
細胞医薬の製造方法
【課題】疾患に対して有効かつ危険性の低い細胞医薬を製造する方法を提供すること。
【解決手段】人工多能性幹細胞にタンパク質発現ベクターを導入する工程;及び該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程を含む、外来タンパク質を発現する貪食細胞の製造方法。
【解決手段】人工多能性幹細胞にタンパク質発現ベクターを導入する工程;及び該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程を含む、外来タンパク質を発現する貪食細胞の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、人工多能性幹細胞を用いた細胞医薬の製造方法に関する。より詳細には、本発明は、人工多能性幹細胞に遺伝的改変を施した後、生体外での分化誘導により貪食能を有する血球系の細胞(マクロファージ)を製造する方法に関する。本発明の方法により製造される細胞医薬(マクロファージ)は、遺伝子導入により特定の分子に対する抗体等を発現するものであり、標的となる分子を特異的に認識し、標的分子の貪食・分解、あるいは標的分子を発現する細胞を特異的に貪食・傷害する等の機能を発揮する。本発明の細胞医薬は、アルツハイマー病、がん、プリオン病、アミロイドーシス等の疾患の治療において有用であると期待される。
【背景技術】
【0002】
人工多能性幹細胞(誘導多能性幹細胞)(induced pluripotent stem cell; iPS細胞)とは、体細胞を初期化することによって得られる多能性を有する細胞である。人工多能性幹細胞の作製は、京都大学の山中伸弥教授らのグループ、マサチューセッツ工科大学のルドルフ・ヤニッシュ(Rudolf Jaenisch)らのグループ、ウイスコンシン大学のジェームズ・トムソン(James Thomson)らのグループ、ハーバード大学のコンラッド・ホッケドリンガー(Konrad Hochedlinger)らのグループなどを含む複数のグループが成功している。人工多能性幹細胞は、拒絶反応や倫理的問題のない理想的な多能性細胞として大きな期待を集めている。例えば、国際公開WO2007/069666号公報には、Octファミリー遺伝子、Klfファミリー遺伝子、及びMycファミリー遺伝子の遺伝子産物を含む体細胞の核初期化因子、並びにOctファミリー遺伝子、Klfファミリー遺伝子、Soxファミリー遺伝子及びMycファミリー遺伝子の遺伝子産物を含む体細胞の核初期化因子が記載されており、さらに体細胞に上記核初期化因子を接触させる工程を含む、体細胞の核初期化により誘導多能性幹細胞を製造する方法が記載されている。
【0003】
人工多能性幹細胞は、健康人・患者を問わず、ヒトの皮膚線維芽細胞や血液細胞などの体細胞を材料として作製することが可能である。人工多能性幹細胞は、胚性幹細胞(ES細胞)と同様に、あらゆる細胞に分化する潜在的能力を秘めた細胞であり、かつ、適切な条件の下で培養することにより無尽蔵に増殖させることが可能な細胞である。さらに、電気穿孔法などの方法を用いることにより、容易に遺伝子導入を施すことが可能な細胞である。一方で、人工多能性幹細胞の作製には、ヒト胚を必要としないため、胚性幹細胞の使用に際して障害となる倫理的な問題を回避できる。今日、人工多能性幹細胞を作製する新たな方法の開発が進んでおり、今後、より効率の良い、かつ、使用に際して危険性の低い人工多能性幹細胞の作製技術の開発が期待される。
【0004】
アルツハイマー病は、脳内の神経細胞に進行性の機能的障害と脱落が生じる疾患であり、多くの認知症症例の原因疾患である。また、加齢に伴って発症リスクが上昇する疾患であり、高齢化が進む各国において、社会的・医療経済的にも今後さらに深刻な問題になることが予想される。今日、この疾患の進行の抑制を期待できる有効な治療法はなく、疾患発症機序の解明に基づく治療法の開発が急務である。アルツハイマー病の剖検例において脳組織に認められる老人斑は、アミロイド前駆タンパク(APP)の限定分解によって生じたβアミロイドペプチド(Aβ1-42)が重合したものを主な成分としている。本疾患の発症機序として、これまでに得られた数々の知見から、オリゴマーを形成したβアミロイドペプチド、あるいは多数のβアミロイドペプチドが重合して不溶化したものが神経細胞の機能的障害・脱落を引き起こす原因物質であるとする説が最も有力である。加齢に伴って疾患発症のリスクが上昇する理由の一つとして、βアミロイドの分解を担っているNeprilysin等のタンパク質分解酵素の局所における発現レベルが加齢と共に低下し、産生と分解のバランスが崩れることが挙げられる。このような疾患発症のメカニズムから、何らかの方法によりβアミロイドペプチドを分解あるいは除去できれば、この疾患に対する有力な治療法になるものと期待されている。βアミロイドを分解あるいはその産生を抑制する薬剤の開発については、精力的な研究が行われているにもかかわらず、現時点では効果が確認されているものはない。
【0005】
免疫反応を介してβアミロイドペプチドを除去する治療法も検討されており、アルツハイマー病モデルマウスを用いた実験では、βアミロイドペプチドをワクチンとして投与し、これに対する免疫応答を誘導することにより、βアミロイドペプチドの蓄積と神経学的症状を抑制できることが示された。この結果に基づき、ワクチン療法の臨床試験が実施されたが、一部の症例で脳炎が惹起されたため試験は中止となった。最近、臨床試験の中止以前にワクチンを投与された症例の追跡調査の結果が報告されたが、このようなワクチン投与例においてもワクチンによる有意な臨床的効果は認められていない。アルツハイマー病モデルマウスを用いた実験において、βアミロイドペプチドに対するモノクローナル抗体を投与することにより、疾患発症を抑制できるとの報告がなされた。この結果に基づき、βアミロイドペプチドに対するモノクローナル抗体を患者に投与する臨床試験が進められているが、現時点では有意な臨床的効果は認められていない。
【0006】
また、アルツハイマー病以外の神経変性疾患においても、異常なタンパク質の凝集体が神経細胞死を引き起こすことが知られている。その例として、プリオン病、パーキンソン病、筋萎縮性側索硬化症(ALS)がある。また、ハンチントン舞踏病をはじめとするいわゆるポリグルタミン病においては、ポリグルタミンタンパク質の凝集によって神経細胞死が引き起こされる。アミロイドーシスは、アミロイドと呼ばれる異常蛋白が沈着して臓器の機能障害をおこす疾患である。アミロイドが全身性に多臓器に沈着する全身性アミロイドーシスの代表的なものとして、家族性アミロイドポリニューロパチーがある。家族性アミロイドポリニューロパチーでは、トランスサイレチン遺伝子の異常に起因して異常トランスサイレチンタンパク質が産生され蓄積することにより、末梢神経障害、自律神経障害などを含む各種臓器障害が発生する。このようなタンパク質の異常な蓄積に起因する疾患群の多くに対して、現在、有効な治療法は存在しないのが現状である。アルツハイマー病の場合と同様に、疾患発症の原因となっている異状な蓄積タンパク質を除去あるいは分解することができれば、有効な治療法になると予測される。
【0007】
また、多発性の転移巣を有する固形癌や白血病等の悪性腫瘍に対しては、外科的切除および放射線療法が有効でなく、効果のある化学療法剤が存在しない場合、治療は不可能である。昨今、がんに対する新たな治療法として、抗体療法、すなわち、がん細胞の表面に発現する分子(がん細胞表面抗原)を特異的に認識するモノクローナル抗体を投与する方法が開発されている。具体的には、乳癌細胞表面に発現するHER2に対するモノクローナル抗体を投与する治療法は、乳癌に対する標準的な治療と一つとなっている。また、悪性リンパ腫患者の70〜80%を占めるB細胞リンパ腫については、B細胞系の細胞に発現するCD20抗原に対するモノクローナル抗体を投与する抗体療法が確立されている。このような抗体療法の作用メカニズムの一つとして、Fcレセプター(抗体の定常領域であるFc部分に結合するレセプター)を高いレベルで発現する免疫細胞(マクロファージあるいはナチュラルキラー細胞)が、細胞表面に抗体が結合したがん細胞を攻撃する抗体依存性細胞性細胞傷害活性(ADCC)が知られている。抗体依存性細胞性細胞傷害活性は、体内に本来存在する免疫細胞の機能に依存しているため、体内の免疫系細胞が機能的にあるいは数的に十分であるかどうかが、治療の有効性を左右する一つの因子となる。一般に、がん患者においては、しばしば免疫系細胞の機能が低下していることが知られている。マクロファージに、がん細胞表面抗原に対する抗体の遺伝子を導入することにより、がん細胞を特異的に認識し、貪食あるいは攻撃するマクロファージを作製することができると考えられる。しかしながら、人体から分離できるマクロファージあるいはその前駆細胞であるモノサイト(単球)の数には限度があり、また、これらの細胞への人為的な遺伝子導入は容易ではない。
【0008】
他方、特許文献2には、霊長類動物の胚性幹細胞から樹状細胞への分化方法、霊長類動物胚性幹細胞から樹状細胞の製造方法、該製造方法により得られる樹状細胞、該樹状細胞、免疫応答を抗原特異的に制御することにより治療効果を得ることができる疾患の治療のための医薬の製造のための該樹状細胞の使用、及び該疾患の治療のための細胞医薬が記載されている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開WO2007/069666号公報
【特許文献2】国際公開WO2006/022330号公報
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、アルツハイマー病、プリオン病、アミロイドーシス、および、転移巣を有する進行がん、化学療法が無効な白血病等、今日、有効な治療法が存在しない疾患に対して、有効かつ危険性の低い細胞医薬を製造する方法を提供することを解決すべき課題とした。
【課題を解決するための手段】
【0011】
本発明者は上記課題を解決するために鋭意検討した結果、人工多能性幹細胞にタンパク質発現ベクターを導入した後に、該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導することによって外来タンパク質を発現する貪食細胞を製造できることを見出し、本発明を完成するに至った。
【0012】
本発明によれば、以下の発明が提供される。
(1) 人工多能性幹細胞にタンパク質発現ベクターを導入する工程;及び該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程を含む、外来タンパク質を発現する貪食細胞の製造方法。
(2) 人工多能性幹細胞が、体細胞に初期化因子を導入することにより得られる細胞である、(1)に記載の方法。
(3) 人工多能性幹細胞が、体細胞にOct3/4遺伝子、Klf4遺伝子、Sox2遺伝子、及びc-Myc遺伝子を導入することにより得られる細胞である、(1)又は(2)に記載の方法。
(4) 体細胞がヒト体細胞である、(2)又は(3)に記載の方法。
【0013】
(5) タンパク質発現ベクターにより発現されるタンパク質が、抗体、タンパク質分解酵素、又は細胞傷害因子である、(1)から(4)の何れかに記載の方法。
(6) タンパク質発現ベクターにより発現されるタンパク質が、βアミロイドペプチドに対する抗体又は腫瘍細胞の表面に発現する分子に対する抗体である、(1)から(5)の何れかに記載の方法。
(7) タンパク質発現ベクターがプラスミドベクターである、(1)から(6)の何れかに記載の方法。
(8) タンパク質発現ベクターが、薬剤耐性遺伝子を含むベクターである、(1)から(7)の何れかに記載の方法。
【0014】
(9) 該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程が、人工多能性幹細胞に由来する細胞を、顆粒球マクロファージコロニー刺激因子(GM-CSF)及び/又はマクロファージコロニー刺激因子(M-CSF)の存在下で培養することを含む、(1)から(8)の何れかに記載の方法。
【0015】
(10) 該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程が、
(A)タンパク質発現ベクターが導入された人工多能性幹細胞を、中胚葉系細胞を含む細胞群Aへ分化させる工程;及び、
(B)前記工程(A)で得られた細胞群Aを、顆粒球マクロファージコロニー刺激因子(GM-CSF)の存在下で共培養して、貪食能力を有するミエロイド系血液細胞(マクロファージ)である細胞群Bを得る工程を含む、
(1)から(9)の何れかに記載の方法。
【0016】
(11) (1)から(10)の何れかに記載の方法により製造される貪食細胞。
(12) (1)から(10)の何れかに記載の方法により製造される貪食細胞を含む、細胞医薬。
(13) 神経変性疾患又は悪性腫瘍の治療及び/又は予防のための細胞医薬である、(12)に記載の細胞医薬。
【発明の効果】
【0017】
本発明による人工多能性幹細胞の樹立および体外での人工多能性幹細胞からの分化誘導による貪食細胞(マクロファージ)の作製は、担がん状態における免疫担当細胞の異常、あるいは、造血能力の低下などの患者の体内環境に左右されない。また、人工多能性幹細胞は、適切な条件で培養して増殖させることにより大量に得ることができ、これを材料として貪食細胞を大量に作製することが可能であり、かつ、遺伝的改変も容易である。したがって、人工多能性幹細胞から貪食細胞(マクロファージ)を作製する技術を応用することにより、人工多能性幹細胞を材料として、アルツハイマー病やがんを含む各種疾患を治療するための有効な細胞医薬を必要に応じて大量に、かつ、安定的に製造することが可能になる。
【図面の簡単な説明】
【0018】
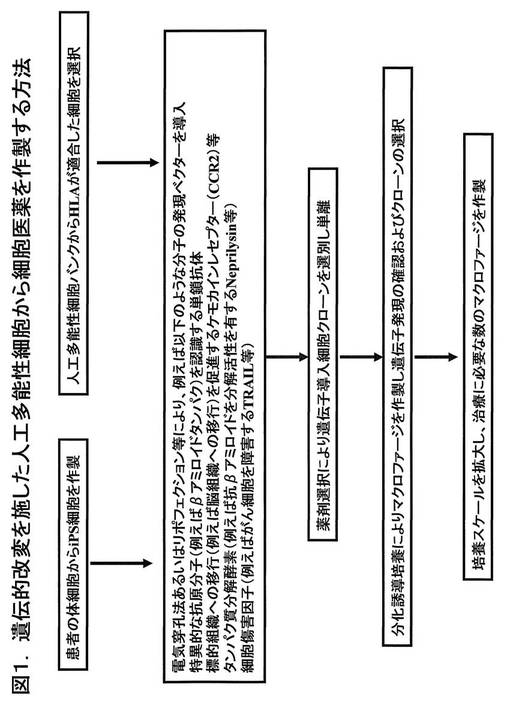
【図1】図1は、本発明による人工多能性幹細胞から各種疾患を治療するための細胞医薬を作製する方法の概要を示す。人工多能性幹細胞は、患者自身の体細胞に由来するものを用いる方法でもよいし、また、将来において設立されるであろう各種のHLA(ヒト白血球組織適合抗原)型をカバーする人工多能性幹細胞バンクから適切な細胞を選択して用いることも可能である。
【図2】図2は、βアミロイドタンパクを認識する単鎖抗体可変領域(scFv)に相当する塩基配列を示す。リーダーペプチド部分を含む、βアミロイドタンパクを認識する単鎖抗体可変領域(scFv)の塩基配列を示す。重鎖および軽鎖の配列は、マウスハイブリドーマクローン3D6の配列に由来する。重鎖と軽鎖の間には、リンカー配列を介在させている。また、軽鎖の下流には、リンカー配列に加えて、遺伝子導入を行った細胞においてフローサイトメトリー解析によりベクターに由来する分子の発現を確認する目的で、cMyc-Tag配列を付加している。
【図3】図3は、免疫グロブリンFc部およびマウスFcレセプター細胞膜貫通-細胞質部位塩基配列を示す。図2に示した塩基配列によってコードされるタンパク質のアミノ酸配列を示す。
【図4】図4は、実施例におけるヒトiPS細胞の作製と遺伝子改変ならびにiPS-MP作製と解析の概略を示す。
