
細胞内薬物送達のためのFAS(アポ−1,CD95)標的化プラットフォーム
Fasリガンドに特異的であるリガンド結合部分、および薬理学的活性剤またはマーカーのためのキャリアを含む、薬理学的活性剤またはマーカーを細胞に送達するための送達媒体。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、薬物送達の分野に関する。より詳細には、本発明は、標的化送達媒体または薬物キャリア、ならびに治療および診断用途におけるこれらの使用に関する。好ましくは、本発明に従う媒体またはキャリアは、それらの標的細胞に対して高い特異性を有し、薬理学的活性剤または検出可能マーカーの効果的な細胞内送達を提供することができる。態様では、本発明は、薬理学的活性剤または検出可能マーカーをFasリガンド発現細胞へ特異的に送達するための手段を提供する。本発明は、組成物、ならびに治療、特に新生物および神経障害の治療または予防における使用にも拡張される。
【背景技術】
【0002】
現在、ほとんどの薬物は全身投与される。ほとんどの治療薬では、薬剤の大部分が全身クリアランスによって体内から排出され、薬剤の僅かな一部のみが標的器官または組織に到達する。さらに、健全な組織および器官の投与薬物への全身曝露は、重篤な毒性を引き起こす可能性がある。毒性のリスクは、薬物の用量が高く体積が多い場合に悪化する可能性があるが、これらは、低い生体利用度の克服、および対象内での適切な分布の提供に必要であることが多い。逆に、標的化薬物送達は、薬剤を対象とする組織または器官に集中させ、同時に全身薬物曝露を最小限に抑えることを目的としている。しかし、標的化薬物送達の既存のシステムは理想的ではなく、現在の薬物治療計画は、全身薬物曝露に付随する毒性の問題への対処が不十分である。これは、癌治療の分野で特にそうであり、この場合、細胞毒性抗癌薬は、それが健全組織に与えうる損傷の結果としての重篤な副作用を起こす恐れがある。従って、標的組織または器官へ薬物を選択的に送達する標的化薬物送達計画が切実に求められている。そのような計画であれば、薬物に関連する毒性のリスクを最小限に抑えながら、薬物の治療指数が高められることによって薬物治療の効力が改善されるはずである。
【0003】
さらに、RNA干渉(非特許文献1、非特許文献2)またはペプチドに基づく細胞内修飾因子(非特許文献3)などの新しい治療計画と合わせて、ゲノミクス、エピゲノミクス、およびプロテオミクスの知識が豊富となったことで、疾患治療の可能性が変化しつつある。特に、個別の細胞型の治療、または例えば癌で見られるような異常細胞の破壊を選択的に行う目的で、細胞内標的に作用する能力を有する小分子またはより大きい生物学的薬物を製剤することが望ましい。しかし、細胞内活性薬物の候補は、その細胞内標的分子と相互作用を起こすための細胞膜の貫通ができないために、薬理学的に有用となり得ないことが多い。細胞膜は、脂質の二重層であり、これは通常、細胞内部環境を細胞外部(または外側)環境から分離する半透過性バリアとして作用する。細胞膜を通して薬物を運搬するための現在の送達システムは、想定されるほど効率的でも信頼性が高いわけでもない。従って、効率的な細胞内薬物送達システムが求められている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Mello & Conte,2004
【非特許文献2】Grimm,2009
【非特許文献3】Sawyer TK,2009
【発明の概要】
【0005】
本発明の第一の局面によると、薬理学的活性剤またはマーカーを細胞に送達するための、Fasリガンドに特異的なリガンド結合部分および薬理学的活性剤またはマーカーのためのキャリアを含む送達媒体が提供される。
【0006】
この送達媒体は、Fasリガンドに特異的なリガンド結合部分および薬物のためのキャリアを含む薬物送達媒体であってよい。リガンド結合部分は、本発明に従う送達媒体をFasリガンド発現細胞へ標的化することができる。従って、送達媒体は、薬理学的活性剤、マーカー、または薬物の細胞内送達を提供することができる。薬理学的活性剤、マーカー、または薬物は、好ましくは、Fasリガンドと特異的に結合する能力を持つ薬剤ではない。
【0007】
本発明に従うキャリアは、マイクロ粒子、ナノ粒子、マイクロカプセル、マイクロスフェア、ミセル、またはリポソームであってよい。好ましくは、キャリアは、マイクロ粒子である。マイクロ粒子は、0.01μm、0.05μm、0.1μm、0.2μm、0.5μm、1μm、2μm、5μm、または10μmまでの平均径を有していてよい。マイクロ粒子は、0.1μmから10μm、好ましくは0.2μmから5μm、より好ましくは0.3μmから2μm、さらにより好ましくは0.4μmから1.5μm、最も好ましくは0.5μmから2μmの平均径を有していてよい。マイクロ粒子は、乳酸−グリコール酸共重合体(PLGA)マトリックスを含んでいてよい。
【0008】
Fasリガンドに特異的である媒体のリガンド結合部分は、Fas受容体またはその誘導体を含んでいてよい。それはまた、完全長Fasタンパク質であっても、またはその断片であってもよい。リガンド結合部分は、Fasタンパク質の細胞外ドメインもしくはその断片を含むか、またはそれから成っていてよく、好ましくは、Fasタンパク質のリガンド結合ドメインを含むか、またはそれから成っている。媒体のリガンド結合部分は、ペプチド、タンパク質、アプタマー、抗体、抗体断片、融合タンパク質、またはキメラタンパク質であってよい。リガンド結合部分は、免疫グロブリンの結晶化可能断片領域(Fc領域)と融合してキメラ融合タンパク質、すなわちFas−Fc、を形成したFasタンパク質、またはその断片であってよい。好ましくは、免疫グロブリンは、IgG1などのヒト免疫グロブリンである。媒体は、Fasリガンドに特異的な複数のリガンド結合部分を含んでいてよく、後者の各々は、前述の例のいずれによっても実現することができる。
【0009】
この、または各々のリガンド結合部分は、どのような由来のものであってもよいが、好ましくは、ヒトもしくはマウス、またはその組み合わせである。
【0010】
この、または各々のリガンド結合部分は、特にマイクロ粒子の場合、表面吸収、吸着、化学結合、もしくはマトリックス内組み込みによってキャリアとカップリングしていてよく、または、特にキャリアがマイクロ粒子の場合、キャリアと共有結合もしくは非共有結合によって会合していてよい。この、または各々のリガンド結合部分は、連結分子を介してキャリアとカップリングしていてよい。連結分子は、免疫グロブリン、好ましくはIgG1などのヒト免疫グロブリンの結晶化可能断片領域(Fc)であってよい。
【0011】
別の選択肢として、アビジン−ビオチンなどの連結系を用いて、Fasリガンドに特異的なリガンド結合部分をキャリアと間接的にカップリングさせてもよい。従って、連結分子は、アビジンまたはビオチンであってよい。Fasリガンドに特異的なリガンド結合部分は、これをビオチン化して、アビジン−コーティングしたキャリア表面にカップリングさせてよい。別の連結分子としては、ブドウ球菌プロテインAであってよい。
【0012】
好ましい態様では、薬理学的活性剤、マーカー、または薬物は、好ましくは標的細胞にて、もしくはその内部で放出されるように、キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている。
【0013】
本発明の第一の局面の特に好ましい態様では、送達媒体は、薬理学的活性物質を含有するマイクロ粒子であり、ここで、マイクロ粒子は、Fas、その誘導体、もしくはFasリガンドに対する特異的結合分子と共有結合または非共有結合によって会合している。
【0014】
薬理学的活性剤は、タンパク質、ペプチド、ポリペプチド、ポリヌクレオチド、ポリサッカリド、脂質、小分子薬物、またはその他のいずれの生物学的活性物質であってもよい。薬理学的活性剤は、細胞内で作用してよく、または標的細胞内部の成分に対して特異的であってもよい。薬理学的活性剤は、細胞毒性剤、または細胞分裂阻害剤であってよい。また、薬理学的活性剤としては、細胞毒性放射性核種、化学毒素、またはタンパク質毒素も挙げることができる。好ましくは、薬理学的活性剤は、抗癌剤、特にドキソルビシンまたはパクリタキセルである。
【0015】
マーカーは、蛍光マーカー、放射性核種、または造影剤であってよい。従って、マーカーは、検出可能マーカー、またはイメージングマーカーであってよい。
【0016】
薬理学的活性剤、マーカー、または薬物は、特にキャリアがマイクロ粒子である場合、キャリアによって含有されるかまたはカプセル化されていてよい。活性剤、マーカー、または薬物は、化学結合によってキャリアもしくはマイクロ粒子と結合しているか、またはその内部にあってよく、または、キャリアを形成する物質のマトリックス内に物理的に組み込まれていてもよい。
【0017】
第二の局面では、本発明は、医療に、好ましくは治療目的に用いられる、本発明の第一の局面に従う送達媒体を提供する。本局面では、薬理学的活性剤、マーカー、または薬物は、キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されていることが好ましい。
【0018】
治療目的とは、新生物または神経障害に関連する疾患または医学的病状の治療であってよい。
【0019】
本発明はまた、さらなる局面において、疾患または医学的病状の治療方法も提供し、その方法は、本発明の第一の局面に従う送達媒体を対象へ投与することを含み、ここで、送達媒体は、薬理学的活性剤または薬物の効果量を含む。
【0020】
疾患または病状は、脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、またはFasリガンドが腫瘍細胞中で発現される癌の種類もしくは腫瘍であってよい。好ましくは、疾患または病状は、卵巣癌または髄芽腫である。
【0021】
疾患または病状は、運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、および末梢神経または脊髄損傷を含む神経疾患であってよい。
【0022】
本発明のさらなる局面によると、Fasリガンド発現細胞の検出を含む診断方法に用いるための、本発明の第一の局面に従う送達媒体が提供され、ここで、マーカーが、キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている。
【0023】
別の局面では、本発明は、本発明に従う送達媒体を対象へ投与することによってFasリガンド発現細胞を検出することを含む診断の方法を提供し、ここで、マーカーが、キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている。
【0024】
好ましくは、診断方法、または診断の方法は、新生物または神経障害を診断する方法である。
【0025】
本発明はまた、本発明に従う送達媒体、および1もしくは2つ以上の生理学的または薬理学的に許容されるキャリア、賦形剤、または安定化剤を含む医薬組成物も提供する。そのような組成物は、本明細書で述べる用途および方法のいずれにも用いることができる。
【0026】
別の局面では、本発明は、本発明に従う送達媒体を作製するための方法を提供し、その方法は、薬理学的活性剤、マーカー、もしくは薬物を含有するかまたはカプセル化するマイクロ粒子を形成する工程、およびFasリガンド特異的リガンド結合部分を前記マイクロ粒子に結合させる工程を含む。
【0027】
発明者らは、驚くべきことに、Fas/Fasリガンド系が、マイクロ粒子の取り込みを高めることができることを見出した。この手法を用いることで、発明者らは、Fasとカップリングしたマイクロ粒子により薬物を標的細胞へ送達することができることを示した。この分子標的化の方法により、Fasリガンド発現細胞によるマイクロ粒子の細胞内取り込みが高められる。
【0028】
本特許出願において、発明者らは、薬物送達粒子(例:乳酸−グリコール酸共重合体マトリックス(PLGA)、ポリ乳酸(PLA)、ポリ−ε−カプロラクトン(PCL)、ポリドロキシブチレート(PHB)、またはキトサン生分解性マイクロスフェア、ケイ素系粒子、高分子電解質カプセル、リポソームなど)を修飾してこれらの取り込みの向上、ならびにニューロン、癌細胞、および/または種々のFasリガンド(FasL、アポ−1L、CD95L)発現細胞を含む特定の細胞への特異化を行うための、Fas(アポ−1、CD95とも称される)、Fasの融合タンパク質(例:キメラ融合タンパク質:免疫グロブリンの結晶化可能断片領域(Fc領域)と融合したFas(FasFc)など)、および/またはFasの役割を再現する能力を有する同等部分(例:ペプチド、タンパク質、アプタマーなど)の新規な使用を初めて報告する。本発明から、発明者らはまた、治療的送達のための薬物接合体としてのFas(アポ−1、CD95)、Fasの融合タンパク質(例:FasFcなど)、および/またはFasの役割を再現する能力を有する同等部分(例:ペプチド、タンパク質、アプタマーなど)の使用の可能性についても報告する。従って、本発明は、薬物およびその他の物質の生物細胞への細胞内送達を可能とし、生物医学的研究および治療の分野に適用することができる。
【0029】
Fas/Fasリガンド系は、発明者らの知る限りにおいて、アポトーシスおよび非免疫組織における種々の新しい役割について広く研究されてきたが、標的化、および細胞における薬物の細胞内取り込みの向上のためのFas(アポ−1、CD95)修飾薬物充填マイクロ粒子(例:PLGA、PCL、高分子電解質カプセル、またはリポソーム)、またはFas(アポ−1、CD95)接合薬物の使用に関する先行技術はない。さらに、本発明は、非自明性を有し、ならびに、癌、神経病状、およびその他の疾患を患う患者への治療製剤の送達において、ならびに/または生物医学および創薬における研究ツールとして、著しい産業上の利用性を有する。
【0030】
細胞内薬物送達システムは、末梢部位での不要な副作用を低減するものであり、数々の技術が現在研究されており、例えば、標的化ナノ粒子(Farokhzad et al.,2006;Gu et al.,2009;Faraji et al.,2009)である。また、細胞内薬物送達にマイクロ粒子を用いることも可能であり、これは、薬物の効力向上および持続放出のために充填容量を大きくできるという利点がある。
【0031】
しかし、マイクロ粒子を用いた薬物送達に関する研究はあまりなく、それらは、本質的に、マクロファージなどのプロフェッショナル貪食細胞(professional phagocytes)に関するものである(Walter et al.,2001;Brandhonneur et al.,2009)。発明者らおよび他の研究者らは、非プロフェッショナル貪食細胞(non−professional phagocytes)、すなわちニューロンの貪食能を研究し、直径が0.5ミクロンより大きい生物粒子および合成粒子の摂取を実証した(Esselens et al.,2004;Bowen et al.,2007)。この特性はニューロンについては広く認識はされていなかったが、多くの非プロフェッショナル貪食細胞が、線維芽細胞および上皮細胞を含む比較的大きな粒子を摂取する能力を有することは多く報告されている(Rabinovitch,1995)。非プロフェッショナル貪食細胞における貪食を媒介するメカニズムの理解を深めることは、これらの細胞を標的とする薬物送達粒子設計の手助けとなり得る。例えば、海馬ニューロンの場合のテレンセファリンなどの特定の細胞表面受容体によって取り込みが調節され得ることが最近示された(Esselens et al.,2004;国際公開公報第/2006/030013号「The modulation of phagocytosis in neurons」)。
【0032】
発明者らは、驚いたことに、貪食に関与することがこれまでは知られていなかったFasリガンドが、FasおよびFas接合体の細胞内取り込みを可能とすることが見出した。本発明において、Fas(アポ−1、CD95)タンパク質が、薬物充填マイクロ粒子の修飾に基づく細胞内薬物送達システムに用いられる。Fas、Fasの融合タンパク質、および/またはFasの役割を再現する能力を有する同等部分による薬物充填粒子の表面修飾により、ニューロン、癌細胞、および/または種々のFasリガンド(FasL、アポ−1L、CD95L)発現細胞を含む特定の細胞によるマイクロ粒子のインターナリゼーションが大きく高められる。
【0033】
発明者らは、Fas(アポ−1、CD95)表面修飾マイクロ粒子が、未修飾粒子、またはその他のリガンドで修飾された粒子と比較して、多くの細胞型によって選択的に摂取されることを示す多くのデータを有する(実施例で示すように)。発明者らはまた、薬剤含有マイクロ粒子として作られた生分解性Fas(アポ−1、CD95)表面修飾PLGA系マイクロ粒子が、細胞によって取り込まれ、充填された薬剤を研究対象細胞内部へ送達することも示す。実施例で示されるように、発明者らは、Fas/Fasリガンド−補助貪食を介して、約0.5〜1.5μmの平均径のものを含むマイクロ粒子が細胞内へ効率的に取り込まれることを示した。さらに、実施例では、生物細胞が抗癌薬を充填したマイクロ粒子を取り込んだ場合の機能的効果も示す。従って、実施例に示したデータは、Fas(アポ−1、CD95)の使用に基づいた治療プラットフォームの新規性、進歩性、および有用性を示すものである。
【0034】
従って、本発明に従う送達媒体を用いて、投与薬物の総量を大きく低減し、同時に標的部位での薬物濃度を高めることができる。従って、これらを用いて、新生物および神経障害などの広範囲にわたる疾患における細胞毒性剤およびその他の薬物の機能を改善することができ、毒性を低下させると同時に薬物効力を向上させる可能性を有する。これらはまた、現時点で開発可能であると見なされていない有望な治療薬に対する送達オプションを提供することもできる。薬理学的活性剤または薬物がキャリアまたはマイクロ粒子でカプセル化される本発明の態様は、この観点で特に効果的であり、それは、これらによって、その標的に到達するまでに生物学的に利用可能となり得る薬剤または薬物の量が低減されるからである。
【0035】
当業者には公知であるように、Fas(アポ−1、CD95としても知られる)は、TNF/NGF受容体スーパーファミリーのメンバーであり、細胞膜受容体として、または可溶性の形態で発現される。ヒトFas(アポ−1、CD95)遺伝子の完全配列および転写物は、ホモサピエンスおよびその他のいくつかの生物種のEnsemblデータベースに登録されている(http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000026103)。マウスFas遺伝子の完全長配列および転写物は、マウスゲノム(Mus musculus)のEnsemblデータベースに登録されており(http://www.ensembl.org/Mus_musculus/Gene/Summary?db=core;g=ENSMUSG00000024778)、Watanabe−Fukunaga et al.,1992、に報告されている。Fasは、アポ−1LおよびCD95Lとしても知られるそのリガンドであるFasリガンド(FasL)との結合を介して、選択された細胞のアポトーシスの誘発を媒介する。Fasリガンドは、TNFスーパーファミリーのメンバーであり、選択された細胞においてFasと結合することでプログラム細胞死を誘発する。Fas/Fasリガンド系は、ほとんどの場合アポトーシスに関連して研究されてきており(Nagata,1997)、Fas/Fasリガンド系の異常調節は、免疫恒常性の破壊を伴う(Nagata and Suda,1995;Lettau et al.,2008)。しかし、免疫系組織における単一の役割を超えて、中枢神経系などの種々の組織におけるFas/Fasリガンド系の機能を提案する報告があり、そこでは、FasおよびFasリガンドは、非免疫細胞によって発現される(Choi and Benveniste,2004)。これらとしては、例えば、パーキンソン病(Landau et al.,2005)およびアルツハイマー病(Ethell et al.,2002)における新規な役割、ニューロンの枝分かれの制御(Zuliani et al.,2006)、ならびに脊髄損傷における神経保護(Ackery et al.,2006)が挙げられる。
【0036】
さらに、中枢神経系(Choi and Benveniste,2004)、眼(Ferguson and Griffith,2006)、および腫瘍(O’Connell et al.,2001;Ryan et al.,2005)を含む特定の組織または器官の細胞におけるFasリガンドの発現が、活性化されたFas陽性免疫細胞の死滅を可能とすることで、これらの組織または器官に免疫特権状態を付与するものであることが強く主張されている(Flugel et al.,2000;Green and Ferguson,2001)。Fasリガンドを発現する腫瘍細胞としては、共培養実験にてT細胞を死滅させる能力が示されたヒト肺癌が挙げられ(Niehans et al.,1997);この能力は、融合タンパク質FasFcを培養物へ添加すると阻害された。これらとしてはまた、神経膠芽腫および髄芽腫などの脳腫瘍(Gratas et al., 1997,Weller et al.,1998)、インビボトランスフェクションによるFasリガンドの下方制御が腫瘍浸潤リンパ球の数の増加に起因して腫瘍サイズの減少をもたらした(Ryan et al.,2005)結腸癌(O’Connell et al.,1998)、およびFasリガンドの誘発がT−リンパ球のアポトーシスを増加させた(Meng et al.,2004)卵巣癌(Abrahams et al.2003)も挙げられる。
【0037】
キャリアまたはマイクロ粒子は、ポリアルキレン、ポリカーボネート、ポリ(ジオキサノン)、ポリ酸無水物、ポリヒドロキシ酸、ポリフマレート、ポリカプロラクトン、ポリアミド、ポリアセタール、ポリエーテル、ポリエステル、ポリ(オルソエステル)、ポリヒドロキシブチレート、ポリビニルアルコール、ポリウレタン、ポリホスファゼン、ポリアクリレート、ポリメタクリレート、ポリシアノアクリレート、ポリウレア、ポリスチレン、ポリアミン、ポリ(アリレート)、ポリカーボネート、ポリ(プロピレンフマレート)、ポリヒドロキシアルカノエート、ポリケタール、ポリエステルアミド、ポリヒドロキシバリレート、ポリオルソカーボネート、ポリ(ビニルピロリドン)、ポリアルキレンオキサレート、ポリアルキレンスクシネート、ポリ(リンゴ酸)、ポリ(メチルビニルエーテル)、およびポリ(無水マレイン酸)から成る群より選択されるポリマーを含んでいてよい。
【0038】
キャリアまたはマイクロ粒子は、PLGA(乳酸−グリコール酸共重合体)マトリックス、PLA(ポリ乳酸)、PCL(ポリ−ε−カプロラクトン)、もしくはPHB(ポリヒドロキシブチレート)を含んでよく、または、キトサン生分解性マイクロスフェア、ケイ素もしくはケイ素系の粒子、高分子電解質カプセル、デンドリマー、もしくはリポソームである。キャリアまたはマイクロ粒子は、非ポリマーであってもよい。好ましくは、マイクロ粒子はPLGAマトリックスを含む。
【0039】
本発明に従うマイクロ粒子の生分解性は、インビトロにて適切ないずれの培地によるインキュベーションによっても測定または試験することができる。生分解性はまた、皮下または筋肉内を例とするマイクロ粒子の非経口注射、および時間の関数としての組織の組織学的試験によって調べることもできる。非経口投与後の生分解性マイクロ粒子は、体内で溶解し、内因性物質を、最終的には例えば乳酸を形成することができる。
【0040】
本発明に従うマイクロ粒子の生体適合性は、皮下または筋肉内を例とするマイクロ粒子の非経口投与、および組織の組織学的評価によって試験することができる。
【0041】
マイクロ粒子に関連するほとんどの研究では、乳酸−グリコール酸共重合体(PLGA)マトリックス内に製剤された薬物が考慮されてきた。PLGAは、臨床での使用に関してUS FDA(米国食品医薬品局)を含む監督機関によって認可された生分解性ポリマーであり、整形インプラントおよび全身薬物送達デポーシステム(例:Trelstar(登録商標)、Lupron Depot(登録商標)、Risperdal Consta(登録商標))に一般的に用いられている。生物活性タンパク質(Giteau et al.,2008)および核酸(Patil and Panyam,2009)を含むPLGAマイクロ粒子への薬物の充填は、通常、合成の過程で行われる。さらに、PLGA薬物キャリアは、その標的特異性を高めるために、単純な吸着または化学結合のいずれかを介して適切なリガンドにより表面官能化することができる。Farokhzad and colleagues(2006)は、前立腺癌細胞を標的とする目的で、ドセタキセル充填ペグ化PLGAナノ粒子へ官能化されたアプタマーを用いてそのような手法を例証した。また、ポリ−ε−カプロラクトン(Sinha et al.,2004)を含むその他の種類の生分解性マイクロ粒子内、および高分子電解質マイクロカプセル(Sukhorukov et al.,2007;Munoz Javier et al.,2008)またはリポソーム(Huwyler et al.,2008)などの広範囲の様々なシステム内に細胞内送達用の薬物を閉じ込めることも可能である。
【0042】
薬理学的活性剤は、抗腫瘍剤、抗生物質、抗炎症剤、抗ヒスタミン剤、鎮静剤、筋弛緩剤、抗てんかん剤、抗うつ剤、抗アレルギー剤、気管支拡張剤、強心剤、抗不整脈剤、血管拡張剤、抗糖尿病剤、抗凝固剤、止血剤、麻酔剤(narcotic)、およびステロイド剤であってよい。
【0043】
薬理学的活性剤は、細胞毒性剤、細胞分裂阻害剤、またはその他の薬物であってよい。細胞毒性剤、細胞分裂阻害剤、または薬物は、プラチン(誘導体)およびタキソールの類であってよい。細胞分裂阻害薬または薬物は、例えば、シスプラチン、サトラプラチン、オキサリプラチン、カルボプラチン、ネダプラチン、クロラムブシル、シクロホスファミド、メファラン(mephalan)、アザチオプリン、フルオロウラシル、メルカプトプリン、メトレキサート(methrexat)、ナンドロロン、アミノグルテミド(aminogluthemid)、メドロキシプロゲステロン、酢酸メゲストロール、プロカルバジン、ドセタキセル、パクリタキセル、エピポドフィロトキシン、ポドフィロトキシン、ビンクリスチン、ドセタキセル、ダウノマイシン、ドキソルビシン、ミトキサントロン、トポテカン、ブレオマイシン、ゲムシタビン、フルダラビン、および5−FUDRから成る群より選択することができる。好ましくは、生物活性剤は、抗癌剤、特にドキソルビシンまたはパクリタキセルである。
