
第VIII因子変異体および使用の方法
モジュレーターのアミノ酸配列がB−ドメインに存在するか、またはモジュレーターのアミノ酸配列がB−ドメインの一部または全てのアミノ酸配列を置換している第VIII因子を含む第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを記載している。本発明の融合タンパク質および融合ヘテロダイマーをコードする核酸、融合タンパク質および融合ヘテロダイマーを生産するための方法、医薬組成物、および凝固欠乏症を本発明の融合分子で処置する方法も記載されている。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2009年3月24日に出願された米国仮出願番号61/162,986号(この内容は、その全体において出典明示により本明細書に包含させる)の優先権を主張する。
【0002】
技術分野
本発明は、変異体第VIII因子(FVIII)タンパク質に関する。本発明は、また、変異体FVIIIタンパク質をコードする核酸およびこのような核酸を同定するための方法に関する。本発明は、変異体FVIIIタンパク質を作製および使用する方法に関する。
【背景技術】
【0003】
発明の背景
血液の凝固は、接触活性化経路(以前は内因性経路として知られていた)または組織因子経路(以前は外因性経路として知られていた)のいずれかにより起こり、それによって、特定の血液タンパク質がタンパク質分解活性化のカスケードに相互作用し、最終的に可溶性フィブリノーゲンを不溶性フィブリンに変換する。これらのフィブリンの繊維状物は架橋して、凝血の足場を形成する。すなわち、フィブリン形成なしに、凝固が起こることができない。
【0004】
接触活性化経路は、以下のいくつかの工程からなる、(1)第XII因子のタンパク質分解活性化、(2)活性化された第XII因子が第XI因子を開裂して、それを活性化し、(3)活性化された第XI因子が第IX因子を開裂し、それによりそれを活性化し、(4)活性化された第IX因子が活性化されたFVIIIと相互作用して、第X因子を開裂および活性化し、(5)活性化された第X因子が膜表面上で活性化された第V因子に結合し、この複合体がタンパク質分解的にプロトロンビンを開裂して、トロンビンを形成し、(6)トロンビンがタンパク質分解的にフィブリノーゲンを開裂して、フィブリンを形成し、(7)フィブリンモノマーが線維に集合し、次にこれが第XIII因子により架橋される。
【0005】
組織因子経路は、以下の工程からなる、(1)血管の破裂時に、第VII因子が血管系の外側の組織に存在する組織因子であるリポタンパク質と結合し、(2)第VII因子が、タンパク質分解的切断により、第VIIa因子に活性化され、および(3)第VIIa因子−組織因子複合体が、第X因子を開裂し、活性化させる。その後、組織因子経路は接触活性化経路と同一であり、すなわち、2つの経路は上記最後の3つの工程を共有している。
【0006】
FVIIIの生合成、細胞内プロセッシングおよび分泌、ならびに次に血漿中で活性化されるメカニズムは、当該分野でよく知られている(例えば、Lentingら., Blood 92:3983-3996, 1998; Thompson, Seminars in 止血 29:11-22, 2003; Grawら., Nature Reviews: Genetics 6:489-501, 2005、参照)。ヒトFVIIIは、最初に、2351個のアミノ酸の一本鎖ポリペプチド(配列番号:1)として翻訳され、最初の19個のアミノ酸は、ER内でシグナルペプチダーゼにより除去されるシグナルペプチドを定義する。したがって、成熟ヒトFVIIIは、ドメイン構造A1−a1−A2−a2−B−a3−A3−C1−C2を有する2332個のアミノ酸からなる(図1A)。FVIIIは、Bドメインのカルボキシ−末端付近(B−a3接合点のArg−1648)の切断により、分泌前に細胞内でグリコシル化され、プロセッシングされ、Bドメイン内、主にArg−1313後で可変的に切断され、90−210kDaの重鎖および80kDaの軽鎖を生産する(図1B)。FVIIIは、その後、1つの重鎖および1つの軽鎖からなるヘテロダイマー糖タンパク質として分泌される。
【0007】
血漿糖タンパク質FVIIIは、フォン・ヴィレブランド因子(vWf)にしっかりと非共有結合的に結合した、血液中で不活性な前駆体として循環する。FVIIIは、3つのArg−Serペプチド結合、すなわちArg−372、Arg−740およびArg−1689後でトロンビンまたは第Xa因子による切断によりタンパク質分解的に活性化され、それはvWfから分離され、カスケードにおけるその凝血促進機能を活性化される。得られるヘテロトリマーはFVIIIaとなる(図1C)。
【0008】
その活性型(すなわち、FVIIIa)において、FVIIIは、血液凝固の接触活性化経路において第X因子活性化の酵素複合体に対する補因子として機能し、それは血友病Aを有する患者において減少しているか、または非機能的である。FVIII活性の減少レベルは、疾患の重症度に正比例する。したがって、FVIII欠乏の、またはFVIIIに対する抗体を有する人々は、FVIIIで処置されない限り広範で深刻な症状を引き起こし得る非制御の内出血に罹患する。症状は、関節における炎症応答から早期死亡の範囲である。FVIIIの古典的な定義は、実際には、血友病Aを有する個体由来の血漿における凝固欠損を正す正常血漿中に存在する物質である。vWfの欠損は、また、vWfが機能性FVIIIの重要な成分であるため、表現型血友病Aを引き起こし得る。これらの場合において、血漿中のFVIIIの循環半減期は、血液凝固におけるその特定の機能をもはや行うことができない程度に減少している。血友病Aの現在の処置は、血漿由来または組換えFVIIIの投与による欠損タンパク質の補充からなる。
【0009】
FVIIIの活性を阻害する抗体(「インヒビター」または「阻害抗体」)の発現は、血友病Aを有する患者の管理において深刻な合併症である。自己抗体は、FVIIIの治療的注入に応答して、血友病Aを有する患者の約20%において発現する。インヒビターを発現する以前に処置されていない血友病Aを有する患者において、インヒビターは、通常、処置から1年以内に発現する。さらに、FVIIIを不活性化する自己抗体は、以前に正常FVIIIレベルを有する個体において時折発現する。インヒビター力価が十分に低いとき、患者はFVIIIの用量を増加させることにより管理することができる。しかしながら、しばしば、インヒビター力価は、FVIIIにより圧倒されることができないほど高い。別の戦略は、第IX因子複合体調製物または組換えヒト第VIIa因子を使用して正常な止血中のFVIIIの必要性を迂回することである。さらに、ブタFVIIIは、通常、ヒトFVIIIよりもインヒビターと実質的にあまり反応性を有さないため、部分的に精製されたブタFVIII調製物が使用され得る。ヒトFVIIIに対して阻害抗体を発現した多数の患者は、ブタFVIIIで成功裏に処置され、長期間のこのような処置に耐える。しかしながら、ブタFVIIIの投与は、1以上の注入後に、ブタFVIIIに対するインヒビターが発現し得るため、完全な解決ではない。したがって、組換えヒトFVIIIまたは部分的に精製されたブタFVIIIの使用は、全ての問題を解決していない。
【0010】
阻害抗体に加えて、問題は、また、FVIIIが、静脈内に投与されたとき、循環中に比較的に短期の半減期(ヒトにおいて13時間)を有することで生じ、頻繁な注入が必要であり、患者の投与コンプライアンスにおいて困難性をもたらす。したがって、週1回の投与またはさらに月1回の投与用の長期間作用性FVIIIは、満たされていない医学的要望である(Dargaudら., Expert Opinion on Biological Therapy 7:651-663, 2007)。より長期の保護は、FVIII半減期を延長することにより達成される。多くのFVIIIの生物工学的アプローチが、より長期の保護を生じる目的のために探索される(Baruら., Thromb. Haemost. 93:1061-1068, 2005; Pipe, J. Thromb. Haemost. 3:1692-1701, 2005; Saenkoら., Haemophilia 12(Suppl 3):42-51, 2006)。
【発明の概要】
【0011】
本発明は、修飾された活性および/または修飾された薬物動態学的特性(例えば、より長い循環半減期)を示すFVIII変異体に関する。1つの例として、FVIII変異体は、アミノ酸配列(例えば、モジュレーター)がFVIIIタンパク質のB−ドメイン部分もしくはB−ドメインのいずれかにおいて挿入されているか、またはB−ドメインの一部が該アミノ酸配列で置換されている融合またはヘテロダイマータンパク質であり得る。該挿入/置換アミノ酸配列はFVIIIの翻訳後プロセッシングを中断させず、該FVIII変異体は凝固因子としての活性を有する。これらのFVIII変異体は、血友病Aを処置するために使用され得、例えば、より長い循環半減期によって、より少ない頻度の投与をもたらし得る。より少ない頻度の投与により、本発明のFVIII変異体は、患者のコンプライアンスを改善させ、FVIIIが投与されるためにFVIIIに対する免疫応答を発現する患者の可能性を減少させ得る。
【0012】
発明の概要
本発明は、FVIII融合タンパク質およびその発現産物に関する(本明細書においてFVIII融合ヘテロダイマーとしても称される)。本発明は、さらに、ハイブリッドFVIII融合ヘテロダイマーおよびマルチマーFVIII融合ヘテロダイマーに関する。1つの態様において、FVIII融合ヘテロダイマーは、FVIIIタンパク質またはポリペプチドおよびアミノ酸配列(本明細書においてモジュレーターとして称される)を含む。別の態様において、モジュレーター配列は、FVIII Bドメインに挿入される。さらなる態様において、少なくともBドメインの一部は、モジュレーター配列により欠失および置換されている。
【0013】
本発明は、また、FVIII融合ヘテロダイマーをコードする核酸配列に関する。1つの態様において、核酸配列は、モジュレーター配列がBドメインに存在するか、またはモジュレーター配列がBドメインの一部または全てのアミノ酸配列を置換している、FVIIIタンパク質を含むFVIII融合ヘテロダイマーをコードする。FVIII融合ヘテロダイマーをコードする核酸配列は、発現カセットに作動可能に連結し得る。本発明は、また、FVIII融合ヘテロダイマーを作製する方法を含む。例えば、FVIII融合ヘテロダイマーをコードする発現カセットは、すでに発現ベクターの一部でないとき、発現ベクターに導入され、次にFVIII融合ヘテロダイマーの組換え生産のために適当な宿主細胞に導入される。生産された融合ヘテロダイマーは、インビトロおよびインビボでFVIII活性を有し、例えば、インビボでの循環半減期の増加を示し得る。
【0014】
本発明のさらなる態様において、FVIII融合ヘテロダイマーは、配列番号:1、3または5のいずれか1つのアミノ酸20−764に対応する第1のアミノ酸配列、配列番号:1、3または5のいずれか1つのアミノ酸1656−2351に対応する第2のアミノ酸配列、およびモジュレーター配列を含み、(1)該モジュレーター配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合しているか、または(2)該モジュレーター配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、該モジュレーター配列が、第2のアミノ酸配列に共有結合していない。
【0015】
本発明の別の態様において、核酸配列は、FVIII融合ヘテロダイマーをコードし、該FVIII融合ヘテロダイマーは、配列番号:1、3または5のいずれか1つのアミノ酸20−764に対応する第1のアミノ酸配列、配列番号:1、3または5のいずれか1つのアミノ酸1656−2351に対応する第2のアミノ酸配列、およびモジュレーター配列を含み、該モジュレーター配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合している。加えて、本発明は、また、ベクター、宿主細胞、融合ヘテロダイマーを生産する方法および凝固欠乏症を処置する方法に関する。
【図面の簡単な説明】
【0016】
【図1】図1Aは、以下のドメイン:S(シグナルペプチド)、A1、a1、A2、a2、B、a3、A3、C1およびC2をN−末端からC−末端に含む全長ヒトFVIIIの構造を説明する。図1Bは、ヘテロダイマーヒト第VIII因子の重鎖および軽鎖の構造を説明する。重鎖のサイズは、B−ドメイン内の可変タンパク質分解的切断の結果として変化する。図1Cは、活性ヒトFVIII(すなわち、FVIIIa)のサブユニットの構造を説明する。
【0017】
【図2】図2は、実施例の部において記載されている本発明の第VIII因子融合ヘテロダイマーの3つの典型的な態様を説明する。3つの態様は、「BDDFc+ヒンジ」、「BDDFc−ヒンジ」、および「BDDFc」(これらは、所望により、単離を容易にするために異種ペプチドタグを含み得る)を示す。3つの典型的な態様は、ダイマー(これらのFc部分を介して)またはタンパク質凝集体を形成するそれらの能力において、およびFcRnに対するそれらの結合親和性において異なる。
【0018】
【図3】図3は、実施例1および2にしたがって生産される第VIII因子融合タンパク質の構造ドメインを記載している。具体的には、マウスFc領域(ヒンジ有りまたは無し)をB−ドメイン欠失(BDD)第VIII因子タンパク質の特定の部位(N−745からS−1637)に挿入し、B−ドメインの欠失部分を置換する。マウスFc領域のN−末端およびC−末端側上の非欠失B−ドメイン部分のアミノ酸配列を示す。
【0019】
【図4】図4は、実施例5にしたがって生産されるモノシストロン性BDD.mFcモノマー構築物を説明する。
【0020】
【図5】図5は、活性アッセイによる高い発現クローンの同定を説明する。BDDFc+ヒンジを発現するHKB11の安定な細胞系を、FVIII aPPT凝固アッセイによりスクリーニングした。クローン(4、8、12、18、27および33)は、500−3500mIU/mLの範囲である高い凝固活性を示した。
【0021】
【図6】図6は、ELISAアッセイによる高い発現クローンの同定を説明する。BDDFc+ヒンジを発現するHKB11の安定な細胞系を抗−FVIII捕捉ELISAによりスクリーニングした。3つのクローン(クローン8、18および27)は、〜1ug/mLでBDDFc+ヒンジ融合を発現する。
【0022】
【図7】図7は、BDDFc+ヒンジ融合タンパク質のタンパク質精製の結果を示す。還元ゲルにおいて、BDDFc+ヒンジは、80−kDa軽鎖(L)、115−kDa重鎖(H)、および195−kDa未処理一本鎖(U)(レーン8)として分離された。非還元ゲルにおいて、BDDFc+ヒンジは、80−、115−、195−kDaバンド(レーン8)に加えて390−kDaバンド(ダイマー)を生じた。
【0023】
【図8】図8は、血友病A(HemA)マウスにおけるBDDFc−ヒンジ(「FVIII−Fc」)およびBDD−FVIIIの回収を示す。9匹のHemAマウスに、5%のアルブミンを含む製剤バッファー中の50μg/kg(400IU/kg)(●)のBDDFc−ヒンジを与えた。さらなるHemAマウスに、200IU/kg(□)のBDDFc−ヒンジ由来の第VIII因子変異体であるBDD−FVIIIを与えた。BDD−FVIIIの減衰曲線と比較して、BDDFc−ヒンジは、迅速な分配相と二相減衰を示した。BDDFc−ヒンジのベータ相半減期は、50μg/kgで11.9hrあり、ベータ相半減期が6.03hrである修飾されていないBDD−FVIIIと比較して約2倍に改善された。
【0024】
【図9】図9Aは、BDDFc+ヒンジのBDD−Fcキメラ鎖をウエスタンブロット分析において115kDaバンドとして検出したことを説明する。一過性トランスフェクタント(trans)および安定なプール(sp)の両方からのサンプルを5倍濃縮し、次に還元条件下で10%のNuPAGE(登録商標)ゲル上に流した。レーン:1)分子量マーカー;2)標準として精製されたBDDタンパク質;3−5)pSK207ベクター、pSK207BDD、およびpSK207BDDFc+ヒンジ、それぞれで一過性でトランスフェクトされたHKB11細胞からの濃縮馴化培地;7−9)pSK207ベクター、pSK207BDD、pSK207BDDFc+ヒンジ、それぞれで安定にトランスフェクトされたHKB11細胞の安定なプールからの濃縮馴化培地。ブロットは、HRP−接合抗−マウスIgG(H+L)で探査した。未処理一本鎖形態のBDDFc+ヒンジ(「sc BDD−Fc」)は、195kDaバンドとして検出され、BDDFc+ヒンジのヘテロダイマー型は、115kDaの第VIII因子重鎖およびFc(「重鎖Fc」)のキメラを含んだ。HRP−接合抗−マウスIgGにより結合しないため、ヘテロダイマーBDDFc+ヒンジの軽鎖に関するバンドは現れない。図9Bは、BDDFc軽鎖がウエスタンブロット分析において80kDaバンドとして検出されたことを示す。タンパク質サンプルを還元条件下で10%のNuPAGE(登録商標)ゲル上に流した。レーン:1)分子量マーカー;2)標準として精製されたBDDタンパク質;3−5)pSK207ベクター、pSK207BDD、およびpSK207BDDFc+ヒンジ、それぞれで一過性でトランスフェクトされたHKB11細胞からの濃縮馴化培地。ブロットは、FVIII軽鎖特異的抗体で探査した。
【0025】
【図10】図10は、第VIII因子活性アッセイの結果を示す。pSK207BDDFc+ヒンジ(「Fc+ヒンジ sup」)およびpSK207BDDFc−ヒンジ(「Fc−ヒンジ sup」)で一過性でトランスフェクトされたHKB11細胞からの馴化培地を回収し、Coatest(登録商標)アッセイおよびaPPT凝固アッセイの両方においてFVIII活性に対して試験した。コントロールとして、修飾されていない第VIII因子タンパク質をコードするベクターpSK207およびpSK207BDD(「BDD sup」)を、トランスフェクションならびに活性アッセイにおいて使用した。
【0026】
【図11】図11は、第VIII因子融合ヘテロダイマーに対する第VIII因子活性を示す。HKB11細胞[(BDDFc+ヒンジ一過性トランスフェクタント(Tr)および安定なプール(Sp)]からの馴化培地を、ウサギ−抗−マウスFc抗体であらかじめ被覆された96−ウェルプレート上に負荷した。室温で2時間インキュベーション後、プレートをPBS/Tween(登録商標)−20/BSAで3回洗浄し、Coatest(登録商標)アッセイの前に非特異的結合を除去した。
【0027】
【図12】図12は、BDDFc+ヒンジがダイマーを形成し、BDDFc−ヒンジがモノマーであることを証明する。ウエスタンブロット分析を、pSK207BDDFc+ヒンジまたはpSK207BDDFc−ヒンジ発現ベクターでトランスフェクトされたHKB11細胞からの5倍濃縮馴化培地を使用して行った。サンプルを、還元および非還元条件下で4−12%のNuPAGE(登録商標)ゲル上に流した。ブロットは、ウサギモノクローナル抗−FVIII軽鎖抗体(Epitomics、Burlingame、CA)、次にHRP−接合抗−ウサギIgG二次抗体で探査した。未処理一本鎖BDDおよびBDDFcは、それぞれ170−kDaおよび195−kDaバンドとして検出された。レーン:BDD−−精製されたBDDタンパク質;+H−−BDDFc+ヒンジ;−H−−BDDFc−ヒンジ;およびV−−pSK207ベクター単独。
【0028】
【図13】図13は、固定されたマウスFcRnに結合するマウスFcRn結合エピトープを組み込むBDDFc+ヒンジおよびBDDFc−ヒンジ(「BDDFc−H」)の能力を測定するBiacoreTM試験の結果を示す。BDDFc+ヒンジ(「BDDFc+H」)、BDDFc−ヒンジ(「BDDFc−H」)、BDD、および全長組換え第VIII因子(「FVIII」)。検出可能な結合は、BDDまたは全長第VIII因子で見られなかった。BDDFc+ヒンジおよびBDDFc−ヒンジ融合タンパク質は、mFcRnに対する強い結合をnMの親和性で示した。
【0029】
【図14】図14は、固定されたヒトフォン・ヴィレブランド因子(vWF)に結合するBDDFc+ヒンジおよびBDDFc−ヒンジの能力を測定するBiacoreTM試験の結果を示す。マウスFcRnを、アミンカップリングによりCM−5チップ上に固定した。BDDFc+ヒンジ(「BDDFc+H」)、BDDFc−ヒンジ(「BDDFc−H」)、BDD、および全長組換え第VIII因子(「FVIII」)は、vWFに対するナノモル以下の親和性を示す。
【0030】
【図15】図15は、BDDFc−ヒンジがHemAマウスの尾静脈切断出血モデルにおいて有効であることを示す。BDDFc−ヒンジは、インビボで出血を処置する機能性があるか否かを決定するために、1つの側部尾静脈の切断の48時間前に、HemAマウスにBDDFc−ヒンジ、BDD−FVIIIまたは輸送媒体コントロールを尾静脈を介して注射した。損傷24時間後10%のみ生存する輸送媒体コントロール群(▲)と比較して、12IU/kg(●)および60IU/kg(■)のBDDFc−ヒンジは、それぞれ25%および80%の生存を成し遂げた。FVIII−Fc−ヒンジの有効性を、BDD−FVIIIと比較して40IU/kgで60%生存となる(◆)と評価した。
【発明を実施するための形態】
【0031】
本発明の説明
本発明は、記載されている特定の方法論、プロトコール、細胞系、動物種または属、構築物、および試薬に限定されず、それ自体変化し得ると理解すべきである。また、本明細書において使用される用語は特定の態様のみを記載するための目的であり、本発明の範囲を限定する意図はなく、特許請求の範囲のみにより限定されると理解すべきである。
【0032】
文脈が明白に他に指示されていない限り、本明細書および特許請求の範囲において使用されるとき、単数形「1つ(a、an)」および「その(the)」は、複数形を含むことに注意すべきである。したがって、例えば、「1つのタンパク質」の言及は、1つ以上のタンパク質の言及であり、当業者に知られているものと同等物などを含む。
【0033】
他に定義のない限り、本明細書において使用される全ての技術および学術用語は、本発明が属する当業者に一般的に理解されるものと同じ意味を有する。本明細書に記載されているものと同様または同等のあらゆる方法、デバイスおよび物質が本発明の実施または試験において使用することができるが、好ましい方法、デバイスおよび物質はここに記載されている。
【0034】
本明細書に記載されている全ての文献および特許は、本願発明に関連して使用され得る刊行物において記載されている、例えば、構築物および方法論を記載および開示する目的のために、出典明示により本明細書に包含させる。上記および明細書中の刊行物は、単に本特許出願の出願時前の開示のために提供される。本発明者らが以前の発明によってこのような開示に先行する権利がないと認めると、本明細書において解釈されるべきでない。
【0035】
本明細書において使用される、種々の用語は以下に定義されている。
【0036】
「核酸」は、一本鎖または二本鎖型のいずれかのデオキシリボヌクレオチドまたはリボヌクレオチドおよびそれらのポリマーを示す。具体的に限定されていないかぎり、該用語は、参照核酸と同様の結合特性を有し、天然ヌクレオチドと同様の様式において代謝される、既知の天然ヌクレオチドの類似体を含む核酸を含む。他に記載のない限り、特定の核酸配列は、また、保存的に修飾されたそれらの変異体(例えば、縮重コドン置換)および相補的配列ならびに明白に示されている配列を暗に含む。縮重コドン置換は、1つ以上の選択された(またはすべての)コドンの第3の位置が混合塩基および/またはデオキシイノシン残基で置換されている配列を産生することにより達成され得る。核酸なる用語は、文脈に依存して、遺伝子、cDNAおよび遺伝子によってコードされるmRNAと互換的に使用される。
【0037】
「遺伝子由来の核酸」は、合成遺伝子またはその部分配列が最終的に鋳型として使用されている核酸を示す。したがって、mRNA、mRNAから逆転写されるcDNA、cDNAから転写されるRNA、cDNAから増幅されるDNA、増幅されたDNAから転写されるRNAなどは遺伝子由来であり、このような由来産物の検出は、サンプル中の元の遺伝子および/または遺伝子転写産物の存在および/または存在量を示す。
【0038】
核酸配列は、別の核酸配列と機能的な関係に置かれているとき、「作動可能に連結」または「作動可能に挿入」されている。例えば、プロモーターまたはエンハンサーは、コード配列に作動可能に連結していてよい。作動可能に連結された核酸配列は、2つのタンパク質コード領域で隣接および/または結合していてもよい。いくつかの核酸配列は、作動可能に連結しているが、隣接していないかもしれない。核酸配列の連結は、制限酵素認識部位でのライゲーションにより達成され得る。このような部位が存在しないとき、合成オリゴヌクレオチドアダプターまたはリンカーは、慣用の実施にしたがって使用され得る。
【0039】
生物学的活性を有する第1のポリペプチドは、少なくとも最小限のレベルの生物学的活性が第1のポリペプチドおよび第2のポリペプチドの両方により維持されているような、第2のポリペプチドとの機能的な関係に置かれているとき、生物学的活性を有する第2のポリペプチドに「作動可能に連結」されている。ポリペプチドの文脈において、作動可能な連結は、第1および第2のポリペプチドが隣接することを必ずしも意味しない。当業者が理解できるとおり、生物学的活性の維持は、ペプチドリンカーの包含により促進され得る。
【0040】
ポリペプチド、核酸、または他の成分は、通常、関連している成分(他のペプチド、ポリペプチド、タンパク質(天然配列に伴って起こり得る複合体、例えば、ポリメラーゼおよびリボソームを含む)、核酸、細胞、合成試薬、細胞汚染物、細胞成分など)、例えば、通常、元の由来する細胞と関連している他の成分から部分的に、または完全に分離されているとき、「単離」されている。ポリペプチド、核酸または他の成分は、組成物、混合物または成分の回収物に存在する優勢な種類である(すなわち、モル基準において、組成物中の他のあらゆる個々の種類よりも豊富である)ように、自然環境の他の成分から部分的に、または完全に回収されるか、または分離されているとき、単離されている。場合によっては、調製物は、約60%、70%または75%以上、一般的に約80%以上、または約90%以上の単離された種類からなる。
【0041】
「実質的に純粋な」核酸(例えば、RNAまたはDNA)、ポリペプチド、タンパク質、または組成物は、また、対象の種類(例えば、核酸またはポリペプチド)が、組成物中に存在する全ての分子の種類の少なくとも約50、60、70、80、90または95重量パーセントを含むことを意味する。対象の種類を、また、十分な同質性に精製することができ(汚染の種類が慣用の検出方法により組成物中で検出することができない)、組成物が本質的に単一の高分子の種類の誘導体からなる。
【0042】
「精製」なる用語は、一般的に、核酸、ポリペプチドまたはタンパク質が、少なくとも約50%純粋、60%純粋、70%純粋、75%純粋、85%純粋、および99%純粋であることを意味する。
【0043】
「組換え」なる用語は、例えば、一般的に、細胞、ポリヌクレオチド、ベクター、タンパク質またはポリペプチドに関して使用されるとき、細胞、ポリヌクレオチドまたはベクターが異種(または外来)核酸の導入または天然の核酸の変化により修飾されているか、またはタンパク質またはポリペプチドが異種アミノ酸の導入により修飾されているか、または細胞がそのように修飾されている細胞由来であることを示す。組換え細胞は、細胞の天然(非組換え)型において見出されないであろう核酸配列を発現するか、または天然の核酸配列を発現するが、異常に発現するか、あまり発現しないか、または全く発現しない。「組換え」なる用語は、細胞を基準にして使用されるとき、細胞が異種核酸を複製するか、または異種核酸によってコードされるポリペプチドを発現することを示す。組換え細胞は、細胞の天然(非組換え)型内に見られないコード配列を含み得る。組換え細胞は、また、人工的手段により、修飾され、細胞に再導入されている、細胞の天然型において見られるコード配列を含み得る。該用語は、また、細胞から核酸を除去することなく修飾されている細胞に対して内在的な核酸を含む細胞を包含し、このような修飾は、遺伝子置換、部位特異的変異、再結合、および関連技術により得られるものを含む。
【0044】
「組換え的に生産」なる用語は、核酸セグメントの再帰的な配列再結合もしくはヌクレオチドの他の多様な産生方法(例えば、シャッフリング)のいずれかの化学合成手段、または、例えば、当業者に知られている遺伝子操作技術による、核酸の単離されたセグメントの操作により通常、達成される人工的組合せを示す。「組換え的に発現」は、一般的に、インビトロでの組換え核酸の生産およびインビボ、インビトロまたはエキソビボでの組換え核酸の発現または増殖され得る細胞への移動のための技術を示す。
【0045】
「組換え発現カセット」または単に「発現カセット」は、このような配列に適合する宿主において構造タンパク質をコードする核酸の発現に影響することができる核酸因子と共に、組換え的に、または合成的に産生される核酸構築物を示す。発現カセットは、必ずしも転写されるべき核酸(例えば、所望のポリペプチドをコードする核酸)、およびプロモーターを含まない。発現の影響において必要な、もしくは補助的なさらなる成分も、また、本明細書に記載されているとおりに使用され得る。例えば、発現カセットは、また、宿主細胞から発現されたタンパク質の分泌を指向する選別シグナル(例えば、シグナルペプチドまたは分泌リーダー配列)をコードするヌクレオチド配列を含み得る。転写終結シグナル、エンハンサー、および遺伝子発現に影響する他の核酸配列も、また、発現カセットに含まれ得る。本発明の目的のために、「第VIII因子融合遺伝子を含む発現カセット」は、発現カセットにより発現される所望のタンパク質が、さらに以下で定義されている「第VIII因子融合タンパク質」であることを示す。
【0046】
「ベクター」なる用語は、文脈に依存して、クローニングベクター、発現ベクター、またはその両方を示し得る。ベクターなる用語および「プラスミド」なる用語は互換的に使用される。
【0047】
「発現ベクター」または「発現プラスミド」なる用語は、宿主を形質転換し、導入された配列の発現(例えば、転写および翻訳)を促進するように、発現カセットを宿主細胞に導入することができる輸送媒体を示す。
【0048】
「発現する」および「発現」なる用語は、遺伝子またはDNA配列における情報を明らかにする、または明らかに誘導する、例えば、対応する遺伝子またはDNA配列の転写および翻訳に関与する細胞機能を活性化することによりタンパク質を生産することを意味する。DNA配列は、細胞において、または細胞により発現され、「発現産物」、例えば、タンパク質を形成する。発現産物それ自体、例えば、得られたタンパク質も、また、「発現」されたと言う。発現産物は、細胞内、細胞外、または分泌として特徴付けることができる。
【0049】
「アミノ酸修飾」は、所定のアミノ酸配列のアミノ酸配列における変化を示す。典型的な修飾は、アミノ酸置換、挿入および/または欠失を含む。
【0050】
「アミノ酸挿入」は、少なくとも1つのアミノ酸の所定のアミノ酸配列への取り込みを示す。挿入は、1つまたは2つのアミノ酸残基の挿入またはより長い挿入からなり得る。挿入される残基は、上記のとおり天然または非天然であり得る。
【0051】
「アミノ酸欠失」は、所定のアミノ酸配列から少なくとも1つのアミノ酸残基の除去を示す。
【0052】
「アミノ酸置換」は、所定のアミノ酸配列における少なくとも1つの存在するアミノ酸残基の、別の異なる「置換」アミノ酸残基での置換を示す。置換残基は、「天然アミノ酸残基」(すなわち、遺伝子コードによってコードされる)であり得、アラニン(Ala);アルギニン(Arg);アスパラギン(Asn);アスパラギン酸(Asp);システイン(Cys);グルタミン(Gln);グルタミン酸(Glu);グリシン(Gly);ヒスチジン(His);イソロイシン(Ile):ロイシン(Leu);リジン(Lys);メチオニン(Met);フェニルアラニン(Phe);プロリン(Pro):セリン(Ser);スレオニン(Thr);トリプトファン(Trp);チロシン(Tyr);およびバリン(Val)からなる群から選択される。1つまたはそれ以上の非天然アミノ酸残基での置換も、また、本明細書においてアミノ酸置換の定義により包含される。「非天然アミノ酸残基」は、ポリペプチド鎖において隣接アミノ酸残基に共有結合することができる上記天然アミノ酸残基以外の残基を示す。非天然アミノ酸残基の例は、ノルロイシン、オルニチン、ノルバリン、ホモセリン、および他のアミノ酸残基類似体、例えば、Ellmanら. (Meth. Enzym. 202:301-336, 1991)に記載されているものを含む。このような非天然アミノ酸残基を産生するために、Norenら. (Science 244:182, 1989)およびEllmanら., 1991の方法を使用され得る。簡潔には、これらの方法は、非天然アミノ酸残基でのサプレッサーtRNAの化学的活性化、次にインビトロでのRNAの転写および翻訳を含む。最後に、当業者は、例えば、タンパク質の領域のアミノ酸置換が1つの工程、または2つの工程(例えば、アミノ酸欠失、次にアミノ酸挿入により、または逆もまた同様)において達成されることができることを認識している。
【0053】
特定のポリペプチドまたはタンパク質の「変異体」は、本明細書で定義されている少なくとも1つの「アミノ酸修飾」によって特定のポリペプチドまたはタンパク質のアミノ酸配列と異なるアミノ酸配列を含む。「変異体」は、所望の活性を示すポリペプチドまたはタンパク質のフラグメント、例えば、FcRnへ結合し、凝固因子と結合しているとき、循環半減期を改善するIgGのFc領域のフラグメントを含む。
【0054】
「融合ポリペプチド」は、天然に同じポリペプチドにおいて生じることが見られない少なくとも2つの別々のペプチド部分を含むポリペプチドを示す。
【0055】
「融合タンパク質」は、少なくとも1つの融合ポリペプチドを含むタンパク質を示す。したがって、マルチ−サブユニットタンパク質は、サブユニットの1つのみが融合ポリペプチドであるときでさえ、融合タンパク質として示される。
【0056】
「FVIII」、「第VIII因子」または「第VIII因子タンパク質」なる用語は、機能的対立遺伝子変異体を含む野生型第VIII因子タンパク質、または第VIII因子の生物学的活性を有するそのあらゆる誘導体、変異体もしくは類似体を包含することを意図する。この定義の目的のために、「第VIII因子の生物学的活性」は、血液凝固の内因性経路に参加することができる能力を示す。一般的に、この生物学的活性は、市販されている第VIII因子アッセイ(Coatest(登録商標)、diaPharma(登録商標)、West Chester, Ohio)または当該分野における他のアッセイを使用して、血漿由来の第VIII因子標準を基準にして決定され得る。
【0057】
第VIII因子ドメインについて記載されているとき、「ドメイン」は、当業者に知られている第VIII因子の隣接領域を示すために使用される。ヒト第VIII因子に対して、異なる第VIII因子ドメインに対するアミノ酸番号付けは、図1において示されている。
【0058】
「第VIII因子融合遺伝子」は、さらに以下で定義されている「第VIII因子融合タンパク質」をコードし、Bドメインコード部分に対応する第VIII因子タンパク質コード配列内の位置で、モジュレーターをコードする核酸の第VIII因子タンパク質をコードする核酸への作動可能な挿入により生産され得る非天然核酸構築物を示す。1つの例として、Bドメインコード配列の少なくとも1部分が、モジュレーターをコードする核酸により欠失および置換されている。当業者により理解されるとおり、「作動可能な挿入」は、モジュレーターをコードする1部およびモジュレーターをコードする核酸の上流および下流にある第VIII因子の1部をコードする核酸が全て適当なリーディングフレーム内にある核酸構築物を生産するモジュレーターをコードする核酸の挿入を包含することを意図するのみである。第VIII因子融合遺伝子は、ペプチドリンカーまたは多重合配列をコードするさらなる核酸配列をさらに含み得る。最後に、上記定義の目的のために、「遺伝子」は、他に、転写、翻訳、または適当な翻訳後プロセッシングを可能にするために必要とされる任意の核酸配列(すなわち、プロモーター、エンハンサー、シグナルペプチド、分泌リーダー配列など)の存在を意味することを意図しない。