【図5】図5は、ヒトiPS細胞に由来するマクロファージ(iPS-MP)の位相差顕微鏡画像を示す。図5Aは、scFv遺伝子を導入していないiPS-MPの形態を示す。図5Bは、Cre発現ベクターの導入により初期化因子であるc-mycおよびSox2遺伝子を除去した後にβアミロイドタンパクに対する単鎖抗体(scFv)の遺伝子を導入したiPS-MPの形態を示す。
【図6】図6は、iPS-MPの細胞表面分子の発現をフローサイトメトリーにより解析した結果を示す。図6Aは、scFv遺伝子を導入していないヒトiPS細胞由来のiPS-MPの細胞表面上のマーカー分子の発現を示す。図6Bは、βアミロイドタンパクに対するscFv遺伝子を導入したヒト人工多能性幹細胞に由来するiPS-MPの細胞表面上のマーカー分子の発現を示す。蛍光色素で標識したマクロファージマーカー分子(CD11b, CD14, CD68)に対するモノクローナル抗体を用いてiPS-MPを染色した後に、フローサイトメトリーによる解析を行った。ヒストグラムの横軸が蛍光の強度を縦軸が細胞数を示す。各々の分子に特異的なモノクローナル抗体で染色した結果(太い実線)とそれらの抗体と同じアイソタイプを有する無関係の抗体(アイソタイプ対照抗体)による染色の結果(細い点線)を示している。
【図7】図7は、βアミロイドタンパクに対する単鎖抗体を発現するiPS-MPによるβアミロイドをコートした蛍光粒子の貪食を示す。ヒトβアミロイドタンパクに対する単鎖抗体を発現するiPS-MPを、βアミロイドをコーティングした(右:ヒトβアミロイドタンパクとアルブミンをコート)、あるいはしていない(左:アルブミンのみコート)蛍光粒子と反応させ、iPS-MPによる蛍光粒子の貪食の程度をフローサイトメトリーにより解析した結果を示す。蛍光粒子にβアミロイドをコーティングすることにより、より多くの細胞がビーズを貪食(25.5→60.3%)し、かつ、細胞あたりに貪食するビーズ数が多くなっている(平均蛍光強度 781→1348)ことがわかる。このようなβアミロイドに特異的な貪食活性の上昇は、 βアミロイドに特異的な単鎖抗体を発現させたiPS-MPにおいてのみ認められた。
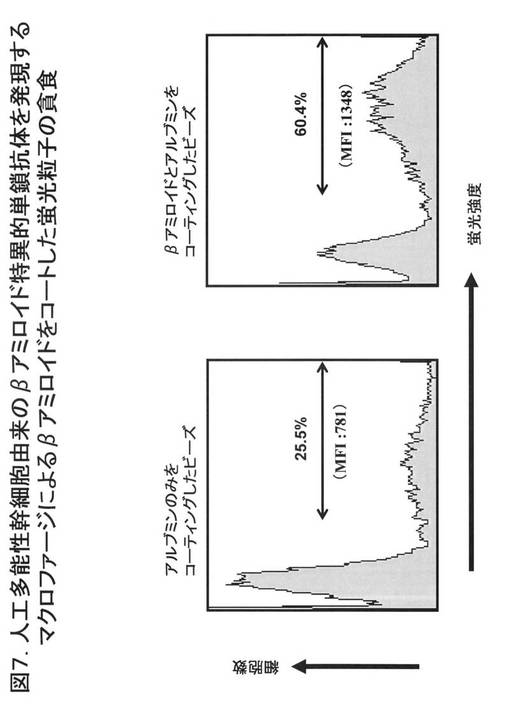
【図8】図8は、βアミロイドをコートした蛍光ビーズを貪食したβアミロイドに対する単鎖抗体を発現するiPS-MPの顕微鏡はゾウを示す。左側の写真は、位相差レンズを用いて撮影した明視野像であり、iPS-MPの形態を示す。右側の写真は、蛍光撮影像であり、蛍光ビーズの局在が示され、iPS-MPの細胞内に蛍光ビーズがとりこまれていることが示されている。
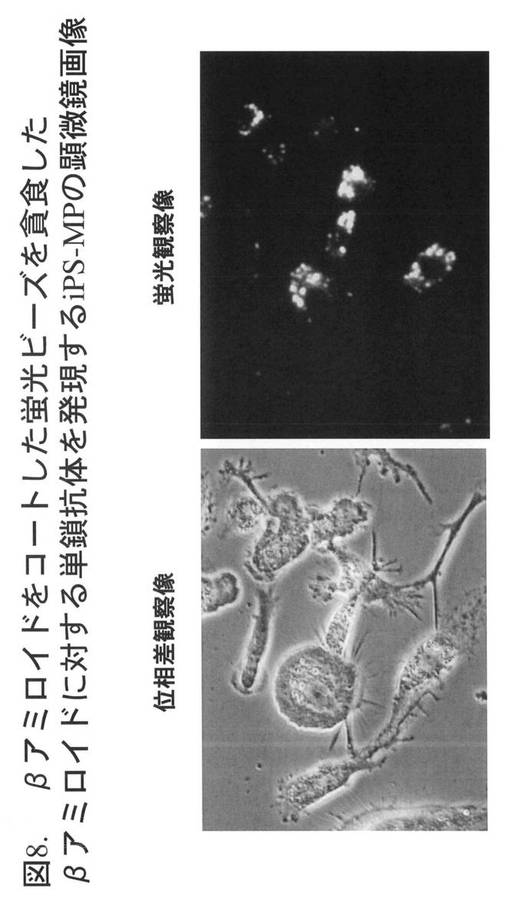
【図9】図9は、βアミロイドに対する単鎖抗体を発現するiPS-MPによるβアミロイドをコートしたビーズの特異的貪食(フローサイトメトリーによる解析)を示す。左側のパネルは、βアミロイドをコーティングしていないビーズを加えたiPS-MP、右側のパネルは、βアミロイドをコーティングしたビーズを加えたiPS-MPの解析結果を示す。
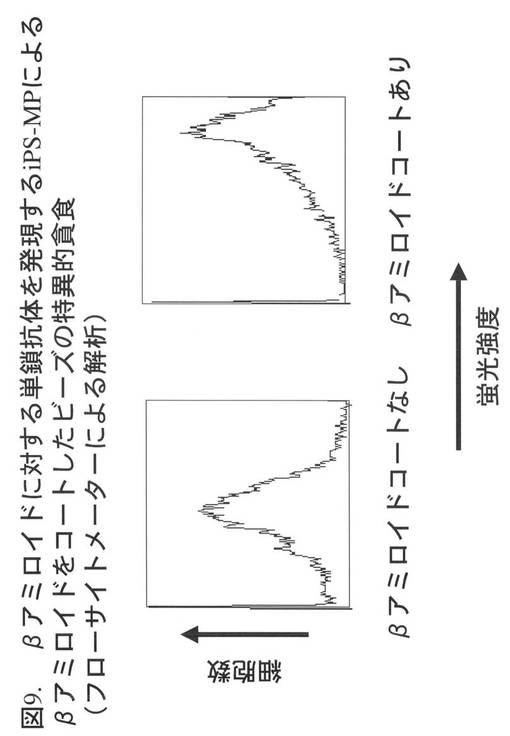
【図10】図10は、付着状態における遺伝的改変iPS-MPによるβアミロイドコートビーズの特異的貪食(フローサイトメトリーによる解析)を示し、iPS-MPに加えるビーズの濃度を変化させた場合のiPS-MPの蛍光強度の平均値をグラフにしたものである。白丸は、βアミロイドをコーティングしていないビーズを加えた場合、黒丸は、βアミロイドをコーティングしたビーズを加えた場合を示す。グラフの横軸は、iPS-MPに加えたビーズの濃度、縦軸は、回収後の細胞の蛍光強度を示す。
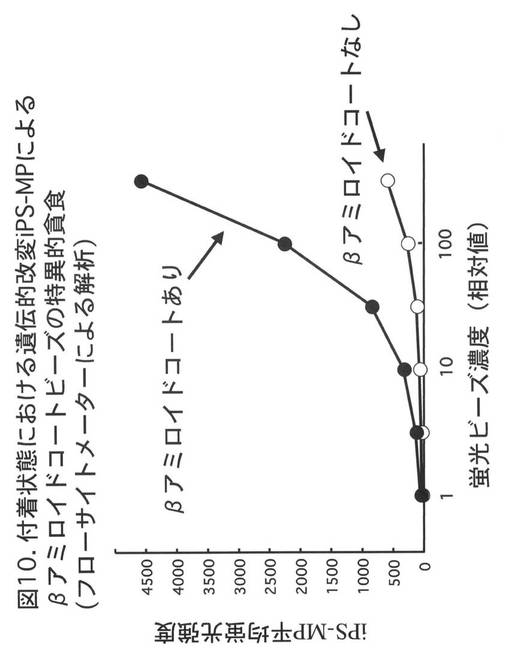
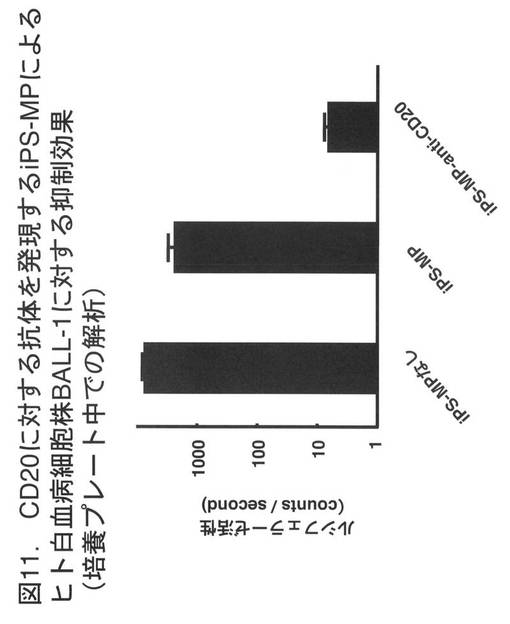
【図11】図11は、CD20に対する抗体を発現するiPS-MPによるヒト白血病細胞株BALL-1に対する抑制効果を示す。BALL-1細胞のみを培養した場合に比べ、あるいは、これにCD20に特異的な単鎖抗体を発現しないiPS-MPを加えて培養した場合に比較し、CD20に特異的な単鎖抗体を発現するiPS-MPを加えて培養した場合、ルシフェラーゼの活性は、1/100以下となった。
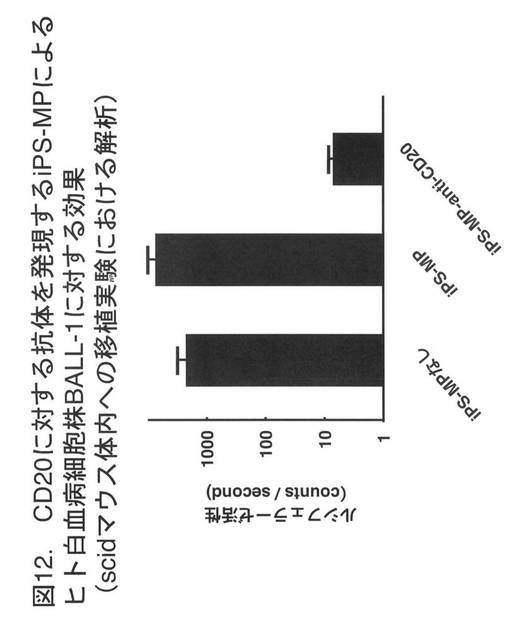
【図12】図12は、CD20に対する抗体を発現するiPS-MPによるヒト白血病細胞株BALL-1に対する効果(scidマウス体内への移植実験における解析)を示す。BALL-1細胞のみを注射したグループ、あるいは、それに加えてCD20に特異的な単鎖抗体を発現しないiPS-MPを加えて培養したグループに比較し、CD20に特異的な単鎖抗体を発現するiPS-MPを加えて培養したグループでは大網組織のルシフェラーゼ活性が1/100以下となった。
【発明を実施するための形態】
【0019】
以下、本発明について更に詳細に説明する。
本明細書の以下に示す実施例においては、人工多能性幹細胞に特定の分子に対する抗体の遺伝子を導入し、引き続いて、生体外での分化誘導によりマクロファージ系の貪食細胞を製造した。製造される貪食細胞は、特定の分子に対する抗体等を発現するものであり、標的となる分子を特異的に認識し貪食する活性を有している。特異的抗体に加えて、タンパク質分解酵素などを同時に発現させることにより、高い効率で標的となる分子を貪食し分解する、あるいは標的分子を発現する細胞を特異的に貪食・傷害する等の機能を発揮する貪食細胞(マクロファージ)を作製することができる。図1に、本発明による人工多能性幹細胞から細胞医薬を製造する方法の概要を示している。
【0020】
(1)人工多能性幹細胞
本発明で用いる人工多能性幹細胞は、体細胞を初期化することにより製造することができる。ここで用いる体細胞の種類は特に限定されず、任意の体細胞を用いることができる。即ち、本発明で言う体細胞とは、生体を構成する細胞のうち生殖細胞以外の全ての細胞を包含し、分化した体細胞でもよいし、未分化の幹細胞でもよい。体細胞の由来は、哺乳動物、鳥類、魚類、爬虫類、両生類の何れでもよく特に限定されないが、好ましくは哺乳動物(例えば、マウスなどのげっ歯類、またはヒトなどの霊長類)であり、特に好ましくはヒトである。また、ヒトの体細胞を用いる場合、胎児、新生児又は成人の何れの体細胞を用いてもよい。本発明の方法で製造される人工多能性幹細胞を再生医療など疾患の治療に用いる場合には、該疾患を患う患者自身から分離した体細胞を用いることが好ましい。
【0021】
本発明で言う人工多能性幹細胞とは、所定の培養条件下(例えば、ES細胞を培養する条件下)において長期にわたって自己複製能を有し、また所定の分化誘導条件下において外胚葉、中胚葉及び内胚葉への多分化能を有する幹細胞のことを言う。また、本発明における人工多能性幹細胞はマウスなどの試験動物に移植した場合にテラトーマを形成する能力を有する幹細胞でもよい。
【0022】
体細胞から人工多能性幹細胞を製造するためには、まず、少なくとも1種類以上の初期化遺伝子を体細胞に導入する。初期化遺伝子とは、体細胞を初期化して人工多能性幹細胞とする作用を有する初期化因子をコードする遺伝子である。本発明では、少なくとも1種類以上の初期化遺伝子を使用する。本発明で使用する初期化遺伝子の組み合わせの具体例としては、以下の組み合わせをあげることができるが、これらに限定されるものではない。
(i)Oct遺伝子、Klf遺伝子、Sox遺伝子、Myc遺伝子
(ii)Oct遺伝子、Sox遺伝子、NANOG遺伝子、LIN28遺伝子
(iii)Oct遺伝子、Klf遺伝子、Sox遺伝子、Myc遺伝子、hTERT遺伝子、SV40 large T遺伝子
(iv)Oct遺伝子、Klf遺伝子、Sox遺伝子
【0023】
Oct遺伝子、Klf遺伝子、Sox遺伝子及びMyc遺伝子にはそれぞれ、複数のファミリー遺伝子が含まれている。それぞれのファミリー遺伝子の具体例としては、国際公開WO2007/069666号公報の明細書の第11頁から第13頁に記載されているものを用いることができる。具体的には、以下の通りである。
【0024】
Oct遺伝子に属する遺伝子の具体例としては、Oct3/4(NM_002701)、Oct1A(NM_002697)、及びOct6(NM_002699)などを挙げることができる(括弧内は、ヒト遺伝子のNCBI accession 番号を示す)。好ましくはOct3/4である。Oct3/4はPOUファミリーに属する転写因子であり、未分化マーカーとして知られており、また多能性維持に関与しているとの報告もある。
【0025】
Klf遺伝子に属する遺伝子の具体例としては、Klf1(NM_006563)、Klf2(NM_016270)、Klf4(NM_004235)、及びKlf5(NM_001730)などを挙げることができる(括弧内は、ヒト遺伝子のNCBI accession 番号を示す)。好ましくはKlf4である。Klf4(Kruppel like factor-4)は腫瘍抑制因子として報告されている。
【0026】
Sox遺伝子に属する遺伝子の具体例としては、例えば、Sox1(NM_005986)、Sox2(NM_003106)、Sox3(NM_005634)、Sox7(NM_031439)、Sox15(NM_006942)、Sox17(NM_0022454)、及びSox18(NM_018419)を挙げることができる(括弧内は、ヒト遺伝子のNCBI accession 番号を示す)。好ましくはSox2である。Sox2は初期発生過程で発現し、転写因子をコードする遺伝子である。
【0027】
Myc遺伝子に属する遺伝子の具体例としては、c-Myc(NM_002467)、N-Myc(NM_005378)、及びL-Myc(NM_005376)などを挙げることができる(括弧内は、ヒト遺伝子のNCBI accession 番号を示す)。好ましくは、c-Myc である。c-Mycは細胞の分化及び増殖に関与する転写制御因子であり、多能性維持に関与しているとの報告がある。
【0028】
上記した遺伝子は、ヒトを含む哺乳類動物において共通して存在する遺伝子であり、本発明において任意の哺乳類動物由来(例えばヒト、マウス、ラット、サルなどの哺乳類動物由来)の遺伝子を用いることができる。また、野生型の遺伝子に対して、数個(例えば1〜30個、好ましくは1〜20、より好ましくは1〜10個、さらに好ましくは1〜5個、特に好ましくは1から3個)の塩基が置換、挿入及び/又は欠失した変異遺伝子であって、野生型の遺伝子と同様の機能を有する遺伝子を使用することもできる。
【0029】
本発明では特に好ましくは、初期化遺伝子として、Oct3/4遺伝子、Klf4遺伝子、Sox2遺伝子、及びc-Myc遺伝子の組み合わせを用いることができる。
【0030】
初期化遺伝子を体細胞に導入する方法は、導入された初期化遺伝子が発現して体細胞の初期化を達成できる限り特に限定されない。例えば、少なくとも1種類以上の初期化遺伝子を含む発現ベクターを用いて該初期化遺伝子を体細胞に導入することができる。ベクターを用いて2種類以上の初期化遺伝子を体細胞に導入する場合には、一つの発現ベクターに2種類以上の初期化遺伝子を組み込んで、該発現ベクターを体細胞に導入してもよいし、1種類の初期化遺伝子を組み込んだ発現ベクターを2種類以上用意して、それらを体細胞に導入してもよい。
【0031】
発現ベクターの種類は特に限定されず、ウイルスベクターでもプラスミドベクターでもよいが、好ましくはウイルスベクターであり、特に好ましくは導入した初期化遺伝子が体細胞の染色体に組み込まれるようなウイルスベクターである。本発明で使用できるウイルスベクターとしては、レトロウィルスベクター(レンチウィルスベクターを含む)、アデノウイルスベクター、アデノ随伴ウイルスベクターなどを挙げることができる。上記の中でも好ましくはレトロウィルスベクターであり、特に好ましくはレンチウィルスベクターである。
【0032】
組み換えウイルスベクターを作製するために用いるパッケージング細胞としては、ウイルスのパッケージングに必要なタンパク質をコードする遺伝子の少なくとも1つを欠損している組換えウイルスベクタープラスミドの該欠損するタンパク質を補給できる細胞であれば任意の細胞を用いることができる。例えばヒト腎臓由来のHEK293細胞、マウス繊維芽細胞NIH3T3に基づくパッケージング細胞を用いることができる。
【0033】
組換えウイルスベクタープラスミドをパッケージング細胞に導入することで組換えウイルスベクターを生産することができる。上記パッケージング細胞への上記ウイルスベクタープラスミドの導入法は、特に限定されず、リン酸カルシウム法、リポフェクション法又はエレクトロポレーション法などの公知の遺伝子導入法で行うことができる。
【0034】
ES細胞の未分化性及び多能性を維持可能な培地は当業界で公知であり、適当な培地を組み合わせて用いることにより、本発明の人工多能性幹細胞を分離及び培養することができる。即ち、本発明の人工多能性幹細胞を培養するための培地としては、ES培地、ES培地に10ng/ml FGF-2を添加後にマウス胚性繊維芽細胞を24時間培養した上清であるMEF馴化ES培地(以下MEF馴化ES培地)などをあげることができる。本発明の人工多能性幹細胞を培養するための培地には、各種の成長因子、サイトカイン、ホルモンなど(例えば、FGF-2、TGFb-1、アクチビンA、ノギン(Nanoggin)、BDNF、NGF、NT-1、NT-2、NT-3等のヒトES細胞の増殖・維持に関与する成分)を添加してもよい。また、分離された人工多能性幹細胞の分化能及び増殖能は、ES細胞について知られている確認手段を利用することにより確認することができる。
【0035】
(2)タンパク質発現ベクターの人工多能性幹細胞への導入
本発明では、人工多能性幹細胞にタンパク質発現ベクターを導入する。タンパク質発現ベクターにより発現されるタンパク質は、特に限定はされないが、例えば、特異的な抗原分子(例えばβアミロイドタンパク、CD20分子)を認識する抗体(例えば、単鎖抗体)、標的組織への移行(例えば脳組織への移行)を促進するケモカインレセプター(CCR2等)、タンパク質分解酵素(例えばβアミロイドを分解活性を有するNeprilysin、トリプシノーゲン等)、細胞傷害因子(例えばがん細胞を障害するTRAIL等)などを挙げることができる。抗体としては、治療のターゲット対象となるタンパク質又はペプチドに対する抗体を用いることができ、例えば、アルツハイマー病におけるβアミロイドペプチド、神経変性疾患における異常なタンパク質の凝集体、癌細胞に特異的に発現するタンパク質(腫瘍抗原タンパク質)、腫瘍抗原タンパク質の一部であるペプチド等に対する抗体が挙げられる。
【0036】
タンパク質発現ベクターは、薬剤耐性遺伝子を含むことが好ましい。薬剤耐性遺伝子としては、G418、ピューロマイシン、ハイグロマイシン等の選択薬剤に対する耐性を付与する遺伝子を用いることができる。遺伝子導入後は、上記の選択薬剤を含む培養液中で培養し、遺伝子が導入された細胞を選択し、遺伝子導入細胞のクローンを単離することができる。
【0037】
