
第IX因子/第IXa因子の抗体および抗体誘導体
【課題】活性化された第IX因子(FIXa)の凝血促進活性を増大する、第IX因子/活性化された第IX因子に対する抗体または抗体誘導体を提供すること。
【解決手段】本願発明に従って、活性化された第IX因子(FIXa)の凝血促進活性を増大する、第IX因子/活性化された第IX因子に対する抗体または抗体誘導体が提供される。本発明の第IX因子/第IXa因子−活性化抗体または抗体誘導体の作用は、インヒビター(例えば、第VIII因子/第VIIIa因子に対するインヒビター)の存在によっては反対方向に影響されないが、代わりに、この場合は第IXa因子の凝血促進活性がまた増加される。本発明に従う調製物の投与は、FVIIIを抑制する患者の場合でさえも、第VIII因子または第VIIIa因子の非存在においてでも迅速な血液凝固を可能とする。
【解決手段】本願発明に従って、活性化された第IX因子(FIXa)の凝血促進活性を増大する、第IX因子/活性化された第IX因子に対する抗体または抗体誘導体が提供される。本発明の第IX因子/第IXa因子−活性化抗体または抗体誘導体の作用は、インヒビター(例えば、第VIII因子/第VIIIa因子に対するインヒビター)の存在によっては反対方向に影響されないが、代わりに、この場合は第IXa因子の凝血促進活性がまた増加される。本発明に従う調製物の投与は、FVIIIを抑制する患者の場合でさえも、第VIII因子または第VIIIa因子の非存在においてでも迅速な血液凝固を可能とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、第IX因子/第IXa因子−抗体および抗体誘導体に関する。
【背景技術】
【0002】
血餅(血栓)は、凝固カスケードとして呼ばれる一連のチモーゲン活性化によって形成される。この酵素的カスケードの過程において、このようなチモーゲン(因子といわれる)の各々の活性化形態は、次のチモーゲンの活性化を触媒する。血栓は、血管壁の表面上の血液成分の蓄積物であり、そして主に、凝集した血小板および不溶性の架橋したフィブリンからなる。フィブリンの形成は、フィブリノーゲンの制限されたタンパク分解によって、トロンビンによってもたらされる。トロンビンは、凝固カスケードの最終生成物である(K.G.Mann,Blood,1990,Vol.76,pp.1−16)。
【0003】
活性化された第IX因子(FIXa)および活性化された第VIII因子(FVIIIa)の複合体による第X因子の活性化は、凝固における重要な工程である。この複合体の成分の非存在またはその機能の妨害は、血友病と呼ばれる血液凝固障害に関連する(J.E.Sadler&E.W.Davie:Hemophilia A,Hemophilia B and von Willebrand’s disease,G.Stamatoyannopoulosら(編):The molecular basis of blood diseases.W.B.Saunders Co.,Philadelphia,1987,pp.576−602)。血友病Aは、第VIII因子活性の(機能的)欠如を示すが、血友病Bは、第IX因子活性の欠如によって特徴付けられる。現在は、血友病Aの処置は、第VIII因子濃縮物の投与による補充療法を介してもたらされる。しかし、約20〜30%の血友病Aの患者は、第VIII因子インヒビター(すなわち、第VIII因子に対する抗体)を発生させ、それによって投与された第VIII因子調製物の効果が阻害される。第VIII因子を抑制する患者の処置は、非常に困難かつ危険性を含み、そして従来はこれらの患者を処置するために限定された数の処置しか存在しなかった。
【0004】
低いFVIIIインヒビターレベルを有する患者の場合において、高用量の第VIII因子をこのような患者に投与して、従って第VIII因子に対する抗体を中和することは、高価であるが可能である。次いで、インヒビター抗体を中和するために必要な量を超える量の第VIII因子は、うっ血作用を有する。多くの場合において、脱感作がもたらされ得、次いでその上に、標準的な第VIII因子処置を再び適用し得る。しかし、大量の第VIII因子を必要とするこのような高用量第VIII因子処置は多大な時間を必要とし、そして重篤なアナフィラキシーの副反応を伴い得る。あるいは、この処置は、ブタの第VIII因子分子を用いて行われ得る。
【0005】
さらなる高コストの方法は、免疫グロブリン(プロテインA、プロテインG)または固定化された第VIII因子に結合するレクチン上の、特別な体の免疫吸着(extra corporeal immunoadsorption)を介して第VIII因子インヒビターを除去する工程を包含する。この処置の間、患者はアフェレーシス器械に連結されなければならないので、この処置はまた、患者への大きな負担となる。この方法においてはまた、急性の出血を処置することはできない。
【0006】
現在、最上の治療は、活性化されたプロトロンビン複合体濃縮物(APCC)(例えば、FEIBA(登録商標)およびAUTOPLEX(登録商標))を投与することであり、これは、高いインヒビター力価を有する患者においてでさえも、急性出血の処置に適切である(DE 31 27 318)。
【0007】
血液凝固の脈管内系において、最後の段階は第X因子の活性化である。この反応は、第VIIIa因子の第IXa因子への結合、ならびに第IXa因子、第VIIIa因子、第X因子およびリン脂質からなる「テナーゼ(tenase)」複合体の形成によって刺激される。FVIIIaの結合なしでは、FIXaは酵素活性を示さないか、またはFXと比較してほんのわずかの酵素活性しか示さない。
【0008】
最近の数年間にわたって、第IXa因子に対する第VIIIa因子の可能な多くの結合部位が特徴付けられ、これらの領域に結合する抗体またはペプチドが、FIXaの活性を阻害することが示されている(Fayら、J.Biol.Chem.,1994,Vol.269,pp.20522−20527,Lentingら、J.Biol.Chem.,1996,Vol.271,pp.1935−1940,Jorqueraら、Circulation,1992,Vol.86,抄録2725)。第IX因子のような凝固因子の阻害はまた、血栓症の形成の予防の目的で、モノクローナル抗体の使用を介して達成される(WO
97/26010)。
【0009】
逆の効果(すなわち、第X因子の第IXa因子媒介活性化における増大)は、第IX因子への第VIII因子ペプチド(アミノ酸698〜712)の結合を介して、Liles D.K.ら、(Blood,1997,Vol.90,補遺1,2054)によって記載された。けれども、この効果は、第VIIIa因子の非存在においてのみ生じ、一方、第VIIIaの存在下では、第X因子の第IXa因子/第VIIIa因子媒介切断は、このペプチドによって阻害される。
【発明の開示】
【発明が解決しようとする課題】
【0010】
(発明の要旨)
血友病患者の処置において生じ得る可能な危険性および副作用の観点から、FVIIIを抑制する患者の有効な処置を可能にする治療の必要性が存在する。そのため、第VIII因子を抑制する患者についての特定の利点を有する、血液凝固障害の処置のための調製物を提供することが本発明の目的である。
【課題を解決するための手段】
【0011】
従って、本発明によって、以下が提供される:
(項目1) 第IX因子/第IXa因子に対する抗体または抗体誘導体であって、FIXaの凝血促進活性を増大させる、抗体または抗体誘導体。
(項目2) 前記抗体または抗体誘導体が、FVIIIインヒビターの存在下でFIXaの凝血促進活性を増大させる、項目1に記載の抗体または抗体誘導体。
(項目3) 前記抗体が、IgG抗体、IgM抗体、IgA抗体およびIgE抗体からなる群から選択される、項目1に記載の抗体。
(項目4) 項目1に記載の抗体または抗体誘導体であって、ここで、該抗体または抗体誘導体は、モノクローナル抗体、抗体フラグメント、キメラ抗体、ヒト化抗体、単鎖抗体、二重特異性抗体、ダイアボディー、およびそれらのダイマー、オリゴマー、またはマルチマーからなる群から選択される、抗体または抗体誘導体。
(項目5) 前記抗体誘導体が、相補的決定領域(CDR)ペプチドを含む、項目1に記載の抗体誘導体。
(項目6) 前記CDRペプチドがCDR3ペプチドである、項目5に記載の抗体誘導体。
(項目7) 項目6に記載の抗体誘導体であって、ここで、前記CDR3ペプチドが、以下:
Tyr−Gly−Asn−Ser−Pro−Lys−Gly−Phe−Ala−Tyr;
Cys−X−X−Tyr−Gly−Asn−Ser−Pro−Lys−Gly−Phe−Ala−Tyr−X−X−Cys;
Tyr−Gly−Asn−Ser−Pro−Lys−Gly−Phe−Ala−Tyr;
Asp−Gly−Gly−His−Gly−Tyr−Gly−Ser−Ser−Phe−Asp−Tyr;および
Phe−Arg−Asn−Arg−Gly−Met−Thr−Ala−Leu−Leu−Lys−Val−Ser−Ser−Cys−Asp、
からなる群から選択されるアミノ酸配列を含み、ここで、
Xは、任意の所望のアミノ酸であり得る、抗体誘導体。
(項目8) 項目1に記載の抗体または抗体誘導体であって、ここで、該抗体または抗体誘導体の可変領域が、図14に記載のアミノ酸1〜357および/またはアミノ酸403〜726を含む、抗体または抗体誘導体。
(項目9) 前記抗体または抗体誘導体が、人工的なリンカー配列をさらに含む、項目8に記載の抗体または抗体誘導体。
(項目10) 項目1に記載の抗体または抗体誘導体であって、ここで、該抗体または抗体誘導体の可変領域が、図15に記載のアミノ酸1〜363および/またはアミノ酸409〜747を含む、抗体または抗体誘導体。
(項目11) 前記抗体または抗体誘導体が、人工的なリンカー配列をさらに含む、項目10に記載の抗体または抗体誘導体。
(項目12) 項目1に記載の抗体または抗体誘導体であって、ここで、該抗体または抗体誘導体の可変領域が、図16に記載のアミノ酸1〜366および/またはアミノ酸412〜747を含む、抗体または抗体誘導体。
(項目13) 前記抗体または抗体誘導体が、人工的なリンカー配列をさらに含む、項目12に記載の抗体または抗体誘導体。
(項目14) 項目1に記載の、第IX因子/第IXa因子に対する抗体または抗体誘導体を発現する、ハイブリドーマ細胞株。
(項目15) 項目14に記載のハイブリドーマ細胞株であって、ここで、該細胞株が、#196/AF1、#196/AF2、#193/AD3、#193/K2−1、#198/AC1/1、#198/AM1、#198/A1、#198/B1、#198/AP1、198/A1、198/B1、198/BB1、198/A1、198/B1、198/BB1からなる群から選択される、ハイブリドーマ細胞株。
(項目16) 項目14に記載のハイブリドーマ細胞株によって発現される、項目1に記載の抗体または抗体誘導体。
(項目17) DNA分子であって、該DNA分子が、項目1に記載の抗体または抗体誘導体をコードする、DNA分子。
(項目18) 項目1に記載の抗体または抗体誘導体および薬学的に受容可能なキャリアを含有する、薬学的調製物。
(項目19) 第IXaα因子および/または第IXaβ因子をさらに含む、項目18に記載の調製物。
(項目20) 血液凝固障害に罹患した患者を処置するための方法であって、薬学的有効量の項目18に記載の調製物を該患者に投与する工程を包含する、方法。
(項目21) 項目20に記載の方法であって、ここで、前記血液凝固障害が、血友病Aおよび出血性素質を含む群から選択される、方法。
(項目22) 血友病を抑制する患者を選択する工程をさらに包含する、項目21に記載の方法。
(項目23) 第IX因子/第IXa因子と相互作用し、そして第IXa因子の凝血促進活性を増大させる抗体または抗体誘導体を得る方法であって、以下の工程:
− FIX、FIXaα、FIXaβまたはそれらのフラグメントからなる群から選択される抗原で、免疫応答性マウスを免疫する工程、
− 免疫されたマウスの脾細胞を単離する工程、
− ハイブリドーマクローンを産生する工程、
− 第IXa因子の凝血促進活性における増大について、ハイブリドーマ細胞の上清をスクリーニングし、第IXa因子の凝固促進活性における増大を示すハイブリドーマ細胞の上清から、該抗体または抗体誘導体を単離および精製する工程、
を包含する、方法。
(項目24) 第IXa因子のアミド分解活性を増大させるための、項目1に記載の抗体または抗体誘導体の使用。
【0012】
本発明に従って、この目的は、第VIIIa因子補因子活性または第IXa因子活性化活性を有し、そして第IXa因子の凝血促進活性における増加を導く、第IX因子/第IXa因子に対する抗体または抗体誘導体の使用を通して達成される。驚いたことに、本発明の第IX因子/第IXa因子−活性化抗体または抗体誘導体の作用は、インヒビター(例えば、第VIII因子/第VIIIa因子に対するインヒビター)の存在によっては反対方向に影響されないが、代わりに、この場合は第IXa因子の凝血促進活性がまた増加される。
【0013】
本発明のさらなる利点は、本発明に従う調製物の投与が、FVIIIを抑制する患者の場合でさえも、第VIII因子または第VIIIa因子の非存在においてでも迅速な血液凝固を可能とすることである。驚いたことに、これらの因子はまた、第VIIIa因子の存在下においても有効である。
【0014】
従って、本発明に従う抗体および抗体誘導体は、FVIII補因子様の活性を有し、これは、2時間のインキュベーション後のFVIIIアッセイ(例えば、COATEST(登録商標)アッセイまたはイムノクロム(Immunochrom)試験)において、少なくとも3のバックグラウンド(基本的ノイズ)対測定値の比を示す。この比の計算は、例えば、2時間のインキュベーションの後に、以下のスキームに従って達成され得る:
【0015】
【数1】

