
糖鎖固定化蛍光性ナノ粒子、およびその製造方法
【課題】粒径が均一でありかつ高い水分散性および発光性を有し、かつ容易に糖鎖を固定化することができる、安定的な糖鎖固定化蛍光性ナノ粒子の製造法を提供すること。
【解決手段】糖鎖とリンカー化合物とを結合させたリガンド複合体を、加熱処理した蛍光性ナノ粒子と結合させて、糖鎖を固定化した蛍光性ナノ粒子を得る。糖鎖を固定化した蛍光性ナノ粒子をタンパク質の希釈系列に加えることによって複合体を形成させて、凝集体の沈殿を生じさせる。
【解決手段】糖鎖とリンカー化合物とを結合させたリガンド複合体を、加熱処理した蛍光性ナノ粒子と結合させて、糖鎖を固定化した蛍光性ナノ粒子を得る。糖鎖を固定化した蛍光性ナノ粒子をタンパク質の希釈系列に加えることによって複合体を形成させて、凝集体の沈殿を生じさせる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、糖鎖を固定化した蛍光性ナノ粒子とその製造方法に関する。
【背景技術】
【0002】
近年、ナノ粒子は、ライフサイエンス、医療診断、バイオテクノロジーなどの様々な分野への応用が行われている。例えば、抗体などを固定化したナノ粒子は、抗原抗体反応による免疫染色や、抗原の検出、分離/精製などに有用である。また、薬剤や遺伝子を固定化したナノ粒子は、ドラッグデリバリーシステム(DDS)のキャリアとして有用である。また、標識したナノ粒子は、分析用マーカー、トレーサーなどの分析用試薬として有用である。
【0003】
蛍光性ナノ粒子は、蛍光体成分として有機材料または無機材料を用いて調製される。有機材料は、容易に光退色するので長時間の蛍光イメージングには適さない。一方、無機材料は、有機材料に比べて光退色しにくいので長時間の蛍光イメージングが可能となる。
【0004】
無機材料を用いる蛍光性ナノ粒子の製造法は、これまでに様々な方法が開発されている。一般的には、カドミウムイオンと硫黄やセレン、テルルなどのカルコゲンとを界面活性剤存在下で加熱し、ナノ結晶を形成/成長させることによって調製する(非特許文献1)。この方法では、反応時間によって粒径分布の狭い均一な蛍光性ナノ粒子を調製することができるが、ナノ粒子表面が界面活性剤で覆われており疎水性を示す。疎水性のため、水溶性の溶媒に不溶であるため、ライフサイエンス、医療診断、バイオテクノロジーなどの様々な分野への応用が難しい。
【0005】
疎水性の蛍光性ナノ粒子から親水性の蛍光性ナノ粒子に転換させるために、方法として、デキストランやポリアクリル酸などの親水性ポリマーによる被覆、シランカップリング剤によるシリカ表面処理による親水化(非特許文献2)、リポソーム被覆(非特許文献3、4)などが知られているが、親水性の蛍光性ナノ粒子に不可欠な糖鎖を導入するためには多段階を要する。また導入できたとしても糖鎖本来の機能発現に必要な糖鎖の高密度化は困難である。
【0006】
上記の方法に対して、近年、親水性の蛍光性ナノ粒子を水中で調製する方法が注目されている。テルル化カドミウムの調製法では(非特許文献5)、無機材料成分としてカドミウムイオンとテルル化水素ナトリウムを用いて、安定化剤として金属成分の配位子となる3−メルカプトプロピオン酸などのチオール基とカルボキシル基の二官能性を有する化合物を用いる。安定化剤に使用したチオール配位子は、容易に配位子交換することができるため、目的の分子構造を持つ蛍光性ナノ粒子に変換することができる。しかしながら、テルル化カドミウムの蛍光性ナノ粒子は、テルルが解離しやすく不安定である。また配位子交換によって導入したチオール配位子は、可逆的に固定化されているため、目的分子を長時間保持したナノ粒子の調製は難しい。さらに依然として、導入できたとしても糖鎖本来の機能発現に必要な糖鎖の高密度化は困難である。
【0007】
テルル化カドミウムの蛍光性ナノ粒子の安定性を向上させるため、硫化カドミウムや硫化亜鉛などによる被覆する方法が開発されている(非特許文献6)。しかしながら、これらのナノ粒子に対して、糖鎖分子の固定化は困難である。
【0008】
一方、糖鎖を固定化した蛍光性ナノ粒子(以下、適宜、糖鎖固定化蛍光性ナノ粒子、またはSFNPともいう。)として、硫化カドミウムのナノ粒子に糖鎖を固定化する方法が知られているが(特許文献1)、高い発光性能を有する蛍光性ナノ粒子の合成は達成されていない。
【0009】
近年、毒性が高いCdを含まない蛍光性ナノ粒子の製造法の開発が進められている。Zn1-x(AgIn)xS(ZAIS)(特許文献2)からなる蛍光性ナノ粒子はCdを含まないため注目されているが、その親水化は難しく、糖鎖分子の固定化にいたっては、これまでに達成されていない。
【0010】
また、テルル化カドミウム、硫化カドミウム、ZAIS等の親水化した蛍光性ナノ粒子に対して、リガンド交換反応による糖鎖の固定化を行うと、蛍光性ナノ粒子の発光性が著しく低下する問題点があった。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特表2004−511511号公報(平成16年4月15日公表)
【特許文献2】特開2010−31115号公報(平成22年2月12日公開)
【非特許文献】
【0012】
【非特許文献1】M. Bruchez Jr., et al., Science, 281, 2013 (1998)
【非特許文献2】C. Earhart, et al., Langmuir, 24, 6215 (2008)
【非特許文献3】F. Osaki, et al., J. Am. Chem. Soc., 126, 6520 (2004)
【非特許文献4】X.-L. Sun, et al., ChemBioChem, 5, 1593 (2004)
【非特許文献5】H. Zhang, et al, J. Phys. Chem. B, 107, 8 (2003)
【非特許文献6】H. Peng, et al., J. Lumin, 127, 721 (2007)
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明は、上記の問題点に鑑みてなされたものであり、ライフサイエンス、医療診断、バイオテクノロジーなどの様々な分野への利用に適した、粒径が均一でありかつ高い水分散性および発光性を有し、かつ容易に糖鎖を固定化することができる、安定的な糖鎖固定化蛍光性ナノ粒子の製造法を提供することを目的とする。
【課題を解決するための手段】
【0014】
本発明に係る糖鎖固定化蛍光性ナノ粒子は、還元剤処理した糖鎖リガンド複合体と、加熱処理した蛍光性ナノ粒子を混和することによって製造され、分散液として提供されることを特徴としている。本発明において、上記糖鎖リガンド複合体は水溶性であり、上記蛍光性ナノ粒子は親水化されている。得られた糖鎖固定化蛍光性ナノ粒子は、水溶液中で分散する。すなわち、本発明に係る糖鎖固定化蛍光性ナノ粒子の製造方法は、加熱処理された蛍光性ナノ粒子と、還元剤処理された糖鎖リガンド複合体を混和する工程を包含することを特徴としている。
【0015】
このような構成を採用することによって、表面が糖鎖リガンド複合体で修飾されており、平均粒径が1〜10nmの糖鎖固定化蛍光性ナノ粒子を得ることができる。
【0016】
本発明に係る糖鎖固定化蛍光性ナノ粒子において、上記糖鎖リガンド複合体は、炭化水素鎖または炭化水素誘導鎖を備えているリンカー化合物の一端に存在するアミノ基にて還元末端を有する糖鎖と結合し、該リンカー化合物の他端に存在する硫黄原子を含む炭化水素構造にて任意の金属と結合している。
【0017】
上記炭化水素誘導鎖は、炭素及び水素からなる炭化水素鎖であり、一部の炭素や水素が他の原子や置換基に置き換わっていてもよい。すなわち、上記炭化水素誘導鎖とは、末端にアミノ基を有し、炭化水素鎖の主鎖構造である炭素−炭素結合(C−C結合)の一部が、炭素−窒素結合(C−N結合)、炭素−酸素結合(C−O結合)、アミド結合(CO−NH結合)に置き換わっていてもよいものを指す。
【0018】
また、上記硫黄原子を含む炭化水素構造とは、炭素及び水素からなる炭化水素構造にて、一部の炭素が硫黄に置き換わっているものを意味する。また、この硫黄原子を含む炭化水素構造は、鎖状(直鎖、枝分かれ鎖の両方を含む)であっても、環状であってもよく、また、鎖状構造および環状構造の両方の構造を含んでいてもよい。
【0019】
本発明に係る糖鎖固定化蛍光性ナノ粒子において、上記硫黄原子を含む炭化水素構造は、S−S結合またはSH基を含む炭化水素構造を備えているものであってもよい。つまり、上記硫黄原子を含む炭化水素構造中に、ジスルフィド結合(S−S結合)またはチオール基(SH基)が含まれていてもよい。
【0020】
また、本発明に係る糖鎖固定化蛍光性ナノ粒子において、上記アミノ基は芳香族アミノ基であることが好ましい。還元アミノ化反応の最適条件であるpH3〜4においては、アミノ基がプロトン化されないことが必要である。そのため、芳香族との共役によってpH3〜4でも非共有電子対が窒素原子上に存在する芳香族アミノ基が好ましい。
【0021】
また、本発明に係る糖鎖固定化蛍光性ナノ粒子において、糖鎖リガンド複合体の還元剤処理に用いる還元剤は水素化ホウ素ナトリウムであることが好ましい。
【0022】
上記構成によれば、リンカー化合物内のS−S結合を還元して−SH基に変換し、任意の金属と、金属−硫黄(S)結合、例えばカドミウム−硫黄(Cd−S)結合により結合することができる。これにより、このCd−S結合を介して、糖鎖固定化蛍光性ナノ粒子を提供することができる。このことにより、容易に糖鎖を固定化することができる安定な糖鎖固定化蛍光性ナノ粒子の製造法の提供が可能となった。
【0023】
還元剤処理した水溶性の糖鎖リガンド複合体を蛍光性ナノ粒子に添加することによって、蛍光性ナノ粒子を水溶液中で安定化することが可能となった。本発明に係る糖鎖固定化蛍光性ナノ粒子は、粒径が均一であり、かつ高い水分散性を有する。
【0024】
本発明の抗体固定化蛍光性ナノ粒子は、加熱処理された蛍光性ナノ粒子と、還元剤処理されたリガンド複合体を混和する工程を包含することを特徴としており、蛍光性ナノ粒子に固定化されたリガンド複合体に抗体を結合することによって製造される。本発明において、上記リガンド複合体は水溶性であり、上記蛍光性ナノ粒子は親水化されている。得られた抗体固定化蛍光性ナノ粒子は、水溶液中で分散する。
【0025】
このような構成を採用することによって、表面が抗体リガンド複合体で修飾されており、平均粒径が5〜50nmの抗体固定化蛍光性ナノ粒子を得ることができる。
【0026】
本発明に係る抗体固定化蛍光性ナノ粒子において、上記リガンド複合体は、炭化水素鎖または炭化水素誘導鎖を備えているリンカー化合物の一端にて抗体と直接的または間接的に結合し、該リンカー化合物の他端に存在する硫黄原子を含む炭化水素構造にて任意の金属(蛍光性ナノ粒子)と結合している。
【0027】
上記炭化水素誘導鎖は、上述した糖鎖固定化蛍光性ナノ粒子における炭化水素誘導鎖と同様の構造を有しているが、糖鎖固定化蛍光性ナノ粒子の製造に用いられる炭化水素誘導鎖の一端に存在するアミノ基は他の反応基であってもよく、抗体を直接的または間接的に結合させ得る反応基であれば特に限定されず、糖鎖が結合していても結合していなくてもよい。すなわち、本発明の抗体固定化蛍光性ナノ粒子におけるリガンド複合体は、糖鎖が結合していても結合していなくてもよい。
【0028】
また、上記硫黄原子を含む炭化水素構造は、上述した糖鎖固定化蛍光性ナノ粒子における炭化水素構造と同一の構造であればよい。このような構造を有することによって、糖鎖固定化蛍光性ナノ粒子を製造する際の利点が、抗体固定化蛍光性ナノ粒子の製造の際においてももたらされる。すなわち、本発明の抗体固定化蛍光性ナノ粒子の製造方法を用いれば、蛍光性ナノ粒子に抗体を容易に固定化することができ、かつ安定な抗体固定化蛍光性ナノ粒子の提供が可能となる。さらに、本発明を用いて製造された抗体固定化蛍光性ナノ粒子は、粒径が均一であり、かつ高い水分散性を有する。
【発明の効果】
【0029】
以上のように、還元剤処理した糖鎖リガンド複合体と、加熱処理した親水性の蛍光性ナノ粒子とを混和するだけで、本発明に係る糖鎖固定化蛍光性ナノ粒子を調製することができる。このことは、容易に分解しやすい糖鎖であっても蛍光性ナノ粒子上に固定化できると同時に蛍光性ナノ粒子上に高い安定性を付与し得るという効果を奏する。また、本発明に係る糖鎖固定化蛍光性ナノ粒子を含む溶液に、上記糖鎖固定化蛍光性ナノ粒子の外側に位置する糖鎖を認識するタンパク質を添加するだけで、糖鎖とタンパク質とが相互作用した凝集物を生成し、溶液の蛍光も変化するので、糖鎖−タンパク質相互作用を、標識を用いることなく目視によって容易に測定することができるという、糖鎖−タンパク質相互作用の測定方法を提供し得るという効果を奏する。
【0030】
糖鎖は、細胞内外に糖タンパク質や糖脂質の形で存在しており、細胞間認識やシグナル伝達をはじめとする様々な生体機能を担っている。細胞内外の糖鎖結合性分子を蛍光標識などによって可視化することができれば、糖鎖が関わる生体機能の解明に繋がる。細胞の糖鎖結合性を利用し、本発明の糖鎖固定化蛍光性ナノ粒子を用いて細胞の蛍光標識を行うことによって、蛍光性ナノ粒子が細胞に結合し、糖鎖による細胞の蛍光標識を可能にするという効果を奏する。同様の観測は、既知の蛍光を発する有機化合物を糖鎖に付与することによっても達成されるが、有機化合物の場合は、発光に伴って、有機化合物自体が光のエネルギーによって分解するため、蛍光が突然消失することが起こる。しかし、糖鎖固定化蛍光性ナノ粒子の場合には、このような光による分解は起こらず、長時間の連続測定も可能である。
【0031】
本発明の別の側面によれば、上記構成による本発明の蛍光性ナノ粒子を含む、相互作用検出剤、細胞標識剤、診断剤が提供される。
【図面の簡単な説明】
【0032】
【図1】α−グルコース固定化蛍光性ナノ粒子のコロイド溶液の透過型電子顕微鏡画像を示す図である。
【図2】目視下における糖鎖−タンパク質相互作用解析を示す図である。
【図3】糖鎖固定化蛍光性ナノ粒子を用いたFACS解析を示す図である。
【図4】本発明の合成法または従来法によって得られた蛍光性ナノ粒子の半値幅(a)および蛍光波長(b)を比較したグラフである。
【図5】MALDI−TOF/MSによって本発明の糖鎖固定化蛍光性ナノ粒子における糖鎖の固定化を示すチャート(a)、および本発明の糖鎖固定化蛍光性ナノ粒子の透過型電子顕微鏡画像(b)を示す図である。
【図6】本発明の糖鎖固定化蛍光性ナノ粒子のタンパク質との相互作用を示すグラフである。
【図7】本発明の糖鎖固定化蛍光性ナノ粒子の結合特異性を示すグラフである。
【図8】本発明の糖鎖固定化蛍光性ナノ粒子による細胞認識を示すグラフである。
【図9】本発明の糖鎖固定化蛍光性ナノ粒子による細胞認識を示す蛍光顕微鏡画像である。
【図10】本発明の糖鎖固定化蛍光性ナノ粒子の製造に用いる蛍光性ナノ粒子の特性を示す図である。
【図11】本発明の糖鎖固定化蛍光性ナノ粒子の製造に用いる蛍光性ナノ粒子の特性を示す図である。
【図12】本発明の糖鎖固定化蛍光性ナノ粒子の特性を示す図である。
【図13】本発明の糖鎖固定化蛍光性ナノ粒子による細胞認識を示す蛍光顕微鏡画像である。
【図14】本発明の糖鎖固定化蛍光性ナノ粒子のタンパク質との相互作用および結合特異性を示す図である。
【図15】抗体を固定化した本発明の糖鎖固定化蛍光性ナノ粒子の、SDS−PAGE後の銀染色の結果を示す図である。
【図16】抗体を固定化した本発明の糖鎖固定化蛍光性ナノ粒子の、UV−Visおよび蛍光強度を測定した結果を示す図である。
【図17】抗体を固定化した本発明の糖鎖固定化蛍光性ナノ粒子の、黄色ブドウ球菌との結合性を示す蛍光顕微鏡画像である。
【図18】MALDI−TOF/MSによって本発明の抗体固定化蛍光性ナノ粒子におけるリンカー分子の固定化を示すチャートである。
【図19】本発明の抗体固定化蛍光性ナノ粒子の、SDS−PAGE後の銀染色の結果を示す図である。
【図20】抗体固定化蛍光性ナノ粒子を用いたFACS解析を示す図である。
【発明を実施するための形態】
【0033】
本発明の実施の形態について説明すれば以下のとおりであるが、本発明はこれに限定されるものではない。以下、本発明に係る糖鎖固定化蛍光性ナノ粒子の製造方法、糖鎖−タンパク質相互作用の測定方法、細胞の糖鎖結合性を利用して細胞を蛍光標識する方法、および抗体/糖鎖固定化蛍光性ナノ粒子の製造方法とそれを用いた細菌の検出方法について詳述する。
【0034】
〔1.糖鎖固定化蛍光性ナノ粒子の製造法〕
本発明は、糖鎖を固定化した蛍光性ナノ粒子(すなわち、糖鎖固定化蛍光性ナノ粒子)を製造する方法を提供する。本発明の方法によって製造された蛍光性ナノ粒子は水溶液中で分散する特徴を有している。すなわち、本発明の方法は、糖鎖固定化蛍光性ナノ粒子のコロイド溶液を調製する方法でもある。
【0035】
本明細書において、「糖鎖固定化蛍光性ナノ粒子」とは、以下に詳述する糖鎖リガンド複合体と、任意の蛍光性ナノ粒子(蛍光性の金属ナノ粒子ともいう。)とを結合させてなるものである。なお、蛍光性ナノ粒子は後述するように製造してもよいし、市販のものを使用してもよい。
【0036】
なお、本明細書において、「金属ナノ粒子」は、無機金属成分を含むナノ粒子であれば特に限定されず、本発明において好適に用いられる金属成分としては、Si、Ge、Cd、Zn、Cu、Ag、Ga、As、In、Te、Sなどが挙げられるがこれらに限定されない。
【0037】
また、本明細書中において、「ナノ粒子」は水溶液中で分散してコロイド溶液を形成するものが意図される。よって、ナノ粒子の平均粒子径は、0.5〜400nmの範囲内であることが好ましく、0.5nm〜100nmの範囲内であることがより好ましく、1nm〜10nmの範囲内であることがさらに好ましい。平均粒子径は0.5nm未満であってもよいが、そのような粒子の製造は高コストであって実用的でなく、400nmを超えると、粒子の分散安定性が経時的に変化しやすいので好ましくない。
【0038】
また、本発明に係る糖鎖固定化蛍光性ナノ粒子の構成要素である糖鎖リガンド複合体は、任意の蛍光性ナノ粒子と結合することのできるリンカー化合物と、分析対象となるタンパク質などと特異的に相互作用することができる糖鎖とから構成されている。そのため、上記糖鎖リガンド複合体は、タンパク質等の物質と疎水性に基づく非特異的な相互作用を形成しないことが必要とされる。好ましくは、上記糖鎖リガンド複合体は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えたリンカー化合物および糖鎖からなり、上記リンカー化合物の主鎖は、その一端に糖鎖と結合したアミノ基を有し、その他端に硫黄原子を含む炭化水素構造を備えている。
【0039】
本発明に利用されるリガンド複合体として好適なものは、WO2005/077965に示されている化合物が挙げられる。具体的には、一般式(1)
【0040】
【化1】

【0041】
(式中、p,qはそれぞれ独立して0以上6以下の整数)にて表される構造を備え、上記Xとして、末端に芳香族アミノ基を有するとともに、主鎖に炭素−窒素結合を有していてもよい炭化水素誘導鎖を、1鎖又は2鎖又は3鎖含んでなる構造を備え、上記Yとして、硫黄原子又は硫黄原子を含む炭化水素構造を備え、上記Zとして、炭素−炭素結合又は炭素−酸素結合を持つ直鎖構造を備えているリンカー化合物と、還元末端を有する糖とが、上記芳香族アミノ基を介して結合している構造を有していることが好ましく、上記Xは、一般式(2)、または一般式(3)、または一般式(4)
【0042】
【化2】
【0043】
【化3】
【0044】
【化4】
【0045】
(式中、m1〜m5はそれぞれ独立して0以上6以下の整数、R’は水素(H)またはR)にて表される構造を備え、上記Rは糖鎖由来化合物であることがより好ましく、上記Zは、式(5)または式(6)
【0046】
【化5】
【0047】
【化6】
【0048】
(式中、n1,n2はそれぞれ1以上6以下の整数)であってもよい。より好ましくは、本発明に利用可能なリガンド複合体は、一般式(7)、または、一般式(8)、または、一般式(9)、または、一般式(10)、または、一般式(11)
【0049】
【化7】
【0050】
【化8】
【0051】
【化9】
【0052】
【化10】
【0053】
【化11】
【0054】
にて表される構造を有するリンカー化合物の芳香族アミノ基に、還元末端を有する糖が導入された構造を有するものが挙げられる。このようなリガンド複合体は、例えば、一般式(7)〜(11)にて表される構造を有するリンカー化合物と、還元末端を有する糖とを用いて還元アミノ化反応を行うことによって製造することができ、
【0055】
【化12】
【0056】
【化13】
【0057】
【化14】
【0058】
【化15】
【0059】
【化16】
【0060】
(式中、m1,m2,m3,m4,m5はそれぞれ独立して0以上6以下の整数、n1,n2はそれぞれ独立して1以上6以下の整数、qは0以上6以下の整数、Rは還元末端を有する糖、R’は水素(H)またはR)にて表される構造を有し得る。
【0061】
また、本発明に利用可能なリガンド複合体は、上記一般式(7)、(8)、(9)または(10)にて表されるリンカー化合物は、例えば、チオクト酸と、芳香族アミノ基末端が保護基によって保護されたアミン化合物との縮合反応を行い、上記芳香族アミノ基末端の保護基を脱保護することによって製造される。また、上記一般式(11)にて表されるリンカー化合物は、例えば、γ−メルカプト酪酸の2量体と、2分子の芳香族アミノ基末端が保護基によって保護されたアミン化合物との縮合反応を行い、上記芳香族アミノ基末端の保護基を脱保護することによって製造される。
