
細菌細胞及び哺乳動物細胞における抗体発現のための二重発現ベクター系
本発明は、二重発現ベクター、その使用、真核細胞における目的の全長ポリペプチドの発現及び分泌、並びに細菌におけるポリペプチドの可溶性ドメイン又は断片の発現及び分泌のための方法を提供する。細菌において発現させた場合、第1イントロン内の細菌プロモーターから転写し、第2イントロン内の停止コドンにおいて終結することによって、ポリペプチドの断片、例えばFabフラグメントが発現する。これに対し哺乳動物細胞においては、スプライシングにより2つのイントロン内に位置する細菌調節配列が除去され、哺乳動物シグナル配列が生成され、これにより全長ポリペプチド、例えばIgG重鎖又は軽鎖ポリペプチドが発現する。本発明の二重発現ベクター系は、新規なモノクローナル抗体を選択及びスクリーニングし、並びに目的の抗原分子に対する結合についてモノクローナル抗体を最適化するために用いることができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、細菌におけるFabフラグメントの発現及び分泌、並びに哺乳動物細胞における対応する全長IgGの発現及び分泌のための二重発現ベクター及びその使用方法を提供する。このベクターは、目的のポリペプチド、例えばIgG重鎖又は軽鎖の調節配列及びコード配列を含み、細菌プロモーター及びシグナル配列が、ポリペプチド、例えばIgG重鎖又は軽鎖遺伝子のシグナル配列内に位置する第1イントロン内に含まれ、目的のタンパク質が、例えばIg重鎖が複数のイントロンを有する場合、細菌停止コドンが、第2イントロン、例えば重鎖遺伝子のCH1ドメインとヒンジ領域の間のイントロン内に含まれる。このベクターは、哺乳動物プロモーター、細菌細胞と哺乳動物細胞の両方の複製起点、及び場合によっては、1つ又は複数の選択マーカーも含む。したがって、細菌において発現させた場合、細菌プロモーターから転写し、第2イントロン内の停止コドンにおいて終結することによって、ポリペプチドフラグメント、例えばFabフラグメントが発現する。これに対し、哺乳動物細胞においては、スプライシングによりイントロン内に位置する細菌調節配列が除去され、哺乳動物シグナル配列が生成され、これにより全長ポリペプチド、例えばIgG重鎖又は軽鎖ポリペプチドが発現する。この二重ベクター発現系は、新規なモノクローナル抗体を選択及びスクリーニングし、並びに目的の抗原分子に対する結合についてモノクローナル抗体を最適化するために用いることができる。この系を用いると、大腸菌(E. coli)でFab(又はscFv)を発現させることにより、最初のスクリーニング又は選択のステップが実現され、機能試験用に所望のアイソタイプの二価IgG分子として、得られたFab結合分子を容易に発現させることができる。
【背景技術】
【0002】
組換え発現系は、現在の抗体工学技術開発の鍵となっている。哺乳動物細胞におけるIgM又はIgGのクローン化された軽鎖及び重鎖遺伝子の共発現が示されると、ヒトの定常領域を含んでいるキメラMabが迅速に生成され、試験された(Ochi et al., 1983、Proc. Natl. Acad. Sci. U.S.A. 80:6351-6355;Oi et al., 1983、Proc. Natl. Acad. Sci. U.S.A. 80:825-829;Morrison et al., 1984、Proc. Natl. Acad. Sci. U.S.A. 81(21):6851-5)。続いて、アビディティを低下させずにヒトの配列をマウスイムノグロブリン可変領域に導入して、ヒト被験者内での潜在的な免疫原性が非常に低い抗体を得る方法が開発された(Jones et al., 1986、Nature 321:522-525;Queen et al., 1989、Proc. Natl. Acad. Sci. U.S.A. 86:10029-10033)。ヒトでの試験用及び最終的な販売用の高度に精製された物質を調製するため、CHO細胞又はマウス骨髄腫細胞内でこのような組換え抗体を大量に発現させる再現可能な方法が開発された。癌治療用のリツキサン及びハーセプチン、RSV感染防止用のシナジス、慢性関節リウマチ治療用のレミケード、並びに移植片拒絶防止用のゼナパックスを含めて(Reff et al., 1994、Blood 83:435-445;Carter et al., 1992、Proc. Natl. Acad. Sci. U.S.A. 89(10):4285-9;Johnson et al., 1997、J. Infect. Dis. 176:1215-1224;Queen et al., 1989、Proc. Natl. Acad. Sci. U.S.A. 86:10029-10033;表1を参照のこと)、現在多数のこのようなMabがヒトでの使用に承認され市販されている。
【0003】
同様に、Fv、一本鎖Fv、又はFab分子を細菌の系でうまく発現させることができたことが実証されると、この発現技術を利用して、VH及びVL配列の多様なライブラリーを利用する方法が急速に開発された(Skerra et al., 1988、Science 240:1038-1041;Bird et al., 1988、Science 242:423-426;Huston et al., 1988、Proc. Natl. Acad. Sci. U.S.A. 85:5879-5883)。VL及びVH配列のコンビナトリアルライブラリーは、最初にバクテリオファージλから発現させ、プラークリフトアッセイを用いて特定の組合せと抗原との結合についてスクリーニングしていた(Huse et al., 1989、Science 246:1275-81;Huse et al., 1992、Biotechnology 24:517-523)。糸状バクテリオファージの表面上にscFv又はFabのどちらか一方を固定することにより、パニング技術を用いて、そのゲノム内に結合領域の遺伝子を含むファージと結合するかどうかで選択することが可能になった(McCafferty et al., 1990、Nature 348:552-554;Hoogenboom et al., 1991、19: 4133-4137;Bird et al., 1988、Science 242:423-426;Kang et al., 1991、Proc. Natl. Acad. Sci. U.S.A. 88:4363-4366)。スクリーニングではなく選択することができることにより、まれな結合物を同定し単離するために開発された、個々のメンバーが109以上という大きなライブラリーに含まれる莫大な多様性が可能となった。したがって、現在大きく多様なバクテリオファージライブラリーから、ほとんどどんなタンパク質抗原でも適度なアフィニティで結合する抗体フラグメントを単離することが可能である。CDRの突然変異誘発を何ラウンドも反復実施することによって抗体フラグメントのアフィニティを改善し、抗原との改善された結合についてスクリーニング又は選択を行う方法も開発されている(Schier et al., 1996、J. Mol. Biol. 263: 551-567;Wu et al., 1998、Proc. Natl. Acad. Sci. U.S.A. 95:6037-6042)。
【0004】
Fab又はscFv発現系を用いて新規な特異性を有するものの同定及びアフィニティ成熟を行うことにおける大きな進歩があったにも関わらず、全長IgG分子はいくつかの利点を提供する。IgGの重要な特徴の1つは、その二価構造である。抗原との結合におけるIgGの2本のFab腕間の共同性により、一価のFabと比べて二価IgGのアビディティがより高くなる。抗原もまた多価である又は表面に結合している場合には、個々のFab腕のアフィニティとIgGのアビディティとの差異の程度は最も顕著である。抗原がより高い密度で存在している場合には、共同性の総計はより顕著となるが、Fab腕が高アフィニティのMabでは顕著ではなくなる。実際的な表現では、このことは、特定の抗原密度の閾値を超えると、アフィニティは高いが共同性が低いMabが、アフィニティは中程度であるが共同性が高いFab腕を有するMabと同じアビディティを有することを意味する。この後者のMabは、前者のMabと比べて抗原の密度が高い領域に対して選択的であるはずである。どちらかのMabが有利であるかという場合を想像することができる。例えば、真の癌抗原、すなわち腫瘍細胞上にしか発現していない抗原は極めて少数である。大部分は、より高密度で腫瘍細胞上に発現しているが、同様に他の細胞型にも発現している。したがって、アビディティは高いがアフィニティが中程度であるMabは、より低密度で抗原を発現している正常細胞より腫瘍細胞に対して選択的である可能性がある。同様に、ウイルス感染過程において、抗原は、ウイルス上に存在し、ウイルス感染細胞上に存在し、遊離型で分泌されている可能性がある。抗原密度がより高い領域に選択的な中和Mabは、遊離抗原、又は抗原密度が低い他の領域ではなくウイルス及び感染細胞を標的とすることができ、したがって高アフィニティMabと比べて同等又はより高い効力を有する可能性がある。Fab又はscFv単量体と連結する様々な手法を用いて、アビディティがより高いフラグメントを選択する方法が開発されている(Hudson et al., 1999、J Immunol. Methods 231:177-189)。この構築物は有用であるが、Fcを用いてFab腕を連結することによってもたらされたアビディティを正確に複製できない可能性がある。さらに、最初に特定の結合物を同定し、次いで共同性がより高いものを同定することが望まれることがある。例えば、上記の例で、ウイルスを中和するかどうかでスクリーニングしたいが、大部分の一価Fabがほとんど活性がないことが判明することがある。全長IgGに転換することにより、アビディティの増大に起因して中和活性を有するかどうかで選択を行うことが可能となる。
【0005】
他の場合では、エフェクター機能が、結合分子の最適な効力を得るのに必要となる可能性がある。イムノグロブリン分子のFc部分と特定の細胞表面受容体との相互作用により、抗原結合とエフェクター細胞機能の組合せが可能になる。
【0006】
ヒト及びげっ歯類に存在するIgGのFc受容体は3クラスあり、それらはRI、RII及びRIIIと名付けられている(Ravetch and Bolland、2001、Annu. Rev. Immunol. 19: 275 290)。RIは、単球及びマクロファージ上に存在し、単量体IgGと高いアフィニティで結合する。RIIは、B細胞、血小板、顆粒球、マクロファージ及び単球を含めた多種多様な細胞上に存在し、中程度のアフィニティで多量体IgG(免疫複合体の又は凝集したIgG)と結合する。2種の形態のRIIが発現し、これらは、受容体の細胞内部分に活性化型(ITAM)RIIaドメイン又は抑制型(ITIM)RIIbドメインが存在するかどうかによって異なる。所与の細胞上の活性化型及び抑制型受容体の相対レベルによって、免疫複合体に対する反応が決まる。B細胞は抑制型のみを発現している。RIIIは、RIIと同様に、中程度のアフィニティで多量体IgG(免疫複合体の又は凝集したIgG)と結合する。RIIIにも2種の形態が存在する。結合したγ鎖上のITAMドメインは、RIによるシグナル伝達も媒介し、RIIIa及びFcE受容体によるシグナル伝達も媒介する。シグナル伝達分子RIIIaは、NK細胞、単球、マクロファージ、及び特定のT細胞上でITAMを含むγ鎖と結合する。NK細胞上では、RIIIaによるシグナル伝達にTCRζ鎖も関与する。RIIIbは、非シグナル伝達型であり、GPI結合分子として(ヒト)好中球上で発現している。
【0007】
体内で、RI部位は、一般に単量体IgGによって占有されているが、RII及びRIII受容体は、占有されておらず、免疫複合体との相互作用に使用可能である。抗体抗原複合体によって活性化型Fc受容体が架橋されると、病原体を貪食し、直接の細胞傷害性によって外来細胞及び形質転換細胞を死滅させ、毒性物質を排除し、炎症反応を開始することができる。さらに、Fcは補体成分と相互作用する部位を含んでいる(Tao et al., 1993、J Exp Med 178:661-667)。最後に、Fcは、MHC関連FcRn受容体との特異的な相互作用によって、in vivoでIgGの半減期を長くする役割を果たしている(Ghetie and Ward、2002、Immunol Res. 25:97-113)。
【0008】
明らかに、標的が細菌又は癌細胞の場合、結合するだけでなく排除し又は死滅させる作用因子を試験すると有利であるはずである。その場合、IgGは、試験するのに好ましい分子であるりうる。例えば、キメラ抗CD20 Mabであるリツキサンは、それがヒトB細胞に対する強いADCC活性を有することに基づいて選択された(Reff et al., 1994、Blood 83:435-445)。さらに、抗HER2抗体であるハーセプチンは、腫瘍細胞上のEGF様受容体と結合し、それを介するシグナル伝達を遮断するが、最近の研究から、腫瘍の防御が主としてFcを媒介するものであることが示唆されている(Clynes et al., 2000、Nat. Med. 6:443-446)。
【0009】
scFv又はFabの系を用いた抗体の発現、選択及び改良についての関心は高い。しかし、現在使用可能な技術を用いても、さらなる試験及び開発のために、得られたscFv又はFabフラグメントを、糖鎖付加した全長Mabを発現させるためのベクター中に再クローン化しなければならない。このステップにより、この段階で試験することができるMabの数が厳格に制限される。したがって、その技術におけるこのような関心があるにも関わらず、ヒト治療薬として有用な全長Mab分子を選択し改良する有効な系は、現在まで開発されていない。
【0010】
本明細書における文献の引用又は考察を、そのようなものが本発明に対する従来技術であると認めるものと解釈すべきでない。
【発明の開示】
【0011】
本発明は、最適化されたモノクローナル抗体の選択、スクリーニング及び発現のためのベクター系、及びその使用方法を提供する。このベクター系を用いて、細菌内、好ましくは大腸菌内でFabフラグメントを、真核細胞内、好ましくは哺乳動物細胞内で対応する全長抗体(例えば、IgG)を発現させ選択することができる。この系を用いると、最初の結合分子のスクリーニング及び/又は選択を、Fab又はscFvを発現した大腸菌を用いて実現することができ、得られた結合分子を、機能試験用に所望のアイソタイプの二価抗体として迅速に発現させることができる。
【0012】
本発明は、細菌において細胞膜周辺腔へと抗体フラグメントを発現させ分泌し、哺乳動物細胞において全長IgGを発現させ分泌することができる二重発現ベクター系の出願人による発見及び開発に部分的に基づいている。この新規なベクターでは、細菌内でFabフラグメントを発現させ分泌するのに必要な調節エレメントは、哺乳動物細胞におけるIgG重鎖及び軽鎖RNA転写物の適当なプロセシングにも、IgG重鎖及び軽鎖ポリペプチドを分泌するのにも必要な配列エレメントと重複する。具体的には、細菌プロモーター及びシグナル配列は、哺乳動物IgG重鎖又は軽鎖遺伝子のシグナル配列をコードする配列内に位置するイントロン内に含まれ、細菌停止コドンは、重鎖遺伝子のCH1とヒンジ領域の間にある他のイントロンに(又は、別の実施形態ではヒンジ領域とCH2ドメインの間に位置するイントロンに)含まれる。したがって、細菌において発現させた場合、細菌プロモーターから転写し、第2イントロン内の停止コドンにおいて終結することによって、細菌の細胞膜周辺腔でFabフラグメントが発現する。これに対し哺乳動物細胞においては、スプライシングにより細菌プロモーター及びシグナル配列が除去され、哺乳動物シグナル配列が再生成され、これにより全長IgG重鎖又は軽鎖ポリペプチドが発現する。このベクターの重要な特徴は、細菌の及び哺乳動物の調節配列エレメント、すなわち哺乳動物シグナル配列及びスプライス受容部位、並びに細菌プロモーター及びシグナル配列が、この配列エレメント4種すべての機能が維持されるように、構造的にかつ機能的に重複していることである。したがって、このベクターの構築において、この重複した領域内でどんな変化が生じても、この配列エレメント4種の機能が維持されることが重要である。
【0013】
この改良系は、治療薬として使用するのに最良の機能性Mabの同定を促進し合理化する。細菌細胞において発現させた場合、細菌プロモーターは、Fab(又はscFv)の発現を制御し、それによって細菌細胞において抗原と結合するかどうかで選択しスクリーニングすることが可能になる。しかし、哺乳動物細胞において発現させた場合、スプライシングにより哺乳動物シグナル配列内のイントロン、及び重鎖のCH1とヒンジ領域との間のイントロンが除去され、したがって細菌プロモーター、細菌シグナル配列、及び停止コドンが除去され、哺乳動物シグナル配列が再構築される。哺乳動物プロモーター、例えばCMVプロモーターは、哺乳動物シグナル配列の開始部位の5’に位置し、IgG分子の重鎖及び軽鎖をコードするヌクレオチド配列の転写を誘導し、それによって全長の重鎖又は軽鎖IgG分子が哺乳動物細胞内で発現する。
【0014】
一実施形態では、本発明は、哺乳動物細胞においてIgG重鎖又は軽鎖を、細菌において重鎖又は軽鎖のFabフラグメント部分を発現させるためのベクターを包含し、前記ベクターは、(a)細菌性複製起点、(b)哺乳動物複製起点、及び(c)細胞内発現のための哺乳動物プロモーターを含み、そして前記重鎖又は前記軽鎖をコードするヌクレオチド配列と機能的に結合しており、前記ヌクレオチド配列は、(i)第1イントロンを含む哺乳動物シグナル配列であって、第1イントロンが、細菌細胞において細菌プロモーター及びシグナル配列が前記重鎖又は前記軽鎖のFabフラグメントの細胞膜周辺腔への発現及び分泌を誘導し、哺乳動物細胞において前記哺乳動物プロモーター及びシグナル配列が前記重鎖又は前記軽鎖の発現及び分泌を誘導するように、前記重鎖又は前記軽鎖のFabドメインをコードする配列と機能的に結合した細菌プロモーター及び細菌シグナル配列を含むものである哺乳動物シグナル配列と、(ii)前記ベクターが前記重鎖をコードし、第2イントロンが前記重鎖配列のCH1とヒンジ領域の間に含まれる場合、細菌における翻訳が、前記ヒンジ領域の配列の後で終わるように、好ましくはイントロンの5’末端に近接して停止コドンを含む第2イントロンとを含む。他の実施形態では、本発明は、細菌プロモーターがlacPO配列を含む、上記に記載のベクターを提供する。特定の実施形態では、本発明は、細菌シグナル配列がpelBシグナル配列であるそのようなベクターを提供する。他の特定の実施形態では、本発明は、エピトープタグ又はアフィニティ標識をコードする配列を含むように、軽鎖配列を遺伝子改変した、上記に記載のベクターを提供する。他の実施形態では、本発明は、エピトープタグがFd鎖のC末端にあるHSVタグである、上記に記載のベクターを提供する。他の実施形態では、このベクターのアフィニティタグは、Fd鎖のC末端にあるヘキサヒスチジンタグである。
【0015】
他の特定の実施形態では、このベクターは、重鎖及び軽鎖の両方をコードする配列を含み、それぞれが哺乳動物及び細菌のプロモーター及びシグナル配列と機能的に連結している。他の特定の実施形態では、重鎖又は軽鎖は、キメラ重鎖又は軽鎖である。他の特定の実施形態では、重鎖又は軽鎖配列は、ヒト又はヒト化重鎖又は軽鎖配列である。
【0016】
他の実施形態では、本発明は、上記に記載のベクターを含む細菌細胞を提供する。特定の実施形態では、その細菌細胞は、大腸菌細胞である。
【0017】
他の特定の実施形態では、本発明は、上記に記載のベクターを含む哺乳動物細胞を提供する。特定の実施形態では、その哺乳動物細胞はヒト又はネズミ細胞であり、好ましくは、骨髄腫細胞、CHO細胞、HEK細胞、NSO細胞、NS1細胞、BHK細胞、COS細胞、293細胞、又は3T3細胞である。
【0018】
他の特定の実施形態では、本発明は、重鎖及び軽鎖の両方を発現する、上記に記載のベクターを含む細胞を提供する。本発明のこの態様の他の特定の実施形態では、重鎖及び軽鎖は、同じ細胞内で異なるベクターから発現し、その少なくとも1つ(好ましくは両方)は、上記に記載のベクターである。