本発明に従う抗体は、好ましくは少なくとも5日、より好ましくは少なくとも10日のインビボ半減期を有するが、少なくとも20日の半減期を有するのがより好ましい。
【0016】
本発明のさらなる局面は、第IX因子/第IXa因子に対する抗体および/または抗体誘導体、ならびに薬学的に受容可能なキャリア物質を含む調製物である。さらに、本発明に従う調製物は、第IX因子および/または第IXa因子をさらに含み得る。
【0017】
本発明のさらなる局面は、第IXa因子のアミド分解(amidolytic)活性を増加するための、この抗体または抗体誘導体の使用である。
【0018】
(抗体および抗体誘導物)
本発明はまた、本発明の抗体および抗体誘導物、発現ベクター、ハイブリドーマ細胞株をコードする核酸、ならびにこれらを産生するための方法を包含する。
【0019】
抗体は、免疫グロブリン分子の合成(または、それぞれ、その免疫原)を誘発する抗原にのみ結合する、あるいはその抗原に大変類似する抗原(または免疫原)にのみ結合する、特異的なアミノ酸配列を有する免疫グロブリン分子である。各免疫グロブリン分子は、2つの型のポリペプチド鎖からなる。各分子は、大きな、同一の重鎖(H鎖)、および2つの軽い、同一でもある鎖(L鎖)からなる。ポリペプチドは、ジスルフィド架橋および非共有結合によって結合する。インビボでは、重鎖および軽鎖は、異なるリボソーム上で形成され、細胞内で組み立てられ、そしてインタクトな免疫グロブリンとして分泌される(Immunology、第2版、1989内のRoitt I.ら)。
【0020】
本発明の抗体、および抗体誘導物、ならびにこれらから誘導された有機化合物は、ヒトおよび動物のモノクローナル抗体またはそれらのフラグメント、一本鎖抗体およびそれらのフラグメントならびにミニ抗体、二重特異的(bispecific)抗体、二重抗体(diabody)、三重抗体(triabody)、またはそれらのダイマー、オリゴマーもしくはマルチマー(multimer)を含む。本発明に従う抗体から誘導されたペプチドミメティックス(peptidomimetics)またはペプチド(例えば、これらは、1つまたはいくつかのCDR領域、好ましくはCDR3領域を含む)もまた含まれる。
【0021】
さらに、構造的活性関連に基づいて、人工的モデリングプロセスによって産生されるヒトモノクローナル抗体およびペプチド配列も含まれる(Greer J.ら、J.Med.Chem.、1994、Vol.37、pp.1035−1054)。
【0022】
用語第IX/IXa因子活性化抗体および抗体誘導物はまた、宿主細胞内における、変更された、免疫グロブリンコード領域の発現によって産生されるタンパク質(例えば、合成抗体、キメラ抗体またはヒト化抗体のような「技術的改変抗体」、またはそれらの混合物、あるいは例えば、Fv、Fab、Fab’またはF(ab)’2などの定常領域を部分的もしくは完全に欠損する抗体フラグメントを含み得る。これらの技術的改変抗体において、例えば、軽鎖および/または重鎖の一部分またはいくらかの部分は、置換され得る。このような分子は、例えば、ヒト化重鎖および未改変軽鎖(またはキメラ軽鎖)からなる抗体を含み得、逆も同様である。用語Fv、Fc、Fd、Fab、Fab’またはF(ab)’2は、先行技術(Harlow E.およびLane D.、「抗体、実験室マニュアル(Antibodies、A Laboratory Manual)」、Cold Spring Harbor Laboratory、1988)に記載されるように用いられる。
【0023】
本発明はまた、モノクローナル抗体(mAb)から誘導されるFabフラグメントまたはF(ab)2フラグメントの使用を含み、これらのフラグメントは、第IX因子/第IXa因子に対して指向され、そして第IXa因子のプロコアギュラント(procoagulant)活性の増加を引き起こす。好ましくは、異種のフレームワーク領域および定常領域は、ヒト免疫グロブリンクラスおよびアイソタイプ(例えば、IgG(サブタイプ1〜4)、IgM、IgAおよびIgE)から選択される。免疫応答の過程において、免疫グロブリンのクラス変更(例えば、IgMからIgGへの変更;ここでは、定常領域が(例えば、μからγへ)変換される)が起こり得る。先行技術(Esser C.およびRadbruch A.、Annu.Rev.Immunol.、1990、Vol.8、pp.717−735)から公知のように、クラス変更はまた、指向された様式で、遺伝子操作方法(「指向されたクラス変更組換え」)によって引き起こされ得る。しかし、本発明に従う抗体および抗体誘導物は、免疫グロブリンタンパク質のヒト配列を特別に含む必要はない。
【0024】
ある特定の実施形態において、ヒト化抗体は、マウスモノクローナル抗体からの補体決定領域(CDR)を含み、この領域は、選択されたヒト抗体配列のフレームワーク領域に挿入される。しかし、ヒトCDR領域もまた使用され得る。好ましくは、ヒト軽鎖および重鎖における可変領域は、1以上のCDR変換によって技術的に変えられる。6つの全てのCDR、または6未満のCDRの種々の組み合わせを使用することもまた可能である。
【0025】
本発明に従うヒト化抗体は、好ましくは、ヒト抗体の構造、またはそのフラグメントの構造を有し、そして治療的適用(例えば、患者(好ましくは、第VIII因子を抑制する患者)における凝固障害の処置)のために必要な特徴の組み合わせを含む。
【0026】
キメラ抗体は、ヒト免疫グロブリンからの重鎖および軽鎖の両方の鎖の定常領域と組み合わせて、非ヒト起原の重鎖および軽鎖のフレームワーク領域を含む、可変領域全体を含むという点で、ヒト化抗体と異なる。例えば、マウス配列およびヒト配列からなるキメラ抗体が産生され得る。本発明に従うと、抗体および抗体誘導物はまた、一本鎖抗体、またはミニ抗体(例えば、プロリンリッチ配列およびオリゴマー化(oligomerisation)ドメインに結合するscFvフラグメント(例えば、Pluckthun A.およびPack P.、Immunotechnology、1997、Vol.3、pp.83−105)、もしくは一本鎖Fv(sFv)(これは、一本のポリペプチド鎖内の抗体結合領域全体を組み込む)であり得る。例えば、一本鎖抗体は、オリゴヌクレオチドにV遺伝子を結合することによって形成され得、このオリゴヌクレオチドは、リンカー配列として構築されており、そして第1のV領域のC末端を第2のV領域のN末端と、例えば、VH−リンカー−VLまたはVL−リンカー−VHの配置で結合し;従って、両方とも(VHおよびVL)は、N末端ドメインを表し得る(Huston JSら、Int.Rev.Immunol.、1993、Vol.10、pp.195−217;Raag R.およびWhitlow M.、FASEB J.、1995、Vol.9、pp.73−80)。リンカー配列として使用され得るタンパク質は、例えば、150Åまで、好ましくは80Åまで、そしてより好ましくは40Åまでの長さを有する。グリシンおよびセリンを含むリンカー配列は、これらの可撓性のために特に好ましく、または、グルタミンおよびリジンを含むリンカー配列は、それぞれ、これらの可溶性のために好ましい。アミノ酸の選択は、免疫原性および安定性の基準に従ってなされ、これらの一本鎖抗体が、生理学的適用または工業的適用(例えば、免疫アフィニティクロマトグラフィー)に適し得るか否かにも依存する。この一本鎖抗体はまた、凝集体として(例えば、トリマー、オリゴマー、またはマルチマーとして)存在し得る。しかし、リンカー配列はまた失われ、そしてVH鎖およびVL鎖の結合が直接起こり得る。
【0027】
二重特異性抗体は、1つの単一分子内に2つの異なった結合特異性を有する、高分子のヘテロ二機能性架橋である。この群には、例えば、二重特異性(bs)IgG、bs IgM−IgA、bs IgA−二量体、bs(Fab’)2、bs(scFv)2、ダイアボディー(diabodies)、およびbs bis Fab Fcが属する(Cao Y.およびSuresh M.R.,Bioconjugate Chem.,1998,第9巻、635−644頁)。
【0028】
ペプチド模倣物によって、天然ペプチド成分の構造を模倣するかまたはその鋳型が隣接したペプチド配列における特異的構造形成を誘導する低分子のタンパク質成分が理解される(Kemp DS,Trends Biotechnol.,1990,249−255頁)。ペプチド模倣物は、例えば、CDR3ドメインに由来し得る。所定のペプチド配列の系統的な変異分析、すなわち、アラニンまたはグルタミン酸走査変異分析によって、凝血促進性活性に重要なペプチド残基の同定を可能にする。特定のペプチド配列の活性を改善するための別の可能性は、高い処理能力のスクリーニングと組み合わせたペプチドライブラリーの使用である。
【0029】
用語抗体および抗体誘導体はまた、構造−活性関係に関連するデータの分析によって獲得される因子を含み得る。これらの化合物はまた、ペプチド模倣物として使用され得る(Grassy G.ら、Nature Biotechnol.1998,第16巻、748−752頁;Greer J.ら、J.Med.Chem.,1994,第37巻、1035−1054頁)。
【0030】
本発明に従って、抗体または抗体誘導体を発現するハイブリドーマ細胞の例としては、ブタペスト条約に従って、番号99090924(#198/A1)、99090925(#198/B1)および99090926(#198/BB1)下で1999年9月9日に、そして、番号99121614(#193/A0)、99121615(#196/C4)、99121616(#198/D1)、99121617(198/T2)、99121618(#198/G2)、99121619(#198/AC1)および99121620(#198/U2)下で1999年12月16日に寄託された。
【0031】
(産生の方法)
本発明の抗体は、先行技術から公知の方法によって調製され得る(例えば、慣例的なハイブリドーマ技術によってかまたはファージディスプレイ遺伝子ライブラリー、免疫グロブリン鎖混合の方法もしくはヒト化技術によって)(Harlow E.およびLane D.,Antibodies,A Laboratory Manual,Cold Spring Harbor Laboratory,1988)。本発明の抗体および抗体誘導体の産生は、例えば、慣例的なハイブリドーマ技術によって行なわれる(Antibodies,A Laboratory Manual,Cold Spring Harbor Laboratory,1988,HarlowおよびLane編、148−242頁)。本発明に従って、ヒトおよび非ヒト種(例えば、ウシ、ブタ、サル、ニワトリおよびげっ歯類(マウス、ラット))はまた、ハイブリドーマ技術のために使用され得る。通常、免疫競合Balb/cマウスまたはFIX欠損マウスは、使用され得る(第IX因子欠損マウスは、ノースキャロライナ大学、Chapel HillのDarrel Stafford博士より分譲され得る)。免疫化は、例えば、第IX因子、第IXaα因子または完全に活性化された第IXaβ因子、またはそのフラグメントで影響され得る。
【0032】
ハイブリドーマ細胞の上清における抗体および抗体誘導体が第IX因子/第IXa因子に結合し、そして、第IXa因子の凝血促進性活性の増加の原因となるという事実を望んで、ハイブリドーマは選択される。凝血促進性活性の増加は、例えば、第VIII因子様活性の測定のための先行技術から公知のアッセイ方法(例えば、色素形成(chromogenic)アッセイ)によって証明され得る。
【0033】
あるいは、本発明の抗体および抗体誘導体はまた、組換え産生方法によって産生され得る。そうする際、本発明に従う抗体のDNA配列は、公知の技術によって決定され得、そして、抗体DNA全体またはその一部は、適切な系で発現され得る。ファージディスプレイ、合成および天然ライブラリー、公知の発現系における抗体タンパク質の発現、またはトランスジェニック動物における発現を含むような組換え産生方法は、使用され得る(Jonesら、Nature,1986、第321巻、522−525頁;Phage Display of Peptides and Proteins,A Laboratory Manual,1996,Kayら編、127−139頁;米国特許第4,873,316号;Vaughan T.J.ら、Nature Biotechnology,1998,535−539頁;Persic Lら、Gene,1997,9−18頁;Ames R.S.ら、J.Immunol.Methods,1995,177−186頁)。
【0034】
組換え的に産生された抗体の発現は、慣例的な発現ベクターの方法(例えば、細菌ベクター(例えば、pBr322およびその誘導体)、pSKFまたは真核生物ベクター(例えば、pMSGおよびSV40ベクター))によって影響され得る。抗体をコードするこれらの配列は、複製、発現および宿主細胞からの分泌を調節する制御配列を提供され得る。これらの制御配列は、プロモーター(例えば、CMVまたはSV40)およびシグナル配列を含む。
【0035】
発現ベクターはまた、ジヒドロ葉酸レダクターゼ遺伝子(DHFR)、ハイグロマイシン−B−ホスホトランスフェラーゼ、チミジンキナーゼなどの選択および増幅マーカーを含み得る。
【0036】
使用されるベクターの成分(例えば、選択マーカー、レプリコン、エンハンサーなど)は、慣例的な方法の手段によって商業的に獲得され得るかまたは調製され得る。このベクターは、例えば、哺乳動物細胞(CHO、COS、線維芽細胞など)、昆虫細胞、酵母または細菌(E.coliなど)についての種々の細胞培養物における発現のために構築され得る。好ましくは、発現タンパク質の最適なグリコシル化を可能にするこれらの細胞が使用される。特に好ましいのは、CHO細胞またはSK−Hepにおいて発現されるベクターpBax(参考、図17)である。
FabフラグメントまたはF(ab)2フラグメントの産生は、先行技術から公知の方法に従って影響され得る(例えば、パパインおよび/またはペプシンなどのタンパク分解性酵素を使用して、mAbを切断することによってかまたは組換え法によって)。これらのFabおよびF(ab)2フラグメントはまた、ファージディスプレイ遺伝子ライブラリーの方法によって調製され得る(Winterら、1994,Ann.Rev.Immunol.,12:433−455)。
【0037】
抗体誘導体はまた、先行技術から公知の方法の手段によって調製され得る(例えば、Grassy G.ら、Nature Biotechnol.,1998,第16巻、748−752、またはGreer J.ら、J.Med.Chem.,第37巻、1035−1054頁、またはRees A.ら、「Pretein Structure Prediction;A practical approach」Sternberg M.J.E.編、IRL出版、1996、第7〜10章、141−261頁からの分子モデリングによって)。
【0038】
本発明の抗体および抗体誘導体の精製はまた、先行技術で記載された方法によって行なわれ得る(例えば、硫酸アンモニウム沈殿、アフィニティー精製(プロテインGセファロース)、イオン交換クロマトグラフィー、またはゲルクロマトグラフィーによって)。本発明の抗体および抗体誘導体が、第IX因子/第IXa因子に結合し、第IXa因子の凝血促進性活性を増加するかまたは第VIII因子様活性を有することを示すための試験方法として、以下の方法は使用され得る:一工程凝血試験(MikaelassonおよびOswaldson,Scand.J.Haematol.,Suppl.,33,79−86頁、1984)または色素形成試験(COATEST VIII:C(登録商標)(Chromogenix)またはImmunochrom(IMMUNO)など)。原則的には、第VIII因子活性を決定するために使用される全ての方法が使用され得る。測定のコントロールブランク値として、例えば、非特定的マウスIgG抗体が使用され得る。
【0039】
本発明の抗体および抗体誘導体は、例えば、血友病Aの場合において、第VIII因子を抑制する患者などのための凝血障害の処置における治療的使用に適している。投与は、患者に治療学的因子を効果的に投与するのに適切な任意の方法によって、影響され得る(例えば、経口投与、皮下投与、筋内投与、静脈内投与または鼻腔内投与によって)。
【0040】
本発明に従う治療学的因子は、薬学的に受容可能なキャリア物質中に活性な因子として十分な量の抗体または抗体誘導体を含む調製物として産生され得る。これらの因子は、液体または粉末形態のいずれかで存在し得る。さらに、本発明に従う調製物はまた、異なった抗体、その誘導体および/またはそれらから誘導された有機化合物の混合物、ならびに、抗体および第IX因子および/または第IXa因子からなる混合物を含み得る。第IXa因子は、第IXaα因子および/または第IXaβ因子として存在し得る。水性キャリア物質の例は、例えば、生理食塩水である。溶液は滅菌性で、滅菌は慣例的な方法によって影響される。
【0041】
本発明に従う抗体または抗体誘導体は、貯蔵のために凍結乾燥形態で存在し得、そして、投与の前に適切な溶媒で懸濁され得る。この方法は、一般的に慣例的な免疫グロブリンについて有利であることが証明され、そして、公知の凍結乾燥および再構成方法は、この場合で適用され得る。
【0042】
さらに、本発明に従う抗体および抗体誘導体はまた、工業的適用に使用され得る(例えば、アフィニティークロマトグラフィーの方法による第IX因子/第IXa因子の精製のためか、または検出方法(例えば、ELISAアッセイ)の成分としてか、または標的タンパク質の機能的ドメインの同定および相互作用のための因子として)。
【0043】
本発明は、以下の実施例および図面によって、より詳細に記載されるが、本発明はそれに対して制限されるべきではない。
【実施例】
【0044】
(実施例1:免疫競合マウスの免疫化および抗FIX/IXa抗体分泌ハイブリドーマ細胞の産生)
1〜3匹の正常免疫競合5〜8週齢Balb/cマウスを個体内(i.p.)経路を介して100μgの抗原(100μl用量)で免疫化した。代表的な実験では、マウスを組換えヒト凝血因子(F)IX(BenefixTM)、ヒト活性化FIXaα(Enzyme Research Laboratories,Lot:FIXaα 1190L)または水酸化アルミニウムもしくはKFAで補助したヒトFIXaβ(Enzyme Research Laboratories,Lot:HFIXaα 1332 AL)のいずれかで接種した。
【0045】
個々のマウスを100μgの抗原(100μl用量、i.p.)で種々の時間にブーストし、そして、2日後に屠殺した。脾臓細胞を取り出し、そして、Laneら、1985(J.Immunol.Methods,第81巻、223−228頁)に基本的に記載されるように、P3 X63−Ag86.5.3骨髄腫細胞に融合させた。それぞれの融合実験を個々に番号付けした(すなわち、#193、195、196または198)。
【0046】
ハイブリドーマ細胞をマクロファージフィーダー層上の96ウェルプレート上で増殖し(約105細胞/ml)そしてHAT培地(抗生物質、10%FCS、ピルビン酸ナトリウム、L−グルタミン、2−メルカプトエタノールおよびHAT(HAT 100×:H2O中の1.0×10-2Mヒポキサンチン(136.1mg/100ml H2O)、H2O中の4.0×10-5Mアミノプテリン(1.76mg/100ml H2O)およびH2O中の1.6×10-3Mチミジン(38.7mg/100ml H2O))で補充したRPMI−1640培地)中で選択した。培地を6日後に最初に交換し、その後1週間に2回交換した。2〜3週間後、HAT培地をHT培地(抗生物質、10%FCS、ピルビン酸ナトリウム、L−グルタミン、2−メルカプトエタノールおよびHTで補充したRpMI−1640)に変更し、その後(さらに1〜2週間後)通常の増殖培地(10%FCS、ピルビン酸ナトリウム、L−グルタミンおよび2−メルカプトエタノールで補充したRPMI−1640培地)(HYBRIDOMA TECHNIQUES、EMBO,SKMB Course 1980,Baselを参照のこと)に変更した。
【0047】
別のセットの実験において、FIX欠損C57B16マウス(Linら、1997、Blood,90:3962)を免疫化および後に続くハイブリドーマ産生のために使用した。FIXノックアウト(k.o.)マウスは、内因性FIXを発現しないので、達成可能な抗(a)−FIX抗体スペクトルは、正常なBalb/cマウスと比較して異なっていることが推定される(耐性の欠如に起因して)。
【0048】
(実施例2:抗FIX/FIXa抗体分泌ハイブリドーマ細胞の上清におけるFVIII様活性のためのアッセイ)
ハイブリドーマ細胞によって分泌される抗FIXa抗体のFVIII様活性をアッセイするために、市販されている試験キットCOATEST VIII:C/4(登録商標)(Chromogenix)を使用した。アッセイは、以下の改変を使用して基本的に製造業者によって記載されるように行なった:
高い処理能力のスクリーニングを可能にするために、アッセイをマイクロタイタープレート型式に小規模化した。簡潔には、ハイブリドーマ上清の25μlのアリコートを、マイクロタイタープレート(Costar,#3598)ウェルに移し、そして、37℃に温めた。色素形成基質(S−2222)、合成トロンビンインヒビター(I−2581)、因子(F)IXaおよびFXを滅菌水中で再構築し、そして、FIXa/FXを、製造業者のプロトコルに従って、リン脂質で混合した。反応液当り、50μlのリン脂質/FIXa/FX溶液を25μlの塩化カルシウム(25mM)および50μlの基質/インヒビター反応混液と組み合わせた。反応を開始するために、125μlのプレミックスをマイクロタイタープレート中のハイブリドーマ上清に添加し、そして、37℃でインキュベートした。サンプルの405nmおよび490nmの吸光度を、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーで、試薬ブランクに対して(MLW、ハイブリドーマ上清の代わりの細胞培養培地)種々の時間(30分〜12時間)測定した。サンプルのFVIII様活性を、GENESISTMソフトウェアを用いて、希釈したFVIII参照規準(IMMUNO AG#5T4AR00)の吸光度に対してサンプルの吸光度を比較することによって計算した。
ハイブリドーマ細胞培養物上清におけるFVIII様活性のスクリーニングの結果を図1に示す。融合実験#193、#195および#196由来の選択される前のクローン(上記を参照のこと)を記載されるように、色素形成FVIIIアッセイで調査した。クローン193/M1、193/N1および193/P1は、マスタークローン193/C0由来のサブクローンであった(以下を参照のこと)。マスタークローン195/10は融合実験#195に由来し、そして、クローン196/A0、196/B0および196/C0は、融合実験#196由来であった。代表的なスクリーニング実験において、単一の融合実験由来の約1000クローン(96ウェル中)は、FVIII様活性についてスクリーニング前と同じであった。続いて、選択されたクローンを大規模に増殖し(3〜5mlの上清)、そして、色素形成アッセイで再分析した。陰性コントロールとして、細胞培養培地をそれぞれのプレート(MLW)上でアッセイした。
【0049】
高いFVIII様活性または有意なFVIII様活性のどちらかを示すウェルをサブクローニング手順に供した。選択およびサブクローニングプロセスは、細胞株(すなわち、193/C0)を産生するIgGのスクリーニングおよびサブクローニングについて例示されるが、クローンを産生するIgM(すなわち、196/C0、以下を参照のこと、図5)について同一の方法を正確に行なった。
【0050】
選択プロセスを、10の96ウェルプレート上の単一融合実験由来の全てのハイブリドーマ細胞クローンを最初に播種することによって行ない、これによって、いわゆる「マスタープレート」を作製した。マスタープレート上の別々の位置(ウェル)は、通常、1つ以上のハイブリドーマ細胞クローン(通常3〜15の異なったクローン)を含んだ。続いて、数千の細胞のみによって分泌された抗体を試験した。これらの細胞を、抗体産生に最適以下の条件下で増殖し、この条件は、瀕死の細胞において最も公知である。それで、上清における予想された特異的な抗FIX抗体濃度は、10-12〜10-14Mの範囲であり得る。このことは、なぜインキュベーション期間が、標準的FVIIIアッセイと比較して延長されなければならないかを説明する。
【0051】
マスタープレートのハイブリドーマ細胞培養物上清におけるIgG媒介性FVIII様活性についてのスクリーニングの結果を、図2に示す。上清を、色素形成FVIIIアッセイにおいて試験した。融合実験番号#193(FIXaαで免疫したBalb/cマウス)の5番目のマスタープレートから誘導される結果を示す。吸光度を、37℃でのインキュベーションの4時間後に読み取った。ES位を、ブランク(MLW)より有意に高いFVIII様活性を示すと同定した。この細胞プールを、193/C0と名付け、そしてさらにサブクローニングした(図3)。マスタープレートの各ウェルは、1つより多いハイブリドーマ細胞クローンを含むので、単一の陽性ウェルの細胞を、2〜0.2細胞/ウェルの計算された細胞密度で、96ウェルプレートにおいて、伸長およびプレートした。再度、その上清を、FVIII様活性について試験し、そして陽性位置を、サブクローニングのさらなるラウンドに供した。代表的に、均一な細胞集団を得るために、FVIII様活性を示す各クローンを使用して、3〜4ラウンドのサブクローニングを実施した。ここで、193/C0サブクローンの色素形成アッセイの結果を示す。吸光度を、37℃での4時間のインキュベーション期間の後に、読み取った。A6位およびD5位は、かなりのFVIII様活性を示し、そしてそれぞれ、193/M1および193/P1と命名した。これら2つのクローンを、さらなるラウンドのサブクローニングに供した。ネガティブコントロールとして、単純細胞培養培地を、各プレートにおいてアッセイした(MLW(H1))。
【0052】
小スケール(3ml)ハイブリドーマ培養物における色素形成FVIII様活性とFIX−ELISA反応性との比較を、図4に示す。マスタークローン(またはサブクローン)がさらにサブクローニングされるべきか否かを決定する前に、クローンを3〜5mlのスケールで増殖し、そしてその上清を、再度チェックした。このグラフは、マスタークローン193/C0、および陽性と同定され、そして再チェックされた全てのそのサブクローンの、FIX特異的ELISAの結果およびFVIII様色素形成活性を示す。本明細書中に記載されるELISAおよび色素形成アッセイの読み取りの場合には、ブランク(色素形成試薬自体の吸光度)を減算した。クローン193/M1をサブクローニングし、そしてクローン193/V2、193/M2および193/U2を得た。193/P1、193/AB2および193/P2由来の第2ラウンドの他のクローンを、サブクローニングした。193/AF3、193/AB3および193/AE3は、193/AB2のサブクローンである。第3ラウンドの他のクローンが、193/P2から生じた。最後に、193/AF3(→193/AF4)、AE3(→193/AE4、193/AL4、193/AN4および193/AO4)ならびに193/AD3(→193/AG4、193/AH4、193/AD4、193/AI4、193/AK4)を、サブクローニングした。
【0053】
各融合実験から、いくつかの(5〜15)マスタークローン(マスタープレートから選択される)を同定し、そしてサブクローニングに供した。3ラウンドのサブクローニングの後に、大部分の細胞株は、ELISAおよび色素形成活性分析(図4を参照のこと)ならびにcDNA配列分析により示されるように、均一であった。特定のマスタークローンおよびその全てのサブクローンは、同じFIX/FIXa結合抗体を産生する。しかし、異なるマスタークローン由来のクローンの抗体タンパク質配列には、大きな差異が存在する(実施例11を参照のこと)。大部分のハイブリドーマ細胞株は、IgGサブクラスから抗体を発現する(すなわち、クローン#193、#198から、198/A1、198/B1、198/BB1のような)。しかし、本発明者らはまた、IgM抗体を発現するいくつかのクローンを、選択し得た。
【0054】
いくつかの重要なマスタークローンおよびサブクローンのハイブリドーマ上清の色素形成活性を、決定した。吸光度を、37℃での1時間30分および3時間30分のインキュベーション期間の後に、測定した(図5)。193番目の融合物由来の全てのクローンとは対照的に、クローン196/C0およびそのサブクローン196/AP2は、FIX/FIXa特異的IgM抗体を産生した。この抗体は、短時間のインキュベーションの後でさえも、強力な色素形成活性を示した。
【0055】
以下の細胞株を、ブダペスト条約に従って、European Collection of Cell Cultures(ECACC)に寄託した:98/B1(ECACC No.99090925);198/A1(ECACC No.99090924);198/BB1(ECACC No.99090926);193/AO(ECACC No.99121614);196/C4(ECACC No.99121615);198/D1(ECACC No.99121616);198/T2(ECACC No.99121617);198/G2(ECACC No.99121618);198/AC1(ECACC No.99121619);および198/U2(ECACC No.99121620)。
【0056】
特定の抗体の生物学的特性の、より深い分析を行うために、FVIII様活性を有する異なる抗体を発現する均一なハイブリドーマ細胞株を伸長し、そして目的の抗体をより大きなスケール(100〜1000ml)で発現するために使用した。これらの抗体をアフィニティー精製し(実施例3を参照のこと)、その後、さらなる実験に使用した。
【0057】
(実施例3:FIX/FIXa活性化活性を示す抗体の第IX因子/FIXa(α,β)結合特性)
第IX因子ならびにFIXの2つの活性化形態(FIXaαおよびFIXaβ)(FIX/FIXa(α,β))を、2g/mlの最終濃度まで、TBS(25mM Tris HCl、150mM NaCl、pH7.5)に希釈した。Nunc Maxisorp ELISAプレートを、標準的な手順(4℃、一晩)に従って、100μlのFIX/FIXa(α,β)溶液でコートし、そしてTBST(TBS、0.1%(v/v)Tween 20)で数回洗浄した。50μlのハイブリドーマ上清を、50μl TBST/2% BSAで1:1に希釈し、コートしたELISAプレートに添加した。室温(RT)で2時間のインキュベーション期間の後に、プレートをTBSTで4回洗浄し、そして100μl/ウェルの抗マウスIgG(Fc特異的)ペルオキシダーゼ結合体化抗体(Sigma,#A−0168)の1:25000希釈物(TBST/1% BSA中)とともにインキュベートした(2時間、RT)。ウェルを、TBSTで5回洗浄し、そして最後に、100μlの新たに調製した染色溶液(100μl OPD(60mg OPD/ml)および10μl 30% H2O2を補充した10mlの50mMクエン酸ナトリウム(pH5))で染色した。この反応を、50mlのH2SO4の添加により停止させ、そしてその光学密度を、492nmおよび620nmにおいて、GENESISTMソフトウェアを使用して、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーで記録した。
【0058】
特定の場合においては、抗マウスIgG ELISAの代わりに、抗マウスIgM ELISAを実施した。
【0059】
(ハイブリドーマ細胞培養物上清からのマウスIgGの精製)
ハイブリドーマ上清(100〜500ml)に、200mM Tris/HCl緩衝液(pH7.0)および固体NaClを補充して、それぞれ20mM Trisおよび3M NaClの最終濃度を得た。次いで、この上清を、5500×gにおける10分間の遠心分離によって、清澄化した。1mlのプロテインGアフィニティークロマトグラフィーカラム(Protein G Sepharose Fast Flow,Amersham−Pharmacia)を、15mlの20mM Tris/Cl(pH7.0)で洗浄し、そしてその後、3M NaClを含む10mlの20mM Tris/Cl緩衝液(pH7.0)で平衡化した。次いで、この3M NaClを含むハイブリドーマ上清を、重力によってこのカラムに装填した。このカラムを、3M NaClを含む15mlの20mM Tris/Cl緩衝液(pH7.0)で洗浄した。結合したIgGを、12mlのグリシン/HCl緩衝液(pH2.8)でさらに溶出し、そして1mlの画分を収集した。中和のために、100μlの1M Tris(pH9.0)を、各画分に添加した。IgGを含む画分を、マイクロプレートのウェル内で、50μlを150μlの染色溶液(BioRad濃縮物、水で1:5に希釈)と混合することによって、同定した。陽性の画分を「プール」し、限外濾過濃縮器デバイス(Centricon Plus 20,Amicon)によって、製造業者に従って、1mlに濃縮した。この濃縮物を、19ml TBS(150mM NaClを含む20mM Tris/Cl緩衝液(pH7.0))で希釈し、そして再度、1mlに濃縮した。IgGをTBSにするために、この希釈−濃縮工程を、さらに2回繰り返した。
【0060】
(ハイブリドーマ細胞上清からのマウスIgMの精製)
100〜500mlのハイブリドーマ細胞培養物上清を、限外濾過濃縮器デバイス(Centricon Plus 20,Amicon)を用いて製造業者に従ってか、または硫酸アンモニウム沈殿(40%飽和、0℃)およびこの沈殿物の5〜10mlのTBSへの再溶解によってかのいずれかで、5〜10mlに濃縮した。いずれの場合においても、この濃縮物を、1.25M NaClを含む20mM Tris Cl緩衝液(pH7.4)に対して透析し、そしてさらに、Centricon Plus 20(Amicon)限外濾過デバイスにおいて1mlに濃縮した。IgMを、ImmunoPure IgM Purification Kit(Pierce)を用いて、製造業者に従って、この濃縮物から精製した。マルトース結合タンパク質カラムからの溶出の間に収集された画分を、IgMに関して試験し、プールし、濃縮し、そしてIgGに関して記載したように、TBSにした。
【0061】
(精製した調製物におけるIgG濃度の決定)
適切な希釈物の、全IgG内容物の280nm消光を、測定した。E280=1.4は、1mg/mlのタンパク質に対応する。
【0062】
(第IXa因子特異的IgG(定量的ELISA))
マイクロプレート(Nunc Maxisorp)のウェルを、TBS(150mM NaClを含む25mM Tris/HCl(pH7.5))に希釈された2μg/mlの第IXa因子とともに、4℃で一晩インキュベートした。ウェルを、TBST(150mM NaClおよび0.1%(v/v)Tween
20を含む25mM Tris/HCl(pH7.5))で4回洗浄した。標準モノクローナルABとして、HIX1抗FIX(精密)を使用した。標準およびサンプルを、2%(w/v)BSAを含むTBST中で希釈した。標準希釈系列およびサンプルの適切な希釈物を、ELISAプレート上で室温で2時間インキュベートした。プレートを、TBSTで4回洗浄し、そして抗マウスIgG(Fc特異的)ペルオキシダーゼ結合体化抗体(Sigma、#A−0168)FIXaの100μl/ウェルの1:25000の希釈物(TBST/1% BSA中)とともにインキュベートした(2時間、RT)。ウェルを、TBSTで5回洗浄し、そして最後に、100μlの新たに調製した染色溶液(100μlのOPD(60mg OPD/ml)および10μlの30% H2O2を補充した10ml 50mMクエン酸ナトリウム(pH5))で染色した。この反応を、50ml H2SO4の添加により停止させ、そして30分後、光学密度を、492nmおよび620nmにおいて、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーによって、GENESISTMソフトウェアを使用して、記録した。
【0063】
(実施例4:色素形成FVIIIアッセイにおいてFVIII様活性を示す抗FIX/FIXa抗体)
いくつかの抗FIX/FIXa抗体産生ハイブリドーマクローンを、4回までサブクローニングし、そして得られるモノクローナルハイブリドーマ細胞株を使用して、モノクローナル抗体含有上清を産生した。これらの上清由来のIgGアイソタイプ抗体を、アフィニティーカラムで精製し、そしてTBSに対して透析した(上記を参照のこと)。IgM抗体を、精製していない上清画分として使用した。以下の実験を、以下の2セットの代表的な抗体を使用して行った:193/AD3および198/AC1/1(IgGアイソタイプ、抗体198/AC1/1は、親198/AC1ハイブリドーマクローンからの調製物であり、すなわち、198/AC1細胞を含む(凍結した)ウイルスが、培養され、そして抗体が産生される。次いで、この上清を、これらの実験のために使用する)ならびに196/AF2および196/AF1(IgMアイソタイプ)(図6Aおよび図6B)。簡単に言えば、モノクローナル抗体を含むサンプル(精製されていないハイブリドーマ上清、または示される場合には、特定の量のFIX特異的抗体)の25μlのアリコートを、マイクロタイタープレートウェルに移し、そして37℃に昇温させた。色素形成基質(S−2222)、合成トロンビンインヒビター(I−2581)、第IXa因子(FIXa)およびFXを、滅菌水中で再形成し、そしてFIXa/FXを、供給者のプロトコルに従って、リン脂質と混合した。1反応あたり、50μlのリン脂質/FIXa/FX溶液を、25μlのCaCl2(25mM)および50μlの基質/インヒビターカクテルと混合した。反応を開始させるために、125μlのプレミックス(premix)を、マイクロタイタープレート中のモノクローナル抗体溶液に添加し、そして37℃でインキュベートした。405nmおよび490nmにおける、サンプルの吸光度を、種々の時点において(5分〜6時間)、試薬ブランク(ハイブリドーマ上清の代わりに細胞培養培地)に対して、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーによって、GENESISTMソフトウェアを使用して、読み取った。
【0064】
ヒトFVIII(12および16mU/ml)、TBSならびに細胞培養培地と比較した、モノクローナル抗体193/AD3(IgGアイソタイプ)および196/AF2(IgMアイソタイプ)により示されるFVIII様活性の時間経過を、図6Aに示す。405nmの波長において測定可能な光学密度の増加により判断する場合に、誘導期の後に、両方の抗体が、色素形成基質切断を生じる。
【0065】
ヒトFVIII(16mU/ml)および10μg/mlのマウスIgGと比較した、モノクローナル抗体198/AC1/1(IgGアイソタイプ、10μg/ml)および196/AF1(IgMアイソタイプ、精製されていない上清)により示されるFVIII様活性の時間経過を、図6Bに示す。405nmの波長において測定可能な光学密度の増加により判断する場合に、誘導期の後に、両方の抗体が、色素形成基質切断を生じる。
