
糸状菌におけるプロテアーゼ抑制剤及びその変異体の発現
本発明は糸状菌におけるプロテアーゼ抑制剤及びその変異体の発現に関する。本発明は融合核酸、ベクター、融合タンパク質及びプロテアーゼ抑制剤、及びその生産方法を開示する。本発明による生産方法により大量のタンパク質治療薬の生産が可能となる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は糸状菌におけるプロテアーゼ抑制剤及びその変異体の発現の方法に関する。本発明は融合核酸、ベクター、融合ポリペプチド及びプロテアーゼ抑制剤を得る方法を開示する。
【背景技術】
【0002】
プロテアーゼは広範囲の生物学的プロセスに関わっている。プロテアーゼとプロテアーゼ抑制剤のバランスの崩壊はしばしば組織の病理的破壊に関係して来る。
【0003】
組織損傷におけるプロテアーゼの役割に焦点をあてた多くの研究がされており、プロテアーゼとプロテアーゼ抑制剤のバランスが組織の統一性を維持する主要決定要素であると考えられている。好中球を含み、炎症を起こした細胞のセリーン プロテアーゼは肺気腫、関節炎、アトピー性皮膚炎、及び疥癬等の種々の炎症傷害に関わっている。
【0004】
プロテアーゼはまたある種の癌の進展に役割をも果たしていると考えられる。正常な細胞は、細胞外マトリクス(exteracellular matrix, ECM)と呼ばれる複雑なタンパク質ネットワークと常時接触を保っている。ECMは細胞の移動に対するバリアーであり、癌細胞は転移するためにはその付着を立ち切り、分解し、そしてECMを通じ移動する手段を作り出さねばならない。プロテアーゼは他のタンパク質を分解する酵素であり、長い間、ECMを噛み千切ることによってその元の位置から腫瘍細胞を解放することを助ける酵素であると考えられていた。最近の研究によれば、プロテアーゼはプロテアーゼにより活性化する受容体−2(Protease-Activated Receptor-2, PAR2)と呼ばれる腫瘍細胞膜に含まれるタンパク質の活性化により細胞の形を変化させ、またその運動を促進することが分かった。これにより細胞の運動組織を活性化する多くの細胞内反応が起こる。したがって、腫瘍の転移における第一ステップの一つは、細胞の形が移動の方向に面しているその一端が明確に突起した形になるように再編されることであると仮定されている。そして細胞は血管壁を通り、そして遠く離れた場所に移動し、結局再付着して転移腫瘍を作り出す。例えば、ヒトの前立腺表皮細胞は構造的に前立腺特異抗原(PSA)、精漿(seminal plasma)の通常の構成要素であるカリクレイン(kallikrein)様のセリンプロテアーゼを分泌する。プロテアーゼは細胞外マトリクスを分解し、癌状細胞の侵入を容易にするように作用する。
【0005】
合成及び天然プロテアーゼ抑制剤はインビボ及びインビトロにおいて腫瘍の進行を抑制することが分かっている。これまでの研究、調査によると、セリンプロテアーゼ抑制剤、あるいはセルピン(SERPINS)として分類される、構造的に関連するタンパク質のファミリーに属するある種のプロテアーゼ抑制剤は、好中菌エラスターゼ(neutrophil elastase)のみならず、トリプシン(trypsin)、カテプシンG(cathpsin G)、トロンビン(thrombin)、組織カリクレイン(kallikrein)を含むいくつかのプロテアーゼを抑制することが知られている。セルピン(SERPINS)はインビトロのカルシノゲン(carcinogen)誘発形質転換、及び動物モデル系でのカルシノゲン変異(carcinogenesis)の予防及び抑制に極めて有効である。精製プロテアーゼ抑制剤を計画的に与えることにより、関節の炎症並びに軟骨及び骨の破壊も減らすことができる。
【0006】
プロテアーゼ抑制剤を局所的に投与する場合として、よく見られる皮膚の炎症症状であるアトピー性皮膚炎があるが、これは身体の一部の数箇所に留まる場合と、または身体の広い範囲に拡がる場合がある。プロテアーゼ抑制剤に脱色活性及び紫外線で誘発される色素沈着を防止する力があることは、インビトロ及びインビボで実証されている。Paine et al., Journal of Investigative Dermatology 116, 587-595(2001)。プロテアーゼ抑制剤はまた傷の治癒を助けることが分かっている(http://www.sciencedaily.com/releases/2000/10/001002071718.htm)。分泌白血球プロテアーゼ抑制剤は組織破壊を逆転させ、局所的に適用された場合は瑕の治癒プロセスを促進することが実証された。さらに、セリンプロテアーゼ抑制剤はまた紅斑性狼瘡(lupus erythematosus)患者の痛みを和らげることができる(米国特許番号6537968参照)。
【0007】
上記で見たように、プロテアーゼ抑制剤はプロテアーゼの活動に干渉する。自然発生のプロテアーゼ抑制剤は、穀物(オート、大麦、メイズ)芽キャベツ(Brussel sprout)、玉ねぎ、ビートの根、小麦、シコクビエ(finger millet)及びピーナツの様に種々の食物に存在する。興味あるソースに大豆がある。大豆での平均含有水準は、二つの最も重要なプロテアーゼ抑制剤であるKunitz及びBowman-Birkで、各々約1.4%及び0.6%である。この様に含有水準が低いため、診療的に応用するには自然のプロテアーゼ抑制剤を単離することは現実的とはいえない。
【0008】
したがって、血液感染性媒体が哺乳類の組織培養細胞で作り出される場合には、血液感染性媒体に関連したリスクも減らし、または除去するプロテアーゼ抑制剤及びその変異体を大量に生産する方法が求められる。本明細書記載の生産方法は大量のタンパク質治療剤の生産を可能にする。
【発明の開示】
【0009】
発明の簡単な概要
本明細書は核酸、細胞及びプロテアーゼ抑制剤及びその変異体の生産方法を提供する。
【0010】
第一の実施の形態においては、機能的(functional)プロテアーゼ抑制剤をコードする核酸を提供する。一つの特徴として第一、第二、第三及び第四の核酸配列と動作可能にリンクされた(operably linked)調節塩基配列を含む核酸が提供される。第4の核酸配列に続き終了配列が提供される。
【0011】
第二の特徴として、第一の核酸配列は第一の糸状菌においてシグナルポリペプチド機能部位(functional)を分泌配列としてコードし、第二の核酸は分泌ポリペプチド、または通常の第一または第二の糸状菌から分泌されるその機能部位をコードし、第三の核酸は切断可能なリンカーをコードし、そして第四の核酸はプロテアーゼ抑制剤、またはその断片をコードする。
【0012】
第三の特徴として、プロテアーゼ抑制剤をコードする核酸配列を含む発現カセットを提供する。
【0013】
第四の特徴として、本発明はプロテアーゼ抑制剤変異体をコードするポリヌクレオチドに関する。ポリヌクレオチドは、少なくとも一つのループが変えられたボウマン ビルク(Bowman-Birk)抑制剤変異体をコードすることもある。ポリヌクレオチドは、少なくとも一つのループが変えられた大豆トリプシン抑制剤(Soybean Trypsin Inhibitor)変異体をコードすることもある。
【0014】
第二の実施の形態においては、機能的プロテアーゼ抑制剤またはその変異体の発現方法が提供される。一つの特徴として、宿主細胞は(i)プロテアーゼ抑制剤またはその変異体をコードする核酸配列を含む発現カセットにより形質転換され、また(ii)プロテアーゼ抑制剤またはその変異体を発現させる適当な条件下で培養されることである。任意的に、この方法はさらにプロテアーゼ抑制剤またはその変異体を回収することを含む。
【0015】
第二の特徴として、宿主細胞は(i)プロテアーゼ抑制剤またはその変異体をコードする核酸配列を含む第一の発現カセットにより形質転換され、(ii)シャペロンをコードする核酸配列を含む第二の発現カセットで形質転換され(iii) プロテアーゼ抑制剤またはその変異体を発現させる適当な条件の下で培養されることである。任意的に、プロテアーゼ抑制剤またはその変異体が回収されることもある。一つの特徴として、プロテアーゼ抑制剤またはその変異体は融合タンパク質として発現される。任意的にこの方法はさらにプロテアーゼ抑制剤またはその変異体を回収する方法を含む。
【0016】
第三の実施の形態においては、プロテアーゼ抑制剤またはその変異体を発現させることのできる細胞を提供する。宿主細胞はプロテアーゼ抑制剤及びその変異体をコードする発現カセットにより形質転換される。宿主細胞はアスペルギルス(Aspergillus)及びトリコデルマ(Trichoderma)からなるグループから選択されることもある。
【0017】
第四の実施の形態においては、機能的プロテアーゼ抑制剤及びその変異体が提供される。一つの特徴として、機能的プロテアーゼ抑制剤及びその変異体は、グルコアミラーゼシグナル配列、プロ配列(prosequence)、触媒ドメイン及び成熟グルコアミラーゼのアミノ酸配列502までのリンカー領域、それに続くアミノ酸NVISKR、及び成熟プロテアーゼ抑制剤及びその変異体よりなる融合タンパク質として発現される。
【0018】
第二の特徴として、発現したタンパク質は融合タンパク質からプロテアーゼ抑制剤及びその変異体を解放するためプロテアーゼで処理されることである。第三の特徴として、本発明はプロテアーゼ抑制活性を持つポリペプチドを提供する、それは下記よりなるグループから選択される;
Bowman-Birk 抑制変異体
Soybean Trypsin抑制変異体
Bowman-Birk抑制剤
Soybean Trypsin 抑制剤 及び
少なくとも一つの変異体配列を含む骨格(scaffold)
本発明の他の目的、特徴及び利益は以下に述べる詳細な説明により明らかになろう。しかし、詳細な説明及び特徴的な実施例は本発明の好ましい実施形態を示しているが、これらは説明のために取り上げられているものであることを理解する必要がある。この詳細な説明の記載より本発明の範囲及び思想に属する種々の変更、及び修飾をすることは当業者に明らかであるからである。
【0019】
発明の詳細な説明
本発明は以下の定義、及び実施例を参照として用い詳細に説明する。すべての特許及び刊行物は、それらの特許及び刊行物に開示される配列を含み、本明細書において参照されているものは引用により明確に本明細書に組み入れられる。
【0020】
他の意味に解釈されるよう記載がない限り、本明細書で用いられる全ての技術、科学用語は本発明の属する技術分野の当業者により通常理解される意味と同じ意味を持つ。Singleton et al., DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOGY 2nd ED., John Wiley and Sons, New York (1994), and Hale & Marham, THE HARPER COLLINS DICTIONARY OF BIOLOGY, Harper Perennial, NY (1991)は、当業者に本発明で使用されている用語の多くを説明している一般的な辞書を提供する。これらに記載の方法及び材料で本明細書に記載のものと同一または類似のものは、本発明の実施または試験のために用いることができるが、本明細書には好ましい方法、及び材料が記載されている。数値の範囲は範囲を規定する数値を含む。他の意味に解される様に示されていない限り、核酸は、左から右方向に向けて、5’から 3’の方向に、またアミノ酸配列は、左から右方向に向けて、アミノ基からカルボキシ基の方向となっている。開業医師は、定義と用語については特にSambrook et al., 1989, 及び Ausubel FM et al., 1993を参照することを奨める。本発明は、記載された特定の方法、プロトコール、及び試薬に限定されるものではない。これらのものは内容が種々変わりうるからである。
【0021】
本明細書に記載の各見出し(heading)は本発明の実施の態様に限定するものではない。本発明は明細書全体を参照することにより明らかとなるからである。したがって、以下に引き続き定義される用語は、明細書全体を参照することにより十分な定義となる。
【0022】
定義
「発現カセット」(expression cassette)または「発現ベクター」(expression vector)は、標的細胞において特定の核酸の転写を許す特定の一連の核酸要素により、遺伝子組み換え法または合成法によって生成される核酸構築(construct)をいう。組み換え発現カセットはプラスミド、染色体、ミトコンドリアDNA、プラスチドDNA(plastid DNA)、ビールス、または核酸断片に組み入れることができる。典型的には、発現ベクターの組み換え発現カセット部分は、特に転写される核酸配列及びプロモータを含む。発現カセットはDNA構築及びその文法的な同等物と代替可能に使用することができる。
【0023】
本明細書で用いる「ベクター」(vector)は、核酸配列を細胞に転写するようにデザインされた核酸構築をいう。「発現ベクター」は異質細胞(foreign cell)に異種(heterologous)DNA断片を組み込み、そして発現させる能力を持つベクターをいう。多くの原核生物発現ベクター及び真核生物発現ベクターは市場で入手可能である。適当な発現ベクターを選択することは当技術分野の当業者の知識の範囲に属する。
【0024】
本明細書で用いる「プラスミド」(plasmid)はクローンベクターとして用いられ、そしてある真核生物において染色体外の自己複製遺伝子要素を形成し、または宿主染色体に合体する環状二重鎖(double-stranded;ds)DNA構築をいう。
【0025】
「核酸分子」(nucleic acid molecule)または「核酸配列」(nucleic acid sequence)はRNA, DNA及びcDNA分子を含む。遺伝子コードの縮重の結果、あるタンパク質をコードする多数のヌクレオチド配列が作り出されることがあることが理解されるであろう。
【0026】
本明細書に記載の「融合DNA配列」(fusion DNA sequence)は、5’から 3’方向に向け、第一、第二、第三、及び第四のDNA配列を含む。
【0027】
本明細書に記載の「第一の核酸配列」(a first nucleic acid sequence)または「第一のDNA配列」(first DNA sequence)は第一の糸状菌においてシグナルポリペプチド機能部位(functional)を分泌配列としてコードする。そのようなシグナル配列は、以下の配列を含む。すなわち、牛のキモシン(bovine chymosin), ヒトの組織プラスミノゲン活性剤(human tissue plasminogen activator),ヒトのインターフェロン(human interferon)から得られるシグナル配列を含む真核生物のシグナル配列、及びGwynne et al.,(1987) Bio/Technology 5, 713- 719に記載の合成コンセンサス真核生物(synthetic consensus eukaryotic)シグナル配列に加えて、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger variant awamori)、 アスペルギルス ニジェール(Aspergillus niger), アスペルギルス オリザ(Aspergillus oryzae)から得られるグルコアミラーゼ(glucoamylase), α-アミラーゼ(α-amylase)及びアスパルチル プロテアーゼ(aspartyl protease)のシグナル配列, トリコデルマ(Trichoderma)から得るセロビオヒドロラーゼ I(cellobiohydrolase I), セロビオヒドロラーゼ II(cellobiohydrolase II), エンドグルカナーゼI (endoglucanase I), エンドグルカナーゼIII(endoglucanase III)のシグナル配列、ノイロスポラ(Neurospora)及びフミコラ(Humicola)から得るグルコアミラーゼ(glucoamylase)のシグナル配列である。特に好ましいシグナル配列は、融合タンパク質を発現及び分泌させるために用いられる発現宿主により分泌されるポリペプチドに由来する配列である。例えば、アスペルギルス ニジェール(Aspergillus niger)から融合タンパク質を発現させ分泌させる場合、アスペルギルスニジェールから得られるグルコアミラーゼ(glucoamylase)のシグナル配列が好ましい。本明細書に記載の第一のアミノ酸配列は糸状菌において機能的である分泌配列に対応する。そのようなアミノ酸配列は、規定した第一のDNA配列によりコードされる。
【0028】
本明細書に記載の「第二のDNA配列」(second DNA sequence)は通常糸状菌から発現した「分泌ポリペプチド」(secreted polypeptide)をコードする。そのような分泌ポリペプチはアスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)、及び アスペルギルス ニジェール(Aspergillus niger), アスペルギルス オリザ(Aspergillus oryzae)から得られるグルコアミラーゼ、α-アミラーゼ、及びアスパルチル プロテアーゼ(aspartyl proteases)、およびトリコデルマ(Trichoderma)から得るセロビオヒドロラーゼ I(cellobiohydrolase I),セロビオヒドロラーゼ II(cellobiohydrolase II), エンドグルカナーゼI (endoglucanase I), エンドグルカナーゼIII(endoglucanase III)、ノイロスポラ(Neurospora)種及びフミコラ(Humicola)種から得られるグルコアミラーゼ(glucoamylase)を含む。第一のDNA配列の場合と同様に、望ましい分泌ポリペプチは、糸状菌発現宿主により自然に分泌されるポリペプチドである。したがって、例えば、アスペルギルスニジェール(Aspergillus niger)を用いる場合、望ましい分泌ポリペプチはアスペルギルス ニジェールから得られるグルコアミラーゼ(glucoamylase)及びαアミラーゼ、最も好ましくはグルコアミラーゼである。一つの特徴的な場合においては、グルコアミラーゼはアスペルギルス グルコアミラーゼと95%、96%、97%、98%、または99%以上相同である。
【0029】
アスペルギルス グルコアミラーゼが第二のDNA配列によりコードされる分泌ポリペプチドである場合は、任意的にプロ配列(prosequence)を含み、タンパク質全体、またはその一部が用いられる。したがって、切断リンカーポリペプチドはアミノ酸残基の468から509間の何れの位置においてもグルコアミラーゼに融合してことがある。他のアミノ酸残基は融合部位かもしれないが、上記の残基を用いることは特に有益である。
【0030】
「分泌ポリペプチドの機能する部分」(functional portion of a secreted polypeptide)または文法的な同等物(grammatical equivalent)は、端が切断されているとしても、正常な立体配置に折り畳まれる能力を維持している端の切断された分泌ポリペプチを意味する。例えば、アスペルギルス グルコアミラーゼ変異体アワモリによる牛のキモシン生成の場合において、成熟グルコアミラーゼの11番目のアミノ酸に続くプロキモシン(prochymosin)の融合はプロキモシン(prochymosin)の生成に比べて利点をもたらさないことが示された(米国特許5,364,770)。USSN 08/318,494において、プレプログルコアミラーゼ(preproglucoamylase)のC末端において成熟グルコアミラーゼ(glucoamylase)の297番目までのアミノ酸にプロキモシン(prochymosin)を融合させ、それに加えて成熟グルコアミラーゼ(glucoamylase)のアミノ酸1−11を繰り返すことは、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)において分泌されるキモシン(chymosin)を作り出さないことが示されている。後のケースにおいては、融合タンパク質に存在するグルコアミラーゼ(glucoamylase)触媒ドメインの部分(約63%)は正しく折り畳まれることが困難と思われ、したがって、細胞によって分泌され得なかったであろう異常な、誤った折り畳み方をし(misfolded)及び/または不安定な融合タンパク質が生成されたのかも知れない。部分的触媒ドメインが正しく折り畳むことができないことは付着キモシン(chymosin)の折り畳みの妨げとなったかも知れない。したがって、そのポリペプチドが付着している所望するポリペプチドから独立にその通常の立体配置に従い折り畳むことができるように、自然分泌のポリペプチドのドメインに十分な数の残基が存在している必要があると思われる。
【0031】
殆どの場合、分泌ポリペプチドの部分は正しく折り畳まれ、そしてまた折り畳みがない場合に比べて分泌が増加する。
【0032】
同様に殆どの場合、分泌ポリペプチドの先端を切り取ることは、機能的部分が生物学的機を維持することを意味する。好ましい実施の形態においては、例えば、基質結合ドメインのような他の機能的ドメインが用いられることもあるが、分泌ポリペプチドの触媒ドメインが用いられる。アスペルギルス ニジェール(Aspergillus niger)及びアスペルギルス ニジェール変異体アワモリ グルコアミラーゼ(Aspergillus niger var. awamori glucoamylase)の場合、好ましい機能部位は酵素の触媒ドメイン部分を保持し、アミノ酸1から471を含む。さらに、好ましい実施の態様においては、触媒ドメイン及び全てのまたは一部のリンカー領域を用いる。代替的に、グルコアミラーゼ(glucoamylase)のでんぷん結合領域が使用される場合もあり、それはアスペルギルス ニジェール変異体アワモリグルコアミラーゼ(Aspergillus niger var. awamori glucoamylase)のアミノ酸509−616 を含む。
【0033】
本明細書に記載の「第3のDNA配列」(third nucleic sequence)は切断リンカーポリペプチドをコードするDNA配列を含む。そのような配列はグルコアミラーゼ(glucoamylase)のプロ配列、牛のキモシン(chymosin)のプロ配列、スブチリシン(subtilisin)のプロ配列、ヒトの免疫不全ウィルスプロテアーゼを含むレトロウィルスプロテアーゼ(retroviral protease)のプロ配列含み、またトリプシン(trypsin), Xa因子コラゲナーゼ(factor Xa collagenase)、クロストリピン(clostripin)、スブチリシン(subtilisin), キモシン(chymosin),イーストKEX2 プロテアーゼ(yeast KEX2 protease)、アスペルギルスKEXB(Aspergillus KEXB)等により認識され、かつ切断されるアミノ酸配列をコードするDNA配列を含む。例えば、Marston, F. A.O. (1986) Biol. Chem J. 240,1-12を参照願いたい。そのような第3のDNA配列はまた、臭化シアン(cyanogen bromide)により選択的に切断されるアミノ酸メチオニン(methionine)をコードすることもある。第3のDNA配列は融合ポリペプチドの切断をもたらす特定の酵素、または化学物質により認識されることの必要なアミノ酸配列をコードするのみで足りることは理解されるべきである。このように、例えば、全体のグルコアミラーゼ(glucoamylase)、キモシン(chymosin)またはスブチリシン(subtilisin)の全体のプロ配列を使用することは必要ない。むしろ、適当な酵素により認識され、また切断されるのに必要なプロ配列の部分のみが必要になる。
【0034】
第3の核酸は、融合ポリペプチド質の切断をもたらす特定の酵素、または化学物質により認識されるのに必要なアミノ酸配列をコードするのみでよいことは理解すべきである。
【0035】
特に好ましい切断リンカーは、天然のアスペルギルスKEX2類似のプロテアーゼ(Aspergillus KEX2-like protease)により切断され得るKEX2プロテアーゼ認識部位(Lys 及びArg)、Lys 及びArgのトリプシン プロテアーゼ(trypsin protease)認識部位、及びエンドプロテナーゼ-Lys-C(endo- proteinase-Lys-C)切断認識部位である。
【0036】
本明細書に記載の「第4のDNA配列」(forth DNA sequence)は「所望されるポリペプチド」(desired polypeptide)をコードする。そのような所望されるポリペプチドはプロテアーゼ抑制剤及びその変異体を含む。
【0037】
対応する4つのアミノ酸配列をコードする上記規定の4つのDNA配列は組み合わされて「融合DNA配列」(fusion DNA sequence)を形成する。そのような融合DNA配列は5’末端から3’末端に掛けて第1,2、3及び第4のDNA配列の順に適切な読み枠(reading frame)に組み立てられる。そのように組み立てられて、DNA配列は糸状菌のシグナルペプチド機能部位を分泌配列として、通常糸状菌から分泌される分泌ポリペプチまたはその部分、切断リンカーポリペプチド及び所望のポリペプチドをコードする「融合ポリペプチド」(fusion polypeptide),「融合タンパク質」(fusion protein)または「融合類似体」(fusion analog)をアミノ末端からコードする。
【0038】
本明細書に記載の「所望するタンパク質」(desired protein)または「所望するポリペプチド」(desired protein)は、分泌を増大させる構築に融合されていない熟成した形のポリペプチドまたはタンパク質をいう。したがって、「所望するタンパク質」または「所望するポリペプチド」は融合されていない形の宿主細胞により発現され、また分泌されるタンパク質をいう。
【0039】
本明細書に記載の「融合ポリペプチド」(fusion polypeptide),「融合タンパク質」(fusion protein)または「融合類似体」(fusion analog)は、アミノ末端から、宿主細胞においてシグナルペプチド機能部位を分泌配列機能として、通常宿主細胞から分泌される分泌ポリペプチまたはその部分、切断リンカーポリペプチド及び所望のポリペプチドをコードする。融合タンパク質は、融合タンパク質において他のタンパク質配列から自由である所望するタンパク質を生み出すために、例えば、プロテアーゼなどの宿主細胞酵素によりプロセスされるかもしれない。本明細書に記載の「融合類似体」(fusion analog)または「融合ポリペプチド」(fusion polypeptide),または「融合タンパク質」(fusion protein)は相互に交換可能に用いられることもある。
【0040】
本明細書に記載の「プロモータ配列」(promoter sequence)は、発現の目的で特定の糸状菌により認識されるDNA配列をいう。これは上記に定義された融合タンパク質をコードするDNA配列に動作可能にリンクされている。そのようなリンクすることには、融合DNA配列をコードするDNA配列の翻訳開始コドンとの関係でプロモータの位置を決めることを含む。プロモータ配列は融合DNA配列の発現を仲介する転写及び翻訳制御配列を含む。その例として、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)またはアスペルギルス ニジェール グルコアミラーゼ(Aspergillus niger glucoamylase)遺伝子(Nunberg, J. H. et al. (1984) Mol. Cell Biol, 4, 2306-2315; Boel, E. et al. (1984)EMBO J.3, 1581-1585)、アスペルギルス オリザ(Aspergillus oryzae)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori) またはアスペルギルス ニジェール(Aspergillus niger)、アルファアミラーゼ(alpha amylase)遺伝子、リゾムコール ミーヘイカルボキシルプロテアーゼ(Rhizomucor miehei carboxyl protease)遺伝子、トリコデルマ リーセイ セロビオヒドロラーゼ I (Trichoderma reesei cellobiohydrolase I)遺伝子 (Shoemaker, S. P. et al. (1984) 欧州特許出願番号 EPO0137280A1), アスペルギルス ニジュランス trpC (Aspergillus nidulans trp C)遺伝子 (Yelton, M. et al. (1984) Proc. Natl. Acad. Sci. USA 81, 1470-1474; Mullaney, E. J. et al. Mol. Gen. Genet. 199, 37-45) 、アスペルギルス ニジュランス alcA (Aspergillus nidulans alcA )遺伝子(Lockington, R. A. et al. (1986) Gene 33 137-149)、アスペルギルス ニジュランス amdS (Aspergillus nidulans amdS)遺伝子 (McKnight, G. L. et al. (1986) Cell 46, 143-147), アスペルギルス ニジュランスamdS (Aspergillus nidulans amdS)遺伝子 (Hynes, M. J. et al. (1983) Mol. Cell Biol 3 1430-1439), 及びSV40初期プロモータのようなより高度の真核プロモータ(Barclay, S. L. and E. Meller (1983) Molecular and Cellular Biology 3, 2117-2130)がある。
【0041】
同様に、「終了配列」は、転写を終了させる発現宿主により認識されるDNA配列である。これは発現される融合ポリペプチドをコードする融合DNAの3’末端に動作可能にリンクされている。これらの例には、アスペルギルス ニジュランス trpC (Aspergillus nidulans trp C)遺伝子 (Yelton, M. et al. (1984) Proc. Natl. Acad. Sci. USA 81, 1470-1474; Mullaney, E. J. et al. (1985)Mol. Gen. Genet. 199, 37-45)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)またはアスペルギルス ニジェール グルコアミラーゼ(Aspergillus niger glucoamylase)遺伝子 (Nunberg, J. H. et al. (1984) Mol. Cell. Biol 4、2306-253; Boel, E. et al. (1984), EMBO J. 3, 1581-1585 )、アスペルギルスオリザ(Aspergillus oryzae)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori) またはアスペルギルス ニジェール(Aspergillus niger)、アルファアミラーゼ(alpha amylase)遺伝子、リゾムコール ミーヘイ カルボキシルプロテアーゼ(Rhizomucor miehei carboxyl protease)遺伝子、(EPO 公開番号 0215 594)がル。ただし、本発明においては、どの糸状ターミネ-タ(terminator)も機能すると思われる。
【0042】
「ポリアデニレイション配列」(polyadenylation sequence)は、転写されるときに、転写されるmRNAにポリアデノシン残基を加える際に発現宿主により認識されるDNA 配列をいう。それは発現される融合ポリペプチドをコードする融合DNAの3’末端に動作可能にリンクされている。この例には,上記で述べた様に、アスペルギルス ニジュランス trpC (Aspergillus nidulans trp C)遺伝子 (Yelton, M. et al. (1984) Proc. Natl. Acad. Sci. USA 81, 1470-1474; Mullaney, E. J. et al. (1985)Mol. Gen. Genet. 199, 37-45)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)またはアスペルギルス ニジェールグルコアミラーゼ(Aspergillus niger glucoamy- lase)遺伝子 (Nunberg, J. H. et al. (1984) Mol. Cell Biol 4 2306-2315) (Boel, E. et al. (1984), EMBO J. 3, 1581-1585 )、アスペルギルスオリザ(Aspergillus oryzae)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori) またはアスペルギルス ニジェール(Aspergillus niger)、アルファアミラーゼ(alpha amylase)遺伝子、及びリゾムコール ミーヘイ カルボキシルプロテアーゼ(Rhizomucor miehei carboxyl protease)遺伝子を含む。しかしながら、本発明においてはどの糸状菌ポリアデニレイション配列も機能的であると思われる。
【0043】
本明細書に記載の「選択可能なマーカーをコードするヌクレオチド配列」(selectable marker-encoding nucleic sequence)は、糸状菌細胞で発現することができ、そして選択可能なマーカーの発現により、発現遺伝子を含む細胞に対して、対応するある選択された条件下において成長する能力を与えるヌクレオチド配列をいう。
【0044】
核酸が他の核酸配列と機能する関係におかれた場合、「動作可能にリンクされる」(operably linked))という。例えば、分泌リーダーをコードするDNAは、ポリペプチドの分泌に参加するプレタンパク質(preprotein)として発現する場合は、ポリペプチドのDNAに動作可能にリンクされ、プロモータまたはエンハンサー(enhancer)は、配列の転写に影響を与える場合はコード配列に動作可能にリンクされ、リボソーム結合部位は、翻訳を促進するように位置されている場合はコード配列に動作可能にリンクされている。一般に「動作可能にリンクされる」とは、リンクされるDNA配列が連続しており、分泌リーダーの場合には、連続しているとともに読取フェーズにあることをいう。しかしながら、エンハンサーは連続している必要はない。リンクは適当な制限部位において結紮によりなされる。もしそのような部位が存在しない場合は、通常の用法に従いオリゴヌクレオチド アダプターまたはリンカーが用いられる。
【0045】
本明細書に記載の「組み換えの」(recombinant)は、異種(hetero-)核酸配列の導入により修飾され、または細胞がそのように修飾された細胞に由来する細胞またはベクターについていう。したがって、例えば、組み換え細胞は天然の形(非組み換え)細胞の範囲内において同一の形で見出されない遺伝子を発現させ、またはそうでなければ、人の意図的な介在により発現されまたは発現がされなかった、またはそうでなければ異常に発現されるであろう天然遺伝子を発現させるのである。