【0019】
他の実施形態では、本発明は、細菌細胞においてベクターを発現させた場合にFd遺伝子VIII又はFd遺伝子III融合物が産生されるように、糸状ファージ遺伝子VIII又は遺伝子IIIタンパク質のコード領域をコードする配列と機能的に連結した(好ましくは融合した)、IgG重鎖又は軽鎖のFd(VH CH1)セグメントをコードするヌクレオチド配列を含む、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクターを提供する。他の実施形態では、このベクターは、相補的な重鎖遺伝子又は軽鎖遺伝子をコードするヌクレオチド配列を含み、これはファージ配列と機能的に連結していない。
【0020】
本発明の他の態様では、治療薬として使用するMabを同定する方法が提供される。この方法は、(a)対照IgGをコードする、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクターを含む対照細胞を用意するステップと、(b)対照IgGと比べて遺伝子改変されたIgGをコードする、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクターをそれぞれ発現する試験細胞のライブラリーと抗原とを接触させるステップと、(c)対照細胞の周辺質抽出物と前記抗原の結合アフィニティと比較して試験細胞の周辺質抽出物と前記抗原の結合アフィニティを測定するステップとを含む。この方法の特定の実施形態では、この方法はさらに、ステップ(c)の後に、(d)哺乳動物細胞においてステップ(c)の試験細胞から単離したベクターを発現させるステップと、(e)その哺乳動物細胞と前記抗原とを接触させるステップと、(f)対照IgGの結合アフィニティと比較して、その哺乳動物細胞内で発現した遺伝子改変IgGの結合アフィニティを測定するステップとを含み、試験細胞の周辺質抽出物と前記抗原の結合アフィニティが対照細胞の周辺質抽出物と前記抗原の結合アフィニティより大きい場合に治療薬として有用なMabを発現している細胞が同定される。
【0021】
本発明の他の態様では、治療薬として使用するMabを同定するファージディスプレイスクリーニング法が提供される。この方法は、(a)fd−糸状ファージ−遺伝子IIIの融合物又はfd−糸状ファージ−遺伝子VIIIの融合物をコードする対照ファージを発現する細菌細胞を用意するステップと、(b)対照ファージと比べて改変された、軽鎖及びfd(VH CH1)遺伝子III融合物又はfd(VH CH1)遺伝子VIII融合物をコードする試験ファージを産生する複数の試験細胞を含むファージライブラリーのメンバーと抗原とを接触させるステップと、(c)対照ファージと前記抗原の結合アフィニティと比較して試験ファージと前記抗原の結合アフィニティを測定するステップとを含み、試験ファージと前記抗原の結合アフィニティが対照ファージと前記抗原の結合アフィニティより大きい場合に治療薬として有用なMabを発現している細胞が同定される。この方法の特定の実施形態では、この方法はさらに、ステップ(c)の後に、(d)哺乳動物細胞においてステップ(c)の試験細胞から単離したベクターを発現させるステップと、(e)その哺乳動物細胞と前記抗原とを接触させるステップと、(f)対照IgGの結合アフィニティと比較して、その哺乳動物細胞内で発現した遺伝子改変IgGの結合アフィニティを測定するステップとを含む。
【0022】
本発明はまた、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクターを含む、Fabポリペプチドを発現する複数の細菌細胞を含む組成物をも提供する。
【0023】
さらに本発明はまた、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクター、及びFabポリペプチドを発現する糸状ファージを含む、複数の細菌細胞を含む組成物をも提供する。特定の実施形態では、その細菌細胞は大腸菌細胞であり、その糸状ファージはfdファージである。
【0024】
他の実施形態では、本発明はさらに、複数の標的に対する受動治療薬の迅速な開発に特に有用なMabカクテルの作製を包含する。本明細書に記載のベクターを用いて、臨床的に重要なMabを単離するために、未感作の及び免疫感作されたヒト被験体に由来するFab発現大腸菌及びファージのライブラリーを作製することができる。
【0025】
さらに、細菌においてFabフラグメントを、哺乳動物細胞において全長IgGを発現させるためのベクターを設計する際に使用する原理を適用して、細菌において特定のタンパク質の一部を、哺乳動物細胞において全長タンパク質を発現させるためのベクターを作製することができる。例えば、本発明は、哺乳動物細胞において分泌型又は膜結合型ポリペプチドを、細菌において前記ポリペプチドの可溶性フラグメントを発現させるためのベクターを提供し、前記ベクターは、(a)細菌性複製起点、(b)哺乳動物複製起点、及び(c)前記分泌型又は膜結合型ポリペプチドをコードするヌクレオチド配列と機能的に結合した哺乳動物プロモーターを含み、前記ヌクレオチド配列は、イントロンを少なくとも1つ含む哺乳動物シグナル配列を含むものであり、前記イントロンは、前記細菌プロモーター及び細菌シグナル配列が細菌細胞において前記ポリペプチドの前記可溶性ドメインの細胞膜周辺腔への発現及び分泌を誘導し、及び前記哺乳動物プロモーター前記哺乳動物シグナル配列が哺乳動物細胞において前記ポリペプチドの発現及び分泌を誘導するように、前記ポリペプチドの前記可溶性ドメインをコードする配列と機能的に結合した細菌プロモーター及び細菌シグナル配列を含み、前記哺乳動物プロモーターは、前記ポリペプチドの前記可溶性ドメインをコードする前記ヌクレオチド配列と機能的に結合している。
【0026】
本明細書において、「抗体」(antibody及びantibodies)という用語は、モノクローナル抗体、ヒト化抗体、キメラ抗体、一本鎖Fv(scFv)、一本鎖抗体、Fabフラグメント、F(ab’)フラグメント、ジスルフィド結合Fv(sdFv)、及び抗イディオタイプ(抗Id)抗体(例えば、対象とする抗体に対する抗Id抗体を含む)、並びに上記のいずれかのエピトープ結合性フラグメントを指す。具体的には、抗体には、イムノグロブリン分子、及びイムノグロブリン分子の免疫活性フラグメント、すなわち抗原結合部位を含む分子が含まれる。イムノグロブリン分子は、どのタイプ(例えば、IgG、IgE、IgM、IgD、IgA及びIgY)でもよく、どのクラス(例えば、IgG1、IgG2、IgG3、IgG4、IgA1及びIgA2)でもよく、どのサブクラスでもよい。
【0027】
本明細書において、「二重発現ベクター系」という用語は、真核細胞において、好ましくは哺乳動物細胞において対象とするポリペプチドを発現させ、細菌細胞において細胞膜周辺腔へとそのポリペプチドフラグメントを発現させるベクター系を指す。好ましい実施形態では、対象とするポリペプチドは抗体鎖であり、そのフラグメント又はドメインは、Fabフラグメント又はscFvフラグメントである。最も好ましくは、対象とするポリペプチドは、IgGの重鎖又は軽鎖、及びIgGのFabフラグメントである。したがって、本明細書においてこの用語は、「抗体二重発現ベクター系」及び「IgG二重発現ベクター系」という用語も指している。
【0028】
本明細書において、「二重発現ベクターカセット」又は「二重発現ベクターポリヌクレオチドカセット」という用語は、本明細書で同義に用いられ、対象とするポリヌクレオチドのコード配列、並びに真核細胞において、好ましくは哺乳動物細胞において対象とするポリペプチドの発現及び分泌に、細菌細胞において対象とするポリペプチドのフラグメント又はドメインの細胞膜周辺腔への発現及び分泌に必要な調節配列を含むポリヌクレオチドを指す。このような調節配列は、真核生物のシグナル配列内に、好ましくは哺乳動物のシグナル配列内にイントロンを含み、この配列は、真核細胞において、好ましくは哺乳動物細胞において対象とするポリペプチドの発現及び分泌を、細菌細胞において(対象とするポリペプチドをコードする配列の第2イントロン内に停止コドンを置くことで決定する)対象とするポリペプチドのフラグメント又はドメインの細胞膜周辺腔への発現及び分泌を可能にするように、本明細書で詳細に説明するように特定の形で位置する細菌プロモーター及び細菌シグナル配列をイントロン内に含む。「抗体発現ベクターポリヌクレオチドカセット」という用語は、本発明の好ましい実施形態を指し、この場合、対象とするポリペプチドは抗体鎖であり、そのフラグメント又はドメインは、Fabフラグメント又はscFvフラグメントである。同様に、「IgG発現ベクターポリヌクレオチドカセット」という用語は、特定の実施形態を指し、この場合、対象とするポリペプチドは、IgGの重鎖又は軽鎖、及びIgGのFabフラグメントである。
【0029】
本発明のこれらの態様及び他の態様は、添付の図面及び以下の詳細な説明の項を参照することによってより理解されるであろう。
【発明を実施するための最良の形態】
【0030】
本発明は、細菌細胞、例えば大腸菌細胞においてFabフラグメントを発現させ分泌し、真核細胞、好ましくは哺乳動物細胞において、しかし例えば昆虫細胞又は鳥類細胞においてもIgG重鎖又は軽鎖ポリペプチドを発現させるための新規な二重発現ベクター系、並びに特定の結合性を有するモノクローナル抗体のスクリーニング及び最適化にそれを使用する方法に関する。上記で論じたように、本発明は、細菌において細胞膜周辺腔へと抗体フラグメントを発現させ分泌し、哺乳動物細胞において全長抗体を発現させ分泌することができる二重発現ベクター系の出願人による開発に部分的に基づいている。この新規なベクターでは、細菌プロモーター及びシグナル配列は、哺乳動物IgG重鎖又は軽鎖遺伝子のシグナル配列内に位置するイントロン内に含まれ、細菌停止コドンは、重鎖遺伝子のCH1とヒンジ領域の間にある他のイントロンに含まれる。したがって、細菌において発現させた場合、細菌プロモーターから転写し、第2イントロン内の停止コドンにおいて終結することによってFabフラグメントが発現する。これに対し哺乳動物細胞においては、スプライシングにより細菌性制御エレメントが除去され、哺乳動物シグナル配列が生成され、これにより全長IgG重鎖又は軽鎖ポリペプチドが発現する。したがって、この二重の目的のベクターは、細菌性の及び哺乳動物の調節配列エレメント、すなわち哺乳動物シグナル配列及びスプライス受容部位、並びに細菌プロモーター及びシグナル配列の構造及び機能を維持するように設計する。
【0031】
この新規な二重発現ベクター系の構築及び使用に関する組成物及び方法について以下に説明する。具体的には、第5.1節で、真核細胞において対象とするポリペプチドが、細菌においてそのフラグメントが二重に発現するように設計されたDNAカセット、二重発現ベクターポリヌクレオチドカセットを含むベクター、このようなカセット及びベクターを含む宿主細胞、並びにこのようなカセット、ベクター、及び宿主細胞を含むキットを含めた本発明の組成物について説明する。第5.2節及び第5.3節で、新規な抗体の同定にこの新規なベクター配列を使用する方法、真核細胞の系と細菌の系の両方において、最適化されたモノクローナル抗体を選択しスクリーニングする方法、並びにこの原理を適用して、対象とする任意の膜結合型又は分泌型タンパク質用の二重発現ベクター系をも構築する一般化した方法を含めた、本発明の使用方法について説明する。
【0032】
5.1.二重発現ベクター
上記で概略を述べた配列及び機能エレメントを有する二重発現ベクター系は、当技術分野で使用可能な様々な技術を用いて構築することができる。好ましい実施形態では、ベクターは、
(1)哺乳動物プロモーター、
(2)IgG重鎖又は軽鎖をコードするヌクレオチド配列であって、(a)以下の(i)及び(ii)を含む哺乳動物シグナル配列を含む上記ヌクレオチド配列:
(i)細菌プロモーター及びシグナル配列が細菌細胞において前記重鎖又は前記軽鎖のFabドメインの細胞膜周辺腔への発現及び分泌を誘導し、前記哺乳動物プロモーター及びシグナル配列が哺乳動物細胞において前記重鎖又は前記軽鎖の発現及び分泌を誘導するように、前記細菌プロモーター及びシグナル配列が第1スプライス受容部位と重複し、IgG重鎖又は軽鎖コード配列と機能的に連結するように前記細菌プロモーター及びシグナル配列を含む第1イントロンと、(ii)前記ベクターが前記重鎖をコードする場合には、CH1をコードする配列と重鎖遺伝子のヒンジ領域との間にあり、細菌停止コドンを含む第2イントロン、
(3)細菌性複製起点、並びに
(4)哺乳動物複製起点
を含む。前記の配列成分に加えて、ベクターは、細菌細胞においてベクターをクローン化し増殖させ、細菌細胞と真核細胞の両方でベクターを有する細胞を増殖させ選択するための選択マーカー、他のヌクレオチド配列を付加するためのマルチクローニング部位配列、並びに対象とする他の配列をさらに含むことができる。この配列エレメントを、本明細書において以下に詳細に説明する。
【0033】
5.1.1.二重発現ベクターカセット配列
対象とするポリペプチド、例えばIgG重鎖又は軽鎖をコードするヌクレオチド配列は、第1イントロンを含む哺乳動物シグナル配列、及びその配列が重鎖をコードする場合は第2イントロンを含むCH1からヒンジまでの領域を用いて設計する。第1及び第2イントロンは、細菌細胞においてポリペプチドの発現及び分泌を誘導し、二重発現ベクターが哺乳動物細胞において発現する場合、スプライシングによって除去される細菌調節配列を含むように設計する。二重発現ベクターの具体的な配列組成及び構造について、本明細書において詳細に説明する。
【0034】
第1イントロンは、対象とするポリペプチドの哺乳動物シグナル配列内に位置するように設計する。哺乳動物シグナル配列が天然にイントロンを有さない場合、第1イントロンは、当技術分野で知られている任意の組換えDNA法を用いて構築する。第1イントロンは、スプライス受容部位と重複する細菌プロモーター及びシグナル配列を含む。細菌プロモーター及びシグナル配列は、ポリペプチド配列と、例えばIgG重鎖配列又は軽鎖配列と「機能的に連結する」ように構築する。すなわち、細菌細胞においてFabフラグメント又はscFv配列の転写を誘導するように、細菌プロモーターを配置し、細菌細胞において細胞膜周辺腔へとポリペプチドが分泌されるようにスプライス受容部位と重複するように、細菌シグナル配列を配置する。
【0035】
細菌プロモーター、細菌シグナル配列、哺乳動物シグナル配列及びスプライス受容部位の機能を維持するため、第1イントロンのヌクレオチド配列は、当技術分野で周知であり、第6節で提示する実施例において示すプロモーターコンセンサス配列、シグナル配列コンセンサス配列及びスプライス部位コンセンサス配列を用いて設計することができる。例えば、対象とするポリペプチド、例えばIgGなどの抗体を標的として細菌の周辺質膜へと誘導するいずれのシグナル配列も使用することができる。細菌シグナル配列は、由来が天然のものでもよく、合成のものでもよい。天然に周辺質に存在することが定められているタンパク質と結合しているリーダー配列は、例えば、外来タンパク質の周辺質への分泌を誘導することが知られている(MacIntyre et al., 1990、Mol. Gen. Genet. 221:466-474)。好ましい実施形態では、シグナル配列は、pelB配列及びOmpAタンパク質リーダー配列(Hobom et al., 1995、Dev. Biol. Stand. 84:255-262)をコードする。それだけに限らないが、大腸菌PhoA(Oka et al., 1985、Proc. Natl. Acad. Sci 82:7212-16)、OmpT(Johnson et al., 1996、Protein Expression 7:104-113)、LamB及びOmpF(Hoffman & Wright、1985、Proc. Natl. Acad. Sci. USA 82:5107-5111)、β−ラクタマーゼ(Kadonaga et al., 1984、J. Biol. Chem. 259:2149-54)、エンテロトキシン(Morioka-Fujimoto et al., 1991、J. Biol. Chem. 266:1728-32)、黄色ブドウ球菌(Staphylococcus aureus)のプロテインA(Abrahmsen et al., 1986、Nucleic Acids Res. 14:7487-7500)、枯草菌(B. subtilis)のエンドグルカナーゼ(Lo et al., Appl. Environ. Microbiol. 54:2287-2292)由来のリーダー配列、並びに人工及び合成シグナル配列(MacIntyre et al., 1990、Mol. Gen. Genet. 221:466-74;Kaiser et al., 1987、Science、235:312-317)を含めた他のシグナル配列も考えられる。
【0036】
Fabフラグメント又はscFvの細菌の周辺質への分泌は、最初のベクターを作製した後にベクターの配列を変更することによって改善することができる。例えば、シグナル配列内に可変性を導入した一本鎖fv(scFv)のファージディスプレイを用いて、分泌が改善した変異体を選択することができる。さらに、他のシグナルペプチドのコード配列を改変し試験することもできる。この場合、シグナルペプチドのコード配列のデータベースを作成して所望のスプライス部位と比較することができる。次いで、最も相同なセグメントを必要なら改変して、大腸菌においてFabを分泌させ、HEK293細胞においてMabの発現及び分泌を保持することができる。
【0037】
すべての配列エレメントの機能、すなわち、細菌と哺乳動物の両方の宿主での転写、哺乳動物宿主での転写物のスプライシング、細菌での周辺質への分泌及び哺乳動物宿主での分泌が維持される条件で、個々のエレメントの機能を高めるのに適した変化を加えることにより、この領域内の個々の配列成分を最適化することができる。
【0038】
改変した原核細胞シグナルペプチドが依然として好ましいスプライス切断部位を保持しているかどうかを予測するために、神経ネットワークアルゴリズムを用いるシグナルP(SignalP)プログラムによって配列を分析することができる(Nielsen et al., 1997、Int. J. Neural Sys. 8、581 599)。バークレーショウジョウバエゲノムプロジェクト(Berkeley Drosophila Genome Project)ウェブサイトのスプライス部位予測(Splice Site Prediction)プログラムを用いて、スプライス部位の潜在的機能を評価することができる(Reese et al., J. Comput. Biol.、1997、4(3):311 23を参照のこと)。このプログラムも、ヒト遺伝子向けの神経ネットワークアルゴリズムを用いている。
【0039】
精製又はスクリーニングのプロトコルの間に、エピトープ/アフィニティタグを付加して、ポリペプチド、例えばFabフラグメント、scFv、或いは軽鎖又は重鎖の精製又は同定を改善することが望ましいこともある。確実に分泌後にタンパク質上にタグをさらすために、GGGGSなどの柔軟なリンカー配列を、機能ドメインとエピトープ/アフィニティタグ配列の間に導入する。ペプチドタグには、タグ付きのポリペプチド又はフラグメントの同定を容易にするが、タグ付きのポリペプチド又はフラグメントの機能を阻害し又はそれに干渉する可能性が低い方法/試薬が存在するタグが含まれ得る。タグは、対応する結合用試薬との結合が可能であるが、mRNAと結合しているタグ付きタンパク質を干渉しない任意の長さでよい。好ましい実施形態では、タグの長さは、約8、10、12、15、18又は20アミノ酸であり、15、20、25、30、40又は50アミノ酸以下であるが、100、150、200、300、400アミノ酸又は500アミノ酸以上でもよい。タグは、(1)対象とする細胞、又は(2)対象とするポリソーム調製物、又は(3)タグと結合する試薬と接触している、対象とするいずれの細胞分画、のどんな成分とも結合しない試薬と特異的に結合することができる。分子タグには、一例としてであり限定するものではないが、プロテインAフラグメント、mycエピトープ(Evan et al., Mol. Cell Biol. 5(12):3610-3616)、Btag(Wang et al., 1996、Gene 169(1): 53-58)及びポリヒスチジン束(Bornhorst et al., 2000、Methods Enzymol 326:245-54)が含まれ得る。他の好ましいタグには、以下のものがあるが、それだけに限らない:
(1)インフルエンザウイルス血球凝集素タンパク質の一部(Tyr−Pro−Tyr−Asp−Val−Pro−Asp−Tyr−Ala、配列番号1)。精製に用いる試薬は、タグ付きタンパク質を認識するモノクローナル抗体(12CA5)である(Wilson et al., 1984、Cell 37(3):767 78)。
【0040】
(2)ヒトc myc遺伝子の一部(Glu−Gln−Lys−Leu−Ile−Ser−Glu−Glu−Asp−Leu、配列番号2)。精製に用いる試薬は、タグ付きタンパク質を認識するモノクローナル抗体(9E10)である(Evan et al., 1985、Mol Cell Biol. 5(12):3610 6)。
【0041】
(3)ブルータングウイルス(bluetongue virus)VP7タンパク質の一部(Gln−Tyr−Pro−Ala−Leu−Thr、配列番号3)。精製に用いる試薬は、タグ付きタンパク質を認識するモノクローナル抗体(D11及び/又はF10)である(Wang et al., Gene. 1996 Feb 22;169(1):53-8)。
【0042】
(4)FLAGペプチド(例えば、Asp−Tyr−Lys−Asp−Asp−Asp−Asp−Lys、配列番号4)。精製に用いる試薬は、タグ付きタンパク質を認識するモノクローナル抗体(例えばM1及び/又はM2)(Sigma)である(Hopp et al., 「Synthesis of protein with an identification peptide」という名称の、1987年10月27日発行の米国特許第4,703,004号;Brizzard et al., 1994、Biotechniques. Apr;16(4):730-5;Knappik et al., 1994、Biotechniques 17(4):754-761)。
【0043】
(5)Strepタグペプチド(例えば、Ala Trp Arg His Pro Gln Phe Gly Gly、配列番号5)。好ましい実施形態では、strepタグペプチドを用いる。精製に用いる試薬は、タグ付きタンパク質を認識するいくつかの最適化された形のストレプトアビジンのうちの1つ(IBA GmbH)である(Skerraら、「Fusion peptides with binding activity for streptavidin」という名称の、1996年4月9日発行の米国特許第5,506,121号;Skerra et al., 1999、Biomol. Eng. 16(14):79-86;Skerra et al., 2000、Methods Enzymol. 2000;326:271-304)。
【0044】
細菌において発現されるポリペプチドの可溶性ドメイン/フラグメントが、対象とする全長ポリペプチドと共通の末端でない場合、細菌停止コドンを含む第2イントロンを、対象とするポリペプチド内に含める。このイントロンは、可溶性ドメイン/フラグメントの終結が所望されるポリペプチドの領域内に位置する。イントロンが、対象とするタンパク質の所望の位置に天然に存在しない場合、当技術分野で周知の組換え又は合成DNA技術を用いて、所望の位置に適当なイントロン配列を導入することができる。例えば、好ましい実施形態では、IgG重鎖の配列は、CH1ドメインとヒンジ領域の間に位置する第2イントロンを含む。細菌においてこの部位で翻訳が終結する結果、Fab1フラグメントが発現する。或いは、Fab2フラグメントの産生が所望される場合、ヒンジとCH2ドメインの間のポリペプチドをコードする配列内に、停止コドンを含んでいるイントロンを含めることもできる。これによって、より大きなFab2フラグメントが産生されるはずである。
【0045】
スプライス受容部位及び細菌シグナル配列の機能が確実に維持されているかどうか、二重発現ベクターカセット配列を試験する。少なくともスクリーニングの目的について、機能に関するわずかな影響なら許容することができるが、哺乳動物細胞から分泌される全長IgGの能力に負の影響を与えるどんな配列変化も有用でないはずである。
【0046】
当技術分野で知られている標準的な方法を用いて、IgG及びFab発現配列を組み込んだ環状ベクターを構築することができる(Sambrook et al., 1989、上記;Ausubel et al., Current Protocols in Molecular Biology、Greene Publishing Associates and Wiley Interscience、New Yorkを参照のこと)。例えば、合成又は組換えDNA技術を用いることができる。一実施形態では、ポリメラーゼ連鎖反応(「PCR」)増幅によって、二重発現ベクターカセット配列を含むベクターを作製する。この方法では、その5’末端に制限酵素部位を、その3’末端にIgGの調節配列とコード配列の境界配列と相補的なPCRプライマー配列を含むようにオリゴヌクレオチドを合成する。次いでそのオリゴヌクレオチドをPCR増幅反応でプライマーとして使用して、IgGの調節配列及びコード配列の領域を増幅する。次いで、標準的な分子生物学技術を用いて、哺乳動物の及び細菌性複製起点、並びに適当な選択マーカー配列を含むベクター中にこの増幅した領域をクローン化する(例えば、Methods in Enzymology、1987、Volume 154、Academic Press;Sambrook et al., 1989、上記;及びAusubel et al., 上記を参照のこと)。次いで、増幅のためにこの環状産物で大腸菌を形質転換して、大量のベクターを得る。
【0047】
好ましくは、下記に詳細に論じるように、このベクターは、細菌性複製起点、哺乳動物複製起点、及び1つ又は複数の選択マーカーを含む。本明細書に記載の二重発現ベクター系が哺乳動物宿主と細菌宿主のどちらにも使用されるように設計されているので、様々な細菌株又は細胞系統を使用することができる。複製起点配列など特定のベクター配列の選択は、宿主の選択に依存し、対象とする特定のポリペプチド又は抗体の発現、分泌、及びスクリーニング又は選択に必要とするファクターなど様々なファクターにも依存する可能性がある。
【0048】
5.1.2.抗体の製造方法
抗原と免疫特異的に結合する抗体は、抗体を合成するための当技術分野で公知の任意の方法により、特に化学合成又は好ましくは組換え発現技術により製造することができる。
【0049】
モノクローナル抗体は、ハイブリドーマ技術、組換え技術、及びファージディスプレイ技術、又はそれらの組合せの利用を含む、当技術分野で公知の多種多様な技術を用いて調製することができる。例えば、モノクローナル抗体は、当技術分野で公知であって、例えば、Harlow et al., 「Antibodies: A Laboratory Manual」, (Cold Spring Harbor Laboratory Press, 第2版 1988);Hammerling et al., 「Monoclonal Antibodies and T-Cell Hybridomas」 563-681に収載(Elsevier, N. Y., 1981)(前記参考文献は、参照によりその全文が組み入れられる)に教示される方法をはじめとするハイブリドーマ技術を利用して製造することができる。本明細書に用いる用語「モノクローナル抗体」は、ハイブリドーマ技術によって製造される抗体に限定されるものではない。用語「モノクローナル抗体」は、それを製造する方法ではなく、任意の真核生物、原核生物、又はファージクローンを含む単一クローンから得られる抗体を意味する。
【0050】
ハイブリドーマ技術を用いて特異的抗体を製造しスクリーニングする方法は、当技術分野では日常的に利用されかつ公知である。簡単に説明すると、マウスを、非マウス抗原を用いて免疫感作することができ、そして免疫応答が一度検出されれば(例えば、その抗原に特異的な抗体がマウス血清中に検出されれば)、マウス脾臓を採取して脾細胞を単離する。次いでその脾細胞を公知の技術により任意の適当な骨髄腫細胞、例えばATCCから入手しうる培養細胞株SP20由来の細胞と、融合する。ハイブリドーマを選択し、限定希釈によりクローン化する。次いで、ハイブリドーマクローンを、当技術分野で公知の方法により、本発明のポリペプチドと結合できる抗体を分泌する細胞についてアッセイする。一般的に高レベルの抗体を含有する腹水液は、マウスを陽性ハイブリドーマクローンを用いて免疫感作することにより生成することができる。
【0051】
従って、本発明は、抗体を分泌するハイブリドーマ細胞を培養することを含むモノクローナル抗体を作製する方法、並びにその方法により製造される抗体を提供するものであり、好ましくは、上記方法におけるハイブリドーマは、非マウス抗原を用いて免疫感作したマウスから単離した脾細胞を骨髄腫細胞と融合し、次いで融合から得たハイブリドーマを、その抗原と結合しうる抗体を分泌するハイブリドーマクローンについてスクリーニングすることにより作製されるものである。
【0052】
特異的な特定のエピトープを認識する抗体フラグメントは、当業者に公知のいかなる技術により作製してもよい。例えば、Fab及びF(ab’)2フラグメントは、パパイン(Fabフラグメントを生成する)又はペプシン(F(ab’)2フラグメントを生成する)などの酵素を用いて、免疫グロブリン分子のタンパク質分解切断により生成することができる。F(ab’)2フラグメントは可変領域、軽鎖定常領域及び重鎖のCH1ドメインを含有する。さらに、本発明の抗体はまた、当技術分野で公知の様々なファージディスプレイ法を用いて作製することができる。
【0053】
ヒトにおける抗体のin vivo使用、及びin vitro検出アッセイを含む複数の用途について、ヒト又はキメラ抗体を使用することが好ましい場合がある。完全なヒト抗体は、ヒト被験者の治療のためには特に望ましい。ヒト抗体は、ヒト免疫グロブリン配列由来の抗体ライブラリーを用いる上記のファージディスプレイ法を含む、当技術分野で公知の様々な方法により作製することができる。米国特許第4,444,887号及び第4,716,111号、;並びにPCT公報WO98/46645号、WO98/50433号、WO98/24893号、W098/16654号、WO96/34096号、WO96/33735号、及びWO91/10741号も参照すること;これらはそれぞれ参照によりその全文が本明細書に組み入れられる。
【0054】
ヒト抗体はまた、機能性の内因性免疫グロブリンを発現できないがヒト免疫グロブリン遺伝子を発現できるトランスジェニックマウスを用いて製造することもできる。例えば、ヒト重鎖及び軽鎖免疫グロブリン遺伝子複合体を、無作為に又は相同組換えによりマウス胚幹細胞中に導入してもよい。あるいは、ヒト重鎖及び軽鎖遺伝子に加えて、ヒト可変領域、定常領域、及び多様性領域を、マウス胚性幹細胞中に導入してもよい。相同組換えによりヒト免疫グロブリン遺伝子座を導入して、マウス重鎖及び軽鎖免疫グロブリン遺伝子を別々に又は同時に非機能化してもよい。特に、JH領域のホモ接合性欠失は内因性抗体産生を妨げる。改変した胚幹細胞を増殖させ、胚盤胞中にマイクロインジェクションしてキメラマウスを作る。次いでキメラマウスを交配し、ヒト抗体を発現するホモ接合性子孫を作る。そのトランスジェニックマウスを、通常の方式で、選択した抗原、例えば目的のポリペプチドの全体若しくは部分を用いて、免疫感作する。その抗原に対するモノクローナル抗体を、免疫感作したトランスジェニックマウスから通常のハイブリドーマ技術を利用して得ることができる。トランスジェニックマウスによって保持されるヒト免疫グロブリントランスジーンはB細胞分化の際に再配列し、その後、クラススイッチ及び体細胞突然変異を受ける。従って、かかる技術を利用して、治療上有用なIgG、IgA、IgM及びIgE抗体を製造することが可能である。ヒト抗体を製造するためのこの技術の総括については、Lonberg and Huszar(1995, Int. Rev. Immunol. 13:65-93)を参照すること。ヒト抗体及びヒトモノクローナル抗体を製造するためのこの技術並びにかかる抗体を製造するためのプロトコルの詳細な考察については、例えば、参照によりその全文が本明細書に組み入れられる、PCT公報WO98/24893号、WO96/34096号、及びWO96/33735号;及び米国特許第5,413,923号、第5,625,126号、第5,633,425号、第5,569,825号、第5,661,016号、第5,545,806号、第5,814,318号、及び第5,939,598号を参照すること。さらに、Abgenix, Inc.(Freemont、CA)及びGenpharm(San Jose、CA)などの会社と、選択した抗原に対するヒト抗体を上記と類似の技術を利用して提供する契約を結ぶことができる。
【0055】
キメラ抗体は、抗体の異なる部分が異なる免疫グロブリンに由来する分子、例えばヒト抗体由来の可変領域及び非ヒト免疫グロブリンの定常領域を有する抗体である。キメラ抗体を製造する方法は当技術分野で公知である。例えば、Morrison, 1985, Science 229:1202;Oi et al., 1986, BioTechniques 4:214;Gillies et al., 1989, J. Immunol. Methods 125:191-202;並びに米国特許第5,807,715号、及び第4,816,567号、第4,816,397号(これらは本明細書に参照によりその全文が組み入れられる)を参照すること。ヒト種からの1以上のCDRと非ヒト免疫グロブリン分子からのフレームワーク領域を含むキメラ抗体は、当技術分野で公知の種々の技法を用いて作製しうる、例えば、CDRグラフト(欧州特許EP239,400;PCT公報WO91/09967号;及び米国特許第5,225,539号、第5,530,101号、第5,585,089号)、ベニアリング(veneering)又は再表面形成(resurfacing)(欧州特許EP592,106;EP519,596;Padlan, 1991, Molecular Immunology 28 (4/5):489-498;Studnicka et al., 1994, Protein Engineering 7 (6):805-814;及びRoguska et al., 1994, PNAS 91:969-973)、及びチェーンシャッフリング(chain shuffling)(米国特許第5,565,332号)が挙げられる。好ましい実施形態において、キメラ抗体は、表2に例示したCDR3のいずれか1つのアミノ酸配列を有するヒトCDR3と非ヒトフレームワーク領域を含む。しばしば、フレームワーク領域のフレームワーク残基を、CDRドナー抗体由来の対応する残基で置換して、抗原結合を改変し、好ましくは改良する。これらのフレームワーク置換は、当技術分野で公知の方法によって、例えば、CDRとフレームワーク残基の相互作用をモデル化して抗原結合に重要なフレームワーク残基を同定し、さらに配列比較により特定位置の異常なフレームワーク残基を同定することによって確認する(例えば、本明細書に参照によりその全文が組み入れられる、Queen et al., 米国特許第5,585,089号;及びRiechmann et al., 1988, Nature 332:323を参照)。
【0056】
さらに、目的の抗体は、続いて、当業者に周知の技術を用いて抗原を「模倣」する抗イディオタイプ抗体を作製するために用いることができる(例えば、Greenspan & Bona, 1989, FASEB J. 7(5):437-444;及びNissinoff, 1991, J. Immunol. 147(8):2429-2438参照)。例えば、目的の抗原と結合し、その宿主細胞受容体への結合を競合的に阻害する抗体(当技術分野で周知の及び上述のアッセイにより測定しうる)を用いて、目的の結合ドメインの抗原を「模倣」し、その結果として該抗原及び/又はその宿主細胞受容体と結合し、中和する抗イディオタイプを作製することができる。かかる中和抗イディオタイプ又はかかる抗イディオタイプのFabフラグメントは、抗原を中和するために治療方法において用いることができる。例えば、かかる抗イディオタイプ抗体は、目的の抗原と結合するため及び/又はその宿主細胞受容体と結合するために用いることができる。
【0057】
5.1.3.組換え発現及びタンパク質産生
目的のポリペプチドをコードするカセット配列(例えば適当に設計したイントロン配列を有する抗体分子をコードするカセット配列)を含む二重発現ベクターを構築した後、本発明の二重発現ベクターは、当技術分野で周知の技術を利用して組換えDNA技術により作製することができる。例えば、本明細書に参照によりその全文が組み入れられる、米国特許第6,331,415号を参照すること。
【0058】
例えば、好ましい実施形態においては、当業者に周知の方法を利用して、適当な転写及び翻訳制御シグナルを含有する二重発現ベクターカセットを構築することができる。これらの方法としては、例えば、in vitro組換えDNA技術、合成技術、及びin vivo遺伝子組換えが挙げられる。本発明は、従って、抗体分子、抗体の重鎖若しくは軽鎖、抗体の重鎖若しくは軽鎖可変ドメイン又はそれらの部分、あるいは重鎖若しくは軽鎖のCDRをコードし、プロモーターと機能的に連結されたヌクレオチド配列を含む発現ベクターを提供する。かかるベクターは、抗体分子の定常領域をコードするヌクレオチド配列(例えば、PCT公報WO86/05807号;PCT公報WO89/01036号;及び米国特許第5,122,464号を参照)を含んでもよく、また抗体の可変ドメインをかかるベクター中にクローニングして重鎖全体、軽鎖全体、又は重鎖全体と軽鎖全体の両方を発現させてもよい。
【0059】
二重発現ベクターを通常の技術により宿主細胞に導入し、次いでトランスフェクトした細胞を通常の技術により培養して、目的の抗体を製造する。従って、本発明は、目的の抗体又はそのフラグメント、あるいはその重鎖若しくは軽鎖又はそれらの一部分、あるいは本発明の一本鎖抗体をコードし、異種プロモーターと機能的に連結されたポリヌクレオチドを含有する宿主細胞を含む。二本鎖抗体を発現させるための好ましい実施形態においては、以下に詳しく説明するように、重鎖及び軽鎖の両方をコードするベクターを宿主細胞において共発現させ、免疫グロブリン分子全体を発現させることができる。
【0060】
様々な宿主系を利用して、本発明の二重発現ベクターを発現させることができる(例えば、米国特許第5,807,715号を参照)。かかる宿主系は、適当なヌクレオチドコード配列を用いて形質転換又はトランスフェクトすると、目的の抗体分子をin situで発現することができる細胞も意味する。これらとしては、限定されるものでないが、細菌などの微生物(例えば、大腸菌(E.coli)及び枯草菌(B.subtilis));二重発現ベクターを用いて形質転換した酵母(例えば、サッカロミセス・ピキア(Saccharomyces Pichia));組換えウイルス発現ベクター(例えば、バキュロウイルス)に感染した昆虫細胞系;二重発現カセットで形質転換した組換えウイルス発現ベクター(例えば、カリフラワーモザイクウイルス、CaMV;タバコモザイクウイルス、TMV)に感染した植物細胞系;又は、哺乳動物細胞のゲノム由来のプロモーター(例えば、メタロチオネインプロモーター)若しくは哺乳動物ウイルス由来のプロモーター(例えば、アデノウイルス後期プロモーター;ワクシニアウイルス7.5Kプロモーター)を含有する組換え発現構築物を保持する哺乳動物細胞系(例えば、COS、HEK、CHO、BHK、293、NSO、及び3T3細胞)が挙げられる。
【0061】
