
糖タンパク質の試料調製方法および分析方法
【課題】 糖タンパク質含有試料から糖鎖を遊離することなく糖タンパク質を分離する糖タンパク質の試料調製方法を提供する。
【解決手段】糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とからなる、糖タンパク質の試料調製方法である。
【解決手段】糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とからなる、糖タンパク質の試料調製方法である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、糖鎖が付加されたペプチドの分析方法に関し、より詳細には、糖タンパク質から糖鎖を遊離せずに糖タンパク質の試料を調製する方法、およびこのようにして得られた糖タンパク質を分析する方法などに関する。
【背景技術】
【0002】
プロテオミクスやグライコミクスなどの発展に伴い、MALDI−TOF MSやLC/MSによる分析が活発化し、種々のタンパク質や糖鎖、その他の分析結果がデータベース化されている。試料タンパク質や糖質のMALDI−TOF MSやLC/MSのデータとデータベースに収納されたデータとを対比させることで、短時間に試料タンパク質や糖質を同定することができる。
【0003】
このようなMALDI−TOF MSやLC/MSによって分析しうる試料として、糖タンパク質がある。糖タンパク質の生合成において、糖鎖は、タンパク質が翻訳される際に、または翻訳後に付加されるため、糖タンパク質の糖鎖構造が疾患や加齢、遺伝的背景による影響を受ける可能性があり、実際、腫瘍と特定の糖タンパク質との関係なども見出されている。このため、糖鎖をシグナルとして、診断や治療へ応用されることが期待されている。糖タンパク質には、糖鎖がアスパラギンに結合したN−結合型、糖鎖がセリンやスレオニン、ヒドロキシリジンなどに結合したO−結合型などが存在する。
【0004】
このような糖タンパク質をMALDI−TOF MSやLC/MSで分析する方法として、糖タンパク質からエンド型グリコシダーゼを使用して糖鎖を遊離させ、遊離した糖鎖を解析する方法がある(特許文献1)。未精製糖タンパク質をSDS変性ポリアクリルアミドゲル電気泳動によって分離し、泳動後のゲルから該当糖タンパク質部分を分取し、ペプチド:N−グリカナーゼ処理して糖鎖をタンパク質部分と切断して遊離させる。糖鎖はアルデヒド基を有するため、遊離糖鎖とヒドラジド基を官能基とするポリマー粒子とを反応させると、アルデヒド基とヒドラジド基とのヒドラゾン結合によって糖鎖がポリマー粒子に結合され、ついで、N−アミノオキシアセチルトリプトファニルアルギニンメチルエステルを反応させるとポリマー粒子のヒドラジド基に結合していたアルデヒド基がN−アミノオキシアセチルトリプトファニルアルギニンメチルエステルのアミノオキシル基に結合するというものである。特許文献1では、このようにして得た糖鎖をマトリックスと混合し、MALDI−TOF MS分析を行っている。なお、上記特許文献1で使用するペプチド:N−グリカナーゼ(PNGase)は、糖タンパク質や糖ペプチドからN型糖鎖を遊離させる酵素である。従って、N−結合型糖タンパク質の糖鎖の分析に適し、O−結合型糖タンパク質の糖鎖を分析することはできない。
【0005】
一方、このような糖タンパク質としてコラーゲンがあり、タンパク質を構成するアミノ酸としてガラクトシルヒドロキシリジン(galactosyl hydroxylysine: GHL)や、グルコシルガラクトシルヒドロキシリジン(glucosylgalactosyl hydroxylysine:GGHL)を含むものが存在する。コラーゲンの型、組織、生理的条件によって糖付加の割合は変化し、骨形成不全症での糖鎖の増加など、疾患との関係が報告されている。コラーゲンの糖鎖は、ヒドロキシリジンに結合するO−結合型糖鎖であり、このようなO−結合型糖タンパク質を簡便に解析する要請も高い。
【0006】
このような、タンパク質のセリンやスレオニン残基に糖鎖が結合するO−結合型糖鎖を分析する方法として、酢酸酸性条件で糖鎖を遊離させる方法がある(特許文献2)。実施例では糖タンパク質を2%酢酸/アセトニトリル中で80℃で1時間反応させて糖タンパク質から糖鎖を遊離させ、遊離した糖鎖の還元末端のアルデヒド基をそのままポリマー粒子のヒドラジド基と結合させ、ついで、ポリマー粒子にO−ベンジルヒドロキシルアミンを加えて2%酢酸/アセトニトリル中で80℃で1時間加熱し、ヒドラゾン−オキシム交換反応によって糖鎖の還元末端のアルデヒド基をO−ベンジルヒドロキシルアミンのアミノオキシル基に結合させている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2009−156587号
【特許文献2】特開2009−216609号
【発明の概要】
【発明が解決しようとする課題】
【0008】
糖タンパク質は、生体内において細胞同士の認識や化学物質の輸送などの役割を担い、細胞膜の構成に不可欠な要素であって、その機能解明は様々な病気が起きる仕組みを明らかにしたり治療法を開発するうえで手がかりになる。細胞内のリボソームで糖タンパク質を構成するタンパク質が合成され、さらにゴルジ体に運ばれ分泌される間に糖が運び込まれて糖タンパク質となり機能するものであるから、糖鎖の解析と共にタンパク質部分の解析も重要である。しかしながら現在のところ、コラーゲンの配列解析においては、糖鎖修飾部位はペプチドシークエンサでは検出することができないため、cDNA配列情報、アミノ酸分析、糖鎖分析などの結果を組合わせてその部位を推定しているに過ぎない。
【0009】
たとえば、特許文献1記載の方法は、SDS変性ポリアクリルアミドゲル電気泳動で糖タンパク質を精製したのちにN−グリコシダーゼ処理して糖鎖を遊離させ、遊離した糖鎖をマトリックスと混合しMALDI−TOF MS分析するものである。遊離した糖鎖の解析が目的であって、糖タンパク質やタンパク質部分を解析するものではない。
【0010】
また、特許文献2記載の方法も、特許文献1と同様に、糖タンパク質から糖鎖のみを分離し解析するものである。特に、特許文献2は、O−結合型糖鎖の切り出しに使用する無水ヒドラジンを使用したヒドラジン分解は毒性が強いこと、ヒドラジン分解で遊離された糖鎖試料には莢雑物が含まれており、ハイスループットで糖鎖を精製できる方法としてなされたものである。酢酸酸性条件で糖タンパク質から糖鎖を遊離すること、および遊離した糖鎖を酢酸酸性条件で前記ヒドラジド基含有ポリマー粒子と反応させると、糖鎖をポリマー粒子に結合させうることが特徴である。従って、当初から、糖鎖を遊離させることを目的とするものであって、特定の糖ペプチドを精製したり、タンパク質部分を解析するものではない。
【0011】
一方、糖タンパク質のタンパク質部分を解析するには、タンパク質部分を精製する必要がある。しかしながら、糖タンパク質は、N−結合型とO−結合型とによってタンパク質部分の分離方法や分離能が異なる。たとえば、上記した特許文献1では、N−結合型糖タンパク質からN−グリコシダーゼ処理によって糖鎖を遊離するものであり、特許文献2では、O−結合型糖タンパク質を酢酸酸性条件で反応させて糖鎖を遊離するものである。従って、糖タンパク質からタンパク質部分を遊離させて分析する方法は、N−結合型とO−結合型とによってタンパク質部分の分離方法を変える必要があり、操作が煩雑である。
【0012】
また、糖タンパク質をMALDI−TOF MSやLC/MSで分析するためには、目的とする糖タンパク質を精製する必要がある。上記特許文献1では、糖鎖を精製するために、糖鎖にヒドラジド基含有ポリマー粒子を結合させ、前記ポリマー粒子を洗浄することで莢雑物を除去しているが、糖タンパク質自体は、SDS変性ポリアクリルアミドゲル電気泳動で他と分離されるに過ぎない。特許文献2でも、糖タンパク質から遊離した糖鎖をヒドラジド基含有ポリマー粒子と結合させ、前記ポリマー粒子を洗浄することで莢雑物を除去しているが、糖タンパク質の分離は行っていない。
【0013】
糖タンパク質は、発現率その他において、疾患と特定の関係が観察される化合物である。従って、健常者と疾患者由来の糖タンパク質を解析することで、疾患の診断ができれば便利である。特に、糖タンパク質の解析のみならず、定量できることがより好ましい。
【0014】
上記現状に鑑み、本発明は、糖タンパク質を精製する方法を提供することを目的とする。
また、本発明は、精製した糖タンパク質をMALDI−TOF MSおよび/またはLC/MSで同定する方法を提供することを目的とする。
【0015】
加えて、上記分析方法によって、疾患の診断方法を提供することを目的とする。
【課題を解決するための手段】
【0016】
本発明者は、コラーゲンに含まれる糖タンパク質の分析方法について詳細に検討した結果、糖タンパク質を構成する糖鎖をガラクトースオキシダーゼ処理すると、糖タンパク質に含まれるガラクトース6位の−CH2OHが−CHOに酸化され、この酸化糖タンパク質とヒドラジド基を有するポリマー粒子とを反応させると、糖タンパク質−ポリマー粒子結合体が形成されること、糖タンパク質−ポリマー粒子結合体を洗浄した後に糖タンパク質とヒドラジド基との結合を切断すれば、糖タンパク質を遊離させ、糖タンパク質を高純度に精製できること、この糖タンパク質は、MALDI−TOF MSおよび/またはLC/MSにて糖タンパク質のまま分析でき、データベースと比較することで試料を同定できること、精製した糖タンパク質を標識化合物と反応させた後にLC/MSやMALDI−TOF MSで分析することで、糖タンパク質の定量が可能となること、健常者由来の糖タンパク質と特定疾患由来の糖タンパク質とを用いてそれぞれ糖タンパク質を精製し、それぞれに異なる標識化合物と反応させた後にMALDI−TOF MSおよび/またはLC/MSで分析すれば、健常者と特定疾患由来の糖タンパク質の含有量を評価し、疾患を診断できることを見出し、本発明を完成させた。
【0017】
すなわち本発明は、糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とからなる、糖タンパク質の試料調製方法を提供するものである。
【0018】
本発明は、前記糖タンパク質含有試料が、O−結合型糖タンパク質含有試料である、上記糖タンパク質の試料調製方法を提供するものである。
本発明は、前記O−結合型糖タンパク質が、コラーゲン由来であることを特徴とする、上記糖タンパク質の試料調製方法を提供するものである。
【0019】
本発明は、前記ポリマー粒子が、ヒドラジド基含有ポリマー粒子であることを特徴とする、上記糖タンパク質の試料調製方法を提供するものである。
本発明は、上記糖タンパク質の試料調製方法で得た糖タンパク質をMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法を提供するものである。
【0020】
本発明は、上記糖タンパク質の試料調製方法で得た糖タンパク質に標識化合物を結合させ、ついでMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法を提供するものである。
【0021】
本発明は、上記糖タンパク質の試料調製方法で得た糖タンパク質をイオン化促進標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法を提供するものである。
【0022】
本発明は、前記糖タンパク質含有試料が、健常者由来の糖タンパク質と特定疾患由来の糖タンパク質であり、上記糖タンパク質の試料調製方法で得た糖タンパク質を標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析するものであり、精製した健常者由来の糖タンパク質または特定疾患由来の糖タンパク質のいずれかに標識化合物を結合させ、他方に前記標識化合物に対応する安定同位体標識化合物を結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析し、前記標識化合物と前記安定同位体標識化合物との質量数の差から健常者と特定疾患由来の糖タンパク質の含有量を評価することを特徴とする、診断方法を提供するものである。
