
細胞浸透効率を向上させた輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質及びその用途
本発明は、機能を有する蛋白質またはペプチドをより高い効率で細胞内に導入させる融合蛋白質とその用途に関するものであって、より詳しくは6ないし12個のアミノ酸残基で構成され、アルギニンまたはリシン残基を3/4以上含む輸送ドメインが目標蛋白質のアミノ基とカルボキシル基に共有結合された、細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質とその用途に関するものである。本発明は、蛋白質の細胞内導入能と効率及び細胞内活性を容易に分析するための緑色蛍光蛋白質(Green fluorescence protein)と生体防御メカニズムにおいて重要な役割を行うものに知られているCu,Zn−スーパーオキシドジスムタ−ゼ(SOD)を目標蛋白質に使用した。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は機能を有する蛋白質又はペプチドをより高い効率で細胞内に導入させる融合蛋白質とその用途に関するものである。
【背景技術】
【0002】
最近、様々な疾患が細胞蛋白質の非正常的な活性に起因するという事実が知られてきているため、これら蛋白質の活性を調節することにより致命的な人体疾患を治療できる薬剤の開発が全世界的関心の対象となっている。
【0003】
その一例として、活性酸素種と関連されたス−パ−オキシドジスムタ−ゼ(Superoxide Dismutase)を細胞蛋白質の例として挙げる(但し、本発明の意図する範囲はこの蛋白質だけを目標蛋白質に限定するものではない。)。
【0004】
活性酸素種(reactive oxygen species)は、エネルギ−を得る全ての生命体から、酸素を利用して多くの細胞内代謝過程の副産物として必然的に生成される。このような活性酸素種は、細胞内の蛋白質、核酸及び脂肪のような生体高分子に損傷を与え、人体の多くの疾病の進行過程と深く関連されている。特に発癌過程、脳卒中、関節炎、動脈硬化、放射線損傷及び炎症反応に関与し、正常的な老化過程においても老化を促進させる重要な要因として作用する(非特許文献1〜3)。
【非特許文献1】Floyd,R.A.(1990)FASEB J.4,2587−2597.
【非特許文献2】Anderson,W.F.(1988)Human gene therapy.Nature392,25−30.
【非特許文献3】Halliwell B.andGutteridge J.M.C.(1999)Freeradicals inbiology and medicine,Oxford University Press,Oxford.
【0005】
Cu,Zn−ス−パ−オキシドジスムタ−ゼと関連された疾病等を簡略に検討すれば表1のとおりである。
【0006】
【表1】
【0007】
公知の活性酸素種としては1O2、OH、O2、H2O2等があり、これらは生体内で各種の酵素反応によって生成され、各種の生理活性物質の生合成、免疫機能、薬物の代謝等において大変重要な役割をするが、外部からの放射線、紫外線、公害及び各種のストレス等によって過剰生成される場合には、反って生体に損傷を来すことがある。そこで、生体は防御機能として、SOD、カタラ−ゼ(Catalase)、ペルオキシダ−ゼ(Peroxidase)等の酵素を有するが、老化が始まると皮膚の均衡が壊れるようになり、各種の活性酸素類に対する皮膚保護能力が劣るようになる。
【0008】
よって、このような活性酸素類等から細胞を保護する必要性が強くなってきており、化粧品原料としての活性酸素除去剤として、SOD、ラクトフエリン、酸化防止剤等が使用ないし開発されている。しかしながら、SODは、活性酸素除去能を有するにもかかわらず、酵素蛋白質という性質により地質に対し難溶性であり、安定性が悪く、分子量が30,000ダルトン(Dalton)以上で皮膚角質層の透過が不可能であるという問題点があるため、化粧料組成物として有用に利用されなかった。
【0009】
Cu,Zu−ス−パ−オキシドジスムタ−ゼは、自由ラジカルの害毒と酸素の損傷から細胞の損傷を防止する細胞内重要な防御酵素である(非特許文献4)。全ての生体内高分子等は、このような酸素ラジカルの有害な作用に常に晒されているため、多くの疾患においてCu,Zn−ス−パ−オキシドジスムタ−ゼを治療目的に利用しようとする関心が非常に高くなっている。
【非特許文献4】Fridovich,I.(1995)Annu.Rev.Biochem.64,97−112.
【0010】
最近、Cu,Zn−ス−パ−オキシドジスムタ−ゼを臨床的に応用するために多くの方法が示されている。現在まで開発されたCu,Zn−ス−パ−オキシドジスムタ−ゼの生体内運搬方法は大きく3種に区分できる。
【0011】
第1に、Cu,Zn−ス−パ−オキシドジスムタ−ゼにポリエチレングリコ−ル(polyethylene glycol)、フイコ−ル(Ficoll)、レシチン(lecithin)、アルブミン(albumin)等を集合させる方法がある。
【非特許文献5】Del Zoppo,g.J.et al.(1997)Drugs 54,9−38
【非特許文献6】Muzykantov, V.R.et al.(1996)Proc.Natl.Acad.Sci.USA93,5213−5218.
【0012】
第2に、リポソ−ム(liposome)を利用してCu,Zn−ス−パ−オキシドジスムタ−ゼをカプセル化させる方法がある。
【非特許文献7】Perdereau,B.et al.(1994)Bull.Cancer 81,659−669.
【0013】
第3に、Cu,Zn−ス−パ−オキシドジスムタ−ゼ遺伝子を形質導入して細胞内で酵素の過多発現を誘導する遺伝子治療等の試みが示されている。
【非特許文献8】Okumura,K. et al.(1997)Pharm.Res.14,1223−1227
【非特許文献9】Lehmann,T.G.et al.(2000)Transplantation 69,1051−1057.
【非特許文献10】Liu,R.et al.(1997)Hum.Gene Ther.8,585−595.
【0014】
これらのうち、もっとも注目を受けているものは、遺伝子治療であり、Cu,Zn−ス−パ−オキシドジスムタ−ゼ遺伝子治療を疾病に適用しようとする多くの研究が行われてきた。しかし、遺伝子治療は遺伝子の細胞内への運搬が容易でなく、標的細胞における発現率が低く、発現される細胞で蛋白質が発現される期間が短く、標的細胞から発現される蛋白質の量を人為的に調節するのが非常に難しい等、様々な問題点を有している。
【非特許文献11】Verma,I.M.et al(1997)Nature389,239−242.
【0015】
治療のための薬物や蛋白質を細胞内に移動させることに関しては、他の方法として、目標蛋白質を細胞膜を経て直接伝達する方法が考えられる。しかし、治療用薬物や蛋白質は、そのサイズ又は多くの生化学的性質のため細胞膜を通過することが大変難しい。一般的に分子量600以上の物質は細胞膜を通過するのが殆ど不可能であることが知られている。
【0016】
最近になって、ヒト免疫欠乏ビ−ルス(Human Immunodeficiency Virus type−1)蛋白質の一種であるTat(transactivator of transcription)蛋白質が、効率的に細胞膜を通過して細胞質内に容易に移動するということが明らかになった。このような機能はTat蛋白質の中間部位である蛋白質形質導入部位(Protein Transduction Domain)の特性のため表れ、未だその正確なメカニズムは知られていない状態である(非特許文献12〜15)。しかし、Tat蛋白質の細胞膜通過には特定受容体や運搬体が関与しないことが見出されており、Tat蛋白質の蛋白質形質導入部位が、直接膜の地質二重層と作用することにより起こるものと理解されている(非特許文献16、17)。
【非特許文献12】Frankel,A.D.andPabo,C.O.(1988)Cell 55,1189−1193.
【非特許文献13】Green,M. and Loewenstein,P.M.(1988)Cell55,1179−1188.
【非特許文献14】Ma,M. and Nath,A.(1997),J.Virol.71,2495−2499.
【非特許文献15】Vives,E.,Brodin,P.and Lebleu,B.(1997),J.Biol.Chem.272,16010−16017.
【非特許文献16】Vives,E.et al.(1997)J.Biol.Chem.272,16010−16017.
【非特許文献17】Derossi,D.et al.(1996),J.Biol.Chem.271,18188−18193.
【0017】
最近の研究において,オバルブミン(ovalbumin)、β−ガラクトシダ−ゼ(galactosidase)、セイヨウワサビ ペルオキシダ−ゼ(horseradish peroxidase)のような異形蛋白質をHIV−1 Tat蛋白質と融合させて投与した際、生体の各組織及び培養された細胞内に直接運搬されるとのことが発表された(非特許文献18〜20)。このような実験結果は、Tat蛋白質がそれ自身だけではなく、他の種類の巨大な蛋白質も細胞内に一緒に運搬できる能力を有することを意味している。
【非特許文献18】Fawell,S.et al.(1994),Proc.Natl.Acad.Sci.USA91,664−668.
【非特許文献19】Scwartze,S.R.et al.(1999),Science.285,1569−1572.
【非特許文献20】Watson,K.and Edward,R.J.(1999),Biochem.Pharmacol.58,1521−1528.
【0018】
しかしながら、実際には、全ての蛋白質がTat蛋白質によって運搬されるものではない。また、Tatによって細胞内に運搬された全ての蛋白質が生物学的活性を表すかも未だ明らかになっていないのが現状である。
【0019】
本発明者は、HIV Tat 49−57輸送ドメインの外にもリシン(lycine)が6ないし12個結合されたオリゴリシン輸送ドメイン、HIV Tat 48−57残基のうち、2、3個の残基が欠如した塩基性輸送ドメインについて、アミノ基に共有結合された融合蛋白質への細胞透過実験を行っており、これらが円滑な細胞透過能を有すること示している
【特許文献1】韓国特許公報第2002−100446号
【特許文献2】韓国特許公報第2002−67108号
【発明の開示】
【発明が解決しようとする課題】
【0020】
本発明の目的は、目標蛋白質を細胞内に導入又は発現させる際に、従来技術の問題点を解決し、高効率で細胞内に導入させ、細胞内で活性を有するようにしようとするものである。
【0021】
また、本発明はス−パ−オキシドジスムタ−ゼを細胞内に導入又は発現させる際に、従来技術の問題点を解決し、高効率で細胞内に導入させ、細胞内で活性を有するようにしようとするものである。
【0022】
また、本発明は、輸送ドメインと融合されたス−パ−オキシドジスムタ−ゼを利用して皮膚表皮、真皮層、皮下脂肪層まで浸透して卓越した活性酸素種の除去能を有する化粧料を提供するものである。
【課題を解決するための手段】
【0023】
上記、課題を解決するために、本発明者らは、輸送ドメインとヒトCu,Zn−ス−パ−オキシドジスムタ−ゼを融合させ、該融合蛋白質が効果的に細胞内に透過されるかをHeLa細胞を利用した実験において確認した。また、輸送ドメイン−ス−パ−オキシドジスムタ−ゼ輸送ドメイン融合蛋白質を多量に生産する方法及び精製する方法を開発した。
【0024】
本発明は、機能を有する蛋白質又はぺプチドをより効率的に細胞内に運搬し、細胞内でもより高い活性を有するようにするために、輸送ドメインと融合させた融合蛋白質と該融合蛋白質を利用した薬剤組成物及び化粧料組成物を提供する。
【0025】
本発明の輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質は遺伝子組み替え方法の他にも、一般的な化学結合方法で製造することができる。
【0026】
本発明は、6ないし12個のアミノ酸残基から構成され、アルギニン又はリシン(lycine)残基を3/4以上含む輸送ドメインが目標蛋白質のアミノ基とカルボキシル基に共有結合された、細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質に関するものである。目標蛋白質の両末端に位置する輸送ドメインは必ず同一な残基からなっていなければならないものではない。
【0027】
また、本発明は、上記輸送ドメインが9個のアミノ酸残基で構成される細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質に関するものである。
【0028】
また、本発明は、上記輸送ドメインがHIV tat 49−57残基、オリゴリシン、オリゴアルギニン又はオリゴ(リシン,アルギニン)中の1種以上であることを特徴とする。
【0029】
更に、本発明は、上記目標蛋白質が治療分子、予防分子、診断分子から構成されたグル−プから選ばれたことを特徴とする。
【0030】
また、本発明は、上記目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物であることを特徴とする。
【0031】
