
細胞の自然突然変異率の低減
【課題】細胞または生物の自然突然変異の頻度を低下させる方法、ならびにそのような細胞および生物を産生する方法と、自然突然変異の頻度が低下している細胞および/または生物と、自然突然変異の頻度が低下している細胞を用いることによってタンパク質の発現系を産生する方法、タンパク質を産生する方法、および発酵産物を産生する方法の提供。
【解決手段】少なくとも2つの変異を細胞または生物に導入することによって上記細胞または生物における自然突然変異の頻度を低下させる方法を提供することによって、この技術的課題を解決し、その際、上記少なくとも2つの変異は、それらの複合作用によって、少なくとも2つの細胞性DNA修復機構の強化に導くものである。自然発生した突然変異を修正する細胞性DNA修復機構の能力が大幅に強化され、細胞で安定的に遺伝する変異の頻度の低下に導かれ、それによって、変異率の総合的な低下に導かれる。
【解決手段】少なくとも2つの変異を細胞または生物に導入することによって上記細胞または生物における自然突然変異の頻度を低下させる方法を提供することによって、この技術的課題を解決し、その際、上記少なくとも2つの変異は、それらの複合作用によって、少なくとも2つの細胞性DNA修復機構の強化に導くものである。自然発生した突然変異を修正する細胞性DNA修復機構の能力が大幅に強化され、細胞で安定的に遺伝する変異の頻度の低下に導かれ、それによって、変異率の総合的な低下に導かれる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、細胞または生物の自然突然変異の頻度を低下させる方法、ならびにそのような細胞および生物を産生する方法と、自然突然変異の頻度が低下している細胞および/または生物と、自然突然変異の頻度が低下している細胞を用いることによって、タンパク質の発現系を産生する方法、タンパク質を産生する方法、および発酵産物を産生する方法とに関する。
【背景技術】
【0002】
変異は、生体システムの特徴であり、自然選択の材料を提供する。細菌などの生物では絶えず自然突然変異が起きている。変異の発生率は、生物、例えば特定の遺伝子のサイズ、および塩基対置換による不活性化に対する遺伝子の感受性によって大きく異なる。大腸菌(Escherichia coli)では、1ゲノム複製あたり、1ゲノムあたりの変異の値がμg=0.0025であり、ゲノムサイズ4.6×106塩基対の中での1複製あたり、1塩基対あたりの変異率がμb=5.4×10−10である。G−C→A−Tなど、詳細が明らかにされている単一の経路の発生率でさえ、単一遺伝子内の異なった部位では、2000倍を越える相違を有することがあり、これは、いまなお大部分が謎である、局所的DNA配列およびDNA構造の影響によるものと考えられている。変異の種類、およびそれらを生成する過程は、両方とも多様であり、部分的にしか発見されていない。
【0003】
変異は多細胞生物でも重要である。ここではしかし、遺伝物質の変化における2つの異なった種類、すなわち配偶子の変異、換言すれば生殖細胞系変異と、体細胞の変化、換言すれば体細胞変異とを区別しなければならない。生殖細胞系変異はその生物の子孫へと引き継がれるが、体細胞変異は引き継がれない。しかし、体細胞変異は癌の発生に寄与するので、重要である可能性がある。生殖細胞系変異の検出、特にヒトの生殖細胞系変異の検出、およびヒトにおける変異率の測定には、二倍性の問題がある。ほとんどの前進変異は劣性であり、したがって、接合体が2コピーの変異対立遺伝子を得ない限り検出されないであろう。復帰変異の頻度は、通常、それよりはるかに低いが、それは、遺伝子に変異導入する方法が、既存の変異を元に戻す方法よりもはるかに多く存在するためである。ヒト細胞をin vitroで用いたin vivo研究から、総合的なヒト変異率は、原核および真核の様々な微生物のものに極めて類似していると推算されている。
【0004】
遺伝物質の遺伝的変化など、変異は、大域的変化、詳細には染色体レベルでの変化である場合も、あるいは細胞学的な異常として視覚化されないこともある点変異である場合もある。点変異には、トランジションおよびトランスバージョン、ならびにフレームシフト変異などの塩基対置換が含まれる。遺伝子のタンパク質コード領域における塩基置換変異の結果は、置換のタイプとその位置に依存する。そのような塩基対置換はサイレント、すなわちタンパク質配列中で新規のアミノ酸残基をもたらさないこともある。しかし、それらはアミノ酸置換をもたらすこともある。そのようなミスセンス変異は、鎌状赤血球貧血の場合のように、極めて重大な結果をもたらすことも、軽度の結果をもたらすことも、全く何の結果ももたらさないこともある。最後に、タンパク質コード領域における塩基置換は、アミノ酸コドンを終止コドンに変異させることもあれば、その逆もある。前者のタイプは、未熟な状態に短縮されたタンパク質をもたらし、ナンセンス変異と呼ばれる。塩基置換変異は、遺伝子のプロモーターもしくは5’調節領域、またはイントロンなど、遺伝子の発現の調節に関与する配列で起こることもあり、さらに、それらの転写、翻訳またはスプライシングに影響を与えることもある。β−サラセミアの多くが、グロビン遺伝子の発現レベルに影響を与えるこのタイプの非構造変異の結果である。フレームシフト突然変異は、遺伝子のコード領域における1つまたは複数(ただし3の倍数ではない)のヌクレオチドの挿入または欠失から生じる。これがリーディングフレームの変更を引き起こす。この種類の変異は下流のすべてのアミノ酸を改変し、産物が正常なタンパク質から大きく異なる場合があるので、機能しない産物を生成する可能性が非常に高い。
【0005】
自然突然変異は、細胞内での自然の過程の結果起こり得るものであり、外部因子、すなわち変異原とのDNAの相互作用の結果起こる変異である誘導性変異と区別することができる。自然突然変異の重要な発生源は、例えば、DNAポリメラーゼの不正確な作用による、DNA複製における誤りである。DNAポリメラーゼが誤りをおかす頻度は、この自然突然変異の頻度に影響を与えるであろう。そして、それによって、異なったDNAポリメラーゼに、精度の相違があることが観測されている。DNAポリメラーゼの精度に影響を与える重要な一要因は、ポリメラーゼによって挿入された誤対合塩基を除去するプルーフリーディング3’−5’エキソヌクレアーゼの存在である。この3’−5’エキソヌクレアーゼの機能は、DNA複製中の誤取込みを防止し、変異を防止することである。
【0006】
自然突然変異の別の主要な発生源は、互変異性化と呼ばれる核酸の塩基の構造変化である。塩基は、2つの形で存在でき、それらの間で相互変換する。例えば、グアニンはケト型およびエノール型で存在できる。様々な互変異性体型の塩基は異なった対合特性を有する。DNA複製中にGがエノール型である場合、DNAポリメラーゼは、それの向かいに正常なCではなくTを付加する。したがって、互変異性化はトランジション変異の原因となる。細胞で起こる別の変異原性の過程は、自発性の塩基分解である。細胞内では、シトシンからウラシルへの脱アミノ化が有意な発生率で起こる。脱アミノ化は、DNA中に通常存在しないウラシルを検出する特異的な修復過程によって修復することができ、それが起こらない場合、Uがその向かい側にAの挿入を引き起こし、DNA複製の際にC:GからT:Aへのトランジションを引き起こすことができる。発生頻度の高い自発性DNA損傷の第3のタイプは、酸素のフリーラジカルによる塩基の損傷である。これらは、細胞内で酸化代謝の結果起こり、また、放射線照射などの物理的作用因子によっても形成される。重要な酸化生成物の1つに8−ヒドロキシグアニンがあり、これはアデニンとの誤対合を起こし、その結果、G:CからT:Aへのトランスバージョンをもたらす。さらに別のタイプの自発性DNA損傷が、アルキル化、すなわちDNAの塩基またはバックボーンへのアルキル基の付加である。アルキル化は、S−アデノシルメチオニンなどの化合物の、DNAとの反応を介して起こり得る。アルキル化された塩基は、自発性の分解または誤対合の対象となる。
【0007】
さらに、DNA複製中における、鋳型鎖と新規に合成された鎖との間の「スリップ誤対合(slipped mispairing)」と呼ばれる機構によって、自発性のフレームシフト変異が生じることもある。
【0008】
自然突然変異はゲノムのいかなる部位でもランダムに起こり、そのため、自然発生した突然変異の大部分が、影響を受ける生物に有害であり、有利な変異が起こる確率は非常に低くなっている。したがって、生物は、過度の変異率から自らを防御する機構を進化させている。これらの防御機構は、DNAの複製または自発性の脱アミノ化の結果、DNA中に生じたミスマッチを認識および修正し、外因性または内因性の変異原とのDNAの反応の結果生じた潜在的に変異原性である変化を認識し除去する。
【0009】
特に、発酵工程などの生物工学の工程では、変異が極めて望ましくない。変異は、発酵細胞のゲノム中のいかなる部分にも生じ得るので、発酵産物の生成または組成に影響を与えることがある。例えば、発酵産物がタンパク質である場合、そのタンパク質をコードする核酸配列の変異は、改変されたアミノ酸配列を有するタンパク質変種に導くことがある。発酵細胞のわずかな部分のみが変異の影響を受けた場合にさえ、これによって、その変種に汚染された最終タンパク質調製物を生じる結果となり得る。これは、そのタンパク質がヒトの疾患の治療に用いられることとなっている場合、例えば、タンパク質の抗原特性が改変された場合に、予見できない劇的な結果をもたらす可能性がある。発酵細胞集団内での変異の発生も、その変異が、例えば、発酵産物、または所望の発酵産物の発現を制御する調節ユニットの形成に関与する酵素などの上流の調節経路に影響を与える場合、産物生成の速度に影響を与えることがある。
【0010】
さらに、変異が、発酵細胞集団のわずかな部分にのみ影響を与える希少な事象である場合でさえ、それらが、影響を受けた細胞に、変異導入されていない細胞と比較して選択有利性を与える場合、そのような集団でそれらが迅速に広まることがある。特に、定常状態を維持するために定常的に細胞を除去する連続培養では、これによって、培養時間の進行に伴う、変異導入された細胞と比較した、変異導入されていない細胞の漸進的減少に導かれる。
【0011】
特に発酵時間中の変異が極めて望ましくない別の理由が、いわゆる定常期変異の現象であるが、これは、適応変異とも呼ばれる。発酵の定常期中に行われる選択など、致死的でない選択に微生物集団が曝露された場合、選択圧を軽減する変異が高頻度で発生する(Cairnsら、Genetics、128(1991年)、695−701)。当初は、有用な変異のみが現れると思われたが、選択された変異には、選択されていない変異も伴うこと、すなわちこの過程が有用遺伝子へと方向付けられていないことが、現在では明らかである(Foster、J.Bacteriol.、179(1997年)、1550−1554)。適応変異に関するほとんどの研究は、ラクトースを利用することができない(Lac−)が、ラクトースが唯一の炭素源である場合に容易にラクトース利用性(Lac+)に復帰する大腸菌株に関して集中的に行われた。適応変異を生成する過程は、正常成長中にLac+変異を生成する過程と同じものではない。成長に依存した変異とは異なり、ほとんどすべての適応型のLac+変異は、大腸菌のRecBCD二本鎖切断(DSB)修復系の相同組換え機能など、組換え機能に依存している(Cairnsら、Genetics、128(1991年)、695−701;Foster、Annu.Rev.Microbiol.、47(1993年)、467−504;Harrisら、Science、264(1994年)、258−260)。
【先行技術文献】
【非特許文献】
【0012】
【非特許文献1】Cairnsら、Genetics、128(1991年)、695−701
【非特許文献2】Foster、J.Bacteriol.、179(1997年)、1550−1554
【非特許文献3】Cairnsら、Genetics、128(1991年)、695−701
【非特許文献4】Foster、Annu.Rev.Microbiol.、47(1993年)、467−504
【非特許文献5】Harrisら、Science、264(1994年)、258−260
【発明の概要】
【発明が解決しようとする課題】
【0013】
したがって、本発明の根底にある技術的課題は、細胞または生物によって、タンパク質などの発酵産物を産生する方法および手段を提供し、その際、自然発生した突然変異、中でも発酵工程の定常期中に発生する変異に対して、産生を行う細胞または生物が保護および/または安定化され、かつ発酵産物、詳細にはタンパク質の形成速度および組成の両方が長期的に保障および保護されるようにすることである。
【課題を解決するための手段】
【0014】
本発明は、少なくとも2つの変異を細胞または生物に導入することによって上記細胞または生物における自然突然変異の頻度を低下させる方法を提供することによって、この技術的課題を解決し、その際、上記少なくとも2つの変異は、それらの複合作用によって、少なくとも2つの細胞性DNA修復機構の強化に導くものである。
【0015】
本発明は、自然突然変異の頻度が低下している細胞または生物を、少なくとも2つの変異を導入し、さらにその生物が多細胞生物である場合にはそれから生物を再生することによって産生する方法を提供することによっても、その根底にある技術的課題を解決し、その際、上記少なくとも2つの変異は、それらの複合作用によって少なくとも2つの細胞性DNA修復機構の強化に導くものである。
【0016】
本発明によれば、自然発生した突然変異を修正する細胞性DNA修復機構の能力が大幅に強化され、それによって、異なった修復系に影響を与える少なくとも2つの異なった変異が細胞に導入される。本発明による、少なくとも2つの異なった変異の導入によって得られた、自然発生した突然変異を修正する細胞性DNA修復機構の能力の強化によって、細胞で安定的に遺伝する変異の頻度の低下に導かれ、それによって、変異率の総合的な低下に導かれると有利である。
【0017】
したがって、本発明によれば、驚くべきことに、特定の細胞性DNA修復機構を改変することによって、詳細には自然発生した突然変異を修復するこれらのDNA修復系の能力をより効率的に強化する特定の変異を導入することによって、野生型細胞の自然突然変異率を劇的に低下できることが見出された。例えば、本発明によれば、驚くべきことに、MutSタンパク質の過剰発現によって、増殖期に、宿主細胞の変異性の有意な低下に導かれることが見出された。これはしかし、現在の技術において記載されている結果と対照的である。米国特許第6656736号によれば、酵母または他の生物の野生型または変異型MMRタンパク質の過剰発現の結果、ミスマッチ修復系の欠損が生じ、それによって、そのようなミスマッチ修復系の欠陥を有する酵母細胞は高変異性となる。さらに、dinB10の欠失と、抗変異原アレルdnaE911とを保持する細菌株は、対応する野生型株と比較して10分の1に低下した変異性を示す。ミスマッチ修復系に関与するMutLタンパク質を追加して過剰発現すると、変異性の低下が50分の1にまで達する。別の系では、特定のフレームシフト変異の復帰変異の低下が1000分の1に達する可能性さえあると示されるはずである。このようにして、成長に依存した変異の発生率だけでなく、適応変異中の変異発生率、すなわち定常期に依存した変異の発生率も有意に低下させることができる。観測された、宿主細胞の自然突然変異発生率に対するいくつかの変異の影響は、驚くべきことに、相加的ではなく、相乗的に共働しているようである。
【0018】
さらに、本発明によれば、驚くべきことに、これらの変異の導入は、自然発生した突然変異を修正する細胞性DNA修復機構の能力の強化に導くだけではなく、有利なことに、細胞生存率も大幅に増大させることが見出された。例えば、発酵細菌株におけるMutLタンパク質の過剰発現によって、約103倍の細胞生存率の増大が観測された。
【0019】
自然発生した突然変異を修正する細胞性DNA修復機構の能力を増強するいくつかの変異を導入することによる、本発明の細胞内自然突然変異率の低減および本発明の細胞生存率の増大は、タンパク質などの発酵産物の発現および/または産生に使用される細胞または株に特に大きな価値を有する。そのような細胞の使用によって、例えば、得られたタンパク質産物のアミノ酸配列を、産生を行う細胞または生物で何代にもわたって、変化させずに維持することができる。そのような細胞の使用によって得られたタンパク質調製物は、変異によるタンパク質変種によって汚染されておらず、したがって、治療薬としての適用において何の問題も起こさず、また、受容する身体に対する望ましくない影響も導かない。細胞性DNA修復機構の能力が強化されているそのような本発明の細胞の使用は、組換え体タンパク質の産生に特に有用である。
【0020】
しかし、細胞性DNA修復機構の能力が強化されている本発明の細胞の使用は、発酵細胞によるタンパク質産生に限定されない。原則として、本発明の細胞は、抗生物質、有機酸など、あらゆる種類の発酵産物用に使用することができる。宿主細胞内での変異率の低下によって、産物生成速度の有効性を制御する因子も、安定かつ不変に維持されるであろうから、これらの細胞における自然突然変異率の大幅な低下によって、産物組成の完全性の長期的な維持のみでなく、産物生成速度の高さの維持も提供される。
【図面の簡単な説明】
【0021】
【図1】プラスミドpmutLの物理構造を示す図である。このプラスミドは、原則的にpmutSプラスミドと同じ構造を有する。
【図2】野生型、dinB10、dnaE911、dinB10 dnaE911、mutL−、mutL− dinB10、mutL− dnaE911、およびmutL− dinB10 dnaE911株のリファンピシン耐性への自然突然変異の頻度の値のグラフ表示である。図が示すように、MutLの発現によって変異性が強く抑制される。データは、SMF値として示す。プラスミドpmutLは、プラスミドpTrcHis2/lacZのmutL+派生物である。全てのセットの遺伝子操作に野生型株MG1655(wt)を選択した。関連する遺伝子型が示されている。棒グラフは三つ組みに分割されており、各三つ組みは、同一の遺伝子型を示し、かつa)最初の棒グラフ:細菌株、b)プラスミドpTrcHis2/lacZを保持する対応する細菌株、およびc)プラスミドpmutLを保持する対応する細菌株から構成されている。
【図3】野生型、dinB10、dnaE911、dinB10 dnaE911、mutS−、mutS− dinB10、mutS− dnaE911、およびmutS− dinB10 dnaE911株のリファンピシン抵抗性への自然突然変異の頻度の値のグラフ表示である。この図は、mutS−バックグランドにおける、dinB10、dnaE911、およびdinB10 dnaE911による変異性の抑制を示す。データは、SMF値として示す。関連する遺伝子型が示されている。
【図4】プラスミドpdapAIF(A)およびプラスミドpSU40dapA(B)の物理構造を示す図である。
【図5】プラスミドpSU40mutLを産生するストラテジーを示す図である。
【図6】菌株Top10(野生型;dapA+)、MG1655 dinB10 dapA::kn(dapA−)、AT997(dapA15、dapA−)、AT997 pdapAlF pTrc(dapA+)、AT997 pdapAlF pmutL(dapA+)、AT997 pdapA+1 pTrc(dapA−)、AT997 pdapA+1 pmutL(dapA−)、MG1655 dinB10 dapA::kn pdapA15 pSU40(dapA−)、MG1655 dinB10 dapA::kn pdapA15 pSU40mutL(dapA−)におけるdapA点突然変異の復帰変異のグラフ表示である。この図は、MutLの発現による変異性の抑制を示す。DAPに対する耐性を与える変異の値は、生存細胞の総数で割ることによって表される。プラスミドpmutLは、プラスミドpTrcHis2/lacZまたはpSU40のmutL+派生物である。全てのセットの遺伝子操作に野生型株MG1655(wt)を選択した。関連する遺伝子型が示されている。図は、対照の三つ組みの1つと、検査する株の3つの二つ組みとに分割されている。各二つ組みは、a)最初の棒グラフ:親ベクターを保持する細菌株、およびb)mutL+派生物を保持する細菌株で構成されている。使用されたすべての細菌株は染色体の野生型MutLを発現する。
【図7】プラスミドpXX7の派生物を示す図である。A)pXX7へのクロラムフェニコールカセットのクローニング。それによってプラスミドpXX7cmを得た。得られたクローンのうち9クローンを、EcoRIおよびNcoI(予測された断片:1677bpおよび3292bp)による制限消化によって分析した。試験されたクローンのすべてが正しい移動パターンを示した。B)プラスミドpXX7へのサイレントtetA遺伝子のクローニング。それによってプラスミドpXX7tetを得た。得られたクローンのうち24クローンを、EcoRI(予測された断片:4141bpおよび3113bp)による制限消化によって分析した。試験されたクローンのうち1クローンが正しい移動パターンを示した。
【図8】菌株JM83 pXX7、JM83 pXX7 pTrc、およびJM83 pXX7 pmutLにおけるリファンピシン抵抗性への自然突然変異の頻度を示す図である。プラスミドpmutLは、プラスミドpTrcのmutL+派生物である。SMF値は、生存細胞の総数に対するリファンピシン耐性として与えられる。野生型株JM83は、染色体の野生型MutLを発現する。
【図9】菌株JM83 pXX7、JM83 pXX7 pTrc、およびJM83 pXX7 pmutLにおける、ホスホマイシン耐性への自然突然変異の頻度の値を示す図である。プラスミドpmutLは、プラスミドpTrcのmutL+派生物である。SMF値は、生存細胞の総数に対するホスホマイシン耐性として与えられる。野生型株JM83は、染色体の野生型MutLを発現する。
【図10】菌株JM83 pXX7cm、JM83 pXX7cm pTrc、およびJM83 pXX7cm pmutLのクロラムフェニコール耐性レポータ遺伝子における自然突然変異の頻度によるcmRコロニーの数を示す図である。プラスミドpXX7cmは、pXX7のcm+派生物である。プラスミドpmutLは、pTrcのmutL+派生物である。生存細胞の総数が割った、クロラムフェニコールに対する感受性を導入する変異の値が示されている。対照株であるJM83pXX7、JM83pXX7 pTrc、およびJM83pXX7 pmutLは、クロラムフェニコールに対する、予測通りの感受性を示す(クロラムフェニコール耐性遺伝子を保持していない)。
【図11】菌株JM83 pXX7tet、JM83 pXX7tet pTrc、およびJM83 pXX7tet pmutLのサイレントtetAレポータ遺伝子における自然突然変異の頻度によるtetRコロニーの数を示す図である。プラスミドpXX7tetは、サイレントtetAを保持するpXX7派生物である。プラスミドpmutLは、pTrcのmutL+派生物である。生存細胞の総数が割った、テトラサイクリンに対する耐性を導入する変異の値が示されている。対照株であるJM83pXX7,JM83pXX7 pTrc、およびJM83pXX7 pmutLは、テトラサイクリンに対する、予測通りの感受性を示す(テトラサイクリン耐性遺伝子を保持していない)。JM83 pXX7およびJM83 pXX7tetの2株では、変異性の値が0.43または0.39であった(Δ1.1)。MutLの発現によって、変異性の値が0.25(Δ1.72)に低下する。
【図12】「発酵状態」中における、改変された産生株JM83 pXX7cmの変異性を示す図である。G−CSFが抑制(A)または(B)発現された際の、細胞におけるクロラムフェニコール耐性の頻度が示されている。試験したそれぞれのケースで、MutLが染色体から発現された。対照株であるJM83 pXX7、JM83 pXX7pTrc、およびJM83 pXX7pTrc pmutLは、ケース(A)およびケース(B)でクロラムフェニコールに対する予測通りの感受性を示す。
【図13】「発酵状態」中における、改変された産生株JM83 pXX7tetの変異性を示す図である。図は、G−CSFが抑制(A)または(B)発現された際の、細胞のテトラサイクリン耐性の頻度を示す。試験したそれぞれのケースで、MutLが染色体から発現された。
【発明を実施するための形態】
【0022】
本発明のコンテクストにおいて、「細胞性DNA修復機構の能力の強化に導く変異」という用語は、細胞のゲノムにおける、少なくとも1つの特定のDNA修復機構に関与するタンパク質または酵素をコードする部分の任意の遺伝的改変、あるいはDNAの複製もしくは自発性の脱アミノ化、または自然発生の突然変異に導く任意の他の自然過程の結果としてDNAで生じた細胞内または生物内でのゲノム規模の誤りの認識および修正の強化に導く、細胞性DNA修復機構の構成要素の発現調節に関与する、ゲノムの部分の任意の遺伝的改変を意味する。したがって、「細胞性DNA修復機構の能力の強化に導く変異」は、所与の細胞または生物のゲノム内の自然発生した誤りの、より効率的かつより正確な修正を提供する。
【0023】
したがって、「細胞性DNA修復機構の能力の強化に導く変異」は、集団内で安定的に遺伝する変異の頻度の低下、すなわちその集合内での変異頻度の低下に導く。本発明のコンテクストでは、「変異の頻度」を、集団の個体総数に対する変異体数の比率として定義する。「細胞性DNA修復機構の能力の強化に導く変異」は、所与の遺伝子が複製中に変異し、かつ該遺伝子のこの変異が安定的に遺伝するであろう確率として定義される変異発生率の低下も導く。さらに、本発明のコンテクストにおいて、細胞性DNA修復機構の能力の強化に導く変異は、トランスポゾンに媒介された変異導入の低下も導く。
【0024】
したがって、細胞または生物は、細胞性修復機構の能力の強化に導く変異の存在によって、非常に低い自然突然変異率を示し、それは、詳細には、その変異を有しない相当する細胞または生物よりかなり低いものである。細胞性DNA修復機構の能力の強化に導く変異には、細胞性DNA修復に関与する酵素をコードする構造遺伝子の欠失、例えば、誤りがちな(error−prone)DNAポリメラーゼの構造遺伝子の欠失;細胞性DNA修復に関与する酵素をコードする構造遺伝子における、塩基の置換、欠失、逆位、および/または付加、例えば、抗変異原表現型またはDNAポリメラーゼの忠実度の改善に導くことができるもの、細胞または生物の成長、分化、または増殖における特定のフェーズで律速的になるタンパク質の過剰発現、タンパク質、例えば、誤りがちなDNAポリメラーゼの発現の低下などが含まれるが、これらに限定されない。しかし、本発明のコンテクストでは、細胞性DNA修復機構の能力の強化に導く個々の変異それぞれが、第2または第3の細胞性DNA修復機構の能力の強化も導くことがある。さらに、細胞性DNA修復機構の能力の強化に導く個々の変異それぞれが、細胞生存率の増強も導くことがある。
【0025】
本発明のコンテクストにおいて、「2つの変異の複合作用によって少なくとも2つの細胞性DNA修復機構に導かれる」という用語は、両方の変異が少なくとも2つの異なったDNA修復機構に影響を与え、それによって、変異導入されていないDNA修復系と比較して、自然発生したゲノム規模の変異をより効率的かつより正確に修復するこれらのDNA修復系の能力が強化されることを意味する。
【0026】
本発明のコンテクストにおいて、「細胞性DNA修復機構」という用語は、細胞または生物が、ゲノム内の、DNA複製によって生じた誤りならびに/または塩基変化および塩基損傷による誤りを、それによって認識および修正できる酵素機構または酵素系を意味する。本発明の好ましい実施形態では、これらの細胞性DNA修復機構に、ミスマッチ修復系、複製後(組換え)修復系、およびSOS修復系が含まれる。ミスマッチ修復の強化に導く変異は、ミスマッチ修復に関与するタンパク質のうちの1つが律速的となる状況を打開する変異である。
【0027】
細胞内の全修復事象の99%が「ミスマッチ修復系」(MMR)によるものである。この修復系は、大腸菌の複製忠実度に対する貢献が最も大きい系であり、それによって、その成分タンパク質が転写共役DNA修復および超短鎖パッチ修復(very short patch repair)に用いられることによって、他のDNA修復経路でも作用する。MMRは、それなしではゲノムの再編成を引き起こし得る、不正確な相同性を有するもの相互の組換えを抑制することによって、遺伝的安定性の強制も行う。ミスマッチ修復は、トランスポゾンの切り出しを編集することによっても、遺伝的安定性を増強する。大腸菌MMRタンパク質の相同体は、他の細菌、酵母、マウス、およびヒトで同様の機能を行う(Modrich、Science、266(1994年)、1959−1960;Radmanら、Phil.Trans.R.Soc.London、347(1995年)、97−103)。大腸菌では、mutH、mutL、mutS、およびmutUの遺伝子産物がMMRに関与している。この系は、メチル化のため、親鎖ではなく新規に合成された鎖を認識する。mutS遺伝子産物は、DNA塩基ミスマッチを認識し、結合する。mutL遺伝子産物は、ミスマッチ結合の後にMutSと相互作用し、MutSをMutHと組み合わせると考えられている。その後、MutHエキソヌクレアーゼが、メチル化されていない新規のDNA鎖にニックをいれる。MutUヘリカーゼは、一本鎖ニックの箇所でDNAに進入し、ニックの入った鎖を除去する。ミスマッチ修復の強化に導く変異は、ミスマッチ修復に関与するタンパク質の1つが律速的となる状況を打開する変異である。
【0028】
複製後修復系は、後に続く複製ラウンドでDNA中の損傷を修復する系である。娘鎖中の損傷したDNAの複製によって、ギャップが作られる。欠失している遺伝情報は、組み換えによって、親鎖の対応するDNA領域によって補填される。損傷を含むDNAの複製は、損傷乗り越え合成とも呼ばれる過程であり、点変異の主要な発生源である。最近、関与しているDNAポリメラーゼが同定されたため、この過程に関する多くの理解が得られた(Friedbergら、Proc.Natl.Acad.Sci.USA、97(2000年)、5681−5683)。
【0029】
SOS応答は、概してDNA複製を妨害するものである様々な遺伝毒性ストレスおよび代謝ストレスへの細胞の曝露によって誘導される1セットの細胞反応である。SOS系の調節は、LexAおよびRecAタンパク質によって媒介される。LexAは、recAおよびlexAを含めた約30の様々な遺伝子のリプレッサーとして作用する。SOSを誘導するシグナルは、一本鎖DNAであり、これにRecAが結合して、コプロテアーゼ(RecA*)として活性化される。RecA*は、LexAレプレッサーおよび一部のファージリプレッサーのタンパク分解性自己切断を促進し、それによって、SOSレギュロンを抑制解除する。大腸菌は、ポリメラーゼII(Pol II)、ポリメラーゼIV(Pol IV)、およびポリメラーゼV(Pol V)の、少なくとも3つのSOS誘導性ポリメラーゼを有する。
【0030】
本発明によれば、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから、少なくとも2つの変異が選択される。本発明のコンテクストにおいて、「抗変異原アレル」は、対応する野生型細胞と比較して、細胞または生物の自然突然変異の頻度の低下を引き起こすアレルを意味する。
【0031】
本発明の好ましい実施形態では、MutLまたはその相同タンパク質の発現、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、ベクターを細胞内に導入することによって実現され、上記ベクターは、それぞれ、mutL遺伝子またはMutLの相同タンパク質をコードする遺伝子、およびmutS遺伝子またはMutSの相同タンパク質をコードする遺伝子を、それぞれのMutタンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に含む。上記ベクターは、多コピープラスミドであることが好ましい。上記調節ユニットは、誘導性または構成的プロモーターであることが好ましい。
