
細胞培養方法
【課題】マルトース結合タンパク質(MBP)を使用した生理活性ポリペプチドの固定化方法、および前記方法によって生理活性ポリペプチドが固定された生物活性固体基質を提供する。
【解決手段】マルトース結合タンパク質(MBP)のカルボキシル末端に、生理活性ポリペプチドが融合された融合タンパク質を得る工程、および前記融合タンパク質を、マルトース結合タンパク質の表面に露出した疎水性ドメインを含むアミノ末端と固体基質の疎水性表面との物理的吸着によって、前記疎水性表面に固定させる工程を含む、生理活性ポリペプチドを固体基質に固定化させる方法。および前記方法によって生理活性ポリペプチドが固定された、生物活性固体基質を提供する。
【解決手段】マルトース結合タンパク質(MBP)のカルボキシル末端に、生理活性ポリペプチドが融合された融合タンパク質を得る工程、および前記融合タンパク質を、マルトース結合タンパク質の表面に露出した疎水性ドメインを含むアミノ末端と固体基質の疎水性表面との物理的吸着によって、前記疎水性表面に固定させる工程を含む、生理活性ポリペプチドを固体基質に固定化させる方法。および前記方法によって生理活性ポリペプチドが固定された、生物活性固体基質を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、マルトース結合タンパク質(maltose binding protein、MBP)をリンカーに使用して生理活性ポリペプチドを固体基質の疎水性表面に固定させる方法、および前記方法によって生理活性ポリペプチドが固定された生物活性固体基質に関するものである。
【背景技術】
【0002】
タンパク質の固定は、診断やバイオセンサーあるいは酵素反応器などのために非常に有用に使用されている。タンパク質の固定化方法では、グルタルアルデヒド(glutaraldehyde)のような架橋剤を使用した化学的架橋、疎水性結合を使用した物理的吸着、アミン反応性基を使用した化学的カップリングなどが知られている。このような方法の最大の問題点としては、固定させようとするタンパク質あるいはポリペプチドの活性を維持することにより化学的架橋やカップリングする方法では、新しい化学的結合が形成されたり標的タンパク質の化学的性質が変化したりすることによって活性が阻害され得ることがあり、物理的吸着方法では、標的タンパク質が直接疎水性表面に吸着される場合に、タンパク質構造の変化を伴って変性を誘発し得るという問題点がある。このような問題点を解決するために、固定させようとするポリペプチドにリンカーを結合させて、このリンカーを媒介にする固定化方法が報告されている(非特許文献1〜3)。リンカーを生理活性ポリペプチドに結合させる代表的な方法は、遺伝工学技術を使用して組換え融合タンパク質を合成するものである。
【0003】
例えば、グルタチオン−S−転移酵素(glutathione−S−transferase;GST)のような酵素を使用して、表面に酵素を認識するグルタチオン分子をあらかじめ結合させて、エリサ法(ELISA:enzyme−linked immunosorbent assay)に活用する方法が知られている(非特許文献4)。しかし前記方法は、ポリスチレンのように反応性がない疎水性表面には、グルタチオンのような物質を結合しにくいという短所がある。
【0004】
一方、疎水性ドメインを有するリンカーを使用してタンパク質の効率的な吸着と同時に変性を防いで、吸着後にも目的タンパク質の生物学的機能が維持され、それを細胞培養用表面として使用する方法が開発された。疎水性結合による融合タンパク質の固定化のためのリンカーとしては、免疫グロブリンのFcドメイン(非特許文献5、非特許文献3)、ハイドロフォビン(hydrophobin)のような疎水性が強いタンパク質などを使用する(非特許文献6)。前記方法は、吸着させようとする表面に別途の前処理をしないで、簡単に使用することができるという長所を有している。
【0005】
例えば、EGF−Fcあるいはカドヘリン(cadherin)−Fcをポリスチレン培養容器に固定化して、溶液性EGFあるいはゼラチンが吸着した表面で上皮腫瘍細胞または胚芽幹細胞を培養すると、これら細胞は生化学的および細胞生物学的に異なった挙動を示すということが報告されている(非特許文献5、非特許文献3)。しかし、このようにFcを使用する場合には、Fcのカルボキシル末端は疎水性表面との物理的吸着に必要とされ、Fcのアミノ末端に標的とする生理活性ポリペプチドを結合させなければならないので、生理活性ポリペプチドのカルボキシル末端が活性に必須な場合には使用が制限される。その他にも、生理活性ポリペプチドのカルボキシル基末端を、連結部位に使用する多様な結合方法も開発された。一例として、特許文献1は、ハイドロフォビンを使用して酵素などを疎水性表面に結合させるタンパク質固定化方法を開示している。しかし、前記で言及された疎水性結合を誘導する融合タンパク質のリンカー部分は、大部分が動物およびカビ由来のものであり、大腸菌で発現させて精製する場合は、封入体(inclusion body)の形成など多くの短所を有している。
【0006】
以上のことに鑑みて、本発明者等は、前記の従来技術の問題点を解決するために鋭意研究努力した結果、大腸菌で発現されるタンパク質として、マルトースとの優れた結合能力と発現および精製が容易であるという長所を有する、マルトース結合タンパク質を使用して生理活性ポリペプチドとの融合タンパク質を製造して、前記融合タンパク質をマルトース結合タンパク質の疎水性ドメイン(非特許文献7)を使用して、ポリスチレンのような疎水性表面に単純な物理的吸着によって固定させた後、固定された融合タンパク質の生理活性ポリペプチド部分が、生物学的活性を維持していることを確認することにより、本発明を完成した。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許公開第20040235050号
【非特許文献】
【0008】
【非特許文献1】Lee J.M.等,Anal.Chem.,2007年,第79巻,2680−2687頁
【非特許文献2】Ogiwara K.等,Biochem.Biophy.Res.Comm.,2006年,第345巻,255−259頁
【非特許文献3】Nagaoka M.等,PLoS ONE、2006年,第1巻,e15頁
【非特許文献4】Sehr P.等,J.Immuno.Methods.,2001年,第253巻,153−162頁
【非特許文献5】Ogiwara K.等,Biotech.Letters,2005年,第27巻,1633−1637頁
【非特許文献6】Qin M.等,Colloids Surfaces,2007年,第60巻,243−249頁
【非特許文献7】Fox J.D.等,Protein Science,2001年,第10巻,622−630頁
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明の目的は、大腸菌での発現および精製が容易であり、疎水性ドメインを有するマルトース結合タンパク質を使用して、特定の成長因子またはサイトカインのような生理活性ポリペプチドを、前処理過程なしに簡単な物理的吸着によって固体基質の疎水性表面に固定化する方法、および前記方法によって製造された生理活性ポリペプチドが固定された生物活性固体基質を提供することである。
【課題を解決するための手段】
【0010】
前記目的を達成するために本発明は、
1)マルトース結合タンパク質(MBP)のカルボキシル末端に、生理活性ポリペプチドが融合された融合タンパク質を得る工程、および
2)前記融合タンパク質を、マルトース結合タンパク質の表面に露出した疎水性ドメインを含むアミノ末端と固体基質の疎水性表面との物理的吸着によって、前記疎水性表面に固定させる工程とを含む、生理活性ポリペプチドを固体基質に固定化させる方法を提供する。
【0011】
また本発明は、前記固定化方法によって生理活性ポリペプチドが、マルトース結合タンパク質をリンカーに使用して固体基質の疎水性表面に固定されている、生物活性固体基質を提供する。
【0012】
同時に本発明は、マルトース結合タンパク質の、前記生物活性固体基質の製造のための使用を提供する。
以下、本発明を詳しく説明する。
本発明は、
1)マルトース結合タンパク質(MBP)のカルボキシル末端に、生理活性ポリペプチドが融合された融合タンパク質を得る工程、および
2)前記融合タンパク質を、マルトース結合タンパク質の表面に露出した疎水性ドメインを含むアミノ末端と固体基質の疎水性表面との物理的吸着によって、前記疎水性表面に固定させる工程とを含む、生理活性ポリペプチドを固体基質に固定化させる方法を提供する。
【0013】
前記方法において、工程1)は、マルトース結合タンパク質をコードする塩基配列のカルボキシル末端に生理活性ポリペプチドをコードする塩基配列を融合した後、その融合遺伝子断片を大腸菌(Escherichia coli)で形質転換して発現させた後、精製することにより、マルトース結合タンパク質−生理活性ポリペプチドの融合タンパク質を得る工程である。
【0014】
前記マルトース結合タンパク質(MBP)は、大腸菌の細胞膜を横切って原形質膜空間に位置したタンパク質で、細胞内でマルトースまたはマルトデキストリン(maltodextrin)のような糖類の移動に関与するペリプラズム(periplasm)タンパク質である。マルトース結合タンパク質は、有用な外来タンパク質を融合タンパク質形態に生産するために主に使用され、細胞内の遺伝子malEから解読されて作られ、クローニングされたmalE遺伝子の下流に外来タンパク質の遺伝子を挿入して細胞内で発現させると、2個のタンパク質が結合した融合タンパク質を手軽に大量生産することができる。特に、発現させようとするタンパク質の大きさが小さい場合や、他の宿主細胞内ではその安全性が低下する外来タンパク質の場合、このようにマルトース結合タンパク質を使用して融合タンパク質形態で細胞内で発現させることが有利である。前記のようにmalE遺伝子が結合された遺伝子から発現される外来タンパク質は、マルトース結合タンパク質がマルトースに結合親和力を有するという特性を利用して分離することができる。例えば、マルトースが多重化された形態であるアミロース(amylose)がコーティングされた樹脂と細胞破砕液とを反応させて、反応させた樹脂を数回洗浄して汚染した他のタンパク質を除去した後、高濃度のマルトースを添加して競争させることによって、所望のタンパク質のみを簡単に溶出させることができる。このように、マルトース結合タンパク質を使用すると、所望するタンパク質を細胞内で大量発現させた後、非常に容易に分離精製することができるので、マルトース結合タンパク質と結合した融合タンパク質を発現させるシステムは、全世界的に目的とする外来タンパク質を生産するのに多く使用されている。
【0015】
前記方法において、工程1)は、前記のようなマルトース結合タンパク質の特性を使用して、マルトース結合タンパク質と目的とする生理活性ポリペプチドとの融合タンパク質を生産する工程であり、具体的に(i)目的とする生理活性ポリペプチドが、マルトース結合タンパク質のカルボキシル末端に融合された融合タンパク質を、暗号化する融合遺伝子断片を得る工程、(ii)前記融合遺伝子断片を含む発現ベクターを製造する工程、(iii)前記発現ベクターで形質転換された形質転換微生物を製造する工程、および(iv)前記形質転換微生物から融合タンパク質を発現および精製する工程を含むことができる。
【0016】
まず、マルトース結合タンパク質をコードする塩基配列のカルボキシル末端に、目的とする生理活性ポリペプチドをコードする塩基配列を融合して、融合遺伝子断片を製造する。
【0017】
本発明によると、マルトース結合タンパク質のカルボキシル末端には、融合タンパク質の製造のために、目的とする生理活性ポリペプチドが結合される一方、表面に露出した疎水性ドメインを含むアミノ末端は、以後の工程で疎水性表面への物理的吸着に使用される。
【0018】
本発明に適合した生理活性ポリペプチドの適用範囲は、特異的な生物学的活性を有したり、目的とする一つ以上の分子に結合したりすることができる任意のポリペプチドであり、そのアミノ末端が生物学的活性に必須ではないポリペプチドを含むことができる。このようなタンパク質としては、抗原、抗体、酵素、構造タンパク質(structural protein)、付着タンパク質(adhesion protein)または調節タンパク質(regulatory protein)などを含むことができるが、これに限定されるものではない。
【0019】
生理活性ポリペプチド遺伝子は、人体医学的または産業的に重要性があり、組換え生産の必要性がある人体を含んだ多様な動植物および微生物来由の遺伝子から分離されたり化学的に合成されたりするものであり、前記のポリペプチドをコードする遺伝子である。このような生理活性ポリペプチドをコードする遺伝子で、これらの生物学的活性を示す一部または全体塩基配列が、マルトース結合タンパク質のカルボキシル末端の転写解読枠(open reading frame、ORF)部位に結合されて、融合遺伝子断片が製造される。
【0020】
前記のように製造された融合遺伝子断片を発現させるために、これを大腸菌用発現ベクターに挿入して組換え発現ベクターを製造する。前記大腸菌用発現ベクターには、外来遺伝子を大腸菌内で発現させることができるベクターなら、制限なしに使用することができる。
【0021】
本発明の実施例では、生理活性ポリペプチドで血管内皮細胞成長因子(vascular endothelial growth factor、VEGF)を重合酵素連鎖反応(PCR)で増幅した後、増幅されたVEGF遺伝子をマルトース結合タンパク質遺伝子を有するベクターにクローニングして、マルトース結合タンパク質のカルボキシル末端にVEGF遺伝子が連結された融合遺伝子を含む発現ベクターを製造する。
【0022】
本発明による組換え発現ベクターを使用して大腸菌を形質転換させた後、形質転換された大腸菌を培養してマルトース結合タンパク質−生理活性ポリペプチドの融合タンパク質を発現させる。ここで培養は、培養液のOD600値が0.3〜0.6になった時に、IPTGを最終濃度2〜4mMで添加して、2〜6時間程度さらに培養することが好ましい。
その後、発現された融合タンパク質を大腸菌培養液から分離精製する。ここで、使用することができる分離精製方法としては、マルトースを特異的に結合することができる物質、例えばアミロース樹脂を使用したアフィニティークロマトグラフィー(affinity chromatography)を使用することができる。
【0023】
