
糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する耐熱性酵素
【課題】耐熱性で、広い基質特異性を有する糖ヌクレオチド合成活性およびリン酸化糖異性化活性を有する酵素タンパク質を新たに提供し、糖鎖合成の基質となる糖ヌクレオチドおよびリン酸化糖異性体を安定的に合成する。
【解決手段】超好熱古細菌パイロコッカス、ホリコシイのゲノムから糖ヌクレオチド合成活性及びリン酸化糖異性化活性を有する酵素の遺伝子を見出し、該遺伝子を用いて遺伝子工学的手段により、耐熱性の糖ヌクレオチド合成活性およびリン酸化糖異性化活性を有する酵素タンパク質を製造するとともに、これを用いて糖ヌクレオチドおよびリン酸化糖異性体を安定的に合成する。
【解決手段】超好熱古細菌パイロコッカス、ホリコシイのゲノムから糖ヌクレオチド合成活性及びリン酸化糖異性化活性を有する酵素の遺伝子を見出し、該遺伝子を用いて遺伝子工学的手段により、耐熱性の糖ヌクレオチド合成活性およびリン酸化糖異性化活性を有する酵素タンパク質を製造するとともに、これを用いて糖ヌクレオチドおよびリン酸化糖異性体を安定的に合成する。
【発明の詳細な説明】
【技術分野】
【0001】
本願発明は、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する耐熱性蛋白質、該蛋白質をコードするDNA、該DNAを含有する組み換え体DNA、該組み換え体DNAを保有する形質転換体、該形質転換体を用いた糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質の製造法、および該蛋白質あるいは該形質転換体を用いた糖ヌクレオチドの製造法および異性化リン酸化糖の製造法に関する。
【背景技術】
【0002】
糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する酵素としては、サルモネラ菌(Salmonella enterica) (非特許文献1参照)、豚の肝臓 (非特許文献2参照)、マイコバクテリア菌(Mycobacterium smegmatis)(非特許文献3参照)由来の糖ヌクレオチド合成活性を示す酵素であるGDP-mannose pyrophosphorylase(GMP)、及びヘリコバクター菌(Helicobacter pylori)(非特許文献4参照)のリン酸化糖異性化活性を示す酵素の詳しい性質がすでに報告されている。GMPはGDP-マンノース合成に必須な酵素であり、マンノース−1−リン酸とグアノシン三リン酸(GTP)を基質としてGDP-マンノースを生産する。また、リン酸化糖異性化活性を示す酵素であるPhosphomannose isomerase(PMI)は、マンノース−6−リン酸を基質としてフルクトース−6−リン酸を生産する反応と、フルクトース−6−リン酸を基質としてマンノース−6−リン酸を生産する反応と両方向に働き、マンノース−6−リン酸の異性化に必須の酵素である。その他にも、ヒトや酵母、病原菌からも糖ヌクレオチド合成反応並びにリン酸化糖の異性化反応を触媒する酵素は見出されているが、その多くが常温生物由来のため室温以上では極めて不安定で、活性は80℃程度の加熱処理により速やかに失活する。このため、使用時に反応系を滅菌する等の処理が必要であったり、低温での注意深い保存が必要であった。
【0003】
【非特許文献1】Elling L, RitterJE, Verseck S. “Expression, purification andcharacterization of recombinant phosphomannomutase and GDP-alpha-D-mannosepyrophosphorylase from Salmonella enterica, group B, for the synthesis ofGDP-alpha-D-mannose from D-mannose” (1996) Glycobiology, 6, 591-597.
【非特許文献2】Ning B, Elbein AD.“Cloning, expression and characterization of the pigliver GDP-mannose pyrophosphorylase. Evidence that GDP-mannose and GDP-Glcpyrophosphorylases are different proteins” (2000) European Journal of Biochemistry, 267,6866-6874.
【非特許文献3】Ning B, Elbein AD.“Purification and properties of mycobacterialGDP-mannose pyrophosphorylase” (1999) Archives of Biochemistry and Biophysics,362(2),339-345.
【非特許文献4】Wu B, Zhang Y,Zheng R, Guo C, Wang PG. “Bifunctional phosphomannoseisomerase/GDP-D-mannose pyrophosphorylase is the point of control forGDP-D-mannose biosynthesis in Helicobacter pylori” (2002) FEBS Letter 519,87-92.
【発明の開示】
【発明が解決しようとする課題】
【0004】
糖ヌクレオチドを合成する活性を有し、且つ耐熱性を有する酵素は、糖鎖合成の基質となる糖ヌクレオチドを安定に合成することを可能にするなど極めて有用である。また、糖鎖合成の際の基質として必要な様々な種類の糖を生産する際には、糖の異性化反応は有効な反応である。このリン酸化糖異性化反応活性を有し、且つ耐熱性を有する酵素は、様々な種類の糖を安定に合成することを可能にするなど極めて有用である。すなわち、これら両酵素活性を併せ持つ耐熱性酵素が見出されれば、糖鎖合成の基質となる様々な種類の糖ヌクレオチドを安定に生産することが可能となる。また、これらの反応を一つの酵素が行うことで、様々な種類のリン酸化糖を安定に合成することと、それらを用いて糖ヌクレオチドを合成することを共役させることが出来、効率的な反応として進めることが可能になる。そこで、両酵素活性を有する両反応性酵素で、且つ熱安定性を有する酵素は渇望されていた。
【0005】
したがって、本発明の課題は、様々な糖ヌクレオシド三リン酸を基質として糖ヌクレオチド合成が可能で、且つリン酸化糖を基質として異なる種類の異性化されたリン酸化糖の合成が可能な、2種類の活性を有し、かつ熱安定な耐熱性を有する新規酵素を提供することにある。
【課題を解決するための手段】
【0006】
本発明は、以上のような課題を解決すべく、90−95℃で生育する超好熱古細菌Pyrococcus horikoshii OT3に着目し、そのゲノム中に本酵素活性を有する遺伝子を見いだした。さらに、大腸菌を使ってその遺伝子がコードする該酵素を生産し、この酵素が高温(90℃)で安定に存在し、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を示すことを確認し、さらに、本酵素を用いることにより様々な糖ヌクレオチドを生産すること並びに種々の糖リン酸の異性体を生産することも見出して、本発明を完成するに至ったものである。
【0007】
即ち、本発明は、以下の(1)〜(15)に係るものである。
(1) 配列番号1に記載のアミノ酸配列を有するか、あるいは、配列番号1に記載のアミノ酸配列において1乃至数個のアミノ酸残基が欠失、置換、挿入又は付加されたアミノ酸配列を有し、かつ糖ヌクレオチド合成活性及びリン酸化糖異性化活性を有することを特徴とする蛋白質。
(2) 上記(1)に記載の蛋白質をコードするDNA。
(3) 上記(2)に記載の塩基配列を有することを特徴とするDNA。
(4) 配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズし、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質をコードすることを特徴とするDNA。
(5) 上記(2)〜(4)のいずれかに記載のDNAがベクターに組み込まれていることを特徴とする組み換え体DNA。
(6) 上記(5)に記載の組み換え体DNAが宿主細胞に導入されていることを特徴とする形質転換体。
(7) 上記(6)に記載の形質転換体を培地に培養し、培養物から、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質を採取することを特徴とする、糖ヌクレオチド合成活性及びリン酸化糖異性化活性を有する蛋白質の製造法。
(8) 糖−1−リン酸及びヌクレオシド三リン酸に、上記(1)に記載の蛋白質を作用させ、糖ヌクレオチドに変換することを特徴とする糖ヌクレオチドの製造方法。
(9) 糖−6−リン酸に、上記(1)に記載の蛋白質を作用させ、糖リン酸を異性化させることを特徴とする異性化された糖−6−リン酸の製造方法。
(10) 糖−1−リン酸及びヌクレオシド三リン酸に、上記(6)に記載の形質転換体の培養液あるいは培養物の処理物を作用させ、糖ヌクレオチドに変換することを特徴とする、糖ヌクレオチドの製造方法。
(11) 糖−6−リン酸に、上記(6)に記載の形質転換体の培養液あるいは培養物の処理物を作用させ、糖−6−リン酸を異性化させることを特徴とする、異性化された糖−6−リン酸の製造方法。
(12) 糖−1−リン酸及びヌクレオシド三リン酸に、上記(3)又は(4)に記載のDNAにコードされる蛋白質を作用させ、糖ヌクレオチドに変換することを特徴とする、糖ヌクレオチドの製造方法。
(13) 糖−6−リン酸に、上記(3)又は(4)に記載のDNAにコードされる蛋白質を作用させ、糖リン酸を異性化させることを特徴とする、異性化された糖−6−リン酸の製造方法。
(14) リン酸化糖の異性化反応、分子内リン酸基転移反応及び糖−ヌクレオチド合成反応を行い、糖−ヌクレオチドを製造する方法において、上記リン酸化糖の異性化反応およ糖−ヌクレオチド合成反応を、請求項1に記載の蛋白質を用いて行うことを特徴とする、上記方法。
(15) 請求項1に記載のタンパク質を含有する反応系において、カルシウムイオン、亜鉛イオンまたは銅イオンのうちいずれかを選択して含有させることにより、該タンパクの酵素作用を、糖ヌクレオチド合成作用とリン酸化糖異性化作用との間で切り替えることを特徴とする、酵素反応の切り替え方法。
【発明の効果】
【0008】
本発明により、試験管内での様々な種類の糖ヌクレオチドを合成する事が可能でかつ熱等に安定で、さらに試験管内での様々な種類のリン酸化糖異性体を合成することが可能でかつ熱等に安定な新規な両反応を触媒する酵素が提供できた。その結果、新規な糖ヌクレオチド及びリン酸化糖の合成が可能になった。一方、本酵素により生産が可能となるリン酸化糖異性体や糖ヌクレオチドは糖蛋白質、糖脂質、多糖類の糖合成に糖提供体として機能するものであり、これらの糖鎖合成は、癌転移、器官発生あるいは細胞性免疫等に密接に関与するものとして近年注目されており、本発明はこれらの研究の発展において、その貢献度はきわめて大きい。
【発明を実施するための最良の形態】
【0009】
以下に、本願発明を具体的に説明する。
本発明で使用した超好熱古細菌は、超好熱古細菌パイロコッカス、ホリコシイ(JCM登録番号JCM9974)である。本発明者等は超好熱古細菌染色体上の本酵素活性を示す遺伝子領域を、PCR反応で増幅・抽出し、蛋白質発現プラスミドpET28aに挿入後、そのプラスミドにより形質転換した大腸菌を用いて本酵素の生産をおこなった。生産された酵素は加熱処理およびカラムクロマトグラムで単離精製した。精製された酵素は分子量が約53,000の蛋白質で様々な糖ヌクレオチドを合成する活性を有する酵素であることが判明した。
この酵素は50mM MOPS緩衝液(pH 7.6)中で、80℃30分以上の加熱後も活性を有し、高い耐熱性を示した。また、該タンパク質溶液の金属塩の種類を変更することにより、該タンパクの酵素作用が、糖ヌクレオチド合成作用とリン酸化糖異性化作用との間で切り替わるという驚くべき性質を有する。
この超好熱古細菌パイロコッカス、ホリコシイが有するGMP/PMI(PHGMP/PMI)のアミノ酸配列およびその遺伝子DNA(PH0925)の塩基配列を、それぞれ配列表の配列番号1および2に示す。
【0010】