【0046】
本明細書に記載の「発現」(expression)はポリペプチドが遺伝子の核酸配列に基づき生産されるプロセスをいう。このプロセスは、転写及び翻訳の両者を含む。したがって、「プロテアーゼ抑制剤発現」(protease inhibitor expression)は、その生成物にRNA前駆体、mRNA, ポリペプチド、翻訳後プロセスされるポリペプチド(post-translation processed polypeptide), 及びその派生物を含む、発現されるべき特定のプロテアーゼ抑制剤及びその変異体遺伝子の転写及び翻訳についていう。同様に、「プロテアーゼ抑制剤発現」はプロテアーゼ抑制剤及びその変異体を、図6に例示されているような形に転写、翻訳、及び組み立てることに関していう。例えば、プロテアーゼ抑制剤発現のアッセイには、適当な条件に置かれた場合の菌群の検査、ノーザンブロット分析(northern blot analysis)は勿論のこと、プロテアーゼ抑制剤タンパク質のウエスターンブロット(western blot)、及びプロテアーゼ抑制剤mRNAの逆転写酵素ポリメラーゼ連鎖反応(RT-PCR)アッセイを含む。
【0047】
本明細書に記載の「グリコシド化された」(glycosylated)は、オリゴ糖分子がタンパク質の特定のアミノ酸残基に加えられていることをいう。「脱グリコシド化された」(de-glycosylated)タンパク質はタンパク質からオリゴ糖分子を部分的にまたは完全に除去するように処理されたタンパク質である。「非グリコシ化された」(a-glycosylated)タンパク質は、タンパク質にオリゴ糖分子が付加されていないタンパク質である。これはオリゴ糖の付加を妨げるタンパク質における変異のためである。
【0048】
「グリコシド化されていない」(non-glycosylated)タンパク質はタンパク質にオリゴ糖が付着されていないタンパク質である。これには、オリゴ糖のタンパク質への付加に必要な酵素が存在しないなど種々の理由があるが、これに限定されるものではない。「グリコシド化されていない」(non-glycosy- lated)は、オリゴ糖がタンパク質に付加されなかったタンパク質と、オリゴ糖は付加されたが、結局除去されたタンパク質の両者を含む。「非グリコシ化された」(a-glycosylated)タンパク質は「グリコシド化されていない」(non- glycosylated)タンパク質であるかもしれない。「グリコシド化されていない」(non-glycosylated)タンパク質は、「非グリコシ化された」(a-glycosylated)タンパク質、または「脱グリコシド化された」(de-glycosylated)タンパク質であるかもしれない。
【0049】
本明細書に記載の「単離された」(isolated)または「精製された」(purified)は、それが自然状態において関係した少なくとも一つの成分から除去された核酸またはポリペプチドについていう。
【0050】
「実質的に含んでいない」(substantially free)は、約20%(乾燥重量)以下の他のタンパク質(すなわち、夾雑タンパク質)、約10%以下の他のタンパク質、約5%以下の他のタンパク質、約1%以下の他のタンパク質を含む所望するポリペプチドの製剤を含む場合をいう。
【0051】
「実質的に純粋」(substantially pure)は、本発明のタンパク質またはその断片に適用された場合、そのタンパク質が意図される用途に現実にまた適切に用いられる程度に、本質的に他の物質を含んでいないことを意味する。特にタンパク質が、例えば、タンパク質配列の決定、または製剤の生成において有用であるために十分純粋であり、また宿主細胞の生物学的構成物をほとんど含んでいないほどに純粋であることをいう。
【0052】
本明細書に記載の「標的タンパク質」(target protein)は、例えば、変異体抑制剤の結合によりその活動がブロックされる酵素、ホルモン等のタンパク質をいう。
【0053】
「変異体配列」(variant sequence(s))は、野生型プロテアーゼ抑制剤または他の骨格の結合ループを置換する短いポリペプチド配列をいう。変異体配列は、それが骨格において置換する結合ループ配列と同じ長さである必要はない。
【0054】
「骨格」(scaffold)は、変異体配列が導入される野生型タンパク質配列をいう。ある実施の形態においては、骨格は、例えば、ループのような置換される部分をもつであろう。例えば、本明細書に記載のSTI及び BBI配列は変異体配列の骨格といえよう。
【0055】
プロテアーゼ抑制剤
2つのタンパク質プロテアーゼ抑制剤が大豆から単離された。クニッツ型トリプシン抑制剤(Kunitz-type trypsin inhibitor)(大豆トリプシン抑制剤、STI)及びボウマンービルク プロテアーゼ抑制剤(Bowman-Birk inhibitor)(BBI) である。例えば、Birk, Int. J. Pept. Protein Res. 25: 113-131 (1985) 及びKennedy, Am. J. Clin. Neutr. 68: 1460S-1412S (1998)を参照願いたい。これらの抑制剤は変異体配列の骨格としての役割をする。
【0056】
さらに変異体配列を含む骨格における変更に加え、本明細書で用いられる他の所望するタンパク質はN末端において3つのグリシン残基、及び/またはC末端において6つのヒスチジン(histidine)残基を含む(図3および4を参照願いたい)。
【0057】
大豆トリプシン抑制剤(STI)
STIは安定した化学量論的合成物の形成によりトリプシンのタンパク質分解活性を抑制する(Liu, K., Chemistry and Nutritional value of soybean components. In : Soybeans, chemistry, technology and utilization. pp. 32-35 (Aspen publishers, Gaithersburg, Md., 1999を参照のこと)。STIは2つのジスルフィド架橋をもつ181のアミノ酸残基よりなり、おおよそ球形をしている(例えば、Song et al., J. Mol. Biol. 275: 347-63 (1998)を参照のこと)。2つのジスルフィド架橋は以下に記載のBBIのループと類似の2つの結合ループを形成する。
【0058】
クニッツ型トリプシン抑制剤(Kunitz-type trypsin inhibitor)(STI)は、プロテナーゼの初期の研究において中心の役割を果たし、生化学上及び運動作用における主要な基質として用いられ、プロテアーゼ抑制剤の活性の標準的メカニズムについての定義を導くものであった。
【0059】
ボウマン−ビルク抑制剤(Bowman-Birk Inhibitor(BBI)
BBI タンパク質はマメ科(leguminous)の種から単離された小さなタンパク質(60-90の残基)の動力学的にまた構造的に特徴のあるファミリーである。これは各々が独立した結合ループを含む2つの三環ドメインの対称構造を持つ。ループ1は特にトリプシンを抑制し、ループIIはキモトリプシン(chymotrypsin)を抑制する(Chen et al., J. Biol Chem. (1992) 267: 1990-1994; Werner & Wemmer Lin, et al., Eur. J. Biochem. (1993) 212: 549-555; Voss et al., Eur. J. Biochem. 242: 122-131)。これらの結合領域は各々「カノニカル ループ」(canonical loop)構造を含み、それは種々のセリンプロテアーゼ抑制剤中に見出される基調(motif)である (Bode & Huber, Eur. J. Biochem. (1992) 204: 433-451)。
【0060】
BBI はそれぞれ別個の反応部位においてプロテアーゼ トリプシン及びチモトリプシンを抑制する8 k-Daタンパク質である(Billings et al., Pro. Natl. Acad. Sci. 89:3120-3124(1992)を参照のこと)。STI及びBBIは大豆の種にのみ存在し、その植物の他のどの部分にも見出されていない(例えば、Birk, Int. J. Pept. Protein Res. 25: 113-131 (1985)を参照のこと)。
【0061】
数多くのBBIのイソ型(isoform)の特徴が明らかになっているが、配列番号7(図3)は約71のアミノ酸残基を持つ本明細書に記載の BBIの骨格のアミノ酸配列を示している。さらに、BBIは、NまたはC末端の何れかにおいて最大10個のアミノ酸残基が除去されることにより先端が切断されて(truncated)いることもある。例えば、種を乾燥させると、BBIはC末端の9または10のアミノ酸が除去されることがある。したがって、タンパク質分解は最初のジスルフィド結合以前、及び最終のジスルフィド結合直後においては大いに許容されるべきであり、その結果により通常標的となるタンパク質に対する結合に不利となることは起こらない。しかし、イソ型(isoform)または切断型の何れかを用いることが望ましい。
【0062】
プロテアーゼ抑制剤変異体
上記より理解されるように、STI及びBBIプロテアーゼ抑制剤はプロテアーゼを抑制する結合ループを持っている。本発明のプロテアーゼ抑制剤変異体はループI, ループII またはその両者に改変がなされている。ある実施の形態では、ループは標的タンパク質と相互に作用する配列に置換されている。
【0063】
ループはC2, C3, C4, またはC5抑制剤、コットン結合タンパク質(cotton binding protein)、コムプスタチン(Compstatin)等のような補体活性化経路抑制剤であるVEGF結合タンパク質に由来する配列により置換することができる。代替的に、変異体配列は、例えば、ファージ提示法、または他のスクリーニング法などの他の種々の公知の方法により選択することもできる。例えば、ランダムペプチド遺伝子ライブラリーはファージPIII遺伝子と融合され、ペプチドライブラリーはファージの表面に提示される。その結果、ファージ提示ライブラリーは標的タンパク質に曝されて、非特異的結合を除去するために緩衝液で洗浄される(このプロセスは、パニング(panning)と呼ばれることもある)。最終的に結合ファージ及びPCRでコードされるペプチドのDNA 配列は単離される。
【0064】
通常ループは変異体配列により置換される、すなわち、ペプチドで長さがアミノ酸の数で3−14、5−10が望ましい。より長い配列は、それらが所望する結合及び/または抑制を可能にする限り用いることができる。さらに、結合ループの代替としての用いるのに適したぺプチドは拘束ループ中に抑制されている場合、すなわち、二つのシステイン残基の間のジスルフィド結合の存在により形成されているループの場合には、そのペプチドは機能的コンフォーメイションをとる必要がある。特定の実施の形態においては、ペプチドはその長さがアミノ酸数で7から9である。この置換配列はまたプロテアーゼの抑制または標的タンパク質に対する結合をもたらす。
【0065】
あるケースにおいては、一つのアミノ酸を変えることが有利なことがある。特に、野生型STIまたはBBIの残基13番のアラニン(Alanine)をセリン(Serine)、グリシン(Glycine)またはグルタミン(Glutamine)に変えることもできる。
【0066】
融合タンパク質
各プロテアーゼ抑制剤及びその変異体は、宿主菌細胞により融合タンパク質として発現される。融合ポリペプチドを切断することにより、所望のタンパク質を解放することは有益であるが、それは必要ではない。融合タンパク質として発現され、分泌されるプロテアーゼ抑制剤及びその変異体は驚異的にその機能を保持する。
【0067】
対応する4つのアミノ酸配列をコードする上記規定の4つのDNA配列は、組み合わされて「融合DNA配列」を形成する。そのような融合DNA配列は、5’末端から3’末端に掛けて適切な読み枠に、第1、第2、第3、及び第4のDNA配列の順に組み立てられる。そのように組み立てられたDNAは、糸状菌のシグナルペプチド機能部位(functional)を分泌配列として、通常,糸状菌から分泌される分泌ポリペプチドまたはその部分、切断リンカーペプチド、及び例えばプロテアーゼ抑制剤及びその変異体のような所望のポリペプチドをコードする融合ポリペプチドをアミノ端末からコードする。
【0068】
融合タンパク質の生産は、例えば、米国特許5,411,873, 5,429,950, 及び5,679,543に開示されている方法を用いて実現することができる。その他の方法は技術分野でよく知れらている。
【0069】
組み換えプロテアーゼ抑制剤の発現
本発明の融合タンパク質の生産に関する範囲においては、本発明は組み換え遺伝子学分野の通常の技術に基づいている。本発明で用いられる一般的な使用方法を開示する基礎的文献は、Sambrook et al., Molecular Cloning, A Laboratory Manual (2nd ed. 1989); Kriegler, Gene Transfer and Expression : A Laboratory Manual (1990); 及び Ausubel et al., eds., Current Protocols in Molecular Biology (1994)を含む。
【0070】
本発明はプロテアーゼ抑制剤をコードする核酸配列を含む発現ベクターにより、導入され、形質転換され、または移入された糸状菌宿主細胞を提供する。温度、pH等の培養条件は、導入、形質転換、または移入までに親宿主細胞に適用される条件と同じであリ当業者には明らかであろう。
【0071】
基本的には、融合タンパク質をコードする核酸配列は宿主細胞中のプロモータ配列に動作可能なようにリンクされている。このプロモータ遺伝子単位は、その後、典型的には、複製及び/または発現のために宿主細胞に形質転換される前に中間ベクターにクローンされる。この中間ベクターは典型的にはプラスミド、またはシャトルベクターなどの原核生物ベクター(prokaryotic vector)である。
【0072】
一つのアプローチとして、糸状菌細胞株は、宿主細胞株で機能するプロモータ、生物学的に活性を持つプロモータ断片、または一または二以上の(例えば、シリーズ)エンハンサーを持つ発現ベクターにより移入されたものであり、プロテアーゼ抑制剤をコードする核酸配列に動作可能にリンクされており、そのためプロテアーゼは細胞株で発現する。好ましい実施の形態においては、DNA配列はプロテアーゼ抑制剤及びその変異体をコードする。他の好ましい実施の形態においては、プロモータは制御可能なものである。
【0073】
A. コドンの最適化
十分発現しない遺伝子による十分発現する遺伝子でのコドンの使用を最適化することは公知の技術である。Barnett et al., GB2200118 及びBergquist et Extremophiles (2002) 6: 177-184を参照のこと。本明細書に記載のコドンの最適化は、アスペルギルス(Aspergillus)で十分発現された異種タンパク質(heterologous protein)と十分発現していない異種タンパク質に分泌される天然タンパク質を比較することに基づいている。表1を参照願いたい。
【表1】

【0074】
発現タンパク質で用いられず、または度々は用いられなかったコドンが選択され、度々用いられるコドンに変えられるであろう。したがって、我々はコドンのサブセットのみを変えた。
【0075】
B.核酸の構築/発現ベクター
プロテアーゼ抑制剤をコードする天然または合成ポリヌクレオチド断片(「PIコード核酸配列」PI encoding nucleic acid sequence)は、異種核酸構築またはベクターに組み込むことができ、糸状菌細胞に導入され、そして複製されることが可能となる。本明細書に記載のベクター及び方法は、宿主細胞においてプロテアーゼ抑制剤及びその変異体の発現に用いるのに適したものである。どのベクターでもそれが導入された細胞内で複製され成長可能である限り使用することができる。多数の適当なベクター及びプロモータが当業者に知られており、商業的に入手可能である。糸状菌細胞で用いるのに適したクローニング及び発現のためのベクターは、またSambrook et al., 1989 及び Ausubel FM et al., 1989に記載されているが、これらは引用により本明細書に組み入れられる。適切なDNA配列は種々の手順でプラスミドまたはベクター(集合的に本明細書では、「ベクター」という)に挿入される。一般的には、DNA配列は、標準の手順により適切な制限酵素(restriction endo-nuclease)部位に挿入される。そのような手順及び関連するサブクローニング手順は当業者の知識の範囲にあるとみなされる。
【0076】
適切なベクターは、典型的には、終了配列のような選択可能なマーカーをコードする核酸配列、挿入部位、及び適当なコントロール要素を備えている。ベクターは、例えば、イントロン(intron)及びコントロール(control)要素、すなわち、プロモータ、ターミネータ要素、または5’及び/または3’非翻訳領域のような非コード配列を含む調節塩基配列を含むこともある。それらは宿主細胞(及び/または修飾された可溶性タンパク質をコードする配列が通常発現しないベクターまたは宿主細胞環境)においてコード配列の発現に効果的であり、コード配列と動作可能にリンクされる。多くの適当なベクター及びプロモータが当業者に知られており、その多くは商業的に入手可能であり、及び/またはSambrook et al.,(上記)に記載されている。
【0077】
代表的なプロモータは構成的プロモータ(constitutive promoter)及び誘導プロモータ(inducible promoter)を共に含む。その例には、CMVプロモータ、SV40早期プロモータ、RSVプロモータ、EF-1αプロモータ、tet-on またはtet-offシステム(Clon Tech and BASF)のtet応答素子(TRE)を含むプラスミド、ベータアクチン(beta actin)プロモータ、及びある金属塩を追加することにより上昇させることのできる(upregulate)メタロチオネイン(metallothionein)プロモータを含む。ある実施の形態においてはglaAプロモータが用いられる。このプロモータはマルトースの存在下で誘導される。その様なプロモータは当業者に周知である。
【0078】
天然のプロモータはその機能を変えることなく、一または二以上の核酸を代替、置換、追加または除去することにより修飾することができることは当業者に知られている。本発明の実施においては、プロモータのそのような変更を含むが、それらの変更に拘束されるものではない。
【0079】
遺伝子構築におけるプロモータの選択は、当業者の知識の範囲に属する。
【0080】
適切な選択可能なマーカーの選択は、宿主細胞により決められ、異なる宿主に適しているマーカーは当技術分野でよく知られている。典型的な選択可能なマーカー遺伝子は、(a)抗生物質または他の毒素、例えば、アンピシリン(ampicillin), メトトレキサート(methotrexate)、テトラサイクリン(tetracycline)、ネオマイシン(neomycin)(Southern and Berg, J., 1982),ミコフェノール酸(mycophenolic acid)(Mulligan and Berg, 1980), プロマイシン(puromycin),ゼオマイシン(zeomycin),またはヒグロマイシン(hygromycin)(Sugden et 1985) に抵抗力を与え、または (b)宿主株において栄養要求体変異または自然発生の栄養不足を補うタンパク質をコードする。好ましい実施の態様においては、pyrG菌遺伝子は選択可能なマーカーとして用いられる (Ballance D. J.et al., 1983, Biochem Biophys. Res. Commun. 112: 284-289)。 他の好ましい実施の態様においては、amdS菌遺伝子が選択可能なマーカーとして用いられる (Tilburn, J. et al., 1983, Gene 26: 205-221)。
【0081】
選択されたPIコード配列は良く知られている組み換え技術により、適当なベクターに挿入され、細胞株をPI発現が可能なように形質転換するのに用いられる。遺伝子コードの固有の縮退により、実質的に同じ,または機能的に同等なアミノ酸配列をコードする他の核酸配列が、上記で詳細記載したように特定のプロテアーゼ抑制剤をクローンし、また発現させるために用いられる。したがって、コード領域においてその様な置換をすることは、本発明による配列変異の範囲に属することに留意する必要がある。如何なるまたはすべてのこれらの配列の変異は、親PIをコードする核酸配列について本明細書に記載されていると同様方法で用いることができる。当業者は異なるPIは異なる核酸配列によりコードされることに気付くであろう。
【0082】
所望の形のプロテアーゼ抑制核酸配列、その相同体、変異体または断片が得られたならば、種々の方法で修飾することができる。配列がノンコード隣接領域を含む場合は、隣接領域は切除、変異等を受けるかもしれない。したがって、転位、転換、欠失、挿入が自然発生の配列において行われることもある。
【0083】
異種核酸構築は、プロテアーゼ抑制剤、その変異体、断片、またはその接合変異体の以下のコード配列を含むことがある。すなわち、(i) 単離されている、(ii)融合タンパク質またはシグナルペプチドコード配列のような追加的コード配列と組み合わされている場合であり、その場合はPIコード配列は主要コード配列である、(iii)イントロン(intron)、及びプロモータ、ターミネーター要素、または5’及び/または3’非翻訳領域のようなコントロール(control)要素が、適当な宿主細胞においてコード配列の発現に効果的であるような非コード配列と組合されている、及び/または、(iv)PIコード配列が異種遺伝子であるベクターまたは宿主環境にある配列である。
【0084】
適当な核酸をコードする配列を含む異種核酸は、上で述べたように、適当なプロモータ及びコントロール配列と共に、糸状菌細胞がプロテアーゼ抑制剤及びその変異体を発現させるように細胞を形質転換するのに用いられることもある。
【0085】
本発明の一つの特徴として、異種核酸構築はPIコード核酸配列をインビトロで細胞に移転するのに用いられるが、その場合確立した細胞株であることが望ましい。好ましくは、生産宿主として用いられる細胞株は本発明の核酸配列が安定的に統合されているものがよい。したがって、どの様なものであれ、安定した形質転換細胞を生み出すための効果的な方法であれば、本発明を実施するのに用いることができる。
【0086】
本発明の一つ特徴として、第一、及び第2の発現カセットは単一のベクター、または別個のベクターに存在してもよい。
【0087】
特に断りのない限り、本発明においては、当業者の技術の範囲内である分子生物学、微生物学、及び組み換えDNAでの通常の技術を用いる。そのような技術は、文献に詳しく説明されており、例えば、"Molecular Cloning : A Laboratory Manual", Second Edition (Sambrook, Fritsch & Maniatis, 1989), Animal Cell Culture" (R. I. Freshney, ed., 1987); 及びCurrent Protocols in Molecular Biology”(F.M. Ausubel et al., eds 1987) を参照願いたい。以上または以下に記載のすべての特許、特許出願、記事、刊行物は引用により本明細書に組み入れられる。
【0088】
プロモータ配列に加え発現カセットもまた、効果的な終了を提供するための構造遺伝子の転写終了領域下流を含むべきである。終了領域は、プロモータ配列と同じ遺伝子、または当業者の知識の範囲内にある異なる遺伝子から得られることもある。
【0089】
細胞に遺伝情報を運搬するために用いられる特定の発現ベクターは特に決定的要素ではない。真核細胞または原核細胞における発現に用いられるどのような通常のベクターであれ用いることができるであろう。標準的なバクテリア発現ベクターにはpBR322ベースのプラスミド、pSKF, pET23Dは勿論、バクテリオファージλ及びM13、及びMBP, GST 及びLacZのような融合発現システムを含む。例えば、c-mycの様に、便利な単離の方法を提供するために組み換えタンパク質にエピトープタッグ(epitope tag)を追加することもできる。
【0090】
また発現ベクターに典型的に含まれる要素には、組み換えプラスミドを宿すバクテリアの選択を許す抗生物質への抵抗をコードする遺伝子であるレプリコン(replicon)、及び異種配列の挿入を許すプラスミドの非必須領域におけるユニーク制限部位を含む。特定の抗生物質への抵抗遺伝子を選ぶことは決定的ではない。技術分野で知られている多くの抵抗遺伝子は適性がある。
【0091】
C.宿主細胞及び培養条件
本発明は、プロテアーゼ抑制剤及びその変異体の発現にとって効果的な方法によって、修飾され、選択されそして培養された細胞を含む細胞株を提供する。
【0092】
PI発現のために処理され、および/または修飾される親細胞株は、これに限定されるものではないが、糸状菌細胞を含む。本発明を実施するために用いられる適当な初代細胞タイプは、これに限定されるものではないが、アスペルギルス(Aspergillus)及びトリコデルマ(Trichoderma)を含む。
【0093】
細胞発現プロテアーゼ抑制剤は,典型的には親細胞株を培養するのに用いられる条件で培養される。細胞は通常、標準のRPMI, MEM, IMEM, またはDMEMに、典型的には牛の胎児の血清のような、5−10%の血清を加えた生理食塩水 及び栄養素を含む標準的な媒体中で培養される。培養条件は、また標準的なものである。すなわち、培養は所望のプロテアーゼ抑制剤発現の水準が達成されるまで37℃で静止またはローラ培養で増殖される。
【0094】
与えられた細胞株に対する好ましい培養条件は、科学文献及び/またはAmerican Type Culture Collection (ATCC;"http ://www.atcc.org/")の様な細胞株のソースから得ることができる。典型的には、細胞の生育がなされたら、細胞はプロテアーゼ抑制剤及びその変異体に発現を起こさせ、または抑制するの効果的な条件に置かれる。
【0095】
PIコード配列が誘導プロモータ(inducible promoter)の管理下に置かれるような好ましい実施の形態においては、誘発剤(inducing agent),例えば、炭水化物, 金属塩、または抗生物質がプロテアーゼ抑制剤の発現を誘導するために効果的な濃度で媒体に加えられる。
【0096】
D.プロテアーゼ抑制剤をコードする核酸配列の宿主細胞への誘入
形質転換の方法により、形質転換ベクターの全部またはその一部を糸状菌のゲノムに安定的に統合させることができるかもしれない。しかし、自己複製する、染色体外の形質転換ベクターの維持をすることとなる形質転換も考慮される。
【0097】
本発明はさらに、外来的に供給されるPIコード核酸配列を含む様遺伝子を修飾された細胞及び細胞組成物を提供する。親細胞または細胞株はクローンベクター、または発現ベクターにより遺伝子的に修飾される〈すなわち、変換され、転換され、または移入される〉。ベクターは、上記のように、例えば、プラスミド、ウイルスの粒子、ファージなどの形であるかも知れない。好ましい実施の形態においては、プラスミドは糸状菌細胞に核酸を導入するのに用いられることもある。形質転換は逐次(sequential)に行われるか、または同時の形質転換(co-tranformation)である。
【0098】
インビトロで細胞に発現ベクターを送るために種々の方法が用いられる。異種核酸配列の発現のために核酸を細胞導入する方法は、これらに限定されるものではないが、通常の熟練者に知られており、それらには、電気穿孔法、核ミクロインジェクション(nuclear microinjection)または単一細胞に直接ミクロインジェクションする、無傷細胞(intact cell)による原型質体融合(protoplast fusion)、例えば、ポリブレン(polybrene),またはポリオルニチン(polyornithine)の様なポリカチオン(polycation)の使用、PEG, リポソーム、リポフェクタミン(lipofectamine)またはリポフェクション媒介の核酸注入(lipofaction-mediated transfaction)による膜融合,DNA被覆のミクロ投射物(microprojectile)による高速爆撃(bombardment),リン酸カルシウム−DNA 沈殿物による増殖、DEAE-Dextran媒介の核酸移入、修飾されたウイルス核酸による感染、アグロバクテリア媒介のDNA転位(Agrobacterium - mediated DNA transfer)等を含む。さらにPIコード核酸配列を含む異種核酸構築はインビトロで転写され、その結果のRNAは周知の方法、例えば、注射により宿主細胞に導入されうる。
【0099】
プロテアーゼ抑制剤のコード配列を含む異種の核酸構築を導入するのに続き、遺伝子的に修飾された細胞はプロモータを活性化し、形質転換細胞を選択し、PIコード核酸配列の発現を増幅するのに適するように修飾された通常の栄養媒体中で培養することができる。温度、pH等の培養条件は、以前に発現のため選択された宿主細胞に既に用いられた条件と同じであり、当業者には明らかである。
【0100】
そのような異種核酸構築が導入された細胞の子孫(progeny)は通常、異種核酸構築に見出されるPIコード核酸配列を含んでいると考えられている。
【0101】
E.菌の発現
適当な宿主細胞は糸状菌細胞を含む。本発明の「糸状菌」は、発現宿主として、また第1及び第2の核酸としての役割をするが、これは真核微生物であり、Eumycotina subdivision の 全ての線状体を含む(Alexopoulos, C. J. (1962), Introductory Mycology, New York: Wiley)。これらの菌はチティン(chitin)、グルカン(glucan)及び他の複合多糖よりなる細胞壁を持つ栄養菌糸(vegetative mycelium)であることに特徴がある。本発明の糸状菌は形態学的に、生理学的に、及び遺伝学的にイースト菌とはまったく別個のものである。糸状菌の栄養増殖は菌糸伸長によるものである。対照的に、出芽酵母(saccharomyces cerevisiae)のようなイースト菌による栄養増殖は、単細胞性葉状体の発芽によるものである。出芽酵母と糸状菌の違いには、出芽酵母はアスペルギルス(Aspergillus )及びトリコデルマ(Trichoderma)イントロン(introns)を処理する能力がなく、糸状菌の多くの転写制御因子を認識する能力がないことを含む(Innis, M. A. et al. (1985) Science, 228, 21-26)。
【0102】
種々の糸状菌種が以下の属を含み発現宿主として用いられる:アスペルギルス(Aspergillus)、トリコデルマ(Trichoderma), ニュロスポラ (Neurospora), ペニシリウム(Penicillium), セファロスポリウム(Cephalosporium)、 アキラ(Achlya), ファネロチート(Phanerochaete), ポドスポラ(Podospora),エンドチナ(Endothi), ムコール(Mucor), フサリウム(Fusarium), フミコラ(Humicola) 及びクリソスポリウム(Chrysosporium)である。特定発現宿主はアスペルギルス ニジュランス(Aspergillus nidulans (Yelton, M., et al., Proc. Acad. Sci. USA, 81,1470-1474 ; Mullaney, E. J.et al., (1985) Mol. Gen. Genet. 199,37-45 ; John, M. A. 及び J. F. Peberdy (1984) Enzyme Microb. Technol. 6,386-389 ; Tilburn,et al., (1982) Gene 26, 205-221; Ballance, D. J. et al.,(1983) Biochem. Biophys. Res. Comm. 112,284-289 ; Johnson, I. L.(1985) EMBO J. 4,1307-1311)、 アスペルギルス ニジェール(Aspergillus niger) (Kelly, J. M. 及び M. Hynes (1985) EMBO 4, 475-479), アスペルギルスニジェール変異体アワモリ(Aspergillus niger var. awamori)例えば、NRRL 3112, ATCC 22342, ATCC 44733, ATCC 1433-1,及びUVK-143f株、アスペルギルス オリザ(Aspergillus oryzae), 例えば、 ATCC 11490, ニュロスポラ クラッサ(Neurospora crassa) (Case, M. E. et al.,(1979) Proc. Natl. Acad. Sci. USA 76,5259-5263; Lambowitz U. S. Patent No. 4,486, 553; Kinsey, J. A. 及び J. A. Rambosek (1984) Molecular and Cellular Biology 4, 117-122; Bull, J. 及び J. C. Wooton (1984) Nature 310,701-704), トリコデルマ レーセイ(Trichoderma reesei)、例えば、NRRL 15709, ATCC 13631,56764, 56765,56466, 56767, 及び トリコデルマ ビリド(Trichoderma viride)、例えば、ATCC 32098 及び32086。好ましい発現宿主は主な分泌アスパルチル(aspartyl)プロテアーゼをコードする遺伝子が削除されているアスペルギルスニジェール変異体アワモリ(Aspergillus niger var. awamori)である。この好ましい発現宿主の生産については米国特許出願番号214,237、出願日1988年7月1日に記載されており、ここに引用により本明細書に組み入れられる。
【0103】
真核生物である糸状菌の分泌プロセスにおいて、分泌タンパク質は細胞質(cytoplasm)から細胞膜を超えて小胞体(endoplasmic reticulum)(ER)の内腔(lumen)に入る。ここで、タンパク質は折り畳まれ、ジスルフィド結合が形成される。BiPのようなシャペロン(chaperone)及びタンパク質ジスルフィド イソメラーゼのようなタンパク質はこのプロセスを助ける。またこの段階において、糖鎖が、グリコシド化された(glycosylated)タンパク質を生み出すためにタンパク質に付着される。砂糖は、典型的には、N-リンクグリコシル化(glycosylation)としてアスパラギン残基に加えられ、またはO-リンクグリコシル化としてセリン(serine)またはトレオニン(threonine)残基に加えられる。正しく折り畳まれ、グリコシル化されたタンパク質はER(小胞体)から糖鎖が修飾され、またイースト及び菌のKEX2またはKEXBプロテアーゼが存在するゴルジ体(Golgi apparatus)に運ばれる。菌で生産された分泌タンパク質に付加されるN-リンクのグリコシル化は、哺乳類細胞により付加されるものと異なっている。
