、溶媒−イオン、イオン−イオン間の相互作用の増大によって電解質が解離しにくくなり、同時に電解液の粘度が増加するためと考えられている。電解質濃度がさらに増加するとそれ以上解離できなくなり、電解質濃度が飽和する。したがって電解質濃度を高めようとした場合には電解質が溶解しにくくなるといった問題があった。また高濃度の電解質を溶解させた電解液を低温環境下で使用すると塩の析出が生じ、電解液の電気伝導性が悪くなってしまうといった問題も生じる。電解質の解離度を高めるには通常高誘電率溶媒が好まれ、プロピレンカーボネート、エチレンカーボネート、γ−ブチロラクトン等が使われてきた。また電解質にはテトラエチルアンモニウムテトラフルオロボレートやトリエチルメチルアンモニウムテトラフルオロボレート等が好適に用いられてきたが、これらの電解質は高誘電率溶媒には比較的溶解するものの、常温において2M程度が限界であり、それ以上の濃度、又は低温域では結晶の析出が生じるといった不具合があった。また低誘電率溶媒にはほとんど溶解せず、電解液としては使用できないレベルであった。
【0004】
また高電圧を求める使用において、溶媒にプロピレンカーボネート、エチレンカーボネート、γ−ブチロラクトン等を使用した場合、電解質を高耐電圧タイプに変換しても溶媒の分解電圧に支配されてしまい、従来のキャパシタの動作電圧は2.5V程度が上限であった。2.5Vを超える電圧で動作させると電解液(主に溶媒)の電気化学的分解が起こり性能の著しい劣化、ガス発生等の好ましくない現象が発生する。ハイブリッド自動車、電気自動車のような移動体のエネルギーストレージデバイスとしてのキャパシタの応用においてはエネルギー密度の向上が求められており、動作電圧の向上はエネルギー密度を向上させる有効な手段であるが、従来の電解液では耐電圧を向上することが不可能であり、より耐電圧の高い、電解質、溶媒が求められていた。より耐電圧の高い溶媒として鎖状カーボネート系が挙げられるが、誘電率の低いこれらの溶媒には従来のテトラエチルアンモニウムテトラフルオロボレートやトリエチルメチルアンモニウムテトラフルオロボレート等といった電解質は溶解度が低く、電解液としては使用できないレベルであった。
【0005】
近年、融点を常温近傍にもつ塩、或いは融点が常温以下である塩(常温溶融塩)が見出されている。こうした塩は常温において固体であっても通常の電解質に比べて高濃度に有機溶媒に溶解することが知られている。また常温溶融塩は特定の有機溶媒とは任意の割合でまざり合う。それゆえ、従来の固体状電解質を有機溶媒に溶解しても達成できなかった高濃度の電解液が得られ、しかも高濃度でありながら低温環境下でも塩が析出するといった問題が生じにくい。さらに常温溶融塩は塩そのものが液体であるため、塩単体を電解液として使用することも可能である。
【0006】
一方、常温溶融塩は液体でありながら、イオンのみからなることから蒸気圧が低く難燃性であることが知られている。それゆえ、常温溶融塩を有機溶媒に高濃度に溶解することにより、電解液を難燃化することが可能である。
【0007】
代表的な常温溶融塩として1−エチル−3メチルイミダゾリウムテトラフロオロボレート(EMI・BF4)が挙げられる。EMI・BF4は高い電気伝導性をもちリチウム二次電池や電気二重層キャパシタをはじめとする電気化学デバイスへの応用が検討されてきた。しかしながら、イミダゾリウム塩の電気化学的安定性は4V程度であり、電気二重層キャパシタへ適用した場合、動作電圧は2.5V程度が上限となり、応用の幅が広がらない状況である。
【0008】
近年、より広い電位範囲で安定な常温溶融塩が検討されている。例えば、特許文献2(特許第2981545号公報)に示されるようなカチオン成分に脂肪族アンモニウム系の骨格を持つ常温溶融塩は5.8V以上の耐電圧を有しリチウム二次電池への適用が可能であるとされている。しかしながら、脂肪族アンモニウム骨格をカチオン成分にもつ常温溶融塩は一般に粘性が高く、電気伝導度が低いという欠点がある。有機溶媒と混合することによって電気伝導性の改善は見られるが、従来の固体状電解質を有機溶媒に溶解した電解液が示す電気伝導性には及んでいない。
【0009】
特許文献3(WO 02/076924号公報)には、アルコキシアルキル基を導入した脂肪族アンモニウム塩は非水系有機溶媒への溶解性に優れ、低温時における塩の析出が起こりにくいことが記載されているが、より有機溶媒に対する溶解性が高く、耐電圧、電気伝導性の高い電解質が求められている。
特許文献3に記載されているジエチルメチルメトキシエチルアンモニウムをカチオンとする常温溶融塩を有機溶媒に溶解させた場合においても、従来の固体状電解質(例えば、トリエチルメチルアンモニウムテトラフルオロボレートなど)を有機溶媒に溶解した電解液が示す電気伝導度には及んでいない。また鎖状カーボネートへの溶解性も満足できるものではなく、より有機溶媒に対する溶解性が高く、耐電圧、電気伝導度の高い電解質が求められている。
【特許文献1】特開平3−58526号公報
【特許文献2】特許第2981545号公報
【特許文献3】WO 02/076924号公報
【0010】
本発明の課題は高い電気伝導性、耐電圧を有する第4級アンモニウム塩を提供することにある。
本発明の課題は有機溶媒に対する溶解性が高く、耐電圧、電気伝導度の高い電解質を提供することにある。
本発明の課題は耐電圧、電気伝導性が高い電解液を提供することにある。
本発明の課題は溶媒に溶解した場合には、高濃度な電解質を含んだ電解液を提供することができ、その結果、高電圧、高放電容量、大電流放電性能を有する電気化学デバイスを提供することにある。
【課題を解決するための手段】
【0011】
本発明は式(1)で表される第4級アンモニウム塩に係る。
【0012】
【化1】
(式中、R1〜R2は、共にメチル基を示す。X−は、BF4−を示す。)
【0013】
本発明者らは、電気伝導性の向上という課題を解決できる新規化学物質を開発すべく鋭意研究を重ねた結果、式(1)で表されるN,O−アセタール骨格構造を分子内に持つカチオンが、高い電気伝導性を持ち、中でも特にピロリジン骨格とN,O−アセタール基を持つアンモニウムカチオンが電気伝導性、耐電圧、有機溶媒に対する溶解性が高いことを見出した。
【発明の効果】
【0014】
本発明の電解質を使用すれば、耐電圧、電気伝導性が高い電解液を提供することができ、溶媒に溶解した場合には、従来の固体状電解質を有機溶媒に溶解した電解液をしのぐ電気伝導性をもち、高濃度な電解質を含んだ電解液を提供することができ、その結果、高電圧、高放電容量、大電流放電性能を有する電気化学デバイスが得られる。また鎖状カーボネートへの溶解性にも優れ、高い耐電圧を要する用途にも好適に使用できる。
【発明を実施するための最良の形態】
【0015】
以下に本発明の実施の形態を説明する。
本発明は式(1)で表される第4級アンモニウム塩であり、第4級アンモニウムカチオンとBF4−アニオンとから構成される。第4級アンモニウムカチオンの具体例としてはN−メチル−N−メトキシメチルピロリジニウムカチオン(N−メトキシメチル−N−メチルピロリジニウムカチオン)である。
【0016】
本発明で得られる第4級アンモニウム塩は、常温で液状を示す常温溶融塩として、該塩そのものを電解液として用いることができる。
本発明で得られる第4級アンモニウム塩を電解質として使用する場合は、適当な有機溶媒に混合して用いてもよく、有機溶媒としては、環状炭酸エステル、鎖状炭酸エステル、リン酸エステル、環状エーテル、鎖状エーテル、ラクトン化合物、鎖状エステル、ニトリル化合物、アミド化合物、スルホン化合物などが挙げられる。例えば、以下の化合物が挙げられるがこれらに限定されるものではない。
【0017】
環状炭酸エステルとしては、エチレンカーボネート、プロピレンカーボネート、ブチレンカーボネートなどが挙げられ、好ましくは、プロピレンカーボネートが良い。
鎖状炭酸エステルとしては、ジメチルカーボネート、エチルメチルカーボネート、ジエチルカーボネートなどが挙げられ、好ましくは、ジメチルカーボネート、エチルメチルカーボネートが良い。
【0018】
リン酸エステルとしては、リン酸トリメチル、リン酸トリエチル、リン酸エチルジメチル、リン酸ジエチルメチルなどが挙げられる。
環状エーテルとしては、テトラヒドロフラン、2−メチルテトラヒドロフランなどが挙げられる。
鎖状エーテルとしては、ジメトキシエタンなどが挙げられる。
ラクトン化合物としては、γ−ブチロラクトンなどが挙げられる。
鎖状エステルとしては、メチルプロピオネート、メチルアセテート、エチルアセテート、メチルホルメートなどが挙げられる。
ニトリル化合物としては、アセトニトリルなどが挙げられる。
アミド化合物としては、ジメチルホルムアミドなどが挙げられる。
スルホン化合物としては、スルホラン、メチルスルホランなどが挙げられる。
好ましくは、環状炭酸エステル、鎖状炭酸エステル、ニトリル化合物、スルホン化合物が良い。
【0019】
これらの溶媒は1種類でも2種類以上を混合してもよい。好ましい混合有機溶媒としては、環状炭酸エステルと鎖状炭酸エステル、例えば、エチレンカーボネートとジメチルカーボネート、エチレンカーボネートとエチルメチルカーボネート、エチレンカーボネートとジエチルカーボネート、プロピレンカーボネートとジメチルカーボネート、プロピレンカーボネートとエチルメチルカーボネート、プロピレンカーボネートとジエチルカーボネート、鎖状炭酸カーボネート同士、例えば、ジメチルカーボネートとエチルメチルカーボネート、スルホラン化合物同士、例えば、スルホランとメチルスルホランが良い。更に好ましくは、エチレンカーボネートとエチルメチルカーボネート、プロピレンカーボネートとエチルメチルカーボネート、ジメチルカーボネートとエチルメチルカーボネートが良い。
【0020】
本発明の第4級アンモニウム塩を電解質として使用する場合、電解質濃度は0.1M以上であることが好ましく、より好ましくは0.5M以上であり、さらに好ましいのは1M以上である。0.1Mに満たない場合には電気伝導性が低くなり、電気化学デバイスの性能を低下させてしまう。上限濃度は、常温で液体の塩に関しては、分離する濃度とする。分離しない場合は100%とする。また、常温で固体の塩に関しては、塩が飽和する濃度を上限濃度とする。
【0021】
本発明の電解質は本発明以外の電解質と混合使用することができる。本発明の電解質と混合して使用する電解質としては、たとえばアルカリ金属塩、4級アンモニウム塩、4級ホスホニウム塩などが挙げられ、これらの電解質のうち1種類でも、2種類以上を併用し、混合して使用してもよい。アルカリ金属塩としては、リチウム塩、ナトリウム塩、カリウム塩が挙げられ、例えば、6フッ化リン酸リチウム、硼フッ化リチウム、過塩素酸リチウム、トリフロロメタンスルホン酸リチウム、スルホニルイミドリチウム、スルホニルメチドリチウムなどが挙げられるがこれらに限定するものではない。ナトリウム塩としては6フッ化リン酸ナトリウム、硼フッ化ナトリウム、過塩素酸ナトリウム、トリフルオロメタンスルホン酸ナトリウム、スルホニルイミドナトリウム、スルホニルメチドナトリウムなどが挙げられる。カリウム塩としては6フッ化リン酸カリウム、硼フッ化カリウム、過塩素酸カリウム、トリフルオロスルホン酸カリウム、スルホニルイミドカリウム、スルホニルメチドカリウムなどが挙げられるがこれらに限定するものではない。
【0022】
4級アンモニウム塩としては、テトラアルキルアンモニウム塩、イミダゾリウム塩、ピラゾリウム塩、ピリジニウム塩、トリアゾリウム塩、ピリダジニウム塩などが挙げられるがこの限りではない。テトラアルキルアンモニウム塩としては、テトラエチルアンモニウムテトラフルオロボレート、テトラメチルアンモニウムテトラフルオロボレート、テトラプロピルアンモニウムテトラフルオロボレート、テトラブチルアンモニウムテトラフルオロボレート、トリエチルメチルアンモニウムテトラフルオロボレート、トリメチルエチルアンモニウムテトラフルオロボレート、ジメチルジエチルアンモニウムテトラフルオロボレート、トリメチルプロピルアンモニウムテトラフルオロボレート、トリメチルブチルアンモニウムテトラフルオロボレート、ジメチルエチルプロピルアンモニウムテトラフルオロボレート、メチルエチルプロピルブチルアンモニウムテトラフルオロボレート、N,N−ジメチルピロリジニウムテトラフルオロボレート、N−エチル−N−メチルピロリジニウムテトラフルオロボレート、N−メチル−N−プロピルピロリジニウムテトラフルオロボレート、N−エチル−N−プロピルピロリジニウムテトラフルオロボレート、N,N−ジメチルピペリジニウムテトラフルオロボレート、N−メチル−N−エチルピペリジニウムテトラフルオロボレート、N−メチル−N−プロピルピペリジニウムテトラフルオロボレート、N−エチル−N−プロピルピペリジニウムテトラフルオロボレート、N,N−ジメチルモルホリニウムテトラフルオロボレート、N−メチル−N−エチルモルホリニウムテトラフルオロボレート、N−メチル−N−プロピルモルホリニウムテトラフルオロボレート、N−エチル−N−プロピルモルホリニウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。イミダゾリウム塩としては、1,3−ジメチルイミダゾリウムテトラフルオロボレート、1−エチル−3−メチルイミダゾリウムテトラフルオロボレート、1,3−ジエチルイミダゾリウムテトラフルオロボレート、1,2−ジメチル−3−エチルイミダゾリウムテトラフルオロボレート、1,2−ジメチル−3−プロピルイミダゾリウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。ピラゾリウム塩としては1,2−ジメチルピラゾリウムテトラフルオロボレート、1−メチル−2−エチルピラゾリウムテトラフルオロボレート、1−プロピル−2−メチルピラゾリウムテトラフルオロボレート、1−メチル−2−ブチルピラゾリウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。ピリジニウム塩としてはN−メチルピリジニウムテトラフルオロボレート、N−エチルピリジニウムテトラフルオロボレート、N−プロピルピリジニウムテトラフルオロボレート、N−ブチルピリジニウムテトラフルオロボレートなどが挙げらるがこれらの限りではない。トリアゾリウム塩としては、1−メチルトリアゾリウムテトラフルオロボレート、1−エチルトリアゾリウムテトラフルオロボレート、1−プロピルトリアゾリウムテトラフルオロボレート、1−ブチルトリアゾリウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。ピリダジニウム塩としては1−メチルピリダジニウムテトラフルオロボレート、1−エチルピリダジニウムテトラフルオロボレート、1−プロピルピリダジニウムテトラフルオロボレート、1−ブチルピリダジニウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。
【0023】
4級ホスホニウム塩としては、テトラエチルホスホニウムテトラフルオロボレート、テトラメチルホスホニウムテトラフルオロボレート、テトラプロピルホスホニウムテトラフルオロボレート、テトラブチルホスホニウムテトラフルオロボレート、トリエチルメチルホスホニウムテトラフルオロボレート、トリメチルエチルホスホニウムテトラフルオロボレート、ジメチルジエチルホスホニウムテトラフルオロボレート、トリメチルプロピルホスホニウムテトラフルオロボレート、トリメチルブチルホスホニウムテトラフルオロボレート、ジメチルエチルプロピルホスホニウムテトラフルオロボレート、メチルエチルプロピルブチルホスホニウムテトラフルオロボレートなどが挙げられるがこれらに限定するものではない。これらは1種でも2種以上を併用してもよい。
【0024】
尚、上記テトラフルオロボレートをビストリフルオロメタンスルホニルイミド、ヘキサフルオロホスフェート、トルフルオロアセテートに代えたものも含まれる。
【0025】
本発明の電解質と上記電解質とを混合し、電解質として使用する際、混合使用する上記電解質の上限濃度は電解質の析出あるいは分離を生じる濃度とする。混合使用する電解質の下限濃度は適用する電気化学デバイスの種類に拠る。例えば、電気二重層キャパシタの電解質として使用する際は、本発明の第4級アンモニウム塩だけでも使用可能なので、混合使用する電解質の下限濃度は0Mである。リチウム電池に使用する際は、少なくとも前記載リチウム塩を混合使用する。リチウム塩濃度は0.1M以上、2.0M以下であることが好ましく、より好ましくは0.15M以上、1.5M以下であることが好ましく、さらにより好ましいのは0.2M以上、1.2M以下である。特に好ましいのは0.3M以上、1.0M以下である。
【0026】
本発明の第4級アンモニウム塩(1)は種々の方法で製造される。その代表的な合成方法を下記反応式−1及び反応式−2で示す。
【0027】
【化2】
【0028】
式(7)のアルキルピロリジン(R1は上記と同じ)と式(8)の化合物(R2は上記と同じでYはCl、Br、Iなどを示す)とを反応させることにより、式(5)で表される第4級アンモニウム塩が製造され、次いで得られる式(5)で表される第4級アンモニウム塩と式(9)で表される化合物とを反応させることにより、XがY以外のXを示す式(1)で表される第4級アンモニウム塩が製造される。式(9)においてMで示される原子はH又はNa、K、Li等のアルカリ金属原子、Ca、Mg、Ba等のアルカリ土類金属原子、Ag等の金属原子を含む。XはBF4である。
【0029】
式(7)のアルキルピロリジンと式(8)の化合物とを反応させることにより、本発明の式(5)で表される第4級アンモニウム塩が製造される。
出発原料として用いられる式(7)のアルキルピロリジンと式(8)で表される化合物はいずれも公知物質である。式(7)のアルキルピロリジンとしてはメチルピロリジンが挙げられる。式(8)の化合物としてはクロロメチルメチルエーテル、ブロモメチルメチルエーテル、ヨードメチルメチルエーテルが挙げられ、両者の反応は適当な溶媒中で行われる。
【0030】
用いられる溶媒としては、式(7)のアルキルピロリジン及び式(8)の化合物を溶解し得、反応に悪影響を及ぼさない溶媒である限り、公知のものを広く使用できる。このような溶媒としては、例えば、ベンゼン、トルエン、キシレン等の芳香族炭化水素、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素、メタノール、エタノール、イソプロパノール、n−ブタノール、tert−ブタノール等の低級アルコール、アセトン、メチルエチルケトン等のケトン、ジエチルエーテル、ジイソプロピルエーテル等のエーテル、n−ヘキサン、n−ヘプタン等の脂肪族炭化水素、シクロヘキサン等の脂肪族炭化水素等が挙げられる。これらの中でも、トルエン等の芳香族炭化水素、クロロホルム等のハロゲン化炭化水素、アセトン等のケトンが好ましい。斯かる溶媒は、1種単独で又は2種以上混合して使用できる。また溶媒は無水溶媒(水分1000ppm以下)としての使用が特に好ましい。
【0031】
式(7)のアルキルピロリジンと式(8)の化合物との使用割合としては、通常前者1モルに対して後者を通常0.5〜5モル、好ましくは0.9〜1.2モル使用する。
式(7)のアルキルピロリジンと式(8)の化合物との反応は、通常−30〜100℃において行われ、更に詳しくは−10〜40℃にて行われる。一般に数時間〜24時間程度で完結する。
【0032】
上記で得られる式(5)で表される第4級アンモニウム塩と式(9)の化合物との反応は、通常塩交換反応により行われる。
出発原料として用いられる式(9)の化合物は公知化合物であり、例えば、HBF4、LiBF4、NaBF4、KBF4、AgBF4などが挙げられる。
【0033】
この反応は適当な溶媒中で行われる。使用される溶媒としては、式(5)の第4級アンモニウム塩及び式(9)の化合物を溶解し得、反応に悪影響を及ぼさない溶媒である限り、公知のものを広く使用できる。このような溶媒としては、例えば、水又はジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素、メタノール、エタノール、イソプロパノール、n−ブタノール、tert−ブタノール等の低級アルコール、アセトン、メチルエチルケトン等のケトン、酢酸エチル、酢酸ブチル等のエステル、ジメチルスルホキシド、ジメチルホルムアミド等の非プロトン性極性溶媒が挙げられる。これらの中でも、メタノール等の低級アルコール類、クロロホルム等のハロゲン化炭化水素、水が好ましい。これらの溶媒は、1種単独で又は2種以上混合して使用できる。
【0034】
式(5)の第4級アンモニウム塩と式(9)の化合物との使用割合としては、通常前者1モルに対して後者を通常0.3〜5モル、好ましくは0.9〜1.2モル使用する。
式(5)の第4級アンモニウム塩と式(9)の化合物との反応は、通常速やかに進行するので、例えば、両者を溶媒に溶解した溶液を5℃〜150℃で10分〜2時間程度反応させる。
【0035】
上記各反応で得られる目的物は、通常の分離手段、例えば、遠心分離、濃縮、洗浄、有機溶媒抽出、クロマトグラフィー、再結晶等の慣用の単離及び精製手段により、反応混合物から容易に単離、精製される。
【0036】
またハロゲンの混入を嫌う用途の場合、一度ハロゲン塩を中和、塩交換し、ハロゲンを系外に除いた後、更に目的に応じた塩に変換することで、ハロゲンの混入を削減することも出来る。中和剤としては各種アルカリ金属塩、アルカリ土類金属塩、有機アルカリ金属塩、銀塩等が挙げられる。具体的には炭酸ナトリウム、炭酸カリウム、炭酸リチウム、炭酸カルシウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウム、炭酸水素カルシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム、過塩素酸ナトリウム、過塩素酸カリウム、過塩素酸リチウム、酢酸ナトリウム、酢酸カリウム、硫酸銀、硝酸銀、過塩素酸銀等が挙げられる。反応形式は先の式(1)で表される第4級アンモニウム塩を合成する手法にて行うことができ、脱ハロゲン中間体としては、式(6)で表すことができる。
【0037】
【化3】
(式中、R1〜R2は、メチル基を示す。Z−は、1/2CO32−、HCO3−、1/2SO42−、ClO4−、CH3CO2−、OH−を示す。)
【0038】
具体的には、1−メトキシメチル−1−メチルピロリジニウムカーボネート、1−メトキシメチル−1−メチルピロリジニウムヒドロキシド、1−メトキシメチル−1−メチルピロリジニウムスルホネート、1−メトキシメチル−1−メチルピロリジニウムパークロレート、1−メトキシメチル−1−メチルピロリジニウムアセテート、1−メトキシメチル−1−メチルピロリジニウムハイドロカーボネート等が挙げられる。更に続く目的に応じた塩へ変換する手法も式(1)で表される第4級アンモニウム塩を合成する手法を適用できる。
【0039】
式(5)の第4級アンモニウム塩からXがBF4を示す式(1)の第4級アンモニウム塩を製造する場合の反応条件を具体的に示すと、式(5)の第4級アンモニウム塩を上記低級アルコールに溶解し、この溶液に所定量(例えば、硼フッ化水素酸濃度70wt%以下)のメタノール硼フッ化水素酸、硼フッ化銀等のフッ化硼素塩を添加し、5℃〜150℃で30分程度反応させる。反応により生成するハロゲン化水素を留去し、またハロゲン化銀等のハロゲン塩を濾別し、濾液を減圧濃縮し、乾燥することにより、目的化合物を単離することができる。尚、ハロゲン化水素の留去には、例えば、遠心分離、熱時下N2バブリング(例えば、60℃〜150℃)による留去、減圧による留去等を適用できる。上記方法で得られた本発明の第4級アンモニウム塩を電解質として使用する際には、水分がデバイス性能に悪影響を与えるため、水分を十分に取り除く必要がある。水分は、熱時下N2バブリングによる留去、減圧による留去等を適用できるがこれらの手法に限定されるわけではない。含水分量は100ppm以下であることが好ましく、より好ましくは50ppm以下、さらに好ましいのは30ppm以下であり、特に好ましいのは10ppm以下である。
【0040】
またハロゲンの混入を嫌う用途の一般式(1)の第4級アンモニウム塩を製造する場合の反応条件を具体的に示すと、一般式(5)の第4級アンモニウム塩をメタノール又は水等に溶解し、この溶液に所定量の炭酸ナトリウムや硫酸銀等のハロゲン以外の金属塩を添加し、0〜50℃で1時間程度反応させる。溶媒を減圧濃縮、真空乾燥した後、反応により生成したハロゲン化金属塩が不溶で、第4級アンモニウム塩が可溶な溶媒、例えば、ジクロロメタン等のハロゲン溶媒やイソプロパノール、ブタノール等のアルコール類等に再溶解し、ハロゲン塩を濾別する。濾液を減圧濃縮し、乾燥することにより、大部分のハロゲンを除いた第4級アンモニウム塩を製造することができる。
【0041】
【化4】
【0042】
式(10)のアルコキシピロリジン(R2は上記と同じ)と式(11)の化合物(R1及びYは上記と同じ)とを反応させることにより、式(5)で表される第4級アンモニウム塩が製造され、次いで得られる式(5)で表される第4級アンモニウム塩と式(9)で表される化合物(M及びXは上記と同じ)とを反応させることにより、XがY以外のXを示す式(1)で表される第4級アンモニウム塩が製造される。
【0043】
式(10)のアルコキシピロリジンと式(11)の化合物とを反応させることにより、本発明の式(5)で表される第4級アンモニウム塩が製造される。
出発原料として用いられる式(10)のアルコキシピロリジンは公知の手法によって合成できる。例えばC.M.McLeod
und G.M.Robinson,J.Chem.Soc.119,1470(1921). G.M.Robinson und R.Robinson,J.Chem.Soc.123,532(1923). Stewert,T.D;Bradly,W.E.J.Am.Chem.Soc.1932,54,4172−4183.に例示されている。
【0044】
式(10)のアルコキシピロリジンの一般的な合成方法は原料にピロリジン、ホルムアルデヒドあるいはパラホルムアルデヒド、アルコール、炭酸アルカリを用いて合成する。使用割合はピロリジン1モルに対し、ホルムアルデヒドあるいはパラホルムアルデヒドを0.5〜3.0モル、好ましくは0.6〜1.5モル使用し、アルコールを0.5〜3.0モル、好ましくは2.0〜3.0モル使用し、炭酸アルカリを0.2〜3.0モル、好ましくは0.4〜1.0モル使用して行う。反応温度は−5〜100℃で、反応時間は数時間〜24時間程度で終了する。目的物は抽出、精留により単離できる。