タンパク質発現ベクターの導入は、慣用の方法、例えば、エレクトロポレーション法(電気穿孔法)、リポフェクション法等により行うことができる。上記の中でも、エレクトロポレーション法が望ましい。
【0038】
発現ベクターは、マクロファージを含む哺乳動物細胞において高い効率で遺伝子発現がなされるものを用いることが好ましく、例えば、プラスミドベクター、ファージベクターなどを用いることができる。発現ベクターは、必要に応じ、各種プロモーター、エンハンサー、ターミネーター等の転写促進に有効なエレメントを含有してもよい。
【0039】
(3)人工多能性幹細胞から貪食細胞への分化誘導
本発明では、生体外での分化誘導法により、貪食細胞への分化誘導を行う。
即ち、タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程は、貪食細胞への分化誘導を行うことができる限り特に限定されない。例えば、人工多能性幹細胞を、血液細胞の分化と増殖とを誘導する性質を有する細胞(フィーダー細胞)と一緒に培養する共培養法によって貪食細胞を含む血液細胞への分化誘導を行うことができるが、特にこのフィーダー細胞共培養法には限定されない。例えば、フィブロネクチン等で表面をコーティングした細胞接着性培養容器内で培養することによりフィーダー細胞を使用せず分化誘導を行うことも可能である。また、人工多能性幹細胞の細胞塊を浮遊状態で培養することにより胚様体を形成させ、しかる後に、貪食細胞への分化誘導を行ってもよい。細胞医薬として人体へ投与することを考慮すると、フィーダー細胞を使用せず分化誘導を行う方法がより好ましい。
【0040】
人工多能性幹細胞から貪食細胞への分化誘導の際に用いられる培養液としては、哺乳動物細胞の培養に適した培養液であればよく、用いられる人工多能性幹細胞の種類などに応じて適宜選択され得、例えば、αMEM、DMEM、IMDM(イスコブ改変ダルベッコ培地)等が挙げられる。
【0041】
分化誘導培養において用いる培養液は、血清含有培養液又は無血清培養液であり得る。無血清培養液としては、ヒトあるいはヒト以外の動物の血清由来の物質を全く含まないものもあるが、精製された血清由来の特定の成分(例えばアルブミンなど)が添加された培養液もこれに含まれる。血清由来の物質を全く含まない無血清培養液の場合は、遺伝子組み換えにより作製されたインスリンなどのタンパク質、脂肪酸、微量元素、界面活性剤を適宜含有するものでありうる。細胞医薬として人体へ投与することを考慮すると、無血清培養液を用いることがより好ましい。
【0042】
本発明において、人工多能性幹細胞と、前記フィーダー細胞との共培養の際、前記フィーダー細胞の作製に用いられうる培地としては、付着性の哺乳動物細胞の培養に適した培地であればよく、フィーダー細胞として用いる細胞の種類などに応じて適宜選択され得、例えば、αMEM、DMEM〔ダルベッコ改変イーグル培地(培養液)〕等が挙げられる。
【0043】
人工多能性幹細胞から貪食細胞への分化誘導の際に用いられる培養容器は、分化誘導を行うことが可能な培養容器であればよい。例えば、ゼラチン、フィブロネクチン等により表面をコーティングし、細胞接着性を増強したポリスチレン製の培養容器を用いて行なわれうるが、必ずしもこれに限定されない。例えば、無菌的に大量の細胞を調整することを意図して、細胞培養用のガス透過性バッグなどを用いることも可能である。
【0044】
前記フィーダー細胞の培養条件としては、フィーダー細胞として用いる細胞の種類などに応じて適宜設定されうる。例えば、ST2細胞、OP9細胞等の場合、0.1重量%ゼラチン溶液等でコーティングされた培養容器上、10容量%ウシ胎仔血清を添加した培地で37℃、5体積%CO2で培養する条件等が挙げられる。
【0045】
培養気相の条件は、用いられる人工多能性幹細胞の種類、培養液の組成等に応じて、適宜設定されうるが、例えば、37℃前後(特に、37℃)、5体積%CO2等の条件が挙げられる。
【0046】
人工多能性幹細胞から貪食細胞への分化誘導するためには、好ましくは、人工多能性幹細胞を、顆粒球マクロファージコロニー刺激因子(GM-CSF)あるいはマクロファージコロニー刺激因子(M-CSF)の存在下で培養することができる。培地中における顆粒球マクロファージコロニー刺激因子(GM-CSF)の含有量は、50〜200ng/mlの範囲が望ましい。
【0047】
上記方法により、貪食細胞の性質(例えば、CD11b、CD14、CD68の発現)を示す貪食細胞を得ることができる。本発明の方法により製造される貪食細胞は、抗原タンパク質を貪食する活性を有する。
【0048】
(4)本発明の貪食細胞及び細胞医薬
本発明はさらに、上記した方法により製造される貪食細胞、並びにそれを含む細胞医薬に関する。本発明の貪食細胞によれば、アルツハイマー病、プリオン病、アミロイドーシス、および、転移巣を有する進行がん、化学療法が無効な白血病等の疾患に対して、有効かつ危険性の低い細胞医薬を提供することができる。
【0049】
本発明の細胞医薬の製造の際には、本発明の貪食細胞を安定に保持しうる助剤、例えば、培地等を適宜用いてもよい。
【0050】
また、本発明によれば、被験体に前記製造方法により得られる貪食細胞の治療有効量を投与することを特徴とする、被験体における上記疾患の治療方法が提供される。
【0051】
治療有効量とは、前記製造方法により得られる貪食細胞を被験体に投与した場合に、該貪食細胞を投与していない被験体と比較して前記のような疾患に対して治療効果を得ることができる該貪食細胞の量である。具体的な治療有効量としては、貪食細胞の投与形態、投与方法、使用目的および被験体の年齢、体重、症状等によって適宜設定されればよく、一概には決定されないが、例えば、前記貪食細胞の細胞数で、ヒト(例えば成人)の1回の治療あたり、200,000〜1,000,000個/kg体重が好ましい。
【0052】
本発明の細胞医薬の投与方法としては、例えば、皮下注射、リンパ節内注射、静脈内注射、腹腔内注射、胸腔内注射あるいは悪性腫瘍の局所への直接注射などが挙げられるが、これらに限定されない。
【0053】
以下の実施例により本発明をさらに具体的に説明するが、本発明は実施例によって限定されるものではない。
【実施例】
【0054】
実施例1.:ヒトβアミロイドペプチドを認識する単鎖抗体(scFv)発現ベクターの作製
(1)可変領域塩基配列の作製
ヒトβアミロイドペプチドを認識するモノクローナル抗体(マウスハイブリドーマクローン: 3D6)の重鎖(heavy chain)および軽鎖(light chain)の可変領域の塩基配列(特許文献2007−0238154に記載)に基づき、βアミロイドペプチドを認識する単鎖抗体可変領域の塩基配列を設計した。また、図2に塩基配列を、また、図3にこの塩基配列にコードされるタンパク質のアミノ酸配列を示す。設計した塩基配列に基づき、GeneScript Corporation社(米国 ニュージャージー州)に依頼してDNAの人工合成を行い、さらに、プラスミドベクターへの挿入を行った。
【0055】
(2)免疫グロブリン(マウスIgG2a)Fc部およびマウスFcレセプター(FcγRI)細胞膜貫通-細胞質部位の作製
C57BL/6マウスの脾臓組織より抽出したRNAに由来するcDNAを鋳型として、PCR法により作製した。
【0056】
(3)βアミロイドペプチドを認識する単鎖抗体と免疫グロブリンFc部およびFcレセプター(Fcγ細胞膜貫通-細胞質部位)との融合タンパク質の発現ベクターの作製
前記の単鎖抗体可変領域のDNA断片と免疫グロブリンFc部およびマウスFcレセプター細胞膜貫通-細胞質部位のDNA断片をPCR法を用いて融合し、プラスミドベクター(pENTR-D-TOPO, Invitrogen社製)へ挿入し、大腸菌を用いてクローニングを行った。
【0057】
クローニングを行ったプラスミドDNAの塩基配列をシークエンス解析により確認した後、DNA組換え酵素LR clonase (Invitrogen社製)を用いて哺乳動物発現ベクターへ導入した。哺乳動物発現ベクターとしては、CAGプロモーターにより作動し、IRES (internal ribosomal entry site)とピューロマイシン耐性遺伝子が組込まれたベクターであるpCAGGS-IRES-Puroを用いた。
【0058】
実施例2:ヒト人工多能性幹細胞の作製および培養
健常人腹部の皮膚組織に由来する線維芽細胞に、レンチウイルスベクターを用いてヒトのOct3/4, Sox2, Klf4, c-Mycの発現ベクターを導入することにより、ヒト人工多能性幹細胞の作製を行った。
【0059】
具体的には、ヒト腹部の皮膚片を採取し、細胞培養用プレート中で培養液(DMEM / 10%牛血清)を用いて培養した。培養開始1週間目以降より、皮膚片からの線維芽細胞の遊走と増殖が認められたので、トリプシンとEDTAを用いて、適宜、線維芽細胞を回収し凍結保存した。
【0060】
初期化因子であるヒトのOct3/4, Sox2, Klf4, c-MycのcDNAは、ヒトES細胞(KhES-1)に由来するcDNA、あるいはゲノムDNAを鋳型として、PCR法により作製し、プラスミドベクター(pENTR-D-TOPO, Gibco-Invitrogen社)へ挿入した。クローニングを行ったプラスミドDNAの塩基配列をシークエンス解析により確認の後、LR clonase (Gibco-Invitrogen社)を用いて、レンチウイルスベクター(CSII-EF-RfAl, 理化学研究所 三好博士より分与)へ導入した。Sox2とc-Mycについては、CSII-EF-RfAlへ挿入後にcDNA両端へのLOX配列の付加を行った。
【0061】
Creの構造遺伝子を鋳型として、PCR法により遺伝子断片を作製し、CAGプロモーターにより作動し、IRES (internal ribosomal entry site)とネオマイシン耐性遺伝子が組込まれたベクターであるpCAGGS-IRES-Neoへ挿入し、Cre酵素とネオマイシン耐性遺伝子を同時に発現するベクターを作製した。
【0062】
リポフェクション法(Lipofectamine 2000, Invitrogen社)を用いて、上記で作製したCSII-EFに導入したリプログラミング因子遺伝子の各々とパッケージングコンストラクト、およびエンベロープおよびRevのコンストラクトを293T細胞へ遺伝子導入した。遺伝子導入3日後に、細胞培養液を回収し、0.45μmのフィルターを通した後、遠心分離法(50,000G、2時間)によりウイルス粒子を沈澱させ回収した。回収した組換えウイルス粒子は、DMEM溶液に懸濁した後、凍結チューブに分注し、使用時まで冷凍庫(-80℃)にて保存した。
【0063】
凍結保存しておいたヒト線維芽細胞を解凍し、数日間、再度培養した後、培養プレート中においてウイルス懸濁液を加えることにより、感染させ、遺伝子導入を行った。遺伝子導入の4-6日後、トリプシン-EDTAを用いて感染細胞を回収し、事前に準備しておいた、マイトマイシンC処理により増殖を停止させたマウス胎児由来線維芽細胞(フィーダー細胞)との共培養を開始した。その翌日より、培養液をヒトES細胞用の培養液に置換し、培養を継続した。
【0064】
初期化因子の遺伝子を導入してから、20-30日の後、一部の細胞については顕微鏡観察の下で、マイクロチップを用いてES細胞様の形態を示すコロニーをiPS細胞クローンとして単離し、別に準備しておいたマウス胎児由来フィーダー細胞との共培養を行った。その後、細胞の増殖に応じて、培養容器を拡大しつつ培養を継続した(図4Aの経路)。また、初期化因子の遺伝子を導入した細胞集団のうち別の一部のものについては、トリプシン-EDTAを用いて回収した後、Cre発現ベクターの導入を行った(図4Bの経路)。
【0065】
Cre発現ベクターの導入
図4Bの経路におけるCre発現ベクターの導入は、電気穿孔法により行った。Cre発現ベクターが挿入された細胞を選別する目的で、電気穿孔法施行後、24−48時間後より、培養液中にG418 (Gibco- Invitrogen社)を加え、培養を継続した。そして、電気穿孔法施行後15-25日後に、顕微鏡観察の下で、ES細胞様の形態を示すG418耐性の細胞コロニーをマイクロチップを用いて単離し、別に準備しておいたマウス胎児由来フィーダー細胞との共培養を行った。その後、細胞の増殖に応じて、培養容器を拡大しつつ培養を継続した。
【0066】
ヒトiPS細胞の維持培養は、ヒト胚性幹細胞培養液(DMEM-F12(Wako Chemicals)/20% KSR(Gibco-Invitrogen)/bFGF (basic fibroblast growth factor: 10 ng/ml)/2-ME (2-メルカプトエタノール, 50μM))を用いて、マイトマイシンC処理マウス胎児繊維芽細胞をフィーダー細胞としてポリスチレン製培養皿にて行った。
【0067】
実施例3:ヒトiPS細胞への単鎖抗体発現ベクターの導入
実施例1で作製した単鎖抗体(scFv)発現ベクター(プラスミドベクター)DNAは、大腸菌からアルカリ-SDS法で抽出した後、陰イオン交換カラム精製(Qiagen Plasmid Maxiキット, Qiagen社製)を行った。そして、制限酵素Pvu Iを用いて切断することにより直線状DNAとした。さらに、フェーノールとクロロホルムを用いた精製を行った後に、イソプロパノール沈澱により回収し、70%エタノールによる洗浄を行った上で、リン酸干渉生理的食塩水(PBS)に溶解した。
【0068】
維持培養を行っているヒトiPS細胞(図4Bの経路により作製した細胞クローン)をトリプシン-EDTA (0.25%トリプシン/1 mM EDTA)を用いて培養容器より回収し、DMEM溶液に懸濁(1x107細胞/0.4 ml)し、PBSに溶解したプラスミドベクター(30μg/0.1 ml)と混和した後、遺伝子導入装置(ジーンパルサー、BioRad社製)を用いて電気穿孔法による遺伝子導入(キュベットの電極間距離:4 mm, 通電:200V, 800μF, 1回)を行った。
【0069】
電気穿孔法施行後、細胞の生存率を高めるために、24時間はRoh結合キナーゼ阻害剤(Y-27632,和光純薬製)存在下に培養を行い、また、48時間後より培養液中にピューロマイシン (Sigma社)を加え、培養を継続した。
【0070】
その後、断続的にピューロマイシン (5-15μg/ml)を加え、培養を継続した。そして、電気穿孔法施行後15-18日後に、倒立顕微鏡による観察を行いつつ、ピューロマイシン耐性の細胞コロニーをマイクロチップを用いて単離し、別に準備しておいたマウス胎児由来フィーダー細胞との共培養を行った。
【0071】
その後、細胞の増殖に応じて、培養容器を拡大しつつ培養を継続し、一部はそのまま凍結保存し、一部は後述のようにマクロファージへの分化誘導を行った。
【0072】
実施例4:ヒト人工多能性細胞のマクロファージへの分化誘導
(1)OP9フィーダー細胞の調整
マウス由来の培養細胞株OP9をマイトマイシンCにて処理(0.01 mg/ml、60分)したものを、ゼラチンコートしたディッシュに播種し、翌日以降に使用した。
【0073】
(2)分化誘導培養