【0044】
細胞毒性核種または放射線治療用同位体元素は、225Ac、211At、212Bi、213Bi、212Pb、224Ra、または223Raなどのアルファ線放射性同位体元素であってよい。別の選択肢として、細胞毒性放射性核種は、186Rh、188Rh、177Lu、90Y、131I、67Cu、64Cu、153Sm、または166Hoなどのベータ線放射性同位体元素であってよい。別の選択肢として、細胞毒性放射性核種は、オージェ電子および低エネルギー電子を放射するものであってもよく、125I、123I、または77Brの同位体元素が挙げられる。
【0045】
適切な検出可能またはイメージングマーカーとしては、これらに限定されないが、Molecular Probes(Handbook of fluorescent probes and research products)に記載のものなどの蛍光分子が挙げられ、ローダミン、フルオレセイン(フルオレセインイソチオシアネート(FTIC)など)、テキサスレッド、アクリジンオレンジ、アレクサフルオー(Alexa Fluor)(種々)、アロフィコシアニン、7−アミノアクチノマイシンD、BOBO−I、BODIPY(種々)、カルシエン(Calcien)、カルシウムクリムゾン(Calcium Crimson)、カルシウムグリーン、カルシウムオレンジ、6−カルボキシローダミン6G、カスケードブルー(Cascade blue)、カスケードイエロー、DAPI、DiA、DiD、DiI、DiO、DiR、ELF97、エオシン、ERトラッカーブルー−ホワイト(ER Tracker Blue−White)、EthD−1、エチジウムブロミド、Fluo−3、Fluo4、FM1−43>FM4−64、Fura−2、フラレッド(Fura Red)、ヘキスト33258、ヘキスト33342、7−ヒドロキシ−4−メチルクマリン、Indo−1、JC−I、JC−9、JOE色素、リサミンローダミンB、ルシファーイエローCH、リソセンサーブルーDND−167(LysoSensor Blue DND−167)、リソセンサーグリーン、リソセンサーイエロー/ブルー、リソトラッカーグリーンFM(Lysotracker Green FM)、マグネシウムグリーン、マリーナブルー、ミトトラッカーグリーンFM(Mitotracker Green FM)、ミトトラッカーオレンジCMTMRos、ミトトラッカーレッドCMXRos、モノブロモビマン、NBDアミン、ネルオトレース500/525グリーン(NeruoTrace 500/525 green)、ナイルレッド、オレゴングリーン(Oregon Green)、パシフィックブルー(Pacific Blue)、POP−I、プロピジウムヨージド、ローダミン110、ローダミンレッド、R−ファイコエリトルム(R−Phycoerythrm)、レゾルフム(Resorfm)、RH414、Rhod−2、ローダミングリーン、ローダミン123、ROX色素、ナトリウムグリーン、SYTOブルー(種々)、SYTOグリーン(種々)、SYTOオレンジ(種々)、SYTOXブルー、SYTOXグリーン、SYTOXオレンジ、テトラメチルローダミンB、TOT−I、TOT−3、X−rhod−1、YOYO−1、またはYOYO−3などである。
【0046】
さらに、放射性核種は、イメージング剤として用いることができる。適切な放射性核種としては、Fe(III)、Fe(II)、Cu(II)、Mg(II)、Ca(II)、およびZn(II)、インジウム、ガリウム、およびテクネチウムの放射性種を挙げることができる。その他の適切な造影剤としては、常磁性T1強調またはT2強調MRI造影剤におけるキレート化に一般的に用いられる金属イオンを挙げることができ、銅、クロム、鉄、ガドリニウム、マンガン、エルビウム、ユーロピウム、ジスプロシウム、およびホルミウムなどの二価および三価カチオンが挙げられる。キレート化が可能であり、放射性核種イメージングに用いることができる金属イオンとしては、ガリウム、ゲルマニウム、コバルト、カルシウム、インジウム、インジウム、ルビジウム、イットリウム、ルテニウム、イットリウム、テクネチウム、レニウム、白金、タリウム、およびサマリウムなどの金属を挙げることができる。さらに、中性子捕獲放射線治療に有用であることが公知である金属イオンとしては、ホウ素および核断面の大きいその他の金属が挙げられる。適するものとしてはまた、超音波造影およびX線造影組成物に有用である金属イオンも挙げることができる。その他の適切な造影剤の例としては、気体または気体発生化合物であって、放射線不透過性であるものを挙げることができる。
【0047】
Fasリガンド発現細胞は、当業者に公知の標準的なイメージングまたは検出技術を用いて検出することができる。例えば、顕微鏡観察、フローサイトメトリー、医療用超音波検査、X線検査(投影X線検査および蛍光透視法など)、核医学イメージング(シンチレーションカメラなど)、磁気共鳴画像法(MRI)、光音響イメージング、デジタル赤外イメージングサーモグラフィ、またはトモグラフィである。
【0048】
様々な条件下にて種々の種類およびサイズの粒子を作製することができる多くのマイクロカプセル化技術が存在する。方法には通常、温度の変化、溶媒の留去、または化学架橋剤の添加による乳化されたポリマー液滴の固化が含まれる。
【0049】
マイクロカプセル化プロセスによって均一なサイズ範囲の粒子が作製されない場合は、篩いまたはろ過などの標準的な技術を用いて粒子を分離し、所望されるサイズ範囲の粒子集団を得ることができる。粒子はすべて、光学顕微鏡、Coulter Multisizer(ベックマンコールター、動的光散乱(Malvern Zetasizer)、透過型電子顕微鏡(TEM)、走査型電子顕微鏡(SEM)、および準弾性光散乱(QELS)などの標準的な技術により、粒子サイズ分布という観点から特徴づけられる。本発明のマイクロ粒子は、平均径が0.01μm、0.05μm、0.1μm、0.2μm、0.5μm、1μm、2μm、5μm、または10μmまでのものであってよい。
【0050】
一般的なマイクロカプセル化技術としては、界面縮重合、スプレードライ、ホットメルトマイクロカプセル化、および相分離技術(溶媒除去および溶媒留去)が挙げられる。このような技術は、米国特許第2001020011号,Mathiowitz and Langer 1987;Mathiowitz,et al.,1987,1988,1990,1992;Benita,et al.,1984、に記載されている。
【0051】
界面縮重合を用いることで、以下のようにしてコア物質(本発明の薬理学的活性剤またはマーカーなど)をマイクロカプセル化することができる。1つのモノマーおよびコア物質を溶媒に溶解する。第二のモノマーを、第一の溶媒と不混和性の第二の溶媒(通常は水性)に溶解する。第一の溶液を攪拌しながら第二の溶液中に懸濁させることにより、エマルジョンが形成される。エマルジョンが安定したところで、水相に開始剤を添加し、エマルジョンの各液滴の界面で界面重合を引き起こす。
【0052】
スプレードライは、通常、1〜10ミクロンのサイズのマイクロスフェアを作製するためのプロセスであり、ここでは、含有またはカプセル化されるコア物質(本発明の薬理学的活性剤またはマーカーなど)がポリマー溶液(通常は水性)に分散または溶解され、この溶液または分散液は、ポンプ送液によって圧縮ガス流で駆動される微粒子化ノズルを通され、得られたエアロゾルは、空気加熱サイクロン中に浮遊され、溶媒がマイクロ液滴から蒸発される。固化された粒子は第二のチェンバーに送られ、回収フラスコ中に捕集される。
【0053】
ホットメルトマイクロカプセル化は、コア物質(本発明の薬理学的活性剤またはマーカーなど)が溶融ポリマーへ添加される方法である。この混合物は、ポリマーの融点よりも10℃高い温度まで加熱しておいたポリマーに対する非溶媒(多くの場合油系)中に溶融液滴として懸濁される。このエマルジョンは激しい攪拌を維持され、同時に非溶媒浴はポリマーのガラス転移点未満まで急冷され、溶融液滴の固化およびコア物質の閉じ込めが引き起こされる。この技術によって作製されたマイクロスフェアは、通常、直径50ミクロンから2mmのサイズ範囲である。
【0054】
溶媒蒸発マイクロカプセル化では、通常、ポリマーが水不混和性有機溶媒中に溶解され、含有またはカプセル化される物質(本発明の薬理学的活性剤またはマーカーなど)が、有機溶媒中の懸濁液または溶液としてこのポリマー溶液に添加される。この懸濁液または溶液がビーカー中で激しく攪拌される水(エマルジョンの安定化のために界面活性剤を含有することが多い)へ添加されることで、エマルジョンが形成される。攪拌を継続した状態で有機溶媒が蒸発される。蒸発によってポリマーが析出し、コア物質を含有する固体マイクロカプセルが形成される。
【0055】
別の選択肢としての溶媒蒸発プロセスは、マイクロシーブを用いることを含む。ポリマーは、通常、水不混和性有機溶媒中に溶解され、含有またはカプセル化される物質(本発明の薬理学的活性剤またはマーカーなど)は、ジクロロメタンなどの有機溶媒中の懸濁液または溶液としてこのポリマー溶液へ添加される。次に、この懸濁液または溶液は、PTFEフィルターを通してろ過される。その後、ポリマーは、表面一帯に均一な細孔を有する微細加工された膜であるマイクロシーブ膜(Nanomi BV,オランダ、など)を通して、乳化剤を含有する水溶液中へ乳化される。得られたエマルジョンは、室温にて少なくとも3時間攪拌され、溶媒が留去される。固化されたマイクロスフェアは、ろ過によって濃縮され、繰り返し洗浄される。次に、粒子は、凍結乾燥され、評価まで低温(例:−20℃)で保存された。
【0056】
溶媒除去マイクロカプセル化では、ポリマーは、通常、油混和性有機溶媒中に溶解され、含有またはカプセル化される物質(本発明の薬理学的活性剤またはマーカーなど)は、有機溶媒中の懸濁液または溶液としてこのポリマー溶液へ添加される。この懸濁液または溶液がビーカー中で激しく攪拌される油へ添加されることで、エマルジョンが形成され、ここで、この油はポリマーに対する非溶媒であり、ポリマー/溶媒溶液は油に不混和性である。有機溶媒は、攪拌を継続した状態で、油相中へ拡散されることで除去される。溶媒除去によってポリマーが析出し、コア物質を含有する固体マイクロカプセルが形成される。
【0057】
薬理学的活性剤またはマーカーは、媒体またはマイクロ粒子の約25重量/重量%もしくはそれ以上を占めていてよい。好ましくは、薬理学的活性剤またはマーカーは、媒体またはマイクロ粒子の0.01%、0.1%、1%、5%、10%、15%、20%、25%、30%、または40%(重量/重量)までを占めている。
【0058】
マイクロ粒子は、約0.5〜1.5μmの平均径を有していてよく、上述のプロセスのいずれによって作製されてもよい。
【0059】
Fasタンパク質、その誘導体、またはそのキメラタンパク質を例とする、Fasリガンドに特異的なリガンド結合部分は、好ましくは、キャリアまたはマイクロ粒子の表面に吸着または化学結合されている。また、キャリアまたはマイクロ粒子のマトリックス中に組み込まれていてもよい。当業者であれば、この工程が、単純な吸着、化学結合技術、またはマイクロ粒子のマトリックス中への組み込みなどの標準的な技術によって行うことができることは理解される。Fasリガンドに特異的なリガンド結合部分は、好ましくは、キャリア(マイクロ粒子など)の表面と結合または会合している。結合部分は、媒体またはマイクロ粒子の約0.01重量/重量%、0.1重量/重量%、1重量/重量%、5重量/重量%、10重量/重量%、15重量/重量%、20重量/重量%、25重量/重量%、または30重量/重量%までを占めていてよい。
【0060】
従って、本発明は、薬理学的活性物質を含有するマイクロ粒子の作製方法を提供し、ここで、マイクロ粒子は、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と共有結合または非共有結合で会合しており、この方法は、表面吸収、吸着、化学結合、またはマトリックス内組み込みによって、マイクロ粒子を、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と会合させることを含む。
【0061】
適切な薬理学的に許容されるキャリア、賦形剤、または安定化剤は、無毒性であることが好ましく、活性成分の効果または生物活性に干渉しない。薬理学的に許容されるキャリアは、ヒトまたはその他の脊椎動物への投与に適する、1つ以上の適合性を有する固体もしくは液体充填剤、希釈剤、またはカプセル化物質であってよい。薬理学的に許容されるキャリアは、有機または無機成分、天然または合成物であってよく、適用を補助するために活性成分がこれらと組み合わされる。
【0062】
医薬組成物は、薬理学的に使用することができる製剤への活性化合物の処理を促進する賦形剤および助剤を含む1もしくは2つ以上の生理学的に許容されるキャリアを用いた従来の方法で製剤することができる。適切な製剤は、選択された投与経路に依存する。典型的な注射用製剤は、滅菌生理食塩水またはリン酸緩衝生理食塩水などのキャリアを含む。粘度調節剤および保存剤も添加される場合が多い。適切な薬理学的に許容される賦形剤としては、これらに限定されないが、希釈剤、バインダー、滑沢剤、崩壊剤、着色剤、安定化剤、および界面活性剤が挙げられる。
【0063】
本発明の送達媒体または組成物が、新生物もしくは神経障害の治療、寛解、または予防のための公知の療法の補助として、またはそれらと組み合わせて用いることができることは理解される。本発明に従う送達媒体または組成物は、組成物が使用される方法に特に依存する数多くの種々の形態を有する組成物に組み合わせることができる。
【0064】
Fasリガンド発現細胞は、脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、および癌の種類で見られる。Fasリガンド発現細胞はまた、神経疾患、運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、ならびに末梢神経および脊髄損傷に関連する細胞にも見られる。
【0065】
対象は、脊椎動物、哺乳類、または飼育動物であってよい。従って、本発明に従う送達媒体または組成物は、家畜(例:ウマ)、ペットを例とするいずれの哺乳類の治療にも用いることができ、またはその他の獣医学的用途に用いることもできる。最も好ましくは、対象はヒトである。
【0066】
本発明の送達媒体または組成物の効果量は、効果量の薬理学的活性剤の標的への到達を可能とし、それによって対象において治療効果を発生させる量であってよい。薬理学的活性剤を含む送達媒体または組成物の実際の効果量は、用いられる具体的な薬剤、物質、またはそれらの組み合わせ、Fasリガンドに特異的なリガンド結合部分の密度および/または性質、カプセル化薬理学的活性剤の放出特性、製剤された特定の組成物、投与モード、ならびに治療される対象の年齢、体重、病態、さらには投与経路および疾患または障害を含む因子に応じて様々であり得る。
【0067】
本発明に従う送達媒体または組成物は、非経口投与に適するものであってよい。非経口投与は、皮下、静脈内、筋肉内、関節内、滑膜内、腹腔内、胸骨内、くも膜下腔内、肝臓内、病巣内、腫瘍内、および頭蓋内注射または注入技術であってよい。非経口投与される場合、医薬組成物は、皮下または静脈内投与されることが好ましい。
【0068】
しかし、本発明に従う送達媒体または組成物の投与は、送達媒体または組成物の効果量がその標的へ到達することを可能とする許容されるいかなる方法で行なってもよい。選択される特定のモードは、特定の製剤、治療される対象の状態の重篤度、および効果的な治療を誘発するために要する用量などの因子に依存する。
【0069】
本明細書(添付の請求項、要約、および図面を含む)で述べる特徴のすべて、および/またはそのように開示されるいずれの方法の工程のすべても、そのような特徴および/または工程の少なくともいくつかが互いに相容れない場合を除いて、上記の局面のいずれといずれの組み合わせで組み合わせてもよい。ここで、本発明の理解を深め、その態様を効果的に実施する方法を示すために、例として添付の図面を参照する。
【図面の簡単な説明】
【0070】
【図1】図1は、未修飾合成粒子の一次感覚ニューロンでの取り込みを示す。共焦点顕微鏡により、β3チューブリン標識後根神経節ニューロン(核は細胞の中央に見られる)における合成粒子(丸形状のマイクロスフェア)の細胞内取り込みが示される。(A、B)1μmポリスチレンマイクロスフェア。(C、D)蛍光色素(FITC)結合ウシ血清アルブミンを充填した2μm高分子電解質カプセル。粒子は、細胞体および神経突起の両方で見られた。
【図2】図2は、未修飾合成粒子の一次感覚ニューロンでの取り込みを示す。後根神経節ニューロンの共焦点顕微鏡観察(Bowen et al.,2007より引用)。(A)明視野イメージ。(B)β3−チューブリンに対する抗体で染色したラット後根神経節ニューロンを1μmマイクロスフェア(丸形状のマイクロスフェア)とインキュベートした後の共焦点顕微鏡観察。XY平面の白色点線に沿ったzスタック断面(z−stack cross−sections)を示す上側および横側のパネルから、マイクロスフェアがインターナライズされていることが分かる。(C、E)白色線でハイライトしたニューロン内部(C)の緑色および赤色チャネルの蛍光強度プロファイル(E)は、0.33μmの光学的スライス像(optical slice)の中でマイクロスフェアとβ3−チューブリン標識とが共存していることを示す。(D、F)白色線でハイライトしたニューロン外部(D)の緑色および赤色チャネルの蛍光強度プロファイル(F)は、共存が存在しないことを示す。
【図3】図3は、Daoy髄芽腫細胞における未修飾合成粒子の取り込みを示す。(A−B)フローサイトメトリー散布図(左)およびゲーティングされたDaoy細胞に対するゲーティングプロット(右):(A)(右)濃度の上昇に従って、0.5μmマイクロ粒子を有する細胞の集団が増加することを示す(右上の四半分へ向けてのシフトが大きくなっている)。(左)高い濃度では、散布図の側方散乱の増加が見られる。コントロール細胞はマイクロ粒子を有しておらず、これをネガティブコントロールとして用いる;(B)(右)濃度が上昇すると、1.0μmマイクロ粒子を有する細胞集団の同様の増加を示す。これは、側方散乱の増加を伴っている(左)。同一濃度では、1.0μmマイクロスフェアを有する細胞集団は、0.5μmマイクロ粒子を有する細胞よりも明確である。SSC−H 側方散乱細胞;FSC−H=前方散乱細胞(サイズ);FL1−H=ポリスチレンドラゴングリーンマイクロ粒子を有するFITC検出細胞(FITC registering cells)のチャネル。(C)種々の濃度およびサイズにおけるND7/23細胞によるマイクロ粒子の取り込みを示すグラフ(フローサイトメトリーデータからまとめたもの)。いずれの場合でも、同一濃度ではサイズ間に取り込みの有意差はない(p>0.05)(ns)(サイズ依存性なし)。0.5μmの濃度1×107および1×108の間では取り込みに有意差はないが、濃度1×108および1×109の間の差は有意である(p<0.001)***。1.0μmマイクロ粒子では、濃度1×107および1×108の間(p<0.05)*、および濃度1×108および1×109の間(p<0.001)***で取り込みに有意差がある。エラーバーはn=3でのSEMを示す。
【図4】図4は、ND7/23感覚ニューロン細胞株における未修飾合成粒子の取り込みを示す。(A−B)フローサイトメトリー散布図(左)およびND7/23細胞に対するゲーティングプロット(右):(A)(右)濃度の上昇に従って、0.5μmマイクロ粒子を有する細胞の集団が増加することを示す(右上の四半分へ向けてのシフトが大きくなっている)。(左)高い濃度では、散布図の側方散乱の増加が見られる。コントロール細胞はマイクロ粒子を有しておらず、これをネガティブコントロールとして用いる;(B)(右)濃度が上昇すると、1.0μmマイクロ粒子を有する細胞集団の同様の増加を示す。これは、側方散乱の増加を伴っている(左)。同一濃度では、1.0μmマイクロ粒子を有する細胞集団は(淡色矢印)、0.5μmマイクロ粒子を有する細胞(黒色矢印)よりも明確である。SSC−H=側方散乱細胞;FSC−H=前方散乱細胞(サイズ);FL1−H=ドラゴングリーンマイクロ粒子を有するFITC検出細胞のチャネル。(C)種々の濃度およびサイズにおけるND7/23細胞によるマイクロ粒子の取り込みを示すグラフ(フローサイトメトリーデータからまとめたもの)。いずれの場合でも、同一濃度ではサイズ間に取り込みの有意差はない(p>0.05)(ns)(サイズ依存性なし)。0.5μmの濃度1×107および1×108の間では取り込みに有意差はないが、濃度1×108および1×109の間の差は有意である(p<0.001)***。1.0μmマイクロ粒子では、濃度1×107および1×108の間(p<0.05)*、ならびに濃度1×108および1×109の間(p<0.001)***で取り込みに有意差がある。エラーバーはn=3でのSEMを示す。
【図5】図5は、一次皮質ニューロンおよびその他の細胞型における未修飾合成粒子の取り込みを示す。このフローサイトメトリーのグラフは、24時間にわたるポリスチレンマイクロ粒子の添加後の、マウス脳由来一次皮質培養物のFITC(FL1−H)に対するPE(FL2−H)の蛍光をプロットしたものである。CD90.2−PE陽性細胞(皮質ニューロン)は右上と左上の四半分に見られ、その他の細胞型(主としてグリア細胞)は下側の四半分に見られる。マイクロ粒子を摂取した細胞は、右上および右下の四半分に見られる。このグラフは、皮質ニューロンおよびその他の細胞型が、培養中に未修飾粒子も摂取することを示している。
【図6】図6は、成体ラット由来の一次感覚ニューロンによる粒子の取り込み、およびその生存能を示す。(A)DRGニューロンおよびその他の細胞型の数は、培養物への添加マイクロ粒子の濃度を上昇させても、変化していない。このことは、マイクロ粒子が細胞に対して毒性を引き起こすものではないことを示唆している。(B)マイクロ粒子濃度を上昇させた場合の健全なニューロンの例(×20 OM)。
【図7】図7は、電子顕微鏡による研究から、超微細構造レベルでの毒性が示されないことを示している。(A〜F)透過型電子顕微鏡を用いたニューロンにおけるマイクロ粒子(MP)の研究に対する選択基準:(A)ニューロンは特徴的な長く薄い突起を有する。隣接するグリア細胞が短く厚い突起を有することに注目されたい(丸で囲んだ)。(B)膜結合MPを白色矢印で表す。Bには小さいMPが存在する(矢印)。(C)食胞膜と結合しないMP(矢印)は定量されなかった。食胞膜と結合したMPも存在している(矢印)。(D)ニューロン突起中の空の食胞(矢印)。E、F:二重膜によって結合した空の食胞(*)のさらなる例(矢印)。Aのバーは10μm、B〜Fのバーは0.5μmである。(G〜L)後根神経節ニューロンにおける食胞膜有りおよび無しのマイクロ粒子の透過型電子顕微鏡写真:(G)二重膜食胞内の0.5μmのサイズのマイクロ粒子(矢印で示す)。(H、J、K)食胞内の1μmマイクロ粒子。(I、L)食胞と結合していない0.5μmマイクロ粒子。バー=0.5μm。(M〜O)マイクロ粒子および後根神経節の走査型電子顕微鏡写真:(M)ニューロン膜はほとんど融合して、マイクロ粒子の周囲に小胞を形成した(矢印)。(N)2つのマイクロスフェアの基部周囲のニューロン膜の突起。(O)マイクロ粒子が取り除かれた可能性のある食作用カップ(phagocytic cup)(矢印)と考えられるものの形成。バー=1μm。
【図8】図8は、Daoyヒト髄芽腫細胞株による粒子の取り込み、およびその生存能を示す。このグラフは、種々の実験グループにおける総細胞死を示す。X軸はマイクロ粒子サイズおよび濃度である。1.0μmマイクロ粒子は、0.5μmのマイクロ粒子と比較してより多い細胞死を伴っていた。マイクロ粒子の無い培養でも、ある程度の細胞死が見られる。H2O2ポジティブコントロールのデータは含まれていない。エラーバーはn=3におけるSEMを示す。このデータは、粒子の濃度が非常に高い(実用的な使用を超える)場合であっても、細胞毒性は25%未満に限定されることを示している。
【図9】図9は、ND7/23感覚細胞株による粒子の取り込み、およびその生存能を示す。このグラフは、種々の実験グループにおける総細胞死を示す。X軸はマイクロ粒子サイズおよび濃度である。この細胞株では、マイクロ粒子を含まないコントロール培養ですでに細胞死が高く、マイクロ粒子の添加によって悪化はしていない。このグラフは、総細胞死が種々の実験グループ間で比較的一定であることを示している。エラーバーはn=3におけるSEMを示す。
【図10】図10は、一次DRGニューロンへ添加されたFasFc修飾ポリスチレン粒子を示す。DRGニューロンによる1μmドラゴングリーンポリスチレンマイクロ粒子の取り込みに対するFasFc修飾の効果を、蛍光顕微鏡で調べた。Rat血清、フィブロネクチン、ビトロネクチン、およびFasFc修飾マイクロ粒子はすべて、コントロール1と同じ密度で播種した(ウェルあたり1×105マイクロ粒子)。コントロールおよびその他の種類のリガンドと比較して、FasFc修飾は、ニューロンによる相当量のマイクロスフェアの取り込みを誘発した。
【図11】図11は、DRGニューロンを用いた未修飾およびFasFc修飾粒子のTEM研究を示す。DRGニューロンによる1μmドラゴングリーンポリスチレンマイクロ粒子の取り込みに対するFasFc修飾の効果をTEMによって調べた。未修飾およびFasFc修飾マイクロ粒子を培養物に24時間添加した(ウェルあたり2×106マイクロ粒子)。摂取されたマイクロ粒子は、細胞質内の電子密度の高い(暗色)直径約1ミクロンの正球形として見ることができる(矢印)。コントロールと比較して、FasFc修飾は、ニューロンによる相当量のマイクロスフェアの取り込みを誘発した。
