
細胞膜タンパク質の立体構造を認識する抗体を産生するハイブリドーマの作製方法
【課題】 膜タンパク質の立体構造を認識する抗体を発現するハイブリドーマの作製方法の提供。
【解決手段】 本発明は、目的のタンパク質を抗原として用いる免疫により獲得されたリンパ球集団と、細胞表面に当該タンパク質を提示するミエローマ細胞とを混合し;当該リンパ球集団内の当該タンパク質に特異的な膜結合型モノクローナル抗体を提示するリンパ球を当該ミエローマ細胞に対し、当該膜結合型抗体と当該ミエローマ細胞表面に存在する当該膜タンパク質との特異的な結合を介し、選択的に近寄らせ;そして両細胞を融合させる、ステップを含むことを特徴とする、当該タンパク質に特異的なモノクローナル抗体を産生するハイブリドーマの作製方法、を提供する。
【解決手段】 本発明は、目的のタンパク質を抗原として用いる免疫により獲得されたリンパ球集団と、細胞表面に当該タンパク質を提示するミエローマ細胞とを混合し;当該リンパ球集団内の当該タンパク質に特異的な膜結合型モノクローナル抗体を提示するリンパ球を当該ミエローマ細胞に対し、当該膜結合型抗体と当該ミエローマ細胞表面に存在する当該膜タンパク質との特異的な結合を介し、選択的に近寄らせ;そして両細胞を融合させる、ステップを含むことを特徴とする、当該タンパク質に特異的なモノクローナル抗体を産生するハイブリドーマの作製方法、を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、タンパク質、特に細胞膜タンパク質の立体構造を認識するモノクローナル抗体を産生する目的のハイブリドーマを効率良く産生する方法に関する。
【0002】
これまでに種々の抗原を認識するモノクローナル抗体は多数単離されており、医薬、診断薬だけではなく、各種の実験などに広く利用されている。
モノクローナル抗体は、動物由来のリンパ球とミエローマ細胞を融合することで作製されており、融合させる方法としてはポリエチレングリコール(PEG)法が一般的に利用されている。一般的なPEG法では、目的抗体を発現する細胞とミエローマ細胞との融合だけではなく、目的抗体を発現していないリンパ球とミエローマ細胞との融合、リンパ球同士の融合、ミエローマ細胞同士の融合などもおこり、得られるハイブリドーマ中の目的ハイブリドーマの割合は数%程度である。しかしながら、一般的な研究・実験(たとえば、ELISA、ウェスタンブロッティングなど)に使用するモノクローナル抗体の作製であれば、この方法でも特に大きな問題なく抗体を十分に単離することができる。他の融合方法として、電気的に細胞を融合させる方法(たとえば、はじめに交流下で細胞同士を近接させた後に、直流パルスをかけて細胞を融合させる方法)も広く利用されている。かかる方法も原理的には、PEG法と同様、リンパ球とミエローマ細胞を非特異的に融合させることから、目的抗体のスクリーニングには煩雑な作業を必要とする。
【0003】
上記細胞融合法の欠点を克服するため、下記に示す方法が報告されている。ここに示す方法のポイントは、リンパ球の細胞表面にそのリンパ球が発現する抗体が提示されていることを極めて巧妙に利用する点にある。以下に示す二つの細胞(1)、(2)を準備する。(1)あらかじめ作製しておいた抗原−アビジン複合体をリンパ球と反応させ、抗原結合能を有した抗体を発現しているリンパ球だけを、抗原−アビジン複合体で標識する。(2)ミエローマ細胞の表面をビオチンで修飾した細胞を準備する。その後、(1)と(2)の細胞を混合すると、ビオチン-アビジンの極めて強力な結合力により、リンパ球−抗原−アビジン−ビオチン−ミエローマ細胞という細胞の複合体を形成させることができる。その後、電気パルスをかけることで、複合体を形成した細胞を選択的に融合させることが可能となる。この方法によると、目的抗体を発現していないリンパ球がミエローマ細胞と融合する確立が極めて低くなる点が特徴となる。この方法でハイブリドーマを作製すると、得られるハイブリドーマ中の目的ハイブリドーマの割合は、多い場合には60%を超える極めて高い値を示すことが報告されており、非常に優れた方法である(冨田昌弘、Bioベンチャー、Vol.1、108(2001))。
【0004】
しかし、抗原が膜タンパク質である場合には上記冨田らの方法を利用するのは困難である。その理由としては、高純度の十分な量(数mg)の膜タンパク質を精製すること自体一般に困難であることや、その膜タンパク質を高次構造を保持した状態でアビジンと結合体を作製するのが極めて難しいことにある。つまり、膜タンパク質を認識する抗体を単離する場合には、冨田らの方法を採用することができず、通常の細胞融合法、即ちリンパ球とミエローマ細胞を非特異的に融合させる方法を介し、抗体を単離する方法しかなかった。
【0005】
通常の細胞融合法で膜タンパク質を認識する抗体の単離を試みる場合、ウェスタンブロッティングなど、タンパク質を変性させた条件下で膜タンパク質に結合する抗体をスクリーニングする操作を繰り返す必要がある。しかしながら、細胞膜上に存在する状態の膜タンパク質を認識できる抗体の単離はきわめて困難である。その理由は、抗原に結合する抗体のうち、細胞膜タンパク質の立体構造を認識できる抗体の存在率が低いことによる。上記でも述べたように、通常の細胞融合法では、得られるハイブリドーマ中の目的抗体の割合は多くても数%程度と低く、さらにその中から目的の機能を持った抗体を探し出す必要がある。
【0006】
このように、細胞膜上に存在する膜タンパク質の立体構造を認識する抗体は、単離することが難しいというのが現状であった。さらに、膜タンパク質がホルモンなどの生理活性物質の何らかの受容体である場合などにおいては、それを特異的に認識する抗体は、通常の研究用途として重要であるだけではなく、医薬品としての利用も考えられる。しかし、そのような抗体を単離するには、従来から報告されている細胞融合法を繰り返し、膨大な数のハイブリドーマの評価を行い、目的の抗体単離を試みる必要があったが、目的の抗体を単離するには偶然に頼るしかなかった。
【0007】
【非特許文献1】冨田昌弘、Bioベンチャー、Vol.1、108(2001)
【発明の開示】
【発明が解決しようとする課題】
【0008】
そこで、本発明者らは、膜タンパク質を認識する抗体を発現するハイブリドーマの作製法について鋭意検討した結果、本発明に到達した。
【課題を解決するための手段】
【0009】
本発明は、目的のタンパク質を抗原として用いる免疫により獲得されたリンパ球集団と、細胞表面に当該タンパク質を提示するミエローマ細胞とを混合し;
当該リンパ球集団内の当該タンパク質に特異的な膜結合型モノクローナル抗体を提示するリンパ球を当該ミエローマ細胞に対し、当該膜結合型抗体と当該ミエローマ細胞表面に存在する当該膜タンパク質との特異的な結合を介し、選択的に近寄らせ;そして
両細胞を融合させる、
ステップを含むことを特徴とする、当該タンパク質に特異的なモノクローナル抗体を産生するハイブリドーマの作製方法、を提供する。好ましくは、目的のタンパク質は細胞の表層上に提示されるいわゆる膜タンパク質である。
【0010】
具体的には、遺伝子操作などによって高次構造又は立体構造を保持したタンパク質を細胞表層上において提示して有するミエローマ細胞(以下、単に「抗原提示ミエローマ細胞」と称する場合がある)を作製する。それとは別に、タンパク質を抗原として用いて免疫した動物に由来するリンパ球集団を準備する。このリンパ球集団は、目的のタンパク質の立体構造を特異的に認識する抗体を細胞表層上に提示し、当該抗体を産生する能力の備わったリンパ球(以下、単に「抗体産生リンパ球」と称する場合がある)を含有すると予想される。両者の細胞を混合すると、抗原抗体反応に基づき、抗体産生リンパ球上に存在する当該タンパク質に特異的な抗体が、抗原提示ミエローマ細胞上のタンパク質に結合することで、抗体産生リンパ球-細胞表面抗体-細胞表面タンパク質-抗原提示ミエローマ細胞、という細胞の複合体を形成させることができる。その後、電気パルスを負荷することにより両細胞の選択的融合を行い、目的のタンパク質に特異的なモノクローナル抗体を産生するハイブリドーマを作製する方法である。
【0011】
本発明によれば、細胞膜上のタンパク質を認識できない抗体を発現したリンパ球は、上記で示す複合体を抗原提示ミエローマ細胞と形成することができない。つまり、細胞膜上に存在するタンパク質を認識できる抗体を発現している抗体産生リンパ球だけを選択的にハイブリドーマにすることができる。
【発明の効果】
【0012】
本方法に従うと、得られるハイブリドーマは細胞膜上に存在するタンパク質を認識する抗体を発現することになる。たとえば、タンパク質が何らかの生理活性物質の受容体である場合などは、受容体のアゴニストあるいはアンタゴニスト型の抗体を、これまでの方法と比較してはるかに容易に単離できることが予想され、研究用途用の抗体のみならず、医薬品用の抗体としての展開も考えられる。
【発明を実施するための最良の形態】
【0013】
以下で本発明について詳細に説明する。
【0014】
本発明は、タンパク質、特に細胞膜上で提示される膜タンパク質を特異的に認識するモノクローナル抗体を産生するハイブリドーマの作製に好適に利用される。膜タンパク質とは、細胞表面上に存在するタンパク質をいい、膜を貫通する回数および分子量は問わない。一般的に、膜タンパク質の膜貫通回数が増加するに伴い、従来法では膜タンパク質を認識する抗体を単離することが困難になっていくため、そのような膜タンパク質を認識する抗体を単離する場合に本発明は特に有効であると推測されるが、特にそれらに限定されない。従来法では膜タンパク質を認識する抗体を単離するのが困難なのは以下の理由による。たとえば、一回膜貫通型の膜タンパク質の場合、膜タンパク質の細胞外領域だけを遺伝子組換え法などを用い作製した場合でも、リガンドの結合能は失われない場合が多い。たとえばインターロイキン-6(IL-6)の受容体は、一回膜貫通型のタンパク質であるが、細胞外領域だけを遺伝子組換え型で発現させたとしても、細胞外領域だけでもIL-6 との結合能は保持している。しかしながら、多回膜貫通型のタンパク質、たとえば7回膜貫通型のタンパク質である甲状腺刺激ホルモン受容体(Thyroid stimulation hormone receptor: TSHR)などの場合、細胞外領域のみを発現させたとしても、抗原結合能を保持しないか、あったとしても極めて弱い結合しか示さない。このことは、TSHRの細胞外領域が、10アミノ酸程度からなる細胞外ループ(TSHRでは3つ存在)と何らかの相互作用があって初めて抗原結合能が発現することを意味している。
