
細胞接着を阻害するため、またはRGD結合部位に診断用薬もしくは治療薬を送達するための組成物および方法
化合物は、R−G−システイン酸(すなわちR−G−NH−CH(CH2−SO3H)COOHまたはArg−Gly−NH−CH(CH2−SO3H)COOH)、並びに医薬として許容される塩、水和物、立体異性体、多量体、環状型、直鎖状型、薬物複合体、プロドラッグおよびそれらの誘導体を含めたそれらの誘導体を含む。また、ヒトもしくは動物の対象において、RGD結合部位への細胞接着を阻害するか、またはRGD結合部位に他の診断用薬または治療薬を送達する方法を含む、そのような化合物を製造および使用する方法が開示される。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、概して化学および医学の分野に関連し、より具体的には、Arg−Gly−Asp(「RGD」トリペプチド)結合部位への細胞接着を阻害するために使用可能な組成物および方法、並びに関連する、炎症、創傷治癒、瘢痕形成の防止、血栓症、癌転移および腫瘍、増殖性または非増殖性糖尿病性網膜症、硝子体液の液化、後部硝子体網膜剥離(PVD:posterior vitreo−retinal detachment)の誘発、硝子体網膜疾患、例えば、飛蚊症(floaters)、特発性黄斑円孔、硝子体黄斑牽引、加齢性黄斑変性、滲出型黄斑変性などの病因、脈絡膜血管新生、硝子体網膜手術、静脈閉塞、角膜血管新生、虚血性視神経、虹彩血管新生(rubiosis iridis)、および緑内障手術における瘢痕形成の防止のような疾患の治療に関する。
【背景技術】
【0002】
RGDトリペプチド配列は、多くのタンパク質中に見られ、そこで細胞接着に関与する。RGDトリペプチド配列が存在するタンパク質の例としては、コラーゲン、フィブロネクチン、ビトロネクチン、フォンヴィレブランド因子(VWF:von Willebrand factor)、特定のジスインテグリン、および特定のジスコイジンが挙げられる。
【0003】
インテグリンは、露出したRGD配列を有する配位子に結合することによって細胞と細胞外マトリックス(ECM:extracellular matrix)との間の接着を媒介するヘテロ二量体性細胞表面受容体である。正常なインテグリン−RGD結合は、細胞の成長、移動および生存に関わる遺伝子発現に関与すると考えられている。そのような細胞の成長、移動および生存の不完全な調節は、血栓形成、炎症および癌を含む多くの病態をもたらし得る。したがって、RGDペプチドは、細胞接着タンパク質の可能な模倣体として、またインテグリンに結合するそれらの能力について、アポトーシス、血管新生、および腫瘍形成の阻害のような治療目的のため、内部放射線治療薬並びに癌造影剤としてのそれらの多量体形態における使用のため、並びにそれらの抗癌剤輸送能力について、研究されてきた。
【0004】
眼において、インテグリンは、眼発生(ocular development)、細胞移動、治癒およびいくつかの病的過程を含む多くの過程に影響を与える。インテグリンはまた、眼組織において、炎症および血栓形成を調節し得る。硝子体内に注入されたRGDペプチドはまた動物モデルにおいて後方硝子体網膜剥離を引き起こすことが報告されており、したがって、特定の網膜障害の治療および/または硝子体切除術における硝子体の除去を容易にするため有用であり得る(非特許文献1参照)。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】オリベラ、エル.ビー.(Olivera,L.B.)ら、RGD Peptide−Assisted Vitrectomy to Facilitate Induction of a Posterior Vitreous Detachment:a New Principle in Pharmacological Vitreolysis、Current Eye Research(8):第333−40頁(2002年12月25日)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明はR−G−システイン酸(すなわちR−G−NH−CH(CH2−SO3H)COOHまたはArg−Gly−NH−CH(CH2−SO3H)COOH)およびその誘導体を含む(医薬として許容される塩、水和物、立体異性体、多量体、環状型、直鎖状型、薬物複合体、プロドラッグおよびそれらの誘導体を含む)新規な化合物を提供する。
【課題を解決するための手段】
【0007】
本発明は、ヒトまたは動物の対象において、対象に対してR−G−システイン酸ペプチドまたはその誘導体(医薬として許容される塩、水和物、立体異性体、多量体、環状型、直鎖状型(linear forms)、薬物複合体、プロドラッグおよびそれらの誘導体を含む)を含有する有効量の組成物を投与することによって、RGD結合部位への細胞接着を阻害するため、またはRGD結合部位に他の診断用薬または治療薬を送達するための組成物および方法を提供する。
【0008】
本発明のR−G−システイン酸ペプチドの特定の例としては、直鎖状型のArg−Gly−NH−CH(CH2−SO3H)COOH(本願で化合物1と称される例)および環状型のArg−Gly−NH−CH(CH2−SO3H)COOH(本願で化合物2と称される例)が挙げられる。
【0009】
本発明のR−G−システイン酸誘導体の一般式としては、下記のような一般式I〜VIIを有する化合物が挙げられるが、これらに限定されるものではない。
一般式I:
【0010】
【化1】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択され、かつY=OHまたはNH2である。
【0011】
一般式II:
【0012】
【化2】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択される。
【0013】
一般式III:
【0014】
【化3】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択され、かつZはHまたはSO3Hから選択される。
【0015】
一般式IV:
【0016】
【化4】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択され、YはOHまたはNH2から選択される。
【0017】
一般式V:
【0018】
【化5】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択される。
【0019】
一般式VI:
【0020】
【化6】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択される。
【0021】
一般式VII:
X1−R−G−システイン酸−X
前記式中、XおよびX1は環状または直鎖状の−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはArg、Gly、システイン酸(Cysteic)、Phe、Val、Ala、Leu、Pro、ThrのD−異性体またはL−異性体の任意の組み合わせの任意の塩から選択される。
【0022】
一般式VIIの環状型の例としては
【0023】
【化7】
が挙げられるが、必ずしもこれに限定されるものではない。
【0024】
前記式中、X’はH、C1−C6アルキル、Ph、またはSO3Hから選択され、ZはHまたはMeから選択され、YはOHまたはNH2から選択される。
スルホン酸は対応するカルボン酸より強力な酸である。このスルホン酸基のより高い極性はより強力な分子間結合をもたらす。例えば、より分極したO−H結合を有するR−G−システイン酸は、比較的分極に劣るO−H結合を有するR−G−アスパラギン酸(RGDペプチド)よりも強力な水素結合をインテグリン結合部位のタンパク質のアミド基と形成し、かつ/または、インテグリン結合部位において錯体を形成する金属イオンとのより強力な相互作用を有し得る。
【0025】
本明細書においてより詳細に別記するように、一般式VIIの1つの特定の例であるグリシニル−アルギニル−グリシニル−システイン酸−トレオニル−プロリン−COOH(Glycinyl−Arginyl−Glycinyl−Cysteic−Threonyl−Proline−COOH)(GRDシステイン酸TP、下記で化合物1と称する)を合成して動物において試験したところ、該化合物は、インテグリン−細胞外マトリックス(ECM)相互作用を阻害することにより、網膜表面からの後部硝子体剥離(PVD)を誘発するのに有効であることが判明した。また本明細書においてより詳細に別記するように、化合物1は、ヒト臍静脈内皮細胞(HUVEC)を用いた創傷治癒のモデルにおいて試験したところ、環状RGDペプチドによる40%の阻害と比較して、12時間で細胞接着を74%阻害することが示された。これらの研究は、グリシニル−アルギニル−グリシニル−システイン酸−トレオニル−プロリン−COOHが、インテグリンに対して、RGDペプチド自身よりも、さらにより強力に結合し得る論理的根拠を示唆および確証する。
【0026】
RGシステイン酸ペプチド配列は、L型またはD型のいずれかであり得、インテグリン−ECM相互作用の競合的阻害剤である。RGシステイン酸ペプチド配列は、プロテアーゼ耐性誘導体もしくは環状誘導体のもの、またはプロドラッグ誘導体、もしくは薬物送達システムに関連するもの、またはモノクローナル抗体のものであり得る。
【0027】
本発明の組成物は血管新生を阻害するために使用可能であり、血管新生の阻害は、炎症、創傷治癒、血栓症、癌転移および腫瘍を治療するために有用であり得る。さらに有用な用途は、増殖性または非増殖性糖尿病性網膜症、硝子体の液化(liquefaction of the vitreous)、後部硝子体網膜剥離(PVD)の誘発、例えば特発性黄斑円孔、硝子体黄斑牽引、加齢性黄斑変性、滲出型黄斑変性などの硝子体網膜疾患の病因、脈絡膜血管新生、硝子体網膜手術、静脈閉塞、並びに緑内障手術における瘢痕形成の防止を含む眼科学に見られ得る。さらに、本発明の多量体性かつ/または放射性標識された組成物は、腫瘍の検出のための診断用薬/造影剤、および腫瘍の治療のための放射線治療薬として、並びにそれらの腫瘍指向性による抗癌剤担体として、使用可能である。
【0028】
RGCペプチドを組込む生体材料はまた、細胞の長期生存および増殖に対して、および組織工学および再生医療における応用のための生体組織工学に対して、合成接着微小環境(synthetic adhesive microenvironment)を提供することができる。RGCペプチドは、インテグリン接着受容体を結合するそれらの特性によって、生体材料またはスキャフォールド上に拘束されると、接着促進信号を提供することができる。RGCに基づく物質は、細胞接着、細胞の拡張および移動を媒介する。さらに、インテグリン媒介細胞接着(integrin−mediated cell adhesion)は細胞増殖および生存を促進し、多細胞構造および組織の集合および構築において重要な役割を果たす。
【0029】
ルセンティス(Lucentis)およびアバスチン(Avastin)のような黄斑変性の治療のための薬剤は、他の場合には新たな血管の成長、血管新生を引き起こし、従って黄斑浮腫の一因となるVEGFを阻害することに基づく。小さなペプチドRGDは、ブルックスらに付与された米国特許第6,500,924号に示されているように、競合的結合によって細胞外マトリックス1への細胞付着を阻害することにより、アポトーシスを誘発し得ることが知られている。
【0030】
RGDペプチド結合または認識モチーフは、RGD接着エピトープを認識することによって細胞の細胞内細胞骨格(intracellular cytoskeleton)をECMと結び付ける細胞外マトリックスおよびインテグリンのタンパク質中に見られ得る。例えば、フース、アール ワイ.(Foos,R Y.),Invest.Opthalmol.Vis.Sci.(1972年)第11巻、第801−808頁を参照されたい。細胞は、通常、ECMへ付着することなく、アポトーシスを受ける。
【0031】
一般に、線維芽細胞と細胞外マトリックスの糖タンパク質成分との間の相互作用は、主に細胞表面インテグリン上で相互作用するRGD含有アミノ酸配列によって媒介される深刻な瘢痕形成を引き起こす。また、RGD配列は炎症反応およびホメオスタシス反応の間の細胞−ECM相互作用に関与し(ヘルシュコビッツ、エス.エム.(Hershkoviz,S.M.)ら、Invest.Ophthalmol.Vis.Sci.、(1994年)、第35巻、第2585−2591頁参照)、インテグリンは、創傷治癒または病的過程における細胞移動、並びに炎症および血栓形成の調節に重要な役割を果たすことも知られている。したがって、RGDペプチドのような強力なインテグリンアンタゴニストは、抗炎症剤、抗転移剤、または抗血栓剤のような薬物として非常に有用であり得る(エルナー、エス.ジー.(Elner,S.G.)およびエルナー、ヴィー.エム.(Elner,V.M.)、IOVS(1996)第37巻:第696−701頁参照)。ヒアルロン酸の受容体であるCD44は、ヒアルロン酸に対するその親和性によって、おそらくまたオステオポンチン、コラーゲンおよびマトリックスメタロプロテアーゼ(MMP)のような他の配位子に対するその親和性によって、細胞−細胞および細胞−基質相互作用を媒介することも文献に報告されている。
【0032】
ヒアルロナンとの接着は、細胞移動、腫瘍の成長および進行に重要な役割を果たし、またリンパ球の活性化、再循環、およびホーミング、並びに造血にも関与する。発現変化(altered expression)または機能不全は多くの病理表現型を引き起こす(例えば、ジャン、ディー.(Jiang D.)、Annu.Rev.Cell.Dev.Biol.(2007年)第23巻:第435−461頁;およびナドソン、ダブリュ.(Knudson,W.)ら、Matrix Bio.(2002年)、第21巻:第15−23号参照)。
【0033】
最近、CD44と細胞マトリックス成分(例えばHA)との相互作用が様々な炎症性疾患の発症に大きな役割を果しており、また、ヒアルロナン−CD44相互作用の断絶は脈絡膜血管新生の改善をもたらすことが示された(ヒロシ モチマル(Hiroshi Mochimaru)ら、Invest.Ophthalmol.Vis.Sci.(2009年)第50巻:第4410−4415頁参照)。
【0034】
これらの証拠は、RGDペプチドまたはCD44のような接着分子が細胞−細胞および細胞−ECM相互作用において多くの病原性疾患(pathogenic diseases)の発症に重要な役割を果たし、前記相互作用の阻害がその疾患を治療および治癒する上における新規な治療標的であり得ることを示している。
【0035】
合成ペプチドもまたインテグリンおよび成長因子に結合することが示されている。RGD配列を含む環状ペンタペプチドは、ビトロネクチンのαvβ3インテグリン(integin)への結合(マイケル エイ.デチャンストレイター(Michael A.Dechantsreiter)ら、J.Med.Chem.(1999年)第42巻:第3033−3040頁参照)、およびビトロネクチンおよびフィブロネクチンの双方のαvβ3およびαIIbβ3インテグリンへの結合(ローランド ハウブナー(Roland Haubner)ら、J.Am.Chem.Soc,(1996年)第118巻:第7461−7472頁参照)を阻害することが明らかとなった。この阻害は、多くの関連のない疾患の治療に有用であるこが示された。ハムスターによる研究において、環状ペンタペプチドは、対照動物と比較して、腫瘍の成長およびメタセシスを遅延させた(エム.エイ.バークル(M.A.Buerkle)ら、British J.Cancer (2002年)第86巻:第788−795頁参照)。ペンタペプチドはまた、鎌状赤血球細胞の血管内皮への結合を低減し、血行動態挙動を改善したことが示されている(アイリーン エム.フィンネガン(Eileen M.Finnegan)ら、Am.J.Physiol.Heart Circ.Physiol.,(2007年)第293巻:第H1038−H1045頁参照)。RGD配列を含む別の環状ペプチドは、炎症反応性および免疫反応における白血球結合(leucocyte binding)に関与することが知られているインテグリンであるα4β1に強力に結合することが示された。(ピーナ エム.カルダレッリ(Pina M.Cardarelli)ら、J.Biol.Chem.(1994年)第269巻:18668−18673頁参照)。合成硫酸化テトラペプチド(synthetic,sulfated tetrapeptide)はVEGFに強力に結合することが示されている(メイナード、ジェイ.エイ.ハベル(Maynard,J.A.Hubbell)、Acta Biomaterialia(2005年)第1巻:第451−459頁参照)。
【0036】
さらに、重要で有用な用途において、二量体RGDペプチド−薬物複合体は、腫瘍を標的とするためのインテグリン標的薬物送達に有用であることが証明されている(チェン(Chen)ら、J.Med.Chem.,(2005年)第48(4)巻:第1098−1106頁参照)。
【0037】
別の等しく重要かつ有用な用途において、多量体放射性標識RGDペプチドは、腫瘍検出のための診断用薬/造影剤として、並びにインテグリンαvβ3を標的とすることによる腫瘍に特異的なターゲティングおよび治療のための放射線治療薬として有用であることが証明されている(ツィ−ボ リ(Zi−Bo Li)ら、J.Nucl.Medicine,(2007年)第48巻:第1162−1171頁参照)。
【0038】
眼科学において、線維芽細胞による創傷治癒における瘢痕形成は、特に緑内障において、主な問題のうちの1つである。これは線維芽細胞とECMの糖タンパク質成分と間の相互作用に起因する。ECM糖タンパク質の認識は、Arg−Gly−Asp(すなわちRGD)配列のような、接着エピトープに特異的な細胞表面インテグリンを介して起こる。フィブロネクチン(FN)およびビトロネクチン(VN)を含む血漿たんぱくのいくつかのマトリックス中に存在するRGD配列は、炎症反応およびホメオスタシス反応の間に生じる細胞−ECM相互作用に関係する。線維芽細胞とECMの糖タンパク質との間の相互作用の抑制は瘢痕形成を緩和した(ラミ ヘルシュコビッツ(Rami Hershkoviz)ら、Invest.Ophthalmol.Vis.Sci.(1994年)第35巻:第2585−2591頁参照)。
【0039】
後部硝子体皮質の膠原細線維は、硝子体網膜界面、特に網膜表面の内境界層(inner limiting lamina)に付着する(セバーグ ジェイ.(Sebag J.)、Eye(1992年)、第6巻:第541−552頁参照)。硝子体基部、視神経円板(optic disc)における、主要な網膜血管に沿った、後極全体に対して面での(in facial manner)付着は、特発性黄斑円孔、硝子体黄斑牽引、増殖性糖尿病性網膜症などのような硝子体網膜疾患の病因において重要な役割を果たす(アキバ ジェイ.(Akiba J.) キロス エム.エイ.(Quiroz M.A.)ら、Ophthalmol.(1990年)第97巻、第1619−1655頁)。
【0040】
血管新生抑制は、ともに眼血管の過剰成長に起因する糖尿病性網膜症および黄斑変性について早期に有望であることが示されている。これらの疾患において、前記血管は眼内の正常組織に干渉したり、またはそれらの血管は光が眼の後部へ到達するのを遮断したりする。新たな血管はそれら自身が主な病的状態であり、血管成長の停止が失明を防止し得る。したがって、血管抑制(angioinhibition)は、単に病気の治療だけではなく、治癒になり得る。更に、網膜からの硝子体の分離は黄斑の牽引を緩和して、黄斑円孔の形成の危険性を低減し得ると仮定されてきた。従って、硝子体網膜界面上のインテグリンに結合するフィブロネクチンおよびラミニンの抑制による後部硝子体剥離は、糖尿病性網膜症および網膜静脈閉塞を有する眼における網膜血管新生を防止し得る(アキバ ジェイ.(Akiba J.)、キロス エムエイ(Quiroz MA)、 Ophthalmol.(1990年)第97巻、第1619−1655頁;ケリー エヌ.イー.(Kelly N.E.)ら、Ophthalmol.(1991年)第109巻、第654−659頁;およびカド エム(Kado M)ら、Am.J.Ophthalmol.(1988年)第105巻:第20−24頁参照)。
【0041】
近年、硝子体外科手術は、硝子体網膜牽引を軽減し、かつ網膜浮腫を低減するために非常に改善されてきた。外科技術および機器における継続的な改善にもかかわらず、網膜裂孔、網膜剥離および網膜神経繊維損傷などのような合併症により、特に一部の糖尿病性網膜症の患者および小児患者において、硝子体皮質の非外傷性除去を達成することは依然として困難なままである(セバーグ ジェイ.(Sebag J.)、Arch.Clin.Exp.Ophthalmol.(1987年)第225巻:第89−93頁;ハン ディ.ピー.(Han D.P.)ら、Arch.Ophthalmol.(1998年)第106巻:998−1000頁参照)。したがって、網膜を損なうことなく、硝子体界面を選択的に切り裂く外傷の少ないアプローチは非常に望ましい。近年、硝子体網膜界面の分離のための多くの薬物に関する報告が文献に現れてきている(トレーズ エム.ティ.(Trese M.T.)、Eye.(2002年)第16巻:第365−368頁;ガンドルファー エイ.(Gandorfer A.)ら、Am.J.Ophthalmol.(2002年)第133巻:第156−159頁;およびヘッセ エル.(Hesse L)ら、Eye Res.(2000年)第70巻:第31−39頁参照)。ヒアルロニダーゼ(カラジョージアン(Karageozian)らに付与された米国特許第6,863,886号参照)および自己由来プラスミン(サクマ ティ(Sakuma T)ら、日本眼科学会雑誌(2003年)、第107巻:第709−718頁)のような酵素を用いた薬理学的硝子体索切断は、細胞外マトリックスの消化(digestion)を促進し、後部硝子体剥離を誘発することがこれまでに研究されてきた。しかしながら、用いられた酵素による隣接組織の非特異的な破壊が、それらの治療適用の成功を妨げている。ここ数年において、尿素のような非酵素薬物(non−enzymatic pharmacologic agents)(ニッカーソン、シー.(Nickerson,C.)ら、J.Biomechanics、(2008年)第41巻:第1840−1846頁、およびMacromol.Symp.(2005年):第183−189頁参照)およびRGDペプチド(レオナルド ビー.オリビエラ(Leonardo B.Oliviera)ら、Curr.Eye Res.(2002年)第25巻:第333−340頁参照)を用いた新規なアプローチが、硝子体網膜界面の分離に注目することによって研究されてきている。RGDペプチドの合成類似体は、インビトロ(ウィリアムズ ジェイ.エイ.(Williams J.A.)、Pathol.Bio.(1992年)第40巻:第813−821頁;ゲールセン ケイ.アール(Gehlsen K.R.)ら、J.Cell.Biol.(1988年)第106巻:第925−930頁;ピアシュバッヘル、エム.ディ.(Pierschbacher,M.D.)ら、J.Biol.CHem.,(1987年)第262巻:第17294−17298頁;およびゾーン エル.エル.(Zhon L.L.)ら、IOVS.(1996年)第37巻:第104−113頁)およびインビボで、ECMタンパク質のRGDモチーフがインテグリン−ECM相互作用を妨害し、かつ付着を緩めるのに競合することが示されている。従って、可溶性RGDペプチドの硝子体内注入は、ウサギモデルにおいて、網膜表面の不溶性ECMタンパク質からのRGDエピトープの解放をもたらし、従って非酵素PVDを容易にした。明らかに、これらの結果は、硝子体網膜界面が、ECMのRGDモチーフへのインテグリン接続、並びに硝子体皮質コラーゲンの内境界層(ILL:inner limiting lamella)への接着に関与することを示している。RGDペプチドおよびその誘導体は、創傷における上皮細胞の移動を促進し(ピー.エム.メルツ(P.M. Mertz)ら、J.Burn Care Res.(1996年)第17巻:第199−206頁参照)、ヒドロゲル(エムピー ルットルフ(MP Lutolf)ら、Proc.Nat.Acad.Sci.(2003年)第100巻:第5413−5418頁:エムピー ルットルフ(MP Lutolf)ら、Nature Biotechnol.(2003年)第21巻:第513−518頁参照)、他のポリマーマトリクス(ホーン−バン リン(Horng−Ban Lin)ら、J.Biomed.Material.Res.(2004年)第28巻:第329−342頁参照)のような合成生体材料に組み込まれた場合、または固い物質(hard substance)上の表面膜として(ディ.エム.フェリス(D.M.Ferris)ら、Biomaterials(1999年)第20巻:第2323−2331頁)、それらの生物活性を維持する。RGDペプチドはまた、ペプチド配列で被覆された人工血管(ケイ.ワルシェク(K.Walluscheck)ら、Eur.J.Vascular and Endovascular Surgery(1996年)第12巻:第321−330頁参照)および他の人工臓器(ジェスケ、ブリゲッテ(Jeschke,Brigette)、Biomaterials(2002年)第23巻:第3455−3463頁)への上皮細胞または内皮細胞の増大した接着を促進し、神経再生を支持することが示されている(エム.ラフィウディン アフマッド(M.Rafiuddin Ahmed)ら、Brain Res.(2003年)第993巻:第208−216頁参照)。前記人工器官の生物活性表面は合成樹脂繊維またはポリマーを含むことができる。
