
細胞膜改変
本発明は、細胞の表面を改変するための方法及び組成物であって、標的化部分及び/又は粒子が細胞膜に結合されている方法に関する。該粒子は、薬物送達用に治療薬をさらに含む。本明細書に記載の組成物は、組織再生、薬物送達、又は両方の組合せのために細胞を標的化することにより、疾患組織又は損傷組織を治療するのに有用である。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2008年4月29日に出願された米国特許仮出願第61/048,773号の35 U.S.C.§119(e)に基づく利益を主張するものであり、その内容は、その全体が参照により本明細書に組み込まれる。
【0002】
本発明は、再生促進用の標的細胞送達の分野に関する。
【背景技術】
【0003】
細胞は、組織特異的薬物送達を含む様々な生物医学的応用のための重要な治療用ツールである。例えば、種々の抗癌剤(ドキソルビシン、プラチンなど)を負荷した細菌細胞が、癌細胞に特異的に標的化されている1。送達媒体としての細胞の別の例は、分子工学技術によりヒト欠乏症および疾患を治療するための、遺伝学的に形質導入されたヒト間葉系幹細胞(MSC)の使用、並びに適切な化学療法剤の送達である。インターフェロン−β(IFN−β)を保持するアデノウイルス発現ベクターにより形質導入されたMSCは、静脈内又は皮下のいずれかでIFN−βを悪性癌細胞に送達して腫瘍増殖を阻害するために使用されてきた2。同様に、CRAd(条件付複製アデノウイルス)は、マウス異種移植モデルの使用により、MSCを介して悪性腫瘍細胞へと標的化された3。これらの結果は、CRAd形質導入幹細胞が、CRAd単独より効果的に腫瘍部位にホーミングされたことを示す。MSCは、硬骨4および他の幾つかの組織5へのホーミング能力を有することも知られている。
【0004】
マクロファージは、腫瘍部位に集中する能力、腫瘍細胞を死滅させる能力、及び腫瘍増殖を阻害する能力のため、例えば癌治療において魅力的な細胞に基づく担体の別の例であり、例えば、CY2BP6を発現するように形質導入されたマクロファージは、超毒性環境で腫瘍球状体に浸潤した際に腫瘍細胞を死滅させることができた6。腫瘍特異的T細胞(例えば、腫瘍浸潤性リンパ球)も、治療目的で腫瘍へのホーミングに使用されており、例えば、MOv−γ遺伝子で形質導入されたマウスT細胞が、ヒト卵巣癌細胞を有するマウスに経皮内投与された7。しかし、これらの細胞応用が成功したにもかかわらず、多くの研究により、適切な標的化が多くの場合に達成されていないことも示されている8。標的とされていない非特異的部位において治療剤が制御されずにT細胞から放出されることは、患者の健康に対する相当程度の毒性及びリスクの原因となる。同様に、腫瘍部位におけるあるマクロファージの腫瘍形成的役割が報告されている9。
【0005】
細胞は、細胞工学的応用における再生プロセスに不可欠な部分である。遺伝子組換え細胞及び/又は適切な遺伝子の使用は、組織再生にますます重要な役割を果たす。遺伝子組換えポリマーマトリクス(ポリマー性放出及び基質媒介性放出の両方による)は、組織再生プロセスを支援するための特定の遺伝学的情報を放出する10。増殖因子で補完されたスキャフォールドでの組織の操作に細胞を使用することは、組織の再生を増強する11、12。
【0006】
しかしながら、これらの方法に伴う問題には以下のものが含まれる:制御されない放出、不適当なレベルの遺伝子発現、及び異常組織増殖。指定組織再生用の適切な遺伝子の送達は、非ウイルスベクターまたはウイルスベクターを使用して所望の遺伝子を細胞に移動させ、続いて組織再生用の細胞を送達することによっても達成される13。MSCの全身性投与による自己細胞療法は、組織再生の強力な治療用ツールである14〜16。しかしながら、ex vivoで培養されたMSCは、遺伝子発現の欠如により、脾臓及び骨髄へのホーミング能力を欠如する14。
【0007】
細胞とその環境との間の特定の相互作用を明確にすることは、治療をうまく機能させるために積極的に研究されている領域である。細胞膜の構造及び機能性は、かなりのクロストークを示す種々の相互作用により、細胞が細胞外マトリックス(ECM)と相互作用することを可能にし、細胞接着、増殖、遊走、分化、マトリックス産生、プロテアーゼ分泌、又はアポトーシスの原因となる可能性がある。その結果として、細胞膜は、細胞とその周囲との相互作用の媒介に重要である。細胞膜(例えば、原形質膜)は、多種多様な生体分子、主として無数の細胞性プロセスに関与するタンパク質及び脂質を含有し、細胞内の細胞骨格の結合点としても機能する半透過性脂質二分子層である。細胞膜は、ECMとの相互作用に加えて、イオン、小分子、及びウイルスなどのより大型物質の出入口として機能する。マウス白血病ウイルス(MLV)及びヒト免疫不全ウイルス(HIV)粒子に関する研究は、ウイルス粒子が最初に細胞表面と相互作用し結合することを示している17。
【0008】
細胞膜は、細胞への出入口を提供することに加えて、周囲との相互作用を能動的に媒介する。例えば、細胞外マトリックスを遊走可能な細胞は、典型的には細胞膜を介してMMPなどのプロテアーゼを分泌し、ある細胞タイプは、細胞膜内にMMPを含有する(膜型マトリックスメタロプロテアーゼ1(MT1−MMP))18。プロテアーゼの分泌が増強された癌細胞は、細胞外マトリックスに侵入する高度な能力も示す19。プロテアーゼ20又はタンパク質分解に関与する細胞表面受容体21の産生を増強するように遺伝子組換えにより修飾された細胞は、侵入能力を増強させたことを明らかに示している。これは、細胞表面を適切に修飾することにより、細胞とその環境との相互作用を調節することが可能であることを示す。
【0009】
細胞表面は、外部環境との相互作用を媒介し、複雑な相互作用を包含する基質であるため、細胞膜媒介性相互作用のより高度な制御を示して生体特異的効果をもたらすことが望まれている。
【発明の概要】
【発明が解決しようとする課題】
【0010】
一般的に、本明細書に記載の発明は、種々の技術により細胞表面を機能化するための方法、及び様々な応用に使用するための方法を提供する。ナノ粒子による標的薬物送達とは異なり、及び生体材料に基づくビーズ上の又はその内部の未修飾細胞の送達とは異なり、細胞表面を様々な機能性で修飾することは、様々な応用用に細胞性事象を操作するための制御の強化を容易にする新しい手法である。機能化された細胞表面は、実質的にあらゆる生物系を制御するために効果的に使用することができる。より具体的には、本明細書に記載の組成物及び方法は、再生用に細胞を組織に標的化するのに有用であるか、又はその代わりに作用剤を特定組織へと細胞に基づいて送達するのに有用である。本明細書に記載の方法に有用な細胞は、粒子並びにリガンドの付加により修飾することができる。該粒子は、タンパク質、小分子、RNA干渉分子、薬物、ビタミン、治療薬、診断薬、栄養補助剤、化粧用の特性を有する作用剤、又は核酸などの作用剤をさらに含んでいてもよい。加えて、本明細書に記載の組成物は、例えば、特に、脳卒中、臓器再生、癌、骨折、及び虚血性心疾患を含む広範囲の疾患又は創傷を治療するために、当業者によって個別化することができる。
【課題を解決するための手段】
【0011】
1つの態様では、本発明は、細胞、該細胞の表面に結合する膜結合リガンド、及び該細胞の表面に結合する粒子を含む、単離された改変細胞組成物に関する。
【0012】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、該細胞は、幹細胞又は前駆細胞である。
【0013】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該該幹細胞は、再プログラムされた細胞である。
【0014】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、分化細胞である。
【0015】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、治療薬を発現するように遺伝子操作されている。
【0016】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、1000〜8000nmである。或いは、該粒子は、500〜1000nm又は1〜500nmである。
【0017】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、リンカー分子を介して結合している。
【0018】
同様に、この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、リンカー分子を介して結合している。或いは、別の実施形態では、該粒子は、リンカー分子を介さずに結合している。
【0019】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、共有結合で結合している。或いは、別の実施形態では、該膜結合リガンドは、非共有結合で結合している。
【0020】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、共有結合で結合している。或いは、別の実施形態では、該粒子は、非共有結合で結合している。
【0021】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、組織における該細胞の蓄積をもたらす。
【0022】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、薬物、栄養補助剤、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される。
【0023】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、2つ以上の膜結合リガンドが、該細胞に結合している。
【0024】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞の機能を増強する作用剤を含む。或いは、該粒子は、組織の機能を増強する作用剤を含む。
【0025】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、脂質、ナノ粒子、マイクロ粒子、又はデンドリマーからなる群から選択される。
【0026】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は、細胞成長、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着からなる群から選択される機能を増強する。
【0027】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は、小分子、成長因子(growth factor)、サイトカイン、RNA干渉分子、増殖因子(proliferation factor)、ビタミン、栄養補助剤、化粧用の特性を有する作用剤、治療薬、診断薬、ケモカイン、標的化作用剤、又は分化因子からなる群から選択される。
【0028】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、作用剤を含む粒子用の組織特異的担体として使用される。
【0029】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な基質若しくは注射可能な基質内に捕捉若しくは基質に結合されている。
【0030】
1つの態様では、本発明は、細胞、及び該細胞の表面に組み込まれた自己集合分子に結合した膜結合リガンドを含む、単離された改変細胞組成物に関する。
【0031】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該自己集合分子は、両親媒性である。
【0032】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、該自己集合両親媒性分子に共有結合で結合している。或いは、膜結合リガンドは、該自己集合両親媒性分子に非共有結合で結合している。
【0033】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、少なくとも1つの粒子が、該細胞に結合している。或いは、1〜10個以上の、例えば2、3、4、5、6、7、8、9、又は10個以上の粒子が、該細胞に結合されている。
【0034】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、標的化部分であるリガンドを含む。
【0035】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な基質若しくは注射可能な基質内に捕捉若しくは基質に結合されている。
【0036】
単離された改変細胞組成物を形成するための方法であって、(a)膜結合リガンドを自己集合分子に結合させて、修飾分子を形成するステップと、(b)該修飾分子を用いて小胞を形成するステップと、(c)該小胞を細胞と融合させるステップとを含む方法も、本明細書に記載されている。
【0037】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該単離された改変細胞組成物は、in vivoで形成される。
【0038】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、細胞凝集塊を形成するために使用される。
【0039】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、細胞凝集塊を形成するために使用されるリガンドを含む。
【0040】
単離された改変細胞組成物を形成するための方法であって、(a)膜結合リガンドを自己集合分子に結合させて、修飾分子を形成するステップと、(b)該修飾分子を用いてミセルを形成するステップと、(c)該ミセルを該細胞と融合させるステップとを含む方法も、本明細書に記載されている。
【0041】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、細胞膜へ組み込み前に該自己集合分子との結合を介して該細胞に結合される。或いは、該粒子は、細胞膜への組み込み後に該自己集合分子との結合を介して該細胞に結合される。
【0042】
本発明の別の態様は、細胞、及び該細胞の表面に結合された膜結合リガンドを含む単離された改変細胞組成物に関し、前記膜結合リガンドは、該細胞の表面の第1の部分に結合されており、該細胞の表面の第2の部分には該リガンドがない。
【0043】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、粒子をさらに含む。
【0044】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、細胞凝集塊を形成するために使用される。
【0045】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞凝集塊は、in vivoで形成される。
【0046】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な基質若しくは注射可能な基質内に捕捉若しくは基質に結合されている。
【0047】
本明細書に記載の別の態様は、標的組織再生を必要とする個体を治療する方法であって、(a)標的細胞を、該細胞の表面に膜結合リガンドを結合させることにより形成し、前記膜結合リガンドが、治療される組織の中に該標的細胞の蓄積をもたらすステップと、(b)該標的細胞の表面に粒子を結合させることにより二重機能性細胞を形成し、該粒子が、該二重機能性細胞の機能を増強する作用剤を含むステップと、(c)標的組織再生を必要とする個体に該二重機能性細胞を投与するステッとを含む方法である。
【0048】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、標的組織再生を必要とする個体から単離される。或いは、該細胞は、標的組織再生を必要とする個体以外の個体から単離される。
【0049】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、該膜結合リガンドを結合させる前に、ex vivo培養環境で増殖される。
【0050】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、標的組織再生の部位で放出される。
【0051】
本明細書に記載の別の態様は、対象体にワクチン接種をするための方法であって、(a)膜結合リガンドを樹状細胞の表面に結合させることにより、標的化樹状細胞を形成し、該膜結合リガンドが、リンパ組織での該標的化樹状細胞の蓄積をもたらすステップと、(b)該標的化樹状細胞を抗原と接触させることにより活性化標的化樹状細胞を形成するステップと、(c)ワクチンを必要とする対象体に該活性化標的化樹状細胞を投与するステップとを含む方法である。
【0052】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該抗原は、ウイルス抗原を含む。或いは、該抗原は、細菌抗原又は癌関連抗原を含む。
【0053】
細胞を修飾するためのキットであって、(a)結合部分を有する自己集合分子と、(b)細胞を修飾するための方法を含む説明書と、(c)それらの包装材料とを含むキットも、本明細書に記載されている。該キットに含まれる説明書には、単離された改変細胞組成物を形成するための方法であって、(a)結合部分を有する自己集合分子を用いて小胞又はミセルを形成するステップと、(b)該小胞又はミセルを目的細胞と融合させるステップとを含む方法が記述されている。
【0054】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該結合部分は、膜結合リガンドである。或いは、該結合部分は粒子である。1つの実施形態では、該キットは、第2の部分に加えて粒子も含むことができる。
【0055】
(a)細胞と、(b)該細胞の表面に結合した粒子と、(c)該細胞に結合しない粒子結合リガンドとを含む単離された改変細胞組成物も、本明細書に記載されている。そのような実施形態では、該粒子上に存在するリガンドは該細胞と直接的に相互作用せず、むしろ該粒子がリガンドを、内部的に(例えば、該粒子内に)含むか、外部的に(例えば、該粒子表面上に)含むか、又は該粒子に組み込まれているか(例えば、該細胞表面の構成要素になる修飾両親媒性分子)のいずれかであることが好ましい。該リガンドは、該粒子を該細胞に結合するリンカー分子として機能しないことが好ましい。
【0056】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、粒子は作用剤を含み、幾つかの場合には、該作用剤は該粒子の構成要素である。これらの実施形態では、該粒子のおよそ1%から実質的に全てが、目的の作用剤で構成されていてもよい。
【0057】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、粒径が1nm〜5000nmである。
【0058】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞に内部移行されない。
【0059】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、血漿又は血清との接触による不活性化に感受性である作用剤をさらに含む。この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該不活性化に感受性である作用剤は、RNA干渉分子である。
【0060】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は、該細胞の輸送を増強する。
【0061】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は治療薬を含む。或いは、該作用剤は診断薬を含む。
【0062】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は、宿主細胞及び/又は組織の機能を増強する。
【0063】
本明細書に記載の別つの態様は、(a)細胞と、(b)該細胞の表面に組み込まれた脂質分子に結合した膜結合リガンドとを含む、単離された改変細胞組成物である。
【0064】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該脂質分子は、単一の尾部を含む。或いは、該脂質分子は、複数の尾部を含むこともできる。
【0065】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該脂質分子は、複数の電荷を帯びている。
【0066】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該脂質分子は、リピドイド(lipidoid)などの改変脂質である。
【0067】
本明細書に記載の別の態様は、該リガンドの部分(例えば、該リガンドの少なくとも3分の1)が、修飾後の少なくとも2日間は該細胞表面上で安定している細胞組成物を調製するための方法であって、(a)ビオチン及びリガンドの供給源を含む脂質小胞を調製するステップと、(b)細胞(例えば、ヒト間葉系幹細胞)を前記小胞と接触させるステップとを含み、該リガンドが前記接触ステップ後の少なくとも2日間は該細胞上に存在するように、細胞組成物が形成される方法に関する。
【0068】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該リガンドは、ビオチンを含む。
【0069】
幹細胞の特徴を損なわずに前駆細胞又は幹細胞をリガンドで修飾するための方法であって、該細胞表面を共有結合で修飾するステップを含み、該細胞組成物が、前駆細胞又は幹細胞の特徴を喪失せずに形成される方法も本明細書に記載されている。
【0070】
幹細胞の特徴を損なわずに前駆体幹細胞をリガンドで修飾するための方法であって、(a)細胞をビオチンの供給源と接触させるステップと、(b)ステップ(a)の前記細胞を、ストレプトアビジン及びリガンドと接触させるステップとを含み、幹細胞組成物が、幹細胞の特徴を損なわずに形成される方法も本明細書に記載されている。
【0071】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、該幹細胞の特徴には、多系統分化、生存能、増殖、パラクリン因子の分泌、in vivo経内皮遊走、及び/又は接着が含まれる。
【0072】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、該リガンドは、シアリルルイスXを含む。
【0073】
接着リガンドを細胞の表面に結合するための方法であって、(a)細胞をビオチンの供給源と接触させるステップと、(b)ステップ(a)の前記細胞を、ストレプトアビジン及び強力な接着を促進する接着リガンドと接触させるステップとを含み、細胞組成物が、該細胞表面上に接着リガンドを含んで形成され、前記接着リガンドが強力な接着を可能にする方法も本明細書に記載されている。
【0074】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、複数の接着リガンドが、細胞の表面に結合されている。
【0075】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該接着リガンドは、PSGL−1又はP−セレクチン抗体を含む。
【0076】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該接着リガンドは、1.9ダイン/cm2まで、2μm/秒未満の速度で回転することを可能にする。
【0077】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該接着リガンドは、組織内局在化を増強する。
【0078】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該組織は骨髄である。
【0079】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該リガンドは、特定の物理的機能性(負電荷、正電荷、脂質、抗体など)又は化学的機能性(NHS基、ストレプトアビジンなど)を含む。
【0080】
細胞の表面に粒子を結合するための方法であって、(a)リガンドを粒子に結合させて機能化粒子を調製するステップと、(b)前記機能化粒子を細胞と接触させるステップとを含み、前記機能化粒子が前記細胞の表面に結合されている方法も、本明細書に記載されている。
【0081】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該機能化粒子は、PLGAを含む。
【0082】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該機能化粒子は、エマルション法を使用して調製される。
【0083】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該リガンドは、前記細胞の組織内局在化を可能にする。
【0084】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該機能化粒子は、内部移行される。
【0085】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該機能化粒子は、内部移行されない。
【0086】
該脂質(該細胞表面に結合されている)に結合されたリガンドの少なくとも3分の1が、修飾の2日後に安定している方法であって、(a)1,2−ジオレオイル−sn−グリセロ−3−ホスホエタノールアミン−N−(ビオチニル)ナトリウム塩から小胞を調製するステップと、(b)引き続き、該小胞をhMSCと共に30分間インキュベーションするステップとを含み、hMSC上で安定化されたリガンドがビオチンである方法。
【0087】
強力な接着を促進する接着リガンドを結合させるための方法であって、(a)ビオチニル−N−ヒドロキシ−スクシンイミドのN−ヒドロキシ−スクシンイミド基で細胞を修飾(ビオチン化)するステップと、(b)ストレプトアビジン及び接着リガンドで処理するステップとを含む方法も、本明細書に記載されている。
【0088】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、該リガンドは、P−セレクチン抗体を含む。
【0089】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該回転は、1.9ダイン/cm2まで、2μm/秒未満である。
【0090】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該回転は、組織内局在化を増強する。
【0091】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該組織は骨髄である。
【0092】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞の表面に結合したままである。
【0093】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞表面上で安定化された後、該細胞内に内部移行される。
【0094】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該表面結合粒子は、全身性注射又は局所投与された後、該細胞により特定部位へと運搬される。
【0095】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞の表面に結合したままであるか、又は該細胞表面から分離しているか、又は該標的部位で内部移行される。
【0096】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該内部移行粒子は、全身性注射又は局所投与された後、該細胞により特定部位へと運搬される。
【図面の簡単な説明】
【0097】
【図1】細胞膜修飾を示す概略図である。1)特異的細胞表面官能基を導入するために細胞膜が機能化される。2)官能化された基に治療薬を含有する粒子をリンカーを介して結合し、該粒子は第2の官能基を含有する。3)該細胞表面上の官能化された基に粒子をリンカーを介して結合させる。4)細胞表面上の官能化された基に粒子を結合させる。5)細胞表面上の官能化された基を介して粒子が細胞に結合され、粒子及び細胞は官能リガンドを含有する。6)細胞膜を多官能性作用剤で機能化する。7)脂質分子又は脂質に基づく小胞の結合により細胞膜を機能化する。8)物理的な(非共有結合)相互作用又は化学的相互作用(共有結合)のいずれかにより、機能化粒子を細胞に結合する(細胞膜を機能化せずに)。9)機能化粒子をリンカーを介して細胞に結合する(細胞膜に遊離官能基を結合せずに)。10)機能化粒子をリンカーを介して細胞に結合し(細胞膜を遊離官能基で機能化せずに)、粒子はリガンドを含有する。
【図2】蛍光顕微鏡画像で観察した、4、8、及び12時間の時点での、粒子が結合されているヒト間葉系幹細胞(hMSC)のパーセントを示す棒グラフである。
【図3】様々な表面特徴および様々な粒径を有する粒子の内部移行を経時的に示す棒グラフである。図3Aは、y−z、x−z、及びx−y平面のZスタック共焦点画像で観察された、4、8、及び12時間の時点での、hMSCによって内部移行された様々な表面特徴を有する粒子のパーセントを示す。共焦点顕微鏡の場合、hMSCをヨウ化プロピジウムで染色し、粒子はDiD封入されている。図3Bは、4、8、及び12時間の時点での、hMSCに結合されたCD90抗体コーティングPLGA粒子の内部移行に対する粒径の効果を示しており、より大型粒子と比較して、3μm未満の粒子がより高い効率で内部移行されることが示されている。
【図4】hMSCに結合された負荷電PLGA粒子の詳細な分析を示す一連のグラフである。図4Aは、0時間及び36時間(左)の時点で、粒子が結合されている細胞のパーセントを示す。図4Bは、0時間及び36時間の時点で、1個の粒子、2個の粒子、及び3個以上の粒子を有する細胞のパーセントを示す。このパーセントは、粒子を有する細胞の総数に基づく。図4Cは、負荷電PLGA粒子のhMSCへの結合に対するトリプシン処理の影響を示す。トリプシン処理直後並びにその後の2日目及び4日目における、粒子が結合されている細胞のパーセント並びに観察視野にある細胞の総個数;図4Dは、0日目(トリプシン処理日)、2日目、及び4日目の、1個の粒子、2個の粒子、及び3個以上の粒子を有する細胞のパーセントを示す。このパーセントは、結合した粒子を含有する細胞の総数に基づく。
【図5】例示的な修飾細胞の細胞及び粒子特徴を示す一連のグラフである。図5Aは、ビオチン及びストレプトアビジン相互作用によりhMSCに結合された粒子の、結合直後の数を示しており、ビオチン化hMSCが、ストレプトアビジンをコーティングした粒子に特異的に結合することを示す。図5Bは、0時間及び48時間後の、PLGA結合hMSC(ビオチン−ストレプトアビジンによる)の生存能を示す。図5Cは、10、30、及び90分の時点での、組織培養表面に接着したPLGA結合hMSC(ビオチン−ストレプトアビジンによる)のパーセントを示す。図5Dは、8日間にわたるPLGA結合hMSC(ビオチンーストレプトアビジンによる)の増殖を示す。
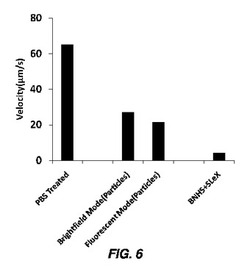
【図6】Strep−PLGA粒子で修飾されたビオチン化細胞の回転を示す棒グラフであり、細胞及び粒子は、P−セレクチン上のSLeXで機能化されている。平行平板フローチャンバーアッセイ(parallel plate flow chamber assay)において0.36ダイン/cm2で、ビオチン−ストレプトアビジンによりPLGA粒子と結合されたhMSC、及びP−セレクチンでコーティングされた基質上のビオチン化シアリルルイスX(SLeX)の回転応答が示されている。対照群は、未修飾hMSC、つまりPBS処理hMSCを含む(回転アッセイは、明視野モード及び蛍光モードの両方で実施した)。
【図7】細胞表面へのPLGA粒子の結合(PEGリンカーによる、及び細胞を機能化せず)を示す棒グラフである。hMSCに結合されたポリエチレングリコール(PEG)リンカーを有する及び有していない、N−ヒドロキシスクシンイミド(NHS)基を有するPLGA粒子の平均数(Cl、C2、及びC3は、NHS活性化PLGA粒子を機能化するのに使用されたPEGリンカーの濃度を表す)が示されており、より高い濃度のPEGリンカーで粒子を機能化する時、より多数の粒子が細胞に結合されることを示している。
【図8】骨髄における修飾及び未修飾間葉系幹細胞(MSC)の局在化を示す棒グラフである。図8Aは、3つの別々の実験において細胞を尾部に静脈内注射した2時間後に骨髄に局在化した、未修飾及び修飾MSC(MSCは、ビオチン−N−ヒドロキシスクシイミドで修飾され、その後ストレプトアビジン及びビオチン化シアリルルイスX、SLeXで修飾されている)の数を示す。未修飾MSCと比較して、結合したSLeXを有するMSCは、骨髄に局在化する数が増加する。図8Bは、細胞を尾部に静脈内注射した24時間後に、骨髄内皮から溢出した未修飾及び未修飾のMSCの数を示す。
【図9】37℃、24時間の時点での、PBS処理細胞及びSLeX修飾MSCによるパラクリン因子SDF−1、IGF−1、及びPGE2の細胞培養上清への分泌を示す一連の棒グラフである。
【図10】フローチャンバーアッセイのフロー条件下で、P−セレクチン表面上の修飾及び未修飾MSCの接着性相互作用を示す一連のグラフである。図1OAは、P−セレクチン表面上の未修飾MSCが、フロー条件下では表面との接着性相互作用を示さず、細胞速度は0.36ダイン/cm2のせん断応力で70μm/秒であることを示し、あらゆる修飾が無ければ、MSCがP−セレクチンと特異的に相互作用しないことを示している。図1OBは、Ab−Pセレクチン(P−セレクチンの抗体)で修飾したMSCの80%超が、強力な接着又は10ダイン/cm2まで3μm/秒未満の速度で回転することのいずれかにより、10ダイン/cm2のせん断応力まで基質と相互作用することを示す。これは、接着リガンド(この場合、P−セレクチンの抗体)の結合が、せん断条件下で細胞とP−セレクチンとの接着性相互作用を誘導することを示す。
【図11】ビオチン−HABA−アビジンアッセイにより測定された、接着モード及び懸濁モードでビオチン−N−ヒドロキシサクシニミドによりビオチン化されたhMSCSの表面上のビオチンリガンドの定量化を示す棒グラフである。
【図12】接着モードで色素(DiD)封入自己集合ファイバーを用いて修飾した細胞から得た蛍光強度(FI)を示す棒グラフである。ファイバーは、両親媒性物質の自己集合により生成され、自己集合プロセス中に蛍光色素(DiD)が封入された。これは接着モードで実施され、細胞表面の半分が修飾された。
【図13】懸濁モードで色素(DiD)封入自己集合ファイバーを用いて修飾した細胞から得た蛍光強度(FI)を示す棒グラフである。ファイバーは、両親媒性物質の自己集合により生成され、自己集合プロセス中に蛍光色素(DiD)が封入された。これは「懸濁モード」で実施され、全細胞表面が修飾された。
【図14】修飾細胞の蛍光強度を示す一連の棒グラフである。図14Aは、接着モードでの小胞によるhMSCの修飾を示す。ビオチン化脂質小胞をhMSCに付加し、その後ローダミン結合ストレプトアビジンを付加した。hMSCの修飾を、ローダミン結合ストレプトアビジンの蛍光シグナルの関数として、結合直後及び7日目に測定した。小胞で修飾されたhMSCの蛍光は、これらの方法により、ビオチン化脂質が細胞上に組み込まれ、7日目まで細胞と結合していることを示す。図14Bは、7日間にわたってMSCに結合されたビオチン脂質小胞に付加されたローダミン結合ストレプトアビジンの蛍光シグナルによって測定した、hMSC表面上のビオチン(ビオチン化脂質小胞によりhMSCに結合された)の安定性及び接近可能性を示し、MSCは、0日目にビオチン化され、ローダミン−ストレプトアビジンの蛍光シグナルは、0、2、4、及び7日目にローダミン−ストレプトアビジンを付加することにより測定した。蛍光強度は、細胞表面上のビオチンの少なくとも3分の1が、修飾のために2日目まで接近可能であり、ビオチンの約4分の1は、さらなる修飾のために依然として表面上で接近可能であることを示している。
【図15】フローチャンバーアッセイにおける、P−セレクチンでコーティングした基質上での小胞修飾hMSCの回転相互作用を示す図である。図15Aは、0.5ダイン/cm2のせん断応力における、未修飾表面上での、および小胞修飾法によるP−セレクチン処理表面上での、SLeX結合hMSCの速度を示す。図15Bは、小胞修飾hMSCの回転速度に対するせん断応力の効果を示す。
【発明を実施するための形態】
【0098】
本発明は、種々の技術により細胞表面を機能化するための方法、及び様々な応用における使用に関する。本明細書に記載の組成物及び方法は、再生用に細胞を組織に標的化するのに有用であるか、又はその代わりに作用剤を目的の特定組織へ細胞に基づいて送達するのに有用である。本明細書に記載の方法に有用な細胞は、粒子並びにリガンドの付加により修飾することができる。粒子は、タンパク質、小分子、RNA干渉分子、薬物、ビタミン、治療薬、診断薬、栄養補助剤、化粧用の特性を有する作用剤、又は核酸などの作用剤をさらに含んでいてもよい。加えて、本明細書に記載の組成物は、例えば、特に脳卒中、臓器再生、癌、骨折、及び虚血性心疾患を含む広範囲の疾患又は創傷を治療するために、当業者によって個別化することができる。
【0099】
細胞及び細胞の内部機構は、細胞が外因的に付加されるか又は本体内で標的とされるかにかかわらず、多くの生物学的応用に不可欠な構成要素である。特定細胞及び特定部位へのマイクロ粒子及びナノ粒子に基づく標的送達は、少なからぬ注目を集めてきた。細胞特異的相互作用を利用することによる本発明の方法を使用して、粒子に基づく手法により細胞を標的化することは、特定組織に薬物及び他の因子を送達するのに有用である。本発明者らは、局所的投与又は全身性投与によって、機能化粒子を細胞に標的化することができることを示した。
【0100】
本明細書中に記載の方法及び組成物には、細胞膜と細胞外マトリックス間の代替的相互作用を模倣するか又は改変するかのいずれかにより、細胞膜を機能化して細胞機能に指令を与えるか又は微小環境シグナルを制御するための様々な手法が含まれる。これは、特定の機能化を有する粒子及び/又はポリマー鎖を結合させることにより細胞を改変することによって達成される。細胞の機能化は、粒子を化学的に結合させることによって、又は非粒子コーティングにより細胞膜を直接的に機能化することによって達成することができる。結合性及び/又は可溶性作用剤を送達して細胞の遺伝子(及び/又はタンパク質)発現を変更することによって、細胞の遺伝的機能化を含むようにこの方法を拡張することができる。例えば、これは、ウイルス性及び非ウイルス性遺伝子治療、特にsiRNA送達により達成することができる。
【0101】
本発明の方法及び組成物の応用には、以下が含まれるがそれらに限定されない:生理学的及び病理学的状態下での指向性細胞遊走/侵入、細胞外マトリックス内での又は全身性標的化による生物活性剤(成長因子、酵素など)の標的化及び/又は制御放出、細胞表現型(例えば、受容体発現、細胞の追跡及び/又は画像化)の変化を検出するため細胞センサー応用。
【0102】
本明細書では、「単離された」及び、「単離する」という用語は、哺乳動物由来の生体試料から、選択された細胞タイプを分離する過程を記述するために使用される。細胞単離の方法は当業者に周知であり、一般的に、酵素反応(例えば、所望の組織又は生体試料(例えば、血液)から細胞を分離するコラゲナーゼ)、遠心分離、及び/又は組織培養皿での細胞播種が伴う。例えば、本明細書に記載の方法及び組成物に好適な細胞単離法は、米国特許第6,475,764号明細書、第5,424,208号明細書、第7,217,568号明細書、第6,991,897号明細書、又は第6,627,759号明細書に見出すことができ、それらは参照によりそれらの全体が本明細書に組み込まれる。本明細書に記載の方法及び組成物の実施に、細胞の均質集団又は細胞の異種性(例えば、複数の細胞タイプ)集団が使用できることは、本明細書において明確に意図されている。
【0103】
本明細書では、「機能化」又は「機能化された」という用語は、細胞が、例えば処置しようとする組織への標的化又は薬物送達などの所望の機能を有することを可能にする、細胞膜に対する修飾を記述するために使用される。細胞の機能化は、例えば、ポリマー、リンカー、粒子、標的化部分、化学的側鎖基、リガンド、又はこれらの任意の組合せを結合させることを包含し得る。機能化は、例えば、細胞表面上のリンカー分子に粒子を結合させることも包含する。加えて、細胞は、一部分を細胞膜に結合させることにより、又は薬物送達用に作用剤を細胞に負荷することにより機能化することができる。特に所望の場合には、2つの異なる目的、例えば組織及び受容体媒介性取込みの標的化、又は組織及び薬物送達の標的化のために細胞を機能化することもできる。そのような細胞は、本明細書では「二重機能の」細胞と呼ばれる。幾つかの場合では、細胞の機能化は、例えば、細胞表面を改変するために、自己集合両親媒性分子で形成された小胞を組織に注射することにより、in vivoで起こり得る(例えば、細胞表面は、in situ又はin vivoで修飾され得る)。
【0104】
本明細書では、「リガンド」又は「膜結合リガンド」という用語は、特に受容体、細胞表面ポリペプチド、膜、又は糖質への結合などの生物学的作用又はそのような作用の可能性を有する細胞膜に結合された外来性部分を記述するために使用される。「外来性」とは、リガンドが生物又は系内で合成されないことを意味する。幾つか場合には、リガンドは、標的組織再生(例えば、損傷組織の修復)のために、細胞を特定の臓器又は組織に向けることを可能にすることにより、標的化部分として機能することができる。標的化部分には、例えば、薬物、受容体、抗体、抗体断片、アプタマー、ペプチド、ビタミン、糖質、タンパク質、接着分子、糖タンパク質、糖残基若しくはグリコサミノグリカン、治療薬、薬物、又はこれらの組合せが含まれていてもよい。当業者であれば、細胞を修飾するための種々の標的化部分を、その細胞の意図された目的に基づいて容易に設計することができる。
【0105】
本明細書では、「粒子」という用語は、作用剤又は作用剤の混合物を送達するために又は細胞に機能性を提供するために使用することができる、細胞膜に結合されている部分を記述するために使用される。「粒子」という用語は、例えば、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、ナノ粒子、マイクロ粒子、脂質、又はデンドリマーを包含する。粒子の大きさは、粒径がおよそ0.1〜10,000nmと幅広く異なっていてもよいが、好ましくは粒子は、粒径がおよそ1〜8000nmである。粒子は、粒径がおよそl〜999nmである場合にナノ粒子とみなされ、または粒径がおよそ1000nm〜8000nmである場合にマイクロ粒子とみなされる。当業者であれば、細胞に結合させる種々の粒子を、その粒子の意図された目的に基づいて容易に設計することができる。
【0106】
本明細書では、「作用剤」という用語は、細胞効果又は組織効果を誘導することができる生物活性分子又は生物活性分子の前駆体を記述するために使用されるか、又はその代わりに送達される細胞組成物を記述するために使用される。作用剤は、小分子、薬物、成長因子、サイトカイン、酵素、RNA干渉分子(例えば、siRNA、shRNA、又はmiRNA)、増殖因子、プロドラッグ、酵素前駆体、ビタミン、栄養補助剤、治療薬、診断薬、ケモカイン、脱分化因子、又は分化因子であってもよい。作用剤の投与により調節することができる機能には、例えば、細胞成長、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着が含まれる。例えば、組織再生のために骨組織を標的としたい場合は、本明細書の実施例セクションに記載されているように、細胞を作用剤(又は複数の作用剤)で処置して骨形成系統へ分化を誘導することができる可能性がある。そのような作用剤には、これらに限定されないが、デキサメタゾン、β−グリセロリン酸、L−アスコルビン酸−2−ホスフェートが含まれる。
【0107】
本明細書では、「標的組織再生」という用語は、損傷又は疾患を軽減するための臓器又は組織の治療を記述するために使用される。「疾患の損傷の軽減」は、疾患の重症度の低減、症状の低減、疾患の完全寛解、又は医学分野の当業者に公知であるような疾患に関連する他の測定可能なパラメーターの任意の変化により測定することができる。標的組織再生は、作用剤(又は複数の作用剤)を損傷組織又は患部組織に送達すること、並びに損傷組織又は患部組織を再定植させるために細胞を投与することを包含する。このようにして治療することができる疾患又は障害には、例えば、ALS、クローン病、脊髄傷害、心臓病、脳卒中、自閉症、狼瘡、眼疾患、多発性硬化症、慢性閉塞性肺疾患、関節炎、糖尿病、自己免疫障害、虚血性心疾患、癌、創傷治癒、熱傷、及びパーキンソン病が含まれる。本質的にいかなる組織又は臓器も、本明細書で開示されている方法及び組成物による治療の標的にすることができ、それらの組織又は臓器には、脳、心臓、脈管系、肺系、腎系、脾臓系、リンパ系、骨髄、硬骨、骨筋肉系、免疫系、生殖器系、皮膚、軟骨、神経系、消化器系、肝臓、膵臓系、造血系、ホルモン系が特に含まれる。
【0108】
本明細書では、「小胞」という用語は、両親媒性二分子層を含む球状の脂質構造を指し、生物活性剤をさらに含むことができる。そのような球状構造は、本明細書では「リポソーム」とも呼ばれる。対照的に、「ミセル」という用語は、両親媒性分子(例えば、リン脂質)の単層がエネルギー的に有利である立体構造を含む球状の脂質構造を指す。一般的に、ミセルは、外側親水性球体及び内部疎水性領域を含む。ミセルを使用する生物活性剤の送達も、本明細書で意図されている。本明細書では、「両親媒性分子」という用語は、一方の端部に親水性領域を含み、反対側の端部に疎水性領域を含む分子(例えば、リン脂質)を指す。「両親媒性分子」という用語は、本明細書では、「脂質分子」という用語も包含する。「リピドイド」という用語は、RNA干渉分子の全身性送達のためのナノ粒子製剤を指し、Akinc et al., Nature Biotechnology advance online publication, 27 April 2008 (DOI:10.1038/nbt1402)に記載されている。
【0109】
「RNA干渉分子」は、本明細書中では、RNA干渉(RNAi)により標的遺伝子又はゲノム配列の発現に干渉するか又は阻害する任意の作用剤と定義される。そのようなRNA干渉剤には、これらに限定されないが、標的遺伝子又はゲノム配列に相同性のRNA分子を含む核酸分子又はその断片、低分子干渉RNA(siRNA)、低分子ヘアピン又は小分子ヘアピンRNA(shRNA)、マイクロRNA(miRNA)、及びRNA干渉(RNAi)により標的遺伝子の発現に干渉するか又は阻害する小分子が含まれる。
【0110】
本明細書では、「ビオチンの供給源」という用語は、細胞表面又は粒子表面のビオチン化を可能にするビオチン部分を含む化合物を指す。ビオチンの供給源は、当技術分野で周知である。1つの実施形態では、ビオチンの供給源は、1,2−ジオレオイル−sn−グリエルコ−3−ホスホエタノールアミン−N−(ビオチニル)ナトリウム塩(1,2-Dioleoyl-sn-Glyerco-3-Phosphoethanolamine-N-(Biotinyl) sodium salt)である。別の実施形態では、ビオチンの供給源は、ビオチニル−N−ヒドロキシ−スクシンイミドを含む。
【0111】
本明細書では、「含んでいる」又は「含む」という用語は、本発明に必須であるが、必須であるかどうかに関わらず不特定要素の包含の余地がある組成物、方法、及びその対応する構成要素(複数可)を指すために使用される。
【0112】
本明細書では、「から本質的になる」という用語は、所与の実施形態に必要な要素を指す。この用語は、本発明のその実施形態の基本的な及び新規的又は機能的な特徴に実質的な影響を及ぼさない要素の存在を許容する。
【0113】
「からなる」という用語は、本明細書に記載の組成物、方法、及びその対応する構成要素を指し、実施形態のその説明で列挙されていないあらゆる要素を除外する。
【0114】
本明細書及び添付の特許請求の範囲では、単数形「a」、「an」、及び「the」は、状況が明白にそうでないと示さない限り、複数の参照を含む。したがって、例えば、「本方法」への言及は、本明細書に記載されているタイプの及び/又は本開示などを読めば当業者に明白になるだろう1つ又は複数の方法及び/又はステップを含む。
【0115】
利点及び使用
細胞表面を修飾するために使用されている1つの手法は、細胞膜上に存在するシアル酸残基のビオチン化を含む26。この手法は、明確な基質上に細胞をパターン化するために使用された。同様に、シアル酸残基は、ビオチン化ホスフィンに基づくリンカーを化学的に結合させるために使用された27。前駆細胞は、軟骨損傷部位への幹細胞又は前駆細胞の接着を容易にするために、細胞膜をマトリックス分子に対する抗体で「ペイントする」ことにより、体中の特定領域へと標的化されている28。この戦略は、まず、脂質付加プロテインG(疎水性基、パルミチン酸による28)の細胞膜への挿入を可能にさせ、次に、軟骨基質抗原に対する抗体を溶液中でインキュベーションすることにより、細胞の外部表面上のプロテインGに対する抗体との結合を可能にさせる2段階工程に依存する。細胞表面改変の他の例には以下が含まれる:グリカンのシアル酸の非天然N−アシル置換29、アセトアミド糖のケトン基の反応30、チオール基によるシアル酸の誘導体化31、シアル酸の過ヨウ素酸酸化32、細胞内タンパク質の化学的修飾(例えば、AGT)33。RGDによる細胞表面の電気活性修飾は、細胞表面を修飾するための外部電場の使用例である34。これらの結果は、細胞膜表面を化学的及び/又は物理的に修飾できることを示す。本明細書に記載の方法及び組成物は、その微小環境との細胞相互作用に影響を与えるためのプラットフォーム技術として、粒子及び/又は膜に基づく技術により細胞表面を改変させて細胞表面を機能化するためにさらに拡張される。
【0116】
表面に結合した磁性ナノ粒子を細胞単離に使用することが示されており35、36、磁性ナノ粒子は細胞追跡にも使用されている37。これらのデータは、造血幹細胞に対していかなる有害作用も及ぼさずに、より大型のナノ粒子(例えば、0.9〜1.0μmと大型)を取り込むことができ、より低分解能での検出が可能であることを示す。これらのビーズは、典型的には、種々の種又はビオチンに由来する一次抗体に対する二次抗体であらかじめコーティングされており、選択した一次抗体を使用して細胞単離系を容易に構築することが可能になる。ビーズが細胞を捕捉した後で、ビーズを含有するチューブを磁性粒子濃縮器に配置する。結合している細胞はチューブ壁面に迅速に引き寄せられ、選択した方法に依存して上清を新しいチューブに移すか又は破棄することができる。特定のビーズは、切断部位も含有しており、粒子を細胞表面から容易に放出することが可能である。Dynabeadを用いた研究では、より高いビーズ濃度は、細胞表面上のビーズ数の増加の原因となりが、その結果として毒性がもたらされる場合があることを示している36。さらに、磁性ナノ粒子38は、例えば、内部移行を防止するためにペプチド及び/又は親水性ポリマー(PEG)で容易に修飾することができる39。細胞表面上の受容分子の挙動を追跡する研究では、受容体の移動性及び動的結合をモニターするために、標識PE−IgG粒子の結合を使用できることが示されている40。
【0117】
ポリマー又は粒子による細胞の機能化は、本明細書に記載のように、細胞機能の制御及び細胞微小環境の制御を含む生物医学的応用に多くの利点を提供する。
【0118】
遊離流動性の機能化粒子及び/又はポリマーで細胞を標的化することとは対照的に41、この方法は、特定の応用のために機能化細胞の生成を可能にする。さらに、標的送達系(粒子を標的化して、特定の細胞に作用剤を放出させる)42と比較して、この方法は、細胞を機能化して所望の作用を行なわせるため、より良好な制御を可能にする。これらのタイプの機能化により達成することができる特性を様々に組合せると、細胞が複数のタスクを行うことが可能になり、制御された所望の様式でそれらのタスクを行うように細胞に指図することが可能になるだろう。
【0119】
機能化細胞の使用は、各種の応用を有する。例えば、特定部位に対して細胞を標的送達することができる。機能化細胞を、再生用に、又は特定の腫瘍部位における作用剤の送達、例えば様々なベクター送達用に細胞を必要とする部位に向けて正確に標的化することができる。特定の指向性で遊走するか又は3Dマトリックス内でより高い侵襲能力を達成するように細胞をプログラムすることにより、細胞を機能化することもできる。例えば、機能化細胞は、細胞表面上に結合された機能的粒子により細胞外ポリマーマトリクスを分解することができ、したがって遊走速度が増加する。この方法は、組織、例えば腫瘍内の細胞分布を達成するために、及び特定の作用剤を送達するために有用であり得る。特定の応用目的(例えば、組織再生、特定の細胞を死滅させる癌治療など)で、作用剤又は複数の作用剤、つまり薬物成長因子、又は酵素を送達するために機能化細胞を使用することもできる。特定の作用剤を産生するように細胞を遺伝子組換えすることもでき、したがって機能化を使用して、特定の環境内にこれら作用剤を送達するように細胞に指図することができる。作用剤を細胞に直接送達することにより、投薬を繰り返す必要性を低減することができる。食作用(例えば、マクロファージ)による細胞の破壊を防止することにより、細胞の生存能をさらに増加させることができる。作用剤、例えばRNA干渉分子(例えば、siRNA、shRNA、miRNA)の長期間にわたる細胞への放出制御に、細胞を使用することができる。これにより、周囲の微小環境と実質的に相互作用させずに(つまり、分解を回避して)、薬物を細胞に向けることができるため、治療に必要なsiRNAの量が著しく低減されるだろう。作用剤を細胞から遠ざかるように向かわせて、細胞に初期的及び/又は直接的影響を与えずに微小環境を改変することができる。粒子は、体内又はin vitroモデル系内の特定の組織又は他の細胞に、細胞を接着させる機能(細胞固定化)を果たすことができる。
【0120】
本発明の応用は広範囲に及んでおり、様々な疾患状態に適用可能である。応用及び疾患状態の幾つかの非限定的な例には、以下のものが含まれる:免疫細胞(例えば、T細胞)の標的化、骨粗しょう症(予防及び治療)、骨形成不全症、炎症性疾患(例えば、クローン病(Chrohn's disease)、移植片と宿主との拒絶反応、関節炎、セリアック病など)、老化(例えば、変性の減少及び再生の増加)、虚血組織(例えば、心筋梗塞及び関連疾患、並びに一般的な筋変性)、心血管疾患(例えば、抹消動脈疾患)、癌、急性放射線症候群、肺疾患、心臓病、糖尿病、肝不全及び腎不全、脳卒中、禿頭症、創傷治癒、脳疾患/損害、及び神経疾患又は損害。本明細書に記載の方法及び組成物を疾患の療法に適用することは、十分に当業者の能力内にある。
【0121】
細胞
本質的にあらゆる細胞を、本明細書に記載の方法及び組成物に使用することができる。動物使用の場合には、細胞は動物由来であることが好ましく、ヒト使用の場合には、細胞はヒト細胞であることが好ましく、どちら場合も自己由来細胞供給源が好ましい。細胞は、初代細胞、例えば初代肝細胞、初代ニューロン細胞、初代筋芽細胞、初代間葉系幹細胞、初代前駆細胞であってもよく、又は細胞は樹立細胞系の細胞であってもよい。細胞は細胞分裂を起こすことが可能である必要はなく、最終分化細胞を本明細書に記載の方法に使用することができる。この状況では、細胞は、これらに限定されないが、上皮細胞、内皮細胞、ニューロン細胞、脂肪細胞、心臓細胞、骨格筋細胞、線維芽細胞、免疫細胞、肝細胞、脾細胞、肺細胞、循環血液細胞、生殖細胞、消化管細胞、腎細胞、骨髄細胞、及び膵細胞を含む任意の細胞タイプであってもよい。細胞は、これらに限定されないが、脳、肝臓、肺、腸、胃、脂肪、筋肉、精巣、子宮、卵巣、皮膚、脾臓、内分泌器官、及び硬骨などを含む任意の組織から単離された細胞系、幹細胞、又は初代細胞であってもよい。
【0122】
細胞がin vitro条件下で維持される場合、従来の組織培養条件及び方法を使用することができ、それらは当業者に公知である。種々の細胞の単離及び培養方法は、十分に当業者の知識内にある。
【0123】
特に所望の場合、リガンド及び/又は粒子による機能化の前に、細胞を処理することができる。全身性送達前に細胞を種々の作用剤で前処理して、細胞表面上の特定受容体の発現を促進させるか、又は細胞のホーミング及び生着を増強するために細胞が特定の因子を産生するように促進させるか、又はその代わり特定の細胞機能を促進させることができる。例えば、治療のために対象体へと送達する前に、細胞を誘導して細胞遊走を増強させることができる。
【0124】
加えて、異種性細胞集団及び均質細胞集団は両方とも、本明細書に記載の方法及び組成物と共に使用されることが意図されている。加えて、細胞の凝集塊、粒子に結合された又は粒子内に封入された細胞、ヒドロゲルなどの注射可能な送達媒体内の細胞、及びスキャフォールドを含む移植可能な基質に結合された細胞は、本明細書に記載の方法及び組成物と共に使用されることが意図されている。
【0125】
リガンド