【0045】
式(11)で表させる化合物は公知物質であり、例えばメチルクロライド、メチルブロマイド、メチルアイオダイド、エチルアイオダイド、エチルブロマイド、n−プロピルクロライド、n−プロピルブロマイド、n−プロピルアイオダイド、iso−プロピルクロライド、iso−プロピルブロマイド、iso−プロピルアイオダイド、n−ブチルクロライド、n−ブチルブロマイド、n−ブチルアイダイド、iso−ブチルクロライド、iso−ブチルブロマイド、iso−ブチルアイオダイド、tert−ブチルクロライド、tert−ブチルブロマイド、tert−ブチルアイオダイドなどが挙げられる。式(10)のアルキルピロリジンと式(11)の化合物との反応は適当な溶媒中で行われる。
【0046】
用いられる溶媒としては、式(10)のアルコキルピロリジン及び式(11)の化合物を溶解し得、反応に悪影響を及ぼさない溶媒である限り、公知のものを広く使用できる。このような溶媒としては、例えば、ベンゼン、トルエン、キシレン等の芳香族炭化水素、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素、メタノール、エタノール、イソプロパノール、n−ブタノール、tert−ブタノール等の低級アルコール、アセトン、メチルエチルケトン等のケトン、ジエチルエーテル、ジイソプロピルエーテル等のエーテル、n−ヘキサン、n−ヘプタン等の脂肪族炭化水素、シクロヘキサン等の脂肪族炭化水素等が挙げられる。これらの中でも、アセトン等のケトン、トルエン等の芳香族炭化水素、クロロホルム等のハロゲン化炭化水素が好ましい。斯かる溶媒は、1種単独で又は2種以上混合して使用できる。また溶媒は無水溶媒(水分1000ppm以下)としての使用が特に好ましい。
【0047】
式(10)のアルコキシピロリジンと式(11)の化合物との使用割合としては、通常前者1モルに対して後者を通常0.5〜5モル、好ましくは0.9〜1.2モル使用する。
式(10)のアルコキシピロリジンと式(11)の化合物との反応は、通常、0〜150℃において行われ、一般に24時間〜72時間程度で完結する。低沸点のハロゲン化アルキルで4級化する場合オートクレイブを使用することが好ましい。
上記で得られる式(5)で表される第4級アンモニウム塩と式(9)の化合物との反応は、通常塩交換反応により行われる。
【0048】
この反応は適当な溶媒中で行われる。使用される溶媒としては、式(5)の第4級アンモニウム塩及び式(9)の化合物を溶解し得、反応に悪影響を及ぼさない溶媒である限り、公知のものを広く使用できる。このような溶媒としては、例えば、水又はジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素、メタノール、エタノール、イソプロパノール、n−ブタノール、tert−ブタノール等の低級アルコール、アセトン、メチルエチルケトン等のケトン、酢酸エチル、酢酸ブチル等のエステル、ジメチルスルホキシド、ジメチルホルムアミド等の非プロトン性極性溶媒が挙げられる。これらの中でも、メタノール等の低級アルコール類、クロロホルム等のハロゲン化炭化水素、水が好ましい。これらの溶媒は、1種単独で又は2種以上混合して使用される。
【0049】
式(5)の第4級アンモニウム塩と式(9)の化合物との使用割合としては、通常前者1モルに対して後者を通常0.3〜5モル、好ましくは0.9〜1.2モル使用する。
式(5)の4級アンモニウム塩と式(9)の化合物との反応は、通常速やかに進行するので、例えば、両者を溶媒に溶解した溶液を5℃〜150℃付近で10分〜2時間程度反応させる。
【0050】
上記各反応で得られる目的物は、通常の分離手段、例えば、遠心分離、濃縮、洗浄、有機溶媒抽出、クロマトグラフィー、再結晶等の慣用の単離及び精製手段により、反応混合物から容易に単離、精製される。
またハロゲンの混入を嫌う用途の場合、一度ハロゲン塩を中和、塩交換し、ハロゲンを系外に除いた後、更に目的に応じた塩に変換することで、ハロゲンの混入を削減することも出来る。中和剤としては各種アルカリ金属塩、アルカリ土類金属塩、有機アルカリ金属塩、銀塩等が挙げられる。具体的には炭酸ナトリウム、炭酸カリウム、炭酸リチウム、炭酸カルシウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウム、炭酸水素カルシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム、過塩素酸ナトリウム、過塩素酸カリウム、過塩素酸リチウム、酢酸ナトリウム、酢酸カリウム、硫酸銀、硝酸銀、過塩素酸銀等が挙げられる。反応形式は先の式(1)で表される第4級アンモニウム塩を合成する手法にて行うことができ、脱ハロゲン中間体としては、式(6)で表すことができる。
【0051】
【化5】
(式中、R1〜R2は、メチル基を示す。Z−は、1/2CO32−、HCO3−、1/2SO42−、ClO4−、CH3CO2−、OH−を示す。)
【0052】
具体的には、1−メトキシメチル−1−メチルピロリジニウムカーボネート、1−メトキシメチル−1−メチルピロリジニウムヒドロキシド、1−メトキシメチル−1−メチルピロリジニウムスルホネート、1−メトキシメチル−1−メチルピロリジニウムパークロレート、1−メトキシメチル−1−メチルピロリジニウムアセテート、1−メトキシメチル−1−メチルピロリジニウムハイドロカーボネート等が挙げられる。更に続く目的に応じた塩へ変換する手法も式(1)で表される第4級アンモニウム塩を合成する手法を適用できる。
またアルコキシピロリジンと酸エステル等を反応することで、ハロゲンを含まないハロゲンフリー中間体を製造できる。酸エステルとしては炭酸エステル、硫酸エステル、アルキルエステル、リン酸エステル等が挙げられ、中でも炭酸エステルが好ましい。炭酸エステルとしてはジメチルカーボネート、ジエチルカーボネート、ジプロピルカーボネート、ジイソプロピルカーボネート等が挙げられ、反応は通常オートクレイブ中50〜160℃にて行うことが効率的である。反応時間は数時間〜48時間程度で十分である。
ハロゲンフリー中間体としては、式(12)で表すことができる。
【0053】
【化6】
(式中、R1〜R2は、メチルを示す。Q−はR1OCO2−を示す。)
【0054】
具体的には1−メトキシメチル−1−メチルピロリジニウムメチルカーボネート等が挙げられる。
式(5)の第4級アンモニウム塩からXがBF4を示す式(1)の第4級アンモニウム塩を製造する場合の反応条件を具体的に示すと、式(5)の第4級アンモニウム塩を上記低級アルコールに溶解し、この溶液に所定量(例えば、硼フッ化水素酸濃度70wt%以下)のメタノール硼フッ化水素酸、硼フッ化銀等のフッ化硼素塩を添加し、5℃〜150℃で30分程度反応させる。反応により生成するハロゲン化水素を留去し、またハロゲン化銀等のハロゲン塩を濾別し、濾液を減圧減圧濃縮し、乾燥することにより、目的化合物を単離することができる。尚、ハロゲン化水素の留去には、例えば、遠心分離、熱時下N2バブリング(例えば、60℃〜150℃)による留去、減圧による留去等を適用できる。上記方法で得られた本発明の第4級アンモニウム塩を電解質として使用する際には、水分がデバイス性能に悪影響を与えるため、水分を十分に取り除く必要がある。水分は、熱時下N2バブリングによる留去、減圧による留去等を適用できるがこれらの手法に限定されるわけではない。含水分量は100ppm以下であることが好ましく、より好ましくは50ppm以下、さらに好ましいのは30ppm以下であり、特に好ましいのは10ppm以下である。
【0055】
またハロゲンの混入を嫌う用途の一般式(1)の第4級アンモニウム塩を製造する場合の反応条件を具体的に示すと、一般式(5)の第4級アンモニウム塩をメタノール又は水等に溶解し、この溶液に所定量の炭酸ナトリウムや硫酸銀等のハロゲン以外の金属塩を添加し、0〜50℃で1時間程度反応させる。溶媒を減圧濃縮、真空乾燥した後、反応により生成したハロゲン化金属塩が不溶で、第4級アンモニウム塩が可溶な溶媒、例えば、ジクロロメタン等のハロゲン溶媒やイソプロパノール、ブタノール等のアルコール類等に再溶解し、ハロゲン塩を濾別する。濾液を減圧濃縮し、乾燥することにより、大部分のハロゲンを除いた第4級アンモニウム塩を製造することができる。
【0056】
上記のように、本発明の第4級アンモニウム塩や該塩を有機溶媒に溶解した溶液は、電気二重層キャパシタや二次電池などの電気化学デバイス用電解液として使用することができる。
第4級アンモニウム塩を有機溶媒に溶解した溶液を電気化学デバイス用電解液として使用する場合、電解質濃度は0.1M以上であることが好ましく、より好ましくは0.5M以上であり、さらに好ましいのは1M以上である。0.1Mに満たない場合には電気伝導性が低くなり、電気化学デバイスの性能を低下させてしまう。上限濃度は、常温で液体の塩に関しては、有機溶媒と分離する濃度とする。分離しない場合は100%とする。また、常温で固体の塩に関しては、塩が有機溶媒に飽和する濃度を上限濃度とする。
【0057】
本発明の第4級アンモニウム塩を用いて電気化学デバイス用電解液を好適に調製できる。本発明で得られる電解液は電気エネルギーを物理的な作用或いは化学的な作用により蓄積できる電気化学デバイスに使用できるが、電気二重層キャパシタやリチウム電池に好適に使用できる。
【0058】
以下、本発明の第4級アンモニウム塩を用いた電気二重層キャパシタ用電解液の調製方法を説明する。本発明の第4級アンモニウム塩は塩単体が液体の場合はそれ自身、電解液として使用できるが、適当な有機溶媒と混合して使用してもよい。合成した第4級アンモニウム塩を扱う場合、或いは有機溶媒と混合する場合、作業をおこなう環境としては、水分が電気二重層キャパシタの性能に悪影響を与えるため、大気が混入しない環境であれば特に限定はしないが、アルゴンや窒素などの不活性雰囲気のグローブボックス内において調製作業することが好ましい。作業環境の水分は露点計で管理することができ、マイナス60℃以下であることが好ましい。マイナス60℃以上になると、作業時間が長くなる場合、電解液が雰囲気中の水分を吸収するため電解液中の水分が上昇してしまう。電解液中の水分はカールフィッシャー計で測定することができる。
【0059】
本発明の第4級アンモニウム塩を有機溶媒に溶解した溶液を電気化学デバイス用電解液として使用する場合、前記載の通り電解質濃度は電解液の電気伝導性の観点から0.1M以上であれば電解質の分離を生じない限り限定はしないが、好ましいのは0.5M以上であり、さらに好ましいのは1M以上である。上限濃度は電解質の析出および分離を生じない限り限定しない。使用する有機溶媒としては前記載の通り種々溶媒が挙げられるが、溶媒種によって誘電率や粘性、融点等の物性が異なるため、溶媒種と混合使用する本発明の第4級アンモニウム塩の組み合わせに応じて、混合組成を決定するのが好ましい。例えば、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとプロピレンカーボネートからなる電解液の場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は10〜80重量%が好ましく、より好ましくは15〜70重量%、更に好ましくは20〜60重量%である。N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとアセトニトリルからなる電解液の場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は10〜90重量%が好ましく、より好ましくは20〜70重量%、更に好ましくは30〜60重量%である。N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとジメチルカーボネートからなる電解液の場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は40〜90重量%が好ましく、更に好ましくは60〜80重量%である。N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとエチルメチルカーボネートからなる電解液の場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は65〜90重量%が好ましく、更に好ましくは65〜80重量%である。また有機溶媒を混合使用することもでき、ジメチルカーボネートとエチルメチルカーボネートの混合溶媒を使用する場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は40〜80重量%が好ましい。
【0060】
本発明の第4級アンモニウム塩はリチウム電池用電解液に使用することもできる。電気二重層キャパシタ用電解液の調製時と同様に、水分がリチウム電池特性に悪影響を与えるため、調製作業をおこなう作業環境としては、露点が管理されたグローブボックス内が好ましい。
【0061】
本発明の第4級アンモニウム塩は塩単体が液体の場合は第4級アンモニウム塩にリチウム塩を溶解すれば電解液として使用できる。或いは本発明の第4級アンモニウム塩は適当な有機溶媒と混合し、リチウム塩を溶解すれば電解液として使用できる。使用するリチウム塩は前記載の通り種々の塩が挙げられるが、塩の析出が生じなければ特に限定はしない。リチウム塩濃度は0.1M以上、2.0M以下であることが好ましく、より好ましくは0.15M以上、1.5M以下であることが好ましく、さらにより好ましいのは0.2M以上、1.2M以下である。特に好ましいのは0.3M以上、1.0M以下である。リチウム塩濃度が0.1Mに満たない場合には充放電レートが大きい場合に電極近傍においてリチウムイオンの枯渇が生じ、充放電特性が低下する。またリチウムイオン濃度が2.0Mを超えると電解液の粘度が高くなり、電気伝導性が低くなってしまう。なお、本発明の第4級アンモニウム塩およびリチウム塩を形成するアニオンのうちいずれか一方にはBF4−が含まれていることが好ましい。その理由は定かではないが、テトラフルオロボレートを含む場合には正極集電体として使用されるアルミニウムの表面に不働態皮膜が形成され、アルミニウムの溶出を抑制できるためではないかと考えられる。BF4−の含有量イオン数が電解液中の全アニオン数の0.5%以上になるように調製するのが好ましく、より好ましくは0.8%以上になるように調製するのがよい。上限濃度は、BF4−の含有イオン数が電解液中の全アニオン数の100%とする。
【0062】
また本電解液は有機溶媒に希釈して使用することもできる。使用できる有機溶媒としては環状炭酸エステル、鎖状炭酸エステル、環状エーテル、鎖状エーテル、ニトリル化合物、及びスルホン化合物等があげられ、環状炭酸エステルではエチレンカーボネートやプロピレンカーボネート等が、鎖状炭酸エステルではジメチルカーボネート、エチルメチルカーボネート等があげられ、環状エーテルではテトラヒドロフラン、ヘキサヒドロピラン等が、鎖状エーテルでは1,2−ジメトキシエタン等が、ニトリル化合物ではアセトニトリル等が、スルホン化合物ではスルホラン等があげられる。またこれらの溶媒は混合して用いることができ、エチレンカーボネートとジメチルカーボネート、エチレンカーボネートとエチルメチルカーボネート、エチレンカーボネートとプロピレンカーボネート、エチレンカーボネートとテトラヒドロフラン等があげられる。
【0063】
なお本発明で使用される電解液には特定の有機添加剤を含むことが好ましい。特定の有機添加剤とは下記(式A)、(式B)、(式C)で表される化合物を指す。その理由は、該有機添加剤を含むことにより、リチウム電池負極表面にSEI(Solid Electrolyte Interface)として知られるリチウムイオン選択的透過膜が形成され、常温溶融塩を形成するアンモニウムカチオンの分解や負極材料への挿入を抑制できるためと考えられる。その結果、安定した充放電特性が得られる。これらの有機添加剤は種類によっては希釈有機溶媒としての機能も併せ持つ物質もある。(式A)で表される構造を有するものとして例えばエチレンカーボネート、ビニレンカーボネート、ブチレンカーボネートなどが挙げれられ、(式B)の構造を有するものとして例えばエチレントリチオカーボネート、ビニレントリチオカーボネートなどが挙げられ、(式C)の構造を有するものとしては例えばエチレンサルファイトなどが挙げられるが、これらに限定されるわけではない。これらの添加剤は1種類でも2種類以上を混合してもよい。これら(式A)、(式B)、(式C)で表される有機添加剤の含有量は、使用する有機添加剤がすべて(式A)、(式B)、(式C)で表される構造を有する有機添加剤であってもよいが、好ましいのは全電解液重量に対するこれらの有機添加剤の割合が1重量%以上、40重量%以下であり、さらに好ましいのは1重量%以上、30重量%以下であり、特に好ましいのは1重量%以上、20重量%以下であり、さらに最適な濃度は1重量%以上、10重量%以下である。1重量%以下の場合には負極表面に十分な皮膜が形成されないため、常温溶融塩の分解や挿入が生じてしまう。
【0064】
R3−O(CO)−O−R4 (式A)
R5−S−(CS)−S−R6 (式B)
R7−O(SO)−O−R8 (式C)
(R3〜R8は炭素数が1〜3の飽和炭化水素基或いは不飽和炭化水素基を表し、R3とR4、R5とR6、R7とR8が単結合或いは2重結合或いは3重結合で互いに結びつき環を形成していてもよい。)
【0065】
以上のようにして得られる本発明の電解液を用いて電気二重層キャパシタを好適に作製できる。この電気二重層キャパシタの一例としては、例えば、図7に示すようなものを挙げることができる。以下、図7と共に説明を続ける。
【0066】
図7は、本発明の電気二重層キャパシタの断面を示す図面である。図中、41は電気二重層キャパシタ、42は第一の容器体、43は第一の電極、44は第二の容器体、45は第二の電極、46は隔膜、47は非電導性材料、48は電解液を示す。
第一の容器体42及び第一の電極43、第二の容器体44及び第二の電極45はそれぞれ電気的に接続する。しかし、第一の電極42と第二の電極45の間は、隔膜46により隔離されている。この第一の電極43及び第二の電極45は、対向配置されていることが好ましい。
【0067】
上記第一の容器体42及び第二の容器体44は、電解液48により腐食されない電導性の物質であれば良く、例えば、アルミニウム、ステンレス鋼等の材料が使用される。一方、これと電気的に接続する第一の電極43及び第二の電極45は、電導性のものであれば良いが、高容量を得るためにその表面積が大きい多孔性電極であることが望ましく、例えば、電導性物質の粉末をバインダーと混合して成型したものが好ましく用いられる。また、電導性物質の粉末をバインダーとともにピロリドン等の有機溶剤に混合し、ペースト状にしたものをアルミニウム箔等の集電体に塗工後、乾燥して得たシート状電極が好ましく用いられる。電導性物質としては、活性炭粉末、活性炭繊維等の炭素材料;貴金属酸化物材料;導電性高分子材料等が用いられるが、このうち炭素材料が安価であるため好ましい。更に、第一の電極43と第二の電極45の間に挟み込まれ、これらを隔離する隔膜46としては、電解液が通過しやすく、電子伝導に関しては絶縁体で、化学的に安定な材質であれば特に限定はないが、レーヨン系抄紙、ポリオレフィン系多孔質フィルム、ポリエチレン不織布、ポリプロピレン不織布、セルロース等が好適に用いられる。
【0068】
本発明の電気二重層キャパシタは、上記した第一の容器体42と第二の容器体44の間を電解液48で満たし、更に非電導性材料47で電気的に接続しないように密封することにより製造される。
【0069】
電解液48としては、前記したものが使用されるが、その充填は、充填される容器体等を真空乾燥した後、不活性ガスを満たしたグローブボックス内で電解液8を注液し、エージングを施すことにより行うことが好ましい。なお真空乾燥は、120〜300℃で加熱して行うことが好ましく、その時間は、キャパシタのサイズにもよるが、5〜100時間程度行うことが好ましい。また、エージングは、電極、特に活性炭等で製造された多孔性電極の細孔深部までイオンを吸着させ、微量に含まれる不純物を電気的に分解させるために行われるものであり、室温にて、2〜3Vの範囲で、5〜100時間程度の充電を行うことが好ましい。最後に好ましくは減圧脱泡し、本発明の電気二重層キャパシタを完成させる。
【0070】
以上のようにして製作される本発明の電気二重層キャパシタは、第一の容器体42及び第二の容器体44が、それぞれの内面側で第一の電極43と第二の電極45の集電体となると共に、それぞれの外面側を第一の電極43と第二の電極45の接続端子として用いることができる。
【0071】
以上のようにして得られる本発明の電解液を用いてリチウム二次電池を好適に作成できる。本発明のリチウム二次電池の形態は、コイン型、円筒型、角型、ラミネート等挙げられるが、特に限定されるわけではない。本発明のリチウム二次電池の一例としては、例えば、図9に示すコイン型セルの形態を挙げることができる。以下、図9と共に説明を続ける。コイン型セルは、正極・負極の電極がセパレータを介して配置され、これら正極・負極の活物質層およびセパレータに電解液が含浸されている。1対の正極・負極およびセパレータが図9に示すようにスペーサー、スプリングとともに正極缶および負極缶内部にガスケットを介してかしめられ、密閉される。
【0072】
本発明で使用する正極としては、活物質として、例えばLiCoO2、LiNiO2、LiNi1−xCoxO2、LiNi1−x−yCoxMny、LiNi0.5Mn0.5O2、LiMnO2、LiMn2O4、LiNi0.5Mn1.5O4等のリチウムと遷移金属との複合酸化物、TiO2、V2O5等の酸化物、TiS2、FeS等の硫化物などが挙げられるが、電池容量・エネルギー密度の観点からリチウムと遷移金属との複合酸化物が好ましい。これらの正極活物質を正極として成型する際には、公知の導電助剤や結着剤とともに加圧成型でき、または正極活物質を公知の導電助剤や結着剤とともにピロリドン等の有機溶剤に混合し、ペースト状にしたものをアルミニウム箔等の集電体に塗工後、乾燥して得ることができる。
【0073】
また本発明で使用する負極としては、リチウム金属、リチウム金属と他金属との合金、リチウムイオンが挿入脱離する材料が使用される。リチウム金属と他金属との合金としてはLi−Al、Li−Sn、Li−Zn、Li−Siなどが挙げられ、またリチウムイオンが挿入脱離する材料としては樹脂やピッチ等を焼成したカーボン材料やこれらのカーボン材料にホウ素化合物を添加したカーボン材料、天然黒鉛などを使用できる。これらの負極材料は単独で用いても、2種以上を混合して使用することもできる。これらの負極材料を負極として成型する際には、公知の導電助剤や結着剤とともに加圧成型でき、または負極活物質を公知の導電助剤や結着剤とともにピロリドン等の有機溶剤に混合し、ペースト状にしたものを銅箔等の集電体に塗工後、乾燥して得ることができる。
【0074】
また、本発明で使用するセパレータとしては、電解液が通過しやすく、絶縁体で、化学的に安定な材質であれば特に限定はない。
本発明の第4級アンモニウム塩およびこれを含有する電解液は耐電圧、電気伝導性が高く、有機溶媒に対する溶解性が高く、電気化学デバイスの電解液として好適である。電気化学デバイスとしては、例えば、電気二重層キャパシタ、二次電池、色素増感型太陽電池、エレクトロクロミック素子、コンデンサなどが例示されるがこの限りではない。特に好適な電気化学デバイスは電気二重層キャパシタ、二次電池である。
【実施例】
【0075】
以下に参考例、実施例、試験例を挙げ、本発明を具体的に説明するが、何らこれに限定されるものではない。
【0076】
実施例1
N−メチル−N−メトキシメチルピロリジニウムクロライド(N−メトキシメチル−N−メチルピロリジニウムクロライド)の合成
N−メチルピロリジン(試薬:東京化成製)30.0gを120gのトルエンに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製)31.2gを1時間で滴下した。5℃にて1時間攪拌し、徐々に昇温、室温にて10時間攪拌し、反応を終了した。反応液を濾別し、得られた固体を150gのトルエン、150gのアセトンにて洗浄した。減圧乾燥し53.7gの目的物(白色固体)を得た。
1H−NMR(D2O)δppm:
2.08(br 4H),2.96(s 3H),3.31(m 2H),3.47(m 2H),3.55(s 3H),4.50(s 2H)
【0077】
実施例2
N−メチル−N−メトキシメチルピロリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート)の合成
実施例1で製造したN−メチル−N−メトキシメチルピロリジニウムクロライド(N−メトキシメチル−N−メチルピロリジニウムクロライド)15.0gをMeOH35gに溶解し、30%HBF4のメタノール溶液27.83gを添加した。減圧下、塩化水素と過剰のHBF4を除き目的物(薄黄色液体)19.6gを得た。
1H−NMR(d−DMSO)δppm:
2.07(br 4H),3.00(s 3H),3.42(m 4H),3.60(s 3H),4.62(s 2H)
【0078】
実施例3
実施例2で製造したN−メチル−N−メトキシメチルピロリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート)の電気伝導度、耐電圧の測定をおこなった。
電気伝導度の測定にはRadiometer社製電気伝導度メーターを使用した。測定セルにはRadiometer社製CDC641Tを使用した。
耐電圧の測定には3極式電気化学セルを使用した。作用極として、φ1.0mm、電極面積0.0079cm−2のグラシーカーボン電極(BAS株式会社製)、参照極としてφ0.5mmの銀ワイヤー(株式会社ニラコ製、純度99.99%)、対極としてφ0.5mm×50mmの白金電極(BAS株式会社製、11−2233)を使用した。リニアスイープボルタンメトリーをおこない、酸化電流密度および還元電流密度が0.5mAcm−2になる電位を別々に調べた。これらの電位の差を耐電圧とした。なお電位の挿引印加速度は50mVs−1とした。電気化学測定には北斗電工製、HZ−3000を使用した。測定結果を表1に示した。