【0104】
糸状菌宿主細胞において生産されるプロテアーゼ抑制剤及びその変異体は「グリコシド化された」(glycosylated)か、または「グリコシド化されていない」(non-glycosylated)(すなわち、「非グリコシ化された」(a-glycosylated)または「脱グリコシド化された」(de-glycosylatedである)。菌のグリコシド化のパターンは哺乳類細胞により生み出されるパターンと異なるため、プロテアーゼ抑制剤はプロテアーゼ抑制剤を「脱グリコシド化する」(de-glycosylate)ために酵素で処理されることもある。そのようなN-リンク「脱グリコシド化」に有用な酵素は、エンドグリコシダーゼ H(endoglycosidase H), エンドグリコシダーゼ F1, エンドグリコシダーゼ F2, エンドグリコシダーゼA, PNガーゼ F(PNGase F), PNガーゼ A, PNガーゼ Atがある。そのようなO-リンク「脱グリコシド化」に有用な酵素に、エクソグリコシダーゼ (exoglycosidase)、特にアルファ-マンノシダーゼ(alpha-manodisases)(例えば、アルファ-マンノシダーゼ(alpha-manodisases)(アスペルギルス サイト(Aspergillus saito, iGKX-5009),アルファ (1-2,3, 6)- マンノシダーゼ (alpha (1-2,3, 6) - Mannosidase )(Jack bean, GKX-5010), アルファ- マンノシダーゼ/MANase VI (alpha - Mannosidase/MANase ) (ザントモナス マニホチの組み換え、GKX80070)(Xanthomonas manihoti, GKX80070) これらすべてはGlyko (Prozyme), San Leandro, California)から得られる。
【0105】
驚くことには自然分泌のタンパク質に融合された菌において、高い水準のプロテアーゼ抑制剤及びその変異体を得ることができることが分かった。上記の情報から、プロテアーゼ抑制剤及びその変異体は、グルコアミラーゼがまだN末端に付着している場合にはERで組み立てられることが明らかに予測される。これにより56kDよりも大きなタンパク質を生み出すこととなろう。グルコアミラーゼが、さらに修飾されることなくゴルジ体を通る場合には、所望のタンパク質から切り裂かれることはないと思われる。
【0106】
本発明の方法及び宿主細胞を用いて、我々は驚くべき高い発現レベルを達成した。本明細書に記載のシステムによりプロテアーゼ抑制剤0.5g/l以上の発現及び分泌レベルを達成した。
【0107】
細胞にベクターが導入された後に、感染された(transfected)細胞は所望のタンパク質をコードする遺伝子の発現に有利な条件で培養される。形質転換された多くのバッチ数の細胞は上記のように培養することができる。最後に生成物は公知の技術を用いて培養体から回収される。
【0108】
シャペロン(CHAPERON)
上に述べた通り、ERにおける分泌タンパク質の折り畳み、グリコシル化(glycosylation)は、シャペロン(chaperon)と呼ばれる多くのER居住タンパク質により助けられる。Bip, (GRP78), GRP94 またはLhs1pイーストのようなシャペロンは、表面に出ている疎水領域に折り畳まれない状態で結合することにより分泌タンパク質の折り畳みを助け、不利な相互作用を阻止する(Blond- Elguindi et al., 1993, Cell 75: 717-728)。シャペロンはまた、ER細胞膜を通してタンパク質が異動するのに重要な役割を果たす。タンパク質 ジスルフィド イソメラーゼ(pdi)、その同属体、及びポリルーペプチド シスートランス イソメラーゼ(prolyl-peptidyl cis-trans isomerase)のようなフォルダーゼタンパク質(foldase protein) は、ジスルフィド架橋の形成、及びプロリン残基(prolien residue) に隣接するペプチド鎖の正しいコンフォーメイションの生成をそれぞれ助けている。
【0109】
発明の一つの特徴として、宿主細胞はシャペロンをコードする発現ベクターにより転換される。シャペロンはpdiA 及びprpAからなる群から選択される。
【0110】
発酵パラメータ(FERMENTATION PARAMETER)
本発明は菌類の培養のための発酵手順に基づくものである。異種タンパク質を生産する発酵手続は、それ自身技術分野で良く知られている。例えば、タンパク質はバッチ、フェドバッチ(fed-batch),及び連続フロー工程を含む、固形、または液内培養により生産することができる。
【0111】
培養は、水性ミネラル塩媒体、有機的成長因子、炭素及びエネルギー源媒体、分子酸素及び当然ながら、一または二以上の使用される特定微生物種の開始接種材料を含む成長媒体において成される。
【0112】
炭素及びエネルギー源、酸素、同化窒素、及び微生物の接種材料に加え、微生物の適切な成長を確保し、微生物変換プロセスにおける細胞による炭素及びエネルギー源の同化を最大にし、発酵媒体中の最大細胞密度で最大細胞生成を達成するために、それに応じた適量のミネラル栄養分を供給することが必要である。
【0113】
水性ミネラル培体の組成は広範囲にわたり、技術分野で知られているように、使用される微生物、及び基質に一部依存する。ミネラル媒体は、窒素に加え、適量のリン、マグネシウム、カルシウム、カリ、硫黄、及びナトリウムを、適当な可溶性同化イオン、及び組合された形で含むことが必要であり、また、当技術分野で知られているように、また好ましくは適当な可溶性同化の形の銅、マンガン、モリブデン、亜鉛、鉄,ボロン、沃素及び他の要素のような、ある微量元素も存在するべきである。
【0114】
発酵反応は、必要とされる分子酸素が、空気、酸素富化空気またはさらには実質的に純粋分子酸素も含む分子酸素含有ガスにより供給される好気性(aerobic)プロセスである。ただし、微生物種が効果的に繁殖できるように発酵容器内容物を適切な酸素分圧に維持することが必要である。つまり、含酸素炭化水素基質を用いることにより、微生物の成長に必要な酸素量が減少することになる。しかしながら、基質の同化、及びそれに対応する微生物の成長は一部は燃焼プロセスであるため、成長のためには分子酸素が供給される必要がある。
【0115】
通気速度は非常に広範囲に変わりうるが、通気は通常は0.5から10、好ましくは0.5から7の範囲でなされ、これは分当りの発酵槽の単位液体容量に含まれる酸素含有ガスの容量(その時の圧力、及び温度は25℃)である。この量は、反応搭に送られる通常の酸素量の空気に基づくものであり、純粋酸素に換算すると各々0.1から1.7、好ましくは0.1から1.3であり、分当りの発酵槽の単位液体容量に含まれる酸素の容量(その時の圧力、及び温度25℃)である。
【0116】
微生物変換プロセスにおける圧力は広範囲に変わりうる。一般的には、0から50psigであり、ここでは好ましい圧力は約0から30psig、より好ましくは、少なくとも大気圧より僅か高い圧力である。酸素溶解度に対する設備機器と操業コストのバランスが達成できるからである。大気圧より高い場合には、水生発酵体中の酸素溶解濃度が増すため、細胞の成長率が増し有利になる。しかしながらその場合には同時に、高い大気圧は設備機器及び操業コストを増す結果をもたらす。
【0117】
発酵温度は幾らかは変えることができる、しかし、アスペルギルスニジェール変異体アワモリ(Aspergillus niger var. awamori)のような糸状菌では、選択される微生物株にもよるが、温度は通常約20℃から40℃の範囲内であり、一般的には好ましくは28℃から37℃の範囲である。
【0118】
微生物はまた同化窒素源を必要とする。同化窒素源は微生物による代謝利用に適した形の窒素を放出することのできる窒素含有化合物であればどのようなものでもよい。タンパク質加水分解物のような種々の有機窒素化合物をソースとして用いることができるが、通常は、アンモニア、水酸化アンモニウム(ammonium hydroxide)、尿素のような安価な窒素含有化合物、及びリン酸アンモニウム(ammonium phosphate)、硫酸アンモニウム(ammonium sulfate)、ピロリン酸アンモニウム(ammonium pyrophosphate),塩化アンモニウム(ammonium chloride)の様な種々のアンモニア塩、または他のアンモニア化合物を用いることができる。アンモニアガス自体は規模が大きな操業に便宜であり、適量の水性発酵(発酵媒体)による場合のバブリングの形で用いることができる。アンモニアはまたpHコントロールを助けるのにも用いることができる。
【0119】
水微生物発酵(発酵混合剤)のpHは典型的な場合として2.0から8.0の範囲であるべきである。糸状菌では、通常pHは2.5から8.0の範囲であり、アスペルギルスニジェール変異体アワモリ(Aspergillus niger var. awamori)では、通常pHは4.5から5.5の範囲である。ある微生物に適しているpHの範囲は微生物の特徴、並びにある程度使用される媒体によって決まり、したがって媒体により幾分変わってくるがこれは当業者は容易に選定が可能である。
【0120】
発酵槽中の発酵混合剤の平均保持時間は、ある程度発酵温度、及び使用される培養体に依存するが、大きく変わる。通常は約24から500時間の範囲であり、ここでは好ましくは約24から400時間である。
【0121】
好ましくは発酵は炭素含有基質が制限要因としてコントロールされ、炭素含有基質の細胞への転換がスムーズになされ、大量の非転換基質により細菌の汚染が生じない様に行われるのが望ましい。後者については水溶性基質については問題とならない。残留痕は直ぐに洗い流されるからである。しかし、非水溶性基質では問題となり、適当な洗浄ステップにより生成物を処理する製品の追加処理工程が必要となる。
【0122】
上記に記載の通り、この様な制限基質のレベルに達するまでの時間は決定的な要素ではなく、微生物によって及び実施される発酵プロセスにより変わりうる。しかし、発酵媒体における炭素源の濃度、及び所望の炭素源のレベルが達成されたかを否か決定する方法は当技術分野でよく知られている。
【0123】
発酵はバッチ、または連続操業によることができるが、管理の容易さ、生成物の質の均一性、及び設備機器が最も経済的に使用できるという理由により、通常フェドバッチ(fed-batch)操業が好まれる。
【0124】
もし望むなら、培養槽に水性ミネラル媒体を加える前に、一部または全部の炭素及びエネルギー源材料、及び/またはアンモニアの様な同化窒素源の一部を加えることができる。
【0125】
反応搭に導入される各流液は、各事前に定められた数値により、または炭素及びエネルギー基質の濃度、pH, 溶解酸素、発酵槽から出る排気ガス中の酸素または二酸化炭素、透光度により測定される細胞密度等をモニターすることによりコントロールされるのが望ましい。各種材料の供給率は、効果的に炭素及びエネルギー源を活用して、可能な限りの細胞成長を促進することができるよう、また基質の投入に対して可能な限りの高い微生物細胞を得ることができるよう変えることができるが、より大事なことは、単位容量当り所望するタンパク質を最も多く生産できることである。
【0126】
バッチ、または好ましいフェドバッチ(fed- batch)操業のいずれににおいても、すべての機器、反応搭、または発酵手段、容器、またはコンテナー、パイプ、循環または冷却装置の付属物等は、まず最初に殺菌する必要があり、これは通常は約121℃の蒸気を使って少なくとも15分間行う。そして殺菌された反応搭に、酸素、及び炭素含有基質を含む必要なすべての栄養素が揃っている中で選択された微生物の培養体が植えつけられる。使用される発酵槽は決定的な要素ではないが、ここで操業に好ましいのは15L Biolafitte (Saint- Germain-en-Laye, France)である。
【0127】
タンパク質の分離
所望されるタンパク質が発現されると、選択的に所望するタンパク質の分泌の回集が必要となる。本発明は所望するタンパク質を融合類似物から分離する方法を提供する。本明細書に記載の方法は、融合類似物からプロテアーゼ抑制剤及びその変異体を分離するのに役立つよう特に考慮されている。
【0128】
発酵培養液から所望するタンパク質の収集及び精製は、また技術分野においてそれ自体で知られている手順により行うことができる。発酵培養液は通常、所望のタンパク質のみならず、細胞、種々の浮遊物、及び他のバイオマス汚染物質(biomass contaminant)を含む細胞破片を含んでおり、これらはその技術分野において知られている手段により、発酵培養液より除去されるのが望ましい。
【0129】
そのような除去に適したプロセスは、例えば、遠心分離、ろ過、透析(dialysis)、精密ろ過(microfiltration),ロータリー真空ろ過(rotary vacuum filtration)、または他の既知のプロセスの様な細胞を含まないろ過液を得るための通常の固体―液体分離技術を含む。結晶化させる前に限外ろ過(ultrafiltration)、蒸発または沈殿などの技術を用いて発酵培養液または細胞を含まないろ過液をさらに濃縮することが望ましい。
【0130】
浮遊物またはろ過液のタンパク質成分は、例えば硫酸アンモニウムの様な塩を用いることにより、またはpHを2から3に調整し、沈殿させることができる。その後、培養液を80℃で2時間熱処理し、続いて、例えば、イオン交換クロマトグラフィー、親和性クロマトグラフィーのような種々のクロマトグラフィーによる手順、または類似の認められた方法により精製する。
【0131】
発現された所望のポリペプチドが分泌される場合、ポリペプチドは成長媒体から精製されることもある。発現宿主細胞は、ポリペプチドの精製の前に培体から除去する(例えば、遠心分離により)のが好ましい。
【0132】
発現組み換えの所望のタンパク質が宿主細胞から分泌されない場合は、好ましくは宿主細胞が破壊され、ポリペプチドが精製の第一段階である水性「抽出物」(extract)中に解放されるのが望ましい。発現宿主細胞は細胞が破壊される前に媒体から回収されるのが好ましい(例えば、遠心分離により)。
【0133】
細胞破壊は、例えば、リゾチーム、またはベータグルカナーゼ分解により、または細胞を高圧により圧迫するなどの従来技術により行うことができる。細胞破壊に関するさらに詳細な記載はRobert K. Scobes, Protein Purifi- cation, Second edition, Springer-Verlagを参照願いたい。
【0134】
6つのヒスチジン残基、すなわち、His Tag、をC末端に追加することは所望のタンパク質及びその融合類似体の精製を助ける。His tag を精製のための協力者として用いることは公知の技術である。例えば、Hengen (1995) TIBS 20 (7): 285-286を参照願いたい。6つのHis tagの付いたタンパク質は固定化金属イオン親和クロマトグラフィー(Immobilized Metal ion Affinity Chromato−graphy (IMAC))を使い容易に精製される。
【0135】
プロテアーゼ抑制剤及びその変異体は、疎水負荷誘導クロマトグラフィー(hydrophobic charge induction chromatography (HCIC))を組み合わせて用い、例えば、全細胞発酵培養液または浄化培養液のような水性タンパク質溶液から精製することができることは特に考察するに値する。HCICは所望するタンパク質を培養液及びその融合類似体から分離することができる。
【0136】
ユーティリティー
いくつかの所望するタンパク質の応用例においては、例えば、99%以上の純度を持つというように、プロテアーゼ抑制剤はきわめて純粋であることが非常に重要である。これは所望のタンパク質が治療目的に用いられる場合に特に当てはまるが、他に応用される場合にもまた必要である。本明細書に記載の方法は、本質的に純粋な所望のタンパク質を得る方法を提供するものである。本明細書に記載の所望のタンパク質は製薬及び個人医療の薬の組成として有用である。
【0137】
以下に続けて開示する実施例においては以下の略記号を用いる:eq (equivalents) ; M (Molar) ;μM (micromolar) ; N(Normal) ; mol (moles) ; mmol (millimoles) ; μmol(micromoles) ; nmol (nanomoles) ; g (grams); mg (milligrams) ; kg (kilograms); μg (micrograms); L (liters) ; ml (milliliters) ; μl(microliters) ; cm (centimeters); mm (millimeters) ; μm (micrometers); nm (nanometers); ℃ (degrees Centigrade); h (hours); min (minutes); sec (seconds); msec (milliseconds); Ci (Curies) mCi (milliCuries) ; μCi(microCuries); TLC (thin layer achromatography); Ts (tosyl); Bn (benzyl) ; Ph (phenyl) ; Ms (mesyl) ; Et (ethyl), Me (methyl). PI (proteinase inhibitor), BBI (Bowman-Birk inhibitor), STI (Soybean Trypsin inhibitor).
実施例
本発明はその実施例においてさらに詳細が記載されるが、いかなる意味においても、それにより特許請求の範囲に記載の発明の範囲が制限されるものと解してはならない。添付の図面は本明細書及び発明の不可欠の一部である。本明細書に参考として引用されたものは、その全てが引用により本明細書に組み入れられる。以下の実施例は請求の範囲の発明の説明のために記載されるものであるが、それにより請求の範囲の発明を限定するものではない。
【0138】
実施例1 大豆トリプシン抑制剤をコードするDNAのクローニング
本実施例はSTIの発現ベクターの発育について明らかにする。
【0139】
通常、所望するタンパク質をコードする遺伝子は、N末端のNhel制限酵素部位を経て加工されたkexB切断部位(NVISKR)及びSTI終了コドン、TAGに続くC末端のBstEll 制限酵素部位を持つグルコアミラーゼ(glucoamylase)のリンカー領域をコードするDNAに融合されていた。大豆のSTIをコードする遺伝子はkexB切断部位、及び3つのグリシン残基をN末端に待ち、6つのヒスチジン残基をC端末に持つ二つの制限部位を含むDNA断片としてインビトロでMCLAB(South San Francisco, California)により合成された(配列番号3、図2に示す遺伝子)ここで用いられるPCRによる生成DNA断片はすべて当初pCRII-TOPOベクターにクローンされた(Invitrogen, Carlsbad, CA)。大腸菌(One Shot TOP10 cells from Invitrogen)は通常のプラスミド単離及びプラスミド保持に用いられた。Nhel 及びBstEll部位はpCRII-TOPOベクターからPCR製品を切り取るために用いられ、そして結果物のDNA断片は発現ベクターpSL1180- GAMpR-2に結紮された (図5参照)。発現ベクターpSL1180-GAMpR-2は、アスペルギルス ニジェール グルコアミラーゼ(Aspergillus niger glucoamylase)プロモータ、グルコアミラーゼ触媒ドメインおよび終了領域(terminator region)を含む。発現プラスミドはまた選択マーカーとしてアスペルギルス ニジェール pyrG(Aspergillus niger pyrG)を含む。したがって、発現カセットによる形質転換細胞はウリジン(uridine)が欠乏する媒体における成長により検知する。
【0140】
STIペプチドをコードする遺伝子(アミノ酸配列番号10、図4A, ヌクレオチド配列番号6、図2)はMCLABにより合成され、pCRII-TOPOベクターにクローンされた(Invitrogen)。Nhel からBstEllの断片は制限酵素分解によりプラスミドから解放され、発現プラスミドpSLGAMpR2―SBTI/nonpti(Q110)を作り出すために, DNA断片は寒天ゲルから取り出され、pSLGAMpR2 、グルコアミラーゼキモシン発現ベクター(pSLGAMpR2、glucoamylase-chymosin expression vector)にクローンされた。詳細はWO 9831821に記載されている。
【0141】
発現プラスミドはdgr246ΔGAP: pyr2-に転換された。この株はpepA遺伝子が欠失した遺伝子dgr246 P2に由来し、pyrGマイナスであり、異種遺伝子製品の改良された生産方法により、何ラウンドかの変異、スクリーニング及び選択を経ている(Ward, M. et al.,1993, Appl. Microbiol. Biotech. 39: 738-743 及びその中の参照例)。dgr246ΔGAP: pyr2株作るために、まったく同じ欠失プラスミド(pΔGAM NB-Pyr)を用いて、またファウラー(Fowler, T. et al (1990) Curr. Genet. 18: 537-545)により報告されている手順に従い、glaA(グルコアミラーゼ)遺伝子がdgr246 P2から取り除かれた。簡単に言えば、除去は, 両端にglaA フランキング配列(flanking sequence)を持つリニアDNA断片及びプロモータの一部により転換されることにより達成され、glaA遺伝子のコード領域が選択マーカーとして、アスペルギルス ニジュランスpyrG(Aspergillus nidulans pryG)遺伝子により置換されているものである。glaA フランキング配列(flanking sequence)及びpyrG遺伝子を含むリニアー断片が染色体glaA遺伝子座で統合されている形質転換細胞は、サザ゛―ンブロット(Southern blot) 分析により同定された。この変化は形質転換株dgr246ΔGAPで起きていた。この形質転換細胞の胞子はフッ化オロチン酸(fluororotic acid)を含む媒体に塗られ、van Hartingsveldt, W. et al. (1987) Mol. Gen. Genet. 206: 71-75に記載のように、自然発生的耐性変異株が得られた。これらの一つである、dgr246ΔGAP: pyr2は、野生型pryG 遺伝子を持つプラスミドにより転換されることで補完されうるウリジン栄養要求株(uridine auxotroph)であることが示された。
【0142】
アスペルギルス(Aspergillus)形質転換プロトコールはキャンペル法(Campell method)の修正である(Campell et al. (1989). Curr. Genet. 16: 53-56)。すべての溶液及び媒体は加圧滅菌され、または0.2ミクロンフィルターでろ過殺菌された。アスペルギルス ニジェール 変異体 アワモリ(Aspergillus niger variant awamori)の胞子は寒天媒体複合体(complex media ager, CMA)プレートから集菌された。CMAは20g/lデクストロース、20g/l Difco ブランド麦芽エキス、1g/l Bacto Peptpone, 20g/l Bacto 寒天、100mg/mlのアルギニン20ml/l、及び100mg/mlのウリジン20ml/lを含む。約1.5 cm四角の寒天胞子の一切れを使い液体100 mlのCMA(処方は、Bacto が除かれる以外はCMAと同じである)を植えつけた。フラスコは、温度37℃、250−275rpmで一夜振動培養された。菌糸(mycelia)が殺菌されたMilacloth(Calbiochem. San Diego, CA, USA)で集菌され、50mlの溶液A(Solution A)(pH5.8のリン酸ナトリウム10mM中0.8M のMgSO4)で洗浄された。洗浄された菌糸(mycelia)は、20ml溶液A中300mgのβ―D−グルカナーゼ(beta-D- glucanase (Interspex Products, San Mateo, CA)の殺菌液中に置かれた。これを殺菌した250ml プラスチックびん(Corning Inc, Corning, New York)中で28℃から30℃、200rpmで2時間培養した。培養の後、このプロトプラスト(protoplasting)溶液は殺菌されたMiraclothを通してろ過され、殺菌した円錐形チューブ(Sarstedt, USA)に入れられた。 結果物のプロトプラスト(protoplasting)を含む液は2つの50ml円錐形チューブに等量に分けられた。40mlの溶液B(1.2M ソルビトール、50mM CaCl2, 10mM Tris, pH7.5)がそれぞれのチューブに加えられ、卓上臨床
遠心分離器(Damon IEC HN Sill centrifuge)によりフル回転で5分遠心分離された。各チューブからの上澄みは取り除かれ、20mlの新たな溶液Bが1つのチューブに加えられ、混合され、そして次のチューブに注がれ、全ての粒(pellet)が再懸濁されるまで混合された。その後チューブは5分間遠心分離器に掛けられた。上澄みは取り除かれ、20ml の新たな溶液Bがチューブに加えられ5分間遠心分離された。洗浄されたプロトプラストを濃度0.5-10x107 protoplast /100ulの溶液B中で再懸濁する前に、最終の洗浄を行った。殺菌した15ml円錐形チューブ(Sarstedt, USA)の各プロトプラスト100ulに、10ul の形質転換プラスミドDNAが加えられた。これに、12.5ulの溶液C(50% PEG 4000, 50mM CaCl2, 10mM Tris, pH7.5)が加えられ、チューブは氷の上に20分間置かれた。1mlの溶液Cが加えられ、そしてチューブは氷上から除かれて室温に置かれ、静かに振られた。2mlの溶液Bが直ちに加えられ溶液Cが希釈された。転換ミックスが等分に3つのチューブの溶解したMMS重層(6 g/l NaN03, 0.52g/l KCl, 1.52g/l KH2PO4, 218.5g/l D-sorbitol, 1.0ml/l 微量元素-LW(trace element-LW), 10g/l SeaPlaque 寒天(FMC Bioproducts, Rook1 and, Maine, USA)、 20m/l 50% グルコース, 2.5 ml/l 20% MgSO4・7H20, NAOHによりpH6.5にした)に加えられ、45℃の温浴槽に保存された。微量元素-LW (Trace elements-LW)は1g/l FeSO4・7H20, 8.8 g/l ZnSO4・7H20, 0.4g/l CuSO4・5H20, 0.15 g/l MnSO4・4H20, 0.1 g Na2B4O7・10H2O, 50mg/l (NH4)6Mo7O24・4H20, 250 mls H20, 200 ul/l 濃縮HCLよりなる。形質転換ミックスを加えた溶解重層は直ちに、寒天のプレート上に直接加えられた333ul/プレートの100mg/mlのアルギニンで補充されていた3MMSプレート(10g/lのSeaPlaque アガロースに代えて、20g/l Bacto寒天が用いられる以外はMMS重層と同じ調合である)に振りかけられた。アガロースが固まった後に、形質転換細胞が成長するまでプレートは30℃で培養された。
【0143】
萌芽形成形質転換細胞は殺菌した楊枝を使い、最少培地(mimimal media (MM))プラス グルコースのプレートに採られた。MMは6 g/l NaN03, 0.52 g/l KCl, 1.52 g/l KH2PO4, 1 ml/l微量元素-LW(Trace elements-LW), 20 g/l Bacto寒天, NaOHによりpH6.5 にされ、25 ml/l の40%グルコース, 2.5 ml/l の 20% MgSO4・7H20 及び20 ml/l の 100 mg/ml アルギニンよりなる。形質転換細胞がMM上で成長すると、それらはCMAプレートに移された。
【0144】
各形質転換細胞のプレート培養体から1.5cm四角の寒天胞子の一片(agar plug)が250mlの振盪フラスコ中でPromosoy specialと呼ばれる50mlの生産媒体に加えられた。この媒体は以下の組成を持つ。すなわち、70g/l クエン酸ナトリウム(sodium citrate), 15 g/l (NH4)2S04,1 g/l NaH2PO4・H2O, 1g/l MgSO4, 1 ml Tween 80, NaOHによりpH 6.2にされ、2 ml/l Mazu DF60-P, 45 g/l Promosoy 100 (Central Soya, Fort Wayne, IN), 120 g/l マルトースである。生産媒体フラスコは30℃、200rpm で5日間培養され、上澄みサンプルが集菌された。形質転換細胞は, 生産されたタンパク質の量に基づき形質転換細胞を選択するために、SDSゲル上でタンパク質生産のアッセイが行われた。最上の形質転換細胞の培養液につき、そのトリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0145】
各形質転換細胞のプレート培養体から1.5cm四角の寒天胞子の一片(agar plug)が250mlの振盪フラスコ中で修飾CSS(modified CSS)と呼ばれる50mlの生産媒体に加えられた。この媒体は以下の組成を持つ。すなわち、50g/l Corn Street Solids, 1g/l NaH2PO4・H2O, 0.5 g/l MgSO4 (無水), 50 g/l Staley 7350 (55%) 及び8g/lクエン酸ナトリウム(Na Citrate)である。生産媒体フラスコは36℃で、200rpm で3日間培養され、上澄みサンプルが集菌され, SDSゲル上でタンパク質生産のアッセイが行われた。最上の形質転換細胞の培養液につき、そのトリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0146】
実施例2 大豆トリプシン抑制剤をコードするDNAのコドンの最適化
以下の実施例はSTIをコードするDNAが、糸状菌で最適に発現するようどのように改変されたかを詳説するものである。
【0147】
合成遺伝子(MCLAにより合成された実施例1の出発物質)コドンは、アスペルギルス(Aspergillus)で高度に発現したタンパク質でのコドンの使用法に従い最適化されたものである。基本的には、グルコアミラーゼ、アルファアミラーゼ、及びプロキモシン(prochymosin)等のよく発現したタンパク質と、ヒトのNEP 及びDPP4などの様なアスペルギルス(Aspergillus)で十分発現しなかったタンパク質と比較した。表1を参照願いたい。アスペルギルス(Aspergillus)における両タイプのタンパク質発現にもちいたのコドンの使用を表IIに示す。
【表2】
【0148】
【0149】
【0150】
よく発現した遺伝子においては、コドンは多くは使用されず、またはそれほどしばしば使用されなかったことは明らかである。これらのコドンは、発現の良くなかった遺伝子(表IIの星印*で示す)においてはよりしばしば見出される。よく発現するタンパク質では使われない、またはそれほどしばしばは使用されることのないコドンをSTI遺伝子において同定し、それらのコドンはよく発現するタンパク質において、よりしばしば用いられるコドンに変えられた。表III及びIVを参照願いたい。
【表3】
【表4】
【0151】
最適化されたDNAはインビトロでMCLAB (South San Francisco) により、3つの制限部位(遺伝子の5’末端にNhel、3’末端にXhol及びBstEll), kexB切断部位、及び3つのグリシン残基をN末端に持ち、6つのヒスチジン残基をC末端に持つ(配列番号3)DNA断片として合成された。この最適化された遺伝子はpCRII-TOPOベクターにクローンされた。上記実施例1に記載の手順に従い、Nhel からBstEllへの断片はプラスミドから制限分解により解放され、DNA断片はアガロースゲル上で精製され、そしてそこから引き抜かれ、pSLGAMpR2-SBTI2(Q107)発現プラスミドを作るためpSLGAMpR2にクローンされた。
【0152】
発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1に記載の通りである。31の形質転換細胞がアッセイされ、SDSゲルがタンパク質発現レベルチェックのために用いられた。最上の6個の形質転換細胞の培養液について、トリプシン抑制活性がアッセイされた。
【0153】
実施例3 アスペルギルス(Aspergillus )でのボウマンービルク(Bowman-Birk)抑制剤及びその変異体の発現
a. kexB部位、N末端に3つのグリシン、及びC末端に6つのヒスチジン残基を持つグルコアミラーゼへのBBIの融合
上記の実施例2の手順に従い、BBIをコードするDNAは最適化され、本実施例で使用された。DNAはインビトロでMCLABにより、3つの制限部位(遺伝子の5’末端にNhel、3’末端にXhol及びBstEll), kexB切断部位、及び3つのグリシン残基をN末端に持ち、6つのヒスチジン残基をC末端に持つ(配列番号54)DNA断片として合成された。この遺伝子はpCRII-TOPOベクターにクローンされた(Invitrogen)。上記実施例1に記載の手順に従って、Nhel からBstEllへの断片はプラスミドから制限分解により解放され、DNA断片はアガロースゲルから引き抜かれ、pSLGAMpR2-BBIkex+(Q104)発現プラスミドを作るためにpSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1の場合と同様である。28の形質転換細胞が生産され、25の細胞のアッセイが振盪フラスコでおこなわれた。SDSゲルがタンパク質発現レベルチェックに用いられた。最上の形質転換細胞の培養液について、トリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0154】
b. 6つのヒスチジン残基をC末端に持つグルコアミラーゼへのBBIの融合
上記の実施例2の手順に従い、BBIをコードするDNAは最適化され、本実施例で使用された。DNAはインビトロでMCLABにより、3つの制限部位(遺伝子の5’末端にNhel、3’末端にXhol及びBstEll), 及び6つのヒスチジン残基をC末端に持つDNA断片として合成された(配列番号42は以下の通り)。
【0155】
(配列番号42:
GCTAGCGACGATGAGAGCTCTAAGCCCTGTTGCGATCAGTGCGCGTGTACCAAATCGAACCCTCCGCAGTGTCGCTGCTCCGATATGCGTCTGAATTCCTGTCATAGCGCATGCAA,GAGCTGTATCTGCGCCCTGAGCTACCCCGCGCAGTGTTTCTGCGTCGACATCACGGACTTCTGCTACGAGCCGTGTAAGCCCAGCGAGGACGATAAGGAGAACCATCATCACCATCACCATTAGCTCGAGGGTGACC).