本発明における酵素タンパク質は、上記配列番号1に示されるアミノ酸配列を有するもののみに限定されず、該アミノ酸配列において、1乃至数個のアミノ酸残基が欠失、置換、挿入又は付加されたアミノ酸配列であっても、このアミノ酸配列を有する蛋白質が、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を示すものも含む。また、本発明のこれら酵素遺伝子DNAについても、上記同配列番号2に示す塩基配列を有するもののみに限定されず、上記アミノ酸配列をコードするものを包含する。さらに上記配列2に示されるDNAにストリンジェントな条件下でハイブリダイズし、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質をコードするDNAも包含する。このストリンジェントな条件とは、ハイブリダイゼーション溶液1リットル中に52.59gNaCl、26.46gクエン酸ナトリウム、1gフィコール(Type 400)、1gポリビニルピロリドン、1gウシ血清アルブミン、5gSDS、1g断片化鮭精子DNA、500ml ホルムアミドを含み、温度42℃で行う。その後の洗浄は、洗浄用溶液1リットル中に17.53gNaCl、8.82gクエン酸ナトリウム、5gSDSを含み、温度68℃で行う条件である。
【0011】
本発明の酵素タンパク質を得るには、通常の遺伝子工学的手法が適用でき、上記酵素遺伝子DNAを、例えばpET28a、pHY481等の蛋白質発現プラスミドベクター等に挿入して組み換えベクターを作製し、該組み換えベクターを用いて宿主細胞を形質転換し、該形質転換体を培地で培養し、培養物、培養処理物あるいはこれら培養物から分離回収された形質転換体から、酵素タンパク質を通常の蛋白質精製手段により精製し単離する。上記宿主細胞としては、大腸菌・枯草菌等が利用可能である。
【0012】
本発明においては、さらにこの酵素タンパク質を用いて、糖ヌクレオチドを合成するが、この合成においては、糖−1−リン酸とヌクレオシド三リン酸を含有する溶液に該酵素を添加し、反応温度60〜95℃で反応させ糖ヌクレオチドを得る。
糖−1−リン酸としては、例えば、マンノース−1−リン酸、グルコース−1−リン酸等が挙げられ、ヌクレオシド三リン酸としては、例えばグアノシン三リン酸(GTP)、アデノシン三リン酸(ATP)酸等が挙げられ、さらに、本発明の酵素はデオキシリボヌクレオシド三リン酸も基質とすることができる。
【0013】
この糖ヌクレオチド合成活性の反応式として、マンノース−1−リン酸とGTPからGDP-マンノースを合成する場合について以下に示す。
【化1】

【0014】
また、本発明においては、さらにこの酵素を用いて、リン酸化糖異性体を合成するが、この合成においては、糖−6−リン酸に該酵素を添加し、反応温度60〜95℃で反応させ糖−6−リン酸異性体を得る。
糖−6−リン酸としては、例えば、マンノース−6−リン酸、フルクトース−6−リン酸等が挙げられる。
【0015】
このリン酸化糖異性化活性の反応式として、マンノース−6−リン酸からフルクトース−6−リン酸を合成する場合について以下に示す。
【化2】
【0016】
また、この反応においては、上記精製した酵素のみならず、粗酵素であってもよい。例えば、宿主として枯草菌等分泌型の系を用いる場合には、培養液中に本酵素が生成蓄積され、大腸菌等の非分泌型の系を用いる場合には、菌体内に生成されるので、本酵素を含有する培養液あるいはその処理物、もしくは菌体破砕物等の培養処理物を用いて、糖ヌクレオチドあるいは糖リン酸異性体を合成してもよい。
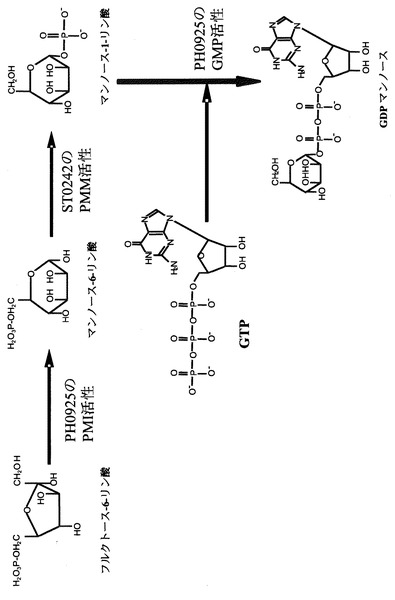
本発明の酵素タンパク質は、他の酵素と組み合わせることにより、一連の酵素反応系からなる糖ヌクレオチド合成系を構築することができる。図11は、本発明の酵素タンパク質と、本発明者等の先の出願(特願2004−236321号)に係るリン酸化糖のリン酸基分子内転移酵素とを組み合わせることにより、フルクトース−6−リン酸からGDP−マンノースを生成する合成系の構築例である。
上記リン酸基分子内転移酵素は、好酸性好気性超好熱古細菌スルフォロバス、トーコーダイイ(JCM登録番号JCM10545)由来の酵素であり、そのアミノ酸配列及び遺伝子の塩基配列は配列番号5、6に示される。使用する糖リン酸のリン酸基分子内転移酵素としては、配列番号5に示すアミノ酸配列を有するものの他、該アミノ酸配列において、1乃至数個のアミノ酸残基が欠失、置換、挿入または付加されたアミノ酸配列を有するものであって、かつ糖リン酸化糖のリン酸基分子内転移活性を有するものも使用できる。
該酵素は、マンノース−6−リン酸、グルコース−6−リン酸等の糖−6−リン酸のリン酸基を分子内転移させて、それぞれマンノース−1−リン酸、グルコース−1−リン酸等の糖−1−リン酸を生成する。この酵素は60〜95℃で反応可能であり、上記酵素を組み合わせた反応系は耐熱性である。
図11に示されるGDP−マンノース合成系においては、まず、本発明の酵素タンパク質の糖異性化作用を利用して、フルクトース−6−リン酸からマンノース−6−リン酸を生成させ、次いで、上記リン酸基分子内転移酵素を用いて、生成したマンノース−6−リン酸のリン酸基を分子内転移させて、マンノース−1−リン酸を生成させる。この後、本発明の酵素タンパク質の糖−ヌクレオチド合成作用を利用して、マンノース−1−リン酸とGTPからGDP−マンノースを生成させる。
この糖−ヌクレオチド合成系においては、該合成系の各反応工程において、各酵素溶液を順次基質に接触させて各反応を行ってもよいが、本発明の酵素タンパク質と上記リン酸基分子内転移酵素とを共に含有する酵素反応系を用いることにより、異性化反応、分子内リン酸基転移反応及び糖−ヌクレオチド合成反応を一つの反応工程で行い、糖−ヌクレオチドを合成することも可能である。
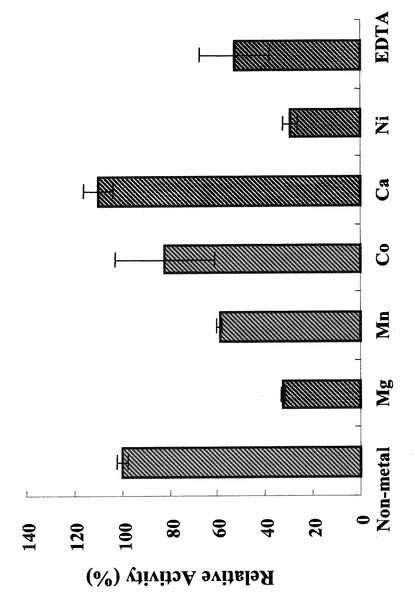
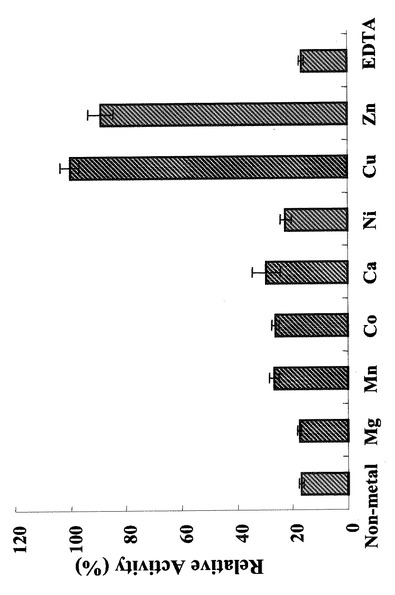
また、図12及び図13に示されるように、本発明の酵素タンパク質は、酵素反応系に含有させる金属イオンによって、その酵素作用を切り替えることが可能である。図12は、各金属イオンを2mM濃度で酵素反応系に含有させた場合の、本酵素タンパク質のイソメラーゼ活性を測定した結果を示す図であり、図13は、同糖−ヌクレオチド合成活性を示す図である。図12の結果によれば、本酵素タンパク質の糖リン酸化糖の異性化活性は、Caイオンを含有させた場合に高いことが分かる。また、亜鉛イオン、銅イオンを含有させた場合は、糖リン酸化糖の異性化活性は低く、検出限界以下であった。一方、図13の結果によれば、同糖−ヌクレオチド合成活性は、亜鉛イオン、銅イオンを含有させた場合は高く、カルシウムを含有させた場合には低い。すなわち、本発明の酵素タンパク質の反応系においては、カルシウムイオンを含有させる場合は、リン酸化糖の異性化活性を主に示し、亜鉛イオンあるいは銅イオンを含有させた場合は糖−ヌクレオチド合成活性を示す。
したがって、本発明においては、本発明の酵素タンパク質の作用を含有させる金属イオン種の種類により変更可能であり、このことは、本酵素タンパク質の異なる作用を利用する上記糖−ヌクレオチド合成系に応用可能である。なお、本発明の酵素タンパク質は、上記金属イオンを含有させない場合においては、上記リン酸化糖の異性化活性と上記糖−ヌクレオチド合成活性を併せて有する。
以下に、本発明の実施例を示すが、本発明実施例により限定されるものではない。
【実施例】
【0017】
[実施例1] 糖ヌクレオチド合成及びリン酸化糖異性化酵素の製造
(1) 菌の培養
好酸性好気性超好熱性古細菌パイロコッカス、ホリコシイJCM9974は次の方法で培養した。
13.85gのNaCl、3.5gのMgSO4・7H2O、2.75gのMgCl2・6H2O、0.325gのKCl、0.05gのNaBr、15mgのH3BO3、7.5mgのSrCl2・6H2O、0.05mgのKI、0.75gのCaCl2、0.5gのKH2PO4、2.0mgの(KH4))2Ni(SO4)2・6H2Oを1Lの蒸留水に溶かしSalt base solutionを作製した。1.5gのNitrilotriacetic acidを1Lの蒸留水に溶かし、この溶液のpHを6.5にKOH溶液で調製した。その溶液に3.0gのMgSO4・7H2O、0.5gのMnSO4・xH2O、1.0gのNaCl、0.1gのFeSO4・7H2O、0.1gのCoSO4・7H2O、0.1gのCaCl2・2H2O、0.1gのZnSO4・7H2O、0.01gのCuSO4・5H2O、0.01gのAlK(SO4)2、0.01gのH3BO3、0.01gのNa2MoO4・2H2Oを加え、この溶液のpHを7.0にKOH溶液で調製し、Trace mineralsを作製した。1.0LのSalt bace solution、10.0mLのTrace minerals、5.0mLのクエン酸、1.0gのYeast extract、5.0gのBacto peptone、30.0gの硫黄、1.0mgのレサズリンを作製し、この溶液のpHを6.5にH2SO4溶液で調製した。この溶液を窒素下でフィルター滅菌し、1日に3時間硫黄蒸気に当てることを連続3日間行った。5%のNaS・9H2Oを窒素下でオートクレーブし、無菌かつ嫌気下で培地に加え、JCM9974を植菌した。この培養液を98℃で培養し、その後遠心分離し集菌した。
【0018】
(2) 染色体DNAの調製
JCM9974の染色体DNAは以下の方法により調製した。
培養終了後5000rpm、10分間の遠心分離により菌体を集菌する。菌体を10mMEDTA(pH 6.0)溶液で洗浄後、50mM Tris/HCl-50mM EDTA(pH8.5)溶液を加えて細胞を溶解させる。さらに、0.5% Na-lauroylsarcosinate、1mg/mlプロテアーゼKとなるように各々を加えた後、50℃で3時間保温する。フェノール処理を3回行った後、溶液を10mM EDTA(pH8.0)溶液に対して透析する。37℃で30分間のRNaseによるRNAの分解後、フェノールクロロフォルム溶液で処理した後、10mM Tris-1mM EDTA(pH8.0)で透析を行った。
【0019】
(3)発現プラスミドの構築
上記(2)で得た染色体DNAを鋳型とし、下記Upper primerとLower primerを使用して PCRを行った。PCRで得られたDNAは、目的の構造遺伝子領域の前後に制限酵素(NdeIとBamHI)サイトを有する。
【0020】
Upper primer、
5’- GGGAATTCCATATGAAGGCTTTGATTCTAG -3’ (配列番号3)
(下線部はNdeIサイトを示す)
Lower primer、
5’- CGCGGATCCTCACCTCCCGTAATCGTCTTG -3’ (配列番号4)
(下線部はBamHIサイトを示す)
【0021】
PCR反応後、制限酵素(NdeIとBamHI)で完全分解(37℃で2時間)した後、目的の構造遺伝子領域断片を精製した。該遺伝子領域(PH0925)の塩基配列は配列番号2に示される。
制限酵素NdeIとBamHIで切断後精製したpET28a(Novagen社製)と上記の構造遺伝子領域断片とをT4リガーゼを用いて16℃、2時間反応させることによって連結した。連結したDNAの一部を大腸菌DH5αのコンピテントセルに導入し、形質転換体のコロニーを得た。得られたコロニーからプラスミドをQIAprep Spin Miniprep Kit(QIAGEN社製)で精製し、塩基配列を確認して発現プラスミド、pET28a/PH0925を得た。発現プラスミドpET28a/PH0925を用いるとPHGMP/PMIはN末端にヒスチジンタグが付加された融合蛋白質として生産される。
【0022】
(6)組み換え遺伝子の発現
大腸菌 BL21-CodonPlus(DE3)-RIL、(Novagen社製)のコンピテントセルを融解して、ファルコンチューブに0.1ml移す。その中に上記の発現プラスミド10ng分に相当する溶液を加え氷中に30分間放置した後、42℃でヒートショックを30秒間おこない、そこにSOC培地0.9mlを加え、37℃で1時間振とう培養する。その後、カナマイシンを含むLB寒天プレート上に適量まき、37℃で一晩培養し、形質転換体大腸菌BL21-CodonPus(DE3)-RIL/pET28a/PH0925を得た。
【0023】