【0079】
実施例4
実施例2で製造したN−メチル−N−メトキシメチルピロリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート)とプロピレンカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)を種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表2に示した。
【0080】
実施例5
実施例2で製造したN−メチル−N−メトキシメチルピロリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート)とアセトニトリル(キシダ化学株式会社製、リチウムバッテリーグレード)を種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表3に示した。
【0081】
比較例1
N−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレートの合成
N−メチルピロリジン(試薬:東京化成製)31.10gを124.30gのトルエンに溶解し、窒素置換した。27℃下ブロモエチルメチルエーテル(試薬:アルドリッチ製)61.22gを1時間で滴下した。徐々に昇温し、60〜70℃にて37時間攪拌し、反応を終了した。室温まで冷却し、生じた固体を窒素下濾別した。70gのトルエンにて洗浄した後、減圧乾燥した(茶褐色固体 78.99g)。得られた固体は200gのアセトンに懸濁し、室温にて攪拌洗浄、窒素下濾別した(×2回繰り返した)。減圧乾燥し、収量62.64gを得た。着色があった為、水131.83gに溶解し、活性炭〔カルボラフィン 武田薬品工業(株)製〕6.00gを加え12時間90〜95℃にて攪拌処理した。室温まで、冷却し、活性炭を濾別した。減圧濃縮、減圧乾燥し、収量58.34gを得た。アセトン200.48g、クロロホルム27.22gの混合溶媒に加熱溶解し、再結晶した。生成した白色固体は窒素下濾別し、アセトン50gにて洗浄、減圧乾燥し、N−メトキシエチル−N−メチルピロリジニウムブロマイド34.10gを得た。
1H−NMR(CD3OD)δppm:
2.24(m 4H),3.15(s 3H),3.40(s 3H),3.65(m 6H),3.83(m 2H)
続いて、上記で製造したN−メトキシエチル−N−メチルピロリジニウムブロマイド40.0gをMeOH40.0gに溶解し、30wt%HBF4のメタノール溶液54.0gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4を除き目的物(白色固体)39.9gを得た。
1H−NMR(CD3OD)δppm:
2.22(m 4H),3.10(S 3H),3.39(S 3H),3.58(m 6H),3.81(m 2H)
上記で製造したN−メチル−N−メトキシエチルピロリジニウムテトラフルオロボレート(N−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレート)を、実施例4と同様な測定をおこなった。測定結果を表1に示した。
【0082】
比較例2
N−メトキシメチル−N−メチルピペリジニウムテトラフルオロボレートの合成
N−メチルピペリジン(試薬:東京化成製)54.50gを脱水アセトン(試薬:和光純薬製)700gに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製を蒸留精製)44.30gを1時間で滴下した。滴下終了後は15℃以下にて5時間攪拌し、反応を終了した。5℃まで冷却し、生成した固体を窒素下濾別した。400gのアセトンにて洗浄した後、減圧乾燥した。得られた白色固体はアセトン550gに懸濁し、還流下30分攪拌した。濾過し、アセトン300gにて洗浄した(2回繰り返した)。減圧乾燥し、目的物(N−メトキシメチル−N−メチルピペリジニウムクロライド)66.0gを得た。
1H−NMR(CD3OD)δppm:
1.60〜1.96(m 6H),3.05(s 3H),3.35(m 4H),3.69(s 3H),4.65(s 2H)
続いて、上記で製造したN−メトキシメチル−N−メチルピペリジニウムクロライド35.0gをMeOH35.0gに溶解し、30wt%HBF4のメタノール溶液59.9gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4およびメタノールを除き目的物43.7gを得た。
1H−NMR(CD3OD)δppm:
1.55〜2.00(m 6H),3.04(s 3H),3.34(m 4H),3.67(s 3H),4.62(s 2H)
上記で製造したN−メチル−N−メトキシメチルピペリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピペリジニウムテトラフルオロボレート)を、実施例3と同様な測定をおこなった。測定結果を表1に示した。
【0083】
比較例3
N−メトキシメチル−N−メチルモルホリニウムテトラフルオロボレートの合成
N−メチルモルホリン(試薬:東京化成製)92.13gを脱水2−ブタノン(試薬:和光純薬製)670gに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製を蒸留精製)76.47gを1時間で滴下した。滴下終了後は15℃以下にて2時間攪拌し、反応を終了した。5℃まで冷却し、生成した固体を窒素下濾別した。500mlの2−ブタノンにて洗浄した後、減圧乾燥した。得られた白色固体はアセトン500mlに懸濁し、還流下30分攪拌した。濾過し、アセトン500mlにて洗浄した(2回繰り返した)。減圧乾燥し、目的物(N−メトキシメチル−N−メチルモルホリニウムクロライド)150.46gを得た。
1H−NMR(CD3OD)δppm:
3.22(s 3H),3.36〜3.42(m 2H),3.52〜3.61(m 2H),3.71(s 3H),4.01(m 4H),4.77(s 2H)
続いて、上記で製造したN−メトキシメチル−N−メチルモルホリニウムクロライド30.0gをMeOH30.0gに溶解し、30wt%HBF4のメタノール溶液50.8gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4およびメタノールを除き目的物37.2gを得た。
1H−NMR(CD3OD)δppm:
3.19(s 3H),3.31(m 2H),3.52(m 2H),3.70(s 3H),4.00(m 4H),4.72(s 2H)
上記で製造したN−メチル−N−メトキシメチルモルフォリニウムテトラフルオロボレート(N−メトキシメチル−N−メチルモルフォリニウムテトラフルオロボレート)を用い、1Mのプロピレンカーボネート溶液を調製した。実施例3と同様な測定法で耐電圧を測定した。測定結果を表1に示した。
【0084】
比較例4
N−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムテトラフルオロボレートの合成
エチルジメチルアミン(試薬:東京化成製)47.50gを脱水アセトン(試薬:和光純薬製)300gに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製を蒸留精製)52.30gを1時間で滴下した。滴下終了後は15℃以下にて5時間攪拌し、反応を終了した。5℃まで冷却し、生成した固体を窒素下濾別した。150gのアセトンにて洗浄した後、減圧乾燥した。N−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムクロライド(白色固体)85.90gを得た。
1H−NMR(CD3OD)δppm:
1.35(m 3H),3.03(s 6H),3.40(q 2H),3.68(s 3H),4.61(s 2H)
続いて、上記で製造したN−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムクロライド40.0gをMeOH40.0gに溶解し、30wt%HBF4のメタノール溶液80.0gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4およびメタノールを除き目的物51.6gを得た。
1H−NMR(CD3OD)δppm:
1.34(m 3H),3.00(s 6H),3.38(q 2H),3.66(s 3H),4.57(s 2H)
上記で製造したジメチルエチルメトキシメチルアンモニウムテトラフルオロボレート(N−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムテトラフルオロボレート)を用い、実施例3と同様な測定をおこなった。測定結果を表1に示した。
【0085】
比較例5
N,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレートの合成
ジエチルメチルアミン(試薬:東京化成製)35.53gを161.37gのトルエンに溶解し、窒素置換した。27℃下ブロモエチルメチルエーテル(試薬:アルドリッチ製)68.00gを1時間で滴下した。徐々に昇温し、60〜70℃にて44時間攪拌し、反応を終了した。室温まで冷却し、生じた固体を窒素下濾別した。70gのトルエンにて洗浄した後、減圧乾燥した(茶褐色固体 67.30g)。着色が激しい為、水131.52gに溶解し、活性炭〔カルボラフィン 武田薬品工業(株)製〕7.02gを加え12時間90〜95℃にて攪拌処理した。室温まで、冷却し、活性炭を濾別した。減圧濃縮、減圧乾燥し、収量58.34gを得た。アセトン200.48g、クロロホルム27.22gの混合溶媒に加熱溶解し、再結晶した。生成した白色固体は窒素下濾別し、アセトン50gにて洗浄、減圧乾燥し、目的物(N,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムブロマイド)47.58gを得た。
1H−NMR(CD3OD)δppm:
1.35(m 6H),3.07(s 3H),3.39(s 3H),3.40〜3.57(m 6H),3.80(m 2H)
続いて、上記で製造したN,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムブロマイド30.0gをMeOH30.0gに溶解し、30wt%HBF4のメタノール溶液40.8gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4およびメタノールを除き目的物30.2gを得た。
1H−NMR(CD3OD)δppm:
1.33(m 6H),3.03(s 3H),3.38(s 3H),3.39〜3.52(m 6H),3.77(m 2H)
上記で製造したジエチルメチルメトキシエチルアンモニウムテトラフルオロボレート(N,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレート)を用い、実施例3と同様な測定をおこなった。測定結果を表1に示した。
【0086】
比較例6
N,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレート(TEMA)
トリエチルメチルアンモニウムクロライド(試薬:東京化成製)100gをメタノール100gに溶解し、30wt%HBF4のメタノール溶液200.0gを添加した。30分攪拌するとトリエチルメチルアンモニウムテトラフルオロボレートの結晶が析出した。溶液を濾過後、結晶をイソプロピルアルコールで洗浄してから、130℃の加熱下、窒素気流中にて乾燥し、副生した塩化水素と過剰のHBF4およびメタノール、イソプロピルアルコールを除き目的物(白色固体)127.1gを得た。
1H−NMR(CD3OD)δppm:
1.31(m 9H),2.95(S 3H),3.34(q 6H)
上記で製造したトリエチルメチルアンモニウムテトラフルオロボレート(N,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレート)を、1Mのプロピレンカーボネート溶液を調整した。実施例3と同様な測定法で耐電圧を測定した。測定結果を表1に示した。
【0087】
比較例7
上記で製造したN−メチル−N−メトキシエチルピロリジニウムテトラフルオロボレート(N−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレート)とプロピレンカーボネートを種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表4に示した。
【0088】
比較例8
上記で製造したN−メチル−N−メトキシエチルピロリジニウムテトラフルオロボレート(N−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレート)とアセトニトリルを種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表5に示した。
【0089】
比較例9
上記で製造したジエチルメチルメトキシエチルアンモニウムテトラフルオロボレート(N,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレート)とプロピレンカーボネートを種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表6に示した。
【0090】
比較例10
上記で製造したトリエチルメチルアンモニウムテトラフルオロボレート(N,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレート)とプロピレンカーボネートを種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表7に示した。
【0091】
【表1】
【0092】
【表2】
【0093】
【表3】
【0094】
【表4】
【0095】
【表5】
【0096】
【表6】
【0097】
【表7】
【0098】
実施例6
N−メトキシメチル−N−メチルピロリジニウムクロライドの合成
N−メチルピロリジン(試薬:東京化成製を精留により精製 ピロリジン、水含有量共に0.1%以下)50.0gを292.0gの脱水アセトン(水含有量0.1%以下)に溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製を蒸留精製)47.3gを1時間で滴下した。5℃にて1時間攪拌し、5〜15℃以下にて4時間攪拌し、反応を終了した。反応液を濾別し、得られた固体を120gのアセトンにて洗浄した。減圧乾燥し92.5gの目的物(白色固体)を得た。
1H−NMR(CD3OD)δppm:
2.22(m 4H),3.11(s 3H),3.46(m 2H),3.60(m 2H),3.67(s 3H),4.65(s 2H)
【0099】
実施例7
N−メトキシメチル−N−メチルピロリジニウムクロライドの合成
N−メチルピロリジン(試薬:東京化成製)30.0gを150gのトルエンに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製)31.2gを1時間で滴下した。5℃にて1時間攪拌し、徐々に昇温、室温にて10時間攪拌し、反応を終了した。反応液を濾別し、得られた固体を150gのアセトンにて洗浄した。減圧乾燥し53.7g白色固体を得た。得られた第4級アンモニウム塩をアセトン150gに懸濁し5時間攪拌した。濾過、洗浄、乾燥し白色固体48.3gを得た。続き得られた第4級アンモニウム塩はクロロホルム/アセトン(1/6(W/W))420gで再結晶、減圧乾燥し36.2gの目的物(白色固体)を得た。
1H−NMR(CD3OD)δppm:
2.22(m 4H),3.11(s 3H),3.46(m 2H),3.60(m 2H),3.67(s 3H),4.65(s 2H)
【0100】
実施例8
N−メトキシメチル−N−メチルピロリジニウムブロマイドの合成
N−メチルピロリジン17.0gを160gの脱水アセトン(水0.1%以下)に溶解し、窒素置換した。5℃下ブロモメチルメチルエーテル(試薬:東京化成製)24.6gを1.5時間で滴下した。5〜15℃以下にて4時間攪拌し、反応を終了した。反応液を濾別し、得られた固体を160gのアセトンにて洗浄した。減圧乾燥し30.9gの目的物(白色固体)を得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.11(s 3H),3.48(m 2H),3.60(m 2H),3.67(s 3H),4.65(s 2H)
【0101】
実施例9
N−メトキシメチル−N−メチルピロリジニウムアイオダイドの合成
N−メチルピロリジン(試薬:東京化成製を精留により精製 ピロリジン、水含有量共に0.1%以下)2.46gを21.74gの脱水2−ブタノン(水0.1%以下)に溶解し、窒素置換した。5℃下ヨードメチルメチルエーテル(試薬:Aldrich製を精留により精製)5.07gを1.5時間で滴下した。5〜15℃にて5時間攪拌し反応を終了した。反応液を濾別し、濾液を減圧乾燥し6.40gの目的物(薄赤褐色液体)を得た。
1H−NMR(CD3OD)δppm:
2.23(m 4H),3.13(s 3H),3.50(m 2H),3.62(m 2H),3.68(s 3H),4.68(s 2H)
【0102】
実施例10
N−メトキシメチル−N−メチルピロリジニウムアイオダイドの合成
パラホルムアルデヒド(試薬:和光純薬製)25.3gと炭酸カリウム58.3gをメタノール81.0gに懸濁させ、ピロリジン(試薬:東京化成製)60.0gを室温下1時間かけて滴下した。滴下終了後は昇温し、70℃で3時間反応した。反応終了後、室温まで冷却し濾過した。濾液は精留し無色透明液体メトキシメチルピロリジン68.9gを得た。得られたメトキシメチルピロリジンはアセトン600gに溶解し、ヨウ化メチル93.6gを添加し、窒素置換したオートクレイブ中80℃にて3日間攪拌した。濾過し、濾液を減圧乾燥することで目的物(赤褐液体)107.3gを得た。
メトキシメチルピロリジンの1H−NMRデータ
1H−NMR(CDCl3)δppm:
1.77(m 4H),2.76(m 4H),3.31(s 3H),4.14(s 2H)
N−メトキシメチル−N−メチルピロリジニウムアイオダイドの1H−NMRデータ
1H−NMR(CD3OD)δppm:
2.23(m 4H),3.13(s 3H),3.50(m 2H),3.62(m 2H),3.68(s 3H),4.68(s 2H)
【0103】
実施例11
N−メトキシメチル−N−メチルピロリジニウムカーボネートの合成
炭酸ナトリウム(和光純薬製)1.60gを脱イオン水18gに溶解し、N−メトキシメチル−N−メチルピロリジニウムクロライド5.01gを添加した。室温で0.5時間反応し反応を終了した。濃縮真空乾燥した後、残さにエチルアルコール100mlを加え不溶の塩化ナトリウムを除いた。ジクロロメタンに溶解し、メンブランフィルターによる再濾過を行い、減圧濃縮、乾燥し目的物5.41gを得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.11(s 3H),3.47(m 2H),3.59(m 2H),3.67(s 3H),4.64(s 2H)
【0104】
実施例12
N−メトキシメチル−N−メチルピロリジニウムスルホネートの合成
硫酸銀(和光純薬製)3.14gを脱イオン水400mlに溶解し、N−メトキシメチル−N−メチルピロリジニウムクロライド3.34gを添加した。室温で0.5時間反応し反応を終了した。生じた塩化銀を濾過し、濃縮真空乾燥した。メタノールに溶解し、メンブランフィルターによる再濾過を行い、減圧濃縮、乾燥し目的物3.99gを得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.11(s 3H),3.48(m 2H),3.61(m 2H),3.67(s 3H),4.65(s 2H)
【0105】
実施例13
N−メトキシメチル−N−メチルピロリジニウムパークロレートの合成
過塩素酸ナトリウム(和光純薬製)5.91gをエチルアルコール77gに溶解し、N−メトキシメチル−N−メチルピロリジニウムクロライド7.99gを添加した。室温で1.5時間反応し反応を終了した。生じた塩化ナトリウムを濾過し、濃縮真空乾燥した。ジクロロメタンに溶解し、メンブランフィルターによる再濾過を行い、減圧濃縮、乾燥し目的物10.94gを得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.10(s 3H),3.46(m 2H),3.58(m 2H),3.66(s 3H),4.61(s 2H)
【0106】
実施例14
N−メトキシメチル−N−メチルピロリジニウムフルオライドの合成
フッ化カリウム(和光純薬製)0.44gを脱イオン水11gに溶解し、N−メトキシメチル−N−メチルピロリジニウムパークロレート1.74gを添加した。室温で1.5時間反応し反応を終了した。メタノール100mlを添加し濾過した。濃縮真空乾燥した後、ジクロロメタンに溶解しメンブランフィルターによる再濾過を行った。濃縮乾燥し目的物1.05gを得た。
1H−NMR(CD3OD)δppm:
2.20(m 4H),3.09(s 3H),3.46(m 2H),3.58(m 2H),3.66(s 3H),4.60(s 2H)
【0107】
実施例15
N−メトキシメチル−N−メチルピロリジニウムメチルカーボネートの合成
メトキシメチルピロリジン10.00gと炭酸ジメチル117.39gをオートクレイブに添加した。120℃にて24時間反応した。生じた固体を濾過し、炭酸ジメチルにて洗浄した。減圧乾燥し、目的物10.70gを得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.10(s 3H),3.34(s 3H),3.45(m 2H),3.58(m 2H),3.66(s 3H),4.62(s 2H)
【0108】
実施例16
N−メトキシメチル−N−メチルピロリジニウムアセテートの合成
酢酸ナトリウム(和光純薬製)9.46gをメタノール95gに溶解し、N−メトキシメチル−N−メチルピロリジニウムクロライド19.10gを添加した。室温で1.5時間反応し反応を終了した。濾過した後、濃縮真空乾燥した。残さにジクロロメタン100mlを加えメンブランフィルターによる濾過を行い、減圧濃縮、乾燥し目的物20.38gを得た。
1H−NMR(CD3OD)δppm:
1.89(s 3H),2.20(m 4H),3.10(s 3H),3.44(m 2H),3.60(m 2H),3.66(s 3H),4.61(s 2H)
【0109】
実施例17
N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの合成
実施例6で製造したN−メトキシメチル−N−メチルピロリジニウムクロライド50.0gをMeOH120gに溶解し、30%HBF4のメタノール溶液92.8gを添加した。130℃の加熱下、N2バブリングをおこない、塩化水素と過剰のHBF4を除き目的物(微黄色液体)65.2gを得た。
1H−NMR(CD3OD)δppm:
2.19(m 4H),3.08(s 3H),3.43(m 2H),3.56(m 2H),3.65(s 3H),4.59(s 2H)
【0110】
比較例11
テトラエチルアンモニウムテトラフルオロボレート(TEA)
テトラエチルアンモニウムブロマイド(試薬:東京化成製)120gをメタノール120gに溶解し、30wt%HBF4のメタノール溶液172gを添加した。30分攪拌するとテトラエチルアンモニウムテトラフルオロボレートの結晶が析出した。溶液を濾過後、結晶をイソプロピルアルコールで洗浄してから、130℃の加熱下、窒素気流中にて乾燥し、副生した臭化水素と過剰のHBF4およびメタノール、イソプロピルアルコールを除き目的物(白色固体)118gを得た。
1H−NMR(CD3OD)δppm:
1.28(m 12H),3.29(q 8H)
【0111】
<電気二重層キャパシタ用電解液の調製>
実施例18(MMMP−BF4/PC)
実施例17で製造したN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとプロピレンカーボネート(PC)(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表8に掲げる通りとした。
【0112】
実施例19(MMMP−BF4/DMC)
実施例17で製造したN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとジメチルカーボネート(DMC)(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表9に掲げる通りとした。
【0113】
実施例20(MMMP−BF4/EMC)
実施例17で製造したN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとエチルメチルカーボネート(EMC)(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表10に掲げる通りとした。
【0114】
実施例21(MMMP−BF4/DMC+EMC)
実施例17で製造したN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとジメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)およびエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表11に掲げる通りとした。
【0115】
比較例12(TEMA−BF4/PC)
比較例6で製造したN,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレートとプロピレンカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表12に掲げる通りとした。
