
細胞培養物の寿命および由来するタンパク質の収率を増加させるための方法および組成物
【課題】細胞培養物の寿命を増加させ、タンパク質、抗体、ペプチド、酵素、成長因子、インターロイキン、インターフェロン、ホルモン、およびワクチンなどの組換えタンパク質、の産生を増加させる組成物および方法を開示する。
【解決手段】細胞のアポトーシス阻害遺伝子またはベクターでのトランスフェクションにより、培養物中の細胞はより長く生存することができ、それにより、タンパク質生合成の状態および収率が増大する。細胞内でのアポトーシスインヒビターの発現により、これが細胞を死滅させないので、細胞またはその増加したフラクションをより長い期間培養物中で維持することが可能である
【解決手段】細胞のアポトーシス阻害遺伝子またはベクターでのトランスフェクションにより、培養物中の細胞はより長く生存することができ、それにより、タンパク質生合成の状態および収率が増大する。細胞内でのアポトーシスインヒビターの発現により、これが細胞を死滅させないので、細胞またはその増加したフラクションをより長い期間培養物中で維持することが可能である
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2004年7月23日出願の米国特許出願番号60/590,349号の優
先権を主張する。本出願は、上記仮出願で開示された主題のみを主張し、それにより、新
規事項を示さない。
【背景技術】
【0002】
特に、巨大なバイオリアクターにおけるインビトロでの細胞培養は、多数のバイオテク
ノロジー産物の産生の基本であり、支持体へのこれらのタンパク質産物の細胞による作製
(elaboration)を含み、これらの産物を単離し、臨床的使用の前にさらに処
理する。培養物中で増殖した細胞からの長期にわたるタンパク質の産生量は、例えば、細
胞密度、細胞周期の段階、細胞のタンパク質生合成率、細胞の生存度および増殖を支持す
るために使用される培地の条件、ならびに培養物中の細胞の寿命(プログラム細胞死(す
なわち、アポトーシス)のために死滅するまでの期間)などの多数の要因に依存する。例
えば、栄養素、細胞密度、酸素および二酸化炭素の含有量、乳酸デヒドロゲナーゼ、pH
、浸透圧、異化生成物などの調節による所望のタンパク質の生産性を増加させる方法と共
に、培養物中の細胞の生存度および寿命に対する種々の改良方法が開発されている。例え
ば、細胞密度の増加により、過程の生産性をより高くすることができるが、培養物中の細
胞の寿命も短縮され得る。したがって、最大密度が達成された場合、できるだけ長く最も
生産性の高い状態で細胞集団を維持するために、培養物中のこのような細胞の増殖速度を
低下させることが望ましい。これにより、その産生ピークでのバイオリアクターサイクル
が増加または延長され、所望のタンパク質産物が長期間で作製され、これにより、バイオ
リアクターサイクル由来の収率がより高くなる。
【0003】
細胞増殖を支持する培地の調整、一定の成長促進因子の添加、およびタンパク質合成に
影響を与えない細胞増殖の阻害などのバイオリアクターのサイクル時間を増加させるため
の多数の異なるアプローチが追求されている。1つの特定のアプローチは、細胞周期標的
に影響を与えるための遺伝子またはアンチセンスオリゴヌクレオチドの使用による細胞周
期の調節によって培養細胞の寿命を増加させることを目的とし、それにより、細胞周期の
進行を阻害していわゆる偽老化状態を誘導するベクターでのトランスフェクション、形質
転換、または感染によって細胞を偽老化状態に誘導し、さらに、細胞分裂を遮断し、培養
物中の細胞のタンパク質合成能を拡大する。言い換えれば、細胞周期インヒビターを発現
するベクターでの細胞の形質転換によって偽老化状態を誘導することができる(Bucc
iarelli et al.,US Patent 2002/0160450 A1
;idem.,WO 02/16590 A2)。後者の方法は、Goldsteinお
よびSingal(Exp Cell Res 88,359−64,1974;Bre
nner et al.,Oncogene 17:199−205,1998)に記載
のように、細胞複製の阻害により、細胞を細胞培養の寿命を延長することができる状態に
なるように図り、アポトーシスに耐性を示し得る(Chang et al.,Proc
Natl Acad Sci USA 97,4291−6,2000;Javela
nd et al.,Oncogene 19,61−8,2000)。
【0004】
さらに別のアプローチは、アデノウイルスE1遺伝子でのトランスフェクション後に初
代二倍体ヒト細胞または無制限に増殖するその誘導体を樹立するステップを含む。機能的
なAd5E1AおよびE1B遺伝子産物を発現する新規の細胞株(そのうちの1つは、P
ER.C6(ECACC寄託番号96022940)である)は、遺伝子療法およびワク
チンならびにヒト成長因子およびヒト抗体などの組換え治療タンパク質の産生のためにデ
ザインされた組換えアデノウイルスおよび他のウイルス(例えば、インフルエンザウイル
ス、単純ヘルペスウイルス、ロタウイルス、麻疹ウイルス)を産生することができる(V
ogels et al.,WO02/40665A2)。
【0005】
他のアプローチは、細胞中のアポトーシスの防止または遅延のためのカスパーゼインヒ
ビターの使用に注目している。例えば、米国特許第6,586,206号を参照のこと。
さらに他のアプローチにより、細胞中でのアポトーシス防止または遅延のためのBcl−
2ファミリーのメンバーなどのアポトーシスインヒビターの使用が試みされている。Ar
den et al.,Bioprocessing Journal,3:23−28
(2004)を参照のこと。これらのアプローチにより、予想不可能な結果が得られてい
る。例えば、ある研究では、Bcl−2発現により細胞の生存度が増加するが、タンパク
質産生は増加しなかった。Tey et al.Biotechnol.Bioeng.
68:31−43(2000)を参照のこと。別の例にはCHO細胞のアポトーシスを遅
延させるためのBcl−2タンパク質の過剰発現が記載されているが、Bcl−xLはタ
ンパク質産生を増加させるのに対して、Bcl−2はタンパク質産生を減少させた(WO
03/083093を参照のこと)。さらなる例には、培養物中のSp2/0−Ag14
(ATCC番号CRL−1581、以後Sp2/0と呼ぶ)細胞の生存を延長するために
Bcl−2タンパク質の発現を使用した実験が記載されているが、Bcl−2発現クロー
ンの細胞密度はその親培養物の細胞密度より20〜50%低く、生物医薬品産業における
その実用的な適用への関心が高まっている(WO03/040374を参照のこと)。し
たがって、組換えタンパク質の高レベル発現のための改良宿主細胞および宿主細胞におけ
る組換えタンパク質産生、特に抗体および抗体フラグメント、多重特異性抗体、フラグメ
ント、および一本鎖構築物、ペプチド、酵素、成長因子、ホルモン、インターロイキン、
インターフェロン、およびワクチンの産生を確実に増加させる方法が非常に望ましいこと
が明らかである。
【発明の概要】
【0006】
したがって、本発明の目的は、老化を阻害するか細胞生存を促進する薬剤(例えば、抗
アポトーシス剤)の細胞への導入による、細胞培養物の寿命および組換えタンパク質の収
率を増加させる改良された宿主細胞および方法を提供することである。このような薬剤の
使用により、所望の組換えタンパク質の産生のために使用された培養物中の細胞の寿命お
よび生存度を優先的に増加させ、それに伴ってこのような培養物中の細胞の生産性が増加
し、それにより、所望のタンパク質の収率が最適になる。好ましくは、本発明の方法で使
用されるアポトーシスインヒビターには、Bcl−2およびそのファミリーメンバーが含
まれるが、これらに限定されない。あるいは、細胞クローンの寿命および組換えタンパク
質の収率を、p53およびRbなどの細胞内アポトーシス促進性タンパク質レベルをダウ
ンレギュレートするか、Bcl−2などの細胞内抗アポトーシス性タンパク質をアップレ
ギュレートする薬剤を細胞に導入することによって改良することができる。好ましくは、
本発明の方法で使用される調節剤には、ヒト乳頭腫ウイルス16型(HPV−16)の腫
瘍性タンパク質E6およびE7ならびにその組み合わせが含まれるが、これらに限定され
ない。さらに、本明細書中に記載のカスパーゼインヒビターはまた、アポトーシスの遮断
または減少に寄与し、それにより、細胞生存が増加し、培養物中の細胞による組換えタン
パク質産生が増加し得る。これらの培養物中で組換えタンパク質の産生を増強するために
使用することができる抗アポトーシス剤のさらなるクラスには、エリスロポエチン(EP
O)などのサイトカインI型スーパーファミリーの特定のメンバーが含まれる。このクラ
スの代表的な分子としてのEPOは、赤血球だけでなく複数の細胞型のアポトーシスの主
な修飾因子であり、それにより、内皮細胞、心筋細胞、腎臓の尿細管上皮細胞、皮膚、お
よびニューロンなどにおいてより一般的な細胞保護機能を有する[P.Ghezzi a
nd M.Brines,Cell Death and Differentiati
on 11(suppl.1),s37−s44,July 2004の概説を参照のこ
と]。
【0007】
本発明はまた、細胞培養物の寿命が増加し、および/または最適になり、所望の組換え
タンパク質の収率が増加する細胞培養条件をもたらすための因子(トランスフェクション
ベクター、所望の性質を有する細胞クローンの分泌および選択、細胞培養培地、増殖条件
、バイオリアクターの配置、および細胞型が含まれるが、これらに限定されない)の新規
の組み合わせを組み込んだ細胞培養法を教示する。これらの細胞培養方法には、懸濁産生
法、灌流産生法、および流加産生法が含まれる。Tey et al.,J.Biote
chnol.79:147−159(2000);Zhang et al.,J.Ch
em.Technol.Biotechnol.79:171−181(2004);Z
hou et al.,Biotechnol.Bioeng.55:783−792(
1997)を参照のこと。
【0008】
他で定義しない限り、本発明で使用した全ての技術用語および科学用語は、当業者に一
般に理解されている意味を有する。さらに、本明細書中に引用した全ての特許および他の
引例の内容全体が、本明細書中に参照することにより組み込まれる。
【図面の簡単な説明】
【0009】
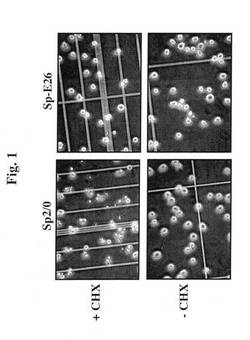
【図1】シクロヘキシミドで処置(+CHX)、又は未処置(−CHX)のSp2/0細胞およびSp−E26細胞の画像を示す。
【図2】CHX処置に対する耐性がより高いHPV E6/E7形質導入細胞のスクリーニングの結果を示す。全部で55クローンをスクリーニングし、第1の実験では、31クローンをスクリーニングし(上のパネル)、第2の実験では、24クローンをスクリーニングした(下のパネル)。各クローンの健康な細胞を、2つの等しい部分に分割した。一方をCHXで2時間処置し、他方を未処置のままにした。次いで、これら2つの培養物中の生存細胞を、MTTアッセイによって測定し、処置した生存細胞集団(CHX+):非処置生存細胞集団(CHX-)比をプロットした。上のパネルに示すように、CHX処置により、Sp2/0細胞の生存度が30%減少する一方で、Sp−E26細胞では6%しか減少しなかった。スクリーニングした31クローンのうちの7クローン(*で示す)は、Sp2/0よりも有意に良好であったが(生存度の減少は20%未満)、Sp−E26ほどではなかった。第2の実験でスクリーニングした24クローンについて(下のパネル)、CHX処置により、Sp2/0細胞の生存度が約50%減少し、Sp−E26の生存度の減少が約20%未満であった。スクリーニングした24クローンのうちの10クローン(*または**で示す)は、Sp2/0よりも有意に良好であり(減少は30%未満)、これらのうちの6クローン(**で示す)は、Sp−E26に匹敵するかこれより良好であった(20%未満)。E28およびE36は2つのさらなるコントロールクローンであり、Sp2/0よりも良好であるが、Sp−E26ほどではない。
【図3】GuavaネキシンVアッセイのドットプロットを示す。初期アポトーシス細胞(ネキシンV陽性および7−AAD陰性)の比率を、右下の四分円に示す。
【図4】CHXによって処置されたSp2/0のDNA断片化を示す。対照的に、Sp−E26細胞は、この処置に耐性を示す。
【図5】Tフラスコ中でのSp2/0細胞およびSp−E26細胞の増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度200,000/mlでTフラスコに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬(Guava technologies,Inc.)およびPCA装置(Guava Technologies,Inc.)を使用して毎日計数した。NH4+および乳酸塩の蓄積もモニタリングした。
【図6】3Lバイオリアクター中でのバッチ培養について測定したSp2/0細胞およびSp−E26細胞の増殖プロフィールを比較する。健康な細胞(生存度95%超)を、初期細胞密度250,000/mlでバイオリアクターに播種した。トリパンブルーおよび顕微鏡によって細胞を毎日計数した。
【図7】Bcl−2−EEE発現についてクローンをスクリーニングするためにBcl−2(100)抗体(Santa CruzBiotech.)で染色し、増強された化学発光を使用して発色させた代表的免疫ブロットを示す。
【図8】Guava Expressを使用したフローサイトメトリーの結果のグラフを示す。細胞を固定し、透過処理を行い、その後、フィコエリトリン抱合抗Bcl−2抗体(Santa Cruz Biotechnology,Inc.)で染色した。いくつかのサブクローンを比較する。
【図9】Guava Expressを使用したフローサイトメトリーの結果のグラフを示す。細胞を固定し、透過処理を行い、その後、フィコエリトリン抱合抗Bcl−2抗体(Santa Cruz Biotechnology,Inc.)で染色した。Sp2/0細胞、Raji細胞、およびDaudi細胞を、Bcl−2−EEEクローンと比較した。
【図10】665.B4.1C1細胞、Sp2/0細胞、Raji細胞、Daudi細胞、Sp−EEE細胞(87〜29クローン)、およびSp−EEE細胞(7〜16クローン)の溶解物の免疫ブロット分析の結果を示す。A.ヒトBcl−2特異的抗体(Santa Cruz Biotechnology,Inc.)を使用して染色したブロット。B.マウスBcl−2およびヒトBcl−2を認識する抗Bcl−2抗体(Santa Cruz Biotechnology,Inc.)を使用して染色したブロット。
【図11】10%ウシ胎児血清を補足した培地で増殖したSp2/0細胞と比較したSp−EEEクローンの増殖曲線(A)および生存度(B)を示す。
【図12】1%ウシ胎児血清を補足した培地で増殖したSp2/0細胞と比較したSp−EEEクローンの増殖曲線(A)および生存度(B)を示す。
【図13】無血清培地で増殖したSp2/0細胞と比較したSp−EEEクローンの増殖曲線(A)および生存度(B)を示す。
【図14】Sp−EEE細胞(87〜29クローン)のメトトレキセート死滅曲線を示す。
【図15】1mg/mlゼオシンの存在下または非存在下で増殖したSp−EEEクローンを含むGuava Expressを使用したフローサイトメトリーの結果のグラフを示す。細胞を固定し、透過処理を行い、その後、フィコエリトリン抱合抗Bcl−2抗体(Santa Cruz Biotechnology,Inc.)で染色した。
【図16】Sp2/0細胞をトランスフェクトして、ヒト化抗体配列、SV40プロモーター配列、およびエンハンサー配列を有する665.2B9クローンを得るために使用したpdHL2ベクターのマップを示す。
【図17】クローン665.2B9のトランスフェクションに使用したBcl−2遺伝子を組み込んだDNAプラスミドのマップを示す。
【図18】Bcl−2トランスフェクションクローン665.2B9#4、Bcl−2陰性クローン、および非トランスフェクションコントロールの増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度400,000/mlで24ウェルプレートに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図19】Bcl−2トランスフェクションクローン665.2B9#4、Bcl−2陰性クローン、および非トランスフェクションコントロールの増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度400,000/mlで24ウェルプレートに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図20】異なるMTX濃度でのBcl−2トランスフェクションクローン665.2B9#4およびBcl−2陰性クローンの増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度100,000/mlでTフラスコに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図21】異なるMTX濃度でのBcl−2トランスフェクションクローン665.2B9#4およびBcl−2陰性クローンの増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度100,000/mlでTフラスコに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図22】ウェスタンブロッティングによって検出した漸増濃度のMTXおよびクローン#13中でクローン665.2B9#4によって発現したヒトBcl−2レベルを示す。
【図23】急増L−グルタミンおよびグルコースを含むか含まない0.6μMまたは1μMのMTX中で培養したクローン665.2B9#4ならびに0.3μM MTX中で培養したBcl−2陰性クローンの細胞生存度および生存細胞密度のプロフィールをそれぞれ示す。健康な細胞(生存度95%超)を、初期細胞密度200,000/mlでローラーボトルに播種した。2日目および4日目に(矢印で示す)、グルコースおよびL−グルタミンを含む栄養補足溶液を、「急増」培養物に添加した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図24】急増L−グルタミンおよびグルコースを含むか含まない0.6μMまたは1μMのMTX中で培養したクローン665.2B9#4ならびに0.3μM MTX中で培養したBcl−2陰性クローンの細胞生存度および生存細胞密度のプロフィールをそれぞれ示す。健康な細胞(生存度95%超)を、初期細胞密度200,000/mlでローラーボトルに播種した。2日目および4日目に(矢印で示す)、グルコースおよびL−グルタミンを含む栄養補足溶液を、「急増」培養物に添加した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図25】バイオリアクター供給ストラテジーについての過程の略図を示す。
【図26】組換えインスリンを供給しない過程番号1、過程番号1に基づいて修飾リノール酸および脂質供給スケジュールを使用し、インスリンをさらに供給した過程番号2による665.2B9.1E4細胞株および665.B4.1C1細胞株の増殖曲線(VCDおよび生存度)を示す。
【図27】過程番号1および2における665.2B9.1E4細胞株および665.B4.1C1細胞株の抗体収率を示す。665.2B9.1E4細胞の最終収率は、過程番号1では0.42g/Lであり、過程番号2では0.55g/Lであった。比較のために、665.B4.1C1細胞は、両方の過程において1.5g/Lのより高い最終収率が実現された。
【図28】1日の特異的抗体産生性を示す(細胞基準あたり)。図に示すように、一連の培養による両過程についての665.2B9.1E4細胞の1日あたりの平均Q[MAb]は約15pg/細胞/日であった。過程番号2における最も高いVCDでの増殖日数の追加により、最終抗体濃度がより高くなった。
【発明を実施するための形態】
【0010】
本発明は、改良された組成物(宿主細胞株を含む)およびこのような細胞株中での組換
えタンパク質の産生を増強する方法を提供する。1つまたは複数の抗アポトーシス性遺伝
子を構成的に発現し、この抗アポトーシス性遺伝子の発現によって培養物中のトランスフ
ェクション細胞の生存が延長されて目的のタンパク質またはペプチドの収率が増強される
、目的のタンパク質またはペプチドをコードする発現構築物でトランスフェクトすること
ができる細胞株を作製した。
【0011】
詳細には、本発明者らは、Sp2/0骨髄腫細胞株から、バッチ培養における生存が増
強されたSp−E26およびSp−EEEと呼ばれる2つの新規の細胞株を作製した。S
p−E26およびSp−EEEは、それぞれ、HPV−16のE6およびE7タンパク質
ならびにBcl−2変異体(Bcl−2−EEEと呼ばれる)を構成的に発現する。さら
に、目的の組換えタンパク質のための発現ベクターでのSp−E26またはSp−EEE
のいずれかのトランスフェクションの際に、組換えタンパク質産生、特に、組換え抗体お
よび抗体フラグメントの産生を改良することができる。E6/E7またはBcl−2−E
EEタンパク質は、宿主細胞におけるアポトーシスの誘導を遅延させ、宿主細胞における
組換えタンパク質産生を増強する。1つまたは複数のカスパーゼインヒビター(例えば、
カスパーゼ1インヒビターおよび/またはカスパーゼ3インヒビター)の添加(Bin
Yang et al. Nephron Experimental Nephrol
ogy 2004;96:e39−e51)および/またはサイトカインI型スーパーフ
ァミリーの1つまたは複数のメンバー(エリスロポエチン(EPO)など)の細胞の増殖
培地への添加の添加によって、タンパク質産生をなおさらに高めることができる。これに
関して、pan−カスパーゼインヒビターが特に有効である。
【0012】
本発明者らはまた、アポトーシスインヒビター(Bcl−2など)の同時発現によって
宿主細胞中で抗体または抗体フラグメントなどの組換えタンパク質の産生を有意に増強す
ることができることを見出した。特に、抗体または抗体フラグメントをコードする発現ベ
クターで安定にトランスフェクトされ、アポトーシスインヒビター(Bcl−2など)を
コードする発現ベクターで同時トランスフェクトした骨髄腫細胞株(Sp2/0など)に
おいて、タンパク質産生が有意に増強される。E6/E7遺伝子でトランスフェクトした
宿主細胞で抗体産生を増加させることもできる。細胞の増殖培地への1つまたは複数のカ
スパーゼインヒビターの添加によって、組換えタンパク質産生をいっそう高めることがで
きる。これに関して、pan−カスパーゼインヒビターが特に有効である。また、細胞培
養培地へのEPO(すなわち、別の抗アポトーシス性サイトカイン)の供給によって、組
換えタンパク質の産生を増強することができる。
【0013】
アポトーシスと呼ばれる生理学的細胞死(すなわち、プログラム細胞死)(Kerr
et al.,Br J Cancer.,26:239−257,1972)は、適切
な組織の発達および維持に不可欠であり、進化過程で保存されている内因性遺伝子プログ
ラムによって調節される(Ellis et al.,Annu Rev Cell B
iol,7,663−698,1991)。したがって、人工的環境で(エクスビボ培養
物などで)細胞が増殖する場合、この遺伝子の素質によって寿命に限度がある。したがっ
て、医薬品および産業ならびに研究で使用されるタンパク質の産生のためのこのような細
胞培養物の有用性は、このような培養物がアポトーシス機構によって死滅する前に寿命(
すなわち、周期)が長期間維持されることに依存する。
【0014】
アポトーシス効果からの細胞周期の区別によって、細胞増殖および細胞死事象と無関係
に作用する方法および薬剤を発見した。Bcl−2は、周知のアポトーシスの細胞内調節
因子であり(Vaux et al.,Nature 335,440−2,1988)
、細胞周期に入ることに対するその阻害的影響とは遺伝的に異なる抗アポトーシス効果を
有することが見出されているがん原遺伝子である(Huang et al.,EMBO
J 16,4628−38,1997)。Bcl−2、Bcl−xL、およびBcl−
wの2つのホモログも細胞生存を延長させるが、Bcl−2ファミリーの他のメンバー(
BaxおよびBakなど)はアポトーシス促進性を示す(Oltvai et al.,
Cell 74,609−19,1993;Chittenden et al.,Na
ture 374,733−6,1995;Farrow et al.,Nature
374,731−3,1995;Kiefer et al.,Nature 374
,736−9,1995)。他の抗アポトーシス性遺伝子には、Bcl−6およびMcl
−1が含まれる。
【0015】
したがって、Bcl−2および特定のそのファミリーメンバーは、アポトーシスに対し
て保護効果を発揮し、それにより、それは、タンパク質産生のために使用される培養物中
で特定の宿主細胞の寿命を増加させ、それによって産生および単離されるタンパク質量が
増強される方法と仮定される。Bリンパ球、特に、骨髄腫細胞によって抗体が産生される
ので、抗アポトーシス性Bcl−2ファミリーメンバー(Bcl−2、Bcl−xL、B
cl−w、またはこれらのタンパク質の変異体など)の過剰発現によってアポトーシスが
阻害され、それにより、細胞密度が増加し、培養物の生存が長くなる。したがって、他で
提案されているように(同節)、抗アポトーシス性Bcl−2ファミリー遺伝子のトラン
スフェクションにより、細胞周期自体の妨害によって細胞培養を延長する必要性が回避さ
れる。同様に、Bcl−2遺伝子での線維芽細胞のトランスフェクションによってこれら
の細胞中でBcl−2が過剰発現され、再度アポトーシスに拮抗作用し、これらの細胞の
寿命が増加し、それに伴って組換えタンパク質の産生および単離が増加する。サイトカイ
ンの使用中止の際に、インターロイキン−6(IL−6)依存性マウス骨髄腫細胞は、ア
ポトーシスを受けているかのように期限が切れることも認められた。このような細胞中の
IL−6−受容体をBcl−2またはBcl−xLによって調節してアポトーシスを延長
することができることも見出された(Schwarz et al.,Cancer R
es 55:2262−5,1995)。
【0016】
最近の文献もまた、3箇所の点変異(T69E、S70E、およびS87E)を有する
変異Bcl−2が、野生型または1箇所の点変異体と比較して、有意により高い抗アポト
ーシス活性を示すことを証明している(Deng et al.,PNAS(101)1
53−158,2004)。したがって、本発明は、Bcl−2−EEE三重変異体の発
現ベクターの構築を教示し、これを使用してSp2/0細胞をトランスフェクトして、寿
命および組換えタンパク質産生が改善されたSp−EEEクローンおよびサブクローンを
作製する。
【0017】
腫瘍ウイルスなどの他の作用因子はまた、その細胞不死化誘発の一部としてアポトーシ
スを妨害し、最終的には極めて有害な形質転換(高リスク型HPV癌タンパク質E6およ
びE7など)を完了することができる(Finzer et al.,Cancer L
ett 188,15−24,2002)。例えば、ウイルスE6タンパク質は、紫外線
に対する上皮アポトーシス応答を有効に遮断する(Storey,Trends Mol
Med 8,417−21,2002)。状況証拠から、ヒト乳頭腫ウイルスによって
扁平上皮癌(基底細胞癌ではない)におけるアポトーシスを減少させることができる(J
ackson et al.,Br J Cancer 87,319−23,2002
)。しかし、全ての乳頭腫ウイルスの癌タンパク質が抗アポトーシス効果を有するわけで
はない。例えば、他の研究では、ウシ種の乳頭腫ウイルスE6タンパク質が細胞にアポト
ーシス感受性を与えると報告されており(Liu et al.,Virology 2
95,230−7,2002)、これは、HPV−16 E7遺伝子が、一定の刺激によ
って誘導されたアポトーシスから星状細胞を防御することを示す他の研究(Lee et
al.,Yonsei Med J 42,471−9,2001)と対照的である。
E6結合タンパク質アプタマーの使用により、HPV E6癌タンパク質がHPV陽性腫
瘍細胞において抗アポトーシス活性を有するという直接的な実験による証拠が得られた(
Butz et al.,Proc Natl Acad Sci USA 97,66
93−7,2000)。しかし、他のHPV癌タンパク質は逆の効果を有することができ
、E2タンパク質は他のHPVタンパク質の非存在下でアポトーシスを誘導する(Web
ster et al.,J Biol Chem 275,87−94,2000)。
E6およびE7タンパク質の連続的発現には、子宮頸癌細胞の最適な増殖および2つのウ
イルスタンパク質が細胞生存に対して異なる効果を発揮することが必要であることが知ら
れている(DeFilippis et al.,J Virol 77,1551−6
3,2003)。HPV−16 E6に寄与する主な細胞内標的はp53である。E6は
、p53および細胞ユビキチンリガーゼと三重複合体(E6AP)を形成し、それにより
、プロテオソーム経路およびp53の不活化によってp53をユビキチン化して分解する
。他方では、HPV−16 E7タンパク質は、腫瘍抑制タンパク質Rbと相互作用して
不安定にする。さらに、アポトーシスおよび細胞周期経路に関与する種々の他の細胞内タ
ンパク質のレベルは、E6およびE7形質転換(Bcl−2、Bcl−xL、p73、M
DM2、p21、サイクリンおよびcdc、cdkタンパク質など)によって調節される
ことが報告されていた。これらのタンパク質発現の変化は、細胞の生理学的特性に非常に
影響を与える。したがって、本発明者らは、培養物中でのHPV−16 E6およびE7
による細胞のトランスフェクションが加齢培養条件誘導性アポトーシスに耐性を示す遺伝
子操作されたクローンの生成に非常に有効であり、それにより、細胞培養物の寿命が延長
されるという仮説を立てた。HPV−16癌タンパク質E7またはE6のみのいずれかの
細胞への導入が、加齢培養条件誘導性アポトーシス耐性が改良された遺伝子操作されたク
ローンの生成に十分であり得るとも仮定した。細胞が組換えタンパク質産生クローンであ
る場合、生理学的性質の改良により、全タンパク質生産性が増強される。
【0018】
<ウイルス抗アポトーシス遺伝子を発現する新規の宿主細胞の生成>
ウイルス抗アポトーシス性遺伝子(HPV−16 E6およびE7タンパク質など)を
構成的に発現する宿主細胞(骨髄腫宿主細胞など)を生成することができる。これらの宿
主細胞を、目的の組換えタンパク質をコードする発現ベクターでトランスフェクトするこ
とができ、抗アポトーシス性遺伝子の同時発現により、組換えタンパク質の産生が有意に
増加する。
【0019】
宿主細胞は、本質的に、ウイルス抗アポトーシス性遺伝子で安定に形質転換することが
できる組換えタンパク質の産生に適切な任意の宿主細胞であり得る。多数の組換えタンパ
