
第VIII族および第VIB族金属を含み、酢酸およびC1−C4ジアルキルスクシナートを有する、水素化処理において使用可能な触媒および調製
本発明は、水素化処理方法において用いられ得る触媒であって、アルミナをベースとする無定形担体と、リンと、C1−C4ジアルキルスクシナートと、酢酸と、第VIII族の少なくとも1種の元素および第VIB族の少なくとも1種の元素を含み、好ましくは、コバルトおよびモリブデンからなる水素化脱水素基とを含み、触媒のラマンスペクトルは、ケギンヘテロポリアニオン(974および/または990cm−1)、およびC1−C4ジアルキルスクシナート、および酢酸(896cm−1)の特徴である最も強いバンドを含む、触媒に関する。好ましくは、用いられるジアルキルスクシナートは、ジメチルスクシナートであり、その主要バンドは853cm−1である。本発明はまた、前記触媒を調製する方法であって、触媒前駆体は、第VIB族および第VIII族の元素、特に、モリブデン−コバルトの対と、リンとを含み、これらは含浸によって導入され、次いで、180℃未満温度で乾燥させられ、ジアルキルスクシナート、酢酸およびリン化合物(このものがすでに事前に完全には導入されなかった場合)を含浸させられ、次いで、成熟の後、180℃より低い温度に乾燥させられ、その後、場合によっては硫化される、方法に関する。本発明はさらに、任意の水素化処理方法における前記触媒の使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、触媒、その調製方法、および水素化処理分野におけるその使用に関する。
【0002】
通常、炭化水素留分水素化処理触媒の目的は、炭化水素留分に含まれる硫黄または窒素の化合物を除去し、石油生成物が、所与の適用(車の燃料、ガソリンまたはディーゼル燃料、暖房油、ジェット燃料)について要求される規格(例えば、硫黄含有量、芳香族化合物含有量等)に適合するようにさせることにある。目的はまた、この供給原料を種々の転化方法、例えば、改質、真空蒸留物水素化分解、接触分解、常圧または真空の残渣の水素化転化方法等に付してその物理化学的特性を改変する前にこれを予備処理して、それが含む不純物を除去することであり得る。水素化処理触媒の組成および使用は、書物Catalysis Science and Technology, Vol.11(1996), Springer-VerlagからのB. S. Clausen, H. T. Topsoe および F. E. Massothによる論文において特によく記載されている。硫化後、担体上に複数の表面種が存在し、これは、全てが、所望の反応について良好な性能を示すわけではない。これらの種は、特に、1984年のCatalysis Review Science and Engineering 第26号, pp.395-420において出版されたTopsoeらによる出版物においてよく記載されている。
【0003】
欧州共同体における自動車汚染基準の厳格化により(Official Journal of the European Union, L76, 22 March 2003, Directive 2003/70/CE, pp.L76/10-L76/19)、精製業者は、ディーゼル燃料およびガソリン中の硫黄含有量を相当低減させるように強要されている(2005年1月1日において50重量ppmであるのに対して2009年1月1日において硫黄最大10重量ppm)。加えて、精製業者は、一方では、原油がますます重質であり、したがって、ますます多くの不純物を含有するため、他方で、精製所における転化方法の増加のため、水素化処理方法に対してますます難処理性である供給原料を使用するように強要される。実際に、常圧蒸留に直接的に由来する留分より水素化処理し難い留分が生じている。その例は、その高い芳香族化合物含有量に関連して、接触分解から得られたディーゼル留分(LCO(Light Cycle Oil)とも呼ばれる)である。これらの留分は、常圧蒸留から得られたディーゼル留分と共処理される;それらは、芳香族化合物含有量を低減させて規格に従う密度およびセタン価を得るために、従来の触媒に対して高度に改善された水素化脱硫および水素化の機能を有する触媒を必要とする。
【0004】
さらに、接触分解または水素化分解等の転化方法には、酸基を有する触媒が使用される。酸基は、それらを、窒素不純物、特に塩基性窒素化合物の存在に対して特に感受性にする。したがって、これらの供給原料を予備処理してこれらの化合物を除去するための触媒を用いることが必要である。これらの水素化処理触媒はまた、第一の水素化脱窒段階がC−N結合に隣接する芳香環の水素化の段階として知られている限りにおいて、改善された水素化基を要求する。
【0005】
したがって、改善された性能を有する新しい触媒を得ることを可能にする水素化処理触媒を調製する手段を見出すことは興味深い。
【背景技術】
【0006】
有機化合物を水素化処理触媒に添加して、それらの活性を改善することは、今や、当業者に周知である。多くの特許は、種々の範囲の有機化合物、例えば、エーテル化されてもよい、モノ−、ジー、またはポリアルコールの使用を保護する(特許文献1〜5)。C2−C14モノエステルにより改変された触媒は、特許出願(特許文献6〜7)において記載されているが、しかしながら、これらの改変は、燃料の硫黄含有量に対する規格を満たすように触媒の性能を十分に増強することを常に可能にするというわけではなく、このことは、精製業者をますます制限することとなった。
【0007】
これを克服するために、TOTAL Companyによって出願された特許文献8には、第VIB族および第VIII族金属、担体としての耐火性酸化物、および有機化合物を含む触媒であって、該有機化合物は、式R1−O−CO−R2−CO−O−R1またはR1−CO−O−R2−O−CO−R1(式中、各R1は、独立して、C1−C18アルキル基、C2−C18アルケニル基、C6−C18アリール基、C3−C8シクロアルキル基、C7−C20アルキルアリールまたはアリールアルキル基を示すか、または、2つの基R1は、一緒に、C2−C18の二価基を形成し、R2は、C1−C18アルキレン基、C6−C18アリーレン基、C3−C7シクロアルキレン基を示すか、またはその組合せであり、R1およびR2によって示される有炭化水素基の炭素鎖は、N、SおよびOの中から選択される1個以上のヘテロ原子を含むか有し得、各基R1およびR2は、式−C(=O)O−R1または−O−C(=O)−R1(式中、R1は、前述の意味を有する)の1個以上の置換基を有し得る)の少なくとも2個のカルボン酸エステル基を含む、触媒を用いることが提案される。好ましい態様では、C1−C4ジアルキルスクシナート、特に、例示されるジメチルスクシナートが用いられる。これらの化合物は、溶媒(かなりの溶媒のリストが言及される)またはカルボン酸の存在下に導入され得る。特に言及される約30の酸の中で、酢酸があるが、しかしながら、これは、10の好ましい酸の中で言及されない。クエン酸が好ましいことはすでに留意され得る。
【0008】
特許文献8において記載された触媒調製方法は、数日まで、例えば49〜115日続き得る成熟および熱処理段階を含み、このことは、これらの触媒の製造を大きく制限するだろうし、したがって、改良を要求するだろう。
【0009】
従来技術の他の特許により、水素化処理触媒上の有機酸またはアルコールの組み合わされた使用と関連する活性増大が記載される。それ故に、株式会社ジャパンエナジーによって出願された特許文献9には、
− 発明の第一の好ましい態様に従って、触媒前駆体、周期律表の第VI族および第VIII族からの1種以上の金属および有機酸を含有する溶液を調製する段階;発明の第二の好ましい態様に従って、この溶液はリン前駆体も含む、
− 200〜400℃で熱処理を行う段階、
− 金属のモル当たり0.1〜2の比で有機酸またはアルコールによって上記の得られた触媒の含浸を行う段階
からなる解決方法が提供される。
【0010】
発明の好ましい態様の一つは、200℃未満の温度で乾燥させる段階を含むのに対して、その発明の第二の好ましい態様は、400℃以上の温度での最終熱処理を含む。
【0011】
これらの触媒は、精製業者に利用可能な水素がますます欠乏する供給原料の面で新たな環境基準を満たす十分な活性を有しないことが観察された。
【0012】
同様に、特許文献10は、第VIII族および第VIB族の金属酸化物を含む、再生触媒上に存在するCoMoO4タイプの結晶化相の比率を減少させることを概要的に可能にする活性化法であって、再生触媒を、酸および有機添加物と接触させる工程を含む、方法を特許請求している。したがって、クエン酸(CA)−ポリエチレングリコール(PEG)の組合せが、多くの実施例において再生触媒上で用いられた。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】国際公開第96/41848号パンフレット
【特許文献2】国際公開第01/76741号パンフレット
【特許文献3】米国特許第4012340号明細書
【特許文献4】米国特許第3954673号明細書
【特許文献5】欧州特許出願公開第601722号明細書
【特許文献6】欧州特許出願公開第466568号明細書
【特許文献7】欧州特許出願公開第1046424号明細書
【特許文献8】国際公開第06/077326号パンフレット
【特許文献9】特開平07−136523号公報
【特許文献10】国際公開第05/035691号パンフレット
【発明の概要】
【課題を解決するための手段】
【0014】
(発明の概要)
本発明は、触媒およびその調製方法に関し、触媒は、水素化処理に使用可能であり、従来技術の触媒に対して触媒性能(特に触媒活性)を改良することを可能にする。実際に、C1−C4ジアルキルスクシナート(特にジメチル)および酢酸から構成される対を乾燥触媒前駆体上で使用することにより、驚くべきことに、対の化合物のそれぞれ一つと比較して顕著に改良された触媒活性が導かれることが示された。
【0015】
より正確には、本発明は、アルミナベースの無定形担体と、リンと、少なくとも1種のC1−C4ジアルキルスクシナートと、酢酸と、少なくとも1種の第VIII族元素および少なくとも1種の第VIB族元素を含む水素化脱水素基(function)とを含む触媒であって、そのラマンスペクトルは、少なくとも1種のケギンヘテロポリアニオンの特徴である990および/または974cm−1のバンド、前記スクシナートの特徴であるバンドおよび酢酸の特徴である896cm−1の主要バンドを含む、触媒に関する。水素化脱水素基は、好ましくは、コバルトおよびモリブデンからなる。それは、水素化脱水素基がコバルトおよびモリブデンからなる以外は、少なくとも1種の第VIII族元素および少なくとも1種の第VIB族元素も含み得る。
【0016】
得られた触媒は、以下に分類する特徴的なラマンスペクトルを有する;
1)ケギンPXY11O40x−および/またはPY12O40x−タイプのヘテロポリアニオン(単数または複数)の特徴であるバンド(式中Yは第VIB族金属でありXは第VIII族金属である)。
【0017】
Griboval, Blanchard, Payen, Fournier, DuboisのCatalysis Today 45(1998) 277 Fig. 3 e)によると、PCoMo11O40x−構造の主要バンドは、乾燥触媒上において、232、366、943、974cm−1にあり、M. T. Popeの“Heteropoly and Isopoly oxometalates”, Springer Verlag, p 8によると、これらのバンドは、元素XまたはYの性質の特徴ではなく、HPAの構造の特徴である。このタイプの空隙ケギンHPAの特徴である最も強いバンドは974cm−1にある。
【0018】
Griboval、 Blanchard、 Gengembre、 Payen、 Fournier、 Dubois、 BernardのJournal of Catalysis 188(1999) 102, Fig. 1 a)によると、PMo12O40x−の主要バンドは、例えばカウンターイオンとしてのコバルトとのHPAの塊状態において、251、603、902、970、990cm−1にある。このケギンHPAの特徴である最も強いバンドは、990cm−1にある。M. T. Popeの“Heteropoly and Isopoly oxometalates”, Springer Verlag, p 8も、我々にこれらのバンドは元素XまたはYの性質の特徴でなく、完全な、空隙性または置換されたケギンHPAの構造の特徴であることをを教示する。
【0019】
2)用いられるジアルキルスクシナート(単数または複数)の特徴であるバンド。ジメチルスクシナートのラマンスペクトルは、この分子の一意のフィンガープリントである。300−1800cm−1のスペクトルゾーンにおいて、このスペクトルは、以下の一連のバンド(最も強いバンドのみが記録される,cm−1)によって特徴付けられる:391、853(最も強いバンド)、924、964、1739cm−1。ジエチルスクシナートのスペクトルは、考慮されるスペクトルゾーンにおいて、以下の主要バンドを含む;861(最も強いバンド)、1101、1117cm−1。同様に、ジブチルスクシナートについて:843、1123、1303、1439、1463cm−1およびジイソプロピルスクシナートについて:833、876、1149、1185、1469(最も強いバンド)、1733cm−1。
【0020】
3)酢酸の特徴であるバンド。以下の主要なバンドを有する:448、623、896cm−1。最も強いバンドは896cm−1である。
【0021】
バンドの正確な位置、それらの形状およびそれらの相対強度は、スペクトル記録条件に応じて所定の範囲で変動し得るが、この分子の特徴はそのままである。有機化合物のラマンスペクトルは、ラマンスペクトルデータベース(例えば、Spectral Database for the organic compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi参照)において、または、生成物の供給者(例えばwww.sigmaaldrich.comを参照)によってさらによく実証されている。
【0022】
ラマンスペクトルは、イオン化アルゴンレーザ(514nm)を備えた分散ラマンタイプの分光計により得られる。レーザビームは、50倍長作動距離対物レンズを備えた顕微鏡によってサンプルに焦点を合わされる。サンプルのレベルにおけるレーザの出力は、1mW程度である。サンプルによって放出されるラマンシグナルは、同じ対物レンズによって集められ、1800rpmネットワークによって分散させられ、次いで、CCD検出器によって集められる。得られたスペクトルの分解能は、0.5cm−1程度である。記録されたスペクトルゾーンは300〜1800cm−1である。捕捉時間は、記録される各ラマンスペクトルのために120秒に設定される。
【0023】
好ましくは、用いられるジアルキルスクシナートはジメチルスクシナートであり、触媒は、そのスペクトルにおいて、ケギンヘテロポリアニオン(単数または複数)の特徴である990および/または974cm−1、ジメチルスクシナートの特徴である853cm−1、および酢酸の特徴である896cm−1に位置する主要ラマンバンドを有する。
【0024】
好ましくは、本発明の触媒は、アルミナまたはシリカ−アルミナからなる担体を含む。
【0025】
本発明による触媒はまた、ホウ素および/またはフッ素および/またはケイ素を含み得る。
【0026】
また、本発明による触媒の調製方法についても記載されている。本発明の調製方法は、180℃未満の温度で乾燥させられ、かつ、少なくともリンおよび水素化脱水素基、ならびに無定形担体を少なくとも含む触媒前駆体に、酢酸およびC1−C4ジアルキルスクシナートの組み合わせを含む含浸溶液を含浸させる少なくとも1回の含浸段階と、その後の前記含浸させられた触媒前駆体の成熟段階と、その後の180℃未満の温度での乾燥段階とを含み、その後に焼成段階(空気中での熱処理)は行われない。得られた触媒は、好ましくは、硫化段階に付される。
【0027】
水素化脱水素基は、少なくとも1種の第VIII族元素および少なくとも1種の第VIB族元素を含む。好ましくは、水素化脱水素基は、コバルトおよびモリブデンからなる。
【0028】
数時間を超えない単位段階を用いる簡単かつ迅速な調製方法により、従って、従来技術の方法と比較して、工業規模で、より高い生産性を得ることが可能となる。
【0029】
より正確には、本発明による水素化処理触媒の調製方法は、以下に詳述されることになる、連続する段階を含む:
a)アルミナをベースとする無定形担体に、水素化脱水素基の元素およびリンを含む少なくとも1種の溶液を含浸させる、少なくとも1回の含浸段階(得られた生成物は「触媒前駆体」と称される);
b)180℃未満の温度での乾燥段階であって、その後に焼成は行われない、段階(得られた生成物は「乾燥触媒前駆体」と称される);
c)少なくとも1種のC1−C4ジアルキルスクシナートと、酢酸と、少なくとも1種のリン化合物(段階a)において、リン化合物が完全には導入されていない場合)とを含む含浸溶液による、少なくとも1回の含浸段階(得られた生成物は「含浸乾燥触媒前駆体」と称される);
d)成熟段階;
e)180℃未満の温度での乾燥段階であって、その後に焼成段階は行われない、段階(得られた生成物は「触媒」と称される)。
【0030】
好ましくは、段階e)の終わりに得られた生成物は、硫化段階f)に付される。
【0031】
以下に記載されるように、本発明による方法は好ましくは、以下の様式を単独または組み合わせて用いて行われる:担体はアルミナまたはシリカ−アルミナからなり;水素化基の全てが段階a)において導入され;リンの全てが段階a)において導入され;ジアルキルスクシナートはジメチルスクシナートであり;段階c)は溶媒の非存在下に行われ;段階d)は17〜50℃の範囲の温度で行われ;段階e)は80〜160℃の範囲の温度で行われる。
【0032】
より好ましくは、本発明による方法は、以下の連続する段階を含む:
a)前記担体に、水素化脱水素基の元素の全てとリンの全てとを含む溶液を乾式含浸させる、少なくとも1回の段階、
b)75〜130℃の範囲の温度での乾燥段階であって、その後に焼成は行われない、段階、
c)ジメチルスクシナートおよび酢酸を含む含浸溶液による、少なくとも1回の乾式含浸段階、
d)17〜50℃での成熟段階、
e)80〜160℃の範囲の温度で行われる乾燥段階(好ましくは窒素下)であって、その後に焼成段階は行われない、段階。
【0033】
水素化脱水素基と、アルミナをベースとする無定形担体とを含む触媒前駆体、ならびに、その調製様式は以下に記載される。
【発明を実施するための形態】
【0034】
(詳細な説明)
本発明による方法の段階a)の終わりに得られる前記触媒前駆体は、殆どの場合当業者に公知のあらゆる方法によって調製され得る。
【0035】
前記触媒前駆体は、水素化脱水素基を含み、前記触媒前駆体は、ドーパントとしてのリンおよび/またはホウ素および/またはフッ素、ならびに無定形担体を含む。水素化脱水素基は、少なくとも1種の第VIB族元素と少なくとも1種の第VIII族元素とを含む。好ましくは、水素化脱水素基は、コバルトおよびモリブデンからなる。
【0036】
前記触媒前駆体の無定形担体は、アルミナをベースとし、すなわち、それは、50%超のアルミナを含み、それは、一般的には、アルミナのみを含むか、または、以下に規定されるようなシリカ−アルミナを含み、場合によっては、含浸の外側で導入された(例えば、担体の調製、混練、解膠等またはその成形の間に導入された)、金属(単数または複数)および/またはドーパント(単数または複数)を含む。担体は、成形(例えば、押出)および一般に300〜600℃での焼成の後に得られる。
【0037】
好ましくは、担体は、アルミナからなり、好ましくは押し出されたアルミナからなる。アルミナは、好ましくはガンマアルミナであり、前記無定形担体は、好ましくはガンマアルミナからなる。
【0038】
別の好ましい場合において、それは、少なくとも50%のアルミナを含むシリカ−アルミナである。担体中のシリカの割合は、最大50重量%、ほとんどの場合45重量%以下、好ましくは40重量%以下である。
【0039】
ケイ素源は、当業者に周知である。ケイ素源の例としては、ケイ酸、粉末シリカ、コロイド形態のシリカ(シリカゾル)、オルトケイ酸テトラエチルSi(OEt)4が挙げられる。
【0040】
前記触媒前駆体の水素化脱水素基は、少なくとも1種の第VIB族元素および少なくとも1種の第VIII族元素によって提供される。コバルトおよびモリブデンから構成される対が好ましい。水素化脱水素元素の全割合は、有利には、触媒の全重量に対して酸化物6重量%超である。好ましい第VIB族元素は、モリブデンおよびタングステンであり、一般的にモリブデンである。好ましい第VIII族元素は、非貴金属元素であり、特にコバルトおよびニッケルである。
【0041】
有利には、水素化基は、以下の元素の組合せから構成される群より選択される:コバルト−モリブデン、ニッケル−モリブデン、ニッケル−コバルト−モリブデン、またはニッケル−モリブデン−タングステン。
【0042】
高い水素化脱硫、もしくは水素化脱窒、および芳香族の水素化の活性が望まれる場合、水素化脱水素基は、有利にはニッケルとモリブデンの組合せによって果たされる;モリブデンの存在下での、ニッケルとタングステンの組合せも有利であり得る。真空蒸留物またはより重質なタイプの供給原料の場合、コバルト-ニッケル−モリブデンのタイプの組合せが、有利に用いられ得る。
【0043】
使用され得るモリブデン前駆体もまた、当業者に周知である。例えば、モリブデン源の中から、酸化物および水酸化物、モリブデン酸およびこれらの塩、特に、アンモニウム塩、例えば、モリブデン酸アンモニウム、七モリブデン酸アンモニウム、リンモリブデン酸(H3PMo12O40)、およびこれらの塩、ならびに、場合によるケイモリブデン酸(H4SiMo12O40)およびその塩が使用可能である。モリブデン源はまたあらゆるヘテロポリ化合物であり得、例えば、ケギン型、空隙ケギン型、置換ケギン型、ドーソン(Dawson)型、アンダーソン(Anderson)型、ストランベルグ(Strandberg)型である。三酸化モリブデン、ならびに、ストランベルグ型、ケギン型、空隙ケギン型、または置換ケギン型のヘテロポリ化合物(ヘテロポリアニオン)が、好ましくは使用される。
【0044】
使用され得るタングステン前駆体もまた、当業者に周知である。例えば、使用され得るタングステン源は、酸化物および水酸化物、タングステン酸およびそれらの塩、特にアンモニウム塩、例えば、タングステン酸アンモニウム、メタタングステン酸アンモニウム、リンタングステン酸、およびそれらの塩、ならびに場合によるケイタングステン酸(H4SiW12O40)およびその塩である。タングステン源はまた、あらゆるヘテロポリ化合物であり得、例えば、ケギン型、空隙ケギン型、置換ケギン型、ドーソン型のヘテロポリ化合物である。好ましくは、アンモニウム酸化物および塩、例えば、メタタングステン酸アンモニウム、もしくは、ケギン型、空隙ケギン型、または置換ケギン型のヘテロポリアニオンが用いられる。
【0045】