また、本発明は、上記細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とすることを特徴とする化粧料組成物に関するものである。
【0032】
また、本発明は、上記目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物であることを特徴とする化粧料組成物に関するものである。
【0033】
また、本発明は、上記化粧料組成物が化粧水、ジエル、水溶性リキッド、水中油(O/W)型又は油中水(W/O)型であることを特徴とする。
【0034】
のみならず、本発明は上記輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とし、薬学的に許与される担体を含むことを特徴とする薬剤学的組成物に関するものである。
【0035】
また、本発明は、上記目標蛋白質がCu,Zn−ス−パ−ジスムタ−ゼ、又は、その機能的等価物であることを特徴とする。
【0036】
輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分として含有する薬剤学的組成物は薬剤学的分野で通常的に許容される担体と共に配合して通常的な方法によって経口又は注射形態に成型することができる。経口用組成物としては、例えば、錠製及びゼラチンカプセルがあり、これらは活性成分以外にも必要によって、希釈剤(例:ラクト−ス、デキストロ−ス、蔗糖、マンニト−ル、ソルビト−ル、セルロ−ス及び/又はグリシン)、滑沢剤(例:シリカ、タルク、ステアリン酸及びそのマグネシウム又はカルシウム塩及び/又はポリエチレングリコ−ル)を含有し、錠剤はまた結合剤(例:マグネシウムアルミニウムシリケ−ト、澱粉ペ−スト、ゼラチン、メチルセルロ−ス、ナトリウムカルボキシメチルセルロ−ス及び/又はポリビニルピロリドン)を含有し、場合によって、崩解剤(例:澱粉、寒天、アルギン酸、又はそのナトリウム塩)又は沸騰混合物及び/又は吸水剤、着色剤、抗味剤及び甘味剤を含有することが好ましい。注射用組成物は、等張性水溶液又は懸濁液が好ましく、これらの組成物は滅菌され、必要によって補助剤(例:防腐剤、安定化剤、湿潤剤、又は乳化剤溶液促進剤、浸透圧調節のための塩及び/又は緩衝剤)を含有する。又、これらはその他治療的に有用な物質を含有することができる。
【0037】
このように製造された薬剤学的製剤は、その目的により、経口投与するか、非経口方式、即ち、静脈内、皮下、腹腔内投与又は局所適用することができ、容量は1日の投与量が0.0001〜100mg/kgの量を1ないし数回に分けて投与することができる。特定患者に対する投与容量水準は、患者の体重、年齢、性別、健康状態、投与時間、投与方法、排泄率、疾患の重症度等によって変化し得る。
【0038】
また、本発明の化粧料組成物は、クリ−ム、軟膏、ジエル、ロ−ション、化粧水等で製品化されて使用できる、どんな製品として使用するかは、本発明の属する技術分野の当業者によって極めて容易に決定され得る。
【0039】
また、本発明の輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とするロ−ション、ジェル、エセンス、クリ−ム、化粧水等はそれぞれ、通常的な製造方法によって、どんな形態でも容易に製造することができ、基礎化粧品等に適宜に添加して使用することができる。
【0040】
その一例として、クリ−ムを製造するにおいては、一般的な水中油型(O/W)又は油中水型(W/O)のクリ−ムベ−スに本発明の融合蛋白質を含有させ、ここに香料、キレ−ト剤、色素、酸化防止剤、防腐剤等を使用する一方、物性改善を目的に蛋白質、ミネラル、ビタミン等合成又は天然素材を併用することができる。
【0041】
本明細書及び請求範囲の”目標蛋白質(target protein)”は、上記HIV−1 Tat輸送ドメインと共有結合をなして細胞内に導入され活性を示す治療分子、予防分子及び診断分子等を意味し、実際には、純粋な蛋白質にだけ限定されるものでなく、ペプチド、ポリペプチド、糖類と結合された糖蛋白質、ペプチドグリカン等を通称する表現に使用される。
【0042】
また、本発明において”輸送ドメイン“はペプチド、蛋白質、オリゴペプチド、糖類と結合された糖蛋白質、ペプチドグリカン又はポリペプチド等と共有結合をなして別途の受容体や運搬体、エネルギ−を必要としないで上記有機化合物等を細胞内へ導入させることができるドメインであって、6〜12個のアミノ酸残基からなっており、その中、3/4以上がアルギニン又はリシン残基からなっていることをいい、代表的な例を挙げるとすれば、HIV−1 Tat(48−57)、オリゴリシン、オリゴアルギニン、オリゴ(リシン、アルギニン)等をいう。さらに好ましくは、9個のアミノ酸残基からなっており、そのうち、3/4以上がアルギニン又はリシン残基からなっているものを本発明の輸送ドメインの例に挙げることができる。
【0043】
また、本明細書及び請求範囲では蛋白質、ペプチド、糖蛋白質、オリゴペプチド、ポリペプチド等を細胞内に”導入“させることに対して、”運搬”、”浸透”、”輸送“、”伝達”、”透過“、”通過”するという表現と混合して使用している。
【発明を実施するための最良の形態】
【0044】
以下では、本発明の構成を実施例を通じて詳しく説明する。
【0045】
実施例1:輸送ドメイン融合蛋白質発現ベクタ−の製造
機能を有する蛋白質又はぺプチドを細胞内に浸透させる技術を開発するために細胞内に目標蛋白質を伝達できる融合蛋白質発現べクタ−を製造した。
【0046】
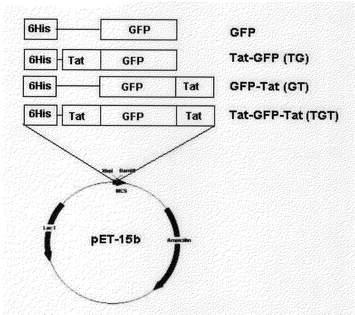
本実施例では、輸送ドメイン蛋白質を細胞内に伝達する能力を簡易に分析するために、緑色蛍光蛋白質(Green Fluorescence Protein;本明細書において“GFP”と略称する)を目標蛋白質に選んだ。GFPの塩基配列に該当するDNA切片をプラスミドpEGFP−C2(ク−ロンテック)を利用してPfu DNA重合酵素で重合酵素連鎖反応(PCR)させ、GFPの完全な配列を増幅した。正方向開始体(sense primer)は5‘−CTCGAGGTGAGCAAGGGCGAGGAGCTG−3’(配列番号1)であり、逆方向開始体(antisense primer)の配列は5‘−GGATCCTTACTTGTACAGCTCGTCC ATGCCGAG−3’(配列番号2)である。PCR産物はXhoI−BamHIで切られ、pET15b(Invitrogen、Carlsbad、CA)のXhoI−BamHI座にサブク−ロンされ、HIV−1 Tatの塩基性ドメインがないGFP融合蛋白質を発現させるpGFPを製造した。約0.7kbの挿入体を有するク−ロンはXhol−BamHI制限酵素分析で選ばれ、配列決定を通じて分析された。
【0047】
GFPと融合されたHIV−1 Tatの塩基性ドメイン(アミノ酸48−57)を発現させるpTat−GFPは、次の方法で製造された。
【0048】
第1の方法:二つのオリゴヌクレオチドが製造され、HIV−1 Tatの塩基性ドメインの9個のアミノ酸をコ−デイングする二重鎖オリゴヌクレオチドでアニ−ルされた。HIV−1 Tatの塩基性ドメインの9個のアミノ酸をコ−デイングする配列は上方の鎖(top strand)二重鎖オリゴヌクレオチドでアニ−ルされた。HIV−1 Tatの塩基性ドメイン5‘−TAGGAAGAAGCGGAGACAGCGACGAAGAC−3’(配列番号3)と下方鎖(bottom strand)5‘−TAGGAAGAAGCGGAGACAGCGACGAAGAC−3’(配列番号4)である。二重鎖オリゴヌクレオチドは直接pGFPのNdeI−XhoIで消化された部位に連結される。そして、インフレ−ム(in frame)で6個のヒスチジンオ−プンリディングフレ−ムと連結されたTat−GFP発現プラスミドであるpTat−GFPが製造された(図1)。
【0049】
また、Tat塩基性ドメインGFP両側末端に融合されたpTat−GFP−TatはpTat−GFPにTat塩基性ドメイン9個のアミノ酸をコ−デイングする二重鎖オリゴヌクレオチドがアニ−ルされ、カルボキシル末端に挿入され、Tat塩基性末端がカルボキシル末端に融合されたpGFP−TatはpGFPにTat塩基性ドメイン9個のアミノ酸をコ−デイングする二重鎖オリゴマヌクレオチドがアニ−ルされてカルボキシル末端に挿入された(図1)。
【0050】
プラスミドにク−ロンされたオリゴヌクレオチドの配列は蛍光自動配列分析器(fluorescence−based automated sequencer,モデル373A,Applied Biosystems,Inc.)で確認した。
【0051】
実施例2:Tat−GFP、GFP−Tat及びTat−GFP−Tat融合蛋白質の発現及び精製(Native、denatured)
pGFP及びpTat−GFP、pGFP−TatとpTat−GFP−Tat等で形質転換されたE.coli BL 21(パ−マシア)を選んだ後、コロニ−を100μg/mlのアンピシリンが含有されたLB培地に接種し、37℃で一晩培養した。培養した培養液は新鮮なLB培地で10倍希釈して250rpmで撹伴して培養した。培養液内のバクテリア濃度が0.D6001.0を示したとき、IPTGを培地内に添加して最終濃度が0.5mMになるようにした後、4時間さらに培養した。
【0052】
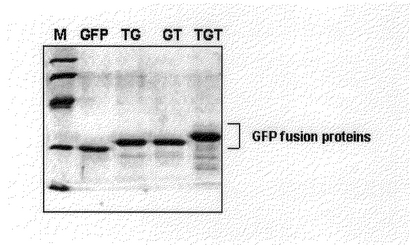
変性された輸送ドメイン−GFP融合蛋白質を得るために、培養された細胞に6Mウレアとプロテア−ゼ抑制剤(20mg/mlソイビントリップシン抑制剤、2mg/mlアプロチ二ン、5mg/mlリュペップチン、100mg/ml PMSF)が含まれた結合緩衝溶液(binding buffer;5mMイミダゾ−ル、0.5M NaCl、20mM Tris−HCl、pH7.9)を入れて超音波処理(sonication)して収獲し、溶菌した。緩衝液A(buffer A;6Mウレア、20mM HEPES、pH8.0、100mM NaCl)に入れ超音波処理して細胞を破砕させた後、遠心分離を2回(16、500rpm×30min、40,000rpm×30min、4℃)連続して行って、不溶性の細胞破片(cell debris)を除去した。その後、溶菌液(lysate)をNi+++−IDAコラムを通じて精製した。コラムは先ず6Mウレアがない結合緩衝液で洗浄した後、洗浄緩衝溶液(80mMイミダゾ−ル、0.5M NaCl、20mM Tris−HCl、pH7.9)で洗浄した。蛋白質は溶出緩衝液(1mM イミダゾ−ル、0.5M NaCl、20mM Tris−HCL、pH7.9)で溶出した後、PD−10コラムクロマトグラフィ−(Amersham)を利用して蛋白質に含まれた塩分を除去した(図2)。精製されたGFP融合蛋白質は約30kDaのサイズに発現、精製され、Tat−GFP、GFP−Tat融合蛋白質はTat輸送ドメインのサイズ(約1kDa)ほどより大きいサイズに発現、精製され、Tat−GFP−Tat融合蛋白質は両側にTat輸送ドメインが融合されているので、Tat−GFP、GFP−Tat融合蛋白質より一つのTat輸送ドメインのサイズほどもっと大きい分子量で発現され精製された。
【0053】
変性されない自然状態の輸送ドメイン−GFP融合蛋白質は上記言及された変性剤なしで同一の過程で得られた。各分画の蛋白質濃度は、SDS−PAGEに分離した後、密度測定分析方法で牛胎血清アルブミン(BSA)をスタンダ−ドとして定量した。蛋白質濃度はブレドフオ−ド蛋白質アッセイ(Biorad)で測定した。精製された融合蛋白質等は、20%グリセロ−ルが含まれたPBSに溶解され−80℃で貯蔵された。
【0054】
実施例3:細胞培養と融合蛋白質の細胞内伝達実験
HeLa細胞は37℃で20mM HEPES/NaOH(pH7.4)、5mM NaHCO3、10%牛胎血清(fetal bovine serum;FBS)及び抗生剤(100μg/ml ストレプトマイシン、100U/ml ペニシリン)が含まれたDMEM(Dulbecco’S Modified Eagle’s Medium)において培養した。
【0055】
9個のアミノ酸(49−57)からなるHIV−1 Tat蛋白質輸送部分をアミノ酸末端とカルボキシル末端に結合させた変異体輸送ドメインに対する化物分子細胞透過能を観察するために次のように実験した。即ち、輸送ドメイン−GFP、GFP−輸送ドメイン、輸送ドメイン−GFP−輸送ドメイン融合蛋白質等の細胞内透過を観察するために、HeLa細胞を6−ウエルプレ−トで4〜6時間養った後、10%FBSが含まれた新鮮なDMEM培養液で交替し、多くの濃度のGFP融合蛋白質を培養液内に処理した。37℃で1時間培養した後、細胞をりん酸緩衝液生理食塩水(phosphate buffered saline;PBS)で十分に洗浄し、10分間トリプシン−EDTA(Gibco BRL)で処理した。細胞を粉砕した後、細胞内に透過された輸送ドメイン−GFP融合蛋白質の量を次の実施例のようにウエスタン−ブロット分析で測定した。
【0056】
実施例4:ウエスタン−ブロット分析
Tat輸送ドメインがアミノ酸末端に一つが融合されている蛋白質と両側に融合されている蛋白質間の細胞内移動効率を分析するために、ウエスタン−ブロット実験方法を行った。先ず、蛋白質等を変性された状態で精製した。準備されたHeLa動物細胞に1Mの変性された融合蛋白質を処理した後、1時間後に、細胞等だけを集めてウエスタン−ブロットを行った。細胞を溶菌して得られた細胞溶菌液を溶菌緩衝液(125mM Tris−HCl pH6.8、2% SDS、10% v/vグリセロ−ル)がある6−ウエル プレ−トに入れた。全体細胞溶菌液各15μgずつをSDS−12%ポリアクリルアミドジェル上で電気移動させた。