ク質について、CHOおよびCOS細胞などの宿主細胞が有利である一方で、抗体などの
他のタンパク質については、骨髄腫細胞およびCHO細胞などの宿主細胞を一般的に選択
する。遺伝子が構成的または誘導的に発現される任意の適切な方法(すなわち、宿主細胞
の染色体に遺伝子が安定に組み込まれる一方で、遺伝子が発現される任意の方法)によっ
て、ウイルス遺伝子を宿主細胞に導入することができる。目的の遺伝子で宿主細胞を安定
に形質転換する方法は、当分野で周知である。特に有利な方法は、ウイルス抗アポトーシ
ス性遺伝子をコードするレトロウイルスベクターを使用することである。適切なベクター
には、LSXNベクターが含まれる(Miller et al.Biotechniq
ues 7,980−90,1989)。
【0020】
有利には、宿主細胞をトランスフェクトするために使用されるベクターは、ベクターを
含む細胞が選択される選択マーカーを含む。適切な選択マーカー(トランスフェクション
細胞に抗生物質耐性を付与する酵素など)は、当分野で周知である。トランスフェクショ
ン後、選択剤(抗生物質など)を含む培地中に細胞を維持し、マーカー耐性についてスク
リーニングする。細胞を選択し、従来の方法を使用した限界希釈によってクローニングす
ることができる。
【0021】
ウイルス抗アポトーシス性遺伝子が細胞生存度を増加させる能力を、アポトーシスを誘
導する薬剤(シクロヘキシミド(CHX)など)での細胞の攻撃誘発によって試験するこ
とができる。ウイルス抗アポトーシス性遺伝子を発現しない細胞が有意なアポトーシスの
発症を示す傾向があるのに対して、この遺伝子を発現する細胞がアポトーシス活性を劇的
に減少させる。アポトーシスの検出方法は当分野で周知であり、例えば、細胞表面FIT
C−アネキシンV結合アッセイ、DNAラダリングアッセイ、およびTUNELアッセイ
が含まれる。ウイルス抗アポトーシス遺伝子を発現する適切な細胞の選択の際、細胞を、
最適な組換えタンパク質をコードする発現ベクターでトランスフェクトすることができる
。発現ベクターは、一過性発現に適切なベクターにすることができ、真核生物複製起点を
含むエピソームベクターまたは安定に組み込まれ、その後に発現カセットの遺伝子が増幅
する増幅ベクターとすることができる。適切なベクターは当分野で周知であり、例えば、
特に、抗体および抗体フラグメントの産生に適切なpdHL2ベクターが含まれる。増幅
発現カセットを使用する場合、レトロウイルスベクター中で使用される選択マーカーと異
なり、トランスフェクション細胞が選択される選択マーカーを含むことが有利である。再
度、適切にトランスフェクトされた細胞を選択し、その後に限界希釈によってクローニン
グすることができる。
【0022】
適切なクローンの選択の際、細胞を適切な培地に入れ、培養して、目的の所望のタンパ
ク質を産生することができる。培地は、血清を含むことができるが、好ましくは、無血清
である。さらに、細胞の寿命およびタンパク質産生を、培養培地への1つまたは複数のカ
スパーゼインヒビター(例えば、カスパーゼ1または3)の添加によって増加させること
ができる。好ましくは、カスパーゼインヒビターは、1つまたは複数のカスパーゼ3、カ
スパーゼ9、および/またはカスパーゼ12を阻害するように作用する。細胞透過性カス
パーゼインヒビターを使用することが有利であり、pan−カスパーゼインヒビターが特
に有利である。Z−VAD−fmkおよびAc−DEVD−choなどの適切なインヒビ
ターが当分野で周知である。あるいは、カスパーゼインヒビター(AvenまたはXIA
Pなど)を発現するように細胞株をさらにトランスフェクトして、アポトーシスに影響を
及ぼすことによってその増殖特性を増強することができる。これに関して、サイトカイン
I型スーパーファミリーの特定のメンバー(EPOなど)はまた、抗アポトーシス性作用
および細胞保護作用を有することによって細胞生存を増加させることができる。上記の方
法により、本質的に任意の所望の遺伝子によるトランスフェクションに使用することがで
きる細胞株を生成することができる。しかし、当業者は、所望のタンパク質、特に、組換
えタンパク質を構成的に発現する樹立細胞株を、その後にウイルスまたはBcl−2ファ
ミリー抗アポトーシス性遺伝子をコードする適切なベクターで形質転換することができる
ことを認識している。以下の実施例2を参照のこと。
【0023】
目的のタンパク質は、本質的に、宿主細胞中で検出可能な量で産生することができる任
意のタンパク質であり得る。例には、伝統的なIgG型抗体、F(ab’)2またはFa
bフラグメント、scFv、ダイアボディ(diabody)、IgG−scFvまたは
Fab−scFv融合抗体、IgG−またはFab−ペプチド毒素融合タンパク質、また
はワクチン(例えば、A型肝炎、B型肝炎、またはC型肝炎;HIV、インフルエンザウ
イルス、呼吸器合胞体ウイルス、乳頭腫ウイルス、疱疹ウイルス、ハンターンウイルス、
エボラウイルス、ロタウイルス、サイトメガロウイルス、リーシュマニアRNAウイルス
、SARS、マラリア、結核(放線菌)、炭疽、天然痘、野兎病、およびwww.vac
cines.org(参照することによりその全体が本明細書に組み込まれる)に列挙さ
れている他のワクチンが含まれるが、これらに限定されない)が含まれる。本明細書中に
記載の宿主細胞は、実施例1および2に記載の骨髄種細胞株中での抗体および抗体フラグ
メントならびに組換え成長因子(例えば、EPO、G−CSF、GM−CSF、EGF、
VEGF、トロンボポエチン)、ホルモン、インターロイキン(例えば、IL−1〜IL
−31)、インターフェロン(例えば、α、β、γ、およびコンセンサス)、ならびに酵
素の高度に有効な産生に特に適切である。これらの方法を、組換えタンパク質の産生で使
用される任意の多数の細胞株(他の骨髄腫細胞株(マウスのNSOまたはラットYB2/
0など);上皮株(CHOおよびHEK 293など);間葉細胞株(線維芽細胞株CO
S−1またはCOS−7など);およびニューロン細胞(網膜細胞、膠細胞、および神経
膠種細胞など)が含まれる)に適用することができる。
【0024】
<アポトーシス性インヒビターを発現する細胞中での組換え抗体発現>
以前の研究では、キメラ抗体を産生する組換えCHO細胞中で同時発現するBcl−2
(天然に存在するアポトーシスインヒビター)の効果が記載されている。Tey et
al.,Biotechnol.Bioeng.68:31−43(2000)を参照の
こと。細胞培養物の寿命の増加が認められるにもかかわらず、Bcl−2発現を欠いた等
価な細胞では抗体産生は増加しなかった。しかし、本発明者らは、細胞がBcl−2も発
現する場合、骨髄腫細胞からの組換え抗体の産生が有意に増加することを見出した。
【0025】
有利には、骨髄腫細胞株を、抗体または抗体フラグメントをコードする発現カセットで
安定にトランスフェクトする。適切な発現カセットは、上記の選択マーカーと共に抗体の
重鎖および軽鎖の発現(scFvの場合、一本鎖)を調節する1つまたは複数のプロモー
ターを含む。特に有用なベクターは、選択マーカー酵素をコードするDNA配列に作動可
能に連結されたプロモーターを含む選択マーカー遺伝子、目的のタンパク質をコードする
DNA配列に作動可能に連結されたプロモーターを含む転写単位、第1のエンハンサーの
非存在下での選択マーカー遺伝子および第1の転写単位の両方の転写と比較して、選択マ
ーカー遺伝子および第1の転写単位の両方の転写を刺激する、選択マーカー遺伝子と転写
単位との間のエンハンサーエレメントを含むpdHL2である。ベクターはまた、第1の
エンハンサーと選択マーカー遺伝子との間に存在するプロモーターを有し、選択マーカー
遺伝子の転写刺激を選択的に軽減させる遮断エレメントを含む。VH配列およびVL配列を
、pdHL2にライゲーションすることができ、pdHL2は、ヒト軽鎖定常領域、重鎖
定常領域、増幅可能なdhfr遺伝子(それぞれ、別のプロモーターによって調節される
)の配列を含む増幅可能なベクターである。Leung et al.,Tumor T
argeting 2:184,(1996)およびLosman et al.,Ca
ncer 80:2660−2667,(1997)を参照のこと。このベクターを、例
えば、エレクトロポレーションによって細胞にトランスフェクトすることができる。培養
培地への0.1μMまたは適切な濃度のメトトレキセート(MTX)の添加によって選択
することができる。3μMまでまたはそれを超える漸増濃度のMTXを使用した段階的様
式で増幅することができる。したがって、発現カセットで安定にトランスフェクトされ、
目的の抗体を構成的に発現する細胞を得ることができ、当分野で周知の方法を使用して特
徴づけることができる。以下の実施例4も参照のこと。選択およびクローニング後、抗体
発現細胞株を、Bcl−2などの抗アポトーシス性遺伝子をコードする発現ベクターでト
ランスフェクトすることができる。例えば、SV40プロモーターに融合したBcl−2
遺伝子を含むベクターpZeoSV(Invitrogen,Carlsbad,CA)
を、エレクトロポレーションなどの適切な方法を使用して細胞にトランスフェクトし、必
要に応じて選択および遺伝子の増幅を行うことができる。上記のように得られた細胞株を
使用して抗体を産生し、アポトーシス性インヒビターを発現しない細胞における産生と比
較することができる。
【0026】
本方法は、抗体または抗体フラグメントを発現する細胞株の最初の調製およびその後の
Bcl−2または類似のインヒビターを発現するベクターでのトランスフェクションを記
載する。しかし、当業者は、Bcl−2または他の抗アポトーシス性タンパク質を構成的
に発現する細胞株を樹立し、その後に抗体または抗体フラグメントをコードする適切なベ
クターにより形質転換することができることを認識する。本発明を説明する代表例を以下
に示す。実施例1は、HPV−16 E6/E7のSp2/0細胞への組み込みによって
アポトーシスを減少/遅延することを特徴とする改良された細胞クローンSp−E26が
得られることを記載している。実施例2は、HPV−16 E7エレメントのみの過剰発
現によって宿主細胞株を改良する方法を記載している。実施例3は、組換えAbを産生す
る細胞クローンを開発するための宿主として改良された細胞Sp−E26の使用を記載し
ている。実施例4は、E6/E7エレメントを同時発現する抗体産生細胞株で認められた
Mab産生の増強を説明している。実施例5は、3つの点変異を有する変異Bcl−2(
Bcl−2−EEE)を構成的に発現し、寿命が改良された修飾Sp2/0細胞株の生成
および特徴付けを記載している。実施例6は、Bcl−2を発現する抗体産生細胞株の増
殖特性の改良を記載している。実施例7は、実施例6のBcl−2発現細胞株で認められ
たMAb産生の増強を記載している。実施例8は、細胞中のBcl−2発現の導入による
低レベル組換えタンパク質を産生する細胞クローンの改良方法を記載している。実施例9
は、細胞中でのBcl−2発現の導入によるSp−E26の改良方法を記載している。実
施例10は、組換えAbを産生する細胞クローンを開発するための宿主としての改良され
た細胞株(Sp−EEE)の使用を記載している。実施例11は、収率を最適にするため
の流加反応器のプロフィールおよび供給スケジュールの使用を記載している。
【実施例1】
【0027】
<HPV−16 E6およびE7遺伝子の安定な発現によるアポトーシス耐性細胞クロー
ンの生成>
<CHX処理に耐性を示す細胞クローンの選択>
Sp2/0細胞を、HPV−16 E6およびE7遺伝子の発現カセットを含むLXS
Nレトロウイルスベクターによって10:1のMOI(感染多重度)で形質導入した。2
4時間後の回収後、感染細胞を、G418(1000μg/ml)中で10日間選択した
。G418耐性細胞を、限界希釈(0.5細胞/ウェル)によって96ウェル細胞培養プ
レート中でクローニングした。安定な感染細胞を、シクロヘキシミド(CHX)(強力な
アポトーシス誘導剤)による処理に対しての耐性についてスクリーニングした。簡単に述
べれば、健康な細胞(生存度95%超、図1CおよびD)を、25μg/mlのCHXを
含む培地中でインキュベートし、顕微鏡下で細胞の形態を調査した。2〜3時間のインキ
ュベーション後に50%を超える親Sp2/0細胞の形態が変化し、断片化されるように
なり(図1A)、いくつかのE6/E7トランスフェクションクローンの形態変化の範囲
が狭く、アポトーシス耐性を示す。Sp−E26と呼ばれる最良のクローンは、4時間の
処理で明らかな形態の変化は認められなかった(図1B)。
【0028】
面倒な目視試験を回避するために、MTTアッセイを使用して、生存細胞集団の変化を
得ることができる。健康な細胞を、CHXを含むか含まない通常の培養条件下で2〜3時
間インキュベートした後、MTT色素をウェルに添加した。さらなる2時間のインキュベ
ーション後、SDSおよびHClを含む溶解緩衝液の添加によって細胞を可溶化した。プ
レートを、37℃で一晩インキュベートし、ELISAプレートリーダーを使用して59
0nmのODを読み取った。図2に示すように、Sp2/0細胞をCHXで処理した場合
に生存細胞集団がわずかに減少した。比較により、同一の処理条件下で(CHX濃度およ
び時間)、Sp−E26細胞は、CHX処理に対してより耐性を示した。これに関して、
さらなる分析のために多数のクローンをスクリーニングし、選択した(図2)。
【0029】
<Sp−E26の抗アポトーシス特性>
Sp−E26および親Sp2/0細胞におけるCHX誘導性アポトーシスを、アネキシ
ンV染色およびDNA断片化アッセイによって評価した。25μg/mlのCHXを含む
培地中でのインキュベーション後、細胞を回収し、Guava Nexin試薬(アネキ
シンV染色の等価物)で染色し、Guavaパーソナル細胞分析システム(Guava
Technologies,Inc.)を使用して分析した。図3は、約1.5時間CH
X処理に曝露した場合に30%を超えるSp2/0細胞がアネキシンV陽性になる(アポ
トーシスを示す)一方で、Sp−E26は健康なままであり、初期アポトーシス細胞中で
増加しないことを示す。
【0030】
細胞内オリゴヌクレオソームDNAフラグメント(アポトーシスの特質)形成の分析に
よって、CHXによるアポトーシスの誘導を明らかにすることができる。細胞DNAを、
CHX処理および未処理Sp−E26およびSp2/0細胞から抽出し、DNAラダリン
グアッセイを行った。Sp2/0細胞をCHXで処理し、多数のDNA断片化が検出され
た(図4)。対照的に、同一の処理条件下で、Sp−E26のゲノムDNAは依然として
無傷のままであり、DNA断片化の外観は認められなかった(図4)。
【0031】
<Sp−E26中のHPV E6およびE7遺伝子の存在>
E6およびE7遺伝子がSp−E26細胞のゲノム中に安定に存在することを確認する
ために、E6およびE7遺伝子に特異的なオリゴヌクレオチドプライマーをデザインし、
テンプレートしてSp−E26から抽出したゲノムDNAとのPCR反応で使用し、約7
00bpのDNAフラグメントを得た。PCR産物をクローニングし、DNA配列決定に
よってE6およびE7遺伝子であることを確認した。親Sp2/0細胞中にE6およびE
7遺伝子が検出された。
【0032】
<Sp−E26の増殖特性の改良>
Tフラスコ(図5)および3Lバッチバイオリアクター(図6)Sp−E26の増殖特
性を評価した。Sp−E26は、バッチ培養において親Sp2/0細胞よりも改良された
増殖特性を示し、より高い最大細胞密度およびより長い生存期間が達成された。
【実施例2】
【0033】
<HPV16 E7遺伝子の安定な過剰発現によるアポトーシス耐性細胞クローンの生成
>
クローンSp−E26のゲノムに組み込まれたポリシストロン性HPV16 E6およ
びE7遺伝子の構造を、プライマー対E6−N8+(5’−ATG TTT CAG G
AC CCA CAG GAG CGA−3’)およびE7−C8-(5’−TTA T
GG TTT CTG AGA ACA GAT GGG−3’)を使用したPCRおよ
びDNA配列決定によって分析した。プライマーE6−N8+およびE7−C8-の配列は
、E6のN末端8アミノ酸残基のコード配列およびE7のC末端8コドンの相補配列とそ
れぞれ適合するので、全長E6およびE7のアンプリコンは、約850bpであると予想
される。しかし、E6−N8+およびE7−C8-を使用したSp−E26細胞から調製し
たゲノムDNAの増幅により、約700bpのPCRフラグメントしか得られなかった。
700bpのPCR産物のDNA配列決定により、E6遺伝子からの182ポリヌクレオ
チドフラグメントの欠失が明らかとなった。欠損E6遺伝子は、スプライシングに起因す
る可能性が高く、N末端43アミノ酸残基を有する短縮E6ペプチドをコードする。E6
による主な生理活性がp53発現のダウンレギュレート能力であることを考慮すると、S
p−E26中のp53レベルがSp2/0よりも安定であることが見出されているので、
短縮E6タンパク質は、おそらく、十分に機能的ではない。
【0034】
したがって、HPV−16 E7遺伝子のみで抗アポトーシス効果を有し、Sp2/0
細胞の増殖特性を改良するのに十分であるかどうかを評価するために、HPV−16 E
7でのSp2/0細胞のトランスフェクションを、以下のように行った。
(i)E7をコードするDNA配列を、RT−PCRによってSp−E26細胞からク
ローニングする。哺乳動物ベクターpRc/CMV(Invitrogen)への遺伝子
のライゲーションを容易にするために、適切な制限部位を導入する。E7pRcと命名さ
れたベクター内でのウイルス遺伝子の転写は、CMVプロモーター−エンハンサー配列か
ら誘導される。ベクターはまた、SV40プロモーターから転写されるネオマイシン耐性
を付与する遺伝子を含む。
(ii)Sp2/0細胞を、HPV−16E7遺伝子の発現カセットを含む発現ベクタ
ーによってトランスフェクトする。簡単に述べれば、5μgのE7pRcを、ScaIに
よって線状化し、エレクトロポレーションによって細胞にトランスフェクトする。
(iii)24時間後の回収後、トランスフェクション細胞を、G418(1000μ
g/ml)中で10日間選択する。
(iv)次いで、G418耐性細胞を、限界希釈(0.5細胞/ウェル)によって96
ウェル細胞培養プレート中でクローニングする。安定なトランスフェクタントを選択し、
シクロヘキシミド(CHX)(強力なアポトーシス誘導剤)によって治療耐性についてス
クリーニングする。
(v)健康な細胞(生存度95%超)を、通常の培養条件下で25μg/mlのCHX
を含むまたは含まない培地中で3〜4時間のインキュベーション後、MTT色素をウェル
に添加する。さらなる2時間のインキュベーション後、SDSおよびHClを含む溶解緩
衝液の添加によって細胞を可溶化する。プレートを、37℃で一晩インキュベートし、E
LISAプレートリーダーを使用して590nmのODを読み取る。さらなる分析のため
にCHX処理に耐性を示す細胞クローンを選択し、拡大する。
(vi)E7トランスフェクション細胞の抗アポトーシス性を、アネキシンV染色およ
びDNA断片化アッセイによって評価する。アネキシンVアッセイでは、25μg/ml
のCHXを含む培地中でのインキュベーション後、細胞を回収し、Guava Nexi
n試薬(アネキシンV染色の等価物)で染色し、Guavaパーソナル細胞分析システム
(Guava Technologies,Inc.)を使用して分析する。DNA断片
化アッセイでは、細胞DNAを、CHX処理および未処理E7トランスフェクタントおよ
びSp2/0細胞から抽出し、アガロースゲル電気泳動で分析する。
(vii)E7トランスフェクタント中のウイルス癌遺伝子の発現を、サザンブロット
分析(ゲノムレベル)、ノーザンブロット分析(mRNAレベル)、および免疫ブロット
分析(タンパク質レベル)によって評価する。アポトーシス過程に関与し、E7タンパク
質に影響を受ける細胞内タンパク質の発現を、免疫ブロッティング分析によって試験する
。
(viii)選択されたE7トランスフェクタントの増殖特性を、T−フラスコ中およ
び3Lバッチバイオリアクター中で評価する。トランスフェクタントは、バッチ培養にお
いて親Sp2/0細胞よりも改良された増殖特性を示し(より高い最大細胞密度およびよ
り長い生存期間が達成され)、より良好な宿主細胞と見なされる。
【実施例3】
【0035】
<Sp−E26中でのhLL2 IgGの高レベル発現>
この実施例では、Sp−E26を、hLL2(NHL患者および自己免疫疾患患者を治
療するために開発されたヒト化抗CD22Ab)を産生する細胞クローンを生成するため
の宿主として使用する。hLL2産生クローン(87−2−C9)は、以前に宿主として
Sp2/0細胞を使用することによって生成され(Losman et al.,Can
cer 80,2660−2666,1997)、この場合、トランスフェクション後に
1つの陽性クローンのみが同定され(約2.5×10-7の頻度)、増幅前のT−フラスコ
におけるコンディション最終培養培地中の抗体濃度として定義されたhLL2産生クロー
ンのみの最大生産性(Pmax)は、1.4mg/Lであった。Losman et al
.(Cancer 80,2660−2666,1997)によって記載された類似の方
法の使用による同一のhLL2pdHL2ベクターでのSp−E26細胞のトランスフェ
クションにより、200個を超える安定なhLL2産生クローンが得られた(10-4を超
える頻度)。12個の無作為に選択したクローンのPmaxを評価し、13mg/Lと17
0mg/Lとの間であり、平均50mg/Lであることが見出された。これらのクローン
の生産性を、MTXでの遺伝子増幅によってさらに増強することができる。この実施例に
より、組換えタンパク質を産生する細胞クローンの開発のための宿主としてSp−E26
を使用することがその親Sp2/0細胞よりも有利であることが証明された。
【実施例4】
【0036】
<HPV16 E6およびE7遺伝子の安定な発現によるAb産生細胞株の改良>
607−3u−8細胞をトランスフェクションによってSp2/0から最初に生成し、
ヒト化モノクローナルAbを産生した。最大(Ab)生産性を150mg/Lまで増強す
る(培養培地中の血清補足のウィーニング(weaning)後に約100mg/Lに減
少した)ために、(MTXでの)遺伝子の増幅およびサブクローニングによってクローン
を開発した。無血清条件下でより高い抗体生産性を得るために、HPV−16のE6/E
7遺伝子を、607−3u−8に導入し、Ab生産性に対するE6/E7の効果を以下の
ように評価した。10%FBSおよび3μMのMTXを補足したHSFM中で維持した6
07−3u−8細胞を、HPV−16 E6およびE7遺伝子の発現カセットを含むLX
SNレトロウイルスベクターで10:1のMOIで形質導入した。24時間後の回収後、
安定にトランスフェクトされた細胞を、G418(400μg/ml)中で10日間選択
した。G418耐性細胞を、限界希釈(0.5細胞/ウェル)によって96ウェル細胞培
養プレート中でサブクローニングした。評価のために607E1C12と命名した生存ク
ローンを得た。E6/E7トランスフェクションを行っていない607−3u−8−7G
7および607−3u−8−2D10と命名された607−3u−8の2つのサブクロー
ンも選択した。これら3つのクローンのPmaxを決定し、有意差はなかった(表1)。こ
れらの結果により、細胞へのE6/E7遺伝子の導入によって細胞のAb産生能力は変化
しないことが示唆される。次に、607E1C12、607−3u−8−7G7、および
607−3u−8−2D10を、無血清培地中で増殖するように適合させ、これらのクロ
ーンの生産性を決定した。全細胞を、無血清培地を含む増殖ウェル中で増殖させた。クロ
ーン607E1C12の最終抗体生産性は150mg/Lに維持される一方で、E6/E
7を含まない2つのクローンでは実質的に減少した。さらに、607E1C12の生産性
は、凍結融解後に(低温保存に対して)安定であった(表1)。
【0037】
【表1】

【実施例5】
【0038】
<変異Bcl−2を構成的に発現する遺伝子操作されたSp2/0細胞株の生成および特
徴付け>
証拠により、3箇所の点変異(T69E、S70E、およびS87E)を有する変異B
cl−2が、野生型または1箇所の点変異体と比較して、有意により高い抗アポトーシス
活性を示すことが示唆される(Deng et al.,PNAS 101:153−1
58,2004)。したがって、この三重変異体(Bcl−2−EEEと命名する)のた
めの発現ベクターを構築し、これを使用して、特にバイオリアクター中の生存および生産
性を増加させるためにSp2/0細胞をトランスフェクトした。クローンを単離し、Bc
l−2−EEE発現レベル、増殖、およびアポトーシス特性について評価した。Bcl−
2−EEEの核酸配列を、配列番号3に示し、対応するBcl−2−EEEタンパク質の
アミノ酸配列を、配列#4に示す。
【0039】
ヒトBcl−2のアミノ酸残基64〜101のコード配列に基づいて、116bpの合
成DNA二重鎖をデザインした。残基69、70、および87のコドン全てを、グルタミ
ン酸のコドン(E)に変更した。全配列は並外れてGCがリッチであり、多数のポリGお
よびポリC連続物(run)が存在する。GおよびC連続物を破壊するためにいくつかの
コドンを保存的に変化させ、全GC含有量を減少させた。
【0040】
組み合わせ、長さが116bpの配列であり、その3’末端が22bp重複した2つの
80量体オリゴヌクレオチドを合成した(配列番号5および6を参照のこと)。オリゴヌ
クレオチドをアニーリングし、TaqDNAポリメラーゼを使用したプライマー伸長によ
って二重鎖DNAを生成した。PCRプライマー、Bcl−2−EEE PCR左(5’
−TATATGGACCCGGTCGCCAGAGAAG−3’)およびBcl−2−E
EE PCR右(5’−TTAATCGCCGGCCTGGCGGAGGGTC−3’)
を使用して、二重鎖を増幅させた。
【0041】
次いで、126bpの増幅物を、pGemT PCRクローニングベクターにクローニ
ングした。Bcl−2−EEE−pGemT構築物を、TthIおよびNgoMI制限エ
ンドヌクレアーゼで消化し、150bpフラグメントをゲルで単離し、TthIおよびN
goMIで消化したhBcl−2−puc19ベクター(ATCC 79804)でライ
ゲーションして、hBcl−2(EEE)−puc19を生成した。この構築物の配列を
確認した。
【0042】
948bpインサートフラグメントを、EcoRIを使用してhBcl−2(EEE)
−puc19から切り出し、EcoRIで消化したpZeoSV2+ベクターとライゲー
ションし、アルカリホスファターゼで処理した。得られた構築物は、hBcl−2(EE
E)−pZeoSV2+である。
【0043】
次いで、Sp2/0細胞についての標準的なプロトコールにしたがったエレクトロポレ
ーションによって、Sp2/0細胞(5.6×106)を、60μgのhBcl−2(E
EE)−pZeoSV2+でトランスフェクトした。細胞を、6つの96ウェルプレート
にプレートし、選択せずに48時間インキュベートした。2日後、800μg/mlのゼ
オシンを培地に添加した。
【0044】
40ウェル由来の細胞を、24ウェルプレートに拡大し、抗hBcl−2および抗βア
クチンを使用したウェスタンブロットによって分析した。40個のうちの5個を除けば、
中レベル〜高レベルのBcl−2−EEE発現を示した。4つのゲルのうちの1つの結果
を、図7に示す。事前に野生型Bcl−2でトランスフェクトしたSp2/0由来のhM
N14細胞株(クローン664.B4)を、正のコントロール(+)として使用した。D
eng et al.で証明されているように、SDS−PAGEにおいて、Bcl−2
−EEEは、野生型Bcl−2よりもわずかにゆっくり移動する。
【0045】
さらなる評価およびサブクローニングのために、3つの強い陽性のウェル(番号7、番
号25、および番号87)を選択した。限界希釈プレーティングにより、96ウェルプレ
ートあたりの陽性ウェルは20未満であり、これにより、各ウェル中の細胞が実際にクロ
ーニングされる非常に高い確率が示される(99%超)。最初に、3つの元のウェル由来
の23個のサブクローンを、抗hBcl−2−PEを使用したGuava Expres
sによって分析した(図8)。結果により、元のウェルが細胞クローンの混合物を含んで
いたことが確認された。ウェル番号7から最も強いシグナルを有するクローンが得られ、
ウェル番号25から最も低いシグナルを有するクローンが得られた。さらなる分析のため
に、クローン7−12、7−16、87−2および87−10を拡大した。その後、最初
に増殖の遅いサブクローンを同様に分析し、1つのクローン(87−29)から任意の他
のクローンよりも20%高いシグナルが得られ、さらなる分析のためにこれを拡大した。
2つの高発現SP−EEEクローン(87−29および7−16)を、非トランスフェク
ションSp2/0細胞、Raji細胞、およびDaudi細胞と比較した(図9)。Sp
−EEEクローンは、推定上正常な細胞レベルでBcl−2を発現することが知られてい
るRaji細胞およびDaudi細胞より約20倍発現する。Sp2/0細胞は、陰性で
あった。これを、抗Bcl−2免疫ブロットによってさらに検証した(図10)。ヒトB
cl−2特異的抗体を使用してSp2/0細胞中にBcl−2は検出されず、高タンパク
質負荷(50K細胞)および長期X線フィルム曝露でさえも検出されなかった。マウス、
ラット、およびヒトのBcl−2を認識する抗Bcl−2 MAb(C−2、Santa
Cruz Biotech.)を使用した免疫ブロット分析によって、非トランスフェ
クションSp2/0細胞からいかなるBcl−2も検出されず、高タンパク質負荷(10
0K細胞)および長期X線フィルム曝露でさえも検出されなかった(図10B)。Sp2
/0細胞中で任意のBcl−2が発現される場合、クローン87−29中のBcl−2−
EEEより満たない2桁を超えるレベルである。5つのSp−EEEサブクローンおよび
Sp2/0細胞の増殖曲線を比較した。3つのSp−EEEサブクローンは、Sp2/0
細胞よりも明確な利点を示した。これら3つ(7−12、7−16、および87−29)
はまた、最も高いレベルのBcl−2−EEEを発現する。7−12および7−16は、
同起源のウェルに由来し、ほぼ同一の性質を有し(Bcl−2−EEEレベルおよび増殖
曲線)、これらは、同一の元のクローンに由来する可能性が高い。最良の2つのSP−E
EEサブクローン(7−16および87−29)を、さらなる評価のために使用した。
【0046】
10%、1%、または0%の血清(ウィーニング(weaning)せず)を補足した
培地中にクローンをプレートし、細胞密度および生存度をモニタリングした。10%血清
では、87−29は高密度まで増殖し、Sp2/0細胞と比較して4日間を超えて生存が
延長された(図11)。1%血清では、全細胞は、10%血清で達成された密度の約35
〜40%に増殖し、Bcl−2−EEEトランスフェクタントは、Sp2/0に類似の延
命効果を有していた(図12)。無血清培地に直接移した場合、Sp2/0細胞は600
K細胞/mlしか増殖せず、87−29細胞は2倍の密度で増殖した(図13)。各血清