【0032】
本発明の別の好ましい実施形態では、MutLまたはその相同タンパク質の発現、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、宿主細胞または宿主生物における天然のMutタンパク質の発現を指示する調節ユニットを、天然のMutタンパク質が、細胞で通常観測されるよりも多くの量で、細胞内で生成されるように改変することによって実現される。これは、染色体に存在する、それぞれのMutタンパク質をコードする天然のヌクレオチド配列から相補的なRNA配列への転写、および/またはそのようにして得られたコードRNA配列からMutタンパク質のポリペプチド鎖への翻訳を指示する調節ユニットが、他の適当な相同または異種の調節ユニットによって、対応する野生型の細胞または生物で観測されるものより高い天然Mutタンパク質の産生が得られるように、変異導入または置換されることを意味する。
【0033】
本発明の別の実施形態では、それぞれのmut遺伝子を1コピーまたは複数コピー追加して、適当な調節ユニットの機能的制御下に宿主細胞の染色体に導入することによって、それぞれのMutタンパク質の過剰発現が実現され、その際、追加のmut遺伝子は、別の種に由来する天然遺伝子か、または異種遺伝子のいずれかであり得る。
【0034】
本発明の好ましい実施形態では、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレルがdinB10であり、これは、事実上dinB遺伝子の欠失である。dinBによってコードされているDNAポリメラーゼIV(Pol IV)は、最近同定された、誤りがちなDNAポリメラーゼのYファミリー、すなわちUmuC/DinBヌクレオチジルトランスフェラーゼファミリーに属する。UmuC、DinB、Rad30、およびRev1サブファミリーがこのファミリーを代表する(Gerlachら、Proc.Natl.Acad.Sci.USA、96(1999年)、11922−11927)。dinBによってコードされている大腸菌のDNA ポリメラーゼIVは、自然突然変異生成と、翻訳合成(TLS)と呼ばれている過程であり、点突然変異の主要な発生源である、損傷を含有するDNAの複製とに関与することが示されている。DNAポリメラーゼVと同様に、Pol IVも、DNA損傷に対するSOS応答の一部として誘導される。
【0035】
本発明の別の好ましい実施形態では、DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルが使用され、それは、dnaE911であることが好ましい。細胞のDNA合成の90%超がDNAポリメラーゼIIIホロ酵素(Pol III HE)によるものであり、また、この酵素は、主要な複製後ミスマッチ修正経路にも必要である。ポリメラーゼIIIホロ酵素は、無塩基部位を通過する損傷乗り越えDNA合成を効果的に実行することが示されている。大腸菌のdnaE遺伝子は、DNAポリメラーゼIIIホロ酵素のαサブユニットをコードし、このαサブユニットはポリメラーゼ活性を提供する。したがって、αサブユニットは、忠実度の主要な決定因子の1つである。FijalkowskaおよびSchaaper、J.Bact.、177(1995年)、5979−5986から、いくつかのdnaE抗変異原アレルが知られており、それらは、ミスマッチ修復欠損性のmutL株における高変異性を抑制する。
【0036】
本発明によれば、dinB10変異およびdnaE911変異の複合作用によって、細胞の自然突然変異の頻度が、野生型細胞または野生型生物と比較して少なくとも10分の1に低下する。本発明の別の好ましい実施形態では、dinB10、dnaE911、および過剰発現したMutLタンパク質の複合作用によって、細胞内の自然突然変異の頻度が、野生型細胞または野生型生物と比較して、少なくとも50分の1に低下する。
【0037】
細胞性DNA修復機構の増強および/または細胞生存率の増強がそれらの複合作用によって導かれる少なくとも2つの変異は、任意の知られている方法によって、詳細には、細胞性DNA修復系の1つに関与していることが知られている遺伝子に変異導入することを目的として所与の細胞に変異導入することによって、あるいは既知の変異またはアレルをその細胞内に導入することによって、細胞に導入することができる。
【0038】
限定されるものではないが、ランダム変異導入、部位特異的変異導入、オリゴヌクレオチドカセット変異導入、または誤りがちなPCRによる点変異導入を含めた多数の異なった変異導入法が存在する。ランダム変異導入は、例えば、亜硝酸、ヒドラジンなどの化学物質で処理することによって、クローニングされたDNA断片中にランダムに分布した多数のヌクレオチド置換変異の生成を引き起こす。誤りがちなPCRは、クローニングされた遺伝子にランダムな点変異を導入するために開発された。PCR反応の忠実度を低下させる改変には、MgCl2濃度の増大、MnCl2の添加、または4種類のdNTPの相対濃度の改変が含まれる。これらの伝統的な変異導入法は、別々の選択可能な表現型を有する個々の遺伝子の改変に焦点をしぼって行われる。通常の戦略は、遺伝子のクローニングを行い、その遺伝子および/またはその機能をモニターできるアッセイを確立し、遺伝子内の選択された位置に変異導入し、そして、その遺伝子の機能が改変されている遺伝子変種を選択するというものである。その後、機能が改変された変種を所望の細胞型に導入して、発現させることができる。その遺伝子の所望の機能を得るために、変異導入法のサイクルを反復して実行することができる。遺伝子の機能を改変する別の手法は、十分に確立されている多数の系のうちの1つを用いた組換えによるものである。
【0039】
当然ながら、既存のアレルまたは変異を本発明の目的に用いることもできる。そのような既存のアレルまたは変異は、任意の知られている方法によって細胞に導入することができる。本発明によれば、細胞内への少なくとも2つの変異の導入は、限定されるものではないが、形質転換、接合、形質導入、伴性導入、感染、および/またはエレクトロポレーションを含めた任意の知られている適切な方法によって実施できる。
【0040】
本発明のコンテクストにおいて、「形質転換」という用語は、細胞、例えば微生物細胞による、環境からの、単離された核酸分子、好ましくは精製された核酸分子の取込みを意味する。「接合」は、細胞間接触を介した、細菌プラスミドの、ある細菌細胞から別の細菌細胞の中へのプラスミド媒介性の移入を意味する。関与する移入機構は、通常、プラスミドまたは接合トランスポゾンによってコードされている。そのようなプラスミドの例は、接合性プラスミドまたはヘルパープラスミドである。接合は、原核生物細胞の異なった表現形グループ間、および原核生物と真核生物との間の遺伝子交換の主要経路の1つである。「形質導入」は、バクテリオファージによる、ある細菌細胞から別の細菌細胞の中への核酸分子の移入を意味し、それは、一方の細胞からのバクテリオファージの放出と、それに続く、もう一方の細胞の感染とを含む。2つのタイプの形質導入がある。特殊形質導入は、溶原バクテリオファージの溶原生活環中に起こることがあり、それによって、細菌の遺伝物質が、バクテリオファージゲノムの一部を置換することがある。細菌DNAのこの小片は、ファージゲノムの一部として複製し、そのファージによって別の受容細胞の中に移入されることがある。普遍形質導入の場合には、溶菌ファージの全ゲノムが細菌DNAによって置換される可能性を有する。「エレクトロポレーション」は、細胞を核酸分子と混合して、その後、高電位の短いパルスに曝露する過程である。宿主細胞の細胞膜が貫通可能となり、それによって、外来核酸が宿主細胞内に入るのが可能となる。
【0041】
自然突然変異率を低下させる本発明の方法、および自然突然変異率が低下している細胞または生物を産生する本発明の方法で用いられる細胞は、いかなる原核細胞でも、真核細胞でもよい。
【0042】
「真核細胞」および「真核細胞宿主細胞」という用語には、膜に結合した核、細胞小器官、および染色体に組織化された遺伝物質を有し、染色体において、DNAがヒストンに結合しているいかなる細胞も含まれる。細胞骨格は、真核生物に特有な別の特徴であり、これは、細胞膜に繋ぎ止められて、細胞辺縁を縦横に走るタンパク質フィラメント、主としてアクチンおよびチューブリンのネットワークである。「真核生物」は、分類学上の原生生物界、菌類界、植物界、および動物界を含む。真核生物は、原生生物、例えば、ゾウリムシおよびアメーバなどの単細胞生物である場合も、様々な真菌、動物、および植物などの多細胞生物である場合もある。自然突然変異率を低下させる本発明の方法の好ましい実施形態では、動物細胞、植物細胞、または真菌細胞が使用されている。
【0043】
「原核細胞」および「原核細胞の宿主細胞」という用語には、ゲノムが環状構造として細胞質中に遊離して存在している細胞、すなわちゲノムが核膜によって囲まれていない細胞のいかなるものも含まれる。原核細胞はさらに、それが必ずしも酸素に依存していないこと、およびそのリボソームが真核細胞のものより小さいことを特徴とする。原核細胞には、古細菌および真正細菌が含まれる。細胞壁の組成物に応じて、真正細菌を、グラム陽性菌、グラム陰性菌、およびラン細菌に分類することができる。自然突然変異率を低下させるか、あるいはそれに対応する生物を産生する本発明の方法の好ましい実施形態では、使用される原核細胞が古細菌または真正細菌の細胞であり、上記原核細胞が、グラム陰性細菌、グラム陽性菌、またはラン細菌であることが特に好ましい。
【0044】
本発明は、自然突然変異の頻度の低下および/または細胞生存率の増強を有する細胞にも関し、これは、細胞または生物の自然突然変異の頻度を低下させる本発明の方法、または自然突然変異の頻度が低下している細胞もしくは生物を生成させる本発明の方法によって取得可能であり、上記細胞は、少なくとも2つの細胞性DNA修復機構の増強が、それらの複合作用によって導かれる少なくとも2つの変異を含む。本発明の好ましい実施形態では、上記細胞が細菌細胞、例えば、大腸菌などのグラム陰性細菌、もしくは枯草菌(Bacillus subtilis)などのグラム陽性菌、真菌細胞、植物細胞、または昆虫細胞もしくは哺乳類細胞などの動物細胞である。
【0045】
本発明の好ましい特定の実施形態では、上記細菌が、プラスミドpmutLを含有する大腸菌MG1655 dinB10、プラスミドpmutLを含有する大腸菌MG1655 dinB10 mutL::tet、プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm、プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm mutL::tet、大腸菌MG1655 dinB10 dnaE zae::cm大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet、プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm、またはプラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm mutL::tetである。
【0046】
本発明は、自然突然変異率が低下している生物を産生するための、自然突然変異率の低下および/または細胞生存率の増強を有する本発明の細胞の使用、タンパク質発現用の宿主細胞としての本発明の細胞の使用、発酵産物産生用の発酵生物としての本発明の細胞の使用、候補薬物の作用を研究するためのモデル系としての本発明の細胞の使用、ヒト、動物、または植物の病気を研究するためのモデル系としての本発明の細胞の使用、およびトランスジェニック生物の産生、特に育成または培養のための本発明の細胞の使用にも関する。
【0047】
当業者は、完全な多細胞生物、特に植物または動物を単一細胞から産生することのできる多数の方法を知っている。本発明の細胞から産生された多細胞生物も、同様に、総合的な自然突然変異率が低下していることを特徴とする。
【0048】
本発明は、自然突然変異の頻度が低下している生物にも関し、これは、細胞または生物内の自然突然変異の頻度を低下させる本発明の方法、または自然突然変異の頻度が低下している細胞または生物を産生する本発明の方法によって取得可能であり、上記生物の細胞は少なくとも2つの変異を含み、上記少なくとも2つの変異の複合作用によって、少なくとも2つの細胞性DNA修復機構の強化および/または細胞生存率の増強に導かれる。本発明の好ましい実施形態では、上記多細胞生物が、出芽酵母(Saccharomyces cerevisiae)、偽巣性コウジ菌(Aspergillus nidulans)などの真菌、植物、特にトウモロコシ(Zea mays)などの農業有用性のある植物、または動物、特に哺乳動物、例えば、所与の疾患、または所与の疾患を治療するための薬物候補の作用を研究するためのモデル系として使用できる哺乳動物である。
【0049】
本発明は、経済的、医学的、または農業的に重要なタンパク質など、タンパク質を発現および/または産生するための宿主としての本発明の生物の使用、トランスジェニック生物を育成または培養するための本発明の生物の使用、疾患を研究するためのモデル系としての本発明の生物の使用、および候補薬物の作用を研究するためのモデル系としての本発明の生物の使用にも関する。
【0050】
本発明の根底にある技術的課題は、タンパク質の発現系を産生する方法を提供することによっても解決され、その際、上記タンパク質のアミノ酸配列は、自然発生した突然変異に対して安定化されており、上記方法は、
a) 少なくとも2つの変異を含有するタンパク質をコードする核酸配列を、上記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、宿主細胞のゲノムに挿入する工程であって、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる工程、または
b) 上記タンパク質をコードする核酸配列を、上記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下にベクターに挿入し、さらに、少なくとも2つの変異を含有するベクターを宿主細胞内に移入する工程であって、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる工程と、
c) 適当な培地中で上記宿主細胞を培養および/または維持する工程と
を含む。
【0051】
タンパク質の発現系を産生する本発明の方法は、特に、タンパク質の発現をその中で行うことができ、自然突然変異率が非常に低く、特に他の既知の宿主細胞より低いことを特徴とする宿主細胞の産生を対象とするものである。したがって、本発明の方法によって産生された発現系は、所与のタンパク質をコードするヌクレオチド配列が変異によって変化し、それによって、そのタンパク質のアミノ酸配列が変化する確率が大幅に低下するのを確実にするものである。したがって、本発明の方法によって提供され、低自然突然変異率および増強された細胞生存率を有する本発明の宿主細胞に基づく発現系は、その完全性が長期的に保障されるタンパク質の発現および産生に用いることができる。本発明の発現系は、特に、治療目的で使用されることになっているタンパク質であって、そのアミノ酸配列のいかなる変化も、例えば、そのタンパク質の生物活性の変化に導くか、あるいはそのタンパク質の抗原特性を改変する変化も、そのタンパク質の治療効果に有害な影響を与える恐れがあるタンパク質に使用できる。したがって、発現系を産生する本発明の方法は、産生されたタンパク質調製物における、変異導入された異常なタンパク質による最小限の汚染さえも回避するのに有用である。本発明の方法によって提供され、低自然突然変異率を有する本発明の宿主細胞に基づく発現系は、特定のタンパク質をコードするヌクレオチド配列の長期的な維持に使用でき、それによって、ヌクレオチド配列が改変しているか否かを判定するために、シーケンシングによって定期的に検査する必要性を取り除くものが有利である。
【0052】
本発明のコンテクストにおいて、「発現系」は、特に、細胞における所与のタンパク質の効率的発現、すなわちそのタンパク質の大量生産を可能にする細胞システムを意味する。詳細には、「発現系」は、所望のタンパク質をコードするヌクレオチド配列から相補的なRNA配列に転写し、非コード配列部分を除去するために、該RNA配列のスプライシングを行い、そして、コードRNA配列部分を翻訳し、それによって、該タンパク質のポリペプチド鎖を取得するのを可能にする細胞環境に関する。「発現系」は、タンパク質のグリコシル化、またはタンパク質からの既存のリーダー配列の除去など、翻訳後プロセシング工程を可能にする細胞環境にも関するものである場合がある。したがって、「発現系の産生」は、コードされている遺伝子産物の発現を所与の細胞環境で制御および指示する適当な調節ユニットに、所望のタンパク質をコードするヌクレオチド配列が機能的に連結されていなければならないこと、および該タンパク質の最適な発現が実現されるように、使用される調節ユニットが機能するであろう適当な宿主細胞が提供されなければならないことを意味する。
【0053】
本発明によれば、タンパク質を発現するための系で使用される宿主細胞における自然突然変異率の低下および生存率の増強が、少なくとも2つの変異を使用することによって得られ、上記少なくとも2つの変異の複合作用によって、少なくとも2つの細胞性DNA修復機構の能力の強化に導かれ、それによって、宿主細胞内に存在するいずれの核酸で自然発生した突然変異も修復される。詳細には、これらの少なくとも2つの変異によって、自然発生した突然変異を修復する、ミスマッチ修復系、プルーフリーディング機能、および/またはSOS修復系の能力が強化される。本発明の好ましい実施形態では、これらの変異が、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから選択される。
【0054】
本発明の一実施形態において、MutLまたはその相同タンパク質の発現の上方制御、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、ベクターを宿主細胞に導入することによって実現することができ、上記ベクターは、それぞれ、mutL遺伝子またはその相同タンパク質をコードする遺伝子、およびmutS遺伝子またはその相同タンパク質をコードする遺伝子を、対応する野生型宿主細胞との比較において、それぞれのMutタンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に含む。MutLまたはMutSのいずれかのタンパク質の過剰発現に用いるベクターは、1つまたは複数の調節ユニットの機能的制御下にそれぞれのmut遺伝子を含む多コピープラスミドであることが好ましい。本発明の好ましい実施形態では、それぞれのmut遺伝子の過剰発現を制御する調節ユニットは誘導性プロモーターでも、構成的プロモーターでもよい。
【0055】
本発明の別の実施形態では、MutLまたはその相同タンパク質の発現の上方制御、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、細胞の染色体にそれぞれのmut遺伝子を1コピーまたは複数コピー追加して導入することによって、あるいは対応する野生型細胞と比較して、細胞内におけるそれぞれのMutタンパク質のより高い産生がもたらされるように、天然の染色体に位置するmut遺伝子の転写を指示する調節ユニットに1つまたは複数の変異を導入することによって実現することができる。
【0056】
本発明の好ましい実施形態では、DNAポリメラーゼIVをコードする遺伝子の抗変異原アレルがdinB10である。別の好ましい実施形態では、DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルがdnaE911である。
【0057】
本発明によれば、産生された発現系は、いかなるタンパク質の発現にも用いることができる。本発明のコンテクストにおいて、「タンパク質」という用語は、アミド連結で連結された少なくとも2つのアミノ酸を含む分子である。したがって本発明によれば、「タンパク質」という用語には、ペプチド、例えば、オリゴペプチド、ポリペプチド、またはドメインなどの天然に存在するタンパク質の一部が含まれる。発現されるタンパク質は、そのアミノ酸配列が天然に存在するタンパク質の配列であってもよく、すなわち野生型の配列を有するものでもよい。当然ながら、上記の発現系によって発現されるタンパク質は、野生型タンパク質と比較して、例えば、異なったアミノ酸組成および/または異なった長さによって改変されたアミノ酸配列を有するものでもよい。したがって、発現されるタンパク質は、野生型タンパク質と比較して、1つまたは複数の位置に異なったアミノ酸残基を有するものでもよい。発現されるタンパク質は、野生型タンパク質と比較して、先端切除されていても、延長されていてもよい。さらに、発現されるタンパク質は、対応する野生型タンパク質と異なる特徴を有するものでもよい。これらの異なる特徴は、改変された熱安定性、異なった基質特異性、異なった活性、改変された触媒部位、または新規の触媒部位などに関するものでよいが、これらに限定されない。発現されるタンパク質は、2つ以上の個別のポリペプチドを含む融合タンパク質でも、第2のポリペプチドの1つまたは複数のドメインを追加して含む融合タンパク質でもよい。タンパク質のそのような改変または変異は、当技術分野で知られているタンパク質をコードするヌクレオチド配列を適切に操作し、その後、このタンパク質をコードするヌクレオチド配列を、自然突然変異率が低下している宿主細胞のゲノムに挿入するか、あるいはベクターに挿入して、その後、それをその宿主細胞に導入することによって得ることができる。本発明の好ましい実施形態では、発現されるタンパク質が組換え体タンパク質、すなわち遺伝工学的手法および/またはDNA組換え技法によって改変されたタンパク質である。
【0058】
本発明の好ましい実施形態では、発現されるタンパク質が、天然化合物および非天然化合物の工業生産に利用できる酵素であってもよい。酵素、または酵素の補助によって生産それらの化合物は、薬物、化粧品、食品などの生産に用いることができる。発現されるタンパク質は、ヒトおよび動物の健康に関する分野で治療応用を有する物質でもよい。医学的に重要なタンパク質の重要なクラスには、例えば、サイトカインおよび成長因子が含まれる。
【0059】
本発明の方法によれば、タンパク質をコードする核酸配列を、1つまたは複数の調節ユニットの機能的制御下に、ゲノムまたはベクターのいずれかに挿入する。遺伝子および遺伝子産物の発現をそれぞれ制御および指示する調節ユニットには、プロモーター、リボゾーム結合部位、エンハンサー、サイレンサー、ポリアデニル化部位、および/または3’転写終結区が含まれるが、これらに限定されない。調節ユニットは、宿主細胞の所与の区画、または細胞の外に向けたタンパク質の発現を指示するリーダー配列またはシグナル配列を含んでもよい。そのような調節ユニットの使用は、使用される宿主細胞のタイプに依存する。所与の原核宿主細胞または真核宿主細胞で使用できる調節ユニットは、周知である(例えば、Sambrookら、「Molecular cloning:A Laboratory Handbook」、第2版(1989年)、Cold Spring Harbor Laboratory Press社、NY,USAを参照のこと)。
【0060】
好ましい実施形態では、タンパク質をコードするヌクレオチド配列をベクターにクローニングする。プラスミド、バクテリオファージ、ウイルス、またはコスミドをベクターとして使用するのが好ましい。当業者は、タンパク質をコードするヌクレオチド配列を所与の宿主細胞に導入するのに使用できる、真核宿主細胞用または原核宿主細胞用の適当なベクターを多数知っている。
【0061】
さらに、当業者は、調節ユニットに機能的に連結された、タンパク質をコードするヌクレオチド配列をクローニングする方法、およびヌクレオチド配列を宿主細胞に導入する方法を多数知っている(例えば、Sambrookら、1989年を参照)。
【0062】
本発明のさらに別の態様は、タンパク質の産生する方法に関し、上記タンパク質のアミノ酸配列は、自然発生した突然変異に対して安定化されており、この方法は、
a) 少なくとも2つの変異を含有するタンパク質をコードする核酸配列を、上記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、宿主細胞のゲノムに挿入する工程であって、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる工程、または
b) 上記タンパク質をコードする核酸配列を、上記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下にベクターに挿入し、さらに、少なくとも2つの変異を含有するベクターを宿主細胞内に移入する工程であって、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる工程と、
c) 適当な培地中で、タンパク質の発現を可能にする条件下に宿主細胞を培養する工程と、
d) 発現された上記タンパク質を単離工程と
を含む。
【0063】
上記タンパク質をコードする核酸配列がリーダー配列またはシグナル配列に機能的に連結されているかどうかに応じて、発現されたタンパク質は、特定の細胞小器官、特定の細胞内区画、細胞外間隙、または細胞が培養されている培地中に輸送される。したがって、タンパク質は、細胞内に蓄積されるか、あるいは細胞から分泌されるかのいずれかであり得る。タンパク質を産生する本発明の方法の好ましい実施形態では、それゆえ、タンパク質が培地から単離される。タンパク質を産生する本発明の方法の別の好ましい実施形態では、タンパク質を宿主細胞から、例えば、特定の細胞小器官から抽出する。また、当業者は、細胞、細胞小器官、または培地からタンパク質を単離する多数のプロトコールを知っている。
【0064】
本発明によれば、いかなる真核細胞も、あるいは原核細胞も、タンパク質を発現または産生するための宿主細胞として用いることができる。特に好ましい例には、真菌細胞、動物細胞、植物細胞、古細菌、ラン細菌、グラム陽性菌、およびグラム陰性菌が含まれる。
【0065】
本発明は、タンパク質を産生する本発明の方法によって取得可能なタンパク質にも関する。
【0066】
本発明の別の態様は、発酵産物、および/または培地中での発酵産物の生成に関与する少なくとも1つの酵素を産生する細胞を培養することによって発酵産物を産生する方法に関し、上記細胞のゲノムは、少なくとも2つの変異によって自然発生の配列変化に対して安定化されており、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる。
【0067】
したがって、本発明の方法は、細胞の、発酵を目的とした使用、すなわち発酵産物を生産するための使用を対象とし、その際、上記細胞のゲノムは、自然発生の配列変化に対して安定化されており、それによって、非常に低い自然突然変異率を示す。さらに、使用される細胞は、改善された細胞生存率を有する。自然突然変異率が低下している細胞を使用することによって、本発明の方法は、所与のタンパク質をコードするヌクレオチド配列が変異によって変化し、それによって、そのタンパク質のアミノ酸配列が変化する確率が大幅に低下するのを確実にする。これらの細胞は、細胞生存率の大幅な増強を示すので、これらの細胞の使用は、さらに、発酵工程中に撹乱が起こらないこと、および発酵産物の高い生産性が得られることを確実にする。
【0068】
本発明のコンテクストにおいて、「発酵」という用語には、微生物、真菌細胞、植物細胞、または動物細胞の嫌気的または好気的代謝によって、あるいはそのような細胞からの酵素を使用することによって特定の産物を産生するいかなる過程も含まれ、上記酵素は、単離、精製、および/または固定することができる。「発酵」には、微生物細胞、植物細胞、真菌細胞、および/または動物細胞の1つまたは複数の酵素の作用によって引き起こされる、有機基質のあらゆる酵素的化学的変成が含まれる。所望の発酵産物は、細胞それ自体の中で、例えば、タンパク質の発現によって、または細胞の代謝の結果として産生されることがあり、それによって、発酵産物が細胞内に蓄積されることもあれば、細胞から環境中に輸送されることもあり、あるいは細胞によって1つまたは複数の酵素が産生され、それが、細胞外に分泌された際に、培地中に存在する基質から、所望の発酵産物への変換を触媒する。発酵産物が核酸、または核酸によってコードされたタンパク質である場合には、本発明の方法は、変異による発酵産物の最小限の汚染さえも回避し、それによって、発酵産物の長期的な完全性を保障するのに有用である。発酵産物の産生が、細胞によって産生された1つまたは複数の酵素の作用による場合には、本発明の方法は、それぞれの酵素の完全性を維持し、それによって、発酵産物の産生に導く代謝過程の特異性および有効性を維持するのに有用である。
【0069】
発酵産物を産生する本発明の方法は、ある経路に関与する遺伝子クラスター、および/またはそのような遺伝子クラスターによってコードされた遺伝子産物の完全性を維持し、それによって、そのような遺伝子クラスターによってコードされた遺伝子産物が関与する経路の特異性および有効性を維持するのにも有用であり得る。そのような遺伝子クラスターの例には、ポリケチドシンターゼ(PKS)クラスターが含まれるが、これに限定されない。ポリケチド代謝産物は、放線菌および粘液細菌などの細菌ならびに真菌によって産生される、様々な構造および生物活性を有する天然物の大きなグループである。複合ポリケチドは、動物の長鎖脂肪酸合成に類似した機構を用いた多機能性PKSによって産生される。サッカロポリスポラエリスラエア(Saccharopolyspora erythraea)のエリスロマイシンPKSの研究は、モジュール構成を明らかにした。複合ポリケチド合成は、前進性反応機序に従い、PKS内の各モジュールは、ほぼ直線的な順序の反応を触媒する、ひと続きになった3つから6つの酵素ドメインを有する。
【0070】
本発明によれば、発酵を目的として使用される細胞における自然突然変異率の低下、および高い細胞生存率が、少なくとも2つの変異を使用することによって得られ、上記細胞内に存在するいかなる核酸に自然発生した突然変異も修復する少なくとも2つの細胞性DNA修復機構の能力の強化に、上記少なくとも2つの変異の複合作用によって導かれる。詳細には、これらの少なくとも2つの変異は、上記細胞の自然発生した突然変異を修復するミスマッチ修復系、プルーフリーディング機能、および/またはSOS修復系の能力を強化する。本発明の好ましい実施形態では、これらの変異が、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから選択される。
【0071】
本発明の一実施形態において、MutLまたはその相同タンパク質の発現の上方制御、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、発酵に使用される細胞にベクターを導入することによって実現でき、上記ベクターは、それぞれ、mutL遺伝子またはその相同タンパク質をコードする遺伝子、およびmutS遺伝子またはその相同タンパク質をコードする遺伝子を、対応する野生型宿主細胞との比較における、それぞれのmutタンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に含む。MutLまたはMutSいずれかのタンパク質の過剰発現に用いるベクターは、1つまたは複数の調節ユニットの機能的制御下にそれぞれのmut遺伝子を含む多コピープラスミドであることが好ましい。本発明の好ましい実施形態では、それぞれのmut遺伝子の過剰発現を制御する調節ユニットは誘導性プロモーターでも、構成的プロモーターでもよい。