細菌宿主での発現のために、二重発現ベクターは、プラスミドベクターの複製及び増幅のために必要とされる複製起点を含む。大腸菌におけるクローニング及び増幅のために、大腸菌の複製起点、例えば当技術分野で周知のものを用いる(Miller, 1992, A Short Course in Bacterial Genetics, Cold Spring Harbor Laboratory Press, NY、及びそれに含まれる参考文献を参照)。容易に入手可能なプラスミドの複製起点の例としては、限定されるものではないが、ColE1由来の複製起点(Bolivar et al., 1977, Gene 2:95-113;Sambrook et al., 1989、前掲を参照)、pACYC184などのプラスミド上に存在するp15A起点(Chang and Cohen, 1978, J. Bacteriol. 134:1141-56;Miller, 1992, p.10.4-10.11も参照)、及び低コピープラスミド発現のために利用可能なpSC101起点が当技術分野で周知である。
【0062】
例えば、一実施形態において、高コピープラスミド、例えば、ColE1由来複製起点を含有するプラスミドなどに由来する複製起点を用いることができ、この例は当技術分野で周知である(Sambrook et al., 1989、前掲参照;Miller, 1992, A Short Course in Bacterial Genetics, Cold Spring Harbor Laboratory Press, NY、及びそれに含まれる参考文献も参照)。一例としては、pUC19由来の起点及びその誘導体がある(Yanisch-Perron et al., 1985, Gene 33:103-119)。pUCベクターは、細胞当たり300〜500コピーのレベルで存在し、外来遺伝子の挿入のための都合のよいクローニング部位を有している。非常に高い発現のため、λベクター、例えばλgt11(Huynh et al., 1984, "DNA Cloning Techniques: VolI: A Practical Approach", D. Glover編, pp.49-78, IRL Press, Oxford)、又はT7及びSp6ポリメラーゼ発現系を含有する細胞におけるT7若しくはSP6ファージプロモーター(Studier et al., 1990, Methods Enzymol. 185:60-89)を使用することも可能である。
【0063】
低レベルの発現が望まれる場合には、中程度又は低コピーに由来する複製起点を使用しうる。中程度コピープラスミドは当技術分野で周知であり、例えばColE1由来の複製起点を有し、細胞当たり20〜100コピーであるpBR322(Bolivar et al., 1977, Gene 2:95-113;Sambrook et al., 1989、前掲を参照)、又はpACYC100系のプラスミドの1つであり、p15A複製起点を有し、細胞当たり10〜12コピーで存在するpACYC184(Chang and Cohen, 1978, J. Bacteriol. 134:1141-56;またMiller, 1992, p.10.4-10.11参照)がある。低コピープラスミドもまた当技術分野で周知であり、例えば、pSC101起点を有し、細胞当たり約5コピーで存在するpSC101がある。pACYC及びpSC101プラスミドベクターは両方とも都合の良いクローニング部位を有し、同じ細胞においてpBR及びpUCプラスミドとして共存しうる。これは、これらが適合可能な複製起点を有し、特有の選択的抗生物質マーカーを有するためである。他の好適なプラスミド複製起点としては、λ又はファージP1レプリコンに基づくプラスミド、例えばLorist系が挙げられる(Gibson et al., 1987, Gene 53:283-286)。
【0064】
さらに低い発現が望まれる場合には、複製起点は、細菌染色体から得てもよい(Miller, 1992、前掲;Niedhardt, F.C.編、1987, Escherichia coli and Salmonella typhimurium, American Society for Microbiology, Washington, D.C.;Yarmolinsky, M.B. & Sternberg, N., 1988, pp.291-438, The Bacteriophages, Vol.1、R. Calendar編、Plenum Press, New York)。さらに、合成の複製起点、細菌プロモーター、又は細菌シグナル配列を用いることも可能である。
【0065】
哺乳動物宿主細胞において、二重発現ベクター配列は、エピソームとして哺乳動物宿主細胞中に存在するように設計してもよいし、あるいは例えばベクターを線状化して設計することなどにより、宿主のゲノムDNAへの組込みを促進し、安定な細胞系が作製されるように設計を行ってもよい。かかるベクターは当技術分野で公知である。
【0066】
例えば、哺乳動物宿主細胞においては、様々なウイルスに基づく発現系を使用することができる。アデノウイルスを発現ベクターとして利用する場合、二重発現ベクターのカセット配列を、アデノウイルス転写/翻訳制御複合体、例えば、後期プロモーター及びトリパータイト(tripartite)リーダー配列と連結しうる。次いでこのキメラ遺伝子を、in vitro又はin vivo組換えによりアデノウイルスゲノムに挿入することができる。ウイルスゲノムの非必須領域(例えば、領域E1又はE3)への挿入により、感染宿主において生存可能でかつIgG遺伝子産物を発現しうる組換えウイルスを得ることができる(例えば、Logan and Shenk, 1984, Proc. Natl. Acad. Sci. USA 8 1:3655-3659を参照)。特定の開始シグナルも、挿入された発現ベクターカセット配列の効率的な翻訳に必要でありうる。これらのシグナルとしてはATG開始コドン及び隣接配列が挙げられる。IgG重鎖又は軽鎖を、それ自体の開始コドン及び隣接配列を含めて、適当な発現ベクターに挿入する場合には、さらに翻訳制御シグナルは必要ない。しかしながら、IgGコード配列の一部のみを挿入する場合には、おそらくATG開始コドンを含む外因性の翻訳制御シグナルを提供する必要がある。さらに、全インサートの翻訳を保証するため開始コドンは所望のコード配列のリーディングフレームと一致している必要がある。これらの外因性翻訳制御シグナル及び開始コドンは、天然及び合成の両方の様々な起源のものであってよい。発現効率は、適当な転写エンハンサーエレメント、転写ターミネーターなど(例えば、Bittner et al., 1987, Methods in Enzymol. 153:51-544を参照)を組み入れることにより増強することができる。
【0067】
さらに、挿入された配列の発現をモジュレートするか、又は所望の特定の様式で遺伝子産物を修飾及びプロセシングする宿主細胞株を選択することができる。タンパク質産物のかかる修飾(例えば、グリコシル化)及びプロセシング(例えば、切断)は、タンパク質の機能にとって重要でありうる。異なる宿主細胞は、タンパク質及び遺伝子産物の翻訳後プロセシング及び修飾のための特有かつ特定の機構を有する。適当な細胞系又は宿主系を選んで、発現された外来タンパク質の正しい修飾とプロセシングを保証することができる。この目的のために、適切な一次転写産物のプロセシング、遺伝子産物のグリコシル化及びリン酸化のための細胞機構を持つ宿主真核細胞を使用することができる。かかる哺乳動物宿主細胞としては、限定されるものでないが、CHO、VERY、BHK、Hela、COS、MDCK、293、3T3、W138、BT483、Hs578T、HTB2、BT20及びT47D、NSO(内因的に全く免疫グロブリン鎖を産生しないマウス骨髄腫培養細胞株)が挙げられる。
【0068】
組換えタンパク質の長期間にわたる高収量の生産のためには、安定した発現が好ましい。例えば、安定して抗体分子を発現する培養細胞株を遺伝子操作によって作ることができる。ウイルスの複製起点を含有する発現ベクターを用いるのではなしに、宿主細胞を、適当な発現制御エレメント(例えば、プロモーター、エンハンサー、配列、転写ターミネーター、ポリアデニル化部位など)により制御したDNA、及び選択マーカーを用いて形質転換することができる。外来DNAの導入後、遺伝子操作した細胞を、1〜2日間富栄養培地で増殖させ、次いで選択培地に切り換えてもよい。組換えプラスミド中の選択マーカーは、選択に対する耐性を付与し、細胞がプラスミドをその染色体中に安定的に組み込み、増殖して細胞巣を形成することを可能にするので、これを次にクローニングして培養細胞株に拡大することができる。この方法を利用して抗体分子を発現する細胞系を遺伝子操作により有利に作製することができる。かかる遺伝子操作で作製した培養細胞株は、抗体分子と直接又は間接に相互作用する組成物をスクリーニングしたり評価したりするのに特に有用である。
【0069】
様々な選択系を利用することができ、限定されるものでないが、例えば単純ヘルペスウイルスチミジンキナーゼ(Wigler et al., 1977, Cell 11:223)、ヒポキサンチン−グアニン・ホスホリボシルトランスフェラーゼ(Szybalska & Szybalski, 1992, Proc. Natl. Acad. Sci. USA 48:202)、及びアデニン・ホスホリボシルトランスフェラーゼ(Lowy et al., 1980, Cell 22:8-17)遺伝子が挙げられ、これらはそれぞれtk−、hgprt−又はaprt−細胞において使用することができる。また、以下の遺伝子については代謝拮抗物質耐性を選択の基礎として利用することができる:メトトレキセート耐性を付与するdhfr(Wigler et al., 1980, Natl. Acad. Sci. USA 77:357;O'Hare et al., 1981, Proc. Natl. Acad. Sci. USA 78:1527);ミコフェノール酸耐性を付与するgpt(Mulligan & Berg, 1981, Proc. Natl. Acad. Sci. USA 78:2072);アミノグリコシドG−418耐性を付与するneo(Wu and Wu, 1991, Biotherapy 3:87-95;Tolstoshev, 1993, Ann. Rev. Pharmacol. Toxicol. 32:573-596;Mulligan, 1993, Science 260:926-932;並びにMorgan and Anderson, 1993, Ann. Rev. Biochem. 62:191-217;May, 1993, TIB TECH 11 (5):155-215);並びにハイグロマイシン耐性を付与するhygro(Santerre et al., 1984, Gene 30:147)。当技術分野で公知の組換えDNA技法を慣用的に適用して所望の組換えクローンを選択することができ、かかる方法は、例えば、Ausubel et al.(編), 「Current Protocols in Molecular Biology」, John Wiley & Sons, NY (1993);Kriegler, 「Gene Transfer and Expression, A Laboratory Manual」, Stockton Press, NY (1990);及びDracopoli et al.(編), 「Current Protocols in Human Genetics」, John Wiley & Sons, NY (1994)の第12章及び第13章;Colberre-Garapin et al., 1981, J. Mol. Biol. 150:1(これらは参照により本明細書にその全文が組み入れられる)に記載されている。
【0070】
細菌における選択のために、好ましくは抗生物質耐性マーカー、例えば、Tn903由来のカナマイシン耐性遺伝子(Friedrich and Soriano, 1991, Genes Dev. 5:1513-1523)、又は他のアミノグリコシド(限定されるものではないが、ジヒドロストレプトマイシン、ゲンタマイシン、ネオマイシン、パロマイシン及びストレプトマイシンなど)に対する耐性を付与する遺伝子、ペニシリン(限定されるものではないが、アンピシリン、カルベニシリン、メチシリン、ペニシリンN、ペニシリンO、及びペニシリンV)に対する耐性を付与するTn9由来のTEM−1 β−ラクタマーゼ遺伝子を用いる。他の選択可能な遺伝子配列としては、限定されるものではないが、ゼオシン耐性を付与するポリペプチドをコードする遺伝子配列が含まれる(Hegedus et al., 1998, Gene 207:241-249)。使用しうる他の抗生物質は、アムフェニコール類、例えばクロラムフェニコールなどに対する耐性を付与する遺伝子、例えば、クロラムフェニコールトランスアセチラーゼ(CAT)のコード配列を使用することができる(Eikmanns et al., 1991, Gene 102:93-98)。当業者により理解されているように、プラスミドの維持について選択するための他の非抗生物質方法を用いることも可能であり、例えば、種々の栄養要求性マーカーなどである(Sambrook et al., 1989、前掲;Ausubel et al.、前掲)。
【0071】
抗体分子の発現レベルは、ベクター増幅により増加させることができる(総説については、Bebbington and Hentschel, 「The use of vectors based on gene amplification for the expression of cloned genes in mammalian cells in DNA cloning」, Vol.3. (Academic Press, New York, 1987)を参照)。抗体を発現するベクター系中のマーカーが増幅可能であれば、宿主細胞培養中に存在するインヒビターレベルの増加は、マーカー遺伝子のコピー数を増加させうる。増幅される領域は、抗体遺伝子と関連しているので、抗体の産生も増加しうる(Crouse et al., 1983, Mol. Cell. Biol. 3:257)。
【0072】
宿主細胞を、本発明の2つの二重発現ベクター、すなわち、重鎖由来のポリペプチドをコードする第1ベクターと軽鎖由来のポリペプチドをコードする第2ベクターとを用いて共トランスフェクトすることができる。その2つのベクターは、哺乳動物細胞においては重鎖ポリペプチドと軽鎖ポリペプチドの、また細菌細胞においてはFab又はscFvポリペプチドの同等の発現を可能にする同一の選択マーカーを含有してもよい。あるいは、重鎖ポリペプチドと軽鎖ポリペプチドの両方をコードしかつ発現できる単一ベクターを利用してもよい。かかる状況では重鎖の前に軽鎖を配置して、毒性である遊離の重鎖が過剰になることを避けなければならない(Proudfoot, 1986, Nature 322:52;及びKohler, 1980, Proc. Natl. Acad. Sci. USA 77:2 197)。重鎖と軽鎖のコード配列はcDNA又はゲノムDNAを含んでいてもよい。
【0073】
ポリペプチドは、標準組換えDNA技術により、製造することができる。例えば、遺伝子断片のPCR増幅を、2つの連続した遺伝子断片の間を相補的に重複するようなアンカープライマーを用いて実施し、次いでそれらをアニーリングして再増幅することによりキメラ遺伝子配列を作製することができる(例えば、「Current Protocols in Molecular Biology」, Ausubel et al.編, John Wiley & Sons, 1992を参照)。さらに、生物活性分子をコードする核酸を、Fcドメイン又はそのフラグメントを含有する発現ベクター中にクローニングして、その生物活性分子がFcドメイン又はFcドメインのフラグメントとイン・フレームとなるように連結することができる。
【0074】
ポリペプチドを抗体の定常領域に融合又は複合体化する方法は、当技術分野では公知である。例えば、米国特許第5,336,603号、第5,622,929号、第5,359,046号、第5,349,053号、第5,447,851号、第5,723,125号、第5,783,181号、第5,908,626号、第5,844,095号、及び第5,112,946号;欧州特許EP307,434;EP367,166;EP394,827;PCT公報WO91/06570号、WO96/04388号、WO96/22024号、WO97/34631号、及びWO99/04813号;Ashkenazi et al., 1991, Proc. Natl. Acad. Sci. USA 88:10535-10539;Traunecker et al., 1988, Nature, 331:84-86;Zheng et al., 1995, J. Immunol. 154:5590-5600;及びVil et al., 1992, Proc. Natl. Acad. Sci. USA 89:11337-11341(これらは本明細書に参照によりその全文が組み入れられる)を参照すること。
【0075】
生物活性分子及びFcドメイン又はそのフラグメントをコードするヌクレオチド配列は、当業者が利用しうる任意の情報から(すなわち、Genbank、文献から、又は慣用的なクローニングにより)得ることができる。ポリペプチド又は融合タンパク質をコードするヌクレオチド配列を、二重発現ベクター中に挿入してもよい。
【0076】
真核細胞におけるポリペプチドの発現は、当技術分野で公知の任意のプロモーター若しくはエンハンサーエレメントにより制御することができる。融合タンパク質をコードする遺伝子の発現を制御するのに利用しうるプロモーターとしては、限定されるものでないが、SV40初期プロモーター領域(Bernoist and Chambon, 1981, Nature 290:304-310)、ラウス肉腫ウイルスの3’長末端反復配列に含有されるプロモーター(Yamamoto et al., 1980, Cell 22:787-797)、ヘルペスチミジンキナーゼプロモーター(Wagner et al., 1981, Proc. Natl. Acad. Sci. U. S. A. 78:1441-1445)、メタロチオネイン遺伝子の調節配列(Brinster et al., 1982, Nature 296:39-42)、テトラサイクリン(Tet)プロモーター(Gossen et al., 1995, Proc. Nat. Acad. Sci. USA 89:5547-5551);細菌においては、原核生物プロモーター、例えばβ−ラクタマーゼ・プロモーター(Villa-Kamaroff et al., 1978, Proc. Natl. Acad. Sci. U. S. A. 75:3727-3731)若しくはtacプロモーター(DeBoer et al., 1983, Proc. Natl. Acad. Sci. U. S. A. 80:21-25などの原核生物発現ベクター;また「Useful proteins from recombinant bacteria」, Scientific American, 1980,242: 74-94も参照)も参照;植物細胞においては、ノパリンシンセターゼプロモーター領域(Herrera-Estrella et al., Nature 303:209-213)又はカリフラワーモザイクウイルス35SRNAプロモーター(Gardner et al., 1981, Nucl. Acids Res. 9:2871)を含む植物発現ベクター、及び光合成酵素リブロース二リン酸カルボキシラーゼのプロモーター(Herrera-Estrella et al., 1984, Nature 310:115-120);Gal4プロモーターなどの酵母又は他の真菌由来のプロモーターエレメント、ADC(アルコールデヒドロゲナーゼ)プロモーター、PGK(ホスホグリセロールキナーゼ)プロモーター、及びアルカリホスファターゼプロモーターが挙げられる。
【0077】