第VIB族元素(単数または複数)の前駆体(単数または複数)の割合は、触媒前駆体の全質量に対して有利には第VIB族酸化物5〜40重量%、好ましくは8〜35重量%、より好ましくは10〜30重量%である。
【0046】
使用され得る第VIII族元素(単数または複数)の前駆体は、有利には、酸化物、水酸化物、ヒドロキシ炭酸塩、炭酸塩および硝酸塩の中から選択され、例えば、ヒドロキシ炭酸ニッケル、炭酸コバルト、または水酸化コバルトが好ましくは使用される。
【0047】
第VIII族元素(単数または複数)の前駆体(単数または複数)の割合は、触媒前駆体の全質量に対して第VIII族酸化物有利には1〜10重量%、好ましくは1.5〜9重量%、より好ましくは2〜8重量%である。
【0048】
前記触媒前駆体の水素化脱水素基は、調製の種々のレベルにおいて、かつ種々の方法で、触媒に導入され得る。前記水素化脱水素基は、常に、成形された担体の含浸によって、少なくとも部分的に、好ましくは完全に導入される。それはまた、前記無定形担体を成形する際に、部分的に導入され得る。
【0049】
水素化脱水素基が、前記無定形担体の成形の際に、部分的に導入される場合、それは、マトリクスとして選択されたアルミナゲルとの混練の際にのみ、部分的に導入され得(例えば最大10%の第VIB族元素が、例えば混練を通じて導入される)、その後、水素化元素(単数または複数)の残りは、後に導入される。好ましくは、水素化脱水素基が混練の際に部分的に導入される場合、この段階中に導入される第VIB族元素(単数または複数)の割合は、最終触媒上に導入される第VIB族元素(単数または複数)の全量の5%未満である。いかなる導入様式が用いられるにせよ、好ましくは、少なくとも1種の第VIB族元素(またはそれらの全て)が、少なくとも1種の第VIII族元素(またはそれらの全て)と同時に導入される。元素の導入のためのこれらの方法および割合は、特に、水素化脱水素基がCoMoからなる場合に用いられる。
【0050】
前記無定形担体の成形後に、水素化脱水素基が、少なくとも部分的に、好ましくは完全に導入される場合、無定形担体上への前記水素化脱水素基の導入は、有利には、成形されかつ焼成された担体上での1回以上の過剰溶液含浸によって、あるいは、好ましくは、1回以上の乾式含浸によって、より好ましくは、前記成形されかつ焼成された担体の乾式含浸によって、金属前駆体塩を含む溶液を用いて行われ得る。更により好ましくは、水素化脱水素基は、前記無定形担体の成形後、金属前駆体塩を含む含浸溶液を用いる前記担体の乾式含浸によって完全に導入される。前記水素化脱水素基の導入はまた、有利には、活性相の前駆体(単数または複数)の溶液よる、成形されかつ焼成された担体の1回以上の含浸によって行われ得る。元素が、対応する前駆体塩の数回の含浸において導入される場合、中間の触媒乾燥段階が一般的には行われ、その際の温度は、50〜180℃、好ましくは60〜150℃、より好ましくは75〜130℃である。
【0051】
リンも触媒に導入される。別の触媒ドーパントも導入され得、これは、ホウ素、およびフッ素の中から単独でまたは混合物として選択される。ドーパントは、それ自体は触媒的特徴を有しない添加元素であるが、それは、金属(単数または複数)の触媒活性を高める。
【0052】
前記ドーパントは、有利には、単独で、あるいは、水素化脱水素基の少なくとも1種の元素との混合物として、導入され得る。
【0053】
それはまた、担体の合成と同じ時期の早期に導入され得る。
【0054】
それはまた、選択されたマトリクスの解膠の直前または直後に導入され得る。選択されたマトリクスは、例えばかつ好ましくは、アルミナ前駆体またはオキシ水酸化アルミニウム(ベーマイト)である。
【0055】
前記ドーパントはまた、有利には、水素化脱水素基の前駆体(単数または複数)との混合物として、成形された無定形担体(好ましくは押出物形態のアルミナまたはシリカ−アルミナ)上に完全にまたは部分的に、金属前駆体塩およびドーパント(単数または複数)の前駆体(単数または複数)を含む溶液を用いる前記無定形担体の乾式含浸により導入される。
【0056】
ホウ素源は、ホウ酸、好ましくは、オルトホウ酸H3BO3、二ホウ酸アンモニウム、五ホウ酸アンモニウム、酸化ホウ素、ホウ酸エステルであり得る。ホウ素は、例えば、水/アルコール混合物中または水/エタノールアミン混合物中のホウ酸の溶液によって、導入され得る。
【0057】
好ましいリン源は、オルトリン酸H3PO4であるが、その塩及びエステル、例えばリン酸アンモニウムもまた適している。リンはまた、ケギン型、空隙ケギン型、置換ケギン型、またはストランドベルグ型のヘテロポリアニオンの形態で、第VIB族元素と同時に導入され得る。
【0058】
使用され得るフッ素源は、当業者に周知である。例えば、フッ化物アニオンが、フッ化水素酸またはその塩の形態で導入され得る。これらの塩は、アルカリ金属、アンモニウム、または有機化合物を用いて形成される。有機化合物の場合、塩は、有利には、有機化合物とフッ化水素酸との間の反応によって、反応混合物中に形成される。フッ素は、例えば、フッ化水素酸、フッ化アンモニウム、または二フッ化アンモニウムの水溶液の含浸によって導入され得る。
【0059】
ドーパントは、有利には、以下の、触媒に対する前記ドーパントの酸化物の割合で触媒前駆体に導入される:
− 前記ドーパントがホウ素である場合、0〜40%、好ましくは0〜30%、より好ましくは0〜20%、一層より好ましくは0〜15%、最も好ましくは0〜10%であり;ホウ素が存在する場合、最小限の量は、好ましく0.1重量%であり、
− 前記ドーパントがリンである場合、0.1〜20%、好ましくは0.1〜15%、一層より好ましくは0.1〜10%であり、
− 前記ドーパントがフッ素である場合、0〜20%、好ましくは0〜15%、一層より好ましくは0〜10%であり;フッ素が存在する場合、最小限の量は、好ましくは0.1重量%である。
【0060】
リンは、常に存在する。リンは、段階a)における触媒前駆体および場合による段階c)における乾燥触媒前駆体上での含浸によって、少なくとも部分的に(好ましくは完全に)導入される。同様のことが、好ましくは、他のドーパントについても適用される。しかし、上に記載のように、ドーパントは、担体の調製(成形を含む)時に部分的に、または完全に(リンを除く)、導入され得る。
【0061】
前記水素化脱水素基および場合によるドーパントが、成形された焼成担体の中または上に導入された後、有利には、乾燥段階b)が行われ、ここで、金属(単数または複数)酸化物(単数または複数)の前駆体として使用される金属塩の溶媒(溶媒は、一般的に水である)が、50〜180℃、好ましくは60〜150℃または65〜145℃、より好ましくは70〜140℃または75〜130℃の温度で除去される。このようにして得られた「乾燥触媒前駆体」を乾燥させる段階の後に、200℃超の温度での空気中の焼成の段階は決して行われない。有利には、それは、これらの温度範囲において、最高150℃の温度で行われ、その後に180℃超の温度での焼成は行われない。
【0062】
好ましくは、本発明による方法の段階a)において、前記「触媒前駆体」は、水素化脱水素基の前駆体(単数または複数)とリンとを含む溶液を、成形されかつ焼成された、アルミナをベースとする無定形担体上に乾式含浸させることによって得られる。この後に、前記「触媒前駆体」は、180℃未満、好ましくは50〜180℃、より好ましくは60〜150℃、最も好ましくは75〜130℃での温度で乾燥させられる。
【0063】
「乾燥触媒前駆体」は、段階b)の終わりにこのようにして得られる。
【0064】
本発明による方法の段階a)において、ホウ素およびフッ素の中から選択される少なくとも1種のドーパントを単独でまたは混合物として含む含浸溶液を調製することが可能である。
【0065】
より好ましくは、本発明による方法の段階a)における「触媒前駆体」は、リン前駆体の存在下に、水素化脱水素基の各元素の少なくとも1種の前駆体を含む含浸溶液を用いて調製される。無定形担体は、アルミナまたはシリカ−アルミナからなる。
【0066】
本発明の方法の段階c)によると、前記乾燥触媒前駆体は、少なくとも1種のC1−C4ジアルキルスクシナート(特に、ジメチルスクシナート)と酢酸とを含む含浸溶液によって含浸させられる。
【0067】
前記化合物は、有利には、本発明による方法の段階c)の含浸溶液中に、以下に相当する割合で、導入される:
− 触媒前駆体に含浸させられた第VIB族元素(単数または複数)当たりのジアルキルスクシナート(例えば、ジメチルスクシナート)のモル比は、0.15〜2モル/モル、好ましくは0.3〜1.8モル/モル、より好ましくは0.5〜1.5モル/モル、最も好ましくは0.8〜1.2モル/モルであり、
− 触媒前駆体に含浸させられた第VIB族元素(単数または複数)当たりの酢酸のモル比は、0.1〜5モル/モル、好ましくは0.5〜4モル/モル、より好ましくは1.3〜3モル/モル、最も好ましくは1.5〜2.5モル/モルである。このことは、水素化脱水素基がCoMoからなる場合に特に当てはまる。
【0068】
本発明の方法の段階c)によると、ジアルキルスクシナートと酢酸の組合せは、少なくとも1回の含浸段階、好ましくは含浸溶液を前記乾燥触媒前駆体上に含浸させる単一の段階によって、乾燥触媒前駆体上に導入される。
【0069】
前記組合せは、スラリー含浸または過剰含浸、乾式含浸、あるいは当業者に公知のあらゆる他の手段のいずれかによる、1つ以上の段階において、有利には沈着させられ得る。
【0070】
本発明の調製方法の段階c)の好ましい実施形態によると、段階c)は、単一の乾式含浸段階である。
【0071】
本発明の方法の段階c)によると、段階c)の含浸溶液は、少なくとも、C1−C4ジアルキル(特に、ジメチル)スクシナートと酢酸の組合せを含む。
【0072】
本発明の方法の段階c)において使用される含浸溶液は、当業者に公知のあらゆる非プロトン性溶媒によって補足され得るが、特に、トルエン、キシレンが挙げられる。
【0073】
本発明による方法段階c)において使用される含浸溶液は、当業者に公知のあらゆる極性溶媒によって補足され得る。使用される前記極性溶媒は、有利には、メタノール、エタノール、水、フェノール、シクロヘキサノールから構成される群より、単独でまたは混合物として選択される。本発明による方法の段階c)において使用される前記極性溶媒はまた、有利には、炭酸プロピレン、DMSO(dimethyl sulfoxide:ジメチルスルホキシド)、またはスルホランから構成される群より、単独でまたは混合物として選択され得る。好ましくは、極性プロトン性溶媒が使用される。通常の極性溶媒およびそれらの誘電率(dielectric constant)のリストが、著書“Solvents and Solvent Effects in Organic Chemistry”, C. Reichardt, Wiley-VCH, 3rd edition, 2003, pages 472-474に見出され得る。好ましくは、使用される溶媒はエタノールである。
【0074】
好ましくは、本発明による方法の段階c)において使用される含浸溶液中に、溶媒は存在しない。このことにより、工業規模での実施が促進させられる。それは、好ましくは、ジアルキルスクシナートと酢酸のみを含む。
【0075】
使用されるジアルキルスクシナートは、好ましくは、ジメチルスクシナート、ジエチルスクシナート、ジプロピルスクシナート、ジイソプロピルスクシナート、およびジブチルスクシナートからなる群に含まれている。好ましくは、使用されるC1−C4ジアルキルスクシナートは、ジメチルスクシナートまたはジエチルスクシナートである。より好ましくは、使用されるC1−C4ジアルキルスクシナートは、ジメチルスクシナートである。少なくとも1種のC1−C4ジアルキルスクシナートが使用され、好ましくは1種のみが使用され、好ましくはジメチルスクシナートが使用される。
【0076】
本発明の調製方法の段階d)によると、段階c)からの含浸触媒前駆体は、成熟段階に付される。それは、有利には、大気圧下、17〜50℃の範囲の温度で行われ、一般的に成熟時間は、10分〜48時間、好ましくは30分〜5時間で十分である。これより長い時間は除外されない。成熟時間を調整する簡単な方法は、ラマン分光法により、本発明による方法の段階c)からの含浸乾燥触媒前駆体におけるケギンヘテロポリアニオンの形成を特徴付けることである。好ましくは、改質ヘテロポリアニオンの量を改変することなく生産性を向上させるためには、成熟時間は、30分〜4時間の範囲である。より好ましくは、成熟時間は30分〜3時間の範囲である。
【0077】
本発明の調製方法の段階e)によると、段階d)からの触媒前駆体は、180℃未満の温度での乾燥段階に付され、その後に200℃超の温度での焼成段階は行われない。
【0078】
この段階の目的は、搬送可能、貯蔵可能で、かつ、操作可能な触媒、特に、水素化処理装置に装填するための触媒を得ることにある。それは、有利には、選択された本発明の実施形態によると、C1−C4ジアルキル(特にジメチル)スクシナートと酢酸の組合せの導入を可能にした任意の溶媒の全てまたは一部を除去することからなる。あらゆる場合において、特に、C1−C4ジアルキル(特にジメチル)スクシナートと酢酸の組合せのみが用いられる場合において、搬送、貯蔵、操作、または充填の段階中に押出物が互いにくっつくのを防ぐために、触媒には、乾燥した局面が与えられることとなる。
【0079】
本発明による方法乾燥段階e)は、有利には、当業者に公知のあらゆる技術を用いて行われる。それは、有利には、大気圧下または減圧下で行われる。好ましくは、この段階は大気圧下で行われる。
【0080】
この段階e)は、有利には、50〜180℃、好ましくは60〜170℃、より好ましくは80〜160℃の範囲の温度で行われる。有利には、それは、これらの温度範囲内において、最高160℃の温度で行われ(好ましい範囲は80〜180℃である)、その後に180℃超の温度での焼成は行われない。
【0081】
それは、有利には、空気または他のあらゆる高温ガスを用いて、横断床(traversed bed)内で行われる。好ましくは、乾燥が固定床内で行われる場合、用いられるガスは、空気、または不活性ガス、例えば、アルゴンまたは窒素等のいずれかである。より好ましくは、乾燥は、横断床内で窒素の存在下に行われる。
【0082】
この段階は、好ましくは30分〜4時間、より好ましくは1〜3時間の間にわたって続く。
【0083】
本発明による方法の段階e)の終わりに、乾燥触媒が得られるが、この触媒は、その後に、空気中、例えば200℃超の温度での焼成段階に付されない。
【0084】
段階d)または段階e)の終わりに得られた触媒は、990、974cm−1における最も強いバンド(ケギン型のヘテロポリアニオン)と、スクシナートに相当するバンド(ジメチルスクシナートについて、最も強いバンドは853cm−1に位置する)と、酢酸の特徴であるバンド(最も強いバンドは896cm−1に位置する)とを含むラマンスペクトルを有する。
【0085】
使用される前に、乾燥したあるいは焼成された触媒は、有利には、硫化触媒に転化され、その活性種が形成される。この活性化または硫化の段階は、当業者に公知の方法を用いて、有利には、スルホ−還元雰囲気中、水素および硫化水素の存在下に行われる。
【0086】
本発明による方法の段階e)の終わりに、得られた前記乾燥触媒はこのように、有利には、硫化段階f)に付され、中間の焼成段階は行われない。
【0087】
前記乾燥触媒は、有利には、現場外で(ex situ)であるいは現場で(in situ)硫化される。硫化剤は、H2Sガス、または、触媒を硫化する目的で炭化水素供給原料の活性化のために用いられる硫黄を含むあらゆる他の化合物である。前記硫黄含有化合物は、有利には、アルキルジスルフィド、例えば、ジメチルジスルフィド(DMDS)、アルキルスルフィド、例えば、ジメチルスルフィド、n−ブチルメルカプタン、第三ノニルポリスルフィド(tertiononylpolysulfide)タイプのポリスルフィド化合物、例えば、ARKEMA Companyによって販売されているTPS-37またはTPS-54、あるいは触媒の良好な硫化を得ることを可能とする、当業者に公知のあらゆる他の化合物の中から選択される。好ましくは、触媒は、硫化剤および炭化水素供給原料の存在下に現場硫化される。より好ましくは、触媒は、炭化水素供給原料にジメチルジスルフィドを添加したものの存在下に現場硫化される。
【0088】
最後に、本発明の別の目的は、水素化処理方法において、特に、水素化脱硫、水素化脱窒、水素化脱金属、芳香族化合物の水素化、および石油留分の水素化転化等のプロセスにおいて、本発明による触媒を使用することである。
【0089】
本発明による方法によって得られ、かつ、好ましくは事前に硫化段階f)に付された、乾燥触媒は、有利には、炭化水素供給原料、例えば、石油留分、石炭からの留分または天然ガスから製造された炭化水素の水素化処理反応のために、より具体的には、水素化、水素化脱窒、水素化脱金属、水素化脱硫、水素化脱金属または炭化水素供給原料の水素化転化の反応のために用いられる。
【0090】
そのような使用において、本発明による方法によって得られ、かつ、好ましくは事前に硫化段階f)に付された触媒は、従来技術の触媒に対して向上した活性を示す。これらの触媒はまた、有利には、接触分解供給原料の予備処理または残油の水素化脱硫、あるいはディーゼル燃料(ULSD:超低硫黄ディーゼル(Ultra Low Sulfur Diesel))の深度水素化脱硫の間に使用され得る。
【0091】
水素化処理方法において使用される供給原料としては、例えば、ガソリン、ガスオイル、真空ガスオイル、常圧残油、真空残油、常圧蒸留物、真空蒸留物、重質燃料オイル、オイル、ワックスおよびパラフィン、使用済石油、脱アスファルト化された、残渣または原油、熱的または触媒的転化方法に由来する供給原料が、単独でまたは混合物で使用される。処理される供給原料、特に上記の供給原料は、一般的に、ヘテロ原子、例えば硫黄、酸素、および窒素を含み、重質供給原料について、それらはほとんどの場合金属も含む。
【0092】
上記の炭化水素供給原料の水素化処理反応を実施する方法において適用される操作条件は、一般的に以下の通りである:温度は、有利には180〜450℃、好ましくは250〜440℃であり、圧力は、有利には0.5〜30MPa、好ましくは1〜18MPaであり、毎時空間速度は、有利には0.1〜20h−1、好ましくは0.2〜5h−1であり、液体供給原料の体積当たりの標準温度および圧力条件下に測定される水素の体積で表される水素/供給原料比は、有利には、50〜2000L/Lである。
【0093】
本明細書の以降における実施例には、従来技術の触媒に対する、本発明方法に従って調製された触媒についての相当な活性増強が示されているが、実施例により、本発明が明らかにされるが、しかしながら、本発明の限定するものではない。
【0094】
(実施例1:焼成触媒C1A(NiMoP/アルミナ)、クエン酸(citric acid:CA)とポリエチレングリコール(polyethylene glycol:PEG)とが添加された乾燥触媒C1E(NiMoP/アルミナ)(C1AおよびC1Eは、本発明に合致しない)、ならびに、酢酸とジメチルスクシナートとが添加された、乾燥触媒C1Bおよび乾燥触媒C1F(NiMoP/アルミナ)(C1BおよびC1Fは、本発明に合致する)の調製)
Condea Chemie GmbHによって販売されている、超微細平板状ベーマイトまたはアルミナゲルからなるマトリクスを使用した。このゲルを、66%の硝酸を含む水溶液(乾燥ゲルの重量(g)あたり酸7重量%)と混合し、次いで15分間にわたり混練した。混練後、得られた練粉状物(dough)を、径1.6mmの円筒状オリフィスを有するダイに通した。次いで、押出物を、120℃で終夜乾燥させ、次いで、600℃で2時間にわたり、乾燥空気の重量(kg)当たり50kgの水を含む湿潤空気下に焼成した。このようにして、比表面積が300m2/gである担体押出物を得た。X線回折の分析により、担体は、低結晶性の立方晶系のガンマアルミナのみからなることが示された。
【0095】
ニッケル、モリブデンおよびリンを、「押出物」の形態とされた上記アルミナ担体上に添加した。含浸溶液の調製は、酸化モリブデンおよびヒドロキシ炭酸ニッケルを、水溶液中のリン酸溶液中に高温溶解させることによる、最終触媒の乾燥物質の量に対する、ニッケル、モリブデンの酸化物の重量%および無水リン酸の重量%で表される約4/22.5/4の配合が得られた。最終触媒の乾燥物質の量に対する酸化ニッケルの重量%、酸化モリブデンの重量%、および無水リン酸の重量%で表される配合は、約4/22.5/4であった。乾式含浸後、押出物を、水飽和雰囲気中12時間にわたり成熟させるよう静置した後、90℃で終夜乾燥させた。このようにして得られた乾燥触媒前駆体を、C1と表示する。450℃での2時間にわたるC1の焼成により、焼成触媒C1Aが得られるに至った。酸化物の形態で表される、触媒C1およびC1Aの最終組成は、以下の通りである:MoO3=22.4±0.2(重量%)、NiO=4.1±0.1(重量%)、およびP2O5=4.0±0.1(重量%)。
【0096】
触媒C1Eの調製は、乾燥触媒前駆体C1に、クエン酸(CA)およびポリエチレングリコール(PEG)をエタノール溶液中に含有する溶液を含浸させることにより行った。含浸容積の体積は、乾燥触媒前駆体C1の細孔容積に等しくなるようにされた。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、共に10重量%であった。
【0097】
本発明に合致する触媒C1Bの調製は、乾燥触媒前駆体C1から、ジメチルスクシナートおよび酢酸の混合物を含有するエタノール溶液を乾式含浸させることにより行った。最終触媒上に10重量%の酢酸および10重量%のジメチルスクシナートが得られた。
【0098】
本発明に合致する触媒C1Fの調製は、同様の方法であるが、エタノールの非存在下に行われた。最終割合として、13重量%の酢酸および20重量%のジメチルスクシナートを得た。
【0099】
触媒は、次いで、空気中20℃での3時間の成熟段階を経て、その後、横断床タイプのオーブン内において、110℃での3時間にわたる熱処理を行った。
【0100】
(実施例2:蒸留ディーゼル燃料の水素化処理についての、触媒C1A(NiMoP/アルミナ)(本発明に合致しない)、触媒C1E(本発明に合致しない)、触媒C1B(本発明に合致する)の評価)
触媒(粒子サイズ0.8mmのSiC 10cm3と混合された押出物形態の30cm3の触媒)の硫化を、50バール、毎時空間速度2h−1、入口H2/HC比(体積流量)400標準L/Lで行った。硫化供給原料(Arkema CompanyからのDMDS 放出(Evolution)2%が添加されたディーゼル燃料)を、反応器が150℃に達した時にH2下に反応器に供給した。150℃での1時間の後、温度は、25℃/hの昇温率で220℃まで上昇し、次いで、12℃/hの昇温率で350℃の平坦域まで達し、この平坦温度を12時間にわたって維持した。
【0101】
硫化の後、330℃に温度を下げ、試験供給原料を注入した。触媒試験は、全圧50バールで、廃棄水素を基礎とし、毎時空間速度2h−1、入口H2/HC比400標準L/L(H2流量=24標準L/L、供給原料の流量=60cm3/h)、330℃および350℃で行われた。