細胞組成物の表面にリガンドを結合するための方法が、本明細書に記載されている。リガンドとは、細胞がin vivoで組織との所望の生物学的相互作用を示すことを可能にする細胞表面に結合された部分である。リガンドは、抗体、抗体断片、アプタマー、ペプチド、ビタミン、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、薬物、グリコサミノグリカン、又はこれらの任意の組合せであってもよい。例えば、リガンドは、癌細胞特異性抗原を認識する抗体であってもよく、したがって細胞は、腫瘍細胞と優先的に相互作用して、作用剤の腫瘍特異的送達を可能にする。一般的に、本明細書に記載の方法及び組成物の場合、リガンド(又は複数のリガンド)は、外来性である(つまり、生物内又は系内で合成されない)。好ましいリガンドは、処置される組織の外側表面上の標的分子と相互作用することが可能であるため、リガンドは、処置される組織に細胞組成物が蓄積する能力を付与することができる。他の組織との交差反応性が限定されているリガンドが、一般的に好ましい。ステルスリガンド(stealth ligand)とは、目的の組織に達するまで露出されない(例えば、粒子内に封入されているか又は捕捉されている)リガンドである。ステルスリガンドを使用する利点は、リガンド(例えば、薬物)を用いて全身性治療する場合に生じる可能性のある任意の非特異的効果又は副作用を限定することである。
【0126】
幾つかの場合には、リガンドは、細胞が特定の組織を標的とすることを可能にする標的化部分として機能することができる。そのような好適な標的化部分には、例えば特異的結合対の任意のメンバー、抗体、モノクローナル抗体、又はその誘導体若しくは類似体、限定ではないが、Fv断片、単鎖Fv(scFv)断片、Fab’断片、F(ab’)2断片、単一ドメイン抗体、ラクダ化抗体及び抗体断片、ヒト化抗体及び抗体断片、並びに先述の多価性型を含む、;限定ではないが、典型的には共有結合で結合されているか、又はそうでなければ安定化されている(つまり、ロイシンジッパー又はヘリックス安定化)scFv断片である、ジスルフィド安定化Fv断片、scFvタンデム((ScFv)2断片)、ダイアボディ、トリボディ(tribody)、又はテトラボディ(tetrabody)などの単一特異性又は二重特異性抗体を含む多価性結合試薬が含まれていてもよく、他の標的化部分には、例えば、アプタマー、受容体、リガンド、及び融合タンパク質が含まれる。
【0127】
当業者であれば、細胞の目的に基づいてあらゆる標的化部分を選択することができ、例えば、タモキシフェンなどのエストロゲン受容体リガンドは、細胞表面上のエストロゲン受容体数の増加を示すエストロゲン依存性乳癌細胞に対して細胞を標的できる。リガンド/受容体相互作用の他の非限定的な例には、以下のものが含まれる:CCR1(例えば、関節リウマチ及び/又は多発性硬化症の炎症関節組織又は脳の治療用)、CCR7、CCR8(例えば、リンパ節組織の標的化)、CCR6、CCR9、CCR10(例えば、腸組織の標的化用)、CCR4、CCR1O(例えば、皮膚の標的化用)、CXCR4(例えば、一般的な遊出増強用)、HCELL(例えば、炎症及び炎症性障害、骨髄の治療用)、アルファ4ベータ7(例えば、腸粘膜標的化用)、VLA−4/VCAM−1(例えば、内皮の標的化)。一般的に、標的化(例えば、癌転移)に関与するあらゆる受容体を、本明細書に記載の方法及び組成物での使用に利用することができる。幾つかの実施形態では、修飾リガンドは、ポリ(エチレングリコール)、ヒアルロン酸、デキストラン、キトシン(chitosin)、又はポリ(エチレンオキシド)などのステルスリガンドを含む。
【0128】
粒子
細胞に結合された粒子の特性は、例えば、それらの粒径、形態、組成、表面電荷、多孔度、表面性状、機能的ドメインの濃度又はドメインのタイプ、分解特性、粒子が1つ又は複数の作用剤(成長因子、磁石、サイトカイン、接着性作用剤、毒素、タンパク質、ペプチド、酵素、核酸、抗体、細胞受容体、又はそれらの断片を含む)を含有しているかどうか、そのような作用剤の位置(例えば、表面上又は内部)などを含む多数のパラメーターに関して、粒子のタイプ間で異なる場合があり、又は単一粒子内でさえ異なる場合がある。特に所望の場合、粒子は作用剤で構成されていてもよく、その場合およそ1%から実質的に粒子全体(つまり、およそ100%)が所望の作用剤である。潜在的な磁性剤を含有することに加えて、粒子はコーティング磁石であってもよく又は非コーティング磁石であってもよい。
【0129】
粒子は、細胞膜との直接的相互作用により細胞表面に結合されていてもよい。粒子上に存在する機能性は、ポリマー性であってもよく、非ポリマー性であってもよく、又はオリゴマー性であってもよい。粒子上の結合部位は、粒子が細胞表面と相互作用することができれば、イオン性(陽イオン性及び陰イオン性)であってもよく、又は非イオン性であってもよい。細胞に粒子を結合させる手法は、種々の化学的方法及び/又は物理的方法により表面があらかじめ機能化されている「ボトムアップ」手法を使用して実施することができる。その後、機能化された細胞表面を使用してポリマー粒子を結合させ、機能化細胞を作製することができる。
【0130】
粒子の粒径及び形状は、標的化における粒子の運命を決定するために重要である。一般的に、200nmを超える粒子は細胞によって内部移行され、したがって細胞膜に吸着される22。最近の研究では、棒状の粒子は、球状微粒子と比較してそれほど効果的に内部移行されないことが示されている23。物質の特性も、内部移行に大きな影響を及ぼす22。例えば、疎水性で(例えば、ポリスチレン粒子)、それほど接着性でない(例えば、PVA)粒子コーティングを使用して、細胞の取り込みを阻害することができる24。さらに、ポリマー粒子の表面電荷も、粒子の内部移行の運命を決定するために重要である25。これらの研究は、粒子を内部移行させずに細胞表面を機能的に修飾できる可能性が高いことを示すが、細胞をそのように機能化する方法は報告されていない。
【0131】
粒子の内部移行の可能性を回避するために、粒子と細胞表面との間にスペーサー分子を導入することにより、粒子の寸法を増加させることができる。これにより、非腫瘍細胞と比較して、ペグ化されたナノ粒子の取り込み低下を示す腫瘍細胞で観察されるように、内部移行が低減されるだろう。同様に、官能化されたスペーサー(又はリンカー)分子を介して粒子を細胞表面に結合させることができる。リンカーは、任意の物理化学的相互作用により直接的又は間接的に粒子に結合可能な分子であり、本明細書中の発明を実施するための形態にさらに記載されている。粒子及び結合剤は、それらが介在構造なしで共有結合又は非共有結合によって互いに結合されている場合、「直接的に結合」されている。粒子及び結合剤は、それらがリンカーを介して互いに結合されている場合、「間接的に結合」されると言う。
【0132】
細胞表面が最初に機能化され、その後リンカー及び機能化粒子が結合される別の「ボトムアップ」手法を使用することができる。リンカー分子の選択は、一方の端部があらかじめ機能化された細胞に接着し、他方の端部が、機能化粒子に結合するようなものであろう。結合剤(あらかじめ機能化された細胞上の)又はリンカーは、粒子上の官能基に結合される。或いは、粒子又はリンカーは、結合剤の官能基に結合される。
【0133】
別の粒子に基づく手法は、異なる特徴を有する異種性(例えば、ヤヌス)粒子を使用して達成される。粒子の一方の半分は、粒子が細胞表面と相互作用することを可能にするだろう細胞接着機能性を有していてもよく(つまり、陽イオン性ポリマーは、優先的に細胞膜と相互作用することが示されている)、その一方で粒子の他方の半分は、本明細書に記載の方法の所望の応用、例えば薬物送達用に設計されるだろう。
【0134】
粒子特性は、これらに限定されないが、大きさ、直径、形状、組成、表面電荷、分解特性、粒子が1つ又は複数の作用剤を含有するかどうか、又はそのような作用剤の位置(例えば、表面上又は内部)を含む多数のパラメーターに関して、互いに異なってもよく(例えば、異種性集団)、又は単一粒子集団内で異なってもよい。
【0135】
「異種性粒子」の修飾の1つは、作用剤を送達する粒子の一方の半分(細胞に結合される)を標的とすることを含み、他方の半分は、これらに限定されないが、特に指向性細胞遊走、指向性細胞結合、及び標的送達などの応用を含む特定の機能を果たすように機能化される。
【0136】
別のタイプの機能化は、リンカー分子によって分離されている、2つの異なる機能性を含有する物質を使用することにより達成することができる。2つの機能性のうちの1つは、細胞と特異的に相互作用する(細胞膜又は細胞質内で優先的に)が、他方の機能性は、記載されている方法の所望の応用、例えば、薬物送達のために外部環境中に存在する。細胞に結合された他方の機能性は、細胞膜を介して内部移行される場合があり、細胞のセンサー及び/又はマーカーとして機能することができるか、又は異なる手法によって細胞膜の表面に結合させることができる。
【0137】
別の技術には、ポリマー鎖を構築して、ポリマー鎖と細胞膜間の適切な相互作用によって細胞表面をコーティングすることを伴う。
【0138】
機能化(例えば、NHS、ペプチド、エポキシ、イミドエステルなど)ポリマーを使用して、機能化ポリマーが細胞表面と相互作用するように細胞膜を包み込むことができる。この技術は、当業者に公知の連続ポリマー吸着に又はエマルジョン技術によって適用することができる。
【0139】
ポリマーを、ポリマーが細胞膜上に層を形成するように、連続して細胞膜に適用することができる。加えて、例えば連続吸着により又は細胞表面上にあらかじめ吸着されているポリマーに機能化粒子を結合させることにより、所望の応用のために機能化粒子をポリマー層に接着することができる。
【0140】
これらの技術は両方とも、粒子に基づく手法と比較してより効率的な様式で、細胞膜の機能化を可能にするだろう。膜に基づく手法は、適切な結合部位を介してポリマーを結合することにより細胞表面を修飾するという同じ概念を利用する。結合相互作用は、物理的、例えば荷電ポリマーのイオン性相互作用、抗体−抗原相互作用などであってもよい。同様に、相互作用は、ポリマー機能性(アミン、カルボキシル、硫化物など)に依存して化学的であってもよい。
【0141】
結合部位の粒子及び/又はリンカーは、生物学的な事象に応答して切断されるか、又は外部的に制御される切断部位を含有することもできる。これら粒子は、組織へと拡散してもよく、又は細胞近傍に留まっていてもよい。
【0142】
粒子を使用して、移植された(注射又は埋め込まれた)細胞の局在化を増強することもでき、例えば、細胞に結合された反応基を使用して、ある組織又は物質内又は特定の組織又は物質上に細胞を固定化することができる。加えて、より高度の弾性を有する粒子(例えば、軟質粒子)を使用して、生物学的障壁(例えば、血管内皮)を越えて輸送することにより、粒子の局在化能力を増強することができる。
【0143】
機能化粒子は、in vitroで又はin vivoで細胞に結合させることができる(in vivoでの結合には、最初に粒子を体内の特定細胞へと標的化することを伴っていてもよく、粒子は局所的に又は全身性に送達することができる)。幾つかの実施形態では、粒子は、ポリ(エチレングリコール)、ヒアルロン酸、デキストラン、キトサン、又はポリ(エチレンオキシド)などのステルスリガンドを含有する。
【0144】
修飾することができる粒子の特性には、これらに限定されないが、形状、表面電荷、多孔度、化学的組成、相対的疎水性/親水性、機械的特性、及び表面性状が含まれる。粒子は、生物学的分子(例えば、抗体、ペプチド、ヌクレオチド)、又は合成分子(例えば、小分子、アプタマー)を結合させることにより修飾されていてもよい。同様の技術を使用して、活性のタイミング又は位置を制御することもできる。加えて、粒子は、1つ又は複数の作用剤をさらに含んでいてもよい。作用剤は、粒子内(例えば、粒子の細孔内又はチャネル内)及び/又は粒子の外部表面上に位置して(例えば、組み込まれて)いてもよい。幾つかの実例では、粒子には、1つ又は複数の作用剤があらかじめ負荷されている。粒子がリガンドを含有する場合、リガンドは細胞と直接的に相互作用しないが、むしろリガンド相互作用が粒子内のみで生じることが好ましい。
【0145】
作用剤
様々な異なる医薬品/治療薬を、本明細書に記載の方法と共に使用することができ、それらには、小分子、タンパク質、抗体、ペプチド、及び核酸が含まれるがこれらに限定されない。一般的に、本発明により投与することができる生物活性剤には、限定ではないが以下のものが含まれる:抗生物質及び抗ウイルス物質などの抗感染剤;化学療法剤(つまり、抗癌剤);拒絶反応抑制剤;鎮痛薬及び鎮痛薬の組合せ;抗炎症剤;ホルモン(例えば、ステロイド);成長因子(例えば、骨形態形成タンパク質(つまり、BMP1〜7)、上皮成長因子(EGF)、線維芽細胞増殖因子(つまり、FGF1〜9)、血小板由来増殖因子(PDGF)、インスリン様増殖因子(IGF−I及びIGF−II)、形質転換増殖因子(つまり、TGF−β−III)、血管内皮増殖因子(VEGF));エンドスタチンなどの抗血管新生タンパク質、及び他の天然由来又は遺伝子組換えタンパク質、ポリサッカライド、糖タンパク質、又はリポタンパク質。加えて、本明細書に記載の粒子を使用して、例えば薬剤、ビタミン、鎮静薬、ステロイド、催眠薬、抗生物質、化学療法剤、プロスタグランジン、及び放射性医薬品などの任意のタイプの分子化合物を送達することができる。本明細書に記載の細胞組成物は、これらに限定されないが、タンパク質、ペプチド、ヌクレオチド、糖質、単糖、細胞、遺伝子、抗血栓剤、抗代謝剤、増殖因子阻害剤、成長促進物質、抗凝血剤、抗有糸分裂剤、線維素溶解剤、抗炎症ステロイド、薬物、及びモノクローナル抗体を含む上記物質及びその他の送達に好適である。
【0146】
本明細書に記載の方法で使用するのに好適な他の生物活性剤の例には、これらに限定されないが、以下のものが含まれる:コラーゲン、エラスチン、フィブロネクチン、ビトロネクチン、ラミニン、プロテオグリカン、又は細胞結合に影響を及ぼすことが知られている既知のインテグリン結合ドメイン、例えば「RGD」インテグリン結合配列を含有するペプチド若しくはその変異体などの細胞結合媒介物質(Schaffner P & Dard 2003 Cell Mol Life Sci. Jan;60(1): 119-32; Hersel U. et al. 2003 Biomaterials Nov;24(24):4385-415);生物活性リガンド;及び特定の種類の細胞内部成長又は組織内部成長を増強又は排除する物質。
【0147】
幾つかの実施形態では、粒子は、ビスホスホネートに基づく薬物、エストロゲン受容体モジュレーター、又はホルモンに基づく療法などの骨粗しょう症の治療用薬物を含む。ある実施形態では、粒子は、ラロキシフェン、AAR494、又はE−64などの破骨細胞再吸収を阻害する薬物を含む。幾つかの実施形態では、本方法は、骨減少症を治療するための方法をさらに含む。幾つかの実施形態では、本方法は、骨癌を治療するための方法をさらに含む。幾つかの実施形態では、粒子は、果粒球コロニー刺激因子又は骨髄特異的表面膜受容体などの硬骨標的化因子を含む。幾つかの実施形態では、硬骨標的化因子は、骨粗鬆症の硬骨を標的とするペントシジンなどの因子である。幾つかの実施形態では、本方法は、骨髄空間を標的とするための方法をさらに含む。
【0148】
幾つかの実施形態では、作用剤は、骨成長を促進するための作用剤を含む。ある実施形態では、作用剤は、レプチン、ソルチリン、トランスグルタミナーゼ、プロスタグランジンE、1,25−ジヒドロキシビタミンD3、アスコルビン酸、β−グリセリンリン酸、TAK−778、スタチン、IL−3及びIL−6などのインターロイキン、成長ホルモン、造血幹細胞因子(SF)、アクチビンA(ACT)、レチノイン酸(RA)、上皮成長因子(EGF)、骨形態形成タンパク質(BMP)、血小板由来成長因子(PDGF)、肝細胞成長因子、インスリン様成長因子(IGF)I及びII、造血成長因子、ペプチド成長因子、エリトロポイエチン、インターロイキン、腫瘍壊死因子、インターフェロン、コロニー刺激因子、ヘパリン結合性成長因子(HBGF)、TGF−β1などのアルファ又はベータ形質転換成長因子(α又はβ−TGF)、線維芽細胞成長因子、血管内皮成長因子(VEGF)、神経成長因子(NGF)、及び筋肉形態形成因子(MMP)などの成長因子又はサイトカインを含む。
【0149】
幾つかの実施形態では、粒子は、プロコラーゲンなどのコラーゲン又はアスコルビン酸の産生又は構築を促進する作用剤を含む。ある実施形態では、粒子は、NFκBリガンド(Rank−L)の受容体活性化因子などの、硬骨へ粒子再吸収を促進するための再吸収因子、デキサメタゾンなどのコルチトステロイド(cortitosteroid)、副甲状腺ホルモン、マクロファージコロニー刺激因子(M−CSF)、又は形質転換成長因子−β1(TGF−β1)を含む。幾つかの実施形態では、粒子は、EDTAに基づく作用剤などの、血液由来の無機質をキレートする作用剤、ポリ(ビスホスホネート)、ポリ(ホスフェート)、カルシウム及び/又はリン酸塩を核とする生物学的又は非生物学的物質、アスパラギン酸、オステオポンチン、又は骨シアロタンパク質を含む。
【0150】
細胞組成物それ自体は、幾つかの態様では、生物活性剤を送達するための担体として考えることができるが、本明細書に記載の細胞組成物の生物活性剤/治療薬/医薬品は、処置される組織だけでなく細胞組成物の微小環境にも影響を与えることもでき、そのため細胞組成物それ自体が生物活性剤になる。例えば、細胞組成物の増殖特徴を増強する目的のために、生物活性剤を粒子に供給することができる。このタイプの送達は、細胞組成物が損傷組織を再定植させるために使用される標的組織再生に特に有用である。
【0151】
機能化方法
幾つかの機能化方法を、本明細書に記載の方法と共に使用することができる。幾つかの例示的な実施形態には、(1)細胞表面上の糖残基にある種々の官能性が関与する反応、(2)細胞表面上のペプチド残基にある官能基が関与する反応、(3)抗原−抗体相互作用の使用、(4)細胞表面と粒子間のイオン性相互作用、及び(5)細胞表面と粒子間の疎水性相互作用を含む。
【0152】
粒子に基づく細胞表面の修飾は、異なる機能性を有する粒子を細胞膜に結合させることにより達成することができる。細胞表面に存在する反応部位及び表面部分を、様々な化学を使用することにより化学的に修飾することができる。
【0153】
細胞表面の物理的特性は、細胞とECM間の相互作用を変更するために使用される化学的修飾と同じように操作することができる。細胞表面は、異なる物理的特性(例えば、電荷)を示す複雑な構造を有する。粒子又はポリマー鎖の結合に使用することができる細胞表面上の様々な官能性には、これらに限定されないが以下のものが含まれる:特に、極性(NH2/NH3+)末端基、及びリン脂質の他の官能性、ヒドロキシル(OH)、及び糖質基の他の官能性、カルボン酸基(COOH)、チオール(SH)、及び種々のタンパク質、及び糖タンパク質、抗原。粒子(中実又は中空)は、細胞膜に接着するように異なるイオン特徴に調製することができる。他のタイプの物理的接着は、細胞表面上の抗原−抗体相互作用を制御することにより可能である。抗体は、特定の応用のために機能化することができる。
【0154】
陽イオン性結合剤の例には、これらに限定されないが、特にキトサン及びポリアミンが含まれる。陰イオン性結合剤の例には、これらに限定されないが、特にポリスルホネート、ポリホスフェート、DNA、ヘパリン、及びPMAMデンドリマーが含まれる。非イオン結合には、例えば、カルボキシル、アミン、又はスルフィド官能性との結合が含まれる。
【0155】
本明細書に記載の1つの手法には、ポリマー鎖及び粒子の使用による化学的修飾が伴う。この技術には、有機ポリマー及び/又はオリゴマー粒子を使用することができるが、同様に種々の無機粒子にも拡張することができる。粒子(中空又は中実)表面と細胞膜間の化学的相互作用は、種々の官能性(例えば、ビオチン化、N−ヒドロキシサクシニミド(NHS)、エポキシ、ペプチド、イミドエステル、マレイミド、アジド、ハロアセチル、ピリジルジスルフィド、カルボジイミド、ヒドラジド)により達成することができ、文献に記載されている細胞表面修飾の化学的経路を使用することができ、それらは当業者に公知である。物理的相互作用は、例えば、荷電ポリマー;自己集合性荷電粒子、抗体−抗原、又は疎水性相互作用の使用により実施することができる。
【0156】
この手法では、中実粒子及び中空粒子の両方を使用することができ、それらは、生物医学的プロセス(例えば、細胞遊走、接着、増殖、分化、生存、マトリックス産生)を制御/モニターするための、重要な機能的作用剤(例えば、成長因子、薬物、酵素、蛍光性部分)を含有することができる。粒子を「コーティング」または「修飾」するための当業者に公知の方法により、粒子がそれで構成されている巨大分子とは異なる分子を、粒子の外側表面に結合することができる。例えば、リンカーの使用により(下記で検討)、分子を、粒子の外側表面に直接的又は間接的に結合することができる。これらの分子は、結合を容易にし、受容体媒介性を増強し、エンドサイトーシス又は破壊の回避を提供するなどの目的のために結合される。例えば、リン脂質などの生体分子を粒子表面に結合させて、エンドソームによるエンドサイトーシスを防止することができ、受容体、抗体、又はホルモンを表面に結合させて、体内の所望の臓器、組織、又は細胞に対する粒子の結合を促進又は容易にすることができ、グルカンなどのポリサッカライド、又はポリビニルピロリドン及びPEGなど他のポリマーを粒子の外側表面に結合させて、マクロファージによる取り込みを増強又は回避することができる。
【0157】
溶液中での細胞機能化を使用すると、均質な機能化を容易にする及び/又は細胞凝集を刺激することができる一方で、表面上での細胞の機能化は、細胞の1つの表面を優先的に機能化するのに有用であり得る。
【0158】
細胞への粒子の追加は、静的又は動的条件下で達成することができ、マイクロ流体チャネルを含むバイオリアクター及び/又はBioMEMSデバイスの使用を含んでいてもよい。粒子を有する細胞は、粒子を有する細胞の肺送達用に吸入剤として使用することができる。
【0159】
物質選択
本明細書に記載のように、粒子を結合させて細胞組成物を形成するための物質の選択は、当業者による所望の応用に必要と見なされる修飾のタイプに依存する。合成、天然、並びに半合成ポリマーを、ポリマー粒子の合成に使用することができる。様々な合成ポリマーには、例えば、ヒドロゲルポリマー(PEG、PVAなど)又はアクリレートが含まれる。これらポリマーは、当業者の必要性に従って直鎖状であってもよく又は架橋されていてもよい。使用することができる天然ポリマーには、これらに限定されないが、ヒアルロン酸、ゼラチン、キチンなどが含まれる。物理的相互作用の場合、例えば、ポリエチレンイミン(PEI)、ポリ(リジン)、キトサン、又はセルロースを含む幾つかのポリマーを、電荷に基づく細胞表面への接着に使用することができる。使用することができるポリマーのリストには、これらに限定されないが、以下のものが含まれる:ポリ(ラクチド)(PLA)、ポリ(グリコリド)(PGA)、ポリ(ラクチド−コ−グリコリド)(PLGA)、ポリ(カプロラクトン)(PCL)、ポリカーボネート、ポリアミド、ポリ無水物、ポリホスファゼン、ポリアミノ酸、ポリオルトエステル、ポリアセタール、ポリシアノアクリレート、及び生分解性ポリウレタンなどの生分解性ポリマー;ポリアクリレート、エチレン酢酸ビニールポリマー、及び他のアシル置換酢酸セルロース、並びにそれらの誘導体などの非生物分解性ポリマー;ポリウレタン、ポリスチレン、ポリ塩化ビニル、ポリフッ化ビニル、ポリ(ビニルイミダゾール)、クロロスルホン化ポリオレフィン、及びポリエチレンオキシド。生分解性天然ポリマーの例には、以下のものが含まれる:アルブミン、コラーゲン、合成ポリアミノ酸、及びプロラミンなどのタンパク質;アルギナート、ヘパリンなどのポリサッカライド;及び他の天然に存在する糖ユニットの生分解性ポリマー。或いは、前述のポリマーの組合せを使用することができる。
【0160】
使用することができる無機粒子の例には、これらに限定されないが以下のものが含まれる:特に、二酸化チタン、炭酸カルシウム、リン酸カルシウム、ケイ酸カルシウム、銀及び金ナノ粒子、及び磁性粒子。本明細書に記載の方法に有用である広範囲の幾何学的形状を有する様々なタイプの粒子を使用することができる。粒子形状の非限定的なリストには、例えば、コアシェル物質、中空粒子、ケージ様粒子が特に含まれる。
【0161】
リンカー
リンカーには、ヒドロキシル基、第一級又は第二級アミノ基、リン酸基若しくはその置換誘導体、又はカルボン酸基などの官能基が含まれていてもよい。アシルカルニチン、アシル化カルニチン、スフィンゴシン、セラミド、ホスファチジルコリン、ホスファチジルグリセロール、ホスファチジルエタノールアミン、ホスファチジルイノシトール、ホスファチジルセリン、カルディオリピン、及びホスファチジン酸などの極性脂質も、リンカーとして機能することができる。極性脂質分子は、官能基を有していてもよく又は有していなくともよい有機スペーサー分子に共有結合で随意に結合されてもよい。他のリンカーには、ヘテロ二官能性架橋剤が含まれる。例えば、サクシニミジル−4−(N−マレイミドメチル)シクロヘキサン−1−(カルボキシ−6−アミノカプロエート)は、LC−SMCCとしても知られており、スルフヒドリル基及びアミン基と反応するヘテロ二官能性架橋剤である。リンカーの非限定的な例には、上述されているポリマー性の陰イオン性及び陽イオン性、非イオン性作用剤が含まれる。
【0162】
粒子の製作は、様々な技術により実施することができる。エマルジョンの溶媒蒸発又は噴霧乾燥を応用して、粒子を製作することができる。中実粒子が望ましいか又は中空粒子が望ましいかに依存して、複数エマルジョン技術を使用することができる。化学的修飾は、様々な化学反応の使用により実施することができ、それは必要とされる側鎖基又は官能性に依存する。
【0163】
幾つかの実施形態では、リンカーは、ポリ(エチレングリコール)、ヒアルロン酸、デキストラン、キトシン、又はポリ(エチレンオキシド)などのステルスリガンドを含む。
【0164】
細胞への架橋粒子及び/又はリガンド
細胞膜を研究するために使用される方法
細胞膜分子の構造的組成、分布、細胞結合、及び動力学は、様々な化学を使用するにより研究することができる。様々な化学的架橋剤を、細胞膜及び細胞小器官膜を膜の外側表面及び膜に囲まれた空間内の両方に架橋することができる。架橋剤は、表面受容体又はそれらのリガンドを識別するために使用されることが多い。膜不透過性の架橋剤は、細胞表面特異的な架橋を保証する。不水溶性の架橋剤を、制御された量の試薬及び制御された反応時間で使用すると、膜透過及び内膜タンパク質との反応を低減することができる。NHSエステルのサクシニミジル環に結合されたスルホニル基により、水溶性で膜不透過性であり、内膜タンパク質と反応しない架橋剤がその結果としてもたらされる。したがって、スルフォ−NHS−エステルを使用する場合、反応時間及び架橋剤の量はそれほど重要ではない。
【0165】
細胞膜の構造研究では、細胞の脂質二分子膜内の位置及び環境を決定するために、様々な疎水性の試薬が必要とされる。蛍光タグは、膜の内側及び外側のタンパク質、脂質、又は他の分子を位置付けるために使用される。スペーサーアームの長さが様々である種々の架橋剤を使用して、タンパク質を膜内の結合分子に架橋し、分子間の距離を決定することができる。より短い架橋剤で架橋が成功する場合は、2つの分子がある様式で相互作用していることを強く示す。一団のより短い架橋剤では架橋が得られないが、より長い試薬を使用すると結合が得られる場合は、一般的に、分子が膜の同一部分に位置するが相互作用していないことを示す。ホモ二官能性のNHS−エステル、イミドテソル(imidotesor)ヘテロ二官能性のNHS−エステル/光活性化可能なフェニルアジドは、これらの手順で一般的に使用されている。イミドエステル架橋剤(イミデート)は水溶性であるが、やはり膜を透過することができる。スルフヒドリル反応性架橋剤は、システインを有する分子を膜内の他の分子に対して標的化するのに有用であり得る。EDC、不水溶性ジシクロヘキシルカルボジイミド、DCC、及び他の水溶性/水不溶性カップリング試薬対は、膜及び細胞構造、タンパク質サブユニット構造及び配置、酵素:基質相互作用、並びに細胞表面及び膜受容体を研究するために使用される。EDCが親水性であるという特徴は、その結果として、膜及びサブユニット研究において、DCCなどの疎水性カルボジイミドよりももっと異なる架橋パターンをもたらすことができる。1つの実施形態では、水溶性及び水不溶性のカルボジイミドと架橋させて、関与するタンパク質:タンパク質相互作用の空間的配置の全体像を取得することができる。細胞表面上の機能化粒子及び/又はリガンドの位置及び相互作用を試験するためのこれら架橋法は、本明細書に記載の組成物に有用である。
【0166】
細胞表面上で架橋反応を実施するための方法
架橋剤を使用して、試料中のタンパク質の構造及び組成を研究することができる。幾つかのタンパク質は、pH又は塩条件が異なると異なる立体構造で存在するため、研究するのが難しい。立体構造の変化を回避する1つの方法は、サブユニットを架橋することである。アミン、カルボキシル、又はスルフヒドリル反応性試薬が、特定のアミノ酸の特定又はサブユニットの数、位置、及び大きさの測定に使用される。分子内架橋が所望の場合は、短〜中程度の長さのスペーサーアーム架橋剤が選択される。スペーサーアームが長すぎると、分子間架橋が生じ得る。
【0167】
アミン反応性又は光活性化可能なアミン反応性架橋剤などの長さが短い結合試薬と一緒になって、スペーサーアームを結果としてもたらさないカルボジイミドは、至適濃度及び条件で使用されれば、外来性分子と架橋することなくサブユニット間で架橋する。
【0168】
わずかにより長い架橋剤は、サブユニット間も架橋することができるが、分子間結合がその結果としてもたらされる場合がある。試薬量及びタンパク濃度を調整することにより、分子間架橋を制御することができる。ホモ二官能性の架橋剤を使用する場合、希釈タンパク質溶液及び高濃度架橋剤が、分子内架橋に有利である。
【0169】
三次元構造を決定又は確認する場合は、長さがより増加したスペーサーアームを有する切断可能な架橋剤を使用して、サブユニット間の距離を決定することができる。様々な反応基を有する架橋剤を使用した実験により、特定のアミノ酸の位置を示すことができる。結合させた後、タンパク質を二次元電気泳動にかける。第1の次元では、タンパク質は非還元条件を使用して分離され、分子量が記録される。幾つかのサブユニットは架橋されていない場合があリ、それらの個々の分子量により分離されるだろう。結合されたサブユニットは、合計の分子量により分離されるだろう。その後ゲルの第2の次元を、架橋サブユニットを切断する条件を使用して実施する。架橋サブユニットの個々の分子量を決定することができる。還元されなかった架橋サブユニットは、対角線パターンをもたらすはずだが、切断されたサブユニットは、対角線からはずれるだろう。個々のサブユニットの分子量は、還元SDSポリアクリルアミドゲル電気泳動を使用して、タンパク質サブユニットの所定の分子量と比較されるべきである。この架橋技術は、必要な生体特異的応用のために種々の機能性を有する細胞表面を作り出すことを可能にする。
【0170】
より好ましい実施形態では、ホモ二官能性スルフォ−NHS−エステル、ヘテロ二官能性スルフォ−NHS−エステル、及び光反応性フェニルアジドが、細胞表面上のタンパク質を架橋するために使用される。特定のタンパク質が表面上に位置するか又は膜内部分に位置するかの決定は、水溶性又は水不溶性架橋剤を使用して、既知のタンパク質又は放射性標識に対する細胞膜調製物の結合反応を実施することにより達成することができる。結合させた後、細胞を洗浄し、可溶化し、SDSポリアクリルアミドゲル電気泳動(PAGE)により特徴付け、目的タンパク質が結合したかどうかを決定することができる。内在性膜タンパク質は、水不溶性架橋剤の存在下で結合体を形成するが、水溶性架橋剤の存在下では形成しないだろう。表面の膜タンパク質は、水溶性及び不水溶性の架橋剤の存在下で結合することができる。ホモ二官能性の光活性化可能なフェニルアジドは、タンパク質相互作用及び結合を研究するためのより汎用性の高い架橋剤の1つである。これは、切断可能であり、125Iで放射性標識することができる。切断した後、解離された分子は両方とも依然としてヨウ素化されているだろう。この架橋剤上の両反応基は非特異性であるため、架橋はアミノ酸組成に依存せず、結合が成功する。
【0171】
細胞投与
細胞を対象体に投与するための様々な手段が、当業者に公知である。そのような方法には、全身性注射、例えばi.v.注射、又は対象体の標的部位への細胞の埋め込みが含まれていてもよい。細胞は、対象体への注射又は埋め込みによる導入を容易にする送達デバイスに挿入されてもよい。そのような送達デバイスには、受容対象体の体内に細胞及び液体を注入するためのチューブ、例えばカテーテルが含まれていてもよい。1つの好ましい実施形態では、チューブは、本発明の細胞を所望の位置で対象体へ導入することができる針、例えば注射器をさらに有している。細胞は、様々な異なる形態での送達用に調製することができる。例えば、細胞は、溶液又はゲルに懸濁されてもよく、又はそのような送達デバイスに含有される場合は支持マトリックスに埋め込まれていてもよい。細胞は、本発明の細胞が依然として生存可能である、薬学的に許容される担体又は希釈剤と混合されていてもよい。薬学的に許容される担体及び希釈剤には、生理食塩水、緩衝水溶液、溶媒、及び/又は分散媒質が含まれる。そのような担体及び希釈剤の使用は、当技術分野で周知である。溶液は、好ましくは無菌及び流動性である。好ましくは、溶液は、製造及び保管条件下で安定しており、例えばパラベン、クロロブタノール、フェノール、アスコルビン酸、及びチメロサールなどの使用により、細菌及び真菌などの微生物の汚染作用に対して保存されている。本発明の溶液は、本明細書に記載の細胞を、薬学的に許容される担体又は希釈剤、並びに必要に応じて上記に列挙されている他の成分に組み込むことにより調製され、その後滅菌ろ過されていてもよい。
【0172】
細胞投与経路は、比較的非侵入性であることが好ましく、例えば静脈注射、吸入による肺送達、経口送達、口腔内投与、直腸内投与、膣内投与、局所的投与、又は鼻腔内投与による。しかしながら、細胞投与経路は、治療される組織に依存するはずであり、埋め込みを含んでいてもよい。細胞送達用の方法は当業者に公知であり、医学分野の当業者であれば、本明細書に記載の方法及び組成物に使用するために推定することができる。
【0173】
細胞投与用の直接注射技術を使用して、脈管構造全体を介した遊出、又は特定の臓器、例えば肝臓又は腎臓又は他の任意の臓器の脈管構造への遊出を刺激することもできる。これは、脈管構造の非特異的標的化を含む。例えば肝臓門脈などの特定の注射部位を選択することにより、任意の臓器を標的とすることができる。或いは、注射は、体中の任意の静脈へと全身性に実施することができる。この方法は、高齢患者の幹細胞数を増強するのに有用である。加えて、細胞は、空白の幹細胞ニッチに定植するか、又は臓器を補充する新しい幹細胞を生成するために機能し、それにより臓器機能を向上させることができる。例えば、細胞は、脈管構造内の周皮細胞位置を占めることができる。
【0174】
細胞送達を使用して、活性血管新生部位を標的とすることもできる。例えば、内皮前駆細胞又は間葉系幹細胞若しくは前駆細胞の送達は、創傷部位での血管新生応答を増強することができる。血管新生の標的化は、薬物を腫瘍に標的化するための媒体として細胞を使用するのにも有用である。
【0175】
特に所望の場合、哺乳動物又は対象体を、作用剤であらかじめ処置してもよく、例えば、組織への細胞の標的化を増強するために作用剤(例えば、ホーミング因子)を投与し、それをその部位に設置して、所望の組織への細胞の標的化を容易にすることができる。例えば、ホーミング因子の組織への直接注射は、リガンド標的細胞の全身性送達に先立って実施することができる。
【0176】
本明細書及び添付の特許請求の範囲では、単数形「a」、「an」、及び「the」は、状況が明白にそうでないと示さない限り、複数の参照を含む。したがって、例えば、「本方法」への言及は、本明細書に記載されている及び/又は本開示などを読めば当業者に明白になるだろうタイプの1つ又は複数の方法及び/又はステップを含む。加えて、「細胞」という用語は、性質が異種性又は均質性のいずれでもあってもよく、細胞の凝集塊とも呼ばれる細胞集団として解釈することができる。
【0177】
記載されている技術は、胚の生着効率を向上させるために応用することができ、例えば体外受精中に、特定の組織(例えば、子宮)への結合を増強することができる接着リガンドで、胚を修飾することができる。
【0178】
先述の詳細な説明及び以下の例は単なる例示に過ぎず、本発明の範囲の限定として受け取られるべきではないことが理解される。開示された実施形態の種々の変更及び改変は、当業者であれば明白であり、本発明の趣旨及び範囲から逸脱することなく行うことができる。さらに、特許、特許出願、及び特定されている出版物は全て、例えば、本発明に関して使用し得るような出版物に記載の方法を説明及び開示するために、参照により明示的に本明細書に組み込まれる。これら出版物は、もっぱら本出願の出願日前にそれらが開示されているために提供される。これに関してはいかなるものも、本発明者らが、先行発明であるため又は他のいかなる理由でもそのような開示に優先する資格がないことを承認すると解釈されるべきではない。日付に関する宣言又はこれら文書の内容に関する提示は全て、本出願者らに利用可能な情報に基づいており、これら文書の日付又は内容の正確さに関していかなる承認も構成しない。
【実施例】
【0179】
実施例1
細胞をどのように修飾して細胞標的化を向上させることができるかに関する非限定的な例を、本明細書に記載する。具体的には、標的化作用剤を細胞表面に結合し、これにより、in vitro実験で実証されたように細胞の回転を誘導することができる。
【0180】
シアリルルイスX(SLeX)によるヒト間葉系幹細胞(hMSC)の修飾
hMSCの修飾を実施して、細胞膜の表面にSLeXを化学的に結合させた。ビオチンストレプトアビジン結合を利用して、細胞表面のSLeX部分を化学的に組み込んだ。細胞表面上に存在する遊離アミン基を、ビオチニル−N−ヒドロキシ−スクシンイミドのN−ヒドロキシ−スクシンイミド基と反応させて、細胞表面をビオチン化した。引き続いてこのステップの後、ストレプトアビジン分子と細胞表面のビオチン部分を反応させた。ビオチンとストレプトアビジンとの強力な相互作用は、ストレプトアビジン分子の細胞表面上への固定化を可能にする。その後、ストレプトアビジンをビオチン化SLeX(シアリル−Lex−PAA−ビオチン)と反応させて、細胞表面上にSLeXを導入する。hMSCの表面上にSLeXなどの標的化作用剤が存在しないことは、細胞の非回転性特徴の要因であるため、SLeXを細胞上に固定化することにより、hMSCに回転性特徴を誘導することができる。ビオチン化ステップを向上させるために、スルホン化ビオチニル―N−ヒドロキシ−スクシンイミド(スルフォ−NHS−ビオチン、BNHS)を使用した。BNHSは水溶性であり、細胞膜を透過せず、したがってBNHSのN−ヒドロキシ−スクシンイミド基が、細胞膜と優先的に相互作用することが可能になる。
【0181】
一般的方法
典型的には、約5000個の細胞を、96ウエルプレートの各ウエルの200μL hMSC細胞培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)に、24〜48時間の期間添加して、その結果細胞は約80〜90%コンフルエントだった。
【0182】
細胞を処理するために、培地をウエルから吸引し、細胞を各ウエルの中で、200μLのリン酸緩衝生理食塩水(PBS、pH7.4、Ca/Mg非含有)を用いて穏やかに室温で洗浄した。Ca/Mg非含有のPBS、pH7.4中1mMの100μL BNHS溶液を、各ウエルに室温で20〜25分間添加した。指定の期間後、細胞を、200μLのリン酸緩衝生理食塩水(PBS、pH7.4、Ca/Mg非含有)で2回室温で洗浄した。その後、50μLのストレプトアビジン溶液(Ca/Mg非含有のPBS、pH7.4中50μg/mL)を各ウエルに添加し、室温で20分間インキュベートした。指定の期間後、細胞を、200μLのリン酸緩衝液生理食塩水(PBS、pH7.4、Ca/Mg非含有)で2回室温で洗浄し、その後50μLの4μg/mL SLex(Ca/Mg非含有PBS、pH7.4中)を室温で添加した。
【0183】
BNHS及びストレプトアビジンによる細胞修飾を評価するために、蛍光性ローダミンを結合したストレプトアビジン(SR)を、ビオチン化細胞に付加し、SR結合の蛍光強度を測定した。この実験に使用した対照は、SRのみで処理した細胞、ビオチン及びSRで処理した細胞、及び未処理細胞だった。蛍光強度を7日後に再び測定し、PBS中0.1%の100μLトリトンX溶液で処理した後、細胞を4%ホルムアルデヒド溶液で固定し、100μLのDAPI溶液(PBS中1μg/mL)で染色した。ビオチン類似体で処理した細胞の蛍光強度を、機能化後の0日目及び7日目に測定した。BNHS−SR結合細胞の蛍光強度は、B−SR又はSR処理のみの蛍光強度より高かった。報告した結果は全て、三重反復で行われた実験の結果である。
【0184】
細胞の生存率
細胞の生存率は、トリパンブルー排除法を使用して実施した。手短に言えば、細胞を12ウエルプレートに播種し、終夜放置して接着させ、細胞を上述のようにBNHS溶液で処理した。すすきの後、細胞を37℃及び5%CO2で48時間インキュベートした。その後、培地を吸引し、200μLの細胞解離溶液を使用することにより、細胞をウエルから分離した。300μLの培地を添加し、合計500μLの細胞懸濁液を収集した。この懸濁液からの10μL細胞懸濁液を、4%トリパンブルー溶液を使用することにより1:1に希釈し、細胞を血球計で計数して、生存可能(未染色)及び生存不能(青染色)な細胞数を決定した。三重反復実験から計算した群中の平均細胞生存率に差異はなかった。この実験の対照には、未処理細胞(しかし、実験中にPBSを添加し、室温で維持した)、並びにNHSを用いずにビオチン及びSRで処理した細胞が含まれていた。
【0185】
BNHSで修飾した細胞の生存率は、BHSによる処理が、修飾の48時間後でさえ細胞生存率を変更しないことを示す。未処理細胞は、48時間後で85%生存可能であり、一方でビオチン処理細胞は76%生存可能であり、BNHS処理細胞は75%生存可能だった。細胞は修飾の48時間後で生存可能だったため、これは、BNHSによる細胞の修飾が細胞に対して毒性ではないことを示す。
【0186】
ビオチン化細胞上のビオチンの安定性及び接近可能度
細胞表面上のビオチンの安定性を分析するために、細胞を上述のようにビオチン化した。NHSを用いずにビオチン処理した細胞を対照として使用した。0、2、4、及び7日目に、200μLのSR溶液(Ca/Mg非含有のPBS、pH7.4中50μg/mL)を、ビオチン処理又はBNHS処理細胞のいずれかに室温で20分間添加して、細胞表面上のビオチンの安定性を評価した。SRを付加した後で、PBS中細胞を4%ホルムアルデヒド溶液で固定し、0.1%の100μLトリトンX溶液で処理した後、100μLのDAPI溶液(PBS中1μg/mL)で染色した。BNHS処理及びビオチン処理細胞の両方の蛍光顕微鏡画像を、蛍光強度を測定することにより分析した。ビオチン官能性の安定性を、付加したローダミン−ストレプトアビジンの蛍光強度を分析することにより測定した。
【0187】
細胞表面上のビオチンの安定性は、各時点でローダミン−ストレプトアビジンを付加し、画像の蛍光強度を直ちに測定することにより分析した。7日間にわたるBNHS機能化の安定性を、NHSを用いていないビオチン(対照)と比較して試験した。NHS処理により、7日間にわたって細胞膜上に安定的なビオチン基が提供されることが示された。
【0188】
BNHSで機能化された細胞の蛍光強度は、0日目(機能化の当日)では、B(ビオチン)及びSRと比較してより高く、細胞がNHS官能性により機能化されたことを示す。7日後に強度を再検査し、BNHS+SR処理の強度が、B+SR及びSR対照と比較して著しく高くなったことが見出され、ビオチン化細胞へのローダミン結合が依然として存在したことが示された。代表的な7日間の光学顕微鏡画像及び同じ観察視野の対応する蛍光画像は、細胞が、NHS化学により効果的にビオチン化され、1日目に付加した結合ストレプトアビジンが、7日間の培養の間安定的であったことを示す。
【0189】
細胞接着
細胞接着は、細胞のビオチン化後に組織培養ウエル上の接着細胞数を測定することにより分析した。T25フラスコ中の80%のコンフルエント細胞を、上述のようにビオチン化した。ストレプトアビジン結合の後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、その後5000個の細胞を、96ウエルプレートの各ウエルに10、30、及び90分間播種した。その後、PBSで2回すすぐことにより非接着細胞を除去し、その後接着細胞を固定し、トルイジンブルー溶液で細胞を染色した。接着細胞を、1O×倍率で3つのウエルの6つの観察視野で計数して、接着細胞数を決定した。表面に接着した細胞のパーセントを、細胞の初期播種密度に基づいて計算した。この実験の対照は、未処理細胞を含んでいた。
【0190】
BNHS処理細胞と対照との間の接着細胞パーセントに差異はなく、BNHSによる細胞の修飾は、細胞の接着特徴を変更しないことを示す。修飾細胞は、組織培養プラスチック上でそれぞれ10、30、及び90分間接着させた後の未処理細胞と比較して、著しく異なる接着特徴を示さない。これは、細胞の接着特徴の要因である細胞表面受容体(例えば、インテグリン)が、NHSビオチン細胞表面修飾により影響を受けないことを明白に示す。
【0191】
修飾細胞の増殖アッセイ
細胞増殖アッセイを使用して、T25フラスコ上の細胞数を測定することにより細胞増殖を分析した。T25フラスコ中の80%コンフルエント細胞を、1mLの1mM BNHSで処理し、その後1mLの50μg/mLストレプトアビジン溶液で20分間処理した。その後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、50,000個の細胞を3つのT25フラスコに播種した。細胞数を、3個のフラスコの1O×倍率の10観察視野で、播種後の1日目、2日目、4日目、及び6日目に評価した。BNHSにより修飾された細胞の増殖特徴を示す増殖曲線は、増殖が最終的に、両群においてコンフルエント単層化の原因となること示している。
【0192】
SLeXによる細胞の機能化及びその結果生じる回転の特徴付け
T25フラスコ中の80%コンフルエント細胞を処理することにより、細胞の修飾を実施した。細胞を1mLの1mM BNHSで処理し、その後1mLの50μg/mLストレプトアビジン溶液で20分間処理した。細胞を1mLのPBSで2回洗浄し、その後1mLの4μg/mL SLeX(Ca/Mg非含有PBS、pH7.4中)を、室温で20分間添加した。細胞を、細胞解離溶液を使用してフラスコから分離し、遠心分離後に培地に再分散した。フローチャンバー実験用の細胞濃度は、典型的には1×105細胞/mlだった。細胞の回転特徴は、ガスケットの厚さが127μmで長さが6cmの長方形の平行平板フローチャンバー実験より評価した。ガラス表面上へのP−セレクチン固定化を、700μLのP−セレクチン溶液(5μg/mL)をスライドガラス上で18時間インキュベートすることにより実施し、フロー実験は、スライドガラス上にチャンバーを設置することにより実施した。0.094ダイン/cm2の壁面せん断応力に対応する20μL/分の流速を使用した。細胞の回転をモニターするために位相差顕微鏡法を使用し、画像を1O倍の観察視野で記録し、およそ10秒毎に手動で取り込んだ。細胞の速度は、10秒間わたって移動した細胞の距離を測定することにより計算した。対照は、未処理細胞、及び細胞表面上に物理的に吸着されたSLeXを有する細胞だった。SLx(SLeX)又はBNHS−SLxで修飾した細胞は、速度の低下及び回転特徴の増加を示す。せん断応力及び細胞に付加したBNHSの濃度の影響を評価するため、細胞の回転速度及びフラックスを、複数の流速(それぞれ0.366、0.73、及び1.89ダイン/cm2に対応する20、40、及び100μL/分)で測定した。
【0193】
培養したhMSCは、それらの表面上にリガンドを発現して細胞回転を可能にしないことが明確に記述されており、これらの結果は未処理hMSCで確認されていた。BNHSで修飾しその後SLeXで修飾した細胞は、0.366ダイン/cm2の壁面せん断応力において、未処理細胞、及びSLeXが物理的に細胞表面上に吸着した細胞より著しく低い速度示す。未処理細胞と比較してSLeX固定化の速度が著しくより低いことは、SLeXとP−セレクチン間の相互作用が、0.366ダイン/cm2の壁面せん断応力で、速度を約2μm/sに減少させることを示す。これは、ビオチン−ストレプトアビジン、その後にSLeX−ビオチンによる細胞修飾が、この細胞タイプに本来備わっていないhMSCの回転特徴を誘導することを示す。
【0194】
修飾細胞の回転特徴に対する流速及びしたがって様々なせん断応力の影響を測定した。せん断応力が0.366ダイン/cm2から1.88ダイン/cm2に増加すると共に、修飾細胞の速度は増加するが、フラックス(基質と相互作用する細胞の数)は、ほとんど不変のままである(1mMのBNHS定濃度において)。未修飾細胞は、19.5細胞/mm2秒の一定のフラックスを示した。これは、SLeXで修飾した細胞が、認識された細胞回転に基づく接着現象により、表面上のP−セレクチンと特異的に相互作用することを示す。
【0195】
0.366ダイン/cm2の一定のせん断応力における、修飾細胞の回転特徴に対するBNHS濃度の影響(これは、本質的には細胞表面上の修飾リガンドの密度対照である)も評価した。1mMから0.01mMまでBNHS濃度を減少させると、速度は増加するが、修飾細胞のフラックスはほとんど不変である(及び19.5細胞/mm2秒のフラックスを示した未修飾細胞のフラックスより高い)。これは、細胞表面上のリガンド濃度を変更することにより、回転応答を制御する能力を示す。低濃度のBNHSが使用される場合でさえ、細胞は、フラックスデータで観察されるようにP−セレクチンと相互作用することができる。
【0196】
BNHS結合並びにその後のストレプトアビジン及びSLex結合による細胞修飾を、組織培養プレート上の接着細胞で実施した。この細胞表面修飾では、表面と結合していない細胞の部分のみが修飾され、したがってリガンドと接触して結合し、組織培養プレートに接着している細胞表面は、修飾にさらされない。これは、細胞表面の半分がSLexにより修飾されることの原因となる。したがって、P−セレクチン表面に対するSLex修飾細胞の接着は、回転速度より顕著である。これにより、様々な条件のせん断応力及びBNHS濃度でも、細胞のフラックス値が変わらない理由が説明される。
【0197】
修飾細胞の分化能
ビオチン/ストレプトアビジン結合により修飾した細胞のin vitro骨形成及び脂肪生成分化能を、下記に記載のように評価した。
【0198】
骨形成分化
hMSCを24ウエルプレートの4つのウエルに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。細胞修飾を2段階法により実施した。典型的には、細胞を1mMのBNHSでビオチン化し、その後PBS中のストレプトアビジン(50μg/mL)を室温で結合させた。その後、デキサメタゾン、β−グリセロリン酸、L‐アスコルビン酸−2−ホスフェート、及びα−MEMを含有する骨形成誘導培地(Lonza社製、hMSC骨形成Single Quoteキット)中で23日間細胞を培養することにより、骨形成分化を誘導した。陽性対照として、未処理(未修飾)細胞を有する同数のウエルを骨形成誘導培地中で維持した。両群の培地を3日毎に交換した。骨形成は、アルカリホスファターゼアッセイ及びフォンコッサ染色により評価した。
【0199】
アルカリホスファターゼアッセイは、培地を吸引し、蒸留水で細胞を洗浄することにより実施した。細胞を3.7%ホルムアルデヒド溶液で15分間室温で固定し、その後蒸留水で2回洗浄した。それに、(DMF及びナフトールAS MX−P04)を含有するTris HCl及び蒸留水中0.06%のRed Violet LB食塩溶液を添加した。プレートを45分間インキュベートし、その後ウエルを蒸留水で3回すすいだ。
【0200】
ビオチン及びストレプトアビジンにより修飾した細胞の骨形成分化能を、アルカリホスファターゼ染色により測定した。修飾細胞用のアルカリホスファターゼ染色は、陽性染色を示し、未修飾細胞と同様の結果を示す。これは、細胞のビオチン−ストレプトアビジン修飾が、細胞の骨形成能を妨害せず、修飾細胞が骨形成系統に分化することができることを示す。
【0201】
脂肪生成分化
hMSCを24ウエルプレートの4つのウエルに播種し、100%コンフルエンスに達するまで細胞増殖培地で培養した。細胞修飾を2段階法により実施した。典型的には、細胞を1mMのBNHSでビオチン化し、その後PBS中のストレプトアビジン(50μg/mL)を室温で結合させた。その後、脂肪生成誘導培地(Lonza社製、h−インスリン(組換え)、L−グルタミン、デキサメタゾン、インドメタシン、IBMX(3−イソブチル−1−メチル−キサンチン)、Pen/Strepを含有するhMSC脂肪生成Single Quoteキット)、及び脂肪生成維持培地(Lonza社製、h−インスリン(組換え)L−グルタミン、Pen/Strepを含有するhMSC脂肪生成Single Quoteキット)中で細胞を23日間培養することにより、脂肪生成分化を誘導した。陽性対照として、未処理(未修飾)細胞を有する同数のウエルを、脂肪生成誘導培地中で維持した。誘導培地及び維持培地と周期的に接触させるLonza社のプロトコールに従って、両群の培地を3日毎に交換した。
脂肪生成は、オイルレッドO染色により評価した。
【0202】
使用したオイルレッドO染色プロトコールは以下の通りである:細胞から培地を完全に吸引し、PBSで1回洗浄し、PBSを3.7%ホルムアルデヒドに室温で30分間置換して細胞を固定し、ホルムアルデヒドを蒸留水に数分間置換し、イソプロパノールをオイルレッドO作業用溶液(30mlのオイルレッド0.5%イソプロパノール溶液を20mlの蒸留水で希釈することにより作製)に置換し、5分後にオイルレッドO溶液を蒸留水で2回洗浄した。1mlのヘマトキシリン(Sigma−Aldrich社製)をウエルに1分間添加し、その後吸引し、ウエルを蒸留水で洗浄した。倒立位相差顕微鏡を使用してウエルを観察した。脂質は赤色見え、核は青色に見えた。
【0203】
ビオチン及びストレプトアビジンで修飾した細胞の脂肪生成分化能を、オイルレッドO及びヘマトキシリン染色により評価し、その結果は、修飾細胞が陽性染色を示し、未修飾細胞と同様の結果を示すことを表している。これは、細胞のビオチン−ストレプトアビジン修飾が、細胞の脂肪生成能を妨害せず、修飾細胞が脂肪生成系統に分化することができることを示す。
【0204】
実施例2
本明細書の実施例2に記載の方法は、例えば、幹細胞及び分化細胞を含む任意の細胞タイプの標的送達に使用することができる。特異的抗原を提示する活性化樹状細胞(DC)をリンパ節に標的化して、ワクチン接種戦略を向上させることができる43。加えて、T細胞又は他の免疫細胞の標的送達を、本明細書に記載の方法により実施することができる。薬物又は薬物送達デバイス(例えば、粒子)を細胞表面に封入することは、細胞微小環境を制御するのに、及び薬物を細胞に直接的に送達する(長期間にわたって)のにも有用である。これは、血漿又は他の生物学的実体との相互作用により迅速に除去又は不活化される薬物に特に有用である。徐放性薬物送達は、本明細書に記載されている細胞表面への共有結合性固定化によって、又は非共有結合的方法による組み込みによって達成することができる。
【0205】
この方法は、本明細書で開示されている方法の1つの実施形態を記述しており、ヒト間葉系幹細胞の共有結合的機能化を伴っており、任意の細胞タイプに適用することができる。
【0206】
この実施形態では、細胞機能化は、細胞表面に共有結合で直接結合させずに達成される。このためには、以下のビオチン化脂質を使用することができる:中性、陽イオン性、及び陰イオン性など電荷が異なる様々な頭部基を有する脂質;様々な長さの糖鎖を有する脂質(表1を参照);及び糖鎖の不飽和度が様々である脂質。
【0207】
このプラットフォーム手法は、既存の機能化方法より優れており、以下を含む複数の利点を提供する:i)簡単な調製方法、ii)穏やかな反応条件、iii)高価な/複雑なタンパク質発現ステップの回避、iv)時間の短縮及びキットで使用することができる細胞操作。
【0208】
同じ目標、つまり特異的リガンドによる細胞の機能化を達成するために、2つの異なる手法を使用することができる。1つの手法では、ビオチン化脂質を有する小胞にストレプトアビジンを付加し、ストレプトアビジンでコーティングした細胞にシアリルルイスXを付加する。その後、修飾しようとする細胞の二分子層に小胞を融合させる。第2の手法では、最初に修飾しようとする細胞をビオチン化脂質小胞と融合させて、その後ストレプトアビジン、次にシアリルルイスXを細胞に付加する。両手法の最終産物は本質的に同一である。
【0209】
方法1
方法1では、最初に単層状又は/及び多重層状の小胞を、リン酸緩衝生理食塩水(PBS、pH7.4)又は他の任意の水溶液中で、「ビオチン化脂質」のみ又は「異なる比率で支持脂質を有するビオチン化脂質」のいずれかを使用して製作することができ、それをストレプトアビジン溶液(ビオチンに結合することができる)に添加し、室温で5〜30分間インキュベートし、小胞溶液を10,000rpmで2分間遠心分離し、その後上清を除去する。ペレット(小胞を含有する)をPBSで洗浄して、未結合ストレプトアビジンを除去した。ビオチン化シアリルイスX(biotinylated Sialy Lewis X)(SiLeX)をペレットに添加し、5〜30分間インキュベートし、その後遠心分離及びPBS洗浄ステップを繰り返して小胞を取得し、それらの脂質頭部基をビオチン−ストレプトアビジン−ビオチン−SiLeX基でコーティングする。最後に、小胞を、hMSC及び/又は別の細胞タイプと共に1〜15分間インキュベートした。小胞と細胞膜との融合が生じ、細胞表面をビオチン−ストレプトアビジン−ビオチン−SiLeX基でコーティングすることができ、これらはセレクチン(この場合P−セレクチン)との排他的な相互作用に感受性である。
【0210】