【0015】
つまり、膜貫通回数が多くなるにつれて、細胞膜上に存在する形態と、それを免疫している状態とが異なることが予想され、細胞融合後に得られる抗体の中で、細胞膜上に存在するタンパク質を認識できる抗体の割合が低くなってゆくことが予想される。つまり、本発明はこれまでに非常に単離が困難であった多回膜貫通型のタンパク質が細胞膜上に存在する状態を認識する抗体の単離方法として利用できる可能性が高い。
【0016】
本発明の方法において対象となる膜タンパク質には、上記TSHRのほか、特に限定することなく、たとえば下記の膜タンパク質が挙げられる:
ニコチン、GABA、グリシンなどのイオンチャンネル内蔵型受容体、またはアルファー1、ヒスタミン1(H1)、ブラジキニン(B2)、ムスカリン1(M1)、アンジオテンシンII(AT1)などの受容体でイノシトールリン酸を分解する受容体、ベータ、ヒスタミン2(H2)、プロスタサイクリン(IP)、グルカゴンなどの受容体、TSH、FSH、LHなどの各種ペプチドホルモンなどの受容体でcAMPを合成し、GTP結合蛋白質共役型の受容体、またはインシュリン、EGF、FGF、PDGFなどのチロシンキナーゼ活性を有するチロシンキナーゼ型受容体。
【0017】
免疫方法
目的とするタンパク質に特異的な抗体を産生するリンパ球を獲得するために必要となる動物の免疫方法については特に限定されず、どのような免疫方法も採用することができる。たとえば、抗原の免疫量、免疫回数、抗原の作製方法、免疫方法、免疫動物、免疫場所、アジュバントの種類、リンパ球の摘出場所などについては、一般的に実施されている方法でかまわない。例えば、免疫動物には哺乳動物、例えばラット、マウス、モルモット、ウサギなどが使用され、抗原の動物1匹当たりの投与量は、アジュバントを用いないときは例えば0.1〜10mg、アジュバントを用いるときは10〜1000μgであるが、それらに限定されることはない。アジュバントとしては、フロイントコンプリートアジュバント、フロイントインコンプリートアジュバント、水酸化アルミニウムアジュバント等が挙げられる。免疫は、主として静脈内、皮下又は腹腔内等に注入することにより行われる。また、免疫の間隔は特に限定されず、数日から数週間間隔、好ましくは2〜5週間間隔で、1〜10回、好ましくは2〜8回免疫を行う。
また、タンパク質をコードするDNAを免疫する方法も利用することができる。本免疫方法は、好ましくは立体構造を保持した膜タンパク質を動物体内で発現させ、それに対して抗体が作製されるため、本発明と組み合わせることで非常に効率よく膜タンパク質を認識する抗体を単離できることが期待される。
また、動物のリンパ球集団を取り出した後に、in vitroで免疫する生体外免疫法なども適用することが出来る。
【0018】
ミエローマ細胞
本発明で使用するミエローマ細胞は、動物から単離したリンパ球と融合させることで、リンパ球を不死化させ、さらにリンパ球が持っている抗体産生能を維持できる細胞であれば特に限定されない。一般的には、マウス由来のミエローマ細胞がよく使用され、一例をあげると、SP2/0、PAI、P3x63Ag8.653などが例示されるが、特に限定されない。また、リンパ球を単離する動物種により、融合に利用するミエローマ細胞もそれに適した細胞種に馴化したものを使用することができる。
【0019】
ミエローマ細胞膜上へのタンパク質の存在形態
本発明の目的は、細胞膜上に存在するタンパク質、特に膜タンパク質の立体構造を認識する抗体を単離することにあり、細胞膜上に当該タンパク質を提示するミエローマ細胞の調製方法は、その細胞膜上に当該タンパク質が機能を持った形で存在させることができればどのような方法でもかまわない。一般的に考えられる方法は、遺伝子組換え法により遺伝子を外部から導入し、ミエローマ細胞膜上に、目的のタンパク質を遺伝子組換え法で発現させることにより、比較的容易に目的膜タンパク質が発現したミエローマ細胞を得ることができる(例えば、Takao、M. et al.,J.Biochem.,118、265−270(1995)参照)。単離したい抗体が認識する領域は、一般的にはタンパク質やペプチド構造の領域が多いと考えられるが、膜タンパク質の糖鎖の部分を認識する抗体でもよい。また、本来、ミエローマ細胞膜上に存在している膜タンパク質に対する抗体を単離したい場合などは、ミエローマ細胞をそのまま使用することができる。
その他の方法としては、ミエローマ細胞をリンパ球と融合する前に、ある目的タンパク質を過剰発現した細胞またはそのタンパク質を発現することができるウィルスなどをあらかじめミエローマ細胞と融合あるいは感染させておくことで、ミエローマ細胞膜表面上に目的タンパク質を存在させることができるようになる。
【0020】
目的タンパク質を細胞膜上に発現したミエローマ細胞(抗原提示ミエローマ細胞)の作製方法
本発明に係る抗原提示ミエローマ細胞の作製は、通常知られている遺伝子組換え操作に従い、発現の所望される膜タンパク質をコードする遺伝子を導入した動物細胞用発現ベクターを構築し、その発現ベクターをミエローマ細胞に導入することで成し遂げられる。遺伝子の導入方法は特に限定されず、通常実施されている方法であればどのような方法でもよく、リン酸カルシウム法、リポフェクチン法、セルフェクチン法、エレクトロポレーション法などで導入することができる。本発明中の実施例では、BCMGSneoという発現ベクターを使用しているが、このベクターに限定されるものではなく、一般的に入手できる発現ベクターでも作製することができる。また、一過性発現用あるいは定常発現用のどちらの発現ベクターでもかまわない。
【0021】
抗体産生リンパ球と抗原提示ミエローマ細胞の結合方法(近接方法)
本発明において、抗体産生リンパ球と当該タンパク質を細胞表面上に提示するミエローマ細胞を結合させる際には、通常の抗原抗体反応で、両細胞を近接させる。従って、その結合条件は、細胞へのダメージが掛からず、通常の抗原抗体反応が生ずる条件であれば特に限定されることはない。結合させる際の溶液は、たとえば、通常細胞を生育させる培地、たとえばRPMI-1640培地、DMEM培地中で、またはPBSなどの緩衝液中で実施することができる。抗体産生リンパ球を含有すると予想されるリンパ球集団と抗原提示ミエローマ細胞の細胞数の混合比は特に限定されるものではないが、たとえばリンパ球集団:抗原提示ミエローマ細胞を10:1〜1:1、好ましくは3:1〜1:1、より好ましくは1:1の割合で混合してよい。リンパ球集団と抗原提示ミエローマ細胞の反応時間は、抗体産生リンパ球上に存在する抗体の性質および、抗原提示ミエローマ細胞膜上に存在している膜タンパク質の数および性質により異なるため一概に規定できないが、通常2℃から37℃、好ましくは4℃から10℃の範囲で15分から2時間程度反応させることで成し遂げられる。特に接触時間を短くすれば、親和性の高い抗体を単離する際には好ましい方法である。
【0022】
抗体産生リンパ球と抗原提示ミエローマ細胞の融合方法
上記のとおりにして調製したミエローマ細胞とリンパ球との複合体を、次に、好適な態様において、電気パルスにより選択的に電気融合させる。電気パルスの条件は、通常行われている方法を用いることで成し遂げられる。一般的には、グルコース、シュークロース、マンニトールなどの糖で浸透圧を調整した、低塩濃度の緩衝液で細胞融合を行う。たとえば、250mMシュークロース、2mMリン酸緩衝液(pH7.2)、0.1mM塩化カルシウム、0.1mM塩化マグネシウムの緩衝液を例示できるが、浸透圧を調整するための糖の種類と濃度、緩衝能を持たせるための緩衝液の種類と濃度、pH、塩化カルシウムと塩化マグネシウムの濃度は、細胞融合する細胞の性質により最適値を求め実施される。
【0023】
電気パルスの条件は、細胞および緩衝液の状態により一概に規定できないが、通常の電気パルスを用いた細胞融合で使用される程度のパルス強度でよい。たとえば、電気パルスの条件は、1kV/cm〜4kV/cmの間で一般的に実施され、好ましくは2kV/cm〜3kV/cm、より好ましくは約2kV/cmの強さで行ってよい。さらにパルスの回数は、一般に1回から10回程度で実施される。電気パルスは、たとえばelectro square porator ECM2001(BTX社製)を用いて行うことができる。
【0024】
抗体産生リンパ球と抗原提示ミエローマ細胞の融合は、上記で示した電気パルス法が非特異的な細胞の融合を低く抑えることができるため好ましい方法である。しかし、両細胞を近接させた後にポリエチレングリコール(PEG)等の細胞融合促進剤を利用して行うこともできる。この場合、細胞融合は、血清を含まないDMEM、RPMI-1640倍地などの動物細胞培養用培地中で、抗体産生リンパ球を含有すると予想されるリンパ球集団と抗原提示ミエローマ細胞と適当な混合比(たとえばリンパ球集団:ミエローマ細胞との細胞比が10:1〜1:1)で混合し、細胞融合促進剤存在のもとで融合反応を行う。細胞融合促進剤として、平均分子量1,000〜6,000ダルトンのPEGを使用することが好ましい。
【0025】
細胞融合後のハイブリドーマの選別方法