【0102】
触媒のHDS性能を評価することができるように、かつ、H2Sの存在を克服することができるように、受入タンクを、10Lh−1の割合の窒素でストリッピングした。
【0103】
ここで使用されたディーゼル燃料は、アラビアンヘビー原油(Arabian heavy crude)に由来する。それは、0.89重量%の硫黄と、100重量ppmの窒素を含んでおり、そのPMT[(T5+2T50+4T95)/7]は324℃であり、その密度は0.848g/cm3であった。
【0104】
HDS活性は、以下の式によるHDS転化率から測定された:
【0105】
【数1】

【0106】
HDS転化率(%HDS)は、以下の式により与えられる:
【0107】
【数2】
【0108】
試験中、各温度で得られた流出物の密度を15℃で測定した。密度の進展を図1に示す。このグラフにより所与の密度を有するために要求される温度を決めることが可能となり、精製業者は、この性能を最も低い温度で提供することになる触媒を使用するために適切にアドバイスされる。図1において、本発明による触媒により、流出物の等密度条件下に、従来技術の触媒C1Aに対して約15℃操作温度を下げることが可能となることが理解され得る。この試験中の水素化脱硫について得られた結果を、以下の表に示す:
【0109】
【表1A】
【0110】
得られた結果により、ディーゼル燃料の水素化処理の場合、水素化脱粒並びに水素化脱芳香族(これは、流出物密度の進展に翻訳される)に関して、ジメチルスクシナートを酢酸と組み合わせて本発明による触媒に添加することが興味深いことが示される。実際、上記の表に示されているように、本発明による触媒では、得られたHDS活性は、高温で126(ULSDの範囲に相当する、すなわち、10重量ppmに近似する硫黄含有量)であって、一方、焼成触媒は100(基準)、従来技術である触媒C1Eでは106であった。
【0111】
(実施例3:水素化分解予備処理タイプの適用のための真空蒸留物水素化脱窒における触媒C1A(NiMoP/アルミナ)(本発明に合致しない)、C1F(本発明に合致する)の評価)
用いられた真空蒸留物の主要な特徴は、以下に与えられる;
20℃での密度: 0.9365
硫黄: 2.92重量%
全窒素: 1400重量ppm
模擬蒸留:
初留点(IP): 361℃
10%: 430℃
50%: 492℃
90%: 567℃
終点(EP): 598℃
試験は、横断固定床(traversed fixed bed)を備えた等温パイロット反応器で行われ、流体は上方に流通していた。直留ディーゼル燃料に2重量%のジメチルジスルフィドを添加したものによる、圧力下のプラント内の350℃での現場硫化の後、水素化処理試験を、以下の操作条件下に行った。
【0112】
全圧: 12MPa
触媒の体積: 40cm3
温度: 380℃
水素の流量: 40L/h
供給原料の流量: 40cm3/h
試験された触媒の触媒性能は、以下の表に与えられる。それらは、触媒C1Aの活性が100であると仮定し、かつ、それらは1.5次であると考えた、相対活性で表される。活性と水素化脱硫の転化率(%HDS)とを結び付ける関係式は以下の通りである:
【0113】
【数3】
【0114】
同じ関係式が、水素化脱窒(%HDNおよびAHDN)に適用される。
【0115】
加えて、各触媒により得られる、380℃未満の沸点を有するフラクションに対する総転化率も評価される。それは、模擬蒸留結果(ASTM D86法)から以下の式により表される:
【0116】
【数4】
【0117】
下記の表は、3種の触媒について得られた試験結果を与える。
【0118】
【表1B】
【0119】
得られた触媒結果により、水素化分解予備処理タイプの適用の場合、本発明による触媒は、本発明による触媒が水素化脱硫および水素化脱窒並びに転化率の増大を可能にする限りにおいて、焼成NiMoP触媒より効果的であることが示され、これはより驚くべきことである。
【0120】
(実施例4:触媒C2A(NiMoP/焼成シリカ−アルミナ)(本発明に合致しない)および触媒C2B(酢酸およびジメチルスクシナートが添加された乾燥NiMoP/シリカアルミナ)(本発明に合致する)の調製)
3.6/18/1.6の配合を有する2種のNiMoP触媒の調製がSASOLによって販売されるSIRALOXタイプのシリカ−アルミナ上に行われ、シリカ含有量は25%であった。NiMoP/シリカ−アルミナタイプの乾燥触媒前駆体の調製は、H3PO4によって可溶化させられたMoO3およびNi(OH)2前駆体から、2時間にわたり90℃で還流加熱することによって行われた。透明な溶液は、次いで、水の蒸発によって濃縮され、含浸容積に達し、次いで、周囲温度でシリカ−アルミナ上に含浸させられる。このように含浸させられた担体押出物は、水飽和閉鎖封入装置中で終夜成熟段階を経、次いで、それらは、ストーブ中120℃で24時間にわたって乾燥させられる。この触媒前駆体は、次いで、2つのバッチに分けられる;
− 第1のバッチ:このものは、触媒C2A(本発明に合致しない)を得るように、450℃で2時間にわたって空気中、横断固定床において焼成される;
− 第2のバッチ:このものは、本発明の手順に従って、酢酸およびジメチルスクシナートをジメチルスクシナート/酢酸のモル比0.58で含有する溶液を、初期の湿気の出現(これは、全ての細孔が満たされたことを示す)まで滴下して含浸させることによって用いられる。触媒は、次いで、成熟させるために3時間にわたって静置され、それは、次いで、125℃での2時間にわたる熱処理に付されて、本発明に合致する触媒C2Bが得られる。
【0121】
(実施例5:触媒C2A(焼成NiMoP/シリカ−アルミナ(本発明に合致しない)およびC2B(酢酸およびジメチルスクシナートが添加された乾燥NiMoP/シリカ−アルミナ(本発明に合致する)のアニリンの存在下でのトルエン水素化および真空蒸留物の穏やかな(mild)水素化分解の評価)
アニリン存在下でのトルエン水素化試験(「HTA」試験)の目的は、H2Sの存在下および水素圧下での担持型または塊状型の硫化触媒の水素化(HYDrogenizing:HYD)活性を評価することにある。シリカ−アルミナ上に担持された触媒の酸機能を特徴付ける異性化および分解は、NH3(アニリンの分解に由来する)の存在によって阻害される。アニリンおよび/またはNH3は、それ故に、酸−塩基反応を介して、担体の酸点(acid site)と反応することとなる。提示された全ての試験は、複数のミクロ反応器を並列に含むプラントにおいて行われる。「HTA」試験の間、触媒の硫化のためおよび適正な触媒試験段階のために同一の供給原料が用いられる。装填の前に、触媒は、調節処理され、すなわち、粉砕されかつサンプルの粒子サイズが2〜4mmの範囲にわたるように分別された。4cm3の粉砕された触媒が、4cm3のカーボランダム(SiC,500μm)と混合され、このものは、反応器に供給された。
【0122】
この試験のために使用された供給原料は、以下の通りである。
【0123】
トルエン: 20重量%
シクロヘキサン: 73.62重量%
DMDS(ジメチルジスルフィド): 5.88重量%(3.8重量% S)
アニリン: 0.5重量%(750ppm N)
触媒は、反応器に、その乾燥させられた、不活性の形態で供給される。活性化(硫化)は、プラント内で同一の供給原料により行われる。それは、DMDSの分解後に形成されたH2Sであり、酸化物相を硫化する。供給原料中に存在するアニリンの量は、分解後に、約750ppmのNH3を得るように選択される。
【0124】
トルエン水素化試験の操作条件は以下の通りである:
P=6MPa
HSV=2h−1(供給原料の流量=8cm3/h)
H2/HC=450NL/L,(H2流量=3.6NL/L)
T=350℃
転化されたトルエンの百分率が評価され、反応について一次を仮定して、活性は、以下の関係式によって推定される:
【0125】
【数5】
【0126】
(%HYDtoluene=転化されたトルエンの百分率)
触媒C2A(本発明に合致しない)は、0.52の活性を有し、触媒C2B(本発明に合致する)の活性度は、0.93であり、これは、相当な増大を表しており、穏やかな水素化分解についてのシリカ−アルミナ上のNiMoPタイプの触媒の水素化活性を増加させるための酢酸とジメチルスクシナートの組合せの興味が示されている。真空蒸留物タイプの供給原料に関する水素化処理試験は、転化率および水素化脱硫の増大を定量するために行われる。
【0127】
用いられる供給原料は、真空蒸留物タイプの供給原料であり、その主要な特徴は、以下の表において与えられる。
【0128】
【表1C】
【0129】
2〜4mmの範囲の長さの押出物の断片が試験される。4cm3の触媒が、反応器に、それらの酸化物である不活性な形態で供給される。活性化(硫化)は、プラント内で、試験を開始する前に、硫化供給原料と呼ばれる供給原料により行われる(直留ディーゼル燃料+2重量%のDMDS)。それは、DMDSの分解後に形成されたH2Sであり、触媒を硫化する。
【0130】
試験の間に適用される操作条件は以下の通りである:
P=6MPa
HSV=0.6h−1
出口H2/HC=480NL/L
T=380℃
この試験は、370+フラクションの370−への総転化率を評価することによって、触媒の分類を得ることを可能にする:370−への総転化率=370℃−流出物の重量%。
【0131】
触媒結果は、以下の表にまとめられる。本発明に合致する触媒C2Bは、触媒C2A(本発明に合致しない)に対する5%の転化率の増大および特にHDSの増大を可能にする。酢酸と組み合わされたジメチルスクシナートが本発明の手順に従って用いられる触媒に加えられる時に液体流出物のS含有量が60ppmから32ppmに変化しているからである。
【0132】
【表1D】
【0133】
これらの結果により、水素化の増大に加えて、本発明に合致する触媒は、類似する配合を有する焼成された従来の触媒に対して著しく穏やかな水素化分解の増大を得ることを可能にすることができることが示される。
【0134】
(実施例6:焼成触媒C3A(CoMoNiP/アルミナ)(本発明に合致しない)、乾燥触媒C3B(ジメチルスクシナートおよび酢酸が添加されたCoMoNiP/アルミナ)(本発明に合致する)および乾燥触媒C3C(ジメチルスクシナートが添加されたCoMoNiP/アルミナ)(本発明に合致しない)の調製)
実施例1において用いられたアルミナが、ここでも、式NiCoMoP/アルミナの「乾燥触媒前駆体」を調製するために用いられる。用いられた前駆体は、三酸化モリブデン、炭酸コバルト、ヒドロキシ炭酸ニッケルおよびリン酸である。含浸溶液は、単一段階において、これらの前駆体を還流下に加熱することによって調製される。ターゲットは、乾燥触媒(550℃での強熱減量後)に対する酸化物の重量%で表される含有量に対応する:NiO/CoO/MoO3/P2O5 1/2.3/15/4.4。含浸段階の終わりに、押出物は、成熟させるために終夜水飽和雰囲気中で静置され、次いで、2時間にわたって120℃でストーブ中に置かれる。その後、乾燥触媒前駆体が得られ、実施例4と同様に、3つのバッチに分けられる。
【0135】
− 第1のバッチは、450℃で3時間にわたって焼成され、触媒C3A(本発明に合致しない)が得られる;
− 第2のバッチは、本発明の手順に従って、酢酸およびジメチルスクシナートを含有する溶液を含浸させられる:溶液中のジメチルスクシナート/酢酸の比は0.58であり、乾燥触媒前駆体は、触媒前駆体の細孔がジメチルスクシナートおよび酢酸を含有する溶液により満たされたことを示す初期の湿気が出現するまで、この溶液によって含浸させられる。次いで2時間の成熟段階が行われた後、140℃での熱処理が1時間にわたって行われる。こうして得られた触媒は、触媒C3B(本発明に合致する)である。
【0136】
− 第3のバッチは、触媒前駆体の細孔がジメチルスクシナート溶液により満たされたことを示す初期の湿気が出現するまでジメチルスクシナートを含浸させられる。次いで、2時間の成熟段階が行われた後、140℃での熱処理が1時間にわたって行われる。このようにして得られた触媒は、触媒C3C(本発明に合致しない)である。
【0137】
(実施例7:焼成触媒C3A(CoMoNiP/アルミナ)(本発明に合致しない)、乾燥触媒C3B(ジメチルスクシナートおよび酢酸が添加されたCoMoNiP/アルミナ)(本発明に合致する)および乾燥触媒C3C(ジメチルスクシナートが添加されたCoMoNiP/アルミナ)(本発明に合致しない)の、トルエン水素化モデル分子試験における評価)
真空蒸留物および残渣の水素化処理等の適用において、水素化脱水素基は、これらの供給原料中の芳香族化合物の高い割合を考慮して、重要な役割を果たす。トルエン水素化試験は、それ故に、接触分解予備処理または残渣の水素化脱硫等の適用を意図する触媒の利益を知るために用いられた。
【0138】
上記実施例6において記載された触媒は、動力学的条件下に、Microcatタイプのパイロットプラントの横断固定床管型反応器(Vinciにより製造)において現場硫化され、流体は下方に流通する。水素化活性の測定は、硫化の直後に圧力下に空気中に戻すことなく行われ、炭化水素供給原料が触媒の硫化のために用いられた。
【0139】
硫化および試験の供給原料は、5.8%のジメチルジスルフィド(DMDS)、20%のトルエンおよび74.2%のシクロヘキサンからなっている(重量による)。
【0140】
硫化は、周囲温度で350℃までで行われ、温度上昇は2℃/分であり、HSV=4h−1であり、H2/HC=450NL/Lである。触媒試験は、350℃で行われ、HSV=2h−1であり、H2/HCは、硫化の同比と等価であり、最小限4個のサンプルが利用され、ガスクロマトグラフィーにより分析される。
【0141】
トルエン水素化反応における、等しい体積の触媒による安定化された触媒活性は、こうして測定される。
【0142】
詳細な活性の測定条件は以下の通りである:
全圧: 6.0MPa
トルエンの圧力: 0.37MPa
シクロヘキサンの圧力: 1.42MPa
メタンの圧力: 0.22MPa
水素の圧力: 3.68MPa
H2Sの圧力: 0.22MPa
触媒の体積: 4cm3(2〜4mm長の押出物)
毎時空間速度: 2h−1
硫化および試験の温度: 350℃
液体流出物サンプルが、ガスクロマトグラフィーによって分析される。未転化トルエンのモル濃度(T)およびその水素化生成物(メチルシクロヘキサン(MCC6)、エチルシクロペンタン(EtCC5)およびジメチルシクロペンタン(DMCC5))の濃度の測定は、以下によって定義されるトルエン水素化率XHYDを計算することを可能にする。
【0143】
【数6】
【0144】
トルエンの水素化反応は、適用される試験条件下に一次であり、反応器は、理想的なピストン反応器のように振る舞い、触媒の水素化活性AHYDは、以下の式を適用することによって計算される。
【0145】
【数7】
【0146】
以下の表により、実施例6において調製された触媒の水素化活性を比較することができる。
【0147】
【表1E】
【0148】
これらの触媒結果により、CoMoNiP/アルミナ上の酢酸(acetic acid:AA)とジメチルスクシナート(dimethyl succinate:DMSU)の組合せの水素化活性に関して、従来技術の焼成CoMoNiP/アルミナ触媒に対する乾燥CoMoNiP/アルミナ触媒前駆体(本発明に合致する)についての特定の効果が示される。この水素化活性の増大は、接触分解予備処理または残渣の水素化脱硫等の重質供給原料タイプの適用に特に有利である。
【0149】
ラマンスペクトルは、イオン化アルゴンレーザ(514nm)を備えた分散ラマンタイプの分光計により得られる。レーザビームは、50倍長作動距離対物レンズを備えた顕微鏡によってサンプルに焦点を合わされる。サンプルのレベルにおけるレーザの出力は、1mW程度である。サンプルによって放出されるラマンシグナルは、同じ対物レンズによって集められ、1800rpmネットワークによって分散させられ、次いで、CCD検出器によって集められる。得られたスペクトルの分解能は、0.5cm−1程度である。記録されたスペクトルゾーンは300〜1800cm−1である。捕捉時間は、記録される各ラマンスペクトルのために120秒に設定される。
【0150】
ラマン分析は、触媒C16〜C19について行われ、本発明に合致する触媒について、ラマンスペクトルにおいて、ケギンHPAの特徴である最も強いバンド、ジメチルスクシナートおよび酢酸の存在が示された。バンドの正確な位置、それらの形状およびそれらの相対強度は、スペクトル記録条件に応じて所定の程度で変動し得るが、この分子の特徴は残っている。有機化合物のラマンスペクトルは、なおまた、ラマンスペクトルデータベース(例えば、Spectral Database for Organic Compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi参照)において、または、生成物の供給者(例えば、www.sigmaaldrich.com参照)によって十分に実証される。
【0151】
ラマンスペクトルは、触媒C3B(DMSU+AA)およびC3C(高純度DMSU)について記録され、それらは、図2に与えられる。各測定は、押出物の3つの異なるゾーンにおいて繰り返された。触媒C3Bの場合、ケギンヘテロポリアニオンの特徴である990および971cm−1において2つのバンドの存在が見られ得る。より弱い強度のバンド(これもこれらの種に起因し得る)は、ケギンヘテロポリアニオンについての970、902、602cm−1におけるバンドである。これらの2つのスペクトルにおいて851cm−1におけるジメチルスクシナートの強いバンドの存在も観察され得る。他方、896cm−1におけるバンドは、本発明に合致する触媒についてのみ存在する。
【0152】
要するに、本発明に合致する触媒C3Bのラマンスペクトルは、ケギンヘテロポリアニオン、ジメチルスクシナートおよび酢酸の特徴であるバンドを呈する一方で、触媒C3Cは、ケギンヘテロポリアニオンおよびジメチルスクシナートの特徴であるバンドのみを呈する。
【0153】
(実施例8:触媒C1、C2、C3、C4(CoMoP/アルミナ)(本発明に合致しない)の調製)
Condea Chemie GmbH Companyによって販売される超微細平板状ベーマイトまたはアルミナゲルからなるマトリクスが用いられる。このゲルは、66%の硝酸を含有する水溶液と混合され(乾燥ゲルの重量(g)当たり7重量%の酸)、次いで、15分間にわたって混練される。混練の後、得られた練粉状物は、1.6mm径の円筒状オリフィスを有するダイに通される。押出物は、次いで、120℃で終夜乾燥させられ、次いで、600℃で2時間にわたって、乾燥空気の重量(kg)当たり水50kgを含有する湿潤空気下に焼成される。比表面積300m2/gを有する担体押出物がこのようにして得られる。X線回折分析により、担体は、低結晶性の立方晶系のガンマアルミナのみからなることが示される。
【0154】
コバルト、モリブデンおよびリンが、「押出物」形態とされる上記のアルミナ担体上に加えられる。含浸溶液は、酸化モリブデン(24.34g)および水酸化コバルト(5.34g)の水溶液中のリン酸溶液(7.47g)中の高温溶解によって調製される。乾式含浸の後、押出物は、水飽和雰囲気中に12時間にわたって成熟させるために静置され、次いで、それらは、90℃で終夜乾燥させられる。このようにして得られた乾燥触媒前駆体は、C1で表示される。450℃での2時間にわたる触媒前駆体C1の焼成により、焼成触媒C2が得られる。酸化物の形態で表される触媒C1およびC2の最終組成は以下の通りである:MoO3=22.5±0.2(重量%)、CoO=4.1±0.1(重量%)およびP2O5=4.0±0.1(重量%)。
【0155】
焼成触媒C2は、横断床装置に供給され、直留ディーゼル燃料に2重量%のジメチルジスルフィドが添加されたものによって硫化される。次いで、直留ディーゼル燃料および接触分解からのディーゼル燃料の混合物についてのHDS試験が、300時間にわたって行われる。試験後、用いられた触媒は、取り出され、集められ、および、還流下にトルエンにより洗浄され、2つのバッチに分離される。第1のバッチは、各温度工程につき酸素量を増加させて導入することによって制御された燃焼オーブン中で再生され、これは、コークス燃焼と関連する発熱状態(exothermy)を制限することを可能にする。最終の再生工程は450℃である。こうして再生された触媒は、XRDによって分析される。結晶化CoMoO4の存在の特徴である26°におけるラインの欠如が観察され得る。この触媒は、以降においてC3により表示される。洗浄された使用触媒の第2のバッチは、マッフル炉において400℃でコークス燃焼発熱状態制御なく再生される。再生後に行われたXRD分析により、結晶化CoMoO4の存在の特徴である26°において鋭いラインの存在が示される。さらに、C4によって表示されるこの触媒の色も非常に明るい青色である。
【0156】
(実施例9:触媒C5、C6、C7、C8、C9、C10、C11、C12(添加物を有するCoMoP/アルミナ)(本発明に合致しない)の調製)
触媒C5の調製は、乾燥触媒前駆体C1に、エタノール中にクエン酸(CA)およびポリエチレングリコール(PEG)を含有する溶液を含浸させることにより行われた。含浸溶液の体積は、乾燥触媒前駆体C1の細孔容積に等しくなるようにした。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、両方10重量%である。
【0157】
触媒C6の調製は、乾燥触媒前駆体C1に、エタノール中にクエン酸(CA)およびポリエチレングリコール(PEG)を含有する溶液を含浸させることにより行われた。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、それぞれ、4および10重量%である。
【0158】
触媒C7の調製は、焼成触媒C2に、エタノール中にクエン酸およびポリエチレングリコールを含有する溶液を含浸させることにより行った。含浸溶液の体積は、焼成触媒C2の細孔容積と等しくなるようにした。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、両方10重量%である。
【0159】
触媒C8の調製は、焼成触媒C2に、エタノール中にクエン酸およびポリエチレングリコールを含有する溶液を含浸させることにより行った。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、それぞれ、4および10重量%である。
【0160】
触媒C9の調製は、焼成触媒C2に、エタノール中に酢酸(AA)およびジメチルスクシナート(DMSU)を含有する溶液を含浸させることにより行った。酢酸(AA)およびジメチルスクシナート(DMSU)の所望の割合は、それぞれ、4および10重量%である。
【0161】
触媒C10の調製は、CoMoO4タイプの耐火性相を含まない再生触媒C3に、エタノール中にクエン酸(CA)およびポリエチレングリコール(PEG)を含有する溶液を含浸させることにより行った。含浸溶液の体積は、C3の細孔容積に等しくなるようにした。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、両方10重量%である。