特定の実施形態においては、ポリペプチドの発現は構成的プロモーター(CMVプロモーターなど)により調節される。他の実施形態においては、ポリペプチドの発現は誘導性プロモーターにより調節される。
【0078】
ポリペプチドをコードする遺伝子のインサートを含有する発現ベクターは、3つの一般的手法:(a)核酸ハイブリダイゼーション、(b)「マーカー」遺伝子機能の存在若しくは非存在、及び(c)挿入した配列の発現により、同定することができる。第1の手法においては、発現ベクター中のポリペプチドをコードする遺伝子の存在を、ポリペプチドをコードする挿入した遺伝子と相同的な配列を含むプローブを用いて核酸ハイブリダイゼーションを行うことにより検出することができる。第2の手法においては、組換えベクター/宿主系は、ポリペプチドをコードするヌクレオチド配列のベクター中への挿入により生じる、ある特定の「マーカー」遺伝子機能(例えば、チミジンキナーゼ活性、抗生物質耐性、形質転換表現型、バキュロウイルス中の封入体(occlusion body)形成など)の存在又は非存在に基づいて同定しかつ選択することができる。例えば、融合タンパク質をコードするヌクレオチド配列がベクターのマーカー遺伝子配列内に挿入される場合には、融合タンパク質インサートをコードする遺伝子を含有する組換え体は、マーカー遺伝子機能の不在により同定することができる。第3の手法においては、組換え発現ベクターは、組換え体が発現する遺伝子産物(例えば、融合タンパク質)をアッセイすることにより同定することができる。かかるアッセイは、例えば、in vitroアッセイ系における融合タンパク質の物理的又は機能的特性、例えば、抗生物活性分子抗体との結合に基づくものでもよい。
【0079】
DNA断片のベクターへの挿入について既に記載されている方法の任意のものを用いて、適当な転写/翻訳制御シグナル及びタンパク質コード配列を含有するキメラ遺伝子を含む発現ベクターを構築しうる。これらの方法としては、in vitro組換えDNA及び合成技法、並びにin vivo組換え体(遺伝的組換え)が含まれる。哺乳動物細胞におけるIgG重鎖又は軽鎖の発現は、IgG重鎖又は軽鎖が組換えDNA分子で形質転換された宿主において発現されるように、第2の核酸配列により調節することができる。例えば、IgG重鎖又は軽鎖の発現は、当技術分野で公知の任意のプロモーター/エンハンサーエレメントにより制御されうる。
【0080】
好ましくは、Fabフラグメント又は全長IgGの細菌発現は誘導性プロモーターにより制御される。広範囲の発現をもたらす誘導性発現は、種々の誘導性調節配列を用いることにより達成しうる。一実施形態において、例えば、lacI遺伝子及びその無償性の誘導剤IPTGを用いて、ポリペプチドをコードする配列がlacOP調節配列を介して転写される場合には、大腸菌におけるFabフラグメントの誘導性の高レベル発現を得ることができる。また、使用可能である多様な他の誘導性プロモーター系が当業者に周知である。IgG二重発現ベクター系からの発現レベルもまた種々の強度のプロモーターで変更することができる。
【0081】
使用可能である他の調節される発現系としては、限定されるものではないが、アラビノース(AraC)により誘導されうるaraCプロモーター(例えば、Schleif, 2000, Trends Genet. 16:559-565参照)、TET系(Geissendorfer and Hillen, 1990, Appl. Microbiol. Biotechnol. 33:657-663)、ファージλ温度のpLプロモーター及び誘導性λリプレッサーCI857(Pirrotta, 1975, Nature 254:114-117;Petrenko et al., 1989, Gene 78:85-91)、trpプロモーター及びtrpリプレッサー系(Bennett et al., 1976, Proc. Natl. Acad. Sci USA 73:2351-55;Wame et al., 1986, Gene 46:103-112)、lacUV5プロモーター(Gilbert and Maxam, 1973, Proc. Natl. Acad. Sci. USA 70:1559-63)、lpp(Nokamura et al., 1982, J. Mol. Appl. Gen. 1:289-299)、T7遺伝子10プロモーター、phoA(アルカリホスファターゼ)、recA(Horii et al., 1980, Proc. Natl. Acad. Sci. USA 77:313-7)、及びIPTGにより誘導されるtacプロモーター、trp−lac融合プロモーター(Amann et al., 1983, Gene 25:167-78)は全て一般的に使用される強力なプロモーターであり、その結果、各プロモーターによりそのレベルが制御されるタンパク質について約1〜10%の総細胞タンパク質のレベルが蓄積する。より強力なプロモーターが望まれる場合には、tacプロモーターはlacUV5よりも約10倍強力であるが、基底レベルの発現が高く、過剰発現が必要とされる場合にのみ使用すべきである。より弱いプロモーターが必要な場合には、例えば、マルトース、ガラクトース、又は他の所望のプロモーターなど(かかるプロモーターの配列はGenBankから入手可能である)の、他の細菌プロモーターが当技術分野で周知である(Burks et al., 1991, Nucl. Acids Res. 19:2227-2230)。
【0082】
全長IgG重鎖又は軽鎖の真核生物における発現については、ベクターは、RNAポリメラーゼの認識、結合及び転写開始に十分な特異的配列を含む、真核生物特異的複製起点及びプロモーター領域を含みうる。さらに、プロモーター領域は、RNAポリメラーゼの認識、結合及び転写開始活性をモジュレートする配列を含んでもよい。かかる配列としては、cic作用性因子であるか、又はtrans作用性因子に応答するものでありうる。調節の性質に応じて、プロモーターは構成的であるか又は調節型でありうる。TnpI発現を制御するために用い得るプロモーターとしては、限定されるものではないが、SV40初期プロモーター領域(Benoist and Chambon, 1981, Nature 290:304-310)、ラウス肉腫ウイルスの3’長末端リピートに含まれるプロモーター(Yamamoto et al., 1980, Cell 22:787-797)、ヘルペスチミジンキナーゼプロモーター(Wagner et al., 1981, Proc. Natl. Acad. Sci. U.S.A. 78:1441-1445)、メタロチオネイン遺伝子の調節配列(Brinster et al., 1982, Nature 296:39-42)、ノパリンシンテターゼプロモーター領域を含む植物発現ベクター(Herrera-Estrella et al., 1984, Nature 303:209-213)、又はカリフラワーモザイクウイルス35S RNAプロモーター(Gardner et al., 1981, Nucl. Acids Res. 9:2871)、及び光合成酵素リブロース二リン酸カルボキシラーゼのプロモーター(Herrera-Estrella et al., 1984, Nature 310:115-120)、酵母又は他の真菌由来のプロモーターエレメント、例えばGal4プロモーター、又はADC(アルコールデヒドロゲナーゼ)プロモーターが挙げられる。
【0083】
プロモーター、及びポリヌクレオチドを機能的に連結しうるクローニング部位を含有するベクターは当技術分野で周知である。かかるベクターは、in vitroで又はin vivoでRNAを転写可能であり、Stratagene(La Jolla, Calif.)及びPromega Biotech(Madison, Wis.)などの供与元から市販されている。発現及び/又はin vitro転写を最適化するために、クローン化DNAの5’及び/又は3’非翻訳部分を除去し、付加し又は改変して、さらなる潜在的な不適当な別の翻訳開始コドン、又は転写若しくは翻訳レベルのいずれかで発現を妨害若しくは低減しうるほかの配列を排除する必要がありうる。あるいは、コンセンサスリボソーム結合部位を開始コドンのすぐ5’側に挿入して発現を増強することができる(例えばKozak, 1991, J. Biol. Chem. 266:19867参照)。同様に、同じアミノ酸をコードする別のコドンをコード配列と置換して、翻訳を増強してもよい(例えば、宿主細胞のコドン最適性を採用して、G−Cリッチドメインの存在を低減する、等)。
【0084】
ベクターはまた、タンパク質発現、操作又は挿入された標的DNAの維持のための目的のヌクレオチド配列を含み得る。例えば、プロモーター配列、エンハンサー配列、翻訳配列(例えばシャイン及びダルガーノ配列)、転写因子認識部位、Kozakコンセンサス配列、及び終結シグナルを、ベクターの適当な位置に含有させてもよい。
【0085】
ベクターはまた、天然又は合成由来のシグナル配列を含む必要がある。ポリペプチド(例えば抗体、IgGなど)を細胞膜内に標的化しうるシグナル配列を用いることができる。天然に周辺質に存在することが定められたタンパク質と結合しているリーダー配列は、例えば、外来タンパク質の周辺質への分泌を誘導することが知られている(MacIntyre et al., 1990、Mol. Gen. Genet. 221:466-474)。好ましい実施形態では、シグナル配列は、OmpAタンパク質リーダー配列(Hobom et al., 1995、Dev. Biol. Stand. 84:255-262)をコードする。他のシグナル配列もまた可能であり、限定されるものではないが、大腸菌PhoA(Oka et al., 1985、Proc. Natl. Acad. Sci 82:7212-16)、OmpT(Johnson et al., 1996、Protein Expression 7:104-113)、LamB及びOmpF(Hoffman and Wright、1985、Proc. Natl. Acad. Sci. USA 82:5107-5111)由来のリーダー、β−ラクタマーゼ(Kadonaga et al., 1984、J. Biol. Chem. 259:2149-54)、エンテロトキシン(Morioka-Fujimoto et al., 1991、J. Biol. Chem. 266:1728-32)、黄色ブドウ球菌(Staphylococcus aureus)のプロテインA(Abrahmsen et al., 1986、Nucleic Acids Res. 14:7487-7500)、枯草菌(B. subtilis)のエンドグルカナーゼ(Lo et al., Appl. Environ. Microbiol. 54:2287-2292)由来のリーダー配列、並びに人工及び合成シグナル配列(MacIntyre et al., 1990、Mol. Gen. Genet. 221:466-74;Kaiser et al., 1987、Science、235:312-317)が挙げられる。
【0086】
二重発現ベクターカセット配列を含むDNA調製物を宿主細胞に送達するための当技術分野で公知の方法の任意のものが上記方法と共に使用するのに適している。かかる方法は当技術分野で公知であり、限定されるものではないが、細胞のエレクトロポレーション、塩化カルシウム又はルビジウムを用いたコンピテント細胞の調製、及びウイルス粒子にパッケージングした標的DNAを用いたDNAの形質導入が挙げられる。真核細胞については、限定されるものではないが、エレクトロポレーション、DNAのリン酸カルシウム沈殿によるトランスフェクション、及びウイルスパッケージングなどの方法がある。好ましい実施形態においては、エレクトロポレーションを使用する。細胞は、標準的な方法によりエレクトロポレーションにコンピテントとなるように処理する(Ausubel et al., Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley Interscience, New York参照)。好ましくは、エレクトロコンピテント細胞の標準調製物約50μlを標準的な手順によりエレクトロポレーションに用いる。線状又は環状ベクターの形質転換を必要とする実験においては、好ましくは0.3g以上のベクターを使用する。IgG DNAを含有するDNA調製物の形質転換を必要とする実験においては、好ましくは0.3μg以上のベクターを使用する。共形質転換実験においては、DNAはエレクトロポレーションの前に混合することが好ましい。エレクトロポレーション後、細胞を培養培地に希釈し、約1時間半の回復期間にわたりインキュベートし、その後選択マーカー遺伝子により付与される表現型変化を同定する条件下で培養することが好ましい。
【0087】
好適には、表現型の変化は抗生物質に対する耐性であり、細胞は対応する抗生物質を含有するプレート上で培養する。この場合、抗生物質耐性コロニーは終夜培養後に出現するが、これは大部分が所望のサブクローニング産物を含有する。選択マーカーに関して、好ましくは抗生物質耐性マーカー、例えば、Tn903由来のカナマイシン耐性遺伝子(Friedrich and Soriano, 1991, Genes Dev. 5:1513-1523)、又は他のアミノグリコシド(限定されるものではないが、ジヒドロストレプトマイシン、ゲンタマイシン、ネオマイシン、パロマイシン及びストレプトマイシンなど)に対する耐性を付与する遺伝子、ペニシリン(限定されるものではないが、アンピシリン、カルベニシリン、メチシリン、ペニシリンN、ペニシリンO、及びペニシリンV)に対する耐性を付与するTn9由来のTEM−1 β−ラクタマーゼ遺伝子を用いる。他の選択可能な遺伝子配列としては、限定されるものではないが、ゼオシン耐性を付与するポリペプチドをコードする遺伝子配列が含まれる(Hegedus et al., 1998, Gene 207:241-249)。使用しうる他の抗生物質は、アムフェニコール類、例えばクロラムフェニコールなどに対する耐性を付与する遺伝子、例えば、クロラムフェニコールトランスアセチラーゼ(CAT)のコード配列を使用することができる(Eikmanns et al., 1991, Gene 102:93-98)。当業者により理解されているように、プラスミドの維持について選択するための他の非抗生物質方法を用いることも可能であり、例えば、種々の栄養要求性マーカーなどである(Sambrook et al., 1989、前掲;Ausubel et al.、前掲を参照)。
【0088】
別の実施形態において、DNAは、ファージ粒子にパッケージングされたDNAの形質導入により宿主細胞に送達する。P1又はλ形質導入及びパッケージングプロトコールは当技術分野で公知である。λパッケージング抽出物は市販されている(例えば、Promega, Madison, WI製)。
【0089】
目的の抗体分子を組換え発現により産生させた後、免疫グロブリン分子を精製するための当技術分野で公知の任意の方法、例えば、クロマトグラフィー(例えば、イオン交換、アフィニティー、特に、プロテインAの後の特異的抗原に対するアフィニティ、及びサイズ分離カラムクロマトグラフィ)により、遠心分離、溶解度差により、又はタンパク質を精製するための他の任意の標準技術により、それらを精製することができる。さらに、本発明の抗体又はそのフラグメントは、精製を容易にするために、本明細書に記載の又は当技術分野で公知の異種ポリペプチド配列と融合してもよい。
【0090】
5.2.抗体の選択及びスクリーニング
5.2.1.抗体の選択及び特性決定
本発明の全長IgG及びFabフラグメントを様々な方法で特徴決定することができる。特に、全長IgG及びFabフラグメントを目的の抗原と免疫特異的に結合する能力についてアッセイすることができる。かかるアッセイは、溶液中で(例えば、Houghten, 1992, Bio/Techniques 13:412-421)、マイクロタイターディッシュなどの固相支持体上で、ビーズ上で(Lam, 1991, Nature 354:82-84)、チップ上で(Fodor, 1993, Nature 364:555-556)、細菌上で(米国特許第5,223,409号)、胞子上で(米国特許第5,571,698号、第5,403,484号及び第5,223,409号)、プラスミド上で(Cull et al., 1992, Proc. Natl. Acad. Sci. USA 89:1865-1869)又はファージ上で(Scott and Smith, 1990, Science 249:386-390;Devlin, 1990, Science 249: 404-406;Cwirla et al., 1990, Proc. Natl. Acad. Sci. USA 87:6378-6382;並びにFelici, 1991, J. Mol. Biol. 222:301-310)(これらの参考文献はそれぞれ参照により本明細書にその全文が組み入れられる)実施することができる。目的の抗原又はその断片と免疫特異的に結合すると同定されている抗体又はそのフラグメントを、次いで目的の抗原に対する特異性とアフィニティについて試験することができる。
【0091】
目的の抗体又はそのフラグメントを、当技術分野で公知のいずれかの方法により、目的の抗原に対する免疫特異的結合及び他の抗原との交差反応性についてアッセイすることができる。免疫特異的結合と交差反応性を分析するために利用できるイムノアッセイとしては、限定されるものでないが、いくつかを列挙すれば、ウェスタンブロット、ラジオイムノアッセイ、ELISA(酵素結合免疫吸着アッセイ)、「サンドイッチ」イムノアッセイ、免疫沈降アッセイ、沈降反応、ゲル拡散沈降反応、免疫拡散アッセイ、凝集アッセイ、補体結合試験、免疫放射線測定アッセイ、蛍光イムノアッセイ、プロテインAイムノアッセイを利用する競合及び非競合アッセイ系が挙げられる。かかるアッセイは日常的に行われかつ当技術分野では公知である(例えば、Ausubel et al.編, 1994, 「Current Protocols in Molecular Biology」, Vol.1, John Wiley & Sons, Inc., New York、これは参照により本明細書にその全文が組み入れられる)。例示のイムノアッセイを簡単に以下に(限定を意図するのではないが)記載する。
【0092】
免疫沈降プロトコルは一般的に、細胞の集団を、タンパク質ホスファターゼ及び/又はプロテアーゼ阻害剤(例えば、EDTA、PMSF、アプロチニン、バナジン酸ナトリウム)を添加したRIPAバッファー(1%NP−40又はTritonX−100、1%デオキシコール酸ナトリウム、0.1%SDS、0.15M NaCl、0.01Mリン酸ナトリウム、pH7.2、1%トラジロール)などの溶解バッファーに溶解し、目的の抗体を該細胞溶解物に加え、ある時間(例えば、1〜4時間)40℃にてインキュベートし、プロテインA及び/又はプロテインGセファロースビーズを細胞溶解液に加え、約1時間以上40℃にてインキュベートし、ビーズを溶解バッファー中で洗浄し、そしてSDS/サンプルバッファーに再懸濁することを含む。目的の抗体の特定抗原と免疫沈降する能力は、例えばウェスタンブロット分析により評価することができる。当業者であれば、抗体の抗原との結合を増加しかつバックグラウンドを減少するように改変しうるパラメーター(例えば、セファロースビーズによる細胞溶解液の前清澄化)を理解しているであろう。免疫沈降プロトコルに関するさらなる考察については、例えば、Ausubel et al.編, 1994, 「Current Protocols in Molecular Biology」, Vol.1, John Wiley & Sons, Inc., New Yorkの10.16.1を参照されたい。
【0093】