【0066】
(実施例5:抗FIX/FIXa−抗体により示されるFVIII様活性は、第Xa因子を産生し、そしてリン脂質、FIXa/FXおよびCa2+に依存性である。)
第VIII因子活性は、通常、色素形成アッセイおよび/またはAPTTに基づく凝固アッセイを用いて、決定される。両方の型のアッセイは、FVIIIa/FIXaにより媒介される第Xa因子産生に依存する。色素形成FVIIIアッセイの場合には、産生される第Xa因子は、引き続いて色素形成基質と反応し、これは、分光学的に(例えば、ELISAリーダーにおいて)モニタリングされ得る。APTTに基づく凝集アッセイにおいては、遊離の第Xa因子が、リン脂質表面におけるFVaと組み合わさって、いわゆるプロトロンビナーゼ複合体となり、そしてプロトロンビンをトロンビンへと活性化させる。トロンビンは、次に、フィブリン産生を生じ、そして最終的に、凝固の形成を生じる。これら2つのアッセイ系の中心は、FVIIIa/FIXa複合体による第Xa因子の産生である。
【0067】
抗FIX/FIXa−抗体により示されるFVIII様活性が実際に第Xa因子を産生することを実証するために、以下の実験を実施した。精製されていないハイブリドーマ上清196/AF2(IgMアイソタイプ)のいくつかの25μlのアリコートを、マイクロタイタープレートウェルに移し、そして37℃まで昇温させた。ポジティブコントロールとして、16mUのRecombinateTMを、ハイブリドーマ培地(196 HM 007/99)中に希釈し、そしてハイブリドーマ上清と全く同じ様式で処理した。ネガティブコントロールとして、純ハイブリドーマ培地を使用した。色素形成基質(S−2222)合成トロンビンインヒビター(I−2581)第IXa因子およびFXを、滅菌水中で再形成し、そしてFIXa/FXを、供給者のプロトコルに従って、リン脂質と混合した。Pefabloc Xa(登録商標)(第Xa因子特異的プロテイナーゼインヒビター)(Pentapharm,LTD)を、水で再形成して、最終濃度を1mM/lとした。1反応あたり50μlのリン脂質/FIXa/FX溶液を、25μlのCaCl2(25mM)および50μlの基質/トロンビンインヒビターカクテルと混合した。反応を開始させるために、125μlのプレミックスを、マイクロタイタープレート中のサンプルに添加し、そして37℃でインキュベートした。示される場合には、35μMのPefabloc Xa(登録商標)を添加した。405nmおよび490nmにおける吸光度を、種々の時点において(5分ごと〜6時間ごと)、試薬ブランク(細胞培養培地)に対して、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーによって、GENESISTMソフトウェアを使用して、読み取った。
【0068】
色素形成基質S−2222の容易に測定可能な切断により判断する場合の、IgM抗FIX/FIXa抗体196/AF2により示されるFVIII様活性による、第Xa因子の産生における第IXa因子刺激の結果(「16mU FVIII」と「196/AF2」とを比較のこと)を、図7Aに示す。第Xa因子活性は、FXa特異的インヒビター「Pefabloc Xa(登録商標)」により効果的にブロックされ(「196/AF2」と「196/AF2 35μM Pefabloc Xa(登録商標)」とを比較のこと)、このことは、実際にFXaが産生されたことを示す。
【0069】
同じ実験を、クローン198/AM1の精製したIgG調製物を使用して、実施した(図7B)。精製されたIgGをTBSで希釈し、最終濃度を0,4mg/mlおよび25μl(すなわち、合計10μg)とし、マイクロタイタープレートウェルに移し、そして37℃に昇温させた。ポジティブコントロールとして、6mUの血漿由来FVIIIを使用した。10μgの非特異的マウスIgG(Sigma,I−5381)は、ネガティブコントロールとして作用した。このアッセイを、上記のように実施した。
【0070】
さらなる実験は、容易に測定可能な色素形成基質S−2222の切断により判断する場合に、IgG抗FIX/FIXa抗体198/AM1により示されるFVIII様活性による第IXa因子の刺激が、第Xa因子を産生することを示す(図7B)。再度、第VIII因子および抗体198/AM1は、FXaを産生し、これは、FXa特異的インヒビター「Pefabloc Xa(登録商標)」によって効果的にブロックされる。ネガティブコントロールとして、非特異的マウスIgG(Sigma,I5381)をアッセイした。
【0071】
別のセットの実験において、精製した抗FIX/FIXa抗体(IgM、図8A)または細胞培養物上清由来の精製していない抗体(IgG、図8B)のいずれかのFVIII様活性の、リン脂質(PL)、FIXa/FXおよびCa2+の存在への依存を、実証した。マウスIgGを、コントロールとして使用した(図8C)。第VIII因子様活性を、本質的に上に記載のように、アッセイした。示される場合には、FIXa/FX混合物、PLまたはCa2+のいずれかを、その反応から省略した。405nmおよび490nmにおける、サンプルの吸光度を、種々の時点において、試薬ブランク(精製された抗体の代わりに緩衝液)に対して、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーで読み取った。その結果を図8A、図8Bおよび図8Cに示す。
【0072】
精製された抗FIXa抗体198/AC1/1(IgGアイソタイプ、このアッセイを通して使用された濃度は10μg/mlであった)の、リン脂質(PL)、FIXa/FX、およびCa2+の存在下におけるFVIII様活性の依存性をさらに、図8Aに示す。容易に認識可能であるように、競合的アッセイ(抗体、PL、Ca2+およびFIXa/FXを含む)のみが、穏当なFXa生成を引き起こす。リン脂質、FIXa/FX、およびCa2+の存在下における不純IgMアイソタイプ抗FIX/FIXa抗体(196/AF1)を含む細胞培養上清のFVIII様活性の依存性を、図8Bに示す。
【0073】
重ねて、精製されたIgG調製物について既に示されたように(図8A)、抗体198/AC1/1は、競合的アッセイ(PL、Ca2+およびFIXa/FXを含む)のみが、穏当な量のFXa生成を引き起こす。反応の特異性を実証するために、正常なマウス血漿から調製された総IgGを、上記と同じ条件下でアッセイした。この結果を、図8Cに示す。いかなるFVIII様活性も検出され得なかった。予期されたように、リン脂質、FIXa/FX、およびCa2+の不在下において検出可能な活性は存在しない。すべての実験をマイクロタイタープレートにおいて実施し、そしてOD405を、6時間にわたって5分毎に走査した。
【0074】
(実施例6:特定の抗FIX/FIXa抗体は、FIXaの存在下において凝血原である)
正常なホメオーシスの間、FIXはまず、組織因子(TF)/第VIIa因子経路によってか、または後に活性化第XI因子(FIXa)によってかのいずれかで活性化となる。その活性化後、FIXaは、活性化FVIIIとの膜結合複合体において、血小板表面上で会合する。第IXa因子は単独では、FXに対する酵素活性をほとんどまたは全く有さないが、FVIIIaの存在下では、高度に活性となる。特定の抗FIX/FIXa抗体が、FVIII様活性を有し、従って、FVIIIを欠損したヒト血漿における凝血原であることを実証するために、以下の実験を行った。異なる量の抗体193/AD3またはマウスIgG(コントロールとして)を、標準的なaPTTに基づく一段階凝固アッセイにおいて使用した。簡潔には、100μlの抗体含有サンプルを、100μlのFVIII欠損血漿(DP)および100μlのDAPTTIN(活性化されたトロンボプラスチンの時間を決定するためのPTT試薬;IMMUNO AG)試薬と共に、KC10A凝固分析装置においてインキュベートした。示された場合では、総量50ngの活性化FIXを、反応混合物中に含ませた。4分間のインキュベーション後に、100μlのCaCl2(25mM)を添加することによって、反応を開始した。この結果を、表1および図9に示す。
【0075】
【表1】
表1:50ngの活性化FIX(0.01UFIX)の存在下で、種々の量の凝血原(193/AD3)およびコントロール抗体(マウスIgG)を使用するAPTTに基づく凝固アッセイにおけるFVIII欠損血漿の凝固時間。反応および活性化FIXにおける抗体のモル比は、10:1である。抗体と総FIX(FIXおよびFIXa、ヒトFVIII欠損血漿が1U(5μg)のFIXを含むと仮定)との間のモル比は、6:1(反応における9μgの抗体)と1:6(反応における0.23μgの抗体)との間で変動する。凝固時間の最適な短縮化に際して、抗体と総FIXとの間のモル比は、1:1である。FIXaの添加を伴わない凝固時間は、120秒間の範囲である。
【0076】
図9は、50ngの活性化FIXの存在下で、種々の量の凝血原(193/AD3)およびコントロール(マウスIgG)抗体を使用するaPTTに基づく凝固アッセイにおけるFVIII欠損血漿の凝固時間のグラフ表示である。抗体193/AD3を補充したサンプルには、明らかな凝固時間の用量依存的減少が存在する。これらの結果は、抗体193/AD3が、FIXaの存在下において凝血原であることを意味する。
【0077】
(実施例7:抗FIX/FIXa抗体は、FVIIIインヒビターおよびFIXaの存在下において凝血原である)
標準的なFVIII置換療法の重篤な合併症が、FVIIIに対する同種抗体の発達であり、これは、FVIIIの中和へと導き、そして患者の血液が凝固しないという状態に導く。
【0078】
特定の抗FIXa抗体が、FVIIIインヒビターの存在下においてさえ、FVIII様活性を有するということを実証するために、以下の実験を行った。異なる量の抗体193/AD3またはコントロールとしてのマウスIgGを、標準的なAPTTに基づく一段階凝固アッセイにおいて使用した。簡潔には、100μlの抗体サンプルを、100μlのFVIII欠損血漿(図10A)またはFVIIIインヒビター血漿(インヒビター効力400BU/ml)(図10B)のいずれか、および100μlのDAPTTIN試薬と共に、KC10A凝固分析装置においてインキュベートした。さらに、総量50ngの活性化FIXaを、反応混合物に含ませた。4分間のインキュベーション後に、100μlのCaCl2(25mM)を添加することによって、反応を開始した。等価な条件を確実にするために、FVIII欠損血漿およびFVIIIインヒビター血漿を使用する実験を、並置して実施した。この結果を、図10Aおよび10Bに示す。実施例6において既に示したように、FVIIIインヒビターの存在下で抗体193/AD3を補充したサンプルには、明らかな凝固時間の用量依存的減少が存在する。
【0079】
(実施例8:抗FIX/FIXa抗体は、欠損性FVIIIおよびFIXaの存在下において凝血原である)
特定の抗FIXa抗体が、欠損性FVIIIの存在下においてFVIII様活性を有することを実証するために、以下の実験を実施し得る。漸増量の抗体193/AD3、またはコントロールとしてのマウスIgGを、標準的なaPTTに基づく一段階凝固アッセイにおいて使用する。この凝固アッセイでは、欠損性FVIII(DF8)の存在に起因して非常に低い凝固活性を有する血友病Aの患者の血漿を、使用する。簡潔には、100μlの抗体サンプルを、100μlのDF8血漿またはFVIII欠損性血漿のいずれか、および100μlのDAPTTIN試薬と共に、KC10A凝固分析装置においてインキュベートする。さらに、総量50ngの活性化FIXaを、反応混合物に含ませた。短期間のインキュベーション後に、100μlのCaCl2(25mM)を添加することによって、反応を開始した。等価な条件を確実にするために、FVIII欠損血漿およびDF8血漿を使用する実験を、並置して実施する。
【0080】
(実施例9:FIXaの存在下において凝血原活性を有する抗FIX/FIXa抗体は、ヒトFIXaとウシFIXaとの間を識別する)
198回目の融合実験から選択されたFIX/FIXa特異的モノクローナル抗体を、個々のハイブリドーマ上清から精製し、そして実施例3に記載のように定量した。これらの抗体を、改変された一段階凝固アッセイ(実施例6に記載のような)において分析した。そしていくつかが、凝血原活性を示した。
【0081】
これらの抗体調製物の色素形成活性を、以下のFXa生成動力学的アッセイにおいて測定した:10μgのモノクローナル抗体(25μl中)を、マイクロタイタープレートウェルに移し、そして37℃まで加温した。色素形成基質(S−2222)、合成トロンビンインヒビター(I−2581)、第IXa因子、およびFXを、滅菌水中で再構築し、そしてFIXa/FX(両方ともウシ)を、供給業者のプロトコールに従って、リン脂質と混合した。反応につき、50μlのリン脂質/FIXa/FX溶液を、25μlのCaCl2(25mM)および50μlの基質/インヒビター反応混液と組み合わせた。反応を開始するために、125μlのプレミックスを、マイクロタイタープレート中におけるモノクローナル抗体溶液に添加し、そして37℃でインキュベートした。サンプルの405nmおよび490nmでの吸光度を、GENESISTMソフトウェアを使用するLabsystems iEMS Reader MFTMマイクロタイタープレートリーダーにおいて、試薬ブランク(モノクローナル抗体の代わりに、25ml TBS)に対して種々の時点(5分間〜2時間)で読み取った。並行して、反応あたり50ngのヒトFIXaを添加したことを除いて、同じ反応を実施した。凝血原活性を示したこれらの抗体は、ウシFIXの場合では色素形成活性を有さなかったが、ヒトFIXaが存在した場合では、高い活性を示した。
【0082】
図11は、50ngのヒトFIXaβの添加あり(+)および添加なし(−)でモノクローナル抗体198/A1、198/B1、および198/AP1によって示されたFVIII様活性の時間経過を示す。非特異的ポリクローナルマウスIgGを、コントロールとして使用した。198/A1および198/B1は、凝血原活性を示す(実施例6における198/AD3と類似)が、198/AP1は示さない。抗体198/BB1は、同じ活性パターンを有した(データは示さず)。
【0083】
198回目の融合実験から選択された、さらなるモノクローナル抗体としては、198/D1(ECACC番号 99121616)、198/T2(ECACC番号 99121617)、198/G2(ECACC番号 9912118)、198/U2(ECACC番号 99121620)が挙げられる。
【0084】
(実施例10:抗FIX/FIXa抗体から誘導された抗体誘導体の構造および凝血原活性;ハイブリドーマ細胞株193/AD3、193/K2、198/A1および198/B1(クローンAB2)からの抗体可変ドメインのサブクローニング)
クローニング手順:「QickPrep(登録商標)Micro mRNA Purification Kit」(Pharmacia)を製造業者の指示書に従い使用して、メッセンジャーRNAを、個々の細胞株(193/AD3、193/K2、198/A1または198/B1(クローンAB2)のいずれか)の1×106個のハイブリドーマ細胞から調製した。対応するcDNAを、「Ready−To−Go−You−Prime−First−Strand Beads kit」(Pharmacia)を製造業者の指示書に従い使用して、mRNAのレトロ(retro)転写によって生成した。重鎖および軽鎖をコードする配列を、プライマーセットを使用して、対応するcDNAに変換した。重鎖特異的mRNA(VH)を逆転写するために、以下のオリゴヌクレオチドの等モル混合物を使用した(RTmix1):MOCG1−2FOR(5’ CTC AAT TTT CTT GTC CAC CTT GGT GC 3’)(配列番号1)、MOCG3FOR(5’ CTC GAT TCT CTT GAT CAA CTC AGT CT 3’)(配列番号2)および、MOCMFOR(5’ TGG AAT GGG CAC ATG CAG ATC TCT 3’)(配列番号3)。同じ反応チューブにおいて、軽鎖特異的cDNA(VL)を、プライマーMOCKFOR−(5’ CTC ATT CCT GTT GAA GCT CTT GAC 3’)(配列番号4)を使用して合成した。
【0085】
VHについてのコード配列を、図12に示したプライマーセットおよびテンプレートとして上記の逆転写混合物(RTmix1)由来の特異的cDNAを用いるPCRによって増幅した。VK鎖遺伝子を、図13に示したプライマーセットを使用し、そしてまたテンプレートとしてRtmix1を用いて増幅した。VH−PCR産物を、SfiI−AscIで切断し、そしてSfiI−AscIで消化したベクターpDAP2(GeneBank登録番号:U35316)に挿入した。これにより得られたpDAP2−VH構築物を、それぞれ、pDAP2−193AD3/VH、pDAP2−198A1/VH、pDAP2−198AB2/VH(抗体198/B1由来)およびpDAP2−193/K2/VHと命名した。引き続いて、プラスミドをAscI−NotIで切断し、そして対応するAscI−NotI消化されたVK遺伝子のPCR産物を挿入した。結果として生じたベクターをpDAP2−193/AD3scFv、pDAP2−198/AlscFv、pDAP2−198/AB2scFv(抗体198/B1由来)およびpDAP2−193/K2scFvと命名した。これらは、モノクローナル抗体193/AD3、198/A1、198/AB2(抗体198/B1由来)および193/K2のVH遺伝子およびVL遺伝子をコードする。重鎖および軽鎖を、人工の可撓性リンカー(G4SGGRASG4S;Engelhardtら,1994)についてのコード配列に連結し、そして個々の抗体のscFv改変体の発現を可能にする。
【0086】
図14では、ハイブリドーマ細胞株193/AD3由来のscFvのDNAおよび推定タンパク質配列を示す。ヌクレオチド1〜357は、重鎖可変ドメインをコードし、ヌクレオチド358〜402は、人工の可撓性リンカーをコードし、そしてヌクレオチド403〜726は、軽鎖可変領域をコードする。重鎖のCDR3領域のタンパク質配列は、配列YGNSPKGFAY(配列番号5)を有し、そしてボールドの文字で提供される。人工のリンカー配列(G4SGGRASG4S)を示す。
【0087】
図15では、ハイブリドーマ細胞株193/K2由来のscFvのDNAおよび推定タンパク質配列を示す。ヌクレオチド1〜363は、重鎖可変ドメインをコードし、ヌクレオチド364〜408は、人工の可撓性リンカーをコードし、そしてヌクレオチド409〜747は、軽鎖可変領域をコードする。重鎖のCDR3のタンパク質配列は、配列DGGHGYGSSFDY(配列番号6)を有し、そしてボールドの文字で提供される。人工のリンカー配列(G4SGGRASG4S)を示す。
【0088】
図16では、ハイブリドーマ細胞株198/AB2(抗体198/B1由来)由来のscFvのDNAおよび推定タンパク質配列を示す。ヌクレオチド1〜366は、重鎖可変ドメインをコードし、ヌクレオチド367〜411は、人工の可撓性リンカーをコードし、そしてヌクレオチド412〜747は、軽鎖可変領域をコードする。重鎖のCDR3領域のタンパク質配列は、配列EGGGFTVNWYFDV(配列番号7)を有し、そしてボールドの文字で提供される。人工のリンカー配列(G4SGGRASG4S)もまた示す。
【0089】
図17では、ハイブリドーマ細胞株198/A1由来のscFvのDNAおよび推定タンパク質配列を示す。ヌクレオチド1〜366は、重鎖可変ドメインをコードし、ヌクレオチド367〜411は、人工の可撓性リンカーをコードし、そしてヌクレオチド412〜747は、軽鎖可変領域をコードする。重鎖のCDR3領域のタンパク質配列は、配列EGGGYYVNWYFDV(配列番号8)を有し、そしてボールドの文字で提供される。人工のリンカー配列(G4SGGRASG4S)もまた示す。
【0090】
(実施例11:抗FIX/FIXa抗体のCDR3領域由来のペプチドの凝血原活性)
原理上、抗体分子は、三次元空間におけるペプチドエレメントのコンビナトリアルアレイの提示のための生物学的デバイスとして想定され得る(Gaoら,1999,PNAS,96:6025を参照のこと)。従って、抗体(または抗体誘導体(例えば、scFv、Fabなど))を特定の標的タンパク質の機能的に重要なドメインの検出のためのツールとして、または他方で、特定の相互作用を特異的に媒介する(すなわち、生物学的基質FXに対するFIXaの活性を活性化または増強する)アミノ酸配列を描写(delineation)するためのいずれにも使用され得る。後者のプロセスは、FIXa増強剤としての、多数の重鎖CDR3領域(CDR3H)由来ペプチド配列の評価へと導いた。
【0091】
このような活性を示すペプチドの凝血原活性の増強は、FIXa活性の増強を媒介するために重要なペプチド領域における配列改変を通して達成され得る。増強された凝血原活性を有するペプチド配列への可能な工程として、FIXa分子上の抗体の結合部位(すなわち、198/A1または198/B1)を、配列比較分析、競合的結合アッセイ、ウェスタンブロット分析および競合的ELISA分析を使用することによってマッピングする。FIXの結晶構造が既知であるので、引き続いて、分子モデリングを使用して、すなわち、ヒトFIXa上の198/B1結合部位における198/B1由来ペプチドの適合を改善する。
【0092】
他方で、例えば、「アラニンスキャニング変異分析(alanine scanning mutational analysis)」によって、所定のペプチド配列(例えば、198/A1または198/B1 CDR3H由来のペプチド配列)の秩序的(methodical)変異分析によって、凝血原活性に重要なペプチド残基の同定が可能となる。特定のペプチド配列の活性を改善するための別の方法は、高スループットスクリーニングと組み合わせたペプチドライブラリーの使用である。
【0093】
抗体の抗原結合部位は、VL−HLダイマー(またはFv領域)のN末端の近位する6つの「相補的決定領域(complement determining region)(CDR)」に由来する。所定の抗原に対する抗体特異性に対する単一のCDRの寄与は、非常に多様であり得るが、一般に、重鎖のCDR3領域(CDR3H)が特に影響する、すなわち、CDR3H領域の特定のタンパク質配列が抗原認識に非常に重要であり得ると考えられる。CDR3H領域の長さは、非常に多様であることが報告されており、4〜25アミノ酸の範囲である(Borrebaeck、16頁)。
【0094】
ペプチド配列の系統的な変異分析の例を以下に示す。抗FIX/FIXa抗体のCD3領域由来のペプチドの可溶性/凝血促進性(procoagulant)効率を改善するために、N末端およびC末端のアミノ酸配列を変化させた。さらに、一連の変異ペプチドを構築しそして分析した。
【0095】
このような研究の原理を、抗体198/A1および198B/1のCDR3H領域由来の一連のペプチドによって例示する。それぞれ、元のペプチドA1(表2を参照のこと)は、抗体198/A1のCDR3H領域に由来し、そしてペプチドB1は、抗体198B/1のCDR3H領域由来に由来する(実施例10、図16および17を参照のこと)。用語「スクランブルバージョン(scrambled version)」とは、ペプチドが、同じアミノ酸をランダムな順序で有することを意味する。
【0096】
【表2】
(表2)
一連の抗体198/A1由来ペプチドのリスト。ペプチドの長さ(aa.アミノ酸数)、算出した分子量(MW、ダルトン(D))および統計学的等電点(pI)をリストする。D−Argは、Rdと省略する。
【0097】
第1のシリーズの実験において、本発明者らは、元のCDR3HペプチドのC末端Val残基を除去し、そしてN末端およびC末端にいくつかの荷電残基を付加することによって、その元のCDR3Hペプチド配列(A1;EGGGYYVNWYFDV)の可溶性を改善した。得られたペプチドA1/2(酸性のpI)、A1/3(塩基性のpI)およびこれらのそれぞれのスクランブルバージョンA1/4、A1/5およびA1/3scr3は、生理学的pHで種々の緩衝液系において容易に可溶性であった。
【0098】
これらのペプチドのFVIII様(FIXa活性化)活性を分析するために、市販のFVIIIアッセイに基づくアッセイ系を開発した(実施例2および4を参照のこと)。この基本的原理は、FIXaが、補因子なしで、その天然の基質FXに対する非常に限定された活性を有することである。FIXa活性化特性を有する基質(すなわち、FVIIIまたはFVIII様活性を示す基質)の存在下でのみ、実質的な量のFXaが、FIXa/アクチベーター複合体を介するFXの切断によって生成される。生成されたFXaの量を、色素形成性の基質の切断によってモニターする。この改正した色素形成アッセイの原理を、2つの代表的なペプチドA1/3およびA1/5(表2)について記載する。簡単に言うと、ペプチドストック溶液(イミダゾール緩衝液(IZ)(50mM イミダゾール、100mM NaCl、pH7.2)中)の25μlのアリコートを、マイクロタイタープレートウェルに移し、そして37℃に温めた。供給者のプロトコルに従って、色素形成性のFXa基質(S−2222)、合成トロンビンインヒビター(I−2581)、ウシFIXaおよびウシFXを、滅菌水中で再構成し、そしてFIXa/FXを、リン脂質と混合した。これらのペプチドは、ウシFIXaとは反応しないので、(Testキット中のウシFXとの混合物として生じる)2.9nM(多くの場合では、2.3nM)のヒトFIXa(ERL)を添加した(実施例11、図19を参照のこと)。反応あたり、50μlのこのリン脂質/FIXa/FX溶液を、25μlのCaCl2(25mM)および50μlの基質/インヒビター混液と合わせた。反応を開始するために、この125μlのプレミックスを、マイクロタイタープレート中のペプチド溶液に添加し、37℃でインキュベートした。サンプルの405nmおよび490nmの吸光度を、GENESISTMソフトウェアを使用するLabsystems iMES
Reader MFTMマイクロタイタープレートリーダーにおいて、試薬ブランクに対して種々の時点で(5分〜2時間)で読み取った。この実験の結果を、実施例11、図18に示す。ペプチドA1/3は、2.9nMのヒトFIXaの存在下で容易に測定可能なFXa生成を誘導したが、そのスクランブルバージョンA1/5は、不活性であった。さらに、酸性ペプチドA1/2およびそのスクランブルバージョンA1/4およびA1/3−scr3は、匹敵する条件下で試験した場合に、有意な色素形成活性を付与しなかった(データは示さず)。このペプチドA1/3様の親抗体198/A1が、ウシFIXaおよびFXと反応しないことを証明するために、図19に示す実験を行った。ペプチドA1/3を、2.3nMのヒトFIXa(hFIXa)と共に(A1/3(24μM)、+hFIXa)、または伴わずに(A1/3(24μM)、w/o hFIXa)、上記のようにインキュベートした。コントロール実験において、本発明者らは、反応混合液に、2.3nMのhFIXaを補充したそのままの希釈緩衝液(IZ)を添加した。図19に示されるように、反応は、ヒトFIXaの存在下でのみ生じる。
【0099】
図18は、2.9nMのヒトFIXa(hFIXa)の存在下でのペプチドA1/3の色素形成性FVIII様活性を実証する。ペプチドA1/3のスクランブルバージョンであるペプチドA1/5は、FXaの生成を生じない。図19は、ヒトFIXa(hFIXa)の存在に対する、ペプチドA1/3の色素形成性のFVIII様活性の依存性を実証する。ヒトFIXaの非存在下で、ペプチドA1/3は、FXaの生成を生じない。緩衝液コントロールであるそのままのイミダゾール緩衝液をIZと示す。
【0100】
これらのペプチドをまた、FVIII欠乏血漿において凝固時間を減少するその能力について分析した。aPTTベースの1段階の凝固アッセイを、本質的には、記載(実施例6を参照のこと)のように行った。凝固時間(反応が開始してから「凝固」が形成するまでの時間)を、FVIII、緩衝液コントロール(IZ)またはコントロールペプチド(スクランブルバージョン)のいずれかに対して比較した。2つの異なるaPTT試薬(DAPTTINおよびPathromtin SL)を用いて行った、2つの代表的な凝固実験の結果を、表3Aおよび表3Bに示す。
【0101】
【表3】
表3A.2.2nMのヒトFIXaの存在下または非存在下(w/o)のいずれかにおけるFVIII欠乏血漿におけるペプチドA1/3およびA1/3−scr(A1/3のスクランブルバージョン)の凝固活性。2つの独立した代表的な実験(実験1および実験2)を示す。全ての凝固実験を2連で行った。個々の実験についての凝固時間を取り、平均凝固時間を秒(sec.)で表す。表3Aに示す実験は、aPTT試薬DAPTTIN(Baxter Hyland Immuno)を使用して行った。緩衝液コントロール(IZ、イミダゾール緩衝液)に比べて、ペプチドA1/3は、凝固時間における用量依存性の減少を生じた。この凝固時間の減少は、この反応混合液に対する2.2nMの活性化ヒトFIXの添加によってさらにより明らかとなった。ペプチドA1/3のスクランブルバージョンA1/3−scr3は、凝固時間の減少を示さなかった。実際に、2.5μMよりも高い濃度では、このスクランブルペプチドは、阻害性になり、従って、凝固時間を延長した。ペプチドA1/1、A1/2、A1/4およびA1/5は、凝固時間の減少を生じず、これは、これらが、凝血促進活性を欠いていることを示す(データは示さず)。
【0102】
【表4】
表3B.Pathromtin SL(DADE Behring)をaPTT試薬として使用する場合のFVIII欠乏血漿におけるペプチドA1/3の凝固活性。実験を、2.2nMのヒトFIXaの存在下または非存在下(w/o)のいずれかで、2連で行った。個々の実験についての凝固時間を取り、平均凝固時間を秒(sec.)で表す。第VIII因子およびイミダゾール緩衝液(IZ)を、それぞれ、ポジティブコントロールおよびネガティブコントロールとして含んだ。
【0103】
表3Aに示した実験と対照的に、表3Bに示した実験は、aPTT試薬Pathromtin SLを使用して行った。FIXaの存在下で、ペプチドA1/3は、凝固時間の用量依存性の減少を生じたが、FIXaの非存在下では、凝固時間の減少は検出されなかった。
【0104】
別のシリーズの実験において、本発明者らは、ペプチドA1/3の血漿安定性(例えば、タンパク質分解からの保護)を改善することを行った。1つのアプローチは、N末端およびC末端のL−Arg残基のD−Arg残基での置換である(ペプチドA1/3−rdおよびA1/3−Rd−srmbによって例示される)。次いで、ペプチドA1/3−rdおよびA1/3−Rd−srmb(このペプチドA1/3−rdのスクランブルバージョン)を、色素形成性およびaPTTベースの凝固アッセイにおいて分析した。これらの実験は、末端L−Arg残基のD−Arg残基での交換が、色素形成アッセイにおいて測定されたようなFVIII様活性を変化しないことを示し、これは、このArg残基のキラリティーが、色素形成活性において主な役割を果たしていないことを示す(図20)。さらに、aPTTベースの1段階の凝固活性は、幾分減少されたが、なお容易に検出可能であった(表4)。
【0105】
【表5】
表4 ペプチドA1/3、A1/3−RdおよびA1/3−Rd−srmb(配列は、表2を参照のこと)の1段階の凝固活性。IZ(緩衝液コントロール)。
【0106】
図20は、ペプチドA1/3−Rdの変化されない色素形成活性を実証する。12μMの最終濃度のペプチドまたは緩衝液コントロール(IZ)を、2.3nMのヒトFIXa(+)の存在下でインキュベートした。ペプチドA1/3およびA1/3−Rdの色素形成活性は、実質的に変化されないことが示され、そしてこの色素形成アッセイにおいてほぼ同一の結果を生じた。A1/3のスクランブルバージョン、A1/5および緩衝液は、有意なFXa生成を生じなかった。
【0107】
次のシリーズの実験において、本発明者らは、各々の残基のアミノ酸アラニンでの置換による、このペプチドコア配列の任意のアミノ酸の個々の役割を決定することを行った(表5)。
【0108】
【表6】
表5.ペプチドコア配列(E1G2G3G4Y5Y6V7N8W9Y10F11D12)内の任意の単一のアミノ酸の役割を解明するために設計されたペプチドを列挙する。下の枠の番号は、そのペプチド内のアミノ酸の位置を記載する。アラニン(荷電していない小さなアミノ酸)で、各アミノ酸を置換した(「アラニンスキャン」)。ペプチドの長さ(アミノ酸数)、分子量計算値(MW(ダルトン(D))および統計的等電点(pI)もまた列挙される。
【0109】
各ペプチドを、それぞれイミダゾール緩衝液(50mM イミダゾール、100mM NaCl、pH 7.2)に溶解し、続いて凝固緩衝液(50mM イミダゾール、100mM NaCl、1% ヒトアルブミン、pH 7.4)中所望の最終濃度まで希釈した。これらのペプチドをその色素形成活性について、およびそのFVIII欠損血漿における凝固時間を減少する能力について分析した。一段階凝固アッセイを本質的に記載されるように(実施例6を参照のこと)行った。凝固時間(反応の開始から「凝塊」形成までの時間)を、緩衝液コントロールまたはコントロールペプチドのいずれかに対して比較した(スクランブルバージョン)。「アラニンスキャン」のいくつかの結果をペプチドA1/3−2およびA1/3−3について示す。ペプチドA1/3−2で例示される場合のG3−A3の変化は、2.2nMのヒトFIXaの存在下で、高い色素形成活性および一段階凝固時間の強い減少(12.5μMの濃度で34秒)を生じる。ペプチドA1/3−3(G4−A4)は、より高いかまたはより低い濃度での減少した活性と共に、12μM付近の最終濃度での最適の色素形成活性を示す。このペプチドは、FIXaの非存在下でのより高い濃度(12.5μM)での一段階凝固アッセイにおいていくらか抑制性であるが、2.2nMのFIXaの存在下では強く活性になる(31秒、12.5μM)。
【0110】
次の一連の実験において、本発明者らは、各コア残基をアミノ酸であるグルタミン酸(E)で置換することにより、ペプチドコア配列の任意のアミノ酸の個々の役割を決定するように設計した(表6を参照のこと)。
【0111】
【表7】
表6.ペプチドコア配列(E1G2G3G4Y5Y6V7N8W9Y10F11D12)内の任意の単一のアミノ酸の役割を解明するために設計されたペプチドを列挙する。下の枠の番号は、そのペプチド内のアミノ酸の位置を記載する。グルタミン酸(負に荷電した大きなアミノ酸)で、このコア配列の各アミノ酸を置換した(「グルタミン酸スキャン」)。ペプチドの長さ(アミノ酸数)、分子量計算値(MW(ダルトン(D))および統計的等電点(pI)もまた列挙される。
【0112】
各ペプチドを、それぞれイミダゾール緩衝液(50mM イミダゾール、100mM NaCl、pH 7.2)に溶解し、続いて凝固緩衝液(50mM イミダゾール、100mM NaCl、1% ヒトアルブミン、pH 7.4)中所望の最終濃度まで希釈した。一連の「グルタミン酸スキャン」由来のペプチドをその色素形成FVIII様活性について、およびそのFVIII欠損血漿における凝固時間を減少する能力について分析した。一段階凝固アッセイを本質的に記載されるように(実施例6を参照のこと)行った。
【0113】
ペプチドA1/3−24は、いくつかの興味深い特性を示した。この分子は、高色素形成FV−III様活性を6.5μM〜12μMの間の濃度で示したが、より高い濃度(24μMまで)では失活した。このペプチドは、ヒトFIXaの非存在下では凝血促進活性を有していなかったが、2.2nMのhFIXaの存在下では強く活性であった。
【0114】
第2の一連の実験において、本発明者らは、ペプチド配列B1由来の抗体198/B1 CDR3Hの凝血促進活性を改善するように設計した。第1の工程において、本発明者らは、元のペプチド配列(B1;EGGGFTVNWYFDV)の溶解性を、C末端Val残基を除去し、そしていくつかの荷電した残基をこのペプチドのN末端およびC末端に付加することにより改善した。得られたペプチドB1/4、B1/6(酸性 pI)、B1/7(塩基性 pI)およびそのスクランブルされたバージョンB1/5、B1/7scr3は、生理学的pHにおいて種々の緩衝液系に容易に可溶である。
【0115】
【表8】
表7は、一連の抗体198/B1誘導ペプチドのリストである。ペプチドの長さ(aa、アミノ酸数)、分子量計算値(MW、ダルトン(D))および静止等電点(pI))が列挙される。
【0116】
ペプチドB1/4およびB1/5は、50mMTris、100mM NaCl、pH=6.5に可溶であった。両方のペプチドを、色素形成FVIIIアッセイにおいて分析した。ペプチドB1/4は、いくらかの色素形成活性(データは示されず)を有することが見出されたが、スクランブルバージョンB1/5では見いだされなかった。
【0117】
続いて、ペプチドB1/6、B1/7およびB1/7scr3を分析した。各ペプチドを、それぞれ50mM イミダゾール、100mM NaCl、pH 7.2に溶解し、そして続いて凝固緩衝液(50mM イミダゾール、100mM NaCl、1%ヒトアルブミン、pH 7.4)またはイミダゾール緩衝液のいずれか中に所望の最終濃度まで希釈した。これらのペプチドをその色素形成活性およびFVIII欠損血漿における凝固時間を減少する能力について分析した(表8および9)。一段階凝固アッセイを、本質的に記載されるとおりに(実施例6を参照のこと)行った。凝固時間(反応の開始から「凝塊」形成までの時間)を、緩衝液コントロールまたはコントロールペプチドのいずれかに対して比較した(スクランブルバージョン)。
【0118】
ペプチドB1/7由来のFIXa活性化活性(FVIII補因子様活性)を、最初に上記の色素形成アッセイにおいて測定した。
【0119】
図21に示されるように、2.4μMペプチドB1/7の反応混合物への添加は、十分に測定可能なFXaの生成をもたらした。対照的に、35μMのPefablock Xa(FXaプロテアーゼ活性の特異的インヒビタ−)の添加は、色素形成基質切断反応の有意な減少(図22)を生じ、それにより実際にペプチド−FIXa媒介FXa生成をもたらした。FIXaおよびFXの反応混合物への添加がない場合、FXaは合成されなかった(図22)。ペプチドB1/6およびコントロールペプチドB1/5およびB1/7scr3は、活性を示さなかった(データは示さず)。
【0120】
図21は、ペプチドB1/7の色素形成活性を実証する。最終濃度2.4μMでのペプチドまたは緩衝液コントロール(IZ)を2.3nMのヒトFIXaの存在下でインキュベートした。
【0121】
図22において、最終濃度2.4μMでのペプチドB1/7または緩衝液コントロール(IZ)でのペプチドを2.3nMのヒトFIXaの存在下でインキュベートした(「+2.3nM hFIXa」、または「+」のいずれかとして示される)。ペプチドB1/7の色素形成活性は、FIXaおよびFXの存在に依存することが見出された。なぜならば、FIXaおよびFXが反応から離される場合(w/o FIXa/FX)、反応が全く検出できないからである。ペプチドB1/7が実際にFXa生成を媒介することを立証するために、FXa特異的プロテアーゼインヒビタ−Pefabloc Xaを反応混合物に添加した(35μM Pefabloc Xa)。第2の一連の実験において、ペプチドB1/6、B1/7およびB1/7scr3の凝血促進効果を、aPTTベースの1工程凝固アッセイにおいて試験した。これらの実験を本質的に実施例6に記載の通りに行った。結果を表8および9に示す。