未分化なiPS細胞(図4の経路AあるいはBのもの)を、CTK液(コラゲナーゼ-トリプシン-KSR液、末盛ら Biochemical and Biophysical Research Communications 345: 926-932, 2006年)を用いて5-10分間処理し、FCS(牛胎児血清、GIBCO-Invitrogen社製)入りの培養液で回収した。細胞を遠心し、a-MEM/20%FCSに懸濁し、OP9フィーダー細胞上へ播種した。以降、3日に1度、培養液(a-MEM/20%FCS)を交換しつつ培養を継続した。
【0074】
分化誘導開始から15-18日後、トリプシン-EDTA-コラゲナーゼ液を用いて細胞を処理(37℃ 60分)して解離させて回収し、ピペッティング操作により細胞浮遊液を作製した。そして、ディッシュ1枚由来の細胞を10 ml のDMEM/10% FCSに懸濁し、フィーダー細胞なし、ゼラチンコートなしの90-mmディッシュ2枚にまいた。2-5時間後、ディッシュに付着しなかった細胞を回収し、100ミクロンのメッシュ(BD Falcon社製 セルストレイナー)を通過させることにより、細胞塊を除いた細胞集団を得た。この細胞を、a-MEM / 20%FCS / ヒト GM-CSF (100 ng/ml、ぺプロテック社製) / ヒト M-CSF (50 ng/ml、ぺプロテック社製) / 2-ME (0.05 mM)に浮遊させ、再度、OP9フィーダー細胞上へ播種し培養を行った。
【0075】
7〜9日経過した後、ピペッティング操作により細胞を回収し、RPMI-1640 / 10%FCS/ M-CSF (100 ng/ml) に懸濁して、培養皿(BD FALCON社製)へ播種して3-5日間培養を行いマクロファージ(iPS-MP)を作製した。図5は、位相差顕微鏡レンズを用いてディシュ内のiPS-MPを撮影した画像である。図5Aは、図4のAの経路、すなわち、Cre発現ベクターとscFv発現ベクターの導入を行っていないiPS細胞に由来するiPS-MPである。図5Bは、図4のBの経路、すなわち、Cre発現ベクターとscFv発現ベクターを導入したiPS細胞に由来するiPS-MPである。
【0076】
実施例5:フローサイトメトリーによるiPS-MPの細胞表面上のマーカー分子の発現の検討
上記のように作製したiPS-MPをピペッティング操作により回収し、抗体の非特異的な結合を阻害する目的で、まずFcレセプターブロック試薬(Miltenyi Biotec社製)を用いて10分間処理した。
【0077】
その後、以下のようなフィコエリスロシン(PE)結合モノクローナル抗体あるいはフルオレセインイソチオシアネート(FITC)結合モノクローナル抗体を用いて室温中で40分間染色した。PE-抗ヒト/マウスCD11b(クローンM1/70、ラットIgG2b)、PE-抗ヒトCD14(クローンTUK4、マウスIgG2a)、FITC-抗ヒトCD68(クローン Y1/82A、マウスIgG2b)。また、アイソタイプ適合対照抗体として、PE-ラットIgG2b(クローン LO-DNP-11)、PE-マウスIgG2a(クローンG155−178)とFITC-マウスIgG2b(クローン 27-35)を用いて染色した。
【0078】
その後、細胞を、PBS/2% FCSで2回洗浄した。
洗浄後の細胞について、CellQuest ソフトウェアを備えたフローサイトメーター解析装置(商品名:FACScan,Becton Dickinson社製)を用いて分析した。
【0079】
図6に抗体染色後のフローサイトメーター解析の結果を示す。図6Aは、図4のAの経路、すなわち、Cre発現ベクターとscFv発現ベクターの導入を行っていないiPS細胞に由来するiPS-MPである。図6Bは、図4のBの経路、すなわち、Cre発現ベクターとscFv発現ベクターを導入したiPS細胞に由来するiPS-MPである。いずれのiPS-MPも、細胞表面上にCD11b、CD14、CD68を発現しており、生理的に存在するヒトマクロファージと同様のマーカー分子を発現していた。
【0080】
実施例6:βアミロイドペプチドに対する単鎖抗体を発現するiPS-MPによるβアミロイドペプチドをコートした蛍光粒子の結合能の浮遊状態での解析
(1)蛍光粒子へのβアミロイドペプチドのコーティング
蛍光粒子(Sulfate Microspheres, Yellow-green, #F8828, Molecular Probes社製)にβアミロイドペプチド(Amyloid beta-Protein 1-42, Wako Chemicals社製)を添加(終濃度0.03mg/ml)し25℃にて震盪しつつ12時間反応させた。その後、0.1%の牛血清アルブミンを含有するPBS(PBS-BSA)を2倍容量加え、さらに、25℃にて震盪しつつ2時間反応させた。アルブミンのみをコーティングした蛍光粒子は、PBS-BSA中で、25℃にて震盪しつつ14時間反応させることにより作製した。
その後、蛍光粒子をPBSで2回洗浄した後、実験に供するまでの間、遮光して冷蔵保存した。
【0081】
(2)iPS-MPによる蛍光粒子の結合能の解析
βアミロイドに特異的な単鎖抗体の遺伝子を導入したiPS-MPを培養皿から回収し洗浄した後、βアミロイドをコーティングした蛍光粒子(直径0.2μm)とプラスチック製試験管内で混和し、37℃にて90分静置した。
その後、PBS-FCS で2回洗浄した後、フローサイトメーターを用いて、iPS-MPの蛍光強度、すなわち、蛍光ビーズの貪食を定量化した。
【0082】
図7に、フローサイトメトリーによる解析の結果を示す。蛍光ビーズにβアミロイドをコーティングすることにより、より多くの細胞がビーズを結合(25.5→60.3%)し、かつ、細胞あたりに貪食するビーズ数が多くなっている(平均蛍光強度 781→1348)。
【0083】
実施例7:βアミロイドペプチドに対する単鎖抗体を発現するiPS-MPによる、付着培養状態でのβアミロイドペプチドをコートした蛍光粒子の貪食能の解析
(1)蛍光粒子へのβアミロイドペプチドのコーティング
蛍光粒子(Sulfate Microspheres, Yellow-green, #F8828, Molecular Probes社製)にβアミロイドペプチド(Amyloid beta-Protein 1-42, Wako Chemicals社製)を添加(終濃度0.03mg/ml)し25℃にて震盪しつつ24時間反応させた。その後、2%の牛血清アルブミンを含有するPBS(PBS-BSA)を等量加え、さらに、25℃にて震盪しつつ24時間反応させた。対照実験に用いた、アルブミンのみをコーティングした蛍光粒子は、1%PBS-BSA中で、25℃にて震盪しつつ48時間反応させることにより作製した。
その後、蛍光粒子をPBSで2回洗浄した後、実験に供するまでの間、遮光して冷蔵保存した。
【0084】
(2)iPS-MPによる蛍光粒子の貪食能の解析
細胞培養容器(24穴培養プレート)にβアミロイドに特異的な単鎖抗体の遺伝子を導入したiPS-MPを播種した。細胞が培養プレートに付着した後、βアミロイドをコーティングした、あるいは、βアミロイドをコーティングしていない蛍光ビーズ(直径0.2μm)を加え、37℃にて16時間培養した。
【0085】
図8に示すように、蛍光顕微鏡を用いてiPS-MPが蛍光ビーズを貪食していることを確認した。この図の左側の写真は、位相差レンズを用いて撮影した明視野像であり、iPS-MPの形態を示す。この図の右側の写真は、蛍光撮影像であり、蛍光ビーズの局在が示され、iPS-MPの細胞内に蛍光ビーズがとりこまれていることが示されている。
【0086】
培養プレートからiPS-MPを回収し、フローサイトメーターによりiPS-MPが貪食したビーズの量を定量化した。まず、培養プレートのウェルの中に存在するiPS-MPを全て回収した。次に、強く付着している細胞を回収する目的で、0.25%トリプシン/1mMEDTA溶液を加えて30分間保温した後、ピペットを用いて細胞を回収した。その後、フローサイトメーターを用いて、iPS-MPの蛍光強度、すなわち、iPS-MP1個あたりの蛍光ビーズの貪食量を定量化した。
【0087】
図9に、フローサイトメーターによる解析結果を示す。左側のパネルは、βアミロイドをコーティングしていないビーズを加えたiPS-MP、右側のパネルは、βアミロイドをコーティングしたビーズを加えたiPS-MPの解析結果を示す。アミロイドをコーティングしていないビーズよりも、βアミロイドをコーティングしたビーズを加えた場合の方が、iPS-MPの蛍光強度が強い、すなわち、1個のiPS-MPあたりより多くのビーズを貪食している。この結果から、βアミロイドに特異的な単鎖抗体を発現するiPS-MPは、βアミロイドをコーティングしていないビーズよりも、βアミロイドをコーティングしたビーズをより効率良く貪食することが示された。
【0088】
図10は、iPS-MPに加えるビーズの濃度を変化させた場合のiPS-MPの蛍光強度の平均値をグラフにしたものである。白丸は、βアミロイドをコーティングしていないビーズを加えた場合、黒丸は、βアミロイドをコーティングしたビーズを加えた場合を示す。グラフの横軸は、iPS-MPに加えたビーズの濃度、縦軸は、回収後の細胞の蛍光強度を示す。この図に示された結果から、加えるビーズの濃度に依存してiPS-MPが貪食するビーズの量は変化することが示された。そして、グラフに示す相対的濃度10以上のいずれのビーズの濃度においても、βアミロイドに特異的な単鎖抗体を発現するiPS-MPは、βアミロイドをコーティングしていないビーズよりも、βアミロイドをコーティングしたビーズをより効率良く貪食した。
【0089】
実施例8:
ヒトのCD20分子に対する単鎖抗体を発現するiPS-MPによる、CD20分子を発現するヒトBリンパ球性白血病細胞株BALL-1に対するin vitroでの効果を検討した。細胞培養容器(96穴平底培養プレート)にCD20に特異的な単鎖抗体の遺伝子を導入した、あるいは、単鎖抗体の遺伝子を導入していないiPS-MP(2.5 x 104個/ウェル)を播種した。翌日、遺伝子導入により発光タンパク質である蛍由来のルシフェラーゼを発現させたBALL-1細胞(2.5 x 103個/ウェル)を加え培養した。培養液は、RPMI-1640 medium / 10%牛血清 / M-CSF (50 ng/ml) / GM-CSF (100 ng/ml) / インターフェロンガンマ (500単位/ml)を用いた。
【0090】
さらに3日間した後、各々のウェルをピペッティング操作によりよく撹拌した後、浮遊した細胞を含む培養液0.1mlを発光測定用プレート(B&W Isoplate、 Wallac社)へ回収した。さらに、ルシフェラーゼ基質液(Steadyliteplus, PerkinElmer社)を加え、ルミノメーター(Tristar LB941, Berthold Technologies)を用いてルシフェラーゼの活性すなわち、サンプル中に含まれるBALL-1細胞の数を測定した。
【0091】
図11に、実験結果を示す。BALL-1細胞のみを培養した場合に比べ、あるいは、単鎖抗体を発現しないiPS-MPを加えて培養した場合に比較し、CD20に特異的な単鎖抗体を発現するiPS-MPを加えて培養した場合、ルシフェラーゼの活性は、1/100以下となった。この結果より、CD20に特異的な単鎖抗体を発現するiPS-MPは、培養容器内においてBALL-1細胞の増殖を抑制、あるいは、BALL-1細胞を死滅させる効果を有することが示された。
【0092】
実施例9:
ヒトのCD20分子に対する単鎖抗体を発現するiPS-MPによる、CD20分子を発現するヒトBリンパ球性白血病細胞株BALL-1に対する生体内での効果を検討した。Tリンパ球およびBリンパ球を欠損する重症複合型免疫不全マウス(scidマウス)の腹空内に、ルシフェラーゼを発現するBALL-1細胞(5 x 106個/マウス)とインターフェロンガンマ (2万単位/マウス)を注射した。さらに、一群のマウスには、これらに加えて、単鎖抗体を発現しないiPS-MP(2 x 107個/マウス)を注射した。さらに、一群のマウスには、これらに加えて、CD20に特異的な単鎖抗体を発現するiPS-MP(2 x 107個/マウス)を注射した。3日間経過した後、マウスをと殺し、大網組織を摘出した。そして、大網組織の抽出液のルシフェラーゼの活性を測定することにより、大網組織内に存在するBALL-1細胞の数を測定した。
【0093】
図12に、実験結果を示す。BALL-1細胞のみを注射したグループ、あるいは、それに加えて単鎖抗体を発現しないiPS-MPを加えて培養したグループに比較し、CD20に特異的な単鎖抗体を発現するiPS-MPを加えて培養したグループでは大網組織のルシフェラーゼ活性が1/100以下となった。この結果より、CD20に特異的な単鎖抗体を発現するiPS-MPは、免疫不全状態のマウスの腹空内組織においてBALL-1細胞の増殖を抑制、あるいは、BALL-1細胞を死滅させる効果を有することが示された。
【技術分野】
【0001】
本発明は、人工多能性幹細胞を用いた細胞医薬の製造方法に関する。より詳細には、本発明は、人工多能性幹細胞に遺伝的改変を施した後、生体外での分化誘導により貪食能を有する血球系の細胞(マクロファージ)を製造する方法に関する。本発明の方法により製造される細胞医薬(マクロファージ)は、遺伝子導入により特定の分子に対する抗体等を発現するものであり、標的となる分子を特異的に認識し、標的分子の貪食・分解、あるいは標的分子を発現する細胞を特異的に貪食・傷害する等の機能を発揮する。本発明の細胞医薬は、アルツハイマー病、がん、プリオン病、アミロイドーシス等の疾患の治療において有用であると期待される。
【背景技術】
【0002】
人工多能性幹細胞(誘導多能性幹細胞)(induced pluripotent stem cell; iPS細胞)とは、体細胞を初期化することによって得られる多能性を有する細胞である。人工多能性幹細胞の作製は、京都大学の山中伸弥教授らのグループ、マサチューセッツ工科大学のルドルフ・ヤニッシュ(Rudolf Jaenisch)らのグループ、ウイスコンシン大学のジェームズ・トムソン(James Thomson)らのグループ、ハーバード大学のコンラッド・ホッケドリンガー(Konrad Hochedlinger)らのグループなどを含む複数のグループが成功している。人工多能性幹細胞は、拒絶反応や倫理的問題のない理想的な多能性細胞として大きな期待を集めている。例えば、国際公開WO2007/069666号公報には、Octファミリー遺伝子、Klfファミリー遺伝子、及びMycファミリー遺伝子の遺伝子産物を含む体細胞の核初期化因子、並びにOctファミリー遺伝子、Klfファミリー遺伝子、Soxファミリー遺伝子及びMycファミリー遺伝子の遺伝子産物を含む体細胞の核初期化因子が記載されており、さらに体細胞に上記核初期化因子を接触させる工程を含む、体細胞の核初期化により誘導多能性幹細胞を製造する方法が記載されている。
【0003】