この手法では、以下の方法によりビオチン化脂質を使用して小胞を調製した:a)ストレパタビシン(strepatavidin)溶液をこの混合物と共に5〜30分間インキュベートし、その後遠心分離し、過剰の未結合ストレプトアビジンを除去することにより、表面上にビオチン−ストレプトアビジン機能化を有する小胞の調製が可能になった。B)さらなるビオチン化SiLeXを小胞に添加し、5〜30分間インキュベートし、遠心分離及び過剰(未融合)小胞の除去を実施した。残りの小胞は、表面上にビオチン−ストレプトアビジン−ビオチン−SiLeX基を含有する。C)そのような小胞を、ヒト間葉系幹細胞(hMSC)(又は他の細胞タイプ)と共に1〜15分間インキュベートし、その結果生じた小胞と細胞膜との融合により、ビオチン−ストレプトアビジン−ビオチン−SiLeX基による細胞表面の機能化が引き起こされた。この機能化はセレクチン(この場合P−セレクチン)との排他的な相互作用に感受性である。
【0211】
方法2
方法2では、ビオチン化脂質を使用して小胞を調製し、hMSC(又は他の任意の細胞タイプ)の懸濁物と共にPBS中で1〜15分間インキュベートし、その後細胞を10,000rpmで3分間遠心分離し、上清を除去した。その結果生じたペレットは、ビオチンでコーティングされた細胞を含有する。ビオチンでコーティングされた細胞を、ストレプトアビジン溶液と共に5〜30分間インキュベートし、その後遠心分離して未結合ストレプトアビジンを除去した。これにより、ビオチン−ストレプトアビジン基による細胞表面のコーティングが可能になる。前のステップと同様に、ビオチン化SiLeXと共に細胞を5〜30分間インキュベートし、遠心分離を繰り返し過剰な試薬を除去し、このようにして表面上にビオチン−ストレプトアビジン−ビオチン−SiLeX基を有する細胞を生成する。
【0212】
この手法では、以下のプロトコールによりビオチン化脂質を使用して小胞を調製した:a)hMSC又は他の任意の細胞タイプのいずれかと共に1〜15分間インキュベートし、その後細胞を10,000rpmで3分間遠心分離し、その後上清を除去した。その結果生じたペレットはビオチンでコーティングされた細胞を含有する。b)それらの細胞に、ストレプトアビジン溶液を添加し、5〜30分間インキュベートし、その後未結合ストレプトアビジンを遠心分離で除去した。この方法により、ビオチン−ストレプトアビジン基による細胞表面のコーティングが可能だった。C)ビオチン化SiLeXを添加し、5〜30分間インキュベートし、細胞を遠心分離にかけ過剰な試薬を除去した。この方法は、表面上にビオチン−ストレプトアビジン−ビオチン−SiLeX基を有する細胞の調製の原因となる。同様のステップは、培養プレート上の接着細胞にも実施することができる。
【0213】
所望の小胞特性に依存して、様々な頭部基及び様々な電荷(例えば、陽イオン性、陰イオン性、中性、及び双極イオン性)を有する種々のリン脂質を使用することができる。例えば、ホスファチジルコリン、ホスファチジルセリン、及びホスファチジルエタノールアミンに基づく脂質を、小胞の製作に使用することができる。様々な疎水性鎖、例えばグリセロール骨格に結合された対称性及び非対称性アシル基も、本明細書に記載の方法で使用するために意図されている。表1には、本明細書で意図される非対称性アシル基及び対称性アシル基が列挙されている。
【0214】
この方法は、ビオチン化脂質のみに限定されておらず、むしろ細胞機能化は、任意の「修飾脂質」を用いて実施することができる。所望の官能基/分子/ナノ粒子/ビーズを脂質頭部基に結合させ、その後任意のタイプの細胞表面に挿入することができる。加えて、所望の薬物/分子/成長因子/粒子/ビーズを、機能化脂質により調製された小胞に封入することができ、したがって封入された物質を細胞内に送達し、同時に機能化脂質で表面をコーティングすることができる。
【0215】
実施例3:懸濁状態の細胞の細胞表面機能化
懸濁モードでのシアリルルイスX(SLeX)によるヒト間葉系幹細胞(hMSC)の修飾
懸濁状態のビオチニル−N−ヒドロキシ−スクシンイミドのN−ヒドロキシ−スクシンイミド基を反応させることによるhMSCの共有結合性修飾は、細胞の表面全体を修飾する機会を提供するが、接着モードでは細胞表面のおよそ半分が表面修飾中に露出する。したがって、修飾が懸濁状態の細胞上で実施される場合、ビオチン及び結合部分の分布は、細胞膜全体にわたってより均一である。これにより、細胞の標的化及びホーミング効率を増加させることができる。具体的には、細胞が懸濁状態にある間にSLeXを結合させることにより、接着細胞上で実施された修飾と比較して(試薬が同じ濃度で使用された場合)、よりロバストな回転応答が付与される。
【0216】
典型的には、hMSCを、hMSC増殖培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)中で90%コンフルエンスまでT75フラスコで培養し、その後細胞を、1×トリプシン−EDTA溶液を使用してトリプシン処理し、次にリン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して、培地及びトリプシンを除去した。その後、細胞ペレットを15〜20分間1mLの1mM BNHS溶液に分散した。その後、細胞を遠心分離及び遠心沈降させて、BNHS溶液を除去した。細胞を再懸濁しその後遠心分離することにより、細胞ペレットをPBSで2回洗浄した。遠心分離後、細胞ペレットを、1mLのストレプトアビジン溶液(Ca/Mg非含有PBS、pH7.4中50μg/mL)に15〜20分間再懸濁した。非結合ストレプトアビジンを遠心分離により除去した。
【0217】
BNHS及びストレプトアビジンによる細胞修飾を評価するために、蛍光性ローダミンを結合したストレプトアビジン(SR)を、ビオチン化細胞に付加し、結合したSRの蛍光強度を測定した。この実験に使用した対照は、SRのみで処理した細胞、ビオチン及びSRで処理した細胞、及び未処理細胞だった。蛍光強度を7日後に再び測定し、細胞を4%ホルムアルデヒド溶液で固定し、PBS中0.1%の100μLトリトンX溶液で処理した後で、100μLのDAPI溶液(PBS中1μg/mL)で染色した。報告した結果は全て、三重反復で実施した実験の結果である。修飾の当日(0日目)に、BNHS+SR基は著しくより高い蛍光を示し、蛍光は7日目にさらに増加した(対照SR及びB+SRと比較して)。これは、BNHSが特異的に及び共有結合で細胞表面に結合し、細胞表面の恒久的修飾の原因となることを示す。
【0218】
ビオチン化細胞上のビオチンの安定性及び接近可能度
細胞表面上のビオチンの安定性を分析するために、細胞を上述のようにビオチン化した。NHS非含有のビオチン処理細胞を対照として使用した。細胞を96ウエルプレートに24時間接着させた。指定の期間後、1、2、4、及び7日目に、200μLのSR溶液(Ca/Mg非含有PBS、pH7.4中50μg/mL)を、ビオチン処理細胞又はBNHS処理細胞のいずれかに室温で20分間添加して、細胞表面上のビオチンの安定性を評価した。SRを添加した後、細胞を4%ホルムアルデヒド溶液で固定し、PBS中0.1%の100μLトリトンX溶液で処理した後で、100μLのDAPI溶液(PBS中1μg/mL)で染色した。BNHS処理細胞及びビオチン処理細胞の両蛍光顕微鏡画像を、蛍光強度を測定することにより分析した。ビオチン機能性の安定性を、付加したローダミン−ストレプトアビジンの蛍光強度を分析することにより測定した。7日間にわたるBNHS機能化の安定性を、NHS非含有のビオチン(対照)と比較して試験し、細胞膜上で7日間にわたって安定的なビオチン基が提供された。
【0219】
BNHSにより修飾された細胞の生存率
懸濁状態で修飾した細胞の生存率を、修飾直後(0時間)にトリパンブルーを使用して試験した。結果は、修飾ステップ後で約69%の細胞が生存可能であることを示す。この実験の対照は、ビオチン処理細胞、及びいかなる処理もしないが同様の実験条件下で懸濁及び遠心分離した細胞だった。対照細胞は約80%の生存率を示し、一方でビオチンをコーティングした細胞は約73%の生存率を示した。これは、BNHS修飾が生存率を著しくは減少させないことを示す。生存率が低かったのは、生存率を減少させる修飾工程のステップが原因である。
【0220】
懸濁状態におけるBNHS修飾の効果を評価するため、細胞を懸濁状態で修飾し、同じ対照を共に使用して48時間後に生存率を試験した。BNHS修飾細胞は、48時間後に80%生存可能だった(90%の生存細胞及び85%の生存可能ビオチン対照と比較して)が、それは修飾が細胞に対していかなる実質的な毒性効果も誘導しないことを示す。
【0221】
修飾細胞の接着
細胞をビオチン化した後、96ウエルプレートの細胞培養ウエル上の接着細胞数を測定することにより、細胞接着を分析した。細胞を、上述のように懸濁状態でビオチン化した。ストレプトアビジンを結合した後、細胞を洗浄し、細胞培地に再懸濁した。細胞を血球計で計数し、その後およそ5000個の細胞を、96ウエルプレートの各ウエルに10、30、及び90分間播種した。その後、PBSで2回すすぐことにより非接着細胞を除去し、その後接着細胞を固定し、トルイジンブルー溶液で細胞を染色した。接着細胞を、1O×倍率で3つのウエルの6つの観察視野で計数して、接着細胞数を決定した。
【0222】
懸濁状態で修飾した細胞の接着を、10、30、及び90分直後に試験したところ、修飾細胞の約25%が最初の10分で接着し、その後30分でおよそ40%、及び90分でおよそ68%が接着することが示される。細胞の接着がより低いのは、細胞表面全体にわたる細胞修飾に起因し、そのため細胞は接着分子の一部を失う。同様の実験条件下で懸濁及び遠心分離処理した対照細胞は、10、30、及び90分で、それぞれ約36%、60%、及び75%の接着を示す。これは、細胞の接着特徴がより低いことが、主としてBNHSによる修飾によるのではなく、修飾のステップに依存することを示す。
【0223】
修飾細胞の増殖評価
懸濁状態で修飾した細胞の増殖を、8日間試験した。細胞を懸濁状態でBNHSにより修飾した後、およそ50,000個の細胞を、T25フラスコに入れ、8日間の期間にわたって計数した。増殖曲線は、最終的にコンフルエントな単層化に結びつく増殖を示す。同様の実験条件下で懸濁及び遠心分離処理した対照細胞は、懸濁状態での細胞修飾が、修飾細胞の増殖能を損なわないことを示す。
【0224】
懸濁状態で修飾したhMSCの回転特徴
ビオチン化SLeXを使って懸濁状態で修飾したhMSCは、未修飾細胞(PBS処理)より著しく低い速度を示す。懸濁状態でBNHSにより修飾したhMSCの速度は、0.37ダイン/cm2のせん断速度でフローチャンバーを通って流れ、細胞は0.55μm/秒の速度で回転していた。修飾細胞と比較して、細胞を修飾しなかった対照は、75μm/秒の速度で移動した。これは、懸濁モードでBNHSを使用したビオチン−ストレプトアビジン結合によるSLexの導入は、P−セレクチンでコーティングした表面上におけるより効果的な細胞の回転を誘導することを示す。
【0225】
修飾細胞の回転特徴に対する流速及びしたがって様々なせん断応力の効果を評価した。懸濁状態でBNHSにより修飾したhMSCの速度に対するせん断応力の効果は、せん断応力の増加が速度を増加させることを示す。せん断応力を0.37ダイン/cm2から1.8ダイン/cm2に変化させることにより、0.55μm/秒から1.5μm/秒の速度の増加が観察される。これは、より高いせん断応力レベルでは、修飾細胞が、自然に回転する細胞と同様のより低い速度の回転特徴を示すことを表す。懸濁状態でBNHSにより修飾したhMSCのフラックスに対するせん断応力の影響は、せん断応力の増加がフラックスを減少させることを示す。0.37ダイン/cm2から1.8ダイン/cm2に速度を変化させることにより、155細胞/(mm2×秒)から118細胞/mm2秒へのフラックス減少が観察される(20細胞/(mm2×秒)のフラックスを示す未修飾細胞と比べて)。これは、より高いせん断応力レベルで、修飾細胞がP−セレクチン表面に接着することができることを示す。
【0226】
実施例4:ビオチン−ストレプトアビジン結合を使用したPLGA粒子によるhMSCの修飾
ストレプトアビジン結合PLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。ストレプトアビジンを、標準的なカルボジイミドカップリング技術を使用して、PLGA粒子のカルボン酸基に共有結合で結合させた。PLGA(Strep−PLGA粒子)粒子に共有結合で結合されたストレプトアビジンを3回洗浄して、物理的に吸着したストレプトアビジンをPLGA粒子の表面から除去した。
【0227】
hMSC表面へのStrep−PLGA粒子の結合
hMSCを、PBS中で1mMスルフォ−NHS−ビオチン(BNHS)により室温でビオチン化し、その後ストレプトアビジン結合PLGA(Strep−PLGA)粒子を結合させた。2つの陰性対照をこの実験に使用した。一方の対照セットでは、Strep−PLGAを未修飾細胞に付加し、つまり細胞はビオチン化されていなかった。第2の対照セットでは、細胞はビオチン化されていたが、未修飾PLGA粒子、つまりカルボン酸で末端化されたPLGA粒子を付加した。
【0228】
Strep−PLGA粒子を有する細胞(及び対照)の画像を計数したところ、1個の細胞当たり最も多くの平均粒子を有する細胞は、BNHS/Strep/PGLAを使用して修飾したそれらの細胞だったことが示されている。結果は、Strep−PLGAビーズが、特異的なビオチン−ストレプトアビジン相互作用により、ビオチン化hMSCに結合することを示す。実験群(BNHS+Strep−PLGA)と陰性対照群(Strep−PLGA及びBNHS−PLGA)との間に著しい差異が存在する。非常に多くのStrep−PLGA粒子が、共有結合でビオチン化されたhMSCに結合したが、これは粒子が細胞と特異的に結合することを示す。
【0229】
Strep−PLGA粒子で修飾したhMSCの生存率
細胞の生存率を、トリパンブルー排除法を使用して測定した。手短に言えば、細胞を12ウエルプレートに播種し、終夜放置して接着させ、細胞をBNHSでその後Strep−PLGA粒子で上述のように処理した。異なるウエルで観察された浮遊細胞の数は少なく、群間で同様であった。すすいだ後、200μLの細胞解離溶液を使用して、細胞をウエルから分離した。それに、300μLの培地を添加し、合計500μLの細胞懸濁液をエッペンドルフチューブに収集した。この懸濁液からの10μL細胞懸濁液を、4%トリパンブルー溶液を使用して1:1に希釈し、細胞を血球計で計数して、生存可能な(未染色)及び生存不能な(青染色)細胞数を決定した。三重反復実験の平均として報告した結果を決定したところ、群中の生存率に変化は検出されなかった。この実験の対照には、未処理細胞(しかし、実験中にPBSを添加し、室温で維持した)が含まれていた。48時間後の生存率を試験するため、修飾細胞を、37℃及び5%CO2で48時間インキュベートした。その後、培地を吸引し、細胞解離溶液を使用することにより、細胞をウエルから分離した。生存率を、本明細書に前述されているのと同じように試験した。
【0230】
Strep−PLGA粒子により修飾したhMSCの接着
細胞をビオチン化した後、96ウエルプレートの細胞培養ウエル上の接着細胞数を測定することにより、細胞接着を分析した。T25フラスコ中の80%コンフルエント細胞を、上述のようにビオチン化した。Strep−PLGA結合の後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、およそ5000個の細胞を、96ウエルプレートの各ウエルに10、30、及び90分間播種した。その後、非接着細胞をPBSで2回すすぐことにより除去し、その後接着細胞を固定し、トルイジンブルー溶液で細胞を染色した。接着細胞を、1O×倍率で3つのウエルの6つの観察視野で計数して、接着細胞数を決定した。接着細胞のパーセントを、細胞の初期播種密度に基づいて計算した。この実験の対照は、未処理細胞を含んでいた。
【0231】
Strep−PLGA粒子により修飾したhMSCの増殖アッセイ
細胞の増殖アッセイを、T25フラスコ上の細胞数を測定することにより分析した。T25フラスコ中の80%コンフルエント細胞を、BNHSで処理し、その後Strep−PLGA粒子溶液で20分間処理した。その後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、およそ50,000個の細胞を、3個のT25フラスコに播種した。細胞数を、3個のフラスコの10×倍率の10観察視野で、播種後の1日目、2日目、4日目、6日目、及び8日目に計数し、細胞数を決定した。この実験のための対照は、PBSで処理した細胞である。
【0232】
P−セレクチン表面上でのStrep−PLGA修飾細胞の回転
T25フラスコ中の80%コンフルエント細胞を処理することにより、細胞の修飾を実施した。フラスコをBNHSで処理し、その後strept−PLGA粒子で処理した。その後、細胞をPBSで2回洗浄し、1mLの4μg/mL SLeX(Ca/Mg非含有PBS、pH7.4中)を、室温で20分間添加した。細胞解離溶液を使用して細胞をフラスコから分離し、遠心分離後に培地に再分散した。フローチャンバー実験用の細胞濃度は、典型的には105細胞/mlだった。細胞の回転特徴は、ガスケットの厚さが127μmで長さが6cmの長方形の平行平板フローチャンバー実験より評価した。ガラス表面上のP−セレクチン固定化を、700μLのP−セレクチン溶液(5μg/mL)をスライドガラス上で18時間インキュベートすることにより実施し、フロー実験は、スライドガラス上にチャンバーを設置することにより実施した。0.366ダイン/cm2の壁面せん断応力に対応する20μL/分の流速を使用した。細胞の回転をモニターするために位相差顕微鏡法を使用し、画像を10X観察視野で記録し、およそ10秒毎に手動で取り込んだ。細胞の速度は、10秒間わたって移動した細胞の距離を測定することにより計算した。
【0233】
細胞−粒子結合体の回転特徴を、明視野及び蛍光の両モードで(PLGA粒子は蛍光性だったため)、0.366ダイン/cm2のせん断応力での回転速度及びフラックスを決定することにより測定した。細胞及び/又は粒子結合体の速度は、細胞への粒子結合後で速度がわずかに速いことを示す。これは、粒子が、立体障害により速度の低下を引き起こしていることを示す。しかしながら、strep−PLGA修飾細胞の速度は、未修飾細胞の速度(約70μm/秒)より著しく低く、細胞がP―セレクチンと特異的に相互作用し、したがって回転に基づく接着を示すことを表している。
【0234】
表1は、本明細書に記載の方法に使用することができるアシル基のリストを示す。
【表1】

【0235】
実施例5:非共有結合的方法を使用したPLGA粒子によるhMSCの修飾
負荷電PLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して、負荷電カルボン酸により機能性に末端化されたPLGAから製作した。
【0236】
正荷電PLGA粒子の調製
負荷電PLGA粒子を、過剰濃度(PBS中0.5mg/mL)の正荷電ポリ−L−リジンと共に室温で2時間インキュベートした。PLGA粒子表面の負電荷とポリ‐L‐リジンの正電荷との電荷相互作用は、その結果として粒子表面の過剰正電荷をもたらす。
【0237】
脂質をコーティングしたPLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。ストレプトアビジンを、標準的なカルボジイミドカップリング技術を使用して、PLGA粒子のカルボン酸基に共有結合で結合させた。PLGA(Strep−PLGA粒子)粒子に共有結合で結合されたストレプトアビジンをPBSで3回洗浄して、物理的に吸着したストレプトアビジンをPLGA粒子の表面から除去した。ストレプトアビジン結合PLGA粒子を、ビオチン化脂質、1、2−ジパルミトイル−sn−グリセロ−3−ホスホエタノールアミン−N−(ビオチニル)(ナトリウム塩)溶液(PBS中0.1mg/mL)中で2時間室温でインキュベートし、その後PBSで洗浄して過剰な物理的に吸着した脂質分子を除去した。
【0238】
抗体をコーティングしたPLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。ストレプトアビジンを、標準的なカルボジイミドカップリング技術を使用して、PLGA粒子のカルボン酸基に共有結合で結合させた。PLGA(Strep−PLGA粒子)粒子に共有結合で結合されたストレプトアビジンをPBSで3回洗浄して、物理的に吸着したストレプトアビジンをPLGA粒子の表面から除去した。ストレプトアビジン結合PLGA粒子を、ビオチン化CD90抗体(PBS中0.01mg/mL)溶液中で2時間室温でインキュベートし、その後PBSで洗浄して過剰な物理的に吸着した抗体を除去した。
【0239】
非共有結合によるPLGA粒子のhMSCへの結合
種々の表面特徴を有するPLGA粒子を、組織培養プレート上の接着hMSCに添加し、その後洗浄して非接着粒子を細胞から除去した。特徴付けのため、インキュベーションの4時間、8時間、及び12時間後に、細胞を洗浄し、4%ホルムアルデヒドで固定し、その後ヨウ化プロピジウム(PI)(PBS中10μg/mL)で10分間染色し、PBSで2回洗浄した。PI染色細胞及びDiO封入PLGA粒子の場合は、細胞−粒子結合体を、蛍光顕微鏡を使用して画像化した。粒子と結合した細胞数を、4つの異なる表面特徴について計算し、図2に示されているように、粒子を有する細胞のパーセントとして表す。結果は、どの表面機能性を有する粒子もMSCに結合したことを示しており、正荷電粒子及びCD90抗体をコーティングした粒子は最も効率的に結合したが、脂質でコーティングした粒子及び負荷電粒子は8時間まで結合が最も非効率的だった。脂質でコーティングした粒子の結合は、12時間のインキュベーション後に70%まで増加した。
【0240】
非共有結合的方法によりhMSCに結合されたPLGA粒子の取り込み
hMSC表面に結合されたPLGA粒子の安定性を、レーザー共焦点顕微鏡法で、細胞(赤色、PI染色)粒子(DiO封入、緑色)のZスタック画像を検査することにより視覚化した。共焦点顕微鏡画像を、インキュベーションの4時間、8時間、及び12時間後に取得した。y−z、x−z、及びx−y平面のZスタック共焦点画像を分析して、粒子の内部移行を評価した。観察視野にはあるが細胞と結合していない粒子は考慮しなかった。内部移行のパーセントを、細胞に結合された粒子の合計数及び内部移行された粒子数から計算し、図3Aに示す。CD90抗体でコーティングした粒子は最も効果的に内部移行されたが、脂質でコーティングした粒子は、4時間又は8時間後で内部移行されなかった。脂質でコーティングしたPLGA粒子の内部移行は、12時間のインキュベーション後に80%まで増加した。負荷電粒子及び正荷電粒子の場合、細胞による粒子取り込みは、4時間から8時間までで、それぞれ20%から60%に増加した。これは、細胞表面上で機能化された脂質が8時間まで安定していることを示す。粒径の効果も、図3Bに示されているように粒子の内部移行に影響を及ぼす。結果は、3μmより小さな抗体コーティングPLGA粒子の場合、より大型の粒子と比較して、より高い効率性で内部移行されることを示す。3μmより大きな粒子の50%は12時間で内部移行され、粒径のより大きな粒子は、より短い時間で細胞表面上に安定化されることを示す。
【0241】
培養中にhMSCに結合された負荷電PLGA粒子の分析、及び粒子の安定性に対するトリプシン処理の効果
負荷電PLGA粒子の細胞への結合は、細胞の80%が結合直後に結合粒子を含有しており、細胞の60%がインキュベーションの36時間後に結合粒子を含有することを示す(図4A、4B)。 細胞1個当たりの粒子数がこのように変化するのは、細胞増殖による可能性が高い。負荷電PLGA粒子と共に細胞を36時間インキュベートした後、細胞を、1×トリプシン−EDTA溶液を使用してトリプシン処理し、その後リン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して培地及びトリプシンを除去し、6ウエルプレートに播種した。図4Cは、トリプシン処理後(0日目)及び2日目及び4日目の、負荷電PLGA粒子と結合している細胞のパーセントを示す。これは、トリプシン処理後に粒子が細胞と結合していることを示し、粒子を有する細胞のトリプシン処理は、細胞表面上の粒子の安定性に影響を及ぼさないことを示す。トリプシン処理後に1、2、及び3個以上の粒子と結合していた細胞のパーセントは、図4Dに示されている。
【0242】
hMSCに結合した負荷電PLGA粒子による骨形成分化の誘導
デキサメタゾン(hMSCの骨形成分化因子)をPLGA粒子内に封入した。hMSCを、アスコルビン酸及びβ−グリセリンリン酸の存在下でデキサメタゾン含有PLGA粒子と共に培地中でインキュベートし、21日後にアルカリホスファターゼ及びフォンコッサ染色により、骨形成を評価した。(80%の細胞がデキサメタゾン含有PLGA粒子を含有しており、70%を超える細胞が3個以上の粒子を有していた)。陰性対照群には、増殖培地中にアスコルビン酸及びβ−グリセリンリン酸を有し、デキサメタゾンを含有しないPLGA粒子と結合されたhMSC;増殖培地中にアスコルビン酸及びβ−グリセリンリン酸を有し粒子を含有しないhMSC、及び増殖培地中のhMSCが含まれる。陽性対照には、増殖培地中にデキサメタゾン、アスコルビン酸、及びβ−グリセリンリン酸を有するhMSCが含まれる。実験群、つまりアスコルビン酸及びβ−グリセリンリン酸存在下のデキサメタゾン含有PLGA粒子は、対照群と比較して著しい陽性骨形成染色を示し、PLGA内に封入されたデキサメタゾンが、細胞の骨形成分化を誘導することができることを示す。これは、hMSCに非共有結合的に結合されたPLGA粒子が、粒子内に封入された因子を介して骨形成分化を特異的に誘導することができることを示す。
【0243】
培地を吸引し、蒸留水で細胞を洗浄することにより、アルカリホスファターゼアッセイを実施した。細胞を3.7%ホルムアルデヒド溶液で15分間室温で固定し、その後蒸留水で2回洗浄した。それに、(DMF及びナフトールAS MX−P04)を含有するTris HCl及び蒸留水中の0.06%Red Violet LB塩水を添加した。プレートを45分間インキュベートし、その後ウエルを蒸留水で3回すすいだ。アルカリホスフェートアッセイ(alkaline phosphate assay)後、フォンコッサ染色用に2.5%硝酸銀溶液で、細胞を30分間暗所でインキュベートした。インキュベーション後、ウエルを、蒸留水で3回すすいだ。
【0244】
ヒト間葉系幹細胞(hMSC)に結合された負荷電PLGA粒子のin vivo経内皮遊走
負荷電蛍光性PLGA粒子(DiO封入、平均粒径1〜2μm)を、実験前に90%コンフルエントT75フラスコ中で24時間hMSCに結合させた。細胞をトリプシン処理し、PBSで1回洗浄してトリプシンを除去した。細胞を、DiDで蛍光標識した。典型的には、200μLのPBS中500,000個の細胞(DiO粒子で修飾したDiD標識hMSC)を、尾部静脈に注射した。この実験の対照は、同様の数の未修飾hMSCの注射だった。生体内in vivo共焦点顕微鏡法を使用して、注射の24時間後にZスタック画像を取得し(重複がないことを保証するために)、頭蓋内の骨髄にある細胞を視覚化した。骨髄中の血管を、画像化前に蛍光標識した。粒子を有するhMSCの骨髄内皮を越えた血管外遊出を、赤色蛍光(DiD標識hMSCからの)及び緑色蛍光(DiO封入粒子からの)の共局在化を使用して観察したところ、hMSCに結合された負荷電PLGA粒子が内皮を越えて遊走することができることが示された。
【0245】
実施例6:細胞を直接的にビオチン化し、その後ビオチン−ストレプトアビジン架橋を含有するPLGA粒子を付加することによるhMSCの修飾
ストレプトアビジン結合PLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。ストレプトアビジンを、標準的なカルボジイミドカップリング技術を使用して、PLGA粒子のカルボン酸基に共有結合で結合させた。PLGA(Strep−PLGA粒子)粒子に共有結合で結合されたストレプトアビジンを3回洗浄して、物理的に吸着したストレプトアビジンをPLGA粒子の表面から除去した。
【0246】
hMSC表面へのStrep−PLGA粒子の結合
図5Aに示されているように、hMSCを、PBS中で1mMスルフォ−NHS−ビオチン(BNHS)により室温でビオチン化し、その後ストレプトアビジン結合PLGA(Strep−PLGA)粒子を結合させた。2つの陰性対照をこの実験に使用した。一方の対照セットでは、Strep−PLGAを未修飾細胞に付加し、つまり細胞はビオチン化されなかった。第2の対照セットでは、細胞はビオチン化されていたが、未修飾PLGA粒子、つまりカルボン酸で末端化されたPLGA粒子を付加した。
【0247】
結果(図5A)は、Strep−PLGAビーズが、特異的なビオチン−ストレプトアビジン相互作用により、ビオチン化hMSCに結合することを示す。実験群(BNHS+Strep−PLGA)と陰性対照群(Strep−PLGA及びBNHS−PLGA)との間に著しい差異が存在する。著しく多数のStrep−PLGA粒子が、共有結合でビオチン化されたhMSCに結合した。これは粒子が細胞と特異的に結合することを示す。
【0248】
Strep−PLGA粒子で修飾したhMSCの生存率
細胞の生存率を、トリパンブルー排除法を使用して測定した。手短に言えば、細胞を12ウエルプレートに播種し、終夜放置して、接着させ、細胞を上述のようにBNHSでその後Strep−PLGA粒子で処理した。異なるウエルで観察された浮遊細胞の数は少なく、群間で同様であった。すすいだ後、200μLの細胞解離溶液を使用することにより、細胞をウエルから分離した。それに、300μLの培地を添加し、合計500μLの細胞懸濁液をエッペンドルフチューブに収集した。この懸濁液からの10μL細胞懸濁液を、4%トリパンブルー溶液を使用して1:1に希釈し、細胞を血球計で計数して、生存可能な(未染色)及び生存不能な(青染色)細胞数を決定した。三重反復実験の平均として報告した結果は、図5Bに示されている。この実験の対照には、未処理細胞(しかし、実験中にPBSを添加し、室温で維持した)が含まれていた。48時間後の生存率を試験するため、修飾細胞を、37℃及び5%CO2で48時間インキュベートした。その後、培地を吸引し、細胞解離溶液を使用して、細胞をウエルから分離した。生存率を、本明細書で前述されているのと同じように試験した。
【0249】
Strep−PLGA粒子で修飾したhMSCの接着動力学
細胞のビオチン化後、96ウエルプレートの組織培養ウエル上の接着細胞数を測定することにより、細胞接着を分析した。T25フラスコ中の80%コンフルエント細胞を、上述のようにビオチン化した。Strep−PLGAを結合した後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、およそ5000個の細胞を、96ウエルプレートの各ウエルに10、30、及び90分間播種した。その後、非接着細胞をPBSで2回すすぐことにより除去し、その後接着細胞を固定し、トルイジンブルー溶液で細胞を染色した。接着細胞を、1O×倍率で3つのウエルの6つの観察視野で計数して、接着細胞数を決定した。接着細胞のパーセントを、細胞の初期播種密度に基づいて計算し、結果を図5Cに示す。この実験の対照は、未処理細胞を含んでいた。
【0250】
Strep−PLGA粒子で修飾したhMSCの増殖アッセイ
細胞の増殖アッセイを、T25フラスコ上の細胞数を測定することにより分析した。T25フラスコ中の80%コンフルエント細胞を、BNHSで処理し、その後Strep−PLGA粒子溶液で20分間処理した。細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、およそ50,000個の細胞を、3個のT25フラスコに播種した。細胞数を、1O×倍率で3個のフラスコの10観察視野で、播種後の1日目、2日目、4日目、6日目、及び8日目に計数し、細胞の数を査定した。結果は図5Dに示されている。この実験のための対照は、PBSで処理した細胞である。
【0251】
Strep−PLGA粒子で修飾したhMSCの分化能
Strep−PLGA結合で修飾した細胞のin vitro骨形成及び脂肪生成分化能を、下記に記載のように評価した。
【0252】
骨形成分化
hMSCを24ウエルプレートの4つのウエルに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。細胞修飾を2段階法により実施した。典型的には、細胞を1mM BNHSでビオチン化し、その後strep−PLGA粒子を結合した。その後、デキサメタゾン、β−グリセロリン酸、L‐アスコルビン酸−2−ホスフェート、及びα−MEMを含有する骨形成誘導培地(Lonza社製、hMSC骨形成Single Quoteキット)中で21日間細胞を培養することにより、骨形成分化を誘導した。陽性対照として、未処理(未修飾)細胞を有する同数のウエルを、骨形成誘導培地中で維持した。両群の培地は3日毎に交換した。アルカリホスファターゼアッセイ及びフォンコッサ染色により、骨形成を評価した。
【0253】
培地を吸引し、蒸留水で細胞を洗浄することにより、アルカリホスファターゼアッセイを実施した。細胞を3.7%ホルムアルデヒド溶液で15分間室温で固定し、その後蒸留水で2回洗浄した。それに、(DMF及びナフトールAS MX−P04)を含有するTris HCl及び蒸留水中の0.06%Red Violet LB塩水を添加した。プレートを45分間インキュベートし、その後ウエルを蒸留水で3回すすいだ。
【0254】
strep−PLGA粒子で修飾された細胞の骨形成分化能を、修飾細胞用のアルカリホスファターゼ染色により観察した。粒子で修飾された細胞は、陽性染色を示し、未修飾細胞と同様の結果を示した。これは、細胞の粒子修飾が、細胞の骨形成能を妨害せず、修飾細胞が骨形成系統に分化することができることを示す。
【0255】
脂肪生成分化
hMSCを24ウエルプレートの4つのウエルに播種し、100%コンフルエンスに達するまで細胞増殖培地で培養した。細胞修飾を2段階法により実施した。典型的には、細胞を1mMのBNHSでビオチン化し、その後PBS中のストレプトアビジン(50μg/mL)と室温で結合させた。その後、脂肪生成誘導培地(Lonza社製、h−インスリン(組換え)、L−グルタミン、デキサメタゾン、インドメタシン、IBMX(3−イソブチル−1−メチル−キサンチン)、Pen/Strepを含有するhMSC脂肪生成Single Quoteキット)、及び脂肪生成維持培地(Lonza社製、h−インスリン(組換え)L−グルタミン、Pen/Strepを含有するhMSC脂肪生成Single Quoteキット)中で細胞を23日間培養することにより、脂肪生成分化を誘導した。陽性対照として、未処理(未修飾)細胞を有する同数のウエルを、脂肪生成誘導培地中で維持した。誘導培地及び維持培地と周期的に接触させるLonza社のプロトコールに従って、両群の培地を3日毎に交換した。
脂肪生成は、オイルレッドO染色により評価した。
【0256】
使用したオイルレッドO染色プロトコールは以下の通りである:細胞から培地を完全に吸引し、PBSで1回洗浄し、PBSを3.7%ホルムアルデヒドに室温で30分間置換して細胞を固定し、ホルムアルデヒドを蒸留水で数分間置換し、イソプロパノールをオイルレッドO作業用溶液(30mlの0.5%オイルレッドイソプロパノール溶液を20mlの蒸留水で希釈することにより製作)で置換し、5分後にオイルレッドO溶液を蒸留水で2回洗浄した。1mlのヘマトキシリン(Sigma−Aldrich社製)をウエルに1分間添加し、その後吸引し、ウエルを蒸留水で洗浄した。倒立位相差顕微鏡を使用してウエルを観察した。脂質は赤色に見え、核は青色に見える。
【0257】
Strep−PLGA粒子で修飾した細胞の脂肪生成分化能を、オイルレッドO及びヘマトキシリン染色により観察した。粒子で修飾された細胞は、陽性染色を示し、未修飾細胞と同様の結果を示した。これは、細胞の粒子修飾が、細胞の脂肪生成能を妨害せず、修飾細胞が脂肪生成系統に分化することができることを示す。
【0258】
細胞及び粒子がSLeXで機能化されているStrep−PLGA粒子で修飾したビオチン化細胞の回転
細胞修飾は、BNHSで細胞をビオチン化し、その後ストレプトアビジン溶液の存在下でstrep−PLGA粒子を結合させることにより実施した。その後、strep−PLGA粒子が結合された細胞を処理して、細胞表面上及び粒子表面上の両方にビオチン化SLeXを結合させた。粒子表面上及び細胞表面上にSLeXが存在することにより、P−セレクチン表面上での回転相互作用を誘導することができる。 T25フラスコ中の80%コンフルエント細胞を処理することにより、細胞修飾を実施した。フラスコをBNHSで処理し、その後ストレプトアビジン溶液(Ca/Mg非含有PBS中50μg/mL)の存在下でstrep−PLGA粒子で処理した。その後、細胞をPBSで2回洗浄し、1mLの4μg/mL SLeX(Ca/Mg非含有PBS、pH7.4中)を、室温で20分間添加した。細胞解離溶液を使用して細胞をフラスコから分離し、遠心分離後に培地に再分散した。フローチャンバー実験用の細胞濃度は、典型的には105細胞/mlだった。ガスケットの厚さが127μmで長さが6cmの長方形の平行平板フローチャンバー実験より、細胞の回転特徴を評価した。ガラス表面上へのP−セレクチン固定化を、700μLのP−セレクチン溶液(5μg/mL)をスライドガラス上で18時間インキュベートすることにより実施し、フロー実験は、スライドガラス上にチャンバーを設置することにより実施した。0.366ダイン/cm2の壁面せん断応力に対応する20μL/分の流速を使用した。細胞の回転をモニターするために位相差顕微鏡法を使用して、画像を1O×倍の観察視野で記録し、およそ10秒毎に手動で取り込んだ。細胞の速度は、10秒間わたって移動する細胞の距離を測定することにより計算した。
【0259】
細胞−粒子結合体の回転特徴を、図6に示されているように、明視野及び蛍光の両モードで(PLGA粒子は蛍光性だったため)、0.366ダイン/cm2のせん断応力での回転速度及びフラックスを決定することにより測定した。細胞及び/又は粒子結合体の速度は、粒子の結合が、接着に基づく回転応答を依然として可能にすることを示す。
【0260】
hMSCにしたStrep−PLGA粒子による骨形成分化の誘導
ビオチンストレプトアビジンを介してhMSCに結合したStrep−PLGA粒子は、細胞の運命を特異的に制御することができる。具体的には、デキサメタゾン(hMSCの骨形成分化因子)を、PLGA粒子内に封入した。デキサメタゾン含有PLGA粒子を、カルボジイミドカップリングによりストレプトアビジンに結合させた。hMSCを、1mMスルフォ−NHS−ビオチン(BNHS)によりPBS中で室温でビオチン化し、その後デキサメタゾンを含有するストレプトアビジン結合PLGA(Strep−PLGA)粒子を結合させた。ビオチン−ストレプトアビジンを介してデキサメタゾン含有PLGA粒子と結合したhMSCを、培地中のアスコルビン酸及びβ−グリセリンリン酸の存在下で培養し、アルカリホスファターゼ及びフォンコッサ染色により、骨形成分化を21日後に評価した。陰性対照群には、培地中にアスコルビン酸及びβ−グリセリンリン酸を有しデキサメタゾンを含有しないPLGA粒子と結合されたhMSC;培地中にアスコルビン酸及びβ−グリセリンリン酸を有するhMSC、及び増殖培地中のhMSCが含まれる。陽性対照には、培地中にデキサメタゾン、アスコルビン酸、及びβ−グリセリンリン酸を有するhMSCが含まれる。実験群、つまりアスコルビン酸及びβ−グリセリンリン酸存在下のデキサメタゾン含有PLGA粒子を、対照と比較して著しい陽性骨形成染色を示し、PLGA内に封入されたデキサメタゾンが、細胞の骨形成分化を誘導することができることを示す。これは、ビオチン−ストレプトアビジンを介してhMSCと共有結合的に結合されたPLGA粒子が、粒子内に封入された因子を介して細胞の骨形成分化を特異的に誘導することができることを示す。
【0261】
実施例7:N−ヒドロキシスクシンイミド(NHS)官能基及びPEGリンカーと結合したNHS基を使用したPLGA粒子によるhMSCの修飾
PEGリンカーを有する及び有しないNHS官能化PLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。PLGA粒子のカルボン酸官能基を、1−エチル−3−(3−ジメチルアミノプロピル)−カルボジイミド(EDC)及びNHSカップリングにより、N−ヒドロキシスクシンイミド(NHS)基に変換した。NHS官能化PLGA粒子を、二官能性ポリエチレングリコール(カルボン酸及び第一級アミンで官能化、MW:7500)とNHS官能化PLGA粒子のNHS基との反応によりPEGに結合させた。PEGのカルボキシ末端基を、EDC及びNHSでさらに官能化した。
【0262】
PEGリンカーを有する及び有しないNHS官能化PLGA粒子の結合
官能化された粒子をhMSCと共にインキュベーションすることにより、PEGリンカーを有する及び有しないNHS官能化PLGA粒子にhMSCを結合させた。図7は、NHS基は細胞表面と反応して粒子を結合させるが、いかなる官能基も有していないPLGA粒子は細胞に結合しないことを示す。これは、細胞表面官能性(細胞表面上のアミン基)と粒子の官能基(つまり、PEGリンカーを有する及び有しないNHS基)間の共有結合反応を使用して、細胞表面上に粒子を結合させることができることを示す。PLGA粒子上のPEGリンカーの数は、二官能性ポリエチレングリコール(カルボン酸及び第一級アミンで官能化)とHNS官能化PLGA粒子のNHS基との反応中に、二官能性PEG分子の濃度を変更することにより変化させることができる。具体的には、NHS活性化PLGA粒子を官能化するために、1、0.1、及び0.01mg/mLのPEG溶液を使用した。図7の結果は、PEGリンカーの濃度を変化させることにより、細胞に結合するPLGA粒子(PEGにより官能化され、その後NHSで活性化)の数を変化させることができることを示す。図7は、PEGリンカー濃度が増加すると共に、より多数の粒子が細胞に結合することを示す。
【0263】
PEGリンカーを有するNHSを介してhMSCに結合したPLGA粒子による骨形成分化の誘導
PEGリンカーを有するNHS基を介してhMSCに結合したPLGA粒子は、細胞の運命を特異的に制御することができる。具体的には、デキサメタゾン(hMSCの骨形成分化成分)を、PLGA粒子内に封入した。デキサメタゾン含有PLGA粒子を、カルボジイミドカップリングによりPEG及びNHSで機能化した。デキサメタゾン含有PLGA粒子に結合したhMSCを、培地中のアスコルビン酸及びβ−グリセリンリン酸の存在下で培養し、アルカリホスファターゼ及びフォンコッサ染色により、骨形成分化を21日後に評価した。陰性対照群には、培地中にアスコルビン酸及びβ−グリセリンリン酸を有しデキサメタゾンを含有しないPLGA粒子と結合されたhMSC;培地中にアスコルビン酸及びβ−グリセリンリン酸を有するhMSC、及び増殖培地中のhMSCが含まれる。陽性対照には、培地中にデキサメタゾン、アスコルビン酸、及びβ−グリセリンリン酸を有するhMSCが含まれる。実験群、つまりアスコルビン酸及びβ−グリセリンリン酸存在下のデキサメタゾン含有PLGA粒子は、対照と比較して著しい陽性骨形成染色を示し、PLGA内に封入されたデキサメタゾンが、細胞の骨形成分化を誘導することができることを示す。これは、PEGリンカーを有するNHS官能性を介してhMSCに共有結合的に結合されたPLGA粒子が、粒子内に封入された因子を介して細胞の骨形成分化を特異的に誘導することができることを示す。
【0264】
実施例8:シアリルルイスX(SLeX)で修飾したヒト間葉系幹細胞(hMSC)を用いたin vivo実験
ビオチン及びストレプトアビジンを介してSLeXで修飾したhMSCを、尾部静脈注射によりマウスに注射した。手短に言えば、hMSCを、1mLの1mM BNHS溶液で処理し、その後1mLの50μg/mLストレプトアビジン溶液、及び1mLの5μg/mLビオチン化SLeXにより室温で処理した。注射後に細胞を画像化するため、修飾細胞をDiDで蛍光標識した。典型的には、200μLのPBS中の500,000個の細胞(SLeXで修飾したDiD標識hMSC)を、尾部静脈に注射した。この実験の対照は、同様の数を注射した未修飾hMSC。生体内in vivo共焦点顕微鏡法を使用して、注射の2時間及び24時間後に、頭蓋内の骨髄にある細胞を視覚化した。骨髄中の血管を、画像化前に蛍光標識した。図8Aは、3つの別々の実験、#1、2、3において細胞を尾部静脈内注射した2時間後に骨髄に局在化した、未修飾及び修飾MSC(MSCは、ビオチン−N−ヒドロキシスクシイミドで修飾され、その後ストレプトアビジン及びビオチン化シアリルルイスX、SLeXで修飾されている)の数を示す。未修飾MSCと比較してより多数の修飾MSCが、注射の2時間後に骨髄に局在化した。図8Bは、注射の24時間後に骨髄内皮を越えて遊走した血管外遊出MSCの数を示す。24時間後の経内皮遊走能力に修飾MSC及び未修飾MSC間の差異はなかった。これは、細胞表面の共有結合的修飾及びビオチン−ストレプトアビジン架橋を介したSLeXの結合が、MSCの遊出能力を損なわないことを示す。
【0265】
実施例9:ストレプトアビジン−ビオチン架橋を介してシアリルルイスX(SLeX)で共有結合的に修飾されたヒト間葉系幹細胞(hMSC)のパラクリン因子分泌の検査
hMSCを、1mLの1mM BNHS溶液で処理し、その後1mlの50μg/mLストレプトアビジン溶液、及び1mLの5μg/mLビオチン化SLeXで処理することにより室温で修飾した。修飾後、細胞増殖培地を有する24ウエルプレートに修飾細胞を播種し、37℃で24時間インキュベートした。ELISAアッセイを実施して、細胞培養上清中のSDF−1、IGF−1、及びPGE−2の発現レベルを検査した。この実験の対照には、未修飾hMSCが含まれている。図9は、SLeXによるhMSCの修飾が、24時間後に未修飾細胞と比較して修飾hMSCにより分泌されたパラクリン因子レベルを変化させないことを示す。IGF−1及びPGE2は、15%血清を含有する増殖培地で検出可能ではなかったが、SDF−1は、24時間後にMSCから観察された約5%の量で検出された。
【0266】
実施例10:P−セレクチン抗体(Ab)によるヒト間葉系幹細胞(hMSC)の修飾
典型的には、hMSCを、hMSC増殖培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)中で90%コンフルエンスまでT75フラスコ中で培養し、その後細胞を、1×トリプシン−EDTA溶液を使用してトリプシン処理し、次にリン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して、培地及びトリプシンを除去した。その後、細胞ペレットを1mLの1mM BNHS溶液に分散した。その後、細胞を遠心分離及び遠心沈降させて、BNHS溶液を除去した。細胞を再懸濁しその後遠心分離することにより、細胞ペレットをPBSで2回洗浄した。遠心分離後、細胞ペレットを、1mLのストレプトアビジン溶液(Ca/Mg非含有PBS、pH7.4中50μg/mL)に再懸濁した。非結合ストレプトアビジンを遠心分離により除去した。細胞ペレットを、1mLのビオチン化P−セレクチン抗体溶液(Ca/Mg非含有PBS、pH7.4中5μg/mL)に再懸濁した。非結合ストレプトアビジンを遠心分離により除去した。
【0267】
ビオチン化P−セレクチン抗体により懸濁状態で修飾されたhMSCは、修飾細胞が、フローチャンバーアッセイにおいて、P−セレクチンをコーティングした基質と相互作用することを示す。未修飾細胞は、10ダイン/cm2まで、フローチャンバーにおいて回転相互作用又は強力な接着のいずれかを示したが、未修飾細胞は相互作用を示さなかった。これは、P−セレクチン抗体結合hMSCが、フロー条件下でP−セレクチンと特異的に相互作用することを示す(図10)。回転相互作用を示したP−セレクチン抗体修飾細胞の速度は、3μm/秒の平均速度でP−セレクチン表面上を回転することができた。
【0268】
実施例11:ビオチン−HABA−アビジンアッセイによる、ビオチン化ヒト間葉系幹細胞(hMSC)表面上のビオチンの定量化
ビオチン化hMSC(BNHSによるビオチン化)の表面に存在するビオチン部分の数を、製造業者のプロトコール(Thermo scientific社、米国インディアナ州)に従ってビオチン−HABA−アビジンアッセイを使用することにより定量化した。具体的には、2つの技術、接着モード及び懸濁モードを使用して細胞をビオチン化した。接着モードでは、T25フラスコ上の80〜90%コンフルエントのhMSCの単層を、1mLの1mM BNHSで10分間処理し、その後PBSで3回洗浄した。その後、1mLの非酵素性細胞解離溶液を使用して、細胞をプレートから分離し、ビオチン−HABA−アビジンアッセイのプロトコールに従って分析し、細胞上に存在するビオチンリガンドの数を定量化した。懸濁モードでは、hMSC増殖培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)中で90%コンフルエンスまでのT75フラスコを、1×トリプシン−EDTA溶液を使用してトリプシン処理し、その後リン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して、培地及びトリプシンを除去した。その後、細胞ペレットを1mLの1mM BNHS溶液に分散した。その後、細胞を遠心分離及び遠心沈降させて、BNHS溶液を除去した。細胞を再懸濁しその後遠心分離することにより、細胞ペレットをPBSで2回洗浄し、ビオチン−HABA−アビジンアッセイのプロトコールに従って分析して、細胞上に存在するビオチンリガンドの数を定量化した。図11は、接着モードで修飾したhMSCの表面に存在するビオチンの数が、懸濁モードで修飾したhMSCより少ないことを示す。これは、接着モードでは細胞表面の一部が培養皿に付着しているため、細胞表面の一部だけが修飾を受けるが、懸濁モードでは細胞表面全体が修飾に露出していることを示す。
【0269】
実施例12:接着モードでの自己集合ファイバーによるhMSCの修飾
色素封入自己集合ファイバーの調製
典型的には、4〜8mgの小分子両親媒性物質(サリシンデカノアート)をねじ蓋ガラスバイアルに取り、その中に180μLのPBS及び20μLのDMSO中DiD(色素)を添加し、蓋をしたバイアルを、物質が融解するまで60〜80℃まで加熱し、その後室温に静置して冷却させた。バイアルが室温に達した時、試料はヒドロゲルを形成していた。その後、ヒドロゲルを300μLのPBSを使用して希釈し、エッペンドルフに移し、10,000rpmで7分間遠心分離した。上清を除去して未結合色素を洗浄し、ファイバーを1mLのPBSに再分散した。
【0270】
色素封入自己集合ファイバーのhMSCへの負荷
hMSCを24ウエルプレートの4つのウエルに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。色素封入自己集合ファイバーを、組織培養プレート上の接着hMSCに添加し、30分間インキュベートし、その後洗浄して細胞から非接着自己集合ファイバーを除去した。DiDが赤色発光を示す蛍光顕微鏡法を使用して、細胞−ファイバー結合体を画像化した。細胞上の色素を定量化することにより、細胞に結合したファイバーの推定量がもたらされるだろう。対照実験(未修飾細胞)で測定したバックグラウンド蛍光を、ファイバー結合細胞の蛍光強度から差し引き、値を図12にプロットした。同様に、蛍光強度を、様々な時点:0日目、1日後、及び2日後に評価した(図12)。結果は、蛍光強度が2日後でさえ著しくは減少しなかったことを示す。
【0271】
別の対照として、遊離色素を自己集合ファイバーに封入せずに使用して、色素が非特異的に細胞に結合した可能性を除外した。これらの実験では、自己集合ファイバーを使用した以外は、同様の手順を本明細書に記載のように使用した。結果は図12に要約されている。
【0272】
実施例13:懸濁モードでの自己集合ファイバーによるhMSCの修飾
色素封入自己集合ファイバーのhMSC上への負荷
懸濁モードでは、hMSC増殖培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)中で90%コンフルエンスまでのT75フラスコを、1×トリプシン−EDTA溶液を使用してトリプシン処理し、その後リン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して、培地及びトリプシンを除去した。その後、細胞ペレットを、1mLの色素封入自己集合ファイバー含有PBSに分散し、室温で30分間インキュベートした。その後、細胞を遠心分離及び遠心沈降させて非結合ファイバーを除去した。しかしながら、残留量のファイバーが、細胞と共に遠心沈降された。細胞を再懸濁しその後遠心分離することにより、細胞ペレットをPBSで2回洗浄し、最後に、細胞を24ウエルプレートに播種した。細胞が培養フラスコに接着した後(3時間)、プレートをPBSで3回洗浄して残留未結合ファイバーを除去した。DiDが赤色発光を示す蛍光顕微鏡法を使用して、細胞−ファイバー結合体を画像化した。細胞上の色素を定量化することにより、細胞に結合したファイバーの推定量がもたらされるだろう。対照実験(未修飾細胞)で測定したバックグラウンド蛍光を、ファイバー結合細胞の蛍光強度から差し引き、値を図13にプロットした。同様に、本発明者らは、様々な時点:0日目、1日後、及び2日後の蛍光強度を追跡した、(図13)。結果は、蛍光強度が2日後でさえ著しくは減少しなかったことを示す。
【0273】
別の対照として、遊離色素を自己集合ファイバーに封入せずに使用して、色素が非特異的に細胞上に結合した可能性を除外した。これらの実験では、本発明者らは、自己集合ファイバーを使用した以外は、00026に記載されている同様の手順を使用した。結果は図13に要約されている。
【0274】
実施例14:ビオチン化小胞によるhMSCの修飾
ビオチン化脂質を使用した小胞の調製
1,2−ジオレオイル−sn−グリセロ−3−ホスホエタノールアミン−N−(ビオチニル)ナトリウム塩のクロロホルム溶液(1mgの脂質)を、ガラスバイアルに取り、クロロホルムをゆっくりと高真空で蒸発させて無水薄膜を製作した。薄膜を、5mLのPBSで24時間水和させ、その後5分間超音波処理し、氷中で5分間冷却した。同様に、凍結及び解凍を3回繰り返して小胞を生成した。その結果生じた小胞を透過型電子顕微鏡で検査し、小胞が、形状が球状であり、性質が単層であることを見出した。
【0275】
ビオチン化小胞によるhMSCの修飾
hMSCを96ウエルプレートの細胞培養ウエルに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。0.5mLの小胞溶液を、組織培養プレート上の接着hMSCに添加し、30分間インキュベートし、その後洗浄して過剰な小胞/脂質を除去した。ウエルを300μLの培地で3回すすいで、過剰な小胞/脂質を除去した。その後、ローダミン−ストレプトアビジン溶液(Ca/Mg非含有PBS中50μg/mL)を添加し、5分間インキュベートした。その後、細胞をPBSで2回洗浄し、未結合ローダミン−ストレプトアビジンを除去した。ローダミン−ストレプトアビジンが赤色蛍光を示す蛍光顕微鏡法を使用して、細胞を画像化した。対照実験として、上述した同様の手順(小胞溶液の代りにビオチンのみを使用した以外は)を使用して、hMSCをビオチンのみ(脂質分子/小胞形態を用いずに)で処理した。結果は図14Aに要約されている。結果は、ビオチン化脂質が7日目まで細胞と結合したままであることを示す(図14A)。
【0276】
hMSC表面上のビオチン−脂質の接近可能度
小胞で細胞を修飾した後の細胞表面上のビオチン化脂質/小胞の接近可能度を特徴付けるため、200μLのSR溶液(Ca/Mg非含有PBS、pH7.4中50μg/mL)を、0、2、4、及び7日後に小胞修飾細胞に室温で20分間添加した。過剰なストレプトアビジンを除去した後、蛍光強度を定量化した(図14B)。結果は、脂質を細胞挿入2日後で、ビオチン化脂質の一部が、ストレプトアビジンをさらに結合させるために接近可能だったことを示す。
【0277】
実施例15:間葉系幹細胞回転応答の誘導
物質及び方法:
物質
同意を得た健康なドナーのヒト骨髄から単離した初代ヒトMSCを、チューレーン大学の遺伝子治療センターから取得した。P−セレクチンは、R&D systems社(ミネアポリス、米国ミネソタ州)から購入し、ビオチン化シアリルルイス(x)−ポリ(アクリルアミド)(シアリル−ルイスX−PAA−ビオチン、BSLeX)はGlycotech社(ゲーサーズバーグ、米国メリーランド州)から購入した。α−MEM、L−グルタミン、及びPenn−Strepは、Invitrogen社から購入した。スルホン化ビオチニル−N−ヒドロキシ−スクシンイミド、BNHSは、Thermo Fisher Scientific社(Piercenet社製、ロックフォード、米国イリノイ州)から購入し、ウシ胎仔血清はAtlanta Biologicals社から購入した。ビオチン−4−フルオレセインは、Molecular Probes社(ユージーン、米国オレゴン州)から購入した。抗ヒト皮膚リンパ球抗原抗体(HECA−452)、二次抗体(FITCマウス抗ラットIgM)、FITC CD90及びPE−Cy5抗体は、BD Biosciences社から購入した。FACS緩衝液は、1%FBSを有するPBSである。他の化学薬品及び試薬は全て、Sigma Aldrich社(セントルイス、米国ミズーリ州)から購入し、指定が無い限りさらなる精製をせずに使用した。
【0278】
間葉系幹細胞の培養
初代ヒトMSCを、15%ウシ胎仔血清(これがMSCを増殖させる能力を持つため選択した)、1%(容積/容積)のL−グルタミン、1%(容積/容積)Penn−Strep、及びα−MEMで構成される増殖培地中で維持した。実験は全て、継代数4〜6のMSCを使用して実施し、フローサイトメトリー分析で観察したところ、細胞は高レベルのMSCマーカーCD90及びCD29を発現し(99%を超える細胞)、造血性マーカーCD34又はCD45を発現しなかった(0%の細胞)(結果は非表示)。
