
糖鎖構造解析方法
【課題】構造未知の糖鎖についてCID−MSn測定を行い、得られたデータをすでに取得されている参照データと比較することにより該糖鎖の構造解析を行う方法において、多量のサンプルを準備して多数のデータを取得する必要がない方法を提供すること。
【解決手段】(a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求め、(c)該相関と、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を比較することを特徴とする糖鎖構造解析方法。
【解決手段】(a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求め、(c)該相関と、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を比較することを特徴とする糖鎖構造解析方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、構造未知の糖鎖についてCID−MSn測定を行い、得られたデータをすでに取得されている参照データと比較することにより該糖鎖の構造解析を行う方法に関する。
【背景技術】
【0002】
糖鎖は主に細胞表面に糖タンパク質や糖脂質の一部として存在し、発生、分化誘導、受精、免疫、癌化、感染症等、様々な生命現象に深く関与している。このように多彩な機能を有する糖鎖の研究は近年盛んになっている。また、糖鎖は、タンパク質の翻訳後修飾の重要な部分を占め、今後の大きな研究対象である。糖タンパク質や糖脂質の機能は糖鎖により制御される場合があり、糖鎖の構造解析を極微量において達成する技術が必要である。しかし、糖鎖は生物工学的手法により増幅ができない分子種であるため、微量の物質のみで構造解析を達成することを可能とする新たな構造解析技術の開発が必要である。タンパク質の配列はゲノム配列が判明している場合、対応するタンパク質のアミノ酸配列も取得できるため、目的のタンパク質のアミノ酸配列は質量分析(例えば、MS/MS法)により解析することができる。しかし、被修飾タンパク質の配列解析技術については今後の課題として残されている。特に、糖鎖によるタンパク質の修飾は極めて大きな分子の多様性を生み出している。したがって、糖鎖の構造解析技術の開発は必要不可欠である。
【0003】
糖鎖は、核酸やタンパク質とは異なり配列以外の要因による構造異性体群を形成している。この理由の基本は、糖鎖を形成する単糖には反応点となる水酸基が複数存在するため結合位置異性体を形成する性質を有し、かつ、単糖間の結合の際にはアノメリック位の立体異性によるアノマー異性体を形成する性質を有しているためである。単糖間の結合は、生物体内においては糖鎖の合成に関わる酵素群の連続反応により行われるため、必ずしも組み合わせの原理に基づく糖鎖群を形成しているわけではない。しかし、酵素反応は副反応を伴うことが知られており、このような現象の生物における意味は解明されておらず、このような場合には遺伝情報によることのない全く新しい概念に基づく構造解析法が必要である。もちろん、遺伝情報が解明されていない、また、糖鎖の生合成経路が解明されていない生物種における糖鎖構造の解析についても新しい構造解析法が必要である。
【0004】
現在用いられている糖鎖の構造解析技術としては、(1)核磁気共鳴分光法、(2)質量分析法、(3)多次元クロマトグラフィーによるマッピング法(非特許文献1を参照)、(4)加水分解酵素による特異的部分加水分解法(非特許文献2を参照)、(5)レクチンによるマッピング法(非特許文献2を参照)、及び(6)メチル化−加水分解-ガスクロマトグラフィーによる組成分析法(非特許文献2を参照)をあげることができ、通常はこれらを組み合わせて糖鎖の解析が行われている。
【0005】
上記の中でも特に有効な糖鎖の解析手段としては、核磁気共鳴分光法及び質量分析法をあげることができるが、前者の問題点は解析に必要な量がμg以上であることであり、後者の問題点は立体異性体の解析が不可能なことである。これらの方法を含めいずれの解析法を用いるにせよ、糖鎖の構造解析は、得られた天然糖鎖構造の構造解析を直接行わなければならない(非特許文献3を参照)。
【0006】
一方、異なる糖鎖を質量分析することにより、その結果が異なることが示されている。例えば、ステロイドの水酸基の立体異性体のCID−MS/MS測定によるフラグメント化を行うと、各フラグメント強度が異なること(非特許文献4を参照)や、合成された硫酸化糖鎖の構造異性体のCID−MS/MS測定を行って、特定イオンの強度を測定すると、その値に差があること(非特許文献5を参照)が示されている。しかし、これらはいずれも具体的に糖鎖の構造解析方法を示すものではなかった。
【0007】
近年、この情報を基にスペクトルの比較による構造解析法の研究が報告されている(非特許文献6〜8を参照)が、従来知られている構造の特定を行おうとするものであり新規物質の構造解析を可能とするものではなかった。
【0008】
また、特許文献1には、(a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、得られた特定のm/zのプロダクトイオンの総イオンカウント数とCIDエネルギーとの関係を示すCIDエネルギー依存曲線を作成し、(c)該CIDエネルギー依存曲線と、構造既知の参照糖鎖から得られた、上記フラグメントイオンと同一のm/zのフラグメントイオンをCID−MS/MS測定することにより得られた、上記プロダクトイオンと同一のm/zのプロダクトイオンのCIDエネルギー依存曲線とを比較することを特徴とする糖鎖構造解析方法が記載されている。この方法は、天然に存在する構造未知の糖鎖を、置換基が異なるものや立体異性体等も含めその構造、またはその一部を決定又は推定することができ、また極微量の解析が行える点で有用であるが、多数のデータを取得する必要があり、多量のサンプルを準備する必要があった。
【0009】
【非特許文献1】Royle, L. et al., Anal. Biochem., 2002,304,70-90
【非特許文献2】Chaplin, M.F et al., Carbohydrate Analysis, A Practical Approach, IRL Press, Oxford,1994
【非特許文献3】Dell, A., Adv. Carbohydr. Chem. Biochem. 1987, 45, 19-72
【非特許文献4】Faretto, D. et al., Mass Spectrom., 1991,5,240-244
【非特許文献5】Kurono, S. et al., J. Mass Spectrom., 1998, 33,35-44
【非特許文献6】Y. Takegawa, K. Deguchi, S. Ito, S. Yoshioka, A. Sano, et al., Anal. Chem. 76, 7294-7303 (2004).
【非特許文献7】A. Kameyama, N. Kikuchi, S. Nakaya, H. Ito, T. Sato, et al., Anal. Chem. 77, 4719-4725 (2005).
【非特許文献8】D. Ashline, S. Singh, A. Hanneman, V. N. Reinhold, Anal. Chem. 77, 6250-6262 (2005).
【特許文献1】特開2006−145519号公報
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明の目的は、構造未知の糖鎖についてCID−MSn測定を行い、得られたデータをすでに取得されている参照データと比較することにより該糖鎖の構造解析を行う方法において、多量のサンプルを準備して多数のデータを取得する必要がない方法を提供することである。
【課題を解決するための手段】
【0011】
本発明者らは、上記課題を達成するために鋭意検討を進めた結果、目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求め、該相関と、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を比較することによって糖鎖の構造を同定できることを見出し、本発明を完成するに至った。
【0012】
すなわち本発明によれば、以下の発明が提供される。
(1) (a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求め、(c)該相関と、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を比較することを特徴とする糖鎖構造解析方法。
【0013】
(2) (a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とをプロットしてグラフを作成し、(c)該グラフと、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率をプロットして得られたグラフとを比較することを特徴とする糖鎖構造解析方法。
【0014】
(3) 工程(b)において、少なくとも1回以上のCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とを少なくとも1点以上プロットする、(2)に記載の糖鎖構造解析方法。
【0015】
(4) 工程(b)において、少なくとも2以上の異なるCIDエネルギーを用いてCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とを少なくとも2点以上プロットする、(2)又は(3)に記載の糖鎖構造解析方法。
(5) 工程(b)において作成したグラフを直線で近似することを特徴とする、(2)から(4)の何れかに記載の糖鎖構造解析方法。
【0016】
(6) 工程(b)において作成したグラフを直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率を算出し;工程(c)において構造既知の参照糖鎖について得られたグラフも直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率を算出し;上記工程(b)及び工程(c)において算出した百分率を比較することを特徴とする、(5)に記載の糖鎖構造解析方法。
【0017】
(7) 工程(b)において作成したグラフを直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率と、上記の直線の傾きを算出し;工程(c)において構造既知の参照糖鎖について得られたグラフも直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率と、上記直線の傾きを算出し;上記工程(b)及び工程(c)において算出した百分率及び傾きを比較することを特徴とする、(5)又は(6)に記載の糖鎖構造解析方法。
【発明の効果】
【0018】
本発明の糖鎖解析方法は、構造未知の糖鎖についてCID−MSn測定を行ない、得られたCIDエネルギー依存曲線を構造既知の糖鎖から得られた参照データと比較することにより構造決定を行う方法である。特に本発明の糖鎖解析方法は、多量のサンプルを準備して多数のデータを取得する必要がないという利点を有している。本発明の方法は、天然に存在する構造未知の糖鎖を、置換基が異なるものや立体異性体等も含めその構造、またはその一部を決定又は推定することができ、また極微量の解析が行える点で非常に有用である。また該方法は、糖鎖の構造解析という次世代生命科学研究の基盤技術となるのみならず、糖鎖の異常を伴う疾病や感染症を引き起こすきっかけとなる糖鎖レセプターの特定等、診断医療に貢献する技術となるものである。さらに、本発明の純度検定方法により、検体の詳細なデータ解析をおこなう以前にその必要性を簡便に判定することが可能となる。
【0019】
本発明においては、プレカーサーイオンとプロダクトイオンの比が衝突エネルギーによらず一定であるということが判明した。本発明によれば、(1)直線近似が可能なため、スペクトル間の一致による構造決定が容易であり、(2)特定の衝突エネルギーで測定したMS/MSスペクトルを如何なる衝突エネルギーで測定したスペクトルとも定量的に比較することが可能であり、(3)従来より少ない測定回数で定量的データを取得することができるようになり、(4)MS/MS測定時における衝突エネルギーの検討が不要なため測定の自動化が可能であり、(5)イメージングマスにおけるMS/MS測定に利用でき、(5)質量分析法による構造解析に重要なERMS法を必要最小限のサンプル消費量(従来のMS/MS法と同程度)で達成可能となるという効果が得られる。本発明の方法は、特に溶液反応(化学反応、酵素反応)のモニタリング(特に生成物の異性化を伴う場合)に有効である。
【0020】
グリコシダーゼは、糖鎖、あるいは、配糖体を基質とし加水分解する酵素であり、エキソ型とエンド型に分類される。それらは、それぞれ立体保持型と立体反転型に分類される。生成物はアノマー遊離の単糖、あるいは、オリゴ糖であり水溶液中でα-とβ-アノマー(ヘミアセタール)は開環したアルデヒド体を経由して平衡に至る。立体保持型と立体反転型の酵素においては、その過程において初期生成物が異なる。一方、これらの酵素は、任意のアルコールを添加しておくとこれを受容体として新たなグリコシド化合物を生成することが知られている。この過程は、トランスグリコシル化と呼ばれる。本発明による糖鎖解析方法は、上記したような酵素反応の解析に応用することもできる。
【発明を実施するための最良の形態】
【0021】
以下、本発明を更に詳細に説明するが、以下の構成要件の説明は、本発明の実施態様の代表例であり、本発明はこれらの内容のみに特定されるものではない。
以下の説明において、「CID−MSn測定」とは、CID(Collision induced dissociation:衝突誘起解離)による多段マススペクトロメトリーの取得を行うことを示し、「フラグメントイオン」とは、上記CID−MSn測定や物理化学的分解等により得られる各m/zを有するイオンを示し、「m/z」とは、質量数(m)と電荷(z)の比を示し、「プロダクトイオン」とは、フラグメントイオンを上記CID−MSn測定することにより得られる各m/zを有するフラグメントイオンを示し、「総イオンカウント数」とは、各m/zを有するすべてのフラグメントイオンのイオン強度の総和を示し、また、「CIDエネルギー」とは、CIDを起こすときに加えるエネルギーを一般的に示し、実際にはイオンを振動させるためのある周波数の電場の電圧を示す。さらに、「構造解析方法」とは、構造決定方法および/または構造推定方法を意味する。
【0022】
(1)構造解析方法の概略
本明は、構造未知の糖鎖(本明細書中では、これを「目的糖鎖」と称することがある)を特定のm/zのフラグメントイオン(以下、これを「プレカーサーイオン」又は「プレカーサーフラグメントイオン」と称することがある)が得られるまでCID−MSn測定を行い、得られたプレカーサーフラグメントイオンについてさらにCID−MS/MS測定を行う。これにより得られる、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求める。この際、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とをプロットしてグラフを作成してもよい。また、上記で作成したグラフは直線で近似してもよい。
【0023】
一方、参照データとして、構造既知の糖鎖(本明細書中では、これを「参照糖鎖」と称することがある)またはそのライブラリーについて、特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、得られたフラグメントイオンについてさらにCID−MS/MS測定を行う。これにより得られる構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求める。この際、プレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関は、直線で近似してもよい。
【0024】
かくして得られた目的糖鎖のプレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関(目的糖鎖のデータ)と、参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関(参照データ)とを比較し、目的糖鎖のデータと一致する参照データがあった場合に、その参照データが得られた参照糖鎖と目的糖鎖の一部の構造が一致すると判断することができる。目的糖鎖のデータと参照データとの比較は、作成したグラフ同士を、パターン認識ソフトを用いて比較することができる。
【0025】
作成したグラフを直線で近似する場合は、直線同士を直接比較してもよいし、あるいは直線の切片の値を求め、それらを比較してもよいし、直線の切片の値と傾きを求め、それらを比較してもよい。ここで言う直線の切片の値とは、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率に相当する値である。
【0026】
また、目的糖鎖のデータと参照データが一致しない場合でも、目的糖鎖のパラメータが参照糖鎖のパラメータの統計的解析結果の範囲内であれば、部分構造を推定することができる。本発明には、このような構造の推定を行う方法も含まれる。