人工多能性幹細胞は、健康人・患者を問わず、ヒトの皮膚線維芽細胞や血液細胞などの体細胞を材料として作製することが可能である。人工多能性幹細胞は、胚性幹細胞(ES細胞)と同様に、あらゆる細胞に分化する潜在的能力を秘めた細胞であり、かつ、適切な条件の下で培養することにより無尽蔵に増殖させることが可能な細胞である。さらに、電気穿孔法などの方法を用いることにより、容易に遺伝子導入を施すことが可能な細胞である。一方で、人工多能性幹細胞の作製には、ヒト胚を必要としないため、胚性幹細胞の使用に際して障害となる倫理的な問題を回避できる。今日、人工多能性幹細胞を作製する新たな方法の開発が進んでおり、今後、より効率の良い、かつ、使用に際して危険性の低い人工多能性幹細胞の作製技術の開発が期待される。
【0004】
アルツハイマー病は、脳内の神経細胞に進行性の機能的障害と脱落が生じる疾患であり、多くの認知症症例の原因疾患である。また、加齢に伴って発症リスクが上昇する疾患であり、高齢化が進む各国において、社会的・医療経済的にも今後さらに深刻な問題になることが予想される。今日、この疾患の進行の抑制を期待できる有効な治療法はなく、疾患発症機序の解明に基づく治療法の開発が急務である。アルツハイマー病の剖検例において脳組織に認められる老人斑は、アミロイド前駆タンパク(APP)の限定分解によって生じたβアミロイドペプチド(Aβ1-42)が重合したものを主な成分としている。本疾患の発症機序として、これまでに得られた数々の知見から、オリゴマーを形成したβアミロイドペプチド、あるいは多数のβアミロイドペプチドが重合して不溶化したものが神経細胞の機能的障害・脱落を引き起こす原因物質であるとする説が最も有力である。加齢に伴って疾患発症のリスクが上昇する理由の一つとして、βアミロイドの分解を担っているNeprilysin等のタンパク質分解酵素の局所における発現レベルが加齢と共に低下し、産生と分解のバランスが崩れることが挙げられる。このような疾患発症のメカニズムから、何らかの方法によりβアミロイドペプチドを分解あるいは除去できれば、この疾患に対する有力な治療法になるものと期待されている。βアミロイドを分解あるいはその産生を抑制する薬剤の開発については、精力的な研究が行われているにもかかわらず、現時点では効果が確認されているものはない。
【0005】
免疫反応を介してβアミロイドペプチドを除去する治療法も検討されており、アルツハイマー病モデルマウスを用いた実験では、βアミロイドペプチドをワクチンとして投与し、これに対する免疫応答を誘導することにより、βアミロイドペプチドの蓄積と神経学的症状を抑制できることが示された。この結果に基づき、ワクチン療法の臨床試験が実施されたが、一部の症例で脳炎が惹起されたため試験は中止となった。最近、臨床試験の中止以前にワクチンを投与された症例の追跡調査の結果が報告されたが、このようなワクチン投与例においてもワクチンによる有意な臨床的効果は認められていない。アルツハイマー病モデルマウスを用いた実験において、βアミロイドペプチドに対するモノクローナル抗体を投与することにより、疾患発症を抑制できるとの報告がなされた。この結果に基づき、βアミロイドペプチドに対するモノクローナル抗体を患者に投与する臨床試験が進められているが、現時点では有意な臨床的効果は認められていない。
【0006】
また、アルツハイマー病以外の神経変性疾患においても、異常なタンパク質の凝集体が神経細胞死を引き起こすことが知られている。その例として、プリオン病、パーキンソン病、筋萎縮性側索硬化症(ALS)がある。また、ハンチントン舞踏病をはじめとするいわゆるポリグルタミン病においては、ポリグルタミンタンパク質の凝集によって神経細胞死が引き起こされる。アミロイドーシスは、アミロイドと呼ばれる異常蛋白が沈着して臓器の機能障害をおこす疾患である。アミロイドが全身性に多臓器に沈着する全身性アミロイドーシスの代表的なものとして、家族性アミロイドポリニューロパチーがある。家族性アミロイドポリニューロパチーでは、トランスサイレチン遺伝子の異常に起因して異常トランスサイレチンタンパク質が産生され蓄積することにより、末梢神経障害、自律神経障害などを含む各種臓器障害が発生する。このようなタンパク質の異常な蓄積に起因する疾患群の多くに対して、現在、有効な治療法は存在しないのが現状である。アルツハイマー病の場合と同様に、疾患発症の原因となっている異状な蓄積タンパク質を除去あるいは分解することができれば、有効な治療法になると予測される。
【0007】
また、多発性の転移巣を有する固形癌や白血病等の悪性腫瘍に対しては、外科的切除および放射線療法が有効でなく、効果のある化学療法剤が存在しない場合、治療は不可能である。昨今、がんに対する新たな治療法として、抗体療法、すなわち、がん細胞の表面に発現する分子(がん細胞表面抗原)を特異的に認識するモノクローナル抗体を投与する方法が開発されている。具体的には、乳癌細胞表面に発現するHER2に対するモノクローナル抗体を投与する治療法は、乳癌に対する標準的な治療と一つとなっている。また、悪性リンパ腫患者の70〜80%を占めるB細胞リンパ腫については、B細胞系の細胞に発現するCD20抗原に対するモノクローナル抗体を投与する抗体療法が確立されている。このような抗体療法の作用メカニズムの一つとして、Fcレセプター(抗体の定常領域であるFc部分に結合するレセプター)を高いレベルで発現する免疫細胞(マクロファージあるいはナチュラルキラー細胞)が、細胞表面に抗体が結合したがん細胞を攻撃する抗体依存性細胞性細胞傷害活性(ADCC)が知られている。抗体依存性細胞性細胞傷害活性は、体内に本来存在する免疫細胞の機能に依存しているため、体内の免疫系細胞が機能的にあるいは数的に十分であるかどうかが、治療の有効性を左右する一つの因子となる。一般に、がん患者においては、しばしば免疫系細胞の機能が低下していることが知られている。マクロファージに、がん細胞表面抗原に対する抗体の遺伝子を導入することにより、がん細胞を特異的に認識し、貪食あるいは攻撃するマクロファージを作製することができると考えられる。しかしながら、人体から分離できるマクロファージあるいはその前駆細胞であるモノサイト(単球)の数には限度があり、また、これらの細胞への人為的な遺伝子導入は容易ではない。
【0008】
他方、特許文献2には、霊長類動物の胚性幹細胞から樹状細胞への分化方法、霊長類動物胚性幹細胞から樹状細胞の製造方法、該製造方法により得られる樹状細胞、該樹状細胞、免疫応答を抗原特異的に制御することにより治療効果を得ることができる疾患の治療のための医薬の製造のための該樹状細胞の使用、及び該疾患の治療のための細胞医薬が記載されている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】国際公開WO2007/069666号公報
【特許文献2】国際公開WO2006/022330号公報
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、アルツハイマー病、プリオン病、アミロイドーシス、および、転移巣を有する進行がん、化学療法が無効な白血病等、今日、有効な治療法が存在しない疾患に対して、有効かつ危険性の低い細胞医薬を製造する方法を提供することを解決すべき課題とした。
【課題を解決するための手段】
【0011】
本発明者は上記課題を解決するために鋭意検討した結果、人工多能性幹細胞にタンパク質発現ベクターを導入した後に、該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導することによって外来タンパク質を発現する貪食細胞を製造できることを見出し、本発明を完成するに至った。
【0012】
本発明によれば、以下の発明が提供される。
(1) 人工多能性幹細胞にタンパク質発現ベクターを導入する工程;及び該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程を含む、外来タンパク質を発現する貪食細胞の製造方法。
(2) 人工多能性幹細胞が、体細胞に初期化因子を導入することにより得られる細胞である、(1)に記載の方法。
(3) 人工多能性幹細胞が、体細胞にOct3/4遺伝子、Klf4遺伝子、Sox2遺伝子、及びc-Myc遺伝子を導入することにより得られる細胞である、(1)又は(2)に記載の方法。
(4) 体細胞がヒト体細胞である、(2)又は(3)に記載の方法。
【0013】
(5) タンパク質発現ベクターにより発現されるタンパク質が、抗体、タンパク質分解酵素、又は細胞傷害因子である、(1)から(4)の何れかに記載の方法。
(6) タンパク質発現ベクターにより発現されるタンパク質が、βアミロイドペプチドに対する抗体又は腫瘍細胞の表面に発現する分子に対する抗体である、(1)から(5)の何れかに記載の方法。
(7) タンパク質発現ベクターがプラスミドベクターである、(1)から(6)の何れかに記載の方法。
(8) タンパク質発現ベクターが、薬剤耐性遺伝子を含むベクターである、(1)から(7)の何れかに記載の方法。
【0014】
(9) 該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程が、人工多能性幹細胞に由来する細胞を、顆粒球マクロファージコロニー刺激因子(GM-CSF)及び/又はマクロファージコロニー刺激因子(M-CSF)の存在下で培養することを含む、(1)から(8)の何れかに記載の方法。
【0015】
(10) 該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程が、
(A)タンパク質発現ベクターが導入された人工多能性幹細胞を、中胚葉系細胞を含む細胞群Aへ分化させる工程;及び、
(B)前記工程(A)で得られた細胞群Aを、顆粒球マクロファージコロニー刺激因子(GM-CSF)の存在下で共培養して、貪食能力を有するミエロイド系血液細胞(マクロファージ)である細胞群Bを得る工程を含む、
(1)から(9)の何れかに記載の方法。
【0016】
(11) (1)から(10)の何れかに記載の方法により製造される貪食細胞。
(12) (1)から(10)の何れかに記載の方法により製造される貪食細胞を含む、細胞医薬。
(13) 神経変性疾患又は悪性腫瘍の治療及び/又は予防のための細胞医薬である、(12)に記載の細胞医薬。
【発明の効果】
【0017】
本発明による人工多能性幹細胞の樹立および体外での人工多能性幹細胞からの分化誘導による貪食細胞(マクロファージ)の作製は、担がん状態における免疫担当細胞の異常、あるいは、造血能力の低下などの患者の体内環境に左右されない。また、人工多能性幹細胞は、適切な条件で培養して増殖させることにより大量に得ることができ、これを材料として貪食細胞を大量に作製することが可能であり、かつ、遺伝的改変も容易である。したがって、人工多能性幹細胞から貪食細胞(マクロファージ)を作製する技術を応用することにより、人工多能性幹細胞を材料として、アルツハイマー病やがんを含む各種疾患を治療するための有効な細胞医薬を必要に応じて大量に、かつ、安定的に製造することが可能になる。
【図面の簡単な説明】
【0018】
【図1】図1は、本発明による人工多能性幹細胞から各種疾患を治療するための細胞医薬を作製する方法の概要を示す。人工多能性幹細胞は、患者自身の体細胞に由来するものを用いる方法でもよいし、また、将来において設立されるであろう各種のHLA(ヒト白血球組織適合抗原)型をカバーする人工多能性幹細胞バンクから適切な細胞を選択して用いることも可能である。
【図2】図2は、βアミロイドタンパクを認識する単鎖抗体可変領域(scFv)に相当する塩基配列を示す。リーダーペプチド部分を含む、βアミロイドタンパクを認識する単鎖抗体可変領域(scFv)の塩基配列を示す。重鎖および軽鎖の配列は、マウスハイブリドーマクローン3D6の配列に由来する。重鎖と軽鎖の間には、リンカー配列を介在させている。また、軽鎖の下流には、リンカー配列に加えて、遺伝子導入を行った細胞においてフローサイトメトリー解析によりベクターに由来する分子の発現を確認する目的で、cMyc-Tag配列を付加している。
【図3】図3は、免疫グロブリンFc部およびマウスFcレセプター細胞膜貫通-細胞質部位塩基配列を示す。図2に示した塩基配列によってコードされるタンパク質のアミノ酸配列を示す。
【図4】図4は、実施例におけるヒトiPS細胞の作製と遺伝子改変ならびにiPS-MP作製と解析の概略を示す。
【図5】図5は、ヒトiPS細胞に由来するマクロファージ(iPS-MP)の位相差顕微鏡画像を示す。図5Aは、scFv遺伝子を導入していないiPS-MPの形態を示す。図5Bは、Cre発現ベクターの導入により初期化因子であるc-mycおよびSox2遺伝子を除去した後にβアミロイドタンパクに対する単鎖抗体(scFv)の遺伝子を導入したiPS-MPの形態を示す。
【図6】図6は、iPS-MPの細胞表面分子の発現をフローサイトメトリーにより解析した結果を示す。図6Aは、scFv遺伝子を導入していないヒトiPS細胞由来のiPS-MPの細胞表面上のマーカー分子の発現を示す。図6Bは、βアミロイドタンパクに対するscFv遺伝子を導入したヒト人工多能性幹細胞に由来するiPS-MPの細胞表面上のマーカー分子の発現を示す。蛍光色素で標識したマクロファージマーカー分子(CD11b, CD14, CD68)に対するモノクローナル抗体を用いてiPS-MPを染色した後に、フローサイトメトリーによる解析を行った。ヒストグラムの横軸が蛍光の強度を縦軸が細胞数を示す。各々の分子に特異的なモノクローナル抗体で染色した結果(太い実線)とそれらの抗体と同じアイソタイプを有する無関係の抗体(アイソタイプ対照抗体)による染色の結果(細い点線)を示している。
【図7】図7は、βアミロイドタンパクに対する単鎖抗体を発現するiPS-MPによるβアミロイドをコートした蛍光粒子の貪食を示す。ヒトβアミロイドタンパクに対する単鎖抗体を発現するiPS-MPを、βアミロイドをコーティングした(右:ヒトβアミロイドタンパクとアルブミンをコート)、あるいはしていない(左:アルブミンのみコート)蛍光粒子と反応させ、iPS-MPによる蛍光粒子の貪食の程度をフローサイトメトリーにより解析した結果を示す。蛍光粒子にβアミロイドをコーティングすることにより、より多くの細胞がビーズを貪食(25.5→60.3%)し、かつ、細胞あたりに貪食するビーズ数が多くなっている(平均蛍光強度 781→1348)ことがわかる。このようなβアミロイドに特異的な貪食活性の上昇は、 βアミロイドに特異的な単鎖抗体を発現させたiPS-MPにおいてのみ認められた。
【図8】図8は、βアミロイドをコートした蛍光ビーズを貪食したβアミロイドに対する単鎖抗体を発現するiPS-MPの顕微鏡はゾウを示す。左側の写真は、位相差レンズを用いて撮影した明視野像であり、iPS-MPの形態を示す。右側の写真は、蛍光撮影像であり、蛍光ビーズの局在が示され、iPS-MPの細胞内に蛍光ビーズがとりこまれていることが示されている。
【図9】図9は、βアミロイドに対する単鎖抗体を発現するiPS-MPによるβアミロイドをコートしたビーズの特異的貪食(フローサイトメトリーによる解析)を示す。左側のパネルは、βアミロイドをコーティングしていないビーズを加えたiPS-MP、右側のパネルは、βアミロイドをコーティングしたビーズを加えたiPS-MPの解析結果を示す。
【図10】図10は、付着状態における遺伝的改変iPS-MPによるβアミロイドコートビーズの特異的貪食(フローサイトメトリーによる解析)を示し、iPS-MPに加えるビーズの濃度を変化させた場合のiPS-MPの蛍光強度の平均値をグラフにしたものである。白丸は、βアミロイドをコーティングしていないビーズを加えた場合、黒丸は、βアミロイドをコーティングしたビーズを加えた場合を示す。グラフの横軸は、iPS-MPに加えたビーズの濃度、縦軸は、回収後の細胞の蛍光強度を示す。