【0057】
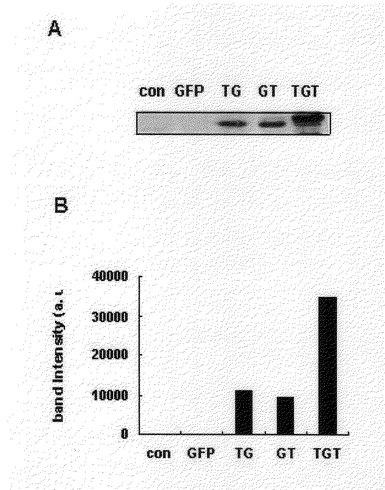
電気移動で分離された蛋白質をニトロセルロ−スメンブレイン(nitrocellulose membrane;Amersham,UK)へ移動させた。蛋白質が移動されたニトロセルロ−スメンブレインを10%粉乳を含むPBSでブロッキングした。次いで、メンブレインはラビット抗−GFPポリク−ロン抗体(polyclonal antibody;Clontech、USA、1:1,000)で処理した。その後、セイヨウワサビペルオキシダ−ゼ−(horseradish peroxidase)結合されたゴ−ト抗−ラビット(goat anti−rabbit)IgG抗体(Sigma、1:10,000希釈)と反応させた。結合された抗体等は、化学発光にて確認することができた(ECL;Amersham)。その結果、図3で現したように、Tat輸送ドメインがアミノ酸末端にのみある融合蛋白質と、Tat輸送ドメインがカルボキシル末端にある融合蛋白質は、同様に細胞内に移動した。しかし、Tat輸送ドメインが両側の末端に融合されている蛋白質は、一つのTat輸送ドメインが連結されている融合蛋白質より効果的に細胞内に移動した。この結果は、Tat輸送ドメインが融合蛋白質のアミノ酸末端とカルボキシル末端のいずれ側に融合されてあってもその効果は類似するが、Tat輸送ドメインが両側に融合されておれば、細胞内への移動効果が向上することを示す。
【0058】
実施例5:共焦点蛍光分析(confocal microscopy)による細胞導入分析
目標細胞に融合蛋白質を伝達しようとするとき、伝達される細胞の割合がいくらになるかは重要である。また、細胞内に透過された融合蛋白質がその固有な活性を維持しなければ、このような蛋白質伝達技術が応用できない。そこで、本実験において、GFP融合蛋白質が伝達される細胞の割合や伝達された融合蛋白質の活性の有無を分析した。変性されたGFP融合蛋白質を処理したHeLa細胞からGFPが現れる蛍光度を蛍光顕微鏡で観察した。
【0059】
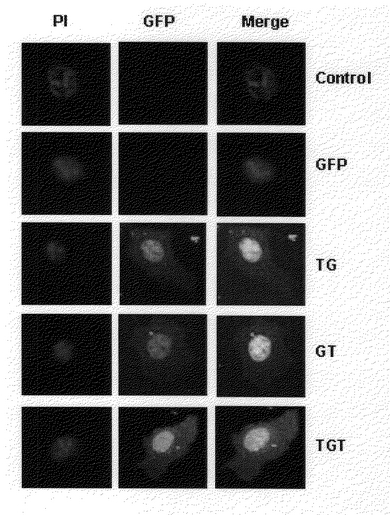
カバ−スリップ上で50〜70%のコンフルエンシ(confluency)になるように細胞を培養した次の両側末端に輸送ドメインが結合されているGFP−融合蛋白質を1M処理し、15分間培養した。HeLa細胞等はPBSで2回洗浄した後、トリプシン処理し、室温で5分間ホルムアルデヒドが含有されたPBSで固定された。細胞等はさらにPBSで洗浄した後、核だけを染色するために2g/mlのプロピジウムアイオダイド(propidium iodied;PI)を15分間処理した。細胞等はさらにPBSで洗浄した後、マウンテイング溶液(mounting solution;90%グリセロ−ル、0.1%フエニレンジアミンが含まれたりん酸緩衝液食塩水(PBS))を処理し、カバ−ガラスを被せて共焦点蛍光顕微鏡を利用して488nmと545nm蛍光フイルタ−で細胞内の蛍光の有無を観察、蛋白質の細胞内分布を分析した(Eric et al.,1997)。その結果、Tat輸送ドメインがアミノ酸末端に融合されている蛋白質とカルボキシル末端に融合されている蛋白質は殆ど細胞質のみならず核まで到達した(図4)。これは、ウエスタン−ブロットの結果と同じく、Tat輸送ドメインが蛋白質のどの部分に融合されていても、その効果が類似することを示す。その反面、Tat輸送ドメインが両側に付いた融合蛋白質は、一方にのみ存在する融合蛋白質より明るい蛍光を現した。加えて、Tat輸送ドメインが両側に付いた融合蛋白質は、より多くの核内に移動していることが確認された。
【0060】
実施例6:FACS(flow cytometry)分析による細胞導入分析
9個のアミノ酸(49−57)からなるHIV−1 Tat蛋白質輸送部分をアミノ酸末端とカルボキシル末端両側に結合させた変異体輸送ドメインに対する化物分子細胞透過能を観察するために次のように実験した(図5)。即ち、輸送ドメイン−GFP−輸送ドメイン融合蛋白質の細胞内透過を観察するために、HeLa細胞を6−ウエル プレ−トにおいて4〜6時間培養した後、10%FBSが分布された新鮮なDMEM培養液で交替し、多数濃度のGFP融合蛋白質を培養液内に処理した。37℃で30分間培養した後、細胞をりん酸緩衝液生理食塩水(phosphate buffered saline;PBS)で十分に洗浄し、10分間トリプシン−EDTA(Gibco BRL)で処理した。細胞を集めて、さらにPBSで2回洗浄した後、4%パラホルムアルデヒド(paraformaldehyed)で1時間固定させた。固定させた細胞に対してFACSを利用して細胞内蛍光を分析した。FACS分析結果、図5のような曲線を得ることができた。上記の結果と同様、Tat輸送ドメインが一方にだけ融合されている蛋白質は似た量で細胞内に導入される反面、両側にTat輸送ドメインがある融合蛋白質は多くの量で細胞内に導入されることを分かることができた。平均蛍光度(Mean Fluorescence intensity)は、輸送ドメイン−GFPが23.86、GFP−輸送ドメインが17.21、輸送ドメイン−Tat−輸送ドメインは204.52であって、Tat−GFP−Tat融合蛋白質が特に高い数値を示した。その後、Tat輸送ドメインを両側に融合された蛋白質の量によって細胞内に導入されるメカニズムを分析するために多様な濃度で処理して30分間培養した後、FACS分析を行った(図6).この結果、Tat輸送ドメインを両側に有する融合蛋白質は、その濃度と共に平均蛍光度が増加することが確認できた。
【0061】
実施例7:FACS(flow cytometry)分析とウエスタン−ブロットによる自然状態の融合蛋白質と変性された融合蛋白質の細胞導入比較分析
自然状態で輸送ドメイン−GFPを精製して細胞内に導入したとき、変性させ精製した輸送ドメイン−GFPより導入されるときと比較して、その効果が制限される。輸送ドメインが両側の末端に存在するGFP融合蛋白質は変性して精製されたとき、一方にだけ存在するGFPより大きな効果を示すことから、輸送ドメイン−GFP−輸送ドメイン融合蛋白質が自然状態で精製されたとき、細胞内に導入される効果について検討すべく実験を行った。まず、変性された蛋白質と自然状態に精製した蛋白質をウエスタン−ブロットで分析した結果、事前に精製された蛋白質も輸送ドメインが一つ存在するGFP融合蛋白質と同じ数値で細胞内に導入されたときの結果を示した(図7)。また、自然状態に分離した輸送ドメイン−GFP−輸送ドメイン融合蛋白質の細胞内導入効果を知るためにFACS分析法を行った。それぞれ自然状態と変性された状態で精製された蛋白質をそれぞれの濃度に処理して1時間培養した。その結果図8のように変性された状態の輸送ドメイン−GFP−輸送ドメイン融合蛋白質は、1M処理したとき平均蛍光度が117である反面、自然状態に精製した輸送ドメイン−GFP−輸送ドメインは54である数値を示した。これは、変性された状態の融合蛋白質の細胞内導入効果より劣っているが、図7と比較すると、変性して精製した輸送ドメイン−GFP融合蛋白質程度の細胞内導入能力を有しているのが分かった。結果的に、Tat輸送ドメインを一方の末端に有する融合蛋白質よりTat輸送ドメインを両側に有しており、変性して(denatured)精製した融合蛋白質が効果的に細胞内浸透をすることができ、自然状態(native)に精製した輸送ドメイン−GFP−融合蛋白質もまた細胞内浸透が可能であることを結論づけることができた。
【0062】
実施例8:組み替えSOD融合蛋白質等の発現ベクタ−製造
機能を有する蛋白質又はペプチドを細胞内に浸透させる技術を開発するために細胞内に目標蛋白質を伝達し得る融合蛋白質発現ベクタ−を製造し、輸送ドメインが蛋白質を細胞内に伝達する能力を容易に分析するために、ヒトCu,Zn−ス−パ−オキシドジスムタ−ゼ(Cu,Zn−superoxide dismutase;以下、本明細書ではSODと略称する)蛋白質を選んだ。
【0063】
組み替え融合蛋白質を過大に発現させるために、SOD、HIV−1Tatの形質導入部(Tat 49−57)及び6個のヒスチジンに対するcDNAが連続的に含まれているTat−SOD、SOD−Tat、Tat−SOD−Tat発現ベクタ−を製造した(図9)。融合蛋白質に対する対照蛋白質であるSODを過大に発現させるために、Tatの形質導入部位だけ含まれておらず、残りの部位は同一なpET−SOD発現ベクタ−を製造した。
【0064】
Tat基本ドメインに該当する2種類のオリゴヌクレオチド[上位鎖(top strand)、5‘−TAGGAAGAAGCGGAGACAGCGACGAAGAC−3’(配列番号3)と下位鎖5‘−TCGAGTCTTCGTCGCTGCTCCGCTTCTTCC−3’(配列番号4)]をNdeI−XhoI制限酵素で切ったpET−15bベクタ−に結さつ(ligation)して挿入した後、ヒトSODのcDNAの配列を基本として2種類のオリゴヌクレオチドを合成した。正方向プライマ−(Forward Primer)はXho I 制限部位を有しており、逆方向プライマ−(reverse primer)はBamH I制限部位を有している。重合酵素連鎖反応(PCR)を行なった後、反応物を分離し、これをTAクロニングベクタ−に結さつして形質変換させプラスミドを分離した。ヒトSOD cDNAがTAベクタ−をXho I とBamH Iで切断した後、pET−15b及びpET−15b−Tat発現ベクターに挿入した。
【0065】
また、Tat塩基性ドメインがSOD両側末端に融合されたpTat−SOD−TatはpTat−SODにTat塩基性ドメイン9個アミノ酸をコ−デイングする2重鎖オリゴヌクレオチドがアニ−ルされ、カルボキシル末端に挿入され、Tat塩基性末端がカルボキシル末端に融合されたpSOD−TatはpSODにTat塩基性ドメイン9個のアミノ酸をコ−デイングする2重鎖オリゴヌクレオチドがアニ−ルされ、カルボキシル末端に挿入された(図9)。
【0066】
実施例9:組み替えSOD融合蛋白質等の発現及び精製
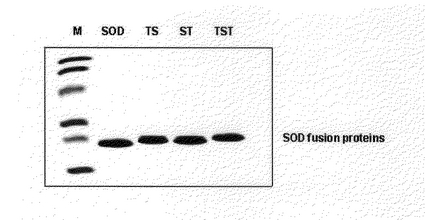
本研究室で製造したヒトCu,Zn−ス−パ−オキシドジスムタ−ゼcDNAが含まれているE.coli BL21(DE3)細胞(pSOD及びpTat−SOD、pSOD−Tat、pTat−SOD−Tat)をアンピシリンが含まれているLB培地に入れ37℃で200rpmに撹拌して培養した。培養液内のバクテリア濃度が0.D600=0.5〜1.0を表す時、IPTGを培地内に添加して最終濃度が0.5mMになるようにした後、3〜4時間さらに培養した。培養した細胞を遠心分離して集めた後、6M Ureaが添加された結合緩衝溶液(binding buffer;5mM imidazole、0.5M NaCl、20mM Tris−HCl、pH7.9)5ml入れ超音波粉砕器で粉砕(sonication)した。遠心分離して上層液を直ちにNi−ニトリロトリアセチックアクシド セパロズス−パ−フロウ(Ni−nitrilotriacetic acid sepharose super flow)コラムに負荷し、10倍容積の結合緩衝溶液(binding buffer)と6倍容積の洗浄緩衝溶液(washing buffer;60mM imidazole、0.5M NaCl、20mM Tris−HCl、pH7.9)で洗浄した後、溶出緩衝溶液(elition buffer;1M imidazole、0.5M NaCl、20mM Tris−HCl、pH7.9)で融合蛋白質を溶出した。次いで、融合蛋白質が含まれた分画等を集めてPD−10コラムクロマトグラフイ−(column chromatography)を行なって分画中に含まれた塩分を除去した。融合蛋白質は6個のヒスチジンを含んでいるため、金属イオン−キレ−ト親和クロマトグラフイ−単一段階に融合蛋白質を純粋に(純度>90%)精製された(図10)。
【0067】
分画の蛋白質濃度は牛血清アルブミンを標準物質に使用してブレッドフオ−ド(Bradford)方法で測定した。
【0068】
実施例10:HeLa細胞培養及び組み替え融合蛋白質等の細胞内透過
HeLa細胞は37℃で95%空気と5%CO2とを供給し、20mMHEPES/NaOH(pH7.4)、5mM NaHCO3、10%牛胎血清(fetal bovine serum;FBS)及び抗生剤(100μg/ml ストレフトマイシン、 100U/ml ペニシリン)が含まれたDMEM(Dulbecco‘s Modified Eagle’s Medium)で培養した。
【0069】
9個のアミノ酸(49−57)からなるHIV−1 Tat蛋白質輸送部分をアミノ酸末端とカルボキシル末端に結合させた変異体輸送ドメインに対する化物分子細胞透過能を観察するために次のように実験した。即ち、Tat−SOD、SOD−Tat、Tat−SOD−Tat融合蛋白質等の細胞内透過を観察するために、HeLa細胞を6−ウエルプレ−トで4〜6時間培養した後、10%FBSが含まれた新鮮なDMEM培養液に交換し、2M SOD融合蛋白質を培養液内に処理した。1時間後、細胞をトリプシン−EDTA(Gibco BRL)で処理し、リン酸緩衝液生理食塩水(phosphate buffered saline;PBS)で十分に洗浄した。細胞を粉砕した後、細胞内に透過されたSODの量と活性度をSOD活性度分析とウエスタン−ブロッド分析で測定した。
【0070】