【0279】
回転リガンドによるMSCの表面修飾
ビオチン−ストレプトアビジンを介したMSC表面へのBSLeX結合を、リン酸緩衝液生理食塩水(PBS、pH7.4、Ca/Mg非含有)中で室温で実施した。典型的には、80〜90%コンフルエントT75フラスコから培地を吸引し、1×トリプシン−EDTA溶液を使用して細胞をトリプシン処理し、遠心分離してペレットにし、PBSで2回洗浄した。その結果生じた細胞ペレットを、スルホン化ビオチニル−N−ヒドロキシ−スクシンイミド、BNHS溶液(1mM、1mL)に分散し、それを1、5、又は10分間室温でインキュベートさせた(反応ステップ1)。その後、細胞をペレットにし、遠心分離によりPBSで2回洗浄して、未結合の及び/又は物理的に吸着したBNHSを細胞表面から除去した。その後、ストレプトアビジン溶液(PBS中50μg/mL、1mL)を使用して、細胞を1、5、又は10分間室温で処理した(反応ステップ2)。その後、細胞をペレットにし、PBSで洗浄した。ストレプトアビジン結合細胞に、BSLeX溶液(PBS中5μg/mL、1mL)を添加し、上清を5、10、又は60分間室温でインキュベートさせた(反応ステップ3)。最後に、細胞をペレットにし、PBSで洗浄した。
【0280】
フローサイトメトリー
フローサイトメトリー分析を、空冷式15mWの488nmアルゴンイオンレーザー、並びに530nm、585nm、及び661nmバンドパスフィルターを備えた光電子増倍管を装備したBD FACSCaliburアナライザー(BD Biosciences社製、サンホセ、カリフォルニア州、アメリカ合衆国)を使用して実施した。データは、ソフトウェアBD CellQuest及びWinMDIを使用して分析した。生存細胞を選択し、蛍光をヒストグラムで表示した(検査した細胞数対相対蛍光強度の対数)。1測定当たり1万事象を記録した。PBSで処理した細胞を使用して、バックグラウンド蛍光を正規化した。
【0281】
フローサイトメトリーを使用した細胞修飾の分析及び向上
色素と結合した試薬(つまり、ビオチン−4−フルオレセイン)をビオチン−ストレプトアビジン架橋用の代表的なビオチン化リガンドとして使用して、フローサイトメトリーにより、化学的結合効率をモニターした。インキュベーション時間を変えることにより結合工程を至適化して、各反応ステップにおける最大カップリング効率(つまり、部位密度)を決定した(表1)。ステップ3におけるビオチン−4−フルオレセインのインキュベーション時間が、結合効率にどのような影響を与えるかを最初に検討した。対照には、ステップ1及び2のPBSインキュベーション後の吸着ビオチン−4−フルオレセインが含まれていた。ステップ3における効果的な反応条件を決定した後、ステップ2におけるストレプトアビジン用のインキュベーション時間を変化させることにより、フルオレセインシグナルがどのように影響をうけるかを次に検討した。対照には、実験群で使用したのと同じステップ3がその後に続く吸着ストレプトアビジン(BNHS無し)が含まれていた。ステップ2の為の効果的な反応条件を決定した後、最後に、ステップ1におけるBNHSの反応時間を検討した。
【0282】
表2:カップリング効率を評価するために使用した実験条件。
【表2】
【0283】
部位密度の決定
MSC表面上のビオチン化SLeXリガンドの部位密度を、Bangs Laboratories社製(フィッシャーズ、米国インディアナ州)のQuantum(商標)Simply Cellular(登録商標)キットを指示通りに使用して決定した。(マウス抗体)結合部位の濃度が明確であるビーズを使用して、記録した蛍光を抗体数に関連付ける検量線を作成した。精製ラット抗ヒト皮膚リンパ球抗原抗体(HECA−452)及び二次抗体(FITCマウス抗ラットIgM)を使用して、細胞表面上に固定化されたSLeXを検出した。未修飾のPBS処理細胞を、一次及び二次抗体と共にインキュベートし、それを対照として使用した。この対照群からの抗体結合能(ABC)値を修飾細胞の値から差し引いて、データを正規化した。3つの細胞試料を、BNHSで10分間、ストレプトアビジンで1分間、及びBSLeXで4分間のインキュベーションを含んでいた上述の至適化修飾反応を用いて修飾した。SLeX部位密度を、HECA−452抗体に対するSLeXの結合比率が1対1であり、一次抗体と二次抗体との結合比率が1対1であると仮定して決定した。MSCの直径は、20μLの細胞溶液(細胞増殖培地中)を用いて、自動細胞計数器Cellometer(登録商標)Auto(Nexcelom Biosciences社製、米国マサチューセッツ州)により、使い捨ての血球計算板で測定した。MSCの平均細胞直径を、Cellometer(登録商標)AUTOのソフトウェア分析から取得した。
【0284】
細胞表面上に共有結合で結合したビオチンの安定性及び接近可能度
NHS化学により細胞表面に結合したビオチンの潜在的な安定性及び接近可能度を特徴付けるために、経時的な蛍光シグナルを検査して、ローダミン結合ストレプトアビジン(SR)のBNHS修飾細胞への結合を追跡した。PBS処理細胞又はビオチン処理細胞とのSRインキュベーションを、対照として使用した。SRと共にインキュベーションした後で、細胞をホルムアルデヒド溶液(PBS中4%)で固定し、トリトンX溶液(PBS中0.1%、100μL)で処理し、DAPI(PBS中1μg/mL、100μL)で染色した。ニコン社製定量ソフトウェア(NIS−Elements バージョン3)を使用して個々の細胞の強度を検査することにより顕微鏡画像の蛍光強度を分析し、データを細胞の単位面積当たりの蛍光強度として表した。
【0285】
生存率、接着動力学、及び増殖分析
BSLeX修飾MSC及び対照PBS処理MSCの生存率、接着動力学、及び増殖を検査した。手短に言えば、細胞の生存率を、修飾直後(時点0)に、及び細胞を6ウエルプレート内で48時間インキュベートした後で、トリパンブルー排除法アッセイを使用して検査した。細胞接着動力学を、10、30、90、及び150分後に組織培養表面上の接着細胞数を測定することにより定量化した。増殖を、T25フラスコ内の細胞数を、10倍の光学顕微鏡を用いて10のランダム観察視野について7日間の間計数することにより定量化した。
【0286】
細胞の分化
BSLeX修飾細胞及びPBS処理細胞の多系統分化能を、骨形成誘導培地及び脂肪生成誘導培地で細胞をインキュベートし、その後それぞれの比色定量組織学的染色を行うことにより検査した。それぞれ細胞膜結合アルカリホスファターゼ活性及びオイルレッドO染色を使用して、細胞を骨形成分化及び脂肪生成分化について分析した。
【0287】
パラクリン因子の分泌:SDF−1、IGF−1、及びPGE−2の定量化
酵素免疫吸着測定法(ELISA)キット(R&D systems社製、米国ミネソタ州)を使用して、BSLeX修飾MSC又はPBS処理MSCによる間質細胞由来増殖因子−1(SDF−1)、インスリン様増殖因子−1(IGF−1)、及びプロスタグランジンE2(PGE2)の産生を定量化した。48ウエルプレートのウエル内に播種した1×104個の細胞の培養から、細胞上清を収集した。標準的ELISAプロトコール(製造業者による)を、500μLのMSC増殖培地を用いて37℃及び5%CO2で細胞を24時間インキュベーションした後で、培養液上清中のSDF−1、IGF−1、及びPGE2を測定するために使用した。試料は全て、2つの独立実験で重複して実行した。15%血清を含有する増殖培地中のパラクリン因子のレベルも検査した。
【0288】
化学的修飾後のMSC表面マーカー発現の検査
MSC表面マーカーの発現に対する化学的修飾の効果を、時点0(修飾直後)及び24時間後のCD90、CD29、及びCD49dの抗体染色のフローサイトメトリーによって検査した。CD90及びCD29はMSCのマーカーであり、CD49d(VLA−4)はMSCの表面上のホーミングリガンドであると考えられている31。化学的に修飾されたMSC用の表面マーカー発現の変化を、未修飾(PBS処理)細胞に対する相対蛍光で表した。8つのT−150フラスコの細胞をトリプシン処理し、混合し、6群に分割して全群の抗原密度が同じであることを保証した。3群を上述の至適化条件で処理し、対照には、PBSのみで処理した(全ステップで)3群の細胞が含まれていた。修飾直後に、各群からの100,000個の細胞を100μLのFACS緩衝液に再懸濁し、1μLのCD90、4μLのCD29、又は4μLのCD49d抗体溶液で20分間氷上で染色した。FACS緩衝液で1回洗浄した後、細胞を300μlのFACS緩衝液に再懸濁し、フローサイトメトリーで分析した。BNHS又はPBSで(しかし抗体によってではなく)処理した細胞を播種し、増殖培地中で24時間インキュベートした。その後、細胞をトリプシン処理し、各群から100,000個の細胞を収集し、上述したのと同じプロトコール後に抗体で染色した。
【0289】
P−セレクチン表面の調製
6ウエルプレート内のウエル表面を、P−セレクチン溶液(PBS中5μg/mL、1mL)により、室温で18時間プレート振とう器上でコーティングした。P−セレクチン表面は全て、フローチャンバーアッセイの前に新たに調製した。
【0290】
フローチャンバーアッセイ
フローチャンバーによる細胞速度の分析の場合、細胞を、フローチャンバーアッセイ用のMSC増殖培地に懸濁した(約105細胞/mL)。ガスケット厚さが127μmで幅が2.5mmの円形平行平板フローチャンバー(Glycotech社製、ゲーサーズバーグ、米国メリーランド州)を使用した。細胞回転をモニターするために、位相差顕微鏡(DS−Qil Monochrome Cooledデジタルカメラを備えたニコン社製TE2000−U型倒立顕微鏡)を使用し、画像を10×倍率の観察視野で10秒間隔で記録した。細胞の速度は、10秒間隔内に移動した細胞の距離を測定することにより計算した。細胞は、観察視野にある間に10秒間回転した場合、及び非相互作用細胞の自由流動速度の計算値の50%未満の平均速度で移動した場合、回転として分類した。フラックスは、基質と相互作用し観察視野に10秒間留まる細胞の数に基づいて、手作業で計算した。強力に接着した細胞及び回転細胞を両方とも、フラックス計算用に考慮した。せん断速度の影響を評価するために、回転速度及びフラックスを、0.36、0.72、及び1.89ダイン/cm2を含むせん断応力で測定した。
【0291】
統計分析:
多重対比較の場合、両側スチューデントt検定を、ボンフェローニ補正と共に使用した。
【0292】
結果:
細胞表面上のビオチンの安定性及び接近可能度
細胞培養中でMSC上に固定化したビオチン及びローダミン結合ストレプトアビジン(SR)の安定性及び接近可能度を検査するために、BNHS及びSR処理の直後(0日目)及びBNHS及びSR処理の7日後に、SRの蛍光シグナルを検査した。BNHSで共有結合的に修飾した細胞は、修飾後の0日目及び7日目に、対照(SRのみ(SR)と共にインキュベートしたMSC、及びビオチン(B)及びSR(B+SR)で処理したMSC)と比較して、有意により高いローダミンシグナルを示す。これは、MSC上に共有結合で結合したビオチンが、少なくとも7日間安定していたことを示唆する。したがって、MSC表面上に共有結合で結合したビオチンの経時的な安定性及び接近可能度を、BNHS処理後の1、2、4、及び7日間遅延後の、細胞に付加されているSRの蛍光シグナルを定量化することにより検査した。蛍光強度は7日間まで安定化及び保持されており、対照より有意に高かった(P<0.01)。これらのデータは、BNHS処理によるビオチンの共有結合固定化が、結果として細胞表面上の安定的で接近可能なビオチンをもたらしたことを示す。
【0293】
フローサイトメトリーを使用した機能化効率の分析及び至適化
修飾工程の3番目のステップでのビオチン−4−フルオレセインのインキュベーション時間の効果を決定した。 ビオチン−4−フルオレセインのインキュベーション時間の5分間を超える増加は、結果として実験群の蛍光に有意な増加をもたらさなかった。対照的に、対照は、ビオチン−4−フルオレセインのインキュベーションを増加させると共に、蛍光を増加させた。
【0294】
ストレプトアビジンと共にBNHS修飾MSCをインキュベーションする(ステップ2)時間は、そのすぐ後のステップで結合されるビオチン−4−フルオレセインの量に有意な影響を及ぼした。具体的には、ストレプトアビジンとの1分間インキュベーションは、その結果として、鋭いピークにより示されたより強い全体的な蛍光をもたらした。ストレプトアビジンとのより長いインキュベーション時間(5分及び10分)は、BNHS修飾MSCをストレプトアビジンと共に1分間インキュベートした群と比較して、蛍光の減少及びより幅の広いピークを結果としてもたらした。対照群細胞(PBS処理MSC、その後ストレプトアビジン及びビオチン−4−フルオレセインと共にインキュベーションした)の蛍光は、ストレプトアビジンのインキュベーション時間を増加させると共に増加した。
【0295】
(ステップ1)のBNHSとMSCとの反応時間も、その後ストレプトアビジンを結合させ、次にビオチン−4−フルオレセインを結合させることにより導入される相対的蛍光に影響を及ぼす。蛍光シグナルは、BNHS処理後約10分でプラトーに達し、その後は反応時間が増加しても有意な増加を示さなかった。
【0296】
さらなる実験には、細胞集団全体にわたって最も強く及び最も均一な染色に結び付いた条件を使用した。実験群のピークはこれらの条件を表す:10分間のBNHS、1分間のストレプトアビジン、及び5分間のビオチン−4−フルオレセイン。10分間のBNHS、1分間のストレプトアビジン、及び4分間のビオチン−4−フルオレセインで構成される反応条件を使用してMSC表面上に固定化したBSLeXのABCは、約27201±13786であると決定された。このABCは、部位密度が約10±5 SLeX部分/μm2で平均細胞直径が15±4μmに相当する。
【0297】
細胞表現型の特徴付け
BSLeX修飾細胞の細胞生存率は、細胞修飾後48時間以内に観察したところ、PBS処理細胞と比較して、有意な影響を受けていなかった。具体的には、SLeX修飾細胞の88±4%がカップリング直後に生存可能であり、それと比較してPBS修飾細胞の生存率は92±3%だった。48時間の培養後、90±2%のSLeX修飾細胞が生存可能であり、それと比較してPBS処理細胞は89±2%だった。これは、SLeXによるMSCの修飾が、いかなる実質的な毒性も誘導しなかったことを示す。10、30、90、及び150分時点の、組織培養ポリスチレン皿上のSLeX修飾MSCの接着動力学を、PBS処理細胞と比較した。修飾細胞は、対照と比較して同様の接着動力学を示した。BSLeX修飾MSC及びPBS処理細胞の増殖速度、又は7日後にコンフルエント単層に達するそれらの能力に差異は検出されなかった。
【0298】
修飾細胞の分化能
BSLeX修飾MSCの分化能を、骨形成培養条件下及び脂肪生成培養条件下で細胞の分化を誘導することにより検査し、対照としてのPBS処理細胞と比較した。具体的には、アルカリホスファターゼ(ALP)活性及びオイルレッド0(ORO)染色を、それぞれ骨形成分化及び脂肪生成分化の指標として使用した。BSLeX修飾細胞及びPBS処理細胞間の、ALP又はORO染色における有意差は、骨形成分化及び脂肪生成分化誘導後に観察されなかった。さらに、MSC増殖媒地中で増殖させたBSLeX修飾細胞は、ORO又はALPで陽性染色されなかった。これらの結果は、MSC表面の共有結合修飾が、脂肪生成分化も骨形成分化も誘導せず、細胞の多系統分化能も損なわないことを示す。
【0299】
パラクリン因子の放出
BSLeX修飾MSCによるパラクリン因子の分泌を、ELISAアッセイを使用して、24時間の時間枠にわたって、培地中へのSDF−1、IGF−1、及びPGE2の分泌を定量化することにより検査した。PBS処理細胞と比較して、BSLeX修飾MSCのパラクリン因子の発現のレベル間には、有意差は観察されなかった。IGF−1及びPGE2は、15%血清を含有する増殖培地で検出可能ではなかったが、SDF−1は、24時間後にMSCから観察された量の約5%で検出された。
【0300】
MSC表面マーカー発現に対する化学的修飾後の影響
MSC表面上のCD90及びCD29受容体の発現は、PBS処理細胞と比較して、BNHS、ストレプトアビジン、及びBSLeXによる3段階修飾後に低減した。具体的には、修飾直後に、BSLeX結合MSCは、PBS処理細胞のCD90、CD29、及びCD49dと比較して、それぞれ84%、75%、及び40%の相対的蛍光を示す。24時間後には、修飾MSC及びPBS処理細胞の蛍光は類似しており、CD90、CD29、及びCD49d表面受容体が24時間後に回復し、これら受容体の発現レベルが未修飾MSC対照に類似していたことを示した。
【0301】
P−セレクチン上での修飾MSCの回転
P−セレクチンを有するMSC表面上に結合したBSLeX部分の、回転に基づく接着相互作用をフローチャンバーアッセイで分析した。修飾MSCのSLeXとP−セレクチン間の特異的相互作用は、0.36ダイン/cm2の壁面せん断応力において、速度を約70μm/sから約0.5μm/sに低下させた。10秒間隔内に相互作用する細胞の数は、PBS処理MSCの場合の20細胞/mm2から、SLeX修飾細胞の場合の150細胞/mm2に増加した。せん断応力を0.36ダイン/cm2から1.89ダイン/cm2に増加させると、修飾細胞の速度は、0.5μm/sから2μm/sまで中程度に増加し、フラックスは、150細胞/mm2と140細胞/mm2との間でほとんど一定のままだった。以前の研究では、0.72ダイン/cm2を超えるせん断応力において、速度は著しく増加した。
【0302】
実施例16:小胞を介してSLEXを有する修飾間葉系幹細胞の回転応答
hMSCをT25フラスコに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。2−ジオレオイル−sn−グリセロ−3−ホスホエタノールアミン−N−(ビオチニル)ナトリウム塩の1mL(1mM)小胞溶液を、接着hMSCに添加し、10分間インキュベートし、その後洗浄して過剰な小胞/脂質を除去した。フラスコを1mLの培地で3回すすいで、過剰な小胞/脂質を除去した。その後、1mLのストレプトアビジン溶液(Ca/Mg非含有PBS中50μg/mL)を添加し、5分間インキュベートした。その後、細胞をPBSで2回洗浄し、未結合ストレプトアビジンを除去した。その後、1mLのビオチン化シアリルルイスX(SLeX)溶液(Ca/Mg非含有PBS中5μg/mL)を添加し、5分間インキュベートした。細胞をPBSで2回洗浄し、未結合SLeXを除去した。1mLの非酵素性細胞解離溶液を使用して、細胞をフラスコから分離し、1mLのPBSで1回洗浄して細胞解離溶液を除去した。その後、細胞を2mLの細胞増殖培地に分散し、回転相互作用をフローチャンバーアッセイを使用して分析した。対照実験として、PBS処理hMSC(未修飾細胞)をフローチャンバーアッセイに使用した。結果は図15A〜15Bに要約されている。結果は、接着モードにおいて小胞を介してSLeXにより修飾したhMSCが、P−セレクチンでコーティングした基質上での回転相互作用を示すことを表す。小胞修飾細胞は、0.5ダイン/cm2のせん断応力において7μm/sの速度を示したが、未修飾細胞は、60μm/sの速度を示した。図15Bは、小胞を介したSLeX修飾hMSCに対するせん断応力増加の効果を示す。
【0303】
(文献)
以下に、並びに本明細書及び実施例の全体にわたって引用された全ての文献は、その全体が参照により本明細書に組み込まれる。
1. MacDiarmid, J. A., et al., Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell, 2007. 11(5): p. 431-45.
2. Studeny, M., et al., Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res, 2002. 62(13): p. 3603-8.
3. Stoff-Khalili, M.A., et al., Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res Treat, 2007. 105(2): p. 157-67.
4. Kumar, S. and S. Ponnazhagan, Bone homing of mesenchymal stem cells by ectopic {alpha}4 integrin expression. Faseb J, 2007.
5. Chamberlain, G., et al., Mesenchymal Stem Cells: their Phenotype, Differentiation Capacity, Immunological Features and Potential for Homing. Stem Cells, 2007.
6. Griffiths, L., et al., The macrophage - a novel system to deliver gene therapy to pathological hypoxia. Gene Ther, 2000. 7(3): p. 255-62.
7. Hwu, P., et al., In vivo antitumor activity of T cells redirected with chimeric antibody/T- cell receptor genes. Cancer Res, 1995. 55(15): p. 3369-73.
8. Harrington, K., et al., Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery? Hum Gene Ther, 2002. 13(11): p. 1263-80.
9. Elgert, K.D., D. G. Alleva, and D.W. Mullins, Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol, 1998. 64(3): p. 275-90.
10. Jang, J. H., T. L. Houchin, and L.D. Shea, Gene delivery from polymer scaffolds for tissue engineering. Expert Rev Med Devices, 2004. 1(1): p. 127-38.
11. Hill, E., T. Boontheekul, and DJ. Mooney, Regulating activation of transplanted cells controls tissue regeneration. Proc Natl Acad Sci U S A, 2006. 103(8): p. 2494-9.
12. Leo, AJ. and D. A. Grande, Mesenchymal stem cells in tissue engineering. Cells Tissues Organs, 2006. 183(3): p. 112-22.
13. Sun X-D, L.I.-S., Gene technology in tissue engineering. American Journal of Biochemistry and Biotechnology, 2006. 2(2): p. 66-72.
14. Kemp, K.C., J. Hows, and C. Donaldson, Bone marrow-derived mesenchymal stem cells. Leuk Lymphoma, 2005. 46(11): p. 1531-44.
15. Gafni, Y., et al., Stem cells as vehicles for orthopedic gene therapy. Gene Ther, 2004. 11(4): p. 417-26.
16. Nakamura, K., et al., Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther, 2004. 11(14): p. 1155-64.
17. Pizzato, M., et al., Initial binding of murine leukemia virus particles to cells does not require specific Env-receptor interaction. J Virol, 1999. 73(10): p. 8599-611.
18. Zhuge, Y. and J. Xu, Racl mediates type I collagen-dependent MMP-2 activation, role in cell invasion across collagen barrier. J Biol Chem, 2001. 276(19): p. 16248-56.
19. Cheung, L.W., P.C. Leung, and A. S. Wong, Gonadotropin-releasing hormone promotes ovarian cancer cell invasiveness through c-Jun NH2-terminal kinase-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9. Cancer Res, 2006. 66(22): p. 10902- 10.
20. Fujiuchi, Y., et al., Effect of hepatocyte growth factor on invasion of prostate cancer cell lines. Oncol Rep, 2003. 10(4): p. 1001-6.
21. Mohanam, S., et al., Increased invasion of neuroglioma cells transfected with urokinase plasminogen activator receptor cDNA. Int J Oncol, 1998. 13(6): p. 1285-90.
22. Farokhzad, O. C, J.M. Karp, and R. Langer, Nanoparticle-aptamer bioconjugates for cancer targeting. Expert Opin Drug Deliv, 2006. 3(3): p. 311-24.
23. Chithrani, B.D., A.A. Ghazani, and W.C. Chan, Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett, 2006. 6(4): p. 662-8.
24. Win, K.Y. and S. S. Feng, Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials, 2005. 26(15): p. 2713-22.
25. Schneider, G. B., et al., The effect of hydrogel charge density on cell attachment. Biomaterials, 2004. 25(15): p. 3023-8.
26. Sinclair, J. and A. K. Salem, Rapid localized cell trapping on biodegradable polymers using cell surface derivatization and microfluidic networking. Biomaterials, 2006. 27(9): p. 2090-4.
27. Saxon, E. and CR. Bertozzi, Cell surface engineering by a modified Staudinger reaction. Science, 2000. 287(5460): p. 2007-10.
28. Huang, A., L. Huang, and SJ. Kennel, Monoclonal antibody covalently coupled with fatty acid. A reagent for in vitro liposome targeting. J Biol Chem, 1980. 255(17): p. 8015- 8.
29. Dube, D. H. and CR. Bertozzi, Metabolic oligosaccharide engineering as a tool for glycobiology. Curr Opin Chem Biol, 2003. 7(5): p. 616-25.
30. Hang, H. C. and CR. Bertozzi, Ketone isosteres of 2-N-acetamidosugars as substrates for metabolic cell surface engineering. J Am Chem Soc, 2001. 123(6): p. 1242-3.
31. Sampathkumar, S. G., M.B. Jones, and KJ. Yarema, Metabolic expression of thiol- derivatized sialic acids on the cell surface and their quantitative estimation by flow cytometry. Nat Protoc, 2006. 1(4): p. 1840-51.
32. De Bank, P.A., et al., Surface engineering of living myoblasts via selective periodate oxidation. Biotechnol Bioeng, 2003. 81(7): p. 800-8.
33. Johnsson, N., N. George, and K. Johnsson, Protein chemistry on the surface of living cells. Chembiochem, 2005. 6(1): p. 47-52.
34. Toriello, N.M., E. S. Douglas, and R.A. Mathies, Microfluidic device for electric field- driven single-cell capture and activation. Anal Chem, 2005. 77(21): p. 6935-41.
35. Albretsen, C, et al., Applications of magnetic beads with covalently attached oligonucleotides in hybridization: isolation and detection of specific measles virus mRNA from a crude cell lysate. Anal Biochem, 1990. 189(1): p. 40-50.
36. Tiwari, A., et al., Magnetic beads (Dynabead) toxicity to endothelial cells at high bead concentration: implication for tissue engineering of vascular prosthesis. Cell Biol Toxicol, 2003. 19(5): p. 265-72.
37. Hinds, K.A., et al., Highly efficient endosomal labeling of progenitor and stem cells with large magnetic particles allows magnetic resonance imaging of single cells. Blood, 2003. 102(3): p. 867-72.
38. Lewin, M., et al., Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol, 2000. 18(4): p. 410-4.
39. Lacava, L.M., et al., Magnetic resonance of a dextran-coated magnetic fluid intravenously administered in mice. Biophys J, 2001. 80(5): p. 2483-6.
40. Wilson, K.M., et al., Single particle tracking of cell-surface HLA-DR molecules using R- phycoerythrin labeled monoclonal antibodies and fluorescence digital imaging. J Cell Sci, 1996. 109 ( Pt 8): p. 2101-9.
41. Mistry, S.C., Iksoo;Messina, Darin Jeffrey; Gosiewska, Anna, Microparticles for cell delivery. 2005, JOHNSON & JOHNSON USA.
42. Halbert, G.W., Microparticles. 2007: USA.
43. Robert C, Klein, C, Cheng, G., Kogan, A., Mulligan, R.C., von Andrian, U.H., Cooper, T. S, Gene therapy to target dendritic cells from blood to lymph nodes. Gene Therapy 2003. 10(17):1479-86.
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2008年4月29日に出願された米国特許仮出願第61/048,773号の35 U.S.C.§119(e)に基づく利益を主張するものであり、その内容は、その全体が参照により本明細書に組み込まれる。
【0002】
本発明は、再生促進用の標的細胞送達の分野に関する。
【背景技術】
【0003】
細胞は、組織特異的薬物送達を含む様々な生物医学的応用のための重要な治療用ツールである。例えば、種々の抗癌剤(ドキソルビシン、プラチンなど)を負荷した細菌細胞が、癌細胞に特異的に標的化されている1。送達媒体としての細胞の別の例は、分子工学技術によりヒト欠乏症および疾患を治療するための、遺伝学的に形質導入されたヒト間葉系幹細胞(MSC)の使用、並びに適切な化学療法剤の送達である。インターフェロン−β(IFN−β)を保持するアデノウイルス発現ベクターにより形質導入されたMSCは、静脈内又は皮下のいずれかでIFN−βを悪性癌細胞に送達して腫瘍増殖を阻害するために使用されてきた2。同様に、CRAd(条件付複製アデノウイルス)は、マウス異種移植モデルの使用により、MSCを介して悪性腫瘍細胞へと標的化された3。これらの結果は、CRAd形質導入幹細胞が、CRAd単独より効果的に腫瘍部位にホーミングされたことを示す。MSCは、硬骨4および他の幾つかの組織5へのホーミング能力を有することも知られている。
【0004】
マクロファージは、腫瘍部位に集中する能力、腫瘍細胞を死滅させる能力、及び腫瘍増殖を阻害する能力のため、例えば癌治療において魅力的な細胞に基づく担体の別の例であり、例えば、CY2BP6を発現するように形質導入されたマクロファージは、超毒性環境で腫瘍球状体に浸潤した際に腫瘍細胞を死滅させることができた6。腫瘍特異的T細胞(例えば、腫瘍浸潤性リンパ球)も、治療目的で腫瘍へのホーミングに使用されており、例えば、MOv−γ遺伝子で形質導入されたマウスT細胞が、ヒト卵巣癌細胞を有するマウスに経皮内投与された7。しかし、これらの細胞応用が成功したにもかかわらず、多くの研究により、適切な標的化が多くの場合に達成されていないことも示されている8。標的とされていない非特異的部位において治療剤が制御されずにT細胞から放出されることは、患者の健康に対する相当程度の毒性及びリスクの原因となる。同様に、腫瘍部位におけるあるマクロファージの腫瘍形成的役割が報告されている9。
【0005】
細胞は、細胞工学的応用における再生プロセスに不可欠な部分である。遺伝子組換え細胞及び/又は適切な遺伝子の使用は、組織再生にますます重要な役割を果たす。遺伝子組換えポリマーマトリクス(ポリマー性放出及び基質媒介性放出の両方による)は、組織再生プロセスを支援するための特定の遺伝学的情報を放出する10。増殖因子で補完されたスキャフォールドでの組織の操作に細胞を使用することは、組織の再生を増強する11、12。
【0006】
しかしながら、これらの方法に伴う問題には以下のものが含まれる:制御されない放出、不適当なレベルの遺伝子発現、及び異常組織増殖。指定組織再生用の適切な遺伝子の送達は、非ウイルスベクターまたはウイルスベクターを使用して所望の遺伝子を細胞に移動させ、続いて組織再生用の細胞を送達することによっても達成される13。MSCの全身性投与による自己細胞療法は、組織再生の強力な治療用ツールである14〜16。しかしながら、ex vivoで培養されたMSCは、遺伝子発現の欠如により、脾臓及び骨髄へのホーミング能力を欠如する14。
【0007】
細胞とその環境との間の特定の相互作用を明確にすることは、治療をうまく機能させるために積極的に研究されている領域である。細胞膜の構造及び機能性は、かなりのクロストークを示す種々の相互作用により、細胞が細胞外マトリックス(ECM)と相互作用することを可能にし、細胞接着、増殖、遊走、分化、マトリックス産生、プロテアーゼ分泌、又はアポトーシスの原因となる可能性がある。その結果として、細胞膜は、細胞とその周囲との相互作用の媒介に重要である。細胞膜(例えば、原形質膜)は、多種多様な生体分子、主として無数の細胞性プロセスに関与するタンパク質及び脂質を含有し、細胞内の細胞骨格の結合点としても機能する半透過性脂質二分子層である。細胞膜は、ECMとの相互作用に加えて、イオン、小分子、及びウイルスなどのより大型物質の出入口として機能する。マウス白血病ウイルス(MLV)及びヒト免疫不全ウイルス(HIV)粒子に関する研究は、ウイルス粒子が最初に細胞表面と相互作用し結合することを示している17。
【0008】
細胞膜は、細胞への出入口を提供することに加えて、周囲との相互作用を能動的に媒介する。例えば、細胞外マトリックスを遊走可能な細胞は、典型的には細胞膜を介してMMPなどのプロテアーゼを分泌し、ある細胞タイプは、細胞膜内にMMPを含有する(膜型マトリックスメタロプロテアーゼ1(MT1−MMP))18。プロテアーゼの分泌が増強された癌細胞は、細胞外マトリックスに侵入する高度な能力も示す19。プロテアーゼ20又はタンパク質分解に関与する細胞表面受容体21の産生を増強するように遺伝子組換えにより修飾された細胞は、侵入能力を増強させたことを明らかに示している。これは、細胞表面を適切に修飾することにより、細胞とその環境との相互作用を調節することが可能であることを示す。
【0009】
細胞表面は、外部環境との相互作用を媒介し、複雑な相互作用を包含する基質であるため、細胞膜媒介性相互作用のより高度な制御を示して生体特異的効果をもたらすことが望まれている。
【発明の概要】
【発明が解決しようとする課題】
【0010】
一般的に、本明細書に記載の発明は、種々の技術により細胞表面を機能化するための方法、及び様々な応用に使用するための方法を提供する。ナノ粒子による標的薬物送達とは異なり、及び生体材料に基づくビーズ上の又はその内部の未修飾細胞の送達とは異なり、細胞表面を様々な機能性で修飾することは、様々な応用用に細胞性事象を操作するための制御の強化を容易にする新しい手法である。機能化された細胞表面は、実質的にあらゆる生物系を制御するために効果的に使用することができる。より具体的には、本明細書に記載の組成物及び方法は、再生用に細胞を組織に標的化するのに有用であるか、又はその代わりに作用剤を特定組織へと細胞に基づいて送達するのに有用である。本明細書に記載の方法に有用な細胞は、粒子並びにリガンドの付加により修飾することができる。該粒子は、タンパク質、小分子、RNA干渉分子、薬物、ビタミン、治療薬、診断薬、栄養補助剤、化粧用の特性を有する作用剤、又は核酸などの作用剤をさらに含んでいてもよい。加えて、本明細書に記載の組成物は、例えば、特に、脳卒中、臓器再生、癌、骨折、及び虚血性心疾患を含む広範囲の疾患又は創傷を治療するために、当業者によって個別化することができる。
【課題を解決するための手段】
【0011】
1つの態様では、本発明は、細胞、該細胞の表面に結合する膜結合リガンド、及び該細胞の表面に結合する粒子を含む、単離された改変細胞組成物に関する。
【0012】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、該細胞は、幹細胞又は前駆細胞である。
【0013】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該該幹細胞は、再プログラムされた細胞である。
【0014】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、分化細胞である。
【0015】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、治療薬を発現するように遺伝子操作されている。
【0016】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、1000〜8000nmである。或いは、該粒子は、500〜1000nm又は1〜500nmである。
【0017】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、リンカー分子を介して結合している。
【0018】
同様に、この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、リンカー分子を介して結合している。或いは、別の実施形態では、該粒子は、リンカー分子を介さずに結合している。
【0019】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、共有結合で結合している。或いは、別の実施形態では、該膜結合リガンドは、非共有結合で結合している。
【0020】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、共有結合で結合している。或いは、別の実施形態では、該粒子は、非共有結合で結合している。
【0021】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、組織における該細胞の蓄積をもたらす。
【0022】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、薬物、栄養補助剤、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される。
【0023】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、2つ以上の膜結合リガンドが、該細胞に結合している。
【0024】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞の機能を増強する作用剤を含む。或いは、該粒子は、組織の機能を増強する作用剤を含む。
【0025】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、脂質、ナノ粒子、マイクロ粒子、又はデンドリマーからなる群から選択される。
【0026】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は、細胞成長、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着からなる群から選択される機能を増強する。
【0027】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は、小分子、成長因子(growth factor)、サイトカイン、RNA干渉分子、増殖因子(proliferation factor)、ビタミン、栄養補助剤、化粧用の特性を有する作用剤、治療薬、診断薬、ケモカイン、標的化作用剤、又は分化因子からなる群から選択される。
【0028】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、作用剤を含む粒子用の組織特異的担体として使用される。
【0029】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な基質若しくは注射可能な基質内に捕捉若しくは基質に結合されている。
【0030】
1つの態様では、本発明は、細胞、及び該細胞の表面に組み込まれた自己集合分子に結合した膜結合リガンドを含む、単離された改変細胞組成物に関する。
【0031】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該自己集合分子は、両親媒性である。
【0032】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、該自己集合両親媒性分子に共有結合で結合している。或いは、膜結合リガンドは、該自己集合両親媒性分子に非共有結合で結合している。
【0033】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、少なくとも1つの粒子が、該細胞に結合している。或いは、1〜10個以上の、例えば2、3、4、5、6、7、8、9、又は10個以上の粒子が、該細胞に結合されている。
【0034】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、標的化部分であるリガンドを含む。
【0035】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な基質若しくは注射可能な基質内に捕捉若しくは基質に結合されている。
【0036】
単離された改変細胞組成物を形成するための方法であって、(a)膜結合リガンドを自己集合分子に結合させて、修飾分子を形成するステップと、(b)該修飾分子を用いて小胞を形成するステップと、(c)該小胞を細胞と融合させるステップとを含む方法も、本明細書に記載されている。
【0037】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該単離された改変細胞組成物は、in vivoで形成される。
【0038】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、細胞凝集塊を形成するために使用される。
【0039】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、細胞凝集塊を形成するために使用されるリガンドを含む。
【0040】
単離された改変細胞組成物を形成するための方法であって、(a)膜結合リガンドを自己集合分子に結合させて、修飾分子を形成するステップと、(b)該修飾分子を用いてミセルを形成するステップと、(c)該ミセルを該細胞と融合させるステップとを含む方法も、本明細書に記載されている。
【0041】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、細胞膜へ組み込み前に該自己集合分子との結合を介して該細胞に結合される。或いは、該粒子は、細胞膜への組み込み後に該自己集合分子との結合を介して該細胞に結合される。
【0042】
本発明の別の態様は、細胞、及び該細胞の表面に結合された膜結合リガンドを含む単離された改変細胞組成物に関し、前記膜結合リガンドは、該細胞の表面の第1の部分に結合されており、該細胞の表面の第2の部分には該リガンドがない。
【0043】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、粒子をさらに含む。
【0044】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該膜結合リガンドは、細胞凝集塊を形成するために使用される。
【0045】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞凝集塊は、in vivoで形成される。
【0046】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な基質若しくは注射可能な基質内に捕捉若しくは基質に結合されている。
【0047】
本明細書に記載の別の態様は、標的組織再生を必要とする個体を治療する方法であって、(a)標的細胞を、該細胞の表面に膜結合リガンドを結合させることにより形成し、前記膜結合リガンドが、治療される組織の中に該標的細胞の蓄積をもたらすステップと、(b)該標的細胞の表面に粒子を結合させることにより二重機能性細胞を形成し、該粒子が、該二重機能性細胞の機能を増強する作用剤を含むステップと、(c)標的組織再生を必要とする個体に該二重機能性細胞を投与するステッとを含む方法である。
【0048】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、標的組織再生を必要とする個体から単離される。或いは、該細胞は、標的組織再生を必要とする個体以外の個体から単離される。
【0049】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該細胞は、該膜結合リガンドを結合させる前に、ex vivo培養環境で増殖される。
【0050】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、標的組織再生の部位で放出される。
【0051】
本明細書に記載の別の態様は、対象体にワクチン接種をするための方法であって、(a)膜結合リガンドを樹状細胞の表面に結合させることにより、標的化樹状細胞を形成し、該膜結合リガンドが、リンパ組織での該標的化樹状細胞の蓄積をもたらすステップと、(b)該標的化樹状細胞を抗原と接触させることにより活性化標的化樹状細胞を形成するステップと、(c)ワクチンを必要とする対象体に該活性化標的化樹状細胞を投与するステップとを含む方法である。
【0052】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該抗原は、ウイルス抗原を含む。或いは、該抗原は、細菌抗原又は癌関連抗原を含む。
【0053】
細胞を修飾するためのキットであって、(a)結合部分を有する自己集合分子と、(b)細胞を修飾するための方法を含む説明書と、(c)それらの包装材料とを含むキットも、本明細書に記載されている。該キットに含まれる説明書には、単離された改変細胞組成物を形成するための方法であって、(a)結合部分を有する自己集合分子を用いて小胞又はミセルを形成するステップと、(b)該小胞又はミセルを目的細胞と融合させるステップとを含む方法が記述されている。
【0054】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該結合部分は、膜結合リガンドである。或いは、該結合部分は粒子である。1つの実施形態では、該キットは、第2の部分に加えて粒子も含むことができる。
【0055】
(a)細胞と、(b)該細胞の表面に結合した粒子と、(c)該細胞に結合しない粒子結合リガンドとを含む単離された改変細胞組成物も、本明細書に記載されている。そのような実施形態では、該粒子上に存在するリガンドは該細胞と直接的に相互作用せず、むしろ該粒子がリガンドを、内部的に(例えば、該粒子内に)含むか、外部的に(例えば、該粒子表面上に)含むか、又は該粒子に組み込まれているか(例えば、該細胞表面の構成要素になる修飾両親媒性分子)のいずれかであることが好ましい。該リガンドは、該粒子を該細胞に結合するリンカー分子として機能しないことが好ましい。
【0056】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、粒子は作用剤を含み、幾つかの場合には、該作用剤は該粒子の構成要素である。これらの実施形態では、該粒子のおよそ1%から実質的に全てが、目的の作用剤で構成されていてもよい。
【0057】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、粒径が1nm〜5000nmである。
【0058】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞に内部移行されない。
【0059】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、血漿又は血清との接触による不活性化に感受性である作用剤をさらに含む。この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該不活性化に感受性である作用剤は、RNA干渉分子である。
【0060】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は、該細胞の輸送を増強する。
【0061】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は治療薬を含む。或いは、該作用剤は診断薬を含む。
【0062】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該作用剤は、宿主細胞及び/又は組織の機能を増強する。
【0063】
本明細書に記載の別つの態様は、(a)細胞と、(b)該細胞の表面に組み込まれた脂質分子に結合した膜結合リガンドとを含む、単離された改変細胞組成物である。
【0064】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該脂質分子は、単一の尾部を含む。或いは、該脂質分子は、複数の尾部を含むこともできる。
【0065】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該脂質分子は、複数の電荷を帯びている。
【0066】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該脂質分子は、リピドイド(lipidoid)などの改変脂質である。
【0067】
本明細書に記載の別の態様は、該リガンドの部分(例えば、該リガンドの少なくとも3分の1)が、修飾後の少なくとも2日間は該細胞表面上で安定している細胞組成物を調製するための方法であって、(a)ビオチン及びリガンドの供給源を含む脂質小胞を調製するステップと、(b)細胞(例えば、ヒト間葉系幹細胞)を前記小胞と接触させるステップとを含み、該リガンドが前記接触ステップ後の少なくとも2日間は該細胞上に存在するように、細胞組成物が形成される方法に関する。
【0068】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該リガンドは、ビオチンを含む。
【0069】
幹細胞の特徴を損なわずに前駆細胞又は幹細胞をリガンドで修飾するための方法であって、該細胞表面を共有結合で修飾するステップを含み、該細胞組成物が、前駆細胞又は幹細胞の特徴を喪失せずに形成される方法も本明細書に記載されている。
【0070】
幹細胞の特徴を損なわずに前駆体幹細胞をリガンドで修飾するための方法であって、(a)細胞をビオチンの供給源と接触させるステップと、(b)ステップ(a)の前記細胞を、ストレプトアビジン及びリガンドと接触させるステップとを含み、幹細胞組成物が、幹細胞の特徴を損なわずに形成される方法も本明細書に記載されている。
【0071】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、該幹細胞の特徴には、多系統分化、生存能、増殖、パラクリン因子の分泌、in vivo経内皮遊走、及び/又は接着が含まれる。
【0072】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、該リガンドは、シアリルルイスXを含む。
【0073】
接着リガンドを細胞の表面に結合するための方法であって、(a)細胞をビオチンの供給源と接触させるステップと、(b)ステップ(a)の前記細胞を、ストレプトアビジン及び強力な接着を促進する接着リガンドと接触させるステップとを含み、細胞組成物が、該細胞表面上に接着リガンドを含んで形成され、前記接着リガンドが強力な接着を可能にする方法も本明細書に記載されている。
【0074】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、複数の接着リガンドが、細胞の表面に結合されている。
【0075】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該接着リガンドは、PSGL−1又はP−セレクチン抗体を含む。
【0076】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該接着リガンドは、1.9ダイン/cm2まで、2μm/秒未満の速度で回転することを可能にする。
【0077】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該接着リガンドは、組織内局在化を増強する。
【0078】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該組織は骨髄である。
【0079】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該リガンドは、特定の物理的機能性(負電荷、正電荷、脂質、抗体など)又は化学的機能性(NHS基、ストレプトアビジンなど)を含む。