当該形質転換体をカナマイシン含有LB培地(2リットル)中で一晩37℃において振とう培養した後、IPTG(Isopropyl-β-D-thiogalacopyranoside)を1mMになるように加え、さらに30℃で5時間振とう培養した。培養後、遠心分離機(6000rpm、20分間)により集菌をおこなった。
【0024】
(7)PHGMP/PMI酵素の精製
2リットル培養液から集菌した菌体に2倍量の20mMリン酸ナトリウム塩緩衝液(pH7.4)、菌体1gあたり2mgのLysozyme(Wako社製)、0.1μgのDNaseI(Takara社製)を加え懸濁液を得た。氷上で1時間静置後、超音波破砕し、遠心分離(20000g 30分間)により上清液を得た。得られた上清液を80℃で30分間加熱した後、遠心分離(20000g 30分間)により上清液を得た。この上清液を20mMリン酸ナトリウム塩緩衝液(pH7.4)0.5M NaClで平衡化したHiTrap phenyl sepharose(ファルマシア社製)カラムに吸着させ、同緩衝液中のイミダゾール濃度を0.0 Mから0.5 Mに増加させることにより目的蛋白質の溶出を行った。
【0025】
【0026】
[実施例2] 糖ヌクレオチド合成
(1) 糖ヌクレオチド合成反応(糖とヌクレオチドの結合反応)
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、1mM マンノース−1−リン酸、0.05mM GTPからなる酵素反応溶液10μL中に実施例1で得られた精製酵素1 ngを加えた。この酵素反応液を85℃で保温することにより、反応させた。5分後に100μlの500mM KH2PO4溶液を加える事により反応を停止させた。この反応により、マンノースがGDPと結合したGDPマンノースが生成する。
【0027】
(2)糖ヌクレオチド合成反応(糖とヌクレオチドの結合反応)の測定
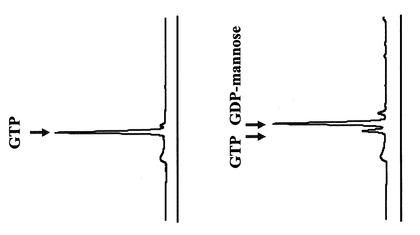
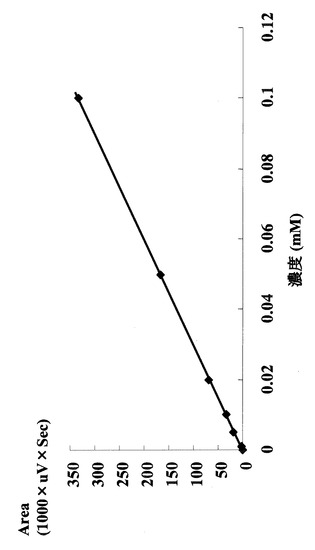
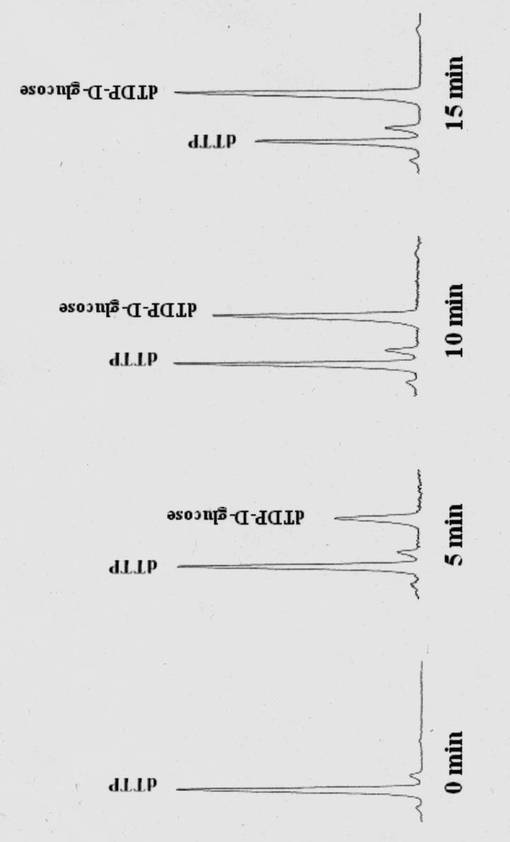
HPLCを用いて反応生成物であるGDPマンノースの量をヌクレオチド部分の紫外線の吸収を指標に測定した。図2に示すように標準物質であるGDP及びGDPマンノースは、HPLCにおいて溶出位置が異なる。さらに、図3に示すように標準サンプル添加量を変化させたときのピークの面積と標準物質量は正確な比例関係にあり、この検量線を用いることにより反応生成物を定量できることが示された。
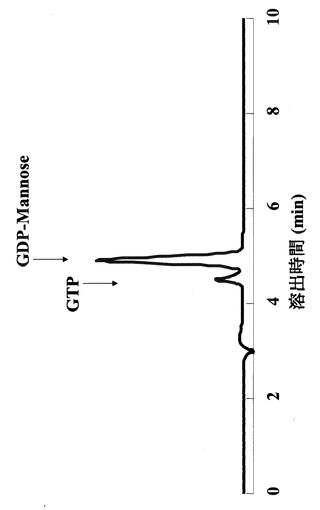
そこで、上記(1)で反応させたサンプルに関しても、HPLCで同様の反応の解析を行った。その結果、図4に示すように、確かにGDPマンノースが見出されたことから、該酵素は、糖ヌクレオチドを確かに生成している事が確認された。
【0028】
[実施例3] リン酸化糖異性体合成
(1)リン酸化糖異性化反応
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、1mM NADP(ニコチンアミドアデニンジヌクレオチドリン酸酸化型)、1unit グルコース−6−リン酸異性化酵素、1unit グルコース−6−リン酸脱水素酵素、0.4mMマンノース−6−リン酸、からなる酵素反応溶液2,000μL中に実施例1で得られた精製酵素2.0μgを加えた。この酵素反応液を65℃で保温することにより、反応させた。この反応により、マンノース−6−リン酸が異性化されたフルクトース−6−リン酸が生成する。
【0029】
(2)糖リン酸異性化反応の測定
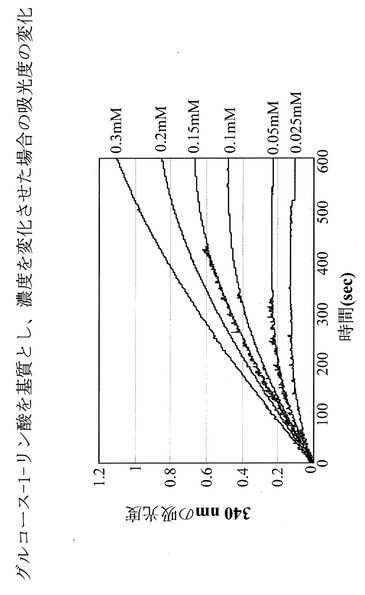
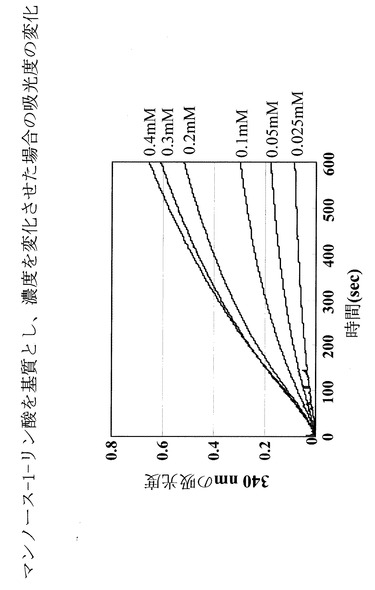
反応生成物であるフルクトース−6−リン酸を、グルコース−6−リン酸異性化酵素によりグルコース−6−リン酸に変換し、このグルコース−6−リン酸をグルコース−6−リン酸脱水素酵素により6−リン酸グルコノラクトンに変換する際の副反応であるNADPの酸化型から還元型への変化量を紫外・可視分光光度計によって波長340nmにおける吸光度の変化を指標に測定した。
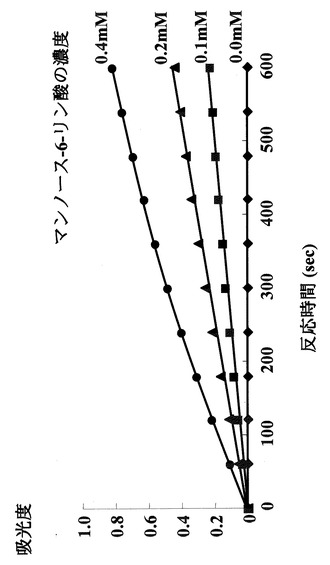
NADPの酸化型と還元型では340nmの吸光度が全く異なる。さらに、図5に示すように、酵素反応溶液中の基質であるマンノース−6−リン酸濃度を変化させたときの、吸光度の計時変化とNADP還元型の量は正確な比例関係にあり、この検量線を用いることにより反応生成物を定量できることが示された。
そこで、(1)で反応させたサンプルに関しても、紫外・可視分光光度計による340nmの吸光度を用いて(1)の反応の解析を行った。その結果、図6に示すように、確かに吸光度の増加が見出されたことから、該酵素は、マンノース−6−リン酸を異性化しフルクトース−6−リン酸を確かに生成している事が確認された。
【0030】
[実施例4] 酵素の性質
(1) 蛋白質化学的性質
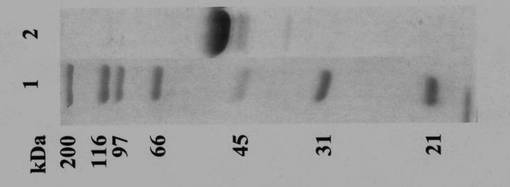
当該酵素は上記の精製プロセスで完全に精製され、SDS-PAGEで分子量約53,000Daであった(図1)。当該酵素は464アミノ酸残基より構成され(配列番号1)、そのアミノ酸配列から予測された分子量は52,888.64 Daであった。また、ヘリコバクター菌のGMP/PMIとの相同性も見出され、(pyrophosphorylase signature)活性中心、金属イオン結合、糖認識に関与するモチーフが保存されていた(図1)。
【0031】
(2)糖ヌクレオチド合成活性(糖ヌクレオチドの結合活性)
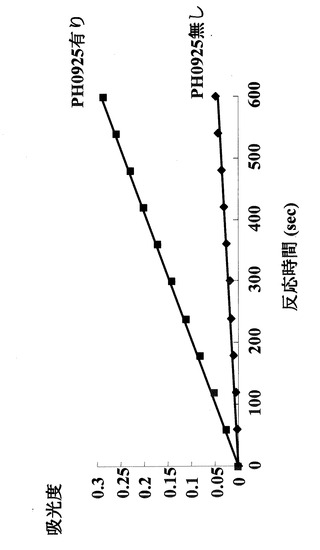
実施例1により精製された該酵素を用いて、マンノース−1−リン酸とGTPとの結合活性を実施例2に従って測定した。その結果、図4にあるように時間に依存したGDPマンノースの合成が85℃の反応温度で検出された。従って耐熱性の高い糖ヌクレオチド合成活性を示すことが確認された。
【0032】
(3)pH依存性
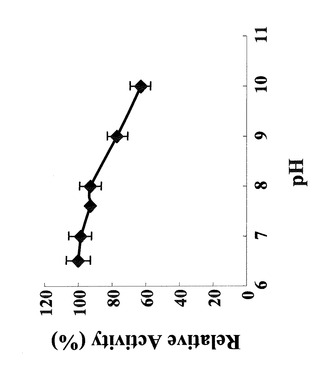
2mM MgSO4、1mM Mannose-1-リン酸、0.05mM GTPからなる酵素反応溶液10μ 中の50mM緩衝液をリン酸ナトリウム塩緩衝液pH6.0、pH6.5、MOPS緩衝液pH7.0、Tris-HCl緩衝液pH8.0、グリシン-NaOH緩衝液pH9.0、pH10.0に変化させ実施例1で得られた精製酵素1ngを加えた。この酵素反応液を85℃で5分間保温することにより反応させた後に、100μLの500mM KH2PO4溶液を加えることにより反応を停止させた。反応の進行は、実施例2の(2)にあるようにHPLCで測定した。図8に示すように、本酵素の活性はいずれのpHにおいても高い活性を示した。
【0033】
(4)ヌクレオシド三リン酸基質の多様性
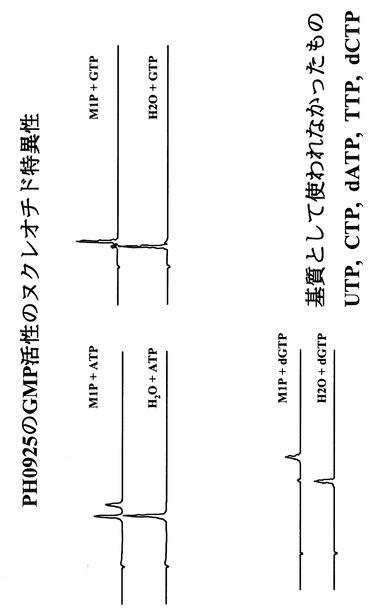
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、1mM Mannose-1-リン酸及び0.05mM GTPまたは0.05mM ATP等からなる酵素反応溶液10μL中に実施例1で得られた精製酵素1 ngを加えた。この酵素反応液を85℃で5分間保持することにより反応させた後に、100μLの500mM KH2PO4溶液を加えることにより反応を停止させた。結果を図9に示す。図9から分かるように、本酵素はGTP以外にATP、dGTPを基質として利用できる事が示された。
【0034】
(5)糖−1−リン酸基質の多様性
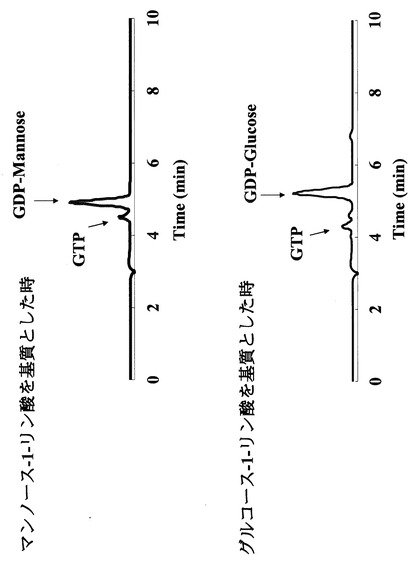
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、0.05mM dGTP及び1mM マンノース−1−リン酸、または1mM グルコース−1−リン酸からなる酵素反応溶液10μL中に実施例1で得られた精製酵素1 ngを加えた。この酵素反応液を85℃で5分間保持することにより反応させた後に、100μLの500mM KH2PO4溶液を加えることにより反応を停止させた。結果を図10に示す。図10から分かるように、本酵素はマンノース−1−リン酸以外にグルコース−1−リン酸を基質として利用できる事が示された。
【0035】
(6)リン酸化糖異性化活性
実施例1により精製された該酵素を用いて、マンノース−6−リン酸の糖分子を異性化することでフルクトース−6−リン酸に変化させる活性を実施例3に従って測定した。その結果、図6に有るように、基質であるフルクトース−6−リン酸が反応時間に従って生産される活性が65℃で検出された。この事から、本酵素はリン酸化糖異性化活性を有する熱に安定な酵素であることが示された。
【0036】
[参考例]
以下に、本発明の糖−ヌクレオチド生産に用いる、特願2004−236321号に係るリン酸化糖のリン酸基分子内転移酵素の製造例及び該酵素の酵素作用の試験結果を参考例1、2として示す。
【0037】
[参考例1]
(1) 菌の培養
好酸性好気性超好熱性古細菌スルフォロバス、トーコーダイイJCM10545は次の方法で培養した。
1.3g の(NH4)2SO4、0.28gのKH2PO4、0.25gのMgSO4・7H2O、0.07gのCaCl2・2H2O、0.02gのFeCl3・6H2O、1.8mgのMnCl2・4H2O、4.5mgのNa2B4O7・10H2O、0.22mgのZnSO4・7H2O、0.05mgのCuCl2・2H2O、0.03mgのNa2MoO4・2H2O、0.03mgのVoSO4・xH2O、0.01mgのCoSO4・7H2O、1.0gの酵母エキスを1リッターの蒸留水に溶かし、この溶液のpHを3.5に10規定H2SO4溶液で調製した。加圧殺菌した後、JCM10545を植菌した。この培養液を80℃で1〜2日培養し、その後遠心分離し集菌した。
【0038】
(2) 染色体DNAの調製
JCM10545の染色体DNAは以下の方法により調製した。
培養終了後5,000rpm、10分間の遠心分離により菌体を集菌する。菌体を10mM EDTA(pH 6.0)溶液で洗浄後、50mM Tris/HCl-50mM EDTA(pH8.5)溶液を加えて細胞を溶解させる。さらに、0.5% Na-lauroylsarcosinate、1mg/mL プロテアーゼKとなるように各々を加えた後、50℃で3時間保温する。フェノール処理を3回行った後、溶液を10mM EDTA (pH8.0)溶液に対して透析する。37℃で30分間のRNaseによるRNAの分解後、フェノールクロロフォルム溶液で処理した後、10mM Tris-1mM EDTA(pH8.0)で透析を行った。
【0039】
(3) 染色体DNAを含むショットガンライブラリークローンの作製
上記(2)の工程で得られた染色体DNAを超音波処理することにより断片化した後、アガロースゲル電気泳動により1kb及び2kb長のDNA断片を回収した。この断片をプラスミドベクターpUC118のHincII制限酵素部位に挿入したショットガンライブラリーを作製した。各ショットガンクローンの末端塩基配列を、ABI社製自動塩基配列読み取り装置377を用いて解読していった。
【0040】
(4) STPMM/PGM遺伝子の同定
上記手法で決定された好酸性好気性超好熱古細菌スルフォロバス、トーコーダイイのゲノム塩基配列の大型計算機による解析を行い、phosphomannomutase/phosphoglucomutase酵素の機能を含むであろう蛋白質をコードする遺伝子(ST0242)を同定した。この超好熱性古細菌スルフォロバス、トーコーダイイのST0242遺伝子の開始コドンはATGで、455アミノ酸残基の蛋白質(配列番号5)をコードする遺伝子として同定された(配列番号6)。
【0041】
(5)発現プラスミドの構築
構造遺伝子領域の前後に制限酵素(NdeIとXhoI)サイトを構築する目的でDNAプライマーを合成し、PCRでその遺伝子の前後に制限酵素サイトを導入した。
【0042】
Upper primer、
5’- TTAATTCCATATGGGTAAGCTTTTTGGTACTGAC -3’ (配列番号7)
(下線部はNdeIサイトを示す)
Lower primer、
5’- ATATACTCGAGTCATTTACCCTCTACAATC -3’ (配列番号8)
(下線部はXhoIサイトを示す)
【0043】
Upper primerとLower primerを組み合わせたPCR反応後、制限酵素(NdeIとXhoI)で完全分解(37℃で2時間)した後、その構造遺伝子領域断片を精製した。