濃度では、87−29細胞は、Sp2/0細胞よりも4〜6日間長く生存した。
【0047】
87−29のメトトレキセート(MTX)感受性を決定した(図14)。データにより
、MTX耐性クローンの最初の選択には最小濃度(0.04μM)のMTXで十分である
ことが示唆される。したがって、Sp2/0細胞で使用した同一の選択および増幅プロト
コールを、SP−EEE細胞に使用することができる。
【0048】
Bcl−2は、生存促進性/抗アポトーシス性タンパク質である。いくつかのグループ
により、可動性ループドメイン(FLD)を欠くBcl−2欠失変異体はアポトーシス阻
害能力が増強されることが証明されている(Figueroa et al.,2001
,Biotechnology and Bioengineering,73,211
−222;Chang et al.,1997,EMBO J.,16,968−97
7)。より最近では、リン酸化を模倣するBcl−2のFLD中の1〜3つのS/T残基
のグルタミン酸への変異により、その抗アポトーシス能力が有意に増強されることが証明
されている(Deng et al.2004,PNAS,101,153−158)。
三重変異体(T69E、S70E、およびS87E)は、最も有意に生存が増強された。
ここで、本発明は、Sp2/0細胞を安定にトランスフェクトするために使用される類似
のBcl−2三重変異構築物(Bcl−2−EEE)の生成を教示する。
【0049】
上記の全実験は、Bcl−2−EEEの発現によってSp2/0細胞におけるアポトー
シス率が減少することを示す。この効果は、クローンの発現レベルが高いほどより低いレ
ベルのクローンより長く生存するという点で、非常に用量に依存した。最良のクローン(
87−29)は、非トランスフェクションSp2/0細胞と比較して、15〜20%高い
細胞密度で増殖し、さらに4〜6日間長く生存する。
【0050】
クローン(87−29)中のBcl−2−EEEレベルは、Daudi細胞またはRa
ji細胞中の通常のレベルの約20倍である。非トランスフェクションSp2/0細胞で
はBcl−2発現は検出されなかった。実施例6に記載するように、hMN−14発現S
p2/0細胞を、野生型Bcl−2発現のための類似の構築物でトランスフェクトし、例
外的な増殖特性および生産性が増強したクローンを単離した。このクローン(664.B
4)を、MTXを使用してさらに増幅させた場合、Bcl−2レベルが有意に増加した。
最終的に、増幅された(3μM MTX)細胞株をサブクローニングし、1つのクローン
(664.B4.1C1)のBcl−2レベルは、664.B4の2倍であった。この特
定のサブクローンは、生産性および増殖特性が優れている。87−29中のBcl−2−
EEEレベルは、増幅された664.B4.1C1中のBcl−2レベルの約2倍である
。87−29細胞は、Sp2/0細胞に匹敵する増殖率を有し、見かけ上さらに1日間増
殖し続け、Sp2/0よりも15〜20%高い最大密度に到達し得る。E6/E7発現S
p−E26細胞株について類似の性質が見出された。Bcl−2−EEE発現87−29
クローンは、親Sp2/0細胞よりもさらに4〜6日間長く生存し、1日だけ長く生存す
るSp−E26クローンより優れている。
【0051】
87−29クローンとして示したSp−EEE細胞株は、組換えタンパク質の遺伝子を
含む適切なベクターでのトランスフェクションの際の組換えタンパク質の発現のためのア
ポトーシス耐性宿主として有用である。この細胞株を有用にするために、この細胞株は、
トランスフェクションおよび増幅後ならびに培養物の拡大中にそのBcl−2−EEE発
現および延命効果を維持しなければならない。安定にトランスフェクトされたBcl−2
−EEE遺伝子はその後のトランスフェクション中に失われる可能性が低いので、生存特
性が損なわれるはずがない。MTX増幅はBcl−2タンパク質発現の増加によって産生
クローンの生存を実に改良することが可能である。実際、このことは、野生型Bcl−2
でトランスフェクトしたhMN14 664.B4細胞株でもそうであった。増幅および
サブクローニング後、Bcl−2レベルは数倍に増加し、細胞生存は有意に改良された。
【実施例6】
【0052】
<ヒトBcl−2遺伝子の安定な発現による静置バッチ培養におけるAb産生細胞生存の
改良>
<Bcl−2トランスフェクション細胞クローンの生成>
トランスフェクションによってSp2/0から細胞クローン665.2B9を最初に生
成し、ヒト化モノクローナル抗CEA Abを産生した(Qu et al.、未発表の
結果)。hMN14pdHL2と命名されたベクターを使用してSp2/0細胞をトラン
スフェクトし、細胞クローン665.2B9を得た。pdHL2ベクターは、Gilli
es et al.によって最初に記載され、メトトレキセート処理によってその後に選
択および増幅される増幅可能なマウスdhfr遺伝子を有していた(Gillies e
t al.,J.Immunol.Methods 125:191(1989))。一
般に、pdHL2ベクターはIgG重鎖および軽鎖の遺伝子両方を発現し、これらは、2
つのメタロチオネインプロモーターおよびIgHエンハンサーによって独立して調節され
る。hMN14pdHL2ベクターの図を、図16に示す。配列番号1は、このベクター
の配列を示し、配列番号2は、エンハンサー配列として定義された72bp配列を示し、
プロモーター配列は、hMN14pdHL2のnt2908〜2979に対応する。
【0053】
Sp2/0細胞を、一般に、エレクトロポレーションによって、本実施例で使用される
hMN14pdHL2などの線状化pdHL2ベクターでトランスフェクトすることがで
きる。0.05〜0.1μM MTXを含む培地との細胞のインキュベーションによって
、トランスフェクションから48時間後に選択を開始することができる。5μMまでの濃
度のMTXの段階的増加によって、挿入された抗体配列を増幅させる。
【0054】
クローンを、0.3μMまでのMTXの段階的増加を使用した遺伝子増幅に供し、この
時点で、抗体の最大生産性(Pmax)は約100mg/Lに増加した。細胞増殖特性を
改良するために、665.2B9細胞を、エレクトロポレーションによって、ヒトBcl
−2遺伝子を含むプラスミド発現ベクター(図17)でトランスフェクトした。Bcl−
2遺伝子を、EcoRI部位を使用してATCCから購入したpB4プラスミド(pB4
、カタログ番号79804)から切り出し、同一の制限酵素を使用して、哺乳動物発現ベ
クターpZeoSV(+)のMCSに挿入した。ゼオシン耐性遺伝子がベクターの一部で
あるので、トランスフェクション細胞を、50〜30μg/mLの範囲のゼオシンを含む
培地に入れた。300mg/mlゼオシンを含む培地から安定なクローンを選択し、0.
5細胞/100μL/ウェルの密度での96ウェルプレートへのプレーティングによって
ゼオシンを含まない培地中でサブクローニングした。その後、ゼオシンを含まない培地を
使用した。ウェル中のクローンの形成を、顕微鏡下での目視によって確認した。1細胞ク
ラスターのみを含むウェル由来の細胞を拡大した。各96ウェルプレートから約30クロ
ーンが産生され、そのうちの14クローンをさらなる研究のために無作為に選択した。こ
れらのクローンの増殖特性を、1日の細胞計数ならびにViaCount試薬およびGu
avaPCAを使用した生存度の測定によって評価した。24ウェルプレート中で評価し
た14クローンから(図18、19)、増殖特性が改良された(細胞密度がより高くなり
、細胞生存が延長された)1つのBcl−2トランスフェクションクローンを同定し、6
65.2B9#4(またはクローン#4)と命名した。親665.2B9クローンと比較
して、クローン#4は、Tフラスコ中でより高い細胞密度(約1.7倍)で増殖し、4〜
6日間長く生存し(図20、21)、より良好な増殖の結果として、ELISA滴定およ
びプロテインAカラム精製によって決定したところ、クローン#4のPmaxは、約170
mg/Lに増加した。
【0055】
<665.2B9#4におけるBcl−2発現>
665.2B9#4の増殖特性の改良がBcl−2のトランスフェクションに起因する
ことを確認するために、ヒトBcl−2タンパク質の細胞内レベルを、Guava Ex
press試薬およびGuava PCA装置の使用によって測定した。簡単に述べれば
、1.5mlスピンチューブに入れた4×105細胞を、1500rpmで5分間遠心分
離し、1×PBSで3回洗浄した。上清を慎重に吸引した。Santa Cruz Bi
otechnology(SCB)Inc.の固定液(10×、60μL)(カタログ番
号sc−3622)を細胞ペレットに15分間添加し、氷上でインキュベートした。固定
液を、4℃の4×1mL PBSを使用して除去し、記載の時間スピンした。−20℃の
透過処理緩衝液(0.5mL)(SCBカタログ番号sc−3623)をボルテックスし
ながら滴下し、その後、氷上で15分間インキュベートした。次いで、細胞をスピンし、
0.5mL FCM洗浄緩衝液(SCBカタログ番号sc−3624)で2回洗浄した。
最終細胞ペレットを、100μLのFCM洗浄緩衝液に再懸濁し、10μLのPEにコン
ジュゲートした抗Bcl−2マウスモノクローナル抗体(SCBから入手)にてBcl−
2細胞内タンパク質を染色した。室温の暗所で1時間インキュベートした。その後に、0
.5mLのFCM洗浄緩衝液で2回洗浄した。最終細胞ペレットを、0.4mLのFCM
洗浄緩衝液に再懸濁し、GuavaPCにて細胞を分析した。各クローンについての蛍光
強度の平均値(MFI)を、PEとコンジュゲートした非特異的アイソタイプマウスIg
G1でのコントロール染色と比較した。表2にまとめた結果により、クローン665.2
B9#4が親細胞株と比較して高レベルのBcl−2タンパク質を発現することが確認さ
れる。親665.2B9と類似の増殖プロフィールを示したゼオシン耐性クローン(#1
3)は、Bcl−2染色に対して陰性であり、増殖の改良にBcl−2発現が必要である
ことが確認された。
【0056】
【表2】
【0057】
Guava Express分析を使用して、Bcl−2レベルに対応する蛍光染色強
度がクローン665.2B9#4のMTX増幅と共に上昇することが見出され、Bcl−
2のdhfr遺伝子との同時増幅が示唆された。増幅細胞の細胞内Bcl−2レベルを比
較するために、抗ヒトBcl−2抗体を使用して、クローン665.2B9#4(Bcl
−2陽性)およびクローン#13(Bcl−2陰性)の細胞溶解物に対してウェスタンブ
ロッティング分析を行った。濃度測定評価により、1.0μM MTX中で増殖したクロ
ーン665.2B9#4のBcl−2シグナルが0.6μM MTX中の細胞よりも2倍
強いことが示された。クローン#13の溶解物は、Bcl−2タンパク質の存在を示さな
かった(図22)。
【実施例7】
【0058】
<バッチ培養条件下でのクローン665.2B9#4のAb産生の改良>
細胞培養物における末期付近の栄養素消費のモニタリングにより、グルコースおよびL
−グルタミンが最初に消費される栄養素であることが見出された。これらの制限された栄
養の補足によって最終抗体収率が改良されるかどうかを決定するための実験を行った。以
下の2つの培養型を開始した:これらの制限成分をその消費時に補足する急増流加培養お
よび栄養素を補足しない非流加培養。0.6μMおよび1μMのMTXを含む培地中で増
殖するBcl−2陽性クローン665.2B9#4ならびに0.3μM MTX中で増殖
するBcl−2陰性クローン#13を試験した。図23および24は、両培養型が末期に
達するまでの両培養型における細胞生存度および細胞密度のプロフィールを示す。mg/
Lで示すタンパク質収率を、表3に示す。この実験の結果により、全培養物において、栄
養素の急増によって抗体の産生全収率が約2倍に改良されることが示唆される。
【0059】
【表3】
【実施例8】
【0060】
<低レベルの組換えタンパク質を産生する細胞株へのBcl−2遺伝子の導入>
トランスフェクションによってSp2/0から細胞クローン482.2C4Aを最初に
生成し、IgG型(抗CEA)および2つのscFv(抗DTPA)型(それぞれ、Ig
G重鎖のC末端に共有結合されている)の二重特異性Abを産生した(Leung et
al.,J.Nuc.Med.41:270P,2000;Hayes et al.
,Proc.Am.Asso.Cancer.Res.43:969,2002)。クロ
ーンを遺伝子増幅に供し、その最終生産性は約20mg/Lであった。増殖特性および最
終的なAb生産性を改良するために、実施例6に記載のように、482.2C4A細胞を
、エレクトロポレーションによってヒトBcl−2遺伝子を含むプラスミド発現ベクター
でトランスフェクトした。3週間後、750μg/mlのゼオシンを含む培地中でトラン
スフェクタントを選択した。
【0061】
ゼオシン耐性細胞を25μg/mlのCHXで5時間処理して、アポトーシス感受性細
胞を排除した。処理細胞を新鮮な培養培地で2回洗浄してCHXを除去し、新鮮な増殖培
地中に再懸濁した。24時間後の回収後、生存細胞を、限界希釈(0.5細胞/ウェル)
によって、96ウェル細胞培養プレートにクローニングした。クローンが2週間でウェル
中に出現し、これを、Ab産生、CHX誘導性アポトーシス耐性、および増殖プロフィー
ルについてスクリーニングした。全局面で親482.2C4Aよりも能力の高いクローン
を選択し、さらに特徴づける。親482.2C4A細胞と比較して、最良のクローンは、
ストレス条件下で培養した場合により頑強であり、老化培養条件誘導性アポトーシス耐性
を示し、最大Ab生産性がより高い(約150%またはそれ以上)と予想される。
【実施例9】
【0062】
<細胞増殖特性のさらなる改良のためのSp−E26へのBcl−2遺伝子の導入>
実施例5に記載のように、Sp−E26細胞を、エレクトロポレーションによって、ヒ
トBcl−2−EEE遺伝子を含むプラスミド発現ベクターでトランスフェクトする。3
週間後に、500μg/mlのゼオシンを含む培地中でトランスフェクタントを選択する
。
【0063】
ゼオシン耐性細胞を25μg/mlのCHXで5時間処理して、アポトーシス感受性細
胞を排除する。処理細胞を新鮮な培養培地で2回洗浄してCHXを除去し、新鮮な増殖培
地中に再懸濁する。24時間後の回収後、生存細胞を、限界希釈(0.5細胞/ウェル)
によって、96ウェル細胞培養プレートにクローニングする。クローンが2週間でウェル
中に出現し、これを、CHX誘導性アポトーシス耐性および増殖プロフィールについてス
クリーニングする。Sp−EEEと同様に、全局面で親Sp−E26よりも能力の高いク
ローンを選択し、さらに特徴づける。親Sp−E26およびSp/EEE細胞と比較して
、HPV−16 E6/E7およびBcl−2−EEEを含む最良のクローンは、ストレ
ス条件下で培養した場合により頑強であり、老化培養条件誘導性アポトーシス耐性を示す
と予想され、それにより、組換えタンパク質産生のためのより良好な宿主細胞である。
【実施例10】
【0064】
<Sp−EEE細胞株を使用した組換えタンパク質産生の改良>
組換えタンパク質の産生のために生存度が増強された細胞株を開発する場合に取ること
ができる経路が2つ存在する。実施例6に記載のように、非常に首尾よく達成された1つ
の方法は、生存促進性遺伝子(Bcl−2など)での既に産生されている細胞株の安定な
トランスフェクションを含む。しかし、この方法は、さらなるトランスフェクションステ
ップ、選択ステップ、およびクローニングステップが必要であり、それにより、細胞株の
開発過程が少なくとも2ヶ月、おそらくそれ以上長期化する。さらに、各クローンに対し
て多数のパラメーター(増殖/生存、Bcl−2発現レベル、および生産性が含まれる)
を決定する必要があるので、「最良の」クローンのスクリーニングがむしろ必要である。
したがって、少数のクローンしか評価することができない。生産性が最も高いクローンの
生存がより優れていないかもしれない可能性およびその逆の可能性が非常に高い。本明細
書で使用した別のストラテジーは、より優れた増殖特性および生存特性を有する親細胞株
を開発し、その後に所望のタンパク質の産生のための発現ベクターでトランスフェクトす
ることである。
【0065】
Sp2/0細胞と比較して、Sp−EEE細胞は、培養液中でさらに1日増殖し続け、
15〜20%高い最大密度に達し、さらに4〜6日間長く生存する。組換えタンパク質(
IgG、抗体フラグメント、および融合タンパク質など)、成長因子(G−CSF、GM
−CFS、EPO、EGF、VEGFなど)、サイトカイン(インターロイキンファミリ
ーメンバー(IL−1〜IL−31)またはインターフェロンファミリーメンバー(α、
β、またはγインターフェロンなど)など)、オリゴヌクレオチド、ペプチド、ホルモン
、酵素、またはワクチン(例えば、A型肝炎、B型肝炎、またはC型肝炎、および上記の
他のワクチン)の産生のための遺伝子でその後にトランスフェクトした場合、細胞はその
増強された増殖特性および生存特性を保持する。
【0066】
組換えタンパク質(IgGなど)のための1つまたは複数の発現カセットを含むDNA
ベクター(pdHL2など)を使用して、標準的な方法(エレクトロポレーションなど)
によってSp−EEE細胞をトランスフェクトする。トランスフェクタントを、96ウェ
ルプレートに入れ、クローンを、ELISAまたはBiacoreなどの樹立された技術
によってタンパク質産生について分析する。生産性の高いクローンを、培養培地中で数ヵ
月にわたって漸増濃度のMTXに供し、遺伝子コピー数を増幅させる。Bcl−2陰性S
p2/0細胞中に生成されたクローンと比較して、Bcl−2−EEE発現クローンが約
20%高い細胞密度で増殖し、少なくともさらに4日間長く生存し、前者は、標準的なフ
ラスコまたはローラーボトル培養において、少なくとも20%より多くの組み合わせタン
パク質を生成する。懸濁、灌流、または流加バイオリアクター培養でさらにより高い増加
が実現される。
【実施例11】
【0067】
<バイオリアクター中でのBcl−2トランスフェクションクローン665.B4.1C
1のAb産生の改良>
実施例6の665.2B9#4および親クローン665.2B9両方を、無血清培地に
ウィーニングさせた(weaned)。細胞を、Tフラスコ中で数ヵ月の継続的継代培養
によって3μM MTXを含むハイブリドーマ無血清培地(HSFM)(Immunom
edics PN 10070)のカスタマイズされた形成に適合させた。適合させた細
胞を、保存のためにTフラスコからローラーボトルに掻き取った。45%コンディション
培地(対数増殖期における培養物の遠心分離後に上清として回収した培地)、10%DM
SO、および45%HSFMから構成される無FBS低温保存液を使用した1mLバイア
ル中で、1×10-7生存細胞を使用して、各細胞株についてのマスターセルバンク(MC
B)を作製した。MCB細胞株を、それぞれ、665.2B9.1E4(Bcl−2遺伝
子なし)および665.B4.1C1(Bcl−2遺伝子を含む)と命名した。これら2
つのクローンの増殖特性および抗体産生を、バッチ培養条件下で比較した。
【0068】
MCBから拡大された上記細胞を使用して、3Lのベンチスケールバイオリアクター中
で実験を行った。3Lバイオリアクターシステムは、2500L cGMPバイオリアク
ターシステムの縮小モデルである。したがって、評価の結果は、商業的大量製造へのこれ
らの細胞株の適合性を支持する。
【0069】
MCB(Immunomedics PN 10070)の作製で使用したものと同一
の増殖HSFMを使用して、細胞株を維持し、接種材料を調製した。基本HSFM(特別
に修正した増殖HSFMに基づいた特別な配合物(formulation))(Imm
unomedics PN 10194)を、3L流加バイオリアクター過程で使用した
。両培地は、微量タンパク質としてのみインスリンおよびトランスフェリンを含む。撹拌
および曝気による剪断から細胞を保護するために、さらに0.1%のPluronic
F68を配合物に組み込んだ。この培地も3μMのMTXを含んだ。
【0070】
連続供給溶液およびパルス供給溶液の特徴を、以下の表4および5に示す。
【0071】
【表4】
【0072】
【表5】
【0073】
可動範囲2Lで3L Bellco撹拌フラスコバイオリアクターシステム(Bell
co glasses,Vineland,NJ)中で流加試験を行った。バイオリアク
ターの温度、pH、および溶存酸素(DO)をモニタリングし、単ループ制御装置によっ
て制御した。反応器の温度を、加熱ブランケットによって37℃に制御した。培養物のp
Hを、CO2または6%Na2CO3の添加によってpH7.3に制御した。円筒形焼結(
sintered)スパージャーによって10ml/分の曝気を行った。培地へのO2の
間欠的噴霧によって、DOを40%を超える空気飽和(air saturation)
に制御した。50〜60rpmの一定の撹拌速度を培養中に使用した。
【0074】
MCB由来の凍結バイアルを融解し、約1〜2週間Tフラスコに回収した。次いで、細
胞をTフラスコからローラーボトルに拡大し、バイオリアクターでインキュベートした。
細胞を、37℃、5%CO2雰囲気下で培養し、拡大過程中は対数増殖期を維持した。
【0075】
インキュベーション前に、1.2リットルの基本HSFMを、ポンプにて無菌でバイオ
リアクターに移した。培地の空気を飽和させて、溶存酸素(DO)プローブを較正した。
また、pHプローブを較正するために、培地サンプルを採取した。一旦pHプローブおよ
びDOプローブが較正されると、両制御装置を、AUTOモードに設定した。一旦システ
ムが、pH(7.3)および温度(37℃)の定値に達すると、計算された量の接種材料
が、ローラーボトルからバイオリアクターに移された。接種後生存細胞密度(VCD)は
、約2×105バイアル細胞/mlであった。
【0076】
供給ストラテジーを以下に示す。培養中、濃縮栄養溶液をバイオリアクターに供給して
、細胞に必要且つ過剰でない栄養素を細胞に供給した(全過程の略図については、図25
を参照のこと)。濃縮栄養溶液を、連続供給およびパルス供給によって培養物に送達させ
た。連続供給溶液を、蠕動ポンプ(Watson−Marlow 101U/R)を使用
して、反応器に移した。パルス供給溶液を、1日1回培養物にパルス供給した。
【0077】
2つの流加ストラテジーを開発し、両細胞株に適用した。過程番号1は、培養中に組換
えインスリンを供給しない。過程番号2を、修飾リノール酸、脂質供給スケジュール、お
よびインスリンのさらなる供給を使用して過程番号1に基づいてデザインする。
【0078】
以下の表は、両細胞株の両過程の供給をまとめている。
【0079】
【表6】
【0080】
【表7】
【0081】
【表8】
【0082】
【表9】
【0083】
培養中、オフライン分析のためにバイオリアクターサンプルを定期的に採取した。生存
細胞密度(VCD)および細胞生存度を、0.4%トリパンブルー色素での染色後の血球
計算板を使用した顕微鏡での計数によって測定した。グルコース、乳酸塩、グルタミン、
アンモニアの濃度を、Nova Bioprofile 200を使用して測定した。抗
体濃度を、プロテインAアフィニティクロマトグラフィカラムを使用したHPLC(Ap
plied Biosystems,P/N2−1001−00)によって決定した。
【0084】
以下のように、産生培養抗体を培養物中の全生存細胞の時間積分で割ることによって、
特異的抗体生産性を計算した。
【0085】
【数1】
(ここで、
【数2】
は台形公式
【数3】
によって概算する)
【0086】
図26は、過程番号1および過程番号2による両細胞株の増殖曲線(VCDおよび生存
度)を示す。過程番号1により、665.2B9.1E4細胞は、6日目に生存度86%
で1×107生存細胞/mlの最大VCDに増殖した。6日後、VCDおよびV%は急速
に減少し、8日目に培養物を回収した。過程番号2は、1.2×107生存細胞/mlの
より高いVCDに到達し、1日長く維持されるのに役立つ。
【0087】
665.2B9.1E4細胞と比較して、665.B4.1C1細胞は、両過程でさら
により良好に増殖した。過程番号1では、そのVCDは、7日目に生存度97%で2×1
07生存細胞/mlに達した。培養物はまた、このVCDおよびV%をさらに2日間維持
し、その後減少し始めた。11日目に培養物を回収した。過程番号2では、665.B4
.1C1細胞は、過程番号1と類似の増殖プロフィールを示した。より詳細には、細胞は
、2.3×107生存細胞/mlの最も高いVCDに到達し、生存度はわずかに遅く、1
1日目に回収した。この所見は、過程番号2で増殖における利点が証明された665.2
B9.1E4細胞株と幾らか異なっていた。
【0088】
過程番号1および過程番号2における2つの細胞株の抗体収率を、図27で比較した。
665.2B9.1E4細胞の最終収率は、過程番号1で0.42g/L、過程番号2で
0.55g/Lであった。比較のために、665.B4.1C1細胞は、両過程において
、1.5g/Lのより高い最終収率であった。
【0089】
1日の特異的抗体生産性(細胞基準あたり)を計算し、図28に示す。図に示すように
、665.2B9.1E4細胞の1日あたりの平均Q[MAb]は、両過程の一連の培養を通
して約15pg/細胞/日であった。過程番号2における最も高いVCDでのより長い増
殖日数により、より高い最終抗体濃度が得られた。
【0090】
665.B4.1C1細胞は、両過程において類似の1日あたりの特異的抗体生産性プ
ロフィールを示し、過程番号1の生産性はわずかに高かった。1日のQ[MAb]は、9日目
まで20〜25pg/細胞/日に維持された。その後、生産性は低下した。
【0091】
665.2B9.1E4細胞株と比較して、665.B4.1C1細胞株は、15pg
/細胞/日と比較して、20〜25pg/細胞/日のより高い特異的抗体生産性を示した
。そのより良好な増殖と組み合わせて、665.B4.1C1細胞株の最終抗体収率は、
665.2B9.1E4細胞株によって達成された0.55g/Lと比較して、1.5g
/Lと3倍になった。これらの結果は、組換えタンパク質(この場合、臨床用抗体)の商
業的大量調製のためにバイオリアクター中の無血清培地中でその増殖および抗体収率を増
強するために宿主細胞にBcl−2遺伝子を組み込むことが有利であることを証明する。
当業者は、本明細書中に開示の方法および過程を必要に応じて修正することができる。本
明細書中に含まれる全ての刊行物、特許、特許出願、および引例は、その全体が本明細書
中で参照することにより組み込まれる。
【技術分野】
【0001】
本出願は、2004年7月23日出願の米国特許出願番号60/590,349号の優
先権を主張する。本出願は、上記仮出願で開示された主題のみを主張し、それにより、新
規事項を示さない。
【背景技術】
【0002】
特に、巨大なバイオリアクターにおけるインビトロでの細胞培養は、多数のバイオテク
ノロジー産物の産生の基本であり、支持体へのこれらのタンパク質産物の細胞による作製
(elaboration)を含み、これらの産物を単離し、臨床的使用の前にさらに処
理する。培養物中で増殖した細胞からの長期にわたるタンパク質の産生量は、例えば、細
胞密度、細胞周期の段階、細胞のタンパク質生合成率、細胞の生存度および増殖を支持す
るために使用される培地の条件、ならびに培養物中の細胞の寿命(プログラム細胞死(す
なわち、アポトーシス)のために死滅するまでの期間)などの多数の要因に依存する。例
えば、栄養素、細胞密度、酸素および二酸化炭素の含有量、乳酸デヒドロゲナーゼ、pH
、浸透圧、異化生成物などの調節による所望のタンパク質の生産性を増加させる方法と共
に、培養物中の細胞の生存度および寿命に対する種々の改良方法が開発されている。例え
ば、細胞密度の増加により、過程の生産性をより高くすることができるが、培養物中の細
胞の寿命も短縮され得る。したがって、最大密度が達成された場合、できるだけ長く最も
生産性の高い状態で細胞集団を維持するために、培養物中のこのような細胞の増殖速度を
低下させることが望ましい。これにより、その産生ピークでのバイオリアクターサイクル
が増加または延長され、所望のタンパク質産物が長期間で作製され、これにより、バイオ
リアクターサイクル由来の収率がより高くなる。
【0003】
細胞増殖を支持する培地の調整、一定の成長促進因子の添加、およびタンパク質合成に
影響を与えない細胞増殖の阻害などのバイオリアクターのサイクル時間を増加させるため
の多数の異なるアプローチが追求されている。1つの特定のアプローチは、細胞周期標的
に影響を与えるための遺伝子またはアンチセンスオリゴヌクレオチドの使用による細胞周
期の調節によって培養細胞の寿命を増加させることを目的とし、それにより、細胞周期の
進行を阻害していわゆる偽老化状態を誘導するベクターでのトランスフェクション、形質
転換、または感染によって細胞を偽老化状態に誘導し、さらに、細胞分裂を遮断し、培養
物中の細胞のタンパク質合成能を拡大する。言い換えれば、細胞周期インヒビターを発現
するベクターでの細胞の形質転換によって偽老化状態を誘導することができる(Bucc
iarelli et al.,US Patent 2002/0160450 A1
;idem.,WO 02/16590 A2)。後者の方法は、Goldsteinお
よびSingal(Exp Cell Res 88,359−64,1974;Bre
nner et al.,Oncogene 17:199−205,1998)に記載
のように、細胞複製の阻害により、細胞を細胞培養の寿命を延長することができる状態に
なるように図り、アポトーシスに耐性を示し得る(Chang et al.,Proc
Natl Acad Sci USA 97,4291−6,2000;Javela
nd et al.,Oncogene 19,61−8,2000)。
【0004】
さらに別のアプローチは、アデノウイルスE1遺伝子でのトランスフェクション後に初
代二倍体ヒト細胞または無制限に増殖するその誘導体を樹立するステップを含む。機能的
なAd5E1AおよびE1B遺伝子産物を発現する新規の細胞株(そのうちの1つは、P
ER.C6(ECACC寄託番号96022940)である)は、遺伝子療法およびワク
チンならびにヒト成長因子およびヒト抗体などの組換え治療タンパク質の産生のためにデ
ザインされた組換えアデノウイルスおよび他のウイルス(例えば、インフルエンザウイル
ス、単純ヘルペスウイルス、ロタウイルス、麻疹ウイルス)を産生することができる(V
ogels et al.,WO02/40665A2)。
【0005】
他のアプローチは、細胞中のアポトーシスの防止または遅延のためのカスパーゼインヒ
ビターの使用に注目している。例えば、米国特許第6,586,206号を参照のこと。
さらに他のアプローチにより、細胞中でのアポトーシス防止または遅延のためのBcl−
2ファミリーのメンバーなどのアポトーシスインヒビターの使用が試みされている。Ar
den et al.,Bioprocessing Journal,3:23−28
(2004)を参照のこと。これらのアプローチにより、予想不可能な結果が得られてい
る。例えば、ある研究では、Bcl−2発現により細胞の生存度が増加するが、タンパク
質産生は増加しなかった。Tey et al.Biotechnol.Bioeng.