【図11】図11は、CD20に対する抗体を発現するiPS-MPによるヒト白血病細胞株BALL-1に対する抑制効果を示す。BALL-1細胞のみを培養した場合に比べ、あるいは、これにCD20に特異的な単鎖抗体を発現しないiPS-MPを加えて培養した場合に比較し、CD20に特異的な単鎖抗体を発現するiPS-MPを加えて培養した場合、ルシフェラーゼの活性は、1/100以下となった。
【図12】図12は、CD20に対する抗体を発現するiPS-MPによるヒト白血病細胞株BALL-1に対する効果(scidマウス体内への移植実験における解析)を示す。BALL-1細胞のみを注射したグループ、あるいは、それに加えてCD20に特異的な単鎖抗体を発現しないiPS-MPを加えて培養したグループに比較し、CD20に特異的な単鎖抗体を発現するiPS-MPを加えて培養したグループでは大網組織のルシフェラーゼ活性が1/100以下となった。
【発明を実施するための形態】
【0019】
以下、本発明について更に詳細に説明する。
本明細書の以下に示す実施例においては、人工多能性幹細胞に特定の分子に対する抗体の遺伝子を導入し、引き続いて、生体外での分化誘導によりマクロファージ系の貪食細胞を製造した。製造される貪食細胞は、特定の分子に対する抗体等を発現するものであり、標的となる分子を特異的に認識し貪食する活性を有している。特異的抗体に加えて、タンパク質分解酵素などを同時に発現させることにより、高い効率で標的となる分子を貪食し分解する、あるいは標的分子を発現する細胞を特異的に貪食・傷害する等の機能を発揮する貪食細胞(マクロファージ)を作製することができる。図1に、本発明による人工多能性幹細胞から細胞医薬を製造する方法の概要を示している。
【0020】
(1)人工多能性幹細胞
本発明で用いる人工多能性幹細胞は、体細胞を初期化することにより製造することができる。ここで用いる体細胞の種類は特に限定されず、任意の体細胞を用いることができる。即ち、本発明で言う体細胞とは、生体を構成する細胞のうち生殖細胞以外の全ての細胞を包含し、分化した体細胞でもよいし、未分化の幹細胞でもよい。体細胞の由来は、哺乳動物、鳥類、魚類、爬虫類、両生類の何れでもよく特に限定されないが、好ましくは哺乳動物(例えば、マウスなどのげっ歯類、またはヒトなどの霊長類)であり、特に好ましくはヒトである。また、ヒトの体細胞を用いる場合、胎児、新生児又は成人の何れの体細胞を用いてもよい。本発明の方法で製造される人工多能性幹細胞を再生医療など疾患の治療に用いる場合には、該疾患を患う患者自身から分離した体細胞を用いることが好ましい。
【0021】
本発明で言う人工多能性幹細胞とは、所定の培養条件下(例えば、ES細胞を培養する条件下)において長期にわたって自己複製能を有し、また所定の分化誘導条件下において外胚葉、中胚葉及び内胚葉への多分化能を有する幹細胞のことを言う。また、本発明における人工多能性幹細胞はマウスなどの試験動物に移植した場合にテラトーマを形成する能力を有する幹細胞でもよい。
【0022】
体細胞から人工多能性幹細胞を製造するためには、まず、少なくとも1種類以上の初期化遺伝子を体細胞に導入する。初期化遺伝子とは、体細胞を初期化して人工多能性幹細胞とする作用を有する初期化因子をコードする遺伝子である。本発明では、少なくとも1種類以上の初期化遺伝子を使用する。本発明で使用する初期化遺伝子の組み合わせの具体例としては、以下の組み合わせをあげることができるが、これらに限定されるものではない。
(i)Oct遺伝子、Klf遺伝子、Sox遺伝子、Myc遺伝子
(ii)Oct遺伝子、Sox遺伝子、NANOG遺伝子、LIN28遺伝子
(iii)Oct遺伝子、Klf遺伝子、Sox遺伝子、Myc遺伝子、hTERT遺伝子、SV40 large T遺伝子
(iv)Oct遺伝子、Klf遺伝子、Sox遺伝子
【0023】
Oct遺伝子、Klf遺伝子、Sox遺伝子及びMyc遺伝子にはそれぞれ、複数のファミリー遺伝子が含まれている。それぞれのファミリー遺伝子の具体例としては、国際公開WO2007/069666号公報の明細書の第11頁から第13頁に記載されているものを用いることができる。具体的には、以下の通りである。
【0024】
Oct遺伝子に属する遺伝子の具体例としては、Oct3/4(NM_002701)、Oct1A(NM_002697)、及びOct6(NM_002699)などを挙げることができる(括弧内は、ヒト遺伝子のNCBI accession 番号を示す)。好ましくはOct3/4である。Oct3/4はPOUファミリーに属する転写因子であり、未分化マーカーとして知られており、また多能性維持に関与しているとの報告もある。
【0025】
Klf遺伝子に属する遺伝子の具体例としては、Klf1(NM_006563)、Klf2(NM_016270)、Klf4(NM_004235)、及びKlf5(NM_001730)などを挙げることができる(括弧内は、ヒト遺伝子のNCBI accession 番号を示す)。好ましくはKlf4である。Klf4(Kruppel like factor-4)は腫瘍抑制因子として報告されている。
【0026】
Sox遺伝子に属する遺伝子の具体例としては、例えば、Sox1(NM_005986)、Sox2(NM_003106)、Sox3(NM_005634)、Sox7(NM_031439)、Sox15(NM_006942)、Sox17(NM_0022454)、及びSox18(NM_018419)を挙げることができる(括弧内は、ヒト遺伝子のNCBI accession 番号を示す)。好ましくはSox2である。Sox2は初期発生過程で発現し、転写因子をコードする遺伝子である。
【0027】
Myc遺伝子に属する遺伝子の具体例としては、c-Myc(NM_002467)、N-Myc(NM_005378)、及びL-Myc(NM_005376)などを挙げることができる(括弧内は、ヒト遺伝子のNCBI accession 番号を示す)。好ましくは、c-Myc である。c-Mycは細胞の分化及び増殖に関与する転写制御因子であり、多能性維持に関与しているとの報告がある。
【0028】
上記した遺伝子は、ヒトを含む哺乳類動物において共通して存在する遺伝子であり、本発明において任意の哺乳類動物由来(例えばヒト、マウス、ラット、サルなどの哺乳類動物由来)の遺伝子を用いることができる。また、野生型の遺伝子に対して、数個(例えば1〜30個、好ましくは1〜20、より好ましくは1〜10個、さらに好ましくは1〜5個、特に好ましくは1から3個)の塩基が置換、挿入及び/又は欠失した変異遺伝子であって、野生型の遺伝子と同様の機能を有する遺伝子を使用することもできる。
【0029】
本発明では特に好ましくは、初期化遺伝子として、Oct3/4遺伝子、Klf4遺伝子、Sox2遺伝子、及びc-Myc遺伝子の組み合わせを用いることができる。
【0030】
初期化遺伝子を体細胞に導入する方法は、導入された初期化遺伝子が発現して体細胞の初期化を達成できる限り特に限定されない。例えば、少なくとも1種類以上の初期化遺伝子を含む発現ベクターを用いて該初期化遺伝子を体細胞に導入することができる。ベクターを用いて2種類以上の初期化遺伝子を体細胞に導入する場合には、一つの発現ベクターに2種類以上の初期化遺伝子を組み込んで、該発現ベクターを体細胞に導入してもよいし、1種類の初期化遺伝子を組み込んだ発現ベクターを2種類以上用意して、それらを体細胞に導入してもよい。
【0031】
発現ベクターの種類は特に限定されず、ウイルスベクターでもプラスミドベクターでもよいが、好ましくはウイルスベクターであり、特に好ましくは導入した初期化遺伝子が体細胞の染色体に組み込まれるようなウイルスベクターである。本発明で使用できるウイルスベクターとしては、レトロウィルスベクター(レンチウィルスベクターを含む)、アデノウイルスベクター、アデノ随伴ウイルスベクターなどを挙げることができる。上記の中でも好ましくはレトロウィルスベクターであり、特に好ましくはレンチウィルスベクターである。
【0032】
組み換えウイルスベクターを作製するために用いるパッケージング細胞としては、ウイルスのパッケージングに必要なタンパク質をコードする遺伝子の少なくとも1つを欠損している組換えウイルスベクタープラスミドの該欠損するタンパク質を補給できる細胞であれば任意の細胞を用いることができる。例えばヒト腎臓由来のHEK293細胞、マウス繊維芽細胞NIH3T3に基づくパッケージング細胞を用いることができる。
【0033】
組換えウイルスベクタープラスミドをパッケージング細胞に導入することで組換えウイルスベクターを生産することができる。上記パッケージング細胞への上記ウイルスベクタープラスミドの導入法は、特に限定されず、リン酸カルシウム法、リポフェクション法又はエレクトロポレーション法などの公知の遺伝子導入法で行うことができる。
【0034】
ES細胞の未分化性及び多能性を維持可能な培地は当業界で公知であり、適当な培地を組み合わせて用いることにより、本発明の人工多能性幹細胞を分離及び培養することができる。即ち、本発明の人工多能性幹細胞を培養するための培地としては、ES培地、ES培地に10ng/ml FGF-2を添加後にマウス胚性繊維芽細胞を24時間培養した上清であるMEF馴化ES培地(以下MEF馴化ES培地)などをあげることができる。本発明の人工多能性幹細胞を培養するための培地には、各種の成長因子、サイトカイン、ホルモンなど(例えば、FGF-2、TGFb-1、アクチビンA、ノギン(Nanoggin)、BDNF、NGF、NT-1、NT-2、NT-3等のヒトES細胞の増殖・維持に関与する成分)を添加してもよい。また、分離された人工多能性幹細胞の分化能及び増殖能は、ES細胞について知られている確認手段を利用することにより確認することができる。
【0035】
(2)タンパク質発現ベクターの人工多能性幹細胞への導入
本発明では、人工多能性幹細胞にタンパク質発現ベクターを導入する。タンパク質発現ベクターにより発現されるタンパク質は、特に限定はされないが、例えば、特異的な抗原分子(例えばβアミロイドタンパク、CD20分子)を認識する抗体(例えば、単鎖抗体)、標的組織への移行(例えば脳組織への移行)を促進するケモカインレセプター(CCR2等)、タンパク質分解酵素(例えばβアミロイドを分解活性を有するNeprilysin、トリプシノーゲン等)、細胞傷害因子(例えばがん細胞を障害するTRAIL等)などを挙げることができる。抗体としては、治療のターゲット対象となるタンパク質又はペプチドに対する抗体を用いることができ、例えば、アルツハイマー病におけるβアミロイドペプチド、神経変性疾患における異常なタンパク質の凝集体、癌細胞に特異的に発現するタンパク質(腫瘍抗原タンパク質)、腫瘍抗原タンパク質の一部であるペプチド等に対する抗体が挙げられる。
【0036】
タンパク質発現ベクターは、薬剤耐性遺伝子を含むことが好ましい。薬剤耐性遺伝子としては、G418、ピューロマイシン、ハイグロマイシン等の選択薬剤に対する耐性を付与する遺伝子を用いることができる。遺伝子導入後は、上記の選択薬剤を含む培養液中で培養し、遺伝子が導入された細胞を選択し、遺伝子導入細胞のクローンを単離することができる。
【0037】
タンパク質発現ベクターの導入は、慣用の方法、例えば、エレクトロポレーション法(電気穿孔法)、リポフェクション法等により行うことができる。上記の中でも、エレクトロポレーション法が望ましい。
【0038】
発現ベクターは、マクロファージを含む哺乳動物細胞において高い効率で遺伝子発現がなされるものを用いることが好ましく、例えば、プラスミドベクター、ファージベクターなどを用いることができる。発現ベクターは、必要に応じ、各種プロモーター、エンハンサー、ターミネーター等の転写促進に有効なエレメントを含有してもよい。
【0039】
(3)人工多能性幹細胞から貪食細胞への分化誘導
本発明では、生体外での分化誘導法により、貪食細胞への分化誘導を行う。
即ち、タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程は、貪食細胞への分化誘導を行うことができる限り特に限定されない。例えば、人工多能性幹細胞を、血液細胞の分化と増殖とを誘導する性質を有する細胞(フィーダー細胞)と一緒に培養する共培養法によって貪食細胞を含む血液細胞への分化誘導を行うことができるが、特にこのフィーダー細胞共培養法には限定されない。例えば、フィブロネクチン等で表面をコーティングした細胞接着性培養容器内で培養することによりフィーダー細胞を使用せず分化誘導を行うことも可能である。また、人工多能性幹細胞の細胞塊を浮遊状態で培養することにより胚様体を形成させ、しかる後に、貪食細胞への分化誘導を行ってもよい。細胞医薬として人体へ投与することを考慮すると、フィーダー細胞を使用せず分化誘導を行う方法がより好ましい。
【0040】
人工多能性幹細胞から貪食細胞への分化誘導の際に用いられる培養液としては、哺乳動物細胞の培養に適した培養液であればよく、用いられる人工多能性幹細胞の種類などに応じて適宜選択され得、例えば、αMEM、DMEM、IMDM(イスコブ改変ダルベッコ培地)等が挙げられる。
【0041】
分化誘導培養において用いる培養液は、血清含有培養液又は無血清培養液であり得る。無血清培養液としては、ヒトあるいはヒト以外の動物の血清由来の物質を全く含まないものもあるが、精製された血清由来の特定の成分(例えばアルブミンなど)が添加された培養液もこれに含まれる。血清由来の物質を全く含まない無血清培養液の場合は、遺伝子組み換えにより作製されたインスリンなどのタンパク質、脂肪酸、微量元素、界面活性剤を適宜含有するものでありうる。細胞医薬として人体へ投与することを考慮すると、無血清培養液を用いることがより好ましい。
【0042】
本発明において、人工多能性幹細胞と、前記フィーダー細胞との共培養の際、前記フィーダー細胞の作製に用いられうる培地としては、付着性の哺乳動物細胞の培養に適した培地であればよく、フィーダー細胞として用いる細胞の種類などに応じて適宜選択され得、例えば、αMEM、DMEM〔ダルベッコ改変イーグル培地(培養液)〕等が挙げられる。
【0043】
人工多能性幹細胞から貪食細胞への分化誘導の際に用いられる培養容器は、分化誘導を行うことが可能な培養容器であればよい。例えば、ゼラチン、フィブロネクチン等により表面をコーティングし、細胞接着性を増強したポリスチレン製の培養容器を用いて行なわれうるが、必ずしもこれに限定されない。例えば、無菌的に大量の細胞を調整することを意図して、細胞培養用のガス透過性バッグなどを用いることも可能である。
【0044】
前記フィーダー細胞の培養条件としては、フィーダー細胞として用いる細胞の種類などに応じて適宜設定されうる。例えば、ST2細胞、OP9細胞等の場合、0.1重量%ゼラチン溶液等でコーティングされた培養容器上、10容量%ウシ胎仔血清を添加した培地で37℃、5体積%CO2で培養する条件等が挙げられる。
【0045】