細胞融合後の細胞の取り扱いについては、通常のハイブリドーマを作製した後の取り扱いと特に変わるところはない。細胞融合後の細胞をHAT培地での選別後にHT培地で培養し、ハイブリドーマを樹立する。培養上清中に存在する抗体の性能は通常行われている方法でスクリーニングすることができる。
【0026】
モノクローナル抗体の採取
樹立したハイブリドーマからモノクローナル抗体を採取する方法として、通常の細胞培養法又は腹水形成法等を採用することができる。細胞培養法においては、ハイブリドーマを通常用いられる培地、たとえば10%ウシ胎児血清含有RPMI−1640培地、MEM培地又は無血清培地中で、通常の培養条件(たとえば37℃、5% CO2濃度)で7〜14日間培養し、その培養上清から抗体を取得する。腹水形成法の場合は、ミエローマ細胞由来の哺乳動物と同種系動物の腹腔内にハイブリドーマをたとえば約1×107個の投与量で投与し、ハイブリドーマを大量に増殖させる。そして、1〜2週間後に腹水を採取する。抗体の採取方法において抗体の精製が必要とされる場合は、硫安塩析法、イオン交換クロマトグラフィー、ゲル濾過、アフィニティークロマトグラフィーなどの公知の方法を適宜選択して、又はこれらを組み合わせることにより精製することができる。
【実施例】
【0027】
以下に本発明の限定でない実施例を提供する。
【0028】
実施例1
TSHR発現動物細胞の構築
TSHRの全長をコードする遺伝子を2種類の発現ベクター(BCMGSneo、pSV)に導入し、それぞれの発現ベクターを、マウスミエローマ細胞(SP2/0)、およびCHO細胞(DXB11)に導入し、TSHRを発現細胞しているそれぞれの細胞を得た(SP56、CHO55)。詳細な発現ベクターの構築方法および細胞の特性については、下記の論文に詳細な記載がある。Takao、M. et al.,J.Biochem.,118、265−270(1995)。
【0029】
実施例2
TSHR細胞外領域発現ベクターの作製
TSHRをコードする遺伝子の全長を含むプラスミド(pUC118 TSHR)を出発材料とし、オリゴマー、5’−CAATTCTCAGGAATTCTTAGTAGCCCATTATGTC−3’(配列番号1)用いることで、TSHRの細胞外領域に終始コドンとEcoRIサイトを導入し、得られたプラスミドを材料に、オリゴマー、5’−GACGAACACCCCATGATATCCGCCCAGGTCCCTG−3’(配列番号2)を使うことで、シグナルペプチドの直下にEcoRVサイトを導入した。なお、どちらの変異の導入も、アマシャム社から販売されている、in vitroのミュータゲネシスキットを用いた。その後、得られたプラスミドから、EcoRIとEcoRVでTSHRの細胞外領域をコードする遺伝子フラグメントを切り出し、マルトーバイインディングプロテインとの融合発現ベクター(pMAL-c2:ニューイングランド レイブス社)のEcoRIとXmnIサイトに導入した。
【0030】
実施例3
免疫抗原の大腸菌による作製
大腸菌RB791を、実施例2で作製した発現ベクターで形質転換した。その後、アンピシリンを含むLB培地1000ml中で増殖させ、ODが0.5になるまで37℃で増殖させた。その後、30℃まで冷却後、IPTGを終濃度0.1mMになるように添加し、融合タンパク質の発現を誘導し、その後、30℃で4時間培養を継続した。大腸菌を遠心分離(8000rpm、20分)で回収し、細胞破砕液(1mM PMSF、10mM EDTAを含むPBS)を100ml添加し、超音波で大腸菌を破砕した。その後、アミロースカラムで精製を行い目的の分子量のところにタンパク質が発現していることを確認した(図1)。目的のバンドが検出されたフラクション番号4、5、6番を集めた。
【0031】
実施例4
免疫抗原のマウスへの免疫
生後8週齢のマウス(BALB/c)に対し、毎回50マイクログラムずつの実施例3で作製したタンパク質を抗原として用い、1回目はフロイントのコンプリートアジュバントと共に、2回目以降はフロイントのインコンプリートアジュバントと共に、2週間おきに6回免疫を行った。なお細胞融合3日前に、同量のタンパク質をPBSに溶解したもの、あるいは、5x107個のCHO-55細胞の懸濁液を腹腔へ免疫した。
【0032】
実施例5
SP56細胞およびCHO55細胞の培養
(SP56細胞の継代)
通常の継代は、T−25培養フラスコの底にはりついたSP56細胞を4〜5回軽くたたいて剥がし、RPMI-1640完全培地で5〜10倍に希釈して行った。細胞融合を行う際には、大量のSP56細胞を必要とするため、細胞融合の1週間前からスケールアップを行った。まず、SP56細胞をセルカウントし、1×105細胞/mlの濃度でT−75培養フラスコを用いて培養した。融合2日前には、1×105細胞/mlの濃度に希釈したSP56細胞をT−150培養フラスコ2個とT−75培養フラスコ1個にスケールアップして培養した。なお、融合2日前にRPMI-1640完全培地の抗生物質をジェネティシンから硫酸カナマイシン(100μg/ml)に替えた培地(RPMI-1640完全培地)を使用した。以降、融合後もすべてRPMI-1640完全培地を用いた。
【0033】
(CHO55細胞の継代)
CHO55細胞の培養は、RPMI-1640完全培地を用いて行った。
CHO55細胞は培養フラスコの底に貼り付き、自然には剥がれないため、CHO55細胞がフラスコ底面を覆うまで、T−25培養フラスコの上清を毎日入れ替えて培養を行った。CHO55細胞がフラスコ底面を完全に覆った場合は、生細胞を培養フラスコから剥がすために、トリプシン/EDTA溶液を用いた。この操作は、ほぼ1週間に1回の割合で行った。まず、PBS 10mlを加えてフラスコの底に貼り付いたCHO55細胞を2回洗浄した。2mlのトリプシン/EDTA溶液を加えて5〜10分、37℃、5%炭酸ガスインキュベーター内に放置し、完全にCHO55細胞が剥がれた後、130gで5分間遠心分離し、上清を除去した細胞沈殿を5〜10倍希釈し、RPMI-1640完全培地に懸濁し継代を行った。
【0034】
また、CHO55細胞をELISA法(後述)に基づく目的のモノクローナル抗体産生ハイブリドーマのスクリーニングに用いる場合は、トリプシン/EDTA溶液処理後5〜10倍希釈したCHO55細胞を96穴培養プレートに3〜4滴(350μl)ずつ滴下し、2〜3日37℃、5%炭酸ガスインキュベーター内で培養して用いた。
【0035】
実施例6
マウス脾細胞の調製
細胞融合日の3日前に最終免疫が終了したマウスの腹腔内に、0.1g/mlの無菌泡水クロラールを100〜200μl注射し、マウスが完全に麻酔にかかったことを確認した後、解剖用ハサミで心臓が見えるまで開胸し、すばやく心臓から採血した。心臓採血後のマウスは、70%エタノールが入った200mlのビーカー内に浸して無菌化した後、クリーンベンチ内に入れて解剖した。
【0036】
クリーンベンチ内にて解剖用ハサミでマウスより無菌的に脾臓を摘出した。摘出した脾臓は、10mlの抗生物質(100μg/ml硫酸カナマイシン)入りRPMI-1640を入れたシャーレで数回洗浄し、周りの脂肪をハサミで切り除いた後、ステンレスメッシュ上に置いてラバーポリスマンで穏やかに砕いた。ステンレスメッシュを30〜40mlの抗生物質入りRPMI-1640で洗浄し、懸濁液を2,000rpm(800g)で5分間遠心分離後、細胞沈殿を5mlの赤血球破砕緩衝液(SIGMA社、R7757)で懸濁し、氷中で5分間静置することにより赤血球を溶血させた。その後、抗生物質入りRPMI-1640で洗浄し、最終的に5mlの抗生物質入りRPMI-1640に懸濁し、生細胞数をカウントした。
【0037】
実施例7
細胞融合
(SP56細胞の調製)
スケールアップ培養したSP56細胞を回収し、130gで5分間遠心分離し、沈殿を10mlの抗生物質入りRPMI-1640に懸濁した。これを再び5分間遠心分離し、5mlの抗生物質入りRPMI-1640で懸濁し、生細胞数をカウントした。
【0038】
(リンパ球−SP56細胞複合体の作製と電気融合)
実施例6で調製した脾細胞懸濁液と上述で調製したSP56細胞を1:1の割合で混合し、4℃または37℃にて2時間ゆっくりと混和した。その後、1、000rpm(200g)で10分間遠心分離し、沈殿を1ml抗生物質入りRPMI-1640に懸濁した。400〜500rpm(〜50g)で1〜2分間遠心後、5%炭酸ガスインキュベーター内で30分間放置した。さらに30分間、5%炭酸ガスインキュベーター内でゆっくりとローテーションした。ローテーション後、再び200gで10分間遠心分離し、2mlのアイソトニックシュークロース緩衝液 [0.25M シュークロース+2mM リン酸ナトリウム緩衝液(pH7.2)+0.1mM MgCl2+0.1mM CaCl2]に懸濁した。これをプレパラート型電極上に0.5mlずつ加え、electro square porator ECM2001(BTX社製)により、2kV/cmと3kV/cmの条件で電気融合を行った。電気融合後、20mlのRPMI-1640完全培地に融合細胞懸濁液を静かに加え、30分間静置し、96穴プレートに0.2ml/wellになるように分注した。その後、速やかに37℃、5%炭酸ガスインキュベーター内で培養した。次の日に、プレートの培養上清を0.1ml/wellずつ除去し、RPMI-1640完全培地で50倍に希釈したHAT培地を0.1ml/wellずつ加えた。
細胞融合後の翌日から、2〜3日ごとに96穴プレート中の培養上清をHAT培地で0.1ml/wellずつ交換した。2週間経過した後、HAT培地をHT培地に替えて、同様に2週間行い、それ以降はRPMI-1640完全培地により培地交換を行った。
【0039】
実施例8
培養上清の評価方法
(CHO55細胞を用いたELISA法)