【図面の簡単な説明】
【0042】
【図1】創傷治癒の動態における、3つのペプチド、すなわち、環状RGDペプチド、RGCペプチド(化合物1)、RGEペプチドの効果を示す図。
【図2】RGCペプチド(化合物1)のHPLCクロマトグラムを示す図。
【図3】RGCペプチド(化合物1)のエレクトロスプレー質量クロマトグラムを示す図。
【図4】環状RGCペプチド(化合物2)のHPLCクロマトグラムを示す図。
【図5】環状RGCペプチド(化合物2)のエレクトロスプレー質量クロマトグラムを示す図。
【発明を実施するための形態】
【0043】
本発明は、上記の一般式I〜VIIの化合物を含む新規な化合物を提供する。本発明の特定の例としては、直鎖状型のArg−Gly−NH−CH(CH2−SO3H)COOH(本願では化合物1と称される例)および環状型のArg−Gly−NH−CH(CH2−SO3H)COOH(本願では化合物2と称される例)、並びに、医薬として許容される塩、水和物、立体異性体、多量体(mutimers)、環状型、直鎖状型、多量体型(multimeric forms)、薬物複合体、プロドラッグおよびそれらの誘導体を含む、それらの誘導体が挙げられる。
【0044】
化合物1および2の合成
当業者に既知の従来の固相ペプチド合成(SPPS:solid−phase peptide synthesis、アール.ビー.メリフィールド(R.B.Merrifield)、J.Am.Chem.Soc.(1963年)第85(14)巻:第2149−2154頁)が実施され得る。SPPSは高収率なので好ましい合成の方法である。一般に、固相ペプチド合成技術の第1段階は、高分子支持体(polymeric support)上における保護されたアミノ酸誘導体によるペプチド鎖集合から成る。前記技術の第2段階は、粗製自由ペプチドを与える、すべての側鎖保護基の同時切断による樹脂支持体からのペプチドの切断である。SPPSの一般的な原理は、カップリング−脱保護の繰返しサイクルのうちの1つである。固相付着ペプチドの自由N末端アミンは、単一のN−保護アミノ酸ユニットに結合される。次に、このユニットは脱保護され、新たなN末端基アミンを表す。前記N末端基アミンにはさらにアミノ酸が付着され得る。合成、保護および脱保護の方法については、Asymmetric Synthesis、フォン ジー.エム.コッポラ(Von G.M.Coppola)およびH.F.シュースター(H.F.Schuster)著、ジョン・ワイリー・アンド・サンズ(John Wiley & Sons)、ニューヨーク、1987年を、保護および脱保護の方法については、Greene’s Protective Groups in Organic Synthesis、ピーター ジー.エム.ウッツ(G.M.Wuts)およびシアドーラ ダブリュ.グリーン(Theodora W.Greene)著(第二版)、ジェイ.ワイリー・アンド・サンズ(J.Wiley & Sons)(1991年)を参照されたい。
【0045】
固相ペプチド合成の2つの主に用いられる形態、すなわちFmoc(9−フルオレニルメチルオキシカルボニル;塩基に不安定な(base labile)アルファ−アミノ保護基)およびt−Boc(t−ブチルオキシカルボニル;酸に不安定な保護基)のうち、本ペプチドの合成には、好ましくはFmocが用いられ得る。各方法は、異なる樹脂およびアミノ酸側鎖保護、並びに結果として生ずる切断/脱保護工程を伴う。前記樹脂からの切断後、ペプチドは、通常、C−18、C−8およびC−4のようなカラムを用いた逆相HPLCによって精製される。
【0046】
固相ペプチド合成の例
下記は、塩基に不安定な9−フルオレニルメチルオキシカルボニル(Fmoc)保護基を用いて、固体保持体としてのワング樹脂(Wang resin)上におけるペプチド合成のための合成工程の概要である。
【0047】
Fmoc脱保護
0.08mmolのFmoc−Pro−ワング樹脂を、プラスチックキャップを装備したフリットカラム(fritted column)内に充填する。前記樹脂を2×3mL量のDMF(ジメチルホルムアミド)で各々1分間にわたって洗浄する。次に、約3mLの20%ピペリジン/DMF溶液を添加し、Fmoc脱保護を15分間にわたって継続させる。この時間の間、カラムを優しく回すか、または撹拌して、確実に完全に混合する。反応が完了した後(約15分)、反応カラムを流出させて、前記樹脂をDMF(4×3mL)で再び洗浄する。
【0048】
アミド結合カップリング
次に、所望のFmoc保護アミノ酸であるFmoc−Thr−tBu(3当量:供給者によって表示された樹脂充填量に対して)およびDIEA(6当量)のDCM溶液(アミノ酸に対して0.5M)を前記樹脂に添加する。その混合物を−20°Cで20分間にわたって冷却する。次に、ベンゾトリアゾール−1−イル−オキシトリピロリジノホスホニウムヘキサフルオロホスファート(PyBOP)、固相ペプチド合成に用いられるペプチドカップリング試薬(3当量)を前記反応に添加する。−20°Cで8時間にわたって振盪した後、反応混合物を流出させて、前記樹脂をDCM(3×)で洗浄する。
【0049】
20%ピペリジン/DMF溶液を用いたFmoc脱保護(15分)およびDMF(3×)による洗浄の後、次のFmoc保護アミノ酸(3当量;樹脂充填量に対して)であるPyBopを上記と同じ方法でカップリングする。
【0050】
切断
遊離酸型のペプチドを得るために、エステル結合をTFA(トリフルオロ酢酸)のような強い酸性条件を用いて切断する。前記樹脂を2〜3mLのトリフルオロ酢酸および水の95:5の溶液で処理する。その樹脂を25分間にわたって優しく撹拌する。次に、カラムを流出させて、濾液をガラス収集容器に慎重に収集にする。
【0051】
化合物1(GRGシステイン酸TP)の合成:
【0052】
【化8】
工程1. 樹脂の充填:プロリンで事前に充填されたo−クロロトリチル樹脂を出発材料として用いる。
【0053】
工程2. ペプチド集合:Fmoc合成を用いてペプチドを集合させる。保護されたアミノ酸をPyBOPで活性化し、末端Fmoc基を20%ピペリジン/DMF溶液で除去する。下記の保護されたアミノ酸は、それらが現われる順番で、順番に用いられる:
a.Fmoc−Thr−tBu(Fmocトレオニン−t−ブチルエステル)
b.Fmoc−システイン酸−Pfp(Fmocシステイン酸−ペンタフルオロフェニルエステル)
c.Fmoc−Gly(Fmocグリシン)
d.Fmoc−Arg−Pbf(Nα−Fmoc−Nω−(2,2,4,6,7ペンタメチルジヒドロベンゾフラン−5−スルホニル)−L−アルギニン)
e.Fmoc−Gly(Fmocグリシン)
工程3. 樹脂からのペプチド切断:結果として生じたペプチドは固体支持体から切断され、保護基は85.5%のTFA、5%のフェノール、2%の水、5%のチオアニソールおよび2.5%のエタンジチオールの溶液によって除去される。
【0054】
工程4. 精製:高性能液体クロマトグラフィー(HPLC)を用いて、生じたRGCペプチドを精製する。
上述のように調製した一定量の化合物1は、高性能液体クロマトグラフィーによって、純度>98%area/areaであると分析された[HPLC条件: バッファーA:0.1%のトリフルオロ酢酸(TFA)水溶液、バッファーB:0.1%のTFAアセトニトリル溶液、移動相(MP)A:97%のバッファーAおよび3%のバッファーB、移動相B:79%のバッファーAおよび21%のバッファーB、移動相C:50%のバッファーAおよび50%のバッファーB、勾配については、下記の表1を参照のこと、流速:1.0mL/分、カラム:ウォーターズ シンメトリー(登録商標)C18、5μ、4.6×250mm、カラム温度:30°C、検出器:UV@220nm、試料注入体積:20.0μL、試料調製:20μLの試料を1.0mLの移動相Aで希釈(約0.5mg/mL)]。対応するHPLCクロマトグラムを図2に示す。さらに、ペプチド配列の合成のための対応するアミノ酸の段階的な添加に基づいて、精製した化合物1の分子量は、エレクトロスプレイ質量分析法によって638.3amuであると測定され(理論質量:637.7amu)、化合物1の同一性を確認した。化合物1のエレクトロスプレー質量スペクトログラムを図3に示す。
【0055】
表1:HPLCによる化合物1を検出するためのポンプ勾配プログラム
【0056】
【表1】
化合物2(シクロ−RGシステイン酸fN(CH3)V)の合成:
【0057】
【化9】
工程1. 樹脂の充填:o−クロロトリチル樹脂を出発材料として用いる。Fmoc−N −メチル−L−Valを樹脂に付着させる。
【0058】
工程2. ペプチド集合:2.Fmoc合成をペプチド集合に用いる。保護されたアミノ酸をPyBOPで活性化し、末端Fmoc基を20%ピペリジン/DMF溶液で除去する。下記の保護されたアミノ酸は、それらが現われる順番で用いられる:
a.Fmoc−Phe(Fmoc−フェニルアラニン)
b.Fmoc−システイン酸−PfP(Fmocシステイン酸ペンタフルオロフェニルエステル)
c.Fmoc−Gly(Fmoc−グリシン)
d.Fmoc−Arg−Pbf(Nα−Fmoc−Nω−(2,2,4,6,7ペンタメチルジヒドロベンゾフラン−5−スルホニル)−L−アルギニン)
工程3. 樹脂からのペプチド切断:ペプチドを酢酸/TFA/DCM(1:3:3)を用いて固体支持体から切断する。
【0059】
工程4. 所望の環状ペプチドへの環化および脱保護:高希釈下のジフェニルホスホリルアジド(diphenylphosphorylazide)および重炭酸ナトリウムを用いたインサイチュー活性化による環化。側鎖を、85.5%のTFA、5%のフェノール、2%の水、5%のチオアニソール、2.5%のエタンジチオールを用いて脱保護する。
【0060】
工程5.精製:HPLCを精製に用いる。
上述のように調製した一定量の化合物2は、高性能液体クロマトグラフィーによって、純度>99%area/areaであると分析された(HPLC条件: 移動相A:0.1%のトリフルオロ酢酸(TFA)水溶液、B:(80%のアセトニトリル+20%の水)中に0.1%のTFA、20分間で26%から36%のBへ勾配、流速:1.0mL/分、カラム:フェノメネクス(Phenomenex)C18(2)4.6×150mm、5μ、100A、検出器:UV@220nm、試料注入体積:100.0μL)。対応するHPLCクロマトグラムを図4に示す。さらに、ペプチド配列の合成のための対応するアミノ酸の段階的な添加に基づいて、精製した化合物2の分子量は、エレクトロスプレー質量分析法によって625.3amuであると測定され(理論質量:625.77amu)、化合物2の同一性を確認した。化合物2のエレクトロスプレー質量スペクトログラムを図5に示す。
【0061】
細胞接着を阻害するための方法
本発明はまた、対象に有効量のR−G−システイン酸(すなわち、直鎖状型のR−G−NH−CH(CH2−SO3H)COOHまたは環状型のR−G−NH−CH(CH2−SO3H)COOH)およびその誘導体(医薬として許容される塩、水和物、立体異性体、多量体、環状型、直鎖状型、薬物複合体、プロドラッグおよびそれらの誘導体を含む)を投与することによって、ヒトまたは動物の対象におけるRGD結合部位への細胞接着を阻害する方法を提供する。
【0062】
本出願人は、ECM成分への細胞付着を競合的に阻害することにより、本発明の合成RGシステイン酸ペプチドがアポトーシスを誘発することを発見した。従って、本発明の合成RGシステイン酸ペプチドおよびその誘導体は、血管新生、炎症、癌転移、血栓形成、瘢痕形成の防止および治療に対する治療薬、並びに薬学的硝子体索切断剤として、強力なインテグリンアンタゴニストとして用いることができる。さらに本発明の重要な態様において、腫瘍検出(診断用薬または造影剤)および腫瘍治療用の内部放射線治療薬として使用するための、多量体化され、かつ放射性標識されたRGCペプチドを用いたα,βインテグリンの改善されたターゲティングが想定される。別の重要な態様において、改善されたRGCペプチド複合体または多量体RGCペプチド複合体は、薬剤担体として、例えば、効率的な腫瘍ターゲティングのための抗癌剤担体として機能する。
【0063】
本願において別記するように、RGCペプチド中のスルホン酸はRDGペプチド中の対応するカルボン酸よりも強力な酸である。このスルホン酸基のより高い極性は、より強力な分子間結合をもたらす。例えば、より分極したO−H結合を有するR−G−システイン酸は、比較的分極に劣るO−H結合を有するR−G−アスパラギン酸よりも強力な水素結合をインテグリン結合部位のタンパク質のアミド基および/またはアミノ酸の側鎖と形成し、かつ/またはインテグリン結合部位において錯体を形成する金属イオンとのより強力な相互作用を有し得る。従って、本発明の新規なRGCペプチドおよびその誘導体は、インテグリンレセプター認識および結合において、対応するRGDペプチドに対して、改善された化合物および組成物を提供する。
【0064】
加えて、高いIOPに関連する慢性高血糖症を有する糖尿病患者において、開放隅角緑内障は、特に、小柱網組織におけるフィブロネクチンの蓄積に関係することが報告されており、過剰のフィブロネクチンは房水流出を阻害すると考えられる(オシタリ、ティ(Oshitari,T)ら、Am.J.Ophthalmol.(2007年)第143巻:第363−365頁参照)。小柱網での細胞−ECM相互作用におけるフィブロネクチンの関与は、原発性開放隅角緑内障において示されている(マーク エス.フィラ(Mark S.Filla)ら、Invest.Ophthalmol.Vis.Sci.(2002年)第43巻:第151−161頁参照;およびチェリール アール.ハン(Cheryl R.Hann)ら、Ophthalmic Res.(2001年)第33巻:第314−324頁参照)。シュレム管の内皮細胞は、流出能に影響を及ぼすのに細胞外マトリックスと相互作用することも報告されている(シンディー ケイ.バーレー(Cindy K.Bahler)ら、Invest.Ophthalmol.Vis.Sci.(2004年)第45巻:第2246−2254頁参照)。フィブロネクチンのような細胞外マトリックス成分の過剰の存在による房水流出の抑制を踏まえると、糖尿病患者の高いIOPを治療するRGシステイン酸ペプチドおよびその誘導体の適用は、糖尿病患者にとって非常に有益であろう。
【0065】
好ましいRGシステイン酸ペプチドは、融合ポリペプチド、環状または直鎖状ポリペプチド、薬物送達システムまたは例えば抗癌剤のような他の薬剤で誘導された、またはそれらと関連付けられた、またはそれらと結合されたRGシステイン酸ペプチドを含む誘導ポリペプチド、多量体化RGCペプチド、インテグリン結合部位またはその機能的フラグメントと免疫反応するRGシステイン酸配列を含むモノクローナル抗体であり得る。
【0066】
ポリペプチドを含むRGシステイン酸は、フィブリノゲン、フィブロネクチン、ビトロネクチン、フォンヴィレブランド因子、ラミニン、トロンボスポンジンおよび同様な配位子中に存在するもののような天然インテグリン接着結合領域のアミノ酸残基配列に対応する配列を有し得る。
【0067】
本ペプチド配列は、末端グアニジノ基、スルホン基、およびカルボン酸基を有する3つのアミノ酸と、ペプチドフラグメント、糖タンパク質、およびPEG、プルロニックおよび他のポリマー基のようなポリマー基、並びにリポソームおよびナノ粒子を含む薬物送達システムに結合および/または関連付けられたそれらの誘導体とから成る。様々な病理学的疾患の治療のためにそれらを含む医薬組成物は、注入可能な剤形、ゲル剤形、懸濁液剤形、軟膏剤形、固体剤形および液体剤形を備える。
【0068】
フィブロネクチン、ビトロネクチン、ラミニン、フィブリノゲン、トロンボスポンジンおよびフォンヴィレブランド因子のような細胞接着モチーフに関連付けられたインテグリン受容体は、RGシステイン酸ペプチドおよびその誘導体の標的エピトープである。トリペプチドのRGシステイン酸は、細胞結合ドメインによって認識可能な最小アミノ酸配列として発見された。この配列はまた、インテグリンに無関係な免疫機能に干渉することもできる。よって、合成RGシステイン酸配列はRGD細胞結合ドメインを模倣するはずであり、アスパラギン酸のα−炭素上おける置換は、標的インテグリンへのより強力な結合親和性を与えることが発見された。RGCペプチド中のスルホン酸は、RDGペプチド中の対応するカルボン酸より強力な酸である。このスルホン酸基のより高い極性はより強力な分子間結合をもたらす。例えば、より分極したO−H結合を有するR−G−システイン酸は、比較的分極に劣るO−H結合を有するR−G−アスパラギン酸よりも強力な水素結合をインテグリン結合部位のアミド基および/またはアミノ酸の側鎖と形成し、かつ/または、インテグリン結合部位において錯体を形成する金属イオンとのより強力な相互作用を有し得る。
【0069】
本発明のRGシステイン酸配列用の最も一般的な式は、以下の通りである。
式A:
【0070】
【化10】
前記式中、X=−CH(R1)−S(=O)2−Y、−CH(R1)−SH、−CH(R1)−OZ、−CH(R1)S(=O)Y、−CH(R1)−O−S(=O)2−OX1、および−CH(R1)−O−P(=O)2OX1であり、かつ
前記式中、Y=OX1、NH2であり、X1=−H、C1−C6直鎖アルキル、フェニルであり;
R1=H、C1−C6直鎖アルキル、フェニルまたはSO3Hであり;
Z=H、SO3Hである。
【0071】
式B:
【0072】
【化11】
前記式中、X=−CH(R1)−S(=O)2−Y、−CH(R1)−SH、−CH(R1)−OZ、−CH(R1)S(=O)Y、−CH(R1)−O−S(=O)2−OX1、および−CH(R1)−O−P(=O)2OX1であり、かつ
前記式中、Y=OX1、NH2であり、X1=−H、C1−C6直鎖アルキル、フェニルであり、R1=H、C1−C6直鎖アルキル、フェニルまたはSO3Hであり、Z=H、SO3Hであり、かつ、A2は、−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはそれらの塩もしくはN−アルキル化誘導体のうちから選択される。Arg、Gly、システイン酸、Phe、Val、Ala、Leu、Pro、ThrのD型またはL型、並びに上記配列の環状型の任意の組み合わせを用いることができる。
【0073】
環状型の例としては下記式Cが挙げられる。
式C:
【0074】
【化12】
前記式中、X’は、H、C1−C6アルキル、Ph、SO3Hのうちから選択され、Y=OH、NH2であり、かつZ=H、CH3である。
【0075】
環状型としては、ペンタペプチドおよびヘプタペプチド、例えば下記に示される一般式Cの特定な化合物、すなわち、化合物2(シクロRGシステイン酸fN(CH3)V)も挙げられる。
【0076】
【化13】
式Aは、本願に別記する一般式I〜VIを包含する。式Bは、本願に別記する一般式VIIを包含する。
【0077】
式D:
A1−Arg−Gly−NH−CH(X)−CO−A2
前記式中、X=−CH(R1)−S(=O)2−Y、−CH(R1)−SH、−CH(R1)−OZ、−CH(R1)S(=O)Y、−CH(R1)−O−S(=O)2−OX1、および−CH(R1)−O−P(=O)2OX1であり、かつ
前記式中、Y=OX1、NH2であり、X1=−H、C1−C6直鎖アルキル、フェニルであり、R1=H、C1−C6直鎖アルキル、フェニルまたはSO3Hであり、Z=H、SO3Hであり、かつ、A1およびA2は、−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはそれらの塩もしくはN−アルキル化誘導体のうちから選択される。Arg、Gly、システイン酸、Phe、Val、Ala、Leu、Pro、ThrのD型またはL型、並びに上記配列の環状型の任意の組み合わせを用いることができる。
【0078】
置換RGシステイン酸配列としては、環状RGシステイン酸類似体が挙げられる。
RGシステイン酸およびその誘導体の投与は、薬物送達システムまたは任意の医薬として許容される剤形の注入製剤または固体製剤または軟膏製剤を用いることにより、皮下に、皮膚科学的に、眼科的に、および全身的に行うことができる。
【0079】
本発明の化合物は、経口、直腸、静脈内、動脈内、皮内、皮下、筋肉内、鞘内、舌下、口腔内、鼻腔内、経粘膜、経皮的、局所的、眼内、硝子体内、他の腸内、他の非経口、および/または他の可能な投与経路を含むが、これらに限定されない、意図した治療効果をもたらすのに適当な任意の経路によって投与され得る。
【0080】
本発明の化合物は、副作用または有毒作用を回避しながら、意図した治療効果を提供するいかなる投与量で投与されてもよい。本発明の化合物のヒト対象に投与されてもよい典型的な投与量は、約1ng/kg〜約1.0g/kgの範囲にある。
【0081】
可能かつ適当な場合には、本発明の化合物は、任意でリポソームまたはナノ粒子(例えばナノカプセル)の形態で調製されていてもよい。リポソームの形成および使用は、一般に当業者には知られている。リポソームは、それらが多重ラメラ小胞(MLVs:multilamellar vesicles)と称される場合がある多重ラメラ同心状二層小胞(multilamellar concentric bilayer vesicles)を自発的に形成するように、水性媒体中に分散されたリン脂質から形成される。MLVは、典型的には25nm〜4μmの直径を有する。超音波で処理した場合、MLVは、水溶液を含む核を有する、直径約200〜500オングストロームの小さな単層小胞(SUVs:small unilamellar vesicles)を形成する。一般に、リン脂質は、水性媒体中に分散された場合、脂質の水に対するモル比に応じて、リポソーム以外の様々な構造を形成することができる。脂質対水のモル比が低い場合には、リポソームが形成するであろう。リポソームの物理的特性は、pH、張度、および2価カチオンの有無に依存する。リポソームは、1)飲食作用(例えば、マクロファージおよび好中球のような細胞によるリポソームの食作用)、細胞表面への吸着、2)細胞表面成分との相互作用、3)原形質膜へのリポソームの脂質二重層の挿入による原形質細胞膜との融合、または4)リポソーム脂質の細胞膜または細胞内膜への移動またはその逆の移動、を含む様々な機構によって細胞と相互作用し得る。リポソーム製剤を変更することによって、同リポソームが副鼻腔、鼻粘膜などにおいて細胞と相互作用する機構を変更することができる。
【0082】
ナノカプセルは、外殻と、所望の物質が配置され得る空間とから成る任意のナノ粒子である。ナノカプセルを形成するための技術は当該技術分野において知られている。高分子ナノカプセルは特定の寸法および形状で製造され得る。前記高分子ナノカプセルは、物理的および化学的性質を正確に定めた単分散粒子として生成され得、したがって、pH、粘液の流れ、あるいはデバイスが植え込まれる副鼻腔内または耳、鼻もしくは咽喉の他の領域内に存在する他の条件のような特定の二分子トリガー機構(bimolecular triggering mechanisms)に応答して、治療用または診断用物質の放出を容易にするように作られ得る。ナノカプセルは、本発明において、特定の標的細胞(例えば、癌細胞または炎症状態に関連する細胞)に結合する特異的な化学的受容体または結合部位を有する「スマートドラッグ」として用いることができる。
【0083】
下記は、本発明の化合物を含有する医薬品のための処方の非限定的な例である。また、細胞接着の阻害においてRGCペプチドまたは誘導体を用いて示された安全性および/または有効性の例が含まれている。本願において、「RGシステイン酸ペプチド」、「RGCペプチド」、「RGCys−ペプチド」および、「本発明の化合物」という用語は、本明細書に記載される一般式I〜VIIおよび化合物1および3〜5によって定義されるものを含むが、これらに限定されるものではない、R−G−システイン酸配列を含む組成物およびそれらの誘導体を同義的に意味するものとする。
【実施例1】
【0084】
医薬製剤
下記は、本明細書に記載される一般式I〜VIIによって定義されるもののいずれか、または化合物1〜5のうちのいずれかのような本発明のR−G−システイン酸ペプチドを含有する医薬製剤I〜Xの実施例である。
【0085】
【表2】
【実施例2】
【0086】
ウサギにおけるRGDペプチドおよびグリシル−アルギニル−グリシル−システイン酸−トレオニル−プロリン−COOH(GRGシステイン酸TP;化合物1)のPVD誘発効果の比較
この実施例では、RGDペプチドおよびグリシル−アルギニル−グリシル−システイン酸−トレオニル−プロリン−COOH(RGシステイン酸ペプチド;GRGシステイン酸TP;化合物1)のPVD誘発効果をウサギにおいて比較した。この研究のための手順は以下の通りであった。
【0087】
手順:
動物モデル
a)20匹の雄および雌のウサギ
b)体重 約1.5〜2.5kg
c)2つのグループに分ける。
【0088】
i)10匹のウサギに、pH=6.5の2.5%RGD溶液を硝子体内に注入した。
a)10匹の右眼に2.5%RGD溶液を注入した。
b)5匹の左眼はBSS対照として用いた。
【0089】
c)5匹の左眼にpH=6.5の2.5%RGD+0.02%EDTAを注入した。