実施例11:ウエスタン−ブロッド分析
Tat蛋白質がアミノ酸末端に1個が融合されている蛋白質と、両側に融合された蛋白質の細胞内移動効率を分析するためにウエスタン−ブロット方法を行なった。先ず、蛋白質等は変性された状態で精製した。準備されたHeLa細胞に2Mの変性された融合蛋白質を処理した後、1時間後に細胞等だけを集めてウエスタン−ブロットを行なった。細胞を溶菌して得られた細胞溶菌液を溶菌緩衝液(125mM Tris−HCl pH6.8、2% SDS、10% v/v グリセロ−ル)を有する6−ウエルプレ−トに入れた。全体細胞溶菌液各15μgずつをSDS−12%ポリアクリルアミドジェル上で電気移動させた。
【0071】
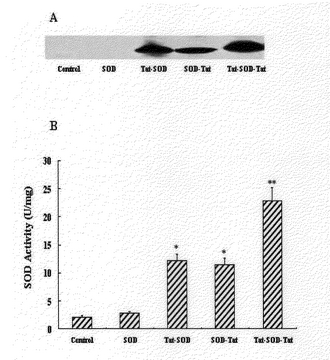
電気移動で分離された蛋白質をニトロセルロ−スメンブレイン(nitrocellulose membrane;Amersham,UK)で移動させた。蛋白質が移動されたニトロセルロ−スメンブレインを5%脱粉乳(non−dry milk)が入っているPBSでブロキングした。次いで、メンブレインはゴト抗−SODポリク−ロン抗体(polyclonalantibody;Santacruze、USA、1:1,000)で処理した。その後セイヨウワサビペルオキシダ−ゼ(horse radish peroxidase)−結合されたマウス抗−ゴ−ト(mouse anti−rabbit)IgG抗体(Sigma、1:10,000希釈)と反応させた。結合された抗体等は化学発光で確認することができた(ECL;Amersham)。その結果、図11aのようにTat蛋白質がアミノ酸末端にだけある融合蛋白質とTat蛋白質がカルボキシル末端にある融合蛋白質は共に細胞内に透過された。しかし、Tat蛋白質が両側の末端に融合されている融合蛋白質は1個のTat蛋白質が連結されている融合蛋白質より効果的に細胞内に移動した。この結果は、Tat蛋白質が融合蛋白質のアミノ酸末端とカルボキシル末端のいずれかの側に融合されているものの効果は類似する、Tat蛋白質が両側に融合されているものが細胞内移動効果が大きく向上されていることを示す(図11a)。
【0072】
実施例12:SOD酵素活性度測定
本研究において、SODの活性度は、McCordとFridovichの方法(1969)によってキサンチン/キサンチンオキシダ−ゼ(xanthine/xanthine oxidase)反応によるペリシトクロム(ferricytochrome)c還元の抑制程度を分光光度計で観察することにより測定した(McCord、JM and Fridovich、I.(1969)J.Biol.Chem.244、6049−6055)。
【0073】
標準分析方法は25℃で2mlの0.1mM EDTAが含まれた50mMリン酸カリウム緩衝溶液(pH7.8)において行なった。反応混合液には10uM ferricytochrome c、50uMキサンチン(xanthine)及び十分な量のキサンチンオキシダ−ゼ(xanthine oxidase)を含有しており、550nmにおいてフエリシトクロム(ferricytochrome)cの還元程度を測定した。上記の条件下においてシトクロム(cytochrome)cを50%減少させるス−パ−オキシドジスムタ−ゼの量を1単位に定義した。
【0074】
細胞内に透過された溶融蛋白質は、その固有な活性を維持しなければ蛋白質治療に有用することができない。よって、細胞内に透過された融合蛋白質が生物学的な活性を有することは大変重要な問題である。図11bは2Mの融合蛋白質等を1時間HeLa細胞培地に透過したとき、細胞内のSOD酵素活性の変化を表した結果である。溶融蛋白質を投与しなかった細胞からSOD活性度は2.46±0.39U/mg蛋白質であり、Tat−SODとSOD−Tat融合蛋白質を投与した時にSOD活性度は12.68±1.44U/mg蛋白質として有意に上昇し、Tat−SOD−Tat融合蛋白質を投与した時にSOD活性度は23.75±2.35U/mg蛋白質でTat−SODとSOD−Tat融合蛋白質より2倍以上高いSOD活性度を表した(図11b)。よって、Tat−SOD−Tat融合蛋白質の細胞内透過及び活性がTat−SODとSOD−Tat融合蛋白質より2倍以上大きく増加されることを示した(図11)。
【0075】
実施例13:アルギニン9−GFP−アルギニン9融合蛋白質の細胞浸透活性測定
上記実施例1ないし3の方法と類似な方法で、9個のアルギニン残基で構成された輸送ドメインが緑色蛍光蛋白質のアミノ基及びカロボキシ基に共有結合されたオリゴリシン−GFPオリゴリシン融合蛋白質を製造した。
【0076】
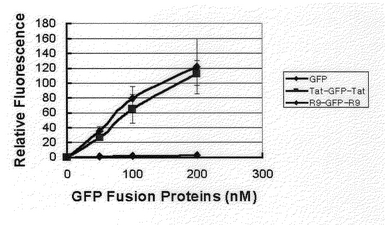
Tat−GFP−Tat融合蛋白質とアルギニン9−GFP−アルギニン9(R9−GFP−R9)融合蛋白質の細胞浸透効率を比較するために、各融合蛋白質50、100、200nMずつをHeLa細胞が入っている24ウエルプレ−トに加え、1時間が経過した後、細胞をトリプシン処理し、PBSで数回洗浄した後、蛍光をフルオロメ−タ−で測定した。結果は図12のとおりである。
【0077】
実施例14:リシン9−GFP−リシン9融合蛋白質の細胞浸透活性測定
実施例1ないし3の方法と類似の方法で9個のリシン残基で構成された輸送ドメインが緑色蛍光蛋白質のアミノ基及びカロボキシ基に共有結合されたオリゴリシン−GFPオリゴリシン融合蛋白質を製造した。
【0078】
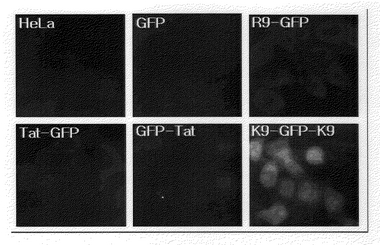
Tat−GFP融合蛋白質、GFP−Tat融合蛋白質、アルギニン9−GFP融合蛋白質とリシン9−GFP−リシン9(K9−GFP−K9)融合蛋白質の細胞浸透効率を比較するために、各融合蛋白質200nMずつをHeLa細胞が入っている24ウエルプレ−トに加えた後、1時間が経過した後、細胞をトリプシン処理し、PBSで数回洗浄した後、蛍光を蛍光顕微鏡で測定した。結果は図13のとおりである。
【産業上の利用可能性】
【0079】
本発明は機能を有する蛋白質を蛋白質水準で細胞内に透過させるにおいて、既存の塩基性輸送ドメインを1側に結合させたものよりもっと高い透過効率と、細胞内活性を有する溶融蛋白質を提供することができる。
【0080】
また、本発明の融合蛋白質を有効成分とする薬剤学的組成物及び化粧品等を提供することができる。
【0081】
更に、本発明はヒトGu,Zn−ス−パ−オキシド ジスムタ−ゼ(Cu,Zn−SOD)を蛋白質水準で細胞内に直接透過させるもので、特に細胞内に投与された−輸送ドメイン−SOD−輸送ドメイン融合蛋白質はSOD活性度を回復して蛋白質治療に効率的に活用され得ることを提示する。
【0082】
活性酸素種は、細胞内の生体高分子に損傷を加え、報告によれば、100余種の疾病の進行過程と深く関連されている。よって、本発明はこのような活性酸素種を除去する際に主な役割を担当するSODを細胞内に投与することにより、疾患を治療する蛋白質治療に本発明の輸送ドメイン−SOD−輸送ドメイン融合蛋白質が効果的に活用され得る。
【0083】
本技術を基にして抗酸化酵素の一種であるSODを細胞内に直接伝達して我らの人体に有害な活性酸素を除去することができ、多くの疾病以外にも化粧品及び健康食品産業等に本技術を広範囲に活用することができる。
【図面の簡単な説明】
【0084】
【図1】Tat−GFP、GFP−Tat、Tat−GFP−Tat、GFP発現ベクタ−の概略図である。合成されたTat輸送ドメインはGFPのアミノ末端やカルボキシル末端、又は、両側末端に融合してベクタ−を製造した。GFP発現ベクタ−pGFPはGFPのコ−デイング配列をpET15bに挿入して構成された。
【図2】精製された輸送ドメイン−GFP融合蛋白質を12%SDS−PAGEに分離した後、クマシ−ブル−(Coomassie blue)で染色したものである。レイン M:SDS Markerレイン GFP:GFPレイン TG:Tat−GFPレイン GT:GFP−Tatレイン TGT:Tat−GFP−Tat
【図3】A:変性されたTat−GFP、GFP−Tat、Tat−GFP−Tat融合蛋白質の細胞内導入分析結果であって、1M Tat融合蛋白質等を1時間HeLa細胞内に導入させた時、細胞内に導入された蛋白質をウエスタン−ブロット(western blot)法で分析したものである。B:細胞内に導入された蛋白質をウエスタン−ブロットした結果を基に、結果に表れた蛋白質等線の強さを図式化したものである。
【図4】細胞内に導入されたTat−GFP、GFP−Tat、Tat−GFP−Tat融合蛋白質を公焦点蛍光顕微鏡で可視化したものである。各セクションは、それぞれ次のとおりである。Control:何も処理しない細胞GFP:対照群 GFP 1MTG: Tat−GFP 1MGT:GFP−Tat 1MTGT:Tat−GFP−Tat 1M
【図5】FACS分析法によってGFP融合蛋白質の細胞内導入効率を比較分析したものである。各融合蛋白質等は2M濃度で30分間処理した。TG:Tat−GFPGT:GFP−TatTGT:Tat−GFP−Tat
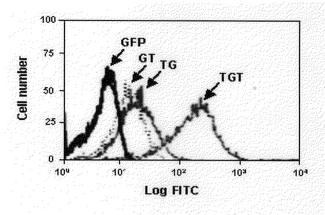
【図6】Tat−GFP−Tat融合蛋白質を濃度別に処理して30分間培養した結果をFACS分析法によって分析した結果。対照群GFPは最高濃度である2Mで処理した。
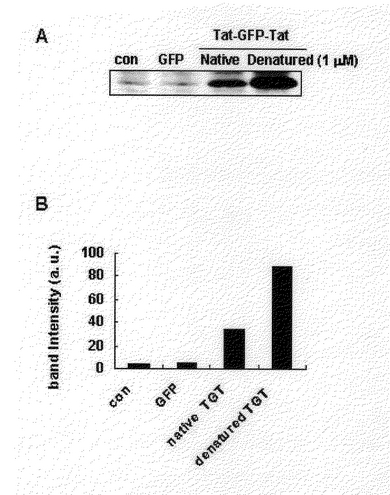
【図7】A:Tat−GFP−Tat融合蛋白質を自然状態と変性された状態で精製して細胞内導入効率を分析するためにウエスタン−ブロットである。各融合蛋白質等は1Mで処理して1時間培養した。レインcon:処理しない細胞レインGFP:対照群 GFPレインnative:自然状態に精製したTat−GFP−Tatレインdenatured:変性された状態に精製したTat−GFP−TatB: 図7Aの結果に表れた線の強さを図表で模式化したものである。
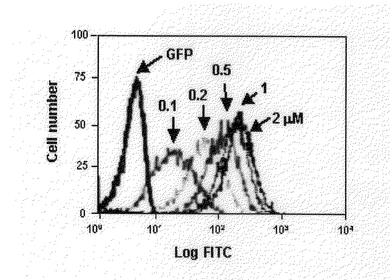
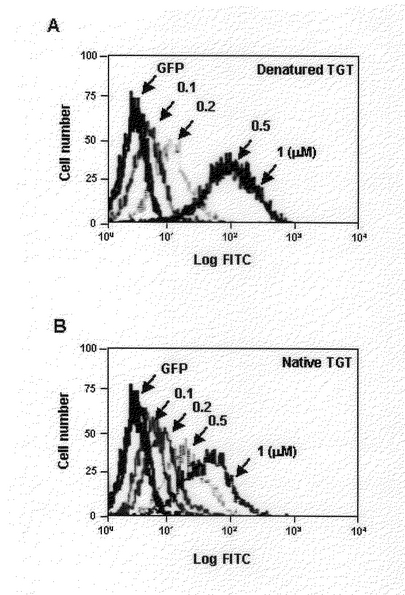
【図8】FACS分析法によって自然状態に精製されたTat−GFP−Tatと変性された状態に精製されたTat−GFP−Tatの細胞内導入効果を濃度別に1時間処理して分析したものである。A:変性された状態に精製されたTat−GFP−Tatを0.1M、0.2M、0.5M、1M処理して分析した結果である。B:自然状態に精製されたTat−GFP−Tatを0.1M、0.2M、0.5M、1M処理して分析した結果である。
【図9】Tat−SOD、SOD−Tat、Tat−SOD−Tat、SOD発現ベクタ−の概略図である. 合成されたTat蛋白質をSODのアミノ末端やカルボキル末端、又は両側末端に融合してベクタ−を製造した。SOD発現ベクタ−であるpSODはSODのコ−デイング配列をpET−15bベクタ−に挿入して製造した。
【図10】精製された輸送ドメイン−SOD融合蛋白質等を12%SDS−PAGEに分離した後、クマシ−ブル−で染色したものである。レイン M:SDS Markerレイン SOD:SODレイン TS:Tat−SODレイン ST:SOD−Tatレイン TST:Tat−SOD−Tat
【図11】A:変性された状態に精製したTat−SOD、SOD−Tat、Tat−SOD−Tat融合蛋白質の細胞内導入分析結果であって、2M Tat融合蛋白質を1時間HeLa細胞内に導入させたとき、細胞内の導入された蛋白質をウエスタン−ブロット法で分析したものである。B:2M融合蛋白質等を1時間HeLa細胞に投与したとき、細胞内に導入されたSOD酵素の活性度変化を示したグラフである。
【図12】Tat−GFP−Tat融合蛋白質とアルギニン9−GFP−アルギニン9(R9−GFP−R9)融合蛋白質の細胞浸透効率を蛍光を測定して図式化したグラフである。
【図13】GFP、Tat−GFP融合蛋白質、GFP−Tat融合蛋白質、アルギニン9−GFP融合蛋白質とリシン9−GFP−リシン9(K9−GFP−K9)融合蛋白質の細胞浸透効率を比較するために、各融合蛋白質200nMずつをHeLa細胞に加えて1時間が経過した後、蛍光顕微鏡で観察した写真である。
【配列表】
【技術分野】
【0001】
本発明は機能を有する蛋白質又はペプチドをより高い効率で細胞内に導入させる融合蛋白質とその用途に関するものである。
【背景技術】
【0002】
最近、様々な疾患が細胞蛋白質の非正常的な活性に起因するという事実が知られてきているため、これら蛋白質の活性を調節することにより致命的な人体疾患を治療できる薬剤の開発が全世界的関心の対象となっている。
【0003】
その一例として、活性酸素種と関連されたス−パ−オキシドジスムタ−ゼ(Superoxide Dismutase)を細胞蛋白質の例として挙げる(但し、本発明の意図する範囲はこの蛋白質だけを目標蛋白質に限定するものではない。)。
【0004】
活性酸素種(reactive oxygen species)は、エネルギ−を得る全ての生命体から、酸素を利用して多くの細胞内代謝過程の副産物として必然的に生成される。このような活性酸素種は、細胞内の蛋白質、核酸及び脂肪のような生体高分子に損傷を与え、人体の多くの疾病の進行過程と深く関連されている。特に発癌過程、脳卒中、関節炎、動脈硬化、放射線損傷及び炎症反応に関与し、正常的な老化過程においても老化を促進させる重要な要因として作用する(非特許文献1〜3)。
【非特許文献1】Floyd,R.A.(1990)FASEB J.4,2587−2597.