【0122】
【表9】
表8:FVIII欠損血漿を、ペプチドB1/6、B1/7src3またはB1/7のいずれかと共に活性化ヒトFIXの非存在下でインキュベートした。ネガティブコントロールとして、純粋な緩衝液を欠損血漿に添加した。種々の組合せについての凝固時間を示す。これらの条件下で、その最高濃度(12.5μM)におけるペプチドB1/7は、157秒という延長した凝固時間により示されるように、凝固プロセスに対して抑制性になる。
【0123】
【表10】
表9:FVIII欠損血漿を、ペプチドB1/6、B1/7scr3またはB1/7のいずれかと共に、活性化ヒトFIXの存在下でインキュベートした。ネガティブコントロールとして、純粋な緩衝液をこの欠損血漿に添加した。凝固時間を種々の組合せについて示す。FIXaの存在下において、ペプチドB1/7は、減少した凝固時間(スクランブルペプチドについて102秒、および緩衝液コントロールについて100秒と比較して83秒)により示されるように凝血促進性になる。
【0124】
(実施例12:FVIIIインヒビタ−血漿における抗FIX/FIXa抗体のCDR3領域から得られるペプチド誘導体の凝血促進活性)
FVIIIインヒビター血漿におけるペプチドA1/3の凝血促進活性についてアッセイするために、以下の実験を実行した。本発明者らは、一段階凝固アッセイをベースとする標準aPTTを行ったが、FVIII欠損血漿の代わりに、本発明者らはFVIIIインヒビター血漿を採用した。血漿の阻害性能力は、1mlあたり8.1Bethesdaユニットであった。
【0125】
【表11】
表10:種々の量のペプチドA1/3(12.5μM〜1.25μM)をFVIIIインヒビター血漿に(2.2nMのFIXaの存在下(FIXa)または非存在下(w/o FIXa)のいずれかで)添加した。ネガティブコントロールとして、純粋な緩衝液を血漿に添加した(IZ)。実験を2回行い、そして平均(aver.)を算出した。凝固時間(秒)を種々の組合せについて示す。ペプチドA1/3が、(用量依存性の様式で)FVIIIインヒビタ−血漿の凝固時間を、FIXaの存在下で、たとえ非常に少ない程度でも、またFIXaの非存在下でも減少させることは容易に理解できる。
【0126】
(実施例13:196/C4 IgMのIgG1への変換)
いくつかのIgM抗体は高FVIII様活性を色素形成アッセイにおいて示すので、このようなIgM抗体をIgG抗体に(Fab、F(ab)2、scFvなどのような抗体誘導体もまた産生され得るが)変換させようと試みた。IgM可変領域遺伝子のレスキューが以下に詳細に記載される。発現ベクターpBax−IgG1(図23)を、ベクターpSI(Promega)およびpEF/Bsd(Invitrogen)から複数のクローニング工程を介して最初に構築した。ドナーのBリンパ球を血液から精製し、そして成熟mRNAを「micro−mRNA purification−kit」(Pharmacia)を使用してこれらの細胞から精製した。ヒトκ鎖およびヒトγ1鎖のcDNAを、「you−primefirst−strand−cDNA−kit」(Pharmacia)を採用して特異的プライマーを使用して調製した。
【0127】
ヒトκ軽鎖定常ドメインのコード配列を、PCRにより特的プライマーを使用してcDNAから増幅する。
【0128】
ヒトγ1鎖定常領域(CH1−ヒンジ−CH2−CH3)の遺伝子を、PCRにより特異的プライマーを使用して増幅する。
【0129】
軽鎖定常ドメインの軽鎖のPCR産物を、XbaIおよびNheIを用いて消化し、そして消化されたpSI中に挿入した。得られたベクターをEcoRIおよびXbaIを用いて切断し、そしてアニーリングされたオリゴヌクレオチドを挿入し、ベクターpSI−Cκを生じた。アニーリングされたオリゴヌクレオチドは、κ鎖可変領域挿入のためのリーダー部位およびSacI−XbaI部位を提供する。ヒトγ1鎖定常領域のPCR産物を、SpeIおよびBamHIを用いて消化し、そして消化されたpSIに挿入する。得られたベクターをSpeIおよびNotIで切断し、そしてアニーリングされたオリゴヌクレオチドを挿入して、ベクターpSI−Cγを生じる。アニーリングされたオリゴヌクレオチドは、重鎖可変領域の挿入のためのリーダー部位およびXhoI−BstEI部位を提供する。ベクターpEF/BsdをNheIおよびSfiIで消化し、Klenow処理により平滑末端にし、そしてpSI−Cκ全体の発現カセット(BglIIおよびBamH1を用いて切除される)を(Klenow処理の後に)挿入する。得られたベクターをEcoRIおよびHindIIIを用いて消化し、そしてKlenow処理する。pSI−Cγの全体の発現カセットをBglIIおよびBamH1で切除し、そして挿入する(Klenow処理後)。得られたベクターをpBax−IgG1と称する。
【0130】
軽鎖可変領域をSacI−XbaI部位の間に挿入し得、κ軽鎖の完全コード配列を得る。重鎖可変領域を、XhoI−BstEI部位の間にクローニングし得、完全IgG1重鎖遺伝子を生じる。両方のオープンリーディングフレームは、SV40プロモーターの制御下で発現され、シグナルペプチドのコード配列を、遺伝子の5’末端において小胞体への重鎖および軽鎖の分泌のために含む。COS細胞へのトランスフェクションは、親IgMと同じ結合特性を有するIgG1の発現を可能にする。プラスミドpBax−196/C4の構築は、特異的プライマーを使用するPCRにより196/C4 scFvのVH(実験10において記載されるようにサブクローニングされる)を増幅することによりさらに達成され得る。PCR産物を、XhoIおよびBstEIIを用いて消化し、そしてXhoIおよびBstEII消化pBax IgG1に挿入する。196/C4 scFvのVLを、特異的プライマーを使用するPCRにより増幅する。PCR産物をSacIおよびXbaIを用いて消化し、そしてSacIおよびXbaI消化pBax IgG1−VHに挿入する。得られたベクター(pBax−196/C4)をエレクトロポレーションによりCOS細胞にトランスフェクトし、そして親IgMと同じ特異性を有するハイブリッドIgG1分子(マウス可変領域およびヒト定常領域)が発現される。
【0131】
(実施例14:抗FIXa抗体によるFIXaアミド分解活性の活性化:)
手短に言うと、20μlの第IXa因子(200mUのFIXa(Stago)を含む)を、200μlの反応緩衝液(50mM TrisHCl pH7.4,100mM NaCl,5mM CaCl2および40%エチレングリコール)、25μlのFIXa基質(CH3SO2−D−CHG−Gly−Arg−pNA,AcOH,10μM/ml,Pentapharm LTD)と共に、種々の量の抗FIX抗体198/B1(IgGアイソタイプ)または196/AF1(IgMアイソタイプ)の存在下または非存在下で、37℃でインキュベートした。FIXa基質の特異的切断を、ELISAリーダーにおいて405nmでモニターした。抗FIX抗体の存在は、FIXaのアミド分解活性を少なくとも2倍増大した。図24は、抗体198/B1(図24A)および抗体198/AF1(図24B)の存在下でのFIXaのアミド分解活性の増加を示す。
【0132】
(実施例15:抗FIX/FIXa−抗体由来のFabフラグメントによって示されるFVIII様活性)
抗FIX/FIXa抗体のFabフラグメントを、標準的プロトコルに従って調製し、そして精製した。手短に言うと、1mlの抗体198/A1(50mMのイミダゾール中に4mg/ml,100mM NaCl,pH7.4)を、87μlの細分緩衝液(1M Naアセテート,10mM EDTA 67.5mg/ml L−システイン)および0.25mgのパパイン(アガロースビーズ上に固定化)と共に一晩37℃でインキュベートした。この調製物を濾過してパパインを除去した。L−ヒスチジンを添加し(最終濃度50mM)、そしてその後pHを7.0に調製した。最後に、固体のNaClを添加して、1Mの最終濃度を得た。引き続いて、198/A1 Fabフラグメントを、プロテインLに結合させることによって精製した:本発明者らは、PHARMACIA XK 16/20カラム(ゲル容量:2ml)中のImmunoPure Immobilized PROTEIN L Plus(Pierce)を使用し、クロマトグラフィーのための緩衝液は以下のようであった:1)平衡緩衝液:50mM L−ヒスチジン pH7.0;1M NaCl;0,1%(w/v)NaN3;2)洗浄緩衝液:50mM L−ヒスチジン pH7.0;0.1%(w/v)NaN3;3)溶出緩衝液:100mM グリシン pH2.5;0.1%(w/v)NaN3;および4)中和緩衝液:2M Tris/Cl pH8,0;
クロマトグラフィーは、本質的に表11に記載される以下の工程1〜7によって行った。溶出緩衝液の低いpHを中和するために、「画分チューブ(Fraction−tube)」を、0.2ml 2M Tris pH8.0と共に予めロードした。
【0133】
【表12】
最後の198/A1 Fab調製物を、50mMイミダゾール、100mM NaCl、pH7.4に対して透析し、そして上記のような色素形成(chromogenic)FVIIIアッセイにおいて分析した(図25)。インタクトな抗体と比較して、この198/A1 Fabフラグメントはある程度低い活性を有していたが;このFabフラグメントは依然としてFIX依存性FXa産生を生じた。図25は、2.3nMのヒトFIXaの存在下での抗体198/A1
Fabフラグメントの色素形成FVIII様活性を実証する。ポジティブコントロールとして、本発明者らはインタクトな抗体198/A1ならびに7.5pMのFVIIIを使用した。198/A1 FabフラグメントまたはFVIIIの代わりに、緩衝液コントロール(IZ)をネガティブコントロールとして使用した。
【0134】
(実施例16:抗FIX/FIXa抗体のおよびE.coliアルカリホスファターゼのscFvフラグメント間の融合タンパク質によって示されるFVIII様活性)
抗体198/B1(サブクローンAB2)の単鎖Fvフラグメント(実施例10を参照のこと)を、pDAP2ベクター系を用いてE.coliアルカリホスファターゼのN末端に融合した(Kerschbaumerら、1996)。2つの同一のクローンを単離し、そしてpDAP2−198AB2#1およびpDAP2−198AB2#100と名付けた(図26)。得られた融合タンパク質をE.coli中で発現し、金属親和性クロマトグラフィー(Kerschbaumerら、1997)によって精製し、そして標準的な色素形成アッセイにおいて分析した(図27)。図27は、2.3nMのヒトFIXaの存在下での2つの抗体198/B1(サブクローンAB2)scFvフラグメント−アルカリホスファターゼ融合タンパク質(198AB2#1および198AB2#100)の色素形成FVIII様活性を実証する。ポジティブコントロールとして、本発明者らは7.5pM FVIIIを使用した。
【0135】
(実施例17:二価のミニ抗体によって示されるFVIII様活性)
二価のミニ抗体を得るために、抗体198/B1(サブクローンAB2)のscFvフラグメントを、pZip1ベクター系を用いて両親媒性らせん構造に融合した(Kerschbaumerら(Analytical Biochemistry 249,219−227,1997))。手短に言うと、198/B1 scFvフラグメントの遺伝子を、SfiIおよびNotIでの消化によってプラスミドpDAP−198AB2#100(実施例16)から単離した。このDNAフラグメントをゲル精製し、そしてSfiI/NotI消化ベクターpZip1に挿入した。得られたプラスミドを配列決定し、そしてpZip−198AB2#102と名付けた(図28)。並行して、本発明者らは、#8860と呼ばれる無関係のモノクローナル抗体からミニ抗体の異形を構築した。最初の段階において、抗体#8860の単鎖Fvフラグメントを、ベクターpDAP2内に構築した。このクローニングを、本質的には実施例10に記載されるように行った。この構築物をpDAP2−8860scFv#11と名付けた(図29)。pDAP2−8860scFv#11内に含まれるscFvフラグメントの、プラスミドpZip1(上記を参照のこと)へのサブクローニングは、ミニ抗体構築物p8860−Zip#1.2を生じた(図30)。抗体#8860はFIX/FIXaとは反応しないので(ウエスタンブロットおよびELISA分析により判断)、これは適切なネガティブコントロールを表す。その後、このミニ抗体タンパク質をE.coliにおいて発現させ、そして以下のプロトコルに従うプロテインLへの結合によって細菌の上清から精製した:
アフィニティークロマトグラフィーについて、本発明者らは、4mlのゲル容量を有するPHARMACIA XK 16/20カラムにおいて、ImmunoPure Immobilized PROTEIN L Plus(Pierce)を使用した。使用した緩衝液は以下のようであった:1)平衡緩衝液:50mM L−ヒスチジン pH7.0,1M NaCl,0.1%(w/v)NaN3;洗浄緩衝液:50mM L−ヒスチジン pH7.0,0.1%(w/v)NaN3;溶出緩衝液:100mMグリシン pH2.5,0.1%(w/v)NaN3;および中和緩衝液:2M Tris/Cl pH8.0。
【0136】
サンプルを以下のように調製した:細菌培養物の上清を、細菌発現培養物の遠心分離(11,000×g、4℃、10分)によって得た。470gの硫酸アンモニウムを1リットルの上清に加え、そしてこの溶液を氷上で1時間攪拌してタンパク質を沈殿させた。この沈殿を、2℃、14,000×gの35分間によってペレット化し、そして100mlの20mM Tris pH7.0に再溶解させた。その後、この濃縮物を20mMのTris pH7.0に対して透析し、50mMの最終濃度までL−ヒスチジンを添加し、そしてpHを7.0に調節した。最後に、固体のNaClを添加し、1Mの最終濃度を得た。カラムにロードする前に、サンプルをまず室温、16,000×gで15分間遠心分離し、次いで0.45μmの滅菌フィルタを通して濾過した。
【0137】
クロマトグラフィーを、表12に記載される以下の工程1〜7によって本質的に行った。溶出緩衝液の低いpHを中和するために、「画分チューブ(Fraction−tube)」を、0.2ml 2M Tris pH8.0と共に予めロードした。
【0138】
【表13】
表12。最後の198/B1(サブクローンAB2)ミニ抗体調製物(198AB−Zip#102と名付けた)およびネガティブコントロールの8860−Zip#1.2を、50mMイミダゾール、100mM NaCl、pH7.4に対して透析し、そして上記のように色素形成FVIIIアッセイにおいて分析した(図31)。
【0139】
図31に見られ得るように、ミニ抗体構築物198AB−Zip#102は、実質的なFXa産生を生じたが(FVIIIと比較して)、一方、ネガティブコントロールミニ抗体8860−Zip#1.2は生じなかった。図31は、2.3nMのヒトFIXaの存在下での、198/B1(サブクローンAB2)ミニ抗体198AB−Zip#102の色素形成FVIII様活性を実証する。ポジティブコントロールとして、本発明者らは4.8pMのFVIIIを使用し、一方、無関係のミニ抗体(8860−Zip#1.2)および単純反応緩衝液(plain reaction buffer)(IZ)はネガティブコントロールとして役立った。
【0140】
(実施例18:抗FIXa/FIX抗体scFvフラグメントによって示されるFVIII様活性)
抗体198/B1(サブクローンAB2)の単鎖Fvフラグメントならびに抗体#8860のscFvフラグメントを、pMycHis6ベクター系を用いて発現させた。ベクターpMycHis6(図32&33)を、ベクターpCOCK(Engelhardtら,1994,Biotechniques,17:44−46)をNotIおよびEcoRIで切断し、そして以下のオリゴヌクレオチドの挿入によって構築した:mychis6−co:5’ggccgcagaacaaaaactcatctcagaagaggatctgaatggggcggcacatcaccatcaccatcactaataag3’(配列番号79)およびmycchis−ic:5’aattcttattagtgatggtgatggtgatgtgccgccccattcagatcctcttctgagatgagtttttgttctgc3’(配列番号80)。図32は、プラスミドpMycHis6の略図を示す。c−myc−タグ配列は、ELISAまたはウエスタンブロット分析においてscFvフラグメントを検出するために使用される(Evanら,Mol.Cell.Biol.,1985,5(12),pp.3610−6)。His6−タグ配列は、金属イオンクロマトグラフィーによるscFフラグメントの精製を容易にするために含まれる(Hochuliら,1988.Biotechnology,6:1321−1325)。このプラスミドは、lacZ遺伝子プロモーター(PlacZ)、PelB−リーダー配列(図26の説明文を参照のこと)、E.coli複製起源(colE1ori)およびM13ファージ複製起源(M13ori)を含む。特異的選択を可能にするために、このプラスミドはまた、抗生物質アンピシリンに対する耐性を媒介する酵素β−ラクタマーゼのための遺伝子(AmpR)を保有する。198/B1(クローンAB2)−scFvの遺伝子を、SfiIおよびNotIで消化して、そしてSfiI/NotI切断されたpMycHis6に挿入することによって、プラスミドpDAP2−198AB2#100(実施例16)からレスキューした。得られたプラスミドをpMycHis−198AB2#102と名付けた。図34は、198AB2 scFvのヌクレオチド配列およびアミノ酸配列(c−myc−タグおよびHis6−タグに連結した)を示す:発現ベクターの得られたORFを、pMycHis6−198AB2#102と名付けた。ベクターpMycHis6を、ベクターpCOCK(Engelhardt O.ら,BioTechniques 17,44−46,1994)NotI−EcoRIを切断し、そして以下のアニールされたオリゴヌクレオチドを挿入することによって構築した:(5’−GGCCGCAGAACAAAAACTCATCTCAGAAGAGGATCTGAATGGGGCGGCACATCACCATCACCATCACTAATAAG−3’(配列番号103)および5’−TTATTAGTGATGGTGATGGTGATGTGCCGCCCCATTCAGATCCTCTTCTGAGATGAGTTTTTGTTCTGC−3’(配列番号104))。得られたベクター(pMycHis6と名付けた)は切断されたSfiI−NotIであり、そしてscFv 198AB2の遺伝子を、ベクターpDAP2−198AB2#100からこのベクターに交換した。198AB2構築物に対する類似性において、#8860scFvフラグメントを、pDAP2−8860scFvクローン11と名付けたプラスミドからクローンした。#8860の純粋なscFvタンパク質を、8860−M/H#4cと名付けた(プラスミドp8860−M/H#4c、図35)。scFvタンパク質をE.coli中で発現させ、そしてプロテインLカラム上の細菌の上清から親和性精製した(実施例17を参照のこと)。最後のMycHis−198AB2#102および8860−M/H#4c調製物を、50mMイミダゾール、100mM NaCl、pH7.4に対して透析し、そして上記のように色素形成FVIIIアッセイにおいて分析した(図36)。
【0141】
図36において見られ得るように、scFv構築物MycHis−198AB2#102は、実質的なFXaの産生を生じるが、一方、ネガティブコントロール8860−M/H#4cおよび単純反応緩衝液(IZ)は生じない。図36は、2.3nMのヒトFIXaの存在下での、198/B1(サブクローンAB2)scFvフラグメント(MycHis−198AB2#102)の色素形成FVIII様活性を実証する。ポジティブコントロールとして、本発明者らは4.8pMのFVIIIを使用し、一方、無関係のscFv(8860−M/H#4c)および単純反応緩衝液(IZ)はネガティブコントロールとして役立った。
【図面の簡単な説明】
【0142】
【図1】図1は、FVIII様活性についての、ハイブリドーマ細胞培養からの上清のスクリーニングの結果を示す。融合実験から予め選択されたクローン(#193、#195および#196)を、色素形成アッセイで試験した。
【図2】図2は、マスタープレートのハイブリドーマ細胞培養における上清のIgG媒介第VIII因子様活性についてのスクリーニングの結果を示す。
【図3】図3は、クローン193/C0のサブクローニング、すなわち、一回目のクローニングの結果を示す。
【図4】図4は、開始クローン193/C0由来のハイブリドーマ培養物の色素形成FVIII様活性および第IX因子−ELISA反応性の比較を示す。
【図5】図5は、いくつかのマスタークローンおよびサブクローンの色素形成活性の測定結果を示す。
【図6A】図6Aは、ヒトFVIII、TBS緩衝液および細胞培養培地と比較した、抗FIX/FIXa抗体193/AD3および196/AF2のFVIII様活性を示す。遅滞期の後に、両方の抗体は、増加する光学密度によって判断されるように、色素形成基質の切断を生じた。
【図6B】図6Bは、第VIII因子、196/AF1、198/AC1/1およびマウスIgGの色素形成活性の比較を示す。
【図7A】図7Aは、第Xa因子特異的インヒビターの添加を伴った、または伴わない、第VIII因子および196/AF2による第Xa因子の生成の速度論の比較を示す。
【図7B】図7Bは、第Xa因子特異的インヒビターであるPefabloc Xa(登録商標)の添加を伴った、または伴わない、第VIII因子、マウスIgG、および抗第IX/IXa因子抗体198/AM1による第Xa因子の生成の速度論の比較を示す。
【図8A】図8Aは、リン脂質、FIXa/FXおよびカルシウムイオンの存在下、および非存在下における、精製された抗第IX/IXa因子抗体198/AC1/1の第VIII因子様活性の依存性の測定を示す。
【図8B】図8Bは、リン脂質、Ca2+およびFIXa/FXの存在下における、抗FIXa抗体196/AF1によるFXa生成の依存性の測定を示す。
【図8C】図8Cは、非特異的マウスIgG抗体によるFXaの生成を示す。
【図9】図9は、種々の濃度の抗第IX/IXa因子抗体193/AD3を用いることによるAPTTアッセイにおける第VIII因子欠乏血漿の凝固時間のグラフ表示である。
【図10A】図10Aは、因子IXaの存在下において、抗体193/AD3が、第VIII因子欠乏血漿の凝固時間を短縮することを示す。
【図10B】図10Bは、第IXa因子インヒビターおよび第VIII因子インヒビターの存在下における、抗体193/AD3による凝固時間の、用量依存性短縮を示す。
【図11】図11は、ヒトFIXaβの存在下、および非存在下における、抗体198/A1、198/B1および198/AP1の色素形成活性を示す。
【図12−1】図12は、マウス抗体の可変重鎖の遺伝子増幅のためのプライマー配列を示す。
【図12−2】図12−1の続き。
【図13−1】図13は、マウス抗体の可変軽(κ)鎖の遺伝子増幅のためのプライマー配列を示す。
【図13−2】図13−1の続き。
【図14−1】図14は、ハイブリドーマ細胞株193/AD3由来のscFvのDNAおよび誘導タンパク質の配列を示す(配列番号81および配列番号82)。
【図14−2】図14−1の続き。
【図15−1】図15は、ハイブリドーマ細胞株193/K2由来のscFvのDNAおよび誘導タンパク質配列を示す(配列番号83および配列番号84)。
【図15−2】図15−1の続き。
【図16−1】図16は、ハイブリドーマ細胞株198/AB2(198/B1のサブクローン)由来のscFvのDNAおよび誘導タンパク質配列を示す(配列番号85および配列番号86)。
【図16−2】図16−1の続き。
【図17−1】図17は、細胞株198/A1由来のscFvのDNAおよび推論されるタンパク質配列を示す(配列番号87および配列番号88)。
【図17−2】図17−1の続き。
【図18】図18は、2.9nMのヒトFIXaの存在下における、ペプチドA1/3の色素形成FVIII様活性を示す。ペプチドA1/3のスクランブルバージョンペプチドA1/5は、いずれのFXa生成も起こさない。
【図19】図19は、ヒトFIXaの存在に対する、ペプチドA1/3の色素形成FVIII様活性の依存性を示す。ヒトFIXaの非存在下において、ペプチドA1/3は、いずれのFXaの生成も起こさない。緩衝液コントロールである単体のイミダゾール緩衝液は、IZと表される。
【図20】図20は、Arg残基のキラリティーが、ペプチドA1/3−rdおよびA1/3−Rd−srmbの色素形成活性に対して有意な役割を果たさないことを示す。
【図21】図21は、反応混合物への2.4μMのペプチドB1/7の添加が、Fxaの測定可能な生成を引き起こしたことを示す。
【図22】図22は、FX特異的インヒビターの添加が、反応に有意な減少を引き起こすことを示す。FIXaが全く存在せず、かつFXが反応混合物に添加される場合、FXaは合成されなかった。
【図23】図23は、ベクターpBax−IgG1を示す。
【図24】図24は、抗体198/B1(図24A)およびIgM抗体198/AF1(図24B)の存在下における、FIXaのアミド分解活性の増加を示す。
【図25】図25は、2.3nMのヒトFIXaの存在下における抗体198/A1 Fabフラグメントの色素形成FVIII様活性を示す。ポジティブコントロールとして、インタクトな抗体198/A1および7.5pMのFVIIIを使用した。緩衝液コントロール(IZ)を、ネガティブコントロールとして用いた。
【図26−1】図26は、198AB2 scFv−アルカリホスファターゼ融合タンパク質のヌクレオチド配列およびアミノ酸配列を示す(発現ベクターpDAP2−198AB2#100のORF、(配列番号89および配列番号90)。抗体198/AB2(198/AB2は、198/B1の同一サブクローンである)のVLおよびVHドメインに対する遺伝子は、実施例10に記載されるように、対応するハイブリドーマ細胞に由来した。VH遺伝子のPCR産物は、消化SfiI−AscIであり、そしてVL遺伝子のPCR産物は消化AscIおよびNotIであった。VH遺伝子およびVL遺伝子は、AscI部位を介して結合し、SfiI−NotI消化ベクターpDAP2に挿入された(Kerschbaumer R.J.ら、Immunotechnology 2、145−150、1996;GeneBank 受託番号:U35316)。PelBリーダー:Erwinia carotovoraペクテートリアーゼBのリーダー配列、Hisタグ、金属イオンクロマトグラフィーのためのヒスチジン(Histidinee)タグ。
【図26−2】図26−1の続き。
【図26−3】図26−2の続き。
【図26−4】図26−3の続き。
【図26−5】図26−4の続き。
【図27】図27は、2.3nMのヒトFIXaの存在下における、2つの抗体198/B1(サブクローンAB2)scFvフラグメントアルカリホスファターゼ融合タンパク質(198AB2#1および198AB2#100)の色素形成FVIII様活性を示す。ポジティブコントロールとして、7.5pMのFVIIIを使用した。
【図28−1】図28は、pZip198AB2#102のアミノ酸およびヌクレオチド配列を示す(配列番号91および配列番号92)。
【図28−2】図28−1の続き。
【図29−1】図29は、mAB#8860scFvアルカリホスファターゼ融合タンパク質(ベクターpDAP2−8860scFv#11)のヌクレオチド配列およびアミノ酸配列を示す(配列番号93および94)。抗体#8860のVLドメインおよびVHドメインに対する遺伝子は、実施例10に記載されるように、対応するハイブリドーマ細胞から誘導された。VH遺伝子のPCR産物は、消化SfiI−AscIであり、そしてVL遺伝子のPCR産物は、消化AscIおよびNotIであった。VH遺伝子およびVL遺伝子は、AscI部位を介して結合し、そしてSfiI−NotI消化ベクターpDAP2に挿入された(Kerschbaumer R.J.ら、Immunotechnology 2、145−150、1996;GeneBank 受託番号:U35316)。
【図29−2】図29−1の続き。
【図29−3】図29−2の続き。
【図29−4】図29−3の続き。
【図29−5】図29−4の続き。
【図30−1】図30は、mAB#8860 scFv−ロイシンジッパー融合タンパク質のヌクレオチド配列およびアミノ酸配列を示す(ミニ抗体(miniantibody);ベクターp8860−Zip#1.2(配列番号95および配列番号96)。scFvフラグメントの遺伝子は、mAB#8860から誘導され、そしてベクターpDAP2−8860scFv#11からSfiI−NotI消化プラスミドpZip1へスワップされた(Kerschbaumer R.J.ら、Analytical Biochemistry 249、219−227、1997;GeneBank 受託番号:U94951)。
【図30−2】図30−1の続き。
【図30−3】図30−2の続き。
【図31】図31は、2.3nMのヒトFIXaの存在下における、198/B1(サブクローンAB2)ミニ抗体198AB−Zip#102のの色素形成FVIII様活性を示す。ポジティブコントロールとして、4.8pMのFVIIIが用いられ、一方、ネガティブコントロールとして、無関係のミニ抗体(8860−Zip#1.2)および単体反応緩衝液(IZ)が利用された。
【図32】図32は、プラスミドpMycHis6の概略図を示す。
【図33】図33は、ベクターpCOCKと異なるプラスミドpMycHis6の一部分のヌクレオチド配列およびアミノ酸配列を示す(配列番号97および配列番号98)。ベクターpMycHis6は、NotIおよびEcoRIによって、ベクターpCOCK(Engelhardtら、1994、Biotechniques、17:44−46)を切断し、そしてオリゴヌクレオチド:mychis6−co:5’ggccgcagaacaaaaactcatctcagaagaggatctgaatggggcggcacatcaccatcaccatcactaataag3’(配列番号79)およびmycchis−ic:5’aattcttattagtgatggtgatggtgatgtgccgccccattcagatcctcttctgagatgagtttttgttctgc(配列番号80)を挿入することによって構築された。
【図34−1】図34は、(c−myc−タグおよびHis6−タグに結合した)198AB2 scFv:発現ベクターpMycHis6−198AB2#102のORFのヌクレオチド配列およびアミノ酸配列を示す。ベクターpMycHis6は、ベクターpCOCK(Engelhardt O.ら、BioTechniques 17、44−46、1994)をNotI−EcoRIで切断し、そして以下のアニーリングしたオリゴヌクレオチド:(5’−GGCCGCAGAACAAAAACTCATCTCAGAAGAGGATCTGAATGGGGCGGCACATCACCATCACCATCACTAATAAG−3’(配列番号103)、および5’−TTATTAGTGATGGTGATGGTGATGTGCCGCCCCATTCAGATCCTCTTCTGAGATGAGTTTTTGTTCTGC−3’(配列番号104))を挿入することによって構築された。その結果得られたベクター(pMycHis6と命名される)は、切断されたSfiI−NotIであり、そしてscFv198AB2の遺伝子は、このベクターへ、pDAP2−198AB2#100ベクターからスワップされた。
【図34−2】図34−1の続き。
【図35−1】図35は、c−myc−タグおよびHis6−タグに結合したmAB#8860scFvのヌクレオチド配列およびアミノ酸配列を示す(ベクターp8860−M/H#4c、配列番号101および配列番号102)。プラスミドpMycHis6は、SfiIおよびNotIによって切断され、そしてscFv 8860#11タンパク質をコードするDNA配列は、pDAP2−8860scFv#11(図29を参照)から挿入され、プラスミドp8860−M/H#4を与えた。
【図35−2】図35−1の続き。
【図36】図36は、2.3nMのヒトFIXaの存在下における、198/B1(サブクローンAB2)scFvフラグメント(MycHis−198AB2#102)の色素形成FVIII様活性を示す。ポジティブコントロールとして、4.8pMのFVIIIを使用し、一方、ネガティブコントロールとして、無関係のscFv(8860−M/H#4c)および単体の反応緩衝液(IZ)が利用された。
【技術分野】
【0001】
本発明は、第IX因子/第IXa因子−抗体および抗体誘導体に関する。
【背景技術】
【0002】
血餅(血栓)は、凝固カスケードとして呼ばれる一連のチモーゲン活性化によって形成される。この酵素的カスケードの過程において、このようなチモーゲン(因子といわれる)の各々の活性化形態は、次のチモーゲンの活性化を触媒する。血栓は、血管壁の表面上の血液成分の蓄積物であり、そして主に、凝集した血小板および不溶性の架橋したフィブリンからなる。フィブリンの形成は、フィブリノーゲンの制限されたタンパク分解によって、トロンビンによってもたらされる。トロンビンは、凝固カスケードの最終生成物である(K.G.Mann,Blood,1990,Vol.76,pp.1−16)。
【0003】
活性化された第IX因子(FIXa)および活性化された第VIII因子(FVIIIa)の複合体による第X因子の活性化は、凝固における重要な工程である。この複合体の成分の非存在またはその機能の妨害は、血友病と呼ばれる血液凝固障害に関連する(J.E.Sadler&E.W.Davie:Hemophilia A,Hemophilia B and von Willebrand’s disease,G.Stamatoyannopoulosら(編):The molecular basis of blood diseases.W.B.Saunders Co.,Philadelphia,1987,pp.576−602)。血友病Aは、第VIII因子活性の(機能的)欠如を示すが、血友病Bは、第IX因子活性の欠如によって特徴付けられる。現在は、血友病Aの処置は、第VIII因子濃縮物の投与による補充療法を介してもたらされる。しかし、約20〜30%の血友病Aの患者は、第VIII因子インヒビター(すなわち、第VIII因子に対する抗体)を発生させ、それによって投与された第VIII因子調製物の効果が阻害される。第VIII因子を抑制する患者の処置は、非常に困難かつ危険性を含み、そして従来はこれらの患者を処置するために限定された数の処置しか存在しなかった。
【0004】
低いFVIIIインヒビターレベルを有する患者の場合において、高用量の第VIII因子をこのような患者に投与して、従って第VIII因子に対する抗体を中和することは、高価であるが可能である。次いで、インヒビター抗体を中和するために必要な量を超える量の第VIII因子は、うっ血作用を有する。多くの場合において、脱感作がもたらされ得、次いでその上に、標準的な第VIII因子処置を再び適用し得る。しかし、大量の第VIII因子を必要とするこのような高用量第VIII因子処置は多大な時間を必要とし、そして重篤なアナフィラキシーの副反応を伴い得る。あるいは、この処置は、ブタの第VIII因子分子を用いて行われ得る。
【0005】
さらなる高コストの方法は、免疫グロブリン(プロテインA、プロテインG)または固定化された第VIII因子に結合するレクチン上の、特別な体の免疫吸着(extra corporeal immunoadsorption)を介して第VIII因子インヒビターを除去する工程を包含する。この処置の間、患者はアフェレーシス器械に連結されなければならないので、この処置はまた、患者への大きな負担となる。この方法においてはまた、急性の出血を処置することはできない。
【0006】
現在、最上の治療は、活性化されたプロトロンビン複合体濃縮物(APCC)(例えば、FEIBA(登録商標)およびAUTOPLEX(登録商標))を投与することであり、これは、高いインヒビター力価を有する患者においてでさえも、急性出血の処置に適切である(DE 31 27 318)。
【0007】
血液凝固の脈管内系において、最後の段階は第X因子の活性化である。この反応は、第VIIIa因子の第IXa因子への結合、ならびに第IXa因子、第VIIIa因子、第X因子およびリン脂質からなる「テナーゼ(tenase)」複合体の形成によって刺激される。FVIIIaの結合なしでは、FIXaは酵素活性を示さないか、またはFXと比較してほんのわずかの酵素活性しか示さない。
【0008】
最近の数年間にわたって、第IXa因子に対する第VIIIa因子の可能な多くの結合部位が特徴付けられ、これらの領域に結合する抗体またはペプチドが、FIXaの活性を阻害することが示されている(Fayら、J.Biol.Chem.,1994,Vol.269,pp.20522−20527,Lentingら、J.Biol.Chem.,1996,Vol.271,pp.1935−1940,Jorqueraら、Circulation,1992,Vol.86,抄録2725)。第IX因子のような凝固因子の阻害はまた、血栓症の形成の予防の目的で、モノクローナル抗体の使用を介して達成される(WO
97/26010)。
【0009】
逆の効果(すなわち、第X因子の第IXa因子媒介活性化における増大)は、第IX因子への第VIII因子ペプチド(アミノ酸698〜712)の結合を介して、Liles D.K.ら、(Blood,1997,Vol.90,補遺1,2054)によって記載された。けれども、この効果は、第VIIIa因子の非存在においてのみ生じ、一方、第VIIIaの存在下では、第X因子の第IXa因子/第VIIIa因子媒介切断は、このペプチドによって阻害される。
【発明の開示】
【発明が解決しようとする課題】
【0010】
(発明の要旨)
血友病患者の処置において生じ得る可能な危険性および副作用の観点から、FVIIIを抑制する患者の有効な処置を可能にする治療の必要性が存在する。そのため、第VIII因子を抑制する患者についての特定の利点を有する、血液凝固障害の処置のための調製物を提供することが本発明の目的である。
【課題を解決するための手段】
【0011】
従って、本発明によって、以下が提供される:
(項目1) 第IX因子/第IXa因子に対する抗体または抗体誘導体であって、FIXaの凝血促進活性を増大させる、抗体または抗体誘導体。
(項目2) 前記抗体または抗体誘導体が、FVIIIインヒビターの存在下でFIXaの凝血促進活性を増大させる、項目1に記載の抗体または抗体誘導体。
(項目3) 前記抗体が、IgG抗体、IgM抗体、IgA抗体およびIgE抗体からなる群から選択される、項目1に記載の抗体。