ii)10匹のウサギに、pH=6.5の2.5%RGシステイン酸溶液を硝子体内に注入した。
【0090】
a)10匹の右眼にpH=6.5の2.5%RGシステイン酸溶液を注入した。
b)10匹の左眼にpH=6.5の2.5%RGシステイン酸溶液+0.02%EDTAを注入した。
【0091】
活性薬品
d)EDTAナトリウム−99.0〜100.5%、スペクトラム ケミカル コーポレイション(Spectrum Chemical Corp.)より入手
e)RGシステイン酸−cGMP供給者(純度>98%)
f)RGD−cGMP供給者(純度>98%)
g)BSS溶液
RGシステイン酸、RGDの双方、RGシステイン酸+ナトリウムEDTA、RGD+ナトリウムEDTAおよびBS溶液は、手術の24時間前に硝子体腔内に注入した。ウサギ(10mg/kg体重)は、キシラジン(100mg/mL)および塩酸ケタミン(100mg/mL)の1:1の合剤2.0mLの筋肉内注入によって麻酔した。瞳は、局所的な塩酸シクロペントラート1%および塩酸フェニレフリン10%で拡張される。
【0092】
先在する硝子体網膜異常を有する動物を除外するために、すべての動物を、最初に細隙灯生体顕微鏡検査および間接検眼によって検査した。0.10ccの硝子体内注入は、1.0cc注入器に取り付けられた30ゲージ針を用いて、鼻上象限(supranasal quadrant)における縁に対して2mm後方に投与された。水晶体または網膜への損傷を避けるために注意しなければならない。
【0093】
注入後24時間かつ機械的硝子体切除の開始直前に、後部硝子体の状態、また硝子体の液化の状態を測定するために、B−スキャン超音波検査を行った。2ポート経毛様体扁平部硝子体切除術(two−port pars plana vitrectomy)は、注入光ファイバー、および硝子体切除ユニットに取り付けられた硝子体カッターを用いて行なった。30秒の中心部硝子体切除術に続いて、硝子体カッターを乳頭周囲網膜の表面に導き、そこで、低吸引(<30mmHg)を用いて、網膜表面からの後部硝子体皮質の分離を4象限において試みた。強膜切開を縫合し、各象限に見られる任意のPVDの存在および範囲を測定するために術後B−スキャン超音波検査を行なった。前記動物は心臓内ペントバルビタールナトリウム注射によって安楽死させ、直ちに眼を摘出した。
【0094】
術後B−スキャン超音波検査に基づいて、PVDの範囲を評価するために、この格付けシステムに従って、硝子体の液化およびPVDの等級を格付けした。
等級0. a)後部硝子体の剥離は観察されない。
【0095】
b)硝子体液化
等級1. a)硝子体が2つ以下の象限において分離されている眼から成る。
b)硝子体液化
等級2. a)硝子体が3つ以上の象限において分離されているが、放射組織(medullary rays)に沿って局所的な付着(focal attachments)が残存する眼から成る。
【0096】
b) 硝子体液化
等級3. a)硝子体が網膜表面から完全に分離されている眼から成る。
b)硝子体液化
すべての眼は、固定剤の迅速な浸透を保証するために、摘出直後に、毛様体扁平部に隣接した(sub−adjacent)上極(superior pole)において鋭利なカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるように注意した。その眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒドに摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球(posterior calotte)を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。
【0097】
結果
注入:2.5%RGシステイン酸
グループ1.ベースラインにおいて、すべての動物は両眼にPVDを有していない。
【0098】
【表3】
注入:2.5%のRGD
グループ2.ベースラインにおいて、すべての動物は両眼にPVDを有していない。
【0099】
【表4】
注入:2.5%RGシステイン酸+0.02%NaEDTA
グループ3.ベースラインにおいて、すべての動物は両眼にPVDを有していない。
【0100】
【表5】
注入:2.5%RGD+0.02%NaEDTA
グループ4.ベースラインにおいて、すべての動物は両眼にPVDを有していない。
【0101】
【表6】
この研究の実施例4における動態研究の結果は、RGDおよびRGシステイン酸(GRGシステイン酸TP;化合物1)が同様の特性を有し、2.5%RGシステイン酸の硝子体内への注入は、24時間で網膜からの硝子体の完全な分離を生じ、さらにRGDのウサギおよびRGシステイン酸のウサギの双方の硝子体は、完全に液化されることを示している。
【0102】
全体にわたって、RGシステイン酸の活性は、ウサギでの完全なPVDの誘発および硝子体の液化において、RGDのそれと同等であるか、または若干良好である。これは、恐らく、インテグリン細胞外マトリックス相互作用の結合部位に対して、RGDと比較して、より強力な競合的結合能力による。本明細書で別記するように、スルホン酸は対応するカルボン酸より強力な酸である。このスルホン酸基のより高い極性はより強力な分子間結合をもたらす。例えば、より分極したO−H結合を有するR−G−システイン酸は、比較的分極に劣るO−H結合を有するR−G−アスパラギン酸よりも強力な水素結合をインテグリン結合部位のアミド基および/またはアミノ酸の側鎖と形成し、かつ/または、インテグリン結合部位において錯体を形成する金属イオンとのより強力な相互作用を有し得る。
【0103】
前記結果はまた、これらの化合物が0.02%のエデト酸ナトリウムと共に投与される場合、RGDおよびRGシステイン酸の双方の活性は変化しないことを示している。
前記結果はまた、RGシステイン酸化合物の化合物1またはRGD化合物の硝子体内注入による副作用または有害な安全性効果はなかったことを示している。
【実施例3】
【0104】
ウサギの眼におけるRGCペプチド化合物1(GRGシステイン酸TP)の多数回注入の安全性の検討
この実施例では、RGCペプチド化合物1の多数回の注入を、体重約1.5〜2.5kgの5匹の雄および4匹の雌のニュージーランドウサギの眼に投与し、その眼を下記に記載の通りに試験した。
【0105】
研究手順は以下の通りであった。
A)ベースライン試験:ベースラインにおいて、9匹のウサギのすべての右眼および左眼を細隙灯生体顕微鏡検査法および間接検眼法で検査して、前記動物が先在する硝子体網膜異常を有していないことを確認した。さらに、9匹の動物すべての左眼および右眼に対して、β−スキャン超音波検査並びにERGスキャンを行って、ベースライン読み取り値を得た。
【0106】
B)実験的治療:次に、9匹のウサギすべてに、RGC溶液または生理食塩水(対照)のいずれかの硝子体内注入を受けさせた。治療溶液は以下のように調製した。
RGC溶液:0.02mgのEDTA二ナトリウム+0.80mgの塩化ナトリウムと、pH6.5に調整されたUSP滅菌注入用蒸留水(USP sterile Water for injection)とを含有する、2.5mg/100μLのRGC化合物1の溶液。
【0107】
生理食塩水(対照):pH6.5に調整されたUSP無菌等張食塩水液(USP Isotonic sterile saline solution)。
投薬は以下の通りに進めた。
【0108】
1)9匹の各ウサギの右眼に、100μLの2.5mg/100μL RGC溶液(投与量=2.5mgの化合物1を送達)を硝子体内に注入した。
2)9匹の各ウサギの左眼に、100μLの生理食塩水(対照)を硝子体内に注入した。
【0109】
3)最初の硝子体内注入後1日に、前記ウサギのうちのいずれかが前記注入による何らかの副作用を有していないかをチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、9匹のウサギすべての右眼および左眼を検査した。
【0110】
4)1回目の硝子体内注入後の7日目に、前記ウサギのうちのいずれかが前記注入による副作用を呈していないかを判定するために、細隙灯生体顕微鏡検査および間接検眼によって、9匹のウサギすべての右眼および左眼を再び検査した。さらに、ベースラインからの何らかの変化があったかを判定するために、すべての動物の右眼および左眼のすべてについてERGスキャンを実施した。
【0111】
5)次に、901番、904番および909番の3匹のウサギのグループを無作為に選択し、後部硝子体の状態を判定するために、これらの3匹の選択した動物の右眼および左眼に対して機械的硝子体切除を実施した。
【0112】
6)3匹の無作為に選択した動物(901番、904番、および909番)を心臓内ペントバルビタールナトリウム注入によって安楽死させて、直ちに眼を摘出した。前記眼はすべて、固定剤の迅速な浸透を保証するために、摘出直後に毛様体扁平部に隣接する上極において鋭利な鋭いカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるために注意した。前記眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒド(gluteraldehyde)中に摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。他の試料は病理組織検査に供した。
【0113】
902番、903番、905番、906番、907番、および908番の残りの6匹のウサギには、最初の注入後7日に第2回目を注入した。
1)6匹の各ウサギの右眼に、100μLの2.5mg/100μL RGC溶液(2回目の化合物1の2.5mg投与量を送達)を硝子体内に再び注入した。
【0114】
2)6匹の各ウサギの左眼に、100μLの生理食塩水(対照)を再び硝子体内に注入した。
3)2回目の硝子体内注入後1日に、前記ウサギのうちのいずれかが前記注入による何らかの副作用を有していないかをチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、6匹の残りのウサギすべての右眼および左眼を検査した。
【0115】
4)2回目の硝子体内注入後7日目に、前記注入による何らかの副作用をチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、6匹の残りのウサギすべての右眼および左眼を再び検査した。さらに、ベースラインからの何らかの変化があったかを判定するために、すべての動物の左眼および右眼のERGスキャンを実施した。
【0116】
5)次に、残りの6匹から902番、903番および907番の3匹のウサギのグループを無作為に選択し、後部硝子体の状態を判定するために、これらの3匹の無作為に選択した動物の右眼および左眼に対して機械的硝子体切除を実施した。
【0117】
6)902番、903番、および907番の3匹の無作為に選択した動物を心臓内ペントバルビタールナトリウム注入によって安楽死させて、直ちに眼を摘出した。前記眼はすべて、固定剤の迅速な浸透を保証するために、摘出直後に毛様体扁平部に隣接する上極において鋭利な鋭いカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるように注意した。前記眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒド(gluteraldehyde)に摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。他の試料は病理組織検査に供した。
【0118】
次に、残りのグループの905番、906番、および908番の3匹のウサギには、最初の注入後14日に3回目の注入を以下の通りに行った。
1)3匹の残りの各ウサギの右眼に、100μLの2.5mg/100μL RGC溶液を硝子体内に再び注入した(3回目の化合物1の2.5mg投与量を送達)。
【0119】
2)3匹の残りの各ウサギの左眼に、100μLの生理食塩水(対照)を硝子体内に再び注入した。
3)3回目の硝子体内注入後1日に、前記ウサギのうちのいずれかが前記注入による何らかの副作用を有していないかをチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、6匹の残りのウサギすべての右眼および左眼を検査した。
【0120】
4)3回目の硝子体内注入後7日目に、前記注入による何らかの副作用をチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、3匹の残りのウサギすべての右眼および左眼を再び検査した。さらに、ベースラインからの何らかの変化があったかを判定するために、すべての動物の左眼および右眼のERGスキャンを実施した。
【0121】
5)後部硝子体の状態を判定するために、3匹の残りの動物(905番、906番、および908番)の右眼および左眼に対して機械的硝子体切除を行った。
6)3匹の残りの動物(905番、906番、および908番)を心臓内ペントバルビタールナトリウム注入によって安楽死させて、直ちに眼を摘出した。前記眼はすべて、固定剤の迅速な浸透を保証するために、摘出直後に毛様体扁平部に隣接する上極において鋭利な鋭いカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるように注意した。前記眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒドに摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。他の試料は病理組織検査に供した。
【0122】
3)活性薬品
この研究に用いた活性薬品は以下の通りであった。
a.EDTA二ナトリウム−99.0〜100.5%、スペクトル ケミカル コーポレイションより
b.RGCペプチド(化合物1)
c.USP無菌等張食塩水液
4)研究製剤
a)RGC溶液:0.02mgのEDTA二ナトリウム+0.80mgの塩化ナトリウムおよびpH6.5に調整されたUSP滅菌注入用蒸留水を含有する、2.5mg/100μL RGC化合物1の溶液。0.22μフィルタを通して2.0mLバイアルへ無菌濾過。
【0123】
生理食塩水(対照):pHを6.5に調整されたUSP無菌等張食塩水。0.22μフィルタを通して無菌バイアル内へ滅菌濾過。
5)注入準備のための麻酔
a.2.0mLのキシラジン(100mg/mL)および塩酸ケタミン(100mg/mL)の1:1の合剤の筋肉内注入。
【0124】
b.瞳は、局所的な塩酸シクロペントラート1%および塩酸フェニレフリン10%によって拡張した。
6)硝子体内注入の準備:
2.5mg/100μLを含むRGC溶液およびpHが6.5に調整された無菌等張食塩液を収容する無菌バイアルを用意した。
【0125】
注入前に、研究者は、1.0ccの注入器内に0.10cc(100マイクロリットル)の溶液が存在することを確認した。
7)注入手順:
硝子体内注入がウサギの正常な日常活動を妨げるのに十分なレベルの視覚障害を生じないので、これは、視覚眼科学協会(Association for Research in Vision and Ophthalmology)ガイドラインの動物決議に従ってメイジャー・サバイバル・プロシージャ(major survival procedure)とは見なされない。
【0126】
細隙灯生体顕微鏡検査法、検眼鏡検査法およびERGのベースライン試験が前記ウサギに対して完了した後、RGC溶液並びに無菌食塩液の双方を硝子体腔内に注入した。前記ウサギ(10mg/kg体重)をキシラジン(100mg/mL)および塩酸ケタミン(100mg/mL)の1:1の合剤2.0mLの筋肉内注入によって麻酔した。瞳は、局所的な塩酸シクロペントラート1%および塩酸フェニレフリン10%で拡張させた。
【0127】
先在する硝子体網膜異常を備えたいかなる動物も除外するために、動物はすべて、最初に細隙灯生体顕微鏡検査および間接検眼(indirect opthalmoscopy)によって検査した。0.10ccの硝子体内注入は、1.0ccの注入器に取り付けられた30ゲージ針を用いて、鼻上象限(supronasal quadrent)における縁に対して2mm後方に投与された。水晶体または網膜への損傷を避けるために注意した。
【0128】
注入後7日に、前記動物に対して機械的硝子体切除を行なった。2ポート経毛様体扁平部硝子体切除術は、注入光ファイバー、および硝子体切除ユニットに取り付けられた硝子体カッターを用いて行なった。30秒の中心部硝子体切除術に続いて、硝子体カッターを乳頭周囲網膜の表面に導き、そこで、低吸引(<30mmHg)を用いて、4象限において網膜表面からの後部硝子体皮質の分離を試みた。前記動物を心臓内ペントバルビタールナトリウム注入で安楽死させ、直ちに眼を摘出した。
【0129】
前記眼はすべて、固定剤の迅速な浸透を保証するために、摘出直後に毛様体扁平部に隣接する上極において鋭利な鋭いカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるように注意した。前記眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒドに摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。
【0130】
結果の分析
RGC溶液および無菌食塩液で処理した眼における安全性に関するデータを、下記技術、すなわち、i)細隙灯生体顕微鏡検査法、ii)検眼鏡検査法、iii)ERG、iv)組織病理学、および、v)電子顕微鏡法を用いて安全性について分析した。
【0131】
安全性プロフィール:
901番、902番、903番、904番、905番、906番、907番、908番、909番の9匹のウサギのグループへの100μLの2.5%RGC溶液の1回目の硝子体内投与は全時点においていかなる有意な毒性にも関係していなかった。2.5%RGCグループと等張食塩水液グループとの間には報告された副作用において有意な差異はなかった。この毒性の欠如は、臨床検査、間接検眼法、および超音波β−スキャンおよび機械的硝子体切除によって判定された。
【0132】
細隙灯生体顕微鏡検査法は、すべての治験検診(study visits)において行なわれ、100μLの2.5%RGC溶液および等張食塩水液のグループの硝子体内注入に対して炎症反応がほぼ全くないことを示す炎症の兆候について、結膜および強膜、角膜、内皮の変更、前眼房反応、虹彩、水晶体および眼窩(capsule)、並びに前部硝子体に焦点が当てられた。すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な毒性の兆候があるようには見えなかった。
【0133】
有意な網膜毒性が存在しなかったことを保証する検討を通じて、後部(posterior segment)の臨床評価も続けられた。間接検眼並びに細隙灯眼底評価は、各評価時点において、網膜毒性の何らかの兆候に対して特に注意して行なった。後部は、硝子体密度、硝子体液化、硝子体付着および出血の可能性(possible hemorrhage)の変化について評価した。網膜は、RPE毒性、網膜血管障害性網膜出血、滲出、網膜裂孔、破壊または剥離の兆候について評価した。治療前のベースラインにおけるRPEの変化はなく、すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な後部変化の兆候があるようには見えなかった。硝子体内注入前に9匹の動物に対して行なったERGスキャン、並びに注入後1日および7日にすべての前記動物に対して行ったERGスキャンは、被験物質によって誘発された有意な変化または毒性のいかなる兆候も生じないようであったことに注目することは重要である。
【0134】
902番、903番、905番、906番、907番、908番の6匹のウサギのグループへの100μLの2.5%RGC溶液の2回目の硝子体内投与は全時点においていかなる有意な毒性にも関係していなかった。2.5%RGCグループと等張食塩水液グループとの間に報告された副作用における有意な差異はなかった。この毒性の欠如は、臨床検査、間接検眼法、および超音波β−スキャンおよび機械的硝子体切除によって判定した。
【0135】
細隙灯生体顕微鏡検査法は、すべての治験検診において行なわれ、100μLの2.5%のRGC溶液および等張食塩水液のグループの硝子体内注入に対して炎症反応がほぼ全くないことを示す炎症の兆候について、結膜および強膜、角膜、内皮の変更、前眼房反応、虹彩、水晶体および眼窩、並びに前部硝子体に焦点が当てられた。すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な毒性の兆候があるようには見えなかった。
【0136】
有意な網膜毒性が存在しなかったことを保証する検討の全体にわたって、後部の臨床評価も続けられた。間接検眼並びに細隙灯眼底評価は、各評価時点において、網膜毒性の何らかの兆候に対して特に注意して行なった。後部は、硝子体密度、硝子体液化、硝子体付着および出血の可能性の変化について評価した。網膜は、RPE毒性、網膜血管障害性網膜出血、滲出、網膜裂孔、破壊または剥離の兆候について評価した。2回目の治療の7日前のベースラインにおけるRPEの変化はなく、すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な後部変化の兆候があるようには見えなかった。硝子体内注入後2回目の6匹の動物に対して行なったERGスキャン、並びに注入後8日および14日にすべての前記動物に対して行ったERGスキャンは、被験物質によって誘発された有意な変化または毒性の任意の兆候を生じなかったようであることに注目することは重要である。
【0137】
905番、906番、908番の6匹のウサギのグループへの100μLの2.5%RGC溶液の3回目の硝子体内投与は全時点においていかなる有意な毒性にも関係していなかった。2.5%RGCグループと等張食塩水液グループとの間には、報告された副作用において有意な差異はなかった。この毒性の欠如は、臨床検査、間接検眼法、および超音波β−スキャンおよび機械的硝子体切除によって判定された。
【0138】
細隙灯生体顕微鏡検査法は、すべての治験検診において行なわれ、100μLの2.5%RGC溶液および等張食塩水液のグループの硝子体内注入に対して炎症反応がほぼ全くないことを示す炎症の兆候について、結膜および強膜、角膜、内皮の変更、前眼房反応、虹彩、水晶体および眼窩、並びに前部硝子体に焦点が当てられた。すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な毒性の兆候があるようには見えなかった。
【0139】
有意な網膜毒性が存在しなかったことを保証する検討の全体にわたって、後部の臨床評価も続けられた。間接検眼並びに細隙灯眼底評価は、各評価時点において、網膜毒性の何らかの兆候に対して特に注意して行なった。後部は、硝子体密度、硝子体液化、硝子体付着および出血の可能性の変化について評価した。網膜は、RPE毒性、網膜血管障害性網膜出血、滲出、網膜裂孔、破壊または剥離の兆候について評価した。3回目の処理前14日のベースラインにおけるRPEの変化はなく、すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な後部変化の兆候はあるようには見えなかった。硝子体内注入後3回目の3匹の動物に対して行なったERGスキャン、並びに注入後15日および21日にすべての前記動物に対して行ったERGスキャンは、被験物質によって誘発された有意な変化または毒性の任意の兆候を生じなかったようであることに注目することは重要である。
【実施例4】
【0140】
RGCペプチドの抗接着性:化合物1(GRGシステイン酸TP)、環状−RGDおよびRGEによる創傷治癒の動態研究
この実施例では、創傷治癒のモデルにおいて、RGCペプチドは抗接着性を有し、従って、多くの病理学的硝子体網膜疾患の発症を防止することができ、またヒト黒色腫および結腸癌細胞における転移を阻害し得ることが示された。
【0141】
RGCペプチドの抗接着性をインビトロで試験するために、ヒト臍帯静脈内皮細胞(HUVEC:human umbilical vein endothelial cells)によって創傷治癒アッセイを実施した。HUVECをフィブロネクチン被覆面上に播種して、コンフルエントな単層を成長させた。前記HUVEC単層上で小さなピペットチップを引き摺ることによって、創傷(引っ掻き傷(scratch rift))を形成した。次に、前記細胞を、RGCペプチド(化合物1、10mM)を含有する新鮮な増殖培地中でインキュベートし、創傷閉鎖の動態を判定するために、創傷領域を5つの異なる範囲において様々な時点(0、4、8、12、16、20、24時間)で撮像した。創傷閉鎖のレベルは、細胞の接着、移動および増殖を通じてHUVECによって再び占有された元の創傷領域の画分の測定によって定量した。
【0142】
対照試験では、以下のペプチド、すなわち、環状RGDペプチド(1mM、陽性対照)およびRGEペプチド(1mM、陰性対照)をRGC(化合物1)の代わりに用いた。
創傷治癒の動態に対するペプチドの効果を図1に示す。
【0143】
結果は元の面積のパーセンテージとして示されている。エラーバーは、2〜6回の独立試行における創傷サイズの標準偏差に対応する。