前記方法において、工程2)は、得たマルトース結合タンパク質−生理活性ポリペプチドの融合タンパク質を疎水性表面に固定させる工程であり、前記固定化は、融合タンパク質を、マルトース結合タンパク質のアミノ末端に位置した表面に露出した疎水性ドメインをリンカーに使用した、疎水性表面との物理的吸着によって達成される。
【0024】
具体的に、前記融合タンパク質を適合した緩衝溶液、例えばリン酸塩緩衝溶液(buffered phosphate saline、PBS)、ツイーン20/PBS、トリス−HCl緩衝溶液、重炭酸塩緩衝溶液(bicarbonate buffer)などに1ng/ml〜0.5mg/mlの濃度に希釈した後、この希釈液を疎水性表面に添加して、4〜25℃で1〜24時間反応させて、マルトース結合タンパク質のアミノ末端に位置した疎水性ドメインと前記疎水性表面との物理的吸着によって、融合タンパク質を疎水性表面に固定させる。
【0025】
本発明に適合した疎水性表面は、シラン化された表面(silanized surface)、カーボンナノチューブ(CNT)表面、炭化水素でコーティングされた表面(hydrocarbon coated surface)、高分子(例えば、ポリスチレン、ポリカーボネート、ポリプロピレン、ポリエチレン、テフロン(登録商標)、ポリテトラフルオロエチレン、ポリエステル含有生分解性高分子など)表面、または金属(例えば、ステンレススチール、チタン、金、白金など)表面であることが可能であるが、これらに限定されない。
【0026】
このように疎水性表面に固定された融合タンパク質は、細胞認識に重要な生理活性ポリペプチド部分が外側に露出しているので、細胞膜に存在する受容体に容易に結合して細胞の機能を調節するのに重要な役割を担当することができる。また、疎水性表面に固定された融合タンパク質の生理活性ポリペプチドは、本来の生物学的活性、例えば酵素活性、触媒活性、抗原特性または調節機能をそのまま維持している。本発明で「生物学的活性を維持する」と言うのは、マルトース結合タンパク質に融合された生理活性ポリペプチドが融合された後にも、本来の生物学的活性または機能を50%以上、好ましくは60%以上、さらに好ましくは70%以上維持することを意味する。融合された生理活性ポリペプチドは、本来の生物学的活性または機能の80%以上を維持することがさらに好ましく、最も好ましいのは90%以上を維持することである。
【0027】
本発明の実施例では、マルトース結合タンパク質との融合タンパク質形態で発現、精製された生理活性ポリペプチドが、本来の生物学的活性または機能を99%以上維持する。
【0028】
また本発明は、前記固定化方法によって、生理活性ポリペプチドが、マルトース結合タンパク質をリンカーに使用して固体基質の疎水性表面に固定されている、生物活性固体基質を提供する。
【0029】
同時に本発明は、マルトース結合タンパク質の前記生物活性固体基質の製造のための使用を提供する。
【0030】
前記生物活性固体基質は、特定生物学的活性を有する生理活性ポリペプチドが前記固体基質の表面の外部に露出していて、目的とする分子または細胞受容体との相互作用が容易いなだけではなく、前記表面にどのような生物学的活性を有する生理活性ペプチドを固定させるのかによって、多様な目的に使用することができる。本発明による生物活性固体基質の最も一般的な用途は、細胞培養に使用することである。すなわち、前記固定化方法によって、特定細胞の培養に求められる生理活性ポリペプチドを、マルトース結合タンパク質を使用して疎水性材質の細胞培養容器基質に固定させて、前記培養容器に細胞を培養すると、細胞と生理活性ポリペプチドとの相互作用が直接的に成立して、培養を円滑に成立させることができる。このように、生理活性ポリペプチドが固定された生物活性固体基質に細胞を培養するようになれば、融合された生理活性ポリペプチドの生物学的活性によって細胞内信号伝達の変化をもたらし、細胞の形態および機能変化の誘導が可能なだけでなく、幹細胞の分化研究、組織工学のような再生医療分野、細胞センサーまたは細胞チップ研究に有用に使用することができる。
【0031】
また、本発明による生物活性固体基質を使用した細胞培養方法は、マルトース結合タンパク質によって疎水性表面に固定されて外部に露出した生理活性ポリペプチドが、直接細胞接着に関与するようにしたり、コラーゲン、フィブロネクチンおよびラミニンなどのような天然細胞の外基質(extracellular matrix、ECM)を一緒に吸着させて細胞接着に使用したり、固定された生理活性ポリペプチドを細胞の信号伝逹に使用することなどに活用することができる。
【0032】
また、本発明の生物活性固体基質は、診断装備などに適用することができる。例えば、診断しようとする生理活性ポリペプチドを疎水性材質からなるストリップ(strips)またはマイクロタイター(microtiter)の表面にマルトース結合タンパク質をリンカーに使用して固定させると、バイオセンサー(biosensor)として有用に使用することができる。
【0033】
前述したように、マルトース結合タンパク質を使用して生理活性ポリペプチドを固定することで生物活性固体基質を製造することは、固定された生理活性ポリペプチドと異なるポリペプチドとの相互作用に対する研究を可能にする。したがって、高性能スクリーニング(high−throughput screening)、固相抽出(solid phase extraction)またはクロマトグラフィー精製などに適用することができる。
【発明の効果】
【0034】
本発明による生理活性ポリペプチドの固定化方法は、原形質膜空間タンパク質であるマルトース結合タンパク質の表面に露出した疎水性ドメインをリンカーに使用して、生理活性ポリペプチドの生物学的活性はそのまま維持しながら、簡単な物理的吸着によって固体基質の疎水性表面に固定させることができ、このような生理活性ポリペプチドが固定された固体基質は、多様な細胞の培養、幹細胞の分化研究、組織工学のような再生医療分野、細胞センサーまたは細胞チップ研究などに非常に有用に使用することができるという長所を有する。
【図面の簡単な説明】
【0035】
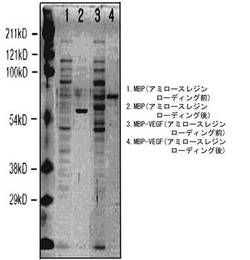
【図1】本発明によって組換えられたマルトース結合タンパク質(MBP)と血管内皮細胞成長因子(VEGF)とのMBP−VEGF融合タンパク質をSDS−PAGEで分析した結果である。
【図2】本発明のMBP−VEGF融合タンパク質で処理された293/KDR細胞で処理濃度による細胞の形態変化を観察した結果である。
【図3】本発明のMBP−VEGF融合タンパク質で処理された293/KDR細胞でリン酸化シグナルの変化を観察した結果である。
【図4】本発明のMBP−VEGF融合タンパク質がポリスチレン疎水性表面に固定化される程度を生化学的に分析した結果である。
【図5】本発明のMBP−VEGF融合タンパク質がポリスチレン疎水性表面に固定化される程度を物理学的に分析した結果である。
【図6】本発明のMBP−VEGF融合タンパク質が固定されたポリスチレン疎水性表面でBSA存在下で培養された293/KDR細胞の形態変化を観察した結果である。
【図7】本発明のMBP−VEGF融合タンパク質が固定されたポリスチレン疎水性表面でBSA不在下で培養された293/KDR細胞の形態変化を観察した結果である。
【発明を実施するための形態】
【0036】
以下、実施例を通じて本発明をより詳しく説明する。
【0037】
これら実施例は、単に本発明をより具体的に説明するためのものであって、本発明の要旨にしたがって、本発明の範囲がこれら実施例によって制限されないということは、通常の知識を有する当業者にとって自明であろう。
<参考例1>制限酵素を使用したDNAの切断および切片の回収
使用した制限酵素と緩衝溶液は、エンザイノミックス(Enzynomics)社から購入したし滅菌された1.5mlエッペンドルフチューブ(eppendorf tube)に、普通反応体積が20〜30μlになるようにして、37℃の反応温度で4〜5時間反応させた。制限酵素反応に使用した10×緩衝溶液の組成を、下記に示す:
1)10×エンザイノミックス緩衝溶液EzバッファーI:100mM トリス−HCl(pH7.5、25℃)、50mM NaCl、10mM MgCl2、0.025% トリトンX−100、および
2)10×エンザイノミックス緩衝溶液EzバッファーII:10mM トリス−HCl(pH7.5、25℃)、50mM NaCl、10mM MgCl2、1mM ジチオトレイトール(dithiothreitol)。
【0038】
DNA切片の回収は、電気泳動されたアガロースゲルをUV透過照明装置(transilluminator、Avegene)で照射して所望する切片を切断した後、ゲル抽出キット(gel extraction kit、Qiagene)を使用して分離した。
<参考例2>細菌性アルカリフォスファターゼ(bacterial alkaline phosphatase)処理
細菌性アルカリフォスファターゼ(BAP)処理時に使用したBAP溶液は、フェルメンタス(Fermentas)社から購入し、滅菌された1.5mlエッペンドルフチューブに普通反応体積が50μlになるようにして、60〜65℃の反応温度で1時間反応させた。BAP反応には、1M トリス−HCl緩衝溶液(pH8.0)(Bioneer)を使用した。
<参考例3>ライゲーション反応(ligation reaction)
ライゲーション反応は、DNAライゲーションキット(DNA Ligation Kit Ver2.1、タカラ)を使用してベクター(vector)と挿入物(insert)の割合を1:3程度で混合して反応体積を10〜20μlになるようにして、16℃で少なくとも12時間以上反応させた。
<参考例4>大腸菌形質転換
形質転換のための大腸菌宿主細胞には、E.coli K12 TB1(New England Biolabs)を使用した。前記細胞を60mlの液体培地(10g/lバクト・トリプトン[bacto−tryptone]、5g/l 酵母抽出物、10g/l NaCl)に接種した後、吸光度(OD600)値が0.4〜0.6になるまで37℃振盪培養した。前記培養された細胞を1.5mlエッペンドルフチューブに分注して、遠心分離を通じて細胞を得た。得られた細胞に50mM CaCl2を300μlずつ添加して弱くボルテキシング(vortexing)した後、再度遠心分離を通じて細胞を収穫した。前記得られた細胞に、50mM CaCl2を300μlずつ添加して細胞を均一に分散させた後、0℃で30分間静置させた。前記静置液を再度遠心分離して上澄み液は捨てた後、50mM CaCl2に15%グリセロールを添加した冷たい溶液を、残った細胞に150μlずつ添加して均一に分散させて冷凍保管した。
<参考例5>オリゴヌクレオチドの合成
目的とする生理活性ポリペプチドをコードする遺伝子を増幅するために、重合酵素連鎖反応(PCR)に使用されるプライマー対は、バイオニア(Bioneer)社のオリゴヌクレオチド合成サービスを使用して製作した。
<参考例6>重合酵素連鎖反応
鋳型50ngと正方向および逆方向プライマーそれぞれ10μMに蒸留水を添加して、総体積が10μlになるようにした後、ホットスタートPCRプレミックス(Hot Start PCR Premix、Bioneer)に添加した。温度循環器(T−gradient thermo block、Applied Biometra)を使用して、前記反応混合物を95℃で1分間変性させた後、94℃で30秒、55℃で30秒、および68℃で1分間の反応を31回反復して、最終的に72℃で5分間増幅させた。前記増幅された反応産物をPCR精製キット(PCR purification kit、Bioneer)を使用して精製して、アガロースゲルに電気泳動した後、UVトランスイルミネーター(transilluminator)(Avegene)を照射して所望する切片を切断した。回収されたゲル切片からゲル抽出キット(gel extraction kit、Qiagene)を使用して増幅されたDNAを分離した。
<参考例7>細胞培養
HEK293ヒト胚芽腎臓細胞から由来した293/KDR細胞は、ヒトVEGFR−2(KDR/Flk1)受容体を発現するように作られた細胞である。293/KDR細胞はシブテック(Sibtech)社から5回継代培養された状態で購入した。細胞培養には、DMEM液体培地(Dulbecco’s Modified Eagle’s Medium、ウェルジン)( D−グルコース 4500mg/l 、L−グルタミン、 ピルビン酸ナトリウム 110mg/l 、10%FBS)に、0.375μg/mlのピュロマイシン(Puromysin、Sigma)を添加した培地を使用して、37℃で5%のCO2を添加しながらインキュベーター(Thermo)で培養した。培地は、2日間隔で新しいものと交換して、細胞がT−フラスコに70〜80%程度生育した後に継代した。
<参考例8>7.5%ポリアクリルアミドゲル(polyacrylamide gel)の製造
ミニプロテアン3電気泳動セット(Mini−protean 3 Electrophoresis Set)(Bio−rad)を使用して、7.5%ポリアクリルアミドゲルを製造した。まず、1.0mmのガラスプレートをフレームに固定させて、キャスティングフレーム(casting frame)を形成した。50mlのコニカルチューブに蒸留水4.94mlと1.5M トリス−HCl緩衝溶液を2.5ml、30%アクリルアミド溶液を2.5ml、10%過硫酸アンモニウム(ammonium persulfate、APS)50μlおよびTEMED(N,N,N’、N’−テトラメチルエチレンジアミン)10μlを添加してよく混合した後、前記1.0mmガラスプレートに4.5mlずつ入れて解像度ゲル(resolution gel)を作製した。その後、ゲルが乾かないように500μlの蒸留水を注入した後、解像度ゲルが完全に固まるとゲルの上の蒸留水を除去した。スタッキングゲル(stacking gel)を製造するために、50mlのコニカルチューブに蒸留水3.05ml、0.5M トリス−HCl緩衝溶液 1.25ml、30%アクリルアミド溶液 0.67ml、10%APS 25μlおよびTEMED 5μlを添加してよく混合した後、1.0mmガラスプレートに最後まで注いで、15ウェル(20μl)鋳型を挿入した後、固めた。ポリアクリルアミドゲルに使用された試薬の組成を下記に示す:
1)1.5M トリス−HCl緩衝溶液:トリス塩基 18.17g(Invitrogen)、20% SDS(Amersham Pharmacia Biotech)2 ml、蒸留水 80ml,(pH8.8)、