【0027】
このようにして目的糖鎖の一部の構造が決定または推定された場合、これらを目的糖鎖の生合成経路を参照する等して全体の構造を決定または推定していくことができる。
【0028】
(2)目的プレカーサーフラグメントイオンの調製
本発明に用いられる目的糖鎖は、以下に説明する方法により分解するなどしてCID−MSn測定を行い得るものであればいかなるものであってもよい。また、構造解析を目的として本発明の方法に供するものであるので、その全部または一部の構造が未知のものが好ましい。目的糖鎖は、生体組織又は細胞等から得られたものでもよいし、合成されたタンパク質に結合したものから得られたものでもよく、またそれを酸加水分解又は酵素分解したり、さらにHPLC等で分離精製したもの等を用いることができる。また、化学合成された糖鎖を用いることもできる。
【0029】
本発明では、目的糖鎖と参照糖鎖から得られる同一のm/zを有するプレカーサーイオンについて、CID−MSn測定して得られるプロダクトイオン強度を比較するので、参照データを誘導するプレカーサーイオン(以下、これを「参照プレカーサーフラグメントイオン」と称することがある)と同一のm/zを有するフラグメントイオン(以下、これを「目的プレカーサーフラグメントイオン」と称することがある)が得られるまで何らかの方法で分解する必要がある。分解の方法は、それ自体公知の常法を用いることができるが、例えば、酸加水分解、酵素分解等により得られる糖鎖を液体クロマトグラフィー、キャピラリー電気泳動、ゲル電気泳動等により分離精製して用いることもでき、あるいは後述するCID−MSn測定等により得られる任意のフラグメントイオンをm/zにより分離して用いることができる。参照糖鎖として3糖ライブラリーを用いる場合は、目的糖鎖を1〜3糖に分解することが望ましい。また、目的糖鎖が置換基を有する場合、これを除去しても、また参照データに置換基を含むものがあれば付いたままでも用いることができる。最終的に得られた目的糖鎖分解物は、それぞれ質量分析することによりm/zを確認し、この質量とその他の情報、例えば生合成経路などの情報から配列を決定することもできるが、本発明の方法は、ここで決定することのできなかったフラグメントイオンについて用いることができる。
【0030】
(3)目的プレカーサーフラグメントイオンのCID−MSn測定
上記で得られた目的プレカーサーフラグメントイオンは、これをCID−MSn測定する(CID−MSn測定により得られた目的プレカーサーフラグメントの場合は、これをCID−MS/MS測定する)。
【0031】
イオン化法としては、FAB(高速原子衝撃法)、CI(化学イオン化法)、ESI(エレクトロスプレーイオン化法)、MALDI(マトリクス支援レーザー脱離イオン化法)、APCI(大気圧化学イオン化法)等が用いられるが、本発明においてはサンプル調製が容易で、かつ、マトリックス由来の夾雑イオンの影響がないESI法を用いることが好ましい。しかし、上記CID−MSn測定が可能であればイオン化法はこれらに限定されるものではない。また、ESI法にはマイクロスプレー法とナノスプレー法があるが、サンプル使用量の面から本発明においてはナノスプレー法が好ましく用いられる。
【0032】
上記目的糖鎖またはその分解物をイオン化する場合、微液滴を真空下溶媒を蒸発させることでイオン化するため水/メタノールあるいは水/アセトニトリル(1:1)を溶媒として用いることができるが、物質の性質に従い選択が可能でありこれに限られることはない。目的糖鎖またはその分解物は、この溶媒に対して0.01〜100nmol/ml、より好ましくは0.5〜5nmol/ml、さらに好ましくは1nmol/mlの濃度に溶解することが好ましい。
【0033】
また、CID−MSn測定を行なう装置は、これが可能であれば、機種を問うものではないが、本発明においては、CID−MSn測定が可能な四重極イオントラップ型質量分析計を用いることが好ましい。このような装置としては、具体的には例えば、esquire 3000 plus(ブルカーダルトニクス社製)、LCQ DECA(サーモフィニガン社製)、AXIMA QIT(島津社製)、LCMS-IT-TOF(島津社製)等が挙げられる。測定方法としては、各プレカーサーフラグメントイオンから解離するプロダクトイオンのイオン強度が測定できる方法であればいかなるものでもよいが、具体的には、例えば、熱キャピラリー温度20〜365℃、キャピラリー電圧0〜6.0kV、CIDエネルギーが0〜25V、ポジティブモードまたはネガティブモードのいずれか等で測定することができる。ポジティブモードを用いる場合には、プロトン、ナトリウム、カリウム、リチウム、カルシウム、アンモニウム等の陽イオンを選択して用いることができる。
【0034】
上述のCID−MS/MS測定により得られたデータに基づいて、プレカーサーイオンと、特定のm/zのプロダクトイオンとの比率をプロットしてプレカーサーイオンとプロダクトイオンとの比率を求める。
【0035】
<<測定するコリジョンエネルギーの決定法>>
<標準品の分解エネルギーの測定>
マルトオリゴ糖([Glc]n:n= 3-12)などの標準品を用いERMSを行いプレカーサーイオンの半分が分解するエネルギー(V50)を各々のプレカーサーについて取得し、これを質量に対する分解エネルギー(V50)の“ものさし”と位置づける。
<ERMS測定するコリジョンエネルギーの中心の決定>
MSn測定を行い当該するイオンが得られた段階でERMS測定を行う。このとき、当該イオンのm/zを上述の“ものさし”に参照して該当するV50(コリジョンエネルギーの中心)を求める。
【0036】
<ERMS相関法の測定>
上記の様にして求めたコリジョンエネルギーの中心、および、コリジョンエネルギーの中心から±0.1, 0.2, 0.3Vのコリジョンエネルギーを選択し、これら7つの設定条件からMS/MSを最低1回、好ましくは、7回おこない、得られるフラグメントイオンの強度を上述の算式により標準化(相対値化)し、プレカーサーイオンをX軸としプロダクトイオンをY軸としてプロットする。その後、直線近似を行ってY切片(通常ERMSのプラトー値)を求める。
【0037】
<ERMS相関からERMSへの変換>
ERMS相関において使用したデータポイント(各々のコリジョンエネルギーを表す)から求めた近似曲線上へ垂線をひく。この点を理想のデータとし、スプライン補完をおこなって理想データを求める。求めた理想データは元々の実験データに対応するコリジョンエネルギーからスプライン補間法により与えられているので、各々の理想データポイントはコリジョンエネルギーの項目を有している。求まった一連の理想データ群をコリジョンエネルギーに対するプレカーサーイオン、及び、各フラグメントイオンの相対強度としてプロットし直すと理想化された通常のERMSを与える。このグラフからV50などのプレカーサーイオンの分解に要するエネルギー該当項目を正確に導くことができる。
【0038】
(4)参照プレカーサーフラグメントイオンの調製
参照データを取得する目的で、参照糖鎖について上記と同様にプレカーサーフラグメントイオンを調製する。参照糖鎖は、目的糖鎖の全部または一部の構造を本発明の方法を用いて決定し得るものであればいかなるものであってもよい。具体的には、目的糖鎖またはその一部と構造が一致する可能性があり、構造が明らかであるものが用いられる。このような参照糖鎖は、天然のものでも合成されたものでもよく、また置換基がついていてもよい。糖鎖の長さは、1〜13個の糖が連結したものが用いられる。
【0039】
この参照糖鎖として、ある特定の集団であるライブラリーを用いることによれば、より網羅的、組織的に目的糖鎖の構造決定を行なうことができる。このようなライブラリーとしては、例えば、2〜25個の糖が連結した構造を有しており、ライブラリーを構成する各糖鎖は、天然糖鎖を構成する全ての種類の単糖の順列組み合わせの配列を有するもので、かつ立体異性も含むもの等が好ましく用いられる。また、非天然の単糖を含んでいてもよい。
【0040】
また、糖鎖構造を一般化すれば、非還元末端糖、還元末端糖、およびそれらの中間に存在する糖からなるポリマー性糖化合物ということができ、このポリマーの最小単位は3糖が連結した糖鎖である。このような3糖からなる天然糖鎖を構成する全ての種類の単糖の順列組み合わせの配列を有するもので、かつ立体異性も含む3糖ライブラリーは、生体に存在するあらゆる直鎖構造を有する糖鎖の部分構造を含んでいるため、本発明の参照糖鎖のライブラリーとしてより好ましい。
【0041】
上記の天然糖鎖を構成する単糖とは、例えば、グルコース、ガラクトース、マンノース、N-アセチルグルコサミン、N-アセチルガラクトサミン、キシロース、シアル酸、グルクロン酸、フコース等が挙げられる。また、これらは修飾基を有していても良い。
【0042】
参照糖鎖および参照糖鎖ライブラリーは、これを化学合成することが好ましい。合成の方法としては、通常用いられるそれ自体公知の方法を用いることができる。化学合成には、上述のような単糖各々を適当に保護した合成ユニットが必要である。さらに、立体異性体から成る糖鎖ライブラリーを効率良く合成するためには、立体選択的な合成ではα−グリコシドを得るためには隣接基関与のない保護基、例えばベンジル基(Bn)を、β−グリコシドを得るためには、隣接基関与のある保護基、例えばアセチル基等を用いる必要がある。これらの立体制御のための保護基の他に通して用いる保護基を用いる必要性から、単糖合成ユニット数は多くなり、合成経路の複雑化につながる。従って、2位の保護基は隣接基関与のない保護基とし、合成経路の簡略化を、例えばPCT/JP2004/009523号明細書に記載の方法等に従って行なうことが好ましい。
【0043】
かくして得られた参照糖鎖または参照糖鎖ライブラリーは、これを目的糖鎖と同様に特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行なう。特定のm/zのフラグメントイオンとは、目的糖鎖のプレカーサーフラグメントイオンが有するm/zと同一のものであればいかなるものでもよい。また、参照糖鎖がライブラリーの場合には、該ライブラリーの各構成要素から得られる何れの解裂しえるフラグメントイオンでもよく、全ての解裂し得るフラグメントイオンでもよい。
【0044】
(5)参照プレカーサーフラグメントイオンのCID−MSn測定
上記(4)で取得された参照プレカーサーフラグメントイオンは、これを目的プレカーサーフラグメントイオンと同様にCID−MSn測定する(CID−MSn測定により取得された参照プレカーサーフラグメントの場合は、これをCID−MS/MS測定する)。イオン化法、溶媒、糖鎖の調製、測定装置などは全て目的糖鎖およびプレカーサーフラグメントイオンと同様のものを用いることができる。
【0045】
上述のCID−MS/MS測定により得られたデータに基づいて、プレカーサーイオンと、特定のm/zのプロダクトイオンとの比率をプロットしてプレカーサーイオンとプロダクトイオンとの比率を求める。
【0046】
(6)目的糖鎖の構造決定
目的プレカーサーフラグメントイオンから得られたプレカーサーイオンとプロダクトイオンとの比率と、これに対応する参照データが一致した場合、参照データが誘導されたもとの参照プレカーサーフラグメントイオンの構造と目的プレカーサーフラグメントイオンの構造が同じであると決定することができる。ここで、それぞれのデータが一致するとは、プレカーサーイオンの百分率が0%の場合における、特定のm/zのプロダクトイオンの百分率値の差が±2%以内であることを示す。また、目的プレカーサーフラグメントイオンに置換基が結合していた場合には、置換基が結合した参照プレカーサーフラグメントイオンから得られた参照データと比較して、一致した場合には、同一の置換基を有していると判断することができる。このとき、Z検定を判断基準とすることもできる。
【0047】
かくして本発明の方法により目的糖鎖の一部が決定された場合、これらの全配列を決定するために、例えば、生合成経路に当てはまるものを参照してつなぎ合わせたり、初めに目的プレカーサーフラグメントイオンを産生するために分解した場合には、その分解の方法から再構築する等の方法が用いられる。
【実施例】
【0048】
以下、実施例を挙げて本発明を詳細に説明するが、本発明の範囲はこれらの実施例により限定されるものではない。
【0049】
(A)材料及び方法
(a)合成
実施例で使用した化合物は、以下のスキーム(化合物S6−S8の合成)に従って合成した。
【0050】
【化1】

【0051】
Bnはベンジル、Phはフェニル、Bocはt-ブトキシカルボニル、NISはN-ヨードスクシンイミド、TfOHはトリフルオロメタンスルホン酸、AW-300はモレキュラーシーブAW-300、DCEはジクロロエタン、Acはアセチル、Meはメチル、Etはエチル、HEPESは4-(2-ヒドロキシエチル)ピペラジン-1-エタンスルホン酸、APはアルカリホスファターゼ、KPB bufferはリン酸カリウム緩衝液、DMFはN,N-ジメチルホルムアミドを示す。
【0052】
(a−1)一般的方法
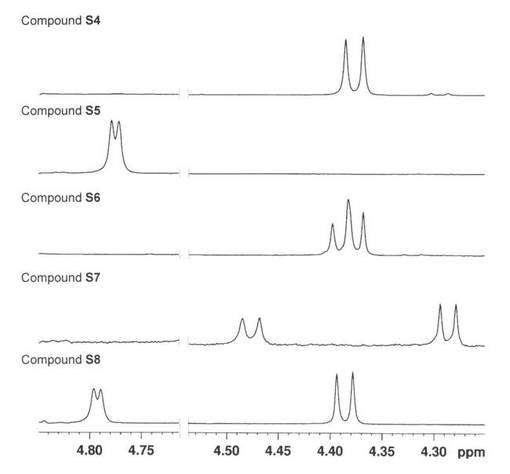
薄層クロマトグラフィ(TLC)は、Kieselgel 60 F254 (E. Merck) プレートで行い、1% Ce(SO4)2-1.5% (NH4)6MoO24・4H2O-10% H2SO4 で焦がすことによって検出した。シリカゲルカラムクロマトグラフィーは、Wakogel C-300 (Wako Pure Chemical Industries, Ltd.)で行った。 Sep-Pak C18 はWaters Corpから得た。UDP-Gal+2Na 及び pNP-β-Gal はSigma-Aldrich Corpから購入した。アルカリホスファターゼはNacalai Tesque, Incから購入した。β1,4-GalT'ase はCalbiochem Corpから購入した。β1,3-Gal'aseはKatsumi Ajisaka 教授(Niigata University of Pharmacy and Applied Life Sciences)から供与された。1H NMR (500 MHz)スペクトルは、参照としての溶媒ピーク(residual CHD2OD; 3.31 ppm)とともにAVANCE 500スペクトロメーター (Bruker Biospin Inc.)で記録した。1H NMRスペクトルのアノマー領域は図5に示す。
【0053】
(a−2)4-(N-Boc-アミノ)ブチル2-アセタミド-2-デオキシ-β-D-グルコピラノシド(S4)及び4-(N-Boc-アミノ)ブチル2-アセタミド-2-デオキシ-α-D-グルコピラノシド(S5).
DCE (2.0 mL)中のフェニル2-アジド-3,4,6-トリ-O-ベンジル-2-デオキシ-1-チオ-β-D-グルコピラノシド(S1, 120 mg, 0.12 mmol), 4-(N-Boc-アミノ)ブタノール(S2,81 mg, 0.43 mmol) 及びAW-300 (ca. 0.5 g) の混合物を窒素雰囲気下25℃で1時間攪拌した。この混合物に、NIS (72 mg, 0.32 mmol)及びTfOH (2.0μL, 0.21 mmol) を0℃で添加し、1時間攪拌した。反応を飽和NaHCO3 で停止し、混合物をDCMで希釈し、セライトパッドでろ過した。ろ液をNa2S2O4を含む飽和NaHCO3に注ぎ、DCMで抽出し、水で洗浄し、MgSO4上で乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(n-ヘキサン: EtOAc = 5:1-2:1)に供し、4-(N-Boc-アミノ)ブチル2-アジド-3,4,6-トリ-O-ベンジル-2-デオキシ-D-グルコピラノシドS3 (80 mg, 58%, α/β = 4/6)を得た。S3 (80 mg, 0.12 mmol, α/β = 4/6)のEtOAc (1 mL)溶液にPd(OH)2/C (20%, ca. 10 mg), Ac2O (1.0 mL) 及びMeOH (1.0 mL)を添加した。混合物を水素雰囲気下で47時間攪拌し、ろ過し、減圧濃縮して4-(N-Boc-アミノ)ブチルGlcNAc 誘導体S4 (β-グリコシド) 及びS5 (α-グリコシド) (23 mg, 47%, S4/S5 = 1.5)を得た。これらのアノマー異性体をシリカゲルカラムクロマトグラフィー(DCM : MeOH = 19:1-8.5:1)によって分離した。
【0054】
S4: 1H NMR (CD3OD): δ4.38 (d, 1H, J1,2 8.4, Hz, H-1), 3.92-3.87 (m, 1H, OCH2(CH2)2CH2N), 3.88 (dd, 1H, J5,6a 2.0 Hz, J6a,6b 12.0 Hz, H-6a), 3.68 (dd, 1H, J5,6b 5.8 Hz, H-6b), 3.64 (dd, 1H, J2,3 9.2 Hz, H-2), 3.50-3.45 (m, 1H, OCH2(CH2)2CH2N), 3.44 (dd, 1H, J3,4 8.7 Hz, H-3), 3.31 (dd, 1H, J4,5, 9.7 Hz, H-4), 3.26 (ddd, 1H, H-5), 3.04 (t, 2H, J 6.4 Hz, OCH2(CH2)2CH2N), 1.97 (s, 3H, NHAc), 1.58-1.48 (m, 4H, OCH2(CH2)2CH2N), 1.43 (s, 9H, tBu).