【0059】
「第VIII因子融合タンパク質」は、翻訳後プロセッシングをまだ受けていない第VIII因子融合遺伝子の転写および翻訳により生産される全長ポリペプチドを示す。翻訳後プロセッシング中に、第VIII因子融合タンパク質は、以下で定義されている「第VIII因子融合ヘテロダイマー」に変換される。
【0060】
「第VIII因子融合ヘテロダイマー」は、第VIII因子の生物学的活性を有し、第VIII因子融合遺伝子の転写および翻訳、およびそれにより生産された第VIII因子融合タンパク質の翻訳後修飾(タンパク質分解プロセッシングを含む)の結果として生産されるヘテロダイマータンパク質を示す。したがって、第VIII因子融合ヘテロダイマーは、血漿中の循環が見られる野生型第VIII因子のヘテロダイマー型(すなわち、重鎖および軽鎖を含む)に類似している。本発明の第VIII因子融合ヘテロダイマーは、例えば、「修飾された重鎖」(すなわち、モジュレーターを含み、また、B−ドメインの少なくとも1つの部分の欠失を有し得る第VIII因子重鎖)から構成されるという点において起源の第VIII因子タンパク質のヘテロダイマー型と異なっていてよい。本発明の第VIII因子融合ヘテロダイマーは、融合ヘテロダイマーが起源の第VIII因子タンパク質と比較して、例えば、循環半減期の増加を示し得る。「第VIII因子融合ヘテロダイマー」は、また、以下でさらに定義されている「マルチマー」および「ハイブリッド」第VIII因子融合ヘテロダイマーを包含し得る。
【0061】
「マルチマー第VIII因子融合ヘテロダイマー」は、少なくとも2つの第VIII因子融合ヘテロダイマーを含むタンパク質を示す。マルチマー第VIII因子融合ヘテロダイマーは、第1の第VIII因子融合ヘテロダイマーに存在するモジュレーターのアミノ酸、ペプチド、またはポリペプチド部分が、第2の第VIII因子融合ヘテロダイマーに存在するモジュレーターの同種または異種部分との非共有結合性または共有結合性会合を仲介することができるとき、生じ得る。例えば、IgGのFc部分のヒンジ領域は、第VIII因子融合ヘテロダイマーが、同一のアミノ酸配列(マルチマー第VIII因子融合ヘテロダイマーが「ホモ−マルチマー第VIII因子融合ヘテロダイマー」として称することができる場合)または異なるアミノ酸配列(マルチマー第VIII因子融合ヘテロダイマーが「ヘテロ−マルチマー第VIII因子融合ヘテロダイマー」として称することができる場合)を有するかにかかわらず、2つの第VIII因子融合ヘテロダイマー間の共有結合性会合を仲介することができる。マルチマー第VIII因子融合ヘテロダイマーは、また、モジュレーターをコードする核酸に加えて、第VIII因子融合遺伝子が、同種アミノ酸、ペプチド、またはポリペプチド(「ホモ−多重合配列」として以下で称される)、または、異種ペプチドまたはポリペプチド(「ヘテロ−多重合配列」として以下で称される)との非共有結合性または共有結合性会合を仲介することができるアミノ酸、ペプチド、またはポリペプチドをコードする作動可能に連結している核酸を含むとき、生じ得る。当業者は、第1の第VIII因子融合遺伝子のみがヘテロ−多重合配列を含むとき、第2の異なる第VIII因子融合遺伝子がマルチマー第VIII因子融合ヘテロダイマーを生産するために必要とされ得ることを認識している。例えば、当業者は、米国特許第5,807,706号に記載されている「空洞への隆起(protuberance-into-cavity)」手法を利用するために、2つの第VIII因子融合遺伝子を入手することを認識する。マルチマー第VIII因子融合ヘテロダイマーの組換え生産に関して、当業者は、ホモ−マルチマー型は単一の組換え宿主細胞により生産され得るが、ヘテロ−マルチマー型は単一の宿主細胞内での共発現または複数の宿主細胞(同じか、または異なっている細胞培養系における)における別々の発現により生産され得ることを認識している。現在既知の手法に限定することを意図しないが、以下の非限定的な文献において教示されているマルチマーポリペプチドを生産するための一般的な手法は、マルチマー第VIII因子融合ヘテロダイマーの生産における使用に適している:米国特許公報第2007/0287170号;米国特許第7,183,076号において記載されている「multimerization domain」手法、例えば、免疫グロブリン部分を使用するもの;米国特許第5,932,448号において使用されているFos and Jun leucine zippersの使用;および米国特許第6,833,441号において使用されている「heterodimerization sequence」手法
【0062】
「ハイブリッド第VIII因子融合ヘテロダイマー」は、少なくとも1つの他のポリペプチドと共有結合性または非共有結合性会合する第1の第VIII因子融合ヘテロダイマーのみを含む本発明のあらゆる組換えタンパク質を示す。当業者は、モジュレーターが、ダイマーまたはマルチマー(例えば、免疫グロブリンのダイマーFc領域)を形成することができるとき、マルチマー第VIII因子融合ヘテロダイマー(上記定義のとおり)を生産することができることを認識している。しかしながら、当業者は、マルチマー半減期モジュレーターのすべてのポリペプチドが、第VIII因子融合タンパク質として発現される必要性がないことを認識している。例えば、Fc領域がモジュレーターとして使用されるとき、Fc領域のみ(またはアフィニティータグまたは非第VIII因子ペプチド、タンパク質またはタンパク質フラグメントに作動可能に連結したFc領域)をコードする発現カセットが、第VIII因子融合遺伝子を含む発現カセットを含む同じまたは異なっている宿主細胞に導入され得る。当業者は、また、モジュレーターがダイマーまたはマルチマーを形成することができないときでさえ、ハイブリッド第VIII因子融合ヘテロダイマーが設計され得ることを認識している。具体的には、ホモ−またはヘテロダイマー配列は、第VIII因子融合遺伝子内にモジュレーターをコードする核酸に対して5’(発現するときにN−末端)または3’(すなわち、発現するときにC−末端)のいずれかで位置し得る。
【0063】
「モジュレーター」なる用語は、タンパク質(例えば、第VIII因子)の挿入または置換されたとき、例えば、タンパク質の活性および/または薬物動態学的特性を修飾するあらゆるポリペプチド、タンパク質、タンパク質フラグメント、またはそれらの変異体(1つ以上のポリペプチドサブユニットから構成される)を示す。1つの例として、「半減期モジュレーター」は、起源のタンパク質と比較して、タンパク質(例えば、該挿入または置換の結果として生産される、第VIII因子融合ヘテロダイマー、ハイブリッド第VIII因子融合ヘテロダイマー、またはマルチマー第VIII因子融合ヘテロダイマー)の循環半減期を増加または減少させ得る。半減期モジュレーターは、例えば、タンパク質(例えば、第VIII因子融合ヘテロダイマー、ハイブリッド第VIII因子融合ヘテロダイマー、またはマルチマー第VIII因子融合ヘテロダイマー)の循環半減期を、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも40%、少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、または少なくとも100%増加させ得る。1つの態様において、半減期モジュレーターは、第VIII因子融合ヘテロダイマー、ハイブリッド第VIII因子融合ヘテロダイマー、またはマルチマー第VIII因子融合ヘテロダイマーの循環半減期を、起源の第VIII因子タンパク質と比較して、少なくとも約2倍増加させ得、さらなる態様において、循環半減期を、少なくとも約2.5倍、少なくとも約3倍またはそれ以上増加させる。別の態様において、半減期モジュレーターは、第VIII因子タンパク質のあらゆる内因性因子、例えば、限定はしないがB−ドメインを含まない。
【0064】
ペプチド薬物を患者へ投与する文脈において本明細書において使用される「循環半減期」、「血漿半減期」、「血清半減期」または「t[1/2]」なる用語は、患者における薬物の血漿濃度が2分の1にまで減少するために必要な時間として定義され得る。多数のクリアランスメカニズム、再分配、および当該分野でよく知られている他のメカニズムに依存して、ペプチド薬物と関連する1つ以上の半減期があり得る。通常、アルファおよびベータ半減期は、アルファ相は再分配と関連し、ベータ相はクリアランスと関連するように定義される。しかしながら、大部分、血流に留まっているタンパク質薬物で、少なくとも2つのクリアランス半減期があり得る。本発明の目的のために、ベータ半減期は、投与後に適当に選択された時点で血漿タンパク質レベル(例えば、抗原ELISAを使用して)を測定すること、または適当に選択された時点で凝固活性(例えば、Coatestアッセイを使用して)を測定することにより計算されうる。「半減期」のさらなる説明は、Pharmaceutical Biotechnology(1997, DFA Crommelin and RD Sindelar, eds., Harwood Publishers, Amsterdam, pp 101-120)において見いだされ得る。
【0065】
第VIII因子融合遺伝子の構築
本発明の1つの局面は、第VIII因子融合遺伝子に関する。第VIII因子融合遺伝子は、RNAまたはDNAのいずれかであり得る。上記のとおり、第VIII因子融合遺伝子は、第VIII因子融合タンパク質をコードする核酸分子である。第VIII因子融合遺伝子は、第VIII因子コード配列、モジュレーターをコードする核酸、および、所望により、モジュレーターをコードする核酸と異なるホモもしくはヘテロ−多重合配列をコードする核酸由来である。第VIII因子融合遺伝子のこれらの因子は以下でさらに詳細に記載されているが、第VIII因子融合遺伝子の構築が、よく知られている方法を使用してこれらの別々の因子をコードする核酸から合成することができることを当業者は理解している。本発明における使用で見いだされ得る種々の方法は、Molecular Cloning - A Laboratory Manual, 3rd Ed. (Maniatis, Cold Spring Harbor Laboratory Press, New York, 2001)、およびCurrent Protocols in Molecular Biology (John Wiley & Sons)に記載されている。
【0066】
第VIII因子をコードする核酸の選択
本発明の組換え第VIII因子融合タンパク質およびヘテロダイマーは、野生型第VIII因子、存在し、ある個体から別の個体で発生し得る第VIII因子の天然対立遺伝子変異体、キメラ第VIII因子(例えば、ヒト/ブタ)、または、他の点で修飾されており、凝血促進機能をなお維持している変異体第VIII因子、例えば、野生型第VIII因子または第VIIIa因子タンパク質の特性、例えば、グリコシル化部位およびパターン、抗原性、比活性、循環半減期、タンパク質分泌、第IXa因子および/または第X因子に対する親和性、変更された第VIII因子−不活性化切断部位、活性化された第VIIIa因子形態の安定性、免疫原性、保存期間などに影響するように修飾されている変異体をコードする核酸を修飾することにより製造され得る。本発明にしたがって修飾され得る適当な変異体第VIII因子配列は、野生型第VIII因子と関連する凝血促進機能を有する以前に知られているか、または後に同定される任意のあらゆる変異体第VIII因子配列を含み得る。
【0067】
本発明にしたがって修飾することができる適当な野生型第VIII因子は、限定はしないが、哺乳動物、例えば、ヒト(例えば、GenBankアクセッション番号AAA52484(アミノ酸)(配列番号:1)およびK01740(ヌクレオチド)(配列番号:2)、GenBankアクセッション番号AAA52485(アミノ酸)(配列番号:3)およびM14113(ヌクレオチド)(配列番号:4)、およびGenBank受入番号AAA52420(アミノ酸)(配列番号:5)、参照);ラット(例えば、GenBankアクセッション番号AAQ21580(アミノ酸)およびAY362193(ヌクレオチド)、参照);マウス(see、例えば、GenBankアクセッション番号AAA37385(アミノ酸)およびL05573(ヌクレオチド)、参照);イヌ(例えば、GenBankアクセッション番号AAB87412(アミノ酸)およびAF016234(ヌクレオチド)、参照);コウモリ(例えば、GenBankアクセッション番号ACC68917(アミノ酸)およびDP000725(ヌクレオチド)、参照);ニワトリ(例えば、GenBankアクセッション番号AAO33367(アミノ酸)およびAF465272(ヌクレオチド)、参照);チンパンジー(例えば、GenBankアクセッション番号XP_529212(アミノ酸)およびXM_529212(ヌクレオチド)、参照);ブタ(例えば、GenBankアクセッション番号.NP_999332(アミノ酸)およびNM_214167(ヌクレオチド)、参照);ウサギ(例えば、GenBankアクセッション番号ACA42556(アミノ酸)およびEU447260(ヌクレオチド)、参照);ネコ、サル、モルモット、オランウータン、ウシ、ウマ、ヒツジ、ヤギまたは他の哺乳動物種を含む種々の動物由来であり得る。ヒト、ブタ、マウスおよびイヌに対する配列は、また、Haemophilia A Mutation, Structure, Test and Resource Site(またはHAMSTeRS)を介して電子的に利用でき、これは、ヒト、ブタ、マウスおよびイヌの第VIII因子タンパク質のアラインメントをさらに提供する。当業者が理解できるとおり、哺乳動物の第VIII因子タンパク質中の保存および相同性はよく知られている。
【0068】
本発明にしたがって修飾され得る適当な変異体第VIII因子の1つの非限定的な例は、天然ヒトFVIIIのB−ドメインの14個のアミノ酸(SFSQNPPVLKRHQR、配列番号:6)の他全ての欠失を含むアミノ酸配列を有することにより特徴付けられるB−ドメイン欠失第VIII因子(「BDD第VIII因子」)である(Lindら., Eur. J. Biochem. 232:19-27, 1995)。このBDD第VIII因子は、配列番号:7のアミノ酸配列を有する。
【0069】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、ヒト第VIII因子の抗原性に関与するヒトアミノ酸残基に対する置換基として1つ以上の動物アミノ酸残基を含むキメラヒト/動物第VIII因子である(例えば、米国特許第5,364,771;5,663,060;および5,888,974号、参照)。例えば、動物(例えば、ブタ)残基置換は、1つ以上のR484A、R488G、P485A、L486S、Y487L、Y487A、S488A、S488L、R489A、R489S、R490G、L491S、P492L、P492A、K493A、G494S、V495A、K496M、H497L、L498S、K499M、D500A、F501A、P502L、1503M、L504M、P505A、G506A、E507G、1508M、1508A、M2199I、F2200L、L2252F、V2223A、K2227E、および/またはL2251を含み得るが、これらに限定されない(例えば、米国特許第5,859,204および6,770,744号ならびに米国特許公報第2003/0166536号、参照)。
【0070】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、融合A2およびA3ドメインによって活性化された第VIII因子のより良い安定性により特徴付けられる第VIII因子である。例えば、第VIII因子は、位置664および1826でシステイン残基に置換することにより修飾され、A2およびA3ドメインに共有結合するCys664−Cys1826ジスルフィド結合を含む変異体第VIII因子をもたらし得る(Galeら., J. Thromb. Haemost. 1:1966-1971, 2003)。
【0071】
本発明にしたがって修飾され得る適当な変異体第VIII因子のさらなる非限定的な例は、変更された不活性化切断部位を有する第VIII因子である(例えば、Amanoら., Thromb. Haemost. 79:557-63, 1998; Thornburgら., Blood 102:299, 2003、参照)。これらの変化は、通常、野生型第VIII因子を不活性化する切断酵素に対する変異体第VIII因子の感受性を減少させるために使用され得る。
【0072】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、第IXa因子(例えば、Fayら., J. Biol. Chem. 269:20522-20527, 1994); Bajajら., J. Biol. Chem. 276:16302-16309, 2001;およびLentingら., J. Biol. Chem. 271:1935-1940, 1996、参照)および/または第X因子(例えば、Lapanら., J. Biol. Chem. 272:2082-2088, 1997、参照)に対する親和性が増強されている第VIII因子である。
【0073】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、第VIII因子の分泌を増強するように修飾されている第VIII因子である(例えば、Swaroopら., J. Biol. Chem. 272:24121-24124, 1997、参照)。
【0074】
本発明にしたがって修飾され得る適当な変異体第VIII因子のさらなる非限定的な例は、増加した循環半減期を有する第VIII因子である。これらの変異体第VIII因子タンパク質は、限定はしないが、ヘパラン硫酸と減少した相互作用(Sarafanovら., J. Biol. Chem. 276:11970-11979, 2001)および/または低比重リポタンパク質受容体関連タンパク質(「LRP」)と減少した相互作用を有するとして特徴付けることができる(例えば、WO00/28021;WO00/71714;Saenkoら., J. Biol. Chem. 274:37685-37692, 1999;およびLentingら., J. Biol. Chem. 274:23734-23739, 1999、参照)。
【0075】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、既知の、既存のエピトープ内のアミノ酸をコードし、アスパラギン残基でグリコシル化のための認識配列を生産するように修飾されたヌクレオチド配列によってコードされる修飾された第VIII因子である(例えば、米国特許第6,759,216号、参照)。該例の変異体第VIII因子は、既存の阻害抗体による検出を逃れ(低い抗原性の第VIII因子)、阻害抗体を発現する可能性を減少させる(低い免疫原性の第VIII因子)修飾された第VIII因子の提供において有用であり得る。本発明にしたがって修飾され得る変異体第VIII因子のこの型の1つの態様は、N−連結グリコシル化のためのコンセンサスアミノ酸配列を有するように変異されている第VIII因子である。このようなコンセンサス配列の1つの例は、N−−X−−S/T(式中、Nはアスパラギンであり、Xは任意のアミノ酸であり、S/Tはセリンまたはスレオニンを示す)である(例えば、米国特許第6,759,216号、参照)。
【0076】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、種々の変異を有する凝血促進活性な第VIII因子である(例えば、米国特許第6,838,437号および米国特許出願第2004/0092442号、参照)。この態様の1つの例は、(i)フォン・ヴィレブランド因子結合部位を欠失する、(ii)Arg740に変異を加える、および(iii)A2およびA3−ドメイン間にアミノ酸配列スペーサーを加えるように修飾されている変異体第VIII因子に関し、ここで、該アミノ酸スペーサーは、活性化時に、凝血促進活性な第VIII因子タンパク質がヘテロダイマーになるように十分な長さである(例えば、米国特許公報第2004/0092442号;Pittmanら., PNAS 85:2429-2433, 1988、参照:コファクター活性を産生するために、Arg740での切断が必須ではないと記載している)。
【0077】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、1つ以上の位置に挿入された短縮(truncated)第IX因子イントロン1を有するヌクレオチド配列によってコードされる変異体第VIII因子である(例えば、米国特許第6,800,461および6,780,614号、参照)。この変異体第VIII因子は、インビトロでの組換え第VIII因子のより高い生産を得るために、ならびに遺伝子治療のためのトランスファーベクターにおいて使用され得る(例えば、米国特許第6,800,461号、参照)。この態様の1つの例において、変異体第VIII因子は、2つの位置に挿入された短縮第IX因子イントロン1、および造血細胞系および血小板における発現を駆動するために適当であるプロモーターを有するヌクレオチド配列によってコードされ得る(例えば、米国特許第6,780,614号、参照)。
【0078】
本発明にしたがって修飾され得る適当な変異体第VIII因子のさらなる非限定的な例は、阻害抗体により阻害の減少を示す変異体第VIII因子である(例えば、米国特許第5,859,204;6,180,371;6,458,563;および7,122,634号、参照)。
【0079】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、第VIII因子の半減期および/または比活性の増加の影響を有するA2ドメインにおいて1つ以上のアミノ酸置換を有する変異体第VIII因子である(例えば、米国特許第7,211,559号、参照)。
【0080】
本発明にしたがって修飾され得る適当な変異体第VIII因子のさらなる非限定的な例は、比活性の増加を示す変異体第VIII因子である(例えば、米国特許公報第2007/0265199号、参照)。
【0081】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、1つ以上の生体適合性ポリマーにあらかじめ定義された部位で共有結合しているFVIII変異タンパク質である(例えば、米国特許公報第2006/0115876号、参照)。
【0082】
モジュレーターをコードする核酸の選択
本発明の第VIII因子融合遺伝子は、モジュレーターをコードする核酸を含む。例えば、治療的タンパク質と融合されたとき、融合されていない治療的タンパク質と比較して血清半減期を延長する効果を有する多数のタンパク質(およびそれらをコードする核酸)は、当該分野で知られている。これらの参考文献において教示されている任意のモジュレーターをコードする核酸は、本発明の第VIII因子融合遺伝子を構築するために潜在的に使用され得る。例えば、第VIII因子融合ヘテロダイマーの循環半減期を潜在的に増加させ得る(それが由来する第VIII因子タンパク質と比較して)候補モジュレーターを選択するために、(1)モジュレーターの循環半減期が修飾のために選択される第VIII因子タンパク質の循環半減期よりも大きくなるべきであること;および(2)融合タンパク質の免疫原性を考慮する。第2の考慮に関して、第VIII因子融合ヘテロダイマーで処置されることが意図される個体群(例えば、ヒト)において天然に発現されるか、または該群において天然に発現されるタンパク質に由来するモジュレーターを使用することが好ましいかもしれない。例えば、モジュレーターは、処置されることが意図される個体群の血清中に天然に存在する(例えば、ヒトが意図される処置個体群であるとき、ヒトFc領域の使用)。
【0083】
本発明の1つの態様において、免疫グロブリン定常領域はモジュレーターとして使用され得る。したがって、本発明の第VIII因子融合遺伝子の構築において使用されるモジュレーターコード核酸配列は、免疫グロブリン(Ig)のFc領域をコードするポリヌクレオチドまたはそのフラグメントおよび/または変異体、およびFcRn結合ペプチドをコードするポリヌクレオチドまたはその変異体であり得る。1つの態様において、使用される核酸は、ヒトIgG1、IgG2、IgG3、IgG4、IgE、IgD、もしくはIgM、またはマウスIgG1、IgG2a、IgG2b、IgG3、IgA、もしくはIgMから得られる免疫グロブリンのFc領域またはそのフラグメントおよび/または変異体であるモジュレーターをコードする。別の態様において、使用される核酸は、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有する(ヒンジ領域中のシステイン残基の置換または欠失による)ヒトもしくはマウスIgGのFc領域の変異体、または、ヒトもしくはマウスIgGのFc領域の非ヒンジ部分であるモジュレーターをコードする。さらなる態様において、使用される核酸は、マウスIgG1またはヒトIgG1のFc領域、または、ヒトIgG1またはマウスIgG1のFc領域の非ヒンジ部分であるモジュレーターをコードする。さらなる態様において、使用される核酸は、ヒトIgG1のFc領域、非機能的ヒンジを有する(システイン残基の置換または欠失による)ヒトIgG1の変異体Fc領域、ヒトIgG1のFc領域の非ヒンジ部分であるモジュレーターをコードする。
【0084】
免疫グロブリンのFc領域のフラグメントについて、モジュレーターをコードする核酸は、新生児のFc受容体(FcRn)により結合されるエピトープを定義するFc領域の少なくとも1つのアミノ酸セグメントをコードし得、さらにFc領域のヒンジ部分に対応するセグメントをコードし得る。あるいは、モジュレーターをコードする核酸は、少なくとも1つのFcRn結合ペプチドをコードし得る。限定はしないが、適当なFcRn結合ペプチドの例は配列PKNSSMISNTP(配列番号:24)を含み、さらにHQSLGTQ(配列番号:25)、HQNLSDGK(配列番号:26)、HQNISDGK(配列番号:27)、またはVISSHLGQ(配列番号:28)から選択される配列を含み得る(例えば、米国特許第5,739,277号、参照)。
【0085】
本発明の1つの態様において、モジュレーターは、配列番号:9(ヒトIgG1のFc領域)、配列番号:11(ヒトIgG2のFc領域)、配列番号:13(ヒトIgG3のFc領域)、配列番号:15(ヒトIgG4のFc領域)、配列番号:29(マウスIgG1のFc領域)、配列番号:17(ヒトIgG1のFc領域の非ヒンジ部分)、配列番号:19(ヒトIgG2のFc領域の非ヒンジ部分)、配列番号:21(ヒトIgG3のFc領域の非ヒンジ部分)、配列番号:23(ヒトIgG4のFc領域の非ヒンジ部分)、または配列番号:30(マウスIgG1のFc領域の非ヒンジ部分)と少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、または少なくとも約95%のアミノ酸同一性で同一であるか、または共有するアミノ酸配列をコードする核酸配列によってコードされ得る。上記の1つをコードする核酸の特定の例は、配列番号:8(ヒトIgG1のFc領域)、配列番号:10(ヒトIgG2のFc領域)、配列番号:12(ヒトIgG3のFc領域)、配列番号:14(ヒトIgG4のFc領域)、配列番号:47(マウスIgG1のFc領域)、配列番号:16(ヒトIgG1のFc領域の非ヒンジ部分)、配列番号:18(ヒトIgG2のFc領域の非ヒンジ部分)、配列番号:20(ヒトIgG3のFc領域の非ヒンジ部分)、配列番号:22(ヒトIgG4のFc領域の非ヒンジ部分)、および配列番号:48(マウスIgG1のFc領域の非ヒンジ部分)を含む。
【0086】
モジュレーターをコードする核酸を同定するための方法
他のポリペプチドまたはタンパク質は、本明細書に記載されている方法論の使用により、適当なモジュレーターとして同定され得る。候補モジュレーター(例えば、第VIII因子融合遺伝子および第VIII因子融合タンパク質の創造において潜在的に有用であり得るポリペプチド)は、治療的タンパク質への融合により非第VIII因子治療的タンパク質またはペプチドの血清半減期を延長させることが示されている該モジュレーターのペプチドおよびタンパク質を含む。例えば、第VIII因子タンパク質のモジュレーターを同定するための1つの方法は、血友病Aの動物モデル、例えば、血友病A(HemA)のマウスにおけるモジュレーターを含む第VIII因子融合ヘテロダイマーの薬物動態学を試験することである。
【0087】
モジュレーターをコードする核酸の挿入部位
本発明の第VIII因子融合遺伝子は、モジュレーターをコードする核酸を含む。モジュレーターをコードする核酸は、第VIII因子遺伝子のB−ドメイン部分内に挿入され得る。例えば、第VIII因子遺伝子のB−ドメインをコードする核酸の少なくとも1部分は、モジュレーターをコードする核酸の挿入の前または後に欠失され得る(例えば、N−745からS−1637のBドメインの部分をコードする第VIII因子遺伝子の少なくとも1部分が欠失)。あるいは、部位特異的再結合は、モジュレーターをコードする核酸を同時に挿入し、核酸をコードするB−ドメイン領域の部分を欠失するために使用され得る。挿入、欠失および部位特異的再結合を達成するための組換え方法は、当該分野でよく知られている。
【0088】
1つの態様において、第VIII因子融合遺伝子は、第VIII因子融合タンパク質をコードする核酸配列を含み、該第VIII因子融合タンパク質は、モジュレーターのアミノ酸配列がB−ドメインに存在するか、またはモジュレーターのアミノ酸配列がB−ドメインの一部または全てのアミノ酸配列と置き換わっている第VIII因子タンパク質を含む。第2の態様において、第VIII因子融合遺伝子は、配列番号:1または5のいずれか1つのアミノ酸20−764に対応する第1のアミノ酸配列、配列番号:1または5のいずれか1つのアミノ酸1656−2351に対応する第2のアミノ酸配列、および半減期モジュレーターアミノ酸配列がそのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合しているモジュレーターアミノ酸配列を含む、第VIII因子融合タンパク質をコードする核酸配列を含む。
【0089】
分泌前に、Bドメインは、Arg1648(すなわち、B−a3接合点)で開裂され、主にArg1313後で、B−ドメインにおいて可変的に開裂される(例えば、Thompson, Semin. Thromb. Hemost. 29:11-22, 2003、参照)。したがって、当業者は、B−ドメイン領域における挿入、欠失および/または置換において、B−a3ドメイン接合点で起こる切断部位が第VIII因子融合ヘテロダイマーへの第VIII因子融合タンパク質の適当な翻訳後プロセッシングのために維持されるべきであることを理解できる。同様に、B−ドメイン領域における挿入のために、モジュレーターをコードする核酸は、Arg1313をコードする核酸に対して5’であるB−ドメインをコードする核酸内の部位で挿入されるべきである。あるいは、B−ドメイン内の切断部位(B−a3接合点での切断部位を除いて)は、第VIII因子融合タンパク質の翻訳後プロセッシング中にN−末端(「重鎖」)部分からモジュレーターの切断(したがって分離)を防止するために変異され得る。
【0090】
a2−Bドメイン接合点での切断が第VIII因子のコファクター活性を産生するために必須でないことが知られている(Pittmanら., PNAS 85:2429-2433, 1988)。ヒト第VIII因子において、a2−Bドメイン接合点はArg740で生じる。本発明の第VIII因子融合遺伝子は、a2−Bドメイン接合点で切断を受ける第VIII因子融合タンパク質をコードする遺伝子ならびに切断を防止するa2−Bドメイン接合点でアミノ酸修飾を有する第VIII因子融合タンパク質をコードする遺伝子を含む。1つの例として、本発明の第VIII因子融合ヘテロダイマーの活性化時の該接合点での切断が、第VIII因子融合ヘテロダイマー由来の第VIII因子タンパク質の活性化時に生産される第VIIIa因子タンパク質と同一である(したがって、同じ生物学的活性を有する)第VIIIa因子タンパク質の形成をもたらすとき、a2−Bドメイン接合点切断部位は、無傷なままであり得る。
【0091】
当業者は、第VIII因子融合ヘテロダイマーにおけるa2−Bドメイン接合点での切断の程度または割合が、それが由来する第VIII因子タンパク質において見られるものよりも小さいとき、恐らくモジュレーターによる立体障害によると理解する。したがって、a2−Bドメイン接合点と半減期モジュレーター間のペプチドリンカー(すなわち、スペーサー)、例えば、ペプチドリンカーDDDDK(配列番号:49)およびGGGGSGGGGSGGGGS(配列番号:50)の形態においてさらなるアミノ酸を含むことが望ましいかもしれない。
【0092】
ホモもしくはヘテロ−多重合配列をコードする核酸の選択および挿入部位
本発明の第VIII因子融合遺伝子は、所望により、モジュレーターをコードする核酸と異なるホモもしくはヘテロ−多重合配列をコードする核酸を含む。ホモもしくはヘテロ−多重合配列をコードする核酸の包含は、第1の第VIII因子融合ヘテロダイマーにおいて使用されるモジュレーターが、第2の第VIII因子融合ヘテロダイマーに存在するモジュレーターの同種または異種部分と非共有結合性または共有結合性会合を介在することができないとき、マルチマー第VIII因子融合ヘテロダイマーを生産することが望ましいかもしれない。あるいは、ホモもしくはヘテロ−多重合配列をコードする核酸の包含は、第1の第VIII因子融合ヘテロダイマーにおいて使用されるモジュレーターが、単一のポリペプチドからなるとき、ハイブリッド第VIII因子融合ヘテロダイマーを生産することが望ましいかもしれない。
【0093】
当業者により理解されているとおり、ホモもしくはヘテロ−多重合配列をコードする核酸の選択は、利用されている特定の多重合アプローチにより決定される。以下の非限定的な文献において教示されるマルチマーポリペプチドを生産するために一般的なアプローチにおいて使用されるホモもしくはヘテロ−多重合配列をコードする核酸配列は、本発明の第VIII因子融合遺伝子に包含することができる。米国特許公報第2007/0287170号、米国特許第7,183,076号に記載されている「多重合ドメイン」アプローチ、例えば、免疫グロブリン部分を使用するもの、米国特許第5,932,448号において使用されているとおりのFosおよびJunロイシンジッパーの使用、および米国特許第6,833,441号に記載されている「ヘテロ二量化配列」アプローチ、および米国特許第5,807,706号に記載されている「空洞への隆起」アプローチ。
【0094】
当業者は、第1の第VIII因子融合遺伝子のみがヘテロ−多重合配列を含むとき、第2の異なる第VIII因子融合遺伝子がマルチマー第VIII因子融合ヘテロダイマーまたはハイブリッド第VIII因子融合ヘテロダイマーを生産するために必要であり得ることを理解している。例えば、当業者は、米国特許第5,807,706号に記載されている「空洞への隆起」アプローチを利用するために、それぞれ異なるヘテロ−多重合配列を含む2つの第VIII因子融合遺伝子が必要であることを認識している。
【0095】
当業者は、第VIII因子融合遺伝子内のホモもしくはヘテロ−多重合配列をコードする核酸が、モジュレーターに対して5’(すなわち、発現されたときN−末端と関連する)または3’(すなわち、発現されたときC−末端と関連する)のいずれかで第VIII因子融合遺伝子内に位置し得る(ただし、その位置が、他に、第VIII因子融合ヘテロダイマーの形成のために必要である転写、翻訳または翻訳後修飾を妨げない)ことを理解している。モジュレーターをコードする核酸のような多重合配列をコードする核酸は、Bドメインをコードする第VIII因子遺伝子の領域の少なくとも1部分内に挿入されるか、または該部分と置き換えられ得る。
【0096】
発現カセットおよび発現ベクター