【0116】
比較例13(TEA−BF4/PC)
比較例11で製造したテトラエチルアンモニウムテトラフルオロボレートとプロピレンカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、テトラエチルアンモニウムテトラフルオロボレートの濃度が0.8Mになるように、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表13に掲げる通りとした。尚テトラエチルアンモニウムテトラフルオロボレートとジメチルカーボネート及びエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で混合したが不溶であった。
【0117】
比較例14(EMI−BF4/PC)
1−エチル−3−メチルイミダゾリウムテトラフルオロボレート(EMI−BF4)とプロピレンカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表14に掲げる通りとした。
【0118】
比較例15(TEMA−BF4/EMC)
比較例6で製造したN,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表15に掲げる通りとした。
【0119】
比較例16(TEMA−BF4/DMC)
比較例6で製造したN,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレートとジメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表16に掲げる通りとした。
【0120】
比較例17(EMI−BF4/EMC)
1−エチル−3−メチルイミダゾリウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表17に掲げる通りとした。
【0121】
比較例18(EMI−BF4/DMC)
1−エチル−3−メチルイミダゾリウムテトラフルオロボレートとジメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表18に掲げる通りとした。
【0122】
比較例19
比較例4で製造したN−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表19に掲げる通りとした。
【0123】
比較例20
比較例5で製造したN,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表20に掲げる通りとした。
【0124】
比較例21(MMEP−BF4/EMC)
比較例1で製造したN−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表21に掲げる通りとした。
【0125】
比較例22(MMMPI−BF4/EMC)
比較例2で製造したN−メトキシメチル−N−メチルピペリジニウムテトラフルオロボレート(MMMPI−BF4)とエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表22に掲げる通りとした。
【0126】
比較例23(MMMM−BF4/EMC)
比較例3で製造したN−メトキシメチル−N−メチルモルホリニウムテトラフルオロボレート(MMMM−BF4)とエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表23に掲げる通りとした。
【0127】
<混合状態の観察>
実施例18〜実施例21および比較例12〜比較例23で得られた各種組成物を、ドライボックス内で4ccずつ、スクリュー栓が付いたガラス容器に移し、ドライボックスの外に取り出した。各種溶液が入ったガラス容器を恒温槽に浸漬し、25℃、0℃、−30℃でそれぞれ5時間保持し、目視で状態を確認した。結果を表8〜表23に示した。表において、「−」は二層分離を、「固」は固体状態を示す。
【0128】
<電気伝導度の測定>
混合状態の観察後、分離していない、或いは固化していない液体状態の組成溶液について、再度ドライボックス内から溶液を取り出し電気伝導度を測定した。電気伝導度の測定には、導電率計(CDM210 Radiometer社製)を使用した。測定セルにはXE−100(Radiometer社製)を使用した。結果を、表8〜表23に示した。
【0129】
【表8】
【0130】
【表9】
【0131】
【表10】
【0132】
【表11】
【0133】
【表12】
【0134】
【表13】
【0135】
【表14】
【0136】
【表15】
【0137】
【表16】
【0138】
【表17】
【0139】
【表18】
【0140】
【表19】
【0141】
【表20】
【0142】
【表21】
【0143】
【表22】
【0144】
【表23】
【0145】
実施例22〜23および比較例24
<円筒型電気二重層キャパシタの製造等>
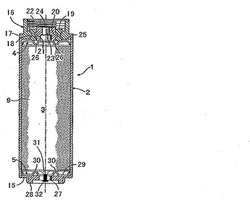
図1において、円筒型電気二重層キャパシタ1は円筒状密閉容器2を有し、その密閉容器2内に電極巻回体3、2つの円盤状集電体4、5および電解液が収容されている。
図2に示すように、電極巻回体3は、帯状正極6、帯状負極7およびそれらの一方、実施例では帯状正極6を挟む2つの帯状セパレータ8、9よりなる重合せ物を、帯状正極6の外側に在る一方の帯状セパレータ8が最も内側に位置するようにAl製巻心10を中心に渦巻状に巻回したものである。この場合、帯状正、負極6、間に存する他方の帯状セパレータ9は、外周の帯状負極7を覆うべく略一巻分だけ帯状負極7の終端より延出している。
帯状正極6は、それぞれ帯状集電体11およびその帯状集電体11の両面に積層形成された一対の分極性電極12を有する。帯状正極6において、集電体11両面の長手方向一側縁部は電極不存在領域であって正極端子との接続部13として機能する。帯状負極7は帯状正極6とは点対称の関係にあり、その集電体11両面の長手方向他側縁部は電極不存在領域であって負極端子との接続部14として機能する。
図1に示すように、密閉容器2はAl製有底筒形本体15と、その開口を閉鎖する蓋板16よりなる。その蓋板16は本体15に溶接されたAl製環状外周板17と、その外周板17の内周縁に外周溝18を嵌着させた電気絶縁性樹脂製環状中間板19と、その中間板19の内周溝20に外周凸部21を嵌着させたAl合金製筒状正極端子22を有する。
一方のAl合金製円盤状集電体4において,その中心に在るボス23は筒状正極端子22の中心孔24に嵌合してそれに溶接されている。また円盤部25は、放射状に配列されて下方に突出する複数のV形凸条26を有し、それら凸条26の底部に図2に示す帯状正極6の接続部13が溶接されている。
他方のAl合金製円盤状集電体5において、その中心に在るボス27は負極端子28である有底筒形本体15の底壁に溶接されている。また円盤部29は、放射状に配列されて上方へ突出する複数の山形凸条30を有し、それら凸条30の稜線部に図2に示す帯状負極7の接続部14が溶接されている。
電解液は、負極側円盤状集電体5のボス27に形成された注入孔31から密閉容器2内に注入され、その後、注入孔31はゴム栓32により封鎖される。
図2に示す各分極性電極12は活性炭、導電フィラおよびバインダよりなる。
原料として活性炭を80wt%,ケッチェンブラックEC 10wt%およびテフロン(登録商標)6J(商品名、三井デュポンフロロケミカル社製)10wt%を混練し、次いで混練物を用いて圧延を行い、厚さ150μmの電極シートを製作した。電極シートから幅103mm、長さ1400mmの複数の帯状分極性電極12を切出し、次いで2枚の分極性電極12と、幅109mm、長さ1400mm、厚さ30μmのアルミ箔よりなる帯状集電体11とを一対の加圧ローラを用い、線圧6tにて圧着して帯状正極6を製作した。また同様の方法で帯状負極7を製作した。
帯状正極6、帯状負極7および帯状正極6を挟む両帯状セパレータ8、9よりなる重合せ物を、帯状正極6の外側に在る一方の帯状セパレータ8が最も内側に位置するようにAl製巻心10を中心に渦巻き状に巻回して、外径D1が38.5mmで、長さが115mmの電極巻回体3を製造した。
電極巻回体3を、内径D2が39.5mmで、長さが120mmの有底筒形本体15内に入れ、次いでその本体15内に実施例59〜60および比較例18の電解液を注入した。その後,注入孔31をゴム栓32により封鎖した。
上記のようにして作成された円筒型電気二重層キャパシタの耐久信頼性を確認すべく、45℃の環境下にて、連続的に2.5Vの電圧を印加した際の性能変化を下記に示す。
【0146】
<円筒型電気二重層キャパシタによる結果>
上記のようにして作成された円筒型電気二重層キャパシタの耐久信頼性を確認すべく、45℃の環境下にて、1000時間の間、連続的に2.5Vの電圧を印加した際の1000時間後の性能を下表に示す。
表24、および図3〜5において、静電容量、抵抗、発生ガス量の値は比較例24の値を100とする相対値で表されている。
【0147】
【表24】
【0148】
<電気二重層キャパシタの製造A>
実施例24
実施例18で調製された電解液を使用して図6の構造を有する電気二重層キャパシタAを製造した。電極33及び電極34は、活性炭を主成分とする電導性物質、バインダー、N−メチルピロリドンとともに混練して得られたペーストをアルミニウム箔に150μmの厚さで塗工後、乾燥して得られたシート状電極を円板状に切り出したものである。容器体36、容器体37、スペーサー、スプリングは共にステンレス鋼製であり、セパレータは、ポリプロピレン不織布である。電気二重層キャパシタの組み立てはアルゴンガスを満たしたグローブボックス内でおこなった。電極33、電極34、容器体36、容器体37、スプリング、スペーサーは120℃の加熱下、24時間真空乾燥した後、グローブボックス内に持ち込んだ。実施例18の電気二重層キャパシタ用電解液を電極33、電極34、セパレータに含浸させ、図6の構成で容器体36と容器体37をガスケットを介してかしめることによって電気二重層キャパシタを得た。
【0149】
比較例25
実施例24において、実施例18で調製した電解液に代えて、比較例12で調製した電解液を使用した以外は、実施例24と同様にして電気二重層キャパシタを得た。
【0150】
比較例26
実施例24において、実施例18で調製した電解液に代えて、比較例13で調製した電解液を使用した以外は、実施例24と同様にして電気二重層キャパシタを得た。
測定例
実施例24及び比較例25、比較例26で作製されたコイン型電気二重層キャパシタに関し、内部抵抗および静電容量を25℃、−30℃において測定した。コイン型セルを専用のホルダにセットした後、低温恒温槽に浸漬してコイン型セルの温度を一定に保つようにした。この際、ホルダ全体をビニール袋で覆い、コイン型セルが恒温槽内の冷媒に接液しないようにした。所定の温度に設定した恒温槽に、コイン型セルを浸漬し、4時間保持した後、電気ニ重層キャパシタの充放電を開始した。電流密度が2.0mAの定電流充電をおこない、電圧が2.5Vに達した時点で定電圧充電に切り替えた。2.5Vで120分保持した後、2.0mAの定電流放電をおこない、電圧が0Vに達した時点で低電圧放電に切り替え0Vで120分間保持した。放電の際の電気エネルギーの積算値から静電容量を算出した。また、放電直後の電圧降下の値と放電電流の値とからセルの内部抵抗を算出した。実施例24の25℃と−30℃における内部抵抗と静電容量を100とした際の比較例25と比較例26の結果を表25に示した。
【0151】
【表25】
【0152】
<電気二重層キャパシタの製造B>
実施例25〜27
次に、実施例19〜21で調製された電解液を、図7の構造を有する容器に充填し、電気二重層キャパシタBを製造した。なお、第一の電極43及び第二の電極45は、いずれも活性炭を主成分とする電導性物質をバインダーと混練して、円板状に成形したものであり、第一の容器42及び第二の容器44は共にアルミニウム製であり、第一の電極43及び第二の電極45は、それぞれ電導性接着剤等により接着されている。また、隔離膜46は、レーヨン系抄紙である。
電気二重層キャパシタは、図7の構造、上記の材質の容器を150℃で、5時間真空乾燥した後、アルゴンガスを満たしたグローブボックス内で、実施例19〜21で調製した電解液を注入することにより製造した。
【0153】
比較例27〜比較例30
実施例25において、実施例19〜21で調製した電解液に代えて、比較例12、14、17および18で調製した電解液を使用した以外は、実施例25と同様にして電気二重層キャパシタを得た。
<電気二重層キャパシタの反応電流値の測定>
実施例25及び比較例27で製造された電気二重層キャパシタに対し、充放電試験装置を用いて段階的に電圧を印加し、各電圧毎に、電解液の分解によって生じる反応電流を測定することにより、実施例19〜21と比較例12、14、17および18の電解液の耐電圧を求めた。
すなわち、まず25℃にて2.4Vまで定電流にて充電を行った後、2.4Vにて2時間保持充電した際の電解液の分解による反応電流値を測定した。次いで、定電流にて所定電圧(0.1V)まで放電し、2.6Vまで定電流にて充電を行った後、2.6Vにて2時間保持充電した際の反応電流値を測定した。以後同様にして、4.0Vまで0.2Vずつ段階的に電圧を上昇させていき、その度に反応電流値を測定した。反応電流値が初めて0.1mAを超える電圧を耐電圧値とする。結果を図8及び表26に示す。なお、この電気二重層キャパシタは、実際には約2.5Vで使用するが、この耐電圧値が高いほど長期の耐久性が優れていることが分かっている。
【0154】
【表26】
【0155】
実施例25〜27で製造された電気二重層キャパシタは、3.2〜3.4Vまで、反応電流が発生しなかったが、比較例27〜30で製造された電気二重層キャパシタでは、3.0Vを超えると反応電流が発生した。また、実施例25〜27で製造された電気二重層キャパシタの方が、全ての電圧で反応電流が小さかった。
以上より、実施例19〜21の電解液(MMMP−BF4/DMC、MMMP−BF4/EMC、MMMP−BF4/DMC+EMC)の方が、従来の電気二重層キャパシタ用電解液として一般的に用いられている比較例12および14の電解液(TEMA−BF4/PC、EMI−BF4/PC)に比較して、耐電圧が高く、耐久性に優れていることが判った。更に、高い電圧においても反応電流値が小さいことより、高電圧で作動させても静電容量の低下が小さく、長期信頼性が高いことが判った。
【0156】
<リチウム二次電池用電解液の調製>
実施例28
実施例17で得られたN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート(MMMP−BF4)にリチウムテトラフルオロボレート(LiBF4)を0.6Mの濃度で混合した。露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。
【0157】
参考例1
N−メトキシメチル−N−メチルピロリジニウムビス(トリフルオロメタンスルホニル)イミド(MMMP−TFSI)にリチウムビス(トリフルオロメタンスルホニル)イミド(LiTFSI)を0.6Mの濃度で混合した。露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。
【0158】
比較例31
比較例5で得られたN,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレート(DEMME−BF4)にリチウムテトラフルオロボレート(LiBF4)を0.6Mの濃度で混合した。露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。
【0159】
<電気伝導度の測定>
実施例28および比較例31の電解液の電気伝導度を測定した。電気伝導度の測定には、導電率計(CDM210 Radiometer社製)を使用した。測定セルにはXE−100(Radiometer社製)を使用した。結果を、表27に示した。
【0160】
【表27】
【0161】
<リチウム二次電池の作製>
参考例2
図9に示すようなコイン型リチウム二次電池を作製した。図9において、49は正極、50は負極、51はセパレーター、52は正極缶、53は負極缶、54はガスケット、55はスペーサー、56はスプリングである。図9に示すリチウム二次電池を以下に示す手順で作成した。正極缶52、負極缶53、スペーサー55およびスプリング56はステンレス製のものを使用した。負極50には厚さ200μmのリチウム金属箔を円形状に切り出したものを使用した。次に正極49の作製を示す。LiCoO2粉末と導電助剤のアセチレンブラックと結着剤のPVdFを85:10:5の重量比で混合し、N−メチルピロリドンを加えペースト状にした。これを厚さ30μmのアルミニウム箔上に電極塗工用アプリケーターで均一に塗工した。これを120℃で8時間、真空乾燥した後、電極打ち抜き機で円形状に切り出し正極49を得た。セパレータおよび切り出した正極に参考例1で得られた電解液を含浸させておく。正極を正極缶52の底面に載せ、その上にセパレーターを載置した後、ガスケット54を挿入した。その後、負極50とさらにその上に、スペーサー55とスプリング56と負極缶53とを順々にセパレーター上に載置し、コイン形電池かしめ機を使用して、正極缶52の開口部分を内方へ折曲することにより封口しリチウム二次電池を作成した。
【図面の簡単な説明】
【0162】
【図1】本発明の電気二重層キャパシタの断面図である。
【図2】本発明の電気二重層キャパシタの構成図である。
【図3】連続的に2.5Vの電圧を1000時間印加した際の円筒型電気二重層キャパシタの静電容量の変化を示すグラフである。
【図4】連続的に2.5Vの電圧を1000時間印加した際の円筒型電気二重層キャパシタの抵抗の変化を示すグラフである。
【図5】連続的に2.5Vの電圧を1000時間印加した際の円筒型電気二重層キャパシタのガス発生量の変化を示すグラフである。
【図6】他の構造を有する電気二重層キャパシタの断面図である。
【図7】更に他の構造を有する電気二重層キャパシタの断面図である。
【図8】電気二重層キャパシタに印加した電圧と反応電流の関係を示すグラフである。
【図9】コイン型リチウム二次電池の断面図である。
【符号の説明】
【0163】
1…円筒型電気二重層キャパシタ、2…円筒状密閉容器、3…電極巻回体、4,5…円盤状集電体、6…帯状正極、7…帯状負極、8,9…帯状セパレータ、10…Al製巻心、11…帯状集電体、12…分極性電極、13…正極端子との接続部、14…負極端子との接続部14、15…Al製有底筒形本体、16…蓋板、17…Al製環状外周板、18…外周溝、19…電気絶縁性樹脂製環状中間板、20…内周溝、21…外周凸部、22…Al合金製筒状正極端子、23…ボス、24…中心孔、25…円盤部、26…V形凸条、27…ボス、28…負極端子、29…円盤部、30…山形凸条、31…注入孔、32…ゴム栓、33,34…電極、36,37…容器体、41…電気二重層キャパシタ、42…第一の容器体、43…第一の電極、44…第二の容器体、45…第二の電極、46…隔膜、47…非電導性材料、48…電解液、49…正極、50…負極、51…セパレーター、52…正極缶、53…負極缶、54…ガスケット、55…スペーサー、56…スプリング
【技術分野】
【0001】
本発明は、第4級アンモニウム塩および電解質、電解液並びに電気化学デバイスに係る。詳しくは、有機溶媒に対する溶解性が高く、耐電圧、電気伝導性が高い電解質として使用できる機能性材料に関する。
【背景技術】
【0002】
近年、バッテリーやキャパシタをはじめとする電気化学デバイスの出力密度、エネルギー密度向上の要求が高まっており、耐電圧性の観点から電解液は水系よりも有機系が多用されてきている。有機電解液としてはプロピレンカーボネートなどの有機溶媒にアルカリ金属塩や固体アンモニウム塩を溶解させた例が挙げられ、前者はリチウムイオン電池用の電解液として、後者は電気二重層キャパシタ用の電解液として使用されている。有機電解液は水系に比べて電気伝導性が劣っており、電気伝導性を向上するために有機溶媒や電解質に関する研究が数多くおこなわれてきた。この結果、特許文献1(特開平3−58526号公報)では電気二重層キャパシタの電解質として非対称型アンモニウム塩が示されている。テトラアルキルアンモニウム塩の種類と電気伝導性に関してはUe et al., J. Electrochem. Soc. 141(2989) 1994で詳細に検討されており、テトラエチルアンモニウムテトラフルオロボレートやトリエチルメチルアンモニウムテトラフルオロボレートが用いられているのが一般的である。
【発明の開示】
【発明が解決しようとする課題】
【0003】
こうした固体状電解質を溶媒に溶解させた非水電解液では、電解液の電気伝導性は電解質の濃度とともに変化する。濃度の上昇とともに電解液中のイオン濃度が増加することによって電気伝導度が増加するがやがて極大点に達する。電気伝導度が極大点に達し減少し始めるのは電解液中にイオンの数が増すにつれて、溶媒−イオン、イオン−イオン間の相互作用の増大によって電解質が解離しにくくなり、同時に電解液の粘度が増加するためと考えられている。電解質濃度がさらに増加するとそれ以上解離できなくなり、電解質濃度が飽和する。したがって電解質濃度を高めようとした場合には電解質が溶解しにくくなるといった問題があった。また高濃度の電解質を溶解させた電解液を低温環境下で使用すると塩の析出が生じ、電解液の電気伝導性が悪くなってしまうといった問題も生じる。電解質の解離度を高めるには通常高誘電率溶媒が好まれ、プロピレンカーボネート、エチレンカーボネート、γ−ブチロラクトン等が使われてきた。また電解質にはテトラエチルアンモニウムテトラフルオロボレートやトリエチルメチルアンモニウムテトラフルオロボレート等が好適に用いられてきたが、これらの電解質は高誘電率溶媒には比較的溶解するものの、常温において2M程度が限界であり、それ以上の濃度、又は低温域では結晶の析出が生じるといった不具合があった。また低誘電率溶媒にはほとんど溶解せず、電解液としては使用できないレベルであった。
【0004】
また高電圧を求める使用において、溶媒にプロピレンカーボネート、エチレンカーボネート、γ−ブチロラクトン等を使用した場合、電解質を高耐電圧タイプに変換しても溶媒の分解電圧に支配されてしまい、従来のキャパシタの動作電圧は2.5V程度が上限であった。2.5Vを超える電圧で動作させると電解液(主に溶媒)の電気化学的分解が起こり性能の著しい劣化、ガス発生等の好ましくない現象が発生する。ハイブリッド自動車、電気自動車のような移動体のエネルギーストレージデバイスとしてのキャパシタの応用においてはエネルギー密度の向上が求められており、動作電圧の向上はエネルギー密度を向上させる有効な手段であるが、従来の電解液では耐電圧を向上することが不可能であり、より耐電圧の高い、電解質、溶媒が求められていた。より耐電圧の高い溶媒として鎖状カーボネート系が挙げられるが、誘電率の低いこれらの溶媒には従来のテトラエチルアンモニウムテトラフルオロボレートやトリエチルメチルアンモニウムテトラフルオロボレート等といった電解質は溶解度が低く、電解液としては使用できないレベルであった。
【0005】
近年、融点を常温近傍にもつ塩、或いは融点が常温以下である塩(常温溶融塩)が見出されている。こうした塩は常温において固体であっても通常の電解質に比べて高濃度に有機溶媒に溶解することが知られている。また常温溶融塩は特定の有機溶媒とは任意の割合でまざり合う。それゆえ、従来の固体状電解質を有機溶媒に溶解しても達成できなかった高濃度の電解液が得られ、しかも高濃度でありながら低温環境下でも塩が析出するといった問題が生じにくい。さらに常温溶融塩は塩そのものが液体であるため、塩単体を電解液として使用することも可能である。
【0006】
一方、常温溶融塩は液体でありながら、イオンのみからなることから蒸気圧が低く難燃性であることが知られている。それゆえ、常温溶融塩を有機溶媒に高濃度に溶解することにより、電解液を難燃化することが可能である。
【0007】
代表的な常温溶融塩として1−エチル−3メチルイミダゾリウムテトラフロオロボレート(EMI・BF4)が挙げられる。EMI・BF4は高い電気伝導性をもちリチウム二次電池や電気二重層キャパシタをはじめとする電気化学デバイスへの応用が検討されてきた。しかしながら、イミダゾリウム塩の電気化学的安定性は4V程度であり、電気二重層キャパシタへ適用した場合、動作電圧は2.5V程度が上限となり、応用の幅が広がらない状況である。
【0008】
近年、より広い電位範囲で安定な常温溶融塩が検討されている。例えば、特許文献2(特許第2981545号公報)に示されるようなカチオン成分に脂肪族アンモニウム系の骨格を持つ常温溶融塩は5.8V以上の耐電圧を有しリチウム二次電池への適用が可能であるとされている。しかしながら、脂肪族アンモニウム骨格をカチオン成分にもつ常温溶融塩は一般に粘性が高く、電気伝導度が低いという欠点がある。有機溶媒と混合することによって電気伝導性の改善は見られるが、従来の固体状電解質を有機溶媒に溶解した電解液が示す電気伝導性には及んでいない。
【0009】
特許文献3(WO 02/076924号公報)には、アルコキシアルキル基を導入した脂肪族アンモニウム塩は非水系有機溶媒への溶解性に優れ、低温時における塩の析出が起こりにくいことが記載されているが、より有機溶媒に対する溶解性が高く、耐電圧、電気伝導性の高い電解質が求められている。
特許文献3に記載されているジエチルメチルメトキシエチルアンモニウムをカチオンとする常温溶融塩を有機溶媒に溶解させた場合においても、従来の固体状電解質(例えば、トリエチルメチルアンモニウムテトラフルオロボレートなど)を有機溶媒に溶解した電解液が示す電気伝導度には及んでいない。また鎖状カーボネートへの溶解性も満足できるものではなく、より有機溶媒に対する溶解性が高く、耐電圧、電気伝導度の高い電解質が求められている。
【特許文献1】特開平3−58526号公報
【特許文献2】特許第2981545号公報
【特許文献3】WO 02/076924号公報
【0010】
本発明の課題は高い電気伝導性、耐電圧を有する第4級アンモニウム塩を提供することにある。
本発明の課題は有機溶媒に対する溶解性が高く、耐電圧、電気伝導度の高い電解質を提供することにある。
