
糖ヌクレオチド合成酵素変異体
【課題】耐熱性UDP-GlcNAc合成酵素は、その活性が常温微生物由来の酵素に比して比較的低かった。そこで、耐熱性を維持し、かつ活性を上昇させたUDP-GlcNAc合成酵素を提供し、糖鎖合成の基質となるUDP-GlcNAcを効率的に合成する。
【解決手段】
超好熱古細菌 Sulfolobus tokodaii 由来のUDP-GlcNAc合成活性を有する耐熱性ST0452酵素タンパク質中のアミノ酸を置換し、耐熱性を維持しながらUDP-GlcNAc合成活性が向上した新たな変異体蛋白質を得、これを用いてUDP-GlcNAcを安定的に合成する。
【解決手段】
超好熱古細菌 Sulfolobus tokodaii 由来のUDP-GlcNAc合成活性を有する耐熱性ST0452酵素タンパク質中のアミノ酸を置換し、耐熱性を維持しながらUDP-GlcNAc合成活性が向上した新たな変異体蛋白質を得、これを用いてUDP-GlcNAcを安定的に合成する。
【発明の詳細な説明】
【技術分野】
【0001】
本願発明は、耐熱性の低下を伴わずに糖ヌクレオチド合成活性が促進された耐熱性変異体蛋白質、該蛋白質をコードするDNA、該DNAを含有する組換え体DNA、該組換え体DNAを保有する形質転換体、該形質転換体を用いた糖ヌクレオチド合成活性を有する蛋白質の製造法、および該蛋白質あるいは該形質転換体を用いた糖ヌクレオチドの製造法に関する。
【背景技術】
【0002】
糖ヌクレオチド(UDP-GlcNAc)合成活性を有する酵素としては大腸菌(Escherichia coli)(非特許文献1参照)やストレプトコッカス菌(Streptococcus pneumoniae) (非特許文献2参照)、ネイセリア菌(Neiseia gonorrhoeae) (非特許文献3参照)由来のGlmU (N-Acetyl-D-Glucosamine-1-phosphate uridylyltransferase)の詳しい性質がすでに報告されている。GlmUは様々な糖鎖合成に必須な構成要素であるN-アセチル-D-グルコサミン(N-Acetyl-D-Glucosamine (GlcNAc))の活性体ウリジン二リン酸-N-アセチル-D-グルコサミン(UDP-GlcNAc)を合成する酵素であり、N-アセチル-D-グルコサミン-1-リン酸(N-Acetyl-D-Glucosamine-1-phosphate (GlcNAc-1-P))とウリジン三リン酸(UTP)を基質として、ウリジン二リン酸-N-アセチル-D-グルコサミン(UDP-GlcNAc)を生産する。上記の様に、幾つかの微生物からUDP-GlcNAcを合成する酵素が見出されているが、それらは常温生物由来のため室温以上では極めて不安定で、活性は80℃程度の加熱処理により速やかに失活する。このため、使用時の滅菌等の処理が必要であったり、低温での注意深い保存が必要であった。また、それら常温微生物由来酵素と比較すると、本酵素活性を有する耐熱性酵素ST0452蛋白質の活性は同等か若干低いものであった。そこで、有利な耐熱性を有する酵素ST0452蛋白質の本酵素活性の促進が求められた。
【非特許文献1】Dominique Mengin-Lecreulx and Lean van Heijenoort “Identification ofthe GlmU Gene Encoding N-Acetylglucosamine-1-PhosphateUridylyltransferase in Escherichia coli “ (1993) Journal of Bacteriology, 175, 6150-6157.
【非特許文献2】Dirk Kostrewa, Allan D’Arcy, Bela Takacs and Markus Kamber “CrystalStructure of Streptococcus pneumoniae N-Acetyl-glucosamine-1-phosphate Uridylyltransferase,GlmU, in Apo Form at 2.33 Å Resolution and in Complex withUDP-N-Acetylglucosamine and Mg2+ at 1.96 Å Resolution" (2001) Journal ofMolecular Biology, 305, 279-289.
【非特許文献3】Joachim Ullrich and Jos P. M. van Putten "Identification of theGonococcal glmU Gene Encoding the Enzyme N-Acetylglucosamine1-Phosphate Uridylyltransferase Involved in the Synthesis of UDP-GlcNAc" (1995) Journal of Bacteriology, 177, 6902-6909.
【発明の開示】
【発明が解決しようとする課題】
【0003】
糖ヌクレオチドUDP-GlcNAcを合成する活性を有する耐熱性酵素を我々は既にスルフォロバス・トーコーダイイから見出してきた。本耐熱性酵素では、常温微生物が有しているUDP-GlcNAc合成活性に加えてdTDP-GlcNAc合成活性も有しており、活性が半減するためには80度で160分の加熱処理を必要とする程、その耐熱性は高い。しかし、この耐熱性酵素が有するUDP-GlcNAc合成活性は、既に詳しく解析されている常温微生物が有する類似酵素の活性と同等か少し劣るものであった。そこで、本耐熱性酵素が有する特徴的な活性や高い耐熱性を有効に利用するためには、さらに高いUDP-GlcNAc合成活性を有する安定な酵素が渇望されていた。
【0004】
したがって本発明の課題は、耐熱性を維持したまま、既存のN-アセチル-D-グルコサミン-1-リン酸(GlcNAc-1-P)等の糖一リン酸、及びウリジン三リン酸(UTP)等のヌクレオシド三リン酸を基質として、UDP-GlcNAc等の糖ヌクレオチドを合成する活性を促進した新規酵素を提供することにある。
【課題を解決するための手段】
【0005】
本発明者は、以上のような課題を解決すべく、75 - 80℃で生育する超好熱古細菌 Sulfolobus tokodaii strain7由来ST0452蛋白質に着目し、その配列中のアミノ酸を変化させた変異体を作成した。さらに、大腸菌を使ってそれらの遺伝子から変異体酵素を生産し、この酵素が高温(80℃)での安定性に変化なく、かつ促進された糖ヌクレオチド合成活性を示すものが存在することを確認した。さらに、本酵素を用いることにより様々な種類の糖ヌクレオチドを生産することもできることを見いだし、本発明を完成するに至ったものである。
【0006】
即ち、本発明は、以下の(1)〜(8)に係るものである。
(1) 配列番号2〜7のいずれかに示されるアミノ酸配列を有することを特徴とする、糖ヌクレオチド合成活性を有する蛋白質。
(2)上記(1)に記載の蛋白質をコードするDNA。
(3)配列番号9〜14のいずれかに示される塩基配列を有することを特徴とするDNA。
(4)上記(2)または(3)に記載のDNAがベクターに組み込まれていることを特徴とする組換え体DNA。
(5)上記(4)に記載の組換え体DNAが宿主細胞に導入されていることを特徴とする形質転換体。
(6)上記(5)に記載の形質転換体を培地に培養し、培養物から糖ヌクレオチド合成活性を有する蛋白質を採取することを特徴とする、糖ヌクレオチド合成活性を有する蛋白質の製造方法。
(7) 糖一リン酸及びヌクレオシド三リン酸に、上記(1)に記載の蛋白質を作用させることを特徴とする、糖ヌクレオチドの製造方法。
(8) 糖一リン酸及びヌクレオシド三リン酸に、上記(5)に記載の形質転換体の培養液あるいは培養物の処理物を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【発明の効果】
【0007】