【0080】
細胞の表面に粒子を結合するための方法であって、(a)リガンドを粒子に結合させて機能化粒子を調製するステップと、(b)前記機能化粒子を細胞と接触させるステップとを含み、前記機能化粒子が前記細胞の表面に結合されている方法も、本明細書に記載されている。
【0081】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該機能化粒子は、PLGAを含む。
【0082】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該機能化粒子は、エマルション法を使用して調製される。
【0083】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該リガンドは、前記細胞の組織内局在化を可能にする。
【0084】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該機能化粒子は、内部移行される。
【0085】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該機能化粒子は、内部移行されない。
【0086】
該脂質(該細胞表面に結合されている)に結合されたリガンドの少なくとも3分の1が、修飾の2日後に安定している方法であって、(a)1,2−ジオレオイル−sn−グリセロ−3−ホスホエタノールアミン−N−(ビオチニル)ナトリウム塩から小胞を調製するステップと、(b)引き続き、該小胞をhMSCと共に30分間インキュベーションするステップとを含み、hMSC上で安定化されたリガンドがビオチンである方法。
【0087】
強力な接着を促進する接着リガンドを結合させるための方法であって、(a)ビオチニル−N−ヒドロキシ−スクシンイミドのN−ヒドロキシ−スクシンイミド基で細胞を修飾(ビオチン化)するステップと、(b)ストレプトアビジン及び接着リガンドで処理するステップとを含む方法も、本明細書に記載されている。
【0088】
この態様及び本明細書に記載の他の全ての態様の1つの実施形態では、該リガンドは、P−セレクチン抗体を含む。
【0089】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該回転は、1.9ダイン/cm2まで、2μm/秒未満である。
【0090】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該回転は、組織内局在化を増強する。
【0091】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該組織は骨髄である。
【0092】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞の表面に結合したままである。
【0093】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞表面上で安定化された後、該細胞内に内部移行される。
【0094】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該表面結合粒子は、全身性注射又は局所投与された後、該細胞により特定部位へと運搬される。
【0095】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該粒子は、該細胞の表面に結合したままであるか、又は該細胞表面から分離しているか、又は該標的部位で内部移行される。
【0096】
この態様及び本明細書に記載の他の全ての態様の別の実施形態では、該内部移行粒子は、全身性注射又は局所投与された後、該細胞により特定部位へと運搬される。
【図面の簡単な説明】
【0097】
【図1】細胞膜修飾を示す概略図である。1)特異的細胞表面官能基を導入するために細胞膜が機能化される。2)官能化された基に治療薬を含有する粒子をリンカーを介して結合し、該粒子は第2の官能基を含有する。3)該細胞表面上の官能化された基に粒子をリンカーを介して結合させる。4)細胞表面上の官能化された基に粒子を結合させる。5)細胞表面上の官能化された基を介して粒子が細胞に結合され、粒子及び細胞は官能リガンドを含有する。6)細胞膜を多官能性作用剤で機能化する。7)脂質分子又は脂質に基づく小胞の結合により細胞膜を機能化する。8)物理的な(非共有結合)相互作用又は化学的相互作用(共有結合)のいずれかにより、機能化粒子を細胞に結合する(細胞膜を機能化せずに)。9)機能化粒子をリンカーを介して細胞に結合する(細胞膜に遊離官能基を結合せずに)。10)機能化粒子をリンカーを介して細胞に結合し(細胞膜を遊離官能基で機能化せずに)、粒子はリガンドを含有する。
【図2】蛍光顕微鏡画像で観察した、4、8、及び12時間の時点での、粒子が結合されているヒト間葉系幹細胞(hMSC)のパーセントを示す棒グラフである。
【図3】様々な表面特徴および様々な粒径を有する粒子の内部移行を経時的に示す棒グラフである。図3Aは、y−z、x−z、及びx−y平面のZスタック共焦点画像で観察された、4、8、及び12時間の時点での、hMSCによって内部移行された様々な表面特徴を有する粒子のパーセントを示す。共焦点顕微鏡の場合、hMSCをヨウ化プロピジウムで染色し、粒子はDiD封入されている。図3Bは、4、8、及び12時間の時点での、hMSCに結合されたCD90抗体コーティングPLGA粒子の内部移行に対する粒径の効果を示しており、より大型粒子と比較して、3μm未満の粒子がより高い効率で内部移行されることが示されている。
【図4】hMSCに結合された負荷電PLGA粒子の詳細な分析を示す一連のグラフである。図4Aは、0時間及び36時間(左)の時点で、粒子が結合されている細胞のパーセントを示す。図4Bは、0時間及び36時間の時点で、1個の粒子、2個の粒子、及び3個以上の粒子を有する細胞のパーセントを示す。このパーセントは、粒子を有する細胞の総数に基づく。図4Cは、負荷電PLGA粒子のhMSCへの結合に対するトリプシン処理の影響を示す。トリプシン処理直後並びにその後の2日目及び4日目における、粒子が結合されている細胞のパーセント並びに観察視野にある細胞の総個数;図4Dは、0日目(トリプシン処理日)、2日目、及び4日目の、1個の粒子、2個の粒子、及び3個以上の粒子を有する細胞のパーセントを示す。このパーセントは、結合した粒子を含有する細胞の総数に基づく。
【図5】例示的な修飾細胞の細胞及び粒子特徴を示す一連のグラフである。図5Aは、ビオチン及びストレプトアビジン相互作用によりhMSCに結合された粒子の、結合直後の数を示しており、ビオチン化hMSCが、ストレプトアビジンをコーティングした粒子に特異的に結合することを示す。図5Bは、0時間及び48時間後の、PLGA結合hMSC(ビオチン−ストレプトアビジンによる)の生存能を示す。図5Cは、10、30、及び90分の時点での、組織培養表面に接着したPLGA結合hMSC(ビオチン−ストレプトアビジンによる)のパーセントを示す。図5Dは、8日間にわたるPLGA結合hMSC(ビオチンーストレプトアビジンによる)の増殖を示す。
【図6】Strep−PLGA粒子で修飾されたビオチン化細胞の回転を示す棒グラフであり、細胞及び粒子は、P−セレクチン上のSLeXで機能化されている。平行平板フローチャンバーアッセイ(parallel plate flow chamber assay)において0.36ダイン/cm2で、ビオチン−ストレプトアビジンによりPLGA粒子と結合されたhMSC、及びP−セレクチンでコーティングされた基質上のビオチン化シアリルルイスX(SLeX)の回転応答が示されている。対照群は、未修飾hMSC、つまりPBS処理hMSCを含む(回転アッセイは、明視野モード及び蛍光モードの両方で実施した)。
【図7】細胞表面へのPLGA粒子の結合(PEGリンカーによる、及び細胞を機能化せず)を示す棒グラフである。hMSCに結合されたポリエチレングリコール(PEG)リンカーを有する及び有していない、N−ヒドロキシスクシンイミド(NHS)基を有するPLGA粒子の平均数(Cl、C2、及びC3は、NHS活性化PLGA粒子を機能化するのに使用されたPEGリンカーの濃度を表す)が示されており、より高い濃度のPEGリンカーで粒子を機能化する時、より多数の粒子が細胞に結合されることを示している。
【図8】骨髄における修飾及び未修飾間葉系幹細胞(MSC)の局在化を示す棒グラフである。図8Aは、3つの別々の実験において細胞を尾部に静脈内注射した2時間後に骨髄に局在化した、未修飾及び修飾MSC(MSCは、ビオチン−N−ヒドロキシスクシイミドで修飾され、その後ストレプトアビジン及びビオチン化シアリルルイスX、SLeXで修飾されている)の数を示す。未修飾MSCと比較して、結合したSLeXを有するMSCは、骨髄に局在化する数が増加する。図8Bは、細胞を尾部に静脈内注射した24時間後に、骨髄内皮から溢出した未修飾及び未修飾のMSCの数を示す。
【図9】37℃、24時間の時点での、PBS処理細胞及びSLeX修飾MSCによるパラクリン因子SDF−1、IGF−1、及びPGE2の細胞培養上清への分泌を示す一連の棒グラフである。
【図10】フローチャンバーアッセイのフロー条件下で、P−セレクチン表面上の修飾及び未修飾MSCの接着性相互作用を示す一連のグラフである。図1OAは、P−セレクチン表面上の未修飾MSCが、フロー条件下では表面との接着性相互作用を示さず、細胞速度は0.36ダイン/cm2のせん断応力で70μm/秒であることを示し、あらゆる修飾が無ければ、MSCがP−セレクチンと特異的に相互作用しないことを示している。図1OBは、Ab−Pセレクチン(P−セレクチンの抗体)で修飾したMSCの80%超が、強力な接着又は10ダイン/cm2まで3μm/秒未満の速度で回転することのいずれかにより、10ダイン/cm2のせん断応力まで基質と相互作用することを示す。これは、接着リガンド(この場合、P−セレクチンの抗体)の結合が、せん断条件下で細胞とP−セレクチンとの接着性相互作用を誘導することを示す。
【図11】ビオチン−HABA−アビジンアッセイにより測定された、接着モード及び懸濁モードでビオチン−N−ヒドロキシサクシニミドによりビオチン化されたhMSCSの表面上のビオチンリガンドの定量化を示す棒グラフである。
【図12】接着モードで色素(DiD)封入自己集合ファイバーを用いて修飾した細胞から得た蛍光強度(FI)を示す棒グラフである。ファイバーは、両親媒性物質の自己集合により生成され、自己集合プロセス中に蛍光色素(DiD)が封入された。これは接着モードで実施され、細胞表面の半分が修飾された。
【図13】懸濁モードで色素(DiD)封入自己集合ファイバーを用いて修飾した細胞から得た蛍光強度(FI)を示す棒グラフである。ファイバーは、両親媒性物質の自己集合により生成され、自己集合プロセス中に蛍光色素(DiD)が封入された。これは「懸濁モード」で実施され、全細胞表面が修飾された。
【図14】修飾細胞の蛍光強度を示す一連の棒グラフである。図14Aは、接着モードでの小胞によるhMSCの修飾を示す。ビオチン化脂質小胞をhMSCに付加し、その後ローダミン結合ストレプトアビジンを付加した。hMSCの修飾を、ローダミン結合ストレプトアビジンの蛍光シグナルの関数として、結合直後及び7日目に測定した。小胞で修飾されたhMSCの蛍光は、これらの方法により、ビオチン化脂質が細胞上に組み込まれ、7日目まで細胞と結合していることを示す。図14Bは、7日間にわたってMSCに結合されたビオチン脂質小胞に付加されたローダミン結合ストレプトアビジンの蛍光シグナルによって測定した、hMSC表面上のビオチン(ビオチン化脂質小胞によりhMSCに結合された)の安定性及び接近可能性を示し、MSCは、0日目にビオチン化され、ローダミン−ストレプトアビジンの蛍光シグナルは、0、2、4、及び7日目にローダミン−ストレプトアビジンを付加することにより測定した。蛍光強度は、細胞表面上のビオチンの少なくとも3分の1が、修飾のために2日目まで接近可能であり、ビオチンの約4分の1は、さらなる修飾のために依然として表面上で接近可能であることを示している。
【図15】フローチャンバーアッセイにおける、P−セレクチンでコーティングした基質上での小胞修飾hMSCの回転相互作用を示す図である。図15Aは、0.5ダイン/cm2のせん断応力における、未修飾表面上での、および小胞修飾法によるP−セレクチン処理表面上での、SLeX結合hMSCの速度を示す。図15Bは、小胞修飾hMSCの回転速度に対するせん断応力の効果を示す。
【発明を実施するための形態】
【0098】
本発明は、種々の技術により細胞表面を機能化するための方法、及び様々な応用における使用に関する。本明細書に記載の組成物及び方法は、再生用に細胞を組織に標的化するのに有用であるか、又はその代わりに作用剤を目的の特定組織へ細胞に基づいて送達するのに有用である。本明細書に記載の方法に有用な細胞は、粒子並びにリガンドの付加により修飾することができる。粒子は、タンパク質、小分子、RNA干渉分子、薬物、ビタミン、治療薬、診断薬、栄養補助剤、化粧用の特性を有する作用剤、又は核酸などの作用剤をさらに含んでいてもよい。加えて、本明細書に記載の組成物は、例えば、特に脳卒中、臓器再生、癌、骨折、及び虚血性心疾患を含む広範囲の疾患又は創傷を治療するために、当業者によって個別化することができる。
【0099】
細胞及び細胞の内部機構は、細胞が外因的に付加されるか又は本体内で標的とされるかにかかわらず、多くの生物学的応用に不可欠な構成要素である。特定細胞及び特定部位へのマイクロ粒子及びナノ粒子に基づく標的送達は、少なからぬ注目を集めてきた。細胞特異的相互作用を利用することによる本発明の方法を使用して、粒子に基づく手法により細胞を標的化することは、特定組織に薬物及び他の因子を送達するのに有用である。本発明者らは、局所的投与又は全身性投与によって、機能化粒子を細胞に標的化することができることを示した。
【0100】
本明細書中に記載の方法及び組成物には、細胞膜と細胞外マトリックス間の代替的相互作用を模倣するか又は改変するかのいずれかにより、細胞膜を機能化して細胞機能に指令を与えるか又は微小環境シグナルを制御するための様々な手法が含まれる。これは、特定の機能化を有する粒子及び/又はポリマー鎖を結合させることにより細胞を改変することによって達成される。細胞の機能化は、粒子を化学的に結合させることによって、又は非粒子コーティングにより細胞膜を直接的に機能化することによって達成することができる。結合性及び/又は可溶性作用剤を送達して細胞の遺伝子(及び/又はタンパク質)発現を変更することによって、細胞の遺伝的機能化を含むようにこの方法を拡張することができる。例えば、これは、ウイルス性及び非ウイルス性遺伝子治療、特にsiRNA送達により達成することができる。
【0101】
本発明の方法及び組成物の応用には、以下が含まれるがそれらに限定されない:生理学的及び病理学的状態下での指向性細胞遊走/侵入、細胞外マトリックス内での又は全身性標的化による生物活性剤(成長因子、酵素など)の標的化及び/又は制御放出、細胞表現型(例えば、受容体発現、細胞の追跡及び/又は画像化)の変化を検出するため細胞センサー応用。
【0102】
本明細書では、「単離された」及び、「単離する」という用語は、哺乳動物由来の生体試料から、選択された細胞タイプを分離する過程を記述するために使用される。細胞単離の方法は当業者に周知であり、一般的に、酵素反応(例えば、所望の組織又は生体試料(例えば、血液)から細胞を分離するコラゲナーゼ)、遠心分離、及び/又は組織培養皿での細胞播種が伴う。例えば、本明細書に記載の方法及び組成物に好適な細胞単離法は、米国特許第6,475,764号明細書、第5,424,208号明細書、第7,217,568号明細書、第6,991,897号明細書、又は第6,627,759号明細書に見出すことができ、それらは参照によりそれらの全体が本明細書に組み込まれる。本明細書に記載の方法及び組成物の実施に、細胞の均質集団又は細胞の異種性(例えば、複数の細胞タイプ)集団が使用できることは、本明細書において明確に意図されている。
【0103】
本明細書では、「機能化」又は「機能化された」という用語は、細胞が、例えば処置しようとする組織への標的化又は薬物送達などの所望の機能を有することを可能にする、細胞膜に対する修飾を記述するために使用される。細胞の機能化は、例えば、ポリマー、リンカー、粒子、標的化部分、化学的側鎖基、リガンド、又はこれらの任意の組合せを結合させることを包含し得る。機能化は、例えば、細胞表面上のリンカー分子に粒子を結合させることも包含する。加えて、細胞は、一部分を細胞膜に結合させることにより、又は薬物送達用に作用剤を細胞に負荷することにより機能化することができる。特に所望の場合には、2つの異なる目的、例えば組織及び受容体媒介性取込みの標的化、又は組織及び薬物送達の標的化のために細胞を機能化することもできる。そのような細胞は、本明細書では「二重機能の」細胞と呼ばれる。幾つかの場合では、細胞の機能化は、例えば、細胞表面を改変するために、自己集合両親媒性分子で形成された小胞を組織に注射することにより、in vivoで起こり得る(例えば、細胞表面は、in situ又はin vivoで修飾され得る)。
【0104】
本明細書では、「リガンド」又は「膜結合リガンド」という用語は、特に受容体、細胞表面ポリペプチド、膜、又は糖質への結合などの生物学的作用又はそのような作用の可能性を有する細胞膜に結合された外来性部分を記述するために使用される。「外来性」とは、リガンドが生物又は系内で合成されないことを意味する。幾つか場合には、リガンドは、標的組織再生(例えば、損傷組織の修復)のために、細胞を特定の臓器又は組織に向けることを可能にすることにより、標的化部分として機能することができる。標的化部分には、例えば、薬物、受容体、抗体、抗体断片、アプタマー、ペプチド、ビタミン、糖質、タンパク質、接着分子、糖タンパク質、糖残基若しくはグリコサミノグリカン、治療薬、薬物、又はこれらの組合せが含まれていてもよい。当業者であれば、細胞を修飾するための種々の標的化部分を、その細胞の意図された目的に基づいて容易に設計することができる。
【0105】
本明細書では、「粒子」という用語は、作用剤又は作用剤の混合物を送達するために又は細胞に機能性を提供するために使用することができる、細胞膜に結合されている部分を記述するために使用される。「粒子」という用語は、例えば、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、ナノ粒子、マイクロ粒子、脂質、又はデンドリマーを包含する。粒子の大きさは、粒径がおよそ0.1〜10,000nmと幅広く異なっていてもよいが、好ましくは粒子は、粒径がおよそ1〜8000nmである。粒子は、粒径がおよそl〜999nmである場合にナノ粒子とみなされ、または粒径がおよそ1000nm〜8000nmである場合にマイクロ粒子とみなされる。当業者であれば、細胞に結合させる種々の粒子を、その粒子の意図された目的に基づいて容易に設計することができる。
【0106】
本明細書では、「作用剤」という用語は、細胞効果又は組織効果を誘導することができる生物活性分子又は生物活性分子の前駆体を記述するために使用されるか、又はその代わりに送達される細胞組成物を記述するために使用される。作用剤は、小分子、薬物、成長因子、サイトカイン、酵素、RNA干渉分子(例えば、siRNA、shRNA、又はmiRNA)、増殖因子、プロドラッグ、酵素前駆体、ビタミン、栄養補助剤、治療薬、診断薬、ケモカイン、脱分化因子、又は分化因子であってもよい。作用剤の投与により調節することができる機能には、例えば、細胞成長、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着が含まれる。例えば、組織再生のために骨組織を標的としたい場合は、本明細書の実施例セクションに記載されているように、細胞を作用剤(又は複数の作用剤)で処置して骨形成系統へ分化を誘導することができる可能性がある。そのような作用剤には、これらに限定されないが、デキサメタゾン、β−グリセロリン酸、L−アスコルビン酸−2−ホスフェートが含まれる。
【0107】
本明細書では、「標的組織再生」という用語は、損傷又は疾患を軽減するための臓器又は組織の治療を記述するために使用される。「疾患の損傷の軽減」は、疾患の重症度の低減、症状の低減、疾患の完全寛解、又は医学分野の当業者に公知であるような疾患に関連する他の測定可能なパラメーターの任意の変化により測定することができる。標的組織再生は、作用剤(又は複数の作用剤)を損傷組織又は患部組織に送達すること、並びに損傷組織又は患部組織を再定植させるために細胞を投与することを包含する。このようにして治療することができる疾患又は障害には、例えば、ALS、クローン病、脊髄傷害、心臓病、脳卒中、自閉症、狼瘡、眼疾患、多発性硬化症、慢性閉塞性肺疾患、関節炎、糖尿病、自己免疫障害、虚血性心疾患、癌、創傷治癒、熱傷、及びパーキンソン病が含まれる。本質的にいかなる組織又は臓器も、本明細書で開示されている方法及び組成物による治療の標的にすることができ、それらの組織又は臓器には、脳、心臓、脈管系、肺系、腎系、脾臓系、リンパ系、骨髄、硬骨、骨筋肉系、免疫系、生殖器系、皮膚、軟骨、神経系、消化器系、肝臓、膵臓系、造血系、ホルモン系が特に含まれる。
【0108】
本明細書では、「小胞」という用語は、両親媒性二分子層を含む球状の脂質構造を指し、生物活性剤をさらに含むことができる。そのような球状構造は、本明細書では「リポソーム」とも呼ばれる。対照的に、「ミセル」という用語は、両親媒性分子(例えば、リン脂質)の単層がエネルギー的に有利である立体構造を含む球状の脂質構造を指す。一般的に、ミセルは、外側親水性球体及び内部疎水性領域を含む。ミセルを使用する生物活性剤の送達も、本明細書で意図されている。本明細書では、「両親媒性分子」という用語は、一方の端部に親水性領域を含み、反対側の端部に疎水性領域を含む分子(例えば、リン脂質)を指す。「両親媒性分子」という用語は、本明細書では、「脂質分子」という用語も包含する。「リピドイド」という用語は、RNA干渉分子の全身性送達のためのナノ粒子製剤を指し、Akinc et al., Nature Biotechnology advance online publication, 27 April 2008 (DOI:10.1038/nbt1402)に記載されている。
【0109】
「RNA干渉分子」は、本明細書中では、RNA干渉(RNAi)により標的遺伝子又はゲノム配列の発現に干渉するか又は阻害する任意の作用剤と定義される。そのようなRNA干渉剤には、これらに限定されないが、標的遺伝子又はゲノム配列に相同性のRNA分子を含む核酸分子又はその断片、低分子干渉RNA(siRNA)、低分子ヘアピン又は小分子ヘアピンRNA(shRNA)、マイクロRNA(miRNA)、及びRNA干渉(RNAi)により標的遺伝子の発現に干渉するか又は阻害する小分子が含まれる。
【0110】
本明細書では、「ビオチンの供給源」という用語は、細胞表面又は粒子表面のビオチン化を可能にするビオチン部分を含む化合物を指す。ビオチンの供給源は、当技術分野で周知である。1つの実施形態では、ビオチンの供給源は、1,2−ジオレオイル−sn−グリエルコ−3−ホスホエタノールアミン−N−(ビオチニル)ナトリウム塩(1,2-Dioleoyl-sn-Glyerco-3-Phosphoethanolamine-N-(Biotinyl) sodium salt)である。別の実施形態では、ビオチンの供給源は、ビオチニル−N−ヒドロキシ−スクシンイミドを含む。
【0111】
本明細書では、「含んでいる」又は「含む」という用語は、本発明に必須であるが、必須であるかどうかに関わらず不特定要素の包含の余地がある組成物、方法、及びその対応する構成要素(複数可)を指すために使用される。
【0112】
本明細書では、「から本質的になる」という用語は、所与の実施形態に必要な要素を指す。この用語は、本発明のその実施形態の基本的な及び新規的又は機能的な特徴に実質的な影響を及ぼさない要素の存在を許容する。
【0113】
「からなる」という用語は、本明細書に記載の組成物、方法、及びその対応する構成要素を指し、実施形態のその説明で列挙されていないあらゆる要素を除外する。
【0114】
本明細書及び添付の特許請求の範囲では、単数形「a」、「an」、及び「the」は、状況が明白にそうでないと示さない限り、複数の参照を含む。したがって、例えば、「本方法」への言及は、本明細書に記載されているタイプの及び/又は本開示などを読めば当業者に明白になるだろう1つ又は複数の方法及び/又はステップを含む。
【0115】
利点及び使用
細胞表面を修飾するために使用されている1つの手法は、細胞膜上に存在するシアル酸残基のビオチン化を含む26。この手法は、明確な基質上に細胞をパターン化するために使用された。同様に、シアル酸残基は、ビオチン化ホスフィンに基づくリンカーを化学的に結合させるために使用された27。前駆細胞は、軟骨損傷部位への幹細胞又は前駆細胞の接着を容易にするために、細胞膜をマトリックス分子に対する抗体で「ペイントする」ことにより、体中の特定領域へと標的化されている28。この戦略は、まず、脂質付加プロテインG(疎水性基、パルミチン酸による28)の細胞膜への挿入を可能にさせ、次に、軟骨基質抗原に対する抗体を溶液中でインキュベーションすることにより、細胞の外部表面上のプロテインGに対する抗体との結合を可能にさせる2段階工程に依存する。細胞表面改変の他の例には以下が含まれる:グリカンのシアル酸の非天然N−アシル置換29、アセトアミド糖のケトン基の反応30、チオール基によるシアル酸の誘導体化31、シアル酸の過ヨウ素酸酸化32、細胞内タンパク質の化学的修飾(例えば、AGT)33。RGDによる細胞表面の電気活性修飾は、細胞表面を修飾するための外部電場の使用例である34。これらの結果は、細胞膜表面を化学的及び/又は物理的に修飾できることを示す。本明細書に記載の方法及び組成物は、その微小環境との細胞相互作用に影響を与えるためのプラットフォーム技術として、粒子及び/又は膜に基づく技術により細胞表面を改変させて細胞表面を機能化するためにさらに拡張される。
【0116】
表面に結合した磁性ナノ粒子を細胞単離に使用することが示されており35、36、磁性ナノ粒子は細胞追跡にも使用されている37。これらのデータは、造血幹細胞に対していかなる有害作用も及ぼさずに、より大型のナノ粒子(例えば、0.9〜1.0μmと大型)を取り込むことができ、より低分解能での検出が可能であることを示す。これらのビーズは、典型的には、種々の種又はビオチンに由来する一次抗体に対する二次抗体であらかじめコーティングされており、選択した一次抗体を使用して細胞単離系を容易に構築することが可能になる。ビーズが細胞を捕捉した後で、ビーズを含有するチューブを磁性粒子濃縮器に配置する。結合している細胞はチューブ壁面に迅速に引き寄せられ、選択した方法に依存して上清を新しいチューブに移すか又は破棄することができる。特定のビーズは、切断部位も含有しており、粒子を細胞表面から容易に放出することが可能である。Dynabeadを用いた研究では、より高いビーズ濃度は、細胞表面上のビーズ数の増加の原因となりが、その結果として毒性がもたらされる場合があることを示している36。さらに、磁性ナノ粒子38は、例えば、内部移行を防止するためにペプチド及び/又は親水性ポリマー(PEG)で容易に修飾することができる39。細胞表面上の受容分子の挙動を追跡する研究では、受容体の移動性及び動的結合をモニターするために、標識PE−IgG粒子の結合を使用できることが示されている40。
【0117】
ポリマー又は粒子による細胞の機能化は、本明細書に記載のように、細胞機能の制御及び細胞微小環境の制御を含む生物医学的応用に多くの利点を提供する。
【0118】
遊離流動性の機能化粒子及び/又はポリマーで細胞を標的化することとは対照的に41、この方法は、特定の応用のために機能化細胞の生成を可能にする。さらに、標的送達系(粒子を標的化して、特定の細胞に作用剤を放出させる)42と比較して、この方法は、細胞を機能化して所望の作用を行なわせるため、より良好な制御を可能にする。これらのタイプの機能化により達成することができる特性を様々に組合せると、細胞が複数のタスクを行うことが可能になり、制御された所望の様式でそれらのタスクを行うように細胞に指図することが可能になるだろう。
【0119】
機能化細胞の使用は、各種の応用を有する。例えば、特定部位に対して細胞を標的送達することができる。機能化細胞を、再生用に、又は特定の腫瘍部位における作用剤の送達、例えば様々なベクター送達用に細胞を必要とする部位に向けて正確に標的化することができる。特定の指向性で遊走するか又は3Dマトリックス内でより高い侵襲能力を達成するように細胞をプログラムすることにより、細胞を機能化することもできる。例えば、機能化細胞は、細胞表面上に結合された機能的粒子により細胞外ポリマーマトリクスを分解することができ、したがって遊走速度が増加する。この方法は、組織、例えば腫瘍内の細胞分布を達成するために、及び特定の作用剤を送達するために有用であり得る。特定の応用目的(例えば、組織再生、特定の細胞を死滅させる癌治療など)で、作用剤又は複数の作用剤、つまり薬物成長因子、又は酵素を送達するために機能化細胞を使用することもできる。特定の作用剤を産生するように細胞を遺伝子組換えすることもでき、したがって機能化を使用して、特定の環境内にこれら作用剤を送達するように細胞に指図することができる。作用剤を細胞に直接送達することにより、投薬を繰り返す必要性を低減することができる。食作用(例えば、マクロファージ)による細胞の破壊を防止することにより、細胞の生存能をさらに増加させることができる。作用剤、例えばRNA干渉分子(例えば、siRNA、shRNA、miRNA)の長期間にわたる細胞への放出制御に、細胞を使用することができる。これにより、周囲の微小環境と実質的に相互作用させずに(つまり、分解を回避して)、薬物を細胞に向けることができるため、治療に必要なsiRNAの量が著しく低減されるだろう。作用剤を細胞から遠ざかるように向かわせて、細胞に初期的及び/又は直接的影響を与えずに微小環境を改変することができる。粒子は、体内又はin vitroモデル系内の特定の組織又は他の細胞に、細胞を接着させる機能(細胞固定化)を果たすことができる。
【0120】
本発明の応用は広範囲に及んでおり、様々な疾患状態に適用可能である。応用及び疾患状態の幾つかの非限定的な例には、以下のものが含まれる:免疫細胞(例えば、T細胞)の標的化、骨粗しょう症(予防及び治療)、骨形成不全症、炎症性疾患(例えば、クローン病(Chrohn's disease)、移植片と宿主との拒絶反応、関節炎、セリアック病など)、老化(例えば、変性の減少及び再生の増加)、虚血組織(例えば、心筋梗塞及び関連疾患、並びに一般的な筋変性)、心血管疾患(例えば、抹消動脈疾患)、癌、急性放射線症候群、肺疾患、心臓病、糖尿病、肝不全及び腎不全、脳卒中、禿頭症、創傷治癒、脳疾患/損害、及び神経疾患又は損害。本明細書に記載の方法及び組成物を疾患の療法に適用することは、十分に当業者の能力内にある。
【0121】
細胞
本質的にあらゆる細胞を、本明細書に記載の方法及び組成物に使用することができる。動物使用の場合には、細胞は動物由来であることが好ましく、ヒト使用の場合には、細胞はヒト細胞であることが好ましく、どちら場合も自己由来細胞供給源が好ましい。細胞は、初代細胞、例えば初代肝細胞、初代ニューロン細胞、初代筋芽細胞、初代間葉系幹細胞、初代前駆細胞であってもよく、又は細胞は樹立細胞系の細胞であってもよい。細胞は細胞分裂を起こすことが可能である必要はなく、最終分化細胞を本明細書に記載の方法に使用することができる。この状況では、細胞は、これらに限定されないが、上皮細胞、内皮細胞、ニューロン細胞、脂肪細胞、心臓細胞、骨格筋細胞、線維芽細胞、免疫細胞、肝細胞、脾細胞、肺細胞、循環血液細胞、生殖細胞、消化管細胞、腎細胞、骨髄細胞、及び膵細胞を含む任意の細胞タイプであってもよい。細胞は、これらに限定されないが、脳、肝臓、肺、腸、胃、脂肪、筋肉、精巣、子宮、卵巣、皮膚、脾臓、内分泌器官、及び硬骨などを含む任意の組織から単離された細胞系、幹細胞、又は初代細胞であってもよい。
【0122】
細胞がin vitro条件下で維持される場合、従来の組織培養条件及び方法を使用することができ、それらは当業者に公知である。種々の細胞の単離及び培養方法は、十分に当業者の知識内にある。
【0123】
特に所望の場合、リガンド及び/又は粒子による機能化の前に、細胞を処理することができる。全身性送達前に細胞を種々の作用剤で前処理して、細胞表面上の特定受容体の発現を促進させるか、又は細胞のホーミング及び生着を増強するために細胞が特定の因子を産生するように促進させるか、又はその代わり特定の細胞機能を促進させることができる。例えば、治療のために対象体へと送達する前に、細胞を誘導して細胞遊走を増強させることができる。
【0124】
加えて、異種性細胞集団及び均質細胞集団は両方とも、本明細書に記載の方法及び組成物と共に使用されることが意図されている。加えて、細胞の凝集塊、粒子に結合された又は粒子内に封入された細胞、ヒドロゲルなどの注射可能な送達媒体内の細胞、及びスキャフォールドを含む移植可能な基質に結合された細胞は、本明細書に記載の方法及び組成物と共に使用されることが意図されている。
【0125】
リガンド
細胞組成物の表面にリガンドを結合するための方法が、本明細書に記載されている。リガンドとは、細胞がin vivoで組織との所望の生物学的相互作用を示すことを可能にする細胞表面に結合された部分である。リガンドは、抗体、抗体断片、アプタマー、ペプチド、ビタミン、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、薬物、グリコサミノグリカン、又はこれらの任意の組合せであってもよい。例えば、リガンドは、癌細胞特異性抗原を認識する抗体であってもよく、したがって細胞は、腫瘍細胞と優先的に相互作用して、作用剤の腫瘍特異的送達を可能にする。一般的に、本明細書に記載の方法及び組成物の場合、リガンド(又は複数のリガンド)は、外来性である(つまり、生物内又は系内で合成されない)。好ましいリガンドは、処置される組織の外側表面上の標的分子と相互作用することが可能であるため、リガンドは、処置される組織に細胞組成物が蓄積する能力を付与することができる。他の組織との交差反応性が限定されているリガンドが、一般的に好ましい。ステルスリガンド(stealth ligand)とは、目的の組織に達するまで露出されない(例えば、粒子内に封入されているか又は捕捉されている)リガンドである。ステルスリガンドを使用する利点は、リガンド(例えば、薬物)を用いて全身性治療する場合に生じる可能性のある任意の非特異的効果又は副作用を限定することである。
【0126】
幾つかの場合には、リガンドは、細胞が特定の組織を標的とすることを可能にする標的化部分として機能することができる。そのような好適な標的化部分には、例えば特異的結合対の任意のメンバー、抗体、モノクローナル抗体、又はその誘導体若しくは類似体、限定ではないが、Fv断片、単鎖Fv(scFv)断片、Fab’断片、F(ab’)2断片、単一ドメイン抗体、ラクダ化抗体及び抗体断片、ヒト化抗体及び抗体断片、並びに先述の多価性型を含む、;限定ではないが、典型的には共有結合で結合されているか、又はそうでなければ安定化されている(つまり、ロイシンジッパー又はヘリックス安定化)scFv断片である、ジスルフィド安定化Fv断片、scFvタンデム((ScFv)2断片)、ダイアボディ、トリボディ(tribody)、又はテトラボディ(tetrabody)などの単一特異性又は二重特異性抗体を含む多価性結合試薬が含まれていてもよく、他の標的化部分には、例えば、アプタマー、受容体、リガンド、及び融合タンパク質が含まれる。
【0127】
当業者であれば、細胞の目的に基づいてあらゆる標的化部分を選択することができ、例えば、タモキシフェンなどのエストロゲン受容体リガンドは、細胞表面上のエストロゲン受容体数の増加を示すエストロゲン依存性乳癌細胞に対して細胞を標的できる。リガンド/受容体相互作用の他の非限定的な例には、以下のものが含まれる:CCR1(例えば、関節リウマチ及び/又は多発性硬化症の炎症関節組織又は脳の治療用)、CCR7、CCR8(例えば、リンパ節組織の標的化)、CCR6、CCR9、CCR10(例えば、腸組織の標的化用)、CCR4、CCR1O(例えば、皮膚の標的化用)、CXCR4(例えば、一般的な遊出増強用)、HCELL(例えば、炎症及び炎症性障害、骨髄の治療用)、アルファ4ベータ7(例えば、腸粘膜標的化用)、VLA−4/VCAM−1(例えば、内皮の標的化)。一般的に、標的化(例えば、癌転移)に関与するあらゆる受容体を、本明細書に記載の方法及び組成物での使用に利用することができる。幾つかの実施形態では、修飾リガンドは、ポリ(エチレングリコール)、ヒアルロン酸、デキストラン、キトシン(chitosin)、又はポリ(エチレンオキシド)などのステルスリガンドを含む。
【0128】
粒子
細胞に結合された粒子の特性は、例えば、それらの粒径、形態、組成、表面電荷、多孔度、表面性状、機能的ドメインの濃度又はドメインのタイプ、分解特性、粒子が1つ又は複数の作用剤(成長因子、磁石、サイトカイン、接着性作用剤、毒素、タンパク質、ペプチド、酵素、核酸、抗体、細胞受容体、又はそれらの断片を含む)を含有しているかどうか、そのような作用剤の位置(例えば、表面上又は内部)などを含む多数のパラメーターに関して、粒子のタイプ間で異なる場合があり、又は単一粒子内でさえ異なる場合がある。特に所望の場合、粒子は作用剤で構成されていてもよく、その場合およそ1%から実質的に粒子全体(つまり、およそ100%)が所望の作用剤である。潜在的な磁性剤を含有することに加えて、粒子はコーティング磁石であってもよく又は非コーティング磁石であってもよい。
【0129】
粒子は、細胞膜との直接的相互作用により細胞表面に結合されていてもよい。粒子上に存在する機能性は、ポリマー性であってもよく、非ポリマー性であってもよく、又はオリゴマー性であってもよい。粒子上の結合部位は、粒子が細胞表面と相互作用することができれば、イオン性(陽イオン性及び陰イオン性)であってもよく、又は非イオン性であってもよい。細胞に粒子を結合させる手法は、種々の化学的方法及び/又は物理的方法により表面があらかじめ機能化されている「ボトムアップ」手法を使用して実施することができる。その後、機能化された細胞表面を使用してポリマー粒子を結合させ、機能化細胞を作製することができる。
【0130】
粒子の粒径及び形状は、標的化における粒子の運命を決定するために重要である。一般的に、200nmを超える粒子は細胞によって内部移行され、したがって細胞膜に吸着される22。最近の研究では、棒状の粒子は、球状微粒子と比較してそれほど効果的に内部移行されないことが示されている23。物質の特性も、内部移行に大きな影響を及ぼす22。例えば、疎水性で(例えば、ポリスチレン粒子)、それほど接着性でない(例えば、PVA)粒子コーティングを使用して、細胞の取り込みを阻害することができる24。さらに、ポリマー粒子の表面電荷も、粒子の内部移行の運命を決定するために重要である25。これらの研究は、粒子を内部移行させずに細胞表面を機能的に修飾できる可能性が高いことを示すが、細胞をそのように機能化する方法は報告されていない。
【0131】
粒子の内部移行の可能性を回避するために、粒子と細胞表面との間にスペーサー分子を導入することにより、粒子の寸法を増加させることができる。これにより、非腫瘍細胞と比較して、ペグ化されたナノ粒子の取り込み低下を示す腫瘍細胞で観察されるように、内部移行が低減されるだろう。同様に、官能化されたスペーサー(又はリンカー)分子を介して粒子を細胞表面に結合させることができる。リンカーは、任意の物理化学的相互作用により直接的又は間接的に粒子に結合可能な分子であり、本明細書中の発明を実施するための形態にさらに記載されている。粒子及び結合剤は、それらが介在構造なしで共有結合又は非共有結合によって互いに結合されている場合、「直接的に結合」されている。粒子及び結合剤は、それらがリンカーを介して互いに結合されている場合、「間接的に結合」されると言う。
【0132】
細胞表面が最初に機能化され、その後リンカー及び機能化粒子が結合される別の「ボトムアップ」手法を使用することができる。リンカー分子の選択は、一方の端部があらかじめ機能化された細胞に接着し、他方の端部が、機能化粒子に結合するようなものであろう。結合剤(あらかじめ機能化された細胞上の)又はリンカーは、粒子上の官能基に結合される。或いは、粒子又はリンカーは、結合剤の官能基に結合される。
【0133】
別の粒子に基づく手法は、異なる特徴を有する異種性(例えば、ヤヌス)粒子を使用して達成される。粒子の一方の半分は、粒子が細胞表面と相互作用することを可能にするだろう細胞接着機能性を有していてもよく(つまり、陽イオン性ポリマーは、優先的に細胞膜と相互作用することが示されている)、その一方で粒子の他方の半分は、本明細書に記載の方法の所望の応用、例えば薬物送達用に設計されるだろう。
【0134】
粒子特性は、これらに限定されないが、大きさ、直径、形状、組成、表面電荷、分解特性、粒子が1つ又は複数の作用剤を含有するかどうか、又はそのような作用剤の位置(例えば、表面上又は内部)を含む多数のパラメーターに関して、互いに異なってもよく(例えば、異種性集団)、又は単一粒子集団内で異なってもよい。
【0135】
「異種性粒子」の修飾の1つは、作用剤を送達する粒子の一方の半分(細胞に結合される)を標的とすることを含み、他方の半分は、これらに限定されないが、特に指向性細胞遊走、指向性細胞結合、及び標的送達などの応用を含む特定の機能を果たすように機能化される。
【0136】
別のタイプの機能化は、リンカー分子によって分離されている、2つの異なる機能性を含有する物質を使用することにより達成することができる。2つの機能性のうちの1つは、細胞と特異的に相互作用する(細胞膜又は細胞質内で優先的に)が、他方の機能性は、記載されている方法の所望の応用、例えば、薬物送達のために外部環境中に存在する。細胞に結合された他方の機能性は、細胞膜を介して内部移行される場合があり、細胞のセンサー及び/又はマーカーとして機能することができるか、又は異なる手法によって細胞膜の表面に結合させることができる。
【0137】
別の技術には、ポリマー鎖を構築して、ポリマー鎖と細胞膜間の適切な相互作用によって細胞表面をコーティングすることを伴う。
【0138】
機能化(例えば、NHS、ペプチド、エポキシ、イミドエステルなど)ポリマーを使用して、機能化ポリマーが細胞表面と相互作用するように細胞膜を包み込むことができる。この技術は、当業者に公知の連続ポリマー吸着に又はエマルジョン技術によって適用することができる。
【0139】
ポリマーを、ポリマーが細胞膜上に層を形成するように、連続して細胞膜に適用することができる。加えて、例えば連続吸着により又は細胞表面上にあらかじめ吸着されているポリマーに機能化粒子を結合させることにより、所望の応用のために機能化粒子をポリマー層に接着することができる。
【0140】
これらの技術は両方とも、粒子に基づく手法と比較してより効率的な様式で、細胞膜の機能化を可能にするだろう。膜に基づく手法は、適切な結合部位を介してポリマーを結合することにより細胞表面を修飾するという同じ概念を利用する。結合相互作用は、物理的、例えば荷電ポリマーのイオン性相互作用、抗体−抗原相互作用などであってもよい。同様に、相互作用は、ポリマー機能性(アミン、カルボキシル、硫化物など)に依存して化学的であってもよい。
【0141】
結合部位の粒子及び/又はリンカーは、生物学的な事象に応答して切断されるか、又は外部的に制御される切断部位を含有することもできる。これら粒子は、組織へと拡散してもよく、又は細胞近傍に留まっていてもよい。
【0142】
粒子を使用して、移植された(注射又は埋め込まれた)細胞の局在化を増強することもでき、例えば、細胞に結合された反応基を使用して、ある組織又は物質内又は特定の組織又は物質上に細胞を固定化することができる。加えて、より高度の弾性を有する粒子(例えば、軟質粒子)を使用して、生物学的障壁(例えば、血管内皮)を越えて輸送することにより、粒子の局在化能力を増強することができる。
【0143】
機能化粒子は、in vitroで又はin vivoで細胞に結合させることができる(in vivoでの結合には、最初に粒子を体内の特定細胞へと標的化することを伴っていてもよく、粒子は局所的に又は全身性に送達することができる)。幾つかの実施形態では、粒子は、ポリ(エチレングリコール)、ヒアルロン酸、デキストラン、キトサン、又はポリ(エチレンオキシド)などのステルスリガンドを含有する。
【0144】
修飾することができる粒子の特性には、これらに限定されないが、形状、表面電荷、多孔度、化学的組成、相対的疎水性/親水性、機械的特性、及び表面性状が含まれる。粒子は、生物学的分子(例えば、抗体、ペプチド、ヌクレオチド)、又は合成分子(例えば、小分子、アプタマー)を結合させることにより修飾されていてもよい。同様の技術を使用して、活性のタイミング又は位置を制御することもできる。加えて、粒子は、1つ又は複数の作用剤をさらに含んでいてもよい。作用剤は、粒子内(例えば、粒子の細孔内又はチャネル内)及び/又は粒子の外部表面上に位置して(例えば、組み込まれて)いてもよい。幾つかの実例では、粒子には、1つ又は複数の作用剤があらかじめ負荷されている。粒子がリガンドを含有する場合、リガンドは細胞と直接的に相互作用しないが、むしろリガンド相互作用が粒子内のみで生じることが好ましい。
【0145】
作用剤
様々な異なる医薬品/治療薬を、本明細書に記載の方法と共に使用することができ、それらには、小分子、タンパク質、抗体、ペプチド、及び核酸が含まれるがこれらに限定されない。一般的に、本発明により投与することができる生物活性剤には、限定ではないが以下のものが含まれる:抗生物質及び抗ウイルス物質などの抗感染剤;化学療法剤(つまり、抗癌剤);拒絶反応抑制剤;鎮痛薬及び鎮痛薬の組合せ;抗炎症剤;ホルモン(例えば、ステロイド);成長因子(例えば、骨形態形成タンパク質(つまり、BMP1〜7)、上皮成長因子(EGF)、線維芽細胞増殖因子(つまり、FGF1〜9)、血小板由来増殖因子(PDGF)、インスリン様増殖因子(IGF−I及びIGF−II)、形質転換増殖因子(つまり、TGF−β−III)、血管内皮増殖因子(VEGF));エンドスタチンなどの抗血管新生タンパク質、及び他の天然由来又は遺伝子組換えタンパク質、ポリサッカライド、糖タンパク質、又はリポタンパク質。加えて、本明細書に記載の粒子を使用して、例えば薬剤、ビタミン、鎮静薬、ステロイド、催眠薬、抗生物質、化学療法剤、プロスタグランジン、及び放射性医薬品などの任意のタイプの分子化合物を送達することができる。本明細書に記載の細胞組成物は、これらに限定されないが、タンパク質、ペプチド、ヌクレオチド、糖質、単糖、細胞、遺伝子、抗血栓剤、抗代謝剤、増殖因子阻害剤、成長促進物質、抗凝血剤、抗有糸分裂剤、線維素溶解剤、抗炎症ステロイド、薬物、及びモノクローナル抗体を含む上記物質及びその他の送達に好適である。
【0146】
本明細書に記載の方法で使用するのに好適な他の生物活性剤の例には、これらに限定されないが、以下のものが含まれる:コラーゲン、エラスチン、フィブロネクチン、ビトロネクチン、ラミニン、プロテオグリカン、又は細胞結合に影響を及ぼすことが知られている既知のインテグリン結合ドメイン、例えば「RGD」インテグリン結合配列を含有するペプチド若しくはその変異体などの細胞結合媒介物質(Schaffner P & Dard 2003 Cell Mol Life Sci. Jan;60(1): 119-32; Hersel U. et al. 2003 Biomaterials Nov;24(24):4385-415);生物活性リガンド;及び特定の種類の細胞内部成長又は組織内部成長を増強又は排除する物質。
【0147】
幾つかの実施形態では、粒子は、ビスホスホネートに基づく薬物、エストロゲン受容体モジュレーター、又はホルモンに基づく療法などの骨粗しょう症の治療用薬物を含む。ある実施形態では、粒子は、ラロキシフェン、AAR494、又はE−64などの破骨細胞再吸収を阻害する薬物を含む。幾つかの実施形態では、本方法は、骨減少症を治療するための方法をさらに含む。幾つかの実施形態では、本方法は、骨癌を治療するための方法をさらに含む。幾つかの実施形態では、粒子は、果粒球コロニー刺激因子又は骨髄特異的表面膜受容体などの硬骨標的化因子を含む。幾つかの実施形態では、硬骨標的化因子は、骨粗鬆症の硬骨を標的とするペントシジンなどの因子である。幾つかの実施形態では、本方法は、骨髄空間を標的とするための方法をさらに含む。
【0148】
幾つかの実施形態では、作用剤は、骨成長を促進するための作用剤を含む。ある実施形態では、作用剤は、レプチン、ソルチリン、トランスグルタミナーゼ、プロスタグランジンE、1,25−ジヒドロキシビタミンD3、アスコルビン酸、β−グリセリンリン酸、TAK−778、スタチン、IL−3及びIL−6などのインターロイキン、成長ホルモン、造血幹細胞因子(SF)、アクチビンA(ACT)、レチノイン酸(RA)、上皮成長因子(EGF)、骨形態形成タンパク質(BMP)、血小板由来成長因子(PDGF)、肝細胞成長因子、インスリン様成長因子(IGF)I及びII、造血成長因子、ペプチド成長因子、エリトロポイエチン、インターロイキン、腫瘍壊死因子、インターフェロン、コロニー刺激因子、ヘパリン結合性成長因子(HBGF)、TGF−β1などのアルファ又はベータ形質転換成長因子(α又はβ−TGF)、線維芽細胞成長因子、血管内皮成長因子(VEGF)、神経成長因子(NGF)、及び筋肉形態形成因子(MMP)などの成長因子又はサイトカインを含む。
【0149】
幾つかの実施形態では、粒子は、プロコラーゲンなどのコラーゲン又はアスコルビン酸の産生又は構築を促進する作用剤を含む。ある実施形態では、粒子は、NFκBリガンド(Rank−L)の受容体活性化因子などの、硬骨へ粒子再吸収を促進するための再吸収因子、デキサメタゾンなどのコルチトステロイド(cortitosteroid)、副甲状腺ホルモン、マクロファージコロニー刺激因子(M−CSF)、又は形質転換成長因子−β1(TGF−β1)を含む。幾つかの実施形態では、粒子は、EDTAに基づく作用剤などの、血液由来の無機質をキレートする作用剤、ポリ(ビスホスホネート)、ポリ(ホスフェート)、カルシウム及び/又はリン酸塩を核とする生物学的又は非生物学的物質、アスパラギン酸、オステオポンチン、又は骨シアロタンパク質を含む。
【0150】
細胞組成物それ自体は、幾つかの態様では、生物活性剤を送達するための担体として考えることができるが、本明細書に記載の細胞組成物の生物活性剤/治療薬/医薬品は、処置される組織だけでなく細胞組成物の微小環境にも影響を与えることもでき、そのため細胞組成物それ自体が生物活性剤になる。例えば、細胞組成物の増殖特徴を増強する目的のために、生物活性剤を粒子に供給することができる。このタイプの送達は、細胞組成物が損傷組織を再定植させるために使用される標的組織再生に特に有用である。
【0151】
機能化方法
幾つかの機能化方法を、本明細書に記載の方法と共に使用することができる。幾つかの例示的な実施形態には、(1)細胞表面上の糖残基にある種々の官能性が関与する反応、(2)細胞表面上のペプチド残基にある官能基が関与する反応、(3)抗原−抗体相互作用の使用、(4)細胞表面と粒子間のイオン性相互作用、及び(5)細胞表面と粒子間の疎水性相互作用を含む。
【0152】
粒子に基づく細胞表面の修飾は、異なる機能性を有する粒子を細胞膜に結合させることにより達成することができる。細胞表面に存在する反応部位及び表面部分を、様々な化学を使用することにより化学的に修飾することができる。
【0153】
細胞表面の物理的特性は、細胞とECM間の相互作用を変更するために使用される化学的修飾と同じように操作することができる。細胞表面は、異なる物理的特性(例えば、電荷)を示す複雑な構造を有する。粒子又はポリマー鎖の結合に使用することができる細胞表面上の様々な官能性には、これらに限定されないが以下のものが含まれる:特に、極性(NH2/NH3+)末端基、及びリン脂質の他の官能性、ヒドロキシル(OH)、及び糖質基の他の官能性、カルボン酸基(COOH)、チオール(SH)、及び種々のタンパク質、及び糖タンパク質、抗原。粒子(中実又は中空)は、細胞膜に接着するように異なるイオン特徴に調製することができる。他のタイプの物理的接着は、細胞表面上の抗原−抗体相互作用を制御することにより可能である。抗体は、特定の応用のために機能化することができる。
【0154】
陽イオン性結合剤の例には、これらに限定されないが、特にキトサン及びポリアミンが含まれる。陰イオン性結合剤の例には、これらに限定されないが、特にポリスルホネート、ポリホスフェート、DNA、ヘパリン、及びPMAMデンドリマーが含まれる。非イオン結合には、例えば、カルボキシル、アミン、又はスルフィド官能性との結合が含まれる。
【0155】
本明細書に記載の1つの手法には、ポリマー鎖及び粒子の使用による化学的修飾が伴う。この技術には、有機ポリマー及び/又はオリゴマー粒子を使用することができるが、同様に種々の無機粒子にも拡張することができる。粒子(中空又は中実)表面と細胞膜間の化学的相互作用は、種々の官能性(例えば、ビオチン化、N−ヒドロキシサクシニミド(NHS)、エポキシ、ペプチド、イミドエステル、マレイミド、アジド、ハロアセチル、ピリジルジスルフィド、カルボジイミド、ヒドラジド)により達成することができ、文献に記載されている細胞表面修飾の化学的経路を使用することができ、それらは当業者に公知である。物理的相互作用は、例えば、荷電ポリマー;自己集合性荷電粒子、抗体−抗原、又は疎水性相互作用の使用により実施することができる。
【0156】
この手法では、中実粒子及び中空粒子の両方を使用することができ、それらは、生物医学的プロセス(例えば、細胞遊走、接着、増殖、分化、生存、マトリックス産生)を制御/モニターするための、重要な機能的作用剤(例えば、成長因子、薬物、酵素、蛍光性部分)を含有することができる。粒子を「コーティング」または「修飾」するための当業者に公知の方法により、粒子がそれで構成されている巨大分子とは異なる分子を、粒子の外側表面に結合することができる。例えば、リンカーの使用により(下記で検討)、分子を、粒子の外側表面に直接的又は間接的に結合することができる。これらの分子は、結合を容易にし、受容体媒介性を増強し、エンドサイトーシス又は破壊の回避を提供するなどの目的のために結合される。例えば、リン脂質などの生体分子を粒子表面に結合させて、エンドソームによるエンドサイトーシスを防止することができ、受容体、抗体、又はホルモンを表面に結合させて、体内の所望の臓器、組織、又は細胞に対する粒子の結合を促進又は容易にすることができ、グルカンなどのポリサッカライド、又はポリビニルピロリドン及びPEGなど他のポリマーを粒子の外側表面に結合させて、マクロファージによる取り込みを増強又は回避することができる。
【0157】
溶液中での細胞機能化を使用すると、均質な機能化を容易にする及び/又は細胞凝集を刺激することができる一方で、表面上での細胞の機能化は、細胞の1つの表面を優先的に機能化するのに有用であり得る。
【0158】
細胞への粒子の追加は、静的又は動的条件下で達成することができ、マイクロ流体チャネルを含むバイオリアクター及び/又はBioMEMSデバイスの使用を含んでいてもよい。粒子を有する細胞は、粒子を有する細胞の肺送達用に吸入剤として使用することができる。
【0159】
物質選択
本明細書に記載のように、粒子を結合させて細胞組成物を形成するための物質の選択は、当業者による所望の応用に必要と見なされる修飾のタイプに依存する。合成、天然、並びに半合成ポリマーを、ポリマー粒子の合成に使用することができる。様々な合成ポリマーには、例えば、ヒドロゲルポリマー(PEG、PVAなど)又はアクリレートが含まれる。これらポリマーは、当業者の必要性に従って直鎖状であってもよく又は架橋されていてもよい。使用することができる天然ポリマーには、これらに限定されないが、ヒアルロン酸、ゼラチン、キチンなどが含まれる。物理的相互作用の場合、例えば、ポリエチレンイミン(PEI)、ポリ(リジン)、キトサン、又はセルロースを含む幾つかのポリマーを、電荷に基づく細胞表面への接着に使用することができる。使用することができるポリマーのリストには、これらに限定されないが、以下のものが含まれる:ポリ(ラクチド)(PLA)、ポリ(グリコリド)(PGA)、ポリ(ラクチド−コ−グリコリド)(PLGA)、ポリ(カプロラクトン)(PCL)、ポリカーボネート、ポリアミド、ポリ無水物、ポリホスファゼン、ポリアミノ酸、ポリオルトエステル、ポリアセタール、ポリシアノアクリレート、及び生分解性ポリウレタンなどの生分解性ポリマー;ポリアクリレート、エチレン酢酸ビニールポリマー、及び他のアシル置換酢酸セルロース、並びにそれらの誘導体などの非生物分解性ポリマー;ポリウレタン、ポリスチレン、ポリ塩化ビニル、ポリフッ化ビニル、ポリ(ビニルイミダゾール)、クロロスルホン化ポリオレフィン、及びポリエチレンオキシド。生分解性天然ポリマーの例には、以下のものが含まれる:アルブミン、コラーゲン、合成ポリアミノ酸、及びプロラミンなどのタンパク質;アルギナート、ヘパリンなどのポリサッカライド;及び他の天然に存在する糖ユニットの生分解性ポリマー。或いは、前述のポリマーの組合せを使用することができる。
【0160】
使用することができる無機粒子の例には、これらに限定されないが以下のものが含まれる:特に、二酸化チタン、炭酸カルシウム、リン酸カルシウム、ケイ酸カルシウム、銀及び金ナノ粒子、及び磁性粒子。本明細書に記載の方法に有用である広範囲の幾何学的形状を有する様々なタイプの粒子を使用することができる。粒子形状の非限定的なリストには、例えば、コアシェル物質、中空粒子、ケージ様粒子が特に含まれる。
【0161】
リンカー
リンカーには、ヒドロキシル基、第一級又は第二級アミノ基、リン酸基若しくはその置換誘導体、又はカルボン酸基などの官能基が含まれていてもよい。アシルカルニチン、アシル化カルニチン、スフィンゴシン、セラミド、ホスファチジルコリン、ホスファチジルグリセロール、ホスファチジルエタノールアミン、ホスファチジルイノシトール、ホスファチジルセリン、カルディオリピン、及びホスファチジン酸などの極性脂質も、リンカーとして機能することができる。極性脂質分子は、官能基を有していてもよく又は有していなくともよい有機スペーサー分子に共有結合で随意に結合されてもよい。他のリンカーには、ヘテロ二官能性架橋剤が含まれる。例えば、サクシニミジル−4−(N−マレイミドメチル)シクロヘキサン−1−(カルボキシ−6−アミノカプロエート)は、LC−SMCCとしても知られており、スルフヒドリル基及びアミン基と反応するヘテロ二官能性架橋剤である。リンカーの非限定的な例には、上述されているポリマー性の陰イオン性及び陽イオン性、非イオン性作用剤が含まれる。