制限酵素NdeIとXhoIで切断後精製したpET28a(Novagen社製)と上記の構造遺伝子(ST0242)領域断片とT4リガーゼを用いて16℃、2時間反応させることによって連結した。連結したDNAの一部を大腸菌DH5αのコンピテントセルに導入し形質転換体のコロニーを得た。得られたコロニーからプラスミドをQIAprep Spin Miniprep Kit(QIAGEN社製)で精製し、塩基配列を確認して発現プラスミド、pET28a/ST0242を得た。発現プラスミドpET28a/ST0242を用いるとSTPMM/PGMはN末端にヒスチジンタグが付加された融合蛋白質として生産される。
【0044】
(6)組み換え遺伝子の発現
大腸菌 BL21-CodonPlus(DE3)-RIL、(Novagen社製)のコンピテントセルを融解して、ファルコンチューブに0.1ml移す。その中に上記の発現プラスミド10ng分に相当する溶液を加え氷中に30分間放置した後、42℃でヒートショックを30秒間おこない、そこにSOC培地0.9mlを加え、37℃で1時間振とう培養する。その後、カナマイシンを含むLB寒天プレート上に適量まき、37℃で一晩培養し、形質転換体大腸菌BL21-CodonPus(DE3)-RIL/pET28a/ST0242を得た。
【0045】
当該形質転換体をカナマイシン含有LB培地(2リットル)中で一晩37℃において振とう培養した後、IPTG(Isopropyl-β-D-thiogalacopyranoside)を1mMになるように加え、さらに30℃で5時間振とう培養した。培養後、遠心分離(6000rpm、20分間)により集菌をおこなった。
【0046】
【0047】
[参考例2]
リン酸化糖のリン酸基分子内転移反応の測定
a.グルコース−1−リン酸のリン酸基分子内転移反応
(1)グルコース−1−リン酸のリン酸基分子内転移反応の酵素反応溶液
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、10μM グルコース 1,6-ビスリン酸、1mM NADP(ニコチンアミドアデニンジヌクレオチドリン酸酸化型)、1unit グルコース−6−リン酸脱水素酵素、0.4mMグルコース−1−リン酸、からなる酵素反応溶液2,000μL中に参考例1で得られた精製酵素3.5μgを加えた。この酵素反応液を65℃で保温することにより、反応させた。この反応により、グルコース−1−リン酸のリン酸基が分子内転移しグルコース−6−リン酸が生成する。
【0048】
(2) グルコース−1−リン酸のリン酸基分子内転移反応の測定
本酵素を用いた反応の反応生成物であるグルコース−6−リン酸をグルコース−6−リン酸脱水素酵素により6−リン酸グルコノラクトンに変換する際に、副反応としてニコチンアミドアデニンジヌクレオチドリン酸(NADP)が酸化型から還元型へ変化する。その変化量を340nmにおける吸光度の変化として紫外・可視分光光度計を用いて測定した(Klingenberg M “Methods of Enzymatic Analysis” (1974) Weinheim-Academic Press, New York, vol. 4, pp. 2045参照)。NADPの酸化型と還元型では波長に依存した吸光度プロファイルが全く異なる。特に340nmの吸光度が全く異なる事から、この波長の吸光度を利用して生成された還元型NADPの量を定量する。
【0049】
そこで、(1)で反応させたサンプルに関しても、紫外・可視分光光度計による340nmの吸光度を用いて(1)の反応の解析を行った。図14に示すように、基質であるグルコース−1−リン酸の濃度に依存した活性が65℃で検出された。
実施例1により精製された該酵素を用いて、グルコース−1−リン酸及びマンノース−1リン酸が有するリン酸基の分子内転移活性を実施例3,4に従って測定した。その結果、図14に有るように、基質であるグルコース−1−リン酸の濃度に依存した活性が65℃で検出された。また、図15に有るように、同様の活性がマンノース−1−リン酸を基質としたときにも検出されたことから、該酵素は耐熱性PGM/PMM活性を有する事が確認された。また、グルコース−6−リン酸を基質とした場合にも、図16に示されたように、確かに分子内リン酸基が一位に転移している事からTDPグルコースの生産が確認された。
【0050】
b.マンノース−1−リン酸のリン酸基分子内転移反応
(1) マンノース−1−リン酸のリン酸基分子内転移反応の酵素反応溶液
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、10μM グルコース 1,6-ビスリン酸、1mM NADP、1unit グルコース−6−リン酸脱水素酵素、1unit マンノース−6−リン酸異性化酵素、1unit グルコース−6−リン酸異性化酵素、0.4mMマンノース−1−リン酸、からなる酵素反応溶液2,000μL中に実施例1で得られた精製酵素7.0μgを加えた。 この酵素反応液を65℃で保温することにより、反応させた。この反応により、マンノース−1−リン酸のリン酸基が分子内転移しマンノース−6−リン酸が生成する。
【0051】
(2)マンノース−6−リン酸化合物のリン酸基分子内転移反応の測定
本酵素を用いた反応の反応生成物であるマンノース−6−リン酸をマンノース−6−リン酸異性化酵素によりフルクトース−6−リン酸に変換し、フルクトース−6−リン酸をグルコース−6−リン酸異性化酵素によりグルコース−6−リン酸に変換し、グルコース−6−リン酸脱水素酵素により6−リン酸グルコノラクトンに変換する際に、副反応としてニコチンアミドアデニンジヌクレオチドリン酸(NADP)が酸化型から還元型へ変化する。その変化量を340nmにおける吸光度の変化として紫外・可視分光光度計を用いて測定した(非特許文献4参照)。NADPの酸化型と還元型では波長に依存した吸光度プロファイルが全く異なる。特に340nmの吸光度が全く異なる事から、この波長の吸光度を利用して生成された還元型NADPの量を定量する。
そこで、(1)で反応させたサンプルに関しても、紫外・可視分光光度計による340nmの吸光度を用いて(1)の反応の解析を行った。図15に示すように、基質であるマンノース−1−リン酸の濃度に依存した活性が65℃で検出された。
【0052】
c.グルコース−6−リン酸のリン酸基分子内転移反応
(1)グルコース−6−リン酸のリン酸基分子内転移反応の酵素反応溶液
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、10μl グルコース1,6ビスリン酸、0.4mM TTP、2.5μg スルフォロバス・トーコーダイイ由来のRmlA(glucose-1-phosphate thymidylyltransferase )、1mMグルコース−6−リン酸からなる酵素反応溶液2,000μL中に、実施例1で得られた精製酵素2.5μgを加えた。この酵素反応液を80℃で30分間保温することにより反応させた。反応後、100μLの500mM リン酸二水素カリウム溶液を加えることにより反応を停止させた。この反応により、グルコース−6−リン酸のリン酸基が分子内転移しグルコース−1−リン酸が生成する。
【0053】
(2)グルコース−6−リン酸のリン酸基分子内転移反応の測定
HPLCを用いて、反応性生物であるグルコース−1−リン酸をRmlAによりTDP-グルコースに変換し、TDP-グルコースの量をヌクレオチド部分の紫外線の吸収を目安に測定した。図2からも明らかなように、標準物質であるTTPおよびTDP-グルコースは、HPLCにおいて溶出位置がまったく異なる。そこで、上記(1)で反応させたサンプルに関しても、HPLCで同様の解析をおこなった。その結果、図16に示すように、確かにTDP-グルコースが見出されたことから、該酵素は、グルコース−6−リン酸の分子内リン酸基を転移させたグルコース−1−リン酸を確かに生成している事が確認された。
【図面の簡単な説明】
【0054】
【図1】精製されたPHGMP/PMI蛋白質のSDS-PAGEパターンを示す図である。
【図2】HPLCによるGTP及びGDP-Mannoseの分離パターンを測定した図である。
【図3】GDPマンノース標準物質の添加量とピーク面積変化の関係を示す図である。
【図4】PHGMP/PMI蛋白質の85℃における糖ヌクレオチド合成活性を示す図である。
【図5】マンノース−6−リン酸を基質とし、その濃度を変化させたときの吸光度の経時変化を示す図である。
【図6】PHGMP/PMI蛋白質による波長340nmにおける吸光度の経時変化を示す図である。
【図7】PHGMP/PMI蛋白質とへリコバクター菌のGMP/PMI蛋白質との相同性を示す図である。
【図8】PHGMP/PMI蛋白質のpHの違いによる活性変化を示す図である。
【図9】PHGMP/PMI蛋白質のヌクレオシド三リン酸基質の多様性を示す図である。
【図10】PHGMP/PMI蛋白質の糖リン酸基質の多様性を示す図である。
【図11】本発明の酵素タンパク質および特願2004−236321号の酵素を用いてフルクトース−6−リン酸からGDP−マンノースを合成する反応工程を示す概略図である。
【図12】本発明の酵素のリン酸化糖の異性化活性に対する各金属イオン種の影響を示す図である。
【図13】本発明の酵素の糖−ヌクレオチド合成活性に対する各金属イオン種の影響を示す図である。
【図14】参考例2において、グルコース−1−リン酸を基質とし、その濃度を変化させた場合の波長340nmにおける吸光度の経時変化を示す図である。
【図15】参考例2において、マンノース−1−リン酸を基質とし、その濃度を変化させた場合の波長340nmにおける吸光度の経時変化を示す図である。
【図16】参考例2において、グルコース−6−リン酸を基質とした場合において、TDP-グルコース生成の経時変化を示す図である。
【技術分野】
【0001】
本願発明は、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する耐熱性蛋白質、該蛋白質をコードするDNA、該DNAを含有する組み換え体DNA、該組み換え体DNAを保有する形質転換体、該形質転換体を用いた糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質の製造法、および該蛋白質あるいは該形質転換体を用いた糖ヌクレオチドの製造法および異性化リン酸化糖の製造法に関する。
【背景技術】
【0002】
糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する酵素としては、サルモネラ菌(Salmonella enterica) (非特許文献1参照)、豚の肝臓 (非特許文献2参照)、マイコバクテリア菌(Mycobacterium smegmatis)(非特許文献3参照)由来の糖ヌクレオチド合成活性を示す酵素であるGDP-mannose pyrophosphorylase(GMP)、及びヘリコバクター菌(Helicobacter pylori)(非特許文献4参照)のリン酸化糖異性化活性を示す酵素の詳しい性質がすでに報告されている。GMPはGDP-マンノース合成に必須な酵素であり、マンノース−1−リン酸とグアノシン三リン酸(GTP)を基質としてGDP-マンノースを生産する。また、リン酸化糖異性化活性を示す酵素であるPhosphomannose isomerase(PMI)は、マンノース−6−リン酸を基質としてフルクトース−6−リン酸を生産する反応と、フルクトース−6−リン酸を基質としてマンノース−6−リン酸を生産する反応と両方向に働き、マンノース−6−リン酸の異性化に必須の酵素である。その他にも、ヒトや酵母、病原菌からも糖ヌクレオチド合成反応並びにリン酸化糖の異性化反応を触媒する酵素は見出されているが、その多くが常温生物由来のため室温以上では極めて不安定で、活性は80℃程度の加熱処理により速やかに失活する。このため、使用時に反応系を滅菌する等の処理が必要であったり、低温での注意深い保存が必要であった。
【0003】
【非特許文献1】Elling L, RitterJE, Verseck S. “Expression, purification andcharacterization of recombinant phosphomannomutase and GDP-alpha-D-mannosepyrophosphorylase from Salmonella enterica, group B, for the synthesis ofGDP-alpha-D-mannose from D-mannose” (1996) Glycobiology, 6, 591-597.