【発明の効果】
【0023】
本発明によれば、N−結合型糖タンパク質であるか、O−結合型糖タンパク質であるかを問わず、糖タンパク質を高収率で精製し、糖タンパク質の試料を調製することができる。
【0024】
本発明によれば、糖鎖を温和な条件で酸化して、糖タンパク質に含まれる−CH2OHを−CHOに酸化できるため、安全性および再現性に優れる。
本発明によれば、既存のデータと対比することで、試料糖タンパク質の同定を簡便に行うことができる。これにより、糖鎖のみならず、タンパク質部分も分析することができる。
【0025】
本発明によれば、健常者のデータと疾患由来の糖タンパク質の分析データとを比較することで、疾患の診断を行うことができる。
【図面の簡単な説明】
【0026】
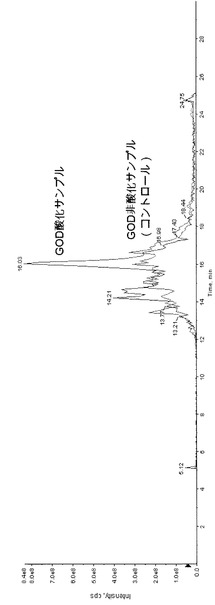
【図1】図1は、実施例3のGOD酸化サンプルとGOD非酸化サンプル(コントロール)との全イオン電流クロマトグラムのチャートである。
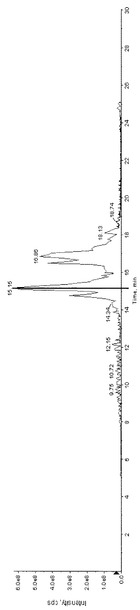
【図2】図2は、実施例3のGOD酸化サンプルの全イオン電流クロマトグラムのチャートである。
【図3】図3は、実施例3の15.081分のMSのチャートである。
【図4】図4は、実施例3の15.081分のMS/MSのチャートである。
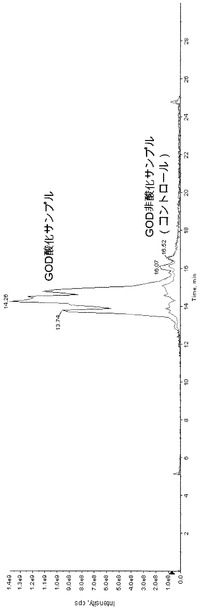
【図5】図5は、実施例4のGOD酸化サンプルとGOD非酸化サンプル(コントロール)との全イオン電流クロマトグラムのチャートである。
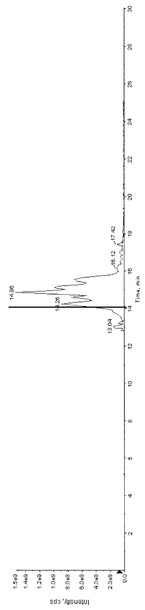
【図6】図6は、実施例4のGOD酸化サンプルの全イオン電流クロマトグラムのチャートである。
【図7】図7は、実施例4の14.078分のMSのチャートである。
【図8】図8は、実施例4の14.078分のMS/MSのチャートである。
【発明を実施するための形態】
【0027】
本発明の第一は、糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とからなる、糖タンパク質の試料調製方法である。
【0028】
糖タンパク質含有試料に含まれる莢雑物を除去し、分析対象の糖タンパク質を分離して試料を調製する。タンパク質や核酸、脂質など糖鎖以外の生体内物質にはアルデヒド基が含まれていないため、アルデヒド基と特異的に反応するポリマー粒子と結合させることで糖タンパク質のみを選択的に捕捉し、ポリマー粒子を洗浄することで莢雑物を除去し、その後、ポリマー粒子から糖タンパク質を分離することで、糖タンパク質の試料を調製する。得られた糖タンパク質試料は、MALDI−TOF MSやLC/MSなどで分析することができる。以下、本発明を詳細に説明する。
【0029】
(1)糖タンパク質
本発明において、糖タンパク質とは、タンパク質を構成するアミノ酸の一部に糖鎖が結合したものをいう。糖鎖がアスパラギンのアミノ基に糖が結合するN−結合型や、セリンやスレオニン、ヒドロキシリジンなどの水酸基に結合するO−結合型などが存在するが、いずれであってもよい。従来は、N−結合型糖タンパク質、O−結合型糖タンパク質について、それぞれ糖タンパク質から結合型の相違に由来した異なる方法で糖鎖を遊離させ、ついで遊離糖鎖を分析するものであったが、本発明は、糖タンパク質から糖鎖を遊離させることなく糖タンパク質のまま分析するため、いずれの結合型であるかを問わず、試料とすることができる。
【0030】
一方、好ましくは、O−結合型糖タンパク質である。従来の糖鎖を遊離する方法では、糖タンパク質から糖鎖を遊離することが困難であるため、O−結合型糖タンパク質の解析は、N−結合型糖タンパク質よりも遅滞していた。本発明によれば、N−結合型糖タンパク質と同様に簡便に分析できるため、特にO−結合型糖タンパク質に有効である。
【0031】
本発明で好適に使用できるO−結合型糖タンパク質としては、I〜XXIX型コラーゲン;アグリカン、パールカン、シンデカン、デコリンなどのプロテオグリカン;カドヘリン;ムチンなどを例示することができる。特に好ましくは、I〜XXIX型コラーゲンである。糖鎖の精製や濃縮にはレクチンがよく使用されるが、コラーゲンの糖鎖に結合するレクチンは知られておらず、コラーゲン由来の糖タンパク質の精製方法や濃縮方法は存在しなかった。一方、コラーゲンは、体内に多量に存在するタンパク質であり、単一のタンパク質として精製する方法が確立しており、および糖鎖が1糖または2糖と単純であってその構造も判明しており、データベースの分析結果と対比することで、同定が容易だからである。
【0032】
対象とする糖タンパク質の糖鎖の長さも限定されるものではない。一般には、糖が1〜20個、好ましくは1〜12個、特に好ましくは1〜8個、更に好ましくは1〜5個結合した糖鎖を有する糖タンパク質の精製に好適である。
【0033】
また、糖タンパク質のタンパク質部分の長さも限定されない。ただし、MALDI−TOF MSやLC/MSにおいて、既存のデータと対比する場合には、タンパク質部分は対象とする糖タンパク質を特定できる限度で、短いことが好ましい。一般には、糖タンパク質をトリプシンなどによって切断されるものを好適に使用することができる。
【0034】
(2)ポリマー粒子
本発明は、糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化し、当該−CHOを介してポリマー粒子と反応させるものである。従って、ポリマー粒子としては、−CHOと結合しうる反応基を有するポリマー粒子を広く使用することができ、例えば、ヒドラジド基含有ポリマー粒子、アミノ基含有ポリマー粒子、オキシルアミノ基含有ポリマー粒子、セミチオカルバジド基含有ポリマー粒子などを好ましく使用することができる。オキシルアミノ基とアルデヒド基との反応によって生じるオキシム結合やヒドラジド基とアルデヒド基との反応によって生じるヒドラゾン結合は、酸処理などによって容易に切断されるため、ポリマー粒子によって糖タンパク質を捕捉したのち、簡便に糖タンパク質をポリマー粒子から簡単に切り離すことができる点で好ましい。
【0035】
前記ポリマー粒子は、固体やゲル粒子であってもよい。ポリマー粒子に糖タンパク質を捕捉させたのち、遠心分離やろ過などの手段によって他と分離して回収することができる。また、ポリマー粒子をカラムに充填して用いてもよい。カラムに充填して用いる方法は、特に連続操作化に好適である。
【0036】
ポリマー粒子の形状は特に限定しないが、球状またはそれに類する形状が好ましい。ポリマー粒子が球状の場合、平均粒径は好ましくは0.05〜1000μmであり、より好ましくは0.05〜200μmであり、さらに好ましくは0.1〜200μmであり、最も好ましくは0.1〜100μmである。この範囲で通液性に優れ、かつ糖タンパク質の捕捉性に優れるからである。
【0037】
ポリマー粒子は、遠心分離やろ過などの手段で回収する時にのみ固体粒子あるいはゲル粒子であってもよい。たとえば温度、pHなどの環境変化によって溶解性が変化するポリマーを用い、溶媒に溶解した状態の該ポリマーに糖鎖の−CHOと特異的に反応する官能基を介して糖鎖を捕捉させ、溶解性を変化させて該ポリマーを沈殿させ、回収してもよい。環境によって溶解性が変化するポリマーとして、たとえばポリ(N−イソプロピルアクリルアミド)を挙げることができる。ポリ(N−イソプロピルアクリルアミド)分子の少なくとも一部に糖タンパク質の−CHOと特異的に反応する官能基を導入することで、上記のような糖タンパク質の捕捉が可能となる。
【0038】
このようなポリマー粒子は、市販品であってもよい。例えば、住友ベークライト株式会社製のBlotGlyco、BIO−RAD社製のAffiGel HZ Hydrazide Gelなどを使用することができる。
【0039】
(3)糖タンパク質含有試料
本発明で使用する糖タンパク質含有試料は、上記したN−結合型糖タンパク質やO−結合型糖タンパク質それ自体でもよいが、タンパク質部分の一次構造の解析が進んでいるO−結合型糖タンパク質のコラーゲンを分析する場合で説明する。なお、コラーゲンのタンパク質部分を構成するアミノ酸には、ヒドロキシリジンにガラクトシル基が結合したガラクトシルヒドロキシリジン(galactosyl hydroxylysine: GHL)、グルコシル−ガラクトシル基が結合したグルコシルガラクトシルヒドロキシリジン(glucosylgalactosyl hydroxylysine:GGHL)が存在することが知られている。
【0040】
コラーゲンは、現在、I〜XXIX型が知られており、上記GHLやGGHLの含有率は、コラーゲン型によって大きく異なる。本発明で使用するコラーゲンとしてはいずれであってもよく、新たに見出されるコラーゲンであってもよい。コラーゲンは、魚や豚、牛などの生皮、腱、骨などを形成する主要タンパク質であるが、生体内に含まれる大部分は水に不溶性である。本発明では、動物の皮や骨等の原料に含まれるコラーゲンをプロテアーゼなどの酵素を添加して可溶化し、酸やアルカリを添加して可溶化したものを使用することができる。
【0041】
更に、糖タンパク質含有試料としては、コラーゲンを、例えば、トリプシンやキモトリプシンなどでタンパク質部分をより短鎖に切断したものを使用することができる。コラーゲンはアミノ酸配列が解明されているため、タンパク質部分を分析すれば、トリプシンなどで切断した糖タンパク質がコラーゲンのいずれの部位であるかを、容易に特定することができる。更に、例えばSDS−PAGE(SDS変性ポリアクリルアミドゲル電気泳動)や、等電点電気泳動とSDS−PAGEを組み合わせた二次元電気泳動によって分離したものを糖タンパク質含有試料とすることもできる。また、泳動後のゲルを例えばクーマシーブリリアントブルーなどの染色剤を用いて染色したのち、ニトロセルロース、PVDF(ポリフッ化ビニリデン)などのメンブレンに転写してもよい。転写は一般的なブロッティング装置を用いて行うことができる。染色はメンブレンへの転写の後に行ってもよい。
(4)糖タンパク質の試料調製方法
本発明では、上記糖タンパク質含有試料を使用し、
糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、
前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、
前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、
前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とによって糖タンパク質試料を調製することができる。
【0042】
(i)酸化工程
上記糖タンパク質を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化し、これに前記ポリマー粒子を反応させる。N−結合型の糖タンパク質にN−グリコシダーゼを作用させて糖鎖を分離すると、遊離した糖鎖の還元末端にアルデヒド基が生じ、このアルデヒド基を介して上記ポリマー粒子と糖鎖とが結合する。しかしながら、本発明では、糖タンパク質に含まれる6位の−CH2OHを−CHOに酸化させるため、糖タンパク質から糖鎖を遊離させることなく、このアルデヒド基を介してポリマー粒子と糖タンパク質とを結合させることができる。
【0043】
−CH2OHを−CHOに酸化させる方法に限定は無いが、ガラクトースオキシダーゼを使用することが安全かつ簡便である。一般には、37℃で6〜24時間反応させる。
(ii)糖タンパク質−ポリマー粒子結合工程
次いで、前記酸化糖タンパク質含有試料を、−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る。ポリマー粒子の添加量は、ポリマー粒子に結合される−CHOと結合しうる反応基の量によって異なり、使用するポリマー粒子の前記反応基量に応じて適宜選択することができる。一般には、室温で3〜24時間、反応させればよい。
【0044】