【0062】
上記チオクト酸は、
【0063】
【化17】
【0064】
にて表される構造を備えており、上記アミン化合物は、保護基によって保護された芳香族アミノ基末端を有するものであれば特に限定されるものではない。
【0065】
本発明の一実施形態において、上記一般式(9)にて表される構造においてn1およびqが0であるリンカー化合物
【0066】
【化18】
【0067】
に、還元末端を有する糖が導入されたリガンド複合体が用いられる。このようなリガンド複合体の合成手順もまたWO2005/077965に開示されているが、例えば、リガンド複合体(化合物30)の合成の手順を、式(35)にしたがって簡単に説明すると以下のとおりである。
【0068】
【化19】
【0069】
化合物28(2.47mg,8.24μmol)とシアル酸含有三糖(化合物29、5.11mg,7.57μmol)をジメチルアセトアミド/水(1:1,1.0ml)に溶解し、酢酸(100μl)を加え、37℃で10時間放置した。反応液にシアノ水素化ホウ素ナトリウム(1.55mg,24.7μmol)を加え、37℃でさらに72時間放置した。反応液にアセトン3mlを加え、未反応のシアノ化ホウ素ナトリウムをクエンチした後、濃縮した。得られた残渣は、Chromatorex ODSを用いたカラムクロマトグラフィー、HPLC(カラム:DAISO SP−120−5−ODS−BP)、Sephadex G−25を用いたクロマトグラフィーを順に行って精製した。
【0070】
なお、化合物30は白色の固体として得られ、収量は2.31mg(32%)であり、化合物30の1H−NMRスペクトル(600MHz、D2O)測定を行い、またMALDI−TOF−MS測定を行ったところ、m/z 977.5[(M+Na)+]であった。これらによって化合物30の構造を確認することができた(分子質量は954.34)。
【0071】
以下の式(36)〜(51)にて表されるリガンド複合体を同様の手順で合成し、これらのリガンド複合体の1H−NMRスペクトルを測定し、得られたチャートより、リガンド複合体の構造が確認されている。これらもまた、本実施形態において好適に用いられ得る。なお、後述する実施例にて示される、Glcα1−4Glc−monoおよびGalβ1−4Glc−monoは、上記手順に従って合成されている。
【0072】
【化20】
【0073】
【化21】
【0074】
【化22】
【0075】
【化23】
【0076】
【化24】
【0077】
【化25】
【0078】
【化26】
【0079】
【化27】
【0080】
【化28】
【0081】
【化29】
【0082】
【化30】
【0083】
【化31】
【0084】
【化32】
【0085】
【化33】
【0086】
【化34】
【0087】
【化35】
【0088】
このように本発明に利用可能なリンカー化合物は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えており、主鎖の一末端にアミノ基を有している。さらに、上記リンカー化合物は、末端にアミノ基を有していることによって、糖鎖分子を簡便に導入することができる。なお、上記アミノ基は、修飾されているアミノ基(例えばアセチル基、メチル基やホルミル基等で修飾されたアミノ基)や、芳香族アミノ基であってもよいし、未修飾のアミノ基であってもよい。
【0089】
糖鎖リガンド複合体には、硫黄原子(S)が含まれており、この硫黄原子(S)は、例えば、蛍光性ナノ粒子に含まれるカドミウム(Cd)と、金属−硫黄結合(Cd−S結合)を形成し、蛍光性ナノ粒子に結合することができる。
【0090】
本発明に利用可能なリンカー化合物は、金属−硫黄結合(例えばCd−S結合)を容易に形成することができるという点で、S−S結合またはSH基が含まれている炭化水素構造を主鎖の他端に備えていることが好ましい。これによって、上記リンカー化合物は、蛍光性ナノ粒子上に糖鎖分子を集合化して配列することができる。ジスルフィド結合(S−S結合)またはSH基中の硫黄(S)は、例えば、蛍光性ナノ粒子上に存在するカドミウム(Cd)と、金属−硫黄結合(Cd−S結合)を形成し、金属との結合を強固にすることができる。
【0091】
そして、上記糖鎖リガンド複合体には、上記リンカー化合物のアミノ基に、還元末端を有する糖鎖が導入されている。言い換えれば、上記糖鎖リガンド複合体は、上記リンカー化合物と、還元末端を有する糖鎖とが、アミノ基を介して結合している構造を有している。この糖鎖の導入は、例えば、上記リンカー化合物のアミノ基(−NH2基)と糖鎖との還元アミノ化反応によって行うことができる。つまり、平衡によって生じる糖鎖中のアルデヒド基(−CHO基)またはケトン基(−CRO基、Rは炭化水素基)と、上記リンカー化合物が有するアミノ基とが反応する。そして、この反応によって形成されたシッフ塩基を引き続き還元することによって、アミノ基に容易に糖鎖を導入することができる。
【0092】
なお、上記「還元末端を有する糖鎖」とは、アノマー炭素原子が置換を受けていない単糖、オリゴ糖鎖、多糖鎖である。つまり、上記還元末端を有する糖鎖とは、還元糖鎖である。上記還元末端を有する糖鎖としては、市販のものであっても天然のものであってもよく、あるいは、市販および天然の多糖鎖を分解して調製したものを用いることができる。
【0093】
上記還元末端を有する糖鎖としてより具体的には、グルコース、ガラクトース、マンノース、マルトース、イソマルトース、ラクトース、パノース、セロビオース、メリビオース、マンノオリゴ糖鎖、キトオリゴ糖鎖、ラミナリオリゴ糖鎖、グルコサミン、N−アセチルグルコサミン、グルクロン酸、ヘパリン、ヘパラン硫酸、コンドロイチン硫酸、デルマタン硫酸などが挙げられるが、これに限定されることはない。
【0094】
上記リガンド複合体に含まれるリンカー化合物は、金属に結合可能な硫黄原子と、オリゴ糖鎖等の糖鎖分子に結合可能なアミノ基とを有している。従って、例えばCd−S結合などの金属−硫黄結合により上記リガンド複合体が蛍光性ナノ粒子上の金属に固定されるので、上記リンカー化合物を介して、本発明に係る蛍光性ナノ粒子に糖鎖分子を強固にかつ簡単に結合させることができるとともに、蛍光性ナノ粒子をコロイド状態で安定化することができる。また、上記リガンド複合体の固定化は、還元剤処理した上記リガンド複合体と蛍光性ナノ粒子を含む溶液を混和するだけで行うことができるので、非常に容易に糖鎖を固定化することができる。
【0095】
また、上記リガンド複合体は、タンパク質との非特異的な相互作用の影響をほとんど無視することができる。それゆえ、上記リンカー化合物を有する上記リガンド複合体を用いることによって、上記糖鎖とタンパク質との相互作用を再現性よく評価することが可能になる。
【0096】
上記のリガンド複合体は、リンカー化合物と糖鎖分子とを含んでなっているので、リンカー化合物内のS−S結合にて、金属−硫黄(S)結合、例えばカドミウム−硫黄(Cd−S)結合によって、任意の金属と結合することができる。これにより、このCd−S結合を介して、蛍光性ナノ粒子上に糖鎖分子が固定化されてなる糖鎖固定化蛍光性ナノ粒子を提供することができる。上記任意の金属としては、上記リガンド複合体と結合可能なものであればよく、上記カドミウムの他、亜鉛、銅、銀、インジウム等の金属を用いることができるが、特にカドミウム、亜鉛を用いることが好ましい。
【0097】
なお、上記リガンド複合体に導入されている糖鎖は、還元末端を有するものであれば単糖でもよいし、同一の単糖分子からなる単一オリゴ糖鎖または多糖鎖であってもよいし、種々の単糖分子やその誘導体からなる複合糖鎖・多糖鎖であってもよい。また、上記糖鎖は、いずれも、自然界から単離/精製して得られる種々の天然の糖鎖であってもよく、人工的に合成された糖鎖であってもよい。また、上記糖鎖は、多糖鎖を分解して得られたものであってもよい。
【0098】
本発明に係る糖鎖固定化蛍光性ナノ粒子は、加熱処理した親水性の蛍光性ナノ粒子を含む溶液と、還元剤処理した上記リガンド複合体との混和によって得ることができ、リガンド複合体のS−S結合の各S原子が、ナノ粒子上の金属と金属−硫黄結合によって結合する。具体的には、例えば、蛍光性ナノ粒子の溶液に、還元剤処理した上記リガンド複合体を含む溶液を添加することによって、上記リガンド複合体のS−S結合を、ナノ粒子上の金属−硫黄結合に変換して、糖鎖固定化蛍光性ナノ粒子を得ることができる。
【0099】
なお、親水性の蛍光性ナノ粒子の加熱処理の条件は、特に限定されるものではないが、チオール安定化剤の存在下にて、50〜200℃で行われることが好ましく、70〜180℃がより好ましく、100℃以上であることがさらに好ましい。チオール安定化剤としては、特に限定されないが、チオアセトアミド、3−メルカプトプロピオン酸(3−MPA)、チオグリコール酸(TGA)、4−メルカプトブタン酸、システイン、シスタミンなどのチオ化合物および塩類が挙げられる。
【0100】
還元剤処理に用いられる還元剤としては、特に限定されるものではないが、例えば、水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウムなどの塩類および陽イオン成分が異なる塩類等を挙げることができる。
【0101】
蛍光性ナノ粒子およびリガンド複合体を含む溶液に用いる溶媒としては、特に限定されるものではないが、例えば、水、メタノール、エタノール、プロパノール、これらの混合溶媒等を挙げることができる。また、上記混和によって得られた糖鎖固定化蛍光性ナノ粒子を遠心濾過し、低分子の塩などの成分を除くことによって、溶液状態で安定な糖鎖固定化蛍光性ナノ粒子を得ることができる。
【0102】
糖鎖固定化蛍光性ナノ粒子を調製するために用いる金属ナノ粒子、上記リガンド複合体、還元剤の混合比は、特に限定されるものではないが、金属成分としてカドミウムが含まれる場合は、溶液中のカドミウム濃度が、最終濃度で0.1mM〜1mMであることが好ましい。
【0103】
上記リガンド複合体の濃度は、溶液中の最終濃度として0.1mM〜10mMであることが好ましい。
【0104】
また、用いられる還元剤の濃度は、溶液中の最終濃度としてリガンド複合体濃度の10倍モル濃度であることが好ましい。
【0105】
このように、好ましい実施形態において、本発明の糖鎖固定化蛍光性ナノ粒子の製造方法は、加熱処理された蛍光性ナノ粒子と、還元剤処理された糖鎖リガンド複合体を混和する工程を包含し、上記蛍光性ナノ粒子は、第一および第二の金属成分からなる粒子コアが第一および第三の金属成分からなるシェル層によって被覆されており、上記糖鎖リガンド複合体は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えたリンカー化合物および糖鎖からなり、上記リンカー化合物の主鎖は、その一端に糖鎖と結合したアミノ基を有しており、その他端に硫黄原子を含む炭化水素構造を備えており、還元剤処理された上記炭化水素構造が上記層に固定化されて、糖鎖固定化蛍光性ナノ粒子の分散液が得られる。
【0106】
本実施形態の糖鎖固定化蛍光性ナノ粒子の構成要素である蛍光性ナノ粒子は、第一および第二の金属成分からなる粒子(コア)が第一および第三の金属成分からなる層(シェル)によって被覆されている「コア/シェル」構造を有していることが好ましい。蛍光性ナノ粒子が加熱処理されていることによって、シェル層の表面が均質化されて、上記糖鎖リガンド複合体を効率よく結合させることができ、その結果、蛍光性ナノ粒子上に高い安定性を付与し得る。
【0107】
第一の金属成分は、Cd、Zn、Ag、InおよびSからなる群より選択されることが好ましく、第二の金属成分が、TeおよびSからなる群より選択されることが好ましい。また、第三の金属成分は、Cd、SおよびZnからなる群より選択されることが好ましく、後述するチオール安定化剤として提供されてもよい。本発明に使用可能な「コア/シェル」構造の蛍光性ナノ粒子としては、CdTe/CdS、ZAIS/ZnSが挙げられるがこれらに限定されない。
【0108】
1つの局面において、本実施形態の製造方法は、第一の金属成分を含む第一溶液と、第二の金属成分を含む第二溶液とを加熱条件下にて混合し、第一溶液と第二溶液との混合溶液を室温に冷却し、第一および第二の金属成分からなる粒子を混合溶液から精製することによって行われ、第一工程において混合された溶液が第三の金属成分をさらに含んでいることによって、第一および第二の金属成分からなる粒子が、第一および第三の金属成分からなる層によって被覆される。
【0109】
別の局面において、本実施形態の製造方法は、第一の金属成分を含む第一溶液と、第二の金属成分を含む第二溶液とを加熱条件下にて混合し、第一溶液と第二溶液との混合溶液を室温に冷却し、第一および第二の金属成分からなる粒子を混合溶液から精製し、精製した粒子および第三の金属成分を含む水溶液を加熱処理することによって、第一および第二の金属成分からなる粒子が、第一および第三の金属成分からなる層によって被覆される。
【0110】
粒子コアを精製した後に加熱処理を行うことによって、シェル層の表面の均質化がより一層改善されて、上記糖鎖リガンド複合体の結合が著しく向上する。この場合、精製の前に行われる加熱処理は、30分間〜8時間にわたって行われることが好ましい。これにより、粒径が均一でありかつ高い水分散性を有する糖鎖固定化蛍光性ナノ粒子が得られる。
【0111】
なお、本明細書において、「室温」は通常の実験室内での温度であり、20〜30℃が好ましいが、加熱処理における反応を停止することができる温度であれば特に限定されず、いわゆる低温室(例えば4℃)であってもよい。
【0112】
本実施形態において、第一溶液は、第一の金属成分の塩または錯塩が溶解した溶液であり、第二溶液は、第二の金属成分の塩または錯塩が溶解した溶液であっても、親水化された第二の金属成分が溶解した溶液であってもよい。
【0113】
このような製造方法によって製造された糖鎖固定化蛍光性ナノ粒子は、その表面上に抗体を容易に固定化し得るので、生体組織に対する特異性が向上した抗体固定化蛍光性ナノ粒子を容易に提供し得る。
【0114】
本明細書において、「生体組織」は、生物体外に取り出された組織であっても、生物体内の組織であってもよく、臓器または器官であっても、これらに由来する単離された細胞であっても、培養細胞であってもよく、細胞を構成するタンパク質であってもよい。例えば、タンパク質−タンパク質の相互作用を検出するために用いられても、細胞を検出可能に標識するために用いられてもよく、生体内での物質動態を画像化するために用いられてもよい。
【0115】
このように、本発明の糖鎖固定化蛍光性ナノ粒子および抗体固定化蛍光性ナノ粒子を用いれば、生体組織の標識を容易に行うことができる。すなわち、本発明の糖鎖固定化蛍光性ナノ粒子を含む組成物は、生体組織を標識するための組成物であり得る。
【0116】
〔2.糖鎖−タンパク質相互作用の測定方法〕
本発明に係る糖鎖−タンパク質相互作用の測定方法は、糖鎖固定化蛍光性ナノ粒子を含む溶液と、上記糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖を認識するタンパク質とを混和することによって、糖鎖とタンパク質とを相互作用させ、糖鎖−タンパク質複合体を生成させる工程を含むものである。
【0117】
上記「糖鎖固定化蛍光性ナノ粒子を含む溶液」とは、本発明に係る糖鎖固定化蛍光性ナノ粒子が液体に分散したコロイド溶液が意図される。糖鎖固定化蛍光性ナノ粒子を含んでいれば、他に塩などが含まれていてもよい。上記液体としては、例えば水や緩衝液等を用いることができる。
【0118】
上記タンパク質としては、特に限定されるものではなく、上記糖鎖固定化蛍光性ナノ粒子の外側に位置する糖鎖を認識することができるものであればよい。例えば、糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖がグルコースの場合は、グルコースを認識することができるタンパク質であるコンカナバリンA(ConA)、レンチルレクチン(LCA)、ピーナッツレクチン(PSA)等を用いることができる。
【0119】
同様に、糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖がガラクトースである場合は、ガラクトースを認識するタンパク質であるヒママメレクチン(RCA120)等を用いることができる。また、糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖がN−アセチルグルコサミンである場合は、N−アセチルグルコサミンを認識するタンパク質である小麦胚芽レクチン等を用いることができる。
【0120】
糖鎖固定化蛍光性ナノ粒子を含む溶液と、上記糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖を認識するタンパク質とを混和する方法は、特に限定されるものではなく、糖鎖とタンパク質とを近接させて相互作用が可能な状況を提供し得るものであればよい。例えば、マイクロプレートやエッペンドルフチューブなどにタンパク質の希釈系列を作製し、糖鎖固定化蛍光性ナノ粒子を含む溶液を添加して放置することにより混和を行うことができる。
【0121】
糖鎖とタンパク質との相互作用(糖鎖−タンパク質相互作用)としては、水素結合、イオン結合、静電気的相互作用、ファンデルワールス力等を挙げることができる。すなわち、上記糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖をタンパク質が認識し、その結果、水素結合等の糖鎖−タンパク質相互作用が生じる。
【0122】
なお、上記「認識」とは、タンパク質が分子内に有する糖鎖結合部位(糖鎖認識部位)によって糖鎖に結合することをいう。
【0123】
上記「糖鎖−タンパク質複合体」とは、糖鎖とタンパク質とが相互作用して特異的に結合し、その結果生成される凝集物を意味する。本発明に係る糖鎖−タンパク質相互作用の測定方法は、糖鎖−タンパク質相互作用を凝集物の生成として目視で確認することができる。また、その際の溶液の蛍光色が変化する。糖鎖とタンパク質とが相互作用しない場合、またはタンパク質の糖鎖結合部位がタンパク質あたり1つしかない場合(単価タンパク質)には、凝集物は形成されない。
【0124】
ここで、凝集反応を確認することによって物質間の相互作用を測定する方法としては、例えば抗原抗体反応を用いたラテックス凝集法等を挙げることができる(「バイオ診断薬の開発・評価と企業」、CMCテクニカルライブラリー146、シーエムシー出版、P92−97,P109−113)。上記ラテックス凝集法は、ラテックス表面に抗体を固定化させておき、96穴のマイクロプレートを用いて試料抗原の希釈系列を作り、凝集を生じる最大希釈倍率を求め標準溶液と比較して測定するという方法である。結果は一定波長の光で吸光度として測定される。
【0125】
コロイドについて凝集反応を確認することによって物質間の相互作用を測定する方法としては、上記蛍光性ナノ粒子の呈する赤褐色をもって結果を判断するという手法が考えられる。したがって、コロイドにおいても糖鎖とタンパク質とを相互作用させて凝集物を生成させ、当該凝集物の生成を確認することによって結果を判定することができる。本発明に係る方法は、従来の方法よりも非常に簡便であり、蛍光を使用することから感度の高い、有用な方法であるということができる。
【0126】
また、本発明に係る方法を用いれば、未標識の状態で糖鎖−タンパク質相互作用を測定することができるので、標識を要する方法のような前処理を必要としない点で簡便である。さらに、標識効果が測定のばらつきに大きく影響するという問題も存在せず、再現性のよい測定を行うことができる。また、糖鎖−タンパク質相互作用を目視で確認することができるので、特別な装置も必要なく、非常に安価かつ容易に糖鎖−タンパク質相互作用を測定することができる。すなわち、本発明の糖鎖固定化蛍光性ナノ粒子は、相互作用検出に利用可能であり、そのような用途に利用される組成物を構成し得る。
【0127】
したがって、本発明に係る方法は、糖鎖やタンパク質の機能解析や、臨床的な検査および診断などに用いることが可能である。すなわち、本発明の糖鎖固定化蛍光性ナノ粒子は、臨床的な検査に用いられる生体イメージング、近赤外増感などに利用可能であり、そのような用途に利用される組成物を構成し得る。
【0128】
なお、本発明に係る糖鎖−タンパク質相互作用の測定方法は、糖鎖固定化蛍光性ナノ粒子を含む溶液と、上記糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖を認識するタンパク質とを混和することによって、糖鎖とタンパク質とを相互作用させ、糖鎖−タンパク質複合体を生成させる工程を含んでいればよい。したがって、例えば、上述したように、糖鎖−タンパク質複合体(凝集体)の生成を目視で確認するだけでもよいし、より詳細な測定を行う場合は、一定波長の紫外可視吸光スペクトルを測定する工程を含んでいてもよい。また、一定波長の光で励起した後に蛍光スペクトルを測定する工程を含んでいてもよい。
【0129】
〔3.細胞の糖鎖結合性を利用して細胞を蛍光標識する方法〕
細胞は多量の糖鎖成分を含んでいる。細胞に存在するタンパク質は、細胞の内外に存在する糖鎖を認識し、細胞間の認識やシグナル伝達などの機能の調節を行っている。言い換えると、細胞の糖鎖結合性を利用し、糖鎖固定化蛍光性ナノ粒子と細胞を結合させ、蛍光標識することができる。
【0130】
蛍光標識した細胞は、フローサイトメトリーおよび蛍光顕微鏡等の機器を用いて解析する。すなわち、本発明の糖鎖固定化蛍光性ナノ粒子は、細胞標識、ウイルス標識、細胞イメージング、生体イメージング、近赤外増感などに利用可能であり、そのような用途に利用される組成物を構成し得る。