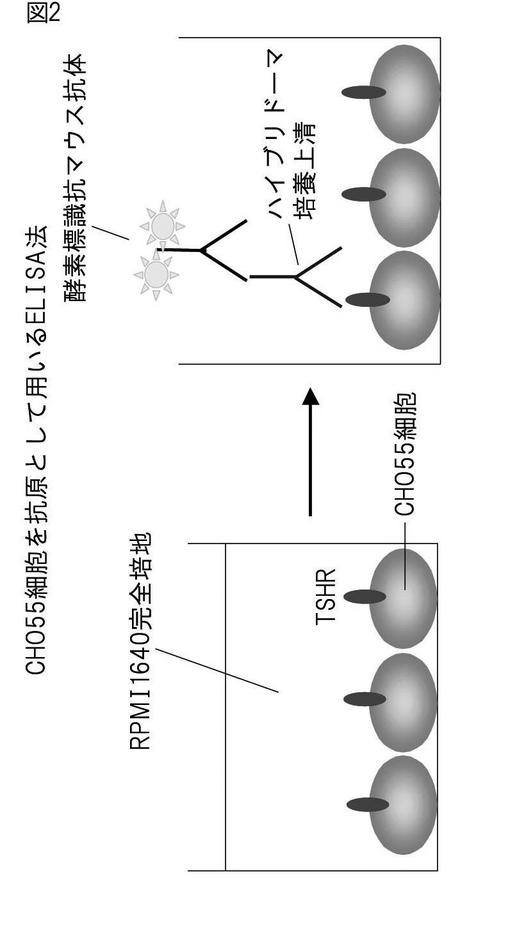
概念図を図2に示した。実施例5の方法で96穴培養プレートにて37℃、5%炭酸ガスインキュベーター内で2〜3日間培養することにより、CHO55細胞を培養プレートの底に吸着させた。それと同時に2〜3日間培養することにより、RPMI-1640完全培地に含まれる10% FCSによって96穴ウェルのブロッキングも行った。次に、CHO55細胞培養上清を回収し、一次抗体としてハイブリドーマ培養上清、または100倍希釈したマウス血清を100μl加え、37℃で1時間インキュベートした。アイソトニックシュークロース緩衝液で2回洗浄後、同緩衝液で10,000倍に希釈した二次抗体[HRP標識された抗マウスIgG(H+L)抗体]を50μl加え、37℃、1時間インキュベートした。最後に、アイソトニックシュークロース緩衝液で3〜5回洗浄後、発色剤[0.1Mクエン酸ナトリウム緩衝液(pH5.2)にオルトフェニレンジアミン(1mg/ml)と0.02%過酸化水素を溶解させたもの]を100μl加えて37℃、10分間インキュベートして発色させ、1M硫酸を50μl加えて反応を止めた。この上清を96穴アッセイプレートに移しマイクロプレートリーダーを用い490nmの吸光度の測定を行った。
【0040】
(競合アッセイ法に基づくELISA法)
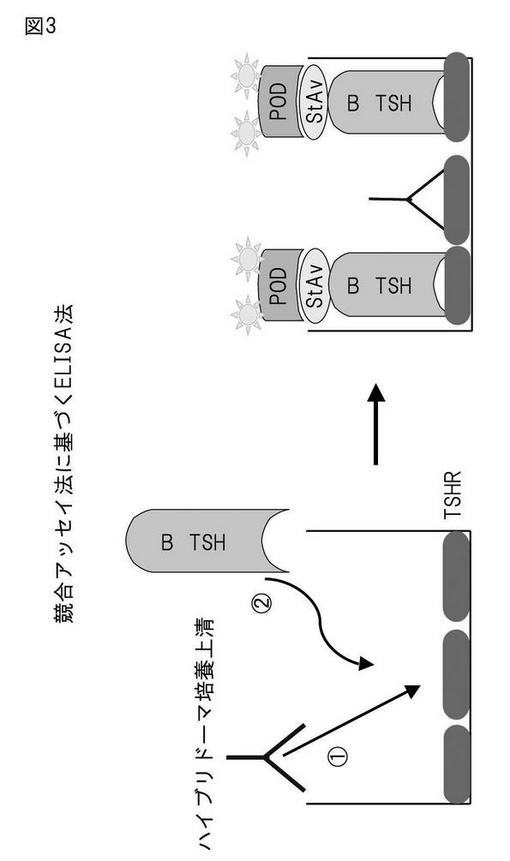
概念図を図3に示した。競合ELISA法に基づく検出用キット[TRAb ELISA「コスミック」]を用いて抗TSHRモノクローナル抗体の評価を行った。具体的には、TSHRが予め固相化されたストリップウェルを必要な数だけ取り出し、少なくとも30分間、25℃に放置した。ウェルに開始緩衝液を50μl加え、ハイブリドーマ培養上清、陰性、陽性コントロールを100μl加え、25℃で2時間インキュベートした。各ウェルを洗浄後、ビオチン化TSHを100μl加え、25℃、40分間インキュベートし、再び各ウェルを洗浄後、ストレプトアビジンペルオキシタ-ゼ100μlを加え、25℃、20分間インキュベートした。最後に、洗浄用緩衝液で2回、精製水で1回洗浄後、基質液を100μl加えて遮光し、25℃、20分間インキュベートして発色させ、停止液を50μl加えて反応を止めた。これをマイクロプレートリーダーを用い、450nmの吸光度の測定を行った。
【0041】
(CHO55細胞を用いたDELFIA法)
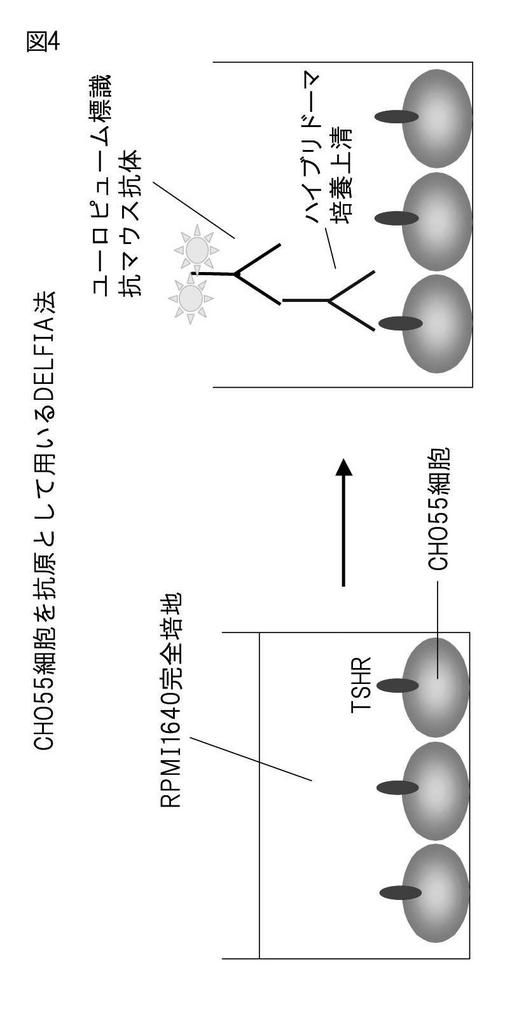
概念図を図4に示した。24ウェルプレートにCHO55細胞を増殖させたあとに、ハイブリドーマの培養上清を、0.25%BSAを含むアイソトニックシュークロース緩衝液5倍に希釈したサンプルを250μl/ウェルで加え、37℃で1時間反応させた。その後、同緩衝液で3回洗浄し、同緩衝液で1、000倍に希釈した 二次抗体[ユーロピューム標識された抗マウス抗体 ]を250μl加え、37℃、1時間インキュベートした。BSAを含まない、アイソトニックシュークロース緩衝液で3回洗浄後、発色剤(エンハンスメント溶液:パーキンエルマー社)を200μl/ウェルで添加し、ユーロピュームの遅延蛍光を測定できるプレートリーダーで測定した。
【0042】
実施例9
細胞融合の結果
表1および表2に示す結果が得られた。最終免疫にタンパク質を使用した場合、平均して約20%の効率でハイブリドーマを得ることができた。ELISA陽性率は全ハイブリドーマ中、約24%であった。これに対して、最終免疫にCHO細胞を使用した場合、平均して約42%の高い効率でハイブリドーマを得ることができたが、全ハイブリドーマ中、ELISA陽性率は約13%の低い値となった。しかしながら融合効率およびELISA陽性率の両面から判断して、本実験において、目的の抗体産生ハイブリドーマ作製における免疫方法による大きな差異はないと考えられた。いずれにせよ、リンパ球とミエローマ細胞との非特異的融合による従来技術の融合方法では目的とするハイブリドーマは獲得できなかったため、この結果は驚くべきものである。また、新技術によるハイブリドーマ出現効率を比較すると、2kV/cmと3kV/cmにおいて顕著な差は認められなかったが、ELISA陽性率に関しては、2kV/cmの方が高い値を示した。さらに、TSHR発現ミエローマ細胞を用いた高次構造特異抗体発現リンパ球の選択温度は、4℃の方が、37℃より適していると推測された。
【0043】
表1 最終免疫を実施例3で作製したTSHRで行った場合の結果
【表1】

【0044】
表2 最終免疫をCHO55で行った場合の結果
【表2】
【0045】
実施例10
モノクローナル抗体産生ハイブリドーマの検証
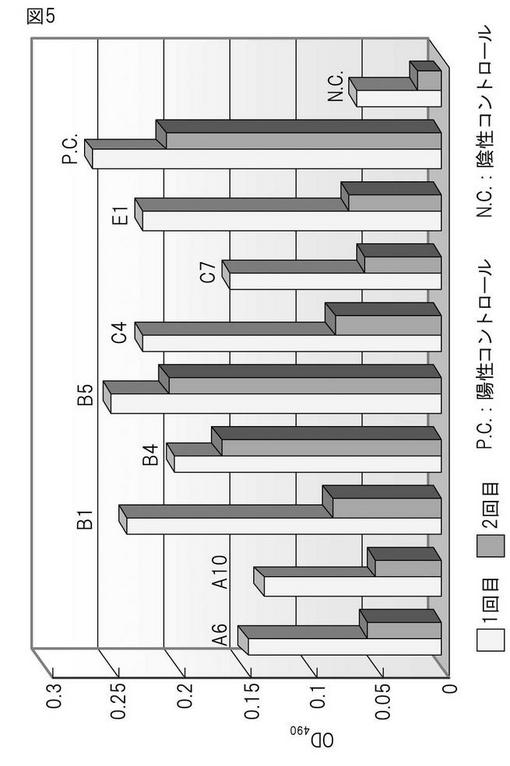
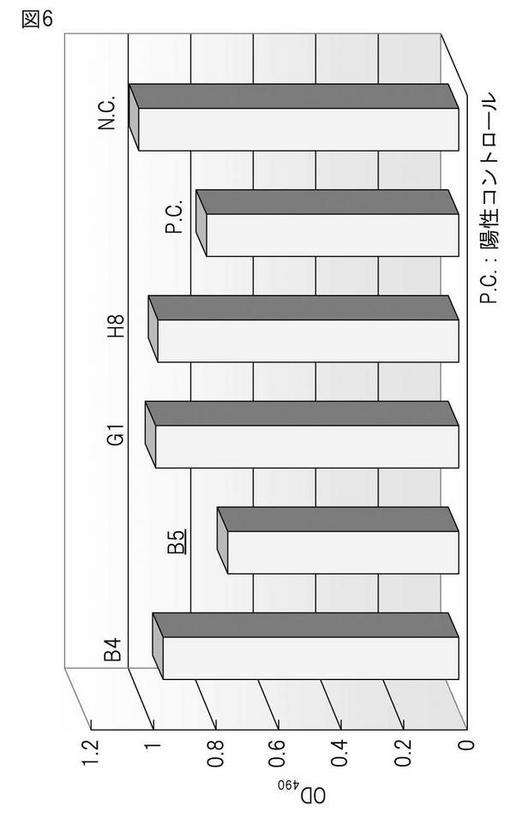
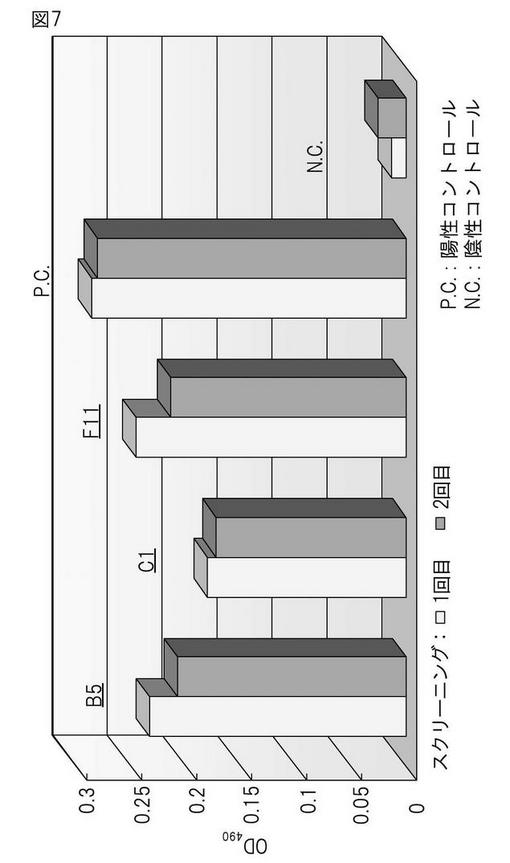
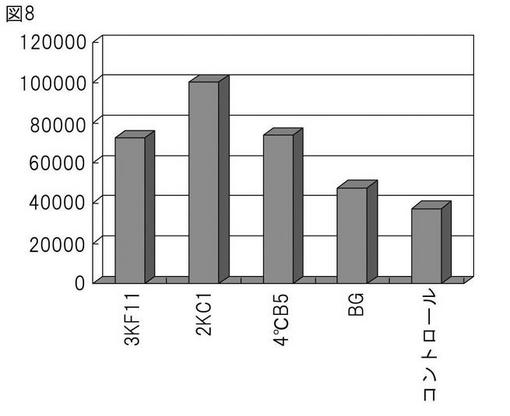
本発明によって得られたハイブリドーマによって産生されるモノクローナル抗体の親和性を検証するため、CHO55細胞を抗原とするELISA法(図2)および競合アッセイ法に基づくELISA法(図3)の2種の方法を用い検討した。まず初めに、CHO55細胞を抗原とするELISA法(図2)による解析の結果、ハイブリドーマB5(8/12日細胞融合)によって産生されるモノクローナル抗体が高い抗体価を示し、またその再現性も認められた(図5)。さらに、競合アッセイ法に基づくELISA法(図3)に基づき解析を行った結果、B5(8/12日細胞融合)においては、陽性コントロールに近い値を示し、高い抗体価を有することが確認できた(図6)。すべての融合プレートにおいて、CHO55細胞を抗原とするELISA法 による分析の結果、ハイブリドーマB5(8/12日細胞融合)およびハイブリドーマC1、F11(10/23日細胞融合)によって産生されるモノクローナル抗体が、CHO55細胞に対して再現性よく高い親和性を示すことが明らかとなった(図7)。また、遅延蛍光による評価でも、優位な結合が確認さらた(図8)。
【0046】
実施例11
共焦点レーザー蛍光顕微鏡による可視化解析
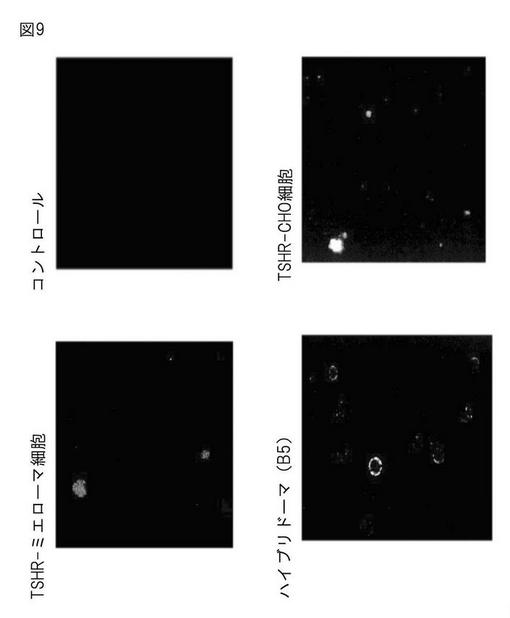
CHO55細胞、SP56細胞、ELISA陽性ハイブリドーマ(B5)をそれぞれT−75培養フラスコで培養した後、130gで5分間、遠心分離し、沈殿をRPMI-1640 0.5mlに懸濁した。生体内免疫によって得られたマウスの血清(ポリクローナル抗体)の原液を10μl加え、4℃で2時間ローテーションした後、ハンクス平衡塩10mlで洗浄し、沈殿をハンクス平衡塩1mlに懸濁した。Alexa Fluor 488にて蛍光標識された抗マウスIgG(H+L)抗体を6μl加え、遮光条件下にて37℃で30分間、ゆっくりローテーションを行った。ハンクス平衡塩10mlで洗浄後、細胞をハンクス平衡塩0.5mlに懸濁して、共焦点レーザー顕微鏡で観察した。その結果、すべての細胞において特異的蛍光が観察されたことから、それら細胞の膜表面にTSHRが高次構造を保持し発現されていることが確認された(図9)。
【0047】
これらの結果に基づき、本発明により目的の細胞膜上に存在するTSHR認識モノクローナル抗体産生ハイブリドーマの作出に成功したと結論づける。
【産業上の利用可能性】
【0048】
本方法によれば、従来では単離することが困難であった、細胞膜上に存在する膜タンパク質を認識する抗体を、非常に効率良く認識することができる。膜タンパク質のなかでも、特に単離することが難しい多回膜貫通型のタンパク質を認識する抗体を単離できるようになる。
【図面の簡単な説明】
【0049】
【図1】融合タンパク質精製した際のSDSポリアクリルアミドの図。
【図2】CHO55細胞を抗原とするELISA法の原理の概説図。
【図3】競合アッセイに基づくELISA法の原理の概説図。
【図4】CHO55細胞を抗原とするDELFIAの原理の概説図。
【図5】CHO55細胞を抗原とするELISAスクリーニングの結果。
【図6】競合アッセイに基づくELISAスクリーニングの結果。
【図7】CHO55細胞を抗原とするELISAスクリーニングの再現性。
【図8】CHO55細胞を抗原とするDELFIAの結果。コントロールは、まったく違う抗原(エストラジオール)を認識する抗体を示す。
【図9】共焦点レーザー蛍光顕微鏡による可視化解析の結果。
【技術分野】
【0001】
本発明は、タンパク質、特に細胞膜タンパク質の立体構造を認識するモノクローナル抗体を産生する目的のハイブリドーマを効率良く産生する方法に関する。
【0002】
これまでに種々の抗原を認識するモノクローナル抗体は多数単離されており、医薬、診断薬だけではなく、各種の実験などに広く利用されている。
モノクローナル抗体は、動物由来のリンパ球とミエローマ細胞を融合することで作製されており、融合させる方法としてはポリエチレングリコール(PEG)法が一般的に利用されている。一般的なPEG法では、目的抗体を発現する細胞とミエローマ細胞との融合だけではなく、目的抗体を発現していないリンパ球とミエローマ細胞との融合、リンパ球同士の融合、ミエローマ細胞同士の融合などもおこり、得られるハイブリドーマ中の目的ハイブリドーマの割合は数%程度である。しかしながら、一般的な研究・実験(たとえば、ELISA、ウェスタンブロッティングなど)に使用するモノクローナル抗体の作製であれば、この方法でも特に大きな問題なく抗体を十分に単離することができる。他の融合方法として、電気的に細胞を融合させる方法(たとえば、はじめに交流下で細胞同士を近接させた後に、直流パルスをかけて細胞を融合させる方法)も広く利用されている。かかる方法も原理的には、PEG法と同様、リンパ球とミエローマ細胞を非特異的に融合させることから、目的抗体のスクリーニングには煩雑な作業を必要とする。