S5: 1H NMR (CD3OD): δ4.78 (d, 1H, J1,2 3.5, Hz, H-1), 3.88 (dd, 1H, J2,3 10.7 Hz, H-2), 3.82 (dd, 1H, J5,6a 2.2 Hz, J6a,6b 11.9 Hz, H-6a), 3.76-3.71 (m, 1H, OCH2(CH2)2CH2N), 3.68 (dd, 1H, J5,6b 6.0 Hz, H-6b), 3.67 (t, 1H, J3,4 10.6 Hz, H-3), 3.58 (ddd, 1H, J4,5 9.8 Hz, H-5), 3.42-3.37 (m, 1H, OCH2(CH2)2CH2N), 3.34 (t, 1H, H-4), 3.07 (t, 2H, J 6.6 Hz, OCH2(CH2)2CH2N), 1.99 (s, 3H, NHAc), 1.66-1.53 (m, 4H, OCH2(CH2)2CH2N), 1.43 (s, 9H, tBu).
【0055】
(a−3)4-(N-Boc-アミノ)ブチルβ-ラクトサミニド(S6)
化合物S4 (3.1 mg, 7.9μmol), UDP-Gal+2Na (7.2 mg, 12μmol) 及びアルカリホスファターゼ(20-300 U/mg, 1.0 mg) をHEPES緩衝液(0.5μL, 0.10 M, pH 7.0, 40 mM MnCl2を含有)に溶解した。この混合物に、HEPES緩衝液(0.5μL, 0.10 M, pH 7.0, 40 mM MnCl2を含有)中のβ1,4-GalT'ase (1U, 牛乳由来)を添加し、37℃で20時間インキュベートした。反応混合物をろ過し、Sep-Pak C18に供し、水で洗浄した。4-(N-Boc-アミノ)ブチルグリコシド及びS4を含有する生成物をMeOHで溶出した。混合物をシリカゲルカラムクロマトグラフィー(DCM : MeOH = 9:1-6:4)で精製して、4-(N-Boc-アミノ)ブチルβ-ラクトサミニドS6 (2.1 mg, 36%) を得て、S4 (1.6 mg, 52%)を回収した。
【0056】
S6: 1H NMR (CD3OD): δ4.39 (d, 1H, J1,2 7.5, Hz, H-1), 4.38 (d, 1H, J1',2' 7.5 Hz, H-1'), 3.91 (dd, 1H, J5,6a 2.4 Hz, J6a,6b 12.2 Hz, H-6a), 3.91-3.87 (m, 1H, OCH2(CH2)2CH2N), 3.86 (dd, 1H, J5,6b 4.2 Hz, H-6b), 3.81 (d, 1H, J3',4' 2.9 Hz, H-4'), 3.76 (dd, 1H, J5',6'a 7.6 Hz, J6'a,6'b 11.5 Hz, H-6' a), 3.74-3.69 (m, 1H, H-2), 3.68 (dd, 1H, J5',6'b 4.5 Hz, H-6' b), 3.64-3.57 (m, 3H, H-3,4,5'), 3.54 (dd, 1H, J2',3' 9.7Hz, H-2'), 3.50-3.44 (m, 1H, OCH2(CH2)2CH2N), 3.48 (dd, 1H, H-3'), 3.41-3.37 (m, 1H, H-5), 3.03 (t, 2H, J 6.4 Hz, OCH2(CH2)2CH2N), 1.97 (s, 3H, NHAc), 1.58-1.49 (m, 4H, OCH2(CH2)2CH2N), 1.43 (s, 9H, tBu).
【0057】
(a−4)4-(N-Boc-アミノ)ブチル2-アセタミド-2-デオキシ-3-O-β-D-ガラクトピラノシル-β-D-グルコピラノシド(S7)
化合物S4 (5.0 mg, 13 μmol), pNP-β-Gal (3.8 mg, 13 μmol), リン酸緩衝液 (0.16 μL, 0.10 M, pH 6.0, 0.15 M NaClを含有)、DMF (40 μL)及びβ1,3-Gal'ase (20 μL, 13U/mL)の混合物を37℃で3時間インキュベートした。溶液を蒸発させ、残渣をSep-Pak C18に供し、水で洗浄した。4-(N-Boc-アミノ)ブチルグリコシド及びS4を含む生成物をMeOHで溶出した。混合物をシリカゲルカラムクロマトグラフィー(EtOAc : MeOH = 9:1-2:1 then EtOAc : MeOH : H2O = 12:3:1)で精製して、化合物S7 (0.8 mg, 11%) 及びS4 (3.5 mg, 70%)を得た。
S7: 1H NMR (CD3OD): δ 4.46 (d, 1H, J1,2 8.3, Hz, H-1), 4.27 (d, 1H, J1',2' 7.6 Hz, H-1'), 3.93-3.88 (m, 1H, OCH2(CH2)2CH2N), 3.89 (dd, 1H, J5,6a 1.9 Hz, J6a,6b 11.7 Hz, H-6'a), 3.80 (d, 1H, J3',4' 3.0 Hz, H-4'), 3.76 (dd, 1H, J5,6a 7.7 Hz, J6a'6b 11.5 Hz, H-6a), 3.77-3.65 (m, 4H, H-2,3,6b,6'b), 3.57-3.44 (m, 4H, H-2',3',5', OCH2(CH2)2CH2N), 3.29-3.35 (m, 1H, H-5), 3.04 (t, 2H, J 6.9 Hz, OCH2(CH2)2CH2N), 1.97 (s, 3H, NHAc), 1.59-1.48 (m, 4H, OCH2(CH2)2CH2N), 1.43 (s, 9H, tBu).
【0058】
(a−5)4-(N-Boc-アミノ)ブチル2-アセタミド-2-デオキシ-3-O-β-D-ガラクトピラノシル-α-D-グルコピラノシド(S8)
化合物S5 (6.5 mg, 17μmol), pNP-β-Gal (5.0 mg, 17μmol), リン酸緩衝液(0.16 mL, 0.10 M, pH 6.0, 0.15 M NaClを含有), DMF (40μL) 及びβ1,3-Gal'ase (20μL, 13 U/mL)の混合物を37℃で3時間インキュベートした。溶液を蒸発させ、残渣をSep-Pak C18に供し、水で洗浄した。4-(N-Boc-アミノ)ブチルグリコシド及びS5 を含む生成物をMeOHで溶出した。混合物をシリカゲルカラムクロマトグラフィー(DCM : MeOH = 9:1-7:3)で精製して、化合物S8 (2.8 mg, 30%) 及びS5 (3.6 mg, 55%)を得た。
S8: 1H NMR (CD3OD):δ4.78 (d, 1H, J1,2 3.3 Hz, H-1), 4.37 (d, 1H, J1',2' 7.6 Hz, H-1'), 4.06 (dd, 1H, J2,3 10.6 Hz, H-2), 3.85-3.67 (m, 7H, H-3,4',6a,6'a,6b,6'b, OCH2(CH2)2CH2N), 3.62 (ddd, 1H, J4,5 9.8 Hz, J5,6a 1.9 Hz, J5,6b 5.4 Hz, H-5), 3.56 (dd, 1H, J5',6'a 4.5 Hz, J5',6'b 7.5 Hz, 7.5 Hz, H-5'), 3.53 (dd, 1H, J2',3' 9.5 Hz, H-2'), 3.48-3.41 (m, 3H, H-3',4, OCH2(CH2)2CH2N), 3.07 (t, 2H, J 6.5 Hz, OCH2(CH2)2CH2N), 1.98 (s, 3H, NHAc), 1.68-1.53 (m, 4H, OCH2(CH2)2CH2N), 1.44 (s, 9H, tBu).
【0059】
(b)質量分析の実験方法
(b−1)材料
GD1a 及びGD1bはSeikagaku Biobusiness Corp(東京)から購入した。α-enrichedラクトン及びβ-enriched ラクトンの試料は、SIGMA, St. Louis, MOから得た。質量分析計を用いた解析のために、HPLCグレードの溶媒を使用した。MeOH及び無水ジメチルスルホキシド (Wako Pure Chemical Industries, Ltd, Osaka, Japan)。重水素化したメチルスルホキシド-d6 (NMRグレード) (Acros Organics, Geel, Belgium) をNMR実験に使用した。
【0060】
(b−2)装置及びデータ収集
試料は、エレクトロスプレーインターフェースを連結した四重極イオントラップ質量分析計(Esquire 3000 plus, Bruker Daltonics GmbH, Bremen, Germany)で解析した。無水DMSO (1 pmol/μL)に溶解した試料を、注入(流速120μL/h)によってイオン源に導入した。分析のパラメーターは以下の通りである。(1) "乾燥温度", 250 °C; (2) 噴霧ガス(N2), 10 psi; (3) 乾燥ガス(N2), 4.0 L/min; (4) "スマートフラグ", オフ (5) 走査範囲, m/z 50-600 (ラクトース、合成)及びm/z 50-400 (ラクトース, 市販品); (6) 化合物安定性, 300%; (7) ICC ターゲット, 8000; (8) 最大捕捉時間, 200 ms; (9) 平均, 10 スペクトル; 及び(10) "カットオフ", 対応するプレカーサーイオンの27.6% (合成ラクトース) 27.1% (市販のラクトース)
【0061】
本MSn 実験では、端部キャップrf振幅は、プレカーサーイオンが検出できなくなるまでに0.02-V増大した(全イオン電流の0.9%未満の安定状態)。端部キャップrf振幅のみをCID実験中で調節した。He 圧力は4.86×10-6 mbarとし、CID時間40 msとした。(m−4)個のスペクトルの平均をCID測定のために使用した(m = 15-28: ここで、m は実験中に得たスペクトルの数である)。不正確さを回避するために、rf振幅工程における、定常状態への一過性期間に関連する最初と最後の2個ずつのデータは使用しなかった。[Ii + 1] 及び[Ii + 2](In はフラグメントイオンを示す)を有する同位体ピークは、計算に含めた(「データの取り扱い」も参照)。プロダクトイオンの単離のために、m/z ± 2 (w = 2)を単離し、CID実験に供し、同位体を含めた。
【0062】
(b−3)データの取り扱い
ERMSのグラフを得るために、以下の等式を用いた。イオン"IP"が一連のプロダクトイオンI1, I2, I3,... Iiを生成する場合、各々のイオンについての相対イオン電流を、以下の式で定義した。
【0063】
【化2】
【0064】
式中、relCは、観測されたイオンのうちの所定のイオンのイオン電流(%)を示す。CIiは、 焦点での観測されたイオン電流を示す。CIPは、プレカーサーイオンのイオン電流を示す。計算は、DSUM関数に基づき、考慮すべき同位体範囲(W)(本実験ではW=2)を選ぶようにプログラムした、エクセル(Excel 2000 (Microsoft Co.))を用いて作成したプログラムを用いて行った。
【0065】
(b−4)シグモイド曲線フィッティング
質量分析機の複数のrf振幅(端部キャップ)で得たMSnデータのセットを、Excelを用いて分析した。一定のm/z 値を有するピークのセットを一連のデータとして扱った。個々のシグナルについて全イオン電流について相対強度を各振幅において取得した(式S1)。データをPrism 4ソフトウエア(GraphPad Software, Inc.)を用いて分析した。個々のデータを非線形回帰分析によるボルツマン・シグモイド関数を用いてフィッティングした[式S2 (growth) 及びS3 (decay)].