【0131】
本発明の製造方法で細胞が特異的に認識する糖鎖を固定化した糖鎖固定化蛍光性ナノ粒子を製造したのち、糖鎖固定化蛍光性ナノ粒子と細胞含有溶液と混合させるだけで、細胞の表面のレセプターを介して結合し、糖鎖固定化蛍光性ナノ粒子による標識が行われる。
【0132】
〔4.抗体固定化蛍光性ナノ粒子の製造法〕
本発明は、抗体を固定化した蛍光性ナノ粒子(すなわち、抗体固定化蛍光性ナノ粒子)を製造する方法を提供する。本発明の方法によって製造された蛍光性ナノ粒子は水溶液中で分散する特徴を有している。すなわち、本発明の方法は、抗体固定化蛍光性ナノ粒子のコロイド溶液を調製する方法でもある。
【0133】
本発明の方法によって製造される抗体固定化蛍光性ナノ粒子の製造に用いられるリガンド複合体は、上述した一般式(1)にて表される構造を備える化合物であっても、例えば、下記式(I)または(II)
【0134】
【化36】
【0135】
【化37】
【0136】
(式中、n1,n2はそれぞれ独立して1以上6以下の整数、qは0以上6以下の整数)にて表される構造を備える化合物であってもよい。本発明の抗体固定化蛍光性ナノ粒子を製造する際に、上記式(I)および(II)の構造を備える化合物をリガンド複合体として用いる場合もまた、上述した糖鎖固定化蛍光性ナノ粒子を製造する方法に準じて行えばよいことを、本明細書を読んだ当業者は容易に理解する。
【0137】
なお、本発明は以上説示した各構成に限定されるものではなく、特許請求の範囲に示した範囲内で種々の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0138】
本発明について、実施例に基づいてより具体的に説明するが、本発明はこれに限定されるものではない。
【0139】
〔実施例1:α−グルコース固定化蛍光性ナノ粒子の調製〕
非特許文献5および6に記載の方法を改変して、塩化カドミウムを含む水溶液に3−MPAを添加し、次いでNaHTeの溶液を加え、混合溶液を1時間還流した後にチオアセトアミドを加えることによって、金属ナノ粒子を含む溶液を調製した。
【0140】
5mMのカドミウムを含む金属ナノ粒子を含む溶液1mLを分注し、マイクロコン(Millipore、Microcon、10000NMWL)を用いて遠心濾過(3500rpm、5分間)し、続いて、ナノ粒子を500μLの水に懸濁した。別途で、250μLの、α−グルコースが結合した糖鎖リガンド複合体(Glcα1−4Glc−mono;式(46)にて表される化合物)の水溶液(10mM)、および250μLの、水素化ホウ素ナトリウム水溶液(100mM)を混合した。その後、この混合溶液500μLとナノ粒子500μLを遮光下にて混和および撹拌して糖鎖固定化蛍光性ナノ粒子の粗コロイド溶液を調製した。図1に、調製したナノ粒子の透過型電子顕微鏡画像を示す。
【0141】
次に、上記粗コロイド溶液を再び遠心濾過によって精製し、α−グルコースが固定化した蛍光性ナノ粒子のコロイド溶液を得た。
【0142】
〔実施例2:糖鎖−タンパク質相互作用の測定〕
ConAまたはBSAのPBS溶液(各2mg/mL)を調製し、96ウェルプレートに1レーンずつConAおよびBSAの溶液を2倍段階希釈して50μLずつ添加した(図2、右側から1〜11:左が高濃度で右が低濃度、最左:ブランク)。すべてのウェルに実施例1で調製したα−グルコースを固定化した蛍光性ナノ粒子のコロイド溶液を50μL添加して混合した。約2時間放置した後、目視による相互作用の確認を行った。
【0143】
図2の写真は、目視下での糖鎖−タンパク質相互作用の解析の結果を示す。結合性タンパク質が添加されているレーンにおいて特異的な凝集反応が見られた。
【0144】
〔実施例3:糖鎖結合性を利用して細胞を蛍光標識する方法〕
5mMのカドミウムを含む金属ナノ粒子を含む溶液1mLを分注し、マイクロコン(Millipore、Microcon、10000NMWL)を用いて遠心濾過(3500rpm、5分間)し、続いて、ナノ粒子を500μLの水に懸濁した。別途で、250μLの、β−ガラクトースが結合した糖鎖リガンド複合体(Galβ1−4Glc−mono;式(39)にて表される化合物)の水溶液(10mM)、および250μLの、水素化ホウ素ナトリウム水溶液(100mM)を混合した。その後、この混合溶液500μLとナノ粒子500μLを遮光下にて混和および撹拌して糖鎖固定化蛍光性ナノ粒子の粗コロイド溶液を調製した。
【0145】
次に、上記粗コロイド溶液を再び遠心濾過によって精製し、β−ガラクトース固定化した蛍光性ナノ粒子のコロイド溶液を得た。
【0146】
引き続いて、調製した蛍光性ナノ粒子の細胞結合性をフローサイトメトリー(FACS)を用いて解析した。細胞にはマウスのマクロファージ系の細胞であるJ774を用いた。細胞数を1×106個/mLに調整した後、調製したナノ粒子を加え、1時間インキュベートした。その後、遠心分離、および細胞の洗浄を行い、細胞をホルマリンで固定化した。細胞を再び洗浄した後、FACS解析を行った。
【0147】
図3は解析結果のグラフを示す。横軸は蛍光強度、縦軸は細胞数を示す。また黒色線は細胞のみ(コントロール)を、黒色破線はα−グルコースを固定化した蛍光性ナノ粒子を加えたもの、灰色線はβ−ガラクトースを固定化した蛍光性ナノ粒子を加えたものをそれぞれ示す。細胞に対して蛍光性ナノ粒子が結合した場合、ピークはコントロールよりも右側にシフトする。測定の結果、α−グルコースまたはβ−ガラクトースを固定化した蛍光性ナノ粒子のどちらも、右側にピークがシフトしている。このことから、これらナノ粒子による細胞標識が可能であることが分かった。
【0148】
〔実施例4:蛍光性ナノ粒子の調製1〕
非特許文献5および6に記載の方法は、いわゆるワンポットでの合成法であり、この方法に従えば、CdTeをコアとしかつCdSをシェルとする構造の蛍光性ナノ粒子(CdTe/CdS)が得られ、この構造に基づいて安定性や蛍光強度が高くなることが報告されている。
【0149】
しかし、このような方法では、得られた蛍光性ナノ粒子の蛍光スペクトル幅が広く、固定化する糖鎖に応じて蛍光波長(すなわち、蛍光の色)を変えることは困難であることがわかった。
【0150】
本発明者らによる創意工夫の結果、上記問題点を解消した方法を完成するに至った。このような蛍光性ナノ粒子の調製方法は以下の通りであり、別々に調製した2種類の溶液を混合し、反応によって形成した粒子を一旦精製し、再度反応させる、ツーポットでの合成法である。
<溶液1の調製>:Te(粉末、0.125mmol(シグマ・アルドリッチより購入)および水素化ホウ素ナトリウム(0.0500mmol)を、アルゴン雰囲気下にてアルゴンガスをバブリングした水(2mL)に溶解し90分間撹拌してテルル化水素ナトリウム水溶液を調製した。
<溶液2の調製>:塩化カドミウム(0.0500mmol)とチオグリコール酸(TGA;0.0265mmol)を、アルゴン雰囲気下にて、アルゴンガスをバブリングした水(10mL)に溶解し、得られた溶液を水酸化ナトリウム水溶液(1M)でpHを9に調整した後、アルゴンガスでさらにバブリングした。
<ナノ粒子の調製>:溶液2を105℃に加熱し、空気雰囲気下にて、激しく撹拌しながら溶液1を加えた。混合溶液を遮光下にて1時間撹拌した後に室温に戻して反応を停止させた。その後、混合溶液に2−プロパノール(蛍光性ナノ粒子溶液量に対して3倍量)を加え、遠心分離(9500rpm、5分間)を行った。上清を除去した後、沈殿を2−プロパノールに懸濁し、さらに遠心分離を行った後に、上清を除去した。この操作を再度行ってナノ粒子を洗浄した後、沈殿を水(10mL)に溶解した。得られた溶液を4℃で10時間保存した後、チオアセトアミド(0.27μL)を加え、105℃で再加熱を行った。遮光下にて適度な時間にわたって加熱撹拌を行った後、室温に冷却して反応を停止させた。蛍光性ナノ粒子濃度は吸光度から見積り7μMであることがわかった。
【0151】
本発明における、ツーポットでの合成法で得られた蛍光性ナノ粒子と、従来法のワンポット得られた蛍光性ナノ粒子の半値幅を比較したグラフ(図4(a))、および蛍光波長(図4(b))を示す。半値幅(半値全幅)とは最大蛍光強度の1/2の値を示す波長の幅である。示されるように、ワンポット合成法よりもツーポット合成法を用いる方が、蛍光性ナノ粒子の半値幅が狭い。また、チオール安定化剤として従来用いられる3−MPAよりもTGAを用いる方が、蛍光性ナノ粒子の半値幅が狭いことがわかった。このように、表面配位子としてTGAを用い、CdTeコアへのCdSシェルの付加にツーポット合成法を用いることによって、スペクトル幅が狭く、かつ多色同時蛍光発光が可能な蛍光性ナノ粒子を得られることがわかった。
【0152】
〔実施例5:糖鎖固定化蛍光性ナノ粒子の調製1〕
上述したツーポット合成法によって得られた蛍光性ナノ粒子(CdTe/CdS)を用いて、糖鎖固定化蛍光性ナノ粒子(SFNP)を製造した。
【0153】
α−グルコースが結合した糖鎖リガンド複合体(Glcα1−4Glc−mono)(125μL、10mM)と水素化ホウ素ナトリウム(125μL、100mM)を混合し、2時間静置して、糖鎖リガンド複合体溶液(複合体の最終濃度2.5mM)を調製した。蛍光性ナノ粒子溶液(500μL、7μM)を、マイクロコン(Millipore、Microcon、10000NMWL)を用いて遠心濾過(3500rpm、5分間)した後、濾過物を再度250μLの水に懸濁した。この蛍光性ナノ粒子の懸濁液と上記糖鎖リガンド複合体溶液(250μL)を混合し、遮光下にて24時間反応させた。過剰の糖鎖リガンド溶液を遠心濾過によって除去し、沈査をPBSに分散させることによって水中での分散性が良好なSFNP(αGlc−FNP)を得た。
【0154】
糖鎖の固定化について、上記αGlc−FNPを、MALDI−TOF/MSによって糖鎖リガンド複合体の分子質量に相当するピークが測定されたことから確認した(図5(a))。また、透過型電子顕微鏡によって、αGlc−FNPの粒径を測定したところ、粒径が約5nmであり、比較的均一度の高い粒子であることがわかった(図5(b))。さらに、アントロン−硫酸法によって糖の定量を行った。換算すると、最終濃度2.5mMの糖鎖リガンド複合体を蛍光性ナノ粒子と反応させた際に、ナノ粒子1つに約130個の糖鎖が固定化されていることがわかった。
【0155】
〔実施例6:糖鎖固定化蛍光性ナノ粒子のタンパク質との相互作用1〕
上述したように調製したSFNP(2.5mM)と、糖鎖と特異的に結合するタンパク質との相互作用を調べた。5μMのタンパク質溶液を、96ウェルのマイクロプレートにそれぞれ100μL加え、次いで各ウェルにα−グルコースを固定化したαGlc−FNPを100μL加えた。プレートを遮光下にて18時間放置した後、各ウェルにおける上清の蛍光スペクトルを測定した。タンパク質には、コンカナバリンA(ConA)、ヒママメレクチン(RCA120)、ウシ血清アルブミン(BSA)を用いた。α−グルコースと特異的に結合することが知られているConAを加えた時にのみ、蛍光強度の減少が観察された(図6)。UV−VISよりも感度が高い蛍光において変化が観測されたことは、用いる糖鎖の量が微量であっても、タンパク質との特異的結合を測定することができることを示し、本発明の方法によって得られたSFNPが従来の方法によって得られる糖鎖固定化金ナノ粒子よりもかなり優れた性質を有していることがわかった。
【0156】
〔実施例7:糖鎖固定化蛍光性ナノ粒子の結合特異性1〕
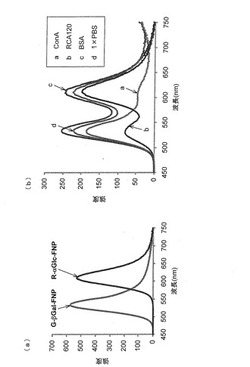
実施例4の手順に従ってβ−ガラクトースが結合した糖鎖リガンド複合体(Galβ1−4Glc−mono)調製し、これを用いて、実施例5の手順に従って水中での分散性が良好なSFNP(βGal−FNP)を得た。2種類のSFNP(αGlc−FNPおよびβGal−FNP)を用いて、タンパク質との選択的結合活性を調べた。
【0157】
ConAと特異的に結合するα−グルコースを固定化したαGlc−FNPは、橙色の蛍光(Ex=620nm)を示す(図7(a)中、R−αGlc−FNPと示す。)。と特異的に結合するβ−ガラクトースを固定化したβGal−FNPは、橙色の蛍光(Ex=530nm)を示す(図7(a)中、G−βGal−FNPと示す。)。
【0158】
これらのFSNPを1μMに希釈し、各50μLずつを混合した。混合溶液を入れたチューブに5μMのタンパク溶液(ConA、RCA120またはBSA)をそれぞれ100μL加え、撹拌した後に一晩放置した。ConAを加えたチューブには、ConAとαGlc−FNPとの相互作用による橙色の蛍光を示す凝集体が生成し、上清は、ConAと相互作用しないβGal−FNPが残ることによって緑色になった(図7(b))。一方、RCA120を加えたチューブには、RCA120とβGal−FNPとの相互作用による緑の蛍光を示す凝集体が生じ、上清は、RCA120と相互作用しない結合しないα−グルコースを固定化したαGlc−FNPが残ることによって橙色になった(図7(b))。なお、BSAを加えたチューブでは、凝集は生成せず、チューブ内の溶液は2つのSFNPの混合溶液の色であった(図7(b))。このように、複数のタンパク質との相互作用を目視にて観察することができた。
【0159】
〔実施例8:糖鎖固定化蛍光性ナノ粒子による細胞認識1〕
SFNPの細胞結合活性をフローサイトメトリー(FACS)により解析した。SFNPとして、αGlc−FNPおよびβGal−FNPを用い、コントロールとしてテトラエチレングリコール(TEG)を固定化したナノ粒子(TEG−FNP)を用いた。細胞には、肝癌由来の細胞であるHepG2細胞を用いた。
【0160】
細胞を1×106個/mLに希釈した後、SFNPまたはTEG−FNPを加えて12時間培養した。SFNPまたはTEG−FNPを含む培地を除去した後、PBSで2回洗浄し、セルスクレイパーで細胞をプレートから剥がし、細胞を含むPBS溶液を調製した。この溶液を、FACS解析に供した。
【0161】
SFNPでは、右側への大きなピークシフトが観察された(図8)。特に、βGal−FNPにおいてピークシフトは顕著であった。HepG2細胞は、β−ガラクトース親和性があることが知られており、この結果は、HepG2細胞のβ−ガラクトース親和性が明確に示されたことになる。また、αGlc−FNPにおいてもピークシフトが観察された。このことは、HepG2細胞が、β−ガラクトースほどではないがα−グルコースに対しても高い親和性があることを示す。コントロールとして用いたTEG−FNPでは、右側へのピークシフトがほとんど見られなかった。これは、TEG−FNPに糖鎖が結合していないことに起因するといえる。
【0162】
さらに、プレートから剥がす前の細胞を蛍光顕微鏡(HSオールインワン蛍光顕微鏡BZ−9000、BIOREVO, KEYENCE)で観察した。コントロールとして用いたTEG−FNPでは、細胞にて蛍光がほとんど見られなかったが、αGlc−FNPおよびβGal−FNPでは、細胞にて蛍光が観察された(図9)。
【0163】
〔実施例9:蛍光性ナノ粒子の調製2〕
αGlc−FNPおよびβGal−FNPは、蛍光性ナノ粒子(CdTe/CdS)を用いて製造されたものであり、毒性のCdを含むものである。毒性を低減させることができれば、応用範囲をより広げることができる。そこで、特許文献2に記載の手順を改変して、Cdよりも毒性が低いZAIS(ZnS−AgInS2)を合成し、ZAISの親水化と糖鎖の固定化を行いSFNPを調製した。
<溶液1’>:N,N−ジエチルカルバミド酸ナトリウム(2.5mmol)を水(50mL)に溶解した。
<溶液2’>:AgNO3(11.25mM)、In(NO3)3・3H2O(11.25mM)、Zn(NO3)2・6H2O(2.5mM)になるように水(50mL)に溶解した。
<ナノ粒子の調製>:遮光下にて室温で撹拌した溶液1’に、溶液2’を穏やかに加えた。5分間攪拌した後に、遠心分離(3000rpm、10分間)を行った。沈殿に超純水を加えて再度遠心分離(3000rpm、5分間)を行い、上清を廃棄して沈殿を回収した。この操作を4回行った。次に、沈殿にメタノールを加えて遠心分離(3000rpm、5分間)を行い、上清を廃棄して沈殿を回収した。この操作を2回行い、得られた沈殿を減圧下にて乾燥させることによってN,N−ジエチルカルバミド酸錯体を得た。得られた錯体(50mg)を試験管に移し、アルゴン置換した後に、オイルバスにて180℃で30分間加熱攪拌した。放冷後に、アルゴン雰囲気下にて、オレイルアミン(3mL)を加え、オイルバスにて105℃で5分間加熱撹拌した。遠心分離(3500rpm、10分間)した後、上清をメンブレン(0.45μm、PTFE)で濾過し、得られた濾液にメタノール(3mL)を加え、沈殿を生じさせた。濾液全体の遠心分離(3500rpm、10分間)を行った後に、上清を廃棄して沈殿を回収した。得られた沈殿にオレイルアミン(2mL)を加えて溶解させ、メンブレン濾過した後に、濾液にメタノール(2mL)を加えて再度沈殿を生じさせた。濾液全体の遠心分離(3500rpm、10分間)を行った後に、上清を廃棄して沈殿を回収した。得られた沈殿に再度オレイルアミン2mLを加え、アルゴン置換した後に、オイルバスにて180℃で30分間加熱攪拌した。放冷後に、メタノール(2mL)を加えて沈殿を生じさせた。さらに、遠心分離(3500rpm、10分間)を行った後に、上清を廃棄して沈殿を回収した。得られた沈殿をクロロホルム(2mL)に再度溶解してZAIS溶液を得た。
【0164】
ZAISの製造の際に、各試薬の濃度を図10(a)のように変更することによって、組成比が異なりかつ蛍光スペクトルが異なるナノ粒子を製造することができた(図10(b)および(c))。以下の実験には、組成比0.4の用いた例を説明する。
【0165】
ZAISのオレイルアミン溶液(2mL)に酢酸亜鉛無水和物(0.562μmol)、チオアセトアミド(0.562μmol)を加え、アルゴン置換した後に、遮光下にて、オイルバスにて180℃で30分間加熱撹拌した。放冷後に、メタノールを加えて遠心分離(3500 rpm、5分間)を行い、上清を廃棄して沈殿を回収した。得られた沈殿をクロロホルム(1mL)に溶解して、ZAISをコアとしかつZnSをシェルとする構造の蛍光性ナノ粒子(ZAIS/ZnS)のクロロホルム溶液を得た。
【0166】
ZAIS/ZnSのクロロホルム溶液(1mL)にクロロホルム(1mL)を加え、0℃に冷却し、攪拌下、3−MPAのエタノール溶液(0.2M、1mL)を加えた。続いて、水酸化カリウムのエタノール溶液(0.3M、1mL)を加え、遮光下にて12時間攪拌した。遠心分離(3500rpm、5分間)を行い、上清を廃棄して沈殿を回収した。得られた沈殿を水(1mL)に溶解し、遠心分離(3500rpm、5分間)を行い、得られた上清をメンブラン濾過(0.45μm)を行って、3−MPA被覆された親水性ZAIS/ZnS(水溶液)を得た。
【0167】
親水性ZAIS/ZnSに以下の処理を行って、より強い蛍光を発する親水化量子ドットを調製した。酢酸亜鉛無水和物(0.1124mmol)を水(2mL)に溶解し、チオアセアミド(0.1124mmol)、TGAまたは3−MPA(0.2248mmol)を加えた。水酸化ナトリウム水溶液(1M)を用いてpHを9に調整した後に、3−MPA−ZAIS/ZnS水溶液(2mL)を加え、遮光下にて、オイルバスにて80℃で加熱撹拌した。放冷後に、メタノールを加え、遠心分離(3500rpm、5分間)を行い、上清を廃棄して沈殿を回収した。得られた沈殿を水(1mL)に溶解し、メンブラン濾過(0.22μm)を行い、不溶部分を除去して、親水化ZAIS/ZnS水溶液を得た。チオール安定化剤としてTGAを用いて製造したZAIS/ZnSの方が、3−MPAを用いて製造したものよりも強い蛍光を発した(図11)。
【0168】
〔実施例10:糖鎖固定化蛍光性ナノ粒子の調製2〕
チオール安定化剤としてTGAまたは3−MPAを用いて製造した蛍光性ナノ粒子(ZAIS/ZnS)を用いて、糖鎖固定化蛍光性ナノ粒子(SFNP)を製造した。
【0169】
Glcα1−4Glc−mono(125μL、10mM)と水素化ホウ素ナトリウム(125μL、100mM)を混合し、2時間静置して、糖鎖リガンド複合体溶液(複合体の最終濃度2.5mM)を調製した。親水化したZAIS/ZnS水溶液(500μL、濃度7μM)を、マイクロコンを用いて遠心濾過(3500rpm、5分間)した後、濾過物を再度250μLの水に懸濁した。この蛍光性ナノ粒子の懸濁液と上記糖鎖リガンド複合体溶液(250μL)を混合し、遮光下にて24時間反応させた。過剰の糖鎖リガンド溶液を遠心濾過によって除去し、沈査をPBSに分散させることによって水中での分散性が良好なSFNP(αGlc−ZAIS/ZnS)を得た。
【0170】
チオール安定化剤としてTGAを用いて、還流を5時間行って得たZAIS/ZnSに固定化したαGlc−ZAIS/ZnSの蛍光は、3−MPAを用いて製造したαGlc−ZAIS/ZnSの蛍光よりも強く、しかも、室温で数週間安定であった(図12(a)および(b))。
【0171】
〔実施例11:糖鎖固定化蛍光性ナノ粒子による細胞認識2〕
還流を5時間行って得たZAIS/ZnSに固定化したαGlc−ZAIS/ZnSによる細胞結合活性を検討した。細胞には、肝癌由来の細胞であるHepG2細胞を用いた。比較対照として、CdTe/CdSに固定化したαGlc−FNP(αGlc−CdTe/CdS)を用いた。
【0172】