【0162】
粒子の製作は、様々な技術により実施することができる。エマルジョンの溶媒蒸発又は噴霧乾燥を応用して、粒子を製作することができる。中実粒子が望ましいか又は中空粒子が望ましいかに依存して、複数エマルジョン技術を使用することができる。化学的修飾は、様々な化学反応の使用により実施することができ、それは必要とされる側鎖基又は官能性に依存する。
【0163】
幾つかの実施形態では、リンカーは、ポリ(エチレングリコール)、ヒアルロン酸、デキストラン、キトシン、又はポリ(エチレンオキシド)などのステルスリガンドを含む。
【0164】
細胞への架橋粒子及び/又はリガンド
細胞膜を研究するために使用される方法
細胞膜分子の構造的組成、分布、細胞結合、及び動力学は、様々な化学を使用するにより研究することができる。様々な化学的架橋剤を、細胞膜及び細胞小器官膜を膜の外側表面及び膜に囲まれた空間内の両方に架橋することができる。架橋剤は、表面受容体又はそれらのリガンドを識別するために使用されることが多い。膜不透過性の架橋剤は、細胞表面特異的な架橋を保証する。不水溶性の架橋剤を、制御された量の試薬及び制御された反応時間で使用すると、膜透過及び内膜タンパク質との反応を低減することができる。NHSエステルのサクシニミジル環に結合されたスルホニル基により、水溶性で膜不透過性であり、内膜タンパク質と反応しない架橋剤がその結果としてもたらされる。したがって、スルフォ−NHS−エステルを使用する場合、反応時間及び架橋剤の量はそれほど重要ではない。
【0165】
細胞膜の構造研究では、細胞の脂質二分子膜内の位置及び環境を決定するために、様々な疎水性の試薬が必要とされる。蛍光タグは、膜の内側及び外側のタンパク質、脂質、又は他の分子を位置付けるために使用される。スペーサーアームの長さが様々である種々の架橋剤を使用して、タンパク質を膜内の結合分子に架橋し、分子間の距離を決定することができる。より短い架橋剤で架橋が成功する場合は、2つの分子がある様式で相互作用していることを強く示す。一団のより短い架橋剤では架橋が得られないが、より長い試薬を使用すると結合が得られる場合は、一般的に、分子が膜の同一部分に位置するが相互作用していないことを示す。ホモ二官能性のNHS−エステル、イミドテソル(imidotesor)ヘテロ二官能性のNHS−エステル/光活性化可能なフェニルアジドは、これらの手順で一般的に使用されている。イミドエステル架橋剤(イミデート)は水溶性であるが、やはり膜を透過することができる。スルフヒドリル反応性架橋剤は、システインを有する分子を膜内の他の分子に対して標的化するのに有用であり得る。EDC、不水溶性ジシクロヘキシルカルボジイミド、DCC、及び他の水溶性/水不溶性カップリング試薬対は、膜及び細胞構造、タンパク質サブユニット構造及び配置、酵素:基質相互作用、並びに細胞表面及び膜受容体を研究するために使用される。EDCが親水性であるという特徴は、その結果として、膜及びサブユニット研究において、DCCなどの疎水性カルボジイミドよりももっと異なる架橋パターンをもたらすことができる。1つの実施形態では、水溶性及び水不溶性のカルボジイミドと架橋させて、関与するタンパク質:タンパク質相互作用の空間的配置の全体像を取得することができる。細胞表面上の機能化粒子及び/又はリガンドの位置及び相互作用を試験するためのこれら架橋法は、本明細書に記載の組成物に有用である。
【0166】
細胞表面上で架橋反応を実施するための方法
架橋剤を使用して、試料中のタンパク質の構造及び組成を研究することができる。幾つかのタンパク質は、pH又は塩条件が異なると異なる立体構造で存在するため、研究するのが難しい。立体構造の変化を回避する1つの方法は、サブユニットを架橋することである。アミン、カルボキシル、又はスルフヒドリル反応性試薬が、特定のアミノ酸の特定又はサブユニットの数、位置、及び大きさの測定に使用される。分子内架橋が所望の場合は、短〜中程度の長さのスペーサーアーム架橋剤が選択される。スペーサーアームが長すぎると、分子間架橋が生じ得る。
【0167】
アミン反応性又は光活性化可能なアミン反応性架橋剤などの長さが短い結合試薬と一緒になって、スペーサーアームを結果としてもたらさないカルボジイミドは、至適濃度及び条件で使用されれば、外来性分子と架橋することなくサブユニット間で架橋する。
【0168】
わずかにより長い架橋剤は、サブユニット間も架橋することができるが、分子間結合がその結果としてもたらされる場合がある。試薬量及びタンパク濃度を調整することにより、分子間架橋を制御することができる。ホモ二官能性の架橋剤を使用する場合、希釈タンパク質溶液及び高濃度架橋剤が、分子内架橋に有利である。
【0169】
三次元構造を決定又は確認する場合は、長さがより増加したスペーサーアームを有する切断可能な架橋剤を使用して、サブユニット間の距離を決定することができる。様々な反応基を有する架橋剤を使用した実験により、特定のアミノ酸の位置を示すことができる。結合させた後、タンパク質を二次元電気泳動にかける。第1の次元では、タンパク質は非還元条件を使用して分離され、分子量が記録される。幾つかのサブユニットは架橋されていない場合があリ、それらの個々の分子量により分離されるだろう。結合されたサブユニットは、合計の分子量により分離されるだろう。その後ゲルの第2の次元を、架橋サブユニットを切断する条件を使用して実施する。架橋サブユニットの個々の分子量を決定することができる。還元されなかった架橋サブユニットは、対角線パターンをもたらすはずだが、切断されたサブユニットは、対角線からはずれるだろう。個々のサブユニットの分子量は、還元SDSポリアクリルアミドゲル電気泳動を使用して、タンパク質サブユニットの所定の分子量と比較されるべきである。この架橋技術は、必要な生体特異的応用のために種々の機能性を有する細胞表面を作り出すことを可能にする。
【0170】
より好ましい実施形態では、ホモ二官能性スルフォ−NHS−エステル、ヘテロ二官能性スルフォ−NHS−エステル、及び光反応性フェニルアジドが、細胞表面上のタンパク質を架橋するために使用される。特定のタンパク質が表面上に位置するか又は膜内部分に位置するかの決定は、水溶性又は水不溶性架橋剤を使用して、既知のタンパク質又は放射性標識に対する細胞膜調製物の結合反応を実施することにより達成することができる。結合させた後、細胞を洗浄し、可溶化し、SDSポリアクリルアミドゲル電気泳動(PAGE)により特徴付け、目的タンパク質が結合したかどうかを決定することができる。内在性膜タンパク質は、水不溶性架橋剤の存在下で結合体を形成するが、水溶性架橋剤の存在下では形成しないだろう。表面の膜タンパク質は、水溶性及び不水溶性の架橋剤の存在下で結合することができる。ホモ二官能性の光活性化可能なフェニルアジドは、タンパク質相互作用及び結合を研究するためのより汎用性の高い架橋剤の1つである。これは、切断可能であり、125Iで放射性標識することができる。切断した後、解離された分子は両方とも依然としてヨウ素化されているだろう。この架橋剤上の両反応基は非特異性であるため、架橋はアミノ酸組成に依存せず、結合が成功する。
【0171】
細胞投与
細胞を対象体に投与するための様々な手段が、当業者に公知である。そのような方法には、全身性注射、例えばi.v.注射、又は対象体の標的部位への細胞の埋め込みが含まれていてもよい。細胞は、対象体への注射又は埋め込みによる導入を容易にする送達デバイスに挿入されてもよい。そのような送達デバイスには、受容対象体の体内に細胞及び液体を注入するためのチューブ、例えばカテーテルが含まれていてもよい。1つの好ましい実施形態では、チューブは、本発明の細胞を所望の位置で対象体へ導入することができる針、例えば注射器をさらに有している。細胞は、様々な異なる形態での送達用に調製することができる。例えば、細胞は、溶液又はゲルに懸濁されてもよく、又はそのような送達デバイスに含有される場合は支持マトリックスに埋め込まれていてもよい。細胞は、本発明の細胞が依然として生存可能である、薬学的に許容される担体又は希釈剤と混合されていてもよい。薬学的に許容される担体及び希釈剤には、生理食塩水、緩衝水溶液、溶媒、及び/又は分散媒質が含まれる。そのような担体及び希釈剤の使用は、当技術分野で周知である。溶液は、好ましくは無菌及び流動性である。好ましくは、溶液は、製造及び保管条件下で安定しており、例えばパラベン、クロロブタノール、フェノール、アスコルビン酸、及びチメロサールなどの使用により、細菌及び真菌などの微生物の汚染作用に対して保存されている。本発明の溶液は、本明細書に記載の細胞を、薬学的に許容される担体又は希釈剤、並びに必要に応じて上記に列挙されている他の成分に組み込むことにより調製され、その後滅菌ろ過されていてもよい。
【0172】
細胞投与経路は、比較的非侵入性であることが好ましく、例えば静脈注射、吸入による肺送達、経口送達、口腔内投与、直腸内投与、膣内投与、局所的投与、又は鼻腔内投与による。しかしながら、細胞投与経路は、治療される組織に依存するはずであり、埋め込みを含んでいてもよい。細胞送達用の方法は当業者に公知であり、医学分野の当業者であれば、本明細書に記載の方法及び組成物に使用するために推定することができる。
【0173】
細胞投与用の直接注射技術を使用して、脈管構造全体を介した遊出、又は特定の臓器、例えば肝臓又は腎臓又は他の任意の臓器の脈管構造への遊出を刺激することもできる。これは、脈管構造の非特異的標的化を含む。例えば肝臓門脈などの特定の注射部位を選択することにより、任意の臓器を標的とすることができる。或いは、注射は、体中の任意の静脈へと全身性に実施することができる。この方法は、高齢患者の幹細胞数を増強するのに有用である。加えて、細胞は、空白の幹細胞ニッチに定植するか、又は臓器を補充する新しい幹細胞を生成するために機能し、それにより臓器機能を向上させることができる。例えば、細胞は、脈管構造内の周皮細胞位置を占めることができる。
【0174】
細胞送達を使用して、活性血管新生部位を標的とすることもできる。例えば、内皮前駆細胞又は間葉系幹細胞若しくは前駆細胞の送達は、創傷部位での血管新生応答を増強することができる。血管新生の標的化は、薬物を腫瘍に標的化するための媒体として細胞を使用するのにも有用である。
【0175】
特に所望の場合、哺乳動物又は対象体を、作用剤であらかじめ処置してもよく、例えば、組織への細胞の標的化を増強するために作用剤(例えば、ホーミング因子)を投与し、それをその部位に設置して、所望の組織への細胞の標的化を容易にすることができる。例えば、ホーミング因子の組織への直接注射は、リガンド標的細胞の全身性送達に先立って実施することができる。
【0176】
本明細書及び添付の特許請求の範囲では、単数形「a」、「an」、及び「the」は、状況が明白にそうでないと示さない限り、複数の参照を含む。したがって、例えば、「本方法」への言及は、本明細書に記載されている及び/又は本開示などを読めば当業者に明白になるだろうタイプの1つ又は複数の方法及び/又はステップを含む。加えて、「細胞」という用語は、性質が異種性又は均質性のいずれでもあってもよく、細胞の凝集塊とも呼ばれる細胞集団として解釈することができる。
【0177】
記載されている技術は、胚の生着効率を向上させるために応用することができ、例えば体外受精中に、特定の組織(例えば、子宮)への結合を増強することができる接着リガンドで、胚を修飾することができる。
【0178】
先述の詳細な説明及び以下の例は単なる例示に過ぎず、本発明の範囲の限定として受け取られるべきではないことが理解される。開示された実施形態の種々の変更及び改変は、当業者であれば明白であり、本発明の趣旨及び範囲から逸脱することなく行うことができる。さらに、特許、特許出願、及び特定されている出版物は全て、例えば、本発明に関して使用し得るような出版物に記載の方法を説明及び開示するために、参照により明示的に本明細書に組み込まれる。これら出版物は、もっぱら本出願の出願日前にそれらが開示されているために提供される。これに関してはいかなるものも、本発明者らが、先行発明であるため又は他のいかなる理由でもそのような開示に優先する資格がないことを承認すると解釈されるべきではない。日付に関する宣言又はこれら文書の内容に関する提示は全て、本出願者らに利用可能な情報に基づいており、これら文書の日付又は内容の正確さに関していかなる承認も構成しない。
【実施例】
【0179】
実施例1
細胞をどのように修飾して細胞標的化を向上させることができるかに関する非限定的な例を、本明細書に記載する。具体的には、標的化作用剤を細胞表面に結合し、これにより、in vitro実験で実証されたように細胞の回転を誘導することができる。
【0180】
シアリルルイスX(SLeX)によるヒト間葉系幹細胞(hMSC)の修飾
hMSCの修飾を実施して、細胞膜の表面にSLeXを化学的に結合させた。ビオチンストレプトアビジン結合を利用して、細胞表面のSLeX部分を化学的に組み込んだ。細胞表面上に存在する遊離アミン基を、ビオチニル−N−ヒドロキシ−スクシンイミドのN−ヒドロキシ−スクシンイミド基と反応させて、細胞表面をビオチン化した。引き続いてこのステップの後、ストレプトアビジン分子と細胞表面のビオチン部分を反応させた。ビオチンとストレプトアビジンとの強力な相互作用は、ストレプトアビジン分子の細胞表面上への固定化を可能にする。その後、ストレプトアビジンをビオチン化SLeX(シアリル−Lex−PAA−ビオチン)と反応させて、細胞表面上にSLeXを導入する。hMSCの表面上にSLeXなどの標的化作用剤が存在しないことは、細胞の非回転性特徴の要因であるため、SLeXを細胞上に固定化することにより、hMSCに回転性特徴を誘導することができる。ビオチン化ステップを向上させるために、スルホン化ビオチニル―N−ヒドロキシ−スクシンイミド(スルフォ−NHS−ビオチン、BNHS)を使用した。BNHSは水溶性であり、細胞膜を透過せず、したがってBNHSのN−ヒドロキシ−スクシンイミド基が、細胞膜と優先的に相互作用することが可能になる。
【0181】
一般的方法
典型的には、約5000個の細胞を、96ウエルプレートの各ウエルの200μL hMSC細胞培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)に、24〜48時間の期間添加して、その結果細胞は約80〜90%コンフルエントだった。
【0182】
細胞を処理するために、培地をウエルから吸引し、細胞を各ウエルの中で、200μLのリン酸緩衝生理食塩水(PBS、pH7.4、Ca/Mg非含有)を用いて穏やかに室温で洗浄した。Ca/Mg非含有のPBS、pH7.4中1mMの100μL BNHS溶液を、各ウエルに室温で20〜25分間添加した。指定の期間後、細胞を、200μLのリン酸緩衝生理食塩水(PBS、pH7.4、Ca/Mg非含有)で2回室温で洗浄した。その後、50μLのストレプトアビジン溶液(Ca/Mg非含有のPBS、pH7.4中50μg/mL)を各ウエルに添加し、室温で20分間インキュベートした。指定の期間後、細胞を、200μLのリン酸緩衝液生理食塩水(PBS、pH7.4、Ca/Mg非含有)で2回室温で洗浄し、その後50μLの4μg/mL SLex(Ca/Mg非含有PBS、pH7.4中)を室温で添加した。
【0183】
BNHS及びストレプトアビジンによる細胞修飾を評価するために、蛍光性ローダミンを結合したストレプトアビジン(SR)を、ビオチン化細胞に付加し、SR結合の蛍光強度を測定した。この実験に使用した対照は、SRのみで処理した細胞、ビオチン及びSRで処理した細胞、及び未処理細胞だった。蛍光強度を7日後に再び測定し、PBS中0.1%の100μLトリトンX溶液で処理した後、細胞を4%ホルムアルデヒド溶液で固定し、100μLのDAPI溶液(PBS中1μg/mL)で染色した。ビオチン類似体で処理した細胞の蛍光強度を、機能化後の0日目及び7日目に測定した。BNHS−SR結合細胞の蛍光強度は、B−SR又はSR処理のみの蛍光強度より高かった。報告した結果は全て、三重反復で行われた実験の結果である。
【0184】
細胞の生存率
細胞の生存率は、トリパンブルー排除法を使用して実施した。手短に言えば、細胞を12ウエルプレートに播種し、終夜放置して接着させ、細胞を上述のようにBNHS溶液で処理した。すすきの後、細胞を37℃及び5%CO2で48時間インキュベートした。その後、培地を吸引し、200μLの細胞解離溶液を使用することにより、細胞をウエルから分離した。300μLの培地を添加し、合計500μLの細胞懸濁液を収集した。この懸濁液からの10μL細胞懸濁液を、4%トリパンブルー溶液を使用することにより1:1に希釈し、細胞を血球計で計数して、生存可能(未染色)及び生存不能(青染色)な細胞数を決定した。三重反復実験から計算した群中の平均細胞生存率に差異はなかった。この実験の対照には、未処理細胞(しかし、実験中にPBSを添加し、室温で維持した)、並びにNHSを用いずにビオチン及びSRで処理した細胞が含まれていた。
【0185】
BNHSで修飾した細胞の生存率は、BHSによる処理が、修飾の48時間後でさえ細胞生存率を変更しないことを示す。未処理細胞は、48時間後で85%生存可能であり、一方でビオチン処理細胞は76%生存可能であり、BNHS処理細胞は75%生存可能だった。細胞は修飾の48時間後で生存可能だったため、これは、BNHSによる細胞の修飾が細胞に対して毒性ではないことを示す。
【0186】
ビオチン化細胞上のビオチンの安定性及び接近可能度
細胞表面上のビオチンの安定性を分析するために、細胞を上述のようにビオチン化した。NHSを用いずにビオチン処理した細胞を対照として使用した。0、2、4、及び7日目に、200μLのSR溶液(Ca/Mg非含有のPBS、pH7.4中50μg/mL)を、ビオチン処理又はBNHS処理細胞のいずれかに室温で20分間添加して、細胞表面上のビオチンの安定性を評価した。SRを付加した後で、PBS中細胞を4%ホルムアルデヒド溶液で固定し、0.1%の100μLトリトンX溶液で処理した後、100μLのDAPI溶液(PBS中1μg/mL)で染色した。BNHS処理及びビオチン処理細胞の両方の蛍光顕微鏡画像を、蛍光強度を測定することにより分析した。ビオチン官能性の安定性を、付加したローダミン−ストレプトアビジンの蛍光強度を分析することにより測定した。
【0187】
細胞表面上のビオチンの安定性は、各時点でローダミン−ストレプトアビジンを付加し、画像の蛍光強度を直ちに測定することにより分析した。7日間にわたるBNHS機能化の安定性を、NHSを用いていないビオチン(対照)と比較して試験した。NHS処理により、7日間にわたって細胞膜上に安定的なビオチン基が提供されることが示された。
【0188】
BNHSで機能化された細胞の蛍光強度は、0日目(機能化の当日)では、B(ビオチン)及びSRと比較してより高く、細胞がNHS官能性により機能化されたことを示す。7日後に強度を再検査し、BNHS+SR処理の強度が、B+SR及びSR対照と比較して著しく高くなったことが見出され、ビオチン化細胞へのローダミン結合が依然として存在したことが示された。代表的な7日間の光学顕微鏡画像及び同じ観察視野の対応する蛍光画像は、細胞が、NHS化学により効果的にビオチン化され、1日目に付加した結合ストレプトアビジンが、7日間の培養の間安定的であったことを示す。
【0189】
細胞接着
細胞接着は、細胞のビオチン化後に組織培養ウエル上の接着細胞数を測定することにより分析した。T25フラスコ中の80%のコンフルエント細胞を、上述のようにビオチン化した。ストレプトアビジン結合の後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、その後5000個の細胞を、96ウエルプレートの各ウエルに10、30、及び90分間播種した。その後、PBSで2回すすぐことにより非接着細胞を除去し、その後接着細胞を固定し、トルイジンブルー溶液で細胞を染色した。接着細胞を、1O×倍率で3つのウエルの6つの観察視野で計数して、接着細胞数を決定した。表面に接着した細胞のパーセントを、細胞の初期播種密度に基づいて計算した。この実験の対照は、未処理細胞を含んでいた。
【0190】
BNHS処理細胞と対照との間の接着細胞パーセントに差異はなく、BNHSによる細胞の修飾は、細胞の接着特徴を変更しないことを示す。修飾細胞は、組織培養プラスチック上でそれぞれ10、30、及び90分間接着させた後の未処理細胞と比較して、著しく異なる接着特徴を示さない。これは、細胞の接着特徴の要因である細胞表面受容体(例えば、インテグリン)が、NHSビオチン細胞表面修飾により影響を受けないことを明白に示す。
【0191】
修飾細胞の増殖アッセイ
細胞増殖アッセイを使用して、T25フラスコ上の細胞数を測定することにより細胞増殖を分析した。T25フラスコ中の80%コンフルエント細胞を、1mLの1mM BNHSで処理し、その後1mLの50μg/mLストレプトアビジン溶液で20分間処理した。その後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、50,000個の細胞を3つのT25フラスコに播種した。細胞数を、3個のフラスコの1O×倍率の10観察視野で、播種後の1日目、2日目、4日目、及び6日目に評価した。BNHSにより修飾された細胞の増殖特徴を示す増殖曲線は、増殖が最終的に、両群においてコンフルエント単層化の原因となること示している。
【0192】
SLeXによる細胞の機能化及びその結果生じる回転の特徴付け
T25フラスコ中の80%コンフルエント細胞を処理することにより、細胞の修飾を実施した。細胞を1mLの1mM BNHSで処理し、その後1mLの50μg/mLストレプトアビジン溶液で20分間処理した。細胞を1mLのPBSで2回洗浄し、その後1mLの4μg/mL SLeX(Ca/Mg非含有PBS、pH7.4中)を、室温で20分間添加した。細胞を、細胞解離溶液を使用してフラスコから分離し、遠心分離後に培地に再分散した。フローチャンバー実験用の細胞濃度は、典型的には1×105細胞/mlだった。細胞の回転特徴は、ガスケットの厚さが127μmで長さが6cmの長方形の平行平板フローチャンバー実験より評価した。ガラス表面上へのP−セレクチン固定化を、700μLのP−セレクチン溶液(5μg/mL)をスライドガラス上で18時間インキュベートすることにより実施し、フロー実験は、スライドガラス上にチャンバーを設置することにより実施した。0.094ダイン/cm2の壁面せん断応力に対応する20μL/分の流速を使用した。細胞の回転をモニターするために位相差顕微鏡法を使用し、画像を1O倍の観察視野で記録し、およそ10秒毎に手動で取り込んだ。細胞の速度は、10秒間わたって移動した細胞の距離を測定することにより計算した。対照は、未処理細胞、及び細胞表面上に物理的に吸着されたSLeXを有する細胞だった。SLx(SLeX)又はBNHS−SLxで修飾した細胞は、速度の低下及び回転特徴の増加を示す。せん断応力及び細胞に付加したBNHSの濃度の影響を評価するため、細胞の回転速度及びフラックスを、複数の流速(それぞれ0.366、0.73、及び1.89ダイン/cm2に対応する20、40、及び100μL/分)で測定した。
【0193】
培養したhMSCは、それらの表面上にリガンドを発現して細胞回転を可能にしないことが明確に記述されており、これらの結果は未処理hMSCで確認されていた。BNHSで修飾しその後SLeXで修飾した細胞は、0.366ダイン/cm2の壁面せん断応力において、未処理細胞、及びSLeXが物理的に細胞表面上に吸着した細胞より著しく低い速度示す。未処理細胞と比較してSLeX固定化の速度が著しくより低いことは、SLeXとP−セレクチン間の相互作用が、0.366ダイン/cm2の壁面せん断応力で、速度を約2μm/sに減少させることを示す。これは、ビオチン−ストレプトアビジン、その後にSLeX−ビオチンによる細胞修飾が、この細胞タイプに本来備わっていないhMSCの回転特徴を誘導することを示す。
【0194】
修飾細胞の回転特徴に対する流速及びしたがって様々なせん断応力の影響を測定した。せん断応力が0.366ダイン/cm2から1.88ダイン/cm2に増加すると共に、修飾細胞の速度は増加するが、フラックス(基質と相互作用する細胞の数)は、ほとんど不変のままである(1mMのBNHS定濃度において)。未修飾細胞は、19.5細胞/mm2秒の一定のフラックスを示した。これは、SLeXで修飾した細胞が、認識された細胞回転に基づく接着現象により、表面上のP−セレクチンと特異的に相互作用することを示す。
【0195】
0.366ダイン/cm2の一定のせん断応力における、修飾細胞の回転特徴に対するBNHS濃度の影響(これは、本質的には細胞表面上の修飾リガンドの密度対照である)も評価した。1mMから0.01mMまでBNHS濃度を減少させると、速度は増加するが、修飾細胞のフラックスはほとんど不変である(及び19.5細胞/mm2秒のフラックスを示した未修飾細胞のフラックスより高い)。これは、細胞表面上のリガンド濃度を変更することにより、回転応答を制御する能力を示す。低濃度のBNHSが使用される場合でさえ、細胞は、フラックスデータで観察されるようにP−セレクチンと相互作用することができる。
【0196】
BNHS結合並びにその後のストレプトアビジン及びSLex結合による細胞修飾を、組織培養プレート上の接着細胞で実施した。この細胞表面修飾では、表面と結合していない細胞の部分のみが修飾され、したがってリガンドと接触して結合し、組織培養プレートに接着している細胞表面は、修飾にさらされない。これは、細胞表面の半分がSLexにより修飾されることの原因となる。したがって、P−セレクチン表面に対するSLex修飾細胞の接着は、回転速度より顕著である。これにより、様々な条件のせん断応力及びBNHS濃度でも、細胞のフラックス値が変わらない理由が説明される。
【0197】
修飾細胞の分化能
ビオチン/ストレプトアビジン結合により修飾した細胞のin vitro骨形成及び脂肪生成分化能を、下記に記載のように評価した。
【0198】
骨形成分化
hMSCを24ウエルプレートの4つのウエルに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。細胞修飾を2段階法により実施した。典型的には、細胞を1mMのBNHSでビオチン化し、その後PBS中のストレプトアビジン(50μg/mL)を室温で結合させた。その後、デキサメタゾン、β−グリセロリン酸、L‐アスコルビン酸−2−ホスフェート、及びα−MEMを含有する骨形成誘導培地(Lonza社製、hMSC骨形成Single Quoteキット)中で23日間細胞を培養することにより、骨形成分化を誘導した。陽性対照として、未処理(未修飾)細胞を有する同数のウエルを骨形成誘導培地中で維持した。両群の培地を3日毎に交換した。骨形成は、アルカリホスファターゼアッセイ及びフォンコッサ染色により評価した。
【0199】
アルカリホスファターゼアッセイは、培地を吸引し、蒸留水で細胞を洗浄することにより実施した。細胞を3.7%ホルムアルデヒド溶液で15分間室温で固定し、その後蒸留水で2回洗浄した。それに、(DMF及びナフトールAS MX−P04)を含有するTris HCl及び蒸留水中0.06%のRed Violet LB食塩溶液を添加した。プレートを45分間インキュベートし、その後ウエルを蒸留水で3回すすいだ。
【0200】
ビオチン及びストレプトアビジンにより修飾した細胞の骨形成分化能を、アルカリホスファターゼ染色により測定した。修飾細胞用のアルカリホスファターゼ染色は、陽性染色を示し、未修飾細胞と同様の結果を示す。これは、細胞のビオチン−ストレプトアビジン修飾が、細胞の骨形成能を妨害せず、修飾細胞が骨形成系統に分化することができることを示す。
【0201】
脂肪生成分化
hMSCを24ウエルプレートの4つのウエルに播種し、100%コンフルエンスに達するまで細胞増殖培地で培養した。細胞修飾を2段階法により実施した。典型的には、細胞を1mMのBNHSでビオチン化し、その後PBS中のストレプトアビジン(50μg/mL)を室温で結合させた。その後、脂肪生成誘導培地(Lonza社製、h−インスリン(組換え)、L−グルタミン、デキサメタゾン、インドメタシン、IBMX(3−イソブチル−1−メチル−キサンチン)、Pen/Strepを含有するhMSC脂肪生成Single Quoteキット)、及び脂肪生成維持培地(Lonza社製、h−インスリン(組換え)L−グルタミン、Pen/Strepを含有するhMSC脂肪生成Single Quoteキット)中で細胞を23日間培養することにより、脂肪生成分化を誘導した。陽性対照として、未処理(未修飾)細胞を有する同数のウエルを、脂肪生成誘導培地中で維持した。誘導培地及び維持培地と周期的に接触させるLonza社のプロトコールに従って、両群の培地を3日毎に交換した。
脂肪生成は、オイルレッドO染色により評価した。
【0202】
使用したオイルレッドO染色プロトコールは以下の通りである:細胞から培地を完全に吸引し、PBSで1回洗浄し、PBSを3.7%ホルムアルデヒドに室温で30分間置換して細胞を固定し、ホルムアルデヒドを蒸留水に数分間置換し、イソプロパノールをオイルレッドO作業用溶液(30mlのオイルレッド0.5%イソプロパノール溶液を20mlの蒸留水で希釈することにより作製)に置換し、5分後にオイルレッドO溶液を蒸留水で2回洗浄した。1mlのヘマトキシリン(Sigma−Aldrich社製)をウエルに1分間添加し、その後吸引し、ウエルを蒸留水で洗浄した。倒立位相差顕微鏡を使用してウエルを観察した。脂質は赤色見え、核は青色に見えた。
【0203】
ビオチン及びストレプトアビジンで修飾した細胞の脂肪生成分化能を、オイルレッドO及びヘマトキシリン染色により評価し、その結果は、修飾細胞が陽性染色を示し、未修飾細胞と同様の結果を示すことを表している。これは、細胞のビオチン−ストレプトアビジン修飾が、細胞の脂肪生成能を妨害せず、修飾細胞が脂肪生成系統に分化することができることを示す。
【0204】
実施例2
本明細書の実施例2に記載の方法は、例えば、幹細胞及び分化細胞を含む任意の細胞タイプの標的送達に使用することができる。特異的抗原を提示する活性化樹状細胞(DC)をリンパ節に標的化して、ワクチン接種戦略を向上させることができる43。加えて、T細胞又は他の免疫細胞の標的送達を、本明細書に記載の方法により実施することができる。薬物又は薬物送達デバイス(例えば、粒子)を細胞表面に封入することは、細胞微小環境を制御するのに、及び薬物を細胞に直接的に送達する(長期間にわたって)のにも有用である。これは、血漿又は他の生物学的実体との相互作用により迅速に除去又は不活化される薬物に特に有用である。徐放性薬物送達は、本明細書に記載されている細胞表面への共有結合性固定化によって、又は非共有結合的方法による組み込みによって達成することができる。
【0205】
この方法は、本明細書で開示されている方法の1つの実施形態を記述しており、ヒト間葉系幹細胞の共有結合的機能化を伴っており、任意の細胞タイプに適用することができる。
【0206】
この実施形態では、細胞機能化は、細胞表面に共有結合で直接結合させずに達成される。このためには、以下のビオチン化脂質を使用することができる:中性、陽イオン性、及び陰イオン性など電荷が異なる様々な頭部基を有する脂質;様々な長さの糖鎖を有する脂質(表1を参照);及び糖鎖の不飽和度が様々である脂質。
【0207】
このプラットフォーム手法は、既存の機能化方法より優れており、以下を含む複数の利点を提供する:i)簡単な調製方法、ii)穏やかな反応条件、iii)高価な/複雑なタンパク質発現ステップの回避、iv)時間の短縮及びキットで使用することができる細胞操作。
【0208】
同じ目標、つまり特異的リガンドによる細胞の機能化を達成するために、2つの異なる手法を使用することができる。1つの手法では、ビオチン化脂質を有する小胞にストレプトアビジンを付加し、ストレプトアビジンでコーティングした細胞にシアリルルイスXを付加する。その後、修飾しようとする細胞の二分子層に小胞を融合させる。第2の手法では、最初に修飾しようとする細胞をビオチン化脂質小胞と融合させて、その後ストレプトアビジン、次にシアリルルイスXを細胞に付加する。両手法の最終産物は本質的に同一である。
【0209】
方法1
方法1では、最初に単層状又は/及び多重層状の小胞を、リン酸緩衝生理食塩水(PBS、pH7.4)又は他の任意の水溶液中で、「ビオチン化脂質」のみ又は「異なる比率で支持脂質を有するビオチン化脂質」のいずれかを使用して製作することができ、それをストレプトアビジン溶液(ビオチンに結合することができる)に添加し、室温で5〜30分間インキュベートし、小胞溶液を10,000rpmで2分間遠心分離し、その後上清を除去する。ペレット(小胞を含有する)をPBSで洗浄して、未結合ストレプトアビジンを除去した。ビオチン化シアリルイスX(biotinylated Sialy Lewis X)(SiLeX)をペレットに添加し、5〜30分間インキュベートし、その後遠心分離及びPBS洗浄ステップを繰り返して小胞を取得し、それらの脂質頭部基をビオチン−ストレプトアビジン−ビオチン−SiLeX基でコーティングする。最後に、小胞を、hMSC及び/又は別の細胞タイプと共に1〜15分間インキュベートした。小胞と細胞膜との融合が生じ、細胞表面をビオチン−ストレプトアビジン−ビオチン−SiLeX基でコーティングすることができ、これらはセレクチン(この場合P−セレクチン)との排他的な相互作用に感受性である。
【0210】
この手法では、以下の方法によりビオチン化脂質を使用して小胞を調製した:a)ストレパタビシン(strepatavidin)溶液をこの混合物と共に5〜30分間インキュベートし、その後遠心分離し、過剰の未結合ストレプトアビジンを除去することにより、表面上にビオチン−ストレプトアビジン機能化を有する小胞の調製が可能になった。B)さらなるビオチン化SiLeXを小胞に添加し、5〜30分間インキュベートし、遠心分離及び過剰(未融合)小胞の除去を実施した。残りの小胞は、表面上にビオチン−ストレプトアビジン−ビオチン−SiLeX基を含有する。C)そのような小胞を、ヒト間葉系幹細胞(hMSC)(又は他の細胞タイプ)と共に1〜15分間インキュベートし、その結果生じた小胞と細胞膜との融合により、ビオチン−ストレプトアビジン−ビオチン−SiLeX基による細胞表面の機能化が引き起こされた。この機能化はセレクチン(この場合P−セレクチン)との排他的な相互作用に感受性である。
【0211】
方法2
方法2では、ビオチン化脂質を使用して小胞を調製し、hMSC(又は他の任意の細胞タイプ)の懸濁物と共にPBS中で1〜15分間インキュベートし、その後細胞を10,000rpmで3分間遠心分離し、上清を除去した。その結果生じたペレットは、ビオチンでコーティングされた細胞を含有する。ビオチンでコーティングされた細胞を、ストレプトアビジン溶液と共に5〜30分間インキュベートし、その後遠心分離して未結合ストレプトアビジンを除去した。これにより、ビオチン−ストレプトアビジン基による細胞表面のコーティングが可能になる。前のステップと同様に、ビオチン化SiLeXと共に細胞を5〜30分間インキュベートし、遠心分離を繰り返し過剰な試薬を除去し、このようにして表面上にビオチン−ストレプトアビジン−ビオチン−SiLeX基を有する細胞を生成する。
【0212】
この手法では、以下のプロトコールによりビオチン化脂質を使用して小胞を調製した:a)hMSC又は他の任意の細胞タイプのいずれかと共に1〜15分間インキュベートし、その後細胞を10,000rpmで3分間遠心分離し、その後上清を除去した。その結果生じたペレットはビオチンでコーティングされた細胞を含有する。b)それらの細胞に、ストレプトアビジン溶液を添加し、5〜30分間インキュベートし、その後未結合ストレプトアビジンを遠心分離で除去した。この方法により、ビオチン−ストレプトアビジン基による細胞表面のコーティングが可能だった。C)ビオチン化SiLeXを添加し、5〜30分間インキュベートし、細胞を遠心分離にかけ過剰な試薬を除去した。この方法は、表面上にビオチン−ストレプトアビジン−ビオチン−SiLeX基を有する細胞の調製の原因となる。同様のステップは、培養プレート上の接着細胞にも実施することができる。
【0213】
所望の小胞特性に依存して、様々な頭部基及び様々な電荷(例えば、陽イオン性、陰イオン性、中性、及び双極イオン性)を有する種々のリン脂質を使用することができる。例えば、ホスファチジルコリン、ホスファチジルセリン、及びホスファチジルエタノールアミンに基づく脂質を、小胞の製作に使用することができる。様々な疎水性鎖、例えばグリセロール骨格に結合された対称性及び非対称性アシル基も、本明細書に記載の方法で使用するために意図されている。表1には、本明細書で意図される非対称性アシル基及び対称性アシル基が列挙されている。
【0214】
この方法は、ビオチン化脂質のみに限定されておらず、むしろ細胞機能化は、任意の「修飾脂質」を用いて実施することができる。所望の官能基/分子/ナノ粒子/ビーズを脂質頭部基に結合させ、その後任意のタイプの細胞表面に挿入することができる。加えて、所望の薬物/分子/成長因子/粒子/ビーズを、機能化脂質により調製された小胞に封入することができ、したがって封入された物質を細胞内に送達し、同時に機能化脂質で表面をコーティングすることができる。
【0215】
実施例3:懸濁状態の細胞の細胞表面機能化
懸濁モードでのシアリルルイスX(SLeX)によるヒト間葉系幹細胞(hMSC)の修飾
懸濁状態のビオチニル−N−ヒドロキシ−スクシンイミドのN−ヒドロキシ−スクシンイミド基を反応させることによるhMSCの共有結合性修飾は、細胞の表面全体を修飾する機会を提供するが、接着モードでは細胞表面のおよそ半分が表面修飾中に露出する。したがって、修飾が懸濁状態の細胞上で実施される場合、ビオチン及び結合部分の分布は、細胞膜全体にわたってより均一である。これにより、細胞の標的化及びホーミング効率を増加させることができる。具体的には、細胞が懸濁状態にある間にSLeXを結合させることにより、接着細胞上で実施された修飾と比較して(試薬が同じ濃度で使用された場合)、よりロバストな回転応答が付与される。
【0216】
典型的には、hMSCを、hMSC増殖培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)中で90%コンフルエンスまでT75フラスコで培養し、その後細胞を、1×トリプシン−EDTA溶液を使用してトリプシン処理し、次にリン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して、培地及びトリプシンを除去した。その後、細胞ペレットを15〜20分間1mLの1mM BNHS溶液に分散した。その後、細胞を遠心分離及び遠心沈降させて、BNHS溶液を除去した。細胞を再懸濁しその後遠心分離することにより、細胞ペレットをPBSで2回洗浄した。遠心分離後、細胞ペレットを、1mLのストレプトアビジン溶液(Ca/Mg非含有PBS、pH7.4中50μg/mL)に15〜20分間再懸濁した。非結合ストレプトアビジンを遠心分離により除去した。
【0217】
BNHS及びストレプトアビジンによる細胞修飾を評価するために、蛍光性ローダミンを結合したストレプトアビジン(SR)を、ビオチン化細胞に付加し、結合したSRの蛍光強度を測定した。この実験に使用した対照は、SRのみで処理した細胞、ビオチン及びSRで処理した細胞、及び未処理細胞だった。蛍光強度を7日後に再び測定し、細胞を4%ホルムアルデヒド溶液で固定し、PBS中0.1%の100μLトリトンX溶液で処理した後で、100μLのDAPI溶液(PBS中1μg/mL)で染色した。報告した結果は全て、三重反復で実施した実験の結果である。修飾の当日(0日目)に、BNHS+SR基は著しくより高い蛍光を示し、蛍光は7日目にさらに増加した(対照SR及びB+SRと比較して)。これは、BNHSが特異的に及び共有結合で細胞表面に結合し、細胞表面の恒久的修飾の原因となることを示す。
【0218】
ビオチン化細胞上のビオチンの安定性及び接近可能度
細胞表面上のビオチンの安定性を分析するために、細胞を上述のようにビオチン化した。NHS非含有のビオチン処理細胞を対照として使用した。細胞を96ウエルプレートに24時間接着させた。指定の期間後、1、2、4、及び7日目に、200μLのSR溶液(Ca/Mg非含有PBS、pH7.4中50μg/mL)を、ビオチン処理細胞又はBNHS処理細胞のいずれかに室温で20分間添加して、細胞表面上のビオチンの安定性を評価した。SRを添加した後、細胞を4%ホルムアルデヒド溶液で固定し、PBS中0.1%の100μLトリトンX溶液で処理した後で、100μLのDAPI溶液(PBS中1μg/mL)で染色した。BNHS処理細胞及びビオチン処理細胞の両蛍光顕微鏡画像を、蛍光強度を測定することにより分析した。ビオチン機能性の安定性を、付加したローダミン−ストレプトアビジンの蛍光強度を分析することにより測定した。7日間にわたるBNHS機能化の安定性を、NHS非含有のビオチン(対照)と比較して試験し、細胞膜上で7日間にわたって安定的なビオチン基が提供された。
【0219】
BNHSにより修飾された細胞の生存率
懸濁状態で修飾した細胞の生存率を、修飾直後(0時間)にトリパンブルーを使用して試験した。結果は、修飾ステップ後で約69%の細胞が生存可能であることを示す。この実験の対照は、ビオチン処理細胞、及びいかなる処理もしないが同様の実験条件下で懸濁及び遠心分離した細胞だった。対照細胞は約80%の生存率を示し、一方でビオチンをコーティングした細胞は約73%の生存率を示した。これは、BNHS修飾が生存率を著しくは減少させないことを示す。生存率が低かったのは、生存率を減少させる修飾工程のステップが原因である。
【0220】
懸濁状態におけるBNHS修飾の効果を評価するため、細胞を懸濁状態で修飾し、同じ対照を共に使用して48時間後に生存率を試験した。BNHS修飾細胞は、48時間後に80%生存可能だった(90%の生存細胞及び85%の生存可能ビオチン対照と比較して)が、それは修飾が細胞に対していかなる実質的な毒性効果も誘導しないことを示す。
【0221】
修飾細胞の接着
細胞をビオチン化した後、96ウエルプレートの細胞培養ウエル上の接着細胞数を測定することにより、細胞接着を分析した。細胞を、上述のように懸濁状態でビオチン化した。ストレプトアビジンを結合した後、細胞を洗浄し、細胞培地に再懸濁した。細胞を血球計で計数し、その後およそ5000個の細胞を、96ウエルプレートの各ウエルに10、30、及び90分間播種した。その後、PBSで2回すすぐことにより非接着細胞を除去し、その後接着細胞を固定し、トルイジンブルー溶液で細胞を染色した。接着細胞を、1O×倍率で3つのウエルの6つの観察視野で計数して、接着細胞数を決定した。
【0222】
懸濁状態で修飾した細胞の接着を、10、30、及び90分直後に試験したところ、修飾細胞の約25%が最初の10分で接着し、その後30分でおよそ40%、及び90分でおよそ68%が接着することが示される。細胞の接着がより低いのは、細胞表面全体にわたる細胞修飾に起因し、そのため細胞は接着分子の一部を失う。同様の実験条件下で懸濁及び遠心分離処理した対照細胞は、10、30、及び90分で、それぞれ約36%、60%、及び75%の接着を示す。これは、細胞の接着特徴がより低いことが、主としてBNHSによる修飾によるのではなく、修飾のステップに依存することを示す。
【0223】
修飾細胞の増殖評価
懸濁状態で修飾した細胞の増殖を、8日間試験した。細胞を懸濁状態でBNHSにより修飾した後、およそ50,000個の細胞を、T25フラスコに入れ、8日間の期間にわたって計数した。増殖曲線は、最終的にコンフルエントな単層化に結びつく増殖を示す。同様の実験条件下で懸濁及び遠心分離処理した対照細胞は、懸濁状態での細胞修飾が、修飾細胞の増殖能を損なわないことを示す。
【0224】
懸濁状態で修飾したhMSCの回転特徴
ビオチン化SLeXを使って懸濁状態で修飾したhMSCは、未修飾細胞(PBS処理)より著しく低い速度を示す。懸濁状態でBNHSにより修飾したhMSCの速度は、0.37ダイン/cm2のせん断速度でフローチャンバーを通って流れ、細胞は0.55μm/秒の速度で回転していた。修飾細胞と比較して、細胞を修飾しなかった対照は、75μm/秒の速度で移動した。これは、懸濁モードでBNHSを使用したビオチン−ストレプトアビジン結合によるSLexの導入は、P−セレクチンでコーティングした表面上におけるより効果的な細胞の回転を誘導することを示す。
【0225】
修飾細胞の回転特徴に対する流速及びしたがって様々なせん断応力の効果を評価した。懸濁状態でBNHSにより修飾したhMSCの速度に対するせん断応力の効果は、せん断応力の増加が速度を増加させることを示す。せん断応力を0.37ダイン/cm2から1.8ダイン/cm2に変化させることにより、0.55μm/秒から1.5μm/秒の速度の増加が観察される。これは、より高いせん断応力レベルでは、修飾細胞が、自然に回転する細胞と同様のより低い速度の回転特徴を示すことを表す。懸濁状態でBNHSにより修飾したhMSCのフラックスに対するせん断応力の影響は、せん断応力の増加がフラックスを減少させることを示す。0.37ダイン/cm2から1.8ダイン/cm2に速度を変化させることにより、155細胞/(mm2×秒)から118細胞/mm2秒へのフラックス減少が観察される(20細胞/(mm2×秒)のフラックスを示す未修飾細胞と比べて)。これは、より高いせん断応力レベルで、修飾細胞がP−セレクチン表面に接着することができることを示す。
【0226】
実施例4:ビオチン−ストレプトアビジン結合を使用したPLGA粒子によるhMSCの修飾
ストレプトアビジン結合PLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。ストレプトアビジンを、標準的なカルボジイミドカップリング技術を使用して、PLGA粒子のカルボン酸基に共有結合で結合させた。PLGA(Strep−PLGA粒子)粒子に共有結合で結合されたストレプトアビジンを3回洗浄して、物理的に吸着したストレプトアビジンをPLGA粒子の表面から除去した。
【0227】
hMSC表面へのStrep−PLGA粒子の結合
hMSCを、PBS中で1mMスルフォ−NHS−ビオチン(BNHS)により室温でビオチン化し、その後ストレプトアビジン結合PLGA(Strep−PLGA)粒子を結合させた。2つの陰性対照をこの実験に使用した。一方の対照セットでは、Strep−PLGAを未修飾細胞に付加し、つまり細胞はビオチン化されていなかった。第2の対照セットでは、細胞はビオチン化されていたが、未修飾PLGA粒子、つまりカルボン酸で末端化されたPLGA粒子を付加した。
【0228】
Strep−PLGA粒子を有する細胞(及び対照)の画像を計数したところ、1個の細胞当たり最も多くの平均粒子を有する細胞は、BNHS/Strep/PGLAを使用して修飾したそれらの細胞だったことが示されている。結果は、Strep−PLGAビーズが、特異的なビオチン−ストレプトアビジン相互作用により、ビオチン化hMSCに結合することを示す。実験群(BNHS+Strep−PLGA)と陰性対照群(Strep−PLGA及びBNHS−PLGA)との間に著しい差異が存在する。非常に多くのStrep−PLGA粒子が、共有結合でビオチン化されたhMSCに結合したが、これは粒子が細胞と特異的に結合することを示す。
【0229】
Strep−PLGA粒子で修飾したhMSCの生存率
細胞の生存率を、トリパンブルー排除法を使用して測定した。手短に言えば、細胞を12ウエルプレートに播種し、終夜放置して接着させ、細胞をBNHSでその後Strep−PLGA粒子で上述のように処理した。異なるウエルで観察された浮遊細胞の数は少なく、群間で同様であった。すすいだ後、200μLの細胞解離溶液を使用して、細胞をウエルから分離した。それに、300μLの培地を添加し、合計500μLの細胞懸濁液をエッペンドルフチューブに収集した。この懸濁液からの10μL細胞懸濁液を、4%トリパンブルー溶液を使用して1:1に希釈し、細胞を血球計で計数して、生存可能な(未染色)及び生存不能な(青染色)細胞数を決定した。三重反復実験の平均として報告した結果を決定したところ、群中の生存率に変化は検出されなかった。この実験の対照には、未処理細胞(しかし、実験中にPBSを添加し、室温で維持した)が含まれていた。48時間後の生存率を試験するため、修飾細胞を、37℃及び5%CO2で48時間インキュベートした。その後、培地を吸引し、細胞解離溶液を使用することにより、細胞をウエルから分離した。生存率を、本明細書に前述されているのと同じように試験した。
【0230】
Strep−PLGA粒子により修飾したhMSCの接着
細胞をビオチン化した後、96ウエルプレートの細胞培養ウエル上の接着細胞数を測定することにより、細胞接着を分析した。T25フラスコ中の80%コンフルエント細胞を、上述のようにビオチン化した。Strep−PLGA結合の後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、およそ5000個の細胞を、96ウエルプレートの各ウエルに10、30、及び90分間播種した。その後、非接着細胞をPBSで2回すすぐことにより除去し、その後接着細胞を固定し、トルイジンブルー溶液で細胞を染色した。接着細胞を、1O×倍率で3つのウエルの6つの観察視野で計数して、接着細胞数を決定した。接着細胞のパーセントを、細胞の初期播種密度に基づいて計算した。この実験の対照は、未処理細胞を含んでいた。
【0231】
Strep−PLGA粒子により修飾したhMSCの増殖アッセイ
細胞の増殖アッセイを、T25フラスコ上の細胞数を測定することにより分析した。T25フラスコ中の80%コンフルエント細胞を、BNHSで処理し、その後Strep−PLGA粒子溶液で20分間処理した。その後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、およそ50,000個の細胞を、3個のT25フラスコに播種した。細胞数を、3個のフラスコの10×倍率の10観察視野で、播種後の1日目、2日目、4日目、6日目、及び8日目に計数し、細胞数を決定した。この実験のための対照は、PBSで処理した細胞である。
【0232】
P−セレクチン表面上でのStrep−PLGA修飾細胞の回転
T25フラスコ中の80%コンフルエント細胞を処理することにより、細胞の修飾を実施した。フラスコをBNHSで処理し、その後strept−PLGA粒子で処理した。その後、細胞をPBSで2回洗浄し、1mLの4μg/mL SLeX(Ca/Mg非含有PBS、pH7.4中)を、室温で20分間添加した。細胞解離溶液を使用して細胞をフラスコから分離し、遠心分離後に培地に再分散した。フローチャンバー実験用の細胞濃度は、典型的には105細胞/mlだった。細胞の回転特徴は、ガスケットの厚さが127μmで長さが6cmの長方形の平行平板フローチャンバー実験より評価した。ガラス表面上のP−セレクチン固定化を、700μLのP−セレクチン溶液(5μg/mL)をスライドガラス上で18時間インキュベートすることにより実施し、フロー実験は、スライドガラス上にチャンバーを設置することにより実施した。0.366ダイン/cm2の壁面せん断応力に対応する20μL/分の流速を使用した。細胞の回転をモニターするために位相差顕微鏡法を使用し、画像を10X観察視野で記録し、およそ10秒毎に手動で取り込んだ。細胞の速度は、10秒間わたって移動した細胞の距離を測定することにより計算した。
【0233】
細胞−粒子結合体の回転特徴を、明視野及び蛍光の両モードで(PLGA粒子は蛍光性だったため)、0.366ダイン/cm2のせん断応力での回転速度及びフラックスを決定することにより測定した。細胞及び/又は粒子結合体の速度は、細胞への粒子結合後で速度がわずかに速いことを示す。これは、粒子が、立体障害により速度の低下を引き起こしていることを示す。しかしながら、strep−PLGA修飾細胞の速度は、未修飾細胞の速度(約70μm/秒)より著しく低く、細胞がP―セレクチンと特異的に相互作用し、したがって回転に基づく接着を示すことを表している。
【0234】
表1は、本明細書に記載の方法に使用することができるアシル基のリストを示す。
【表1】
【0235】
実施例5:非共有結合的方法を使用したPLGA粒子によるhMSCの修飾
負荷電PLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して、負荷電カルボン酸により機能性に末端化されたPLGAから製作した。
【0236】
正荷電PLGA粒子の調製
負荷電PLGA粒子を、過剰濃度(PBS中0.5mg/mL)の正荷電ポリ−L−リジンと共に室温で2時間インキュベートした。PLGA粒子表面の負電荷とポリ‐L‐リジンの正電荷との電荷相互作用は、その結果として粒子表面の過剰正電荷をもたらす。
【0237】
脂質をコーティングしたPLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。ストレプトアビジンを、標準的なカルボジイミドカップリング技術を使用して、PLGA粒子のカルボン酸基に共有結合で結合させた。PLGA(Strep−PLGA粒子)粒子に共有結合で結合されたストレプトアビジンをPBSで3回洗浄して、物理的に吸着したストレプトアビジンをPLGA粒子の表面から除去した。ストレプトアビジン結合PLGA粒子を、ビオチン化脂質、1、2−ジパルミトイル−sn−グリセロ−3−ホスホエタノールアミン−N−(ビオチニル)(ナトリウム塩)溶液(PBS中0.1mg/mL)中で2時間室温でインキュベートし、その後PBSで洗浄して過剰な物理的に吸着した脂質分子を除去した。
【0238】
抗体をコーティングしたPLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。ストレプトアビジンを、標準的なカルボジイミドカップリング技術を使用して、PLGA粒子のカルボン酸基に共有結合で結合させた。PLGA(Strep−PLGA粒子)粒子に共有結合で結合されたストレプトアビジンをPBSで3回洗浄して、物理的に吸着したストレプトアビジンをPLGA粒子の表面から除去した。ストレプトアビジン結合PLGA粒子を、ビオチン化CD90抗体(PBS中0.01mg/mL)溶液中で2時間室温でインキュベートし、その後PBSで洗浄して過剰な物理的に吸着した抗体を除去した。
【0239】
非共有結合によるPLGA粒子のhMSCへの結合
種々の表面特徴を有するPLGA粒子を、組織培養プレート上の接着hMSCに添加し、その後洗浄して非接着粒子を細胞から除去した。特徴付けのため、インキュベーションの4時間、8時間、及び12時間後に、細胞を洗浄し、4%ホルムアルデヒドで固定し、その後ヨウ化プロピジウム(PI)(PBS中10μg/mL)で10分間染色し、PBSで2回洗浄した。PI染色細胞及びDiO封入PLGA粒子の場合は、細胞−粒子結合体を、蛍光顕微鏡を使用して画像化した。粒子と結合した細胞数を、4つの異なる表面特徴について計算し、図2に示されているように、粒子を有する細胞のパーセントとして表す。結果は、どの表面機能性を有する粒子もMSCに結合したことを示しており、正荷電粒子及びCD90抗体をコーティングした粒子は最も効率的に結合したが、脂質でコーティングした粒子及び負荷電粒子は8時間まで結合が最も非効率的だった。脂質でコーティングした粒子の結合は、12時間のインキュベーション後に70%まで増加した。
【0240】
非共有結合的方法によりhMSCに結合されたPLGA粒子の取り込み
hMSC表面に結合されたPLGA粒子の安定性を、レーザー共焦点顕微鏡法で、細胞(赤色、PI染色)粒子(DiO封入、緑色)のZスタック画像を検査することにより視覚化した。共焦点顕微鏡画像を、インキュベーションの4時間、8時間、及び12時間後に取得した。y−z、x−z、及びx−y平面のZスタック共焦点画像を分析して、粒子の内部移行を評価した。観察視野にはあるが細胞と結合していない粒子は考慮しなかった。内部移行のパーセントを、細胞に結合された粒子の合計数及び内部移行された粒子数から計算し、図3Aに示す。CD90抗体でコーティングした粒子は最も効果的に内部移行されたが、脂質でコーティングした粒子は、4時間又は8時間後で内部移行されなかった。脂質でコーティングしたPLGA粒子の内部移行は、12時間のインキュベーション後に80%まで増加した。負荷電粒子及び正荷電粒子の場合、細胞による粒子取り込みは、4時間から8時間までで、それぞれ20%から60%に増加した。これは、細胞表面上で機能化された脂質が8時間まで安定していることを示す。粒径の効果も、図3Bに示されているように粒子の内部移行に影響を及ぼす。結果は、3μmより小さな抗体コーティングPLGA粒子の場合、より大型の粒子と比較して、より高い効率性で内部移行されることを示す。3μmより大きな粒子の50%は12時間で内部移行され、粒径のより大きな粒子は、より短い時間で細胞表面上に安定化されることを示す。
【0241】
培養中にhMSCに結合された負荷電PLGA粒子の分析、及び粒子の安定性に対するトリプシン処理の効果
負荷電PLGA粒子の細胞への結合は、細胞の80%が結合直後に結合粒子を含有しており、細胞の60%がインキュベーションの36時間後に結合粒子を含有することを示す(図4A、4B)。 細胞1個当たりの粒子数がこのように変化するのは、細胞増殖による可能性が高い。負荷電PLGA粒子と共に細胞を36時間インキュベートした後、細胞を、1×トリプシン−EDTA溶液を使用してトリプシン処理し、その後リン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して培地及びトリプシンを除去し、6ウエルプレートに播種した。図4Cは、トリプシン処理後(0日目)及び2日目及び4日目の、負荷電PLGA粒子と結合している細胞のパーセントを示す。これは、トリプシン処理後に粒子が細胞と結合していることを示し、粒子を有する細胞のトリプシン処理は、細胞表面上の粒子の安定性に影響を及ぼさないことを示す。トリプシン処理後に1、2、及び3個以上の粒子と結合していた細胞のパーセントは、図4Dに示されている。
【0242】
hMSCに結合した負荷電PLGA粒子による骨形成分化の誘導