この遺伝子はpCRII-TOPOベクターにクローンされた。上記実施例1の手順に従い、Nhel からBstEllへの断片はプラスミドから制限分解により解放され、精製され、そしてアガロースゲルから引き抜かれ、pSLGAMpR2-BBlkex (Q105) 発現プラスミドを作るためにpSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1と同じである。38の形質転換細胞が生産され、25の細胞のアッセイが振盪フラスコでおこなわれ。SDSゲルがタンパク質発現レベルチェックのためにも用いられた。最上の形質転換細胞の培養液について、トリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0156】
c. kexB部位、及び3つのグリシン残基をN末端に持つグルコアミラーゼへのBBI融合
インビトロでMCLABにより合成され(配列番号5)、pCRII-TOPOベクターにクローンされたプラスミドDNAはPCR増幅のDNA鋳型(template)として用いられた。2つのプライマーは以下の様にデザインされた:
5’GGG CTA GCA ACG TCA TST CCA AG3’(配列番号43)
5’ GTC ACC TAG TTC TCC TTA TCG TCC TCG CTG3’ (配列番号44)
DNAはプライマーの存在下で以下の条件で増幅された:DNAはTris-EDTA緩衝液により10から100倍に希釈された。10μlの希釈されたDNAが、各ヌクレオチド(A, G, C 及びT)0.2mM、1反応緩衝液、0.5から0.6μgのプライマー1(配列番号43)及びプライマー2(配列番号44)を含む全100μlのエッペンドルフ管の反応混合物に加えられた。混合物は100℃で5分熱し、反応混合物に2.5単位のTaq DNAポリメラーゼが加えられた。PCR反応は95℃で1分行われ、プライマーは50℃で1分間鋳型にアニールされ、さらに72℃で1分の延長がされた。このサイクルは30回繰り返えされ、さらに延長サイクルとして68℃で7分おこなわれ、その後の使用に備え4℃で保管された。PCR断片は寒天ゲルにより検出され、その後プラスミドベクターpCRII-TOPOにクローンされた(Invitrogen)。得られたPCR断片は、6つのヒスチジン残基およびXhoL制限部位をコードするヌクレオチドが除去されていることを除いては、配列番号54のものと同一の配列を含む。上記実施例1記載の手続きに従い、PCR断片は、制限酵素Nhel 及びBstEllにより分解された。分解されたDNA断片はエタノールで沈殿させ、histag 無しのpSLGAMpR2-BBl (Q108) 発現プラスミドを作り出すため、pSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1と同じである。57の形質転換細胞が生産され、25の細胞のアッセイが振盪フラスコでおこなわれた。SDSゲルがタンパク質発現レベルチェックのために用いられた。最上の形質転換細胞の培養液について、トリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0157】
d. kexB部位を持つグルコアミラーゼへのBBIの融合
インビトロでMCLAB (配列番号1) により合成され、pCRII-TOPOベクターにクローンされたプラスミドDNAがPCR増幅のDNA鋳型(template)として使用された。2つのプライマーは以下の様にデザインされた:
5’GGG CTA ACC TAG TTC TCC TTA TCG TCC TCG CTG 3’(配列番号44)
5’GGG CTA GCA ACG TCA TCT CCA AGC GCG ACG ATG AGA GCT CTA AG3’ (配列番号45)
得られたPCR断片は、3つのグリシン残基、6つのヒスチジン残基およびXhoL制限部位をコードするヌクレオチドが除去されていることを除いては、配列番号54(図1C)と同一配列を含む。上記実施例1記載の手続きに従い、PCR断片は、制限酵素Nhel 及びBstEllにより分解された。分解されたDNA断片はエタノールで沈殿させ、3G 及びhistag(Q109) 無しのpSLGAMpR2-BBl 発現プラスミドを作り出すため、pSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1と同じである。127の形質転換細胞が生産され、42の形質転換細胞のアッセイが振盪フラスコでおこなわれた。SDSゲルがタンパク質発現レベルチェックのために用いられた。最上の形質転換細胞の培養液について、トリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0158】
実施例4 アスペルギルス(Aspergillus )でのボウマンービルク(Bowman-Birk) 抑制剤及びその変異体(他のバインダーによるループの置換)の発現
変異体配列が、その技術分野で知られている標準的手順を用いて、一又は両方のループに導入された。変異体配列は、製作者指示に従い商業的に入手可能なファージ ペプチド ライブラリーPhD C7C(New England Biolabs, Beverly, MA)を、標的タンパク質または基質に対して3ラウンドのパンニング(panning)をするか、または公知の活性を持つ配列を用いて決定された。以下に記載の配列では、ループヌクレオチド配列に導入された変更は、下側のケースのヌクレオチドにより示される。
【0159】
a. ループIにaーVEGF(CK37281)を持つBBI
インビトロでMCLAB (配列番号1) により合成され、pCRII-TOPOベクターにクローンされたプラスミドDNAはPCR増幅のDNA鋳型(template)として用いられた。VEGF(a-VEGFと表す)を結合し、VEGF機能を抑制するためのペプチド配列を導入するために2つのプライマーは以下の様にデザインされた:
5' GTTGCGATCAGTGCGCGTGTtacaatctgtatggctggaccTGTCGCTGCT 3' (配列番号46) 及び
5' CGCATATCGGAGCAGCGACAggtccagccatacagattgtaACACGCGCAC 3'. (配列番号47)
PCRが実行され、まず94℃で2分間混合物を熱し、94℃で30秒間、63℃で30秒間、72℃で30秒間のサイクルを30サイクル行った。30サイクルの後に、混合物は72℃で4分間培養され、その後4℃で保存された。置換結合ループはDNA配列により確認された。ループ1(Q117)においてpSLGAMpR2-BBl (CK37281) 発現プラスミドを作り出すために、NhelからBstEllのDNA断片は制限分解によりプラスミドから解放され、精製され、そしてpSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1と同じである。30以上の形質転換細胞が生産され、42の形質転換細胞のアッセイが振盪フラスコでおこなわれた。SDSゲルがタンパク質発現レベルチェックのために用いられた。
【0160】
b. ループIIにaーVEGF(CK37281)を持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、以下の2つのプライマーが用いられた以外は上記実施例の場合と同じ手順に従った:
5' CATGCAAGAGCTGTATCTGCtacaatctgtatggctggaccCAGTGTTTCTG 3' (配列番号48)
5' GATGTCGACGCAGAAACACTGggtccagccatacagattgtaGCAGATACAG 3'. (配列番号49)
【0161】
c. ループI及びIIにaーVEGF(CK37281)ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、以下の4つのプライマーが用いられた以外は上記と同じ手順に従った:
5' GTTGCGATCAGTGCGCGTGTtacaatctgtatggctggaccTGTCGCTGCT 3' (配列番号46)
5' CGCATATCGGAGCAGCGACAggtccagccatacagattgtaACACGCGCAC 3' (配列番号47)
5' CATGCAAGAGCTGTATCTGCtacaatctgtatggctggaccCAGTGTTTCTG 3' (配列番号48)
5' GATGTCGACGCAGAAACACTGggtccagccatacagattgtaGCAGATACAG 3'. (配列番号49)
【0162】
d. ループI に a-補体タンパク質c2ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、c2(a-c2と表す)を結合しc2機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCAGCTGTagctgcggcaggaagatccccatccagtgcTGTCGCTGCTCCGATATGCGTC 3' (配列番号50)
5'GAGCAGCGACAgcactggatggggatcttcctgccgcagctACAGCTGCACTGATCGCAACAGGGCTTA 3'(配列番号51) ・
【0163】
e. ループI に a-補体タンパク質c3ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、c3( a-c3と表す)を結合し、c3機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCGGCTGTgccaggagcaacctcgacgagTGTCGCTGCTCCGATATGCGTC 3' (配列番号52)
5'GAGCAGCGACActcgtcgaggttgctcctggcACAGCCGCACTGATCGCAACAGGGCTTA 3' (配列番号53)
【0164】
f. ループI に a-補体タンパク質c4ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、c4( a-c4と表す)を結合し、c4機能を抑制するためのペプチド配列を導入するため以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCGCGTGTcagagggccctccccatcctcTGTCGCTGCTCCGATATGCGTC 3' (配列番号55)
5'GAGCAGCGACAgaggatggggagggccctctgACACGCGCACTGATCGCAACAGGGCTTA 3' (配列番号56) ・
【0165】
g. ループI に a-補体タンパク質c5ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、c5(a- c5と表す)を結合し、c5機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCCAGTGTggcaggctccacatgaagaccTGTCGCTGCTCCGATATGCGTC 3' (配列番号57)
5'GAGCAGCGACAggtcttcatgtggagcctgccACACTGGCACTGATCGCAACAGGGCTTAGA 3' (配列番号58)
【0166】
h. ループI に ヒトのa-補体因子B ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、因子B(a-因子Bと表す)を結合し、因子B機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCCAGTGTaagaggaagatcgtcctcgacTGTCGCTGCTCCGATATGCGTC 3' (配列番号59)
5'GAGCAGCGACAgtcgaggacgatcttcctcttACACTGGCACTGATCGCAACAGGGCTTAGA 3' (配列番号60)
【0167】
i. ループI に a-膜メタロプロテアーゼ2(MMP2)ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、MMP2( a-MMP2と表す)を結合し、MMP2機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5' CAGTGCGCGTGTgccgccatgttcggccccgccTGTCGCTGCTCCGATATGCGTC 3' (配列番号61)
5'GAGCAGCGACAggcggggccgaacatggcggcACACGCGCACTGATCGCAACAG 3;(配列番号62)
【0168】
j. ループI にa-膜メタロプロテアーゼ12(MMP12)ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、MMP12( a-MMP12と表す)を結合し、MMP2機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5' CAGTGCGCGTGTggcgccctcggcctcttcggcTGTCGCTGCTCCGATATGCGTC 3' (配列番号63)
5'GAGCAGCGACAgccgaagaggccgagggcgccACACGCGCACTGATCGCAACAG 3' (配列番号64)
【0169】
k. ループI にコットン結合(cotton binding)ペプチド2314を持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、コットンと結合するペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5' GTTGCGATCAGTGCGCGTGTgagcccctgatccaccagcgcTGTCGCTGCT 3' (配列番号65)
5' CGCATATCGGAGCAGCGACAgcgctggtggatcaggggctcACACGCGCAC 3' (配列番号66)
【0170】
l. ループIにコットン結合(cotton binding)ペプチド2317を持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、コットンと結合するペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GTTGCGATCAGTGCGCGTGTagcgccttccgcggccccaccTGTCGCTGCT3' (配列番号67)
5' CGCATATCGGAGCAGCGACAggtggggccgcggaaggcgctACACGCGCAC 3' (配列番号68)
【0171】
m. ループI にコンプスタチン(compstatin)ループを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、コンプスタチン(compstatin)ペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GTTGCGATCAGTGCGCGTGTgttgttcaggactggggccaccaccgcTGTCGCTGCT (配列番号6 9)
5'CGCATATCGGAGCAGCGACAgcggtggtggccccagtcctgaacaacACACGCGCAC (配列番号70)
この場合BBIトリプシン結合ループの7つのアミノ酸は、コンプスタチン(compstatin)結合ループの9つのアミノ酸により代替された。
【0172】
n. ループII にコンプスタチン(compstatin)ループを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、コンプスタチン(compstatin)ペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5' CATGCAAGAGCTGTATCTGCgttgttcaggactggggccaccaccgcTGTTTCTGCG (配列番号71)
5'GTGATGTCGACGCAGAAACAgcggtggtggccccagtcctgaacaacGCAGATACAG (配列番号72)
この場合BBIトリプシン結合ループの7つのアミノ酸は、コンプスタチン(compstatin)結合ループの9つのアミノ酸により代替された。
【0173】
実施例5 トリコデルマ リーセイ(Trichoderma reesei)でのボウマンービルク(Bowman-Birk) 抑制剤及びその変異体の発現
上記例2に記載の手順に従い、BBIをコードするDNAが最適化され、本実施例において用いられた。2つのプライマーはループI 及びIIのプラスミドpSLGAMpR2-BBIまたはaーVEGF(CK37281)ペプチドを持つpSLGAMpR2-BBIを鋳型として用いDNAを増幅するようにデザインされた:
5, GGA CTA GTA AGC GCG ACG ATG AGA GCT CT 3' (配列番号73)
5' AAG GCG CGC CTA GTT CTC CTT ATC GTP CT 3' (配列番号74)
第3のプライマーは、また上記の第2のプライマー(配列番号74)と連動して使用される場合は、BBIタンパク質のN末端に3つのグリシン残基を含むPCR断片を作るために用いられた。
【0174】
5' GGA CTA GTA AGC GCG GCG GTG GCG ACG ATG AGA GCT CT 3' (配列番号75).
上記実施例2に記載と同様の手順に従い、BBIをコードするDNAが最適化され、本実施例において用いられた。PCR断片は制限酵素Spel 及びAscl により切断され、トリコデルマ(Trichoderma)発現プラスミド、pTrex4(図8)に結紮された。pTrex4はpTREX2(図9)の修飾形であり、これはまたpTEXの修飾形である。pTEXベクターの生成のための完全な記述についてはPCT公報番号 WO 96/23928を参照願いたい。当該資料は発現プラスミドを作り出すための遺伝子発現のCBHIプロモータ、ターミネータ、及び形質転換細胞の選択マーカーとしてのトリコデルマ(Trichoderma)pry4遺伝子を含んでおり、引用により本明細書に組み入れられる。pTrex4プラスミドにおいては、BBI遺伝子はCBHIコアーのC末端及びトリコデルマ リーセイ(Trichoderma reesei)のリンカーに融合された。菌の形質転換の間、アスペルギルス ニジュランス(Aspergillus nidulans)のamdS遺伝子が選択マーカーとして用いられた。発現プラスミドはトリコデルマ リーセイ(Trichoderma reesei)に転換された。安定している形質転換細胞は、アセトアミドを窒素源としてトリコデルマ(Trichoderma)最小板(minimal plate)上で単離された。形質転換細胞はamd マイナスプレートで生育された。プレートは1ml/l 1000x塩(salt)、 20g/l Noble Agar, 1.68g/l CsCl, 20g/l グルコース, 15g/l KH2PO4, 0.6g/l MgSO4・7H2O, 0.6g/l CaCl2,・2H2O 及び0.6g/l アセトアミドを含む。pHは最終的に4.5に調整された。1000x塩(salts)は5g/l FeSO4, 1.6g/l MnSO4, 1.4g/l ZnSO4, 及び1g/l CoCl2 を含み、フィルターろ過殺菌された。3日間28℃で培養した後、形質転換細胞は新しいamdマイナスプレートに移され、さらに28℃で3日間育成された。
【0175】
形質転換細胞はその後250mlの振盪フラスコのトリコデルマ リーセイ(Trichoderma reesei)プロフロ媒体(proflo medium)(各形質転換細胞に対し50ml)に植えつけられた。トリコデルマ リーセイ(Trichoderma reesei)プロフロ媒体(proflo medium)は、30g/l アルファラクト−ス, 6.5g/l (NH4)2SO4, 2g/l KH2PO4, 0.3g/l MgSO4・7H2O, 0.2g/l CaCl2, 1ml/l 1000xTris Trace Salts, 2ml/l 10% Tween 80, 22.5g/l Proflo 及び 0.72g/l CaC03. を含む。100xTris Trace Salts は5g/l FeS04・7H20, 1.6g/l. MnS04・H20 及び 1.4g/l ZnS04・7H20.を含む。2日間30℃で育成した後、4ml の培養液が合成培地に移された。合成培地は5g/l (NH4)2SO4, 33g/l PIPPS 緩衝液, 9g/l カサミノ酸(CASAMINO ACIDS), 4.5g/l KH2PO4, 1g/l CACL2, 1g/l MgSO4・7H2O, 5ml/l MAZU 及び 2.5ml/l 400x トリコデルマ リーセイ(Trichoderma reesei)TRACE.を含む。pHは5.5に調整され、殺菌の後に40ml/l 40% のラクトースが加えられた。400x トリコデルマ リーセイ(Trichoderma reesei)TRACEは175g/l クエン酸(無水), 200g/l FeS04・7H20, 16g/l ZnS04 7H20, 3.2g/l CuSO4・5H20, 1.4g/l MnS04 ・ H20 及び0.8g/l H3BO3. (ホウ酸、Boric Acid)をふくむ。
【0176】
プレートで約40個の形質転換細胞が生成され、20個が振盪フラスコでアッセイされた。培養液の上澄みはSDS-PAGE分析に使われ、トリプシン、またはキモトリプシン(chymotrypsin)の抑制活性がアッセイされた。ウエスターン ブロット(Western blot)はまた、融合した形(Cbhl-BBI)及びBBI単独の両者が存在することを示した。
【0177】
実施例6 アスペルギルス(Aspergillus)におけるボウマンービルク(Bowman- Birk)抑制剤及び分泌シャペロン(Secretory Chaperones)の同時発現
以下の実施例はどの様に分泌が増進されるかを詳細に示すものである。STIタンパク質は2つのジスルヒド結合を含み、BBIはその三次構造(tertiary structure)に7つのジスルヒド結合を含み、これらのジスルヒド結合はその機能にとり重要である。ジスルヒド結合を持つタンパク質の折り畳みにはタンパク質ジスルヒドイソメラーゼ(PDI)またはERに他のシャペロンが必要であることは知られている。
【0178】
STIまたはBBIの発現の増進について、一つのプラスミドがSTIまたはBBI発現カセットを含み、他の一つがPDI遺伝子またはシャペロン遺伝子を含む2つのプラスミドの同時発現、または逐次発現すること調査・検討した。まず、3G及びヒスタッグ(histag)(Q109)を持たないpSLGAMpR2-BBIプラスミドを、アスペルギルス ニジェール(Aspergillus Niger)のpdiA遺伝子領域をカバーする4.6kbゲノムDNAを持つプラスミドQ51により、ベクターpUC219で同じ菌株(dgr246ΔGAP : pyr2)に同時転換させた。51の形質転換細胞が得られ、47が振盪フラスコでスクリーニングされた。形質転換細胞14番が、SDSゲルデータ(gel data)による最も高いBBIタンパク質を生成したため選択された。BBIタンパク質の発現レベルは、同時形質転換される場合の方が3G及びヒスタッグ(histag)(Q109)を持たないpSLGAMpR2-BBIプラスミドのみを含む場合よりも高い。図7は増強されたBBIの発現をしめす。この菌株はまた、胞子精製され、再度振盪フラスコでテストされた。
【0179】
上記の手順に従い、我々はヒスタッグ(histag)(Q108)を持たないpSLGAMpR2-BBIプラスミドを、pdiA遺伝子(上記に同じ)を含むプラスミドQ51と同じ菌株(dgr246ΔGAP : pyr2)に同時形質転換させることとした。34の形質転換細胞が振盪フラスコでスクリーニングされた。SDSゲルデータに基づき最高レベルのBBIタンパク質生成能力を持つ1つの形質転換細胞が選択された。BBIタンパク質の発現レベルはヒスタッグ(histag)(Q108)を持たないpSLGAMpR2-BBIプラスミドのみを含む菌株よりも高い。
【0180】
上記の手順に従い、我々はまた3G及びヒスタッグ(histag)(Q109)を持たないpSLGAMpR2-BBIプラスミドと、アスペルギルス ニジェール(Aspergillus Niger)の遺伝子prpAの領域をカバーする1623bpゲノムDNAを含むプラスミドQ124とをベクターpUC219で同じ菌株(dgr246ΔGAP : pyr2)に転換させた。28の形質転換細胞が振盪フラスコでスクリーニングされた。SDSゲルデータ(gel data)による最も高いBBIタンパク質を生成能力を持つ1つの形質転換細胞が選択された。BBIタンパク質の発現レベルは、同時形質転換された菌株の場合の方が、単に3G及びヒスタッグ(histag)(Q109)を持たないpSLGAMpR2-BBIプラスミドのみを含む菌株よりも高い。図7は増強された発現を表す(レーン15対レーン3)。この菌株はまた、胞子精製され、再度振盪フラスコでテストされた。
【0181】
実施例7 組み換えプロテアーゼ抑制剤変異体保持活性(Recombinant Protease Inhibitor Veriants Retain Actvity)
上記の方法により生産されたSTI、BBI 及びその変異体はその活性、例えばプロテアーゼ活性の抑制がテストされた。
【0182】
a.プロテアーゼ抑制
950μlのTris 緩衝食塩水+0.02% Tween 20が20μlのプロテアーゼ(牛のトリプシンまたはキモトリプシン)1mM HCL中100μg/ml )及び20μlサンプルと組み合わされる。液は混合され、室温で30分間培養される。10μl基質(トリプシンに対しサクシニル(succinyl)-ala-ala-pro-arg-パラニトロアニリド(paranitroanilide), DMSO 中10mg/ml、及びキモトリプシン(chymotrypsin)に対しサクシニル(succinyl)-ala-ala-pro-phe-パラニトロアニリド(paranitroanilide), DMSO 中10mg/ml)を加え、液を混合する。405nmで吸光度を測定し、レート(A405/分)が決められる。プロテアーゼのコントロールブランクサンプルに対する活性抑制比率を対比し、次の式に従い算出される:
(A405/分(サンプル))/(A405/分(ブランク))*100μg/ml(プロテアーゼ)*
(MW抑制剤)/(MWプロテアーゼ)=[抑制剤] μg/ml
b.a VEGFペプチドによるHUVEC増殖の抑制
HUVE細胞(Cambrex, East Rutherford, NJ)を1−5回通過させ、製造者指示に従い保持された。HUVECの成長は0.03から20ng/mlのVEGFにより刺激され、最高の増殖は10ng/ml VEGF165 (R&D Systems)の場合であった。この濃度はそれ以降の実験においても用いられた。0.00052μMから25μMの一連のa-VEGFペプチド(実施例4参照)及びアンチVEGF MAb コントロール(R&D Systems)が,96ウエルプレートに3重に播種されたHUVECsに加えられる前に、10ng/ml VEGFと混合された。細胞の増殖が3H-thymidine により測定された。極めて大きな抑制効果が観察された(データは示されていない)。
【0183】
本明細書に記載の実施例、実施の形態は説明のために記載したものであり、当業者に対してはそれらの種々の改変、変更を示唆することができ、それらはすべて本出願の精神及び趣旨の範囲、及び本願の請求の範囲に含まれる。本明細書に引用したすべての刊行物、特許、特許出願は、そのすべては引用により本明細書に組み入れられる。
【図面の簡単な説明】
【0184】
【図1】図1は大豆Bowman-Birk タイプ プロテアーゼ抑制剤(BBI)のヌクレオチド配列(配列番号1)に最適化されたコドンである。この配列は、NVISKR(点線の下線部分)、融合タンパク質の切断部位及び発現プラスミドにクローンされる3つの制限酵素部位を含む。5’ 末端のNhel部位及び3’末端のXhol部位は下線及び標識を付してある。3’末端のBstEll部位は♯記号を付して示す。終止コドンは*(asterisk)で示す。融合タンパク質として発現するため、開始コドンは存在しない。成熟BBIコード配列は二重下線により示す(配列番号2)。成熟BBIコード配列に先立ち3つのグリシン(図1B)残基をコードするヌクレオチドの追加は、図2(配列番号5)に示す3つのグリシン残基をコードする配列を用い行うことができる。図1Cは、BBIをコードするヌクレオチド配列、3つの制限部位、kex2部位、N末端に3つのグリシン残基及びC末端に6つのヒスチジン(histidine)基を示す(配列番号54)。
【図2】図2は大豆トリプシン抑制剤(Soybean Trypsin Inhibitor, STI)に最適化されたコドン、クーニッツタイプ(Kunitz type)プロテアーゼ抑制剤(配列番号3)を示す。この配列はヌクレオチドをコードするNVISKR(点線の下線)(配列番号4)である。融合タンパク質の切断部位、及びC末端に6つのヒスチジン(histidine)基を持つ(点線で示す)。発現プラスミドにクローンする3つの制限酵素部位(5’末端のNhel、3’末端のXhol 及びBstEllは図1に示す通り)も含まれる。kex2部位(NVISKR)の後の3つのグリシン残基は太字で示す。成熟STIをコードするヌクレオチド配列は破線の下線で示す(配列番号6)。
【図3】図3AはBBIの成熟アミノ酸配列(配列番号7)を示す。図3BはN末端に3つのグリシン残基を持つBBIである(配列番号8)。図3CはN末端に3つのグリシン残基を持ち、C末端に6つのヒスチジン残基を持つBBIである(配列番号9)。図3A-CのループIは下線を付したアミノ酸残基で示し、ループIIのアミノ酸残基は太字で示す。
【図4】図4AはN末端に3つのグリシン残基を持ち、C末端に6つのヒスチジン残基を持つ成熟STIである(配列番号10)。図4BはN末端に3つのグリシン残基を持つSTIである(配列番号11)。図4CはSTIの成熟アミノ酸配列(配列番12)を示す。ループ1は下線を付したアミノ酸残基で示す(配列番号13)。ループIIのアミノ酸残基は太字で示す(配列番号14)。
【図5】図5は発現プラスミド pSLGAMpR2-BBIのダイアグラムである。このプラスミドはアスペルギルス ニジェール(Aspergillus niger)グルコアミラーゼ プロモータ、触媒コアー及びターミネータ、マーカー遺伝子(Aspergillus Niger pyrG)及び牛のプロキモシン(prochymosin)遺伝子を挿入することによるpSL1180に由来のpSLGAMpR2をベースにしている。pSL1180プラスミドはAmersham Biosciences (Piscataway, NJ)から入手可能である。pSLGAMpR2プラスミドは、pSLGAMpR2-BBIに示した位置と同じ相対位置に上記リストした要素が挿入されている。ただし、牛のプロキモシン(prochymosin)遺伝子はBBI遺伝子があるところに存在する。したがって、BBI遺伝子はpSLGAMpR2-BBIを作り出すためpSLGAMpR2中のプロキモシン(prochymosin)遺伝子に代替する。
【図6】図6は野生型BBIアミノ酸配列(配列番号7)及び精選されBBIの変異体である(配列番号15から29)。野生型BBIは下線を付したループを持つ。野生型との相違は、太字、下線(ループI)または太字(ループII)で示す。変異体によっては、例えば、C2,C3,C4, C5 及びファクターB,位置13のアラニン(2つのシステインの間)はまた、「セリン」「グリシン」または「グルタミン」の何れかに変えられた。またコンプスタチン(compstatin)ペプチドは7個のアミノ酸に代えて9個持っている。また変異体配列も示す(配列番号30から40)。
【図7】図7はタンパク質SDSゲルの写真である。レーン(Lane)1は分子量マーカーを含む。レーン2は形質転換されていない親株である。レーン3はBBIをコードするDNAにより同時形質転換された親株である。レーン4はBBIをコードするベクター及びシャペロン(pdiA)をコードするベクターで形質転換された親株である。レーン15はBBIをコードするベクター及びシャペロン(prpA)をコードするベクターで同時形質転換された親株である。所望されるタンパク質、例えば、BBIの発現はシャペロンの存在により増進された。
【図8】図8はプラスミドpTrex4のダイアグラムである。
【図9】図9A−DはpTrex2の核酸配列である(配列番号41)。
【図1A】
【図1B】
【図1C】
【技術分野】
【0001】
本発明は糸状菌におけるプロテアーゼ抑制剤及びその変異体の発現の方法に関する。本発明は融合核酸、ベクター、融合ポリペプチド及びプロテアーゼ抑制剤を得る方法を開示する。
【背景技術】
【0002】
プロテアーゼは広範囲の生物学的プロセスに関わっている。プロテアーゼとプロテアーゼ抑制剤のバランスの崩壊はしばしば組織の病理的破壊に関係して来る。
【0003】
組織損傷におけるプロテアーゼの役割に焦点をあてた多くの研究がされており、プロテアーゼとプロテアーゼ抑制剤のバランスが組織の統一性を維持する主要決定要素であると考えられている。好中球を含み、炎症を起こした細胞のセリーン プロテアーゼは肺気腫、関節炎、アトピー性皮膚炎、及び疥癬等の種々の炎症傷害に関わっている。
【0004】
プロテアーゼはまたある種の癌の進展に役割をも果たしていると考えられる。正常な細胞は、細胞外マトリクス(exteracellular matrix, ECM)と呼ばれる複雑なタンパク質ネットワークと常時接触を保っている。ECMは細胞の移動に対するバリアーであり、癌細胞は転移するためにはその付着を立ち切り、分解し、そしてECMを通じ移動する手段を作り出さねばならない。プロテアーゼは他のタンパク質を分解する酵素であり、長い間、ECMを噛み千切ることによってその元の位置から腫瘍細胞を解放することを助ける酵素であると考えられていた。最近の研究によれば、プロテアーゼはプロテアーゼにより活性化する受容体−2(Protease-Activated Receptor-2, PAR2)と呼ばれる腫瘍細胞膜に含まれるタンパク質の活性化により細胞の形を変化させ、またその運動を促進することが分かった。これにより細胞の運動組織を活性化する多くの細胞内反応が起こる。したがって、腫瘍の転移における第一ステップの一つは、細胞の形が移動の方向に面しているその一端が明確に突起した形になるように再編されることであると仮定されている。そして細胞は血管壁を通り、そして遠く離れた場所に移動し、結局再付着して転移腫瘍を作り出す。例えば、ヒトの前立腺表皮細胞は構造的に前立腺特異抗原(PSA)、精漿(seminal plasma)の通常の構成要素であるカリクレイン(kallikrein)様のセリンプロテアーゼを分泌する。プロテアーゼは細胞外マトリクスを分解し、癌状細胞の侵入を容易にするように作用する。
【0005】
合成及び天然プロテアーゼ抑制剤はインビボ及びインビトロにおいて腫瘍の進行を抑制することが分かっている。これまでの研究、調査によると、セリンプロテアーゼ抑制剤、あるいはセルピン(SERPINS)として分類される、構造的に関連するタンパク質のファミリーに属するある種のプロテアーゼ抑制剤は、好中菌エラスターゼ(neutrophil elastase)のみならず、トリプシン(trypsin)、カテプシンG(cathpsin G)、トロンビン(thrombin)、組織カリクレイン(kallikrein)を含むいくつかのプロテアーゼを抑制することが知られている。セルピン(SERPINS)はインビトロのカルシノゲン(carcinogen)誘発形質転換、及び動物モデル系でのカルシノゲン変異(carcinogenesis)の予防及び抑制に極めて有効である。精製プロテアーゼ抑制剤を計画的に与えることにより、関節の炎症並びに軟骨及び骨の破壊も減らすことができる。
【0006】
プロテアーゼ抑制剤を局所的に投与する場合として、よく見られる皮膚の炎症症状であるアトピー性皮膚炎があるが、これは身体の一部の数箇所に留まる場合と、または身体の広い範囲に拡がる場合がある。プロテアーゼ抑制剤に脱色活性及び紫外線で誘発される色素沈着を防止する力があることは、インビトロ及びインビボで実証されている。Paine et al., Journal of Investigative Dermatology 116, 587-595(2001)。プロテアーゼ抑制剤はまた傷の治癒を助けることが分かっている(http://www.sciencedaily.com/releases/2000/10/001002071718.htm)。分泌白血球プロテアーゼ抑制剤は組織破壊を逆転させ、局所的に適用された場合は瑕の治癒プロセスを促進することが実証された。さらに、セリンプロテアーゼ抑制剤はまた紅斑性狼瘡(lupus erythematosus)患者の痛みを和らげることができる(米国特許番号6537968参照)。
【0007】
上記で見たように、プロテアーゼ抑制剤はプロテアーゼの活動に干渉する。自然発生のプロテアーゼ抑制剤は、穀物(オート、大麦、メイズ)芽キャベツ(Brussel sprout)、玉ねぎ、ビートの根、小麦、シコクビエ(finger millet)及びピーナツの様に種々の食物に存在する。興味あるソースに大豆がある。大豆での平均含有水準は、二つの最も重要なプロテアーゼ抑制剤であるKunitz及びBowman-Birkで、各々約1.4%及び0.6%である。この様に含有水準が低いため、診療的に応用するには自然のプロテアーゼ抑制剤を単離することは現実的とはいえない。
【0008】
したがって、血液感染性媒体が哺乳類の組織培養細胞で作り出される場合には、血液感染性媒体に関連したリスクも減らし、または除去するプロテアーゼ抑制剤及びその変異体を大量に生産する方法が求められる。本明細書記載の生産方法は大量のタンパク質治療剤の生産を可能にする。
【発明の開示】
【0009】
発明の簡単な概要
本明細書は核酸、細胞及びプロテアーゼ抑制剤及びその変異体の生産方法を提供する。
【0010】
第一の実施の形態においては、機能的(functional)プロテアーゼ抑制剤をコードする核酸を提供する。一つの特徴として第一、第二、第三及び第四の核酸配列と動作可能にリンクされた(operably linked)調節塩基配列を含む核酸が提供される。第4の核酸配列に続き終了配列が提供される。
【0011】
第二の特徴として、第一の核酸配列は第一の糸状菌においてシグナルポリペプチド機能部位(functional)を分泌配列としてコードし、第二の核酸は分泌ポリペプチド、または通常の第一または第二の糸状菌から分泌されるその機能部位をコードし、第三の核酸は切断可能なリンカーをコードし、そして第四の核酸はプロテアーゼ抑制剤、またはその断片をコードする。
【0012】
第三の特徴として、プロテアーゼ抑制剤をコードする核酸配列を含む発現カセットを提供する。
【0013】
第四の特徴として、本発明はプロテアーゼ抑制剤変異体をコードするポリヌクレオチドに関する。ポリヌクレオチドは、少なくとも一つのループが変えられたボウマン ビルク(Bowman-Birk)抑制剤変異体をコードすることもある。ポリヌクレオチドは、少なくとも一つのループが変えられた大豆トリプシン抑制剤(Soybean Trypsin Inhibitor)変異体をコードすることもある。
【0014】
第二の実施の形態においては、機能的プロテアーゼ抑制剤またはその変異体の発現方法が提供される。一つの特徴として、宿主細胞は(i)プロテアーゼ抑制剤またはその変異体をコードする核酸配列を含む発現カセットにより形質転換され、また(ii)プロテアーゼ抑制剤またはその変異体を発現させる適当な条件下で培養されることである。任意的に、この方法はさらにプロテアーゼ抑制剤またはその変異体を回収することを含む。
【0015】
第二の特徴として、宿主細胞は(i)プロテアーゼ抑制剤またはその変異体をコードする核酸配列を含む第一の発現カセットにより形質転換され、(ii)シャペロンをコードする核酸配列を含む第二の発現カセットで形質転換され(iii) プロテアーゼ抑制剤またはその変異体を発現させる適当な条件の下で培養されることである。任意的に、プロテアーゼ抑制剤またはその変異体が回収されることもある。一つの特徴として、プロテアーゼ抑制剤またはその変異体は融合タンパク質として発現される。任意的にこの方法はさらにプロテアーゼ抑制剤またはその変異体を回収する方法を含む。