【化3】
【0066】
式中、(各々、最大応答、最大応答の半値、及び傾斜因子を示す)パラメーター"a", "b", 及び "c"は各曲線から得た。
本実験で使用した一連のデータにおいて、シグモイド曲線及びパラメータは、上記エクセルプログラムから得た全データを処理し、回帰曲線をプロットすることによって得た。
【0067】
(c)変旋光
(c−1)試料の調製
α-及びβ-enrichedラクトースの水溶液(10 mg/mL, 25 ℃)を全実験のために調製し、4時間までの指定時間に0.1 mLずつ分画した。分画は液体窒素を用いて直ちに凍結し、凍結乾燥した。各試料を無水DMSO 及びDMSO-d6 に溶解し、MS 及びNMRで解析した。DMSO を実験のための溶媒として使用した。非プロトン性溶媒はアノマー化を妨げるからである (B. Casu, M. Reggiani, Tetrahedron Lett. 39, 2839-2843 (1964))。
【0068】
(c−2)MSに基づいた測定についての試料の消費
DMSO (1 pmol/μL) 中のα- 及びβ-enrichedラクトースの溶液を、MSに基づいた分析のために使用した。12.3μLを注入条件下で使用した(120μL/h, 6 min)。全量123 pmolの各化合物を実験で消費した。
【0069】
(c−3)NMR 分析
1H NMR (500 MHz)スペクトルを、AVANCE 500分光測定器を用いて記録した。DMSO-d6中のα-ラクトースのβ-ラクトースに対する比率を、各1H NMRスペクトルにおけるアノマー部位(グルコース成分)におけるヒドロキシル部の積分により測定した(図6)。
【0070】
(c−4)旋光度
α- 及びβ-enrichedラクトースの水溶液(10 mg/mL) を迅速に10 cmのセルに注入し、旋光度を示した時間で25℃でSEPA-200旋光度計 (HORIBA, Ltd. Kyoto, Japan)を用いて測定した。
【0071】
(d)酵素反応
(d−1)試薬
ウシ・ラクトシルセラミドをCALBIOCHEM : EMD Biosciences, Inc., La Jolla, CAから購入した。Rhodococcus sp.から組み換えエンドグリコセラミダーゼII [EC 3. 2. 1. 123]をTakara Shuzo Co., Ltd., Shiga, Japanから購入した。
【0072】
(d−2)エンドグリコセラミダーゼ反応
ラクトシルセラミド(50 nmol)を、0.3%のコール酸ナトリウム及び BSA (10μg)を含む50μLの50 mM 酢酸アンモニウム溶液(pH 6.0) に溶解し、2mUの酵素とともに37℃で15又は60分間インキュベートした。試料溶液を、示した反応時間の直後にSep-Pak C18カトリッジカラムで処理し、水で溶出させた画分(4 mL) を回収した。各画分を直ちに凍結乾燥し、DMSO (1mL)に溶解し、ERMSで分析した。
【0073】
(d−3)ナトリウム付加したヘミアセタール分子はCID条件下でアノマー化しないという知見の一般性
ナトリウム付加したヘミアセタールはCID条件下でアノマー化しないという事実は、本実験の重要な基礎である。この現象の一般性を確認するために、β-Gal-(1→4)-β-GlcNAc-O(CH2)4NHBoc (S6), β-Gal-(1→3)-β-GlcNAc-O(CH2)4NHBoc (S7), 及びβ-Gal-(1→3)-α-GlcNAc-O(CH2)4NHBoc (S8)などの一連のBoc-保護されたアミノブチルグリコシドを用いて、上記と同様の実験を行った。これらの化合物は全て、ナトリウムを付加したヘミアセタールイオン種をMS3 段階で与え、これをMS4 及びMS5でERMS分析に供した(図7)。図7に示す通り、[β-Gal-(1→3)-α-GlcNAc-OH+Na]+ (図7A)及び[β-Gal-(1→3)- β-GlcNAc-OH+Na]+ (図7C) のERMSは異なっていたが、MS4 (図7A, C, E) 及びMS5 (図73 C, D, F)について得たERMSは区別できなかった。
【0074】
(B)結果
(B−1)ERMS相関法
本発明者らはナトリウム付加分子とフラグメントの様々なフラグメント化過程について、四重極イオントラップ質量分析計(QIT-MS)を使用して観測してきた。この結果、フラグメント化過程はほとんどの場合S字形のプロフィールであることが判明した。GD1aとGD1bの開裂を例にとると、ERMSは、フラグメント化過程に関する定量的情報のみならず、CIDのエネルギー依存性をも提供する。したがって、開裂に必要な活性化エネルギーは、GD1bについてより小さいことが分かる(図1 C,D)。この情報は、ERMSによってのみ入手可能である。ここで、生成イオンはそれらの前駆体から、互いに独立の関係で、直接生成したように見える。これは、各々の生成物イオンの前駆体イオンに対するERMS相関において強い直線相関が観察されたことから明らかとなった(図1 E,F)。この結果は、印加されるCIDエネルギーとは無関係に、個々のフラグメントイオンがそれぞれにそれらの共通前駆体に由来することを初めて示すものである。これにより2つの重要な点が明らかとなった。すなわち、1)個々のフラグメントイオンの比率は、CIDエネルギーに無関係に一定である、2)個々のフラグメントイオンの比率に関する定量的データ、あるいは、ERMSにおけるプラトー値を一般的なCID実験において使用するサンプル量による実験(データポイント)からERMS相関図におけるy切片から推定できる。
【0075】
本発明者らは遊離糖のアノマー立体異性を決定するためにERMS相関を利用した。また、このために変旋光のプロセスの分析と、グリコシダーゼの作用機序を推定した。ヘミアセタール種の分析には、以下の基準を満たす必要がある。すなわち、1)個々のアノマーを区別する、2)気相でのアノマー化の否定。図2Aは、前駆体1と前駆体2が同じである場合、それらが同じMSnスペクトルを与えなければならないという仮説を示している。ここでの目的は、アノマー化に関する実験のためのセットアップである。前駆体1がα-アノマーであり、かつ、アノマー化が起こるとき、前駆体2はα-とβ-アノマーの混合物となり各々のアノマーから識別可能である可能性がある。この実験のためには、α-、β-各々のアノマーが異なるスペクトルを与えることを確認することが必要である。そこで、前駆体1を一連の生成物イオン(生成物1)を生ずるフラグメンテーション反応にかける、このとき前駆体1と同じm/z値のイオン種(おそらく残留する前駆体)を単離し(前駆体2)、同じ状況の下で再びCIDに供する。前駆体1と2の構造が同一の場合、得られるフラグメント化プロフィールは同じとなる。
【0076】
まず、α-とβ-ヘミアセタール体が判別可能であるかどうか(基準1)について述べる。ヘミアセタール構造以外の結合が開裂する条件下におけるヘミアセタール構造の反応を検討するためラクトース(2糖)を用いた。ナトリウムの付与された4-アミノブチルβ-ラクトシド(2β)がCID状況の下でナトリウムの付与されたラクトース(β-配置と考えられる)を与え、GM3ガングリオシドに由来する同一のm/zのイオンとのとERMSの比較から、ヘミアセタールイオン種が構造解析に役立つことを明らかにしている(19)。しかし、α-アノマーのERMSは報告されていなかった。そこで、両アノマー(図2B)の気相における合成をおこないERMS解析をおこなった。ERMS相関解析により、形成されたイオンが異なるスペクトルを与えることを確認した。(図2 C,D)(K. Suzuki, S. Daikoku, T. Ako, A. Kurimoto, A. Ohtake, et al., Anal. Chem. 79, 9022-9029 (2007))
【0077】
全ての考察はアノマー化がCID条件下で起こらないという仮定に依存するため、3α-、および、β-体と考えられるラクトース(3α/β)を用い検討した(基準2)。
両方のイオンについて、前駆体の約25%が消費される[0.86V(α)と0.92V(β)] CIDエネルギーでMS5の段数でERMS実験をおこなった。それぞれ、MS4で得られたスペクトルとMS5のそれらは各々のα-とβ-アノマーのそれとよく一致し、(図2 E,F)MS5の際、残留した前駆体イオンは初期段階(MS4)のものと同じ構造であったことが判明した。(図2 G,H)もし平衡が存在するならば、MSnとMSn+1で観測されるα-とβ-アノマーのスペクトルは異なり、平衡化混合物のスペクトルに収束する方向に変化すると考えられる。実験では、しかしながら、MS4とMS5で得られたスペクトルは、α-、および、β-ラクトース共に見分けがつかなかった。これはアノマー(少なくともナトリウム付加体)の平衡が気相において起こらない、あるいは、検出不可能なレベルであったことを示唆する。ここで、様々なフラグメントイオンが生じている点に留意する必要がある。このことは、異性化のために必要な充分なエネルギー(もし可能であれば)が前駆体に印加されていたことを示す。このようにして、ヘミアセタール(ナトリウム付加体)はCID条件下において気相ではアノマー化しないことが初めて示された。
【0078】
(B−2)旋光度の測定
以上の結果から、ラクトースのナトリウム付加体のアノマー異性に関する議論が可能となり、また、ヘミアセタールのα-とβ-両アノマーが識別可能であることを証明した。結果に基づいて、水溶液(3A図)で起こっているラクトースの変旋光を検討した。それぞれα-体とβ-体の比率を高めたラクトース[α:β=約98:2、および、約30:70]の水溶液を25度でインキュベートし、各々の溶液の一定量をERMSに基づいて分析した。観察されたフラグメントとイオンについてERMS相関図から推定されたy切片は、質量分析法が変旋光の測定に用いることができることを示した。(図3 B)これは、旋光計によって確認された(3C図)。質量分析法によれば、平衡状態の3つのフラグメントの個々のシグナル強度は、16%(m/z 203)、56%(m/z 305)、28%(m/z 347)である。アノマーの比率(α/β)は、核磁気共鳴(NMR)を使用することで42:58であると推定された。このようにしてERMS相関法が変旋光を測定することに非常に役立つということを証明した(3C図)。
【0079】
(B−3)グリコシダーゼ反応機構の解析
本方法の有用性を拡大するために酵素反応メカニズムを明らかにする実験をおこなった。グリコシダーゼの機序の研究は、グリコシド交換反応により医学的に重要な化合物を合成しうるため重要である。エンドグリコセラミダーゼは、ガングリオシド等の糖脂質を基質とし、アノマー中心の立体化学の保持でグリカンとセラミドを切断する加水分解酵素の1つである。グリコシダーゼの反応機構の研究は、通常、NMRやX線結晶解析により達成される。本法を用いてRhodococcus sp.由来のエンドグリコセラミダーゼIIの反応機構を分析した。ラクトシルセラミドの酵素反応を15、60分間おこなった(4A図)。反応混合物の一部を取り出し生成したラクトースを回収、ERMS相関によって分析、平衡状態にあるラクトースのデータと比較した。(SI)ここで初期生成物は経時的にアノマー化するため、α-、β-ラクトースで観察される共通イオン(m/z 203、305と347によるイオン)のy切片の微妙な違いを観察することとなる。予想通り、15分(丸数字1)後に得られる混合物と60分(丸数字2)は、異なる値を与えた。特に、反応の初期段階(15分)においては平衡状態にあるラクトースの値(丸数字4)から離れて値を持つことが判明した。また、これらの値は、β-アノマー(丸数字3)によって示される平均値)のそれらに近いことがわかった(図3B)。このことは、初めに形成されたラクトースがβ-配置を有し、アノマー化したことを示している。このように、エンドグリコセラミダーゼIIがβ-保持メカニズムであることが本法により確認された。この質量分析法に基づく方法がグリコシダーゼのメカニズム研究に有効であることを初めて示した。
【図面の簡単な説明】
【0080】
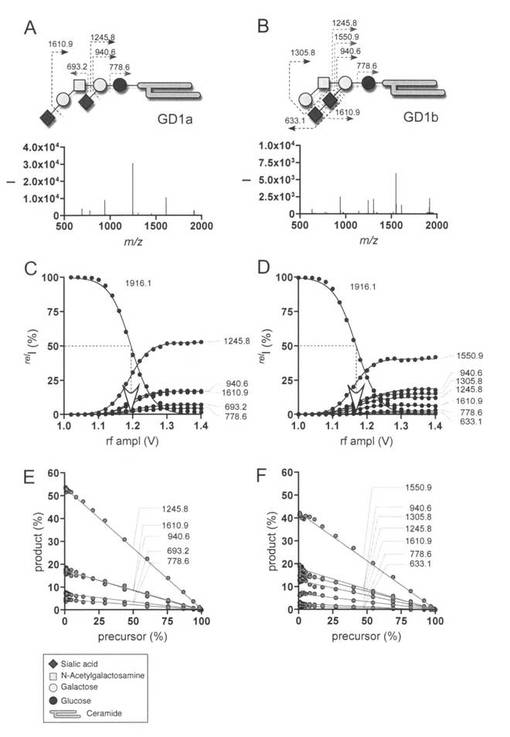
【図1】図1のAは、ガングリオシドGD1aとフラグメンテーションパターンの模式図と、MS/MSスペクトルを示す。ナトリウム付加した分子を分析した。ガングリオシドのメチルエステルをCID分析のために使用した。図1のBは、GD1bの構造及びMS/MS スペクトルを示す。図1のCは、GD1aのERMSを示す。プレカーサーイオンの50%消費に必要なエネルギーを矢印で示す。図1のDは、GD1bのERMSを示す。プレカーサーイオンの50%消費に必要なエネルギーを矢印で示す。図1のEは、GD1a.のERMS 相関、図1のFは、GD1bのERMS 相関を示す。菱形はシアル酸(Sia); 灰色の○はガラクトース(Gal); 四角はN-アセチルガラクトサミン(GalNAc); 黒の○はグルコース(Glc);
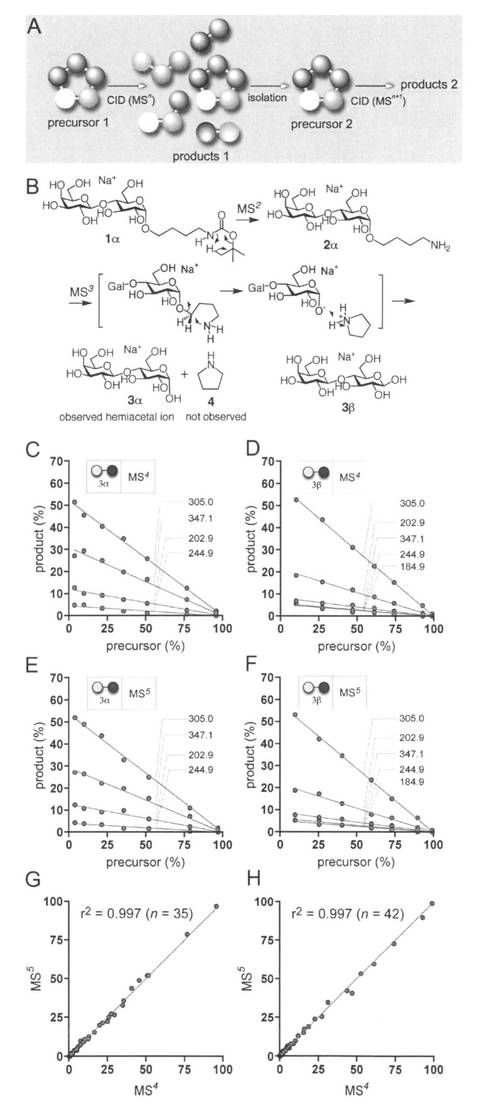
【図2】図2のAは、CID条件下での所定のイオンの構造変化を調べる概念を示す。プレカーサー1のCID上に、プロダクト1が生成する。CID過程のある時点で、前のときと同じm/z値を有するプレカーサー2を単離し、これを2回目のCIDに供する。その後、2つのプレカーサーの特性を、フラグメンテーションプロファイルを比較することによって測定する。図2のBは、ナトリウム付加したヘミアセタール種の気相合成及びα- (3α) 及びβ-ラクトース (3β)の構造を示す。図2のCは、ナトリウム付加したα-ラクトース3α (m/z 365)のERMS相関プロット(MS4)を示す。図2のDは、ナトリウム付加したβ-lactose 3β (m/z 365)のERMS相関プロット(MS4)を示す。図2のEは、ナトリウム付加した3αのERMS相関プロット(MS5)を示す。図2のFは、ナトリウム付加した3βのERMS相関プロット(MS5)を示す。G,Hは、MS4 及びMS5においてERMSについて得た相対プロダクト強度の相関を示す。理想曲線は、y = xである。図2C−Fにおいて、灰色の○はガラクトース(Gal); 黒の○はグルコース(Glc)。
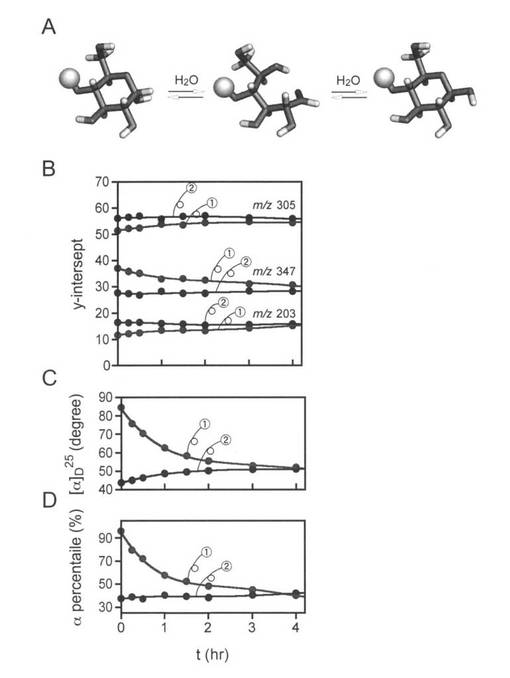
【図3】図3のAは、ラクトースのアノマー化の模式図を示す。図3のBは、ナトリウム付加したα-enriched (5) 及びβ-enriched (6) ラクトースの平衡の観察結果を示す。両方の反応に見られた共通のフラグメントの相対強度(ERMSカウンタープロットにおいて各プロダクトについて見積もったy-切片を用いた)をプロットした。図3のCは、偏光計で得たデータを示す。図4Dは、1H NMRで得たデータを示す。アノマー性プロトンについて計算したα/β比を用いた。
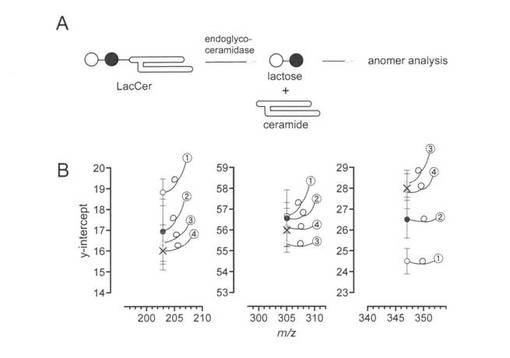
【図4】図4のAは、エンドグリコセラミダーゼがグリコスフィンゴ脂質を加水分解してグリカンとセラミドを生成することを示す。ラクトシルセラミド(LacCer)を基質として使用した。白丸(ガラクトース)、黒丸(グルコース)。図4のBは、エンドグリコセラミダーゼで生成したラクトースのアノマー立体配置の分析結果を示す。m/z 203, 305及び347のイオンについてのERMS相関プロットから得たy切片の見積もりを示す。反応は、15分後(丸数字1)及び60分後(丸数字2)に停止させた。丸数字4は60分後の反応培地のラクトース、丸数字3は平衡化したラクトース(水中)。
【図5】図5は、4-(Boc-アミノ)ブチルグリコシドの1H-NMRを示す。
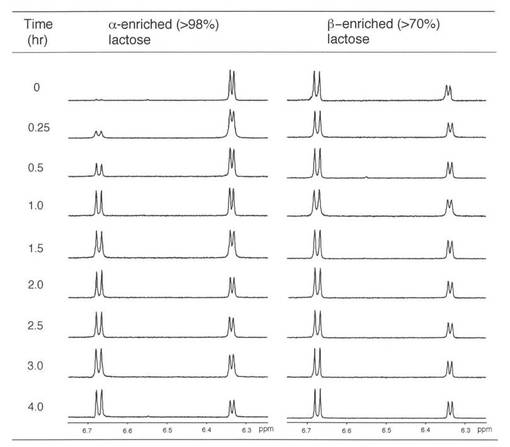
【図6】図6は、DMSO-d6中のラクトン溶液の1H NMRスペクトルを示す。溶液の一部を溶液試料から取り出し、凍結乾燥後に測定した。スペクトルの一部を(OH-1)を示す。δ6.33 (OH-1α) 及び6.67 (OH-1β)。
【図7】図7は、幾つかのナトリウム付加したヘミアセタール種のERMSを示す。個々のヘミアセタール種は、MS3でCID条件下で生成させ、MS4 及びMS5でERMS解析に供した。
【技術分野】
【0001】
本発明は、構造未知の糖鎖についてCID−MSn測定を行い、得られたデータをすでに取得されている参照データと比較することにより該糖鎖の構造解析を行う方法に関する。
【背景技術】
【0002】