【図12】図12は、Daoyヒト髄芽腫細胞株に添加したFasFc修飾ポリスチレン粒子を示す。このグラフは、Daoy細胞について、取り込みに対するFasFcマイクロ粒子の表面修飾の効果を、未コーティングのマイクロ粒子、または他のリガンドでコーティングした粒子と比較して示す。未処理マイクロ粒子(培養物に直接適用)、およびバッファー(リガンド無し)によるコーティングプロセスを受けた未コーティングマイクロ粒子の間に取り込みの有意差はなかった。0.5μmマイクロ粒子については、未コーティングおよびIgGオプソニン化マイクロ粒子の間に取り込みの有意差はないが(p>0.05)、フィブロネクチンと未コーティングの間(p<0.05)*、およびFasFcと未コーティングの間(p<0.05)*には有意差があった。1.0μmマイクロ粒子については、未コーティングコントロールと比較したIgGコーティングマイクロ粒子(p<0.05)*、未コーティングコントロールと比較したフィブロネクチンコーティング(p<0.001)***、ならびに未コーティングコントロールと比較したFasFc(p<0.001)***の取り込みに有意差があった。グラフはまた、サイズ間の取り込みの違いも示す。結果はまた、FcリッチであるIgGが、FasFcコーティングマイクロ粒子と同程度にはマイクロ粒子の取り込みを増加させておらず、従って、観察された増加には、融合タンパク質のFas部分が重要であることも示している。エラーバーはn=3におけるSEMを示す。60mmペトリ皿あたり1×107のマイクロ粒子を添加した。
【図13】図13は、ND7/23感覚ニューロン細胞株へ添加されたFasFc修飾ポリスチレン粒子を示す。このグラフは、未コーティングのマイクロ粒子と比較した、ND7/23細胞による取り込みに対するFasFcマイクロ粒子の表面修飾の効果を示す。右上の四半分(Q2)に示されるように、FasFcコーティングした0.5μmマイクロ粒子は、未コーティングマイクロ粒子(3.4%)と比較して、より高い割合の細胞(18%)によって取り込まれた。35mmペトリ皿あたり1×105のマイクロ粒子を添加した。
【図14】図14は、Daoy/皮質ニューロン共培養物へ添加されたFasFc修飾ポリスチレン粒子を示す。これらのグラフは、Daoyヒト髄芽腫細胞株とマウス由来の一次皮質ニューロンとの間で比較した、24時間の培養後のFasFc表面修飾マイクロ粒子(1μm)の取り込みを示す。培養中、Daoy癌細胞は、皮質ニューロンおよびその他の細胞型(グリア細胞など)よりも多くの粒子を摂取している。実験は、P7皮質ニューロンで行い(A)、P14皮質ニューロンで反復した(B)。60mmペトリ皿あたり1×107のマイクロ粒子を添加した。
【図15】図15は、DRG一次ニューロンに添加したFasFc修飾粒子を用いた細胞内染色剤送達を示す。(A)マージしたイメージ(×63 OM)、(B)ヘキスト染色核、(C)βIIIチューブリンニューロンマーカー、および(D)粒子が存在しないためにニューロン細胞質が染色されていないエチジウムホモダイマー充填PLGA粒子(直径<1.2μm)、(E)マージしたイメージ(×63 OM)、(F)ヘキスト染色核、(G)βIIIチューブリンニューロンマーカー、および(H)48時間後に核酸染色剤の放出および細胞質核酸の標識が見られたエチジウムホモダイマー充填PLGA粒子。
【図16】図16は、ND7/23感覚ニューロン細胞株に添加したFasFc修飾粒子を用いた細胞内薬物送達を示す。FasFc修飾粒子をND7/23細胞培養物へ一晩添加し、FACSによってドキソルビシン充填粒子を摂取した細胞を分離した後(左上の図)、コントロール細胞(右上のイメージ)は、そこから2週間にわたって正常に増殖を続けたが(顕微鏡下で観察)、一方ドキソルビシン充填粒子を摂取した細胞は(左上のグラフ;ドキソルビシン充填粒子の蛍光を示す右下のイメージを含む下側のイメージ)、まったく増殖しなかった(分離後2週間にわたって顕微鏡下で観察)。これらの観察結果は、有糸分裂阻害薬ドキソルビシンを充填した本発明を用いることによる、ND7/23感覚ニューロン細胞株での機能的薬物送達を示している。
【図17】図17は、Daoyヒト髄芽腫細胞株に添加したFasFc修飾粒子を用いた細胞内薬物送達を示す。(A)1μmパクリタキセル充填PLGAマイクロ粒子の添加後、7AAD陽性細胞と測定されたDaoy細胞のフローサイトメトリー散布図(左)。7AAD処理なしのコントロール細胞は、FL3−Hチャネルでの検出は高くない(R1領域)。薬物やプラセボ処理をまったく行わないコントロール細胞は、7AADアッセイ後の細胞死は最小限である。過酸化水素処理コントロール細胞は、7AADアッセイ後、高いパーセントの細胞死を示す。プラセボ充填と比較してパクリタキセル充填PLGAマイクロ粒子では、第1日および3日に機能的効果が見られ、細胞死が発生し、それはパクリタキセル単独の処理と同等である。SSC−H 側方散乱;FL3−H=7AAD検出細胞のチャネル。(B)7AADアッセイ後に測定された細胞死のパーセント(フローサイトメトリーデータからまとめたもの)。データは、プラセボと比較してパクリタキセル充填粒子では、第1日および3日に細胞死が増加していることを示している。このデータは、アポトーシス促進性薬物であるパクリタキセルを充填した本発明を用いることによる、Daoy髄芽腫細胞株における機能的薬物送達を実証するものである。
【図18】図18は、FasFc(CD95−Fc)修飾により、パクリタキセル充填マイクロ粒子のインビボでの効力が高められることを示す。実験では、未充填(プラセボ)または25重量/重量%パクリタキセル充填とした乳酸−グリコール酸共重合体(PLGA)生分解性マイクロ粒子(直径約1.5μm)を用いた。次に、マイクロ粒子に、CD95−Fcによる表面コーティングまたは偽コーティング(−CD95−Fc)を施した。(A)侵襲性増殖をする髄芽腫皮下異種移植片について、腫瘍体積の変化を示す。(B)第7日、単一の腫瘍内注射後、腫瘍の増殖は、パクリタキセル単独と比較して、+CD95 PLGAプラセボ、−CD95 PLGAパクリタキセル、および+CD95 PLGAパクリタキセルの場合でより効率的に阻害された。平均±SEM、n=4。(C)腹膜卵巣癌内転移のマウスモデル(IGROV1ルシフェラーゼ発現癌細胞)では、Taxolとして製剤されたパクリタキセルの等用量(Cremophor ELに溶解)と比較して、+CD95−Fc PLGAパクリタキセルは、4回の毎週の処理投与後、腫瘍バイオルミネセンスで>65倍の違いである抗腫瘍効力を示す。平均±SEM、n=5。処理の中断後、+CD95−Fc PLGAパクリタキセルのグループでは、腫瘍の再増殖は比較的遅い。プラセボグループはいずれも、疾患の広がりの度合いのために、第28日までに屠殺しなければならず;第35日までに、−CD95−Fc PLGAパクリタキセルのグループの1匹が屠殺され;第48日までには、−CD95−Fc PLGAパクリタキセルのさらに2匹、パクリタキセルの2匹、および+CD95−Fc PLGAパクリタキセルのグループの1匹が屠殺された。統計的比較(両側t検定)を、第35日について示す:パクリタキセル対+CD95−Fc PLGAパクリタキセル:*P=0.012;第35日:パクリタキセル対−CD95−Fc PLGAパクリタキセル:P=ns;第41日:パクリタキセル対+CD95−Fc PLGAパクリタキセル:*P=0.03;第41日:パクリタキセル対−CD95−Fc PLGAパクリタキセル:P=ns;;第48日:パクリタキセル対+CD95−Fc PLGAパクリタキセル:**P=0.0093;第48日:パクリタキセル対−CD95−Fc PLGAパクリタキセル:*P=0.02。(D)卵巣癌研究の生体イメージングの例。
【実施例】
【0071】
物質と方法
重要な物質
FasFcキメラ − カタログ番号:F8799−50ug;供給業者:シグマアルドリッチ(英国)
ポリスチレン蛍光マイクロ粒子:ドラゴングリーン0.5μm − カタログ番号:FS03F/5069;ドラゴングリーン1.0μm − カタログ番号:FS03F/7220;供給業者:バングズラボラトリーズ(Bangs Laboratories)(米国)
乳酸−グリコール酸共重合体(PLGA) − カタログ番号 Resomer(登録商標)RG502H;供給業者:アルファケミカルズ(Alfa Chemicals)(英国)
【0072】
細胞株および一次培養物
DaoyおよびND7/23細胞株を、10%ウシ胎仔血清(FBS)および1%ペニシリン/ストレプトマイシン(P/S)を添加したDMEM(グルタミン含有)培地中、37℃および5%CO2にて培養した。細胞は、60mmのペトリ皿に播種し、実験グループあたり3つのペトリ皿とした。ND7/23細胞株は、培養中、フローサイトメトリーの5日前に神経成長因子(NGF)で分化させた。Daoy細胞は、NGFを添加せず、分化に必要である追加の成長因子なしで培養した。
【0073】
後根神経節(DRG)感覚ニューロンをメスの成体ウィスターラット(2〜4月齢、>180g)から切除した。DRGニューロンは、1%BSA、N2サプリメント、NGF、および供給業者が示す濃度でのペニシリン−ストレプトマイシンを含有する培地で培養した。
【0074】
C57BL/6系統のマウスを用いて、皮質ニューロンを得た(内務省の法令に従う)。皮質ニューロンを生後0〜3日のマウスの脳から切除した。皮質ニューロンは、1%ウマ血清、1%ペニシリン−ストレプトマイシン、および2%B−27サプリメントを含有するニューロベーサル培地(neurobasal media)中にて培養した。
【0075】
マイクロ粒子を培養物へ添加し、さらなる分析を行う前に、細胞株と共に24、48、72、または244時間のインキュベーションを行った。
【0076】
免疫蛍光および共焦点顕微鏡分析
免疫細胞化学的に標識した細胞を、顕微鏡を用いてウェル培養スライドから直接観察した。Leica DMRD顕微鏡(レイカ(Leica)、英国)を、蛍光顕微鏡観察のみに用い、共焦点分析にはZeiss LSM510顕微鏡(Zeiss(ゼイス)、英国)を用いた。顕微鏡に付属のソフトウェアおよびAdobe Photoshop 7.0を用いてイメージの取り込みと表示を行った。
【0077】
電子顕微鏡
透過型電子顕微鏡については、リン酸緩衝した4%グルタルアルデヒド中で細胞を1時間固定し、緩衝液中にて一晩静置した。細胞は、1%四酸化オスミウムで30分間の後固定を行った。それぞれ、Durcupan溶液の一連の勾配(50、70、90、100、および100%)、100%Durcupanと包埋媒体との混合物、ならびに純包埋媒体によって脱水を行った。70〜80nmの超薄切片を銅グリッド上に回収し、酢酸ウラニルおよびクエン酸鉛で染色し、その後、加速電圧80kVでの観察を行った。
【0078】
走査型電子顕微鏡については、カバーグラス上の細胞をリン酸緩衝液で洗浄し、リン酸緩衝1.5%グルタルアルデヒドで少なくとも2時間固定した。細胞は、1%四酸化オスミウムで1時間の後固定を行い、一連のメタノール勾配で脱水した。次に、細胞をヘキサメチジサリジン(hexamethydisalizine)に曝露し、室温にて一晩乾燥させた。カバーグラスを取り、アルミニウム製スタブに載せ、金コーティングした後、10kVの電圧下にて観察した。
【0079】
フローサイトメトリー
本研究には、セルクエストソフトウェア付きのFACScanフローサイトメーター(ベクトンディクソン(Beckton Dixon))を用いた。各実験グループからの細胞をプロトコルごとに調製し、ラベル付きフローサイトメトリーチューブに移して分析した。適切なゲーティングおよびコントロールを用いた。細胞は、適切な抗体を用いて検出した。インターナリゼーションの研究では、ポリスチレン粒子はFITCチャネル中で蛍光性であった。
【0080】
7AAD細胞死アッセイ
死細胞集団を、7AAD(7−アミノアクチンミオシンD)(7−Aminoactinmyosin D)アッセイを用いて識別した。H2O2をポジティブコントロールとして用い、これを、37℃で4時間、最終濃度100mMにて培養物へ添加した。浮遊する細胞を、各ペトリ皿からファルコンチューブへ移し、遠心分離に掛けてフローサイトメトリー用に細胞を調製した。7AADアッセイについては、10μlの7AADを各フローチューブに添加し、サンプルを分析して細胞死を評価した。
【0081】
FasFcによるマイクロ粒子修飾
マイクロ粒子を、フィブロネクチン、ビトロネクチン、ラット血清、IgG、またはFasFcを用いた単純吸着によって表面修飾した。これらを10μg/200μlの対応するリガンド中に少なくとも90分間懸濁させ、一方、未コーティングマイクロスフェアを、同体積のリン酸緩衝生理食塩水中に懸濁させた。この懸濁液を30分ごとにボルテックス攪拌し、マイクロスフェアの適切なコーティングを確保した。また、種々の化学結合技術を用いてマイクロ粒子を修飾することも可能であり、または、リガンドを生分解性マイクロ粒子のマトリックス中に組み込むこともできる。
【0082】
薬物充填マイクロ粒子
ドキソルビシンまたはパクリタキセル充填PLGAマイクロ粒子を、標準的な二重エマルジョン技術を用いて合成した。PLGA(RG502H、ベーリンガーインゲルハイム(Boehringer Ingelheim)、ドイツ)プラセボ(未充填)マイクロスフェアを、単一エマルジョン溶媒蒸発技術、マイクロシーブ乳化によって作製した。乳化の前に、ジクロロメタン中7重量/体積%のPLGA溶液を0.2μmPTFEフィルターを通してろ過した。その後、このPLGAを、表面一帯に均一な細孔を有する微細加工された膜であるマイクロシーブ膜(Nanomi BV、オランダ)を通して、乳化剤を含有する水溶液中へ乳化した。得られたエマルジョンを、室温にて少なくとも3時間攪拌し、溶媒を蒸発させた。固化されたマイクロスフェアを、ろ過によって濃縮し、繰り返し洗浄した。次に、粒子を凍結乾燥し、評価まで−20℃で保存した。パクリタキセル充填PLGA(RG502H、ベーリンガーインゲルハイム、ドイツ)マイクロスフェアについては、パクリタキセルを、ジクロロメタン中6重量/体積%のPLGA溶液に添加、溶解し、25重量/重量%の最終マイクロ粒子薬物濃度を得た。この溶液を、0.2μmPTFEフィルターを通してろ過し、シリコンマイクロシーブを通して乳化した。乳化剤を含有する超純水を連続相として用いた。エマルジョンをマグネティックスターラーにより室温にて少なくとも3時間攪拌し、ジクロロメタンを蒸発させた。固化後、マイクロスフェアをやはりろ過によって回収し、繰り返し洗浄した。次に、粒子を凍結乾燥し、評価まで−20℃で保存した。均一なサイズのパクリタキセル充填およびプラセボマイクロ粒子(約1.5μm)がNanomi BV(オランダ)から得られた。
【0083】
卵巣癌異種移植片
5×106 IGROV1−ルシフェラーゼ細胞を、第1日にメスBalb C nu/nuマウスへIP接種した。パクリタキセル(20mg/kg)およびPLGAマイクロスフェアを、週1回(第7、14、21、および28日)IP投与した。バイオルミネセンスイメージングでは、マウスに125mg/kg D−ルシフェリン(キャリパーライフサイエンスイズ(Calliper Life Sciences)、英国)をIP注射し、次に麻酔した(2%イソフランの吸入による)。5分後、依然として麻酔下のマウスを、加温ステージ上(37℃)の遮光チャンバーに配置し、腹側表面上の定めた対象領域からの発光を、Xenogen IVIS Imaging System 100のシステム(アラメダ、カリフォルニア州、米国)でイメージングした。データは、Living Imageソフトウェア(これもゼノゲン(Xenogen)、アラメダ、カリフォルニア州、米国)を用いて分析し、相対ラジアンスで表す(平均ラジアンス 光子/s/cm2/srから算出)。
【0084】
統計分析
平均値の標準誤差(SEM)を用いて各実験グループ間の整合性を評価した。ボンフェローニポストテストと共に一元配置ANOVAを用いてグループ間の差を評価した。両側t検定を用いて2つのグループを比較した。
【0085】
結果
未修飾ポリスチレン粒子の非プロフェッショナル貪食細胞による取り込み
発明者らは、ニューロンがマイクロ粒子およびデブリをインビトロおよびインビボで取り込む能力を有することをこれまでに実証してきた(Bowen et al.,2007)。図(1および2)は、ポリスチレン粒子の場合の一次感覚ニューロン培養物におけるこのことの例を示しており、また、別の薬物送達システム;高分子電解質カプセルについてのこれまでに未発表のデータも含まれている。発明者らはまた、フローサイトメトリー実験から得られた、Daoyヒト髄芽腫細胞株(図3)、ND7/23感覚ニューロン細胞株(図4)、および一次皮質ニューロン(図5)を含むその他の細胞による未修飾合成粒子の取り込みも示す。
【0086】
未修飾ポリスチレン粒子の毒性研究
この研究では、未修飾1μmポリスチレン(PS)マイクロスフェアを添加することによる、種々の細胞型の生存能に対する影響について調べた。マイクロスフェアは濃度を上昇させながら添加し、24時間後、視野あたりのDRGニューロン、その他の細胞の数を定量した(図6)。このモデルについての重要な観察結果は、極めて高い濃度であってもマイクロスフェアの存在下にて細胞数の減少が見られなかったということである。このことは、これらの培養物における粒子の取り込みが、有意な毒性をまったくもたらさないことを示唆している。このことはまた、透過型および走査型電子顕微鏡観察による詳細な超微細構造の研究を含む独立した反復実験でも確認された(図7)。Daoyヒト髄芽腫細胞株(図8)およびND7/23感覚ニューロン細胞株(図9)を用いたフローサイトメトリー実験では、7−AAD細胞死アッセイにより細胞生存能への効果について調べた。
【0087】
FasFc修飾ポリスチレン粒子を用いた取り込みの研究
FasFc融合タンパク質による粒子の表面修飾により、特定の種類の神経および癌細胞による粒子の取り込みが大きく増加する結果となった。図10は、この増加を、後根神経節一次ニューロンについて、コントロールおよび他のリガンドによる修飾と比較して示している。これらの結果は、本発明で達成されるニューロンによる粒子の取り込みの改善を強く示すものである。透過型電子顕微鏡を用いた研究により(図11)、後根神経節一次ニューロンでのこれらの結果が確認された。Daoyヒト髄芽腫細胞株(図12)およびND7/23感覚ニューロン細胞株(図13)でのさらなる実例から、FasFc修飾粒子を用いることによって、特定の細胞型による取り込みを増加させることができることが示された。さらに、Daoyヒト髄芽腫細胞のマウス皮質ニューロンとの共培養において、皮質ニューロンと比較したDaoy細胞による粒子の選択的な取り込みが見られ、このことは、脳腫瘍の治療での有用性を示している(図14)。
【0088】
FasFc修飾PLGA粒子を用いた細胞質内薬物送達
乳酸−グリコール酸共重合体(PLGA)生分解性粒子を、確立された二重エマルジョン法を用いて合成し、またはNanomi BV(オランダ)からその商標であるマイクロシーブ(商標)技術(www.nanomi.com)を用いて粒子を合成した。合成の過程で、小分子、ペプチド、タンパク質、および核酸を含む種々の薬理学的活性剤またはマーカーを粒子に組み込むことができる。発明者らは、エチジウムホモダイマー核酸染色剤、ならびに抗癌薬ドキソルビシンおよびパクリタキセルを組み込んだマイクロ粒子を用いて、本発明を例証した。発明者らは、FasFc修飾したエチジウムホモダイマー充填PLGAが、取り込み、およびそれに続く薬剤の送達を高め(図15H)、粒子の摂取のないコントロール細胞では細胞質核酸の染色が見られないこと(図15D)を示した。未修飾コントロール粒子をコントロールに添加した場合、取り込みおよび薬剤送達の例は見られなかった。同様に、発明者らは、ドキソルビシン充填PLGA粒子をND7/23細胞に添加したところ、FACSによる細胞分離後、ドキソルビシン充填粒子を摂取した細胞の増殖が阻害された(図16)。パクリタキセル充填粒子をDaoyヒト髄芽腫細胞に添加した場合、未充填プラセボ粒子と比較して、パクリタキセル充填粒子では、薬物単独と同様の機能的効果が第1日および3日に見られた(図17)。これらの結果は、本発明の薬物送達用途での有用性を示している。
【0089】
FasFc(CD95−Fc)修飾によるパクリタキセル充填マイクロ粒子のインビボでの効力向上
より臨床上関連のあるシナリオへ進み、発明者らは、同所性卵巣癌モデルを用い、腹腔内注射によって、コンパートメント空間内のCD95L発現IGROV1−ルシフェラーゼ細胞をその他の競合細胞型(例:マクロファージ)の存在下にて標的とした(図18C、D)。生体イメージングより、臨床標準治療であるTaxol(Cremophor ELに溶解したパクリタキセル)の等用量と比較した+CD95−Fc PLGAパクリタキセル処理のグループにおいて、第4週までの腫瘍バイオルミネセンスの>65倍の減少が示された(図18C)。未処理(−CD95−Fc)PLGAパクリタキセルは、Taxolと同等であった。このモデルでは、いずれのプラセボも(+CD95−Fcおよび−CD95−Fc)効果がなかった。+CD95−Fc PLGAパクリタキセル処理における著しい腫瘍減少効果は、処理中断後も持続された(図18C)。このグループのマウスはすべて第48日まで生存し、第62日の終了時には、80%の動物が無症状のままであった(データ示さず)。これまでの研究は、高用量にて組成変更されたパクリタキセルを用いることによる腫瘍増殖阻害の可能性を明らかにした。また、死亡リスクの25%の低減が報告されたことから、臨床状況において、静脈内治療から抗卵巣癌薬の腹腔内送達への転換も行われる。しかし、本明細書で報告されるように、通常の用量で腫瘍量の減少が見られることは稀である。データは、CD95修飾薬物充填マイクロ粒子が、卵巣癌における標的化細胞内薬物送達を向上させる重要な役割を有することを強く支持している。これは、進行期の卵巣癌での治療は成功させることが困難であることから、臨床上必要とされている重要な領域である。
【0090】
参考文献
Abrahams VM,Straszewski SL,Kamsteeg M,Hanczaruk B,Schwartz PE,Rutherford TJ and Mor G.Epithelial ovarian cancer cells secrete functional Fas ligand.Cancer Res 2003;63:5573−5581
Ackery A,Robins S,and Fehlings MG.Inhibition of Fas−mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury.J Neurotrauma 2006;23:604−616
Benita,et al.,J.Pharm.Sci.73,1721−1724(1984)
Bowen S,Ateh DD,Deinhardt K,Bird MM,Price KM,Baker CS,Robson JC,Swash M,Shamsuddin W,Kawar S,El−Tawil T,Roos J,Hoyle A,Nickols CD,Knowles CH,Pullen AH,Luthert PJ,Weller RO,Hafezparast M,Franklin RJM,Revesz T,King RHM,Berninghausen O,Fisher EMC,Schiavo G,Martin JE.The phagocytic capacity of neurons.Eur J of Neurosci 2007;25:2947−2955
Brandhonneur N,Chevanne F,Vie V,Frisch B,Primault R,Le Potier MF,Le Corre P.Specific and non−specific phagocytosis of ligand−grafted PLGA microspheres by macrophages.Eur J Pharm Sci 2009;36:474−85
Choia C and Benveniste EN.Fas ligand/Fas system in the brain:regulator of immune and apoptotic responses.Brain Res Rev.2004;44:65−81
Esselens C,Oorschot V,Baert V,Raemaekers T,Spittaels K,Serneels L,Zheng H,Saftig P,De Strooper B,Klumperman J and Annaert W.Presenilin 1 mediates the turnover of telencephalin in hippocampal neurones via autophagic degradative pathway.J Cell biol 2004;166:1041−1054
Ethell DW,Kinloch R and Green DR.Metalloproteinase shedding of Fas ligand regulates beta−amyloid neurotoxicity.Curr Biol 2002;12:1595−1600
Faraji AH and Wipf P.Nanoparticles in cellular drug delivery.Bioorganic Med Chem 2009;17:2950−2962
Farokhzad OC,Cheng J,Teply BA,Sherifi I,Jon S,Kantoff PW,Richie JP and Langer R.Targeted nanoparticle−aptamer bioconjugates for cancer chemotherapy in vivo.PNAS 2006;103:6315−6320
Ferguson TA and Griffith TS.A vision of cell death:Fas ligand and immune privilege 10 years later.Immunol Rev.2006;213:228−238
Flugel A,Schwaiger FW,Neumann H,Medana I,Willem M,Wekerle H,Kreutzberg GW and Graeber,MB.Neuronal FasL induces cell death of encephalitogenic T lymphocytes.Brain Pathol 2000;10:353−364.