糖タンパク質を特異的に捕捉するポリマー粒子によって糖タンパク質を捕捉する際の反応系のpHは、好ましくは2〜9、より好ましくは2〜7であり、さらに好ましくは2〜6である。pH調整のためには、各種緩衝液を用いることができる。糖鎖捕捉時の温度は、好ましくは4〜60℃、より好ましくは10〜50℃、さらに好ましくは15〜40℃である。反応時間は適宜設定することができる。ポリマー粒子をカラムに充填して試料溶液を通過させてもよい。
【0045】
(iii)洗浄工程
糖タンパク質−ポリマー粒子結合体を洗浄することで、糖タンパク質含有試料に含まれる糖タンパク質以外の成分を除去することができる。洗浄液は、糖タンパク質−ポリマー粒子結合体から糖タンパク質を切断せず、かつ莢雑物と親和性を有するものを広く使用することができる。たとえば、水、緩衝液、有機溶媒、界面活性剤を含む水または緩衝液などを適宜組み合わせて用いることが好ましい。特に好ましい形態は、界面活性剤を含む水または緩衝液で十分に洗浄したのち、有機溶剤で洗浄し、最後に水で洗浄する方法である。これらの洗浄により、非特異的吸着物がポリマー粒子表面から除去される。
【0046】
なお、ポリマー粒子に存在する−CHOと結合しうる反応基をキャップすることが好ましい。例えば、ポリマー粒子が−CHOと結合しうる反応基としてヒドラジド基を有する場合には、ポリマー粒子を洗浄した後に、無水酢酸を添加し、室温で30分間反応させる。その後、再度、ポリマー粒子を洗浄することが好ましい。
【0047】
(iv)糖タンパク質遊離工程
糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離させることで、糖タンパク質を精製し、純度の高い糖タンパク質試料とすることができる。
【0048】
遊離方法は、ポリマー粒子の前記反応基の種類に応じて適宜選択することができるが、糖タンパク質とポリマー粒子との結合以外を切断するものであってはならない。本発明では、揮発性酸処理により糖タンパク質をポリマー粒子から分離することができる。このような揮発性酸によれば、糖タンパク質をポリマー粒子から分離できると共に、脱塩等の必要なくそのままMALDI−TOF MSおよび/またはLC/MS分析を行うことができるからである。本発明で使用できる揮発性酸としては、ギ酸、酢酸、トリフルオロ酢酸、塩酸などを例示することができる。ギ酸処理の場合は、例えば、濃度0.1〜1質量%のギ酸溶液で温度60〜80℃で、0.5〜2時間、より好ましくは0.1〜1時間反応させる。
【0049】
(5)LC/MS分析
本発明では、上記で調製した糖タンパク質試料をLC/MS分析し、糖タンパク質を同定することができる。LC/MSの分析結果を、各種サンプルの分析結果を集積したデータベースと比較することで、糖鎖修飾部位を推定することができる。
【0050】
さらに、上記糖タンパク質試料をそのまま使用するほか、例えば糖タンパク質の前記−CHOにヒドラジン(H2NNH2)や重水素処理ヒドラジン(D2NND2)を結合して標識すれば、質量数が2異なる位置にピークが出現することを利用し、サンプル間での比較定量を行うことができる。このように、一方に標識化合物を、他方に前記標識化合物に対応する安定同位体標識化合物を結合することで、質量数の違いによるサンプル間の比較定量を行う事ができる。安定同位体としては、重水素(D)のほか、13Cや、15N、18O等を例示することができる。
【0051】
また、標識化合物として、ヒドラジンのほか、ヒドラジド基を含む物質として、ジラール試薬P(ピリジニウム−1−アセトヒドラジド・クロリド)、ジラール試薬T(ベタインヒドラジド・ヒドロクロライド)、5−ジメチルアミノナフタレン−1−スルフォニルヒドラジン(ダンシルヒドラジン);2−ヒドラジノピリジン;9−フルオレニルメチルカルバゼート(Fmoc ヒドラジン);ベンジルヒドラジン;4,4−ジフルオロ−5,7−ジメチル−4−ボラ−3a,4a−ジアザ−s−インダセン−3−プロピオノ酸,ヒドラジド;2−(6,8−ジフルオロ−7−ヒドロキシ−4−メチルクマリン)アセトヒドラジン;7−ジエチルアミノクマリン−3−カルボキシリック酸;ヒドラジン(DCCH);フェニルヒドラジン;1−ナフタレンアセトヒドラジン;2−ヒドラジノベンゾイックアシド;フェニルアセチックヒドラジン;ビオチンヒドラジンなどがある。また、−CHOと反応する反応基として、アミノオキシ基がある。アミノオキシ基を含む物質として、O−ベンジルヒドロキシアミン;O−フェニルヒドロキシルアミン;O−(2,3,4,5,6−ペンタフルオロベンジル)ヒドロキシルアミン;O−(4−ニトロベンジル)ヒドロキシルアミン;2−アミノオキシピリジン;2−アミノオキシメチルピリジン;4−[(アミノオキシアセチル)アミノ]ベンゾイックアシドメチルエステル;4−[(アミノオキシアセチル)アミノ]ベンゾイック酸エチルエステル;4−[(アミノオキシアセチル)アミノ]ベンズアミノ酸n−ブチルエステル;アミkノオキシ−ビオチンなどを例示することができる。
【0052】
更に、このような標識として、ジラール試薬Pや重水素処理ジラール試薬Pなどの標識化合物を使用すれば、ヒドラジンと同様に−CHOと反応して糖タンパク質に結合する標識となり、上記と同様に質量数が5異なる位置にピークが出現することを利用してサンプル間で比較定量を行うことができる。かつベンゼン環の中に4級アミンが含まれるためMSでのイオン化が促進され、感度を向上させることもできる。
【0053】
このような標識化合物は、糖タンパク質の1.5質量倍以上、より好ましくは3質量倍以上、さらに好ましくは5質量倍以上である。pH調整のためには、各種緩衝液を用いることができる。反応系の温度は,好ましくは4〜90℃、より好ましくは4〜70℃、さらに好ましくは30〜80℃であり、最も好ましくは40〜80℃である。
【0054】
(6)MALDI−TOF MS分析
本発明では、上記した糖タンパク質の試料調製方法で得た糖タンパク質をそのまま用いてMALDI−TOF MS分析を行うことができる。また、LC/MS分析の試料と同様に、例えば糖タンパク質の前記−CHOにヒドラジン(H2NNH2)や重水素処理ヒドラジン(D2NND2)を結合して標識して試料とすることができる。更に、ジラール試薬Pや重水素処理ジラール試薬P、その他の標識化合物を付加して試料とすることができる。これにより、高感度にMALDI−TOF MS分析を行うことができる。
【0055】
本発明では、上記で調製した糖タンパク質試料をMALDI−TOF MS分析し、糖タンパク質を同定することができる。MALDI−TOF MSの分析結果を、各種サンプルの分析結果を集積したデータベースと比較することで、糖鎖修飾部位を推定することができる。
【0056】
(7)診断方法
本発明の診断方法は、糖タンパク質含有試料を使用し、健常者由来の糖タンパク質と特定疾患由来の糖タンパク質との質量の差から、糖タンパク質を定量し、疾患を診断するものである。
【0057】
上記した糖タンパク質の試料調製方法で得た糖タンパク質を標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析するものであり、精製した健常者由来の糖タンパク質または特定疾患由来の糖タンパク質のいずれかに標識化合物を結合させ、他方に前記標識化合物に対応する安定同位体標識化合物を結合させる。得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析すると、標識化合物と前記標識化合物に対応する安定同位体標識化合物との質量数の差から健常者と特定疾患由来の糖タンパク質の含有量を評価することができる。
【実施例】
【0058】
次に実施例を挙げて本発明を具体的に説明するが、これらの実施例は何ら本発明を制限するものではない。
LC/MS測定条件
1.装置
高速液体クロマトグラフ:1200 Series(Agilent Technologies)、
質量分析装置:3200QTRAP(AB Sciex)
2.GHL、GGHLの測定
(i)HPLC条件
分析カラム:ZIC−HILIC 5μm, 2.1mmi.d.×150mm(SeQuant)、
移動相:A液;0.1%酢酸、5mM 酢酸アンモニウム、
B液;100%アセトニトリル、
グラジエント条件:0−5分:A液10%、5−10分:A液10−50%、10−25分:A液50−80%、25.1−30分:A液10%、
流速:0.2mL/min、
カラム温度:40℃、
注入量:10μl、
(ii)質量分析条件
イオン化:ESI、ポジティブ、
分析モード:Multiple Reaction Monitoring(MRM)モード、
イオンスプレー電圧:4.5kV、
イオンソース温度:600℃、
検出チャンネル(m/z):GHL 325.1→163.2、酸化GHL 323.1→163.2、
GGHL 487.2→163.3、酸化GGHL 485.2→163.3、
3.コラーゲン糖タンパク質の測定
(i)HPLC条件
分析カラム:Ascentis Express 2.7mm, 2.1mmi.d.×150mm(SUPELCO)、
移動相:A液;0.1%ギ酸、B液;100%アセトニトリル、
グラジエント条件:0−5分:A液98%、5−20分:A液98−50%、20.1−25分:A液10%、25.1−30分:A液98%、
流速:0.2mL/min、
カラム温度:25℃、
注入量:10μl
(ii)質量分析条件
イオン化:ESI、ポジティブ、
分析モード:Information Dependent Acquisition(IDA)モード、
イオンスプレー電圧:5.5kV、
イオンソース温度:600℃、
データベース検索ソフト:ProteinPilotソフトウェア3.0(AB Sciex)
(実施例1)
ガラクトシルヒドロキシリジン(GHL)10nmolを、100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液に溶解し、ついで、ガラクトースオキシダーゼ(SIGMA社製、商品名「galactose oxidase」)を50単位添加し、37℃で24時間処理した。この一部を採取し、溶液Aとした。
【0059】
その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)を250mg添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。この一部を採取し、溶液Bとした。
【0060】
一方、ガラクトシルヒドロキシリジン(GHL)10nmolを、100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液に溶解し、37℃で24時間処理した。この一部を採取し、対照溶液Aとした。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)を250mg添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。この一部を採取し、対照溶液Bとした。
【0061】
得られた溶液A、溶液B、対照溶液A、対照溶液BをそれぞれLC/MS(AB Sciex社製、商品名「3200QTRAP」)にて、GHLと酸化GHLの強度を上記測定条件にて測定した。GHLと酸化GHLの強度および対照溶液Aに対する%、溶液Aに対する%の結果を表1に示す。
【0062】
表1より、対照溶液AのGHL強度に対する溶液AのGHL強度は、1.6%であり、ガラクトースオキシダーゼによる酸化反応率は、98.4%(=100−1.6)となった。また、溶液Aの酸化GHLの強度に対する溶液Bの酸化GHLの強度で示される、酸化GHLの回収率は、401000×100/922000=43.5%であった。
【0063】
【表1】

(実施例2)
グルコシルガラクトシルヒドロキシリジン(GGHL)10nmolを、100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液に溶解し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。この一部を採取し、溶液Aとした。
【0064】
その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)を250mg添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。この一部を採取し、溶液Bとした。