2)0.5M トリス−HCl緩衝溶液:トリス塩基 6.06g(Invitrogen)、20% SDS(Amersham Pharmacia Biotech)2ml、蒸留水 80ml,(pH6.8)、および
3)30% アクリルアミド溶液:29% アクリルアミド(Sigma)、1% ビス−アクリルアミド(Sigma)。
<参考例9>ビオチンカップリング(biotin coupling)
タンパク質のビオチンカップリングは、スルホ−NHS−ビオチン化キット(sulfo−NHS−biotinylation kit、Pierce)を使用して行った。反応体積が0.5〜2ml、タンパク質の濃度が1〜10mg/mlになるようにリン酸塩緩衝溶液(phosphate buffered saline、PBS)でタンパク質を希釈した後、10mM スルホ−NHS−ビオチン溶液をビオチン処理されるタンパク質の分子量によって計算して添加して、1時間常温で反応させた。一方、スルホ−NHS−ビオチン化キット内に含まれている脱塩スピンカラム(desalt spin column)に15mlのコニカルチューブを結合させて、1000×gで2分間遠心分離(ハニル)を行った後、コニカルチューブに集められた溶液を除去した。その後、脱塩スピンカラムにPBS 2.5mlを添加して、1000×gで2分間遠心分離をして脱塩スピンカラムを洗浄した。前記洗浄過程を2回繰り返し実施した。このように準備した脱塩スピンカラムに新しい15mlのコニカルチューブを結合させた後、前記ビオチン処理された反応物を添加して、1000×gで2分間遠心分離を行ってビオチンが結合されたタンパク質を分離した。
【実施例】
【0039】
<実施例1>組換えタンパク質を発現するプラスミドpMAL−c2X−VEGFの製造<1−1>VEGF遺伝子がクローニングされたプラスミドの製造
VEGF遺伝子がクローニングされたプラスミドを製造するため、配列番号1の塩基配列を有する正方向プライマーVEGF−F(EcoRI)および配列番号2の塩基配列を有する逆方向プライマーVEGF−R(HindIII)を合成した。前記プライマー対を使用して、ヒト血管平滑筋(vascular smooth muscle、VSM)細胞から抽出した全体遺伝子を鋳型に、重合酵素連鎖反応(PCR)を行って、血管内皮細胞成長因子(vascular endothelial growth factor、VEGF)165部分のみを増幅した。
【0040】
pGEM−TベクターシステムI(Promega)に含まれている2×ライゲーション緩衝溶液(rapid ligation buffer)5μlに、前記で増幅されたVEGF遺伝子断片4ng、pGEM−Tベクター50ngおよびT4 DNAリガーゼ(ligase)1μlを添加した後、総体積が10μlになるように蒸留水を添加して、常温で1時間放置した後、続いて16℃で12時間反応させた。反応が終結した後、連結物を大腸菌E.coli K12 TB1に形質転換させて、それから目的とする遺伝子がクローニングされた組換えプラスミドを選別して、pGEM−VEGFと命名した。
<1−2>MBP−VEGF融合遺伝子がクローニングされたプラスミドの製造
リンカーとしてマルトース結合タンパク質(MBP)とVEGF遺伝子とを融合させるために、前記実施例<1−1>で製造された組換えプラスミドpGEM−VEGFを、エンザイノミックス緩衝溶液EzバッファーI、緩衝溶液EzバッファーII中で、制限酵素EcoRIとHindIIIで切断した後、アガロースゲル上でVEGF遺伝子切片を分離した。以後のライゲーション反応を容易に行うために、分離したVEGF遺伝子をBAPで処理した。BAP処理は、緩衝溶液(1M トリス−HCl、pH8.0、Bioneer)7.5μlおよびBAP溶液(Fermentas)1μlを入れた後、最終体積が50μlになるようにVEGF遺伝子を添加して、65℃で1時間反応させた。反応物をアガロースゲルで電気泳動した後、UVトランスイルミネーター(Avegene)で照射して所望する部分を切断した後、ゲル抽出キット(gel extraction kit、Qiagene)を使用してVEGF遺伝子断片を分離した。
【0041】
一方、ライゲーション反応に使用されるMBP遺伝子を有しているベクターpMAL−c2X(New England Biolabs)を、エンザイノミックス緩衝溶液EzバッファーI、緩衝溶液EzバッファーIIで制限酵素EcoRIとHindIIIで切断した後、アガロースゲル上でMBP遺伝子含有ベクター切片を分離した。
【0042】
前記で分離したVEGF遺伝子9μl、切断したベクター切片pMAL−c2X 3μlおよびDNAライゲーションキット(DNA Ligation Kit Ver2.1、タカラ)に含まれた酵素溶液I(enzyme solution I)12μlを混合して、総体積が20μlになるように蒸留水を加えた後、16℃で16時間反応させた。反応が終結した後、接合物を大腸菌E.coli K12 TB1に形質転換させて、それからMBPとVEGFの融合遺伝子がクローニングされた組換えプラスミドをスクリーニングして、pMAL−c2X−VEGFと命名した。
<実施例2>MBP−VEGF融合タンパク質の発現および精製
<2−1>MBP−VEGF融合タンパク質の発現誘導
MBP−VEGF融合タンパク質を発現する組換えプラスミドpMAL−c2X−VEGFを、大腸菌E.coli K12 TB1に形質転換させて、37℃のLB(Luria−Bertani)固体培地で一日の間培養した。翌日、培地の上に形成されたコロニー(colony)を採取して、60μg/ml濃度のアンピシリンを含む3mlのRB(rich medium+グルコース)液体培地に接種して、37℃で約2時間培養した。そこに、IPTG(イソプロピル−β−D−チオガラクトピラノシド)を最終濃度3mMになるように添加して、再び37℃で2時間さらに培養した。培養が終わった後、1mlの培養液を採取して、遠心分離機を通じて細胞沈殿物を得た。前記細胞沈殿物に20μlの1×試料ローディング緩衝溶液(sample loading buffer)を添加してよく混合した。前記混合物を95℃で5分間沸かしてから常温に冷却させた後、15μlを取って10%SDS−ポリアクリルアミドゲル電気泳動を行った。電気泳動が終結した後、ポリアクリルアミドゲルをクマシブルー(coomassie brilliant blue)で染色して、抗−MBP抗血清(New England Biolabs)を使用したウエスタンブロットを通じて、MBP−VEGF融合タンパク質の発現有無を観察した。
<2−2>水溶性融合タンパク質の発現および精製
前記実施例<2−1>で組換えプラスミドpMAL−c2X−VEGFに形質転換された大腸菌細胞を、アンピシリンが60μg/ml添加されたRB培地に接種した後、37℃で一晩中培養した。前記培養液10mlを1lのRB培地に添加して、37℃で継続振盪培養し、培養液の吸光度が650nmの波長で約0.6程度になった時、3mMの最終濃度でIPTGを添加した。IPTGを添加して約2時間後に培養を停止した後、培養液を4000×gで20分間遠心分離(Combi−514R、ハニル)して細胞沈殿物を集めた。前記細胞沈殿物に緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml)50mlを添加して懸濁させた後、最終濃度が1mMになるようにEDTA(ethylenediaminetetraacetic acid)とPMSF(phenylmethanesulphonyl fluoride)を添加した。前記細胞混合物の精製に先立って、−20℃での冷凍および解凍を繰り返して細胞破砕が容易に起きるようにした。その後、細胞混合物を冷却浴内で超音波粉砕機(Fisher Scientific Model 500 Sonic Dismembrator)を使用して、10%出力で約10秒間超音波処理して細胞を破砕した後、30秒間冷却浴内に放置した。前記のような過程を2回反復して細胞を完全に破砕した。このように得た細胞均質液を、9000×gで1時間遠心分離(Combi−514R、ハニル)して、可溶性タンパク質が溶解している上澄み液を得た後、緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA、2ml)で5倍希釈した。
【0043】
一方、大腸菌形質転換体から発現されたMBP−VEGF融合タンパク質を分離するために、アミロース樹脂(amylose resin、New England Biolabs)を使用したアフィニティークロマトグラフィーを準備した。このカラムをあらかじめ約8×ベッドボリューム(bed volume)の緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml)で洗浄して平衡化させた。平衡化されたアミロース樹脂親和性クロマトグラフィーに、前記で得た可溶性タンパク質が溶解している上澄み液を分当たり1mlの速度でローディング(loading)した。続いて、12×ベッドボリューム以上の緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml)をカラムに流して、樹脂に吸着されないタンパク質を除去した。その次に、10mM マルトース溶出緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml、10mM マルトース)を添加して、樹脂に吸着されていたタンパク質を分当たり1mlの速度で溶出させて回収した。その後、回収されたタンパク質を10%ポリアクリルアミドゲルに電気泳動(Bio−rad)して、精製されたタンパク質の概算の分子量と純度を確認した。その結果、精製されたタンパク質の純度は95%以上で、概算の分子量は60,000Da程度と示された。
【0044】
前記タンパク質試料を、希釈膜(MWCO12−14,000)(dialysis membrane、Spectrum laboratories、Inc.)に入れてPBSで3日間希釈させた後、マルトースが除去されたタンパク質のみを収得した。その後、遠心分離ろ過器(Centrifugal Filter、Amicon Ultra−15 MWCO 5,000、Millipore)を使用して、4000×gで45分間遠心分離(Combi−514R、ハニル)してタンパク質を濃縮した。前記過程によって最終的に濃縮されたタンパク質を、MBP−VEGFと命名した。
【0045】
図1は、アミロースレジンにローディングする前のMBP単独(ライン1)およびMBP−VEGF融合タンパク質(ライン3)と、アミロースレジンにローディングした後に精製されたMBP単独(ライン2)およびMBP−VEGF融合タンパク質(ライン4)のSDS−PAGE結果を示したもので、MBP−VEGF融合タンパク質が、MBP単独より高い分子量を有することが確認されて、大腸菌形質転換体から融合タンパク質が発現されたことが分かった。
<実施例3>MBP−VEGF融合タンパク質の活性測定
<3−1>MBP−VEGF融合タンパク質による293/KDRのリン酸化
MBP−VEGF融合タンパク質の活性を測定するために、まず293/KDRを培養した後、6−ウェルに5×105の細胞濃度で移植した。293/KDR細胞は、293HEK(human embryonic kidney cell)にVEGFR2(VEGF受容体)を過発現させた細胞で、VEGFのリン酸化試験などに使用されている。細胞移植6時間後、0.05%のFBS(fetal bovine serum、ウェルジン)が添加されたDMEM液体培地(Dulbecco’s Modified Eagle’s Medium、ウェルジン)に培地を交換した。それから16時間経過後、前記培地を分析培地(assay medium、DMEM、ウェルジン、25mM HEPES pH7.2、Sigma、5mM Na3VO4、Sigma、0.05%BSA(bovine serum albumin、Sigma))に交換して、10分間培養器(Thermo)で培養した。
【0046】
一方、293/KDR細胞を刺激するためにVEGF165(vascular endothelial cell growth factor 165、R&D Systems)および製造されたMBP−VEGF融合タンパク質を、それぞれ0、1、5、10、50および100ng/mlの濃度で分析培地に希釈した。続いて希釈されたVEGFおよびMBP−VEGF融合タンパク質を、前記で培養された293/KDR細胞に1.5mlずつ濃度別にそれぞれのグループに添加した後、37℃培養器(Thermo)で10分間培養した。VEGFおよびMBP−VEGF融合タンパク質によって刺激された細胞を氷に浸した後、冷たいPBSで2回洗浄した。続いて細胞を破砕するために、RIPA(Radio−Immunoprecipitation Assay、Pierce)緩衝溶液に1mM オルトバナジン酸塩ナトリウム(sodium orthovanadate、Sigma)、5mM ピロリン酸塩ナトリウム(sodium pyrophosphate、Sigma)および25mM フッ化ナトリウム(sodium fluoride、Sigma)を混合した後、100μlの混合液を前記細胞反応液に添加してスクラッパー(scraper、SPL)を使用して細胞を回収した。回収された各グループの細胞を1時間氷に浸した後、15,000rpmで30分間遠心分離(micro 17TR、ハニル)して上澄み液のみを採取して、BCA(Pierce)タンパク質測定方法を使用して定量した。それぞれの試料を5×試料緩衝溶液(0.6ml 1M トリス−HCl、pH6.8、5ml 50% グリセロール、2ml 10% SDS、0.5ml 2−メルカプトエタノール、1ml 1%ブロモフェノールブルー、0.9ml 蒸留水)に添加して、蒸留水で各試料の最終濃度を同一に合わせた後、1.5ml エッペンドルフチューブに入れて4℃で冷蔵保管した。
【0047】
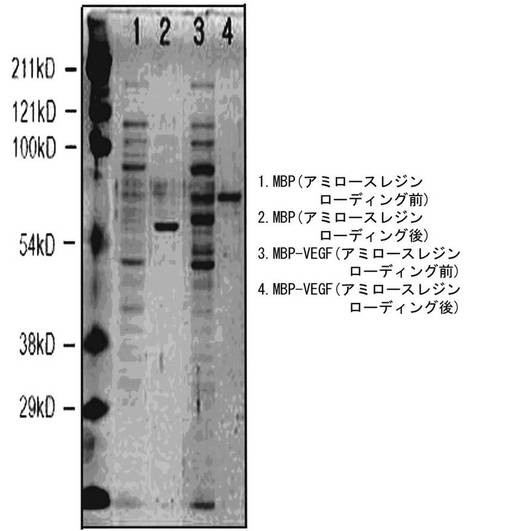
図2は、細胞を破送する前の細胞形態を観察した結果で、図2のAは細胞に分析培地を添加する前の細胞形態で、Bは分析培地を、CはMBP(100ng/ml)を、DはMBP−VEGF(150ng/ml)を、EはVEGF(50ng/ml)を10分間処理した後の細胞形態である。分析培地とMBPを処理した群(BおよびC)では、細胞形態が処理する前の形態と類似に伸長した状態であるが、MBP−VEGFとVEGFを処理した群(DおよびE)では細胞が丸い形態に変化したことが分かった。このような結果は、本発明によってVEGFがMBPとの融合タンパク質形態に発現されて精製されても、本来のVEGF活性をそのまま維持していることを示すものである。