68:31−43(2000)を参照のこと。別の例にはCHO細胞のアポトーシスを遅
延させるためのBcl−2タンパク質の過剰発現が記載されているが、Bcl−xLはタ
ンパク質産生を増加させるのに対して、Bcl−2はタンパク質産生を減少させた(WO
03/083093を参照のこと)。さらなる例には、培養物中のSp2/0−Ag14
(ATCC番号CRL−1581、以後Sp2/0と呼ぶ)細胞の生存を延長するために
Bcl−2タンパク質の発現を使用した実験が記載されているが、Bcl−2発現クロー
ンの細胞密度はその親培養物の細胞密度より20〜50%低く、生物医薬品産業における
その実用的な適用への関心が高まっている(WO03/040374を参照のこと)。し
たがって、組換えタンパク質の高レベル発現のための改良宿主細胞および宿主細胞におけ
る組換えタンパク質産生、特に抗体および抗体フラグメント、多重特異性抗体、フラグメ
ント、および一本鎖構築物、ペプチド、酵素、成長因子、ホルモン、インターロイキン、
インターフェロン、およびワクチンの産生を確実に増加させる方法が非常に望ましいこと
が明らかである。
【発明の概要】
【0006】
したがって、本発明の目的は、老化を阻害するか細胞生存を促進する薬剤(例えば、抗
アポトーシス剤)の細胞への導入による、細胞培養物の寿命および組換えタンパク質の収
率を増加させる改良された宿主細胞および方法を提供することである。このような薬剤の
使用により、所望の組換えタンパク質の産生のために使用された培養物中の細胞の寿命お
よび生存度を優先的に増加させ、それに伴ってこのような培養物中の細胞の生産性が増加
し、それにより、所望のタンパク質の収率が最適になる。好ましくは、本発明の方法で使
用されるアポトーシスインヒビターには、Bcl−2およびそのファミリーメンバーが含
まれるが、これらに限定されない。あるいは、細胞クローンの寿命および組換えタンパク
質の収率を、p53およびRbなどの細胞内アポトーシス促進性タンパク質レベルをダウ
ンレギュレートするか、Bcl−2などの細胞内抗アポトーシス性タンパク質をアップレ
ギュレートする薬剤を細胞に導入することによって改良することができる。好ましくは、
本発明の方法で使用される調節剤には、ヒト乳頭腫ウイルス16型(HPV−16)の腫
瘍性タンパク質E6およびE7ならびにその組み合わせが含まれるが、これらに限定され
ない。さらに、本明細書中に記載のカスパーゼインヒビターはまた、アポトーシスの遮断
または減少に寄与し、それにより、細胞生存が増加し、培養物中の細胞による組換えタン
パク質産生が増加し得る。これらの培養物中で組換えタンパク質の産生を増強するために
使用することができる抗アポトーシス剤のさらなるクラスには、エリスロポエチン(EP
O)などのサイトカインI型スーパーファミリーの特定のメンバーが含まれる。このクラ
スの代表的な分子としてのEPOは、赤血球だけでなく複数の細胞型のアポトーシスの主
な修飾因子であり、それにより、内皮細胞、心筋細胞、腎臓の尿細管上皮細胞、皮膚、お
よびニューロンなどにおいてより一般的な細胞保護機能を有する[P.Ghezzi a
nd M.Brines,Cell Death and Differentiati
on 11(suppl.1),s37−s44,July 2004の概説を参照のこ
と]。
【0007】
本発明はまた、細胞培養物の寿命が増加し、および/または最適になり、所望の組換え
タンパク質の収率が増加する細胞培養条件をもたらすための因子(トランスフェクション
ベクター、所望の性質を有する細胞クローンの分泌および選択、細胞培養培地、増殖条件
、バイオリアクターの配置、および細胞型が含まれるが、これらに限定されない)の新規
の組み合わせを組み込んだ細胞培養法を教示する。これらの細胞培養方法には、懸濁産生
法、灌流産生法、および流加産生法が含まれる。Tey et al.,J.Biote
chnol.79:147−159(2000);Zhang et al.,J.Ch
em.Technol.Biotechnol.79:171−181(2004);Z
hou et al.,Biotechnol.Bioeng.55:783−792(
1997)を参照のこと。
【0008】
他で定義しない限り、本発明で使用した全ての技術用語および科学用語は、当業者に一
般に理解されている意味を有する。さらに、本明細書中に引用した全ての特許および他の
引例の内容全体が、本明細書中に参照することにより組み込まれる。
【図面の簡単な説明】
【0009】
【図1】シクロヘキシミドで処置(+CHX)、又は未処置(−CHX)のSp2/0細胞およびSp−E26細胞の画像を示す。
【図2】CHX処置に対する耐性がより高いHPV E6/E7形質導入細胞のスクリーニングの結果を示す。全部で55クローンをスクリーニングし、第1の実験では、31クローンをスクリーニングし(上のパネル)、第2の実験では、24クローンをスクリーニングした(下のパネル)。各クローンの健康な細胞を、2つの等しい部分に分割した。一方をCHXで2時間処置し、他方を未処置のままにした。次いで、これら2つの培養物中の生存細胞を、MTTアッセイによって測定し、処置した生存細胞集団(CHX+):非処置生存細胞集団(CHX-)比をプロットした。上のパネルに示すように、CHX処置により、Sp2/0細胞の生存度が30%減少する一方で、Sp−E26細胞では6%しか減少しなかった。スクリーニングした31クローンのうちの7クローン(*で示す)は、Sp2/0よりも有意に良好であったが(生存度の減少は20%未満)、Sp−E26ほどではなかった。第2の実験でスクリーニングした24クローンについて(下のパネル)、CHX処置により、Sp2/0細胞の生存度が約50%減少し、Sp−E26の生存度の減少が約20%未満であった。スクリーニングした24クローンのうちの10クローン(*または**で示す)は、Sp2/0よりも有意に良好であり(減少は30%未満)、これらのうちの6クローン(**で示す)は、Sp−E26に匹敵するかこれより良好であった(20%未満)。E28およびE36は2つのさらなるコントロールクローンであり、Sp2/0よりも良好であるが、Sp−E26ほどではない。
【図3】GuavaネキシンVアッセイのドットプロットを示す。初期アポトーシス細胞(ネキシンV陽性および7−AAD陰性)の比率を、右下の四分円に示す。
【図4】CHXによって処置されたSp2/0のDNA断片化を示す。対照的に、Sp−E26細胞は、この処置に耐性を示す。
【図5】Tフラスコ中でのSp2/0細胞およびSp−E26細胞の増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度200,000/mlでTフラスコに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬(Guava technologies,Inc.)およびPCA装置(Guava Technologies,Inc.)を使用して毎日計数した。NH4+および乳酸塩の蓄積もモニタリングした。
【図6】3Lバイオリアクター中でのバッチ培養について測定したSp2/0細胞およびSp−E26細胞の増殖プロフィールを比較する。健康な細胞(生存度95%超)を、初期細胞密度250,000/mlでバイオリアクターに播種した。トリパンブルーおよび顕微鏡によって細胞を毎日計数した。
【図7】Bcl−2−EEE発現についてクローンをスクリーニングするためにBcl−2(100)抗体(Santa CruzBiotech.)で染色し、増強された化学発光を使用して発色させた代表的免疫ブロットを示す。
【図8】Guava Expressを使用したフローサイトメトリーの結果のグラフを示す。細胞を固定し、透過処理を行い、その後、フィコエリトリン抱合抗Bcl−2抗体(Santa Cruz Biotechnology,Inc.)で染色した。いくつかのサブクローンを比較する。
【図9】Guava Expressを使用したフローサイトメトリーの結果のグラフを示す。細胞を固定し、透過処理を行い、その後、フィコエリトリン抱合抗Bcl−2抗体(Santa Cruz Biotechnology,Inc.)で染色した。Sp2/0細胞、Raji細胞、およびDaudi細胞を、Bcl−2−EEEクローンと比較した。
【図10】665.B4.1C1細胞、Sp2/0細胞、Raji細胞、Daudi細胞、Sp−EEE細胞(87〜29クローン)、およびSp−EEE細胞(7〜16クローン)の溶解物の免疫ブロット分析の結果を示す。A.ヒトBcl−2特異的抗体(Santa Cruz Biotechnology,Inc.)を使用して染色したブロット。B.マウスBcl−2およびヒトBcl−2を認識する抗Bcl−2抗体(Santa Cruz Biotechnology,Inc.)を使用して染色したブロット。
【図11】10%ウシ胎児血清を補足した培地で増殖したSp2/0細胞と比較したSp−EEEクローンの増殖曲線(A)および生存度(B)を示す。
【図12】1%ウシ胎児血清を補足した培地で増殖したSp2/0細胞と比較したSp−EEEクローンの増殖曲線(A)および生存度(B)を示す。
【図13】無血清培地で増殖したSp2/0細胞と比較したSp−EEEクローンの増殖曲線(A)および生存度(B)を示す。
【図14】Sp−EEE細胞(87〜29クローン)のメトトレキセート死滅曲線を示す。
【図15】1mg/mlゼオシンの存在下または非存在下で増殖したSp−EEEクローンを含むGuava Expressを使用したフローサイトメトリーの結果のグラフを示す。細胞を固定し、透過処理を行い、その後、フィコエリトリン抱合抗Bcl−2抗体(Santa Cruz Biotechnology,Inc.)で染色した。
【図16】Sp2/0細胞をトランスフェクトして、ヒト化抗体配列、SV40プロモーター配列、およびエンハンサー配列を有する665.2B9クローンを得るために使用したpdHL2ベクターのマップを示す。
【図17】クローン665.2B9のトランスフェクションに使用したBcl−2遺伝子を組み込んだDNAプラスミドのマップを示す。
【図18】Bcl−2トランスフェクションクローン665.2B9#4、Bcl−2陰性クローン、および非トランスフェクションコントロールの増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度400,000/mlで24ウェルプレートに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図19】Bcl−2トランスフェクションクローン665.2B9#4、Bcl−2陰性クローン、および非トランスフェクションコントロールの増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度400,000/mlで24ウェルプレートに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図20】異なるMTX濃度でのBcl−2トランスフェクションクローン665.2B9#4およびBcl−2陰性クローンの増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度100,000/mlでTフラスコに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図21】異なるMTX濃度でのBcl−2トランスフェクションクローン665.2B9#4およびBcl−2陰性クローンの増殖プロフィールを示す。健康な細胞(生存度95%超)を、初期細胞密度100,000/mlでTフラスコに播種した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図22】ウェスタンブロッティングによって検出した漸増濃度のMTXおよびクローン#13中でクローン665.2B9#4によって発現したヒトBcl−2レベルを示す。
【図23】急増L−グルタミンおよびグルコースを含むか含まない0.6μMまたは1μMのMTX中で培養したクローン665.2B9#4ならびに0.3μM MTX中で培養したBcl−2陰性クローンの細胞生存度および生存細胞密度のプロフィールをそれぞれ示す。健康な細胞(生存度95%超)を、初期細胞密度200,000/mlでローラーボトルに播種した。2日目および4日目に(矢印で示す)、グルコースおよびL−グルタミンを含む栄養補足溶液を、「急増」培養物に添加した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図24】急増L−グルタミンおよびグルコースを含むか含まない0.6μMまたは1μMのMTX中で培養したクローン665.2B9#4ならびに0.3μM MTX中で培養したBcl−2陰性クローンの細胞生存度および生存細胞密度のプロフィールをそれぞれ示す。健康な細胞(生存度95%超)を、初期細胞密度200,000/mlでローラーボトルに播種した。2日目および4日目に(矢印で示す)、グルコースおよびL−グルタミンを含む栄養補足溶液を、「急増」培養物に添加した。生存細胞および死滅細胞を、Guava ViaCount試薬およびPCA装置を使用して毎日計数した。
【図25】バイオリアクター供給ストラテジーについての過程の略図を示す。
【図26】組換えインスリンを供給しない過程番号1、過程番号1に基づいて修飾リノール酸および脂質供給スケジュールを使用し、インスリンをさらに供給した過程番号2による665.2B9.1E4細胞株および665.B4.1C1細胞株の増殖曲線(VCDおよび生存度)を示す。
【図27】過程番号1および2における665.2B9.1E4細胞株および665.B4.1C1細胞株の抗体収率を示す。665.2B9.1E4細胞の最終収率は、過程番号1では0.42g/Lであり、過程番号2では0.55g/Lであった。比較のために、665.B4.1C1細胞は、両方の過程において1.5g/Lのより高い最終収率が実現された。
【図28】1日の特異的抗体産生性を示す(細胞基準あたり)。図に示すように、一連の培養による両過程についての665.2B9.1E4細胞の1日あたりの平均Q[MAb]は約15pg/細胞/日であった。過程番号2における最も高いVCDでの増殖日数の追加により、最終抗体濃度がより高くなった。
【発明を実施するための形態】
【0010】
本発明は、改良された組成物(宿主細胞株を含む)およびこのような細胞株中での組換
えタンパク質の産生を増強する方法を提供する。1つまたは複数の抗アポトーシス性遺伝
子を構成的に発現し、この抗アポトーシス性遺伝子の発現によって培養物中のトランスフ
ェクション細胞の生存が延長されて目的のタンパク質またはペプチドの収率が増強される
、目的のタンパク質またはペプチドをコードする発現構築物でトランスフェクトすること
ができる細胞株を作製した。
【0011】
詳細には、本発明者らは、Sp2/0骨髄腫細胞株から、バッチ培養における生存が増
強されたSp−E26およびSp−EEEと呼ばれる2つの新規の細胞株を作製した。S
p−E26およびSp−EEEは、それぞれ、HPV−16のE6およびE7タンパク質
ならびにBcl−2変異体(Bcl−2−EEEと呼ばれる)を構成的に発現する。さら
に、目的の組換えタンパク質のための発現ベクターでのSp−E26またはSp−EEE
のいずれかのトランスフェクションの際に、組換えタンパク質産生、特に、組換え抗体お
よび抗体フラグメントの産生を改良することができる。E6/E7またはBcl−2−E
EEタンパク質は、宿主細胞におけるアポトーシスの誘導を遅延させ、宿主細胞における
組換えタンパク質産生を増強する。1つまたは複数のカスパーゼインヒビター(例えば、
カスパーゼ1インヒビターおよび/またはカスパーゼ3インヒビター)の添加(Bin
Yang et al. Nephron Experimental Nephrol
ogy 2004;96:e39−e51)および/またはサイトカインI型スーパーフ
ァミリーの1つまたは複数のメンバー(エリスロポエチン(EPO)など)の細胞の増殖
培地への添加の添加によって、タンパク質産生をなおさらに高めることができる。これに
関して、pan−カスパーゼインヒビターが特に有効である。
【0012】
本発明者らはまた、アポトーシスインヒビター(Bcl−2など)の同時発現によって
宿主細胞中で抗体または抗体フラグメントなどの組換えタンパク質の産生を有意に増強す
ることができることを見出した。特に、抗体または抗体フラグメントをコードする発現ベ
クターで安定にトランスフェクトされ、アポトーシスインヒビター(Bcl−2など)を
コードする発現ベクターで同時トランスフェクトした骨髄腫細胞株(Sp2/0など)に
おいて、タンパク質産生が有意に増強される。E6/E7遺伝子でトランスフェクトした
宿主細胞で抗体産生を増加させることもできる。細胞の増殖培地への1つまたは複数のカ
スパーゼインヒビターの添加によって、組換えタンパク質産生をいっそう高めることがで
きる。これに関して、pan−カスパーゼインヒビターが特に有効である。また、細胞培
養培地へのEPO(すなわち、別の抗アポトーシス性サイトカイン)の供給によって、組
換えタンパク質の産生を増強することができる。
【0013】
アポトーシスと呼ばれる生理学的細胞死(すなわち、プログラム細胞死)(Kerr
et al.,Br J Cancer.,26:239−257,1972)は、適切
な組織の発達および維持に不可欠であり、進化過程で保存されている内因性遺伝子プログ
ラムによって調節される(Ellis et al.,Annu Rev Cell B
iol,7,663−698,1991)。したがって、人工的環境で(エクスビボ培養
物などで)細胞が増殖する場合、この遺伝子の素質によって寿命に限度がある。したがっ
て、医薬品および産業ならびに研究で使用されるタンパク質の産生のためのこのような細
胞培養物の有用性は、このような培養物がアポトーシス機構によって死滅する前に寿命(
すなわち、周期)が長期間維持されることに依存する。
【0014】
アポトーシス効果からの細胞周期の区別によって、細胞増殖および細胞死事象と無関係
に作用する方法および薬剤を発見した。Bcl−2は、周知のアポトーシスの細胞内調節
因子であり(Vaux et al.,Nature 335,440−2,1988)
、細胞周期に入ることに対するその阻害的影響とは遺伝的に異なる抗アポトーシス効果を
有することが見出されているがん原遺伝子である(Huang et al.,EMBO
J 16,4628−38,1997)。Bcl−2、Bcl−xL、およびBcl−
wの2つのホモログも細胞生存を延長させるが、Bcl−2ファミリーの他のメンバー(
BaxおよびBakなど)はアポトーシス促進性を示す(Oltvai et al.,
Cell 74,609−19,1993;Chittenden et al.,Na
ture 374,733−6,1995;Farrow et al.,Nature
374,731−3,1995;Kiefer et al.,Nature 374
,736−9,1995)。他の抗アポトーシス性遺伝子には、Bcl−6およびMcl
−1が含まれる。
【0015】
したがって、Bcl−2および特定のそのファミリーメンバーは、アポトーシスに対し
て保護効果を発揮し、それにより、それは、タンパク質産生のために使用される培養物中
で特定の宿主細胞の寿命を増加させ、それによって産生および単離されるタンパク質量が
増強される方法と仮定される。Bリンパ球、特に、骨髄腫細胞によって抗体が産生される
ので、抗アポトーシス性Bcl−2ファミリーメンバー(Bcl−2、Bcl−xL、B
cl−w、またはこれらのタンパク質の変異体など)の過剰発現によってアポトーシスが
阻害され、それにより、細胞密度が増加し、培養物の生存が長くなる。したがって、他で
提案されているように(同節)、抗アポトーシス性Bcl−2ファミリー遺伝子のトラン
スフェクションにより、細胞周期自体の妨害によって細胞培養を延長する必要性が回避さ
れる。同様に、Bcl−2遺伝子での線維芽細胞のトランスフェクションによってこれら
の細胞中でBcl−2が過剰発現され、再度アポトーシスに拮抗作用し、これらの細胞の
寿命が増加し、それに伴って組換えタンパク質の産生および単離が増加する。サイトカイ
ンの使用中止の際に、インターロイキン−6(IL−6)依存性マウス骨髄腫細胞は、ア
ポトーシスを受けているかのように期限が切れることも認められた。このような細胞中の
IL−6−受容体をBcl−2またはBcl−xLによって調節してアポトーシスを延長
することができることも見出された(Schwarz et al.,Cancer R
es 55:2262−5,1995)。
【0016】
最近の文献もまた、3箇所の点変異(T69E、S70E、およびS87E)を有する
変異Bcl−2が、野生型または1箇所の点変異体と比較して、有意により高い抗アポト
ーシス活性を示すことを証明している(Deng et al.,PNAS(101)1
53−158,2004)。したがって、本発明は、Bcl−2−EEE三重変異体の発
現ベクターの構築を教示し、これを使用してSp2/0細胞をトランスフェクトして、寿
命および組換えタンパク質産生が改善されたSp−EEEクローンおよびサブクローンを
作製する。
【0017】
腫瘍ウイルスなどの他の作用因子はまた、その細胞不死化誘発の一部としてアポトーシ
スを妨害し、最終的には極めて有害な形質転換(高リスク型HPV癌タンパク質E6およ
びE7など)を完了することができる(Finzer et al.,Cancer L
ett 188,15−24,2002)。例えば、ウイルスE6タンパク質は、紫外線
に対する上皮アポトーシス応答を有効に遮断する(Storey,Trends Mol
Med 8,417−21,2002)。状況証拠から、ヒト乳頭腫ウイルスによって
扁平上皮癌(基底細胞癌ではない)におけるアポトーシスを減少させることができる(J
ackson et al.,Br J Cancer 87,319−23,2002
)。しかし、全ての乳頭腫ウイルスの癌タンパク質が抗アポトーシス効果を有するわけで
はない。例えば、他の研究では、ウシ種の乳頭腫ウイルスE6タンパク質が細胞にアポト
ーシス感受性を与えると報告されており(Liu et al.,Virology 2
95,230−7,2002)、これは、HPV−16 E7遺伝子が、一定の刺激によ
って誘導されたアポトーシスから星状細胞を防御することを示す他の研究(Lee et
al.,Yonsei Med J 42,471−9,2001)と対照的である。
E6結合タンパク質アプタマーの使用により、HPV E6癌タンパク質がHPV陽性腫
瘍細胞において抗アポトーシス活性を有するという直接的な実験による証拠が得られた(
Butz et al.,Proc Natl Acad Sci USA 97,66
93−7,2000)。しかし、他のHPV癌タンパク質は逆の効果を有することができ
、E2タンパク質は他のHPVタンパク質の非存在下でアポトーシスを誘導する(Web
ster et al.,J Biol Chem 275,87−94,2000)。
E6およびE7タンパク質の連続的発現には、子宮頸癌細胞の最適な増殖および2つのウ
イルスタンパク質が細胞生存に対して異なる効果を発揮することが必要であることが知ら
れている(DeFilippis et al.,J Virol 77,1551−6
3,2003)。HPV−16 E6に寄与する主な細胞内標的はp53である。E6は
、p53および細胞ユビキチンリガーゼと三重複合体(E6AP)を形成し、それにより
、プロテオソーム経路およびp53の不活化によってp53をユビキチン化して分解する
。他方では、HPV−16 E7タンパク質は、腫瘍抑制タンパク質Rbと相互作用して
不安定にする。さらに、アポトーシスおよび細胞周期経路に関与する種々の他の細胞内タ