【0065】
一方、グルコシルガラクトシルヒドロキシリジン(GGHL)10nmolを、100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液に溶解し、37℃で24時間処理した。この一部を採取し、対照溶液Aとした。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)を250mg添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。この一部を採取し、対照溶液Bとした。
【0066】
得られた溶液A、溶液B、対照溶液A、対照溶液BをそれぞれLC/MS(AB Sciex社製、商品名「3200QTRAP」)にて、GGHLと酸化GGHLの強度を上記条件で測定した。GGHLと酸化GGHLの強度および対照溶液Aに対する%、溶液Aに対する%の結果を表2に示す。
【0067】
表2より、対照溶液AのGGHL強度に対する溶液AのGGHL強度は、88.9%であり、ガラクトースオキシダーゼによる酸化反応率は、11.1%(=100−88.9)となった。
【0068】
また、溶液Aの酸化GGHLの強度に対する溶液Bの酸化GGHLの強度で示される、酸化GGHLの回収率は、22900×100/52900=43.3%であった。
【0069】
【表2】
(実施例3)
(1)脱脂した牛皮に40倍量(w/v)の0.5M酢酸を添加し、2〜3時間攪拌させて牛皮を十分に膨潤させ、ポリトロンホモジナイザーで牛皮をホモジナイズした。ペプシン量が組織の1/20量になるように、組織の10倍量のペプシン−0.5M酢酸を加えて一晩保持した後、遠心分離した。上清に終濃度1Mとなるように塩化ナトリウムを加えて塩析させ、遠心分離により沈殿を回収した。沈殿を最初に水、次に5mM酢酸に対して透析した後、リン酸緩衝液でpH7.5に調整して等電点沈殿させた。さらに沈殿を5 mM酢酸に対して透析し、フィルター濾過してI型コラーゲンを得た。
【0070】
このI型コラーゲン1mgを100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液で60℃、30分加熱処理して変性させ、トリプシン(SIGMA社製、商品名「Trypsin」)50μgを投与し、37℃、16時間処理した。トリプシン処理後、トリプシンインヒビター(SIGMA社製、商品名「Trypsin−chymotrypsin inhibitor」)を100μg添加し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)250mgを添加し、室温で6時間、転倒攪拌した。
【0071】
ついで、ヒドラジドゲルを、100mM酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液、1.5M NaCl、メタノール、蒸留水の順に洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。
【0072】
得られた反応液をLC/MSにて上記条件で分析した。全イオン電流クロマトグラムを図1に示す。なお、ガラクトースオキシダーゼで酸化しないサンプルについて同様に操作した。GOD酸化サンプルとGOD非酸化サンプル(コントロール)とを同一チャートに示す。
【0073】
また、GOD酸化サンプルの全イオン電流クロマトグラムを図2に示す。図2の15.081分のピークのMS分析の結果を図3に、MS/MS分析の結果を図4に示す。MS/MSの結果から、データベース検索ソフト(ProteinPilotソフトウェア3.0(AB Sciex))で検索したところ、下記配列がヒットした。この同定された糖鎖修飾部位は、これまでに報告されている部位と一致するものである。なお、配列表の配列番号1において、右から4番目のリジンがGHLとして同定された。
【0074】
(実施例4)
ウシの大腿骨から軟骨をメスで削りとり、10%塩化ナトリウムで洗浄した後、組織の9.9倍量の0.1N塩酸に浸して一晩置いた。氷冷しながらポリトロンホモジナイザーでホモジナイズし、ペプシン量が組織の1/100量となるように組織と等量のペプシン0.1N塩酸を加え、一晩攪拌して可溶化処理を行った。遠心分離により得た上清を1N水酸化ナトリウムでpH8.0に調整して等電点沈殿させ、遠心分離して沈殿を回収した。得られた沈殿の量に対して10倍量の0.01N塩酸を加えて溶解し、そこに10%濃度となるように塩化ナトリウムを加えて塩析させ、遠心分離により沈殿を回収した。沈殿を最初に水、次に5mM酢酸に対して透析した後、フィルター濾過してII型コラーゲンを得た。
【0075】
このII型コラーゲン1mgを100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液で60℃、30分加熱処理して変性させ、トリプシン50μgを投与し、37℃、16時間処理した。トリプシン処理後、トリプシンインヒビター(SIGMA社製、商品名「Trypsin−chymotrypsin inhibitor」)を100μg添加し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)250mgを添加し、室温で6時間、転倒攪拌した。
【0076】
ついで、ヒドラジドゲルを、100mM酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液、1.5M NaCl、メタノール、蒸留水の順にした後、1%ギ酸200μlを添加し、80℃で1時間反応した。
【0077】
得られた反応液をLC/MSにて上記条件で分析した。全イオン電流クロマトグラムを図5に示す。なお、ガラクトースオキシダーゼで酸化しないサンプルについて同様に操作した。GOD酸化サンプルとGOD非酸化サンプル(コントロール)とを同一チャートに示す。
【0078】
また、GOD酸化サンプルの全イオン電流クロマトグラムを図6に示す。図6の14.078分のピークのMS分析の結果を図7に、MS/MS分析の結果を図8に示す。MS/MSの結果から、データベース検索ソフト(ProteinPilotソフトウェア3.0(AB Sciex))で検索したところ、下記配列がヒットした。この同定された糖鎖修飾部位は、従来知られているものではなかった。なお、配列表の配列番号2において、左から9番目のリジンがGHLとして同定された。
【0079】
(実施例5)
実施例3と同様に操作してI型コラーゲンを得た。実施例3と同様に、このI型コラーゲン等電点沈殿物1mgを100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液で60℃、30分加熱処理して変性させ、トリプシン50μgを投与し、37℃、16時間処理した。トリプシン処理後、トリプシンインヒビター(SIGMA社製、商品名「Trypsin−chymotrypsin inhibitor」)を100μg添加し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)250mgを添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液、1.5M NaCl、メタノール、蒸留水の順に洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。
【0080】
次いで、上記反応液液に、0.1mMジラール試薬P、または重水素処理ジラール試薬Pを加え、80℃で1時間反応させた。さらに、形成されたヒドラゾン結合を1mMシアノ水素化ホウ素ナトリウムにより室温で1時間還元することにより、アミンの形にして安定化させた。得られた反応液を100mMリン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH 7.2溶液に対して透析後、LC−MS(AB Sciex社製、商品名「3200QTRAP」)で分析した。
【0081】
(実施例6)
健常者由来コラーゲンおよび骨形成不全症患者由来コラーゲンを使用した。実施例3と同様に、コラーゲン1mgを100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液で60℃、30分加熱処理して変性させ、トリプシン50μgを投与し、37℃、16時間処理した。トリプシン処理後、トリプシンインヒビター(SIGMA社製、商品名「Trypsin−chymotrypsin inhibitor」)を100μg添加し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)250mgを添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。
【0082】
健常者由来サンプルには0.1mMジラール試薬P、骨形成不全症患者由来サンプルには0.1mM重水素処理ジラール試薬Pを加え、80℃で1時間反応させた。さらに、形成されたヒドラゾン結合を1mMシアノ水素化ホウ素ナトリウムにより室温で1時間還元することにより、アミンの形にして安定化させた。得られた反応液を100mMリン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH 7.2溶液に対して透析後、LC−MS(AB Sciex社製、商品名「3200QTRAP」)で分析した。
【産業上の利用可能性】
【0083】
本発明によれば、簡便な操作で糖タンパク質を解析することができ、有用である。
【技術分野】
【0001】
本発明は、糖鎖が付加されたペプチドの分析方法に関し、より詳細には、糖タンパク質から糖鎖を遊離せずに糖タンパク質の試料を調製する方法、およびこのようにして得られた糖タンパク質を分析する方法などに関する。
【背景技術】
【0002】
プロテオミクスやグライコミクスなどの発展に伴い、MALDI−TOF MSやLC/MSによる分析が活発化し、種々のタンパク質や糖鎖、その他の分析結果がデータベース化されている。試料タンパク質や糖質のMALDI−TOF MSやLC/MSのデータとデータベースに収納されたデータとを対比させることで、短時間に試料タンパク質や糖質を同定することができる。
【0003】
このようなMALDI−TOF MSやLC/MSによって分析しうる試料として、糖タンパク質がある。糖タンパク質の生合成において、糖鎖は、タンパク質が翻訳される際に、または翻訳後に付加されるため、糖タンパク質の糖鎖構造が疾患や加齢、遺伝的背景による影響を受ける可能性があり、実際、腫瘍と特定の糖タンパク質との関係なども見出されている。このため、糖鎖をシグナルとして、診断や治療へ応用されることが期待されている。糖タンパク質には、糖鎖がアスパラギンに結合したN−結合型、糖鎖がセリンやスレオニン、ヒドロキシリジンなどに結合したO−結合型などが存在する。
【0004】
このような糖タンパク質をMALDI−TOF MSやLC/MSで分析する方法として、糖タンパク質からエンド型グリコシダーゼを使用して糖鎖を遊離させ、遊離した糖鎖を解析する方法がある(特許文献1)。未精製糖タンパク質をSDS変性ポリアクリルアミドゲル電気泳動によって分離し、泳動後のゲルから該当糖タンパク質部分を分取し、ペプチド:N−グリカナーゼ処理して糖鎖をタンパク質部分と切断して遊離させる。糖鎖はアルデヒド基を有するため、遊離糖鎖とヒドラジド基を官能基とするポリマー粒子とを反応させると、アルデヒド基とヒドラジド基とのヒドラゾン結合によって糖鎖がポリマー粒子に結合され、ついで、N−アミノオキシアセチルトリプトファニルアルギニンメチルエステルを反応させるとポリマー粒子のヒドラジド基に結合していたアルデヒド基がN−アミノオキシアセチルトリプトファニルアルギニンメチルエステルのアミノオキシル基に結合するというものである。特許文献1では、このようにして得た糖鎖をマトリックスと混合し、MALDI−TOF MS分析を行っている。なお、上記特許文献1で使用するペプチド:N−グリカナーゼ(PNGase)は、糖タンパク質や糖ペプチドからN型糖鎖を遊離させる酵素である。従って、N−結合型糖タンパク質の糖鎖の分析に適し、O−結合型糖タンパク質の糖鎖を分析することはできない。