<3−2>MBP−VEGF融合タンパク質による293/KDRのリン酸化シグナル変化
前記実施例<3−1>で準備した試料を95℃で10分間加熱した後、15,000rpmで1分間遠心分離(micro 17TR、ハニル)して、ふたに気化された試料を集めた。ウエスタンブロット(western blotting)分析のために7.5% ポリアクリルアミドゲルを製造して、前記ゲルに前記試料をそれぞれ20μlずつローディングした後、電気泳動(Bio−rad社)を実施した。一方、ニトロセルロース膜とろ過紙を10×トリス/グリシン緩衝溶液(25mM トリス、192mM グリシン、pH8.3、Bio−rad)20mlとMeOH 40mlおよび蒸留水140mlを混合した溶液に20分間浸けて活性化させた。
【0048】
その後、半乾燥伝達システム(Semi−Dry Transfer System、Bio−rad)を使用して、前記で電気泳動されたポリアクリルアミドゲルをニトロセルロース膜に伝達した。前記ニトロセルロース膜を5%BSA(bovine serum albumin、Sigma)で4℃で8時間処理して遮断させた後、TBS−T(Tris buffered saline−NaCl 8g、KCl 0.2g、トリス 3g、ツイーン20 0.5ml)緩衝溶液で10分間3回洗浄した。その後、1次抗体であるホスホチロシン:ビオチン(phosphotyrosine:biotin、BD Biosciences)と単一クローン抗−ホスホチロシン(monoclonal anti−phosphotyrosine、Sigma)を5%BSA(bovine serum albumin、Sigma)に1:2000に希釈した後、前記洗浄されたニトロセルロース膜に10mlずつ添加して、8時間4℃で反応させた。前記ニトロセルロース膜をTBS−T緩衝溶液で10分間3回洗浄した後、5%BSA(bovine serum albumin、Sigma)に1:10000に希釈された2次抗体(Mouse−Pierce)と、ストレプタビジン(streptavidin、Sigma)とを10mlずつ添加して、1時間常温で反応させた。前記ニトロセルロース膜をTBS−T緩衝溶液で10分間3回洗浄した後、ウェストフェムト最大敏感性基質(West Femto maximum sensitivity substrate、Pierce)に含まれているルミノール/エンハンサー溶液(West Femto Luminol/Enhancer Solution)と、過酸化溶液(West Femto Stable Peroxide Solution)とを、それぞれ500μlずつ混合した溶液を添加して、常温で5分間反させた。反応が終結した後、前記ニトロセルロース膜をイメージ分析機(image analyzer、Las 3000、フジフィルム)で分析して、MBP−VEGF融合タンパク質による293/KDR細胞のリン酸化シグナルを確認した。
【0049】
その結果、図3に示したように、MBP−VEGF融合タンパク質で処理した細胞では、VEGFだけで処理した細胞群と類似して220KDa付近から濃度依存的にリン酸化が起きることを分かった。このようなリン酸化様相は、VEGFによって処理された293/KDR細胞で本実施例に使用された抗体によって示されるリン酸化様相と同一である(Backer MV等、Biomaterials,2006年,第27巻,5452−5458頁)。その他にも、130KDa付近で細胞内のリン酸化が濃度依存的に増加することを確認することができた。
<実施例4>ポリスチレン表面でのMBP−VEGF融合タンパク質の固定化に対する生化学的分析
本発明によるMBP−VEGF融合タンパク質が、ポリスチレン(polystyrene)のような疎水性表面に固定化される程度を調べるため、まず、下記のようにMBP−VEGF融合タンパク質をビオチン(biotin)で処理した。具体的に、1mg/mlのMBP−VEGF融合タンパク質185μlに、10mM スルホ−NHS−ビオチン溶液16μlを入れた後、最終体積が0.5mlになるようにPBSを添加して、1時間常温で反応させた。一方、<参考例10>に記述した方法で脱塩スピンカラム(Pierce)を洗浄した後、前記反応物を15mlコニカルチューブを結合した洗浄された脱塩スピンカラムに入れて、1000×gで2分間遠心分離してビオチン処理されたMBP−VEGF融合タンパク質を得た。
【0050】
10〜10,000ng/ml濃度のビオチン処理されたMBP−VEGF融合タンパク質を100μlずつ、ポリスチレン製の96ウェル(非−組織培養用、Falcon)に加えて、常温で4時間処理して、PBS200μlで3回洗浄した。その後、ポリスチレン表面にMBP−VEGF融合タンパク質の非特異的結合を防止するために、1%BSA(bovine serum albumin、Sigma)200μlを入れて常温で2時間処理した後、0.05% ツイーン20(Amersham Pharmacia Biotech)含有PBS200μlで5回洗浄した。
【0051】
前記のMBP−VEGFが固定された96ウェルに、ストレプタビジンペルオキシダーゼ(streptavidin−peroxidase、Sigma)がPBSに1:10,000に希釈された溶液を100μl添加して、常温で1時間反応させた後、0.05%ツイーン20含有PBS200μlで5回洗浄した。続いて、前記96ウェルに安定化された過酸化水素(stabilized hydrogen peroxide、R&D Systems)と安定化されたテトラメチルベンジジン(stabilized tetramethylbenzidine、R&D Systems)を1:1で混合した溶液100μlを添加してアルミホイルで包んだ後、20分間反応させた。20分後、2N硫酸溶液50μlを前記ウェルに添加して反応を停止させた。前記96ウェルをマイクロプレートリーダー(microplate reader、Molecular Device)を使用して450nmで吸光度を測定して、MBP−VEGF融合タンパク質がポリスチレン表面に固定化された程度を確認した。
【0052】
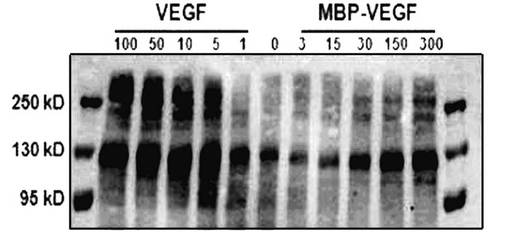
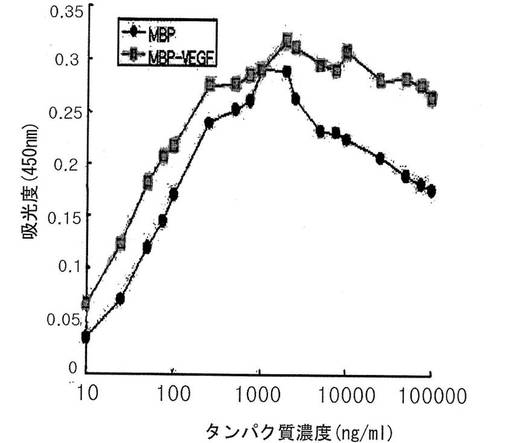
その結果、図4に示されたように、MBPおよびMBP−VEGFが10〜1,000ng/mlの範囲で、濃度依存的に疎水性ポリスチレン表面に吸着されることが分かった。しかし、前記範囲を逸脱した高い濃度では、このような生化学的方法で固定化された程度を分析するのに限界があった。
<実施例5>ポリスチレン表面でのMBP−VEGF融合タンパク質の固定化に対する物理学的分析
前記実施例4の生化学的方法では、高濃度でMBP−VEGF融合タンパク質の固定化程度を測定するのに限界があることを確認して、物理学的方法である水晶振動子微量秤(quartz crystal microbalance、QCM)を使用して、高濃度で前記融合タンパク質が疎水性表面に吸着する程度を調査した。QCMは、水晶振動子(quartz crystal)に物質が吸着した時に振動数の減少が生じて、このような振動数の減少を検出して吸着されたタンパク質の量を測定する方法である。本実験で水晶振動子は、0.5%ポリスチレン/トルエン溶液をスピンコーティングした後、使用した。試料は、MBPおよびMBP−VEGFを、それぞれ1、10、50、100および500μg/mlの濃度で緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml)に希釈して準備した。水晶振動子に前記緩衝溶液を1時間流して平衡化させた後、準備したそれぞれの指標を15分間流して水晶振動子にMBPおよびMBP−VEGFが吸着するようにした後、再び緩衝溶液を流して吸着されないタンパク質を除去した。その後、タンパク質吸着前と比べて吸着後の水晶振動子の振動数減少を測定して、吸着した重さをソルベリー式(Sauerbrey equation、Δf=−ΔmC/n)(Hook F、Rodahl M.等、Langmuir.,1998年,第14巻,729−734頁)に代入して算出した。
【0053】
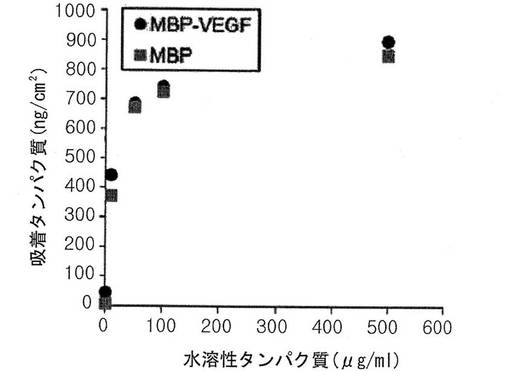
その結果、図5に示されように、本発明によるMBP−VEGF融合タンパク質が、1〜500μg/mlの高濃度でも濃度依存的に疎水性ポリスチレン表面に吸着して、このような濃度によるタンパク質の吸着がログ関数的増加を示すラングミュアタイプ(Langmuir−type)の吸着挙動と類似に単層に吸着することを確認した。
<実施例6>MBP−VEGF融合タンパク質による293/KDR細胞の形態変化
MBP−VEGF融合タンパク質による293/KDR細胞の形態変化を調査するため、まず、前記<実施例2>で抽出したMBP−VEGF融合タンパク質を無菌作業台(Sanyo)で0.22μm注射器フィルター(Millex GV、Millipore)を使用してろ過した後、0.1、1および10μg/mlの濃度で100μlずつポリスチレン材質の96ウェルプレート(非−組織培養用、Falcon)に添加した後、プレート表面にコーティングされるように無菌作業台中で4時間放置した。その後、前記96ウェルをPBS200μlで3回洗浄し、293/KDR細胞とMBP−VEGF融合タンパク質がコーティングされたウェル表面との非特異的結合を防止するために、一部のウェルには1%BSA(bovine serum albumin、Sigma)200μlを添加して無菌作業台内で2時間処理した後、PBS200μlで3回洗浄した。
【0054】
MBP−VEGF融合タンパク質がコーティングされた96ウェルに、ウェル当たり293/KDR細胞を2×104の濃度で移植した。ここで、使用された培地は、DMEM(ウェルジン)無血清培地を使用した。移植された細胞は、37℃培養器(Thermo)で45時間培養した後、位相差顕微鏡(ニコン社)を使用してこれらの形態変化を観察した。
【0055】
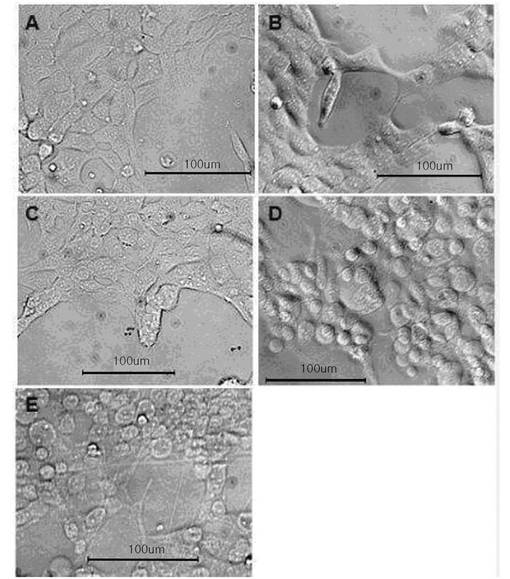
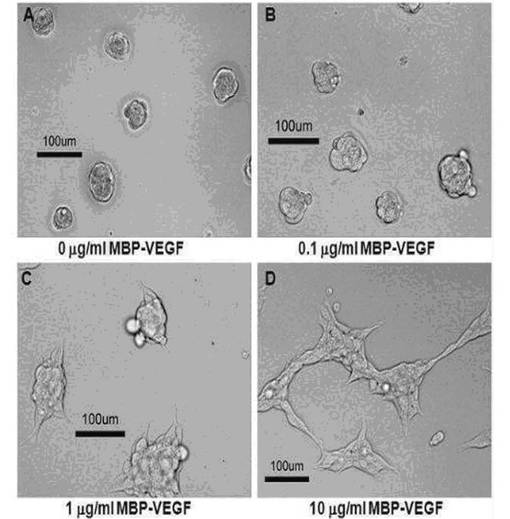
図6および図7は、それぞれ本発明のMBP−VEGF融合タンパク質が固定されたポリスチレン疎水性表面において、BSA存在下および不在下に培養された293/KDR細胞の形態変化を観察した結果である。図6に示されたように、細胞の非特異的な結合を防止するためにMBP−VEGFが吸着された表面にBSAを処理した場合に、MBP−VEGFが吸着されない表面(図6のA)では細胞が凝固した丸い(spheroid)形態を維持していて、吸着されたMBP−VEGFの濃度が増加するに比例して細胞に偽足が形成されながら細長い形態に生育することを確認することができた(図6のB〜D)。
【0056】
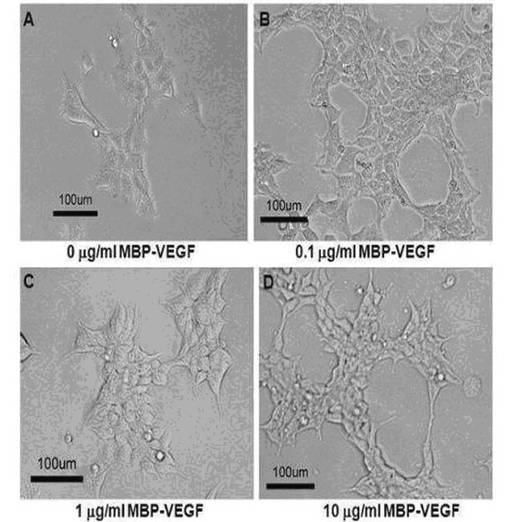
一方、図7に示されたように、MBP−VEGFが吸着した表面にBSAを処理しない場合に、MBP−VEGFが吸着されない表面(図7のA)では細胞がよく接着して広くひろがった形態に生育したが、MBP−VEGFが吸着した表面では図6のB〜Dと類似に細胞が細長に偽足を生成しながら生育することを確認した。このような結果は、本発明によるMBP−VEGF融合タンパク質のVEGFが、細胞の形態形成に影響を及ぼしていることを示すものである。
【0057】
以上、本発明の具体的な内容を詳しく記述したように、通常の知識を有する当業者において、このような具体的技術は単に好ましい実施様態であるだけであり、これによって本発明の範囲が制限されるものではない点は明白であろう。したがって、本発明の実質的な範囲は、添付された請求項とそれらの均等物によって定義される。
【技術分野】
【0001】
本発明は、マルトース結合タンパク質(maltose binding protein、MBP)をリンカーに使用して生理活性ポリペプチドを固体基質の疎水性表面に固定させる方法、および前記方法によって生理活性ポリペプチドが固定された生物活性固体基質に関するものである。
【背景技術】
【0002】
タンパク質の固定は、診断やバイオセンサーあるいは酵素反応器などのために非常に有用に使用されている。タンパク質の固定化方法では、グルタルアルデヒド(glutaraldehyde)のような架橋剤を使用した化学的架橋、疎水性結合を使用した物理的吸着、アミン反応性基を使用した化学的カップリングなどが知られている。このような方法の最大の問題点としては、固定させようとするタンパク質あるいはポリペプチドの活性を維持することにより化学的架橋やカップリングする方法では、新しい化学的結合が形成されたり標的タンパク質の化学的性質が変化したりすることによって活性が阻害され得ることがあり、物理的吸着方法では、標的タンパク質が直接疎水性表面に吸着される場合に、タンパク質構造の変化を伴って変性を誘発し得るという問題点がある。このような問題点を解決するために、固定させようとするポリペプチドにリンカーを結合させて、このリンカーを媒介にする固定化方法が報告されている(非特許文献1〜3)。リンカーを生理活性ポリペプチドに結合させる代表的な方法は、遺伝工学技術を使用して組換え融合タンパク質を合成するものである。
【0003】
例えば、グルタチオン−S−転移酵素(glutathione−S−transferase;GST)のような酵素を使用して、表面に酵素を認識するグルタチオン分子をあらかじめ結合させて、エリサ法(ELISA:enzyme−linked immunosorbent assay)に活用する方法が知られている(非特許文献4)。しかし前記方法は、ポリスチレンのように反応性がない疎水性表面には、グルタチオンのような物質を結合しにくいという短所がある。
【0004】
一方、疎水性ドメインを有するリンカーを使用してタンパク質の効率的な吸着と同時に変性を防いで、吸着後にも目的タンパク質の生物学的機能が維持され、それを細胞培養用表面として使用する方法が開発された。疎水性結合による融合タンパク質の固定化のためのリンカーとしては、免疫グロブリンのFcドメイン(非特許文献5、非特許文献3)、ハイドロフォビン(hydrophobin)のような疎水性が強いタンパク質などを使用する(非特許文献6)。前記方法は、吸着させようとする表面に別途の前処理をしないで、簡単に使用することができるという長所を有している。