(項目4) 項目1に記載の抗体または抗体誘導体であって、ここで、該抗体または抗体誘導体は、モノクローナル抗体、抗体フラグメント、キメラ抗体、ヒト化抗体、単鎖抗体、二重特異性抗体、ダイアボディー、およびそれらのダイマー、オリゴマー、またはマルチマーからなる群から選択される、抗体または抗体誘導体。
(項目5) 前記抗体誘導体が、相補的決定領域(CDR)ペプチドを含む、項目1に記載の抗体誘導体。
(項目6) 前記CDRペプチドがCDR3ペプチドである、項目5に記載の抗体誘導体。
(項目7) 項目6に記載の抗体誘導体であって、ここで、前記CDR3ペプチドが、以下:
Tyr−Gly−Asn−Ser−Pro−Lys−Gly−Phe−Ala−Tyr;
Cys−X−X−Tyr−Gly−Asn−Ser−Pro−Lys−Gly−Phe−Ala−Tyr−X−X−Cys;
Tyr−Gly−Asn−Ser−Pro−Lys−Gly−Phe−Ala−Tyr;
Asp−Gly−Gly−His−Gly−Tyr−Gly−Ser−Ser−Phe−Asp−Tyr;および
Phe−Arg−Asn−Arg−Gly−Met−Thr−Ala−Leu−Leu−Lys−Val−Ser−Ser−Cys−Asp、
からなる群から選択されるアミノ酸配列を含み、ここで、
Xは、任意の所望のアミノ酸であり得る、抗体誘導体。
(項目8) 項目1に記載の抗体または抗体誘導体であって、ここで、該抗体または抗体誘導体の可変領域が、図14に記載のアミノ酸1〜357および/またはアミノ酸403〜726を含む、抗体または抗体誘導体。
(項目9) 前記抗体または抗体誘導体が、人工的なリンカー配列をさらに含む、項目8に記載の抗体または抗体誘導体。
(項目10) 項目1に記載の抗体または抗体誘導体であって、ここで、該抗体または抗体誘導体の可変領域が、図15に記載のアミノ酸1〜363および/またはアミノ酸409〜747を含む、抗体または抗体誘導体。
(項目11) 前記抗体または抗体誘導体が、人工的なリンカー配列をさらに含む、項目10に記載の抗体または抗体誘導体。
(項目12) 項目1に記載の抗体または抗体誘導体であって、ここで、該抗体または抗体誘導体の可変領域が、図16に記載のアミノ酸1〜366および/またはアミノ酸412〜747を含む、抗体または抗体誘導体。
(項目13) 前記抗体または抗体誘導体が、人工的なリンカー配列をさらに含む、項目12に記載の抗体または抗体誘導体。
(項目14) 項目1に記載の、第IX因子/第IXa因子に対する抗体または抗体誘導体を発現する、ハイブリドーマ細胞株。
(項目15) 項目14に記載のハイブリドーマ細胞株であって、ここで、該細胞株が、#196/AF1、#196/AF2、#193/AD3、#193/K2−1、#198/AC1/1、#198/AM1、#198/A1、#198/B1、#198/AP1、198/A1、198/B1、198/BB1、198/A1、198/B1、198/BB1からなる群から選択される、ハイブリドーマ細胞株。
(項目16) 項目14に記載のハイブリドーマ細胞株によって発現される、項目1に記載の抗体または抗体誘導体。
(項目17) DNA分子であって、該DNA分子が、項目1に記載の抗体または抗体誘導体をコードする、DNA分子。
(項目18) 項目1に記載の抗体または抗体誘導体および薬学的に受容可能なキャリアを含有する、薬学的調製物。
(項目19) 第IXaα因子および/または第IXaβ因子をさらに含む、項目18に記載の調製物。
(項目20) 血液凝固障害に罹患した患者を処置するための方法であって、薬学的有効量の項目18に記載の調製物を該患者に投与する工程を包含する、方法。
(項目21) 項目20に記載の方法であって、ここで、前記血液凝固障害が、血友病Aおよび出血性素質を含む群から選択される、方法。
(項目22) 血友病を抑制する患者を選択する工程をさらに包含する、項目21に記載の方法。
(項目23) 第IX因子/第IXa因子と相互作用し、そして第IXa因子の凝血促進活性を増大させる抗体または抗体誘導体を得る方法であって、以下の工程:
− FIX、FIXaα、FIXaβまたはそれらのフラグメントからなる群から選択される抗原で、免疫応答性マウスを免疫する工程、
− 免疫されたマウスの脾細胞を単離する工程、
− ハイブリドーマクローンを産生する工程、
− 第IXa因子の凝血促進活性における増大について、ハイブリドーマ細胞の上清をスクリーニングし、第IXa因子の凝固促進活性における増大を示すハイブリドーマ細胞の上清から、該抗体または抗体誘導体を単離および精製する工程、
を包含する、方法。
(項目24) 第IXa因子のアミド分解活性を増大させるための、項目1に記載の抗体または抗体誘導体の使用。
【0012】
本発明に従って、この目的は、第VIIIa因子補因子活性または第IXa因子活性化活性を有し、そして第IXa因子の凝血促進活性における増加を導く、第IX因子/第IXa因子に対する抗体または抗体誘導体の使用を通して達成される。驚いたことに、本発明の第IX因子/第IXa因子−活性化抗体または抗体誘導体の作用は、インヒビター(例えば、第VIII因子/第VIIIa因子に対するインヒビター)の存在によっては反対方向に影響されないが、代わりに、この場合は第IXa因子の凝血促進活性がまた増加される。
【0013】
本発明のさらなる利点は、本発明に従う調製物の投与が、FVIIIを抑制する患者の場合でさえも、第VIII因子または第VIIIa因子の非存在においてでも迅速な血液凝固を可能とすることである。驚いたことに、これらの因子はまた、第VIIIa因子の存在下においても有効である。
【0014】
従って、本発明に従う抗体および抗体誘導体は、FVIII補因子様の活性を有し、これは、2時間のインキュベーション後のFVIIIアッセイ(例えば、COATEST(登録商標)アッセイまたはイムノクロム(Immunochrom)試験)において、少なくとも3のバックグラウンド(基本的ノイズ)対測定値の比を示す。この比の計算は、例えば、2時間のインキュベーションの後に、以下のスキームに従って達成され得る:
【0015】
【数1】
本発明に従う抗体は、好ましくは少なくとも5日、より好ましくは少なくとも10日のインビボ半減期を有するが、少なくとも20日の半減期を有するのがより好ましい。
【0016】
本発明のさらなる局面は、第IX因子/第IXa因子に対する抗体および/または抗体誘導体、ならびに薬学的に受容可能なキャリア物質を含む調製物である。さらに、本発明に従う調製物は、第IX因子および/または第IXa因子をさらに含み得る。
【0017】
本発明のさらなる局面は、第IXa因子のアミド分解(amidolytic)活性を増加するための、この抗体または抗体誘導体の使用である。
【0018】
(抗体および抗体誘導物)
本発明はまた、本発明の抗体および抗体誘導物、発現ベクター、ハイブリドーマ細胞株をコードする核酸、ならびにこれらを産生するための方法を包含する。
【0019】
抗体は、免疫グロブリン分子の合成(または、それぞれ、その免疫原)を誘発する抗原にのみ結合する、あるいはその抗原に大変類似する抗原(または免疫原)にのみ結合する、特異的なアミノ酸配列を有する免疫グロブリン分子である。各免疫グロブリン分子は、2つの型のポリペプチド鎖からなる。各分子は、大きな、同一の重鎖(H鎖)、および2つの軽い、同一でもある鎖(L鎖)からなる。ポリペプチドは、ジスルフィド架橋および非共有結合によって結合する。インビボでは、重鎖および軽鎖は、異なるリボソーム上で形成され、細胞内で組み立てられ、そしてインタクトな免疫グロブリンとして分泌される(Immunology、第2版、1989内のRoitt I.ら)。
【0020】
本発明の抗体、および抗体誘導物、ならびにこれらから誘導された有機化合物は、ヒトおよび動物のモノクローナル抗体またはそれらのフラグメント、一本鎖抗体およびそれらのフラグメントならびにミニ抗体、二重特異的(bispecific)抗体、二重抗体(diabody)、三重抗体(triabody)、またはそれらのダイマー、オリゴマーもしくはマルチマー(multimer)を含む。本発明に従う抗体から誘導されたペプチドミメティックス(peptidomimetics)またはペプチド(例えば、これらは、1つまたはいくつかのCDR領域、好ましくはCDR3領域を含む)もまた含まれる。
【0021】
さらに、構造的活性関連に基づいて、人工的モデリングプロセスによって産生されるヒトモノクローナル抗体およびペプチド配列も含まれる(Greer J.ら、J.Med.Chem.、1994、Vol.37、pp.1035−1054)。
【0022】
用語第IX/IXa因子活性化抗体および抗体誘導物はまた、宿主細胞内における、変更された、免疫グロブリンコード領域の発現によって産生されるタンパク質(例えば、合成抗体、キメラ抗体またはヒト化抗体のような「技術的改変抗体」、またはそれらの混合物、あるいは例えば、Fv、Fab、Fab’またはF(ab)’2などの定常領域を部分的もしくは完全に欠損する抗体フラグメントを含み得る。これらの技術的改変抗体において、例えば、軽鎖および/または重鎖の一部分またはいくらかの部分は、置換され得る。このような分子は、例えば、ヒト化重鎖および未改変軽鎖(またはキメラ軽鎖)からなる抗体を含み得、逆も同様である。用語Fv、Fc、Fd、Fab、Fab’またはF(ab)’2は、先行技術(Harlow E.およびLane D.、「抗体、実験室マニュアル(Antibodies、A Laboratory Manual)」、Cold Spring Harbor Laboratory、1988)に記載されるように用いられる。
【0023】
本発明はまた、モノクローナル抗体(mAb)から誘導されるFabフラグメントまたはF(ab)2フラグメントの使用を含み、これらのフラグメントは、第IX因子/第IXa因子に対して指向され、そして第IXa因子のプロコアギュラント(procoagulant)活性の増加を引き起こす。好ましくは、異種のフレームワーク領域および定常領域は、ヒト免疫グロブリンクラスおよびアイソタイプ(例えば、IgG(サブタイプ1〜4)、IgM、IgAおよびIgE)から選択される。免疫応答の過程において、免疫グロブリンのクラス変更(例えば、IgMからIgGへの変更;ここでは、定常領域が(例えば、μからγへ)変換される)が起こり得る。先行技術(Esser C.およびRadbruch A.、Annu.Rev.Immunol.、1990、Vol.8、pp.717−735)から公知のように、クラス変更はまた、指向された様式で、遺伝子操作方法(「指向されたクラス変更組換え」)によって引き起こされ得る。しかし、本発明に従う抗体および抗体誘導物は、免疫グロブリンタンパク質のヒト配列を特別に含む必要はない。
【0024】
ある特定の実施形態において、ヒト化抗体は、マウスモノクローナル抗体からの補体決定領域(CDR)を含み、この領域は、選択されたヒト抗体配列のフレームワーク領域に挿入される。しかし、ヒトCDR領域もまた使用され得る。好ましくは、ヒト軽鎖および重鎖における可変領域は、1以上のCDR変換によって技術的に変えられる。6つの全てのCDR、または6未満のCDRの種々の組み合わせを使用することもまた可能である。
【0025】
本発明に従うヒト化抗体は、好ましくは、ヒト抗体の構造、またはそのフラグメントの構造を有し、そして治療的適用(例えば、患者(好ましくは、第VIII因子を抑制する患者)における凝固障害の処置)のために必要な特徴の組み合わせを含む。
【0026】
キメラ抗体は、ヒト免疫グロブリンからの重鎖および軽鎖の両方の鎖の定常領域と組み合わせて、非ヒト起原の重鎖および軽鎖のフレームワーク領域を含む、可変領域全体を含むという点で、ヒト化抗体と異なる。例えば、マウス配列およびヒト配列からなるキメラ抗体が産生され得る。本発明に従うと、抗体および抗体誘導物はまた、一本鎖抗体、またはミニ抗体(例えば、プロリンリッチ配列およびオリゴマー化(oligomerisation)ドメインに結合するscFvフラグメント(例えば、Pluckthun A.およびPack P.、Immunotechnology、1997、Vol.3、pp.83−105)、もしくは一本鎖Fv(sFv)(これは、一本のポリペプチド鎖内の抗体結合領域全体を組み込む)であり得る。例えば、一本鎖抗体は、オリゴヌクレオチドにV遺伝子を結合することによって形成され得、このオリゴヌクレオチドは、リンカー配列として構築されており、そして第1のV領域のC末端を第2のV領域のN末端と、例えば、VH−リンカー−VLまたはVL−リンカー−VHの配置で結合し;従って、両方とも(VHおよびVL)は、N末端ドメインを表し得る(Huston JSら、Int.Rev.Immunol.、1993、Vol.10、pp.195−217;Raag R.およびWhitlow M.、FASEB J.、1995、Vol.9、pp.73−80)。リンカー配列として使用され得るタンパク質は、例えば、150Åまで、好ましくは80Åまで、そしてより好ましくは40Åまでの長さを有する。グリシンおよびセリンを含むリンカー配列は、これらの可撓性のために特に好ましく、または、グルタミンおよびリジンを含むリンカー配列は、それぞれ、これらの可溶性のために好ましい。アミノ酸の選択は、免疫原性および安定性の基準に従ってなされ、これらの一本鎖抗体が、生理学的適用または工業的適用(例えば、免疫アフィニティクロマトグラフィー)に適し得るか否かにも依存する。この一本鎖抗体はまた、凝集体として(例えば、トリマー、オリゴマー、またはマルチマーとして)存在し得る。しかし、リンカー配列はまた失われ、そしてVH鎖およびVL鎖の結合が直接起こり得る。
【0027】
二重特異性抗体は、1つの単一分子内に2つの異なった結合特異性を有する、高分子のヘテロ二機能性架橋である。この群には、例えば、二重特異性(bs)IgG、bs IgM−IgA、bs IgA−二量体、bs(Fab’)2、bs(scFv)2、ダイアボディー(diabodies)、およびbs bis Fab Fcが属する(Cao Y.およびSuresh M.R.,Bioconjugate Chem.,1998,第9巻、635−644頁)。
【0028】
ペプチド模倣物によって、天然ペプチド成分の構造を模倣するかまたはその鋳型が隣接したペプチド配列における特異的構造形成を誘導する低分子のタンパク質成分が理解される(Kemp DS,Trends Biotechnol.,1990,249−255頁)。ペプチド模倣物は、例えば、CDR3ドメインに由来し得る。所定のペプチド配列の系統的な変異分析、すなわち、アラニンまたはグルタミン酸走査変異分析によって、凝血促進性活性に重要なペプチド残基の同定を可能にする。特定のペプチド配列の活性を改善するための別の可能性は、高い処理能力のスクリーニングと組み合わせたペプチドライブラリーの使用である。
【0029】
用語抗体および抗体誘導体はまた、構造−活性関係に関連するデータの分析によって獲得される因子を含み得る。これらの化合物はまた、ペプチド模倣物として使用され得る(Grassy G.ら、Nature Biotechnol.1998,第16巻、748−752頁;Greer J.ら、J.Med.Chem.,1994,第37巻、1035−1054頁)。
【0030】
本発明に従って、抗体または抗体誘導体を発現するハイブリドーマ細胞の例としては、ブタペスト条約に従って、番号99090924(#198/A1)、99090925(#198/B1)および99090926(#198/BB1)下で1999年9月9日に、そして、番号99121614(#193/A0)、99121615(#196/C4)、99121616(#198/D1)、99121617(198/T2)、99121618(#198/G2)、99121619(#198/AC1)および99121620(#198/U2)下で1999年12月16日に寄託された。
【0031】
(産生の方法)
本発明の抗体は、先行技術から公知の方法によって調製され得る(例えば、慣例的なハイブリドーマ技術によってかまたはファージディスプレイ遺伝子ライブラリー、免疫グロブリン鎖混合の方法もしくはヒト化技術によって)(Harlow E.およびLane D.,Antibodies,A Laboratory Manual,Cold Spring Harbor Laboratory,1988)。本発明の抗体および抗体誘導体の産生は、例えば、慣例的なハイブリドーマ技術によって行なわれる(Antibodies,A Laboratory Manual,Cold Spring Harbor Laboratory,1988,HarlowおよびLane編、148−242頁)。本発明に従って、ヒトおよび非ヒト種(例えば、ウシ、ブタ、サル、ニワトリおよびげっ歯類(マウス、ラット))はまた、ハイブリドーマ技術のために使用され得る。通常、免疫競合Balb/cマウスまたはFIX欠損マウスは、使用され得る(第IX因子欠損マウスは、ノースキャロライナ大学、Chapel HillのDarrel Stafford博士より分譲され得る)。免疫化は、例えば、第IX因子、第IXaα因子または完全に活性化された第IXaβ因子、またはそのフラグメントで影響され得る。
【0032】
ハイブリドーマ細胞の上清における抗体および抗体誘導体が第IX因子/第IXa因子に結合し、そして、第IXa因子の凝血促進性活性の増加の原因となるという事実を望んで、ハイブリドーマは選択される。凝血促進性活性の増加は、例えば、第VIII因子様活性の測定のための先行技術から公知のアッセイ方法(例えば、色素形成(chromogenic)アッセイ)によって証明され得る。
【0033】
あるいは、本発明の抗体および抗体誘導体はまた、組換え産生方法によって産生され得る。そうする際、本発明に従う抗体のDNA配列は、公知の技術によって決定され得、そして、抗体DNA全体またはその一部は、適切な系で発現され得る。ファージディスプレイ、合成および天然ライブラリー、公知の発現系における抗体タンパク質の発現、またはトランスジェニック動物における発現を含むような組換え産生方法は、使用され得る(Jonesら、Nature,1986、第321巻、522−525頁;Phage Display of Peptides and Proteins,A Laboratory Manual,1996,Kayら編、127−139頁;米国特許第4,873,316号;Vaughan T.J.ら、Nature Biotechnology,1998,535−539頁;Persic Lら、Gene,1997,9−18頁;Ames R.S.ら、J.Immunol.Methods,1995,177−186頁)。
【0034】
組換え的に産生された抗体の発現は、慣例的な発現ベクターの方法(例えば、細菌ベクター(例えば、pBr322およびその誘導体)、pSKFまたは真核生物ベクター(例えば、pMSGおよびSV40ベクター))によって影響され得る。抗体をコードするこれらの配列は、複製、発現および宿主細胞からの分泌を調節する制御配列を提供され得る。これらの制御配列は、プロモーター(例えば、CMVまたはSV40)およびシグナル配列を含む。
【0035】
発現ベクターはまた、ジヒドロ葉酸レダクターゼ遺伝子(DHFR)、ハイグロマイシン−B−ホスホトランスフェラーゼ、チミジンキナーゼなどの選択および増幅マーカーを含み得る。
【0036】
使用されるベクターの成分(例えば、選択マーカー、レプリコン、エンハンサーなど)は、慣例的な方法の手段によって商業的に獲得され得るかまたは調製され得る。このベクターは、例えば、哺乳動物細胞(CHO、COS、線維芽細胞など)、昆虫細胞、酵母または細菌(E.coliなど)についての種々の細胞培養物における発現のために構築され得る。好ましくは、発現タンパク質の最適なグリコシル化を可能にするこれらの細胞が使用される。特に好ましいのは、CHO細胞またはSK−Hepにおいて発現されるベクターpBax(参考、図17)である。
FabフラグメントまたはF(ab)2フラグメントの産生は、先行技術から公知の方法に従って影響され得る(例えば、パパインおよび/またはペプシンなどのタンパク分解性酵素を使用して、mAbを切断することによってかまたは組換え法によって)。これらのFabおよびF(ab)2フラグメントはまた、ファージディスプレイ遺伝子ライブラリーの方法によって調製され得る(Winterら、1994,Ann.Rev.Immunol.,12:433−455)。
【0037】
抗体誘導体はまた、先行技術から公知の方法の手段によって調製され得る(例えば、Grassy G.ら、Nature Biotechnol.,1998,第16巻、748−752、またはGreer J.ら、J.Med.Chem.,第37巻、1035−1054頁、またはRees A.ら、「Pretein Structure Prediction;A practical approach」Sternberg M.J.E.編、IRL出版、1996、第7〜10章、141−261頁からの分子モデリングによって)。
【0038】
本発明の抗体および抗体誘導体の精製はまた、先行技術で記載された方法によって行なわれ得る(例えば、硫酸アンモニウム沈殿、アフィニティー精製(プロテインGセファロース)、イオン交換クロマトグラフィー、またはゲルクロマトグラフィーによって)。本発明の抗体および抗体誘導体が、第IX因子/第IXa因子に結合し、第IXa因子の凝血促進性活性を増加するかまたは第VIII因子様活性を有することを示すための試験方法として、以下の方法は使用され得る:一工程凝血試験(MikaelassonおよびOswaldson,Scand.J.Haematol.,Suppl.,33,79−86頁、1984)または色素形成試験(COATEST VIII:C(登録商標)(Chromogenix)またはImmunochrom(IMMUNO)など)。原則的には、第VIII因子活性を決定するために使用される全ての方法が使用され得る。測定のコントロールブランク値として、例えば、非特定的マウスIgG抗体が使用され得る。
【0039】
本発明の抗体および抗体誘導体は、例えば、血友病Aの場合において、第VIII因子を抑制する患者などのための凝血障害の処置における治療的使用に適している。投与は、患者に治療学的因子を効果的に投与するのに適切な任意の方法によって、影響され得る(例えば、経口投与、皮下投与、筋内投与、静脈内投与または鼻腔内投与によって)。
【0040】
本発明に従う治療学的因子は、薬学的に受容可能なキャリア物質中に活性な因子として十分な量の抗体または抗体誘導体を含む調製物として産生され得る。これらの因子は、液体または粉末形態のいずれかで存在し得る。さらに、本発明に従う調製物はまた、異なった抗体、その誘導体および/またはそれらから誘導された有機化合物の混合物、ならびに、抗体および第IX因子および/または第IXa因子からなる混合物を含み得る。第IXa因子は、第IXaα因子および/または第IXaβ因子として存在し得る。水性キャリア物質の例は、例えば、生理食塩水である。溶液は滅菌性で、滅菌は慣例的な方法によって影響される。
【0041】
本発明に従う抗体または抗体誘導体は、貯蔵のために凍結乾燥形態で存在し得、そして、投与の前に適切な溶媒で懸濁され得る。この方法は、一般的に慣例的な免疫グロブリンについて有利であることが証明され、そして、公知の凍結乾燥および再構成方法は、この場合で適用され得る。
【0042】
さらに、本発明に従う抗体および抗体誘導体はまた、工業的適用に使用され得る(例えば、アフィニティークロマトグラフィーの方法による第IX因子/第IXa因子の精製のためか、または検出方法(例えば、ELISAアッセイ)の成分としてか、または標的タンパク質の機能的ドメインの同定および相互作用のための因子として)。
【0043】
本発明は、以下の実施例および図面によって、より詳細に記載されるが、本発明はそれに対して制限されるべきではない。
【実施例】
【0044】
(実施例1:免疫競合マウスの免疫化および抗FIX/IXa抗体分泌ハイブリドーマ細胞の産生)
1〜3匹の正常免疫競合5〜8週齢Balb/cマウスを個体内(i.p.)経路を介して100μgの抗原(100μl用量)で免疫化した。代表的な実験では、マウスを組換えヒト凝血因子(F)IX(BenefixTM)、ヒト活性化FIXaα(Enzyme Research Laboratories,Lot:FIXaα 1190L)または水酸化アルミニウムもしくはKFAで補助したヒトFIXaβ(Enzyme Research Laboratories,Lot:HFIXaα 1332 AL)のいずれかで接種した。
【0045】
個々のマウスを100μgの抗原(100μl用量、i.p.)で種々の時間にブーストし、そして、2日後に屠殺した。脾臓細胞を取り出し、そして、Laneら、1985(J.Immunol.Methods,第81巻、223−228頁)に基本的に記載されるように、P3 X63−Ag86.5.3骨髄腫細胞に融合させた。それぞれの融合実験を個々に番号付けした(すなわち、#193、195、196または198)。
【0046】
ハイブリドーマ細胞をマクロファージフィーダー層上の96ウェルプレート上で増殖し(約105細胞/ml)そしてHAT培地(抗生物質、10%FCS、ピルビン酸ナトリウム、L−グルタミン、2−メルカプトエタノールおよびHAT(HAT 100×:H2O中の1.0×10-2Mヒポキサンチン(136.1mg/100ml H2O)、H2O中の4.0×10-5Mアミノプテリン(1.76mg/100ml H2O)およびH2O中の1.6×10-3Mチミジン(38.7mg/100ml H2O))で補充したRPMI−1640培地)中で選択した。培地を6日後に最初に交換し、その後1週間に2回交換した。2〜3週間後、HAT培地をHT培地(抗生物質、10%FCS、ピルビン酸ナトリウム、L−グルタミン、2−メルカプトエタノールおよびHTで補充したRpMI−1640)に変更し、その後(さらに1〜2週間後)通常の増殖培地(10%FCS、ピルビン酸ナトリウム、L−グルタミンおよび2−メルカプトエタノールで補充したRPMI−1640培地)(HYBRIDOMA TECHNIQUES、EMBO,SKMB Course 1980,Baselを参照のこと)に変更した。
【0047】
別のセットの実験において、FIX欠損C57B16マウス(Linら、1997、Blood,90:3962)を免疫化および後に続くハイブリドーマ産生のために使用した。FIXノックアウト(k.o.)マウスは、内因性FIXを発現しないので、達成可能な抗(a)−FIX抗体スペクトルは、正常なBalb/cマウスと比較して異なっていることが推定される(耐性の欠如に起因して)。
【0048】
(実施例2:抗FIX/FIXa抗体分泌ハイブリドーマ細胞の上清におけるFVIII様活性のためのアッセイ)
ハイブリドーマ細胞によって分泌される抗FIXa抗体のFVIII様活性をアッセイするために、市販されている試験キットCOATEST VIII:C/4(登録商標)(Chromogenix)を使用した。アッセイは、以下の改変を使用して基本的に製造業者によって記載されるように行なった:
高い処理能力のスクリーニングを可能にするために、アッセイをマイクロタイタープレート型式に小規模化した。簡潔には、ハイブリドーマ上清の25μlのアリコートを、マイクロタイタープレート(Costar,#3598)ウェルに移し、そして、37℃に温めた。色素形成基質(S−2222)、合成トロンビンインヒビター(I−2581)、因子(F)IXaおよびFXを滅菌水中で再構築し、そして、FIXa/FXを、製造業者のプロトコルに従って、リン脂質で混合した。反応液当り、50μlのリン脂質/FIXa/FX溶液を25μlの塩化カルシウム(25mM)および50μlの基質/インヒビター反応混液と組み合わせた。反応を開始するために、125μlのプレミックスをマイクロタイタープレート中のハイブリドーマ上清に添加し、そして、37℃でインキュベートした。サンプルの405nmおよび490nmの吸光度を、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーで、試薬ブランクに対して(MLW、ハイブリドーマ上清の代わりの細胞培養培地)種々の時間(30分〜12時間)測定した。サンプルのFVIII様活性を、GENESISTMソフトウェアを用いて、希釈したFVIII参照規準(IMMUNO AG#5T4AR00)の吸光度に対してサンプルの吸光度を比較することによって計算した。
ハイブリドーマ細胞培養物上清におけるFVIII様活性のスクリーニングの結果を図1に示す。融合実験#193、#195および#196由来の選択される前のクローン(上記を参照のこと)を記載されるように、色素形成FVIIIアッセイで調査した。クローン193/M1、193/N1および193/P1は、マスタークローン193/C0由来のサブクローンであった(以下を参照のこと)。マスタークローン195/10は融合実験#195に由来し、そして、クローン196/A0、196/B0および196/C0は、融合実験#196由来であった。代表的なスクリーニング実験において、単一の融合実験由来の約1000クローン(96ウェル中)は、FVIII様活性についてスクリーニング前と同じであった。続いて、選択されたクローンを大規模に増殖し(3〜5mlの上清)、そして、色素形成アッセイで再分析した。陰性コントロールとして、細胞培養培地をそれぞれのプレート(MLW)上でアッセイした。
【0049】
高いFVIII様活性または有意なFVIII様活性のどちらかを示すウェルをサブクローニング手順に供した。選択およびサブクローニングプロセスは、細胞株(すなわち、193/C0)を産生するIgGのスクリーニングおよびサブクローニングについて例示されるが、クローンを産生するIgM(すなわち、196/C0、以下を参照のこと、図5)について同一の方法を正確に行なった。
【0050】
選択プロセスを、10の96ウェルプレート上の単一融合実験由来の全てのハイブリドーマ細胞クローンを最初に播種することによって行ない、これによって、いわゆる「マスタープレート」を作製した。マスタープレート上の別々の位置(ウェル)は、通常、1つ以上のハイブリドーマ細胞クローン(通常3〜15の異なったクローン)を含んだ。続いて、数千の細胞のみによって分泌された抗体を試験した。これらの細胞を、抗体産生に最適以下の条件下で増殖し、この条件は、瀕死の細胞において最も公知である。それで、上清における予想された特異的な抗FIX抗体濃度は、10-12〜10-14Mの範囲であり得る。このことは、なぜインキュベーション期間が、標準的FVIIIアッセイと比較して延長されなければならないかを説明する。
【0051】
マスタープレートのハイブリドーマ細胞培養物上清におけるIgG媒介性FVIII様活性についてのスクリーニングの結果を、図2に示す。上清を、色素形成FVIIIアッセイにおいて試験した。融合実験番号#193(FIXaαで免疫したBalb/cマウス)の5番目のマスタープレートから誘導される結果を示す。吸光度を、37℃でのインキュベーションの4時間後に読み取った。ES位を、ブランク(MLW)より有意に高いFVIII様活性を示すと同定した。この細胞プールを、193/C0と名付け、そしてさらにサブクローニングした(図3)。マスタープレートの各ウェルは、1つより多いハイブリドーマ細胞クローンを含むので、単一の陽性ウェルの細胞を、2〜0.2細胞/ウェルの計算された細胞密度で、96ウェルプレートにおいて、伸長およびプレートした。再度、その上清を、FVIII様活性について試験し、そして陽性位置を、サブクローニングのさらなるラウンドに供した。代表的に、均一な細胞集団を得るために、FVIII様活性を示す各クローンを使用して、3〜4ラウンドのサブクローニングを実施した。ここで、193/C0サブクローンの色素形成アッセイの結果を示す。吸光度を、37℃での4時間のインキュベーション期間の後に、読み取った。A6位およびD5位は、かなりのFVIII様活性を示し、そしてそれぞれ、193/M1および193/P1と命名した。これら2つのクローンを、さらなるラウンドのサブクローニングに供した。ネガティブコントロールとして、単純細胞培養培地を、各プレートにおいてアッセイした(MLW(H1))。
【0052】
小スケール(3ml)ハイブリドーマ培養物における色素形成FVIII様活性とFIX−ELISA反応性との比較を、図4に示す。マスタークローン(またはサブクローン)がさらにサブクローニングされるべきか否かを決定する前に、クローンを3〜5mlのスケールで増殖し、そしてその上清を、再度チェックした。このグラフは、マスタークローン193/C0、および陽性と同定され、そして再チェックされた全てのそのサブクローンの、FIX特異的ELISAの結果およびFVIII様色素形成活性を示す。本明細書中に記載されるELISAおよび色素形成アッセイの読み取りの場合には、ブランク(色素形成試薬自体の吸光度)を減算した。クローン193/M1をサブクローニングし、そしてクローン193/V2、193/M2および193/U2を得た。193/P1、193/AB2および193/P2由来の第2ラウンドの他のクローンを、サブクローニングした。193/AF3、193/AB3および193/AE3は、193/AB2のサブクローンである。第3ラウンドの他のクローンが、193/P2から生じた。最後に、193/AF3(→193/AF4)、AE3(→193/AE4、193/AL4、193/AN4および193/AO4)ならびに193/AD3(→193/AG4、193/AH4、193/AD4、193/AI4、193/AK4)を、サブクローニングした。
【0053】
各融合実験から、いくつかの(5〜15)マスタークローン(マスタープレートから選択される)を同定し、そしてサブクローニングに供した。3ラウンドのサブクローニングの後に、大部分の細胞株は、ELISAおよび色素形成活性分析(図4を参照のこと)ならびにcDNA配列分析により示されるように、均一であった。特定のマスタークローンおよびその全てのサブクローンは、同じFIX/FIXa結合抗体を産生する。しかし、異なるマスタークローン由来のクローンの抗体タンパク質配列には、大きな差異が存在する(実施例11を参照のこと)。大部分のハイブリドーマ細胞株は、IgGサブクラスから抗体を発現する(すなわち、クローン#193、#198から、198/A1、198/B1、198/BB1のような)。しかし、本発明者らはまた、IgM抗体を発現するいくつかのクローンを、選択し得た。
【0054】
いくつかの重要なマスタークローンおよびサブクローンのハイブリドーマ上清の色素形成活性を、決定した。吸光度を、37℃での1時間30分および3時間30分のインキュベーション期間の後に、測定した(図5)。193番目の融合物由来の全てのクローンとは対照的に、クローン196/C0およびそのサブクローン196/AP2は、FIX/FIXa特異的IgM抗体を産生した。この抗体は、短時間のインキュベーションの後でさえも、強力な色素形成活性を示した。
【0055】
以下の細胞株を、ブダペスト条約に従って、European Collection of Cell Cultures(ECACC)に寄託した:98/B1(ECACC No.99090925);198/A1(ECACC No.99090924);198/BB1(ECACC No.99090926);193/AO(ECACC No.99121614);196/C4(ECACC No.99121615);198/D1(ECACC No.99121616);198/T2(ECACC No.99121617);198/G2(ECACC No.99121618);198/AC1(ECACC No.99121619);および198/U2(ECACC No.99121620)。
【0056】
特定の抗体の生物学的特性の、より深い分析を行うために、FVIII様活性を有する異なる抗体を発現する均一なハイブリドーマ細胞株を伸長し、そして目的の抗体をより大きなスケール(100〜1000ml)で発現するために使用した。これらの抗体をアフィニティー精製し(実施例3を参照のこと)、その後、さらなる実験に使用した。
【0057】
(実施例3:FIX/FIXa活性化活性を示す抗体の第IX因子/FIXa(α,β)結合特性)
第IX因子ならびにFIXの2つの活性化形態(FIXaαおよびFIXaβ)(FIX/FIXa(α,β))を、2g/mlの最終濃度まで、TBS(25mM Tris HCl、150mM NaCl、pH7.5)に希釈した。Nunc Maxisorp ELISAプレートを、標準的な手順(4℃、一晩)に従って、100μlのFIX/FIXa(α,β)溶液でコートし、そしてTBST(TBS、0.1%(v/v)Tween 20)で数回洗浄した。50μlのハイブリドーマ上清を、50μl TBST/2% BSAで1:1に希釈し、コートしたELISAプレートに添加した。室温(RT)で2時間のインキュベーション期間の後に、プレートをTBSTで4回洗浄し、そして100μl/ウェルの抗マウスIgG(Fc特異的)ペルオキシダーゼ結合体化抗体(Sigma,#A−0168)の1:25000希釈物(TBST/1% BSA中)とともにインキュベートした(2時間、RT)。ウェルを、TBSTで5回洗浄し、そして最後に、100μlの新たに調製した染色溶液(100μl OPD(60mg OPD/ml)および10μl 30% H2O2を補充した10mlの50mMクエン酸ナトリウム(pH5))で染色した。この反応を、50mlのH2SO4の添加により停止させ、そしてその光学密度を、492nmおよび620nmにおいて、GENESISTMソフトウェアを使用して、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーで記録した。
【0058】
特定の場合においては、抗マウスIgG ELISAの代わりに、抗マウスIgM ELISAを実施した。
【0059】
(ハイブリドーマ細胞培養物上清からのマウスIgGの精製)
ハイブリドーマ上清(100〜500ml)に、200mM Tris/HCl緩衝液(pH7.0)および固体NaClを補充して、それぞれ20mM Trisおよび3M NaClの最終濃度を得た。次いで、この上清を、5500×gにおける10分間の遠心分離によって、清澄化した。1mlのプロテインGアフィニティークロマトグラフィーカラム(Protein G Sepharose Fast Flow,Amersham−Pharmacia)を、15mlの20mM Tris/Cl(pH7.0)で洗浄し、そしてその後、3M NaClを含む10mlの20mM Tris/Cl緩衝液(pH7.0)で平衡化した。次いで、この3M NaClを含むハイブリドーマ上清を、重力によってこのカラムに装填した。このカラムを、3M NaClを含む15mlの20mM Tris/Cl緩衝液(pH7.0)で洗浄した。結合したIgGを、12mlのグリシン/HCl緩衝液(pH2.8)でさらに溶出し、そして1mlの画分を収集した。中和のために、100μlの1M Tris(pH9.0)を、各画分に添加した。IgGを含む画分を、マイクロプレートのウェル内で、50μlを150μlの染色溶液(BioRad濃縮物、水で1:5に希釈)と混合することによって、同定した。陽性の画分を「プール」し、限外濾過濃縮器デバイス(Centricon Plus 20,Amicon)によって、製造業者に従って、1mlに濃縮した。この濃縮物を、19ml TBS(150mM NaClを含む20mM Tris/Cl緩衝液(pH7.0))で希釈し、そして再度、1mlに濃縮した。IgGをTBSにするために、この希釈−濃縮工程を、さらに2回繰り返した。