【0005】
一方、このような糖タンパク質としてコラーゲンがあり、タンパク質を構成するアミノ酸としてガラクトシルヒドロキシリジン(galactosyl hydroxylysine: GHL)や、グルコシルガラクトシルヒドロキシリジン(glucosylgalactosyl hydroxylysine:GGHL)を含むものが存在する。コラーゲンの型、組織、生理的条件によって糖付加の割合は変化し、骨形成不全症での糖鎖の増加など、疾患との関係が報告されている。コラーゲンの糖鎖は、ヒドロキシリジンに結合するO−結合型糖鎖であり、このようなO−結合型糖タンパク質を簡便に解析する要請も高い。
【0006】
このような、タンパク質のセリンやスレオニン残基に糖鎖が結合するO−結合型糖鎖を分析する方法として、酢酸酸性条件で糖鎖を遊離させる方法がある(特許文献2)。実施例では糖タンパク質を2%酢酸/アセトニトリル中で80℃で1時間反応させて糖タンパク質から糖鎖を遊離させ、遊離した糖鎖の還元末端のアルデヒド基をそのままポリマー粒子のヒドラジド基と結合させ、ついで、ポリマー粒子にO−ベンジルヒドロキシルアミンを加えて2%酢酸/アセトニトリル中で80℃で1時間加熱し、ヒドラゾン−オキシム交換反応によって糖鎖の還元末端のアルデヒド基をO−ベンジルヒドロキシルアミンのアミノオキシル基に結合させている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2009−156587号
【特許文献2】特開2009−216609号
【発明の概要】
【発明が解決しようとする課題】
【0008】
糖タンパク質は、生体内において細胞同士の認識や化学物質の輸送などの役割を担い、細胞膜の構成に不可欠な要素であって、その機能解明は様々な病気が起きる仕組みを明らかにしたり治療法を開発するうえで手がかりになる。細胞内のリボソームで糖タンパク質を構成するタンパク質が合成され、さらにゴルジ体に運ばれ分泌される間に糖が運び込まれて糖タンパク質となり機能するものであるから、糖鎖の解析と共にタンパク質部分の解析も重要である。しかしながら現在のところ、コラーゲンの配列解析においては、糖鎖修飾部位はペプチドシークエンサでは検出することができないため、cDNA配列情報、アミノ酸分析、糖鎖分析などの結果を組合わせてその部位を推定しているに過ぎない。
【0009】
たとえば、特許文献1記載の方法は、SDS変性ポリアクリルアミドゲル電気泳動で糖タンパク質を精製したのちにN−グリコシダーゼ処理して糖鎖を遊離させ、遊離した糖鎖をマトリックスと混合しMALDI−TOF MS分析するものである。遊離した糖鎖の解析が目的であって、糖タンパク質やタンパク質部分を解析するものではない。
【0010】
また、特許文献2記載の方法も、特許文献1と同様に、糖タンパク質から糖鎖のみを分離し解析するものである。特に、特許文献2は、O−結合型糖鎖の切り出しに使用する無水ヒドラジンを使用したヒドラジン分解は毒性が強いこと、ヒドラジン分解で遊離された糖鎖試料には莢雑物が含まれており、ハイスループットで糖鎖を精製できる方法としてなされたものである。酢酸酸性条件で糖タンパク質から糖鎖を遊離すること、および遊離した糖鎖を酢酸酸性条件で前記ヒドラジド基含有ポリマー粒子と反応させると、糖鎖をポリマー粒子に結合させうることが特徴である。従って、当初から、糖鎖を遊離させることを目的とするものであって、特定の糖ペプチドを精製したり、タンパク質部分を解析するものではない。
【0011】
一方、糖タンパク質のタンパク質部分を解析するには、タンパク質部分を精製する必要がある。しかしながら、糖タンパク質は、N−結合型とO−結合型とによってタンパク質部分の分離方法や分離能が異なる。たとえば、上記した特許文献1では、N−結合型糖タンパク質からN−グリコシダーゼ処理によって糖鎖を遊離するものであり、特許文献2では、O−結合型糖タンパク質を酢酸酸性条件で反応させて糖鎖を遊離するものである。従って、糖タンパク質からタンパク質部分を遊離させて分析する方法は、N−結合型とO−結合型とによってタンパク質部分の分離方法を変える必要があり、操作が煩雑である。
【0012】
また、糖タンパク質をMALDI−TOF MSやLC/MSで分析するためには、目的とする糖タンパク質を精製する必要がある。上記特許文献1では、糖鎖を精製するために、糖鎖にヒドラジド基含有ポリマー粒子を結合させ、前記ポリマー粒子を洗浄することで莢雑物を除去しているが、糖タンパク質自体は、SDS変性ポリアクリルアミドゲル電気泳動で他と分離されるに過ぎない。特許文献2でも、糖タンパク質から遊離した糖鎖をヒドラジド基含有ポリマー粒子と結合させ、前記ポリマー粒子を洗浄することで莢雑物を除去しているが、糖タンパク質の分離は行っていない。
【0013】
糖タンパク質は、発現率その他において、疾患と特定の関係が観察される化合物である。従って、健常者と疾患者由来の糖タンパク質を解析することで、疾患の診断ができれば便利である。特に、糖タンパク質の解析のみならず、定量できることがより好ましい。
【0014】
上記現状に鑑み、本発明は、糖タンパク質を精製する方法を提供することを目的とする。
また、本発明は、精製した糖タンパク質をMALDI−TOF MSおよび/またはLC/MSで同定する方法を提供することを目的とする。
【0015】
加えて、上記分析方法によって、疾患の診断方法を提供することを目的とする。
【課題を解決するための手段】
【0016】
本発明者は、コラーゲンに含まれる糖タンパク質の分析方法について詳細に検討した結果、糖タンパク質を構成する糖鎖をガラクトースオキシダーゼ処理すると、糖タンパク質に含まれるガラクトース6位の−CH2OHが−CHOに酸化され、この酸化糖タンパク質とヒドラジド基を有するポリマー粒子とを反応させると、糖タンパク質−ポリマー粒子結合体が形成されること、糖タンパク質−ポリマー粒子結合体を洗浄した後に糖タンパク質とヒドラジド基との結合を切断すれば、糖タンパク質を遊離させ、糖タンパク質を高純度に精製できること、この糖タンパク質は、MALDI−TOF MSおよび/またはLC/MSにて糖タンパク質のまま分析でき、データベースと比較することで試料を同定できること、精製した糖タンパク質を標識化合物と反応させた後にLC/MSやMALDI−TOF MSで分析することで、糖タンパク質の定量が可能となること、健常者由来の糖タンパク質と特定疾患由来の糖タンパク質とを用いてそれぞれ糖タンパク質を精製し、それぞれに異なる標識化合物と反応させた後にMALDI−TOF MSおよび/またはLC/MSで分析すれば、健常者と特定疾患由来の糖タンパク質の含有量を評価し、疾患を診断できることを見出し、本発明を完成させた。
【0017】
すなわち本発明は、糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とからなる、糖タンパク質の試料調製方法を提供するものである。
【0018】
本発明は、前記糖タンパク質含有試料が、O−結合型糖タンパク質含有試料である、上記糖タンパク質の試料調製方法を提供するものである。
本発明は、前記O−結合型糖タンパク質が、コラーゲン由来であることを特徴とする、上記糖タンパク質の試料調製方法を提供するものである。
【0019】
本発明は、前記ポリマー粒子が、ヒドラジド基含有ポリマー粒子であることを特徴とする、上記糖タンパク質の試料調製方法を提供するものである。
本発明は、上記糖タンパク質の試料調製方法で得た糖タンパク質をMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法を提供するものである。
【0020】
本発明は、上記糖タンパク質の試料調製方法で得た糖タンパク質に標識化合物を結合させ、ついでMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法を提供するものである。
【0021】
本発明は、上記糖タンパク質の試料調製方法で得た糖タンパク質をイオン化促進標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法を提供するものである。
【0022】
本発明は、前記糖タンパク質含有試料が、健常者由来の糖タンパク質と特定疾患由来の糖タンパク質であり、上記糖タンパク質の試料調製方法で得た糖タンパク質を標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析するものであり、精製した健常者由来の糖タンパク質または特定疾患由来の糖タンパク質のいずれかに標識化合物を結合させ、他方に前記標識化合物に対応する安定同位体標識化合物を結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析し、前記標識化合物と前記安定同位体標識化合物との質量数の差から健常者と特定疾患由来の糖タンパク質の含有量を評価することを特徴とする、診断方法を提供するものである。
【発明の効果】
【0023】
本発明によれば、N−結合型糖タンパク質であるか、O−結合型糖タンパク質であるかを問わず、糖タンパク質を高収率で精製し、糖タンパク質の試料を調製することができる。
【0024】
本発明によれば、糖鎖を温和な条件で酸化して、糖タンパク質に含まれる−CH2OHを−CHOに酸化できるため、安全性および再現性に優れる。
本発明によれば、既存のデータと対比することで、試料糖タンパク質の同定を簡便に行うことができる。これにより、糖鎖のみならず、タンパク質部分も分析することができる。
【0025】
本発明によれば、健常者のデータと疾患由来の糖タンパク質の分析データとを比較することで、疾患の診断を行うことができる。
【図面の簡単な説明】
【0026】
【図1】図1は、実施例3のGOD酸化サンプルとGOD非酸化サンプル(コントロール)との全イオン電流クロマトグラムのチャートである。
【図2】図2は、実施例3のGOD酸化サンプルの全イオン電流クロマトグラムのチャートである。
【図3】図3は、実施例3の15.081分のMSのチャートである。
【図4】図4は、実施例3の15.081分のMS/MSのチャートである。
【図5】図5は、実施例4のGOD酸化サンプルとGOD非酸化サンプル(コントロール)との全イオン電流クロマトグラムのチャートである。
【図6】図6は、実施例4のGOD酸化サンプルの全イオン電流クロマトグラムのチャートである。
【図7】図7は、実施例4の14.078分のMSのチャートである。
【図8】図8は、実施例4の14.078分のMS/MSのチャートである。
【発明を実施するための形態】
【0027】
本発明の第一は、糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とからなる、糖タンパク質の試料調製方法である。
【0028】
糖タンパク質含有試料に含まれる莢雑物を除去し、分析対象の糖タンパク質を分離して試料を調製する。タンパク質や核酸、脂質など糖鎖以外の生体内物質にはアルデヒド基が含まれていないため、アルデヒド基と特異的に反応するポリマー粒子と結合させることで糖タンパク質のみを選択的に捕捉し、ポリマー粒子を洗浄することで莢雑物を除去し、その後、ポリマー粒子から糖タンパク質を分離することで、糖タンパク質の試料を調製する。得られた糖タンパク質試料は、MALDI−TOF MSやLC/MSなどで分析することができる。以下、本発明を詳細に説明する。
【0029】
(1)糖タンパク質
本発明において、糖タンパク質とは、タンパク質を構成するアミノ酸の一部に糖鎖が結合したものをいう。糖鎖がアスパラギンのアミノ基に糖が結合するN−結合型や、セリンやスレオニン、ヒドロキシリジンなどの水酸基に結合するO−結合型などが存在するが、いずれであってもよい。従来は、N−結合型糖タンパク質、O−結合型糖タンパク質について、それぞれ糖タンパク質から結合型の相違に由来した異なる方法で糖鎖を遊離させ、ついで遊離糖鎖を分析するものであったが、本発明は、糖タンパク質から糖鎖を遊離させることなく糖タンパク質のまま分析するため、いずれの結合型であるかを問わず、試料とすることができる。
【0030】