糖鎖は主に細胞表面に糖タンパク質や糖脂質の一部として存在し、発生、分化誘導、受精、免疫、癌化、感染症等、様々な生命現象に深く関与している。このように多彩な機能を有する糖鎖の研究は近年盛んになっている。また、糖鎖は、タンパク質の翻訳後修飾の重要な部分を占め、今後の大きな研究対象である。糖タンパク質や糖脂質の機能は糖鎖により制御される場合があり、糖鎖の構造解析を極微量において達成する技術が必要である。しかし、糖鎖は生物工学的手法により増幅ができない分子種であるため、微量の物質のみで構造解析を達成することを可能とする新たな構造解析技術の開発が必要である。タンパク質の配列はゲノム配列が判明している場合、対応するタンパク質のアミノ酸配列も取得できるため、目的のタンパク質のアミノ酸配列は質量分析(例えば、MS/MS法)により解析することができる。しかし、被修飾タンパク質の配列解析技術については今後の課題として残されている。特に、糖鎖によるタンパク質の修飾は極めて大きな分子の多様性を生み出している。したがって、糖鎖の構造解析技術の開発は必要不可欠である。
【0003】
糖鎖は、核酸やタンパク質とは異なり配列以外の要因による構造異性体群を形成している。この理由の基本は、糖鎖を形成する単糖には反応点となる水酸基が複数存在するため結合位置異性体を形成する性質を有し、かつ、単糖間の結合の際にはアノメリック位の立体異性によるアノマー異性体を形成する性質を有しているためである。単糖間の結合は、生物体内においては糖鎖の合成に関わる酵素群の連続反応により行われるため、必ずしも組み合わせの原理に基づく糖鎖群を形成しているわけではない。しかし、酵素反応は副反応を伴うことが知られており、このような現象の生物における意味は解明されておらず、このような場合には遺伝情報によることのない全く新しい概念に基づく構造解析法が必要である。もちろん、遺伝情報が解明されていない、また、糖鎖の生合成経路が解明されていない生物種における糖鎖構造の解析についても新しい構造解析法が必要である。
【0004】
現在用いられている糖鎖の構造解析技術としては、(1)核磁気共鳴分光法、(2)質量分析法、(3)多次元クロマトグラフィーによるマッピング法(非特許文献1を参照)、(4)加水分解酵素による特異的部分加水分解法(非特許文献2を参照)、(5)レクチンによるマッピング法(非特許文献2を参照)、及び(6)メチル化−加水分解-ガスクロマトグラフィーによる組成分析法(非特許文献2を参照)をあげることができ、通常はこれらを組み合わせて糖鎖の解析が行われている。
【0005】
上記の中でも特に有効な糖鎖の解析手段としては、核磁気共鳴分光法及び質量分析法をあげることができるが、前者の問題点は解析に必要な量がμg以上であることであり、後者の問題点は立体異性体の解析が不可能なことである。これらの方法を含めいずれの解析法を用いるにせよ、糖鎖の構造解析は、得られた天然糖鎖構造の構造解析を直接行わなければならない(非特許文献3を参照)。
【0006】
一方、異なる糖鎖を質量分析することにより、その結果が異なることが示されている。例えば、ステロイドの水酸基の立体異性体のCID−MS/MS測定によるフラグメント化を行うと、各フラグメント強度が異なること(非特許文献4を参照)や、合成された硫酸化糖鎖の構造異性体のCID−MS/MS測定を行って、特定イオンの強度を測定すると、その値に差があること(非特許文献5を参照)が示されている。しかし、これらはいずれも具体的に糖鎖の構造解析方法を示すものではなかった。
【0007】
近年、この情報を基にスペクトルの比較による構造解析法の研究が報告されている(非特許文献6〜8を参照)が、従来知られている構造の特定を行おうとするものであり新規物質の構造解析を可能とするものではなかった。
【0008】
また、特許文献1には、(a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、得られた特定のm/zのプロダクトイオンの総イオンカウント数とCIDエネルギーとの関係を示すCIDエネルギー依存曲線を作成し、(c)該CIDエネルギー依存曲線と、構造既知の参照糖鎖から得られた、上記フラグメントイオンと同一のm/zのフラグメントイオンをCID−MS/MS測定することにより得られた、上記プロダクトイオンと同一のm/zのプロダクトイオンのCIDエネルギー依存曲線とを比較することを特徴とする糖鎖構造解析方法が記載されている。この方法は、天然に存在する構造未知の糖鎖を、置換基が異なるものや立体異性体等も含めその構造、またはその一部を決定又は推定することができ、また極微量の解析が行える点で有用であるが、多数のデータを取得する必要があり、多量のサンプルを準備する必要があった。
【0009】
【非特許文献1】Royle, L. et al., Anal. Biochem., 2002,304,70-90
【非特許文献2】Chaplin, M.F et al., Carbohydrate Analysis, A Practical Approach, IRL Press, Oxford,1994
【非特許文献3】Dell, A., Adv. Carbohydr. Chem. Biochem. 1987, 45, 19-72
【非特許文献4】Faretto, D. et al., Mass Spectrom., 1991,5,240-244
【非特許文献5】Kurono, S. et al., J. Mass Spectrom., 1998, 33,35-44
【非特許文献6】Y. Takegawa, K. Deguchi, S. Ito, S. Yoshioka, A. Sano, et al., Anal. Chem. 76, 7294-7303 (2004).
【非特許文献7】A. Kameyama, N. Kikuchi, S. Nakaya, H. Ito, T. Sato, et al., Anal. Chem. 77, 4719-4725 (2005).
【非特許文献8】D. Ashline, S. Singh, A. Hanneman, V. N. Reinhold, Anal. Chem. 77, 6250-6262 (2005).
【特許文献1】特開2006−145519号公報
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明の目的は、構造未知の糖鎖についてCID−MSn測定を行い、得られたデータをすでに取得されている参照データと比較することにより該糖鎖の構造解析を行う方法において、多量のサンプルを準備して多数のデータを取得する必要がない方法を提供することである。
【課題を解決するための手段】
【0011】
本発明者らは、上記課題を達成するために鋭意検討を進めた結果、目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求め、該相関と、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を比較することによって糖鎖の構造を同定できることを見出し、本発明を完成するに至った。
【0012】
すなわち本発明によれば、以下の発明が提供される。
(1) (a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求め、(c)該相関と、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を比較することを特徴とする糖鎖構造解析方法。
【0013】
(2) (a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とをプロットしてグラフを作成し、(c)該グラフと、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率をプロットして得られたグラフとを比較することを特徴とする糖鎖構造解析方法。
【0014】
(3) 工程(b)において、少なくとも1回以上のCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とを少なくとも1点以上プロットする、(2)に記載の糖鎖構造解析方法。
【0015】
(4) 工程(b)において、少なくとも2以上の異なるCIDエネルギーを用いてCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とを少なくとも2点以上プロットする、(2)又は(3)に記載の糖鎖構造解析方法。
(5) 工程(b)において作成したグラフを直線で近似することを特徴とする、(2)から(4)の何れかに記載の糖鎖構造解析方法。
【0016】
(6) 工程(b)において作成したグラフを直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率を算出し;工程(c)において構造既知の参照糖鎖について得られたグラフも直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率を算出し;上記工程(b)及び工程(c)において算出した百分率を比較することを特徴とする、(5)に記載の糖鎖構造解析方法。
【0017】
(7) 工程(b)において作成したグラフを直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率と、上記の直線の傾きを算出し;工程(c)において構造既知の参照糖鎖について得られたグラフも直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率と、上記直線の傾きを算出し;上記工程(b)及び工程(c)において算出した百分率及び傾きを比較することを特徴とする、(5)又は(6)に記載の糖鎖構造解析方法。
【発明の効果】
【0018】
本発明の糖鎖解析方法は、構造未知の糖鎖についてCID−MSn測定を行ない、得られたCIDエネルギー依存曲線を構造既知の糖鎖から得られた参照データと比較することにより構造決定を行う方法である。特に本発明の糖鎖解析方法は、多量のサンプルを準備して多数のデータを取得する必要がないという利点を有している。本発明の方法は、天然に存在する構造未知の糖鎖を、置換基が異なるものや立体異性体等も含めその構造、またはその一部を決定又は推定することができ、また極微量の解析が行える点で非常に有用である。また該方法は、糖鎖の構造解析という次世代生命科学研究の基盤技術となるのみならず、糖鎖の異常を伴う疾病や感染症を引き起こすきっかけとなる糖鎖レセプターの特定等、診断医療に貢献する技術となるものである。さらに、本発明の純度検定方法により、検体の詳細なデータ解析をおこなう以前にその必要性を簡便に判定することが可能となる。
【0019】
本発明においては、プレカーサーイオンとプロダクトイオンの比が衝突エネルギーによらず一定であるということが判明した。本発明によれば、(1)直線近似が可能なため、スペクトル間の一致による構造決定が容易であり、(2)特定の衝突エネルギーで測定したMS/MSスペクトルを如何なる衝突エネルギーで測定したスペクトルとも定量的に比較することが可能であり、(3)従来より少ない測定回数で定量的データを取得することができるようになり、(4)MS/MS測定時における衝突エネルギーの検討が不要なため測定の自動化が可能であり、(5)イメージングマスにおけるMS/MS測定に利用でき、(5)質量分析法による構造解析に重要なERMS法を必要最小限のサンプル消費量(従来のMS/MS法と同程度)で達成可能となるという効果が得られる。本発明の方法は、特に溶液反応(化学反応、酵素反応)のモニタリング(特に生成物の異性化を伴う場合)に有効である。
【0020】
グリコシダーゼは、糖鎖、あるいは、配糖体を基質とし加水分解する酵素であり、エキソ型とエンド型に分類される。それらは、それぞれ立体保持型と立体反転型に分類される。生成物はアノマー遊離の単糖、あるいは、オリゴ糖であり水溶液中でα-とβ-アノマー(ヘミアセタール)は開環したアルデヒド体を経由して平衡に至る。立体保持型と立体反転型の酵素においては、その過程において初期生成物が異なる。一方、これらの酵素は、任意のアルコールを添加しておくとこれを受容体として新たなグリコシド化合物を生成することが知られている。この過程は、トランスグリコシル化と呼ばれる。本発明による糖鎖解析方法は、上記したような酵素反応の解析に応用することもできる。
【発明を実施するための最良の形態】
【0021】
以下、本発明を更に詳細に説明するが、以下の構成要件の説明は、本発明の実施態様の代表例であり、本発明はこれらの内容のみに特定されるものではない。
以下の説明において、「CID−MSn測定」とは、CID(Collision induced dissociation:衝突誘起解離)による多段マススペクトロメトリーの取得を行うことを示し、「フラグメントイオン」とは、上記CID−MSn測定や物理化学的分解等により得られる各m/zを有するイオンを示し、「m/z」とは、質量数(m)と電荷(z)の比を示し、「プロダクトイオン」とは、フラグメントイオンを上記CID−MSn測定することにより得られる各m/zを有するフラグメントイオンを示し、「総イオンカウント数」とは、各m/zを有するすべてのフラグメントイオンのイオン強度の総和を示し、また、「CIDエネルギー」とは、CIDを起こすときに加えるエネルギーを一般的に示し、実際にはイオンを振動させるためのある周波数の電場の電圧を示す。さらに、「構造解析方法」とは、構造決定方法および/または構造推定方法を意味する。
【0022】
(1)構造解析方法の概略
本明は、構造未知の糖鎖(本明細書中では、これを「目的糖鎖」と称することがある)を特定のm/zのフラグメントイオン(以下、これを「プレカーサーイオン」又は「プレカーサーフラグメントイオン」と称することがある)が得られるまでCID−MSn測定を行い、得られたプレカーサーフラグメントイオンについてさらにCID−MS/MS測定を行う。これにより得られる、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求める。この際、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とをプロットしてグラフを作成してもよい。また、上記で作成したグラフは直線で近似してもよい。
【0023】
一方、参照データとして、構造既知の糖鎖(本明細書中では、これを「参照糖鎖」と称することがある)またはそのライブラリーについて、特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、得られたフラグメントイオンについてさらにCID−MS/MS測定を行う。これにより得られる構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求める。この際、プレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関は、直線で近似してもよい。
【0024】
かくして得られた目的糖鎖のプレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関(目的糖鎖のデータ)と、参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関(参照データ)とを比較し、目的糖鎖のデータと一致する参照データがあった場合に、その参照データが得られた参照糖鎖と目的糖鎖の一部の構造が一致すると判断することができる。目的糖鎖のデータと参照データとの比較は、作成したグラフ同士を、パターン認識ソフトを用いて比較することができる。
【0025】
作成したグラフを直線で近似する場合は、直線同士を直接比較してもよいし、あるいは直線の切片の値を求め、それらを比較してもよいし、直線の切片の値と傾きを求め、それらを比較してもよい。ここで言う直線の切片の値とは、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率に相当する値である。
【0026】
また、目的糖鎖のデータと参照データが一致しない場合でも、目的糖鎖のパラメータが参照糖鎖のパラメータの統計的解析結果の範囲内であれば、部分構造を推定することができる。本発明には、このような構造の推定を行う方法も含まれる。
【0027】
このようにして目的糖鎖の一部の構造が決定または推定された場合、これらを目的糖鎖の生合成経路を参照する等して全体の構造を決定または推定していくことができる。
【0028】
(2)目的プレカーサーフラグメントイオンの調製
本発明に用いられる目的糖鎖は、以下に説明する方法により分解するなどしてCID−MSn測定を行い得るものであればいかなるものであってもよい。また、構造解析を目的として本発明の方法に供するものであるので、その全部または一部の構造が未知のものが好ましい。目的糖鎖は、生体組織又は細胞等から得られたものでもよいし、合成されたタンパク質に結合したものから得られたものでもよく、またそれを酸加水分解又は酵素分解したり、さらにHPLC等で分離精製したもの等を用いることができる。また、化学合成された糖鎖を用いることもできる。