【0003】
上記細胞融合法の欠点を克服するため、下記に示す方法が報告されている。ここに示す方法のポイントは、リンパ球の細胞表面にそのリンパ球が発現する抗体が提示されていることを極めて巧妙に利用する点にある。以下に示す二つの細胞(1)、(2)を準備する。(1)あらかじめ作製しておいた抗原−アビジン複合体をリンパ球と反応させ、抗原結合能を有した抗体を発現しているリンパ球だけを、抗原−アビジン複合体で標識する。(2)ミエローマ細胞の表面をビオチンで修飾した細胞を準備する。その後、(1)と(2)の細胞を混合すると、ビオチン-アビジンの極めて強力な結合力により、リンパ球−抗原−アビジン−ビオチン−ミエローマ細胞という細胞の複合体を形成させることができる。その後、電気パルスをかけることで、複合体を形成した細胞を選択的に融合させることが可能となる。この方法によると、目的抗体を発現していないリンパ球がミエローマ細胞と融合する確立が極めて低くなる点が特徴となる。この方法でハイブリドーマを作製すると、得られるハイブリドーマ中の目的ハイブリドーマの割合は、多い場合には60%を超える極めて高い値を示すことが報告されており、非常に優れた方法である(冨田昌弘、Bioベンチャー、Vol.1、108(2001))。
【0004】
しかし、抗原が膜タンパク質である場合には上記冨田らの方法を利用するのは困難である。その理由としては、高純度の十分な量(数mg)の膜タンパク質を精製すること自体一般に困難であることや、その膜タンパク質を高次構造を保持した状態でアビジンと結合体を作製するのが極めて難しいことにある。つまり、膜タンパク質を認識する抗体を単離する場合には、冨田らの方法を採用することができず、通常の細胞融合法、即ちリンパ球とミエローマ細胞を非特異的に融合させる方法を介し、抗体を単離する方法しかなかった。
【0005】
通常の細胞融合法で膜タンパク質を認識する抗体の単離を試みる場合、ウェスタンブロッティングなど、タンパク質を変性させた条件下で膜タンパク質に結合する抗体をスクリーニングする操作を繰り返す必要がある。しかしながら、細胞膜上に存在する状態の膜タンパク質を認識できる抗体の単離はきわめて困難である。その理由は、抗原に結合する抗体のうち、細胞膜タンパク質の立体構造を認識できる抗体の存在率が低いことによる。上記でも述べたように、通常の細胞融合法では、得られるハイブリドーマ中の目的抗体の割合は多くても数%程度と低く、さらにその中から目的の機能を持った抗体を探し出す必要がある。
【0006】
このように、細胞膜上に存在する膜タンパク質の立体構造を認識する抗体は、単離することが難しいというのが現状であった。さらに、膜タンパク質がホルモンなどの生理活性物質の何らかの受容体である場合などにおいては、それを特異的に認識する抗体は、通常の研究用途として重要であるだけではなく、医薬品としての利用も考えられる。しかし、そのような抗体を単離するには、従来から報告されている細胞融合法を繰り返し、膨大な数のハイブリドーマの評価を行い、目的の抗体単離を試みる必要があったが、目的の抗体を単離するには偶然に頼るしかなかった。
【0007】
【非特許文献1】冨田昌弘、Bioベンチャー、Vol.1、108(2001)
【発明の開示】
【発明が解決しようとする課題】
【0008】
そこで、本発明者らは、膜タンパク質を認識する抗体を発現するハイブリドーマの作製法について鋭意検討した結果、本発明に到達した。
【課題を解決するための手段】
【0009】
本発明は、目的のタンパク質を抗原として用いる免疫により獲得されたリンパ球集団と、細胞表面に当該タンパク質を提示するミエローマ細胞とを混合し;
当該リンパ球集団内の当該タンパク質に特異的な膜結合型モノクローナル抗体を提示するリンパ球を当該ミエローマ細胞に対し、当該膜結合型抗体と当該ミエローマ細胞表面に存在する当該膜タンパク質との特異的な結合を介し、選択的に近寄らせ;そして
両細胞を融合させる、
ステップを含むことを特徴とする、当該タンパク質に特異的なモノクローナル抗体を産生するハイブリドーマの作製方法、を提供する。好ましくは、目的のタンパク質は細胞の表層上に提示されるいわゆる膜タンパク質である。
【0010】
具体的には、遺伝子操作などによって高次構造又は立体構造を保持したタンパク質を細胞表層上において提示して有するミエローマ細胞(以下、単に「抗原提示ミエローマ細胞」と称する場合がある)を作製する。それとは別に、タンパク質を抗原として用いて免疫した動物に由来するリンパ球集団を準備する。このリンパ球集団は、目的のタンパク質の立体構造を特異的に認識する抗体を細胞表層上に提示し、当該抗体を産生する能力の備わったリンパ球(以下、単に「抗体産生リンパ球」と称する場合がある)を含有すると予想される。両者の細胞を混合すると、抗原抗体反応に基づき、抗体産生リンパ球上に存在する当該タンパク質に特異的な抗体が、抗原提示ミエローマ細胞上のタンパク質に結合することで、抗体産生リンパ球-細胞表面抗体-細胞表面タンパク質-抗原提示ミエローマ細胞、という細胞の複合体を形成させることができる。その後、電気パルスを負荷することにより両細胞の選択的融合を行い、目的のタンパク質に特異的なモノクローナル抗体を産生するハイブリドーマを作製する方法である。
【0011】
本発明によれば、細胞膜上のタンパク質を認識できない抗体を発現したリンパ球は、上記で示す複合体を抗原提示ミエローマ細胞と形成することができない。つまり、細胞膜上に存在するタンパク質を認識できる抗体を発現している抗体産生リンパ球だけを選択的にハイブリドーマにすることができる。
【発明の効果】
【0012】
本方法に従うと、得られるハイブリドーマは細胞膜上に存在するタンパク質を認識する抗体を発現することになる。たとえば、タンパク質が何らかの生理活性物質の受容体である場合などは、受容体のアゴニストあるいはアンタゴニスト型の抗体を、これまでの方法と比較してはるかに容易に単離できることが予想され、研究用途用の抗体のみならず、医薬品用の抗体としての展開も考えられる。
【発明を実施するための最良の形態】
【0013】
以下で本発明について詳細に説明する。
【0014】
本発明は、タンパク質、特に細胞膜上で提示される膜タンパク質を特異的に認識するモノクローナル抗体を産生するハイブリドーマの作製に好適に利用される。膜タンパク質とは、細胞表面上に存在するタンパク質をいい、膜を貫通する回数および分子量は問わない。一般的に、膜タンパク質の膜貫通回数が増加するに伴い、従来法では膜タンパク質を認識する抗体を単離することが困難になっていくため、そのような膜タンパク質を認識する抗体を単離する場合に本発明は特に有効であると推測されるが、特にそれらに限定されない。従来法では膜タンパク質を認識する抗体を単離するのが困難なのは以下の理由による。たとえば、一回膜貫通型の膜タンパク質の場合、膜タンパク質の細胞外領域だけを遺伝子組換え法などを用い作製した場合でも、リガンドの結合能は失われない場合が多い。たとえばインターロイキン-6(IL-6)の受容体は、一回膜貫通型のタンパク質であるが、細胞外領域だけを遺伝子組換え型で発現させたとしても、細胞外領域だけでもIL-6 との結合能は保持している。しかしながら、多回膜貫通型のタンパク質、たとえば7回膜貫通型のタンパク質である甲状腺刺激ホルモン受容体(Thyroid stimulation hormone receptor: TSHR)などの場合、細胞外領域のみを発現させたとしても、抗原結合能を保持しないか、あったとしても極めて弱い結合しか示さない。このことは、TSHRの細胞外領域が、10アミノ酸程度からなる細胞外ループ(TSHRでは3つ存在)と何らかの相互作用があって初めて抗原結合能が発現することを意味している。
【0015】
つまり、膜貫通回数が多くなるにつれて、細胞膜上に存在する形態と、それを免疫している状態とが異なることが予想され、細胞融合後に得られる抗体の中で、細胞膜上に存在するタンパク質を認識できる抗体の割合が低くなってゆくことが予想される。つまり、本発明はこれまでに非常に単離が困難であった多回膜貫通型のタンパク質が細胞膜上に存在する状態を認識する抗体の単離方法として利用できる可能性が高い。
【0016】
本発明の方法において対象となる膜タンパク質には、上記TSHRのほか、特に限定することなく、たとえば下記の膜タンパク質が挙げられる:
ニコチン、GABA、グリシンなどのイオンチャンネル内蔵型受容体、またはアルファー1、ヒスタミン1(H1)、ブラジキニン(B2)、ムスカリン1(M1)、アンジオテンシンII(AT1)などの受容体でイノシトールリン酸を分解する受容体、ベータ、ヒスタミン2(H2)、プロスタサイクリン(IP)、グルカゴンなどの受容体、TSH、FSH、LHなどの各種ペプチドホルモンなどの受容体でcAMPを合成し、GTP結合蛋白質共役型の受容体、またはインシュリン、EGF、FGF、PDGFなどのチロシンキナーゼ活性を有するチロシンキナーゼ型受容体。
【0017】
免疫方法
目的とするタンパク質に特異的な抗体を産生するリンパ球を獲得するために必要となる動物の免疫方法については特に限定されず、どのような免疫方法も採用することができる。たとえば、抗原の免疫量、免疫回数、抗原の作製方法、免疫方法、免疫動物、免疫場所、アジュバントの種類、リンパ球の摘出場所などについては、一般的に実施されている方法でかまわない。例えば、免疫動物には哺乳動物、例えばラット、マウス、モルモット、ウサギなどが使用され、抗原の動物1匹当たりの投与量は、アジュバントを用いないときは例えば0.1〜10mg、アジュバントを用いるときは10〜1000μgであるが、それらに限定されることはない。アジュバントとしては、フロイントコンプリートアジュバント、フロイントインコンプリートアジュバント、水酸化アルミニウムアジュバント等が挙げられる。免疫は、主として静脈内、皮下又は腹腔内等に注入することにより行われる。また、免疫の間隔は特に限定されず、数日から数週間間隔、好ましくは2〜5週間間隔で、1〜10回、好ましくは2〜8回免疫を行う。