ウェスタンブロット分析は一般的に、タンパク質サンプルを調製し、タンパク質サンプルをポリアクリルアミド中で電気泳動し(例えば、抗原の分子量に基づいて8%〜20%SDS−PAGE)、タンパク質サンプルをポリアクリルアミドゲルからニトロセルロース、PVDF又はナイロンなどのメンブランに移し、ブロッキング溶液(例えば、3%BSA又は無脂肪乳入りのPBS)中でメンブランをブロッキングし、メンブランを洗浄バッファー(例えば、PBS−Tween20)で洗浄し、メンブランをブロッキング溶液に希釈した一次抗体(例えば、目的の抗体)とともにインキュベートし、メンブランを洗浄バッファーで洗浄し、メンブランをブロッキング溶液に希釈した酵素基質(例えば、西洋ワサビペルオキシダーゼ又はアルカリホスファターゼ)若しくは放射性分子(例えば32P又は125I)と複合した二次抗体(一次抗体を認識するもの、例えば抗ヒト抗体)と共にインキュベートし、メンブランを洗浄バッファーで洗浄し、そして抗原の存在を検出することを含む。当業者であれば、検出されるシグナルを増加しかつバックグラウンドノイズを減少するように改変することができるパラメーターを理解しているであろう。ウェスタンブロットプロトコルに関するさらなる考察については、例えば、Ausubel et al.編, 1994, 「Current Protocols in Molecular Biology」, Vol.1, John Wiley & Sons, Inc., New Yorkの10.8.1を参照されたい。
【0094】
ELISAは、抗原を調製し、96ウエルマイクロタイタープレートのウエルを該抗原によりコーティングし、酵素基質(例えば、西洋ワサビペルオキシダーゼ又はアルカリホスファターゼ)などの検出可能な化合物と複合した目的の抗体をウエルに加え、そしてある時間インキュベートして、該抗原の存在を検出することを含む。ELISAにおいて、目的の抗体を検出可能な化合物と必ずしも複合させる必要はなく;代わりに検出可能な化合物と複合した二次抗体(目的の抗体を認識する抗体)をウエルに加えてもよい。さらに、ウエルを抗原でコーティングする代わりに、抗体をウエルにコーティングしてもよい。この場合、コーティングしたウエルに目的の抗原を加えた後、検出可能な化合物と複合した二次抗体を加えてもよい。当業者であれば、検出されるシグナルを増加するように改変することができるパラメーターだけでなく当技術分野で公知のELISAの他の変法も理解しているであろう。ウェスタンブロットプロトコルに関するさらなる考察については、例えば、Ausubel et al.編, 1994, 「Current Protocols in Molecular Biology」, Vol.1, John Wiley & Sons, Inc., New Yorkの11.2.1を参照されたい。
【0095】
抗体の抗原との結合アフィニティ及び抗体−抗原相互作用の解離速度(off-rate)は、競合結合アッセイにより測定することができる。競合結合アッセイの一例はラジオイムノアッセイであって、これは、増加する量の非標識抗原の存在のもとでの、標識(例えば、3H又は125I)した抗原を目的の抗体と共にインキュベーション、及び標識した抗原と結合した抗体の検出を含む。本発明の抗体又はそのフラグメントの目的の抗原に対するアフィニティ及び解離速度は、データからスキャッチャードプロット)分析により測定することができる。二次抗体との競合をラジオイムノアッセイを用いて測定することもできる。この場合、非標識の二次抗体の増加する量の存在のもとで、目的の抗原を標識(例えば、3H又は125I)した化合物と複合した本発明の抗体又はそのフラグメントとともにインキュベートする。
【0096】
好ましい実施形態においては、BIAcore動力学分析を用いて抗体又はそのフラグメントの目的の抗原との会合及び解離速度を決定することができる。BIAcore動力学分析は、その表面上に抗体又はそのフラグメントを固定したチップからの、目的の抗原の結合及び解離を分析することを含む(後述の実施例を参照)。
【0097】
抗体又はそのフラグメントはまた、目的の抗原のその宿主細胞受容体への結合を阻害するその能力について、当技術分野で公知の方法を用いてアッセイしうる。例えば、目的の抗原に対する受容体を発現する細胞を、抗体又はそのフラグメント(すなわちFabフラグメント)の存在下又は不在下にて抗原と接触させ、該抗体又はそのフラグメントが目的の抗原の結合を阻害する能力を、例えばフローサイトメトリー又はシンチレーションアッセイにより測定しうる。目的の抗原又は抗体若しくは抗体フラグメントは、検出可能な化合物、例えば放射性標識(例えば32P、35S、及び125I)、又は蛍光標識(例えば、フルオレセインイソチオシアネート、ローダミン、フィコエリスリン、フィコシアニン、アロフィコシアニン、o−フタルデヒド及びフルオレスカミン)で標識して、目的の抗原とその宿主細胞受容体との相互作用の検出を可能にしてもよい。あるいは、抗体又はそのフラグメントが目的の抗原のその受容体との結合を阻害する能力は、細胞非含有アッセイにおいて測定しうる。例えば、目的の抗原を抗体又はFabフラグメントと接触させ、該抗体又は抗体フラグメントが目的の抗原のその宿主細胞との結合を阻害する能力を測定しうる。好ましくは、抗体又はFabフラグメントを固相支持体に固定化し、目的の抗原を検出可能な化合物で標識する。あるいは、目的の抗原を固相支持体に固定化し、抗体又はFabフラグメントを検出可能な化合物で標識する。目的の抗原は、完全に又は部分的に精製されたものであってもよいし(例えば部分的又は完全に他のポリペプチドを含まない)、あるいは細胞溶解物の一部であってもよい。さらに、抗原は、抗原及びドメイン(結合ドメインなど)を含む融合タンパク質であってもよい。あるいは、抗原は、当業者に周知の技法を用いてビオチニル化されていてもよい(例えば、ビオチニル化キット、Pierce Chemicals, Rockford, IL)。
【0098】
目的の抗体又はそのフラグメントはまた、抗原の活性を阻害又はダウンレギュレートするその能力について、当業者に公知の技法を用いてアッセイしてもよい。本発明のベクター系により作製される抗体又はFabフラグメントは、抗原ポリペプチドの発現を阻害又はダウンレギュレートするそれらの能力についてアッセイすることも可能である。当業者に公知の技術としては、限定されるものではないが、ウエスタンブロット分析、ノーザンブロット分析、及びRT−PCRを用いてタンパク質発現を測定することができる。
【0099】
本発明のベクター系により作製される抗体又はFabフラグメントは、好ましくはin vitroで試験し、その後、ヒトに使用する前に、所望の治療的又は予防的活性についてin vivoで試験しうる。例えば、特異的抗体又は本発明の組成物の投与が適応か否かを判定するために用い得るin vitroアッセイとしては、被験体の組織サンプルを培養中で増殖させ、本発明の抗体又は組成物に曝露し又はそれを投与し、かかる本発明の抗体又は組成物の組織サンプルに対する効果を観察することを含むin vitro細胞培養アッセイが挙げられる。種々の特定の実施形態において、in vitroアッセイは、本発明の抗体又は組成物が特定の細胞型に対して所望の効果を有するか否かを判定するために行いうる。好ましくは、本発明のベクター系により作製された抗体又はFabフラグメントはまた、in vitroアッセイ及び動物モデル系においてヒトに投与する前に試験しうる。さらに、この実施形態に従って、犠牲にしたラット由来の組織を組織学的変化について試験してもよい。
【0100】
本発明においては、本発明のベクター系により作製された抗体又はFabフラグメントの予防的及び/又は治療的効力を証明するために、ヒト被験者を用いた臨床試験を行う必要はない。抗体又はそのフラグメントを用いたin vitro及び動物モデル試験からヒトについても推定することができ、またこれらの試験は該抗体又は抗体フラグメントの予防用途及び/又は治療用途を証明するために十分である。
【0101】
治療に使用するための本発明の抗体又は組成物は、適当な動物モデル系(限定されるものではないが、ラット、マウス、ウシ、サル、及びウサギなど)においてその毒性を試験することができる。抗体又は組成物の毒性のin vivo試験については、当技術分野で公知の任意の動物モデル系を用いることができる。
【0102】
ウイルス感染の治療又は予防における効力は、本発明のベクター系により作製される抗体又はFabフラグメントが、病原体の感染を阻害する、又は抗原と関連する1以上の症候を予防、改善若しくは緩和する能力を検出することにより証明することができる。例えば1以上の症候の改善、又は目的の抗体若しくは組成物の投与後の死亡率及び/若しくは疾患率の低減がある場合には、その処置は治療的と考えられる。さらに、本発明のベクター系により作製され、1以上の抗原と免疫特異的に結合する1以上の抗体又はFabフラグメントの投与後の免疫応答の増大がある場合には、その処置は治療的と考えられる。
【0103】
目的の抗体又は組成物は、IFN−α、IFN−β、IFN−γ、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、IL−8、IL−9、IL−10、IL−12及びIL−15などのサイトカインの発現を誘導する能力についてin vitro及びin vivoにおいて試験しうる。当業者に公知の技法を用いて、サイトカインの発現レベルを測定しうる。例えば、サイトカインの発現レベルは、例えばRT−PCR及びノーザンブロット分析によりサイトカインのRNAのレベルを分析することで、例えば免疫沈降の後にウエスタンブロット分析及びELISAを行うことによりサイトカインのレベルを分析することで、測定することができる。好ましい実施形態において、本発明のベクター系により作製される抗体又はFabフラグメントは、IFN−γの発現を誘導するその能力について試験する。
【0104】
目的の抗体又は組成物は、それらが免疫細胞、好ましくはヒト免疫細胞(例えばT細胞、B細胞、及びナチュラルキラー細胞)の生物学的活性をモジュレートする能力についてin vitro及びin vivoで試験しうる。目的のベクター系により作製された抗体又はFabフラグメントが免疫細胞の生物学的活性をモジュレートする能力は、抗原の発現を検出し、免疫細胞の増殖を検出し、シグナル伝達分子の活性化を検出し、免疫細胞のエフェクター機能を検出し、あるいは免疫細胞の分化を検出することにより、評価しうる。これらの活性を測定するために、当業者に公知の技法を使用することができる。例えば、細胞増殖は、3Hチミジン取り込みアッセイ及びトリパンブルー細胞計数によりアッセイしうる。抗原の発現は、例えば、限定されるものではないが、ウエスタンブロット、免疫組織化学的ラジオイムノアッセイ、ELISA(酵素結合免疫吸着アッセイ)、「サンドイッチ」イムノアッセイ、免疫沈降アッセイ、沈降素反応、ゲル拡散沈降素反応、免疫拡散アッセイ、凝集アッセイ、補体結合アッセイ、免疫放射アッセイ、蛍光イムノアッセイ、プロテインAイムノアッセイ、及びFACS分析等の技法を用いる競合及び非競合アッセイ系などのイムノアッセイによりアッセイしうる。シグナル伝達分子の活性化は、例えばキナーゼアッセイ及び電気泳動シフトアッセイ(EMSA)によりアッセイしうる。
【0105】
目的の抗体又は組成物はまた、ウイルス複製を阻害又はウイルス負荷を低減する能力について、in vitro、ex vivo及びin vivoアッセイにおいて試験しうる。本発明のベクター系により作製される抗体又はFabフラグメントはまた、感染の時間経過を低減する能力について試験しうる。本発明のベクター系により作製される抗体又はFabフラグメントはまた、感染症に罹患したヒトの生存期間を少なくとも25%、好ましくは少なくとも50%、少なくとも60%、少なくとも75%、少なくとも85%、少なくとも95%又は少なくとも99%で増大する能力について試験しうる。さらに、本発明のベクター系により作製される抗体又はFabフラグメントは、感染症に罹患したヒトの入院期間を少なくとも60%、好ましくは少なくとも75%、少なくとも85%、少なくとも95%又は少なくとも99%で低減する能力について試験しうる。当業者に公知の方法を用いて、目的の抗体又は組成物の機能をin vivoで分析することも可能である。
【0106】
5.2.2.ファージディスプレイを用いたスクリーニング方法
当業者であれば理解できるように、個々のタンパク質又は他の化合物、並びにタンパク質又は他の化合物の大きなライブラリー(例えばファージディスプレイライブラリー)をスクリーニングして、目的の特定の抗原と結合する分子を同定する方法がいくつかある。
【0107】
ファージディスプレイ法においては、機能性抗体ドメインが、同ドメインをコードするポリヌクレオチド配列を保持するファージ粒子の表面に提示される。特に、VH及びVLドメインをコードするDNA配列を、動物cDNAライブラリー(例えば、リンパ組織のヒト又はマウスcDNAライブラリー)から増幅する。VH及びVLドメインをコードするDNAをPCRによりscFvリンカーと一緒に組換えて、ファージミドベクター(例えば、pCANTAB6又はpComb3 HSS)中にクローニングする。そのベクターを大腸菌(E.coli)中にエレクトロポレーションして大腸菌(E.coli)をヘルパーファージに感染させる。これらの方法で使用されるファージは、典型的には、fd及びM13を含む繊維状ファージであり、VH及びVLドメインを通常、組換え法によりファージ遺伝子III又は遺伝子VIIIのいずれかに融合させる。目的の抗原と結合する抗原結合ドメインを発現するファージを、抗原により、例えば、標識した抗原又は固体表面若しくはビーズに結合若しくは捕獲された抗原を用いて、選択又は同定することができる。本発明の抗体を作製するために利用しうるファージディスプレイ法の例としては、Brinkman et al., 1995, J. Immunol. Methods 182:41-50;Ames et al., 1995, J. Immunol. Methods 184:177-186;Kettleborough et al., 1994, Eur. J. Immunol. 24:952-958;Persic et al., 1997, Gene 187:9-18;Burton et al., 1994, Advances in Immunology 57:191-280;PCT出願第PCT/GB91/01134号;国際公報WO90/02809号、WO91/10737号、WO92/01047号、WO92/18619号、WO93/11236号、WO95/15982号、WO95/20401号、及びW097/13844号;並びに米国特許第5,698,426号、第5,223,409号、第5,403,484号、第5,580,717号、第5,427,908号、第5,750,753号、第5,821,047号、第5,571,698号、第5
,427,908号、第5,516,637号、第5,780,225号、第5,658,727号、第5,733,743号、及び第5,969,108号に開示された方法が挙げられ、これらはそれぞれ参照によりその全文が本明細書に組み入れられる。
【0108】
ファージディスプレイライブラリの例は以下の文献に記載されている:Scott and Simith, 1990, Science 249:386-390;Devlin et al., 1990, Science, 249:404-406;Christian et al., 1992, J. Mol. Biol. 227:711-718;Lenstra, 1992, J. Immunol. Meth. 152:149-157;Kay et al., 1993, Gene 128:58-65;及びPCT公報WO94/18318(1994年8月18日出願)。
【0109】
以上の参考文献に記載の通り、ファージ選択の後、ファージからの抗体コード領域を単離し、さらにそれを使用して、ヒト抗体を含む完全抗体、又は任意の他の所望の抗原結合フラグメントを作製し、それらを、例えば以下に記載のようにして、哺乳動物細胞、昆虫細胞、植物細胞、酵母、及び細菌を含む任意の所望の宿主にて発現させることができる。組換え法によりFab、Fab'及びF(ab')2フラグメントを製造する技術はまた、PCT公報WO92/22324号;Mullinax et al., 1992, BioTechniques 12 (6):864-869;Sawai et al., 1995, AJRI 34:26-34;並びにBetter et al., 1988, Science 240:1041-1043(上記参考文献は参照によりその全文が組み入れられる)に開示された方法などの当技術分野で公知の方法を用いて、使用することができる。
【0110】
完全抗体を作製するために、VH又はVLヌクレオチド配列、制限酵素切断部位、及び制限酵素切断部位を保護するフランキング配列を含むPCRプライマーを用いて、scFvクローン中のVH又はVL配列を増幅することができる。当業者に公知のクローニング技術を利用して、PCR増幅されたVHドメインをVH定常領域、例えば、ヒトγ4定常領域を発現するベクター中にクローニングし、また、PCR増幅したVLドメインをVL定常領域、例えばヒトκ若しくはλ定常領域を発現するベクター中にクローニングすることができる。好ましくは、VH又はVLドメインを発現するベクターは、EF−1αプロモーター、分泌シグナル、可変ドメイン、定常ドメイン及びネオマイシンなどの選択マーカーに対するクローニング部位を含む。VH及びVLドメインを、必要な定常領域を発現する1つのベクター中にクローニングすることもできる。次いで、重鎖変換ベクター及び軽鎖変換ベクターを培養細胞株中に同時トランスフェクトし、当業者に公知の技術を用いて、全長抗体、例えばIgGを発現する安定した一過性の培養細胞株を作製する。
【0111】
5.2.3.抗体配列の最適化方法
新規抗体の試験及び特性決定についての上記用途に加えて、二重発現ベクターは、所望の結合特性又は治療特性について既存の抗体を最適化するために用いることが可能である。本発明のこの態様において、既知のIgG重鎖又は軽鎖配列をコードするヌクレオチド配列を二重発現ベクター系にクローニングし、化学的、合成又は遺伝的突然変異誘発を行い、そのヌクレオチド配列を改変することができる。続いて、配列変異体は、目的の特性の変化について細菌及び/又はヒト細胞においてスクリーニングしうる。
【0112】
抗体をコードするポリヌクレオチドを得て、当技術分野で公知のいずれかの方法によりヌクレオチド配列を決定することができる。所望の抗原に対して免疫特異的な抗体のヌクレオチド配列は、例えば、文献又はGenBankなどのデータベースから得ることができる。VITAXINTMのアミノ酸配列は既知であるので、この抗体をコードするヌクレオチド配列を、当技術分野で公知の方法を利用して(すなわち、特定のアミノ酸をコードすることが知られるヌクレオチドコドンを、抗体をコードする核酸を生成するように組み立てて)決定することができる。抗体をコードするかかるポリヌクレオチドは、化学合成したオリゴヌクレオチドから組み立てることができ(例えば、Kutmeier et al., 1994, BioTechniques 17:242に記載の通り)、これは、簡単に説明すると、抗体をコードする配列の一部分を含有する重複オリゴヌクレオチドの合成、これらのオリゴヌクレオチドのアニーリング及びライゲーション、次いでライゲートしたオリゴヌクレオチドのPCRによる増幅を伴う。
【0113】
あるいは、抗体をコードするポリヌクレオチドを、好適な起源由来の核酸から作製することができる。特定の抗体をコードする核酸を含有するクローンが入手できなくても、その抗体分子の配列が既知であれば、その免疫グロブリンをコードする核酸は、化学的に合成することができるし、あるいは、適当な起源(例えば、抗体cDNAライブラリー若しくはそれから作製したcDNAライブラリー、それから単離した核酸、好ましくはポリA+RNA、目的の抗体を発現するように選択されたハイブリドーマ細胞などの、抗体を発現する任意の組織若しくは細胞)からその配列の3’及び5’末端にハイブリダイズする合成プライマーを用いるPCR増幅によるか、又は、特定の遺伝子配列に特異的なオリゴヌクレオチドプローブを用いて、例えばcDNAライブラリーから前記抗体をコードするcDNAクローンを同定してクローニングすることにより、取得することができる。PCRにより作製された増幅核酸は、次いで、当技術分野で公知の方法を用いて複製可能なクローニングベクター中にクローニングすることができる。