【0029】
本発明では、目的糖鎖と参照糖鎖から得られる同一のm/zを有するプレカーサーイオンについて、CID−MSn測定して得られるプロダクトイオン強度を比較するので、参照データを誘導するプレカーサーイオン(以下、これを「参照プレカーサーフラグメントイオン」と称することがある)と同一のm/zを有するフラグメントイオン(以下、これを「目的プレカーサーフラグメントイオン」と称することがある)が得られるまで何らかの方法で分解する必要がある。分解の方法は、それ自体公知の常法を用いることができるが、例えば、酸加水分解、酵素分解等により得られる糖鎖を液体クロマトグラフィー、キャピラリー電気泳動、ゲル電気泳動等により分離精製して用いることもでき、あるいは後述するCID−MSn測定等により得られる任意のフラグメントイオンをm/zにより分離して用いることができる。参照糖鎖として3糖ライブラリーを用いる場合は、目的糖鎖を1〜3糖に分解することが望ましい。また、目的糖鎖が置換基を有する場合、これを除去しても、また参照データに置換基を含むものがあれば付いたままでも用いることができる。最終的に得られた目的糖鎖分解物は、それぞれ質量分析することによりm/zを確認し、この質量とその他の情報、例えば生合成経路などの情報から配列を決定することもできるが、本発明の方法は、ここで決定することのできなかったフラグメントイオンについて用いることができる。
【0030】
(3)目的プレカーサーフラグメントイオンのCID−MSn測定
上記で得られた目的プレカーサーフラグメントイオンは、これをCID−MSn測定する(CID−MSn測定により得られた目的プレカーサーフラグメントの場合は、これをCID−MS/MS測定する)。
【0031】
イオン化法としては、FAB(高速原子衝撃法)、CI(化学イオン化法)、ESI(エレクトロスプレーイオン化法)、MALDI(マトリクス支援レーザー脱離イオン化法)、APCI(大気圧化学イオン化法)等が用いられるが、本発明においてはサンプル調製が容易で、かつ、マトリックス由来の夾雑イオンの影響がないESI法を用いることが好ましい。しかし、上記CID−MSn測定が可能であればイオン化法はこれらに限定されるものではない。また、ESI法にはマイクロスプレー法とナノスプレー法があるが、サンプル使用量の面から本発明においてはナノスプレー法が好ましく用いられる。
【0032】
上記目的糖鎖またはその分解物をイオン化する場合、微液滴を真空下溶媒を蒸発させることでイオン化するため水/メタノールあるいは水/アセトニトリル(1:1)を溶媒として用いることができるが、物質の性質に従い選択が可能でありこれに限られることはない。目的糖鎖またはその分解物は、この溶媒に対して0.01〜100nmol/ml、より好ましくは0.5〜5nmol/ml、さらに好ましくは1nmol/mlの濃度に溶解することが好ましい。
【0033】
また、CID−MSn測定を行なう装置は、これが可能であれば、機種を問うものではないが、本発明においては、CID−MSn測定が可能な四重極イオントラップ型質量分析計を用いることが好ましい。このような装置としては、具体的には例えば、esquire 3000 plus(ブルカーダルトニクス社製)、LCQ DECA(サーモフィニガン社製)、AXIMA QIT(島津社製)、LCMS-IT-TOF(島津社製)等が挙げられる。測定方法としては、各プレカーサーフラグメントイオンから解離するプロダクトイオンのイオン強度が測定できる方法であればいかなるものでもよいが、具体的には、例えば、熱キャピラリー温度20〜365℃、キャピラリー電圧0〜6.0kV、CIDエネルギーが0〜25V、ポジティブモードまたはネガティブモードのいずれか等で測定することができる。ポジティブモードを用いる場合には、プロトン、ナトリウム、カリウム、リチウム、カルシウム、アンモニウム等の陽イオンを選択して用いることができる。
【0034】
上述のCID−MS/MS測定により得られたデータに基づいて、プレカーサーイオンと、特定のm/zのプロダクトイオンとの比率をプロットしてプレカーサーイオンとプロダクトイオンとの比率を求める。
【0035】
<<測定するコリジョンエネルギーの決定法>>
<標準品の分解エネルギーの測定>
マルトオリゴ糖([Glc]n:n= 3-12)などの標準品を用いERMSを行いプレカーサーイオンの半分が分解するエネルギー(V50)を各々のプレカーサーについて取得し、これを質量に対する分解エネルギー(V50)の“ものさし”と位置づける。
<ERMS測定するコリジョンエネルギーの中心の決定>
MSn測定を行い当該するイオンが得られた段階でERMS測定を行う。このとき、当該イオンのm/zを上述の“ものさし”に参照して該当するV50(コリジョンエネルギーの中心)を求める。
【0036】
<ERMS相関法の測定>
上記の様にして求めたコリジョンエネルギーの中心、および、コリジョンエネルギーの中心から±0.1, 0.2, 0.3Vのコリジョンエネルギーを選択し、これら7つの設定条件からMS/MSを最低1回、好ましくは、7回おこない、得られるフラグメントイオンの強度を上述の算式により標準化(相対値化)し、プレカーサーイオンをX軸としプロダクトイオンをY軸としてプロットする。その後、直線近似を行ってY切片(通常ERMSのプラトー値)を求める。
【0037】
<ERMS相関からERMSへの変換>
ERMS相関において使用したデータポイント(各々のコリジョンエネルギーを表す)から求めた近似曲線上へ垂線をひく。この点を理想のデータとし、スプライン補完をおこなって理想データを求める。求めた理想データは元々の実験データに対応するコリジョンエネルギーからスプライン補間法により与えられているので、各々の理想データポイントはコリジョンエネルギーの項目を有している。求まった一連の理想データ群をコリジョンエネルギーに対するプレカーサーイオン、及び、各フラグメントイオンの相対強度としてプロットし直すと理想化された通常のERMSを与える。このグラフからV50などのプレカーサーイオンの分解に要するエネルギー該当項目を正確に導くことができる。
【0038】
(4)参照プレカーサーフラグメントイオンの調製
参照データを取得する目的で、参照糖鎖について上記と同様にプレカーサーフラグメントイオンを調製する。参照糖鎖は、目的糖鎖の全部または一部の構造を本発明の方法を用いて決定し得るものであればいかなるものであってもよい。具体的には、目的糖鎖またはその一部と構造が一致する可能性があり、構造が明らかであるものが用いられる。このような参照糖鎖は、天然のものでも合成されたものでもよく、また置換基がついていてもよい。糖鎖の長さは、1〜13個の糖が連結したものが用いられる。
【0039】
この参照糖鎖として、ある特定の集団であるライブラリーを用いることによれば、より網羅的、組織的に目的糖鎖の構造決定を行なうことができる。このようなライブラリーとしては、例えば、2〜25個の糖が連結した構造を有しており、ライブラリーを構成する各糖鎖は、天然糖鎖を構成する全ての種類の単糖の順列組み合わせの配列を有するもので、かつ立体異性も含むもの等が好ましく用いられる。また、非天然の単糖を含んでいてもよい。
【0040】
また、糖鎖構造を一般化すれば、非還元末端糖、還元末端糖、およびそれらの中間に存在する糖からなるポリマー性糖化合物ということができ、このポリマーの最小単位は3糖が連結した糖鎖である。このような3糖からなる天然糖鎖を構成する全ての種類の単糖の順列組み合わせの配列を有するもので、かつ立体異性も含む3糖ライブラリーは、生体に存在するあらゆる直鎖構造を有する糖鎖の部分構造を含んでいるため、本発明の参照糖鎖のライブラリーとしてより好ましい。
【0041】
上記の天然糖鎖を構成する単糖とは、例えば、グルコース、ガラクトース、マンノース、N-アセチルグルコサミン、N-アセチルガラクトサミン、キシロース、シアル酸、グルクロン酸、フコース等が挙げられる。また、これらは修飾基を有していても良い。
【0042】
参照糖鎖および参照糖鎖ライブラリーは、これを化学合成することが好ましい。合成の方法としては、通常用いられるそれ自体公知の方法を用いることができる。化学合成には、上述のような単糖各々を適当に保護した合成ユニットが必要である。さらに、立体異性体から成る糖鎖ライブラリーを効率良く合成するためには、立体選択的な合成ではα−グリコシドを得るためには隣接基関与のない保護基、例えばベンジル基(Bn)を、β−グリコシドを得るためには、隣接基関与のある保護基、例えばアセチル基等を用いる必要がある。これらの立体制御のための保護基の他に通して用いる保護基を用いる必要性から、単糖合成ユニット数は多くなり、合成経路の複雑化につながる。従って、2位の保護基は隣接基関与のない保護基とし、合成経路の簡略化を、例えばPCT/JP2004/009523号明細書に記載の方法等に従って行なうことが好ましい。
【0043】
かくして得られた参照糖鎖または参照糖鎖ライブラリーは、これを目的糖鎖と同様に特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行なう。特定のm/zのフラグメントイオンとは、目的糖鎖のプレカーサーフラグメントイオンが有するm/zと同一のものであればいかなるものでもよい。また、参照糖鎖がライブラリーの場合には、該ライブラリーの各構成要素から得られる何れの解裂しえるフラグメントイオンでもよく、全ての解裂し得るフラグメントイオンでもよい。
【0044】
(5)参照プレカーサーフラグメントイオンのCID−MSn測定
上記(4)で取得された参照プレカーサーフラグメントイオンは、これを目的プレカーサーフラグメントイオンと同様にCID−MSn測定する(CID−MSn測定により取得された参照プレカーサーフラグメントの場合は、これをCID−MS/MS測定する)。イオン化法、溶媒、糖鎖の調製、測定装置などは全て目的糖鎖およびプレカーサーフラグメントイオンと同様のものを用いることができる。
【0045】
上述のCID−MS/MS測定により得られたデータに基づいて、プレカーサーイオンと、特定のm/zのプロダクトイオンとの比率をプロットしてプレカーサーイオンとプロダクトイオンとの比率を求める。
【0046】
(6)目的糖鎖の構造決定
目的プレカーサーフラグメントイオンから得られたプレカーサーイオンとプロダクトイオンとの比率と、これに対応する参照データが一致した場合、参照データが誘導されたもとの参照プレカーサーフラグメントイオンの構造と目的プレカーサーフラグメントイオンの構造が同じであると決定することができる。ここで、それぞれのデータが一致するとは、プレカーサーイオンの百分率が0%の場合における、特定のm/zのプロダクトイオンの百分率値の差が±2%以内であることを示す。また、目的プレカーサーフラグメントイオンに置換基が結合していた場合には、置換基が結合した参照プレカーサーフラグメントイオンから得られた参照データと比較して、一致した場合には、同一の置換基を有していると判断することができる。このとき、Z検定を判断基準とすることもできる。
【0047】
かくして本発明の方法により目的糖鎖の一部が決定された場合、これらの全配列を決定するために、例えば、生合成経路に当てはまるものを参照してつなぎ合わせたり、初めに目的プレカーサーフラグメントイオンを産生するために分解した場合には、その分解の方法から再構築する等の方法が用いられる。
【実施例】
【0048】
以下、実施例を挙げて本発明を詳細に説明するが、本発明の範囲はこれらの実施例により限定されるものではない。
【0049】
(A)材料及び方法
(a)合成
実施例で使用した化合物は、以下のスキーム(化合物S6−S8の合成)に従って合成した。
【0050】
【化1】
【0051】
Bnはベンジル、Phはフェニル、Bocはt-ブトキシカルボニル、NISはN-ヨードスクシンイミド、TfOHはトリフルオロメタンスルホン酸、AW-300はモレキュラーシーブAW-300、DCEはジクロロエタン、Acはアセチル、Meはメチル、Etはエチル、HEPESは4-(2-ヒドロキシエチル)ピペラジン-1-エタンスルホン酸、APはアルカリホスファターゼ、KPB bufferはリン酸カリウム緩衝液、DMFはN,N-ジメチルホルムアミドを示す。
【0052】
(a−1)一般的方法
薄層クロマトグラフィ(TLC)は、Kieselgel 60 F254 (E. Merck) プレートで行い、1% Ce(SO4)2-1.5% (NH4)6MoO24・4H2O-10% H2SO4 で焦がすことによって検出した。シリカゲルカラムクロマトグラフィーは、Wakogel C-300 (Wako Pure Chemical Industries, Ltd.)で行った。 Sep-Pak C18 はWaters Corpから得た。UDP-Gal+2Na 及び pNP-β-Gal はSigma-Aldrich Corpから購入した。アルカリホスファターゼはNacalai Tesque, Incから購入した。β1,4-GalT'ase はCalbiochem Corpから購入した。β1,3-Gal'aseはKatsumi Ajisaka 教授(Niigata University of Pharmacy and Applied Life Sciences)から供与された。1H NMR (500 MHz)スペクトルは、参照としての溶媒ピーク(residual CHD2OD; 3.31 ppm)とともにAVANCE 500スペクトロメーター (Bruker Biospin Inc.)で記録した。1H NMRスペクトルのアノマー領域は図5に示す。
【0053】
(a−2)4-(N-Boc-アミノ)ブチル2-アセタミド-2-デオキシ-β-D-グルコピラノシド(S4)及び4-(N-Boc-アミノ)ブチル2-アセタミド-2-デオキシ-α-D-グルコピラノシド(S5).
DCE (2.0 mL)中のフェニル2-アジド-3,4,6-トリ-O-ベンジル-2-デオキシ-1-チオ-β-D-グルコピラノシド(S1, 120 mg, 0.12 mmol), 4-(N-Boc-アミノ)ブタノール(S2,81 mg, 0.43 mmol) 及びAW-300 (ca. 0.5 g) の混合物を窒素雰囲気下25℃で1時間攪拌した。この混合物に、NIS (72 mg, 0.32 mmol)及びTfOH (2.0μL, 0.21 mmol) を0℃で添加し、1時間攪拌した。反応を飽和NaHCO3 で停止し、混合物をDCMで希釈し、セライトパッドでろ過した。ろ液をNa2S2O4を含む飽和NaHCO3に注ぎ、DCMで抽出し、水で洗浄し、MgSO4上で乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(n-ヘキサン: EtOAc = 5:1-2:1)に供し、4-(N-Boc-アミノ)ブチル2-アジド-3,4,6-トリ-O-ベンジル-2-デオキシ-D-グルコピラノシドS3 (80 mg, 58%, α/β = 4/6)を得た。S3 (80 mg, 0.12 mmol, α/β = 4/6)のEtOAc (1 mL)溶液にPd(OH)2/C (20%, ca. 10 mg), Ac2O (1.0 mL) 及びMeOH (1.0 mL)を添加した。混合物を水素雰囲気下で47時間攪拌し、ろ過し、減圧濃縮して4-(N-Boc-アミノ)ブチルGlcNAc 誘導体S4 (β-グリコシド) 及びS5 (α-グリコシド) (23 mg, 47%, S4/S5 = 1.5)を得た。これらのアノマー異性体をシリカゲルカラムクロマトグラフィー(DCM : MeOH = 19:1-8.5:1)によって分離した。
【0054】
S4: 1H NMR (CD3OD): δ4.38 (d, 1H, J1,2 8.4, Hz, H-1), 3.92-3.87 (m, 1H, OCH2(CH2)2CH2N), 3.88 (dd, 1H, J5,6a 2.0 Hz, J6a,6b 12.0 Hz, H-6a), 3.68 (dd, 1H, J5,6b 5.8 Hz, H-6b), 3.64 (dd, 1H, J2,3 9.2 Hz, H-2), 3.50-3.45 (m, 1H, OCH2(CH2)2CH2N), 3.44 (dd, 1H, J3,4 8.7 Hz, H-3), 3.31 (dd, 1H, J4,5, 9.7 Hz, H-4), 3.26 (ddd, 1H, H-5), 3.04 (t, 2H, J 6.4 Hz, OCH2(CH2)2CH2N), 1.97 (s, 3H, NHAc), 1.58-1.48 (m, 4H, OCH2(CH2)2CH2N), 1.43 (s, 9H, tBu).