本発明によれば、耐熱性を維持したままで活性の比較的低かったST0452酵素のUDP-GlcNAc合成活性を1.5倍から2倍程度促進した酵素を提供でき、これにより、試験管内でのUDP-GlcNAc合成の際に、熱に安定でかつ効率的に目的とするUDP-GlcNAcを合成することができる。
一方、UDP-GlcNAcは、糖タンパク質、糖脂質、多糖類の糖鎖合成の際にGlcNAc供与体として機能するものであり、これらの糖鎖合成は、癌転移、器官発生あるいは細胞性免疫等に密接に関連するものとして近年注目されており、本発明は、これら研究の発展において、その貢献度は極めて大きい。
【発明を実施するための最良の形態】
【0008】
以下に、本願発明を具体的に説明する。
本発明の酵素タンパク質は、好酸性好気性超好熱古細菌スルフォロバス・トーコーダイイ(JCM登録番号JCM10545)由来のST0452UDP-GlcNAc合成活性を有するタンパク質(以下、ST0452タンパク質という場合がある。配列番号1)に変異を導入したものであり、該変異体酵素タンパク質は、耐熱性が低下することなく、上記ST0452UDP-GlcNAc合成活性が向上したものである。また、本発明のDNAは、上記変異体酵素タンパク質をコードするDNAである。
【0009】
本発明者は、これまでに類似酵素についての立体構造解析結果等を勘案して、上記スルフォロバス、トーコーダイイ(JCM登録番号JCM10545)由来のST0452UDP-GlcNAc合成活性を有するタンパク質における9個のアミノ酸残基と1つの領域を変異導入の対象として選択し、これらの変異が導入された変異体酵素を作成した。
すなわち、後記する表2に示す変異を導入したプライマーを、同表3に示した組み合わせでPCR増幅を行い、変異を導入したDNA断片を得た。
この断片を、蛋白質発現プラスミドpET21bに挿入後、そのプラスミドにより形質転換した大腸菌を用いて変異体酵素の生産をおこなった。得られた変異体酵素は、各変異の導入位置に従って、G9A、R13A、RegionI、K23A、T80A、T80L、Y97A、Y97F、D99A、E146A、K147A、D208Aと名づけた。このうち、本発明の変異体酵素は、G9A、T80A、Y97A、Y97F、K14A及びD208A である(なお、これらの命名記号における、数字はST0452タンパク質におけるアミノ酸置換位置を、その左側のアルファベットは置換前のアミノ酸を、及び右側のアルファベットは置換後のアミノ酸をそれぞれ表す。)。
上記12種の変異体酵素は、いずれもST0452タンパク質の活性中心に変異を導入したものである。この理由は、活性中心は酵素の内側に位置するから、この位置のアミノ酸を置換させても、酵素タンパク質の耐熱性に影響を与える酵素の全体構造の変化は少ないと考えたからであるが、活性中心に変異を導入する場合は酵素活性は低下するのが普通である。しかし、本発明における、上記G9A、T80A、Y97A、Y97F、K14A及びD208Aの各変異体酵素は、全く意外にも、ST0452UDP-GlcNAc合成活性がむしろ向上している点で極めてユニークである。
【0010】
これら6種の変異体酵素のアミノ酸配列およびこれをコードするDNAの塩基配列は配列表の配列番号2〜7及び9〜14にそれぞれ示される。これらの関係を分かりやすくするため以下の表1にまとめて示す。
【表1】

【0011】
本発明における上記変異体酵素の優れた特性は、以下の実験により確かめられている。
すなわち、本変異体酵素を、50mトリス塩酸緩衝液(pH7.5)中で、80℃、20分間加熱処理を行った後に、SDSポリアクリルアミドゲル電気泳動で確認したところ、全ての変異体が可溶性タンパク質として確認されたことから、全ての変異体タンパク質は元のST0452タンパク質と同等の耐熱性を維持していることが示された。
これら作成した12種類の変異体ST0452タンパク質について、UDP-GlcNAc合成活性の各基質に関するKm値及び反応のKcat値を求めた。各変異体のKm及びKcat値を表4にまとめた。その結果表4に示されているように、全ての変異体において基質との結合の強さを示すKm値は低下していたが、変異体G9A、Y97F、K147A等では変異を導入する前のST0452タンパク質よりも反応の進む程度を示すKcat値が上昇していることが明らかとなった。
【0012】
上記結果から、基質との結合は弱くなっているが、反応は進みやすくなっている変異体が存在していることが明らかとなった。そこで、基質濃度を上げることで、変異を導入する前のST0452酵素よりも高い活性を見出せることが推定された。そこで、ヌクレオチド基質であるUTP(ウリジン三リン酸)の濃度を20倍、GlcNAc-1-P(N-アセチル-D-グルコサミン-1-リン酸)の濃度を200倍高くして活性を測定したところ、図6の塗りつぶしたバーのように、G9A、T80A、Y97A、Y97F、K174Aの各変異体酵素では、変異を導入していないST0452タンパク質の活性よりも高い活性が検出された。
【0013】
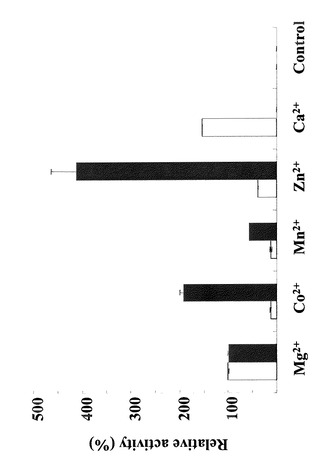
さらに、D208A変異体酵素においては、反応液に加える金属イオンを変えて反応を行った。その結果、図7に示す様に亜鉛イオンを加えた場合には、変異を導入していないST0452タンパク質の活性の3倍もの活性を見出すことができた。
【0014】
すなわち、本発明のG9A、T80A、Y97A、Y97F、K174Aの各変異体タンパク質は、反応液中の基質濃度を上げることにより、また、同D208A変異体酵素は亜鉛イオンを加えることにより、糖ヌクレオチド合成活性を促進させることができ、これにより糖ヌクレオチドの生産性が向上する。またこれらの変異体酵素は、80℃、20分間の熱処理でも変性しないという優れた耐熱性を有する。
G9A、T80A、Y97A、Y97F、K174Aの各変異体酵素を使用する場合における、ヌクレオシド三リン酸の濃度は50〜100 uM、糖一リン酸の濃度は2〜10 mMが好ましい。また、同D208A変異体酵素を使用する場合の亜鉛イオン濃度は1〜2 mMが好ましい。
【0015】
本発明の酵素を得るには、通常の遺伝子工学的手法が適用でき、本発明の上記各種変異体酵素をコードするDNAを、例えば、pET21b、pHY481等の蛋白質発現プラスミドベクター等に挿入して組み換えベクターを作製し、該組み換えベクターを用いて宿主細胞を形質転換し、該形質転換体を培地で培養し、培養物、培養処理物あるいはこれら培養物から分離回収された形質転換体から、酵素を常法の蛋白質精製手段により精製し単離する。上記宿主細胞としては、大腸菌・枯草菌等が利用可能である。
【0016】
本発明においては、さらに上記変異体酵素を用いて、糖ヌクレオチドを合成するが、この合成においては、例えば、糖一リン酸とヌクレオシド三リン酸を含有する溶液に、該酵素を添加し、反応温度60℃〜95℃で反応させることで、目的の糖ヌクレオチドを得ることができる。
また、ヌクレオシド三リン酸としては、UTP以外にTTP(チミジン三リン酸)も用いることができる。
【0017】
以下に、UTP(ウリジン三リン酸)とGlcNAc-1-P(N-アセチル-D-グルコサミン-1-リン酸)からUDP-GlcNAc(ウリジン二リン酸-N-アセチル-D-グルコサミン)を合成する場合の反応式を示す。
【化1】
【0018】
この反応においては、上記精製した酵素のみならず、粗酵素であってもよい。例えば、宿主として枯草菌等分泌型の系を用いる場合には、培養液中に本酵素が生成蓄積され、大腸菌等の非分泌型の系を用いる場合には、菌体内に生成されるので、本酵素を含有する培養液あるいはその処理物、もしくは菌体破砕物等の培養処理物を用いて、UDP-GlcNAcを合成してもよい。
以下に、本発明の実施例を示すが、本発明実施例により限定されるものではない。
【実施例1】
【0019】
糖ヌクレオチド合成酵素変異体の作成
(1)菌の培養