ンパク質のレベルは、E6およびE7形質転換(Bcl−2、Bcl−xL、p73、M
DM2、p21、サイクリンおよびcdc、cdkタンパク質など)によって調節される
ことが報告されていた。これらのタンパク質発現の変化は、細胞の生理学的特性に非常に
影響を与える。したがって、本発明者らは、培養物中でのHPV−16 E6およびE7
による細胞のトランスフェクションが加齢培養条件誘導性アポトーシスに耐性を示す遺伝
子操作されたクローンの生成に非常に有効であり、それにより、細胞培養物の寿命が延長
されるという仮説を立てた。HPV−16癌タンパク質E7またはE6のみのいずれかの
細胞への導入が、加齢培養条件誘導性アポトーシス耐性が改良された遺伝子操作されたク
ローンの生成に十分であり得るとも仮定した。細胞が組換えタンパク質産生クローンであ
る場合、生理学的性質の改良により、全タンパク質生産性が増強される。
【0018】
<ウイルス抗アポトーシス遺伝子を発現する新規の宿主細胞の生成>
ウイルス抗アポトーシス性遺伝子(HPV−16 E6およびE7タンパク質など)を
構成的に発現する宿主細胞(骨髄腫宿主細胞など)を生成することができる。これらの宿
主細胞を、目的の組換えタンパク質をコードする発現ベクターでトランスフェクトするこ
とができ、抗アポトーシス性遺伝子の同時発現により、組換えタンパク質の産生が有意に
増加する。
【0019】
宿主細胞は、本質的に、ウイルス抗アポトーシス性遺伝子で安定に形質転換することが
できる組換えタンパク質の産生に適切な任意の宿主細胞であり得る。多数の組換えタンパ
ク質について、CHOおよびCOS細胞などの宿主細胞が有利である一方で、抗体などの
他のタンパク質については、骨髄腫細胞およびCHO細胞などの宿主細胞を一般的に選択
する。遺伝子が構成的または誘導的に発現される任意の適切な方法(すなわち、宿主細胞
の染色体に遺伝子が安定に組み込まれる一方で、遺伝子が発現される任意の方法)によっ
て、ウイルス遺伝子を宿主細胞に導入することができる。目的の遺伝子で宿主細胞を安定
に形質転換する方法は、当分野で周知である。特に有利な方法は、ウイルス抗アポトーシ
ス性遺伝子をコードするレトロウイルスベクターを使用することである。適切なベクター
には、LSXNベクターが含まれる(Miller et al.Biotechniq
ues 7,980−90,1989)。
【0020】
有利には、宿主細胞をトランスフェクトするために使用されるベクターは、ベクターを
含む細胞が選択される選択マーカーを含む。適切な選択マーカー(トランスフェクション
細胞に抗生物質耐性を付与する酵素など)は、当分野で周知である。トランスフェクショ
ン後、選択剤(抗生物質など)を含む培地中に細胞を維持し、マーカー耐性についてスク
リーニングする。細胞を選択し、従来の方法を使用した限界希釈によってクローニングす
ることができる。
【0021】
ウイルス抗アポトーシス性遺伝子が細胞生存度を増加させる能力を、アポトーシスを誘
導する薬剤(シクロヘキシミド(CHX)など)での細胞の攻撃誘発によって試験するこ
とができる。ウイルス抗アポトーシス性遺伝子を発現しない細胞が有意なアポトーシスの
発症を示す傾向があるのに対して、この遺伝子を発現する細胞がアポトーシス活性を劇的
に減少させる。アポトーシスの検出方法は当分野で周知であり、例えば、細胞表面FIT
C−アネキシンV結合アッセイ、DNAラダリングアッセイ、およびTUNELアッセイ
が含まれる。ウイルス抗アポトーシス遺伝子を発現する適切な細胞の選択の際、細胞を、
最適な組換えタンパク質をコードする発現ベクターでトランスフェクトすることができる
。発現ベクターは、一過性発現に適切なベクターにすることができ、真核生物複製起点を
含むエピソームベクターまたは安定に組み込まれ、その後に発現カセットの遺伝子が増幅
する増幅ベクターとすることができる。適切なベクターは当分野で周知であり、例えば、
特に、抗体および抗体フラグメントの産生に適切なpdHL2ベクターが含まれる。増幅
発現カセットを使用する場合、レトロウイルスベクター中で使用される選択マーカーと異
なり、トランスフェクション細胞が選択される選択マーカーを含むことが有利である。再
度、適切にトランスフェクトされた細胞を選択し、その後に限界希釈によってクローニン
グすることができる。
【0022】
適切なクローンの選択の際、細胞を適切な培地に入れ、培養して、目的の所望のタンパ
ク質を産生することができる。培地は、血清を含むことができるが、好ましくは、無血清
である。さらに、細胞の寿命およびタンパク質産生を、培養培地への1つまたは複数のカ
スパーゼインヒビター(例えば、カスパーゼ1または3)の添加によって増加させること
ができる。好ましくは、カスパーゼインヒビターは、1つまたは複数のカスパーゼ3、カ
スパーゼ9、および/またはカスパーゼ12を阻害するように作用する。細胞透過性カス
パーゼインヒビターを使用することが有利であり、pan−カスパーゼインヒビターが特
に有利である。Z−VAD−fmkおよびAc−DEVD−choなどの適切なインヒビ
ターが当分野で周知である。あるいは、カスパーゼインヒビター(AvenまたはXIA
Pなど)を発現するように細胞株をさらにトランスフェクトして、アポトーシスに影響を
及ぼすことによってその増殖特性を増強することができる。これに関して、サイトカイン
I型スーパーファミリーの特定のメンバー(EPOなど)はまた、抗アポトーシス性作用
および細胞保護作用を有することによって細胞生存を増加させることができる。上記の方
法により、本質的に任意の所望の遺伝子によるトランスフェクションに使用することがで
きる細胞株を生成することができる。しかし、当業者は、所望のタンパク質、特に、組換
えタンパク質を構成的に発現する樹立細胞株を、その後にウイルスまたはBcl−2ファ
ミリー抗アポトーシス性遺伝子をコードする適切なベクターで形質転換することができる
ことを認識している。以下の実施例2を参照のこと。
【0023】
目的のタンパク質は、本質的に、宿主細胞中で検出可能な量で産生することができる任
意のタンパク質であり得る。例には、伝統的なIgG型抗体、F(ab’)2またはFa
bフラグメント、scFv、ダイアボディ(diabody)、IgG−scFvまたは
Fab−scFv融合抗体、IgG−またはFab−ペプチド毒素融合タンパク質、また
はワクチン(例えば、A型肝炎、B型肝炎、またはC型肝炎;HIV、インフルエンザウ
イルス、呼吸器合胞体ウイルス、乳頭腫ウイルス、疱疹ウイルス、ハンターンウイルス、
エボラウイルス、ロタウイルス、サイトメガロウイルス、リーシュマニアRNAウイルス
、SARS、マラリア、結核(放線菌)、炭疽、天然痘、野兎病、およびwww.vac
cines.org(参照することによりその全体が本明細書に組み込まれる)に列挙さ
れている他のワクチンが含まれるが、これらに限定されない)が含まれる。本明細書中に
記載の宿主細胞は、実施例1および2に記載の骨髄種細胞株中での抗体および抗体フラグ
メントならびに組換え成長因子(例えば、EPO、G−CSF、GM−CSF、EGF、
VEGF、トロンボポエチン)、ホルモン、インターロイキン(例えば、IL−1〜IL
−31)、インターフェロン(例えば、α、β、γ、およびコンセンサス)、ならびに酵
素の高度に有効な産生に特に適切である。これらの方法を、組換えタンパク質の産生で使
用される任意の多数の細胞株(他の骨髄腫細胞株(マウスのNSOまたはラットYB2/
0など);上皮株(CHOおよびHEK 293など);間葉細胞株(線維芽細胞株CO
S−1またはCOS−7など);およびニューロン細胞(網膜細胞、膠細胞、および神経
膠種細胞など)が含まれる)に適用することができる。
【0024】
<アポトーシス性インヒビターを発現する細胞中での組換え抗体発現>
以前の研究では、キメラ抗体を産生する組換えCHO細胞中で同時発現するBcl−2
(天然に存在するアポトーシスインヒビター)の効果が記載されている。Tey et
al.,Biotechnol.Bioeng.68:31−43(2000)を参照の
こと。細胞培養物の寿命の増加が認められるにもかかわらず、Bcl−2発現を欠いた等
価な細胞では抗体産生は増加しなかった。しかし、本発明者らは、細胞がBcl−2も発
現する場合、骨髄腫細胞からの組換え抗体の産生が有意に増加することを見出した。
【0025】
有利には、骨髄腫細胞株を、抗体または抗体フラグメントをコードする発現カセットで
安定にトランスフェクトする。適切な発現カセットは、上記の選択マーカーと共に抗体の
重鎖および軽鎖の発現(scFvの場合、一本鎖)を調節する1つまたは複数のプロモー
ターを含む。特に有用なベクターは、選択マーカー酵素をコードするDNA配列に作動可
能に連結されたプロモーターを含む選択マーカー遺伝子、目的のタンパク質をコードする
DNA配列に作動可能に連結されたプロモーターを含む転写単位、第1のエンハンサーの
非存在下での選択マーカー遺伝子および第1の転写単位の両方の転写と比較して、選択マ
ーカー遺伝子および第1の転写単位の両方の転写を刺激する、選択マーカー遺伝子と転写
単位との間のエンハンサーエレメントを含むpdHL2である。ベクターはまた、第1の
エンハンサーと選択マーカー遺伝子との間に存在するプロモーターを有し、選択マーカー
遺伝子の転写刺激を選択的に軽減させる遮断エレメントを含む。VH配列およびVL配列を
、pdHL2にライゲーションすることができ、pdHL2は、ヒト軽鎖定常領域、重鎖
定常領域、増幅可能なdhfr遺伝子(それぞれ、別のプロモーターによって調節される
)の配列を含む増幅可能なベクターである。Leung et al.,Tumor T
argeting 2:184,(1996)およびLosman et al.,Ca
ncer 80:2660−2667,(1997)を参照のこと。このベクターを、例
えば、エレクトロポレーションによって細胞にトランスフェクトすることができる。培養
培地への0.1μMまたは適切な濃度のメトトレキセート(MTX)の添加によって選択
することができる。3μMまでまたはそれを超える漸増濃度のMTXを使用した段階的様
式で増幅することができる。したがって、発現カセットで安定にトランスフェクトされ、
目的の抗体を構成的に発現する細胞を得ることができ、当分野で周知の方法を使用して特
徴づけることができる。以下の実施例4も参照のこと。選択およびクローニング後、抗体
発現細胞株を、Bcl−2などの抗アポトーシス性遺伝子をコードする発現ベクターでト
ランスフェクトすることができる。例えば、SV40プロモーターに融合したBcl−2
遺伝子を含むベクターpZeoSV(Invitrogen,Carlsbad,CA)
を、エレクトロポレーションなどの適切な方法を使用して細胞にトランスフェクトし、必
要に応じて選択および遺伝子の増幅を行うことができる。上記のように得られた細胞株を
使用して抗体を産生し、アポトーシス性インヒビターを発現しない細胞における産生と比
較することができる。
【0026】
本方法は、抗体または抗体フラグメントを発現する細胞株の最初の調製およびその後の
Bcl−2または類似のインヒビターを発現するベクターでのトランスフェクションを記
載する。しかし、当業者は、Bcl−2または他の抗アポトーシス性タンパク質を構成的
に発現する細胞株を樹立し、その後に抗体または抗体フラグメントをコードする適切なベ
クターにより形質転換することができることを認識する。本発明を説明する代表例を以下
に示す。実施例1は、HPV−16 E6/E7のSp2/0細胞への組み込みによって
アポトーシスを減少/遅延することを特徴とする改良された細胞クローンSp−E26が
得られることを記載している。実施例2は、HPV−16 E7エレメントのみの過剰発
現によって宿主細胞株を改良する方法を記載している。実施例3は、組換えAbを産生す
る細胞クローンを開発するための宿主として改良された細胞Sp−E26の使用を記載し
ている。実施例4は、E6/E7エレメントを同時発現する抗体産生細胞株で認められた
Mab産生の増強を説明している。実施例5は、3つの点変異を有する変異Bcl−2(
Bcl−2−EEE)を構成的に発現し、寿命が改良された修飾Sp2/0細胞株の生成
および特徴付けを記載している。実施例6は、Bcl−2を発現する抗体産生細胞株の増
殖特性の改良を記載している。実施例7は、実施例6のBcl−2発現細胞株で認められ
たMAb産生の増強を記載している。実施例8は、細胞中のBcl−2発現の導入による
低レベル組換えタンパク質を産生する細胞クローンの改良方法を記載している。実施例9
は、細胞中でのBcl−2発現の導入によるSp−E26の改良方法を記載している。実
施例10は、組換えAbを産生する細胞クローンを開発するための宿主としての改良され
た細胞株(Sp−EEE)の使用を記載している。実施例11は、収率を最適にするため
の流加反応器のプロフィールおよび供給スケジュールの使用を記載している。
【実施例1】
【0027】
<HPV−16 E6およびE7遺伝子の安定な発現によるアポトーシス耐性細胞クロー
ンの生成>
<CHX処理に耐性を示す細胞クローンの選択>
Sp2/0細胞を、HPV−16 E6およびE7遺伝子の発現カセットを含むLXS
Nレトロウイルスベクターによって10:1のMOI(感染多重度)で形質導入した。2
4時間後の回収後、感染細胞を、G418(1000μg/ml)中で10日間選択した
。G418耐性細胞を、限界希釈(0.5細胞/ウェル)によって96ウェル細胞培養プ
レート中でクローニングした。安定な感染細胞を、シクロヘキシミド(CHX)(強力な
アポトーシス誘導剤)による処理に対しての耐性についてスクリーニングした。簡単に述
べれば、健康な細胞(生存度95%超、図1CおよびD)を、25μg/mlのCHXを
含む培地中でインキュベートし、顕微鏡下で細胞の形態を調査した。2〜3時間のインキ
ュベーション後に50%を超える親Sp2/0細胞の形態が変化し、断片化されるように
なり(図1A)、いくつかのE6/E7トランスフェクションクローンの形態変化の範囲
が狭く、アポトーシス耐性を示す。Sp−E26と呼ばれる最良のクローンは、4時間の
処理で明らかな形態の変化は認められなかった(図1B)。
【0028】
面倒な目視試験を回避するために、MTTアッセイを使用して、生存細胞集団の変化を
得ることができる。健康な細胞を、CHXを含むか含まない通常の培養条件下で2〜3時
間インキュベートした後、MTT色素をウェルに添加した。さらなる2時間のインキュベ
ーション後、SDSおよびHClを含む溶解緩衝液の添加によって細胞を可溶化した。プ
レートを、37℃で一晩インキュベートし、ELISAプレートリーダーを使用して59
0nmのODを読み取った。図2に示すように、Sp2/0細胞をCHXで処理した場合
に生存細胞集団がわずかに減少した。比較により、同一の処理条件下で(CHX濃度およ
び時間)、Sp−E26細胞は、CHX処理に対してより耐性を示した。これに関して、
さらなる分析のために多数のクローンをスクリーニングし、選択した(図2)。
【0029】
<Sp−E26の抗アポトーシス特性>
Sp−E26および親Sp2/0細胞におけるCHX誘導性アポトーシスを、アネキシ
ンV染色およびDNA断片化アッセイによって評価した。25μg/mlのCHXを含む
培地中でのインキュベーション後、細胞を回収し、Guava Nexin試薬(アネキ
シンV染色の等価物)で染色し、Guavaパーソナル細胞分析システム(Guava
Technologies,Inc.)を使用して分析した。図3は、約1.5時間CH
X処理に曝露した場合に30%を超えるSp2/0細胞がアネキシンV陽性になる(アポ
トーシスを示す)一方で、Sp−E26は健康なままであり、初期アポトーシス細胞中で
増加しないことを示す。
【0030】
細胞内オリゴヌクレオソームDNAフラグメント(アポトーシスの特質)形成の分析に
よって、CHXによるアポトーシスの誘導を明らかにすることができる。細胞DNAを、
CHX処理および未処理Sp−E26およびSp2/0細胞から抽出し、DNAラダリン
グアッセイを行った。Sp2/0細胞をCHXで処理し、多数のDNA断片化が検出され
た(図4)。対照的に、同一の処理条件下で、Sp−E26のゲノムDNAは依然として
無傷のままであり、DNA断片化の外観は認められなかった(図4)。
【0031】
<Sp−E26中のHPV E6およびE7遺伝子の存在>
E6およびE7遺伝子がSp−E26細胞のゲノム中に安定に存在することを確認する
ために、E6およびE7遺伝子に特異的なオリゴヌクレオチドプライマーをデザインし、
テンプレートしてSp−E26から抽出したゲノムDNAとのPCR反応で使用し、約7
00bpのDNAフラグメントを得た。PCR産物をクローニングし、DNA配列決定に
よってE6およびE7遺伝子であることを確認した。親Sp2/0細胞中にE6およびE
7遺伝子が検出された。
【0032】
<Sp−E26の増殖特性の改良>
Tフラスコ(図5)および3Lバッチバイオリアクター(図6)Sp−E26の増殖特
性を評価した。Sp−E26は、バッチ培養において親Sp2/0細胞よりも改良された
増殖特性を示し、より高い最大細胞密度およびより長い生存期間が達成された。
【実施例2】
【0033】
<HPV16 E7遺伝子の安定な過剰発現によるアポトーシス耐性細胞クローンの生成
>
クローンSp−E26のゲノムに組み込まれたポリシストロン性HPV16 E6およ
びE7遺伝子の構造を、プライマー対E6−N8+(5’−ATG TTT CAG G
AC CCA CAG GAG CGA−3’)およびE7−C8-(5’−TTA T
GG TTT CTG AGA ACA GAT GGG−3’)を使用したPCRおよ
びDNA配列決定によって分析した。プライマーE6−N8+およびE7−C8-の配列は
、E6のN末端8アミノ酸残基のコード配列およびE7のC末端8コドンの相補配列とそ
れぞれ適合するので、全長E6およびE7のアンプリコンは、約850bpであると予想
される。しかし、E6−N8+およびE7−C8-を使用したSp−E26細胞から調製し
たゲノムDNAの増幅により、約700bpのPCRフラグメントしか得られなかった。
700bpのPCR産物のDNA配列決定により、E6遺伝子からの182ポリヌクレオ
チドフラグメントの欠失が明らかとなった。欠損E6遺伝子は、スプライシングに起因す
る可能性が高く、N末端43アミノ酸残基を有する短縮E6ペプチドをコードする。E6
による主な生理活性がp53発現のダウンレギュレート能力であることを考慮すると、S
p−E26中のp53レベルがSp2/0よりも安定であることが見出されているので、
短縮E6タンパク質は、おそらく、十分に機能的ではない。
【0034】
したがって、HPV−16 E7遺伝子のみで抗アポトーシス効果を有し、Sp2/0
細胞の増殖特性を改良するのに十分であるかどうかを評価するために、HPV−16 E
7でのSp2/0細胞のトランスフェクションを、以下のように行った。
(i)E7をコードするDNA配列を、RT−PCRによってSp−E26細胞からク
ローニングする。哺乳動物ベクターpRc/CMV(Invitrogen)への遺伝子
のライゲーションを容易にするために、適切な制限部位を導入する。E7pRcと命名さ
れたベクター内でのウイルス遺伝子の転写は、CMVプロモーター−エンハンサー配列か
ら誘導される。ベクターはまた、SV40プロモーターから転写されるネオマイシン耐性
を付与する遺伝子を含む。
(ii)Sp2/0細胞を、HPV−16E7遺伝子の発現カセットを含む発現ベクタ
ーによってトランスフェクトする。簡単に述べれば、5μgのE7pRcを、ScaIに
よって線状化し、エレクトロポレーションによって細胞にトランスフェクトする。
(iii)24時間後の回収後、トランスフェクション細胞を、G418(1000μ
g/ml)中で10日間選択する。
(iv)次いで、G418耐性細胞を、限界希釈(0.5細胞/ウェル)によって96
ウェル細胞培養プレート中でクローニングする。安定なトランスフェクタントを選択し、
シクロヘキシミド(CHX)(強力なアポトーシス誘導剤)によって治療耐性についてス
クリーニングする。
(v)健康な細胞(生存度95%超)を、通常の培養条件下で25μg/mlのCHX
を含むまたは含まない培地中で3〜4時間のインキュベーション後、MTT色素をウェル
に添加する。さらなる2時間のインキュベーション後、SDSおよびHClを含む溶解緩
衝液の添加によって細胞を可溶化する。プレートを、37℃で一晩インキュベートし、E
LISAプレートリーダーを使用して590nmのODを読み取る。さらなる分析のため
にCHX処理に耐性を示す細胞クローンを選択し、拡大する。
(vi)E7トランスフェクション細胞の抗アポトーシス性を、アネキシンV染色およ
びDNA断片化アッセイによって評価する。アネキシンVアッセイでは、25μg/ml
のCHXを含む培地中でのインキュベーション後、細胞を回収し、Guava Nexi
n試薬(アネキシンV染色の等価物)で染色し、Guavaパーソナル細胞分析システム
(Guava Technologies,Inc.)を使用して分析する。DNA断片
化アッセイでは、細胞DNAを、CHX処理および未処理E7トランスフェクタントおよ
びSp2/0細胞から抽出し、アガロースゲル電気泳動で分析する。
(vii)E7トランスフェクタント中のウイルス癌遺伝子の発現を、サザンブロット
分析(ゲノムレベル)、ノーザンブロット分析(mRNAレベル)、および免疫ブロット
分析(タンパク質レベル)によって評価する。アポトーシス過程に関与し、E7タンパク
質に影響を受ける細胞内タンパク質の発現を、免疫ブロッティング分析によって試験する
。
(viii)選択されたE7トランスフェクタントの増殖特性を、T−フラスコ中およ
び3Lバッチバイオリアクター中で評価する。トランスフェクタントは、バッチ培養にお
いて親Sp2/0細胞よりも改良された増殖特性を示し(より高い最大細胞密度およびよ
り長い生存期間が達成され)、より良好な宿主細胞と見なされる。
【実施例3】
【0035】
<Sp−E26中でのhLL2 IgGの高レベル発現>
この実施例では、Sp−E26を、hLL2(NHL患者および自己免疫疾患患者を治
療するために開発されたヒト化抗CD22Ab)を産生する細胞クローンを生成するため
の宿主として使用する。hLL2産生クローン(87−2−C9)は、以前に宿主として
Sp2/0細胞を使用することによって生成され(Losman et al.,Can
cer 80,2660−2666,1997)、この場合、トランスフェクション後に
1つの陽性クローンのみが同定され(約2.5×10-7の頻度)、増幅前のT−フラスコ
におけるコンディション最終培養培地中の抗体濃度として定義されたhLL2産生クロー
ンのみの最大生産性(Pmax)は、1.4mg/Lであった。Losman et al
.(Cancer 80,2660−2666,1997)によって記載された類似の方
法の使用による同一のhLL2pdHL2ベクターでのSp−E26細胞のトランスフェ
クションにより、200個を超える安定なhLL2産生クローンが得られた(10-4を超
える頻度)。12個の無作為に選択したクローンのPmaxを評価し、13mg/Lと17
0mg/Lとの間であり、平均50mg/Lであることが見出された。これらのクローン
の生産性を、MTXでの遺伝子増幅によってさらに増強することができる。この実施例に
より、組換えタンパク質を産生する細胞クローンの開発のための宿主としてSp−E26
を使用することがその親Sp2/0細胞よりも有利であることが証明された。
【実施例4】
【0036】
<HPV16 E6およびE7遺伝子の安定な発現によるAb産生細胞株の改良>
607−3u−8細胞をトランスフェクションによってSp2/0から最初に生成し、
ヒト化モノクローナルAbを産生した。最大(Ab)生産性を150mg/Lまで増強す