培養気相の条件は、用いられる人工多能性幹細胞の種類、培養液の組成等に応じて、適宜設定されうるが、例えば、37℃前後(特に、37℃)、5体積%CO2等の条件が挙げられる。
【0046】
人工多能性幹細胞から貪食細胞への分化誘導するためには、好ましくは、人工多能性幹細胞を、顆粒球マクロファージコロニー刺激因子(GM-CSF)あるいはマクロファージコロニー刺激因子(M-CSF)の存在下で培養することができる。培地中における顆粒球マクロファージコロニー刺激因子(GM-CSF)の含有量は、50〜200ng/mlの範囲が望ましい。
【0047】
上記方法により、貪食細胞の性質(例えば、CD11b、CD14、CD68の発現)を示す貪食細胞を得ることができる。本発明の方法により製造される貪食細胞は、抗原タンパク質を貪食する活性を有する。
【0048】
(4)本発明の貪食細胞及び細胞医薬
本発明はさらに、上記した方法により製造される貪食細胞、並びにそれを含む細胞医薬に関する。本発明の貪食細胞によれば、アルツハイマー病、プリオン病、アミロイドーシス、および、転移巣を有する進行がん、化学療法が無効な白血病等の疾患に対して、有効かつ危険性の低い細胞医薬を提供することができる。
【0049】
本発明の細胞医薬の製造の際には、本発明の貪食細胞を安定に保持しうる助剤、例えば、培地等を適宜用いてもよい。
【0050】
また、本発明によれば、被験体に前記製造方法により得られる貪食細胞の治療有効量を投与することを特徴とする、被験体における上記疾患の治療方法が提供される。
【0051】
治療有効量とは、前記製造方法により得られる貪食細胞を被験体に投与した場合に、該貪食細胞を投与していない被験体と比較して前記のような疾患に対して治療効果を得ることができる該貪食細胞の量である。具体的な治療有効量としては、貪食細胞の投与形態、投与方法、使用目的および被験体の年齢、体重、症状等によって適宜設定されればよく、一概には決定されないが、例えば、前記貪食細胞の細胞数で、ヒト(例えば成人)の1回の治療あたり、200,000〜1,000,000個/kg体重が好ましい。
【0052】
本発明の細胞医薬の投与方法としては、例えば、皮下注射、リンパ節内注射、静脈内注射、腹腔内注射、胸腔内注射あるいは悪性腫瘍の局所への直接注射などが挙げられるが、これらに限定されない。
【0053】
以下の実施例により本発明をさらに具体的に説明するが、本発明は実施例によって限定されるものではない。
【実施例】
【0054】
実施例1.:ヒトβアミロイドペプチドを認識する単鎖抗体(scFv)発現ベクターの作製
(1)可変領域塩基配列の作製
ヒトβアミロイドペプチドを認識するモノクローナル抗体(マウスハイブリドーマクローン: 3D6)の重鎖(heavy chain)および軽鎖(light chain)の可変領域の塩基配列(特許文献2007−0238154に記載)に基づき、βアミロイドペプチドを認識する単鎖抗体可変領域の塩基配列を設計した。また、図2に塩基配列を、また、図3にこの塩基配列にコードされるタンパク質のアミノ酸配列を示す。設計した塩基配列に基づき、GeneScript Corporation社(米国 ニュージャージー州)に依頼してDNAの人工合成を行い、さらに、プラスミドベクターへの挿入を行った。
【0055】
(2)免疫グロブリン(マウスIgG2a)Fc部およびマウスFcレセプター(FcγRI)細胞膜貫通-細胞質部位の作製
C57BL/6マウスの脾臓組織より抽出したRNAに由来するcDNAを鋳型として、PCR法により作製した。
【0056】
(3)βアミロイドペプチドを認識する単鎖抗体と免疫グロブリンFc部およびFcレセプター(Fcγ細胞膜貫通-細胞質部位)との融合タンパク質の発現ベクターの作製
前記の単鎖抗体可変領域のDNA断片と免疫グロブリンFc部およびマウスFcレセプター細胞膜貫通-細胞質部位のDNA断片をPCR法を用いて融合し、プラスミドベクター(pENTR-D-TOPO, Invitrogen社製)へ挿入し、大腸菌を用いてクローニングを行った。
【0057】
クローニングを行ったプラスミドDNAの塩基配列をシークエンス解析により確認した後、DNA組換え酵素LR clonase (Invitrogen社製)を用いて哺乳動物発現ベクターへ導入した。哺乳動物発現ベクターとしては、CAGプロモーターにより作動し、IRES (internal ribosomal entry site)とピューロマイシン耐性遺伝子が組込まれたベクターであるpCAGGS-IRES-Puroを用いた。
【0058】
実施例2:ヒト人工多能性幹細胞の作製および培養
健常人腹部の皮膚組織に由来する線維芽細胞に、レンチウイルスベクターを用いてヒトのOct3/4, Sox2, Klf4, c-Mycの発現ベクターを導入することにより、ヒト人工多能性幹細胞の作製を行った。
【0059】
具体的には、ヒト腹部の皮膚片を採取し、細胞培養用プレート中で培養液(DMEM / 10%牛血清)を用いて培養した。培養開始1週間目以降より、皮膚片からの線維芽細胞の遊走と増殖が認められたので、トリプシンとEDTAを用いて、適宜、線維芽細胞を回収し凍結保存した。
【0060】
初期化因子であるヒトのOct3/4, Sox2, Klf4, c-MycのcDNAは、ヒトES細胞(KhES-1)に由来するcDNA、あるいはゲノムDNAを鋳型として、PCR法により作製し、プラスミドベクター(pENTR-D-TOPO, Gibco-Invitrogen社)へ挿入した。クローニングを行ったプラスミドDNAの塩基配列をシークエンス解析により確認の後、LR clonase (Gibco-Invitrogen社)を用いて、レンチウイルスベクター(CSII-EF-RfAl, 理化学研究所 三好博士より分与)へ導入した。Sox2とc-Mycについては、CSII-EF-RfAlへ挿入後にcDNA両端へのLOX配列の付加を行った。
【0061】
Creの構造遺伝子を鋳型として、PCR法により遺伝子断片を作製し、CAGプロモーターにより作動し、IRES (internal ribosomal entry site)とネオマイシン耐性遺伝子が組込まれたベクターであるpCAGGS-IRES-Neoへ挿入し、Cre酵素とネオマイシン耐性遺伝子を同時に発現するベクターを作製した。
【0062】
リポフェクション法(Lipofectamine 2000, Invitrogen社)を用いて、上記で作製したCSII-EFに導入したリプログラミング因子遺伝子の各々とパッケージングコンストラクト、およびエンベロープおよびRevのコンストラクトを293T細胞へ遺伝子導入した。遺伝子導入3日後に、細胞培養液を回収し、0.45μmのフィルターを通した後、遠心分離法(50,000G、2時間)によりウイルス粒子を沈澱させ回収した。回収した組換えウイルス粒子は、DMEM溶液に懸濁した後、凍結チューブに分注し、使用時まで冷凍庫(-80℃)にて保存した。
【0063】
凍結保存しておいたヒト線維芽細胞を解凍し、数日間、再度培養した後、培養プレート中においてウイルス懸濁液を加えることにより、感染させ、遺伝子導入を行った。遺伝子導入の4-6日後、トリプシン-EDTAを用いて感染細胞を回収し、事前に準備しておいた、マイトマイシンC処理により増殖を停止させたマウス胎児由来線維芽細胞(フィーダー細胞)との共培養を開始した。その翌日より、培養液をヒトES細胞用の培養液に置換し、培養を継続した。
【0064】
初期化因子の遺伝子を導入してから、20-30日の後、一部の細胞については顕微鏡観察の下で、マイクロチップを用いてES細胞様の形態を示すコロニーをiPS細胞クローンとして単離し、別に準備しておいたマウス胎児由来フィーダー細胞との共培養を行った。その後、細胞の増殖に応じて、培養容器を拡大しつつ培養を継続した(図4Aの経路)。また、初期化因子の遺伝子を導入した細胞集団のうち別の一部のものについては、トリプシン-EDTAを用いて回収した後、Cre発現ベクターの導入を行った(図4Bの経路)。
【0065】
Cre発現ベクターの導入
図4Bの経路におけるCre発現ベクターの導入は、電気穿孔法により行った。Cre発現ベクターが挿入された細胞を選別する目的で、電気穿孔法施行後、24−48時間後より、培養液中にG418 (Gibco- Invitrogen社)を加え、培養を継続した。そして、電気穿孔法施行後15-25日後に、顕微鏡観察の下で、ES細胞様の形態を示すG418耐性の細胞コロニーをマイクロチップを用いて単離し、別に準備しておいたマウス胎児由来フィーダー細胞との共培養を行った。その後、細胞の増殖に応じて、培養容器を拡大しつつ培養を継続した。
【0066】
ヒトiPS細胞の維持培養は、ヒト胚性幹細胞培養液(DMEM-F12(Wako Chemicals)/20% KSR(Gibco-Invitrogen)/bFGF (basic fibroblast growth factor: 10 ng/ml)/2-ME (2-メルカプトエタノール, 50μM))を用いて、マイトマイシンC処理マウス胎児繊維芽細胞をフィーダー細胞としてポリスチレン製培養皿にて行った。
【0067】
実施例3:ヒトiPS細胞への単鎖抗体発現ベクターの導入
実施例1で作製した単鎖抗体(scFv)発現ベクター(プラスミドベクター)DNAは、大腸菌からアルカリ-SDS法で抽出した後、陰イオン交換カラム精製(Qiagen Plasmid Maxiキット, Qiagen社製)を行った。そして、制限酵素Pvu Iを用いて切断することにより直線状DNAとした。さらに、フェーノールとクロロホルムを用いた精製を行った後に、イソプロパノール沈澱により回収し、70%エタノールによる洗浄を行った上で、リン酸干渉生理的食塩水(PBS)に溶解した。
【0068】
維持培養を行っているヒトiPS細胞(図4Bの経路により作製した細胞クローン)をトリプシン-EDTA (0.25%トリプシン/1 mM EDTA)を用いて培養容器より回収し、DMEM溶液に懸濁(1x107細胞/0.4 ml)し、PBSに溶解したプラスミドベクター(30μg/0.1 ml)と混和した後、遺伝子導入装置(ジーンパルサー、BioRad社製)を用いて電気穿孔法による遺伝子導入(キュベットの電極間距離:4 mm, 通電:200V, 800μF, 1回)を行った。
【0069】
電気穿孔法施行後、細胞の生存率を高めるために、24時間はRoh結合キナーゼ阻害剤(Y-27632,和光純薬製)存在下に培養を行い、また、48時間後より培養液中にピューロマイシン (Sigma社)を加え、培養を継続した。
【0070】
その後、断続的にピューロマイシン (5-15μg/ml)を加え、培養を継続した。そして、電気穿孔法施行後15-18日後に、倒立顕微鏡による観察を行いつつ、ピューロマイシン耐性の細胞コロニーをマイクロチップを用いて単離し、別に準備しておいたマウス胎児由来フィーダー細胞との共培養を行った。
【0071】
その後、細胞の増殖に応じて、培養容器を拡大しつつ培養を継続し、一部はそのまま凍結保存し、一部は後述のようにマクロファージへの分化誘導を行った。
【0072】
実施例4:ヒト人工多能性細胞のマクロファージへの分化誘導
(1)OP9フィーダー細胞の調整
マウス由来の培養細胞株OP9をマイトマイシンCにて処理(0.01 mg/ml、60分)したものを、ゼラチンコートしたディッシュに播種し、翌日以降に使用した。
【0073】
(2)分化誘導培養
未分化なiPS細胞(図4の経路AあるいはBのもの)を、CTK液(コラゲナーゼ-トリプシン-KSR液、末盛ら Biochemical and Biophysical Research Communications 345: 926-932, 2006年)を用いて5-10分間処理し、FCS(牛胎児血清、GIBCO-Invitrogen社製)入りの培養液で回収した。細胞を遠心し、a-MEM/20%FCSに懸濁し、OP9フィーダー細胞上へ播種した。以降、3日に1度、培養液(a-MEM/20%FCS)を交換しつつ培養を継続した。
【0074】
分化誘導開始から15-18日後、トリプシン-EDTA-コラゲナーゼ液を用いて細胞を処理(37℃ 60分)して解離させて回収し、ピペッティング操作により細胞浮遊液を作製した。そして、ディッシュ1枚由来の細胞を10 ml のDMEM/10% FCSに懸濁し、フィーダー細胞なし、ゼラチンコートなしの90-mmディッシュ2枚にまいた。2-5時間後、ディッシュに付着しなかった細胞を回収し、100ミクロンのメッシュ(BD Falcon社製 セルストレイナー)を通過させることにより、細胞塊を除いた細胞集団を得た。この細胞を、a-MEM / 20%FCS / ヒト GM-CSF (100 ng/ml、ぺプロテック社製) / ヒト M-CSF (50 ng/ml、ぺプロテック社製) / 2-ME (0.05 mM)に浮遊させ、再度、OP9フィーダー細胞上へ播種し培養を行った。
【0075】
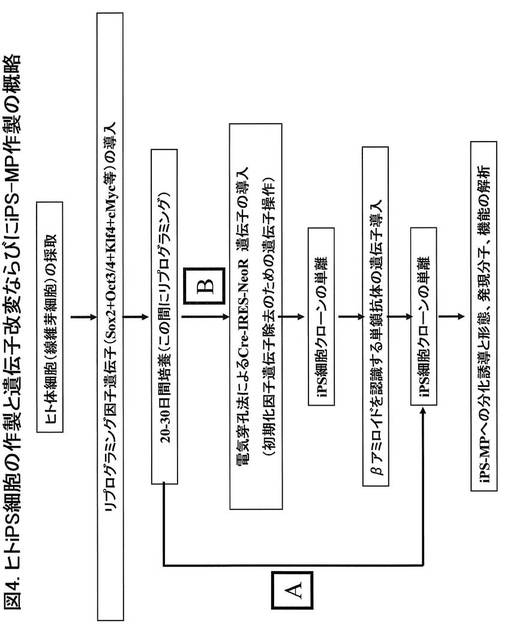
7〜9日経過した後、ピペッティング操作により細胞を回収し、RPMI-1640 / 10%FCS/ M-CSF (100 ng/ml) に懸濁して、培養皿(BD FALCON社製)へ播種して3-5日間培養を行いマクロファージ(iPS-MP)を作製した。図5は、位相差顕微鏡レンズを用いてディシュ内のiPS-MPを撮影した画像である。図5Aは、図4のAの経路、すなわち、Cre発現ベクターとscFv発現ベクターの導入を行っていないiPS細胞に由来するiPS-MPである。図5Bは、図4のBの経路、すなわち、Cre発現ベクターとscFv発現ベクターを導入したiPS細胞に由来するiPS-MPである。
【0076】
実施例5:フローサイトメトリーによるiPS-MPの細胞表面上のマーカー分子の発現の検討
上記のように作製したiPS-MPをピペッティング操作により回収し、抗体の非特異的な結合を阻害する目的で、まずFcレセプターブロック試薬(Miltenyi Biotec社製)を用いて10分間処理した。
【0077】
その後、以下のようなフィコエリスロシン(PE)結合モノクローナル抗体あるいはフルオレセインイソチオシアネート(FITC)結合モノクローナル抗体を用いて室温中で40分間染色した。PE-抗ヒト/マウスCD11b(クローンM1/70、ラットIgG2b)、PE-抗ヒトCD14(クローンTUK4、マウスIgG2a)、FITC-抗ヒトCD68(クローン Y1/82A、マウスIgG2b)。また、アイソタイプ適合対照抗体として、PE-ラットIgG2b(クローン LO-DNP-11)、PE-マウスIgG2a(クローンG155−178)とFITC-マウスIgG2b(クローン 27-35)を用いて染色した。
【0078】
その後、細胞を、PBS/2% FCSで2回洗浄した。
洗浄後の細胞について、CellQuest ソフトウェアを備えたフローサイトメーター解析装置(商品名:FACScan,Becton Dickinson社製)を用いて分析した。
【0079】
図6に抗体染色後のフローサイトメーター解析の結果を示す。図6Aは、図4のAの経路、すなわち、Cre発現ベクターとscFv発現ベクターの導入を行っていないiPS細胞に由来するiPS-MPである。図6Bは、図4のBの経路、すなわち、Cre発現ベクターとscFv発現ベクターを導入したiPS細胞に由来するiPS-MPである。いずれのiPS-MPも、細胞表面上にCD11b、CD14、CD68を発現しており、生理的に存在するヒトマクロファージと同様のマーカー分子を発現していた。