Giteau A,Venier−Julienne MC,Aubert−Pouessel A,Benoit JP.How to achieve sustained and complete protein release from PLGA−based microparticles? Int J Pharm 2008;350:14−26
Gratas C,Tohma Y,Van Meir EG,Kleinz M,Tenans M,Ishii N,Tachibana O,Kleihues P and Ohgaki H.Fas ligand expression in glioblastoma cell lines and primary astrocytic brain tumors.Brain Pathol 1997;7:863−869
Green DR and Ferguson TA.The role of Fas ligand in immune privilege.Nat Rev Mol Cell Biol.2001;2:917−24
Grimm D.Small silencing RNAs:state−of−the−art.Adv Drug Deliv Rev 2009;61:672−703
Gu F,Langer R and Farokhzad OC.Formulation/Preparation of functionalized nanoparticles for in vivo targeted drug delivery.Methods Mol Biol 2009;544:589−98
Huwyler J,Drewe J and Krahenbuhl S.Tumor targeting using liposomal antineoplastic drugs.Int J Nanomedicine 2008;3:21−9
Landau AM,Luk KC,Jones ML,Siegrist−Johnstone R,Young YK,Kouassi E,Rymar VV,Dagher A,Sadikot AF and Desbarats J.Defective Fas expression exacerbates neurotoxicity in a model of Parkinson’s disease.J Exp Med 2005;202:575−581
Lettau M,Paulsen M,Kabelitz D,Janssen O.Storage,expression and function of Fas ligand,the key death factor of immune cells.Curr Med Chem.2008;15:1684−1696
Mello CC and Conte Jr D.Revealing the world of RNA interference.Nature 2004;431:338−342
Meng Y,Graves L,Do T−V,So J,Fishman DA.Upregulation of FasL by LPA on ovarian cancer cell surface leads to apoptosis of activated lymphocytes.Gynecologic Oncology 2004;95:488−495
Mathiowitz and Langer,J.Controlled Release 5,13−22(1987)
Mathiowitz,et al.,Reactive Polymers 6,275−283(1987)
Mathiowitz,et al.,J.Appl.Polymer Sci.35,755−774(1988)
Mathiowitz,et al.,Scanning Microscopy 4,329−340(1990)
Mathiowitz,et al.,J.Appl.Polymer Sci.45,125−134(1992)
Munoz Javier A,del Pino P,Bedard MF,Ho D,Skirtach AG,Sukhorukov GB,Plank C and Parak WJ.Photoactivated release of cargo from the cavity of polyelectrolyte capsules to the cytosol of cells.Langmuir 2008;24:12517−12520
Nagata S and Suda T.Fas and Fas ligand:Ipr and gld mutations.Immunol Today.1995;16:39−43
Nagata S.Apoptosis by Death Factor.Cell 1997;88:355−365
Niehans GA,Brunner T,Frizelle SP,Liston JC,Salerno CT,Knapp DJ,Green DR,and Kratzke RA.Human Lung Carcinomas Express Fas Ligand.Cancer Res 1997;57:1007−1012
O’Connell J,Bennett MW,O’SULLIVAN GC,Roche D,Kelly J,J.Collins K and Fergus S.Fas ligand expression in primary colon adenocarcinomas:evidence that the Fas counterattack is a prevalent mechanism of immune evasion in human colon cancer.J Pathol 1998;186:240−246
O’Connell J,Houston A,Bennett MW,O’Sullivan GC and Shanahan F.Immune privilege or inflammation? Insights into the Fas ligand enigma.Nature Med.2001;7:271−274(Commentary)
Patil Y and Panyam J.Polymeric nanoparticles for siRNA delivery and gene silencing.Int J Pharm 2009;367:195−203
Rabinovitch M.Professional and non professional phagocytes:an introduction.Trends in cell biology 1995;5:85−87
Ryan AE,Shanahan F,O’Connell J and Houston AM.Addressing the“Fas counterattack”controversy: blocking fas ligand expression suppresses tumor immune evasion of colon cancer in vivo.Cancer Res.2005;65:9817−9823
Sawyer TK.AILERON therapeutics.Chem Biol Drug Des(R&D Spotlight)2009;73:3−6
Sinha VR,Bansal K,Kaushik R,Kumria R and Trehan A.Poly−ε−caprolactone microspheres and nanospheres:an overview.Int J Pharm 2004;278:1−23
Sukhorukov GB,Rogach AL,Garstka M,Springer S,Parak WJ,MuCoz−Javier A,Kreft O,Skirtach AG,Susha AS,Racane Y,Palankar R and Winterhalter M.Multifunctionalized polymer microcapsules:Novel tools for biological and pharmacological applications. Small 2007;3:944−955
Watanabe−Fukunaga et al.,The cDNA structure,expression,and chromosomal assignment of the mouse Fas antigen.Journal of Immunology 1992;148:1274−1279
Walter E,Dreher D,Kok M,Thiele L,Kiama SG,Gehr P and Merkle HP.Hydrophilic poly(DL−lactide−co−glycolide)microspheres for the delivery of DNA to human−derived macrophages and dendritic cells.J Control Release 2001;76:149−168
Weller M,Schuster M,Pietsch T,and Schabet M.CD95 ligand−induced apoptosis of human medulloblastoma cells.Cancer Lett 1998;128:121−126
Zuliani C,Kleber S,Klussmann S,Wenger T,Kenzelmann M,Schreglmann N,Martinez A,del Rio JA,Soriano E,Vodrazka P,Kuner R,Groene H−J,Herr I,Krammer PH and Martin−Villalba A.Cell Death Diff.2006;13:31−40
【図1A】

【図1B】
【図1C】
【図1D】
【技術分野】
【0001】
本発明は、薬物送達の分野に関する。より詳細には、本発明は、標的化送達媒体または薬物キャリア、ならびに治療および診断用途におけるこれらの使用に関する。好ましくは、本発明に従う媒体またはキャリアは、それらの標的細胞に対して高い特異性を有し、薬理学的活性剤または検出可能マーカーの効果的な細胞内送達を提供することができる。態様では、本発明は、薬理学的活性剤または検出可能マーカーをFasリガンド発現細胞へ特異的に送達するための手段を提供する。本発明は、組成物、ならびに治療、特に新生物および神経障害の治療または予防における使用にも拡張される。
【背景技術】
【0002】
現在、ほとんどの薬物は全身投与される。ほとんどの治療薬では、薬剤の大部分が全身クリアランスによって体内から排出され、薬剤の僅かな一部のみが標的器官または組織に到達する。さらに、健全な組織および器官の投与薬物への全身曝露は、重篤な毒性を引き起こす可能性がある。毒性のリスクは、薬物の用量が高く体積が多い場合に悪化する可能性があるが、これらは、低い生体利用度の克服、および対象内での適切な分布の提供に必要であることが多い。逆に、標的化薬物送達は、薬剤を対象とする組織または器官に集中させ、同時に全身薬物曝露を最小限に抑えることを目的としている。しかし、標的化薬物送達の既存のシステムは理想的ではなく、現在の薬物治療計画は、全身薬物曝露に付随する毒性の問題への対処が不十分である。これは、癌治療の分野で特にそうであり、この場合、細胞毒性抗癌薬は、それが健全組織に与えうる損傷の結果としての重篤な副作用を起こす恐れがある。従って、標的組織または器官へ薬物を選択的に送達する標的化薬物送達計画が切実に求められている。そのような計画であれば、薬物に関連する毒性のリスクを最小限に抑えながら、薬物の治療指数が高められることによって薬物治療の効力が改善されるはずである。
【0003】
さらに、RNA干渉(非特許文献1、非特許文献2)またはペプチドに基づく細胞内修飾因子(非特許文献3)などの新しい治療計画と合わせて、ゲノミクス、エピゲノミクス、およびプロテオミクスの知識が豊富となったことで、疾患治療の可能性が変化しつつある。特に、個別の細胞型の治療、または例えば癌で見られるような異常細胞の破壊を選択的に行う目的で、細胞内標的に作用する能力を有する小分子またはより大きい生物学的薬物を製剤することが望ましい。しかし、細胞内活性薬物の候補は、その細胞内標的分子と相互作用を起こすための細胞膜の貫通ができないために、薬理学的に有用となり得ないことが多い。細胞膜は、脂質の二重層であり、これは通常、細胞内部環境を細胞外部(または外側)環境から分離する半透過性バリアとして作用する。細胞膜を通して薬物を運搬するための現在の送達システムは、想定されるほど効率的でも信頼性が高いわけでもない。従って、効率的な細胞内薬物送達システムが求められている。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Mello & Conte,2004
【非特許文献2】Grimm,2009
【非特許文献3】Sawyer TK,2009
【発明の概要】
【0005】
本発明の第一の局面によると、薬理学的活性剤またはマーカーを細胞に送達するための、Fasリガンドに特異的なリガンド結合部分および薬理学的活性剤またはマーカーのためのキャリアを含む送達媒体が提供される。
【0006】
この送達媒体は、Fasリガンドに特異的なリガンド結合部分および薬物のためのキャリアを含む薬物送達媒体であってよい。リガンド結合部分は、本発明に従う送達媒体をFasリガンド発現細胞へ標的化することができる。従って、送達媒体は、薬理学的活性剤、マーカー、または薬物の細胞内送達を提供することができる。薬理学的活性剤、マーカー、または薬物は、好ましくは、Fasリガンドと特異的に結合する能力を持つ薬剤ではない。
【0007】
本発明に従うキャリアは、マイクロ粒子、ナノ粒子、マイクロカプセル、マイクロスフェア、ミセル、またはリポソームであってよい。好ましくは、キャリアは、マイクロ粒子である。マイクロ粒子は、0.01μm、0.05μm、0.1μm、0.2μm、0.5μm、1μm、2μm、5μm、または10μmまでの平均径を有していてよい。マイクロ粒子は、0.1μmから10μm、好ましくは0.2μmから5μm、より好ましくは0.3μmから2μm、さらにより好ましくは0.4μmから1.5μm、最も好ましくは0.5μmから2μmの平均径を有していてよい。マイクロ粒子は、乳酸−グリコール酸共重合体(PLGA)マトリックスを含んでいてよい。
【0008】
Fasリガンドに特異的である媒体のリガンド結合部分は、Fas受容体またはその誘導体を含んでいてよい。それはまた、完全長Fasタンパク質であっても、またはその断片であってもよい。リガンド結合部分は、Fasタンパク質の細胞外ドメインもしくはその断片を含むか、またはそれから成っていてよく、好ましくは、Fasタンパク質のリガンド結合ドメインを含むか、またはそれから成っている。媒体のリガンド結合部分は、ペプチド、タンパク質、アプタマー、抗体、抗体断片、融合タンパク質、またはキメラタンパク質であってよい。リガンド結合部分は、免疫グロブリンの結晶化可能断片領域(Fc領域)と融合してキメラ融合タンパク質、すなわちFas−Fc、を形成したFasタンパク質、またはその断片であってよい。好ましくは、免疫グロブリンは、IgG1などのヒト免疫グロブリンである。媒体は、Fasリガンドに特異的な複数のリガンド結合部分を含んでいてよく、後者の各々は、前述の例のいずれによっても実現することができる。
【0009】
この、または各々のリガンド結合部分は、どのような由来のものであってもよいが、好ましくは、ヒトもしくはマウス、またはその組み合わせである。
【0010】
この、または各々のリガンド結合部分は、特にマイクロ粒子の場合、表面吸収、吸着、化学結合、もしくはマトリックス内組み込みによってキャリアとカップリングしていてよく、または、特にキャリアがマイクロ粒子の場合、キャリアと共有結合もしくは非共有結合によって会合していてよい。この、または各々のリガンド結合部分は、連結分子を介してキャリアとカップリングしていてよい。連結分子は、免疫グロブリン、好ましくはIgG1などのヒト免疫グロブリンの結晶化可能断片領域(Fc)であってよい。
【0011】
別の選択肢として、アビジン−ビオチンなどの連結系を用いて、Fasリガンドに特異的なリガンド結合部分をキャリアと間接的にカップリングさせてもよい。従って、連結分子は、アビジンまたはビオチンであってよい。Fasリガンドに特異的なリガンド結合部分は、これをビオチン化して、アビジン−コーティングしたキャリア表面にカップリングさせてよい。別の連結分子としては、ブドウ球菌プロテインAであってよい。
【0012】
好ましい態様では、薬理学的活性剤、マーカー、または薬物は、好ましくは標的細胞にて、もしくはその内部で放出されるように、キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている。
【0013】
本発明の第一の局面の特に好ましい態様では、送達媒体は、薬理学的活性物質を含有するマイクロ粒子であり、ここで、マイクロ粒子は、Fas、その誘導体、もしくはFasリガンドに対する特異的結合分子と共有結合または非共有結合によって会合している。
【0014】
薬理学的活性剤は、タンパク質、ペプチド、ポリペプチド、ポリヌクレオチド、ポリサッカリド、脂質、小分子薬物、またはその他のいずれの生物学的活性物質であってもよい。薬理学的活性剤は、細胞内で作用してよく、または標的細胞内部の成分に対して特異的であってもよい。薬理学的活性剤は、細胞毒性剤、または細胞分裂阻害剤であってよい。また、薬理学的活性剤としては、細胞毒性放射性核種、化学毒素、またはタンパク質毒素も挙げることができる。好ましくは、薬理学的活性剤は、抗癌剤、特にドキソルビシンまたはパクリタキセルである。
【0015】
マーカーは、蛍光マーカー、放射性核種、または造影剤であってよい。従って、マーカーは、検出可能マーカー、またはイメージングマーカーであってよい。
【0016】
薬理学的活性剤、マーカー、または薬物は、特にキャリアがマイクロ粒子である場合、キャリアによって含有されるかまたはカプセル化されていてよい。活性剤、マーカー、または薬物は、化学結合によってキャリアもしくはマイクロ粒子と結合しているか、またはその内部にあってよく、または、キャリアを形成する物質のマトリックス内に物理的に組み込まれていてもよい。
【0017】
第二の局面では、本発明は、医療に、好ましくは治療目的に用いられる、本発明の第一の局面に従う送達媒体を提供する。本局面では、薬理学的活性剤、マーカー、または薬物は、キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されていることが好ましい。
【0018】
治療目的とは、新生物または神経障害に関連する疾患または医学的病状の治療であってよい。
【0019】
本発明はまた、さらなる局面において、疾患または医学的病状の治療方法も提供し、その方法は、本発明の第一の局面に従う送達媒体を対象へ投与することを含み、ここで、送達媒体は、薬理学的活性剤または薬物の効果量を含む。
【0020】
疾患または病状は、脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、またはFasリガンドが腫瘍細胞中で発現される癌の種類もしくは腫瘍であってよい。好ましくは、疾患または病状は、卵巣癌または髄芽腫である。
【0021】
疾患または病状は、運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、および末梢神経または脊髄損傷を含む神経疾患であってよい。
【0022】
本発明のさらなる局面によると、Fasリガンド発現細胞の検出を含む診断方法に用いるための、本発明の第一の局面に従う送達媒体が提供され、ここで、マーカーが、キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている。
【0023】
別の局面では、本発明は、本発明に従う送達媒体を対象へ投与することによってFasリガンド発現細胞を検出することを含む診断の方法を提供し、ここで、マーカーが、キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている。
【0024】
好ましくは、診断方法、または診断の方法は、新生物または神経障害を診断する方法である。
【0025】
本発明はまた、本発明に従う送達媒体、および1もしくは2つ以上の生理学的または薬理学的に許容されるキャリア、賦形剤、または安定化剤を含む医薬組成物も提供する。そのような組成物は、本明細書で述べる用途および方法のいずれにも用いることができる。
【0026】
別の局面では、本発明は、本発明に従う送達媒体を作製するための方法を提供し、その方法は、薬理学的活性剤、マーカー、もしくは薬物を含有するかまたはカプセル化するマイクロ粒子を形成する工程、およびFasリガンド特異的リガンド結合部分を前記マイクロ粒子に結合させる工程を含む。
【0027】
発明者らは、驚くべきことに、Fas/Fasリガンド系が、マイクロ粒子の取り込みを高めることができることを見出した。この手法を用いることで、発明者らは、Fasとカップリングしたマイクロ粒子により薬物を標的細胞へ送達することができることを示した。この分子標的化の方法により、Fasリガンド発現細胞によるマイクロ粒子の細胞内取り込みが高められる。
【0028】
本特許出願において、発明者らは、薬物送達粒子(例:乳酸−グリコール酸共重合体マトリックス(PLGA)、ポリ乳酸(PLA)、ポリ−ε−カプロラクトン(PCL)、ポリドロキシブチレート(PHB)、またはキトサン生分解性マイクロスフェア、ケイ素系粒子、高分子電解質カプセル、リポソームなど)を修飾してこれらの取り込みの向上、ならびにニューロン、癌細胞、および/または種々のFasリガンド(FasL、アポ−1L、CD95L)発現細胞を含む特定の細胞への特異化を行うための、Fas(アポ−1、CD95とも称される)、Fasの融合タンパク質(例:キメラ融合タンパク質:免疫グロブリンの結晶化可能断片領域(Fc領域)と融合したFas(FasFc)など)、および/またはFasの役割を再現する能力を有する同等部分(例:ペプチド、タンパク質、アプタマーなど)の新規な使用を初めて報告する。本発明から、発明者らはまた、治療的送達のための薬物接合体としてのFas(アポ−1、CD95)、Fasの融合タンパク質(例:FasFcなど)、および/またはFasの役割を再現する能力を有する同等部分(例:ペプチド、タンパク質、アプタマーなど)の使用の可能性についても報告する。従って、本発明は、薬物およびその他の物質の生物細胞への細胞内送達を可能とし、生物医学的研究および治療の分野に適用することができる。
【0029】
Fas/Fasリガンド系は、発明者らの知る限りにおいて、アポトーシスおよび非免疫組織における種々の新しい役割について広く研究されてきたが、標的化、および細胞における薬物の細胞内取り込みの向上のためのFas(アポ−1、CD95)修飾薬物充填マイクロ粒子(例:PLGA、PCL、高分子電解質カプセル、またはリポソーム)、またはFas(アポ−1、CD95)接合薬物の使用に関する先行技術はない。さらに、本発明は、非自明性を有し、ならびに、癌、神経病状、およびその他の疾患を患う患者への治療製剤の送達において、ならびに/または生物医学および創薬における研究ツールとして、著しい産業上の利用性を有する。
【0030】
細胞内薬物送達システムは、末梢部位での不要な副作用を低減するものであり、数々の技術が現在研究されており、例えば、標的化ナノ粒子(Farokhzad et al.,2006;Gu et al.,2009;Faraji et al.,2009)である。また、細胞内薬物送達にマイクロ粒子を用いることも可能であり、これは、薬物の効力向上および持続放出のために充填容量を大きくできるという利点がある。
【0031】
しかし、マイクロ粒子を用いた薬物送達に関する研究はあまりなく、それらは、本質的に、マクロファージなどのプロフェッショナル貪食細胞(professional phagocytes)に関するものである(Walter et al.,2001;Brandhonneur et al.,2009)。発明者らおよび他の研究者らは、非プロフェッショナル貪食細胞(non−professional phagocytes)、すなわちニューロンの貪食能を研究し、直径が0.5ミクロンより大きい生物粒子および合成粒子の摂取を実証した(Esselens et al.,2004;Bowen et al.,2007)。この特性はニューロンについては広く認識はされていなかったが、多くの非プロフェッショナル貪食細胞が、線維芽細胞および上皮細胞を含む比較的大きな粒子を摂取する能力を有することは多く報告されている(Rabinovitch,1995)。非プロフェッショナル貪食細胞における貪食を媒介するメカニズムの理解を深めることは、これらの細胞を標的とする薬物送達粒子設計の手助けとなり得る。例えば、海馬ニューロンの場合のテレンセファリンなどの特定の細胞表面受容体によって取り込みが調節され得ることが最近示された(Esselens et al.,2004;国際公開公報第/2006/030013号「The modulation of phagocytosis in neurons」)。
【0032】
発明者らは、驚いたことに、貪食に関与することがこれまでは知られていなかったFasリガンドが、FasおよびFas接合体の細胞内取り込みを可能とすることが見出した。本発明において、Fas(アポ−1、CD95)タンパク質が、薬物充填マイクロ粒子の修飾に基づく細胞内薬物送達システムに用いられる。Fas、Fasの融合タンパク質、および/またはFasの役割を再現する能力を有する同等部分による薬物充填粒子の表面修飾により、ニューロン、癌細胞、および/または種々のFasリガンド(FasL、アポ−1L、CD95L)発現細胞を含む特定の細胞によるマイクロ粒子のインターナリゼーションが大きく高められる。
【0033】
発明者らは、Fas(アポ−1、CD95)表面修飾マイクロ粒子が、未修飾粒子、またはその他のリガンドで修飾された粒子と比較して、多くの細胞型によって選択的に摂取されることを示す多くのデータを有する(実施例で示すように)。発明者らはまた、薬剤含有マイクロ粒子として作られた生分解性Fas(アポ−1、CD95)表面修飾PLGA系マイクロ粒子が、細胞によって取り込まれ、充填された薬剤を研究対象細胞内部へ送達することも示す。実施例で示されるように、発明者らは、Fas/Fasリガンド−補助貪食を介して、約0.5〜1.5μmの平均径のものを含むマイクロ粒子が細胞内へ効率的に取り込まれることを示した。さらに、実施例では、生物細胞が抗癌薬を充填したマイクロ粒子を取り込んだ場合の機能的効果も示す。従って、実施例に示したデータは、Fas(アポ−1、CD95)の使用に基づいた治療プラットフォームの新規性、進歩性、および有用性を示すものである。
【0034】
従って、本発明に従う送達媒体を用いて、投与薬物の総量を大きく低減し、同時に標的部位での薬物濃度を高めることができる。従って、これらを用いて、新生物および神経障害などの広範囲にわたる疾患における細胞毒性剤およびその他の薬物の機能を改善することができ、毒性を低下させると同時に薬物効力を向上させる可能性を有する。これらはまた、現時点で開発可能であると見なされていない有望な治療薬に対する送達オプションを提供することもできる。薬理学的活性剤または薬物がキャリアまたはマイクロ粒子でカプセル化される本発明の態様は、この観点で特に効果的であり、それは、これらによって、その標的に到達するまでに生物学的に利用可能となり得る薬剤または薬物の量が低減されるからである。
【0035】
当業者には公知であるように、Fas(アポ−1、CD95としても知られる)は、TNF/NGF受容体スーパーファミリーのメンバーであり、細胞膜受容体として、または可溶性の形態で発現される。ヒトFas(アポ−1、CD95)遺伝子の完全配列および転写物は、ホモサピエンスおよびその他のいくつかの生物種のEnsemblデータベースに登録されている(http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000026103)。マウスFas遺伝子の完全長配列および転写物は、マウスゲノム(Mus musculus)のEnsemblデータベースに登録されており(http://www.ensembl.org/Mus_musculus/Gene/Summary?db=core;g=ENSMUSG00000024778)、Watanabe−Fukunaga et al.,1992、に報告されている。Fasは、アポ−1LおよびCD95Lとしても知られるそのリガンドであるFasリガンド(FasL)との結合を介して、選択された細胞のアポトーシスの誘発を媒介する。Fasリガンドは、TNFスーパーファミリーのメンバーであり、選択された細胞においてFasと結合することでプログラム細胞死を誘発する。Fas/Fasリガンド系は、ほとんどの場合アポトーシスに関連して研究されてきており(Nagata,1997)、Fas/Fasリガンド系の異常調節は、免疫恒常性の破壊を伴う(Nagata and Suda,1995;Lettau et al.,2008)。しかし、免疫系組織における単一の役割を超えて、中枢神経系などの種々の組織におけるFas/Fasリガンド系の機能を提案する報告があり、そこでは、FasおよびFasリガンドは、非免疫細胞によって発現される(Choi and Benveniste,2004)。これらとしては、例えば、パーキンソン病(Landau et al.,2005)およびアルツハイマー病(Ethell et al.,2002)における新規な役割、ニューロンの枝分かれの制御(Zuliani et al.,2006)、ならびに脊髄損傷における神経保護(Ackery et al.,2006)が挙げられる。