【0072】
本発明の別の実施形態では、MutLまたはその相同タンパク質の発現の上方制御、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、細胞の染色体にそれぞれのmut遺伝子を1コピーまたは複数コピー追加して導入することによって、あるいは対応する野生型細胞と比較して、細胞内におけるそれぞれのMutタンパク質のより高い産生がもたらされるように、天然の染色体に位置するmut遺伝子の転写を指示する調節ユニットに1つまたは複数の変異を導入することによって実現することができる。
【0073】
本発明の方法の好ましい実施形態では、DNAポリメラーゼIVをコードする遺伝子の抗変異原アレルがdinB10である。別の好ましい実施形態では、DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルがdnaE911である。
【0074】
本発明によれば、発酵を目的として使用する細胞の中にdinB10およびdnaE911が存在すると、その細胞の自然突然変異の頻度が、対応する野生型宿主細胞と比較して少なくとも10分の1に低下する。mutLタンパク質をさらに過剰発現する細胞の中にdinB10およびdnaE911が存在すると、その細胞の自然突然変異の頻度が、野生型宿主細胞と比較して少なくとも50分の1に低下する。
【0075】
本発明の方法によって得られる発酵産物は、細胞の一次代謝物である場合も、二次代謝物である場合もある。一次代謝物は、生細胞によって合成され、かつ細胞構造の形成および代謝過程を維持するために、細胞に必ず必要とされる物質である。一次代謝物と対照的に、二次代謝物は、細胞によって合成され、細胞の機能の維持には必ずしも必要でない化合物、特に低分子化合物である。通常、二次代謝物は、特定の環境における選択有利性をその細胞または生物に与えることができる。本発明の方法によって得られる発酵産物は、古典的な意味での発酵産物、すなわちアルコール発酵、乳酸発酵、プロピオン酸発酵等の産物など、もっぱら微生物によって行われる、酸素を用いた糖質の酵素的分解である場合もある。
【0076】
発酵産物の例には、核酸、ヌクレオシド、ヌクレオチド、タンパク質、アミノ酸、有機酸、アルコール、糖質、ビタミン、抗生物質、およびアルカロイドが含まれるが、これらに限定されない。
【0077】
本発明の好ましい実施形態では、発酵産物が核酸であり、詳細には、治療上の有用性がある核酸、例えばワクチンとして使用されることになっている核酸である。
【0078】
本発明のコンテクストにおいて、「核酸」は、ホスホジエステル結合によって連結された少なくとも2つのヌクレオチドを含む分子である。したがって、「核酸」という用語は、オリゴヌクレオチドも意味する。核酸は、DNAまたはRNAのいずれかであり得る。この核酸は、一本鎖でも、二本鎖でもよい。
【0079】
本発明の好ましい実施形態では、発酵産物がタンパク質であり、詳細には、治療上の有用性がある酵素もしくはタンパク質、またはドメインなど、それらの部分である。好ましい酵素の例。好ましい酵素の例には、プロテアーゼ、アミラーゼ、ペクチナーゼ、およびグルコースイソメラーゼが含まれるが、これらに限定されない。治療上の有用性があるタンパク質の好ましい例には、インターフェロン、インターロイキンなどのサイトカイン、成長因子、血液凝固因子、抗体などが含まれるが、これらに限定されない。
【0080】
有機酸の好ましい例には、クエン酸、乳酸、または酢酸が含まれるが、これらに限定されない。アルコールの好ましい例には、エタノール、プロパノール、およびブタノールが含まれるが、これらに限定されない。糖質の好ましい例には、ショ糖、マルトース、もしくはパラチノーゼなどの糖、キシリトールもしくはマルチトールなどの糖アルコールが含まれるが、これらに限定されない。ビタミンの好ましい例には、ビタミンB12もしくはリボフラビンが含まれるが、これらに限定されない。抗生物質の好ましい例には、ペニシリン、セファロスポリン、ストレプトマイシン、エリスロマイシンなどのポリケチド抗生物質が含まれるが、これらに限定されない。
【0081】
発酵産物は、発酵細胞、または培養に使用された培地のいずれかから単離することができる。
【0082】
したがって、本発明は、発酵産物を産生する本発明の方法によって取得可能な発酵産物にも関する。
【0083】
発酵産物を産生する本発明の方法では、真核細胞および原核細胞の両方が使用可能である。発酵産物を産生する本発明の方法の好ましい実施形態では、使用される真核細胞が、真菌細胞、動物細胞、または植物細胞である。発酵産物を産生する本発明の方法で使用される原核細胞は、ラン細菌、古細菌、グラム陽性菌、またはグラム陰性細菌であることが好ましい。
【0084】
当然ながら、対応する野生型宿主細胞と比較して、自然突然変異率が、好ましくは大幅に、低下しているいかなる細胞も、発酵工程を産生する本発明の方法で使用することが可能である。詳細には、自然突然変異率を低下させる本発明の方法によってその自然突然変異の頻度が低下している細胞または生物、あるいは自然突然変異の頻度が低下している細胞または生物を産生する本発明の方法によって産生された細胞または生物の使用が好ましい。
【0085】
本発明の方法の好ましい実施形態では、適当な液体培地中で細胞を培養する。当業者は、発酵産物を産生するために、細胞、例えば原核細胞または真核細胞を、その中で培養できる多数の液体培地を知っている。本発明によれば、細胞を連続培養またはバッチ培養のいずれかで培養することができる。バッチ培養は、細胞を播種された液体培地から始まり、培養中、培地の交換が連続的に行われない培養であって、場合によっては、酸素などの気体のみが導入される。産物生成の異化抑制を回避するために、バッチ培養を流加培養で行ってもよい。流加培養の場合には、したがって、所与の糖など、基質の消費がその基質の追加分量を配送することによって補填される。連続培養は、1回だけ細胞で播種され、その後、細胞量の増大または産物の蓄積に応じて、消費済みの培養培地を新たな培地で連続的に置換し、それによって、消費済みの培地と共に、場合によってはさらに細胞と共に、発酵産物を培養から同時に取り出す培養である。連続培養は発酵槽で行うことができる。
【0086】
本発明の好ましい実施形態では、細胞が、固定された形態で培養中に存在する場合がある。本発明の方法の別の実施形態では、細胞を固体または半固体の培地上で培養する。
【0087】
好ましい実施形態では、本発明は、生物、詳細には原核生物、最も好ましくは大腸菌株に関し、これは、
プラスミドpmutLを含有する大腸菌MG1655 dinB10(DSM17016)、
プラスミドpmutLを含有する大腸菌MG1655 dinB10 mutL::tet(DSM17017)、
プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm(DSM17018)、
プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm mutL::tet(DSM17019)、
大腸菌MG1655 dinB10 dnaE zae::cm(DSM17015)、
大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet(DSM17014)、
プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm(DSM17020)、および
プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet(DSM17021)
からなる群から選択される。
【0088】
上記に特定された微生物は、ブダペスト条約に従って、2005年1月3日に、上記に特定したDSM番号の下に、DSMZ(独国、Braunschweig所在の独国微生物細胞培養収集館(Deutsche Sammlung fur Mikroorganismen und Zellkulturen GmbH))に寄託した。
【0089】
本発明を、以下の配列表、図、および実施例によって例示する。
【0090】
配列番号1および2は、それぞれ、MutLタンパク質をコードするDNA断片を増幅するためのプライマーMutLNcoIhinおよびMutLXhoIherの配列を示す。
【0091】
配列番号3および4は、それぞれ、MutSタンパク質をコードするDNA断片を増幅するためのプライマーMutSBam−HIhinおよびMutSXhoIherの配列を示す。
【0092】
配列番号5から配列番号10は、それぞれ、DapAタンパク質をコードするDNA断片を増幅するためのプライマーdapABamHIIF、dapABamH1+1、dapABamH1+2、dapAXhoI、dapAEcoRIhin、およびdapAHindIIIherの配列を示す。
【0093】
配列番号11および12は、それぞれ、クロラムフェニコール耐性をコードするDNA断片を増幅するためのプライマーcmBbvCIhinおよびcmAhdIherの配列を示す。
【実施例】
【0094】
実施例1
(様々な大腸菌株のリファンピシン耐性に対する自然突然変異の頻度(SMF)の測定)
通常、細菌である大腸菌(Escherichia coli)の細胞は、抗生物質リファンピシンに対して感受性であって、すなわちそれらは、この抗生物質を含有する培地で増殖できない。しかし、リファンピシンに対する耐性を与える変異は、大腸菌ゲノムで低頻度で自然に発生する。したがって、リファンピシンを含有する培地上に、多数の細胞(108)をプレーティングした場合、ほとんどの細胞は抗生物質によって殺されるであろうが、いくつかのコロニーが成長するであろう。これは、リファンピシンに対する耐性を与える変異が、細胞ゲノム中で自然に発生するために可能である。それらの変異は、その次に、元の変異体の子孫によって引き継がれる。この変異は、リファンピシンが存在する環境においてのみ有益であり、おそらく、他のほとんどの条件下においては、それは、その変種に何の利点も与えないことに留意するのが重要である。リファンピシンは、主として、結核およびライ病、および場合によっては他の疾患を治療するのに使用される抗生物質である。耐性は、膜透過性の変化、またはmRNAポリメラーゼをコードするrpoB遺伝子における変異によるものである。これらの機構は両方とも、染色体突然変異を介して起動する。しかし、大腸菌の染色体には、リファンピシンに対する細菌の耐性を与え得るいくつかの標的遺伝子が存在する。
【0095】
自然突然変異の頻度(SMF)を計算するために、リファンピシンに対する耐性を与える自然突然変異の頻度を、培養中の生存細胞の総数で割って求めた。
【0096】
【数1】

【0097】
A) 強度の変異誘発性表現型(mutLおよび/またはdnaQの染色体欠失)を示す大腸菌株におけるMutLの過剰発現および抗生物質リファンピシン上への該細胞のプレーティング
(実験の概要)
(1.MutLおよびMutSの過剰発現プラスミドの構築)
a) pTrcHis2/lacZへのmutLのクローニング
このプラスミドは、大腸菌におけるMutLの高レベルで、かつ「制御された」発現を実現するために構築した。
【0098】
大腸菌株MG1655から調製されたゲノムDNAを使用して、プライマー1(配列番号1)および2(配列番号2)(表1も参照)によって、MutLタンパク質をコードする1848bpのDNA断片を増幅した。このPCR断片を消化し、NcoI−XhoI断片として、プラスミドpTrcHis2/lacZにクローニングして、プラスミドpmutLを産生した。プラスミドpmutLの物理構造を図1に示す。PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.50;72/2.5)35(72/10);(Taq/Pfu)の比率(5/1)であった。
【0099】
得られたクローンのうち6クローンを単離し、EcoRVによる制限消化によって分析した(予測された断片:871bpおよび5373bp)。6クローンすべてが正しい移動パターンを示した。
【0100】
b) pTrcHis2/lacZへのmutSのクローニング
このプラスミドは、大腸菌におけるMutSの高レベルで、かつ「制御された」発現を実現するために構築した。
【0101】
大腸菌株MG1655から調製されたゲノムDNAを使用して、プライマー3(配列番号3)および4(配列番号4)(表1を参照)によって、MutSタンパク質をコードする2562bpのDNA断片を増幅した。このPCR断片を消化し、BamHI−XhoI断片として、プラスミドpTrcHis2/lacZにクローニングして、プラスミドpmutSを産生させた。プラスミドpmutSは、図1に示すプラスミドpmutLと原則的に同じ物理構造を有する。
【0102】
PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.58;72/3)35(72/10);Herculaseであった。
【0103】
得られたクローンのうち6クローンを単離し、EcoRVによる制限消化によって分析した(予測された断片:1186bpおよび5775bp)。6クローンすべてが正しい移動パターンを示した。
【0104】
【表1】
【0105】
(2.ウェスタンブロット分析)
a) MutLの検出
ES568(mutL13;Mut−) pmutLの終夜培養物20μlを0.4%グルコースおよびアンピシリン(100μg/m)を含有するLB10mlに播種し、ODが約0.5に達するまで培養した。グルコースを除去するために、細胞を遠心分離し、必要な抗生物質を含有するLB5mlに懸濁した。最終濃度1mMのIPTGを添加することによって、PlacからのpmutLの発現を誘導した。この培養物を37℃で16時間インキュベートし、その後、遠心分離し、SDS−PAGE分析用に調製した。ウェスタンブロット分析は、AP結合抗His−tac抗体を用いて行った(データは示されていない)。
【0106】
b) MutSの検出
構築されたプラスミドpmutSで、菌株Top10の野生型細胞を形質転換し、得られた形質転換体を精製した。これらの細菌の終夜培養物20μlを、アンピシリン(100μg/ml)および0.4%グルコースを含有するLB10mlに播種し、ODが約0.5に達するまで培養した。グルコースを除去するために、細胞を遠心分離し、必要な抗生物質を含有するLB5mlに懸濁した。最終濃度1mMのIPTGを添加することによって、PlacからのpmutSの発現を誘導した。この培養物を37℃で16時間インキュベートし、その後、遠心分離し、SDS−PAGE分析用に調製した。ウェスタンブロット分析は、AP結合抗His−tac抗体を用いて行った(データは示されていない)。
【0107】
(3.増殖研究)
菌株ES568(mutL13;Mut−)、MG1655 dnaQ::kn(Mut−)、および野生型株MG1655(Mut+)を、プラスミドpmutLまたは親プラスミドpTrcHis2/lacZで形質転換した。アンピシリン100μg/mlを含有するLB寒天プレート上で、得られた単一コロニーを精製し、その後、アンピシリン100μg/mlを含有するLBに播種し、37℃で終夜培養した。
【0108】
これらの細菌の終夜培養物20μlを、0.4%グルコースと、必要な抗生物質(アンピシリン100μg/ml;カナマイシン50μg/ml)とを含有するLB5mlに播種し、ODが約0.5に達するまで培養した。グルコースを除去するために、細胞を遠心分離し、対応する抗生物質を含有するLB5mlに懸濁した。最終濃度1mMのIPTGを添加することによって、PlacからのpmutLの発現を誘導した。この培養物を、37℃で終夜インキュベートし、細胞を遠心分離し、培地容積を1/10(v/v)にまで減少させた。
【0109】
最後に、培養物を希釈して、100μg/mlアンピシリン、または100μg/mlアンピシリン+100μg/mlリファンピシンを含有する寒天プレート上にプレーティングし、30℃で3日間インキュベートした。
【0110】
ウェスタンブロット分析によって、MutLの発現を確認した。
【0111】
要約および結論:(mutL+バックグランドにおける)MutLの過剰発現によって、自然突然変異の頻度が約20分の1に低下することを観測した。高い突然変異率を示すmutL−細胞またはdnaQ−細胞でも、同様の効果が観測された(データは示されていない)。
【0112】
B) 様々な組合せのdinB欠失および/またはdnaE911抗変異原アレルを保持する大腸菌株におけるMutLの過剰発現
(1.菌株の構築)
a) 大腸菌株MG1655およびMG1655dinB10の細菌細胞を感染させるために、大腸菌株ES1484(mutL218::Tn10)からP1vir溶解物を調製した。
【0113】
(P1virの調製)
大腸菌株ES1484(mutL218::tet)の終夜培養物のアリコートを、CaCl2(5mM)を含有する37℃のLBに播種した。指数関数的増殖期に、(野生型株MG1655の)P1virを添加した。感染した培養物を37℃で約4h、細胞溶解が観察されるまでインキュベートした。CHCl3を添加し、溶解物を遠心分離し、CHCl3を含有する新しいチューブに上清を移して、4℃で保存した。
【0114】
(P1形質導入)
宿主株MG1655およびMG1655 dinB10の終夜培養物のアリコートをLBに播種し、指数関数的増殖期に達するまで37℃で培養した。細胞を遠心分離し、CaCl2(cE=5mM)を含有するMgSO4(cE=10mM)に再懸濁し、調製されたP1vir溶解物を添加し、細胞混合物をRTで30分間インキュベートした。(ファージ感染を停止させるために)クエン酸ナトリウムを添加し、栄養源としてSOCを添加した。細胞を37℃で1hインキュベートした後、テトラサイクリン(12.5μg/ml)を含有する寒天プレート上にアリコートをプレーティングした。得られた単一コロニーを精製し、試験し、単離された株を保存した。
【0115】
b) 大腸菌株MG1655、MG1655 dinB10、MG1655 mutL::tet、およびMG1655 dinB10 mutL::tetの細菌細胞を感染させるために、dnaE株NR10171(dnaE911 zae::cm)から、P1vir溶解物を調製した。得られた単一コロニーを精製し、試験し、単離された株を保存した。
【0116】
(2.増殖研究)
細菌株MG1655、MG1655 dinB10、MG1655 dnaE911 zae::cm、MG1655 dinB10 dnaE911 zae::cm、MG1655 mutL::tet、MG1655 dinB10 mutL::tet、MG1655 dinB10 dnaE911 zae::cm mutL::tet、およびMG1655 dinB10 dnaE911 zae::cm mutL::tetを、プラスミドpTrcHis2AまたはpmutLで形質転換した。得られた単一コロニーを精製し、100μg/mlアンピシリンを含有するLBに播種し、30℃で終夜培養した。
【0117】
これらの細菌の終夜培養物20μlを、100μg/mlアンピシリンを含有する30℃のLB5mlに播種した。指数関数的増殖期に、PlacからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加した。この培養物を1時間、30℃に維持し、その後、温度を37℃に変化させ、細胞を37℃で終夜インキュベートした。
【0118】
細胞を遠心分離し、培地の容積を1/10(v/v)にまで減少させた。
【0119】
最後に、培養物を希釈して、100μg/mlアンピシリン、または100μg/mlアンピシリン+100μg/mlリファンピシンを含有する寒天プレート上にプレーティングし、30℃で3日間インキュベートした。
【0120】
リファンピシン耐性への自然突然変異の頻度(SMF)の値を表2に示す。図2はMutLの発現による変異性の抑制を示す。
【0121】
【表2】
【0122】
dinB10(欠失)またはdnaE911(アレル)などの抗変異原の作用を介して、変異性(SMF値)がそれぞれ4または3分の1に低下する。さらに、それらの株でMutLを発現させることによって、変異性がさらに低下する(2から7分の1)ことを観測した。dinB10およびdnaE911の両方の抗変異原を保持する細菌株は、野生型株MG1655と比較して、変異性の10分の1の低下を示す。MutLを追加して発現させても、変異性に変化は見られない。
【0123】
mutL遺伝子の染色体欠失は、宿主株の変異性を、強烈に約103倍に増大させる。この変異性は、抗変異原dinB10(5分の1)、dnaE911(3分の1)、dinB10およびdnaE911(10分の1)を介して低下した。MutLを追加して誘導することによって、変異性が50分の1にまで(MG1655 mutL::tetでは50分の1、MG1655 dinB10 mutL::tetでは22分の1、MG1655 dnaE911 zae::cm mutL::tetでは20分の1、そして、MG1655 dinB10 dnaE911 zae::cm mutL::tetでは11分の1)低下する。
【0124】
要約および結論:抗変異原dinB10およびdnaE911は、自然突然変異の頻度を3から4分の1で低下させる。これらの菌株でMutLを発現させることによって、この頻度がさらに2から7分の1で低下した。
【0125】
C) 様々な組合せのdinB欠失および/またはdnaE911抗変異原を保持するmutS−株の変異性
前述の結果を確認するために、dinB10(ポリメラーゼIVの欠失)および/またはdnaE911(ポリメラーゼIIIのαサブユニットのコード配列内のアレル)の抗変異原効果をmutS−バックグランドで試験した。
【0126】
(1.菌株の構築)
大腸菌株MG1655、MG1655 dinB10、MG1655 dnaE911 zae::cm、およびMG1655 dinB10 dnaE911 zae::cmの細菌細胞を感染させるために、P1vir溶解物を大腸菌株ES1481(mutL215::Tn10)から調製した。得られた単一コロニーを精製し、試験し、その菌株を保存した。
【0127】
(2.増殖研究)
これらの細菌の終夜培養物10μlを、12:5μg/mlテトラサイクリンを含有する30℃のLB10mlに播種した。指数関数的増殖期(8hの増殖の後)に、温度を37℃に変化させ、細胞を37℃で終夜インキュベートした。
【0128】
細胞を遠心分離し、培地の容積を1/10(v/v)にまで減少させた。
【0129】
最後に、培養物を希釈して、リファンピシン(100μg/ml)の存在下および非存在下に寒天プレートにプレーティングし、30℃で5日間インキュベートした。
【0130】
リファンピシン耐性への自然突然変異の頻度(SMF)の値を表3に示す。図3は、mutS−バックグランドにおける変異性の抑制を示す。
【0131】
【表3】
【0132】
dinB10またはdnaE911などの抗変異原の存在下では、野生型と比較して、変異性(SMF値)が低下している(約1.5分の1)。さらに、抗変異原dinB10およびdnaE911を保持する細菌株は、野生型MG1655と比較して、変異性の4.5分の1の低下を示す。
【0133】
mutS遺伝子の染色体欠失は、宿主株の変異性を劇的に増大させる。この変異性は、抗変異原dinB10(12.5分の1)、dnaE911(10分の1)、dinB10およびdnaE911(60分の1)によって低下する。
【0134】
結論および要約:dinB10またはdnaE911によって引き起こされた抗変異原効果は、mutSの染色体欠失によって引き起こされたMut−表現型を部分的に補完する。
【0135】
実施例2
(dapA変異の復帰変異)
特定の標的遺伝子における点変異およびフレームシフト変異の復帰変異を研究するために、大腸菌において栄養要求変異株表現型を有する系(dapA−株)を用いた。
【0136】
dapA遺伝子はジヒドロジピコリン酸シンターゼをコードし、この酵素はピンポン機構を介してL−アスパラギン酸−β−セミアルデヒドおよびピルビン酸からジヒドロピコリン酸への縮合を触媒するが、その機構では、ピルビン酸が、リジン残基とシッフ塩基を形成することによって、この酵素に結合する。機能しない遺伝子産物に導くdapA遺伝子変異を有する細菌細胞は、DL−ジアミノピメリン酸(DAP)を含有しない培地では増殖できない。
【0137】
dapA−バックグランド(dapA15(DapA−表現型を有する点突然変異)またはdapA::kn)において、dapAIF(wt ORF)、dapA+1(フレームシフト変異)、dapA+2、またはdapA15(点突然変異)をコードするプラスミドの存在下に、MutLを発現させる。復帰変異体は、DL−ジアミノピメリン酸を欠失したプレート上で検出する。
【0138】
(実験の概要)
(1.プラスミドの構築)
a) dapAのクローニング
(pTrcHis2/lacZへのdapA(dapA15)、dapA+1(dapA15+1)、dapA+2(dapA15+2)のクローニング)
大腸菌株MG1655(wt)またはAT997(dapA15)から調製されたゲノムDNAを使用し、プライマー5(配列番号5)、6(配列番号6)、7(配列番号7)、および8(配列番号8)(表4を参照)によって、DapAタンパク質をコードする732bpのDNA断片を増幅した。これらのPCR断片を消化し、BamHI−XhoI断片として、プラスミドpTrcHis2/lacZにクローニングして、プラスミドpdapAIF、pdapA+1、およびpdapA+2を産生させた。プラスミドpdapAIFの物理構造を図4Aに示す。PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.50;72/2)35(72/10);Herculaseであった。
【0139】
それぞれの場合について、得られたクローンのうち8クローンをBstEIIによる制限消化によって分析した(予測された断片:1314bpおよび3964bp)。試験された合計24クローンのうち23クローンが正しい移動パターンを示した。
【0140】
(pSU19およびpSU40へのdapA15のクローニング)
大腸菌株AT997から調製されたゲノムDNAを使用して、プライマー9(配列番号9)および10(配列番号10)(表4も参照)によって、DapAタンパク質をコードする732bpのDNA断片を増幅した。このPCR断片を消化し、HindIII−EcoRI断片として、プラスミドpSU19およびpSU40にクローニングして、プラスミドpSU19dapA15およびpSU40dapA15を産生させた。プラスミドpSU40dapAの物理構造を図4Bに示す。
【0141】
PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.50;72/2)35(72/10);(Taq/Pfu)の比率(5/1)であった。
【0142】
得られたクローンのうち10クローンを単離し、NheI(予測された断片:pSU40では1122bpおよび2557bp)またはApoI(予測された断片:478bpおよび3192bp)による制限消化によって分析した。すべてのクローンが正しい移動パターンを示した。
【0143】
2) mutLのクローニング
a) pSU40へのmutLのサブクローニング
プラスミドpmutLをSpHI−XmnI消化し、3629bpの断片をゲル電気泳動によって単離した。プラスミドpSU40をSphI−HincII消化し、2680bpの断片をゲル電気泳動によって単離した。精製された2つの断片を粘着端−平滑端連結して、プラスミドpSU40mutLを産生させた。プラスミドpSU40mutLを産生するストラテジーを図5に示す。
【0144】
得られたクローンのうち6クローンを、NcoI(予測された断片:2675bpおよび3634bp)、PvuI(予測された断片:1368bpおよび4941bp)、およびBglI(予測された断片:1828bpおよび4481bp)による制限消化によって分析した。試験された6クローンのうち3クローンが正しい移動パターンを示した。
【0145】
b) プラスミドpmutLspec: specによるblaの置換
(プラスミドpmutLにおける、スペクトマイシン耐性遺伝子によるアンピシリン耐性遺伝子の置換)
プラスミドpIC156から調製されたDNAをFspI−XmnI消化し、ゲル電気泳動を用いて1304bpの断片を単離し、プラスミドpmutLから調製されたDNAをScaI−XmnI消化し、ゲル電気泳動を用いて5443bpの断片を単離した。精製された2つの断片を平滑端−平滑端連結して、プラスミドmutLspecを産生させた。
【0146】
得られたクローンのうち12クローンをEcoRI(予測された断片:1291bpおよび5456bp)による制限消化によって分析した。試験された12クローンのうち2クローンが正しいサイズを示した。
【0147】
【表4】
【0148】
(2.ウェスタンブロット分析)
a) DapA/DapA15の検出
菌株dapA::knを、pdapAIF、pdapA+1、pdapA+2、pdapA15IF、pdapA15+1、およびpdapA15+2で形質転換し、観察された形質転換体を精製した。
【0149】
これらの細菌の終夜培養物20μlを、0.4%グルコース、DL−ジアミノピメリン酸(100μg/ml)、アンピシリン(100μg/ml)、およびカナマイシン(50μg/ml)を含有するLB10mlに播種し、ODが約0.5に達するまで培養した。グルコースを除去するために、細胞の一部を遠心分離し、必要な抗生物質を含有するLB5mlに懸濁した。最終濃度1mMのIPTGを添加することによって、Placからの様々なpTrcHis2/lacZ派生物の発現を誘導した。この培養物を37℃で16時間インキュベートし、その後、遠心分離し、SDS−PAGE分析用に調製した。ウェスタンブロット分析は、AP結合抗His−tac抗体を用いて行った(データは示されていない)。
【0150】
b) MutLの検出
野生型株Top10をプラスミドpmutLspecで形質転換し、得られた形質転換体を精製した。これらの細菌の終夜培養物20μlを、スペクトマイシン(75μg/ml)を含有するLB10mlに播種し、ODが約0.5に達するまで培養した。この培養物を5mlアリコートに分割し、プロモーターPlacからのMutLの発現を誘導するために、最終濃度1mMのIPTGを1回、添加した。この培養物を37℃で4時間インキュベートし、その後、遠心分離し、SDS−PAGE分析用に調製した。MutLの発現を証明するために、AP結合抗His−tac抗体を用いてウェスタンブロット分析を行った(データは示されていない)。
【0151】
(3.菌株の構築)
大腸菌株MG1655、MG1655 dinB10、MG1655 dnaE911 zae::cm、MG1655 dinB10 dnaE911 zae::cm、MG1655 mutL::tet、MG1655 dinB10 mutL::tet、MG1655 dnaE911 zae::cm mutL::tet、MG1655 dinB10 dnaE911 zae::cm mutL::tetの細菌細胞を感染させるために、P1vir溶解物を大腸菌株dapA::knから調製した。得られた単一コロニーを精製し、試験し、単離された株を保存した。
【0152】
(4.増殖研究)
a) 点突然変異の復帰変異:
細菌株AT997(dapA15)およびMG1655 dinB10 dapA::knを、プラスミドpTrcHis2/lacZおよびpSU19dapA15、またはプラスミドpmutLおよびpSU19dapA15で形質転換した。得られた単一コロニーを精製し、100μg/mlアンピシリン、30μg/mlクロラムフェニコール、および50μg/ml DAPを含有するLBに播種して、37℃で終夜培養した。
【0153】
b) フレームシフト変異の復帰変異:
細菌株AT997(dapA15;DapA−)を、プラスミドpSU40およびpdapAIF、プラスミドpSU40mutLおよびpdapAIF、プラスミドpSU40およびpdapA+1、プラスミドpSU40mutLおよびpdapA+1、プラスミドpSU40およびpdapA+2、またはプラスミドpSU40mutLおよびpdapA+2で形質転換した。得られた単一コロニーを精製し、100μg/mlアンピシリン、50μg/mlカナマイシン、および50μg/ml DAPを含有するLBに播種して、37℃で終夜培養した。
【0154】
細菌細胞の終夜培養物20μlを、100μg/mlアンピシリン、50μg/mlクロラムフェニコール、および50μg/ml DAPを含有する30℃のLB10mlに播種した。指数関数的増殖期に、PlacプロモーターからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加した。この培養物を1時間、30℃に維持し、その後、温度を37℃に変化させ、細胞を37℃で終夜インキュベートした。
【0155】
最後に、培養物を希釈して、9mlの培地を取り除き、残っている細胞を希釈し、100μg/mlアンピシリン+30μg/mlクロラムフェニコール、または100μg/mlアンピシリン+30μg/mlクロラムフェニコール+50μg/ml DAPを含有する寒天プレート上にプレーティングし、30℃で5日間インキュベートした。
【0156】
野生株およびMutL過剰発現株におけるdapA変異の復帰変異の値を表5に示す。図6は、MutLの発現による変異性の抑制を示す。
【0157】
【表5】
【0158】
対照株(1〜3)であるTop10(wt;DapA+)、AT997(dapA15;DapA−)、およびMG1655dapA::kn(DapA−)は、予測通りのdap−/dap+値、すなわち野生型株では約1(DL−ジアミノピメリン酸非存在下で増殖する)、そして、dapA−株では0(DL−ジアミノピメリン酸非存在下で増殖しない)を示す。
【0159】
機能するdapA遺伝子がプラスミドpdapAIFに存在するので、AT997(dapA15) pdapAIF pTrc(4)、およびAT997(dapA15) pdapAIF pmutL(5)の2株は、予測通りの野生型表現型を示す。dapA−宿主中のプラスミドにコードされているdapAのフレームシフト変異体(6、8)または点突然変異(10)は、DAPの非存在下における細胞増殖を抑制するはずであり、一方、導入された変異の復帰変異(7、11)は、DAPの非存在下における増殖を可能にする。
【0160】
要約および結論:いずれの場合(フレームシフトおよび点突然変異)も、変異からの復帰が、MutLの非存在下でより高い割合で観察された。フレームシフト変異では、dap−/dap+値が1000分の1にまで低下し、点突然変異では10分の1にまで低下した。これは、MutLの発現が、変異からの復帰を抑制し、それによって、染色体における自然突然変異の修復に対抗して作用することを意味する。
【0161】
実施例3
(「発酵状態」中におけるMutLの発現)
トリプトファンによって制御されたG−CSF発現ベクターを保持する産生株JM83 pXX7の変異性を、A)細胞をリファンピシリン上にプレーティングすることによる自然突然変異の発生率の計算、B)細胞をホスホマイシン上にプレーティングすることによる自然突然変異の発生率の計算、およびC)産生プラスミドpXX7(G−CSFをコードする)へのレポータ遺伝子の導入によって分析した。
【0162】
B)に向けて、実験条件下では、ホスホマイシンに対する耐性が大腸菌でより迅速に発生するので、リファンピシリンをホスホマイシンで置換した。ホスホマイシンは、無併発性の下部尿路感染の治療に主として使用される細胞壁阻害剤である。
【0163】
(実験の概要)
(1.クローニング)
a) pXX7へのクロラムフェニコールカセットのクローニング:
プラスミドpSUI9から調製されたDNAを使用して、プライマー11(配列番号11)および12(配列番号12)(表6)によって、クロラムフェニコール耐性をコードするDNA断片を増幅した。これらのPCR断片を消化し、BbvcI−AhdI断片として、プラスミドpXX7にクローニングして、プラスミドpXX7cmを産生させた。プラスミドpXX7cmを産生するストラテジーを図7Aに示す。選択過程を単純化するために、「シャイン−ダルガルノ」配列の−10領域のみをクローニングした。
【0164】
PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.50;72/1)35(72/10);Herculaseである。
【0165】
【表6】
【0166】
b) プラスミドpXX7へのサイレントtetA遺伝子のクローニング:
プラスミドpGBG1の選択カートリッジを単離し、MslI−SmaI断片として、プラスミドpXX7にクローニングして、プラスミドpXX7tetを産生させた(平滑端−平滑端連結)。プラスミドpXX7tetを産生するストラテジーを図7Bに示す。
【0167】
プラスミドpGBG1は、様々なグラム陰性菌における移動性エレメントおよび他の自然突然変異の単離および特性分析を目的として構築された(Schneiderら、Plasmid 44、201−207(2000年))。選択カートリッジは、変異導入の標的を含有し、バクテリオファージλのPRプロモーターの制御下にあるサイレントtetA遺伝子で構成されており、このPRプロモーターはλCIリプレッサーで抑制される。cIを不活性化するか、あるいはCIの結合部位を除去する自然突然変異(点突然変異、欠失、および挿入)はPRを抑制解除し、それによって、tetA発現の引き金となる。変異導入の標的は、約1kbの長さである。この場合、MutLの発現は、変異の発生率を低下させ、それによって、所与の宿主株におけるテトラサイクリン耐性を減少させるはずである。
【0168】
(2.増殖研究)
A) 細胞をリファンピシリン上にプレーティングすることによる大腸菌の自然突然変異の頻度(SMF)の測定
細菌株JM83 pXX7を、プラスミドpTrcHis2/lacZ(対照プラスミド)およびpmutLで形質転換した。得られた単一コロニーを精製し、その細胞を、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有するLBに播種し、30℃で終夜培養した。これらの細菌の終夜培養物3mlを、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有する30℃のRMG100mlに播種した。指数関数的増殖期に、プラスミドpmutLのPlacプロモーターからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加し、さらに、プラスミドpXX7からのG−CSFの発現を誘導するために、最終濃度100μg/mlのトリプトファンを添加した。誘導の1時間後に、播種温度を30℃から37℃に変化させ、その後、細胞を終夜、37℃に維持した。
【0169】
細胞を遠心分離して、培地容積を10mlまで減少させた。
【0170】
最後に、培養物を希釈して、
a) 100μg/mlアンピシリン、および必要に応じて50μg/mlカナマイシン、
b) 100μg/mlアンピシリン、ならびに、必要に応じて50μg/mlカナマイシンおよび100μg/mlリファンピシン
を含有する、A) LBAプレート、B) RMGプレート、およびC) M9プレート上にプレーティングし、30℃で3日間インキュベートした。
【0171】
菌株JM83 pXX7、JM83 pXX7 pTrc、およびJM83 pXX7 pmutLのリファンピシン耐性への自然突然変異の頻度の値を図8に示す。産生株JM83 pXX7は約0.45のSMF値を示す。このSMF値は、産生株に「空」のベクターであるpTrcを追加して導入した場合でも維持される。しかし、さらにpmutLをJM83 pXX7に導入すると、MutLが過剰発現された場合、SMF値の約2分の1の低下が引き起こされる。
【0172】
B) 細胞をホスホマイシン上にプレーティングすることによる大腸菌の自然突然変異の頻度(SMF)の測定
細菌株JM83 pXX7を、プラスミドpTrcHis2AおよびpmutLで形質転換した。得られた単一コロニーを精製し、それぞれの株(JM83 pXX7、JM83 pXX7 pTrcA、およびJM83 pXX7 pmutL)につき20の独立かつ特有の細胞を、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有するLBに播種し、30℃で終夜培養した。
【0173】
これらの細菌の終夜培養物10μlを、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有する30℃のRMG10mlに播種した。指数関数的増殖期に、プラスミドpmutLのPlacからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加し、さらに、プラスミドpXX7のPtrpからのG−CSFの発現を誘導するために、最終濃度100μg/mlのトリプトファンを添加した。誘導の1時間後に、播種温度を30℃から37℃に変化させ、その後、細胞を終夜、37℃に維持した。
【0174】
細胞を遠心分離して、培地容積を1mlにまで減少させた。
【0175】
最後に、培養物を希釈して、
a) 100μg/mlアンピシリン、および必要に応じて50μg/mlカナマイシン、
b) 100μg/mlアンピシリン、ならびに、必要に応じて50μg/mlカナマイシンおよび30μg/mlホスホマイシン
を含有するLB寒天プレート上にプレーティングし、30℃で3日間インキュベートした。
【0176】
菌株JM83 pXX7、JM83 pXX7 pTrc、およびJM83 pXX7 pmutLのホスホマイシン耐性への自然突然変異の頻度の値を図9に示す。産生株JM83 pXX7は、約4.9のSMF値を示す。このSMF値は、産生株に「空」のベクターであるpTrcを追加して導入した場合でも維持される。しかし、さらにpmutLをJM83 pXX7に導入すると、MutLが過剰発現された場合、SMF値の約3分の1の低下が引き起こされる。
【0177】
要約および結論:MutLの過剰発現は、G−CSF産生株の変異性を約3分の1に低下させ、さらに、細胞生存率を103倍にまで増強する。
【0178】
C) レポータ遺伝子の導入による大腸菌株JM83pXX7の自然突然変異の測定
a) G−CSFの発現
b) G−CSFの発現および抑制
G−CSF活性をモニターすることができなかったので、プラスミドpXX7にレポータ遺伝子を導入した。
【0179】
変異生成を2つの異なった系でアッセイした。第1の系では、クロラムフェニコール耐性遺伝子を使用し、これを弱いプロモーターによって制御する。cm遺伝子の発現を不活性化する点変異、欠失、および挿入などの自然突然変異は、クロラムフェニコール耐性を低減する。MutLの不在は、培養中の生存細胞の総数に対する、クロラムフェニコール耐性を与える自然突然変異の割合の減少として報告されるはずである。第2の系では、バクテリオファージλのPRプロモーターおよびλcIリプレッサーの制御下にサイレントtetA遺伝子を含有するカセットを使用し、λcIリプレッサーはtetAの発現を阻止する。cIを不活性化するか、あるいはCIの結合部位を除去する、点突然変異、欠失、および挿入などの自然突然変異は、PRの抑制解除に導き、それによって、tetA発現の引き金となる。MutLの発現は、培養中の生存細胞の総数に対する、テトラサイクリン耐性を与える自然突然変異の割合の減少として報告されるはずである。
【0180】
a) G−CSFの発現
細菌株JM83をプラスミドpXX7cm、pXX7cm pTrcHis2/lacZ、pXX7cm pmutL、pXX7tet、pXX7tet pTrcHis2/lacZ、およびpXX7tet pmutLで形質転換した。得られた単一コロニーを精製し、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有するLBに細胞を播種し、30℃で終夜培養した。
【0181】
これらの細菌の終夜培養物10μlを、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有する30℃のRMG10mlに播種した。指数関数的増殖期に、プラスミドpmutLのPlacからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加し、さらに、プラスミドpXX7のPtrpからのG−CSFの発現を誘導するために、最終濃度100μg/mlのトリプトファンを添加した。誘導の1時間後に、播種温度を30℃から37℃に変化させ、その後、細胞を終夜、37℃に維持した。
【0182】
細胞を遠心分離して、培地容積を1mlにまで減少させた。
【0183】
最後に、培養物を希釈して、様々な抗生物質(表7に示す)を含有するLB寒天プレート上にプレーティングし、30℃で4日間インキュベートした。
【0184】
【表7−1】
【表7−2】
【0185】
菌株JM83 pXX7cm、JM83 pXX7cm pTrc、およびJM83 pXX7cm pmutLのクロラムフェニコール耐性レポータ遺伝子における自然突然変異の頻度の値を図10に示す。JM83 pXX7cmおよびJM83 pXX7cm pTrcの2株では、変異性の値が0.28または0、35(Δ1.25)であった。MutLの発現によって、変異性の値が0.49(Δ1.75)に低下する。これは、MutLの発現が、クロラムフェニコール耐性からクロラムフェニコール感受性への転換を抑制し、それによって、染色体における自然突然変異の修復に対抗して作用することを意味する。
【0186】
菌株JM83 pXX7tet、JM83 pXX7tet pTrc、およびJM83 pXX7tet pmutLのサイレントtetAレポータ遺伝子における自然突然変異の頻度の値を図11に示す。JM83 pXX7tetおよびJM83 pXX7tet pTrcの2株では、変異性の値が0.43または0.39(Δ1.1)であった。MutLの発現によって、変異性の値が0.25(Δ1.72)に低下する。これは、MutLの発現が、テトラサイクリン感受性からテトラサイクリン耐性への転換を抑制し、それによって、染色体における自然突然変異の修復に対抗して作用することを意味する。
【0187】
b) G−CSFの発現および抑制
野生型細菌株JM83を、プラスミドpXX7cm、pXX7cm pTrcHis2/lacZ、pXX7cm pmutL、pXX7tet、pXX7tet pTrcHis2/lacZ、およびpXX7tet pmutLで形質転換した。得られた単一コロニーを精製し、その細胞を、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有するLBに播種し、30℃で終夜培養した。
【0188】
これらの細菌の終夜培養物10μlを、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有する30℃のRMG10mlに播種した。指数関数的増殖期に、プラスミドpmutLのPtrcプロモーターからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加した。この培養物を2等分割し、プラスミドpXX7のPtrpプロモーターからのG−CSFの発現を誘導するために、最終濃度100μg/mlのトリプトファンを1回添加した。誘導の1時間後に、播種温度を30℃から37℃に変化させ、その後、細胞を終夜、37℃に維持した。
【0189】
細胞を遠心分離して、培地容積を1mlにまで減少させた。
【0190】
最後に、培養物を希釈して、様々な抗生物質(表7参照)を含有するLB寒天プレート上にプレーティングし、37℃で終夜インキュベートした。
【0191】
発酵状態中における、改変された産生株JM83 pXXcmの変異性を図12に示す。G−CSFの非存在下では、JM83 pXX7cm、JM83 pXX7cm pTrc、およびJM83 pXX7cm pmutLの、測定されたクロラムフェニコール耐性の頻度が50と55との間を移行する。G−CSFの誘導によって、JM83 pXX7tetおよびJM83 pXX7tet pTrcの値が3分の1に低下する。しかし、菌株JM83 pXX7 pmutLでは、G−CSFが発現された際に、クロラムフェニコール耐性が維持されている。
【0192】
MutLの発現は、クロラムフェニコール耐性からクロラムフェニコール感受性への変換を抑制する。MutLは、染色体における自然突然変異の修復に対抗して作用する。
【0193】
図13は、発酵状態中における、改変された産生株JM83 pXX7の変異性が有意に増大していることを示す。しかし、プラスミドによってコードされたMutLを発現する菌株JM83 pXX7 pmutLでは、G−CSFが発現された際にテトラサイクリン耐性が一定に保たれている。MutLの発現は、テトラサイクリン耐性からテトラサイクリン感受性への転換を抑制し、染色体における自然突然変異の修復に対抗して作用する。
【0194】
結論および要約:得られた結果は、MutLの過剰発現が、G−CSF産生中のG−CSF発酵株の変異性を約3分の1に低下させることを示す。さらに、MutLの過剰発現が細胞生存率を103倍にまで増強することも見出された。
【0195】
G−CSFの誘導は、試験された産生株の自然突然変異の発生率を約2から3倍に上昇させる。
【技術分野】
【0001】
本発明は、細胞または生物の自然突然変異の頻度を低下させる方法、ならびにそのような細胞および生物を産生する方法と、自然突然変異の頻度が低下している細胞および/または生物と、自然突然変異の頻度が低下している細胞を用いることによって、タンパク質の発現系を産生する方法、タンパク質を産生する方法、および発酵産物を産生する方法とに関する。
【背景技術】
【0002】
変異は、生体システムの特徴であり、自然選択の材料を提供する。細菌などの生物では絶えず自然突然変異が起きている。変異の発生率は、生物、例えば特定の遺伝子のサイズ、および塩基対置換による不活性化に対する遺伝子の感受性によって大きく異なる。大腸菌(Escherichia coli)では、1ゲノム複製あたり、1ゲノムあたりの変異の値がμg=0.0025であり、ゲノムサイズ4.6×106塩基対の中での1複製あたり、1塩基対あたりの変異率がμb=5.4×10−10である。G−C→A−Tなど、詳細が明らかにされている単一の経路の発生率でさえ、単一遺伝子内の異なった部位では、2000倍を越える相違を有することがあり、これは、いまなお大部分が謎である、局所的DNA配列およびDNA構造の影響によるものと考えられている。変異の種類、およびそれらを生成する過程は、両方とも多様であり、部分的にしか発見されていない。
【0003】
変異は多細胞生物でも重要である。ここではしかし、遺伝物質の変化における2つの異なった種類、すなわち配偶子の変異、換言すれば生殖細胞系変異と、体細胞の変化、換言すれば体細胞変異とを区別しなければならない。生殖細胞系変異はその生物の子孫へと引き継がれるが、体細胞変異は引き継がれない。しかし、体細胞変異は癌の発生に寄与するので、重要である可能性がある。生殖細胞系変異の検出、特にヒトの生殖細胞系変異の検出、およびヒトにおける変異率の測定には、二倍性の問題がある。ほとんどの前進変異は劣性であり、したがって、接合体が2コピーの変異対立遺伝子を得ない限り検出されないであろう。復帰変異の頻度は、通常、それよりはるかに低いが、それは、遺伝子に変異導入する方法が、既存の変異を元に戻す方法よりもはるかに多く存在するためである。ヒト細胞をin vitroで用いたin vivo研究から、総合的なヒト変異率は、原核および真核の様々な微生物のものに極めて類似していると推算されている。
【0004】
遺伝物質の遺伝的変化など、変異は、大域的変化、詳細には染色体レベルでの変化である場合も、あるいは細胞学的な異常として視覚化されないこともある点変異である場合もある。点変異には、トランジションおよびトランスバージョン、ならびにフレームシフト変異などの塩基対置換が含まれる。遺伝子のタンパク質コード領域における塩基置換変異の結果は、置換のタイプとその位置に依存する。そのような塩基対置換はサイレント、すなわちタンパク質配列中で新規のアミノ酸残基をもたらさないこともある。しかし、それらはアミノ酸置換をもたらすこともある。そのようなミスセンス変異は、鎌状赤血球貧血の場合のように、極めて重大な結果をもたらすことも、軽度の結果をもたらすことも、全く何の結果ももたらさないこともある。最後に、タンパク質コード領域における塩基置換は、アミノ酸コドンを終止コドンに変異させることもあれば、その逆もある。前者のタイプは、未熟な状態に短縮されたタンパク質をもたらし、ナンセンス変異と呼ばれる。塩基置換変異は、遺伝子のプロモーターもしくは5’調節領域、またはイントロンなど、遺伝子の発現の調節に関与する配列で起こることもあり、さらに、それらの転写、翻訳またはスプライシングに影響を与えることもある。β−サラセミアの多くが、グロビン遺伝子の発現レベルに影響を与えるこのタイプの非構造変異の結果である。フレームシフト突然変異は、遺伝子のコード領域における1つまたは複数(ただし3の倍数ではない)のヌクレオチドの挿入または欠失から生じる。これがリーディングフレームの変更を引き起こす。この種類の変異は下流のすべてのアミノ酸を改変し、産物が正常なタンパク質から大きく異なる場合があるので、機能しない産物を生成する可能性が非常に高い。
【0005】
自然突然変異は、細胞内での自然の過程の結果起こり得るものであり、外部因子、すなわち変異原とのDNAの相互作用の結果起こる変異である誘導性変異と区別することができる。自然突然変異の重要な発生源は、例えば、DNAポリメラーゼの不正確な作用による、DNA複製における誤りである。DNAポリメラーゼが誤りをおかす頻度は、この自然突然変異の頻度に影響を与えるであろう。そして、それによって、異なったDNAポリメラーゼに、精度の相違があることが観測されている。DNAポリメラーゼの精度に影響を与える重要な一要因は、ポリメラーゼによって挿入された誤対合塩基を除去するプルーフリーディング3’−5’エキソヌクレアーゼの存在である。この3’−5’エキソヌクレアーゼの機能は、DNA複製中の誤取込みを防止し、変異を防止することである。
【0006】
自然突然変異の別の主要な発生源は、互変異性化と呼ばれる核酸の塩基の構造変化である。塩基は、2つの形で存在でき、それらの間で相互変換する。例えば、グアニンはケト型およびエノール型で存在できる。様々な互変異性体型の塩基は異なった対合特性を有する。DNA複製中にGがエノール型である場合、DNAポリメラーゼは、それの向かいに正常なCではなくTを付加する。したがって、互変異性化はトランジション変異の原因となる。細胞で起こる別の変異原性の過程は、自発性の塩基分解である。細胞内では、シトシンからウラシルへの脱アミノ化が有意な発生率で起こる。脱アミノ化は、DNA中に通常存在しないウラシルを検出する特異的な修復過程によって修復することができ、それが起こらない場合、Uがその向かい側にAの挿入を引き起こし、DNA複製の際にC:GからT:Aへのトランジションを引き起こすことができる。発生頻度の高い自発性DNA損傷の第3のタイプは、酸素のフリーラジカルによる塩基の損傷である。これらは、細胞内で酸化代謝の結果起こり、また、放射線照射などの物理的作用因子によっても形成される。重要な酸化生成物の1つに8−ヒドロキシグアニンがあり、これはアデニンとの誤対合を起こし、その結果、G:CからT:Aへのトランスバージョンをもたらす。さらに別のタイプの自発性DNA損傷が、アルキル化、すなわちDNAの塩基またはバックボーンへのアルキル基の付加である。アルキル化は、S−アデノシルメチオニンなどの化合物の、DNAとの反応を介して起こり得る。アルキル化された塩基は、自発性の分解または誤対合の対象となる。
【0007】
さらに、DNA複製中における、鋳型鎖と新規に合成された鎖との間の「スリップ誤対合(slipped mispairing)」と呼ばれる機構によって、自発性のフレームシフト変異が生じることもある。
【0008】
自然突然変異はゲノムのいかなる部位でもランダムに起こり、そのため、自然発生した突然変異の大部分が、影響を受ける生物に有害であり、有利な変異が起こる確率は非常に低くなっている。したがって、生物は、過度の変異率から自らを防御する機構を進化させている。これらの防御機構は、DNAの複製または自発性の脱アミノ化の結果、DNA中に生じたミスマッチを認識および修正し、外因性または内因性の変異原とのDNAの反応の結果生じた潜在的に変異原性である変化を認識し除去する。
【0009】
特に、発酵工程などの生物工学の工程では、変異が極めて望ましくない。変異は、発酵細胞のゲノム中のいかなる部分にも生じ得るので、発酵産物の生成または組成に影響を与えることがある。例えば、発酵産物がタンパク質である場合、そのタンパク質をコードする核酸配列の変異は、改変されたアミノ酸配列を有するタンパク質変種に導くことがある。発酵細胞のわずかな部分のみが変異の影響を受けた場合にさえ、これによって、その変種に汚染された最終タンパク質調製物を生じる結果となり得る。これは、そのタンパク質がヒトの疾患の治療に用いられることとなっている場合、例えば、タンパク質の抗原特性が改変された場合に、予見できない劇的な結果をもたらす可能性がある。発酵細胞集団内での変異の発生も、その変異が、例えば、発酵産物、または所望の発酵産物の発現を制御する調節ユニットの形成に関与する酵素などの上流の調節経路に影響を与える場合、産物生成の速度に影響を与えることがある。
【0010】
さらに、変異が、発酵細胞集団のわずかな部分にのみ影響を与える希少な事象である場合でさえ、それらが、影響を受けた細胞に、変異導入されていない細胞と比較して選択有利性を与える場合、そのような集団でそれらが迅速に広まることがある。特に、定常状態を維持するために定常的に細胞を除去する連続培養では、これによって、培養時間の進行に伴う、変異導入された細胞と比較した、変異導入されていない細胞の漸進的減少に導かれる。
【0011】
特に発酵時間中の変異が極めて望ましくない別の理由が、いわゆる定常期変異の現象であるが、これは、適応変異とも呼ばれる。発酵の定常期中に行われる選択など、致死的でない選択に微生物集団が曝露された場合、選択圧を軽減する変異が高頻度で発生する(Cairnsら、Genetics、128(1991年)、695−701)。当初は、有用な変異のみが現れると思われたが、選択された変異には、選択されていない変異も伴うこと、すなわちこの過程が有用遺伝子へと方向付けられていないことが、現在では明らかである(Foster、J.Bacteriol.、179(1997年)、1550−1554)。適応変異に関するほとんどの研究は、ラクトースを利用することができない(Lac−)が、ラクトースが唯一の炭素源である場合に容易にラクトース利用性(Lac+)に復帰する大腸菌株に関して集中的に行われた。適応変異を生成する過程は、正常成長中にLac+変異を生成する過程と同じものではない。成長に依存した変異とは異なり、ほとんどすべての適応型のLac+変異は、大腸菌のRecBCD二本鎖切断(DSB)修復系の相同組換え機能など、組換え機能に依存している(Cairnsら、Genetics、128(1991年)、695−701;Foster、Annu.Rev.Microbiol.、47(1993年)、467−504;Harrisら、Science、264(1994年)、258−260)。
【先行技術文献】
【非特許文献】
【0012】
【非特許文献1】Cairnsら、Genetics、128(1991年)、695−701
【非特許文献2】Foster、J.Bacteriol.、179(1997年)、1550−1554
【非特許文献3】Cairnsら、Genetics、128(1991年)、695−701
【非特許文献4】Foster、Annu.Rev.Microbiol.、47(1993年)、467−504
【非特許文献5】Harrisら、Science、264(1994年)、258−260
【発明の概要】
【発明が解決しようとする課題】
【0013】
したがって、本発明の根底にある技術的課題は、細胞または生物によって、タンパク質などの発酵産物を産生する方法および手段を提供し、その際、自然発生した突然変異、中でも発酵工程の定常期中に発生する変異に対して、産生を行う細胞または生物が保護および/または安定化され、かつ発酵産物、詳細にはタンパク質の形成速度および組成の両方が長期的に保障および保護されるようにすることである。
【課題を解決するための手段】
【0014】
本発明は、少なくとも2つの変異を細胞または生物に導入することによって上記細胞または生物における自然突然変異の頻度を低下させる方法を提供することによって、この技術的課題を解決し、その際、上記少なくとも2つの変異は、それらの複合作用によって、少なくとも2つの細胞性DNA修復機構の強化に導くものである。
【0015】
本発明は、自然突然変異の頻度が低下している細胞または生物を、少なくとも2つの変異を導入し、さらにその生物が多細胞生物である場合にはそれから生物を再生することによって産生する方法を提供することによっても、その根底にある技術的課題を解決し、その際、上記少なくとも2つの変異は、それらの複合作用によって少なくとも2つの細胞性DNA修復機構の強化に導くものである。
【0016】
本発明によれば、自然発生した突然変異を修正する細胞性DNA修復機構の能力が大幅に強化され、それによって、異なった修復系に影響を与える少なくとも2つの異なった変異が細胞に導入される。本発明による、少なくとも2つの異なった変異の導入によって得られた、自然発生した突然変異を修正する細胞性DNA修復機構の能力の強化によって、細胞で安定的に遺伝する変異の頻度の低下に導かれ、それによって、変異率の総合的な低下に導かれると有利である。
【0017】
したがって、本発明によれば、驚くべきことに、特定の細胞性DNA修復機構を改変することによって、詳細には自然発生した突然変異を修復するこれらのDNA修復系の能力をより効率的に強化する特定の変異を導入することによって、野生型細胞の自然突然変異率を劇的に低下できることが見出された。例えば、本発明によれば、驚くべきことに、MutSタンパク質の過剰発現によって、増殖期に、宿主細胞の変異性の有意な低下に導かれることが見出された。これはしかし、現在の技術において記載されている結果と対照的である。米国特許第6656736号によれば、酵母または他の生物の野生型または変異型MMRタンパク質の過剰発現の結果、ミスマッチ修復系の欠損が生じ、それによって、そのようなミスマッチ修復系の欠陥を有する酵母細胞は高変異性となる。さらに、dinB10の欠失と、抗変異原アレルdnaE911とを保持する細菌株は、対応する野生型株と比較して10分の1に低下した変異性を示す。ミスマッチ修復系に関与するMutLタンパク質を追加して過剰発現すると、変異性の低下が50分の1にまで達する。別の系では、特定のフレームシフト変異の復帰変異の低下が1000分の1に達する可能性さえあると示されるはずである。このようにして、成長に依存した変異の発生率だけでなく、適応変異中の変異発生率、すなわち定常期に依存した変異の発生率も有意に低下させることができる。観測された、宿主細胞の自然突然変異発生率に対するいくつかの変異の影響は、驚くべきことに、相加的ではなく、相乗的に共働しているようである。
【0018】