細胞を1×106個/mLに希釈した後、αGlc−ZAIS/ZnSまたはαGlc−CdTe/CdSを加えて12時間培養した。αGlc−ZAIS/ZnSまたはαGlc−CdTe/CdSを含む培地を除去した後、PBSで2回洗浄し、細胞を蛍光顕微鏡(HSオールインワン蛍光顕微鏡BZ−9000、BIOREVO, KEYENCE)で観察した。αGlc−ZAIS/ZnSおよびαGlc−CdTe/CdSのいずれを用いた場合も、細胞にて蛍光が観察されたが、αGlc−ZAIS/ZnSを用いた場合は、細胞の形態が良好に維持されていた(図13)。このことは、ZAISの低毒性効果がSFNPにおいても維持されており、その効果が反映されたことを示す。
【0173】
〔実施例12:糖鎖固定化蛍光性ナノ粒子のタンパク質との相互作用および結合特異性2〕
ZAIS/ZnSにGalβ1−4Glc−monoを用いて、上記手順に従って水中での分散性が良好なSFNP(βGal−ZAIS/ZnS)を得た。βGal−ZAIS/ZnSを用いて、糖鎖と特異的に結合するタンパク質との相互作用を調べた。
【0174】
5μMのタンパク質溶液を、96ウェルのマイクロプレートにそれぞれ100μL加え、次いで各ウェルにβ−ガラクトースを固定化したβGal−FNP(βGal−ZAIS/ZnS)を100μL加えた。プレートを遮光下にて18時間放置した後、各ウェルにおける上清の蛍光スペクトルを測定した。タンパク質には、コンカナバリンA(ConA)、ヒママメレクチン(RCA120)、ウシ血清アルブミン(BSA)を用いた。β−ガラクトースと特異的に結合することが知られているRCA120を加えた時にのみ、βGal−ZAIS/ZnSの柿色の沈殿物が生じたことが目視で観測され(図14(a))、蛍光強度の減少が観察された(図14(b))。RCA120以外のタンパク質を加えた時には、凝集も蛍光強度の変化もなく、また糖鎖を固定化していないTEG−ZAIS/ZnSおよびTGA−ZAIS/ZnSの場合にも、何ら変化が観測されなかった(図14(a)、(c)および(d))。UV−VISよりも感度が高い蛍光において変化が観測されたことは、用いる糖鎖の量が微量であっても、タンパク質との特異的結合を測定することができることを示し、本発明の方法によって得られたSFNPが従来の方法によって得られる糖鎖固定化金ナノ粒子よりもかなり優れた性質を有していることがわかった。
【0175】
〔実施例13:抗体を固定化した糖鎖固定化蛍光性ナノ粒子〕
上述したように、抗体を固定化したナノ粒子は、抗原抗体反応による免疫染色や、抗原の検出、分離/精製に有用である。しかし、蛍光性ナノ粒子はサイズが小さく、抗体のサイズはナノ粒子と同程度に大きいので、抗体を固定化したナノ粒子では、抗体の疎水性ドメインの影響によって水中で分散性が良好であるというナノ粒子の効果が損なわれてしまう。本発明の方法によって得られたSFNPを用いて抗体を固定化すれば、糖が存在していることに起因して良好な分散性を維持することができる。
【0176】
黄色ブドウ球菌(SA)の表面タンパク質であるProtein Aに強く結合することが知られているヒトIgGを固定化した蛍光性ナノ粒子を用いて、水中での分散性が良好な抗体/糖鎖固定化蛍光性ナノ粒子(Ab−SFNP)を作製した。また、このAb−SFNPを用いてSAを標識化し、共焦点レーザー顕微鏡下にてリアルタイムでの観察を行った。
【0177】
Te粉末(40.04mg)およびNaBH4(46.54mg)をアルゴンガスで置換し、脱気水を2mL添加した後に1時間撹拌してNaHTe溶液を調製した。50mLの脱気水にCdCl2(45.83mg)および3−MPA(43.6mL)を加え、1M NaOHでpHを9.0に調整し、脱気した後に溶液を105℃に加温し、さらに。NaHTe溶液を0.5mL加え、遮光下にて1時間還流してCdTeを生成させた。別途、0.4mMチオアセトアミド水溶液を作製し、脱気した後にその1.5mLを上記CdTe溶液に加え、遮光下にて、105℃で18時間還流してCdTe/CdSを調製した。Amicon 10,000MWCOを用いて、CdTe/CdS溶液を限外濾過した(9500rpm×10分間)。沈殿に超純水を加え、9500rpm×10分間遠心分離して沈殿を洗浄した。この操作をさらに2回繰り返して、蛍光性ナノ粒子CdTe/CdSを回収した。得られた蛍光性ナノ粒子を4℃で保存した。
【0178】
10mM NaBH4 125μLに、Glcα1−4Glc−monoまたはGalβ1−4Glc−monoの混合液125μLを加え、遮光下にて室温で10分間撹拌し、さらに、CdTe/CdSの水溶液250μLを加え、遮光下にて室温で24時間撹拌した。Amicon 10,000MWCOを用いて、25℃で限外濾過した(14,000×gにて5分間)。沈殿に超純水500μLを加え、25℃での限外濾過(14,000×gにて5分間)によって沈殿を洗浄した。この操作をさらに2回繰り返して、SFNP(それぞれ、Glcα1−4Glc−mono−FNP、およびGalβ1−4Glc−mono−FNP)を回収した。500μLになるように超純水を添加したSFNPを遮光下にて4℃で保存した。
【0179】
得られたSFNPの懸濁液200μLに10mM ホウ酸緩衝液(pH7.4)300μLを加え、撹拌した後に、0.2mg/mLの濃度に調製したヒトIgG 50μLを加えた。混合溶液を撹拌した後に、さらに水溶性カルボジイミドWSCD・HCl(10mg/mL)を30μL加え、2時間撹拌した。撹拌後の溶液を、4℃で、50000rpm、30分間超遠心分離した。上清を廃棄した後に、得られた沈殿にPBS(pH8)に溶解したエタノールアミン(50mM)を500μL加えた。混合溶液を、10秒間の超音波処理の後に、遮光下にて1時間撹拌した。撹拌後の溶液を、4℃で、50000rpm、30分間超遠心分離した。上清を廃棄した後に、得られた沈殿に50mM ホウ酸緩衝液(pH8.3)200μLを加え、混和した溶液を4℃で、50000rpm、30分間超遠心分離することによって、沈殿を洗浄した。この操作をさらに2回繰り返した後に、0.2%スキンミルク、0.05% Tween−20、0.1% アジ化ナトリウムを含むPBS 200μLに沈殿物を溶解して、Ab−SFNP溶液を得た。この溶液の保存を4℃で行った。
【0180】
調製したAb−SFNPを2−メルカプトエタノールで還元し、SDS−PAGEおよび銀染色を行った。その結果、2種類のAb−SFNP(ヒトIgGを固定化したGlcα1−4Glc−mono−FNPおよびGalβ1−4Glc−mono−FNP)のいずれにおいても、ヒトIgGのH鎖に相当するバンドが検出された(図15)。このことは、ヒトIgGがSFNPに固定化していることを示す。また、UV−Vis測定の結果、ヒトIgGを固定化したことによって吸光度が全体的に下がるものの、スペクトルの形に変化はなかった(図16)。ヒトIgGの固定化前と比較して固定化後では、350nmで励起したときの蛍光強度が約1/3に低減し、ピークトップが610nmから625nmへと長波長側へシフトした。なお、これらの結果について、固定化している糖鎖に起因する差異は認められなかった。
【0181】
調製したAb−SFNPの菌体への結合特性を、2種類の黄色ブドウ球菌(SA)(野生型およびProtein A欠損株)を用いて調べた。2種類の菌を一晩培養し、100μL(1×108 cell/mL)の培養液を遠心分離してSAを回収した。ヒトIgG固定化SFNPを5%スキムミルク/PBSのブロッキング液で10倍希釈し、その100μLを、回収したSAに加え、37℃で30分間インキュベートした。次いで、菌体をPBSで3回洗浄した後に、SAの沈殿にUV光を照射して生じた蛍光を観測した。
【0182】
野生株では、Ab−SFNPによる濃いピンク色の蛍光が呈されたが、Protein A欠損株では蛍光が呈されず、抗体を固定化していないSFNPを用いた場合の結果と同じであった(図17)。これらの結果は、Ab−SFNPのヒトIgGがProtein Aと結合したことを示している。
【0183】
〔実施例14:抗体を固定化した糖鎖固定化蛍光性ナノ粒子2〕
一端をチオクト酸に、他端をカルボン酸にしたテトラエチレングリコール(TEG)誘導体と、実施例4に記載したツーポット法で合成した蛍光性ナノ粒子QD(CdTe/CdS)との付加を行い、さらに、上記TEG誘導体のカルボン酸末端にNα,Nα−ビス(カルボキシメチル)−L−リジン水和物を導入し、さらに、ニッケルを結合させた化合物を生成し、ニッケルに親和性を有するヒスチジンタグが付加されたM13ファージ由来の一本鎖抗体scFvを、上記ニッケルを介して上記化合物に固定化した。このような、一本鎖抗体scFv結合化合物を用いて、この一本鎖抗体scFvとの親和性が高い培養細胞S1T細胞への結合活性を調べた。具体的な手順を以下に示す。
【0184】
テトラエチレングリコール(TEG)の一端にチオクト酸を付加し、他端をOHで修飾したTEG−OH
【0185】
【化38】
【0186】
およびCOOHで修飾したTEG−COOH
【0187】
【化39】
【0188】
を調製した。これらを、両者の合計の最終濃度が20mMになるように、各混合比(TEG−OH/COOH=10/0、9/1、8/2、7/3)にて混合した。得られた各溶液125μLに10mM水素化ホウ素ナトリウム 125μLを入れ、遮光下にて室温で10分間攪拌した。撹拌後の各混合液にCdTe/CdS溶液 250μLを入れ、遮光下にて室温で24時間攪拌した。その後、各サンプルを10Kカット Amicon限外濾過チューブに入れて、14000×gにて15℃で5分間遠心分離した。さらに、得られた濃縮画分に超純水400μLを入れて、14000×gにて15℃で5分間遠心分離する工程を3回繰り返した。得られた濃縮画分を超純水で250μLにフィルアップし、それぞれを、QD−TEG−OH/COOH=10/0、QD−TEG−OH/COOH=9/1、QD−TEG−OH/COOH=8/2、QD−TEG−OH/COOH=7/3とした。
【0189】
上記のように作製した4種類の蛍光性ナノ粒子(QD−TEG−OH/COOH=10/0、QD−TEG−OH/COOH=9/1、QD−TEG−OH/COOH=8/2、QD−TEG−OH/COOH=7/3)各100μLを、それぞれ10Kカット Amicon限外濾過チューブに入れて、400μLの20mM 炭酸水素ナトリウム水溶液(pH8.0)を加え、14000×gにて15℃で5分間遠心分離する工程を3回繰り返した。得られた各サンプル(50μL)に50μLの20mM 炭酸水素ナトリウム溶液(pH8.0)を加えた。さらに、50μLの水溶性の塩酸カルボジイミド(EDC−HCl,最終濃度161mM)と50μLのN−ヒドロキシスクシンイミド(NHS,最終濃度21.6mM)を混合し、遮光下にて室温で30分間攪拌した。これをA液とした。
【0190】
Nα,Nα−ビス(カルボキシメチル)−L−リジン水和物(NTA)と塩化ニッケルを、それぞれの最終濃度が20mMおよび38mMになるように、20mM 炭酸緩衝液(pH8.0)に溶かし、室温下で1時間攪拌した。これをB液とした。
【0191】
上記A液およびB液を、等量ずつ(200μL/サンプル)混合し、遮光下にて室温で2時間攪拌した後、各サンプルを10Kカット Amicon限外濾過チューブに入れて14000×gにて15℃で5分間遠心分離した。得られた濃縮画分に、400μLの20mM炭酸緩衝液(pH8.0)を加え、14000×gにて15℃で5分間遠心分離する工程を3回繰り返した。その後、400μLのDulbeccoリン酸緩衝化生理食塩水を加えて14000×gにて15℃で5分間遠心分離を行い、得られた濃縮画分をQD−TEG−OH/COOH−NTA−Niとした。TEG−COOH−NTA−Niの構造は以下のとおりである。
【0192】
【化40】
【0193】
MALDI−TOF−MSによる質量分析でTEG−COOH−NTA−Niに相当する分子質量が観測されたことから、TEG−COOH−NTA−NiがQDに固定化されていることを確認した(図18)。
【0194】
QD−TEG−COOH−NTA−Niに固定化させる一本鎖抗体scFvを、以下のようにして作製した。文献(The Journal of Biochemistry. 2009; 145 (6): 799-810, Muraoka S et al.)に従い、成人T細胞白血病(ATL)患者由来のS1T細胞に特異的に結合するscFv(S1TA3)を発現する大腸菌株からscFv(S1TA3)タンパク質を抽出しかつ精製した。scFv(S1TA3)を発現する大腸菌HB2151株を、0.1mg/mLアンピシリン、1mM イソプロピル−β−チオガラクトピラノシドを含む2YT培地(100mL)中で24時間培養し、S1TA3の発現を誘導した。大腸菌を含む培地を3000×gにて4℃で20分間遠心分離し、得られた沈殿を2mLのトリス−EDTA緩衝液に溶かした。その後、超音波破砕を行い、15000×gにて4℃で5分間遠心分離して得た上清を、0.45μmミリポアフィルターに通し、粗タンパク抽出とした。得られた粗抽出物をニッケルカラムに通し、ヒスチジンタグの付いたscFv(S1TA3)タンパク質を精製し、蛍光性ナノ粒子に固定化するscFvサンプルとした。
【0195】
scFv(S1TA3)の蛍光性ナノ粒子への固定化を、以下のようにして行った。QD−TEG−OH/COOH−NTA−Ni 各20μLに、上述のようにして得られたS1TA3精製タンパク質溶液 10μLを混合し、遮光下にて4℃で2時間静置した。得られた抗体サンプルscFv(S1TA3)を固定化した蛍光性ナノ粒子(QD−S1TA3)を、SDS−PAGEおよびフローサイトメトリー解析に供した。
【0196】
SDS−PAGEによるscFv固定化QDの確認を、以下のようにして行った。上述のようにして得られたQD−S1TA3(50μL)を、60000rpm,4℃で30分間超遠心分離し、沈殿と上清画分a(sup 1)に分けた。沈殿に20mM 炭酸水素ナトリウム溶液(pH8.0)を加えて容量を50μLとし、同様の操作を繰り返し、上清画分b(sup 2)と沈殿画分(ppt)を得た。上記操作で得られたサンプル画分各10μLと、2−メルカプトエタノールを含むサンプルローディングバッファー 10μLとを混合し、100℃で10分間加熱した。得られたサンプルを12.5%のポリアクリルアミドゲル上に乗せ、40mAで80分間電気泳動した。電気泳動後のゲルを銀染色によって染色した。図19にはそれぞれの画分の分析結果を示す。
【0197】
QD−S1TA3 OH/COOH=10/0では、TEG−COOH−NTA−Niが存在しないため、scFv(S1TA3)タンパク質は固定化されない。そのため、QD−S1TA3 OH/COOH=10/0ではsup 1,sup 2,ppt全てにおいて、scFv(S1TA3)タンパク質の分子質量である27kDaの位置にはバンドは検出されないが、QD−S1TA3 OH/COOH=9/1、QD−S1TA3 OH/COOH=8/2、QD−S1TA3 OH/COOH=7/3では、いずれのpptにも27kDaの位置にバンドが検出された。このことにより、蛍光性ナノ粒子に、TEG−COOH−NTA−Niを介して一本鎖抗体が固定化されていることが確認された。
【0198】
scFv(S1TA3)タンパク質を固定化した蛍光性ナノ粒子の細胞への結合のフローサイトメトリーによる解析を以下のように行った。細胞には、ATL由来のS1T細胞、及び非ATLの白血病細胞であるMOLT4細胞を用いた。各細胞を1サンプルあたり106個に調整後、1サンプルあたり15μLのQD−S1TA3を加え、攪拌した後、遮光下にて4℃で1時間静置した。その後1mLのDulbeccoリン酸緩衝化生理食塩水で1回洗浄し、2000rpm,4℃で5分間遠心分離した。細胞沈殿後に上清を破棄し、1mLのDulbeccoリン酸緩衝化生理食塩水で細胞を攪拌し、フローサイトメトリーに用いるサンプルとした。
【0199】
フローサイトメトリーにはベックマンコールター社製FC500を使用し、解析にはCXP 解析ソフトウェアを使用した。QDの蛍光励起にはアルゴンレーザー(488nm)を使用し、検出にはFL3フィルタ(620nm)を用いた。図20に結果を示すが、特にQD−S1TA3 OH/COOH=8/2、QD−S1TA3 OH/COOH=9/1、QD−S1TA3 OH/COOH=7/3では、MOLT4細胞と比較して、S1T細胞に対して右側への大きなピークシフトが観察された。
【産業上の利用可能性】
【0200】
本発明に係る糖鎖固定化蛍光性ナノ粒子はリガンド複合体と蛍光性ナノ粒子とを結合させたものであるので、容易に糖鎖を固定することができる。そのため、本発明は糖鎖やタンパク質の機能解析や、細胞の標識等に用いることが可能であり、医薬品開発や生命現象の解明に寄与することが期待される。したがって、医薬、バイオ産業等において広く利用することが可能である。
【技術分野】
【0001】
本発明は、糖鎖を固定化した蛍光性ナノ粒子とその製造方法に関する。
【背景技術】
【0002】
近年、ナノ粒子は、ライフサイエンス、医療診断、バイオテクノロジーなどの様々な分野への応用が行われている。例えば、抗体などを固定化したナノ粒子は、抗原抗体反応による免疫染色や、抗原の検出、分離/精製などに有用である。また、薬剤や遺伝子を固定化したナノ粒子は、ドラッグデリバリーシステム(DDS)のキャリアとして有用である。また、標識したナノ粒子は、分析用マーカー、トレーサーなどの分析用試薬として有用である。
【0003】
蛍光性ナノ粒子は、蛍光体成分として有機材料または無機材料を用いて調製される。有機材料は、容易に光退色するので長時間の蛍光イメージングには適さない。一方、無機材料は、有機材料に比べて光退色しにくいので長時間の蛍光イメージングが可能となる。
【0004】
無機材料を用いる蛍光性ナノ粒子の製造法は、これまでに様々な方法が開発されている。一般的には、カドミウムイオンと硫黄やセレン、テルルなどのカルコゲンとを界面活性剤存在下で加熱し、ナノ結晶を形成/成長させることによって調製する(非特許文献1)。この方法では、反応時間によって粒径分布の狭い均一な蛍光性ナノ粒子を調製することができるが、ナノ粒子表面が界面活性剤で覆われており疎水性を示す。疎水性のため、水溶性の溶媒に不溶であるため、ライフサイエンス、医療診断、バイオテクノロジーなどの様々な分野への応用が難しい。
【0005】
疎水性の蛍光性ナノ粒子から親水性の蛍光性ナノ粒子に転換させるために、方法として、デキストランやポリアクリル酸などの親水性ポリマーによる被覆、シランカップリング剤によるシリカ表面処理による親水化(非特許文献2)、リポソーム被覆(非特許文献3、4)などが知られているが、親水性の蛍光性ナノ粒子に不可欠な糖鎖を導入するためには多段階を要する。また導入できたとしても糖鎖本来の機能発現に必要な糖鎖の高密度化は困難である。
【0006】
上記の方法に対して、近年、親水性の蛍光性ナノ粒子を水中で調製する方法が注目されている。テルル化カドミウムの調製法では(非特許文献5)、無機材料成分としてカドミウムイオンとテルル化水素ナトリウムを用いて、安定化剤として金属成分の配位子となる3−メルカプトプロピオン酸などのチオール基とカルボキシル基の二官能性を有する化合物を用いる。安定化剤に使用したチオール配位子は、容易に配位子交換することができるため、目的の分子構造を持つ蛍光性ナノ粒子に変換することができる。しかしながら、テルル化カドミウムの蛍光性ナノ粒子は、テルルが解離しやすく不安定である。また配位子交換によって導入したチオール配位子は、可逆的に固定化されているため、目的分子を長時間保持したナノ粒子の調製は難しい。さらに依然として、導入できたとしても糖鎖本来の機能発現に必要な糖鎖の高密度化は困難である。
【0007】
テルル化カドミウムの蛍光性ナノ粒子の安定性を向上させるため、硫化カドミウムや硫化亜鉛などによる被覆する方法が開発されている(非特許文献6)。しかしながら、これらのナノ粒子に対して、糖鎖分子の固定化は困難である。
【0008】
一方、糖鎖を固定化した蛍光性ナノ粒子(以下、適宜、糖鎖固定化蛍光性ナノ粒子、またはSFNPともいう。)として、硫化カドミウムのナノ粒子に糖鎖を固定化する方法が知られているが(特許文献1)、高い発光性能を有する蛍光性ナノ粒子の合成は達成されていない。
【0009】
近年、毒性が高いCdを含まない蛍光性ナノ粒子の製造法の開発が進められている。Zn1-x(AgIn)xS(ZAIS)(特許文献2)からなる蛍光性ナノ粒子はCdを含まないため注目されているが、その親水化は難しく、糖鎖分子の固定化にいたっては、これまでに達成されていない。
【0010】
また、テルル化カドミウム、硫化カドミウム、ZAIS等の親水化した蛍光性ナノ粒子に対して、リガンド交換反応による糖鎖の固定化を行うと、蛍光性ナノ粒子の発光性が著しく低下する問題点があった。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特表2004−511511号公報(平成16年4月15日公表)
【特許文献2】特開2010−31115号公報(平成22年2月12日公開)
【非特許文献】
【0012】
【非特許文献1】M. Bruchez Jr., et al., Science, 281, 2013 (1998)
【非特許文献2】C. Earhart, et al., Langmuir, 24, 6215 (2008)
【非特許文献3】F. Osaki, et al., J. Am. Chem. Soc., 126, 6520 (2004)
【非特許文献4】X.-L. Sun, et al., ChemBioChem, 5, 1593 (2004)