S5: 1H NMR (CD3OD): δ4.78 (d, 1H, J1,2 3.5, Hz, H-1), 3.88 (dd, 1H, J2,3 10.7 Hz, H-2), 3.82 (dd, 1H, J5,6a 2.2 Hz, J6a,6b 11.9 Hz, H-6a), 3.76-3.71 (m, 1H, OCH2(CH2)2CH2N), 3.68 (dd, 1H, J5,6b 6.0 Hz, H-6b), 3.67 (t, 1H, J3,4 10.6 Hz, H-3), 3.58 (ddd, 1H, J4,5 9.8 Hz, H-5), 3.42-3.37 (m, 1H, OCH2(CH2)2CH2N), 3.34 (t, 1H, H-4), 3.07 (t, 2H, J 6.6 Hz, OCH2(CH2)2CH2N), 1.99 (s, 3H, NHAc), 1.66-1.53 (m, 4H, OCH2(CH2)2CH2N), 1.43 (s, 9H, tBu).
【0055】
(a−3)4-(N-Boc-アミノ)ブチルβ-ラクトサミニド(S6)
化合物S4 (3.1 mg, 7.9μmol), UDP-Gal+2Na (7.2 mg, 12μmol) 及びアルカリホスファターゼ(20-300 U/mg, 1.0 mg) をHEPES緩衝液(0.5μL, 0.10 M, pH 7.0, 40 mM MnCl2を含有)に溶解した。この混合物に、HEPES緩衝液(0.5μL, 0.10 M, pH 7.0, 40 mM MnCl2を含有)中のβ1,4-GalT'ase (1U, 牛乳由来)を添加し、37℃で20時間インキュベートした。反応混合物をろ過し、Sep-Pak C18に供し、水で洗浄した。4-(N-Boc-アミノ)ブチルグリコシド及びS4を含有する生成物をMeOHで溶出した。混合物をシリカゲルカラムクロマトグラフィー(DCM : MeOH = 9:1-6:4)で精製して、4-(N-Boc-アミノ)ブチルβ-ラクトサミニドS6 (2.1 mg, 36%) を得て、S4 (1.6 mg, 52%)を回収した。
【0056】
S6: 1H NMR (CD3OD): δ4.39 (d, 1H, J1,2 7.5, Hz, H-1), 4.38 (d, 1H, J1',2' 7.5 Hz, H-1'), 3.91 (dd, 1H, J5,6a 2.4 Hz, J6a,6b 12.2 Hz, H-6a), 3.91-3.87 (m, 1H, OCH2(CH2)2CH2N), 3.86 (dd, 1H, J5,6b 4.2 Hz, H-6b), 3.81 (d, 1H, J3',4' 2.9 Hz, H-4'), 3.76 (dd, 1H, J5',6'a 7.6 Hz, J6'a,6'b 11.5 Hz, H-6' a), 3.74-3.69 (m, 1H, H-2), 3.68 (dd, 1H, J5',6'b 4.5 Hz, H-6' b), 3.64-3.57 (m, 3H, H-3,4,5'), 3.54 (dd, 1H, J2',3' 9.7Hz, H-2'), 3.50-3.44 (m, 1H, OCH2(CH2)2CH2N), 3.48 (dd, 1H, H-3'), 3.41-3.37 (m, 1H, H-5), 3.03 (t, 2H, J 6.4 Hz, OCH2(CH2)2CH2N), 1.97 (s, 3H, NHAc), 1.58-1.49 (m, 4H, OCH2(CH2)2CH2N), 1.43 (s, 9H, tBu).
【0057】
(a−4)4-(N-Boc-アミノ)ブチル2-アセタミド-2-デオキシ-3-O-β-D-ガラクトピラノシル-β-D-グルコピラノシド(S7)
化合物S4 (5.0 mg, 13 μmol), pNP-β-Gal (3.8 mg, 13 μmol), リン酸緩衝液 (0.16 μL, 0.10 M, pH 6.0, 0.15 M NaClを含有)、DMF (40 μL)及びβ1,3-Gal'ase (20 μL, 13U/mL)の混合物を37℃で3時間インキュベートした。溶液を蒸発させ、残渣をSep-Pak C18に供し、水で洗浄した。4-(N-Boc-アミノ)ブチルグリコシド及びS4を含む生成物をMeOHで溶出した。混合物をシリカゲルカラムクロマトグラフィー(EtOAc : MeOH = 9:1-2:1 then EtOAc : MeOH : H2O = 12:3:1)で精製して、化合物S7 (0.8 mg, 11%) 及びS4 (3.5 mg, 70%)を得た。
S7: 1H NMR (CD3OD): δ 4.46 (d, 1H, J1,2 8.3, Hz, H-1), 4.27 (d, 1H, J1',2' 7.6 Hz, H-1'), 3.93-3.88 (m, 1H, OCH2(CH2)2CH2N), 3.89 (dd, 1H, J5,6a 1.9 Hz, J6a,6b 11.7 Hz, H-6'a), 3.80 (d, 1H, J3',4' 3.0 Hz, H-4'), 3.76 (dd, 1H, J5,6a 7.7 Hz, J6a'6b 11.5 Hz, H-6a), 3.77-3.65 (m, 4H, H-2,3,6b,6'b), 3.57-3.44 (m, 4H, H-2',3',5', OCH2(CH2)2CH2N), 3.29-3.35 (m, 1H, H-5), 3.04 (t, 2H, J 6.9 Hz, OCH2(CH2)2CH2N), 1.97 (s, 3H, NHAc), 1.59-1.48 (m, 4H, OCH2(CH2)2CH2N), 1.43 (s, 9H, tBu).
【0058】
(a−5)4-(N-Boc-アミノ)ブチル2-アセタミド-2-デオキシ-3-O-β-D-ガラクトピラノシル-α-D-グルコピラノシド(S8)
化合物S5 (6.5 mg, 17μmol), pNP-β-Gal (5.0 mg, 17μmol), リン酸緩衝液(0.16 mL, 0.10 M, pH 6.0, 0.15 M NaClを含有), DMF (40μL) 及びβ1,3-Gal'ase (20μL, 13 U/mL)の混合物を37℃で3時間インキュベートした。溶液を蒸発させ、残渣をSep-Pak C18に供し、水で洗浄した。4-(N-Boc-アミノ)ブチルグリコシド及びS5 を含む生成物をMeOHで溶出した。混合物をシリカゲルカラムクロマトグラフィー(DCM : MeOH = 9:1-7:3)で精製して、化合物S8 (2.8 mg, 30%) 及びS5 (3.6 mg, 55%)を得た。
S8: 1H NMR (CD3OD):δ4.78 (d, 1H, J1,2 3.3 Hz, H-1), 4.37 (d, 1H, J1',2' 7.6 Hz, H-1'), 4.06 (dd, 1H, J2,3 10.6 Hz, H-2), 3.85-3.67 (m, 7H, H-3,4',6a,6'a,6b,6'b, OCH2(CH2)2CH2N), 3.62 (ddd, 1H, J4,5 9.8 Hz, J5,6a 1.9 Hz, J5,6b 5.4 Hz, H-5), 3.56 (dd, 1H, J5',6'a 4.5 Hz, J5',6'b 7.5 Hz, 7.5 Hz, H-5'), 3.53 (dd, 1H, J2',3' 9.5 Hz, H-2'), 3.48-3.41 (m, 3H, H-3',4, OCH2(CH2)2CH2N), 3.07 (t, 2H, J 6.5 Hz, OCH2(CH2)2CH2N), 1.98 (s, 3H, NHAc), 1.68-1.53 (m, 4H, OCH2(CH2)2CH2N), 1.44 (s, 9H, tBu).
【0059】
(b)質量分析の実験方法
(b−1)材料
GD1a 及びGD1bはSeikagaku Biobusiness Corp(東京)から購入した。α-enrichedラクトン及びβ-enriched ラクトンの試料は、SIGMA, St. Louis, MOから得た。質量分析計を用いた解析のために、HPLCグレードの溶媒を使用した。MeOH及び無水ジメチルスルホキシド (Wako Pure Chemical Industries, Ltd, Osaka, Japan)。重水素化したメチルスルホキシド-d6 (NMRグレード) (Acros Organics, Geel, Belgium) をNMR実験に使用した。
【0060】
(b−2)装置及びデータ収集
試料は、エレクトロスプレーインターフェースを連結した四重極イオントラップ質量分析計(Esquire 3000 plus, Bruker Daltonics GmbH, Bremen, Germany)で解析した。無水DMSO (1 pmol/μL)に溶解した試料を、注入(流速120μL/h)によってイオン源に導入した。分析のパラメーターは以下の通りである。(1) "乾燥温度", 250 °C; (2) 噴霧ガス(N2), 10 psi; (3) 乾燥ガス(N2), 4.0 L/min; (4) "スマートフラグ", オフ (5) 走査範囲, m/z 50-600 (ラクトース、合成)及びm/z 50-400 (ラクトース, 市販品); (6) 化合物安定性, 300%; (7) ICC ターゲット, 8000; (8) 最大捕捉時間, 200 ms; (9) 平均, 10 スペクトル; 及び(10) "カットオフ", 対応するプレカーサーイオンの27.6% (合成ラクトース) 27.1% (市販のラクトース)
【0061】
本MSn 実験では、端部キャップrf振幅は、プレカーサーイオンが検出できなくなるまでに0.02-V増大した(全イオン電流の0.9%未満の安定状態)。端部キャップrf振幅のみをCID実験中で調節した。He 圧力は4.86×10-6 mbarとし、CID時間40 msとした。(m−4)個のスペクトルの平均をCID測定のために使用した(m = 15-28: ここで、m は実験中に得たスペクトルの数である)。不正確さを回避するために、rf振幅工程における、定常状態への一過性期間に関連する最初と最後の2個ずつのデータは使用しなかった。[Ii + 1] 及び[Ii + 2](In はフラグメントイオンを示す)を有する同位体ピークは、計算に含めた(「データの取り扱い」も参照)。プロダクトイオンの単離のために、m/z ± 2 (w = 2)を単離し、CID実験に供し、同位体を含めた。
【0062】
(b−3)データの取り扱い
ERMSのグラフを得るために、以下の等式を用いた。イオン"IP"が一連のプロダクトイオンI1, I2, I3,... Iiを生成する場合、各々のイオンについての相対イオン電流を、以下の式で定義した。
【0063】
【化2】
【0064】
式中、relCは、観測されたイオンのうちの所定のイオンのイオン電流(%)を示す。CIiは、 焦点での観測されたイオン電流を示す。CIPは、プレカーサーイオンのイオン電流を示す。計算は、DSUM関数に基づき、考慮すべき同位体範囲(W)(本実験ではW=2)を選ぶようにプログラムした、エクセル(Excel 2000 (Microsoft Co.))を用いて作成したプログラムを用いて行った。
【0065】
(b−4)シグモイド曲線フィッティング
質量分析機の複数のrf振幅(端部キャップ)で得たMSnデータのセットを、Excelを用いて分析した。一定のm/z 値を有するピークのセットを一連のデータとして扱った。個々のシグナルについて全イオン電流について相対強度を各振幅において取得した(式S1)。データをPrism 4ソフトウエア(GraphPad Software, Inc.)を用いて分析した。個々のデータを非線形回帰分析によるボルツマン・シグモイド関数を用いてフィッティングした[式S2 (growth) 及びS3 (decay)].