【非特許文献2】Ning B, Elbein AD.“Cloning, expression and characterization of the pigliver GDP-mannose pyrophosphorylase. Evidence that GDP-mannose and GDP-Glcpyrophosphorylases are different proteins” (2000) European Journal of Biochemistry, 267,6866-6874.
【非特許文献3】Ning B, Elbein AD.“Purification and properties of mycobacterialGDP-mannose pyrophosphorylase” (1999) Archives of Biochemistry and Biophysics,362(2),339-345.
【非特許文献4】Wu B, Zhang Y,Zheng R, Guo C, Wang PG. “Bifunctional phosphomannoseisomerase/GDP-D-mannose pyrophosphorylase is the point of control forGDP-D-mannose biosynthesis in Helicobacter pylori” (2002) FEBS Letter 519,87-92.
【発明の開示】
【発明が解決しようとする課題】
【0004】
糖ヌクレオチドを合成する活性を有し、且つ耐熱性を有する酵素は、糖鎖合成の基質となる糖ヌクレオチドを安定に合成することを可能にするなど極めて有用である。また、糖鎖合成の際の基質として必要な様々な種類の糖を生産する際には、糖の異性化反応は有効な反応である。このリン酸化糖異性化反応活性を有し、且つ耐熱性を有する酵素は、様々な種類の糖を安定に合成することを可能にするなど極めて有用である。すなわち、これら両酵素活性を併せ持つ耐熱性酵素が見出されれば、糖鎖合成の基質となる様々な種類の糖ヌクレオチドを安定に生産することが可能となる。また、これらの反応を一つの酵素が行うことで、様々な種類のリン酸化糖を安定に合成することと、それらを用いて糖ヌクレオチドを合成することを共役させることが出来、効率的な反応として進めることが可能になる。そこで、両酵素活性を有する両反応性酵素で、且つ熱安定性を有する酵素は渇望されていた。
【0005】
したがって、本発明の課題は、様々な糖ヌクレオシド三リン酸を基質として糖ヌクレオチド合成が可能で、且つリン酸化糖を基質として異なる種類の異性化されたリン酸化糖の合成が可能な、2種類の活性を有し、かつ熱安定な耐熱性を有する新規酵素を提供することにある。
【課題を解決するための手段】
【0006】
本発明は、以上のような課題を解決すべく、90−95℃で生育する超好熱古細菌Pyrococcus horikoshii OT3に着目し、そのゲノム中に本酵素活性を有する遺伝子を見いだした。さらに、大腸菌を使ってその遺伝子がコードする該酵素を生産し、この酵素が高温(90℃)で安定に存在し、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を示すことを確認し、さらに、本酵素を用いることにより様々な糖ヌクレオチドを生産すること並びに種々の糖リン酸の異性体を生産することも見出して、本発明を完成するに至ったものである。
【0007】
即ち、本発明は、以下の(1)〜(15)に係るものである。
(1) 配列番号1に記載のアミノ酸配列を有するか、あるいは、配列番号1に記載のアミノ酸配列において1乃至数個のアミノ酸残基が欠失、置換、挿入又は付加されたアミノ酸配列を有し、かつ糖ヌクレオチド合成活性及びリン酸化糖異性化活性を有することを特徴とする蛋白質。
(2) 上記(1)に記載の蛋白質をコードするDNA。
(3) 上記(2)に記載の塩基配列を有することを特徴とするDNA。
(4) 配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズし、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質をコードすることを特徴とするDNA。
(5) 上記(2)〜(4)のいずれかに記載のDNAがベクターに組み込まれていることを特徴とする組み換え体DNA。
(6) 上記(5)に記載の組み換え体DNAが宿主細胞に導入されていることを特徴とする形質転換体。
(7) 上記(6)に記載の形質転換体を培地に培養し、培養物から、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質を採取することを特徴とする、糖ヌクレオチド合成活性及びリン酸化糖異性化活性を有する蛋白質の製造法。
(8) 糖−1−リン酸及びヌクレオシド三リン酸に、上記(1)に記載の蛋白質を作用させ、糖ヌクレオチドに変換することを特徴とする糖ヌクレオチドの製造方法。
(9) 糖−6−リン酸に、上記(1)に記載の蛋白質を作用させ、糖リン酸を異性化させることを特徴とする異性化された糖−6−リン酸の製造方法。
(10) 糖−1−リン酸及びヌクレオシド三リン酸に、上記(6)に記載の形質転換体の培養液あるいは培養物の処理物を作用させ、糖ヌクレオチドに変換することを特徴とする、糖ヌクレオチドの製造方法。
(11) 糖−6−リン酸に、上記(6)に記載の形質転換体の培養液あるいは培養物の処理物を作用させ、糖−6−リン酸を異性化させることを特徴とする、異性化された糖−6−リン酸の製造方法。
(12) 糖−1−リン酸及びヌクレオシド三リン酸に、上記(3)又は(4)に記載のDNAにコードされる蛋白質を作用させ、糖ヌクレオチドに変換することを特徴とする、糖ヌクレオチドの製造方法。
(13) 糖−6−リン酸に、上記(3)又は(4)に記載のDNAにコードされる蛋白質を作用させ、糖リン酸を異性化させることを特徴とする、異性化された糖−6−リン酸の製造方法。
(14) リン酸化糖の異性化反応、分子内リン酸基転移反応及び糖−ヌクレオチド合成反応を行い、糖−ヌクレオチドを製造する方法において、上記リン酸化糖の異性化反応およ糖−ヌクレオチド合成反応を、請求項1に記載の蛋白質を用いて行うことを特徴とする、上記方法。
(15) 請求項1に記載のタンパク質を含有する反応系において、カルシウムイオン、亜鉛イオンまたは銅イオンのうちいずれかを選択して含有させることにより、該タンパクの酵素作用を、糖ヌクレオチド合成作用とリン酸化糖異性化作用との間で切り替えることを特徴とする、酵素反応の切り替え方法。
【発明の効果】
【0008】
本発明により、試験管内での様々な種類の糖ヌクレオチドを合成する事が可能でかつ熱等に安定で、さらに試験管内での様々な種類のリン酸化糖異性体を合成することが可能でかつ熱等に安定な新規な両反応を触媒する酵素が提供できた。その結果、新規な糖ヌクレオチド及びリン酸化糖の合成が可能になった。一方、本酵素により生産が可能となるリン酸化糖異性体や糖ヌクレオチドは糖蛋白質、糖脂質、多糖類の糖合成に糖提供体として機能するものであり、これらの糖鎖合成は、癌転移、器官発生あるいは細胞性免疫等に密接に関与するものとして近年注目されており、本発明はこれらの研究の発展において、その貢献度はきわめて大きい。
【発明を実施するための最良の形態】
【0009】
以下に、本願発明を具体的に説明する。
本発明で使用した超好熱古細菌は、超好熱古細菌パイロコッカス、ホリコシイ(JCM登録番号JCM9974)である。本発明者等は超好熱古細菌染色体上の本酵素活性を示す遺伝子領域を、PCR反応で増幅・抽出し、蛋白質発現プラスミドpET28aに挿入後、そのプラスミドにより形質転換した大腸菌を用いて本酵素の生産をおこなった。生産された酵素は加熱処理およびカラムクロマトグラムで単離精製した。精製された酵素は分子量が約53,000の蛋白質で様々な糖ヌクレオチドを合成する活性を有する酵素であることが判明した。
この酵素は50mM MOPS緩衝液(pH 7.6)中で、80℃30分以上の加熱後も活性を有し、高い耐熱性を示した。また、該タンパク質溶液の金属塩の種類を変更することにより、該タンパクの酵素作用が、糖ヌクレオチド合成作用とリン酸化糖異性化作用との間で切り替わるという驚くべき性質を有する。
この超好熱古細菌パイロコッカス、ホリコシイが有するGMP/PMI(PHGMP/PMI)のアミノ酸配列およびその遺伝子DNA(PH0925)の塩基配列を、それぞれ配列表の配列番号1および2に示す。
【0010】
本発明における酵素タンパク質は、上記配列番号1に示されるアミノ酸配列を有するもののみに限定されず、該アミノ酸配列において、1乃至数個のアミノ酸残基が欠失、置換、挿入又は付加されたアミノ酸配列であっても、このアミノ酸配列を有する蛋白質が、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を示すものも含む。また、本発明のこれら酵素遺伝子DNAについても、上記同配列番号2に示す塩基配列を有するもののみに限定されず、上記アミノ酸配列をコードするものを包含する。さらに上記配列2に示されるDNAにストリンジェントな条件下でハイブリダイズし、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質をコードするDNAも包含する。このストリンジェントな条件とは、ハイブリダイゼーション溶液1リットル中に52.59gNaCl、26.46gクエン酸ナトリウム、1gフィコール(Type 400)、1gポリビニルピロリドン、1gウシ血清アルブミン、5gSDS、1g断片化鮭精子DNA、500ml ホルムアミドを含み、温度42℃で行う。その後の洗浄は、洗浄用溶液1リットル中に17.53gNaCl、8.82gクエン酸ナトリウム、5gSDSを含み、温度68℃で行う条件である。
【0011】
本発明の酵素タンパク質を得るには、通常の遺伝子工学的手法が適用でき、上記酵素遺伝子DNAを、例えばpET28a、pHY481等の蛋白質発現プラスミドベクター等に挿入して組み換えベクターを作製し、該組み換えベクターを用いて宿主細胞を形質転換し、該形質転換体を培地で培養し、培養物、培養処理物あるいはこれら培養物から分離回収された形質転換体から、酵素タンパク質を通常の蛋白質精製手段により精製し単離する。上記宿主細胞としては、大腸菌・枯草菌等が利用可能である。
【0012】
本発明においては、さらにこの酵素タンパク質を用いて、糖ヌクレオチドを合成するが、この合成においては、糖−1−リン酸とヌクレオシド三リン酸を含有する溶液に該酵素を添加し、反応温度60〜95℃で反応させ糖ヌクレオチドを得る。
糖−1−リン酸としては、例えば、マンノース−1−リン酸、グルコース−1−リン酸等が挙げられ、ヌクレオシド三リン酸としては、例えばグアノシン三リン酸(GTP)、アデノシン三リン酸(ATP)酸等が挙げられ、さらに、本発明の酵素はデオキシリボヌクレオシド三リン酸も基質とすることができる。
【0013】
この糖ヌクレオチド合成活性の反応式として、マンノース−1−リン酸とGTPからGDP-マンノースを合成する場合について以下に示す。
【化1】
【0014】
また、本発明においては、さらにこの酵素を用いて、リン酸化糖異性体を合成するが、この合成においては、糖−6−リン酸に該酵素を添加し、反応温度60〜95℃で反応させ糖−6−リン酸異性体を得る。
糖−6−リン酸としては、例えば、マンノース−6−リン酸、フルクトース−6−リン酸等が挙げられる。
【0015】
このリン酸化糖異性化活性の反応式として、マンノース−6−リン酸からフルクトース−6−リン酸を合成する場合について以下に示す。
【化2】
【0016】
また、この反応においては、上記精製した酵素のみならず、粗酵素であってもよい。例えば、宿主として枯草菌等分泌型の系を用いる場合には、培養液中に本酵素が生成蓄積され、大腸菌等の非分泌型の系を用いる場合には、菌体内に生成されるので、本酵素を含有する培養液あるいはその処理物、もしくは菌体破砕物等の培養処理物を用いて、糖ヌクレオチドあるいは糖リン酸異性体を合成してもよい。
本発明の酵素タンパク質は、他の酵素と組み合わせることにより、一連の酵素反応系からなる糖ヌクレオチド合成系を構築することができる。図11は、本発明の酵素タンパク質と、本発明者等の先の出願(特願2004−236321号)に係るリン酸化糖のリン酸基分子内転移酵素とを組み合わせることにより、フルクトース−6−リン酸からGDP−マンノースを生成する合成系の構築例である。