HUVEC創傷閉鎖の動態の結果は、RGCペプチドは24時間後にHUVEC創傷治癒を70%阻害し、一方、環状RGD(RGDに基づくペプチド;N−メチル化環状−RGDf−N(Me)V;シレンジタイド(Cilengitide))は、24時間後、HUVEC創傷治癒を45%阻害することを示している。これらは双方とも、24時間後にHUVEC創傷治癒を0%阻害した陰性対照RGEペプチドと比較した。RGC(化合物1)の効果は、インテグリン結合活性の定評のある阻害剤であるRGDに基づくペプチドの活性に定量的に匹敵する。さらに、RGCペプチドは、HUVEC細胞のアポトーシスを生じることなく、RGDに基づくペプチドの活性に類似した特性を示す。
【0144】
本明細書に別記するように、硝子体と網膜と間の強力な接着は、硝子体黄斑牽引、増殖性糖尿病性網膜症、黄斑円孔、加齢性黄斑変性および飛蚊症のような多くの病理学的硝子体網膜疾患の結果的な発症の原因となり得る。したがって、内部網膜表面からの硝子体の機械的分離以外に、後部硝子体剥離を達成する非外傷性の非侵襲性アプローチは非常に望ましい(テゼル、ティ.エイチ.(Tezel,T.H.)ら、Retina(1998年)18:第7−15頁、およびヴェルシュトレーテン、ティ.シー.(Verstraeten.T.C.)ら、Arch. Ophthalmol.(1993年)第111巻:第849−854頁参照)。
【0145】
本明細書に別記するように、ECM成分、特に硝子体皮質の膠原細線維は、内境界層(ILL:(inner limiting lamina)において、インテグリン結合部位を介して網膜の内面に固定されると考えられている(フース、アール.ワイ.(Foos,R.Y.)、Invest.Ophthalmol.Vis.Sci.(1972年)第11巻:第801−808頁参照)。フィブロネクチンおよびラミニンのような眼内のILLの主な接着性糖タンパク質は、RGD(Arg−Gly−Asp)配列を介してインテグリンに対して強く結びつけられ(カーチス、ティ.エム.(Curtis,T.M.)ら、Am.J.Physiol.、(1995年)第269巻:第L248−L260頁、エルナー、エス.ジー.(Elner, S.G.)ら、IOVS(1996年)第37巻:第696−701頁、およびホルマン、エス.エム.(Horman,S.M)ら、Am.J.Physiol.(1995年)第269巻:第L248−L260頁参照)、いくつかのインテグリンはECMタンパク質中に存在するRGDモチーフを介して結合することも知られている。さらに、ビトロネクチンはαvβ3−特異的であるが、フィブロネクチンはαvβ3に加えて他のいくつかのインテグリンに結合することが知られている。
【0146】
インテグリンのECMへの主な接続はArg−Gly−Asp(RGD)配列を含み、そのRGD配列は、インテグリンヘッドのα−サブユニットとβ−サブユニットとの間に位置する浅い間隙へ結合する(ション(Xiong)ら、Science(2002年)第296巻:第151−155頁参照)。そのような結合は、細胞接着、移動、分化、血管新生および創傷治癒を含む様々な細胞シグナリング経路を調節するのを助ける(ルオスラーチ、イー.(Ruoslahti,E.)ら、Science(1987)第238巻:第491−497頁;およびJ.Clin.Invest.(1991年)第87巻:第1−5頁参照)。
【0147】
硝子体細胞外マトリックス、例えば、膠原細線維は、インテグリン結合部位によって細胞網膜(cellular retina)に接続されるので、RGCペプチド(オリゴペプチド)の硝子体内注入は、同一のインテグリン受容体部位への競合的結合によって、硝子体細胞外マトリックス(vitreous extracellular matrix)のRGDモチーフを細胞網膜から解放し得る。
【0148】
数人の研究者(ルオスラーチ、イー.(Ruoslahti,E.)ら、Science(1987)第238巻:第491−497頁;ハインズ、アール.エイ.(Hynes,R.A.)ら、Cell(1992年)第68巻:第303−322頁;およびハンフリーズ、エム.ジェイ.(Humphries,M.J.)ら、J.Cell Sci.,(1990年)第97巻:第585−592頁参照)は、多くのインテグリン(αvβ3、α5β1、α11β3など)はRGD配列モチーフを有する小さなペプチドによって阻害され得ることを示した。また、それらのαvβ3およびα5β1インテグリンは、ビトロネクチンおよびフィブロネクチンと同様に、ヒトメラノーマ細胞(ニップ、ジェイ.(Nip,J.)、J.Clin.Invest.,(1992年)第90巻:第1406−1413頁参照)、ヒト乳癌細胞(ロン、エル.(Rong,L.)ら、Invest.Ophthalmol.Vis.Sci.(2009年)第50巻:第5988−5996頁参照)、およびヒト網膜色素上皮細胞(ピーター シー.ブルックス(Peter C.Brooks)ら、J.Clin.Invest.,(1995年)第96巻:第1815−1822頁)のような腫瘍においてアップレギュレートされることも文献で裏付けられている。したがって、ヒトメラノーマ細胞の転移能と、αvβ3インテグリン受容体を介したメラノーマ細胞のリンパ節ビトロネクチンへの接着との間には良好な相関があり、前記接着はRGD含有ペプチドによって阻害されたことが示されている(ニップ、ジェイ.J.Clin.Invest.,(1992年)第90巻:第1406−1413頁)。これは、RGDペプチドが重要な抗血管新生薬(anti−angiogenic agents)であり得ることを示している。
【0149】
さらに、ヒト結腸癌細胞系において、細胞接着に有意な増大がある場合、そのとき増大した転移活性があることが示されている(レーマン、エム.(Lehmann,M.)、Cancer Res.,(1994年)、第54巻:第2102−2107頁)。従って、細胞接着を阻害する薬剤は、結腸癌および黒色腫が転移するのを効果的に阻害する。
【0150】
RGCが細胞接着を阻害することが示された創傷治癒の検討の結果に基づき、また黒色腫および結腸癌のモデルにおけるRGDの転移能から推定すると、RGCペプチドおよびその誘導体は、例えば黒色腫および結腸癌において、腫瘍転移を効果的に阻害することができる。
【実施例5】
【0151】
腫瘍に薬剤を誘導または送達するためのRGCペプチドの使用。
この実施例では、下記に示す二量体RGCペプチドパクリタキセル複合体(化合物3)が提供される。この組成物は抗腫瘍薬として有用である。二量体RGCペプチドは、特定の癌細胞において高度に発現されるインテグリン受容体に選択的に結合し、細胞接着を阻害することによって、例えば転移性乳癌のような特定の転移癌を治療するのに有用である。
【0152】
化合物3:
【0153】
【化14】
化合物3の対応するRGD類似体の合成および作用機序、生物分散、並びに腫瘍選択性は、チェン、エックス.(Chen,X.)ら、Synthesis and Biological Evaluation of Dimeric RGD Peptide−Paclitaxel Conjugate as a Model for Integrin−Targeted Drug Delivery、J.Med.Chem.、(2005年)第48(4)巻:第1098−1106頁に記載されている通りである。
【0154】
化合物3は、二量体RGCペプチドに結合された特定の抗腫瘍薬剤パクリタキセルを含むが、本発明のこの態様は、腫瘍または他のインテグリン含有組織もしくは構造の診断、撮像、または治療に有用であり得る任意の実現可能な診断用薬または治療薬に結合されたRGCペプチドの単量体型または多量体型すべてを含むことが理解されるはずである。本発明による単量体または多量体RGCペプチドに結合され得る抗腫瘍物質の例としては、抗腫瘍薬(例えば癌化学療法剤、生体応答調節剤、血管新生阻害剤、ホルモン受容体遮断薬、凍結療法薬(cryotherapeutic agents)、または新生組織形成もしくは腫瘍形成を破壊または阻害する他の薬剤)、例えば、それらのDNAを攻撃することにより癌細胞を直接殺すアルキル化剤または他の薬剤(例えばシクロホスファミド、イソホスファミド)、細胞内DNAの修復に必要な変化を阻害することによって癌細胞を殺すニトロソウレアまたは他の薬剤(例えば、カルマスティン(carmustine)(BCNU)およびロムスチン(lomustine)(CCNU))、特定の細胞機能、通常はDNA合成、に干渉することによって癌細胞増殖を阻止する代謝拮抗物質および他の薬剤(例えば6−メルカプトプリンおよび5−フルオロウラシル(5FU))、DNAを結合するか、またはインターカレートし、RNA合成を妨げることによって作用する抗腫瘍抗生物質および他の化合物(例えばドキソルビシン、ダウノルビシン、エピルビシン(epirubicin)、イダルビシン(idarubicin)、マイトマイシン−C、およびブレオマイシン)、植物から誘導された植物(ビンカ)アルカロイドおよび他の抗腫瘍薬(例えばビンクリスチンおよびビンブラスチン)、ホルモン感受性癌の増殖に影響を与えるステロイドホルモン、ホルモン阻害剤、ホルモン受容体アンタゴニストおよび他の薬剤(例えばタモキシフェン、ハーセプチン、アミノグルテチミド(aminoglutethamide)およびホルメスタンのようなアロマターゼ阻害薬、レトロゾールおよびアナストロゾール(anastrazole)のようなトリアゾール阻害剤、エキセメスタンのようなステロイド阻害剤)、抗脈管形成タンパク質、小分子、遺伝子治療薬、および/または腫瘍の血管形成または血管新生を阻害する他の薬剤(例えばmeth−1,meth−2,サリドマイド)、ベバシズマブ(bevacizumab)(アバスチン(R))、スクアラミン、エンドスタチン、アンジオスタチン、アンギオザイム(Angiozyme)、AE−941(ネオバスタット(Neovastat)(R))、CC−5013(レビミド(Revimid)(R))、medi−522(ビタキシン(Vitaxin)(R))、2−メトキシエストラジオール(2ME2、パンゼム(Panzem)(R))、カルボキシアミドトリアゾール(CAI)、コンブレタスタチンA4プロドラッグ(CA4P:combretastatin A4 prodrug)、SU6668、SU11248、BMS−275291、COL−3、EMD121974、IMC−1C11、IM862、TNP−470、セレコキシブ(セレブレックス(Celebrex)(R))、ロフェコキシブ(バイオックス(Vioxx)(R))、インターフェロンアルファ、インターロイキン−12(IL−12)、または参照によって本願に明示的に援用されるScience第289巻、第1197−1201頁、2000年8月17日において特定された化合物のうちのいずれか、生体応答調節剤(例えばインターフェロン、カルメット−ゲラン桿菌(BCG)、モノクローナル抗体、インタールケン2(interluken 2)、顆粒球コロニー刺激因子(GCSF)など)、PGDF受容体アンタゴニスト、ハーセプチン、アスパラギナーゼ、ブスルファン、カルボプラチン、シスプラチン、カルマスティン、クロラムブシル(cchlorambucil)、シタラビン、ダカルバジン、エトポシド、フルカルバジン(flucarbazine)、フルロウラシル(flurouracil)、ゲムシタビン (gemcitabine)、ヒドロキシウレア、イホスファミド(ifosphamide)、イリノテカン、ロムスチン、メルファラン、メルカプトプリン、メトトレキサート、チオグアニン、チオテパ、トミュデックス(tomudex)、トポテカン(topotecan)、トレオスルファン(treosulfan)、ビンブラスチン、ビンクリスチン、ミトアジトロン(mitoazitrone)、オキサリプラチン、プロカルバジン、ストレプトシン(streptocin)、タキソール、タキソテール(taxotere)、そのような化合物の類似体/同族体および誘導体、並びにここに列記されない他の抗腫瘍薬などが挙げられ得る。
【実施例6】
【0155】
インテグリン表出性腫瘍の画像化用64CU標識多量体RGCペプチド
この実施例において、下記に示す本発明の64CU標識四量体および八量体RGCペプチド(それぞれ化合物4および5)は、画像化および診断の目的(例えば、PET走査のための腫瘍の放射性同位元素標識)のため、並びにαvβ3インテグリンを発現する腫瘍のようなインテグリンを発現する腫瘍または他の細胞に治療薬を誘導または送達するための放射線治療薬として有用である。
【0156】
化合物4:
【0157】
【化15】
化合物5:
【0158】
【化16】
化合物4および5の対応するRGD類似体の合成および作用機序、生物分散、腫瘍選択性およびPETに関連する使用は、リ、ゼット.(Li,Z.)ら、64CU−Labeled Tetrameric and Octameric RGD Peptides for Small−Animal PET of Tumor αvβ3 Integrin Expression、J.Nucl.Med.第48(7)巻、第1162−1171頁(2007年)に記載されている通りである。
【0159】
本明細書において使用される場合、疾患または疾患の治療または治療法に対するいかなる言及も、疾患または障害が生じる前または検知される前のそれらの予防または予防法、並びに疾患または障害が生じた後または検出された後のそれらの治療を含むと解釈されるものとする。
【0160】
本発明は本願において本発明の特定の実施例または実施形態に関して上記に記載してきたが、それらの実施例および実施形態に対して、本発明の意図した趣旨および範囲から逸脱することなく、様々な追加、削除、変更および変更がなされてもよいことが理解されるはずである。例えば、一実施形態または実施例の任意の要素または特質は、そうすることがその実施形態または実施例をその目的の用途に適さないようにする場合について他に特に規定がない限り、別の実施形態または実施例に組み込まれてもよいし、またはそれらと共に用いられてもよい。また方法または過程の工程が特定の順序で記載または列記されている場合、特に他に規定されていない限り、またはそうすることが前記方法および過程をその意図した目的に対して実行不可能にしない限り、そのような工程の順序は変更されてもよい。適当な追加、削除、変更および変更はすべて、記載された実施例および実施形態の均等物と見なされ、以下の特許請求の範囲内に含まれる。本願において引用された刊行物および特許文献はすべて、これにより、参照によって、すべての目的について、あたかも各々が個々にそのように示されたのと同じ程度に余すところなく援用される。
【技術分野】
【0001】
本発明は、概して化学および医学の分野に関連し、より具体的には、Arg−Gly−Asp(「RGD」トリペプチド)結合部位への細胞接着を阻害するために使用可能な組成物および方法、並びに関連する、炎症、創傷治癒、瘢痕形成の防止、血栓症、癌転移および腫瘍、増殖性または非増殖性糖尿病性網膜症、硝子体液の液化、後部硝子体網膜剥離(PVD:posterior vitreo−retinal detachment)の誘発、硝子体網膜疾患、例えば、飛蚊症(floaters)、特発性黄斑円孔、硝子体黄斑牽引、加齢性黄斑変性、滲出型黄斑変性などの病因、脈絡膜血管新生、硝子体網膜手術、静脈閉塞、角膜血管新生、虚血性視神経、虹彩血管新生(rubiosis iridis)、および緑内障手術における瘢痕形成の防止のような疾患の治療に関する。
【背景技術】
【0002】
RGDトリペプチド配列は、多くのタンパク質中に見られ、そこで細胞接着に関与する。RGDトリペプチド配列が存在するタンパク質の例としては、コラーゲン、フィブロネクチン、ビトロネクチン、フォンヴィレブランド因子(VWF:von Willebrand factor)、特定のジスインテグリン、および特定のジスコイジンが挙げられる。
【0003】
インテグリンは、露出したRGD配列を有する配位子に結合することによって細胞と細胞外マトリックス(ECM:extracellular matrix)との間の接着を媒介するヘテロ二量体性細胞表面受容体である。正常なインテグリン−RGD結合は、細胞の成長、移動および生存に関わる遺伝子発現に関与すると考えられている。そのような細胞の成長、移動および生存の不完全な調節は、血栓形成、炎症および癌を含む多くの病態をもたらし得る。したがって、RGDペプチドは、細胞接着タンパク質の可能な模倣体として、またインテグリンに結合するそれらの能力について、アポトーシス、血管新生、および腫瘍形成の阻害のような治療目的のため、内部放射線治療薬並びに癌造影剤としてのそれらの多量体形態における使用のため、並びにそれらの抗癌剤輸送能力について、研究されてきた。
【0004】
眼において、インテグリンは、眼発生(ocular development)、細胞移動、治癒およびいくつかの病的過程を含む多くの過程に影響を与える。インテグリンはまた、眼組織において、炎症および血栓形成を調節し得る。硝子体内に注入されたRGDペプチドはまた動物モデルにおいて後方硝子体網膜剥離を引き起こすことが報告されており、したがって、特定の網膜障害の治療および/または硝子体切除術における硝子体の除去を容易にするため有用であり得る(非特許文献1参照)。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】オリベラ、エル.ビー.(Olivera,L.B.)ら、RGD Peptide−Assisted Vitrectomy to Facilitate Induction of a Posterior Vitreous Detachment:a New Principle in Pharmacological Vitreolysis、Current Eye Research(8):第333−40頁(2002年12月25日)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明はR−G−システイン酸(すなわちR−G−NH−CH(CH2−SO3H)COOHまたはArg−Gly−NH−CH(CH2−SO3H)COOH)およびその誘導体を含む(医薬として許容される塩、水和物、立体異性体、多量体、環状型、直鎖状型、薬物複合体、プロドラッグおよびそれらの誘導体を含む)新規な化合物を提供する。
【課題を解決するための手段】
【0007】
本発明は、ヒトまたは動物の対象において、対象に対してR−G−システイン酸ペプチドまたはその誘導体(医薬として許容される塩、水和物、立体異性体、多量体、環状型、直鎖状型(linear forms)、薬物複合体、プロドラッグおよびそれらの誘導体を含む)を含有する有効量の組成物を投与することによって、RGD結合部位への細胞接着を阻害するため、またはRGD結合部位に他の診断用薬または治療薬を送達するための組成物および方法を提供する。
【0008】
本発明のR−G−システイン酸ペプチドの特定の例としては、直鎖状型のArg−Gly−NH−CH(CH2−SO3H)COOH(本願で化合物1と称される例)および環状型のArg−Gly−NH−CH(CH2−SO3H)COOH(本願で化合物2と称される例)が挙げられる。
【0009】
本発明のR−G−システイン酸誘導体の一般式としては、下記のような一般式I〜VIIを有する化合物が挙げられるが、これらに限定されるものではない。
一般式I:
【0010】
【化1】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択され、かつY=OHまたはNH2である。
【0011】
一般式II:
【0012】
【化2】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択される。
【0013】
一般式III:
【0014】
【化3】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択され、かつZはHまたはSO3Hから選択される。
【0015】
一般式IV:
【0016】
【化4】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択され、YはOHまたはNH2から選択される。
【0017】
一般式V:
【0018】
【化5】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択される。
【0019】
一般式VI:
【0020】
【化6】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択される。
【0021】
一般式VII:
X1−R−G−システイン酸−X
前記式中、XおよびX1は環状または直鎖状の−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはArg、Gly、システイン酸(Cysteic)、Phe、Val、Ala、Leu、Pro、ThrのD−異性体またはL−異性体の任意の組み合わせの任意の塩から選択される。
【0022】
一般式VIIの環状型の例としては
【0023】
【化7】
が挙げられるが、必ずしもこれに限定されるものではない。
【0024】
前記式中、X’はH、C1−C6アルキル、Ph、またはSO3Hから選択され、ZはHまたはMeから選択され、YはOHまたはNH2から選択される。
スルホン酸は対応するカルボン酸より強力な酸である。このスルホン酸基のより高い極性はより強力な分子間結合をもたらす。例えば、より分極したO−H結合を有するR−G−システイン酸は、比較的分極に劣るO−H結合を有するR−G−アスパラギン酸(RGDペプチド)よりも強力な水素結合をインテグリン結合部位のタンパク質のアミド基と形成し、かつ/または、インテグリン結合部位において錯体を形成する金属イオンとのより強力な相互作用を有し得る。
【0025】
本明細書においてより詳細に別記するように、一般式VIIの1つの特定の例であるグリシニル−アルギニル−グリシニル−システイン酸−トレオニル−プロリン−COOH(Glycinyl−Arginyl−Glycinyl−Cysteic−Threonyl−Proline−COOH)(GRDシステイン酸TP、下記で化合物1と称する)を合成して動物において試験したところ、該化合物は、インテグリン−細胞外マトリックス(ECM)相互作用を阻害することにより、網膜表面からの後部硝子体剥離(PVD)を誘発するのに有効であることが判明した。また本明細書においてより詳細に別記するように、化合物1は、ヒト臍静脈内皮細胞(HUVEC)を用いた創傷治癒のモデルにおいて試験したところ、環状RGDペプチドによる40%の阻害と比較して、12時間で細胞接着を74%阻害することが示された。これらの研究は、グリシニル−アルギニル−グリシニル−システイン酸−トレオニル−プロリン−COOHが、インテグリンに対して、RGDペプチド自身よりも、さらにより強力に結合し得る論理的根拠を示唆および確証する。
【0026】
RGシステイン酸ペプチド配列は、L型またはD型のいずれかであり得、インテグリン−ECM相互作用の競合的阻害剤である。RGシステイン酸ペプチド配列は、プロテアーゼ耐性誘導体もしくは環状誘導体のもの、またはプロドラッグ誘導体、もしくは薬物送達システムに関連するもの、またはモノクローナル抗体のものであり得る。
【0027】
本発明の組成物は血管新生を阻害するために使用可能であり、血管新生の阻害は、炎症、創傷治癒、血栓症、癌転移および腫瘍を治療するために有用であり得る。さらに有用な用途は、増殖性または非増殖性糖尿病性網膜症、硝子体の液化(liquefaction of the vitreous)、後部硝子体網膜剥離(PVD)の誘発、例えば特発性黄斑円孔、硝子体黄斑牽引、加齢性黄斑変性、滲出型黄斑変性などの硝子体網膜疾患の病因、脈絡膜血管新生、硝子体網膜手術、静脈閉塞、並びに緑内障手術における瘢痕形成の防止を含む眼科学に見られ得る。さらに、本発明の多量体性かつ/または放射性標識された組成物は、腫瘍の検出のための診断用薬/造影剤、および腫瘍の治療のための放射線治療薬として、並びにそれらの腫瘍指向性による抗癌剤担体として、使用可能である。
【0028】
RGCペプチドを組込む生体材料はまた、細胞の長期生存および増殖に対して、および組織工学および再生医療における応用のための生体組織工学に対して、合成接着微小環境(synthetic adhesive microenvironment)を提供することができる。RGCペプチドは、インテグリン接着受容体を結合するそれらの特性によって、生体材料またはスキャフォールド上に拘束されると、接着促進信号を提供することができる。RGCに基づく物質は、細胞接着、細胞の拡張および移動を媒介する。さらに、インテグリン媒介細胞接着(integrin−mediated cell adhesion)は細胞増殖および生存を促進し、多細胞構造および組織の集合および構築において重要な役割を果たす。
【0029】
ルセンティス(Lucentis)およびアバスチン(Avastin)のような黄斑変性の治療のための薬剤は、他の場合には新たな血管の成長、血管新生を引き起こし、従って黄斑浮腫の一因となるVEGFを阻害することに基づく。小さなペプチドRGDは、ブルックスらに付与された米国特許第6,500,924号に示されているように、競合的結合によって細胞外マトリックス1への細胞付着を阻害することにより、アポトーシスを誘発し得ることが知られている。
【0030】
RGDペプチド結合または認識モチーフは、RGD接着エピトープを認識することによって細胞の細胞内細胞骨格(intracellular cytoskeleton)をECMと結び付ける細胞外マトリックスおよびインテグリンのタンパク質中に見られ得る。例えば、フース、アール ワイ.(Foos,R Y.),Invest.Opthalmol.Vis.Sci.(1972年)第11巻、第801−808頁を参照されたい。細胞は、通常、ECMへ付着することなく、アポトーシスを受ける。