【0005】
例えば、EGF−Fcあるいはカドヘリン(cadherin)−Fcをポリスチレン培養容器に固定化して、溶液性EGFあるいはゼラチンが吸着した表面で上皮腫瘍細胞または胚芽幹細胞を培養すると、これら細胞は生化学的および細胞生物学的に異なった挙動を示すということが報告されている(非特許文献5、非特許文献3)。しかし、このようにFcを使用する場合には、Fcのカルボキシル末端は疎水性表面との物理的吸着に必要とされ、Fcのアミノ末端に標的とする生理活性ポリペプチドを結合させなければならないので、生理活性ポリペプチドのカルボキシル末端が活性に必須な場合には使用が制限される。その他にも、生理活性ポリペプチドのカルボキシル基末端を、連結部位に使用する多様な結合方法も開発された。一例として、特許文献1は、ハイドロフォビンを使用して酵素などを疎水性表面に結合させるタンパク質固定化方法を開示している。しかし、前記で言及された疎水性結合を誘導する融合タンパク質のリンカー部分は、大部分が動物およびカビ由来のものであり、大腸菌で発現させて精製する場合は、封入体(inclusion body)の形成など多くの短所を有している。
【0006】
以上のことに鑑みて、本発明者等は、前記の従来技術の問題点を解決するために鋭意研究努力した結果、大腸菌で発現されるタンパク質として、マルトースとの優れた結合能力と発現および精製が容易であるという長所を有する、マルトース結合タンパク質を使用して生理活性ポリペプチドとの融合タンパク質を製造して、前記融合タンパク質をマルトース結合タンパク質の疎水性ドメイン(非特許文献7)を使用して、ポリスチレンのような疎水性表面に単純な物理的吸着によって固定させた後、固定された融合タンパク質の生理活性ポリペプチド部分が、生物学的活性を維持していることを確認することにより、本発明を完成した。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許公開第20040235050号
【非特許文献】
【0008】
【非特許文献1】Lee J.M.等,Anal.Chem.,2007年,第79巻,2680−2687頁
【非特許文献2】Ogiwara K.等,Biochem.Biophy.Res.Comm.,2006年,第345巻,255−259頁
【非特許文献3】Nagaoka M.等,PLoS ONE、2006年,第1巻,e15頁
【非特許文献4】Sehr P.等,J.Immuno.Methods.,2001年,第253巻,153−162頁
【非特許文献5】Ogiwara K.等,Biotech.Letters,2005年,第27巻,1633−1637頁
【非特許文献6】Qin M.等,Colloids Surfaces,2007年,第60巻,243−249頁
【非特許文献7】Fox J.D.等,Protein Science,2001年,第10巻,622−630頁
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明の目的は、大腸菌での発現および精製が容易であり、疎水性ドメインを有するマルトース結合タンパク質を使用して、特定の成長因子またはサイトカインのような生理活性ポリペプチドを、前処理過程なしに簡単な物理的吸着によって固体基質の疎水性表面に固定化する方法、および前記方法によって製造された生理活性ポリペプチドが固定された生物活性固体基質を提供することである。
【課題を解決するための手段】
【0010】
前記目的を達成するために本発明は、
1)マルトース結合タンパク質(MBP)のカルボキシル末端に、生理活性ポリペプチドが融合された融合タンパク質を得る工程、および
2)前記融合タンパク質を、マルトース結合タンパク質の表面に露出した疎水性ドメインを含むアミノ末端と固体基質の疎水性表面との物理的吸着によって、前記疎水性表面に固定させる工程とを含む、生理活性ポリペプチドを固体基質に固定化させる方法を提供する。
【0011】
また本発明は、前記固定化方法によって生理活性ポリペプチドが、マルトース結合タンパク質をリンカーに使用して固体基質の疎水性表面に固定されている、生物活性固体基質を提供する。
【0012】
同時に本発明は、マルトース結合タンパク質の、前記生物活性固体基質の製造のための使用を提供する。
以下、本発明を詳しく説明する。
本発明は、
1)マルトース結合タンパク質(MBP)のカルボキシル末端に、生理活性ポリペプチドが融合された融合タンパク質を得る工程、および
2)前記融合タンパク質を、マルトース結合タンパク質の表面に露出した疎水性ドメインを含むアミノ末端と固体基質の疎水性表面との物理的吸着によって、前記疎水性表面に固定させる工程とを含む、生理活性ポリペプチドを固体基質に固定化させる方法を提供する。
【0013】
前記方法において、工程1)は、マルトース結合タンパク質をコードする塩基配列のカルボキシル末端に生理活性ポリペプチドをコードする塩基配列を融合した後、その融合遺伝子断片を大腸菌(Escherichia coli)で形質転換して発現させた後、精製することにより、マルトース結合タンパク質−生理活性ポリペプチドの融合タンパク質を得る工程である。
【0014】
前記マルトース結合タンパク質(MBP)は、大腸菌の細胞膜を横切って原形質膜空間に位置したタンパク質で、細胞内でマルトースまたはマルトデキストリン(maltodextrin)のような糖類の移動に関与するペリプラズム(periplasm)タンパク質である。マルトース結合タンパク質は、有用な外来タンパク質を融合タンパク質形態に生産するために主に使用され、細胞内の遺伝子malEから解読されて作られ、クローニングされたmalE遺伝子の下流に外来タンパク質の遺伝子を挿入して細胞内で発現させると、2個のタンパク質が結合した融合タンパク質を手軽に大量生産することができる。特に、発現させようとするタンパク質の大きさが小さい場合や、他の宿主細胞内ではその安全性が低下する外来タンパク質の場合、このようにマルトース結合タンパク質を使用して融合タンパク質形態で細胞内で発現させることが有利である。前記のようにmalE遺伝子が結合された遺伝子から発現される外来タンパク質は、マルトース結合タンパク質がマルトースに結合親和力を有するという特性を利用して分離することができる。例えば、マルトースが多重化された形態であるアミロース(amylose)がコーティングされた樹脂と細胞破砕液とを反応させて、反応させた樹脂を数回洗浄して汚染した他のタンパク質を除去した後、高濃度のマルトースを添加して競争させることによって、所望のタンパク質のみを簡単に溶出させることができる。このように、マルトース結合タンパク質を使用すると、所望するタンパク質を細胞内で大量発現させた後、非常に容易に分離精製することができるので、マルトース結合タンパク質と結合した融合タンパク質を発現させるシステムは、全世界的に目的とする外来タンパク質を生産するのに多く使用されている。
【0015】
前記方法において、工程1)は、前記のようなマルトース結合タンパク質の特性を使用して、マルトース結合タンパク質と目的とする生理活性ポリペプチドとの融合タンパク質を生産する工程であり、具体的に(i)目的とする生理活性ポリペプチドが、マルトース結合タンパク質のカルボキシル末端に融合された融合タンパク質を、暗号化する融合遺伝子断片を得る工程、(ii)前記融合遺伝子断片を含む発現ベクターを製造する工程、(iii)前記発現ベクターで形質転換された形質転換微生物を製造する工程、および(iv)前記形質転換微生物から融合タンパク質を発現および精製する工程を含むことができる。
【0016】
まず、マルトース結合タンパク質をコードする塩基配列のカルボキシル末端に、目的とする生理活性ポリペプチドをコードする塩基配列を融合して、融合遺伝子断片を製造する。
【0017】
本発明によると、マルトース結合タンパク質のカルボキシル末端には、融合タンパク質の製造のために、目的とする生理活性ポリペプチドが結合される一方、表面に露出した疎水性ドメインを含むアミノ末端は、以後の工程で疎水性表面への物理的吸着に使用される。
【0018】
本発明に適合した生理活性ポリペプチドの適用範囲は、特異的な生物学的活性を有したり、目的とする一つ以上の分子に結合したりすることができる任意のポリペプチドであり、そのアミノ末端が生物学的活性に必須ではないポリペプチドを含むことができる。このようなタンパク質としては、抗原、抗体、酵素、構造タンパク質(structural protein)、付着タンパク質(adhesion protein)または調節タンパク質(regulatory protein)などを含むことができるが、これに限定されるものではない。
【0019】
生理活性ポリペプチド遺伝子は、人体医学的または産業的に重要性があり、組換え生産の必要性がある人体を含んだ多様な動植物および微生物来由の遺伝子から分離されたり化学的に合成されたりするものであり、前記のポリペプチドをコードする遺伝子である。このような生理活性ポリペプチドをコードする遺伝子で、これらの生物学的活性を示す一部または全体塩基配列が、マルトース結合タンパク質のカルボキシル末端の転写解読枠(open reading frame、ORF)部位に結合されて、融合遺伝子断片が製造される。
【0020】
前記のように製造された融合遺伝子断片を発現させるために、これを大腸菌用発現ベクターに挿入して組換え発現ベクターを製造する。前記大腸菌用発現ベクターには、外来遺伝子を大腸菌内で発現させることができるベクターなら、制限なしに使用することができる。
【0021】
本発明の実施例では、生理活性ポリペプチドで血管内皮細胞成長因子(vascular endothelial growth factor、VEGF)を重合酵素連鎖反応(PCR)で増幅した後、増幅されたVEGF遺伝子をマルトース結合タンパク質遺伝子を有するベクターにクローニングして、マルトース結合タンパク質のカルボキシル末端にVEGF遺伝子が連結された融合遺伝子を含む発現ベクターを製造する。
【0022】
本発明による組換え発現ベクターを使用して大腸菌を形質転換させた後、形質転換された大腸菌を培養してマルトース結合タンパク質−生理活性ポリペプチドの融合タンパク質を発現させる。ここで培養は、培養液のOD600値が0.3〜0.6になった時に、IPTGを最終濃度2〜4mMで添加して、2〜6時間程度さらに培養することが好ましい。
その後、発現された融合タンパク質を大腸菌培養液から分離精製する。ここで、使用することができる分離精製方法としては、マルトースを特異的に結合することができる物質、例えばアミロース樹脂を使用したアフィニティークロマトグラフィー(affinity chromatography)を使用することができる。
【0023】
前記方法において、工程2)は、得たマルトース結合タンパク質−生理活性ポリペプチドの融合タンパク質を疎水性表面に固定させる工程であり、前記固定化は、融合タンパク質を、マルトース結合タンパク質のアミノ末端に位置した表面に露出した疎水性ドメインをリンカーに使用した、疎水性表面との物理的吸着によって達成される。
【0024】
具体的に、前記融合タンパク質を適合した緩衝溶液、例えばリン酸塩緩衝溶液(buffered phosphate saline、PBS)、ツイーン20/PBS、トリス−HCl緩衝溶液、重炭酸塩緩衝溶液(bicarbonate buffer)などに1ng/ml〜0.5mg/mlの濃度に希釈した後、この希釈液を疎水性表面に添加して、4〜25℃で1〜24時間反応させて、マルトース結合タンパク質のアミノ末端に位置した疎水性ドメインと前記疎水性表面との物理的吸着によって、融合タンパク質を疎水性表面に固定させる。
【0025】
本発明に適合した疎水性表面は、シラン化された表面(silanized surface)、カーボンナノチューブ(CNT)表面、炭化水素でコーティングされた表面(hydrocarbon coated surface)、高分子(例えば、ポリスチレン、ポリカーボネート、ポリプロピレン、ポリエチレン、テフロン(登録商標)、ポリテトラフルオロエチレン、ポリエステル含有生分解性高分子など)表面、または金属(例えば、ステンレススチール、チタン、金、白金など)表面であることが可能であるが、これらに限定されない。
【0026】
このように疎水性表面に固定された融合タンパク質は、細胞認識に重要な生理活性ポリペプチド部分が外側に露出しているので、細胞膜に存在する受容体に容易に結合して細胞の機能を調節するのに重要な役割を担当することができる。また、疎水性表面に固定された融合タンパク質の生理活性ポリペプチドは、本来の生物学的活性、例えば酵素活性、触媒活性、抗原特性または調節機能をそのまま維持している。本発明で「生物学的活性を維持する」と言うのは、マルトース結合タンパク質に融合された生理活性ポリペプチドが融合された後にも、本来の生物学的活性または機能を50%以上、好ましくは60%以上、さらに好ましくは70%以上維持することを意味する。融合された生理活性ポリペプチドは、本来の生物学的活性または機能の80%以上を維持することがさらに好ましく、最も好ましいのは90%以上を維持することである。
【0027】
本発明の実施例では、マルトース結合タンパク質との融合タンパク質形態で発現、精製された生理活性ポリペプチドが、本来の生物学的活性または機能を99%以上維持する。
【0028】
また本発明は、前記固定化方法によって、生理活性ポリペプチドが、マルトース結合タンパク質をリンカーに使用して固体基質の疎水性表面に固定されている、生物活性固体基質を提供する。
【0029】
同時に本発明は、マルトース結合タンパク質の前記生物活性固体基質の製造のための使用を提供する。
【0030】
前記生物活性固体基質は、特定生物学的活性を有する生理活性ポリペプチドが前記固体基質の表面の外部に露出していて、目的とする分子または細胞受容体との相互作用が容易いなだけではなく、前記表面にどのような生物学的活性を有する生理活性ペプチドを固定させるのかによって、多様な目的に使用することができる。本発明による生物活性固体基質の最も一般的な用途は、細胞培養に使用することである。すなわち、前記固定化方法によって、特定細胞の培養に求められる生理活性ポリペプチドを、マルトース結合タンパク質を使用して疎水性材質の細胞培養容器基質に固定させて、前記培養容器に細胞を培養すると、細胞と生理活性ポリペプチドとの相互作用が直接的に成立して、培養を円滑に成立させることができる。このように、生理活性ポリペプチドが固定された生物活性固体基質に細胞を培養するようになれば、融合された生理活性ポリペプチドの生物学的活性によって細胞内信号伝達の変化をもたらし、細胞の形態および機能変化の誘導が可能なだけでなく、幹細胞の分化研究、組織工学のような再生医療分野、細胞センサーまたは細胞チップ研究に有用に使用することができる。