【0036】
さらに、中枢神経系(Choi and Benveniste,2004)、眼(Ferguson and Griffith,2006)、および腫瘍(O’Connell et al.,2001;Ryan et al.,2005)を含む特定の組織または器官の細胞におけるFasリガンドの発現が、活性化されたFas陽性免疫細胞の死滅を可能とすることで、これらの組織または器官に免疫特権状態を付与するものであることが強く主張されている(Flugel et al.,2000;Green and Ferguson,2001)。Fasリガンドを発現する腫瘍細胞としては、共培養実験にてT細胞を死滅させる能力が示されたヒト肺癌が挙げられ(Niehans et al.,1997);この能力は、融合タンパク質FasFcを培養物へ添加すると阻害された。これらとしてはまた、神経膠芽腫および髄芽腫などの脳腫瘍(Gratas et al., 1997,Weller et al.,1998)、インビボトランスフェクションによるFasリガンドの下方制御が腫瘍浸潤リンパ球の数の増加に起因して腫瘍サイズの減少をもたらした(Ryan et al.,2005)結腸癌(O’Connell et al.,1998)、およびFasリガンドの誘発がT−リンパ球のアポトーシスを増加させた(Meng et al.,2004)卵巣癌(Abrahams et al.2003)も挙げられる。
【0037】
キャリアまたはマイクロ粒子は、ポリアルキレン、ポリカーボネート、ポリ(ジオキサノン)、ポリ酸無水物、ポリヒドロキシ酸、ポリフマレート、ポリカプロラクトン、ポリアミド、ポリアセタール、ポリエーテル、ポリエステル、ポリ(オルソエステル)、ポリヒドロキシブチレート、ポリビニルアルコール、ポリウレタン、ポリホスファゼン、ポリアクリレート、ポリメタクリレート、ポリシアノアクリレート、ポリウレア、ポリスチレン、ポリアミン、ポリ(アリレート)、ポリカーボネート、ポリ(プロピレンフマレート)、ポリヒドロキシアルカノエート、ポリケタール、ポリエステルアミド、ポリヒドロキシバリレート、ポリオルソカーボネート、ポリ(ビニルピロリドン)、ポリアルキレンオキサレート、ポリアルキレンスクシネート、ポリ(リンゴ酸)、ポリ(メチルビニルエーテル)、およびポリ(無水マレイン酸)から成る群より選択されるポリマーを含んでいてよい。
【0038】
キャリアまたはマイクロ粒子は、PLGA(乳酸−グリコール酸共重合体)マトリックス、PLA(ポリ乳酸)、PCL(ポリ−ε−カプロラクトン)、もしくはPHB(ポリヒドロキシブチレート)を含んでよく、または、キトサン生分解性マイクロスフェア、ケイ素もしくはケイ素系の粒子、高分子電解質カプセル、デンドリマー、もしくはリポソームである。キャリアまたはマイクロ粒子は、非ポリマーであってもよい。好ましくは、マイクロ粒子はPLGAマトリックスを含む。
【0039】
本発明に従うマイクロ粒子の生分解性は、インビトロにて適切ないずれの培地によるインキュベーションによっても測定または試験することができる。生分解性はまた、皮下または筋肉内を例とするマイクロ粒子の非経口注射、および時間の関数としての組織の組織学的試験によって調べることもできる。非経口投与後の生分解性マイクロ粒子は、体内で溶解し、内因性物質を、最終的には例えば乳酸を形成することができる。
【0040】
本発明に従うマイクロ粒子の生体適合性は、皮下または筋肉内を例とするマイクロ粒子の非経口投与、および組織の組織学的評価によって試験することができる。
【0041】
マイクロ粒子に関連するほとんどの研究では、乳酸−グリコール酸共重合体(PLGA)マトリックス内に製剤された薬物が考慮されてきた。PLGAは、臨床での使用に関してUS FDA(米国食品医薬品局)を含む監督機関によって認可された生分解性ポリマーであり、整形インプラントおよび全身薬物送達デポーシステム(例:Trelstar(登録商標)、Lupron Depot(登録商標)、Risperdal Consta(登録商標))に一般的に用いられている。生物活性タンパク質(Giteau et al.,2008)および核酸(Patil and Panyam,2009)を含むPLGAマイクロ粒子への薬物の充填は、通常、合成の過程で行われる。さらに、PLGA薬物キャリアは、その標的特異性を高めるために、単純な吸着または化学結合のいずれかを介して適切なリガンドにより表面官能化することができる。Farokhzad and colleagues(2006)は、前立腺癌細胞を標的とする目的で、ドセタキセル充填ペグ化PLGAナノ粒子へ官能化されたアプタマーを用いてそのような手法を例証した。また、ポリ−ε−カプロラクトン(Sinha et al.,2004)を含むその他の種類の生分解性マイクロ粒子内、および高分子電解質マイクロカプセル(Sukhorukov et al.,2007;Munoz Javier et al.,2008)またはリポソーム(Huwyler et al.,2008)などの広範囲の様々なシステム内に細胞内送達用の薬物を閉じ込めることも可能である。
【0042】
薬理学的活性剤は、抗腫瘍剤、抗生物質、抗炎症剤、抗ヒスタミン剤、鎮静剤、筋弛緩剤、抗てんかん剤、抗うつ剤、抗アレルギー剤、気管支拡張剤、強心剤、抗不整脈剤、血管拡張剤、抗糖尿病剤、抗凝固剤、止血剤、麻酔剤(narcotic)、およびステロイド剤であってよい。
【0043】
薬理学的活性剤は、細胞毒性剤、細胞分裂阻害剤、またはその他の薬物であってよい。細胞毒性剤、細胞分裂阻害剤、または薬物は、プラチン(誘導体)およびタキソールの類であってよい。細胞分裂阻害薬または薬物は、例えば、シスプラチン、サトラプラチン、オキサリプラチン、カルボプラチン、ネダプラチン、クロラムブシル、シクロホスファミド、メファラン(mephalan)、アザチオプリン、フルオロウラシル、メルカプトプリン、メトレキサート(methrexat)、ナンドロロン、アミノグルテミド(aminogluthemid)、メドロキシプロゲステロン、酢酸メゲストロール、プロカルバジン、ドセタキセル、パクリタキセル、エピポドフィロトキシン、ポドフィロトキシン、ビンクリスチン、ドセタキセル、ダウノマイシン、ドキソルビシン、ミトキサントロン、トポテカン、ブレオマイシン、ゲムシタビン、フルダラビン、および5−FUDRから成る群より選択することができる。好ましくは、生物活性剤は、抗癌剤、特にドキソルビシンまたはパクリタキセルである。
【0044】
細胞毒性核種または放射線治療用同位体元素は、225Ac、211At、212Bi、213Bi、212Pb、224Ra、または223Raなどのアルファ線放射性同位体元素であってよい。別の選択肢として、細胞毒性放射性核種は、186Rh、188Rh、177Lu、90Y、131I、67Cu、64Cu、153Sm、または166Hoなどのベータ線放射性同位体元素であってよい。別の選択肢として、細胞毒性放射性核種は、オージェ電子および低エネルギー電子を放射するものであってもよく、125I、123I、または77Brの同位体元素が挙げられる。
【0045】
適切な検出可能またはイメージングマーカーとしては、これらに限定されないが、Molecular Probes(Handbook of fluorescent probes and research products)に記載のものなどの蛍光分子が挙げられ、ローダミン、フルオレセイン(フルオレセインイソチオシアネート(FTIC)など)、テキサスレッド、アクリジンオレンジ、アレクサフルオー(Alexa Fluor)(種々)、アロフィコシアニン、7−アミノアクチノマイシンD、BOBO−I、BODIPY(種々)、カルシエン(Calcien)、カルシウムクリムゾン(Calcium Crimson)、カルシウムグリーン、カルシウムオレンジ、6−カルボキシローダミン6G、カスケードブルー(Cascade blue)、カスケードイエロー、DAPI、DiA、DiD、DiI、DiO、DiR、ELF97、エオシン、ERトラッカーブルー−ホワイト(ER Tracker Blue−White)、EthD−1、エチジウムブロミド、Fluo−3、Fluo4、FM1−43>FM4−64、Fura−2、フラレッド(Fura Red)、ヘキスト33258、ヘキスト33342、7−ヒドロキシ−4−メチルクマリン、Indo−1、JC−I、JC−9、JOE色素、リサミンローダミンB、ルシファーイエローCH、リソセンサーブルーDND−167(LysoSensor Blue DND−167)、リソセンサーグリーン、リソセンサーイエロー/ブルー、リソトラッカーグリーンFM(Lysotracker Green FM)、マグネシウムグリーン、マリーナブルー、ミトトラッカーグリーンFM(Mitotracker Green FM)、ミトトラッカーオレンジCMTMRos、ミトトラッカーレッドCMXRos、モノブロモビマン、NBDアミン、ネルオトレース500/525グリーン(NeruoTrace 500/525 green)、ナイルレッド、オレゴングリーン(Oregon Green)、パシフィックブルー(Pacific Blue)、POP−I、プロピジウムヨージド、ローダミン110、ローダミンレッド、R−ファイコエリトルム(R−Phycoerythrm)、レゾルフム(Resorfm)、RH414、Rhod−2、ローダミングリーン、ローダミン123、ROX色素、ナトリウムグリーン、SYTOブルー(種々)、SYTOグリーン(種々)、SYTOオレンジ(種々)、SYTOXブルー、SYTOXグリーン、SYTOXオレンジ、テトラメチルローダミンB、TOT−I、TOT−3、X−rhod−1、YOYO−1、またはYOYO−3などである。
【0046】
さらに、放射性核種は、イメージング剤として用いることができる。適切な放射性核種としては、Fe(III)、Fe(II)、Cu(II)、Mg(II)、Ca(II)、およびZn(II)、インジウム、ガリウム、およびテクネチウムの放射性種を挙げることができる。その他の適切な造影剤としては、常磁性T1強調またはT2強調MRI造影剤におけるキレート化に一般的に用いられる金属イオンを挙げることができ、銅、クロム、鉄、ガドリニウム、マンガン、エルビウム、ユーロピウム、ジスプロシウム、およびホルミウムなどの二価および三価カチオンが挙げられる。キレート化が可能であり、放射性核種イメージングに用いることができる金属イオンとしては、ガリウム、ゲルマニウム、コバルト、カルシウム、インジウム、インジウム、ルビジウム、イットリウム、ルテニウム、イットリウム、テクネチウム、レニウム、白金、タリウム、およびサマリウムなどの金属を挙げることができる。さらに、中性子捕獲放射線治療に有用であることが公知である金属イオンとしては、ホウ素および核断面の大きいその他の金属が挙げられる。適するものとしてはまた、超音波造影およびX線造影組成物に有用である金属イオンも挙げることができる。その他の適切な造影剤の例としては、気体または気体発生化合物であって、放射線不透過性であるものを挙げることができる。
【0047】
Fasリガンド発現細胞は、当業者に公知の標準的なイメージングまたは検出技術を用いて検出することができる。例えば、顕微鏡観察、フローサイトメトリー、医療用超音波検査、X線検査(投影X線検査および蛍光透視法など)、核医学イメージング(シンチレーションカメラなど)、磁気共鳴画像法(MRI)、光音響イメージング、デジタル赤外イメージングサーモグラフィ、またはトモグラフィである。
【0048】
様々な条件下にて種々の種類およびサイズの粒子を作製することができる多くのマイクロカプセル化技術が存在する。方法には通常、温度の変化、溶媒の留去、または化学架橋剤の添加による乳化されたポリマー液滴の固化が含まれる。
【0049】
マイクロカプセル化プロセスによって均一なサイズ範囲の粒子が作製されない場合は、篩いまたはろ過などの標準的な技術を用いて粒子を分離し、所望されるサイズ範囲の粒子集団を得ることができる。粒子はすべて、光学顕微鏡、Coulter Multisizer(ベックマンコールター、動的光散乱(Malvern Zetasizer)、透過型電子顕微鏡(TEM)、走査型電子顕微鏡(SEM)、および準弾性光散乱(QELS)などの標準的な技術により、粒子サイズ分布という観点から特徴づけられる。本発明のマイクロ粒子は、平均径が0.01μm、0.05μm、0.1μm、0.2μm、0.5μm、1μm、2μm、5μm、または10μmまでのものであってよい。
【0050】
一般的なマイクロカプセル化技術としては、界面縮重合、スプレードライ、ホットメルトマイクロカプセル化、および相分離技術(溶媒除去および溶媒留去)が挙げられる。このような技術は、米国特許第2001020011号,Mathiowitz and Langer 1987;Mathiowitz,et al.,1987,1988,1990,1992;Benita,et al.,1984、に記載されている。
【0051】
界面縮重合を用いることで、以下のようにしてコア物質(本発明の薬理学的活性剤またはマーカーなど)をマイクロカプセル化することができる。1つのモノマーおよびコア物質を溶媒に溶解する。第二のモノマーを、第一の溶媒と不混和性の第二の溶媒(通常は水性)に溶解する。第一の溶液を攪拌しながら第二の溶液中に懸濁させることにより、エマルジョンが形成される。エマルジョンが安定したところで、水相に開始剤を添加し、エマルジョンの各液滴の界面で界面重合を引き起こす。
【0052】
スプレードライは、通常、1〜10ミクロンのサイズのマイクロスフェアを作製するためのプロセスであり、ここでは、含有またはカプセル化されるコア物質(本発明の薬理学的活性剤またはマーカーなど)がポリマー溶液(通常は水性)に分散または溶解され、この溶液または分散液は、ポンプ送液によって圧縮ガス流で駆動される微粒子化ノズルを通され、得られたエアロゾルは、空気加熱サイクロン中に浮遊され、溶媒がマイクロ液滴から蒸発される。固化された粒子は第二のチェンバーに送られ、回収フラスコ中に捕集される。
【0053】
ホットメルトマイクロカプセル化は、コア物質(本発明の薬理学的活性剤またはマーカーなど)が溶融ポリマーへ添加される方法である。この混合物は、ポリマーの融点よりも10℃高い温度まで加熱しておいたポリマーに対する非溶媒(多くの場合油系)中に溶融液滴として懸濁される。このエマルジョンは激しい攪拌を維持され、同時に非溶媒浴はポリマーのガラス転移点未満まで急冷され、溶融液滴の固化およびコア物質の閉じ込めが引き起こされる。この技術によって作製されたマイクロスフェアは、通常、直径50ミクロンから2mmのサイズ範囲である。
【0054】
溶媒蒸発マイクロカプセル化では、通常、ポリマーが水不混和性有機溶媒中に溶解され、含有またはカプセル化される物質(本発明の薬理学的活性剤またはマーカーなど)が、有機溶媒中の懸濁液または溶液としてこのポリマー溶液に添加される。この懸濁液または溶液がビーカー中で激しく攪拌される水(エマルジョンの安定化のために界面活性剤を含有することが多い)へ添加されることで、エマルジョンが形成される。攪拌を継続した状態で有機溶媒が蒸発される。蒸発によってポリマーが析出し、コア物質を含有する固体マイクロカプセルが形成される。
【0055】
別の選択肢としての溶媒蒸発プロセスは、マイクロシーブを用いることを含む。ポリマーは、通常、水不混和性有機溶媒中に溶解され、含有またはカプセル化される物質(本発明の薬理学的活性剤またはマーカーなど)は、ジクロロメタンなどの有機溶媒中の懸濁液または溶液としてこのポリマー溶液へ添加される。次に、この懸濁液または溶液は、PTFEフィルターを通してろ過される。その後、ポリマーは、表面一帯に均一な細孔を有する微細加工された膜であるマイクロシーブ膜(Nanomi BV,オランダ、など)を通して、乳化剤を含有する水溶液中へ乳化される。得られたエマルジョンは、室温にて少なくとも3時間攪拌され、溶媒が留去される。固化されたマイクロスフェアは、ろ過によって濃縮され、繰り返し洗浄される。次に、粒子は、凍結乾燥され、評価まで低温(例:−20℃)で保存された。
【0056】
溶媒除去マイクロカプセル化では、ポリマーは、通常、油混和性有機溶媒中に溶解され、含有またはカプセル化される物質(本発明の薬理学的活性剤またはマーカーなど)は、有機溶媒中の懸濁液または溶液としてこのポリマー溶液へ添加される。この懸濁液または溶液がビーカー中で激しく攪拌される油へ添加されることで、エマルジョンが形成され、ここで、この油はポリマーに対する非溶媒であり、ポリマー/溶媒溶液は油に不混和性である。有機溶媒は、攪拌を継続した状態で、油相中へ拡散されることで除去される。溶媒除去によってポリマーが析出し、コア物質を含有する固体マイクロカプセルが形成される。
【0057】
薬理学的活性剤またはマーカーは、媒体またはマイクロ粒子の約25重量/重量%もしくはそれ以上を占めていてよい。好ましくは、薬理学的活性剤またはマーカーは、媒体またはマイクロ粒子の0.01%、0.1%、1%、5%、10%、15%、20%、25%、30%、または40%(重量/重量)までを占めている。
【0058】
マイクロ粒子は、約0.5〜1.5μmの平均径を有していてよく、上述のプロセスのいずれによって作製されてもよい。
【0059】
Fasタンパク質、その誘導体、またはそのキメラタンパク質を例とする、Fasリガンドに特異的なリガンド結合部分は、好ましくは、キャリアまたはマイクロ粒子の表面に吸着または化学結合されている。また、キャリアまたはマイクロ粒子のマトリックス中に組み込まれていてもよい。当業者であれば、この工程が、単純な吸着、化学結合技術、またはマイクロ粒子のマトリックス中への組み込みなどの標準的な技術によって行うことができることは理解される。Fasリガンドに特異的なリガンド結合部分は、好ましくは、キャリア(マイクロ粒子など)の表面と結合または会合している。結合部分は、媒体またはマイクロ粒子の約0.01重量/重量%、0.1重量/重量%、1重量/重量%、5重量/重量%、10重量/重量%、15重量/重量%、20重量/重量%、25重量/重量%、または30重量/重量%までを占めていてよい。
【0060】
従って、本発明は、薬理学的活性物質を含有するマイクロ粒子の作製方法を提供し、ここで、マイクロ粒子は、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と共有結合または非共有結合で会合しており、この方法は、表面吸収、吸着、化学結合、またはマトリックス内組み込みによって、マイクロ粒子を、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と会合させることを含む。
【0061】
適切な薬理学的に許容されるキャリア、賦形剤、または安定化剤は、無毒性であることが好ましく、活性成分の効果または生物活性に干渉しない。薬理学的に許容されるキャリアは、ヒトまたはその他の脊椎動物への投与に適する、1つ以上の適合性を有する固体もしくは液体充填剤、希釈剤、またはカプセル化物質であってよい。薬理学的に許容されるキャリアは、有機または無機成分、天然または合成物であってよく、適用を補助するために活性成分がこれらと組み合わされる。
【0062】
医薬組成物は、薬理学的に使用することができる製剤への活性化合物の処理を促進する賦形剤および助剤を含む1もしくは2つ以上の生理学的に許容されるキャリアを用いた従来の方法で製剤することができる。適切な製剤は、選択された投与経路に依存する。典型的な注射用製剤は、滅菌生理食塩水またはリン酸緩衝生理食塩水などのキャリアを含む。粘度調節剤および保存剤も添加される場合が多い。適切な薬理学的に許容される賦形剤としては、これらに限定されないが、希釈剤、バインダー、滑沢剤、崩壊剤、着色剤、安定化剤、および界面活性剤が挙げられる。
【0063】
本発明の送達媒体または組成物が、新生物もしくは神経障害の治療、寛解、または予防のための公知の療法の補助として、またはそれらと組み合わせて用いることができることは理解される。本発明に従う送達媒体または組成物は、組成物が使用される方法に特に依存する数多くの種々の形態を有する組成物に組み合わせることができる。
【0064】
Fasリガンド発現細胞は、脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、および癌の種類で見られる。Fasリガンド発現細胞はまた、神経疾患、運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、ならびに末梢神経および脊髄損傷に関連する細胞にも見られる。
【0065】
対象は、脊椎動物、哺乳類、または飼育動物であってよい。従って、本発明に従う送達媒体または組成物は、家畜(例:ウマ)、ペットを例とするいずれの哺乳類の治療にも用いることができ、またはその他の獣医学的用途に用いることもできる。最も好ましくは、対象はヒトである。
【0066】
本発明の送達媒体または組成物の効果量は、効果量の薬理学的活性剤の標的への到達を可能とし、それによって対象において治療効果を発生させる量であってよい。薬理学的活性剤を含む送達媒体または組成物の実際の効果量は、用いられる具体的な薬剤、物質、またはそれらの組み合わせ、Fasリガンドに特異的なリガンド結合部分の密度および/または性質、カプセル化薬理学的活性剤の放出特性、製剤された特定の組成物、投与モード、ならびに治療される対象の年齢、体重、病態、さらには投与経路および疾患または障害を含む因子に応じて様々であり得る。
【0067】
本発明に従う送達媒体または組成物は、非経口投与に適するものであってよい。非経口投与は、皮下、静脈内、筋肉内、関節内、滑膜内、腹腔内、胸骨内、くも膜下腔内、肝臓内、病巣内、腫瘍内、および頭蓋内注射または注入技術であってよい。非経口投与される場合、医薬組成物は、皮下または静脈内投与されることが好ましい。
【0068】
しかし、本発明に従う送達媒体または組成物の投与は、送達媒体または組成物の効果量がその標的へ到達することを可能とする許容されるいかなる方法で行なってもよい。選択される特定のモードは、特定の製剤、治療される対象の状態の重篤度、および効果的な治療を誘発するために要する用量などの因子に依存する。
【0069】
本明細書(添付の請求項、要約、および図面を含む)で述べる特徴のすべて、および/またはそのように開示されるいずれの方法の工程のすべても、そのような特徴および/または工程の少なくともいくつかが互いに相容れない場合を除いて、上記の局面のいずれといずれの組み合わせで組み合わせてもよい。ここで、本発明の理解を深め、その態様を効果的に実施する方法を示すために、例として添付の図面を参照する。
【図面の簡単な説明】
【0070】
【図1】図1は、未修飾合成粒子の一次感覚ニューロンでの取り込みを示す。共焦点顕微鏡により、β3チューブリン標識後根神経節ニューロン(核は細胞の中央に見られる)における合成粒子(丸形状のマイクロスフェア)の細胞内取り込みが示される。(A、B)1μmポリスチレンマイクロスフェア。(C、D)蛍光色素(FITC)結合ウシ血清アルブミンを充填した2μm高分子電解質カプセル。粒子は、細胞体および神経突起の両方で見られた。
【図2】図2は、未修飾合成粒子の一次感覚ニューロンでの取り込みを示す。後根神経節ニューロンの共焦点顕微鏡観察(Bowen et al.,2007より引用)。(A)明視野イメージ。(B)β3−チューブリンに対する抗体で染色したラット後根神経節ニューロンを1μmマイクロスフェア(丸形状のマイクロスフェア)とインキュベートした後の共焦点顕微鏡観察。XY平面の白色点線に沿ったzスタック断面(z−stack cross−sections)を示す上側および横側のパネルから、マイクロスフェアがインターナライズされていることが分かる。(C、E)白色線でハイライトしたニューロン内部(C)の緑色および赤色チャネルの蛍光強度プロファイル(E)は、0.33μmの光学的スライス像(optical slice)の中でマイクロスフェアとβ3−チューブリン標識とが共存していることを示す。(D、F)白色線でハイライトしたニューロン外部(D)の緑色および赤色チャネルの蛍光強度プロファイル(F)は、共存が存在しないことを示す。
【図3】図3は、Daoy髄芽腫細胞における未修飾合成粒子の取り込みを示す。(A−B)フローサイトメトリー散布図(左)およびゲーティングされたDaoy細胞に対するゲーティングプロット(右):(A)(右)濃度の上昇に従って、0.5μmマイクロ粒子を有する細胞の集団が増加することを示す(右上の四半分へ向けてのシフトが大きくなっている)。(左)高い濃度では、散布図の側方散乱の増加が見られる。コントロール細胞はマイクロ粒子を有しておらず、これをネガティブコントロールとして用いる;(B)(右)濃度が上昇すると、1.0μmマイクロ粒子を有する細胞集団の同様の増加を示す。これは、側方散乱の増加を伴っている(左)。同一濃度では、1.0μmマイクロスフェアを有する細胞集団は、0.5μmマイクロ粒子を有する細胞よりも明確である。SSC−H 側方散乱細胞;FSC−H=前方散乱細胞(サイズ);FL1−H=ポリスチレンドラゴングリーンマイクロ粒子を有するFITC検出細胞(FITC registering cells)のチャネル。(C)種々の濃度およびサイズにおけるND7/23細胞によるマイクロ粒子の取り込みを示すグラフ(フローサイトメトリーデータからまとめたもの)。いずれの場合でも、同一濃度ではサイズ間に取り込みの有意差はない(p>0.05)(ns)(サイズ依存性なし)。0.5μmの濃度1×107および1×108の間では取り込みに有意差はないが、濃度1×108および1×109の間の差は有意である(p<0.001)***。1.0μmマイクロ粒子では、濃度1×107および1×108の間(p<0.05)*、および濃度1×108および1×109の間(p<0.001)***で取り込みに有意差がある。エラーバーはn=3でのSEMを示す。
【図4】図4は、ND7/23感覚ニューロン細胞株における未修飾合成粒子の取り込みを示す。(A−B)フローサイトメトリー散布図(左)およびND7/23細胞に対するゲーティングプロット(右):(A)(右)濃度の上昇に従って、0.5μmマイクロ粒子を有する細胞の集団が増加することを示す(右上の四半分へ向けてのシフトが大きくなっている)。(左)高い濃度では、散布図の側方散乱の増加が見られる。コントロール細胞はマイクロ粒子を有しておらず、これをネガティブコントロールとして用いる;(B)(右)濃度が上昇すると、1.0μmマイクロ粒子を有する細胞集団の同様の増加を示す。これは、側方散乱の増加を伴っている(左)。同一濃度では、1.0μmマイクロ粒子を有する細胞集団は(淡色矢印)、0.5μmマイクロ粒子を有する細胞(黒色矢印)よりも明確である。SSC−H=側方散乱細胞;FSC−H=前方散乱細胞(サイズ);FL1−H=ドラゴングリーンマイクロ粒子を有するFITC検出細胞のチャネル。(C)種々の濃度およびサイズにおけるND7/23細胞によるマイクロ粒子の取り込みを示すグラフ(フローサイトメトリーデータからまとめたもの)。いずれの場合でも、同一濃度ではサイズ間に取り込みの有意差はない(p>0.05)(ns)(サイズ依存性なし)。0.5μmの濃度1×107および1×108の間では取り込みに有意差はないが、濃度1×108および1×109の間の差は有意である(p<0.001)***。1.0μmマイクロ粒子では、濃度1×107および1×108の間(p<0.05)*、ならびに濃度1×108および1×109の間(p<0.001)***で取り込みに有意差がある。エラーバーはn=3でのSEMを示す。
【図5】図5は、一次皮質ニューロンおよびその他の細胞型における未修飾合成粒子の取り込みを示す。このフローサイトメトリーのグラフは、24時間にわたるポリスチレンマイクロ粒子の添加後の、マウス脳由来一次皮質培養物のFITC(FL1−H)に対するPE(FL2−H)の蛍光をプロットしたものである。CD90.2−PE陽性細胞(皮質ニューロン)は右上と左上の四半分に見られ、その他の細胞型(主としてグリア細胞)は下側の四半分に見られる。マイクロ粒子を摂取した細胞は、右上および右下の四半分に見られる。このグラフは、皮質ニューロンおよびその他の細胞型が、培養中に未修飾粒子も摂取することを示している。
【図6】図6は、成体ラット由来の一次感覚ニューロンによる粒子の取り込み、およびその生存能を示す。(A)DRGニューロンおよびその他の細胞型の数は、培養物への添加マイクロ粒子の濃度を上昇させても、変化していない。このことは、マイクロ粒子が細胞に対して毒性を引き起こすものではないことを示唆している。(B)マイクロ粒子濃度を上昇させた場合の健全なニューロンの例(×20 OM)。
【図7】図7は、電子顕微鏡による研究から、超微細構造レベルでの毒性が示されないことを示している。(A〜F)透過型電子顕微鏡を用いたニューロンにおけるマイクロ粒子(MP)の研究に対する選択基準:(A)ニューロンは特徴的な長く薄い突起を有する。隣接するグリア細胞が短く厚い突起を有することに注目されたい(丸で囲んだ)。(B)膜結合MPを白色矢印で表す。Bには小さいMPが存在する(矢印)。(C)食胞膜と結合しないMP(矢印)は定量されなかった。食胞膜と結合したMPも存在している(矢印)。(D)ニューロン突起中の空の食胞(矢印)。E、F:二重膜によって結合した空の食胞(*)のさらなる例(矢印)。Aのバーは10μm、B〜Fのバーは0.5μmである。(G〜L)後根神経節ニューロンにおける食胞膜有りおよび無しのマイクロ粒子の透過型電子顕微鏡写真:(G)二重膜食胞内の0.5μmのサイズのマイクロ粒子(矢印で示す)。(H、J、K)食胞内の1μmマイクロ粒子。(I、L)食胞と結合していない0.5μmマイクロ粒子。バー=0.5μm。(M〜O)マイクロ粒子および後根神経節の走査型電子顕微鏡写真:(M)ニューロン膜はほとんど融合して、マイクロ粒子の周囲に小胞を形成した(矢印)。(N)2つのマイクロスフェアの基部周囲のニューロン膜の突起。(O)マイクロ粒子が取り除かれた可能性のある食作用カップ(phagocytic cup)(矢印)と考えられるものの形成。バー=1μm。