一方、好ましくは、O−結合型糖タンパク質である。従来の糖鎖を遊離する方法では、糖タンパク質から糖鎖を遊離することが困難であるため、O−結合型糖タンパク質の解析は、N−結合型糖タンパク質よりも遅滞していた。本発明によれば、N−結合型糖タンパク質と同様に簡便に分析できるため、特にO−結合型糖タンパク質に有効である。
【0031】
本発明で好適に使用できるO−結合型糖タンパク質としては、I〜XXIX型コラーゲン;アグリカン、パールカン、シンデカン、デコリンなどのプロテオグリカン;カドヘリン;ムチンなどを例示することができる。特に好ましくは、I〜XXIX型コラーゲンである。糖鎖の精製や濃縮にはレクチンがよく使用されるが、コラーゲンの糖鎖に結合するレクチンは知られておらず、コラーゲン由来の糖タンパク質の精製方法や濃縮方法は存在しなかった。一方、コラーゲンは、体内に多量に存在するタンパク質であり、単一のタンパク質として精製する方法が確立しており、および糖鎖が1糖または2糖と単純であってその構造も判明しており、データベースの分析結果と対比することで、同定が容易だからである。
【0032】
対象とする糖タンパク質の糖鎖の長さも限定されるものではない。一般には、糖が1〜20個、好ましくは1〜12個、特に好ましくは1〜8個、更に好ましくは1〜5個結合した糖鎖を有する糖タンパク質の精製に好適である。
【0033】
また、糖タンパク質のタンパク質部分の長さも限定されない。ただし、MALDI−TOF MSやLC/MSにおいて、既存のデータと対比する場合には、タンパク質部分は対象とする糖タンパク質を特定できる限度で、短いことが好ましい。一般には、糖タンパク質をトリプシンなどによって切断されるものを好適に使用することができる。
【0034】
(2)ポリマー粒子
本発明は、糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化し、当該−CHOを介してポリマー粒子と反応させるものである。従って、ポリマー粒子としては、−CHOと結合しうる反応基を有するポリマー粒子を広く使用することができ、例えば、ヒドラジド基含有ポリマー粒子、アミノ基含有ポリマー粒子、オキシルアミノ基含有ポリマー粒子、セミチオカルバジド基含有ポリマー粒子などを好ましく使用することができる。オキシルアミノ基とアルデヒド基との反応によって生じるオキシム結合やヒドラジド基とアルデヒド基との反応によって生じるヒドラゾン結合は、酸処理などによって容易に切断されるため、ポリマー粒子によって糖タンパク質を捕捉したのち、簡便に糖タンパク質をポリマー粒子から簡単に切り離すことができる点で好ましい。
【0035】
前記ポリマー粒子は、固体やゲル粒子であってもよい。ポリマー粒子に糖タンパク質を捕捉させたのち、遠心分離やろ過などの手段によって他と分離して回収することができる。また、ポリマー粒子をカラムに充填して用いてもよい。カラムに充填して用いる方法は、特に連続操作化に好適である。
【0036】
ポリマー粒子の形状は特に限定しないが、球状またはそれに類する形状が好ましい。ポリマー粒子が球状の場合、平均粒径は好ましくは0.05〜1000μmであり、より好ましくは0.05〜200μmであり、さらに好ましくは0.1〜200μmであり、最も好ましくは0.1〜100μmである。この範囲で通液性に優れ、かつ糖タンパク質の捕捉性に優れるからである。
【0037】
ポリマー粒子は、遠心分離やろ過などの手段で回収する時にのみ固体粒子あるいはゲル粒子であってもよい。たとえば温度、pHなどの環境変化によって溶解性が変化するポリマーを用い、溶媒に溶解した状態の該ポリマーに糖鎖の−CHOと特異的に反応する官能基を介して糖鎖を捕捉させ、溶解性を変化させて該ポリマーを沈殿させ、回収してもよい。環境によって溶解性が変化するポリマーとして、たとえばポリ(N−イソプロピルアクリルアミド)を挙げることができる。ポリ(N−イソプロピルアクリルアミド)分子の少なくとも一部に糖タンパク質の−CHOと特異的に反応する官能基を導入することで、上記のような糖タンパク質の捕捉が可能となる。
【0038】
このようなポリマー粒子は、市販品であってもよい。例えば、住友ベークライト株式会社製のBlotGlyco、BIO−RAD社製のAffiGel HZ Hydrazide Gelなどを使用することができる。
【0039】
(3)糖タンパク質含有試料
本発明で使用する糖タンパク質含有試料は、上記したN−結合型糖タンパク質やO−結合型糖タンパク質それ自体でもよいが、タンパク質部分の一次構造の解析が進んでいるO−結合型糖タンパク質のコラーゲンを分析する場合で説明する。なお、コラーゲンのタンパク質部分を構成するアミノ酸には、ヒドロキシリジンにガラクトシル基が結合したガラクトシルヒドロキシリジン(galactosyl hydroxylysine: GHL)、グルコシル−ガラクトシル基が結合したグルコシルガラクトシルヒドロキシリジン(glucosylgalactosyl hydroxylysine:GGHL)が存在することが知られている。
【0040】
コラーゲンは、現在、I〜XXIX型が知られており、上記GHLやGGHLの含有率は、コラーゲン型によって大きく異なる。本発明で使用するコラーゲンとしてはいずれであってもよく、新たに見出されるコラーゲンであってもよい。コラーゲンは、魚や豚、牛などの生皮、腱、骨などを形成する主要タンパク質であるが、生体内に含まれる大部分は水に不溶性である。本発明では、動物の皮や骨等の原料に含まれるコラーゲンをプロテアーゼなどの酵素を添加して可溶化し、酸やアルカリを添加して可溶化したものを使用することができる。
【0041】
更に、糖タンパク質含有試料としては、コラーゲンを、例えば、トリプシンやキモトリプシンなどでタンパク質部分をより短鎖に切断したものを使用することができる。コラーゲンはアミノ酸配列が解明されているため、タンパク質部分を分析すれば、トリプシンなどで切断した糖タンパク質がコラーゲンのいずれの部位であるかを、容易に特定することができる。更に、例えばSDS−PAGE(SDS変性ポリアクリルアミドゲル電気泳動)や、等電点電気泳動とSDS−PAGEを組み合わせた二次元電気泳動によって分離したものを糖タンパク質含有試料とすることもできる。また、泳動後のゲルを例えばクーマシーブリリアントブルーなどの染色剤を用いて染色したのち、ニトロセルロース、PVDF(ポリフッ化ビニリデン)などのメンブレンに転写してもよい。転写は一般的なブロッティング装置を用いて行うことができる。染色はメンブレンへの転写の後に行ってもよい。
(4)糖タンパク質の試料調製方法
本発明では、上記糖タンパク質含有試料を使用し、
糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、
前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、
前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、
前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とによって糖タンパク質試料を調製することができる。
【0042】
(i)酸化工程
上記糖タンパク質を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化し、これに前記ポリマー粒子を反応させる。N−結合型の糖タンパク質にN−グリコシダーゼを作用させて糖鎖を分離すると、遊離した糖鎖の還元末端にアルデヒド基が生じ、このアルデヒド基を介して上記ポリマー粒子と糖鎖とが結合する。しかしながら、本発明では、糖タンパク質に含まれる6位の−CH2OHを−CHOに酸化させるため、糖タンパク質から糖鎖を遊離させることなく、このアルデヒド基を介してポリマー粒子と糖タンパク質とを結合させることができる。
【0043】
−CH2OHを−CHOに酸化させる方法に限定は無いが、ガラクトースオキシダーゼを使用することが安全かつ簡便である。一般には、37℃で6〜24時間反応させる。
(ii)糖タンパク質−ポリマー粒子結合工程
次いで、前記酸化糖タンパク質含有試料を、−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る。ポリマー粒子の添加量は、ポリマー粒子に結合される−CHOと結合しうる反応基の量によって異なり、使用するポリマー粒子の前記反応基量に応じて適宜選択することができる。一般には、室温で3〜24時間、反応させればよい。
【0044】
糖タンパク質を特異的に捕捉するポリマー粒子によって糖タンパク質を捕捉する際の反応系のpHは、好ましくは2〜9、より好ましくは2〜7であり、さらに好ましくは2〜6である。pH調整のためには、各種緩衝液を用いることができる。糖鎖捕捉時の温度は、好ましくは4〜60℃、より好ましくは10〜50℃、さらに好ましくは15〜40℃である。反応時間は適宜設定することができる。ポリマー粒子をカラムに充填して試料溶液を通過させてもよい。
【0045】
(iii)洗浄工程
糖タンパク質−ポリマー粒子結合体を洗浄することで、糖タンパク質含有試料に含まれる糖タンパク質以外の成分を除去することができる。洗浄液は、糖タンパク質−ポリマー粒子結合体から糖タンパク質を切断せず、かつ莢雑物と親和性を有するものを広く使用することができる。たとえば、水、緩衝液、有機溶媒、界面活性剤を含む水または緩衝液などを適宜組み合わせて用いることが好ましい。特に好ましい形態は、界面活性剤を含む水または緩衝液で十分に洗浄したのち、有機溶剤で洗浄し、最後に水で洗浄する方法である。これらの洗浄により、非特異的吸着物がポリマー粒子表面から除去される。
【0046】
なお、ポリマー粒子に存在する−CHOと結合しうる反応基をキャップすることが好ましい。例えば、ポリマー粒子が−CHOと結合しうる反応基としてヒドラジド基を有する場合には、ポリマー粒子を洗浄した後に、無水酢酸を添加し、室温で30分間反応させる。その後、再度、ポリマー粒子を洗浄することが好ましい。
【0047】
(iv)糖タンパク質遊離工程
糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離させることで、糖タンパク質を精製し、純度の高い糖タンパク質試料とすることができる。
【0048】
遊離方法は、ポリマー粒子の前記反応基の種類に応じて適宜選択することができるが、糖タンパク質とポリマー粒子との結合以外を切断するものであってはならない。本発明では、揮発性酸処理により糖タンパク質をポリマー粒子から分離することができる。このような揮発性酸によれば、糖タンパク質をポリマー粒子から分離できると共に、脱塩等の必要なくそのままMALDI−TOF MSおよび/またはLC/MS分析を行うことができるからである。本発明で使用できる揮発性酸としては、ギ酸、酢酸、トリフルオロ酢酸、塩酸などを例示することができる。ギ酸処理の場合は、例えば、濃度0.1〜1質量%のギ酸溶液で温度60〜80℃で、0.5〜2時間、より好ましくは0.1〜1時間反応させる。
【0049】
(5)LC/MS分析
本発明では、上記で調製した糖タンパク質試料をLC/MS分析し、糖タンパク質を同定することができる。LC/MSの分析結果を、各種サンプルの分析結果を集積したデータベースと比較することで、糖鎖修飾部位を推定することができる。
【0050】
さらに、上記糖タンパク質試料をそのまま使用するほか、例えば糖タンパク質の前記−CHOにヒドラジン(H2NNH2)や重水素処理ヒドラジン(D2NND2)を結合して標識すれば、質量数が2異なる位置にピークが出現することを利用し、サンプル間での比較定量を行うことができる。このように、一方に標識化合物を、他方に前記標識化合物に対応する安定同位体標識化合物を結合することで、質量数の違いによるサンプル間の比較定量を行う事ができる。安定同位体としては、重水素(D)のほか、13Cや、15N、18O等を例示することができる。
【0051】