好酸性好気性超好熱古細菌スルフォロバス・トーコーダイイJCM10545は次の方法で培養した。
1.3gの(NH4)2SO4、0.28gのKH2PO4、0.25gのMgSO4・7H2O、0.07gのCaCl2・2H2O、0.02gのFeCl3・6H2O、1.8mg のMnCl2・4H2O、4.5mgのNa2B4O7・10H2O、0.22mgのZnSO4・7H2O、0.05mgのCuCl2・2H2O、0.03mgのNa2MoO4・2H2O、0.03mgのVOSO4・xH2O、0.01mgのCoSO4・7H2O、1.0gの酵母エキスを1Lの蒸留水に溶かし、この溶液のpHを3.5に10規定H2SO4溶液で調整した。加圧殺菌した後、JCM10545を植菌した。この培養液を80℃で1〜2日培養し、その後遠心分離し集菌した。
【0020】
(2)染色体DNAの調製
JCM10545の染色体DNAは以下の方法により調製した。
培養終了後5000rpm、10分間の遠心分離により菌体を集菌する。菌体を10 mM EDTA(pH 6.0)溶液で洗浄後、50 mM Tris/HCl-50 mM EDTA (pH8.5)溶液を加えて細胞を溶解させる。さらに、0.5% Na-lauroylsarcosinate、1 mg/ml プロテアーゼKとなるように各々を加えた後、50℃で3時間保温する。フェノール処理を3回行った後、溶液を10 mM Tris-10 mM EDTA (pH 8.0)溶液に対して透析する。37℃で30分間のRNaseによるRNAの分解後、フェノールクロロフォルム溶液で処理した後、10 mM Tris-1 mM EDTA(pH 8.0)で透析を行う。
【0021】
(3)染色体DNAを含むショットガンライブラリークローンの作製
(2)で得られた染色体DNAを超音波処理することにより断片化した後、アガロースゲル電気泳動により1kb及び2kb長のDNA断片を回収した。この断片をプラスミドベクターpUC118のHincII制限酵素部位に挿入したショットガンライブラリーを作製した。各ショットガンクローンの末端塩基配列を、ABI社製自動塩基配列読み取り装置377を用いて解読していった。各ショットガンクローンから得られた塩基配列を塩基配列自動連結ソフトSequencherを用いて連結編集し、本菌の全塩基配列を決定していった。
【0022】
(4)ST0452遺伝子中の変異を導入するアミノ酸残基の選択
上記手法で決定された好酸性好気性超好熱古細菌スルフォロバス・トーコーダイイのゲノム塩基配列の大型計算機による解析を行い、UDP-GlcNAcを合成する機能を持つタンパク質をコードする遺伝子はST0452であると推定され、その後の実験で高い熱安定性及び予想された活性の同定がなされた。該遺伝子の塩基配列及び該遺伝子がコードするタンパク質(ST0452タンパク質)のアミノ酸配列をそれぞれ配列番号8及び1にそれぞれ示す。
ST0452タンパク質の高い熱安定性を損なうことなく活性を促進するために、機能に重要な反応中心のアミノ酸残基に変異を導入することとした。酵素の活性中心は、基質を取り込むために比較的酵素の内側に位置していると考えられるので、活性中心のアミノ酸残基を変化させても、耐熱性に影響を与える恐れがある酵素自身の全体の構造には大きな影響を与えないと考えた。
【0023】
そこで、このST0452タンパク質のアミノ酸配列中、活性中心に位置すると考えられるアミノ酸のうち9個のアミノ酸(配列番号1のアミノ酸配列中、9,13、23、80,97、99、146、147、及び208番目に位置するアミノ酸)と1ヶ所の領域(同14番目〜21番目の領域)を、アミノ酸置換を導入する標的として選択した。
【0024】
(5)各変異体タンパク質発現プラスミドの構築
Gly9、Lys23、Tyr97をAla残基に、Tyr97をPhe残基に、Leu14-Glu-Phe-Ile-Thr-His-Thr-Arg21領域の配列をMet-Tyr-Ser-Asp-Leuに変換するために表2のMP01からMP05までの配列を有するプライマーを作成し、表3に有る様に各プライマーとP3プライマー(TCAACTCGAGGACCTTGAAAAACTCACC;配列番号32)によるPCR増幅断片を各制限酵素で切断後、同一の制限酵素で切断した変異の導入されていないST0452タンパク質を発現ベクターpETST0452Hに導入した。
【表2】
【表3】
Arg12、Thr80をAla残基に、Thr80をLeu残基に変換するために表1のMP06からMP08までの配列を有するプライマーを作成し、表3に有る様にP1プライマー (ATAGCATATGAAGGCATTTATTCTTGCTGC;配列番号31)と各プライマーとによるPCR増幅断片を各制限酵素で切断後、同一の制限酵素で切断した変異の導入されていないST0452タンパク質を発現させることができるベクターpETST0452Hに導入した。Asp99、Glu146、Lys147、Asp208をAla残基に変換するためには、表1のMP09からMP16までの配列を有するプライマーを作成し、表3に有る様にP1プライマーとMP09、MP11、MP13、MP15プライマーとによるPCR増幅断片とMP10、MP12、MP14、MP16各プライマーとP3プライマー(TCAACTCGAGGACCTTGAAAAACTCACC;配列番号32)とによるPCR増幅断片をNdeI及びXhoIで切断したpET21bに挿入した。塩基配列を確認して、目的のアミノ酸残基に変異が導入されていることを確認できた発現ベクターを、それぞれpST0452(G9A)H、pST0452(K23A)H、pST0452(Y97A)H、pST0452(Y97F)H、pST0452(RegionI)H、pST0452(R13A)H、pST0452(T80A)H、pST0452(T80L)H、pST0452(D99A)H、pST0452(E146A)H、pST0452(K147A)H、pST0452(D208A)Hと名づけた。合成されたタンパク質のC末端には、ヒスチジン残基をタグとして結合するようにした。
【0025】
(6)組換え遺伝子の発現
大腸菌(E. coli BL21(DE3) CodonPlus RIL,、Novagen社製)のコンピテントセルを融解して、二本のファルコンチューブに各々0.1mlづつ移す。その中に上記発現プラスミド10ng分に相当する溶液を別々に加え氷中に30分間放置した後42℃でヒートショックを30秒間行い、そこにSOC培地0.9mlを加え、37℃で1時間振とう培養する。その後、アンピシリンを含むLB寒天プレート上に適量まき、37℃で一晩培養し、形質転換体大腸菌 BL21(DE3)CodonPlus RIL/pET21b/pST0452(G9A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(K23A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(Y97A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(Y97F)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(RegionI)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(R13A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(T80A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(T80L)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(D99A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(E146A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(K147A)H、及びBL21(DE3)CodonPlus RIL/pET21b/pST0452(D208A)H を得た。