【0016】
第三の実施の形態においては、プロテアーゼ抑制剤またはその変異体を発現させることのできる細胞を提供する。宿主細胞はプロテアーゼ抑制剤及びその変異体をコードする発現カセットにより形質転換される。宿主細胞はアスペルギルス(Aspergillus)及びトリコデルマ(Trichoderma)からなるグループから選択されることもある。
【0017】
第四の実施の形態においては、機能的プロテアーゼ抑制剤及びその変異体が提供される。一つの特徴として、機能的プロテアーゼ抑制剤及びその変異体は、グルコアミラーゼシグナル配列、プロ配列(prosequence)、触媒ドメイン及び成熟グルコアミラーゼのアミノ酸配列502までのリンカー領域、それに続くアミノ酸NVISKR、及び成熟プロテアーゼ抑制剤及びその変異体よりなる融合タンパク質として発現される。
【0018】
第二の特徴として、発現したタンパク質は融合タンパク質からプロテアーゼ抑制剤及びその変異体を解放するためプロテアーゼで処理されることである。第三の特徴として、本発明はプロテアーゼ抑制活性を持つポリペプチドを提供する、それは下記よりなるグループから選択される;
Bowman-Birk 抑制変異体
Soybean Trypsin抑制変異体
Bowman-Birk抑制剤
Soybean Trypsin 抑制剤 及び
少なくとも一つの変異体配列を含む骨格(scaffold)
本発明の他の目的、特徴及び利益は以下に述べる詳細な説明により明らかになろう。しかし、詳細な説明及び特徴的な実施例は本発明の好ましい実施形態を示しているが、これらは説明のために取り上げられているものであることを理解する必要がある。この詳細な説明の記載より本発明の範囲及び思想に属する種々の変更、及び修飾をすることは当業者に明らかであるからである。
【0019】
発明の詳細な説明
本発明は以下の定義、及び実施例を参照として用い詳細に説明する。すべての特許及び刊行物は、それらの特許及び刊行物に開示される配列を含み、本明細書において参照されているものは引用により明確に本明細書に組み入れられる。
【0020】
他の意味に解釈されるよう記載がない限り、本明細書で用いられる全ての技術、科学用語は本発明の属する技術分野の当業者により通常理解される意味と同じ意味を持つ。Singleton et al., DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOGY 2nd ED., John Wiley and Sons, New York (1994), and Hale & Marham, THE HARPER COLLINS DICTIONARY OF BIOLOGY, Harper Perennial, NY (1991)は、当業者に本発明で使用されている用語の多くを説明している一般的な辞書を提供する。これらに記載の方法及び材料で本明細書に記載のものと同一または類似のものは、本発明の実施または試験のために用いることができるが、本明細書には好ましい方法、及び材料が記載されている。数値の範囲は範囲を規定する数値を含む。他の意味に解される様に示されていない限り、核酸は、左から右方向に向けて、5’から 3’の方向に、またアミノ酸配列は、左から右方向に向けて、アミノ基からカルボキシ基の方向となっている。開業医師は、定義と用語については特にSambrook et al., 1989, 及び Ausubel FM et al., 1993を参照することを奨める。本発明は、記載された特定の方法、プロトコール、及び試薬に限定されるものではない。これらのものは内容が種々変わりうるからである。
【0021】
本明細書に記載の各見出し(heading)は本発明の実施の態様に限定するものではない。本発明は明細書全体を参照することにより明らかとなるからである。したがって、以下に引き続き定義される用語は、明細書全体を参照することにより十分な定義となる。
【0022】
定義
「発現カセット」(expression cassette)または「発現ベクター」(expression vector)は、標的細胞において特定の核酸の転写を許す特定の一連の核酸要素により、遺伝子組み換え法または合成法によって生成される核酸構築(construct)をいう。組み換え発現カセットはプラスミド、染色体、ミトコンドリアDNA、プラスチドDNA(plastid DNA)、ビールス、または核酸断片に組み入れることができる。典型的には、発現ベクターの組み換え発現カセット部分は、特に転写される核酸配列及びプロモータを含む。発現カセットはDNA構築及びその文法的な同等物と代替可能に使用することができる。
【0023】
本明細書で用いる「ベクター」(vector)は、核酸配列を細胞に転写するようにデザインされた核酸構築をいう。「発現ベクター」は異質細胞(foreign cell)に異種(heterologous)DNA断片を組み込み、そして発現させる能力を持つベクターをいう。多くの原核生物発現ベクター及び真核生物発現ベクターは市場で入手可能である。適当な発現ベクターを選択することは当技術分野の当業者の知識の範囲に属する。
【0024】
本明細書で用いる「プラスミド」(plasmid)はクローンベクターとして用いられ、そしてある真核生物において染色体外の自己複製遺伝子要素を形成し、または宿主染色体に合体する環状二重鎖(double-stranded;ds)DNA構築をいう。
【0025】
「核酸分子」(nucleic acid molecule)または「核酸配列」(nucleic acid sequence)はRNA, DNA及びcDNA分子を含む。遺伝子コードの縮重の結果、あるタンパク質をコードする多数のヌクレオチド配列が作り出されることがあることが理解されるであろう。
【0026】
本明細書に記載の「融合DNA配列」(fusion DNA sequence)は、5’から 3’方向に向け、第一、第二、第三、及び第四のDNA配列を含む。
【0027】
本明細書に記載の「第一の核酸配列」(a first nucleic acid sequence)または「第一のDNA配列」(first DNA sequence)は第一の糸状菌においてシグナルポリペプチド機能部位(functional)を分泌配列としてコードする。そのようなシグナル配列は、以下の配列を含む。すなわち、牛のキモシン(bovine chymosin), ヒトの組織プラスミノゲン活性剤(human tissue plasminogen activator),ヒトのインターフェロン(human interferon)から得られるシグナル配列を含む真核生物のシグナル配列、及びGwynne et al.,(1987) Bio/Technology 5, 713- 719に記載の合成コンセンサス真核生物(synthetic consensus eukaryotic)シグナル配列に加えて、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger variant awamori)、 アスペルギルス ニジェール(Aspergillus niger), アスペルギルス オリザ(Aspergillus oryzae)から得られるグルコアミラーゼ(glucoamylase), α-アミラーゼ(α-amylase)及びアスパルチル プロテアーゼ(aspartyl protease)のシグナル配列, トリコデルマ(Trichoderma)から得るセロビオヒドロラーゼ I(cellobiohydrolase I), セロビオヒドロラーゼ II(cellobiohydrolase II), エンドグルカナーゼI (endoglucanase I), エンドグルカナーゼIII(endoglucanase III)のシグナル配列、ノイロスポラ(Neurospora)及びフミコラ(Humicola)から得るグルコアミラーゼ(glucoamylase)のシグナル配列である。特に好ましいシグナル配列は、融合タンパク質を発現及び分泌させるために用いられる発現宿主により分泌されるポリペプチドに由来する配列である。例えば、アスペルギルス ニジェール(Aspergillus niger)から融合タンパク質を発現させ分泌させる場合、アスペルギルスニジェールから得られるグルコアミラーゼ(glucoamylase)のシグナル配列が好ましい。本明細書に記載の第一のアミノ酸配列は糸状菌において機能的である分泌配列に対応する。そのようなアミノ酸配列は、規定した第一のDNA配列によりコードされる。
【0028】
本明細書に記載の「第二のDNA配列」(second DNA sequence)は通常糸状菌から発現した「分泌ポリペプチド」(secreted polypeptide)をコードする。そのような分泌ポリペプチはアスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)、及び アスペルギルス ニジェール(Aspergillus niger), アスペルギルス オリザ(Aspergillus oryzae)から得られるグルコアミラーゼ、α-アミラーゼ、及びアスパルチル プロテアーゼ(aspartyl proteases)、およびトリコデルマ(Trichoderma)から得るセロビオヒドロラーゼ I(cellobiohydrolase I),セロビオヒドロラーゼ II(cellobiohydrolase II), エンドグルカナーゼI (endoglucanase I), エンドグルカナーゼIII(endoglucanase III)、ノイロスポラ(Neurospora)種及びフミコラ(Humicola)種から得られるグルコアミラーゼ(glucoamylase)を含む。第一のDNA配列の場合と同様に、望ましい分泌ポリペプチは、糸状菌発現宿主により自然に分泌されるポリペプチドである。したがって、例えば、アスペルギルスニジェール(Aspergillus niger)を用いる場合、望ましい分泌ポリペプチはアスペルギルス ニジェールから得られるグルコアミラーゼ(glucoamylase)及びαアミラーゼ、最も好ましくはグルコアミラーゼである。一つの特徴的な場合においては、グルコアミラーゼはアスペルギルス グルコアミラーゼと95%、96%、97%、98%、または99%以上相同である。
【0029】
アスペルギルス グルコアミラーゼが第二のDNA配列によりコードされる分泌ポリペプチドである場合は、任意的にプロ配列(prosequence)を含み、タンパク質全体、またはその一部が用いられる。したがって、切断リンカーポリペプチドはアミノ酸残基の468から509間の何れの位置においてもグルコアミラーゼに融合してことがある。他のアミノ酸残基は融合部位かもしれないが、上記の残基を用いることは特に有益である。
【0030】
「分泌ポリペプチドの機能する部分」(functional portion of a secreted polypeptide)または文法的な同等物(grammatical equivalent)は、端が切断されているとしても、正常な立体配置に折り畳まれる能力を維持している端の切断された分泌ポリペプチを意味する。例えば、アスペルギルス グルコアミラーゼ変異体アワモリによる牛のキモシン生成の場合において、成熟グルコアミラーゼの11番目のアミノ酸に続くプロキモシン(prochymosin)の融合はプロキモシン(prochymosin)の生成に比べて利点をもたらさないことが示された(米国特許5,364,770)。USSN 08/318,494において、プレプログルコアミラーゼ(preproglucoamylase)のC末端において成熟グルコアミラーゼ(glucoamylase)の297番目までのアミノ酸にプロキモシン(prochymosin)を融合させ、それに加えて成熟グルコアミラーゼ(glucoamylase)のアミノ酸1−11を繰り返すことは、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)において分泌されるキモシン(chymosin)を作り出さないことが示されている。後のケースにおいては、融合タンパク質に存在するグルコアミラーゼ(glucoamylase)触媒ドメインの部分(約63%)は正しく折り畳まれることが困難と思われ、したがって、細胞によって分泌され得なかったであろう異常な、誤った折り畳み方をし(misfolded)及び/または不安定な融合タンパク質が生成されたのかも知れない。部分的触媒ドメインが正しく折り畳むことができないことは付着キモシン(chymosin)の折り畳みの妨げとなったかも知れない。したがって、そのポリペプチドが付着している所望するポリペプチドから独立にその通常の立体配置に従い折り畳むことができるように、自然分泌のポリペプチドのドメインに十分な数の残基が存在している必要があると思われる。
【0031】
殆どの場合、分泌ポリペプチドの部分は正しく折り畳まれ、そしてまた折り畳みがない場合に比べて分泌が増加する。
【0032】
同様に殆どの場合、分泌ポリペプチドの先端を切り取ることは、機能的部分が生物学的機を維持することを意味する。好ましい実施の形態においては、例えば、基質結合ドメインのような他の機能的ドメインが用いられることもあるが、分泌ポリペプチドの触媒ドメインが用いられる。アスペルギルス ニジェール(Aspergillus niger)及びアスペルギルス ニジェール変異体アワモリ グルコアミラーゼ(Aspergillus niger var. awamori glucoamylase)の場合、好ましい機能部位は酵素の触媒ドメイン部分を保持し、アミノ酸1から471を含む。さらに、好ましい実施の態様においては、触媒ドメイン及び全てのまたは一部のリンカー領域を用いる。代替的に、グルコアミラーゼ(glucoamylase)のでんぷん結合領域が使用される場合もあり、それはアスペルギルス ニジェール変異体アワモリグルコアミラーゼ(Aspergillus niger var. awamori glucoamylase)のアミノ酸509−616 を含む。
【0033】
本明細書に記載の「第3のDNA配列」(third nucleic sequence)は切断リンカーポリペプチドをコードするDNA配列を含む。そのような配列はグルコアミラーゼ(glucoamylase)のプロ配列、牛のキモシン(chymosin)のプロ配列、スブチリシン(subtilisin)のプロ配列、ヒトの免疫不全ウィルスプロテアーゼを含むレトロウィルスプロテアーゼ(retroviral protease)のプロ配列含み、またトリプシン(trypsin), Xa因子コラゲナーゼ(factor Xa collagenase)、クロストリピン(clostripin)、スブチリシン(subtilisin), キモシン(chymosin),イーストKEX2 プロテアーゼ(yeast KEX2 protease)、アスペルギルスKEXB(Aspergillus KEXB)等により認識され、かつ切断されるアミノ酸配列をコードするDNA配列を含む。例えば、Marston, F. A.O. (1986) Biol. Chem J. 240,1-12を参照願いたい。そのような第3のDNA配列はまた、臭化シアン(cyanogen bromide)により選択的に切断されるアミノ酸メチオニン(methionine)をコードすることもある。第3のDNA配列は融合ポリペプチドの切断をもたらす特定の酵素、または化学物質により認識されることの必要なアミノ酸配列をコードするのみで足りることは理解されるべきである。このように、例えば、全体のグルコアミラーゼ(glucoamylase)、キモシン(chymosin)またはスブチリシン(subtilisin)の全体のプロ配列を使用することは必要ない。むしろ、適当な酵素により認識され、また切断されるのに必要なプロ配列の部分のみが必要になる。
【0034】
第3の核酸は、融合ポリペプチド質の切断をもたらす特定の酵素、または化学物質により認識されるのに必要なアミノ酸配列をコードするのみでよいことは理解すべきである。
【0035】
特に好ましい切断リンカーは、天然のアスペルギルスKEX2類似のプロテアーゼ(Aspergillus KEX2-like protease)により切断され得るKEX2プロテアーゼ認識部位(Lys 及びArg)、Lys 及びArgのトリプシン プロテアーゼ(trypsin protease)認識部位、及びエンドプロテナーゼ-Lys-C(endo- proteinase-Lys-C)切断認識部位である。
【0036】
本明細書に記載の「第4のDNA配列」(forth DNA sequence)は「所望されるポリペプチド」(desired polypeptide)をコードする。そのような所望されるポリペプチドはプロテアーゼ抑制剤及びその変異体を含む。
【0037】
対応する4つのアミノ酸配列をコードする上記規定の4つのDNA配列は組み合わされて「融合DNA配列」(fusion DNA sequence)を形成する。そのような融合DNA配列は5’末端から3’末端に掛けて第1,2、3及び第4のDNA配列の順に適切な読み枠(reading frame)に組み立てられる。そのように組み立てられて、DNA配列は糸状菌のシグナルペプチド機能部位を分泌配列として、通常糸状菌から分泌される分泌ポリペプチまたはその部分、切断リンカーポリペプチド及び所望のポリペプチドをコードする「融合ポリペプチド」(fusion polypeptide),「融合タンパク質」(fusion protein)または「融合類似体」(fusion analog)をアミノ末端からコードする。
【0038】
本明細書に記載の「所望するタンパク質」(desired protein)または「所望するポリペプチド」(desired protein)は、分泌を増大させる構築に融合されていない熟成した形のポリペプチドまたはタンパク質をいう。したがって、「所望するタンパク質」または「所望するポリペプチド」は融合されていない形の宿主細胞により発現され、また分泌されるタンパク質をいう。
【0039】
本明細書に記載の「融合ポリペプチド」(fusion polypeptide),「融合タンパク質」(fusion protein)または「融合類似体」(fusion analog)は、アミノ末端から、宿主細胞においてシグナルペプチド機能部位を分泌配列機能として、通常宿主細胞から分泌される分泌ポリペプチまたはその部分、切断リンカーポリペプチド及び所望のポリペプチドをコードする。融合タンパク質は、融合タンパク質において他のタンパク質配列から自由である所望するタンパク質を生み出すために、例えば、プロテアーゼなどの宿主細胞酵素によりプロセスされるかもしれない。本明細書に記載の「融合類似体」(fusion analog)または「融合ポリペプチド」(fusion polypeptide),または「融合タンパク質」(fusion protein)は相互に交換可能に用いられることもある。
【0040】
本明細書に記載の「プロモータ配列」(promoter sequence)は、発現の目的で特定の糸状菌により認識されるDNA配列をいう。これは上記に定義された融合タンパク質をコードするDNA配列に動作可能にリンクされている。そのようなリンクすることには、融合DNA配列をコードするDNA配列の翻訳開始コドンとの関係でプロモータの位置を決めることを含む。プロモータ配列は融合DNA配列の発現を仲介する転写及び翻訳制御配列を含む。その例として、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)またはアスペルギルス ニジェール グルコアミラーゼ(Aspergillus niger glucoamylase)遺伝子(Nunberg, J. H. et al. (1984) Mol. Cell Biol, 4, 2306-2315; Boel, E. et al. (1984)EMBO J.3, 1581-1585)、アスペルギルス オリザ(Aspergillus oryzae)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori) またはアスペルギルス ニジェール(Aspergillus niger)、アルファアミラーゼ(alpha amylase)遺伝子、リゾムコール ミーヘイカルボキシルプロテアーゼ(Rhizomucor miehei carboxyl protease)遺伝子、トリコデルマ リーセイ セロビオヒドロラーゼ I (Trichoderma reesei cellobiohydrolase I)遺伝子 (Shoemaker, S. P. et al. (1984) 欧州特許出願番号 EPO0137280A1), アスペルギルス ニジュランス trpC (Aspergillus nidulans trp C)遺伝子 (Yelton, M. et al. (1984) Proc. Natl. Acad. Sci. USA 81, 1470-1474; Mullaney, E. J. et al. Mol. Gen. Genet. 199, 37-45) 、アスペルギルス ニジュランス alcA (Aspergillus nidulans alcA )遺伝子(Lockington, R. A. et al. (1986) Gene 33 137-149)、アスペルギルス ニジュランス amdS (Aspergillus nidulans amdS)遺伝子 (McKnight, G. L. et al. (1986) Cell 46, 143-147), アスペルギルス ニジュランスamdS (Aspergillus nidulans amdS)遺伝子 (Hynes, M. J. et al. (1983) Mol. Cell Biol 3 1430-1439), 及びSV40初期プロモータのようなより高度の真核プロモータ(Barclay, S. L. and E. Meller (1983) Molecular and Cellular Biology 3, 2117-2130)がある。
【0041】
同様に、「終了配列」は、転写を終了させる発現宿主により認識されるDNA配列である。これは発現される融合ポリペプチドをコードする融合DNAの3’末端に動作可能にリンクされている。これらの例には、アスペルギルス ニジュランス trpC (Aspergillus nidulans trp C)遺伝子 (Yelton, M. et al. (1984) Proc. Natl. Acad. Sci. USA 81, 1470-1474; Mullaney, E. J. et al. (1985)Mol. Gen. Genet. 199, 37-45)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)またはアスペルギルス ニジェール グルコアミラーゼ(Aspergillus niger glucoamylase)遺伝子 (Nunberg, J. H. et al. (1984) Mol. Cell. Biol 4、2306-253; Boel, E. et al. (1984), EMBO J. 3, 1581-1585 )、アスペルギルスオリザ(Aspergillus oryzae)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori) またはアスペルギルス ニジェール(Aspergillus niger)、アルファアミラーゼ(alpha amylase)遺伝子、リゾムコール ミーヘイ カルボキシルプロテアーゼ(Rhizomucor miehei carboxyl protease)遺伝子、(EPO 公開番号 0215 594)がル。ただし、本発明においては、どの糸状ターミネ-タ(terminator)も機能すると思われる。
【0042】
「ポリアデニレイション配列」(polyadenylation sequence)は、転写されるときに、転写されるmRNAにポリアデノシン残基を加える際に発現宿主により認識されるDNA 配列をいう。それは発現される融合ポリペプチドをコードする融合DNAの3’末端に動作可能にリンクされている。この例には,上記で述べた様に、アスペルギルス ニジュランス trpC (Aspergillus nidulans trp C)遺伝子 (Yelton, M. et al. (1984) Proc. Natl. Acad. Sci. USA 81, 1470-1474; Mullaney, E. J. et al. (1985)Mol. Gen. Genet. 199, 37-45)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori)またはアスペルギルス ニジェールグルコアミラーゼ(Aspergillus niger glucoamy- lase)遺伝子 (Nunberg, J. H. et al. (1984) Mol. Cell Biol 4 2306-2315) (Boel, E. et al. (1984), EMBO J. 3, 1581-1585 )、アスペルギルスオリザ(Aspergillus oryzae)、アスペルギルス ニジェール変異体アワモリ(Aspergillus niger var. awamori) またはアスペルギルス ニジェール(Aspergillus niger)、アルファアミラーゼ(alpha amylase)遺伝子、及びリゾムコール ミーヘイ カルボキシルプロテアーゼ(Rhizomucor miehei carboxyl protease)遺伝子を含む。しかしながら、本発明においてはどの糸状菌ポリアデニレイション配列も機能的であると思われる。
【0043】
本明細書に記載の「選択可能なマーカーをコードするヌクレオチド配列」(selectable marker-encoding nucleic sequence)は、糸状菌細胞で発現することができ、そして選択可能なマーカーの発現により、発現遺伝子を含む細胞に対して、対応するある選択された条件下において成長する能力を与えるヌクレオチド配列をいう。
【0044】
核酸が他の核酸配列と機能する関係におかれた場合、「動作可能にリンクされる」(operably linked))という。例えば、分泌リーダーをコードするDNAは、ポリペプチドの分泌に参加するプレタンパク質(preprotein)として発現する場合は、ポリペプチドのDNAに動作可能にリンクされ、プロモータまたはエンハンサー(enhancer)は、配列の転写に影響を与える場合はコード配列に動作可能にリンクされ、リボソーム結合部位は、翻訳を促進するように位置されている場合はコード配列に動作可能にリンクされている。一般に「動作可能にリンクされる」とは、リンクされるDNA配列が連続しており、分泌リーダーの場合には、連続しているとともに読取フェーズにあることをいう。しかしながら、エンハンサーは連続している必要はない。リンクは適当な制限部位において結紮によりなされる。もしそのような部位が存在しない場合は、通常の用法に従いオリゴヌクレオチド アダプターまたはリンカーが用いられる。
【0045】
本明細書に記載の「組み換えの」(recombinant)は、異種(hetero-)核酸配列の導入により修飾され、または細胞がそのように修飾された細胞に由来する細胞またはベクターについていう。したがって、例えば、組み換え細胞は天然の形(非組み換え)細胞の範囲内において同一の形で見出されない遺伝子を発現させ、またはそうでなければ、人の意図的な介在により発現されまたは発現がされなかった、またはそうでなければ異常に発現されるであろう天然遺伝子を発現させるのである。
【0046】
本明細書に記載の「発現」(expression)はポリペプチドが遺伝子の核酸配列に基づき生産されるプロセスをいう。このプロセスは、転写及び翻訳の両者を含む。したがって、「プロテアーゼ抑制剤発現」(protease inhibitor expression)は、その生成物にRNA前駆体、mRNA, ポリペプチド、翻訳後プロセスされるポリペプチド(post-translation processed polypeptide), 及びその派生物を含む、発現されるべき特定のプロテアーゼ抑制剤及びその変異体遺伝子の転写及び翻訳についていう。同様に、「プロテアーゼ抑制剤発現」はプロテアーゼ抑制剤及びその変異体を、図6に例示されているような形に転写、翻訳、及び組み立てることに関していう。例えば、プロテアーゼ抑制剤発現のアッセイには、適当な条件に置かれた場合の菌群の検査、ノーザンブロット分析(northern blot analysis)は勿論のこと、プロテアーゼ抑制剤タンパク質のウエスターンブロット(western blot)、及びプロテアーゼ抑制剤mRNAの逆転写酵素ポリメラーゼ連鎖反応(RT-PCR)アッセイを含む。
【0047】
本明細書に記載の「グリコシド化された」(glycosylated)は、オリゴ糖分子がタンパク質の特定のアミノ酸残基に加えられていることをいう。「脱グリコシド化された」(de-glycosylated)タンパク質はタンパク質からオリゴ糖分子を部分的にまたは完全に除去するように処理されたタンパク質である。「非グリコシ化された」(a-glycosylated)タンパク質は、タンパク質にオリゴ糖分子が付加されていないタンパク質である。これはオリゴ糖の付加を妨げるタンパク質における変異のためである。
【0048】
「グリコシド化されていない」(non-glycosylated)タンパク質はタンパク質にオリゴ糖が付着されていないタンパク質である。これには、オリゴ糖のタンパク質への付加に必要な酵素が存在しないなど種々の理由があるが、これに限定されるものではない。「グリコシド化されていない」(non-glycosy- lated)は、オリゴ糖がタンパク質に付加されなかったタンパク質と、オリゴ糖は付加されたが、結局除去されたタンパク質の両者を含む。「非グリコシ化された」(a-glycosylated)タンパク質は「グリコシド化されていない」(non- glycosylated)タンパク質であるかもしれない。「グリコシド化されていない」(non-glycosylated)タンパク質は、「非グリコシ化された」(a-glycosylated)タンパク質、または「脱グリコシド化された」(de-glycosylated)タンパク質であるかもしれない。
【0049】
本明細書に記載の「単離された」(isolated)または「精製された」(purified)は、それが自然状態において関係した少なくとも一つの成分から除去された核酸またはポリペプチドについていう。
【0050】
「実質的に含んでいない」(substantially free)は、約20%(乾燥重量)以下の他のタンパク質(すなわち、夾雑タンパク質)、約10%以下の他のタンパク質、約5%以下の他のタンパク質、約1%以下の他のタンパク質を含む所望するポリペプチドの製剤を含む場合をいう。
【0051】
「実質的に純粋」(substantially pure)は、本発明のタンパク質またはその断片に適用された場合、そのタンパク質が意図される用途に現実にまた適切に用いられる程度に、本質的に他の物質を含んでいないことを意味する。特にタンパク質が、例えば、タンパク質配列の決定、または製剤の生成において有用であるために十分純粋であり、また宿主細胞の生物学的構成物をほとんど含んでいないほどに純粋であることをいう。
【0052】
本明細書に記載の「標的タンパク質」(target protein)は、例えば、変異体抑制剤の結合によりその活動がブロックされる酵素、ホルモン等のタンパク質をいう。
【0053】
「変異体配列」(variant sequence(s))は、野生型プロテアーゼ抑制剤または他の骨格の結合ループを置換する短いポリペプチド配列をいう。変異体配列は、それが骨格において置換する結合ループ配列と同じ長さである必要はない。
【0054】
「骨格」(scaffold)は、変異体配列が導入される野生型タンパク質配列をいう。ある実施の形態においては、骨格は、例えば、ループのような置換される部分をもつであろう。例えば、本明細書に記載のSTI及び BBI配列は変異体配列の骨格といえよう。
【0055】
プロテアーゼ抑制剤
2つのタンパク質プロテアーゼ抑制剤が大豆から単離された。クニッツ型トリプシン抑制剤(Kunitz-type trypsin inhibitor)(大豆トリプシン抑制剤、STI)及びボウマンービルク プロテアーゼ抑制剤(Bowman-Birk inhibitor)(BBI) である。例えば、Birk, Int. J. Pept. Protein Res. 25: 113-131 (1985) 及びKennedy, Am. J. Clin. Neutr. 68: 1460S-1412S (1998)を参照願いたい。これらの抑制剤は変異体配列の骨格としての役割をする。
【0056】
さらに変異体配列を含む骨格における変更に加え、本明細書で用いられる他の所望するタンパク質はN末端において3つのグリシン残基、及び/またはC末端において6つのヒスチジン(histidine)残基を含む(図3および4を参照願いたい)。
【0057】
大豆トリプシン抑制剤(STI)
STIは安定した化学量論的合成物の形成によりトリプシンのタンパク質分解活性を抑制する(Liu, K., Chemistry and Nutritional value of soybean components. In : Soybeans, chemistry, technology and utilization. pp. 32-35 (Aspen publishers, Gaithersburg, Md., 1999を参照のこと)。STIは2つのジスルフィド架橋をもつ181のアミノ酸残基よりなり、おおよそ球形をしている(例えば、Song et al., J. Mol. Biol. 275: 347-63 (1998)を参照のこと)。2つのジスルフィド架橋は以下に記載のBBIのループと類似の2つの結合ループを形成する。
【0058】
クニッツ型トリプシン抑制剤(Kunitz-type trypsin inhibitor)(STI)は、プロテナーゼの初期の研究において中心の役割を果たし、生化学上及び運動作用における主要な基質として用いられ、プロテアーゼ抑制剤の活性の標準的メカニズムについての定義を導くものであった。
【0059】
ボウマン−ビルク抑制剤(Bowman-Birk Inhibitor(BBI)
BBI タンパク質はマメ科(leguminous)の種から単離された小さなタンパク質(60-90の残基)の動力学的にまた構造的に特徴のあるファミリーである。これは各々が独立した結合ループを含む2つの三環ドメインの対称構造を持つ。ループ1は特にトリプシンを抑制し、ループIIはキモトリプシン(chymotrypsin)を抑制する(Chen et al., J. Biol Chem. (1992) 267: 1990-1994; Werner & Wemmer Lin, et al., Eur. J. Biochem. (1993) 212: 549-555; Voss et al., Eur. J. Biochem. 242: 122-131)。これらの結合領域は各々「カノニカル ループ」(canonical loop)構造を含み、それは種々のセリンプロテアーゼ抑制剤中に見出される基調(motif)である (Bode & Huber, Eur. J. Biochem. (1992) 204: 433-451)。
【0060】
BBI はそれぞれ別個の反応部位においてプロテアーゼ トリプシン及びチモトリプシンを抑制する8 k-Daタンパク質である(Billings et al., Pro. Natl. Acad. Sci. 89:3120-3124(1992)を参照のこと)。STI及びBBIは大豆の種にのみ存在し、その植物の他のどの部分にも見出されていない(例えば、Birk, Int. J. Pept. Protein Res. 25: 113-131 (1985)を参照のこと)。
【0061】
数多くのBBIのイソ型(isoform)の特徴が明らかになっているが、配列番号7(図3)は約71のアミノ酸残基を持つ本明細書に記載の BBIの骨格のアミノ酸配列を示している。さらに、BBIは、NまたはC末端の何れかにおいて最大10個のアミノ酸残基が除去されることにより先端が切断されて(truncated)いることもある。例えば、種を乾燥させると、BBIはC末端の9または10のアミノ酸が除去されることがある。したがって、タンパク質分解は最初のジスルフィド結合以前、及び最終のジスルフィド結合直後においては大いに許容されるべきであり、その結果により通常標的となるタンパク質に対する結合に不利となることは起こらない。しかし、イソ型(isoform)または切断型の何れかを用いることが望ましい。
【0062】
プロテアーゼ抑制剤変異体
上記より理解されるように、STI及びBBIプロテアーゼ抑制剤はプロテアーゼを抑制する結合ループを持っている。本発明のプロテアーゼ抑制剤変異体はループI, ループII またはその両者に改変がなされている。ある実施の形態では、ループは標的タンパク質と相互に作用する配列に置換されている。
【0063】
ループはC2, C3, C4, またはC5抑制剤、コットン結合タンパク質(cotton binding protein)、コムプスタチン(Compstatin)等のような補体活性化経路抑制剤であるVEGF結合タンパク質に由来する配列により置換することができる。代替的に、変異体配列は、例えば、ファージ提示法、または他のスクリーニング法などの他の種々の公知の方法により選択することもできる。例えば、ランダムペプチド遺伝子ライブラリーはファージPIII遺伝子と融合され、ペプチドライブラリーはファージの表面に提示される。その結果、ファージ提示ライブラリーは標的タンパク質に曝されて、非特異的結合を除去するために緩衝液で洗浄される(このプロセスは、パニング(panning)と呼ばれることもある)。最終的に結合ファージ及びPCRでコードされるペプチドのDNA 配列は単離される。
【0064】
通常ループは変異体配列により置換される、すなわち、ペプチドで長さがアミノ酸の数で3−14、5−10が望ましい。より長い配列は、それらが所望する結合及び/または抑制を可能にする限り用いることができる。さらに、結合ループの代替としての用いるのに適したぺプチドは拘束ループ中に抑制されている場合、すなわち、二つのシステイン残基の間のジスルフィド結合の存在により形成されているループの場合には、そのペプチドは機能的コンフォーメイションをとる必要がある。特定の実施の形態においては、ペプチドはその長さがアミノ酸数で7から9である。この置換配列はまたプロテアーゼの抑制または標的タンパク質に対する結合をもたらす。
【0065】
あるケースにおいては、一つのアミノ酸を変えることが有利なことがある。特に、野生型STIまたはBBIの残基13番のアラニン(Alanine)をセリン(Serine)、グリシン(Glycine)またはグルタミン(Glutamine)に変えることもできる。
【0066】
融合タンパク質
各プロテアーゼ抑制剤及びその変異体は、宿主菌細胞により融合タンパク質として発現される。融合ポリペプチドを切断することにより、所望のタンパク質を解放することは有益であるが、それは必要ではない。融合タンパク質として発現され、分泌されるプロテアーゼ抑制剤及びその変異体は驚異的にその機能を保持する。
【0067】
対応する4つのアミノ酸配列をコードする上記規定の4つのDNA配列は、組み合わされて「融合DNA配列」を形成する。そのような融合DNA配列は、5’末端から3’末端に掛けて適切な読み枠に、第1、第2、第3、及び第4のDNA配列の順に組み立てられる。そのように組み立てられたDNAは、糸状菌のシグナルペプチド機能部位(functional)を分泌配列として、通常,糸状菌から分泌される分泌ポリペプチドまたはその部分、切断リンカーペプチド、及び例えばプロテアーゼ抑制剤及びその変異体のような所望のポリペプチドをコードする融合ポリペプチドをアミノ端末からコードする。
【0068】
融合タンパク質の生産は、例えば、米国特許5,411,873, 5,429,950, 及び5,679,543に開示されている方法を用いて実現することができる。その他の方法は技術分野でよく知れらている。
【0069】
組み換えプロテアーゼ抑制剤の発現