【非特許文献5】H. Zhang, et al, J. Phys. Chem. B, 107, 8 (2003)
【非特許文献6】H. Peng, et al., J. Lumin, 127, 721 (2007)
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明は、上記の問題点に鑑みてなされたものであり、ライフサイエンス、医療診断、バイオテクノロジーなどの様々な分野への利用に適した、粒径が均一でありかつ高い水分散性および発光性を有し、かつ容易に糖鎖を固定化することができる、安定的な糖鎖固定化蛍光性ナノ粒子の製造法を提供することを目的とする。
【課題を解決するための手段】
【0014】
本発明に係る糖鎖固定化蛍光性ナノ粒子は、還元剤処理した糖鎖リガンド複合体と、加熱処理した蛍光性ナノ粒子を混和することによって製造され、分散液として提供されることを特徴としている。本発明において、上記糖鎖リガンド複合体は水溶性であり、上記蛍光性ナノ粒子は親水化されている。得られた糖鎖固定化蛍光性ナノ粒子は、水溶液中で分散する。すなわち、本発明に係る糖鎖固定化蛍光性ナノ粒子の製造方法は、加熱処理された蛍光性ナノ粒子と、還元剤処理された糖鎖リガンド複合体を混和する工程を包含することを特徴としている。
【0015】
このような構成を採用することによって、表面が糖鎖リガンド複合体で修飾されており、平均粒径が1〜10nmの糖鎖固定化蛍光性ナノ粒子を得ることができる。
【0016】
本発明に係る糖鎖固定化蛍光性ナノ粒子において、上記糖鎖リガンド複合体は、炭化水素鎖または炭化水素誘導鎖を備えているリンカー化合物の一端に存在するアミノ基にて還元末端を有する糖鎖と結合し、該リンカー化合物の他端に存在する硫黄原子を含む炭化水素構造にて任意の金属と結合している。
【0017】
上記炭化水素誘導鎖は、炭素及び水素からなる炭化水素鎖であり、一部の炭素や水素が他の原子や置換基に置き換わっていてもよい。すなわち、上記炭化水素誘導鎖とは、末端にアミノ基を有し、炭化水素鎖の主鎖構造である炭素−炭素結合(C−C結合)の一部が、炭素−窒素結合(C−N結合)、炭素−酸素結合(C−O結合)、アミド結合(CO−NH結合)に置き換わっていてもよいものを指す。
【0018】
また、上記硫黄原子を含む炭化水素構造とは、炭素及び水素からなる炭化水素構造にて、一部の炭素が硫黄に置き換わっているものを意味する。また、この硫黄原子を含む炭化水素構造は、鎖状(直鎖、枝分かれ鎖の両方を含む)であっても、環状であってもよく、また、鎖状構造および環状構造の両方の構造を含んでいてもよい。
【0019】
本発明に係る糖鎖固定化蛍光性ナノ粒子において、上記硫黄原子を含む炭化水素構造は、S−S結合またはSH基を含む炭化水素構造を備えているものであってもよい。つまり、上記硫黄原子を含む炭化水素構造中に、ジスルフィド結合(S−S結合)またはチオール基(SH基)が含まれていてもよい。
【0020】
また、本発明に係る糖鎖固定化蛍光性ナノ粒子において、上記アミノ基は芳香族アミノ基であることが好ましい。還元アミノ化反応の最適条件であるpH3〜4においては、アミノ基がプロトン化されないことが必要である。そのため、芳香族との共役によってpH3〜4でも非共有電子対が窒素原子上に存在する芳香族アミノ基が好ましい。
【0021】
また、本発明に係る糖鎖固定化蛍光性ナノ粒子において、糖鎖リガンド複合体の還元剤処理に用いる還元剤は水素化ホウ素ナトリウムであることが好ましい。
【0022】
上記構成によれば、リンカー化合物内のS−S結合を還元して−SH基に変換し、任意の金属と、金属−硫黄(S)結合、例えばカドミウム−硫黄(Cd−S)結合により結合することができる。これにより、このCd−S結合を介して、糖鎖固定化蛍光性ナノ粒子を提供することができる。このことにより、容易に糖鎖を固定化することができる安定な糖鎖固定化蛍光性ナノ粒子の製造法の提供が可能となった。
【0023】
還元剤処理した水溶性の糖鎖リガンド複合体を蛍光性ナノ粒子に添加することによって、蛍光性ナノ粒子を水溶液中で安定化することが可能となった。本発明に係る糖鎖固定化蛍光性ナノ粒子は、粒径が均一であり、かつ高い水分散性を有する。
【0024】
本発明の抗体固定化蛍光性ナノ粒子は、加熱処理された蛍光性ナノ粒子と、還元剤処理されたリガンド複合体を混和する工程を包含することを特徴としており、蛍光性ナノ粒子に固定化されたリガンド複合体に抗体を結合することによって製造される。本発明において、上記リガンド複合体は水溶性であり、上記蛍光性ナノ粒子は親水化されている。得られた抗体固定化蛍光性ナノ粒子は、水溶液中で分散する。
【0025】
このような構成を採用することによって、表面が抗体リガンド複合体で修飾されており、平均粒径が5〜50nmの抗体固定化蛍光性ナノ粒子を得ることができる。
【0026】
本発明に係る抗体固定化蛍光性ナノ粒子において、上記リガンド複合体は、炭化水素鎖または炭化水素誘導鎖を備えているリンカー化合物の一端にて抗体と直接的または間接的に結合し、該リンカー化合物の他端に存在する硫黄原子を含む炭化水素構造にて任意の金属(蛍光性ナノ粒子)と結合している。
【0027】
上記炭化水素誘導鎖は、上述した糖鎖固定化蛍光性ナノ粒子における炭化水素誘導鎖と同様の構造を有しているが、糖鎖固定化蛍光性ナノ粒子の製造に用いられる炭化水素誘導鎖の一端に存在するアミノ基は他の反応基であってもよく、抗体を直接的または間接的に結合させ得る反応基であれば特に限定されず、糖鎖が結合していても結合していなくてもよい。すなわち、本発明の抗体固定化蛍光性ナノ粒子におけるリガンド複合体は、糖鎖が結合していても結合していなくてもよい。
【0028】
また、上記硫黄原子を含む炭化水素構造は、上述した糖鎖固定化蛍光性ナノ粒子における炭化水素構造と同一の構造であればよい。このような構造を有することによって、糖鎖固定化蛍光性ナノ粒子を製造する際の利点が、抗体固定化蛍光性ナノ粒子の製造の際においてももたらされる。すなわち、本発明の抗体固定化蛍光性ナノ粒子の製造方法を用いれば、蛍光性ナノ粒子に抗体を容易に固定化することができ、かつ安定な抗体固定化蛍光性ナノ粒子の提供が可能となる。さらに、本発明を用いて製造された抗体固定化蛍光性ナノ粒子は、粒径が均一であり、かつ高い水分散性を有する。
【発明の効果】
【0029】
以上のように、還元剤処理した糖鎖リガンド複合体と、加熱処理した親水性の蛍光性ナノ粒子とを混和するだけで、本発明に係る糖鎖固定化蛍光性ナノ粒子を調製することができる。このことは、容易に分解しやすい糖鎖であっても蛍光性ナノ粒子上に固定化できると同時に蛍光性ナノ粒子上に高い安定性を付与し得るという効果を奏する。また、本発明に係る糖鎖固定化蛍光性ナノ粒子を含む溶液に、上記糖鎖固定化蛍光性ナノ粒子の外側に位置する糖鎖を認識するタンパク質を添加するだけで、糖鎖とタンパク質とが相互作用した凝集物を生成し、溶液の蛍光も変化するので、糖鎖−タンパク質相互作用を、標識を用いることなく目視によって容易に測定することができるという、糖鎖−タンパク質相互作用の測定方法を提供し得るという効果を奏する。
【0030】
糖鎖は、細胞内外に糖タンパク質や糖脂質の形で存在しており、細胞間認識やシグナル伝達をはじめとする様々な生体機能を担っている。細胞内外の糖鎖結合性分子を蛍光標識などによって可視化することができれば、糖鎖が関わる生体機能の解明に繋がる。細胞の糖鎖結合性を利用し、本発明の糖鎖固定化蛍光性ナノ粒子を用いて細胞の蛍光標識を行うことによって、蛍光性ナノ粒子が細胞に結合し、糖鎖による細胞の蛍光標識を可能にするという効果を奏する。同様の観測は、既知の蛍光を発する有機化合物を糖鎖に付与することによっても達成されるが、有機化合物の場合は、発光に伴って、有機化合物自体が光のエネルギーによって分解するため、蛍光が突然消失することが起こる。しかし、糖鎖固定化蛍光性ナノ粒子の場合には、このような光による分解は起こらず、長時間の連続測定も可能である。
【0031】
本発明の別の側面によれば、上記構成による本発明の蛍光性ナノ粒子を含む、相互作用検出剤、細胞標識剤、診断剤が提供される。
【図面の簡単な説明】
【0032】
【図1】α−グルコース固定化蛍光性ナノ粒子のコロイド溶液の透過型電子顕微鏡画像を示す図である。
【図2】目視下における糖鎖−タンパク質相互作用解析を示す図である。
【図3】糖鎖固定化蛍光性ナノ粒子を用いたFACS解析を示す図である。
【図4】本発明の合成法または従来法によって得られた蛍光性ナノ粒子の半値幅(a)および蛍光波長(b)を比較したグラフである。
【図5】MALDI−TOF/MSによって本発明の糖鎖固定化蛍光性ナノ粒子における糖鎖の固定化を示すチャート(a)、および本発明の糖鎖固定化蛍光性ナノ粒子の透過型電子顕微鏡画像(b)を示す図である。
【図6】本発明の糖鎖固定化蛍光性ナノ粒子のタンパク質との相互作用を示すグラフである。
【図7】本発明の糖鎖固定化蛍光性ナノ粒子の結合特異性を示すグラフである。
【図8】本発明の糖鎖固定化蛍光性ナノ粒子による細胞認識を示すグラフである。
【図9】本発明の糖鎖固定化蛍光性ナノ粒子による細胞認識を示す蛍光顕微鏡画像である。
【図10】本発明の糖鎖固定化蛍光性ナノ粒子の製造に用いる蛍光性ナノ粒子の特性を示す図である。
【図11】本発明の糖鎖固定化蛍光性ナノ粒子の製造に用いる蛍光性ナノ粒子の特性を示す図である。
【図12】本発明の糖鎖固定化蛍光性ナノ粒子の特性を示す図である。
【図13】本発明の糖鎖固定化蛍光性ナノ粒子による細胞認識を示す蛍光顕微鏡画像である。
【図14】本発明の糖鎖固定化蛍光性ナノ粒子のタンパク質との相互作用および結合特異性を示す図である。
【図15】抗体を固定化した本発明の糖鎖固定化蛍光性ナノ粒子の、SDS−PAGE後の銀染色の結果を示す図である。
【図16】抗体を固定化した本発明の糖鎖固定化蛍光性ナノ粒子の、UV−Visおよび蛍光強度を測定した結果を示す図である。
【図17】抗体を固定化した本発明の糖鎖固定化蛍光性ナノ粒子の、黄色ブドウ球菌との結合性を示す蛍光顕微鏡画像である。
【図18】MALDI−TOF/MSによって本発明の抗体固定化蛍光性ナノ粒子におけるリンカー分子の固定化を示すチャートである。
【図19】本発明の抗体固定化蛍光性ナノ粒子の、SDS−PAGE後の銀染色の結果を示す図である。
【図20】抗体固定化蛍光性ナノ粒子を用いたFACS解析を示す図である。
【発明を実施するための形態】
【0033】
本発明の実施の形態について説明すれば以下のとおりであるが、本発明はこれに限定されるものではない。以下、本発明に係る糖鎖固定化蛍光性ナノ粒子の製造方法、糖鎖−タンパク質相互作用の測定方法、細胞の糖鎖結合性を利用して細胞を蛍光標識する方法、および抗体/糖鎖固定化蛍光性ナノ粒子の製造方法とそれを用いた細菌の検出方法について詳述する。
【0034】
〔1.糖鎖固定化蛍光性ナノ粒子の製造法〕
本発明は、糖鎖を固定化した蛍光性ナノ粒子(すなわち、糖鎖固定化蛍光性ナノ粒子)を製造する方法を提供する。本発明の方法によって製造された蛍光性ナノ粒子は水溶液中で分散する特徴を有している。すなわち、本発明の方法は、糖鎖固定化蛍光性ナノ粒子のコロイド溶液を調製する方法でもある。
【0035】
本明細書において、「糖鎖固定化蛍光性ナノ粒子」とは、以下に詳述する糖鎖リガンド複合体と、任意の蛍光性ナノ粒子(蛍光性の金属ナノ粒子ともいう。)とを結合させてなるものである。なお、蛍光性ナノ粒子は後述するように製造してもよいし、市販のものを使用してもよい。
【0036】
なお、本明細書において、「金属ナノ粒子」は、無機金属成分を含むナノ粒子であれば特に限定されず、本発明において好適に用いられる金属成分としては、Si、Ge、Cd、Zn、Cu、Ag、Ga、As、In、Te、Sなどが挙げられるがこれらに限定されない。
【0037】
また、本明細書中において、「ナノ粒子」は水溶液中で分散してコロイド溶液を形成するものが意図される。よって、ナノ粒子の平均粒子径は、0.5〜400nmの範囲内であることが好ましく、0.5nm〜100nmの範囲内であることがより好ましく、1nm〜10nmの範囲内であることがさらに好ましい。平均粒子径は0.5nm未満であってもよいが、そのような粒子の製造は高コストであって実用的でなく、400nmを超えると、粒子の分散安定性が経時的に変化しやすいので好ましくない。
【0038】
また、本発明に係る糖鎖固定化蛍光性ナノ粒子の構成要素である糖鎖リガンド複合体は、任意の蛍光性ナノ粒子と結合することのできるリンカー化合物と、分析対象となるタンパク質などと特異的に相互作用することができる糖鎖とから構成されている。そのため、上記糖鎖リガンド複合体は、タンパク質等の物質と疎水性に基づく非特異的な相互作用を形成しないことが必要とされる。好ましくは、上記糖鎖リガンド複合体は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えたリンカー化合物および糖鎖からなり、上記リンカー化合物の主鎖は、その一端に糖鎖と結合したアミノ基を有し、その他端に硫黄原子を含む炭化水素構造を備えている。
【0039】
本発明に利用されるリガンド複合体として好適なものは、WO2005/077965に示されている化合物が挙げられる。具体的には、一般式(1)
【0040】
【化1】
【0041】
(式中、p,qはそれぞれ独立して0以上6以下の整数)にて表される構造を備え、上記Xとして、末端に芳香族アミノ基を有するとともに、主鎖に炭素−窒素結合を有していてもよい炭化水素誘導鎖を、1鎖又は2鎖又は3鎖含んでなる構造を備え、上記Yとして、硫黄原子又は硫黄原子を含む炭化水素構造を備え、上記Zとして、炭素−炭素結合又は炭素−酸素結合を持つ直鎖構造を備えているリンカー化合物と、還元末端を有する糖とが、上記芳香族アミノ基を介して結合している構造を有していることが好ましく、上記Xは、一般式(2)、または一般式(3)、または一般式(4)
【0042】
【化2】
【0043】
【化3】
【0044】
【化4】
【0045】
(式中、m1〜m5はそれぞれ独立して0以上6以下の整数、R’は水素(H)またはR)にて表される構造を備え、上記Rは糖鎖由来化合物であることがより好ましく、上記Zは、式(5)または式(6)
【0046】
【化5】
【0047】
【化6】
【0048】
(式中、n1,n2はそれぞれ1以上6以下の整数)であってもよい。より好ましくは、本発明に利用可能なリガンド複合体は、一般式(7)、または、一般式(8)、または、一般式(9)、または、一般式(10)、または、一般式(11)
【0049】
【化7】
【0050】
【化8】
【0051】
【化9】
【0052】
【化10】
【0053】
【化11】
【0054】
にて表される構造を有するリンカー化合物の芳香族アミノ基に、還元末端を有する糖が導入された構造を有するものが挙げられる。このようなリガンド複合体は、例えば、一般式(7)〜(11)にて表される構造を有するリンカー化合物と、還元末端を有する糖とを用いて還元アミノ化反応を行うことによって製造することができ、
【0055】
【化12】
【0056】
【化13】
【0057】
【化14】
【0058】
【化15】
【0059】
【化16】
【0060】
(式中、m1,m2,m3,m4,m5はそれぞれ独立して0以上6以下の整数、n1,n2はそれぞれ独立して1以上6以下の整数、qは0以上6以下の整数、Rは還元末端を有する糖、R’は水素(H)またはR)にて表される構造を有し得る。
【0061】
また、本発明に利用可能なリガンド複合体は、上記一般式(7)、(8)、(9)または(10)にて表されるリンカー化合物は、例えば、チオクト酸と、芳香族アミノ基末端が保護基によって保護されたアミン化合物との縮合反応を行い、上記芳香族アミノ基末端の保護基を脱保護することによって製造される。また、上記一般式(11)にて表されるリンカー化合物は、例えば、γ−メルカプト酪酸の2量体と、2分子の芳香族アミノ基末端が保護基によって保護されたアミン化合物との縮合反応を行い、上記芳香族アミノ基末端の保護基を脱保護することによって製造される。
【0062】
上記チオクト酸は、
【0063】
【化17】
【0064】
にて表される構造を備えており、上記アミン化合物は、保護基によって保護された芳香族アミノ基末端を有するものであれば特に限定されるものではない。
【0065】
本発明の一実施形態において、上記一般式(9)にて表される構造においてn1およびqが0であるリンカー化合物
【0066】
【化18】
【0067】
に、還元末端を有する糖が導入されたリガンド複合体が用いられる。このようなリガンド複合体の合成手順もまたWO2005/077965に開示されているが、例えば、リガンド複合体(化合物30)の合成の手順を、式(35)にしたがって簡単に説明すると以下のとおりである。
【0068】
【化19】
【0069】
化合物28(2.47mg,8.24μmol)とシアル酸含有三糖(化合物29、5.11mg,7.57μmol)をジメチルアセトアミド/水(1:1,1.0ml)に溶解し、酢酸(100μl)を加え、37℃で10時間放置した。反応液にシアノ水素化ホウ素ナトリウム(1.55mg,24.7μmol)を加え、37℃でさらに72時間放置した。反応液にアセトン3mlを加え、未反応のシアノ化ホウ素ナトリウムをクエンチした後、濃縮した。得られた残渣は、Chromatorex ODSを用いたカラムクロマトグラフィー、HPLC(カラム:DAISO SP−120−5−ODS−BP)、Sephadex G−25を用いたクロマトグラフィーを順に行って精製した。
【0070】
なお、化合物30は白色の固体として得られ、収量は2.31mg(32%)であり、化合物30の1H−NMRスペクトル(600MHz、D2O)測定を行い、またMALDI−TOF−MS測定を行ったところ、m/z 977.5[(M+Na)+]であった。これらによって化合物30の構造を確認することができた(分子質量は954.34)。
【0071】
以下の式(36)〜(51)にて表されるリガンド複合体を同様の手順で合成し、これらのリガンド複合体の1H−NMRスペクトルを測定し、得られたチャートより、リガンド複合体の構造が確認されている。これらもまた、本実施形態において好適に用いられ得る。なお、後述する実施例にて示される、Glcα1−4Glc−monoおよびGalβ1−4Glc−monoは、上記手順に従って合成されている。
【0072】
【化20】
【0073】
【化21】
【0074】
【化22】
【0075】
【化23】
【0076】
【化24】
【0077】
【化25】
【0078】
【化26】
【0079】
【化27】
【0080】
【化28】
【0081】
【化29】
【0082】
【化30】
【0083】
【化31】
【0084】
【化32】
【0085】
【化33】
【0086】
【化34】
【0087】
【化35】
【0088】
このように本発明に利用可能なリンカー化合物は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えており、主鎖の一末端にアミノ基を有している。さらに、上記リンカー化合物は、末端にアミノ基を有していることによって、糖鎖分子を簡便に導入することができる。なお、上記アミノ基は、修飾されているアミノ基(例えばアセチル基、メチル基やホルミル基等で修飾されたアミノ基)や、芳香族アミノ基であってもよいし、未修飾のアミノ基であってもよい。
【0089】
糖鎖リガンド複合体には、硫黄原子(S)が含まれており、この硫黄原子(S)は、例えば、蛍光性ナノ粒子に含まれるカドミウム(Cd)と、金属−硫黄結合(Cd−S結合)を形成し、蛍光性ナノ粒子に結合することができる。
【0090】
本発明に利用可能なリンカー化合物は、金属−硫黄結合(例えばCd−S結合)を容易に形成することができるという点で、S−S結合またはSH基が含まれている炭化水素構造を主鎖の他端に備えていることが好ましい。これによって、上記リンカー化合物は、蛍光性ナノ粒子上に糖鎖分子を集合化して配列することができる。ジスルフィド結合(S−S結合)またはSH基中の硫黄(S)は、例えば、蛍光性ナノ粒子上に存在するカドミウム(Cd)と、金属−硫黄結合(Cd−S結合)を形成し、金属との結合を強固にすることができる。
【0091】
そして、上記糖鎖リガンド複合体には、上記リンカー化合物のアミノ基に、還元末端を有する糖鎖が導入されている。言い換えれば、上記糖鎖リガンド複合体は、上記リンカー化合物と、還元末端を有する糖鎖とが、アミノ基を介して結合している構造を有している。この糖鎖の導入は、例えば、上記リンカー化合物のアミノ基(−NH2基)と糖鎖との還元アミノ化反応によって行うことができる。つまり、平衡によって生じる糖鎖中のアルデヒド基(−CHO基)またはケトン基(−CRO基、Rは炭化水素基)と、上記リンカー化合物が有するアミノ基とが反応する。そして、この反応によって形成されたシッフ塩基を引き続き還元することによって、アミノ基に容易に糖鎖を導入することができる。
【0092】
なお、上記「還元末端を有する糖鎖」とは、アノマー炭素原子が置換を受けていない単糖、オリゴ糖鎖、多糖鎖である。つまり、上記還元末端を有する糖鎖とは、還元糖鎖である。上記還元末端を有する糖鎖としては、市販のものであっても天然のものであってもよく、あるいは、市販および天然の多糖鎖を分解して調製したものを用いることができる。
【0093】
上記還元末端を有する糖鎖としてより具体的には、グルコース、ガラクトース、マンノース、マルトース、イソマルトース、ラクトース、パノース、セロビオース、メリビオース、マンノオリゴ糖鎖、キトオリゴ糖鎖、ラミナリオリゴ糖鎖、グルコサミン、N−アセチルグルコサミン、グルクロン酸、ヘパリン、ヘパラン硫酸、コンドロイチン硫酸、デルマタン硫酸などが挙げられるが、これに限定されることはない。