【0162】
触媒C11の調製は、CoMoO4タイプの耐火性相を含まない再生触媒C3に、エタノール中にクエン酸(CA)およびポリエチレングリコール(PEG)を含有する溶液を含浸させることにより行った。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、それぞれ、4および10重量%である。
【0163】
触媒C12の調製は、CoMoO4タイプの耐火性相を含まない再生触媒に、エタノール中に酢酸およびジメチルスクシナートを含有する溶液を含浸させることにより行った。酢酸およびジメチルスクシナートの所望の割合は、それぞれ、4および10重量%である。
【0164】
触媒C13の調製は、CoMoO4を含む再生触媒C4に、エタノール中にクエン酸およびポリエチレングリコールを含有する溶液を含浸させることにより行った。含浸溶液の体積は、C4の細孔容積に等しくなるようにした。クエン酸およびポリエチレングリコールの所望の割合は、両方10重量%である。
【0165】
触媒C14の調製は、CoMoO4を含む再生触媒C4に、エタノール中にクエン酸およびポリエチレングリコールを含有する溶液を含浸させることにより行った。クエン酸およびポリエチレングリコールの所望の割合は、それぞれ、4および10重量%である。
【0166】
触媒C15の調製は、CoMoO4を含む再生触媒C4に、エタノール中に酢酸およびジメチルスクシナートを含有する溶液を含浸させることにより行った。酢酸およびジメチルスクシナートの所望の割合は、それぞれ、4および10重量%である。
【0167】
触媒C5〜C15は、次いで、3時間の成熟段階に付され、その後、窒素下140℃での1時間の熱処理段階(乾燥)が行われる。
【0168】
(実施例10:触媒C16(添加物を有するCoMoP)(本発明に合致する)の調製)
触媒C16の調製は、乾燥触媒前駆体C1に、酢酸、ジメチルスクシナートおよびエタノールを含有する溶液を含浸させることにより行った。酢酸およびジメチルスクシナートの所望の割合は、それぞれ、4および10重量%である。触媒は、次いで、空気中周囲温度での3時間の成熟段階に付され、その後、窒素下140℃での1時間の熱処理段階(乾燥)が行われる。
【0169】
(実施例11:圧力下および硫化水素の存在中のシクロヘキサン中のトルエン水素化についての触媒C1〜C16の比較試験)
上記の触媒は、動力学的条件下に横断固定床管状反応器であるMicrocatタイプのパイロットプラント(Vinciにより製造)において現場硫化され、流体は、下方に流通する。水素化活性の測定は、硫化直後に、圧力下にかつ空気中に戻すことなく行われ、触媒を硫化するために炭化水素供給原料が用いられる。
【0170】
硫化および試験の供給原料は、5.8%のジメチルジスルフィド(DMDS)、20%のトルエンおよび74.2%のシクロヘキサンからなる(重量による)。
【0171】
硫化は、周囲温度から350℃までで行われ、温度上昇は2℃/分であり、HSV=4h−1であり、H2/HC=450NL/Lである。触媒試験は、350℃で行われ、HSV=2h−1であり、H2/HCは、硫化の同比と等しく、最小4つのサンプルが取られ、ガスクロマトグラフィーによって分析される。
【0172】
トルエン水素化反応における、等しい体積の触媒による安定化した触媒活性がそれ故に測定される。
【0173】
詳細な活性の測定条件は以下の通りである:
全圧: 6.0MPa
トルエンの圧力: 0.37MPa
シクロヘキサンの圧力: 1.42MPa
メタンの圧力: 0.22MPa
水素の圧力: 3.68MPa
H2Sの圧力: 0.22MPa
触媒の体積: 4cm3(2〜4mm長の押出物)
毎時空間速度: 2h−1
硫化および実験の温度: 350℃。
【0174】
液体流出物サンプルがガスクロマトグラフィーによって分析される。未転化トルエンのモル濃度(T)およびその水素化生成物(メチルシクロヘキサン(MCC6)、エチルシクロペンタン(EtCC5)およびジメチルシクロペンタン(DMCC5))の濃度の測定は、以下に定義されるトルエン水素化率XHYDを計算することを可能にする:
【0175】
【数8】
【0176】
トルエン水素化反応は、適用される試験条件下に一次であり、反応器は、理想的なピストン反応器のように振る舞うならば、触媒の水素化活性AHYDは、以下の式を適用することによって計算される:
【0177】
【数9】
【0178】
表1により、乾燥触媒前駆体C1(本発明に合致しない)からの添加物を有する触媒に対する水素化活性が比較される。ここでの活性は、基準(活性100%)として利用される最初の焼成触媒C2(本発明に合致しない)の活性に対する添加物を有する触媒の活性の比に等しい。
【0179】
【表1F】
【0180】
表1により、乾燥触媒前駆体C1(本発明に合致しない)は、焼成触媒C2(本発明に合致しない)より低い活性を有することが示される。添加物を有する触媒C5(本発明に合致しない)は、10%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を乾燥触媒C1に添加することによって調製されたものであるが、このものは試験され得なかった。押出物が、乾燥後に塊状に結束するからである。このことにより、初期の触媒が乾燥触媒である場合には過剰の酸および添加物は適していないことが示される。4%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を乾燥触媒C1に添加することによって調製された触媒C6(本発明に合致しない)は、初期乾燥触媒に対して40%の改良された活性を有する。しかしながら、初期乾燥触媒C1は、焼成された従来の触媒C2に対して25%のより低い活性度を有するので、触媒C2に対する触媒C6の相対的な水素化活性は、5%の増大を示すのみであり、これは、この試験の誤差の範囲を示している。したがって、乾燥触媒に、PEG+CAからなる組合せを添加することは興味深くはない。最後に、4%の酢酸(AA)および10%のジメチルスクシナート(DMSU)を添加することによって調製された触媒C16(本発明に合致する)は、初期乾燥触媒C1(本発明に合致しない)に対して84%の増大を呈する。従来から用いられる焼成触媒C1(本発明に合致しない)に対して、この触媒C16(本発明に合致する)の活性は、38%の増大を示し、これは、新しい触媒世代より高い(25〜30%の増大)。これらの触媒結果により、クエン酸(CA)とポリエチレングリコール(PEG)の組合せ(本発明に合致しない)に対する乾燥触媒上の酢酸(AA)とジメチルスクシナート(DMSU)の組合せ(本発明に合致する)の特定のかつ驚くべき効果が示される。
【0181】
同様に、表2により、乾燥触媒C1(本発明に合致しない)から調製された、添加物を有する触媒の相対的な水素化活性が比較される。ここでの水素化活性は、基準(活性100%)として利用される初期の焼成触媒C2(本発明に合致しない)の活性に対する添加物を有する触媒の活性の比に等しい。
【0182】
【表2】
【0183】
表2により、驚くべきことに、10重量%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を焼成触媒C2に添加することによって調製された、添加物を有する触媒C7(本発明に合致しない)の活性は、初期焼成触媒C2(本発明に合致しない)の活性に近く、あるいはこれに等価でさえあることが示され、このことは、初期触媒が乾燥触媒ではなかった場合に適していなかった過剰の酸および添加物は、焼成触媒の場合に有益性に乏しいことを示す。4%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を焼成触媒C2に添加することによって調製された触媒C8(本発明に合致しない)は、初期焼成触媒に対して14%改善された活性を有する。最後に、4%の酢酸(AA)および10%のジメチルスクシナート(DMSU)を添加することによって調製された触媒C9(本発明に合致しない)の活性は、初期焼成触媒C2(本発明に合致しない)の活性に近い(5%増大)。これらの触媒結果により、焼成触媒C2上(本発明に合致しない組合せ)ではなく、乾燥触媒前駆体(C1)のみ上の酢酸(AA)とジメチルスクシナート(DMSU)の組合せ(本発明に合致する組合せ)の特定の興味が示される。
【0184】
同様に、表3により、CoMoO4タイプの耐火性相を含有しない再生触媒C3から調製された、添加物を有する触媒(本発明に合致しない)の相対的な水素化活性が比較される。
【0185】
【表3】
【0186】
表3により、10重量%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を、CoMoO4タイプの耐火物相を含有しない再生触媒C3に添加することによって調製された、添加物を有する触媒C10(本発明に合致しない)の活性は、初期焼成触媒C2(本発明に合致しない)の活性に近く、さらにはこの活性と等価であることが示され、このことは、過剰の酸および添加物は、結晶化CoMoO4相を有しない再生触媒の場合にも有益性に乏しいことを示す。4%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を、CoMoO4タイプの耐火物相を含有しない再生触媒C3に添加することによって調製された触媒C11(本発明に合致しない)は、初期触媒C3(本発明に合致しない)に対して12%改善された活性を有し、これは、それらに、焼成された新規触媒C2に対して9%改善された活性を与える。最後に、4%の酢酸(AA)および10%のジメチルスクシナート(DMSU)を添加することによって調製された触媒C12(本発明に合致しない)の活性は、焼成触媒C2(本発明に合致しない)の活性に近い。これらの触媒結果により、CoMoO4タイプの結晶化相を有しない再生触媒C3(本発明に合致しない)上の酢酸(AA)とジメチルスクシナート(DMSU)の組合せに対する、乾燥触媒C1(本発明に合致する)上の同組合せの特定の興味が示される。
【0187】
同様に、表4により、CoMoO4を含有する再生触媒C4(本発明に合致しない)から調製された、添加物を有する触媒の相対的な水素化活性が比較される。CoMoO4の存在は、XRD分析によって確認される。
【0188】
【表4】
【0189】
表4により、10重量%のクエン酸(CA)および10重量%のポリエチレングリコール(PEG)を、CoMoO4を含有する再生触媒C4に添加することにより調製された、添加物を有する触媒C13(本発明に合致しない)の活性は、初期焼成触媒C2(本発明に合致しない)の活性に近いか、この活性に等価でさえあることが示され、このことにより、過剰の酸および添加物は、結晶化CoMoO4相を有する再生触媒の場合に有益であることが示される。実際に、4%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を、CoMoO4タイプの結晶化相を含有する再生触媒(本発明に合致しない)に添加することによって調製された触媒C14(本発明に合致しない)は、初期触媒C4(本発明に合致しない)に対して不十分に改善された活性を有する。その活性は、焼成された新規な触媒C2の活性より低いままであるからである。最後に、4%の酢酸(AA)および10%のジメチルスクシナート(DMSU)を添加することによって調製された触媒C15(本発明に合致しない)の活性は、触媒C4(本発明に合致しない)の活性に近く(3%増大)、焼成された新規触媒C2に対してはるかにより低い。これらの触媒結果により、CoMoO4タイプの結晶化相を有する再生触媒C4(本発明に合致しない)上の酢酸(AA)とジメチルスクシナート(DMSU)の組合せに対する乾燥触媒C1(本発明に合致する)上の同組合せの特定の興味が示される。この組合せは、クエン酸(CA)とポリエチレングリコール(PEG)の組合せとは異なり、CoMoO4タイプの結晶化耐火物相を含有する触媒について特に効果がない。
【0190】
(実施例12:触媒C17およびC18(本発明に合致しない)、C19(本発明に合致する)の調製および触媒C2(本発明に合致しない)、C17およびC18(本発明に合致しない)、C16およびC19(本発明に合致する)のディーゼル燃料HDSについての比較)
触媒C17の調製は、触媒前駆体C1に高純度のジメチルスクシナート(DMSU)を含浸させることにより行った。これは、最終触媒上に30重量%のジメチルスクシナートを求めるのに等しい。触媒は、次いで、20℃で3時間の成熟段階に付され、その後、空気中140℃での1時間の熱処理が横断床タイプのオーブンにおいて行われる。この熱処理の終わりに得られた触媒は、C17により示される。この触媒は、本発明に合致していない。ジメチルスクシナートとの組合せで酢酸を含有していないからである。
【0191】
触媒C18は、触媒前駆体C1の細孔を酢酸により満たす以外は、同様の方法で調製された。触媒の重量に対して31重量%が得られる。成熟/熱処理段階はC17と同様である。
【0192】
触媒C19の調製は、触媒前駆体C1に、ジメチルスクシナートおよび酢酸の混合物のみを含有する溶液を含浸させることにより行った。ジメチルスクシナート/モリブデンのモル比は1.1であった。これは、ジメチルスクシナートおよび酢酸のそれぞれについて25および18重量%の最終触媒に対する含有量を求めるのに等しい。触媒は、次いで、周囲温度での3時間の成熟段階に付され、その後に、空気中140℃での1時間の熱処理が横断床タイプのオーブンにおいて行われる。この熱処理の終わりに得られた触媒は、C19により示される。この触媒は、本発明に合致する。
【0193】
触媒C2(本発明に合致しない)、C16(本発明に合致する)、C17(本発明に合致しない)およびC19(本発明に合致する)は、ディーゼル燃料HDSについて試験された。
【0194】
用いられたディーゼル燃料供給原料の特徴:
− 15℃での密度: 0.8522
− 硫黄: 1.44重量%
− 模擬蒸留:
・ IP : 155℃
・ 10% : 247℃
・ 50% : 315℃
・ 90% : 392℃
・ EP : 444℃
試験は、横断固定床を備える等温パイロット反応器において行われ、流体は、上方に流通する。試験されるディーゼル燃料に2重量%のジメチルスクシナートが添加されたものによる圧力下のプラント中での350℃での現場(in-situ)硫化の後、水素化脱硫試験は、以下の操作条件下に行われる:
全圧: 7MPa
触媒体積: 30cm3
温度: 340℃
水素流量: 24L/h
供給原料流量: 60cm3/h
試験された触媒の触媒性能は表3において与えられる。それらは、焼成触媒C2の活性が100であると仮定し、それらは1.5次であることを考慮した相対強度で表される。活性と水素化脱硫転化率(%HDS)を結び付ける関係式は以下の通りである:
【0195】
【数10】
【0196】
得られた結果は、表5において与えられる。
【0197】
【表5】
【0198】
表5により、明らかに、乾燥触媒前駆体上の酢酸とジメチルスクシナートの組合せの相乗効果および特定の興味が示される。実際に、触媒C17およびC18(本発明に合致しない)は、本発明に合致する触媒C16およびC19について得られた活性より低い活性を有する。触媒C19の場合に、触媒C16の場合より多量の添加物DMSUが含浸させられたが、しかしながら、実施例1におけるPEGおよびクエン酸の場合に観察されたこととは反対に活性増大は増加することに留意することはさらに興味深い。
【0199】
ラマンスペクトルは、イオン化アルゴンレーザ(514nm)を備えた分散ラマンタイプの分光計により得られる。レーザビームは、50倍長作動距離対物レンズを備えた顕微鏡によってサンプルに焦点を合わされる。サンプルのレベルにおけるレーザの出力は、1mW程度である。サンプルによって放出されるラマンシグナルは、同じ対物レンズによって集められ、1800rpmネットワークによって分散させられ、次いで、CCD検出器によって集められる。得られたスペクトルの分解能は、0.5cm−1程度である。記録されたスペクトルゾーンは300〜1800cm−1である。捕捉時間は、記録される各ラマンスペクトルのために120秒に設定される。
【0200】
ラマン分析は、触媒C16〜C19について行われ、本発明に合致する触媒について、ラマンスペクトルにおいて、ケギンHPA、ジメチルスクシナートおよび酢酸の特徴である最も強いバンドの存在が示された。バンドの正確な位置、それらの形状およびそれらの相対強度は、スペクトル記録条件に応じて所定の範囲で変動し得る一方で、この分子の特徴はそのままである。有機化合物のラマンスペクトルは、ラマンスペクトルデータベース(例えば、Spectral Database for Organic Compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi参照)において、または、生成物の供給者(例えば、www.sigmaaldrich.com参照)によってさらに十分に実証される。
【0201】
任意の理論によって拘束されることなく、非錯体形成有機添加物は、溶液中の触媒の細孔におけるヘテロポリアニオンの再形成を可能にすることが観察された。ジアルキルスクシナートおよび酢酸の混合物を含む溶液の含浸後のヘテロポリアニオンバンドの出現により強調されるのは典型的にはこの現象である。この効果は、「ヘテロポリアニオンの再形成」と呼ばれる。含浸溶液において最初に存在するが、それらは、新たに含浸させられた乾燥触媒前駆体上に存在しないからである;それらは、しかしながら、添加物の含浸後の成熟段階の間に再形成する。クエン酸存在下のPEGの場合、錯形成効果は、XRDによって容易に特徴付けられる結晶化相の消失を可能にするが、ヘテロポリアニオンは再形成されない。
【図面の簡単な説明】
【0202】
【図1】実施例2の試験中、各温度で得られた流出物の密度を15℃で測定し、密度の進展を示す。
【図2】触媒C3B(DMSU+AA)およびC3C(高純度DMSU)についてのラマンスペクトルを示す。
【技術分野】
【0001】
本発明は、触媒、その調製方法、および水素化処理分野におけるその使用に関する。
【0002】
通常、炭化水素留分水素化処理触媒の目的は、炭化水素留分に含まれる硫黄または窒素の化合物を除去し、石油生成物が、所与の適用(車の燃料、ガソリンまたはディーゼル燃料、暖房油、ジェット燃料)について要求される規格(例えば、硫黄含有量、芳香族化合物含有量等)に適合するようにさせることにある。目的はまた、この供給原料を種々の転化方法、例えば、改質、真空蒸留物水素化分解、接触分解、常圧または真空の残渣の水素化転化方法等に付してその物理化学的特性を改変する前にこれを予備処理して、それが含む不純物を除去することであり得る。水素化処理触媒の組成および使用は、書物Catalysis Science and Technology, Vol.11(1996), Springer-VerlagからのB. S. Clausen, H. T. Topsoe および F. E. Massothによる論文において特によく記載されている。硫化後、担体上に複数の表面種が存在し、これは、全てが、所望の反応について良好な性能を示すわけではない。これらの種は、特に、1984年のCatalysis Review Science and Engineering 第26号, pp.395-420において出版されたTopsoeらによる出版物においてよく記載されている。
【0003】
欧州共同体における自動車汚染基準の厳格化により(Official Journal of the European Union, L76, 22 March 2003, Directive 2003/70/CE, pp.L76/10-L76/19)、精製業者は、ディーゼル燃料およびガソリン中の硫黄含有量を相当低減させるように強要されている(2005年1月1日において50重量ppmであるのに対して2009年1月1日において硫黄最大10重量ppm)。加えて、精製業者は、一方では、原油がますます重質であり、したがって、ますます多くの不純物を含有するため、他方で、精製所における転化方法の増加のため、水素化処理方法に対してますます難処理性である供給原料を使用するように強要される。実際に、常圧蒸留に直接的に由来する留分より水素化処理し難い留分が生じている。その例は、その高い芳香族化合物含有量に関連して、接触分解から得られたディーゼル留分(LCO(Light Cycle Oil)とも呼ばれる)である。これらの留分は、常圧蒸留から得られたディーゼル留分と共処理される;それらは、芳香族化合物含有量を低減させて規格に従う密度およびセタン価を得るために、従来の触媒に対して高度に改善された水素化脱硫および水素化の機能を有する触媒を必要とする。
【0004】
さらに、接触分解または水素化分解等の転化方法には、酸基を有する触媒が使用される。酸基は、それらを、窒素不純物、特に塩基性窒素化合物の存在に対して特に感受性にする。したがって、これらの供給原料を予備処理してこれらの化合物を除去するための触媒を用いることが必要である。これらの水素化処理触媒はまた、第一の水素化脱窒段階がC−N結合に隣接する芳香環の水素化の段階として知られている限りにおいて、改善された水素化基を要求する。
【0005】
したがって、改善された性能を有する新しい触媒を得ることを可能にする水素化処理触媒を調製する手段を見出すことは興味深い。
【背景技術】
【0006】
有機化合物を水素化処理触媒に添加して、それらの活性を改善することは、今や、当業者に周知である。多くの特許は、種々の範囲の有機化合物、例えば、エーテル化されてもよい、モノ−、ジー、またはポリアルコールの使用を保護する(特許文献1〜5)。C2−C14モノエステルにより改変された触媒は、特許出願(特許文献6〜7)において記載されているが、しかしながら、これらの改変は、燃料の硫黄含有量に対する規格を満たすように触媒の性能を十分に増強することを常に可能にするというわけではなく、このことは、精製業者をますます制限することとなった。
【0007】
これを克服するために、TOTAL Companyによって出願された特許文献8には、第VIB族および第VIII族金属、担体としての耐火性酸化物、および有機化合物を含む触媒であって、該有機化合物は、式R1−O−CO−R2−CO−O−R1またはR1−CO−O−R2−O−CO−R1(式中、各R1は、独立して、C1−C18アルキル基、C2−C18アルケニル基、C6−C18アリール基、C3−C8シクロアルキル基、C7−C20アルキルアリールまたはアリールアルキル基を示すか、または、2つの基R1は、一緒に、C2−C18の二価基を形成し、R2は、C1−C18アルキレン基、C6−C18アリーレン基、C3−C7シクロアルキレン基を示すか、またはその組合せであり、R1およびR2によって示される有炭化水素基の炭素鎖は、N、SおよびOの中から選択される1個以上のヘテロ原子を含むか有し得、各基R1およびR2は、式−C(=O)O−R1または−O−C(=O)−R1(式中、R1は、前述の意味を有する)の1個以上の置換基を有し得る)の少なくとも2個のカルボン酸エステル基を含む、触媒を用いることが提案される。好ましい態様では、C1−C4ジアルキルスクシナート、特に、例示されるジメチルスクシナートが用いられる。これらの化合物は、溶媒(かなりの溶媒のリストが言及される)またはカルボン酸の存在下に導入され得る。特に言及される約30の酸の中で、酢酸があるが、しかしながら、これは、10の好ましい酸の中で言及されない。クエン酸が好ましいことはすでに留意され得る。