本発明のさらなる局面は、第VIII因子融合遺伝子を含む発現カセットまたは発現ベクターに関する。第VIII因子融合遺伝子を含む発現カセットの組換え生産のために、第VIII因子融合遺伝子を単離し、プロモーターに作動可能に連結させる。該第VIII因子融合遺伝子は、所望によりさらに、転写終結シグナル、シグナルペプチドをコードする核酸、または遺伝子発現または翻訳後プロセッシングに影響する他の核酸配列(例えば、便利に位置された制限酵素認識部位、エンハンサー、分泌リーダー配列など)に作動可能に連結していてもよい。発現カセットの所望の因子(第VIII因子融合遺伝子以外)がすでに複製可能なクローニングベクターまたは発現ベクター内に含まれているとき、第VIII因子融合遺伝子は、当該分野でよく知られている組換え技術により適当な位置に作動可能に挿入されていることのみ必要である。多数のクローニングベクターが市販されており、一般的に1つ以上の以下のもの:シグナル配列、複製起点、エンハンサーエレメント、プロモーター、転写終結配列、および1つ以上の選択遺伝子またはマーカーを含む。多数の発現ベクターも、また、市販されており、第VIII因子融合遺伝子の挿入は、当該分野でよく知られている方法および試薬を使用して達成され得る(例えば、Sambrookら., Molecular Cloning: A Laboratory Manual, Second Edition, Cold Spring Harbor Press, NY (1989); Ausubelら., Current Protocols in Molecular Biology, New York, N.Y.: John Wiley & Sons (1989)、参照)。発現ベクターの選択は、好ましい形質転換技術および形質転換のための標的宿主に依存する。
【0097】
本発明において有用な発現ベクターは、染色体−、エピソーム−およびウイルス−由来のベクター、例えば、細菌プラスミド、バクテリオファージ、酵母エピソーム、酵母染色体エレメント、ウイルス、例えば、バキュロウイルス、パポバウイルス、ワクシニアウイルス、アデノウイルス、家禽ジフテリアウイルス、仮性狂犬病ウイルスおよびレトロウイルス由来のベクター、およびそれらの組合せ由来のベクター、例えば、コスミドおよびファージミドを含むが、これらに限定されない。動物細胞において組換え発現のために適当なウイルスベクターは、当該分野でよく知られている(例えば、米国特許第5,871,986および6,448,046号、参照)。
【0098】
本発明を実施するために適当なベクターは、以下のウイルスベクター、例えば、ラムダベクター系 gt11、gtWES.tB、Charon 4、およびプラスミドベクター、例えば、pCMV、pBR322、pBR325、pACYC177、pACYC184、pUC8、pUC9、pUC18、pUC19、pLG339、pR290、pKC37、pKC101、SV 40、pBluescript II SK +/−またはKS +/−(Stratagene, LaJolla, CA)、pQE、pIH821、pGEX、pETシリーズ(Studierら., Methods Enzymol. 185:60-89, 1990)、およびそれらのあらゆる誘導体を含むが、これらに限定されない。細菌において使用するために適当なベクターは、pQE70、pQE60、およびpQE−9(Qiagen, Valencia, CA);pBS ベクター、Phagescript ベクター、Bluescript ベクター、pNH8A、pNH16a、pNH18A、およびpNH46A(Stratagene, LaJolla, CA);pcDNA3(Invitrogen, Carlsbad, CA);およびpGEX、ptrxfus、ptrc99a、pET−5、pET−9、pKK223−3、pKK233−3、pDR540、およびpRIT5を含む。適当な真核ベクターは、pWLNEO、pSV2CAT、pOG44、pXT1、pBK、およびpSG(Stratagene, LaJolla, CA);およびpSVK3、pBPV、pMSG、およびpSVLである。他の適当なベクターは、当業者に容易に明白である。
【0099】
発現ベクターは、選択できる表現型、例えば、薬物耐性、栄養要求性、細胞毒性剤に対する抵抗性または表面タンパク質の発現を与える選択可能なマーカーをコードする遺伝子を含むものを使用され得る。使用することができる選択可能なマーカー遺伝子の例は、neo、gpt、dhfr、ada、pac(ピューロマイシン)、hyg、およびhisDを含む。
【0100】
第VIII因子融合遺伝子の成功するライゲーション(またはベクターへの挿入)は、当該分野でよく知られている組換え技術(例えば、慣用の手段を使用する単離およりシーケンシングまたは連結部位に特異的に結合することができるオリゴヌクレオチドプローブの使用)により容易に決定され得る。
【0101】
宿主細胞
本発明のさらなる局面は、第VIII因子融合遺伝子を含む宿主細胞に関する。該第VIII因子融合遺伝子は、クローニングベクター、発現ベクター内に存在するか、または宿主細胞ゲノムに統合されていてもよい。1つの態様において、宿主細胞は、DNA分子形態における必要な核酸構築物を安定なプラスミドまたは宿主細胞ゲノムへの安定な挿入もしくは統合のいずれかとして含む。別の態様において、宿主細胞は、発現系においてDNA分子を含むことができる。
【0102】
1つの態様において、本発明の第VIII因子融合遺伝子は、オープンリーディングフレームが選択されたプロモーターのコントロール下でコードされたタンパク質の発現に対して適当に指向するように、センス方向において適当なベクターに組み込まれる。これは、適当な調節エレメントの発現ベクターへの包含を含む。これらは、例えば、ベクターの非翻訳領域、有用なプロモーター、および転写および翻訳を実施するために宿主細胞タンパク質と相互作用する5’および3’非翻訳領域を含み得る。このようなエレメントは、それらの強度および特異性において変化し得る。利用されるベクター系および宿主に依存して、構成的および誘導プロモーターを含むあらゆる適当な転写および翻訳エレメントが使用され得る。構成的プロモーターは、生物体の発生および生存中に遺伝子の発現を指向するプロモーターである。誘導プロモーターは、誘導因子に応答して1つ以上のDNA配列または遺伝子の転写を直接的または間接的に活性化することができるプロモーターである。誘導因子の非存在下で、該DNA配列または遺伝子は転写されない。
【0103】
本発明の発現ベクターは、また、選択されたタンパク質をコードするDNA分子に作動可能に連結した、選択された宿主細胞における発現に対してmRNAの正しい転写終結およびポリアデニル化を提供することができるものの中から選択される、作動可能な3’調節領域を含み得る。
【0104】
宿主細胞において第VIII因子融合ヘテロダイマーを組換え的に生産するために、第VIII因子融合遺伝子は、宿主細胞に組み込まれ得る。クローニングベクター、発現ベクターおよびプラスミドは、当該分野でよく知られている組換え技術を使用して、例えば、形質転換、形質導入、接合、動員、またはエレクトロポレーションを介して細胞に導入され得る。
【0105】
宿主細胞は、哺乳動物細胞、細菌細胞(例えば、大腸菌)、昆虫細胞(例えば、Sf9細胞)、真菌細胞、酵母細胞(例えば、サッカロミセス(Saccharomyces)またはシゾサッカロミセス(SchizoSaccharomyces))、植物細胞(例えば、シロイヌナズナまたはタバコ細胞)、または藻細胞を含み得るが、これらに限定されない。本発明を実施するために適当な哺乳動物細胞は、COS(例えば、ATCC番号CRL1650または1651)、赤んぼのハムスター腎臓(「BHK」)(例えば、ATCC番号CRL6281)、チャイニーズハムスター卵巣(「CHO」)(ATCC番号CCL61)、HeLa(例えば、ATCC番号CCL2)、293(ATCC番号1573)、NSO骨髄腫、CHOP、NS−1およびHKB11(例えば、米国特許第6,136,599号、参照)を含むが、これらに限定されない。
【0106】
哺乳動物細胞における発現を指向するために適当な発現ベクターは、一般的に、プロモーター、ならびに当該分野で知られている他の転写および翻訳コントロール配列を含む。一般的なプロモーターは、SV40、MMTV、メタロチオネイン−1、アデノウイルスEla、CMV、前初期、免疫グロブリン重鎖プロモーターおよびエンハンサー、およびRSV−LTRを含む。当業者は、プロモーターとしての強度に基づいて適当な哺乳動物プロモーターを容易に選択することができる。あるいは、誘導プロモーターは、特定のタンパク質の発現または抑制が望ましいとき、制御の目的のために使用することができる。当業者は、当該分野で知られているものから適当な誘導哺乳動物プロモーターを容易に選択することができる。
【0107】
本発明の第VIII因子融合ヘテロダイマーの組換え生産のために選択される宿主細胞にかかわらず、タンパク質発現の増加は、第VIII因子融合遺伝子における一般的でないコドンをより一般的であるコドンと置き換えることにより達成され得る(例えば、米国特許第6,924,365号、参照)。当業者は、特定の第VIII因子融合遺伝子コドンが「一般的でない」または「一般的である」であるかを決定することが、組換え生産のために選択される宿主細胞の特定のコドン利用に依存することを理解している。
【0108】
第VIII因子融合タンパク質およびヘテロダイマーの生産
本明細書に記載されている組換え科学技術を考慮して、本発明の別の局面は、本発明の第VIII因子融合ヘテロダイマーを生産する方法に関する。この方法は、宿主細胞が第VIII因子融合タンパク質を発現する条件下で本発明の宿主細胞を増殖させることを含む。第VIII因子融合タンパク質の翻訳後修飾後、次に、組換え第VIII因子融合ヘテロダイマーは、精製され単離され得る。本発明の1つの局面は、第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを生産するための方法であって、(a)第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーをコードする発現ベクターで形質転換された宿主細胞を提供し;(b)該細胞を培養し;(c)第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを単離することを含む方法である。さらなる態様において、該宿主細胞は哺乳動物宿主細胞であり得、モジュレーターのアミノ酸配列はグリコシルされていてもよい。
【0109】
マルチマー第VIII因子融合ヘテロダイマーの組換え生産に関して、当業者は、ホモ−マルチマー型が、単一の組換え宿主細胞により生産され得るが、ヘテロ−マルチマー型が単一の宿主細胞内の共発現または複数の宿主細胞における別々の発現(同じか、または異なっている細胞培養系において)により生産され得ることを理解している。ヘテロ−マルチマー型と同様に、当業者は、ハイブリッド第VIII因子融合ヘテロダイマーが、単一の宿主細胞内の共発現または複数の宿主細胞における別々の発現(同じか、または異なっている細胞培養系において)により生産され得ることを理解している。別々の培養系が利用されるとき、それぞれの培養物からの組換えタンパク質産物は単離され、次に当該分野でよく知られている標準技術を使用して再び連合させ得る。マルチマー第VIII因子融合ヘテロダイマーおよびハイブリッド第VIII因子融合ヘテロダイマーの組換え生産のために、宿主細胞は、所望の様式においてマルチマーまたはハイブリッド第VIII因子融合ヘテロダイマーの鎖を組み立てることができるように選択され得る。
【0110】
別々の遺伝子の共発現における代替案として、必要とされるポリペプチド鎖のすべてをコードするモノシストロン性遺伝子が生産され得る。このような遺伝子がどのように設計され得るか特定の実施例、以下の実施例5参照。
【0111】
組換え第VIII因子融合ヘテロダイマーは、実質的に純粋な形態において生産され得る。当該分野でよく知られている方法は、精製された第VIII因子融合ヘテロダイマーの精製および同定のために使用され得る。
【0112】
医薬組成物
本発明の別の局面は、第VIII因子融合ヘテロダイマーおよび薬学的に許容される担体を含む医薬組成物に関する。「薬学的に許容される担体」は、調製物の製剤化を助けるか、または調製物を安定にさせるために活性成分に加えられ得、患者へ有意な有害な毒性効果を与えない物質である。このような担体の例は、当業者によく知られており、水、糖、例えば、マルトースまたはスクロース、アルブミン、塩、例えば、塩化ナトリウムなどを含む。他の担体は、例えば、Remington's Pharmaceutical Sciences by E. W. Martinに記載されている。このような組成物は、有効量の少なくとも1つの第VIII因子融合ヘテロダイマーを含む。
【0113】
薬学的に許容される担体は、滅菌水性溶液または分散媒および滅菌注射可能溶液または分散媒の即時調製物のための滅菌粉末を含む。薬学的に有効な物質に対するこのような媒体および薬剤の使用は、当該分野で知られている。該組成物は、非経口注射のために製剤化され得る。該組成物は、高い薬物濃度に適当な溶液、マイクロエマルジョン、リポソームまたは他の規則構造として製剤化され得る。該担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、および適当なそれらの混合物を含む溶媒または分散媒であり得る。該組成物は、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール、ソルビトールまたは塩化ナトリウムを含み得る。第VIII因子の医薬組成物の例は、例えば、米国特許第5,047,249;5,656,289;5,665,700;5,690,954;5,733,873;5,919,766;5,925,739;6,835,372;および7,087,723号において記載されている。
【0114】
滅菌注射可能溶液は、上記されている成分の1つまたは組合せを有する適当な溶媒中に必要な量の活性化合物を包含し、必要なとき、次に滅菌精密濾過することにより製造され得る。一般的に、分散媒は、活性化合物を塩基性分散媒および必要な他の成分を含む滅菌輸送媒体に包含させることにより製造され得る。滅菌注射可能溶液の調製物に関する滅菌粉末の場合において、製造方法は、あらかじめ滅菌濾過された溶液から活性成分プラス任意のさらなる所望の成分の粉末を得る真空乾燥およびフリーズドライ(凍結乾燥)を含む。
【0115】
処置の方法
本発明の別の局面は、遺伝的および後天的凝固欠乏症、例えば、血友病(例えば、血友病A)を処置する方法に関する。この方法は、血友病Aを示す患者に有効量の本発明の第VIII因子融合ヘテロダイマー(ハイブリッドまたはマルチマー型を含む)を投与し、それにより該患者が血管損傷後の有効な血液凝固を示すことを含む。適当な有効量の第VIII因子融合ヘテロダイマーは、限定はされないが、約10から約50国際ユニット/kg体重からなる。患者は、何らかの哺乳動物(例えば、ヒト)であり得る。
【0116】
本発明の第VIII因子融合ヘテロダイマーは、静脈内に、皮下に、または筋肉内に投与され得る。特定のモジュレーターは、経口投与が考慮され得る。
【0117】
本発明の第VIII因子融合ヘテロダイマーは、阻害抗体を有する、およびそれを有さない血友病患者ならびに阻害抗体の発現によって後天的第VIII因子欠乏を有する患者における第VIII因子欠乏(例えば、関節内、頭蓋内または消化器出血)による非制御出血を処置するために使用され得る。1つの態様において、第VIII因子融合ヘテロダイマーは、単独または医薬組成物(すなわち、安定剤、送達輸送媒体、および/または担体と組み合わせて)の形態において、ヒトまたは動物第VIII因子の注入のために使用されるのと同じ方法にしたがって、患者の静脈内に注入される。
【0118】
あるいは、またはそれらに加えて、第VIII因子融合ヘテロダイマーは、第VIII因子融合遺伝子発現構築物を含むウイルスベクター、例えば、アデノ随伴ウイルス(例えば、Gnatenkoら., Br. J. Haematol. 104:27-36, 1999、参照)を投与することにより、または一般的にこのような細胞を含むデバイスの移植により、第VIII因子融合ヘテロダイマーを生産するように遺伝的に操作された細胞を移植することにより投与され得る。このような移植は、組換え真皮繊維芽細胞(例えば、Rothら., New Engl. J. Med. 344:1735-1742, 2001、参照);骨髄間質細胞(例えば、米国特許第6,991,787号、参照)または造血前駆宿主細胞(例えば、米国特許第7,198,950号、参照)を使用することを含む。血友病Aの遺伝子治療(第VIII因子をコードする核酸を使用する)における使用のために適当なウイルスベクターおよび遺伝子治療におけるそれらの使用は、当該分野で知られている(例えば、米国特許第6,200,560;6,544,771;6,649,375;6,697,669;6,773,709;6,797,505;6,808,905;6,818,439;6,897,045;6,939,862;7,198,950;および7,238,346号、参照)。
【0119】
このような処置の必要な患者に投与されるべき第VIII因子融合ヘテロダイマーの処置用量は、第VIII因子欠乏の重症度に依存して変化する。一般的に、投与レベルは、それぞれの患者の出血症状の重症度および期間としたがって、頻度、期間およびユニットにおいて調節される。したがって、第VIII因子融合ヘテロダイマーは、標準凝固アッセイにより測定されるとき、患者に治療有効量の出血を止めるためのタンパク質を送達するために十分な量における薬学的に許容される担体、送達輸送媒体、または安定剤中に含まれ得る。
【0120】
通常、第VIII因子融合ヘテロダイマーの投与を介して患者中で達成されるべき所望の血漿の第VIII因子の活性レベルは、通常の30−100%の範囲である。1つの態様において、治療的第VIII因子融合ヘテロダイマーの投与は、約5から約50ユニット/kg体重の範囲、約10から約50ユニット/kg体重の範囲における用量、および約20から約40ユニット/kg体重の用量で静脈内に与えられ得;間隔頻度は、約8から24時間の範囲であり得(深刻な血友病患者において);処置の日数は1から10日間の範囲もしくは出血症状が解決するまでであり得、または第VIII因子融合ヘテロダイマーの投与は予防であり得る(例えば、Robertsら., pp 1453-1474, 1460, in Hematology, Williams, W. Jら., ed. (1990)、参照)。ヒトまたは血漿由来第VIII因子での処置におけるとき、注入される治療的組換え第VIII因子の量は、一段階の第VIII因子凝固アッセイにより定義され得、選択された例において、インビボでの回収率は、注入後の患者の血漿中の第VIII因子を測定することにより測定され得る。あらゆる特定の患者に対して、特定の投与レジメンは、必要な個体および組成物を投与する、またはそれの投与を管理する個体の専門的な判断にしたがって時間をかけて調節されるべきであり、本明細書に記載されている濃度範囲は典型的なだけであり、本発明の第VIII因子融合ヘテロダイマーの範囲または実施を限定すると意図すべきでないと理解すべきである。
【0121】
処置は、単回投与または長期間の周期的もしくは連続的投与の形態を取り得、または、必要なとき、処置は、予防目的のために投与され得る。
【0122】
本発明の第VIII因子融合ヘテロダイマーは、由来の第VIII因子タンパク質と比較して循環半減期の増加を示す。より良い循環半減期を有する第VIII因子タンパク質は、より少ない頻度の投与で患者の第VIII因子欠乏を正すことができるため、血友病の処置において有用である。投与の場合におけるこの増加は、処置プロトコールでの患者のコンプライアンスを改善し、それにより凝固障害の症状を減少させ得る。また、投与の頻度の減少は、少ない抗原が投与されるため、第VIII因子に対する免疫応答の発達の可能性を減少させることが期待される。
【0123】
上記記載は、一般的に本発明を記載している。説明のみの目的のために提供され、本発明の範囲を限定することを意図しない以下の実施例を参照することにより、さらなる完全な理解が得ることができる。
【実施例】
【0124】
実施例
本発明を良く理解するために、以下の実施例を記載する。これらの実施例は、説明のみの目的のためであり、いかなる形であれ本発明の範囲を限定すると解釈されるべきでない。本明細書に記載されている全ての文献は、出典明示によりそれら全体を包含させる。
【0125】
実施例1
以下の実施例は、第VIII因子B−ドメイン欠失(BDD)タンパク質をコードする核酸およびマウスFc領域(「mFc+ヒンジ」と示される)をコードする核酸を使用する、第VIII因子融合遺伝子(「BDDmFc+ヒンジ」と示される)を含む哺乳動物発現ベクター(「pM110」または「pSK207BDDFc+ヒンジ」と示される)の構築を記載する。BDDmFc+ヒンジの組換え発現は、図2において示されている第VIII因子融合ヘテロダイマー(「BDDFc+ヒンジ」と示される)の生産をもたらす。機能的免疫グロブリンヒンジ領域の存在によって、BDDFc+ヒンジの2つの分子は、マルチマー第VIII因子融合ヘテロダイマーを形成するためにジスルフィド結合を介して共有結合している。この型の利点は、ダイマーFcがFcRnへの融合タンパク質の高親和性結合をもたらし、その結果、長期循環半減期をもたらすことである。
【0126】
PmeIおよびNheI部位により結合された第VIII因子B−ドメイン欠失(BDD)遺伝子を含むプラスミドpSK207(「pSK207BDD」と示される)は、部位特異的変異誘発キットを使用して変異させた。2つの制限酵素認識部位(bp4490でAvrIIおよびbp4520でAflII)は、変異プライマーCES16(5’−caatgccattgaacctaggagcttctcccagaacccaccagtccttaagcgccatcaacggg−3’)(配列番号:34)およびCES17(5’−cccgttgatggcgcttaaggactggtgggttctgggagaagctcctaggttcaatggcattg−3’)(配列番号:35)を使用して分子に導入された。bp2537でのAflII部位は、変異オリゴCES18(5’−cagtggtcattacactcaagaacatggcttcccatcc−3’)(配列番号:36)およびCES19(5’−ggatgggaagccatgttcttgagtgtaatgaccactg−3’)(配列番号:37)を使用して除去した。得られたプラスミドをpM109と称した。これらの変異事象は全てサイレントであり、BDDに対するアミノ酸変化をもたらさなかった。マウスFc領域の供給源として、ヒト上皮細胞増殖因子受容体に対する全長マウスIgG1抗体を含むプラスミドpGT234を使用した。マウスFc+ヒンジ領域を、鋳型としてpGT234でプライマーCES36(5’−agcttcctaggagcttctcccagaacgtgcccagggattgtggttg−3’)(配列番号:38)およびCES39(5’−agctacttaaggactggtgggttctgggatttaccaggagagtgggagag−3’)(配列番号:39)を使用してPCR増幅させた。得られたフラグメントをAflII/AvrIIで消化し、AflII/AvrIIで消化されたpM109にクローニングし、プラスミドpM117を製造した。bp2537にて元のAflII部位を復帰させるために、pSK207+BDDのNheI/BglIIフラグメントをpM117に挿入して、その同等領域を置き換え、bp2537にAflII部位を含むプラスミドpM115を製造した。pM115のBDD.mFc+ヒンジ遺伝子(5077bpのNheI/PmeIフラグメントに含まれる)を発現ベクターpSS207のPmeI/NheI部位にクローニングし、プラスミドpM110またはpSK207BDDFc+ヒンジを産生した。pSK207BDDFc+ヒンジの第VIII因子融合遺伝子因子(すなわち、BDDmFc+ヒンジ)は、配列番号:31の核酸配列を有する。BDDmFc+ヒンジによりコードされるタンパク質は、図3(「マウスFc」はmFc+ヒンジの位置を示す)において説明される構造ドメイン、および配列番号:32のアミノ酸配列を有する。
【0127】
実施例2
以下の実施例は、第VIII因子B−ドメイン欠失(BDD)タンパク質をコードする核酸およびマウスFc領域のヒンジ部分(「mFc−ヒンジ」と示される)以外の全てをコードする核酸を使用する、第VIII因子融合遺伝子(「BDDmFc−ヒンジ」と示される)を含む哺乳動物発現ベクター(「pM118」または「pSS207BDDFc−ヒンジ」と示される)の構築物を記載する。BDDmFc−ヒンジの組換え発現は、図2において示されている第VIII因子融合ヘテロダイマー(「BDDFc−ヒンジ」と示される)の生産をもたらす。BDDmFc−ヒンジによりコードされるタンパク質は、図3(「マウスFc」はmFc−ヒンジの位置を示す)において説明される構造ドメインおよび配列番号:33のアミノ酸配列を有する。機能的免疫グロブリンヒンジ領域の非存在によって、BDDFc−ヒンジはマルチマー第VIII因子融合ヘテロダイマーを形成しない。この型の不利な点は、非ダイマー化Fc領域がFcRn結合に対する親和性を減少させることである。
【0128】
BDDmFc−ヒンジの構築物は、上記BDDmFc+ヒンジと非常に類似している。mFc−ヒンジ領域を、PCRプライマーCES37(5’−agcttcctaggagcttctcccagaacgtcccagaagtatcatctgtc−3’)(配列番号:40)およびCES39(配列番号:39)を使用してプラスミドpGT234からPCR増幅させ、AvrII/AflIIで消化し、AvrII/AflIIで消化されたpM109プラスミドにおいてクローニングした。得られたプラスミドをpM114(「pSK207.BDD.mFc−ヒンジ」とも称する)と称した。次に、BDD.mFc−ヒンジ遺伝子を含むpM114のNheI/PmeIフラグメントを発現ベクターpSS207にクローニングし、プラスミドpM118(すなわち、pSS207BDDFc−ヒンジ)を産生した。
【0129】
実施例3
以下の実施例は、アミノ末端でFlagタグを有するマウスFc領域(「mFc+ヒンジ」と示される)の発現のためのプラスミド(「pM130」と示される)の構築物を記載する。同じ宿主細胞においてpSS207BDDFc+ヒンジを有するこのプラスミドの共発現は、BDDFc+ヒンジダイマー、mFc+ヒンジダイマーならびにBDDFc+ヒンジおよびmFc+ヒンジのヘテロダイマーの混合物を製造する。Flagタグの包含は、連続分離工程において抗−Flag抗体および抗−第VIII因子抗体を使用する親和性クロマトグラフィーによる図2において示されているヘテロダイマー(「BDDFc」と示される)の単離を容易にする。しかしながら、当業者は、ペプチドタグの提供がなくても、当該分野でよく知られている技術、例えば、サイズ排除クロマトグラフィーを使用してヘテロダイマー形態を分離することが可能であることを理解している。
【0130】
鋳型としてpM110を使用して、5’(アミノ末端)末端にFlagタグを有するマウスFc領域(ヒンジを有する)を、プライマーCES49(5’−atatgatatcgcggccgccgccaccatggtgttgcagacccaggtcttcatttctctgttgctctggatctctggtgcctacggggactacaaagacgatgacgacaaggtgcccagggattgtggttg−3’)(配列番号:41)およびCES50(5’−ttcgatctcgagtcatttaccaggagagtgggagagg−3’)(配列番号:42)を使用してPCR増幅させた。このフラグメントをNotI/XhoIで消化し、NotI/XhoIで消化された発現ベクターpAGE16にライゲートし、プラスミドpM119(すなわち、pAGE16.mFc+ヒンジ.Flag)を製造した。次に、mFc+ヒンジ.Flag領域を含むpM119のHindIII/XhoIフラグメントをHindIII/XhoIで消化された発現プラスミドpEAK flCMV W/GFPにサブクローニングし、pM130と称した。
【0131】
実施例4
以下の実施例は、第VIII因子B−ドメイン欠失(BDD)タンパク質をコードする核酸およびIgG1、IgG2、IgG3もしくはIgG4のヒトFc領域のいずれか1つ、またはIgG1、IgG2、IgG3もしくはIgG4のヒトFc領域の非ヒンジ部分のいずれか1つをコードする核酸を使用する第VIII因子融合遺伝子(「BDD.Human Fc」と示される)の構築物を記載する。1つの例として、第VIII因子融合ヘテロダイマーを、Bドメインを模倣するために、マウスIgG1 Fc由来の227アミノ酸残基または214アミノ酸残基を第FVIII因子の特定の部位(例えば、N−745からS−1637)に挿入することにより産生し得る。
【0132】
BDD.Human Fc(IgG1、IgG2、IgG3もしくはIgG4抗体由来)発現ベクターの構築物は、上記BDD−マウスFc発現構築物と同じ戦略に従う。pM109プラスミドをAvrII/AflIIで消化し、AvrII/AflII結合Fc+ヒンジおよびFc−ヒンジを対応する部位に挿入する。次に、pSK骨格を有する得られたプラスミドをNheIおよびPmeIで消化し、BDDFcフラグメントを安定な細胞クローンの創造のためにpSS207にライゲートした。同様に、pCEP4.human Fcモノマープラスミドを構築する。
【0133】
典型的なヒトIgGのFc領域の核酸コード配列は、配列番号:8(ヒトIgG1のFc領域)、配列番号:10(ヒトIgG2のFc領域)、配列番号:12(ヒトIgG3のFc領域)および配列番号:14(ヒトIgG4のFc領域)を含む。あるいは、配列番号:9(ヒトIgG1のFc領域)、配列番号:11(ヒトIgG2のFc領域)、配列番号:13(ヒトIgG3のFc領域)および配列番号:15(ヒトIgG4のFc領域)と同じアミノ酸配列(または少なくとも90%同一性を有するアミノ酸配列)をコードする核酸配列を使用してもよい。典型的な非ヒンジ部分のヒトIgGのFc領域の核酸コード配列は、配列番号:16(ヒトIgG1のFc領域の非ヒンジ部分)、配列番号:18(ヒトIgG2のFc領域の非ヒンジ部分)、配列番号:20(ヒトIgG3のFc領域の非ヒンジ部分)および配列番号:22(ヒトIgG4のFc領域の非ヒンジ部分)を含む。あるいは、配列番号:17(ヒトIgG1のFc領域の非ヒンジ部分)、配列番号:19(ヒトIgG2のFc領域の非ヒンジ部分)、配列番号:21(ヒトIgG3のFc領域の非ヒンジ部分)および配列番号:23(ヒトIgG4のFc領域の非ヒンジ部分)と同じアミノ酸配列(または少なくとも90%同一性を有するアミノ酸配列)をコードする核酸配列を使用してもよい。
【0134】
実施例5
以下の実施例は、ハイブリッド第VIII因子融合ヘテロダイマーをコードするモノシストロン性第VIII因子融合遺伝子の構築物を記載する。翻訳された第VIII因子融合タンパク質は、全長FVIIIのBドメインの代わりに2つのタンデムmFc+ヒンジ領域を含む。
【0135】
発現プラスミドを以下のとおり構築する:鋳型としてpM117(pSK207+BDD.mFc+ヒンジ)を使用するオリゴの2つのセットでのPCR−第1は、AvrII(CES36:5’−agcttcctaggagcttctcccagaacgtgcccagggattgtggttg−3’)(配列番号:30)およびSacII(CES51:5’−cagttgccgcgggctttaccaggagagtgggagagg−3’)(配列番号:35)により結合するmFcフラグメントを創造するCES36/CES51であり、プライマーの第2のセットは、SacII(CES52:5’−ttcgcccgcggcaagagagactacaaagacgatgacgacaaggtgcccagggattgtggttg−3’)(配列番号:35)およびAflII(CES39:5’−agctacttaaggactggtgggttctgggatttaccaggagagtgggagag−3’)(配列番号:31)により結合するmFcフラグメントを創造するCES52/CES39である。適当に消化およびライゲートされたとき、これらの2つの得られたPCRフラグメントは、順にA1、a1、A2、a2ドメイン、B−ドメインの最初の5個のN−末端アミノ酸、次にmFc+ヒンジ領域、furinコンセンサス配列(Fc領域の末端である最初のリジン(K)を有するKARGKR(配列番号:36))、Flagタグ(DYKDDDDK)(配列番号:37)、mFc+ヒンジ、B−ドメインの最後の12個のC−末端アミノ酸、ならびに最後にFVIIIのa3、A3、C1およびC2ドメイン(図4)を含むモノシストロン性BDD遺伝子となる。上記モノマー遺伝子を構築するために、2つのPCRフラグメントをAvrII/SacIIまたはSacII/AflIIで消化し、トリプルライゲーションを介して、AvrII/AflIIで消化したpM109(pSK207.BDD)にクローニングする。成功したクローンを配列決定し、次に、1つをpSK207骨格由来のNheI/PmeI(pM109の)を介して発現ベクターにクローニングし、pSK207をNheI/PmeIで消化する。タンパク質の合成および分泌中、該分子を最初にFlag−mFc領域の上流のfurin部位およびa3のすぐ上流のプロテアーゼ部位で開裂させる。該分子は、図2において示されているとおりにFc−Fcジスルフィド相互作用(BDDFc)を介してFVIII分子のmFc領域に結合しているFlag mFc分子を有する成熟FVIIIダイマー(BドメインをmFcで置き換えられている)として循環する。ヘテロダイマー産物を、当業者に知られている方法を使用して、上清中に存在するすべてのホモダイマー産物から単離する。
【0136】
実施例6
以下の実施例は、哺乳動物宿主細胞の一過性トランスフェクションのために有用な一般的な処理およびその細胞培養を記載する。HKB11細胞をオービタルシェーカー(100−125rpm)上で5%CO2インキュベーター中で37℃でタンパク質非含有培地中で懸濁培養中で増殖させ、0.25から1.5×106細胞/mLの密度で維持する。トランスフェクションのためのHKB11細胞を1,000rpmで5分遠心分離により回収し、次に、1.1×106細胞/mLでFreeStyleTM293発現培地(Invitrogen Corporation, Carlsbad, CA)に再懸濁する。細胞を6つのウェルプレートに播種し(4.6mL/ウェル)、オービタルローター上で(125rpm)37℃ CO2インキュベーター中でインキュベートする。それぞれのウェルに対して、5μgのプラスミドDNAを0.2mlのOpti−MEM(登録商標)I培地(Invitrogen Corporation, Carlsbad, CA)と混合する。それぞれのウェルに対して、7μLの293FectinTM試薬(Invitrogen Corporation, Carlsbad, CA)を0.2mLのOpti−MEM(登録商標)I培地と穏やかに混合し、室温で5分インキュベートする。希釈された293FectinTMを希釈されたDNA溶液に加え、穏やかに混合し、室温で20−30分インキュベートし、次に、5×106(4.6mL)のHKB11細胞で播種されたそれぞれのウェルに加える。次に、細胞をオービタルローター(125rpm)上でCO2インキュベーター中で37℃で3日間インキュベートし、その後、細胞を1000rpmで5分、遠心分離によりペレット化し、次に、上清を回収し、−80℃で保存する。
【0137】
実施例7
以下の実施例は、ウエスタンブロッティングによる第VIII因子融合ヘテロダイマーの組換え生産を証明するために有用な一般的な処理を記載する。細胞培養物上清を、Centricon(登録商標)(Millipore Corporation, Billerica, MA)により10倍に濃縮するか(二次抗体がプロービングのために使用されないとき)、またはそのまま使用する(二次抗体がプロービングのために使用されるとき)。50μLの上清をDTT(還元)有りまたはDTT(非還元)無しで20μLの4×SDS−PAGEローディング色素と混合し、95℃で5分加熱し、次に10%のNuPAGE(登録商標)ゲル(Invitrogen Corporation, Carlsbad, CA)(還元条件下)または4−20%のNuPAGE(登録商標)ゲル(Bis-Tris-MOPs)(非還元条件下)上に負荷する。タンパク質をニトロセルロース膜に移す。5%ミルク/PBSで60分ブロッキング後、膜をマウスIgG(H+L)に対するセイヨウワサビペルオキシダーゼ(HRP)−標識ウサギポリクローナル抗体またはHRP−接合抗−第VIII因子Cドメイン抗体とインキュベートする。また、抗−ヒト第VIII因子ウサギモノクローナル抗体(Epitomics, CA)を第VIII因子の軽鎖を検出するために使用され得る。次に、膜を抗ウサギIgG−HRP二次抗体と室温で60分インキュベートする。ブロットをPBS/0.1%のTween(登録商標)−20(ポリオキシエチレンモノラウリン酸ソルビタン)で洗浄後、HRP由来のシグナルを化学発光基質(ECL)(Pierce, Rockford, IL)およびx線フィルムへの暴露を使用して検出する