【0026】
当該形質転換体を、アンピシリンを含むLB培地(2リットル)中で一晩37℃において培養した後、IPTG(Isopropyl-b-D-thiogalactopyranoside)を1mMになるように加え、さらに30℃で5時間培養した。培養後遠心分離(6,000 rpm、20min)により集菌を行った。
【0027】
(7)各変異体酵素の精製
0.5リットル培養液から集菌した菌体に2倍量の20mMリン酸緩衝液(pH7.5)、0.5M NaCl、70 unitのDNaseI(タカラ社製)を加え懸濁液を得た。得られた懸濁液を超音波破砕し、遠心分離(11,000 rpm、20分)により上清液を得た。この上清液を用いNi-loaded HiTrap Chelating HPカラム(Amersham Biosciences社製)による親和性クロマトグラムを行った。ここで得られた0.5 Mイミダゾール溶出画分(5 ml)を、11,000 rpm、20分間遠心分離することにより上清液を得た。さらに、これを50mMトリス塩酸緩衝液(pH7.5)で透析し、精製サンプルとした。
【実施例2】
【0028】
耐熱性の確認
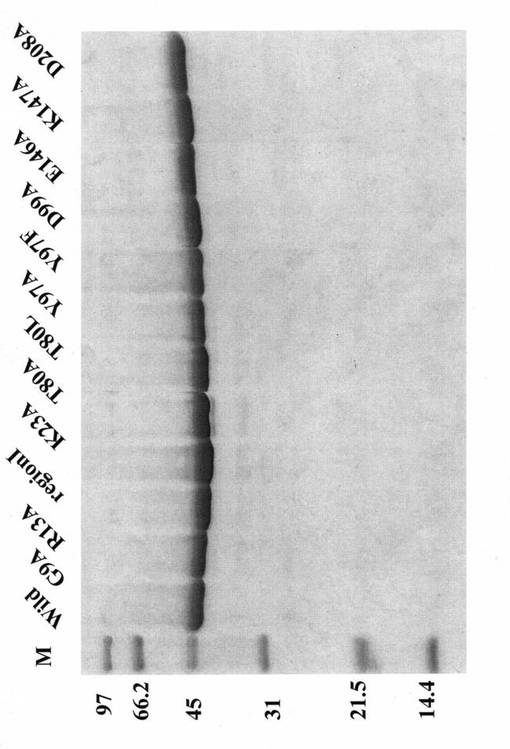
各精製タンパク質を、50mMトリス塩酸緩衝液(pH7.5)中で、20分間80度に加熱処理した後、11,000 rpm、20分間の遠心分離により上清液を得た。この上清をSDS-ポリアクリルアミドゲル電気泳動により、含まれるタンパク質の確認を行った。図1に示すように、全てのタンパク質が元のST0452タンパク質と同様の耐熱性を示した。
【実施例3】
【0029】
UDP-GlcNAcの合成
(1)UDP-GlcNAc合成反応(GlcNAc一リン酸とUTPの結合反応)
50mM Tris緩衝液(pH7.5)、12 mM MgCl2、24 mM GlcNAc-1-phosphate、1μM UTP、1 Uのinorganic pyrophosphataseからなる酵素反応液300μl中に実施例1で得られた精製酵素0.0135 mgを加えた。この酵素反応液を80℃で保温することにより、反応させた。5分、10分、15分、20分、25分後に30μlを分取し、300μlの500 mM KH2PO4溶液に加えることにより反応を停止させた。
【0030】
(2) UDP-GlcNAc合成反応(GlcNAc一リン酸とUTPの結合反応)の測定
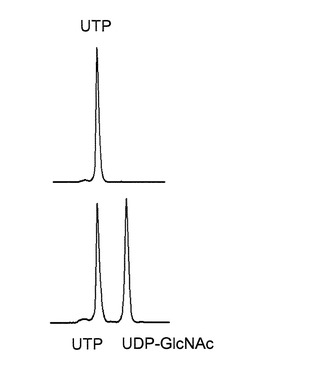
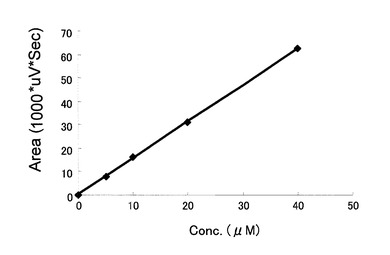
HPLCを用いて、反応生成物であるUDP-GlcNAcの量を、ヌクレオチド部分の紫外線の吸収を目安に測定した。図2に示すように標準物質であるUTP及びUDP-Glucoseは、HPLCにおいて溶出位置が全く異なる。さらに、図3に示すように標準サンプル添加量を変化させたときの、ピークの面積と標準物質量は正確な比例関係にあり、この検量線を用いることにより反応生成物を定量できることが示された。
そこで、上記(1)で反応させたサンプルに関しても、HPLCで同様の解析を行った。
【実施例4】
【0031】
各変異体酵素の酵素学的特性
(1)各変異体の持つ酵素反応パラメータの解析
各変異体酵素は上記の精製プロセスで完全に精製され、SDS-ポリアクリルアミドゲル電気泳動によって単一の酵素だと確認された。そこで、UDP-GlcNAcを合成する活性を測定したところ図4に有る様にほとんどの変異体で活性が低下しているような結果を得た。そこで各変異体酵素に関してKm(ミカエリス常数)及びKcat値を求めた。その結果、表4に有る様に、全ての変異体酵素において、GlcNAc一リン酸基質とのKm値が大幅に上昇していることが明らかとなった。しかし、G9A、T80A、Y97A、Y97F、K147AではKcat値が上昇していることが判明した。このことは、基質との結合力は弱まっているが、一度基質が本変異体酵素に結合すると反応はより早く進むということを示している。この結果から、これらの変異体酵素に十分な基質を与えると、元の酵素よりも早く反応が進むことが予想された。
【表4】
【実施例5】
【0032】
各変異体酵素反応条件の最適化
(1) ウリジン三リン酸基質の濃度条件
各変異体酵素を100μM UTPと50μM GlcNAc一リン酸存在下の反応条件で反応させた。その結果を、図5の網掛けバーで示した。しかし、この条件では高い活性の検出はできなかった。
【0033】
(2) GlcNAc一リン酸基質の濃度条件
各変異体酵素を100μM UTPと10mM GlcNAc一リン酸存在下の反応条件で反応させた。その結果を、図6の塗りつぶしバーで示した。その結果、G9A、T80A、Y97A、Y97F、K147Aでは、同一条件下の元のST0452酵素より、1.15倍、1.5倍、2.1倍、1.9倍、2.0倍高い活性が見出されたことから、本変異により効率的に活性を促進したUDP-GlcNAc合成酵素を作成することができた。
【0034】
(3)金属イオンの違いによる条件
また、D208A変異体で変異を挿入したアミノ酸残基は酵素に結合している金属イオンと結合していることが推定されたので、他の金属イオンを含む反応溶液で活性を測定した。その結果を図7に示す。D208A変異体では最適金属イオンは、元のST0452酵素のカルシウムから亜鉛に変化し、さらに3倍高い活性を示した。さらに、カルシウムを付加した場合には、D208A変異体はまったく活性を示さなくなった。このことは、本D208A変異体の高い活性を、反応液に加える金属イオンの種類によって調節することができることを示している。
【図面の簡単な説明】
【0035】
【図1】精製された各変異体蛋白質溶液を20分間80度に加熱処理した後、得られた各上清液についてSDS-ポリアクリルアミドゲル電気泳動を行った結果を示す写真である。
【図2】HPLCによる、UTP、及びUTPとUDP-GlcNAc混合物の分離パターンを測定したグラフである。
【図3】HPLCを用いたUDP-GlcNAcの検量線を示す図である。
【図4】5μM UTPと50μM GlcNAc一リン酸存在下での各変異体酵素のUDP-GlcNAc合成活性を示すグラフである。
【図5】100μM UTPと50μM GlcNAc一リン酸存在下での各変異体酵素のUDP-GlcNAc合成活性を示すグラフである。
【図6】100μM UTPと10mM GlcNAc一リン酸存在下での各変異体酵素のUDP-GlcNAc合成活性を示すグラフである。
【図7】金属イオンの違いによるUDP-GlcNAc合成活性の違いを示す図である。
【技術分野】
【0001】
本願発明は、耐熱性の低下を伴わずに糖ヌクレオチド合成活性が促進された耐熱性変異体蛋白質、該蛋白質をコードするDNA、該DNAを含有する組換え体DNA、該組換え体DNAを保有する形質転換体、該形質転換体を用いた糖ヌクレオチド合成活性を有する蛋白質の製造法、および該蛋白質あるいは該形質転換体を用いた糖ヌクレオチドの製造法に関する。
【背景技術】
【0002】