【0114】
抗体のヌクレオチド配列が一度決定されると、抗体のヌクレオチド配列を、ヌクレオチド配列の遺伝子操作に関する当技術分野で公知の方法、例えば組換えDNA技術、位置指定突然変異、PCRなど(例えば、Sambrook et al., 1990, 「Molecular Cloning, A Laboratory Manual」, 第2版, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY、及びAusubel et al.編, 1998, 「Current Protocols in Molecular Biology」, John Wiley & Sons, NY、に記載の技術を参照、これらの文献は本明細書に参照によりその全文が組み入れられる)を用いて遺伝子操作して、異なるアミノ酸配列を有する抗体を作製し、例えばアミノ酸置換、欠失、及び/又は挿入を創出することができる。
【0115】
特定の実施形態においては、1以上のCDRを、慣用される組換えDNA技術を利用してフレームワーク領域内に挿入する。フレームワーク領域は、天然又はコンセンサスフレームワーク領域であってもよく、好ましくはヒトフレームワーク領域である(例えば、ヒトフレームワーク領域の列挙については、Chothia et al., 1998, J. Mol. Biol. 278:457-479を参照)。好ましくは、フレームワーク領域とCDRの組合せにより作製されたポリヌクレオチドは、特定の抗原と特異的に結合する抗体をコードする。好ましくは先に考察したように、1以上のアミノ酸置換をフレームワーク領域内で引き起こしてもよく、かつ好ましくはそのアミノ酸置換は抗体のその抗原との結合を改良するものである。さらに、かかる方法を利用して、鎖内ジスルフィド結合に参加する1以上の可変領域システイン残基のアミノ酸置換若しくは欠失を引き起こし、1以上の鎖内ジスルフィド結合を欠く抗体分子を作製してもよい。ポリヌクレオチドに対するその他の改変は本発明により包含されかつ当技術分野の範囲内にある。
【0116】
本発明に従って使用しうる市販の抗体の例を以下の表1に示すが、これに限定されるものではない。
【表1】

【0117】
【0118】
【0119】
【0120】
5.3.本発明と共に使用するための他の方法
5.3.1.他のポリペプチド及びその断片を発現するための二重発現ベクター系の使用
本発明の二重発現ベクター系は、本明細書に開示する抗体を発現するために記載した方法を用いて、哺乳動物細胞において目的の任意のポリペプチドを、及び細菌細胞において該ポリペプチドの断片、好ましくは可能性断片を発現するために用いることができる。目的のポリペプチドは、膜結合ポリペプチド又は分泌ポリペプチドであることが好ましく、細菌細胞の周辺腔領域に発現され分泌された場合にアッセイ可能な活性を保持する可溶性ドメインを有するものである。目的のドメインとしては、限定されるものではないが、DNA結合ドメイン、タンパク質−タンパク質相互作用ドメイン、キナーゼドメイン、又は他の酵素的若しくは機能的タンパク質ドメインが挙げられる。
【0121】
二重発現ベクターは、イントロンを全長ポリペプチドのシグナル配列に挿入することにより構築する。イントロンは、スプライス受容配列と重複し、目的のポリペプチドのコード領域と読み取り枠を合わせた細菌プロモーターとシグナル配列を含み、その結果、細菌プロモーターからの転写が細菌細胞における目的のドメインの発現を指令するように設計する。そのような配列の構築方法は、以下に示す実施例において詳細を説明する。
【0122】
かかる二重発現ベクターの構成要素としては、(a)細菌性複製起点、(b)哺乳動物複製起点、(c)上記分泌又は膜結合ポリペプチドをコードするヌクレオチド配列と機能的に結合した哺乳動物プロモーター及び/又はエンハンサー配列が含まれ、該ヌクレオチド配列は、少なくとも1つのイントロンを含む哺乳動物シグナル配列を含み、該イントロンは該ポリペプチドの可溶性ドメインをコードする配列と機能的に結合された細菌プロモーター及び細菌シグナル配列を含み、その結果、該細菌プロモーター及び細菌シグナル配列が細菌細胞における周辺腔への上記ポリペプチドの可溶性ドメインの発現及び分泌を指令し、上記哺乳動物シグナル配列は該ポリペプチドの発現及び分泌を指令し、ここで哺乳動物細胞における発現のためのプロモーターは、上記ポリペプチドの可溶性ドメインをコードする上記ヌクレオチド配列と機能的に結合している。
【0123】
目的の膜結合又は分泌ポリペプチドの例としては、限定されるものではないが、細胞表面受容体、例えば限定されるものではないが、エリスロポエチン受容体(Epo−R;Noguchi et al., 1991, Blood 78(10):2548-2556)、インスリン受容体(InsR;Ebina et al., 1985, Cell 40:747-758;Ullrich, 1985, Nature 313:756-761)、及び腫瘍壊死因子α受容体(TNFαR;Gray et al., 1990, Proc. Natl. Acad. Sci. USA 87:7380-7384);一回膜貫通チロシン受容体キナーゼ(TRK)様クラス受容体のメンバー(Ullrich & Schlessinger, 1990, Cell 61:203-12;Hunter & Cooper, 1985, Ann. Rev. Biochem. 54:897-930)が挙げられる。このクラスには以下のものが含まれる:表皮増殖因子受容体ファミリー、例えば表皮増殖因子(EGF;Ullrich et al., Nature, 1984, 309:418-25;Schector et al., Nature 278:835-38)、ワクシニア増殖因子(Brown et al., 1985, Nature 313:491-92)、アンフィレギュリン/神経鞘腫由来増殖因子(AR又はSDGF;Schoyab et al., 1989, Science 243:1074-1076)、ヘアピン結合EGF様因子(HB−EGF;Higashiyama et al., 1991, Science 251:936-939)、neu分化因子(NDF;Wen et al., 1992, Cell, 69:559-72)、及びHer2(Coussens et al., 1985, Science 230:1132-39;Santanta et al., 1994, Proc. Natl. Acad. Sci. USA 91:1711-1715)などのヒレギュリン(Holmes et al., 1992, Science 256:1205-10)、インスリン受容体ファミリー、例えばINSR(上記)及びIRR、血小板由来増殖因子(PDGF)受容体ファミリー、例えばα−PDGFR(Potts & Carrington, 1993, Dev. Dyn. 198:14-21)、β−PDGFR(Chi et al., 1997, Oncogene 15:1051-58)、CSF1−R(例えば、Waterfield et al., 1983, Nature 304:35-39)、c−Kit幹細胞因子受容体(Lemmon et al., 1997, J. Biol. Chem. 272:6311-6317)、繊維芽細胞増殖因子受容体(FGFR)、例えばCEK2(Pasquale, 1990, Proc. Natl. Acad. Sci. U.S.A. 87:5812-16)、TRK受容体ファミリー、例えばTRK及びTRK−B;並びにEPH/ECK受容体ファミリー、例えばElf−1及びEck(Cheng & Flanagan, 1994, Cell 79:157-68;Lindberg & Hunter, 1990, Mol. Cell Biol. 10:6316-24)、神経増殖因子受容体(Woo et al., 1998, Protein Sci. 7:1006-1016;Johnson et al., 1986, Cell 47:545-54)、並びにインスリン様増殖因子受容体(Ullrich et al., 1986, EMBO J. 5:2503-12;及びSepp-Lorenzino, 1998, Breast Cancer Res. Treat. 47:235-253)。TK様ファミリーの受容体の他のメンバーもまた用いることが可能である。例えば、van der Greer et al., 1997, Ann. Rev. Cell Biol. 10:251-337;Herz et al., 1997, J. Recept. Signal Transduct. Res. 17:671-776(これらは参照によりその全体を本明細書に援用する)、並びにそれらの参照文献を参照されたい。
【0124】
他の実施形態において、目的のポリペプチドは、7回膜貫通クラスの受容体(例えば、Gタンパク質共役型クラスの受容体;GPCR)のメンバーでありうる:例えば、β3アドレナリン作動性受容体(Emorine et al., 1989, Science 245:1118-21;Huang et al., 1997, J. Recept. Signal Transduct. Res. 17:599-607)、ドーパミン受容体、例えばドーパミンD2受容体(Wilkie et al., 1993, Genomics 18:175-184;Bunzow et al., 1988, Nature 336:783-7)、及びムスカリン作動性アセチルコリン受容体(Strader et al., 1994, Ann. Rev. Biochem. 63:101-32、及びそれに引用された参照文献を参照。参照によりその全体を本明細書に援用する)、イオンチャネル、例えば限定されるものではないが、Kvl.3カリウムチャネル(Kath et al., 1997, Annual Reports in Med. Chem., Hagmann編、32:181-89)、NHEI及びNHE2 Na+/H+交換体(Fafournoux & Pouysseyur, 1994, J. Biol. Chem. 269:2589-96)、電位依存性イオンチャネルファミリーの受容体、例えばK+感受性チャネル及びCa2+感受性チャネル(Hille, B, "Ionic Channels of Excitable Membranes", 1992, Sinauer Associates, Sunderland, MA;Catterall, W.A., 1991, Science 253:1499-1500及びこれらに引用された参考文献を参照。参考によりその全文を本明細書に援用する)、受容体タンパク質−チロシンホスファターゼファミリーのメンバー(すなわちR−PTP)、例えば限定されるものではないが、CD45(すなわちリンパ球共通抗原、LCA)、R−PTPα、β、γ、κ及びその他(例えば、Denu et al., 1996, Cell 87:361-64;Fashena and Zinn, 1995, Curr. Biol. 5:1367-69、それぞれ参照によりその全文を本明細書に援用する)、サイトカイン受容体ファミリーのメンバー、IL−1サイトカイン受容体ファミリー(IL−1α及びIL−1β、例えば、Vigers et al., 1997, Nature 386:190-194参照)、クラスIサイトカインファミリー、特にホモダイマー形成に関与する高度に保存されたシステインを特徴とする造血サイトカイン受容体の成長ホルモン受容体サブファミリー(Watowich et al., Proc. Nat. Acad. Sci., 89:2140-44)。このファミリーには、EPO受容体(Noguchi et al., 1991、前掲)だけではなく、成長ホルモン受容体(Cunningham et al., 1989, Science 243:1330)、プロラクチン受容体(Boutin et al., 1988, Cell 53:69)、CSF、顆粒球−コロニー刺激因子受容体(Seto et al., 1992, J. Immunol. 148(1):259-266)、ソマトトロピン受容体(Leung et al., 1987, Nature 330:537)、グリア由来神経栄養因子(GDNF)受容体、例えばGFRα3(Baloh et al., Proc. Natl. Acad. Sci. 95:5801-06)、及び他多数(Herz et al. 1997、前掲)、並びにクラスIIサイトカイン受容体(インターフェロン)ファミリーメンバー(このファミリーでは、リガンド結合はJAKキナーゼを介して二量体化及び活性化を誘導しうる)(Aguet et al., 1988, Cell 55:273-80;及びUze et al., 1990, Cell 60:225-234)。
【0125】
他の実施形態において、目的のポリペプチドは、核ホルモン受容体スーパーファミリーのメンバーでありうる(例えば、Mangelsdorf et al., 1995, Cell 83:835-39及びそれに引用された参照文献。参照によりその全体を本明細書に援用する):例えば、ステロイド受容体(Beato et al., 1995, Cell 83:851-57及びそれに引用された参照文献。参照によりその全体を本明細書に援用する)、糖質コルチコイド(Hollenberg et al., 1985, Nature, 318:635-41;またEvans, 1989, Recent Prog. Horm. Res. 45:1-22及びそれに引用された参照文献)、アンドロゲン(Tilley et al., Proc. Nat. Acad. Scie. U.S.A., 1989, 86:327-31)、アルドステロン、プロゲステロン、及びエストロゲン受容体(Greene et al., 1986, Nature 320:134-39;Tsai & O'Malley, 1994, Ann. Rev. Biochem. 63:451-86及びそれに引用された参照文献。参照によりその全体を本明細書に援用する)、ヘテロダイマー受容体、例えばチロキシン、ビタミンD、ビタミンA、レチノイド(RAR、RXR)、プロスチノイド受容体(Mangelsdorf & Evans, 1995, Cell 83:841-50及びそれに引用された参照文献。参照によりその全体を本明細書に援用する)、例えば肝核因子HNF4(Sladek et al., 1990, Genes Dev. 4:2353-65)。これらのクラス内のオーファン受容体は特に興味深い配列であり、その推定リガンドが未知であるヘテロダイマー及びホモダイマー受容体のファミリーを表すリガンドを同定するために本発明の方法の一部として用いることができる。
【0126】
別の実施形態において、目的のポリペプチドは、非膜型の非分泌型ポリペプチド、例えば核転写因子タンパク質でありうる。転写因子としては、限定されるものではないが、Fos/Jun(Bohmann et al., Science 238:1386-92;Angel et al., 1988, Nature 332:166-71)、C/EBP(Landshultz et al., 1988, Science, 240:1759-64)、GCN4(例えば、Agre et al., 1989, Science 246:922-926、また本明細書の第9節に示す実施例参照)、ヘリックスループヘリックス(HLH)ドメインタンパク質、例えばMyc(Murre et al., 1989, Cell 56:777-783)及びMyoD並びにE12/E47様タンパク質とin vivoでヘテロオリゴマー化する必要のある他の筋原性HLHタンパク質(Lasser et al., 1991, Cell 66:305-15)、並びに当技術分野で周知の他の転写因子が挙げられる。
【0127】
本明細書に例示するタンパク質に加えて、目的のポリペプチドは、公共データベース、例えばSwiss Protein Data Base(SWISS−PROT;Bairoch & Apweiler, 1998, Nucl. Acids Res. 26:38-42)などに登録された任意の膜結合又は分泌型ポリペプチドに由来するアミノ酸残基を含みうる。
【0128】
5.3.2.抗体の診断用途
本発明のベクター系により作製された抗体又はFabフラグメントを使用し、生物学的サンプル中の抗原レベルを本明細書に記載の又は当業者に公知の古典的免疫組織学的方法を用いて評価することができる(例えば、Jalkanen et al., 1985, J. Cell. Biol. 101:976-985;Jalkanen et al., 1987, J. Cell. Biol. 105:3087-3096を参照)。タンパク質遺伝子発現を検出するために有用な他の抗体に基づく方法は、イムノアッセイ、例えば、酵素結合免疫吸着アッセイ(ELISA)及びラジオイムノアッセイ(RIA)を含む。好適な抗体アッセイ標識は当技術分野で公知であり、酵素標識、例えばグルコースオキシダーゼ;放射性同位体、例えばヨウ素(125I,131I)、炭素(14C)、硫黄(35S)、トリチウム(3H)、インジウム(121In)、及びテクチニウム(99mTC);発光標識、例えばルミノール;及び蛍光標識、例えばフルオレセイン、ローダミン及びビオチンが挙げられる。
【0129】
被験者のサイズと使用するイメージングシステムが、診断イメージを作るために必要なイメージング成分の量を決定しうることは当技術分野で理解されるであろう。ヒト被験者に対する放射性同位体成分の場合、注射される放射能の量は通常約5〜20ミリキューリーの99mTcであろう。標識した抗体又は抗体フラグメントを次いで選択的に特定のタンパク質を含有する細胞の位置に蓄積させる。in vivo腫瘍イメージングは、S. W. Burchiel et al., 「Immunopharmacokinetics of Radiolabeled Antibody and Their Fragments」,Tumor Imaging :The Radiochemical Detection of Cancer, Chapt. 13, S. W. Burchiel and B. A. Rhodes, 編, Masson Publishing Inc. (1982)に記載されている。
【0130】
使用する標識のタイプ及び投与様式を含む複数の変数に依存して、標識した分子が被験者の部位に選択的に濃縮しうるためのかつ無結合の標識した分子がバックグラウンドレベルにクリアされるための投与後の時間間隔は、6〜48時間又は6〜24時間又は6〜12時間である。他の実施形態においては、投与後の間隔は5〜20日又は5〜10日である。
【0131】
被験者中の標識した分子の存在は、in vivo走査に対する当技術分野で公知の方法を用いて検出することができる。これらの方法は、使用する標識のタイプに依存する。当業者は特定標識を検出するための適当な方法を決定することができよう。本発明の診断に利用しうる方法とデバイスは、限定されるものでないが、コンピューター断層撮影(CT)、位置放出断層撮影(PET)などの全身走査、磁気共鳴イメージング(MRI)、及びソノグラフィを含む。
【0132】
特定の実施形態においては、分子を放射性同位体により標識し、患者において放射線応答外科機器(Thurston et al., 米国特許第5,441,050号)を用いて検出する。他の実施形態においては、分子を蛍光化合物により標識し、患者において蛍光応答走査機器を用いて検出する。他の実施形態においては、分子を位置放出性金属により標識し、患者においてポジトロン放出断層撮影を用いて検出する。さらに他の実施形態においては、分子を常磁性標識により標識し、患者において磁気共鳴イメージングを用いて検出する。
【実施例】
【0133】
6.1.抗CD16 Fab及びIgGの発現
大腸菌においてFabを、哺乳動物細胞においてIgGを発現させるための二重発現ベクターの設計の基礎とするため、以下の予備的研究を行った。ベクターを2種構築した。第1のものは、大腸菌においてFab分子を発現させるためのベクターであり、第2のものは、哺乳動物細胞においてIgGを発現させるためのベクターであった。キメラ型又はヒト化型抗CD16 Mabのうちいずれかの重鎖及び軽鎖cDNAを用いて、発現ベクターを検証した。