【非特許文献2】Anderson,W.F.(1988)Human gene therapy.Nature392,25−30.
【非特許文献3】Halliwell B.andGutteridge J.M.C.(1999)Freeradicals inbiology and medicine,Oxford University Press,Oxford.
【0005】
Cu,Zn−ス−パ−オキシドジスムタ−ゼと関連された疾病等を簡略に検討すれば表1のとおりである。
【0006】
【表1】
【0007】
公知の活性酸素種としては1O2、OH、O2、H2O2等があり、これらは生体内で各種の酵素反応によって生成され、各種の生理活性物質の生合成、免疫機能、薬物の代謝等において大変重要な役割をするが、外部からの放射線、紫外線、公害及び各種のストレス等によって過剰生成される場合には、反って生体に損傷を来すことがある。そこで、生体は防御機能として、SOD、カタラ−ゼ(Catalase)、ペルオキシダ−ゼ(Peroxidase)等の酵素を有するが、老化が始まると皮膚の均衡が壊れるようになり、各種の活性酸素類に対する皮膚保護能力が劣るようになる。
【0008】
よって、このような活性酸素類等から細胞を保護する必要性が強くなってきており、化粧品原料としての活性酸素除去剤として、SOD、ラクトフエリン、酸化防止剤等が使用ないし開発されている。しかしながら、SODは、活性酸素除去能を有するにもかかわらず、酵素蛋白質という性質により地質に対し難溶性であり、安定性が悪く、分子量が30,000ダルトン(Dalton)以上で皮膚角質層の透過が不可能であるという問題点があるため、化粧料組成物として有用に利用されなかった。
【0009】
Cu,Zu−ス−パ−オキシドジスムタ−ゼは、自由ラジカルの害毒と酸素の損傷から細胞の損傷を防止する細胞内重要な防御酵素である(非特許文献4)。全ての生体内高分子等は、このような酸素ラジカルの有害な作用に常に晒されているため、多くの疾患においてCu,Zn−ス−パ−オキシドジスムタ−ゼを治療目的に利用しようとする関心が非常に高くなっている。
【非特許文献4】Fridovich,I.(1995)Annu.Rev.Biochem.64,97−112.
【0010】
最近、Cu,Zn−ス−パ−オキシドジスムタ−ゼを臨床的に応用するために多くの方法が示されている。現在まで開発されたCu,Zn−ス−パ−オキシドジスムタ−ゼの生体内運搬方法は大きく3種に区分できる。
【0011】
第1に、Cu,Zn−ス−パ−オキシドジスムタ−ゼにポリエチレングリコ−ル(polyethylene glycol)、フイコ−ル(Ficoll)、レシチン(lecithin)、アルブミン(albumin)等を集合させる方法がある。
【非特許文献5】Del Zoppo,g.J.et al.(1997)Drugs 54,9−38
【非特許文献6】Muzykantov, V.R.et al.(1996)Proc.Natl.Acad.Sci.USA93,5213−5218.
【0012】
第2に、リポソ−ム(liposome)を利用してCu,Zn−ス−パ−オキシドジスムタ−ゼをカプセル化させる方法がある。
【非特許文献7】Perdereau,B.et al.(1994)Bull.Cancer 81,659−669.
【0013】
第3に、Cu,Zn−ス−パ−オキシドジスムタ−ゼ遺伝子を形質導入して細胞内で酵素の過多発現を誘導する遺伝子治療等の試みが示されている。
【非特許文献8】Okumura,K. et al.(1997)Pharm.Res.14,1223−1227
【非特許文献9】Lehmann,T.G.et al.(2000)Transplantation 69,1051−1057.
【非特許文献10】Liu,R.et al.(1997)Hum.Gene Ther.8,585−595.
【0014】
これらのうち、もっとも注目を受けているものは、遺伝子治療であり、Cu,Zn−ス−パ−オキシドジスムタ−ゼ遺伝子治療を疾病に適用しようとする多くの研究が行われてきた。しかし、遺伝子治療は遺伝子の細胞内への運搬が容易でなく、標的細胞における発現率が低く、発現される細胞で蛋白質が発現される期間が短く、標的細胞から発現される蛋白質の量を人為的に調節するのが非常に難しい等、様々な問題点を有している。
【非特許文献11】Verma,I.M.et al(1997)Nature389,239−242.
【0015】
治療のための薬物や蛋白質を細胞内に移動させることに関しては、他の方法として、目標蛋白質を細胞膜を経て直接伝達する方法が考えられる。しかし、治療用薬物や蛋白質は、そのサイズ又は多くの生化学的性質のため細胞膜を通過することが大変難しい。一般的に分子量600以上の物質は細胞膜を通過するのが殆ど不可能であることが知られている。
【0016】
最近になって、ヒト免疫欠乏ビ−ルス(Human Immunodeficiency Virus type−1)蛋白質の一種であるTat(transactivator of transcription)蛋白質が、効率的に細胞膜を通過して細胞質内に容易に移動するということが明らかになった。このような機能はTat蛋白質の中間部位である蛋白質形質導入部位(Protein Transduction Domain)の特性のため表れ、未だその正確なメカニズムは知られていない状態である(非特許文献12〜15)。しかし、Tat蛋白質の細胞膜通過には特定受容体や運搬体が関与しないことが見出されており、Tat蛋白質の蛋白質形質導入部位が、直接膜の地質二重層と作用することにより起こるものと理解されている(非特許文献16、17)。
【非特許文献12】Frankel,A.D.andPabo,C.O.(1988)Cell 55,1189−1193.
【非特許文献13】Green,M. and Loewenstein,P.M.(1988)Cell55,1179−1188.
【非特許文献14】Ma,M. and Nath,A.(1997),J.Virol.71,2495−2499.
【非特許文献15】Vives,E.,Brodin,P.and Lebleu,B.(1997),J.Biol.Chem.272,16010−16017.
【非特許文献16】Vives,E.et al.(1997)J.Biol.Chem.272,16010−16017.
【非特許文献17】Derossi,D.et al.(1996),J.Biol.Chem.271,18188−18193.
【0017】
最近の研究において,オバルブミン(ovalbumin)、β−ガラクトシダ−ゼ(galactosidase)、セイヨウワサビ ペルオキシダ−ゼ(horseradish peroxidase)のような異形蛋白質をHIV−1 Tat蛋白質と融合させて投与した際、生体の各組織及び培養された細胞内に直接運搬されるとのことが発表された(非特許文献18〜20)。このような実験結果は、Tat蛋白質がそれ自身だけではなく、他の種類の巨大な蛋白質も細胞内に一緒に運搬できる能力を有することを意味している。
【非特許文献18】Fawell,S.et al.(1994),Proc.Natl.Acad.Sci.USA91,664−668.
【非特許文献19】Scwartze,S.R.et al.(1999),Science.285,1569−1572.
【非特許文献20】Watson,K.and Edward,R.J.(1999),Biochem.Pharmacol.58,1521−1528.
【0018】
しかしながら、実際には、全ての蛋白質がTat蛋白質によって運搬されるものではない。また、Tatによって細胞内に運搬された全ての蛋白質が生物学的活性を表すかも未だ明らかになっていないのが現状である。
【0019】
本発明者は、HIV Tat 49−57輸送ドメインの外にもリシン(lycine)が6ないし12個結合されたオリゴリシン輸送ドメイン、HIV Tat 48−57残基のうち、2、3個の残基が欠如した塩基性輸送ドメインについて、アミノ基に共有結合された融合蛋白質への細胞透過実験を行っており、これらが円滑な細胞透過能を有すること示している
【特許文献1】韓国特許公報第2002−100446号
【特許文献2】韓国特許公報第2002−67108号
【発明の開示】
【発明が解決しようとする課題】
【0020】
本発明の目的は、目標蛋白質を細胞内に導入又は発現させる際に、従来技術の問題点を解決し、高効率で細胞内に導入させ、細胞内で活性を有するようにしようとするものである。
【0021】
また、本発明はス−パ−オキシドジスムタ−ゼを細胞内に導入又は発現させる際に、従来技術の問題点を解決し、高効率で細胞内に導入させ、細胞内で活性を有するようにしようとするものである。
【0022】
また、本発明は、輸送ドメインと融合されたス−パ−オキシドジスムタ−ゼを利用して皮膚表皮、真皮層、皮下脂肪層まで浸透して卓越した活性酸素種の除去能を有する化粧料を提供するものである。
【課題を解決するための手段】
【0023】
上記、課題を解決するために、本発明者らは、輸送ドメインとヒトCu,Zn−ス−パ−オキシドジスムタ−ゼを融合させ、該融合蛋白質が効果的に細胞内に透過されるかをHeLa細胞を利用した実験において確認した。また、輸送ドメイン−ス−パ−オキシドジスムタ−ゼ輸送ドメイン融合蛋白質を多量に生産する方法及び精製する方法を開発した。
【0024】
本発明は、機能を有する蛋白質又はぺプチドをより効率的に細胞内に運搬し、細胞内でもより高い活性を有するようにするために、輸送ドメインと融合させた融合蛋白質と該融合蛋白質を利用した薬剤組成物及び化粧料組成物を提供する。
【0025】
本発明の輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質は遺伝子組み替え方法の他にも、一般的な化学結合方法で製造することができる。
【0026】
本発明は、6ないし12個のアミノ酸残基から構成され、アルギニン又はリシン(lycine)残基を3/4以上含む輸送ドメインが目標蛋白質のアミノ基とカルボキシル基に共有結合された、細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質に関するものである。目標蛋白質の両末端に位置する輸送ドメインは必ず同一な残基からなっていなければならないものではない。
【0027】
また、本発明は、上記輸送ドメインが9個のアミノ酸残基で構成される細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質に関するものである。
【0028】
また、本発明は、上記輸送ドメインがHIV tat 49−57残基、オリゴリシン、オリゴアルギニン又はオリゴ(リシン,アルギニン)中の1種以上であることを特徴とする。
【0029】
更に、本発明は、上記目標蛋白質が治療分子、予防分子、診断分子から構成されたグル−プから選ばれたことを特徴とする。
【0030】
また、本発明は、上記目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物であることを特徴とする。
【0031】
また、本発明は、上記細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とすることを特徴とする化粧料組成物に関するものである。
【0032】
また、本発明は、上記目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物であることを特徴とする化粧料組成物に関するものである。
【0033】
また、本発明は、上記化粧料組成物が化粧水、ジエル、水溶性リキッド、水中油(O/W)型又は油中水(W/O)型であることを特徴とする。
【0034】
のみならず、本発明は上記輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とし、薬学的に許与される担体を含むことを特徴とする薬剤学的組成物に関するものである。
【0035】
また、本発明は、上記目標蛋白質がCu,Zn−ス−パ−ジスムタ−ゼ、又は、その機能的等価物であることを特徴とする。
【0036】
輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分として含有する薬剤学的組成物は薬剤学的分野で通常的に許容される担体と共に配合して通常的な方法によって経口又は注射形態に成型することができる。経口用組成物としては、例えば、錠製及びゼラチンカプセルがあり、これらは活性成分以外にも必要によって、希釈剤(例:ラクト−ス、デキストロ−ス、蔗糖、マンニト−ル、ソルビト−ル、セルロ−ス及び/又はグリシン)、滑沢剤(例:シリカ、タルク、ステアリン酸及びそのマグネシウム又はカルシウム塩及び/又はポリエチレングリコ−ル)を含有し、錠剤はまた結合剤(例:マグネシウムアルミニウムシリケ−ト、澱粉ペ−スト、ゼラチン、メチルセルロ−ス、ナトリウムカルボキシメチルセルロ−ス及び/又はポリビニルピロリドン)を含有し、場合によって、崩解剤(例:澱粉、寒天、アルギン酸、又はそのナトリウム塩)又は沸騰混合物及び/又は吸水剤、着色剤、抗味剤及び甘味剤を含有することが好ましい。注射用組成物は、等張性水溶液又は懸濁液が好ましく、これらの組成物は滅菌され、必要によって補助剤(例:防腐剤、安定化剤、湿潤剤、又は乳化剤溶液促進剤、浸透圧調節のための塩及び/又は緩衝剤)を含有する。又、これらはその他治療的に有用な物質を含有することができる。
【0037】
このように製造された薬剤学的製剤は、その目的により、経口投与するか、非経口方式、即ち、静脈内、皮下、腹腔内投与又は局所適用することができ、容量は1日の投与量が0.0001〜100mg/kgの量を1ないし数回に分けて投与することができる。特定患者に対する投与容量水準は、患者の体重、年齢、性別、健康状態、投与時間、投与方法、排泄率、疾患の重症度等によって変化し得る。
【0038】
また、本発明の化粧料組成物は、クリ−ム、軟膏、ジエル、ロ−ション、化粧水等で製品化されて使用できる、どんな製品として使用するかは、本発明の属する技術分野の当業者によって極めて容易に決定され得る。