【0094】
上記リガンド複合体に含まれるリンカー化合物は、金属に結合可能な硫黄原子と、オリゴ糖鎖等の糖鎖分子に結合可能なアミノ基とを有している。従って、例えばCd−S結合などの金属−硫黄結合により上記リガンド複合体が蛍光性ナノ粒子上の金属に固定されるので、上記リンカー化合物を介して、本発明に係る蛍光性ナノ粒子に糖鎖分子を強固にかつ簡単に結合させることができるとともに、蛍光性ナノ粒子をコロイド状態で安定化することができる。また、上記リガンド複合体の固定化は、還元剤処理した上記リガンド複合体と蛍光性ナノ粒子を含む溶液を混和するだけで行うことができるので、非常に容易に糖鎖を固定化することができる。
【0095】
また、上記リガンド複合体は、タンパク質との非特異的な相互作用の影響をほとんど無視することができる。それゆえ、上記リンカー化合物を有する上記リガンド複合体を用いることによって、上記糖鎖とタンパク質との相互作用を再現性よく評価することが可能になる。
【0096】
上記のリガンド複合体は、リンカー化合物と糖鎖分子とを含んでなっているので、リンカー化合物内のS−S結合にて、金属−硫黄(S)結合、例えばカドミウム−硫黄(Cd−S)結合によって、任意の金属と結合することができる。これにより、このCd−S結合を介して、蛍光性ナノ粒子上に糖鎖分子が固定化されてなる糖鎖固定化蛍光性ナノ粒子を提供することができる。上記任意の金属としては、上記リガンド複合体と結合可能なものであればよく、上記カドミウムの他、亜鉛、銅、銀、インジウム等の金属を用いることができるが、特にカドミウム、亜鉛を用いることが好ましい。
【0097】
なお、上記リガンド複合体に導入されている糖鎖は、還元末端を有するものであれば単糖でもよいし、同一の単糖分子からなる単一オリゴ糖鎖または多糖鎖であってもよいし、種々の単糖分子やその誘導体からなる複合糖鎖・多糖鎖であってもよい。また、上記糖鎖は、いずれも、自然界から単離/精製して得られる種々の天然の糖鎖であってもよく、人工的に合成された糖鎖であってもよい。また、上記糖鎖は、多糖鎖を分解して得られたものであってもよい。
【0098】
本発明に係る糖鎖固定化蛍光性ナノ粒子は、加熱処理した親水性の蛍光性ナノ粒子を含む溶液と、還元剤処理した上記リガンド複合体との混和によって得ることができ、リガンド複合体のS−S結合の各S原子が、ナノ粒子上の金属と金属−硫黄結合によって結合する。具体的には、例えば、蛍光性ナノ粒子の溶液に、還元剤処理した上記リガンド複合体を含む溶液を添加することによって、上記リガンド複合体のS−S結合を、ナノ粒子上の金属−硫黄結合に変換して、糖鎖固定化蛍光性ナノ粒子を得ることができる。
【0099】
なお、親水性の蛍光性ナノ粒子の加熱処理の条件は、特に限定されるものではないが、チオール安定化剤の存在下にて、50〜200℃で行われることが好ましく、70〜180℃がより好ましく、100℃以上であることがさらに好ましい。チオール安定化剤としては、特に限定されないが、チオアセトアミド、3−メルカプトプロピオン酸(3−MPA)、チオグリコール酸(TGA)、4−メルカプトブタン酸、システイン、シスタミンなどのチオ化合物および塩類が挙げられる。
【0100】
還元剤処理に用いられる還元剤としては、特に限定されるものではないが、例えば、水素化ホウ素ナトリウム、シアノ水素化ホウ素ナトリウムなどの塩類および陽イオン成分が異なる塩類等を挙げることができる。
【0101】
蛍光性ナノ粒子およびリガンド複合体を含む溶液に用いる溶媒としては、特に限定されるものではないが、例えば、水、メタノール、エタノール、プロパノール、これらの混合溶媒等を挙げることができる。また、上記混和によって得られた糖鎖固定化蛍光性ナノ粒子を遠心濾過し、低分子の塩などの成分を除くことによって、溶液状態で安定な糖鎖固定化蛍光性ナノ粒子を得ることができる。
【0102】
糖鎖固定化蛍光性ナノ粒子を調製するために用いる金属ナノ粒子、上記リガンド複合体、還元剤の混合比は、特に限定されるものではないが、金属成分としてカドミウムが含まれる場合は、溶液中のカドミウム濃度が、最終濃度で0.1mM〜1mMであることが好ましい。
【0103】
上記リガンド複合体の濃度は、溶液中の最終濃度として0.1mM〜10mMであることが好ましい。
【0104】
また、用いられる還元剤の濃度は、溶液中の最終濃度としてリガンド複合体濃度の10倍モル濃度であることが好ましい。
【0105】
このように、好ましい実施形態において、本発明の糖鎖固定化蛍光性ナノ粒子の製造方法は、加熱処理された蛍光性ナノ粒子と、還元剤処理された糖鎖リガンド複合体を混和する工程を包含し、上記蛍光性ナノ粒子は、第一および第二の金属成分からなる粒子コアが第一および第三の金属成分からなるシェル層によって被覆されており、上記糖鎖リガンド複合体は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えたリンカー化合物および糖鎖からなり、上記リンカー化合物の主鎖は、その一端に糖鎖と結合したアミノ基を有しており、その他端に硫黄原子を含む炭化水素構造を備えており、還元剤処理された上記炭化水素構造が上記層に固定化されて、糖鎖固定化蛍光性ナノ粒子の分散液が得られる。
【0106】
本実施形態の糖鎖固定化蛍光性ナノ粒子の構成要素である蛍光性ナノ粒子は、第一および第二の金属成分からなる粒子(コア)が第一および第三の金属成分からなる層(シェル)によって被覆されている「コア/シェル」構造を有していることが好ましい。蛍光性ナノ粒子が加熱処理されていることによって、シェル層の表面が均質化されて、上記糖鎖リガンド複合体を効率よく結合させることができ、その結果、蛍光性ナノ粒子上に高い安定性を付与し得る。
【0107】
第一の金属成分は、Cd、Zn、Ag、InおよびSからなる群より選択されることが好ましく、第二の金属成分が、TeおよびSからなる群より選択されることが好ましい。また、第三の金属成分は、Cd、SおよびZnからなる群より選択されることが好ましく、後述するチオール安定化剤として提供されてもよい。本発明に使用可能な「コア/シェル」構造の蛍光性ナノ粒子としては、CdTe/CdS、ZAIS/ZnSが挙げられるがこれらに限定されない。
【0108】
1つの局面において、本実施形態の製造方法は、第一の金属成分を含む第一溶液と、第二の金属成分を含む第二溶液とを加熱条件下にて混合し、第一溶液と第二溶液との混合溶液を室温に冷却し、第一および第二の金属成分からなる粒子を混合溶液から精製することによって行われ、第一工程において混合された溶液が第三の金属成分をさらに含んでいることによって、第一および第二の金属成分からなる粒子が、第一および第三の金属成分からなる層によって被覆される。
【0109】
別の局面において、本実施形態の製造方法は、第一の金属成分を含む第一溶液と、第二の金属成分を含む第二溶液とを加熱条件下にて混合し、第一溶液と第二溶液との混合溶液を室温に冷却し、第一および第二の金属成分からなる粒子を混合溶液から精製し、精製した粒子および第三の金属成分を含む水溶液を加熱処理することによって、第一および第二の金属成分からなる粒子が、第一および第三の金属成分からなる層によって被覆される。
【0110】
粒子コアを精製した後に加熱処理を行うことによって、シェル層の表面の均質化がより一層改善されて、上記糖鎖リガンド複合体の結合が著しく向上する。この場合、精製の前に行われる加熱処理は、30分間〜8時間にわたって行われることが好ましい。これにより、粒径が均一でありかつ高い水分散性を有する糖鎖固定化蛍光性ナノ粒子が得られる。
【0111】
なお、本明細書において、「室温」は通常の実験室内での温度であり、20〜30℃が好ましいが、加熱処理における反応を停止することができる温度であれば特に限定されず、いわゆる低温室(例えば4℃)であってもよい。
【0112】
本実施形態において、第一溶液は、第一の金属成分の塩または錯塩が溶解した溶液であり、第二溶液は、第二の金属成分の塩または錯塩が溶解した溶液であっても、親水化された第二の金属成分が溶解した溶液であってもよい。
【0113】
このような製造方法によって製造された糖鎖固定化蛍光性ナノ粒子は、その表面上に抗体を容易に固定化し得るので、生体組織に対する特異性が向上した抗体固定化蛍光性ナノ粒子を容易に提供し得る。
【0114】
本明細書において、「生体組織」は、生物体外に取り出された組織であっても、生物体内の組織であってもよく、臓器または器官であっても、これらに由来する単離された細胞であっても、培養細胞であってもよく、細胞を構成するタンパク質であってもよい。例えば、タンパク質−タンパク質の相互作用を検出するために用いられても、細胞を検出可能に標識するために用いられてもよく、生体内での物質動態を画像化するために用いられてもよい。
【0115】
このように、本発明の糖鎖固定化蛍光性ナノ粒子および抗体固定化蛍光性ナノ粒子を用いれば、生体組織の標識を容易に行うことができる。すなわち、本発明の糖鎖固定化蛍光性ナノ粒子を含む組成物は、生体組織を標識するための組成物であり得る。
【0116】
〔2.糖鎖−タンパク質相互作用の測定方法〕
本発明に係る糖鎖−タンパク質相互作用の測定方法は、糖鎖固定化蛍光性ナノ粒子を含む溶液と、上記糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖を認識するタンパク質とを混和することによって、糖鎖とタンパク質とを相互作用させ、糖鎖−タンパク質複合体を生成させる工程を含むものである。
【0117】
上記「糖鎖固定化蛍光性ナノ粒子を含む溶液」とは、本発明に係る糖鎖固定化蛍光性ナノ粒子が液体に分散したコロイド溶液が意図される。糖鎖固定化蛍光性ナノ粒子を含んでいれば、他に塩などが含まれていてもよい。上記液体としては、例えば水や緩衝液等を用いることができる。
【0118】
上記タンパク質としては、特に限定されるものではなく、上記糖鎖固定化蛍光性ナノ粒子の外側に位置する糖鎖を認識することができるものであればよい。例えば、糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖がグルコースの場合は、グルコースを認識することができるタンパク質であるコンカナバリンA(ConA)、レンチルレクチン(LCA)、ピーナッツレクチン(PSA)等を用いることができる。
【0119】
同様に、糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖がガラクトースである場合は、ガラクトースを認識するタンパク質であるヒママメレクチン(RCA120)等を用いることができる。また、糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖がN−アセチルグルコサミンである場合は、N−アセチルグルコサミンを認識するタンパク質である小麦胚芽レクチン等を用いることができる。
【0120】
糖鎖固定化蛍光性ナノ粒子を含む溶液と、上記糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖を認識するタンパク質とを混和する方法は、特に限定されるものではなく、糖鎖とタンパク質とを近接させて相互作用が可能な状況を提供し得るものであればよい。例えば、マイクロプレートやエッペンドルフチューブなどにタンパク質の希釈系列を作製し、糖鎖固定化蛍光性ナノ粒子を含む溶液を添加して放置することにより混和を行うことができる。
【0121】
糖鎖とタンパク質との相互作用(糖鎖−タンパク質相互作用)としては、水素結合、イオン結合、静電気的相互作用、ファンデルワールス力等を挙げることができる。すなわち、上記糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖をタンパク質が認識し、その結果、水素結合等の糖鎖−タンパク質相互作用が生じる。
【0122】
なお、上記「認識」とは、タンパク質が分子内に有する糖鎖結合部位(糖鎖認識部位)によって糖鎖に結合することをいう。
【0123】
上記「糖鎖−タンパク質複合体」とは、糖鎖とタンパク質とが相互作用して特異的に結合し、その結果生成される凝集物を意味する。本発明に係る糖鎖−タンパク質相互作用の測定方法は、糖鎖−タンパク質相互作用を凝集物の生成として目視で確認することができる。また、その際の溶液の蛍光色が変化する。糖鎖とタンパク質とが相互作用しない場合、またはタンパク質の糖鎖結合部位がタンパク質あたり1つしかない場合(単価タンパク質)には、凝集物は形成されない。
【0124】
ここで、凝集反応を確認することによって物質間の相互作用を測定する方法としては、例えば抗原抗体反応を用いたラテックス凝集法等を挙げることができる(「バイオ診断薬の開発・評価と企業」、CMCテクニカルライブラリー146、シーエムシー出版、P92−97,P109−113)。上記ラテックス凝集法は、ラテックス表面に抗体を固定化させておき、96穴のマイクロプレートを用いて試料抗原の希釈系列を作り、凝集を生じる最大希釈倍率を求め標準溶液と比較して測定するという方法である。結果は一定波長の光で吸光度として測定される。
【0125】
コロイドについて凝集反応を確認することによって物質間の相互作用を測定する方法としては、上記蛍光性ナノ粒子の呈する赤褐色をもって結果を判断するという手法が考えられる。したがって、コロイドにおいても糖鎖とタンパク質とを相互作用させて凝集物を生成させ、当該凝集物の生成を確認することによって結果を判定することができる。本発明に係る方法は、従来の方法よりも非常に簡便であり、蛍光を使用することから感度の高い、有用な方法であるということができる。
【0126】
また、本発明に係る方法を用いれば、未標識の状態で糖鎖−タンパク質相互作用を測定することができるので、標識を要する方法のような前処理を必要としない点で簡便である。さらに、標識効果が測定のばらつきに大きく影響するという問題も存在せず、再現性のよい測定を行うことができる。また、糖鎖−タンパク質相互作用を目視で確認することができるので、特別な装置も必要なく、非常に安価かつ容易に糖鎖−タンパク質相互作用を測定することができる。すなわち、本発明の糖鎖固定化蛍光性ナノ粒子は、相互作用検出に利用可能であり、そのような用途に利用される組成物を構成し得る。
【0127】
したがって、本発明に係る方法は、糖鎖やタンパク質の機能解析や、臨床的な検査および診断などに用いることが可能である。すなわち、本発明の糖鎖固定化蛍光性ナノ粒子は、臨床的な検査に用いられる生体イメージング、近赤外増感などに利用可能であり、そのような用途に利用される組成物を構成し得る。
【0128】
なお、本発明に係る糖鎖−タンパク質相互作用の測定方法は、糖鎖固定化蛍光性ナノ粒子を含む溶液と、上記糖鎖固定化蛍光性ナノ粒子の末端に位置する糖鎖を認識するタンパク質とを混和することによって、糖鎖とタンパク質とを相互作用させ、糖鎖−タンパク質複合体を生成させる工程を含んでいればよい。したがって、例えば、上述したように、糖鎖−タンパク質複合体(凝集体)の生成を目視で確認するだけでもよいし、より詳細な測定を行う場合は、一定波長の紫外可視吸光スペクトルを測定する工程を含んでいてもよい。また、一定波長の光で励起した後に蛍光スペクトルを測定する工程を含んでいてもよい。
【0129】
〔3.細胞の糖鎖結合性を利用して細胞を蛍光標識する方法〕
細胞は多量の糖鎖成分を含んでいる。細胞に存在するタンパク質は、細胞の内外に存在する糖鎖を認識し、細胞間の認識やシグナル伝達などの機能の調節を行っている。言い換えると、細胞の糖鎖結合性を利用し、糖鎖固定化蛍光性ナノ粒子と細胞を結合させ、蛍光標識することができる。
【0130】
蛍光標識した細胞は、フローサイトメトリーおよび蛍光顕微鏡等の機器を用いて解析する。すなわち、本発明の糖鎖固定化蛍光性ナノ粒子は、細胞標識、ウイルス標識、細胞イメージング、生体イメージング、近赤外増感などに利用可能であり、そのような用途に利用される組成物を構成し得る。
【0131】
本発明の製造方法で細胞が特異的に認識する糖鎖を固定化した糖鎖固定化蛍光性ナノ粒子を製造したのち、糖鎖固定化蛍光性ナノ粒子と細胞含有溶液と混合させるだけで、細胞の表面のレセプターを介して結合し、糖鎖固定化蛍光性ナノ粒子による標識が行われる。
【0132】
〔4.抗体固定化蛍光性ナノ粒子の製造法〕
本発明は、抗体を固定化した蛍光性ナノ粒子(すなわち、抗体固定化蛍光性ナノ粒子)を製造する方法を提供する。本発明の方法によって製造された蛍光性ナノ粒子は水溶液中で分散する特徴を有している。すなわち、本発明の方法は、抗体固定化蛍光性ナノ粒子のコロイド溶液を調製する方法でもある。
【0133】
本発明の方法によって製造される抗体固定化蛍光性ナノ粒子の製造に用いられるリガンド複合体は、上述した一般式(1)にて表される構造を備える化合物であっても、例えば、下記式(I)または(II)
【0134】
【化36】
【0135】
【化37】
【0136】
(式中、n1,n2はそれぞれ独立して1以上6以下の整数、qは0以上6以下の整数)にて表される構造を備える化合物であってもよい。本発明の抗体固定化蛍光性ナノ粒子を製造する際に、上記式(I)および(II)の構造を備える化合物をリガンド複合体として用いる場合もまた、上述した糖鎖固定化蛍光性ナノ粒子を製造する方法に準じて行えばよいことを、本明細書を読んだ当業者は容易に理解する。
【0137】
なお、本発明は以上説示した各構成に限定されるものではなく、特許請求の範囲に示した範囲内で種々の変更が可能であり、異なる実施形態にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施形態についても本発明の技術的範囲に含まれる。
【実施例】
【0138】
本発明について、実施例に基づいてより具体的に説明するが、本発明はこれに限定されるものではない。
【0139】
〔実施例1:α−グルコース固定化蛍光性ナノ粒子の調製〕
非特許文献5および6に記載の方法を改変して、塩化カドミウムを含む水溶液に3−MPAを添加し、次いでNaHTeの溶液を加え、混合溶液を1時間還流した後にチオアセトアミドを加えることによって、金属ナノ粒子を含む溶液を調製した。
【0140】
5mMのカドミウムを含む金属ナノ粒子を含む溶液1mLを分注し、マイクロコン(Millipore、Microcon、10000NMWL)を用いて遠心濾過(3500rpm、5分間)し、続いて、ナノ粒子を500μLの水に懸濁した。別途で、250μLの、α−グルコースが結合した糖鎖リガンド複合体(Glcα1−4Glc−mono;式(46)にて表される化合物)の水溶液(10mM)、および250μLの、水素化ホウ素ナトリウム水溶液(100mM)を混合した。その後、この混合溶液500μLとナノ粒子500μLを遮光下にて混和および撹拌して糖鎖固定化蛍光性ナノ粒子の粗コロイド溶液を調製した。図1に、調製したナノ粒子の透過型電子顕微鏡画像を示す。
【0141】
次に、上記粗コロイド溶液を再び遠心濾過によって精製し、α−グルコースが固定化した蛍光性ナノ粒子のコロイド溶液を得た。
【0142】
〔実施例2:糖鎖−タンパク質相互作用の測定〕
ConAまたはBSAのPBS溶液(各2mg/mL)を調製し、96ウェルプレートに1レーンずつConAおよびBSAの溶液を2倍段階希釈して50μLずつ添加した(図2、右側から1〜11:左が高濃度で右が低濃度、最左:ブランク)。すべてのウェルに実施例1で調製したα−グルコースを固定化した蛍光性ナノ粒子のコロイド溶液を50μL添加して混合した。約2時間放置した後、目視による相互作用の確認を行った。
【0143】
図2の写真は、目視下での糖鎖−タンパク質相互作用の解析の結果を示す。結合性タンパク質が添加されているレーンにおいて特異的な凝集反応が見られた。
【0144】
〔実施例3:糖鎖結合性を利用して細胞を蛍光標識する方法〕
5mMのカドミウムを含む金属ナノ粒子を含む溶液1mLを分注し、マイクロコン(Millipore、Microcon、10000NMWL)を用いて遠心濾過(3500rpm、5分間)し、続いて、ナノ粒子を500μLの水に懸濁した。別途で、250μLの、β−ガラクトースが結合した糖鎖リガンド複合体(Galβ1−4Glc−mono;式(39)にて表される化合物)の水溶液(10mM)、および250μLの、水素化ホウ素ナトリウム水溶液(100mM)を混合した。その後、この混合溶液500μLとナノ粒子500μLを遮光下にて混和および撹拌して糖鎖固定化蛍光性ナノ粒子の粗コロイド溶液を調製した。
【0145】
次に、上記粗コロイド溶液を再び遠心濾過によって精製し、β−ガラクトース固定化した蛍光性ナノ粒子のコロイド溶液を得た。
【0146】
引き続いて、調製した蛍光性ナノ粒子の細胞結合性をフローサイトメトリー(FACS)を用いて解析した。細胞にはマウスのマクロファージ系の細胞であるJ774を用いた。細胞数を1×106個/mLに調整した後、調製したナノ粒子を加え、1時間インキュベートした。その後、遠心分離、および細胞の洗浄を行い、細胞をホルマリンで固定化した。細胞を再び洗浄した後、FACS解析を行った。
【0147】
図3は解析結果のグラフを示す。横軸は蛍光強度、縦軸は細胞数を示す。また黒色線は細胞のみ(コントロール)を、黒色破線はα−グルコースを固定化した蛍光性ナノ粒子を加えたもの、灰色線はβ−ガラクトースを固定化した蛍光性ナノ粒子を加えたものをそれぞれ示す。細胞に対して蛍光性ナノ粒子が結合した場合、ピークはコントロールよりも右側にシフトする。測定の結果、α−グルコースまたはβ−ガラクトースを固定化した蛍光性ナノ粒子のどちらも、右側にピークがシフトしている。このことから、これらナノ粒子による細胞標識が可能であることが分かった。
【0148】
〔実施例4:蛍光性ナノ粒子の調製1〕