【0138】
実施例8
以下の実施例は、ELISAによる細胞培養物上清中の第VIII因子抗原の濃度を測定するために有用である一般的な処理を記載する。細胞培養物上清をPBS/BSA/Tween(登録商標)−20バッファーで希釈し、標準曲線の範囲内のシグナルを達成する。例えば、PBS/BSA/Tween(登録商標)−20で希釈され、精製された(比活性9,700IU/mg)第VIII因子BDDタンパク質を、100ng/mLから0.2ng/mLの標準曲線を創造するために使用され得る。希釈されたサンプルおよび標準物を、ポリクローナル抗−第VIII因子捕捉抗体C2であらかじめ被覆されているELISAプレートに加える。検出抗体としてビオチン化C2を加えた後、プレートを室温で1時間インキュベートし、広範囲に洗浄し、次にキット製造業者(Pierce, Rockford, IL)により記載されているとおりにTMB基質(3,3’,5,5’−テトラメチルベンジジン)を使用して現像する。シグナルをSpectraMax(登録商標)プレートリーダー(SpectraMax(登録商標)340pc, Molecular Devices, Sunnyvale, CA)を使用して450nMで測定され得る。標準曲線を4つのパラメーターモデルに合わせ、未知の値を曲線から推定する。
【0139】
無傷な第VIII因子融合ヘテロダイマーに特異的ではない上記処理の別法として、ELISAアッセイは、捕捉抗体(または検出抗体)として抗−第VIII因子抗体および検出抗体(または捕捉抗体)として半減期モジュレーターに特異的な抗体を利用する。
【0140】
実施例9
以下の実施例は、96ウェル型の市販の発色アッセイキット(Coatest(登録商標) SP4 FVIII, Chromogenix, Lexington, MA)を使用する細胞培養物上清および精製された画分中の第VIII因子融合ヘテロダイマーの活性を測定するために有用な一般的な処理を記載する。トリプリケートサンプルをキットアッセイバッファー(50mMのTris、pH7.3、10mg/Lのシプロフロキサシン(ciprofloxin)および1.0%のBSA)で25μLに希釈し、ウェルに加える。次に、50μLのリン脂質、第IXa因子、第X因子溶液をそれぞれのウェルに加え、水平シェーカー上で37℃で4分インキュベートする。25μLのCaCl2溶液(25mM)を即座にウェルに加え、同じ様式で10分インキュベートする。発色基質溶液(50μL/ウェル)を加え、プレートを10分インキュベートし、カラー現像を25μLの20%酢酸の添加により停止させる。個々のウェルを、405nmでの吸光度を96−ウェルプレートリーダー(SpectraMax(登録商標) 340pc, Molecular Devices, Sunnyvale, CA)で測定する。第VIII因子活性を、未知として、同じバッファーで希釈された500−0.5mIU/mLの範囲である精製された第VIII因子B−ドメイン欠失(BDD)標準に対して定量し、4つのパラメーターモデルに合わせる。比活性(FVIIIのIU/mg)をCoatest(登録商標)および第VIII因子のELISAの結果から計算する。
【0141】
実施例10
以下の実施例は、aPTTアッセイを使用する細胞培養物上清および精製された画分中の第VIII因子融合ヘテロダイマーの凝固活性を測定するために有用な一般的な処理を記載する。第VIII因子の凝固活性は、ElectraTM 1800C 自動凝固分析器(Beckman Coulter Inc., Fullerton, CA)により第VIII因子欠乏ヒト血漿においてaPTTアッセイを使用して測定され得る。簡潔には、凝固希釈剤中の上清サンプルの3つの希釈物を器具により作製し、次に、100μLを100μLのFactorVIII欠乏血漿および100μLの自動aPTT試薬(ウサギ脳リン脂質および微粉化シリカ、bioMerieux, Inc., Durham, NC)と混合する。100μLの25mMのCaCl2溶液の添加後、血塊形成の時間を記録する。標準曲線を、ELISAアッセイにおいて標準として使用されるのと同じ精製された第VIII因子BDDの連続希釈を使用してそれぞれ負荷して作成する。標準曲線は、0.95またはそれ以上の相関係数と直線的であった。標準曲線は、未知のサンプルの第VIII因子の活性を決定するために使用される。
【0142】
実施例11
以下の実施例は、実施例1および2に記載されているベクターを使用する細胞系の安定なトランスフェクションおよび創造を記載する。HKB11細胞を、実施例6に記載されている293FectinTM試薬を使用してプラスミドDNA、pSK207BDDFc+ヒンジまたはpSK207BDDFc−ヒンジでトランスフェクトした。トランスフェクトされた細胞を種々の希釈物(1:100;1:1000;1:10,000)で100mm培養皿に分け、5%のFBSおよび200μg/mLのハイグロマイシン(Invitrogen Corporation, Carlsbad, CA)を補ったDMEM−F12培地に約2週間維持した。個々の単一のコロニーをつつき取り、滅菌クローニングディスク(Scienceware(登録商標), Bel-Art Products, Pequannock, NJ)を使用して6−ウェルプレートに移した。pSK207BDDFc+ヒンジでトランスフェクトされたHKB11細胞の55を越えるクローンが確立され、寄託された。これらのクローンを、図5において示されている第VIII因子の活性アッセイ(上記のCoatest(登録商標)およびaPTTアッセイ)、図6において示されている第VIII因子のELISA(上記)、および増殖アッセイにより第VIII因子融合ヘテロダイマーの高い発現に関してスクリーニングした。非常に高い発現レベルを有する6つの細胞系を図6に示す。BDDFc+ヒンジに関してトップのクローンであるクローン8は、粘着して増殖するとき、〜1μg/mLの融合タンパク質を発現する。クローン8馴化培地由来のBDDFc+ヒンジの比活性は、由来のBDD第VIII因子タンパク質に相当する約5,000−8,000IU/mgであった。同様の安定なトランスフェクションおよび選択処理を使用して、BDDFc−ヒンジに関するクローン(クローンt)は、接着して増殖するとき、〜1μg/mLの融合タンパク質を発現することを決定した。
【0143】
実施例12
以下の実施例は、10L WAVE BioreactorTM(GE Healthcare, Piscataway, NJ)を使用する安定な形質転換体によるタンパク質発現のスケールアップを記載する。クローン8およびクローンt細胞を5%のFBSおよび200μg/mLのハイグロマイシンを補ったDMEM−F12培地に維持した。細胞を3日毎にT75からT225フラスコに1:4で分けた。培養適応のために、12個のT225フラスコの約10億個の細胞を2Lもしくは3L−Erlenmyerフラスコ中の2.5%のFBSを補った無血清である1Lの懸濁培地に移した。2日後、細胞を1.25%のFBSを補った無血清懸濁培地に拡大した。次に、細胞を5%のヒト血漿タンパク質溶液(HPPS)を補った無血清懸濁培地に移した。約100−150億個の細胞を10L WAVE BioreactorTMバッグにおいて培地中に約100万/mlの密度で播種した。3日後、細胞密度は5−600万/mLに到達し、馴化培地を回収した。最初に、粗培地を、蠕動ポンプにより制御されている150mL/分の流速で、Contifuge(登録商標) Stratos(Thermo Fisher Scientific, Waltham, MA)で6,000rpmで連続的な遠心分離により細胞残屑を除去して浄化した。浄化された培地をX−100(ポリエチレングリコール tert−オクチルフェニルエーテル)(0.05%まで)と混合し、10kDa Pellicon 接線流膜(Millipore, Billerica, MA)における限外濾過により約10倍に濃縮した。−80℃で凍結する前にスクロースを1%の濃度に加えた。精製前の組換え的に生産される第VIII因子融合ヘテロダイマーの比活性は、クローン8により生産されるBDDFc+ヒンジについて10,629IU/mgおよびクローンtにより生産されるBDDFc−ヒンジについて11,122IU/mgと測定された。
【0144】
実施例13
以下の実施例は、クローン8のスケールアップ培養物からの第VIII因子融合ヘテロダイマーの精製を記載する。抗−第VIII因子モノクローナル抗体親和性カラム(C7F7)、次に陰イオン交換Q−セファロースTMカラム(GE Healthcare, Piscataway, NJ)を使用して、第VIII因子BDDFc+ヒンジをHKB11細胞馴化培地から精製した。全回収率は約30%であった。10L WAVE BioreactorTM バッグからの凍結濃縮物を解凍し、AEKTATM Purifier システム(Amersham Pharmacia, Uppsala, SW)を使用して1mL/分で免疫親和性カラムに負荷し、次にカラムをバッファー(20mMのイミダゾール、0.01MのCaCl2、0.5MのNaCl、0.01%のTween(登録商標)−80(ポリエチレングリコール ソルビタンオレイン酸モノエステル)、pH7.0)で洗浄した。結合した第VIII因子BDDFc+ヒンジを1.0MのCaCl2を含むバッファーで溶離した。画分をCoatest(登録商標)アッセイにより第VIII因子活性についてアッセイし、活性な画分を貯め、バッファーをHiTrapTM 26/10 脱塩カラム G25M(Amersham Biosciences, Uppsala, SW)でイオン交換ローディングバッファー(20mMのイミダゾール、10mMのCaCl2、200mMのNaCl、0.01%のTween(登録商標)−80、pH7.0)に交換した。タンパク質を1mlのHiTrapTM Q HP カラム(Amersham Biosciences, Uppsala, SW)に負荷し、NaCl勾配(200mM−1000mM)で溶離した。画分をCoatest(登録商標)アッセイにより第VIII因子活性についてアッセイし、ピーク画分を貯めた。タンパク質濃度および比活性を決定した。ベストな画分(すなわち、図7のレーン8の画分5)の純度は、SDS−PAGEおよびSimplyBlueTM染色(Invitrogen, Carlsbad, CA)により概算されるとおり約80%である。ウエスタンブロット分析において抗−Fc抗体により検出されるため、精製された融合タンパク質はFcドメインを含んだ。精製された物質の比活性は、約10,000IU/mgであった。この比活性は、BDDFc+ヒンジが完全に活性であることを示唆する第VIII因子BDD(BDDFc+ヒンジ由来)にほぼ匹敵する。
【0145】
実施例14
以下の実施例は、組換え的に生産される第VIII因子融合ヘテロダイマーにおけるエンドトキシン試験を記載する。精製されたタンパク質溶液のエンドトキシンレベルを、0.005EU/mLの感度で動態発色 Limulus Amebocyte Lysate アッセイ(Endosafe(登録商標)キット)を使用して決定した。BDDFc+ヒンジにおけるエンドトキシンレベルは、5EU/用量をはるかに下回る1.3−2.0EU/mLと見出された。
【0146】
実施例15
以下の実施例は、これらの第VIII因子融合ヘテロダイマーが由来する精製されたBDDFc+ヒンジ、精製されたBDDFc−ヒンジおよび第VIII因子タンパク質(「BDD−FVIII」)を使用する正常マウスの薬物動態学試験を記載する。正常C57オスマウスを、50μg/kg体重でBDDおよび融合タンパク質(BDDFc+ヒンジまたはBDDFc−ヒンジ)の単回投与で静脈内に注射した。血液サンプルをt=注射後0、0.083、0.5、2、4、6、8、24、28、32、48および72時間(時点あたり5匹のマウス)で回収した。血液サンプルにおけるタンパク質レベル(抗原ELISAによる)および凝固活性(Coatest(登録商標)アッセイによる)の両方を薬物動態学分析のために決定した。結果は表1に示されている。
【表1】

【0147】
BDDFc+ヒンジおよびBDDFc−ヒンジのベータ半減期は、正常マウスにおけるBDD−FVIIIと同レベルであった。
【0148】
実施例16
以下の実施例は、精製されたBDDFc−ヒンジ、およびBDDFc−ヒンジが由来する第VIII因子タンパク質(「BDD−FVIII」)を使用する血友病A動物モデル(HemAマウス)における薬物動態学試験を記載する。結果は、BDDFc−ヒンジがBDD−FVIIIと比較してHemAマウスにおいて有意な長期ベータ相半減期を有することを示す。
【0149】
HemAマウスを、5%のアルブミンを含む製剤バッファー中の1.25μg/マウス(50μg/kg)でBDDFc−ヒンジ(「FVIII−Fc」9匹のマウス)を尾静脈(i.v.)を介して注射した。さらなるHemAマウスに、200IU/kgのBDD−FVIII;BDDFc−ヒンジ由来の第VIII因子変異体を投与した。血液を、BDDFc−ヒンジを与えた代わりのマウス(3匹のマウス/時点)から1、24、48、66、72、90、120および148時間目に、およびBDD−FVIIIを与えた代わりのマウス(5匹のマウス/時点)から1、4、8、16、24および32時間目に眼窩後を介してクエン酸塩中に回収した。血漿FVIII活性をCoatest(登録商標) SP FVIIIキット(Instrumentation Laboratory Company, Lexington, MA)を使用して測定した。ベータ相半減期をWinNonlin(登録商標)(Pharsight, Mountain View, CA)における少量のサンプリング(sparse-sampling)および非区画(non-compartment)モデルにより概算した。Coatest(登録商標)アッセイにおいて、標準曲線を作成するためにBDD−FVIIIを使用した。簡潔には、同じ血漿マトリックス中のサンプル、標準、陽性および陰性コントロール(それぞれ25μL)を96−ウェルプレートにデュプリケートで加えた。FIXa、FXおよびリン脂質溶液の混合物(50μL)を加え、37℃で5分インキュベートした。次に、25μLのCaCl2溶液を加え、37℃で5分インキュベートし、次に50μL基質を加え、色が適当な強度に発達するまで37℃で5分インキュベートした。停止溶液(25μL)を加え、プレートをプレートリーダー(SpectraMax(登録商標) 250, Molecular Devices, Sunnyvale, CA)でOD405nmで読んだ。結果は、図8において示されているとおりにSoftMax(登録商標) Pro 4.8(Molecular Devices, Sunnyvale, CA)を使用して計算した。結果は、それぞれの時点で、BDD−FVIIIに対して5匹のマウスおよびFVIII−Fcに対して3匹のマウスからの平均±SDを示した。
【0150】
BDD−FVIIIの減衰曲線と比較して、BDDFc−ヒンジは迅速な分布相で二相崩壊を示した(図8)。BDDFc−ヒンジのベータ相半減期は50μg/kgで11.9時間であり、これはベータ相半減期が6.03時間である修飾されていないBDD−FVIIIと比較して約2倍の改善であった。いくつかの第VIII因子融合ヘテロダイマーが非血友病A動物モデルにおける薬物動態学試験を使用して分析され得ない可能性があり得る。
【0151】
実施例17
以下の実施例は、上記第VIII因子融合遺伝子の発現産物である組換え第VIII因子融合ヘテロダイマーにおけるインビトロ試験を記載する。哺乳動物発現ベクターpSS207BDDFc+ヒンジおよびpSS207BDDFc−ヒンジをHKB11細胞に一過性でトランスフェクトし、馴化培地を上記トランスフェクション72時間後に回収した。図9Aにおいて示されているとおり、還元条件下で濃縮された上清のウエスタンブロット分析は、BDDFc+ヒンジ第VIII因子融合ヘテロダイマーが、最初に抗−Fc抗体により検出されるとおりの(レーン5)〜195kDaの第VIII因子融合タンパク質として発現し、抗−Fc抗体により検出されるとおりの(レーン5)115−kDa重鎖に;図9Bにおいて示されているとおり、第VIII因子軽鎖特異的抗体により検出されるとおりの(レーン5)80−kDa軽鎖に翻訳後プロセッシングされることを示した。比較のために、精製されたBDDタンパク質およびpSK207またはpSK207BDD(BDDFc+ヒンジをコードする第VIII因子融合遺伝子由来のB−ドメイン欠失第VIII因子遺伝子を含む発現ベクター)で一過性でトランスフェクトされたHKB11細胞からの馴化培地は、抗−Fc抗体と反応せず(図9A)、第VIII因子軽鎖抗体を使用すると、精製されたBDDタンパク質またはpSK207BDDで一過性でトランスフェクトされたHKB11細胞からの馴化培地から、予期される80kDa軽鎖を同定した(図9B)。対照的に、pSK207で一過性でトランスフェクトされたHKB11細胞からの馴化培地に軽鎖が存在しなかった(図9B)。これらの結果は、BDDFc+ヒンジをコードする第VIII因子融合遺伝子がB−a3ドメイン接合点で機能的切断部位をまだ維持しているため、軽鎖の分子量が変化することが期待されないため、欠失Bドメイン領域へのFc領域の挿入が翻訳後修飾に影響しないことを示す。
【0152】
第VIII因子活性を、Coatest(登録商標)アッセイおよびaPPT凝固アッセイによりpSK207BDD(コントロール)、pSK207BDDFc+ヒンジおよびpSK207BDDFc−ヒンジトランスフェクタントからの馴化培地で検出した(図10)。第VIII因子活性はpSK207トランスフェクタントからの馴化培地で検出されなかった。BDDFc融合タンパク質(すなわち、BDDFc+ヒンジおよびBDDFc−ヒンジ)の両方の活性範囲は、BDDに匹敵した。データは、使用される特定の部位へのFc領域の挿入が、由来のBDD第VIII因子タンパク質と比較して、第VIII因子融合ヘテロダイマーの翻訳後プロセッシングまたは生物学的活性に影響しないことを示唆した。
【0153】
pSK207BDDFc+ヒンジまたはpSK207BDDで一過性でトランスフェクトされたHKB11細胞から回収された馴化培地を、ウサギ−抗−マウスFc抗体(Pierce、Rockford、IL)であらかじめ被覆された96−ウェルプレートに加え、第VIII因子融合ヘテロダイマーを捕捉する固相Coatest(登録商標)アッセイ。BDDFc+ヒンジ融合タンパク質のみがプレートに結合し、由来の第VIII因子BDDタンパク質は流れる。次に、Coatest(登録商標)アッセイをウェルに固定されたBDDFc+ヒンジに対して直接実施し、図11は、BDDFc+ヒンジがこのアッセイにおいて活性であることを示す。
【0154】
pSK207BDDFc+ヒンジまたはpSK207BDDFc−ヒンジ発現ベクターで一過性でトランスフェクトされたHKB11細胞からの5倍濃縮馴化培地を使用して、分析を実施した。サンプルを還元および非還元条件下で4−12%のNuPAGE(登録商標)ゲルで分離した。ブロットをウサギモノクローナル抗−FVIII軽鎖抗体(Epitomics, Burlingame, CA)、次にHRP−接合抗−ウサギIgG二次抗体でプロービングした。結果は、BDDFc+ヒンジがダイマーを形成するが(すなわち、マルチマー第VIII因子融合ヘテロダイマー)、BDDFc−ヒンジがモノマーであることを示した(図12)。同様の結果がpSK207BDDFc+ヒンジで安定に形質転換された細胞で見られた。
【0155】
実施例18
以下の実施例は、第VIII因子融合ヘテロダイマーに包含されているとき、FcRn結合エピトープがFnRnに結合する能力を維持しているか否かを決定するために、BiacoreTMシステムを使用して実施された機能的試験を記載する。BiacoreTM試験における使用のために、組換えマウスFcRn(mFcRn)タンパク質をCHO−K1細胞で発現させ、マウスIgG−親和性クロマトグラフィーにより精製した。マウスFcRnをアミンカップリングによりCM−5チップ上に固定した。2つの第VIII因子ヘテロダイマー(BDDFc+ヒンジおよびBDDFc−ヒンジ)、BDD(BDDFc+ヒンジおよびBDDFc−ヒンジ由来の第VIII因子タンパク質)、および全長組換え第VIII因子を、種々の濃度(例えば、1.5、3、6、12、25、および50nM)でチップの表面を通過させた。固定されたmFcRnへのBDDFc±ヒンジ第VIII因子融合ヘテロダイマーの結合を検出した(図13)。結合親和性(KD=2.48nM)をBDDFc+ヒンジ(「BDDFc+H」)に対して計算し、BDDFc−ヒンジ(「BDDFc−H」)はBDDFc+ヒンジ(KD=3.75nM)と同様であった。検出可能な結合がBDD(「BDD」)または全長組換え第VIII因子(「FVIII」)で見られなかった。
【0156】
結果は、BDDFc+ヒンジおよびBDDFc−ヒンジ第VIII因子融合ヘテロダイマーがnMの親和性でmFcRnに対して強い結合を示すことを示す。対照的に、BDDも全長組換えFVIIIもmFcRnに結合することができなかった。これは、FcRn結合エピトープを含まないため、予期される。HemAマウスを使用して実施される薬物動態学試験を考慮して、結果は、BDDFc+ヒンジおよびBDDFc−ヒンジが高親和性でmFcRnに結合する機能的FcRn結合エピトープを含み、インビボで長期ベータ相半減期をもたらすことを示唆する。
【0157】
実施例19
循環において、FVIIIは、主に安定な複合体としてフォン・ヴィレブランド因子(vWF)に結合している。トロンビン(第IIa因子)により活性化されると、FVIIIは複合体から分離し、凝固カスケードと相互作用する。活性化FVIIIは、タンパク質分解的にプロセスにおいて不活性化され(活性化タンパク質Cおよび第IXa因子により非常に顕著に)、血流から迅速に除去される。以下の実施例は、第VIII因子融合ヘテロダイマーに包含されるとき、第VIII因子タンパク質がフォン・ヴィレブランド因子(vWF)に結合する能力を維持しているか否かを決定するために、BiacoreTMシステムを使用して実施される機能的試験を記載する。
【0158】
ヒトvWFをアミンカップリングによりCM−5チップ上に固定した。2つの第VIII因子ヘテロダイマー(BDDFc+ヒンジおよびBDDFc−ヒンジ)、BDD(BDDFc+ヒンジおよびBDDFc−ヒンジ由来の第VIII因子タンパク質)、および全長組換え第VIII因子を、種々の濃度(例えば、1、2、4、8、16、および25nM)でチップの表面を通過させた。BDD(「BDD」)および全長組換え第VIII因子(「FVIII」)の両方は、サブナノモル親和性(0.53−0.657nM)でヒトvWFに結合することができ、vWFへのBDDFc+ヒンジ(「BDDFc+H」)またはBDDFc−ヒンジ(「BDDFc−H」)の結合も検出した(図14)。BDDFc+ヒンジおよびBDDFc−ヒンジの結合親和性(KD)は、それぞれ0.465nMおよび0.908nMと計算された。データは、第VIII因子融合ヘテロダイマーBDDFc+ヒンジおよびBDDFc−ヒンジがvWFに対するサブナノモル親和性を有し、モジュレーターとしての免疫グロブリンFc領域の使用がvWFに対するBDDの結合特性をブロックしないことを示す。
【0159】
実施例20
以下の実施例は、BDDFc−ヒンジがHemAマウスの尾静脈切断出血モデルにおいて有効であることを証明する。HemAマウス(8−10週、〜25g)に、1つの側部尾静脈の切断の48時間前に、12もしくは60IU/kgの最終用量で5%のアルブミンを含む製剤バッファー中の100μLのBDDFc−ヒンジ、または40IU/kgで5%のアルブミンを含む製剤バッファー中の100μLのBDD−FVIII、または製剤バッファー単独(輸送媒体)(20匹のマウス/処置群)を尾静脈を介して注射した。マウスを麻酔し(ケタミン/キシラジン)、1つの側部尾静脈を尾の直径が約2.7mmである場所で切断した。次に、凝血するまで尾を37℃にあらかじめ温めた塩水で濯ぎ、出血時間を記録した。次に、マウスを加温パッドの上に紙ベッドを備えた個々のケージに移し、時間単位で最初に損傷9時間後、次に24時間後に観察した。再出血の事象を記録した。統計分析をGraphPad Prism(登録商標)4で実施し、結果を図15に示す。
【0160】
損傷24時間後に10%のみ生存した輸送媒体−コントロール群に対して、BDDFc−ヒンジの12IU/kgおよび60IU/kgは、それぞれ25%および80%の生存を成し遂げた。FVIII−Fc−ヒンジの有効性は、40IU/kgで60%の生存をもたらすBDD−FVIIIと匹敵すると概算される。全ての処置は、有意に改善された(Log−Rank試験による両側 p<0.05)生存曲線 対 輸送媒体コントロールをもたらした。
【0161】
本明細書に記載されている全ての文献および特許は出典明示により本明細書に包含させる。本発明の範囲および精神から逸脱することなく、本発明の記載されている方法の種々の修飾および変化が当業者に明らかである。
【0162】
本発明は特定の態様に関連して記載されているが、本発明がこのような特定の態様に過度に限定されるべきでないと理解すべきである。さらに、生化学または関連分野の当業者に明白である本発明を実施するための上記方法の種々の修飾は、特許請求の範囲内であると意図する。当業者は、わずかな日常の実験を使用して、本明細書に記載されている本発明の特定の態様の多数の均等を認識するか、または確かめることができる。このような均等は特許請求の範囲により包含させることを意図する。
【技術分野】
【0001】
本出願は、2009年3月24日に出願された米国仮出願番号61/162,986号(この内容は、その全体において出典明示により本明細書に包含させる)の優先権を主張する。
【0002】
技術分野
本発明は、変異体第VIII因子(FVIII)タンパク質に関する。本発明は、また、変異体FVIIIタンパク質をコードする核酸およびこのような核酸を同定するための方法に関する。本発明は、変異体FVIIIタンパク質を作製および使用する方法に関する。
【背景技術】
【0003】
発明の背景
血液の凝固は、接触活性化経路(以前は内因性経路として知られていた)または組織因子経路(以前は外因性経路として知られていた)のいずれかにより起こり、それによって、特定の血液タンパク質がタンパク質分解活性化のカスケードに相互作用し、最終的に可溶性フィブリノーゲンを不溶性フィブリンに変換する。これらのフィブリンの繊維状物は架橋して、凝血の足場を形成する。すなわち、フィブリン形成なしに、凝固が起こることができない。
【0004】
接触活性化経路は、以下のいくつかの工程からなる、(1)第XII因子のタンパク質分解活性化、(2)活性化された第XII因子が第XI因子を開裂して、それを活性化し、(3)活性化された第XI因子が第IX因子を開裂し、それによりそれを活性化し、(4)活性化された第IX因子が活性化されたFVIIIと相互作用して、第X因子を開裂および活性化し、(5)活性化された第X因子が膜表面上で活性化された第V因子に結合し、この複合体がタンパク質分解的にプロトロンビンを開裂して、トロンビンを形成し、(6)トロンビンがタンパク質分解的にフィブリノーゲンを開裂して、フィブリンを形成し、(7)フィブリンモノマーが線維に集合し、次にこれが第XIII因子により架橋される。
【0005】
組織因子経路は、以下の工程からなる、(1)血管の破裂時に、第VII因子が血管系の外側の組織に存在する組織因子であるリポタンパク質と結合し、(2)第VII因子が、タンパク質分解的切断により、第VIIa因子に活性化され、および(3)第VIIa因子−組織因子複合体が、第X因子を開裂し、活性化させる。その後、組織因子経路は接触活性化経路と同一であり、すなわち、2つの経路は上記最後の3つの工程を共有している。
【0006】
FVIIIの生合成、細胞内プロセッシングおよび分泌、ならびに次に血漿中で活性化されるメカニズムは、当該分野でよく知られている(例えば、Lentingら., Blood 92:3983-3996, 1998; Thompson, Seminars in 止血 29:11-22, 2003; Grawら., Nature Reviews: Genetics 6:489-501, 2005、参照)。ヒトFVIIIは、最初に、2351個のアミノ酸の一本鎖ポリペプチド(配列番号:1)として翻訳され、最初の19個のアミノ酸は、ER内でシグナルペプチダーゼにより除去されるシグナルペプチドを定義する。したがって、成熟ヒトFVIIIは、ドメイン構造A1−a1−A2−a2−B−a3−A3−C1−C2を有する2332個のアミノ酸からなる(図1A)。FVIIIは、Bドメインのカルボキシ−末端付近(B−a3接合点のArg−1648)の切断により、分泌前に細胞内でグリコシル化され、プロセッシングされ、Bドメイン内、主にArg−1313後で可変的に切断され、90−210kDaの重鎖および80kDaの軽鎖を生産する(図1B)。FVIIIは、その後、1つの重鎖および1つの軽鎖からなるヘテロダイマー糖タンパク質として分泌される。
【0007】
血漿糖タンパク質FVIIIは、フォン・ヴィレブランド因子(vWf)にしっかりと非共有結合的に結合した、血液中で不活性な前駆体として循環する。FVIIIは、3つのArg−Serペプチド結合、すなわちArg−372、Arg−740およびArg−1689後でトロンビンまたは第Xa因子による切断によりタンパク質分解的に活性化され、それはvWfから分離され、カスケードにおけるその凝血促進機能を活性化される。得られるヘテロトリマーはFVIIIaとなる(図1C)。
【0008】
その活性型(すなわち、FVIIIa)において、FVIIIは、血液凝固の接触活性化経路において第X因子活性化の酵素複合体に対する補因子として機能し、それは血友病Aを有する患者において減少しているか、または非機能的である。FVIII活性の減少レベルは、疾患の重症度に正比例する。したがって、FVIII欠乏の、またはFVIIIに対する抗体を有する人々は、FVIIIで処置されない限り広範で深刻な症状を引き起こし得る非制御の内出血に罹患する。症状は、関節における炎症応答から早期死亡の範囲である。FVIIIの古典的な定義は、実際には、血友病Aを有する個体由来の血漿における凝固欠損を正す正常血漿中に存在する物質である。vWfの欠損は、また、vWfが機能性FVIIIの重要な成分であるため、表現型血友病Aを引き起こし得る。これらの場合において、血漿中のFVIIIの循環半減期は、血液凝固におけるその特定の機能をもはや行うことができない程度に減少している。血友病Aの現在の処置は、血漿由来または組換えFVIIIの投与による欠損タンパク質の補充からなる。
【0009】
FVIIIの活性を阻害する抗体(「インヒビター」または「阻害抗体」)の発現は、血友病Aを有する患者の管理において深刻な合併症である。自己抗体は、FVIIIの治療的注入に応答して、血友病Aを有する患者の約20%において発現する。インヒビターを発現する以前に処置されていない血友病Aを有する患者において、インヒビターは、通常、処置から1年以内に発現する。さらに、FVIIIを不活性化する自己抗体は、以前に正常FVIIIレベルを有する個体において時折発現する。インヒビター力価が十分に低いとき、患者はFVIIIの用量を増加させることにより管理することができる。しかしながら、しばしば、インヒビター力価は、FVIIIにより圧倒されることができないほど高い。別の戦略は、第IX因子複合体調製物または組換えヒト第VIIa因子を使用して正常な止血中のFVIIIの必要性を迂回することである。さらに、ブタFVIIIは、通常、ヒトFVIIIよりもインヒビターと実質的にあまり反応性を有さないため、部分的に精製されたブタFVIII調製物が使用され得る。ヒトFVIIIに対して阻害抗体を発現した多数の患者は、ブタFVIIIで成功裏に処置され、長期間のこのような処置に耐える。しかしながら、ブタFVIIIの投与は、1以上の注入後に、ブタFVIIIに対するインヒビターが発現し得るため、完全な解決ではない。したがって、組換えヒトFVIIIまたは部分的に精製されたブタFVIIIの使用は、全ての問題を解決していない。
【0010】
阻害抗体に加えて、問題は、また、FVIIIが、静脈内に投与されたとき、循環中に比較的に短期の半減期(ヒトにおいて13時間)を有することで生じ、頻繁な注入が必要であり、患者の投与コンプライアンスにおいて困難性をもたらす。したがって、週1回の投与またはさらに月1回の投与用の長期間作用性FVIIIは、満たされていない医学的要望である(Dargaudら., Expert Opinion on Biological Therapy 7:651-663, 2007)。より長期の保護は、FVIII半減期を延長することにより達成される。多くのFVIIIの生物工学的アプローチが、より長期の保護を生じる目的のために探索される(Baruら., Thromb. Haemost. 93:1061-1068, 2005; Pipe, J. Thromb. Haemost. 3:1692-1701, 2005; Saenkoら., Haemophilia 12(Suppl 3):42-51, 2006)。
【発明の概要】
【0011】
本発明は、修飾された活性および/または修飾された薬物動態学的特性(例えば、より長い循環半減期)を示すFVIII変異体に関する。1つの例として、FVIII変異体は、アミノ酸配列(例えば、モジュレーター)がFVIIIタンパク質のB−ドメイン部分もしくはB−ドメインのいずれかにおいて挿入されているか、またはB−ドメインの一部が該アミノ酸配列で置換されている融合またはヘテロダイマータンパク質であり得る。該挿入/置換アミノ酸配列はFVIIIの翻訳後プロセッシングを中断させず、該FVIII変異体は凝固因子としての活性を有する。これらのFVIII変異体は、血友病Aを処置するために使用され得、例えば、より長い循環半減期によって、より少ない頻度の投与をもたらし得る。より少ない頻度の投与により、本発明のFVIII変異体は、患者のコンプライアンスを改善させ、FVIIIが投与されるためにFVIIIに対する免疫応答を発現する患者の可能性を減少させ得る。
【0012】
発明の概要
本発明は、FVIII融合タンパク質およびその発現産物に関する(本明細書においてFVIII融合ヘテロダイマーとしても称される)。本発明は、さらに、ハイブリッドFVIII融合ヘテロダイマーおよびマルチマーFVIII融合ヘテロダイマーに関する。1つの態様において、FVIII融合ヘテロダイマーは、FVIIIタンパク質またはポリペプチドおよびアミノ酸配列(本明細書においてモジュレーターとして称される)を含む。別の態様において、モジュレーター配列は、FVIII Bドメインに挿入される。さらなる態様において、少なくともBドメインの一部は、モジュレーター配列により欠失および置換されている。
【0013】
本発明は、また、FVIII融合ヘテロダイマーをコードする核酸配列に関する。1つの態様において、核酸配列は、モジュレーター配列がBドメインに存在するか、またはモジュレーター配列がBドメインの一部または全てのアミノ酸配列を置換している、FVIIIタンパク質を含むFVIII融合ヘテロダイマーをコードする。FVIII融合ヘテロダイマーをコードする核酸配列は、発現カセットに作動可能に連結し得る。本発明は、また、FVIII融合ヘテロダイマーを作製する方法を含む。例えば、FVIII融合ヘテロダイマーをコードする発現カセットは、すでに発現ベクターの一部でないとき、発現ベクターに導入され、次にFVIII融合ヘテロダイマーの組換え生産のために適当な宿主細胞に導入される。生産された融合ヘテロダイマーは、インビトロおよびインビボでFVIII活性を有し、例えば、インビボでの循環半減期の増加を示し得る。
【0014】
本発明のさらなる態様において、FVIII融合ヘテロダイマーは、配列番号:1、3または5のいずれか1つのアミノ酸20−764に対応する第1のアミノ酸配列、配列番号:1、3または5のいずれか1つのアミノ酸1656−2351に対応する第2のアミノ酸配列、およびモジュレーター配列を含み、(1)該モジュレーター配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合しているか、または(2)該モジュレーター配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、該モジュレーター配列が、第2のアミノ酸配列に共有結合していない。
【0015】