さらに、本発明によれば、驚くべきことに、これらの変異の導入は、自然発生した突然変異を修正する細胞性DNA修復機構の能力の強化に導くだけではなく、有利なことに、細胞生存率も大幅に増大させることが見出された。例えば、発酵細菌株におけるMutLタンパク質の過剰発現によって、約103倍の細胞生存率の増大が観測された。
【0019】
自然発生した突然変異を修正する細胞性DNA修復機構の能力を増強するいくつかの変異を導入することによる、本発明の細胞内自然突然変異率の低減および本発明の細胞生存率の増大は、タンパク質などの発酵産物の発現および/または産生に使用される細胞または株に特に大きな価値を有する。そのような細胞の使用によって、例えば、得られたタンパク質産物のアミノ酸配列を、産生を行う細胞または生物で何代にもわたって、変化させずに維持することができる。そのような細胞の使用によって得られたタンパク質調製物は、変異によるタンパク質変種によって汚染されておらず、したがって、治療薬としての適用において何の問題も起こさず、また、受容する身体に対する望ましくない影響も導かない。細胞性DNA修復機構の能力が強化されているそのような本発明の細胞の使用は、組換え体タンパク質の産生に特に有用である。
【0020】
しかし、細胞性DNA修復機構の能力が強化されている本発明の細胞の使用は、発酵細胞によるタンパク質産生に限定されない。原則として、本発明の細胞は、抗生物質、有機酸など、あらゆる種類の発酵産物用に使用することができる。宿主細胞内での変異率の低下によって、産物生成速度の有効性を制御する因子も、安定かつ不変に維持されるであろうから、これらの細胞における自然突然変異率の大幅な低下によって、産物組成の完全性の長期的な維持のみでなく、産物生成速度の高さの維持も提供される。
【図面の簡単な説明】
【0021】
【図1】プラスミドpmutLの物理構造を示す図である。このプラスミドは、原則的にpmutSプラスミドと同じ構造を有する。
【図2】野生型、dinB10、dnaE911、dinB10 dnaE911、mutL−、mutL− dinB10、mutL− dnaE911、およびmutL− dinB10 dnaE911株のリファンピシン耐性への自然突然変異の頻度の値のグラフ表示である。図が示すように、MutLの発現によって変異性が強く抑制される。データは、SMF値として示す。プラスミドpmutLは、プラスミドpTrcHis2/lacZのmutL+派生物である。全てのセットの遺伝子操作に野生型株MG1655(wt)を選択した。関連する遺伝子型が示されている。棒グラフは三つ組みに分割されており、各三つ組みは、同一の遺伝子型を示し、かつa)最初の棒グラフ:細菌株、b)プラスミドpTrcHis2/lacZを保持する対応する細菌株、およびc)プラスミドpmutLを保持する対応する細菌株から構成されている。
【図3】野生型、dinB10、dnaE911、dinB10 dnaE911、mutS−、mutS− dinB10、mutS− dnaE911、およびmutS− dinB10 dnaE911株のリファンピシン抵抗性への自然突然変異の頻度の値のグラフ表示である。この図は、mutS−バックグランドにおける、dinB10、dnaE911、およびdinB10 dnaE911による変異性の抑制を示す。データは、SMF値として示す。関連する遺伝子型が示されている。
【図4】プラスミドpdapAIF(A)およびプラスミドpSU40dapA(B)の物理構造を示す図である。
【図5】プラスミドpSU40mutLを産生するストラテジーを示す図である。
【図6】菌株Top10(野生型;dapA+)、MG1655 dinB10 dapA::kn(dapA−)、AT997(dapA15、dapA−)、AT997 pdapAlF pTrc(dapA+)、AT997 pdapAlF pmutL(dapA+)、AT997 pdapA+1 pTrc(dapA−)、AT997 pdapA+1 pmutL(dapA−)、MG1655 dinB10 dapA::kn pdapA15 pSU40(dapA−)、MG1655 dinB10 dapA::kn pdapA15 pSU40mutL(dapA−)におけるdapA点突然変異の復帰変異のグラフ表示である。この図は、MutLの発現による変異性の抑制を示す。DAPに対する耐性を与える変異の値は、生存細胞の総数で割ることによって表される。プラスミドpmutLは、プラスミドpTrcHis2/lacZまたはpSU40のmutL+派生物である。全てのセットの遺伝子操作に野生型株MG1655(wt)を選択した。関連する遺伝子型が示されている。図は、対照の三つ組みの1つと、検査する株の3つの二つ組みとに分割されている。各二つ組みは、a)最初の棒グラフ:親ベクターを保持する細菌株、およびb)mutL+派生物を保持する細菌株で構成されている。使用されたすべての細菌株は染色体の野生型MutLを発現する。
【図7】プラスミドpXX7の派生物を示す図である。A)pXX7へのクロラムフェニコールカセットのクローニング。それによってプラスミドpXX7cmを得た。得られたクローンのうち9クローンを、EcoRIおよびNcoI(予測された断片:1677bpおよび3292bp)による制限消化によって分析した。試験されたクローンのすべてが正しい移動パターンを示した。B)プラスミドpXX7へのサイレントtetA遺伝子のクローニング。それによってプラスミドpXX7tetを得た。得られたクローンのうち24クローンを、EcoRI(予測された断片:4141bpおよび3113bp)による制限消化によって分析した。試験されたクローンのうち1クローンが正しい移動パターンを示した。
【図8】菌株JM83 pXX7、JM83 pXX7 pTrc、およびJM83 pXX7 pmutLにおけるリファンピシン抵抗性への自然突然変異の頻度を示す図である。プラスミドpmutLは、プラスミドpTrcのmutL+派生物である。SMF値は、生存細胞の総数に対するリファンピシン耐性として与えられる。野生型株JM83は、染色体の野生型MutLを発現する。
【図9】菌株JM83 pXX7、JM83 pXX7 pTrc、およびJM83 pXX7 pmutLにおける、ホスホマイシン耐性への自然突然変異の頻度の値を示す図である。プラスミドpmutLは、プラスミドpTrcのmutL+派生物である。SMF値は、生存細胞の総数に対するホスホマイシン耐性として与えられる。野生型株JM83は、染色体の野生型MutLを発現する。
【図10】菌株JM83 pXX7cm、JM83 pXX7cm pTrc、およびJM83 pXX7cm pmutLのクロラムフェニコール耐性レポータ遺伝子における自然突然変異の頻度によるcmRコロニーの数を示す図である。プラスミドpXX7cmは、pXX7のcm+派生物である。プラスミドpmutLは、pTrcのmutL+派生物である。生存細胞の総数が割った、クロラムフェニコールに対する感受性を導入する変異の値が示されている。対照株であるJM83pXX7、JM83pXX7 pTrc、およびJM83pXX7 pmutLは、クロラムフェニコールに対する、予測通りの感受性を示す(クロラムフェニコール耐性遺伝子を保持していない)。
【図11】菌株JM83 pXX7tet、JM83 pXX7tet pTrc、およびJM83 pXX7tet pmutLのサイレントtetAレポータ遺伝子における自然突然変異の頻度によるtetRコロニーの数を示す図である。プラスミドpXX7tetは、サイレントtetAを保持するpXX7派生物である。プラスミドpmutLは、pTrcのmutL+派生物である。生存細胞の総数が割った、テトラサイクリンに対する耐性を導入する変異の値が示されている。対照株であるJM83pXX7,JM83pXX7 pTrc、およびJM83pXX7 pmutLは、テトラサイクリンに対する、予測通りの感受性を示す(テトラサイクリン耐性遺伝子を保持していない)。JM83 pXX7およびJM83 pXX7tetの2株では、変異性の値が0.43または0.39であった(Δ1.1)。MutLの発現によって、変異性の値が0.25(Δ1.72)に低下する。
【図12】「発酵状態」中における、改変された産生株JM83 pXX7cmの変異性を示す図である。G−CSFが抑制(A)または(B)発現された際の、細胞におけるクロラムフェニコール耐性の頻度が示されている。試験したそれぞれのケースで、MutLが染色体から発現された。対照株であるJM83 pXX7、JM83 pXX7pTrc、およびJM83 pXX7pTrc pmutLは、ケース(A)およびケース(B)でクロラムフェニコールに対する予測通りの感受性を示す。
【図13】「発酵状態」中における、改変された産生株JM83 pXX7tetの変異性を示す図である。図は、G−CSFが抑制(A)または(B)発現された際の、細胞のテトラサイクリン耐性の頻度を示す。試験したそれぞれのケースで、MutLが染色体から発現された。
【発明を実施するための形態】
【0022】
本発明のコンテクストにおいて、「細胞性DNA修復機構の能力の強化に導く変異」という用語は、細胞のゲノムにおける、少なくとも1つの特定のDNA修復機構に関与するタンパク質または酵素をコードする部分の任意の遺伝的改変、あるいはDNAの複製もしくは自発性の脱アミノ化、または自然発生の突然変異に導く任意の他の自然過程の結果としてDNAで生じた細胞内または生物内でのゲノム規模の誤りの認識および修正の強化に導く、細胞性DNA修復機構の構成要素の発現調節に関与する、ゲノムの部分の任意の遺伝的改変を意味する。したがって、「細胞性DNA修復機構の能力の強化に導く変異」は、所与の細胞または生物のゲノム内の自然発生した誤りの、より効率的かつより正確な修正を提供する。
【0023】
したがって、「細胞性DNA修復機構の能力の強化に導く変異」は、集団内で安定的に遺伝する変異の頻度の低下、すなわちその集合内での変異頻度の低下に導く。本発明のコンテクストでは、「変異の頻度」を、集団の個体総数に対する変異体数の比率として定義する。「細胞性DNA修復機構の能力の強化に導く変異」は、所与の遺伝子が複製中に変異し、かつ該遺伝子のこの変異が安定的に遺伝するであろう確率として定義される変異発生率の低下も導く。さらに、本発明のコンテクストにおいて、細胞性DNA修復機構の能力の強化に導く変異は、トランスポゾンに媒介された変異導入の低下も導く。
【0024】
したがって、細胞または生物は、細胞性修復機構の能力の強化に導く変異の存在によって、非常に低い自然突然変異率を示し、それは、詳細には、その変異を有しない相当する細胞または生物よりかなり低いものである。細胞性DNA修復機構の能力の強化に導く変異には、細胞性DNA修復に関与する酵素をコードする構造遺伝子の欠失、例えば、誤りがちな(error−prone)DNAポリメラーゼの構造遺伝子の欠失;細胞性DNA修復に関与する酵素をコードする構造遺伝子における、塩基の置換、欠失、逆位、および/または付加、例えば、抗変異原表現型またはDNAポリメラーゼの忠実度の改善に導くことができるもの、細胞または生物の成長、分化、または増殖における特定のフェーズで律速的になるタンパク質の過剰発現、タンパク質、例えば、誤りがちなDNAポリメラーゼの発現の低下などが含まれるが、これらに限定されない。しかし、本発明のコンテクストでは、細胞性DNA修復機構の能力の強化に導く個々の変異それぞれが、第2または第3の細胞性DNA修復機構の能力の強化も導くことがある。さらに、細胞性DNA修復機構の能力の強化に導く個々の変異それぞれが、細胞生存率の増強も導くことがある。
【0025】
本発明のコンテクストにおいて、「2つの変異の複合作用によって少なくとも2つの細胞性DNA修復機構に導かれる」という用語は、両方の変異が少なくとも2つの異なったDNA修復機構に影響を与え、それによって、変異導入されていないDNA修復系と比較して、自然発生したゲノム規模の変異をより効率的かつより正確に修復するこれらのDNA修復系の能力が強化されることを意味する。
【0026】
本発明のコンテクストにおいて、「細胞性DNA修復機構」という用語は、細胞または生物が、ゲノム内の、DNA複製によって生じた誤りならびに/または塩基変化および塩基損傷による誤りを、それによって認識および修正できる酵素機構または酵素系を意味する。本発明の好ましい実施形態では、これらの細胞性DNA修復機構に、ミスマッチ修復系、複製後(組換え)修復系、およびSOS修復系が含まれる。ミスマッチ修復の強化に導く変異は、ミスマッチ修復に関与するタンパク質のうちの1つが律速的となる状況を打開する変異である。
【0027】
細胞内の全修復事象の99%が「ミスマッチ修復系」(MMR)によるものである。この修復系は、大腸菌の複製忠実度に対する貢献が最も大きい系であり、それによって、その成分タンパク質が転写共役DNA修復および超短鎖パッチ修復(very short patch repair)に用いられることによって、他のDNA修復経路でも作用する。MMRは、それなしではゲノムの再編成を引き起こし得る、不正確な相同性を有するもの相互の組換えを抑制することによって、遺伝的安定性の強制も行う。ミスマッチ修復は、トランスポゾンの切り出しを編集することによっても、遺伝的安定性を増強する。大腸菌MMRタンパク質の相同体は、他の細菌、酵母、マウス、およびヒトで同様の機能を行う(Modrich、Science、266(1994年)、1959−1960;Radmanら、Phil.Trans.R.Soc.London、347(1995年)、97−103)。大腸菌では、mutH、mutL、mutS、およびmutUの遺伝子産物がMMRに関与している。この系は、メチル化のため、親鎖ではなく新規に合成された鎖を認識する。mutS遺伝子産物は、DNA塩基ミスマッチを認識し、結合する。mutL遺伝子産物は、ミスマッチ結合の後にMutSと相互作用し、MutSをMutHと組み合わせると考えられている。その後、MutHエキソヌクレアーゼが、メチル化されていない新規のDNA鎖にニックをいれる。MutUヘリカーゼは、一本鎖ニックの箇所でDNAに進入し、ニックの入った鎖を除去する。ミスマッチ修復の強化に導く変異は、ミスマッチ修復に関与するタンパク質の1つが律速的となる状況を打開する変異である。
【0028】
複製後修復系は、後に続く複製ラウンドでDNA中の損傷を修復する系である。娘鎖中の損傷したDNAの複製によって、ギャップが作られる。欠失している遺伝情報は、組み換えによって、親鎖の対応するDNA領域によって補填される。損傷を含むDNAの複製は、損傷乗り越え合成とも呼ばれる過程であり、点変異の主要な発生源である。最近、関与しているDNAポリメラーゼが同定されたため、この過程に関する多くの理解が得られた(Friedbergら、Proc.Natl.Acad.Sci.USA、97(2000年)、5681−5683)。
【0029】
SOS応答は、概してDNA複製を妨害するものである様々な遺伝毒性ストレスおよび代謝ストレスへの細胞の曝露によって誘導される1セットの細胞反応である。SOS系の調節は、LexAおよびRecAタンパク質によって媒介される。LexAは、recAおよびlexAを含めた約30の様々な遺伝子のリプレッサーとして作用する。SOSを誘導するシグナルは、一本鎖DNAであり、これにRecAが結合して、コプロテアーゼ(RecA*)として活性化される。RecA*は、LexAレプレッサーおよび一部のファージリプレッサーのタンパク分解性自己切断を促進し、それによって、SOSレギュロンを抑制解除する。大腸菌は、ポリメラーゼII(Pol II)、ポリメラーゼIV(Pol IV)、およびポリメラーゼV(Pol V)の、少なくとも3つのSOS誘導性ポリメラーゼを有する。
【0030】
本発明によれば、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから、少なくとも2つの変異が選択される。本発明のコンテクストにおいて、「抗変異原アレル」は、対応する野生型細胞と比較して、細胞または生物の自然突然変異の頻度の低下を引き起こすアレルを意味する。
【0031】
本発明の好ましい実施形態では、MutLまたはその相同タンパク質の発現、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、ベクターを細胞内に導入することによって実現され、上記ベクターは、それぞれ、mutL遺伝子またはMutLの相同タンパク質をコードする遺伝子、およびmutS遺伝子またはMutSの相同タンパク質をコードする遺伝子を、それぞれのMutタンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に含む。上記ベクターは、多コピープラスミドであることが好ましい。上記調節ユニットは、誘導性または構成的プロモーターであることが好ましい。
【0032】
本発明の別の好ましい実施形態では、MutLまたはその相同タンパク質の発現、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、宿主細胞または宿主生物における天然のMutタンパク質の発現を指示する調節ユニットを、天然のMutタンパク質が、細胞で通常観測されるよりも多くの量で、細胞内で生成されるように改変することによって実現される。これは、染色体に存在する、それぞれのMutタンパク質をコードする天然のヌクレオチド配列から相補的なRNA配列への転写、および/またはそのようにして得られたコードRNA配列からMutタンパク質のポリペプチド鎖への翻訳を指示する調節ユニットが、他の適当な相同または異種の調節ユニットによって、対応する野生型の細胞または生物で観測されるものより高い天然Mutタンパク質の産生が得られるように、変異導入または置換されることを意味する。
【0033】
本発明の別の実施形態では、それぞれのmut遺伝子を1コピーまたは複数コピー追加して、適当な調節ユニットの機能的制御下に宿主細胞の染色体に導入することによって、それぞれのMutタンパク質の過剰発現が実現され、その際、追加のmut遺伝子は、別の種に由来する天然遺伝子か、または異種遺伝子のいずれかであり得る。
【0034】
本発明の好ましい実施形態では、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレルがdinB10であり、これは、事実上dinB遺伝子の欠失である。dinBによってコードされているDNAポリメラーゼIV(Pol IV)は、最近同定された、誤りがちなDNAポリメラーゼのYファミリー、すなわちUmuC/DinBヌクレオチジルトランスフェラーゼファミリーに属する。UmuC、DinB、Rad30、およびRev1サブファミリーがこのファミリーを代表する(Gerlachら、Proc.Natl.Acad.Sci.USA、96(1999年)、11922−11927)。dinBによってコードされている大腸菌のDNA ポリメラーゼIVは、自然突然変異生成と、翻訳合成(TLS)と呼ばれている過程であり、点突然変異の主要な発生源である、損傷を含有するDNAの複製とに関与することが示されている。DNAポリメラーゼVと同様に、Pol IVも、DNA損傷に対するSOS応答の一部として誘導される。
【0035】
本発明の別の好ましい実施形態では、DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルが使用され、それは、dnaE911であることが好ましい。細胞のDNA合成の90%超がDNAポリメラーゼIIIホロ酵素(Pol III HE)によるものであり、また、この酵素は、主要な複製後ミスマッチ修正経路にも必要である。ポリメラーゼIIIホロ酵素は、無塩基部位を通過する損傷乗り越えDNA合成を効果的に実行することが示されている。大腸菌のdnaE遺伝子は、DNAポリメラーゼIIIホロ酵素のαサブユニットをコードし、このαサブユニットはポリメラーゼ活性を提供する。したがって、αサブユニットは、忠実度の主要な決定因子の1つである。FijalkowskaおよびSchaaper、J.Bact.、177(1995年)、5979−5986から、いくつかのdnaE抗変異原アレルが知られており、それらは、ミスマッチ修復欠損性のmutL株における高変異性を抑制する。
【0036】
本発明によれば、dinB10変異およびdnaE911変異の複合作用によって、細胞の自然突然変異の頻度が、野生型細胞または野生型生物と比較して少なくとも10分の1に低下する。本発明の別の好ましい実施形態では、dinB10、dnaE911、および過剰発現したMutLタンパク質の複合作用によって、細胞内の自然突然変異の頻度が、野生型細胞または野生型生物と比較して、少なくとも50分の1に低下する。
【0037】
細胞性DNA修復機構の増強および/または細胞生存率の増強がそれらの複合作用によって導かれる少なくとも2つの変異は、任意の知られている方法によって、詳細には、細胞性DNA修復系の1つに関与していることが知られている遺伝子に変異導入することを目的として所与の細胞に変異導入することによって、あるいは既知の変異またはアレルをその細胞内に導入することによって、細胞に導入することができる。
【0038】
限定されるものではないが、ランダム変異導入、部位特異的変異導入、オリゴヌクレオチドカセット変異導入、または誤りがちなPCRによる点変異導入を含めた多数の異なった変異導入法が存在する。ランダム変異導入は、例えば、亜硝酸、ヒドラジンなどの化学物質で処理することによって、クローニングされたDNA断片中にランダムに分布した多数のヌクレオチド置換変異の生成を引き起こす。誤りがちなPCRは、クローニングされた遺伝子にランダムな点変異を導入するために開発された。PCR反応の忠実度を低下させる改変には、MgCl2濃度の増大、MnCl2の添加、または4種類のdNTPの相対濃度の改変が含まれる。これらの伝統的な変異導入法は、別々の選択可能な表現型を有する個々の遺伝子の改変に焦点をしぼって行われる。通常の戦略は、遺伝子のクローニングを行い、その遺伝子および/またはその機能をモニターできるアッセイを確立し、遺伝子内の選択された位置に変異導入し、そして、その遺伝子の機能が改変されている遺伝子変種を選択するというものである。その後、機能が改変された変種を所望の細胞型に導入して、発現させることができる。その遺伝子の所望の機能を得るために、変異導入法のサイクルを反復して実行することができる。遺伝子の機能を改変する別の手法は、十分に確立されている多数の系のうちの1つを用いた組換えによるものである。
【0039】
当然ながら、既存のアレルまたは変異を本発明の目的に用いることもできる。そのような既存のアレルまたは変異は、任意の知られている方法によって細胞に導入することができる。本発明によれば、細胞内への少なくとも2つの変異の導入は、限定されるものではないが、形質転換、接合、形質導入、伴性導入、感染、および/またはエレクトロポレーションを含めた任意の知られている適切な方法によって実施できる。
【0040】
本発明のコンテクストにおいて、「形質転換」という用語は、細胞、例えば微生物細胞による、環境からの、単離された核酸分子、好ましくは精製された核酸分子の取込みを意味する。「接合」は、細胞間接触を介した、細菌プラスミドの、ある細菌細胞から別の細菌細胞の中へのプラスミド媒介性の移入を意味する。関与する移入機構は、通常、プラスミドまたは接合トランスポゾンによってコードされている。そのようなプラスミドの例は、接合性プラスミドまたはヘルパープラスミドである。接合は、原核生物細胞の異なった表現形グループ間、および原核生物と真核生物との間の遺伝子交換の主要経路の1つである。「形質導入」は、バクテリオファージによる、ある細菌細胞から別の細菌細胞の中への核酸分子の移入を意味し、それは、一方の細胞からのバクテリオファージの放出と、それに続く、もう一方の細胞の感染とを含む。2つのタイプの形質導入がある。特殊形質導入は、溶原バクテリオファージの溶原生活環中に起こることがあり、それによって、細菌の遺伝物質が、バクテリオファージゲノムの一部を置換することがある。細菌DNAのこの小片は、ファージゲノムの一部として複製し、そのファージによって別の受容細胞の中に移入されることがある。普遍形質導入の場合には、溶菌ファージの全ゲノムが細菌DNAによって置換される可能性を有する。「エレクトロポレーション」は、細胞を核酸分子と混合して、その後、高電位の短いパルスに曝露する過程である。宿主細胞の細胞膜が貫通可能となり、それによって、外来核酸が宿主細胞内に入るのが可能となる。
【0041】
自然突然変異率を低下させる本発明の方法、および自然突然変異率が低下している細胞または生物を産生する本発明の方法で用いられる細胞は、いかなる原核細胞でも、真核細胞でもよい。
【0042】
「真核細胞」および「真核細胞宿主細胞」という用語には、膜に結合した核、細胞小器官、および染色体に組織化された遺伝物質を有し、染色体において、DNAがヒストンに結合しているいかなる細胞も含まれる。細胞骨格は、真核生物に特有な別の特徴であり、これは、細胞膜に繋ぎ止められて、細胞辺縁を縦横に走るタンパク質フィラメント、主としてアクチンおよびチューブリンのネットワークである。「真核生物」は、分類学上の原生生物界、菌類界、植物界、および動物界を含む。真核生物は、原生生物、例えば、ゾウリムシおよびアメーバなどの単細胞生物である場合も、様々な真菌、動物、および植物などの多細胞生物である場合もある。自然突然変異率を低下させる本発明の方法の好ましい実施形態では、動物細胞、植物細胞、または真菌細胞が使用されている。
【0043】
「原核細胞」および「原核細胞の宿主細胞」という用語には、ゲノムが環状構造として細胞質中に遊離して存在している細胞、すなわちゲノムが核膜によって囲まれていない細胞のいかなるものも含まれる。原核細胞はさらに、それが必ずしも酸素に依存していないこと、およびそのリボソームが真核細胞のものより小さいことを特徴とする。原核細胞には、古細菌および真正細菌が含まれる。細胞壁の組成物に応じて、真正細菌を、グラム陽性菌、グラム陰性菌、およびラン細菌に分類することができる。自然突然変異率を低下させるか、あるいはそれに対応する生物を産生する本発明の方法の好ましい実施形態では、使用される原核細胞が古細菌または真正細菌の細胞であり、上記原核細胞が、グラム陰性細菌、グラム陽性菌、またはラン細菌であることが特に好ましい。
【0044】
本発明は、自然突然変異の頻度の低下および/または細胞生存率の増強を有する細胞にも関し、これは、細胞または生物の自然突然変異の頻度を低下させる本発明の方法、または自然突然変異の頻度が低下している細胞もしくは生物を生成させる本発明の方法によって取得可能であり、上記細胞は、少なくとも2つの細胞性DNA修復機構の増強が、それらの複合作用によって導かれる少なくとも2つの変異を含む。本発明の好ましい実施形態では、上記細胞が細菌細胞、例えば、大腸菌などのグラム陰性細菌、もしくは枯草菌(Bacillus subtilis)などのグラム陽性菌、真菌細胞、植物細胞、または昆虫細胞もしくは哺乳類細胞などの動物細胞である。
【0045】
本発明の好ましい特定の実施形態では、上記細菌が、プラスミドpmutLを含有する大腸菌MG1655 dinB10、プラスミドpmutLを含有する大腸菌MG1655 dinB10 mutL::tet、プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm、プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm mutL::tet、大腸菌MG1655 dinB10 dnaE zae::cm大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet、プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm、またはプラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm mutL::tetである。
【0046】
本発明は、自然突然変異率が低下している生物を産生するための、自然突然変異率の低下および/または細胞生存率の増強を有する本発明の細胞の使用、タンパク質発現用の宿主細胞としての本発明の細胞の使用、発酵産物産生用の発酵生物としての本発明の細胞の使用、候補薬物の作用を研究するためのモデル系としての本発明の細胞の使用、ヒト、動物、または植物の病気を研究するためのモデル系としての本発明の細胞の使用、およびトランスジェニック生物の産生、特に育成または培養のための本発明の細胞の使用にも関する。
【0047】
当業者は、完全な多細胞生物、特に植物または動物を単一細胞から産生することのできる多数の方法を知っている。本発明の細胞から産生された多細胞生物も、同様に、総合的な自然突然変異率が低下していることを特徴とする。
【0048】
本発明は、自然突然変異の頻度が低下している生物にも関し、これは、細胞または生物内の自然突然変異の頻度を低下させる本発明の方法、または自然突然変異の頻度が低下している細胞または生物を産生する本発明の方法によって取得可能であり、上記生物の細胞は少なくとも2つの変異を含み、上記少なくとも2つの変異の複合作用によって、少なくとも2つの細胞性DNA修復機構の強化および/または細胞生存率の増強に導かれる。本発明の好ましい実施形態では、上記多細胞生物が、出芽酵母(Saccharomyces cerevisiae)、偽巣性コウジ菌(Aspergillus nidulans)などの真菌、植物、特にトウモロコシ(Zea mays)などの農業有用性のある植物、または動物、特に哺乳動物、例えば、所与の疾患、または所与の疾患を治療するための薬物候補の作用を研究するためのモデル系として使用できる哺乳動物である。
【0049】
本発明は、経済的、医学的、または農業的に重要なタンパク質など、タンパク質を発現および/または産生するための宿主としての本発明の生物の使用、トランスジェニック生物を育成または培養するための本発明の生物の使用、疾患を研究するためのモデル系としての本発明の生物の使用、および候補薬物の作用を研究するためのモデル系としての本発明の生物の使用にも関する。
【0050】
本発明の根底にある技術的課題は、タンパク質の発現系を産生する方法を提供することによっても解決され、その際、上記タンパク質のアミノ酸配列は、自然発生した突然変異に対して安定化されており、上記方法は、
a) 少なくとも2つの変異を含有するタンパク質をコードする核酸配列を、上記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、宿主細胞のゲノムに挿入する工程であって、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる工程、または
b) 上記タンパク質をコードする核酸配列を、上記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下にベクターに挿入し、さらに、少なくとも2つの変異を含有するベクターを宿主細胞内に移入する工程であって、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる工程と、
c) 適当な培地中で上記宿主細胞を培養および/または維持する工程と
を含む。
【0051】