また、標識化合物として、ヒドラジンのほか、ヒドラジド基を含む物質として、ジラール試薬P(ピリジニウム−1−アセトヒドラジド・クロリド)、ジラール試薬T(ベタインヒドラジド・ヒドロクロライド)、5−ジメチルアミノナフタレン−1−スルフォニルヒドラジン(ダンシルヒドラジン);2−ヒドラジノピリジン;9−フルオレニルメチルカルバゼート(Fmoc ヒドラジン);ベンジルヒドラジン;4,4−ジフルオロ−5,7−ジメチル−4−ボラ−3a,4a−ジアザ−s−インダセン−3−プロピオノ酸,ヒドラジド;2−(6,8−ジフルオロ−7−ヒドロキシ−4−メチルクマリン)アセトヒドラジン;7−ジエチルアミノクマリン−3−カルボキシリック酸;ヒドラジン(DCCH);フェニルヒドラジン;1−ナフタレンアセトヒドラジン;2−ヒドラジノベンゾイックアシド;フェニルアセチックヒドラジン;ビオチンヒドラジンなどがある。また、−CHOと反応する反応基として、アミノオキシ基がある。アミノオキシ基を含む物質として、O−ベンジルヒドロキシアミン;O−フェニルヒドロキシルアミン;O−(2,3,4,5,6−ペンタフルオロベンジル)ヒドロキシルアミン;O−(4−ニトロベンジル)ヒドロキシルアミン;2−アミノオキシピリジン;2−アミノオキシメチルピリジン;4−[(アミノオキシアセチル)アミノ]ベンゾイックアシドメチルエステル;4−[(アミノオキシアセチル)アミノ]ベンゾイック酸エチルエステル;4−[(アミノオキシアセチル)アミノ]ベンズアミノ酸n−ブチルエステル;アミkノオキシ−ビオチンなどを例示することができる。
【0052】
更に、このような標識として、ジラール試薬Pや重水素処理ジラール試薬Pなどの標識化合物を使用すれば、ヒドラジンと同様に−CHOと反応して糖タンパク質に結合する標識となり、上記と同様に質量数が5異なる位置にピークが出現することを利用してサンプル間で比較定量を行うことができる。かつベンゼン環の中に4級アミンが含まれるためMSでのイオン化が促進され、感度を向上させることもできる。
【0053】
このような標識化合物は、糖タンパク質の1.5質量倍以上、より好ましくは3質量倍以上、さらに好ましくは5質量倍以上である。pH調整のためには、各種緩衝液を用いることができる。反応系の温度は,好ましくは4〜90℃、より好ましくは4〜70℃、さらに好ましくは30〜80℃であり、最も好ましくは40〜80℃である。
【0054】
(6)MALDI−TOF MS分析
本発明では、上記した糖タンパク質の試料調製方法で得た糖タンパク質をそのまま用いてMALDI−TOF MS分析を行うことができる。また、LC/MS分析の試料と同様に、例えば糖タンパク質の前記−CHOにヒドラジン(H2NNH2)や重水素処理ヒドラジン(D2NND2)を結合して標識して試料とすることができる。更に、ジラール試薬Pや重水素処理ジラール試薬P、その他の標識化合物を付加して試料とすることができる。これにより、高感度にMALDI−TOF MS分析を行うことができる。
【0055】
本発明では、上記で調製した糖タンパク質試料をMALDI−TOF MS分析し、糖タンパク質を同定することができる。MALDI−TOF MSの分析結果を、各種サンプルの分析結果を集積したデータベースと比較することで、糖鎖修飾部位を推定することができる。
【0056】
(7)診断方法
本発明の診断方法は、糖タンパク質含有試料を使用し、健常者由来の糖タンパク質と特定疾患由来の糖タンパク質との質量の差から、糖タンパク質を定量し、疾患を診断するものである。
【0057】
上記した糖タンパク質の試料調製方法で得た糖タンパク質を標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析するものであり、精製した健常者由来の糖タンパク質または特定疾患由来の糖タンパク質のいずれかに標識化合物を結合させ、他方に前記標識化合物に対応する安定同位体標識化合物を結合させる。得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析すると、標識化合物と前記標識化合物に対応する安定同位体標識化合物との質量数の差から健常者と特定疾患由来の糖タンパク質の含有量を評価することができる。
【実施例】
【0058】
次に実施例を挙げて本発明を具体的に説明するが、これらの実施例は何ら本発明を制限するものではない。
LC/MS測定条件
1.装置
高速液体クロマトグラフ:1200 Series(Agilent Technologies)、
質量分析装置:3200QTRAP(AB Sciex)
2.GHL、GGHLの測定
(i)HPLC条件
分析カラム:ZIC−HILIC 5μm, 2.1mmi.d.×150mm(SeQuant)、
移動相:A液;0.1%酢酸、5mM 酢酸アンモニウム、
B液;100%アセトニトリル、
グラジエント条件:0−5分:A液10%、5−10分:A液10−50%、10−25分:A液50−80%、25.1−30分:A液10%、
流速:0.2mL/min、
カラム温度:40℃、
注入量:10μl、
(ii)質量分析条件
イオン化:ESI、ポジティブ、
分析モード:Multiple Reaction Monitoring(MRM)モード、
イオンスプレー電圧:4.5kV、
イオンソース温度:600℃、
検出チャンネル(m/z):GHL 325.1→163.2、酸化GHL 323.1→163.2、
GGHL 487.2→163.3、酸化GGHL 485.2→163.3、
3.コラーゲン糖タンパク質の測定
(i)HPLC条件
分析カラム:Ascentis Express 2.7mm, 2.1mmi.d.×150mm(SUPELCO)、
移動相:A液;0.1%ギ酸、B液;100%アセトニトリル、
グラジエント条件:0−5分:A液98%、5−20分:A液98−50%、20.1−25分:A液10%、25.1−30分:A液98%、
流速:0.2mL/min、
カラム温度:25℃、
注入量:10μl
(ii)質量分析条件
イオン化:ESI、ポジティブ、
分析モード:Information Dependent Acquisition(IDA)モード、
イオンスプレー電圧:5.5kV、
イオンソース温度:600℃、
データベース検索ソフト:ProteinPilotソフトウェア3.0(AB Sciex)
(実施例1)
ガラクトシルヒドロキシリジン(GHL)10nmolを、100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液に溶解し、ついで、ガラクトースオキシダーゼ(SIGMA社製、商品名「galactose oxidase」)を50単位添加し、37℃で24時間処理した。この一部を採取し、溶液Aとした。
【0059】
その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)を250mg添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。この一部を採取し、溶液Bとした。
【0060】
一方、ガラクトシルヒドロキシリジン(GHL)10nmolを、100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液に溶解し、37℃で24時間処理した。この一部を採取し、対照溶液Aとした。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)を250mg添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。この一部を採取し、対照溶液Bとした。
【0061】
得られた溶液A、溶液B、対照溶液A、対照溶液BをそれぞれLC/MS(AB Sciex社製、商品名「3200QTRAP」)にて、GHLと酸化GHLの強度を上記測定条件にて測定した。GHLと酸化GHLの強度および対照溶液Aに対する%、溶液Aに対する%の結果を表1に示す。
【0062】
表1より、対照溶液AのGHL強度に対する溶液AのGHL強度は、1.6%であり、ガラクトースオキシダーゼによる酸化反応率は、98.4%(=100−1.6)となった。また、溶液Aの酸化GHLの強度に対する溶液Bの酸化GHLの強度で示される、酸化GHLの回収率は、401000×100/922000=43.5%であった。
【0063】
【表1】
(実施例2)
グルコシルガラクトシルヒドロキシリジン(GGHL)10nmolを、100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液に溶解し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。この一部を採取し、溶液Aとした。
【0064】
その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)を250mg添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。この一部を採取し、溶液Bとした。
【0065】
一方、グルコシルガラクトシルヒドロキシリジン(GGHL)10nmolを、100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液に溶解し、37℃で24時間処理した。この一部を採取し、対照溶液Aとした。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)を250mg添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。この一部を採取し、対照溶液Bとした。
【0066】
得られた溶液A、溶液B、対照溶液A、対照溶液BをそれぞれLC/MS(AB Sciex社製、商品名「3200QTRAP」)にて、GGHLと酸化GGHLの強度を上記条件で測定した。GGHLと酸化GGHLの強度および対照溶液Aに対する%、溶液Aに対する%の結果を表2に示す。
【0067】
表2より、対照溶液AのGGHL強度に対する溶液AのGGHL強度は、88.9%であり、ガラクトースオキシダーゼによる酸化反応率は、11.1%(=100−88.9)となった。
【0068】
また、溶液Aの酸化GGHLの強度に対する溶液Bの酸化GGHLの強度で示される、酸化GGHLの回収率は、22900×100/52900=43.3%であった。
【0069】
【表2】
(実施例3)
(1)脱脂した牛皮に40倍量(w/v)の0.5M酢酸を添加し、2〜3時間攪拌させて牛皮を十分に膨潤させ、ポリトロンホモジナイザーで牛皮をホモジナイズした。ペプシン量が組織の1/20量になるように、組織の10倍量のペプシン−0.5M酢酸を加えて一晩保持した後、遠心分離した。上清に終濃度1Mとなるように塩化ナトリウムを加えて塩析させ、遠心分離により沈殿を回収した。沈殿を最初に水、次に5mM酢酸に対して透析した後、リン酸緩衝液でpH7.5に調整して等電点沈殿させた。さらに沈殿を5 mM酢酸に対して透析し、フィルター濾過してI型コラーゲンを得た。
【0070】
このI型コラーゲン1mgを100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液で60℃、30分加熱処理して変性させ、トリプシン(SIGMA社製、商品名「Trypsin」)50μgを投与し、37℃、16時間処理した。トリプシン処理後、トリプシンインヒビター(SIGMA社製、商品名「Trypsin−chymotrypsin inhibitor」)を100μg添加し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)250mgを添加し、室温で6時間、転倒攪拌した。
【0071】
ついで、ヒドラジドゲルを、100mM酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液、1.5M NaCl、メタノール、蒸留水の順に洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。
【0072】
得られた反応液をLC/MSにて上記条件で分析した。全イオン電流クロマトグラムを図1に示す。なお、ガラクトースオキシダーゼで酸化しないサンプルについて同様に操作した。GOD酸化サンプルとGOD非酸化サンプル(コントロール)とを同一チャートに示す。
【0073】