また、タンパク質をコードするDNAを免疫する方法も利用することができる。本免疫方法は、好ましくは立体構造を保持した膜タンパク質を動物体内で発現させ、それに対して抗体が作製されるため、本発明と組み合わせることで非常に効率よく膜タンパク質を認識する抗体を単離できることが期待される。
また、動物のリンパ球集団を取り出した後に、in vitroで免疫する生体外免疫法なども適用することが出来る。
【0018】
ミエローマ細胞
本発明で使用するミエローマ細胞は、動物から単離したリンパ球と融合させることで、リンパ球を不死化させ、さらにリンパ球が持っている抗体産生能を維持できる細胞であれば特に限定されない。一般的には、マウス由来のミエローマ細胞がよく使用され、一例をあげると、SP2/0、PAI、P3x63Ag8.653などが例示されるが、特に限定されない。また、リンパ球を単離する動物種により、融合に利用するミエローマ細胞もそれに適した細胞種に馴化したものを使用することができる。
【0019】
ミエローマ細胞膜上へのタンパク質の存在形態
本発明の目的は、細胞膜上に存在するタンパク質、特に膜タンパク質の立体構造を認識する抗体を単離することにあり、細胞膜上に当該タンパク質を提示するミエローマ細胞の調製方法は、その細胞膜上に当該タンパク質が機能を持った形で存在させることができればどのような方法でもかまわない。一般的に考えられる方法は、遺伝子組換え法により遺伝子を外部から導入し、ミエローマ細胞膜上に、目的のタンパク質を遺伝子組換え法で発現させることにより、比較的容易に目的膜タンパク質が発現したミエローマ細胞を得ることができる(例えば、Takao、M. et al.,J.Biochem.,118、265−270(1995)参照)。単離したい抗体が認識する領域は、一般的にはタンパク質やペプチド構造の領域が多いと考えられるが、膜タンパク質の糖鎖の部分を認識する抗体でもよい。また、本来、ミエローマ細胞膜上に存在している膜タンパク質に対する抗体を単離したい場合などは、ミエローマ細胞をそのまま使用することができる。
その他の方法としては、ミエローマ細胞をリンパ球と融合する前に、ある目的タンパク質を過剰発現した細胞またはそのタンパク質を発現することができるウィルスなどをあらかじめミエローマ細胞と融合あるいは感染させておくことで、ミエローマ細胞膜表面上に目的タンパク質を存在させることができるようになる。
【0020】
目的タンパク質を細胞膜上に発現したミエローマ細胞(抗原提示ミエローマ細胞)の作製方法
本発明に係る抗原提示ミエローマ細胞の作製は、通常知られている遺伝子組換え操作に従い、発現の所望される膜タンパク質をコードする遺伝子を導入した動物細胞用発現ベクターを構築し、その発現ベクターをミエローマ細胞に導入することで成し遂げられる。遺伝子の導入方法は特に限定されず、通常実施されている方法であればどのような方法でもよく、リン酸カルシウム法、リポフェクチン法、セルフェクチン法、エレクトロポレーション法などで導入することができる。本発明中の実施例では、BCMGSneoという発現ベクターを使用しているが、このベクターに限定されるものではなく、一般的に入手できる発現ベクターでも作製することができる。また、一過性発現用あるいは定常発現用のどちらの発現ベクターでもかまわない。
【0021】
抗体産生リンパ球と抗原提示ミエローマ細胞の結合方法(近接方法)
本発明において、抗体産生リンパ球と当該タンパク質を細胞表面上に提示するミエローマ細胞を結合させる際には、通常の抗原抗体反応で、両細胞を近接させる。従って、その結合条件は、細胞へのダメージが掛からず、通常の抗原抗体反応が生ずる条件であれば特に限定されることはない。結合させる際の溶液は、たとえば、通常細胞を生育させる培地、たとえばRPMI-1640培地、DMEM培地中で、またはPBSなどの緩衝液中で実施することができる。抗体産生リンパ球を含有すると予想されるリンパ球集団と抗原提示ミエローマ細胞の細胞数の混合比は特に限定されるものではないが、たとえばリンパ球集団:抗原提示ミエローマ細胞を10:1〜1:1、好ましくは3:1〜1:1、より好ましくは1:1の割合で混合してよい。リンパ球集団と抗原提示ミエローマ細胞の反応時間は、抗体産生リンパ球上に存在する抗体の性質および、抗原提示ミエローマ細胞膜上に存在している膜タンパク質の数および性質により異なるため一概に規定できないが、通常2℃から37℃、好ましくは4℃から10℃の範囲で15分から2時間程度反応させることで成し遂げられる。特に接触時間を短くすれば、親和性の高い抗体を単離する際には好ましい方法である。
【0022】
抗体産生リンパ球と抗原提示ミエローマ細胞の融合方法
上記のとおりにして調製したミエローマ細胞とリンパ球との複合体を、次に、好適な態様において、電気パルスにより選択的に電気融合させる。電気パルスの条件は、通常行われている方法を用いることで成し遂げられる。一般的には、グルコース、シュークロース、マンニトールなどの糖で浸透圧を調整した、低塩濃度の緩衝液で細胞融合を行う。たとえば、250mMシュークロース、2mMリン酸緩衝液(pH7.2)、0.1mM塩化カルシウム、0.1mM塩化マグネシウムの緩衝液を例示できるが、浸透圧を調整するための糖の種類と濃度、緩衝能を持たせるための緩衝液の種類と濃度、pH、塩化カルシウムと塩化マグネシウムの濃度は、細胞融合する細胞の性質により最適値を求め実施される。
【0023】
電気パルスの条件は、細胞および緩衝液の状態により一概に規定できないが、通常の電気パルスを用いた細胞融合で使用される程度のパルス強度でよい。たとえば、電気パルスの条件は、1kV/cm〜4kV/cmの間で一般的に実施され、好ましくは2kV/cm〜3kV/cm、より好ましくは約2kV/cmの強さで行ってよい。さらにパルスの回数は、一般に1回から10回程度で実施される。電気パルスは、たとえばelectro square porator ECM2001(BTX社製)を用いて行うことができる。
【0024】
抗体産生リンパ球と抗原提示ミエローマ細胞の融合は、上記で示した電気パルス法が非特異的な細胞の融合を低く抑えることができるため好ましい方法である。しかし、両細胞を近接させた後にポリエチレングリコール(PEG)等の細胞融合促進剤を利用して行うこともできる。この場合、細胞融合は、血清を含まないDMEM、RPMI-1640倍地などの動物細胞培養用培地中で、抗体産生リンパ球を含有すると予想されるリンパ球集団と抗原提示ミエローマ細胞と適当な混合比(たとえばリンパ球集団:ミエローマ細胞との細胞比が10:1〜1:1)で混合し、細胞融合促進剤存在のもとで融合反応を行う。細胞融合促進剤として、平均分子量1,000〜6,000ダルトンのPEGを使用することが好ましい。
【0025】
細胞融合後のハイブリドーマの選別方法
細胞融合後の細胞の取り扱いについては、通常のハイブリドーマを作製した後の取り扱いと特に変わるところはない。細胞融合後の細胞をHAT培地での選別後にHT培地で培養し、ハイブリドーマを樹立する。培養上清中に存在する抗体の性能は通常行われている方法でスクリーニングすることができる。
【0026】
モノクローナル抗体の採取
樹立したハイブリドーマからモノクローナル抗体を採取する方法として、通常の細胞培養法又は腹水形成法等を採用することができる。細胞培養法においては、ハイブリドーマを通常用いられる培地、たとえば10%ウシ胎児血清含有RPMI−1640培地、MEM培地又は無血清培地中で、通常の培養条件(たとえば37℃、5% CO2濃度)で7〜14日間培養し、その培養上清から抗体を取得する。腹水形成法の場合は、ミエローマ細胞由来の哺乳動物と同種系動物の腹腔内にハイブリドーマをたとえば約1×107個の投与量で投与し、ハイブリドーマを大量に増殖させる。そして、1〜2週間後に腹水を採取する。抗体の採取方法において抗体の精製が必要とされる場合は、硫安塩析法、イオン交換クロマトグラフィー、ゲル濾過、アフィニティークロマトグラフィーなどの公知の方法を適宜選択して、又はこれらを組み合わせることにより精製することができる。
【実施例】
【0027】
以下に本発明の限定でない実施例を提供する。
【0028】
実施例1
TSHR発現動物細胞の構築
TSHRの全長をコードする遺伝子を2種類の発現ベクター(BCMGSneo、pSV)に導入し、それぞれの発現ベクターを、マウスミエローマ細胞(SP2/0)、およびCHO細胞(DXB11)に導入し、TSHRを発現細胞しているそれぞれの細胞を得た(SP56、CHO55)。詳細な発現ベクターの構築方法および細胞の特性については、下記の論文に詳細な記載がある。Takao、M. et al.,J.Biochem.,118、265−270(1995)。
【0029】
実施例2
TSHR細胞外領域発現ベクターの作製
TSHRをコードする遺伝子の全長を含むプラスミド(pUC118 TSHR)を出発材料とし、オリゴマー、5’−CAATTCTCAGGAATTCTTAGTAGCCCATTATGTC−3’(配列番号1)用いることで、TSHRの細胞外領域に終始コドンとEcoRIサイトを導入し、得られたプラスミドを材料に、オリゴマー、5’−GACGAACACCCCATGATATCCGCCCAGGTCCCTG−3’(配列番号2)を使うことで、シグナルペプチドの直下にEcoRVサイトを導入した。なお、どちらの変異の導入も、アマシャム社から販売されている、in vitroのミュータゲネシスキットを用いた。その後、得られたプラスミドから、EcoRIとEcoRVでTSHRの細胞外領域をコードする遺伝子フラグメントを切り出し、マルトーバイインディングプロテインとの融合発現ベクター(pMAL-c2:ニューイングランド レイブス社)のEcoRIとXmnIサイトに導入した。