る(培養培地中の血清補足のウィーニング(weaning)後に約100mg/Lに減
少した)ために、(MTXでの)遺伝子の増幅およびサブクローニングによってクローン
を開発した。無血清条件下でより高い抗体生産性を得るために、HPV−16のE6/E
7遺伝子を、607−3u−8に導入し、Ab生産性に対するE6/E7の効果を以下の
ように評価した。10%FBSおよび3μMのMTXを補足したHSFM中で維持した6
07−3u−8細胞を、HPV−16 E6およびE7遺伝子の発現カセットを含むLX
SNレトロウイルスベクターで10:1のMOIで形質導入した。24時間後の回収後、
安定にトランスフェクトされた細胞を、G418(400μg/ml)中で10日間選択
した。G418耐性細胞を、限界希釈(0.5細胞/ウェル)によって96ウェル細胞培
養プレート中でサブクローニングした。評価のために607E1C12と命名した生存ク
ローンを得た。E6/E7トランスフェクションを行っていない607−3u−8−7G
7および607−3u−8−2D10と命名された607−3u−8の2つのサブクロー
ンも選択した。これら3つのクローンのPmaxを決定し、有意差はなかった(表1)。こ
れらの結果により、細胞へのE6/E7遺伝子の導入によって細胞のAb産生能力は変化
しないことが示唆される。次に、607E1C12、607−3u−8−7G7、および
607−3u−8−2D10を、無血清培地中で増殖するように適合させ、これらのクロ
ーンの生産性を決定した。全細胞を、無血清培地を含む増殖ウェル中で増殖させた。クロ
ーン607E1C12の最終抗体生産性は150mg/Lに維持される一方で、E6/E
7を含まない2つのクローンでは実質的に減少した。さらに、607E1C12の生産性
は、凍結融解後に(低温保存に対して)安定であった(表1)。
【0037】
【表1】
【実施例5】
【0038】
<変異Bcl−2を構成的に発現する遺伝子操作されたSp2/0細胞株の生成および特
徴付け>
証拠により、3箇所の点変異(T69E、S70E、およびS87E)を有する変異B
cl−2が、野生型または1箇所の点変異体と比較して、有意により高い抗アポトーシス
活性を示すことが示唆される(Deng et al.,PNAS 101:153−1
58,2004)。したがって、この三重変異体(Bcl−2−EEEと命名する)のた
めの発現ベクターを構築し、これを使用して、特にバイオリアクター中の生存および生産
性を増加させるためにSp2/0細胞をトランスフェクトした。クローンを単離し、Bc
l−2−EEE発現レベル、増殖、およびアポトーシス特性について評価した。Bcl−
2−EEEの核酸配列を、配列番号3に示し、対応するBcl−2−EEEタンパク質の
アミノ酸配列を、配列#4に示す。
【0039】
ヒトBcl−2のアミノ酸残基64〜101のコード配列に基づいて、116bpの合
成DNA二重鎖をデザインした。残基69、70、および87のコドン全てを、グルタミ
ン酸のコドン(E)に変更した。全配列は並外れてGCがリッチであり、多数のポリGお
よびポリC連続物(run)が存在する。GおよびC連続物を破壊するためにいくつかの
コドンを保存的に変化させ、全GC含有量を減少させた。
【0040】
組み合わせ、長さが116bpの配列であり、その3’末端が22bp重複した2つの
80量体オリゴヌクレオチドを合成した(配列番号5および6を参照のこと)。オリゴヌ
クレオチドをアニーリングし、TaqDNAポリメラーゼを使用したプライマー伸長によ
って二重鎖DNAを生成した。PCRプライマー、Bcl−2−EEE PCR左(5’
−TATATGGACCCGGTCGCCAGAGAAG−3’)およびBcl−2−E
EE PCR右(5’−TTAATCGCCGGCCTGGCGGAGGGTC−3’)
を使用して、二重鎖を増幅させた。
【0041】
次いで、126bpの増幅物を、pGemT PCRクローニングベクターにクローニ
ングした。Bcl−2−EEE−pGemT構築物を、TthIおよびNgoMI制限エ
ンドヌクレアーゼで消化し、150bpフラグメントをゲルで単離し、TthIおよびN
goMIで消化したhBcl−2−puc19ベクター(ATCC 79804)でライ
ゲーションして、hBcl−2(EEE)−puc19を生成した。この構築物の配列を
確認した。
【0042】
948bpインサートフラグメントを、EcoRIを使用してhBcl−2(EEE)
−puc19から切り出し、EcoRIで消化したpZeoSV2+ベクターとライゲー
ションし、アルカリホスファターゼで処理した。得られた構築物は、hBcl−2(EE
E)−pZeoSV2+である。
【0043】
次いで、Sp2/0細胞についての標準的なプロトコールにしたがったエレクトロポレ
ーションによって、Sp2/0細胞(5.6×106)を、60μgのhBcl−2(E
EE)−pZeoSV2+でトランスフェクトした。細胞を、6つの96ウェルプレート
にプレートし、選択せずに48時間インキュベートした。2日後、800μg/mlのゼ
オシンを培地に添加した。
【0044】
40ウェル由来の細胞を、24ウェルプレートに拡大し、抗hBcl−2および抗βア
クチンを使用したウェスタンブロットによって分析した。40個のうちの5個を除けば、
中レベル〜高レベルのBcl−2−EEE発現を示した。4つのゲルのうちの1つの結果
を、図7に示す。事前に野生型Bcl−2でトランスフェクトしたSp2/0由来のhM
N14細胞株(クローン664.B4)を、正のコントロール(+)として使用した。D
eng et al.で証明されているように、SDS−PAGEにおいて、Bcl−2
−EEEは、野生型Bcl−2よりもわずかにゆっくり移動する。
【0045】
さらなる評価およびサブクローニングのために、3つの強い陽性のウェル(番号7、番
号25、および番号87)を選択した。限界希釈プレーティングにより、96ウェルプレ
ートあたりの陽性ウェルは20未満であり、これにより、各ウェル中の細胞が実際にクロ
ーニングされる非常に高い確率が示される(99%超)。最初に、3つの元のウェル由来
の23個のサブクローンを、抗hBcl−2−PEを使用したGuava Expres
sによって分析した(図8)。結果により、元のウェルが細胞クローンの混合物を含んで
いたことが確認された。ウェル番号7から最も強いシグナルを有するクローンが得られ、
ウェル番号25から最も低いシグナルを有するクローンが得られた。さらなる分析のため
に、クローン7−12、7−16、87−2および87−10を拡大した。その後、最初
に増殖の遅いサブクローンを同様に分析し、1つのクローン(87−29)から任意の他
のクローンよりも20%高いシグナルが得られ、さらなる分析のためにこれを拡大した。
2つの高発現SP−EEEクローン(87−29および7−16)を、非トランスフェク
ションSp2/0細胞、Raji細胞、およびDaudi細胞と比較した(図9)。Sp
−EEEクローンは、推定上正常な細胞レベルでBcl−2を発現することが知られてい
るRaji細胞およびDaudi細胞より約20倍発現する。Sp2/0細胞は、陰性で
あった。これを、抗Bcl−2免疫ブロットによってさらに検証した(図10)。ヒトB
cl−2特異的抗体を使用してSp2/0細胞中にBcl−2は検出されず、高タンパク
質負荷(50K細胞)および長期X線フィルム曝露でさえも検出されなかった。マウス、
ラット、およびヒトのBcl−2を認識する抗Bcl−2 MAb(C−2、Santa
Cruz Biotech.)を使用した免疫ブロット分析によって、非トランスフェ
クションSp2/0細胞からいかなるBcl−2も検出されず、高タンパク質負荷(10
0K細胞)および長期X線フィルム曝露でさえも検出されなかった(図10B)。Sp2
/0細胞中で任意のBcl−2が発現される場合、クローン87−29中のBcl−2−
EEEより満たない2桁を超えるレベルである。5つのSp−EEEサブクローンおよび
Sp2/0細胞の増殖曲線を比較した。3つのSp−EEEサブクローンは、Sp2/0
細胞よりも明確な利点を示した。これら3つ(7−12、7−16、および87−29)
はまた、最も高いレベルのBcl−2−EEEを発現する。7−12および7−16は、
同起源のウェルに由来し、ほぼ同一の性質を有し(Bcl−2−EEEレベルおよび増殖
曲線)、これらは、同一の元のクローンに由来する可能性が高い。最良の2つのSP−E
EEサブクローン(7−16および87−29)を、さらなる評価のために使用した。
【0046】
10%、1%、または0%の血清(ウィーニング(weaning)せず)を補足した
培地中にクローンをプレートし、細胞密度および生存度をモニタリングした。10%血清
では、87−29は高密度まで増殖し、Sp2/0細胞と比較して4日間を超えて生存が
延長された(図11)。1%血清では、全細胞は、10%血清で達成された密度の約35
〜40%に増殖し、Bcl−2−EEEトランスフェクタントは、Sp2/0に類似の延
命効果を有していた(図12)。無血清培地に直接移した場合、Sp2/0細胞は600
K細胞/mlしか増殖せず、87−29細胞は2倍の密度で増殖した(図13)。各血清
濃度では、87−29細胞は、Sp2/0細胞よりも4〜6日間長く生存した。
【0047】
87−29のメトトレキセート(MTX)感受性を決定した(図14)。データにより
、MTX耐性クローンの最初の選択には最小濃度(0.04μM)のMTXで十分である
ことが示唆される。したがって、Sp2/0細胞で使用した同一の選択および増幅プロト
コールを、SP−EEE細胞に使用することができる。
【0048】
Bcl−2は、生存促進性/抗アポトーシス性タンパク質である。いくつかのグループ
により、可動性ループドメイン(FLD)を欠くBcl−2欠失変異体はアポトーシス阻
害能力が増強されることが証明されている(Figueroa et al.,2001
,Biotechnology and Bioengineering,73,211
−222;Chang et al.,1997,EMBO J.,16,968−97
7)。より最近では、リン酸化を模倣するBcl−2のFLD中の1〜3つのS/T残基
のグルタミン酸への変異により、その抗アポトーシス能力が有意に増強されることが証明
されている(Deng et al.2004,PNAS,101,153−158)。
三重変異体(T69E、S70E、およびS87E)は、最も有意に生存が増強された。
ここで、本発明は、Sp2/0細胞を安定にトランスフェクトするために使用される類似
のBcl−2三重変異構築物(Bcl−2−EEE)の生成を教示する。
【0049】
上記の全実験は、Bcl−2−EEEの発現によってSp2/0細胞におけるアポトー
シス率が減少することを示す。この効果は、クローンの発現レベルが高いほどより低いレ
ベルのクローンより長く生存するという点で、非常に用量に依存した。最良のクローン(
87−29)は、非トランスフェクションSp2/0細胞と比較して、15〜20%高い
細胞密度で増殖し、さらに4〜6日間長く生存する。
【0050】
クローン(87−29)中のBcl−2−EEEレベルは、Daudi細胞またはRa
ji細胞中の通常のレベルの約20倍である。非トランスフェクションSp2/0細胞で
はBcl−2発現は検出されなかった。実施例6に記載するように、hMN−14発現S
p2/0細胞を、野生型Bcl−2発現のための類似の構築物でトランスフェクトし、例
外的な増殖特性および生産性が増強したクローンを単離した。このクローン(664.B
4)を、MTXを使用してさらに増幅させた場合、Bcl−2レベルが有意に増加した。
最終的に、増幅された(3μM MTX)細胞株をサブクローニングし、1つのクローン
(664.B4.1C1)のBcl−2レベルは、664.B4の2倍であった。この特
定のサブクローンは、生産性および増殖特性が優れている。87−29中のBcl−2−
EEEレベルは、増幅された664.B4.1C1中のBcl−2レベルの約2倍である
。87−29細胞は、Sp2/0細胞に匹敵する増殖率を有し、見かけ上さらに1日間増
殖し続け、Sp2/0よりも15〜20%高い最大密度に到達し得る。E6/E7発現S
p−E26細胞株について類似の性質が見出された。Bcl−2−EEE発現87−29
クローンは、親Sp2/0細胞よりもさらに4〜6日間長く生存し、1日だけ長く生存す
るSp−E26クローンより優れている。
【0051】
87−29クローンとして示したSp−EEE細胞株は、組換えタンパク質の遺伝子を
含む適切なベクターでのトランスフェクションの際の組換えタンパク質の発現のためのア
ポトーシス耐性宿主として有用である。この細胞株を有用にするために、この細胞株は、
トランスフェクションおよび増幅後ならびに培養物の拡大中にそのBcl−2−EEE発
現および延命効果を維持しなければならない。安定にトランスフェクトされたBcl−2
−EEE遺伝子はその後のトランスフェクション中に失われる可能性が低いので、生存特
性が損なわれるはずがない。MTX増幅はBcl−2タンパク質発現の増加によって産生
クローンの生存を実に改良することが可能である。実際、このことは、野生型Bcl−2
でトランスフェクトしたhMN14 664.B4細胞株でもそうであった。増幅および
サブクローニング後、Bcl−2レベルは数倍に増加し、細胞生存は有意に改良された。
【実施例6】
【0052】
<ヒトBcl−2遺伝子の安定な発現による静置バッチ培養におけるAb産生細胞生存の
改良>
<Bcl−2トランスフェクション細胞クローンの生成>
トランスフェクションによってSp2/0から細胞クローン665.2B9を最初に生
成し、ヒト化モノクローナル抗CEA Abを産生した(Qu et al.、未発表の
結果)。hMN14pdHL2と命名されたベクターを使用してSp2/0細胞をトラン
スフェクトし、細胞クローン665.2B9を得た。pdHL2ベクターは、Gilli
es et al.によって最初に記載され、メトトレキセート処理によってその後に選
択および増幅される増幅可能なマウスdhfr遺伝子を有していた(Gillies e
t al.,J.Immunol.Methods 125:191(1989))。一
般に、pdHL2ベクターはIgG重鎖および軽鎖の遺伝子両方を発現し、これらは、2
つのメタロチオネインプロモーターおよびIgHエンハンサーによって独立して調節され
る。hMN14pdHL2ベクターの図を、図16に示す。配列番号1は、このベクター
の配列を示し、配列番号2は、エンハンサー配列として定義された72bp配列を示し、
プロモーター配列は、hMN14pdHL2のnt2908〜2979に対応する。
【0053】
Sp2/0細胞を、一般に、エレクトロポレーションによって、本実施例で使用される
hMN14pdHL2などの線状化pdHL2ベクターでトランスフェクトすることがで
きる。0.05〜0.1μM MTXを含む培地との細胞のインキュベーションによって
、トランスフェクションから48時間後に選択を開始することができる。5μMまでの濃
度のMTXの段階的増加によって、挿入された抗体配列を増幅させる。
【0054】
クローンを、0.3μMまでのMTXの段階的増加を使用した遺伝子増幅に供し、この
時点で、抗体の最大生産性(Pmax)は約100mg/Lに増加した。細胞増殖特性を
改良するために、665.2B9細胞を、エレクトロポレーションによって、ヒトBcl
−2遺伝子を含むプラスミド発現ベクター(図17)でトランスフェクトした。Bcl−
2遺伝子を、EcoRI部位を使用してATCCから購入したpB4プラスミド(pB4
、カタログ番号79804)から切り出し、同一の制限酵素を使用して、哺乳動物発現ベ
クターpZeoSV(+)のMCSに挿入した。ゼオシン耐性遺伝子がベクターの一部で
あるので、トランスフェクション細胞を、50〜30μg/mLの範囲のゼオシンを含む
培地に入れた。300mg/mlゼオシンを含む培地から安定なクローンを選択し、0.
5細胞/100μL/ウェルの密度での96ウェルプレートへのプレーティングによって
ゼオシンを含まない培地中でサブクローニングした。その後、ゼオシンを含まない培地を
使用した。ウェル中のクローンの形成を、顕微鏡下での目視によって確認した。1細胞ク
ラスターのみを含むウェル由来の細胞を拡大した。各96ウェルプレートから約30クロ
ーンが産生され、そのうちの14クローンをさらなる研究のために無作為に選択した。こ
れらのクローンの増殖特性を、1日の細胞計数ならびにViaCount試薬およびGu
avaPCAを使用した生存度の測定によって評価した。24ウェルプレート中で評価し
た14クローンから(図18、19)、増殖特性が改良された(細胞密度がより高くなり
、細胞生存が延長された)1つのBcl−2トランスフェクションクローンを同定し、6
65.2B9#4(またはクローン#4)と命名した。親665.2B9クローンと比較
して、クローン#4は、Tフラスコ中でより高い細胞密度(約1.7倍)で増殖し、4〜
6日間長く生存し(図20、21)、より良好な増殖の結果として、ELISA滴定およ
びプロテインAカラム精製によって決定したところ、クローン#4のPmaxは、約170
mg/Lに増加した。
【0055】
<665.2B9#4におけるBcl−2発現>
665.2B9#4の増殖特性の改良がBcl−2のトランスフェクションに起因する
ことを確認するために、ヒトBcl−2タンパク質の細胞内レベルを、Guava Ex
press試薬およびGuava PCA装置の使用によって測定した。簡単に述べれば
、1.5mlスピンチューブに入れた4×105細胞を、1500rpmで5分間遠心分
離し、1×PBSで3回洗浄した。上清を慎重に吸引した。Santa Cruz Bi
otechnology(SCB)Inc.の固定液(10×、60μL)(カタログ番
号sc−3622)を細胞ペレットに15分間添加し、氷上でインキュベートした。固定
液を、4℃の4×1mL PBSを使用して除去し、記載の時間スピンした。−20℃の
透過処理緩衝液(0.5mL)(SCBカタログ番号sc−3623)をボルテックスし
ながら滴下し、その後、氷上で15分間インキュベートした。次いで、細胞をスピンし、
0.5mL FCM洗浄緩衝液(SCBカタログ番号sc−3624)で2回洗浄した。
最終細胞ペレットを、100μLのFCM洗浄緩衝液に再懸濁し、10μLのPEにコン
ジュゲートした抗Bcl−2マウスモノクローナル抗体(SCBから入手)にてBcl−
2細胞内タンパク質を染色した。室温の暗所で1時間インキュベートした。その後に、0
.5mLのFCM洗浄緩衝液で2回洗浄した。最終細胞ペレットを、0.4mLのFCM
洗浄緩衝液に再懸濁し、GuavaPCにて細胞を分析した。各クローンについての蛍光
強度の平均値(MFI)を、PEとコンジュゲートした非特異的アイソタイプマウスIg
G1でのコントロール染色と比較した。表2にまとめた結果により、クローン665.2
B9#4が親細胞株と比較して高レベルのBcl−2タンパク質を発現することが確認さ
れる。親665.2B9と類似の増殖プロフィールを示したゼオシン耐性クローン(#1
3)は、Bcl−2染色に対して陰性であり、増殖の改良にBcl−2発現が必要である
ことが確認された。
【0056】
【表2】
【0057】
Guava Express分析を使用して、Bcl−2レベルに対応する蛍光染色強
度がクローン665.2B9#4のMTX増幅と共に上昇することが見出され、Bcl−
2のdhfr遺伝子との同時増幅が示唆された。増幅細胞の細胞内Bcl−2レベルを比
較するために、抗ヒトBcl−2抗体を使用して、クローン665.2B9#4(Bcl
−2陽性)およびクローン#13(Bcl−2陰性)の細胞溶解物に対してウェスタンブ
ロッティング分析を行った。濃度測定評価により、1.0μM MTX中で増殖したクロ
ーン665.2B9#4のBcl−2シグナルが0.6μM MTX中の細胞よりも2倍
強いことが示された。クローン#13の溶解物は、Bcl−2タンパク質の存在を示さな
かった(図22)。
【実施例7】
【0058】
<バッチ培養条件下でのクローン665.2B9#4のAb産生の改良>
細胞培養物における末期付近の栄養素消費のモニタリングにより、グルコースおよびL
−グルタミンが最初に消費される栄養素であることが見出された。これらの制限された栄
養の補足によって最終抗体収率が改良されるかどうかを決定するための実験を行った。以
下の2つの培養型を開始した:これらの制限成分をその消費時に補足する急増流加培養お
よび栄養素を補足しない非流加培養。0.6μMおよび1μMのMTXを含む培地中で増
殖するBcl−2陽性クローン665.2B9#4ならびに0.3μM MTX中で増殖
するBcl−2陰性クローン#13を試験した。図23および24は、両培養型が末期に
達するまでの両培養型における細胞生存度および細胞密度のプロフィールを示す。mg/
Lで示すタンパク質収率を、表3に示す。この実験の結果により、全培養物において、栄
養素の急増によって抗体の産生全収率が約2倍に改良されることが示唆される。
【0059】
【表3】
【実施例8】
【0060】
<低レベルの組換えタンパク質を産生する細胞株へのBcl−2遺伝子の導入>
トランスフェクションによってSp2/0から細胞クローン482.2C4Aを最初に
生成し、IgG型(抗CEA)および2つのscFv(抗DTPA)型(それぞれ、Ig
G重鎖のC末端に共有結合されている)の二重特異性Abを産生した(Leung et
al.,J.Nuc.Med.41:270P,2000;Hayes et al.
,Proc.Am.Asso.Cancer.Res.43:969,2002)。クロ
ーンを遺伝子増幅に供し、その最終生産性は約20mg/Lであった。増殖特性および最
終的なAb生産性を改良するために、実施例6に記載のように、482.2C4A細胞を
、エレクトロポレーションによってヒトBcl−2遺伝子を含むプラスミド発現ベクター
でトランスフェクトした。3週間後、750μg/mlのゼオシンを含む培地中でトラン
スフェクタントを選択した。
【0061】
ゼオシン耐性細胞を25μg/mlのCHXで5時間処理して、アポトーシス感受性細
胞を排除した。処理細胞を新鮮な培養培地で2回洗浄してCHXを除去し、新鮮な増殖培
地中に再懸濁した。24時間後の回収後、生存細胞を、限界希釈(0.5細胞/ウェル)
によって、96ウェル細胞培養プレートにクローニングした。クローンが2週間でウェル
中に出現し、これを、Ab産生、CHX誘導性アポトーシス耐性、および増殖プロフィー
ルについてスクリーニングした。全局面で親482.2C4Aよりも能力の高いクローン
を選択し、さらに特徴づける。親482.2C4A細胞と比較して、最良のクローンは、
ストレス条件下で培養した場合により頑強であり、老化培養条件誘導性アポトーシス耐性
を示し、最大Ab生産性がより高い(約150%またはそれ以上)と予想される。
【実施例9】
【0062】
<細胞増殖特性のさらなる改良のためのSp−E26へのBcl−2遺伝子の導入>
実施例5に記載のように、Sp−E26細胞を、エレクトロポレーションによって、ヒ
トBcl−2−EEE遺伝子を含むプラスミド発現ベクターでトランスフェクトする。3
週間後に、500μg/mlのゼオシンを含む培地中でトランスフェクタントを選択する
。
【0063】
ゼオシン耐性細胞を25μg/mlのCHXで5時間処理して、アポトーシス感受性細
胞を排除する。処理細胞を新鮮な培養培地で2回洗浄してCHXを除去し、新鮮な増殖培
地中に再懸濁する。24時間後の回収後、生存細胞を、限界希釈(0.5細胞/ウェル)
によって、96ウェル細胞培養プレートにクローニングする。クローンが2週間でウェル
中に出現し、これを、CHX誘導性アポトーシス耐性および増殖プロフィールについてス