本発明の課題は耐電圧、電気伝導性が高い電解液を提供することにある。
本発明の課題は溶媒に溶解した場合には、高濃度な電解質を含んだ電解液を提供することができ、その結果、高電圧、高放電容量、大電流放電性能を有する電気化学デバイスを提供することにある。
【課題を解決するための手段】
【0011】
本発明は式(1)で表される第4級アンモニウム塩に係る。
【0012】
【化1】
(式中、R1〜R2は、共にメチル基を示す。X−は、BF4−を示す。)
【0013】
本発明者らは、電気伝導性の向上という課題を解決できる新規化学物質を開発すべく鋭意研究を重ねた結果、式(1)で表されるN,O−アセタール骨格構造を分子内に持つカチオンが、高い電気伝導性を持ち、中でも特にピロリジン骨格とN,O−アセタール基を持つアンモニウムカチオンが電気伝導性、耐電圧、有機溶媒に対する溶解性が高いことを見出した。
【発明の効果】
【0014】
本発明の電解質を使用すれば、耐電圧、電気伝導性が高い電解液を提供することができ、溶媒に溶解した場合には、従来の固体状電解質を有機溶媒に溶解した電解液をしのぐ電気伝導性をもち、高濃度な電解質を含んだ電解液を提供することができ、その結果、高電圧、高放電容量、大電流放電性能を有する電気化学デバイスが得られる。また鎖状カーボネートへの溶解性にも優れ、高い耐電圧を要する用途にも好適に使用できる。
【発明を実施するための最良の形態】
【0015】
以下に本発明の実施の形態を説明する。
本発明は式(1)で表される第4級アンモニウム塩であり、第4級アンモニウムカチオンとBF4−アニオンとから構成される。第4級アンモニウムカチオンの具体例としてはN−メチル−N−メトキシメチルピロリジニウムカチオン(N−メトキシメチル−N−メチルピロリジニウムカチオン)である。
【0016】
本発明で得られる第4級アンモニウム塩は、常温で液状を示す常温溶融塩として、該塩そのものを電解液として用いることができる。
本発明で得られる第4級アンモニウム塩を電解質として使用する場合は、適当な有機溶媒に混合して用いてもよく、有機溶媒としては、環状炭酸エステル、鎖状炭酸エステル、リン酸エステル、環状エーテル、鎖状エーテル、ラクトン化合物、鎖状エステル、ニトリル化合物、アミド化合物、スルホン化合物などが挙げられる。例えば、以下の化合物が挙げられるがこれらに限定されるものではない。
【0017】
環状炭酸エステルとしては、エチレンカーボネート、プロピレンカーボネート、ブチレンカーボネートなどが挙げられ、好ましくは、プロピレンカーボネートが良い。
鎖状炭酸エステルとしては、ジメチルカーボネート、エチルメチルカーボネート、ジエチルカーボネートなどが挙げられ、好ましくは、ジメチルカーボネート、エチルメチルカーボネートが良い。
【0018】
リン酸エステルとしては、リン酸トリメチル、リン酸トリエチル、リン酸エチルジメチル、リン酸ジエチルメチルなどが挙げられる。
環状エーテルとしては、テトラヒドロフラン、2−メチルテトラヒドロフランなどが挙げられる。
鎖状エーテルとしては、ジメトキシエタンなどが挙げられる。
ラクトン化合物としては、γ−ブチロラクトンなどが挙げられる。
鎖状エステルとしては、メチルプロピオネート、メチルアセテート、エチルアセテート、メチルホルメートなどが挙げられる。
ニトリル化合物としては、アセトニトリルなどが挙げられる。
アミド化合物としては、ジメチルホルムアミドなどが挙げられる。
スルホン化合物としては、スルホラン、メチルスルホランなどが挙げられる。
好ましくは、環状炭酸エステル、鎖状炭酸エステル、ニトリル化合物、スルホン化合物が良い。
【0019】
これらの溶媒は1種類でも2種類以上を混合してもよい。好ましい混合有機溶媒としては、環状炭酸エステルと鎖状炭酸エステル、例えば、エチレンカーボネートとジメチルカーボネート、エチレンカーボネートとエチルメチルカーボネート、エチレンカーボネートとジエチルカーボネート、プロピレンカーボネートとジメチルカーボネート、プロピレンカーボネートとエチルメチルカーボネート、プロピレンカーボネートとジエチルカーボネート、鎖状炭酸カーボネート同士、例えば、ジメチルカーボネートとエチルメチルカーボネート、スルホラン化合物同士、例えば、スルホランとメチルスルホランが良い。更に好ましくは、エチレンカーボネートとエチルメチルカーボネート、プロピレンカーボネートとエチルメチルカーボネート、ジメチルカーボネートとエチルメチルカーボネートが良い。
【0020】
本発明の第4級アンモニウム塩を電解質として使用する場合、電解質濃度は0.1M以上であることが好ましく、より好ましくは0.5M以上であり、さらに好ましいのは1M以上である。0.1Mに満たない場合には電気伝導性が低くなり、電気化学デバイスの性能を低下させてしまう。上限濃度は、常温で液体の塩に関しては、分離する濃度とする。分離しない場合は100%とする。また、常温で固体の塩に関しては、塩が飽和する濃度を上限濃度とする。
【0021】
本発明の電解質は本発明以外の電解質と混合使用することができる。本発明の電解質と混合して使用する電解質としては、たとえばアルカリ金属塩、4級アンモニウム塩、4級ホスホニウム塩などが挙げられ、これらの電解質のうち1種類でも、2種類以上を併用し、混合して使用してもよい。アルカリ金属塩としては、リチウム塩、ナトリウム塩、カリウム塩が挙げられ、例えば、6フッ化リン酸リチウム、硼フッ化リチウム、過塩素酸リチウム、トリフロロメタンスルホン酸リチウム、スルホニルイミドリチウム、スルホニルメチドリチウムなどが挙げられるがこれらに限定するものではない。ナトリウム塩としては6フッ化リン酸ナトリウム、硼フッ化ナトリウム、過塩素酸ナトリウム、トリフルオロメタンスルホン酸ナトリウム、スルホニルイミドナトリウム、スルホニルメチドナトリウムなどが挙げられる。カリウム塩としては6フッ化リン酸カリウム、硼フッ化カリウム、過塩素酸カリウム、トリフルオロスルホン酸カリウム、スルホニルイミドカリウム、スルホニルメチドカリウムなどが挙げられるがこれらに限定するものではない。
【0022】
4級アンモニウム塩としては、テトラアルキルアンモニウム塩、イミダゾリウム塩、ピラゾリウム塩、ピリジニウム塩、トリアゾリウム塩、ピリダジニウム塩などが挙げられるがこの限りではない。テトラアルキルアンモニウム塩としては、テトラエチルアンモニウムテトラフルオロボレート、テトラメチルアンモニウムテトラフルオロボレート、テトラプロピルアンモニウムテトラフルオロボレート、テトラブチルアンモニウムテトラフルオロボレート、トリエチルメチルアンモニウムテトラフルオロボレート、トリメチルエチルアンモニウムテトラフルオロボレート、ジメチルジエチルアンモニウムテトラフルオロボレート、トリメチルプロピルアンモニウムテトラフルオロボレート、トリメチルブチルアンモニウムテトラフルオロボレート、ジメチルエチルプロピルアンモニウムテトラフルオロボレート、メチルエチルプロピルブチルアンモニウムテトラフルオロボレート、N,N−ジメチルピロリジニウムテトラフルオロボレート、N−エチル−N−メチルピロリジニウムテトラフルオロボレート、N−メチル−N−プロピルピロリジニウムテトラフルオロボレート、N−エチル−N−プロピルピロリジニウムテトラフルオロボレート、N,N−ジメチルピペリジニウムテトラフルオロボレート、N−メチル−N−エチルピペリジニウムテトラフルオロボレート、N−メチル−N−プロピルピペリジニウムテトラフルオロボレート、N−エチル−N−プロピルピペリジニウムテトラフルオロボレート、N,N−ジメチルモルホリニウムテトラフルオロボレート、N−メチル−N−エチルモルホリニウムテトラフルオロボレート、N−メチル−N−プロピルモルホリニウムテトラフルオロボレート、N−エチル−N−プロピルモルホリニウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。イミダゾリウム塩としては、1,3−ジメチルイミダゾリウムテトラフルオロボレート、1−エチル−3−メチルイミダゾリウムテトラフルオロボレート、1,3−ジエチルイミダゾリウムテトラフルオロボレート、1,2−ジメチル−3−エチルイミダゾリウムテトラフルオロボレート、1,2−ジメチル−3−プロピルイミダゾリウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。ピラゾリウム塩としては1,2−ジメチルピラゾリウムテトラフルオロボレート、1−メチル−2−エチルピラゾリウムテトラフルオロボレート、1−プロピル−2−メチルピラゾリウムテトラフルオロボレート、1−メチル−2−ブチルピラゾリウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。ピリジニウム塩としてはN−メチルピリジニウムテトラフルオロボレート、N−エチルピリジニウムテトラフルオロボレート、N−プロピルピリジニウムテトラフルオロボレート、N−ブチルピリジニウムテトラフルオロボレートなどが挙げらるがこれらの限りではない。トリアゾリウム塩としては、1−メチルトリアゾリウムテトラフルオロボレート、1−エチルトリアゾリウムテトラフルオロボレート、1−プロピルトリアゾリウムテトラフルオロボレート、1−ブチルトリアゾリウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。ピリダジニウム塩としては1−メチルピリダジニウムテトラフルオロボレート、1−エチルピリダジニウムテトラフルオロボレート、1−プロピルピリダジニウムテトラフルオロボレート、1−ブチルピリダジニウムテトラフルオロボレートなどが挙げられるがこれらの限りではない。
【0023】
4級ホスホニウム塩としては、テトラエチルホスホニウムテトラフルオロボレート、テトラメチルホスホニウムテトラフルオロボレート、テトラプロピルホスホニウムテトラフルオロボレート、テトラブチルホスホニウムテトラフルオロボレート、トリエチルメチルホスホニウムテトラフルオロボレート、トリメチルエチルホスホニウムテトラフルオロボレート、ジメチルジエチルホスホニウムテトラフルオロボレート、トリメチルプロピルホスホニウムテトラフルオロボレート、トリメチルブチルホスホニウムテトラフルオロボレート、ジメチルエチルプロピルホスホニウムテトラフルオロボレート、メチルエチルプロピルブチルホスホニウムテトラフルオロボレートなどが挙げられるがこれらに限定するものではない。これらは1種でも2種以上を併用してもよい。
【0024】
尚、上記テトラフルオロボレートをビストリフルオロメタンスルホニルイミド、ヘキサフルオロホスフェート、トルフルオロアセテートに代えたものも含まれる。
【0025】
本発明の電解質と上記電解質とを混合し、電解質として使用する際、混合使用する上記電解質の上限濃度は電解質の析出あるいは分離を生じる濃度とする。混合使用する電解質の下限濃度は適用する電気化学デバイスの種類に拠る。例えば、電気二重層キャパシタの電解質として使用する際は、本発明の第4級アンモニウム塩だけでも使用可能なので、混合使用する電解質の下限濃度は0Mである。リチウム電池に使用する際は、少なくとも前記載リチウム塩を混合使用する。リチウム塩濃度は0.1M以上、2.0M以下であることが好ましく、より好ましくは0.15M以上、1.5M以下であることが好ましく、さらにより好ましいのは0.2M以上、1.2M以下である。特に好ましいのは0.3M以上、1.0M以下である。
【0026】
本発明の第4級アンモニウム塩(1)は種々の方法で製造される。その代表的な合成方法を下記反応式−1及び反応式−2で示す。
【0027】
【化2】
【0028】
式(7)のアルキルピロリジン(R1は上記と同じ)と式(8)の化合物(R2は上記と同じでYはCl、Br、Iなどを示す)とを反応させることにより、式(5)で表される第4級アンモニウム塩が製造され、次いで得られる式(5)で表される第4級アンモニウム塩と式(9)で表される化合物とを反応させることにより、XがY以外のXを示す式(1)で表される第4級アンモニウム塩が製造される。式(9)においてMで示される原子はH又はNa、K、Li等のアルカリ金属原子、Ca、Mg、Ba等のアルカリ土類金属原子、Ag等の金属原子を含む。XはBF4である。
【0029】
式(7)のアルキルピロリジンと式(8)の化合物とを反応させることにより、本発明の式(5)で表される第4級アンモニウム塩が製造される。
出発原料として用いられる式(7)のアルキルピロリジンと式(8)で表される化合物はいずれも公知物質である。式(7)のアルキルピロリジンとしてはメチルピロリジンが挙げられる。式(8)の化合物としてはクロロメチルメチルエーテル、ブロモメチルメチルエーテル、ヨードメチルメチルエーテルが挙げられ、両者の反応は適当な溶媒中で行われる。
【0030】
用いられる溶媒としては、式(7)のアルキルピロリジン及び式(8)の化合物を溶解し得、反応に悪影響を及ぼさない溶媒である限り、公知のものを広く使用できる。このような溶媒としては、例えば、ベンゼン、トルエン、キシレン等の芳香族炭化水素、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素、メタノール、エタノール、イソプロパノール、n−ブタノール、tert−ブタノール等の低級アルコール、アセトン、メチルエチルケトン等のケトン、ジエチルエーテル、ジイソプロピルエーテル等のエーテル、n−ヘキサン、n−ヘプタン等の脂肪族炭化水素、シクロヘキサン等の脂肪族炭化水素等が挙げられる。これらの中でも、トルエン等の芳香族炭化水素、クロロホルム等のハロゲン化炭化水素、アセトン等のケトンが好ましい。斯かる溶媒は、1種単独で又は2種以上混合して使用できる。また溶媒は無水溶媒(水分1000ppm以下)としての使用が特に好ましい。
【0031】
式(7)のアルキルピロリジンと式(8)の化合物との使用割合としては、通常前者1モルに対して後者を通常0.5〜5モル、好ましくは0.9〜1.2モル使用する。
式(7)のアルキルピロリジンと式(8)の化合物との反応は、通常−30〜100℃において行われ、更に詳しくは−10〜40℃にて行われる。一般に数時間〜24時間程度で完結する。
【0032】
上記で得られる式(5)で表される第4級アンモニウム塩と式(9)の化合物との反応は、通常塩交換反応により行われる。
出発原料として用いられる式(9)の化合物は公知化合物であり、例えば、HBF4、LiBF4、NaBF4、KBF4、AgBF4などが挙げられる。
【0033】
この反応は適当な溶媒中で行われる。使用される溶媒としては、式(5)の第4級アンモニウム塩及び式(9)の化合物を溶解し得、反応に悪影響を及ぼさない溶媒である限り、公知のものを広く使用できる。このような溶媒としては、例えば、水又はジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素、メタノール、エタノール、イソプロパノール、n−ブタノール、tert−ブタノール等の低級アルコール、アセトン、メチルエチルケトン等のケトン、酢酸エチル、酢酸ブチル等のエステル、ジメチルスルホキシド、ジメチルホルムアミド等の非プロトン性極性溶媒が挙げられる。これらの中でも、メタノール等の低級アルコール類、クロロホルム等のハロゲン化炭化水素、水が好ましい。これらの溶媒は、1種単独で又は2種以上混合して使用できる。
【0034】
式(5)の第4級アンモニウム塩と式(9)の化合物との使用割合としては、通常前者1モルに対して後者を通常0.3〜5モル、好ましくは0.9〜1.2モル使用する。
式(5)の第4級アンモニウム塩と式(9)の化合物との反応は、通常速やかに進行するので、例えば、両者を溶媒に溶解した溶液を5℃〜150℃で10分〜2時間程度反応させる。
【0035】
上記各反応で得られる目的物は、通常の分離手段、例えば、遠心分離、濃縮、洗浄、有機溶媒抽出、クロマトグラフィー、再結晶等の慣用の単離及び精製手段により、反応混合物から容易に単離、精製される。
【0036】
またハロゲンの混入を嫌う用途の場合、一度ハロゲン塩を中和、塩交換し、ハロゲンを系外に除いた後、更に目的に応じた塩に変換することで、ハロゲンの混入を削減することも出来る。中和剤としては各種アルカリ金属塩、アルカリ土類金属塩、有機アルカリ金属塩、銀塩等が挙げられる。具体的には炭酸ナトリウム、炭酸カリウム、炭酸リチウム、炭酸カルシウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウム、炭酸水素カルシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム、過塩素酸ナトリウム、過塩素酸カリウム、過塩素酸リチウム、酢酸ナトリウム、酢酸カリウム、硫酸銀、硝酸銀、過塩素酸銀等が挙げられる。反応形式は先の式(1)で表される第4級アンモニウム塩を合成する手法にて行うことができ、脱ハロゲン中間体としては、式(6)で表すことができる。
【0037】
【化3】
(式中、R1〜R2は、メチル基を示す。Z−は、1/2CO32−、HCO3−、1/2SO42−、ClO4−、CH3CO2−、OH−を示す。)
【0038】
具体的には、1−メトキシメチル−1−メチルピロリジニウムカーボネート、1−メトキシメチル−1−メチルピロリジニウムヒドロキシド、1−メトキシメチル−1−メチルピロリジニウムスルホネート、1−メトキシメチル−1−メチルピロリジニウムパークロレート、1−メトキシメチル−1−メチルピロリジニウムアセテート、1−メトキシメチル−1−メチルピロリジニウムハイドロカーボネート等が挙げられる。更に続く目的に応じた塩へ変換する手法も式(1)で表される第4級アンモニウム塩を合成する手法を適用できる。
【0039】
式(5)の第4級アンモニウム塩からXがBF4を示す式(1)の第4級アンモニウム塩を製造する場合の反応条件を具体的に示すと、式(5)の第4級アンモニウム塩を上記低級アルコールに溶解し、この溶液に所定量(例えば、硼フッ化水素酸濃度70wt%以下)のメタノール硼フッ化水素酸、硼フッ化銀等のフッ化硼素塩を添加し、5℃〜150℃で30分程度反応させる。反応により生成するハロゲン化水素を留去し、またハロゲン化銀等のハロゲン塩を濾別し、濾液を減圧濃縮し、乾燥することにより、目的化合物を単離することができる。尚、ハロゲン化水素の留去には、例えば、遠心分離、熱時下N2バブリング(例えば、60℃〜150℃)による留去、減圧による留去等を適用できる。上記方法で得られた本発明の第4級アンモニウム塩を電解質として使用する際には、水分がデバイス性能に悪影響を与えるため、水分を十分に取り除く必要がある。水分は、熱時下N2バブリングによる留去、減圧による留去等を適用できるがこれらの手法に限定されるわけではない。含水分量は100ppm以下であることが好ましく、より好ましくは50ppm以下、さらに好ましいのは30ppm以下であり、特に好ましいのは10ppm以下である。
【0040】
またハロゲンの混入を嫌う用途の一般式(1)の第4級アンモニウム塩を製造する場合の反応条件を具体的に示すと、一般式(5)の第4級アンモニウム塩をメタノール又は水等に溶解し、この溶液に所定量の炭酸ナトリウムや硫酸銀等のハロゲン以外の金属塩を添加し、0〜50℃で1時間程度反応させる。溶媒を減圧濃縮、真空乾燥した後、反応により生成したハロゲン化金属塩が不溶で、第4級アンモニウム塩が可溶な溶媒、例えば、ジクロロメタン等のハロゲン溶媒やイソプロパノール、ブタノール等のアルコール類等に再溶解し、ハロゲン塩を濾別する。濾液を減圧濃縮し、乾燥することにより、大部分のハロゲンを除いた第4級アンモニウム塩を製造することができる。
【0041】
【化4】
【0042】
式(10)のアルコキシピロリジン(R2は上記と同じ)と式(11)の化合物(R1及びYは上記と同じ)とを反応させることにより、式(5)で表される第4級アンモニウム塩が製造され、次いで得られる式(5)で表される第4級アンモニウム塩と式(9)で表される化合物(M及びXは上記と同じ)とを反応させることにより、XがY以外のXを示す式(1)で表される第4級アンモニウム塩が製造される。
【0043】
式(10)のアルコキシピロリジンと式(11)の化合物とを反応させることにより、本発明の式(5)で表される第4級アンモニウム塩が製造される。
出発原料として用いられる式(10)のアルコキシピロリジンは公知の手法によって合成できる。例えばC.M.McLeod
und G.M.Robinson,J.Chem.Soc.119,1470(1921). G.M.Robinson und R.Robinson,J.Chem.Soc.123,532(1923). Stewert,T.D;Bradly,W.E.J.Am.Chem.Soc.1932,54,4172−4183.に例示されている。
【0044】
式(10)のアルコキシピロリジンの一般的な合成方法は原料にピロリジン、ホルムアルデヒドあるいはパラホルムアルデヒド、アルコール、炭酸アルカリを用いて合成する。使用割合はピロリジン1モルに対し、ホルムアルデヒドあるいはパラホルムアルデヒドを0.5〜3.0モル、好ましくは0.6〜1.5モル使用し、アルコールを0.5〜3.0モル、好ましくは2.0〜3.0モル使用し、炭酸アルカリを0.2〜3.0モル、好ましくは0.4〜1.0モル使用して行う。反応温度は−5〜100℃で、反応時間は数時間〜24時間程度で終了する。目的物は抽出、精留により単離できる。
【0045】
式(11)で表させる化合物は公知物質であり、例えばメチルクロライド、メチルブロマイド、メチルアイオダイド、エチルアイオダイド、エチルブロマイド、n−プロピルクロライド、n−プロピルブロマイド、n−プロピルアイオダイド、iso−プロピルクロライド、iso−プロピルブロマイド、iso−プロピルアイオダイド、n−ブチルクロライド、n−ブチルブロマイド、n−ブチルアイダイド、iso−ブチルクロライド、iso−ブチルブロマイド、iso−ブチルアイオダイド、tert−ブチルクロライド、tert−ブチルブロマイド、tert−ブチルアイオダイドなどが挙げられる。式(10)のアルキルピロリジンと式(11)の化合物との反応は適当な溶媒中で行われる。
【0046】
用いられる溶媒としては、式(10)のアルコキルピロリジン及び式(11)の化合物を溶解し得、反応に悪影響を及ぼさない溶媒である限り、公知のものを広く使用できる。このような溶媒としては、例えば、ベンゼン、トルエン、キシレン等の芳香族炭化水素、ジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素、メタノール、エタノール、イソプロパノール、n−ブタノール、tert−ブタノール等の低級アルコール、アセトン、メチルエチルケトン等のケトン、ジエチルエーテル、ジイソプロピルエーテル等のエーテル、n−ヘキサン、n−ヘプタン等の脂肪族炭化水素、シクロヘキサン等の脂肪族炭化水素等が挙げられる。これらの中でも、アセトン等のケトン、トルエン等の芳香族炭化水素、クロロホルム等のハロゲン化炭化水素が好ましい。斯かる溶媒は、1種単独で又は2種以上混合して使用できる。また溶媒は無水溶媒(水分1000ppm以下)としての使用が特に好ましい。
【0047】
式(10)のアルコキシピロリジンと式(11)の化合物との使用割合としては、通常前者1モルに対して後者を通常0.5〜5モル、好ましくは0.9〜1.2モル使用する。
式(10)のアルコキシピロリジンと式(11)の化合物との反応は、通常、0〜150℃において行われ、一般に24時間〜72時間程度で完結する。低沸点のハロゲン化アルキルで4級化する場合オートクレイブを使用することが好ましい。
上記で得られる式(5)で表される第4級アンモニウム塩と式(9)の化合物との反応は、通常塩交換反応により行われる。
【0048】
この反応は適当な溶媒中で行われる。使用される溶媒としては、式(5)の第4級アンモニウム塩及び式(9)の化合物を溶解し得、反応に悪影響を及ぼさない溶媒である限り、公知のものを広く使用できる。このような溶媒としては、例えば、水又はジクロロメタン、クロロホルム、四塩化炭素等のハロゲン化炭化水素、メタノール、エタノール、イソプロパノール、n−ブタノール、tert−ブタノール等の低級アルコール、アセトン、メチルエチルケトン等のケトン、酢酸エチル、酢酸ブチル等のエステル、ジメチルスルホキシド、ジメチルホルムアミド等の非プロトン性極性溶媒が挙げられる。これらの中でも、メタノール等の低級アルコール類、クロロホルム等のハロゲン化炭化水素、水が好ましい。これらの溶媒は、1種単独で又は2種以上混合して使用される。
【0049】
式(5)の第4級アンモニウム塩と式(9)の化合物との使用割合としては、通常前者1モルに対して後者を通常0.3〜5モル、好ましくは0.9〜1.2モル使用する。
式(5)の4級アンモニウム塩と式(9)の化合物との反応は、通常速やかに進行するので、例えば、両者を溶媒に溶解した溶液を5℃〜150℃付近で10分〜2時間程度反応させる。
【0050】
上記各反応で得られる目的物は、通常の分離手段、例えば、遠心分離、濃縮、洗浄、有機溶媒抽出、クロマトグラフィー、再結晶等の慣用の単離及び精製手段により、反応混合物から容易に単離、精製される。
またハロゲンの混入を嫌う用途の場合、一度ハロゲン塩を中和、塩交換し、ハロゲンを系外に除いた後、更に目的に応じた塩に変換することで、ハロゲンの混入を削減することも出来る。中和剤としては各種アルカリ金属塩、アルカリ土類金属塩、有機アルカリ金属塩、銀塩等が挙げられる。具体的には炭酸ナトリウム、炭酸カリウム、炭酸リチウム、炭酸カルシウム、炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウム、炭酸水素カルシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化カルシウム、過塩素酸ナトリウム、過塩素酸カリウム、過塩素酸リチウム、酢酸ナトリウム、酢酸カリウム、硫酸銀、硝酸銀、過塩素酸銀等が挙げられる。反応形式は先の式(1)で表される第4級アンモニウム塩を合成する手法にて行うことができ、脱ハロゲン中間体としては、式(6)で表すことができる。
【0051】
【化5】
(式中、R1〜R2は、メチル基を示す。Z−は、1/2CO32−、HCO3−、1/2SO42−、ClO4−、CH3CO2−、OH−を示す。)
【0052】