タンパク質の発現系を産生する本発明の方法は、特に、タンパク質の発現をその中で行うことができ、自然突然変異率が非常に低く、特に他の既知の宿主細胞より低いことを特徴とする宿主細胞の産生を対象とするものである。したがって、本発明の方法によって産生された発現系は、所与のタンパク質をコードするヌクレオチド配列が変異によって変化し、それによって、そのタンパク質のアミノ酸配列が変化する確率が大幅に低下するのを確実にするものである。したがって、本発明の方法によって提供され、低自然突然変異率および増強された細胞生存率を有する本発明の宿主細胞に基づく発現系は、その完全性が長期的に保障されるタンパク質の発現および産生に用いることができる。本発明の発現系は、特に、治療目的で使用されることになっているタンパク質であって、そのアミノ酸配列のいかなる変化も、例えば、そのタンパク質の生物活性の変化に導くか、あるいはそのタンパク質の抗原特性を改変する変化も、そのタンパク質の治療効果に有害な影響を与える恐れがあるタンパク質に使用できる。したがって、発現系を産生する本発明の方法は、産生されたタンパク質調製物における、変異導入された異常なタンパク質による最小限の汚染さえも回避するのに有用である。本発明の方法によって提供され、低自然突然変異率を有する本発明の宿主細胞に基づく発現系は、特定のタンパク質をコードするヌクレオチド配列の長期的な維持に使用でき、それによって、ヌクレオチド配列が改変しているか否かを判定するために、シーケンシングによって定期的に検査する必要性を取り除くものが有利である。
【0052】
本発明のコンテクストにおいて、「発現系」は、特に、細胞における所与のタンパク質の効率的発現、すなわちそのタンパク質の大量生産を可能にする細胞システムを意味する。詳細には、「発現系」は、所望のタンパク質をコードするヌクレオチド配列から相補的なRNA配列に転写し、非コード配列部分を除去するために、該RNA配列のスプライシングを行い、そして、コードRNA配列部分を翻訳し、それによって、該タンパク質のポリペプチド鎖を取得するのを可能にする細胞環境に関する。「発現系」は、タンパク質のグリコシル化、またはタンパク質からの既存のリーダー配列の除去など、翻訳後プロセシング工程を可能にする細胞環境にも関するものである場合がある。したがって、「発現系の産生」は、コードされている遺伝子産物の発現を所与の細胞環境で制御および指示する適当な調節ユニットに、所望のタンパク質をコードするヌクレオチド配列が機能的に連結されていなければならないこと、および該タンパク質の最適な発現が実現されるように、使用される調節ユニットが機能するであろう適当な宿主細胞が提供されなければならないことを意味する。
【0053】
本発明によれば、タンパク質を発現するための系で使用される宿主細胞における自然突然変異率の低下および生存率の増強が、少なくとも2つの変異を使用することによって得られ、上記少なくとも2つの変異の複合作用によって、少なくとも2つの細胞性DNA修復機構の能力の強化に導かれ、それによって、宿主細胞内に存在するいずれの核酸で自然発生した突然変異も修復される。詳細には、これらの少なくとも2つの変異によって、自然発生した突然変異を修復する、ミスマッチ修復系、プルーフリーディング機能、および/またはSOS修復系の能力が強化される。本発明の好ましい実施形態では、これらの変異が、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから選択される。
【0054】
本発明の一実施形態において、MutLまたはその相同タンパク質の発現の上方制御、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、ベクターを宿主細胞に導入することによって実現することができ、上記ベクターは、それぞれ、mutL遺伝子またはその相同タンパク質をコードする遺伝子、およびmutS遺伝子またはその相同タンパク質をコードする遺伝子を、対応する野生型宿主細胞との比較において、それぞれのMutタンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に含む。MutLまたはMutSのいずれかのタンパク質の過剰発現に用いるベクターは、1つまたは複数の調節ユニットの機能的制御下にそれぞれのmut遺伝子を含む多コピープラスミドであることが好ましい。本発明の好ましい実施形態では、それぞれのmut遺伝子の過剰発現を制御する調節ユニットは誘導性プロモーターでも、構成的プロモーターでもよい。
【0055】
本発明の別の実施形態では、MutLまたはその相同タンパク質の発現の上方制御、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、細胞の染色体にそれぞれのmut遺伝子を1コピーまたは複数コピー追加して導入することによって、あるいは対応する野生型細胞と比較して、細胞内におけるそれぞれのMutタンパク質のより高い産生がもたらされるように、天然の染色体に位置するmut遺伝子の転写を指示する調節ユニットに1つまたは複数の変異を導入することによって実現することができる。
【0056】
本発明の好ましい実施形態では、DNAポリメラーゼIVをコードする遺伝子の抗変異原アレルがdinB10である。別の好ましい実施形態では、DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルがdnaE911である。
【0057】
本発明によれば、産生された発現系は、いかなるタンパク質の発現にも用いることができる。本発明のコンテクストにおいて、「タンパク質」という用語は、アミド連結で連結された少なくとも2つのアミノ酸を含む分子である。したがって本発明によれば、「タンパク質」という用語には、ペプチド、例えば、オリゴペプチド、ポリペプチド、またはドメインなどの天然に存在するタンパク質の一部が含まれる。発現されるタンパク質は、そのアミノ酸配列が天然に存在するタンパク質の配列であってもよく、すなわち野生型の配列を有するものでもよい。当然ながら、上記の発現系によって発現されるタンパク質は、野生型タンパク質と比較して、例えば、異なったアミノ酸組成および/または異なった長さによって改変されたアミノ酸配列を有するものでもよい。したがって、発現されるタンパク質は、野生型タンパク質と比較して、1つまたは複数の位置に異なったアミノ酸残基を有するものでもよい。発現されるタンパク質は、野生型タンパク質と比較して、先端切除されていても、延長されていてもよい。さらに、発現されるタンパク質は、対応する野生型タンパク質と異なる特徴を有するものでもよい。これらの異なる特徴は、改変された熱安定性、異なった基質特異性、異なった活性、改変された触媒部位、または新規の触媒部位などに関するものでよいが、これらに限定されない。発現されるタンパク質は、2つ以上の個別のポリペプチドを含む融合タンパク質でも、第2のポリペプチドの1つまたは複数のドメインを追加して含む融合タンパク質でもよい。タンパク質のそのような改変または変異は、当技術分野で知られているタンパク質をコードするヌクレオチド配列を適切に操作し、その後、このタンパク質をコードするヌクレオチド配列を、自然突然変異率が低下している宿主細胞のゲノムに挿入するか、あるいはベクターに挿入して、その後、それをその宿主細胞に導入することによって得ることができる。本発明の好ましい実施形態では、発現されるタンパク質が組換え体タンパク質、すなわち遺伝工学的手法および/またはDNA組換え技法によって改変されたタンパク質である。
【0058】
本発明の好ましい実施形態では、発現されるタンパク質が、天然化合物および非天然化合物の工業生産に利用できる酵素であってもよい。酵素、または酵素の補助によって生産それらの化合物は、薬物、化粧品、食品などの生産に用いることができる。発現されるタンパク質は、ヒトおよび動物の健康に関する分野で治療応用を有する物質でもよい。医学的に重要なタンパク質の重要なクラスには、例えば、サイトカインおよび成長因子が含まれる。
【0059】
本発明の方法によれば、タンパク質をコードする核酸配列を、1つまたは複数の調節ユニットの機能的制御下に、ゲノムまたはベクターのいずれかに挿入する。遺伝子および遺伝子産物の発現をそれぞれ制御および指示する調節ユニットには、プロモーター、リボゾーム結合部位、エンハンサー、サイレンサー、ポリアデニル化部位、および/または3’転写終結区が含まれるが、これらに限定されない。調節ユニットは、宿主細胞の所与の区画、または細胞の外に向けたタンパク質の発現を指示するリーダー配列またはシグナル配列を含んでもよい。そのような調節ユニットの使用は、使用される宿主細胞のタイプに依存する。所与の原核宿主細胞または真核宿主細胞で使用できる調節ユニットは、周知である(例えば、Sambrookら、「Molecular cloning:A Laboratory Handbook」、第2版(1989年)、Cold Spring Harbor Laboratory Press社、NY,USAを参照のこと)。
【0060】
好ましい実施形態では、タンパク質をコードするヌクレオチド配列をベクターにクローニングする。プラスミド、バクテリオファージ、ウイルス、またはコスミドをベクターとして使用するのが好ましい。当業者は、タンパク質をコードするヌクレオチド配列を所与の宿主細胞に導入するのに使用できる、真核宿主細胞用または原核宿主細胞用の適当なベクターを多数知っている。
【0061】
さらに、当業者は、調節ユニットに機能的に連結された、タンパク質をコードするヌクレオチド配列をクローニングする方法、およびヌクレオチド配列を宿主細胞に導入する方法を多数知っている(例えば、Sambrookら、1989年を参照)。
【0062】
本発明のさらに別の態様は、タンパク質の産生する方法に関し、上記タンパク質のアミノ酸配列は、自然発生した突然変異に対して安定化されており、この方法は、
a) 少なくとも2つの変異を含有するタンパク質をコードする核酸配列を、上記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、宿主細胞のゲノムに挿入する工程であって、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる工程、または
b) 上記タンパク質をコードする核酸配列を、上記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下にベクターに挿入し、さらに、少なくとも2つの変異を含有するベクターを宿主細胞内に移入する工程であって、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる工程と、
c) 適当な培地中で、タンパク質の発現を可能にする条件下に宿主細胞を培養する工程と、
d) 発現された上記タンパク質を単離工程と
を含む。
【0063】
上記タンパク質をコードする核酸配列がリーダー配列またはシグナル配列に機能的に連結されているかどうかに応じて、発現されたタンパク質は、特定の細胞小器官、特定の細胞内区画、細胞外間隙、または細胞が培養されている培地中に輸送される。したがって、タンパク質は、細胞内に蓄積されるか、あるいは細胞から分泌されるかのいずれかであり得る。タンパク質を産生する本発明の方法の好ましい実施形態では、それゆえ、タンパク質が培地から単離される。タンパク質を産生する本発明の方法の別の好ましい実施形態では、タンパク質を宿主細胞から、例えば、特定の細胞小器官から抽出する。また、当業者は、細胞、細胞小器官、または培地からタンパク質を単離する多数のプロトコールを知っている。
【0064】
本発明によれば、いかなる真核細胞も、あるいは原核細胞も、タンパク質を発現または産生するための宿主細胞として用いることができる。特に好ましい例には、真菌細胞、動物細胞、植物細胞、古細菌、ラン細菌、グラム陽性菌、およびグラム陰性菌が含まれる。
【0065】
本発明は、タンパク質を産生する本発明の方法によって取得可能なタンパク質にも関する。
【0066】
本発明の別の態様は、発酵産物、および/または培地中での発酵産物の生成に関与する少なくとも1つの酵素を産生する細胞を培養することによって発酵産物を産生する方法に関し、上記細胞のゲノムは、少なくとも2つの変異によって自然発生の配列変化に対して安定化されており、上記少なくとも2つの変異の複合作用によって、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化に導かれる。
【0067】
したがって、本発明の方法は、細胞の、発酵を目的とした使用、すなわち発酵産物を生産するための使用を対象とし、その際、上記細胞のゲノムは、自然発生の配列変化に対して安定化されており、それによって、非常に低い自然突然変異率を示す。さらに、使用される細胞は、改善された細胞生存率を有する。自然突然変異率が低下している細胞を使用することによって、本発明の方法は、所与のタンパク質をコードするヌクレオチド配列が変異によって変化し、それによって、そのタンパク質のアミノ酸配列が変化する確率が大幅に低下するのを確実にする。これらの細胞は、細胞生存率の大幅な増強を示すので、これらの細胞の使用は、さらに、発酵工程中に撹乱が起こらないこと、および発酵産物の高い生産性が得られることを確実にする。
【0068】
本発明のコンテクストにおいて、「発酵」という用語には、微生物、真菌細胞、植物細胞、または動物細胞の嫌気的または好気的代謝によって、あるいはそのような細胞からの酵素を使用することによって特定の産物を産生するいかなる過程も含まれ、上記酵素は、単離、精製、および/または固定することができる。「発酵」には、微生物細胞、植物細胞、真菌細胞、および/または動物細胞の1つまたは複数の酵素の作用によって引き起こされる、有機基質のあらゆる酵素的化学的変成が含まれる。所望の発酵産物は、細胞それ自体の中で、例えば、タンパク質の発現によって、または細胞の代謝の結果として産生されることがあり、それによって、発酵産物が細胞内に蓄積されることもあれば、細胞から環境中に輸送されることもあり、あるいは細胞によって1つまたは複数の酵素が産生され、それが、細胞外に分泌された際に、培地中に存在する基質から、所望の発酵産物への変換を触媒する。発酵産物が核酸、または核酸によってコードされたタンパク質である場合には、本発明の方法は、変異による発酵産物の最小限の汚染さえも回避し、それによって、発酵産物の長期的な完全性を保障するのに有用である。発酵産物の産生が、細胞によって産生された1つまたは複数の酵素の作用による場合には、本発明の方法は、それぞれの酵素の完全性を維持し、それによって、発酵産物の産生に導く代謝過程の特異性および有効性を維持するのに有用である。
【0069】
発酵産物を産生する本発明の方法は、ある経路に関与する遺伝子クラスター、および/またはそのような遺伝子クラスターによってコードされた遺伝子産物の完全性を維持し、それによって、そのような遺伝子クラスターによってコードされた遺伝子産物が関与する経路の特異性および有効性を維持するのにも有用であり得る。そのような遺伝子クラスターの例には、ポリケチドシンターゼ(PKS)クラスターが含まれるが、これに限定されない。ポリケチド代謝産物は、放線菌および粘液細菌などの細菌ならびに真菌によって産生される、様々な構造および生物活性を有する天然物の大きなグループである。複合ポリケチドは、動物の長鎖脂肪酸合成に類似した機構を用いた多機能性PKSによって産生される。サッカロポリスポラエリスラエア(Saccharopolyspora erythraea)のエリスロマイシンPKSの研究は、モジュール構成を明らかにした。複合ポリケチド合成は、前進性反応機序に従い、PKS内の各モジュールは、ほぼ直線的な順序の反応を触媒する、ひと続きになった3つから6つの酵素ドメインを有する。
【0070】
本発明によれば、発酵を目的として使用される細胞における自然突然変異率の低下、および高い細胞生存率が、少なくとも2つの変異を使用することによって得られ、上記細胞内に存在するいかなる核酸に自然発生した突然変異も修復する少なくとも2つの細胞性DNA修復機構の能力の強化に、上記少なくとも2つの変異の複合作用によって導かれる。詳細には、これらの少なくとも2つの変異は、上記細胞の自然発生した突然変異を修復するミスマッチ修復系、プルーフリーディング機能、および/またはSOS修復系の能力を強化する。本発明の好ましい実施形態では、これらの変異が、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから選択される。
【0071】
本発明の一実施形態において、MutLまたはその相同タンパク質の発現の上方制御、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、発酵に使用される細胞にベクターを導入することによって実現でき、上記ベクターは、それぞれ、mutL遺伝子またはその相同タンパク質をコードする遺伝子、およびmutS遺伝子またはその相同タンパク質をコードする遺伝子を、対応する野生型宿主細胞との比較における、それぞれのmutタンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に含む。MutLまたはMutSいずれかのタンパク質の過剰発現に用いるベクターは、1つまたは複数の調節ユニットの機能的制御下にそれぞれのmut遺伝子を含む多コピープラスミドであることが好ましい。本発明の好ましい実施形態では、それぞれのmut遺伝子の過剰発現を制御する調節ユニットは誘導性プロモーターでも、構成的プロモーターでもよい。
【0072】
本発明の別の実施形態では、MutLまたはその相同タンパク質の発現の上方制御、およびMutSまたはその相同タンパク質の発現の上方制御は、それぞれ、細胞の染色体にそれぞれのmut遺伝子を1コピーまたは複数コピー追加して導入することによって、あるいは対応する野生型細胞と比較して、細胞内におけるそれぞれのMutタンパク質のより高い産生がもたらされるように、天然の染色体に位置するmut遺伝子の転写を指示する調節ユニットに1つまたは複数の変異を導入することによって実現することができる。
【0073】
本発明の方法の好ましい実施形態では、DNAポリメラーゼIVをコードする遺伝子の抗変異原アレルがdinB10である。別の好ましい実施形態では、DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルがdnaE911である。
【0074】
本発明によれば、発酵を目的として使用する細胞の中にdinB10およびdnaE911が存在すると、その細胞の自然突然変異の頻度が、対応する野生型宿主細胞と比較して少なくとも10分の1に低下する。mutLタンパク質をさらに過剰発現する細胞の中にdinB10およびdnaE911が存在すると、その細胞の自然突然変異の頻度が、野生型宿主細胞と比較して少なくとも50分の1に低下する。
【0075】
本発明の方法によって得られる発酵産物は、細胞の一次代謝物である場合も、二次代謝物である場合もある。一次代謝物は、生細胞によって合成され、かつ細胞構造の形成および代謝過程を維持するために、細胞に必ず必要とされる物質である。一次代謝物と対照的に、二次代謝物は、細胞によって合成され、細胞の機能の維持には必ずしも必要でない化合物、特に低分子化合物である。通常、二次代謝物は、特定の環境における選択有利性をその細胞または生物に与えることができる。本発明の方法によって得られる発酵産物は、古典的な意味での発酵産物、すなわちアルコール発酵、乳酸発酵、プロピオン酸発酵等の産物など、もっぱら微生物によって行われる、酸素を用いた糖質の酵素的分解である場合もある。
【0076】
発酵産物の例には、核酸、ヌクレオシド、ヌクレオチド、タンパク質、アミノ酸、有機酸、アルコール、糖質、ビタミン、抗生物質、およびアルカロイドが含まれるが、これらに限定されない。
【0077】
本発明の好ましい実施形態では、発酵産物が核酸であり、詳細には、治療上の有用性がある核酸、例えばワクチンとして使用されることになっている核酸である。
【0078】
本発明のコンテクストにおいて、「核酸」は、ホスホジエステル結合によって連結された少なくとも2つのヌクレオチドを含む分子である。したがって、「核酸」という用語は、オリゴヌクレオチドも意味する。核酸は、DNAまたはRNAのいずれかであり得る。この核酸は、一本鎖でも、二本鎖でもよい。
【0079】
本発明の好ましい実施形態では、発酵産物がタンパク質であり、詳細には、治療上の有用性がある酵素もしくはタンパク質、またはドメインなど、それらの部分である。好ましい酵素の例。好ましい酵素の例には、プロテアーゼ、アミラーゼ、ペクチナーゼ、およびグルコースイソメラーゼが含まれるが、これらに限定されない。治療上の有用性があるタンパク質の好ましい例には、インターフェロン、インターロイキンなどのサイトカイン、成長因子、血液凝固因子、抗体などが含まれるが、これらに限定されない。
【0080】
有機酸の好ましい例には、クエン酸、乳酸、または酢酸が含まれるが、これらに限定されない。アルコールの好ましい例には、エタノール、プロパノール、およびブタノールが含まれるが、これらに限定されない。糖質の好ましい例には、ショ糖、マルトース、もしくはパラチノーゼなどの糖、キシリトールもしくはマルチトールなどの糖アルコールが含まれるが、これらに限定されない。ビタミンの好ましい例には、ビタミンB12もしくはリボフラビンが含まれるが、これらに限定されない。抗生物質の好ましい例には、ペニシリン、セファロスポリン、ストレプトマイシン、エリスロマイシンなどのポリケチド抗生物質が含まれるが、これらに限定されない。
【0081】
発酵産物は、発酵細胞、または培養に使用された培地のいずれかから単離することができる。
【0082】
したがって、本発明は、発酵産物を産生する本発明の方法によって取得可能な発酵産物にも関する。
【0083】
発酵産物を産生する本発明の方法では、真核細胞および原核細胞の両方が使用可能である。発酵産物を産生する本発明の方法の好ましい実施形態では、使用される真核細胞が、真菌細胞、動物細胞、または植物細胞である。発酵産物を産生する本発明の方法で使用される原核細胞は、ラン細菌、古細菌、グラム陽性菌、またはグラム陰性細菌であることが好ましい。
【0084】
当然ながら、対応する野生型宿主細胞と比較して、自然突然変異率が、好ましくは大幅に、低下しているいかなる細胞も、発酵工程を産生する本発明の方法で使用することが可能である。詳細には、自然突然変異率を低下させる本発明の方法によってその自然突然変異の頻度が低下している細胞または生物、あるいは自然突然変異の頻度が低下している細胞または生物を産生する本発明の方法によって産生された細胞または生物の使用が好ましい。
【0085】
本発明の方法の好ましい実施形態では、適当な液体培地中で細胞を培養する。当業者は、発酵産物を産生するために、細胞、例えば原核細胞または真核細胞を、その中で培養できる多数の液体培地を知っている。本発明によれば、細胞を連続培養またはバッチ培養のいずれかで培養することができる。バッチ培養は、細胞を播種された液体培地から始まり、培養中、培地の交換が連続的に行われない培養であって、場合によっては、酸素などの気体のみが導入される。産物生成の異化抑制を回避するために、バッチ培養を流加培養で行ってもよい。流加培養の場合には、したがって、所与の糖など、基質の消費がその基質の追加分量を配送することによって補填される。連続培養は、1回だけ細胞で播種され、その後、細胞量の増大または産物の蓄積に応じて、消費済みの培養培地を新たな培地で連続的に置換し、それによって、消費済みの培地と共に、場合によってはさらに細胞と共に、発酵産物を培養から同時に取り出す培養である。連続培養は発酵槽で行うことができる。
【0086】
本発明の好ましい実施形態では、細胞が、固定された形態で培養中に存在する場合がある。本発明の方法の別の実施形態では、細胞を固体または半固体の培地上で培養する。
【0087】
好ましい実施形態では、本発明は、生物、詳細には原核生物、最も好ましくは大腸菌株に関し、これは、
プラスミドpmutLを含有する大腸菌MG1655 dinB10(DSM17016)、
プラスミドpmutLを含有する大腸菌MG1655 dinB10 mutL::tet(DSM17017)、
プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm(DSM17018)、
プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm mutL::tet(DSM17019)、
大腸菌MG1655 dinB10 dnaE zae::cm(DSM17015)、
大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet(DSM17014)、
プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm(DSM17020)、および
プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet(DSM17021)
からなる群から選択される。
【0088】
上記に特定された微生物は、ブダペスト条約に従って、2005年1月3日に、上記に特定したDSM番号の下に、DSMZ(独国、Braunschweig所在の独国微生物細胞培養収集館(Deutsche Sammlung fur Mikroorganismen und Zellkulturen GmbH))に寄託した。
【0089】
本発明を、以下の配列表、図、および実施例によって例示する。
【0090】
配列番号1および2は、それぞれ、MutLタンパク質をコードするDNA断片を増幅するためのプライマーMutLNcoIhinおよびMutLXhoIherの配列を示す。
【0091】
配列番号3および4は、それぞれ、MutSタンパク質をコードするDNA断片を増幅するためのプライマーMutSBam−HIhinおよびMutSXhoIherの配列を示す。
【0092】
配列番号5から配列番号10は、それぞれ、DapAタンパク質をコードするDNA断片を増幅するためのプライマーdapABamHIIF、dapABamH1+1、dapABamH1+2、dapAXhoI、dapAEcoRIhin、およびdapAHindIIIherの配列を示す。
【0093】
配列番号11および12は、それぞれ、クロラムフェニコール耐性をコードするDNA断片を増幅するためのプライマーcmBbvCIhinおよびcmAhdIherの配列を示す。
【実施例】
【0094】
実施例1
(様々な大腸菌株のリファンピシン耐性に対する自然突然変異の頻度(SMF)の測定)
通常、細菌である大腸菌(Escherichia coli)の細胞は、抗生物質リファンピシンに対して感受性であって、すなわちそれらは、この抗生物質を含有する培地で増殖できない。しかし、リファンピシンに対する耐性を与える変異は、大腸菌ゲノムで低頻度で自然に発生する。したがって、リファンピシンを含有する培地上に、多数の細胞(108)をプレーティングした場合、ほとんどの細胞は抗生物質によって殺されるであろうが、いくつかのコロニーが成長するであろう。これは、リファンピシンに対する耐性を与える変異が、細胞ゲノム中で自然に発生するために可能である。それらの変異は、その次に、元の変異体の子孫によって引き継がれる。この変異は、リファンピシンが存在する環境においてのみ有益であり、おそらく、他のほとんどの条件下においては、それは、その変種に何の利点も与えないことに留意するのが重要である。リファンピシンは、主として、結核およびライ病、および場合によっては他の疾患を治療するのに使用される抗生物質である。耐性は、膜透過性の変化、またはmRNAポリメラーゼをコードするrpoB遺伝子における変異によるものである。これらの機構は両方とも、染色体突然変異を介して起動する。しかし、大腸菌の染色体には、リファンピシンに対する細菌の耐性を与え得るいくつかの標的遺伝子が存在する。
【0095】
自然突然変異の頻度(SMF)を計算するために、リファンピシンに対する耐性を与える自然突然変異の頻度を、培養中の生存細胞の総数で割って求めた。
【0096】
【数1】
【0097】
A) 強度の変異誘発性表現型(mutLおよび/またはdnaQの染色体欠失)を示す大腸菌株におけるMutLの過剰発現および抗生物質リファンピシン上への該細胞のプレーティング
(実験の概要)
(1.MutLおよびMutSの過剰発現プラスミドの構築)
a) pTrcHis2/lacZへのmutLのクローニング
このプラスミドは、大腸菌におけるMutLの高レベルで、かつ「制御された」発現を実現するために構築した。
【0098】
大腸菌株MG1655から調製されたゲノムDNAを使用して、プライマー1(配列番号1)および2(配列番号2)(表1も参照)によって、MutLタンパク質をコードする1848bpのDNA断片を増幅した。このPCR断片を消化し、NcoI−XhoI断片として、プラスミドpTrcHis2/lacZにクローニングして、プラスミドpmutLを産生した。プラスミドpmutLの物理構造を図1に示す。PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.50;72/2.5)35(72/10);(Taq/Pfu)の比率(5/1)であった。
【0099】
得られたクローンのうち6クローンを単離し、EcoRVによる制限消化によって分析した(予測された断片:871bpおよび5373bp)。6クローンすべてが正しい移動パターンを示した。
【0100】
b) pTrcHis2/lacZへのmutSのクローニング
このプラスミドは、大腸菌におけるMutSの高レベルで、かつ「制御された」発現を実現するために構築した。
【0101】
大腸菌株MG1655から調製されたゲノムDNAを使用して、プライマー3(配列番号3)および4(配列番号4)(表1を参照)によって、MutSタンパク質をコードする2562bpのDNA断片を増幅した。このPCR断片を消化し、BamHI−XhoI断片として、プラスミドpTrcHis2/lacZにクローニングして、プラスミドpmutSを産生させた。プラスミドpmutSは、図1に示すプラスミドpmutLと原則的に同じ物理構造を有する。
【0102】
PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.58;72/3)35(72/10);Herculaseであった。
【0103】
得られたクローンのうち6クローンを単離し、EcoRVによる制限消化によって分析した(予測された断片:1186bpおよび5775bp)。6クローンすべてが正しい移動パターンを示した。
【0104】
【表1】
【0105】
(2.ウェスタンブロット分析)
a) MutLの検出
ES568(mutL13;Mut−) pmutLの終夜培養物20μlを0.4%グルコースおよびアンピシリン(100μg/m)を含有するLB10mlに播種し、ODが約0.5に達するまで培養した。グルコースを除去するために、細胞を遠心分離し、必要な抗生物質を含有するLB5mlに懸濁した。最終濃度1mMのIPTGを添加することによって、PlacからのpmutLの発現を誘導した。この培養物を37℃で16時間インキュベートし、その後、遠心分離し、SDS−PAGE分析用に調製した。ウェスタンブロット分析は、AP結合抗His−tac抗体を用いて行った(データは示されていない)。
【0106】
b) MutSの検出
構築されたプラスミドpmutSで、菌株Top10の野生型細胞を形質転換し、得られた形質転換体を精製した。これらの細菌の終夜培養物20μlを、アンピシリン(100μg/ml)および0.4%グルコースを含有するLB10mlに播種し、ODが約0.5に達するまで培養した。グルコースを除去するために、細胞を遠心分離し、必要な抗生物質を含有するLB5mlに懸濁した。最終濃度1mMのIPTGを添加することによって、PlacからのpmutSの発現を誘導した。この培養物を37℃で16時間インキュベートし、その後、遠心分離し、SDS−PAGE分析用に調製した。ウェスタンブロット分析は、AP結合抗His−tac抗体を用いて行った(データは示されていない)。
【0107】
(3.増殖研究)
菌株ES568(mutL13;Mut−)、MG1655 dnaQ::kn(Mut−)、および野生型株MG1655(Mut+)を、プラスミドpmutLまたは親プラスミドpTrcHis2/lacZで形質転換した。アンピシリン100μg/mlを含有するLB寒天プレート上で、得られた単一コロニーを精製し、その後、アンピシリン100μg/mlを含有するLBに播種し、37℃で終夜培養した。
【0108】