【0039】
また、本発明の輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とするロ−ション、ジェル、エセンス、クリ−ム、化粧水等はそれぞれ、通常的な製造方法によって、どんな形態でも容易に製造することができ、基礎化粧品等に適宜に添加して使用することができる。
【0040】
その一例として、クリ−ムを製造するにおいては、一般的な水中油型(O/W)又は油中水型(W/O)のクリ−ムベ−スに本発明の融合蛋白質を含有させ、ここに香料、キレ−ト剤、色素、酸化防止剤、防腐剤等を使用する一方、物性改善を目的に蛋白質、ミネラル、ビタミン等合成又は天然素材を併用することができる。
【0041】
本明細書及び請求範囲の”目標蛋白質(target protein)”は、上記HIV−1 Tat輸送ドメインと共有結合をなして細胞内に導入され活性を示す治療分子、予防分子及び診断分子等を意味し、実際には、純粋な蛋白質にだけ限定されるものでなく、ペプチド、ポリペプチド、糖類と結合された糖蛋白質、ペプチドグリカン等を通称する表現に使用される。
【0042】
また、本発明において”輸送ドメイン“はペプチド、蛋白質、オリゴペプチド、糖類と結合された糖蛋白質、ペプチドグリカン又はポリペプチド等と共有結合をなして別途の受容体や運搬体、エネルギ−を必要としないで上記有機化合物等を細胞内へ導入させることができるドメインであって、6〜12個のアミノ酸残基からなっており、その中、3/4以上がアルギニン又はリシン残基からなっていることをいい、代表的な例を挙げるとすれば、HIV−1 Tat(48−57)、オリゴリシン、オリゴアルギニン、オリゴ(リシン、アルギニン)等をいう。さらに好ましくは、9個のアミノ酸残基からなっており、そのうち、3/4以上がアルギニン又はリシン残基からなっているものを本発明の輸送ドメインの例に挙げることができる。
【0043】
また、本明細書及び請求範囲では蛋白質、ペプチド、糖蛋白質、オリゴペプチド、ポリペプチド等を細胞内に”導入“させることに対して、”運搬”、”浸透”、”輸送“、”伝達”、”透過“、”通過”するという表現と混合して使用している。
【発明を実施するための最良の形態】
【0044】
以下では、本発明の構成を実施例を通じて詳しく説明する。
【0045】
実施例1:輸送ドメイン融合蛋白質発現ベクタ−の製造
機能を有する蛋白質又はぺプチドを細胞内に浸透させる技術を開発するために細胞内に目標蛋白質を伝達できる融合蛋白質発現べクタ−を製造した。
【0046】
本実施例では、輸送ドメイン蛋白質を細胞内に伝達する能力を簡易に分析するために、緑色蛍光蛋白質(Green Fluorescence Protein;本明細書において“GFP”と略称する)を目標蛋白質に選んだ。GFPの塩基配列に該当するDNA切片をプラスミドpEGFP−C2(ク−ロンテック)を利用してPfu DNA重合酵素で重合酵素連鎖反応(PCR)させ、GFPの完全な配列を増幅した。正方向開始体(sense primer)は5‘−CTCGAGGTGAGCAAGGGCGAGGAGCTG−3’(配列番号1)であり、逆方向開始体(antisense primer)の配列は5‘−GGATCCTTACTTGTACAGCTCGTCC ATGCCGAG−3’(配列番号2)である。PCR産物はXhoI−BamHIで切られ、pET15b(Invitrogen、Carlsbad、CA)のXhoI−BamHI座にサブク−ロンされ、HIV−1 Tatの塩基性ドメインがないGFP融合蛋白質を発現させるpGFPを製造した。約0.7kbの挿入体を有するク−ロンはXhol−BamHI制限酵素分析で選ばれ、配列決定を通じて分析された。
【0047】
GFPと融合されたHIV−1 Tatの塩基性ドメイン(アミノ酸48−57)を発現させるpTat−GFPは、次の方法で製造された。
【0048】
第1の方法:二つのオリゴヌクレオチドが製造され、HIV−1 Tatの塩基性ドメインの9個のアミノ酸をコ−デイングする二重鎖オリゴヌクレオチドでアニ−ルされた。HIV−1 Tatの塩基性ドメインの9個のアミノ酸をコ−デイングする配列は上方の鎖(top strand)二重鎖オリゴヌクレオチドでアニ−ルされた。HIV−1 Tatの塩基性ドメイン5‘−TAGGAAGAAGCGGAGACAGCGACGAAGAC−3’(配列番号3)と下方鎖(bottom strand)5‘−TAGGAAGAAGCGGAGACAGCGACGAAGAC−3’(配列番号4)である。二重鎖オリゴヌクレオチドは直接pGFPのNdeI−XhoIで消化された部位に連結される。そして、インフレ−ム(in frame)で6個のヒスチジンオ−プンリディングフレ−ムと連結されたTat−GFP発現プラスミドであるpTat−GFPが製造された(図1)。
【0049】
また、Tat塩基性ドメインGFP両側末端に融合されたpTat−GFP−TatはpTat−GFPにTat塩基性ドメイン9個のアミノ酸をコ−デイングする二重鎖オリゴヌクレオチドがアニ−ルされ、カルボキシル末端に挿入され、Tat塩基性末端がカルボキシル末端に融合されたpGFP−TatはpGFPにTat塩基性ドメイン9個のアミノ酸をコ−デイングする二重鎖オリゴマヌクレオチドがアニ−ルされてカルボキシル末端に挿入された(図1)。
【0050】
プラスミドにク−ロンされたオリゴヌクレオチドの配列は蛍光自動配列分析器(fluorescence−based automated sequencer,モデル373A,Applied Biosystems,Inc.)で確認した。
【0051】
実施例2:Tat−GFP、GFP−Tat及びTat−GFP−Tat融合蛋白質の発現及び精製(Native、denatured)
pGFP及びpTat−GFP、pGFP−TatとpTat−GFP−Tat等で形質転換されたE.coli BL 21(パ−マシア)を選んだ後、コロニ−を100μg/mlのアンピシリンが含有されたLB培地に接種し、37℃で一晩培養した。培養した培養液は新鮮なLB培地で10倍希釈して250rpmで撹伴して培養した。培養液内のバクテリア濃度が0.D6001.0を示したとき、IPTGを培地内に添加して最終濃度が0.5mMになるようにした後、4時間さらに培養した。
【0052】
変性された輸送ドメイン−GFP融合蛋白質を得るために、培養された細胞に6Mウレアとプロテア−ゼ抑制剤(20mg/mlソイビントリップシン抑制剤、2mg/mlアプロチ二ン、5mg/mlリュペップチン、100mg/ml PMSF)が含まれた結合緩衝溶液(binding buffer;5mMイミダゾ−ル、0.5M NaCl、20mM Tris−HCl、pH7.9)を入れて超音波処理(sonication)して収獲し、溶菌した。緩衝液A(buffer A;6Mウレア、20mM HEPES、pH8.0、100mM NaCl)に入れ超音波処理して細胞を破砕させた後、遠心分離を2回(16、500rpm×30min、40,000rpm×30min、4℃)連続して行って、不溶性の細胞破片(cell debris)を除去した。その後、溶菌液(lysate)をNi+++−IDAコラムを通じて精製した。コラムは先ず6Mウレアがない結合緩衝液で洗浄した後、洗浄緩衝溶液(80mMイミダゾ−ル、0.5M NaCl、20mM Tris−HCl、pH7.9)で洗浄した。蛋白質は溶出緩衝液(1mM イミダゾ−ル、0.5M NaCl、20mM Tris−HCL、pH7.9)で溶出した後、PD−10コラムクロマトグラフィ−(Amersham)を利用して蛋白質に含まれた塩分を除去した(図2)。精製されたGFP融合蛋白質は約30kDaのサイズに発現、精製され、Tat−GFP、GFP−Tat融合蛋白質はTat輸送ドメインのサイズ(約1kDa)ほどより大きいサイズに発現、精製され、Tat−GFP−Tat融合蛋白質は両側にTat輸送ドメインが融合されているので、Tat−GFP、GFP−Tat融合蛋白質より一つのTat輸送ドメインのサイズほどもっと大きい分子量で発現され精製された。
【0053】
変性されない自然状態の輸送ドメイン−GFP融合蛋白質は上記言及された変性剤なしで同一の過程で得られた。各分画の蛋白質濃度は、SDS−PAGEに分離した後、密度測定分析方法で牛胎血清アルブミン(BSA)をスタンダ−ドとして定量した。蛋白質濃度はブレドフオ−ド蛋白質アッセイ(Biorad)で測定した。精製された融合蛋白質等は、20%グリセロ−ルが含まれたPBSに溶解され−80℃で貯蔵された。
【0054】
実施例3:細胞培養と融合蛋白質の細胞内伝達実験
HeLa細胞は37℃で20mM HEPES/NaOH(pH7.4)、5mM NaHCO3、10%牛胎血清(fetal bovine serum;FBS)及び抗生剤(100μg/ml ストレプトマイシン、100U/ml ペニシリン)が含まれたDMEM(Dulbecco’S Modified Eagle’s Medium)において培養した。
【0055】
9個のアミノ酸(49−57)からなるHIV−1 Tat蛋白質輸送部分をアミノ酸末端とカルボキシル末端に結合させた変異体輸送ドメインに対する化物分子細胞透過能を観察するために次のように実験した。即ち、輸送ドメイン−GFP、GFP−輸送ドメイン、輸送ドメイン−GFP−輸送ドメイン融合蛋白質等の細胞内透過を観察するために、HeLa細胞を6−ウエルプレ−トで4〜6時間養った後、10%FBSが含まれた新鮮なDMEM培養液で交替し、多くの濃度のGFP融合蛋白質を培養液内に処理した。37℃で1時間培養した後、細胞をりん酸緩衝液生理食塩水(phosphate buffered saline;PBS)で十分に洗浄し、10分間トリプシン−EDTA(Gibco BRL)で処理した。細胞を粉砕した後、細胞内に透過された輸送ドメイン−GFP融合蛋白質の量を次の実施例のようにウエスタン−ブロット分析で測定した。
【0056】
実施例4:ウエスタン−ブロット分析
Tat輸送ドメインがアミノ酸末端に一つが融合されている蛋白質と両側に融合されている蛋白質間の細胞内移動効率を分析するために、ウエスタン−ブロット実験方法を行った。先ず、蛋白質等を変性された状態で精製した。準備されたHeLa動物細胞に1Mの変性された融合蛋白質を処理した後、1時間後に、細胞等だけを集めてウエスタン−ブロットを行った。細胞を溶菌して得られた細胞溶菌液を溶菌緩衝液(125mM Tris−HCl pH6.8、2% SDS、10% v/vグリセロ−ル)がある6−ウエル プレ−トに入れた。全体細胞溶菌液各15μgずつをSDS−12%ポリアクリルアミドジェル上で電気移動させた。
【0057】
電気移動で分離された蛋白質をニトロセルロ−スメンブレイン(nitrocellulose membrane;Amersham,UK)へ移動させた。蛋白質が移動されたニトロセルロ−スメンブレインを10%粉乳を含むPBSでブロッキングした。次いで、メンブレインはラビット抗−GFPポリク−ロン抗体(polyclonal antibody;Clontech、USA、1:1,000)で処理した。その後、セイヨウワサビペルオキシダ−ゼ−(horseradish peroxidase)結合されたゴ−ト抗−ラビット(goat anti−rabbit)IgG抗体(Sigma、1:10,000希釈)と反応させた。結合された抗体等は、化学発光にて確認することができた(ECL;Amersham)。その結果、図3で現したように、Tat輸送ドメインがアミノ酸末端にのみある融合蛋白質と、Tat輸送ドメインがカルボキシル末端にある融合蛋白質は、同様に細胞内に移動した。しかし、Tat輸送ドメインが両側の末端に融合されている蛋白質は、一つのTat輸送ドメインが連結されている融合蛋白質より効果的に細胞内に移動した。この結果は、Tat輸送ドメインが融合蛋白質のアミノ酸末端とカルボキシル末端のいずれ側に融合されてあってもその効果は類似するが、Tat輸送ドメインが両側に融合されておれば、細胞内への移動効果が向上することを示す。
【0058】
実施例5:共焦点蛍光分析(confocal microscopy)による細胞導入分析
目標細胞に融合蛋白質を伝達しようとするとき、伝達される細胞の割合がいくらになるかは重要である。また、細胞内に透過された融合蛋白質がその固有な活性を維持しなければ、このような蛋白質伝達技術が応用できない。そこで、本実験において、GFP融合蛋白質が伝達される細胞の割合や伝達された融合蛋白質の活性の有無を分析した。変性されたGFP融合蛋白質を処理したHeLa細胞からGFPが現れる蛍光度を蛍光顕微鏡で観察した。
【0059】
カバ−スリップ上で50〜70%のコンフルエンシ(confluency)になるように細胞を培養した次の両側末端に輸送ドメインが結合されているGFP−融合蛋白質を1M処理し、15分間培養した。HeLa細胞等はPBSで2回洗浄した後、トリプシン処理し、室温で5分間ホルムアルデヒドが含有されたPBSで固定された。細胞等はさらにPBSで洗浄した後、核だけを染色するために2g/mlのプロピジウムアイオダイド(propidium iodied;PI)を15分間処理した。細胞等はさらにPBSで洗浄した後、マウンテイング溶液(mounting solution;90%グリセロ−ル、0.1%フエニレンジアミンが含まれたりん酸緩衝液食塩水(PBS))を処理し、カバ−ガラスを被せて共焦点蛍光顕微鏡を利用して488nmと545nm蛍光フイルタ−で細胞内の蛍光の有無を観察、蛋白質の細胞内分布を分析した(Eric et al.,1997)。その結果、Tat輸送ドメインがアミノ酸末端に融合されている蛋白質とカルボキシル末端に融合されている蛋白質は殆ど細胞質のみならず核まで到達した(図4)。これは、ウエスタン−ブロットの結果と同じく、Tat輸送ドメインが蛋白質のどの部分に融合されていても、その効果が類似することを示す。その反面、Tat輸送ドメインが両側に付いた融合蛋白質は、一方にのみ存在する融合蛋白質より明るい蛍光を現した。加えて、Tat輸送ドメインが両側に付いた融合蛋白質は、より多くの核内に移動していることが確認された。