【図8】図8は、Daoyヒト髄芽腫細胞株による粒子の取り込み、およびその生存能を示す。このグラフは、種々の実験グループにおける総細胞死を示す。X軸はマイクロ粒子サイズおよび濃度である。1.0μmマイクロ粒子は、0.5μmのマイクロ粒子と比較してより多い細胞死を伴っていた。マイクロ粒子の無い培養でも、ある程度の細胞死が見られる。H2O2ポジティブコントロールのデータは含まれていない。エラーバーはn=3におけるSEMを示す。このデータは、粒子の濃度が非常に高い(実用的な使用を超える)場合であっても、細胞毒性は25%未満に限定されることを示している。
【図9】図9は、ND7/23感覚細胞株による粒子の取り込み、およびその生存能を示す。このグラフは、種々の実験グループにおける総細胞死を示す。X軸はマイクロ粒子サイズおよび濃度である。この細胞株では、マイクロ粒子を含まないコントロール培養ですでに細胞死が高く、マイクロ粒子の添加によって悪化はしていない。このグラフは、総細胞死が種々の実験グループ間で比較的一定であることを示している。エラーバーはn=3におけるSEMを示す。
【図10】図10は、一次DRGニューロンへ添加されたFasFc修飾ポリスチレン粒子を示す。DRGニューロンによる1μmドラゴングリーンポリスチレンマイクロ粒子の取り込みに対するFasFc修飾の効果を、蛍光顕微鏡で調べた。Rat血清、フィブロネクチン、ビトロネクチン、およびFasFc修飾マイクロ粒子はすべて、コントロール1と同じ密度で播種した(ウェルあたり1×105マイクロ粒子)。コントロールおよびその他の種類のリガンドと比較して、FasFc修飾は、ニューロンによる相当量のマイクロスフェアの取り込みを誘発した。
【図11】図11は、DRGニューロンを用いた未修飾およびFasFc修飾粒子のTEM研究を示す。DRGニューロンによる1μmドラゴングリーンポリスチレンマイクロ粒子の取り込みに対するFasFc修飾の効果をTEMによって調べた。未修飾およびFasFc修飾マイクロ粒子を培養物に24時間添加した(ウェルあたり2×106マイクロ粒子)。摂取されたマイクロ粒子は、細胞質内の電子密度の高い(暗色)直径約1ミクロンの正球形として見ることができる(矢印)。コントロールと比較して、FasFc修飾は、ニューロンによる相当量のマイクロスフェアの取り込みを誘発した。
【図12】図12は、Daoyヒト髄芽腫細胞株に添加したFasFc修飾ポリスチレン粒子を示す。このグラフは、Daoy細胞について、取り込みに対するFasFcマイクロ粒子の表面修飾の効果を、未コーティングのマイクロ粒子、または他のリガンドでコーティングした粒子と比較して示す。未処理マイクロ粒子(培養物に直接適用)、およびバッファー(リガンド無し)によるコーティングプロセスを受けた未コーティングマイクロ粒子の間に取り込みの有意差はなかった。0.5μmマイクロ粒子については、未コーティングおよびIgGオプソニン化マイクロ粒子の間に取り込みの有意差はないが(p>0.05)、フィブロネクチンと未コーティングの間(p<0.05)*、およびFasFcと未コーティングの間(p<0.05)*には有意差があった。1.0μmマイクロ粒子については、未コーティングコントロールと比較したIgGコーティングマイクロ粒子(p<0.05)*、未コーティングコントロールと比較したフィブロネクチンコーティング(p<0.001)***、ならびに未コーティングコントロールと比較したFasFc(p<0.001)***の取り込みに有意差があった。グラフはまた、サイズ間の取り込みの違いも示す。結果はまた、FcリッチであるIgGが、FasFcコーティングマイクロ粒子と同程度にはマイクロ粒子の取り込みを増加させておらず、従って、観察された増加には、融合タンパク質のFas部分が重要であることも示している。エラーバーはn=3におけるSEMを示す。60mmペトリ皿あたり1×107のマイクロ粒子を添加した。
【図13】図13は、ND7/23感覚ニューロン細胞株へ添加されたFasFc修飾ポリスチレン粒子を示す。このグラフは、未コーティングのマイクロ粒子と比較した、ND7/23細胞による取り込みに対するFasFcマイクロ粒子の表面修飾の効果を示す。右上の四半分(Q2)に示されるように、FasFcコーティングした0.5μmマイクロ粒子は、未コーティングマイクロ粒子(3.4%)と比較して、より高い割合の細胞(18%)によって取り込まれた。35mmペトリ皿あたり1×105のマイクロ粒子を添加した。
【図14】図14は、Daoy/皮質ニューロン共培養物へ添加されたFasFc修飾ポリスチレン粒子を示す。これらのグラフは、Daoyヒト髄芽腫細胞株とマウス由来の一次皮質ニューロンとの間で比較した、24時間の培養後のFasFc表面修飾マイクロ粒子(1μm)の取り込みを示す。培養中、Daoy癌細胞は、皮質ニューロンおよびその他の細胞型(グリア細胞など)よりも多くの粒子を摂取している。実験は、P7皮質ニューロンで行い(A)、P14皮質ニューロンで反復した(B)。60mmペトリ皿あたり1×107のマイクロ粒子を添加した。
【図15】図15は、DRG一次ニューロンに添加したFasFc修飾粒子を用いた細胞内染色剤送達を示す。(A)マージしたイメージ(×63 OM)、(B)ヘキスト染色核、(C)βIIIチューブリンニューロンマーカー、および(D)粒子が存在しないためにニューロン細胞質が染色されていないエチジウムホモダイマー充填PLGA粒子(直径<1.2μm)、(E)マージしたイメージ(×63 OM)、(F)ヘキスト染色核、(G)βIIIチューブリンニューロンマーカー、および(H)48時間後に核酸染色剤の放出および細胞質核酸の標識が見られたエチジウムホモダイマー充填PLGA粒子。
【図16】図16は、ND7/23感覚ニューロン細胞株に添加したFasFc修飾粒子を用いた細胞内薬物送達を示す。FasFc修飾粒子をND7/23細胞培養物へ一晩添加し、FACSによってドキソルビシン充填粒子を摂取した細胞を分離した後(左上の図)、コントロール細胞(右上のイメージ)は、そこから2週間にわたって正常に増殖を続けたが(顕微鏡下で観察)、一方ドキソルビシン充填粒子を摂取した細胞は(左上のグラフ;ドキソルビシン充填粒子の蛍光を示す右下のイメージを含む下側のイメージ)、まったく増殖しなかった(分離後2週間にわたって顕微鏡下で観察)。これらの観察結果は、有糸分裂阻害薬ドキソルビシンを充填した本発明を用いることによる、ND7/23感覚ニューロン細胞株での機能的薬物送達を示している。
【図17】図17は、Daoyヒト髄芽腫細胞株に添加したFasFc修飾粒子を用いた細胞内薬物送達を示す。(A)1μmパクリタキセル充填PLGAマイクロ粒子の添加後、7AAD陽性細胞と測定されたDaoy細胞のフローサイトメトリー散布図(左)。7AAD処理なしのコントロール細胞は、FL3−Hチャネルでの検出は高くない(R1領域)。薬物やプラセボ処理をまったく行わないコントロール細胞は、7AADアッセイ後の細胞死は最小限である。過酸化水素処理コントロール細胞は、7AADアッセイ後、高いパーセントの細胞死を示す。プラセボ充填と比較してパクリタキセル充填PLGAマイクロ粒子では、第1日および3日に機能的効果が見られ、細胞死が発生し、それはパクリタキセル単独の処理と同等である。SSC−H 側方散乱;FL3−H=7AAD検出細胞のチャネル。(B)7AADアッセイ後に測定された細胞死のパーセント(フローサイトメトリーデータからまとめたもの)。データは、プラセボと比較してパクリタキセル充填粒子では、第1日および3日に細胞死が増加していることを示している。このデータは、アポトーシス促進性薬物であるパクリタキセルを充填した本発明を用いることによる、Daoy髄芽腫細胞株における機能的薬物送達を実証するものである。
【図18】図18は、FasFc(CD95−Fc)修飾により、パクリタキセル充填マイクロ粒子のインビボでの効力が高められることを示す。実験では、未充填(プラセボ)または25重量/重量%パクリタキセル充填とした乳酸−グリコール酸共重合体(PLGA)生分解性マイクロ粒子(直径約1.5μm)を用いた。次に、マイクロ粒子に、CD95−Fcによる表面コーティングまたは偽コーティング(−CD95−Fc)を施した。(A)侵襲性増殖をする髄芽腫皮下異種移植片について、腫瘍体積の変化を示す。(B)第7日、単一の腫瘍内注射後、腫瘍の増殖は、パクリタキセル単独と比較して、+CD95 PLGAプラセボ、−CD95 PLGAパクリタキセル、および+CD95 PLGAパクリタキセルの場合でより効率的に阻害された。平均±SEM、n=4。(C)腹膜卵巣癌内転移のマウスモデル(IGROV1ルシフェラーゼ発現癌細胞)では、Taxolとして製剤されたパクリタキセルの等用量(Cremophor ELに溶解)と比較して、+CD95−Fc PLGAパクリタキセルは、4回の毎週の処理投与後、腫瘍バイオルミネセンスで>65倍の違いである抗腫瘍効力を示す。平均±SEM、n=5。処理の中断後、+CD95−Fc PLGAパクリタキセルのグループでは、腫瘍の再増殖は比較的遅い。プラセボグループはいずれも、疾患の広がりの度合いのために、第28日までに屠殺しなければならず;第35日までに、−CD95−Fc PLGAパクリタキセルのグループの1匹が屠殺され;第48日までには、−CD95−Fc PLGAパクリタキセルのさらに2匹、パクリタキセルの2匹、および+CD95−Fc PLGAパクリタキセルのグループの1匹が屠殺された。統計的比較(両側t検定)を、第35日について示す:パクリタキセル対+CD95−Fc PLGAパクリタキセル:*P=0.012;第35日:パクリタキセル対−CD95−Fc PLGAパクリタキセル:P=ns;第41日:パクリタキセル対+CD95−Fc PLGAパクリタキセル:*P=0.03;第41日:パクリタキセル対−CD95−Fc PLGAパクリタキセル:P=ns;;第48日:パクリタキセル対+CD95−Fc PLGAパクリタキセル:**P=0.0093;第48日:パクリタキセル対−CD95−Fc PLGAパクリタキセル:*P=0.02。(D)卵巣癌研究の生体イメージングの例。
【実施例】
【0071】
物質と方法
重要な物質
FasFcキメラ − カタログ番号:F8799−50ug;供給業者:シグマアルドリッチ(英国)
ポリスチレン蛍光マイクロ粒子:ドラゴングリーン0.5μm − カタログ番号:FS03F/5069;ドラゴングリーン1.0μm − カタログ番号:FS03F/7220;供給業者:バングズラボラトリーズ(Bangs Laboratories)(米国)
乳酸−グリコール酸共重合体(PLGA) − カタログ番号 Resomer(登録商標)RG502H;供給業者:アルファケミカルズ(Alfa Chemicals)(英国)
【0072】
細胞株および一次培養物
DaoyおよびND7/23細胞株を、10%ウシ胎仔血清(FBS)および1%ペニシリン/ストレプトマイシン(P/S)を添加したDMEM(グルタミン含有)培地中、37℃および5%CO2にて培養した。細胞は、60mmのペトリ皿に播種し、実験グループあたり3つのペトリ皿とした。ND7/23細胞株は、培養中、フローサイトメトリーの5日前に神経成長因子(NGF)で分化させた。Daoy細胞は、NGFを添加せず、分化に必要である追加の成長因子なしで培養した。
【0073】
後根神経節(DRG)感覚ニューロンをメスの成体ウィスターラット(2〜4月齢、>180g)から切除した。DRGニューロンは、1%BSA、N2サプリメント、NGF、および供給業者が示す濃度でのペニシリン−ストレプトマイシンを含有する培地で培養した。
【0074】
C57BL/6系統のマウスを用いて、皮質ニューロンを得た(内務省の法令に従う)。皮質ニューロンを生後0〜3日のマウスの脳から切除した。皮質ニューロンは、1%ウマ血清、1%ペニシリン−ストレプトマイシン、および2%B−27サプリメントを含有するニューロベーサル培地(neurobasal media)中にて培養した。
【0075】
マイクロ粒子を培養物へ添加し、さらなる分析を行う前に、細胞株と共に24、48、72、または244時間のインキュベーションを行った。
【0076】
免疫蛍光および共焦点顕微鏡分析
免疫細胞化学的に標識した細胞を、顕微鏡を用いてウェル培養スライドから直接観察した。Leica DMRD顕微鏡(レイカ(Leica)、英国)を、蛍光顕微鏡観察のみに用い、共焦点分析にはZeiss LSM510顕微鏡(Zeiss(ゼイス)、英国)を用いた。顕微鏡に付属のソフトウェアおよびAdobe Photoshop 7.0を用いてイメージの取り込みと表示を行った。
【0077】
電子顕微鏡
透過型電子顕微鏡については、リン酸緩衝した4%グルタルアルデヒド中で細胞を1時間固定し、緩衝液中にて一晩静置した。細胞は、1%四酸化オスミウムで30分間の後固定を行った。それぞれ、Durcupan溶液の一連の勾配(50、70、90、100、および100%)、100%Durcupanと包埋媒体との混合物、ならびに純包埋媒体によって脱水を行った。70〜80nmの超薄切片を銅グリッド上に回収し、酢酸ウラニルおよびクエン酸鉛で染色し、その後、加速電圧80kVでの観察を行った。
【0078】
走査型電子顕微鏡については、カバーグラス上の細胞をリン酸緩衝液で洗浄し、リン酸緩衝1.5%グルタルアルデヒドで少なくとも2時間固定した。細胞は、1%四酸化オスミウムで1時間の後固定を行い、一連のメタノール勾配で脱水した。次に、細胞をヘキサメチジサリジン(hexamethydisalizine)に曝露し、室温にて一晩乾燥させた。カバーグラスを取り、アルミニウム製スタブに載せ、金コーティングした後、10kVの電圧下にて観察した。
【0079】
フローサイトメトリー
本研究には、セルクエストソフトウェア付きのFACScanフローサイトメーター(ベクトンディクソン(Beckton Dixon))を用いた。各実験グループからの細胞をプロトコルごとに調製し、ラベル付きフローサイトメトリーチューブに移して分析した。適切なゲーティングおよびコントロールを用いた。細胞は、適切な抗体を用いて検出した。インターナリゼーションの研究では、ポリスチレン粒子はFITCチャネル中で蛍光性であった。
【0080】
7AAD細胞死アッセイ
死細胞集団を、7AAD(7−アミノアクチンミオシンD)(7−Aminoactinmyosin D)アッセイを用いて識別した。H2O2をポジティブコントロールとして用い、これを、37℃で4時間、最終濃度100mMにて培養物へ添加した。浮遊する細胞を、各ペトリ皿からファルコンチューブへ移し、遠心分離に掛けてフローサイトメトリー用に細胞を調製した。7AADアッセイについては、10μlの7AADを各フローチューブに添加し、サンプルを分析して細胞死を評価した。
【0081】
FasFcによるマイクロ粒子修飾
マイクロ粒子を、フィブロネクチン、ビトロネクチン、ラット血清、IgG、またはFasFcを用いた単純吸着によって表面修飾した。これらを10μg/200μlの対応するリガンド中に少なくとも90分間懸濁させ、一方、未コーティングマイクロスフェアを、同体積のリン酸緩衝生理食塩水中に懸濁させた。この懸濁液を30分ごとにボルテックス攪拌し、マイクロスフェアの適切なコーティングを確保した。また、種々の化学結合技術を用いてマイクロ粒子を修飾することも可能であり、または、リガンドを生分解性マイクロ粒子のマトリックス中に組み込むこともできる。
【0082】
薬物充填マイクロ粒子
ドキソルビシンまたはパクリタキセル充填PLGAマイクロ粒子を、標準的な二重エマルジョン技術を用いて合成した。PLGA(RG502H、ベーリンガーインゲルハイム(Boehringer Ingelheim)、ドイツ)プラセボ(未充填)マイクロスフェアを、単一エマルジョン溶媒蒸発技術、マイクロシーブ乳化によって作製した。乳化の前に、ジクロロメタン中7重量/体積%のPLGA溶液を0.2μmPTFEフィルターを通してろ過した。その後、このPLGAを、表面一帯に均一な細孔を有する微細加工された膜であるマイクロシーブ膜(Nanomi BV、オランダ)を通して、乳化剤を含有する水溶液中へ乳化した。得られたエマルジョンを、室温にて少なくとも3時間攪拌し、溶媒を蒸発させた。固化されたマイクロスフェアを、ろ過によって濃縮し、繰り返し洗浄した。次に、粒子を凍結乾燥し、評価まで−20℃で保存した。パクリタキセル充填PLGA(RG502H、ベーリンガーインゲルハイム、ドイツ)マイクロスフェアについては、パクリタキセルを、ジクロロメタン中6重量/体積%のPLGA溶液に添加、溶解し、25重量/重量%の最終マイクロ粒子薬物濃度を得た。この溶液を、0.2μmPTFEフィルターを通してろ過し、シリコンマイクロシーブを通して乳化した。乳化剤を含有する超純水を連続相として用いた。エマルジョンをマグネティックスターラーにより室温にて少なくとも3時間攪拌し、ジクロロメタンを蒸発させた。固化後、マイクロスフェアをやはりろ過によって回収し、繰り返し洗浄した。次に、粒子を凍結乾燥し、評価まで−20℃で保存した。均一なサイズのパクリタキセル充填およびプラセボマイクロ粒子(約1.5μm)がNanomi BV(オランダ)から得られた。
【0083】
卵巣癌異種移植片
5×106 IGROV1−ルシフェラーゼ細胞を、第1日にメスBalb C nu/nuマウスへIP接種した。パクリタキセル(20mg/kg)およびPLGAマイクロスフェアを、週1回(第7、14、21、および28日)IP投与した。バイオルミネセンスイメージングでは、マウスに125mg/kg D−ルシフェリン(キャリパーライフサイエンスイズ(Calliper Life Sciences)、英国)をIP注射し、次に麻酔した(2%イソフランの吸入による)。5分後、依然として麻酔下のマウスを、加温ステージ上(37℃)の遮光チャンバーに配置し、腹側表面上の定めた対象領域からの発光を、Xenogen IVIS Imaging System 100のシステム(アラメダ、カリフォルニア州、米国)でイメージングした。データは、Living Imageソフトウェア(これもゼノゲン(Xenogen)、アラメダ、カリフォルニア州、米国)を用いて分析し、相対ラジアンスで表す(平均ラジアンス 光子/s/cm2/srから算出)。
【0084】
統計分析
平均値の標準誤差(SEM)を用いて各実験グループ間の整合性を評価した。ボンフェローニポストテストと共に一元配置ANOVAを用いてグループ間の差を評価した。両側t検定を用いて2つのグループを比較した。
【0085】
結果
未修飾ポリスチレン粒子の非プロフェッショナル貪食細胞による取り込み
発明者らは、ニューロンがマイクロ粒子およびデブリをインビトロおよびインビボで取り込む能力を有することをこれまでに実証してきた(Bowen et al.,2007)。図(1および2)は、ポリスチレン粒子の場合の一次感覚ニューロン培養物におけるこのことの例を示しており、また、別の薬物送達システム;高分子電解質カプセルについてのこれまでに未発表のデータも含まれている。発明者らはまた、フローサイトメトリー実験から得られた、Daoyヒト髄芽腫細胞株(図3)、ND7/23感覚ニューロン細胞株(図4)、および一次皮質ニューロン(図5)を含むその他の細胞による未修飾合成粒子の取り込みも示す。
【0086】
未修飾ポリスチレン粒子の毒性研究
この研究では、未修飾1μmポリスチレン(PS)マイクロスフェアを添加することによる、種々の細胞型の生存能に対する影響について調べた。マイクロスフェアは濃度を上昇させながら添加し、24時間後、視野あたりのDRGニューロン、その他の細胞の数を定量した(図6)。このモデルについての重要な観察結果は、極めて高い濃度であってもマイクロスフェアの存在下にて細胞数の減少が見られなかったということである。このことは、これらの培養物における粒子の取り込みが、有意な毒性をまったくもたらさないことを示唆している。このことはまた、透過型および走査型電子顕微鏡観察による詳細な超微細構造の研究を含む独立した反復実験でも確認された(図7)。Daoyヒト髄芽腫細胞株(図8)およびND7/23感覚ニューロン細胞株(図9)を用いたフローサイトメトリー実験では、7−AAD細胞死アッセイにより細胞生存能への効果について調べた。
【0087】
FasFc修飾ポリスチレン粒子を用いた取り込みの研究
FasFc融合タンパク質による粒子の表面修飾により、特定の種類の神経および癌細胞による粒子の取り込みが大きく増加する結果となった。図10は、この増加を、後根神経節一次ニューロンについて、コントロールおよび他のリガンドによる修飾と比較して示している。これらの結果は、本発明で達成されるニューロンによる粒子の取り込みの改善を強く示すものである。透過型電子顕微鏡を用いた研究により(図11)、後根神経節一次ニューロンでのこれらの結果が確認された。Daoyヒト髄芽腫細胞株(図12)およびND7/23感覚ニューロン細胞株(図13)でのさらなる実例から、FasFc修飾粒子を用いることによって、特定の細胞型による取り込みを増加させることができることが示された。さらに、Daoyヒト髄芽腫細胞のマウス皮質ニューロンとの共培養において、皮質ニューロンと比較したDaoy細胞による粒子の選択的な取り込みが見られ、このことは、脳腫瘍の治療での有用性を示している(図14)。
【0088】
FasFc修飾PLGA粒子を用いた細胞質内薬物送達
乳酸−グリコール酸共重合体(PLGA)生分解性粒子を、確立された二重エマルジョン法を用いて合成し、またはNanomi BV(オランダ)からその商標であるマイクロシーブ(商標)技術(www.nanomi.com)を用いて粒子を合成した。合成の過程で、小分子、ペプチド、タンパク質、および核酸を含む種々の薬理学的活性剤またはマーカーを粒子に組み込むことができる。発明者らは、エチジウムホモダイマー核酸染色剤、ならびに抗癌薬ドキソルビシンおよびパクリタキセルを組み込んだマイクロ粒子を用いて、本発明を例証した。発明者らは、FasFc修飾したエチジウムホモダイマー充填PLGAが、取り込み、およびそれに続く薬剤の送達を高め(図15H)、粒子の摂取のないコントロール細胞では細胞質核酸の染色が見られないこと(図15D)を示した。未修飾コントロール粒子をコントロールに添加した場合、取り込みおよび薬剤送達の例は見られなかった。同様に、発明者らは、ドキソルビシン充填PLGA粒子をND7/23細胞に添加したところ、FACSによる細胞分離後、ドキソルビシン充填粒子を摂取した細胞の増殖が阻害された(図16)。パクリタキセル充填粒子をDaoyヒト髄芽腫細胞に添加した場合、未充填プラセボ粒子と比較して、パクリタキセル充填粒子では、薬物単独と同様の機能的効果が第1日および3日に見られた(図17)。これらの結果は、本発明の薬物送達用途での有用性を示している。
【0089】
FasFc(CD95−Fc)修飾によるパクリタキセル充填マイクロ粒子のインビボでの効力向上
より臨床上関連のあるシナリオへ進み、発明者らは、同所性卵巣癌モデルを用い、腹腔内注射によって、コンパートメント空間内のCD95L発現IGROV1−ルシフェラーゼ細胞をその他の競合細胞型(例:マクロファージ)の存在下にて標的とした(図18C、D)。生体イメージングより、臨床標準治療であるTaxol(Cremophor ELに溶解したパクリタキセル)の等用量と比較した+CD95−Fc PLGAパクリタキセル処理のグループにおいて、第4週までの腫瘍バイオルミネセンスの>65倍の減少が示された(図18C)。未処理(−CD95−Fc)PLGAパクリタキセルは、Taxolと同等であった。このモデルでは、いずれのプラセボも(+CD95−Fcおよび−CD95−Fc)効果がなかった。+CD95−Fc PLGAパクリタキセル処理における著しい腫瘍減少効果は、処理中断後も持続された(図18C)。このグループのマウスはすべて第48日まで生存し、第62日の終了時には、80%の動物が無症状のままであった(データ示さず)。これまでの研究は、高用量にて組成変更されたパクリタキセルを用いることによる腫瘍増殖阻害の可能性を明らかにした。また、死亡リスクの25%の低減が報告されたことから、臨床状況において、静脈内治療から抗卵巣癌薬の腹腔内送達への転換も行われる。しかし、本明細書で報告されるように、通常の用量で腫瘍量の減少が見られることは稀である。データは、CD95修飾薬物充填マイクロ粒子が、卵巣癌における標的化細胞内薬物送達を向上させる重要な役割を有することを強く支持している。これは、進行期の卵巣癌での治療は成功させることが困難であることから、臨床上必要とされている重要な領域である。
【0090】
参考文献
Abrahams VM,Straszewski SL,Kamsteeg M,Hanczaruk B,Schwartz PE,Rutherford TJ and Mor G.Epithelial ovarian cancer cells secrete functional Fas ligand.Cancer Res 2003;63:5573−5581
Ackery A,Robins S,and Fehlings MG.Inhibition of Fas−mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury.J Neurotrauma 2006;23:604−616
Benita,et al.,J.Pharm.Sci.73,1721−1724(1984)
Bowen S,Ateh DD,Deinhardt K,Bird MM,Price KM,Baker CS,Robson JC,Swash M,Shamsuddin W,Kawar S,El−Tawil T,Roos J,Hoyle A,Nickols CD,Knowles CH,Pullen AH,Luthert PJ,Weller RO,Hafezparast M,Franklin RJM,Revesz T,King RHM,Berninghausen O,Fisher EMC,Schiavo G,Martin JE.The phagocytic capacity of neurons.Eur J of Neurosci 2007;25:2947−2955
Brandhonneur N,Chevanne F,Vie V,Frisch B,Primault R,Le Potier MF,Le Corre P.Specific and non−specific phagocytosis of ligand−grafted PLGA microspheres by macrophages.Eur J Pharm Sci 2009;36:474−85
Choia C and Benveniste EN.Fas ligand/Fas system in the brain:regulator of immune and apoptotic responses.Brain Res Rev.2004;44:65−81
Esselens C,Oorschot V,Baert V,Raemaekers T,Spittaels K,Serneels L,Zheng H,Saftig P,De Strooper B,Klumperman J and Annaert W.Presenilin 1 mediates the turnover of telencephalin in hippocampal neurones via autophagic degradative pathway.J Cell biol 2004;166:1041−1054
Ethell DW,Kinloch R and Green DR.Metalloproteinase shedding of Fas ligand regulates beta−amyloid neurotoxicity.Curr Biol 2002;12:1595−1600
Faraji AH and Wipf P.Nanoparticles in cellular drug delivery.Bioorganic Med Chem 2009;17:2950−2962
Farokhzad OC,Cheng J,Teply BA,Sherifi I,Jon S,Kantoff PW,Richie JP and Langer R.Targeted nanoparticle−aptamer bioconjugates for cancer chemotherapy in vivo.PNAS 2006;103:6315−6320
Ferguson TA and Griffith TS.A vision of cell death:Fas ligand and immune privilege 10 years later.Immunol Rev.2006;213:228−238
Flugel A,Schwaiger FW,Neumann H,Medana I,Willem M,Wekerle H,Kreutzberg GW and Graeber,MB.Neuronal FasL induces cell death of encephalitogenic T lymphocytes.Brain Pathol 2000;10:353−364.