クリーニングする。Sp−EEEと同様に、全局面で親Sp−E26よりも能力の高いク
ローンを選択し、さらに特徴づける。親Sp−E26およびSp/EEE細胞と比較して
、HPV−16 E6/E7およびBcl−2−EEEを含む最良のクローンは、ストレ
ス条件下で培養した場合により頑強であり、老化培養条件誘導性アポトーシス耐性を示す
と予想され、それにより、組換えタンパク質産生のためのより良好な宿主細胞である。
【実施例10】
【0064】
<Sp−EEE細胞株を使用した組換えタンパク質産生の改良>
組換えタンパク質の産生のために生存度が増強された細胞株を開発する場合に取ること
ができる経路が2つ存在する。実施例6に記載のように、非常に首尾よく達成された1つ
の方法は、生存促進性遺伝子(Bcl−2など)での既に産生されている細胞株の安定な
トランスフェクションを含む。しかし、この方法は、さらなるトランスフェクションステ
ップ、選択ステップ、およびクローニングステップが必要であり、それにより、細胞株の
開発過程が少なくとも2ヶ月、おそらくそれ以上長期化する。さらに、各クローンに対し
て多数のパラメーター(増殖/生存、Bcl−2発現レベル、および生産性が含まれる)
を決定する必要があるので、「最良の」クローンのスクリーニングがむしろ必要である。
したがって、少数のクローンしか評価することができない。生産性が最も高いクローンの
生存がより優れていないかもしれない可能性およびその逆の可能性が非常に高い。本明細
書で使用した別のストラテジーは、より優れた増殖特性および生存特性を有する親細胞株
を開発し、その後に所望のタンパク質の産生のための発現ベクターでトランスフェクトす
ることである。
【0065】
Sp2/0細胞と比較して、Sp−EEE細胞は、培養液中でさらに1日増殖し続け、
15〜20%高い最大密度に達し、さらに4〜6日間長く生存する。組換えタンパク質(
IgG、抗体フラグメント、および融合タンパク質など)、成長因子(G−CSF、GM
−CFS、EPO、EGF、VEGFなど)、サイトカイン(インターロイキンファミリ
ーメンバー(IL−1〜IL−31)またはインターフェロンファミリーメンバー(α、
β、またはγインターフェロンなど)など)、オリゴヌクレオチド、ペプチド、ホルモン
、酵素、またはワクチン(例えば、A型肝炎、B型肝炎、またはC型肝炎、および上記の
他のワクチン)の産生のための遺伝子でその後にトランスフェクトした場合、細胞はその
増強された増殖特性および生存特性を保持する。
【0066】
組換えタンパク質(IgGなど)のための1つまたは複数の発現カセットを含むDNA
ベクター(pdHL2など)を使用して、標準的な方法(エレクトロポレーションなど)
によってSp−EEE細胞をトランスフェクトする。トランスフェクタントを、96ウェ
ルプレートに入れ、クローンを、ELISAまたはBiacoreなどの樹立された技術
によってタンパク質産生について分析する。生産性の高いクローンを、培養培地中で数ヵ
月にわたって漸増濃度のMTXに供し、遺伝子コピー数を増幅させる。Bcl−2陰性S
p2/0細胞中に生成されたクローンと比較して、Bcl−2−EEE発現クローンが約
20%高い細胞密度で増殖し、少なくともさらに4日間長く生存し、前者は、標準的なフ
ラスコまたはローラーボトル培養において、少なくとも20%より多くの組み合わせタン
パク質を生成する。懸濁、灌流、または流加バイオリアクター培養でさらにより高い増加
が実現される。
【実施例11】
【0067】
<バイオリアクター中でのBcl−2トランスフェクションクローン665.B4.1C
1のAb産生の改良>
実施例6の665.2B9#4および親クローン665.2B9両方を、無血清培地に
ウィーニングさせた(weaned)。細胞を、Tフラスコ中で数ヵ月の継続的継代培養
によって3μM MTXを含むハイブリドーマ無血清培地(HSFM)(Immunom
edics PN 10070)のカスタマイズされた形成に適合させた。適合させた細
胞を、保存のためにTフラスコからローラーボトルに掻き取った。45%コンディション
培地(対数増殖期における培養物の遠心分離後に上清として回収した培地)、10%DM
SO、および45%HSFMから構成される無FBS低温保存液を使用した1mLバイア
ル中で、1×10-7生存細胞を使用して、各細胞株についてのマスターセルバンク(MC
B)を作製した。MCB細胞株を、それぞれ、665.2B9.1E4(Bcl−2遺伝
子なし)および665.B4.1C1(Bcl−2遺伝子を含む)と命名した。これら2
つのクローンの増殖特性および抗体産生を、バッチ培養条件下で比較した。
【0068】
MCBから拡大された上記細胞を使用して、3Lのベンチスケールバイオリアクター中
で実験を行った。3Lバイオリアクターシステムは、2500L cGMPバイオリアク
ターシステムの縮小モデルである。したがって、評価の結果は、商業的大量製造へのこれ
らの細胞株の適合性を支持する。
【0069】
MCB(Immunomedics PN 10070)の作製で使用したものと同一
の増殖HSFMを使用して、細胞株を維持し、接種材料を調製した。基本HSFM(特別
に修正した増殖HSFMに基づいた特別な配合物(formulation))(Imm
unomedics PN 10194)を、3L流加バイオリアクター過程で使用した
。両培地は、微量タンパク質としてのみインスリンおよびトランスフェリンを含む。撹拌
および曝気による剪断から細胞を保護するために、さらに0.1%のPluronic
F68を配合物に組み込んだ。この培地も3μMのMTXを含んだ。
【0070】
連続供給溶液およびパルス供給溶液の特徴を、以下の表4および5に示す。
【0071】
【表4】
【0072】
【表5】
【0073】
可動範囲2Lで3L Bellco撹拌フラスコバイオリアクターシステム(Bell
co glasses,Vineland,NJ)中で流加試験を行った。バイオリアク
ターの温度、pH、および溶存酸素(DO)をモニタリングし、単ループ制御装置によっ
て制御した。反応器の温度を、加熱ブランケットによって37℃に制御した。培養物のp
Hを、CO2または6%Na2CO3の添加によってpH7.3に制御した。円筒形焼結(
sintered)スパージャーによって10ml/分の曝気を行った。培地へのO2の
間欠的噴霧によって、DOを40%を超える空気飽和(air saturation)
に制御した。50〜60rpmの一定の撹拌速度を培養中に使用した。
【0074】
MCB由来の凍結バイアルを融解し、約1〜2週間Tフラスコに回収した。次いで、細
胞をTフラスコからローラーボトルに拡大し、バイオリアクターでインキュベートした。
細胞を、37℃、5%CO2雰囲気下で培養し、拡大過程中は対数増殖期を維持した。
【0075】
インキュベーション前に、1.2リットルの基本HSFMを、ポンプにて無菌でバイオ
リアクターに移した。培地の空気を飽和させて、溶存酸素(DO)プローブを較正した。
また、pHプローブを較正するために、培地サンプルを採取した。一旦pHプローブおよ
びDOプローブが較正されると、両制御装置を、AUTOモードに設定した。一旦システ
ムが、pH(7.3)および温度(37℃)の定値に達すると、計算された量の接種材料
が、ローラーボトルからバイオリアクターに移された。接種後生存細胞密度(VCD)は
、約2×105バイアル細胞/mlであった。
【0076】
供給ストラテジーを以下に示す。培養中、濃縮栄養溶液をバイオリアクターに供給して
、細胞に必要且つ過剰でない栄養素を細胞に供給した(全過程の略図については、図25
を参照のこと)。濃縮栄養溶液を、連続供給およびパルス供給によって培養物に送達させ
た。連続供給溶液を、蠕動ポンプ(Watson−Marlow 101U/R)を使用
して、反応器に移した。パルス供給溶液を、1日1回培養物にパルス供給した。
【0077】
2つの流加ストラテジーを開発し、両細胞株に適用した。過程番号1は、培養中に組換
えインスリンを供給しない。過程番号2を、修飾リノール酸、脂質供給スケジュール、お
よびインスリンのさらなる供給を使用して過程番号1に基づいてデザインする。
【0078】
以下の表は、両細胞株の両過程の供給をまとめている。
【0079】
【表6】
【0080】
【表7】
【0081】
【表8】
【0082】
【表9】
【0083】
培養中、オフライン分析のためにバイオリアクターサンプルを定期的に採取した。生存
細胞密度(VCD)および細胞生存度を、0.4%トリパンブルー色素での染色後の血球
計算板を使用した顕微鏡での計数によって測定した。グルコース、乳酸塩、グルタミン、
アンモニアの濃度を、Nova Bioprofile 200を使用して測定した。抗
体濃度を、プロテインAアフィニティクロマトグラフィカラムを使用したHPLC(Ap
plied Biosystems,P/N2−1001−00)によって決定した。
【0084】
以下のように、産生培養抗体を培養物中の全生存細胞の時間積分で割ることによって、
特異的抗体生産性を計算した。
【0085】
【数1】
(ここで、
【数2】
は台形公式
【数3】
によって概算する)
【0086】
図26は、過程番号1および過程番号2による両細胞株の増殖曲線(VCDおよび生存
度)を示す。過程番号1により、665.2B9.1E4細胞は、6日目に生存度86%
で1×107生存細胞/mlの最大VCDに増殖した。6日後、VCDおよびV%は急速
に減少し、8日目に培養物を回収した。過程番号2は、1.2×107生存細胞/mlの
より高いVCDに到達し、1日長く維持されるのに役立つ。
【0087】
665.2B9.1E4細胞と比較して、665.B4.1C1細胞は、両過程でさら
により良好に増殖した。過程番号1では、そのVCDは、7日目に生存度97%で2×1
07生存細胞/mlに達した。培養物はまた、このVCDおよびV%をさらに2日間維持
し、その後減少し始めた。11日目に培養物を回収した。過程番号2では、665.B4
.1C1細胞は、過程番号1と類似の増殖プロフィールを示した。より詳細には、細胞は
、2.3×107生存細胞/mlの最も高いVCDに到達し、生存度はわずかに遅く、1
1日目に回収した。この所見は、過程番号2で増殖における利点が証明された665.2
B9.1E4細胞株と幾らか異なっていた。
【0088】
過程番号1および過程番号2における2つの細胞株の抗体収率を、図27で比較した。
665.2B9.1E4細胞の最終収率は、過程番号1で0.42g/L、過程番号2で
0.55g/Lであった。比較のために、665.B4.1C1細胞は、両過程において
、1.5g/Lのより高い最終収率であった。
【0089】
1日の特異的抗体生産性(細胞基準あたり)を計算し、図28に示す。図に示すように
、665.2B9.1E4細胞の1日あたりの平均Q[MAb]は、両過程の一連の培養を通
して約15pg/細胞/日であった。過程番号2における最も高いVCDでのより長い増
殖日数により、より高い最終抗体濃度が得られた。
【0090】
665.B4.1C1細胞は、両過程において類似の1日あたりの特異的抗体生産性プ
ロフィールを示し、過程番号1の生産性はわずかに高かった。1日のQ[MAb]は、9日目
まで20〜25pg/細胞/日に維持された。その後、生産性は低下した。
【0091】
665.2B9.1E4細胞株と比較して、665.B4.1C1細胞株は、15pg
/細胞/日と比較して、20〜25pg/細胞/日のより高い特異的抗体生産性を示した
。そのより良好な増殖と組み合わせて、665.B4.1C1細胞株の最終抗体収率は、
665.2B9.1E4細胞株によって達成された0.55g/Lと比較して、1.5g
/Lと3倍になった。これらの結果は、組換えタンパク質(この場合、臨床用抗体)の商
業的大量調製のためにバイオリアクター中の無血清培地中でその増殖および抗体収率を増
強するために宿主細胞にBcl−2遺伝子を組み込むことが有利であることを証明する。
当業者は、本明細書中に開示の方法および過程を必要に応じて修正することができる。本
明細書中に含まれる全ての刊行物、特許、特許出願、および引例は、その全体が本明細書
中で参照することにより組み込まれる。
【特許請求の範囲】
【請求項1】
組換えタンパク質の発現に適切な条件下で適切な培地中で宿主細胞を培養するステップ
を含み、前記宿主細胞が、前記組換えタンパク質をコードする核酸配列を含むことと、前
記宿主細胞が、前記宿主細胞の生存を増強する薬剤によって修飾されていることとを含む
、組換えタンパク質の作製方法。
【請求項2】
前記宿主細胞の生存を増強する薬剤が、アポトーシスを阻害する薬剤である、請求項1
に記載の方法。
【請求項3】
前記宿主細胞が、リンパ球、上皮細胞、間葉細胞、ニューロン細胞、またはこれらの悪
性形態である、請求項1または2に記載の方法。
【請求項4】
前記宿主細胞が、骨髄腫細胞である、請求項3に記載の方法。
【請求項5】
前記骨髄腫細胞が、Sp2/0細胞もしくはその誘導体、マウスNSO細胞、またはラ
ットYB2/0細胞である、請求項4に記載の方法。
【請求項6】
前記宿主細胞が、上皮細胞またはその悪性形態である、請求項3に記載の方法。
【請求項7】
前記上皮細胞が、CHO細胞またはHEK293細胞である、請求項5に記載の方法。
【請求項8】
前記宿主細胞が、間葉細胞またはその悪性形態である、請求項3に記載の方法。
【請求項9】
前記間葉細胞が、線維芽細胞である、請求項8に記載の方法。
【請求項10】
前記線維芽細胞が、COS−1細胞またはCOS−7細胞である、請求項9に記載の方
法。
【請求項11】
前記宿主細胞が、ニューロン細胞、膠細胞、またはこれらの悪性形態である、請求項3
に記載の方法。
【請求項12】
前記ニューロン細胞が、網膜細胞、膠細胞、または神経膠腫細胞である、請求項11に
記載の方法。
【請求項13】
前記宿主細胞の生存を増強する薬剤が、宿主細胞によって発現された異種タンパク質で
ある、請求項3に記載の方法。
【請求項14】
前記異種タンパク質が、前記宿主細胞の染色体DNA中に組み込まれた核酸配列によっ
てコードされる、請求項13に記載の方法。
【請求項15】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE6タンパク質である、請求項
13に記載の方法。
【請求項16】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE7タンパク質である、請求項
13に記載の方法。
【請求項17】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE6タンパク質および乳頭腫ウ
イルスE7タンパク質である、請求項13に記載の方法。
【請求項20】
前記宿主細胞の生存を増強する薬剤が、アポトーシスインヒビターのBcl−2ファミ
リーから選択されるアポトーシスインヒビターである、請求項13に記載の方法。
【請求項21】
前記アポトーシスインヒビターが、Bcl−2、Bcl−xL、Bcl−w、Bcl−
EEE、Bhrf1、KS−Bcl−2、E1B−19K、Bcl−6、およびMcl−
1からなる群から選択される、請求項20に記載の方法。
【請求項22】
前記培地が、少なくとも1つのカスパーゼインヒビターをさらに含む、請求項1または
2に記載の方法。
【請求項23】
前記カスパーゼインヒビターが、カスパーゼ−1インヒビター、カスパーゼ−3インヒ
ビター、カスパーゼ−9インヒビター、カスパーゼ−12インヒビター、およびpan−
カスパーゼインヒビターからなる群から選択される、請求項22に記載の方法。
【請求項24】
前記インヒビターが、Z−VAD−fmk、Ac−DEVD−cho、Aven、およ
びXIAPからなる群から選択される、請求項22に記載の方法。
【請求項25】
前記培地が、アポトーシスを阻害し、および/または細胞保護剤として機能する外因的
に添加した薬剤をさらに含む、請求項1または2に記載の方法。
【請求項26】
前記外因性薬剤が、サイトカインI型スーパーファミリーのメンバーである、請求項2
5に記載の方法。
【請求項27】
前記サイトカインI型スーパーファミリーのメンバーがエリスロポエチンである、請求
項26に記載の方法。
【請求項28】
前記組換えタンパク質が、免疫グロブリン、ペプチド、酵素、成長因子、ホルモン、ワ
クチン、リンフォカイン、およびサイトカインからなる群から選択される、請求項1また
は2に記載の方法。
【請求項29】
前記組換えタンパク質が、抗体、抗体フラグメント、多重特異性抗体、および一本鎖抗
体からなる群から選択される免疫グロブリンである、請求項28に記載の方法。
【請求項30】
前記タンパク質が、エリスロポエチン、G−CSF、GM−CSF、EGF、VEGF
、およびトロンボポエチンからなる群から選択される成長因子である、請求項28に記載
の方法。
【請求項31】
前記タンパク質が、IL−1〜IL−31、αインターフェロン、βインターフェロン
、γインターフェロン、およびコンセンサスインターフェロンからなる群から選択される
リンフォカインである、請求項28に記載の方法。
【請求項32】
前記宿主細胞を、宿主細胞の生存を増強する薬剤で最初に修飾し、前記宿主細胞の生存
を増強する薬剤が、アポトーシスを阻害する薬剤である、請求項1に記載の方法。
【請求項33】
前記宿主細胞が、リンパ球、上皮細胞、間葉細胞、ニューロン細胞、またはこれらの悪
性形態である、請求項32に記載の方法。
【請求項34】
前記宿主細胞が、骨髄腫細胞である、請求項33に記載の方法。
【請求項35】
前記骨髄腫細胞が、Sp2/0細胞もしくはその誘導体、マウスNSO細胞、またはラ
ットYB2/0細胞である、請求項34に記載の方法。
【請求項36】
前記宿主細胞が、上皮細胞またはその悪性形態である、請求項33に記載の方法。
【請求項37】
前記上皮細胞が、CHO細胞またはHEK293細胞である、請求項35に記載の方法
。
【請求項38】
前記宿主細胞が、間葉細胞またはその悪性形態である、請求項33に記載の方法。
【請求項39】
前記間葉細胞が、線維芽細胞である、請求項38に記載の方法。
【請求項40】
前記線維芽細胞が、COS−1細胞またはCOS−7細胞である、請求項39に記載の
方法。
【請求項41】
前記宿主細胞が、ニューロン細胞、膠細胞、またはこれらの悪性形態である、請求項3
3に記載の方法。
【請求項42】
前記ニューロン細胞が、網膜細胞、膠細胞、または神経膠腫細胞である、請求項41に
記載の方法。
【請求項43】
前記宿主細胞の生存を増強する薬剤が、宿主細胞によって発現された異種タンパク質で
ある、請求項32に記載の方法。
【請求項44】
前記異種タンパク質が、前記宿主細胞の染色体DNA中に組み込まれた核酸配列によっ
てコードされる、請求項43に記載の方法。
【請求項45】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE6タンパク質である、請求項
43に記載の方法。
【請求項46】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE7タンパク質である、請求項
43に記載の方法。
【請求項47】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE6タンパク質および乳頭腫ウ
イルスE7タンパク質である、請求項43に記載の方法。
【請求項48】
前記宿主細胞の生存を増強する薬剤が、アポトーシスインヒビターのBcl−2ファミ
リーから選択されるアポトーシスインヒビターである、請求項43に記載の方法。
【請求項49】
前記アポトーシスインヒビターが、Bcl−2、Bcl−xL、Bcl−w、Bcl−
EEE、Bhrf1、KS−Bcl−2、E1B−19K、Bcl−6、およびMcl−
1からなる群から選択される、請求項48に記載の方法。
【請求項50】
前記培地が、少なくとも1つのカスパーゼインヒビターをさらに含む、請求項32に記
載の方法。
【請求項51】
前記カスパーゼインヒビターが、カスパーゼ−1インヒビター、カスパーゼ−3インヒ
ビター、カスパーゼ−9インヒビター、カスパーゼ−12インヒビター、およびpan−
カスパーゼインヒビターからなる群から選択される、請求項50に記載の方法。
【請求項52】
前記インヒビターが、Z−VAD−fmk、Ac−DEVD−cho、Aven、およ
びXIAPからなる群から選択される、請求項50に記載の方法。
【請求項53】
前記培地が、アポトーシスを阻害し、および/または細胞保護剤として機能する外因的
に添加した薬剤をさらに含む、請求項50に記載の方法。
【請求項54】
前記外因性薬剤が、サイトカインI型スーパーファミリーのメンバーである、請求項5
3に記載の方法。
【請求項55】
前記サイトカインI型スーパーファミリーのメンバーがエリスロポエチンである、請求
項54に記載の方法。
【請求項56】
前記組換えタンパク質が、免疫グロブリン、ペプチド、酵素、成長因子、ホルモン、ワ
クチン、リンフォカイン、およびサイトカインからなる群から選択される、請求項54に
記載の方法。
【請求項57】
前記組換えタンパク質が、抗体、抗体フラグメント、多重特異性抗体、および一本鎖抗
体からなる群から選択される免疫グロブリンである、請求項56に記載の方法。
【請求項58】
前記タンパク質が、エリスロポエチン、G−CSF、GM−CSF、EGF、VEGF
、およびトロンボポエチンからなる群から選択される成長因子である、請求項56に記載
の方法。
【請求項59】
前記タンパク質が、IL−1〜IL−31、αインターフェロン、βインターフェロン
、γインターフェロン、およびコンセンサスインターフェロンからなる群から選択される
リンフォカインである、請求項56に記載の方法。
【請求項60】
宿主細胞の生存を増強する組換えタンパク質をコードする外因性核酸配列を含む宿主細
胞。
【請求項61】
前記宿主細胞の生存を増強する組換えタンパク質が、乳頭腫ウイルスE6タンパク質、
乳頭腫ウイルスE7タンパク質、およびアポトーシスインヒビターのBcl−2ファミリ
ーからなる群から選択される、請求項60に記載の宿主細胞。
【請求項62】
前記タンパク質が、Bcl−2、Bcl−xL、Bcl−w、Bcl−EEE、Bhr
f1、KS−Bcl−2、E1B−19K、Bcl−6、およびMcl−1からなる群か
ら選択される、請求項61に記載の宿主細胞。
【請求項63】
前記宿主細胞が、アポトーシス耐性を示す、請求項61に記載の宿主細胞。
【請求項64】
前記宿主細胞が、Sp−E26である、請求項63に記載の宿主細胞。
【請求項65】
前記宿主細胞が、Sp−EEEである、請求項63に記載の宿主細胞。
【請求項66】
前記宿主細胞が、目的の組換えタンパク質をコードする核酸配列をさらに含む、請求項
63に記載の宿主細胞。
【請求項67】
前記組換えタンパク質が、免疫グロブリン、ペプチド、酵素、成長因子、ホルモン、ワ
クチン、リンフォカイン、およびサイトカインからなる群から選択される、請求項66に
記載の宿主細胞。
【請求項68】
前記組換えタンパク質が、抗体、抗体フラグメント、多重特異性抗体、および一本鎖抗
体からなる群から選択される免疫グロブリンである、請求項67に記載の宿主細胞。
【請求項69】
前記タンパク質が、エリスロポエチン、G−CSF、GM−CSF、EGF、VEGF
、およびトロンボポエチンからなる群から選択される成長因子である、請求項67に記載
の宿主細胞。
【請求項70】
前記タンパク質が、IL−1〜IL−31、αインターフェロン、βインターフェロン
、γインターフェロン、およびコンセンサスインターフェロンからなる群から選択される
リンフォカインである、請求項67に記載の宿主細胞。
【請求項71】
前記宿主細胞が、リンパ球、上皮細胞、間葉細胞、ニューロン細胞、またはこれらの悪
性形態からなる群から選択される、請求項63〜70のいずれかに記載の宿主細胞。
【請求項72】