【0031】
また、本発明による生物活性固体基質を使用した細胞培養方法は、マルトース結合タンパク質によって疎水性表面に固定されて外部に露出した生理活性ポリペプチドが、直接細胞接着に関与するようにしたり、コラーゲン、フィブロネクチンおよびラミニンなどのような天然細胞の外基質(extracellular matrix、ECM)を一緒に吸着させて細胞接着に使用したり、固定された生理活性ポリペプチドを細胞の信号伝逹に使用することなどに活用することができる。
【0032】
また、本発明の生物活性固体基質は、診断装備などに適用することができる。例えば、診断しようとする生理活性ポリペプチドを疎水性材質からなるストリップ(strips)またはマイクロタイター(microtiter)の表面にマルトース結合タンパク質をリンカーに使用して固定させると、バイオセンサー(biosensor)として有用に使用することができる。
【0033】
前述したように、マルトース結合タンパク質を使用して生理活性ポリペプチドを固定することで生物活性固体基質を製造することは、固定された生理活性ポリペプチドと異なるポリペプチドとの相互作用に対する研究を可能にする。したがって、高性能スクリーニング(high−throughput screening)、固相抽出(solid phase extraction)またはクロマトグラフィー精製などに適用することができる。
【発明の効果】
【0034】
本発明による生理活性ポリペプチドの固定化方法は、原形質膜空間タンパク質であるマルトース結合タンパク質の表面に露出した疎水性ドメインをリンカーに使用して、生理活性ポリペプチドの生物学的活性はそのまま維持しながら、簡単な物理的吸着によって固体基質の疎水性表面に固定させることができ、このような生理活性ポリペプチドが固定された固体基質は、多様な細胞の培養、幹細胞の分化研究、組織工学のような再生医療分野、細胞センサーまたは細胞チップ研究などに非常に有用に使用することができるという長所を有する。
【図面の簡単な説明】
【0035】
【図1】本発明によって組換えられたマルトース結合タンパク質(MBP)と血管内皮細胞成長因子(VEGF)とのMBP−VEGF融合タンパク質をSDS−PAGEで分析した結果である。
【図2】本発明のMBP−VEGF融合タンパク質で処理された293/KDR細胞で処理濃度による細胞の形態変化を観察した結果である。
【図3】本発明のMBP−VEGF融合タンパク質で処理された293/KDR細胞でリン酸化シグナルの変化を観察した結果である。
【図4】本発明のMBP−VEGF融合タンパク質がポリスチレン疎水性表面に固定化される程度を生化学的に分析した結果である。
【図5】本発明のMBP−VEGF融合タンパク質がポリスチレン疎水性表面に固定化される程度を物理学的に分析した結果である。
【図6】本発明のMBP−VEGF融合タンパク質が固定されたポリスチレン疎水性表面でBSA存在下で培養された293/KDR細胞の形態変化を観察した結果である。
【図7】本発明のMBP−VEGF融合タンパク質が固定されたポリスチレン疎水性表面でBSA不在下で培養された293/KDR細胞の形態変化を観察した結果である。
【発明を実施するための形態】
【0036】
以下、実施例を通じて本発明をより詳しく説明する。
【0037】
これら実施例は、単に本発明をより具体的に説明するためのものであって、本発明の要旨にしたがって、本発明の範囲がこれら実施例によって制限されないということは、通常の知識を有する当業者にとって自明であろう。
<参考例1>制限酵素を使用したDNAの切断および切片の回収
使用した制限酵素と緩衝溶液は、エンザイノミックス(Enzynomics)社から購入したし滅菌された1.5mlエッペンドルフチューブ(eppendorf tube)に、普通反応体積が20〜30μlになるようにして、37℃の反応温度で4〜5時間反応させた。制限酵素反応に使用した10×緩衝溶液の組成を、下記に示す:
1)10×エンザイノミックス緩衝溶液EzバッファーI:100mM トリス−HCl(pH7.5、25℃)、50mM NaCl、10mM MgCl2、0.025% トリトンX−100、および
2)10×エンザイノミックス緩衝溶液EzバッファーII:10mM トリス−HCl(pH7.5、25℃)、50mM NaCl、10mM MgCl2、1mM ジチオトレイトール(dithiothreitol)。
【0038】
DNA切片の回収は、電気泳動されたアガロースゲルをUV透過照明装置(transilluminator、Avegene)で照射して所望する切片を切断した後、ゲル抽出キット(gel extraction kit、Qiagene)を使用して分離した。
<参考例2>細菌性アルカリフォスファターゼ(bacterial alkaline phosphatase)処理
細菌性アルカリフォスファターゼ(BAP)処理時に使用したBAP溶液は、フェルメンタス(Fermentas)社から購入し、滅菌された1.5mlエッペンドルフチューブに普通反応体積が50μlになるようにして、60〜65℃の反応温度で1時間反応させた。BAP反応には、1M トリス−HCl緩衝溶液(pH8.0)(Bioneer)を使用した。
<参考例3>ライゲーション反応(ligation reaction)
ライゲーション反応は、DNAライゲーションキット(DNA Ligation Kit Ver2.1、タカラ)を使用してベクター(vector)と挿入物(insert)の割合を1:3程度で混合して反応体積を10〜20μlになるようにして、16℃で少なくとも12時間以上反応させた。
<参考例4>大腸菌形質転換
形質転換のための大腸菌宿主細胞には、E.coli K12 TB1(New England Biolabs)を使用した。前記細胞を60mlの液体培地(10g/lバクト・トリプトン[bacto−tryptone]、5g/l 酵母抽出物、10g/l NaCl)に接種した後、吸光度(OD600)値が0.4〜0.6になるまで37℃振盪培養した。前記培養された細胞を1.5mlエッペンドルフチューブに分注して、遠心分離を通じて細胞を得た。得られた細胞に50mM CaCl2を300μlずつ添加して弱くボルテキシング(vortexing)した後、再度遠心分離を通じて細胞を収穫した。前記得られた細胞に、50mM CaCl2を300μlずつ添加して細胞を均一に分散させた後、0℃で30分間静置させた。前記静置液を再度遠心分離して上澄み液は捨てた後、50mM CaCl2に15%グリセロールを添加した冷たい溶液を、残った細胞に150μlずつ添加して均一に分散させて冷凍保管した。
<参考例5>オリゴヌクレオチドの合成
目的とする生理活性ポリペプチドをコードする遺伝子を増幅するために、重合酵素連鎖反応(PCR)に使用されるプライマー対は、バイオニア(Bioneer)社のオリゴヌクレオチド合成サービスを使用して製作した。
<参考例6>重合酵素連鎖反応
鋳型50ngと正方向および逆方向プライマーそれぞれ10μMに蒸留水を添加して、総体積が10μlになるようにした後、ホットスタートPCRプレミックス(Hot Start PCR Premix、Bioneer)に添加した。温度循環器(T−gradient thermo block、Applied Biometra)を使用して、前記反応混合物を95℃で1分間変性させた後、94℃で30秒、55℃で30秒、および68℃で1分間の反応を31回反復して、最終的に72℃で5分間増幅させた。前記増幅された反応産物をPCR精製キット(PCR purification kit、Bioneer)を使用して精製して、アガロースゲルに電気泳動した後、UVトランスイルミネーター(transilluminator)(Avegene)を照射して所望する切片を切断した。回収されたゲル切片からゲル抽出キット(gel extraction kit、Qiagene)を使用して増幅されたDNAを分離した。
<参考例7>細胞培養
HEK293ヒト胚芽腎臓細胞から由来した293/KDR細胞は、ヒトVEGFR−2(KDR/Flk1)受容体を発現するように作られた細胞である。293/KDR細胞はシブテック(Sibtech)社から5回継代培養された状態で購入した。細胞培養には、DMEM液体培地(Dulbecco’s Modified Eagle’s Medium、ウェルジン)( D−グルコース 4500mg/l 、L−グルタミン、 ピルビン酸ナトリウム 110mg/l 、10%FBS)に、0.375μg/mlのピュロマイシン(Puromysin、Sigma)を添加した培地を使用して、37℃で5%のCO2を添加しながらインキュベーター(Thermo)で培養した。培地は、2日間隔で新しいものと交換して、細胞がT−フラスコに70〜80%程度生育した後に継代した。
<参考例8>7.5%ポリアクリルアミドゲル(polyacrylamide gel)の製造
ミニプロテアン3電気泳動セット(Mini−protean 3 Electrophoresis Set)(Bio−rad)を使用して、7.5%ポリアクリルアミドゲルを製造した。まず、1.0mmのガラスプレートをフレームに固定させて、キャスティングフレーム(casting frame)を形成した。50mlのコニカルチューブに蒸留水4.94mlと1.5M トリス−HCl緩衝溶液を2.5ml、30%アクリルアミド溶液を2.5ml、10%過硫酸アンモニウム(ammonium persulfate、APS)50μlおよびTEMED(N,N,N’、N’−テトラメチルエチレンジアミン)10μlを添加してよく混合した後、前記1.0mmガラスプレートに4.5mlずつ入れて解像度ゲル(resolution gel)を作製した。その後、ゲルが乾かないように500μlの蒸留水を注入した後、解像度ゲルが完全に固まるとゲルの上の蒸留水を除去した。スタッキングゲル(stacking gel)を製造するために、50mlのコニカルチューブに蒸留水3.05ml、0.5M トリス−HCl緩衝溶液 1.25ml、30%アクリルアミド溶液 0.67ml、10%APS 25μlおよびTEMED 5μlを添加してよく混合した後、1.0mmガラスプレートに最後まで注いで、15ウェル(20μl)鋳型を挿入した後、固めた。ポリアクリルアミドゲルに使用された試薬の組成を下記に示す:
1)1.5M トリス−HCl緩衝溶液:トリス塩基 18.17g(Invitrogen)、20% SDS(Amersham Pharmacia Biotech)2 ml、蒸留水 80ml,(pH8.8)、
2)0.5M トリス−HCl緩衝溶液:トリス塩基 6.06g(Invitrogen)、20% SDS(Amersham Pharmacia Biotech)2ml、蒸留水 80ml,(pH6.8)、および
3)30% アクリルアミド溶液:29% アクリルアミド(Sigma)、1% ビス−アクリルアミド(Sigma)。
<参考例9>ビオチンカップリング(biotin coupling)
タンパク質のビオチンカップリングは、スルホ−NHS−ビオチン化キット(sulfo−NHS−biotinylation kit、Pierce)を使用して行った。反応体積が0.5〜2ml、タンパク質の濃度が1〜10mg/mlになるようにリン酸塩緩衝溶液(phosphate buffered saline、PBS)でタンパク質を希釈した後、10mM スルホ−NHS−ビオチン溶液をビオチン処理されるタンパク質の分子量によって計算して添加して、1時間常温で反応させた。一方、スルホ−NHS−ビオチン化キット内に含まれている脱塩スピンカラム(desalt spin column)に15mlのコニカルチューブを結合させて、1000×gで2分間遠心分離(ハニル)を行った後、コニカルチューブに集められた溶液を除去した。その後、脱塩スピンカラムにPBS 2.5mlを添加して、1000×gで2分間遠心分離をして脱塩スピンカラムを洗浄した。前記洗浄過程を2回繰り返し実施した。このように準備した脱塩スピンカラムに新しい15mlのコニカルチューブを結合させた後、前記ビオチン処理された反応物を添加して、1000×gで2分間遠心分離を行ってビオチンが結合されたタンパク質を分離した。
【実施例】
【0039】
<実施例1>組換えタンパク質を発現するプラスミドpMAL−c2X−VEGFの製造<1−1>VEGF遺伝子がクローニングされたプラスミドの製造
VEGF遺伝子がクローニングされたプラスミドを製造するため、配列番号1の塩基配列を有する正方向プライマーVEGF−F(EcoRI)および配列番号2の塩基配列を有する逆方向プライマーVEGF−R(HindIII)を合成した。前記プライマー対を使用して、ヒト血管平滑筋(vascular smooth muscle、VSM)細胞から抽出した全体遺伝子を鋳型に、重合酵素連鎖反応(PCR)を行って、血管内皮細胞成長因子(vascular endothelial growth factor、VEGF)165部分のみを増幅した。
【0040】
pGEM−TベクターシステムI(Promega)に含まれている2×ライゲーション緩衝溶液(rapid ligation buffer)5μlに、前記で増幅されたVEGF遺伝子断片4ng、pGEM−Tベクター50ngおよびT4 DNAリガーゼ(ligase)1μlを添加した後、総体積が10μlになるように蒸留水を添加して、常温で1時間放置した後、続いて16℃で12時間反応させた。反応が終結した後、連結物を大腸菌E.coli K12 TB1に形質転換させて、それから目的とする遺伝子がクローニングされた組換えプラスミドを選別して、pGEM−VEGFと命名した。
<1−2>MBP−VEGF融合遺伝子がクローニングされたプラスミドの製造
リンカーとしてマルトース結合タンパク質(MBP)とVEGF遺伝子とを融合させるために、前記実施例<1−1>で製造された組換えプラスミドpGEM−VEGFを、エンザイノミックス緩衝溶液EzバッファーI、緩衝溶液EzバッファーII中で、制限酵素EcoRIとHindIIIで切断した後、アガロースゲル上でVEGF遺伝子切片を分離した。以後のライゲーション反応を容易に行うために、分離したVEGF遺伝子をBAPで処理した。BAP処理は、緩衝溶液(1M トリス−HCl、pH8.0、Bioneer)7.5μlおよびBAP溶液(Fermentas)1μlを入れた後、最終体積が50μlになるようにVEGF遺伝子を添加して、65℃で1時間反応させた。反応物をアガロースゲルで電気泳動した後、UVトランスイルミネーター(Avegene)で照射して所望する部分を切断した後、ゲル抽出キット(gel extraction kit、Qiagene)を使用してVEGF遺伝子断片を分離した。