【0008】
特許文献8において記載された触媒調製方法は、数日まで、例えば49〜115日続き得る成熟および熱処理段階を含み、このことは、これらの触媒の製造を大きく制限するだろうし、したがって、改良を要求するだろう。
【0009】
従来技術の他の特許により、水素化処理触媒上の有機酸またはアルコールの組み合わされた使用と関連する活性増大が記載される。それ故に、株式会社ジャパンエナジーによって出願された特許文献9には、
− 発明の第一の好ましい態様に従って、触媒前駆体、周期律表の第VI族および第VIII族からの1種以上の金属および有機酸を含有する溶液を調製する段階;発明の第二の好ましい態様に従って、この溶液はリン前駆体も含む、
− 200〜400℃で熱処理を行う段階、
− 金属のモル当たり0.1〜2の比で有機酸またはアルコールによって上記の得られた触媒の含浸を行う段階
からなる解決方法が提供される。
【0010】
発明の好ましい態様の一つは、200℃未満の温度で乾燥させる段階を含むのに対して、その発明の第二の好ましい態様は、400℃以上の温度での最終熱処理を含む。
【0011】
これらの触媒は、精製業者に利用可能な水素がますます欠乏する供給原料の面で新たな環境基準を満たす十分な活性を有しないことが観察された。
【0012】
同様に、特許文献10は、第VIII族および第VIB族の金属酸化物を含む、再生触媒上に存在するCoMoO4タイプの結晶化相の比率を減少させることを概要的に可能にする活性化法であって、再生触媒を、酸および有機添加物と接触させる工程を含む、方法を特許請求している。したがって、クエン酸(CA)−ポリエチレングリコール(PEG)の組合せが、多くの実施例において再生触媒上で用いられた。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】国際公開第96/41848号パンフレット
【特許文献2】国際公開第01/76741号パンフレット
【特許文献3】米国特許第4012340号明細書
【特許文献4】米国特許第3954673号明細書
【特許文献5】欧州特許出願公開第601722号明細書
【特許文献6】欧州特許出願公開第466568号明細書
【特許文献7】欧州特許出願公開第1046424号明細書
【特許文献8】国際公開第06/077326号パンフレット
【特許文献9】特開平07−136523号公報
【特許文献10】国際公開第05/035691号パンフレット
【発明の概要】
【課題を解決するための手段】
【0014】
(発明の概要)
本発明は、触媒およびその調製方法に関し、触媒は、水素化処理に使用可能であり、従来技術の触媒に対して触媒性能(特に触媒活性)を改良することを可能にする。実際に、C1−C4ジアルキルスクシナート(特にジメチル)および酢酸から構成される対を乾燥触媒前駆体上で使用することにより、驚くべきことに、対の化合物のそれぞれ一つと比較して顕著に改良された触媒活性が導かれることが示された。
【0015】
より正確には、本発明は、アルミナベースの無定形担体と、リンと、少なくとも1種のC1−C4ジアルキルスクシナートと、酢酸と、少なくとも1種の第VIII族元素および少なくとも1種の第VIB族元素を含む水素化脱水素基(function)とを含む触媒であって、そのラマンスペクトルは、少なくとも1種のケギンヘテロポリアニオンの特徴である990および/または974cm−1のバンド、前記スクシナートの特徴であるバンドおよび酢酸の特徴である896cm−1の主要バンドを含む、触媒に関する。水素化脱水素基は、好ましくは、コバルトおよびモリブデンからなる。それは、水素化脱水素基がコバルトおよびモリブデンからなる以外は、少なくとも1種の第VIII族元素および少なくとも1種の第VIB族元素も含み得る。
【0016】
得られた触媒は、以下に分類する特徴的なラマンスペクトルを有する;
1)ケギンPXY11O40x−および/またはPY12O40x−タイプのヘテロポリアニオン(単数または複数)の特徴であるバンド(式中Yは第VIB族金属でありXは第VIII族金属である)。
【0017】
Griboval, Blanchard, Payen, Fournier, DuboisのCatalysis Today 45(1998) 277 Fig. 3 e)によると、PCoMo11O40x−構造の主要バンドは、乾燥触媒上において、232、366、943、974cm−1にあり、M. T. Popeの“Heteropoly and Isopoly oxometalates”, Springer Verlag, p 8によると、これらのバンドは、元素XまたはYの性質の特徴ではなく、HPAの構造の特徴である。このタイプの空隙ケギンHPAの特徴である最も強いバンドは974cm−1にある。
【0018】
Griboval、 Blanchard、 Gengembre、 Payen、 Fournier、 Dubois、 BernardのJournal of Catalysis 188(1999) 102, Fig. 1 a)によると、PMo12O40x−の主要バンドは、例えばカウンターイオンとしてのコバルトとのHPAの塊状態において、251、603、902、970、990cm−1にある。このケギンHPAの特徴である最も強いバンドは、990cm−1にある。M. T. Popeの“Heteropoly and Isopoly oxometalates”, Springer Verlag, p 8も、我々にこれらのバンドは元素XまたはYの性質の特徴でなく、完全な、空隙性または置換されたケギンHPAの構造の特徴であることをを教示する。
【0019】
2)用いられるジアルキルスクシナート(単数または複数)の特徴であるバンド。ジメチルスクシナートのラマンスペクトルは、この分子の一意のフィンガープリントである。300−1800cm−1のスペクトルゾーンにおいて、このスペクトルは、以下の一連のバンド(最も強いバンドのみが記録される,cm−1)によって特徴付けられる:391、853(最も強いバンド)、924、964、1739cm−1。ジエチルスクシナートのスペクトルは、考慮されるスペクトルゾーンにおいて、以下の主要バンドを含む;861(最も強いバンド)、1101、1117cm−1。同様に、ジブチルスクシナートについて:843、1123、1303、1439、1463cm−1およびジイソプロピルスクシナートについて:833、876、1149、1185、1469(最も強いバンド)、1733cm−1。
【0020】
3)酢酸の特徴であるバンド。以下の主要なバンドを有する:448、623、896cm−1。最も強いバンドは896cm−1である。
【0021】
バンドの正確な位置、それらの形状およびそれらの相対強度は、スペクトル記録条件に応じて所定の範囲で変動し得るが、この分子の特徴はそのままである。有機化合物のラマンスペクトルは、ラマンスペクトルデータベース(例えば、Spectral Database for the organic compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi参照)において、または、生成物の供給者(例えばwww.sigmaaldrich.comを参照)によってさらによく実証されている。
【0022】
ラマンスペクトルは、イオン化アルゴンレーザ(514nm)を備えた分散ラマンタイプの分光計により得られる。レーザビームは、50倍長作動距離対物レンズを備えた顕微鏡によってサンプルに焦点を合わされる。サンプルのレベルにおけるレーザの出力は、1mW程度である。サンプルによって放出されるラマンシグナルは、同じ対物レンズによって集められ、1800rpmネットワークによって分散させられ、次いで、CCD検出器によって集められる。得られたスペクトルの分解能は、0.5cm−1程度である。記録されたスペクトルゾーンは300〜1800cm−1である。捕捉時間は、記録される各ラマンスペクトルのために120秒に設定される。
【0023】
好ましくは、用いられるジアルキルスクシナートはジメチルスクシナートであり、触媒は、そのスペクトルにおいて、ケギンヘテロポリアニオン(単数または複数)の特徴である990および/または974cm−1、ジメチルスクシナートの特徴である853cm−1、および酢酸の特徴である896cm−1に位置する主要ラマンバンドを有する。
【0024】
好ましくは、本発明の触媒は、アルミナまたはシリカ−アルミナからなる担体を含む。
【0025】
本発明による触媒はまた、ホウ素および/またはフッ素および/またはケイ素を含み得る。
【0026】
また、本発明による触媒の調製方法についても記載されている。本発明の調製方法は、180℃未満の温度で乾燥させられ、かつ、少なくともリンおよび水素化脱水素基、ならびに無定形担体を少なくとも含む触媒前駆体に、酢酸およびC1−C4ジアルキルスクシナートの組み合わせを含む含浸溶液を含浸させる少なくとも1回の含浸段階と、その後の前記含浸させられた触媒前駆体の成熟段階と、その後の180℃未満の温度での乾燥段階とを含み、その後に焼成段階(空気中での熱処理)は行われない。得られた触媒は、好ましくは、硫化段階に付される。
【0027】
水素化脱水素基は、少なくとも1種の第VIII族元素および少なくとも1種の第VIB族元素を含む。好ましくは、水素化脱水素基は、コバルトおよびモリブデンからなる。
【0028】
数時間を超えない単位段階を用いる簡単かつ迅速な調製方法により、従って、従来技術の方法と比較して、工業規模で、より高い生産性を得ることが可能となる。
【0029】
より正確には、本発明による水素化処理触媒の調製方法は、以下に詳述されることになる、連続する段階を含む:
a)アルミナをベースとする無定形担体に、水素化脱水素基の元素およびリンを含む少なくとも1種の溶液を含浸させる、少なくとも1回の含浸段階(得られた生成物は「触媒前駆体」と称される);
b)180℃未満の温度での乾燥段階であって、その後に焼成は行われない、段階(得られた生成物は「乾燥触媒前駆体」と称される);
c)少なくとも1種のC1−C4ジアルキルスクシナートと、酢酸と、少なくとも1種のリン化合物(段階a)において、リン化合物が完全には導入されていない場合)とを含む含浸溶液による、少なくとも1回の含浸段階(得られた生成物は「含浸乾燥触媒前駆体」と称される);
d)成熟段階;
e)180℃未満の温度での乾燥段階であって、その後に焼成段階は行われない、段階(得られた生成物は「触媒」と称される)。
【0030】
好ましくは、段階e)の終わりに得られた生成物は、硫化段階f)に付される。
【0031】
以下に記載されるように、本発明による方法は好ましくは、以下の様式を単独または組み合わせて用いて行われる:担体はアルミナまたはシリカ−アルミナからなり;水素化基の全てが段階a)において導入され;リンの全てが段階a)において導入され;ジアルキルスクシナートはジメチルスクシナートであり;段階c)は溶媒の非存在下に行われ;段階d)は17〜50℃の範囲の温度で行われ;段階e)は80〜160℃の範囲の温度で行われる。
【0032】
より好ましくは、本発明による方法は、以下の連続する段階を含む:
a)前記担体に、水素化脱水素基の元素の全てとリンの全てとを含む溶液を乾式含浸させる、少なくとも1回の段階、
b)75〜130℃の範囲の温度での乾燥段階であって、その後に焼成は行われない、段階、
c)ジメチルスクシナートおよび酢酸を含む含浸溶液による、少なくとも1回の乾式含浸段階、
d)17〜50℃での成熟段階、
e)80〜160℃の範囲の温度で行われる乾燥段階(好ましくは窒素下)であって、その後に焼成段階は行われない、段階。
【0033】
水素化脱水素基と、アルミナをベースとする無定形担体とを含む触媒前駆体、ならびに、その調製様式は以下に記載される。
【発明を実施するための形態】
【0034】
(詳細な説明)
本発明による方法の段階a)の終わりに得られる前記触媒前駆体は、殆どの場合当業者に公知のあらゆる方法によって調製され得る。
【0035】
前記触媒前駆体は、水素化脱水素基を含み、前記触媒前駆体は、ドーパントとしてのリンおよび/またはホウ素および/またはフッ素、ならびに無定形担体を含む。水素化脱水素基は、少なくとも1種の第VIB族元素と少なくとも1種の第VIII族元素とを含む。好ましくは、水素化脱水素基は、コバルトおよびモリブデンからなる。
【0036】
前記触媒前駆体の無定形担体は、アルミナをベースとし、すなわち、それは、50%超のアルミナを含み、それは、一般的には、アルミナのみを含むか、または、以下に規定されるようなシリカ−アルミナを含み、場合によっては、含浸の外側で導入された(例えば、担体の調製、混練、解膠等またはその成形の間に導入された)、金属(単数または複数)および/またはドーパント(単数または複数)を含む。担体は、成形(例えば、押出)および一般に300〜600℃での焼成の後に得られる。
【0037】
好ましくは、担体は、アルミナからなり、好ましくは押し出されたアルミナからなる。アルミナは、好ましくはガンマアルミナであり、前記無定形担体は、好ましくはガンマアルミナからなる。
【0038】
別の好ましい場合において、それは、少なくとも50%のアルミナを含むシリカ−アルミナである。担体中のシリカの割合は、最大50重量%、ほとんどの場合45重量%以下、好ましくは40重量%以下である。
【0039】
ケイ素源は、当業者に周知である。ケイ素源の例としては、ケイ酸、粉末シリカ、コロイド形態のシリカ(シリカゾル)、オルトケイ酸テトラエチルSi(OEt)4が挙げられる。
【0040】
前記触媒前駆体の水素化脱水素基は、少なくとも1種の第VIB族元素および少なくとも1種の第VIII族元素によって提供される。コバルトおよびモリブデンから構成される対が好ましい。水素化脱水素元素の全割合は、有利には、触媒の全重量に対して酸化物6重量%超である。好ましい第VIB族元素は、モリブデンおよびタングステンであり、一般的にモリブデンである。好ましい第VIII族元素は、非貴金属元素であり、特にコバルトおよびニッケルである。
【0041】
有利には、水素化基は、以下の元素の組合せから構成される群より選択される:コバルト−モリブデン、ニッケル−モリブデン、ニッケル−コバルト−モリブデン、またはニッケル−モリブデン−タングステン。
【0042】
高い水素化脱硫、もしくは水素化脱窒、および芳香族の水素化の活性が望まれる場合、水素化脱水素基は、有利にはニッケルとモリブデンの組合せによって果たされる;モリブデンの存在下での、ニッケルとタングステンの組合せも有利であり得る。真空蒸留物またはより重質なタイプの供給原料の場合、コバルト-ニッケル−モリブデンのタイプの組合せが、有利に用いられ得る。
【0043】
使用され得るモリブデン前駆体もまた、当業者に周知である。例えば、モリブデン源の中から、酸化物および水酸化物、モリブデン酸およびこれらの塩、特に、アンモニウム塩、例えば、モリブデン酸アンモニウム、七モリブデン酸アンモニウム、リンモリブデン酸(H3PMo12O40)、およびこれらの塩、ならびに、場合によるケイモリブデン酸(H4SiMo12O40)およびその塩が使用可能である。モリブデン源はまたあらゆるヘテロポリ化合物であり得、例えば、ケギン型、空隙ケギン型、置換ケギン型、ドーソン(Dawson)型、アンダーソン(Anderson)型、ストランベルグ(Strandberg)型である。三酸化モリブデン、ならびに、ストランベルグ型、ケギン型、空隙ケギン型、または置換ケギン型のヘテロポリ化合物(ヘテロポリアニオン)が、好ましくは使用される。
【0044】
使用され得るタングステン前駆体もまた、当業者に周知である。例えば、使用され得るタングステン源は、酸化物および水酸化物、タングステン酸およびそれらの塩、特にアンモニウム塩、例えば、タングステン酸アンモニウム、メタタングステン酸アンモニウム、リンタングステン酸、およびそれらの塩、ならびに場合によるケイタングステン酸(H4SiW12O40)およびその塩である。タングステン源はまた、あらゆるヘテロポリ化合物であり得、例えば、ケギン型、空隙ケギン型、置換ケギン型、ドーソン型のヘテロポリ化合物である。好ましくは、アンモニウム酸化物および塩、例えば、メタタングステン酸アンモニウム、もしくは、ケギン型、空隙ケギン型、または置換ケギン型のヘテロポリアニオンが用いられる。
【0045】
第VIB族元素(単数または複数)の前駆体(単数または複数)の割合は、触媒前駆体の全質量に対して有利には第VIB族酸化物5〜40重量%、好ましくは8〜35重量%、より好ましくは10〜30重量%である。
【0046】
使用され得る第VIII族元素(単数または複数)の前駆体は、有利には、酸化物、水酸化物、ヒドロキシ炭酸塩、炭酸塩および硝酸塩の中から選択され、例えば、ヒドロキシ炭酸ニッケル、炭酸コバルト、または水酸化コバルトが好ましくは使用される。
【0047】
第VIII族元素(単数または複数)の前駆体(単数または複数)の割合は、触媒前駆体の全質量に対して第VIII族酸化物有利には1〜10重量%、好ましくは1.5〜9重量%、より好ましくは2〜8重量%である。
【0048】
前記触媒前駆体の水素化脱水素基は、調製の種々のレベルにおいて、かつ種々の方法で、触媒に導入され得る。前記水素化脱水素基は、常に、成形された担体の含浸によって、少なくとも部分的に、好ましくは完全に導入される。それはまた、前記無定形担体を成形する際に、部分的に導入され得る。
【0049】
水素化脱水素基が、前記無定形担体の成形の際に、部分的に導入される場合、それは、マトリクスとして選択されたアルミナゲルとの混練の際にのみ、部分的に導入され得(例えば最大10%の第VIB族元素が、例えば混練を通じて導入される)、その後、水素化元素(単数または複数)の残りは、後に導入される。好ましくは、水素化脱水素基が混練の際に部分的に導入される場合、この段階中に導入される第VIB族元素(単数または複数)の割合は、最終触媒上に導入される第VIB族元素(単数または複数)の全量の5%未満である。いかなる導入様式が用いられるにせよ、好ましくは、少なくとも1種の第VIB族元素(またはそれらの全て)が、少なくとも1種の第VIII族元素(またはそれらの全て)と同時に導入される。元素の導入のためのこれらの方法および割合は、特に、水素化脱水素基がCoMoからなる場合に用いられる。
【0050】
前記無定形担体の成形後に、水素化脱水素基が、少なくとも部分的に、好ましくは完全に導入される場合、無定形担体上への前記水素化脱水素基の導入は、有利には、成形されかつ焼成された担体上での1回以上の過剰溶液含浸によって、あるいは、好ましくは、1回以上の乾式含浸によって、より好ましくは、前記成形されかつ焼成された担体の乾式含浸によって、金属前駆体塩を含む溶液を用いて行われ得る。更により好ましくは、水素化脱水素基は、前記無定形担体の成形後、金属前駆体塩を含む含浸溶液を用いる前記担体の乾式含浸によって完全に導入される。前記水素化脱水素基の導入はまた、有利には、活性相の前駆体(単数または複数)の溶液よる、成形されかつ焼成された担体の1回以上の含浸によって行われ得る。元素が、対応する前駆体塩の数回の含浸において導入される場合、中間の触媒乾燥段階が一般的には行われ、その際の温度は、50〜180℃、好ましくは60〜150℃、より好ましくは75〜130℃である。
【0051】
リンも触媒に導入される。別の触媒ドーパントも導入され得、これは、ホウ素、およびフッ素の中から単独でまたは混合物として選択される。ドーパントは、それ自体は触媒的特徴を有しない添加元素であるが、それは、金属(単数または複数)の触媒活性を高める。
【0052】
前記ドーパントは、有利には、単独で、あるいは、水素化脱水素基の少なくとも1種の元素との混合物として、導入され得る。
【0053】
それはまた、担体の合成と同じ時期の早期に導入され得る。
【0054】
それはまた、選択されたマトリクスの解膠の直前または直後に導入され得る。選択されたマトリクスは、例えばかつ好ましくは、アルミナ前駆体またはオキシ水酸化アルミニウム(ベーマイト)である。
【0055】
前記ドーパントはまた、有利には、水素化脱水素基の前駆体(単数または複数)との混合物として、成形された無定形担体(好ましくは押出物形態のアルミナまたはシリカ−アルミナ)上に完全にまたは部分的に、金属前駆体塩およびドーパント(単数または複数)の前駆体(単数または複数)を含む溶液を用いる前記無定形担体の乾式含浸により導入される。
【0056】
ホウ素源は、ホウ酸、好ましくは、オルトホウ酸H3BO3、二ホウ酸アンモニウム、五ホウ酸アンモニウム、酸化ホウ素、ホウ酸エステルであり得る。ホウ素は、例えば、水/アルコール混合物中または水/エタノールアミン混合物中のホウ酸の溶液によって、導入され得る。
【0057】
好ましいリン源は、オルトリン酸H3PO4であるが、その塩及びエステル、例えばリン酸アンモニウムもまた適している。リンはまた、ケギン型、空隙ケギン型、置換ケギン型、またはストランドベルグ型のヘテロポリアニオンの形態で、第VIB族元素と同時に導入され得る。
【0058】
使用され得るフッ素源は、当業者に周知である。例えば、フッ化物アニオンが、フッ化水素酸またはその塩の形態で導入され得る。これらの塩は、アルカリ金属、アンモニウム、または有機化合物を用いて形成される。有機化合物の場合、塩は、有利には、有機化合物とフッ化水素酸との間の反応によって、反応混合物中に形成される。フッ素は、例えば、フッ化水素酸、フッ化アンモニウム、または二フッ化アンモニウムの水溶液の含浸によって導入され得る。
【0059】
ドーパントは、有利には、以下の、触媒に対する前記ドーパントの酸化物の割合で触媒前駆体に導入される:
− 前記ドーパントがホウ素である場合、0〜40%、好ましくは0〜30%、より好ましくは0〜20%、一層より好ましくは0〜15%、最も好ましくは0〜10%であり;ホウ素が存在する場合、最小限の量は、好ましく0.1重量%であり、
− 前記ドーパントがリンである場合、0.1〜20%、好ましくは0.1〜15%、一層より好ましくは0.1〜10%であり、
− 前記ドーパントがフッ素である場合、0〜20%、好ましくは0〜15%、一層より好ましくは0〜10%であり;フッ素が存在する場合、最小限の量は、好ましくは0.1重量%である。
【0060】
リンは、常に存在する。リンは、段階a)における触媒前駆体および場合による段階c)における乾燥触媒前駆体上での含浸によって、少なくとも部分的に(好ましくは完全に)導入される。同様のことが、好ましくは、他のドーパントについても適用される。しかし、上に記載のように、ドーパントは、担体の調製(成形を含む)時に部分的に、または完全に(リンを除く)、導入され得る。
【0061】
前記水素化脱水素基および場合によるドーパントが、成形された焼成担体の中または上に導入された後、有利には、乾燥段階b)が行われ、ここで、金属(単数または複数)酸化物(単数または複数)の前駆体として使用される金属塩の溶媒(溶媒は、一般的に水である)が、50〜180℃、好ましくは60〜150℃または65〜145℃、より好ましくは70〜140℃または75〜130℃の温度で除去される。このようにして得られた「乾燥触媒前駆体」を乾燥させる段階の後に、200℃超の温度での空気中の焼成の段階は決して行われない。有利には、それは、これらの温度範囲において、最高150℃の温度で行われ、その後に180℃超の温度での焼成は行われない。