【0060】
(ハイブリドーマ細胞上清からのマウスIgMの精製)
100〜500mlのハイブリドーマ細胞培養物上清を、限外濾過濃縮器デバイス(Centricon Plus 20,Amicon)を用いて製造業者に従ってか、または硫酸アンモニウム沈殿(40%飽和、0℃)およびこの沈殿物の5〜10mlのTBSへの再溶解によってかのいずれかで、5〜10mlに濃縮した。いずれの場合においても、この濃縮物を、1.25M NaClを含む20mM Tris Cl緩衝液(pH7.4)に対して透析し、そしてさらに、Centricon Plus 20(Amicon)限外濾過デバイスにおいて1mlに濃縮した。IgMを、ImmunoPure IgM Purification Kit(Pierce)を用いて、製造業者に従って、この濃縮物から精製した。マルトース結合タンパク質カラムからの溶出の間に収集された画分を、IgMに関して試験し、プールし、濃縮し、そしてIgGに関して記載したように、TBSにした。
【0061】
(精製した調製物におけるIgG濃度の決定)
適切な希釈物の、全IgG内容物の280nm消光を、測定した。E280=1.4は、1mg/mlのタンパク質に対応する。
【0062】
(第IXa因子特異的IgG(定量的ELISA))
マイクロプレート(Nunc Maxisorp)のウェルを、TBS(150mM NaClを含む25mM Tris/HCl(pH7.5))に希釈された2μg/mlの第IXa因子とともに、4℃で一晩インキュベートした。ウェルを、TBST(150mM NaClおよび0.1%(v/v)Tween
20を含む25mM Tris/HCl(pH7.5))で4回洗浄した。標準モノクローナルABとして、HIX1抗FIX(精密)を使用した。標準およびサンプルを、2%(w/v)BSAを含むTBST中で希釈した。標準希釈系列およびサンプルの適切な希釈物を、ELISAプレート上で室温で2時間インキュベートした。プレートを、TBSTで4回洗浄し、そして抗マウスIgG(Fc特異的)ペルオキシダーゼ結合体化抗体(Sigma、#A−0168)FIXaの100μl/ウェルの1:25000の希釈物(TBST/1% BSA中)とともにインキュベートした(2時間、RT)。ウェルを、TBSTで5回洗浄し、そして最後に、100μlの新たに調製した染色溶液(100μlのOPD(60mg OPD/ml)および10μlの30% H2O2を補充した10ml 50mMクエン酸ナトリウム(pH5))で染色した。この反応を、50ml H2SO4の添加により停止させ、そして30分後、光学密度を、492nmおよび620nmにおいて、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーによって、GENESISTMソフトウェアを使用して、記録した。
【0063】
(実施例4:色素形成FVIIIアッセイにおいてFVIII様活性を示す抗FIX/FIXa抗体)
いくつかの抗FIX/FIXa抗体産生ハイブリドーマクローンを、4回までサブクローニングし、そして得られるモノクローナルハイブリドーマ細胞株を使用して、モノクローナル抗体含有上清を産生した。これらの上清由来のIgGアイソタイプ抗体を、アフィニティーカラムで精製し、そしてTBSに対して透析した(上記を参照のこと)。IgM抗体を、精製していない上清画分として使用した。以下の実験を、以下の2セットの代表的な抗体を使用して行った:193/AD3および198/AC1/1(IgGアイソタイプ、抗体198/AC1/1は、親198/AC1ハイブリドーマクローンからの調製物であり、すなわち、198/AC1細胞を含む(凍結した)ウイルスが、培養され、そして抗体が産生される。次いで、この上清を、これらの実験のために使用する)ならびに196/AF2および196/AF1(IgMアイソタイプ)(図6Aおよび図6B)。簡単に言えば、モノクローナル抗体を含むサンプル(精製されていないハイブリドーマ上清、または示される場合には、特定の量のFIX特異的抗体)の25μlのアリコートを、マイクロタイタープレートウェルに移し、そして37℃に昇温させた。色素形成基質(S−2222)、合成トロンビンインヒビター(I−2581)、第IXa因子(FIXa)およびFXを、滅菌水中で再形成し、そしてFIXa/FXを、供給者のプロトコルに従って、リン脂質と混合した。1反応あたり、50μlのリン脂質/FIXa/FX溶液を、25μlのCaCl2(25mM)および50μlの基質/インヒビターカクテルと混合した。反応を開始させるために、125μlのプレミックス(premix)を、マイクロタイタープレート中のモノクローナル抗体溶液に添加し、そして37℃でインキュベートした。405nmおよび490nmにおける、サンプルの吸光度を、種々の時点において(5分〜6時間)、試薬ブランク(ハイブリドーマ上清の代わりに細胞培養培地)に対して、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーによって、GENESISTMソフトウェアを使用して、読み取った。
【0064】
ヒトFVIII(12および16mU/ml)、TBSならびに細胞培養培地と比較した、モノクローナル抗体193/AD3(IgGアイソタイプ)および196/AF2(IgMアイソタイプ)により示されるFVIII様活性の時間経過を、図6Aに示す。405nmの波長において測定可能な光学密度の増加により判断する場合に、誘導期の後に、両方の抗体が、色素形成基質切断を生じる。
【0065】
ヒトFVIII(16mU/ml)および10μg/mlのマウスIgGと比較した、モノクローナル抗体198/AC1/1(IgGアイソタイプ、10μg/ml)および196/AF1(IgMアイソタイプ、精製されていない上清)により示されるFVIII様活性の時間経過を、図6Bに示す。405nmの波長において測定可能な光学密度の増加により判断する場合に、誘導期の後に、両方の抗体が、色素形成基質切断を生じる。
【0066】
(実施例5:抗FIX/FIXa−抗体により示されるFVIII様活性は、第Xa因子を産生し、そしてリン脂質、FIXa/FXおよびCa2+に依存性である。)
第VIII因子活性は、通常、色素形成アッセイおよび/またはAPTTに基づく凝固アッセイを用いて、決定される。両方の型のアッセイは、FVIIIa/FIXaにより媒介される第Xa因子産生に依存する。色素形成FVIIIアッセイの場合には、産生される第Xa因子は、引き続いて色素形成基質と反応し、これは、分光学的に(例えば、ELISAリーダーにおいて)モニタリングされ得る。APTTに基づく凝集アッセイにおいては、遊離の第Xa因子が、リン脂質表面におけるFVaと組み合わさって、いわゆるプロトロンビナーゼ複合体となり、そしてプロトロンビンをトロンビンへと活性化させる。トロンビンは、次に、フィブリン産生を生じ、そして最終的に、凝固の形成を生じる。これら2つのアッセイ系の中心は、FVIIIa/FIXa複合体による第Xa因子の産生である。
【0067】
抗FIX/FIXa−抗体により示されるFVIII様活性が実際に第Xa因子を産生することを実証するために、以下の実験を実施した。精製されていないハイブリドーマ上清196/AF2(IgMアイソタイプ)のいくつかの25μlのアリコートを、マイクロタイタープレートウェルに移し、そして37℃まで昇温させた。ポジティブコントロールとして、16mUのRecombinateTMを、ハイブリドーマ培地(196 HM 007/99)中に希釈し、そしてハイブリドーマ上清と全く同じ様式で処理した。ネガティブコントロールとして、純ハイブリドーマ培地を使用した。色素形成基質(S−2222)合成トロンビンインヒビター(I−2581)第IXa因子およびFXを、滅菌水中で再形成し、そしてFIXa/FXを、供給者のプロトコルに従って、リン脂質と混合した。Pefabloc Xa(登録商標)(第Xa因子特異的プロテイナーゼインヒビター)(Pentapharm,LTD)を、水で再形成して、最終濃度を1mM/lとした。1反応あたり50μlのリン脂質/FIXa/FX溶液を、25μlのCaCl2(25mM)および50μlの基質/トロンビンインヒビターカクテルと混合した。反応を開始させるために、125μlのプレミックスを、マイクロタイタープレート中のサンプルに添加し、そして37℃でインキュベートした。示される場合には、35μMのPefabloc Xa(登録商標)を添加した。405nmおよび490nmにおける吸光度を、種々の時点において(5分ごと〜6時間ごと)、試薬ブランク(細胞培養培地)に対して、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーによって、GENESISTMソフトウェアを使用して、読み取った。
【0068】
色素形成基質S−2222の容易に測定可能な切断により判断する場合の、IgM抗FIX/FIXa抗体196/AF2により示されるFVIII様活性による、第Xa因子の産生における第IXa因子刺激の結果(「16mU FVIII」と「196/AF2」とを比較のこと)を、図7Aに示す。第Xa因子活性は、FXa特異的インヒビター「Pefabloc Xa(登録商標)」により効果的にブロックされ(「196/AF2」と「196/AF2 35μM Pefabloc Xa(登録商標)」とを比較のこと)、このことは、実際にFXaが産生されたことを示す。
【0069】
同じ実験を、クローン198/AM1の精製したIgG調製物を使用して、実施した(図7B)。精製されたIgGをTBSで希釈し、最終濃度を0,4mg/mlおよび25μl(すなわち、合計10μg)とし、マイクロタイタープレートウェルに移し、そして37℃に昇温させた。ポジティブコントロールとして、6mUの血漿由来FVIIIを使用した。10μgの非特異的マウスIgG(Sigma,I−5381)は、ネガティブコントロールとして作用した。このアッセイを、上記のように実施した。
【0070】
さらなる実験は、容易に測定可能な色素形成基質S−2222の切断により判断する場合に、IgG抗FIX/FIXa抗体198/AM1により示されるFVIII様活性による第IXa因子の刺激が、第Xa因子を産生することを示す(図7B)。再度、第VIII因子および抗体198/AM1は、FXaを産生し、これは、FXa特異的インヒビター「Pefabloc Xa(登録商標)」によって効果的にブロックされる。ネガティブコントロールとして、非特異的マウスIgG(Sigma,I5381)をアッセイした。
【0071】
別のセットの実験において、精製した抗FIX/FIXa抗体(IgM、図8A)または細胞培養物上清由来の精製していない抗体(IgG、図8B)のいずれかのFVIII様活性の、リン脂質(PL)、FIXa/FXおよびCa2+の存在への依存を、実証した。マウスIgGを、コントロールとして使用した(図8C)。第VIII因子様活性を、本質的に上に記載のように、アッセイした。示される場合には、FIXa/FX混合物、PLまたはCa2+のいずれかを、その反応から省略した。405nmおよび490nmにおける、サンプルの吸光度を、種々の時点において、試薬ブランク(精製された抗体の代わりに緩衝液)に対して、Labsystems iEMS Reader MFTMマイクロタイタープレートリーダーで読み取った。その結果を図8A、図8Bおよび図8Cに示す。
【0072】
精製された抗FIXa抗体198/AC1/1(IgGアイソタイプ、このアッセイを通して使用された濃度は10μg/mlであった)の、リン脂質(PL)、FIXa/FX、およびCa2+の存在下におけるFVIII様活性の依存性をさらに、図8Aに示す。容易に認識可能であるように、競合的アッセイ(抗体、PL、Ca2+およびFIXa/FXを含む)のみが、穏当なFXa生成を引き起こす。リン脂質、FIXa/FX、およびCa2+の存在下における不純IgMアイソタイプ抗FIX/FIXa抗体(196/AF1)を含む細胞培養上清のFVIII様活性の依存性を、図8Bに示す。
【0073】
重ねて、精製されたIgG調製物について既に示されたように(図8A)、抗体198/AC1/1は、競合的アッセイ(PL、Ca2+およびFIXa/FXを含む)のみが、穏当な量のFXa生成を引き起こす。反応の特異性を実証するために、正常なマウス血漿から調製された総IgGを、上記と同じ条件下でアッセイした。この結果を、図8Cに示す。いかなるFVIII様活性も検出され得なかった。予期されたように、リン脂質、FIXa/FX、およびCa2+の不在下において検出可能な活性は存在しない。すべての実験をマイクロタイタープレートにおいて実施し、そしてOD405を、6時間にわたって5分毎に走査した。
【0074】
(実施例6:特定の抗FIX/FIXa抗体は、FIXaの存在下において凝血原である)
正常なホメオーシスの間、FIXはまず、組織因子(TF)/第VIIa因子経路によってか、または後に活性化第XI因子(FIXa)によってかのいずれかで活性化となる。その活性化後、FIXaは、活性化FVIIIとの膜結合複合体において、血小板表面上で会合する。第IXa因子は単独では、FXに対する酵素活性をほとんどまたは全く有さないが、FVIIIaの存在下では、高度に活性となる。特定の抗FIX/FIXa抗体が、FVIII様活性を有し、従って、FVIIIを欠損したヒト血漿における凝血原であることを実証するために、以下の実験を行った。異なる量の抗体193/AD3またはマウスIgG(コントロールとして)を、標準的なaPTTに基づく一段階凝固アッセイにおいて使用した。簡潔には、100μlの抗体含有サンプルを、100μlのFVIII欠損血漿(DP)および100μlのDAPTTIN(活性化されたトロンボプラスチンの時間を決定するためのPTT試薬;IMMUNO AG)試薬と共に、KC10A凝固分析装置においてインキュベートした。示された場合では、総量50ngの活性化FIXを、反応混合物中に含ませた。4分間のインキュベーション後に、100μlのCaCl2(25mM)を添加することによって、反応を開始した。この結果を、表1および図9に示す。
【0075】
【表1】
表1:50ngの活性化FIX(0.01UFIX)の存在下で、種々の量の凝血原(193/AD3)およびコントロール抗体(マウスIgG)を使用するAPTTに基づく凝固アッセイにおけるFVIII欠損血漿の凝固時間。反応および活性化FIXにおける抗体のモル比は、10:1である。抗体と総FIX(FIXおよびFIXa、ヒトFVIII欠損血漿が1U(5μg)のFIXを含むと仮定)との間のモル比は、6:1(反応における9μgの抗体)と1:6(反応における0.23μgの抗体)との間で変動する。凝固時間の最適な短縮化に際して、抗体と総FIXとの間のモル比は、1:1である。FIXaの添加を伴わない凝固時間は、120秒間の範囲である。
【0076】
図9は、50ngの活性化FIXの存在下で、種々の量の凝血原(193/AD3)およびコントロール(マウスIgG)抗体を使用するaPTTに基づく凝固アッセイにおけるFVIII欠損血漿の凝固時間のグラフ表示である。抗体193/AD3を補充したサンプルには、明らかな凝固時間の用量依存的減少が存在する。これらの結果は、抗体193/AD3が、FIXaの存在下において凝血原であることを意味する。
【0077】
(実施例7:抗FIX/FIXa抗体は、FVIIIインヒビターおよびFIXaの存在下において凝血原である)
標準的なFVIII置換療法の重篤な合併症が、FVIIIに対する同種抗体の発達であり、これは、FVIIIの中和へと導き、そして患者の血液が凝固しないという状態に導く。
【0078】
特定の抗FIXa抗体が、FVIIIインヒビターの存在下においてさえ、FVIII様活性を有するということを実証するために、以下の実験を行った。異なる量の抗体193/AD3またはコントロールとしてのマウスIgGを、標準的なAPTTに基づく一段階凝固アッセイにおいて使用した。簡潔には、100μlの抗体サンプルを、100μlのFVIII欠損血漿(図10A)またはFVIIIインヒビター血漿(インヒビター効力400BU/ml)(図10B)のいずれか、および100μlのDAPTTIN試薬と共に、KC10A凝固分析装置においてインキュベートした。さらに、総量50ngの活性化FIXaを、反応混合物に含ませた。4分間のインキュベーション後に、100μlのCaCl2(25mM)を添加することによって、反応を開始した。等価な条件を確実にするために、FVIII欠損血漿およびFVIIIインヒビター血漿を使用する実験を、並置して実施した。この結果を、図10Aおよび10Bに示す。実施例6において既に示したように、FVIIIインヒビターの存在下で抗体193/AD3を補充したサンプルには、明らかな凝固時間の用量依存的減少が存在する。
【0079】
(実施例8:抗FIX/FIXa抗体は、欠損性FVIIIおよびFIXaの存在下において凝血原である)
特定の抗FIXa抗体が、欠損性FVIIIの存在下においてFVIII様活性を有することを実証するために、以下の実験を実施し得る。漸増量の抗体193/AD3、またはコントロールとしてのマウスIgGを、標準的なaPTTに基づく一段階凝固アッセイにおいて使用する。この凝固アッセイでは、欠損性FVIII(DF8)の存在に起因して非常に低い凝固活性を有する血友病Aの患者の血漿を、使用する。簡潔には、100μlの抗体サンプルを、100μlのDF8血漿またはFVIII欠損性血漿のいずれか、および100μlのDAPTTIN試薬と共に、KC10A凝固分析装置においてインキュベートする。さらに、総量50ngの活性化FIXaを、反応混合物に含ませた。短期間のインキュベーション後に、100μlのCaCl2(25mM)を添加することによって、反応を開始した。等価な条件を確実にするために、FVIII欠損血漿およびDF8血漿を使用する実験を、並置して実施する。
【0080】
(実施例9:FIXaの存在下において凝血原活性を有する抗FIX/FIXa抗体は、ヒトFIXaとウシFIXaとの間を識別する)
198回目の融合実験から選択されたFIX/FIXa特異的モノクローナル抗体を、個々のハイブリドーマ上清から精製し、そして実施例3に記載のように定量した。これらの抗体を、改変された一段階凝固アッセイ(実施例6に記載のような)において分析した。そしていくつかが、凝血原活性を示した。
【0081】
これらの抗体調製物の色素形成活性を、以下のFXa生成動力学的アッセイにおいて測定した:10μgのモノクローナル抗体(25μl中)を、マイクロタイタープレートウェルに移し、そして37℃まで加温した。色素形成基質(S−2222)、合成トロンビンインヒビター(I−2581)、第IXa因子、およびFXを、滅菌水中で再構築し、そしてFIXa/FX(両方ともウシ)を、供給業者のプロトコールに従って、リン脂質と混合した。反応につき、50μlのリン脂質/FIXa/FX溶液を、25μlのCaCl2(25mM)および50μlの基質/インヒビター反応混液と組み合わせた。反応を開始するために、125μlのプレミックスを、マイクロタイタープレート中におけるモノクローナル抗体溶液に添加し、そして37℃でインキュベートした。サンプルの405nmおよび490nmでの吸光度を、GENESISTMソフトウェアを使用するLabsystems iEMS Reader MFTMマイクロタイタープレートリーダーにおいて、試薬ブランク(モノクローナル抗体の代わりに、25ml TBS)に対して種々の時点(5分間〜2時間)で読み取った。並行して、反応あたり50ngのヒトFIXaを添加したことを除いて、同じ反応を実施した。凝血原活性を示したこれらの抗体は、ウシFIXの場合では色素形成活性を有さなかったが、ヒトFIXaが存在した場合では、高い活性を示した。
【0082】
図11は、50ngのヒトFIXaβの添加あり(+)および添加なし(−)でモノクローナル抗体198/A1、198/B1、および198/AP1によって示されたFVIII様活性の時間経過を示す。非特異的ポリクローナルマウスIgGを、コントロールとして使用した。198/A1および198/B1は、凝血原活性を示す(実施例6における198/AD3と類似)が、198/AP1は示さない。抗体198/BB1は、同じ活性パターンを有した(データは示さず)。
【0083】
198回目の融合実験から選択された、さらなるモノクローナル抗体としては、198/D1(ECACC番号 99121616)、198/T2(ECACC番号 99121617)、198/G2(ECACC番号 9912118)、198/U2(ECACC番号 99121620)が挙げられる。
【0084】
(実施例10:抗FIX/FIXa抗体から誘導された抗体誘導体の構造および凝血原活性;ハイブリドーマ細胞株193/AD3、193/K2、198/A1および198/B1(クローンAB2)からの抗体可変ドメインのサブクローニング)
クローニング手順:「QickPrep(登録商標)Micro mRNA Purification Kit」(Pharmacia)を製造業者の指示書に従い使用して、メッセンジャーRNAを、個々の細胞株(193/AD3、193/K2、198/A1または198/B1(クローンAB2)のいずれか)の1×106個のハイブリドーマ細胞から調製した。対応するcDNAを、「Ready−To−Go−You−Prime−First−Strand Beads kit」(Pharmacia)を製造業者の指示書に従い使用して、mRNAのレトロ(retro)転写によって生成した。重鎖および軽鎖をコードする配列を、プライマーセットを使用して、対応するcDNAに変換した。重鎖特異的mRNA(VH)を逆転写するために、以下のオリゴヌクレオチドの等モル混合物を使用した(RTmix1):MOCG1−2FOR(5’ CTC AAT TTT CTT GTC CAC CTT GGT GC 3’)(配列番号1)、MOCG3FOR(5’ CTC GAT TCT CTT GAT CAA CTC AGT CT 3’)(配列番号2)および、MOCMFOR(5’ TGG AAT GGG CAC ATG CAG ATC TCT 3’)(配列番号3)。同じ反応チューブにおいて、軽鎖特異的cDNA(VL)を、プライマーMOCKFOR−(5’ CTC ATT CCT GTT GAA GCT CTT GAC 3’)(配列番号4)を使用して合成した。
【0085】
VHについてのコード配列を、図12に示したプライマーセットおよびテンプレートとして上記の逆転写混合物(RTmix1)由来の特異的cDNAを用いるPCRによって増幅した。VK鎖遺伝子を、図13に示したプライマーセットを使用し、そしてまたテンプレートとしてRtmix1を用いて増幅した。VH−PCR産物を、SfiI−AscIで切断し、そしてSfiI−AscIで消化したベクターpDAP2(GeneBank登録番号:U35316)に挿入した。これにより得られたpDAP2−VH構築物を、それぞれ、pDAP2−193AD3/VH、pDAP2−198A1/VH、pDAP2−198AB2/VH(抗体198/B1由来)およびpDAP2−193/K2/VHと命名した。引き続いて、プラスミドをAscI−NotIで切断し、そして対応するAscI−NotI消化されたVK遺伝子のPCR産物を挿入した。結果として生じたベクターをpDAP2−193/AD3scFv、pDAP2−198/AlscFv、pDAP2−198/AB2scFv(抗体198/B1由来)およびpDAP2−193/K2scFvと命名した。これらは、モノクローナル抗体193/AD3、198/A1、198/AB2(抗体198/B1由来)および193/K2のVH遺伝子およびVL遺伝子をコードする。重鎖および軽鎖を、人工の可撓性リンカー(G4SGGRASG4S;Engelhardtら,1994)についてのコード配列に連結し、そして個々の抗体のscFv改変体の発現を可能にする。
【0086】
図14では、ハイブリドーマ細胞株193/AD3由来のscFvのDNAおよび推定タンパク質配列を示す。ヌクレオチド1〜357は、重鎖可変ドメインをコードし、ヌクレオチド358〜402は、人工の可撓性リンカーをコードし、そしてヌクレオチド403〜726は、軽鎖可変領域をコードする。重鎖のCDR3領域のタンパク質配列は、配列YGNSPKGFAY(配列番号5)を有し、そしてボールドの文字で提供される。人工のリンカー配列(G4SGGRASG4S)を示す。
【0087】
図15では、ハイブリドーマ細胞株193/K2由来のscFvのDNAおよび推定タンパク質配列を示す。ヌクレオチド1〜363は、重鎖可変ドメインをコードし、ヌクレオチド364〜408は、人工の可撓性リンカーをコードし、そしてヌクレオチド409〜747は、軽鎖可変領域をコードする。重鎖のCDR3のタンパク質配列は、配列DGGHGYGSSFDY(配列番号6)を有し、そしてボールドの文字で提供される。人工のリンカー配列(G4SGGRASG4S)を示す。
【0088】
図16では、ハイブリドーマ細胞株198/AB2(抗体198/B1由来)由来のscFvのDNAおよび推定タンパク質配列を示す。ヌクレオチド1〜366は、重鎖可変ドメインをコードし、ヌクレオチド367〜411は、人工の可撓性リンカーをコードし、そしてヌクレオチド412〜747は、軽鎖可変領域をコードする。重鎖のCDR3領域のタンパク質配列は、配列EGGGFTVNWYFDV(配列番号7)を有し、そしてボールドの文字で提供される。人工のリンカー配列(G4SGGRASG4S)もまた示す。
【0089】
図17では、ハイブリドーマ細胞株198/A1由来のscFvのDNAおよび推定タンパク質配列を示す。ヌクレオチド1〜366は、重鎖可変ドメインをコードし、ヌクレオチド367〜411は、人工の可撓性リンカーをコードし、そしてヌクレオチド412〜747は、軽鎖可変領域をコードする。重鎖のCDR3領域のタンパク質配列は、配列EGGGYYVNWYFDV(配列番号8)を有し、そしてボールドの文字で提供される。人工のリンカー配列(G4SGGRASG4S)もまた示す。
【0090】
(実施例11:抗FIX/FIXa抗体のCDR3領域由来のペプチドの凝血原活性)
原理上、抗体分子は、三次元空間におけるペプチドエレメントのコンビナトリアルアレイの提示のための生物学的デバイスとして想定され得る(Gaoら,1999,PNAS,96:6025を参照のこと)。従って、抗体(または抗体誘導体(例えば、scFv、Fabなど))を特定の標的タンパク質の機能的に重要なドメインの検出のためのツールとして、または他方で、特定の相互作用を特異的に媒介する(すなわち、生物学的基質FXに対するFIXaの活性を活性化または増強する)アミノ酸配列を描写(delineation)するためのいずれにも使用され得る。後者のプロセスは、FIXa増強剤としての、多数の重鎖CDR3領域(CDR3H)由来ペプチド配列の評価へと導いた。
【0091】
このような活性を示すペプチドの凝血原活性の増強は、FIXa活性の増強を媒介するために重要なペプチド領域における配列改変を通して達成され得る。増強された凝血原活性を有するペプチド配列への可能な工程として、FIXa分子上の抗体の結合部位(すなわち、198/A1または198/B1)を、配列比較分析、競合的結合アッセイ、ウェスタンブロット分析および競合的ELISA分析を使用することによってマッピングする。FIXの結晶構造が既知であるので、引き続いて、分子モデリングを使用して、すなわち、ヒトFIXa上の198/B1結合部位における198/B1由来ペプチドの適合を改善する。
【0092】
他方で、例えば、「アラニンスキャニング変異分析(alanine scanning mutational analysis)」によって、所定のペプチド配列(例えば、198/A1または198/B1 CDR3H由来のペプチド配列)の秩序的(methodical)変異分析によって、凝血原活性に重要なペプチド残基の同定が可能となる。特定のペプチド配列の活性を改善するための別の方法は、高スループットスクリーニングと組み合わせたペプチドライブラリーの使用である。
【0093】
抗体の抗原結合部位は、VL−HLダイマー(またはFv領域)のN末端の近位する6つの「相補的決定領域(complement determining region)(CDR)」に由来する。所定の抗原に対する抗体特異性に対する単一のCDRの寄与は、非常に多様であり得るが、一般に、重鎖のCDR3領域(CDR3H)が特に影響する、すなわち、CDR3H領域の特定のタンパク質配列が抗原認識に非常に重要であり得ると考えられる。CDR3H領域の長さは、非常に多様であることが報告されており、4〜25アミノ酸の範囲である(Borrebaeck、16頁)。
【0094】
ペプチド配列の系統的な変異分析の例を以下に示す。抗FIX/FIXa抗体のCD3領域由来のペプチドの可溶性/凝血促進性(procoagulant)効率を改善するために、N末端およびC末端のアミノ酸配列を変化させた。さらに、一連の変異ペプチドを構築しそして分析した。
【0095】
このような研究の原理を、抗体198/A1および198B/1のCDR3H領域由来の一連のペプチドによって例示する。それぞれ、元のペプチドA1(表2を参照のこと)は、抗体198/A1のCDR3H領域に由来し、そしてペプチドB1は、抗体198B/1のCDR3H領域由来に由来する(実施例10、図16および17を参照のこと)。用語「スクランブルバージョン(scrambled version)」とは、ペプチドが、同じアミノ酸をランダムな順序で有することを意味する。
【0096】
【表2】
(表2)
一連の抗体198/A1由来ペプチドのリスト。ペプチドの長さ(aa.アミノ酸数)、算出した分子量(MW、ダルトン(D))および統計学的等電点(pI)をリストする。D−Argは、Rdと省略する。
【0097】
第1のシリーズの実験において、本発明者らは、元のCDR3HペプチドのC末端Val残基を除去し、そしてN末端およびC末端にいくつかの荷電残基を付加することによって、その元のCDR3Hペプチド配列(A1;EGGGYYVNWYFDV)の可溶性を改善した。得られたペプチドA1/2(酸性のpI)、A1/3(塩基性のpI)およびこれらのそれぞれのスクランブルバージョンA1/4、A1/5およびA1/3scr3は、生理学的pHで種々の緩衝液系において容易に可溶性であった。
【0098】
これらのペプチドのFVIII様(FIXa活性化)活性を分析するために、市販のFVIIIアッセイに基づくアッセイ系を開発した(実施例2および4を参照のこと)。この基本的原理は、FIXaが、補因子なしで、その天然の基質FXに対する非常に限定された活性を有することである。FIXa活性化特性を有する基質(すなわち、FVIIIまたはFVIII様活性を示す基質)の存在下でのみ、実質的な量のFXaが、FIXa/アクチベーター複合体を介するFXの切断によって生成される。生成されたFXaの量を、色素形成性の基質の切断によってモニターする。この改正した色素形成アッセイの原理を、2つの代表的なペプチドA1/3およびA1/5(表2)について記載する。簡単に言うと、ペプチドストック溶液(イミダゾール緩衝液(IZ)(50mM イミダゾール、100mM NaCl、pH7.2)中)の25μlのアリコートを、マイクロタイタープレートウェルに移し、そして37℃に温めた。供給者のプロトコルに従って、色素形成性のFXa基質(S−2222)、合成トロンビンインヒビター(I−2581)、ウシFIXaおよびウシFXを、滅菌水中で再構成し、そしてFIXa/FXを、リン脂質と混合した。これらのペプチドは、ウシFIXaとは反応しないので、(Testキット中のウシFXとの混合物として生じる)2.9nM(多くの場合では、2.3nM)のヒトFIXa(ERL)を添加した(実施例11、図19を参照のこと)。反応あたり、50μlのこのリン脂質/FIXa/FX溶液を、25μlのCaCl2(25mM)および50μlの基質/インヒビター混液と合わせた。反応を開始するために、この125μlのプレミックスを、マイクロタイタープレート中のペプチド溶液に添加し、37℃でインキュベートした。サンプルの405nmおよび490nmの吸光度を、GENESISTMソフトウェアを使用するLabsystems iMES
Reader MFTMマイクロタイタープレートリーダーにおいて、試薬ブランクに対して種々の時点で(5分〜2時間)で読み取った。この実験の結果を、実施例11、図18に示す。ペプチドA1/3は、2.9nMのヒトFIXaの存在下で容易に測定可能なFXa生成を誘導したが、そのスクランブルバージョンA1/5は、不活性であった。さらに、酸性ペプチドA1/2およびそのスクランブルバージョンA1/4およびA1/3−scr3は、匹敵する条件下で試験した場合に、有意な色素形成活性を付与しなかった(データは示さず)。このペプチドA1/3様の親抗体198/A1が、ウシFIXaおよびFXと反応しないことを証明するために、図19に示す実験を行った。ペプチドA1/3を、2.3nMのヒトFIXa(hFIXa)と共に(A1/3(24μM)、+hFIXa)、または伴わずに(A1/3(24μM)、w/o hFIXa)、上記のようにインキュベートした。コントロール実験において、本発明者らは、反応混合液に、2.3nMのhFIXaを補充したそのままの希釈緩衝液(IZ)を添加した。図19に示されるように、反応は、ヒトFIXaの存在下でのみ生じる。
【0099】
図18は、2.9nMのヒトFIXa(hFIXa)の存在下でのペプチドA1/3の色素形成性FVIII様活性を実証する。ペプチドA1/3のスクランブルバージョンであるペプチドA1/5は、FXaの生成を生じない。図19は、ヒトFIXa(hFIXa)の存在に対する、ペプチドA1/3の色素形成性のFVIII様活性の依存性を実証する。ヒトFIXaの非存在下で、ペプチドA1/3は、FXaの生成を生じない。緩衝液コントロールであるそのままのイミダゾール緩衝液をIZと示す。
【0100】
これらのペプチドをまた、FVIII欠乏血漿において凝固時間を減少するその能力について分析した。aPTTベースの1段階の凝固アッセイを、本質的には、記載(実施例6を参照のこと)のように行った。凝固時間(反応が開始してから「凝固」が形成するまでの時間)を、FVIII、緩衝液コントロール(IZ)またはコントロールペプチド(スクランブルバージョン)のいずれかに対して比較した。2つの異なるaPTT試薬(DAPTTINおよびPathromtin SL)を用いて行った、2つの代表的な凝固実験の結果を、表3Aおよび表3Bに示す。
【0101】
【表3】
表3A.2.2nMのヒトFIXaの存在下または非存在下(w/o)のいずれかにおけるFVIII欠乏血漿におけるペプチドA1/3およびA1/3−scr(A1/3のスクランブルバージョン)の凝固活性。2つの独立した代表的な実験(実験1および実験2)を示す。全ての凝固実験を2連で行った。個々の実験についての凝固時間を取り、平均凝固時間を秒(sec.)で表す。表3Aに示す実験は、aPTT試薬DAPTTIN(Baxter Hyland Immuno)を使用して行った。緩衝液コントロール(IZ、イミダゾール緩衝液)に比べて、ペプチドA1/3は、凝固時間における用量依存性の減少を生じた。この凝固時間の減少は、この反応混合液に対する2.2nMの活性化ヒトFIXの添加によってさらにより明らかとなった。ペプチドA1/3のスクランブルバージョンA1/3−scr3は、凝固時間の減少を示さなかった。実際に、2.5μMよりも高い濃度では、このスクランブルペプチドは、阻害性になり、従って、凝固時間を延長した。ペプチドA1/1、A1/2、A1/4およびA1/5は、凝固時間の減少を生じず、これは、これらが、凝血促進活性を欠いていることを示す(データは示さず)。
【0102】
【表4】
表3B.Pathromtin SL(DADE Behring)をaPTT試薬として使用する場合のFVIII欠乏血漿におけるペプチドA1/3の凝固活性。実験を、2.2nMのヒトFIXaの存在下または非存在下(w/o)のいずれかで、2連で行った。個々の実験についての凝固時間を取り、平均凝固時間を秒(sec.)で表す。第VIII因子およびイミダゾール緩衝液(IZ)を、それぞれ、ポジティブコントロールおよびネガティブコントロールとして含んだ。
【0103】
表3Aに示した実験と対照的に、表3Bに示した実験は、aPTT試薬Pathromtin SLを使用して行った。FIXaの存在下で、ペプチドA1/3は、凝固時間の用量依存性の減少を生じたが、FIXaの非存在下では、凝固時間の減少は検出されなかった。
【0104】
別のシリーズの実験において、本発明者らは、ペプチドA1/3の血漿安定性(例えば、タンパク質分解からの保護)を改善することを行った。1つのアプローチは、N末端およびC末端のL−Arg残基のD−Arg残基での置換である(ペプチドA1/3−rdおよびA1/3−Rd−srmbによって例示される)。次いで、ペプチドA1/3−rdおよびA1/3−Rd−srmb(このペプチドA1/3−rdのスクランブルバージョン)を、色素形成性およびaPTTベースの凝固アッセイにおいて分析した。これらの実験は、末端L−Arg残基のD−Arg残基での交換が、色素形成アッセイにおいて測定されたようなFVIII様活性を変化しないことを示し、これは、このArg残基のキラリティーが、色素形成活性において主な役割を果たしていないことを示す(図20)。さらに、aPTTベースの1段階の凝固活性は、幾分減少されたが、なお容易に検出可能であった(表4)。
【0105】
【表5】
表4 ペプチドA1/3、A1/3−RdおよびA1/3−Rd−srmb(配列は、表2を参照のこと)の1段階の凝固活性。IZ(緩衝液コントロール)。
【0106】
図20は、ペプチドA1/3−Rdの変化されない色素形成活性を実証する。12μMの最終濃度のペプチドまたは緩衝液コントロール(IZ)を、2.3nMのヒトFIXa(+)の存在下でインキュベートした。ペプチドA1/3およびA1/3−Rdの色素形成活性は、実質的に変化されないことが示され、そしてこの色素形成アッセイにおいてほぼ同一の結果を生じた。A1/3のスクランブルバージョン、A1/5および緩衝液は、有意なFXa生成を生じなかった。
【0107】
次のシリーズの実験において、本発明者らは、各々の残基のアミノ酸アラニンでの置換による、このペプチドコア配列の任意のアミノ酸の個々の役割を決定することを行った(表5)。
【0108】
【表6】
表5.ペプチドコア配列(E1G2G3G4Y5Y6V7N8W9Y10F11D12)内の任意の単一のアミノ酸の役割を解明するために設計されたペプチドを列挙する。下の枠の番号は、そのペプチド内のアミノ酸の位置を記載する。アラニン(荷電していない小さなアミノ酸)で、各アミノ酸を置換した(「アラニンスキャン」)。ペプチドの長さ(アミノ酸数)、分子量計算値(MW(ダルトン(D))および統計的等電点(pI)もまた列挙される。
【0109】