【0080】
実施例6:βアミロイドペプチドに対する単鎖抗体を発現するiPS-MPによるβアミロイドペプチドをコートした蛍光粒子の結合能の浮遊状態での解析
(1)蛍光粒子へのβアミロイドペプチドのコーティング
蛍光粒子(Sulfate Microspheres, Yellow-green, #F8828, Molecular Probes社製)にβアミロイドペプチド(Amyloid beta-Protein 1-42, Wako Chemicals社製)を添加(終濃度0.03mg/ml)し25℃にて震盪しつつ12時間反応させた。その後、0.1%の牛血清アルブミンを含有するPBS(PBS-BSA)を2倍容量加え、さらに、25℃にて震盪しつつ2時間反応させた。アルブミンのみをコーティングした蛍光粒子は、PBS-BSA中で、25℃にて震盪しつつ14時間反応させることにより作製した。
その後、蛍光粒子をPBSで2回洗浄した後、実験に供するまでの間、遮光して冷蔵保存した。
【0081】
(2)iPS-MPによる蛍光粒子の結合能の解析
βアミロイドに特異的な単鎖抗体の遺伝子を導入したiPS-MPを培養皿から回収し洗浄した後、βアミロイドをコーティングした蛍光粒子(直径0.2μm)とプラスチック製試験管内で混和し、37℃にて90分静置した。
その後、PBS-FCS で2回洗浄した後、フローサイトメーターを用いて、iPS-MPの蛍光強度、すなわち、蛍光ビーズの貪食を定量化した。
【0082】
図7に、フローサイトメトリーによる解析の結果を示す。蛍光ビーズにβアミロイドをコーティングすることにより、より多くの細胞がビーズを結合(25.5→60.3%)し、かつ、細胞あたりに貪食するビーズ数が多くなっている(平均蛍光強度 781→1348)。
【0083】
実施例7:βアミロイドペプチドに対する単鎖抗体を発現するiPS-MPによる、付着培養状態でのβアミロイドペプチドをコートした蛍光粒子の貪食能の解析
(1)蛍光粒子へのβアミロイドペプチドのコーティング
蛍光粒子(Sulfate Microspheres, Yellow-green, #F8828, Molecular Probes社製)にβアミロイドペプチド(Amyloid beta-Protein 1-42, Wako Chemicals社製)を添加(終濃度0.03mg/ml)し25℃にて震盪しつつ24時間反応させた。その後、2%の牛血清アルブミンを含有するPBS(PBS-BSA)を等量加え、さらに、25℃にて震盪しつつ24時間反応させた。対照実験に用いた、アルブミンのみをコーティングした蛍光粒子は、1%PBS-BSA中で、25℃にて震盪しつつ48時間反応させることにより作製した。
その後、蛍光粒子をPBSで2回洗浄した後、実験に供するまでの間、遮光して冷蔵保存した。
【0084】
(2)iPS-MPによる蛍光粒子の貪食能の解析
細胞培養容器(24穴培養プレート)にβアミロイドに特異的な単鎖抗体の遺伝子を導入したiPS-MPを播種した。細胞が培養プレートに付着した後、βアミロイドをコーティングした、あるいは、βアミロイドをコーティングしていない蛍光ビーズ(直径0.2μm)を加え、37℃にて16時間培養した。
【0085】
図8に示すように、蛍光顕微鏡を用いてiPS-MPが蛍光ビーズを貪食していることを確認した。この図の左側の写真は、位相差レンズを用いて撮影した明視野像であり、iPS-MPの形態を示す。この図の右側の写真は、蛍光撮影像であり、蛍光ビーズの局在が示され、iPS-MPの細胞内に蛍光ビーズがとりこまれていることが示されている。
【0086】
培養プレートからiPS-MPを回収し、フローサイトメーターによりiPS-MPが貪食したビーズの量を定量化した。まず、培養プレートのウェルの中に存在するiPS-MPを全て回収した。次に、強く付着している細胞を回収する目的で、0.25%トリプシン/1mMEDTA溶液を加えて30分間保温した後、ピペットを用いて細胞を回収した。その後、フローサイトメーターを用いて、iPS-MPの蛍光強度、すなわち、iPS-MP1個あたりの蛍光ビーズの貪食量を定量化した。
【0087】
図9に、フローサイトメーターによる解析結果を示す。左側のパネルは、βアミロイドをコーティングしていないビーズを加えたiPS-MP、右側のパネルは、βアミロイドをコーティングしたビーズを加えたiPS-MPの解析結果を示す。アミロイドをコーティングしていないビーズよりも、βアミロイドをコーティングしたビーズを加えた場合の方が、iPS-MPの蛍光強度が強い、すなわち、1個のiPS-MPあたりより多くのビーズを貪食している。この結果から、βアミロイドに特異的な単鎖抗体を発現するiPS-MPは、βアミロイドをコーティングしていないビーズよりも、βアミロイドをコーティングしたビーズをより効率良く貪食することが示された。
【0088】
図10は、iPS-MPに加えるビーズの濃度を変化させた場合のiPS-MPの蛍光強度の平均値をグラフにしたものである。白丸は、βアミロイドをコーティングしていないビーズを加えた場合、黒丸は、βアミロイドをコーティングしたビーズを加えた場合を示す。グラフの横軸は、iPS-MPに加えたビーズの濃度、縦軸は、回収後の細胞の蛍光強度を示す。この図に示された結果から、加えるビーズの濃度に依存してiPS-MPが貪食するビーズの量は変化することが示された。そして、グラフに示す相対的濃度10以上のいずれのビーズの濃度においても、βアミロイドに特異的な単鎖抗体を発現するiPS-MPは、βアミロイドをコーティングしていないビーズよりも、βアミロイドをコーティングしたビーズをより効率良く貪食した。
【0089】
実施例8:
ヒトのCD20分子に対する単鎖抗体を発現するiPS-MPによる、CD20分子を発現するヒトBリンパ球性白血病細胞株BALL-1に対するin vitroでの効果を検討した。細胞培養容器(96穴平底培養プレート)にCD20に特異的な単鎖抗体の遺伝子を導入した、あるいは、単鎖抗体の遺伝子を導入していないiPS-MP(2.5 x 104個/ウェル)を播種した。翌日、遺伝子導入により発光タンパク質である蛍由来のルシフェラーゼを発現させたBALL-1細胞(2.5 x 103個/ウェル)を加え培養した。培養液は、RPMI-1640 medium / 10%牛血清 / M-CSF (50 ng/ml) / GM-CSF (100 ng/ml) / インターフェロンガンマ (500単位/ml)を用いた。
【0090】
さらに3日間した後、各々のウェルをピペッティング操作によりよく撹拌した後、浮遊した細胞を含む培養液0.1mlを発光測定用プレート(B&W Isoplate、 Wallac社)へ回収した。さらに、ルシフェラーゼ基質液(Steadyliteplus, PerkinElmer社)を加え、ルミノメーター(Tristar LB941, Berthold Technologies)を用いてルシフェラーゼの活性すなわち、サンプル中に含まれるBALL-1細胞の数を測定した。
【0091】
図11に、実験結果を示す。BALL-1細胞のみを培養した場合に比べ、あるいは、単鎖抗体を発現しないiPS-MPを加えて培養した場合に比較し、CD20に特異的な単鎖抗体を発現するiPS-MPを加えて培養した場合、ルシフェラーゼの活性は、1/100以下となった。この結果より、CD20に特異的な単鎖抗体を発現するiPS-MPは、培養容器内においてBALL-1細胞の増殖を抑制、あるいは、BALL-1細胞を死滅させる効果を有することが示された。
【0092】
実施例9:
ヒトのCD20分子に対する単鎖抗体を発現するiPS-MPによる、CD20分子を発現するヒトBリンパ球性白血病細胞株BALL-1に対する生体内での効果を検討した。Tリンパ球およびBリンパ球を欠損する重症複合型免疫不全マウス(scidマウス)の腹空内に、ルシフェラーゼを発現するBALL-1細胞(5 x 106個/マウス)とインターフェロンガンマ (2万単位/マウス)を注射した。さらに、一群のマウスには、これらに加えて、単鎖抗体を発現しないiPS-MP(2 x 107個/マウス)を注射した。さらに、一群のマウスには、これらに加えて、CD20に特異的な単鎖抗体を発現するiPS-MP(2 x 107個/マウス)を注射した。3日間経過した後、マウスをと殺し、大網組織を摘出した。そして、大網組織の抽出液のルシフェラーゼの活性を測定することにより、大網組織内に存在するBALL-1細胞の数を測定した。
【0093】
図12に、実験結果を示す。BALL-1細胞のみを注射したグループ、あるいは、それに加えて単鎖抗体を発現しないiPS-MPを加えて培養したグループに比較し、CD20に特異的な単鎖抗体を発現するiPS-MPを加えて培養したグループでは大網組織のルシフェラーゼ活性が1/100以下となった。この結果より、CD20に特異的な単鎖抗体を発現するiPS-MPは、免疫不全状態のマウスの腹空内組織においてBALL-1細胞の増殖を抑制、あるいは、BALL-1細胞を死滅させる効果を有することが示された。
【特許請求の範囲】
【請求項1】
人工多能性幹細胞にタンパク質発現ベクターを導入する工程;及び該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程を含む、外来タンパク質を発現する貪食細胞の製造方法。
【請求項2】
人工多能性幹細胞が、体細胞に初期化因子を導入することにより得られる細胞である、請求項1に記載の方法。
【請求項3】
人工多能性幹細胞が、体細胞にOct3/4遺伝子、Klf4遺伝子、Sox2遺伝子、及びc-Myc遺伝子を導入することにより得られる細胞である、請求項1又は2に記載の方法。
【請求項4】
体細胞がヒト体細胞である、請求項2又は3に記載の方法。
【請求項5】
タンパク質発現ベクターにより発現されるタンパク質が、抗体、タンパク質分解酵素、又は細胞傷害因子である、請求項1から4の何れかに記載の方法。
【請求項6】
タンパク質発現ベクターにより発現されるタンパク質が、βアミロイドペプチドに対する抗体又は腫瘍細胞の表面に発現する分子に対する抗体である、請求項1から5の何れかに記載の方法。
【請求項7】
タンパク質発現ベクターがプラスミドベクターである、請求項1から6の何れかに記載の方法。
【請求項8】
タンパク質発現ベクターが、薬剤耐性遺伝子を含むベクターである、請求項1から7の何れかに記載の方法。
【請求項9】
該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程が、人工多能性幹細胞に由来する細胞を、顆粒球マクロファージコロニー刺激因子(GM-CSF)及び/又はマクロファージコロニー刺激因子(M-CSF)の存在下で培養することを含む、請求項1から8の何れかに記載の方法。
【請求項10】
該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程が、
(A)タンパク質発現ベクターが導入された人工多能性幹細胞を、中胚葉系細胞を含む細胞群Aへ分化させる工程;及び、
(B)前記工程(A)で得られた細胞群Aを、顆粒球マクロファージコロニー刺激因子(GM-CSF)の存在下で共培養して、貪食能力を有するミエロイド系血液細胞(マクロファージ)である細胞群Bを得る工程を含む、
(1)から(9)の何れかに記載の方法。
【請求項11】
請求項1から10の何れかに記載の方法により製造される貪食細胞。
【請求項12】
請求項1から10の何れかに記載の方法により製造される貪食細胞を含む、細胞医薬。
【請求項13】
神経変性疾患又は悪性腫瘍の治療及び/又は予防のための細胞医薬である、請求項12に記載の細胞医薬。
【請求項1】
人工多能性幹細胞にタンパク質発現ベクターを導入する工程;及び該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程を含む、外来タンパク質を発現する貪食細胞の製造方法。
【請求項2】
人工多能性幹細胞が、体細胞に初期化因子を導入することにより得られる細胞である、請求項1に記載の方法。
【請求項3】
人工多能性幹細胞が、体細胞にOct3/4遺伝子、Klf4遺伝子、Sox2遺伝子、及びc-Myc遺伝子を導入することにより得られる細胞である、請求項1又は2に記載の方法。
【請求項4】
体細胞がヒト体細胞である、請求項2又は3に記載の方法。
【請求項5】
タンパク質発現ベクターにより発現されるタンパク質が、抗体、タンパク質分解酵素、又は細胞傷害因子である、請求項1から4の何れかに記載の方法。
【請求項6】
タンパク質発現ベクターにより発現されるタンパク質が、βアミロイドペプチドに対する抗体又は腫瘍細胞の表面に発現する分子に対する抗体である、請求項1から5の何れかに記載の方法。
【請求項7】
タンパク質発現ベクターがプラスミドベクターである、請求項1から6の何れかに記載の方法。
【請求項8】
タンパク質発現ベクターが、薬剤耐性遺伝子を含むベクターである、請求項1から7の何れかに記載の方法。
【請求項9】
該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程が、人工多能性幹細胞に由来する細胞を、顆粒球マクロファージコロニー刺激因子(GM-CSF)及び/又はマクロファージコロニー刺激因子(M-CSF)の存在下で培養することを含む、請求項1から8の何れかに記載の方法。
【請求項10】
該タンパク質発現ベクターが導入された人工多能性幹細胞を貪食細胞に分化誘導する工程が、
(A)タンパク質発現ベクターが導入された人工多能性幹細胞を、中胚葉系細胞を含む細胞群Aへ分化させる工程;及び、
(B)前記工程(A)で得られた細胞群Aを、顆粒球マクロファージコロニー刺激因子(GM-CSF)の存在下で共培養して、貪食能力を有するミエロイド系血液細胞(マクロファージ)である細胞群Bを得る工程を含む、
(1)から(9)の何れかに記載の方法。
【請求項11】
請求項1から10の何れかに記載の方法により製造される貪食細胞。
【請求項12】
請求項1から10の何れかに記載の方法により製造される貪食細胞を含む、細胞医薬。
【請求項13】
神経変性疾患又は悪性腫瘍の治療及び/又は予防のための細胞医薬である、請求項12に記載の細胞医薬。
【図1】


【図2】

【図3】

【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2010−268789(P2010−268789A)
【公開日】平成22年12月2日(2010.12.2)
【国際特許分類】
【出願番号】特願2009−290152(P2009−290152)
【出願日】平成21年12月22日(2009.12.22)
【出願人】(504159235)国立大学法人 熊本大学 (314)
【Fターム(参考)】
【公開日】平成22年12月2日(2010.12.2)
【国際特許分類】
【出願日】平成21年12月22日(2009.12.22)
【出願人】(504159235)国立大学法人 熊本大学 (314)
【Fターム(参考)】
[ Back to top ]