具体的には、1−メトキシメチル−1−メチルピロリジニウムカーボネート、1−メトキシメチル−1−メチルピロリジニウムヒドロキシド、1−メトキシメチル−1−メチルピロリジニウムスルホネート、1−メトキシメチル−1−メチルピロリジニウムパークロレート、1−メトキシメチル−1−メチルピロリジニウムアセテート、1−メトキシメチル−1−メチルピロリジニウムハイドロカーボネート等が挙げられる。更に続く目的に応じた塩へ変換する手法も式(1)で表される第4級アンモニウム塩を合成する手法を適用できる。
またアルコキシピロリジンと酸エステル等を反応することで、ハロゲンを含まないハロゲンフリー中間体を製造できる。酸エステルとしては炭酸エステル、硫酸エステル、アルキルエステル、リン酸エステル等が挙げられ、中でも炭酸エステルが好ましい。炭酸エステルとしてはジメチルカーボネート、ジエチルカーボネート、ジプロピルカーボネート、ジイソプロピルカーボネート等が挙げられ、反応は通常オートクレイブ中50〜160℃にて行うことが効率的である。反応時間は数時間〜48時間程度で十分である。
ハロゲンフリー中間体としては、式(12)で表すことができる。
【0053】
【化6】
(式中、R1〜R2は、メチルを示す。Q−はR1OCO2−を示す。)
【0054】
具体的には1−メトキシメチル−1−メチルピロリジニウムメチルカーボネート等が挙げられる。
式(5)の第4級アンモニウム塩からXがBF4を示す式(1)の第4級アンモニウム塩を製造する場合の反応条件を具体的に示すと、式(5)の第4級アンモニウム塩を上記低級アルコールに溶解し、この溶液に所定量(例えば、硼フッ化水素酸濃度70wt%以下)のメタノール硼フッ化水素酸、硼フッ化銀等のフッ化硼素塩を添加し、5℃〜150℃で30分程度反応させる。反応により生成するハロゲン化水素を留去し、またハロゲン化銀等のハロゲン塩を濾別し、濾液を減圧減圧濃縮し、乾燥することにより、目的化合物を単離することができる。尚、ハロゲン化水素の留去には、例えば、遠心分離、熱時下N2バブリング(例えば、60℃〜150℃)による留去、減圧による留去等を適用できる。上記方法で得られた本発明の第4級アンモニウム塩を電解質として使用する際には、水分がデバイス性能に悪影響を与えるため、水分を十分に取り除く必要がある。水分は、熱時下N2バブリングによる留去、減圧による留去等を適用できるがこれらの手法に限定されるわけではない。含水分量は100ppm以下であることが好ましく、より好ましくは50ppm以下、さらに好ましいのは30ppm以下であり、特に好ましいのは10ppm以下である。
【0055】
またハロゲンの混入を嫌う用途の一般式(1)の第4級アンモニウム塩を製造する場合の反応条件を具体的に示すと、一般式(5)の第4級アンモニウム塩をメタノール又は水等に溶解し、この溶液に所定量の炭酸ナトリウムや硫酸銀等のハロゲン以外の金属塩を添加し、0〜50℃で1時間程度反応させる。溶媒を減圧濃縮、真空乾燥した後、反応により生成したハロゲン化金属塩が不溶で、第4級アンモニウム塩が可溶な溶媒、例えば、ジクロロメタン等のハロゲン溶媒やイソプロパノール、ブタノール等のアルコール類等に再溶解し、ハロゲン塩を濾別する。濾液を減圧濃縮し、乾燥することにより、大部分のハロゲンを除いた第4級アンモニウム塩を製造することができる。
【0056】
上記のように、本発明の第4級アンモニウム塩や該塩を有機溶媒に溶解した溶液は、電気二重層キャパシタや二次電池などの電気化学デバイス用電解液として使用することができる。
第4級アンモニウム塩を有機溶媒に溶解した溶液を電気化学デバイス用電解液として使用する場合、電解質濃度は0.1M以上であることが好ましく、より好ましくは0.5M以上であり、さらに好ましいのは1M以上である。0.1Mに満たない場合には電気伝導性が低くなり、電気化学デバイスの性能を低下させてしまう。上限濃度は、常温で液体の塩に関しては、有機溶媒と分離する濃度とする。分離しない場合は100%とする。また、常温で固体の塩に関しては、塩が有機溶媒に飽和する濃度を上限濃度とする。
【0057】
本発明の第4級アンモニウム塩を用いて電気化学デバイス用電解液を好適に調製できる。本発明で得られる電解液は電気エネルギーを物理的な作用或いは化学的な作用により蓄積できる電気化学デバイスに使用できるが、電気二重層キャパシタやリチウム電池に好適に使用できる。
【0058】
以下、本発明の第4級アンモニウム塩を用いた電気二重層キャパシタ用電解液の調製方法を説明する。本発明の第4級アンモニウム塩は塩単体が液体の場合はそれ自身、電解液として使用できるが、適当な有機溶媒と混合して使用してもよい。合成した第4級アンモニウム塩を扱う場合、或いは有機溶媒と混合する場合、作業をおこなう環境としては、水分が電気二重層キャパシタの性能に悪影響を与えるため、大気が混入しない環境であれば特に限定はしないが、アルゴンや窒素などの不活性雰囲気のグローブボックス内において調製作業することが好ましい。作業環境の水分は露点計で管理することができ、マイナス60℃以下であることが好ましい。マイナス60℃以上になると、作業時間が長くなる場合、電解液が雰囲気中の水分を吸収するため電解液中の水分が上昇してしまう。電解液中の水分はカールフィッシャー計で測定することができる。
【0059】
本発明の第4級アンモニウム塩を有機溶媒に溶解した溶液を電気化学デバイス用電解液として使用する場合、前記載の通り電解質濃度は電解液の電気伝導性の観点から0.1M以上であれば電解質の分離を生じない限り限定はしないが、好ましいのは0.5M以上であり、さらに好ましいのは1M以上である。上限濃度は電解質の析出および分離を生じない限り限定しない。使用する有機溶媒としては前記載の通り種々溶媒が挙げられるが、溶媒種によって誘電率や粘性、融点等の物性が異なるため、溶媒種と混合使用する本発明の第4級アンモニウム塩の組み合わせに応じて、混合組成を決定するのが好ましい。例えば、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとプロピレンカーボネートからなる電解液の場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は10〜80重量%が好ましく、より好ましくは15〜70重量%、更に好ましくは20〜60重量%である。N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとアセトニトリルからなる電解液の場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は10〜90重量%が好ましく、より好ましくは20〜70重量%、更に好ましくは30〜60重量%である。N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとジメチルカーボネートからなる電解液の場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は40〜90重量%が好ましく、更に好ましくは60〜80重量%である。N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとエチルメチルカーボネートからなる電解液の場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は65〜90重量%が好ましく、更に好ましくは65〜80重量%である。また有機溶媒を混合使用することもでき、ジメチルカーボネートとエチルメチルカーボネートの混合溶媒を使用する場合、N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの組成は40〜80重量%が好ましい。
【0060】
本発明の第4級アンモニウム塩はリチウム電池用電解液に使用することもできる。電気二重層キャパシタ用電解液の調製時と同様に、水分がリチウム電池特性に悪影響を与えるため、調製作業をおこなう作業環境としては、露点が管理されたグローブボックス内が好ましい。
【0061】
本発明の第4級アンモニウム塩は塩単体が液体の場合は第4級アンモニウム塩にリチウム塩を溶解すれば電解液として使用できる。或いは本発明の第4級アンモニウム塩は適当な有機溶媒と混合し、リチウム塩を溶解すれば電解液として使用できる。使用するリチウム塩は前記載の通り種々の塩が挙げられるが、塩の析出が生じなければ特に限定はしない。リチウム塩濃度は0.1M以上、2.0M以下であることが好ましく、より好ましくは0.15M以上、1.5M以下であることが好ましく、さらにより好ましいのは0.2M以上、1.2M以下である。特に好ましいのは0.3M以上、1.0M以下である。リチウム塩濃度が0.1Mに満たない場合には充放電レートが大きい場合に電極近傍においてリチウムイオンの枯渇が生じ、充放電特性が低下する。またリチウムイオン濃度が2.0Mを超えると電解液の粘度が高くなり、電気伝導性が低くなってしまう。なお、本発明の第4級アンモニウム塩およびリチウム塩を形成するアニオンのうちいずれか一方にはBF4−が含まれていることが好ましい。その理由は定かではないが、テトラフルオロボレートを含む場合には正極集電体として使用されるアルミニウムの表面に不働態皮膜が形成され、アルミニウムの溶出を抑制できるためではないかと考えられる。BF4−の含有量イオン数が電解液中の全アニオン数の0.5%以上になるように調製するのが好ましく、より好ましくは0.8%以上になるように調製するのがよい。上限濃度は、BF4−の含有イオン数が電解液中の全アニオン数の100%とする。
【0062】
また本電解液は有機溶媒に希釈して使用することもできる。使用できる有機溶媒としては環状炭酸エステル、鎖状炭酸エステル、環状エーテル、鎖状エーテル、ニトリル化合物、及びスルホン化合物等があげられ、環状炭酸エステルではエチレンカーボネートやプロピレンカーボネート等が、鎖状炭酸エステルではジメチルカーボネート、エチルメチルカーボネート等があげられ、環状エーテルではテトラヒドロフラン、ヘキサヒドロピラン等が、鎖状エーテルでは1,2−ジメトキシエタン等が、ニトリル化合物ではアセトニトリル等が、スルホン化合物ではスルホラン等があげられる。またこれらの溶媒は混合して用いることができ、エチレンカーボネートとジメチルカーボネート、エチレンカーボネートとエチルメチルカーボネート、エチレンカーボネートとプロピレンカーボネート、エチレンカーボネートとテトラヒドロフラン等があげられる。
【0063】
なお本発明で使用される電解液には特定の有機添加剤を含むことが好ましい。特定の有機添加剤とは下記(式A)、(式B)、(式C)で表される化合物を指す。その理由は、該有機添加剤を含むことにより、リチウム電池負極表面にSEI(Solid Electrolyte Interface)として知られるリチウムイオン選択的透過膜が形成され、常温溶融塩を形成するアンモニウムカチオンの分解や負極材料への挿入を抑制できるためと考えられる。その結果、安定した充放電特性が得られる。これらの有機添加剤は種類によっては希釈有機溶媒としての機能も併せ持つ物質もある。(式A)で表される構造を有するものとして例えばエチレンカーボネート、ビニレンカーボネート、ブチレンカーボネートなどが挙げれられ、(式B)の構造を有するものとして例えばエチレントリチオカーボネート、ビニレントリチオカーボネートなどが挙げられ、(式C)の構造を有するものとしては例えばエチレンサルファイトなどが挙げられるが、これらに限定されるわけではない。これらの添加剤は1種類でも2種類以上を混合してもよい。これら(式A)、(式B)、(式C)で表される有機添加剤の含有量は、使用する有機添加剤がすべて(式A)、(式B)、(式C)で表される構造を有する有機添加剤であってもよいが、好ましいのは全電解液重量に対するこれらの有機添加剤の割合が1重量%以上、40重量%以下であり、さらに好ましいのは1重量%以上、30重量%以下であり、特に好ましいのは1重量%以上、20重量%以下であり、さらに最適な濃度は1重量%以上、10重量%以下である。1重量%以下の場合には負極表面に十分な皮膜が形成されないため、常温溶融塩の分解や挿入が生じてしまう。
【0064】
R3−O(CO)−O−R4 (式A)
R5−S−(CS)−S−R6 (式B)
R7−O(SO)−O−R8 (式C)
(R3〜R8は炭素数が1〜3の飽和炭化水素基或いは不飽和炭化水素基を表し、R3とR4、R5とR6、R7とR8が単結合或いは2重結合或いは3重結合で互いに結びつき環を形成していてもよい。)
【0065】
以上のようにして得られる本発明の電解液を用いて電気二重層キャパシタを好適に作製できる。この電気二重層キャパシタの一例としては、例えば、図7に示すようなものを挙げることができる。以下、図7と共に説明を続ける。
【0066】
図7は、本発明の電気二重層キャパシタの断面を示す図面である。図中、41は電気二重層キャパシタ、42は第一の容器体、43は第一の電極、44は第二の容器体、45は第二の電極、46は隔膜、47は非電導性材料、48は電解液を示す。
第一の容器体42及び第一の電極43、第二の容器体44及び第二の電極45はそれぞれ電気的に接続する。しかし、第一の電極42と第二の電極45の間は、隔膜46により隔離されている。この第一の電極43及び第二の電極45は、対向配置されていることが好ましい。
【0067】
上記第一の容器体42及び第二の容器体44は、電解液48により腐食されない電導性の物質であれば良く、例えば、アルミニウム、ステンレス鋼等の材料が使用される。一方、これと電気的に接続する第一の電極43及び第二の電極45は、電導性のものであれば良いが、高容量を得るためにその表面積が大きい多孔性電極であることが望ましく、例えば、電導性物質の粉末をバインダーと混合して成型したものが好ましく用いられる。また、電導性物質の粉末をバインダーとともにピロリドン等の有機溶剤に混合し、ペースト状にしたものをアルミニウム箔等の集電体に塗工後、乾燥して得たシート状電極が好ましく用いられる。電導性物質としては、活性炭粉末、活性炭繊維等の炭素材料;貴金属酸化物材料;導電性高分子材料等が用いられるが、このうち炭素材料が安価であるため好ましい。更に、第一の電極43と第二の電極45の間に挟み込まれ、これらを隔離する隔膜46としては、電解液が通過しやすく、電子伝導に関しては絶縁体で、化学的に安定な材質であれば特に限定はないが、レーヨン系抄紙、ポリオレフィン系多孔質フィルム、ポリエチレン不織布、ポリプロピレン不織布、セルロース等が好適に用いられる。
【0068】
本発明の電気二重層キャパシタは、上記した第一の容器体42と第二の容器体44の間を電解液48で満たし、更に非電導性材料47で電気的に接続しないように密封することにより製造される。
【0069】
電解液48としては、前記したものが使用されるが、その充填は、充填される容器体等を真空乾燥した後、不活性ガスを満たしたグローブボックス内で電解液8を注液し、エージングを施すことにより行うことが好ましい。なお真空乾燥は、120〜300℃で加熱して行うことが好ましく、その時間は、キャパシタのサイズにもよるが、5〜100時間程度行うことが好ましい。また、エージングは、電極、特に活性炭等で製造された多孔性電極の細孔深部までイオンを吸着させ、微量に含まれる不純物を電気的に分解させるために行われるものであり、室温にて、2〜3Vの範囲で、5〜100時間程度の充電を行うことが好ましい。最後に好ましくは減圧脱泡し、本発明の電気二重層キャパシタを完成させる。
【0070】
以上のようにして製作される本発明の電気二重層キャパシタは、第一の容器体42及び第二の容器体44が、それぞれの内面側で第一の電極43と第二の電極45の集電体となると共に、それぞれの外面側を第一の電極43と第二の電極45の接続端子として用いることができる。
【0071】
以上のようにして得られる本発明の電解液を用いてリチウム二次電池を好適に作成できる。本発明のリチウム二次電池の形態は、コイン型、円筒型、角型、ラミネート等挙げられるが、特に限定されるわけではない。本発明のリチウム二次電池の一例としては、例えば、図9に示すコイン型セルの形態を挙げることができる。以下、図9と共に説明を続ける。コイン型セルは、正極・負極の電極がセパレータを介して配置され、これら正極・負極の活物質層およびセパレータに電解液が含浸されている。1対の正極・負極およびセパレータが図9に示すようにスペーサー、スプリングとともに正極缶および負極缶内部にガスケットを介してかしめられ、密閉される。
【0072】
本発明で使用する正極としては、活物質として、例えばLiCoO2、LiNiO2、LiNi1−xCoxO2、LiNi1−x−yCoxMny、LiNi0.5Mn0.5O2、LiMnO2、LiMn2O4、LiNi0.5Mn1.5O4等のリチウムと遷移金属との複合酸化物、TiO2、V2O5等の酸化物、TiS2、FeS等の硫化物などが挙げられるが、電池容量・エネルギー密度の観点からリチウムと遷移金属との複合酸化物が好ましい。これらの正極活物質を正極として成型する際には、公知の導電助剤や結着剤とともに加圧成型でき、または正極活物質を公知の導電助剤や結着剤とともにピロリドン等の有機溶剤に混合し、ペースト状にしたものをアルミニウム箔等の集電体に塗工後、乾燥して得ることができる。
【0073】
また本発明で使用する負極としては、リチウム金属、リチウム金属と他金属との合金、リチウムイオンが挿入脱離する材料が使用される。リチウム金属と他金属との合金としてはLi−Al、Li−Sn、Li−Zn、Li−Siなどが挙げられ、またリチウムイオンが挿入脱離する材料としては樹脂やピッチ等を焼成したカーボン材料やこれらのカーボン材料にホウ素化合物を添加したカーボン材料、天然黒鉛などを使用できる。これらの負極材料は単独で用いても、2種以上を混合して使用することもできる。これらの負極材料を負極として成型する際には、公知の導電助剤や結着剤とともに加圧成型でき、または負極活物質を公知の導電助剤や結着剤とともにピロリドン等の有機溶剤に混合し、ペースト状にしたものを銅箔等の集電体に塗工後、乾燥して得ることができる。
【0074】
また、本発明で使用するセパレータとしては、電解液が通過しやすく、絶縁体で、化学的に安定な材質であれば特に限定はない。
本発明の第4級アンモニウム塩およびこれを含有する電解液は耐電圧、電気伝導性が高く、有機溶媒に対する溶解性が高く、電気化学デバイスの電解液として好適である。電気化学デバイスとしては、例えば、電気二重層キャパシタ、二次電池、色素増感型太陽電池、エレクトロクロミック素子、コンデンサなどが例示されるがこの限りではない。特に好適な電気化学デバイスは電気二重層キャパシタ、二次電池である。
【実施例】
【0075】
以下に参考例、実施例、試験例を挙げ、本発明を具体的に説明するが、何らこれに限定されるものではない。
【0076】
実施例1
N−メチル−N−メトキシメチルピロリジニウムクロライド(N−メトキシメチル−N−メチルピロリジニウムクロライド)の合成
N−メチルピロリジン(試薬:東京化成製)30.0gを120gのトルエンに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製)31.2gを1時間で滴下した。5℃にて1時間攪拌し、徐々に昇温、室温にて10時間攪拌し、反応を終了した。反応液を濾別し、得られた固体を150gのトルエン、150gのアセトンにて洗浄した。減圧乾燥し53.7gの目的物(白色固体)を得た。
1H−NMR(D2O)δppm:
2.08(br 4H),2.96(s 3H),3.31(m 2H),3.47(m 2H),3.55(s 3H),4.50(s 2H)
【0077】
実施例2
N−メチル−N−メトキシメチルピロリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート)の合成
実施例1で製造したN−メチル−N−メトキシメチルピロリジニウムクロライド(N−メトキシメチル−N−メチルピロリジニウムクロライド)15.0gをMeOH35gに溶解し、30%HBF4のメタノール溶液27.83gを添加した。減圧下、塩化水素と過剰のHBF4を除き目的物(薄黄色液体)19.6gを得た。
1H−NMR(d−DMSO)δppm:
2.07(br 4H),3.00(s 3H),3.42(m 4H),3.60(s 3H),4.62(s 2H)
【0078】
実施例3
実施例2で製造したN−メチル−N−メトキシメチルピロリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート)の電気伝導度、耐電圧の測定をおこなった。
電気伝導度の測定にはRadiometer社製電気伝導度メーターを使用した。測定セルにはRadiometer社製CDC641Tを使用した。
耐電圧の測定には3極式電気化学セルを使用した。作用極として、φ1.0mm、電極面積0.0079cm−2のグラシーカーボン電極(BAS株式会社製)、参照極としてφ0.5mmの銀ワイヤー(株式会社ニラコ製、純度99.99%)、対極としてφ0.5mm×50mmの白金電極(BAS株式会社製、11−2233)を使用した。リニアスイープボルタンメトリーをおこない、酸化電流密度および還元電流密度が0.5mAcm−2になる電位を別々に調べた。これらの電位の差を耐電圧とした。なお電位の挿引印加速度は50mVs−1とした。電気化学測定には北斗電工製、HZ−3000を使用した。測定結果を表1に示した。
【0079】
実施例4
実施例2で製造したN−メチル−N−メトキシメチルピロリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート)とプロピレンカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)を種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表2に示した。
【0080】
実施例5
実施例2で製造したN−メチル−N−メトキシメチルピロリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート)とアセトニトリル(キシダ化学株式会社製、リチウムバッテリーグレード)を種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表3に示した。
【0081】
比較例1
N−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレートの合成
N−メチルピロリジン(試薬:東京化成製)31.10gを124.30gのトルエンに溶解し、窒素置換した。27℃下ブロモエチルメチルエーテル(試薬:アルドリッチ製)61.22gを1時間で滴下した。徐々に昇温し、60〜70℃にて37時間攪拌し、反応を終了した。室温まで冷却し、生じた固体を窒素下濾別した。70gのトルエンにて洗浄した後、減圧乾燥した(茶褐色固体 78.99g)。得られた固体は200gのアセトンに懸濁し、室温にて攪拌洗浄、窒素下濾別した(×2回繰り返した)。減圧乾燥し、収量62.64gを得た。着色があった為、水131.83gに溶解し、活性炭〔カルボラフィン 武田薬品工業(株)製〕6.00gを加え12時間90〜95℃にて攪拌処理した。室温まで、冷却し、活性炭を濾別した。減圧濃縮、減圧乾燥し、収量58.34gを得た。アセトン200.48g、クロロホルム27.22gの混合溶媒に加熱溶解し、再結晶した。生成した白色固体は窒素下濾別し、アセトン50gにて洗浄、減圧乾燥し、N−メトキシエチル−N−メチルピロリジニウムブロマイド34.10gを得た。
1H−NMR(CD3OD)δppm:
2.24(m 4H),3.15(s 3H),3.40(s 3H),3.65(m 6H),3.83(m 2H)
続いて、上記で製造したN−メトキシエチル−N−メチルピロリジニウムブロマイド40.0gをMeOH40.0gに溶解し、30wt%HBF4のメタノール溶液54.0gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4を除き目的物(白色固体)39.9gを得た。
1H−NMR(CD3OD)δppm:
2.22(m 4H),3.10(S 3H),3.39(S 3H),3.58(m 6H),3.81(m 2H)
上記で製造したN−メチル−N−メトキシエチルピロリジニウムテトラフルオロボレート(N−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレート)を、実施例4と同様な測定をおこなった。測定結果を表1に示した。
【0082】
比較例2
N−メトキシメチル−N−メチルピペリジニウムテトラフルオロボレートの合成
N−メチルピペリジン(試薬:東京化成製)54.50gを脱水アセトン(試薬:和光純薬製)700gに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製を蒸留精製)44.30gを1時間で滴下した。滴下終了後は15℃以下にて5時間攪拌し、反応を終了した。5℃まで冷却し、生成した固体を窒素下濾別した。400gのアセトンにて洗浄した後、減圧乾燥した。得られた白色固体はアセトン550gに懸濁し、還流下30分攪拌した。濾過し、アセトン300gにて洗浄した(2回繰り返した)。減圧乾燥し、目的物(N−メトキシメチル−N−メチルピペリジニウムクロライド)66.0gを得た。
1H−NMR(CD3OD)δppm:
1.60〜1.96(m 6H),3.05(s 3H),3.35(m 4H),3.69(s 3H),4.65(s 2H)
続いて、上記で製造したN−メトキシメチル−N−メチルピペリジニウムクロライド35.0gをMeOH35.0gに溶解し、30wt%HBF4のメタノール溶液59.9gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4およびメタノールを除き目的物43.7gを得た。
1H−NMR(CD3OD)δppm:
1.55〜2.00(m 6H),3.04(s 3H),3.34(m 4H),3.67(s 3H),4.62(s 2H)
上記で製造したN−メチル−N−メトキシメチルピペリジニウムテトラフルオロボレート(N−メトキシメチル−N−メチルピペリジニウムテトラフルオロボレート)を、実施例3と同様な測定をおこなった。測定結果を表1に示した。
【0083】
比較例3
N−メトキシメチル−N−メチルモルホリニウムテトラフルオロボレートの合成