これらの細菌の終夜培養物20μlを、0.4%グルコースと、必要な抗生物質(アンピシリン100μg/ml;カナマイシン50μg/ml)とを含有するLB5mlに播種し、ODが約0.5に達するまで培養した。グルコースを除去するために、細胞を遠心分離し、対応する抗生物質を含有するLB5mlに懸濁した。最終濃度1mMのIPTGを添加することによって、PlacからのpmutLの発現を誘導した。この培養物を、37℃で終夜インキュベートし、細胞を遠心分離し、培地容積を1/10(v/v)にまで減少させた。
【0109】
最後に、培養物を希釈して、100μg/mlアンピシリン、または100μg/mlアンピシリン+100μg/mlリファンピシンを含有する寒天プレート上にプレーティングし、30℃で3日間インキュベートした。
【0110】
ウェスタンブロット分析によって、MutLの発現を確認した。
【0111】
要約および結論:(mutL+バックグランドにおける)MutLの過剰発現によって、自然突然変異の頻度が約20分の1に低下することを観測した。高い突然変異率を示すmutL−細胞またはdnaQ−細胞でも、同様の効果が観測された(データは示されていない)。
【0112】
B) 様々な組合せのdinB欠失および/またはdnaE911抗変異原アレルを保持する大腸菌株におけるMutLの過剰発現
(1.菌株の構築)
a) 大腸菌株MG1655およびMG1655dinB10の細菌細胞を感染させるために、大腸菌株ES1484(mutL218::Tn10)からP1vir溶解物を調製した。
【0113】
(P1virの調製)
大腸菌株ES1484(mutL218::tet)の終夜培養物のアリコートを、CaCl2(5mM)を含有する37℃のLBに播種した。指数関数的増殖期に、(野生型株MG1655の)P1virを添加した。感染した培養物を37℃で約4h、細胞溶解が観察されるまでインキュベートした。CHCl3を添加し、溶解物を遠心分離し、CHCl3を含有する新しいチューブに上清を移して、4℃で保存した。
【0114】
(P1形質導入)
宿主株MG1655およびMG1655 dinB10の終夜培養物のアリコートをLBに播種し、指数関数的増殖期に達するまで37℃で培養した。細胞を遠心分離し、CaCl2(cE=5mM)を含有するMgSO4(cE=10mM)に再懸濁し、調製されたP1vir溶解物を添加し、細胞混合物をRTで30分間インキュベートした。(ファージ感染を停止させるために)クエン酸ナトリウムを添加し、栄養源としてSOCを添加した。細胞を37℃で1hインキュベートした後、テトラサイクリン(12.5μg/ml)を含有する寒天プレート上にアリコートをプレーティングした。得られた単一コロニーを精製し、試験し、単離された株を保存した。
【0115】
b) 大腸菌株MG1655、MG1655 dinB10、MG1655 mutL::tet、およびMG1655 dinB10 mutL::tetの細菌細胞を感染させるために、dnaE株NR10171(dnaE911 zae::cm)から、P1vir溶解物を調製した。得られた単一コロニーを精製し、試験し、単離された株を保存した。
【0116】
(2.増殖研究)
細菌株MG1655、MG1655 dinB10、MG1655 dnaE911 zae::cm、MG1655 dinB10 dnaE911 zae::cm、MG1655 mutL::tet、MG1655 dinB10 mutL::tet、MG1655 dinB10 dnaE911 zae::cm mutL::tet、およびMG1655 dinB10 dnaE911 zae::cm mutL::tetを、プラスミドpTrcHis2AまたはpmutLで形質転換した。得られた単一コロニーを精製し、100μg/mlアンピシリンを含有するLBに播種し、30℃で終夜培養した。
【0117】
これらの細菌の終夜培養物20μlを、100μg/mlアンピシリンを含有する30℃のLB5mlに播種した。指数関数的増殖期に、PlacからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加した。この培養物を1時間、30℃に維持し、その後、温度を37℃に変化させ、細胞を37℃で終夜インキュベートした。
【0118】
細胞を遠心分離し、培地の容積を1/10(v/v)にまで減少させた。
【0119】
最後に、培養物を希釈して、100μg/mlアンピシリン、または100μg/mlアンピシリン+100μg/mlリファンピシンを含有する寒天プレート上にプレーティングし、30℃で3日間インキュベートした。
【0120】
リファンピシン耐性への自然突然変異の頻度(SMF)の値を表2に示す。図2はMutLの発現による変異性の抑制を示す。
【0121】
【表2】
【0122】
dinB10(欠失)またはdnaE911(アレル)などの抗変異原の作用を介して、変異性(SMF値)がそれぞれ4または3分の1に低下する。さらに、それらの株でMutLを発現させることによって、変異性がさらに低下する(2から7分の1)ことを観測した。dinB10およびdnaE911の両方の抗変異原を保持する細菌株は、野生型株MG1655と比較して、変異性の10分の1の低下を示す。MutLを追加して発現させても、変異性に変化は見られない。
【0123】
mutL遺伝子の染色体欠失は、宿主株の変異性を、強烈に約103倍に増大させる。この変異性は、抗変異原dinB10(5分の1)、dnaE911(3分の1)、dinB10およびdnaE911(10分の1)を介して低下した。MutLを追加して誘導することによって、変異性が50分の1にまで(MG1655 mutL::tetでは50分の1、MG1655 dinB10 mutL::tetでは22分の1、MG1655 dnaE911 zae::cm mutL::tetでは20分の1、そして、MG1655 dinB10 dnaE911 zae::cm mutL::tetでは11分の1)低下する。
【0124】
要約および結論:抗変異原dinB10およびdnaE911は、自然突然変異の頻度を3から4分の1で低下させる。これらの菌株でMutLを発現させることによって、この頻度がさらに2から7分の1で低下した。
【0125】
C) 様々な組合せのdinB欠失および/またはdnaE911抗変異原を保持するmutS−株の変異性
前述の結果を確認するために、dinB10(ポリメラーゼIVの欠失)および/またはdnaE911(ポリメラーゼIIIのαサブユニットのコード配列内のアレル)の抗変異原効果をmutS−バックグランドで試験した。
【0126】
(1.菌株の構築)
大腸菌株MG1655、MG1655 dinB10、MG1655 dnaE911 zae::cm、およびMG1655 dinB10 dnaE911 zae::cmの細菌細胞を感染させるために、P1vir溶解物を大腸菌株ES1481(mutL215::Tn10)から調製した。得られた単一コロニーを精製し、試験し、その菌株を保存した。
【0127】
(2.増殖研究)
これらの細菌の終夜培養物10μlを、12:5μg/mlテトラサイクリンを含有する30℃のLB10mlに播種した。指数関数的増殖期(8hの増殖の後)に、温度を37℃に変化させ、細胞を37℃で終夜インキュベートした。
【0128】
細胞を遠心分離し、培地の容積を1/10(v/v)にまで減少させた。
【0129】
最後に、培養物を希釈して、リファンピシン(100μg/ml)の存在下および非存在下に寒天プレートにプレーティングし、30℃で5日間インキュベートした。
【0130】
リファンピシン耐性への自然突然変異の頻度(SMF)の値を表3に示す。図3は、mutS−バックグランドにおける変異性の抑制を示す。
【0131】
【表3】
【0132】
dinB10またはdnaE911などの抗変異原の存在下では、野生型と比較して、変異性(SMF値)が低下している(約1.5分の1)。さらに、抗変異原dinB10およびdnaE911を保持する細菌株は、野生型MG1655と比較して、変異性の4.5分の1の低下を示す。
【0133】
mutS遺伝子の染色体欠失は、宿主株の変異性を劇的に増大させる。この変異性は、抗変異原dinB10(12.5分の1)、dnaE911(10分の1)、dinB10およびdnaE911(60分の1)によって低下する。
【0134】
結論および要約:dinB10またはdnaE911によって引き起こされた抗変異原効果は、mutSの染色体欠失によって引き起こされたMut−表現型を部分的に補完する。
【0135】
実施例2
(dapA変異の復帰変異)
特定の標的遺伝子における点変異およびフレームシフト変異の復帰変異を研究するために、大腸菌において栄養要求変異株表現型を有する系(dapA−株)を用いた。
【0136】
dapA遺伝子はジヒドロジピコリン酸シンターゼをコードし、この酵素はピンポン機構を介してL−アスパラギン酸−β−セミアルデヒドおよびピルビン酸からジヒドロピコリン酸への縮合を触媒するが、その機構では、ピルビン酸が、リジン残基とシッフ塩基を形成することによって、この酵素に結合する。機能しない遺伝子産物に導くdapA遺伝子変異を有する細菌細胞は、DL−ジアミノピメリン酸(DAP)を含有しない培地では増殖できない。
【0137】
dapA−バックグランド(dapA15(DapA−表現型を有する点突然変異)またはdapA::kn)において、dapAIF(wt ORF)、dapA+1(フレームシフト変異)、dapA+2、またはdapA15(点突然変異)をコードするプラスミドの存在下に、MutLを発現させる。復帰変異体は、DL−ジアミノピメリン酸を欠失したプレート上で検出する。
【0138】
(実験の概要)
(1.プラスミドの構築)
a) dapAのクローニング
(pTrcHis2/lacZへのdapA(dapA15)、dapA+1(dapA15+1)、dapA+2(dapA15+2)のクローニング)
大腸菌株MG1655(wt)またはAT997(dapA15)から調製されたゲノムDNAを使用し、プライマー5(配列番号5)、6(配列番号6)、7(配列番号7)、および8(配列番号8)(表4を参照)によって、DapAタンパク質をコードする732bpのDNA断片を増幅した。これらのPCR断片を消化し、BamHI−XhoI断片として、プラスミドpTrcHis2/lacZにクローニングして、プラスミドpdapAIF、pdapA+1、およびpdapA+2を産生させた。プラスミドpdapAIFの物理構造を図4Aに示す。PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.50;72/2)35(72/10);Herculaseであった。
【0139】
それぞれの場合について、得られたクローンのうち8クローンをBstEIIによる制限消化によって分析した(予測された断片:1314bpおよび3964bp)。試験された合計24クローンのうち23クローンが正しい移動パターンを示した。
【0140】
(pSU19およびpSU40へのdapA15のクローニング)
大腸菌株AT997から調製されたゲノムDNAを使用して、プライマー9(配列番号9)および10(配列番号10)(表4も参照)によって、DapAタンパク質をコードする732bpのDNA断片を増幅した。このPCR断片を消化し、HindIII−EcoRI断片として、プラスミドpSU19およびpSU40にクローニングして、プラスミドpSU19dapA15およびpSU40dapA15を産生させた。プラスミドpSU40dapAの物理構造を図4Bに示す。
【0141】
PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.50;72/2)35(72/10);(Taq/Pfu)の比率(5/1)であった。
【0142】
得られたクローンのうち10クローンを単離し、NheI(予測された断片:pSU40では1122bpおよび2557bp)またはApoI(予測された断片:478bpおよび3192bp)による制限消化によって分析した。すべてのクローンが正しい移動パターンを示した。
【0143】
2) mutLのクローニング
a) pSU40へのmutLのサブクローニング
プラスミドpmutLをSpHI−XmnI消化し、3629bpの断片をゲル電気泳動によって単離した。プラスミドpSU40をSphI−HincII消化し、2680bpの断片をゲル電気泳動によって単離した。精製された2つの断片を粘着端−平滑端連結して、プラスミドpSU40mutLを産生させた。プラスミドpSU40mutLを産生するストラテジーを図5に示す。
【0144】
得られたクローンのうち6クローンを、NcoI(予測された断片:2675bpおよび3634bp)、PvuI(予測された断片:1368bpおよび4941bp)、およびBglI(予測された断片:1828bpおよび4481bp)による制限消化によって分析した。試験された6クローンのうち3クローンが正しい移動パターンを示した。
【0145】
b) プラスミドpmutLspec: specによるblaの置換
(プラスミドpmutLにおける、スペクトマイシン耐性遺伝子によるアンピシリン耐性遺伝子の置換)
プラスミドpIC156から調製されたDNAをFspI−XmnI消化し、ゲル電気泳動を用いて1304bpの断片を単離し、プラスミドpmutLから調製されたDNAをScaI−XmnI消化し、ゲル電気泳動を用いて5443bpの断片を単離した。精製された2つの断片を平滑端−平滑端連結して、プラスミドmutLspecを産生させた。
【0146】
得られたクローンのうち12クローンをEcoRI(予測された断片:1291bpおよび5456bp)による制限消化によって分析した。試験された12クローンのうち2クローンが正しいサイズを示した。
【0147】
【表4】
【0148】
(2.ウェスタンブロット分析)
a) DapA/DapA15の検出
菌株dapA::knを、pdapAIF、pdapA+1、pdapA+2、pdapA15IF、pdapA15+1、およびpdapA15+2で形質転換し、観察された形質転換体を精製した。
【0149】
これらの細菌の終夜培養物20μlを、0.4%グルコース、DL−ジアミノピメリン酸(100μg/ml)、アンピシリン(100μg/ml)、およびカナマイシン(50μg/ml)を含有するLB10mlに播種し、ODが約0.5に達するまで培養した。グルコースを除去するために、細胞の一部を遠心分離し、必要な抗生物質を含有するLB5mlに懸濁した。最終濃度1mMのIPTGを添加することによって、Placからの様々なpTrcHis2/lacZ派生物の発現を誘導した。この培養物を37℃で16時間インキュベートし、その後、遠心分離し、SDS−PAGE分析用に調製した。ウェスタンブロット分析は、AP結合抗His−tac抗体を用いて行った(データは示されていない)。
【0150】
b) MutLの検出
野生型株Top10をプラスミドpmutLspecで形質転換し、得られた形質転換体を精製した。これらの細菌の終夜培養物20μlを、スペクトマイシン(75μg/ml)を含有するLB10mlに播種し、ODが約0.5に達するまで培養した。この培養物を5mlアリコートに分割し、プロモーターPlacからのMutLの発現を誘導するために、最終濃度1mMのIPTGを1回、添加した。この培養物を37℃で4時間インキュベートし、その後、遠心分離し、SDS−PAGE分析用に調製した。MutLの発現を証明するために、AP結合抗His−tac抗体を用いてウェスタンブロット分析を行った(データは示されていない)。
【0151】
(3.菌株の構築)
大腸菌株MG1655、MG1655 dinB10、MG1655 dnaE911 zae::cm、MG1655 dinB10 dnaE911 zae::cm、MG1655 mutL::tet、MG1655 dinB10 mutL::tet、MG1655 dnaE911 zae::cm mutL::tet、MG1655 dinB10 dnaE911 zae::cm mutL::tetの細菌細胞を感染させるために、P1vir溶解物を大腸菌株dapA::knから調製した。得られた単一コロニーを精製し、試験し、単離された株を保存した。
【0152】
(4.増殖研究)
a) 点突然変異の復帰変異:
細菌株AT997(dapA15)およびMG1655 dinB10 dapA::knを、プラスミドpTrcHis2/lacZおよびpSU19dapA15、またはプラスミドpmutLおよびpSU19dapA15で形質転換した。得られた単一コロニーを精製し、100μg/mlアンピシリン、30μg/mlクロラムフェニコール、および50μg/ml DAPを含有するLBに播種して、37℃で終夜培養した。
【0153】
b) フレームシフト変異の復帰変異:
細菌株AT997(dapA15;DapA−)を、プラスミドpSU40およびpdapAIF、プラスミドpSU40mutLおよびpdapAIF、プラスミドpSU40およびpdapA+1、プラスミドpSU40mutLおよびpdapA+1、プラスミドpSU40およびpdapA+2、またはプラスミドpSU40mutLおよびpdapA+2で形質転換した。得られた単一コロニーを精製し、100μg/mlアンピシリン、50μg/mlカナマイシン、および50μg/ml DAPを含有するLBに播種して、37℃で終夜培養した。
【0154】
細菌細胞の終夜培養物20μlを、100μg/mlアンピシリン、50μg/mlクロラムフェニコール、および50μg/ml DAPを含有する30℃のLB10mlに播種した。指数関数的増殖期に、PlacプロモーターからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加した。この培養物を1時間、30℃に維持し、その後、温度を37℃に変化させ、細胞を37℃で終夜インキュベートした。
【0155】
最後に、培養物を希釈して、9mlの培地を取り除き、残っている細胞を希釈し、100μg/mlアンピシリン+30μg/mlクロラムフェニコール、または100μg/mlアンピシリン+30μg/mlクロラムフェニコール+50μg/ml DAPを含有する寒天プレート上にプレーティングし、30℃で5日間インキュベートした。
【0156】
野生株およびMutL過剰発現株におけるdapA変異の復帰変異の値を表5に示す。図6は、MutLの発現による変異性の抑制を示す。
【0157】
【表5】
【0158】
対照株(1〜3)であるTop10(wt;DapA+)、AT997(dapA15;DapA−)、およびMG1655dapA::kn(DapA−)は、予測通りのdap−/dap+値、すなわち野生型株では約1(DL−ジアミノピメリン酸非存在下で増殖する)、そして、dapA−株では0(DL−ジアミノピメリン酸非存在下で増殖しない)を示す。
【0159】
機能するdapA遺伝子がプラスミドpdapAIFに存在するので、AT997(dapA15) pdapAIF pTrc(4)、およびAT997(dapA15) pdapAIF pmutL(5)の2株は、予測通りの野生型表現型を示す。dapA−宿主中のプラスミドにコードされているdapAのフレームシフト変異体(6、8)または点突然変異(10)は、DAPの非存在下における細胞増殖を抑制するはずであり、一方、導入された変異の復帰変異(7、11)は、DAPの非存在下における増殖を可能にする。
【0160】
要約および結論:いずれの場合(フレームシフトおよび点突然変異)も、変異からの復帰が、MutLの非存在下でより高い割合で観察された。フレームシフト変異では、dap−/dap+値が1000分の1にまで低下し、点突然変異では10分の1にまで低下した。これは、MutLの発現が、変異からの復帰を抑制し、それによって、染色体における自然突然変異の修復に対抗して作用することを意味する。
【0161】
実施例3
(「発酵状態」中におけるMutLの発現)
トリプトファンによって制御されたG−CSF発現ベクターを保持する産生株JM83 pXX7の変異性を、A)細胞をリファンピシリン上にプレーティングすることによる自然突然変異の発生率の計算、B)細胞をホスホマイシン上にプレーティングすることによる自然突然変異の発生率の計算、およびC)産生プラスミドpXX7(G−CSFをコードする)へのレポータ遺伝子の導入によって分析した。
【0162】
B)に向けて、実験条件下では、ホスホマイシンに対する耐性が大腸菌でより迅速に発生するので、リファンピシリンをホスホマイシンで置換した。ホスホマイシンは、無併発性の下部尿路感染の治療に主として使用される細胞壁阻害剤である。
【0163】
(実験の概要)
(1.クローニング)
a) pXX7へのクロラムフェニコールカセットのクローニング:
プラスミドpSUI9から調製されたDNAを使用して、プライマー11(配列番号11)および12(配列番号12)(表6)によって、クロラムフェニコール耐性をコードするDNA断片を増幅した。これらのPCR断片を消化し、BbvcI−AhdI断片として、プラスミドpXX7にクローニングして、プラスミドpXX7cmを産生させた。プラスミドpXX7cmを産生するストラテジーを図7Aに示す。選択過程を単純化するために、「シャイン−ダルガルノ」配列の−10領域のみをクローニングした。
【0164】
PCR条件(温度(℃)/時間(分))は以下の通り、すなわち(98/10)(96/0.75;55/0.50;72/1)35(72/10);Herculaseである。
【0165】
【表6】
【0166】
b) プラスミドpXX7へのサイレントtetA遺伝子のクローニング:
プラスミドpGBG1の選択カートリッジを単離し、MslI−SmaI断片として、プラスミドpXX7にクローニングして、プラスミドpXX7tetを産生させた(平滑端−平滑端連結)。プラスミドpXX7tetを産生するストラテジーを図7Bに示す。
【0167】
プラスミドpGBG1は、様々なグラム陰性菌における移動性エレメントおよび他の自然突然変異の単離および特性分析を目的として構築された(Schneiderら、Plasmid 44、201−207(2000年))。選択カートリッジは、変異導入の標的を含有し、バクテリオファージλのPRプロモーターの制御下にあるサイレントtetA遺伝子で構成されており、このPRプロモーターはλCIリプレッサーで抑制される。cIを不活性化するか、あるいはCIの結合部位を除去する自然突然変異(点突然変異、欠失、および挿入)はPRを抑制解除し、それによって、tetA発現の引き金となる。変異導入の標的は、約1kbの長さである。この場合、MutLの発現は、変異の発生率を低下させ、それによって、所与の宿主株におけるテトラサイクリン耐性を減少させるはずである。
【0168】
(2.増殖研究)
A) 細胞をリファンピシリン上にプレーティングすることによる大腸菌の自然突然変異の頻度(SMF)の測定
細菌株JM83 pXX7を、プラスミドpTrcHis2/lacZ(対照プラスミド)およびpmutLで形質転換した。得られた単一コロニーを精製し、その細胞を、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有するLBに播種し、30℃で終夜培養した。これらの細菌の終夜培養物3mlを、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有する30℃のRMG100mlに播種した。指数関数的増殖期に、プラスミドpmutLのPlacプロモーターからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加し、さらに、プラスミドpXX7からのG−CSFの発現を誘導するために、最終濃度100μg/mlのトリプトファンを添加した。誘導の1時間後に、播種温度を30℃から37℃に変化させ、その後、細胞を終夜、37℃に維持した。
【0169】
細胞を遠心分離して、培地容積を10mlまで減少させた。
【0170】
最後に、培養物を希釈して、
a) 100μg/mlアンピシリン、および必要に応じて50μg/mlカナマイシン、
b) 100μg/mlアンピシリン、ならびに、必要に応じて50μg/mlカナマイシンおよび100μg/mlリファンピシン
を含有する、A) LBAプレート、B) RMGプレート、およびC) M9プレート上にプレーティングし、30℃で3日間インキュベートした。
【0171】
菌株JM83 pXX7、JM83 pXX7 pTrc、およびJM83 pXX7 pmutLのリファンピシン耐性への自然突然変異の頻度の値を図8に示す。産生株JM83 pXX7は約0.45のSMF値を示す。このSMF値は、産生株に「空」のベクターであるpTrcを追加して導入した場合でも維持される。しかし、さらにpmutLをJM83 pXX7に導入すると、MutLが過剰発現された場合、SMF値の約2分の1の低下が引き起こされる。
【0172】
B) 細胞をホスホマイシン上にプレーティングすることによる大腸菌の自然突然変異の頻度(SMF)の測定
細菌株JM83 pXX7を、プラスミドpTrcHis2AおよびpmutLで形質転換した。得られた単一コロニーを精製し、それぞれの株(JM83 pXX7、JM83 pXX7 pTrcA、およびJM83 pXX7 pmutL)につき20の独立かつ特有の細胞を、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有するLBに播種し、30℃で終夜培養した。
【0173】
これらの細菌の終夜培養物10μlを、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有する30℃のRMG10mlに播種した。指数関数的増殖期に、プラスミドpmutLのPlacからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加し、さらに、プラスミドpXX7のPtrpからのG−CSFの発現を誘導するために、最終濃度100μg/mlのトリプトファンを添加した。誘導の1時間後に、播種温度を30℃から37℃に変化させ、その後、細胞を終夜、37℃に維持した。
【0174】
細胞を遠心分離して、培地容積を1mlにまで減少させた。
【0175】
最後に、培養物を希釈して、
a) 100μg/mlアンピシリン、および必要に応じて50μg/mlカナマイシン、
b) 100μg/mlアンピシリン、ならびに、必要に応じて50μg/mlカナマイシンおよび30μg/mlホスホマイシン
を含有するLB寒天プレート上にプレーティングし、30℃で3日間インキュベートした。
【0176】
菌株JM83 pXX7、JM83 pXX7 pTrc、およびJM83 pXX7 pmutLのホスホマイシン耐性への自然突然変異の頻度の値を図9に示す。産生株JM83 pXX7は、約4.9のSMF値を示す。このSMF値は、産生株に「空」のベクターであるpTrcを追加して導入した場合でも維持される。しかし、さらにpmutLをJM83 pXX7に導入すると、MutLが過剰発現された場合、SMF値の約3分の1の低下が引き起こされる。
【0177】
要約および結論:MutLの過剰発現は、G−CSF産生株の変異性を約3分の1に低下させ、さらに、細胞生存率を103倍にまで増強する。
【0178】
C) レポータ遺伝子の導入による大腸菌株JM83pXX7の自然突然変異の測定
a) G−CSFの発現
b) G−CSFの発現および抑制
G−CSF活性をモニターすることができなかったので、プラスミドpXX7にレポータ遺伝子を導入した。
【0179】
変異生成を2つの異なった系でアッセイした。第1の系では、クロラムフェニコール耐性遺伝子を使用し、これを弱いプロモーターによって制御する。cm遺伝子の発現を不活性化する点変異、欠失、および挿入などの自然突然変異は、クロラムフェニコール耐性を低減する。MutLの不在は、培養中の生存細胞の総数に対する、クロラムフェニコール耐性を与える自然突然変異の割合の減少として報告されるはずである。第2の系では、バクテリオファージλのPRプロモーターおよびλcIリプレッサーの制御下にサイレントtetA遺伝子を含有するカセットを使用し、λcIリプレッサーはtetAの発現を阻止する。cIを不活性化するか、あるいはCIの結合部位を除去する、点突然変異、欠失、および挿入などの自然突然変異は、PRの抑制解除に導き、それによって、tetA発現の引き金となる。MutLの発現は、培養中の生存細胞の総数に対する、テトラサイクリン耐性を与える自然突然変異の割合の減少として報告されるはずである。
【0180】
a) G−CSFの発現
細菌株JM83をプラスミドpXX7cm、pXX7cm pTrcHis2/lacZ、pXX7cm pmutL、pXX7tet、pXX7tet pTrcHis2/lacZ、およびpXX7tet pmutLで形質転換した。得られた単一コロニーを精製し、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有するLBに細胞を播種し、30℃で終夜培養した。
【0181】
これらの細菌の終夜培養物10μlを、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有する30℃のRMG10mlに播種した。指数関数的増殖期に、プラスミドpmutLのPlacからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加し、さらに、プラスミドpXX7のPtrpからのG−CSFの発現を誘導するために、最終濃度100μg/mlのトリプトファンを添加した。誘導の1時間後に、播種温度を30℃から37℃に変化させ、その後、細胞を終夜、37℃に維持した。
【0182】
細胞を遠心分離して、培地容積を1mlにまで減少させた。
【0183】
最後に、培養物を希釈して、様々な抗生物質(表7に示す)を含有するLB寒天プレート上にプレーティングし、30℃で4日間インキュベートした。
【0184】
【表7−1】
【表7−2】
【0185】
菌株JM83 pXX7cm、JM83 pXX7cm pTrc、およびJM83 pXX7cm pmutLのクロラムフェニコール耐性レポータ遺伝子における自然突然変異の頻度の値を図10に示す。JM83 pXX7cmおよびJM83 pXX7cm pTrcの2株では、変異性の値が0.28または0、35(Δ1.25)であった。MutLの発現によって、変異性の値が0.49(Δ1.75)に低下する。これは、MutLの発現が、クロラムフェニコール耐性からクロラムフェニコール感受性への転換を抑制し、それによって、染色体における自然突然変異の修復に対抗して作用することを意味する。
【0186】
菌株JM83 pXX7tet、JM83 pXX7tet pTrc、およびJM83 pXX7tet pmutLのサイレントtetAレポータ遺伝子における自然突然変異の頻度の値を図11に示す。JM83 pXX7tetおよびJM83 pXX7tet pTrcの2株では、変異性の値が0.43または0.39(Δ1.1)であった。MutLの発現によって、変異性の値が0.25(Δ1.72)に低下する。これは、MutLの発現が、テトラサイクリン感受性からテトラサイクリン耐性への転換を抑制し、それによって、染色体における自然突然変異の修復に対抗して作用することを意味する。
【0187】
b) G−CSFの発現および抑制
野生型細菌株JM83を、プラスミドpXX7cm、pXX7cm pTrcHis2/lacZ、pXX7cm pmutL、pXX7tet、pXX7tet pTrcHis2/lacZ、およびpXX7tet pmutLで形質転換した。得られた単一コロニーを精製し、その細胞を、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有するLBに播種し、30℃で終夜培養した。
【0188】