【化3】
【0066】
式中、(各々、最大応答、最大応答の半値、及び傾斜因子を示す)パラメーター"a", "b", 及び "c"は各曲線から得た。
本実験で使用した一連のデータにおいて、シグモイド曲線及びパラメータは、上記エクセルプログラムから得た全データを処理し、回帰曲線をプロットすることによって得た。
【0067】
(c)変旋光
(c−1)試料の調製
α-及びβ-enrichedラクトースの水溶液(10 mg/mL, 25 ℃)を全実験のために調製し、4時間までの指定時間に0.1 mLずつ分画した。分画は液体窒素を用いて直ちに凍結し、凍結乾燥した。各試料を無水DMSO 及びDMSO-d6 に溶解し、MS 及びNMRで解析した。DMSO を実験のための溶媒として使用した。非プロトン性溶媒はアノマー化を妨げるからである (B. Casu, M. Reggiani, Tetrahedron Lett. 39, 2839-2843 (1964))。
【0068】
(c−2)MSに基づいた測定についての試料の消費
DMSO (1 pmol/μL) 中のα- 及びβ-enrichedラクトースの溶液を、MSに基づいた分析のために使用した。12.3μLを注入条件下で使用した(120μL/h, 6 min)。全量123 pmolの各化合物を実験で消費した。
【0069】
(c−3)NMR 分析
1H NMR (500 MHz)スペクトルを、AVANCE 500分光測定器を用いて記録した。DMSO-d6中のα-ラクトースのβ-ラクトースに対する比率を、各1H NMRスペクトルにおけるアノマー部位(グルコース成分)におけるヒドロキシル部の積分により測定した(図6)。
【0070】
(c−4)旋光度
α- 及びβ-enrichedラクトースの水溶液(10 mg/mL) を迅速に10 cmのセルに注入し、旋光度を示した時間で25℃でSEPA-200旋光度計 (HORIBA, Ltd. Kyoto, Japan)を用いて測定した。
【0071】
(d)酵素反応
(d−1)試薬
ウシ・ラクトシルセラミドをCALBIOCHEM : EMD Biosciences, Inc., La Jolla, CAから購入した。Rhodococcus sp.から組み換えエンドグリコセラミダーゼII [EC 3. 2. 1. 123]をTakara Shuzo Co., Ltd., Shiga, Japanから購入した。
【0072】
(d−2)エンドグリコセラミダーゼ反応
ラクトシルセラミド(50 nmol)を、0.3%のコール酸ナトリウム及び BSA (10μg)を含む50μLの50 mM 酢酸アンモニウム溶液(pH 6.0) に溶解し、2mUの酵素とともに37℃で15又は60分間インキュベートした。試料溶液を、示した反応時間の直後にSep-Pak C18カトリッジカラムで処理し、水で溶出させた画分(4 mL) を回収した。各画分を直ちに凍結乾燥し、DMSO (1mL)に溶解し、ERMSで分析した。
【0073】
(d−3)ナトリウム付加したヘミアセタール分子はCID条件下でアノマー化しないという知見の一般性
ナトリウム付加したヘミアセタールはCID条件下でアノマー化しないという事実は、本実験の重要な基礎である。この現象の一般性を確認するために、β-Gal-(1→4)-β-GlcNAc-O(CH2)4NHBoc (S6), β-Gal-(1→3)-β-GlcNAc-O(CH2)4NHBoc (S7), 及びβ-Gal-(1→3)-α-GlcNAc-O(CH2)4NHBoc (S8)などの一連のBoc-保護されたアミノブチルグリコシドを用いて、上記と同様の実験を行った。これらの化合物は全て、ナトリウムを付加したヘミアセタールイオン種をMS3 段階で与え、これをMS4 及びMS5でERMS分析に供した(図7)。図7に示す通り、[β-Gal-(1→3)-α-GlcNAc-OH+Na]+ (図7A)及び[β-Gal-(1→3)- β-GlcNAc-OH+Na]+ (図7C) のERMSは異なっていたが、MS4 (図7A, C, E) 及びMS5 (図73 C, D, F)について得たERMSは区別できなかった。
【0074】
(B)結果
(B−1)ERMS相関法
本発明者らはナトリウム付加分子とフラグメントの様々なフラグメント化過程について、四重極イオントラップ質量分析計(QIT-MS)を使用して観測してきた。この結果、フラグメント化過程はほとんどの場合S字形のプロフィールであることが判明した。GD1aとGD1bの開裂を例にとると、ERMSは、フラグメント化過程に関する定量的情報のみならず、CIDのエネルギー依存性をも提供する。したがって、開裂に必要な活性化エネルギーは、GD1bについてより小さいことが分かる(図1 C,D)。この情報は、ERMSによってのみ入手可能である。ここで、生成イオンはそれらの前駆体から、互いに独立の関係で、直接生成したように見える。これは、各々の生成物イオンの前駆体イオンに対するERMS相関において強い直線相関が観察されたことから明らかとなった(図1 E,F)。この結果は、印加されるCIDエネルギーとは無関係に、個々のフラグメントイオンがそれぞれにそれらの共通前駆体に由来することを初めて示すものである。これにより2つの重要な点が明らかとなった。すなわち、1)個々のフラグメントイオンの比率は、CIDエネルギーに無関係に一定である、2)個々のフラグメントイオンの比率に関する定量的データ、あるいは、ERMSにおけるプラトー値を一般的なCID実験において使用するサンプル量による実験(データポイント)からERMS相関図におけるy切片から推定できる。
【0075】
本発明者らは遊離糖のアノマー立体異性を決定するためにERMS相関を利用した。また、このために変旋光のプロセスの分析と、グリコシダーゼの作用機序を推定した。ヘミアセタール種の分析には、以下の基準を満たす必要がある。すなわち、1)個々のアノマーを区別する、2)気相でのアノマー化の否定。図2Aは、前駆体1と前駆体2が同じである場合、それらが同じMSnスペクトルを与えなければならないという仮説を示している。ここでの目的は、アノマー化に関する実験のためのセットアップである。前駆体1がα-アノマーであり、かつ、アノマー化が起こるとき、前駆体2はα-とβ-アノマーの混合物となり各々のアノマーから識別可能である可能性がある。この実験のためには、α-、β-各々のアノマーが異なるスペクトルを与えることを確認することが必要である。そこで、前駆体1を一連の生成物イオン(生成物1)を生ずるフラグメンテーション反応にかける、このとき前駆体1と同じm/z値のイオン種(おそらく残留する前駆体)を単離し(前駆体2)、同じ状況の下で再びCIDに供する。前駆体1と2の構造が同一の場合、得られるフラグメント化プロフィールは同じとなる。
【0076】
まず、α-とβ-ヘミアセタール体が判別可能であるかどうか(基準1)について述べる。ヘミアセタール構造以外の結合が開裂する条件下におけるヘミアセタール構造の反応を検討するためラクトース(2糖)を用いた。ナトリウムの付与された4-アミノブチルβ-ラクトシド(2β)がCID状況の下でナトリウムの付与されたラクトース(β-配置と考えられる)を与え、GM3ガングリオシドに由来する同一のm/zのイオンとのとERMSの比較から、ヘミアセタールイオン種が構造解析に役立つことを明らかにしている(19)。しかし、α-アノマーのERMSは報告されていなかった。そこで、両アノマー(図2B)の気相における合成をおこないERMS解析をおこなった。ERMS相関解析により、形成されたイオンが異なるスペクトルを与えることを確認した。(図2 C,D)(K. Suzuki, S. Daikoku, T. Ako, A. Kurimoto, A. Ohtake, et al., Anal. Chem. 79, 9022-9029 (2007))
【0077】
全ての考察はアノマー化がCID条件下で起こらないという仮定に依存するため、3α-、および、β-体と考えられるラクトース(3α/β)を用い検討した(基準2)。
両方のイオンについて、前駆体の約25%が消費される[0.86V(α)と0.92V(β)] CIDエネルギーでMS5の段数でERMS実験をおこなった。それぞれ、MS4で得られたスペクトルとMS5のそれらは各々のα-とβ-アノマーのそれとよく一致し、(図2 E,F)MS5の際、残留した前駆体イオンは初期段階(MS4)のものと同じ構造であったことが判明した。(図2 G,H)もし平衡が存在するならば、MSnとMSn+1で観測されるα-とβ-アノマーのスペクトルは異なり、平衡化混合物のスペクトルに収束する方向に変化すると考えられる。実験では、しかしながら、MS4とMS5で得られたスペクトルは、α-、および、β-ラクトース共に見分けがつかなかった。これはアノマー(少なくともナトリウム付加体)の平衡が気相において起こらない、あるいは、検出不可能なレベルであったことを示唆する。ここで、様々なフラグメントイオンが生じている点に留意する必要がある。このことは、異性化のために必要な充分なエネルギー(もし可能であれば)が前駆体に印加されていたことを示す。このようにして、ヘミアセタール(ナトリウム付加体)はCID条件下において気相ではアノマー化しないことが初めて示された。
【0078】
(B−2)旋光度の測定
以上の結果から、ラクトースのナトリウム付加体のアノマー異性に関する議論が可能となり、また、ヘミアセタールのα-とβ-両アノマーが識別可能であることを証明した。結果に基づいて、水溶液(3A図)で起こっているラクトースの変旋光を検討した。それぞれα-体とβ-体の比率を高めたラクトース[α:β=約98:2、および、約30:70]の水溶液を25度でインキュベートし、各々の溶液の一定量をERMSに基づいて分析した。観察されたフラグメントとイオンについてERMS相関図から推定されたy切片は、質量分析法が変旋光の測定に用いることができることを示した。(図3 B)これは、旋光計によって確認された(3C図)。質量分析法によれば、平衡状態の3つのフラグメントの個々のシグナル強度は、16%(m/z 203)、56%(m/z 305)、28%(m/z 347)である。アノマーの比率(α/β)は、核磁気共鳴(NMR)を使用することで42:58であると推定された。このようにしてERMS相関法が変旋光を測定することに非常に役立つということを証明した(3C図)。
【0079】
(B−3)グリコシダーゼ反応機構の解析
本方法の有用性を拡大するために酵素反応メカニズムを明らかにする実験をおこなった。グリコシダーゼの機序の研究は、グリコシド交換反応により医学的に重要な化合物を合成しうるため重要である。エンドグリコセラミダーゼは、ガングリオシド等の糖脂質を基質とし、アノマー中心の立体化学の保持でグリカンとセラミドを切断する加水分解酵素の1つである。グリコシダーゼの反応機構の研究は、通常、NMRやX線結晶解析により達成される。本法を用いてRhodococcus sp.由来のエンドグリコセラミダーゼIIの反応機構を分析した。ラクトシルセラミドの酵素反応を15、60分間おこなった(4A図)。反応混合物の一部を取り出し生成したラクトースを回収、ERMS相関によって分析、平衡状態にあるラクトースのデータと比較した。(SI)ここで初期生成物は経時的にアノマー化するため、α-、β-ラクトースで観察される共通イオン(m/z 203、305と347によるイオン)のy切片の微妙な違いを観察することとなる。予想通り、15分(丸数字1)後に得られる混合物と60分(丸数字2)は、異なる値を与えた。特に、反応の初期段階(15分)においては平衡状態にあるラクトースの値(丸数字4)から離れて値を持つことが判明した。また、これらの値は、β-アノマー(丸数字3)によって示される平均値)のそれらに近いことがわかった(図3B)。このことは、初めに形成されたラクトースがβ-配置を有し、アノマー化したことを示している。このように、エンドグリコセラミダーゼIIがβ-保持メカニズムであることが本法により確認された。この質量分析法に基づく方法がグリコシダーゼのメカニズム研究に有効であることを初めて示した。
【図面の簡単な説明】
【0080】
【図1】図1のAは、ガングリオシドGD1aとフラグメンテーションパターンの模式図と、MS/MSスペクトルを示す。ナトリウム付加した分子を分析した。ガングリオシドのメチルエステルをCID分析のために使用した。図1のBは、GD1bの構造及びMS/MS スペクトルを示す。図1のCは、GD1aのERMSを示す。プレカーサーイオンの50%消費に必要なエネルギーを矢印で示す。図1のDは、GD1bのERMSを示す。プレカーサーイオンの50%消費に必要なエネルギーを矢印で示す。図1のEは、GD1a.のERMS 相関、図1のFは、GD1bのERMS 相関を示す。菱形はシアル酸(Sia); 灰色の○はガラクトース(Gal); 四角はN-アセチルガラクトサミン(GalNAc); 黒の○はグルコース(Glc);
【図2】図2のAは、CID条件下での所定のイオンの構造変化を調べる概念を示す。プレカーサー1のCID上に、プロダクト1が生成する。CID過程のある時点で、前のときと同じm/z値を有するプレカーサー2を単離し、これを2回目のCIDに供する。その後、2つのプレカーサーの特性を、フラグメンテーションプロファイルを比較することによって測定する。図2のBは、ナトリウム付加したヘミアセタール種の気相合成及びα- (3α) 及びβ-ラクトース (3β)の構造を示す。図2のCは、ナトリウム付加したα-ラクトース3α (m/z 365)のERMS相関プロット(MS4)を示す。図2のDは、ナトリウム付加したβ-lactose 3β (m/z 365)のERMS相関プロット(MS4)を示す。図2のEは、ナトリウム付加した3αのERMS相関プロット(MS5)を示す。図2のFは、ナトリウム付加した3βのERMS相関プロット(MS5)を示す。G,Hは、MS4 及びMS5においてERMSについて得た相対プロダクト強度の相関を示す。理想曲線は、y = xである。図2C−Fにおいて、灰色の○はガラクトース(Gal); 黒の○はグルコース(Glc)。
【図3】図3のAは、ラクトースのアノマー化の模式図を示す。図3のBは、ナトリウム付加したα-enriched (5) 及びβ-enriched (6) ラクトースの平衡の観察結果を示す。両方の反応に見られた共通のフラグメントの相対強度(ERMSカウンタープロットにおいて各プロダクトについて見積もったy-切片を用いた)をプロットした。図3のCは、偏光計で得たデータを示す。図4Dは、1H NMRで得たデータを示す。アノマー性プロトンについて計算したα/β比を用いた。
【図4】図4のAは、エンドグリコセラミダーゼがグリコスフィンゴ脂質を加水分解してグリカンとセラミドを生成することを示す。ラクトシルセラミド(LacCer)を基質として使用した。白丸(ガラクトース)、黒丸(グルコース)。図4のBは、エンドグリコセラミダーゼで生成したラクトースのアノマー立体配置の分析結果を示す。m/z 203, 305及び347のイオンについてのERMS相関プロットから得たy切片の見積もりを示す。反応は、15分後(丸数字1)及び60分後(丸数字2)に停止させた。丸数字4は60分後の反応培地のラクトース、丸数字3は平衡化したラクトース(水中)。
【図5】図5は、4-(Boc-アミノ)ブチルグリコシドの1H-NMRを示す。
【図6】図6は、DMSO-d6中のラクトン溶液の1H NMRスペクトルを示す。溶液の一部を溶液試料から取り出し、凍結乾燥後に測定した。スペクトルの一部を(OH-1)を示す。δ6.33 (OH-1α) 及び6.67 (OH-1β)。
【図7】図7は、幾つかのナトリウム付加したヘミアセタール種のERMSを示す。個々のヘミアセタール種は、MS3でCID条件下で生成させ、MS4 及びMS5でERMS解析に供した。
【特許請求の範囲】
【請求項1】
(a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求め、(c)該相関と、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を比較することを特徴とする糖鎖構造解析方法。
【請求項2】
(a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とをプロットしてグラフを作成し、(c)該グラフと、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率をプロットして得られたグラフとを比較することを特徴とする糖鎖構造解析方法。
【請求項3】
工程(b)において、少なくとも1回以上のCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とを少なくとも1点以上プロットする、請求項2に記載の糖鎖構造解析方法。
【請求項4】
工程(b)において、少なくとも2以上の異なるCIDエネルギーを用いてCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とを少なくとも2点以上プロットする、請求項2又は3に記載の糖鎖構造解析方法。
【請求項5】
工程(b)において作成したグラフを直線で近似することを特徴とする、請求項2から4の何れかに記載の糖鎖構造解析方法。
【請求項6】
工程(b)において作成したグラフを直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率を算出し;工程(c)において構造既知の参照糖鎖について得られたグラフも直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率を算出し;上記工程(b)及び工程(c)において算出した百分率を比較することを特徴とする、請求項5に記載の糖鎖構造解析方法。
【請求項7】
工程(b)において作成したグラフを直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率と、上記の直線の傾きを算出し;工程(c)において構造既知の参照糖鎖について得られたグラフも直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率と、上記直線の傾きを算出し;上記工程(b)及び工程(c)において算出した百分率及び傾きを比較することを特徴とする、請求項5又は6に記載の糖鎖構造解析方法。
【請求項1】
(a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を求め、(c)該相関と、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率との相関を比較することを特徴とする糖鎖構造解析方法。
【請求項2】
(a)目的糖鎖について特定のm/zのフラグメントイオンが得られるまでCID−MSn測定を行い、(b)該フラグメントイオンについてさらにCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とをプロットしてグラフを作成し、(c)該グラフと、構造既知の参照糖鎖のプレカーサーイオン量の総イオン量に対する百分率と特定のm/zのプロダクトイオン量の総イオン量に対する百分率をプロットして得られたグラフとを比較することを特徴とする糖鎖構造解析方法。
【請求項3】
工程(b)において、少なくとも1回以上のCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とを少なくとも1点以上プロットする、請求項2に記載の糖鎖構造解析方法。
【請求項4】
工程(b)において、少なくとも2以上の異なるCIDエネルギーを用いてCID−MS/MS測定を行い、プレカーサーイオン量の総イオン量に対する百分率と、特定のm/zのプロダクトイオン量の総イオン量に対する百分率とを少なくとも2点以上プロットする、請求項2又は3に記載の糖鎖構造解析方法。
【請求項5】
工程(b)において作成したグラフを直線で近似することを特徴とする、請求項2から4の何れかに記載の糖鎖構造解析方法。
【請求項6】
工程(b)において作成したグラフを直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率を算出し;工程(c)において構造既知の参照糖鎖について得られたグラフも直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率を算出し;上記工程(b)及び工程(c)において算出した百分率を比較することを特徴とする、請求項5に記載の糖鎖構造解析方法。
【請求項7】
工程(b)において作成したグラフを直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率と、上記の直線の傾きを算出し;工程(c)において構造既知の参照糖鎖について得られたグラフも直線で近似し、プレカーサーイオン量の総イオン量に対する百分率が0%の場合における特定のm/zのプロダクトイオン量の総イオン量に対する百分率と、上記直線の傾きを算出し;上記工程(b)及び工程(c)において算出した百分率及び傾きを比較することを特徴とする、請求項5又は6に記載の糖鎖構造解析方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2010−133707(P2010−133707A)
【公開日】平成22年6月17日(2010.6.17)
【国際特許分類】
【出願番号】特願2008−157947(P2008−157947)
【出願日】平成20年6月17日(2008.6.17)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年5月16日 第56回質量分析総合討論会にて発表
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(平成15年度独立行政法人新エネルギー・産業技術総合開発機構「基盤技術研究促進事業(民間基盤技術研究支援制度)/糖鎖の極微量構造解析技術開発研究」、平成16年度及び平成17年度独立行政法人新エネルギー・産業技術総合開発機構「基盤技術研究促進事業(民間基盤技術研究支援制度)/糖鎖の極微量構造解析技術開発研究」、産業活力再生特別措置法第30条の適用を受ける特許出願)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
【公開日】平成22年6月17日(2010.6.17)
【国際特許分類】
【出願日】平成20年6月17日(2008.6.17)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年5月16日 第56回質量分析総合討論会にて発表
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(平成15年度独立行政法人新エネルギー・産業技術総合開発機構「基盤技術研究促進事業(民間基盤技術研究支援制度)/糖鎖の極微量構造解析技術開発研究」、平成16年度及び平成17年度独立行政法人新エネルギー・産業技術総合開発機構「基盤技術研究促進事業(民間基盤技術研究支援制度)/糖鎖の極微量構造解析技術開発研究」、産業活力再生特別措置法第30条の適用を受ける特許出願)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
[ Back to top ]