N−メチルモルホリン(試薬:東京化成製)92.13gを脱水2−ブタノン(試薬:和光純薬製)670gに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製を蒸留精製)76.47gを1時間で滴下した。滴下終了後は15℃以下にて2時間攪拌し、反応を終了した。5℃まで冷却し、生成した固体を窒素下濾別した。500mlの2−ブタノンにて洗浄した後、減圧乾燥した。得られた白色固体はアセトン500mlに懸濁し、還流下30分攪拌した。濾過し、アセトン500mlにて洗浄した(2回繰り返した)。減圧乾燥し、目的物(N−メトキシメチル−N−メチルモルホリニウムクロライド)150.46gを得た。
1H−NMR(CD3OD)δppm:
3.22(s 3H),3.36〜3.42(m 2H),3.52〜3.61(m 2H),3.71(s 3H),4.01(m 4H),4.77(s 2H)
続いて、上記で製造したN−メトキシメチル−N−メチルモルホリニウムクロライド30.0gをMeOH30.0gに溶解し、30wt%HBF4のメタノール溶液50.8gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4およびメタノールを除き目的物37.2gを得た。
1H−NMR(CD3OD)δppm:
3.19(s 3H),3.31(m 2H),3.52(m 2H),3.70(s 3H),4.00(m 4H),4.72(s 2H)
上記で製造したN−メチル−N−メトキシメチルモルフォリニウムテトラフルオロボレート(N−メトキシメチル−N−メチルモルフォリニウムテトラフルオロボレート)を用い、1Mのプロピレンカーボネート溶液を調製した。実施例3と同様な測定法で耐電圧を測定した。測定結果を表1に示した。
【0084】
比較例4
N−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムテトラフルオロボレートの合成
エチルジメチルアミン(試薬:東京化成製)47.50gを脱水アセトン(試薬:和光純薬製)300gに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製を蒸留精製)52.30gを1時間で滴下した。滴下終了後は15℃以下にて5時間攪拌し、反応を終了した。5℃まで冷却し、生成した固体を窒素下濾別した。150gのアセトンにて洗浄した後、減圧乾燥した。N−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムクロライド(白色固体)85.90gを得た。
1H−NMR(CD3OD)δppm:
1.35(m 3H),3.03(s 6H),3.40(q 2H),3.68(s 3H),4.61(s 2H)
続いて、上記で製造したN−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムクロライド40.0gをMeOH40.0gに溶解し、30wt%HBF4のメタノール溶液80.0gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4およびメタノールを除き目的物51.6gを得た。
1H−NMR(CD3OD)δppm:
1.34(m 3H),3.00(s 6H),3.38(q 2H),3.66(s 3H),4.57(s 2H)
上記で製造したジメチルエチルメトキシメチルアンモニウムテトラフルオロボレート(N−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムテトラフルオロボレート)を用い、実施例3と同様な測定をおこなった。測定結果を表1に示した。
【0085】
比較例5
N,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレートの合成
ジエチルメチルアミン(試薬:東京化成製)35.53gを161.37gのトルエンに溶解し、窒素置換した。27℃下ブロモエチルメチルエーテル(試薬:アルドリッチ製)68.00gを1時間で滴下した。徐々に昇温し、60〜70℃にて44時間攪拌し、反応を終了した。室温まで冷却し、生じた固体を窒素下濾別した。70gのトルエンにて洗浄した後、減圧乾燥した(茶褐色固体 67.30g)。着色が激しい為、水131.52gに溶解し、活性炭〔カルボラフィン 武田薬品工業(株)製〕7.02gを加え12時間90〜95℃にて攪拌処理した。室温まで、冷却し、活性炭を濾別した。減圧濃縮、減圧乾燥し、収量58.34gを得た。アセトン200.48g、クロロホルム27.22gの混合溶媒に加熱溶解し、再結晶した。生成した白色固体は窒素下濾別し、アセトン50gにて洗浄、減圧乾燥し、目的物(N,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムブロマイド)47.58gを得た。
1H−NMR(CD3OD)δppm:
1.35(m 6H),3.07(s 3H),3.39(s 3H),3.40〜3.57(m 6H),3.80(m 2H)
続いて、上記で製造したN,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムブロマイド30.0gをMeOH30.0gに溶解し、30wt%HBF4のメタノール溶液40.8gを添加した。130℃の加熱下、窒素気流中にて、副生する塩化水素と過剰のHBF4およびメタノールを除き目的物30.2gを得た。
1H−NMR(CD3OD)δppm:
1.33(m 6H),3.03(s 3H),3.38(s 3H),3.39〜3.52(m 6H),3.77(m 2H)
上記で製造したジエチルメチルメトキシエチルアンモニウムテトラフルオロボレート(N,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレート)を用い、実施例3と同様な測定をおこなった。測定結果を表1に示した。
【0086】
比較例6
N,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレート(TEMA)
トリエチルメチルアンモニウムクロライド(試薬:東京化成製)100gをメタノール100gに溶解し、30wt%HBF4のメタノール溶液200.0gを添加した。30分攪拌するとトリエチルメチルアンモニウムテトラフルオロボレートの結晶が析出した。溶液を濾過後、結晶をイソプロピルアルコールで洗浄してから、130℃の加熱下、窒素気流中にて乾燥し、副生した塩化水素と過剰のHBF4およびメタノール、イソプロピルアルコールを除き目的物(白色固体)127.1gを得た。
1H−NMR(CD3OD)δppm:
1.31(m 9H),2.95(S 3H),3.34(q 6H)
上記で製造したトリエチルメチルアンモニウムテトラフルオロボレート(N,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレート)を、1Mのプロピレンカーボネート溶液を調整した。実施例3と同様な測定法で耐電圧を測定した。測定結果を表1に示した。
【0087】
比較例7
上記で製造したN−メチル−N−メトキシエチルピロリジニウムテトラフルオロボレート(N−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレート)とプロピレンカーボネートを種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表4に示した。
【0088】
比較例8
上記で製造したN−メチル−N−メトキシエチルピロリジニウムテトラフルオロボレート(N−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレート)とアセトニトリルを種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表5に示した。
【0089】
比較例9
上記で製造したジエチルメチルメトキシエチルアンモニウムテトラフルオロボレート(N,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレート)とプロピレンカーボネートを種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表6に示した。
【0090】
比較例10
上記で製造したトリエチルメチルアンモニウムテトラフルオロボレート(N,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレート)とプロピレンカーボネートを種々濃度で混合し、混合溶液の電気伝導度を測定した。電気伝導度の測定は実施例3と同様におこなった。測定結果を表7に示した。
【0091】
【表1】
【0092】
【表2】
【0093】
【表3】
【0094】
【表4】
【0095】
【表5】
【0096】
【表6】
【0097】
【表7】
【0098】
実施例6
N−メトキシメチル−N−メチルピロリジニウムクロライドの合成
N−メチルピロリジン(試薬:東京化成製を精留により精製 ピロリジン、水含有量共に0.1%以下)50.0gを292.0gの脱水アセトン(水含有量0.1%以下)に溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製を蒸留精製)47.3gを1時間で滴下した。5℃にて1時間攪拌し、5〜15℃以下にて4時間攪拌し、反応を終了した。反応液を濾別し、得られた固体を120gのアセトンにて洗浄した。減圧乾燥し92.5gの目的物(白色固体)を得た。
1H−NMR(CD3OD)δppm:
2.22(m 4H),3.11(s 3H),3.46(m 2H),3.60(m 2H),3.67(s 3H),4.65(s 2H)
【0099】
実施例7
N−メトキシメチル−N−メチルピロリジニウムクロライドの合成
N−メチルピロリジン(試薬:東京化成製)30.0gを150gのトルエンに溶解し、窒素置換した。5℃下クロロメチルメチルエーテル(試薬:東京化成製)31.2gを1時間で滴下した。5℃にて1時間攪拌し、徐々に昇温、室温にて10時間攪拌し、反応を終了した。反応液を濾別し、得られた固体を150gのアセトンにて洗浄した。減圧乾燥し53.7g白色固体を得た。得られた第4級アンモニウム塩をアセトン150gに懸濁し5時間攪拌した。濾過、洗浄、乾燥し白色固体48.3gを得た。続き得られた第4級アンモニウム塩はクロロホルム/アセトン(1/6(W/W))420gで再結晶、減圧乾燥し36.2gの目的物(白色固体)を得た。
1H−NMR(CD3OD)δppm:
2.22(m 4H),3.11(s 3H),3.46(m 2H),3.60(m 2H),3.67(s 3H),4.65(s 2H)
【0100】
実施例8
N−メトキシメチル−N−メチルピロリジニウムブロマイドの合成
N−メチルピロリジン17.0gを160gの脱水アセトン(水0.1%以下)に溶解し、窒素置換した。5℃下ブロモメチルメチルエーテル(試薬:東京化成製)24.6gを1.5時間で滴下した。5〜15℃以下にて4時間攪拌し、反応を終了した。反応液を濾別し、得られた固体を160gのアセトンにて洗浄した。減圧乾燥し30.9gの目的物(白色固体)を得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.11(s 3H),3.48(m 2H),3.60(m 2H),3.67(s 3H),4.65(s 2H)
【0101】
実施例9
N−メトキシメチル−N−メチルピロリジニウムアイオダイドの合成
N−メチルピロリジン(試薬:東京化成製を精留により精製 ピロリジン、水含有量共に0.1%以下)2.46gを21.74gの脱水2−ブタノン(水0.1%以下)に溶解し、窒素置換した。5℃下ヨードメチルメチルエーテル(試薬:Aldrich製を精留により精製)5.07gを1.5時間で滴下した。5〜15℃にて5時間攪拌し反応を終了した。反応液を濾別し、濾液を減圧乾燥し6.40gの目的物(薄赤褐色液体)を得た。
1H−NMR(CD3OD)δppm:
2.23(m 4H),3.13(s 3H),3.50(m 2H),3.62(m 2H),3.68(s 3H),4.68(s 2H)
【0102】
実施例10
N−メトキシメチル−N−メチルピロリジニウムアイオダイドの合成
パラホルムアルデヒド(試薬:和光純薬製)25.3gと炭酸カリウム58.3gをメタノール81.0gに懸濁させ、ピロリジン(試薬:東京化成製)60.0gを室温下1時間かけて滴下した。滴下終了後は昇温し、70℃で3時間反応した。反応終了後、室温まで冷却し濾過した。濾液は精留し無色透明液体メトキシメチルピロリジン68.9gを得た。得られたメトキシメチルピロリジンはアセトン600gに溶解し、ヨウ化メチル93.6gを添加し、窒素置換したオートクレイブ中80℃にて3日間攪拌した。濾過し、濾液を減圧乾燥することで目的物(赤褐液体)107.3gを得た。
メトキシメチルピロリジンの1H−NMRデータ
1H−NMR(CDCl3)δppm:
1.77(m 4H),2.76(m 4H),3.31(s 3H),4.14(s 2H)
N−メトキシメチル−N−メチルピロリジニウムアイオダイドの1H−NMRデータ
1H−NMR(CD3OD)δppm:
2.23(m 4H),3.13(s 3H),3.50(m 2H),3.62(m 2H),3.68(s 3H),4.68(s 2H)
【0103】
実施例11
N−メトキシメチル−N−メチルピロリジニウムカーボネートの合成
炭酸ナトリウム(和光純薬製)1.60gを脱イオン水18gに溶解し、N−メトキシメチル−N−メチルピロリジニウムクロライド5.01gを添加した。室温で0.5時間反応し反応を終了した。濃縮真空乾燥した後、残さにエチルアルコール100mlを加え不溶の塩化ナトリウムを除いた。ジクロロメタンに溶解し、メンブランフィルターによる再濾過を行い、減圧濃縮、乾燥し目的物5.41gを得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.11(s 3H),3.47(m 2H),3.59(m 2H),3.67(s 3H),4.64(s 2H)
【0104】
実施例12
N−メトキシメチル−N−メチルピロリジニウムスルホネートの合成
硫酸銀(和光純薬製)3.14gを脱イオン水400mlに溶解し、N−メトキシメチル−N−メチルピロリジニウムクロライド3.34gを添加した。室温で0.5時間反応し反応を終了した。生じた塩化銀を濾過し、濃縮真空乾燥した。メタノールに溶解し、メンブランフィルターによる再濾過を行い、減圧濃縮、乾燥し目的物3.99gを得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.11(s 3H),3.48(m 2H),3.61(m 2H),3.67(s 3H),4.65(s 2H)
【0105】
実施例13
N−メトキシメチル−N−メチルピロリジニウムパークロレートの合成
過塩素酸ナトリウム(和光純薬製)5.91gをエチルアルコール77gに溶解し、N−メトキシメチル−N−メチルピロリジニウムクロライド7.99gを添加した。室温で1.5時間反応し反応を終了した。生じた塩化ナトリウムを濾過し、濃縮真空乾燥した。ジクロロメタンに溶解し、メンブランフィルターによる再濾過を行い、減圧濃縮、乾燥し目的物10.94gを得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.10(s 3H),3.46(m 2H),3.58(m 2H),3.66(s 3H),4.61(s 2H)
【0106】
実施例14
N−メトキシメチル−N−メチルピロリジニウムフルオライドの合成
フッ化カリウム(和光純薬製)0.44gを脱イオン水11gに溶解し、N−メトキシメチル−N−メチルピロリジニウムパークロレート1.74gを添加した。室温で1.5時間反応し反応を終了した。メタノール100mlを添加し濾過した。濃縮真空乾燥した後、ジクロロメタンに溶解しメンブランフィルターによる再濾過を行った。濃縮乾燥し目的物1.05gを得た。
1H−NMR(CD3OD)δppm:
2.20(m 4H),3.09(s 3H),3.46(m 2H),3.58(m 2H),3.66(s 3H),4.60(s 2H)
【0107】
実施例15
N−メトキシメチル−N−メチルピロリジニウムメチルカーボネートの合成
メトキシメチルピロリジン10.00gと炭酸ジメチル117.39gをオートクレイブに添加した。120℃にて24時間反応した。生じた固体を濾過し、炭酸ジメチルにて洗浄した。減圧乾燥し、目的物10.70gを得た。
1H−NMR(CD3OD)δppm:
2.21(m 4H),3.10(s 3H),3.34(s 3H),3.45(m 2H),3.58(m 2H),3.66(s 3H),4.62(s 2H)
【0108】
実施例16
N−メトキシメチル−N−メチルピロリジニウムアセテートの合成
酢酸ナトリウム(和光純薬製)9.46gをメタノール95gに溶解し、N−メトキシメチル−N−メチルピロリジニウムクロライド19.10gを添加した。室温で1.5時間反応し反応を終了した。濾過した後、濃縮真空乾燥した。残さにジクロロメタン100mlを加えメンブランフィルターによる濾過を行い、減圧濃縮、乾燥し目的物20.38gを得た。
1H−NMR(CD3OD)δppm:
1.89(s 3H),2.20(m 4H),3.10(s 3H),3.44(m 2H),3.60(m 2H),3.66(s 3H),4.61(s 2H)
【0109】
実施例17
N−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートの合成
実施例6で製造したN−メトキシメチル−N−メチルピロリジニウムクロライド50.0gをMeOH120gに溶解し、30%HBF4のメタノール溶液92.8gを添加した。130℃の加熱下、N2バブリングをおこない、塩化水素と過剰のHBF4を除き目的物(微黄色液体)65.2gを得た。
1H−NMR(CD3OD)δppm:
2.19(m 4H),3.08(s 3H),3.43(m 2H),3.56(m 2H),3.65(s 3H),4.59(s 2H)
【0110】
比較例11
テトラエチルアンモニウムテトラフルオロボレート(TEA)
テトラエチルアンモニウムブロマイド(試薬:東京化成製)120gをメタノール120gに溶解し、30wt%HBF4のメタノール溶液172gを添加した。30分攪拌するとテトラエチルアンモニウムテトラフルオロボレートの結晶が析出した。溶液を濾過後、結晶をイソプロピルアルコールで洗浄してから、130℃の加熱下、窒素気流中にて乾燥し、副生した臭化水素と過剰のHBF4およびメタノール、イソプロピルアルコールを除き目的物(白色固体)118gを得た。
1H−NMR(CD3OD)δppm:
1.28(m 12H),3.29(q 8H)
【0111】
<電気二重層キャパシタ用電解液の調製>
実施例18(MMMP−BF4/PC)
実施例17で製造したN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとプロピレンカーボネート(PC)(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表8に掲げる通りとした。
【0112】
実施例19(MMMP−BF4/DMC)
実施例17で製造したN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとジメチルカーボネート(DMC)(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表9に掲げる通りとした。
【0113】
実施例20(MMMP−BF4/EMC)
実施例17で製造したN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとエチルメチルカーボネート(EMC)(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表10に掲げる通りとした。
【0114】
実施例21(MMMP−BF4/DMC+EMC)
実施例17で製造したN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレートとジメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)およびエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表11に掲げる通りとした。
【0115】
比較例12(TEMA−BF4/PC)
比較例6で製造したN,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレートとプロピレンカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表12に掲げる通りとした。
【0116】
比較例13(TEA−BF4/PC)
比較例11で製造したテトラエチルアンモニウムテトラフルオロボレートとプロピレンカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、テトラエチルアンモニウムテトラフルオロボレートの濃度が0.8Mになるように、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表13に掲げる通りとした。尚テトラエチルアンモニウムテトラフルオロボレートとジメチルカーボネート及びエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で混合したが不溶であった。
【0117】
比較例14(EMI−BF4/PC)
1−エチル−3−メチルイミダゾリウムテトラフルオロボレート(EMI−BF4)とプロピレンカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表14に掲げる通りとした。
【0118】
比較例15(TEMA−BF4/EMC)
比較例6で製造したN,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表15に掲げる通りとした。
【0119】
比較例16(TEMA−BF4/DMC)
比較例6で製造したN,N,N−トリエチル−N−メチルアンモニウムテトラフルオロボレートとジメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表16に掲げる通りとした。
【0120】
比較例17(EMI−BF4/EMC)
1−エチル−3−メチルイミダゾリウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表17に掲げる通りとした。
【0121】
比較例18(EMI−BF4/DMC)
1−エチル−3−メチルイミダゾリウムテトラフルオロボレートとジメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表18に掲げる通りとした。
【0122】
比較例19
比較例4で製造したN−エチル−N−メトキシメチル−N,N−ジメチルアンモニウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表19に掲げる通りとした。
【0123】
比較例20
比較例5で製造したN,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表20に掲げる通りとした。
【0124】
比較例21(MMEP−BF4/EMC)
比較例1で製造したN−メトキシエチル−N−メチルピロリジニウムテトラフルオロボレートとエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表21に掲げる通りとした。
【0125】
比較例22(MMMPI−BF4/EMC)
比較例2で製造したN−メトキシメチル−N−メチルピペリジニウムテトラフルオロボレート(MMMPI−BF4)とエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表22に掲げる通りとした。
【0126】
比較例23(MMMM−BF4/EMC)
比較例3で製造したN−メトキシメチル−N−メチルモルホリニウムテトラフルオロボレート(MMMM−BF4)とエチルメチルカーボネート(キシダ化学株式会社製、リチウムバッテリーグレード)とを、種々濃度で、露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。混合濃度は表23に掲げる通りとした。
【0127】
<混合状態の観察>
実施例18〜実施例21および比較例12〜比較例23で得られた各種組成物を、ドライボックス内で4ccずつ、スクリュー栓が付いたガラス容器に移し、ドライボックスの外に取り出した。各種溶液が入ったガラス容器を恒温槽に浸漬し、25℃、0℃、−30℃でそれぞれ5時間保持し、目視で状態を確認した。結果を表8〜表23に示した。表において、「−」は二層分離を、「固」は固体状態を示す。
【0128】
<電気伝導度の測定>
混合状態の観察後、分離していない、或いは固化していない液体状態の組成溶液について、再度ドライボックス内から溶液を取り出し電気伝導度を測定した。電気伝導度の測定には、導電率計(CDM210 Radiometer社製)を使用した。測定セルにはXE−100(Radiometer社製)を使用した。結果を、表8〜表23に示した。
【0129】
【表8】
【0130】
【表9】
【0131】
【表10】
【0132】
【表11】
【0133】
【表12】
【0134】
【表13】
【0135】
【表14】
【0136】
【表15】
【0137】
【表16】
【0138】
【表17】
【0139】
【表18】
【0140】
【表19】
【0141】
【表20】
【0142】
【表21】
【0143】
【表22】
【0144】
【表23】
【0145】
実施例22〜23および比較例24
<円筒型電気二重層キャパシタの製造等>
図1において、円筒型電気二重層キャパシタ1は円筒状密閉容器2を有し、その密閉容器2内に電極巻回体3、2つの円盤状集電体4、5および電解液が収容されている。