Giteau A,Venier−Julienne MC,Aubert−Pouessel A,Benoit JP.How to achieve sustained and complete protein release from PLGA−based microparticles? Int J Pharm 2008;350:14−26
Gratas C,Tohma Y,Van Meir EG,Kleinz M,Tenans M,Ishii N,Tachibana O,Kleihues P and Ohgaki H.Fas ligand expression in glioblastoma cell lines and primary astrocytic brain tumors.Brain Pathol 1997;7:863−869
Green DR and Ferguson TA.The role of Fas ligand in immune privilege.Nat Rev Mol Cell Biol.2001;2:917−24
Grimm D.Small silencing RNAs:state−of−the−art.Adv Drug Deliv Rev 2009;61:672−703
Gu F,Langer R and Farokhzad OC.Formulation/Preparation of functionalized nanoparticles for in vivo targeted drug delivery.Methods Mol Biol 2009;544:589−98
Huwyler J,Drewe J and Krahenbuhl S.Tumor targeting using liposomal antineoplastic drugs.Int J Nanomedicine 2008;3:21−9
Landau AM,Luk KC,Jones ML,Siegrist−Johnstone R,Young YK,Kouassi E,Rymar VV,Dagher A,Sadikot AF and Desbarats J.Defective Fas expression exacerbates neurotoxicity in a model of Parkinson’s disease.J Exp Med 2005;202:575−581
Lettau M,Paulsen M,Kabelitz D,Janssen O.Storage,expression and function of Fas ligand,the key death factor of immune cells.Curr Med Chem.2008;15:1684−1696
Mello CC and Conte Jr D.Revealing the world of RNA interference.Nature 2004;431:338−342
Meng Y,Graves L,Do T−V,So J,Fishman DA.Upregulation of FasL by LPA on ovarian cancer cell surface leads to apoptosis of activated lymphocytes.Gynecologic Oncology 2004;95:488−495
Mathiowitz and Langer,J.Controlled Release 5,13−22(1987)
Mathiowitz,et al.,Reactive Polymers 6,275−283(1987)
Mathiowitz,et al.,J.Appl.Polymer Sci.35,755−774(1988)
Mathiowitz,et al.,Scanning Microscopy 4,329−340(1990)
Mathiowitz,et al.,J.Appl.Polymer Sci.45,125−134(1992)
Munoz Javier A,del Pino P,Bedard MF,Ho D,Skirtach AG,Sukhorukov GB,Plank C and Parak WJ.Photoactivated release of cargo from the cavity of polyelectrolyte capsules to the cytosol of cells.Langmuir 2008;24:12517−12520
Nagata S and Suda T.Fas and Fas ligand:Ipr and gld mutations.Immunol Today.1995;16:39−43
Nagata S.Apoptosis by Death Factor.Cell 1997;88:355−365
Niehans GA,Brunner T,Frizelle SP,Liston JC,Salerno CT,Knapp DJ,Green DR,and Kratzke RA.Human Lung Carcinomas Express Fas Ligand.Cancer Res 1997;57:1007−1012
O’Connell J,Bennett MW,O’SULLIVAN GC,Roche D,Kelly J,J.Collins K and Fergus S.Fas ligand expression in primary colon adenocarcinomas:evidence that the Fas counterattack is a prevalent mechanism of immune evasion in human colon cancer.J Pathol 1998;186:240−246
O’Connell J,Houston A,Bennett MW,O’Sullivan GC and Shanahan F.Immune privilege or inflammation? Insights into the Fas ligand enigma.Nature Med.2001;7:271−274(Commentary)
Patil Y and Panyam J.Polymeric nanoparticles for siRNA delivery and gene silencing.Int J Pharm 2009;367:195−203
Rabinovitch M.Professional and non professional phagocytes:an introduction.Trends in cell biology 1995;5:85−87
Ryan AE,Shanahan F,O’Connell J and Houston AM.Addressing the“Fas counterattack”controversy: blocking fas ligand expression suppresses tumor immune evasion of colon cancer in vivo.Cancer Res.2005;65:9817−9823
Sawyer TK.AILERON therapeutics.Chem Biol Drug Des(R&D Spotlight)2009;73:3−6
Sinha VR,Bansal K,Kaushik R,Kumria R and Trehan A.Poly−ε−caprolactone microspheres and nanospheres:an overview.Int J Pharm 2004;278:1−23
Sukhorukov GB,Rogach AL,Garstka M,Springer S,Parak WJ,MuCoz−Javier A,Kreft O,Skirtach AG,Susha AS,Racane Y,Palankar R and Winterhalter M.Multifunctionalized polymer microcapsules:Novel tools for biological and pharmacological applications. Small 2007;3:944−955
Watanabe−Fukunaga et al.,The cDNA structure,expression,and chromosomal assignment of the mouse Fas antigen.Journal of Immunology 1992;148:1274−1279
Walter E,Dreher D,Kok M,Thiele L,Kiama SG,Gehr P and Merkle HP.Hydrophilic poly(DL−lactide−co−glycolide)microspheres for the delivery of DNA to human−derived macrophages and dendritic cells.J Control Release 2001;76:149−168
Weller M,Schuster M,Pietsch T,and Schabet M.CD95 ligand−induced apoptosis of human medulloblastoma cells.Cancer Lett 1998;128:121−126
Zuliani C,Kleber S,Klussmann S,Wenger T,Kenzelmann M,Schreglmann N,Martinez A,del Rio JA,Soriano E,Vodrazka P,Kuner R,Groene H−J,Herr I,Krammer PH and Martin−Villalba A.Cell Death Diff.2006;13:31−40
【図1A】
【図1B】
【図1C】
【図1D】
【特許請求の範囲】
【請求項1】
薬理学的活性剤またはマーカーを細胞に送達するための送達媒体であって、Fasリガンドに特異的であるリガンド結合部分、および前記薬理学的活性剤またはマーカーのためのキャリアを含む、送達媒体。
【請求項2】
前記キャリアが、マイクロ粒子、ナノ粒子、マイクロカプセル、マイクロスフェア、ミセル、またはリポソームである、請求項1に記載の送達媒体。
【請求項3】
前記キャリアが、マイクロ粒子である、請求項2に記載の送達媒体。
【請求項4】
前記マイクロ粒子が、0.01μm、0.05μm、0.1μm、0.2μm、0.5μm、1μm、2μm、5μm、または10μmまでの平均径を有するか、または0.1μmから10μm、0.2μmから5μm、0.3μmから2μm、0.4μmから1.5μm、もしくは0.5μmから1μmの平均径を有する、請求項3に記載の送達媒体。
【請求項5】
Fasリガンドに特異的である前記媒体の前記リガンド結合部分が:
(a)Fas受容体もしくはその誘導体;
(b)完全長Fasタンパク質もしくはその断片;
(c)Fasタンパク質の細胞外ドメインもしくはその断片;または、
(d)Fasタンパク質のリガンド結合ドメイン
を含むか、またはそれから成る、請求項1から4のいずれか一項に記載の送達媒体。
【請求項6】
Fasリガンドに特異的である前記媒体の前記リガンド結合部分が:
(a)ペプチド、タンパク質、アプタマー、抗体、抗体断片、融合タンパク質、またはキメラタンパク質;
(b)免疫グロブリンの結晶化可能断片領域(Fc領域)と融合してキメラ融合タンパク質を形成したFasタンパク質、またはその断片、
である、請求項1から5のいずれか一項に記載の送達媒体。
【請求項7】
Fasリガンドに特異的な複数のリガンド結合部分を含む、請求項1から6のいずれか一項に記載の送達媒体。
【請求項8】
前記リガンド結合部分またはその各々が、ヒトまたはマウス由来である、請求項1から7のいずれか一項に記載の送達媒体。
【請求項9】
前記リガンド結合部分またはその各々が、表面吸収、吸着、化学結合、またはマトリックス内組み込みによって前記キャリアとカップリングしている、請求項1から8のいずれか一項に記載の送達媒体。
【請求項10】
前記リガンド結合部分またはその各々が、前記キャリアと共有結合または非共有結合によって会合している、請求項1から9のいずれか一項に記載の送達媒体。
【請求項11】
前記リガンド結合部分またはその各々が、連結分子を介して前記キャリアとカップリングしている、請求項1から10のいずれか一項に記載の送達媒体。
【請求項12】
前記連結分子が、免疫グロブリンの結晶化可能断片領域(Fc)である、請求項12に記載の送達媒体。
【請求項13】
前記薬理学的活性剤、マーカー、または薬物が、好ましくは、Fasリガンドと特異的に結合する能力を持たない剤である、請求項1から12のいずれか一項に記載の送達媒体。
【請求項14】
前記薬理学的活性剤またはマーカーが、好ましくは標的細胞にて、もしくはその内部で放出されるように、前記キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている、請求項1から13のいずれか一項に記載の送達媒体。
【請求項15】
前記送達媒体が、薬理学的物質を含有するマイクロ粒子であり、前記マイクロ粒子は、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と、共有結合または非共有結合によって会合している、請求項1から14のいずれか一項に記載の送達媒体。
【請求項16】
前記薬理学的活性剤が、タンパク質、ペプチド、ポリペプチド、ポリヌクレオチド、ポリサッカリド、脂質、小分子薬物、またはその他のいずれの生物学的活性物質である、請求項1から15のいずれか一項に記載の送達媒体。
【請求項17】
前記薬理学的活性剤が、細胞内で活性であるか、または標的細胞内部の成分に対して特異的である、請求項1から16のいずれか一項に記載の送達媒体。
【請求項18】
前記薬理学的活性剤が、細胞毒性剤、または細胞分裂阻害剤であり、好ましくは、抗癌剤、特にドキソルビシンまたはパクリタキセルである、請求項1から17のいずれか一項に記載の送達媒体。
【請求項19】
前記マーカーが、蛍光マーカー、放射性核種、または造影剤である、請求項1から14のいずれか一項に記載の送達媒体。
【請求項20】
前記薬理学的活性剤またはマーカーが、前記キャリアもしくはマイクロ粒子によって含有されるかまたはカプセル化されている、請求項1から19のいずれか一項に記載の送達媒体。
【請求項21】
前記薬理学的活性剤もしくはマーカーが、化学結合によって前記キャリアもしくはマイクロ粒子と結合、またはその内部に存在しているか、または、前記キャリアもしくはマイクロ粒子を形成する物質のマトリックス内に物理的に組み込まれている、請求項1から20のいずれか一項に記載の送達媒体。
【請求項22】
医療用である、請求項1から21のいずれか一項に記載の送達媒体。
【請求項23】
治療用である、請求項1から22のいずれか一項に記載の送達媒体。
【請求項24】
前記薬理学的活性剤またはマーカーが、前記キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている、請求項22または23に記載の送達媒体。
【請求項25】
新生物もしくは神経障害に関連する疾患または医学的病状の治療のためのものである、請求項22から24のいずれか一項に記載の送達媒体。
【請求項26】
疾患または医学的病状の治療方法であって、請求項1から25のいずれか一項に記載の送達媒体を対象へ投与することを含み、ここで、前記送達媒体は、前記薬理学的活性剤の効果量を含む、方法。
【請求項27】
前記疾患または病状が、脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、またはFasリガンドが腫瘍細胞中で発現される癌の種類もしくは腫瘍である、請求項25に記載の送達媒体、または請求項26に記載の方法。
【請求項28】
前記疾患または病状が、神経疾患であり、場合に応じて運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、または末梢神経もしくは脊髄損傷であってよい、請求項25に記載の送達媒体、または請求項26に記載の方法。
【請求項29】
Fasリガンド発現細胞の検出を含み、ここで、マーカーが前記キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている診断方法に用いるためのものである、請求項1から22のいずれか一項に記載の送達媒体。
【請求項30】
請求項1から22のいずれか一項に記載の送達媒体を対象へ投与することによってFasリガンド発現細胞を検出することを含み、ここで、マーカーが前記キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている、診断の方法。
【請求項31】
前記診断方法、または診断の方法が、新生物または神経障害を診断する方法である、請求項29に記載の送達媒体、または請求項30に記載の方法。
【請求項32】
請求項1から25、27から29、および31のいずれか一項に記載の送達媒体、ならびに1もしくは2つ以上の生理学的にまたは薬理学的に許容されるキャリア、賦形剤、または安定化剤を含む医薬組成物。
【請求項33】
請求項1から25、27から29、および31のいずれか一項に記載の送達媒体を作製するための方法であって、薬理学的活性剤、マーカー、もしくは薬物を含有するかまたはカプセル化するマイクロ粒子を形成する工程、およびFasリガンドに特異的であるリガンド結合部分を前記マイクロ粒子に結合させる工程を含む、方法。
【請求項34】
薬理学的活性剤を含有するマイクロ粒子であって、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と共有結合または非共有結合によって会合している、マイクロ粒子。
【請求項35】
前記マイクロ粒子が、0.01μm、0.05μm、0.1μm、0.2μm、0.5μm、1μm、2μm、5μm、または10μmまでの平均径を有する、請求項34に記載のマイクロ粒子。
【請求項36】
前記キャリアまたはマイクロ粒子が、PLGA(乳酸−グリコール酸共重合体マトリックス)、PLA(ポリ乳酸)、PCL(ポリ−ε−カプロラクトン)、もしくはPHB(ポリヒドロキシブチレート)を含むか、または、キトサン生分解性マイクロスフェア、ケイ素系粒子、高分子電解質カプセル、デンドリマー、もしくはリポソームである、請求項34もしくは請求項35に記載のマイクロ粒子、または請求項1から25、27から29、および31のいずれか一項に記載の送達媒体。
【請求項37】
前記キャリアまたはマイクロ粒子が、ポリアルキレン、ポリカーボネート、ポリ(ジオキサノン)、ポリ酸無水物、ポリヒドロキシ酸、ポリフマレート、ポリカプロラクトン、ポリアミド、ポリアセタール、ポリエーテル、ポリエステル、ポリ(オルソエステル)、ポリヒドロキシブチレート、ポリビニルアルコール、ポリウレタン、ポリホスファゼン、ポリアクリレート、ポリメタクリレート、ポリシアノアクリレート、ポリウレア、ポリスチレン、ポリアミン、ポリ(アリレート)、ポリカーボネート、ポリ(プロピレンフマレート)、ポリヒドロキシアルカノエート、ポリケタール、ポリエステルアミド、ポリヒドロキシバリレート、ポリオルソカーボネート、ポリ(ビニルピロリドン)、ポリアルキレンオキサレート、ポリアルキレンスクシネート、ポリ(リンゴ酸)、ポリ(メチルビニルエーテル)、およびポリ(無水マレイン酸)から成る群より選択されるポリマーを含む、請求項34もしくは請求項35に記載のマイクロ粒子、または請求項1から25、27から29、および31のいずれか一項に記載の送達媒体。
【請求項38】
前記キャリアまたはマイクロ粒子が、非ポリマーである、請求項34から37のいずれか一項に記載のマイクロ粒子、または請求項1から25、27から29、および31のいずれか一項に記載の送達媒体。
【請求項39】
前記マイクロ粒子が、表面吸収、吸着、化学結合、またはマトリックス内組み込みによってFas、その誘導体、またはFasリガンドに対する特異的結合分子と共有結合または非共有結合によって会合している、請求項34から38のいずれか一項に記載のマイクロ粒子。
【請求項40】
前記Fas誘導体が、完全長Fas、FasFc、またはFasの融合タンパク質である、請求項34から39のいずれか一項に記載のマイクロ粒子。
【請求項41】
前記Fasリガンドに対する特異的結合分子が、ペプチド、タンパク質、アプタマー、抗体、または抗体断片である、請求項40に記載のマイクロ粒子。
【請求項42】
医療用である、請求項34から41のいずれか一項に記載のマイクロ粒子。
【請求項43】
脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、ならびにFasリガンドが腫瘍細胞中で発現される癌の種類および腫瘍から成る群より選択される疾患または病状の治療に用いられる、請求項34から42のいずれか一項に記載のマイクロ粒子。
【請求項44】
神経疾患、運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、ならびに末梢神経および脊髄損傷から成る群より選択される疾患または病状の治療に用いられる、請求項34から43のいずれか一項に記載のマイクロ粒子。
【請求項45】
請求項34から41のいずれか一項に記載のマイクロ粒子、または請求項1から25、27から29、および31のいずれか一項に記載の送達媒体を細胞内へ送達するための方法であって、前記送達媒体またはマイクロ粒子を前記細胞へ投与することを含む、方法。