【0030】
実施例3
免疫抗原の大腸菌による作製
大腸菌RB791を、実施例2で作製した発現ベクターで形質転換した。その後、アンピシリンを含むLB培地1000ml中で増殖させ、ODが0.5になるまで37℃で増殖させた。その後、30℃まで冷却後、IPTGを終濃度0.1mMになるように添加し、融合タンパク質の発現を誘導し、その後、30℃で4時間培養を継続した。大腸菌を遠心分離(8000rpm、20分)で回収し、細胞破砕液(1mM PMSF、10mM EDTAを含むPBS)を100ml添加し、超音波で大腸菌を破砕した。その後、アミロースカラムで精製を行い目的の分子量のところにタンパク質が発現していることを確認した(図1)。目的のバンドが検出されたフラクション番号4、5、6番を集めた。
【0031】
実施例4
免疫抗原のマウスへの免疫
生後8週齢のマウス(BALB/c)に対し、毎回50マイクログラムずつの実施例3で作製したタンパク質を抗原として用い、1回目はフロイントのコンプリートアジュバントと共に、2回目以降はフロイントのインコンプリートアジュバントと共に、2週間おきに6回免疫を行った。なお細胞融合3日前に、同量のタンパク質をPBSに溶解したもの、あるいは、5x107個のCHO-55細胞の懸濁液を腹腔へ免疫した。
【0032】
実施例5
SP56細胞およびCHO55細胞の培養
(SP56細胞の継代)
通常の継代は、T−25培養フラスコの底にはりついたSP56細胞を4〜5回軽くたたいて剥がし、RPMI-1640完全培地で5〜10倍に希釈して行った。細胞融合を行う際には、大量のSP56細胞を必要とするため、細胞融合の1週間前からスケールアップを行った。まず、SP56細胞をセルカウントし、1×105細胞/mlの濃度でT−75培養フラスコを用いて培養した。融合2日前には、1×105細胞/mlの濃度に希釈したSP56細胞をT−150培養フラスコ2個とT−75培養フラスコ1個にスケールアップして培養した。なお、融合2日前にRPMI-1640完全培地の抗生物質をジェネティシンから硫酸カナマイシン(100μg/ml)に替えた培地(RPMI-1640完全培地)を使用した。以降、融合後もすべてRPMI-1640完全培地を用いた。
【0033】
(CHO55細胞の継代)
CHO55細胞の培養は、RPMI-1640完全培地を用いて行った。
CHO55細胞は培養フラスコの底に貼り付き、自然には剥がれないため、CHO55細胞がフラスコ底面を覆うまで、T−25培養フラスコの上清を毎日入れ替えて培養を行った。CHO55細胞がフラスコ底面を完全に覆った場合は、生細胞を培養フラスコから剥がすために、トリプシン/EDTA溶液を用いた。この操作は、ほぼ1週間に1回の割合で行った。まず、PBS 10mlを加えてフラスコの底に貼り付いたCHO55細胞を2回洗浄した。2mlのトリプシン/EDTA溶液を加えて5〜10分、37℃、5%炭酸ガスインキュベーター内に放置し、完全にCHO55細胞が剥がれた後、130gで5分間遠心分離し、上清を除去した細胞沈殿を5〜10倍希釈し、RPMI-1640完全培地に懸濁し継代を行った。
【0034】
また、CHO55細胞をELISA法(後述)に基づく目的のモノクローナル抗体産生ハイブリドーマのスクリーニングに用いる場合は、トリプシン/EDTA溶液処理後5〜10倍希釈したCHO55細胞を96穴培養プレートに3〜4滴(350μl)ずつ滴下し、2〜3日37℃、5%炭酸ガスインキュベーター内で培養して用いた。
【0035】
実施例6
マウス脾細胞の調製
細胞融合日の3日前に最終免疫が終了したマウスの腹腔内に、0.1g/mlの無菌泡水クロラールを100〜200μl注射し、マウスが完全に麻酔にかかったことを確認した後、解剖用ハサミで心臓が見えるまで開胸し、すばやく心臓から採血した。心臓採血後のマウスは、70%エタノールが入った200mlのビーカー内に浸して無菌化した後、クリーンベンチ内に入れて解剖した。
【0036】
クリーンベンチ内にて解剖用ハサミでマウスより無菌的に脾臓を摘出した。摘出した脾臓は、10mlの抗生物質(100μg/ml硫酸カナマイシン)入りRPMI-1640を入れたシャーレで数回洗浄し、周りの脂肪をハサミで切り除いた後、ステンレスメッシュ上に置いてラバーポリスマンで穏やかに砕いた。ステンレスメッシュを30〜40mlの抗生物質入りRPMI-1640で洗浄し、懸濁液を2,000rpm(800g)で5分間遠心分離後、細胞沈殿を5mlの赤血球破砕緩衝液(SIGMA社、R7757)で懸濁し、氷中で5分間静置することにより赤血球を溶血させた。その後、抗生物質入りRPMI-1640で洗浄し、最終的に5mlの抗生物質入りRPMI-1640に懸濁し、生細胞数をカウントした。
【0037】
実施例7
細胞融合
(SP56細胞の調製)
スケールアップ培養したSP56細胞を回収し、130gで5分間遠心分離し、沈殿を10mlの抗生物質入りRPMI-1640に懸濁した。これを再び5分間遠心分離し、5mlの抗生物質入りRPMI-1640で懸濁し、生細胞数をカウントした。
【0038】
(リンパ球−SP56細胞複合体の作製と電気融合)
実施例6で調製した脾細胞懸濁液と上述で調製したSP56細胞を1:1の割合で混合し、4℃または37℃にて2時間ゆっくりと混和した。その後、1、000rpm(200g)で10分間遠心分離し、沈殿を1ml抗生物質入りRPMI-1640に懸濁した。400〜500rpm(〜50g)で1〜2分間遠心後、5%炭酸ガスインキュベーター内で30分間放置した。さらに30分間、5%炭酸ガスインキュベーター内でゆっくりとローテーションした。ローテーション後、再び200gで10分間遠心分離し、2mlのアイソトニックシュークロース緩衝液 [0.25M シュークロース+2mM リン酸ナトリウム緩衝液(pH7.2)+0.1mM MgCl2+0.1mM CaCl2]に懸濁した。これをプレパラート型電極上に0.5mlずつ加え、electro square porator ECM2001(BTX社製)により、2kV/cmと3kV/cmの条件で電気融合を行った。電気融合後、20mlのRPMI-1640完全培地に融合細胞懸濁液を静かに加え、30分間静置し、96穴プレートに0.2ml/wellになるように分注した。その後、速やかに37℃、5%炭酸ガスインキュベーター内で培養した。次の日に、プレートの培養上清を0.1ml/wellずつ除去し、RPMI-1640完全培地で50倍に希釈したHAT培地を0.1ml/wellずつ加えた。
細胞融合後の翌日から、2〜3日ごとに96穴プレート中の培養上清をHAT培地で0.1ml/wellずつ交換した。2週間経過した後、HAT培地をHT培地に替えて、同様に2週間行い、それ以降はRPMI-1640完全培地により培地交換を行った。
【0039】
実施例8
培養上清の評価方法
(CHO55細胞を用いたELISA法)
概念図を図2に示した。実施例5の方法で96穴培養プレートにて37℃、5%炭酸ガスインキュベーター内で2〜3日間培養することにより、CHO55細胞を培養プレートの底に吸着させた。それと同時に2〜3日間培養することにより、RPMI-1640完全培地に含まれる10% FCSによって96穴ウェルのブロッキングも行った。次に、CHO55細胞培養上清を回収し、一次抗体としてハイブリドーマ培養上清、または100倍希釈したマウス血清を100μl加え、37℃で1時間インキュベートした。アイソトニックシュークロース緩衝液で2回洗浄後、同緩衝液で10,000倍に希釈した二次抗体[HRP標識された抗マウスIgG(H+L)抗体]を50μl加え、37℃、1時間インキュベートした。最後に、アイソトニックシュークロース緩衝液で3〜5回洗浄後、発色剤[0.1Mクエン酸ナトリウム緩衝液(pH5.2)にオルトフェニレンジアミン(1mg/ml)と0.02%過酸化水素を溶解させたもの]を100μl加えて37℃、10分間インキュベートして発色させ、1M硫酸を50μl加えて反応を止めた。この上清を96穴アッセイプレートに移しマイクロプレートリーダーを用い490nmの吸光度の測定を行った。
【0040】
(競合アッセイ法に基づくELISA法)
概念図を図3に示した。競合ELISA法に基づく検出用キット[TRAb ELISA「コスミック」]を用いて抗TSHRモノクローナル抗体の評価を行った。具体的には、TSHRが予め固相化されたストリップウェルを必要な数だけ取り出し、少なくとも30分間、25℃に放置した。ウェルに開始緩衝液を50μl加え、ハイブリドーマ培養上清、陰性、陽性コントロールを100μl加え、25℃で2時間インキュベートした。各ウェルを洗浄後、ビオチン化TSHを100μl加え、25℃、40分間インキュベートし、再び各ウェルを洗浄後、ストレプトアビジンペルオキシタ-ゼ100μlを加え、25℃、20分間インキュベートした。最後に、洗浄用緩衝液で2回、精製水で1回洗浄後、基質液を100μl加えて遮光し、25℃、20分間インキュベートして発色させ、停止液を50μl加えて反応を止めた。これをマイクロプレートリーダーを用い、450nmの吸光度の測定を行った。
【0041】
(CHO55細胞を用いたDELFIA法)
概念図を図4に示した。24ウェルプレートにCHO55細胞を増殖させたあとに、ハイブリドーマの培養上清を、0.25%BSAを含むアイソトニックシュークロース緩衝液5倍に希釈したサンプルを250μl/ウェルで加え、37℃で1時間反応させた。その後、同緩衝液で3回洗浄し、同緩衝液で1、000倍に希釈した 二次抗体[ユーロピューム標識された抗マウス抗体 ]を250μl加え、37℃、1時間インキュベートした。BSAを含まない、アイソトニックシュークロース緩衝液で3回洗浄後、発色剤(エンハンスメント溶液:パーキンエルマー社)を200μl/ウェルで添加し、ユーロピュームの遅延蛍光を測定できるプレートリーダーで測定した。