各ペプチドを、それぞれイミダゾール緩衝液(50mM イミダゾール、100mM NaCl、pH 7.2)に溶解し、続いて凝固緩衝液(50mM イミダゾール、100mM NaCl、1% ヒトアルブミン、pH 7.4)中所望の最終濃度まで希釈した。これらのペプチドをその色素形成活性について、およびそのFVIII欠損血漿における凝固時間を減少する能力について分析した。一段階凝固アッセイを本質的に記載されるように(実施例6を参照のこと)行った。凝固時間(反応の開始から「凝塊」形成までの時間)を、緩衝液コントロールまたはコントロールペプチドのいずれかに対して比較した(スクランブルバージョン)。「アラニンスキャン」のいくつかの結果をペプチドA1/3−2およびA1/3−3について示す。ペプチドA1/3−2で例示される場合のG3−A3の変化は、2.2nMのヒトFIXaの存在下で、高い色素形成活性および一段階凝固時間の強い減少(12.5μMの濃度で34秒)を生じる。ペプチドA1/3−3(G4−A4)は、より高いかまたはより低い濃度での減少した活性と共に、12μM付近の最終濃度での最適の色素形成活性を示す。このペプチドは、FIXaの非存在下でのより高い濃度(12.5μM)での一段階凝固アッセイにおいていくらか抑制性であるが、2.2nMのFIXaの存在下では強く活性になる(31秒、12.5μM)。
【0110】
次の一連の実験において、本発明者らは、各コア残基をアミノ酸であるグルタミン酸(E)で置換することにより、ペプチドコア配列の任意のアミノ酸の個々の役割を決定するように設計した(表6を参照のこと)。
【0111】
【表7】
表6.ペプチドコア配列(E1G2G3G4Y5Y6V7N8W9Y10F11D12)内の任意の単一のアミノ酸の役割を解明するために設計されたペプチドを列挙する。下の枠の番号は、そのペプチド内のアミノ酸の位置を記載する。グルタミン酸(負に荷電した大きなアミノ酸)で、このコア配列の各アミノ酸を置換した(「グルタミン酸スキャン」)。ペプチドの長さ(アミノ酸数)、分子量計算値(MW(ダルトン(D))および統計的等電点(pI)もまた列挙される。
【0112】
各ペプチドを、それぞれイミダゾール緩衝液(50mM イミダゾール、100mM NaCl、pH 7.2)に溶解し、続いて凝固緩衝液(50mM イミダゾール、100mM NaCl、1% ヒトアルブミン、pH 7.4)中所望の最終濃度まで希釈した。一連の「グルタミン酸スキャン」由来のペプチドをその色素形成FVIII様活性について、およびそのFVIII欠損血漿における凝固時間を減少する能力について分析した。一段階凝固アッセイを本質的に記載されるように(実施例6を参照のこと)行った。
【0113】
ペプチドA1/3−24は、いくつかの興味深い特性を示した。この分子は、高色素形成FV−III様活性を6.5μM〜12μMの間の濃度で示したが、より高い濃度(24μMまで)では失活した。このペプチドは、ヒトFIXaの非存在下では凝血促進活性を有していなかったが、2.2nMのhFIXaの存在下では強く活性であった。
【0114】
第2の一連の実験において、本発明者らは、ペプチド配列B1由来の抗体198/B1 CDR3Hの凝血促進活性を改善するように設計した。第1の工程において、本発明者らは、元のペプチド配列(B1;EGGGFTVNWYFDV)の溶解性を、C末端Val残基を除去し、そしていくつかの荷電した残基をこのペプチドのN末端およびC末端に付加することにより改善した。得られたペプチドB1/4、B1/6(酸性 pI)、B1/7(塩基性 pI)およびそのスクランブルされたバージョンB1/5、B1/7scr3は、生理学的pHにおいて種々の緩衝液系に容易に可溶である。
【0115】
【表8】
表7は、一連の抗体198/B1誘導ペプチドのリストである。ペプチドの長さ(aa、アミノ酸数)、分子量計算値(MW、ダルトン(D))および静止等電点(pI))が列挙される。
【0116】
ペプチドB1/4およびB1/5は、50mMTris、100mM NaCl、pH=6.5に可溶であった。両方のペプチドを、色素形成FVIIIアッセイにおいて分析した。ペプチドB1/4は、いくらかの色素形成活性(データは示されず)を有することが見出されたが、スクランブルバージョンB1/5では見いだされなかった。
【0117】
続いて、ペプチドB1/6、B1/7およびB1/7scr3を分析した。各ペプチドを、それぞれ50mM イミダゾール、100mM NaCl、pH 7.2に溶解し、そして続いて凝固緩衝液(50mM イミダゾール、100mM NaCl、1%ヒトアルブミン、pH 7.4)またはイミダゾール緩衝液のいずれか中に所望の最終濃度まで希釈した。これらのペプチドをその色素形成活性およびFVIII欠損血漿における凝固時間を減少する能力について分析した(表8および9)。一段階凝固アッセイを、本質的に記載されるとおりに(実施例6を参照のこと)行った。凝固時間(反応の開始から「凝塊」形成までの時間)を、緩衝液コントロールまたはコントロールペプチドのいずれかに対して比較した(スクランブルバージョン)。
【0118】
ペプチドB1/7由来のFIXa活性化活性(FVIII補因子様活性)を、最初に上記の色素形成アッセイにおいて測定した。
【0119】
図21に示されるように、2.4μMペプチドB1/7の反応混合物への添加は、十分に測定可能なFXaの生成をもたらした。対照的に、35μMのPefablock Xa(FXaプロテアーゼ活性の特異的インヒビタ−)の添加は、色素形成基質切断反応の有意な減少(図22)を生じ、それにより実際にペプチド−FIXa媒介FXa生成をもたらした。FIXaおよびFXの反応混合物への添加がない場合、FXaは合成されなかった(図22)。ペプチドB1/6およびコントロールペプチドB1/5およびB1/7scr3は、活性を示さなかった(データは示さず)。
【0120】
図21は、ペプチドB1/7の色素形成活性を実証する。最終濃度2.4μMでのペプチドまたは緩衝液コントロール(IZ)を2.3nMのヒトFIXaの存在下でインキュベートした。
【0121】
図22において、最終濃度2.4μMでのペプチドB1/7または緩衝液コントロール(IZ)でのペプチドを2.3nMのヒトFIXaの存在下でインキュベートした(「+2.3nM hFIXa」、または「+」のいずれかとして示される)。ペプチドB1/7の色素形成活性は、FIXaおよびFXの存在に依存することが見出された。なぜならば、FIXaおよびFXが反応から離される場合(w/o FIXa/FX)、反応が全く検出できないからである。ペプチドB1/7が実際にFXa生成を媒介することを立証するために、FXa特異的プロテアーゼインヒビタ−Pefabloc Xaを反応混合物に添加した(35μM Pefabloc Xa)。第2の一連の実験において、ペプチドB1/6、B1/7およびB1/7scr3の凝血促進効果を、aPTTベースの1工程凝固アッセイにおいて試験した。これらの実験を本質的に実施例6に記載の通りに行った。結果を表8および9に示す。
【0122】
【表9】
表8:FVIII欠損血漿を、ペプチドB1/6、B1/7src3またはB1/7のいずれかと共に活性化ヒトFIXの非存在下でインキュベートした。ネガティブコントロールとして、純粋な緩衝液を欠損血漿に添加した。種々の組合せについての凝固時間を示す。これらの条件下で、その最高濃度(12.5μM)におけるペプチドB1/7は、157秒という延長した凝固時間により示されるように、凝固プロセスに対して抑制性になる。
【0123】
【表10】
表9:FVIII欠損血漿を、ペプチドB1/6、B1/7scr3またはB1/7のいずれかと共に、活性化ヒトFIXの存在下でインキュベートした。ネガティブコントロールとして、純粋な緩衝液をこの欠損血漿に添加した。凝固時間を種々の組合せについて示す。FIXaの存在下において、ペプチドB1/7は、減少した凝固時間(スクランブルペプチドについて102秒、および緩衝液コントロールについて100秒と比較して83秒)により示されるように凝血促進性になる。
【0124】
(実施例12:FVIIIインヒビタ−血漿における抗FIX/FIXa抗体のCDR3領域から得られるペプチド誘導体の凝血促進活性)
FVIIIインヒビター血漿におけるペプチドA1/3の凝血促進活性についてアッセイするために、以下の実験を実行した。本発明者らは、一段階凝固アッセイをベースとする標準aPTTを行ったが、FVIII欠損血漿の代わりに、本発明者らはFVIIIインヒビター血漿を採用した。血漿の阻害性能力は、1mlあたり8.1Bethesdaユニットであった。
【0125】
【表11】
表10:種々の量のペプチドA1/3(12.5μM〜1.25μM)をFVIIIインヒビター血漿に(2.2nMのFIXaの存在下(FIXa)または非存在下(w/o FIXa)のいずれかで)添加した。ネガティブコントロールとして、純粋な緩衝液を血漿に添加した(IZ)。実験を2回行い、そして平均(aver.)を算出した。凝固時間(秒)を種々の組合せについて示す。ペプチドA1/3が、(用量依存性の様式で)FVIIIインヒビタ−血漿の凝固時間を、FIXaの存在下で、たとえ非常に少ない程度でも、またFIXaの非存在下でも減少させることは容易に理解できる。
【0126】
(実施例13:196/C4 IgMのIgG1への変換)
いくつかのIgM抗体は高FVIII様活性を色素形成アッセイにおいて示すので、このようなIgM抗体をIgG抗体に(Fab、F(ab)2、scFvなどのような抗体誘導体もまた産生され得るが)変換させようと試みた。IgM可変領域遺伝子のレスキューが以下に詳細に記載される。発現ベクターpBax−IgG1(図23)を、ベクターpSI(Promega)およびpEF/Bsd(Invitrogen)から複数のクローニング工程を介して最初に構築した。ドナーのBリンパ球を血液から精製し、そして成熟mRNAを「micro−mRNA purification−kit」(Pharmacia)を使用してこれらの細胞から精製した。ヒトκ鎖およびヒトγ1鎖のcDNAを、「you−primefirst−strand−cDNA−kit」(Pharmacia)を採用して特異的プライマーを使用して調製した。
【0127】
ヒトκ軽鎖定常ドメインのコード配列を、PCRにより特的プライマーを使用してcDNAから増幅する。
【0128】
ヒトγ1鎖定常領域(CH1−ヒンジ−CH2−CH3)の遺伝子を、PCRにより特異的プライマーを使用して増幅する。
【0129】
軽鎖定常ドメインの軽鎖のPCR産物を、XbaIおよびNheIを用いて消化し、そして消化されたpSI中に挿入した。得られたベクターをEcoRIおよびXbaIを用いて切断し、そしてアニーリングされたオリゴヌクレオチドを挿入し、ベクターpSI−Cκを生じた。アニーリングされたオリゴヌクレオチドは、κ鎖可変領域挿入のためのリーダー部位およびSacI−XbaI部位を提供する。ヒトγ1鎖定常領域のPCR産物を、SpeIおよびBamHIを用いて消化し、そして消化されたpSIに挿入する。得られたベクターをSpeIおよびNotIで切断し、そしてアニーリングされたオリゴヌクレオチドを挿入して、ベクターpSI−Cγを生じる。アニーリングされたオリゴヌクレオチドは、重鎖可変領域の挿入のためのリーダー部位およびXhoI−BstEI部位を提供する。ベクターpEF/BsdをNheIおよびSfiIで消化し、Klenow処理により平滑末端にし、そしてpSI−Cκ全体の発現カセット(BglIIおよびBamH1を用いて切除される)を(Klenow処理の後に)挿入する。得られたベクターをEcoRIおよびHindIIIを用いて消化し、そしてKlenow処理する。pSI−Cγの全体の発現カセットをBglIIおよびBamH1で切除し、そして挿入する(Klenow処理後)。得られたベクターをpBax−IgG1と称する。
【0130】
軽鎖可変領域をSacI−XbaI部位の間に挿入し得、κ軽鎖の完全コード配列を得る。重鎖可変領域を、XhoI−BstEI部位の間にクローニングし得、完全IgG1重鎖遺伝子を生じる。両方のオープンリーディングフレームは、SV40プロモーターの制御下で発現され、シグナルペプチドのコード配列を、遺伝子の5’末端において小胞体への重鎖および軽鎖の分泌のために含む。COS細胞へのトランスフェクションは、親IgMと同じ結合特性を有するIgG1の発現を可能にする。プラスミドpBax−196/C4の構築は、特異的プライマーを使用するPCRにより196/C4 scFvのVH(実験10において記載されるようにサブクローニングされる)を増幅することによりさらに達成され得る。PCR産物を、XhoIおよびBstEIIを用いて消化し、そしてXhoIおよびBstEII消化pBax IgG1に挿入する。196/C4 scFvのVLを、特異的プライマーを使用するPCRにより増幅する。PCR産物をSacIおよびXbaIを用いて消化し、そしてSacIおよびXbaI消化pBax IgG1−VHに挿入する。得られたベクター(pBax−196/C4)をエレクトロポレーションによりCOS細胞にトランスフェクトし、そして親IgMと同じ特異性を有するハイブリッドIgG1分子(マウス可変領域およびヒト定常領域)が発現される。
【0131】
(実施例14:抗FIXa抗体によるFIXaアミド分解活性の活性化:)
手短に言うと、20μlの第IXa因子(200mUのFIXa(Stago)を含む)を、200μlの反応緩衝液(50mM TrisHCl pH7.4,100mM NaCl,5mM CaCl2および40%エチレングリコール)、25μlのFIXa基質(CH3SO2−D−CHG−Gly−Arg−pNA,AcOH,10μM/ml,Pentapharm LTD)と共に、種々の量の抗FIX抗体198/B1(IgGアイソタイプ)または196/AF1(IgMアイソタイプ)の存在下または非存在下で、37℃でインキュベートした。FIXa基質の特異的切断を、ELISAリーダーにおいて405nmでモニターした。抗FIX抗体の存在は、FIXaのアミド分解活性を少なくとも2倍増大した。図24は、抗体198/B1(図24A)および抗体198/AF1(図24B)の存在下でのFIXaのアミド分解活性の増加を示す。
【0132】
(実施例15:抗FIX/FIXa−抗体由来のFabフラグメントによって示されるFVIII様活性)
抗FIX/FIXa抗体のFabフラグメントを、標準的プロトコルに従って調製し、そして精製した。手短に言うと、1mlの抗体198/A1(50mMのイミダゾール中に4mg/ml,100mM NaCl,pH7.4)を、87μlの細分緩衝液(1M Naアセテート,10mM EDTA 67.5mg/ml L−システイン)および0.25mgのパパイン(アガロースビーズ上に固定化)と共に一晩37℃でインキュベートした。この調製物を濾過してパパインを除去した。L−ヒスチジンを添加し(最終濃度50mM)、そしてその後pHを7.0に調製した。最後に、固体のNaClを添加して、1Mの最終濃度を得た。引き続いて、198/A1 Fabフラグメントを、プロテインLに結合させることによって精製した:本発明者らは、PHARMACIA XK 16/20カラム(ゲル容量:2ml)中のImmunoPure Immobilized PROTEIN L Plus(Pierce)を使用し、クロマトグラフィーのための緩衝液は以下のようであった:1)平衡緩衝液:50mM L−ヒスチジン pH7.0;1M NaCl;0,1%(w/v)NaN3;2)洗浄緩衝液:50mM L−ヒスチジン pH7.0;0.1%(w/v)NaN3;3)溶出緩衝液:100mM グリシン pH2.5;0.1%(w/v)NaN3;および4)中和緩衝液:2M Tris/Cl pH8,0;
クロマトグラフィーは、本質的に表11に記載される以下の工程1〜7によって行った。溶出緩衝液の低いpHを中和するために、「画分チューブ(Fraction−tube)」を、0.2ml 2M Tris pH8.0と共に予めロードした。
【0133】
【表12】
最後の198/A1 Fab調製物を、50mMイミダゾール、100mM NaCl、pH7.4に対して透析し、そして上記のような色素形成(chromogenic)FVIIIアッセイにおいて分析した(図25)。インタクトな抗体と比較して、この198/A1 Fabフラグメントはある程度低い活性を有していたが;このFabフラグメントは依然としてFIX依存性FXa産生を生じた。図25は、2.3nMのヒトFIXaの存在下での抗体198/A1
Fabフラグメントの色素形成FVIII様活性を実証する。ポジティブコントロールとして、本発明者らはインタクトな抗体198/A1ならびに7.5pMのFVIIIを使用した。198/A1 FabフラグメントまたはFVIIIの代わりに、緩衝液コントロール(IZ)をネガティブコントロールとして使用した。
【0134】
(実施例16:抗FIX/FIXa抗体のおよびE.coliアルカリホスファターゼのscFvフラグメント間の融合タンパク質によって示されるFVIII様活性)
抗体198/B1(サブクローンAB2)の単鎖Fvフラグメント(実施例10を参照のこと)を、pDAP2ベクター系を用いてE.coliアルカリホスファターゼのN末端に融合した(Kerschbaumerら、1996)。2つの同一のクローンを単離し、そしてpDAP2−198AB2#1およびpDAP2−198AB2#100と名付けた(図26)。得られた融合タンパク質をE.coli中で発現し、金属親和性クロマトグラフィー(Kerschbaumerら、1997)によって精製し、そして標準的な色素形成アッセイにおいて分析した(図27)。図27は、2.3nMのヒトFIXaの存在下での2つの抗体198/B1(サブクローンAB2)scFvフラグメント−アルカリホスファターゼ融合タンパク質(198AB2#1および198AB2#100)の色素形成FVIII様活性を実証する。ポジティブコントロールとして、本発明者らは7.5pM FVIIIを使用した。
【0135】
(実施例17:二価のミニ抗体によって示されるFVIII様活性)
二価のミニ抗体を得るために、抗体198/B1(サブクローンAB2)のscFvフラグメントを、pZip1ベクター系を用いて両親媒性らせん構造に融合した(Kerschbaumerら(Analytical Biochemistry 249,219−227,1997))。手短に言うと、198/B1 scFvフラグメントの遺伝子を、SfiIおよびNotIでの消化によってプラスミドpDAP−198AB2#100(実施例16)から単離した。このDNAフラグメントをゲル精製し、そしてSfiI/NotI消化ベクターpZip1に挿入した。得られたプラスミドを配列決定し、そしてpZip−198AB2#102と名付けた(図28)。並行して、本発明者らは、#8860と呼ばれる無関係のモノクローナル抗体からミニ抗体の異形を構築した。最初の段階において、抗体#8860の単鎖Fvフラグメントを、ベクターpDAP2内に構築した。このクローニングを、本質的には実施例10に記載されるように行った。この構築物をpDAP2−8860scFv#11と名付けた(図29)。pDAP2−8860scFv#11内に含まれるscFvフラグメントの、プラスミドpZip1(上記を参照のこと)へのサブクローニングは、ミニ抗体構築物p8860−Zip#1.2を生じた(図30)。抗体#8860はFIX/FIXaとは反応しないので(ウエスタンブロットおよびELISA分析により判断)、これは適切なネガティブコントロールを表す。その後、このミニ抗体タンパク質をE.coliにおいて発現させ、そして以下のプロトコルに従うプロテインLへの結合によって細菌の上清から精製した:
アフィニティークロマトグラフィーについて、本発明者らは、4mlのゲル容量を有するPHARMACIA XK 16/20カラムにおいて、ImmunoPure Immobilized PROTEIN L Plus(Pierce)を使用した。使用した緩衝液は以下のようであった:1)平衡緩衝液:50mM L−ヒスチジン pH7.0,1M NaCl,0.1%(w/v)NaN3;洗浄緩衝液:50mM L−ヒスチジン pH7.0,0.1%(w/v)NaN3;溶出緩衝液:100mMグリシン pH2.5,0.1%(w/v)NaN3;および中和緩衝液:2M Tris/Cl pH8.0。
【0136】
サンプルを以下のように調製した:細菌培養物の上清を、細菌発現培養物の遠心分離(11,000×g、4℃、10分)によって得た。470gの硫酸アンモニウムを1リットルの上清に加え、そしてこの溶液を氷上で1時間攪拌してタンパク質を沈殿させた。この沈殿を、2℃、14,000×gの35分間によってペレット化し、そして100mlの20mM Tris pH7.0に再溶解させた。その後、この濃縮物を20mMのTris pH7.0に対して透析し、50mMの最終濃度までL−ヒスチジンを添加し、そしてpHを7.0に調節した。最後に、固体のNaClを添加し、1Mの最終濃度を得た。カラムにロードする前に、サンプルをまず室温、16,000×gで15分間遠心分離し、次いで0.45μmの滅菌フィルタを通して濾過した。
【0137】
クロマトグラフィーを、表12に記載される以下の工程1〜7によって本質的に行った。溶出緩衝液の低いpHを中和するために、「画分チューブ(Fraction−tube)」を、0.2ml 2M Tris pH8.0と共に予めロードした。
【0138】
【表13】
表12。最後の198/B1(サブクローンAB2)ミニ抗体調製物(198AB−Zip#102と名付けた)およびネガティブコントロールの8860−Zip#1.2を、50mMイミダゾール、100mM NaCl、pH7.4に対して透析し、そして上記のように色素形成FVIIIアッセイにおいて分析した(図31)。
【0139】
図31に見られ得るように、ミニ抗体構築物198AB−Zip#102は、実質的なFXa産生を生じたが(FVIIIと比較して)、一方、ネガティブコントロールミニ抗体8860−Zip#1.2は生じなかった。図31は、2.3nMのヒトFIXaの存在下での、198/B1(サブクローンAB2)ミニ抗体198AB−Zip#102の色素形成FVIII様活性を実証する。ポジティブコントロールとして、本発明者らは4.8pMのFVIIIを使用し、一方、無関係のミニ抗体(8860−Zip#1.2)および単純反応緩衝液(plain reaction buffer)(IZ)はネガティブコントロールとして役立った。
【0140】
(実施例18:抗FIXa/FIX抗体scFvフラグメントによって示されるFVIII様活性)
抗体198/B1(サブクローンAB2)の単鎖Fvフラグメントならびに抗体#8860のscFvフラグメントを、pMycHis6ベクター系を用いて発現させた。ベクターpMycHis6(図32&33)を、ベクターpCOCK(Engelhardtら,1994,Biotechniques,17:44−46)をNotIおよびEcoRIで切断し、そして以下のオリゴヌクレオチドの挿入によって構築した:mychis6−co:5’ggccgcagaacaaaaactcatctcagaagaggatctgaatggggcggcacatcaccatcaccatcactaataag3’(配列番号79)およびmycchis−ic:5’aattcttattagtgatggtgatggtgatgtgccgccccattcagatcctcttctgagatgagtttttgttctgc3’(配列番号80)。図32は、プラスミドpMycHis6の略図を示す。c−myc−タグ配列は、ELISAまたはウエスタンブロット分析においてscFvフラグメントを検出するために使用される(Evanら,Mol.Cell.Biol.,1985,5(12),pp.3610−6)。His6−タグ配列は、金属イオンクロマトグラフィーによるscFフラグメントの精製を容易にするために含まれる(Hochuliら,1988.Biotechnology,6:1321−1325)。このプラスミドは、lacZ遺伝子プロモーター(PlacZ)、PelB−リーダー配列(図26の説明文を参照のこと)、E.coli複製起源(colE1ori)およびM13ファージ複製起源(M13ori)を含む。特異的選択を可能にするために、このプラスミドはまた、抗生物質アンピシリンに対する耐性を媒介する酵素β−ラクタマーゼのための遺伝子(AmpR)を保有する。198/B1(クローンAB2)−scFvの遺伝子を、SfiIおよびNotIで消化して、そしてSfiI/NotI切断されたpMycHis6に挿入することによって、プラスミドpDAP2−198AB2#100(実施例16)からレスキューした。得られたプラスミドをpMycHis−198AB2#102と名付けた。図34は、198AB2 scFvのヌクレオチド配列およびアミノ酸配列(c−myc−タグおよびHis6−タグに連結した)を示す:発現ベクターの得られたORFを、pMycHis6−198AB2#102と名付けた。ベクターpMycHis6を、ベクターpCOCK(Engelhardt O.ら,BioTechniques 17,44−46,1994)NotI−EcoRIを切断し、そして以下のアニールされたオリゴヌクレオチドを挿入することによって構築した:(5’−GGCCGCAGAACAAAAACTCATCTCAGAAGAGGATCTGAATGGGGCGGCACATCACCATCACCATCACTAATAAG−3’(配列番号103)および5’−TTATTAGTGATGGTGATGGTGATGTGCCGCCCCATTCAGATCCTCTTCTGAGATGAGTTTTTGTTCTGC−3’(配列番号104))。得られたベクター(pMycHis6と名付けた)は切断されたSfiI−NotIであり、そしてscFv 198AB2の遺伝子を、ベクターpDAP2−198AB2#100からこのベクターに交換した。198AB2構築物に対する類似性において、#8860scFvフラグメントを、pDAP2−8860scFvクローン11と名付けたプラスミドからクローンした。#8860の純粋なscFvタンパク質を、8860−M/H#4cと名付けた(プラスミドp8860−M/H#4c、図35)。scFvタンパク質をE.coli中で発現させ、そしてプロテインLカラム上の細菌の上清から親和性精製した(実施例17を参照のこと)。最後のMycHis−198AB2#102および8860−M/H#4c調製物を、50mMイミダゾール、100mM NaCl、pH7.4に対して透析し、そして上記のように色素形成FVIIIアッセイにおいて分析した(図36)。
【0141】
図36において見られ得るように、scFv構築物MycHis−198AB2#102は、実質的なFXaの産生を生じるが、一方、ネガティブコントロール8860−M/H#4cおよび単純反応緩衝液(IZ)は生じない。図36は、2.3nMのヒトFIXaの存在下での、198/B1(サブクローンAB2)scFvフラグメント(MycHis−198AB2#102)の色素形成FVIII様活性を実証する。ポジティブコントロールとして、本発明者らは4.8pMのFVIIIを使用し、一方、無関係のscFv(8860−M/H#4c)および単純反応緩衝液(IZ)はネガティブコントロールとして役立った。
【図面の簡単な説明】
【0142】
【図1】図1は、FVIII様活性についての、ハイブリドーマ細胞培養からの上清のスクリーニングの結果を示す。融合実験から予め選択されたクローン(#193、#195および#196)を、色素形成アッセイで試験した。
【図2】図2は、マスタープレートのハイブリドーマ細胞培養における上清のIgG媒介第VIII因子様活性についてのスクリーニングの結果を示す。
【図3】図3は、クローン193/C0のサブクローニング、すなわち、一回目のクローニングの結果を示す。
【図4】図4は、開始クローン193/C0由来のハイブリドーマ培養物の色素形成FVIII様活性および第IX因子−ELISA反応性の比較を示す。
【図5】図5は、いくつかのマスタークローンおよびサブクローンの色素形成活性の測定結果を示す。
【図6A】図6Aは、ヒトFVIII、TBS緩衝液および細胞培養培地と比較した、抗FIX/FIXa抗体193/AD3および196/AF2のFVIII様活性を示す。遅滞期の後に、両方の抗体は、増加する光学密度によって判断されるように、色素形成基質の切断を生じた。
【図6B】図6Bは、第VIII因子、196/AF1、198/AC1/1およびマウスIgGの色素形成活性の比較を示す。
【図7A】図7Aは、第Xa因子特異的インヒビターの添加を伴った、または伴わない、第VIII因子および196/AF2による第Xa因子の生成の速度論の比較を示す。
【図7B】図7Bは、第Xa因子特異的インヒビターであるPefabloc Xa(登録商標)の添加を伴った、または伴わない、第VIII因子、マウスIgG、および抗第IX/IXa因子抗体198/AM1による第Xa因子の生成の速度論の比較を示す。
【図8A】図8Aは、リン脂質、FIXa/FXおよびカルシウムイオンの存在下、および非存在下における、精製された抗第IX/IXa因子抗体198/AC1/1の第VIII因子様活性の依存性の測定を示す。
【図8B】図8Bは、リン脂質、Ca2+およびFIXa/FXの存在下における、抗FIXa抗体196/AF1によるFXa生成の依存性の測定を示す。
【図8C】図8Cは、非特異的マウスIgG抗体によるFXaの生成を示す。
【図9】図9は、種々の濃度の抗第IX/IXa因子抗体193/AD3を用いることによるAPTTアッセイにおける第VIII因子欠乏血漿の凝固時間のグラフ表示である。
【図10A】図10Aは、因子IXaの存在下において、抗体193/AD3が、第VIII因子欠乏血漿の凝固時間を短縮することを示す。
【図10B】図10Bは、第IXa因子インヒビターおよび第VIII因子インヒビターの存在下における、抗体193/AD3による凝固時間の、用量依存性短縮を示す。
【図11】図11は、ヒトFIXaβの存在下、および非存在下における、抗体198/A1、198/B1および198/AP1の色素形成活性を示す。
【図12−1】図12は、マウス抗体の可変重鎖の遺伝子増幅のためのプライマー配列を示す。
【図12−2】図12−1の続き。
【図13−1】図13は、マウス抗体の可変軽(κ)鎖の遺伝子増幅のためのプライマー配列を示す。
【図13−2】図13−1の続き。
【図14−1】図14は、ハイブリドーマ細胞株193/AD3由来のscFvのDNAおよび誘導タンパク質の配列を示す(配列番号81および配列番号82)。
【図14−2】図14−1の続き。
【図15−1】図15は、ハイブリドーマ細胞株193/K2由来のscFvのDNAおよび誘導タンパク質配列を示す(配列番号83および配列番号84)。
【図15−2】図15−1の続き。
【図16−1】図16は、ハイブリドーマ細胞株198/AB2(198/B1のサブクローン)由来のscFvのDNAおよび誘導タンパク質配列を示す(配列番号85および配列番号86)。
【図16−2】図16−1の続き。
【図17−1】図17は、細胞株198/A1由来のscFvのDNAおよび推論されるタンパク質配列を示す(配列番号87および配列番号88)。
【図17−2】図17−1の続き。
【図18】図18は、2.9nMのヒトFIXaの存在下における、ペプチドA1/3の色素形成FVIII様活性を示す。ペプチドA1/3のスクランブルバージョンペプチドA1/5は、いずれのFXa生成も起こさない。
【図19】図19は、ヒトFIXaの存在に対する、ペプチドA1/3の色素形成FVIII様活性の依存性を示す。ヒトFIXaの非存在下において、ペプチドA1/3は、いずれのFXaの生成も起こさない。緩衝液コントロールである単体のイミダゾール緩衝液は、IZと表される。
【図20】図20は、Arg残基のキラリティーが、ペプチドA1/3−rdおよびA1/3−Rd−srmbの色素形成活性に対して有意な役割を果たさないことを示す。
【図21】図21は、反応混合物への2.4μMのペプチドB1/7の添加が、Fxaの測定可能な生成を引き起こしたことを示す。
【図22】図22は、FX特異的インヒビターの添加が、反応に有意な減少を引き起こすことを示す。FIXaが全く存在せず、かつFXが反応混合物に添加される場合、FXaは合成されなかった。
【図23】図23は、ベクターpBax−IgG1を示す。
【図24】図24は、抗体198/B1(図24A)およびIgM抗体198/AF1(図24B)の存在下における、FIXaのアミド分解活性の増加を示す。
【図25】図25は、2.3nMのヒトFIXaの存在下における抗体198/A1 Fabフラグメントの色素形成FVIII様活性を示す。ポジティブコントロールとして、インタクトな抗体198/A1および7.5pMのFVIIIを使用した。緩衝液コントロール(IZ)を、ネガティブコントロールとして用いた。
【図26−1】図26は、198AB2 scFv−アルカリホスファターゼ融合タンパク質のヌクレオチド配列およびアミノ酸配列を示す(発現ベクターpDAP2−198AB2#100のORF、(配列番号89および配列番号90)。抗体198/AB2(198/AB2は、198/B1の同一サブクローンである)のVLおよびVHドメインに対する遺伝子は、実施例10に記載されるように、対応するハイブリドーマ細胞に由来した。VH遺伝子のPCR産物は、消化SfiI−AscIであり、そしてVL遺伝子のPCR産物は消化AscIおよびNotIであった。VH遺伝子およびVL遺伝子は、AscI部位を介して結合し、SfiI−NotI消化ベクターpDAP2に挿入された(Kerschbaumer R.J.ら、Immunotechnology 2、145−150、1996;GeneBank 受託番号:U35316)。PelBリーダー:Erwinia carotovoraペクテートリアーゼBのリーダー配列、Hisタグ、金属イオンクロマトグラフィーのためのヒスチジン(Histidinee)タグ。
【図26−2】図26−1の続き。
【図26−3】図26−2の続き。
【図26−4】図26−3の続き。
【図26−5】図26−4の続き。
【図27】図27は、2.3nMのヒトFIXaの存在下における、2つの抗体198/B1(サブクローンAB2)scFvフラグメントアルカリホスファターゼ融合タンパク質(198AB2#1および198AB2#100)の色素形成FVIII様活性を示す。ポジティブコントロールとして、7.5pMのFVIIIを使用した。
【図28−1】図28は、pZip198AB2#102のアミノ酸およびヌクレオチド配列を示す(配列番号91および配列番号92)。
【図28−2】図28−1の続き。
【図29−1】図29は、mAB#8860scFvアルカリホスファターゼ融合タンパク質(ベクターpDAP2−8860scFv#11)のヌクレオチド配列およびアミノ酸配列を示す(配列番号93および94)。抗体#8860のVLドメインおよびVHドメインに対する遺伝子は、実施例10に記載されるように、対応するハイブリドーマ細胞から誘導された。VH遺伝子のPCR産物は、消化SfiI−AscIであり、そしてVL遺伝子のPCR産物は、消化AscIおよびNotIであった。VH遺伝子およびVL遺伝子は、AscI部位を介して結合し、そしてSfiI−NotI消化ベクターpDAP2に挿入された(Kerschbaumer R.J.ら、Immunotechnology 2、145−150、1996;GeneBank 受託番号:U35316)。
【図29−2】図29−1の続き。
【図29−3】図29−2の続き。
【図29−4】図29−3の続き。
【図29−5】図29−4の続き。
【図30−1】図30は、mAB#8860 scFv−ロイシンジッパー融合タンパク質のヌクレオチド配列およびアミノ酸配列を示す(ミニ抗体(miniantibody);ベクターp8860−Zip#1.2(配列番号95および配列番号96)。scFvフラグメントの遺伝子は、mAB#8860から誘導され、そしてベクターpDAP2−8860scFv#11からSfiI−NotI消化プラスミドpZip1へスワップされた(Kerschbaumer R.J.ら、Analytical Biochemistry 249、219−227、1997;GeneBank 受託番号:U94951)。
【図30−2】図30−1の続き。
【図30−3】図30−2の続き。
【図31】図31は、2.3nMのヒトFIXaの存在下における、198/B1(サブクローンAB2)ミニ抗体198AB−Zip#102のの色素形成FVIII様活性を示す。ポジティブコントロールとして、4.8pMのFVIIIが用いられ、一方、ネガティブコントロールとして、無関係のミニ抗体(8860−Zip#1.2)および単体反応緩衝液(IZ)が利用された。
【図32】図32は、プラスミドpMycHis6の概略図を示す。
【図33】図33は、ベクターpCOCKと異なるプラスミドpMycHis6の一部分のヌクレオチド配列およびアミノ酸配列を示す(配列番号97および配列番号98)。ベクターpMycHis6は、NotIおよびEcoRIによって、ベクターpCOCK(Engelhardtら、1994、Biotechniques、17:44−46)を切断し、そしてオリゴヌクレオチド:mychis6−co:5’ggccgcagaacaaaaactcatctcagaagaggatctgaatggggcggcacatcaccatcaccatcactaataag3’(配列番号79)およびmycchis−ic:5’aattcttattagtgatggtgatggtgatgtgccgccccattcagatcctcttctgagatgagtttttgttctgc(配列番号80)を挿入することによって構築された。
【図34−1】図34は、(c−myc−タグおよびHis6−タグに結合した)198AB2 scFv:発現ベクターpMycHis6−198AB2#102のORFのヌクレオチド配列およびアミノ酸配列を示す。ベクターpMycHis6は、ベクターpCOCK(Engelhardt O.ら、BioTechniques 17、44−46、1994)をNotI−EcoRIで切断し、そして以下のアニーリングしたオリゴヌクレオチド:(5’−GGCCGCAGAACAAAAACTCATCTCAGAAGAGGATCTGAATGGGGCGGCACATCACCATCACCATCACTAATAAG−3’(配列番号103)、および5’−TTATTAGTGATGGTGATGGTGATGTGCCGCCCCATTCAGATCCTCTTCTGAGATGAGTTTTTGTTCTGC−3’(配列番号104))を挿入することによって構築された。その結果得られたベクター(pMycHis6と命名される)は、切断されたSfiI−NotIであり、そしてscFv198AB2の遺伝子は、このベクターへ、pDAP2−198AB2#100ベクターからスワップされた。
【図34−2】図34−1の続き。
【図35−1】図35は、c−myc−タグおよびHis6−タグに結合したmAB#8860scFvのヌクレオチド配列およびアミノ酸配列を示す(ベクターp8860−M/H#4c、配列番号101および配列番号102)。プラスミドpMycHis6は、SfiIおよびNotIによって切断され、そしてscFv 8860#11タンパク質をコードするDNA配列は、pDAP2−8860scFv#11(図29を参照)から挿入され、プラスミドp8860−M/H#4を与えた。
【図35−2】図35−1の続き。
【図36】図36は、2.3nMのヒトFIXaの存在下における、198/B1(サブクローンAB2)scFvフラグメント(MycHis−198AB2#102)の色素形成FVIII様活性を示す。ポジティブコントロールとして、4.8pMのFVIIIを使用し、一方、ネガティブコントロールとして、無関係のscFv(8860−M/H#4c)および単体の反応緩衝液(IZ)が利用された。
【特許請求の範囲】
【請求項1】
第IX因子または第IXa因子に対する抗体または抗体誘導体であって、凝血促進活性を増大させる、抗体または抗体誘導体。
【請求項1】
第IX因子または第IXa因子に対する抗体または抗体誘導体であって、凝血促進活性を増大させる、抗体または抗体誘導体。
【図1】