【0060】
実施例6:FACS(flow cytometry)分析による細胞導入分析
9個のアミノ酸(49−57)からなるHIV−1 Tat蛋白質輸送部分をアミノ酸末端とカルボキシル末端両側に結合させた変異体輸送ドメインに対する化物分子細胞透過能を観察するために次のように実験した(図5)。即ち、輸送ドメイン−GFP−輸送ドメイン融合蛋白質の細胞内透過を観察するために、HeLa細胞を6−ウエル プレ−トにおいて4〜6時間培養した後、10%FBSが分布された新鮮なDMEM培養液で交替し、多数濃度のGFP融合蛋白質を培養液内に処理した。37℃で30分間培養した後、細胞をりん酸緩衝液生理食塩水(phosphate buffered saline;PBS)で十分に洗浄し、10分間トリプシン−EDTA(Gibco BRL)で処理した。細胞を集めて、さらにPBSで2回洗浄した後、4%パラホルムアルデヒド(paraformaldehyed)で1時間固定させた。固定させた細胞に対してFACSを利用して細胞内蛍光を分析した。FACS分析結果、図5のような曲線を得ることができた。上記の結果と同様、Tat輸送ドメインが一方にだけ融合されている蛋白質は似た量で細胞内に導入される反面、両側にTat輸送ドメインがある融合蛋白質は多くの量で細胞内に導入されることを分かることができた。平均蛍光度(Mean Fluorescence intensity)は、輸送ドメイン−GFPが23.86、GFP−輸送ドメインが17.21、輸送ドメイン−Tat−輸送ドメインは204.52であって、Tat−GFP−Tat融合蛋白質が特に高い数値を示した。その後、Tat輸送ドメインを両側に融合された蛋白質の量によって細胞内に導入されるメカニズムを分析するために多様な濃度で処理して30分間培養した後、FACS分析を行った(図6).この結果、Tat輸送ドメインを両側に有する融合蛋白質は、その濃度と共に平均蛍光度が増加することが確認できた。
【0061】
実施例7:FACS(flow cytometry)分析とウエスタン−ブロットによる自然状態の融合蛋白質と変性された融合蛋白質の細胞導入比較分析
自然状態で輸送ドメイン−GFPを精製して細胞内に導入したとき、変性させ精製した輸送ドメイン−GFPより導入されるときと比較して、その効果が制限される。輸送ドメインが両側の末端に存在するGFP融合蛋白質は変性して精製されたとき、一方にだけ存在するGFPより大きな効果を示すことから、輸送ドメイン−GFP−輸送ドメイン融合蛋白質が自然状態で精製されたとき、細胞内に導入される効果について検討すべく実験を行った。まず、変性された蛋白質と自然状態に精製した蛋白質をウエスタン−ブロットで分析した結果、事前に精製された蛋白質も輸送ドメインが一つ存在するGFP融合蛋白質と同じ数値で細胞内に導入されたときの結果を示した(図7)。また、自然状態に分離した輸送ドメイン−GFP−輸送ドメイン融合蛋白質の細胞内導入効果を知るためにFACS分析法を行った。それぞれ自然状態と変性された状態で精製された蛋白質をそれぞれの濃度に処理して1時間培養した。その結果図8のように変性された状態の輸送ドメイン−GFP−輸送ドメイン融合蛋白質は、1M処理したとき平均蛍光度が117である反面、自然状態に精製した輸送ドメイン−GFP−輸送ドメインは54である数値を示した。これは、変性された状態の融合蛋白質の細胞内導入効果より劣っているが、図7と比較すると、変性して精製した輸送ドメイン−GFP融合蛋白質程度の細胞内導入能力を有しているのが分かった。結果的に、Tat輸送ドメインを一方の末端に有する融合蛋白質よりTat輸送ドメインを両側に有しており、変性して(denatured)精製した融合蛋白質が効果的に細胞内浸透をすることができ、自然状態(native)に精製した輸送ドメイン−GFP−融合蛋白質もまた細胞内浸透が可能であることを結論づけることができた。
【0062】
実施例8:組み替えSOD融合蛋白質等の発現ベクタ−製造
機能を有する蛋白質又はペプチドを細胞内に浸透させる技術を開発するために細胞内に目標蛋白質を伝達し得る融合蛋白質発現ベクタ−を製造し、輸送ドメインが蛋白質を細胞内に伝達する能力を容易に分析するために、ヒトCu,Zn−ス−パ−オキシドジスムタ−ゼ(Cu,Zn−superoxide dismutase;以下、本明細書ではSODと略称する)蛋白質を選んだ。
【0063】
組み替え融合蛋白質を過大に発現させるために、SOD、HIV−1Tatの形質導入部(Tat 49−57)及び6個のヒスチジンに対するcDNAが連続的に含まれているTat−SOD、SOD−Tat、Tat−SOD−Tat発現ベクタ−を製造した(図9)。融合蛋白質に対する対照蛋白質であるSODを過大に発現させるために、Tatの形質導入部位だけ含まれておらず、残りの部位は同一なpET−SOD発現ベクタ−を製造した。
【0064】
Tat基本ドメインに該当する2種類のオリゴヌクレオチド[上位鎖(top strand)、5‘−TAGGAAGAAGCGGAGACAGCGACGAAGAC−3’(配列番号3)と下位鎖5‘−TCGAGTCTTCGTCGCTGCTCCGCTTCTTCC−3’(配列番号4)]をNdeI−XhoI制限酵素で切ったpET−15bベクタ−に結さつ(ligation)して挿入した後、ヒトSODのcDNAの配列を基本として2種類のオリゴヌクレオチドを合成した。正方向プライマ−(Forward Primer)はXho I 制限部位を有しており、逆方向プライマ−(reverse primer)はBamH I制限部位を有している。重合酵素連鎖反応(PCR)を行なった後、反応物を分離し、これをTAクロニングベクタ−に結さつして形質変換させプラスミドを分離した。ヒトSOD cDNAがTAベクタ−をXho I とBamH Iで切断した後、pET−15b及びpET−15b−Tat発現ベクターに挿入した。
【0065】
また、Tat塩基性ドメインがSOD両側末端に融合されたpTat−SOD−TatはpTat−SODにTat塩基性ドメイン9個アミノ酸をコ−デイングする2重鎖オリゴヌクレオチドがアニ−ルされ、カルボキシル末端に挿入され、Tat塩基性末端がカルボキシル末端に融合されたpSOD−TatはpSODにTat塩基性ドメイン9個のアミノ酸をコ−デイングする2重鎖オリゴヌクレオチドがアニ−ルされ、カルボキシル末端に挿入された(図9)。
【0066】
実施例9:組み替えSOD融合蛋白質等の発現及び精製
本研究室で製造したヒトCu,Zn−ス−パ−オキシドジスムタ−ゼcDNAが含まれているE.coli BL21(DE3)細胞(pSOD及びpTat−SOD、pSOD−Tat、pTat−SOD−Tat)をアンピシリンが含まれているLB培地に入れ37℃で200rpmに撹拌して培養した。培養液内のバクテリア濃度が0.D600=0.5〜1.0を表す時、IPTGを培地内に添加して最終濃度が0.5mMになるようにした後、3〜4時間さらに培養した。培養した細胞を遠心分離して集めた後、6M Ureaが添加された結合緩衝溶液(binding buffer;5mM imidazole、0.5M NaCl、20mM Tris−HCl、pH7.9)5ml入れ超音波粉砕器で粉砕(sonication)した。遠心分離して上層液を直ちにNi−ニトリロトリアセチックアクシド セパロズス−パ−フロウ(Ni−nitrilotriacetic acid sepharose super flow)コラムに負荷し、10倍容積の結合緩衝溶液(binding buffer)と6倍容積の洗浄緩衝溶液(washing buffer;60mM imidazole、0.5M NaCl、20mM Tris−HCl、pH7.9)で洗浄した後、溶出緩衝溶液(elition buffer;1M imidazole、0.5M NaCl、20mM Tris−HCl、pH7.9)で融合蛋白質を溶出した。次いで、融合蛋白質が含まれた分画等を集めてPD−10コラムクロマトグラフイ−(column chromatography)を行なって分画中に含まれた塩分を除去した。融合蛋白質は6個のヒスチジンを含んでいるため、金属イオン−キレ−ト親和クロマトグラフイ−単一段階に融合蛋白質を純粋に(純度>90%)精製された(図10)。
【0067】
分画の蛋白質濃度は牛血清アルブミンを標準物質に使用してブレッドフオ−ド(Bradford)方法で測定した。
【0068】
実施例10:HeLa細胞培養及び組み替え融合蛋白質等の細胞内透過
HeLa細胞は37℃で95%空気と5%CO2とを供給し、20mMHEPES/NaOH(pH7.4)、5mM NaHCO3、10%牛胎血清(fetal bovine serum;FBS)及び抗生剤(100μg/ml ストレフトマイシン、 100U/ml ペニシリン)が含まれたDMEM(Dulbecco‘s Modified Eagle’s Medium)で培養した。
【0069】
9個のアミノ酸(49−57)からなるHIV−1 Tat蛋白質輸送部分をアミノ酸末端とカルボキシル末端に結合させた変異体輸送ドメインに対する化物分子細胞透過能を観察するために次のように実験した。即ち、Tat−SOD、SOD−Tat、Tat−SOD−Tat融合蛋白質等の細胞内透過を観察するために、HeLa細胞を6−ウエルプレ−トで4〜6時間培養した後、10%FBSが含まれた新鮮なDMEM培養液に交換し、2M SOD融合蛋白質を培養液内に処理した。1時間後、細胞をトリプシン−EDTA(Gibco BRL)で処理し、リン酸緩衝液生理食塩水(phosphate buffered saline;PBS)で十分に洗浄した。細胞を粉砕した後、細胞内に透過されたSODの量と活性度をSOD活性度分析とウエスタン−ブロッド分析で測定した。
【0070】
実施例11:ウエスタン−ブロッド分析
Tat蛋白質がアミノ酸末端に1個が融合されている蛋白質と、両側に融合された蛋白質の細胞内移動効率を分析するためにウエスタン−ブロット方法を行なった。先ず、蛋白質等は変性された状態で精製した。準備されたHeLa細胞に2Mの変性された融合蛋白質を処理した後、1時間後に細胞等だけを集めてウエスタン−ブロットを行なった。細胞を溶菌して得られた細胞溶菌液を溶菌緩衝液(125mM Tris−HCl pH6.8、2% SDS、10% v/v グリセロ−ル)を有する6−ウエルプレ−トに入れた。全体細胞溶菌液各15μgずつをSDS−12%ポリアクリルアミドジェル上で電気移動させた。
【0071】
電気移動で分離された蛋白質をニトロセルロ−スメンブレイン(nitrocellulose membrane;Amersham,UK)で移動させた。蛋白質が移動されたニトロセルロ−スメンブレインを5%脱粉乳(non−dry milk)が入っているPBSでブロキングした。次いで、メンブレインはゴト抗−SODポリク−ロン抗体(polyclonalantibody;Santacruze、USA、1:1,000)で処理した。その後セイヨウワサビペルオキシダ−ゼ(horse radish peroxidase)−結合されたマウス抗−ゴ−ト(mouse anti−rabbit)IgG抗体(Sigma、1:10,000希釈)と反応させた。結合された抗体等は化学発光で確認することができた(ECL;Amersham)。その結果、図11aのようにTat蛋白質がアミノ酸末端にだけある融合蛋白質とTat蛋白質がカルボキシル末端にある融合蛋白質は共に細胞内に透過された。しかし、Tat蛋白質が両側の末端に融合されている融合蛋白質は1個のTat蛋白質が連結されている融合蛋白質より効果的に細胞内に移動した。この結果は、Tat蛋白質が融合蛋白質のアミノ酸末端とカルボキシル末端のいずれかの側に融合されているものの効果は類似する、Tat蛋白質が両側に融合されているものが細胞内移動効果が大きく向上されていることを示す(図11a)。
【0072】
実施例12:SOD酵素活性度測定
本研究において、SODの活性度は、McCordとFridovichの方法(1969)によってキサンチン/キサンチンオキシダ−ゼ(xanthine/xanthine oxidase)反応によるペリシトクロム(ferricytochrome)c還元の抑制程度を分光光度計で観察することにより測定した(McCord、JM and Fridovich、I.(1969)J.Biol.Chem.244、6049−6055)。
【0073】
標準分析方法は25℃で2mlの0.1mM EDTAが含まれた50mMリン酸カリウム緩衝溶液(pH7.8)において行なった。反応混合液には10uM ferricytochrome c、50uMキサンチン(xanthine)及び十分な量のキサンチンオキシダ−ゼ(xanthine oxidase)を含有しており、550nmにおいてフエリシトクロム(ferricytochrome)cの還元程度を測定した。上記の条件下においてシトクロム(cytochrome)cを50%減少させるス−パ−オキシドジスムタ−ゼの量を1単位に定義した。
【0074】
細胞内に透過された溶融蛋白質は、その固有な活性を維持しなければ蛋白質治療に有用することができない。よって、細胞内に透過された融合蛋白質が生物学的な活性を有することは大変重要な問題である。図11bは2Mの融合蛋白質等を1時間HeLa細胞培地に透過したとき、細胞内のSOD酵素活性の変化を表した結果である。溶融蛋白質を投与しなかった細胞からSOD活性度は2.46±0.39U/mg蛋白質であり、Tat−SODとSOD−Tat融合蛋白質を投与した時にSOD活性度は12.68±1.44U/mg蛋白質として有意に上昇し、Tat−SOD−Tat融合蛋白質を投与した時にSOD活性度は23.75±2.35U/mg蛋白質でTat−SODとSOD−Tat融合蛋白質より2倍以上高いSOD活性度を表した(図11b)。よって、Tat−SOD−Tat融合蛋白質の細胞内透過及び活性がTat−SODとSOD−Tat融合蛋白質より2倍以上大きく増加されることを示した(図11)。
【0075】
実施例13:アルギニン9−GFP−アルギニン9融合蛋白質の細胞浸透活性測定