デキサメタゾン(hMSCの骨形成分化因子)をPLGA粒子内に封入した。hMSCを、アスコルビン酸及びβ−グリセリンリン酸の存在下でデキサメタゾン含有PLGA粒子と共に培地中でインキュベートし、21日後にアルカリホスファターゼ及びフォンコッサ染色により、骨形成を評価した。(80%の細胞がデキサメタゾン含有PLGA粒子を含有しており、70%を超える細胞が3個以上の粒子を有していた)。陰性対照群には、増殖培地中にアスコルビン酸及びβ−グリセリンリン酸を有し、デキサメタゾンを含有しないPLGA粒子と結合されたhMSC;増殖培地中にアスコルビン酸及びβ−グリセリンリン酸を有し粒子を含有しないhMSC、及び増殖培地中のhMSCが含まれる。陽性対照には、増殖培地中にデキサメタゾン、アスコルビン酸、及びβ−グリセリンリン酸を有するhMSCが含まれる。実験群、つまりアスコルビン酸及びβ−グリセリンリン酸存在下のデキサメタゾン含有PLGA粒子は、対照群と比較して著しい陽性骨形成染色を示し、PLGA内に封入されたデキサメタゾンが、細胞の骨形成分化を誘導することができることを示す。これは、hMSCに非共有結合的に結合されたPLGA粒子が、粒子内に封入された因子を介して骨形成分化を特異的に誘導することができることを示す。
【0243】
培地を吸引し、蒸留水で細胞を洗浄することにより、アルカリホスファターゼアッセイを実施した。細胞を3.7%ホルムアルデヒド溶液で15分間室温で固定し、その後蒸留水で2回洗浄した。それに、(DMF及びナフトールAS MX−P04)を含有するTris HCl及び蒸留水中の0.06%Red Violet LB塩水を添加した。プレートを45分間インキュベートし、その後ウエルを蒸留水で3回すすいだ。アルカリホスフェートアッセイ(alkaline phosphate assay)後、フォンコッサ染色用に2.5%硝酸銀溶液で、細胞を30分間暗所でインキュベートした。インキュベーション後、ウエルを、蒸留水で3回すすいだ。
【0244】
ヒト間葉系幹細胞(hMSC)に結合された負荷電PLGA粒子のin vivo経内皮遊走
負荷電蛍光性PLGA粒子(DiO封入、平均粒径1〜2μm)を、実験前に90%コンフルエントT75フラスコ中で24時間hMSCに結合させた。細胞をトリプシン処理し、PBSで1回洗浄してトリプシンを除去した。細胞を、DiDで蛍光標識した。典型的には、200μLのPBS中500,000個の細胞(DiO粒子で修飾したDiD標識hMSC)を、尾部静脈に注射した。この実験の対照は、同様の数の未修飾hMSCの注射だった。生体内in vivo共焦点顕微鏡法を使用して、注射の24時間後にZスタック画像を取得し(重複がないことを保証するために)、頭蓋内の骨髄にある細胞を視覚化した。骨髄中の血管を、画像化前に蛍光標識した。粒子を有するhMSCの骨髄内皮を越えた血管外遊出を、赤色蛍光(DiD標識hMSCからの)及び緑色蛍光(DiO封入粒子からの)の共局在化を使用して観察したところ、hMSCに結合された負荷電PLGA粒子が内皮を越えて遊走することができることが示された。
【0245】
実施例6:細胞を直接的にビオチン化し、その後ビオチン−ストレプトアビジン架橋を含有するPLGA粒子を付加することによるhMSCの修飾
ストレプトアビジン結合PLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。ストレプトアビジンを、標準的なカルボジイミドカップリング技術を使用して、PLGA粒子のカルボン酸基に共有結合で結合させた。PLGA(Strep−PLGA粒子)粒子に共有結合で結合されたストレプトアビジンを3回洗浄して、物理的に吸着したストレプトアビジンをPLGA粒子の表面から除去した。
【0246】
hMSC表面へのStrep−PLGA粒子の結合
図5Aに示されているように、hMSCを、PBS中で1mMスルフォ−NHS−ビオチン(BNHS)により室温でビオチン化し、その後ストレプトアビジン結合PLGA(Strep−PLGA)粒子を結合させた。2つの陰性対照をこの実験に使用した。一方の対照セットでは、Strep−PLGAを未修飾細胞に付加し、つまり細胞はビオチン化されなかった。第2の対照セットでは、細胞はビオチン化されていたが、未修飾PLGA粒子、つまりカルボン酸で末端化されたPLGA粒子を付加した。
【0247】
結果(図5A)は、Strep−PLGAビーズが、特異的なビオチン−ストレプトアビジン相互作用により、ビオチン化hMSCに結合することを示す。実験群(BNHS+Strep−PLGA)と陰性対照群(Strep−PLGA及びBNHS−PLGA)との間に著しい差異が存在する。著しく多数のStrep−PLGA粒子が、共有結合でビオチン化されたhMSCに結合した。これは粒子が細胞と特異的に結合することを示す。
【0248】
Strep−PLGA粒子で修飾したhMSCの生存率
細胞の生存率を、トリパンブルー排除法を使用して測定した。手短に言えば、細胞を12ウエルプレートに播種し、終夜放置して、接着させ、細胞を上述のようにBNHSでその後Strep−PLGA粒子で処理した。異なるウエルで観察された浮遊細胞の数は少なく、群間で同様であった。すすいだ後、200μLの細胞解離溶液を使用することにより、細胞をウエルから分離した。それに、300μLの培地を添加し、合計500μLの細胞懸濁液をエッペンドルフチューブに収集した。この懸濁液からの10μL細胞懸濁液を、4%トリパンブルー溶液を使用して1:1に希釈し、細胞を血球計で計数して、生存可能な(未染色)及び生存不能な(青染色)細胞数を決定した。三重反復実験の平均として報告した結果は、図5Bに示されている。この実験の対照には、未処理細胞(しかし、実験中にPBSを添加し、室温で維持した)が含まれていた。48時間後の生存率を試験するため、修飾細胞を、37℃及び5%CO2で48時間インキュベートした。その後、培地を吸引し、細胞解離溶液を使用して、細胞をウエルから分離した。生存率を、本明細書で前述されているのと同じように試験した。
【0249】
Strep−PLGA粒子で修飾したhMSCの接着動力学
細胞のビオチン化後、96ウエルプレートの組織培養ウエル上の接着細胞数を測定することにより、細胞接着を分析した。T25フラスコ中の80%コンフルエント細胞を、上述のようにビオチン化した。Strep−PLGAを結合した後、細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、およそ5000個の細胞を、96ウエルプレートの各ウエルに10、30、及び90分間播種した。その後、非接着細胞をPBSで2回すすぐことにより除去し、その後接着細胞を固定し、トルイジンブルー溶液で細胞を染色した。接着細胞を、1O×倍率で3つのウエルの6つの観察視野で計数して、接着細胞数を決定した。接着細胞のパーセントを、細胞の初期播種密度に基づいて計算し、結果を図5Cに示す。この実験の対照は、未処理細胞を含んでいた。
【0250】
Strep−PLGA粒子で修飾したhMSCの増殖アッセイ
細胞の増殖アッセイを、T25フラスコ上の細胞数を測定することにより分析した。T25フラスコ中の80%コンフルエント細胞を、BNHSで処理し、その後Strep−PLGA粒子溶液で20分間処理した。細胞を洗浄し、細胞解離溶液を使用してフラスコから分離した。細胞を血球計で計数し、およそ50,000個の細胞を、3個のT25フラスコに播種した。細胞数を、1O×倍率で3個のフラスコの10観察視野で、播種後の1日目、2日目、4日目、6日目、及び8日目に計数し、細胞の数を査定した。結果は図5Dに示されている。この実験のための対照は、PBSで処理した細胞である。
【0251】
Strep−PLGA粒子で修飾したhMSCの分化能
Strep−PLGA結合で修飾した細胞のin vitro骨形成及び脂肪生成分化能を、下記に記載のように評価した。
【0252】
骨形成分化
hMSCを24ウエルプレートの4つのウエルに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。細胞修飾を2段階法により実施した。典型的には、細胞を1mM BNHSでビオチン化し、その後strep−PLGA粒子を結合した。その後、デキサメタゾン、β−グリセロリン酸、L‐アスコルビン酸−2−ホスフェート、及びα−MEMを含有する骨形成誘導培地(Lonza社製、hMSC骨形成Single Quoteキット)中で21日間細胞を培養することにより、骨形成分化を誘導した。陽性対照として、未処理(未修飾)細胞を有する同数のウエルを、骨形成誘導培地中で維持した。両群の培地は3日毎に交換した。アルカリホスファターゼアッセイ及びフォンコッサ染色により、骨形成を評価した。
【0253】
培地を吸引し、蒸留水で細胞を洗浄することにより、アルカリホスファターゼアッセイを実施した。細胞を3.7%ホルムアルデヒド溶液で15分間室温で固定し、その後蒸留水で2回洗浄した。それに、(DMF及びナフトールAS MX−P04)を含有するTris HCl及び蒸留水中の0.06%Red Violet LB塩水を添加した。プレートを45分間インキュベートし、その後ウエルを蒸留水で3回すすいだ。
【0254】
strep−PLGA粒子で修飾された細胞の骨形成分化能を、修飾細胞用のアルカリホスファターゼ染色により観察した。粒子で修飾された細胞は、陽性染色を示し、未修飾細胞と同様の結果を示した。これは、細胞の粒子修飾が、細胞の骨形成能を妨害せず、修飾細胞が骨形成系統に分化することができることを示す。
【0255】
脂肪生成分化
hMSCを24ウエルプレートの4つのウエルに播種し、100%コンフルエンスに達するまで細胞増殖培地で培養した。細胞修飾を2段階法により実施した。典型的には、細胞を1mMのBNHSでビオチン化し、その後PBS中のストレプトアビジン(50μg/mL)と室温で結合させた。その後、脂肪生成誘導培地(Lonza社製、h−インスリン(組換え)、L−グルタミン、デキサメタゾン、インドメタシン、IBMX(3−イソブチル−1−メチル−キサンチン)、Pen/Strepを含有するhMSC脂肪生成Single Quoteキット)、及び脂肪生成維持培地(Lonza社製、h−インスリン(組換え)L−グルタミン、Pen/Strepを含有するhMSC脂肪生成Single Quoteキット)中で細胞を23日間培養することにより、脂肪生成分化を誘導した。陽性対照として、未処理(未修飾)細胞を有する同数のウエルを、脂肪生成誘導培地中で維持した。誘導培地及び維持培地と周期的に接触させるLonza社のプロトコールに従って、両群の培地を3日毎に交換した。
脂肪生成は、オイルレッドO染色により評価した。
【0256】
使用したオイルレッドO染色プロトコールは以下の通りである:細胞から培地を完全に吸引し、PBSで1回洗浄し、PBSを3.7%ホルムアルデヒドに室温で30分間置換して細胞を固定し、ホルムアルデヒドを蒸留水で数分間置換し、イソプロパノールをオイルレッドO作業用溶液(30mlの0.5%オイルレッドイソプロパノール溶液を20mlの蒸留水で希釈することにより製作)で置換し、5分後にオイルレッドO溶液を蒸留水で2回洗浄した。1mlのヘマトキシリン(Sigma−Aldrich社製)をウエルに1分間添加し、その後吸引し、ウエルを蒸留水で洗浄した。倒立位相差顕微鏡を使用してウエルを観察した。脂質は赤色に見え、核は青色に見える。
【0257】
Strep−PLGA粒子で修飾した細胞の脂肪生成分化能を、オイルレッドO及びヘマトキシリン染色により観察した。粒子で修飾された細胞は、陽性染色を示し、未修飾細胞と同様の結果を示した。これは、細胞の粒子修飾が、細胞の脂肪生成能を妨害せず、修飾細胞が脂肪生成系統に分化することができることを示す。
【0258】
細胞及び粒子がSLeXで機能化されているStrep−PLGA粒子で修飾したビオチン化細胞の回転
細胞修飾は、BNHSで細胞をビオチン化し、その後ストレプトアビジン溶液の存在下でstrep−PLGA粒子を結合させることにより実施した。その後、strep−PLGA粒子が結合された細胞を処理して、細胞表面上及び粒子表面上の両方にビオチン化SLeXを結合させた。粒子表面上及び細胞表面上にSLeXが存在することにより、P−セレクチン表面上での回転相互作用を誘導することができる。 T25フラスコ中の80%コンフルエント細胞を処理することにより、細胞修飾を実施した。フラスコをBNHSで処理し、その後ストレプトアビジン溶液(Ca/Mg非含有PBS中50μg/mL)の存在下でstrep−PLGA粒子で処理した。その後、細胞をPBSで2回洗浄し、1mLの4μg/mL SLeX(Ca/Mg非含有PBS、pH7.4中)を、室温で20分間添加した。細胞解離溶液を使用して細胞をフラスコから分離し、遠心分離後に培地に再分散した。フローチャンバー実験用の細胞濃度は、典型的には105細胞/mlだった。ガスケットの厚さが127μmで長さが6cmの長方形の平行平板フローチャンバー実験より、細胞の回転特徴を評価した。ガラス表面上へのP−セレクチン固定化を、700μLのP−セレクチン溶液(5μg/mL)をスライドガラス上で18時間インキュベートすることにより実施し、フロー実験は、スライドガラス上にチャンバーを設置することにより実施した。0.366ダイン/cm2の壁面せん断応力に対応する20μL/分の流速を使用した。細胞の回転をモニターするために位相差顕微鏡法を使用して、画像を1O×倍の観察視野で記録し、およそ10秒毎に手動で取り込んだ。細胞の速度は、10秒間わたって移動する細胞の距離を測定することにより計算した。
【0259】
細胞−粒子結合体の回転特徴を、図6に示されているように、明視野及び蛍光の両モードで(PLGA粒子は蛍光性だったため)、0.366ダイン/cm2のせん断応力での回転速度及びフラックスを決定することにより測定した。細胞及び/又は粒子結合体の速度は、粒子の結合が、接着に基づく回転応答を依然として可能にすることを示す。
【0260】
hMSCにしたStrep−PLGA粒子による骨形成分化の誘導
ビオチンストレプトアビジンを介してhMSCに結合したStrep−PLGA粒子は、細胞の運命を特異的に制御することができる。具体的には、デキサメタゾン(hMSCの骨形成分化因子)を、PLGA粒子内に封入した。デキサメタゾン含有PLGA粒子を、カルボジイミドカップリングによりストレプトアビジンに結合させた。hMSCを、1mMスルフォ−NHS−ビオチン(BNHS)によりPBS中で室温でビオチン化し、その後デキサメタゾンを含有するストレプトアビジン結合PLGA(Strep−PLGA)粒子を結合させた。ビオチン−ストレプトアビジンを介してデキサメタゾン含有PLGA粒子と結合したhMSCを、培地中のアスコルビン酸及びβ−グリセリンリン酸の存在下で培養し、アルカリホスファターゼ及びフォンコッサ染色により、骨形成分化を21日後に評価した。陰性対照群には、培地中にアスコルビン酸及びβ−グリセリンリン酸を有しデキサメタゾンを含有しないPLGA粒子と結合されたhMSC;培地中にアスコルビン酸及びβ−グリセリンリン酸を有するhMSC、及び増殖培地中のhMSCが含まれる。陽性対照には、培地中にデキサメタゾン、アスコルビン酸、及びβ−グリセリンリン酸を有するhMSCが含まれる。実験群、つまりアスコルビン酸及びβ−グリセリンリン酸存在下のデキサメタゾン含有PLGA粒子を、対照と比較して著しい陽性骨形成染色を示し、PLGA内に封入されたデキサメタゾンが、細胞の骨形成分化を誘導することができることを示す。これは、ビオチン−ストレプトアビジンを介してhMSCと共有結合的に結合されたPLGA粒子が、粒子内に封入された因子を介して細胞の骨形成分化を特異的に誘導することができることを示す。
【0261】
実施例7:N−ヒドロキシスクシンイミド(NHS)官能基及びPEGリンカーと結合したNHS基を使用したPLGA粒子によるhMSCの修飾
PEGリンカーを有する及び有しないNHS官能化PLGA粒子の調製
カルボン酸で末端化された1μmのPLGA粒子を、標準的なエマルジョン−溶媒蒸発技術を使用して製作した。PLGA粒子のカルボン酸官能基を、1−エチル−3−(3−ジメチルアミノプロピル)−カルボジイミド(EDC)及びNHSカップリングにより、N−ヒドロキシスクシンイミド(NHS)基に変換した。NHS官能化PLGA粒子を、二官能性ポリエチレングリコール(カルボン酸及び第一級アミンで官能化、MW:7500)とNHS官能化PLGA粒子のNHS基との反応によりPEGに結合させた。PEGのカルボキシ末端基を、EDC及びNHSでさらに官能化した。
【0262】
PEGリンカーを有する及び有しないNHS官能化PLGA粒子の結合
官能化された粒子をhMSCと共にインキュベーションすることにより、PEGリンカーを有する及び有しないNHS官能化PLGA粒子にhMSCを結合させた。図7は、NHS基は細胞表面と反応して粒子を結合させるが、いかなる官能基も有していないPLGA粒子は細胞に結合しないことを示す。これは、細胞表面官能性(細胞表面上のアミン基)と粒子の官能基(つまり、PEGリンカーを有する及び有しないNHS基)間の共有結合反応を使用して、細胞表面上に粒子を結合させることができることを示す。PLGA粒子上のPEGリンカーの数は、二官能性ポリエチレングリコール(カルボン酸及び第一級アミンで官能化)とHNS官能化PLGA粒子のNHS基との反応中に、二官能性PEG分子の濃度を変更することにより変化させることができる。具体的には、NHS活性化PLGA粒子を官能化するために、1、0.1、及び0.01mg/mLのPEG溶液を使用した。図7の結果は、PEGリンカーの濃度を変化させることにより、細胞に結合するPLGA粒子(PEGにより官能化され、その後NHSで活性化)の数を変化させることができることを示す。図7は、PEGリンカー濃度が増加すると共に、より多数の粒子が細胞に結合することを示す。
【0263】
PEGリンカーを有するNHSを介してhMSCに結合したPLGA粒子による骨形成分化の誘導
PEGリンカーを有するNHS基を介してhMSCに結合したPLGA粒子は、細胞の運命を特異的に制御することができる。具体的には、デキサメタゾン(hMSCの骨形成分化成分)を、PLGA粒子内に封入した。デキサメタゾン含有PLGA粒子を、カルボジイミドカップリングによりPEG及びNHSで機能化した。デキサメタゾン含有PLGA粒子に結合したhMSCを、培地中のアスコルビン酸及びβ−グリセリンリン酸の存在下で培養し、アルカリホスファターゼ及びフォンコッサ染色により、骨形成分化を21日後に評価した。陰性対照群には、培地中にアスコルビン酸及びβ−グリセリンリン酸を有しデキサメタゾンを含有しないPLGA粒子と結合されたhMSC;培地中にアスコルビン酸及びβ−グリセリンリン酸を有するhMSC、及び増殖培地中のhMSCが含まれる。陽性対照には、培地中にデキサメタゾン、アスコルビン酸、及びβ−グリセリンリン酸を有するhMSCが含まれる。実験群、つまりアスコルビン酸及びβ−グリセリンリン酸存在下のデキサメタゾン含有PLGA粒子は、対照と比較して著しい陽性骨形成染色を示し、PLGA内に封入されたデキサメタゾンが、細胞の骨形成分化を誘導することができることを示す。これは、PEGリンカーを有するNHS官能性を介してhMSCに共有結合的に結合されたPLGA粒子が、粒子内に封入された因子を介して細胞の骨形成分化を特異的に誘導することができることを示す。
【0264】
実施例8:シアリルルイスX(SLeX)で修飾したヒト間葉系幹細胞(hMSC)を用いたin vivo実験
ビオチン及びストレプトアビジンを介してSLeXで修飾したhMSCを、尾部静脈注射によりマウスに注射した。手短に言えば、hMSCを、1mLの1mM BNHS溶液で処理し、その後1mLの50μg/mLストレプトアビジン溶液、及び1mLの5μg/mLビオチン化SLeXにより室温で処理した。注射後に細胞を画像化するため、修飾細胞をDiDで蛍光標識した。典型的には、200μLのPBS中の500,000個の細胞(SLeXで修飾したDiD標識hMSC)を、尾部静脈に注射した。この実験の対照は、同様の数を注射した未修飾hMSC。生体内in vivo共焦点顕微鏡法を使用して、注射の2時間及び24時間後に、頭蓋内の骨髄にある細胞を視覚化した。骨髄中の血管を、画像化前に蛍光標識した。図8Aは、3つの別々の実験、#1、2、3において細胞を尾部静脈内注射した2時間後に骨髄に局在化した、未修飾及び修飾MSC(MSCは、ビオチン−N−ヒドロキシスクシイミドで修飾され、その後ストレプトアビジン及びビオチン化シアリルルイスX、SLeXで修飾されている)の数を示す。未修飾MSCと比較してより多数の修飾MSCが、注射の2時間後に骨髄に局在化した。図8Bは、注射の24時間後に骨髄内皮を越えて遊走した血管外遊出MSCの数を示す。24時間後の経内皮遊走能力に修飾MSC及び未修飾MSC間の差異はなかった。これは、細胞表面の共有結合的修飾及びビオチン−ストレプトアビジン架橋を介したSLeXの結合が、MSCの遊出能力を損なわないことを示す。
【0265】
実施例9:ストレプトアビジン−ビオチン架橋を介してシアリルルイスX(SLeX)で共有結合的に修飾されたヒト間葉系幹細胞(hMSC)のパラクリン因子分泌の検査
hMSCを、1mLの1mM BNHS溶液で処理し、その後1mlの50μg/mLストレプトアビジン溶液、及び1mLの5μg/mLビオチン化SLeXで処理することにより室温で修飾した。修飾後、細胞増殖培地を有する24ウエルプレートに修飾細胞を播種し、37℃で24時間インキュベートした。ELISAアッセイを実施して、細胞培養上清中のSDF−1、IGF−1、及びPGE−2の発現レベルを検査した。この実験の対照には、未修飾hMSCが含まれている。図9は、SLeXによるhMSCの修飾が、24時間後に未修飾細胞と比較して修飾hMSCにより分泌されたパラクリン因子レベルを変化させないことを示す。IGF−1及びPGE2は、15%血清を含有する増殖培地で検出可能ではなかったが、SDF−1は、24時間後にMSCから観察された約5%の量で検出された。
【0266】
実施例10:P−セレクチン抗体(Ab)によるヒト間葉系幹細胞(hMSC)の修飾
典型的には、hMSCを、hMSC増殖培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)中で90%コンフルエンスまでT75フラスコ中で培養し、その後細胞を、1×トリプシン−EDTA溶液を使用してトリプシン処理し、次にリン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して、培地及びトリプシンを除去した。その後、細胞ペレットを1mLの1mM BNHS溶液に分散した。その後、細胞を遠心分離及び遠心沈降させて、BNHS溶液を除去した。細胞を再懸濁しその後遠心分離することにより、細胞ペレットをPBSで2回洗浄した。遠心分離後、細胞ペレットを、1mLのストレプトアビジン溶液(Ca/Mg非含有PBS、pH7.4中50μg/mL)に再懸濁した。非結合ストレプトアビジンを遠心分離により除去した。細胞ペレットを、1mLのビオチン化P−セレクチン抗体溶液(Ca/Mg非含有PBS、pH7.4中5μg/mL)に再懸濁した。非結合ストレプトアビジンを遠心分離により除去した。
【0267】
ビオチン化P−セレクチン抗体により懸濁状態で修飾されたhMSCは、修飾細胞が、フローチャンバーアッセイにおいて、P−セレクチンをコーティングした基質と相互作用することを示す。未修飾細胞は、10ダイン/cm2まで、フローチャンバーにおいて回転相互作用又は強力な接着のいずれかを示したが、未修飾細胞は相互作用を示さなかった。これは、P−セレクチン抗体結合hMSCが、フロー条件下でP−セレクチンと特異的に相互作用することを示す(図10)。回転相互作用を示したP−セレクチン抗体修飾細胞の速度は、3μm/秒の平均速度でP−セレクチン表面上を回転することができた。
【0268】
実施例11:ビオチン−HABA−アビジンアッセイによる、ビオチン化ヒト間葉系幹細胞(hMSC)表面上のビオチンの定量化
ビオチン化hMSC(BNHSによるビオチン化)の表面に存在するビオチン部分の数を、製造業者のプロトコール(Thermo scientific社、米国インディアナ州)に従ってビオチン−HABA−アビジンアッセイを使用することにより定量化した。具体的には、2つの技術、接着モード及び懸濁モードを使用して細胞をビオチン化した。接着モードでは、T25フラスコ上の80〜90%コンフルエントのhMSCの単層を、1mLの1mM BNHSで10分間処理し、その後PBSで3回洗浄した。その後、1mLの非酵素性細胞解離溶液を使用して、細胞をプレートから分離し、ビオチン−HABA−アビジンアッセイのプロトコールに従って分析し、細胞上に存在するビオチンリガンドの数を定量化した。懸濁モードでは、hMSC増殖培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)中で90%コンフルエンスまでのT75フラスコを、1×トリプシン−EDTA溶液を使用してトリプシン処理し、その後リン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して、培地及びトリプシンを除去した。その後、細胞ペレットを1mLの1mM BNHS溶液に分散した。その後、細胞を遠心分離及び遠心沈降させて、BNHS溶液を除去した。細胞を再懸濁しその後遠心分離することにより、細胞ペレットをPBSで2回洗浄し、ビオチン−HABA−アビジンアッセイのプロトコールに従って分析して、細胞上に存在するビオチンリガンドの数を定量化した。図11は、接着モードで修飾したhMSCの表面に存在するビオチンの数が、懸濁モードで修飾したhMSCより少ないことを示す。これは、接着モードでは細胞表面の一部が培養皿に付着しているため、細胞表面の一部だけが修飾を受けるが、懸濁モードでは細胞表面全体が修飾に露出していることを示す。
【0269】
実施例12:接着モードでの自己集合ファイバーによるhMSCの修飾
色素封入自己集合ファイバーの調製
典型的には、4〜8mgの小分子両親媒性物質(サリシンデカノアート)をねじ蓋ガラスバイアルに取り、その中に180μLのPBS及び20μLのDMSO中DiD(色素)を添加し、蓋をしたバイアルを、物質が融解するまで60〜80℃まで加熱し、その後室温に静置して冷却させた。バイアルが室温に達した時、試料はヒドロゲルを形成していた。その後、ヒドロゲルを300μLのPBSを使用して希釈し、エッペンドルフに移し、10,000rpmで7分間遠心分離した。上清を除去して未結合色素を洗浄し、ファイバーを1mLのPBSに再分散した。
【0270】
色素封入自己集合ファイバーのhMSCへの負荷
hMSCを24ウエルプレートの4つのウエルに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。色素封入自己集合ファイバーを、組織培養プレート上の接着hMSCに添加し、30分間インキュベートし、その後洗浄して細胞から非接着自己集合ファイバーを除去した。DiDが赤色発光を示す蛍光顕微鏡法を使用して、細胞−ファイバー結合体を画像化した。細胞上の色素を定量化することにより、細胞に結合したファイバーの推定量がもたらされるだろう。対照実験(未修飾細胞)で測定したバックグラウンド蛍光を、ファイバー結合細胞の蛍光強度から差し引き、値を図12にプロットした。同様に、蛍光強度を、様々な時点:0日目、1日後、及び2日後に評価した(図12)。結果は、蛍光強度が2日後でさえ著しくは減少しなかったことを示す。
【0271】
別の対照として、遊離色素を自己集合ファイバーに封入せずに使用して、色素が非特異的に細胞に結合した可能性を除外した。これらの実験では、自己集合ファイバーを使用した以外は、同様の手順を本明細書に記載のように使用した。結果は図12に要約されている。
【0272】
実施例13:懸濁モードでの自己集合ファイバーによるhMSCの修飾
色素封入自己集合ファイバーのhMSC上への負荷
懸濁モードでは、hMSC増殖培地(α−MEM中15%ウシ胎仔血清、1%L−グルタミン、1%Penn−Strep)中で90%コンフルエンスまでのT75フラスコを、1×トリプシン−EDTA溶液を使用してトリプシン処理し、その後リン酸緩衝生理食塩水(Ca/Mg非含有PBS、pH7.4)で洗浄して、培地及びトリプシンを除去した。その後、細胞ペレットを、1mLの色素封入自己集合ファイバー含有PBSに分散し、室温で30分間インキュベートした。その後、細胞を遠心分離及び遠心沈降させて非結合ファイバーを除去した。しかしながら、残留量のファイバーが、細胞と共に遠心沈降された。細胞を再懸濁しその後遠心分離することにより、細胞ペレットをPBSで2回洗浄し、最後に、細胞を24ウエルプレートに播種した。細胞が培養フラスコに接着した後(3時間)、プレートをPBSで3回洗浄して残留未結合ファイバーを除去した。DiDが赤色発光を示す蛍光顕微鏡法を使用して、細胞−ファイバー結合体を画像化した。細胞上の色素を定量化することにより、細胞に結合したファイバーの推定量がもたらされるだろう。対照実験(未修飾細胞)で測定したバックグラウンド蛍光を、ファイバー結合細胞の蛍光強度から差し引き、値を図13にプロットした。同様に、本発明者らは、様々な時点:0日目、1日後、及び2日後の蛍光強度を追跡した、(図13)。結果は、蛍光強度が2日後でさえ著しくは減少しなかったことを示す。
【0273】
別の対照として、遊離色素を自己集合ファイバーに封入せずに使用して、色素が非特異的に細胞上に結合した可能性を除外した。これらの実験では、本発明者らは、自己集合ファイバーを使用した以外は、00026に記載されている同様の手順を使用した。結果は図13に要約されている。
【0274】
実施例14:ビオチン化小胞によるhMSCの修飾
ビオチン化脂質を使用した小胞の調製
1,2−ジオレオイル−sn−グリセロ−3−ホスホエタノールアミン−N−(ビオチニル)ナトリウム塩のクロロホルム溶液(1mgの脂質)を、ガラスバイアルに取り、クロロホルムをゆっくりと高真空で蒸発させて無水薄膜を製作した。薄膜を、5mLのPBSで24時間水和させ、その後5分間超音波処理し、氷中で5分間冷却した。同様に、凍結及び解凍を3回繰り返して小胞を生成した。その結果生じた小胞を透過型電子顕微鏡で検査し、小胞が、形状が球状であり、性質が単層であることを見出した。
【0275】
ビオチン化小胞によるhMSCの修飾
hMSCを96ウエルプレートの細胞培養ウエルに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。0.5mLの小胞溶液を、組織培養プレート上の接着hMSCに添加し、30分間インキュベートし、その後洗浄して過剰な小胞/脂質を除去した。ウエルを300μLの培地で3回すすいで、過剰な小胞/脂質を除去した。その後、ローダミン−ストレプトアビジン溶液(Ca/Mg非含有PBS中50μg/mL)を添加し、5分間インキュベートした。その後、細胞をPBSで2回洗浄し、未結合ローダミン−ストレプトアビジンを除去した。ローダミン−ストレプトアビジンが赤色蛍光を示す蛍光顕微鏡法を使用して、細胞を画像化した。対照実験として、上述した同様の手順(小胞溶液の代りにビオチンのみを使用した以外は)を使用して、hMSCをビオチンのみ(脂質分子/小胞形態を用いずに)で処理した。結果は図14Aに要約されている。結果は、ビオチン化脂質が7日目まで細胞と結合したままであることを示す(図14A)。
【0276】
hMSC表面上のビオチン−脂質の接近可能度
小胞で細胞を修飾した後の細胞表面上のビオチン化脂質/小胞の接近可能度を特徴付けるため、200μLのSR溶液(Ca/Mg非含有PBS、pH7.4中50μg/mL)を、0、2、4、及び7日後に小胞修飾細胞に室温で20分間添加した。過剰なストレプトアビジンを除去した後、蛍光強度を定量化した(図14B)。結果は、脂質を細胞挿入2日後で、ビオチン化脂質の一部が、ストレプトアビジンをさらに結合させるために接近可能だったことを示す。
【0277】
実施例15:間葉系幹細胞回転応答の誘導
物質及び方法:
物質
同意を得た健康なドナーのヒト骨髄から単離した初代ヒトMSCを、チューレーン大学の遺伝子治療センターから取得した。P−セレクチンは、R&D systems社(ミネアポリス、米国ミネソタ州)から購入し、ビオチン化シアリルルイス(x)−ポリ(アクリルアミド)(シアリル−ルイスX−PAA−ビオチン、BSLeX)はGlycotech社(ゲーサーズバーグ、米国メリーランド州)から購入した。α−MEM、L−グルタミン、及びPenn−Strepは、Invitrogen社から購入した。スルホン化ビオチニル−N−ヒドロキシ−スクシンイミド、BNHSは、Thermo Fisher Scientific社(Piercenet社製、ロックフォード、米国イリノイ州)から購入し、ウシ胎仔血清はAtlanta Biologicals社から購入した。ビオチン−4−フルオレセインは、Molecular Probes社(ユージーン、米国オレゴン州)から購入した。抗ヒト皮膚リンパ球抗原抗体(HECA−452)、二次抗体(FITCマウス抗ラットIgM)、FITC CD90及びPE−Cy5抗体は、BD Biosciences社から購入した。FACS緩衝液は、1%FBSを有するPBSである。他の化学薬品及び試薬は全て、Sigma Aldrich社(セントルイス、米国ミズーリ州)から購入し、指定が無い限りさらなる精製をせずに使用した。
【0278】
間葉系幹細胞の培養
初代ヒトMSCを、15%ウシ胎仔血清(これがMSCを増殖させる能力を持つため選択した)、1%(容積/容積)のL−グルタミン、1%(容積/容積)Penn−Strep、及びα−MEMで構成される増殖培地中で維持した。実験は全て、継代数4〜6のMSCを使用して実施し、フローサイトメトリー分析で観察したところ、細胞は高レベルのMSCマーカーCD90及びCD29を発現し(99%を超える細胞)、造血性マーカーCD34又はCD45を発現しなかった(0%の細胞)(結果は非表示)。
【0279】
回転リガンドによるMSCの表面修飾
ビオチン−ストレプトアビジンを介したMSC表面へのBSLeX結合を、リン酸緩衝液生理食塩水(PBS、pH7.4、Ca/Mg非含有)中で室温で実施した。典型的には、80〜90%コンフルエントT75フラスコから培地を吸引し、1×トリプシン−EDTA溶液を使用して細胞をトリプシン処理し、遠心分離してペレットにし、PBSで2回洗浄した。その結果生じた細胞ペレットを、スルホン化ビオチニル−N−ヒドロキシ−スクシンイミド、BNHS溶液(1mM、1mL)に分散し、それを1、5、又は10分間室温でインキュベートさせた(反応ステップ1)。その後、細胞をペレットにし、遠心分離によりPBSで2回洗浄して、未結合の及び/又は物理的に吸着したBNHSを細胞表面から除去した。その後、ストレプトアビジン溶液(PBS中50μg/mL、1mL)を使用して、細胞を1、5、又は10分間室温で処理した(反応ステップ2)。その後、細胞をペレットにし、PBSで洗浄した。ストレプトアビジン結合細胞に、BSLeX溶液(PBS中5μg/mL、1mL)を添加し、上清を5、10、又は60分間室温でインキュベートさせた(反応ステップ3)。最後に、細胞をペレットにし、PBSで洗浄した。
【0280】
フローサイトメトリー
フローサイトメトリー分析を、空冷式15mWの488nmアルゴンイオンレーザー、並びに530nm、585nm、及び661nmバンドパスフィルターを備えた光電子増倍管を装備したBD FACSCaliburアナライザー(BD Biosciences社製、サンホセ、カリフォルニア州、アメリカ合衆国)を使用して実施した。データは、ソフトウェアBD CellQuest及びWinMDIを使用して分析した。生存細胞を選択し、蛍光をヒストグラムで表示した(検査した細胞数対相対蛍光強度の対数)。1測定当たり1万事象を記録した。PBSで処理した細胞を使用して、バックグラウンド蛍光を正規化した。
【0281】
フローサイトメトリーを使用した細胞修飾の分析及び向上
色素と結合した試薬(つまり、ビオチン−4−フルオレセイン)をビオチン−ストレプトアビジン架橋用の代表的なビオチン化リガンドとして使用して、フローサイトメトリーにより、化学的結合効率をモニターした。インキュベーション時間を変えることにより結合工程を至適化して、各反応ステップにおける最大カップリング効率(つまり、部位密度)を決定した(表1)。ステップ3におけるビオチン−4−フルオレセインのインキュベーション時間が、結合効率にどのような影響を与えるかを最初に検討した。対照には、ステップ1及び2のPBSインキュベーション後の吸着ビオチン−4−フルオレセインが含まれていた。ステップ3における効果的な反応条件を決定した後、ステップ2におけるストレプトアビジン用のインキュベーション時間を変化させることにより、フルオレセインシグナルがどのように影響をうけるかを次に検討した。対照には、実験群で使用したのと同じステップ3がその後に続く吸着ストレプトアビジン(BNHS無し)が含まれていた。ステップ2の為の効果的な反応条件を決定した後、最後に、ステップ1におけるBNHSの反応時間を検討した。
【0282】
表2:カップリング効率を評価するために使用した実験条件。
【表2】
【0283】
部位密度の決定
MSC表面上のビオチン化SLeXリガンドの部位密度を、Bangs Laboratories社製(フィッシャーズ、米国インディアナ州)のQuantum(商標)Simply Cellular(登録商標)キットを指示通りに使用して決定した。(マウス抗体)結合部位の濃度が明確であるビーズを使用して、記録した蛍光を抗体数に関連付ける検量線を作成した。精製ラット抗ヒト皮膚リンパ球抗原抗体(HECA−452)及び二次抗体(FITCマウス抗ラットIgM)を使用して、細胞表面上に固定化されたSLeXを検出した。未修飾のPBS処理細胞を、一次及び二次抗体と共にインキュベートし、それを対照として使用した。この対照群からの抗体結合能(ABC)値を修飾細胞の値から差し引いて、データを正規化した。3つの細胞試料を、BNHSで10分間、ストレプトアビジンで1分間、及びBSLeXで4分間のインキュベーションを含んでいた上述の至適化修飾反応を用いて修飾した。SLeX部位密度を、HECA−452抗体に対するSLeXの結合比率が1対1であり、一次抗体と二次抗体との結合比率が1対1であると仮定して決定した。MSCの直径は、20μLの細胞溶液(細胞増殖培地中)を用いて、自動細胞計数器Cellometer(登録商標)Auto(Nexcelom Biosciences社製、米国マサチューセッツ州)により、使い捨ての血球計算板で測定した。MSCの平均細胞直径を、Cellometer(登録商標)AUTOのソフトウェア分析から取得した。
【0284】
細胞表面上に共有結合で結合したビオチンの安定性及び接近可能度
NHS化学により細胞表面に結合したビオチンの潜在的な安定性及び接近可能度を特徴付けるために、経時的な蛍光シグナルを検査して、ローダミン結合ストレプトアビジン(SR)のBNHS修飾細胞への結合を追跡した。PBS処理細胞又はビオチン処理細胞とのSRインキュベーションを、対照として使用した。SRと共にインキュベーションした後で、細胞をホルムアルデヒド溶液(PBS中4%)で固定し、トリトンX溶液(PBS中0.1%、100μL)で処理し、DAPI(PBS中1μg/mL、100μL)で染色した。ニコン社製定量ソフトウェア(NIS−Elements バージョン3)を使用して個々の細胞の強度を検査することにより顕微鏡画像の蛍光強度を分析し、データを細胞の単位面積当たりの蛍光強度として表した。
【0285】
生存率、接着動力学、及び増殖分析
BSLeX修飾MSC及び対照PBS処理MSCの生存率、接着動力学、及び増殖を検査した。手短に言えば、細胞の生存率を、修飾直後(時点0)に、及び細胞を6ウエルプレート内で48時間インキュベートした後で、トリパンブルー排除法アッセイを使用して検査した。細胞接着動力学を、10、30、90、及び150分後に組織培養表面上の接着細胞数を測定することにより定量化した。増殖を、T25フラスコ内の細胞数を、10倍の光学顕微鏡を用いて10のランダム観察視野について7日間の間計数することにより定量化した。
【0286】
細胞の分化
BSLeX修飾細胞及びPBS処理細胞の多系統分化能を、骨形成誘導培地及び脂肪生成誘導培地で細胞をインキュベートし、その後それぞれの比色定量組織学的染色を行うことにより検査した。それぞれ細胞膜結合アルカリホスファターゼ活性及びオイルレッドO染色を使用して、細胞を骨形成分化及び脂肪生成分化について分析した。
【0287】
パラクリン因子の分泌:SDF−1、IGF−1、及びPGE−2の定量化
酵素免疫吸着測定法(ELISA)キット(R&D systems社製、米国ミネソタ州)を使用して、BSLeX修飾MSC又はPBS処理MSCによる間質細胞由来増殖因子−1(SDF−1)、インスリン様増殖因子−1(IGF−1)、及びプロスタグランジンE2(PGE2)の産生を定量化した。48ウエルプレートのウエル内に播種した1×104個の細胞の培養から、細胞上清を収集した。標準的ELISAプロトコール(製造業者による)を、500μLのMSC増殖培地を用いて37℃及び5%CO2で細胞を24時間インキュベーションした後で、培養液上清中のSDF−1、IGF−1、及びPGE2を測定するために使用した。試料は全て、2つの独立実験で重複して実行した。15%血清を含有する増殖培地中のパラクリン因子のレベルも検査した。
【0288】
化学的修飾後のMSC表面マーカー発現の検査
MSC表面マーカーの発現に対する化学的修飾の効果を、時点0(修飾直後)及び24時間後のCD90、CD29、及びCD49dの抗体染色のフローサイトメトリーによって検査した。CD90及びCD29はMSCのマーカーであり、CD49d(VLA−4)はMSCの表面上のホーミングリガンドであると考えられている31。化学的に修飾されたMSC用の表面マーカー発現の変化を、未修飾(PBS処理)細胞に対する相対蛍光で表した。8つのT−150フラスコの細胞をトリプシン処理し、混合し、6群に分割して全群の抗原密度が同じであることを保証した。3群を上述の至適化条件で処理し、対照には、PBSのみで処理した(全ステップで)3群の細胞が含まれていた。修飾直後に、各群からの100,000個の細胞を100μLのFACS緩衝液に再懸濁し、1μLのCD90、4μLのCD29、又は4μLのCD49d抗体溶液で20分間氷上で染色した。FACS緩衝液で1回洗浄した後、細胞を300μlのFACS緩衝液に再懸濁し、フローサイトメトリーで分析した。BNHS又はPBSで(しかし抗体によってではなく)処理した細胞を播種し、増殖培地中で24時間インキュベートした。その後、細胞をトリプシン処理し、各群から100,000個の細胞を収集し、上述したのと同じプロトコール後に抗体で染色した。
【0289】
P−セレクチン表面の調製
6ウエルプレート内のウエル表面を、P−セレクチン溶液(PBS中5μg/mL、1mL)により、室温で18時間プレート振とう器上でコーティングした。P−セレクチン表面は全て、フローチャンバーアッセイの前に新たに調製した。
【0290】
フローチャンバーアッセイ
フローチャンバーによる細胞速度の分析の場合、細胞を、フローチャンバーアッセイ用のMSC増殖培地に懸濁した(約105細胞/mL)。ガスケット厚さが127μmで幅が2.5mmの円形平行平板フローチャンバー(Glycotech社製、ゲーサーズバーグ、米国メリーランド州)を使用した。細胞回転をモニターするために、位相差顕微鏡(DS−Qil Monochrome Cooledデジタルカメラを備えたニコン社製TE2000−U型倒立顕微鏡)を使用し、画像を10×倍率の観察視野で10秒間隔で記録した。細胞の速度は、10秒間隔内に移動した細胞の距離を測定することにより計算した。細胞は、観察視野にある間に10秒間回転した場合、及び非相互作用細胞の自由流動速度の計算値の50%未満の平均速度で移動した場合、回転として分類した。フラックスは、基質と相互作用し観察視野に10秒間留まる細胞の数に基づいて、手作業で計算した。強力に接着した細胞及び回転細胞を両方とも、フラックス計算用に考慮した。せん断速度の影響を評価するために、回転速度及びフラックスを、0.36、0.72、及び1.89ダイン/cm2を含むせん断応力で測定した。
【0291】
統計分析:
多重対比較の場合、両側スチューデントt検定を、ボンフェローニ補正と共に使用した。
【0292】
結果:
細胞表面上のビオチンの安定性及び接近可能度
細胞培養中でMSC上に固定化したビオチン及びローダミン結合ストレプトアビジン(SR)の安定性及び接近可能度を検査するために、BNHS及びSR処理の直後(0日目)及びBNHS及びSR処理の7日後に、SRの蛍光シグナルを検査した。BNHSで共有結合的に修飾した細胞は、修飾後の0日目及び7日目に、対照(SRのみ(SR)と共にインキュベートしたMSC、及びビオチン(B)及びSR(B+SR)で処理したMSC)と比較して、有意により高いローダミンシグナルを示す。これは、MSC上に共有結合で結合したビオチンが、少なくとも7日間安定していたことを示唆する。したがって、MSC表面上に共有結合で結合したビオチンの経時的な安定性及び接近可能度を、BNHS処理後の1、2、4、及び7日間遅延後の、細胞に付加されているSRの蛍光シグナルを定量化することにより検査した。蛍光強度は7日間まで安定化及び保持されており、対照より有意に高かった(P<0.01)。これらのデータは、BNHS処理によるビオチンの共有結合固定化が、結果として細胞表面上の安定的で接近可能なビオチンをもたらしたことを示す。
【0293】
フローサイトメトリーを使用した機能化効率の分析及び至適化
修飾工程の3番目のステップでのビオチン−4−フルオレセインのインキュベーション時間の効果を決定した。 ビオチン−4−フルオレセインのインキュベーション時間の5分間を超える増加は、結果として実験群の蛍光に有意な増加をもたらさなかった。対照的に、対照は、ビオチン−4−フルオレセインのインキュベーションを増加させると共に、蛍光を増加させた。
【0294】
ストレプトアビジンと共にBNHS修飾MSCをインキュベーションする(ステップ2)時間は、そのすぐ後のステップで結合されるビオチン−4−フルオレセインの量に有意な影響を及ぼした。具体的には、ストレプトアビジンとの1分間インキュベーションは、その結果として、鋭いピークにより示されたより強い全体的な蛍光をもたらした。ストレプトアビジンとのより長いインキュベーション時間(5分及び10分)は、BNHS修飾MSCをストレプトアビジンと共に1分間インキュベートした群と比較して、蛍光の減少及びより幅の広いピークを結果としてもたらした。対照群細胞(PBS処理MSC、その後ストレプトアビジン及びビオチン−4−フルオレセインと共にインキュベーションした)の蛍光は、ストレプトアビジンのインキュベーション時間を増加させると共に増加した。
【0295】
(ステップ1)のBNHSとMSCとの反応時間も、その後ストレプトアビジンを結合させ、次にビオチン−4−フルオレセインを結合させることにより導入される相対的蛍光に影響を及ぼす。蛍光シグナルは、BNHS処理後約10分でプラトーに達し、その後は反応時間が増加しても有意な増加を示さなかった。
【0296】
さらなる実験には、細胞集団全体にわたって最も強く及び最も均一な染色に結び付いた条件を使用した。実験群のピークはこれらの条件を表す:10分間のBNHS、1分間のストレプトアビジン、及び5分間のビオチン−4−フルオレセイン。10分間のBNHS、1分間のストレプトアビジン、及び4分間のビオチン−4−フルオレセインで構成される反応条件を使用してMSC表面上に固定化したBSLeXのABCは、約27201±13786であると決定された。このABCは、部位密度が約10±5 SLeX部分/μm2で平均細胞直径が15±4μmに相当する。
【0297】
細胞表現型の特徴付け
BSLeX修飾細胞の細胞生存率は、細胞修飾後48時間以内に観察したところ、PBS処理細胞と比較して、有意な影響を受けていなかった。具体的には、SLeX修飾細胞の88±4%がカップリング直後に生存可能であり、それと比較してPBS修飾細胞の生存率は92±3%だった。48時間の培養後、90±2%のSLeX修飾細胞が生存可能であり、それと比較してPBS処理細胞は89±2%だった。これは、SLeXによるMSCの修飾が、いかなる実質的な毒性も誘導しなかったことを示す。10、30、90、及び150分時点の、組織培養ポリスチレン皿上のSLeX修飾MSCの接着動力学を、PBS処理細胞と比較した。修飾細胞は、対照と比較して同様の接着動力学を示した。BSLeX修飾MSC及びPBS処理細胞の増殖速度、又は7日後にコンフルエント単層に達するそれらの能力に差異は検出されなかった。
【0298】
修飾細胞の分化能
BSLeX修飾MSCの分化能を、骨形成培養条件下及び脂肪生成培養条件下で細胞の分化を誘導することにより検査し、対照としてのPBS処理細胞と比較した。具体的には、アルカリホスファターゼ(ALP)活性及びオイルレッド0(ORO)染色を、それぞれ骨形成分化及び脂肪生成分化の指標として使用した。BSLeX修飾細胞及びPBS処理細胞間の、ALP又はORO染色における有意差は、骨形成分化及び脂肪生成分化誘導後に観察されなかった。さらに、MSC増殖媒地中で増殖させたBSLeX修飾細胞は、ORO又はALPで陽性染色されなかった。これらの結果は、MSC表面の共有結合修飾が、脂肪生成分化も骨形成分化も誘導せず、細胞の多系統分化能も損なわないことを示す。
【0299】
パラクリン因子の放出
BSLeX修飾MSCによるパラクリン因子の分泌を、ELISAアッセイを使用して、24時間の時間枠にわたって、培地中へのSDF−1、IGF−1、及びPGE2の分泌を定量化することにより検査した。PBS処理細胞と比較して、BSLeX修飾MSCのパラクリン因子の発現のレベル間には、有意差は観察されなかった。IGF−1及びPGE2は、15%血清を含有する増殖培地で検出可能ではなかったが、SDF−1は、24時間後にMSCから観察された量の約5%で検出された。
【0300】
MSC表面マーカー発現に対する化学的修飾後の影響
MSC表面上のCD90及びCD29受容体の発現は、PBS処理細胞と比較して、BNHS、ストレプトアビジン、及びBSLeXによる3段階修飾後に低減した。具体的には、修飾直後に、BSLeX結合MSCは、PBS処理細胞のCD90、CD29、及びCD49dと比較して、それぞれ84%、75%、及び40%の相対的蛍光を示す。24時間後には、修飾MSC及びPBS処理細胞の蛍光は類似しており、CD90、CD29、及びCD49d表面受容体が24時間後に回復し、これら受容体の発現レベルが未修飾MSC対照に類似していたことを示した。
【0301】
P−セレクチン上での修飾MSCの回転
P−セレクチンを有するMSC表面上に結合したBSLeX部分の、回転に基づく接着相互作用をフローチャンバーアッセイで分析した。修飾MSCのSLeXとP−セレクチン間の特異的相互作用は、0.36ダイン/cm2の壁面せん断応力において、速度を約70μm/sから約0.5μm/sに低下させた。10秒間隔内に相互作用する細胞の数は、PBS処理MSCの場合の20細胞/mm2から、SLeX修飾細胞の場合の150細胞/mm2に増加した。せん断応力を0.36ダイン/cm2から1.89ダイン/cm2に増加させると、修飾細胞の速度は、0.5μm/sから2μm/sまで中程度に増加し、フラックスは、150細胞/mm2と140細胞/mm2との間でほとんど一定のままだった。以前の研究では、0.72ダイン/cm2を超えるせん断応力において、速度は著しく増加した。
【0302】
実施例16:小胞を介してSLEXを有する修飾間葉系幹細胞の回転応答
hMSCをT25フラスコに播種し、90%コンフルエンスに達するまで細胞増殖培地で培養した。2−ジオレオイル−sn−グリセロ−3−ホスホエタノールアミン−N−(ビオチニル)ナトリウム塩の1mL(1mM)小胞溶液を、接着hMSCに添加し、10分間インキュベートし、その後洗浄して過剰な小胞/脂質を除去した。フラスコを1mLの培地で3回すすいで、過剰な小胞/脂質を除去した。その後、1mLのストレプトアビジン溶液(Ca/Mg非含有PBS中50μg/mL)を添加し、5分間インキュベートした。その後、細胞をPBSで2回洗浄し、未結合ストレプトアビジンを除去した。その後、1mLのビオチン化シアリルルイスX(SLeX)溶液(Ca/Mg非含有PBS中5μg/mL)を添加し、5分間インキュベートした。細胞をPBSで2回洗浄し、未結合SLeXを除去した。1mLの非酵素性細胞解離溶液を使用して、細胞をフラスコから分離し、1mLのPBSで1回洗浄して細胞解離溶液を除去した。その後、細胞を2mLの細胞増殖培地に分散し、回転相互作用をフローチャンバーアッセイを使用して分析した。対照実験として、PBS処理hMSC(未修飾細胞)をフローチャンバーアッセイに使用した。結果は図15A〜15Bに要約されている。結果は、接着モードにおいて小胞を介してSLeXにより修飾したhMSCが、P−セレクチンでコーティングした基質上での回転相互作用を示すことを表す。小胞修飾細胞は、0.5ダイン/cm2のせん断応力において7μm/sの速度を示したが、未修飾細胞は、60μm/sの速度を示した。図15Bは、小胞を介したSLeX修飾hMSCに対するせん断応力増加の効果を示す。
【0303】
(文献)
以下に、並びに本明細書及び実施例の全体にわたって引用された全ての文献は、その全体が参照により本明細書に組み込まれる。
1. MacDiarmid, J. A., et al., Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell, 2007. 11(5): p. 431-45.
2. Studeny, M., et al., Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res, 2002. 62(13): p. 3603-8.
3. Stoff-Khalili, M.A., et al., Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res Treat, 2007. 105(2): p. 157-67.
4. Kumar, S. and S. Ponnazhagan, Bone homing of mesenchymal stem cells by ectopic {alpha}4 integrin expression. Faseb J, 2007.
5. Chamberlain, G., et al., Mesenchymal Stem Cells: their Phenotype, Differentiation Capacity, Immunological Features and Potential for Homing. Stem Cells, 2007.
6. Griffiths, L., et al., The macrophage - a novel system to deliver gene therapy to pathological hypoxia. Gene Ther, 2000. 7(3): p. 255-62.
7. Hwu, P., et al., In vivo antitumor activity of T cells redirected with chimeric antibody/T- cell receptor genes. Cancer Res, 1995. 55(15): p. 3369-73.
8. Harrington, K., et al., Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery? Hum Gene Ther, 2002. 13(11): p. 1263-80.
9. Elgert, K.D., D. G. Alleva, and D.W. Mullins, Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol, 1998. 64(3): p. 275-90.
10. Jang, J. H., T. L. Houchin, and L.D. Shea, Gene delivery from polymer scaffolds for tissue engineering. Expert Rev Med Devices, 2004. 1(1): p. 127-38.
11. Hill, E., T. Boontheekul, and DJ. Mooney, Regulating activation of transplanted cells controls tissue regeneration. Proc Natl Acad Sci U S A, 2006. 103(8): p. 2494-9.
12. Leo, AJ. and D. A. Grande, Mesenchymal stem cells in tissue engineering. Cells Tissues Organs, 2006. 183(3): p. 112-22.
13. Sun X-D, L.I.-S., Gene technology in tissue engineering. American Journal of Biochemistry and Biotechnology, 2006. 2(2): p. 66-72.
14. Kemp, K.C., J. Hows, and C. Donaldson, Bone marrow-derived mesenchymal stem cells. Leuk Lymphoma, 2005. 46(11): p. 1531-44.
15. Gafni, Y., et al., Stem cells as vehicles for orthopedic gene therapy. Gene Ther, 2004. 11(4): p. 417-26.
16. Nakamura, K., et al., Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther, 2004. 11(14): p. 1155-64.
17. Pizzato, M., et al., Initial binding of murine leukemia virus particles to cells does not require specific Env-receptor interaction. J Virol, 1999. 73(10): p. 8599-611.