【0031】
一般に、線維芽細胞と細胞外マトリックスの糖タンパク質成分との間の相互作用は、主に細胞表面インテグリン上で相互作用するRGD含有アミノ酸配列によって媒介される深刻な瘢痕形成を引き起こす。また、RGD配列は炎症反応およびホメオスタシス反応の間の細胞−ECM相互作用に関与し(ヘルシュコビッツ、エス.エム.(Hershkoviz,S.M.)ら、Invest.Ophthalmol.Vis.Sci.、(1994年)、第35巻、第2585−2591頁参照)、インテグリンは、創傷治癒または病的過程における細胞移動、並びに炎症および血栓形成の調節に重要な役割を果たすことも知られている。したがって、RGDペプチドのような強力なインテグリンアンタゴニストは、抗炎症剤、抗転移剤、または抗血栓剤のような薬物として非常に有用であり得る(エルナー、エス.ジー.(Elner,S.G.)およびエルナー、ヴィー.エム.(Elner,V.M.)、IOVS(1996)第37巻:第696−701頁参照)。ヒアルロン酸の受容体であるCD44は、ヒアルロン酸に対するその親和性によって、おそらくまたオステオポンチン、コラーゲンおよびマトリックスメタロプロテアーゼ(MMP)のような他の配位子に対するその親和性によって、細胞−細胞および細胞−基質相互作用を媒介することも文献に報告されている。
【0032】
ヒアルロナンとの接着は、細胞移動、腫瘍の成長および進行に重要な役割を果たし、またリンパ球の活性化、再循環、およびホーミング、並びに造血にも関与する。発現変化(altered expression)または機能不全は多くの病理表現型を引き起こす(例えば、ジャン、ディー.(Jiang D.)、Annu.Rev.Cell.Dev.Biol.(2007年)第23巻:第435−461頁;およびナドソン、ダブリュ.(Knudson,W.)ら、Matrix Bio.(2002年)、第21巻:第15−23号参照)。
【0033】
最近、CD44と細胞マトリックス成分(例えばHA)との相互作用が様々な炎症性疾患の発症に大きな役割を果しており、また、ヒアルロナン−CD44相互作用の断絶は脈絡膜血管新生の改善をもたらすことが示された(ヒロシ モチマル(Hiroshi Mochimaru)ら、Invest.Ophthalmol.Vis.Sci.(2009年)第50巻:第4410−4415頁参照)。
【0034】
これらの証拠は、RGDペプチドまたはCD44のような接着分子が細胞−細胞および細胞−ECM相互作用において多くの病原性疾患(pathogenic diseases)の発症に重要な役割を果たし、前記相互作用の阻害がその疾患を治療および治癒する上における新規な治療標的であり得ることを示している。
【0035】
合成ペプチドもまたインテグリンおよび成長因子に結合することが示されている。RGD配列を含む環状ペンタペプチドは、ビトロネクチンのαvβ3インテグリン(integin)への結合(マイケル エイ.デチャンストレイター(Michael A.Dechantsreiter)ら、J.Med.Chem.(1999年)第42巻:第3033−3040頁参照)、およびビトロネクチンおよびフィブロネクチンの双方のαvβ3およびαIIbβ3インテグリンへの結合(ローランド ハウブナー(Roland Haubner)ら、J.Am.Chem.Soc,(1996年)第118巻:第7461−7472頁参照)を阻害することが明らかとなった。この阻害は、多くの関連のない疾患の治療に有用であるこが示された。ハムスターによる研究において、環状ペンタペプチドは、対照動物と比較して、腫瘍の成長およびメタセシスを遅延させた(エム.エイ.バークル(M.A.Buerkle)ら、British J.Cancer (2002年)第86巻:第788−795頁参照)。ペンタペプチドはまた、鎌状赤血球細胞の血管内皮への結合を低減し、血行動態挙動を改善したことが示されている(アイリーン エム.フィンネガン(Eileen M.Finnegan)ら、Am.J.Physiol.Heart Circ.Physiol.,(2007年)第293巻:第H1038−H1045頁参照)。RGD配列を含む別の環状ペプチドは、炎症反応性および免疫反応における白血球結合(leucocyte binding)に関与することが知られているインテグリンであるα4β1に強力に結合することが示された。(ピーナ エム.カルダレッリ(Pina M.Cardarelli)ら、J.Biol.Chem.(1994年)第269巻:18668−18673頁参照)。合成硫酸化テトラペプチド(synthetic,sulfated tetrapeptide)はVEGFに強力に結合することが示されている(メイナード、ジェイ.エイ.ハベル(Maynard,J.A.Hubbell)、Acta Biomaterialia(2005年)第1巻:第451−459頁参照)。
【0036】
さらに、重要で有用な用途において、二量体RGDペプチド−薬物複合体は、腫瘍を標的とするためのインテグリン標的薬物送達に有用であることが証明されている(チェン(Chen)ら、J.Med.Chem.,(2005年)第48(4)巻:第1098−1106頁参照)。
【0037】
別の等しく重要かつ有用な用途において、多量体放射性標識RGDペプチドは、腫瘍検出のための診断用薬/造影剤として、並びにインテグリンαvβ3を標的とすることによる腫瘍に特異的なターゲティングおよび治療のための放射線治療薬として有用であることが証明されている(ツィ−ボ リ(Zi−Bo Li)ら、J.Nucl.Medicine,(2007年)第48巻:第1162−1171頁参照)。
【0038】
眼科学において、線維芽細胞による創傷治癒における瘢痕形成は、特に緑内障において、主な問題のうちの1つである。これは線維芽細胞とECMの糖タンパク質成分と間の相互作用に起因する。ECM糖タンパク質の認識は、Arg−Gly−Asp(すなわちRGD)配列のような、接着エピトープに特異的な細胞表面インテグリンを介して起こる。フィブロネクチン(FN)およびビトロネクチン(VN)を含む血漿たんぱくのいくつかのマトリックス中に存在するRGD配列は、炎症反応およびホメオスタシス反応の間に生じる細胞−ECM相互作用に関係する。線維芽細胞とECMの糖タンパク質との間の相互作用の抑制は瘢痕形成を緩和した(ラミ ヘルシュコビッツ(Rami Hershkoviz)ら、Invest.Ophthalmol.Vis.Sci.(1994年)第35巻:第2585−2591頁参照)。
【0039】
後部硝子体皮質の膠原細線維は、硝子体網膜界面、特に網膜表面の内境界層(inner limiting lamina)に付着する(セバーグ ジェイ.(Sebag J.)、Eye(1992年)、第6巻:第541−552頁参照)。硝子体基部、視神経円板(optic disc)における、主要な網膜血管に沿った、後極全体に対して面での(in facial manner)付着は、特発性黄斑円孔、硝子体黄斑牽引、増殖性糖尿病性網膜症などのような硝子体網膜疾患の病因において重要な役割を果たす(アキバ ジェイ.(Akiba J.) キロス エム.エイ.(Quiroz M.A.)ら、Ophthalmol.(1990年)第97巻、第1619−1655頁)。
【0040】
血管新生抑制は、ともに眼血管の過剰成長に起因する糖尿病性網膜症および黄斑変性について早期に有望であることが示されている。これらの疾患において、前記血管は眼内の正常組織に干渉したり、またはそれらの血管は光が眼の後部へ到達するのを遮断したりする。新たな血管はそれら自身が主な病的状態であり、血管成長の停止が失明を防止し得る。したがって、血管抑制(angioinhibition)は、単に病気の治療だけではなく、治癒になり得る。更に、網膜からの硝子体の分離は黄斑の牽引を緩和して、黄斑円孔の形成の危険性を低減し得ると仮定されてきた。従って、硝子体網膜界面上のインテグリンに結合するフィブロネクチンおよびラミニンの抑制による後部硝子体剥離は、糖尿病性網膜症および網膜静脈閉塞を有する眼における網膜血管新生を防止し得る(アキバ ジェイ.(Akiba J.)、キロス エムエイ(Quiroz MA)、 Ophthalmol.(1990年)第97巻、第1619−1655頁;ケリー エヌ.イー.(Kelly N.E.)ら、Ophthalmol.(1991年)第109巻、第654−659頁;およびカド エム(Kado M)ら、Am.J.Ophthalmol.(1988年)第105巻:第20−24頁参照)。
【0041】
近年、硝子体外科手術は、硝子体網膜牽引を軽減し、かつ網膜浮腫を低減するために非常に改善されてきた。外科技術および機器における継続的な改善にもかかわらず、網膜裂孔、網膜剥離および網膜神経繊維損傷などのような合併症により、特に一部の糖尿病性網膜症の患者および小児患者において、硝子体皮質の非外傷性除去を達成することは依然として困難なままである(セバーグ ジェイ.(Sebag J.)、Arch.Clin.Exp.Ophthalmol.(1987年)第225巻:第89−93頁;ハン ディ.ピー.(Han D.P.)ら、Arch.Ophthalmol.(1998年)第106巻:998−1000頁参照)。したがって、網膜を損なうことなく、硝子体界面を選択的に切り裂く外傷の少ないアプローチは非常に望ましい。近年、硝子体網膜界面の分離のための多くの薬物に関する報告が文献に現れてきている(トレーズ エム.ティ.(Trese M.T.)、Eye.(2002年)第16巻:第365−368頁;ガンドルファー エイ.(Gandorfer A.)ら、Am.J.Ophthalmol.(2002年)第133巻:第156−159頁;およびヘッセ エル.(Hesse L)ら、Eye Res.(2000年)第70巻:第31−39頁参照)。ヒアルロニダーゼ(カラジョージアン(Karageozian)らに付与された米国特許第6,863,886号参照)および自己由来プラスミン(サクマ ティ(Sakuma T)ら、日本眼科学会雑誌(2003年)、第107巻:第709−718頁)のような酵素を用いた薬理学的硝子体索切断は、細胞外マトリックスの消化(digestion)を促進し、後部硝子体剥離を誘発することがこれまでに研究されてきた。しかしながら、用いられた酵素による隣接組織の非特異的な破壊が、それらの治療適用の成功を妨げている。ここ数年において、尿素のような非酵素薬物(non−enzymatic pharmacologic agents)(ニッカーソン、シー.(Nickerson,C.)ら、J.Biomechanics、(2008年)第41巻:第1840−1846頁、およびMacromol.Symp.(2005年):第183−189頁参照)およびRGDペプチド(レオナルド ビー.オリビエラ(Leonardo B.Oliviera)ら、Curr.Eye Res.(2002年)第25巻:第333−340頁参照)を用いた新規なアプローチが、硝子体網膜界面の分離に注目することによって研究されてきている。RGDペプチドの合成類似体は、インビトロ(ウィリアムズ ジェイ.エイ.(Williams J.A.)、Pathol.Bio.(1992年)第40巻:第813−821頁;ゲールセン ケイ.アール(Gehlsen K.R.)ら、J.Cell.Biol.(1988年)第106巻:第925−930頁;ピアシュバッヘル、エム.ディ.(Pierschbacher,M.D.)ら、J.Biol.CHem.,(1987年)第262巻:第17294−17298頁;およびゾーン エル.エル.(Zhon L.L.)ら、IOVS.(1996年)第37巻:第104−113頁)およびインビボで、ECMタンパク質のRGDモチーフがインテグリン−ECM相互作用を妨害し、かつ付着を緩めるのに競合することが示されている。従って、可溶性RGDペプチドの硝子体内注入は、ウサギモデルにおいて、網膜表面の不溶性ECMタンパク質からのRGDエピトープの解放をもたらし、従って非酵素PVDを容易にした。明らかに、これらの結果は、硝子体網膜界面が、ECMのRGDモチーフへのインテグリン接続、並びに硝子体皮質コラーゲンの内境界層(ILL:inner limiting lamella)への接着に関与することを示している。RGDペプチドおよびその誘導体は、創傷における上皮細胞の移動を促進し(ピー.エム.メルツ(P.M. Mertz)ら、J.Burn Care Res.(1996年)第17巻:第199−206頁参照)、ヒドロゲル(エムピー ルットルフ(MP Lutolf)ら、Proc.Nat.Acad.Sci.(2003年)第100巻:第5413−5418頁:エムピー ルットルフ(MP Lutolf)ら、Nature Biotechnol.(2003年)第21巻:第513−518頁参照)、他のポリマーマトリクス(ホーン−バン リン(Horng−Ban Lin)ら、J.Biomed.Material.Res.(2004年)第28巻:第329−342頁参照)のような合成生体材料に組み込まれた場合、または固い物質(hard substance)上の表面膜として(ディ.エム.フェリス(D.M.Ferris)ら、Biomaterials(1999年)第20巻:第2323−2331頁)、それらの生物活性を維持する。RGDペプチドはまた、ペプチド配列で被覆された人工血管(ケイ.ワルシェク(K.Walluscheck)ら、Eur.J.Vascular and Endovascular Surgery(1996年)第12巻:第321−330頁参照)および他の人工臓器(ジェスケ、ブリゲッテ(Jeschke,Brigette)、Biomaterials(2002年)第23巻:第3455−3463頁)への上皮細胞または内皮細胞の増大した接着を促進し、神経再生を支持することが示されている(エム.ラフィウディン アフマッド(M.Rafiuddin Ahmed)ら、Brain Res.(2003年)第993巻:第208−216頁参照)。前記人工器官の生物活性表面は合成樹脂繊維またはポリマーを含むことができる。
【図面の簡単な説明】
【0042】
【図1】創傷治癒の動態における、3つのペプチド、すなわち、環状RGDペプチド、RGCペプチド(化合物1)、RGEペプチドの効果を示す図。
【図2】RGCペプチド(化合物1)のHPLCクロマトグラムを示す図。
【図3】RGCペプチド(化合物1)のエレクトロスプレー質量クロマトグラムを示す図。
【図4】環状RGCペプチド(化合物2)のHPLCクロマトグラムを示す図。
【図5】環状RGCペプチド(化合物2)のエレクトロスプレー質量クロマトグラムを示す図。
【発明を実施するための形態】
【0043】
本発明は、上記の一般式I〜VIIの化合物を含む新規な化合物を提供する。本発明の特定の例としては、直鎖状型のArg−Gly−NH−CH(CH2−SO3H)COOH(本願では化合物1と称される例)および環状型のArg−Gly−NH−CH(CH2−SO3H)COOH(本願では化合物2と称される例)、並びに、医薬として許容される塩、水和物、立体異性体、多量体(mutimers)、環状型、直鎖状型、多量体型(multimeric forms)、薬物複合体、プロドラッグおよびそれらの誘導体を含む、それらの誘導体が挙げられる。
【0044】
化合物1および2の合成
当業者に既知の従来の固相ペプチド合成(SPPS:solid−phase peptide synthesis、アール.ビー.メリフィールド(R.B.Merrifield)、J.Am.Chem.Soc.(1963年)第85(14)巻:第2149−2154頁)が実施され得る。SPPSは高収率なので好ましい合成の方法である。一般に、固相ペプチド合成技術の第1段階は、高分子支持体(polymeric support)上における保護されたアミノ酸誘導体によるペプチド鎖集合から成る。前記技術の第2段階は、粗製自由ペプチドを与える、すべての側鎖保護基の同時切断による樹脂支持体からのペプチドの切断である。SPPSの一般的な原理は、カップリング−脱保護の繰返しサイクルのうちの1つである。固相付着ペプチドの自由N末端アミンは、単一のN−保護アミノ酸ユニットに結合される。次に、このユニットは脱保護され、新たなN末端基アミンを表す。前記N末端基アミンにはさらにアミノ酸が付着され得る。合成、保護および脱保護の方法については、Asymmetric Synthesis、フォン ジー.エム.コッポラ(Von G.M.Coppola)およびH.F.シュースター(H.F.Schuster)著、ジョン・ワイリー・アンド・サンズ(John Wiley & Sons)、ニューヨーク、1987年を、保護および脱保護の方法については、Greene’s Protective Groups in Organic Synthesis、ピーター ジー.エム.ウッツ(G.M.Wuts)およびシアドーラ ダブリュ.グリーン(Theodora W.Greene)著(第二版)、ジェイ.ワイリー・アンド・サンズ(J.Wiley & Sons)(1991年)を参照されたい。
【0045】
固相ペプチド合成の2つの主に用いられる形態、すなわちFmoc(9−フルオレニルメチルオキシカルボニル;塩基に不安定な(base labile)アルファ−アミノ保護基)およびt−Boc(t−ブチルオキシカルボニル;酸に不安定な保護基)のうち、本ペプチドの合成には、好ましくはFmocが用いられ得る。各方法は、異なる樹脂およびアミノ酸側鎖保護、並びに結果として生ずる切断/脱保護工程を伴う。前記樹脂からの切断後、ペプチドは、通常、C−18、C−8およびC−4のようなカラムを用いた逆相HPLCによって精製される。
【0046】
固相ペプチド合成の例
下記は、塩基に不安定な9−フルオレニルメチルオキシカルボニル(Fmoc)保護基を用いて、固体保持体としてのワング樹脂(Wang resin)上におけるペプチド合成のための合成工程の概要である。
【0047】
Fmoc脱保護
0.08mmolのFmoc−Pro−ワング樹脂を、プラスチックキャップを装備したフリットカラム(fritted column)内に充填する。前記樹脂を2×3mL量のDMF(ジメチルホルムアミド)で各々1分間にわたって洗浄する。次に、約3mLの20%ピペリジン/DMF溶液を添加し、Fmoc脱保護を15分間にわたって継続させる。この時間の間、カラムを優しく回すか、または撹拌して、確実に完全に混合する。反応が完了した後(約15分)、反応カラムを流出させて、前記樹脂をDMF(4×3mL)で再び洗浄する。
【0048】
アミド結合カップリング
次に、所望のFmoc保護アミノ酸であるFmoc−Thr−tBu(3当量:供給者によって表示された樹脂充填量に対して)およびDIEA(6当量)のDCM溶液(アミノ酸に対して0.5M)を前記樹脂に添加する。その混合物を−20°Cで20分間にわたって冷却する。次に、ベンゾトリアゾール−1−イル−オキシトリピロリジノホスホニウムヘキサフルオロホスファート(PyBOP)、固相ペプチド合成に用いられるペプチドカップリング試薬(3当量)を前記反応に添加する。−20°Cで8時間にわたって振盪した後、反応混合物を流出させて、前記樹脂をDCM(3×)で洗浄する。
【0049】
20%ピペリジン/DMF溶液を用いたFmoc脱保護(15分)およびDMF(3×)による洗浄の後、次のFmoc保護アミノ酸(3当量;樹脂充填量に対して)であるPyBopを上記と同じ方法でカップリングする。
【0050】
切断
遊離酸型のペプチドを得るために、エステル結合をTFA(トリフルオロ酢酸)のような強い酸性条件を用いて切断する。前記樹脂を2〜3mLのトリフルオロ酢酸および水の95:5の溶液で処理する。その樹脂を25分間にわたって優しく撹拌する。次に、カラムを流出させて、濾液をガラス収集容器に慎重に収集にする。
【0051】
化合物1(GRGシステイン酸TP)の合成:
【0052】
【化8】
工程1. 樹脂の充填:プロリンで事前に充填されたo−クロロトリチル樹脂を出発材料として用いる。
【0053】
工程2. ペプチド集合:Fmoc合成を用いてペプチドを集合させる。保護されたアミノ酸をPyBOPで活性化し、末端Fmoc基を20%ピペリジン/DMF溶液で除去する。下記の保護されたアミノ酸は、それらが現われる順番で、順番に用いられる:
a.Fmoc−Thr−tBu(Fmocトレオニン−t−ブチルエステル)
b.Fmoc−システイン酸−Pfp(Fmocシステイン酸−ペンタフルオロフェニルエステル)
c.Fmoc−Gly(Fmocグリシン)
d.Fmoc−Arg−Pbf(Nα−Fmoc−Nω−(2,2,4,6,7ペンタメチルジヒドロベンゾフラン−5−スルホニル)−L−アルギニン)
e.Fmoc−Gly(Fmocグリシン)
工程3. 樹脂からのペプチド切断:結果として生じたペプチドは固体支持体から切断され、保護基は85.5%のTFA、5%のフェノール、2%の水、5%のチオアニソールおよび2.5%のエタンジチオールの溶液によって除去される。
【0054】
工程4. 精製:高性能液体クロマトグラフィー(HPLC)を用いて、生じたRGCペプチドを精製する。
上述のように調製した一定量の化合物1は、高性能液体クロマトグラフィーによって、純度>98%area/areaであると分析された[HPLC条件: バッファーA:0.1%のトリフルオロ酢酸(TFA)水溶液、バッファーB:0.1%のTFAアセトニトリル溶液、移動相(MP)A:97%のバッファーAおよび3%のバッファーB、移動相B:79%のバッファーAおよび21%のバッファーB、移動相C:50%のバッファーAおよび50%のバッファーB、勾配については、下記の表1を参照のこと、流速:1.0mL/分、カラム:ウォーターズ シンメトリー(登録商標)C18、5μ、4.6×250mm、カラム温度:30°C、検出器:UV@220nm、試料注入体積:20.0μL、試料調製:20μLの試料を1.0mLの移動相Aで希釈(約0.5mg/mL)]。対応するHPLCクロマトグラムを図2に示す。さらに、ペプチド配列の合成のための対応するアミノ酸の段階的な添加に基づいて、精製した化合物1の分子量は、エレクトロスプレイ質量分析法によって638.3amuであると測定され(理論質量:637.7amu)、化合物1の同一性を確認した。化合物1のエレクトロスプレー質量スペクトログラムを図3に示す。
【0055】
表1:HPLCによる化合物1を検出するためのポンプ勾配プログラム
【0056】
【表1】
化合物2(シクロ−RGシステイン酸fN(CH3)V)の合成:
【0057】
【化9】
工程1. 樹脂の充填:o−クロロトリチル樹脂を出発材料として用いる。Fmoc−N −メチル−L−Valを樹脂に付着させる。
【0058】
工程2. ペプチド集合:2.Fmoc合成をペプチド集合に用いる。保護されたアミノ酸をPyBOPで活性化し、末端Fmoc基を20%ピペリジン/DMF溶液で除去する。下記の保護されたアミノ酸は、それらが現われる順番で用いられる:
a.Fmoc−Phe(Fmoc−フェニルアラニン)
b.Fmoc−システイン酸−PfP(Fmocシステイン酸ペンタフルオロフェニルエステル)
c.Fmoc−Gly(Fmoc−グリシン)
d.Fmoc−Arg−Pbf(Nα−Fmoc−Nω−(2,2,4,6,7ペンタメチルジヒドロベンゾフラン−5−スルホニル)−L−アルギニン)
工程3. 樹脂からのペプチド切断:ペプチドを酢酸/TFA/DCM(1:3:3)を用いて固体支持体から切断する。
【0059】
工程4. 所望の環状ペプチドへの環化および脱保護:高希釈下のジフェニルホスホリルアジド(diphenylphosphorylazide)および重炭酸ナトリウムを用いたインサイチュー活性化による環化。側鎖を、85.5%のTFA、5%のフェノール、2%の水、5%のチオアニソール、2.5%のエタンジチオールを用いて脱保護する。
【0060】
工程5.精製:HPLCを精製に用いる。
上述のように調製した一定量の化合物2は、高性能液体クロマトグラフィーによって、純度>99%area/areaであると分析された(HPLC条件: 移動相A:0.1%のトリフルオロ酢酸(TFA)水溶液、B:(80%のアセトニトリル+20%の水)中に0.1%のTFA、20分間で26%から36%のBへ勾配、流速:1.0mL/分、カラム:フェノメネクス(Phenomenex)C18(2)4.6×150mm、5μ、100A、検出器:UV@220nm、試料注入体積:100.0μL)。対応するHPLCクロマトグラムを図4に示す。さらに、ペプチド配列の合成のための対応するアミノ酸の段階的な添加に基づいて、精製した化合物2の分子量は、エレクトロスプレー質量分析法によって625.3amuであると測定され(理論質量:625.77amu)、化合物2の同一性を確認した。化合物2のエレクトロスプレー質量スペクトログラムを図5に示す。
【0061】
細胞接着を阻害するための方法
本発明はまた、対象に有効量のR−G−システイン酸(すなわち、直鎖状型のR−G−NH−CH(CH2−SO3H)COOHまたは環状型のR−G−NH−CH(CH2−SO3H)COOH)およびその誘導体(医薬として許容される塩、水和物、立体異性体、多量体、環状型、直鎖状型、薬物複合体、プロドラッグおよびそれらの誘導体を含む)を投与することによって、ヒトまたは動物の対象におけるRGD結合部位への細胞接着を阻害する方法を提供する。
【0062】