本発明の別の態様において、核酸配列は、FVIII融合ヘテロダイマーをコードし、該FVIII融合ヘテロダイマーは、配列番号:1、3または5のいずれか1つのアミノ酸20−764に対応する第1のアミノ酸配列、配列番号:1、3または5のいずれか1つのアミノ酸1656−2351に対応する第2のアミノ酸配列、およびモジュレーター配列を含み、該モジュレーター配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合している。加えて、本発明は、また、ベクター、宿主細胞、融合ヘテロダイマーを生産する方法および凝固欠乏症を処置する方法に関する。
【図面の簡単な説明】
【0016】
【図1】図1Aは、以下のドメイン:S(シグナルペプチド)、A1、a1、A2、a2、B、a3、A3、C1およびC2をN−末端からC−末端に含む全長ヒトFVIIIの構造を説明する。図1Bは、ヘテロダイマーヒト第VIII因子の重鎖および軽鎖の構造を説明する。重鎖のサイズは、B−ドメイン内の可変タンパク質分解的切断の結果として変化する。図1Cは、活性ヒトFVIII(すなわち、FVIIIa)のサブユニットの構造を説明する。
【0017】
【図2】図2は、実施例の部において記載されている本発明の第VIII因子融合ヘテロダイマーの3つの典型的な態様を説明する。3つの態様は、「BDDFc+ヒンジ」、「BDDFc−ヒンジ」、および「BDDFc」(これらは、所望により、単離を容易にするために異種ペプチドタグを含み得る)を示す。3つの典型的な態様は、ダイマー(これらのFc部分を介して)またはタンパク質凝集体を形成するそれらの能力において、およびFcRnに対するそれらの結合親和性において異なる。
【0018】
【図3】図3は、実施例1および2にしたがって生産される第VIII因子融合タンパク質の構造ドメインを記載している。具体的には、マウスFc領域(ヒンジ有りまたは無し)をB−ドメイン欠失(BDD)第VIII因子タンパク質の特定の部位(N−745からS−1637)に挿入し、B−ドメインの欠失部分を置換する。マウスFc領域のN−末端およびC−末端側上の非欠失B−ドメイン部分のアミノ酸配列を示す。
【0019】
【図4】図4は、実施例5にしたがって生産されるモノシストロン性BDD.mFcモノマー構築物を説明する。
【0020】
【図5】図5は、活性アッセイによる高い発現クローンの同定を説明する。BDDFc+ヒンジを発現するHKB11の安定な細胞系を、FVIII aPPT凝固アッセイによりスクリーニングした。クローン(4、8、12、18、27および33)は、500−3500mIU/mLの範囲である高い凝固活性を示した。
【0021】
【図6】図6は、ELISAアッセイによる高い発現クローンの同定を説明する。BDDFc+ヒンジを発現するHKB11の安定な細胞系を抗−FVIII捕捉ELISAによりスクリーニングした。3つのクローン(クローン8、18および27)は、〜1ug/mLでBDDFc+ヒンジ融合を発現する。
【0022】
【図7】図7は、BDDFc+ヒンジ融合タンパク質のタンパク質精製の結果を示す。還元ゲルにおいて、BDDFc+ヒンジは、80−kDa軽鎖(L)、115−kDa重鎖(H)、および195−kDa未処理一本鎖(U)(レーン8)として分離された。非還元ゲルにおいて、BDDFc+ヒンジは、80−、115−、195−kDaバンド(レーン8)に加えて390−kDaバンド(ダイマー)を生じた。
【0023】
【図8】図8は、血友病A(HemA)マウスにおけるBDDFc−ヒンジ(「FVIII−Fc」)およびBDD−FVIIIの回収を示す。9匹のHemAマウスに、5%のアルブミンを含む製剤バッファー中の50μg/kg(400IU/kg)(●)のBDDFc−ヒンジを与えた。さらなるHemAマウスに、200IU/kg(□)のBDDFc−ヒンジ由来の第VIII因子変異体であるBDD−FVIIIを与えた。BDD−FVIIIの減衰曲線と比較して、BDDFc−ヒンジは、迅速な分配相と二相減衰を示した。BDDFc−ヒンジのベータ相半減期は、50μg/kgで11.9hrあり、ベータ相半減期が6.03hrである修飾されていないBDD−FVIIIと比較して約2倍に改善された。
【0024】
【図9】図9Aは、BDDFc+ヒンジのBDD−Fcキメラ鎖をウエスタンブロット分析において115kDaバンドとして検出したことを説明する。一過性トランスフェクタント(trans)および安定なプール(sp)の両方からのサンプルを5倍濃縮し、次に還元条件下で10%のNuPAGE(登録商標)ゲル上に流した。レーン:1)分子量マーカー;2)標準として精製されたBDDタンパク質;3−5)pSK207ベクター、pSK207BDD、およびpSK207BDDFc+ヒンジ、それぞれで一過性でトランスフェクトされたHKB11細胞からの濃縮馴化培地;7−9)pSK207ベクター、pSK207BDD、pSK207BDDFc+ヒンジ、それぞれで安定にトランスフェクトされたHKB11細胞の安定なプールからの濃縮馴化培地。ブロットは、HRP−接合抗−マウスIgG(H+L)で探査した。未処理一本鎖形態のBDDFc+ヒンジ(「sc BDD−Fc」)は、195kDaバンドとして検出され、BDDFc+ヒンジのヘテロダイマー型は、115kDaの第VIII因子重鎖およびFc(「重鎖Fc」)のキメラを含んだ。HRP−接合抗−マウスIgGにより結合しないため、ヘテロダイマーBDDFc+ヒンジの軽鎖に関するバンドは現れない。図9Bは、BDDFc軽鎖がウエスタンブロット分析において80kDaバンドとして検出されたことを示す。タンパク質サンプルを還元条件下で10%のNuPAGE(登録商標)ゲル上に流した。レーン:1)分子量マーカー;2)標準として精製されたBDDタンパク質;3−5)pSK207ベクター、pSK207BDD、およびpSK207BDDFc+ヒンジ、それぞれで一過性でトランスフェクトされたHKB11細胞からの濃縮馴化培地。ブロットは、FVIII軽鎖特異的抗体で探査した。
【0025】
【図10】図10は、第VIII因子活性アッセイの結果を示す。pSK207BDDFc+ヒンジ(「Fc+ヒンジ sup」)およびpSK207BDDFc−ヒンジ(「Fc−ヒンジ sup」)で一過性でトランスフェクトされたHKB11細胞からの馴化培地を回収し、Coatest(登録商標)アッセイおよびaPPT凝固アッセイの両方においてFVIII活性に対して試験した。コントロールとして、修飾されていない第VIII因子タンパク質をコードするベクターpSK207およびpSK207BDD(「BDD sup」)を、トランスフェクションならびに活性アッセイにおいて使用した。
【0026】
【図11】図11は、第VIII因子融合ヘテロダイマーに対する第VIII因子活性を示す。HKB11細胞[(BDDFc+ヒンジ一過性トランスフェクタント(Tr)および安定なプール(Sp)]からの馴化培地を、ウサギ−抗−マウスFc抗体であらかじめ被覆された96−ウェルプレート上に負荷した。室温で2時間インキュベーション後、プレートをPBS/Tween(登録商標)−20/BSAで3回洗浄し、Coatest(登録商標)アッセイの前に非特異的結合を除去した。
【0027】
【図12】図12は、BDDFc+ヒンジがダイマーを形成し、BDDFc−ヒンジがモノマーであることを証明する。ウエスタンブロット分析を、pSK207BDDFc+ヒンジまたはpSK207BDDFc−ヒンジ発現ベクターでトランスフェクトされたHKB11細胞からの5倍濃縮馴化培地を使用して行った。サンプルを、還元および非還元条件下で4−12%のNuPAGE(登録商標)ゲル上に流した。ブロットは、ウサギモノクローナル抗−FVIII軽鎖抗体(Epitomics、Burlingame、CA)、次にHRP−接合抗−ウサギIgG二次抗体で探査した。未処理一本鎖BDDおよびBDDFcは、それぞれ170−kDaおよび195−kDaバンドとして検出された。レーン:BDD−−精製されたBDDタンパク質;+H−−BDDFc+ヒンジ;−H−−BDDFc−ヒンジ;およびV−−pSK207ベクター単独。
【0028】
【図13】図13は、固定されたマウスFcRnに結合するマウスFcRn結合エピトープを組み込むBDDFc+ヒンジおよびBDDFc−ヒンジ(「BDDFc−H」)の能力を測定するBiacoreTM試験の結果を示す。BDDFc+ヒンジ(「BDDFc+H」)、BDDFc−ヒンジ(「BDDFc−H」)、BDD、および全長組換え第VIII因子(「FVIII」)。検出可能な結合は、BDDまたは全長第VIII因子で見られなかった。BDDFc+ヒンジおよびBDDFc−ヒンジ融合タンパク質は、mFcRnに対する強い結合をnMの親和性で示した。
【0029】
【図14】図14は、固定されたヒトフォン・ヴィレブランド因子(vWF)に結合するBDDFc+ヒンジおよびBDDFc−ヒンジの能力を測定するBiacoreTM試験の結果を示す。マウスFcRnを、アミンカップリングによりCM−5チップ上に固定した。BDDFc+ヒンジ(「BDDFc+H」)、BDDFc−ヒンジ(「BDDFc−H」)、BDD、および全長組換え第VIII因子(「FVIII」)は、vWFに対するナノモル以下の親和性を示す。
【0030】
【図15】図15は、BDDFc−ヒンジがHemAマウスの尾静脈切断出血モデルにおいて有効であることを示す。BDDFc−ヒンジは、インビボで出血を処置する機能性があるか否かを決定するために、1つの側部尾静脈の切断の48時間前に、HemAマウスにBDDFc−ヒンジ、BDD−FVIIIまたは輸送媒体コントロールを尾静脈を介して注射した。損傷24時間後10%のみ生存する輸送媒体コントロール群(▲)と比較して、12IU/kg(●)および60IU/kg(■)のBDDFc−ヒンジは、それぞれ25%および80%の生存を成し遂げた。FVIII−Fc−ヒンジの有効性を、BDD−FVIIIと比較して40IU/kgで60%生存となる(◆)と評価した。
【発明を実施するための形態】
【0031】
本発明の説明
本発明は、記載されている特定の方法論、プロトコール、細胞系、動物種または属、構築物、および試薬に限定されず、それ自体変化し得ると理解すべきである。また、本明細書において使用される用語は特定の態様のみを記載するための目的であり、本発明の範囲を限定する意図はなく、特許請求の範囲のみにより限定されると理解すべきである。
【0032】
文脈が明白に他に指示されていない限り、本明細書および特許請求の範囲において使用されるとき、単数形「1つ(a、an)」および「その(the)」は、複数形を含むことに注意すべきである。したがって、例えば、「1つのタンパク質」の言及は、1つ以上のタンパク質の言及であり、当業者に知られているものと同等物などを含む。
【0033】
他に定義のない限り、本明細書において使用される全ての技術および学術用語は、本発明が属する当業者に一般的に理解されるものと同じ意味を有する。本明細書に記載されているものと同様または同等のあらゆる方法、デバイスおよび物質が本発明の実施または試験において使用することができるが、好ましい方法、デバイスおよび物質はここに記載されている。
【0034】
本明細書に記載されている全ての文献および特許は、本願発明に関連して使用され得る刊行物において記載されている、例えば、構築物および方法論を記載および開示する目的のために、出典明示により本明細書に包含させる。上記および明細書中の刊行物は、単に本特許出願の出願時前の開示のために提供される。本発明者らが以前の発明によってこのような開示に先行する権利がないと認めると、本明細書において解釈されるべきでない。
【0035】
本明細書において使用される、種々の用語は以下に定義されている。
【0036】
「核酸」は、一本鎖または二本鎖型のいずれかのデオキシリボヌクレオチドまたはリボヌクレオチドおよびそれらのポリマーを示す。具体的に限定されていないかぎり、該用語は、参照核酸と同様の結合特性を有し、天然ヌクレオチドと同様の様式において代謝される、既知の天然ヌクレオチドの類似体を含む核酸を含む。他に記載のない限り、特定の核酸配列は、また、保存的に修飾されたそれらの変異体(例えば、縮重コドン置換)および相補的配列ならびに明白に示されている配列を暗に含む。縮重コドン置換は、1つ以上の選択された(またはすべての)コドンの第3の位置が混合塩基および/またはデオキシイノシン残基で置換されている配列を産生することにより達成され得る。核酸なる用語は、文脈に依存して、遺伝子、cDNAおよび遺伝子によってコードされるmRNAと互換的に使用される。
【0037】
「遺伝子由来の核酸」は、合成遺伝子またはその部分配列が最終的に鋳型として使用されている核酸を示す。したがって、mRNA、mRNAから逆転写されるcDNA、cDNAから転写されるRNA、cDNAから増幅されるDNA、増幅されたDNAから転写されるRNAなどは遺伝子由来であり、このような由来産物の検出は、サンプル中の元の遺伝子および/または遺伝子転写産物の存在および/または存在量を示す。
【0038】
核酸配列は、別の核酸配列と機能的な関係に置かれているとき、「作動可能に連結」または「作動可能に挿入」されている。例えば、プロモーターまたはエンハンサーは、コード配列に作動可能に連結していてよい。作動可能に連結された核酸配列は、2つのタンパク質コード領域で隣接および/または結合していてもよい。いくつかの核酸配列は、作動可能に連結しているが、隣接していないかもしれない。核酸配列の連結は、制限酵素認識部位でのライゲーションにより達成され得る。このような部位が存在しないとき、合成オリゴヌクレオチドアダプターまたはリンカーは、慣用の実施にしたがって使用され得る。
【0039】
生物学的活性を有する第1のポリペプチドは、少なくとも最小限のレベルの生物学的活性が第1のポリペプチドおよび第2のポリペプチドの両方により維持されているような、第2のポリペプチドとの機能的な関係に置かれているとき、生物学的活性を有する第2のポリペプチドに「作動可能に連結」されている。ポリペプチドの文脈において、作動可能な連結は、第1および第2のポリペプチドが隣接することを必ずしも意味しない。当業者が理解できるとおり、生物学的活性の維持は、ペプチドリンカーの包含により促進され得る。
【0040】
ポリペプチド、核酸、または他の成分は、通常、関連している成分(他のペプチド、ポリペプチド、タンパク質(天然配列に伴って起こり得る複合体、例えば、ポリメラーゼおよびリボソームを含む)、核酸、細胞、合成試薬、細胞汚染物、細胞成分など)、例えば、通常、元の由来する細胞と関連している他の成分から部分的に、または完全に分離されているとき、「単離」されている。ポリペプチド、核酸または他の成分は、組成物、混合物または成分の回収物に存在する優勢な種類である(すなわち、モル基準において、組成物中の他のあらゆる個々の種類よりも豊富である)ように、自然環境の他の成分から部分的に、または完全に回収されるか、または分離されているとき、単離されている。場合によっては、調製物は、約60%、70%または75%以上、一般的に約80%以上、または約90%以上の単離された種類からなる。
【0041】
「実質的に純粋な」核酸(例えば、RNAまたはDNA)、ポリペプチド、タンパク質、または組成物は、また、対象の種類(例えば、核酸またはポリペプチド)が、組成物中に存在する全ての分子の種類の少なくとも約50、60、70、80、90または95重量パーセントを含むことを意味する。対象の種類を、また、十分な同質性に精製することができ(汚染の種類が慣用の検出方法により組成物中で検出することができない)、組成物が本質的に単一の高分子の種類の誘導体からなる。
【0042】
「精製」なる用語は、一般的に、核酸、ポリペプチドまたはタンパク質が、少なくとも約50%純粋、60%純粋、70%純粋、75%純粋、85%純粋、および99%純粋であることを意味する。
【0043】
「組換え」なる用語は、例えば、一般的に、細胞、ポリヌクレオチド、ベクター、タンパク質またはポリペプチドに関して使用されるとき、細胞、ポリヌクレオチドまたはベクターが異種(または外来)核酸の導入または天然の核酸の変化により修飾されているか、またはタンパク質またはポリペプチドが異種アミノ酸の導入により修飾されているか、または細胞がそのように修飾されている細胞由来であることを示す。組換え細胞は、細胞の天然(非組換え)型において見出されないであろう核酸配列を発現するか、または天然の核酸配列を発現するが、異常に発現するか、あまり発現しないか、または全く発現しない。「組換え」なる用語は、細胞を基準にして使用されるとき、細胞が異種核酸を複製するか、または異種核酸によってコードされるポリペプチドを発現することを示す。組換え細胞は、細胞の天然(非組換え)型内に見られないコード配列を含み得る。組換え細胞は、また、人工的手段により、修飾され、細胞に再導入されている、細胞の天然型において見られるコード配列を含み得る。該用語は、また、細胞から核酸を除去することなく修飾されている細胞に対して内在的な核酸を含む細胞を包含し、このような修飾は、遺伝子置換、部位特異的変異、再結合、および関連技術により得られるものを含む。
【0044】
「組換え的に生産」なる用語は、核酸セグメントの再帰的な配列再結合もしくはヌクレオチドの他の多様な産生方法(例えば、シャッフリング)のいずれかの化学合成手段、または、例えば、当業者に知られている遺伝子操作技術による、核酸の単離されたセグメントの操作により通常、達成される人工的組合せを示す。「組換え的に発現」は、一般的に、インビトロでの組換え核酸の生産およびインビボ、インビトロまたはエキソビボでの組換え核酸の発現または増殖され得る細胞への移動のための技術を示す。
【0045】
「組換え発現カセット」または単に「発現カセット」は、このような配列に適合する宿主において構造タンパク質をコードする核酸の発現に影響することができる核酸因子と共に、組換え的に、または合成的に産生される核酸構築物を示す。発現カセットは、必ずしも転写されるべき核酸(例えば、所望のポリペプチドをコードする核酸)、およびプロモーターを含まない。発現の影響において必要な、もしくは補助的なさらなる成分も、また、本明細書に記載されているとおりに使用され得る。例えば、発現カセットは、また、宿主細胞から発現されたタンパク質の分泌を指向する選別シグナル(例えば、シグナルペプチドまたは分泌リーダー配列)をコードするヌクレオチド配列を含み得る。転写終結シグナル、エンハンサー、および遺伝子発現に影響する他の核酸配列も、また、発現カセットに含まれ得る。本発明の目的のために、「第VIII因子融合遺伝子を含む発現カセット」は、発現カセットにより発現される所望のタンパク質が、さらに以下で定義されている「第VIII因子融合タンパク質」であることを示す。
【0046】
「ベクター」なる用語は、文脈に依存して、クローニングベクター、発現ベクター、またはその両方を示し得る。ベクターなる用語および「プラスミド」なる用語は互換的に使用される。
【0047】
「発現ベクター」または「発現プラスミド」なる用語は、宿主を形質転換し、導入された配列の発現(例えば、転写および翻訳)を促進するように、発現カセットを宿主細胞に導入することができる輸送媒体を示す。
【0048】
「発現する」および「発現」なる用語は、遺伝子またはDNA配列における情報を明らかにする、または明らかに誘導する、例えば、対応する遺伝子またはDNA配列の転写および翻訳に関与する細胞機能を活性化することによりタンパク質を生産することを意味する。DNA配列は、細胞において、または細胞により発現され、「発現産物」、例えば、タンパク質を形成する。発現産物それ自体、例えば、得られたタンパク質も、また、「発現」されたと言う。発現産物は、細胞内、細胞外、または分泌として特徴付けることができる。
【0049】
「アミノ酸修飾」は、所定のアミノ酸配列のアミノ酸配列における変化を示す。典型的な修飾は、アミノ酸置換、挿入および/または欠失を含む。
【0050】
「アミノ酸挿入」は、少なくとも1つのアミノ酸の所定のアミノ酸配列への取り込みを示す。挿入は、1つまたは2つのアミノ酸残基の挿入またはより長い挿入からなり得る。挿入される残基は、上記のとおり天然または非天然であり得る。
【0051】
「アミノ酸欠失」は、所定のアミノ酸配列から少なくとも1つのアミノ酸残基の除去を示す。
【0052】
「アミノ酸置換」は、所定のアミノ酸配列における少なくとも1つの存在するアミノ酸残基の、別の異なる「置換」アミノ酸残基での置換を示す。置換残基は、「天然アミノ酸残基」(すなわち、遺伝子コードによってコードされる)であり得、アラニン(Ala);アルギニン(Arg);アスパラギン(Asn);アスパラギン酸(Asp);システイン(Cys);グルタミン(Gln);グルタミン酸(Glu);グリシン(Gly);ヒスチジン(His);イソロイシン(Ile):ロイシン(Leu);リジン(Lys);メチオニン(Met);フェニルアラニン(Phe);プロリン(Pro):セリン(Ser);スレオニン(Thr);トリプトファン(Trp);チロシン(Tyr);およびバリン(Val)からなる群から選択される。1つまたはそれ以上の非天然アミノ酸残基での置換も、また、本明細書においてアミノ酸置換の定義により包含される。「非天然アミノ酸残基」は、ポリペプチド鎖において隣接アミノ酸残基に共有結合することができる上記天然アミノ酸残基以外の残基を示す。非天然アミノ酸残基の例は、ノルロイシン、オルニチン、ノルバリン、ホモセリン、および他のアミノ酸残基類似体、例えば、Ellmanら. (Meth. Enzym. 202:301-336, 1991)に記載されているものを含む。このような非天然アミノ酸残基を産生するために、Norenら. (Science 244:182, 1989)およびEllmanら., 1991の方法を使用され得る。簡潔には、これらの方法は、非天然アミノ酸残基でのサプレッサーtRNAの化学的活性化、次にインビトロでのRNAの転写および翻訳を含む。最後に、当業者は、例えば、タンパク質の領域のアミノ酸置換が1つの工程、または2つの工程(例えば、アミノ酸欠失、次にアミノ酸挿入により、または逆もまた同様)において達成されることができることを認識している。
【0053】
特定のポリペプチドまたはタンパク質の「変異体」は、本明細書で定義されている少なくとも1つの「アミノ酸修飾」によって特定のポリペプチドまたはタンパク質のアミノ酸配列と異なるアミノ酸配列を含む。「変異体」は、所望の活性を示すポリペプチドまたはタンパク質のフラグメント、例えば、FcRnへ結合し、凝固因子と結合しているとき、循環半減期を改善するIgGのFc領域のフラグメントを含む。
【0054】
「融合ポリペプチド」は、天然に同じポリペプチドにおいて生じることが見られない少なくとも2つの別々のペプチド部分を含むポリペプチドを示す。
【0055】
「融合タンパク質」は、少なくとも1つの融合ポリペプチドを含むタンパク質を示す。したがって、マルチ−サブユニットタンパク質は、サブユニットの1つのみが融合ポリペプチドであるときでさえ、融合タンパク質として示される。
【0056】
「FVIII」、「第VIII因子」または「第VIII因子タンパク質」なる用語は、機能的対立遺伝子変異体を含む野生型第VIII因子タンパク質、または第VIII因子の生物学的活性を有するそのあらゆる誘導体、変異体もしくは類似体を包含することを意図する。この定義の目的のために、「第VIII因子の生物学的活性」は、血液凝固の内因性経路に参加することができる能力を示す。一般的に、この生物学的活性は、市販されている第VIII因子アッセイ(Coatest(登録商標)、diaPharma(登録商標)、West Chester, Ohio)または当該分野における他のアッセイを使用して、血漿由来の第VIII因子標準を基準にして決定され得る。
【0057】
第VIII因子ドメインについて記載されているとき、「ドメイン」は、当業者に知られている第VIII因子の隣接領域を示すために使用される。ヒト第VIII因子に対して、異なる第VIII因子ドメインに対するアミノ酸番号付けは、図1において示されている。
【0058】
「第VIII因子融合遺伝子」は、さらに以下で定義されている「第VIII因子融合タンパク質」をコードし、Bドメインコード部分に対応する第VIII因子タンパク質コード配列内の位置で、モジュレーターをコードする核酸の第VIII因子タンパク質をコードする核酸への作動可能な挿入により生産され得る非天然核酸構築物を示す。1つの例として、Bドメインコード配列の少なくとも1部分が、モジュレーターをコードする核酸により欠失および置換されている。当業者により理解されるとおり、「作動可能な挿入」は、モジュレーターをコードする1部およびモジュレーターをコードする核酸の上流および下流にある第VIII因子の1部をコードする核酸が全て適当なリーディングフレーム内にある核酸構築物を生産するモジュレーターをコードする核酸の挿入を包含することを意図するのみである。第VIII因子融合遺伝子は、ペプチドリンカーまたは多重合配列をコードするさらなる核酸配列をさらに含み得る。最後に、上記定義の目的のために、「遺伝子」は、他に、転写、翻訳、または適当な翻訳後プロセッシングを可能にするために必要とされる任意の核酸配列(すなわち、プロモーター、エンハンサー、シグナルペプチド、分泌リーダー配列など)の存在を意味することを意図しない。
【0059】
「第VIII因子融合タンパク質」は、翻訳後プロセッシングをまだ受けていない第VIII因子融合遺伝子の転写および翻訳により生産される全長ポリペプチドを示す。翻訳後プロセッシング中に、第VIII因子融合タンパク質は、以下で定義されている「第VIII因子融合ヘテロダイマー」に変換される。
【0060】
「第VIII因子融合ヘテロダイマー」は、第VIII因子の生物学的活性を有し、第VIII因子融合遺伝子の転写および翻訳、およびそれにより生産された第VIII因子融合タンパク質の翻訳後修飾(タンパク質分解プロセッシングを含む)の結果として生産されるヘテロダイマータンパク質を示す。したがって、第VIII因子融合ヘテロダイマーは、血漿中の循環が見られる野生型第VIII因子のヘテロダイマー型(すなわち、重鎖および軽鎖を含む)に類似している。本発明の第VIII因子融合ヘテロダイマーは、例えば、「修飾された重鎖」(すなわち、モジュレーターを含み、また、B−ドメインの少なくとも1つの部分の欠失を有し得る第VIII因子重鎖)から構成されるという点において起源の第VIII因子タンパク質のヘテロダイマー型と異なっていてよい。本発明の第VIII因子融合ヘテロダイマーは、融合ヘテロダイマーが起源の第VIII因子タンパク質と比較して、例えば、循環半減期の増加を示し得る。「第VIII因子融合ヘテロダイマー」は、また、以下でさらに定義されている「マルチマー」および「ハイブリッド」第VIII因子融合ヘテロダイマーを包含し得る。
【0061】
「マルチマー第VIII因子融合ヘテロダイマー」は、少なくとも2つの第VIII因子融合ヘテロダイマーを含むタンパク質を示す。マルチマー第VIII因子融合ヘテロダイマーは、第1の第VIII因子融合ヘテロダイマーに存在するモジュレーターのアミノ酸、ペプチド、またはポリペプチド部分が、第2の第VIII因子融合ヘテロダイマーに存在するモジュレーターの同種または異種部分との非共有結合性または共有結合性会合を仲介することができるとき、生じ得る。例えば、IgGのFc部分のヒンジ領域は、第VIII因子融合ヘテロダイマーが、同一のアミノ酸配列(マルチマー第VIII因子融合ヘテロダイマーが「ホモ−マルチマー第VIII因子融合ヘテロダイマー」として称することができる場合)または異なるアミノ酸配列(マルチマー第VIII因子融合ヘテロダイマーが「ヘテロ−マルチマー第VIII因子融合ヘテロダイマー」として称することができる場合)を有するかにかかわらず、2つの第VIII因子融合ヘテロダイマー間の共有結合性会合を仲介することができる。マルチマー第VIII因子融合ヘテロダイマーは、また、モジュレーターをコードする核酸に加えて、第VIII因子融合遺伝子が、同種アミノ酸、ペプチド、またはポリペプチド(「ホモ−多重合配列」として以下で称される)、または、異種ペプチドまたはポリペプチド(「ヘテロ−多重合配列」として以下で称される)との非共有結合性または共有結合性会合を仲介することができるアミノ酸、ペプチド、またはポリペプチドをコードする作動可能に連結している核酸を含むとき、生じ得る。当業者は、第1の第VIII因子融合遺伝子のみがヘテロ−多重合配列を含むとき、第2の異なる第VIII因子融合遺伝子がマルチマー第VIII因子融合ヘテロダイマーを生産するために必要とされ得ることを認識している。例えば、当業者は、米国特許第5,807,706号に記載されている「空洞への隆起(protuberance-into-cavity)」手法を利用するために、2つの第VIII因子融合遺伝子を入手することを認識する。マルチマー第VIII因子融合ヘテロダイマーの組換え生産に関して、当業者は、ホモ−マルチマー型は単一の組換え宿主細胞により生産され得るが、ヘテロ−マルチマー型は単一の宿主細胞内での共発現または複数の宿主細胞(同じか、または異なっている細胞培養系における)における別々の発現により生産され得ることを認識している。現在既知の手法に限定することを意図しないが、以下の非限定的な文献において教示されているマルチマーポリペプチドを生産するための一般的な手法は、マルチマー第VIII因子融合ヘテロダイマーの生産における使用に適している:米国特許公報第2007/0287170号;米国特許第7,183,076号において記載されている「multimerization domain」手法、例えば、免疫グロブリン部分を使用するもの;米国特許第5,932,448号において使用されているFos and Jun leucine zippersの使用;および米国特許第6,833,441号において使用されている「heterodimerization sequence」手法
【0062】
「ハイブリッド第VIII因子融合ヘテロダイマー」は、少なくとも1つの他のポリペプチドと共有結合性または非共有結合性会合する第1の第VIII因子融合ヘテロダイマーのみを含む本発明のあらゆる組換えタンパク質を示す。当業者は、モジュレーターが、ダイマーまたはマルチマー(例えば、免疫グロブリンのダイマーFc領域)を形成することができるとき、マルチマー第VIII因子融合ヘテロダイマー(上記定義のとおり)を生産することができることを認識している。しかしながら、当業者は、マルチマー半減期モジュレーターのすべてのポリペプチドが、第VIII因子融合タンパク質として発現される必要性がないことを認識している。例えば、Fc領域がモジュレーターとして使用されるとき、Fc領域のみ(またはアフィニティータグまたは非第VIII因子ペプチド、タンパク質またはタンパク質フラグメントに作動可能に連結したFc領域)をコードする発現カセットが、第VIII因子融合遺伝子を含む発現カセットを含む同じまたは異なっている宿主細胞に導入され得る。当業者は、また、モジュレーターがダイマーまたはマルチマーを形成することができないときでさえ、ハイブリッド第VIII因子融合ヘテロダイマーが設計され得ることを認識している。具体的には、ホモ−またはヘテロダイマー配列は、第VIII因子融合遺伝子内にモジュレーターをコードする核酸に対して5’(発現するときにN−末端)または3’(すなわち、発現するときにC−末端)のいずれかで位置し得る。
【0063】
「モジュレーター」なる用語は、タンパク質(例えば、第VIII因子)の挿入または置換されたとき、例えば、タンパク質の活性および/または薬物動態学的特性を修飾するあらゆるポリペプチド、タンパク質、タンパク質フラグメント、またはそれらの変異体(1つ以上のポリペプチドサブユニットから構成される)を示す。1つの例として、「半減期モジュレーター」は、起源のタンパク質と比較して、タンパク質(例えば、該挿入または置換の結果として生産される、第VIII因子融合ヘテロダイマー、ハイブリッド第VIII因子融合ヘテロダイマー、またはマルチマー第VIII因子融合ヘテロダイマー)の循環半減期を増加または減少させ得る。半減期モジュレーターは、例えば、タンパク質(例えば、第VIII因子融合ヘテロダイマー、ハイブリッド第VIII因子融合ヘテロダイマー、またはマルチマー第VIII因子融合ヘテロダイマー)の循環半減期を、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも40%、少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、または少なくとも100%増加させ得る。1つの態様において、半減期モジュレーターは、第VIII因子融合ヘテロダイマー、ハイブリッド第VIII因子融合ヘテロダイマー、またはマルチマー第VIII因子融合ヘテロダイマーの循環半減期を、起源の第VIII因子タンパク質と比較して、少なくとも約2倍増加させ得、さらなる態様において、循環半減期を、少なくとも約2.5倍、少なくとも約3倍またはそれ以上増加させる。別の態様において、半減期モジュレーターは、第VIII因子タンパク質のあらゆる内因性因子、例えば、限定はしないがB−ドメインを含まない。
【0064】
ペプチド薬物を患者へ投与する文脈において本明細書において使用される「循環半減期」、「血漿半減期」、「血清半減期」または「t[1/2]」なる用語は、患者における薬物の血漿濃度が2分の1にまで減少するために必要な時間として定義され得る。多数のクリアランスメカニズム、再分配、および当該分野でよく知られている他のメカニズムに依存して、ペプチド薬物と関連する1つ以上の半減期があり得る。通常、アルファおよびベータ半減期は、アルファ相は再分配と関連し、ベータ相はクリアランスと関連するように定義される。しかしながら、大部分、血流に留まっているタンパク質薬物で、少なくとも2つのクリアランス半減期があり得る。本発明の目的のために、ベータ半減期は、投与後に適当に選択された時点で血漿タンパク質レベル(例えば、抗原ELISAを使用して)を測定すること、または適当に選択された時点で凝固活性(例えば、Coatestアッセイを使用して)を測定することにより計算されうる。「半減期」のさらなる説明は、Pharmaceutical Biotechnology(1997, DFA Crommelin and RD Sindelar, eds., Harwood Publishers, Amsterdam, pp 101-120)において見いだされ得る。
【0065】
第VIII因子融合遺伝子の構築
本発明の1つの局面は、第VIII因子融合遺伝子に関する。第VIII因子融合遺伝子は、RNAまたはDNAのいずれかであり得る。上記のとおり、第VIII因子融合遺伝子は、第VIII因子融合タンパク質をコードする核酸分子である。第VIII因子融合遺伝子は、第VIII因子コード配列、モジュレーターをコードする核酸、および、所望により、モジュレーターをコードする核酸と異なるホモもしくはヘテロ−多重合配列をコードする核酸由来である。第VIII因子融合遺伝子のこれらの因子は以下でさらに詳細に記載されているが、第VIII因子融合遺伝子の構築が、よく知られている方法を使用してこれらの別々の因子をコードする核酸から合成することができることを当業者は理解している。本発明における使用で見いだされ得る種々の方法は、Molecular Cloning - A Laboratory Manual, 3rd Ed. (Maniatis, Cold Spring Harbor Laboratory Press, New York, 2001)、およびCurrent Protocols in Molecular Biology (John Wiley & Sons)に記載されている。
【0066】
第VIII因子をコードする核酸の選択
本発明の組換え第VIII因子融合タンパク質およびヘテロダイマーは、野生型第VIII因子、存在し、ある個体から別の個体で発生し得る第VIII因子の天然対立遺伝子変異体、キメラ第VIII因子(例えば、ヒト/ブタ)、または、他の点で修飾されており、凝血促進機能をなお維持している変異体第VIII因子、例えば、野生型第VIII因子または第VIIIa因子タンパク質の特性、例えば、グリコシル化部位およびパターン、抗原性、比活性、循環半減期、タンパク質分泌、第IXa因子および/または第X因子に対する親和性、変更された第VIII因子−不活性化切断部位、活性化された第VIIIa因子形態の安定性、免疫原性、保存期間などに影響するように修飾されている変異体をコードする核酸を修飾することにより製造され得る。本発明にしたがって修飾され得る適当な変異体第VIII因子配列は、野生型第VIII因子と関連する凝血促進機能を有する以前に知られているか、または後に同定される任意のあらゆる変異体第VIII因子配列を含み得る。
【0067】