上記リン酸基分子内転移酵素は、好酸性好気性超好熱古細菌スルフォロバス、トーコーダイイ(JCM登録番号JCM10545)由来の酵素であり、そのアミノ酸配列及び遺伝子の塩基配列は配列番号5、6に示される。使用する糖リン酸のリン酸基分子内転移酵素としては、配列番号5に示すアミノ酸配列を有するものの他、該アミノ酸配列において、1乃至数個のアミノ酸残基が欠失、置換、挿入または付加されたアミノ酸配列を有するものであって、かつ糖リン酸化糖のリン酸基分子内転移活性を有するものも使用できる。
該酵素は、マンノース−6−リン酸、グルコース−6−リン酸等の糖−6−リン酸のリン酸基を分子内転移させて、それぞれマンノース−1−リン酸、グルコース−1−リン酸等の糖−1−リン酸を生成する。この酵素は60〜95℃で反応可能であり、上記酵素を組み合わせた反応系は耐熱性である。
図11に示されるGDP−マンノース合成系においては、まず、本発明の酵素タンパク質の糖異性化作用を利用して、フルクトース−6−リン酸からマンノース−6−リン酸を生成させ、次いで、上記リン酸基分子内転移酵素を用いて、生成したマンノース−6−リン酸のリン酸基を分子内転移させて、マンノース−1−リン酸を生成させる。この後、本発明の酵素タンパク質の糖−ヌクレオチド合成作用を利用して、マンノース−1−リン酸とGTPからGDP−マンノースを生成させる。
この糖−ヌクレオチド合成系においては、該合成系の各反応工程において、各酵素溶液を順次基質に接触させて各反応を行ってもよいが、本発明の酵素タンパク質と上記リン酸基分子内転移酵素とを共に含有する酵素反応系を用いることにより、異性化反応、分子内リン酸基転移反応及び糖−ヌクレオチド合成反応を一つの反応工程で行い、糖−ヌクレオチドを合成することも可能である。
また、図12及び図13に示されるように、本発明の酵素タンパク質は、酵素反応系に含有させる金属イオンによって、その酵素作用を切り替えることが可能である。図12は、各金属イオンを2mM濃度で酵素反応系に含有させた場合の、本酵素タンパク質のイソメラーゼ活性を測定した結果を示す図であり、図13は、同糖−ヌクレオチド合成活性を示す図である。図12の結果によれば、本酵素タンパク質の糖リン酸化糖の異性化活性は、Caイオンを含有させた場合に高いことが分かる。また、亜鉛イオン、銅イオンを含有させた場合は、糖リン酸化糖の異性化活性は低く、検出限界以下であった。一方、図13の結果によれば、同糖−ヌクレオチド合成活性は、亜鉛イオン、銅イオンを含有させた場合は高く、カルシウムを含有させた場合には低い。すなわち、本発明の酵素タンパク質の反応系においては、カルシウムイオンを含有させる場合は、リン酸化糖の異性化活性を主に示し、亜鉛イオンあるいは銅イオンを含有させた場合は糖−ヌクレオチド合成活性を示す。
したがって、本発明においては、本発明の酵素タンパク質の作用を含有させる金属イオン種の種類により変更可能であり、このことは、本酵素タンパク質の異なる作用を利用する上記糖−ヌクレオチド合成系に応用可能である。なお、本発明の酵素タンパク質は、上記金属イオンを含有させない場合においては、上記リン酸化糖の異性化活性と上記糖−ヌクレオチド合成活性を併せて有する。
以下に、本発明の実施例を示すが、本発明実施例により限定されるものではない。
【実施例】
【0017】
[実施例1] 糖ヌクレオチド合成及びリン酸化糖異性化酵素の製造
(1) 菌の培養
好酸性好気性超好熱性古細菌パイロコッカス、ホリコシイJCM9974は次の方法で培養した。
13.85gのNaCl、3.5gのMgSO4・7H2O、2.75gのMgCl2・6H2O、0.325gのKCl、0.05gのNaBr、15mgのH3BO3、7.5mgのSrCl2・6H2O、0.05mgのKI、0.75gのCaCl2、0.5gのKH2PO4、2.0mgの(KH4))2Ni(SO4)2・6H2Oを1Lの蒸留水に溶かしSalt base solutionを作製した。1.5gのNitrilotriacetic acidを1Lの蒸留水に溶かし、この溶液のpHを6.5にKOH溶液で調製した。その溶液に3.0gのMgSO4・7H2O、0.5gのMnSO4・xH2O、1.0gのNaCl、0.1gのFeSO4・7H2O、0.1gのCoSO4・7H2O、0.1gのCaCl2・2H2O、0.1gのZnSO4・7H2O、0.01gのCuSO4・5H2O、0.01gのAlK(SO4)2、0.01gのH3BO3、0.01gのNa2MoO4・2H2Oを加え、この溶液のpHを7.0にKOH溶液で調製し、Trace mineralsを作製した。1.0LのSalt bace solution、10.0mLのTrace minerals、5.0mLのクエン酸、1.0gのYeast extract、5.0gのBacto peptone、30.0gの硫黄、1.0mgのレサズリンを作製し、この溶液のpHを6.5にH2SO4溶液で調製した。この溶液を窒素下でフィルター滅菌し、1日に3時間硫黄蒸気に当てることを連続3日間行った。5%のNaS・9H2Oを窒素下でオートクレーブし、無菌かつ嫌気下で培地に加え、JCM9974を植菌した。この培養液を98℃で培養し、その後遠心分離し集菌した。
【0018】
(2) 染色体DNAの調製
JCM9974の染色体DNAは以下の方法により調製した。
培養終了後5000rpm、10分間の遠心分離により菌体を集菌する。菌体を10mMEDTA(pH 6.0)溶液で洗浄後、50mM Tris/HCl-50mM EDTA(pH8.5)溶液を加えて細胞を溶解させる。さらに、0.5% Na-lauroylsarcosinate、1mg/mlプロテアーゼKとなるように各々を加えた後、50℃で3時間保温する。フェノール処理を3回行った後、溶液を10mM EDTA(pH8.0)溶液に対して透析する。37℃で30分間のRNaseによるRNAの分解後、フェノールクロロフォルム溶液で処理した後、10mM Tris-1mM EDTA(pH8.0)で透析を行った。
【0019】
(3)発現プラスミドの構築
上記(2)で得た染色体DNAを鋳型とし、下記Upper primerとLower primerを使用して PCRを行った。PCRで得られたDNAは、目的の構造遺伝子領域の前後に制限酵素(NdeIとBamHI)サイトを有する。
【0020】
Upper primer、
5’- GGGAATTCCATATGAAGGCTTTGATTCTAG -3’ (配列番号3)
(下線部はNdeIサイトを示す)
Lower primer、
5’- CGCGGATCCTCACCTCCCGTAATCGTCTTG -3’ (配列番号4)
(下線部はBamHIサイトを示す)
【0021】
PCR反応後、制限酵素(NdeIとBamHI)で完全分解(37℃で2時間)した後、目的の構造遺伝子領域断片を精製した。該遺伝子領域(PH0925)の塩基配列は配列番号2に示される。
制限酵素NdeIとBamHIで切断後精製したpET28a(Novagen社製)と上記の構造遺伝子領域断片とをT4リガーゼを用いて16℃、2時間反応させることによって連結した。連結したDNAの一部を大腸菌DH5αのコンピテントセルに導入し、形質転換体のコロニーを得た。得られたコロニーからプラスミドをQIAprep Spin Miniprep Kit(QIAGEN社製)で精製し、塩基配列を確認して発現プラスミド、pET28a/PH0925を得た。発現プラスミドpET28a/PH0925を用いるとPHGMP/PMIはN末端にヒスチジンタグが付加された融合蛋白質として生産される。
【0022】
(6)組み換え遺伝子の発現
大腸菌 BL21-CodonPlus(DE3)-RIL、(Novagen社製)のコンピテントセルを融解して、ファルコンチューブに0.1ml移す。その中に上記の発現プラスミド10ng分に相当する溶液を加え氷中に30分間放置した後、42℃でヒートショックを30秒間おこない、そこにSOC培地0.9mlを加え、37℃で1時間振とう培養する。その後、カナマイシンを含むLB寒天プレート上に適量まき、37℃で一晩培養し、形質転換体大腸菌BL21-CodonPus(DE3)-RIL/pET28a/PH0925を得た。
【0023】
当該形質転換体をカナマイシン含有LB培地(2リットル)中で一晩37℃において振とう培養した後、IPTG(Isopropyl-β-D-thiogalacopyranoside)を1mMになるように加え、さらに30℃で5時間振とう培養した。培養後、遠心分離機(6000rpm、20分間)により集菌をおこなった。
【0024】
(7)PHGMP/PMI酵素の精製
2リットル培養液から集菌した菌体に2倍量の20mMリン酸ナトリウム塩緩衝液(pH7.4)、菌体1gあたり2mgのLysozyme(Wako社製)、0.1μgのDNaseI(Takara社製)を加え懸濁液を得た。氷上で1時間静置後、超音波破砕し、遠心分離(20000g 30分間)により上清液を得た。得られた上清液を80℃で30分間加熱した後、遠心分離(20000g 30分間)により上清液を得た。この上清液を20mMリン酸ナトリウム塩緩衝液(pH7.4)0.5M NaClで平衡化したHiTrap phenyl sepharose(ファルマシア社製)カラムに吸着させ、同緩衝液中のイミダゾール濃度を0.0 Mから0.5 Mに増加させることにより目的蛋白質の溶出を行った。
【0025】
【0026】
[実施例2] 糖ヌクレオチド合成
(1) 糖ヌクレオチド合成反応(糖とヌクレオチドの結合反応)
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、1mM マンノース−1−リン酸、0.05mM GTPからなる酵素反応溶液10μL中に実施例1で得られた精製酵素1 ngを加えた。この酵素反応液を85℃で保温することにより、反応させた。5分後に100μlの500mM KH2PO4溶液を加える事により反応を停止させた。この反応により、マンノースがGDPと結合したGDPマンノースが生成する。
【0027】
(2)糖ヌクレオチド合成反応(糖とヌクレオチドの結合反応)の測定
HPLCを用いて反応生成物であるGDPマンノースの量をヌクレオチド部分の紫外線の吸収を指標に測定した。図2に示すように標準物質であるGDP及びGDPマンノースは、HPLCにおいて溶出位置が異なる。さらに、図3に示すように標準サンプル添加量を変化させたときのピークの面積と標準物質量は正確な比例関係にあり、この検量線を用いることにより反応生成物を定量できることが示された。
そこで、上記(1)で反応させたサンプルに関しても、HPLCで同様の反応の解析を行った。その結果、図4に示すように、確かにGDPマンノースが見出されたことから、該酵素は、糖ヌクレオチドを確かに生成している事が確認された。
【0028】
[実施例3] リン酸化糖異性体合成
(1)リン酸化糖異性化反応
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、1mM NADP(ニコチンアミドアデニンジヌクレオチドリン酸酸化型)、1unit グルコース−6−リン酸異性化酵素、1unit グルコース−6−リン酸脱水素酵素、0.4mMマンノース−6−リン酸、からなる酵素反応溶液2,000μL中に実施例1で得られた精製酵素2.0μgを加えた。この酵素反応液を65℃で保温することにより、反応させた。この反応により、マンノース−6−リン酸が異性化されたフルクトース−6−リン酸が生成する。
【0029】
(2)糖リン酸異性化反応の測定
反応生成物であるフルクトース−6−リン酸を、グルコース−6−リン酸異性化酵素によりグルコース−6−リン酸に変換し、このグルコース−6−リン酸をグルコース−6−リン酸脱水素酵素により6−リン酸グルコノラクトンに変換する際の副反応であるNADPの酸化型から還元型への変化量を紫外・可視分光光度計によって波長340nmにおける吸光度の変化を指標に測定した。
NADPの酸化型と還元型では340nmの吸光度が全く異なる。さらに、図5に示すように、酵素反応溶液中の基質であるマンノース−6−リン酸濃度を変化させたときの、吸光度の計時変化とNADP還元型の量は正確な比例関係にあり、この検量線を用いることにより反応生成物を定量できることが示された。
そこで、(1)で反応させたサンプルに関しても、紫外・可視分光光度計による340nmの吸光度を用いて(1)の反応の解析を行った。その結果、図6に示すように、確かに吸光度の増加が見出されたことから、該酵素は、マンノース−6−リン酸を異性化しフルクトース−6−リン酸を確かに生成している事が確認された。