本出願人は、ECM成分への細胞付着を競合的に阻害することにより、本発明の合成RGシステイン酸ペプチドがアポトーシスを誘発することを発見した。従って、本発明の合成RGシステイン酸ペプチドおよびその誘導体は、血管新生、炎症、癌転移、血栓形成、瘢痕形成の防止および治療に対する治療薬、並びに薬学的硝子体索切断剤として、強力なインテグリンアンタゴニストとして用いることができる。さらに本発明の重要な態様において、腫瘍検出(診断用薬または造影剤)および腫瘍治療用の内部放射線治療薬として使用するための、多量体化され、かつ放射性標識されたRGCペプチドを用いたα,βインテグリンの改善されたターゲティングが想定される。別の重要な態様において、改善されたRGCペプチド複合体または多量体RGCペプチド複合体は、薬剤担体として、例えば、効率的な腫瘍ターゲティングのための抗癌剤担体として機能する。
【0063】
本願において別記するように、RGCペプチド中のスルホン酸はRDGペプチド中の対応するカルボン酸よりも強力な酸である。このスルホン酸基のより高い極性は、より強力な分子間結合をもたらす。例えば、より分極したO−H結合を有するR−G−システイン酸は、比較的分極に劣るO−H結合を有するR−G−アスパラギン酸よりも強力な水素結合をインテグリン結合部位のタンパク質のアミド基および/またはアミノ酸の側鎖と形成し、かつ/またはインテグリン結合部位において錯体を形成する金属イオンとのより強力な相互作用を有し得る。従って、本発明の新規なRGCペプチドおよびその誘導体は、インテグリンレセプター認識および結合において、対応するRGDペプチドに対して、改善された化合物および組成物を提供する。
【0064】
加えて、高いIOPに関連する慢性高血糖症を有する糖尿病患者において、開放隅角緑内障は、特に、小柱網組織におけるフィブロネクチンの蓄積に関係することが報告されており、過剰のフィブロネクチンは房水流出を阻害すると考えられる(オシタリ、ティ(Oshitari,T)ら、Am.J.Ophthalmol.(2007年)第143巻:第363−365頁参照)。小柱網での細胞−ECM相互作用におけるフィブロネクチンの関与は、原発性開放隅角緑内障において示されている(マーク エス.フィラ(Mark S.Filla)ら、Invest.Ophthalmol.Vis.Sci.(2002年)第43巻:第151−161頁参照;およびチェリール アール.ハン(Cheryl R.Hann)ら、Ophthalmic Res.(2001年)第33巻:第314−324頁参照)。シュレム管の内皮細胞は、流出能に影響を及ぼすのに細胞外マトリックスと相互作用することも報告されている(シンディー ケイ.バーレー(Cindy K.Bahler)ら、Invest.Ophthalmol.Vis.Sci.(2004年)第45巻:第2246−2254頁参照)。フィブロネクチンのような細胞外マトリックス成分の過剰の存在による房水流出の抑制を踏まえると、糖尿病患者の高いIOPを治療するRGシステイン酸ペプチドおよびその誘導体の適用は、糖尿病患者にとって非常に有益であろう。
【0065】
好ましいRGシステイン酸ペプチドは、融合ポリペプチド、環状または直鎖状ポリペプチド、薬物送達システムまたは例えば抗癌剤のような他の薬剤で誘導された、またはそれらと関連付けられた、またはそれらと結合されたRGシステイン酸ペプチドを含む誘導ポリペプチド、多量体化RGCペプチド、インテグリン結合部位またはその機能的フラグメントと免疫反応するRGシステイン酸配列を含むモノクローナル抗体であり得る。
【0066】
ポリペプチドを含むRGシステイン酸は、フィブリノゲン、フィブロネクチン、ビトロネクチン、フォンヴィレブランド因子、ラミニン、トロンボスポンジンおよび同様な配位子中に存在するもののような天然インテグリン接着結合領域のアミノ酸残基配列に対応する配列を有し得る。
【0067】
本ペプチド配列は、末端グアニジノ基、スルホン基、およびカルボン酸基を有する3つのアミノ酸と、ペプチドフラグメント、糖タンパク質、およびPEG、プルロニックおよび他のポリマー基のようなポリマー基、並びにリポソームおよびナノ粒子を含む薬物送達システムに結合および/または関連付けられたそれらの誘導体とから成る。様々な病理学的疾患の治療のためにそれらを含む医薬組成物は、注入可能な剤形、ゲル剤形、懸濁液剤形、軟膏剤形、固体剤形および液体剤形を備える。
【0068】
フィブロネクチン、ビトロネクチン、ラミニン、フィブリノゲン、トロンボスポンジンおよびフォンヴィレブランド因子のような細胞接着モチーフに関連付けられたインテグリン受容体は、RGシステイン酸ペプチドおよびその誘導体の標的エピトープである。トリペプチドのRGシステイン酸は、細胞結合ドメインによって認識可能な最小アミノ酸配列として発見された。この配列はまた、インテグリンに無関係な免疫機能に干渉することもできる。よって、合成RGシステイン酸配列はRGD細胞結合ドメインを模倣するはずであり、アスパラギン酸のα−炭素上おける置換は、標的インテグリンへのより強力な結合親和性を与えることが発見された。RGCペプチド中のスルホン酸は、RDGペプチド中の対応するカルボン酸より強力な酸である。このスルホン酸基のより高い極性はより強力な分子間結合をもたらす。例えば、より分極したO−H結合を有するR−G−システイン酸は、比較的分極に劣るO−H結合を有するR−G−アスパラギン酸よりも強力な水素結合をインテグリン結合部位のアミド基および/またはアミノ酸の側鎖と形成し、かつ/または、インテグリン結合部位において錯体を形成する金属イオンとのより強力な相互作用を有し得る。
【0069】
本発明のRGシステイン酸配列用の最も一般的な式は、以下の通りである。
式A:
【0070】
【化10】
前記式中、X=−CH(R1)−S(=O)2−Y、−CH(R1)−SH、−CH(R1)−OZ、−CH(R1)S(=O)Y、−CH(R1)−O−S(=O)2−OX1、および−CH(R1)−O−P(=O)2OX1であり、かつ
前記式中、Y=OX1、NH2であり、X1=−H、C1−C6直鎖アルキル、フェニルであり;
R1=H、C1−C6直鎖アルキル、フェニルまたはSO3Hであり;
Z=H、SO3Hである。
【0071】
式B:
【0072】
【化11】
前記式中、X=−CH(R1)−S(=O)2−Y、−CH(R1)−SH、−CH(R1)−OZ、−CH(R1)S(=O)Y、−CH(R1)−O−S(=O)2−OX1、および−CH(R1)−O−P(=O)2OX1であり、かつ
前記式中、Y=OX1、NH2であり、X1=−H、C1−C6直鎖アルキル、フェニルであり、R1=H、C1−C6直鎖アルキル、フェニルまたはSO3Hであり、Z=H、SO3Hであり、かつ、A2は、−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはそれらの塩もしくはN−アルキル化誘導体のうちから選択される。Arg、Gly、システイン酸、Phe、Val、Ala、Leu、Pro、ThrのD型またはL型、並びに上記配列の環状型の任意の組み合わせを用いることができる。
【0073】
環状型の例としては下記式Cが挙げられる。
式C:
【0074】
【化12】
前記式中、X’は、H、C1−C6アルキル、Ph、SO3Hのうちから選択され、Y=OH、NH2であり、かつZ=H、CH3である。
【0075】
環状型としては、ペンタペプチドおよびヘプタペプチド、例えば下記に示される一般式Cの特定な化合物、すなわち、化合物2(シクロRGシステイン酸fN(CH3)V)も挙げられる。
【0076】
【化13】
式Aは、本願に別記する一般式I〜VIを包含する。式Bは、本願に別記する一般式VIIを包含する。
【0077】
式D:
A1−Arg−Gly−NH−CH(X)−CO−A2
前記式中、X=−CH(R1)−S(=O)2−Y、−CH(R1)−SH、−CH(R1)−OZ、−CH(R1)S(=O)Y、−CH(R1)−O−S(=O)2−OX1、および−CH(R1)−O−P(=O)2OX1であり、かつ
前記式中、Y=OX1、NH2であり、X1=−H、C1−C6直鎖アルキル、フェニルであり、R1=H、C1−C6直鎖アルキル、フェニルまたはSO3Hであり、Z=H、SO3Hであり、かつ、A1およびA2は、−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはそれらの塩もしくはN−アルキル化誘導体のうちから選択される。Arg、Gly、システイン酸、Phe、Val、Ala、Leu、Pro、ThrのD型またはL型、並びに上記配列の環状型の任意の組み合わせを用いることができる。
【0078】
置換RGシステイン酸配列としては、環状RGシステイン酸類似体が挙げられる。
RGシステイン酸およびその誘導体の投与は、薬物送達システムまたは任意の医薬として許容される剤形の注入製剤または固体製剤または軟膏製剤を用いることにより、皮下に、皮膚科学的に、眼科的に、および全身的に行うことができる。
【0079】
本発明の化合物は、経口、直腸、静脈内、動脈内、皮内、皮下、筋肉内、鞘内、舌下、口腔内、鼻腔内、経粘膜、経皮的、局所的、眼内、硝子体内、他の腸内、他の非経口、および/または他の可能な投与経路を含むが、これらに限定されない、意図した治療効果をもたらすのに適当な任意の経路によって投与され得る。
【0080】
本発明の化合物は、副作用または有毒作用を回避しながら、意図した治療効果を提供するいかなる投与量で投与されてもよい。本発明の化合物のヒト対象に投与されてもよい典型的な投与量は、約1ng/kg〜約1.0g/kgの範囲にある。
【0081】
可能かつ適当な場合には、本発明の化合物は、任意でリポソームまたはナノ粒子(例えばナノカプセル)の形態で調製されていてもよい。リポソームの形成および使用は、一般に当業者には知られている。リポソームは、それらが多重ラメラ小胞(MLVs:multilamellar vesicles)と称される場合がある多重ラメラ同心状二層小胞(multilamellar concentric bilayer vesicles)を自発的に形成するように、水性媒体中に分散されたリン脂質から形成される。MLVは、典型的には25nm〜4μmの直径を有する。超音波で処理した場合、MLVは、水溶液を含む核を有する、直径約200〜500オングストロームの小さな単層小胞(SUVs:small unilamellar vesicles)を形成する。一般に、リン脂質は、水性媒体中に分散された場合、脂質の水に対するモル比に応じて、リポソーム以外の様々な構造を形成することができる。脂質対水のモル比が低い場合には、リポソームが形成するであろう。リポソームの物理的特性は、pH、張度、および2価カチオンの有無に依存する。リポソームは、1)飲食作用(例えば、マクロファージおよび好中球のような細胞によるリポソームの食作用)、細胞表面への吸着、2)細胞表面成分との相互作用、3)原形質膜へのリポソームの脂質二重層の挿入による原形質細胞膜との融合、または4)リポソーム脂質の細胞膜または細胞内膜への移動またはその逆の移動、を含む様々な機構によって細胞と相互作用し得る。リポソーム製剤を変更することによって、同リポソームが副鼻腔、鼻粘膜などにおいて細胞と相互作用する機構を変更することができる。
【0082】
ナノカプセルは、外殻と、所望の物質が配置され得る空間とから成る任意のナノ粒子である。ナノカプセルを形成するための技術は当該技術分野において知られている。高分子ナノカプセルは特定の寸法および形状で製造され得る。前記高分子ナノカプセルは、物理的および化学的性質を正確に定めた単分散粒子として生成され得、したがって、pH、粘液の流れ、あるいはデバイスが植え込まれる副鼻腔内または耳、鼻もしくは咽喉の他の領域内に存在する他の条件のような特定の二分子トリガー機構(bimolecular triggering mechanisms)に応答して、治療用または診断用物質の放出を容易にするように作られ得る。ナノカプセルは、本発明において、特定の標的細胞(例えば、癌細胞または炎症状態に関連する細胞)に結合する特異的な化学的受容体または結合部位を有する「スマートドラッグ」として用いることができる。
【0083】
下記は、本発明の化合物を含有する医薬品のための処方の非限定的な例である。また、細胞接着の阻害においてRGCペプチドまたは誘導体を用いて示された安全性および/または有効性の例が含まれている。本願において、「RGシステイン酸ペプチド」、「RGCペプチド」、「RGCys−ペプチド」および、「本発明の化合物」という用語は、本明細書に記載される一般式I〜VIIおよび化合物1および3〜5によって定義されるものを含むが、これらに限定されるものではない、R−G−システイン酸配列を含む組成物およびそれらの誘導体を同義的に意味するものとする。
【実施例1】
【0084】
医薬製剤
下記は、本明細書に記載される一般式I〜VIIによって定義されるもののいずれか、または化合物1〜5のうちのいずれかのような本発明のR−G−システイン酸ペプチドを含有する医薬製剤I〜Xの実施例である。
【0085】
【表2】
【実施例2】
【0086】
ウサギにおけるRGDペプチドおよびグリシル−アルギニル−グリシル−システイン酸−トレオニル−プロリン−COOH(GRGシステイン酸TP;化合物1)のPVD誘発効果の比較
この実施例では、RGDペプチドおよびグリシル−アルギニル−グリシル−システイン酸−トレオニル−プロリン−COOH(RGシステイン酸ペプチド;GRGシステイン酸TP;化合物1)のPVD誘発効果をウサギにおいて比較した。この研究のための手順は以下の通りであった。
【0087】
手順:
動物モデル
a)20匹の雄および雌のウサギ
b)体重 約1.5〜2.5kg
c)2つのグループに分ける。
【0088】
i)10匹のウサギに、pH=6.5の2.5%RGD溶液を硝子体内に注入した。
a)10匹の右眼に2.5%RGD溶液を注入した。
b)5匹の左眼はBSS対照として用いた。
【0089】
c)5匹の左眼にpH=6.5の2.5%RGD+0.02%EDTAを注入した。
ii)10匹のウサギに、pH=6.5の2.5%RGシステイン酸溶液を硝子体内に注入した。
【0090】
a)10匹の右眼にpH=6.5の2.5%RGシステイン酸溶液を注入した。
b)10匹の左眼にpH=6.5の2.5%RGシステイン酸溶液+0.02%EDTAを注入した。
【0091】
活性薬品
d)EDTAナトリウム−99.0〜100.5%、スペクトラム ケミカル コーポレイション(Spectrum Chemical Corp.)より入手
e)RGシステイン酸−cGMP供給者(純度>98%)
f)RGD−cGMP供給者(純度>98%)
g)BSS溶液
RGシステイン酸、RGDの双方、RGシステイン酸+ナトリウムEDTA、RGD+ナトリウムEDTAおよびBS溶液は、手術の24時間前に硝子体腔内に注入した。ウサギ(10mg/kg体重)は、キシラジン(100mg/mL)および塩酸ケタミン(100mg/mL)の1:1の合剤2.0mLの筋肉内注入によって麻酔した。瞳は、局所的な塩酸シクロペントラート1%および塩酸フェニレフリン10%で拡張される。
【0092】
先在する硝子体網膜異常を有する動物を除外するために、すべての動物を、最初に細隙灯生体顕微鏡検査および間接検眼によって検査した。0.10ccの硝子体内注入は、1.0cc注入器に取り付けられた30ゲージ針を用いて、鼻上象限(supranasal quadrant)における縁に対して2mm後方に投与された。水晶体または網膜への損傷を避けるために注意しなければならない。
【0093】
注入後24時間かつ機械的硝子体切除の開始直前に、後部硝子体の状態、また硝子体の液化の状態を測定するために、B−スキャン超音波検査を行った。2ポート経毛様体扁平部硝子体切除術(two−port pars plana vitrectomy)は、注入光ファイバー、および硝子体切除ユニットに取り付けられた硝子体カッターを用いて行なった。30秒の中心部硝子体切除術に続いて、硝子体カッターを乳頭周囲網膜の表面に導き、そこで、低吸引(<30mmHg)を用いて、網膜表面からの後部硝子体皮質の分離を4象限において試みた。強膜切開を縫合し、各象限に見られる任意のPVDの存在および範囲を測定するために術後B−スキャン超音波検査を行なった。前記動物は心臓内ペントバルビタールナトリウム注射によって安楽死させ、直ちに眼を摘出した。
【0094】
術後B−スキャン超音波検査に基づいて、PVDの範囲を評価するために、この格付けシステムに従って、硝子体の液化およびPVDの等級を格付けした。
等級0. a)後部硝子体の剥離は観察されない。
【0095】
b)硝子体液化
等級1. a)硝子体が2つ以下の象限において分離されている眼から成る。
b)硝子体液化
等級2. a)硝子体が3つ以上の象限において分離されているが、放射組織(medullary rays)に沿って局所的な付着(focal attachments)が残存する眼から成る。
【0096】
b) 硝子体液化
等級3. a)硝子体が網膜表面から完全に分離されている眼から成る。
b)硝子体液化
すべての眼は、固定剤の迅速な浸透を保証するために、摘出直後に、毛様体扁平部に隣接した(sub−adjacent)上極(superior pole)において鋭利なカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるように注意した。その眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒドに摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球(posterior calotte)を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。
【0097】
結果
注入:2.5%RGシステイン酸
グループ1.ベースラインにおいて、すべての動物は両眼にPVDを有していない。
【0098】
【表3】
注入:2.5%のRGD
グループ2.ベースラインにおいて、すべての動物は両眼にPVDを有していない。
【0099】
【表4】
注入:2.5%RGシステイン酸+0.02%NaEDTA
グループ3.ベースラインにおいて、すべての動物は両眼にPVDを有していない。
【0100】
【表5】
注入:2.5%RGD+0.02%NaEDTA
グループ4.ベースラインにおいて、すべての動物は両眼にPVDを有していない。
【0101】
【表6】
この研究の実施例4における動態研究の結果は、RGDおよびRGシステイン酸(GRGシステイン酸TP;化合物1)が同様の特性を有し、2.5%RGシステイン酸の硝子体内への注入は、24時間で網膜からの硝子体の完全な分離を生じ、さらにRGDのウサギおよびRGシステイン酸のウサギの双方の硝子体は、完全に液化されることを示している。
【0102】
全体にわたって、RGシステイン酸の活性は、ウサギでの完全なPVDの誘発および硝子体の液化において、RGDのそれと同等であるか、または若干良好である。これは、恐らく、インテグリン細胞外マトリックス相互作用の結合部位に対して、RGDと比較して、より強力な競合的結合能力による。本明細書で別記するように、スルホン酸は対応するカルボン酸より強力な酸である。このスルホン酸基のより高い極性はより強力な分子間結合をもたらす。例えば、より分極したO−H結合を有するR−G−システイン酸は、比較的分極に劣るO−H結合を有するR−G−アスパラギン酸よりも強力な水素結合をインテグリン結合部位のアミド基および/またはアミノ酸の側鎖と形成し、かつ/または、インテグリン結合部位において錯体を形成する金属イオンとのより強力な相互作用を有し得る。
【0103】
前記結果はまた、これらの化合物が0.02%のエデト酸ナトリウムと共に投与される場合、RGDおよびRGシステイン酸の双方の活性は変化しないことを示している。
前記結果はまた、RGシステイン酸化合物の化合物1またはRGD化合物の硝子体内注入による副作用または有害な安全性効果はなかったことを示している。
【実施例3】
【0104】
ウサギの眼におけるRGCペプチド化合物1(GRGシステイン酸TP)の多数回注入の安全性の検討
この実施例では、RGCペプチド化合物1の多数回の注入を、体重約1.5〜2.5kgの5匹の雄および4匹の雌のニュージーランドウサギの眼に投与し、その眼を下記に記載の通りに試験した。
【0105】
研究手順は以下の通りであった。
A)ベースライン試験:ベースラインにおいて、9匹のウサギのすべての右眼および左眼を細隙灯生体顕微鏡検査法および間接検眼法で検査して、前記動物が先在する硝子体網膜異常を有していないことを確認した。さらに、9匹の動物すべての左眼および右眼に対して、β−スキャン超音波検査並びにERGスキャンを行って、ベースライン読み取り値を得た。
【0106】
B)実験的治療:次に、9匹のウサギすべてに、RGC溶液または生理食塩水(対照)のいずれかの硝子体内注入を受けさせた。治療溶液は以下のように調製した。
RGC溶液:0.02mgのEDTA二ナトリウム+0.80mgの塩化ナトリウムと、pH6.5に調整されたUSP滅菌注入用蒸留水(USP sterile Water for injection)とを含有する、2.5mg/100μLのRGC化合物1の溶液。
【0107】
生理食塩水(対照):pH6.5に調整されたUSP無菌等張食塩水液(USP Isotonic sterile saline solution)。
投薬は以下の通りに進めた。
【0108】
1)9匹の各ウサギの右眼に、100μLの2.5mg/100μL RGC溶液(投与量=2.5mgの化合物1を送達)を硝子体内に注入した。
2)9匹の各ウサギの左眼に、100μLの生理食塩水(対照)を硝子体内に注入した。
【0109】
3)最初の硝子体内注入後1日に、前記ウサギのうちのいずれかが前記注入による何らかの副作用を有していないかをチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、9匹のウサギすべての右眼および左眼を検査した。
【0110】
4)1回目の硝子体内注入後の7日目に、前記ウサギのうちのいずれかが前記注入による副作用を呈していないかを判定するために、細隙灯生体顕微鏡検査および間接検眼によって、9匹のウサギすべての右眼および左眼を再び検査した。さらに、ベースラインからの何らかの変化があったかを判定するために、すべての動物の右眼および左眼のすべてについてERGスキャンを実施した。
【0111】
5)次に、901番、904番および909番の3匹のウサギのグループを無作為に選択し、後部硝子体の状態を判定するために、これらの3匹の選択した動物の右眼および左眼に対して機械的硝子体切除を実施した。
【0112】
6)3匹の無作為に選択した動物(901番、904番、および909番)を心臓内ペントバルビタールナトリウム注入によって安楽死させて、直ちに眼を摘出した。前記眼はすべて、固定剤の迅速な浸透を保証するために、摘出直後に毛様体扁平部に隣接する上極において鋭利な鋭いカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるために注意した。前記眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒド(gluteraldehyde)中に摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。他の試料は病理組織検査に供した。
【0113】
902番、903番、905番、906番、907番、および908番の残りの6匹のウサギには、最初の注入後7日に第2回目を注入した。
1)6匹の各ウサギの右眼に、100μLの2.5mg/100μL RGC溶液(2回目の化合物1の2.5mg投与量を送達)を硝子体内に再び注入した。
【0114】
2)6匹の各ウサギの左眼に、100μLの生理食塩水(対照)を再び硝子体内に注入した。
3)2回目の硝子体内注入後1日に、前記ウサギのうちのいずれかが前記注入による何らかの副作用を有していないかをチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、6匹の残りのウサギすべての右眼および左眼を検査した。
【0115】
4)2回目の硝子体内注入後7日目に、前記注入による何らかの副作用をチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、6匹の残りのウサギすべての右眼および左眼を再び検査した。さらに、ベースラインからの何らかの変化があったかを判定するために、すべての動物の左眼および右眼のERGスキャンを実施した。
【0116】
5)次に、残りの6匹から902番、903番および907番の3匹のウサギのグループを無作為に選択し、後部硝子体の状態を判定するために、これらの3匹の無作為に選択した動物の右眼および左眼に対して機械的硝子体切除を実施した。
【0117】
6)902番、903番、および907番の3匹の無作為に選択した動物を心臓内ペントバルビタールナトリウム注入によって安楽死させて、直ちに眼を摘出した。前記眼はすべて、固定剤の迅速な浸透を保証するために、摘出直後に毛様体扁平部に隣接する上極において鋭利な鋭いカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるように注意した。前記眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒド(gluteraldehyde)に摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。他の試料は病理組織検査に供した。
【0118】
次に、残りのグループの905番、906番、および908番の3匹のウサギには、最初の注入後14日に3回目の注入を以下の通りに行った。