本発明にしたがって修飾することができる適当な野生型第VIII因子は、限定はしないが、哺乳動物、例えば、ヒト(例えば、GenBankアクセッション番号AAA52484(アミノ酸)(配列番号:1)およびK01740(ヌクレオチド)(配列番号:2)、GenBankアクセッション番号AAA52485(アミノ酸)(配列番号:3)およびM14113(ヌクレオチド)(配列番号:4)、およびGenBank受入番号AAA52420(アミノ酸)(配列番号:5)、参照);ラット(例えば、GenBankアクセッション番号AAQ21580(アミノ酸)およびAY362193(ヌクレオチド)、参照);マウス(see、例えば、GenBankアクセッション番号AAA37385(アミノ酸)およびL05573(ヌクレオチド)、参照);イヌ(例えば、GenBankアクセッション番号AAB87412(アミノ酸)およびAF016234(ヌクレオチド)、参照);コウモリ(例えば、GenBankアクセッション番号ACC68917(アミノ酸)およびDP000725(ヌクレオチド)、参照);ニワトリ(例えば、GenBankアクセッション番号AAO33367(アミノ酸)およびAF465272(ヌクレオチド)、参照);チンパンジー(例えば、GenBankアクセッション番号XP_529212(アミノ酸)およびXM_529212(ヌクレオチド)、参照);ブタ(例えば、GenBankアクセッション番号.NP_999332(アミノ酸)およびNM_214167(ヌクレオチド)、参照);ウサギ(例えば、GenBankアクセッション番号ACA42556(アミノ酸)およびEU447260(ヌクレオチド)、参照);ネコ、サル、モルモット、オランウータン、ウシ、ウマ、ヒツジ、ヤギまたは他の哺乳動物種を含む種々の動物由来であり得る。ヒト、ブタ、マウスおよびイヌに対する配列は、また、Haemophilia A Mutation, Structure, Test and Resource Site(またはHAMSTeRS)を介して電子的に利用でき、これは、ヒト、ブタ、マウスおよびイヌの第VIII因子タンパク質のアラインメントをさらに提供する。当業者が理解できるとおり、哺乳動物の第VIII因子タンパク質中の保存および相同性はよく知られている。
【0068】
本発明にしたがって修飾され得る適当な変異体第VIII因子の1つの非限定的な例は、天然ヒトFVIIIのB−ドメインの14個のアミノ酸(SFSQNPPVLKRHQR、配列番号:6)の他全ての欠失を含むアミノ酸配列を有することにより特徴付けられるB−ドメイン欠失第VIII因子(「BDD第VIII因子」)である(Lindら., Eur. J. Biochem. 232:19-27, 1995)。このBDD第VIII因子は、配列番号:7のアミノ酸配列を有する。
【0069】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、ヒト第VIII因子の抗原性に関与するヒトアミノ酸残基に対する置換基として1つ以上の動物アミノ酸残基を含むキメラヒト/動物第VIII因子である(例えば、米国特許第5,364,771;5,663,060;および5,888,974号、参照)。例えば、動物(例えば、ブタ)残基置換は、1つ以上のR484A、R488G、P485A、L486S、Y487L、Y487A、S488A、S488L、R489A、R489S、R490G、L491S、P492L、P492A、K493A、G494S、V495A、K496M、H497L、L498S、K499M、D500A、F501A、P502L、1503M、L504M、P505A、G506A、E507G、1508M、1508A、M2199I、F2200L、L2252F、V2223A、K2227E、および/またはL2251を含み得るが、これらに限定されない(例えば、米国特許第5,859,204および6,770,744号ならびに米国特許公報第2003/0166536号、参照)。
【0070】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、融合A2およびA3ドメインによって活性化された第VIII因子のより良い安定性により特徴付けられる第VIII因子である。例えば、第VIII因子は、位置664および1826でシステイン残基に置換することにより修飾され、A2およびA3ドメインに共有結合するCys664−Cys1826ジスルフィド結合を含む変異体第VIII因子をもたらし得る(Galeら., J. Thromb. Haemost. 1:1966-1971, 2003)。
【0071】
本発明にしたがって修飾され得る適当な変異体第VIII因子のさらなる非限定的な例は、変更された不活性化切断部位を有する第VIII因子である(例えば、Amanoら., Thromb. Haemost. 79:557-63, 1998; Thornburgら., Blood 102:299, 2003、参照)。これらの変化は、通常、野生型第VIII因子を不活性化する切断酵素に対する変異体第VIII因子の感受性を減少させるために使用され得る。
【0072】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、第IXa因子(例えば、Fayら., J. Biol. Chem. 269:20522-20527, 1994); Bajajら., J. Biol. Chem. 276:16302-16309, 2001;およびLentingら., J. Biol. Chem. 271:1935-1940, 1996、参照)および/または第X因子(例えば、Lapanら., J. Biol. Chem. 272:2082-2088, 1997、参照)に対する親和性が増強されている第VIII因子である。
【0073】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、第VIII因子の分泌を増強するように修飾されている第VIII因子である(例えば、Swaroopら., J. Biol. Chem. 272:24121-24124, 1997、参照)。
【0074】
本発明にしたがって修飾され得る適当な変異体第VIII因子のさらなる非限定的な例は、増加した循環半減期を有する第VIII因子である。これらの変異体第VIII因子タンパク質は、限定はしないが、ヘパラン硫酸と減少した相互作用(Sarafanovら., J. Biol. Chem. 276:11970-11979, 2001)および/または低比重リポタンパク質受容体関連タンパク質(「LRP」)と減少した相互作用を有するとして特徴付けることができる(例えば、WO00/28021;WO00/71714;Saenkoら., J. Biol. Chem. 274:37685-37692, 1999;およびLentingら., J. Biol. Chem. 274:23734-23739, 1999、参照)。
【0075】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、既知の、既存のエピトープ内のアミノ酸をコードし、アスパラギン残基でグリコシル化のための認識配列を生産するように修飾されたヌクレオチド配列によってコードされる修飾された第VIII因子である(例えば、米国特許第6,759,216号、参照)。該例の変異体第VIII因子は、既存の阻害抗体による検出を逃れ(低い抗原性の第VIII因子)、阻害抗体を発現する可能性を減少させる(低い免疫原性の第VIII因子)修飾された第VIII因子の提供において有用であり得る。本発明にしたがって修飾され得る変異体第VIII因子のこの型の1つの態様は、N−連結グリコシル化のためのコンセンサスアミノ酸配列を有するように変異されている第VIII因子である。このようなコンセンサス配列の1つの例は、N−−X−−S/T(式中、Nはアスパラギンであり、Xは任意のアミノ酸であり、S/Tはセリンまたはスレオニンを示す)である(例えば、米国特許第6,759,216号、参照)。
【0076】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、種々の変異を有する凝血促進活性な第VIII因子である(例えば、米国特許第6,838,437号および米国特許出願第2004/0092442号、参照)。この態様の1つの例は、(i)フォン・ヴィレブランド因子結合部位を欠失する、(ii)Arg740に変異を加える、および(iii)A2およびA3−ドメイン間にアミノ酸配列スペーサーを加えるように修飾されている変異体第VIII因子に関し、ここで、該アミノ酸スペーサーは、活性化時に、凝血促進活性な第VIII因子タンパク質がヘテロダイマーになるように十分な長さである(例えば、米国特許公報第2004/0092442号;Pittmanら., PNAS 85:2429-2433, 1988、参照:コファクター活性を産生するために、Arg740での切断が必須ではないと記載している)。
【0077】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、1つ以上の位置に挿入された短縮(truncated)第IX因子イントロン1を有するヌクレオチド配列によってコードされる変異体第VIII因子である(例えば、米国特許第6,800,461および6,780,614号、参照)。この変異体第VIII因子は、インビトロでの組換え第VIII因子のより高い生産を得るために、ならびに遺伝子治療のためのトランスファーベクターにおいて使用され得る(例えば、米国特許第6,800,461号、参照)。この態様の1つの例において、変異体第VIII因子は、2つの位置に挿入された短縮第IX因子イントロン1、および造血細胞系および血小板における発現を駆動するために適当であるプロモーターを有するヌクレオチド配列によってコードされ得る(例えば、米国特許第6,780,614号、参照)。
【0078】
本発明にしたがって修飾され得る適当な変異体第VIII因子のさらなる非限定的な例は、阻害抗体により阻害の減少を示す変異体第VIII因子である(例えば、米国特許第5,859,204;6,180,371;6,458,563;および7,122,634号、参照)。
【0079】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、第VIII因子の半減期および/または比活性の増加の影響を有するA2ドメインにおいて1つ以上のアミノ酸置換を有する変異体第VIII因子である(例えば、米国特許第7,211,559号、参照)。
【0080】
本発明にしたがって修飾され得る適当な変異体第VIII因子のさらなる非限定的な例は、比活性の増加を示す変異体第VIII因子である(例えば、米国特許公報第2007/0265199号、参照)。
【0081】
本発明にしたがって修飾され得る適当な変異体第VIII因子の別の非限定的な例は、1つ以上の生体適合性ポリマーにあらかじめ定義された部位で共有結合しているFVIII変異タンパク質である(例えば、米国特許公報第2006/0115876号、参照)。
【0082】
モジュレーターをコードする核酸の選択
本発明の第VIII因子融合遺伝子は、モジュレーターをコードする核酸を含む。例えば、治療的タンパク質と融合されたとき、融合されていない治療的タンパク質と比較して血清半減期を延長する効果を有する多数のタンパク質(およびそれらをコードする核酸)は、当該分野で知られている。これらの参考文献において教示されている任意のモジュレーターをコードする核酸は、本発明の第VIII因子融合遺伝子を構築するために潜在的に使用され得る。例えば、第VIII因子融合ヘテロダイマーの循環半減期を潜在的に増加させ得る(それが由来する第VIII因子タンパク質と比較して)候補モジュレーターを選択するために、(1)モジュレーターの循環半減期が修飾のために選択される第VIII因子タンパク質の循環半減期よりも大きくなるべきであること;および(2)融合タンパク質の免疫原性を考慮する。第2の考慮に関して、第VIII因子融合ヘテロダイマーで処置されることが意図される個体群(例えば、ヒト)において天然に発現されるか、または該群において天然に発現されるタンパク質に由来するモジュレーターを使用することが好ましいかもしれない。例えば、モジュレーターは、処置されることが意図される個体群の血清中に天然に存在する(例えば、ヒトが意図される処置個体群であるとき、ヒトFc領域の使用)。
【0083】
本発明の1つの態様において、免疫グロブリン定常領域はモジュレーターとして使用され得る。したがって、本発明の第VIII因子融合遺伝子の構築において使用されるモジュレーターコード核酸配列は、免疫グロブリン(Ig)のFc領域をコードするポリヌクレオチドまたはそのフラグメントおよび/または変異体、およびFcRn結合ペプチドをコードするポリヌクレオチドまたはその変異体であり得る。1つの態様において、使用される核酸は、ヒトIgG1、IgG2、IgG3、IgG4、IgE、IgD、もしくはIgM、またはマウスIgG1、IgG2a、IgG2b、IgG3、IgA、もしくはIgMから得られる免疫グロブリンのFc領域またはそのフラグメントおよび/または変異体であるモジュレーターをコードする。別の態様において、使用される核酸は、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有する(ヒンジ領域中のシステイン残基の置換または欠失による)ヒトもしくはマウスIgGのFc領域の変異体、または、ヒトもしくはマウスIgGのFc領域の非ヒンジ部分であるモジュレーターをコードする。さらなる態様において、使用される核酸は、マウスIgG1またはヒトIgG1のFc領域、または、ヒトIgG1またはマウスIgG1のFc領域の非ヒンジ部分であるモジュレーターをコードする。さらなる態様において、使用される核酸は、ヒトIgG1のFc領域、非機能的ヒンジを有する(システイン残基の置換または欠失による)ヒトIgG1の変異体Fc領域、ヒトIgG1のFc領域の非ヒンジ部分であるモジュレーターをコードする。
【0084】
免疫グロブリンのFc領域のフラグメントについて、モジュレーターをコードする核酸は、新生児のFc受容体(FcRn)により結合されるエピトープを定義するFc領域の少なくとも1つのアミノ酸セグメントをコードし得、さらにFc領域のヒンジ部分に対応するセグメントをコードし得る。あるいは、モジュレーターをコードする核酸は、少なくとも1つのFcRn結合ペプチドをコードし得る。限定はしないが、適当なFcRn結合ペプチドの例は配列PKNSSMISNTP(配列番号:24)を含み、さらにHQSLGTQ(配列番号:25)、HQNLSDGK(配列番号:26)、HQNISDGK(配列番号:27)、またはVISSHLGQ(配列番号:28)から選択される配列を含み得る(例えば、米国特許第5,739,277号、参照)。
【0085】
本発明の1つの態様において、モジュレーターは、配列番号:9(ヒトIgG1のFc領域)、配列番号:11(ヒトIgG2のFc領域)、配列番号:13(ヒトIgG3のFc領域)、配列番号:15(ヒトIgG4のFc領域)、配列番号:29(マウスIgG1のFc領域)、配列番号:17(ヒトIgG1のFc領域の非ヒンジ部分)、配列番号:19(ヒトIgG2のFc領域の非ヒンジ部分)、配列番号:21(ヒトIgG3のFc領域の非ヒンジ部分)、配列番号:23(ヒトIgG4のFc領域の非ヒンジ部分)、または配列番号:30(マウスIgG1のFc領域の非ヒンジ部分)と少なくとも約70%、少なくとも約75%、少なくとも約80%、少なくとも約85%、少なくとも約90%、または少なくとも約95%のアミノ酸同一性で同一であるか、または共有するアミノ酸配列をコードする核酸配列によってコードされ得る。上記の1つをコードする核酸の特定の例は、配列番号:8(ヒトIgG1のFc領域)、配列番号:10(ヒトIgG2のFc領域)、配列番号:12(ヒトIgG3のFc領域)、配列番号:14(ヒトIgG4のFc領域)、配列番号:47(マウスIgG1のFc領域)、配列番号:16(ヒトIgG1のFc領域の非ヒンジ部分)、配列番号:18(ヒトIgG2のFc領域の非ヒンジ部分)、配列番号:20(ヒトIgG3のFc領域の非ヒンジ部分)、配列番号:22(ヒトIgG4のFc領域の非ヒンジ部分)、および配列番号:48(マウスIgG1のFc領域の非ヒンジ部分)を含む。
【0086】
モジュレーターをコードする核酸を同定するための方法
他のポリペプチドまたはタンパク質は、本明細書に記載されている方法論の使用により、適当なモジュレーターとして同定され得る。候補モジュレーター(例えば、第VIII因子融合遺伝子および第VIII因子融合タンパク質の創造において潜在的に有用であり得るポリペプチド)は、治療的タンパク質への融合により非第VIII因子治療的タンパク質またはペプチドの血清半減期を延長させることが示されている該モジュレーターのペプチドおよびタンパク質を含む。例えば、第VIII因子タンパク質のモジュレーターを同定するための1つの方法は、血友病Aの動物モデル、例えば、血友病A(HemA)のマウスにおけるモジュレーターを含む第VIII因子融合ヘテロダイマーの薬物動態学を試験することである。
【0087】
モジュレーターをコードする核酸の挿入部位
本発明の第VIII因子融合遺伝子は、モジュレーターをコードする核酸を含む。モジュレーターをコードする核酸は、第VIII因子遺伝子のB−ドメイン部分内に挿入され得る。例えば、第VIII因子遺伝子のB−ドメインをコードする核酸の少なくとも1部分は、モジュレーターをコードする核酸の挿入の前または後に欠失され得る(例えば、N−745からS−1637のBドメインの部分をコードする第VIII因子遺伝子の少なくとも1部分が欠失)。あるいは、部位特異的再結合は、モジュレーターをコードする核酸を同時に挿入し、核酸をコードするB−ドメイン領域の部分を欠失するために使用され得る。挿入、欠失および部位特異的再結合を達成するための組換え方法は、当該分野でよく知られている。
【0088】
1つの態様において、第VIII因子融合遺伝子は、第VIII因子融合タンパク質をコードする核酸配列を含み、該第VIII因子融合タンパク質は、モジュレーターのアミノ酸配列がB−ドメインに存在するか、またはモジュレーターのアミノ酸配列がB−ドメインの一部または全てのアミノ酸配列と置き換わっている第VIII因子タンパク質を含む。第2の態様において、第VIII因子融合遺伝子は、配列番号:1または5のいずれか1つのアミノ酸20−764に対応する第1のアミノ酸配列、配列番号:1または5のいずれか1つのアミノ酸1656−2351に対応する第2のアミノ酸配列、および半減期モジュレーターアミノ酸配列がそのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合しているモジュレーターアミノ酸配列を含む、第VIII因子融合タンパク質をコードする核酸配列を含む。
【0089】
分泌前に、Bドメインは、Arg1648(すなわち、B−a3接合点)で開裂され、主にArg1313後で、B−ドメインにおいて可変的に開裂される(例えば、Thompson, Semin. Thromb. Hemost. 29:11-22, 2003、参照)。したがって、当業者は、B−ドメイン領域における挿入、欠失および/または置換において、B−a3ドメイン接合点で起こる切断部位が第VIII因子融合ヘテロダイマーへの第VIII因子融合タンパク質の適当な翻訳後プロセッシングのために維持されるべきであることを理解できる。同様に、B−ドメイン領域における挿入のために、モジュレーターをコードする核酸は、Arg1313をコードする核酸に対して5’であるB−ドメインをコードする核酸内の部位で挿入されるべきである。あるいは、B−ドメイン内の切断部位(B−a3接合点での切断部位を除いて)は、第VIII因子融合タンパク質の翻訳後プロセッシング中にN−末端(「重鎖」)部分からモジュレーターの切断(したがって分離)を防止するために変異され得る。
【0090】
a2−Bドメイン接合点での切断が第VIII因子のコファクター活性を産生するために必須でないことが知られている(Pittmanら., PNAS 85:2429-2433, 1988)。ヒト第VIII因子において、a2−Bドメイン接合点はArg740で生じる。本発明の第VIII因子融合遺伝子は、a2−Bドメイン接合点で切断を受ける第VIII因子融合タンパク質をコードする遺伝子ならびに切断を防止するa2−Bドメイン接合点でアミノ酸修飾を有する第VIII因子融合タンパク質をコードする遺伝子を含む。1つの例として、本発明の第VIII因子融合ヘテロダイマーの活性化時の該接合点での切断が、第VIII因子融合ヘテロダイマー由来の第VIII因子タンパク質の活性化時に生産される第VIIIa因子タンパク質と同一である(したがって、同じ生物学的活性を有する)第VIIIa因子タンパク質の形成をもたらすとき、a2−Bドメイン接合点切断部位は、無傷なままであり得る。
【0091】
当業者は、第VIII因子融合ヘテロダイマーにおけるa2−Bドメイン接合点での切断の程度または割合が、それが由来する第VIII因子タンパク質において見られるものよりも小さいとき、恐らくモジュレーターによる立体障害によると理解する。したがって、a2−Bドメイン接合点と半減期モジュレーター間のペプチドリンカー(すなわち、スペーサー)、例えば、ペプチドリンカーDDDDK(配列番号:49)およびGGGGSGGGGSGGGGS(配列番号:50)の形態においてさらなるアミノ酸を含むことが望ましいかもしれない。
【0092】
ホモもしくはヘテロ−多重合配列をコードする核酸の選択および挿入部位
本発明の第VIII因子融合遺伝子は、所望により、モジュレーターをコードする核酸と異なるホモもしくはヘテロ−多重合配列をコードする核酸を含む。ホモもしくはヘテロ−多重合配列をコードする核酸の包含は、第1の第VIII因子融合ヘテロダイマーにおいて使用されるモジュレーターが、第2の第VIII因子融合ヘテロダイマーに存在するモジュレーターの同種または異種部分と非共有結合性または共有結合性会合を介在することができないとき、マルチマー第VIII因子融合ヘテロダイマーを生産することが望ましいかもしれない。あるいは、ホモもしくはヘテロ−多重合配列をコードする核酸の包含は、第1の第VIII因子融合ヘテロダイマーにおいて使用されるモジュレーターが、単一のポリペプチドからなるとき、ハイブリッド第VIII因子融合ヘテロダイマーを生産することが望ましいかもしれない。
【0093】
当業者により理解されているとおり、ホモもしくはヘテロ−多重合配列をコードする核酸の選択は、利用されている特定の多重合アプローチにより決定される。以下の非限定的な文献において教示されるマルチマーポリペプチドを生産するために一般的なアプローチにおいて使用されるホモもしくはヘテロ−多重合配列をコードする核酸配列は、本発明の第VIII因子融合遺伝子に包含することができる。米国特許公報第2007/0287170号、米国特許第7,183,076号に記載されている「多重合ドメイン」アプローチ、例えば、免疫グロブリン部分を使用するもの、米国特許第5,932,448号において使用されているとおりのFosおよびJunロイシンジッパーの使用、および米国特許第6,833,441号に記載されている「ヘテロ二量化配列」アプローチ、および米国特許第5,807,706号に記載されている「空洞への隆起」アプローチ。
【0094】
当業者は、第1の第VIII因子融合遺伝子のみがヘテロ−多重合配列を含むとき、第2の異なる第VIII因子融合遺伝子がマルチマー第VIII因子融合ヘテロダイマーまたはハイブリッド第VIII因子融合ヘテロダイマーを生産するために必要であり得ることを理解している。例えば、当業者は、米国特許第5,807,706号に記載されている「空洞への隆起」アプローチを利用するために、それぞれ異なるヘテロ−多重合配列を含む2つの第VIII因子融合遺伝子が必要であることを認識している。
【0095】
当業者は、第VIII因子融合遺伝子内のホモもしくはヘテロ−多重合配列をコードする核酸が、モジュレーターに対して5’(すなわち、発現されたときN−末端と関連する)または3’(すなわち、発現されたときC−末端と関連する)のいずれかで第VIII因子融合遺伝子内に位置し得る(ただし、その位置が、他に、第VIII因子融合ヘテロダイマーの形成のために必要である転写、翻訳または翻訳後修飾を妨げない)ことを理解している。モジュレーターをコードする核酸のような多重合配列をコードする核酸は、Bドメインをコードする第VIII因子遺伝子の領域の少なくとも1部分内に挿入されるか、または該部分と置き換えられ得る。
【0096】
発現カセットおよび発現ベクター
本発明のさらなる局面は、第VIII因子融合遺伝子を含む発現カセットまたは発現ベクターに関する。第VIII因子融合遺伝子を含む発現カセットの組換え生産のために、第VIII因子融合遺伝子を単離し、プロモーターに作動可能に連結させる。該第VIII因子融合遺伝子は、所望によりさらに、転写終結シグナル、シグナルペプチドをコードする核酸、または遺伝子発現または翻訳後プロセッシングに影響する他の核酸配列(例えば、便利に位置された制限酵素認識部位、エンハンサー、分泌リーダー配列など)に作動可能に連結していてもよい。発現カセットの所望の因子(第VIII因子融合遺伝子以外)がすでに複製可能なクローニングベクターまたは発現ベクター内に含まれているとき、第VIII因子融合遺伝子は、当該分野でよく知られている組換え技術により適当な位置に作動可能に挿入されていることのみ必要である。多数のクローニングベクターが市販されており、一般的に1つ以上の以下のもの:シグナル配列、複製起点、エンハンサーエレメント、プロモーター、転写終結配列、および1つ以上の選択遺伝子またはマーカーを含む。多数の発現ベクターも、また、市販されており、第VIII因子融合遺伝子の挿入は、当該分野でよく知られている方法および試薬を使用して達成され得る(例えば、Sambrookら., Molecular Cloning: A Laboratory Manual, Second Edition, Cold Spring Harbor Press, NY (1989); Ausubelら., Current Protocols in Molecular Biology, New York, N.Y.: John Wiley & Sons (1989)、参照)。発現ベクターの選択は、好ましい形質転換技術および形質転換のための標的宿主に依存する。
【0097】
本発明において有用な発現ベクターは、染色体−、エピソーム−およびウイルス−由来のベクター、例えば、細菌プラスミド、バクテリオファージ、酵母エピソーム、酵母染色体エレメント、ウイルス、例えば、バキュロウイルス、パポバウイルス、ワクシニアウイルス、アデノウイルス、家禽ジフテリアウイルス、仮性狂犬病ウイルスおよびレトロウイルス由来のベクター、およびそれらの組合せ由来のベクター、例えば、コスミドおよびファージミドを含むが、これらに限定されない。動物細胞において組換え発現のために適当なウイルスベクターは、当該分野でよく知られている(例えば、米国特許第5,871,986および6,448,046号、参照)。
【0098】
本発明を実施するために適当なベクターは、以下のウイルスベクター、例えば、ラムダベクター系 gt11、gtWES.tB、Charon 4、およびプラスミドベクター、例えば、pCMV、pBR322、pBR325、pACYC177、pACYC184、pUC8、pUC9、pUC18、pUC19、pLG339、pR290、pKC37、pKC101、SV 40、pBluescript II SK +/−またはKS +/−(Stratagene, LaJolla, CA)、pQE、pIH821、pGEX、pETシリーズ(Studierら., Methods Enzymol. 185:60-89, 1990)、およびそれらのあらゆる誘導体を含むが、これらに限定されない。細菌において使用するために適当なベクターは、pQE70、pQE60、およびpQE−9(Qiagen, Valencia, CA);pBS ベクター、Phagescript ベクター、Bluescript ベクター、pNH8A、pNH16a、pNH18A、およびpNH46A(Stratagene, LaJolla, CA);pcDNA3(Invitrogen, Carlsbad, CA);およびpGEX、ptrxfus、ptrc99a、pET−5、pET−9、pKK223−3、pKK233−3、pDR540、およびpRIT5を含む。適当な真核ベクターは、pWLNEO、pSV2CAT、pOG44、pXT1、pBK、およびpSG(Stratagene, LaJolla, CA);およびpSVK3、pBPV、pMSG、およびpSVLである。他の適当なベクターは、当業者に容易に明白である。
【0099】
発現ベクターは、選択できる表現型、例えば、薬物耐性、栄養要求性、細胞毒性剤に対する抵抗性または表面タンパク質の発現を与える選択可能なマーカーをコードする遺伝子を含むものを使用され得る。使用することができる選択可能なマーカー遺伝子の例は、neo、gpt、dhfr、ada、pac(ピューロマイシン)、hyg、およびhisDを含む。
【0100】
第VIII因子融合遺伝子の成功するライゲーション(またはベクターへの挿入)は、当該分野でよく知られている組換え技術(例えば、慣用の手段を使用する単離およりシーケンシングまたは連結部位に特異的に結合することができるオリゴヌクレオチドプローブの使用)により容易に決定され得る。
【0101】
宿主細胞
本発明のさらなる局面は、第VIII因子融合遺伝子を含む宿主細胞に関する。該第VIII因子融合遺伝子は、クローニングベクター、発現ベクター内に存在するか、または宿主細胞ゲノムに統合されていてもよい。1つの態様において、宿主細胞は、DNA分子形態における必要な核酸構築物を安定なプラスミドまたは宿主細胞ゲノムへの安定な挿入もしくは統合のいずれかとして含む。別の態様において、宿主細胞は、発現系においてDNA分子を含むことができる。
【0102】
1つの態様において、本発明の第VIII因子融合遺伝子は、オープンリーディングフレームが選択されたプロモーターのコントロール下でコードされたタンパク質の発現に対して適当に指向するように、センス方向において適当なベクターに組み込まれる。これは、適当な調節エレメントの発現ベクターへの包含を含む。これらは、例えば、ベクターの非翻訳領域、有用なプロモーター、および転写および翻訳を実施するために宿主細胞タンパク質と相互作用する5’および3’非翻訳領域を含み得る。このようなエレメントは、それらの強度および特異性において変化し得る。利用されるベクター系および宿主に依存して、構成的および誘導プロモーターを含むあらゆる適当な転写および翻訳エレメントが使用され得る。構成的プロモーターは、生物体の発生および生存中に遺伝子の発現を指向するプロモーターである。誘導プロモーターは、誘導因子に応答して1つ以上のDNA配列または遺伝子の転写を直接的または間接的に活性化することができるプロモーターである。誘導因子の非存在下で、該DNA配列または遺伝子は転写されない。
【0103】
本発明の発現ベクターは、また、選択されたタンパク質をコードするDNA分子に作動可能に連結した、選択された宿主細胞における発現に対してmRNAの正しい転写終結およびポリアデニル化を提供することができるものの中から選択される、作動可能な3’調節領域を含み得る。
【0104】
宿主細胞において第VIII因子融合ヘテロダイマーを組換え的に生産するために、第VIII因子融合遺伝子は、宿主細胞に組み込まれ得る。クローニングベクター、発現ベクターおよびプラスミドは、当該分野でよく知られている組換え技術を使用して、例えば、形質転換、形質導入、接合、動員、またはエレクトロポレーションを介して細胞に導入され得る。
【0105】
宿主細胞は、哺乳動物細胞、細菌細胞(例えば、大腸菌)、昆虫細胞(例えば、Sf9細胞)、真菌細胞、酵母細胞(例えば、サッカロミセス(Saccharomyces)またはシゾサッカロミセス(SchizoSaccharomyces))、植物細胞(例えば、シロイヌナズナまたはタバコ細胞)、または藻細胞を含み得るが、これらに限定されない。本発明を実施するために適当な哺乳動物細胞は、COS(例えば、ATCC番号CRL1650または1651)、赤んぼのハムスター腎臓(「BHK」)(例えば、ATCC番号CRL6281)、チャイニーズハムスター卵巣(「CHO」)(ATCC番号CCL61)、HeLa(例えば、ATCC番号CCL2)、293(ATCC番号1573)、NSO骨髄腫、CHOP、NS−1およびHKB11(例えば、米国特許第6,136,599号、参照)を含むが、これらに限定されない。
【0106】
哺乳動物細胞における発現を指向するために適当な発現ベクターは、一般的に、プロモーター、ならびに当該分野で知られている他の転写および翻訳コントロール配列を含む。一般的なプロモーターは、SV40、MMTV、メタロチオネイン−1、アデノウイルスEla、CMV、前初期、免疫グロブリン重鎖プロモーターおよびエンハンサー、およびRSV−LTRを含む。当業者は、プロモーターとしての強度に基づいて適当な哺乳動物プロモーターを容易に選択することができる。あるいは、誘導プロモーターは、特定のタンパク質の発現または抑制が望ましいとき、制御の目的のために使用することができる。当業者は、当該分野で知られているものから適当な誘導哺乳動物プロモーターを容易に選択することができる。
【0107】
本発明の第VIII因子融合ヘテロダイマーの組換え生産のために選択される宿主細胞にかかわらず、タンパク質発現の増加は、第VIII因子融合遺伝子における一般的でないコドンをより一般的であるコドンと置き換えることにより達成され得る(例えば、米国特許第6,924,365号、参照)。当業者は、特定の第VIII因子融合遺伝子コドンが「一般的でない」または「一般的である」であるかを決定することが、組換え生産のために選択される宿主細胞の特定のコドン利用に依存することを理解している。
【0108】
第VIII因子融合タンパク質およびヘテロダイマーの生産
本明細書に記載されている組換え科学技術を考慮して、本発明の別の局面は、本発明の第VIII因子融合ヘテロダイマーを生産する方法に関する。この方法は、宿主細胞が第VIII因子融合タンパク質を発現する条件下で本発明の宿主細胞を増殖させることを含む。第VIII因子融合タンパク質の翻訳後修飾後、次に、組換え第VIII因子融合ヘテロダイマーは、精製され単離され得る。本発明の1つの局面は、第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを生産するための方法であって、(a)第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーをコードする発現ベクターで形質転換された宿主細胞を提供し;(b)該細胞を培養し;(c)第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを単離することを含む方法である。さらなる態様において、該宿主細胞は哺乳動物宿主細胞であり得、モジュレーターのアミノ酸配列はグリコシルされていてもよい。
【0109】
マルチマー第VIII因子融合ヘテロダイマーの組換え生産に関して、当業者は、ホモ−マルチマー型が、単一の組換え宿主細胞により生産され得るが、ヘテロ−マルチマー型が単一の宿主細胞内の共発現または複数の宿主細胞における別々の発現(同じか、または異なっている細胞培養系において)により生産され得ることを理解している。ヘテロ−マルチマー型と同様に、当業者は、ハイブリッド第VIII因子融合ヘテロダイマーが、単一の宿主細胞内の共発現または複数の宿主細胞における別々の発現(同じか、または異なっている細胞培養系において)により生産され得ることを理解している。別々の培養系が利用されるとき、それぞれの培養物からの組換えタンパク質産物は単離され、次に当該分野でよく知られている標準技術を使用して再び連合させ得る。マルチマー第VIII因子融合ヘテロダイマーおよびハイブリッド第VIII因子融合ヘテロダイマーの組換え生産のために、宿主細胞は、所望の様式においてマルチマーまたはハイブリッド第VIII因子融合ヘテロダイマーの鎖を組み立てることができるように選択され得る。
【0110】
別々の遺伝子の共発現における代替案として、必要とされるポリペプチド鎖のすべてをコードするモノシストロン性遺伝子が生産され得る。このような遺伝子がどのように設計され得るか特定の実施例、以下の実施例5参照。
【0111】
組換え第VIII因子融合ヘテロダイマーは、実質的に純粋な形態において生産され得る。当該分野でよく知られている方法は、精製された第VIII因子融合ヘテロダイマーの精製および同定のために使用され得る。
【0112】
医薬組成物