糖ヌクレオチド(UDP-GlcNAc)合成活性を有する酵素としては大腸菌(Escherichia coli)(非特許文献1参照)やストレプトコッカス菌(Streptococcus pneumoniae) (非特許文献2参照)、ネイセリア菌(Neiseia gonorrhoeae) (非特許文献3参照)由来のGlmU (N-Acetyl-D-Glucosamine-1-phosphate uridylyltransferase)の詳しい性質がすでに報告されている。GlmUは様々な糖鎖合成に必須な構成要素であるN-アセチル-D-グルコサミン(N-Acetyl-D-Glucosamine (GlcNAc))の活性体ウリジン二リン酸-N-アセチル-D-グルコサミン(UDP-GlcNAc)を合成する酵素であり、N-アセチル-D-グルコサミン-1-リン酸(N-Acetyl-D-Glucosamine-1-phosphate (GlcNAc-1-P))とウリジン三リン酸(UTP)を基質として、ウリジン二リン酸-N-アセチル-D-グルコサミン(UDP-GlcNAc)を生産する。上記の様に、幾つかの微生物からUDP-GlcNAcを合成する酵素が見出されているが、それらは常温生物由来のため室温以上では極めて不安定で、活性は80℃程度の加熱処理により速やかに失活する。このため、使用時の滅菌等の処理が必要であったり、低温での注意深い保存が必要であった。また、それら常温微生物由来酵素と比較すると、本酵素活性を有する耐熱性酵素ST0452蛋白質の活性は同等か若干低いものであった。そこで、有利な耐熱性を有する酵素ST0452蛋白質の本酵素活性の促進が求められた。
【非特許文献1】Dominique Mengin-Lecreulx and Lean van Heijenoort “Identification ofthe GlmU Gene Encoding N-Acetylglucosamine-1-PhosphateUridylyltransferase in Escherichia coli “ (1993) Journal of Bacteriology, 175, 6150-6157.
【非特許文献2】Dirk Kostrewa, Allan D’Arcy, Bela Takacs and Markus Kamber “CrystalStructure of Streptococcus pneumoniae N-Acetyl-glucosamine-1-phosphate Uridylyltransferase,GlmU, in Apo Form at 2.33 Å Resolution and in Complex withUDP-N-Acetylglucosamine and Mg2+ at 1.96 Å Resolution" (2001) Journal ofMolecular Biology, 305, 279-289.
【非特許文献3】Joachim Ullrich and Jos P. M. van Putten "Identification of theGonococcal glmU Gene Encoding the Enzyme N-Acetylglucosamine1-Phosphate Uridylyltransferase Involved in the Synthesis of UDP-GlcNAc" (1995) Journal of Bacteriology, 177, 6902-6909.
【発明の開示】
【発明が解決しようとする課題】
【0003】
糖ヌクレオチドUDP-GlcNAcを合成する活性を有する耐熱性酵素を我々は既にスルフォロバス・トーコーダイイから見出してきた。本耐熱性酵素では、常温微生物が有しているUDP-GlcNAc合成活性に加えてdTDP-GlcNAc合成活性も有しており、活性が半減するためには80度で160分の加熱処理を必要とする程、その耐熱性は高い。しかし、この耐熱性酵素が有するUDP-GlcNAc合成活性は、既に詳しく解析されている常温微生物が有する類似酵素の活性と同等か少し劣るものであった。そこで、本耐熱性酵素が有する特徴的な活性や高い耐熱性を有効に利用するためには、さらに高いUDP-GlcNAc合成活性を有する安定な酵素が渇望されていた。
【0004】
したがって本発明の課題は、耐熱性を維持したまま、既存のN-アセチル-D-グルコサミン-1-リン酸(GlcNAc-1-P)等の糖一リン酸、及びウリジン三リン酸(UTP)等のヌクレオシド三リン酸を基質として、UDP-GlcNAc等の糖ヌクレオチドを合成する活性を促進した新規酵素を提供することにある。
【課題を解決するための手段】
【0005】
本発明者は、以上のような課題を解決すべく、75 - 80℃で生育する超好熱古細菌 Sulfolobus tokodaii strain7由来ST0452蛋白質に着目し、その配列中のアミノ酸を変化させた変異体を作成した。さらに、大腸菌を使ってそれらの遺伝子から変異体酵素を生産し、この酵素が高温(80℃)での安定性に変化なく、かつ促進された糖ヌクレオチド合成活性を示すものが存在することを確認した。さらに、本酵素を用いることにより様々な種類の糖ヌクレオチドを生産することもできることを見いだし、本発明を完成するに至ったものである。
【0006】
即ち、本発明は、以下の(1)〜(8)に係るものである。
(1) 配列番号2〜7のいずれかに示されるアミノ酸配列を有することを特徴とする、糖ヌクレオチド合成活性を有する蛋白質。
(2)上記(1)に記載の蛋白質をコードするDNA。
(3)配列番号9〜14のいずれかに示される塩基配列を有することを特徴とするDNA。
(4)上記(2)または(3)に記載のDNAがベクターに組み込まれていることを特徴とする組換え体DNA。
(5)上記(4)に記載の組換え体DNAが宿主細胞に導入されていることを特徴とする形質転換体。
(6)上記(5)に記載の形質転換体を培地に培養し、培養物から糖ヌクレオチド合成活性を有する蛋白質を採取することを特徴とする、糖ヌクレオチド合成活性を有する蛋白質の製造方法。
(7) 糖一リン酸及びヌクレオシド三リン酸に、上記(1)に記載の蛋白質を作用させることを特徴とする、糖ヌクレオチドの製造方法。
(8) 糖一リン酸及びヌクレオシド三リン酸に、上記(5)に記載の形質転換体の培養液あるいは培養物の処理物を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【発明の効果】
【0007】
本発明によれば、耐熱性を維持したままで活性の比較的低かったST0452酵素のUDP-GlcNAc合成活性を1.5倍から2倍程度促進した酵素を提供でき、これにより、試験管内でのUDP-GlcNAc合成の際に、熱に安定でかつ効率的に目的とするUDP-GlcNAcを合成することができる。
一方、UDP-GlcNAcは、糖タンパク質、糖脂質、多糖類の糖鎖合成の際にGlcNAc供与体として機能するものであり、これらの糖鎖合成は、癌転移、器官発生あるいは細胞性免疫等に密接に関連するものとして近年注目されており、本発明は、これら研究の発展において、その貢献度は極めて大きい。
【発明を実施するための最良の形態】
【0008】
以下に、本願発明を具体的に説明する。
本発明の酵素タンパク質は、好酸性好気性超好熱古細菌スルフォロバス・トーコーダイイ(JCM登録番号JCM10545)由来のST0452UDP-GlcNAc合成活性を有するタンパク質(以下、ST0452タンパク質という場合がある。配列番号1)に変異を導入したものであり、該変異体酵素タンパク質は、耐熱性が低下することなく、上記ST0452UDP-GlcNAc合成活性が向上したものである。また、本発明のDNAは、上記変異体酵素タンパク質をコードするDNAである。
【0009】
本発明者は、これまでに類似酵素についての立体構造解析結果等を勘案して、上記スルフォロバス、トーコーダイイ(JCM登録番号JCM10545)由来のST0452UDP-GlcNAc合成活性を有するタンパク質における9個のアミノ酸残基と1つの領域を変異導入の対象として選択し、これらの変異が導入された変異体酵素を作成した。
すなわち、後記する表2に示す変異を導入したプライマーを、同表3に示した組み合わせでPCR増幅を行い、変異を導入したDNA断片を得た。
この断片を、蛋白質発現プラスミドpET21bに挿入後、そのプラスミドにより形質転換した大腸菌を用いて変異体酵素の生産をおこなった。得られた変異体酵素は、各変異の導入位置に従って、G9A、R13A、RegionI、K23A、T80A、T80L、Y97A、Y97F、D99A、E146A、K147A、D208Aと名づけた。このうち、本発明の変異体酵素は、G9A、T80A、Y97A、Y97F、K14A及びD208A である(なお、これらの命名記号における、数字はST0452タンパク質におけるアミノ酸置換位置を、その左側のアルファベットは置換前のアミノ酸を、及び右側のアルファベットは置換後のアミノ酸をそれぞれ表す。)。