【0042】
実施例9
細胞融合の結果
表1および表2に示す結果が得られた。最終免疫にタンパク質を使用した場合、平均して約20%の効率でハイブリドーマを得ることができた。ELISA陽性率は全ハイブリドーマ中、約24%であった。これに対して、最終免疫にCHO細胞を使用した場合、平均して約42%の高い効率でハイブリドーマを得ることができたが、全ハイブリドーマ中、ELISA陽性率は約13%の低い値となった。しかしながら融合効率およびELISA陽性率の両面から判断して、本実験において、目的の抗体産生ハイブリドーマ作製における免疫方法による大きな差異はないと考えられた。いずれにせよ、リンパ球とミエローマ細胞との非特異的融合による従来技術の融合方法では目的とするハイブリドーマは獲得できなかったため、この結果は驚くべきものである。また、新技術によるハイブリドーマ出現効率を比較すると、2kV/cmと3kV/cmにおいて顕著な差は認められなかったが、ELISA陽性率に関しては、2kV/cmの方が高い値を示した。さらに、TSHR発現ミエローマ細胞を用いた高次構造特異抗体発現リンパ球の選択温度は、4℃の方が、37℃より適していると推測された。
【0043】
表1 最終免疫を実施例3で作製したTSHRで行った場合の結果
【表1】
【0044】
表2 最終免疫をCHO55で行った場合の結果
【表2】
【0045】
実施例10
モノクローナル抗体産生ハイブリドーマの検証
本発明によって得られたハイブリドーマによって産生されるモノクローナル抗体の親和性を検証するため、CHO55細胞を抗原とするELISA法(図2)および競合アッセイ法に基づくELISA法(図3)の2種の方法を用い検討した。まず初めに、CHO55細胞を抗原とするELISA法(図2)による解析の結果、ハイブリドーマB5(8/12日細胞融合)によって産生されるモノクローナル抗体が高い抗体価を示し、またその再現性も認められた(図5)。さらに、競合アッセイ法に基づくELISA法(図3)に基づき解析を行った結果、B5(8/12日細胞融合)においては、陽性コントロールに近い値を示し、高い抗体価を有することが確認できた(図6)。すべての融合プレートにおいて、CHO55細胞を抗原とするELISA法 による分析の結果、ハイブリドーマB5(8/12日細胞融合)およびハイブリドーマC1、F11(10/23日細胞融合)によって産生されるモノクローナル抗体が、CHO55細胞に対して再現性よく高い親和性を示すことが明らかとなった(図7)。また、遅延蛍光による評価でも、優位な結合が確認さらた(図8)。
【0046】
実施例11
共焦点レーザー蛍光顕微鏡による可視化解析
CHO55細胞、SP56細胞、ELISA陽性ハイブリドーマ(B5)をそれぞれT−75培養フラスコで培養した後、130gで5分間、遠心分離し、沈殿をRPMI-1640 0.5mlに懸濁した。生体内免疫によって得られたマウスの血清(ポリクローナル抗体)の原液を10μl加え、4℃で2時間ローテーションした後、ハンクス平衡塩10mlで洗浄し、沈殿をハンクス平衡塩1mlに懸濁した。Alexa Fluor 488にて蛍光標識された抗マウスIgG(H+L)抗体を6μl加え、遮光条件下にて37℃で30分間、ゆっくりローテーションを行った。ハンクス平衡塩10mlで洗浄後、細胞をハンクス平衡塩0.5mlに懸濁して、共焦点レーザー顕微鏡で観察した。その結果、すべての細胞において特異的蛍光が観察されたことから、それら細胞の膜表面にTSHRが高次構造を保持し発現されていることが確認された(図9)。
【0047】
これらの結果に基づき、本発明により目的の細胞膜上に存在するTSHR認識モノクローナル抗体産生ハイブリドーマの作出に成功したと結論づける。
【産業上の利用可能性】
【0048】
本方法によれば、従来では単離することが困難であった、細胞膜上に存在する膜タンパク質を認識する抗体を、非常に効率良く認識することができる。膜タンパク質のなかでも、特に単離することが難しい多回膜貫通型のタンパク質を認識する抗体を単離できるようになる。
【図面の簡単な説明】
【0049】
【図1】融合タンパク質精製した際のSDSポリアクリルアミドの図。
【図2】CHO55細胞を抗原とするELISA法の原理の概説図。
【図3】競合アッセイに基づくELISA法の原理の概説図。
【図4】CHO55細胞を抗原とするDELFIAの原理の概説図。
【図5】CHO55細胞を抗原とするELISAスクリーニングの結果。
【図6】競合アッセイに基づくELISAスクリーニングの結果。
【図7】CHO55細胞を抗原とするELISAスクリーニングの再現性。
【図8】CHO55細胞を抗原とするDELFIAの結果。コントロールは、まったく違う抗原(エストラジオール)を認識する抗体を示す。
【図9】共焦点レーザー蛍光顕微鏡による可視化解析の結果。
【特許請求の範囲】
【請求項1】
目的のタンパク質を抗原として用いる免疫により獲得されたリンパ球集団と、細胞表面に当該タンパク質を提示するミエローマ細胞とを混合し;
当該リンパ球集団内の当該タンパク質に特異的な膜結合型抗体を提示するリンパ球を当該ミエローマ細胞に対し、当該膜結合型抗体と当該ミエローマ細胞表面に存在する当該タンパク質との特異的な結合を介し、選択的に近寄らせ;そして
両細胞を融合させる、
ステップを含むことを特徴とする、当該タンパク質に特異的なモノクローナル抗体を産生するハイブリドーマの作製方法。
【請求項2】
前記ミエローマ細胞が、前記タンパク質をコードするDNAを含有するDNAベクターにより形質転換されたものである、請求項1記載の方法。
【請求項3】
前記細胞の融合が、両細胞に電気パルスを負荷することにより行われる、請求項1又は2記載の方法。
【請求項4】
前記タンパク質が膜タンパク質である、請求項1〜3のいずれか1項記載の方法。
【請求項5】
前記膜タンパク質が、ニコチン、GABAもしくはグリシンのイオンチャンネル内蔵型受容体、アルファー1ヒスタミン1(H1)、ブラジキニン(B2)、ムスカリン1(M1)もしくはアンジオテンシンII(AT1)の受容体でイノシトールリン酸を分解する受容体、ベータヒスタミン2(H2)、プロスタサイクリン(IP)もしくはグルカゴンの受容体、TSH、FSHもしくはLHの受容体でcAMPを合成しGTP結合蛋白質共役型の受容体、またはインシュリン、EGF、FGFもしくはPDGFのチロシンキナーゼ活性を有するチロシンキナーゼ型受容体である、請求項4記載の方法。
【請求項6】
前記膜タンパク質がTSHRである、請求項5記載の方法。
【請求項7】
請求項1〜6のいずれか1項記載の方法により作製したハイブリドーマを、前記モノクローナル抗体を産生する条件下で培養し、産生されたモノクローナル抗体を回収することを含む、前記目的のタンパク質に特異的なモノクローナル抗体の作製方法。
【請求項1】
目的のタンパク質を抗原として用いる免疫により獲得されたリンパ球集団と、細胞表面に当該タンパク質を提示するミエローマ細胞とを混合し;
当該リンパ球集団内の当該タンパク質に特異的な膜結合型抗体を提示するリンパ球を当該ミエローマ細胞に対し、当該膜結合型抗体と当該ミエローマ細胞表面に存在する当該タンパク質との特異的な結合を介し、選択的に近寄らせ;そして
両細胞を融合させる、
ステップを含むことを特徴とする、当該タンパク質に特異的なモノクローナル抗体を産生するハイブリドーマの作製方法。
【請求項2】
前記ミエローマ細胞が、前記タンパク質をコードするDNAを含有するDNAベクターにより形質転換されたものである、請求項1記載の方法。
【請求項3】
前記細胞の融合が、両細胞に電気パルスを負荷することにより行われる、請求項1又は2記載の方法。
【請求項4】
前記タンパク質が膜タンパク質である、請求項1〜3のいずれか1項記載の方法。
【請求項5】
前記膜タンパク質が、ニコチン、GABAもしくはグリシンのイオンチャンネル内蔵型受容体、アルファー1ヒスタミン1(H1)、ブラジキニン(B2)、ムスカリン1(M1)もしくはアンジオテンシンII(AT1)の受容体でイノシトールリン酸を分解する受容体、ベータヒスタミン2(H2)、プロスタサイクリン(IP)もしくはグルカゴンの受容体、TSH、FSHもしくはLHの受容体でcAMPを合成しGTP結合蛋白質共役型の受容体、またはインシュリン、EGF、FGFもしくはPDGFのチロシンキナーゼ活性を有するチロシンキナーゼ型受容体である、請求項4記載の方法。
【請求項6】
前記膜タンパク質がTSHRである、請求項5記載の方法。
【請求項7】
請求項1〜6のいずれか1項記載の方法により作製したハイブリドーマを、前記モノクローナル抗体を産生する条件下で培養し、産生されたモノクローナル抗体を回収することを含む、前記目的のタンパク質に特異的なモノクローナル抗体の作製方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2006−6250(P2006−6250A)
【公開日】平成18年1月12日(2006.1.12)
【国際特許分類】
【出願番号】特願2004−190340(P2004−190340)
【出願日】平成16年6月28日(2004.6.28)
【出願人】(802000042)株式会社三重ティーエルオー (20)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
【公開日】平成18年1月12日(2006.1.12)
【国際特許分類】
【出願日】平成16年6月28日(2004.6.28)
【出願人】(802000042)株式会社三重ティーエルオー (20)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
[ Back to top ]