本発明の融合タンパク質の生産に関する範囲においては、本発明は組み換え遺伝子学分野の通常の技術に基づいている。本発明で用いられる一般的な使用方法を開示する基礎的文献は、Sambrook et al., Molecular Cloning, A Laboratory Manual (2nd ed. 1989); Kriegler, Gene Transfer and Expression : A Laboratory Manual (1990); 及び Ausubel et al., eds., Current Protocols in Molecular Biology (1994)を含む。
【0070】
本発明はプロテアーゼ抑制剤をコードする核酸配列を含む発現ベクターにより、導入され、形質転換され、または移入された糸状菌宿主細胞を提供する。温度、pH等の培養条件は、導入、形質転換、または移入までに親宿主細胞に適用される条件と同じであリ当業者には明らかであろう。
【0071】
基本的には、融合タンパク質をコードする核酸配列は宿主細胞中のプロモータ配列に動作可能なようにリンクされている。このプロモータ遺伝子単位は、その後、典型的には、複製及び/または発現のために宿主細胞に形質転換される前に中間ベクターにクローンされる。この中間ベクターは典型的にはプラスミド、またはシャトルベクターなどの原核生物ベクター(prokaryotic vector)である。
【0072】
一つのアプローチとして、糸状菌細胞株は、宿主細胞株で機能するプロモータ、生物学的に活性を持つプロモータ断片、または一または二以上の(例えば、シリーズ)エンハンサーを持つ発現ベクターにより移入されたものであり、プロテアーゼ抑制剤をコードする核酸配列に動作可能にリンクされており、そのためプロテアーゼは細胞株で発現する。好ましい実施の形態においては、DNA配列はプロテアーゼ抑制剤及びその変異体をコードする。他の好ましい実施の形態においては、プロモータは制御可能なものである。
【0073】
A. コドンの最適化
十分発現しない遺伝子による十分発現する遺伝子でのコドンの使用を最適化することは公知の技術である。Barnett et al., GB2200118 及びBergquist et Extremophiles (2002) 6: 177-184を参照のこと。本明細書に記載のコドンの最適化は、アスペルギルス(Aspergillus)で十分発現された異種タンパク質(heterologous protein)と十分発現していない異種タンパク質に分泌される天然タンパク質を比較することに基づいている。表1を参照願いたい。
【表1】
【0074】
発現タンパク質で用いられず、または度々は用いられなかったコドンが選択され、度々用いられるコドンに変えられるであろう。したがって、我々はコドンのサブセットのみを変えた。
【0075】
B.核酸の構築/発現ベクター
プロテアーゼ抑制剤をコードする天然または合成ポリヌクレオチド断片(「PIコード核酸配列」PI encoding nucleic acid sequence)は、異種核酸構築またはベクターに組み込むことができ、糸状菌細胞に導入され、そして複製されることが可能となる。本明細書に記載のベクター及び方法は、宿主細胞においてプロテアーゼ抑制剤及びその変異体の発現に用いるのに適したものである。どのベクターでもそれが導入された細胞内で複製され成長可能である限り使用することができる。多数の適当なベクター及びプロモータが当業者に知られており、商業的に入手可能である。糸状菌細胞で用いるのに適したクローニング及び発現のためのベクターは、またSambrook et al., 1989 及び Ausubel FM et al., 1989に記載されているが、これらは引用により本明細書に組み入れられる。適切なDNA配列は種々の手順でプラスミドまたはベクター(集合的に本明細書では、「ベクター」という)に挿入される。一般的には、DNA配列は、標準の手順により適切な制限酵素(restriction endo-nuclease)部位に挿入される。そのような手順及び関連するサブクローニング手順は当業者の知識の範囲にあるとみなされる。
【0076】
適切なベクターは、典型的には、終了配列のような選択可能なマーカーをコードする核酸配列、挿入部位、及び適当なコントロール要素を備えている。ベクターは、例えば、イントロン(intron)及びコントロール(control)要素、すなわち、プロモータ、ターミネータ要素、または5’及び/または3’非翻訳領域のような非コード配列を含む調節塩基配列を含むこともある。それらは宿主細胞(及び/または修飾された可溶性タンパク質をコードする配列が通常発現しないベクターまたは宿主細胞環境)においてコード配列の発現に効果的であり、コード配列と動作可能にリンクされる。多くの適当なベクター及びプロモータが当業者に知られており、その多くは商業的に入手可能であり、及び/またはSambrook et al.,(上記)に記載されている。
【0077】
代表的なプロモータは構成的プロモータ(constitutive promoter)及び誘導プロモータ(inducible promoter)を共に含む。その例には、CMVプロモータ、SV40早期プロモータ、RSVプロモータ、EF-1αプロモータ、tet-on またはtet-offシステム(Clon Tech and BASF)のtet応答素子(TRE)を含むプラスミド、ベータアクチン(beta actin)プロモータ、及びある金属塩を追加することにより上昇させることのできる(upregulate)メタロチオネイン(metallothionein)プロモータを含む。ある実施の形態においてはglaAプロモータが用いられる。このプロモータはマルトースの存在下で誘導される。その様なプロモータは当業者に周知である。
【0078】
天然のプロモータはその機能を変えることなく、一または二以上の核酸を代替、置換、追加または除去することにより修飾することができることは当業者に知られている。本発明の実施においては、プロモータのそのような変更を含むが、それらの変更に拘束されるものではない。
【0079】
遺伝子構築におけるプロモータの選択は、当業者の知識の範囲に属する。
【0080】
適切な選択可能なマーカーの選択は、宿主細胞により決められ、異なる宿主に適しているマーカーは当技術分野でよく知られている。典型的な選択可能なマーカー遺伝子は、(a)抗生物質または他の毒素、例えば、アンピシリン(ampicillin), メトトレキサート(methotrexate)、テトラサイクリン(tetracycline)、ネオマイシン(neomycin)(Southern and Berg, J., 1982),ミコフェノール酸(mycophenolic acid)(Mulligan and Berg, 1980), プロマイシン(puromycin),ゼオマイシン(zeomycin),またはヒグロマイシン(hygromycin)(Sugden et 1985) に抵抗力を与え、または (b)宿主株において栄養要求体変異または自然発生の栄養不足を補うタンパク質をコードする。好ましい実施の態様においては、pyrG菌遺伝子は選択可能なマーカーとして用いられる (Ballance D. J.et al., 1983, Biochem Biophys. Res. Commun. 112: 284-289)。 他の好ましい実施の態様においては、amdS菌遺伝子が選択可能なマーカーとして用いられる (Tilburn, J. et al., 1983, Gene 26: 205-221)。
【0081】
選択されたPIコード配列は良く知られている組み換え技術により、適当なベクターに挿入され、細胞株をPI発現が可能なように形質転換するのに用いられる。遺伝子コードの固有の縮退により、実質的に同じ,または機能的に同等なアミノ酸配列をコードする他の核酸配列が、上記で詳細記載したように特定のプロテアーゼ抑制剤をクローンし、また発現させるために用いられる。したがって、コード領域においてその様な置換をすることは、本発明による配列変異の範囲に属することに留意する必要がある。如何なるまたはすべてのこれらの配列の変異は、親PIをコードする核酸配列について本明細書に記載されていると同様方法で用いることができる。当業者は異なるPIは異なる核酸配列によりコードされることに気付くであろう。
【0082】
所望の形のプロテアーゼ抑制核酸配列、その相同体、変異体または断片が得られたならば、種々の方法で修飾することができる。配列がノンコード隣接領域を含む場合は、隣接領域は切除、変異等を受けるかもしれない。したがって、転位、転換、欠失、挿入が自然発生の配列において行われることもある。
【0083】
異種核酸構築は、プロテアーゼ抑制剤、その変異体、断片、またはその接合変異体の以下のコード配列を含むことがある。すなわち、(i) 単離されている、(ii)融合タンパク質またはシグナルペプチドコード配列のような追加的コード配列と組み合わされている場合であり、その場合はPIコード配列は主要コード配列である、(iii)イントロン(intron)、及びプロモータ、ターミネーター要素、または5’及び/または3’非翻訳領域のようなコントロール(control)要素が、適当な宿主細胞においてコード配列の発現に効果的であるような非コード配列と組合されている、及び/または、(iv)PIコード配列が異種遺伝子であるベクターまたは宿主環境にある配列である。
【0084】
適当な核酸をコードする配列を含む異種核酸は、上で述べたように、適当なプロモータ及びコントロール配列と共に、糸状菌細胞がプロテアーゼ抑制剤及びその変異体を発現させるように細胞を形質転換するのに用いられることもある。
【0085】
本発明の一つの特徴として、異種核酸構築はPIコード核酸配列をインビトロで細胞に移転するのに用いられるが、その場合確立した細胞株であることが望ましい。好ましくは、生産宿主として用いられる細胞株は本発明の核酸配列が安定的に統合されているものがよい。したがって、どの様なものであれ、安定した形質転換細胞を生み出すための効果的な方法であれば、本発明を実施するのに用いることができる。
【0086】
本発明の一つ特徴として、第一、及び第2の発現カセットは単一のベクター、または別個のベクターに存在してもよい。
【0087】
特に断りのない限り、本発明においては、当業者の技術の範囲内である分子生物学、微生物学、及び組み換えDNAでの通常の技術を用いる。そのような技術は、文献に詳しく説明されており、例えば、"Molecular Cloning : A Laboratory Manual", Second Edition (Sambrook, Fritsch & Maniatis, 1989), Animal Cell Culture" (R. I. Freshney, ed., 1987); 及びCurrent Protocols in Molecular Biology”(F.M. Ausubel et al., eds 1987) を参照願いたい。以上または以下に記載のすべての特許、特許出願、記事、刊行物は引用により本明細書に組み入れられる。
【0088】
プロモータ配列に加え発現カセットもまた、効果的な終了を提供するための構造遺伝子の転写終了領域下流を含むべきである。終了領域は、プロモータ配列と同じ遺伝子、または当業者の知識の範囲内にある異なる遺伝子から得られることもある。
【0089】
細胞に遺伝情報を運搬するために用いられる特定の発現ベクターは特に決定的要素ではない。真核細胞または原核細胞における発現に用いられるどのような通常のベクターであれ用いることができるであろう。標準的なバクテリア発現ベクターにはpBR322ベースのプラスミド、pSKF, pET23Dは勿論、バクテリオファージλ及びM13、及びMBP, GST 及びLacZのような融合発現システムを含む。例えば、c-mycの様に、便利な単離の方法を提供するために組み換えタンパク質にエピトープタッグ(epitope tag)を追加することもできる。
【0090】
また発現ベクターに典型的に含まれる要素には、組み換えプラスミドを宿すバクテリアの選択を許す抗生物質への抵抗をコードする遺伝子であるレプリコン(replicon)、及び異種配列の挿入を許すプラスミドの非必須領域におけるユニーク制限部位を含む。特定の抗生物質への抵抗遺伝子を選ぶことは決定的ではない。技術分野で知られている多くの抵抗遺伝子は適性がある。
【0091】
C.宿主細胞及び培養条件
本発明は、プロテアーゼ抑制剤及びその変異体の発現にとって効果的な方法によって、修飾され、選択されそして培養された細胞を含む細胞株を提供する。
【0092】
PI発現のために処理され、および/または修飾される親細胞株は、これに限定されるものではないが、糸状菌細胞を含む。本発明を実施するために用いられる適当な初代細胞タイプは、これに限定されるものではないが、アスペルギルス(Aspergillus)及びトリコデルマ(Trichoderma)を含む。
【0093】
細胞発現プロテアーゼ抑制剤は,典型的には親細胞株を培養するのに用いられる条件で培養される。細胞は通常、標準のRPMI, MEM, IMEM, またはDMEMに、典型的には牛の胎児の血清のような、5−10%の血清を加えた生理食塩水 及び栄養素を含む標準的な媒体中で培養される。培養条件は、また標準的なものである。すなわち、培養は所望のプロテアーゼ抑制剤発現の水準が達成されるまで37℃で静止またはローラ培養で増殖される。
【0094】
与えられた細胞株に対する好ましい培養条件は、科学文献及び/またはAmerican Type Culture Collection (ATCC;"http ://www.atcc.org/")の様な細胞株のソースから得ることができる。典型的には、細胞の生育がなされたら、細胞はプロテアーゼ抑制剤及びその変異体に発現を起こさせ、または抑制するの効果的な条件に置かれる。
【0095】
PIコード配列が誘導プロモータ(inducible promoter)の管理下に置かれるような好ましい実施の形態においては、誘発剤(inducing agent),例えば、炭水化物, 金属塩、または抗生物質がプロテアーゼ抑制剤の発現を誘導するために効果的な濃度で媒体に加えられる。
【0096】
D.プロテアーゼ抑制剤をコードする核酸配列の宿主細胞への誘入
形質転換の方法により、形質転換ベクターの全部またはその一部を糸状菌のゲノムに安定的に統合させることができるかもしれない。しかし、自己複製する、染色体外の形質転換ベクターの維持をすることとなる形質転換も考慮される。
【0097】
本発明はさらに、外来的に供給されるPIコード核酸配列を含む様遺伝子を修飾された細胞及び細胞組成物を提供する。親細胞または細胞株はクローンベクター、または発現ベクターにより遺伝子的に修飾される〈すなわち、変換され、転換され、または移入される〉。ベクターは、上記のように、例えば、プラスミド、ウイルスの粒子、ファージなどの形であるかも知れない。好ましい実施の形態においては、プラスミドは糸状菌細胞に核酸を導入するのに用いられることもある。形質転換は逐次(sequential)に行われるか、または同時の形質転換(co-tranformation)である。
【0098】
インビトロで細胞に発現ベクターを送るために種々の方法が用いられる。異種核酸配列の発現のために核酸を細胞導入する方法は、これらに限定されるものではないが、通常の熟練者に知られており、それらには、電気穿孔法、核ミクロインジェクション(nuclear microinjection)または単一細胞に直接ミクロインジェクションする、無傷細胞(intact cell)による原型質体融合(protoplast fusion)、例えば、ポリブレン(polybrene),またはポリオルニチン(polyornithine)の様なポリカチオン(polycation)の使用、PEG, リポソーム、リポフェクタミン(lipofectamine)またはリポフェクション媒介の核酸注入(lipofaction-mediated transfaction)による膜融合,DNA被覆のミクロ投射物(microprojectile)による高速爆撃(bombardment),リン酸カルシウム−DNA 沈殿物による増殖、DEAE-Dextran媒介の核酸移入、修飾されたウイルス核酸による感染、アグロバクテリア媒介のDNA転位(Agrobacterium - mediated DNA transfer)等を含む。さらにPIコード核酸配列を含む異種核酸構築はインビトロで転写され、その結果のRNAは周知の方法、例えば、注射により宿主細胞に導入されうる。
【0099】
プロテアーゼ抑制剤のコード配列を含む異種の核酸構築を導入するのに続き、遺伝子的に修飾された細胞はプロモータを活性化し、形質転換細胞を選択し、PIコード核酸配列の発現を増幅するのに適するように修飾された通常の栄養媒体中で培養することができる。温度、pH等の培養条件は、以前に発現のため選択された宿主細胞に既に用いられた条件と同じであり、当業者には明らかである。
【0100】
そのような異種核酸構築が導入された細胞の子孫(progeny)は通常、異種核酸構築に見出されるPIコード核酸配列を含んでいると考えられている。
【0101】
E.菌の発現
適当な宿主細胞は糸状菌細胞を含む。本発明の「糸状菌」は、発現宿主として、また第1及び第2の核酸としての役割をするが、これは真核微生物であり、Eumycotina subdivision の 全ての線状体を含む(Alexopoulos, C. J. (1962), Introductory Mycology, New York: Wiley)。これらの菌はチティン(chitin)、グルカン(glucan)及び他の複合多糖よりなる細胞壁を持つ栄養菌糸(vegetative mycelium)であることに特徴がある。本発明の糸状菌は形態学的に、生理学的に、及び遺伝学的にイースト菌とはまったく別個のものである。糸状菌の栄養増殖は菌糸伸長によるものである。対照的に、出芽酵母(saccharomyces cerevisiae)のようなイースト菌による栄養増殖は、単細胞性葉状体の発芽によるものである。出芽酵母と糸状菌の違いには、出芽酵母はアスペルギルス(Aspergillus )及びトリコデルマ(Trichoderma)イントロン(introns)を処理する能力がなく、糸状菌の多くの転写制御因子を認識する能力がないことを含む(Innis, M. A. et al. (1985) Science, 228, 21-26)。
【0102】
種々の糸状菌種が以下の属を含み発現宿主として用いられる:アスペルギルス(Aspergillus)、トリコデルマ(Trichoderma), ニュロスポラ (Neurospora), ペニシリウム(Penicillium), セファロスポリウム(Cephalosporium)、 アキラ(Achlya), ファネロチート(Phanerochaete), ポドスポラ(Podospora),エンドチナ(Endothi), ムコール(Mucor), フサリウム(Fusarium), フミコラ(Humicola) 及びクリソスポリウム(Chrysosporium)である。特定発現宿主はアスペルギルス ニジュランス(Aspergillus nidulans (Yelton, M., et al., Proc. Acad. Sci. USA, 81,1470-1474 ; Mullaney, E. J.et al., (1985) Mol. Gen. Genet. 199,37-45 ; John, M. A. 及び J. F. Peberdy (1984) Enzyme Microb. Technol. 6,386-389 ; Tilburn,et al., (1982) Gene 26, 205-221; Ballance, D. J. et al.,(1983) Biochem. Biophys. Res. Comm. 112,284-289 ; Johnson, I. L.(1985) EMBO J. 4,1307-1311)、 アスペルギルス ニジェール(Aspergillus niger) (Kelly, J. M. 及び M. Hynes (1985) EMBO 4, 475-479), アスペルギルスニジェール変異体アワモリ(Aspergillus niger var. awamori)例えば、NRRL 3112, ATCC 22342, ATCC 44733, ATCC 1433-1,及びUVK-143f株、アスペルギルス オリザ(Aspergillus oryzae), 例えば、 ATCC 11490, ニュロスポラ クラッサ(Neurospora crassa) (Case, M. E. et al.,(1979) Proc. Natl. Acad. Sci. USA 76,5259-5263; Lambowitz U. S. Patent No. 4,486, 553; Kinsey, J. A. 及び J. A. Rambosek (1984) Molecular and Cellular Biology 4, 117-122; Bull, J. 及び J. C. Wooton (1984) Nature 310,701-704), トリコデルマ レーセイ(Trichoderma reesei)、例えば、NRRL 15709, ATCC 13631,56764, 56765,56466, 56767, 及び トリコデルマ ビリド(Trichoderma viride)、例えば、ATCC 32098 及び32086。好ましい発現宿主は主な分泌アスパルチル(aspartyl)プロテアーゼをコードする遺伝子が削除されているアスペルギルスニジェール変異体アワモリ(Aspergillus niger var. awamori)である。この好ましい発現宿主の生産については米国特許出願番号214,237、出願日1988年7月1日に記載されており、ここに引用により本明細書に組み入れられる。
【0103】
真核生物である糸状菌の分泌プロセスにおいて、分泌タンパク質は細胞質(cytoplasm)から細胞膜を超えて小胞体(endoplasmic reticulum)(ER)の内腔(lumen)に入る。ここで、タンパク質は折り畳まれ、ジスルフィド結合が形成される。BiPのようなシャペロン(chaperone)及びタンパク質ジスルフィド イソメラーゼのようなタンパク質はこのプロセスを助ける。またこの段階において、糖鎖が、グリコシド化された(glycosylated)タンパク質を生み出すためにタンパク質に付着される。砂糖は、典型的には、N-リンクグリコシル化(glycosylation)としてアスパラギン残基に加えられ、またはO-リンクグリコシル化としてセリン(serine)またはトレオニン(threonine)残基に加えられる。正しく折り畳まれ、グリコシル化されたタンパク質はER(小胞体)から糖鎖が修飾され、またイースト及び菌のKEX2またはKEXBプロテアーゼが存在するゴルジ体(Golgi apparatus)に運ばれる。菌で生産された分泌タンパク質に付加されるN-リンクのグリコシル化は、哺乳類細胞により付加されるものと異なっている。
【0104】
糸状菌宿主細胞において生産されるプロテアーゼ抑制剤及びその変異体は「グリコシド化された」(glycosylated)か、または「グリコシド化されていない」(non-glycosylated)(すなわち、「非グリコシ化された」(a-glycosylated)または「脱グリコシド化された」(de-glycosylatedである)。菌のグリコシド化のパターンは哺乳類細胞により生み出されるパターンと異なるため、プロテアーゼ抑制剤はプロテアーゼ抑制剤を「脱グリコシド化する」(de-glycosylate)ために酵素で処理されることもある。そのようなN-リンク「脱グリコシド化」に有用な酵素は、エンドグリコシダーゼ H(endoglycosidase H), エンドグリコシダーゼ F1, エンドグリコシダーゼ F2, エンドグリコシダーゼA, PNガーゼ F(PNGase F), PNガーゼ A, PNガーゼ Atがある。そのようなO-リンク「脱グリコシド化」に有用な酵素に、エクソグリコシダーゼ (exoglycosidase)、特にアルファ-マンノシダーゼ(alpha-manodisases)(例えば、アルファ-マンノシダーゼ(alpha-manodisases)(アスペルギルス サイト(Aspergillus saito, iGKX-5009),アルファ (1-2,3, 6)- マンノシダーゼ (alpha (1-2,3, 6) - Mannosidase )(Jack bean, GKX-5010), アルファ- マンノシダーゼ/MANase VI (alpha - Mannosidase/MANase ) (ザントモナス マニホチの組み換え、GKX80070)(Xanthomonas manihoti, GKX80070) これらすべてはGlyko (Prozyme), San Leandro, California)から得られる。
【0105】
驚くことには自然分泌のタンパク質に融合された菌において、高い水準のプロテアーゼ抑制剤及びその変異体を得ることができることが分かった。上記の情報から、プロテアーゼ抑制剤及びその変異体は、グルコアミラーゼがまだN末端に付着している場合にはERで組み立てられることが明らかに予測される。これにより56kDよりも大きなタンパク質を生み出すこととなろう。グルコアミラーゼが、さらに修飾されることなくゴルジ体を通る場合には、所望のタンパク質から切り裂かれることはないと思われる。
【0106】
本発明の方法及び宿主細胞を用いて、我々は驚くべき高い発現レベルを達成した。本明細書に記載のシステムによりプロテアーゼ抑制剤0.5g/l以上の発現及び分泌レベルを達成した。
【0107】
細胞にベクターが導入された後に、感染された(transfected)細胞は所望のタンパク質をコードする遺伝子の発現に有利な条件で培養される。形質転換された多くのバッチ数の細胞は上記のように培養することができる。最後に生成物は公知の技術を用いて培養体から回収される。
【0108】
シャペロン(CHAPERON)
上に述べた通り、ERにおける分泌タンパク質の折り畳み、グリコシル化(glycosylation)は、シャペロン(chaperon)と呼ばれる多くのER居住タンパク質により助けられる。Bip, (GRP78), GRP94 またはLhs1pイーストのようなシャペロンは、表面に出ている疎水領域に折り畳まれない状態で結合することにより分泌タンパク質の折り畳みを助け、不利な相互作用を阻止する(Blond- Elguindi et al., 1993, Cell 75: 717-728)。シャペロンはまた、ER細胞膜を通してタンパク質が異動するのに重要な役割を果たす。タンパク質 ジスルフィド イソメラーゼ(pdi)、その同属体、及びポリルーペプチド シスートランス イソメラーゼ(prolyl-peptidyl cis-trans isomerase)のようなフォルダーゼタンパク質(foldase protein) は、ジスルフィド架橋の形成、及びプロリン残基(prolien residue) に隣接するペプチド鎖の正しいコンフォーメイションの生成をそれぞれ助けている。
【0109】
発明の一つの特徴として、宿主細胞はシャペロンをコードする発現ベクターにより転換される。シャペロンはpdiA 及びprpAからなる群から選択される。
【0110】
発酵パラメータ(FERMENTATION PARAMETER)
本発明は菌類の培養のための発酵手順に基づくものである。異種タンパク質を生産する発酵手続は、それ自身技術分野で良く知られている。例えば、タンパク質はバッチ、フェドバッチ(fed-batch),及び連続フロー工程を含む、固形、または液内培養により生産することができる。
【0111】
培養は、水性ミネラル塩媒体、有機的成長因子、炭素及びエネルギー源媒体、分子酸素及び当然ながら、一または二以上の使用される特定微生物種の開始接種材料を含む成長媒体において成される。
【0112】
炭素及びエネルギー源、酸素、同化窒素、及び微生物の接種材料に加え、微生物の適切な成長を確保し、微生物変換プロセスにおける細胞による炭素及びエネルギー源の同化を最大にし、発酵媒体中の最大細胞密度で最大細胞生成を達成するために、それに応じた適量のミネラル栄養分を供給することが必要である。
【0113】
水性ミネラル培体の組成は広範囲にわたり、技術分野で知られているように、使用される微生物、及び基質に一部依存する。ミネラル媒体は、窒素に加え、適量のリン、マグネシウム、カルシウム、カリ、硫黄、及びナトリウムを、適当な可溶性同化イオン、及び組合された形で含むことが必要であり、また、当技術分野で知られているように、また好ましくは適当な可溶性同化の形の銅、マンガン、モリブデン、亜鉛、鉄,ボロン、沃素及び他の要素のような、ある微量元素も存在するべきである。
【0114】
発酵反応は、必要とされる分子酸素が、空気、酸素富化空気またはさらには実質的に純粋分子酸素も含む分子酸素含有ガスにより供給される好気性(aerobic)プロセスである。ただし、微生物種が効果的に繁殖できるように発酵容器内容物を適切な酸素分圧に維持することが必要である。つまり、含酸素炭化水素基質を用いることにより、微生物の成長に必要な酸素量が減少することになる。しかしながら、基質の同化、及びそれに対応する微生物の成長は一部は燃焼プロセスであるため、成長のためには分子酸素が供給される必要がある。
【0115】
通気速度は非常に広範囲に変わりうるが、通気は通常は0.5から10、好ましくは0.5から7の範囲でなされ、これは分当りの発酵槽の単位液体容量に含まれる酸素含有ガスの容量(その時の圧力、及び温度は25℃)である。この量は、反応搭に送られる通常の酸素量の空気に基づくものであり、純粋酸素に換算すると各々0.1から1.7、好ましくは0.1から1.3であり、分当りの発酵槽の単位液体容量に含まれる酸素の容量(その時の圧力、及び温度25℃)である。
【0116】
微生物変換プロセスにおける圧力は広範囲に変わりうる。一般的には、0から50psigであり、ここでは好ましい圧力は約0から30psig、より好ましくは、少なくとも大気圧より僅か高い圧力である。酸素溶解度に対する設備機器と操業コストのバランスが達成できるからである。大気圧より高い場合には、水生発酵体中の酸素溶解濃度が増すため、細胞の成長率が増し有利になる。しかしながらその場合には同時に、高い大気圧は設備機器及び操業コストを増す結果をもたらす。
【0117】
発酵温度は幾らかは変えることができる、しかし、アスペルギルスニジェール変異体アワモリ(Aspergillus niger var. awamori)のような糸状菌では、選択される微生物株にもよるが、温度は通常約20℃から40℃の範囲内であり、一般的には好ましくは28℃から37℃の範囲である。
【0118】
微生物はまた同化窒素源を必要とする。同化窒素源は微生物による代謝利用に適した形の窒素を放出することのできる窒素含有化合物であればどのようなものでもよい。タンパク質加水分解物のような種々の有機窒素化合物をソースとして用いることができるが、通常は、アンモニア、水酸化アンモニウム(ammonium hydroxide)、尿素のような安価な窒素含有化合物、及びリン酸アンモニウム(ammonium phosphate)、硫酸アンモニウム(ammonium sulfate)、ピロリン酸アンモニウム(ammonium pyrophosphate),塩化アンモニウム(ammonium chloride)の様な種々のアンモニア塩、または他のアンモニア化合物を用いることができる。アンモニアガス自体は規模が大きな操業に便宜であり、適量の水性発酵(発酵媒体)による場合のバブリングの形で用いることができる。アンモニアはまたpHコントロールを助けるのにも用いることができる。
【0119】
水微生物発酵(発酵混合剤)のpHは典型的な場合として2.0から8.0の範囲であるべきである。糸状菌では、通常pHは2.5から8.0の範囲であり、アスペルギルスニジェール変異体アワモリ(Aspergillus niger var. awamori)では、通常pHは4.5から5.5の範囲である。ある微生物に適しているpHの範囲は微生物の特徴、並びにある程度使用される媒体によって決まり、したがって媒体により幾分変わってくるがこれは当業者は容易に選定が可能である。
【0120】
発酵槽中の発酵混合剤の平均保持時間は、ある程度発酵温度、及び使用される培養体に依存するが、大きく変わる。通常は約24から500時間の範囲であり、ここでは好ましくは約24から400時間である。
【0121】
好ましくは発酵は炭素含有基質が制限要因としてコントロールされ、炭素含有基質の細胞への転換がスムーズになされ、大量の非転換基質により細菌の汚染が生じない様に行われるのが望ましい。後者については水溶性基質については問題とならない。残留痕は直ぐに洗い流されるからである。しかし、非水溶性基質では問題となり、適当な洗浄ステップにより生成物を処理する製品の追加処理工程が必要となる。
【0122】
上記に記載の通り、この様な制限基質のレベルに達するまでの時間は決定的な要素ではなく、微生物によって及び実施される発酵プロセスにより変わりうる。しかし、発酵媒体における炭素源の濃度、及び所望の炭素源のレベルが達成されたかを否か決定する方法は当技術分野でよく知られている。
【0123】
発酵はバッチ、または連続操業によることができるが、管理の容易さ、生成物の質の均一性、及び設備機器が最も経済的に使用できるという理由により、通常フェドバッチ(fed-batch)操業が好まれる。
【0124】
もし望むなら、培養槽に水性ミネラル媒体を加える前に、一部または全部の炭素及びエネルギー源材料、及び/またはアンモニアの様な同化窒素源の一部を加えることができる。
【0125】
反応搭に導入される各流液は、各事前に定められた数値により、または炭素及びエネルギー基質の濃度、pH, 溶解酸素、発酵槽から出る排気ガス中の酸素または二酸化炭素、透光度により測定される細胞密度等をモニターすることによりコントロールされるのが望ましい。各種材料の供給率は、効果的に炭素及びエネルギー源を活用して、可能な限りの細胞成長を促進することができるよう、また基質の投入に対して可能な限りの高い微生物細胞を得ることができるよう変えることができるが、より大事なことは、単位容量当り所望するタンパク質を最も多く生産できることである。
【0126】
バッチ、または好ましいフェドバッチ(fed- batch)操業のいずれににおいても、すべての機器、反応搭、または発酵手段、容器、またはコンテナー、パイプ、循環または冷却装置の付属物等は、まず最初に殺菌する必要があり、これは通常は約121℃の蒸気を使って少なくとも15分間行う。そして殺菌された反応搭に、酸素、及び炭素含有基質を含む必要なすべての栄養素が揃っている中で選択された微生物の培養体が植えつけられる。使用される発酵槽は決定的な要素ではないが、ここで操業に好ましいのは15L Biolafitte (Saint- Germain-en-Laye, France)である。
【0127】
タンパク質の分離
所望されるタンパク質が発現されると、選択的に所望するタンパク質の分泌の回集が必要となる。本発明は所望するタンパク質を融合類似物から分離する方法を提供する。本明細書に記載の方法は、融合類似物からプロテアーゼ抑制剤及びその変異体を分離するのに役立つよう特に考慮されている。
【0128】
発酵培養液から所望するタンパク質の収集及び精製は、また技術分野においてそれ自体で知られている手順により行うことができる。発酵培養液は通常、所望のタンパク質のみならず、細胞、種々の浮遊物、及び他のバイオマス汚染物質(biomass contaminant)を含む細胞破片を含んでおり、これらはその技術分野において知られている手段により、発酵培養液より除去されるのが望ましい。
【0129】
そのような除去に適したプロセスは、例えば、遠心分離、ろ過、透析(dialysis)、精密ろ過(microfiltration),ロータリー真空ろ過(rotary vacuum filtration)、または他の既知のプロセスの様な細胞を含まないろ過液を得るための通常の固体―液体分離技術を含む。結晶化させる前に限外ろ過(ultrafiltration)、蒸発または沈殿などの技術を用いて発酵培養液または細胞を含まないろ過液をさらに濃縮することが望ましい。
【0130】
浮遊物またはろ過液のタンパク質成分は、例えば硫酸アンモニウムの様な塩を用いることにより、またはpHを2から3に調整し、沈殿させることができる。その後、培養液を80℃で2時間熱処理し、続いて、例えば、イオン交換クロマトグラフィー、親和性クロマトグラフィーのような種々のクロマトグラフィーによる手順、または類似の認められた方法により精製する。
【0131】
発現された所望のポリペプチドが分泌される場合、ポリペプチドは成長媒体から精製されることもある。発現宿主細胞は、ポリペプチドの精製の前に培体から除去する(例えば、遠心分離により)のが好ましい。
【0132】
発現組み換えの所望のタンパク質が宿主細胞から分泌されない場合は、好ましくは宿主細胞が破壊され、ポリペプチドが精製の第一段階である水性「抽出物」(extract)中に解放されるのが望ましい。発現宿主細胞は細胞が破壊される前に媒体から回収されるのが好ましい(例えば、遠心分離により)。
【0133】
細胞破壊は、例えば、リゾチーム、またはベータグルカナーゼ分解により、または細胞を高圧により圧迫するなどの従来技術により行うことができる。細胞破壊に関するさらに詳細な記載はRobert K. Scobes, Protein Purifi- cation, Second edition, Springer-Verlagを参照願いたい。
【0134】
6つのヒスチジン残基、すなわち、His Tag、をC末端に追加することは所望のタンパク質及びその融合類似体の精製を助ける。His tag を精製のための協力者として用いることは公知の技術である。例えば、Hengen (1995) TIBS 20 (7): 285-286を参照願いたい。6つのHis tagの付いたタンパク質は固定化金属イオン親和クロマトグラフィー(Immobilized Metal ion Affinity Chromato−graphy (IMAC))を使い容易に精製される。
【0135】
プロテアーゼ抑制剤及びその変異体は、疎水負荷誘導クロマトグラフィー(hydrophobic charge induction chromatography (HCIC))を組み合わせて用い、例えば、全細胞発酵培養液または浄化培養液のような水性タンパク質溶液から精製することができることは特に考察するに値する。HCICは所望するタンパク質を培養液及びその融合類似体から分離することができる。
【0136】
ユーティリティー
いくつかの所望するタンパク質の応用例においては、例えば、99%以上の純度を持つというように、プロテアーゼ抑制剤はきわめて純粋であることが非常に重要である。これは所望のタンパク質が治療目的に用いられる場合に特に当てはまるが、他に応用される場合にもまた必要である。本明細書に記載の方法は、本質的に純粋な所望のタンパク質を得る方法を提供するものである。本明細書に記載の所望のタンパク質は製薬及び個人医療の薬の組成として有用である。
【0137】
以下に続けて開示する実施例においては以下の略記号を用いる:eq (equivalents) ; M (Molar) ;μM (micromolar) ; N(Normal) ; mol (moles) ; mmol (millimoles) ; μmol(micromoles) ; nmol (nanomoles) ; g (grams); mg (milligrams) ; kg (kilograms); μg (micrograms); L (liters) ; ml (milliliters) ; μl(microliters) ; cm (centimeters); mm (millimeters) ; μm (micrometers); nm (nanometers); ℃ (degrees Centigrade); h (hours); min (minutes); sec (seconds); msec (milliseconds); Ci (Curies) mCi (milliCuries) ; μCi(microCuries); TLC (thin layer achromatography); Ts (tosyl); Bn (benzyl) ; Ph (phenyl) ; Ms (mesyl) ; Et (ethyl), Me (methyl). PI (proteinase inhibitor), BBI (Bowman-Birk inhibitor), STI (Soybean Trypsin inhibitor).