上記12種の変異体酵素は、いずれもST0452タンパク質の活性中心に変異を導入したものである。この理由は、活性中心は酵素の内側に位置するから、この位置のアミノ酸を置換させても、酵素タンパク質の耐熱性に影響を与える酵素の全体構造の変化は少ないと考えたからであるが、活性中心に変異を導入する場合は酵素活性は低下するのが普通である。しかし、本発明における、上記G9A、T80A、Y97A、Y97F、K14A及びD208Aの各変異体酵素は、全く意外にも、ST0452UDP-GlcNAc合成活性がむしろ向上している点で極めてユニークである。
【0010】
これら6種の変異体酵素のアミノ酸配列およびこれをコードするDNAの塩基配列は配列表の配列番号2〜7及び9〜14にそれぞれ示される。これらの関係を分かりやすくするため以下の表1にまとめて示す。
【表1】
【0011】
本発明における上記変異体酵素の優れた特性は、以下の実験により確かめられている。
すなわち、本変異体酵素を、50mトリス塩酸緩衝液(pH7.5)中で、80℃、20分間加熱処理を行った後に、SDSポリアクリルアミドゲル電気泳動で確認したところ、全ての変異体が可溶性タンパク質として確認されたことから、全ての変異体タンパク質は元のST0452タンパク質と同等の耐熱性を維持していることが示された。
これら作成した12種類の変異体ST0452タンパク質について、UDP-GlcNAc合成活性の各基質に関するKm値及び反応のKcat値を求めた。各変異体のKm及びKcat値を表4にまとめた。その結果表4に示されているように、全ての変異体において基質との結合の強さを示すKm値は低下していたが、変異体G9A、Y97F、K147A等では変異を導入する前のST0452タンパク質よりも反応の進む程度を示すKcat値が上昇していることが明らかとなった。
【0012】
上記結果から、基質との結合は弱くなっているが、反応は進みやすくなっている変異体が存在していることが明らかとなった。そこで、基質濃度を上げることで、変異を導入する前のST0452酵素よりも高い活性を見出せることが推定された。そこで、ヌクレオチド基質であるUTP(ウリジン三リン酸)の濃度を20倍、GlcNAc-1-P(N-アセチル-D-グルコサミン-1-リン酸)の濃度を200倍高くして活性を測定したところ、図6の塗りつぶしたバーのように、G9A、T80A、Y97A、Y97F、K174Aの各変異体酵素では、変異を導入していないST0452タンパク質の活性よりも高い活性が検出された。
【0013】
さらに、D208A変異体酵素においては、反応液に加える金属イオンを変えて反応を行った。その結果、図7に示す様に亜鉛イオンを加えた場合には、変異を導入していないST0452タンパク質の活性の3倍もの活性を見出すことができた。
【0014】
すなわち、本発明のG9A、T80A、Y97A、Y97F、K174Aの各変異体タンパク質は、反応液中の基質濃度を上げることにより、また、同D208A変異体酵素は亜鉛イオンを加えることにより、糖ヌクレオチド合成活性を促進させることができ、これにより糖ヌクレオチドの生産性が向上する。またこれらの変異体酵素は、80℃、20分間の熱処理でも変性しないという優れた耐熱性を有する。
G9A、T80A、Y97A、Y97F、K174Aの各変異体酵素を使用する場合における、ヌクレオシド三リン酸の濃度は50〜100 uM、糖一リン酸の濃度は2〜10 mMが好ましい。また、同D208A変異体酵素を使用する場合の亜鉛イオン濃度は1〜2 mMが好ましい。
【0015】
本発明の酵素を得るには、通常の遺伝子工学的手法が適用でき、本発明の上記各種変異体酵素をコードするDNAを、例えば、pET21b、pHY481等の蛋白質発現プラスミドベクター等に挿入して組み換えベクターを作製し、該組み換えベクターを用いて宿主細胞を形質転換し、該形質転換体を培地で培養し、培養物、培養処理物あるいはこれら培養物から分離回収された形質転換体から、酵素を常法の蛋白質精製手段により精製し単離する。上記宿主細胞としては、大腸菌・枯草菌等が利用可能である。
【0016】
本発明においては、さらに上記変異体酵素を用いて、糖ヌクレオチドを合成するが、この合成においては、例えば、糖一リン酸とヌクレオシド三リン酸を含有する溶液に、該酵素を添加し、反応温度60℃〜95℃で反応させることで、目的の糖ヌクレオチドを得ることができる。
また、ヌクレオシド三リン酸としては、UTP以外にTTP(チミジン三リン酸)も用いることができる。
【0017】
以下に、UTP(ウリジン三リン酸)とGlcNAc-1-P(N-アセチル-D-グルコサミン-1-リン酸)からUDP-GlcNAc(ウリジン二リン酸-N-アセチル-D-グルコサミン)を合成する場合の反応式を示す。
【化1】
【0018】
この反応においては、上記精製した酵素のみならず、粗酵素であってもよい。例えば、宿主として枯草菌等分泌型の系を用いる場合には、培養液中に本酵素が生成蓄積され、大腸菌等の非分泌型の系を用いる場合には、菌体内に生成されるので、本酵素を含有する培養液あるいはその処理物、もしくは菌体破砕物等の培養処理物を用いて、UDP-GlcNAcを合成してもよい。
以下に、本発明の実施例を示すが、本発明実施例により限定されるものではない。
【実施例1】
【0019】
糖ヌクレオチド合成酵素変異体の作成
(1)菌の培養
好酸性好気性超好熱古細菌スルフォロバス・トーコーダイイJCM10545は次の方法で培養した。
1.3gの(NH4)2SO4、0.28gのKH2PO4、0.25gのMgSO4・7H2O、0.07gのCaCl2・2H2O、0.02gのFeCl3・6H2O、1.8mg のMnCl2・4H2O、4.5mgのNa2B4O7・10H2O、0.22mgのZnSO4・7H2O、0.05mgのCuCl2・2H2O、0.03mgのNa2MoO4・2H2O、0.03mgのVOSO4・xH2O、0.01mgのCoSO4・7H2O、1.0gの酵母エキスを1Lの蒸留水に溶かし、この溶液のpHを3.5に10規定H2SO4溶液で調整した。加圧殺菌した後、JCM10545を植菌した。この培養液を80℃で1〜2日培養し、その後遠心分離し集菌した。
【0020】
(2)染色体DNAの調製
JCM10545の染色体DNAは以下の方法により調製した。
培養終了後5000rpm、10分間の遠心分離により菌体を集菌する。菌体を10 mM EDTA(pH 6.0)溶液で洗浄後、50 mM Tris/HCl-50 mM EDTA (pH8.5)溶液を加えて細胞を溶解させる。さらに、0.5% Na-lauroylsarcosinate、1 mg/ml プロテアーゼKとなるように各々を加えた後、50℃で3時間保温する。フェノール処理を3回行った後、溶液を10 mM Tris-10 mM EDTA (pH 8.0)溶液に対して透析する。37℃で30分間のRNaseによるRNAの分解後、フェノールクロロフォルム溶液で処理した後、10 mM Tris-1 mM EDTA(pH 8.0)で透析を行う。
【0021】
(3)染色体DNAを含むショットガンライブラリークローンの作製
(2)で得られた染色体DNAを超音波処理することにより断片化した後、アガロースゲル電気泳動により1kb及び2kb長のDNA断片を回収した。この断片をプラスミドベクターpUC118のHincII制限酵素部位に挿入したショットガンライブラリーを作製した。各ショットガンクローンの末端塩基配列を、ABI社製自動塩基配列読み取り装置377を用いて解読していった。各ショットガンクローンから得られた塩基配列を塩基配列自動連結ソフトSequencherを用いて連結編集し、本菌の全塩基配列を決定していった。