【請求項46】
疾患または病状を治療する方法であって、請求項34から41のいずれか一項に記載のマイクロ粒子を、前記疾患または病状の治療に効果的である量で患者に投与することを含む、方法。
【請求項47】
前記疾患または病状が、脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、ならびにFasリガンドが腫瘍細胞中で発現される癌の種類および腫瘍から成る群より選択される、請求項46に記載の方法。
【請求項48】
前記疾患または病状が、神経疾患、運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、ならびに末梢神経および脊髄損傷から成る群より選択される、請求項46に記載の方法。
【請求項49】
薬理学的活性物質を含有するマイクロ粒子の修飾におけるFas、その誘導体、またはFasリガンドに対する特異的結合分子の使用であって、前記修飾は、表面吸収、吸着、化学結合により、またはFasの前記粒子へのマトリックス内組み込みにより、Fas、その誘導体、もしくはFasリガンドに対する特異的結合分子とマイクロ粒子との共有結合による、または非共有結合による会合をもたらすものである、使用。
【請求項50】
薬理学的活性物質を含有するマイクロ粒子の作製のための方法であって、ここで、前記マイクロ粒子は、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と共有結合、または非共有結合によって会合するものであり、前記マイクロ粒子を、表面吸収、吸着、化学結合によって、またはマトリックス内組み込みによって、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と会合させることを含む、方法。
【請求項51】
前記Fas誘導体が、完全長Fas、FasFc、またはFasの融合タンパク質である、請求項49に記載の使用または請求項50に記載の方法。
【請求項52】
前記Fasリガンドに対する特異的結合分子が、ペプチド、タンパク質、アプタマー、抗体、または抗体断片である、請求項49に記載の使用または請求項50に記載の方法。
【請求項53】
請求項34から41のいずれか一項に記載のマイクロ粒子、および適切な賦形剤を含む、医薬組成物。
【請求項54】
薬物、およびFas、その誘導体、またはFasリガンドに対する特異的結合分子を含む、薬物接合体。
【請求項55】
前記Fas誘導体が、完全長Fas、FasFc、またはFasの融合タンパク質である、請求項54に記載の薬物接合体。
【請求項56】
前記Fasリガンドに対する特異的結合分子が、ペプチド、タンパク質、アプタマー、抗体、または抗体断片である、請求項54に記載の薬物接合体。
【請求項57】
医療用である、請求項54から56のいずれか一項に記載の薬物接合体。
【請求項1】
薬理学的活性剤またはマーカーを細胞に送達するための送達媒体であって、Fasリガンドに特異的であるリガンド結合部分、および前記薬理学的活性剤またはマーカーのためのキャリアを含む、送達媒体。
【請求項2】
前記キャリアが、マイクロ粒子、ナノ粒子、マイクロカプセル、マイクロスフェア、ミセル、またはリポソームである、請求項1に記載の送達媒体。
【請求項3】
前記キャリアが、マイクロ粒子である、請求項2に記載の送達媒体。
【請求項4】
前記マイクロ粒子が、0.01μm、0.05μm、0.1μm、0.2μm、0.5μm、1μm、2μm、5μm、または10μmまでの平均径を有するか、または0.1μmから10μm、0.2μmから5μm、0.3μmから2μm、0.4μmから1.5μm、もしくは0.5μmから1μmの平均径を有する、請求項3に記載の送達媒体。
【請求項5】
Fasリガンドに特異的である前記媒体の前記リガンド結合部分が:
(a)Fas受容体もしくはその誘導体;
(b)完全長Fasタンパク質もしくはその断片;
(c)Fasタンパク質の細胞外ドメインもしくはその断片;または、
(d)Fasタンパク質のリガンド結合ドメイン
を含むか、またはそれから成る、請求項1から4のいずれか一項に記載の送達媒体。
【請求項6】
Fasリガンドに特異的である前記媒体の前記リガンド結合部分が:
(a)ペプチド、タンパク質、アプタマー、抗体、抗体断片、融合タンパク質、またはキメラタンパク質;
(b)免疫グロブリンの結晶化可能断片領域(Fc領域)と融合してキメラ融合タンパク質を形成したFasタンパク質、またはその断片、
である、請求項1から5のいずれか一項に記載の送達媒体。
【請求項7】
Fasリガンドに特異的な複数のリガンド結合部分を含む、請求項1から6のいずれか一項に記載の送達媒体。
【請求項8】
前記リガンド結合部分またはその各々が、ヒトまたはマウス由来である、請求項1から7のいずれか一項に記載の送達媒体。
【請求項9】
前記リガンド結合部分またはその各々が、表面吸収、吸着、化学結合、またはマトリックス内組み込みによって前記キャリアとカップリングしている、請求項1から8のいずれか一項に記載の送達媒体。
【請求項10】
前記リガンド結合部分またはその各々が、前記キャリアと共有結合または非共有結合によって会合している、請求項1から9のいずれか一項に記載の送達媒体。
【請求項11】
前記リガンド結合部分またはその各々が、連結分子を介して前記キャリアとカップリングしている、請求項1から10のいずれか一項に記載の送達媒体。
【請求項12】
前記連結分子が、免疫グロブリンの結晶化可能断片領域(Fc)である、請求項12に記載の送達媒体。
【請求項13】
前記薬理学的活性剤、マーカー、または薬物が、好ましくは、Fasリガンドと特異的に結合する能力を持たない剤である、請求項1から12のいずれか一項に記載の送達媒体。
【請求項14】
前記薬理学的活性剤またはマーカーが、好ましくは標的細胞にて、もしくはその内部で放出されるように、前記キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている、請求項1から13のいずれか一項に記載の送達媒体。
【請求項15】
前記送達媒体が、薬理学的物質を含有するマイクロ粒子であり、前記マイクロ粒子は、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と、共有結合または非共有結合によって会合している、請求項1から14のいずれか一項に記載の送達媒体。
【請求項16】
前記薬理学的活性剤が、タンパク質、ペプチド、ポリペプチド、ポリヌクレオチド、ポリサッカリド、脂質、小分子薬物、またはその他のいずれの生物学的活性物質である、請求項1から15のいずれか一項に記載の送達媒体。
【請求項17】
前記薬理学的活性剤が、細胞内で活性であるか、または標的細胞内部の成分に対して特異的である、請求項1から16のいずれか一項に記載の送達媒体。
【請求項18】
前記薬理学的活性剤が、細胞毒性剤、または細胞分裂阻害剤であり、好ましくは、抗癌剤、特にドキソルビシンまたはパクリタキセルである、請求項1から17のいずれか一項に記載の送達媒体。
【請求項19】
前記マーカーが、蛍光マーカー、放射性核種、または造影剤である、請求項1から14のいずれか一項に記載の送達媒体。
【請求項20】
前記薬理学的活性剤またはマーカーが、前記キャリアもしくはマイクロ粒子によって含有されるかまたはカプセル化されている、請求項1から19のいずれか一項に記載の送達媒体。
【請求項21】
前記薬理学的活性剤もしくはマーカーが、化学結合によって前記キャリアもしくはマイクロ粒子と結合、またはその内部に存在しているか、または、前記キャリアもしくはマイクロ粒子を形成する物質のマトリックス内に物理的に組み込まれている、請求項1から20のいずれか一項に記載の送達媒体。
【請求項22】
医療用である、請求項1から21のいずれか一項に記載の送達媒体。
【請求項23】
治療用である、請求項1から22のいずれか一項に記載の送達媒体。
【請求項24】
前記薬理学的活性剤またはマーカーが、前記キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている、請求項22または23に記載の送達媒体。
【請求項25】
新生物もしくは神経障害に関連する疾患または医学的病状の治療のためのものである、請求項22から24のいずれか一項に記載の送達媒体。
【請求項26】
疾患または医学的病状の治療方法であって、請求項1から25のいずれか一項に記載の送達媒体を対象へ投与することを含み、ここで、前記送達媒体は、前記薬理学的活性剤の効果量を含む、方法。
【請求項27】
前記疾患または病状が、脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、またはFasリガンドが腫瘍細胞中で発現される癌の種類もしくは腫瘍である、請求項25に記載の送達媒体、または請求項26に記載の方法。
【請求項28】
前記疾患または病状が、神経疾患であり、場合に応じて運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、または末梢神経もしくは脊髄損傷であってよい、請求項25に記載の送達媒体、または請求項26に記載の方法。
【請求項29】
Fasリガンド発現細胞の検出を含み、ここで、マーカーが前記キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている診断方法に用いるためのものである、請求項1から22のいずれか一項に記載の送達媒体。
【請求項30】
請求項1から22のいずれか一項に記載の送達媒体を対象へ投与することによってFasリガンド発現細胞を検出することを含み、ここで、マーカーが前記キャリアと結合しているか、またはそれに含有されているか、もしくはカプセル化されている、診断の方法。
【請求項31】
前記診断方法、または診断の方法が、新生物または神経障害を診断する方法である、請求項29に記載の送達媒体、または請求項30に記載の方法。
【請求項32】
請求項1から25、27から29、および31のいずれか一項に記載の送達媒体、ならびに1もしくは2つ以上の生理学的にまたは薬理学的に許容されるキャリア、賦形剤、または安定化剤を含む医薬組成物。
【請求項33】
請求項1から25、27から29、および31のいずれか一項に記載の送達媒体を作製するための方法であって、薬理学的活性剤、マーカー、もしくは薬物を含有するかまたはカプセル化するマイクロ粒子を形成する工程、およびFasリガンドに特異的であるリガンド結合部分を前記マイクロ粒子に結合させる工程を含む、方法。
【請求項34】
薬理学的活性剤を含有するマイクロ粒子であって、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と共有結合または非共有結合によって会合している、マイクロ粒子。
【請求項35】
前記マイクロ粒子が、0.01μm、0.05μm、0.1μm、0.2μm、0.5μm、1μm、2μm、5μm、または10μmまでの平均径を有する、請求項34に記載のマイクロ粒子。
【請求項36】
前記キャリアまたはマイクロ粒子が、PLGA(乳酸−グリコール酸共重合体マトリックス)、PLA(ポリ乳酸)、PCL(ポリ−ε−カプロラクトン)、もしくはPHB(ポリヒドロキシブチレート)を含むか、または、キトサン生分解性マイクロスフェア、ケイ素系粒子、高分子電解質カプセル、デンドリマー、もしくはリポソームである、請求項34もしくは請求項35に記載のマイクロ粒子、または請求項1から25、27から29、および31のいずれか一項に記載の送達媒体。
【請求項37】
前記キャリアまたはマイクロ粒子が、ポリアルキレン、ポリカーボネート、ポリ(ジオキサノン)、ポリ酸無水物、ポリヒドロキシ酸、ポリフマレート、ポリカプロラクトン、ポリアミド、ポリアセタール、ポリエーテル、ポリエステル、ポリ(オルソエステル)、ポリヒドロキシブチレート、ポリビニルアルコール、ポリウレタン、ポリホスファゼン、ポリアクリレート、ポリメタクリレート、ポリシアノアクリレート、ポリウレア、ポリスチレン、ポリアミン、ポリ(アリレート)、ポリカーボネート、ポリ(プロピレンフマレート)、ポリヒドロキシアルカノエート、ポリケタール、ポリエステルアミド、ポリヒドロキシバリレート、ポリオルソカーボネート、ポリ(ビニルピロリドン)、ポリアルキレンオキサレート、ポリアルキレンスクシネート、ポリ(リンゴ酸)、ポリ(メチルビニルエーテル)、およびポリ(無水マレイン酸)から成る群より選択されるポリマーを含む、請求項34もしくは請求項35に記載のマイクロ粒子、または請求項1から25、27から29、および31のいずれか一項に記載の送達媒体。
【請求項38】
前記キャリアまたはマイクロ粒子が、非ポリマーである、請求項34から37のいずれか一項に記載のマイクロ粒子、または請求項1から25、27から29、および31のいずれか一項に記載の送達媒体。
【請求項39】
前記マイクロ粒子が、表面吸収、吸着、化学結合、またはマトリックス内組み込みによってFas、その誘導体、またはFasリガンドに対する特異的結合分子と共有結合または非共有結合によって会合している、請求項34から38のいずれか一項に記載のマイクロ粒子。
【請求項40】
前記Fas誘導体が、完全長Fas、FasFc、またはFasの融合タンパク質である、請求項34から39のいずれか一項に記載のマイクロ粒子。
【請求項41】
前記Fasリガンドに対する特異的結合分子が、ペプチド、タンパク質、アプタマー、抗体、または抗体断片である、請求項40に記載のマイクロ粒子。
【請求項42】
医療用である、請求項34から41のいずれか一項に記載のマイクロ粒子。
【請求項43】
脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、ならびにFasリガンドが腫瘍細胞中で発現される癌の種類および腫瘍から成る群より選択される疾患または病状の治療に用いられる、請求項34から42のいずれか一項に記載のマイクロ粒子。
【請求項44】
神経疾患、運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、ならびに末梢神経および脊髄損傷から成る群より選択される疾患または病状の治療に用いられる、請求項34から43のいずれか一項に記載のマイクロ粒子。
【請求項45】
請求項34から41のいずれか一項に記載のマイクロ粒子、または請求項1から25、27から29、および31のいずれか一項に記載の送達媒体を細胞内へ送達するための方法であって、前記送達媒体またはマイクロ粒子を前記細胞へ投与することを含む、方法。
【請求項46】
疾患または病状を治療する方法であって、請求項34から41のいずれか一項に記載のマイクロ粒子を、前記疾患または病状の治療に効果的である量で患者に投与することを含む、方法。
【請求項47】
前記疾患または病状が、脳腫瘍、卵巣癌、前立腺癌、乳癌、腹腔内腫瘍、卵巣腫瘍、胃腸腫瘍、結腸癌、肺癌、膵臓癌、ならびにFasリガンドが腫瘍細胞中で発現される癌の種類および腫瘍から成る群より選択される、請求項46に記載の方法。
【請求項48】
前記疾患または病状が、神経疾患、運動ニューロン疾患、アルツハイマー病、パーキンソン病、神経因性疼痛症候群、ならびに末梢神経および脊髄損傷から成る群より選択される、請求項46に記載の方法。
【請求項49】
薬理学的活性物質を含有するマイクロ粒子の修飾におけるFas、その誘導体、またはFasリガンドに対する特異的結合分子の使用であって、前記修飾は、表面吸収、吸着、化学結合により、またはFasの前記粒子へのマトリックス内組み込みにより、Fas、その誘導体、もしくはFasリガンドに対する特異的結合分子とマイクロ粒子との共有結合による、または非共有結合による会合をもたらすものである、使用。
【請求項50】
薬理学的活性物質を含有するマイクロ粒子の作製のための方法であって、ここで、前記マイクロ粒子は、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と共有結合、または非共有結合によって会合するものであり、前記マイクロ粒子を、表面吸収、吸着、化学結合によって、またはマトリックス内組み込みによって、Fas、その誘導体、またはFasリガンドに対する特異的結合分子と会合させることを含む、方法。
【請求項51】
前記Fas誘導体が、完全長Fas、FasFc、またはFasの融合タンパク質である、請求項49に記載の使用または請求項50に記載の方法。
【請求項52】
前記Fasリガンドに対する特異的結合分子が、ペプチド、タンパク質、アプタマー、抗体、または抗体断片である、請求項49に記載の使用または請求項50に記載の方法。
【請求項53】
請求項34から41のいずれか一項に記載のマイクロ粒子、および適切な賦形剤を含む、医薬組成物。
【請求項54】
薬物、およびFas、その誘導体、またはFasリガンドに対する特異的結合分子を含む、薬物接合体。
【請求項55】
前記Fas誘導体が、完全長Fas、FasFc、またはFasの融合タンパク質である、請求項54に記載の薬物接合体。
【請求項56】
前記Fasリガンドに対する特異的結合分子が、ペプチド、タンパク質、アプタマー、抗体、または抗体断片である、請求項54に記載の薬物接合体。
【請求項57】
医療用である、請求項54から56のいずれか一項に記載の薬物接合体。
【図2】

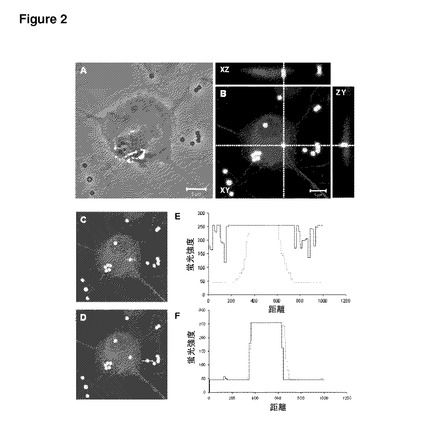
【図3−1】
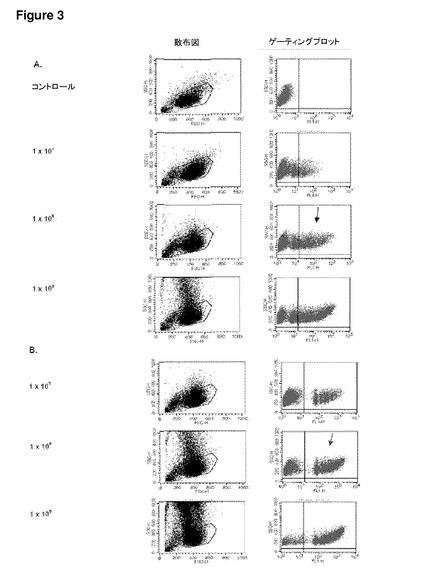
【図3−2】
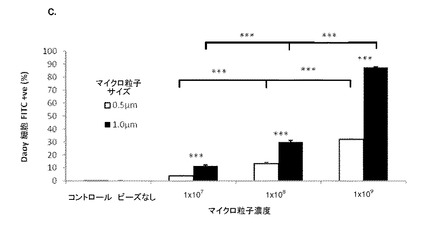
【図4−1】
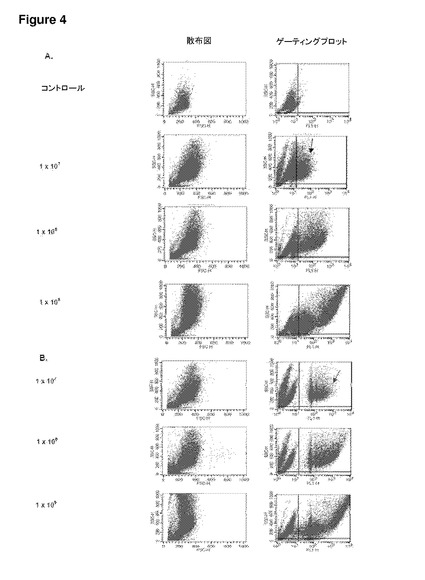
【図4−2】
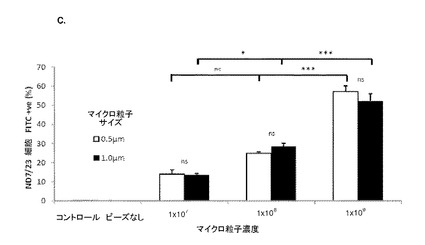
【図5】
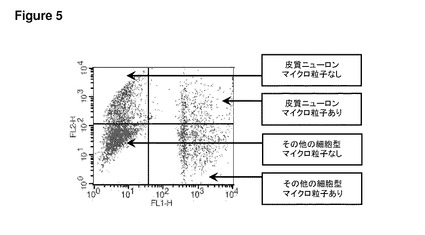
【図6−1】
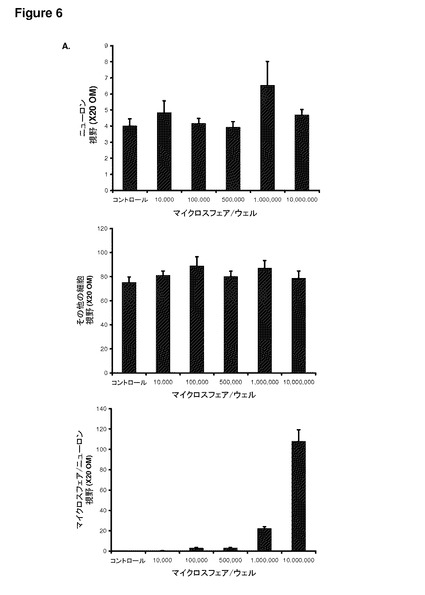
【図6−2】
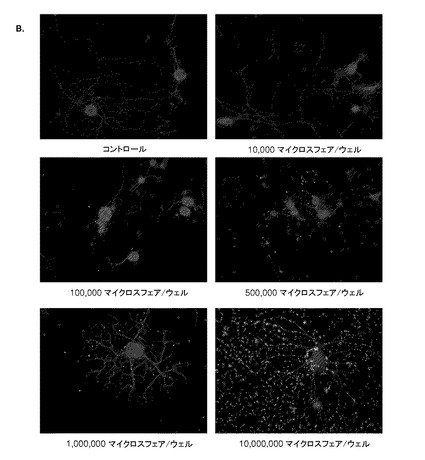
【図7】

【図8】
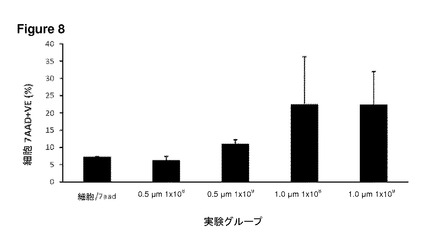
【図9】
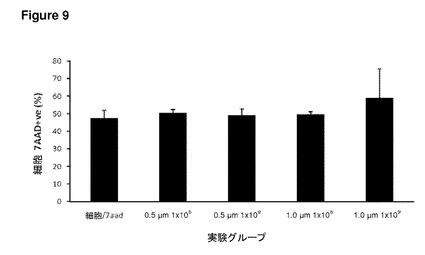
【図10】
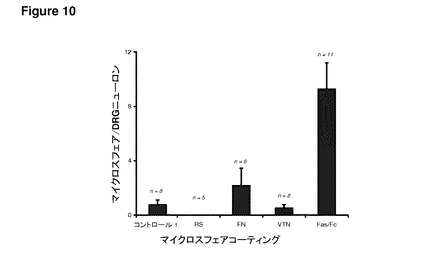
【図11】
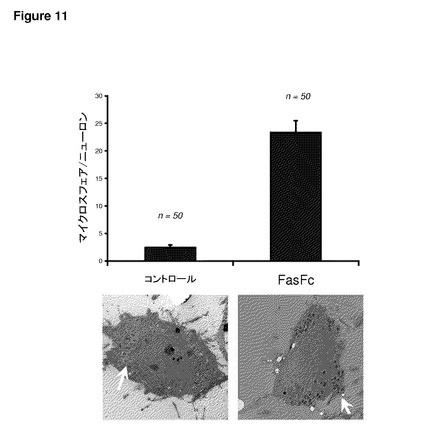
【図12】
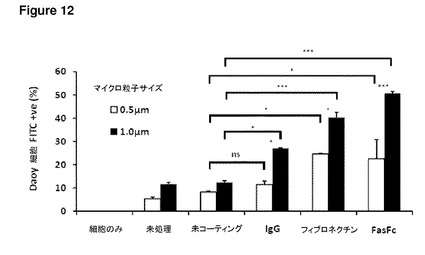
【図13】
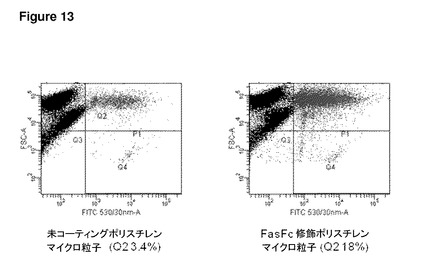
【図14】
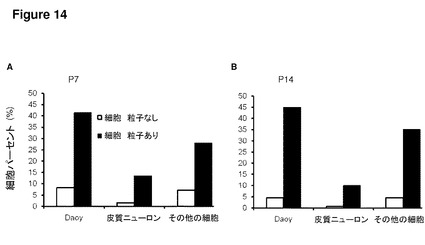
【図15A】

【図15B】

【図15C】

【図15D】

【図15E】
【図15F】

【図15G】
【図15H】

【図16】
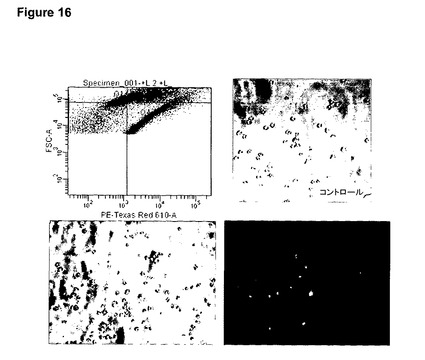
【図17−1】
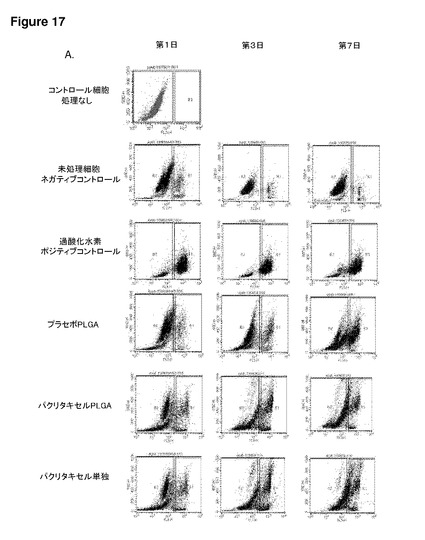
【図17−2】
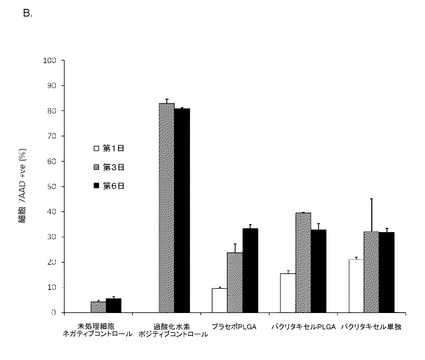
【図18−1】
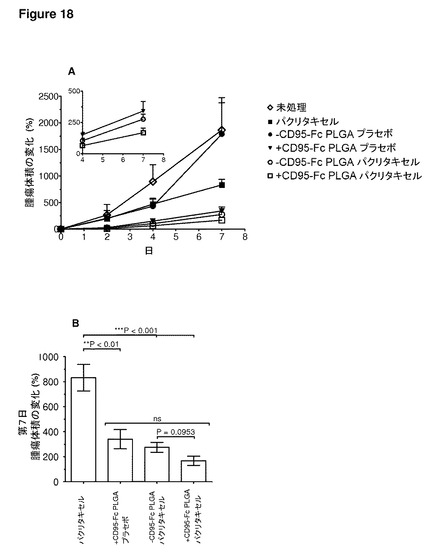
【図18−2】
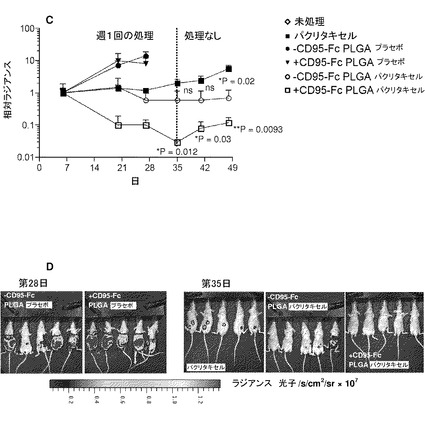
【図3−1】
【図3−2】
【図4−1】
【図4−2】
【図5】
【図6−1】
【図6−2】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15A】
【図15B】
【図15C】
【図15D】
【図15E】
【図15F】
【図15G】
【図15H】
【図16】
【図17−1】
【図17−2】
【図18−1】
【図18−2】
【公表番号】特表2012−533613(P2012−533613A)
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−521104(P2012−521104)
【出願日】平成22年7月21日(2010.7.21)
【国際出願番号】PCT/GB2010/051207
【国際公開番号】WO2011/010156
【国際公開日】平成23年1月27日(2011.1.27)
【出願人】(501008923)クイーン メアリー アンド ウェストフィールド カレッジ (14)
【氏名又は名称原語表記】Queen Mary and Westfield College
【住所又は居所原語表記】Mile End Road,London,U.K.
【Fターム(参考)】
【公表日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願日】平成22年7月21日(2010.7.21)
【国際出願番号】PCT/GB2010/051207
【国際公開番号】WO2011/010156
【国際公開日】平成23年1月27日(2011.1.27)
【出願人】(501008923)クイーン メアリー アンド ウェストフィールド カレッジ (14)
【氏名又は名称原語表記】Queen Mary and Westfield College
【住所又は居所原語表記】Mile End Road,London,U.K.
【Fターム(参考)】
[ Back to top ]