非特許文献5および6に記載の方法は、いわゆるワンポットでの合成法であり、この方法に従えば、CdTeをコアとしかつCdSをシェルとする構造の蛍光性ナノ粒子(CdTe/CdS)が得られ、この構造に基づいて安定性や蛍光強度が高くなることが報告されている。
【0149】
しかし、このような方法では、得られた蛍光性ナノ粒子の蛍光スペクトル幅が広く、固定化する糖鎖に応じて蛍光波長(すなわち、蛍光の色)を変えることは困難であることがわかった。
【0150】
本発明者らによる創意工夫の結果、上記問題点を解消した方法を完成するに至った。このような蛍光性ナノ粒子の調製方法は以下の通りであり、別々に調製した2種類の溶液を混合し、反応によって形成した粒子を一旦精製し、再度反応させる、ツーポットでの合成法である。
<溶液1の調製>:Te(粉末、0.125mmol(シグマ・アルドリッチより購入)および水素化ホウ素ナトリウム(0.0500mmol)を、アルゴン雰囲気下にてアルゴンガスをバブリングした水(2mL)に溶解し90分間撹拌してテルル化水素ナトリウム水溶液を調製した。
<溶液2の調製>:塩化カドミウム(0.0500mmol)とチオグリコール酸(TGA;0.0265mmol)を、アルゴン雰囲気下にて、アルゴンガスをバブリングした水(10mL)に溶解し、得られた溶液を水酸化ナトリウム水溶液(1M)でpHを9に調整した後、アルゴンガスでさらにバブリングした。
<ナノ粒子の調製>:溶液2を105℃に加熱し、空気雰囲気下にて、激しく撹拌しながら溶液1を加えた。混合溶液を遮光下にて1時間撹拌した後に室温に戻して反応を停止させた。その後、混合溶液に2−プロパノール(蛍光性ナノ粒子溶液量に対して3倍量)を加え、遠心分離(9500rpm、5分間)を行った。上清を除去した後、沈殿を2−プロパノールに懸濁し、さらに遠心分離を行った後に、上清を除去した。この操作を再度行ってナノ粒子を洗浄した後、沈殿を水(10mL)に溶解した。得られた溶液を4℃で10時間保存した後、チオアセトアミド(0.27μL)を加え、105℃で再加熱を行った。遮光下にて適度な時間にわたって加熱撹拌を行った後、室温に冷却して反応を停止させた。蛍光性ナノ粒子濃度は吸光度から見積り7μMであることがわかった。
【0151】
本発明における、ツーポットでの合成法で得られた蛍光性ナノ粒子と、従来法のワンポット得られた蛍光性ナノ粒子の半値幅を比較したグラフ(図4(a))、および蛍光波長(図4(b))を示す。半値幅(半値全幅)とは最大蛍光強度の1/2の値を示す波長の幅である。示されるように、ワンポット合成法よりもツーポット合成法を用いる方が、蛍光性ナノ粒子の半値幅が狭い。また、チオール安定化剤として従来用いられる3−MPAよりもTGAを用いる方が、蛍光性ナノ粒子の半値幅が狭いことがわかった。このように、表面配位子としてTGAを用い、CdTeコアへのCdSシェルの付加にツーポット合成法を用いることによって、スペクトル幅が狭く、かつ多色同時蛍光発光が可能な蛍光性ナノ粒子を得られることがわかった。
【0152】
〔実施例5:糖鎖固定化蛍光性ナノ粒子の調製1〕
上述したツーポット合成法によって得られた蛍光性ナノ粒子(CdTe/CdS)を用いて、糖鎖固定化蛍光性ナノ粒子(SFNP)を製造した。
【0153】
α−グルコースが結合した糖鎖リガンド複合体(Glcα1−4Glc−mono)(125μL、10mM)と水素化ホウ素ナトリウム(125μL、100mM)を混合し、2時間静置して、糖鎖リガンド複合体溶液(複合体の最終濃度2.5mM)を調製した。蛍光性ナノ粒子溶液(500μL、7μM)を、マイクロコン(Millipore、Microcon、10000NMWL)を用いて遠心濾過(3500rpm、5分間)した後、濾過物を再度250μLの水に懸濁した。この蛍光性ナノ粒子の懸濁液と上記糖鎖リガンド複合体溶液(250μL)を混合し、遮光下にて24時間反応させた。過剰の糖鎖リガンド溶液を遠心濾過によって除去し、沈査をPBSに分散させることによって水中での分散性が良好なSFNP(αGlc−FNP)を得た。
【0154】
糖鎖の固定化について、上記αGlc−FNPを、MALDI−TOF/MSによって糖鎖リガンド複合体の分子質量に相当するピークが測定されたことから確認した(図5(a))。また、透過型電子顕微鏡によって、αGlc−FNPの粒径を測定したところ、粒径が約5nmであり、比較的均一度の高い粒子であることがわかった(図5(b))。さらに、アントロン−硫酸法によって糖の定量を行った。換算すると、最終濃度2.5mMの糖鎖リガンド複合体を蛍光性ナノ粒子と反応させた際に、ナノ粒子1つに約130個の糖鎖が固定化されていることがわかった。
【0155】
〔実施例6:糖鎖固定化蛍光性ナノ粒子のタンパク質との相互作用1〕
上述したように調製したSFNP(2.5mM)と、糖鎖と特異的に結合するタンパク質との相互作用を調べた。5μMのタンパク質溶液を、96ウェルのマイクロプレートにそれぞれ100μL加え、次いで各ウェルにα−グルコースを固定化したαGlc−FNPを100μL加えた。プレートを遮光下にて18時間放置した後、各ウェルにおける上清の蛍光スペクトルを測定した。タンパク質には、コンカナバリンA(ConA)、ヒママメレクチン(RCA120)、ウシ血清アルブミン(BSA)を用いた。α−グルコースと特異的に結合することが知られているConAを加えた時にのみ、蛍光強度の減少が観察された(図6)。UV−VISよりも感度が高い蛍光において変化が観測されたことは、用いる糖鎖の量が微量であっても、タンパク質との特異的結合を測定することができることを示し、本発明の方法によって得られたSFNPが従来の方法によって得られる糖鎖固定化金ナノ粒子よりもかなり優れた性質を有していることがわかった。
【0156】
〔実施例7:糖鎖固定化蛍光性ナノ粒子の結合特異性1〕
実施例4の手順に従ってβ−ガラクトースが結合した糖鎖リガンド複合体(Galβ1−4Glc−mono)調製し、これを用いて、実施例5の手順に従って水中での分散性が良好なSFNP(βGal−FNP)を得た。2種類のSFNP(αGlc−FNPおよびβGal−FNP)を用いて、タンパク質との選択的結合活性を調べた。
【0157】
ConAと特異的に結合するα−グルコースを固定化したαGlc−FNPは、橙色の蛍光(Ex=620nm)を示す(図7(a)中、R−αGlc−FNPと示す。)。と特異的に結合するβ−ガラクトースを固定化したβGal−FNPは、橙色の蛍光(Ex=530nm)を示す(図7(a)中、G−βGal−FNPと示す。)。
【0158】
これらのFSNPを1μMに希釈し、各50μLずつを混合した。混合溶液を入れたチューブに5μMのタンパク溶液(ConA、RCA120またはBSA)をそれぞれ100μL加え、撹拌した後に一晩放置した。ConAを加えたチューブには、ConAとαGlc−FNPとの相互作用による橙色の蛍光を示す凝集体が生成し、上清は、ConAと相互作用しないβGal−FNPが残ることによって緑色になった(図7(b))。一方、RCA120を加えたチューブには、RCA120とβGal−FNPとの相互作用による緑の蛍光を示す凝集体が生じ、上清は、RCA120と相互作用しない結合しないα−グルコースを固定化したαGlc−FNPが残ることによって橙色になった(図7(b))。なお、BSAを加えたチューブでは、凝集は生成せず、チューブ内の溶液は2つのSFNPの混合溶液の色であった(図7(b))。このように、複数のタンパク質との相互作用を目視にて観察することができた。
【0159】
〔実施例8:糖鎖固定化蛍光性ナノ粒子による細胞認識1〕
SFNPの細胞結合活性をフローサイトメトリー(FACS)により解析した。SFNPとして、αGlc−FNPおよびβGal−FNPを用い、コントロールとしてテトラエチレングリコール(TEG)を固定化したナノ粒子(TEG−FNP)を用いた。細胞には、肝癌由来の細胞であるHepG2細胞を用いた。
【0160】
細胞を1×106個/mLに希釈した後、SFNPまたはTEG−FNPを加えて12時間培養した。SFNPまたはTEG−FNPを含む培地を除去した後、PBSで2回洗浄し、セルスクレイパーで細胞をプレートから剥がし、細胞を含むPBS溶液を調製した。この溶液を、FACS解析に供した。
【0161】
SFNPでは、右側への大きなピークシフトが観察された(図8)。特に、βGal−FNPにおいてピークシフトは顕著であった。HepG2細胞は、β−ガラクトース親和性があることが知られており、この結果は、HepG2細胞のβ−ガラクトース親和性が明確に示されたことになる。また、αGlc−FNPにおいてもピークシフトが観察された。このことは、HepG2細胞が、β−ガラクトースほどではないがα−グルコースに対しても高い親和性があることを示す。コントロールとして用いたTEG−FNPでは、右側へのピークシフトがほとんど見られなかった。これは、TEG−FNPに糖鎖が結合していないことに起因するといえる。
【0162】
さらに、プレートから剥がす前の細胞を蛍光顕微鏡(HSオールインワン蛍光顕微鏡BZ−9000、BIOREVO, KEYENCE)で観察した。コントロールとして用いたTEG−FNPでは、細胞にて蛍光がほとんど見られなかったが、αGlc−FNPおよびβGal−FNPでは、細胞にて蛍光が観察された(図9)。
【0163】
〔実施例9:蛍光性ナノ粒子の調製2〕
αGlc−FNPおよびβGal−FNPは、蛍光性ナノ粒子(CdTe/CdS)を用いて製造されたものであり、毒性のCdを含むものである。毒性を低減させることができれば、応用範囲をより広げることができる。そこで、特許文献2に記載の手順を改変して、Cdよりも毒性が低いZAIS(ZnS−AgInS2)を合成し、ZAISの親水化と糖鎖の固定化を行いSFNPを調製した。
<溶液1’>:N,N−ジエチルカルバミド酸ナトリウム(2.5mmol)を水(50mL)に溶解した。
<溶液2’>:AgNO3(11.25mM)、In(NO3)3・3H2O(11.25mM)、Zn(NO3)2・6H2O(2.5mM)になるように水(50mL)に溶解した。
<ナノ粒子の調製>:遮光下にて室温で撹拌した溶液1’に、溶液2’を穏やかに加えた。5分間攪拌した後に、遠心分離(3000rpm、10分間)を行った。沈殿に超純水を加えて再度遠心分離(3000rpm、5分間)を行い、上清を廃棄して沈殿を回収した。この操作を4回行った。次に、沈殿にメタノールを加えて遠心分離(3000rpm、5分間)を行い、上清を廃棄して沈殿を回収した。この操作を2回行い、得られた沈殿を減圧下にて乾燥させることによってN,N−ジエチルカルバミド酸錯体を得た。得られた錯体(50mg)を試験管に移し、アルゴン置換した後に、オイルバスにて180℃で30分間加熱攪拌した。放冷後に、アルゴン雰囲気下にて、オレイルアミン(3mL)を加え、オイルバスにて105℃で5分間加熱撹拌した。遠心分離(3500rpm、10分間)した後、上清をメンブレン(0.45μm、PTFE)で濾過し、得られた濾液にメタノール(3mL)を加え、沈殿を生じさせた。濾液全体の遠心分離(3500rpm、10分間)を行った後に、上清を廃棄して沈殿を回収した。得られた沈殿にオレイルアミン(2mL)を加えて溶解させ、メンブレン濾過した後に、濾液にメタノール(2mL)を加えて再度沈殿を生じさせた。濾液全体の遠心分離(3500rpm、10分間)を行った後に、上清を廃棄して沈殿を回収した。得られた沈殿に再度オレイルアミン2mLを加え、アルゴン置換した後に、オイルバスにて180℃で30分間加熱攪拌した。放冷後に、メタノール(2mL)を加えて沈殿を生じさせた。さらに、遠心分離(3500rpm、10分間)を行った後に、上清を廃棄して沈殿を回収した。得られた沈殿をクロロホルム(2mL)に再度溶解してZAIS溶液を得た。
【0164】
ZAISの製造の際に、各試薬の濃度を図10(a)のように変更することによって、組成比が異なりかつ蛍光スペクトルが異なるナノ粒子を製造することができた(図10(b)および(c))。以下の実験には、組成比0.4の用いた例を説明する。
【0165】
ZAISのオレイルアミン溶液(2mL)に酢酸亜鉛無水和物(0.562μmol)、チオアセトアミド(0.562μmol)を加え、アルゴン置換した後に、遮光下にて、オイルバスにて180℃で30分間加熱撹拌した。放冷後に、メタノールを加えて遠心分離(3500 rpm、5分間)を行い、上清を廃棄して沈殿を回収した。得られた沈殿をクロロホルム(1mL)に溶解して、ZAISをコアとしかつZnSをシェルとする構造の蛍光性ナノ粒子(ZAIS/ZnS)のクロロホルム溶液を得た。
【0166】
ZAIS/ZnSのクロロホルム溶液(1mL)にクロロホルム(1mL)を加え、0℃に冷却し、攪拌下、3−MPAのエタノール溶液(0.2M、1mL)を加えた。続いて、水酸化カリウムのエタノール溶液(0.3M、1mL)を加え、遮光下にて12時間攪拌した。遠心分離(3500rpm、5分間)を行い、上清を廃棄して沈殿を回収した。得られた沈殿を水(1mL)に溶解し、遠心分離(3500rpm、5分間)を行い、得られた上清をメンブラン濾過(0.45μm)を行って、3−MPA被覆された親水性ZAIS/ZnS(水溶液)を得た。
【0167】
親水性ZAIS/ZnSに以下の処理を行って、より強い蛍光を発する親水化量子ドットを調製した。酢酸亜鉛無水和物(0.1124mmol)を水(2mL)に溶解し、チオアセアミド(0.1124mmol)、TGAまたは3−MPA(0.2248mmol)を加えた。水酸化ナトリウム水溶液(1M)を用いてpHを9に調整した後に、3−MPA−ZAIS/ZnS水溶液(2mL)を加え、遮光下にて、オイルバスにて80℃で加熱撹拌した。放冷後に、メタノールを加え、遠心分離(3500rpm、5分間)を行い、上清を廃棄して沈殿を回収した。得られた沈殿を水(1mL)に溶解し、メンブラン濾過(0.22μm)を行い、不溶部分を除去して、親水化ZAIS/ZnS水溶液を得た。チオール安定化剤としてTGAを用いて製造したZAIS/ZnSの方が、3−MPAを用いて製造したものよりも強い蛍光を発した(図11)。
【0168】
〔実施例10:糖鎖固定化蛍光性ナノ粒子の調製2〕
チオール安定化剤としてTGAまたは3−MPAを用いて製造した蛍光性ナノ粒子(ZAIS/ZnS)を用いて、糖鎖固定化蛍光性ナノ粒子(SFNP)を製造した。
【0169】
Glcα1−4Glc−mono(125μL、10mM)と水素化ホウ素ナトリウム(125μL、100mM)を混合し、2時間静置して、糖鎖リガンド複合体溶液(複合体の最終濃度2.5mM)を調製した。親水化したZAIS/ZnS水溶液(500μL、濃度7μM)を、マイクロコンを用いて遠心濾過(3500rpm、5分間)した後、濾過物を再度250μLの水に懸濁した。この蛍光性ナノ粒子の懸濁液と上記糖鎖リガンド複合体溶液(250μL)を混合し、遮光下にて24時間反応させた。過剰の糖鎖リガンド溶液を遠心濾過によって除去し、沈査をPBSに分散させることによって水中での分散性が良好なSFNP(αGlc−ZAIS/ZnS)を得た。
【0170】
チオール安定化剤としてTGAを用いて、還流を5時間行って得たZAIS/ZnSに固定化したαGlc−ZAIS/ZnSの蛍光は、3−MPAを用いて製造したαGlc−ZAIS/ZnSの蛍光よりも強く、しかも、室温で数週間安定であった(図12(a)および(b))。
【0171】
〔実施例11:糖鎖固定化蛍光性ナノ粒子による細胞認識2〕
還流を5時間行って得たZAIS/ZnSに固定化したαGlc−ZAIS/ZnSによる細胞結合活性を検討した。細胞には、肝癌由来の細胞であるHepG2細胞を用いた。比較対照として、CdTe/CdSに固定化したαGlc−FNP(αGlc−CdTe/CdS)を用いた。
【0172】
細胞を1×106個/mLに希釈した後、αGlc−ZAIS/ZnSまたはαGlc−CdTe/CdSを加えて12時間培養した。αGlc−ZAIS/ZnSまたはαGlc−CdTe/CdSを含む培地を除去した後、PBSで2回洗浄し、細胞を蛍光顕微鏡(HSオールインワン蛍光顕微鏡BZ−9000、BIOREVO, KEYENCE)で観察した。αGlc−ZAIS/ZnSおよびαGlc−CdTe/CdSのいずれを用いた場合も、細胞にて蛍光が観察されたが、αGlc−ZAIS/ZnSを用いた場合は、細胞の形態が良好に維持されていた(図13)。このことは、ZAISの低毒性効果がSFNPにおいても維持されており、その効果が反映されたことを示す。
【0173】
〔実施例12:糖鎖固定化蛍光性ナノ粒子のタンパク質との相互作用および結合特異性2〕
ZAIS/ZnSにGalβ1−4Glc−monoを用いて、上記手順に従って水中での分散性が良好なSFNP(βGal−ZAIS/ZnS)を得た。βGal−ZAIS/ZnSを用いて、糖鎖と特異的に結合するタンパク質との相互作用を調べた。
【0174】
5μMのタンパク質溶液を、96ウェルのマイクロプレートにそれぞれ100μL加え、次いで各ウェルにβ−ガラクトースを固定化したβGal−FNP(βGal−ZAIS/ZnS)を100μL加えた。プレートを遮光下にて18時間放置した後、各ウェルにおける上清の蛍光スペクトルを測定した。タンパク質には、コンカナバリンA(ConA)、ヒママメレクチン(RCA120)、ウシ血清アルブミン(BSA)を用いた。β−ガラクトースと特異的に結合することが知られているRCA120を加えた時にのみ、βGal−ZAIS/ZnSの柿色の沈殿物が生じたことが目視で観測され(図14(a))、蛍光強度の減少が観察された(図14(b))。RCA120以外のタンパク質を加えた時には、凝集も蛍光強度の変化もなく、また糖鎖を固定化していないTEG−ZAIS/ZnSおよびTGA−ZAIS/ZnSの場合にも、何ら変化が観測されなかった(図14(a)、(c)および(d))。UV−VISよりも感度が高い蛍光において変化が観測されたことは、用いる糖鎖の量が微量であっても、タンパク質との特異的結合を測定することができることを示し、本発明の方法によって得られたSFNPが従来の方法によって得られる糖鎖固定化金ナノ粒子よりもかなり優れた性質を有していることがわかった。
【0175】
〔実施例13:抗体を固定化した糖鎖固定化蛍光性ナノ粒子〕
上述したように、抗体を固定化したナノ粒子は、抗原抗体反応による免疫染色や、抗原の検出、分離/精製に有用である。しかし、蛍光性ナノ粒子はサイズが小さく、抗体のサイズはナノ粒子と同程度に大きいので、抗体を固定化したナノ粒子では、抗体の疎水性ドメインの影響によって水中で分散性が良好であるというナノ粒子の効果が損なわれてしまう。本発明の方法によって得られたSFNPを用いて抗体を固定化すれば、糖が存在していることに起因して良好な分散性を維持することができる。
【0176】
黄色ブドウ球菌(SA)の表面タンパク質であるProtein Aに強く結合することが知られているヒトIgGを固定化した蛍光性ナノ粒子を用いて、水中での分散性が良好な抗体/糖鎖固定化蛍光性ナノ粒子(Ab−SFNP)を作製した。また、このAb−SFNPを用いてSAを標識化し、共焦点レーザー顕微鏡下にてリアルタイムでの観察を行った。
【0177】
Te粉末(40.04mg)およびNaBH4(46.54mg)をアルゴンガスで置換し、脱気水を2mL添加した後に1時間撹拌してNaHTe溶液を調製した。50mLの脱気水にCdCl2(45.83mg)および3−MPA(43.6mL)を加え、1M NaOHでpHを9.0に調整し、脱気した後に溶液を105℃に加温し、さらに。NaHTe溶液を0.5mL加え、遮光下にて1時間還流してCdTeを生成させた。別途、0.4mMチオアセトアミド水溶液を作製し、脱気した後にその1.5mLを上記CdTe溶液に加え、遮光下にて、105℃で18時間還流してCdTe/CdSを調製した。Amicon 10,000MWCOを用いて、CdTe/CdS溶液を限外濾過した(9500rpm×10分間)。沈殿に超純水を加え、9500rpm×10分間遠心分離して沈殿を洗浄した。この操作をさらに2回繰り返して、蛍光性ナノ粒子CdTe/CdSを回収した。得られた蛍光性ナノ粒子を4℃で保存した。
【0178】
10mM NaBH4 125μLに、Glcα1−4Glc−monoまたはGalβ1−4Glc−monoの混合液125μLを加え、遮光下にて室温で10分間撹拌し、さらに、CdTe/CdSの水溶液250μLを加え、遮光下にて室温で24時間撹拌した。Amicon 10,000MWCOを用いて、25℃で限外濾過した(14,000×gにて5分間)。沈殿に超純水500μLを加え、25℃での限外濾過(14,000×gにて5分間)によって沈殿を洗浄した。この操作をさらに2回繰り返して、SFNP(それぞれ、Glcα1−4Glc−mono−FNP、およびGalβ1−4Glc−mono−FNP)を回収した。500μLになるように超純水を添加したSFNPを遮光下にて4℃で保存した。
【0179】
得られたSFNPの懸濁液200μLに10mM ホウ酸緩衝液(pH7.4)300μLを加え、撹拌した後に、0.2mg/mLの濃度に調製したヒトIgG 50μLを加えた。混合溶液を撹拌した後に、さらに水溶性カルボジイミドWSCD・HCl(10mg/mL)を30μL加え、2時間撹拌した。撹拌後の溶液を、4℃で、50000rpm、30分間超遠心分離した。上清を廃棄した後に、得られた沈殿にPBS(pH8)に溶解したエタノールアミン(50mM)を500μL加えた。混合溶液を、10秒間の超音波処理の後に、遮光下にて1時間撹拌した。撹拌後の溶液を、4℃で、50000rpm、30分間超遠心分離した。上清を廃棄した後に、得られた沈殿に50mM ホウ酸緩衝液(pH8.3)200μLを加え、混和した溶液を4℃で、50000rpm、30分間超遠心分離することによって、沈殿を洗浄した。この操作をさらに2回繰り返した後に、0.2%スキンミルク、0.05% Tween−20、0.1% アジ化ナトリウムを含むPBS 200μLに沈殿物を溶解して、Ab−SFNP溶液を得た。この溶液の保存を4℃で行った。