【0062】
好ましくは、本発明による方法の段階a)において、前記「触媒前駆体」は、水素化脱水素基の前駆体(単数または複数)とリンとを含む溶液を、成形されかつ焼成された、アルミナをベースとする無定形担体上に乾式含浸させることによって得られる。この後に、前記「触媒前駆体」は、180℃未満、好ましくは50〜180℃、より好ましくは60〜150℃、最も好ましくは75〜130℃での温度で乾燥させられる。
【0063】
「乾燥触媒前駆体」は、段階b)の終わりにこのようにして得られる。
【0064】
本発明による方法の段階a)において、ホウ素およびフッ素の中から選択される少なくとも1種のドーパントを単独でまたは混合物として含む含浸溶液を調製することが可能である。
【0065】
より好ましくは、本発明による方法の段階a)における「触媒前駆体」は、リン前駆体の存在下に、水素化脱水素基の各元素の少なくとも1種の前駆体を含む含浸溶液を用いて調製される。無定形担体は、アルミナまたはシリカ−アルミナからなる。
【0066】
本発明の方法の段階c)によると、前記乾燥触媒前駆体は、少なくとも1種のC1−C4ジアルキルスクシナート(特に、ジメチルスクシナート)と酢酸とを含む含浸溶液によって含浸させられる。
【0067】
前記化合物は、有利には、本発明による方法の段階c)の含浸溶液中に、以下に相当する割合で、導入される:
− 触媒前駆体に含浸させられた第VIB族元素(単数または複数)当たりのジアルキルスクシナート(例えば、ジメチルスクシナート)のモル比は、0.15〜2モル/モル、好ましくは0.3〜1.8モル/モル、より好ましくは0.5〜1.5モル/モル、最も好ましくは0.8〜1.2モル/モルであり、
− 触媒前駆体に含浸させられた第VIB族元素(単数または複数)当たりの酢酸のモル比は、0.1〜5モル/モル、好ましくは0.5〜4モル/モル、より好ましくは1.3〜3モル/モル、最も好ましくは1.5〜2.5モル/モルである。このことは、水素化脱水素基がCoMoからなる場合に特に当てはまる。
【0068】
本発明の方法の段階c)によると、ジアルキルスクシナートと酢酸の組合せは、少なくとも1回の含浸段階、好ましくは含浸溶液を前記乾燥触媒前駆体上に含浸させる単一の段階によって、乾燥触媒前駆体上に導入される。
【0069】
前記組合せは、スラリー含浸または過剰含浸、乾式含浸、あるいは当業者に公知のあらゆる他の手段のいずれかによる、1つ以上の段階において、有利には沈着させられ得る。
【0070】
本発明の調製方法の段階c)の好ましい実施形態によると、段階c)は、単一の乾式含浸段階である。
【0071】
本発明の方法の段階c)によると、段階c)の含浸溶液は、少なくとも、C1−C4ジアルキル(特に、ジメチル)スクシナートと酢酸の組合せを含む。
【0072】
本発明の方法の段階c)において使用される含浸溶液は、当業者に公知のあらゆる非プロトン性溶媒によって補足され得るが、特に、トルエン、キシレンが挙げられる。
【0073】
本発明による方法段階c)において使用される含浸溶液は、当業者に公知のあらゆる極性溶媒によって補足され得る。使用される前記極性溶媒は、有利には、メタノール、エタノール、水、フェノール、シクロヘキサノールから構成される群より、単独でまたは混合物として選択される。本発明による方法の段階c)において使用される前記極性溶媒はまた、有利には、炭酸プロピレン、DMSO(dimethyl sulfoxide:ジメチルスルホキシド)、またはスルホランから構成される群より、単独でまたは混合物として選択され得る。好ましくは、極性プロトン性溶媒が使用される。通常の極性溶媒およびそれらの誘電率(dielectric constant)のリストが、著書“Solvents and Solvent Effects in Organic Chemistry”, C. Reichardt, Wiley-VCH, 3rd edition, 2003, pages 472-474に見出され得る。好ましくは、使用される溶媒はエタノールである。
【0074】
好ましくは、本発明による方法の段階c)において使用される含浸溶液中に、溶媒は存在しない。このことにより、工業規模での実施が促進させられる。それは、好ましくは、ジアルキルスクシナートと酢酸のみを含む。
【0075】
使用されるジアルキルスクシナートは、好ましくは、ジメチルスクシナート、ジエチルスクシナート、ジプロピルスクシナート、ジイソプロピルスクシナート、およびジブチルスクシナートからなる群に含まれている。好ましくは、使用されるC1−C4ジアルキルスクシナートは、ジメチルスクシナートまたはジエチルスクシナートである。より好ましくは、使用されるC1−C4ジアルキルスクシナートは、ジメチルスクシナートである。少なくとも1種のC1−C4ジアルキルスクシナートが使用され、好ましくは1種のみが使用され、好ましくはジメチルスクシナートが使用される。
【0076】
本発明の調製方法の段階d)によると、段階c)からの含浸触媒前駆体は、成熟段階に付される。それは、有利には、大気圧下、17〜50℃の範囲の温度で行われ、一般的に成熟時間は、10分〜48時間、好ましくは30分〜5時間で十分である。これより長い時間は除外されない。成熟時間を調整する簡単な方法は、ラマン分光法により、本発明による方法の段階c)からの含浸乾燥触媒前駆体におけるケギンヘテロポリアニオンの形成を特徴付けることである。好ましくは、改質ヘテロポリアニオンの量を改変することなく生産性を向上させるためには、成熟時間は、30分〜4時間の範囲である。より好ましくは、成熟時間は30分〜3時間の範囲である。
【0077】
本発明の調製方法の段階e)によると、段階d)からの触媒前駆体は、180℃未満の温度での乾燥段階に付され、その後に200℃超の温度での焼成段階は行われない。
【0078】
この段階の目的は、搬送可能、貯蔵可能で、かつ、操作可能な触媒、特に、水素化処理装置に装填するための触媒を得ることにある。それは、有利には、選択された本発明の実施形態によると、C1−C4ジアルキル(特にジメチル)スクシナートと酢酸の組合せの導入を可能にした任意の溶媒の全てまたは一部を除去することからなる。あらゆる場合において、特に、C1−C4ジアルキル(特にジメチル)スクシナートと酢酸の組合せのみが用いられる場合において、搬送、貯蔵、操作、または充填の段階中に押出物が互いにくっつくのを防ぐために、触媒には、乾燥した局面が与えられることとなる。
【0079】
本発明による方法乾燥段階e)は、有利には、当業者に公知のあらゆる技術を用いて行われる。それは、有利には、大気圧下または減圧下で行われる。好ましくは、この段階は大気圧下で行われる。
【0080】
この段階e)は、有利には、50〜180℃、好ましくは60〜170℃、より好ましくは80〜160℃の範囲の温度で行われる。有利には、それは、これらの温度範囲内において、最高160℃の温度で行われ(好ましい範囲は80〜180℃である)、その後に180℃超の温度での焼成は行われない。
【0081】
それは、有利には、空気または他のあらゆる高温ガスを用いて、横断床(traversed bed)内で行われる。好ましくは、乾燥が固定床内で行われる場合、用いられるガスは、空気、または不活性ガス、例えば、アルゴンまたは窒素等のいずれかである。より好ましくは、乾燥は、横断床内で窒素の存在下に行われる。
【0082】
この段階は、好ましくは30分〜4時間、より好ましくは1〜3時間の間にわたって続く。
【0083】
本発明による方法の段階e)の終わりに、乾燥触媒が得られるが、この触媒は、その後に、空気中、例えば200℃超の温度での焼成段階に付されない。
【0084】
段階d)または段階e)の終わりに得られた触媒は、990、974cm−1における最も強いバンド(ケギン型のヘテロポリアニオン)と、スクシナートに相当するバンド(ジメチルスクシナートについて、最も強いバンドは853cm−1に位置する)と、酢酸の特徴であるバンド(最も強いバンドは896cm−1に位置する)とを含むラマンスペクトルを有する。
【0085】
使用される前に、乾燥したあるいは焼成された触媒は、有利には、硫化触媒に転化され、その活性種が形成される。この活性化または硫化の段階は、当業者に公知の方法を用いて、有利には、スルホ−還元雰囲気中、水素および硫化水素の存在下に行われる。
【0086】
本発明による方法の段階e)の終わりに、得られた前記乾燥触媒はこのように、有利には、硫化段階f)に付され、中間の焼成段階は行われない。
【0087】
前記乾燥触媒は、有利には、現場外で(ex situ)であるいは現場で(in situ)硫化される。硫化剤は、H2Sガス、または、触媒を硫化する目的で炭化水素供給原料の活性化のために用いられる硫黄を含むあらゆる他の化合物である。前記硫黄含有化合物は、有利には、アルキルジスルフィド、例えば、ジメチルジスルフィド(DMDS)、アルキルスルフィド、例えば、ジメチルスルフィド、n−ブチルメルカプタン、第三ノニルポリスルフィド(tertiononylpolysulfide)タイプのポリスルフィド化合物、例えば、ARKEMA Companyによって販売されているTPS-37またはTPS-54、あるいは触媒の良好な硫化を得ることを可能とする、当業者に公知のあらゆる他の化合物の中から選択される。好ましくは、触媒は、硫化剤および炭化水素供給原料の存在下に現場硫化される。より好ましくは、触媒は、炭化水素供給原料にジメチルジスルフィドを添加したものの存在下に現場硫化される。
【0088】
最後に、本発明の別の目的は、水素化処理方法において、特に、水素化脱硫、水素化脱窒、水素化脱金属、芳香族化合物の水素化、および石油留分の水素化転化等のプロセスにおいて、本発明による触媒を使用することである。
【0089】
本発明による方法によって得られ、かつ、好ましくは事前に硫化段階f)に付された、乾燥触媒は、有利には、炭化水素供給原料、例えば、石油留分、石炭からの留分または天然ガスから製造された炭化水素の水素化処理反応のために、より具体的には、水素化、水素化脱窒、水素化脱金属、水素化脱硫、水素化脱金属または炭化水素供給原料の水素化転化の反応のために用いられる。
【0090】
そのような使用において、本発明による方法によって得られ、かつ、好ましくは事前に硫化段階f)に付された触媒は、従来技術の触媒に対して向上した活性を示す。これらの触媒はまた、有利には、接触分解供給原料の予備処理または残油の水素化脱硫、あるいはディーゼル燃料(ULSD:超低硫黄ディーゼル(Ultra Low Sulfur Diesel))の深度水素化脱硫の間に使用され得る。
【0091】
水素化処理方法において使用される供給原料としては、例えば、ガソリン、ガスオイル、真空ガスオイル、常圧残油、真空残油、常圧蒸留物、真空蒸留物、重質燃料オイル、オイル、ワックスおよびパラフィン、使用済石油、脱アスファルト化された、残渣または原油、熱的または触媒的転化方法に由来する供給原料が、単独でまたは混合物で使用される。処理される供給原料、特に上記の供給原料は、一般的に、ヘテロ原子、例えば硫黄、酸素、および窒素を含み、重質供給原料について、それらはほとんどの場合金属も含む。
【0092】
上記の炭化水素供給原料の水素化処理反応を実施する方法において適用される操作条件は、一般的に以下の通りである:温度は、有利には180〜450℃、好ましくは250〜440℃であり、圧力は、有利には0.5〜30MPa、好ましくは1〜18MPaであり、毎時空間速度は、有利には0.1〜20h−1、好ましくは0.2〜5h−1であり、液体供給原料の体積当たりの標準温度および圧力条件下に測定される水素の体積で表される水素/供給原料比は、有利には、50〜2000L/Lである。
【0093】
本明細書の以降における実施例には、従来技術の触媒に対する、本発明方法に従って調製された触媒についての相当な活性増強が示されているが、実施例により、本発明が明らかにされるが、しかしながら、本発明の限定するものではない。
【0094】
(実施例1:焼成触媒C1A(NiMoP/アルミナ)、クエン酸(citric acid:CA)とポリエチレングリコール(polyethylene glycol:PEG)とが添加された乾燥触媒C1E(NiMoP/アルミナ)(C1AおよびC1Eは、本発明に合致しない)、ならびに、酢酸とジメチルスクシナートとが添加された、乾燥触媒C1Bおよび乾燥触媒C1F(NiMoP/アルミナ)(C1BおよびC1Fは、本発明に合致する)の調製)
Condea Chemie GmbHによって販売されている、超微細平板状ベーマイトまたはアルミナゲルからなるマトリクスを使用した。このゲルを、66%の硝酸を含む水溶液(乾燥ゲルの重量(g)あたり酸7重量%)と混合し、次いで15分間にわたり混練した。混練後、得られた練粉状物(dough)を、径1.6mmの円筒状オリフィスを有するダイに通した。次いで、押出物を、120℃で終夜乾燥させ、次いで、600℃で2時間にわたり、乾燥空気の重量(kg)当たり50kgの水を含む湿潤空気下に焼成した。このようにして、比表面積が300m2/gである担体押出物を得た。X線回折の分析により、担体は、低結晶性の立方晶系のガンマアルミナのみからなることが示された。
【0095】
ニッケル、モリブデンおよびリンを、「押出物」の形態とされた上記アルミナ担体上に添加した。含浸溶液の調製は、酸化モリブデンおよびヒドロキシ炭酸ニッケルを、水溶液中のリン酸溶液中に高温溶解させることによる、最終触媒の乾燥物質の量に対する、ニッケル、モリブデンの酸化物の重量%および無水リン酸の重量%で表される約4/22.5/4の配合が得られた。最終触媒の乾燥物質の量に対する酸化ニッケルの重量%、酸化モリブデンの重量%、および無水リン酸の重量%で表される配合は、約4/22.5/4であった。乾式含浸後、押出物を、水飽和雰囲気中12時間にわたり成熟させるよう静置した後、90℃で終夜乾燥させた。このようにして得られた乾燥触媒前駆体を、C1と表示する。450℃での2時間にわたるC1の焼成により、焼成触媒C1Aが得られるに至った。酸化物の形態で表される、触媒C1およびC1Aの最終組成は、以下の通りである:MoO3=22.4±0.2(重量%)、NiO=4.1±0.1(重量%)、およびP2O5=4.0±0.1(重量%)。
【0096】
触媒C1Eの調製は、乾燥触媒前駆体C1に、クエン酸(CA)およびポリエチレングリコール(PEG)をエタノール溶液中に含有する溶液を含浸させることにより行った。含浸容積の体積は、乾燥触媒前駆体C1の細孔容積に等しくなるようにされた。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、共に10重量%であった。
【0097】
本発明に合致する触媒C1Bの調製は、乾燥触媒前駆体C1から、ジメチルスクシナートおよび酢酸の混合物を含有するエタノール溶液を乾式含浸させることにより行った。最終触媒上に10重量%の酢酸および10重量%のジメチルスクシナートが得られた。
【0098】
本発明に合致する触媒C1Fの調製は、同様の方法であるが、エタノールの非存在下に行われた。最終割合として、13重量%の酢酸および20重量%のジメチルスクシナートを得た。
【0099】
触媒は、次いで、空気中20℃での3時間の成熟段階を経て、その後、横断床タイプのオーブン内において、110℃での3時間にわたる熱処理を行った。
【0100】
(実施例2:蒸留ディーゼル燃料の水素化処理についての、触媒C1A(NiMoP/アルミナ)(本発明に合致しない)、触媒C1E(本発明に合致しない)、触媒C1B(本発明に合致する)の評価)
触媒(粒子サイズ0.8mmのSiC 10cm3と混合された押出物形態の30cm3の触媒)の硫化を、50バール、毎時空間速度2h−1、入口H2/HC比(体積流量)400標準L/Lで行った。硫化供給原料(Arkema CompanyからのDMDS 放出(Evolution)2%が添加されたディーゼル燃料)を、反応器が150℃に達した時にH2下に反応器に供給した。150℃での1時間の後、温度は、25℃/hの昇温率で220℃まで上昇し、次いで、12℃/hの昇温率で350℃の平坦域まで達し、この平坦温度を12時間にわたって維持した。
【0101】
硫化の後、330℃に温度を下げ、試験供給原料を注入した。触媒試験は、全圧50バールで、廃棄水素を基礎とし、毎時空間速度2h−1、入口H2/HC比400標準L/L(H2流量=24標準L/L、供給原料の流量=60cm3/h)、330℃および350℃で行われた。
【0102】
触媒のHDS性能を評価することができるように、かつ、H2Sの存在を克服することができるように、受入タンクを、10Lh−1の割合の窒素でストリッピングした。
【0103】
ここで使用されたディーゼル燃料は、アラビアンヘビー原油(Arabian heavy crude)に由来する。それは、0.89重量%の硫黄と、100重量ppmの窒素を含んでおり、そのPMT[(T5+2T50+4T95)/7]は324℃であり、その密度は0.848g/cm3であった。
【0104】
HDS活性は、以下の式によるHDS転化率から測定された:
【0105】
【数1】
【0106】
HDS転化率(%HDS)は、以下の式により与えられる:
【0107】
【数2】
【0108】
試験中、各温度で得られた流出物の密度を15℃で測定した。密度の進展を図1に示す。このグラフにより所与の密度を有するために要求される温度を決めることが可能となり、精製業者は、この性能を最も低い温度で提供することになる触媒を使用するために適切にアドバイスされる。図1において、本発明による触媒により、流出物の等密度条件下に、従来技術の触媒C1Aに対して約15℃操作温度を下げることが可能となることが理解され得る。この試験中の水素化脱硫について得られた結果を、以下の表に示す:
【0109】
【表1A】
【0110】
得られた結果により、ディーゼル燃料の水素化処理の場合、水素化脱粒並びに水素化脱芳香族(これは、流出物密度の進展に翻訳される)に関して、ジメチルスクシナートを酢酸と組み合わせて本発明による触媒に添加することが興味深いことが示される。実際、上記の表に示されているように、本発明による触媒では、得られたHDS活性は、高温で126(ULSDの範囲に相当する、すなわち、10重量ppmに近似する硫黄含有量)であって、一方、焼成触媒は100(基準)、従来技術である触媒C1Eでは106であった。
【0111】
(実施例3:水素化分解予備処理タイプの適用のための真空蒸留物水素化脱窒における触媒C1A(NiMoP/アルミナ)(本発明に合致しない)、C1F(本発明に合致する)の評価)
用いられた真空蒸留物の主要な特徴は、以下に与えられる;
20℃での密度: 0.9365
硫黄: 2.92重量%
全窒素: 1400重量ppm
模擬蒸留:
初留点(IP): 361℃
10%: 430℃
50%: 492℃
90%: 567℃
終点(EP): 598℃
試験は、横断固定床(traversed fixed bed)を備えた等温パイロット反応器で行われ、流体は上方に流通していた。直留ディーゼル燃料に2重量%のジメチルジスルフィドを添加したものによる、圧力下のプラント内の350℃での現場硫化の後、水素化処理試験を、以下の操作条件下に行った。
【0112】
全圧: 12MPa
触媒の体積: 40cm3
温度: 380℃
水素の流量: 40L/h
供給原料の流量: 40cm3/h
試験された触媒の触媒性能は、以下の表に与えられる。それらは、触媒C1Aの活性が100であると仮定し、かつ、それらは1.5次であると考えた、相対活性で表される。活性と水素化脱硫の転化率(%HDS)とを結び付ける関係式は以下の通りである:
【0113】
【数3】
【0114】
同じ関係式が、水素化脱窒(%HDNおよびAHDN)に適用される。
【0115】
加えて、各触媒により得られる、380℃未満の沸点を有するフラクションに対する総転化率も評価される。それは、模擬蒸留結果(ASTM D86法)から以下の式により表される:
【0116】
【数4】
【0117】
下記の表は、3種の触媒について得られた試験結果を与える。
【0118】
【表1B】
【0119】
得られた触媒結果により、水素化分解予備処理タイプの適用の場合、本発明による触媒は、本発明による触媒が水素化脱硫および水素化脱窒並びに転化率の増大を可能にする限りにおいて、焼成NiMoP触媒より効果的であることが示され、これはより驚くべきことである。
【0120】
(実施例4:触媒C2A(NiMoP/焼成シリカ−アルミナ)(本発明に合致しない)および触媒C2B(酢酸およびジメチルスクシナートが添加された乾燥NiMoP/シリカアルミナ)(本発明に合致する)の調製)
3.6/18/1.6の配合を有する2種のNiMoP触媒の調製がSASOLによって販売されるSIRALOXタイプのシリカ−アルミナ上に行われ、シリカ含有量は25%であった。NiMoP/シリカ−アルミナタイプの乾燥触媒前駆体の調製は、H3PO4によって可溶化させられたMoO3およびNi(OH)2前駆体から、2時間にわたり90℃で還流加熱することによって行われた。透明な溶液は、次いで、水の蒸発によって濃縮され、含浸容積に達し、次いで、周囲温度でシリカ−アルミナ上に含浸させられる。このように含浸させられた担体押出物は、水飽和閉鎖封入装置中で終夜成熟段階を経、次いで、それらは、ストーブ中120℃で24時間にわたって乾燥させられる。この触媒前駆体は、次いで、2つのバッチに分けられる;
− 第1のバッチ:このものは、触媒C2A(本発明に合致しない)を得るように、450℃で2時間にわたって空気中、横断固定床において焼成される;
− 第2のバッチ:このものは、本発明の手順に従って、酢酸およびジメチルスクシナートをジメチルスクシナート/酢酸のモル比0.58で含有する溶液を、初期の湿気の出現(これは、全ての細孔が満たされたことを示す)まで滴下して含浸させることによって用いられる。触媒は、次いで、成熟させるために3時間にわたって静置され、それは、次いで、125℃での2時間にわたる熱処理に付されて、本発明に合致する触媒C2Bが得られる。
【0121】
(実施例5:触媒C2A(焼成NiMoP/シリカ−アルミナ(本発明に合致しない)およびC2B(酢酸およびジメチルスクシナートが添加された乾燥NiMoP/シリカ−アルミナ(本発明に合致する)のアニリンの存在下でのトルエン水素化および真空蒸留物の穏やかな(mild)水素化分解の評価)
アニリン存在下でのトルエン水素化試験(「HTA」試験)の目的は、H2Sの存在下および水素圧下での担持型または塊状型の硫化触媒の水素化(HYDrogenizing:HYD)活性を評価することにある。シリカ−アルミナ上に担持された触媒の酸機能を特徴付ける異性化および分解は、NH3(アニリンの分解に由来する)の存在によって阻害される。アニリンおよび/またはNH3は、それ故に、酸−塩基反応を介して、担体の酸点(acid site)と反応することとなる。提示された全ての試験は、複数のミクロ反応器を並列に含むプラントにおいて行われる。「HTA」試験の間、触媒の硫化のためおよび適正な触媒試験段階のために同一の供給原料が用いられる。装填の前に、触媒は、調節処理され、すなわち、粉砕されかつサンプルの粒子サイズが2〜4mmの範囲にわたるように分別された。4cm3の粉砕された触媒が、4cm3のカーボランダム(SiC,500μm)と混合され、このものは、反応器に供給された。