図2に示すように、電極巻回体3は、帯状正極6、帯状負極7およびそれらの一方、実施例では帯状正極6を挟む2つの帯状セパレータ8、9よりなる重合せ物を、帯状正極6の外側に在る一方の帯状セパレータ8が最も内側に位置するようにAl製巻心10を中心に渦巻状に巻回したものである。この場合、帯状正、負極6、間に存する他方の帯状セパレータ9は、外周の帯状負極7を覆うべく略一巻分だけ帯状負極7の終端より延出している。
帯状正極6は、それぞれ帯状集電体11およびその帯状集電体11の両面に積層形成された一対の分極性電極12を有する。帯状正極6において、集電体11両面の長手方向一側縁部は電極不存在領域であって正極端子との接続部13として機能する。帯状負極7は帯状正極6とは点対称の関係にあり、その集電体11両面の長手方向他側縁部は電極不存在領域であって負極端子との接続部14として機能する。
図1に示すように、密閉容器2はAl製有底筒形本体15と、その開口を閉鎖する蓋板16よりなる。その蓋板16は本体15に溶接されたAl製環状外周板17と、その外周板17の内周縁に外周溝18を嵌着させた電気絶縁性樹脂製環状中間板19と、その中間板19の内周溝20に外周凸部21を嵌着させたAl合金製筒状正極端子22を有する。
一方のAl合金製円盤状集電体4において,その中心に在るボス23は筒状正極端子22の中心孔24に嵌合してそれに溶接されている。また円盤部25は、放射状に配列されて下方に突出する複数のV形凸条26を有し、それら凸条26の底部に図2に示す帯状正極6の接続部13が溶接されている。
他方のAl合金製円盤状集電体5において、その中心に在るボス27は負極端子28である有底筒形本体15の底壁に溶接されている。また円盤部29は、放射状に配列されて上方へ突出する複数の山形凸条30を有し、それら凸条30の稜線部に図2に示す帯状負極7の接続部14が溶接されている。
電解液は、負極側円盤状集電体5のボス27に形成された注入孔31から密閉容器2内に注入され、その後、注入孔31はゴム栓32により封鎖される。
図2に示す各分極性電極12は活性炭、導電フィラおよびバインダよりなる。
原料として活性炭を80wt%,ケッチェンブラックEC 10wt%およびテフロン(登録商標)6J(商品名、三井デュポンフロロケミカル社製)10wt%を混練し、次いで混練物を用いて圧延を行い、厚さ150μmの電極シートを製作した。電極シートから幅103mm、長さ1400mmの複数の帯状分極性電極12を切出し、次いで2枚の分極性電極12と、幅109mm、長さ1400mm、厚さ30μmのアルミ箔よりなる帯状集電体11とを一対の加圧ローラを用い、線圧6tにて圧着して帯状正極6を製作した。また同様の方法で帯状負極7を製作した。
帯状正極6、帯状負極7および帯状正極6を挟む両帯状セパレータ8、9よりなる重合せ物を、帯状正極6の外側に在る一方の帯状セパレータ8が最も内側に位置するようにAl製巻心10を中心に渦巻き状に巻回して、外径D1が38.5mmで、長さが115mmの電極巻回体3を製造した。
電極巻回体3を、内径D2が39.5mmで、長さが120mmの有底筒形本体15内に入れ、次いでその本体15内に実施例59〜60および比較例18の電解液を注入した。その後,注入孔31をゴム栓32により封鎖した。
上記のようにして作成された円筒型電気二重層キャパシタの耐久信頼性を確認すべく、45℃の環境下にて、連続的に2.5Vの電圧を印加した際の性能変化を下記に示す。
【0146】
<円筒型電気二重層キャパシタによる結果>
上記のようにして作成された円筒型電気二重層キャパシタの耐久信頼性を確認すべく、45℃の環境下にて、1000時間の間、連続的に2.5Vの電圧を印加した際の1000時間後の性能を下表に示す。
表24、および図3〜5において、静電容量、抵抗、発生ガス量の値は比較例24の値を100とする相対値で表されている。
【0147】
【表24】
【0148】
<電気二重層キャパシタの製造A>
実施例24
実施例18で調製された電解液を使用して図6の構造を有する電気二重層キャパシタAを製造した。電極33及び電極34は、活性炭を主成分とする電導性物質、バインダー、N−メチルピロリドンとともに混練して得られたペーストをアルミニウム箔に150μmの厚さで塗工後、乾燥して得られたシート状電極を円板状に切り出したものである。容器体36、容器体37、スペーサー、スプリングは共にステンレス鋼製であり、セパレータは、ポリプロピレン不織布である。電気二重層キャパシタの組み立てはアルゴンガスを満たしたグローブボックス内でおこなった。電極33、電極34、容器体36、容器体37、スプリング、スペーサーは120℃の加熱下、24時間真空乾燥した後、グローブボックス内に持ち込んだ。実施例18の電気二重層キャパシタ用電解液を電極33、電極34、セパレータに含浸させ、図6の構成で容器体36と容器体37をガスケットを介してかしめることによって電気二重層キャパシタを得た。
【0149】
比較例25
実施例24において、実施例18で調製した電解液に代えて、比較例12で調製した電解液を使用した以外は、実施例24と同様にして電気二重層キャパシタを得た。
【0150】
比較例26
実施例24において、実施例18で調製した電解液に代えて、比較例13で調製した電解液を使用した以外は、実施例24と同様にして電気二重層キャパシタを得た。
測定例
実施例24及び比較例25、比較例26で作製されたコイン型電気二重層キャパシタに関し、内部抵抗および静電容量を25℃、−30℃において測定した。コイン型セルを専用のホルダにセットした後、低温恒温槽に浸漬してコイン型セルの温度を一定に保つようにした。この際、ホルダ全体をビニール袋で覆い、コイン型セルが恒温槽内の冷媒に接液しないようにした。所定の温度に設定した恒温槽に、コイン型セルを浸漬し、4時間保持した後、電気ニ重層キャパシタの充放電を開始した。電流密度が2.0mAの定電流充電をおこない、電圧が2.5Vに達した時点で定電圧充電に切り替えた。2.5Vで120分保持した後、2.0mAの定電流放電をおこない、電圧が0Vに達した時点で低電圧放電に切り替え0Vで120分間保持した。放電の際の電気エネルギーの積算値から静電容量を算出した。また、放電直後の電圧降下の値と放電電流の値とからセルの内部抵抗を算出した。実施例24の25℃と−30℃における内部抵抗と静電容量を100とした際の比較例25と比較例26の結果を表25に示した。
【0151】
【表25】
【0152】
<電気二重層キャパシタの製造B>
実施例25〜27
次に、実施例19〜21で調製された電解液を、図7の構造を有する容器に充填し、電気二重層キャパシタBを製造した。なお、第一の電極43及び第二の電極45は、いずれも活性炭を主成分とする電導性物質をバインダーと混練して、円板状に成形したものであり、第一の容器42及び第二の容器44は共にアルミニウム製であり、第一の電極43及び第二の電極45は、それぞれ電導性接着剤等により接着されている。また、隔離膜46は、レーヨン系抄紙である。
電気二重層キャパシタは、図7の構造、上記の材質の容器を150℃で、5時間真空乾燥した後、アルゴンガスを満たしたグローブボックス内で、実施例19〜21で調製した電解液を注入することにより製造した。
【0153】
比較例27〜比較例30
実施例25において、実施例19〜21で調製した電解液に代えて、比較例12、14、17および18で調製した電解液を使用した以外は、実施例25と同様にして電気二重層キャパシタを得た。
<電気二重層キャパシタの反応電流値の測定>
実施例25及び比較例27で製造された電気二重層キャパシタに対し、充放電試験装置を用いて段階的に電圧を印加し、各電圧毎に、電解液の分解によって生じる反応電流を測定することにより、実施例19〜21と比較例12、14、17および18の電解液の耐電圧を求めた。
すなわち、まず25℃にて2.4Vまで定電流にて充電を行った後、2.4Vにて2時間保持充電した際の電解液の分解による反応電流値を測定した。次いで、定電流にて所定電圧(0.1V)まで放電し、2.6Vまで定電流にて充電を行った後、2.6Vにて2時間保持充電した際の反応電流値を測定した。以後同様にして、4.0Vまで0.2Vずつ段階的に電圧を上昇させていき、その度に反応電流値を測定した。反応電流値が初めて0.1mAを超える電圧を耐電圧値とする。結果を図8及び表26に示す。なお、この電気二重層キャパシタは、実際には約2.5Vで使用するが、この耐電圧値が高いほど長期の耐久性が優れていることが分かっている。
【0154】
【表26】
【0155】
実施例25〜27で製造された電気二重層キャパシタは、3.2〜3.4Vまで、反応電流が発生しなかったが、比較例27〜30で製造された電気二重層キャパシタでは、3.0Vを超えると反応電流が発生した。また、実施例25〜27で製造された電気二重層キャパシタの方が、全ての電圧で反応電流が小さかった。
以上より、実施例19〜21の電解液(MMMP−BF4/DMC、MMMP−BF4/EMC、MMMP−BF4/DMC+EMC)の方が、従来の電気二重層キャパシタ用電解液として一般的に用いられている比較例12および14の電解液(TEMA−BF4/PC、EMI−BF4/PC)に比較して、耐電圧が高く、耐久性に優れていることが判った。更に、高い電圧においても反応電流値が小さいことより、高電圧で作動させても静電容量の低下が小さく、長期信頼性が高いことが判った。
【0156】
<リチウム二次電池用電解液の調製>
実施例28
実施例17で得られたN−メトキシメチル−N−メチルピロリジニウムテトラフルオロボレート(MMMP−BF4)にリチウムテトラフルオロボレート(LiBF4)を0.6Mの濃度で混合した。露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。
【0157】
参考例1
N−メトキシメチル−N−メチルピロリジニウムビス(トリフルオロメタンスルホニル)イミド(MMMP−TFSI)にリチウムビス(トリフルオロメタンスルホニル)イミド(LiTFSI)を0.6Mの濃度で混合した。露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。
【0158】
比較例31
比較例5で得られたN,N−ジエチル−N−メトキシエチル−N−メチルアンモニウムテトラフルオロボレート(DEMME−BF4)にリチウムテトラフルオロボレート(LiBF4)を0.6Mの濃度で混合した。露点が−60℃以下の窒素雰囲気ドライボックス内で混合した。混合後の溶液の水分をカールフィッシャー水分計(平沼産業株式会社製、平沼微量水分測定装置AQ−7)で測定し、30ppm以下であることを確認した。
【0159】
<電気伝導度の測定>
実施例28および比較例31の電解液の電気伝導度を測定した。電気伝導度の測定には、導電率計(CDM210 Radiometer社製)を使用した。測定セルにはXE−100(Radiometer社製)を使用した。結果を、表27に示した。
【0160】
【表27】
【0161】
<リチウム二次電池の作製>
参考例2
図9に示すようなコイン型リチウム二次電池を作製した。図9において、49は正極、50は負極、51はセパレーター、52は正極缶、53は負極缶、54はガスケット、55はスペーサー、56はスプリングである。図9に示すリチウム二次電池を以下に示す手順で作成した。正極缶52、負極缶53、スペーサー55およびスプリング56はステンレス製のものを使用した。負極50には厚さ200μmのリチウム金属箔を円形状に切り出したものを使用した。次に正極49の作製を示す。LiCoO2粉末と導電助剤のアセチレンブラックと結着剤のPVdFを85:10:5の重量比で混合し、N−メチルピロリドンを加えペースト状にした。これを厚さ30μmのアルミニウム箔上に電極塗工用アプリケーターで均一に塗工した。これを120℃で8時間、真空乾燥した後、電極打ち抜き機で円形状に切り出し正極49を得た。セパレータおよび切り出した正極に参考例1で得られた電解液を含浸させておく。正極を正極缶52の底面に載せ、その上にセパレーターを載置した後、ガスケット54を挿入した。その後、負極50とさらにその上に、スペーサー55とスプリング56と負極缶53とを順々にセパレーター上に載置し、コイン形電池かしめ機を使用して、正極缶52の開口部分を内方へ折曲することにより封口しリチウム二次電池を作成した。
【図面の簡単な説明】
【0162】
【図1】本発明の電気二重層キャパシタの断面図である。
【図2】本発明の電気二重層キャパシタの構成図である。
【図3】連続的に2.5Vの電圧を1000時間印加した際の円筒型電気二重層キャパシタの静電容量の変化を示すグラフである。
【図4】連続的に2.5Vの電圧を1000時間印加した際の円筒型電気二重層キャパシタの抵抗の変化を示すグラフである。
【図5】連続的に2.5Vの電圧を1000時間印加した際の円筒型電気二重層キャパシタのガス発生量の変化を示すグラフである。
【図6】他の構造を有する電気二重層キャパシタの断面図である。
【図7】更に他の構造を有する電気二重層キャパシタの断面図である。
【図8】電気二重層キャパシタに印加した電圧と反応電流の関係を示すグラフである。
【図9】コイン型リチウム二次電池の断面図である。
【符号の説明】
【0163】
1…円筒型電気二重層キャパシタ、2…円筒状密閉容器、3…電極巻回体、4,5…円盤状集電体、6…帯状正極、7…帯状負極、8,9…帯状セパレータ、10…Al製巻心、11…帯状集電体、12…分極性電極、13…正極端子との接続部、14…負極端子との接続部14、15…Al製有底筒形本体、16…蓋板、17…Al製環状外周板、18…外周溝、19…電気絶縁性樹脂製環状中間板、20…内周溝、21…外周凸部、22…Al合金製筒状正極端子、23…ボス、24…中心孔、25…円盤部、26…V形凸条、27…ボス、28…負極端子、29…円盤部、30…山形凸条、31…注入孔、32…ゴム栓、33,34…電極、36,37…容器体、41…電気二重層キャパシタ、42…第一の容器体、43…第一の電極、44…第二の容器体、45…第二の電極、46…隔膜、47…非電導性材料、48…電解液、49…正極、50…負極、51…セパレーター、52…正極缶、53…負極缶、54…ガスケット、55…スペーサー、56…スプリング
【特許請求の範囲】
【請求項1】
式(1)で表される第4級アンモニウム塩。
【化1】
(式中、R1〜R2は、共にメチル基を示す。X−は、BF4−を示す。)
【請求項2】
請求項1記載の第4級アンモニウム塩と、有機溶媒とを含んでなることを特徴とする組成物(ただしフルオロベンゼンを含まない)。
【請求項3】
有機溶媒が、環状炭酸エステル、鎖状炭酸エステル、ニトリル化合物およびスルホン化合物から選ばれる1種または2種以上の有機溶媒であることを特徴とする請求項2記載の組成物。
【請求項4】
有機溶媒が、プロピレンカーボネート、ジメチルカーボネート、エチルメチルカーボネート、アセトニトリルおよびスルホランから選ばれる1種または2種以上の有機溶媒であることを特徴とする請求項2記載の組成物。
【請求項5】
有機溶媒が、プロピレンカーボネートであることを特徴とする請求項2記載の組成物。
【請求項6】
有機溶媒が、ジメチルカーボネートであることを特徴とする請求項2記載の組成物。
【請求項7】
有機溶媒が、エチルメチルカーボネートであることを特徴とする請求項2記載の組成物。
【請求項8】
有機溶媒が、アセトニトリルであることを特徴とする請求項2記載の組成物。
【請求項9】
有機溶媒が、プロピレンカーボネート、エチレンカーボネート、ジメチルカーボネートおよびエチルメチルカーボネートから選ばれる2種以上の混合有機溶媒であることを特徴とする請求項2記載の組成物。
【請求項10】
有機溶媒が、ジメチルカーボネートおよびエチルメチルカーボネートからなる混合有機溶媒であることを特徴とする請求項2記載の組成物。
【請求項11】
式(3)で表される第4級アンモニウム塩のみからなる電解質。
【化2】
(式中、R1〜R2は、共にメチル基を示す。X−は、BF4−を示す。)
【請求項12】
請求項11記載の電解質を含むことを特徴とする電気化学デバイス用電解液(ただしフルオロベンゼンを含まない)。
【請求項13】
請求項11記載の電解質と、有機溶媒とを含んでなることを特徴とする電気化学デバイス用電解液(ただしフルオロベンゼンを含まない)。
【請求項14】
有機溶媒が、環状炭酸エステル、鎖状炭酸エステル、ニトリル化合物およびスルホン化合物から選ばれる1種または2種以上の有機溶媒であることを特徴とする請求項13に記載の電気化学デバイス用電解液。
【請求項15】
有機溶媒が、プロピレンカーボネート、ジメチルカーボネート、エチルメチルカーボネート、アセトニトリルおよびスルホランから選ばれる1種または2種以上の有機溶媒であることを特徴とする請求項13に記載の電気化学デバイス用電解液。
【請求項16】
有機溶媒が、プロピレンカーボネートであることを特徴とする請求項13記載の電気化学デバイス用電解液。
【請求項17】
有機溶媒が、ジメチルカーボネートであることを特徴とする請求項13記載の電気化学デバイス用電解液。
【請求項18】
有機溶媒が、エチルメチルカーボネートであることを特徴とする請求項13記載の電気化学デバイス用電解液。
【請求項19】
有機溶媒が、アセトニトリルであることを特徴とする請求項13に記載の電気化学デバイス用電解液。
【請求項20】
有機溶媒が、プロピレンカーボネート、エチレンカーボネート、ジメチルカーボネートおよびエチルメチルカーボネートから選ばれる2種以上の混合有機溶媒であることを特徴とする請求項13に記載の電気化学デバイス用電解液。
【請求項21】
有機溶媒が、ジメチルカーボネートおよびエチルメチルカーボネートからなる混合有機溶媒であることを特徴とする請求項13記載の電気化学デバイス用電解液。
【請求項22】
請求項12〜21のいずれかに記載の電解液を用いたことを特徴とする電気化学デバイス。
【請求項23】
請求項12〜21のいずれかに記載の電解液を用いたことを特徴とするキャパシタ。
【請求項24】
請求項12〜21のいずれかに記載の電解液を用いたことを特徴とするリチウム二次電池。
【請求項25】
式(5)で表される第4級アンモニウム塩と式(9)で表される化合物とを反応させる工程を含むことを特徴とする式(1)で表される第4級アンモニウム塩の製造方法。
【化3】
(式中、R1〜R2は、共にメチル基を示す。およびYは、Cl、Br、Iを示す。)
MX (9)
(式中、Mは、水素原子、アルカリ金属原子、アルカリ土類金属原子あるいは金属原子を示す。Xは、BF4を示す。)
【化4】
(式中、R1、R2はおよびXは、上記と同じ。)
【請求項26】
(a)式(7)のアルキルピロリジンと式(8)の化合物とを反応させることにより、式(5)で表される第4級アンモニウム塩を製造する工程、
(b)式(5)で表される第4級アンモニウム塩と式(9)で表される化合物とを反応させる工程、
を含むことを特徴とする式(1)で表される第4級アンモニウム塩の製造方法。
【化5】
(式中、R1は、メチル基を示す。)
【化6】
(式中、R2は、メチル基を示す。Yは、Cl、Br、Iを示す。)
【化7】
(式中、R1、R2およびYは、上記と同じ。)
MX (9)
(式中、Mは、水素原子、アルカリ金属原子、アルカリ土類金属原子あるいは金属原子を示す。Xは、BF4を示す。)
【化8】
(式中、R1、R2はおよびXは、上記と同じ。)
【請求項1】
式(1)で表される第4級アンモニウム塩。
【化1】
(式中、R1〜R2は、共にメチル基を示す。X−は、BF4−を示す。)
【請求項2】
請求項1記載の第4級アンモニウム塩と、有機溶媒とを含んでなることを特徴とする組成物(ただしフルオロベンゼンを含まない)。
【請求項3】
有機溶媒が、環状炭酸エステル、鎖状炭酸エステル、ニトリル化合物およびスルホン化合物から選ばれる1種または2種以上の有機溶媒であることを特徴とする請求項2記載の組成物。
【請求項4】
有機溶媒が、プロピレンカーボネート、ジメチルカーボネート、エチルメチルカーボネート、アセトニトリルおよびスルホランから選ばれる1種または2種以上の有機溶媒であることを特徴とする請求項2記載の組成物。
【請求項5】
有機溶媒が、プロピレンカーボネートであることを特徴とする請求項2記載の組成物。
【請求項6】
有機溶媒が、ジメチルカーボネートであることを特徴とする請求項2記載の組成物。
【請求項7】
有機溶媒が、エチルメチルカーボネートであることを特徴とする請求項2記載の組成物。
【請求項8】
有機溶媒が、アセトニトリルであることを特徴とする請求項2記載の組成物。
【請求項9】
有機溶媒が、プロピレンカーボネート、エチレンカーボネート、ジメチルカーボネートおよびエチルメチルカーボネートから選ばれる2種以上の混合有機溶媒であることを特徴とする請求項2記載の組成物。
【請求項10】
有機溶媒が、ジメチルカーボネートおよびエチルメチルカーボネートからなる混合有機溶媒であることを特徴とする請求項2記載の組成物。
【請求項11】
式(3)で表される第4級アンモニウム塩のみからなる電解質。
【化2】
(式中、R1〜R2は、共にメチル基を示す。X−は、BF4−を示す。)
【請求項12】
請求項11記載の電解質を含むことを特徴とする電気化学デバイス用電解液(ただしフルオロベンゼンを含まない)。
【請求項13】
請求項11記載の電解質と、有機溶媒とを含んでなることを特徴とする電気化学デバイス用電解液(ただしフルオロベンゼンを含まない)。
【請求項14】
有機溶媒が、環状炭酸エステル、鎖状炭酸エステル、ニトリル化合物およびスルホン化合物から選ばれる1種または2種以上の有機溶媒であることを特徴とする請求項13に記載の電気化学デバイス用電解液。
【請求項15】
有機溶媒が、プロピレンカーボネート、ジメチルカーボネート、エチルメチルカーボネート、アセトニトリルおよびスルホランから選ばれる1種または2種以上の有機溶媒であることを特徴とする請求項13に記載の電気化学デバイス用電解液。
【請求項16】
有機溶媒が、プロピレンカーボネートであることを特徴とする請求項13記載の電気化学デバイス用電解液。
【請求項17】
有機溶媒が、ジメチルカーボネートであることを特徴とする請求項13記載の電気化学デバイス用電解液。
【請求項18】
有機溶媒が、エチルメチルカーボネートであることを特徴とする請求項13記載の電気化学デバイス用電解液。
【請求項19】
有機溶媒が、アセトニトリルであることを特徴とする請求項13に記載の電気化学デバイス用電解液。
【請求項20】
有機溶媒が、プロピレンカーボネート、エチレンカーボネート、ジメチルカーボネートおよびエチルメチルカーボネートから選ばれる2種以上の混合有機溶媒であることを特徴とする請求項13に記載の電気化学デバイス用電解液。
【請求項21】
有機溶媒が、ジメチルカーボネートおよびエチルメチルカーボネートからなる混合有機溶媒であることを特徴とする請求項13記載の電気化学デバイス用電解液。
【請求項22】
請求項12〜21のいずれかに記載の電解液を用いたことを特徴とする電気化学デバイス。
【請求項23】
請求項12〜21のいずれかに記載の電解液を用いたことを特徴とするキャパシタ。
【請求項24】
請求項12〜21のいずれかに記載の電解液を用いたことを特徴とするリチウム二次電池。
【請求項25】
式(5)で表される第4級アンモニウム塩と式(9)で表される化合物とを反応させる工程を含むことを特徴とする式(1)で表される第4級アンモニウム塩の製造方法。
【化3】
(式中、R1〜R2は、共にメチル基を示す。およびYは、Cl、Br、Iを示す。)
MX (9)
(式中、Mは、水素原子、アルカリ金属原子、アルカリ土類金属原子あるいは金属原子を示す。Xは、BF4を示す。)
【化4】
(式中、R1、R2はおよびXは、上記と同じ。)
【請求項26】
(a)式(7)のアルキルピロリジンと式(8)の化合物とを反応させることにより、式(5)で表される第4級アンモニウム塩を製造する工程、
(b)式(5)で表される第4級アンモニウム塩と式(9)で表される化合物とを反応させる工程、
を含むことを特徴とする式(1)で表される第4級アンモニウム塩の製造方法。
【化5】
(式中、R1は、メチル基を示す。)
【化6】
(式中、R2は、メチル基を示す。Yは、Cl、Br、Iを示す。)
【化7】
(式中、R1、R2およびYは、上記と同じ。)
MX (9)
(式中、Mは、水素原子、アルカリ金属原子、アルカリ土類金属原子あるいは金属原子を示す。Xは、BF4を示す。)
【化8】
(式中、R1、R2はおよびXは、上記と同じ。)
【図1】

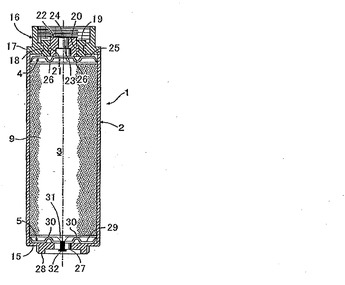
【図2】
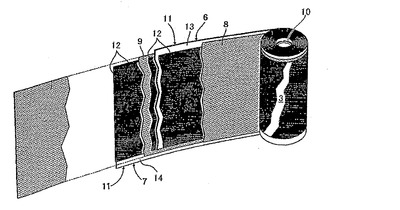
【図3】
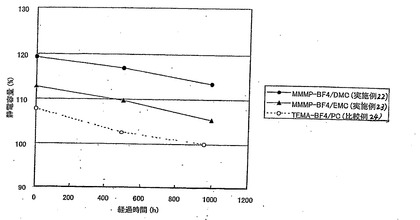
【図4】
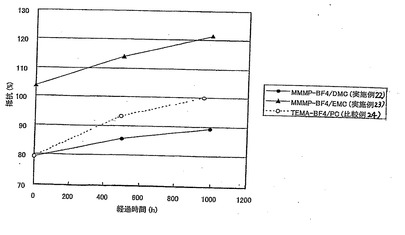
【図5】
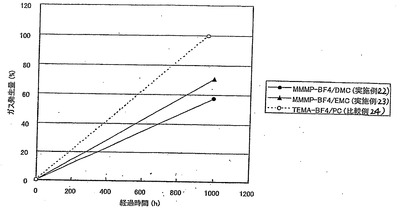
【図6】
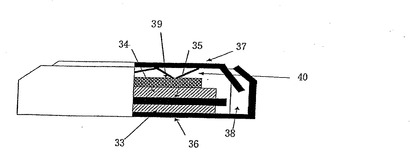
【図7】
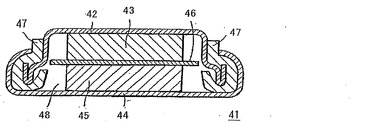
【図8】
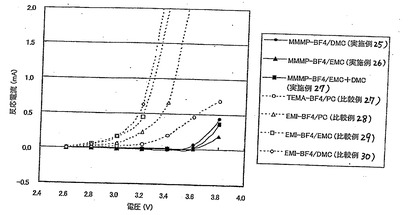
【図9】
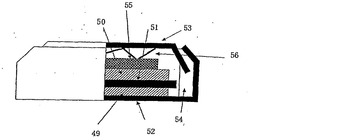
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2007−39460(P2007−39460A)
【公開日】平成19年2月15日(2007.2.15)
【国際特許分類】
【出願番号】特願2006−238217(P2006−238217)
【出願日】平成18年9月1日(2006.9.1)
【分割の表示】特願2005−511389(P2005−511389)の分割
【原出願日】平成16年6月30日(2004.6.30)
【出願人】(302060306)大塚化学株式会社 (88)
【出願人】(000162847)ステラケミファ株式会社 (81)
【Fターム(参考)】
【公開日】平成19年2月15日(2007.2.15)
【国際特許分類】
【出願日】平成18年9月1日(2006.9.1)
【分割の表示】特願2005−511389(P2005−511389)の分割
【原出願日】平成16年6月30日(2004.6.30)
【出願人】(302060306)大塚化学株式会社 (88)
【出願人】(000162847)ステラケミファ株式会社 (81)
【Fターム(参考)】
[ Back to top ]