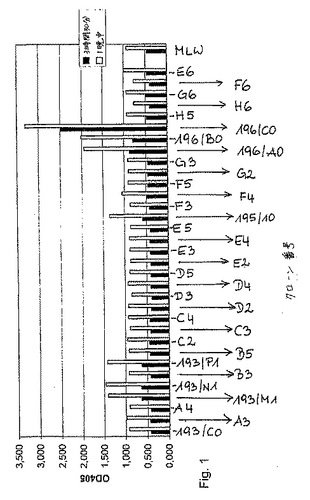
【図2】

【図3】

【図4】
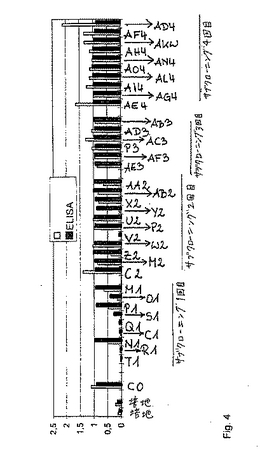
【図5】
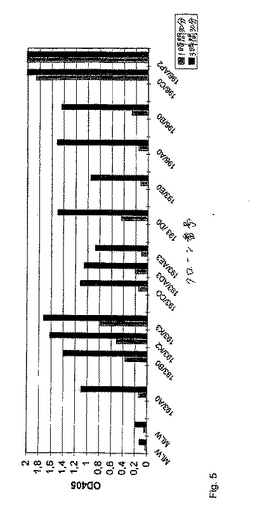
【図6A】
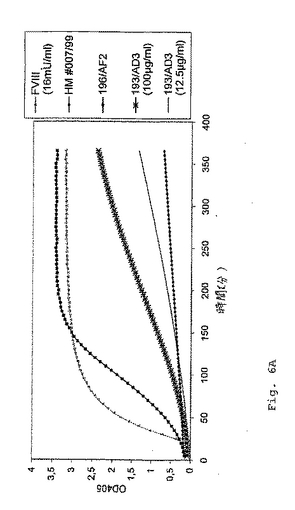
【図6B】
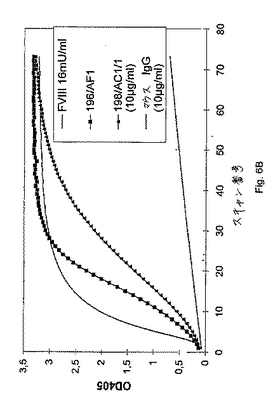
【図7A】
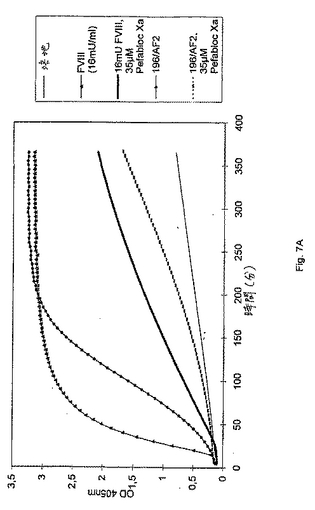
【図7B】
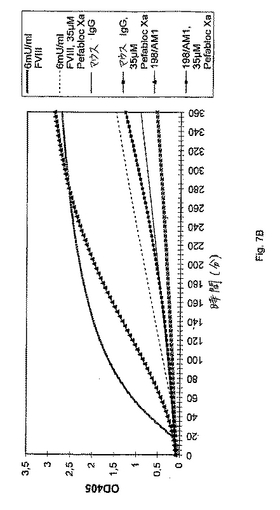
【図8A】
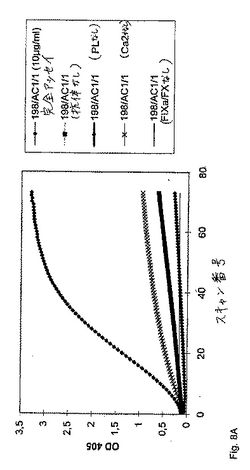
【図8B】
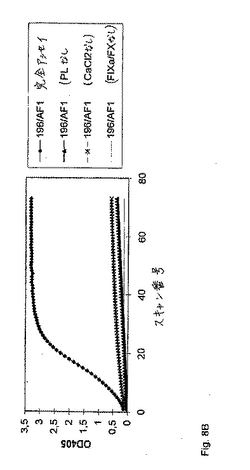
【図8C】
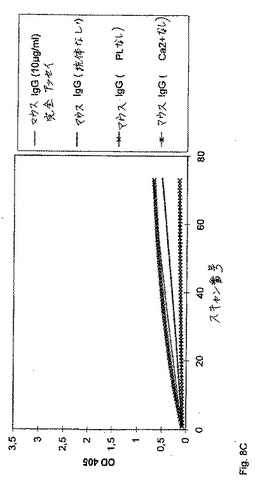
【図9】
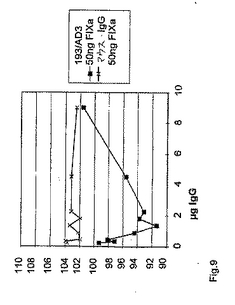
【図10A】
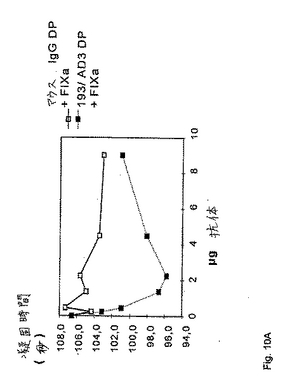
【図10B】
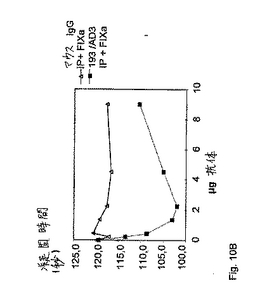
【図11】
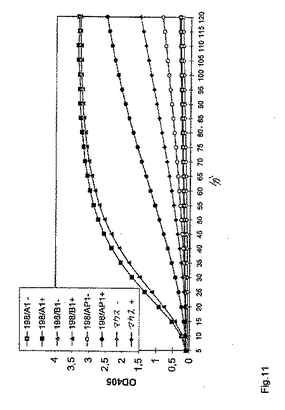
【図12−1】

【図12−2】

【図13−1】

【図13−2】
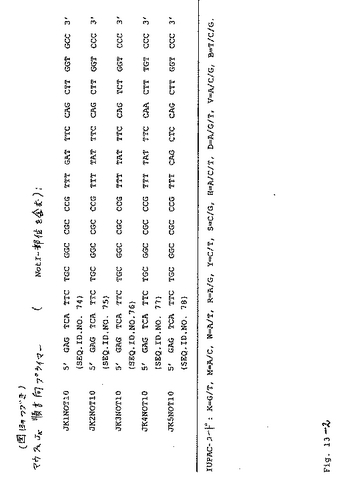
【図14−1】

【図14−2】

【図15−1】

【図15−2】

【図16−1】

【図16−2】

【図17−1】

【図17−2】

【図18】
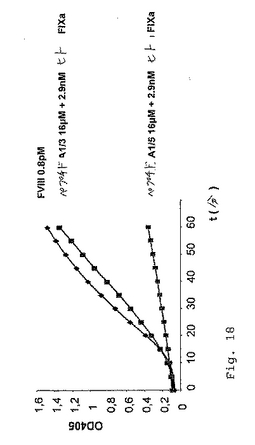
【図19】
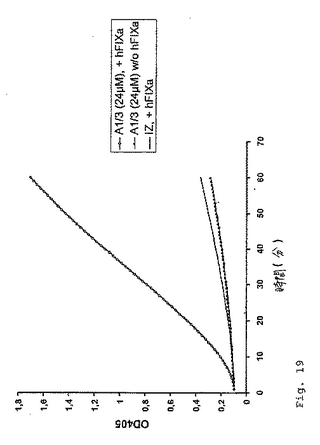
【図20】
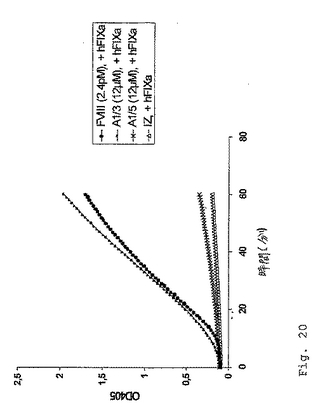
【図21】
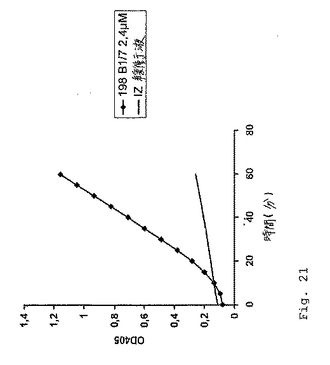
【図22】
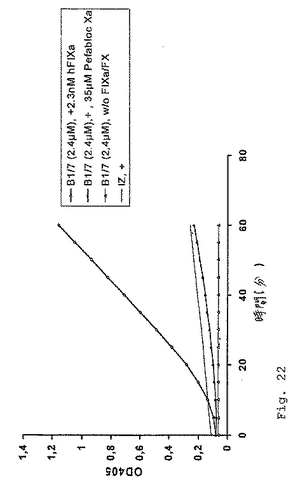
【図23】
【図24】
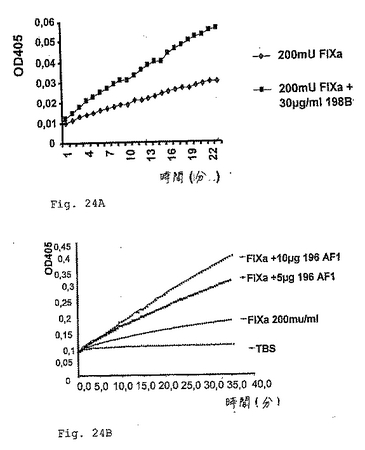
【図25】
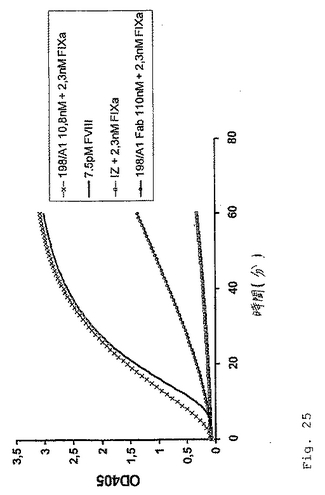
【図26−1】

【図26−2】

【図26−3】

【図26−4】

【図26−5】

【図27】
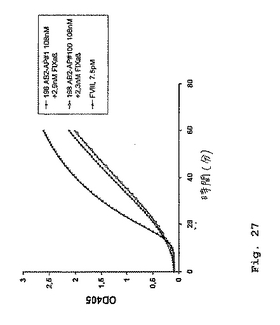
【図28−1】

【図28−2】

【図29−1】

【図29−2】

【図29−3】

【図29−4】

【図29−5】

【図30−1】

【図30−2】

【図30−3】

【図31】
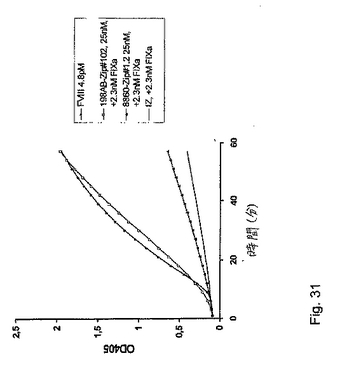
【図32】
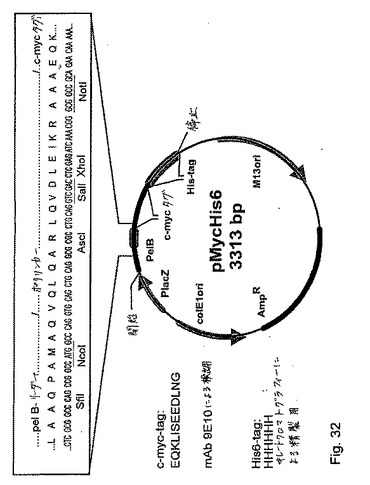
【図33】

【図34−1】

【図34−2】

【図35−1】

【図35−2】

【図36】
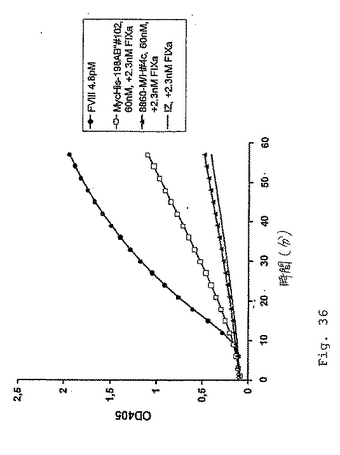
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図7A】
【図7B】
【図8A】
【図8B】
【図8C】
【図9】
【図10A】
【図10B】
【図11】
【図12−1】
【図12−2】
【図13−1】
【図13−2】
【図14−1】
【図14−2】
【図15−1】
【図15−2】
【図16−1】
【図16−2】
【図17−1】
【図17−2】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26−1】
【図26−2】
【図26−3】
【図26−4】
【図26−5】
【図27】
【図28−1】
【図28−2】
【図29−1】
【図29−2】
【図29−3】
【図29−4】
【図29−5】
【図30−1】
【図30−2】
【図30−3】
【図31】
【図32】
【図33】
【図34−1】
【図34−2】
【図35−1】
【図35−2】
【図36】
【公開番号】特開2006−111638(P2006−111638A)
【公開日】平成18年4月27日(2006.4.27)
【国際特許分類】
【出願番号】特願2005−380504(P2005−380504)
【出願日】平成17年12月28日(2005.12.28)
【分割の表示】特願2001−523763(P2001−523763)の分割
【原出願日】平成12年9月13日(2000.9.13)
【出願人】(501056094)バクスター アクチェンゲゼルシャフト (9)
【Fターム(参考)】
【公開日】平成18年4月27日(2006.4.27)
【国際特許分類】
【出願日】平成17年12月28日(2005.12.28)
【分割の表示】特願2001−523763(P2001−523763)の分割
【原出願日】平成12年9月13日(2000.9.13)
【出願人】(501056094)バクスター アクチェンゲゼルシャフト (9)
【Fターム(参考)】
[ Back to top ]