【0122】
この試験のために使用された供給原料は、以下の通りである。
【0123】
トルエン: 20重量%
シクロヘキサン: 73.62重量%
DMDS(ジメチルジスルフィド): 5.88重量%(3.8重量% S)
アニリン: 0.5重量%(750ppm N)
触媒は、反応器に、その乾燥させられた、不活性の形態で供給される。活性化(硫化)は、プラント内で同一の供給原料により行われる。それは、DMDSの分解後に形成されたH2Sであり、酸化物相を硫化する。供給原料中に存在するアニリンの量は、分解後に、約750ppmのNH3を得るように選択される。
【0124】
トルエン水素化試験の操作条件は以下の通りである:
P=6MPa
HSV=2h−1(供給原料の流量=8cm3/h)
H2/HC=450NL/L,(H2流量=3.6NL/L)
T=350℃
転化されたトルエンの百分率が評価され、反応について一次を仮定して、活性は、以下の関係式によって推定される:
【0125】
【数5】
【0126】
(%HYDtoluene=転化されたトルエンの百分率)
触媒C2A(本発明に合致しない)は、0.52の活性を有し、触媒C2B(本発明に合致する)の活性度は、0.93であり、これは、相当な増大を表しており、穏やかな水素化分解についてのシリカ−アルミナ上のNiMoPタイプの触媒の水素化活性を増加させるための酢酸とジメチルスクシナートの組合せの興味が示されている。真空蒸留物タイプの供給原料に関する水素化処理試験は、転化率および水素化脱硫の増大を定量するために行われる。
【0127】
用いられる供給原料は、真空蒸留物タイプの供給原料であり、その主要な特徴は、以下の表において与えられる。
【0128】
【表1C】
【0129】
2〜4mmの範囲の長さの押出物の断片が試験される。4cm3の触媒が、反応器に、それらの酸化物である不活性な形態で供給される。活性化(硫化)は、プラント内で、試験を開始する前に、硫化供給原料と呼ばれる供給原料により行われる(直留ディーゼル燃料+2重量%のDMDS)。それは、DMDSの分解後に形成されたH2Sであり、触媒を硫化する。
【0130】
試験の間に適用される操作条件は以下の通りである:
P=6MPa
HSV=0.6h−1
出口H2/HC=480NL/L
T=380℃
この試験は、370+フラクションの370−への総転化率を評価することによって、触媒の分類を得ることを可能にする:370−への総転化率=370℃−流出物の重量%。
【0131】
触媒結果は、以下の表にまとめられる。本発明に合致する触媒C2Bは、触媒C2A(本発明に合致しない)に対する5%の転化率の増大および特にHDSの増大を可能にする。酢酸と組み合わされたジメチルスクシナートが本発明の手順に従って用いられる触媒に加えられる時に液体流出物のS含有量が60ppmから32ppmに変化しているからである。
【0132】
【表1D】
【0133】
これらの結果により、水素化の増大に加えて、本発明に合致する触媒は、類似する配合を有する焼成された従来の触媒に対して著しく穏やかな水素化分解の増大を得ることを可能にすることができることが示される。
【0134】
(実施例6:焼成触媒C3A(CoMoNiP/アルミナ)(本発明に合致しない)、乾燥触媒C3B(ジメチルスクシナートおよび酢酸が添加されたCoMoNiP/アルミナ)(本発明に合致する)および乾燥触媒C3C(ジメチルスクシナートが添加されたCoMoNiP/アルミナ)(本発明に合致しない)の調製)
実施例1において用いられたアルミナが、ここでも、式NiCoMoP/アルミナの「乾燥触媒前駆体」を調製するために用いられる。用いられた前駆体は、三酸化モリブデン、炭酸コバルト、ヒドロキシ炭酸ニッケルおよびリン酸である。含浸溶液は、単一段階において、これらの前駆体を還流下に加熱することによって調製される。ターゲットは、乾燥触媒(550℃での強熱減量後)に対する酸化物の重量%で表される含有量に対応する:NiO/CoO/MoO3/P2O5 1/2.3/15/4.4。含浸段階の終わりに、押出物は、成熟させるために終夜水飽和雰囲気中で静置され、次いで、2時間にわたって120℃でストーブ中に置かれる。その後、乾燥触媒前駆体が得られ、実施例4と同様に、3つのバッチに分けられる。
【0135】
− 第1のバッチは、450℃で3時間にわたって焼成され、触媒C3A(本発明に合致しない)が得られる;
− 第2のバッチは、本発明の手順に従って、酢酸およびジメチルスクシナートを含有する溶液を含浸させられる:溶液中のジメチルスクシナート/酢酸の比は0.58であり、乾燥触媒前駆体は、触媒前駆体の細孔がジメチルスクシナートおよび酢酸を含有する溶液により満たされたことを示す初期の湿気が出現するまで、この溶液によって含浸させられる。次いで2時間の成熟段階が行われた後、140℃での熱処理が1時間にわたって行われる。こうして得られた触媒は、触媒C3B(本発明に合致する)である。
【0136】
− 第3のバッチは、触媒前駆体の細孔がジメチルスクシナート溶液により満たされたことを示す初期の湿気が出現するまでジメチルスクシナートを含浸させられる。次いで、2時間の成熟段階が行われた後、140℃での熱処理が1時間にわたって行われる。このようにして得られた触媒は、触媒C3C(本発明に合致しない)である。
【0137】
(実施例7:焼成触媒C3A(CoMoNiP/アルミナ)(本発明に合致しない)、乾燥触媒C3B(ジメチルスクシナートおよび酢酸が添加されたCoMoNiP/アルミナ)(本発明に合致する)および乾燥触媒C3C(ジメチルスクシナートが添加されたCoMoNiP/アルミナ)(本発明に合致しない)の、トルエン水素化モデル分子試験における評価)
真空蒸留物および残渣の水素化処理等の適用において、水素化脱水素基は、これらの供給原料中の芳香族化合物の高い割合を考慮して、重要な役割を果たす。トルエン水素化試験は、それ故に、接触分解予備処理または残渣の水素化脱硫等の適用を意図する触媒の利益を知るために用いられた。
【0138】
上記実施例6において記載された触媒は、動力学的条件下に、Microcatタイプのパイロットプラントの横断固定床管型反応器(Vinciにより製造)において現場硫化され、流体は下方に流通する。水素化活性の測定は、硫化の直後に圧力下に空気中に戻すことなく行われ、炭化水素供給原料が触媒の硫化のために用いられた。
【0139】
硫化および試験の供給原料は、5.8%のジメチルジスルフィド(DMDS)、20%のトルエンおよび74.2%のシクロヘキサンからなっている(重量による)。
【0140】
硫化は、周囲温度で350℃までで行われ、温度上昇は2℃/分であり、HSV=4h−1であり、H2/HC=450NL/Lである。触媒試験は、350℃で行われ、HSV=2h−1であり、H2/HCは、硫化の同比と等価であり、最小限4個のサンプルが利用され、ガスクロマトグラフィーにより分析される。
【0141】
トルエン水素化反応における、等しい体積の触媒による安定化された触媒活性は、こうして測定される。
【0142】
詳細な活性の測定条件は以下の通りである:
全圧: 6.0MPa
トルエンの圧力: 0.37MPa
シクロヘキサンの圧力: 1.42MPa
メタンの圧力: 0.22MPa
水素の圧力: 3.68MPa
H2Sの圧力: 0.22MPa
触媒の体積: 4cm3(2〜4mm長の押出物)
毎時空間速度: 2h−1
硫化および試験の温度: 350℃
液体流出物サンプルが、ガスクロマトグラフィーによって分析される。未転化トルエンのモル濃度(T)およびその水素化生成物(メチルシクロヘキサン(MCC6)、エチルシクロペンタン(EtCC5)およびジメチルシクロペンタン(DMCC5))の濃度の測定は、以下によって定義されるトルエン水素化率XHYDを計算することを可能にする。
【0143】
【数6】
【0144】
トルエンの水素化反応は、適用される試験条件下に一次であり、反応器は、理想的なピストン反応器のように振る舞い、触媒の水素化活性AHYDは、以下の式を適用することによって計算される。
【0145】
【数7】
【0146】
以下の表により、実施例6において調製された触媒の水素化活性を比較することができる。
【0147】
【表1E】
【0148】
これらの触媒結果により、CoMoNiP/アルミナ上の酢酸(acetic acid:AA)とジメチルスクシナート(dimethyl succinate:DMSU)の組合せの水素化活性に関して、従来技術の焼成CoMoNiP/アルミナ触媒に対する乾燥CoMoNiP/アルミナ触媒前駆体(本発明に合致する)についての特定の効果が示される。この水素化活性の増大は、接触分解予備処理または残渣の水素化脱硫等の重質供給原料タイプの適用に特に有利である。
【0149】
ラマンスペクトルは、イオン化アルゴンレーザ(514nm)を備えた分散ラマンタイプの分光計により得られる。レーザビームは、50倍長作動距離対物レンズを備えた顕微鏡によってサンプルに焦点を合わされる。サンプルのレベルにおけるレーザの出力は、1mW程度である。サンプルによって放出されるラマンシグナルは、同じ対物レンズによって集められ、1800rpmネットワークによって分散させられ、次いで、CCD検出器によって集められる。得られたスペクトルの分解能は、0.5cm−1程度である。記録されたスペクトルゾーンは300〜1800cm−1である。捕捉時間は、記録される各ラマンスペクトルのために120秒に設定される。
【0150】
ラマン分析は、触媒C16〜C19について行われ、本発明に合致する触媒について、ラマンスペクトルにおいて、ケギンHPAの特徴である最も強いバンド、ジメチルスクシナートおよび酢酸の存在が示された。バンドの正確な位置、それらの形状およびそれらの相対強度は、スペクトル記録条件に応じて所定の程度で変動し得るが、この分子の特徴は残っている。有機化合物のラマンスペクトルは、なおまた、ラマンスペクトルデータベース(例えば、Spectral Database for Organic Compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi参照)において、または、生成物の供給者(例えば、www.sigmaaldrich.com参照)によって十分に実証される。
【0151】
ラマンスペクトルは、触媒C3B(DMSU+AA)およびC3C(高純度DMSU)について記録され、それらは、図2に与えられる。各測定は、押出物の3つの異なるゾーンにおいて繰り返された。触媒C3Bの場合、ケギンヘテロポリアニオンの特徴である990および971cm−1において2つのバンドの存在が見られ得る。より弱い強度のバンド(これもこれらの種に起因し得る)は、ケギンヘテロポリアニオンについての970、902、602cm−1におけるバンドである。これらの2つのスペクトルにおいて851cm−1におけるジメチルスクシナートの強いバンドの存在も観察され得る。他方、896cm−1におけるバンドは、本発明に合致する触媒についてのみ存在する。
【0152】
要するに、本発明に合致する触媒C3Bのラマンスペクトルは、ケギンヘテロポリアニオン、ジメチルスクシナートおよび酢酸の特徴であるバンドを呈する一方で、触媒C3Cは、ケギンヘテロポリアニオンおよびジメチルスクシナートの特徴であるバンドのみを呈する。
【0153】
(実施例8:触媒C1、C2、C3、C4(CoMoP/アルミナ)(本発明に合致しない)の調製)
Condea Chemie GmbH Companyによって販売される超微細平板状ベーマイトまたはアルミナゲルからなるマトリクスが用いられる。このゲルは、66%の硝酸を含有する水溶液と混合され(乾燥ゲルの重量(g)当たり7重量%の酸)、次いで、15分間にわたって混練される。混練の後、得られた練粉状物は、1.6mm径の円筒状オリフィスを有するダイに通される。押出物は、次いで、120℃で終夜乾燥させられ、次いで、600℃で2時間にわたって、乾燥空気の重量(kg)当たり水50kgを含有する湿潤空気下に焼成される。比表面積300m2/gを有する担体押出物がこのようにして得られる。X線回折分析により、担体は、低結晶性の立方晶系のガンマアルミナのみからなることが示される。
【0154】
コバルト、モリブデンおよびリンが、「押出物」形態とされる上記のアルミナ担体上に加えられる。含浸溶液は、酸化モリブデン(24.34g)および水酸化コバルト(5.34g)の水溶液中のリン酸溶液(7.47g)中の高温溶解によって調製される。乾式含浸の後、押出物は、水飽和雰囲気中に12時間にわたって成熟させるために静置され、次いで、それらは、90℃で終夜乾燥させられる。このようにして得られた乾燥触媒前駆体は、C1で表示される。450℃での2時間にわたる触媒前駆体C1の焼成により、焼成触媒C2が得られる。酸化物の形態で表される触媒C1およびC2の最終組成は以下の通りである:MoO3=22.5±0.2(重量%)、CoO=4.1±0.1(重量%)およびP2O5=4.0±0.1(重量%)。
【0155】
焼成触媒C2は、横断床装置に供給され、直留ディーゼル燃料に2重量%のジメチルジスルフィドが添加されたものによって硫化される。次いで、直留ディーゼル燃料および接触分解からのディーゼル燃料の混合物についてのHDS試験が、300時間にわたって行われる。試験後、用いられた触媒は、取り出され、集められ、および、還流下にトルエンにより洗浄され、2つのバッチに分離される。第1のバッチは、各温度工程につき酸素量を増加させて導入することによって制御された燃焼オーブン中で再生され、これは、コークス燃焼と関連する発熱状態(exothermy)を制限することを可能にする。最終の再生工程は450℃である。こうして再生された触媒は、XRDによって分析される。結晶化CoMoO4の存在の特徴である26°におけるラインの欠如が観察され得る。この触媒は、以降においてC3により表示される。洗浄された使用触媒の第2のバッチは、マッフル炉において400℃でコークス燃焼発熱状態制御なく再生される。再生後に行われたXRD分析により、結晶化CoMoO4の存在の特徴である26°において鋭いラインの存在が示される。さらに、C4によって表示されるこの触媒の色も非常に明るい青色である。
【0156】
(実施例9:触媒C5、C6、C7、C8、C9、C10、C11、C12(添加物を有するCoMoP/アルミナ)(本発明に合致しない)の調製)
触媒C5の調製は、乾燥触媒前駆体C1に、エタノール中にクエン酸(CA)およびポリエチレングリコール(PEG)を含有する溶液を含浸させることにより行われた。含浸溶液の体積は、乾燥触媒前駆体C1の細孔容積に等しくなるようにした。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、両方10重量%である。
【0157】
触媒C6の調製は、乾燥触媒前駆体C1に、エタノール中にクエン酸(CA)およびポリエチレングリコール(PEG)を含有する溶液を含浸させることにより行われた。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、それぞれ、4および10重量%である。
【0158】
触媒C7の調製は、焼成触媒C2に、エタノール中にクエン酸およびポリエチレングリコールを含有する溶液を含浸させることにより行った。含浸溶液の体積は、焼成触媒C2の細孔容積と等しくなるようにした。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、両方10重量%である。
【0159】
触媒C8の調製は、焼成触媒C2に、エタノール中にクエン酸およびポリエチレングリコールを含有する溶液を含浸させることにより行った。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、それぞれ、4および10重量%である。
【0160】
触媒C9の調製は、焼成触媒C2に、エタノール中に酢酸(AA)およびジメチルスクシナート(DMSU)を含有する溶液を含浸させることにより行った。酢酸(AA)およびジメチルスクシナート(DMSU)の所望の割合は、それぞれ、4および10重量%である。
【0161】
触媒C10の調製は、CoMoO4タイプの耐火性相を含まない再生触媒C3に、エタノール中にクエン酸(CA)およびポリエチレングリコール(PEG)を含有する溶液を含浸させることにより行った。含浸溶液の体積は、C3の細孔容積に等しくなるようにした。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、両方10重量%である。
【0162】
触媒C11の調製は、CoMoO4タイプの耐火性相を含まない再生触媒C3に、エタノール中にクエン酸(CA)およびポリエチレングリコール(PEG)を含有する溶液を含浸させることにより行った。クエン酸(CA)およびポリエチレングリコール(PEG)の所望の割合は、それぞれ、4および10重量%である。
【0163】
触媒C12の調製は、CoMoO4タイプの耐火性相を含まない再生触媒に、エタノール中に酢酸およびジメチルスクシナートを含有する溶液を含浸させることにより行った。酢酸およびジメチルスクシナートの所望の割合は、それぞれ、4および10重量%である。
【0164】
触媒C13の調製は、CoMoO4を含む再生触媒C4に、エタノール中にクエン酸およびポリエチレングリコールを含有する溶液を含浸させることにより行った。含浸溶液の体積は、C4の細孔容積に等しくなるようにした。クエン酸およびポリエチレングリコールの所望の割合は、両方10重量%である。
【0165】
触媒C14の調製は、CoMoO4を含む再生触媒C4に、エタノール中にクエン酸およびポリエチレングリコールを含有する溶液を含浸させることにより行った。クエン酸およびポリエチレングリコールの所望の割合は、それぞれ、4および10重量%である。
【0166】
触媒C15の調製は、CoMoO4を含む再生触媒C4に、エタノール中に酢酸およびジメチルスクシナートを含有する溶液を含浸させることにより行った。酢酸およびジメチルスクシナートの所望の割合は、それぞれ、4および10重量%である。
【0167】
触媒C5〜C15は、次いで、3時間の成熟段階に付され、その後、窒素下140℃での1時間の熱処理段階(乾燥)が行われる。
【0168】
(実施例10:触媒C16(添加物を有するCoMoP)(本発明に合致する)の調製)
触媒C16の調製は、乾燥触媒前駆体C1に、酢酸、ジメチルスクシナートおよびエタノールを含有する溶液を含浸させることにより行った。酢酸およびジメチルスクシナートの所望の割合は、それぞれ、4および10重量%である。触媒は、次いで、空気中周囲温度での3時間の成熟段階に付され、その後、窒素下140℃での1時間の熱処理段階(乾燥)が行われる。
【0169】
(実施例11:圧力下および硫化水素の存在中のシクロヘキサン中のトルエン水素化についての触媒C1〜C16の比較試験)
上記の触媒は、動力学的条件下に横断固定床管状反応器であるMicrocatタイプのパイロットプラント(Vinciにより製造)において現場硫化され、流体は、下方に流通する。水素化活性の測定は、硫化直後に、圧力下にかつ空気中に戻すことなく行われ、触媒を硫化するために炭化水素供給原料が用いられる。
【0170】
硫化および試験の供給原料は、5.8%のジメチルジスルフィド(DMDS)、20%のトルエンおよび74.2%のシクロヘキサンからなる(重量による)。
【0171】
硫化は、周囲温度から350℃までで行われ、温度上昇は2℃/分であり、HSV=4h−1であり、H2/HC=450NL/Lである。触媒試験は、350℃で行われ、HSV=2h−1であり、H2/HCは、硫化の同比と等しく、最小4つのサンプルが取られ、ガスクロマトグラフィーによって分析される。
【0172】
トルエン水素化反応における、等しい体積の触媒による安定化した触媒活性がそれ故に測定される。
【0173】
詳細な活性の測定条件は以下の通りである:
全圧: 6.0MPa
トルエンの圧力: 0.37MPa
シクロヘキサンの圧力: 1.42MPa
メタンの圧力: 0.22MPa
水素の圧力: 3.68MPa
H2Sの圧力: 0.22MPa
触媒の体積: 4cm3(2〜4mm長の押出物)
毎時空間速度: 2h−1
硫化および実験の温度: 350℃。
【0174】
液体流出物サンプルがガスクロマトグラフィーによって分析される。未転化トルエンのモル濃度(T)およびその水素化生成物(メチルシクロヘキサン(MCC6)、エチルシクロペンタン(EtCC5)およびジメチルシクロペンタン(DMCC5))の濃度の測定は、以下に定義されるトルエン水素化率XHYDを計算することを可能にする:
【0175】
【数8】
【0176】
トルエン水素化反応は、適用される試験条件下に一次であり、反応器は、理想的なピストン反応器のように振る舞うならば、触媒の水素化活性AHYDは、以下の式を適用することによって計算される:
【0177】
【数9】
【0178】
表1により、乾燥触媒前駆体C1(本発明に合致しない)からの添加物を有する触媒に対する水素化活性が比較される。ここでの活性は、基準(活性100%)として利用される最初の焼成触媒C2(本発明に合致しない)の活性に対する添加物を有する触媒の活性の比に等しい。
【0179】
【表1F】
【0180】
表1により、乾燥触媒前駆体C1(本発明に合致しない)は、焼成触媒C2(本発明に合致しない)より低い活性を有することが示される。添加物を有する触媒C5(本発明に合致しない)は、10%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を乾燥触媒C1に添加することによって調製されたものであるが、このものは試験され得なかった。押出物が、乾燥後に塊状に結束するからである。このことにより、初期の触媒が乾燥触媒である場合には過剰の酸および添加物は適していないことが示される。4%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を乾燥触媒C1に添加することによって調製された触媒C6(本発明に合致しない)は、初期乾燥触媒に対して40%の改良された活性を有する。しかしながら、初期乾燥触媒C1は、焼成された従来の触媒C2に対して25%のより低い活性度を有するので、触媒C2に対する触媒C6の相対的な水素化活性は、5%の増大を示すのみであり、これは、この試験の誤差の範囲を示している。したがって、乾燥触媒に、PEG+CAからなる組合せを添加することは興味深くはない。最後に、4%の酢酸(AA)および10%のジメチルスクシナート(DMSU)を添加することによって調製された触媒C16(本発明に合致する)は、初期乾燥触媒C1(本発明に合致しない)に対して84%の増大を呈する。従来から用いられる焼成触媒C1(本発明に合致しない)に対して、この触媒C16(本発明に合致する)の活性は、38%の増大を示し、これは、新しい触媒世代より高い(25〜30%の増大)。これらの触媒結果により、クエン酸(CA)とポリエチレングリコール(PEG)の組合せ(本発明に合致しない)に対する乾燥触媒上の酢酸(AA)とジメチルスクシナート(DMSU)の組合せ(本発明に合致する)の特定のかつ驚くべき効果が示される。
【0181】