本発明の別の局面は、第VIII因子融合ヘテロダイマーおよび薬学的に許容される担体を含む医薬組成物に関する。「薬学的に許容される担体」は、調製物の製剤化を助けるか、または調製物を安定にさせるために活性成分に加えられ得、患者へ有意な有害な毒性効果を与えない物質である。このような担体の例は、当業者によく知られており、水、糖、例えば、マルトースまたはスクロース、アルブミン、塩、例えば、塩化ナトリウムなどを含む。他の担体は、例えば、Remington's Pharmaceutical Sciences by E. W. Martinに記載されている。このような組成物は、有効量の少なくとも1つの第VIII因子融合ヘテロダイマーを含む。
【0113】
薬学的に許容される担体は、滅菌水性溶液または分散媒および滅菌注射可能溶液または分散媒の即時調製物のための滅菌粉末を含む。薬学的に有効な物質に対するこのような媒体および薬剤の使用は、当該分野で知られている。該組成物は、非経口注射のために製剤化され得る。該組成物は、高い薬物濃度に適当な溶液、マイクロエマルジョン、リポソームまたは他の規則構造として製剤化され得る。該担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)、および適当なそれらの混合物を含む溶媒または分散媒であり得る。該組成物は、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール、ソルビトールまたは塩化ナトリウムを含み得る。第VIII因子の医薬組成物の例は、例えば、米国特許第5,047,249;5,656,289;5,665,700;5,690,954;5,733,873;5,919,766;5,925,739;6,835,372;および7,087,723号において記載されている。
【0114】
滅菌注射可能溶液は、上記されている成分の1つまたは組合せを有する適当な溶媒中に必要な量の活性化合物を包含し、必要なとき、次に滅菌精密濾過することにより製造され得る。一般的に、分散媒は、活性化合物を塩基性分散媒および必要な他の成分を含む滅菌輸送媒体に包含させることにより製造され得る。滅菌注射可能溶液の調製物に関する滅菌粉末の場合において、製造方法は、あらかじめ滅菌濾過された溶液から活性成分プラス任意のさらなる所望の成分の粉末を得る真空乾燥およびフリーズドライ(凍結乾燥)を含む。
【0115】
処置の方法
本発明の別の局面は、遺伝的および後天的凝固欠乏症、例えば、血友病(例えば、血友病A)を処置する方法に関する。この方法は、血友病Aを示す患者に有効量の本発明の第VIII因子融合ヘテロダイマー(ハイブリッドまたはマルチマー型を含む)を投与し、それにより該患者が血管損傷後の有効な血液凝固を示すことを含む。適当な有効量の第VIII因子融合ヘテロダイマーは、限定はされないが、約10から約50国際ユニット/kg体重からなる。患者は、何らかの哺乳動物(例えば、ヒト)であり得る。
【0116】
本発明の第VIII因子融合ヘテロダイマーは、静脈内に、皮下に、または筋肉内に投与され得る。特定のモジュレーターは、経口投与が考慮され得る。
【0117】
本発明の第VIII因子融合ヘテロダイマーは、阻害抗体を有する、およびそれを有さない血友病患者ならびに阻害抗体の発現によって後天的第VIII因子欠乏を有する患者における第VIII因子欠乏(例えば、関節内、頭蓋内または消化器出血)による非制御出血を処置するために使用され得る。1つの態様において、第VIII因子融合ヘテロダイマーは、単独または医薬組成物(すなわち、安定剤、送達輸送媒体、および/または担体と組み合わせて)の形態において、ヒトまたは動物第VIII因子の注入のために使用されるのと同じ方法にしたがって、患者の静脈内に注入される。
【0118】
あるいは、またはそれらに加えて、第VIII因子融合ヘテロダイマーは、第VIII因子融合遺伝子発現構築物を含むウイルスベクター、例えば、アデノ随伴ウイルス(例えば、Gnatenkoら., Br. J. Haematol. 104:27-36, 1999、参照)を投与することにより、または一般的にこのような細胞を含むデバイスの移植により、第VIII因子融合ヘテロダイマーを生産するように遺伝的に操作された細胞を移植することにより投与され得る。このような移植は、組換え真皮繊維芽細胞(例えば、Rothら., New Engl. J. Med. 344:1735-1742, 2001、参照);骨髄間質細胞(例えば、米国特許第6,991,787号、参照)または造血前駆宿主細胞(例えば、米国特許第7,198,950号、参照)を使用することを含む。血友病Aの遺伝子治療(第VIII因子をコードする核酸を使用する)における使用のために適当なウイルスベクターおよび遺伝子治療におけるそれらの使用は、当該分野で知られている(例えば、米国特許第6,200,560;6,544,771;6,649,375;6,697,669;6,773,709;6,797,505;6,808,905;6,818,439;6,897,045;6,939,862;7,198,950;および7,238,346号、参照)。
【0119】
このような処置の必要な患者に投与されるべき第VIII因子融合ヘテロダイマーの処置用量は、第VIII因子欠乏の重症度に依存して変化する。一般的に、投与レベルは、それぞれの患者の出血症状の重症度および期間としたがって、頻度、期間およびユニットにおいて調節される。したがって、第VIII因子融合ヘテロダイマーは、標準凝固アッセイにより測定されるとき、患者に治療有効量の出血を止めるためのタンパク質を送達するために十分な量における薬学的に許容される担体、送達輸送媒体、または安定剤中に含まれ得る。
【0120】
通常、第VIII因子融合ヘテロダイマーの投与を介して患者中で達成されるべき所望の血漿の第VIII因子の活性レベルは、通常の30−100%の範囲である。1つの態様において、治療的第VIII因子融合ヘテロダイマーの投与は、約5から約50ユニット/kg体重の範囲、約10から約50ユニット/kg体重の範囲における用量、および約20から約40ユニット/kg体重の用量で静脈内に与えられ得;間隔頻度は、約8から24時間の範囲であり得(深刻な血友病患者において);処置の日数は1から10日間の範囲もしくは出血症状が解決するまでであり得、または第VIII因子融合ヘテロダイマーの投与は予防であり得る(例えば、Robertsら., pp 1453-1474, 1460, in Hematology, Williams, W. Jら., ed. (1990)、参照)。ヒトまたは血漿由来第VIII因子での処置におけるとき、注入される治療的組換え第VIII因子の量は、一段階の第VIII因子凝固アッセイにより定義され得、選択された例において、インビボでの回収率は、注入後の患者の血漿中の第VIII因子を測定することにより測定され得る。あらゆる特定の患者に対して、特定の投与レジメンは、必要な個体および組成物を投与する、またはそれの投与を管理する個体の専門的な判断にしたがって時間をかけて調節されるべきであり、本明細書に記載されている濃度範囲は典型的なだけであり、本発明の第VIII因子融合ヘテロダイマーの範囲または実施を限定すると意図すべきでないと理解すべきである。
【0121】
処置は、単回投与または長期間の周期的もしくは連続的投与の形態を取り得、または、必要なとき、処置は、予防目的のために投与され得る。
【0122】
本発明の第VIII因子融合ヘテロダイマーは、由来の第VIII因子タンパク質と比較して循環半減期の増加を示す。より良い循環半減期を有する第VIII因子タンパク質は、より少ない頻度の投与で患者の第VIII因子欠乏を正すことができるため、血友病の処置において有用である。投与の場合におけるこの増加は、処置プロトコールでの患者のコンプライアンスを改善し、それにより凝固障害の症状を減少させ得る。また、投与の頻度の減少は、少ない抗原が投与されるため、第VIII因子に対する免疫応答の発達の可能性を減少させることが期待される。
【0123】
上記記載は、一般的に本発明を記載している。説明のみの目的のために提供され、本発明の範囲を限定することを意図しない以下の実施例を参照することにより、さらなる完全な理解が得ることができる。
【実施例】
【0124】
実施例
本発明を良く理解するために、以下の実施例を記載する。これらの実施例は、説明のみの目的のためであり、いかなる形であれ本発明の範囲を限定すると解釈されるべきでない。本明細書に記載されている全ての文献は、出典明示によりそれら全体を包含させる。
【0125】
実施例1
以下の実施例は、第VIII因子B−ドメイン欠失(BDD)タンパク質をコードする核酸およびマウスFc領域(「mFc+ヒンジ」と示される)をコードする核酸を使用する、第VIII因子融合遺伝子(「BDDmFc+ヒンジ」と示される)を含む哺乳動物発現ベクター(「pM110」または「pSK207BDDFc+ヒンジ」と示される)の構築を記載する。BDDmFc+ヒンジの組換え発現は、図2において示されている第VIII因子融合ヘテロダイマー(「BDDFc+ヒンジ」と示される)の生産をもたらす。機能的免疫グロブリンヒンジ領域の存在によって、BDDFc+ヒンジの2つの分子は、マルチマー第VIII因子融合ヘテロダイマーを形成するためにジスルフィド結合を介して共有結合している。この型の利点は、ダイマーFcがFcRnへの融合タンパク質の高親和性結合をもたらし、その結果、長期循環半減期をもたらすことである。
【0126】
PmeIおよびNheI部位により結合された第VIII因子B−ドメイン欠失(BDD)遺伝子を含むプラスミドpSK207(「pSK207BDD」と示される)は、部位特異的変異誘発キットを使用して変異させた。2つの制限酵素認識部位(bp4490でAvrIIおよびbp4520でAflII)は、変異プライマーCES16(5’−caatgccattgaacctaggagcttctcccagaacccaccagtccttaagcgccatcaacggg−3’)(配列番号:34)およびCES17(5’−cccgttgatggcgcttaaggactggtgggttctgggagaagctcctaggttcaatggcattg−3’)(配列番号:35)を使用して分子に導入された。bp2537でのAflII部位は、変異オリゴCES18(5’−cagtggtcattacactcaagaacatggcttcccatcc−3’)(配列番号:36)およびCES19(5’−ggatgggaagccatgttcttgagtgtaatgaccactg−3’)(配列番号:37)を使用して除去した。得られたプラスミドをpM109と称した。これらの変異事象は全てサイレントであり、BDDに対するアミノ酸変化をもたらさなかった。マウスFc領域の供給源として、ヒト上皮細胞増殖因子受容体に対する全長マウスIgG1抗体を含むプラスミドpGT234を使用した。マウスFc+ヒンジ領域を、鋳型としてpGT234でプライマーCES36(5’−agcttcctaggagcttctcccagaacgtgcccagggattgtggttg−3’)(配列番号:38)およびCES39(5’−agctacttaaggactggtgggttctgggatttaccaggagagtgggagag−3’)(配列番号:39)を使用してPCR増幅させた。得られたフラグメントをAflII/AvrIIで消化し、AflII/AvrIIで消化されたpM109にクローニングし、プラスミドpM117を製造した。bp2537にて元のAflII部位を復帰させるために、pSK207+BDDのNheI/BglIIフラグメントをpM117に挿入して、その同等領域を置き換え、bp2537にAflII部位を含むプラスミドpM115を製造した。pM115のBDD.mFc+ヒンジ遺伝子(5077bpのNheI/PmeIフラグメントに含まれる)を発現ベクターpSS207のPmeI/NheI部位にクローニングし、プラスミドpM110またはpSK207BDDFc+ヒンジを産生した。pSK207BDDFc+ヒンジの第VIII因子融合遺伝子因子(すなわち、BDDmFc+ヒンジ)は、配列番号:31の核酸配列を有する。BDDmFc+ヒンジによりコードされるタンパク質は、図3(「マウスFc」はmFc+ヒンジの位置を示す)において説明される構造ドメイン、および配列番号:32のアミノ酸配列を有する。
【0127】
実施例2
以下の実施例は、第VIII因子B−ドメイン欠失(BDD)タンパク質をコードする核酸およびマウスFc領域のヒンジ部分(「mFc−ヒンジ」と示される)以外の全てをコードする核酸を使用する、第VIII因子融合遺伝子(「BDDmFc−ヒンジ」と示される)を含む哺乳動物発現ベクター(「pM118」または「pSS207BDDFc−ヒンジ」と示される)の構築物を記載する。BDDmFc−ヒンジの組換え発現は、図2において示されている第VIII因子融合ヘテロダイマー(「BDDFc−ヒンジ」と示される)の生産をもたらす。BDDmFc−ヒンジによりコードされるタンパク質は、図3(「マウスFc」はmFc−ヒンジの位置を示す)において説明される構造ドメインおよび配列番号:33のアミノ酸配列を有する。機能的免疫グロブリンヒンジ領域の非存在によって、BDDFc−ヒンジはマルチマー第VIII因子融合ヘテロダイマーを形成しない。この型の不利な点は、非ダイマー化Fc領域がFcRn結合に対する親和性を減少させることである。
【0128】
BDDmFc−ヒンジの構築物は、上記BDDmFc+ヒンジと非常に類似している。mFc−ヒンジ領域を、PCRプライマーCES37(5’−agcttcctaggagcttctcccagaacgtcccagaagtatcatctgtc−3’)(配列番号:40)およびCES39(配列番号:39)を使用してプラスミドpGT234からPCR増幅させ、AvrII/AflIIで消化し、AvrII/AflIIで消化されたpM109プラスミドにおいてクローニングした。得られたプラスミドをpM114(「pSK207.BDD.mFc−ヒンジ」とも称する)と称した。次に、BDD.mFc−ヒンジ遺伝子を含むpM114のNheI/PmeIフラグメントを発現ベクターpSS207にクローニングし、プラスミドpM118(すなわち、pSS207BDDFc−ヒンジ)を産生した。
【0129】
実施例3
以下の実施例は、アミノ末端でFlagタグを有するマウスFc領域(「mFc+ヒンジ」と示される)の発現のためのプラスミド(「pM130」と示される)の構築物を記載する。同じ宿主細胞においてpSS207BDDFc+ヒンジを有するこのプラスミドの共発現は、BDDFc+ヒンジダイマー、mFc+ヒンジダイマーならびにBDDFc+ヒンジおよびmFc+ヒンジのヘテロダイマーの混合物を製造する。Flagタグの包含は、連続分離工程において抗−Flag抗体および抗−第VIII因子抗体を使用する親和性クロマトグラフィーによる図2において示されているヘテロダイマー(「BDDFc」と示される)の単離を容易にする。しかしながら、当業者は、ペプチドタグの提供がなくても、当該分野でよく知られている技術、例えば、サイズ排除クロマトグラフィーを使用してヘテロダイマー形態を分離することが可能であることを理解している。
【0130】
鋳型としてpM110を使用して、5’(アミノ末端)末端にFlagタグを有するマウスFc領域(ヒンジを有する)を、プライマーCES49(5’−atatgatatcgcggccgccgccaccatggtgttgcagacccaggtcttcatttctctgttgctctggatctctggtgcctacggggactacaaagacgatgacgacaaggtgcccagggattgtggttg−3’)(配列番号:41)およびCES50(5’−ttcgatctcgagtcatttaccaggagagtgggagagg−3’)(配列番号:42)を使用してPCR増幅させた。このフラグメントをNotI/XhoIで消化し、NotI/XhoIで消化された発現ベクターpAGE16にライゲートし、プラスミドpM119(すなわち、pAGE16.mFc+ヒンジ.Flag)を製造した。次に、mFc+ヒンジ.Flag領域を含むpM119のHindIII/XhoIフラグメントをHindIII/XhoIで消化された発現プラスミドpEAK flCMV W/GFPにサブクローニングし、pM130と称した。
【0131】
実施例4
以下の実施例は、第VIII因子B−ドメイン欠失(BDD)タンパク質をコードする核酸およびIgG1、IgG2、IgG3もしくはIgG4のヒトFc領域のいずれか1つ、またはIgG1、IgG2、IgG3もしくはIgG4のヒトFc領域の非ヒンジ部分のいずれか1つをコードする核酸を使用する第VIII因子融合遺伝子(「BDD.Human Fc」と示される)の構築物を記載する。1つの例として、第VIII因子融合ヘテロダイマーを、Bドメインを模倣するために、マウスIgG1 Fc由来の227アミノ酸残基または214アミノ酸残基を第FVIII因子の特定の部位(例えば、N−745からS−1637)に挿入することにより産生し得る。
【0132】
BDD.Human Fc(IgG1、IgG2、IgG3もしくはIgG4抗体由来)発現ベクターの構築物は、上記BDD−マウスFc発現構築物と同じ戦略に従う。pM109プラスミドをAvrII/AflIIで消化し、AvrII/AflII結合Fc+ヒンジおよびFc−ヒンジを対応する部位に挿入する。次に、pSK骨格を有する得られたプラスミドをNheIおよびPmeIで消化し、BDDFcフラグメントを安定な細胞クローンの創造のためにpSS207にライゲートした。同様に、pCEP4.human Fcモノマープラスミドを構築する。
【0133】
典型的なヒトIgGのFc領域の核酸コード配列は、配列番号:8(ヒトIgG1のFc領域)、配列番号:10(ヒトIgG2のFc領域)、配列番号:12(ヒトIgG3のFc領域)および配列番号:14(ヒトIgG4のFc領域)を含む。あるいは、配列番号:9(ヒトIgG1のFc領域)、配列番号:11(ヒトIgG2のFc領域)、配列番号:13(ヒトIgG3のFc領域)および配列番号:15(ヒトIgG4のFc領域)と同じアミノ酸配列(または少なくとも90%同一性を有するアミノ酸配列)をコードする核酸配列を使用してもよい。典型的な非ヒンジ部分のヒトIgGのFc領域の核酸コード配列は、配列番号:16(ヒトIgG1のFc領域の非ヒンジ部分)、配列番号:18(ヒトIgG2のFc領域の非ヒンジ部分)、配列番号:20(ヒトIgG3のFc領域の非ヒンジ部分)および配列番号:22(ヒトIgG4のFc領域の非ヒンジ部分)を含む。あるいは、配列番号:17(ヒトIgG1のFc領域の非ヒンジ部分)、配列番号:19(ヒトIgG2のFc領域の非ヒンジ部分)、配列番号:21(ヒトIgG3のFc領域の非ヒンジ部分)および配列番号:23(ヒトIgG4のFc領域の非ヒンジ部分)と同じアミノ酸配列(または少なくとも90%同一性を有するアミノ酸配列)をコードする核酸配列を使用してもよい。
【0134】
実施例5
以下の実施例は、ハイブリッド第VIII因子融合ヘテロダイマーをコードするモノシストロン性第VIII因子融合遺伝子の構築物を記載する。翻訳された第VIII因子融合タンパク質は、全長FVIIIのBドメインの代わりに2つのタンデムmFc+ヒンジ領域を含む。
【0135】
発現プラスミドを以下のとおり構築する:鋳型としてpM117(pSK207+BDD.mFc+ヒンジ)を使用するオリゴの2つのセットでのPCR−第1は、AvrII(CES36:5’−agcttcctaggagcttctcccagaacgtgcccagggattgtggttg−3’)(配列番号:30)およびSacII(CES51:5’−cagttgccgcgggctttaccaggagagtgggagagg−3’)(配列番号:35)により結合するmFcフラグメントを創造するCES36/CES51であり、プライマーの第2のセットは、SacII(CES52:5’−ttcgcccgcggcaagagagactacaaagacgatgacgacaaggtgcccagggattgtggttg−3’)(配列番号:35)およびAflII(CES39:5’−agctacttaaggactggtgggttctgggatttaccaggagagtgggagag−3’)(配列番号:31)により結合するmFcフラグメントを創造するCES52/CES39である。適当に消化およびライゲートされたとき、これらの2つの得られたPCRフラグメントは、順にA1、a1、A2、a2ドメイン、B−ドメインの最初の5個のN−末端アミノ酸、次にmFc+ヒンジ領域、furinコンセンサス配列(Fc領域の末端である最初のリジン(K)を有するKARGKR(配列番号:36))、Flagタグ(DYKDDDDK)(配列番号:37)、mFc+ヒンジ、B−ドメインの最後の12個のC−末端アミノ酸、ならびに最後にFVIIIのa3、A3、C1およびC2ドメイン(図4)を含むモノシストロン性BDD遺伝子となる。上記モノマー遺伝子を構築するために、2つのPCRフラグメントをAvrII/SacIIまたはSacII/AflIIで消化し、トリプルライゲーションを介して、AvrII/AflIIで消化したpM109(pSK207.BDD)にクローニングする。成功したクローンを配列決定し、次に、1つをpSK207骨格由来のNheI/PmeI(pM109の)を介して発現ベクターにクローニングし、pSK207をNheI/PmeIで消化する。タンパク質の合成および分泌中、該分子を最初にFlag−mFc領域の上流のfurin部位およびa3のすぐ上流のプロテアーゼ部位で開裂させる。該分子は、図2において示されているとおりにFc−Fcジスルフィド相互作用(BDDFc)を介してFVIII分子のmFc領域に結合しているFlag mFc分子を有する成熟FVIIIダイマー(BドメインをmFcで置き換えられている)として循環する。ヘテロダイマー産物を、当業者に知られている方法を使用して、上清中に存在するすべてのホモダイマー産物から単離する。
【0136】
実施例6
以下の実施例は、哺乳動物宿主細胞の一過性トランスフェクションのために有用な一般的な処理およびその細胞培養を記載する。HKB11細胞をオービタルシェーカー(100−125rpm)上で5%CO2インキュベーター中で37℃でタンパク質非含有培地中で懸濁培養中で増殖させ、0.25から1.5×106細胞/mLの密度で維持する。トランスフェクションのためのHKB11細胞を1,000rpmで5分遠心分離により回収し、次に、1.1×106細胞/mLでFreeStyleTM293発現培地(Invitrogen Corporation, Carlsbad, CA)に再懸濁する。細胞を6つのウェルプレートに播種し(4.6mL/ウェル)、オービタルローター上で(125rpm)37℃ CO2インキュベーター中でインキュベートする。それぞれのウェルに対して、5μgのプラスミドDNAを0.2mlのOpti−MEM(登録商標)I培地(Invitrogen Corporation, Carlsbad, CA)と混合する。それぞれのウェルに対して、7μLの293FectinTM試薬(Invitrogen Corporation, Carlsbad, CA)を0.2mLのOpti−MEM(登録商標)I培地と穏やかに混合し、室温で5分インキュベートする。希釈された293FectinTMを希釈されたDNA溶液に加え、穏やかに混合し、室温で20−30分インキュベートし、次に、5×106(4.6mL)のHKB11細胞で播種されたそれぞれのウェルに加える。次に、細胞をオービタルローター(125rpm)上でCO2インキュベーター中で37℃で3日間インキュベートし、その後、細胞を1000rpmで5分、遠心分離によりペレット化し、次に、上清を回収し、−80℃で保存する。
【0137】
実施例7
以下の実施例は、ウエスタンブロッティングによる第VIII因子融合ヘテロダイマーの組換え生産を証明するために有用な一般的な処理を記載する。細胞培養物上清を、Centricon(登録商標)(Millipore Corporation, Billerica, MA)により10倍に濃縮するか(二次抗体がプロービングのために使用されないとき)、またはそのまま使用する(二次抗体がプロービングのために使用されるとき)。50μLの上清をDTT(還元)有りまたはDTT(非還元)無しで20μLの4×SDS−PAGEローディング色素と混合し、95℃で5分加熱し、次に10%のNuPAGE(登録商標)ゲル(Invitrogen Corporation, Carlsbad, CA)(還元条件下)または4−20%のNuPAGE(登録商標)ゲル(Bis-Tris-MOPs)(非還元条件下)上に負荷する。タンパク質をニトロセルロース膜に移す。5%ミルク/PBSで60分ブロッキング後、膜をマウスIgG(H+L)に対するセイヨウワサビペルオキシダーゼ(HRP)−標識ウサギポリクローナル抗体またはHRP−接合抗−第VIII因子Cドメイン抗体とインキュベートする。また、抗−ヒト第VIII因子ウサギモノクローナル抗体(Epitomics, CA)を第VIII因子の軽鎖を検出するために使用され得る。次に、膜を抗ウサギIgG−HRP二次抗体と室温で60分インキュベートする。ブロットをPBS/0.1%のTween(登録商標)−20(ポリオキシエチレンモノラウリン酸ソルビタン)で洗浄後、HRP由来のシグナルを化学発光基質(ECL)(Pierce, Rockford, IL)およびx線フィルムへの暴露を使用して検出する
【0138】
実施例8
以下の実施例は、ELISAによる細胞培養物上清中の第VIII因子抗原の濃度を測定するために有用である一般的な処理を記載する。細胞培養物上清をPBS/BSA/Tween(登録商標)−20バッファーで希釈し、標準曲線の範囲内のシグナルを達成する。例えば、PBS/BSA/Tween(登録商標)−20で希釈され、精製された(比活性9,700IU/mg)第VIII因子BDDタンパク質を、100ng/mLから0.2ng/mLの標準曲線を創造するために使用され得る。希釈されたサンプルおよび標準物を、ポリクローナル抗−第VIII因子捕捉抗体C2であらかじめ被覆されているELISAプレートに加える。検出抗体としてビオチン化C2を加えた後、プレートを室温で1時間インキュベートし、広範囲に洗浄し、次にキット製造業者(Pierce, Rockford, IL)により記載されているとおりにTMB基質(3,3’,5,5’−テトラメチルベンジジン)を使用して現像する。シグナルをSpectraMax(登録商標)プレートリーダー(SpectraMax(登録商標)340pc, Molecular Devices, Sunnyvale, CA)を使用して450nMで測定され得る。標準曲線を4つのパラメーターモデルに合わせ、未知の値を曲線から推定する。
【0139】
無傷な第VIII因子融合ヘテロダイマーに特異的ではない上記処理の別法として、ELISAアッセイは、捕捉抗体(または検出抗体)として抗−第VIII因子抗体および検出抗体(または捕捉抗体)として半減期モジュレーターに特異的な抗体を利用する。
【0140】
実施例9
以下の実施例は、96ウェル型の市販の発色アッセイキット(Coatest(登録商標) SP4 FVIII, Chromogenix, Lexington, MA)を使用する細胞培養物上清および精製された画分中の第VIII因子融合ヘテロダイマーの活性を測定するために有用な一般的な処理を記載する。トリプリケートサンプルをキットアッセイバッファー(50mMのTris、pH7.3、10mg/Lのシプロフロキサシン(ciprofloxin)および1.0%のBSA)で25μLに希釈し、ウェルに加える。次に、50μLのリン脂質、第IXa因子、第X因子溶液をそれぞれのウェルに加え、水平シェーカー上で37℃で4分インキュベートする。25μLのCaCl2溶液(25mM)を即座にウェルに加え、同じ様式で10分インキュベートする。発色基質溶液(50μL/ウェル)を加え、プレートを10分インキュベートし、カラー現像を25μLの20%酢酸の添加により停止させる。個々のウェルを、405nmでの吸光度を96−ウェルプレートリーダー(SpectraMax(登録商標) 340pc, Molecular Devices, Sunnyvale, CA)で測定する。第VIII因子活性を、未知として、同じバッファーで希釈された500−0.5mIU/mLの範囲である精製された第VIII因子B−ドメイン欠失(BDD)標準に対して定量し、4つのパラメーターモデルに合わせる。比活性(FVIIIのIU/mg)をCoatest(登録商標)および第VIII因子のELISAの結果から計算する。
【0141】
実施例10
以下の実施例は、aPTTアッセイを使用する細胞培養物上清および精製された画分中の第VIII因子融合ヘテロダイマーの凝固活性を測定するために有用な一般的な処理を記載する。第VIII因子の凝固活性は、ElectraTM 1800C 自動凝固分析器(Beckman Coulter Inc., Fullerton, CA)により第VIII因子欠乏ヒト血漿においてaPTTアッセイを使用して測定され得る。簡潔には、凝固希釈剤中の上清サンプルの3つの希釈物を器具により作製し、次に、100μLを100μLのFactorVIII欠乏血漿および100μLの自動aPTT試薬(ウサギ脳リン脂質および微粉化シリカ、bioMerieux, Inc., Durham, NC)と混合する。100μLの25mMのCaCl2溶液の添加後、血塊形成の時間を記録する。標準曲線を、ELISAアッセイにおいて標準として使用されるのと同じ精製された第VIII因子BDDの連続希釈を使用してそれぞれ負荷して作成する。標準曲線は、0.95またはそれ以上の相関係数と直線的であった。標準曲線は、未知のサンプルの第VIII因子の活性を決定するために使用される。
【0142】
実施例11
以下の実施例は、実施例1および2に記載されているベクターを使用する細胞系の安定なトランスフェクションおよび創造を記載する。HKB11細胞を、実施例6に記載されている293FectinTM試薬を使用してプラスミドDNA、pSK207BDDFc+ヒンジまたはpSK207BDDFc−ヒンジでトランスフェクトした。トランスフェクトされた細胞を種々の希釈物(1:100;1:1000;1:10,000)で100mm培養皿に分け、5%のFBSおよび200μg/mLのハイグロマイシン(Invitrogen Corporation, Carlsbad, CA)を補ったDMEM−F12培地に約2週間維持した。個々の単一のコロニーをつつき取り、滅菌クローニングディスク(Scienceware(登録商標), Bel-Art Products, Pequannock, NJ)を使用して6−ウェルプレートに移した。pSK207BDDFc+ヒンジでトランスフェクトされたHKB11細胞の55を越えるクローンが確立され、寄託された。これらのクローンを、図5において示されている第VIII因子の活性アッセイ(上記のCoatest(登録商標)およびaPTTアッセイ)、図6において示されている第VIII因子のELISA(上記)、および増殖アッセイにより第VIII因子融合ヘテロダイマーの高い発現に関してスクリーニングした。非常に高い発現レベルを有する6つの細胞系を図6に示す。BDDFc+ヒンジに関してトップのクローンであるクローン8は、粘着して増殖するとき、〜1μg/mLの融合タンパク質を発現する。クローン8馴化培地由来のBDDFc+ヒンジの比活性は、由来のBDD第VIII因子タンパク質に相当する約5,000−8,000IU/mgであった。同様の安定なトランスフェクションおよび選択処理を使用して、BDDFc−ヒンジに関するクローン(クローンt)は、接着して増殖するとき、〜1μg/mLの融合タンパク質を発現することを決定した。
【0143】
実施例12
以下の実施例は、10L WAVE BioreactorTM(GE Healthcare, Piscataway, NJ)を使用する安定な形質転換体によるタンパク質発現のスケールアップを記載する。クローン8およびクローンt細胞を5%のFBSおよび200μg/mLのハイグロマイシンを補ったDMEM−F12培地に維持した。細胞を3日毎にT75からT225フラスコに1:4で分けた。培養適応のために、12個のT225フラスコの約10億個の細胞を2Lもしくは3L−Erlenmyerフラスコ中の2.5%のFBSを補った無血清である1Lの懸濁培地に移した。2日後、細胞を1.25%のFBSを補った無血清懸濁培地に拡大した。次に、細胞を5%のヒト血漿タンパク質溶液(HPPS)を補った無血清懸濁培地に移した。約100−150億個の細胞を10L WAVE BioreactorTMバッグにおいて培地中に約100万/mlの密度で播種した。3日後、細胞密度は5−600万/mLに到達し、馴化培地を回収した。最初に、粗培地を、蠕動ポンプにより制御されている150mL/分の流速で、Contifuge(登録商標) Stratos(Thermo Fisher Scientific, Waltham, MA)で6,000rpmで連続的な遠心分離により細胞残屑を除去して浄化した。浄化された培地をX−100(ポリエチレングリコール tert−オクチルフェニルエーテル)(0.05%まで)と混合し、10kDa Pellicon 接線流膜(Millipore, Billerica, MA)における限外濾過により約10倍に濃縮した。−80℃で凍結する前にスクロースを1%の濃度に加えた。精製前の組換え的に生産される第VIII因子融合ヘテロダイマーの比活性は、クローン8により生産されるBDDFc+ヒンジについて10,629IU/mgおよびクローンtにより生産されるBDDFc−ヒンジについて11,122IU/mgと測定された。
【0144】
実施例13
以下の実施例は、クローン8のスケールアップ培養物からの第VIII因子融合ヘテロダイマーの精製を記載する。抗−第VIII因子モノクローナル抗体親和性カラム(C7F7)、次に陰イオン交換Q−セファロースTMカラム(GE Healthcare, Piscataway, NJ)を使用して、第VIII因子BDDFc+ヒンジをHKB11細胞馴化培地から精製した。全回収率は約30%であった。10L WAVE BioreactorTM バッグからの凍結濃縮物を解凍し、AEKTATM Purifier システム(Amersham Pharmacia, Uppsala, SW)を使用して1mL/分で免疫親和性カラムに負荷し、次にカラムをバッファー(20mMのイミダゾール、0.01MのCaCl2、0.5MのNaCl、0.01%のTween(登録商標)−80(ポリエチレングリコール ソルビタンオレイン酸モノエステル)、pH7.0)で洗浄した。結合した第VIII因子BDDFc+ヒンジを1.0MのCaCl2を含むバッファーで溶離した。画分をCoatest(登録商標)アッセイにより第VIII因子活性についてアッセイし、活性な画分を貯め、バッファーをHiTrapTM 26/10 脱塩カラム G25M(Amersham Biosciences, Uppsala, SW)でイオン交換ローディングバッファー(20mMのイミダゾール、10mMのCaCl2、200mMのNaCl、0.01%のTween(登録商標)−80、pH7.0)に交換した。タンパク質を1mlのHiTrapTM Q HP カラム(Amersham Biosciences, Uppsala, SW)に負荷し、NaCl勾配(200mM−1000mM)で溶離した。画分をCoatest(登録商標)アッセイにより第VIII因子活性についてアッセイし、ピーク画分を貯めた。タンパク質濃度および比活性を決定した。ベストな画分(すなわち、図7のレーン8の画分5)の純度は、SDS−PAGEおよびSimplyBlueTM染色(Invitrogen, Carlsbad, CA)により概算されるとおり約80%である。ウエスタンブロット分析において抗−Fc抗体により検出されるため、精製された融合タンパク質はFcドメインを含んだ。精製された物質の比活性は、約10,000IU/mgであった。この比活性は、BDDFc+ヒンジが完全に活性であることを示唆する第VIII因子BDD(BDDFc+ヒンジ由来)にほぼ匹敵する。
【0145】
実施例14
以下の実施例は、組換え的に生産される第VIII因子融合ヘテロダイマーにおけるエンドトキシン試験を記載する。精製されたタンパク質溶液のエンドトキシンレベルを、0.005EU/mLの感度で動態発色 Limulus Amebocyte Lysate アッセイ(Endosafe(登録商標)キット)を使用して決定した。BDDFc+ヒンジにおけるエンドトキシンレベルは、5EU/用量をはるかに下回る1.3−2.0EU/mLと見出された。
【0146】
実施例15
以下の実施例は、これらの第VIII因子融合ヘテロダイマーが由来する精製されたBDDFc+ヒンジ、精製されたBDDFc−ヒンジおよび第VIII因子タンパク質(「BDD−FVIII」)を使用する正常マウスの薬物動態学試験を記載する。正常C57オスマウスを、50μg/kg体重でBDDおよび融合タンパク質(BDDFc+ヒンジまたはBDDFc−ヒンジ)の単回投与で静脈内に注射した。血液サンプルをt=注射後0、0.083、0.5、2、4、6、8、24、28、32、48および72時間(時点あたり5匹のマウス)で回収した。血液サンプルにおけるタンパク質レベル(抗原ELISAによる)および凝固活性(Coatest(登録商標)アッセイによる)の両方を薬物動態学分析のために決定した。結果は表1に示されている。