上記実施例1ないし3の方法と類似な方法で、9個のアルギニン残基で構成された輸送ドメインが緑色蛍光蛋白質のアミノ基及びカロボキシ基に共有結合されたオリゴリシン−GFPオリゴリシン融合蛋白質を製造した。
【0076】
Tat−GFP−Tat融合蛋白質とアルギニン9−GFP−アルギニン9(R9−GFP−R9)融合蛋白質の細胞浸透効率を比較するために、各融合蛋白質50、100、200nMずつをHeLa細胞が入っている24ウエルプレ−トに加え、1時間が経過した後、細胞をトリプシン処理し、PBSで数回洗浄した後、蛍光をフルオロメ−タ−で測定した。結果は図12のとおりである。
【0077】
実施例14:リシン9−GFP−リシン9融合蛋白質の細胞浸透活性測定
実施例1ないし3の方法と類似の方法で9個のリシン残基で構成された輸送ドメインが緑色蛍光蛋白質のアミノ基及びカロボキシ基に共有結合されたオリゴリシン−GFPオリゴリシン融合蛋白質を製造した。
【0078】
Tat−GFP融合蛋白質、GFP−Tat融合蛋白質、アルギニン9−GFP融合蛋白質とリシン9−GFP−リシン9(K9−GFP−K9)融合蛋白質の細胞浸透効率を比較するために、各融合蛋白質200nMずつをHeLa細胞が入っている24ウエルプレ−トに加えた後、1時間が経過した後、細胞をトリプシン処理し、PBSで数回洗浄した後、蛍光を蛍光顕微鏡で測定した。結果は図13のとおりである。
【産業上の利用可能性】
【0079】
本発明は機能を有する蛋白質を蛋白質水準で細胞内に透過させるにおいて、既存の塩基性輸送ドメインを1側に結合させたものよりもっと高い透過効率と、細胞内活性を有する溶融蛋白質を提供することができる。
【0080】
また、本発明の融合蛋白質を有効成分とする薬剤学的組成物及び化粧品等を提供することができる。
【0081】
更に、本発明はヒトGu,Zn−ス−パ−オキシド ジスムタ−ゼ(Cu,Zn−SOD)を蛋白質水準で細胞内に直接透過させるもので、特に細胞内に投与された−輸送ドメイン−SOD−輸送ドメイン融合蛋白質はSOD活性度を回復して蛋白質治療に効率的に活用され得ることを提示する。
【0082】
活性酸素種は、細胞内の生体高分子に損傷を加え、報告によれば、100余種の疾病の進行過程と深く関連されている。よって、本発明はこのような活性酸素種を除去する際に主な役割を担当するSODを細胞内に投与することにより、疾患を治療する蛋白質治療に本発明の輸送ドメイン−SOD−輸送ドメイン融合蛋白質が効果的に活用され得る。
【0083】
本技術を基にして抗酸化酵素の一種であるSODを細胞内に直接伝達して我らの人体に有害な活性酸素を除去することができ、多くの疾病以外にも化粧品及び健康食品産業等に本技術を広範囲に活用することができる。
【図面の簡単な説明】
【0084】
【図1】Tat−GFP、GFP−Tat、Tat−GFP−Tat、GFP発現ベクタ−の概略図である。合成されたTat輸送ドメインはGFPのアミノ末端やカルボキシル末端、又は、両側末端に融合してベクタ−を製造した。GFP発現ベクタ−pGFPはGFPのコ−デイング配列をpET15bに挿入して構成された。
【図2】精製された輸送ドメイン−GFP融合蛋白質を12%SDS−PAGEに分離した後、クマシ−ブル−(Coomassie blue)で染色したものである。レイン M:SDS Markerレイン GFP:GFPレイン TG:Tat−GFPレイン GT:GFP−Tatレイン TGT:Tat−GFP−Tat
【図3】A:変性されたTat−GFP、GFP−Tat、Tat−GFP−Tat融合蛋白質の細胞内導入分析結果であって、1M Tat融合蛋白質等を1時間HeLa細胞内に導入させた時、細胞内に導入された蛋白質をウエスタン−ブロット(western blot)法で分析したものである。B:細胞内に導入された蛋白質をウエスタン−ブロットした結果を基に、結果に表れた蛋白質等線の強さを図式化したものである。
【図4】細胞内に導入されたTat−GFP、GFP−Tat、Tat−GFP−Tat融合蛋白質を公焦点蛍光顕微鏡で可視化したものである。各セクションは、それぞれ次のとおりである。Control:何も処理しない細胞GFP:対照群 GFP 1MTG: Tat−GFP 1MGT:GFP−Tat 1MTGT:Tat−GFP−Tat 1M
【図5】FACS分析法によってGFP融合蛋白質の細胞内導入効率を比較分析したものである。各融合蛋白質等は2M濃度で30分間処理した。TG:Tat−GFPGT:GFP−TatTGT:Tat−GFP−Tat
【図6】Tat−GFP−Tat融合蛋白質を濃度別に処理して30分間培養した結果をFACS分析法によって分析した結果。対照群GFPは最高濃度である2Mで処理した。
【図7】A:Tat−GFP−Tat融合蛋白質を自然状態と変性された状態で精製して細胞内導入効率を分析するためにウエスタン−ブロットである。各融合蛋白質等は1Mで処理して1時間培養した。レインcon:処理しない細胞レインGFP:対照群 GFPレインnative:自然状態に精製したTat−GFP−Tatレインdenatured:変性された状態に精製したTat−GFP−TatB: 図7Aの結果に表れた線の強さを図表で模式化したものである。
【図8】FACS分析法によって自然状態に精製されたTat−GFP−Tatと変性された状態に精製されたTat−GFP−Tatの細胞内導入効果を濃度別に1時間処理して分析したものである。A:変性された状態に精製されたTat−GFP−Tatを0.1M、0.2M、0.5M、1M処理して分析した結果である。B:自然状態に精製されたTat−GFP−Tatを0.1M、0.2M、0.5M、1M処理して分析した結果である。
【図9】Tat−SOD、SOD−Tat、Tat−SOD−Tat、SOD発現ベクタ−の概略図である. 合成されたTat蛋白質をSODのアミノ末端やカルボキル末端、又は両側末端に融合してベクタ−を製造した。SOD発現ベクタ−であるpSODはSODのコ−デイング配列をpET−15bベクタ−に挿入して製造した。
【図10】精製された輸送ドメイン−SOD融合蛋白質等を12%SDS−PAGEに分離した後、クマシ−ブル−で染色したものである。レイン M:SDS Markerレイン SOD:SODレイン TS:Tat−SODレイン ST:SOD−Tatレイン TST:Tat−SOD−Tat
【図11】A:変性された状態に精製したTat−SOD、SOD−Tat、Tat−SOD−Tat融合蛋白質の細胞内導入分析結果であって、2M Tat融合蛋白質を1時間HeLa細胞内に導入させたとき、細胞内の導入された蛋白質をウエスタン−ブロット法で分析したものである。B:2M融合蛋白質等を1時間HeLa細胞に投与したとき、細胞内に導入されたSOD酵素の活性度変化を示したグラフである。
【図12】Tat−GFP−Tat融合蛋白質とアルギニン9−GFP−アルギニン9(R9−GFP−R9)融合蛋白質の細胞浸透効率を蛍光を測定して図式化したグラフである。
【図13】GFP、Tat−GFP融合蛋白質、GFP−Tat融合蛋白質、アルギニン9−GFP融合蛋白質とリシン9−GFP−リシン9(K9−GFP−K9)融合蛋白質の細胞浸透効率を比較するために、各融合蛋白質200nMずつをHeLa細胞に加えて1時間が経過した後、蛍光顕微鏡で観察した写真である。
【配列表】
【特許請求の範囲】
【請求項1】
6ないし12個のアミノ酸残基から構成され、アルギニン又はリシン残基を3/4以上含む輸送ドメインが目標蛋白質のアミノ基とカルボキシル基に共有結合された細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項2】
輸送ドメインは、9個のアミノ酸残基で構成される請求項1記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項3】
輸送ドメインはHIV tat49−57残基、オリゴリシン、オリゴアルギニン又はオリゴ(リシン,アルギニン)の少なくともいずれか1種である請求項1記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項4】
目標蛋白質は治療分子、予防分子、診断分子で構成されたグル−プから選ばれる請求項1〜請求項3のいずれか1項に記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項5】
目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物である請求項1〜請求項3のいずれか1項に記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項6】
請求項1〜請求項3のいずれか1項に記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とする化粧料組成物。
【請求項7】
目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物である請求項6記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とする化粧料組成物。
【請求項8】
化粧料組成物は化粧水、ジエル、水溶性リキッド、水中油(O/W)型又は油中水(W/O)型である請求項6記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とする化粧料組成物。
【請求項9】
請求項1〜請求項3のいずれか1項記載の輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とし、薬剤学的に許容される担体を含む薬剤学的組成物。
【請求項10】
目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物である請求項9記載の輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とする薬剤学的組成物。
【請求項1】
6ないし12個のアミノ酸残基から構成され、アルギニン又はリシン残基を3/4以上含む輸送ドメインが目標蛋白質のアミノ基とカルボキシル基に共有結合された細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項2】
輸送ドメインは、9個のアミノ酸残基で構成される請求項1記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項3】
輸送ドメインはHIV tat49−57残基、オリゴリシン、オリゴアルギニン又はオリゴ(リシン,アルギニン)の少なくともいずれか1種である請求項1記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項4】
目標蛋白質は治療分子、予防分子、診断分子で構成されたグル−プから選ばれる請求項1〜請求項3のいずれか1項に記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項5】
目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物である請求項1〜請求項3のいずれか1項に記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質。
【請求項6】
請求項1〜請求項3のいずれか1項に記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とする化粧料組成物。
【請求項7】
目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物である請求項6記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とする化粧料組成物。
【請求項8】
化粧料組成物は化粧水、ジエル、水溶性リキッド、水中油(O/W)型又は油中水(W/O)型である請求項6記載の細胞浸透性輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とする化粧料組成物。
【請求項9】
請求項1〜請求項3のいずれか1項記載の輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とし、薬剤学的に許容される担体を含む薬剤学的組成物。
【請求項10】
目標蛋白質がCu,Zn−ス−パ−オキシドジスムタ−ゼ又はその機能的等価物である請求項9記載の輸送ドメイン−目標蛋白質−輸送ドメイン融合蛋白質を有効成分とする薬剤学的組成物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公表番号】特表2006−504770(P2006−504770A)
【公表日】平成18年2月9日(2006.2.9)
【国際特許分類】
【出願番号】特願2004−548119(P2004−548119)
【出願日】平成15年3月13日(2003.3.13)
【国際出願番号】PCT/KR2003/000490
【国際公開番号】WO2004/039846
【国際公開日】平成16年5月13日(2004.5.13)
【出願人】(504117936)ハリム ユニバーシティ (1)
【氏名又は名称原語表記】HALLYM UNIVERSITY
【住所又は居所原語表記】1,Okchon−dong,Chuncheon−si,Kangwon−do 200−702 Republic of Korea
【Fターム(参考)】
【公表日】平成18年2月9日(2006.2.9)
【国際特許分類】
【出願日】平成15年3月13日(2003.3.13)
【国際出願番号】PCT/KR2003/000490
【国際公開番号】WO2004/039846
【国際公開日】平成16年5月13日(2004.5.13)
【出願人】(504117936)ハリム ユニバーシティ (1)
【氏名又は名称原語表記】HALLYM UNIVERSITY
【住所又は居所原語表記】1,Okchon−dong,Chuncheon−si,Kangwon−do 200−702 Republic of Korea
【Fターム(参考)】
[ Back to top ]