1)3匹の残りの各ウサギの右眼に、100μLの2.5mg/100μL RGC溶液を硝子体内に再び注入した(3回目の化合物1の2.5mg投与量を送達)。
【0119】
2)3匹の残りの各ウサギの左眼に、100μLの生理食塩水(対照)を硝子体内に再び注入した。
3)3回目の硝子体内注入後1日に、前記ウサギのうちのいずれかが前記注入による何らかの副作用を有していないかをチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、6匹の残りのウサギすべての右眼および左眼を検査した。
【0120】
4)3回目の硝子体内注入後7日目に、前記注入による何らかの副作用をチェックするために、細隙灯生体顕微鏡検査および間接検眼によって、3匹の残りのウサギすべての右眼および左眼を再び検査した。さらに、ベースラインからの何らかの変化があったかを判定するために、すべての動物の左眼および右眼のERGスキャンを実施した。
【0121】
5)後部硝子体の状態を判定するために、3匹の残りの動物(905番、906番、および908番)の右眼および左眼に対して機械的硝子体切除を行った。
6)3匹の残りの動物(905番、906番、および908番)を心臓内ペントバルビタールナトリウム注入によって安楽死させて、直ちに眼を摘出した。前記眼はすべて、固定剤の迅速な浸透を保証するために、摘出直後に毛様体扁平部に隣接する上極において鋭利な鋭いカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるように注意した。前記眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒドに摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。他の試料は病理組織検査に供した。
【0122】
3)活性薬品
この研究に用いた活性薬品は以下の通りであった。
a.EDTA二ナトリウム−99.0〜100.5%、スペクトル ケミカル コーポレイションより
b.RGCペプチド(化合物1)
c.USP無菌等張食塩水液
4)研究製剤
a)RGC溶液:0.02mgのEDTA二ナトリウム+0.80mgの塩化ナトリウムおよびpH6.5に調整されたUSP滅菌注入用蒸留水を含有する、2.5mg/100μL RGC化合物1の溶液。0.22μフィルタを通して2.0mLバイアルへ無菌濾過。
【0123】
生理食塩水(対照):pHを6.5に調整されたUSP無菌等張食塩水。0.22μフィルタを通して無菌バイアル内へ滅菌濾過。
5)注入準備のための麻酔
a.2.0mLのキシラジン(100mg/mL)および塩酸ケタミン(100mg/mL)の1:1の合剤の筋肉内注入。
【0124】
b.瞳は、局所的な塩酸シクロペントラート1%および塩酸フェニレフリン10%によって拡張した。
6)硝子体内注入の準備:
2.5mg/100μLを含むRGC溶液およびpHが6.5に調整された無菌等張食塩液を収容する無菌バイアルを用意した。
【0125】
注入前に、研究者は、1.0ccの注入器内に0.10cc(100マイクロリットル)の溶液が存在することを確認した。
7)注入手順:
硝子体内注入がウサギの正常な日常活動を妨げるのに十分なレベルの視覚障害を生じないので、これは、視覚眼科学協会(Association for Research in Vision and Ophthalmology)ガイドラインの動物決議に従ってメイジャー・サバイバル・プロシージャ(major survival procedure)とは見なされない。
【0126】
細隙灯生体顕微鏡検査法、検眼鏡検査法およびERGのベースライン試験が前記ウサギに対して完了した後、RGC溶液並びに無菌食塩液の双方を硝子体腔内に注入した。前記ウサギ(10mg/kg体重)をキシラジン(100mg/mL)および塩酸ケタミン(100mg/mL)の1:1の合剤2.0mLの筋肉内注入によって麻酔した。瞳は、局所的な塩酸シクロペントラート1%および塩酸フェニレフリン10%で拡張させた。
【0127】
先在する硝子体網膜異常を備えたいかなる動物も除外するために、動物はすべて、最初に細隙灯生体顕微鏡検査および間接検眼(indirect opthalmoscopy)によって検査した。0.10ccの硝子体内注入は、1.0ccの注入器に取り付けられた30ゲージ針を用いて、鼻上象限(supronasal quadrent)における縁に対して2mm後方に投与された。水晶体または網膜への損傷を避けるために注意した。
【0128】
注入後7日に、前記動物に対して機械的硝子体切除を行なった。2ポート経毛様体扁平部硝子体切除術は、注入光ファイバー、および硝子体切除ユニットに取り付けられた硝子体カッターを用いて行なった。30秒の中心部硝子体切除術に続いて、硝子体カッターを乳頭周囲網膜の表面に導き、そこで、低吸引(<30mmHg)を用いて、4象限において網膜表面からの後部硝子体皮質の分離を試みた。前記動物を心臓内ペントバルビタールナトリウム注入で安楽死させ、直ちに眼を摘出した。
【0129】
前記眼はすべて、固定剤の迅速な浸透を保証するために、摘出直後に毛様体扁平部に隣接する上極において鋭利な鋭いカミソリの侵入を受けた。隣接する網膜および水晶体への損傷を避けるように注意した。前記眼を2%パラホルムアルデヒド+2.5%グルタルアルデヒドに摂氏4度で最短24時間にわたって浸漬した。固有の後部眼球を取り出し、メタノール中で脱水し、二酸化炭素中で臨界点まで乾燥させ、金でスパッタコートして、走査電子顕微鏡を用いて撮影した。
【0130】
結果の分析
RGC溶液および無菌食塩液で処理した眼における安全性に関するデータを、下記技術、すなわち、i)細隙灯生体顕微鏡検査法、ii)検眼鏡検査法、iii)ERG、iv)組織病理学、および、v)電子顕微鏡法を用いて安全性について分析した。
【0131】
安全性プロフィール:
901番、902番、903番、904番、905番、906番、907番、908番、909番の9匹のウサギのグループへの100μLの2.5%RGC溶液の1回目の硝子体内投与は全時点においていかなる有意な毒性にも関係していなかった。2.5%RGCグループと等張食塩水液グループとの間には報告された副作用において有意な差異はなかった。この毒性の欠如は、臨床検査、間接検眼法、および超音波β−スキャンおよび機械的硝子体切除によって判定された。
【0132】
細隙灯生体顕微鏡検査法は、すべての治験検診(study visits)において行なわれ、100μLの2.5%RGC溶液および等張食塩水液のグループの硝子体内注入に対して炎症反応がほぼ全くないことを示す炎症の兆候について、結膜および強膜、角膜、内皮の変更、前眼房反応、虹彩、水晶体および眼窩(capsule)、並びに前部硝子体に焦点が当てられた。すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な毒性の兆候があるようには見えなかった。
【0133】
有意な網膜毒性が存在しなかったことを保証する検討を通じて、後部(posterior segment)の臨床評価も続けられた。間接検眼並びに細隙灯眼底評価は、各評価時点において、網膜毒性の何らかの兆候に対して特に注意して行なった。後部は、硝子体密度、硝子体液化、硝子体付着および出血の可能性(possible hemorrhage)の変化について評価した。網膜は、RPE毒性、網膜血管障害性網膜出血、滲出、網膜裂孔、破壊または剥離の兆候について評価した。治療前のベースラインにおけるRPEの変化はなく、すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な後部変化の兆候があるようには見えなかった。硝子体内注入前に9匹の動物に対して行なったERGスキャン、並びに注入後1日および7日にすべての前記動物に対して行ったERGスキャンは、被験物質によって誘発された有意な変化または毒性のいかなる兆候も生じないようであったことに注目することは重要である。
【0134】
902番、903番、905番、906番、907番、908番の6匹のウサギのグループへの100μLの2.5%RGC溶液の2回目の硝子体内投与は全時点においていかなる有意な毒性にも関係していなかった。2.5%RGCグループと等張食塩水液グループとの間に報告された副作用における有意な差異はなかった。この毒性の欠如は、臨床検査、間接検眼法、および超音波β−スキャンおよび機械的硝子体切除によって判定した。
【0135】
細隙灯生体顕微鏡検査法は、すべての治験検診において行なわれ、100μLの2.5%のRGC溶液および等張食塩水液のグループの硝子体内注入に対して炎症反応がほぼ全くないことを示す炎症の兆候について、結膜および強膜、角膜、内皮の変更、前眼房反応、虹彩、水晶体および眼窩、並びに前部硝子体に焦点が当てられた。すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な毒性の兆候があるようには見えなかった。
【0136】
有意な網膜毒性が存在しなかったことを保証する検討の全体にわたって、後部の臨床評価も続けられた。間接検眼並びに細隙灯眼底評価は、各評価時点において、網膜毒性の何らかの兆候に対して特に注意して行なった。後部は、硝子体密度、硝子体液化、硝子体付着および出血の可能性の変化について評価した。網膜は、RPE毒性、網膜血管障害性網膜出血、滲出、網膜裂孔、破壊または剥離の兆候について評価した。2回目の治療の7日前のベースラインにおけるRPEの変化はなく、すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な後部変化の兆候があるようには見えなかった。硝子体内注入後2回目の6匹の動物に対して行なったERGスキャン、並びに注入後8日および14日にすべての前記動物に対して行ったERGスキャンは、被験物質によって誘発された有意な変化または毒性の任意の兆候を生じなかったようであることに注目することは重要である。
【0137】
905番、906番、908番の6匹のウサギのグループへの100μLの2.5%RGC溶液の3回目の硝子体内投与は全時点においていかなる有意な毒性にも関係していなかった。2.5%RGCグループと等張食塩水液グループとの間には、報告された副作用において有意な差異はなかった。この毒性の欠如は、臨床検査、間接検眼法、および超音波β−スキャンおよび機械的硝子体切除によって判定された。
【0138】
細隙灯生体顕微鏡検査法は、すべての治験検診において行なわれ、100μLの2.5%RGC溶液および等張食塩水液のグループの硝子体内注入に対して炎症反応がほぼ全くないことを示す炎症の兆候について、結膜および強膜、角膜、内皮の変更、前眼房反応、虹彩、水晶体および眼窩、並びに前部硝子体に焦点が当てられた。すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な毒性の兆候があるようには見えなかった。
【0139】
有意な網膜毒性が存在しなかったことを保証する検討の全体にわたって、後部の臨床評価も続けられた。間接検眼並びに細隙灯眼底評価は、各評価時点において、網膜毒性の何らかの兆候に対して特に注意して行なった。後部は、硝子体密度、硝子体液化、硝子体付着および出血の可能性の変化について評価した。網膜は、RPE毒性、網膜血管障害性網膜出血、滲出、網膜裂孔、破壊または剥離の兆候について評価した。3回目の処理前14日のベースラインにおけるRPEの変化はなく、すべての調査項目において、かつすべての調査グループにおいて、被験物質によって誘発された有意な後部変化の兆候はあるようには見えなかった。硝子体内注入後3回目の3匹の動物に対して行なったERGスキャン、並びに注入後15日および21日にすべての前記動物に対して行ったERGスキャンは、被験物質によって誘発された有意な変化または毒性の任意の兆候を生じなかったようであることに注目することは重要である。
【実施例4】
【0140】
RGCペプチドの抗接着性:化合物1(GRGシステイン酸TP)、環状−RGDおよびRGEによる創傷治癒の動態研究
この実施例では、創傷治癒のモデルにおいて、RGCペプチドは抗接着性を有し、従って、多くの病理学的硝子体網膜疾患の発症を防止することができ、またヒト黒色腫および結腸癌細胞における転移を阻害し得ることが示された。
【0141】
RGCペプチドの抗接着性をインビトロで試験するために、ヒト臍帯静脈内皮細胞(HUVEC:human umbilical vein endothelial cells)によって創傷治癒アッセイを実施した。HUVECをフィブロネクチン被覆面上に播種して、コンフルエントな単層を成長させた。前記HUVEC単層上で小さなピペットチップを引き摺ることによって、創傷(引っ掻き傷(scratch rift))を形成した。次に、前記細胞を、RGCペプチド(化合物1、10mM)を含有する新鮮な増殖培地中でインキュベートし、創傷閉鎖の動態を判定するために、創傷領域を5つの異なる範囲において様々な時点(0、4、8、12、16、20、24時間)で撮像した。創傷閉鎖のレベルは、細胞の接着、移動および増殖を通じてHUVECによって再び占有された元の創傷領域の画分の測定によって定量した。
【0142】
対照試験では、以下のペプチド、すなわち、環状RGDペプチド(1mM、陽性対照)およびRGEペプチド(1mM、陰性対照)をRGC(化合物1)の代わりに用いた。
創傷治癒の動態に対するペプチドの効果を図1に示す。
【0143】
結果は元の面積のパーセンテージとして示されている。エラーバーは、2〜6回の独立試行における創傷サイズの標準偏差に対応する。
HUVEC創傷閉鎖の動態の結果は、RGCペプチドは24時間後にHUVEC創傷治癒を70%阻害し、一方、環状RGD(RGDに基づくペプチド;N−メチル化環状−RGDf−N(Me)V;シレンジタイド(Cilengitide))は、24時間後、HUVEC創傷治癒を45%阻害することを示している。これらは双方とも、24時間後にHUVEC創傷治癒を0%阻害した陰性対照RGEペプチドと比較した。RGC(化合物1)の効果は、インテグリン結合活性の定評のある阻害剤であるRGDに基づくペプチドの活性に定量的に匹敵する。さらに、RGCペプチドは、HUVEC細胞のアポトーシスを生じることなく、RGDに基づくペプチドの活性に類似した特性を示す。
【0144】
本明細書に別記するように、硝子体と網膜と間の強力な接着は、硝子体黄斑牽引、増殖性糖尿病性網膜症、黄斑円孔、加齢性黄斑変性および飛蚊症のような多くの病理学的硝子体網膜疾患の結果的な発症の原因となり得る。したがって、内部網膜表面からの硝子体の機械的分離以外に、後部硝子体剥離を達成する非外傷性の非侵襲性アプローチは非常に望ましい(テゼル、ティ.エイチ.(Tezel,T.H.)ら、Retina(1998年)18:第7−15頁、およびヴェルシュトレーテン、ティ.シー.(Verstraeten.T.C.)ら、Arch. Ophthalmol.(1993年)第111巻:第849−854頁参照)。
【0145】
本明細書に別記するように、ECM成分、特に硝子体皮質の膠原細線維は、内境界層(ILL:(inner limiting lamina)において、インテグリン結合部位を介して網膜の内面に固定されると考えられている(フース、アール.ワイ.(Foos,R.Y.)、Invest.Ophthalmol.Vis.Sci.(1972年)第11巻:第801−808頁参照)。フィブロネクチンおよびラミニンのような眼内のILLの主な接着性糖タンパク質は、RGD(Arg−Gly−Asp)配列を介してインテグリンに対して強く結びつけられ(カーチス、ティ.エム.(Curtis,T.M.)ら、Am.J.Physiol.、(1995年)第269巻:第L248−L260頁、エルナー、エス.ジー.(Elner, S.G.)ら、IOVS(1996年)第37巻:第696−701頁、およびホルマン、エス.エム.(Horman,S.M)ら、Am.J.Physiol.(1995年)第269巻:第L248−L260頁参照)、いくつかのインテグリンはECMタンパク質中に存在するRGDモチーフを介して結合することも知られている。さらに、ビトロネクチンはαvβ3−特異的であるが、フィブロネクチンはαvβ3に加えて他のいくつかのインテグリンに結合することが知られている。
【0146】
インテグリンのECMへの主な接続はArg−Gly−Asp(RGD)配列を含み、そのRGD配列は、インテグリンヘッドのα−サブユニットとβ−サブユニットとの間に位置する浅い間隙へ結合する(ション(Xiong)ら、Science(2002年)第296巻:第151−155頁参照)。そのような結合は、細胞接着、移動、分化、血管新生および創傷治癒を含む様々な細胞シグナリング経路を調節するのを助ける(ルオスラーチ、イー.(Ruoslahti,E.)ら、Science(1987)第238巻:第491−497頁;およびJ.Clin.Invest.(1991年)第87巻:第1−5頁参照)。
【0147】
硝子体細胞外マトリックス、例えば、膠原細線維は、インテグリン結合部位によって細胞網膜(cellular retina)に接続されるので、RGCペプチド(オリゴペプチド)の硝子体内注入は、同一のインテグリン受容体部位への競合的結合によって、硝子体細胞外マトリックス(vitreous extracellular matrix)のRGDモチーフを細胞網膜から解放し得る。
【0148】
数人の研究者(ルオスラーチ、イー.(Ruoslahti,E.)ら、Science(1987)第238巻:第491−497頁;ハインズ、アール.エイ.(Hynes,R.A.)ら、Cell(1992年)第68巻:第303−322頁;およびハンフリーズ、エム.ジェイ.(Humphries,M.J.)ら、J.Cell Sci.,(1990年)第97巻:第585−592頁参照)は、多くのインテグリン(αvβ3、α5β1、α11β3など)はRGD配列モチーフを有する小さなペプチドによって阻害され得ることを示した。また、それらのαvβ3およびα5β1インテグリンは、ビトロネクチンおよびフィブロネクチンと同様に、ヒトメラノーマ細胞(ニップ、ジェイ.(Nip,J.)、J.Clin.Invest.,(1992年)第90巻:第1406−1413頁参照)、ヒト乳癌細胞(ロン、エル.(Rong,L.)ら、Invest.Ophthalmol.Vis.Sci.(2009年)第50巻:第5988−5996頁参照)、およびヒト網膜色素上皮細胞(ピーター シー.ブルックス(Peter C.Brooks)ら、J.Clin.Invest.,(1995年)第96巻:第1815−1822頁)のような腫瘍においてアップレギュレートされることも文献で裏付けられている。したがって、ヒトメラノーマ細胞の転移能と、αvβ3インテグリン受容体を介したメラノーマ細胞のリンパ節ビトロネクチンへの接着との間には良好な相関があり、前記接着はRGD含有ペプチドによって阻害されたことが示されている(ニップ、ジェイ.J.Clin.Invest.,(1992年)第90巻:第1406−1413頁)。これは、RGDペプチドが重要な抗血管新生薬(anti−angiogenic agents)であり得ることを示している。
【0149】
さらに、ヒト結腸癌細胞系において、細胞接着に有意な増大がある場合、そのとき増大した転移活性があることが示されている(レーマン、エム.(Lehmann,M.)、Cancer Res.,(1994年)、第54巻:第2102−2107頁)。従って、細胞接着を阻害する薬剤は、結腸癌および黒色腫が転移するのを効果的に阻害する。
【0150】
RGCが細胞接着を阻害することが示された創傷治癒の検討の結果に基づき、また黒色腫および結腸癌のモデルにおけるRGDの転移能から推定すると、RGCペプチドおよびその誘導体は、例えば黒色腫および結腸癌において、腫瘍転移を効果的に阻害することができる。
【実施例5】
【0151】
腫瘍に薬剤を誘導または送達するためのRGCペプチドの使用。
この実施例では、下記に示す二量体RGCペプチドパクリタキセル複合体(化合物3)が提供される。この組成物は抗腫瘍薬として有用である。二量体RGCペプチドは、特定の癌細胞において高度に発現されるインテグリン受容体に選択的に結合し、細胞接着を阻害することによって、例えば転移性乳癌のような特定の転移癌を治療するのに有用である。
【0152】
化合物3:
【0153】
【化14】
化合物3の対応するRGD類似体の合成および作用機序、生物分散、並びに腫瘍選択性は、チェン、エックス.(Chen,X.)ら、Synthesis and Biological Evaluation of Dimeric RGD Peptide−Paclitaxel Conjugate as a Model for Integrin−Targeted Drug Delivery、J.Med.Chem.、(2005年)第48(4)巻:第1098−1106頁に記載されている通りである。
【0154】
化合物3は、二量体RGCペプチドに結合された特定の抗腫瘍薬剤パクリタキセルを含むが、本発明のこの態様は、腫瘍または他のインテグリン含有組織もしくは構造の診断、撮像、または治療に有用であり得る任意の実現可能な診断用薬または治療薬に結合されたRGCペプチドの単量体型または多量体型すべてを含むことが理解されるはずである。本発明による単量体または多量体RGCペプチドに結合され得る抗腫瘍物質の例としては、抗腫瘍薬(例えば癌化学療法剤、生体応答調節剤、血管新生阻害剤、ホルモン受容体遮断薬、凍結療法薬(cryotherapeutic agents)、または新生組織形成もしくは腫瘍形成を破壊または阻害する他の薬剤)、例えば、それらのDNAを攻撃することにより癌細胞を直接殺すアルキル化剤または他の薬剤(例えばシクロホスファミド、イソホスファミド)、細胞内DNAの修復に必要な変化を阻害することによって癌細胞を殺すニトロソウレアまたは他の薬剤(例えば、カルマスティン(carmustine)(BCNU)およびロムスチン(lomustine)(CCNU))、特定の細胞機能、通常はDNA合成、に干渉することによって癌細胞増殖を阻止する代謝拮抗物質および他の薬剤(例えば6−メルカプトプリンおよび5−フルオロウラシル(5FU))、DNAを結合するか、またはインターカレートし、RNA合成を妨げることによって作用する抗腫瘍抗生物質および他の化合物(例えばドキソルビシン、ダウノルビシン、エピルビシン(epirubicin)、イダルビシン(idarubicin)、マイトマイシン−C、およびブレオマイシン)、植物から誘導された植物(ビンカ)アルカロイドおよび他の抗腫瘍薬(例えばビンクリスチンおよびビンブラスチン)、ホルモン感受性癌の増殖に影響を与えるステロイドホルモン、ホルモン阻害剤、ホルモン受容体アンタゴニストおよび他の薬剤(例えばタモキシフェン、ハーセプチン、アミノグルテチミド(aminoglutethamide)およびホルメスタンのようなアロマターゼ阻害薬、レトロゾールおよびアナストロゾール(anastrazole)のようなトリアゾール阻害剤、エキセメスタンのようなステロイド阻害剤)、抗脈管形成タンパク質、小分子、遺伝子治療薬、および/または腫瘍の血管形成または血管新生を阻害する他の薬剤(例えばmeth−1,meth−2,サリドマイド)、ベバシズマブ(bevacizumab)(アバスチン(R))、スクアラミン、エンドスタチン、アンジオスタチン、アンギオザイム(Angiozyme)、AE−941(ネオバスタット(Neovastat)(R))、CC−5013(レビミド(Revimid)(R))、medi−522(ビタキシン(Vitaxin)(R))、2−メトキシエストラジオール(2ME2、パンゼム(Panzem)(R))、カルボキシアミドトリアゾール(CAI)、コンブレタスタチンA4プロドラッグ(CA4P:combretastatin A4 prodrug)、SU6668、SU11248、BMS−275291、COL−3、EMD121974、IMC−1C11、IM862、TNP−470、セレコキシブ(セレブレックス(Celebrex)(R))、ロフェコキシブ(バイオックス(Vioxx)(R))、インターフェロンアルファ、インターロイキン−12(IL−12)、または参照によって本願に明示的に援用されるScience第289巻、第1197−1201頁、2000年8月17日において特定された化合物のうちのいずれか、生体応答調節剤(例えばインターフェロン、カルメット−ゲラン桿菌(BCG)、モノクローナル抗体、インタールケン2(interluken 2)、顆粒球コロニー刺激因子(GCSF)など)、PGDF受容体アンタゴニスト、ハーセプチン、アスパラギナーゼ、ブスルファン、カルボプラチン、シスプラチン、カルマスティン、クロラムブシル(cchlorambucil)、シタラビン、ダカルバジン、エトポシド、フルカルバジン(flucarbazine)、フルロウラシル(flurouracil)、ゲムシタビン (gemcitabine)、ヒドロキシウレア、イホスファミド(ifosphamide)、イリノテカン、ロムスチン、メルファラン、メルカプトプリン、メトトレキサート、チオグアニン、チオテパ、トミュデックス(tomudex)、トポテカン(topotecan)、トレオスルファン(treosulfan)、ビンブラスチン、ビンクリスチン、ミトアジトロン(mitoazitrone)、オキサリプラチン、プロカルバジン、ストレプトシン(streptocin)、タキソール、タキソテール(taxotere)、そのような化合物の類似体/同族体および誘導体、並びにここに列記されない他の抗腫瘍薬などが挙げられ得る。
【実施例6】
【0155】
インテグリン表出性腫瘍の画像化用64CU標識多量体RGCペプチド