【表1】
【0147】
BDDFc+ヒンジおよびBDDFc−ヒンジのベータ半減期は、正常マウスにおけるBDD−FVIIIと同レベルであった。
【0148】
実施例16
以下の実施例は、精製されたBDDFc−ヒンジ、およびBDDFc−ヒンジが由来する第VIII因子タンパク質(「BDD−FVIII」)を使用する血友病A動物モデル(HemAマウス)における薬物動態学試験を記載する。結果は、BDDFc−ヒンジがBDD−FVIIIと比較してHemAマウスにおいて有意な長期ベータ相半減期を有することを示す。
【0149】
HemAマウスを、5%のアルブミンを含む製剤バッファー中の1.25μg/マウス(50μg/kg)でBDDFc−ヒンジ(「FVIII−Fc」9匹のマウス)を尾静脈(i.v.)を介して注射した。さらなるHemAマウスに、200IU/kgのBDD−FVIII;BDDFc−ヒンジ由来の第VIII因子変異体を投与した。血液を、BDDFc−ヒンジを与えた代わりのマウス(3匹のマウス/時点)から1、24、48、66、72、90、120および148時間目に、およびBDD−FVIIIを与えた代わりのマウス(5匹のマウス/時点)から1、4、8、16、24および32時間目に眼窩後を介してクエン酸塩中に回収した。血漿FVIII活性をCoatest(登録商標) SP FVIIIキット(Instrumentation Laboratory Company, Lexington, MA)を使用して測定した。ベータ相半減期をWinNonlin(登録商標)(Pharsight, Mountain View, CA)における少量のサンプリング(sparse-sampling)および非区画(non-compartment)モデルにより概算した。Coatest(登録商標)アッセイにおいて、標準曲線を作成するためにBDD−FVIIIを使用した。簡潔には、同じ血漿マトリックス中のサンプル、標準、陽性および陰性コントロール(それぞれ25μL)を96−ウェルプレートにデュプリケートで加えた。FIXa、FXおよびリン脂質溶液の混合物(50μL)を加え、37℃で5分インキュベートした。次に、25μLのCaCl2溶液を加え、37℃で5分インキュベートし、次に50μL基質を加え、色が適当な強度に発達するまで37℃で5分インキュベートした。停止溶液(25μL)を加え、プレートをプレートリーダー(SpectraMax(登録商標) 250, Molecular Devices, Sunnyvale, CA)でOD405nmで読んだ。結果は、図8において示されているとおりにSoftMax(登録商標) Pro 4.8(Molecular Devices, Sunnyvale, CA)を使用して計算した。結果は、それぞれの時点で、BDD−FVIIIに対して5匹のマウスおよびFVIII−Fcに対して3匹のマウスからの平均±SDを示した。
【0150】
BDD−FVIIIの減衰曲線と比較して、BDDFc−ヒンジは迅速な分布相で二相崩壊を示した(図8)。BDDFc−ヒンジのベータ相半減期は50μg/kgで11.9時間であり、これはベータ相半減期が6.03時間である修飾されていないBDD−FVIIIと比較して約2倍の改善であった。いくつかの第VIII因子融合ヘテロダイマーが非血友病A動物モデルにおける薬物動態学試験を使用して分析され得ない可能性があり得る。
【0151】
実施例17
以下の実施例は、上記第VIII因子融合遺伝子の発現産物である組換え第VIII因子融合ヘテロダイマーにおけるインビトロ試験を記載する。哺乳動物発現ベクターpSS207BDDFc+ヒンジおよびpSS207BDDFc−ヒンジをHKB11細胞に一過性でトランスフェクトし、馴化培地を上記トランスフェクション72時間後に回収した。図9Aにおいて示されているとおり、還元条件下で濃縮された上清のウエスタンブロット分析は、BDDFc+ヒンジ第VIII因子融合ヘテロダイマーが、最初に抗−Fc抗体により検出されるとおりの(レーン5)〜195kDaの第VIII因子融合タンパク質として発現し、抗−Fc抗体により検出されるとおりの(レーン5)115−kDa重鎖に;図9Bにおいて示されているとおり、第VIII因子軽鎖特異的抗体により検出されるとおりの(レーン5)80−kDa軽鎖に翻訳後プロセッシングされることを示した。比較のために、精製されたBDDタンパク質およびpSK207またはpSK207BDD(BDDFc+ヒンジをコードする第VIII因子融合遺伝子由来のB−ドメイン欠失第VIII因子遺伝子を含む発現ベクター)で一過性でトランスフェクトされたHKB11細胞からの馴化培地は、抗−Fc抗体と反応せず(図9A)、第VIII因子軽鎖抗体を使用すると、精製されたBDDタンパク質またはpSK207BDDで一過性でトランスフェクトされたHKB11細胞からの馴化培地から、予期される80kDa軽鎖を同定した(図9B)。対照的に、pSK207で一過性でトランスフェクトされたHKB11細胞からの馴化培地に軽鎖が存在しなかった(図9B)。これらの結果は、BDDFc+ヒンジをコードする第VIII因子融合遺伝子がB−a3ドメイン接合点で機能的切断部位をまだ維持しているため、軽鎖の分子量が変化することが期待されないため、欠失Bドメイン領域へのFc領域の挿入が翻訳後修飾に影響しないことを示す。
【0152】
第VIII因子活性を、Coatest(登録商標)アッセイおよびaPPT凝固アッセイによりpSK207BDD(コントロール)、pSK207BDDFc+ヒンジおよびpSK207BDDFc−ヒンジトランスフェクタントからの馴化培地で検出した(図10)。第VIII因子活性はpSK207トランスフェクタントからの馴化培地で検出されなかった。BDDFc融合タンパク質(すなわち、BDDFc+ヒンジおよびBDDFc−ヒンジ)の両方の活性範囲は、BDDに匹敵した。データは、使用される特定の部位へのFc領域の挿入が、由来のBDD第VIII因子タンパク質と比較して、第VIII因子融合ヘテロダイマーの翻訳後プロセッシングまたは生物学的活性に影響しないことを示唆した。
【0153】
pSK207BDDFc+ヒンジまたはpSK207BDDで一過性でトランスフェクトされたHKB11細胞から回収された馴化培地を、ウサギ−抗−マウスFc抗体(Pierce、Rockford、IL)であらかじめ被覆された96−ウェルプレートに加え、第VIII因子融合ヘテロダイマーを捕捉する固相Coatest(登録商標)アッセイ。BDDFc+ヒンジ融合タンパク質のみがプレートに結合し、由来の第VIII因子BDDタンパク質は流れる。次に、Coatest(登録商標)アッセイをウェルに固定されたBDDFc+ヒンジに対して直接実施し、図11は、BDDFc+ヒンジがこのアッセイにおいて活性であることを示す。
【0154】
pSK207BDDFc+ヒンジまたはpSK207BDDFc−ヒンジ発現ベクターで一過性でトランスフェクトされたHKB11細胞からの5倍濃縮馴化培地を使用して、分析を実施した。サンプルを還元および非還元条件下で4−12%のNuPAGE(登録商標)ゲルで分離した。ブロットをウサギモノクローナル抗−FVIII軽鎖抗体(Epitomics, Burlingame, CA)、次にHRP−接合抗−ウサギIgG二次抗体でプロービングした。結果は、BDDFc+ヒンジがダイマーを形成するが(すなわち、マルチマー第VIII因子融合ヘテロダイマー)、BDDFc−ヒンジがモノマーであることを示した(図12)。同様の結果がpSK207BDDFc+ヒンジで安定に形質転換された細胞で見られた。
【0155】
実施例18
以下の実施例は、第VIII因子融合ヘテロダイマーに包含されているとき、FcRn結合エピトープがFnRnに結合する能力を維持しているか否かを決定するために、BiacoreTMシステムを使用して実施された機能的試験を記載する。BiacoreTM試験における使用のために、組換えマウスFcRn(mFcRn)タンパク質をCHO−K1細胞で発現させ、マウスIgG−親和性クロマトグラフィーにより精製した。マウスFcRnをアミンカップリングによりCM−5チップ上に固定した。2つの第VIII因子ヘテロダイマー(BDDFc+ヒンジおよびBDDFc−ヒンジ)、BDD(BDDFc+ヒンジおよびBDDFc−ヒンジ由来の第VIII因子タンパク質)、および全長組換え第VIII因子を、種々の濃度(例えば、1.5、3、6、12、25、および50nM)でチップの表面を通過させた。固定されたmFcRnへのBDDFc±ヒンジ第VIII因子融合ヘテロダイマーの結合を検出した(図13)。結合親和性(KD=2.48nM)をBDDFc+ヒンジ(「BDDFc+H」)に対して計算し、BDDFc−ヒンジ(「BDDFc−H」)はBDDFc+ヒンジ(KD=3.75nM)と同様であった。検出可能な結合がBDD(「BDD」)または全長組換え第VIII因子(「FVIII」)で見られなかった。
【0156】
結果は、BDDFc+ヒンジおよびBDDFc−ヒンジ第VIII因子融合ヘテロダイマーがnMの親和性でmFcRnに対して強い結合を示すことを示す。対照的に、BDDも全長組換えFVIIIもmFcRnに結合することができなかった。これは、FcRn結合エピトープを含まないため、予期される。HemAマウスを使用して実施される薬物動態学試験を考慮して、結果は、BDDFc+ヒンジおよびBDDFc−ヒンジが高親和性でmFcRnに結合する機能的FcRn結合エピトープを含み、インビボで長期ベータ相半減期をもたらすことを示唆する。
【0157】
実施例19
循環において、FVIIIは、主に安定な複合体としてフォン・ヴィレブランド因子(vWF)に結合している。トロンビン(第IIa因子)により活性化されると、FVIIIは複合体から分離し、凝固カスケードと相互作用する。活性化FVIIIは、タンパク質分解的にプロセスにおいて不活性化され(活性化タンパク質Cおよび第IXa因子により非常に顕著に)、血流から迅速に除去される。以下の実施例は、第VIII因子融合ヘテロダイマーに包含されるとき、第VIII因子タンパク質がフォン・ヴィレブランド因子(vWF)に結合する能力を維持しているか否かを決定するために、BiacoreTMシステムを使用して実施される機能的試験を記載する。
【0158】
ヒトvWFをアミンカップリングによりCM−5チップ上に固定した。2つの第VIII因子ヘテロダイマー(BDDFc+ヒンジおよびBDDFc−ヒンジ)、BDD(BDDFc+ヒンジおよびBDDFc−ヒンジ由来の第VIII因子タンパク質)、および全長組換え第VIII因子を、種々の濃度(例えば、1、2、4、8、16、および25nM)でチップの表面を通過させた。BDD(「BDD」)および全長組換え第VIII因子(「FVIII」)の両方は、サブナノモル親和性(0.53−0.657nM)でヒトvWFに結合することができ、vWFへのBDDFc+ヒンジ(「BDDFc+H」)またはBDDFc−ヒンジ(「BDDFc−H」)の結合も検出した(図14)。BDDFc+ヒンジおよびBDDFc−ヒンジの結合親和性(KD)は、それぞれ0.465nMおよび0.908nMと計算された。データは、第VIII因子融合ヘテロダイマーBDDFc+ヒンジおよびBDDFc−ヒンジがvWFに対するサブナノモル親和性を有し、モジュレーターとしての免疫グロブリンFc領域の使用がvWFに対するBDDの結合特性をブロックしないことを示す。
【0159】
実施例20
以下の実施例は、BDDFc−ヒンジがHemAマウスの尾静脈切断出血モデルにおいて有効であることを証明する。HemAマウス(8−10週、〜25g)に、1つの側部尾静脈の切断の48時間前に、12もしくは60IU/kgの最終用量で5%のアルブミンを含む製剤バッファー中の100μLのBDDFc−ヒンジ、または40IU/kgで5%のアルブミンを含む製剤バッファー中の100μLのBDD−FVIII、または製剤バッファー単独(輸送媒体)(20匹のマウス/処置群)を尾静脈を介して注射した。マウスを麻酔し(ケタミン/キシラジン)、1つの側部尾静脈を尾の直径が約2.7mmである場所で切断した。次に、凝血するまで尾を37℃にあらかじめ温めた塩水で濯ぎ、出血時間を記録した。次に、マウスを加温パッドの上に紙ベッドを備えた個々のケージに移し、時間単位で最初に損傷9時間後、次に24時間後に観察した。再出血の事象を記録した。統計分析をGraphPad Prism(登録商標)4で実施し、結果を図15に示す。
【0160】
損傷24時間後に10%のみ生存した輸送媒体−コントロール群に対して、BDDFc−ヒンジの12IU/kgおよび60IU/kgは、それぞれ25%および80%の生存を成し遂げた。FVIII−Fc−ヒンジの有効性は、40IU/kgで60%の生存をもたらすBDD−FVIIIと匹敵すると概算される。全ての処置は、有意に改善された(Log−Rank試験による両側 p<0.05)生存曲線 対 輸送媒体コントロールをもたらした。
【0161】
本明細書に記載されている全ての文献および特許は出典明示により本明細書に包含させる。本発明の範囲および精神から逸脱することなく、本発明の記載されている方法の種々の修飾および変化が当業者に明らかである。
【0162】
本発明は特定の態様に関連して記載されているが、本発明がこのような特定の態様に過度に限定されるべきでないと理解すべきである。さらに、生化学または関連分野の当業者に明白である本発明を実施するための上記方法の種々の修飾は、特許請求の範囲内であると意図する。当業者は、わずかな日常の実験を使用して、本明細書に記載されている本発明の特定の態様の多数の均等を認識するか、または確かめることができる。このような均等は特許請求の範囲により包含させることを意図する。
【特許請求の範囲】
【請求項1】
モジュレーターのアミノ酸配列がB−ドメインに存在するか、またはモジュレーターのアミノ酸配列がB−ドメインの一部または全てのアミノ酸配列を置換している、第VIII因子タンパク質またはポリペプチドを含む第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項2】
モジュレーターが半減期モジュレーターである、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項3】
モジュレーターのアミノ酸配列がグリコシル化されている、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項4】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項5】
モジュレーターが、ヒトIgG、IgE、IgDもしくはIgMもしくはその変異体、またはマウスIgG、IgA、IgMもしくはその変異体由来の免疫グロブリンのFc領域である、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項6】
第VIII因子タンパク質またはポリペプチドが欠失されているB−ドメインの一部または全てを含む、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項7】
配列番号:1のアミノ酸20−764と同一である第1のアミノ酸配列、配列番号:1のアミノ酸1656−2351と同一である第2のアミノ酸配列およびモジュレーターアミノ酸配列を含む請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーであって、(1)該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合しているか、または(2)該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、該モジュレーターアミノ酸配列が、第2のアミノ酸配列に共有結合していない、第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項8】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項6に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項9】
モジュレーターが、ヒトIgG、IgE、IgDもしくはIgM、またはマウスIgG、IgA、IgM、またはその変異体由来の免疫グロブリンのFc領域である、請求項6に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項10】
第VIII因子融合タンパク質が、モジュレーターのアミノ酸配列がB−ドメインに存在するか、またはモジュレーターのアミノ酸配列がB−ドメインの一部または全てのアミノ酸配列を置換している第VIII因子タンパク質を含む、第VIII因子融合タンパク質をコードする核酸。
【請求項11】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項10に記載の核酸。
【請求項12】
モジュレーターが、ヒトIgG、IgE、IgDもしくはIgM、またはマウスIgG、IgA、IgM、またはその変異体由来の免疫グロブリンのFc領域である、請求項10に記載の核酸。
【請求項13】
第VIII因子タンパク質が欠失されているB−ドメインの一部または全てを有する、請求項10に記載の核酸。
【請求項14】
第VIII因子融合タンパク質が、配列番号:1のアミノ酸20−764と同一である第1のアミノ酸配列、配列番号:1のアミノ酸1656−2351と同一である第2のアミノ酸配列およびモジュレーターアミノ酸配列を含む請求項10に記載の核酸であって、該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合している、核酸。
【請求項15】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項14に記載の核酸。
【請求項16】
モジュレーターが、ヒトIgG、IgE、IgDもしくはIgM、またはマウスIgG、IgA、IgM、またはその変異体由来である免疫グロブリンのFc領域である、請求項15に記載の核酸。
【請求項17】
請求項10に記載の核酸を含むベクター。
【請求項18】
請求項10に記載の核酸を含む宿主細胞。
【請求項19】
(a)第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーをコードする発現ベクターで形質転換された宿主細胞を提供し、(b)該細胞を培養し、(c)第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを単離することを含む、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを生産するための方法。
【請求項20】
宿主細胞が哺乳動物宿主細胞であり、モジュレーターのアミノ酸配列がグリコシル化されている、請求項19に記載の方法。
【請求項21】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項19に記載の方法。
【請求項22】
第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーが、配列番号:1のアミノ酸20−764と同一である第1のアミノ酸配列、配列番号:1のアミノ酸1656−2351と同一である第2のアミノ酸配列およびモジュレーターアミノ酸配列を含む請求項19に記載の方法であって、(1)該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合しているか、または(2)該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、該モジュレーターアミノ酸配列が、第2のアミノ酸配列に共有結合していない、方法。
【請求項23】
請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーおよび薬学的に許容される担体を含む医薬組成物。
【請求項24】
治療有効量の請求項23に記載の医薬組成物を処置を必要とする患者に投与することを含む、遺伝的および後天的凝固欠乏症を処置する方法。
【請求項25】
遺伝的および後天的凝固欠乏症が血友病Aである、請求項24に記載の方法。
【請求項26】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項5に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項27】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項26に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項28】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項9に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項29】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項28に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項30】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項12に記載の核酸。
【請求項31】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項30に記載の核酸。
【請求項32】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項16に記載の核酸。
【請求項33】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項32に記載の核酸。
【請求項34】
請求項31に記載の核酸を含むベクター。
【請求項35】
請求項31に記載の核酸を含む宿主細胞。
【請求項36】
請求項33に記載の核酸を含むベクター。
【請求項37】
請求項33に記載の核酸を含む宿主細胞。
【請求項38】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項21に記載の方法。
【請求項39】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項38に記載の方法。
【請求項40】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項21に記載の方法。
【請求項41】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項22に記載の方法。
【請求項42】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項40に記載の方法。 モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項20に記載の方法。
【請求項43】
モジュレーターが半減期モジュレーターである、請求項3−8のいずれかに記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項44】
モジュレーターが半減期モジュレーターである、請求項10に記載の核酸。
【請求項45】
モジュレーターが半減期モジュレーターである、請求項19に記載の方法。
【請求項1】
モジュレーターのアミノ酸配列がB−ドメインに存在するか、またはモジュレーターのアミノ酸配列がB−ドメインの一部または全てのアミノ酸配列を置換している、第VIII因子タンパク質またはポリペプチドを含む第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項2】
モジュレーターが半減期モジュレーターである、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項3】
モジュレーターのアミノ酸配列がグリコシル化されている、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項4】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項5】
モジュレーターが、ヒトIgG、IgE、IgDもしくはIgMもしくはその変異体、またはマウスIgG、IgA、IgMもしくはその変異体由来の免疫グロブリンのFc領域である、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項6】
第VIII因子タンパク質またはポリペプチドが欠失されているB−ドメインの一部または全てを含む、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項7】
配列番号:1のアミノ酸20−764と同一である第1のアミノ酸配列、配列番号:1のアミノ酸1656−2351と同一である第2のアミノ酸配列およびモジュレーターアミノ酸配列を含む請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーであって、(1)該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合しているか、または(2)該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、該モジュレーターアミノ酸配列が、第2のアミノ酸配列に共有結合していない、第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項8】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項6に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項9】
モジュレーターが、ヒトIgG、IgE、IgDもしくはIgM、またはマウスIgG、IgA、IgM、またはその変異体由来の免疫グロブリンのFc領域である、請求項6に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項10】
第VIII因子融合タンパク質が、モジュレーターのアミノ酸配列がB−ドメインに存在するか、またはモジュレーターのアミノ酸配列がB−ドメインの一部または全てのアミノ酸配列を置換している第VIII因子タンパク質を含む、第VIII因子融合タンパク質をコードする核酸。
【請求項11】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項10に記載の核酸。
【請求項12】
モジュレーターが、ヒトIgG、IgE、IgDもしくはIgM、またはマウスIgG、IgA、IgM、またはその変異体由来の免疫グロブリンのFc領域である、請求項10に記載の核酸。
【請求項13】
第VIII因子タンパク質が欠失されているB−ドメインの一部または全てを有する、請求項10に記載の核酸。
【請求項14】
第VIII因子融合タンパク質が、配列番号:1のアミノ酸20−764と同一である第1のアミノ酸配列、配列番号:1のアミノ酸1656−2351と同一である第2のアミノ酸配列およびモジュレーターアミノ酸配列を含む請求項10に記載の核酸であって、該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合している、核酸。
【請求項15】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項14に記載の核酸。
【請求項16】
モジュレーターが、ヒトIgG、IgE、IgDもしくはIgM、またはマウスIgG、IgA、IgM、またはその変異体由来である免疫グロブリンのFc領域である、請求項15に記載の核酸。
【請求項17】
請求項10に記載の核酸を含むベクター。
【請求項18】
請求項10に記載の核酸を含む宿主細胞。
【請求項19】
(a)第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーをコードする発現ベクターで形質転換された宿主細胞を提供し、(b)該細胞を培養し、(c)第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを単離することを含む、請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーを生産するための方法。
【請求項20】
宿主細胞が哺乳動物宿主細胞であり、モジュレーターのアミノ酸配列がグリコシル化されている、請求項19に記載の方法。
【請求項21】
モジュレーターが、免疫グロブリンのFc領域もしくはその変異体、またはFcRn結合ペプチドもしくはその変異体である、請求項19に記載の方法。
【請求項22】
第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーが、配列番号:1のアミノ酸20−764と同一である第1のアミノ酸配列、配列番号:1のアミノ酸1656−2351と同一である第2のアミノ酸配列およびモジュレーターアミノ酸配列を含む請求項19に記載の方法であって、(1)該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、そのカルボキシル末端で第2のアミノ酸のアミノ末端に共有結合しているか、または(2)該モジュレーターアミノ酸配列が、そのアミノ末端で第1のアミノ酸配列のカルボキシル末端に共有結合しており、該モジュレーターアミノ酸配列が、第2のアミノ酸配列に共有結合していない、方法。
【請求項23】
請求項1に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマーおよび薬学的に許容される担体を含む医薬組成物。
【請求項24】
治療有効量の請求項23に記載の医薬組成物を処置を必要とする患者に投与することを含む、遺伝的および後天的凝固欠乏症を処置する方法。
【請求項25】
遺伝的および後天的凝固欠乏症が血友病Aである、請求項24に記載の方法。
【請求項26】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項5に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項27】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項26に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項28】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項9に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項29】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項28に記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項30】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項12に記載の核酸。
【請求項31】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項30に記載の核酸。
【請求項32】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項16に記載の核酸。
【請求項33】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項32に記載の核酸。
【請求項34】
請求項31に記載の核酸を含むベクター。
【請求項35】
請求項31に記載の核酸を含む宿主細胞。
【請求項36】
請求項33に記載の核酸を含むベクター。
【請求項37】
請求項33に記載の核酸を含む宿主細胞。
【請求項38】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項21に記載の方法。
【請求項39】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項38に記載の方法。
【請求項40】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項21に記載の方法。
【請求項41】
モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項22に記載の方法。
【請求項42】
モジュレーターが、配列番号:9、11、13、15、29、17、19、21、23、30からなる群から選択されるアミノ酸配列、または配列番号:9、11、13、15、29、17、19、21、23、30のいずれかと少なくとも95%のアミノ酸同一性を有する配列を有する、請求項40に記載の方法。 モジュレーターが、ヒトもしくはマウスIgGのFc領域、非機能的ヒンジを有するヒトもしくはマウスIgGのFc領域の変異体(ヒンジ領域中のシステイン残基の置換または欠失による)、またはヒトもしくはマウスIgGのFc領域の非ヒンジ部分である、請求項20に記載の方法。
【請求項43】
モジュレーターが半減期モジュレーターである、請求項3−8のいずれかに記載の第VIII因子融合タンパク質または第VIII因子融合ヘテロダイマー。
【請求項44】
モジュレーターが半減期モジュレーターである、請求項10に記載の核酸。
【請求項45】
モジュレーターが半減期モジュレーターである、請求項19に記載の方法。
【図1】

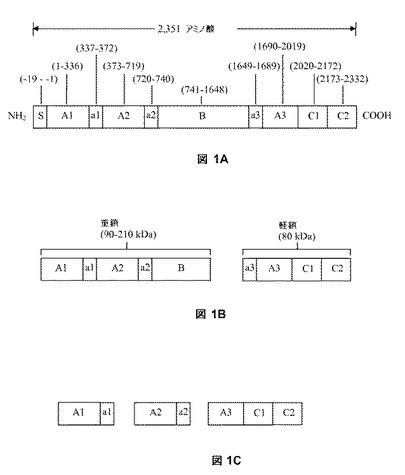
【図2】
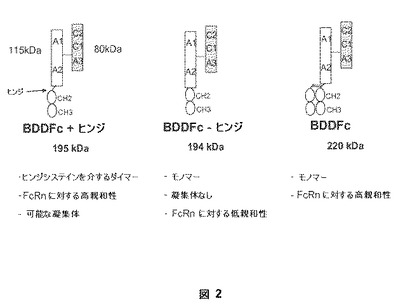
【図3】
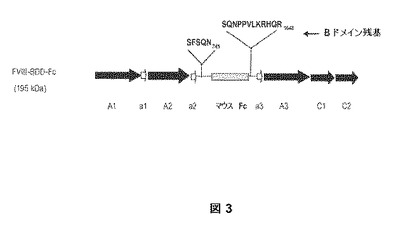
【図4】
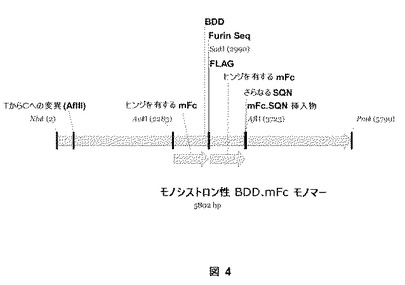
【図5】
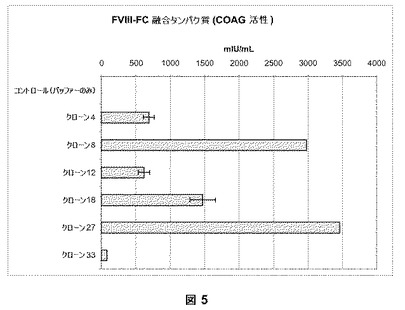
【図6】
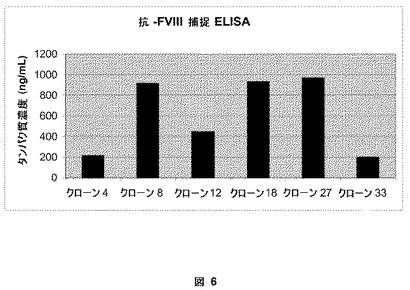
【図7】
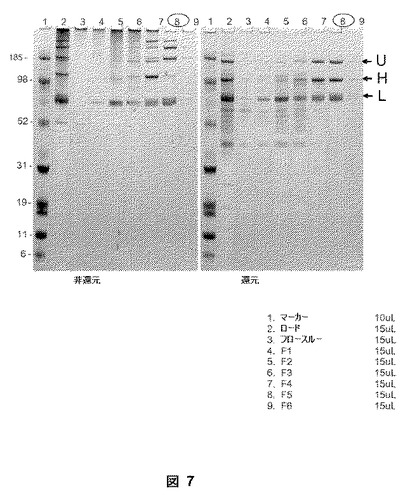
【図8】
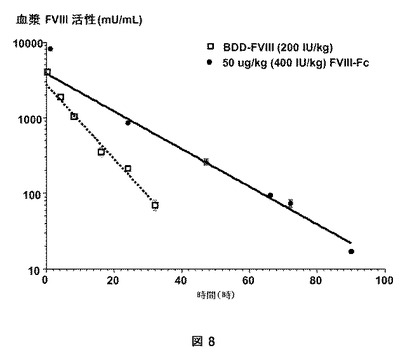
【図9】
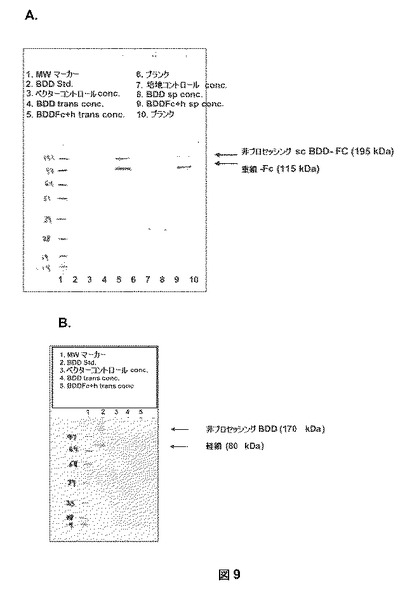
【図10】
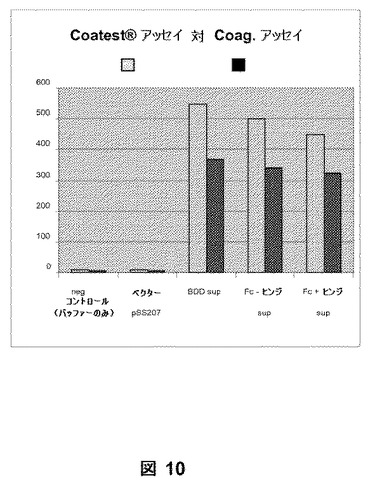
【図11】
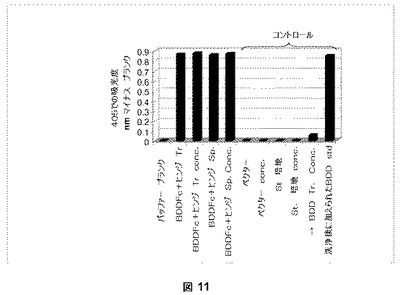
【図12】
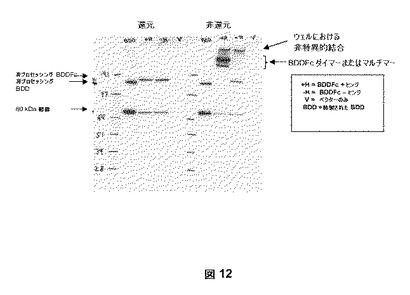
【図13】
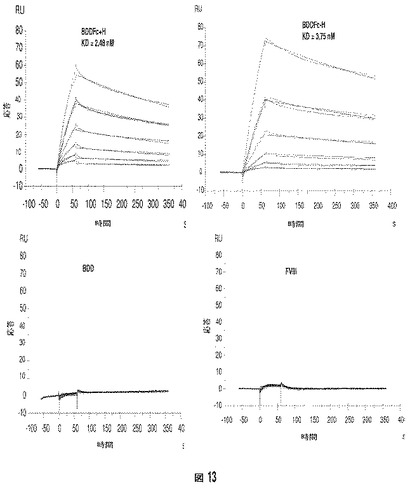
【図14】
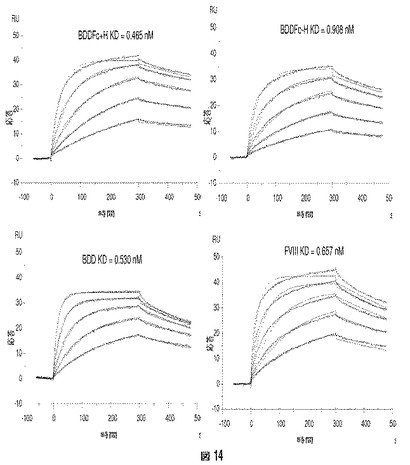
【図15】
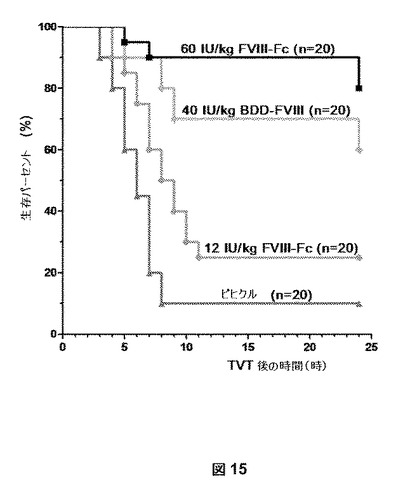
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公表番号】特表2012−522490(P2012−522490A)
【公表日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願番号】特願2012−502213(P2012−502213)
【出願日】平成22年3月24日(2010.3.24)
【国際出願番号】PCT/US2010/028529
【国際公開番号】WO2010/111414
【国際公開日】平成22年9月30日(2010.9.30)
【出願人】(503106111)バイエル・ヘルスケア・エルエルシー (154)
【Fターム(参考)】
【公表日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願日】平成22年3月24日(2010.3.24)
【国際出願番号】PCT/US2010/028529
【国際公開番号】WO2010/111414
【国際公開日】平成22年9月30日(2010.9.30)
【出願人】(503106111)バイエル・ヘルスケア・エルエルシー (154)
【Fターム(参考)】
[ Back to top ]