【0041】
一方、ライゲーション反応に使用されるMBP遺伝子を有しているベクターpMAL−c2X(New England Biolabs)を、エンザイノミックス緩衝溶液EzバッファーI、緩衝溶液EzバッファーIIで制限酵素EcoRIとHindIIIで切断した後、アガロースゲル上でMBP遺伝子含有ベクター切片を分離した。
【0042】
前記で分離したVEGF遺伝子9μl、切断したベクター切片pMAL−c2X 3μlおよびDNAライゲーションキット(DNA Ligation Kit Ver2.1、タカラ)に含まれた酵素溶液I(enzyme solution I)12μlを混合して、総体積が20μlになるように蒸留水を加えた後、16℃で16時間反応させた。反応が終結した後、接合物を大腸菌E.coli K12 TB1に形質転換させて、それからMBPとVEGFの融合遺伝子がクローニングされた組換えプラスミドをスクリーニングして、pMAL−c2X−VEGFと命名した。
<実施例2>MBP−VEGF融合タンパク質の発現および精製
<2−1>MBP−VEGF融合タンパク質の発現誘導
MBP−VEGF融合タンパク質を発現する組換えプラスミドpMAL−c2X−VEGFを、大腸菌E.coli K12 TB1に形質転換させて、37℃のLB(Luria−Bertani)固体培地で一日の間培養した。翌日、培地の上に形成されたコロニー(colony)を採取して、60μg/ml濃度のアンピシリンを含む3mlのRB(rich medium+グルコース)液体培地に接種して、37℃で約2時間培養した。そこに、IPTG(イソプロピル−β−D−チオガラクトピラノシド)を最終濃度3mMになるように添加して、再び37℃で2時間さらに培養した。培養が終わった後、1mlの培養液を採取して、遠心分離機を通じて細胞沈殿物を得た。前記細胞沈殿物に20μlの1×試料ローディング緩衝溶液(sample loading buffer)を添加してよく混合した。前記混合物を95℃で5分間沸かしてから常温に冷却させた後、15μlを取って10%SDS−ポリアクリルアミドゲル電気泳動を行った。電気泳動が終結した後、ポリアクリルアミドゲルをクマシブルー(coomassie brilliant blue)で染色して、抗−MBP抗血清(New England Biolabs)を使用したウエスタンブロットを通じて、MBP−VEGF融合タンパク質の発現有無を観察した。
<2−2>水溶性融合タンパク質の発現および精製
前記実施例<2−1>で組換えプラスミドpMAL−c2X−VEGFに形質転換された大腸菌細胞を、アンピシリンが60μg/ml添加されたRB培地に接種した後、37℃で一晩中培養した。前記培養液10mlを1lのRB培地に添加して、37℃で継続振盪培養し、培養液の吸光度が650nmの波長で約0.6程度になった時、3mMの最終濃度でIPTGを添加した。IPTGを添加して約2時間後に培養を停止した後、培養液を4000×gで20分間遠心分離(Combi−514R、ハニル)して細胞沈殿物を集めた。前記細胞沈殿物に緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml)50mlを添加して懸濁させた後、最終濃度が1mMになるようにEDTA(ethylenediaminetetraacetic acid)とPMSF(phenylmethanesulphonyl fluoride)を添加した。前記細胞混合物の精製に先立って、−20℃での冷凍および解凍を繰り返して細胞破砕が容易に起きるようにした。その後、細胞混合物を冷却浴内で超音波粉砕機(Fisher Scientific Model 500 Sonic Dismembrator)を使用して、10%出力で約10秒間超音波処理して細胞を破砕した後、30秒間冷却浴内に放置した。前記のような過程を2回反復して細胞を完全に破砕した。このように得た細胞均質液を、9000×gで1時間遠心分離(Combi−514R、ハニル)して、可溶性タンパク質が溶解している上澄み液を得た後、緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA、2ml)で5倍希釈した。
【0043】
一方、大腸菌形質転換体から発現されたMBP−VEGF融合タンパク質を分離するために、アミロース樹脂(amylose resin、New England Biolabs)を使用したアフィニティークロマトグラフィーを準備した。このカラムをあらかじめ約8×ベッドボリューム(bed volume)の緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml)で洗浄して平衡化させた。平衡化されたアミロース樹脂親和性クロマトグラフィーに、前記で得た可溶性タンパク質が溶解している上澄み液を分当たり1mlの速度でローディング(loading)した。続いて、12×ベッドボリューム以上の緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml)をカラムに流して、樹脂に吸着されないタンパク質を除去した。その次に、10mM マルトース溶出緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml、10mM マルトース)を添加して、樹脂に吸着されていたタンパク質を分当たり1mlの速度で溶出させて回収した。その後、回収されたタンパク質を10%ポリアクリルアミドゲルに電気泳動(Bio−rad)して、精製されたタンパク質の概算の分子量と純度を確認した。その結果、精製されたタンパク質の純度は95%以上で、概算の分子量は60,000Da程度と示された。
【0044】
前記タンパク質試料を、希釈膜(MWCO12−14,000)(dialysis membrane、Spectrum laboratories、Inc.)に入れてPBSで3日間希釈させた後、マルトースが除去されたタンパク質のみを収得した。その後、遠心分離ろ過器(Centrifugal Filter、Amicon Ultra−15 MWCO 5,000、Millipore)を使用して、4000×gで45分間遠心分離(Combi−514R、ハニル)してタンパク質を濃縮した。前記過程によって最終的に濃縮されたタンパク質を、MBP−VEGFと命名した。
【0045】
図1は、アミロースレジンにローディングする前のMBP単独(ライン1)およびMBP−VEGF融合タンパク質(ライン3)と、アミロースレジンにローディングした後に精製されたMBP単独(ライン2)およびMBP−VEGF融合タンパク質(ライン4)のSDS−PAGE結果を示したもので、MBP−VEGF融合タンパク質が、MBP単独より高い分子量を有することが確認されて、大腸菌形質転換体から融合タンパク質が発現されたことが分かった。
<実施例3>MBP−VEGF融合タンパク質の活性測定
<3−1>MBP−VEGF融合タンパク質による293/KDRのリン酸化
MBP−VEGF融合タンパク質の活性を測定するために、まず293/KDRを培養した後、6−ウェルに5×105の細胞濃度で移植した。293/KDR細胞は、293HEK(human embryonic kidney cell)にVEGFR2(VEGF受容体)を過発現させた細胞で、VEGFのリン酸化試験などに使用されている。細胞移植6時間後、0.05%のFBS(fetal bovine serum、ウェルジン)が添加されたDMEM液体培地(Dulbecco’s Modified Eagle’s Medium、ウェルジン)に培地を交換した。それから16時間経過後、前記培地を分析培地(assay medium、DMEM、ウェルジン、25mM HEPES pH7.2、Sigma、5mM Na3VO4、Sigma、0.05%BSA(bovine serum albumin、Sigma))に交換して、10分間培養器(Thermo)で培養した。
【0046】
一方、293/KDR細胞を刺激するためにVEGF165(vascular endothelial cell growth factor 165、R&D Systems)および製造されたMBP−VEGF融合タンパク質を、それぞれ0、1、5、10、50および100ng/mlの濃度で分析培地に希釈した。続いて希釈されたVEGFおよびMBP−VEGF融合タンパク質を、前記で培養された293/KDR細胞に1.5mlずつ濃度別にそれぞれのグループに添加した後、37℃培養器(Thermo)で10分間培養した。VEGFおよびMBP−VEGF融合タンパク質によって刺激された細胞を氷に浸した後、冷たいPBSで2回洗浄した。続いて細胞を破砕するために、RIPA(Radio−Immunoprecipitation Assay、Pierce)緩衝溶液に1mM オルトバナジン酸塩ナトリウム(sodium orthovanadate、Sigma)、5mM ピロリン酸塩ナトリウム(sodium pyrophosphate、Sigma)および25mM フッ化ナトリウム(sodium fluoride、Sigma)を混合した後、100μlの混合液を前記細胞反応液に添加してスクラッパー(scraper、SPL)を使用して細胞を回収した。回収された各グループの細胞を1時間氷に浸した後、15,000rpmで30分間遠心分離(micro 17TR、ハニル)して上澄み液のみを採取して、BCA(Pierce)タンパク質測定方法を使用して定量した。それぞれの試料を5×試料緩衝溶液(0.6ml 1M トリス−HCl、pH6.8、5ml 50% グリセロール、2ml 10% SDS、0.5ml 2−メルカプトエタノール、1ml 1%ブロモフェノールブルー、0.9ml 蒸留水)に添加して、蒸留水で各試料の最終濃度を同一に合わせた後、1.5ml エッペンドルフチューブに入れて4℃で冷蔵保管した。
【0047】
図2は、細胞を破送する前の細胞形態を観察した結果で、図2のAは細胞に分析培地を添加する前の細胞形態で、Bは分析培地を、CはMBP(100ng/ml)を、DはMBP−VEGF(150ng/ml)を、EはVEGF(50ng/ml)を10分間処理した後の細胞形態である。分析培地とMBPを処理した群(BおよびC)では、細胞形態が処理する前の形態と類似に伸長した状態であるが、MBP−VEGFとVEGFを処理した群(DおよびE)では細胞が丸い形態に変化したことが分かった。このような結果は、本発明によってVEGFがMBPとの融合タンパク質形態に発現されて精製されても、本来のVEGF活性をそのまま維持していることを示すものである。
<3−2>MBP−VEGF融合タンパク質による293/KDRのリン酸化シグナル変化
前記実施例<3−1>で準備した試料を95℃で10分間加熱した後、15,000rpmで1分間遠心分離(micro 17TR、ハニル)して、ふたに気化された試料を集めた。ウエスタンブロット(western blotting)分析のために7.5% ポリアクリルアミドゲルを製造して、前記ゲルに前記試料をそれぞれ20μlずつローディングした後、電気泳動(Bio−rad社)を実施した。一方、ニトロセルロース膜とろ過紙を10×トリス/グリシン緩衝溶液(25mM トリス、192mM グリシン、pH8.3、Bio−rad)20mlとMeOH 40mlおよび蒸留水140mlを混合した溶液に20分間浸けて活性化させた。
【0048】
その後、半乾燥伝達システム(Semi−Dry Transfer System、Bio−rad)を使用して、前記で電気泳動されたポリアクリルアミドゲルをニトロセルロース膜に伝達した。前記ニトロセルロース膜を5%BSA(bovine serum albumin、Sigma)で4℃で8時間処理して遮断させた後、TBS−T(Tris buffered saline−NaCl 8g、KCl 0.2g、トリス 3g、ツイーン20 0.5ml)緩衝溶液で10分間3回洗浄した。その後、1次抗体であるホスホチロシン:ビオチン(phosphotyrosine:biotin、BD Biosciences)と単一クローン抗−ホスホチロシン(monoclonal anti−phosphotyrosine、Sigma)を5%BSA(bovine serum albumin、Sigma)に1:2000に希釈した後、前記洗浄されたニトロセルロース膜に10mlずつ添加して、8時間4℃で反応させた。前記ニトロセルロース膜をTBS−T緩衝溶液で10分間3回洗浄した後、5%BSA(bovine serum albumin、Sigma)に1:10000に希釈された2次抗体(Mouse−Pierce)と、ストレプタビジン(streptavidin、Sigma)とを10mlずつ添加して、1時間常温で反応させた。前記ニトロセルロース膜をTBS−T緩衝溶液で10分間3回洗浄した後、ウェストフェムト最大敏感性基質(West Femto maximum sensitivity substrate、Pierce)に含まれているルミノール/エンハンサー溶液(West Femto Luminol/Enhancer Solution)と、過酸化溶液(West Femto Stable Peroxide Solution)とを、それぞれ500μlずつ混合した溶液を添加して、常温で5分間反させた。反応が終結した後、前記ニトロセルロース膜をイメージ分析機(image analyzer、Las 3000、フジフィルム)で分析して、MBP−VEGF融合タンパク質による293/KDR細胞のリン酸化シグナルを確認した。
【0049】
その結果、図3に示したように、MBP−VEGF融合タンパク質で処理した細胞では、VEGFだけで処理した細胞群と類似して220KDa付近から濃度依存的にリン酸化が起きることを分かった。このようなリン酸化様相は、VEGFによって処理された293/KDR細胞で本実施例に使用された抗体によって示されるリン酸化様相と同一である(Backer MV等、Biomaterials,2006年,第27巻,5452−5458頁)。その他にも、130KDa付近で細胞内のリン酸化が濃度依存的に増加することを確認することができた。
<実施例4>ポリスチレン表面でのMBP−VEGF融合タンパク質の固定化に対する生化学的分析