18. Zhuge, Y. and J. Xu, Racl mediates type I collagen-dependent MMP-2 activation, role in cell invasion across collagen barrier. J Biol Chem, 2001. 276(19): p. 16248-56.
19. Cheung, L.W., P.C. Leung, and A. S. Wong, Gonadotropin-releasing hormone promotes ovarian cancer cell invasiveness through c-Jun NH2-terminal kinase-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9. Cancer Res, 2006. 66(22): p. 10902- 10.
20. Fujiuchi, Y., et al., Effect of hepatocyte growth factor on invasion of prostate cancer cell lines. Oncol Rep, 2003. 10(4): p. 1001-6.
21. Mohanam, S., et al., Increased invasion of neuroglioma cells transfected with urokinase plasminogen activator receptor cDNA. Int J Oncol, 1998. 13(6): p. 1285-90.
22. Farokhzad, O. C, J.M. Karp, and R. Langer, Nanoparticle-aptamer bioconjugates for cancer targeting. Expert Opin Drug Deliv, 2006. 3(3): p. 311-24.
23. Chithrani, B.D., A.A. Ghazani, and W.C. Chan, Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett, 2006. 6(4): p. 662-8.
24. Win, K.Y. and S. S. Feng, Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials, 2005. 26(15): p. 2713-22.
25. Schneider, G. B., et al., The effect of hydrogel charge density on cell attachment. Biomaterials, 2004. 25(15): p. 3023-8.
26. Sinclair, J. and A. K. Salem, Rapid localized cell trapping on biodegradable polymers using cell surface derivatization and microfluidic networking. Biomaterials, 2006. 27(9): p. 2090-4.
27. Saxon, E. and CR. Bertozzi, Cell surface engineering by a modified Staudinger reaction. Science, 2000. 287(5460): p. 2007-10.
28. Huang, A., L. Huang, and SJ. Kennel, Monoclonal antibody covalently coupled with fatty acid. A reagent for in vitro liposome targeting. J Biol Chem, 1980. 255(17): p. 8015- 8.
29. Dube, D. H. and CR. Bertozzi, Metabolic oligosaccharide engineering as a tool for glycobiology. Curr Opin Chem Biol, 2003. 7(5): p. 616-25.
30. Hang, H. C. and CR. Bertozzi, Ketone isosteres of 2-N-acetamidosugars as substrates for metabolic cell surface engineering. J Am Chem Soc, 2001. 123(6): p. 1242-3.
31. Sampathkumar, S. G., M.B. Jones, and KJ. Yarema, Metabolic expression of thiol- derivatized sialic acids on the cell surface and their quantitative estimation by flow cytometry. Nat Protoc, 2006. 1(4): p. 1840-51.
32. De Bank, P.A., et al., Surface engineering of living myoblasts via selective periodate oxidation. Biotechnol Bioeng, 2003. 81(7): p. 800-8.
33. Johnsson, N., N. George, and K. Johnsson, Protein chemistry on the surface of living cells. Chembiochem, 2005. 6(1): p. 47-52.
34. Toriello, N.M., E. S. Douglas, and R.A. Mathies, Microfluidic device for electric field- driven single-cell capture and activation. Anal Chem, 2005. 77(21): p. 6935-41.
35. Albretsen, C, et al., Applications of magnetic beads with covalently attached oligonucleotides in hybridization: isolation and detection of specific measles virus mRNA from a crude cell lysate. Anal Biochem, 1990. 189(1): p. 40-50.
36. Tiwari, A., et al., Magnetic beads (Dynabead) toxicity to endothelial cells at high bead concentration: implication for tissue engineering of vascular prosthesis. Cell Biol Toxicol, 2003. 19(5): p. 265-72.
37. Hinds, K.A., et al., Highly efficient endosomal labeling of progenitor and stem cells with large magnetic particles allows magnetic resonance imaging of single cells. Blood, 2003. 102(3): p. 867-72.
38. Lewin, M., et al., Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol, 2000. 18(4): p. 410-4.
39. Lacava, L.M., et al., Magnetic resonance of a dextran-coated magnetic fluid intravenously administered in mice. Biophys J, 2001. 80(5): p. 2483-6.
40. Wilson, K.M., et al., Single particle tracking of cell-surface HLA-DR molecules using R- phycoerythrin labeled monoclonal antibodies and fluorescence digital imaging. J Cell Sci, 1996. 109 ( Pt 8): p. 2101-9.
41. Mistry, S.C., Iksoo;Messina, Darin Jeffrey; Gosiewska, Anna, Microparticles for cell delivery. 2005, JOHNSON & JOHNSON USA.
42. Halbert, G.W., Microparticles. 2007: USA.
43. Robert C, Klein, C, Cheng, G., Kogan, A., Mulligan, R.C., von Andrian, U.H., Cooper, T. S, Gene therapy to target dendritic cells from blood to lymph nodes. Gene Therapy 2003. 10(17):1479-86.
【特許請求の範囲】
【請求項1】
単離された改変細胞組成物であって、
(a)細胞と、
(b)前記細胞の表面に結合された膜結合リガンドと、
(c)前記細胞の前記表面に結合された粒子とを含む細胞組成物。
【請求項2】
前記細胞が、幹細胞又は前駆細胞である、請求項1に記載の細胞組成物。
【請求項3】
前記幹細胞が、再プログラムされた細胞である、請求項1又は2に記載の細胞組成物。
【請求項4】
前記細胞が分化細胞である、請求項1、2、3、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項5】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項1〜4又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項6】
前記粒子が、1000〜8000nmである、請求項1に記載の細胞組成物。
【請求項7】
前記粒子が、500〜1000nmである、請求項1に記載の細胞組成物。
【請求項8】
前記粒子が、1〜500nmである、請求項1に記載の細胞組成物。
【請求項9】
前記膜結合リガンドが、リンカー分子を介して結合される、請求項1に記載の細胞組成物。
【請求項10】
前記粒子が、リンカー分子結合を介して結合される、請求項1、6〜8に記載の細胞組成物。
【請求項11】
前記粒子が、リンカー分子を介さずに結合される、請求項1、6〜8、10、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項12】
前記膜結合リガンドが、共有結合で結合される、請求項1、9、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項13】
前記膜結合リガンドが、非共有結合で結合される、請求項1、9、12、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項14】
前記粒子が、共有結合で結合される、請求項1、6〜8、10、11、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項15】
前記粒子が、非共有結合で結合される、請求項1、6〜8、10、11、14、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項16】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項11、9、12、13、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項17】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項1、9、12、13、16、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項18】
複数の膜結合リガンドが、前記細胞に結合される、請求項1、9、12、13、16、17、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項19】
前記粒子が、前記細胞の機能を増強する作用剤を含む、請求項1、6〜8、10、11、14、15、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項20】
前記粒子が、組織の機能を増強する作用剤を含む、請求項1、6〜8、10、11、14、15、19、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項21】
前記粒子が、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー性粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、脂質、ナノ粒子、マイクロ粒子、デンドリマー、又はそれらの任意の組合せからなる群から選択される、請求項1、6〜8、10、11、14、15、19、20、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項22】
前記作用剤が、細胞成長、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着からなる群から選択される機能を増強する、請求項19又は20に記載の細胞組成物。
【請求項23】
前記作用剤が、小分子、酵素、酵素前駆体、栄養補助剤、化粧用の特性を有する作用剤、増殖因子、サイトカイン、RNA干渉分子、増殖因子、ビタミン、治療薬、診断薬、ケモカイン、標的化作用剤、又は分化因子からなる群から選択される、請求項19、20、22、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項24】
前記細胞が、作用剤を含む粒子用の組織特異的担体として使用される、請求項1〜4又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項25】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合される、請求項11〜4、24、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項26】
単離された改変細胞組成物であって、
(a)細胞と、
(b)前記細胞の表面に組み込まれた自己集合分子に結合された膜結合リガンドとを含む細胞組成物。
【請求項27】
前記自己集合分子が両親媒性である、請求項26に記載の細胞組成物。
【請求項28】
前記細胞が、幹細胞又は前駆細胞である、請求項26に記載の細胞組成物。
【請求項29】
前記幹細胞が、再プログラムされた細胞である、請求項28に記載の細胞組成物。
【請求項30】
前記細胞が分化細胞である、請求項26又は28に記載の細胞組成物。
【請求項31】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項26、28〜30、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項32】
前記膜結合リガンドが、リンカー分子を介して結合される、請求項26に記載の細胞組成物。
【請求項33】
前記膜結合リガンドが、前記自己集合分子と共有結合で結合される、請求項26又は32に記載の細胞組成物。
【請求項34】
前記膜結合リガンドが、前記自己集合分子と非共有結合で結合される、請求項26、32、33、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項35】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項26、32、33、34、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項36】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項26、32〜35、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項37】
複数の膜結合リガンドが、前記細胞に結合される、請求項26、32〜36、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項38】
少なくとも1つの粒子が前記細胞に結合される、請求項26に記載の細胞組成物。
【請求項39】
前記粒子が、標的化部分であるリガンドを含む、請求項26又は38に記載の細胞組成物。
【請求項40】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合される、請求項26、28〜31に記載の細胞組成物。
【請求項41】
単離された改変細胞組成物を形成する方法であって、
(a)膜結合リガンドを自己集合分子に結合させて修飾分子を形成するステップと、
(b)前記修飾分子を用いて小胞を形成するステップと、
(c)前記小胞を細胞と融合させるステップとを含む方法。
【請求項42】
前記自己集合分子が両親媒性分子である、請求項41に記載の方法。
【請求項43】
前記細胞が、幹細胞又は前駆細胞である、請求項41に記載の方法。
【請求項44】
前記幹細胞が、再プログラムされた細胞である、請求項43に記載の方法。
【請求項45】
前記細胞が分化細胞である、請求項41又は43に記載の方法。
【請求項46】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項41、43〜45、又は前記請求項のいずれか一項に記載の方法。
【請求項47】
前記膜結合リガンドが、リンカー分子結合を介して結合される、請求項41に記載の方法。
【請求項48】
前記膜結合リガンドが、前記自己集合分子と共有結合で結合される、請求項41又は47に記載の方法。
【請求項49】
前記膜結合リガンドが、前記自己集合分子と非共有結合で結合される、請求項41、47、48、又は前記請求項のいずれか一項に記載の方法。
【請求項50】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項41、47〜49、又は前記請求項のいずれか一項に記載の方法。
【請求項51】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、又はグリコサミノグリカンからなる群から選択される、請求項41、47〜50、又は前記請求項のいずれか一項に記載の方法。
【請求項52】
複数の膜結合リガンドが、前記細胞に結合される、請求項41、47〜51、又は前記請求項のいずれか一項に記載の方法。
【請求項53】
前記単離された改変細胞組成物が、in vivoで形成される、請求項41に記載の方法。
【請求項54】
前記膜結合リガンドが、細胞凝集塊を形成するために使用される、請求項41、47〜51、又は前記請求項のいずれか一項に記載の方法。
【請求項55】
前記粒子が、細胞凝集塊を形成するために使用されるリガンドを含む、請求項41に記載の方法。
【請求項56】
単離された改変細胞組成物を形成するための方法であって、
(a)膜結合リガンドを自己集合両親媒性分子に結合させて修飾両親媒性分子を形成するステップと、
(b)前記修飾両親媒性分子を用いてミセルを形成するステップと、
(c)前記ミセルを前記細胞と融合させるステップとを含む方法。
【請求項57】
前記細胞が、幹細胞又は前駆細胞である、請求項56に記載の方法。
【請求項58】
前記幹細胞が、再プログラムされた細胞である、請求項58に記載の方法。
【請求項59】
前記細胞が分化細胞である、請求項56又は57に記載の方法。
【請求項60】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項56〜59、又は前記請求項のいずれか一項に記載の方法。
【請求項61】
前記膜結合リガンドが、リンカー分子を介して結合される、請求項56に記載の方法。
【請求項62】
前記膜結合リガンドが、前記自己集合両親媒性分子と共有結合で結合される、請求項61に記載の方法。
【請求項63】
前記膜結合リガンドが、前記自己集合両親媒性分子と非共有結合で結合される、請求項48に記載の方法。
【請求項64】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項56、61、62、又は前記請求項のいずれか一項に記載の方法。
【請求項65】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、薬物、栄養補助剤、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、薬物、又はグリコサミノグリカンからなる群から選択される、請求項56、61〜64、又は前記請求項のいずれか一項に記載の方法。
【請求項66】
複数の膜結合リガンドが、前記細胞に結合される、請求項56、61〜65、又は前記請求項のいずれか一項に記載の方法。
【請求項67】
前記単離された改変細胞組成物が、in vivoで形成される、請求項56に記載の方法。
【請求項68】
前記細胞が、少なくとも1つの粒子に結合される、請求項56に記載の方法。
【請求項69】
前記粒子が、標的化部分であるリガンドを含む、請求項56又は68に記載の方法。
【請求項70】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合されている、請求項56、57〜59、68、又は前記請求項のいずれか一項に記載の方法。
【請求項71】
前記粒子が、細胞膜への組み込み前に、前記自己集合分子への結合を介して前記細胞に結合される、請求項68又は69に記載の方法。
【請求項72】
前記粒子が、細胞膜への組み込み後に、前記自己集合分子への結合を介して前記細胞に結合される、請求項68、69、71、又は前記請求項のいずれか一項に記載の方法。
【請求項73】
細胞と、前記細胞の表面に結合された膜結合リガンドとを含む単離された改変細胞組成物であって、前記膜結合リガンドが、前記細胞の前記表面の第1の部分に結合されており、前記細胞の前記表面の第2の部分には前記リガンドがないか又は前記リガンドの濃度が著しく低減されている細胞組成物。
【請求項74】
前記細胞が、幹細胞又は前駆細胞である、請求項73に記載の細胞組成物。
【請求項75】
前記幹細胞が、再プログラムされた細胞である、請求項74に記載の細胞組成物。
【請求項76】
前記細胞が分化細胞である、請求項73又は74に記載の細胞組成物。
【請求項77】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項73〜76、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項78】
前記細胞が、粒子をさらに含む、請求項73に記載の細胞組成物。
【請求項79】
前記粒子が、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー性粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、脂質、ナノ粒子、マイクロ粒子、又はデンドリマーからなる群から選択される、請求項73又は78に記載の細胞組成物。
【請求項80】
前記粒子が、前記細胞の機能を増強する作用剤を含む、請求項73、78、79、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項81】
前記粒子が、組織の機能を増強する作用剤を含む、請求項73、78〜80、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項82】
前記作用剤が、細胞成長、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着からなる群から選択される機能を増強する、請求項80又は81に記載の細胞組成物。
【請求項83】
前記作用剤が、小分子、酵素前駆体、酵素、栄養補助剤、化粧用の特性を有する作用剤、増殖因子、サイトカイン、RNA干渉分子、増殖因子、ビタミン、治療薬、診断薬、ケモカイン、標的化作用剤、又は分化因子からなる群から選択される、請求項80〜82、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項84】
前記粒子が、1000〜8000nmである、請求項73、78〜81、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項85】
前記粒子が、500〜1000nmである、請求項73、78〜81、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項86】
前記粒子が、1〜500nmである、請求項73、78〜81、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項87】
前記膜結合リガンドが、リンカー分子を介して結合される、請求項73に記載の細胞組成物。
【請求項88】
前記粒子が、リンカー分子結合を介して結合される、請求項73、78〜81、84〜86、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項89】
前記粒子が、リンカー分子を介さずに結合される、請求項73、78〜81、84〜86、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項90】
前記膜結合リガンドが、共有結合で結合される、請求項73又は87に記載の細胞組成物。
【請求項91】
前記膜結合リガンドが、非共有結合で結合される、請求項73、87、90、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項92】
前記粒子が、共有結合で結合される、請求項73、78〜81、84〜86、89、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項93】
前記粒子が、非共有結合で結合される、請求項73、78〜81、84〜86、89、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項94】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項73、87、90、91、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項95】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、又はグリコサミノグリカンからなる群から選択される、請求項73、87、90〜94、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項96】
複数の膜結合リガンドが、前記細胞に結合される、請求項73、87、90、91、94、95、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項97】
前記細胞が、作用剤を含む粒子の組織特異的担体として使用される、請求項73又は78に記載の細胞組成物。
【請求項98】
前記膜結合リガンドが、細胞凝集塊を形成するために使用される、請求項73、87、90、91、94〜96、又は前記請求項のいずれか一項に記載の方法。
【請求項99】
前記凝集塊がin vivoで形成される、請求項98に記載の細胞組成物。
【請求項100】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合されている、請求項73、87、90、91、94〜96、98、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項101】
標的組織再生を必要とする個体を治療する方法であって、
(a)膜結合リガンドを細胞の表面に結合させることにより標的細胞を形成し、前記膜結合リガンドが、治療される組織における前記標的細胞の蓄積をもたらすステップと、
(b)粒子を前記標的細胞の前記表面に結合させることにより二重機能性細胞を形成し、前記粒子が、前記二重機能性細胞の機能を増強する作用剤を含むステップと、
(c)標的組織再生を必要とする個体に前記二重機能性細胞を投与するステップとを含む方法。
【請求項102】
前記膜結合リガンドが、共有結合で結合される、請求項101に記載の方法。
【請求項103】
前記膜結合リガンドが、非共有結合で結合される、請求項101又は102に記載の方法。
【請求項104】
前記粒子が、共有結合で結合される、請求項101に記載の方法。
【請求項105】
前記粒子が、非共有結合で結合される、請求項101又は104に記載の方法。
【請求項106】
前記細胞が、標的組織再生を必要とする前記個体から単離される、請求項101に記載の方法。
【請求項107】
前記細胞が、標的組織再生を必要とする前記個体以外の個体から単離される、請求項101又は106に記載の方法。
【請求項108】
前記細胞が、前記膜結合リガンドを結合する前に、ex vivo培養環境中で増殖される、請求項101、106、107、又は前記請求項のいずれか一項に記載の方法。
【請求項109】
前記粒子が、前記標的組織再生の部位で放出される、請求項101、104、若しくは105、又は前記請求項のいずれかの一項に記載の方法。
【請求項110】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合されている、請求項101、106、107、108、又は前記請求項のいずれか一項に記載の方法。
【請求項111】
ワクチンを製作するための方法であって、
(a)膜結合リガンドを抗原提示細胞の表面に結合させることにより標的抗原提示細胞を形成し、前記膜結合リガンドが、リンパ組織における前記標的抗原提示細胞の蓄積をもたらすステップと、
(b)前記標的抗原提示細胞を抗原と接触させることにより活性化標的抗原提示細胞を形成し、前記活性化標的抗原提示細胞がワクチンを含むステップとを含む方法。
【請求項112】
前記抗原提示細胞が樹状細胞である、請求項111又は112に記載の方法。
【請求項113】
前記抗原がウイルス抗原を含む、請求項111又は112に記載の方法。
【請求項114】
前記抗原が細菌抗原を含む、請求項111又は112に記載の方法。
【請求項115】
前記抗原が癌関連抗原を含む、請求項111若しくは112、又は前記請求項のいずれか一項に記載の方法。
【請求項116】
細胞を修飾するためのキットであって、
(a)結合部分を有する自己集合分子と、
(b)細胞を修飾するための方法を示す説明書と、
(c)そのための包装材とを含むキット。
【請求項117】
前記自己集合分子が両親媒性である、請求項116に記載のキット。
【請求項118】
前記結合部分が膜結合リガンドである、請求項116に記載のキット。
【請求項119】
前記結合部分が粒子である、請求項116又は118に記載のキット。
【請求項120】
前記粒子が、第2の部分に加えて含まれている、請求項116又は119に記載のキット。
【請求項121】
単離された改変細胞組成物であって、
(a)細胞と、
(b)前記細胞の表面に結合された粒子と、
(c)前記細胞に結合しない粒子結合リガンドとを含む細胞組成物。
【請求項122】
前記粒子結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項121に記載の細胞組成物。
【請求項123】
前記細胞が膜結合リガンドを含む、請求項121に記載の細胞組成物。
【請求項124】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項121又は123に記載の細胞組成物。
【請求項125】
前記粒子が作用剤を含む、請求項121又は122に記載の細胞組成物。
【請求項126】
前記粒子の粒径が1nm〜5000nmである、請求項121、122、125、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項127】
前記粒子が、前記細胞内に内部移行されない、請求項121、122、125、126、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項128】
前記粒子が、作用剤をさらに含む、請求項121、125〜127、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項129】
前記作用剤が、血漿又は血清との接触による不活化に感受性である、請求項125又は128に記載の細胞組成物。
【請求項130】
不活化に感受性である前記作用剤が、RNA干渉分子である、請求項129に記載の細胞組成物。
【請求項131】
前記粒子が、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー性粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、脂質、ナノ粒子、マイクロ粒子、又はデンドリマーからなる群から選択される、請求項121、122、125〜127、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項132】
前記作用剤が、細胞増殖、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着からなる群から選択される機能を増強する、請求項125、128〜130、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項133】
前記作用剤が、前記細胞の前記機能を増強する、請求項125、128〜130、132、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項134】
前記作用剤が、前記細胞の標的化を増強する、請求項125、128〜130、131、133、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項135】
前記作用剤が治療薬を含む、請求項125、128〜130、132〜134、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項136】
前記作用剤が診断薬を含む、請求項125、128〜130、132〜135に記載の細胞組成物。
【請求項137】
前記作用剤が、宿主細胞及び/又は組織の機能を増強する、請求項125、127〜130、132〜136、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項138】
単離された改変細胞組成物であって、
(a)細胞と、
(b)前記細胞の表面に組み込まれた脂質分子に結合された膜結合リガンドとを含む細胞組成物。
【請求項139】
前記脂質分子が単一の尾部を含む、請求項138に記載の細胞組成物。
【請求項140】
前記脂質分子が複数の尾部を含む、請求項138又は139に記載の細胞組成物。
【請求項141】
前記脂質分子が複数の電荷を含む、請求項138〜140、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項142】
前記脂質分子が、リピドイドなどの改変脂質である、請求項138〜141、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項143】
前記膜結合リガンドが、標的化部分ではない、請求項1、26、41、138、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項144】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項138に記載の細胞組成物。
【請求項145】
対象体にワクチン接種するための方法であって、請求項111〜115又は前記請求項のいずれか一項に記載の前記活性化標的抗原提示細胞を投与することを含む方法。
【請求項146】
前記リガンドの部分が、前記細胞表面上で修飾後少なくとも2日間安定している細胞組成物を調製するための方法であって、
(a)リガンドを含む脂質小胞を調製するステップと、
(b)細胞を前記小胞と接触させ、前記接触ステップ後少なくとも2日間、前記リガンドが前記細胞上に存在するように、細胞組成物が形成されるステップとを含む方法。
【請求項147】
前記リガンドがビオチンを含む、請求項146に記載の方法。
【請求項148】
幹細胞の特徴を損なわずに前駆体幹細胞をリガンドで修飾するための方法であって、前記細胞表面を共有結合で修飾するステップを含み、細胞組成物が、前駆体又は幹細胞の特徴を喪失せずに形成される方法。
【請求項149】
前記前駆体又は幹細胞の特徴が、多系統分化、生存能、増殖、パラクリン因子の分泌、in vivo経内皮遊走、及び/又は接着を含む、請求項148に記載の方法。
【請求項150】
前記リガンドがシアリルルイスXを含む、請求項149に記載の方法。
【請求項151】
細胞の表面に接着リガンドを結合させるための方法であって、
(a)細胞をビオチンの供給源と接触させるステップと、
(b)ステップ(a)の前記細胞を、ストレプトアビジン及び接着リガンドと接触させるステップとを含み、
細胞組成物が、前記細胞表面上に接着リガンドを含んで形成され、前記接着リガンドが強力な接着を可能にする方法。
【請求項152】
複数の接着リガンドが、細胞の表面に結合される、請求項151に記載の方法。
【請求項153】
接着リガンドがP−セレクチン抗体を含む、請求項151に記載の方法。
【請求項154】
前記接着リガンドが、PSGL−1を含む、請求項151に記載の方法。
【請求項155】
前記接着リガンドが、1.9ダイン/cm2までで、2μm/秒未満で回転することを可能にする、請求項154に記載の方法。
【請求項156】
前記接着リガンドが、組織内局在化を増強する、請求項154に記載の方法。
【請求項157】
前記組織が骨髄である、請求項156に記載の方法。
【請求項158】
細胞の表面に接着粒子を結合させるための方法であって、
(a)リガンドを粒子に結合させて機能化粒子を調製するステップと、
(b)前記機能化粒子を細胞と接触させるステップとを含み、前記機能化粒子が前記細胞の表面に結合される方法。
【請求項159】
前記機能化粒子がPLGAを含む、請求項158に記載の方法。
【請求項160】
前記機能化粒子が、エマルジョン法を使用して調製される、請求項159に記載の方法。
【請求項161】
前記リガンドが、組織への前記細胞の局在化を可能する、請求項158に記載の方法。
【請求項162】
前記機能化粒子が内部移行される、請求項158に記載の方法。
【請求項163】
前記機能化粒子が内部移行されない、請求項158に記載の方法。
【請求項1】
単離された改変細胞組成物であって、
(a)細胞と、
(b)前記細胞の表面に結合された膜結合リガンドと、
(c)前記細胞の前記表面に結合された粒子とを含む細胞組成物。
【請求項2】
前記細胞が、幹細胞又は前駆細胞である、請求項1に記載の細胞組成物。
【請求項3】
前記幹細胞が、再プログラムされた細胞である、請求項1又は2に記載の細胞組成物。
【請求項4】
前記細胞が分化細胞である、請求項1、2、3、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項5】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項1〜4又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項6】
前記粒子が、1000〜8000nmである、請求項1に記載の細胞組成物。
【請求項7】
前記粒子が、500〜1000nmである、請求項1に記載の細胞組成物。
【請求項8】
前記粒子が、1〜500nmである、請求項1に記載の細胞組成物。
【請求項9】
前記膜結合リガンドが、リンカー分子を介して結合される、請求項1に記載の細胞組成物。
【請求項10】
前記粒子が、リンカー分子結合を介して結合される、請求項1、6〜8に記載の細胞組成物。
【請求項11】
前記粒子が、リンカー分子を介さずに結合される、請求項1、6〜8、10、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項12】
前記膜結合リガンドが、共有結合で結合される、請求項1、9、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項13】
前記膜結合リガンドが、非共有結合で結合される、請求項1、9、12、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項14】
前記粒子が、共有結合で結合される、請求項1、6〜8、10、11、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項15】
前記粒子が、非共有結合で結合される、請求項1、6〜8、10、11、14、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項16】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項11、9、12、13、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項17】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項1、9、12、13、16、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項18】
複数の膜結合リガンドが、前記細胞に結合される、請求項1、9、12、13、16、17、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項19】
前記粒子が、前記細胞の機能を増強する作用剤を含む、請求項1、6〜8、10、11、14、15、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項20】
前記粒子が、組織の機能を増強する作用剤を含む、請求項1、6〜8、10、11、14、15、19、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項21】
前記粒子が、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー性粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、脂質、ナノ粒子、マイクロ粒子、デンドリマー、又はそれらの任意の組合せからなる群から選択される、請求項1、6〜8、10、11、14、15、19、20、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項22】
前記作用剤が、細胞成長、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着からなる群から選択される機能を増強する、請求項19又は20に記載の細胞組成物。
【請求項23】
前記作用剤が、小分子、酵素、酵素前駆体、栄養補助剤、化粧用の特性を有する作用剤、増殖因子、サイトカイン、RNA干渉分子、増殖因子、ビタミン、治療薬、診断薬、ケモカイン、標的化作用剤、又は分化因子からなる群から選択される、請求項19、20、22、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項24】
前記細胞が、作用剤を含む粒子用の組織特異的担体として使用される、請求項1〜4又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項25】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合される、請求項11〜4、24、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項26】
単離された改変細胞組成物であって、
(a)細胞と、
(b)前記細胞の表面に組み込まれた自己集合分子に結合された膜結合リガンドとを含む細胞組成物。
【請求項27】
前記自己集合分子が両親媒性である、請求項26に記載の細胞組成物。
【請求項28】
前記細胞が、幹細胞又は前駆細胞である、請求項26に記載の細胞組成物。
【請求項29】
前記幹細胞が、再プログラムされた細胞である、請求項28に記載の細胞組成物。
【請求項30】
前記細胞が分化細胞である、請求項26又は28に記載の細胞組成物。
【請求項31】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項26、28〜30、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項32】
前記膜結合リガンドが、リンカー分子を介して結合される、請求項26に記載の細胞組成物。
【請求項33】
前記膜結合リガンドが、前記自己集合分子と共有結合で結合される、請求項26又は32に記載の細胞組成物。
【請求項34】
前記膜結合リガンドが、前記自己集合分子と非共有結合で結合される、請求項26、32、33、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項35】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項26、32、33、34、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項36】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項26、32〜35、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項37】
複数の膜結合リガンドが、前記細胞に結合される、請求項26、32〜36、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項38】
少なくとも1つの粒子が前記細胞に結合される、請求項26に記載の細胞組成物。
【請求項39】
前記粒子が、標的化部分であるリガンドを含む、請求項26又は38に記載の細胞組成物。
【請求項40】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合される、請求項26、28〜31に記載の細胞組成物。
【請求項41】
単離された改変細胞組成物を形成する方法であって、
(a)膜結合リガンドを自己集合分子に結合させて修飾分子を形成するステップと、
(b)前記修飾分子を用いて小胞を形成するステップと、
(c)前記小胞を細胞と融合させるステップとを含む方法。
【請求項42】
前記自己集合分子が両親媒性分子である、請求項41に記載の方法。
【請求項43】
前記細胞が、幹細胞又は前駆細胞である、請求項41に記載の方法。
【請求項44】
前記幹細胞が、再プログラムされた細胞である、請求項43に記載の方法。
【請求項45】
前記細胞が分化細胞である、請求項41又は43に記載の方法。
【請求項46】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項41、43〜45、又は前記請求項のいずれか一項に記載の方法。
【請求項47】
前記膜結合リガンドが、リンカー分子結合を介して結合される、請求項41に記載の方法。
【請求項48】
前記膜結合リガンドが、前記自己集合分子と共有結合で結合される、請求項41又は47に記載の方法。
【請求項49】
前記膜結合リガンドが、前記自己集合分子と非共有結合で結合される、請求項41、47、48、又は前記請求項のいずれか一項に記載の方法。
【請求項50】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項41、47〜49、又は前記請求項のいずれか一項に記載の方法。
【請求項51】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、又はグリコサミノグリカンからなる群から選択される、請求項41、47〜50、又は前記請求項のいずれか一項に記載の方法。
【請求項52】
複数の膜結合リガンドが、前記細胞に結合される、請求項41、47〜51、又は前記請求項のいずれか一項に記載の方法。
【請求項53】
前記単離された改変細胞組成物が、in vivoで形成される、請求項41に記載の方法。
【請求項54】
前記膜結合リガンドが、細胞凝集塊を形成するために使用される、請求項41、47〜51、又は前記請求項のいずれか一項に記載の方法。
【請求項55】
前記粒子が、細胞凝集塊を形成するために使用されるリガンドを含む、請求項41に記載の方法。
【請求項56】
単離された改変細胞組成物を形成するための方法であって、
(a)膜結合リガンドを自己集合両親媒性分子に結合させて修飾両親媒性分子を形成するステップと、
(b)前記修飾両親媒性分子を用いてミセルを形成するステップと、
(c)前記ミセルを前記細胞と融合させるステップとを含む方法。
【請求項57】
前記細胞が、幹細胞又は前駆細胞である、請求項56に記載の方法。
【請求項58】
前記幹細胞が、再プログラムされた細胞である、請求項58に記載の方法。
【請求項59】
前記細胞が分化細胞である、請求項56又は57に記載の方法。
【請求項60】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項56〜59、又は前記請求項のいずれか一項に記載の方法。
【請求項61】
前記膜結合リガンドが、リンカー分子を介して結合される、請求項56に記載の方法。
【請求項62】
前記膜結合リガンドが、前記自己集合両親媒性分子と共有結合で結合される、請求項61に記載の方法。
【請求項63】
前記膜結合リガンドが、前記自己集合両親媒性分子と非共有結合で結合される、請求項48に記載の方法。
【請求項64】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項56、61、62、又は前記請求項のいずれか一項に記載の方法。
【請求項65】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、薬物、栄養補助剤、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、薬物、又はグリコサミノグリカンからなる群から選択される、請求項56、61〜64、又は前記請求項のいずれか一項に記載の方法。
【請求項66】
複数の膜結合リガンドが、前記細胞に結合される、請求項56、61〜65、又は前記請求項のいずれか一項に記載の方法。
【請求項67】
前記単離された改変細胞組成物が、in vivoで形成される、請求項56に記載の方法。
【請求項68】
前記細胞が、少なくとも1つの粒子に結合される、請求項56に記載の方法。
【請求項69】
前記粒子が、標的化部分であるリガンドを含む、請求項56又は68に記載の方法。
【請求項70】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合されている、請求項56、57〜59、68、又は前記請求項のいずれか一項に記載の方法。
【請求項71】
前記粒子が、細胞膜への組み込み前に、前記自己集合分子への結合を介して前記細胞に結合される、請求項68又は69に記載の方法。
【請求項72】
前記粒子が、細胞膜への組み込み後に、前記自己集合分子への結合を介して前記細胞に結合される、請求項68、69、71、又は前記請求項のいずれか一項に記載の方法。
【請求項73】
細胞と、前記細胞の表面に結合された膜結合リガンドとを含む単離された改変細胞組成物であって、前記膜結合リガンドが、前記細胞の前記表面の第1の部分に結合されており、前記細胞の前記表面の第2の部分には前記リガンドがないか又は前記リガンドの濃度が著しく低減されている細胞組成物。
【請求項74】
前記細胞が、幹細胞又は前駆細胞である、請求項73に記載の細胞組成物。
【請求項75】
前記幹細胞が、再プログラムされた細胞である、請求項74に記載の細胞組成物。
【請求項76】
前記細胞が分化細胞である、請求項73又は74に記載の細胞組成物。
【請求項77】
前記細胞が、治療薬を発現するように遺伝子操作されている、請求項73〜76、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項78】
前記細胞が、粒子をさらに含む、請求項73に記載の細胞組成物。
【請求項79】
前記粒子が、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー性粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、脂質、ナノ粒子、マイクロ粒子、又はデンドリマーからなる群から選択される、請求項73又は78に記載の細胞組成物。
【請求項80】
前記粒子が、前記細胞の機能を増強する作用剤を含む、請求項73、78、79、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項81】
前記粒子が、組織の機能を増強する作用剤を含む、請求項73、78〜80、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項82】
前記作用剤が、細胞成長、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着からなる群から選択される機能を増強する、請求項80又は81に記載の細胞組成物。
【請求項83】
前記作用剤が、小分子、酵素前駆体、酵素、栄養補助剤、化粧用の特性を有する作用剤、増殖因子、サイトカイン、RNA干渉分子、増殖因子、ビタミン、治療薬、診断薬、ケモカイン、標的化作用剤、又は分化因子からなる群から選択される、請求項80〜82、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項84】
前記粒子が、1000〜8000nmである、請求項73、78〜81、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項85】
前記粒子が、500〜1000nmである、請求項73、78〜81、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項86】
前記粒子が、1〜500nmである、請求項73、78〜81、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項87】
前記膜結合リガンドが、リンカー分子を介して結合される、請求項73に記載の細胞組成物。
【請求項88】
前記粒子が、リンカー分子結合を介して結合される、請求項73、78〜81、84〜86、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項89】
前記粒子が、リンカー分子を介さずに結合される、請求項73、78〜81、84〜86、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項90】
前記膜結合リガンドが、共有結合で結合される、請求項73又は87に記載の細胞組成物。
【請求項91】
前記膜結合リガンドが、非共有結合で結合される、請求項73、87、90、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項92】
前記粒子が、共有結合で結合される、請求項73、78〜81、84〜86、89、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項93】
前記粒子が、非共有結合で結合される、請求項73、78〜81、84〜86、89、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項94】
前記膜結合リガンドが、組織における前記細胞の蓄積をもたらす、請求項73、87、90、91、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項95】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、又はグリコサミノグリカンからなる群から選択される、請求項73、87、90〜94、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項96】
複数の膜結合リガンドが、前記細胞に結合される、請求項73、87、90、91、94、95、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項97】
前記細胞が、作用剤を含む粒子の組織特異的担体として使用される、請求項73又は78に記載の細胞組成物。
【請求項98】
前記膜結合リガンドが、細胞凝集塊を形成するために使用される、請求項73、87、90、91、94〜96、又は前記請求項のいずれか一項に記載の方法。
【請求項99】
前記凝集塊がin vivoで形成される、請求項98に記載の細胞組成物。
【請求項100】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合されている、請求項73、87、90、91、94〜96、98、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項101】
標的組織再生を必要とする個体を治療する方法であって、
(a)膜結合リガンドを細胞の表面に結合させることにより標的細胞を形成し、前記膜結合リガンドが、治療される組織における前記標的細胞の蓄積をもたらすステップと、
(b)粒子を前記標的細胞の前記表面に結合させることにより二重機能性細胞を形成し、前記粒子が、前記二重機能性細胞の機能を増強する作用剤を含むステップと、
(c)標的組織再生を必要とする個体に前記二重機能性細胞を投与するステップとを含む方法。
【請求項102】
前記膜結合リガンドが、共有結合で結合される、請求項101に記載の方法。
【請求項103】
前記膜結合リガンドが、非共有結合で結合される、請求項101又は102に記載の方法。
【請求項104】
前記粒子が、共有結合で結合される、請求項101に記載の方法。
【請求項105】
前記粒子が、非共有結合で結合される、請求項101又は104に記載の方法。
【請求項106】
前記細胞が、標的組織再生を必要とする前記個体から単離される、請求項101に記載の方法。
【請求項107】
前記細胞が、標的組織再生を必要とする前記個体以外の個体から単離される、請求項101又は106に記載の方法。
【請求項108】
前記細胞が、前記膜結合リガンドを結合する前に、ex vivo培養環境中で増殖される、請求項101、106、107、又は前記請求項のいずれか一項に記載の方法。
【請求項109】
前記粒子が、前記標的組織再生の部位で放出される、請求項101、104、若しくは105、又は前記請求項のいずれかの一項に記載の方法。
【請求項110】
前記細胞が、凝集塊の一部であり、粒子に結合若しくは粒子内に封入されているか、又は移植可能な若しくは注射可能な基質内に捕捉若しくは基質に結合されている、請求項101、106、107、108、又は前記請求項のいずれか一項に記載の方法。
【請求項111】
ワクチンを製作するための方法であって、
(a)膜結合リガンドを抗原提示細胞の表面に結合させることにより標的抗原提示細胞を形成し、前記膜結合リガンドが、リンパ組織における前記標的抗原提示細胞の蓄積をもたらすステップと、
(b)前記標的抗原提示細胞を抗原と接触させることにより活性化標的抗原提示細胞を形成し、前記活性化標的抗原提示細胞がワクチンを含むステップとを含む方法。
【請求項112】
前記抗原提示細胞が樹状細胞である、請求項111又は112に記載の方法。
【請求項113】
前記抗原がウイルス抗原を含む、請求項111又は112に記載の方法。
【請求項114】
前記抗原が細菌抗原を含む、請求項111又は112に記載の方法。
【請求項115】
前記抗原が癌関連抗原を含む、請求項111若しくは112、又は前記請求項のいずれか一項に記載の方法。
【請求項116】
細胞を修飾するためのキットであって、
(a)結合部分を有する自己集合分子と、
(b)細胞を修飾するための方法を示す説明書と、
(c)そのための包装材とを含むキット。
【請求項117】
前記自己集合分子が両親媒性である、請求項116に記載のキット。
【請求項118】
前記結合部分が膜結合リガンドである、請求項116に記載のキット。
【請求項119】
前記結合部分が粒子である、請求項116又は118に記載のキット。
【請求項120】
前記粒子が、第2の部分に加えて含まれている、請求項116又は119に記載のキット。
【請求項121】
単離された改変細胞組成物であって、
(a)細胞と、
(b)前記細胞の表面に結合された粒子と、
(c)前記細胞に結合しない粒子結合リガンドとを含む細胞組成物。
【請求項122】
前記粒子結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項121に記載の細胞組成物。
【請求項123】
前記細胞が膜結合リガンドを含む、請求項121に記載の細胞組成物。
【請求項124】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項121又は123に記載の細胞組成物。
【請求項125】
前記粒子が作用剤を含む、請求項121又は122に記載の細胞組成物。
【請求項126】
前記粒子の粒径が1nm〜5000nmである、請求項121、122、125、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項127】
前記粒子が、前記細胞内に内部移行されない、請求項121、122、125、126、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項128】
前記粒子が、作用剤をさらに含む、請求項121、125〜127、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項129】
前記作用剤が、血漿又は血清との接触による不活化に感受性である、請求項125又は128に記載の細胞組成物。
【請求項130】
不活化に感受性である前記作用剤が、RNA干渉分子である、請求項129に記載の細胞組成物。
【請求項131】
前記粒子が、磁性粒子、脂質小胞、ミクロスフェア、リポソーム、ポリマー性粒子、分解性粒子、非分解性粒子、ミセル、ナノチューブ、マイクロチューブ、量子ドット、金属粒子、ナノシェル、無機粒子、脂質、ナノ粒子、マイクロ粒子、又はデンドリマーからなる群から選択される、請求項121、122、125〜127、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項132】
前記作用剤が、細胞増殖、増殖、遊走、細胞分化、脱分化、凝集、マトリックス産生、栄養因子の産生、アポトーシス、ホーミング、動員、又は生着からなる群から選択される機能を増強する、請求項125、128〜130、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項133】
前記作用剤が、前記細胞の前記機能を増強する、請求項125、128〜130、132、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項134】
前記作用剤が、前記細胞の標的化を増強する、請求項125、128〜130、131、133、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項135】
前記作用剤が治療薬を含む、請求項125、128〜130、132〜134、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項136】
前記作用剤が診断薬を含む、請求項125、128〜130、132〜135に記載の細胞組成物。
【請求項137】
前記作用剤が、宿主細胞及び/又は組織の機能を増強する、請求項125、127〜130、132〜136、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項138】
単離された改変細胞組成物であって、
(a)細胞と、
(b)前記細胞の表面に組み込まれた脂質分子に結合された膜結合リガンドとを含む細胞組成物。
【請求項139】
前記脂質分子が単一の尾部を含む、請求項138に記載の細胞組成物。
【請求項140】
前記脂質分子が複数の尾部を含む、請求項138又は139に記載の細胞組成物。
【請求項141】
前記脂質分子が複数の電荷を含む、請求項138〜140、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項142】
前記脂質分子が、リピドイドなどの改変脂質である、請求項138〜141、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項143】
前記膜結合リガンドが、標的化部分ではない、請求項1、26、41、138、又は前記請求項のいずれか一項に記載の細胞組成物。
【請求項144】
前記膜結合リガンドが、抗体、抗体断片、アプタマー、ペプチド、標的化部分、ビタミン、栄養補助剤、薬物、糖質、タンパク質、受容体、接着分子、糖タンパク質、糖残基、治療薬、グリコサミノグリカン、又はそれらの任意の組合せからなる群から選択される、請求項138に記載の細胞組成物。
【請求項145】
対象体にワクチン接種するための方法であって、請求項111〜115又は前記請求項のいずれか一項に記載の前記活性化標的抗原提示細胞を投与することを含む方法。
【請求項146】
前記リガンドの部分が、前記細胞表面上で修飾後少なくとも2日間安定している細胞組成物を調製するための方法であって、
(a)リガンドを含む脂質小胞を調製するステップと、
(b)細胞を前記小胞と接触させ、前記接触ステップ後少なくとも2日間、前記リガンドが前記細胞上に存在するように、細胞組成物が形成されるステップとを含む方法。
【請求項147】
前記リガンドがビオチンを含む、請求項146に記載の方法。
【請求項148】
幹細胞の特徴を損なわずに前駆体幹細胞をリガンドで修飾するための方法であって、前記細胞表面を共有結合で修飾するステップを含み、細胞組成物が、前駆体又は幹細胞の特徴を喪失せずに形成される方法。
【請求項149】
前記前駆体又は幹細胞の特徴が、多系統分化、生存能、増殖、パラクリン因子の分泌、in vivo経内皮遊走、及び/又は接着を含む、請求項148に記載の方法。
【請求項150】
前記リガンドがシアリルルイスXを含む、請求項149に記載の方法。
【請求項151】
細胞の表面に接着リガンドを結合させるための方法であって、
(a)細胞をビオチンの供給源と接触させるステップと、
(b)ステップ(a)の前記細胞を、ストレプトアビジン及び接着リガンドと接触させるステップとを含み、
細胞組成物が、前記細胞表面上に接着リガンドを含んで形成され、前記接着リガンドが強力な接着を可能にする方法。
【請求項152】
複数の接着リガンドが、細胞の表面に結合される、請求項151に記載の方法。
【請求項153】
接着リガンドがP−セレクチン抗体を含む、請求項151に記載の方法。
【請求項154】
前記接着リガンドが、PSGL−1を含む、請求項151に記載の方法。
【請求項155】
前記接着リガンドが、1.9ダイン/cm2までで、2μm/秒未満で回転することを可能にする、請求項154に記載の方法。
【請求項156】
前記接着リガンドが、組織内局在化を増強する、請求項154に記載の方法。
【請求項157】
前記組織が骨髄である、請求項156に記載の方法。
【請求項158】
細胞の表面に接着粒子を結合させるための方法であって、
(a)リガンドを粒子に結合させて機能化粒子を調製するステップと、
(b)前記機能化粒子を細胞と接触させるステップとを含み、前記機能化粒子が前記細胞の表面に結合される方法。
【請求項159】
前記機能化粒子がPLGAを含む、請求項158に記載の方法。
【請求項160】
前記機能化粒子が、エマルジョン法を使用して調製される、請求項159に記載の方法。
【請求項161】
前記リガンドが、組織への前記細胞の局在化を可能する、請求項158に記載の方法。
【請求項162】
前記機能化粒子が内部移行される、請求項158に記載の方法。
【請求項163】
前記機能化粒子が内部移行されない、請求項158に記載の方法。
【図1】

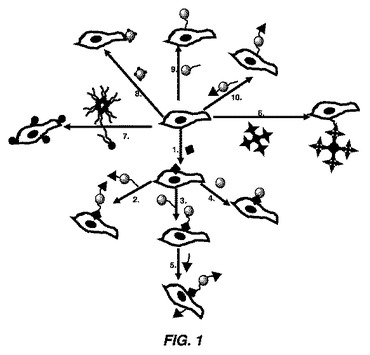
【図2】
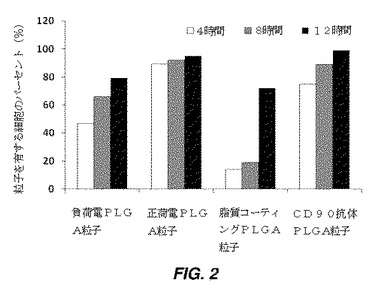
【図3】
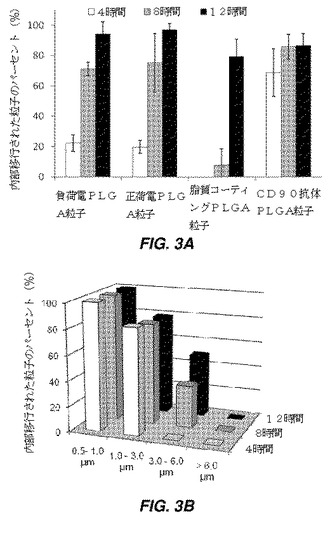
【図4】
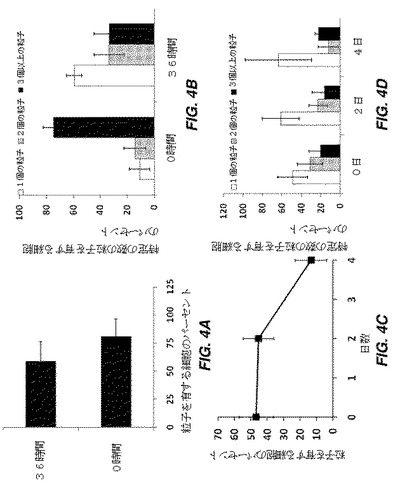
【図5】
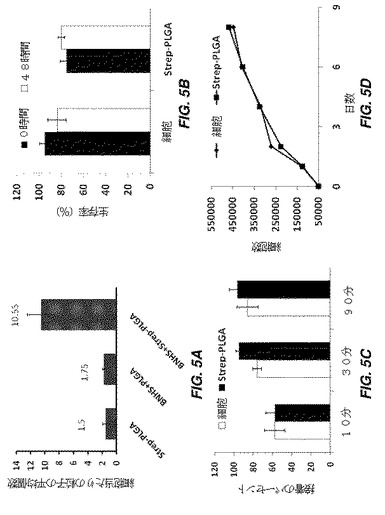
【図6】
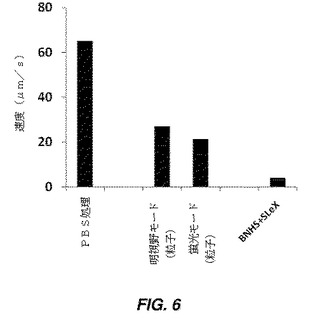
【図7】
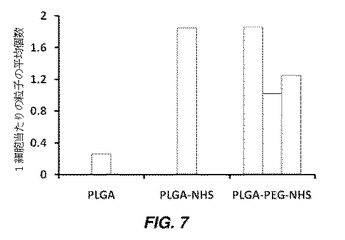
【図8】
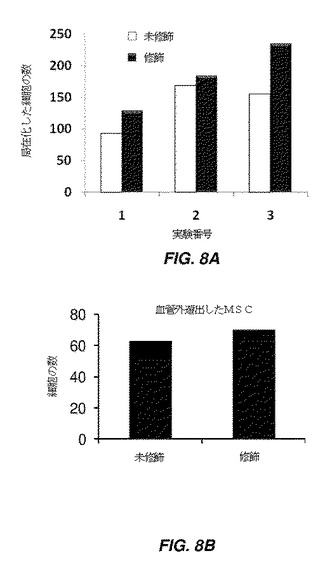
【図9】
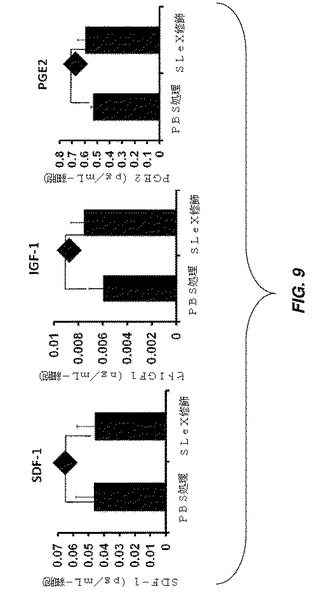
【図10】
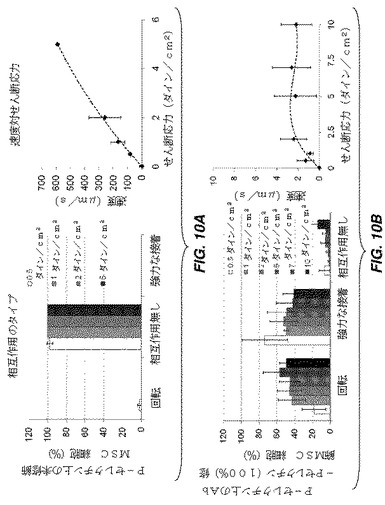
【図11】
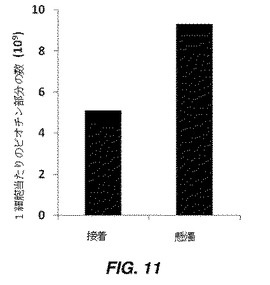
【図12】
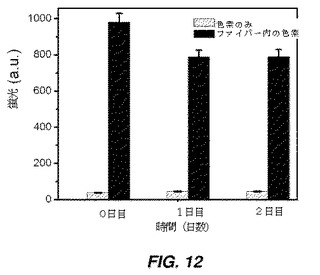
【図13】
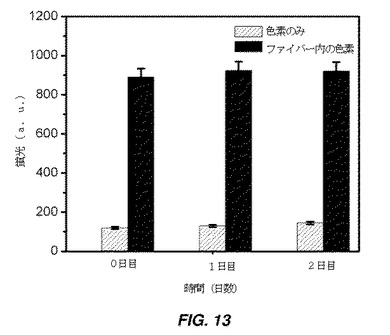
【図14】
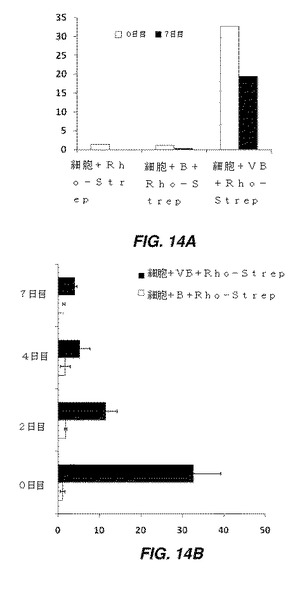
【図15】
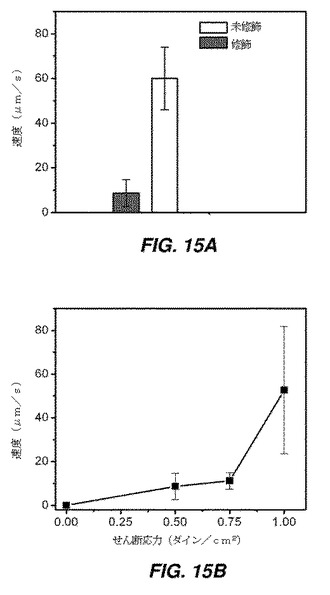
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公表番号】特表2011−518888(P2011−518888A)
【公表日】平成23年6月30日(2011.6.30)
【国際特許分類】
【出願番号】特願2011−507607(P2011−507607)
【出願日】平成21年4月29日(2009.4.29)
【国際出願番号】PCT/US2009/042087
【国際公開番号】WO2009/134866
【国際公開日】平成21年11月5日(2009.11.5)
【出願人】(504412945)ザ ブライハム アンド ウイメンズ ホスピタル, インコーポレイテッド (54)
【Fターム(参考)】
【公表日】平成23年6月30日(2011.6.30)
【国際特許分類】
【出願日】平成21年4月29日(2009.4.29)
【国際出願番号】PCT/US2009/042087
【国際公開番号】WO2009/134866
【国際公開日】平成21年11月5日(2009.11.5)
【出願人】(504412945)ザ ブライハム アンド ウイメンズ ホスピタル, インコーポレイテッド (54)
【Fターム(参考)】
[ Back to top ]