【0022】
(4)ST0452遺伝子中の変異を導入するアミノ酸残基の選択
上記手法で決定された好酸性好気性超好熱古細菌スルフォロバス・トーコーダイイのゲノム塩基配列の大型計算機による解析を行い、UDP-GlcNAcを合成する機能を持つタンパク質をコードする遺伝子はST0452であると推定され、その後の実験で高い熱安定性及び予想された活性の同定がなされた。該遺伝子の塩基配列及び該遺伝子がコードするタンパク質(ST0452タンパク質)のアミノ酸配列をそれぞれ配列番号8及び1にそれぞれ示す。
ST0452タンパク質の高い熱安定性を損なうことなく活性を促進するために、機能に重要な反応中心のアミノ酸残基に変異を導入することとした。酵素の活性中心は、基質を取り込むために比較的酵素の内側に位置していると考えられるので、活性中心のアミノ酸残基を変化させても、耐熱性に影響を与える恐れがある酵素自身の全体の構造には大きな影響を与えないと考えた。
【0023】
そこで、このST0452タンパク質のアミノ酸配列中、活性中心に位置すると考えられるアミノ酸のうち9個のアミノ酸(配列番号1のアミノ酸配列中、9,13、23、80,97、99、146、147、及び208番目に位置するアミノ酸)と1ヶ所の領域(同14番目〜21番目の領域)を、アミノ酸置換を導入する標的として選択した。
【0024】
(5)各変異体タンパク質発現プラスミドの構築
Gly9、Lys23、Tyr97をAla残基に、Tyr97をPhe残基に、Leu14-Glu-Phe-Ile-Thr-His-Thr-Arg21領域の配列をMet-Tyr-Ser-Asp-Leuに変換するために表2のMP01からMP05までの配列を有するプライマーを作成し、表3に有る様に各プライマーとP3プライマー(TCAACTCGAGGACCTTGAAAAACTCACC;配列番号32)によるPCR増幅断片を各制限酵素で切断後、同一の制限酵素で切断した変異の導入されていないST0452タンパク質を発現ベクターpETST0452Hに導入した。
【表2】
【表3】
Arg12、Thr80をAla残基に、Thr80をLeu残基に変換するために表1のMP06からMP08までの配列を有するプライマーを作成し、表3に有る様にP1プライマー (ATAGCATATGAAGGCATTTATTCTTGCTGC;配列番号31)と各プライマーとによるPCR増幅断片を各制限酵素で切断後、同一の制限酵素で切断した変異の導入されていないST0452タンパク質を発現させることができるベクターpETST0452Hに導入した。Asp99、Glu146、Lys147、Asp208をAla残基に変換するためには、表1のMP09からMP16までの配列を有するプライマーを作成し、表3に有る様にP1プライマーとMP09、MP11、MP13、MP15プライマーとによるPCR増幅断片とMP10、MP12、MP14、MP16各プライマーとP3プライマー(TCAACTCGAGGACCTTGAAAAACTCACC;配列番号32)とによるPCR増幅断片をNdeI及びXhoIで切断したpET21bに挿入した。塩基配列を確認して、目的のアミノ酸残基に変異が導入されていることを確認できた発現ベクターを、それぞれpST0452(G9A)H、pST0452(K23A)H、pST0452(Y97A)H、pST0452(Y97F)H、pST0452(RegionI)H、pST0452(R13A)H、pST0452(T80A)H、pST0452(T80L)H、pST0452(D99A)H、pST0452(E146A)H、pST0452(K147A)H、pST0452(D208A)Hと名づけた。合成されたタンパク質のC末端には、ヒスチジン残基をタグとして結合するようにした。
【0025】
(6)組換え遺伝子の発現
大腸菌(E. coli BL21(DE3) CodonPlus RIL,、Novagen社製)のコンピテントセルを融解して、二本のファルコンチューブに各々0.1mlづつ移す。その中に上記発現プラスミド10ng分に相当する溶液を別々に加え氷中に30分間放置した後42℃でヒートショックを30秒間行い、そこにSOC培地0.9mlを加え、37℃で1時間振とう培養する。その後、アンピシリンを含むLB寒天プレート上に適量まき、37℃で一晩培養し、形質転換体大腸菌 BL21(DE3)CodonPlus RIL/pET21b/pST0452(G9A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(K23A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(Y97A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(Y97F)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(RegionI)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(R13A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(T80A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(T80L)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(D99A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(E146A)H、BL21(DE3)CodonPlus RIL/pET21b/pST0452(K147A)H、及びBL21(DE3)CodonPlus RIL/pET21b/pST0452(D208A)H を得た。
【0026】
当該形質転換体を、アンピシリンを含むLB培地(2リットル)中で一晩37℃において培養した後、IPTG(Isopropyl-b-D-thiogalactopyranoside)を1mMになるように加え、さらに30℃で5時間培養した。培養後遠心分離(6,000 rpm、20min)により集菌を行った。
【0027】
(7)各変異体酵素の精製
0.5リットル培養液から集菌した菌体に2倍量の20mMリン酸緩衝液(pH7.5)、0.5M NaCl、70 unitのDNaseI(タカラ社製)を加え懸濁液を得た。得られた懸濁液を超音波破砕し、遠心分離(11,000 rpm、20分)により上清液を得た。この上清液を用いNi-loaded HiTrap Chelating HPカラム(Amersham Biosciences社製)による親和性クロマトグラムを行った。ここで得られた0.5 Mイミダゾール溶出画分(5 ml)を、11,000 rpm、20分間遠心分離することにより上清液を得た。さらに、これを50mMトリス塩酸緩衝液(pH7.5)で透析し、精製サンプルとした。
【実施例2】
【0028】
耐熱性の確認
各精製タンパク質を、50mMトリス塩酸緩衝液(pH7.5)中で、20分間80度に加熱処理した後、11,000 rpm、20分間の遠心分離により上清液を得た。この上清をSDS-ポリアクリルアミドゲル電気泳動により、含まれるタンパク質の確認を行った。図1に示すように、全てのタンパク質が元のST0452タンパク質と同様の耐熱性を示した。
【実施例3】
【0029】
UDP-GlcNAcの合成
(1)UDP-GlcNAc合成反応(GlcNAc一リン酸とUTPの結合反応)
50mM Tris緩衝液(pH7.5)、12 mM MgCl2、24 mM GlcNAc-1-phosphate、1μM UTP、1 Uのinorganic pyrophosphataseからなる酵素反応液300μl中に実施例1で得られた精製酵素0.0135 mgを加えた。この酵素反応液を80℃で保温することにより、反応させた。5分、10分、15分、20分、25分後に30μlを分取し、300μlの500 mM KH2PO4溶液に加えることにより反応を停止させた。