実施例
本発明はその実施例においてさらに詳細が記載されるが、いかなる意味においても、それにより特許請求の範囲に記載の発明の範囲が制限されるものと解してはならない。添付の図面は本明細書及び発明の不可欠の一部である。本明細書に参考として引用されたものは、その全てが引用により本明細書に組み入れられる。以下の実施例は請求の範囲の発明の説明のために記載されるものであるが、それにより請求の範囲の発明を限定するものではない。
【0138】
実施例1 大豆トリプシン抑制剤をコードするDNAのクローニング
本実施例はSTIの発現ベクターの発育について明らかにする。
【0139】
通常、所望するタンパク質をコードする遺伝子は、N末端のNhel制限酵素部位を経て加工されたkexB切断部位(NVISKR)及びSTI終了コドン、TAGに続くC末端のBstEll 制限酵素部位を持つグルコアミラーゼ(glucoamylase)のリンカー領域をコードするDNAに融合されていた。大豆のSTIをコードする遺伝子はkexB切断部位、及び3つのグリシン残基をN末端に待ち、6つのヒスチジン残基をC端末に持つ二つの制限部位を含むDNA断片としてインビトロでMCLAB(South San Francisco, California)により合成された(配列番号3、図2に示す遺伝子)ここで用いられるPCRによる生成DNA断片はすべて当初pCRII-TOPOベクターにクローンされた(Invitrogen, Carlsbad, CA)。大腸菌(One Shot TOP10 cells from Invitrogen)は通常のプラスミド単離及びプラスミド保持に用いられた。Nhel 及びBstEll部位はpCRII-TOPOベクターからPCR製品を切り取るために用いられ、そして結果物のDNA断片は発現ベクターpSL1180- GAMpR-2に結紮された (図5参照)。発現ベクターpSL1180-GAMpR-2は、アスペルギルス ニジェール グルコアミラーゼ(Aspergillus niger glucoamylase)プロモータ、グルコアミラーゼ触媒ドメインおよび終了領域(terminator region)を含む。発現プラスミドはまた選択マーカーとしてアスペルギルス ニジェール pyrG(Aspergillus niger pyrG)を含む。したがって、発現カセットによる形質転換細胞はウリジン(uridine)が欠乏する媒体における成長により検知する。
【0140】
STIペプチドをコードする遺伝子(アミノ酸配列番号10、図4A, ヌクレオチド配列番号6、図2)はMCLABにより合成され、pCRII-TOPOベクターにクローンされた(Invitrogen)。Nhel からBstEllの断片は制限酵素分解によりプラスミドから解放され、発現プラスミドpSLGAMpR2―SBTI/nonpti(Q110)を作り出すために, DNA断片は寒天ゲルから取り出され、pSLGAMpR2 、グルコアミラーゼキモシン発現ベクター(pSLGAMpR2、glucoamylase-chymosin expression vector)にクローンされた。詳細はWO 9831821に記載されている。
【0141】
発現プラスミドはdgr246ΔGAP: pyr2-に転換された。この株はpepA遺伝子が欠失した遺伝子dgr246 P2に由来し、pyrGマイナスであり、異種遺伝子製品の改良された生産方法により、何ラウンドかの変異、スクリーニング及び選択を経ている(Ward, M. et al.,1993, Appl. Microbiol. Biotech. 39: 738-743 及びその中の参照例)。dgr246ΔGAP: pyr2株作るために、まったく同じ欠失プラスミド(pΔGAM NB-Pyr)を用いて、またファウラー(Fowler, T. et al (1990) Curr. Genet. 18: 537-545)により報告されている手順に従い、glaA(グルコアミラーゼ)遺伝子がdgr246 P2から取り除かれた。簡単に言えば、除去は, 両端にglaA フランキング配列(flanking sequence)を持つリニアDNA断片及びプロモータの一部により転換されることにより達成され、glaA遺伝子のコード領域が選択マーカーとして、アスペルギルス ニジュランスpyrG(Aspergillus nidulans pryG)遺伝子により置換されているものである。glaA フランキング配列(flanking sequence)及びpyrG遺伝子を含むリニアー断片が染色体glaA遺伝子座で統合されている形質転換細胞は、サザ゛―ンブロット(Southern blot) 分析により同定された。この変化は形質転換株dgr246ΔGAPで起きていた。この形質転換細胞の胞子はフッ化オロチン酸(fluororotic acid)を含む媒体に塗られ、van Hartingsveldt, W. et al. (1987) Mol. Gen. Genet. 206: 71-75に記載のように、自然発生的耐性変異株が得られた。これらの一つである、dgr246ΔGAP: pyr2は、野生型pryG 遺伝子を持つプラスミドにより転換されることで補完されうるウリジン栄養要求株(uridine auxotroph)であることが示された。
【0142】
アスペルギルス(Aspergillus)形質転換プロトコールはキャンペル法(Campell method)の修正である(Campell et al. (1989). Curr. Genet. 16: 53-56)。すべての溶液及び媒体は加圧滅菌され、または0.2ミクロンフィルターでろ過殺菌された。アスペルギルス ニジェール 変異体 アワモリ(Aspergillus niger variant awamori)の胞子は寒天媒体複合体(complex media ager, CMA)プレートから集菌された。CMAは20g/lデクストロース、20g/l Difco ブランド麦芽エキス、1g/l Bacto Peptpone, 20g/l Bacto 寒天、100mg/mlのアルギニン20ml/l、及び100mg/mlのウリジン20ml/lを含む。約1.5 cm四角の寒天胞子の一切れを使い液体100 mlのCMA(処方は、Bacto が除かれる以外はCMAと同じである)を植えつけた。フラスコは、温度37℃、250−275rpmで一夜振動培養された。菌糸(mycelia)が殺菌されたMilacloth(Calbiochem. San Diego, CA, USA)で集菌され、50mlの溶液A(Solution A)(pH5.8のリン酸ナトリウム10mM中0.8M のMgSO4)で洗浄された。洗浄された菌糸(mycelia)は、20ml溶液A中300mgのβ―D−グルカナーゼ(beta-D- glucanase (Interspex Products, San Mateo, CA)の殺菌液中に置かれた。これを殺菌した250ml プラスチックびん(Corning Inc, Corning, New York)中で28℃から30℃、200rpmで2時間培養した。培養の後、このプロトプラスト(protoplasting)溶液は殺菌されたMiraclothを通してろ過され、殺菌した円錐形チューブ(Sarstedt, USA)に入れられた。 結果物のプロトプラスト(protoplasting)を含む液は2つの50ml円錐形チューブに等量に分けられた。40mlの溶液B(1.2M ソルビトール、50mM CaCl2, 10mM Tris, pH7.5)がそれぞれのチューブに加えられ、卓上臨床
遠心分離器(Damon IEC HN Sill centrifuge)によりフル回転で5分遠心分離された。各チューブからの上澄みは取り除かれ、20mlの新たな溶液Bが1つのチューブに加えられ、混合され、そして次のチューブに注がれ、全ての粒(pellet)が再懸濁されるまで混合された。その後チューブは5分間遠心分離器に掛けられた。上澄みは取り除かれ、20ml の新たな溶液Bがチューブに加えられ5分間遠心分離された。洗浄されたプロトプラストを濃度0.5-10x107 protoplast /100ulの溶液B中で再懸濁する前に、最終の洗浄を行った。殺菌した15ml円錐形チューブ(Sarstedt, USA)の各プロトプラスト100ulに、10ul の形質転換プラスミドDNAが加えられた。これに、12.5ulの溶液C(50% PEG 4000, 50mM CaCl2, 10mM Tris, pH7.5)が加えられ、チューブは氷の上に20分間置かれた。1mlの溶液Cが加えられ、そしてチューブは氷上から除かれて室温に置かれ、静かに振られた。2mlの溶液Bが直ちに加えられ溶液Cが希釈された。転換ミックスが等分に3つのチューブの溶解したMMS重層(6 g/l NaN03, 0.52g/l KCl, 1.52g/l KH2PO4, 218.5g/l D-sorbitol, 1.0ml/l 微量元素-LW(trace element-LW), 10g/l SeaPlaque 寒天(FMC Bioproducts, Rook1 and, Maine, USA)、 20m/l 50% グルコース, 2.5 ml/l 20% MgSO4・7H20, NAOHによりpH6.5にした)に加えられ、45℃の温浴槽に保存された。微量元素-LW (Trace elements-LW)は1g/l FeSO4・7H20, 8.8 g/l ZnSO4・7H20, 0.4g/l CuSO4・5H20, 0.15 g/l MnSO4・4H20, 0.1 g Na2B4O7・10H2O, 50mg/l (NH4)6Mo7O24・4H20, 250 mls H20, 200 ul/l 濃縮HCLよりなる。形質転換ミックスを加えた溶解重層は直ちに、寒天のプレート上に直接加えられた333ul/プレートの100mg/mlのアルギニンで補充されていた3MMSプレート(10g/lのSeaPlaque アガロースに代えて、20g/l Bacto寒天が用いられる以外はMMS重層と同じ調合である)に振りかけられた。アガロースが固まった後に、形質転換細胞が成長するまでプレートは30℃で培養された。
【0143】
萌芽形成形質転換細胞は殺菌した楊枝を使い、最少培地(mimimal media (MM))プラス グルコースのプレートに採られた。MMは6 g/l NaN03, 0.52 g/l KCl, 1.52 g/l KH2PO4, 1 ml/l微量元素-LW(Trace elements-LW), 20 g/l Bacto寒天, NaOHによりpH6.5 にされ、25 ml/l の40%グルコース, 2.5 ml/l の 20% MgSO4・7H20 及び20 ml/l の 100 mg/ml アルギニンよりなる。形質転換細胞がMM上で成長すると、それらはCMAプレートに移された。
【0144】
各形質転換細胞のプレート培養体から1.5cm四角の寒天胞子の一片(agar plug)が250mlの振盪フラスコ中でPromosoy specialと呼ばれる50mlの生産媒体に加えられた。この媒体は以下の組成を持つ。すなわち、70g/l クエン酸ナトリウム(sodium citrate), 15 g/l (NH4)2S04,1 g/l NaH2PO4・H2O, 1g/l MgSO4, 1 ml Tween 80, NaOHによりpH 6.2にされ、2 ml/l Mazu DF60-P, 45 g/l Promosoy 100 (Central Soya, Fort Wayne, IN), 120 g/l マルトースである。生産媒体フラスコは30℃、200rpm で5日間培養され、上澄みサンプルが集菌された。形質転換細胞は, 生産されたタンパク質の量に基づき形質転換細胞を選択するために、SDSゲル上でタンパク質生産のアッセイが行われた。最上の形質転換細胞の培養液につき、そのトリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0145】
各形質転換細胞のプレート培養体から1.5cm四角の寒天胞子の一片(agar plug)が250mlの振盪フラスコ中で修飾CSS(modified CSS)と呼ばれる50mlの生産媒体に加えられた。この媒体は以下の組成を持つ。すなわち、50g/l Corn Street Solids, 1g/l NaH2PO4・H2O, 0.5 g/l MgSO4 (無水), 50 g/l Staley 7350 (55%) 及び8g/lクエン酸ナトリウム(Na Citrate)である。生産媒体フラスコは36℃で、200rpm で3日間培養され、上澄みサンプルが集菌され, SDSゲル上でタンパク質生産のアッセイが行われた。最上の形質転換細胞の培養液につき、そのトリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0146】
実施例2 大豆トリプシン抑制剤をコードするDNAのコドンの最適化
以下の実施例はSTIをコードするDNAが、糸状菌で最適に発現するようどのように改変されたかを詳説するものである。
【0147】
合成遺伝子(MCLAにより合成された実施例1の出発物質)コドンは、アスペルギルス(Aspergillus)で高度に発現したタンパク質でのコドンの使用法に従い最適化されたものである。基本的には、グルコアミラーゼ、アルファアミラーゼ、及びプロキモシン(prochymosin)等のよく発現したタンパク質と、ヒトのNEP 及びDPP4などの様なアスペルギルス(Aspergillus)で十分発現しなかったタンパク質と比較した。表1を参照願いたい。アスペルギルス(Aspergillus)における両タイプのタンパク質発現にもちいたのコドンの使用を表IIに示す。
【表2】
【0148】
【0149】
【0150】
よく発現した遺伝子においては、コドンは多くは使用されず、またはそれほどしばしば使用されなかったことは明らかである。これらのコドンは、発現の良くなかった遺伝子(表IIの星印*で示す)においてはよりしばしば見出される。よく発現するタンパク質では使われない、またはそれほどしばしばは使用されることのないコドンをSTI遺伝子において同定し、それらのコドンはよく発現するタンパク質において、よりしばしば用いられるコドンに変えられた。表III及びIVを参照願いたい。
【表3】
【表4】
【0151】
最適化されたDNAはインビトロでMCLAB (South San Francisco) により、3つの制限部位(遺伝子の5’末端にNhel、3’末端にXhol及びBstEll), kexB切断部位、及び3つのグリシン残基をN末端に持ち、6つのヒスチジン残基をC末端に持つ(配列番号3)DNA断片として合成された。この最適化された遺伝子はpCRII-TOPOベクターにクローンされた。上記実施例1に記載の手順に従い、Nhel からBstEllへの断片はプラスミドから制限分解により解放され、DNA断片はアガロースゲル上で精製され、そしてそこから引き抜かれ、pSLGAMpR2-SBTI2(Q107)発現プラスミドを作るためpSLGAMpR2にクローンされた。
【0152】
発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1に記載の通りである。31の形質転換細胞がアッセイされ、SDSゲルがタンパク質発現レベルチェックのために用いられた。最上の6個の形質転換細胞の培養液について、トリプシン抑制活性がアッセイされた。
【0153】
実施例3 アスペルギルス(Aspergillus )でのボウマンービルク(Bowman-Birk)抑制剤及びその変異体の発現
a. kexB部位、N末端に3つのグリシン、及びC末端に6つのヒスチジン残基を持つグルコアミラーゼへのBBIの融合
上記の実施例2の手順に従い、BBIをコードするDNAは最適化され、本実施例で使用された。DNAはインビトロでMCLABにより、3つの制限部位(遺伝子の5’末端にNhel、3’末端にXhol及びBstEll), kexB切断部位、及び3つのグリシン残基をN末端に持ち、6つのヒスチジン残基をC末端に持つ(配列番号54)DNA断片として合成された。この遺伝子はpCRII-TOPOベクターにクローンされた(Invitrogen)。上記実施例1に記載の手順に従って、Nhel からBstEllへの断片はプラスミドから制限分解により解放され、DNA断片はアガロースゲルから引き抜かれ、pSLGAMpR2-BBIkex+(Q104)発現プラスミドを作るためにpSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1の場合と同様である。28の形質転換細胞が生産され、25の細胞のアッセイが振盪フラスコでおこなわれた。SDSゲルがタンパク質発現レベルチェックに用いられた。最上の形質転換細胞の培養液について、トリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0154】
b. 6つのヒスチジン残基をC末端に持つグルコアミラーゼへのBBIの融合
上記の実施例2の手順に従い、BBIをコードするDNAは最適化され、本実施例で使用された。DNAはインビトロでMCLABにより、3つの制限部位(遺伝子の5’末端にNhel、3’末端にXhol及びBstEll), 及び6つのヒスチジン残基をC末端に持つDNA断片として合成された(配列番号42は以下の通り)。
【0155】
(配列番号42:
GCTAGCGACGATGAGAGCTCTAAGCCCTGTTGCGATCAGTGCGCGTGTACCAAATCGAACCCTCCGCAGTGTCGCTGCTCCGATATGCGTCTGAATTCCTGTCATAGCGCATGCAA,GAGCTGTATCTGCGCCCTGAGCTACCCCGCGCAGTGTTTCTGCGTCGACATCACGGACTTCTGCTACGAGCCGTGTAAGCCCAGCGAGGACGATAAGGAGAACCATCATCACCATCACCATTAGCTCGAGGGTGACC).
この遺伝子はpCRII-TOPOベクターにクローンされた。上記実施例1の手順に従い、Nhel からBstEllへの断片はプラスミドから制限分解により解放され、精製され、そしてアガロースゲルから引き抜かれ、pSLGAMpR2-BBlkex (Q105) 発現プラスミドを作るためにpSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1と同じである。38の形質転換細胞が生産され、25の細胞のアッセイが振盪フラスコでおこなわれ。SDSゲルがタンパク質発現レベルチェックのためにも用いられた。最上の形質転換細胞の培養液について、トリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0156】
c. kexB部位、及び3つのグリシン残基をN末端に持つグルコアミラーゼへのBBI融合
インビトロでMCLABにより合成され(配列番号5)、pCRII-TOPOベクターにクローンされたプラスミドDNAはPCR増幅のDNA鋳型(template)として用いられた。2つのプライマーは以下の様にデザインされた:
5’GGG CTA GCA ACG TCA TST CCA AG3’(配列番号43)
5’ GTC ACC TAG TTC TCC TTA TCG TCC TCG CTG3’ (配列番号44)
DNAはプライマーの存在下で以下の条件で増幅された:DNAはTris-EDTA緩衝液により10から100倍に希釈された。10μlの希釈されたDNAが、各ヌクレオチド(A, G, C 及びT)0.2mM、1反応緩衝液、0.5から0.6μgのプライマー1(配列番号43)及びプライマー2(配列番号44)を含む全100μlのエッペンドルフ管の反応混合物に加えられた。混合物は100℃で5分熱し、反応混合物に2.5単位のTaq DNAポリメラーゼが加えられた。PCR反応は95℃で1分行われ、プライマーは50℃で1分間鋳型にアニールされ、さらに72℃で1分の延長がされた。このサイクルは30回繰り返えされ、さらに延長サイクルとして68℃で7分おこなわれ、その後の使用に備え4℃で保管された。PCR断片は寒天ゲルにより検出され、その後プラスミドベクターpCRII-TOPOにクローンされた(Invitrogen)。得られたPCR断片は、6つのヒスチジン残基およびXhoL制限部位をコードするヌクレオチドが除去されていることを除いては、配列番号54のものと同一の配列を含む。上記実施例1記載の手続きに従い、PCR断片は、制限酵素Nhel 及びBstEllにより分解された。分解されたDNA断片はエタノールで沈殿させ、histag 無しのpSLGAMpR2-BBl (Q108) 発現プラスミドを作り出すため、pSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1と同じである。57の形質転換細胞が生産され、25の細胞のアッセイが振盪フラスコでおこなわれた。SDSゲルがタンパク質発現レベルチェックのために用いられた。最上の形質転換細胞の培養液について、トリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0157】
d. kexB部位を持つグルコアミラーゼへのBBIの融合
インビトロでMCLAB (配列番号1) により合成され、pCRII-TOPOベクターにクローンされたプラスミドDNAがPCR増幅のDNA鋳型(template)として使用された。2つのプライマーは以下の様にデザインされた:
5’GGG CTA ACC TAG TTC TCC TTA TCG TCC TCG CTG 3’(配列番号44)
5’GGG CTA GCA ACG TCA TCT CCA AGC GCG ACG ATG AGA GCT CTA AG3’ (配列番号45)
得られたPCR断片は、3つのグリシン残基、6つのヒスチジン残基およびXhoL制限部位をコードするヌクレオチドが除去されていることを除いては、配列番号54(図1C)と同一配列を含む。上記実施例1記載の手続きに従い、PCR断片は、制限酵素Nhel 及びBstEllにより分解された。分解されたDNA断片はエタノールで沈殿させ、3G 及びhistag(Q109) 無しのpSLGAMpR2-BBl 発現プラスミドを作り出すため、pSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1と同じである。127の形質転換細胞が生産され、42の形質転換細胞のアッセイが振盪フラスコでおこなわれた。SDSゲルがタンパク質発現レベルチェックのために用いられた。最上の形質転換細胞の培養液について、トリプシンまたはキモトリプシン抑制活性がアッセイされた。
【0158】
実施例4 アスペルギルス(Aspergillus )でのボウマンービルク(Bowman-Birk) 抑制剤及びその変異体(他のバインダーによるループの置換)の発現
変異体配列が、その技術分野で知られている標準的手順を用いて、一又は両方のループに導入された。変異体配列は、製作者指示に従い商業的に入手可能なファージ ペプチド ライブラリーPhD C7C(New England Biolabs, Beverly, MA)を、標的タンパク質または基質に対して3ラウンドのパンニング(panning)をするか、または公知の活性を持つ配列を用いて決定された。以下に記載の配列では、ループヌクレオチド配列に導入された変更は、下側のケースのヌクレオチドにより示される。
【0159】
a. ループIにaーVEGF(CK37281)を持つBBI
インビトロでMCLAB (配列番号1) により合成され、pCRII-TOPOベクターにクローンされたプラスミドDNAはPCR増幅のDNA鋳型(template)として用いられた。VEGF(a-VEGFと表す)を結合し、VEGF機能を抑制するためのペプチド配列を導入するために2つのプライマーは以下の様にデザインされた:
5' GTTGCGATCAGTGCGCGTGTtacaatctgtatggctggaccTGTCGCTGCT 3' (配列番号46) 及び
5' CGCATATCGGAGCAGCGACAggtccagccatacagattgtaACACGCGCAC 3'. (配列番号47)
PCRが実行され、まず94℃で2分間混合物を熱し、94℃で30秒間、63℃で30秒間、72℃で30秒間のサイクルを30サイクル行った。30サイクルの後に、混合物は72℃で4分間培養され、その後4℃で保存された。置換結合ループはDNA配列により確認された。ループ1(Q117)においてpSLGAMpR2-BBl (CK37281) 発現プラスミドを作り出すために、NhelからBstEllのDNA断片は制限分解によりプラスミドから解放され、精製され、そしてpSLGAMpR2にクローンされた。発現プラスミドはdgr246ΔGAP: pyr2に形質転換された。形質転換細胞の形質転換及び振盪フラスコテストは実施例1と同じである。30以上の形質転換細胞が生産され、42の形質転換細胞のアッセイが振盪フラスコでおこなわれた。SDSゲルがタンパク質発現レベルチェックのために用いられた。
【0160】
b. ループIIにaーVEGF(CK37281)を持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、以下の2つのプライマーが用いられた以外は上記実施例の場合と同じ手順に従った:
5' CATGCAAGAGCTGTATCTGCtacaatctgtatggctggaccCAGTGTTTCTG 3' (配列番号48)
5' GATGTCGACGCAGAAACACTGggtccagccatacagattgtaGCAGATACAG 3'. (配列番号49)
【0161】
c. ループI及びIIにaーVEGF(CK37281)ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、以下の4つのプライマーが用いられた以外は上記と同じ手順に従った:
5' GTTGCGATCAGTGCGCGTGTtacaatctgtatggctggaccTGTCGCTGCT 3' (配列番号46)
5' CGCATATCGGAGCAGCGACAggtccagccatacagattgtaACACGCGCAC 3' (配列番号47)
5' CATGCAAGAGCTGTATCTGCtacaatctgtatggctggaccCAGTGTTTCTG 3' (配列番号48)
5' GATGTCGACGCAGAAACACTGggtccagccatacagattgtaGCAGATACAG 3'. (配列番号49)
【0162】
d. ループI に a-補体タンパク質c2ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、c2(a-c2と表す)を結合しc2機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCAGCTGTagctgcggcaggaagatccccatccagtgcTGTCGCTGCTCCGATATGCGTC 3' (配列番号50)
5'GAGCAGCGACAgcactggatggggatcttcctgccgcagctACAGCTGCACTGATCGCAACAGGGCTTA 3'(配列番号51) ・
【0163】
e. ループI に a-補体タンパク質c3ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、c3( a-c3と表す)を結合し、c3機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCGGCTGTgccaggagcaacctcgacgagTGTCGCTGCTCCGATATGCGTC 3' (配列番号52)
5'GAGCAGCGACActcgtcgaggttgctcctggcACAGCCGCACTGATCGCAACAGGGCTTA 3' (配列番号53)
【0164】
f. ループI に a-補体タンパク質c4ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、c4( a-c4と表す)を結合し、c4機能を抑制するためのペプチド配列を導入するため以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCGCGTGTcagagggccctccccatcctcTGTCGCTGCTCCGATATGCGTC 3' (配列番号55)
5'GAGCAGCGACAgaggatggggagggccctctgACACGCGCACTGATCGCAACAGGGCTTA 3' (配列番号56) ・
【0165】
g. ループI に a-補体タンパク質c5ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、c5(a- c5と表す)を結合し、c5機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCCAGTGTggcaggctccacatgaagaccTGTCGCTGCTCCGATATGCGTC 3' (配列番号57)
5'GAGCAGCGACAggtcttcatgtggagcctgccACACTGGCACTGATCGCAACAGGGCTTAGA 3' (配列番号58)
【0166】
h. ループI に ヒトのa-補体因子B ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、因子B(a-因子Bと表す)を結合し、因子B機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GCGATCAGTGCCAGTGTaagaggaagatcgtcctcgacTGTCGCTGCTCCGATATGCGTC 3' (配列番号59)
5'GAGCAGCGACAgtcgaggacgatcttcctcttACACTGGCACTGATCGCAACAGGGCTTAGA 3' (配列番号60)
【0167】
i. ループI に a-膜メタロプロテアーゼ2(MMP2)ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、MMP2( a-MMP2と表す)を結合し、MMP2機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5' CAGTGCGCGTGTgccgccatgttcggccccgccTGTCGCTGCTCCGATATGCGTC 3' (配列番号61)
5'GAGCAGCGACAggcggggccgaacatggcggcACACGCGCACTGATCGCAACAG 3;(配列番号62)
【0168】
j. ループI にa-膜メタロプロテアーゼ12(MMP12)ペプチドを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、MMP12( a-MMP12と表す)を結合し、MMP2機能を抑制するためのペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5' CAGTGCGCGTGTggcgccctcggcctcttcggcTGTCGCTGCTCCGATATGCGTC 3' (配列番号63)
5'GAGCAGCGACAgccgaagaggccgagggcgccACACGCGCACTGATCGCAACAG 3' (配列番号64)
【0169】
k. ループI にコットン結合(cotton binding)ペプチド2314を持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、コットンと結合するペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5' GTTGCGATCAGTGCGCGTGTgagcccctgatccaccagcgcTGTCGCTGCT 3' (配列番号65)
5' CGCATATCGGAGCAGCGACAgcgctggtggatcaggggctcACACGCGCAC 3' (配列番号66)
【0170】
l. ループIにコットン結合(cotton binding)ペプチド2317を持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、コットンと結合するペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GTTGCGATCAGTGCGCGTGTagcgccttccgcggccccaccTGTCGCTGCT3' (配列番号67)
5' CGCATATCGGAGCAGCGACAggtggggccgcggaaggcgctACACGCGCAC 3' (配列番号68)
【0171】
m. ループI にコンプスタチン(compstatin)ループを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、コンプスタチン(compstatin)ペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5'GTTGCGATCAGTGCGCGTGTgttgttcaggactggggccaccaccgcTGTCGCTGCT (配列番号6 9)
5'CGCATATCGGAGCAGCGACAgcggtggtggccccagtcctgaacaacACACGCGCAC (配列番号70)
この場合BBIトリプシン結合ループの7つのアミノ酸は、コンプスタチン(compstatin)結合ループの9つのアミノ酸により代替された。
【0172】
n. ループII にコンプスタチン(compstatin)ループを持つBBI
プラスミドを構築し、菌状形質転換細胞を得て、そして振盪フラスコにおいて菌上形質転換細胞をアッセイするために、コンプスタチン(compstatin)ペプチド配列を導入するため、以下の2つのプライマーを用いた以外は上記と同じ手順に従った:
5' CATGCAAGAGCTGTATCTGCgttgttcaggactggggccaccaccgcTGTTTCTGCG (配列番号71)
5'GTGATGTCGACGCAGAAACAgcggtggtggccccagtcctgaacaacGCAGATACAG (配列番号72)
この場合BBIトリプシン結合ループの7つのアミノ酸は、コンプスタチン(compstatin)結合ループの9つのアミノ酸により代替された。
【0173】
実施例5 トリコデルマ リーセイ(Trichoderma reesei)でのボウマンービルク(Bowman-Birk) 抑制剤及びその変異体の発現
上記例2に記載の手順に従い、BBIをコードするDNAが最適化され、本実施例において用いられた。2つのプライマーはループI 及びIIのプラスミドpSLGAMpR2-BBIまたはaーVEGF(CK37281)ペプチドを持つpSLGAMpR2-BBIを鋳型として用いDNAを増幅するようにデザインされた:
5, GGA CTA GTA AGC GCG ACG ATG AGA GCT CT 3' (配列番号73)
5' AAG GCG CGC CTA GTT CTC CTT ATC GTP CT 3' (配列番号74)
第3のプライマーは、また上記の第2のプライマー(配列番号74)と連動して使用される場合は、BBIタンパク質のN末端に3つのグリシン残基を含むPCR断片を作るために用いられた。
【0174】
5' GGA CTA GTA AGC GCG GCG GTG GCG ACG ATG AGA GCT CT 3' (配列番号75).