本発明によるMBP−VEGF融合タンパク質が、ポリスチレン(polystyrene)のような疎水性表面に固定化される程度を調べるため、まず、下記のようにMBP−VEGF融合タンパク質をビオチン(biotin)で処理した。具体的に、1mg/mlのMBP−VEGF融合タンパク質185μlに、10mM スルホ−NHS−ビオチン溶液16μlを入れた後、最終体積が0.5mlになるようにPBSを添加して、1時間常温で反応させた。一方、<参考例10>に記述した方法で脱塩スピンカラム(Pierce)を洗浄した後、前記反応物を15mlコニカルチューブを結合した洗浄された脱塩スピンカラムに入れて、1000×gで2分間遠心分離してビオチン処理されたMBP−VEGF融合タンパク質を得た。
【0050】
10〜10,000ng/ml濃度のビオチン処理されたMBP−VEGF融合タンパク質を100μlずつ、ポリスチレン製の96ウェル(非−組織培養用、Falcon)に加えて、常温で4時間処理して、PBS200μlで3回洗浄した。その後、ポリスチレン表面にMBP−VEGF融合タンパク質の非特異的結合を防止するために、1%BSA(bovine serum albumin、Sigma)200μlを入れて常温で2時間処理した後、0.05% ツイーン20(Amersham Pharmacia Biotech)含有PBS200μlで5回洗浄した。
【0051】
前記のMBP−VEGFが固定された96ウェルに、ストレプタビジンペルオキシダーゼ(streptavidin−peroxidase、Sigma)がPBSに1:10,000に希釈された溶液を100μl添加して、常温で1時間反応させた後、0.05%ツイーン20含有PBS200μlで5回洗浄した。続いて、前記96ウェルに安定化された過酸化水素(stabilized hydrogen peroxide、R&D Systems)と安定化されたテトラメチルベンジジン(stabilized tetramethylbenzidine、R&D Systems)を1:1で混合した溶液100μlを添加してアルミホイルで包んだ後、20分間反応させた。20分後、2N硫酸溶液50μlを前記ウェルに添加して反応を停止させた。前記96ウェルをマイクロプレートリーダー(microplate reader、Molecular Device)を使用して450nmで吸光度を測定して、MBP−VEGF融合タンパク質がポリスチレン表面に固定化された程度を確認した。
【0052】
その結果、図4に示されたように、MBPおよびMBP−VEGFが10〜1,000ng/mlの範囲で、濃度依存的に疎水性ポリスチレン表面に吸着されることが分かった。しかし、前記範囲を逸脱した高い濃度では、このような生化学的方法で固定化された程度を分析するのに限界があった。
<実施例5>ポリスチレン表面でのMBP−VEGF融合タンパク質の固定化に対する物理学的分析
前記実施例4の生化学的方法では、高濃度でMBP−VEGF融合タンパク質の固定化程度を測定するのに限界があることを確認して、物理学的方法である水晶振動子微量秤(quartz crystal microbalance、QCM)を使用して、高濃度で前記融合タンパク質が疎水性表面に吸着する程度を調査した。QCMは、水晶振動子(quartz crystal)に物質が吸着した時に振動数の減少が生じて、このような振動数の減少を検出して吸着されたタンパク質の量を測定する方法である。本実験で水晶振動子は、0.5%ポリスチレン/トルエン溶液をスピンコーティングした後、使用した。試料は、MBPおよびMBP−VEGFを、それぞれ1、10、50、100および500μg/mlの濃度で緩衝溶液(1M トリス−HCl 20ml、pH7.5、NaCl 11.7g、0.5M EDTA 2ml)に希釈して準備した。水晶振動子に前記緩衝溶液を1時間流して平衡化させた後、準備したそれぞれの指標を15分間流して水晶振動子にMBPおよびMBP−VEGFが吸着するようにした後、再び緩衝溶液を流して吸着されないタンパク質を除去した。その後、タンパク質吸着前と比べて吸着後の水晶振動子の振動数減少を測定して、吸着した重さをソルベリー式(Sauerbrey equation、Δf=−ΔmC/n)(Hook F、Rodahl M.等、Langmuir.,1998年,第14巻,729−734頁)に代入して算出した。
【0053】
その結果、図5に示されように、本発明によるMBP−VEGF融合タンパク質が、1〜500μg/mlの高濃度でも濃度依存的に疎水性ポリスチレン表面に吸着して、このような濃度によるタンパク質の吸着がログ関数的増加を示すラングミュアタイプ(Langmuir−type)の吸着挙動と類似に単層に吸着することを確認した。
<実施例6>MBP−VEGF融合タンパク質による293/KDR細胞の形態変化
MBP−VEGF融合タンパク質による293/KDR細胞の形態変化を調査するため、まず、前記<実施例2>で抽出したMBP−VEGF融合タンパク質を無菌作業台(Sanyo)で0.22μm注射器フィルター(Millex GV、Millipore)を使用してろ過した後、0.1、1および10μg/mlの濃度で100μlずつポリスチレン材質の96ウェルプレート(非−組織培養用、Falcon)に添加した後、プレート表面にコーティングされるように無菌作業台中で4時間放置した。その後、前記96ウェルをPBS200μlで3回洗浄し、293/KDR細胞とMBP−VEGF融合タンパク質がコーティングされたウェル表面との非特異的結合を防止するために、一部のウェルには1%BSA(bovine serum albumin、Sigma)200μlを添加して無菌作業台内で2時間処理した後、PBS200μlで3回洗浄した。
【0054】
MBP−VEGF融合タンパク質がコーティングされた96ウェルに、ウェル当たり293/KDR細胞を2×104の濃度で移植した。ここで、使用された培地は、DMEM(ウェルジン)無血清培地を使用した。移植された細胞は、37℃培養器(Thermo)で45時間培養した後、位相差顕微鏡(ニコン社)を使用してこれらの形態変化を観察した。
【0055】
図6および図7は、それぞれ本発明のMBP−VEGF融合タンパク質が固定されたポリスチレン疎水性表面において、BSA存在下および不在下に培養された293/KDR細胞の形態変化を観察した結果である。図6に示されたように、細胞の非特異的な結合を防止するためにMBP−VEGFが吸着された表面にBSAを処理した場合に、MBP−VEGFが吸着されない表面(図6のA)では細胞が凝固した丸い(spheroid)形態を維持していて、吸着されたMBP−VEGFの濃度が増加するに比例して細胞に偽足が形成されながら細長い形態に生育することを確認することができた(図6のB〜D)。
【0056】
一方、図7に示されたように、MBP−VEGFが吸着した表面にBSAを処理しない場合に、MBP−VEGFが吸着されない表面(図7のA)では細胞がよく接着して広くひろがった形態に生育したが、MBP−VEGFが吸着した表面では図6のB〜Dと類似に細胞が細長に偽足を生成しながら生育することを確認した。このような結果は、本発明によるMBP−VEGF融合タンパク質のVEGFが、細胞の形態形成に影響を及ぼしていることを示すものである。
【0057】
以上、本発明の具体的な内容を詳しく記述したように、通常の知識を有する当業者において、このような具体的技術は単に好ましい実施様態であるだけであり、これによって本発明の範囲が制限されるものではない点は明白であろう。したがって、本発明の実質的な範囲は、添付された請求項とそれらの均等物によって定義される。
【特許請求の範囲】
【請求項1】
1)マルトース結合タンパク質(MBP)のカルボキシル末端に、生理活性ポリペプチドが融合された融合タンパク質を得る工程、
2)前記融合タンパク質を、マルトース結合タンパク質の表面に露出した疎水性ドメインを含むアミノ末端と固体基質の疎水性表面との物理的吸着によって、前記疎水性表面に固定させる工程、および、
3)前記融合タンパク質が固定された前記疎水性表面において細胞を培養する工程、
を含む、細胞培養方法。
【請求項2】
工程1)の融合タンパク質が、
(i)目的とする生理活性ポリペプチドが、マルトース結合タンパク質のカルボキシル末端に融合された融合タンパク質を、暗号化する融合遺伝子断片を得る工程、
(ii)前記融合遺伝子断片を含む発現ベクターを製造する工程、
(iii)前記発現ベクターで形質転換された形質転換微生物を製造する工程、および
(iv)前記形質転換微生物から融合タンパク質を発現および精製する工程、
を含む工程によって得られることを特徴とする、請求項1記載の細胞培養方法。
【請求項3】
工程(ii)の発現ベクターが、大腸菌用発現ベクターであることを特徴とする、請求項2記載の細胞培養方法。
【請求項4】
工程(iii)の形質転換微生物が、大腸菌であることを特徴とする、請求項2または3に記載の細胞培養方法。
【請求項5】
工程(iv)の融合タンパク質が、マルトース特異的なアフィニティークロマトグラフィーを使用して精製されることを特徴とする、請求項2〜4のいずれかに記載の細胞培養方法。
【請求項6】
工程2)の疎水性表面が、シラン化表面、カーボンナノチューブ(CNT)表面、炭化水素でコーティングされた表面、高分子表面および金属表面からなる群から選択されるいずれか1種であることを特徴とする、請求項1〜5のいずれかに記載の細胞培養方法。
【請求項7】
高分子が、ポリスチレン、ポリカーボネート、ポリプロピレン、ポリエチレン、テフロン(登録商標)、ポリテトラフルオロエチレン、およびポリエステル含有生分解性高分子からなる群から選択されるいずれか1種であることを特徴とする、請求項6に記載の細胞培養方法。
【請求項8】
金属が、ステンレススチール、チタン、金および白金からなる群から選択されるいずれか1種であることを特徴とする、請求項6に記載の細胞培養方法。
【請求項9】
工程2)の物理的吸着が、融合タンパク質と疎水性表面とを4〜25℃で1〜24時間反応させて成立することを特徴とする、請求項1〜8のいずれかに記載の細胞培養方法。
【請求項10】
工程2)の融合タンパク質の形態において、疎水性表面に固定された生理活性ポリペプチドが、疎水性表面の外部に向くように露出することを特徴とする、請求項1〜9のいずれかに記載の細胞培養方法。
【請求項11】
前記生理活性ポリペプチドは血管内皮細胞成長因子(VEGF)であることを特徴とする、請求項1〜10のいずれかに記載の細胞培養方法。
【請求項1】
1)マルトース結合タンパク質(MBP)のカルボキシル末端に、生理活性ポリペプチドが融合された融合タンパク質を得る工程、
2)前記融合タンパク質を、マルトース結合タンパク質の表面に露出した疎水性ドメインを含むアミノ末端と固体基質の疎水性表面との物理的吸着によって、前記疎水性表面に固定させる工程、および、
3)前記融合タンパク質が固定された前記疎水性表面において細胞を培養する工程、
を含む、細胞培養方法。
【請求項2】
工程1)の融合タンパク質が、
(i)目的とする生理活性ポリペプチドが、マルトース結合タンパク質のカルボキシル末端に融合された融合タンパク質を、暗号化する融合遺伝子断片を得る工程、
(ii)前記融合遺伝子断片を含む発現ベクターを製造する工程、
(iii)前記発現ベクターで形質転換された形質転換微生物を製造する工程、および
(iv)前記形質転換微生物から融合タンパク質を発現および精製する工程、
を含む工程によって得られることを特徴とする、請求項1記載の細胞培養方法。
【請求項3】
工程(ii)の発現ベクターが、大腸菌用発現ベクターであることを特徴とする、請求項2記載の細胞培養方法。
【請求項4】
工程(iii)の形質転換微生物が、大腸菌であることを特徴とする、請求項2または3に記載の細胞培養方法。
【請求項5】
工程(iv)の融合タンパク質が、マルトース特異的なアフィニティークロマトグラフィーを使用して精製されることを特徴とする、請求項2〜4のいずれかに記載の細胞培養方法。
【請求項6】
工程2)の疎水性表面が、シラン化表面、カーボンナノチューブ(CNT)表面、炭化水素でコーティングされた表面、高分子表面および金属表面からなる群から選択されるいずれか1種であることを特徴とする、請求項1〜5のいずれかに記載の細胞培養方法。
【請求項7】
高分子が、ポリスチレン、ポリカーボネート、ポリプロピレン、ポリエチレン、テフロン(登録商標)、ポリテトラフルオロエチレン、およびポリエステル含有生分解性高分子からなる群から選択されるいずれか1種であることを特徴とする、請求項6に記載の細胞培養方法。
【請求項8】
金属が、ステンレススチール、チタン、金および白金からなる群から選択されるいずれか1種であることを特徴とする、請求項6に記載の細胞培養方法。
【請求項9】
工程2)の物理的吸着が、融合タンパク質と疎水性表面とを4〜25℃で1〜24時間反応させて成立することを特徴とする、請求項1〜8のいずれかに記載の細胞培養方法。
【請求項10】
工程2)の融合タンパク質の形態において、疎水性表面に固定された生理活性ポリペプチドが、疎水性表面の外部に向くように露出することを特徴とする、請求項1〜9のいずれかに記載の細胞培養方法。
【請求項11】
前記生理活性ポリペプチドは血管内皮細胞成長因子(VEGF)であることを特徴とする、請求項1〜10のいずれかに記載の細胞培養方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−245013(P2012−245013A)
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願番号】特願2012−205067(P2012−205067)
【出願日】平成24年9月18日(2012.9.18)
【分割の表示】特願2008−272881(P2008−272881)の分割
【原出願日】平成20年10月23日(2008.10.23)
【出願人】(304039548)コリア・インスティテュート・オブ・サイエンス・アンド・テクノロジー (36)
【Fターム(参考)】
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願日】平成24年9月18日(2012.9.18)
【分割の表示】特願2008−272881(P2008−272881)の分割
【原出願日】平成20年10月23日(2008.10.23)
【出願人】(304039548)コリア・インスティテュート・オブ・サイエンス・アンド・テクノロジー (36)
【Fターム(参考)】
[ Back to top ]