また、GOD酸化サンプルの全イオン電流クロマトグラムを図2に示す。図2の15.081分のピークのMS分析の結果を図3に、MS/MS分析の結果を図4に示す。MS/MSの結果から、データベース検索ソフト(ProteinPilotソフトウェア3.0(AB Sciex))で検索したところ、下記配列がヒットした。この同定された糖鎖修飾部位は、これまでに報告されている部位と一致するものである。なお、配列表の配列番号1において、右から4番目のリジンがGHLとして同定された。
【0074】
(実施例4)
ウシの大腿骨から軟骨をメスで削りとり、10%塩化ナトリウムで洗浄した後、組織の9.9倍量の0.1N塩酸に浸して一晩置いた。氷冷しながらポリトロンホモジナイザーでホモジナイズし、ペプシン量が組織の1/100量となるように組織と等量のペプシン0.1N塩酸を加え、一晩攪拌して可溶化処理を行った。遠心分離により得た上清を1N水酸化ナトリウムでpH8.0に調整して等電点沈殿させ、遠心分離して沈殿を回収した。得られた沈殿の量に対して10倍量の0.01N塩酸を加えて溶解し、そこに10%濃度となるように塩化ナトリウムを加えて塩析させ、遠心分離により沈殿を回収した。沈殿を最初に水、次に5mM酢酸に対して透析した後、フィルター濾過してII型コラーゲンを得た。
【0075】
このII型コラーゲン1mgを100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液で60℃、30分加熱処理して変性させ、トリプシン50μgを投与し、37℃、16時間処理した。トリプシン処理後、トリプシンインヒビター(SIGMA社製、商品名「Trypsin−chymotrypsin inhibitor」)を100μg添加し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)250mgを添加し、室温で6時間、転倒攪拌した。
【0076】
ついで、ヒドラジドゲルを、100mM酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液、1.5M NaCl、メタノール、蒸留水の順にした後、1%ギ酸200μlを添加し、80℃で1時間反応した。
【0077】
得られた反応液をLC/MSにて上記条件で分析した。全イオン電流クロマトグラムを図5に示す。なお、ガラクトースオキシダーゼで酸化しないサンプルについて同様に操作した。GOD酸化サンプルとGOD非酸化サンプル(コントロール)とを同一チャートに示す。
【0078】
また、GOD酸化サンプルの全イオン電流クロマトグラムを図6に示す。図6の14.078分のピークのMS分析の結果を図7に、MS/MS分析の結果を図8に示す。MS/MSの結果から、データベース検索ソフト(ProteinPilotソフトウェア3.0(AB Sciex))で検索したところ、下記配列がヒットした。この同定された糖鎖修飾部位は、従来知られているものではなかった。なお、配列表の配列番号2において、左から9番目のリジンがGHLとして同定された。
【0079】
(実施例5)
実施例3と同様に操作してI型コラーゲンを得た。実施例3と同様に、このI型コラーゲン等電点沈殿物1mgを100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液で60℃、30分加熱処理して変性させ、トリプシン50μgを投与し、37℃、16時間処理した。トリプシン処理後、トリプシンインヒビター(SIGMA社製、商品名「Trypsin−chymotrypsin inhibitor」)を100μg添加し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)250mgを添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液、1.5M NaCl、メタノール、蒸留水の順に洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。
【0080】
次いで、上記反応液液に、0.1mMジラール試薬P、または重水素処理ジラール試薬Pを加え、80℃で1時間反応させた。さらに、形成されたヒドラゾン結合を1mMシアノ水素化ホウ素ナトリウムにより室温で1時間還元することにより、アミンの形にして安定化させた。得られた反応液を100mMリン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH 7.2溶液に対して透析後、LC−MS(AB Sciex社製、商品名「3200QTRAP」)で分析した。
【0081】
(実施例6)
健常者由来コラーゲンおよび骨形成不全症患者由来コラーゲンを使用した。実施例3と同様に、コラーゲン1mgを100mM リン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH7.2溶液で60℃、30分加熱処理して変性させ、トリプシン50μgを投与し、37℃、16時間処理した。トリプシン処理後、トリプシンインヒビター(SIGMA社製、商品名「Trypsin−chymotrypsin inhibitor」)を100μg添加し、ついで、ガラクトースオキシダーゼを50単位添加し、37℃で24時間処理した。その後、反応溶液をpH5.5に調整し、ヒドラジドゲル(BIO−RAD社製、商品名「AffiGel HZ Hydrazide Gel」)250mgを添加し、室温で6時間、転倒攪拌した。ついで、ヒドラジドゲルを、100mM 酢酸/酢酸ナトリウム緩衝液、150mM NaCl、pH5.5溶液1mlで3回洗浄した後、1%ギ酸200μlを添加し、80℃で1時間反応した。
【0082】
健常者由来サンプルには0.1mMジラール試薬P、骨形成不全症患者由来サンプルには0.1mM重水素処理ジラール試薬Pを加え、80℃で1時間反応させた。さらに、形成されたヒドラゾン結合を1mMシアノ水素化ホウ素ナトリウムにより室温で1時間還元することにより、アミンの形にして安定化させた。得られた反応液を100mMリン酸二水素ナトリウム/リン酸水素二ナトリウム緩衝液、150mM NaCl、pH 7.2溶液に対して透析後、LC−MS(AB Sciex社製、商品名「3200QTRAP」)で分析した。
【産業上の利用可能性】
【0083】
本発明によれば、簡便な操作で糖タンパク質を解析することができ、有用である。
【特許請求の範囲】
【請求項1】
糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、
前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、
前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、
前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とからなる、糖タンパク質の試料調製方法。
【請求項2】
前記糖タンパク質含有試料が、O−結合型糖タンパク質含有試料である、請求項1記載の糖タンパク質の試料調製方法。
【請求項3】
前記O−結合型糖タンパク質が、コラーゲン由来であることを特徴とする、請求項2記載の糖タンパク質の試料調製方法。
【請求項4】
前記ポリマー粒子が、ヒドラジド基含有ポリマー粒子であることを特徴とする、請求項1〜3のいずれかに記載の糖タンパク質の試料調製方法。
【請求項5】
請求項1〜4のいずれかに記載の糖タンパク質の試料調製方法で得た糖タンパク質をMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法。
【請求項6】
請求項1〜4のいずれかに記載の糖タンパク質の試料調製方法で得た糖タンパク質に標識化合物を結合させ、ついでMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法。
【請求項7】
請求項1〜4のいずれかに記載の糖タンパク質の試料調製方法で得た糖タンパク質をイオン化促進標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法。
【請求項8】
前記糖タンパク質含有試料が、健常者由来の糖タンパク質と特定疾患由来の糖タンパク質であり、
請求項1〜4のいずれかに記載の糖タンパク質の試料調製方法で得た糖タンパク質を標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析するものであり、
精製した健常者由来の糖タンパク質または特定疾患由来の糖タンパク質のいずれかに標識化合物を結合させ、他方に前記標識化合物に対応する安定同位体標識化合物を結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析し、前記標識化合物と前記安定同位体標識化合物との質量数の差から健常者と特定疾患由来の糖タンパク質の含有量を評価することを特徴とする、診断方法。
【請求項1】
糖タンパク質含有試料を酸化して糖タンパク質に含まれる−CH2OHを−CHOに酸化する工程、
前記酸化糖タンパク質含有試料を−CHOと結合しうる反応基を有するポリマー粒子と反応させ、糖タンパク質−ポリマー粒子結合体を得る工程、
前記糖タンパク質−ポリマー粒子結合体を洗浄する工程、
前記洗浄した糖タンパク質−ポリマー粒子結合体から糖タンパク質を遊離する工程とからなる、糖タンパク質の試料調製方法。
【請求項2】
前記糖タンパク質含有試料が、O−結合型糖タンパク質含有試料である、請求項1記載の糖タンパク質の試料調製方法。
【請求項3】
前記O−結合型糖タンパク質が、コラーゲン由来であることを特徴とする、請求項2記載の糖タンパク質の試料調製方法。
【請求項4】
前記ポリマー粒子が、ヒドラジド基含有ポリマー粒子であることを特徴とする、請求項1〜3のいずれかに記載の糖タンパク質の試料調製方法。
【請求項5】
請求項1〜4のいずれかに記載の糖タンパク質の試料調製方法で得た糖タンパク質をMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法。
【請求項6】
請求項1〜4のいずれかに記載の糖タンパク質の試料調製方法で得た糖タンパク質に標識化合物を結合させ、ついでMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法。
【請求項7】
請求項1〜4のいずれかに記載の糖タンパク質の試料調製方法で得た糖タンパク質をイオン化促進標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析することを特徴とする、糖タンパク質の分析方法。
【請求項8】
前記糖タンパク質含有試料が、健常者由来の糖タンパク質と特定疾患由来の糖タンパク質であり、
請求項1〜4のいずれかに記載の糖タンパク質の試料調製方法で得た糖タンパク質を標識化合物と結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析するものであり、
精製した健常者由来の糖タンパク質または特定疾患由来の糖タンパク質のいずれかに標識化合物を結合させ、他方に前記標識化合物に対応する安定同位体標識化合物を結合させ、得られた糖タンパク質−標識化合物をMALDI−TOF MSおよび/またはLC/MSで分析し、前記標識化合物と前記安定同位体標識化合物との質量数の差から健常者と特定疾患由来の糖タンパク質の含有量を評価することを特徴とする、診断方法。
【図1】

【図2】
【図3】

【図4】

【図5】
【図6】
【図7】

【図8】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2012−32298(P2012−32298A)
【公開日】平成24年2月16日(2012.2.16)
【国際特許分類】
【出願番号】特願2010−172700(P2010−172700)
【出願日】平成22年7月30日(2010.7.30)
【出願人】(000135151)株式会社ニッピ (18)
【Fターム(参考)】
【公開日】平成24年2月16日(2012.2.16)
【国際特許分類】
【出願日】平成22年7月30日(2010.7.30)
【出願人】(000135151)株式会社ニッピ (18)
【Fターム(参考)】
[ Back to top ]