同様に、表2により、乾燥触媒C1(本発明に合致しない)から調製された、添加物を有する触媒の相対的な水素化活性が比較される。ここでの水素化活性は、基準(活性100%)として利用される初期の焼成触媒C2(本発明に合致しない)の活性に対する添加物を有する触媒の活性の比に等しい。
【0182】
【表2】
【0183】
表2により、驚くべきことに、10重量%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を焼成触媒C2に添加することによって調製された、添加物を有する触媒C7(本発明に合致しない)の活性は、初期焼成触媒C2(本発明に合致しない)の活性に近く、あるいはこれに等価でさえあることが示され、このことは、初期触媒が乾燥触媒ではなかった場合に適していなかった過剰の酸および添加物は、焼成触媒の場合に有益性に乏しいことを示す。4%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を焼成触媒C2に添加することによって調製された触媒C8(本発明に合致しない)は、初期焼成触媒に対して14%改善された活性を有する。最後に、4%の酢酸(AA)および10%のジメチルスクシナート(DMSU)を添加することによって調製された触媒C9(本発明に合致しない)の活性は、初期焼成触媒C2(本発明に合致しない)の活性に近い(5%増大)。これらの触媒結果により、焼成触媒C2上(本発明に合致しない組合せ)ではなく、乾燥触媒前駆体(C1)のみ上の酢酸(AA)とジメチルスクシナート(DMSU)の組合せ(本発明に合致する組合せ)の特定の興味が示される。
【0184】
同様に、表3により、CoMoO4タイプの耐火性相を含有しない再生触媒C3から調製された、添加物を有する触媒(本発明に合致しない)の相対的な水素化活性が比較される。
【0185】
【表3】
【0186】
表3により、10重量%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を、CoMoO4タイプの耐火物相を含有しない再生触媒C3に添加することによって調製された、添加物を有する触媒C10(本発明に合致しない)の活性は、初期焼成触媒C2(本発明に合致しない)の活性に近く、さらにはこの活性と等価であることが示され、このことは、過剰の酸および添加物は、結晶化CoMoO4相を有しない再生触媒の場合にも有益性に乏しいことを示す。4%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を、CoMoO4タイプの耐火物相を含有しない再生触媒C3に添加することによって調製された触媒C11(本発明に合致しない)は、初期触媒C3(本発明に合致しない)に対して12%改善された活性を有し、これは、それらに、焼成された新規触媒C2に対して9%改善された活性を与える。最後に、4%の酢酸(AA)および10%のジメチルスクシナート(DMSU)を添加することによって調製された触媒C12(本発明に合致しない)の活性は、焼成触媒C2(本発明に合致しない)の活性に近い。これらの触媒結果により、CoMoO4タイプの結晶化相を有しない再生触媒C3(本発明に合致しない)上の酢酸(AA)とジメチルスクシナート(DMSU)の組合せに対する、乾燥触媒C1(本発明に合致する)上の同組合せの特定の興味が示される。
【0187】
同様に、表4により、CoMoO4を含有する再生触媒C4(本発明に合致しない)から調製された、添加物を有する触媒の相対的な水素化活性が比較される。CoMoO4の存在は、XRD分析によって確認される。
【0188】
【表4】
【0189】
表4により、10重量%のクエン酸(CA)および10重量%のポリエチレングリコール(PEG)を、CoMoO4を含有する再生触媒C4に添加することにより調製された、添加物を有する触媒C13(本発明に合致しない)の活性は、初期焼成触媒C2(本発明に合致しない)の活性に近いか、この活性に等価でさえあることが示され、このことにより、過剰の酸および添加物は、結晶化CoMoO4相を有する再生触媒の場合に有益であることが示される。実際に、4%のクエン酸(CA)および10%のポリエチレングリコール(PEG)を、CoMoO4タイプの結晶化相を含有する再生触媒(本発明に合致しない)に添加することによって調製された触媒C14(本発明に合致しない)は、初期触媒C4(本発明に合致しない)に対して不十分に改善された活性を有する。その活性は、焼成された新規な触媒C2の活性より低いままであるからである。最後に、4%の酢酸(AA)および10%のジメチルスクシナート(DMSU)を添加することによって調製された触媒C15(本発明に合致しない)の活性は、触媒C4(本発明に合致しない)の活性に近く(3%増大)、焼成された新規触媒C2に対してはるかにより低い。これらの触媒結果により、CoMoO4タイプの結晶化相を有する再生触媒C4(本発明に合致しない)上の酢酸(AA)とジメチルスクシナート(DMSU)の組合せに対する乾燥触媒C1(本発明に合致する)上の同組合せの特定の興味が示される。この組合せは、クエン酸(CA)とポリエチレングリコール(PEG)の組合せとは異なり、CoMoO4タイプの結晶化耐火物相を含有する触媒について特に効果がない。
【0190】
(実施例12:触媒C17およびC18(本発明に合致しない)、C19(本発明に合致する)の調製および触媒C2(本発明に合致しない)、C17およびC18(本発明に合致しない)、C16およびC19(本発明に合致する)のディーゼル燃料HDSについての比較)
触媒C17の調製は、触媒前駆体C1に高純度のジメチルスクシナート(DMSU)を含浸させることにより行った。これは、最終触媒上に30重量%のジメチルスクシナートを求めるのに等しい。触媒は、次いで、20℃で3時間の成熟段階に付され、その後、空気中140℃での1時間の熱処理が横断床タイプのオーブンにおいて行われる。この熱処理の終わりに得られた触媒は、C17により示される。この触媒は、本発明に合致していない。ジメチルスクシナートとの組合せで酢酸を含有していないからである。
【0191】
触媒C18は、触媒前駆体C1の細孔を酢酸により満たす以外は、同様の方法で調製された。触媒の重量に対して31重量%が得られる。成熟/熱処理段階はC17と同様である。
【0192】
触媒C19の調製は、触媒前駆体C1に、ジメチルスクシナートおよび酢酸の混合物のみを含有する溶液を含浸させることにより行った。ジメチルスクシナート/モリブデンのモル比は1.1であった。これは、ジメチルスクシナートおよび酢酸のそれぞれについて25および18重量%の最終触媒に対する含有量を求めるのに等しい。触媒は、次いで、周囲温度での3時間の成熟段階に付され、その後に、空気中140℃での1時間の熱処理が横断床タイプのオーブンにおいて行われる。この熱処理の終わりに得られた触媒は、C19により示される。この触媒は、本発明に合致する。
【0193】
触媒C2(本発明に合致しない)、C16(本発明に合致する)、C17(本発明に合致しない)およびC19(本発明に合致する)は、ディーゼル燃料HDSについて試験された。
【0194】
用いられたディーゼル燃料供給原料の特徴:
− 15℃での密度: 0.8522
− 硫黄: 1.44重量%
− 模擬蒸留:
・ IP : 155℃
・ 10% : 247℃
・ 50% : 315℃
・ 90% : 392℃
・ EP : 444℃
試験は、横断固定床を備える等温パイロット反応器において行われ、流体は、上方に流通する。試験されるディーゼル燃料に2重量%のジメチルスクシナートが添加されたものによる圧力下のプラント中での350℃での現場(in-situ)硫化の後、水素化脱硫試験は、以下の操作条件下に行われる:
全圧: 7MPa
触媒体積: 30cm3
温度: 340℃
水素流量: 24L/h
供給原料流量: 60cm3/h
試験された触媒の触媒性能は表3において与えられる。それらは、焼成触媒C2の活性が100であると仮定し、それらは1.5次であることを考慮した相対強度で表される。活性と水素化脱硫転化率(%HDS)を結び付ける関係式は以下の通りである:
【0195】
【数10】
【0196】
得られた結果は、表5において与えられる。
【0197】
【表5】
【0198】
表5により、明らかに、乾燥触媒前駆体上の酢酸とジメチルスクシナートの組合せの相乗効果および特定の興味が示される。実際に、触媒C17およびC18(本発明に合致しない)は、本発明に合致する触媒C16およびC19について得られた活性より低い活性を有する。触媒C19の場合に、触媒C16の場合より多量の添加物DMSUが含浸させられたが、しかしながら、実施例1におけるPEGおよびクエン酸の場合に観察されたこととは反対に活性増大は増加することに留意することはさらに興味深い。
【0199】
ラマンスペクトルは、イオン化アルゴンレーザ(514nm)を備えた分散ラマンタイプの分光計により得られる。レーザビームは、50倍長作動距離対物レンズを備えた顕微鏡によってサンプルに焦点を合わされる。サンプルのレベルにおけるレーザの出力は、1mW程度である。サンプルによって放出されるラマンシグナルは、同じ対物レンズによって集められ、1800rpmネットワークによって分散させられ、次いで、CCD検出器によって集められる。得られたスペクトルの分解能は、0.5cm−1程度である。記録されたスペクトルゾーンは300〜1800cm−1である。捕捉時間は、記録される各ラマンスペクトルのために120秒に設定される。
【0200】
ラマン分析は、触媒C16〜C19について行われ、本発明に合致する触媒について、ラマンスペクトルにおいて、ケギンHPA、ジメチルスクシナートおよび酢酸の特徴である最も強いバンドの存在が示された。バンドの正確な位置、それらの形状およびそれらの相対強度は、スペクトル記録条件に応じて所定の範囲で変動し得る一方で、この分子の特徴はそのままである。有機化合物のラマンスペクトルは、ラマンスペクトルデータベース(例えば、Spectral Database for Organic Compounds, http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi参照)において、または、生成物の供給者(例えば、www.sigmaaldrich.com参照)によってさらに十分に実証される。
【0201】
任意の理論によって拘束されることなく、非錯体形成有機添加物は、溶液中の触媒の細孔におけるヘテロポリアニオンの再形成を可能にすることが観察された。ジアルキルスクシナートおよび酢酸の混合物を含む溶液の含浸後のヘテロポリアニオンバンドの出現により強調されるのは典型的にはこの現象である。この効果は、「ヘテロポリアニオンの再形成」と呼ばれる。含浸溶液において最初に存在するが、それらは、新たに含浸させられた乾燥触媒前駆体上に存在しないからである;それらは、しかしながら、添加物の含浸後の成熟段階の間に再形成する。クエン酸存在下のPEGの場合、錯形成効果は、XRDによって容易に特徴付けられる結晶化相の消失を可能にするが、ヘテロポリアニオンは再形成されない。
【図面の簡単な説明】
【0202】
【図1】実施例2の試験中、各温度で得られた流出物の密度を15℃で測定し、密度の進展を示す。
【図2】触媒C3B(DMSU+AA)およびC3C(高純度DMSU)についてのラマンスペクトルを示す。
【特許請求の範囲】
【請求項1】
アルミナをベースとする無定形担体と、リンと、少なくとも1種のC1−C4ジアルキルスクシナートと、酢酸と、少なくとも1種の第VIII族元素および少なくとも1種の第VIB族元素を含む水素化脱水素基とを含む触媒であって、ラマンスペクトルが、少なくとも1種のケギンヘテロポリアニオンの特徴である990および/または974cm−1におけるバンドと、前記スクシナートの特徴であるバンドと、酢酸の特徴である896cm−1における主要バンドとを含む、触媒。
【請求項2】
ジアルキルスクシナートはジメチルスクシナートであり、触媒は、そのスペクトルにおいて、主要ラマンバンドを、ケギンヘテロポリアニオンの特徴である990および/または974cm−1、ジメチルスクシナートの特徴である853cm−1、および酢酸の特徴である896cm−1に有する、請求項1に記載の触媒。
【請求項3】
ジアルキルスクシナートは、ジエチルスクシナート、ジブチルスクシナート、またはジイソプロピルスクシナートである、請求項1に記載の触媒。
【請求項4】
アルミナからなる担体を含む、請求項1〜3のいずれか1つに記載の触媒。
【請求項5】
シリカ−アルミナからなる担体を含む、請求項1〜3のいずれか1つに記載の触媒。
【請求項6】
ホウ素および/またはフッ素を更に含む、請求項1〜5のいずれか1つに記載の触媒。
【請求項7】
水素化脱水素基はコバルトおよびモリブデンからなり、担体はアルミナからなる、請求項1〜4および6のいずれか1つに記載の触媒。
【請求項8】
水素化基は、元素の組み合わせ:ニッケル−モリブデン、ニッケル−コバルト−モリブデン、またはニッケル−モリブデン−タングステンから構成される群より選択される、請求項1〜6および7のいずれか1つに記載の触媒。
【請求項9】
硫化された、請求項1〜8のいずれか1つに記載の触媒。
【請求項10】
以下の連続する段階を含む、請求項1〜9のいずれか1つに記載の触媒を調製する方法:
a) アルミナをベースとする無定形担体に、水素化脱水素基の元素およびリンを含有する少なくとも1種の溶液を含浸させる少なくとも1回の段階、
b) 180℃未満の温度での乾燥段階であって、その後に焼成は行われない、段階、
c) 少なくとも1種のC1−C4ジアルキルスクシナートと、酢酸と、リン化合物が段階a)において完全に導入されていないならば少なくとも1種のリン化合物とを含む含浸溶液を含浸させる少なくとも1回の段階、
d) 成熟段階、
e) 180℃未満の温度での乾燥段階であって、その後に焼成段階は行われない、段階。
【請求項11】
水素化脱水素基は全て、段階a)において導入される、請求項10に記載の方法。
【請求項12】
段階c)は、溶媒の非存在下に行われる、請求項10または11に記載の方法。
【請求項13】
段階c)は、単独でまたは混合物で、メタノール、エタノール、水、フェノール、シクロヘキサノールから構成される群より選択される溶媒の存在下に行われる、請求項10〜12のいずれか1つに記載の方法。
【請求項14】
ジアルキルスクシナートおよび酢酸は、段階c)の含浸溶液に、ジアルキルスクシナート対触媒前駆体に含浸させられた第VIB族元素(単数または複数)のモル比0.15〜2モル/モルおよび酢酸対触媒前駆体に含浸させられた第VIB族元素(単数または複数)のモル比0.1〜5モル/モルに対応する量で導入される、請求項10〜13のいずれか1つに記載の方法。
【請求項15】
段階d)は17〜50℃の範囲の温度で行われる、請求項10〜14のいずれか1つに記載の方法。
【請求項16】
段階e)は、80〜160℃の範囲の温度で行われ、その後に180℃超の温度での焼成は行われない、請求項10〜15のいずれか1つに記載の方法。
【請求項17】
請求項10〜16のいずれか1つに記載の方法であって、以下の連続する段階を含む方法:
a) 前記担体に、水素化脱水素基の元素の全ておよびリンを含有する溶液を乾式含浸させる少なくとも1回の段階、
b) 75〜130℃の範囲の温度での乾燥段階であって、その後に焼成は行われない、段階、
c) ジメチルスクシナートと酢酸とを含む含浸溶液を乾式含浸させる少なくとも1回の段階、
d) 17〜50℃での成熟段階、
e) 80〜160℃の範囲の温度での乾燥段階であって、その後に焼成段階は行われない、段階。
【請求項18】
段階a)において、または、段階a)においてリンが完全には導入されなかったならば段階c)において導入されるリンの量は、触媒に対する酸化物の量で表して0.1〜20重量%、好ましくは0.1〜15重量%、より好ましくは0.1〜10重量%である、請求項10〜17のいずれか1つに記載の方法。
【請求項19】
段階e)は窒素下に行われる、請求項10〜18のいずれか1つに記載の方法。
【請求項20】
段階e)の終わりに得られた生成物は、硫化段階に付される、請求項10〜19のいずれか1つに記載の方法。
【請求項21】
請求項1〜9のいずれか1つに記載の触媒または請求項10〜20のいずれか1つに記載の方法によって調製された触媒の存在下に実施される、炭化水素供給原料の水素化処理方法。
【請求項22】
水素化処理は、水素化脱硫、水素化脱窒、水素化脱金属、芳香族化合物の水素化、または水素化転化である、請求項21に記載の方法。
【請求項23】
水素化処理は、深度ディーゼル燃料水素化脱硫である、請求項22に記載の方法。
【請求項1】
アルミナをベースとする無定形担体と、リンと、少なくとも1種のC1−C4ジアルキルスクシナートと、酢酸と、少なくとも1種の第VIII族元素および少なくとも1種の第VIB族元素を含む水素化脱水素基とを含む触媒であって、ラマンスペクトルが、少なくとも1種のケギンヘテロポリアニオンの特徴である990および/または974cm−1におけるバンドと、前記スクシナートの特徴であるバンドと、酢酸の特徴である896cm−1における主要バンドとを含む、触媒。
【請求項2】
ジアルキルスクシナートはジメチルスクシナートであり、触媒は、そのスペクトルにおいて、主要ラマンバンドを、ケギンヘテロポリアニオンの特徴である990および/または974cm−1、ジメチルスクシナートの特徴である853cm−1、および酢酸の特徴である896cm−1に有する、請求項1に記載の触媒。
【請求項3】
ジアルキルスクシナートは、ジエチルスクシナート、ジブチルスクシナート、またはジイソプロピルスクシナートである、請求項1に記載の触媒。
【請求項4】
アルミナからなる担体を含む、請求項1〜3のいずれか1つに記載の触媒。
【請求項5】
シリカ−アルミナからなる担体を含む、請求項1〜3のいずれか1つに記載の触媒。
【請求項6】
ホウ素および/またはフッ素を更に含む、請求項1〜5のいずれか1つに記載の触媒。
【請求項7】
水素化脱水素基はコバルトおよびモリブデンからなり、担体はアルミナからなる、請求項1〜4および6のいずれか1つに記載の触媒。
【請求項8】
水素化基は、元素の組み合わせ:ニッケル−モリブデン、ニッケル−コバルト−モリブデン、またはニッケル−モリブデン−タングステンから構成される群より選択される、請求項1〜6および7のいずれか1つに記載の触媒。
【請求項9】
硫化された、請求項1〜8のいずれか1つに記載の触媒。
【請求項10】
以下の連続する段階を含む、請求項1〜9のいずれか1つに記載の触媒を調製する方法:
a) アルミナをベースとする無定形担体に、水素化脱水素基の元素およびリンを含有する少なくとも1種の溶液を含浸させる少なくとも1回の段階、
b) 180℃未満の温度での乾燥段階であって、その後に焼成は行われない、段階、
c) 少なくとも1種のC1−C4ジアルキルスクシナートと、酢酸と、リン化合物が段階a)において完全に導入されていないならば少なくとも1種のリン化合物とを含む含浸溶液を含浸させる少なくとも1回の段階、
d) 成熟段階、
e) 180℃未満の温度での乾燥段階であって、その後に焼成段階は行われない、段階。
【請求項11】
水素化脱水素基は全て、段階a)において導入される、請求項10に記載の方法。
【請求項12】
段階c)は、溶媒の非存在下に行われる、請求項10または11に記載の方法。
【請求項13】
段階c)は、単独でまたは混合物で、メタノール、エタノール、水、フェノール、シクロヘキサノールから構成される群より選択される溶媒の存在下に行われる、請求項10〜12のいずれか1つに記載の方法。
【請求項14】
ジアルキルスクシナートおよび酢酸は、段階c)の含浸溶液に、ジアルキルスクシナート対触媒前駆体に含浸させられた第VIB族元素(単数または複数)のモル比0.15〜2モル/モルおよび酢酸対触媒前駆体に含浸させられた第VIB族元素(単数または複数)のモル比0.1〜5モル/モルに対応する量で導入される、請求項10〜13のいずれか1つに記載の方法。
【請求項15】
段階d)は17〜50℃の範囲の温度で行われる、請求項10〜14のいずれか1つに記載の方法。
【請求項16】
段階e)は、80〜160℃の範囲の温度で行われ、その後に180℃超の温度での焼成は行われない、請求項10〜15のいずれか1つに記載の方法。
【請求項17】
請求項10〜16のいずれか1つに記載の方法であって、以下の連続する段階を含む方法:
a) 前記担体に、水素化脱水素基の元素の全ておよびリンを含有する溶液を乾式含浸させる少なくとも1回の段階、
b) 75〜130℃の範囲の温度での乾燥段階であって、その後に焼成は行われない、段階、
c) ジメチルスクシナートと酢酸とを含む含浸溶液を乾式含浸させる少なくとも1回の段階、
d) 17〜50℃での成熟段階、
e) 80〜160℃の範囲の温度での乾燥段階であって、その後に焼成段階は行われない、段階。
【請求項18】
段階a)において、または、段階a)においてリンが完全には導入されなかったならば段階c)において導入されるリンの量は、触媒に対する酸化物の量で表して0.1〜20重量%、好ましくは0.1〜15重量%、より好ましくは0.1〜10重量%である、請求項10〜17のいずれか1つに記載の方法。
【請求項19】
段階e)は窒素下に行われる、請求項10〜18のいずれか1つに記載の方法。
【請求項20】
段階e)の終わりに得られた生成物は、硫化段階に付される、請求項10〜19のいずれか1つに記載の方法。
【請求項21】
請求項1〜9のいずれか1つに記載の触媒または請求項10〜20のいずれか1つに記載の方法によって調製された触媒の存在下に実施される、炭化水素供給原料の水素化処理方法。
【請求項22】
水素化処理は、水素化脱硫、水素化脱窒、水素化脱金属、芳香族化合物の水素化、または水素化転化である、請求項21に記載の方法。
【請求項23】
水素化処理は、深度ディーゼル燃料水素化脱硫である、請求項22に記載の方法。
【図1】

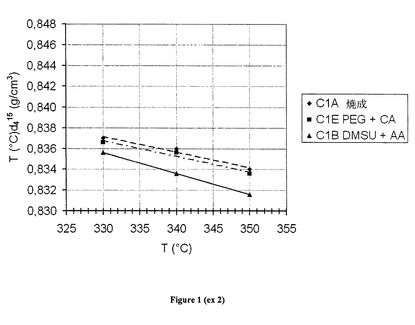
【図2】
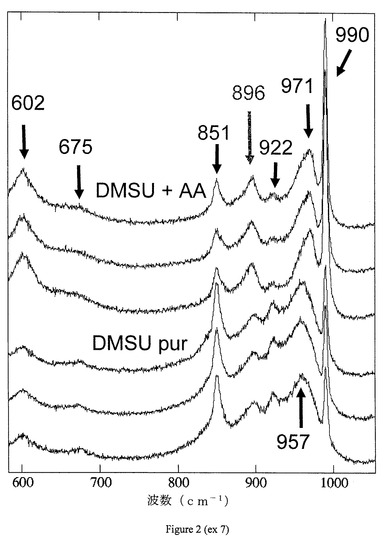
【図2】
【公表番号】特表2013−514169(P2013−514169A)
【公表日】平成25年4月25日(2013.4.25)
【国際特許分類】
【出願番号】特願2012−543861(P2012−543861)
【出願日】平成22年12月8日(2010.12.8)
【国際出願番号】PCT/FR2010/000819
【国際公開番号】WO2011/080407
【国際公開日】平成23年7月7日(2011.7.7)
【出願人】(591007826)イエフペ エネルジ ヌヴェル (261)
【氏名又は名称原語表記】IFP ENERGIES NOUVELLES
【出願人】(512157760)
【Fターム(参考)】
【公表日】平成25年4月25日(2013.4.25)
【国際特許分類】
【出願日】平成22年12月8日(2010.12.8)
【国際出願番号】PCT/FR2010/000819
【国際公開番号】WO2011/080407
【国際公開日】平成23年7月7日(2011.7.7)
【出願人】(591007826)イエフペ エネルジ ヌヴェル (261)
【氏名又は名称原語表記】IFP ENERGIES NOUVELLES
【出願人】(512157760)
【Fターム(参考)】
[ Back to top ]