これらの細菌の終夜培養物10μlを、50μg/mlカナマイシン、そして、必要に応じて100μg/mlアンピシリンを含有する30℃のRMG10mlに播種した。指数関数的増殖期に、プラスミドpmutLのPtrcプロモーターからのMutLの発現を強化するために、最終濃度1mMのIPTGを添加した。この培養物を2等分割し、プラスミドpXX7のPtrpプロモーターからのG−CSFの発現を誘導するために、最終濃度100μg/mlのトリプトファンを1回添加した。誘導の1時間後に、播種温度を30℃から37℃に変化させ、その後、細胞を終夜、37℃に維持した。
【0189】
細胞を遠心分離して、培地容積を1mlにまで減少させた。
【0190】
最後に、培養物を希釈して、様々な抗生物質(表7参照)を含有するLB寒天プレート上にプレーティングし、37℃で終夜インキュベートした。
【0191】
発酵状態中における、改変された産生株JM83 pXXcmの変異性を図12に示す。G−CSFの非存在下では、JM83 pXX7cm、JM83 pXX7cm pTrc、およびJM83 pXX7cm pmutLの、測定されたクロラムフェニコール耐性の頻度が50と55との間を移行する。G−CSFの誘導によって、JM83 pXX7tetおよびJM83 pXX7tet pTrcの値が3分の1に低下する。しかし、菌株JM83 pXX7 pmutLでは、G−CSFが発現された際に、クロラムフェニコール耐性が維持されている。
【0192】
MutLの発現は、クロラムフェニコール耐性からクロラムフェニコール感受性への変換を抑制する。MutLは、染色体における自然突然変異の修復に対抗して作用する。
【0193】
図13は、発酵状態中における、改変された産生株JM83 pXX7の変異性が有意に増大していることを示す。しかし、プラスミドによってコードされたMutLを発現する菌株JM83 pXX7 pmutLでは、G−CSFが発現された際にテトラサイクリン耐性が一定に保たれている。MutLの発現は、テトラサイクリン耐性からテトラサイクリン感受性への転換を抑制し、染色体における自然突然変異の修復に対抗して作用する。
【0194】
結論および要約:得られた結果は、MutLの過剰発現が、G−CSF産生中のG−CSF発酵株の変異性を約3分の1に低下させることを示す。さらに、MutLの過剰発現が細胞生存率を103倍にまで増強することも見出された。
【0195】
G−CSFの誘導は、試験された産生株の自然突然変異の発生率を約2から3倍に上昇させる。
【特許請求の範囲】
【請求項1】
少なくとも2つの変異を細胞または生物に導入することによって、前記細胞または生物における自然突然変異の頻度を低下させる方法であって、その複合作用が少なくとも2つの細胞性DNA修復機構の強化をもたらす、方法。
【請求項2】
少なくとも2つの変異を生物の少なくとも1つの細胞に導入することによって、自然突然変異の頻度が低下している細胞または生物を産生し、前記生物が多細胞生物である場合には、それから前記生物を再生する方法であって、その複合作用が少なくとも2つの細胞性DNA修復機構の強化をもたらす、方法。
【請求項3】
前記細胞が原核細胞である、請求項1または2に記載の方法。
【請求項4】
前記原核細胞が古細菌または真正細菌の細胞である、請求項3に記載の方法。
【請求項5】
前記真正細菌の細胞がグラム陽性細菌またはグラム陰性細菌の細胞である、請求項4に記載の方法。
【請求項6】
前記細胞が真核細胞である、請求項1または2に記載の方法。
【請求項7】
前記真核細胞が、真菌細胞、動物細胞、または植物細胞である、請求項6に記載の方法。
【請求項8】
前記生物が、真菌、動物、または植物である、請求項1、2、または5から7のいずれか一項に記載の方法。
【請求項9】
前記少なくとも2つの変異の複合作用が、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす、請求項1から8のいずれか一項に記載の方法。
【請求項10】
自然発生した突然変異を修復するミスマッチ修復系、複製後修復系、および/またはSOS修復系の能力が強化される、請求項9に記載の方法。
【請求項11】
前記少なくとも2つの変異が、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから選択される、請求項1から10のいずれか一項に記載の方法。
【請求項12】
MutL、MutS、またはそれらの相同タンパク質の発現の上方制御が、ベクターを細胞内に導入することによって実現され、前記ベクターが、MutL、MutS、またはそれらの相同タンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、mutL遺伝子、MutLの相同タンパク質をコードする遺伝子、mutS遺伝子、またはMutSの相同タンパク質をコードする遺伝子を含む、請求項11に記載の方法。
【請求項13】
前記ベクターが多コピープラスミドである、請求項12に記載の方法。
【請求項14】
前記調節ユニットが誘導性または構成的プロモーターである、請求項11または12に記載の方法。
【請求項15】
1コピーまたは複数コピーのそれぞれのmut遺伝子を1つまたは複数の調節ユニットの機能的制御下に宿主細胞の染色体に導入すること、および/または対応する野生型細胞と比較して、それぞれのMutタンパク質の産生が増強されるように、1つまたは複数の変異を、天然のMutタンパク質の発現を制御する調節ユニットに導入することによって、MutL、MutS、またはそれらの相同タンパク質の発現の上方制御が実現される、請求項11に記載の方法。
【請求項16】
DNAポリメラーゼIVをコードする遺伝子の抗変異原アレルがdinB10である、請求項11に記載の方法。
【請求項17】
DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルがdnaE911である、請求項11に記載の方法。
【請求項18】
dinB10およびdnaE911の複合作用によって、自然突然変異の頻度が、野生型細胞または野生型生物と比較して少なくとも10分の1に低下する、請求項1から17のいずれか一項に記載の方法。
【請求項19】
dinB10、dnaE911、および過剰発現したmutLの複合作用によって、自然突然変異の頻度が、野生型細胞または野生型生物と比較して少なくとも50分の1に低下する、請求項1から18のいずれか一項に記載の方法。
【請求項20】
前記少なくとも2つの変異の複合作用によって、細胞生存率の増強に導かれる、請求項1から19のいずれか一項に記載の方法。
【請求項21】
自然突然変異の頻度の低下および/または細胞生存率の増強を有し、請求項1から20のいずれか一項に記載の方法で取得可能な細胞であって、前記細胞が少なくとも2つの変異を含み、その複合作用が少なくとも2つの細胞性DNA修復機構の強化をもたらす、細胞。
【請求項22】
前記細胞が、細菌、真菌、植物、または動物細胞である、請求項21に記載の細胞。
【請求項23】
自然突然変異の頻度が低下しており、請求項1から20のいずれか一項に記載の方法によって取得可能な生物であって、前記生物の細胞が少なくとも2つの変異を含み、その複合作用が少なくとも2つの細胞性DNA修復機構の強化をもたらす、生物。
【請求項24】
プラスミドpmutLを含有する大腸菌MG1655 dinB10(DSM17016)。
【請求項25】
プラスミドpmutLを含有する大腸菌MG1655 dinB10 mutL::tet(DSM17017)。
【請求項26】
プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm(DSM17018)。
【請求項27】
プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm mutL::tet(DSM17019)。
【請求項28】
大腸菌MG1655 dinB10 dnaE zae::cm(DSM17015)。
【請求項29】
大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet(DSM17014)。
【請求項30】
プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm(DSM17020)。
【請求項31】
プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet(DSM17021)。
【請求項32】
タンパク質の発現系を産生する方法であって、前記タンパク質のアミノ酸配列が自然発生した突然変異に対して安定化されており、
a) 少なくとも2つの変異を含有するタンパク質をコードする核酸配列を、前記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、宿主細胞のゲノムに挿入する工程であって、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす工程、または
b) 前記タンパク質をコードする核酸配列を、前記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下にベクターに挿入し、さらに、少なくとも2つの変異を含有するベクターを宿主細胞内に移入する工程であって、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす工程と、
c) 適当な培地中で前記宿主細胞を培養および/または維持する工程と
を含む方法。
【請求項33】
タンパク質を産生する方法であって、前記タンパク質のアミノ酸配列が自然発生した突然変異に対して安定化されており、
a) 少なくとも2つの変異を含有するタンパク質をコードする核酸配列を、前記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、宿主細胞のゲノムに挿入する工程であって、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす工程、または
b) 前記タンパク質をコードする核酸配列を、前記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下にベクターに挿入し、さらに、少なくとも2つの変異を含有するベクターを宿主細胞内に移入する工程であって、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす工程と、
c) 適当な培地中で、前記タンパク質の発現を可能にする条件下に前記宿主細胞を培養する工程と、
d) 発現された前記タンパク質を単離工程と
を含む方法。
【請求項34】
前記タンパク質が培地から単離される、請求項33に記載の方法。
【請求項35】
前記タンパク質が宿主細胞から抽出される、請求項33に記載の方法。
【請求項36】
前記タンパク質が、治療に使用できるタンパク質、詳細にはサイトカインまたは成長因子である、請求項32から35のいずれか一項に記載の方法。
【請求項37】
前記ベクターが、プラスミド、バクテリオファージ、またはコスミドである、請求項32から36のいずれか一項に記載の方法。
【請求項38】
前記調節ユニットが、プロモーター、リボソーム結合部位、エンハンサー、サイレンサー、および/または3’転写ターミネーターである、請求項32から37のいずれか一項に記載の方法。
【請求項39】
前記タンパク質をコードする核酸配列が、細胞小器官、細胞内区画、細胞外間隙、または培地中への、発現されたタンパク質の輸送を指示するリーダー配列に機能的に連結されている、請求項32から38のいずれか一項に記載の方法。
【請求項40】
発酵産物、および/または発酵産物の形成に関与する少なくとも1つの酵素を産生する細胞を培地中で培養することによって、発酵産物を産生する方法であって、少なくとも2つの変異によって前記細胞のゲノムが自然発生の配列変化に対して安定化されており、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす、方法。
【請求項41】
前記発酵産物が、核酸、ヌクレオシド、ヌクレオチド、アミノ酸、タンパク質、酸、糖質、ビタミン、抗生物質、またはアルカロイドである、請求項40に記載の方法。
【請求項42】
自然発生した突然変異を修復するミスマッチ修復系、プルーフリーディング機構、および/またはSOS修復系の能力が強化される、請求項32から41のいずれか一項に記載の方法。
【請求項43】
前記少なくとも2つの変異が、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから選択される、請求項32から42のいずれか一項に記載の方法。
【請求項44】
MutL、MutS、またはそれらの相同タンパク質の発現の上方制御が、細胞内のベクターの存在によるものであり、前記ベクターが、mutL遺伝子、MutLの相同タンパク質をコードする遺伝子、mutS遺伝子、またはMutSの相同タンパク質をコードする遺伝子を、MutL、MutS、またはそれらの相同タンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に含む、請求項43に記載の方法。
【請求項45】
それぞれのmut遺伝子を1コピーまたは複数コピー追加して、1つまたは複数の調節ユニットの機能的制御下に宿主細胞の染色体に導入すること、および/またはそれぞれのMutタンパク質の産生が、対応する野生型細胞と比較して増大するように、天然のMutタンパク質の発現を制御する調節ユニットに1つまたは複数の変異を導入することによって、MutL、MutS、またはそれらの相同タンパク質の発現の上方制御が実現される、請求項43に記載の方法。
【請求項46】
DNAポリメラーゼIVをコードする遺伝子の抗変異原アレルがdinB10である、請求項43に記載の方法。
【請求項47】
DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルがdnaE911である、請求項43に記載の方法。
【請求項48】
dinB10およびdnaE911の複合作用によって、自然突然変異の頻度が、野生型細胞と比較して少なくとも10分の1に低下する、請求項32から47のいずれか一項に記載の方法。
【請求項49】
dinB10、dnaE911、および過剰発現したmutLの複合作用によって、自然突然変異の頻度が、野生型細胞と比較して少なくとも50分の1に低下する、請求項32から48のいずれか一項に記載の方法。
【請求項50】
前記少なくとも2つの変異の複合作用によって、細胞生存率の増強に導かれる、請求項32から49のいずれか一項に記載の方法。
【請求項51】
前記細胞が原核細胞または真核細胞である、請求項32から50のいずれか一項に記載の方法。
【請求項52】
前記細胞がグラム陽性細菌またはグラム陰性細菌の細胞である、請求項51に記載の方法。
【請求項53】
前記細胞が、真菌細胞、動物細胞、または植物細胞である、請求項51に記載の方法。
【請求項54】
前記細胞が、請求項21、22、または24から31のいずれか一項に記載の細胞であるか、あるいは請求項1から20のいずれか一項に記載の方法で取得可能である、請求項32から53のいずれか一項に記載の方法。
【請求項55】
前記細胞が液体培地中で培養される、請求項32から54のいずれか一項に記載の方法。
【請求項56】
前記細胞が連続培養またはバッチ培養で培養される、請求項55に記載の方法。
【請求項57】
前記細胞が固定される、請求項32から56のいずれか一項に記載の方法。
【請求項58】
前記細胞が固体または半固体の培地上で培養される、請求項32から54のいずれか一項に記載の方法。
【請求項59】
前記発酵産物が前記細胞から単離される、請求項40から58のいずれか一項に記載の方法。
【請求項60】
前記発酵産物が培地から単離される、請求項40から58のいずれか一項に記載の方法。
【請求項61】
請求項32から60のいずれか一項に記載の方法で取得可能なタンパク質。
【請求項62】
請求項40から60のいずれか一項に記載の方法で取得可能な発酵産物。
【請求項1】
少なくとも2つの変異を細胞または生物に導入することによって、前記細胞または生物における自然突然変異の頻度を低下させる方法であって、その複合作用が少なくとも2つの細胞性DNA修復機構の強化をもたらす、方法。
【請求項2】
少なくとも2つの変異を生物の少なくとも1つの細胞に導入することによって、自然突然変異の頻度が低下している細胞または生物を産生し、前記生物が多細胞生物である場合には、それから前記生物を再生する方法であって、その複合作用が少なくとも2つの細胞性DNA修復機構の強化をもたらす、方法。
【請求項3】
前記細胞が原核細胞である、請求項1または2に記載の方法。
【請求項4】
前記原核細胞が古細菌または真正細菌の細胞である、請求項3に記載の方法。
【請求項5】
前記真正細菌の細胞がグラム陽性細菌またはグラム陰性細菌の細胞である、請求項4に記載の方法。
【請求項6】
前記細胞が真核細胞である、請求項1または2に記載の方法。
【請求項7】
前記真核細胞が、真菌細胞、動物細胞、または植物細胞である、請求項6に記載の方法。
【請求項8】
前記生物が、真菌、動物、または植物である、請求項1、2、または5から7のいずれか一項に記載の方法。
【請求項9】
前記少なくとも2つの変異の複合作用が、自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす、請求項1から8のいずれか一項に記載の方法。
【請求項10】
自然発生した突然変異を修復するミスマッチ修復系、複製後修復系、および/またはSOS修復系の能力が強化される、請求項9に記載の方法。
【請求項11】
前記少なくとも2つの変異が、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから選択される、請求項1から10のいずれか一項に記載の方法。
【請求項12】
MutL、MutS、またはそれらの相同タンパク質の発現の上方制御が、ベクターを細胞内に導入することによって実現され、前記ベクターが、MutL、MutS、またはそれらの相同タンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、mutL遺伝子、MutLの相同タンパク質をコードする遺伝子、mutS遺伝子、またはMutSの相同タンパク質をコードする遺伝子を含む、請求項11に記載の方法。
【請求項13】
前記ベクターが多コピープラスミドである、請求項12に記載の方法。
【請求項14】
前記調節ユニットが誘導性または構成的プロモーターである、請求項11または12に記載の方法。
【請求項15】
1コピーまたは複数コピーのそれぞれのmut遺伝子を1つまたは複数の調節ユニットの機能的制御下に宿主細胞の染色体に導入すること、および/または対応する野生型細胞と比較して、それぞれのMutタンパク質の産生が増強されるように、1つまたは複数の変異を、天然のMutタンパク質の発現を制御する調節ユニットに導入することによって、MutL、MutS、またはそれらの相同タンパク質の発現の上方制御が実現される、請求項11に記載の方法。
【請求項16】
DNAポリメラーゼIVをコードする遺伝子の抗変異原アレルがdinB10である、請求項11に記載の方法。
【請求項17】
DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルがdnaE911である、請求項11に記載の方法。
【請求項18】
dinB10およびdnaE911の複合作用によって、自然突然変異の頻度が、野生型細胞または野生型生物と比較して少なくとも10分の1に低下する、請求項1から17のいずれか一項に記載の方法。
【請求項19】
dinB10、dnaE911、および過剰発現したmutLの複合作用によって、自然突然変異の頻度が、野生型細胞または野生型生物と比較して少なくとも50分の1に低下する、請求項1から18のいずれか一項に記載の方法。
【請求項20】
前記少なくとも2つの変異の複合作用によって、細胞生存率の増強に導かれる、請求項1から19のいずれか一項に記載の方法。
【請求項21】
自然突然変異の頻度の低下および/または細胞生存率の増強を有し、請求項1から20のいずれか一項に記載の方法で取得可能な細胞であって、前記細胞が少なくとも2つの変異を含み、その複合作用が少なくとも2つの細胞性DNA修復機構の強化をもたらす、細胞。
【請求項22】
前記細胞が、細菌、真菌、植物、または動物細胞である、請求項21に記載の細胞。
【請求項23】
自然突然変異の頻度が低下しており、請求項1から20のいずれか一項に記載の方法によって取得可能な生物であって、前記生物の細胞が少なくとも2つの変異を含み、その複合作用が少なくとも2つの細胞性DNA修復機構の強化をもたらす、生物。
【請求項24】
プラスミドpmutLを含有する大腸菌MG1655 dinB10(DSM17016)。
【請求項25】
プラスミドpmutLを含有する大腸菌MG1655 dinB10 mutL::tet(DSM17017)。
【請求項26】
プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm(DSM17018)。
【請求項27】
プラスミドpmutLを含有する大腸菌MG1655 dnaE zae::cm mutL::tet(DSM17019)。
【請求項28】
大腸菌MG1655 dinB10 dnaE zae::cm(DSM17015)。
【請求項29】
大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet(DSM17014)。
【請求項30】
プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm(DSM17020)。
【請求項31】
プラスミドpmutLを含有する大腸菌MG1655 dinB10 dnaE zae::cm mutL::tet(DSM17021)。
【請求項32】
タンパク質の発現系を産生する方法であって、前記タンパク質のアミノ酸配列が自然発生した突然変異に対して安定化されており、
a) 少なくとも2つの変異を含有するタンパク質をコードする核酸配列を、前記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、宿主細胞のゲノムに挿入する工程であって、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす工程、または
b) 前記タンパク質をコードする核酸配列を、前記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下にベクターに挿入し、さらに、少なくとも2つの変異を含有するベクターを宿主細胞内に移入する工程であって、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす工程と、
c) 適当な培地中で前記宿主細胞を培養および/または維持する工程と
を含む方法。
【請求項33】
タンパク質を産生する方法であって、前記タンパク質のアミノ酸配列が自然発生した突然変異に対して安定化されており、
a) 少なくとも2つの変異を含有するタンパク質をコードする核酸配列を、前記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下に、宿主細胞のゲノムに挿入する工程であって、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす工程、または
b) 前記タンパク質をコードする核酸配列を、前記タンパク質の誘導性または構成的発現を可能にする1つまたは複数の調節ユニットの機能的制御下にベクターに挿入し、さらに、少なくとも2つの変異を含有するベクターを宿主細胞内に移入する工程であって、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす工程と、
c) 適当な培地中で、前記タンパク質の発現を可能にする条件下に前記宿主細胞を培養する工程と、
d) 発現された前記タンパク質を単離工程と
を含む方法。
【請求項34】
前記タンパク質が培地から単離される、請求項33に記載の方法。
【請求項35】
前記タンパク質が宿主細胞から抽出される、請求項33に記載の方法。
【請求項36】
前記タンパク質が、治療に使用できるタンパク質、詳細にはサイトカインまたは成長因子である、請求項32から35のいずれか一項に記載の方法。
【請求項37】
前記ベクターが、プラスミド、バクテリオファージ、またはコスミドである、請求項32から36のいずれか一項に記載の方法。
【請求項38】
前記調節ユニットが、プロモーター、リボソーム結合部位、エンハンサー、サイレンサー、および/または3’転写ターミネーターである、請求項32から37のいずれか一項に記載の方法。
【請求項39】
前記タンパク質をコードする核酸配列が、細胞小器官、細胞内区画、細胞外間隙、または培地中への、発現されたタンパク質の輸送を指示するリーダー配列に機能的に連結されている、請求項32から38のいずれか一項に記載の方法。
【請求項40】
発酵産物、および/または発酵産物の形成に関与する少なくとも1つの酵素を産生する細胞を培地中で培養することによって、発酵産物を産生する方法であって、少なくとも2つの変異によって前記細胞のゲノムが自然発生の配列変化に対して安定化されており、その複合作用が自然発生した突然変異を修復する少なくとも2つの細胞性DNA修復機構の能力の強化をもたらす、方法。
【請求項41】
前記発酵産物が、核酸、ヌクレオシド、ヌクレオチド、アミノ酸、タンパク質、酸、糖質、ビタミン、抗生物質、またはアルカロイドである、請求項40に記載の方法。
【請求項42】
自然発生した突然変異を修復するミスマッチ修復系、プルーフリーディング機構、および/またはSOS修復系の能力が強化される、請求項32から41のいずれか一項に記載の方法。
【請求項43】
前記少なくとも2つの変異が、MutLタンパク質またはその相同タンパク質の発現の上方制御に導く変異、MutSタンパク質またはその相同タンパク質の発現の上方制御に導く変異、DNAポリメラーゼIVまたはその相同タンパク質をコードする遺伝子の抗変異原アレル、およびDNAポリメラーゼIIIのサブユニットまたはその相同タンパク質をコードする遺伝子の抗変異原アレルから選択される、請求項32から42のいずれか一項に記載の方法。
【請求項44】
MutL、MutS、またはそれらの相同タンパク質の発現の上方制御が、細胞内のベクターの存在によるものであり、前記ベクターが、mutL遺伝子、MutLの相同タンパク質をコードする遺伝子、mutS遺伝子、またはMutSの相同タンパク質をコードする遺伝子を、MutL、MutS、またはそれらの相同タンパク質の過剰発現を可能にする1つまたは複数の調節ユニットの機能的制御下に含む、請求項43に記載の方法。
【請求項45】
それぞれのmut遺伝子を1コピーまたは複数コピー追加して、1つまたは複数の調節ユニットの機能的制御下に宿主細胞の染色体に導入すること、および/またはそれぞれのMutタンパク質の産生が、対応する野生型細胞と比較して増大するように、天然のMutタンパク質の発現を制御する調節ユニットに1つまたは複数の変異を導入することによって、MutL、MutS、またはそれらの相同タンパク質の発現の上方制御が実現される、請求項43に記載の方法。
【請求項46】
DNAポリメラーゼIVをコードする遺伝子の抗変異原アレルがdinB10である、請求項43に記載の方法。
【請求項47】
DNAポリメラーゼIIIのサブユニットをコードする遺伝子の抗変異原アレルがdnaE911である、請求項43に記載の方法。
【請求項48】
dinB10およびdnaE911の複合作用によって、自然突然変異の頻度が、野生型細胞と比較して少なくとも10分の1に低下する、請求項32から47のいずれか一項に記載の方法。
【請求項49】
dinB10、dnaE911、および過剰発現したmutLの複合作用によって、自然突然変異の頻度が、野生型細胞と比較して少なくとも50分の1に低下する、請求項32から48のいずれか一項に記載の方法。
【請求項50】
前記少なくとも2つの変異の複合作用によって、細胞生存率の増強に導かれる、請求項32から49のいずれか一項に記載の方法。
【請求項51】
前記細胞が原核細胞または真核細胞である、請求項32から50のいずれか一項に記載の方法。
【請求項52】
前記細胞がグラム陽性細菌またはグラム陰性細菌の細胞である、請求項51に記載の方法。
【請求項53】
前記細胞が、真菌細胞、動物細胞、または植物細胞である、請求項51に記載の方法。
【請求項54】
前記細胞が、請求項21、22、または24から31のいずれか一項に記載の細胞であるか、あるいは請求項1から20のいずれか一項に記載の方法で取得可能である、請求項32から53のいずれか一項に記載の方法。
【請求項55】
前記細胞が液体培地中で培養される、請求項32から54のいずれか一項に記載の方法。
【請求項56】
前記細胞が連続培養またはバッチ培養で培養される、請求項55に記載の方法。
【請求項57】
前記細胞が固定される、請求項32から56のいずれか一項に記載の方法。
【請求項58】
前記細胞が固体または半固体の培地上で培養される、請求項32から54のいずれか一項に記載の方法。
【請求項59】
前記発酵産物が前記細胞から単離される、請求項40から58のいずれか一項に記載の方法。
【請求項60】
前記発酵産物が培地から単離される、請求項40から58のいずれか一項に記載の方法。
【請求項61】
請求項32から60のいずれか一項に記載の方法で取得可能なタンパク質。
【請求項62】
請求項40から60のいずれか一項に記載の方法で取得可能な発酵産物。
【図1】

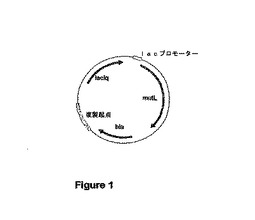
【図2】
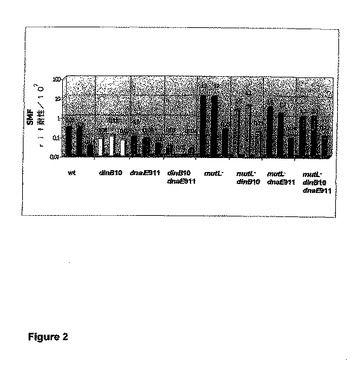
【図3】
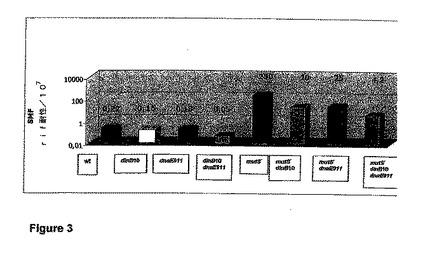
【図4】
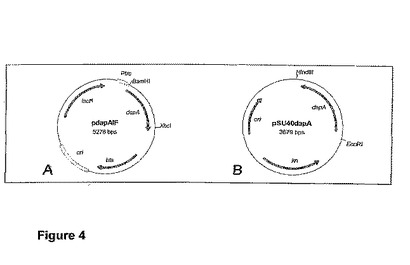
【図5】
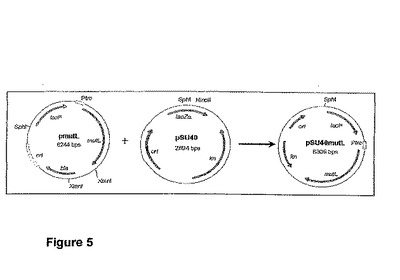
【図6】
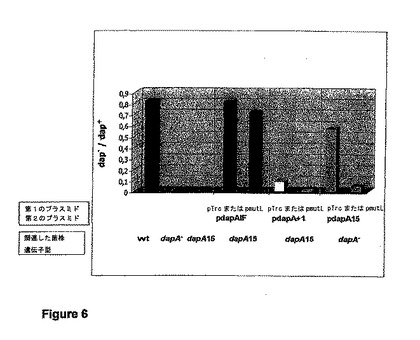
【図7】
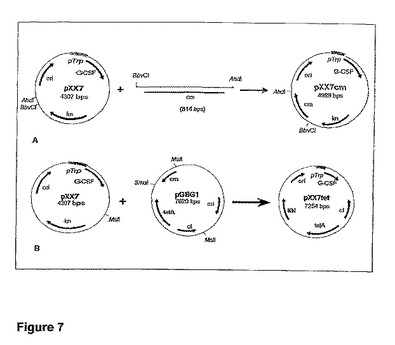
【図8】
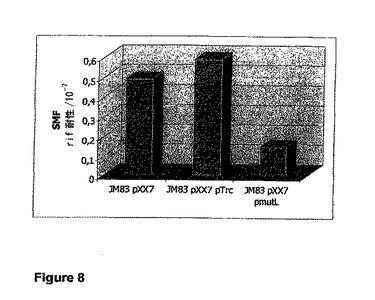
【図9】
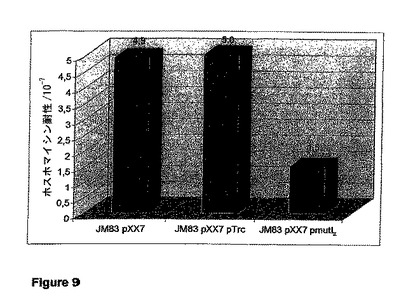
【図10】
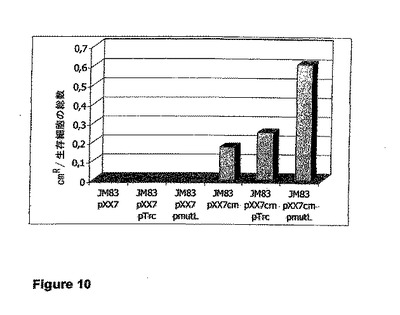
【図11】
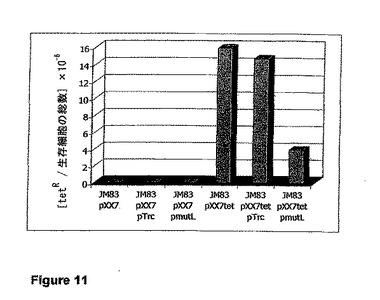
【図12】

【図13】
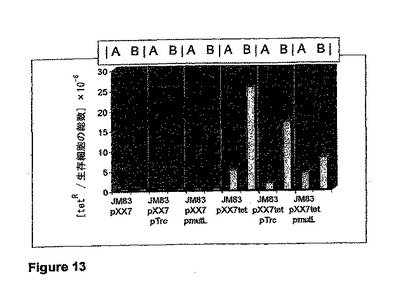
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2012−115276(P2012−115276A)
【公開日】平成24年6月21日(2012.6.21)
【国際特許分類】
【出願番号】特願2012−15735(P2012−15735)
【出願日】平成24年1月27日(2012.1.27)
【分割の表示】特願2007−500178(P2007−500178)の分割
【原出願日】平成17年2月26日(2005.2.26)
【出願人】(504288764)ミクシス フランス ソシエテ アノニム (5)
【Fターム(参考)】
【公開日】平成24年6月21日(2012.6.21)
【国際特許分類】
【出願日】平成24年1月27日(2012.1.27)
【分割の表示】特願2007−500178(P2007−500178)の分割
【原出願日】平成17年2月26日(2005.2.26)
【出願人】(504288764)ミクシス フランス ソシエテ アノニム (5)
【Fターム(参考)】
[ Back to top ]