この実施例において、下記に示す本発明の64CU標識四量体および八量体RGCペプチド(それぞれ化合物4および5)は、画像化および診断の目的(例えば、PET走査のための腫瘍の放射性同位元素標識)のため、並びにαvβ3インテグリンを発現する腫瘍のようなインテグリンを発現する腫瘍または他の細胞に治療薬を誘導または送達するための放射線治療薬として有用である。
【0156】
化合物4:
【0157】
【化15】
化合物5:
【0158】
【化16】
化合物4および5の対応するRGD類似体の合成および作用機序、生物分散、腫瘍選択性およびPETに関連する使用は、リ、ゼット.(Li,Z.)ら、64CU−Labeled Tetrameric and Octameric RGD Peptides for Small−Animal PET of Tumor αvβ3 Integrin Expression、J.Nucl.Med.第48(7)巻、第1162−1171頁(2007年)に記載されている通りである。
【0159】
本明細書において使用される場合、疾患または疾患の治療または治療法に対するいかなる言及も、疾患または障害が生じる前または検知される前のそれらの予防または予防法、並びに疾患または障害が生じた後または検出された後のそれらの治療を含むと解釈されるものとする。
【0160】
本発明は本願において本発明の特定の実施例または実施形態に関して上記に記載してきたが、それらの実施例および実施形態に対して、本発明の意図した趣旨および範囲から逸脱することなく、様々な追加、削除、変更および変更がなされてもよいことが理解されるはずである。例えば、一実施形態または実施例の任意の要素または特質は、そうすることがその実施形態または実施例をその目的の用途に適さないようにする場合について他に特に規定がない限り、別の実施形態または実施例に組み込まれてもよいし、またはそれらと共に用いられてもよい。また方法または過程の工程が特定の順序で記載または列記されている場合、特に他に規定されていない限り、またはそうすることが前記方法および過程をその意図した目的に対して実行不可能にしない限り、そのような工程の順序は変更されてもよい。適当な追加、削除、変更および変更はすべて、記載された実施例および実施形態の均等物と見なされ、以下の特許請求の範囲内に含まれる。本願において引用された刊行物および特許文献はすべて、これにより、参照によって、すべての目的について、あたかも各々が個々にそのように示されたのと同じ程度に余すところなく援用される。
【特許請求の範囲】
【請求項1】
ヒトまたは動物の対象のRGD結合部位への接着を阻害する方法であって、前記方法は、
前記対象に有効量のRGシステイン酸ペプチドまたはその誘導体を投与する工程を含む、方法。
【請求項2】
前記RGシステイン酸ペプチドまたはその誘導体は放射性標識されており、かつ腫瘍を検出するために用いられる、請求項1に記載の方法。
【請求項3】
前記方法は、
血管形成の阻害、血管形成に関連する疾患の治療または予防、細胞の接着、移動および増殖のうちの少なくとも一方の阻害、炎症の治療、癌の治療、転移の治療、治療薬または診断用薬の腫瘍への誘導または送達、血栓症の治療、硝子体索切断の誘起、硝子体網膜剥離の誘起、硝子体切除の性能の助長、創傷治癒の促進、特定のインテグリンを発現する組織の放射性同位体標識、および緑内障の治療のうちから選択される少なくとも1つの治療効果または診断効果をもたらすために行なわれる、請求項1に記載の方法。
【請求項4】
前記方法は、癌、血管奇形、動脈硬化症、血管接着、浮腫性硬化症、角膜移植片新生血管形成、血管新生緑内障、糖尿病性網膜症、翼状片、網膜変性、萎縮型(dry)および滲出型黄斑変性、角膜血管新生、虚血性視神経、虹彩血管新生、中心静脈閉塞、網膜上膜の剥離、水晶体後線維増殖、顆粒性結膜炎、関節リウマチ、全身性エリテマトーデス、甲状腺炎、乾癬、毛細血管拡張、化膿性肉芽腫、脂漏性皮膚炎、およびざ瘡のうちから選択される血管形成に関連する疾患または障害を治療するために行なわれる、請求項1に記載の方法。
【請求項5】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化1】
前記式中、XはH、C1−C6アルキル、PhおよびSO3Hから選択され、YはOHおよびNH2から独立して選択される、請求項1に記載の方法。
【請求項6】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化2】
前記式中、XはH、C1−C6アルキル、PhおよびSO3Hから選択される、請求項1に記載の方法。
【請求項7】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化3】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択され、ZはHおよびSO3Hから選択される、請求項1に記載の方法。
【請求項8】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化4】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択され、YはOHまたはNH2から選択される、請求項1に記載の方法。
【請求項9】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化5】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択される、請求項1に記載の方法。
【請求項10】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化6】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択される、請求項1に記載の方法。
【請求項11】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
X1−R−G−システイン酸−X
前記式中、XおよびX1は環状または直鎖状の−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはArg、Gly、システイン酸、Phe、Val、Ala、Leu、Pro、ThrのD−異性体もしくはL−異性体の任意の組み合わせの任意の塩のうちから選択される、請求項1に記載の方法。
【請求項12】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化7】
前記式中、X’はH、C1−C6アルキル、PhまたはSO3Hから選択され、ZはHまたはMeから選択され、YはOHまたはNH2から選択される、請求項1に記載の方法。
【請求項13】
前記RGシステイン酸ペプチドまたはその誘導体は、多量体RGシステイン酸ペプチドを含む、請求項1に記載の方法。
【請求項14】
前記多量体RGシステイン酸ペプチドは抗腫瘍物質に結合され、該方法は腫瘍の治療のために行なわれる、請求項13に記載の方法。
【請求項15】
前記多量体RGシステイン酸ペプチドは化合物3を含む、請求項14に記載の方法。
【請求項16】
前記抗腫瘍物質は、癌化学療法剤、生体応答調節剤、血管新生阻害剤、ホルモン受容体遮断薬、凍結治療薬;新組織形成または腫瘍形成を破壊または阻害する薬剤;アルキル化薬;それらのDNAを攻撃することにより癌細胞を直接殺す薬剤;シクロホスファミド;イソホスファミド;ニトロソウレア、細胞内DNA修復に必要な変化を阻害することにより癌細胞を殺す薬剤;カルマスティン(BCNU);ロムスチン(CCNU);代謝拮抗物質;DNA合成に干渉することにより癌細胞増殖を阻止する薬剤;6−メルカプトプリン;5−フルオロウラシル(5FU);抗腫瘍抗生物質;DNAに結合するか、またはインターカレートして、RNA合成を妨げることによって作用する化合物;ドキソルビシン;ダウノルビシン;エピルビシン;イダルビシン;マイトマイシン−C;ブレオマイシン;ビンカアルカロイド;ビンクリスチン;ビンブラスチン;ステロイドホルモン;ホルモン阻害剤;ホルモン受容体アンタゴニスト;ホルモン感受性癌の増殖に影響を与える薬剤;タモキシフェン;ハーセプチン;アロマターゼ阻害薬;アミノグルテチミド;ホルメスタン;トリアゾール阻害剤;レトロゾール;アナストロゾール;ステロイド阻害剤;エキセメスタン;抗脈管形成タンパク質;遺伝子療法薬;腫瘍の血管形成または血管新生を阻害する薬剤;meth−1;meth−2;サリドマイド;ベバシズマブ(アバスチン);スクアラミン;エンドスタチン;アンギオスタチン;アンギオザイム;AE−941(ネオバスタット);CC−5013(レビミド);medi−522(ビタキシン);2−メトキシエストラジオール(2ME2、パンゼム);カルボキシアミドトリアゾール(CAI);コンブレタスタチンA4プロドラッグ(CA4P);SU6668;SU11248;BMS−275291;COL−3;EMD121974;IMC−1C11;IM862;TNP−470;セレコキシブ(セレブレックス);ロフェコキシブ(バイオックス);インターフェロンアルファ;インターロイキン−12(IL−2);生体応答調節剤;カルメット−ゲラン桿菌(BCG);モノクローナル抗体;インタールケン2、顆粒球コロニー刺激因子(GCSF);PGDF受容体アンタゴニスト;ハーセプチン;アスパラギナーゼ;ブスルファン;カルボプラチン;シスプラチン;カルマスティン;クロラムブシル;シタラビン;ダカルバジン;エトポシド;フルカルバジン;フルロウラシル;ゲムシタビン;ヒドロキシウレア;イホスファミド;イリノテカン;ロムスチン;メルファラン;メルカプトプリン;メトトレキサート;チオグアニン;チオテパ;トミュデックス;トポテカン;トレオスルファン;ビンブラスチン;ビンクリスチン;ミトアジトロン;オキサリプラチン;プロカルバジン;ストレプトシン;タキソールおよびタキソテールからなる群より選択される、請求項14に記載の方法。
【請求項17】
前記多量体RGシステイン酸ペプチドは放射性標識された化合物に結合され、前記方法は腫瘍組織を放射性標識するために行なわれる、請求項13に記載の方法。
【請求項18】
放射性標識された化合物に結合した前記多量体RGシステイン酸ペプチドは化合物4を含む、請求項17に記載の方法。
【請求項19】
放射性標識された化合物に結合した前記多量体RGシステイン酸ペプチドは化合物5を含む、請求項17に記載の方法。
【請求項20】
RGシステイン酸ペプチドまたはその誘導体を含む組成物。
【請求項21】
下記一般式を有し、
【化8】
前記式中、XはH、C1−C6アルキル、PhおよびSO3Hから選択され、YはOHおよびNH2から独立して選択される、請求項20に記載の組成物。
【請求項22】
下記一般式を有し、
【化9】
前記式中、XはH、C1−C6アルキル、PhおよびSO3Hから選択される、請求項20に記載の組成物。
【請求項23】
下記一般式を有し、
【化10】
XはH、C1−C6アルキル、Ph、またはSO3Hから選択され、ZはHおよびSO3Hから選択される、請求項20に記載の組成物。
【請求項24】
下記一般式を有し、
【化11】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択され、YはOHまたはNH2から選択される、請求項20に記載の組成物。
【請求項25】
下記一般式を有し、
【化12】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択される、請求項20に記載の組成物。
【請求項26】
下記一般式を有し、
【化13】
前記式中、XはH、C1−C6アルキル、PhまたはSOyHから選択され、前記式中、yは2、3または4である、請求項20に記載の組成物。
【請求項27】
下記一般式を有し、
X1−R−G−システイン酸−X
前記式中、XおよびX1は環状または直鎖状の−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはArg、Gly、システイン酸、Phe、Val、Ala、Leu、Pro、ThrのD−異性体またはL−異性体の任意の組み合わせの任意の塩から選択される、請求項20に記載の組成物。
【請求項28】
下記一般式を有し、
【化14】
前記式中、X’はH、C1−C6アルキル、Ph、またはSO3Hから選択され、ZはHまたはMeから選択され、YはOHまたはNH2から選択される、請求項20に記載の組成物。
【請求項29】
組織スキャフォールド、充填材または生体工学材料として使用可能である天然物質または合成物質に組み合わせられたか、または結合された、請求項20に記載の組成物。
【請求項30】
前記天然物質または合成物質は、
ヒドロゲル、コラーゲン、ヒアルロン酸、ポリマー、組織充填剤、および組織充填材のうちから選択される少なくとも1つの物質を含む、請求項29に記載の組成物。
【請求項31】
ヒトまたは動物の対象の身体の所望の位置において、充填、スキャフォールドの形成、または組織成長の促進を行うための方法であって、
前記所望の位置において前記対象の身体に請求項29または30に記載の組成物を導入する工程を含む、方法。
【請求項1】
ヒトまたは動物の対象のRGD結合部位への接着を阻害する方法であって、前記方法は、
前記対象に有効量のRGシステイン酸ペプチドまたはその誘導体を投与する工程を含む、方法。
【請求項2】
前記RGシステイン酸ペプチドまたはその誘導体は放射性標識されており、かつ腫瘍を検出するために用いられる、請求項1に記載の方法。
【請求項3】
前記方法は、
血管形成の阻害、血管形成に関連する疾患の治療または予防、細胞の接着、移動および増殖のうちの少なくとも一方の阻害、炎症の治療、癌の治療、転移の治療、治療薬または診断用薬の腫瘍への誘導または送達、血栓症の治療、硝子体索切断の誘起、硝子体網膜剥離の誘起、硝子体切除の性能の助長、創傷治癒の促進、特定のインテグリンを発現する組織の放射性同位体標識、および緑内障の治療のうちから選択される少なくとも1つの治療効果または診断効果をもたらすために行なわれる、請求項1に記載の方法。
【請求項4】
前記方法は、癌、血管奇形、動脈硬化症、血管接着、浮腫性硬化症、角膜移植片新生血管形成、血管新生緑内障、糖尿病性網膜症、翼状片、網膜変性、萎縮型(dry)および滲出型黄斑変性、角膜血管新生、虚血性視神経、虹彩血管新生、中心静脈閉塞、網膜上膜の剥離、水晶体後線維増殖、顆粒性結膜炎、関節リウマチ、全身性エリテマトーデス、甲状腺炎、乾癬、毛細血管拡張、化膿性肉芽腫、脂漏性皮膚炎、およびざ瘡のうちから選択される血管形成に関連する疾患または障害を治療するために行なわれる、請求項1に記載の方法。
【請求項5】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化1】
前記式中、XはH、C1−C6アルキル、PhおよびSO3Hから選択され、YはOHおよびNH2から独立して選択される、請求項1に記載の方法。
【請求項6】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化2】
前記式中、XはH、C1−C6アルキル、PhおよびSO3Hから選択される、請求項1に記載の方法。
【請求項7】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化3】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択され、ZはHおよびSO3Hから選択される、請求項1に記載の方法。
【請求項8】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化4】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択され、YはOHまたはNH2から選択される、請求項1に記載の方法。
【請求項9】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化5】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択される、請求項1に記載の方法。
【請求項10】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化6】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択される、請求項1に記載の方法。
【請求項11】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
X1−R−G−システイン酸−X
前記式中、XおよびX1は環状または直鎖状の−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはArg、Gly、システイン酸、Phe、Val、Ala、Leu、Pro、ThrのD−異性体もしくはL−異性体の任意の組み合わせの任意の塩のうちから選択される、請求項1に記載の方法。
【請求項12】
前記RGシステイン酸ペプチドまたはその誘導体は下記一般式を有し、
【化7】
前記式中、X’はH、C1−C6アルキル、PhまたはSO3Hから選択され、ZはHまたはMeから選択され、YはOHまたはNH2から選択される、請求項1に記載の方法。
【請求項13】
前記RGシステイン酸ペプチドまたはその誘導体は、多量体RGシステイン酸ペプチドを含む、請求項1に記載の方法。
【請求項14】
前記多量体RGシステイン酸ペプチドは抗腫瘍物質に結合され、該方法は腫瘍の治療のために行なわれる、請求項13に記載の方法。
【請求項15】
前記多量体RGシステイン酸ペプチドは化合物3を含む、請求項14に記載の方法。
【請求項16】
前記抗腫瘍物質は、癌化学療法剤、生体応答調節剤、血管新生阻害剤、ホルモン受容体遮断薬、凍結治療薬;新組織形成または腫瘍形成を破壊または阻害する薬剤;アルキル化薬;それらのDNAを攻撃することにより癌細胞を直接殺す薬剤;シクロホスファミド;イソホスファミド;ニトロソウレア、細胞内DNA修復に必要な変化を阻害することにより癌細胞を殺す薬剤;カルマスティン(BCNU);ロムスチン(CCNU);代謝拮抗物質;DNA合成に干渉することにより癌細胞増殖を阻止する薬剤;6−メルカプトプリン;5−フルオロウラシル(5FU);抗腫瘍抗生物質;DNAに結合するか、またはインターカレートして、RNA合成を妨げることによって作用する化合物;ドキソルビシン;ダウノルビシン;エピルビシン;イダルビシン;マイトマイシン−C;ブレオマイシン;ビンカアルカロイド;ビンクリスチン;ビンブラスチン;ステロイドホルモン;ホルモン阻害剤;ホルモン受容体アンタゴニスト;ホルモン感受性癌の増殖に影響を与える薬剤;タモキシフェン;ハーセプチン;アロマターゼ阻害薬;アミノグルテチミド;ホルメスタン;トリアゾール阻害剤;レトロゾール;アナストロゾール;ステロイド阻害剤;エキセメスタン;抗脈管形成タンパク質;遺伝子療法薬;腫瘍の血管形成または血管新生を阻害する薬剤;meth−1;meth−2;サリドマイド;ベバシズマブ(アバスチン);スクアラミン;エンドスタチン;アンギオスタチン;アンギオザイム;AE−941(ネオバスタット);CC−5013(レビミド);medi−522(ビタキシン);2−メトキシエストラジオール(2ME2、パンゼム);カルボキシアミドトリアゾール(CAI);コンブレタスタチンA4プロドラッグ(CA4P);SU6668;SU11248;BMS−275291;COL−3;EMD121974;IMC−1C11;IM862;TNP−470;セレコキシブ(セレブレックス);ロフェコキシブ(バイオックス);インターフェロンアルファ;インターロイキン−12(IL−2);生体応答調節剤;カルメット−ゲラン桿菌(BCG);モノクローナル抗体;インタールケン2、顆粒球コロニー刺激因子(GCSF);PGDF受容体アンタゴニスト;ハーセプチン;アスパラギナーゼ;ブスルファン;カルボプラチン;シスプラチン;カルマスティン;クロラムブシル;シタラビン;ダカルバジン;エトポシド;フルカルバジン;フルロウラシル;ゲムシタビン;ヒドロキシウレア;イホスファミド;イリノテカン;ロムスチン;メルファラン;メルカプトプリン;メトトレキサート;チオグアニン;チオテパ;トミュデックス;トポテカン;トレオスルファン;ビンブラスチン;ビンクリスチン;ミトアジトロン;オキサリプラチン;プロカルバジン;ストレプトシン;タキソールおよびタキソテールからなる群より選択される、請求項14に記載の方法。
【請求項17】
前記多量体RGシステイン酸ペプチドは放射性標識された化合物に結合され、前記方法は腫瘍組織を放射性標識するために行なわれる、請求項13に記載の方法。
【請求項18】
放射性標識された化合物に結合した前記多量体RGシステイン酸ペプチドは化合物4を含む、請求項17に記載の方法。
【請求項19】
放射性標識された化合物に結合した前記多量体RGシステイン酸ペプチドは化合物5を含む、請求項17に記載の方法。
【請求項20】
RGシステイン酸ペプチドまたはその誘導体を含む組成物。
【請求項21】
下記一般式を有し、
【化8】
前記式中、XはH、C1−C6アルキル、PhおよびSO3Hから選択され、YはOHおよびNH2から独立して選択される、請求項20に記載の組成物。
【請求項22】
下記一般式を有し、
【化9】
前記式中、XはH、C1−C6アルキル、PhおよびSO3Hから選択される、請求項20に記載の組成物。
【請求項23】
下記一般式を有し、
【化10】
XはH、C1−C6アルキル、Ph、またはSO3Hから選択され、ZはHおよびSO3Hから選択される、請求項20に記載の組成物。
【請求項24】
下記一般式を有し、
【化11】
前記式中、XはH、C1−C6アルキル、PhまたはSO3Hから選択され、YはOHまたはNH2から選択される、請求項20に記載の組成物。
【請求項25】
下記一般式を有し、
【化12】
前記式中、XはH、C1−C6アルキル、Ph、またはSO3Hから選択される、請求項20に記載の組成物。
【請求項26】
下記一般式を有し、
【化13】
前記式中、XはH、C1−C6アルキル、PhまたはSOyHから選択され、前記式中、yは2、3または4である、請求項20に記載の組成物。
【請求項27】
下記一般式を有し、
X1−R−G−システイン酸−X
前記式中、XおよびX1は環状または直鎖状の−Phe−Val−Ala、−Phe−Leu−Ala、−Phe−Val−Gly、−Phe−Leu−Gly、−Phe−Pro−Gly、−Phe−Pro−Ala、−Phe−Val、またはArg、Gly、システイン酸、Phe、Val、Ala、Leu、Pro、ThrのD−異性体またはL−異性体の任意の組み合わせの任意の塩から選択される、請求項20に記載の組成物。
【請求項28】
下記一般式を有し、
【化14】
前記式中、X’はH、C1−C6アルキル、Ph、またはSO3Hから選択され、ZはHまたはMeから選択され、YはOHまたはNH2から選択される、請求項20に記載の組成物。
【請求項29】
組織スキャフォールド、充填材または生体工学材料として使用可能である天然物質または合成物質に組み合わせられたか、または結合された、請求項20に記載の組成物。
【請求項30】
前記天然物質または合成物質は、
ヒドロゲル、コラーゲン、ヒアルロン酸、ポリマー、組織充填剤、および組織充填材のうちから選択される少なくとも1つの物質を含む、請求項29に記載の組成物。
【請求項31】
ヒトまたは動物の対象の身体の所望の位置において、充填、スキャフォールドの形成、または組織成長の促進を行うための方法であって、
前記所望の位置において前記対象の身体に請求項29または30に記載の組成物を導入する工程を含む、方法。
【図1】

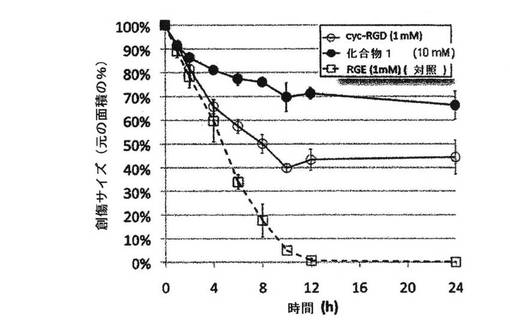
【図2】
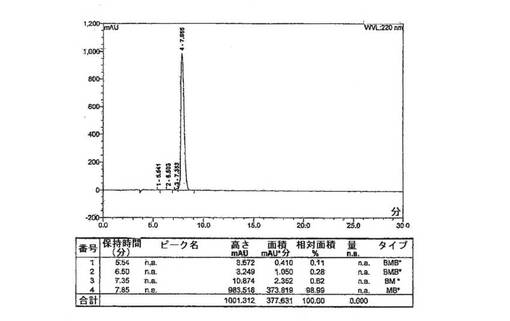
【図3】
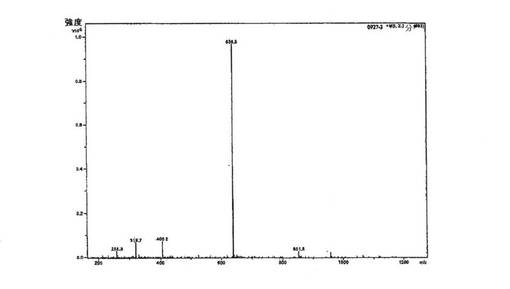
【図4】
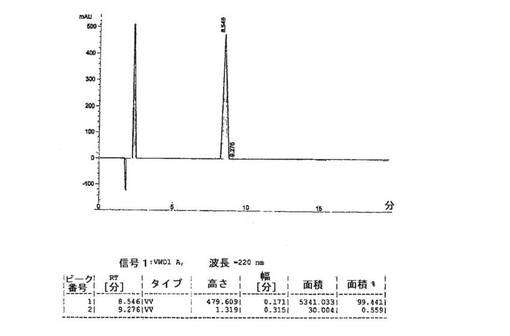
【図5】
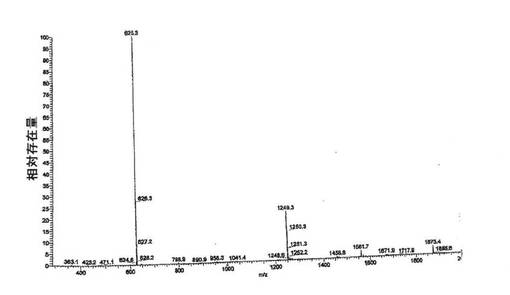
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2013−510863(P2013−510863A)
【公表日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2012−538958(P2012−538958)
【出願日】平成22年11月10日(2010.11.10)
【国際出願番号】PCT/US2010/056277
【国際公開番号】WO2011/060104
【国際公開日】平成23年5月19日(2011.5.19)
【出願人】(512120823)アレグロ ファーマシューティカルズ インコーポレイテッド (1)
【氏名又は名称原語表記】ALLEGRO PHARMACEUTICALS,INC.
【Fターム(参考)】
【公表日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願日】平成22年11月10日(2010.11.10)
【国際出願番号】PCT/US2010/056277
【国際公開番号】WO2011/060104
【国際公開日】平成23年5月19日(2011.5.19)
【出願人】(512120823)アレグロ ファーマシューティカルズ インコーポレイテッド (1)
【氏名又は名称原語表記】ALLEGRO PHARMACEUTICALS,INC.
【Fターム(参考)】
[ Back to top ]