【0030】
[実施例4] 酵素の性質
(1) 蛋白質化学的性質
当該酵素は上記の精製プロセスで完全に精製され、SDS-PAGEで分子量約53,000Daであった(図1)。当該酵素は464アミノ酸残基より構成され(配列番号1)、そのアミノ酸配列から予測された分子量は52,888.64 Daであった。また、ヘリコバクター菌のGMP/PMIとの相同性も見出され、(pyrophosphorylase signature)活性中心、金属イオン結合、糖認識に関与するモチーフが保存されていた(図1)。
【0031】
(2)糖ヌクレオチド合成活性(糖ヌクレオチドの結合活性)
実施例1により精製された該酵素を用いて、マンノース−1−リン酸とGTPとの結合活性を実施例2に従って測定した。その結果、図4にあるように時間に依存したGDPマンノースの合成が85℃の反応温度で検出された。従って耐熱性の高い糖ヌクレオチド合成活性を示すことが確認された。
【0032】
(3)pH依存性
2mM MgSO4、1mM Mannose-1-リン酸、0.05mM GTPからなる酵素反応溶液10μ 中の50mM緩衝液をリン酸ナトリウム塩緩衝液pH6.0、pH6.5、MOPS緩衝液pH7.0、Tris-HCl緩衝液pH8.0、グリシン-NaOH緩衝液pH9.0、pH10.0に変化させ実施例1で得られた精製酵素1ngを加えた。この酵素反応液を85℃で5分間保温することにより反応させた後に、100μLの500mM KH2PO4溶液を加えることにより反応を停止させた。反応の進行は、実施例2の(2)にあるようにHPLCで測定した。図8に示すように、本酵素の活性はいずれのpHにおいても高い活性を示した。
【0033】
(4)ヌクレオシド三リン酸基質の多様性
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、1mM Mannose-1-リン酸及び0.05mM GTPまたは0.05mM ATP等からなる酵素反応溶液10μL中に実施例1で得られた精製酵素1 ngを加えた。この酵素反応液を85℃で5分間保持することにより反応させた後に、100μLの500mM KH2PO4溶液を加えることにより反応を停止させた。結果を図9に示す。図9から分かるように、本酵素はGTP以外にATP、dGTPを基質として利用できる事が示された。
【0034】
(5)糖−1−リン酸基質の多様性
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、0.05mM dGTP及び1mM マンノース−1−リン酸、または1mM グルコース−1−リン酸からなる酵素反応溶液10μL中に実施例1で得られた精製酵素1 ngを加えた。この酵素反応液を85℃で5分間保持することにより反応させた後に、100μLの500mM KH2PO4溶液を加えることにより反応を停止させた。結果を図10に示す。図10から分かるように、本酵素はマンノース−1−リン酸以外にグルコース−1−リン酸を基質として利用できる事が示された。
【0035】
(6)リン酸化糖異性化活性
実施例1により精製された該酵素を用いて、マンノース−6−リン酸の糖分子を異性化することでフルクトース−6−リン酸に変化させる活性を実施例3に従って測定した。その結果、図6に有るように、基質であるフルクトース−6−リン酸が反応時間に従って生産される活性が65℃で検出された。この事から、本酵素はリン酸化糖異性化活性を有する熱に安定な酵素であることが示された。
【0036】
[参考例]
以下に、本発明の糖−ヌクレオチド生産に用いる、特願2004−236321号に係るリン酸化糖のリン酸基分子内転移酵素の製造例及び該酵素の酵素作用の試験結果を参考例1、2として示す。
【0037】
[参考例1]
(1) 菌の培養
好酸性好気性超好熱性古細菌スルフォロバス、トーコーダイイJCM10545は次の方法で培養した。
1.3g の(NH4)2SO4、0.28gのKH2PO4、0.25gのMgSO4・7H2O、0.07gのCaCl2・2H2O、0.02gのFeCl3・6H2O、1.8mgのMnCl2・4H2O、4.5mgのNa2B4O7・10H2O、0.22mgのZnSO4・7H2O、0.05mgのCuCl2・2H2O、0.03mgのNa2MoO4・2H2O、0.03mgのVoSO4・xH2O、0.01mgのCoSO4・7H2O、1.0gの酵母エキスを1リッターの蒸留水に溶かし、この溶液のpHを3.5に10規定H2SO4溶液で調製した。加圧殺菌した後、JCM10545を植菌した。この培養液を80℃で1〜2日培養し、その後遠心分離し集菌した。
【0038】
(2) 染色体DNAの調製
JCM10545の染色体DNAは以下の方法により調製した。
培養終了後5,000rpm、10分間の遠心分離により菌体を集菌する。菌体を10mM EDTA(pH 6.0)溶液で洗浄後、50mM Tris/HCl-50mM EDTA(pH8.5)溶液を加えて細胞を溶解させる。さらに、0.5% Na-lauroylsarcosinate、1mg/mL プロテアーゼKとなるように各々を加えた後、50℃で3時間保温する。フェノール処理を3回行った後、溶液を10mM EDTA (pH8.0)溶液に対して透析する。37℃で30分間のRNaseによるRNAの分解後、フェノールクロロフォルム溶液で処理した後、10mM Tris-1mM EDTA(pH8.0)で透析を行った。
【0039】
(3) 染色体DNAを含むショットガンライブラリークローンの作製
上記(2)の工程で得られた染色体DNAを超音波処理することにより断片化した後、アガロースゲル電気泳動により1kb及び2kb長のDNA断片を回収した。この断片をプラスミドベクターpUC118のHincII制限酵素部位に挿入したショットガンライブラリーを作製した。各ショットガンクローンの末端塩基配列を、ABI社製自動塩基配列読み取り装置377を用いて解読していった。
【0040】
(4) STPMM/PGM遺伝子の同定
上記手法で決定された好酸性好気性超好熱古細菌スルフォロバス、トーコーダイイのゲノム塩基配列の大型計算機による解析を行い、phosphomannomutase/phosphoglucomutase酵素の機能を含むであろう蛋白質をコードする遺伝子(ST0242)を同定した。この超好熱性古細菌スルフォロバス、トーコーダイイのST0242遺伝子の開始コドンはATGで、455アミノ酸残基の蛋白質(配列番号5)をコードする遺伝子として同定された(配列番号6)。
【0041】
(5)発現プラスミドの構築
構造遺伝子領域の前後に制限酵素(NdeIとXhoI)サイトを構築する目的でDNAプライマーを合成し、PCRでその遺伝子の前後に制限酵素サイトを導入した。
【0042】
Upper primer、
5’- TTAATTCCATATGGGTAAGCTTTTTGGTACTGAC -3’ (配列番号7)
(下線部はNdeIサイトを示す)
Lower primer、
5’- ATATACTCGAGTCATTTACCCTCTACAATC -3’ (配列番号8)
(下線部はXhoIサイトを示す)
【0043】
Upper primerとLower primerを組み合わせたPCR反応後、制限酵素(NdeIとXhoI)で完全分解(37℃で2時間)した後、その構造遺伝子領域断片を精製した。
制限酵素NdeIとXhoIで切断後精製したpET28a(Novagen社製)と上記の構造遺伝子(ST0242)領域断片とT4リガーゼを用いて16℃、2時間反応させることによって連結した。連結したDNAの一部を大腸菌DH5αのコンピテントセルに導入し形質転換体のコロニーを得た。得られたコロニーからプラスミドをQIAprep Spin Miniprep Kit(QIAGEN社製)で精製し、塩基配列を確認して発現プラスミド、pET28a/ST0242を得た。発現プラスミドpET28a/ST0242を用いるとSTPMM/PGMはN末端にヒスチジンタグが付加された融合蛋白質として生産される。
【0044】
(6)組み換え遺伝子の発現
大腸菌 BL21-CodonPlus(DE3)-RIL、(Novagen社製)のコンピテントセルを融解して、ファルコンチューブに0.1ml移す。その中に上記の発現プラスミド10ng分に相当する溶液を加え氷中に30分間放置した後、42℃でヒートショックを30秒間おこない、そこにSOC培地0.9mlを加え、37℃で1時間振とう培養する。その後、カナマイシンを含むLB寒天プレート上に適量まき、37℃で一晩培養し、形質転換体大腸菌BL21-CodonPus(DE3)-RIL/pET28a/ST0242を得た。
【0045】
当該形質転換体をカナマイシン含有LB培地(2リットル)中で一晩37℃において振とう培養した後、IPTG(Isopropyl-β-D-thiogalacopyranoside)を1mMになるように加え、さらに30℃で5時間振とう培養した。培養後、遠心分離(6000rpm、20分間)により集菌をおこなった。
【0046】
【0047】
[参考例2]
リン酸化糖のリン酸基分子内転移反応の測定
a.グルコース−1−リン酸のリン酸基分子内転移反応
(1)グルコース−1−リン酸のリン酸基分子内転移反応の酵素反応溶液
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、10μM グルコース 1,6-ビスリン酸、1mM NADP(ニコチンアミドアデニンジヌクレオチドリン酸酸化型)、1unit グルコース−6−リン酸脱水素酵素、0.4mMグルコース−1−リン酸、からなる酵素反応溶液2,000μL中に参考例1で得られた精製酵素3.5μgを加えた。この酵素反応液を65℃で保温することにより、反応させた。この反応により、グルコース−1−リン酸のリン酸基が分子内転移しグルコース−6−リン酸が生成する。
【0048】
(2) グルコース−1−リン酸のリン酸基分子内転移反応の測定
本酵素を用いた反応の反応生成物であるグルコース−6−リン酸をグルコース−6−リン酸脱水素酵素により6−リン酸グルコノラクトンに変換する際に、副反応としてニコチンアミドアデニンジヌクレオチドリン酸(NADP)が酸化型から還元型へ変化する。その変化量を340nmにおける吸光度の変化として紫外・可視分光光度計を用いて測定した(Klingenberg M “Methods of Enzymatic Analysis” (1974) Weinheim-Academic Press, New York, vol. 4, pp. 2045参照)。NADPの酸化型と還元型では波長に依存した吸光度プロファイルが全く異なる。特に340nmの吸光度が全く異なる事から、この波長の吸光度を利用して生成された還元型NADPの量を定量する。
【0049】
そこで、(1)で反応させたサンプルに関しても、紫外・可視分光光度計による340nmの吸光度を用いて(1)の反応の解析を行った。図14に示すように、基質であるグルコース−1−リン酸の濃度に依存した活性が65℃で検出された。
実施例1により精製された該酵素を用いて、グルコース−1−リン酸及びマンノース−1リン酸が有するリン酸基の分子内転移活性を実施例3,4に従って測定した。その結果、図14に有るように、基質であるグルコース−1−リン酸の濃度に依存した活性が65℃で検出された。また、図15に有るように、同様の活性がマンノース−1−リン酸を基質としたときにも検出されたことから、該酵素は耐熱性PGM/PMM活性を有する事が確認された。また、グルコース−6−リン酸を基質とした場合にも、図16に示されたように、確かに分子内リン酸基が一位に転移している事からTDPグルコースの生産が確認された。
【0050】
b.マンノース−1−リン酸のリン酸基分子内転移反応
(1) マンノース−1−リン酸のリン酸基分子内転移反応の酵素反応溶液
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、10μM グルコース 1,6-ビスリン酸、1mM NADP、1unit グルコース−6−リン酸脱水素酵素、1unit マンノース−6−リン酸異性化酵素、1unit グルコース−6−リン酸異性化酵素、0.4mMマンノース−1−リン酸、からなる酵素反応溶液2,000μL中に実施例1で得られた精製酵素7.0μgを加えた。 この酵素反応液を65℃で保温することにより、反応させた。この反応により、マンノース−1−リン酸のリン酸基が分子内転移しマンノース−6−リン酸が生成する。
【0051】