【0180】
調製したAb−SFNPを2−メルカプトエタノールで還元し、SDS−PAGEおよび銀染色を行った。その結果、2種類のAb−SFNP(ヒトIgGを固定化したGlcα1−4Glc−mono−FNPおよびGalβ1−4Glc−mono−FNP)のいずれにおいても、ヒトIgGのH鎖に相当するバンドが検出された(図15)。このことは、ヒトIgGがSFNPに固定化していることを示す。また、UV−Vis測定の結果、ヒトIgGを固定化したことによって吸光度が全体的に下がるものの、スペクトルの形に変化はなかった(図16)。ヒトIgGの固定化前と比較して固定化後では、350nmで励起したときの蛍光強度が約1/3に低減し、ピークトップが610nmから625nmへと長波長側へシフトした。なお、これらの結果について、固定化している糖鎖に起因する差異は認められなかった。
【0181】
調製したAb−SFNPの菌体への結合特性を、2種類の黄色ブドウ球菌(SA)(野生型およびProtein A欠損株)を用いて調べた。2種類の菌を一晩培養し、100μL(1×108 cell/mL)の培養液を遠心分離してSAを回収した。ヒトIgG固定化SFNPを5%スキムミルク/PBSのブロッキング液で10倍希釈し、その100μLを、回収したSAに加え、37℃で30分間インキュベートした。次いで、菌体をPBSで3回洗浄した後に、SAの沈殿にUV光を照射して生じた蛍光を観測した。
【0182】
野生株では、Ab−SFNPによる濃いピンク色の蛍光が呈されたが、Protein A欠損株では蛍光が呈されず、抗体を固定化していないSFNPを用いた場合の結果と同じであった(図17)。これらの結果は、Ab−SFNPのヒトIgGがProtein Aと結合したことを示している。
【0183】
〔実施例14:抗体を固定化した糖鎖固定化蛍光性ナノ粒子2〕
一端をチオクト酸に、他端をカルボン酸にしたテトラエチレングリコール(TEG)誘導体と、実施例4に記載したツーポット法で合成した蛍光性ナノ粒子QD(CdTe/CdS)との付加を行い、さらに、上記TEG誘導体のカルボン酸末端にNα,Nα−ビス(カルボキシメチル)−L−リジン水和物を導入し、さらに、ニッケルを結合させた化合物を生成し、ニッケルに親和性を有するヒスチジンタグが付加されたM13ファージ由来の一本鎖抗体scFvを、上記ニッケルを介して上記化合物に固定化した。このような、一本鎖抗体scFv結合化合物を用いて、この一本鎖抗体scFvとの親和性が高い培養細胞S1T細胞への結合活性を調べた。具体的な手順を以下に示す。
【0184】
テトラエチレングリコール(TEG)の一端にチオクト酸を付加し、他端をOHで修飾したTEG−OH
【0185】
【化38】
【0186】
およびCOOHで修飾したTEG−COOH
【0187】
【化39】
【0188】
を調製した。これらを、両者の合計の最終濃度が20mMになるように、各混合比(TEG−OH/COOH=10/0、9/1、8/2、7/3)にて混合した。得られた各溶液125μLに10mM水素化ホウ素ナトリウム 125μLを入れ、遮光下にて室温で10分間攪拌した。撹拌後の各混合液にCdTe/CdS溶液 250μLを入れ、遮光下にて室温で24時間攪拌した。その後、各サンプルを10Kカット Amicon限外濾過チューブに入れて、14000×gにて15℃で5分間遠心分離した。さらに、得られた濃縮画分に超純水400μLを入れて、14000×gにて15℃で5分間遠心分離する工程を3回繰り返した。得られた濃縮画分を超純水で250μLにフィルアップし、それぞれを、QD−TEG−OH/COOH=10/0、QD−TEG−OH/COOH=9/1、QD−TEG−OH/COOH=8/2、QD−TEG−OH/COOH=7/3とした。
【0189】
上記のように作製した4種類の蛍光性ナノ粒子(QD−TEG−OH/COOH=10/0、QD−TEG−OH/COOH=9/1、QD−TEG−OH/COOH=8/2、QD−TEG−OH/COOH=7/3)各100μLを、それぞれ10Kカット Amicon限外濾過チューブに入れて、400μLの20mM 炭酸水素ナトリウム水溶液(pH8.0)を加え、14000×gにて15℃で5分間遠心分離する工程を3回繰り返した。得られた各サンプル(50μL)に50μLの20mM 炭酸水素ナトリウム溶液(pH8.0)を加えた。さらに、50μLの水溶性の塩酸カルボジイミド(EDC−HCl,最終濃度161mM)と50μLのN−ヒドロキシスクシンイミド(NHS,最終濃度21.6mM)を混合し、遮光下にて室温で30分間攪拌した。これをA液とした。
【0190】
Nα,Nα−ビス(カルボキシメチル)−L−リジン水和物(NTA)と塩化ニッケルを、それぞれの最終濃度が20mMおよび38mMになるように、20mM 炭酸緩衝液(pH8.0)に溶かし、室温下で1時間攪拌した。これをB液とした。
【0191】
上記A液およびB液を、等量ずつ(200μL/サンプル)混合し、遮光下にて室温で2時間攪拌した後、各サンプルを10Kカット Amicon限外濾過チューブに入れて14000×gにて15℃で5分間遠心分離した。得られた濃縮画分に、400μLの20mM炭酸緩衝液(pH8.0)を加え、14000×gにて15℃で5分間遠心分離する工程を3回繰り返した。その後、400μLのDulbeccoリン酸緩衝化生理食塩水を加えて14000×gにて15℃で5分間遠心分離を行い、得られた濃縮画分をQD−TEG−OH/COOH−NTA−Niとした。TEG−COOH−NTA−Niの構造は以下のとおりである。
【0192】
【化40】
【0193】
MALDI−TOF−MSによる質量分析でTEG−COOH−NTA−Niに相当する分子質量が観測されたことから、TEG−COOH−NTA−NiがQDに固定化されていることを確認した(図18)。
【0194】
QD−TEG−COOH−NTA−Niに固定化させる一本鎖抗体scFvを、以下のようにして作製した。文献(The Journal of Biochemistry. 2009; 145 (6): 799-810, Muraoka S et al.)に従い、成人T細胞白血病(ATL)患者由来のS1T細胞に特異的に結合するscFv(S1TA3)を発現する大腸菌株からscFv(S1TA3)タンパク質を抽出しかつ精製した。scFv(S1TA3)を発現する大腸菌HB2151株を、0.1mg/mLアンピシリン、1mM イソプロピル−β−チオガラクトピラノシドを含む2YT培地(100mL)中で24時間培養し、S1TA3の発現を誘導した。大腸菌を含む培地を3000×gにて4℃で20分間遠心分離し、得られた沈殿を2mLのトリス−EDTA緩衝液に溶かした。その後、超音波破砕を行い、15000×gにて4℃で5分間遠心分離して得た上清を、0.45μmミリポアフィルターに通し、粗タンパク抽出とした。得られた粗抽出物をニッケルカラムに通し、ヒスチジンタグの付いたscFv(S1TA3)タンパク質を精製し、蛍光性ナノ粒子に固定化するscFvサンプルとした。
【0195】
scFv(S1TA3)の蛍光性ナノ粒子への固定化を、以下のようにして行った。QD−TEG−OH/COOH−NTA−Ni 各20μLに、上述のようにして得られたS1TA3精製タンパク質溶液 10μLを混合し、遮光下にて4℃で2時間静置した。得られた抗体サンプルscFv(S1TA3)を固定化した蛍光性ナノ粒子(QD−S1TA3)を、SDS−PAGEおよびフローサイトメトリー解析に供した。
【0196】
SDS−PAGEによるscFv固定化QDの確認を、以下のようにして行った。上述のようにして得られたQD−S1TA3(50μL)を、60000rpm,4℃で30分間超遠心分離し、沈殿と上清画分a(sup 1)に分けた。沈殿に20mM 炭酸水素ナトリウム溶液(pH8.0)を加えて容量を50μLとし、同様の操作を繰り返し、上清画分b(sup 2)と沈殿画分(ppt)を得た。上記操作で得られたサンプル画分各10μLと、2−メルカプトエタノールを含むサンプルローディングバッファー 10μLとを混合し、100℃で10分間加熱した。得られたサンプルを12.5%のポリアクリルアミドゲル上に乗せ、40mAで80分間電気泳動した。電気泳動後のゲルを銀染色によって染色した。図19にはそれぞれの画分の分析結果を示す。
【0197】
QD−S1TA3 OH/COOH=10/0では、TEG−COOH−NTA−Niが存在しないため、scFv(S1TA3)タンパク質は固定化されない。そのため、QD−S1TA3 OH/COOH=10/0ではsup 1,sup 2,ppt全てにおいて、scFv(S1TA3)タンパク質の分子質量である27kDaの位置にはバンドは検出されないが、QD−S1TA3 OH/COOH=9/1、QD−S1TA3 OH/COOH=8/2、QD−S1TA3 OH/COOH=7/3では、いずれのpptにも27kDaの位置にバンドが検出された。このことにより、蛍光性ナノ粒子に、TEG−COOH−NTA−Niを介して一本鎖抗体が固定化されていることが確認された。
【0198】
scFv(S1TA3)タンパク質を固定化した蛍光性ナノ粒子の細胞への結合のフローサイトメトリーによる解析を以下のように行った。細胞には、ATL由来のS1T細胞、及び非ATLの白血病細胞であるMOLT4細胞を用いた。各細胞を1サンプルあたり106個に調整後、1サンプルあたり15μLのQD−S1TA3を加え、攪拌した後、遮光下にて4℃で1時間静置した。その後1mLのDulbeccoリン酸緩衝化生理食塩水で1回洗浄し、2000rpm,4℃で5分間遠心分離した。細胞沈殿後に上清を破棄し、1mLのDulbeccoリン酸緩衝化生理食塩水で細胞を攪拌し、フローサイトメトリーに用いるサンプルとした。
【0199】
フローサイトメトリーにはベックマンコールター社製FC500を使用し、解析にはCXP 解析ソフトウェアを使用した。QDの蛍光励起にはアルゴンレーザー(488nm)を使用し、検出にはFL3フィルタ(620nm)を用いた。図20に結果を示すが、特にQD−S1TA3 OH/COOH=8/2、QD−S1TA3 OH/COOH=9/1、QD−S1TA3 OH/COOH=7/3では、MOLT4細胞と比較して、S1T細胞に対して右側への大きなピークシフトが観察された。
【産業上の利用可能性】
【0200】
本発明に係る糖鎖固定化蛍光性ナノ粒子はリガンド複合体と蛍光性ナノ粒子とを結合させたものであるので、容易に糖鎖を固定することができる。そのため、本発明は糖鎖やタンパク質の機能解析や、細胞の標識等に用いることが可能であり、医薬品開発や生命現象の解明に寄与することが期待される。したがって、医薬、バイオ産業等において広く利用することが可能である。
【特許請求の範囲】
【請求項1】
加熱処理された蛍光性ナノ粒子を含む溶液と、還元剤処理された糖鎖リガンド複合体を混和する工程を包含し、
上記蛍光性ナノ粒子は、第一および第二の金属成分からなる粒子コアが第一および第三の金属成分からなる層によって被覆されており、
上記糖鎖リガンド複合体は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えたリンカー化合物および糖鎖からなり、
上記リンカー化合物の主鎖は、その一端に糖鎖と結合したアミノ基を有し、その他端に硫黄原子を含む炭化水素構造を備えており、
上記工程によって上記炭化水素構造が上記層に固定化されて、糖鎖固定化蛍光性ナノ粒子の分散液が得られることを特徴とする糖鎖固定化蛍光性ナノ粒子の製造方法。
【請求項2】
前記加熱処理が、前記蛍光性ナノ粒子または前記粒子コアの精製の後に再度行われることを特徴とする請求項1に記載の製造方法。
【請求項3】
前記精製が、室温下にて行われることを特徴とする請求項2に記載の製造方法。
【請求項4】
前記加熱処理が50℃〜200℃の範囲の温度にて行われることを特徴とする請求項1〜3のいずれか1項に記載の製造方法。
【請求項5】
前記精製の前に行われる前記加熱処理が、30分間〜8時間にわたって行われることを特徴とする請求項2〜4のいずれか1項に記載の製造方法。
【請求項6】
第一の金属成分が、Cd、Zn、Ag、InおよびSからなる群より選択されることを特徴とする請求項1〜5のいずれか1項に記載の製造方法。
【請求項7】
第二の金属成分が、TeおよびSからなる群より選択されることを特徴とする請求項1〜6のいずれか1項に記載の製造方法。
【請求項8】
第三の金属成分が、Cd、SおよびZnからなる群より選択されることを特徴とする請求項1〜7のいずれか1項に記載の製造方法。
【請求項9】
第一溶液が、チオール安定化剤を含んでいることを特徴とする請求項1〜8のいずれか1項に記載の製造方法。
【請求項10】
チオール安定化剤が、チオアセトアミド、3−メルカプトプロピオン酸(3−MPA)、チオグリコール酸(TGA)、4−メルカプトブタン酸、システインまたはシスタミンのチオ化合物およびその塩類からなる群より選択されることを特徴とする請求項9に記載の製造方法。
【請求項11】
請求項1〜10のいずれか1項に記載の製造方法によって製造されたことを特徴とする糖鎖固定化蛍光性ナノ粒子。
【請求項12】
請求項11に記載の糖鎖固定化蛍光性ナノ粒子の表面上に抗体が固定化されていることを特徴とする抗体固定化蛍光性ナノ粒子。
【請求項13】
加熱処理された蛍光性ナノ粒子を含む溶液と、還元剤処理されたリガンド複合体を混和する工程を包含し、
上記蛍光性ナノ粒子は、第一および第二の金属成分からなる粒子コアが第一および第三の金属成分からなる層によって被覆されており、
上記リガンド複合体は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えたリンカー化合物からなり、
上記リンカー化合物の主鎖は、その一端に抗体を直接的または間接的に結合し得る反応基を有し、その他端に硫黄原子を含む炭化水素構造を備えており、
上記工程によって上記炭化水素構造が上記層に固定化されて、抗体固定化蛍光性ナノ粒子の分散液が得られることを特徴とする抗体固定化蛍光性ナノ粒子の製造方法。
【請求項14】
請求項13に記載の製造方法によって製造されたことを特徴とする抗体固定化蛍光性ナノ粒子。
【請求項15】
請求項11に記載の糖鎖固定化蛍光性ナノ粒子あるいは請求項12または14に記載の抗体固定化蛍光性ナノ粒子を含んでいることを特徴とする生体組織を標識するための組成物。
【請求項16】
相互作用検出剤、細胞標識剤、細胞イメージング剤、生体イメージング剤、ウイルス標識剤または近赤外増感剤であることを特徴とする請求項15に記載の組成物。
【請求項1】
加熱処理された蛍光性ナノ粒子を含む溶液と、還元剤処理された糖鎖リガンド複合体を混和する工程を包含し、
上記蛍光性ナノ粒子は、第一および第二の金属成分からなる粒子コアが第一および第三の金属成分からなる層によって被覆されており、
上記糖鎖リガンド複合体は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えたリンカー化合物および糖鎖からなり、
上記リンカー化合物の主鎖は、その一端に糖鎖と結合したアミノ基を有し、その他端に硫黄原子を含む炭化水素構造を備えており、
上記工程によって上記炭化水素構造が上記層に固定化されて、糖鎖固定化蛍光性ナノ粒子の分散液が得られることを特徴とする糖鎖固定化蛍光性ナノ粒子の製造方法。
【請求項2】
前記加熱処理が、前記蛍光性ナノ粒子または前記粒子コアの精製の後に再度行われることを特徴とする請求項1に記載の製造方法。
【請求項3】
前記精製が、室温下にて行われることを特徴とする請求項2に記載の製造方法。
【請求項4】
前記加熱処理が50℃〜200℃の範囲の温度にて行われることを特徴とする請求項1〜3のいずれか1項に記載の製造方法。
【請求項5】
前記精製の前に行われる前記加熱処理が、30分間〜8時間にわたって行われることを特徴とする請求項2〜4のいずれか1項に記載の製造方法。
【請求項6】
第一の金属成分が、Cd、Zn、Ag、InおよびSからなる群より選択されることを特徴とする請求項1〜5のいずれか1項に記載の製造方法。
【請求項7】
第二の金属成分が、TeおよびSからなる群より選択されることを特徴とする請求項1〜6のいずれか1項に記載の製造方法。
【請求項8】
第三の金属成分が、Cd、SおよびZnからなる群より選択されることを特徴とする請求項1〜7のいずれか1項に記載の製造方法。
【請求項9】
第一溶液が、チオール安定化剤を含んでいることを特徴とする請求項1〜8のいずれか1項に記載の製造方法。
【請求項10】
チオール安定化剤が、チオアセトアミド、3−メルカプトプロピオン酸(3−MPA)、チオグリコール酸(TGA)、4−メルカプトブタン酸、システインまたはシスタミンのチオ化合物およびその塩類からなる群より選択されることを特徴とする請求項9に記載の製造方法。
【請求項11】
請求項1〜10のいずれか1項に記載の製造方法によって製造されたことを特徴とする糖鎖固定化蛍光性ナノ粒子。
【請求項12】
請求項11に記載の糖鎖固定化蛍光性ナノ粒子の表面上に抗体が固定化されていることを特徴とする抗体固定化蛍光性ナノ粒子。
【請求項13】
加熱処理された蛍光性ナノ粒子を含む溶液と、還元剤処理されたリガンド複合体を混和する工程を包含し、
上記蛍光性ナノ粒子は、第一および第二の金属成分からなる粒子コアが第一および第三の金属成分からなる層によって被覆されており、
上記リガンド複合体は、主鎖に炭化水素鎖または炭化水素誘導鎖を備えたリンカー化合物からなり、
上記リンカー化合物の主鎖は、その一端に抗体を直接的または間接的に結合し得る反応基を有し、その他端に硫黄原子を含む炭化水素構造を備えており、
上記工程によって上記炭化水素構造が上記層に固定化されて、抗体固定化蛍光性ナノ粒子の分散液が得られることを特徴とする抗体固定化蛍光性ナノ粒子の製造方法。
【請求項14】
請求項13に記載の製造方法によって製造されたことを特徴とする抗体固定化蛍光性ナノ粒子。
【請求項15】
請求項11に記載の糖鎖固定化蛍光性ナノ粒子あるいは請求項12または14に記載の抗体固定化蛍光性ナノ粒子を含んでいることを特徴とする生体組織を標識するための組成物。
【請求項16】
相互作用検出剤、細胞標識剤、細胞イメージング剤、生体イメージング剤、ウイルス標識剤または近赤外増感剤であることを特徴とする請求項15に記載の組成物。
【図4】

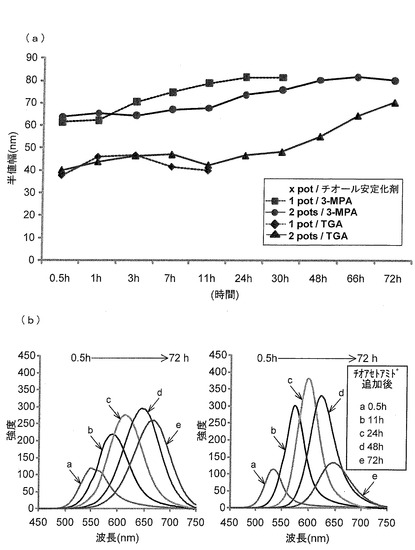
【図6】
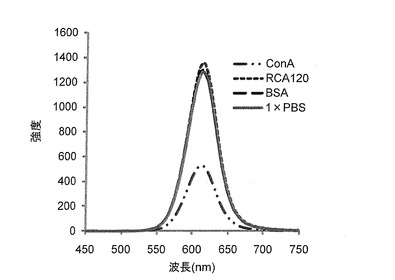
【図7】
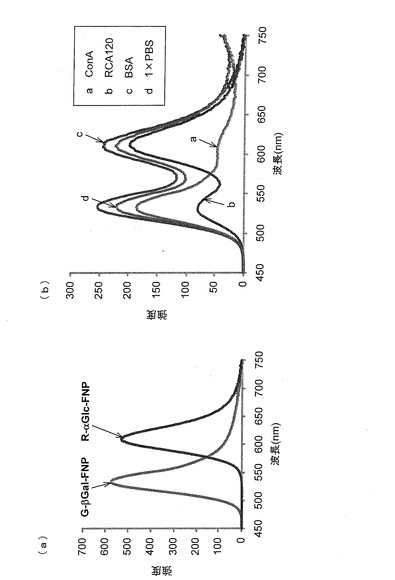
【図8】
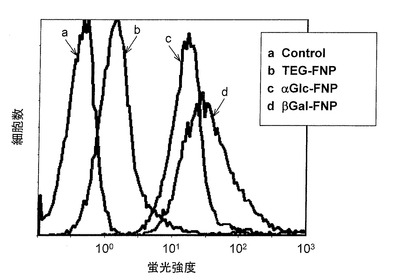
【図10】
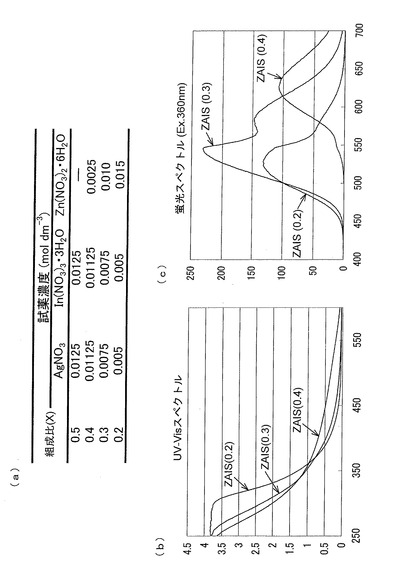
【図11】
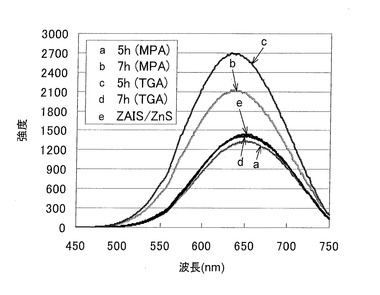
【図12】
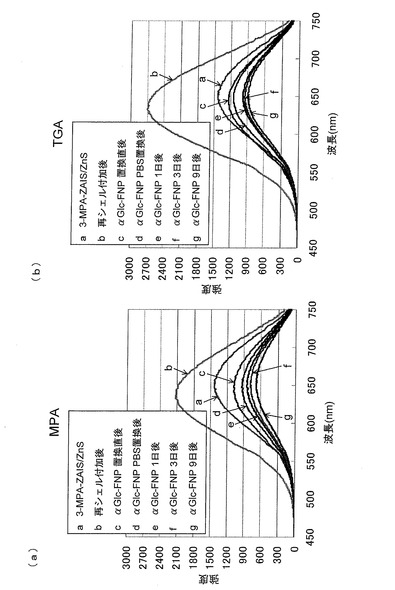
【図16】
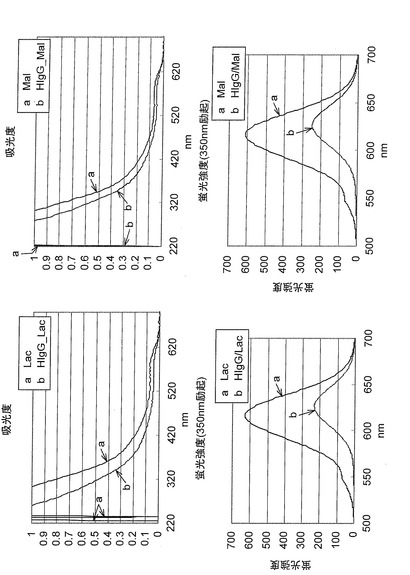
【図18】
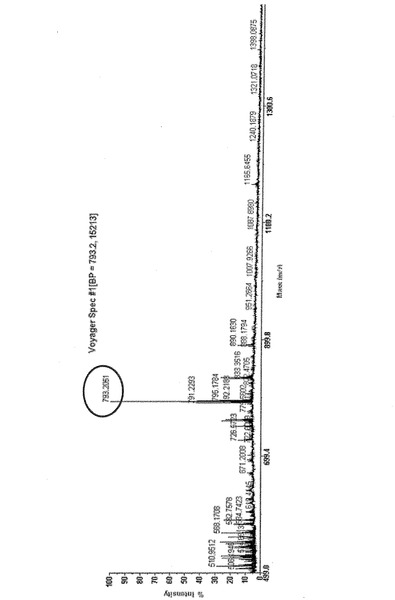
【図20】
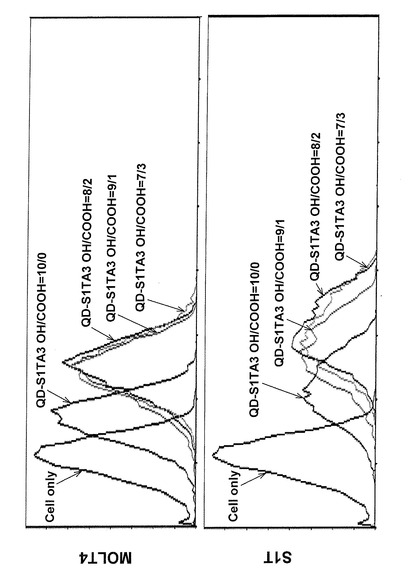
【図1】
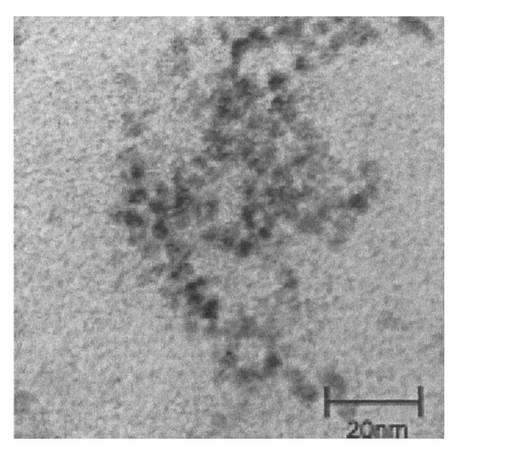
【図2】

【図3】
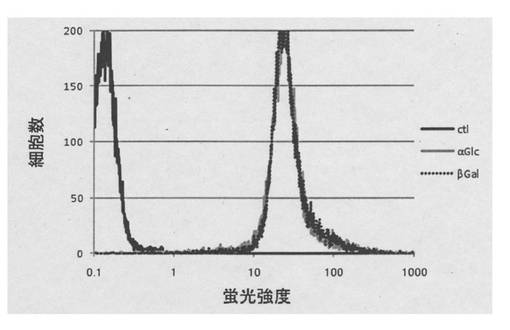
【図5】
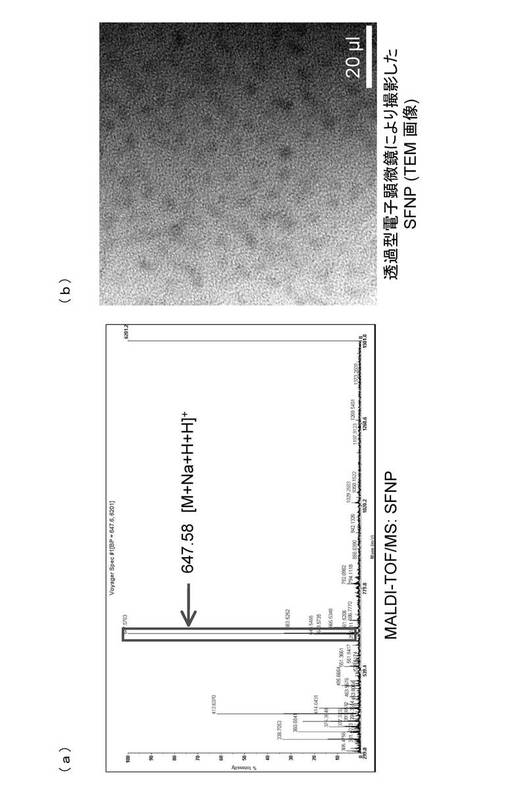
【図9】
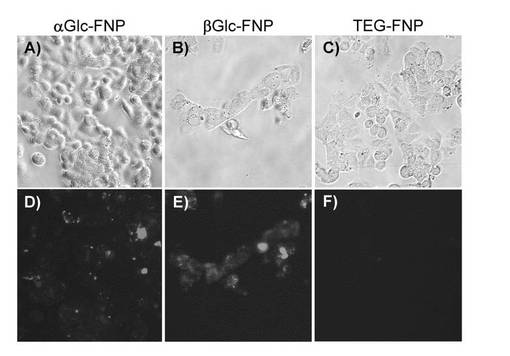
【図13】
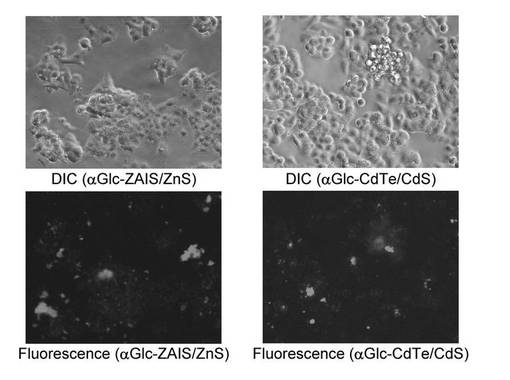
【図14】
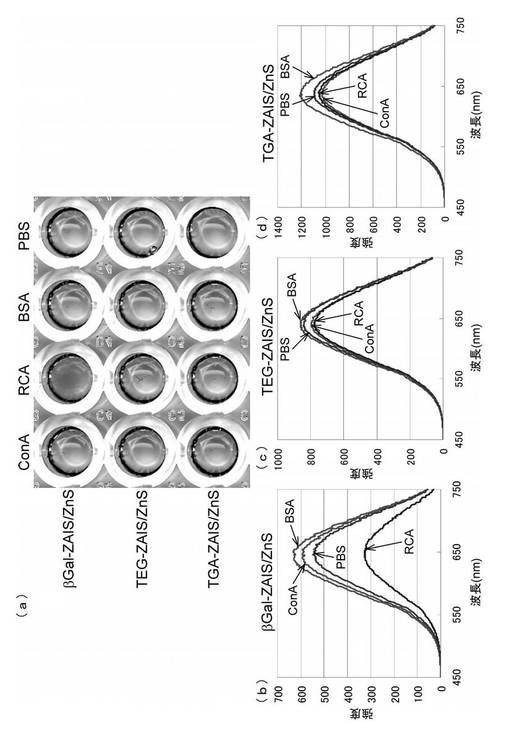
【図15】
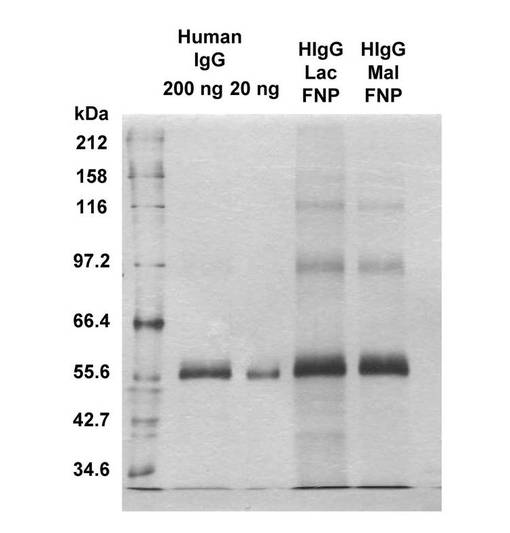
【図17】
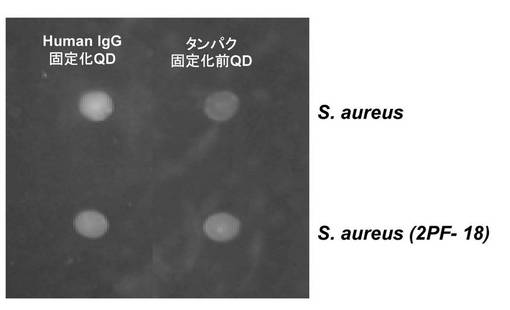
【図19】
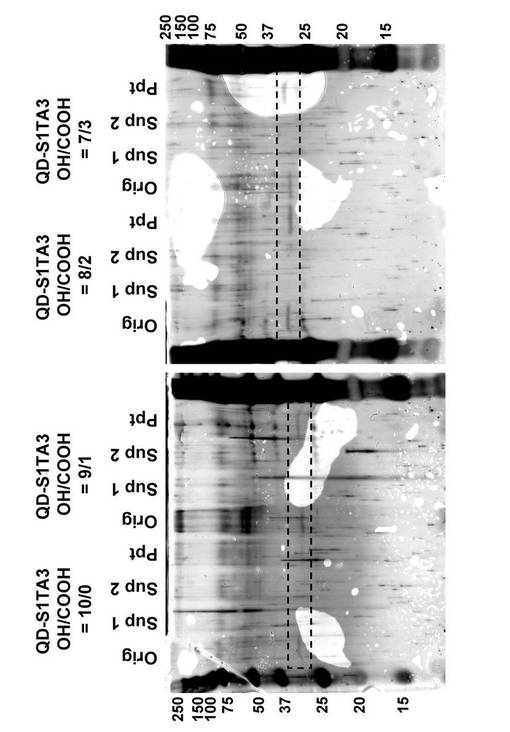
【図6】
【図7】
【図8】
【図10】
【図11】
【図12】
【図16】
【図18】
【図20】
【図1】
【図2】
【図3】
【図5】
【図9】
【図13】
【図14】
【図15】
【図17】
【図19】
【公開番号】特開2011−209282(P2011−209282A)
【公開日】平成23年10月20日(2011.10.20)
【国際特許分類】
【出願番号】特願2011−53453(P2011−53453)
【出願日】平成23年3月10日(2011.3.10)
【出願人】(504258527)国立大学法人 鹿児島大学 (284)
【出願人】(307011381)株式会社スディックスバイオテック (4)
【Fターム(参考)】
【公開日】平成23年10月20日(2011.10.20)
【国際特許分類】
【出願日】平成23年3月10日(2011.3.10)
【出願人】(504258527)国立大学法人 鹿児島大学 (284)
【出願人】(307011381)株式会社スディックスバイオテック (4)
【Fターム(参考)】
[ Back to top ]