【0030】
(2) UDP-GlcNAc合成反応(GlcNAc一リン酸とUTPの結合反応)の測定
HPLCを用いて、反応生成物であるUDP-GlcNAcの量を、ヌクレオチド部分の紫外線の吸収を目安に測定した。図2に示すように標準物質であるUTP及びUDP-Glucoseは、HPLCにおいて溶出位置が全く異なる。さらに、図3に示すように標準サンプル添加量を変化させたときの、ピークの面積と標準物質量は正確な比例関係にあり、この検量線を用いることにより反応生成物を定量できることが示された。
そこで、上記(1)で反応させたサンプルに関しても、HPLCで同様の解析を行った。
【実施例4】
【0031】
各変異体酵素の酵素学的特性
(1)各変異体の持つ酵素反応パラメータの解析
各変異体酵素は上記の精製プロセスで完全に精製され、SDS-ポリアクリルアミドゲル電気泳動によって単一の酵素だと確認された。そこで、UDP-GlcNAcを合成する活性を測定したところ図4に有る様にほとんどの変異体で活性が低下しているような結果を得た。そこで各変異体酵素に関してKm(ミカエリス常数)及びKcat値を求めた。その結果、表4に有る様に、全ての変異体酵素において、GlcNAc一リン酸基質とのKm値が大幅に上昇していることが明らかとなった。しかし、G9A、T80A、Y97A、Y97F、K147AではKcat値が上昇していることが判明した。このことは、基質との結合力は弱まっているが、一度基質が本変異体酵素に結合すると反応はより早く進むということを示している。この結果から、これらの変異体酵素に十分な基質を与えると、元の酵素よりも早く反応が進むことが予想された。
【表4】
【実施例5】
【0032】
各変異体酵素反応条件の最適化
(1) ウリジン三リン酸基質の濃度条件
各変異体酵素を100μM UTPと50μM GlcNAc一リン酸存在下の反応条件で反応させた。その結果を、図5の網掛けバーで示した。しかし、この条件では高い活性の検出はできなかった。
【0033】
(2) GlcNAc一リン酸基質の濃度条件
各変異体酵素を100μM UTPと10mM GlcNAc一リン酸存在下の反応条件で反応させた。その結果を、図6の塗りつぶしバーで示した。その結果、G9A、T80A、Y97A、Y97F、K147Aでは、同一条件下の元のST0452酵素より、1.15倍、1.5倍、2.1倍、1.9倍、2.0倍高い活性が見出されたことから、本変異により効率的に活性を促進したUDP-GlcNAc合成酵素を作成することができた。
【0034】
(3)金属イオンの違いによる条件
また、D208A変異体で変異を挿入したアミノ酸残基は酵素に結合している金属イオンと結合していることが推定されたので、他の金属イオンを含む反応溶液で活性を測定した。その結果を図7に示す。D208A変異体では最適金属イオンは、元のST0452酵素のカルシウムから亜鉛に変化し、さらに3倍高い活性を示した。さらに、カルシウムを付加した場合には、D208A変異体はまったく活性を示さなくなった。このことは、本D208A変異体の高い活性を、反応液に加える金属イオンの種類によって調節することができることを示している。
【図面の簡単な説明】
【0035】
【図1】精製された各変異体蛋白質溶液を20分間80度に加熱処理した後、得られた各上清液についてSDS-ポリアクリルアミドゲル電気泳動を行った結果を示す写真である。
【図2】HPLCによる、UTP、及びUTPとUDP-GlcNAc混合物の分離パターンを測定したグラフである。
【図3】HPLCを用いたUDP-GlcNAcの検量線を示す図である。
【図4】5μM UTPと50μM GlcNAc一リン酸存在下での各変異体酵素のUDP-GlcNAc合成活性を示すグラフである。
【図5】100μM UTPと50μM GlcNAc一リン酸存在下での各変異体酵素のUDP-GlcNAc合成活性を示すグラフである。
【図6】100μM UTPと10mM GlcNAc一リン酸存在下での各変異体酵素のUDP-GlcNAc合成活性を示すグラフである。
【図7】金属イオンの違いによるUDP-GlcNAc合成活性の違いを示す図である。
【特許請求の範囲】
【請求項1】
配列番号2〜7のいずれかに示されるアミノ酸配列を有することを特徴とする、糖ヌクレオチド合成活性を有する蛋白質。
【請求項2】
請求項1記載の蛋白質をコードするDNA。
【請求項3】
配列番号9〜14のいずれかに示される塩基配列を有することを特徴とするDNA。
【請求項4】
請求項2または3に記載のDNAがベクターに組み込まれていることを特徴とする組換え体DNA。
【請求項5】
請求項4に記載の組換え体DNAが宿主細胞に導入されていることを特徴とする形質転換体。
【請求項6】
請求項5に記載の形質転換体を培地に培養し、培養物から糖ヌクレオチド合成活性を有する蛋白質を採取することを特徴とする、糖ヌクレオチド合成活性を有する蛋白質の製造方法。
【請求項7】
糖一リン酸及びヌクレオシド三リン酸に、請求項1に記載の蛋白質を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【請求項8】
糖一リン酸及びヌクレオシド三リン酸に、請求項5に記載の形質転換体の培養液あるいは培養物の処理物を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【請求項1】
配列番号2〜7のいずれかに示されるアミノ酸配列を有することを特徴とする、糖ヌクレオチド合成活性を有する蛋白質。
【請求項2】
請求項1記載の蛋白質をコードするDNA。
【請求項3】
配列番号9〜14のいずれかに示される塩基配列を有することを特徴とするDNA。
【請求項4】
請求項2または3に記載のDNAがベクターに組み込まれていることを特徴とする組換え体DNA。
【請求項5】
請求項4に記載の組換え体DNAが宿主細胞に導入されていることを特徴とする形質転換体。
【請求項6】
請求項5に記載の形質転換体を培地に培養し、培養物から糖ヌクレオチド合成活性を有する蛋白質を採取することを特徴とする、糖ヌクレオチド合成活性を有する蛋白質の製造方法。
【請求項7】
糖一リン酸及びヌクレオシド三リン酸に、請求項1に記載の蛋白質を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【請求項8】
糖一リン酸及びヌクレオシド三リン酸に、請求項5に記載の形質転換体の培養液あるいは培養物の処理物を作用させることを特徴とする、糖ヌクレオチドの製造方法。
【図2】

【図3】
【図4】

【図5】

【図6】

【図7】
【図1】
【図3】
【図4】
【図5】
【図6】
【図7】
【図1】
【公開番号】特開2007−325524(P2007−325524A)
【公開日】平成19年12月20日(2007.12.20)
【国際特許分類】
【出願番号】特願2006−158285(P2006−158285)
【出願日】平成18年6月7日(2006.6.7)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成17年度北海道大学委託研究「遺伝子情報解析に関する研究」、産業活力再生特別措置法第30条の適用を受ける特許出願
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
【公開日】平成19年12月20日(2007.12.20)
【国際特許分類】
【出願日】平成18年6月7日(2006.6.7)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成17年度北海道大学委託研究「遺伝子情報解析に関する研究」、産業活力再生特別措置法第30条の適用を受ける特許出願
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
[ Back to top ]