(2)マンノース−6−リン酸化合物のリン酸基分子内転移反応の測定
本酵素を用いた反応の反応生成物であるマンノース−6−リン酸をマンノース−6−リン酸異性化酵素によりフルクトース−6−リン酸に変換し、フルクトース−6−リン酸をグルコース−6−リン酸異性化酵素によりグルコース−6−リン酸に変換し、グルコース−6−リン酸脱水素酵素により6−リン酸グルコノラクトンに変換する際に、副反応としてニコチンアミドアデニンジヌクレオチドリン酸(NADP)が酸化型から還元型へ変化する。その変化量を340nmにおける吸光度の変化として紫外・可視分光光度計を用いて測定した(非特許文献4参照)。NADPの酸化型と還元型では波長に依存した吸光度プロファイルが全く異なる。特に340nmの吸光度が全く異なる事から、この波長の吸光度を利用して生成された還元型NADPの量を定量する。
そこで、(1)で反応させたサンプルに関しても、紫外・可視分光光度計による340nmの吸光度を用いて(1)の反応の解析を行った。図15に示すように、基質であるマンノース−1−リン酸の濃度に依存した活性が65℃で検出された。
【0052】
c.グルコース−6−リン酸のリン酸基分子内転移反応
(1)グルコース−6−リン酸のリン酸基分子内転移反応の酵素反応溶液
50mM MOPS緩衝液(pH7.6)、2mM MgSO4、10μl グルコース1,6ビスリン酸、0.4mM TTP、2.5μg スルフォロバス・トーコーダイイ由来のRmlA(glucose-1-phosphate thymidylyltransferase )、1mMグルコース−6−リン酸からなる酵素反応溶液2,000μL中に、実施例1で得られた精製酵素2.5μgを加えた。この酵素反応液を80℃で30分間保温することにより反応させた。反応後、100μLの500mM リン酸二水素カリウム溶液を加えることにより反応を停止させた。この反応により、グルコース−6−リン酸のリン酸基が分子内転移しグルコース−1−リン酸が生成する。
【0053】
(2)グルコース−6−リン酸のリン酸基分子内転移反応の測定
HPLCを用いて、反応性生物であるグルコース−1−リン酸をRmlAによりTDP-グルコースに変換し、TDP-グルコースの量をヌクレオチド部分の紫外線の吸収を目安に測定した。図2からも明らかなように、標準物質であるTTPおよびTDP-グルコースは、HPLCにおいて溶出位置がまったく異なる。そこで、上記(1)で反応させたサンプルに関しても、HPLCで同様の解析をおこなった。その結果、図16に示すように、確かにTDP-グルコースが見出されたことから、該酵素は、グルコース−6−リン酸の分子内リン酸基を転移させたグルコース−1−リン酸を確かに生成している事が確認された。
【図面の簡単な説明】
【0054】
【図1】精製されたPHGMP/PMI蛋白質のSDS-PAGEパターンを示す図である。
【図2】HPLCによるGTP及びGDP-Mannoseの分離パターンを測定した図である。
【図3】GDPマンノース標準物質の添加量とピーク面積変化の関係を示す図である。
【図4】PHGMP/PMI蛋白質の85℃における糖ヌクレオチド合成活性を示す図である。
【図5】マンノース−6−リン酸を基質とし、その濃度を変化させたときの吸光度の経時変化を示す図である。
【図6】PHGMP/PMI蛋白質による波長340nmにおける吸光度の経時変化を示す図である。
【図7】PHGMP/PMI蛋白質とへリコバクター菌のGMP/PMI蛋白質との相同性を示す図である。
【図8】PHGMP/PMI蛋白質のpHの違いによる活性変化を示す図である。
【図9】PHGMP/PMI蛋白質のヌクレオシド三リン酸基質の多様性を示す図である。
【図10】PHGMP/PMI蛋白質の糖リン酸基質の多様性を示す図である。
【図11】本発明の酵素タンパク質および特願2004−236321号の酵素を用いてフルクトース−6−リン酸からGDP−マンノースを合成する反応工程を示す概略図である。
【図12】本発明の酵素のリン酸化糖の異性化活性に対する各金属イオン種の影響を示す図である。
【図13】本発明の酵素の糖−ヌクレオチド合成活性に対する各金属イオン種の影響を示す図である。
【図14】参考例2において、グルコース−1−リン酸を基質とし、その濃度を変化させた場合の波長340nmにおける吸光度の経時変化を示す図である。
【図15】参考例2において、マンノース−1−リン酸を基質とし、その濃度を変化させた場合の波長340nmにおける吸光度の経時変化を示す図である。
【図16】参考例2において、グルコース−6−リン酸を基質とした場合において、TDP-グルコース生成の経時変化を示す図である。
【特許請求の範囲】
【請求項1】
配列番号1に記載のアミノ酸配列を有するか、あるいは、配列番号1に記載のアミノ酸配列において1乃至数個のアミノ酸残基が欠失、置換、挿入または付加されたアミノ酸配列を有し、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有することを特徴とする蛋白質。
【請求項2】
請求項1記載の蛋白質をコードするDNA。
【請求項3】
配列番号2に記載の塩基配列を有することを特徴とするDNA。
【請求項4】
配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズし、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有することを特徴とする蛋白質をコードすることを特徴とするDNA。
【請求項5】
請求項2〜4のいずれかに記載のDNAがベクターに組み込まれていることを特徴とする組み換え体DNA。
【請求項6】
請求項5に記載の組み換え体DNAが宿主細胞に導入されていることを特徴とする形質転換体。
【請求項7】
請求項6に記載の形質転換体を培地に培養し、培養物から、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質を採取することを特徴とする、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質の製造法。
【請求項8】
糖−1−リン酸及びヌクレオシド三リン酸に、請求項1に記載の蛋白質を作用させることを特徴とする糖ヌクレオチドの製造方法。
【請求項9】
糖−6−リン酸に、請求項1に記載の蛋白質を作用させることを特徴とする異性化された糖−6−リン酸の製造方法。
【請求項10】
糖−1−リン酸及びヌクレオシド三リン酸に、請求項6に記載の形質転換体の培養液あるいは培養物の処理物を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【請求項11】
糖−6−リン酸に、請求項6に記載の形質転換体の培養液あるいは培養物の処理物を作用させることを特徴とする、異性化された糖−6−リン酸の製造方法。
【請求項12】
糖−1−リン酸及びヌクレオシド三リン酸に、請求項3又は4に記載のDNAにコードされる蛋白質を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【請求項13】
糖−6−リン酸に、請求項3又は4に記載のDNAにコードされる蛋白質を作用させることを特徴とする、異性化された糖−6−リン酸製造方法。
【請求項14】
リン酸化糖の異性化反応、分子内リン酸基転移反応及び糖−ヌクレオチド合成反応を行い、糖−ヌクレオチドを製造する方法において、上記リン酸化糖の異性化反応および糖−ヌクレオチド合成反応を、請求項1に記載の蛋白質を用いて行うことを特徴とする、上記方法。
【請求項15】
請求項1に記載の蛋白質を含有する反応系において、カルシウムイオン、亜鉛イオンまたは銅イオンのうちいずれかを選択して含有させることにより、該タンパク質の酵素作用を、糖ヌクレオチド合成作用とリン酸化糖異性化作用の間で切り替えることを特徴とする、酵素作用の切り替え方法。
【請求項1】
配列番号1に記載のアミノ酸配列を有するか、あるいは、配列番号1に記載のアミノ酸配列において1乃至数個のアミノ酸残基が欠失、置換、挿入または付加されたアミノ酸配列を有し、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有することを特徴とする蛋白質。
【請求項2】
請求項1記載の蛋白質をコードするDNA。
【請求項3】
配列番号2に記載の塩基配列を有することを特徴とするDNA。
【請求項4】
配列番号2に記載のDNAとストリンジェントな条件下でハイブリダイズし、かつ糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有することを特徴とする蛋白質をコードすることを特徴とするDNA。
【請求項5】
請求項2〜4のいずれかに記載のDNAがベクターに組み込まれていることを特徴とする組み換え体DNA。
【請求項6】
請求項5に記載の組み換え体DNAが宿主細胞に導入されていることを特徴とする形質転換体。
【請求項7】
請求項6に記載の形質転換体を培地に培養し、培養物から、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質を採取することを特徴とする、糖ヌクレオチド合成活性並びにリン酸化糖異性化活性を有する蛋白質の製造法。
【請求項8】
糖−1−リン酸及びヌクレオシド三リン酸に、請求項1に記載の蛋白質を作用させることを特徴とする糖ヌクレオチドの製造方法。
【請求項9】
糖−6−リン酸に、請求項1に記載の蛋白質を作用させることを特徴とする異性化された糖−6−リン酸の製造方法。
【請求項10】
糖−1−リン酸及びヌクレオシド三リン酸に、請求項6に記載の形質転換体の培養液あるいは培養物の処理物を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【請求項11】
糖−6−リン酸に、請求項6に記載の形質転換体の培養液あるいは培養物の処理物を作用させることを特徴とする、異性化された糖−6−リン酸の製造方法。
【請求項12】
糖−1−リン酸及びヌクレオシド三リン酸に、請求項3又は4に記載のDNAにコードされる蛋白質を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【請求項13】
糖−6−リン酸に、請求項3又は4に記載のDNAにコードされる蛋白質を作用させることを特徴とする、異性化された糖−6−リン酸製造方法。
【請求項14】
リン酸化糖の異性化反応、分子内リン酸基転移反応及び糖−ヌクレオチド合成反応を行い、糖−ヌクレオチドを製造する方法において、上記リン酸化糖の異性化反応および糖−ヌクレオチド合成反応を、請求項1に記載の蛋白質を用いて行うことを特徴とする、上記方法。
【請求項15】
請求項1に記載の蛋白質を含有する反応系において、カルシウムイオン、亜鉛イオンまたは銅イオンのうちいずれかを選択して含有させることにより、該タンパク質の酵素作用を、糖ヌクレオチド合成作用とリン酸化糖異性化作用の間で切り替えることを特徴とする、酵素作用の切り替え方法。
【図2】

【図3】
【図4】
【図5】
【図6】
【図7】

【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図1】
【図16】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図1】
【図16】
【公開番号】特開2007−174945(P2007−174945A)
【公開日】平成19年7月12日(2007.7.12)
【国際特許分類】
【出願番号】特願2005−375574(P2005−375574)
【出願日】平成17年12月27日(2005.12.27)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成16年度北海道大学委託研究「遺伝子情報解析に関する研究」、産業活力再生特別措置法第30条の適用を受ける特許出願
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
【公開日】平成19年7月12日(2007.7.12)
【国際特許分類】
【出願日】平成17年12月27日(2005.12.27)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成16年度北海道大学委託研究「遺伝子情報解析に関する研究」、産業活力再生特別措置法第30条の適用を受ける特許出願
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
[ Back to top ]