前記宿主細胞が、骨髄腫細胞である、請求項71に記載の宿主細胞。
【請求項73】
前記骨髄腫細胞が、Sp2/0細胞もしくはその誘導体、マウスNSO細胞、またはラ
ットYB2/0細胞である、請求項72に記載の宿主細胞。
【請求項74】
前記宿主細胞が、上皮細胞である、請求項71に記載の宿主細胞。
【請求項75】
前記上皮細胞が、CHO細胞またはHEK293細胞である、請求項74に記載の宿主
細胞。
【請求項76】
前記宿主細胞が、間葉細胞である、請求項71に記載の宿主細胞。
【請求項77】
前記間葉細胞が、線維芽細胞である、請求項76に記載の宿主細胞。
【請求項78】
前記線維芽細胞が、COS−1細胞またはCOS−7細胞である、請求項77に記載の
宿主細胞。
【請求項79】
前記宿主細胞が、ニューロン細胞または膠細胞である、請求項71に記載の宿主細胞。
【請求項80】
前記ニューロン細胞が、網膜細胞、膠細胞、または神経膠腫細胞である、請求項79に
記載の宿主細胞。
【請求項81】
細胞増殖に適切な培地中に請求項63〜70のいずれかに記載の宿主細胞のクローン集
団を含む細胞培養物。
【請求項82】
前記培地が、少なくとも1つのカスパーゼインヒビターを含む、請求項81に記載の細
胞培養物。
【請求項83】
前記カスパーゼインヒビターが、カスパーゼ−1インヒビター、カスパーゼ−3インヒ
ビター、カスパーゼ−9インヒビター、カスパーゼ−12インヒビター、およびpan−
カスパーゼインヒビターからなる群から選択される、請求項82に記載の細胞培養物。
【請求項84】
前記インヒビターが、Z−VAD−fmk、Ac−DEVD−cho、Aven、およ
びXIAPからなる群から選択される、請求項83に記載の細胞培養物。
【請求項85】
前記培地が、アポトーシスを阻害し、および/または細胞保護剤として機能する外因的
に添加した薬剤をさらに含む、請求項81に記載の細胞培養物。
【請求項86】
前記外因性薬剤が、サイトカインI型スーパーファミリーのメンバーである、請求項8
5に記載の細胞培養物。
【請求項87】
前記サイトカインI型スーパーファミリーのメンバーがエリスロポエチンである、請求
項86に記載の細胞培養物。
【請求項88】
前記宿主細胞を、灌流反応器中で培養する、請求項3に記載の方法。
【請求項89】
前記宿主細胞を、灌流反応器中で培養する、請求項32に記載の方法。
【請求項90】
前記宿主細胞を、流加培養で培養する、請求項3に記載の方法。
【請求項91】
前記宿主細胞を、流加培養で培養する、請求項32に記載の方法。
【請求項92】
前記宿主細胞を、懸濁液中で培養する、請求項3に記載の方法。
【請求項93】
前記宿主細胞を、懸濁液中で培養する、請求項32に記載の方法。
【請求項94】
細胞培養物の寿命が少なくとも2日間増加する、請求項88に記載の方法。
【請求項95】
細胞培養物の寿命が少なくとも2日間増加する、請求項90に記載の方法。
【請求項96】
細胞培養物の寿命が少なくとも2日間増加する、請求項92に記載の方法。
【請求項97】
細胞培養物の寿命が少なくとも2日間増加する、請求項89、請求項91、または請求
項93に記載の方法。
【請求項98】
細胞培養物の寿命が少なくとも4日間増加する、請求項88に記載の方法。
【請求項99】
細胞培養物の寿命が少なくとも4日間増加する、請求項90に記載の方法。
【請求項100】
細胞培養物の寿命が少なくとも4日間増加する、請求項92に記載の方法。
【請求項101】
細胞培養物の寿命が少なくとも4日間増加する、請求項89、91、または93に記載
の方法。
【請求項102】
細胞培養物の寿命が少なくとも6日間増加する、請求項88に記載の方法。
【請求項103】
細胞培養物の寿命が少なくとも6日間増加する、請求項90に記載の方法。
【請求項104】
細胞培養物の寿命が少なくとも6日間増加する、請求項92に記載の方法。
【請求項105】
細胞培養物の寿命が少なくとも6日間増加する、請求項89、91、または93に記載
の方法。
【請求項1】
組換えタンパク質の発現に適切な条件下で適切な培地中で宿主細胞を培養するステップ
を含み、前記宿主細胞が、前記組換えタンパク質をコードする核酸配列を含むことと、前
記宿主細胞が、前記宿主細胞の生存を増強する薬剤によって修飾されていることとを含む
、組換えタンパク質の作製方法。
【請求項2】
前記宿主細胞の生存を増強する薬剤が、アポトーシスを阻害する薬剤である、請求項1
に記載の方法。
【請求項3】
前記宿主細胞が、リンパ球、上皮細胞、間葉細胞、ニューロン細胞、またはこれらの悪
性形態である、請求項1または2に記載の方法。
【請求項4】
前記宿主細胞が、骨髄腫細胞である、請求項3に記載の方法。
【請求項5】
前記骨髄腫細胞が、Sp2/0細胞もしくはその誘導体、マウスNSO細胞、またはラ
ットYB2/0細胞である、請求項4に記載の方法。
【請求項6】
前記宿主細胞が、上皮細胞またはその悪性形態である、請求項3に記載の方法。
【請求項7】
前記上皮細胞が、CHO細胞またはHEK293細胞である、請求項5に記載の方法。
【請求項8】
前記宿主細胞が、間葉細胞またはその悪性形態である、請求項3に記載の方法。
【請求項9】
前記間葉細胞が、線維芽細胞である、請求項8に記載の方法。
【請求項10】
前記線維芽細胞が、COS−1細胞またはCOS−7細胞である、請求項9に記載の方
法。
【請求項11】
前記宿主細胞が、ニューロン細胞、膠細胞、またはこれらの悪性形態である、請求項3
に記載の方法。
【請求項12】
前記ニューロン細胞が、網膜細胞、膠細胞、または神経膠腫細胞である、請求項11に
記載の方法。
【請求項13】
前記宿主細胞の生存を増強する薬剤が、宿主細胞によって発現された異種タンパク質で
ある、請求項3に記載の方法。
【請求項14】
前記異種タンパク質が、前記宿主細胞の染色体DNA中に組み込まれた核酸配列によっ
てコードされる、請求項13に記載の方法。
【請求項15】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE6タンパク質である、請求項
13に記載の方法。
【請求項16】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE7タンパク質である、請求項
13に記載の方法。
【請求項17】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE6タンパク質および乳頭腫ウ
イルスE7タンパク質である、請求項13に記載の方法。
【請求項20】
前記宿主細胞の生存を増強する薬剤が、アポトーシスインヒビターのBcl−2ファミ
リーから選択されるアポトーシスインヒビターである、請求項13に記載の方法。
【請求項21】
前記アポトーシスインヒビターが、Bcl−2、Bcl−xL、Bcl−w、Bcl−
EEE、Bhrf1、KS−Bcl−2、E1B−19K、Bcl−6、およびMcl−
1からなる群から選択される、請求項20に記載の方法。
【請求項22】
前記培地が、少なくとも1つのカスパーゼインヒビターをさらに含む、請求項1または
2に記載の方法。
【請求項23】
前記カスパーゼインヒビターが、カスパーゼ−1インヒビター、カスパーゼ−3インヒ
ビター、カスパーゼ−9インヒビター、カスパーゼ−12インヒビター、およびpan−
カスパーゼインヒビターからなる群から選択される、請求項22に記載の方法。
【請求項24】
前記インヒビターが、Z−VAD−fmk、Ac−DEVD−cho、Aven、およ
びXIAPからなる群から選択される、請求項22に記載の方法。
【請求項25】
前記培地が、アポトーシスを阻害し、および/または細胞保護剤として機能する外因的
に添加した薬剤をさらに含む、請求項1または2に記載の方法。
【請求項26】
前記外因性薬剤が、サイトカインI型スーパーファミリーのメンバーである、請求項2
5に記載の方法。
【請求項27】
前記サイトカインI型スーパーファミリーのメンバーがエリスロポエチンである、請求
項26に記載の方法。
【請求項28】
前記組換えタンパク質が、免疫グロブリン、ペプチド、酵素、成長因子、ホルモン、ワ
クチン、リンフォカイン、およびサイトカインからなる群から選択される、請求項1また
は2に記載の方法。
【請求項29】
前記組換えタンパク質が、抗体、抗体フラグメント、多重特異性抗体、および一本鎖抗
体からなる群から選択される免疫グロブリンである、請求項28に記載の方法。
【請求項30】
前記タンパク質が、エリスロポエチン、G−CSF、GM−CSF、EGF、VEGF
、およびトロンボポエチンからなる群から選択される成長因子である、請求項28に記載
の方法。
【請求項31】
前記タンパク質が、IL−1〜IL−31、αインターフェロン、βインターフェロン
、γインターフェロン、およびコンセンサスインターフェロンからなる群から選択される
リンフォカインである、請求項28に記載の方法。
【請求項32】
前記宿主細胞を、宿主細胞の生存を増強する薬剤で最初に修飾し、前記宿主細胞の生存
を増強する薬剤が、アポトーシスを阻害する薬剤である、請求項1に記載の方法。
【請求項33】
前記宿主細胞が、リンパ球、上皮細胞、間葉細胞、ニューロン細胞、またはこれらの悪
性形態である、請求項32に記載の方法。
【請求項34】
前記宿主細胞が、骨髄腫細胞である、請求項33に記載の方法。
【請求項35】
前記骨髄腫細胞が、Sp2/0細胞もしくはその誘導体、マウスNSO細胞、またはラ
ットYB2/0細胞である、請求項34に記載の方法。
【請求項36】
前記宿主細胞が、上皮細胞またはその悪性形態である、請求項33に記載の方法。
【請求項37】
前記上皮細胞が、CHO細胞またはHEK293細胞である、請求項35に記載の方法
。
【請求項38】
前記宿主細胞が、間葉細胞またはその悪性形態である、請求項33に記載の方法。
【請求項39】
前記間葉細胞が、線維芽細胞である、請求項38に記載の方法。
【請求項40】
前記線維芽細胞が、COS−1細胞またはCOS−7細胞である、請求項39に記載の
方法。
【請求項41】
前記宿主細胞が、ニューロン細胞、膠細胞、またはこれらの悪性形態である、請求項3
3に記載の方法。
【請求項42】
前記ニューロン細胞が、網膜細胞、膠細胞、または神経膠腫細胞である、請求項41に
記載の方法。
【請求項43】
前記宿主細胞の生存を増強する薬剤が、宿主細胞によって発現された異種タンパク質で
ある、請求項32に記載の方法。
【請求項44】
前記異種タンパク質が、前記宿主細胞の染色体DNA中に組み込まれた核酸配列によっ
てコードされる、請求項43に記載の方法。
【請求項45】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE6タンパク質である、請求項
43に記載の方法。
【請求項46】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE7タンパク質である、請求項
43に記載の方法。
【請求項47】
前記宿主細胞の生存を増強する薬剤が、乳頭腫ウイルスE6タンパク質および乳頭腫ウ
イルスE7タンパク質である、請求項43に記載の方法。
【請求項48】
前記宿主細胞の生存を増強する薬剤が、アポトーシスインヒビターのBcl−2ファミ
リーから選択されるアポトーシスインヒビターである、請求項43に記載の方法。
【請求項49】
前記アポトーシスインヒビターが、Bcl−2、Bcl−xL、Bcl−w、Bcl−
EEE、Bhrf1、KS−Bcl−2、E1B−19K、Bcl−6、およびMcl−
1からなる群から選択される、請求項48に記載の方法。
【請求項50】
前記培地が、少なくとも1つのカスパーゼインヒビターをさらに含む、請求項32に記
載の方法。
【請求項51】
前記カスパーゼインヒビターが、カスパーゼ−1インヒビター、カスパーゼ−3インヒ
ビター、カスパーゼ−9インヒビター、カスパーゼ−12インヒビター、およびpan−
カスパーゼインヒビターからなる群から選択される、請求項50に記載の方法。
【請求項52】
前記インヒビターが、Z−VAD−fmk、Ac−DEVD−cho、Aven、およ
びXIAPからなる群から選択される、請求項50に記載の方法。
【請求項53】
前記培地が、アポトーシスを阻害し、および/または細胞保護剤として機能する外因的
に添加した薬剤をさらに含む、請求項50に記載の方法。
【請求項54】
前記外因性薬剤が、サイトカインI型スーパーファミリーのメンバーである、請求項5
3に記載の方法。
【請求項55】
前記サイトカインI型スーパーファミリーのメンバーがエリスロポエチンである、請求
項54に記載の方法。
【請求項56】
前記組換えタンパク質が、免疫グロブリン、ペプチド、酵素、成長因子、ホルモン、ワ
クチン、リンフォカイン、およびサイトカインからなる群から選択される、請求項54に
記載の方法。
【請求項57】
前記組換えタンパク質が、抗体、抗体フラグメント、多重特異性抗体、および一本鎖抗
体からなる群から選択される免疫グロブリンである、請求項56に記載の方法。
【請求項58】
前記タンパク質が、エリスロポエチン、G−CSF、GM−CSF、EGF、VEGF
、およびトロンボポエチンからなる群から選択される成長因子である、請求項56に記載
の方法。
【請求項59】
前記タンパク質が、IL−1〜IL−31、αインターフェロン、βインターフェロン
、γインターフェロン、およびコンセンサスインターフェロンからなる群から選択される
リンフォカインである、請求項56に記載の方法。
【請求項60】
宿主細胞の生存を増強する組換えタンパク質をコードする外因性核酸配列を含む宿主細
胞。
【請求項61】
前記宿主細胞の生存を増強する組換えタンパク質が、乳頭腫ウイルスE6タンパク質、
乳頭腫ウイルスE7タンパク質、およびアポトーシスインヒビターのBcl−2ファミリ
ーからなる群から選択される、請求項60に記載の宿主細胞。
【請求項62】
前記タンパク質が、Bcl−2、Bcl−xL、Bcl−w、Bcl−EEE、Bhr
f1、KS−Bcl−2、E1B−19K、Bcl−6、およびMcl−1からなる群か
ら選択される、請求項61に記載の宿主細胞。
【請求項63】
前記宿主細胞が、アポトーシス耐性を示す、請求項61に記載の宿主細胞。
【請求項64】
前記宿主細胞が、Sp−E26である、請求項63に記載の宿主細胞。
【請求項65】
前記宿主細胞が、Sp−EEEである、請求項63に記載の宿主細胞。
【請求項66】
前記宿主細胞が、目的の組換えタンパク質をコードする核酸配列をさらに含む、請求項
63に記載の宿主細胞。
【請求項67】
前記組換えタンパク質が、免疫グロブリン、ペプチド、酵素、成長因子、ホルモン、ワ
クチン、リンフォカイン、およびサイトカインからなる群から選択される、請求項66に
記載の宿主細胞。
【請求項68】
前記組換えタンパク質が、抗体、抗体フラグメント、多重特異性抗体、および一本鎖抗
体からなる群から選択される免疫グロブリンである、請求項67に記載の宿主細胞。
【請求項69】
前記タンパク質が、エリスロポエチン、G−CSF、GM−CSF、EGF、VEGF
、およびトロンボポエチンからなる群から選択される成長因子である、請求項67に記載
の宿主細胞。
【請求項70】
前記タンパク質が、IL−1〜IL−31、αインターフェロン、βインターフェロン
、γインターフェロン、およびコンセンサスインターフェロンからなる群から選択される
リンフォカインである、請求項67に記載の宿主細胞。
【請求項71】
前記宿主細胞が、リンパ球、上皮細胞、間葉細胞、ニューロン細胞、またはこれらの悪
性形態からなる群から選択される、請求項63〜70のいずれかに記載の宿主細胞。
【請求項72】
前記宿主細胞が、骨髄腫細胞である、請求項71に記載の宿主細胞。
【請求項73】
前記骨髄腫細胞が、Sp2/0細胞もしくはその誘導体、マウスNSO細胞、またはラ
ットYB2/0細胞である、請求項72に記載の宿主細胞。
【請求項74】
前記宿主細胞が、上皮細胞である、請求項71に記載の宿主細胞。
【請求項75】
前記上皮細胞が、CHO細胞またはHEK293細胞である、請求項74に記載の宿主
細胞。
【請求項76】
前記宿主細胞が、間葉細胞である、請求項71に記載の宿主細胞。
【請求項77】
前記間葉細胞が、線維芽細胞である、請求項76に記載の宿主細胞。
【請求項78】
前記線維芽細胞が、COS−1細胞またはCOS−7細胞である、請求項77に記載の
宿主細胞。
【請求項79】
前記宿主細胞が、ニューロン細胞または膠細胞である、請求項71に記載の宿主細胞。
【請求項80】
前記ニューロン細胞が、網膜細胞、膠細胞、または神経膠腫細胞である、請求項79に
記載の宿主細胞。
【請求項81】
細胞増殖に適切な培地中に請求項63〜70のいずれかに記載の宿主細胞のクローン集
団を含む細胞培養物。
【請求項82】
前記培地が、少なくとも1つのカスパーゼインヒビターを含む、請求項81に記載の細
胞培養物。
【請求項83】
前記カスパーゼインヒビターが、カスパーゼ−1インヒビター、カスパーゼ−3インヒ
ビター、カスパーゼ−9インヒビター、カスパーゼ−12インヒビター、およびpan−
カスパーゼインヒビターからなる群から選択される、請求項82に記載の細胞培養物。
【請求項84】
前記インヒビターが、Z−VAD−fmk、Ac−DEVD−cho、Aven、およ
びXIAPからなる群から選択される、請求項83に記載の細胞培養物。
【請求項85】
前記培地が、アポトーシスを阻害し、および/または細胞保護剤として機能する外因的
に添加した薬剤をさらに含む、請求項81に記載の細胞培養物。
【請求項86】
前記外因性薬剤が、サイトカインI型スーパーファミリーのメンバーである、請求項8
5に記載の細胞培養物。
【請求項87】
前記サイトカインI型スーパーファミリーのメンバーがエリスロポエチンである、請求
項86に記載の細胞培養物。
【請求項88】
前記宿主細胞を、灌流反応器中で培養する、請求項3に記載の方法。
【請求項89】
前記宿主細胞を、灌流反応器中で培養する、請求項32に記載の方法。
【請求項90】
前記宿主細胞を、流加培養で培養する、請求項3に記載の方法。
【請求項91】
前記宿主細胞を、流加培養で培養する、請求項32に記載の方法。
【請求項92】
前記宿主細胞を、懸濁液中で培養する、請求項3に記載の方法。
【請求項93】
前記宿主細胞を、懸濁液中で培養する、請求項32に記載の方法。
【請求項94】
細胞培養物の寿命が少なくとも2日間増加する、請求項88に記載の方法。
【請求項95】
細胞培養物の寿命が少なくとも2日間増加する、請求項90に記載の方法。
【請求項96】
細胞培養物の寿命が少なくとも2日間増加する、請求項92に記載の方法。
【請求項97】
細胞培養物の寿命が少なくとも2日間増加する、請求項89、請求項91、または請求
項93に記載の方法。
【請求項98】
細胞培養物の寿命が少なくとも4日間増加する、請求項88に記載の方法。
【請求項99】
細胞培養物の寿命が少なくとも4日間増加する、請求項90に記載の方法。
【請求項100】
細胞培養物の寿命が少なくとも4日間増加する、請求項92に記載の方法。
【請求項101】
細胞培養物の寿命が少なくとも4日間増加する、請求項89、91、または93に記載
の方法。
【請求項102】
細胞培養物の寿命が少なくとも6日間増加する、請求項88に記載の方法。
【請求項103】
細胞培養物の寿命が少なくとも6日間増加する、請求項90に記載の方法。
【請求項104】
細胞培養物の寿命が少なくとも6日間増加する、請求項92に記載の方法。
【請求項105】
細胞培養物の寿命が少なくとも6日間増加する、請求項89、91、または93に記載
の方法。
【図2】

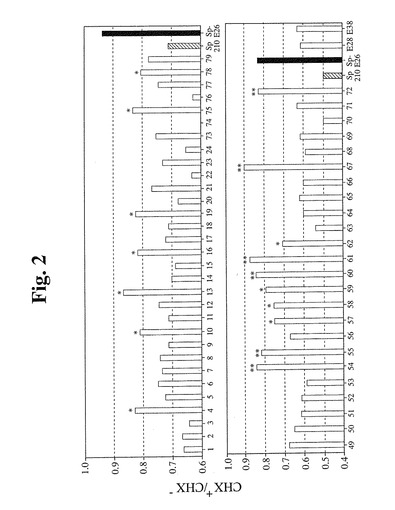
【図3】
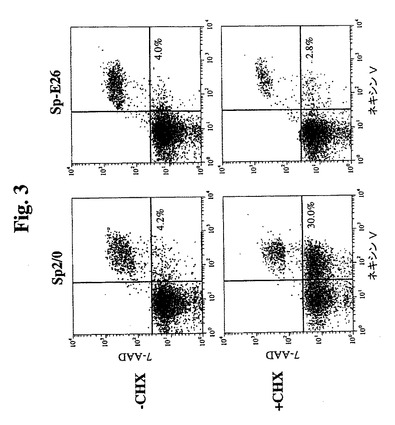
【図5】
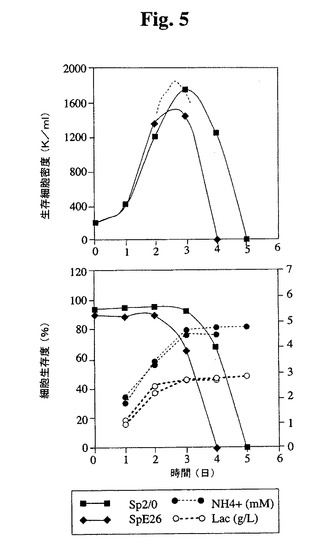
【図6】
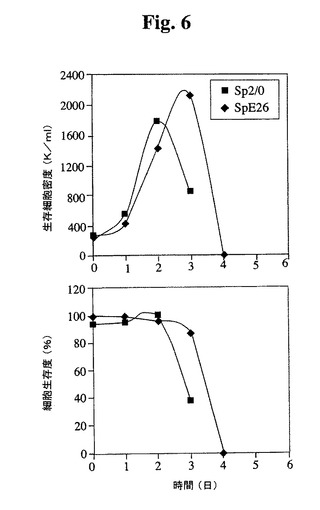
【図7】
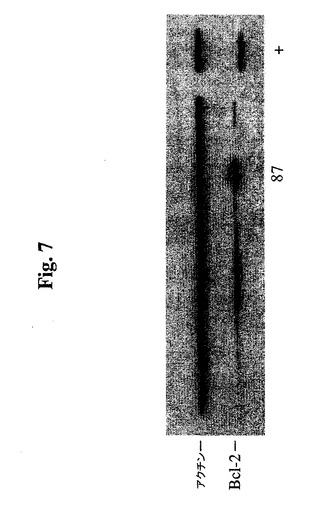
【図8】
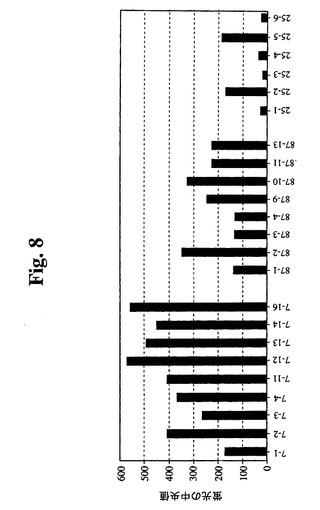
【図9】
【図10】
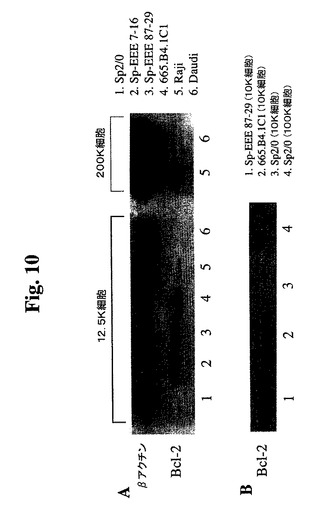
【図11】
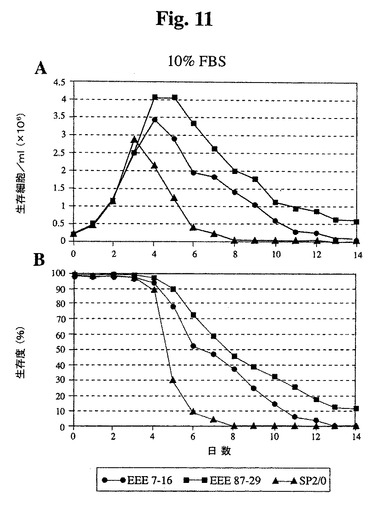
【図12】
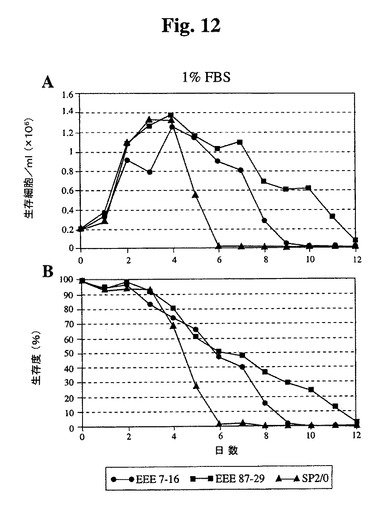
【図13】
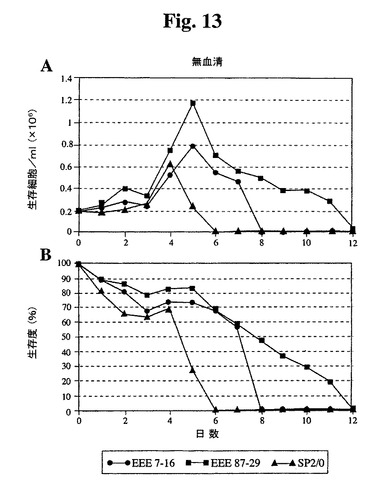
【図14】
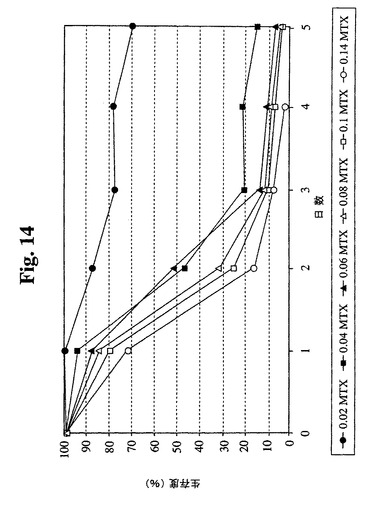
【図15】
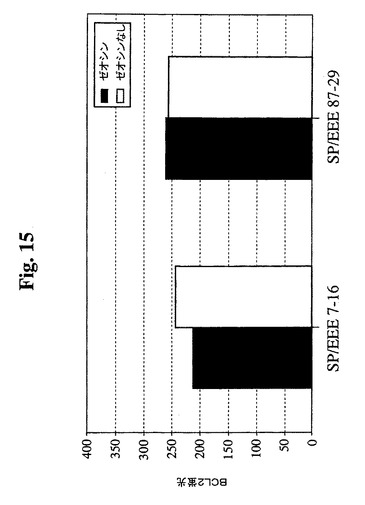
【図16】
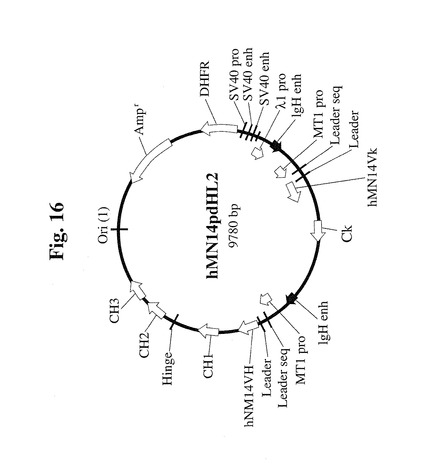
【図17】
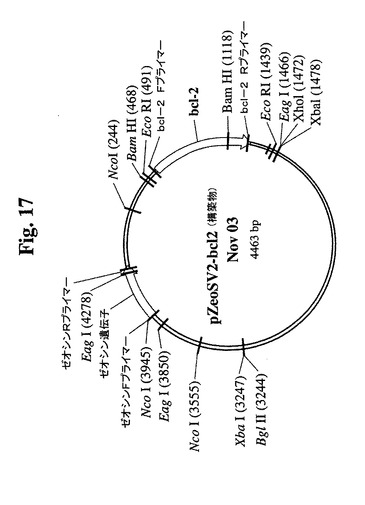
【図18】
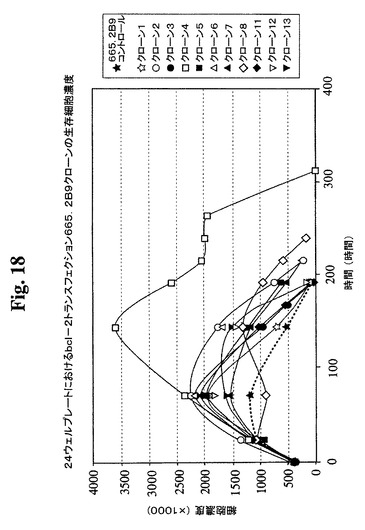
【図19】
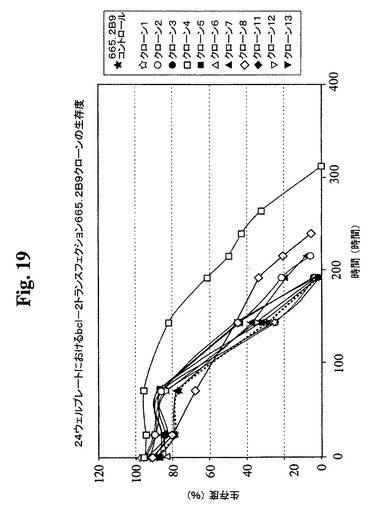
【図20】
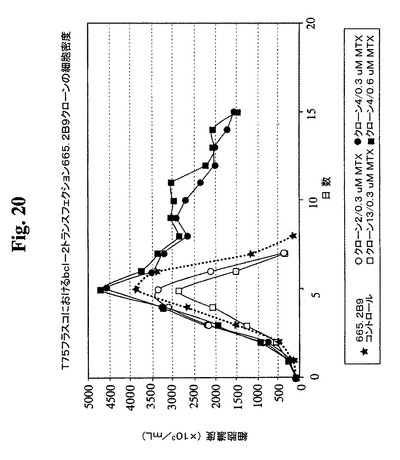
【図21】
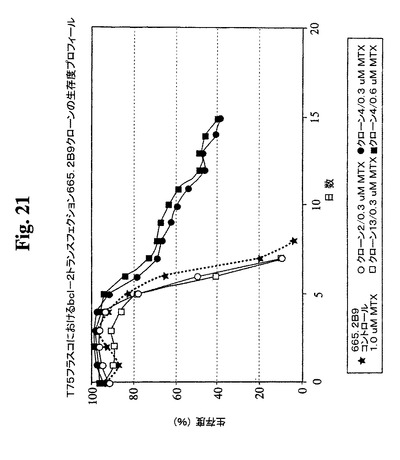
【図22】
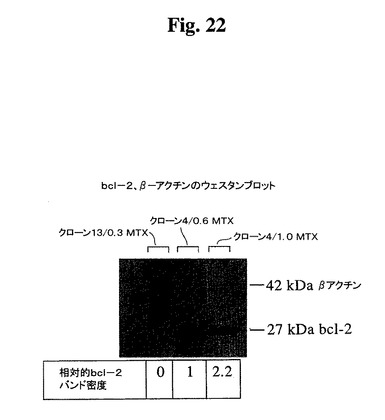
【図23】
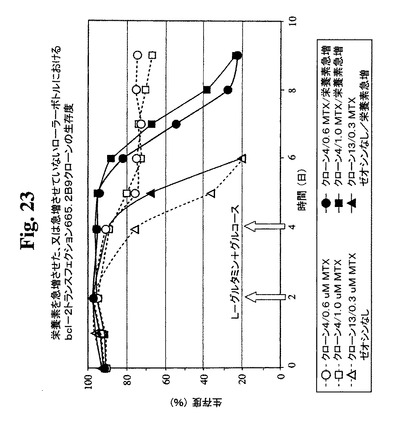
【図24】
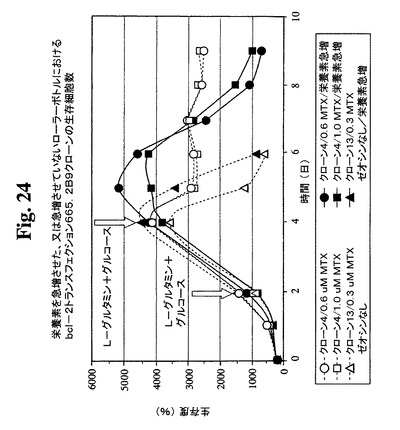
【図25】
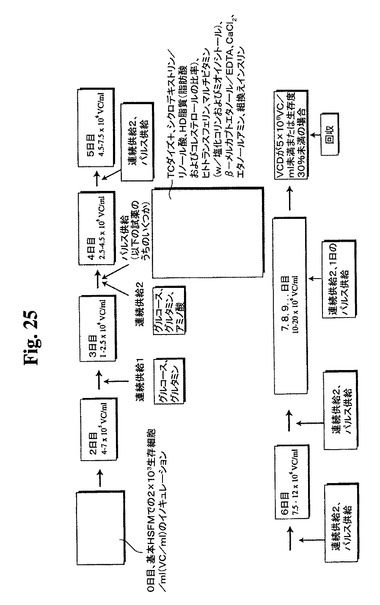
【図26】
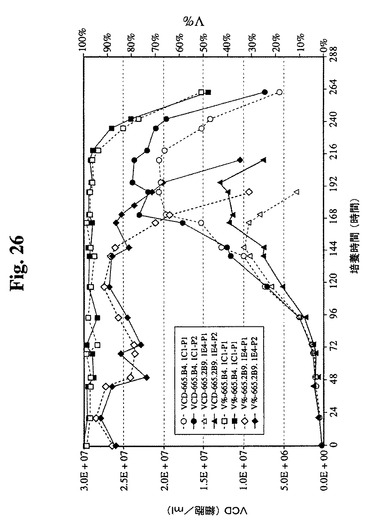
【図27】
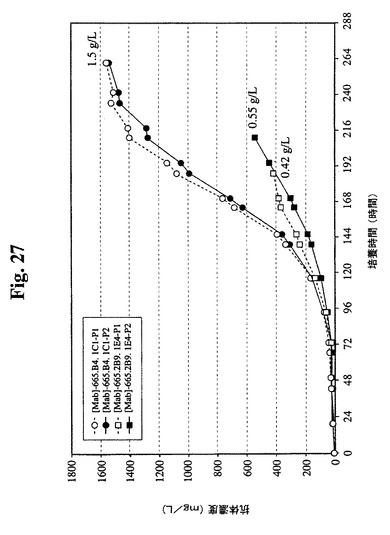
【図28】
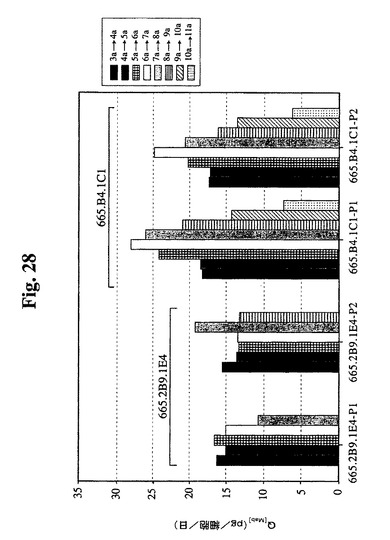
【図1】
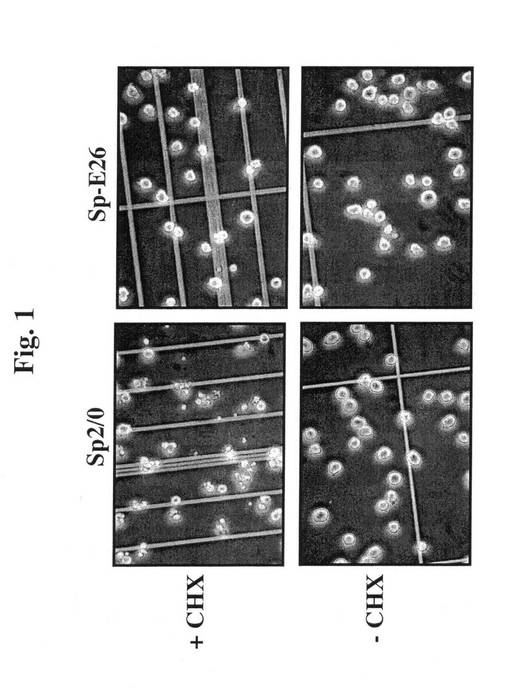
【図4】
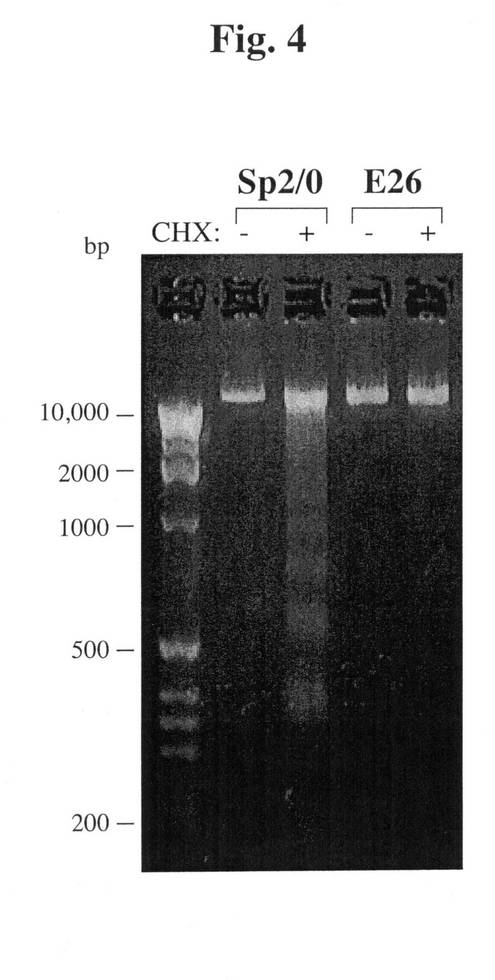
【図3】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図1】
【図4】
【公開番号】特開2012−254084(P2012−254084A)
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−163611(P2012−163611)
【出願日】平成24年7月24日(2012.7.24)
【分割の表示】特願2007−529867(P2007−529867)の分割
【原出願日】平成17年7月25日(2005.7.25)
【出願人】(504149971)イミューノメディクス、インコーポレイテッド (48)
【氏名又は名称原語表記】IMMUNOMEDICS, INC.
【Fターム(参考)】
【公開日】平成24年12月27日(2012.12.27)
【国際特許分類】
【出願番号】特願2012−163611(P2012−163611)
【出願日】平成24年7月24日(2012.7.24)
【分割の表示】特願2007−529867(P2007−529867)の分割
【原出願日】平成17年7月25日(2005.7.25)
【出願人】(504149971)イミューノメディクス、インコーポレイテッド (48)
【氏名又は名称原語表記】IMMUNOMEDICS, INC.
【Fターム(参考)】
[ Back to top ]