上記実施例2に記載と同様の手順に従い、BBIをコードするDNAが最適化され、本実施例において用いられた。PCR断片は制限酵素Spel 及びAscl により切断され、トリコデルマ(Trichoderma)発現プラスミド、pTrex4(図8)に結紮された。pTrex4はpTREX2(図9)の修飾形であり、これはまたpTEXの修飾形である。pTEXベクターの生成のための完全な記述についてはPCT公報番号 WO 96/23928を参照願いたい。当該資料は発現プラスミドを作り出すための遺伝子発現のCBHIプロモータ、ターミネータ、及び形質転換細胞の選択マーカーとしてのトリコデルマ(Trichoderma)pry4遺伝子を含んでおり、引用により本明細書に組み入れられる。pTrex4プラスミドにおいては、BBI遺伝子はCBHIコアーのC末端及びトリコデルマ リーセイ(Trichoderma reesei)のリンカーに融合された。菌の形質転換の間、アスペルギルス ニジュランス(Aspergillus nidulans)のamdS遺伝子が選択マーカーとして用いられた。発現プラスミドはトリコデルマ リーセイ(Trichoderma reesei)に転換された。安定している形質転換細胞は、アセトアミドを窒素源としてトリコデルマ(Trichoderma)最小板(minimal plate)上で単離された。形質転換細胞はamd マイナスプレートで生育された。プレートは1ml/l 1000x塩(salt)、 20g/l Noble Agar, 1.68g/l CsCl, 20g/l グルコース, 15g/l KH2PO4, 0.6g/l MgSO4・7H2O, 0.6g/l CaCl2,・2H2O 及び0.6g/l アセトアミドを含む。pHは最終的に4.5に調整された。1000x塩(salts)は5g/l FeSO4, 1.6g/l MnSO4, 1.4g/l ZnSO4, 及び1g/l CoCl2 を含み、フィルターろ過殺菌された。3日間28℃で培養した後、形質転換細胞は新しいamdマイナスプレートに移され、さらに28℃で3日間育成された。
【0175】
形質転換細胞はその後250mlの振盪フラスコのトリコデルマ リーセイ(Trichoderma reesei)プロフロ媒体(proflo medium)(各形質転換細胞に対し50ml)に植えつけられた。トリコデルマ リーセイ(Trichoderma reesei)プロフロ媒体(proflo medium)は、30g/l アルファラクト−ス, 6.5g/l (NH4)2SO4, 2g/l KH2PO4, 0.3g/l MgSO4・7H2O, 0.2g/l CaCl2, 1ml/l 1000xTris Trace Salts, 2ml/l 10% Tween 80, 22.5g/l Proflo 及び 0.72g/l CaC03. を含む。100xTris Trace Salts は5g/l FeS04・7H20, 1.6g/l. MnS04・H20 及び 1.4g/l ZnS04・7H20.を含む。2日間30℃で育成した後、4ml の培養液が合成培地に移された。合成培地は5g/l (NH4)2SO4, 33g/l PIPPS 緩衝液, 9g/l カサミノ酸(CASAMINO ACIDS), 4.5g/l KH2PO4, 1g/l CACL2, 1g/l MgSO4・7H2O, 5ml/l MAZU 及び 2.5ml/l 400x トリコデルマ リーセイ(Trichoderma reesei)TRACE.を含む。pHは5.5に調整され、殺菌の後に40ml/l 40% のラクトースが加えられた。400x トリコデルマ リーセイ(Trichoderma reesei)TRACEは175g/l クエン酸(無水), 200g/l FeS04・7H20, 16g/l ZnS04 7H20, 3.2g/l CuSO4・5H20, 1.4g/l MnS04 ・ H20 及び0.8g/l H3BO3. (ホウ酸、Boric Acid)をふくむ。
【0176】
プレートで約40個の形質転換細胞が生成され、20個が振盪フラスコでアッセイされた。培養液の上澄みはSDS-PAGE分析に使われ、トリプシン、またはキモトリプシン(chymotrypsin)の抑制活性がアッセイされた。ウエスターン ブロット(Western blot)はまた、融合した形(Cbhl-BBI)及びBBI単独の両者が存在することを示した。
【0177】
実施例6 アスペルギルス(Aspergillus)におけるボウマンービルク(Bowman- Birk)抑制剤及び分泌シャペロン(Secretory Chaperones)の同時発現
以下の実施例はどの様に分泌が増進されるかを詳細に示すものである。STIタンパク質は2つのジスルヒド結合を含み、BBIはその三次構造(tertiary structure)に7つのジスルヒド結合を含み、これらのジスルヒド結合はその機能にとり重要である。ジスルヒド結合を持つタンパク質の折り畳みにはタンパク質ジスルヒドイソメラーゼ(PDI)またはERに他のシャペロンが必要であることは知られている。
【0178】
STIまたはBBIの発現の増進について、一つのプラスミドがSTIまたはBBI発現カセットを含み、他の一つがPDI遺伝子またはシャペロン遺伝子を含む2つのプラスミドの同時発現、または逐次発現すること調査・検討した。まず、3G及びヒスタッグ(histag)(Q109)を持たないpSLGAMpR2-BBIプラスミドを、アスペルギルス ニジェール(Aspergillus Niger)のpdiA遺伝子領域をカバーする4.6kbゲノムDNAを持つプラスミドQ51により、ベクターpUC219で同じ菌株(dgr246ΔGAP : pyr2)に同時転換させた。51の形質転換細胞が得られ、47が振盪フラスコでスクリーニングされた。形質転換細胞14番が、SDSゲルデータ(gel data)による最も高いBBIタンパク質を生成したため選択された。BBIタンパク質の発現レベルは、同時形質転換される場合の方が3G及びヒスタッグ(histag)(Q109)を持たないpSLGAMpR2-BBIプラスミドのみを含む場合よりも高い。図7は増強されたBBIの発現をしめす。この菌株はまた、胞子精製され、再度振盪フラスコでテストされた。
【0179】
上記の手順に従い、我々はヒスタッグ(histag)(Q108)を持たないpSLGAMpR2-BBIプラスミドを、pdiA遺伝子(上記に同じ)を含むプラスミドQ51と同じ菌株(dgr246ΔGAP : pyr2)に同時形質転換させることとした。34の形質転換細胞が振盪フラスコでスクリーニングされた。SDSゲルデータに基づき最高レベルのBBIタンパク質生成能力を持つ1つの形質転換細胞が選択された。BBIタンパク質の発現レベルはヒスタッグ(histag)(Q108)を持たないpSLGAMpR2-BBIプラスミドのみを含む菌株よりも高い。
【0180】
上記の手順に従い、我々はまた3G及びヒスタッグ(histag)(Q109)を持たないpSLGAMpR2-BBIプラスミドと、アスペルギルス ニジェール(Aspergillus Niger)の遺伝子prpAの領域をカバーする1623bpゲノムDNAを含むプラスミドQ124とをベクターpUC219で同じ菌株(dgr246ΔGAP : pyr2)に転換させた。28の形質転換細胞が振盪フラスコでスクリーニングされた。SDSゲルデータ(gel data)による最も高いBBIタンパク質を生成能力を持つ1つの形質転換細胞が選択された。BBIタンパク質の発現レベルは、同時形質転換された菌株の場合の方が、単に3G及びヒスタッグ(histag)(Q109)を持たないpSLGAMpR2-BBIプラスミドのみを含む菌株よりも高い。図7は増強された発現を表す(レーン15対レーン3)。この菌株はまた、胞子精製され、再度振盪フラスコでテストされた。
【0181】
実施例7 組み換えプロテアーゼ抑制剤変異体保持活性(Recombinant Protease Inhibitor Veriants Retain Actvity)
上記の方法により生産されたSTI、BBI 及びその変異体はその活性、例えばプロテアーゼ活性の抑制がテストされた。
【0182】
a.プロテアーゼ抑制
950μlのTris 緩衝食塩水+0.02% Tween 20が20μlのプロテアーゼ(牛のトリプシンまたはキモトリプシン)1mM HCL中100μg/ml )及び20μlサンプルと組み合わされる。液は混合され、室温で30分間培養される。10μl基質(トリプシンに対しサクシニル(succinyl)-ala-ala-pro-arg-パラニトロアニリド(paranitroanilide), DMSO 中10mg/ml、及びキモトリプシン(chymotrypsin)に対しサクシニル(succinyl)-ala-ala-pro-phe-パラニトロアニリド(paranitroanilide), DMSO 中10mg/ml)を加え、液を混合する。405nmで吸光度を測定し、レート(A405/分)が決められる。プロテアーゼのコントロールブランクサンプルに対する活性抑制比率を対比し、次の式に従い算出される:
(A405/分(サンプル))/(A405/分(ブランク))*100μg/ml(プロテアーゼ)*
(MW抑制剤)/(MWプロテアーゼ)=[抑制剤] μg/ml
b.a VEGFペプチドによるHUVEC増殖の抑制
HUVE細胞(Cambrex, East Rutherford, NJ)を1−5回通過させ、製造者指示に従い保持された。HUVECの成長は0.03から20ng/mlのVEGFにより刺激され、最高の増殖は10ng/ml VEGF165 (R&D Systems)の場合であった。この濃度はそれ以降の実験においても用いられた。0.00052μMから25μMの一連のa-VEGFペプチド(実施例4参照)及びアンチVEGF MAb コントロール(R&D Systems)が,96ウエルプレートに3重に播種されたHUVECsに加えられる前に、10ng/ml VEGFと混合された。細胞の増殖が3H-thymidine により測定された。極めて大きな抑制効果が観察された(データは示されていない)。
【0183】
本明細書に記載の実施例、実施の形態は説明のために記載したものであり、当業者に対してはそれらの種々の改変、変更を示唆することができ、それらはすべて本出願の精神及び趣旨の範囲、及び本願の請求の範囲に含まれる。本明細書に引用したすべての刊行物、特許、特許出願は、そのすべては引用により本明細書に組み入れられる。
【図面の簡単な説明】
【0184】
【図1】図1は大豆Bowman-Birk タイプ プロテアーゼ抑制剤(BBI)のヌクレオチド配列(配列番号1)に最適化されたコドンである。この配列は、NVISKR(点線の下線部分)、融合タンパク質の切断部位及び発現プラスミドにクローンされる3つの制限酵素部位を含む。5’ 末端のNhel部位及び3’末端のXhol部位は下線及び標識を付してある。3’末端のBstEll部位は♯記号を付して示す。終止コドンは*(asterisk)で示す。融合タンパク質として発現するため、開始コドンは存在しない。成熟BBIコード配列は二重下線により示す(配列番号2)。成熟BBIコード配列に先立ち3つのグリシン(図1B)残基をコードするヌクレオチドの追加は、図2(配列番号5)に示す3つのグリシン残基をコードする配列を用い行うことができる。図1Cは、BBIをコードするヌクレオチド配列、3つの制限部位、kex2部位、N末端に3つのグリシン残基及びC末端に6つのヒスチジン(histidine)基を示す(配列番号54)。
【図2】図2は大豆トリプシン抑制剤(Soybean Trypsin Inhibitor, STI)に最適化されたコドン、クーニッツタイプ(Kunitz type)プロテアーゼ抑制剤(配列番号3)を示す。この配列はヌクレオチドをコードするNVISKR(点線の下線)(配列番号4)である。融合タンパク質の切断部位、及びC末端に6つのヒスチジン(histidine)基を持つ(点線で示す)。発現プラスミドにクローンする3つの制限酵素部位(5’末端のNhel、3’末端のXhol 及びBstEllは図1に示す通り)も含まれる。kex2部位(NVISKR)の後の3つのグリシン残基は太字で示す。成熟STIをコードするヌクレオチド配列は破線の下線で示す(配列番号6)。
【図3】図3AはBBIの成熟アミノ酸配列(配列番号7)を示す。図3BはN末端に3つのグリシン残基を持つBBIである(配列番号8)。図3CはN末端に3つのグリシン残基を持ち、C末端に6つのヒスチジン残基を持つBBIである(配列番号9)。図3A-CのループIは下線を付したアミノ酸残基で示し、ループIIのアミノ酸残基は太字で示す。
【図4】図4AはN末端に3つのグリシン残基を持ち、C末端に6つのヒスチジン残基を持つ成熟STIである(配列番号10)。図4BはN末端に3つのグリシン残基を持つSTIである(配列番号11)。図4CはSTIの成熟アミノ酸配列(配列番12)を示す。ループ1は下線を付したアミノ酸残基で示す(配列番号13)。ループIIのアミノ酸残基は太字で示す(配列番号14)。
【図5】図5は発現プラスミド pSLGAMpR2-BBIのダイアグラムである。このプラスミドはアスペルギルス ニジェール(Aspergillus niger)グルコアミラーゼ プロモータ、触媒コアー及びターミネータ、マーカー遺伝子(Aspergillus Niger pyrG)及び牛のプロキモシン(prochymosin)遺伝子を挿入することによるpSL1180に由来のpSLGAMpR2をベースにしている。pSL1180プラスミドはAmersham Biosciences (Piscataway, NJ)から入手可能である。pSLGAMpR2プラスミドは、pSLGAMpR2-BBIに示した位置と同じ相対位置に上記リストした要素が挿入されている。ただし、牛のプロキモシン(prochymosin)遺伝子はBBI遺伝子があるところに存在する。したがって、BBI遺伝子はpSLGAMpR2-BBIを作り出すためpSLGAMpR2中のプロキモシン(prochymosin)遺伝子に代替する。
【図6】図6は野生型BBIアミノ酸配列(配列番号7)及び精選されBBIの変異体である(配列番号15から29)。野生型BBIは下線を付したループを持つ。野生型との相違は、太字、下線(ループI)または太字(ループII)で示す。変異体によっては、例えば、C2,C3,C4, C5 及びファクターB,位置13のアラニン(2つのシステインの間)はまた、「セリン」「グリシン」または「グルタミン」の何れかに変えられた。またコンプスタチン(compstatin)ペプチドは7個のアミノ酸に代えて9個持っている。また変異体配列も示す(配列番号30から40)。
【図7】図7はタンパク質SDSゲルの写真である。レーン(Lane)1は分子量マーカーを含む。レーン2は形質転換されていない親株である。レーン3はBBIをコードするDNAにより同時形質転換された親株である。レーン4はBBIをコードするベクター及びシャペロン(pdiA)をコードするベクターで形質転換された親株である。レーン15はBBIをコードするベクター及びシャペロン(prpA)をコードするベクターで同時形質転換された親株である。所望されるタンパク質、例えば、BBIの発現はシャペロンの存在により増進された。
【図8】図8はプラスミドpTrex4のダイアグラムである。
【図9】図9A−DはpTrex2の核酸配列である(配列番号41)。
【図1A】
【図1B】
【図1C】
【特許請求の範囲】
【請求項1】
糸状菌細胞においてプロテアーゼ抑制剤を生産する方法であって、
a) 糸状菌細胞にDNA構築を導入し、前期DNA構築は糸状菌細胞において転写活性を示すプロモータを含み、大豆トリプシン抑制剤(Soybean Trypsin Inhibitor (STI))、ボウマンビルク抑制剤(Bowman-Birk Inhibitor (BBI))またはこれらの変異体に由来するプロテアーゼ抑制剤をコードする異種(heterologous)DNA配列に動作可能にリンクされており、
b) 異種DNA配列を発現させるために適当な培養条件の下で糸状菌細胞を培養し、及び
c) 前期プロテアーゼ抑制剤を生産する
方法。
【請求項2】
さらに前期プロテアーゼ抑制剤を回収することを含む請求項1に記載の方法。
【請求項3】
前期糸状菌細胞がアスペルギルス(Aspergillus )菌株、ペニシリウム(Penicillium)菌株、フサリウム(Fusarium)菌株またはトリコデルマ(Trichoderma)菌株から選択される請求項1に記載の方法。
【請求項4】
前期トリコデルマ(Trichoderma)菌株はトリコデルマ リーセイ(Trichoderma reesei)である請求項3記載の方法。
【請求項5】
前期アスペルギルス(Aspergillus )菌株はアスペルギルス ニジェール(Aspergillus niger)、アスペルギルス ニジュランス(Aspergillus nidulans)、アスペルギルス アワモリ(Aspergillus awamori)、またはアスペルギルス オリザ(Aspergillus oryzae)である請求項3の方法。
【請求項6】
前期プロテアーゼ抑制剤が以下のa)-d)よりなる群から選択される請求項1に記載の方法;
a) 配列番号7と少なくとも90%同一の配列を持つプロテアーゼ抑制剤
b) 配列番号7と少なくとも90%同一の配列を持ち、残基15−21及び/または42−47が変異体配列により置換されているプロテアーゼ抑制剤、
c) 配列番号12と少なくとも90%同一の配列を持つプロテアーゼ抑制剤, 及び
d) 配列番号12と少なくとも90%同一の配列を持ち、残基39−85、及び/または137−144が変異体配列により置換されているプロテアーゼ抑制剤、
【請求項7】
前期プロテアーゼ抑制剤が配列番号7と少なくとも90%同一の配列を持ち、残基15−21、及び/または42−47が変異体配列により置換されている請求項6に記載の方法。
【請求項8】
前期プロテアーゼ抑制剤が配列番号7及び15−29よりなる群から選択される請求項7に記載の方法。
【請求項9】
前記プロテアーゼ抑制剤をコードする前記DNA配列が糸状菌細胞での発現に最適化されているコドンを含む請求項1に記載の方法。
【請求項10】
さらに糸状菌細胞に、シャペロンをコードする第二の核酸配列を導入することを含む請求項1に記載の方法。
【請求項11】
前期シャペロンがpdiA またはprpAである請求項10に記載の方法。
【請求項12】
前期プロテアーゼ抑制剤が融合タンパク質として発現される請求項1に記載の方法。
【請求項13】
前期融合タンパク質がグルコアミラーゼシグナル配列、グルコアミラーゼ触媒ドメイン(catalytic domain)、切断部位、及び前期プロテアーゼ抑制剤を含む請求項12に記載の方法。
【請求項14】
前期融合タンパク質が前期プロテアーゼ抑制剤を解放するプロテアーゼにより処理される請求項12に記載の方法。
【請求項15】
請求項1に記載の方法により生産されるプロテアーゼ抑制剤を含むプロテアーゼ抑制剤組成物。
【請求項16】
標的タンパク質を請求項15のプロテアーゼ抑制剤組成物により接触させ、該標的タンパク質を結合して標的タンパク質の分解活性を抑制することを含む標的タンパク質の分解活性の抑制方法。
【請求項17】
配列番号15−29に規定のポリペプチド配列よりなる群から選択されるプロテアーゼ抑制剤をコードする単離ポリヌクレオチド。
【請求項18】
糸状菌細胞での発現に最適化されたコドンを含む大豆トリプシン抑制剤(Soybean Trypsin Inhibitor(STI)),またはボウマンビルク抑制剤(Bowman-Birk Inhibitor (BBI))をコードする変異野生型(altered wild-type)ポリヌクレオチド。
【請求項19】
(a) 配列番号12と少なくとも90%同一の配列を持ち、残基39−85、及び/または137−144が変異体配列により置換されているプロテアーゼ抑制剤をコードするポリヌクレオチド、または
(b)配列番号7と少なくとも90%同一の配列を持ち、残基15−21、及び/または42−47が変異体配列により置換されているプロテアーゼ抑制剤
を含む発現ベクター。
【請求項20】
さらに5’末端から3’末端に、
糸状菌において分泌配列としてシグナルペプチド機能部位(signalpeptide functional)をコードする第一の核酸配列、と
分泌ポリペプチドまたはその機能部位をコードする第二の核酸配列、と
切断リンカーをコードする第三の核酸配列、と
前記プロテアーゼ抑制剤をコードする前記DNA配列
を含む請求項19の発現ベクター。
【請求項21】
請求項19のベクターにより形質転換された宿主細胞。
【請求項22】
前期宿主細胞がトリコデルマ(Trichoderma)細胞である請求項21の宿主細胞。
【請求項23】
前期宿主細胞がアスペルギルス(Aspergillus)細胞である請求項21の宿主細胞細胞。
【請求項24】
糸状菌細胞においてプロテアーゼ抑制剤の発現を増進する方法であって、
a) 糸状菌細胞において転写活性を示すプロモータを含み、大豆トリプシン抑制剤(Soybean Trypsin Inhibitor (STI))、ボウマンビルク抑制剤(Bowman-Birk Inhibitor (BBI))、またはこれらの変異体に由来するプロテアーゼ抑制剤をコードする異種(heterologous)DNA配列に動作可能にリンクされているDNA構築により糸状菌細胞を形質転換させ、
b) シャペロン遺伝子を含むポリヌクレオチド配列により前期糸状菌細胞を形質転換させ、及び
c) 前記プロテアーゼ抑制剤をコードする前記異種DNA配列の発現及び分泌をさせるために適当な培養条件の下で前記糸状菌細胞を培養する方法であって、
前記a)のステップにより形質転換された対応する糸状菌細胞に比べてプロテアーゼ抑制剤の発現が増進されている方法。
【請求項25】
前期形質転換のステップa)及びb)が同時形質転換である請求項24に記載の方法。
【請求項26】
形質転換のステップa)及びb)が逐次形質転換である請求項24に記載の方法。
【請求項27】
前期糸状菌細胞がアスペルギルス(Aspergillus)細胞、またはトリコデルマ(Trichoderma)細胞である請求項24に記載の方法。
【請求項1】
糸状菌細胞においてプロテアーゼ抑制剤を生産する方法であって、
a) 糸状菌細胞にDNA構築を導入し、前期DNA構築は糸状菌細胞において転写活性を示すプロモータを含み、大豆トリプシン抑制剤(Soybean Trypsin Inhibitor (STI))、ボウマンビルク抑制剤(Bowman-Birk Inhibitor (BBI))またはこれらの変異体に由来するプロテアーゼ抑制剤をコードする異種(heterologous)DNA配列に動作可能にリンクされており、
b) 異種DNA配列を発現させるために適当な培養条件の下で糸状菌細胞を培養し、及び
c) 前期プロテアーゼ抑制剤を生産する
方法。
【請求項2】
さらに前期プロテアーゼ抑制剤を回収することを含む請求項1に記載の方法。
【請求項3】
前期糸状菌細胞がアスペルギルス(Aspergillus )菌株、ペニシリウム(Penicillium)菌株、フサリウム(Fusarium)菌株またはトリコデルマ(Trichoderma)菌株から選択される請求項1に記載の方法。
【請求項4】
前期トリコデルマ(Trichoderma)菌株はトリコデルマ リーセイ(Trichoderma reesei)である請求項3記載の方法。
【請求項5】
前期アスペルギルス(Aspergillus )菌株はアスペルギルス ニジェール(Aspergillus niger)、アスペルギルス ニジュランス(Aspergillus nidulans)、アスペルギルス アワモリ(Aspergillus awamori)、またはアスペルギルス オリザ(Aspergillus oryzae)である請求項3の方法。
【請求項6】
前期プロテアーゼ抑制剤が以下のa)-d)よりなる群から選択される請求項1に記載の方法;
a) 配列番号7と少なくとも90%同一の配列を持つプロテアーゼ抑制剤
b) 配列番号7と少なくとも90%同一の配列を持ち、残基15−21及び/または42−47が変異体配列により置換されているプロテアーゼ抑制剤、
c) 配列番号12と少なくとも90%同一の配列を持つプロテアーゼ抑制剤, 及び
d) 配列番号12と少なくとも90%同一の配列を持ち、残基39−85、及び/または137−144が変異体配列により置換されているプロテアーゼ抑制剤、
【請求項7】
前期プロテアーゼ抑制剤が配列番号7と少なくとも90%同一の配列を持ち、残基15−21、及び/または42−47が変異体配列により置換されている請求項6に記載の方法。
【請求項8】
前期プロテアーゼ抑制剤が配列番号7及び15−29よりなる群から選択される請求項7に記載の方法。
【請求項9】
前記プロテアーゼ抑制剤をコードする前記DNA配列が糸状菌細胞での発現に最適化されているコドンを含む請求項1に記載の方法。
【請求項10】
さらに糸状菌細胞に、シャペロンをコードする第二の核酸配列を導入することを含む請求項1に記載の方法。
【請求項11】
前期シャペロンがpdiA またはprpAである請求項10に記載の方法。
【請求項12】
前期プロテアーゼ抑制剤が融合タンパク質として発現される請求項1に記載の方法。
【請求項13】
前期融合タンパク質がグルコアミラーゼシグナル配列、グルコアミラーゼ触媒ドメイン(catalytic domain)、切断部位、及び前期プロテアーゼ抑制剤を含む請求項12に記載の方法。
【請求項14】
前期融合タンパク質が前期プロテアーゼ抑制剤を解放するプロテアーゼにより処理される請求項12に記載の方法。
【請求項15】
請求項1に記載の方法により生産されるプロテアーゼ抑制剤を含むプロテアーゼ抑制剤組成物。
【請求項16】
標的タンパク質を請求項15のプロテアーゼ抑制剤組成物により接触させ、該標的タンパク質を結合して標的タンパク質の分解活性を抑制することを含む標的タンパク質の分解活性の抑制方法。
【請求項17】
配列番号15−29に規定のポリペプチド配列よりなる群から選択されるプロテアーゼ抑制剤をコードする単離ポリヌクレオチド。
【請求項18】
糸状菌細胞での発現に最適化されたコドンを含む大豆トリプシン抑制剤(Soybean Trypsin Inhibitor(STI)),またはボウマンビルク抑制剤(Bowman-Birk Inhibitor (BBI))をコードする変異野生型(altered wild-type)ポリヌクレオチド。
【請求項19】
(a) 配列番号12と少なくとも90%同一の配列を持ち、残基39−85、及び/または137−144が変異体配列により置換されているプロテアーゼ抑制剤をコードするポリヌクレオチド、または
(b)配列番号7と少なくとも90%同一の配列を持ち、残基15−21、及び/または42−47が変異体配列により置換されているプロテアーゼ抑制剤
を含む発現ベクター。
【請求項20】
さらに5’末端から3’末端に、
糸状菌において分泌配列としてシグナルペプチド機能部位(signalpeptide functional)をコードする第一の核酸配列、と
分泌ポリペプチドまたはその機能部位をコードする第二の核酸配列、と
切断リンカーをコードする第三の核酸配列、と
前記プロテアーゼ抑制剤をコードする前記DNA配列
を含む請求項19の発現ベクター。
【請求項21】
請求項19のベクターにより形質転換された宿主細胞。
【請求項22】
前期宿主細胞がトリコデルマ(Trichoderma)細胞である請求項21の宿主細胞。
【請求項23】
前期宿主細胞がアスペルギルス(Aspergillus)細胞である請求項21の宿主細胞細胞。
【請求項24】
糸状菌細胞においてプロテアーゼ抑制剤の発現を増進する方法であって、
a) 糸状菌細胞において転写活性を示すプロモータを含み、大豆トリプシン抑制剤(Soybean Trypsin Inhibitor (STI))、ボウマンビルク抑制剤(Bowman-Birk Inhibitor (BBI))、またはこれらの変異体に由来するプロテアーゼ抑制剤をコードする異種(heterologous)DNA配列に動作可能にリンクされているDNA構築により糸状菌細胞を形質転換させ、
b) シャペロン遺伝子を含むポリヌクレオチド配列により前期糸状菌細胞を形質転換させ、及び
c) 前記プロテアーゼ抑制剤をコードする前記異種DNA配列の発現及び分泌をさせるために適当な培養条件の下で前記糸状菌細胞を培養する方法であって、
前記a)のステップにより形質転換された対応する糸状菌細胞に比べてプロテアーゼ抑制剤の発現が増進されている方法。
【請求項25】
前期形質転換のステップa)及びb)が同時形質転換である請求項24に記載の方法。
【請求項26】
形質転換のステップa)及びb)が逐次形質転換である請求項24に記載の方法。
【請求項27】
前期糸状菌細胞がアスペルギルス(Aspergillus)細胞、またはトリコデルマ(Trichoderma)細胞である請求項24に記載の方法。
【図2】


【図3A】

【図3B】

【図3C】

【図4A】

【図4B】

【図4C】

【図5】
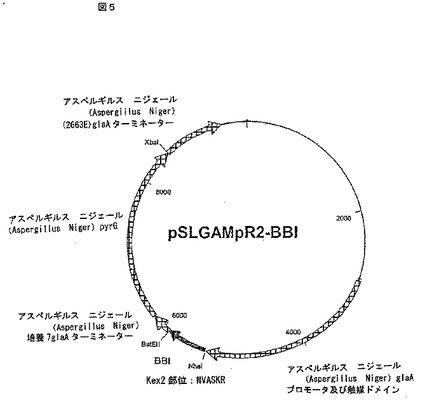
【図6】

【図7】

【図8】
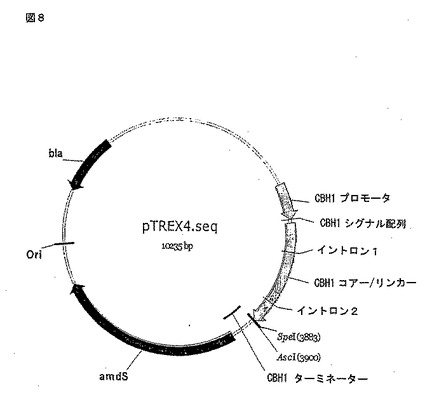
【図9A】

【図9B】

【図9C】

【図9D】

【図3A】
【図3B】
【図3C】
【図4A】
【図4B】
【図4C】
【図5】
【図6】
【図7】
【図8】
【図9A】
【図9B】
【図9C】
【図9D】
【公表番号】特表2007−528212(P2007−528212A)
【公表日】平成19年10月11日(2007.10.11)
【国際特許分類】
【出願番号】特願2006−538148(P2006−538148)
【出願日】平成16年10月22日(2004.10.22)
【国際出願番号】PCT/US2004/035278
【国際公開番号】WO2005/047302
【国際公開日】平成17年5月26日(2005.5.26)
【出願人】(500284580)ジェネンコー・インターナショナル・インク (67)
【Fターム(参考)】
【公表日】平成19年10月11日(2007.10.11)
【国際特許分類】
【出願日】平成16年10月22日(2004.10.22)
【国際出願番号】PCT/US2004/035278
【国際公開番号】WO2005/047302
【国際公開日】平成17年5月26日(2005.5.26)
【出願人】(500284580)ジェネンコー・インターナショナル・インク (67)
【Fターム(参考)】
[ Back to top ]