【0134】
大腸菌においてFab分子を発現させるため、lacプロモーターオペレーター(lacPO)の制御下で個々の軽鎖(LC)配列及びVH CH1(Fd)鎖配列がそれぞれpelBシグナルペプチドコード配列と融合する、Barbasにより記載のもの(Barbaset al., 1991, Proc. Natl. Acad. Sci. U.S.A. 88:7978-7982)と類似するベクターを構築した。Fdセグメント配列はまた、精製用のC末端Hisタグと同定用のHSVエピトープタグの両方をコードする配列を含む。
【0135】
このベクターを使用して、大腸菌(BL21又はXL1 Blue株)の周辺質中にキメラ又はヒト化抗CD16 Fabを約1mg/l分泌させた。この物質に結合活性があるかどうかを試験するために、周辺質抽出物からFabを精製し、サンドイッチELISAでCD16受容体の可溶性部分(sCD16)と結合できるかどうかを、又は免疫複合体とのsCD16の結合を阻害できるかどうかを分析した。図1に示すように、精製したキメラFabは、還元後25kdの単一バンドとしてSDS PAGEゲル上で移動し、又は還元しない場合50kdの単一バンドとして移動する。このことから、この産物がジスルフィド結合していることが示唆される。ウェスタンブロット上で、このバンドは、抗LC抗体にも抗HSVタグ抗体にも反応することから、LC鎖もFd鎖も存在することが示唆される。
【0136】
IgGを発現させるため、全長重鎖(γ1)又は軽鎖(K)遺伝子をコードする配列をネズミVHシグナル配列と融合させ、ベクターpCI neo(Promega)内でCMVieプロモーター/エンハンサーの下流に配置した、個々のベクターを構築した。ネズミVHシグナルペプチドコード領域は、天然に存在するイントロンを含み、多数の抗体の軽鎖配列と重鎖配列をどちらも分泌する信頼性が非常に高いことが認められた。これに対し、分泌のために発現される可変領域由来のシグナルペプチドを含むcDNA配列からは、一貫しない結果が得られた。キメラ又はヒト化抗CD16の軽鎖及び重鎖プラスミドをHEK293細胞に共トランスフェクトした。一般に、3日後に培地中に5〜10μg/ml分泌された。
【0137】
両方のベクターから精製した物質を、結合するかどうかELISAで直接試験し、又はフルオレセインBSAとヒトIgG1キメラ型抗フルオレセインMab4 4 20の間で形成された免疫複合体とsCD16との結合を阻害するかどうかについてアッセイした。免疫複合体とsCD16との結合阻害を図2に示す。このプロトコルは以下の通りである。MaxiSorpイムノプレート(Nunc F96)を、1ウエルにつき500ngの炭酸緩衝液中のBSA FITCで、4℃で一晩被覆した。0.5%BSAのPBST溶液で、このプレートを室温で30分間ブロッキングした。Ch4 4 20(ヒトIgG1)をプレートに1ウエルにつき50ng加え、室温で1時間インキュベートして免疫複合体を形成させた。精製したCh3G8Fab、(陽性対照である)Ch3G8、(陰性対照である)ヒトIgG1を、sCD16 G2ビオチンを0.5μg/ml含む0.5%BSA/PBSTで、図に示した最終濃度まで希釈し、免疫複合体を含んでいるウエルに加えた。次いで、このプレートを室温で2時間インキュベートした。sCD16 G2ビオチンが免疫複合体と結合したことを、1:5000に希釈した西洋ワサビ結合ストレプトアビジン(Pharmacia)によって検出した。室温で30分間インキュベートした後、TMB(BioFX)を検出用の基質として用いた。上記のステップの間で、1×PBS/0.1%Tween20(PBST)でプレートを常に3回洗浄した。発色させるため、このプレートを室温で5〜10分間維持した。この反応を0.18M硫酸で停止し、OD450nmを測定した。
【0138】
図3に示すように、この物質はまた、sCD16Aとの直接の結合においても活性であった。可溶性モノマーCD16は、抗CD16 Mab LNK 16で被覆したイムノプレート上に捕捉された。結合しなかったリガンドを洗浄して除いた後、キメラFabの希釈物をプレートに加え、次いでこのプレートを室温で1時間インキュベートした。次いで、上記に記載のように、ヤギ抗ヒトFab HRP結合物を用い、その後TMBで発色させて、結合したFabを検出した。このsCD16A結合アッセイの結果を図3に示す。
【0139】
6.1.1.哺乳動物細胞においてIgG軽鎖及び重鎖を、大腸菌においてFabを発現させるための発現ベクター
以下の手順を使用して、大腸菌においてFabフラグメントを、哺乳動物細胞においてIgGを発現させ、スクリーニングするための二重発現ベクターを設計し構築した。本発明のこの実施形態では、本発明に従ってどちらの系でも効率のよい発現及び分泌を得るために、2つの条件が課せられた。第1に、大腸菌における発現及び分泌では、分泌されるポリペプチドの前にシグナルペプチドが機能しなければならない。第2に、哺乳動物における発現及び分泌では、メッセージを正しくスプライシングして、シグナル配列をコードするセグメントを結合させなければならない。細菌シグナル配列をコードする領域が、哺乳動物のスプライス受容部位と重複するので、これら2つの構成要素の設計を一緒に考慮しなければならない。
【0140】
この合成セグメントの設計及び構築のための鋳型として、pET25bのpelBシグナルペプチドコード配列を使用した。このコード配列を改変して、シグナルペプチドの中心にある疎水性残基を保持しながら、コンセンサス3’スプライス供与部位との相同性を最大にした。この方法では、ロイシンコドン(CTC)で2つのアラニンコドン(GCC又はGCT)を置き換えて、一続きのピリミジン(ピリミジンストレッチ)を正確なスプライシングに十分な長さにした。さらに、このピリミジンストレッチの上流に、潜在的なスプライシング分枝点をもたらすために、Alaコドン(GCT)を、Ile(ATC)に変化させた。最後に、原核生物と哺乳動物のシグナルペプチドが共有している領域において、−1及び−2位で、どちらの系でも機能的活性を保持する可能性が最も高い残基を選択した。改変型原核生物シグナルペプチドが依然として好ましいスプライス切断部位を保持しているかどうかを予測するために、神経ネットワークアルゴリズムを用いるSignalPプログラム(Nielsen et al., 1997, Int. J. Neural Sys. 8:581-599)によって配列を分析した。バークレーショウジョウバエゲノムプロジェクトウェブサイトのスプライス部位予測プログラムを用いて、スプライス部位の潜在的機能を評価した(Reese et al., J. Comput. Biol., 1997, 4(3):311 23を参照のこと)。このプログラムも、ヒト遺伝子向けの神経ネットワークアルゴリズムを用いている。
【0141】
プラスミドベクターpMGX115は、哺乳動物発現ベクターpCI neo内にヒト化重鎖をコードするミニ遺伝子を含む。このミニ遺伝子では、唯一のイントロンがシグナルペプチドをコードする領域内にある。正確なスプライス結合部位は、Glyコドン内で、シグナルペプチド切断部位の−4位に位置する(図4A〜Bを参照のこと)。図4Aに示す設計されたセグメントを以下のように導入する。最初に、PCRによりpUC18からlacプロモーター及びオペレーター(lacPO)配列を得、BglII−XbaI断片としてこれをpET25b中に導入して、T7プロモーターを置き換え、pMGX102を生成した。次いで、pMGX102を鋳型として用いて、lacPO配列をpelBシグナル配列と一緒にPCRで増幅した。次いで、重複PCRを用いて、シグナル配列の5’側のエキソン(5’スプライス部位を含む)及び重鎖(VH Cγ1)をそれぞれ含むpMGX115由来の2つの断片間にこの断片を配置した。このプロセスの間に、成熟VHをコードするセグメントをlacPO pelB配列を含むセグメントと結合させるのに使用する重複PCRプライマー内に改変を設計することにより、シグナル配列に改変を導入した。得られた断片を発現ベクターpCI Neo中にクローン化して、pMGX121を生成した。
【0142】
イントロン、スプライス結合部位及びシグナルペプチドの改変が発現及び分泌に影響を及ぼすかどうかを決定するために、HEK293細胞においてpMGX121の発現を調べた。抗ヒトFc抗体を用いてヒトIgGを捕捉し、抗ヒト重鎖+軽鎖HRP結合物を用いて検出するELISAで測定したとき、発現に対して有害な影響は認められなかった。
【0143】
大腸菌におけるFd(VH CH1フラグメント)の発現で停止コドンをもたらすために、pMGX121中のCH1とヒンジ領域の間に第2イントロンを導入して、pMGX578を生成した(図4Bを参照のこと)。ゲノムDNAからnested PCRで天然のヒトγ1遺伝子由来のイントロンを増幅し、重複PCRで他のセグメントとこれを結合させた。次いで、部位特異的変異誘発を行ってイントロンの開始部の近くに停止コドンを導入した。さらに、スプライシングに干渉しないような配列を設計し、スプライス部位配列が保持されるかどうかを、上記のプログラムを用いて調べた。次いで、得られたプラスミドpMGX579の配列を決定し、LC(軽鎖)発現プラスミドと共トランスフェクトしたとき、これがHEK293細胞において発現することを確認した。
【0144】
類似のLC発現プラスミドを生成するために、重複PCR手順により、pMGX121由来のlacPO pelB配列を含むシグナルイントロンをヒト化軽鎖コード配列と結合させ、発現カセット全体が切り出されるようにAscI部位がCMVieプロモーターの5’末端及びSV40ポリA部位の3’末端に導入されている以外はpMGX579と同一であるpMGX581中に、この断片をクローン化した。このプラスミドをpMGX582と名付けた。
【0145】
このプラスミドからLCが発現するかどうかについて、以下の通りに試験した。0.5mM IPTGで、pMGX506又はpMGX582(LC)を有する大腸菌株XL 10goldを誘導した。3時間後、細胞を収集し、周辺質分画を単離した。この物質を1/2及び1/10に希釈し、ヤギ抗ヒトFab(Jackson)で被覆したマイクロタイタープレートに加えた。室温で約1時間インキュベートした後、結合しなかった物質を洗浄して除き、HRP結合ヤギ抗ヒトLC(Biosource,Inc.)を用いて、結合した軽鎖(LC)を検出した。室温で1時間インキュベートした後、TMB試薬を用いてプレートを発色させ、約10分後に0.18M H2SO4を用いて発色を停止した。図6に示す結果から、大腸菌においてこのプラスミドからLCが発現することが確認された。LCが大腸菌の周辺質中に明らかに分泌されたことが特に重要であり、このことからシグナル配列が機能的であったことが示される。このプラスミドを重鎖(HC)発現プラスミドpMGX115と共トランスフェクトした後にIgGが発現することも実証された。
【0146】
大腸菌におけるFab、及び哺乳動物細胞におけるIgGの発現を評価するために。プラスミドpMGX583を構築した。pMGX583は、HCとLCの発現カセットをどちらも含み、各カセットにlacPO pelBイントロン配列が含まれている。pMGX583を構築するために、AscIで消化することにより、発現カセット全体であるCMvie lacPO pelB LC SV40pAを切り出し、これを(AscI部位がCMvieプロモーターの5’末端に導入されている以外はpMGX579と同一である)pMGX580に連結した。HEK293細胞にトランスフェクトし、その後ヤギ抗ヒトFc抗体で捕捉するELISAを行うことにより、このプラスミドからIgGが発現することを確認した。プロテインGクロマトグラフィーによってIgGを精製し、SDS−PAGE及びウェスタンブロットで分析した。HEK293細胞においてpMGX583から発現した精製IgGのクーマシーブルー染色及びウェスタンブロットを行った。還元条件下で、SDS−PAGEでタンパク質を分析した。ウェスタンブロットでは、1:5000で希釈したAP結合ヤギ抗ヒトIgG(Fab’)2(Jackson)を使用し、色素原によって発色させた。この結果を図7に示す。
【0147】
大腸菌(BL21又はXL1−blue株)におけるpMGX583によるFab発現も評価した。pMGX121由来のプラスミドがlacリプレッサー遺伝子のコピーを含まないので、プラスミドpLac1(Novagen)を用意した。pLac1はクロラムフェニコール耐性プラスミドであり、lacリプレッサータンパク質をコードし、pMGX121由来のベクターと適合するp15a複製起点を有する。pLac1とpMGX583で大腸菌を共形質転換し、アンピシリン及びクロラムフェニコールでコロニーを選択した。下記のように、形質転換体を増殖させ、1mM IPTGで誘導した。図8に示すように、周辺質抽出物由来のFabをsCD16Aによって捕捉し、HRP結合ヤギ抗ヒトF(ab’)2(Jackson)で検出した。非誘導及び誘導(IPTG、1mM)の周辺質抽出物を同量使用した。市販のヒトIgG1を対照として用いた。図2に示すように、前記の構築物に由来する精製ch3G8Fabの連続希釈物を標準物質として用いた。pMGX583プラスミドによる周辺質におけるFabの推定発現量は、培養物1ml当たり約10ngである。
【0148】
【0149】
上記の予備的研究から、哺乳動物細胞において発現又は分泌を低下させずに、原核生物の転写及び翻訳シグナルをIgG重鎖又は軽鎖構築物のシグナルイントロン内に導入することができることが実証された。
【0150】
LCの及びFdフラグメントのN末端配列を得るのに十分なFabが大腸菌から調製される。非修飾のFab、並びに還元及びアルキル化された鎖に対して質量分析を行う。哺乳動物細胞において一過性に産生されるMabに対しても、同様の分析を行う。
【0151】
細菌において周辺質へのFab分泌レベルを高めるために、細菌シグナル配列領域におけるペプチドのいくつかの改変を設計し構築した。以下に示すように、多数の位置のアミノ酸をpelB配列中に含まれるものに戻した。
【0152】
【0153】
改変シグナルペプチドが依然として好ましいスプライス切断部位を保持しているかを予測するために、SignalPプログラムによって配列を分析した。個々の改変を行い、細菌細胞と哺乳動物細胞の両方の系において試験した。pMGX102由来の元のlacZ pelBセグメント(すなわち野生型pelB)とともにLC cDNAも含むベクターにおけるHC/Fd遺伝子のイントロン内に変異が導入された。このようにして、周辺質抽出物中の分泌Fabによる抗原結合について分析することにより、分泌の増大に対する変異の寄与について評価することができる。哺乳動物での発現では、第2プラスミド上のLC遺伝子を共トランスフェクトすることができる。次いで、最初の分析の結果によって定められるように、変異が組み合わされる。
【0154】
コドン利用も、シグナルペプチドの切断の成功に影響を与えることが示されている。この可能性を利用し、分泌の改善を促進することのできるどんな予測不可能な変異をも捕捉するために、この領域中に無作為な可変性が導入された大腸菌変異体のライブラリーをスクリーニングする。これは、縮重又は付加(doped)オリゴヌクレオチドを用いて構築され、コロニーリフト法及び周辺質抽出物の高速処理スクリーニングの両方によってスクリーニングされる。エピトープタグの組み込みは、このスクリーニングに特に有用である。
【0155】
材料及び方法:
基本的なクローン化法、重複PCR法及び部位特異的変異誘発(Quick change kit、Stratagene)を用いて、ベクターを改変して新規な遺伝的構成要素を導入する。新規構築物のすべてのDNA配列を決定して、構築過程で配列の中に望ましくない変異が導入されていないことを確認する。プラスミドが大腸菌内で安定であることを確認するために、受容株はlacIq+であり、また必要であれば適合するプラスミドに、又は構築物自体にlacIを供給する。さらに、CRPによるlacプロモーターの誘導を防止するため、誘導前にプラスミドを含む細胞を栄養富化培地又はブドウ糖の存在下で増殖させる。次に発現を誘導するための以下のプロトコルを行う。単一コロニー由来の細胞をLブロス(1リットル当たりバクトトリプトン10g、酵母抽出物5g、NaCl10g)中で30℃で一晩増殖させた。一晩培養物をLBで1/100に希釈し、OD600が約0.2になるまで30℃で増殖させた。この時点で、この培養物を3つのフラスコに分け、2つをそれぞれ0.1mM又は1mM IPTG(100mMストック由来)で誘導する。その他のフラスコは、非誘導対照として使用する。誘導してから3時間後、細胞を回収し、浸透圧ショックによって周辺質分画を単離する。得られた分画について、Fabが存在するかどうかELISA及びウェスタンブロットでアッセイする。ELISAアッセイでは、Fabをヤギ抗ヒト軽鎖で捕捉し、マウス抗Fdで、その後ウサギ抗マウスHRP結合物で検出する。予備的な結果についての節で記載した、又は業者から入手した精製Fabを用いて、アッセイ用の標準曲線を作成する。
【0156】
Fabの保持機能を検出するため、捕捉抗原結合ELISAアッセイを適用した。周辺質から精製したFab又は未精製周辺質抽出物をsCD16によって捕捉し、ヤギ抗ヒトF(ab’)2 HRP結合抗体(Jackson)で検出した。市販の精製ヒトIgG1を陰性対照として使用した。
【0157】
哺乳動物細胞の発現分析:
哺乳動物細胞における発現及び分泌レベルを測定するために、LipofectAMINE2000試薬(Invitrogen)を用いたトランスフェクションにより、ヒト胚性腎293細胞(HEK293)において、表1に示した個々の構築物を一過性に発現させた。トランスフェクションの前日に、ポリDリシンで予め被覆した皿(Becton Dickinson)上に1皿(100mm)当たり細胞5×106個で細胞を播いた。各皿の細胞について、総DNA18μlをOPTIMEM I還元血清培地(Invitrogen)1.4mlで希釈した。LipofectAMINE試薬(Invitrogen)54μlをOPTIMEM I還元血清培地1.4mlで希釈し、室温で5分間インキュベートした。次いで希釈したDNA及びLipofectAMINE試薬を混合し、室温で20分間インキュベートして複合体を形成させた。このDNAとLipofectAMINE試薬の複合体を細胞に直接加えた。細胞を37℃でCO2(5%)インキュベーター内で72時間インキュベートして組換えIgGを培地に分泌させた。IgG発現レベルについて、ELISA及びウェスタンブロットで条件培地(conditional medium)をアッセイする。ELISAアッセイでは、(条件培地中の)IgGをヤギ抗ヒトFc抗体(Jackson)で捕捉し、ヤギ抗ヒトIgG(軽鎖+重鎖)HRP結合物で検出する。市販の精製ヒトIgG1を標準曲線に使用した。
【0158】
6.2.ファージディスプレイ及びスクリーニング法
大腸菌によって発現されるFab及び哺乳動物によって発現されるIgGに関与するこのベクターを利用するスクリーニング法は、本発明に包含されている。改変型pET25bを使用してFabを発現させる。この場合、LCはエピトープタグが付加されておらず、Fd鎖は、C末端HSVタグと、その後にヘキサヒスチジンタグが精製用に付加されて発現した。2種類の方法で、この配列を構築物に組み込む。第1に、目下存在するオーカーコドン(TAA)の代わりにアンバー(TAG)停止コドンを使用する。これにより、大腸菌のサプレッサー(supE)株においてリードスルー翻訳が可能となる。このような構築物はファージディスプレイに特に有用であり、この手法は以前に用いられている(Hoogenboom et al., 1991, Nucleic Acids Res. 19: 4133 4137)。アンバーコドンは、XL1 blue(supE44+)などの株で抑制されるはずであり、それによってFabのファージ粒子中への組み込みが可能となるが、Fab発現に好ましい株であるBL21(sup)では抑制されない。
【0159】
【0160】
或いは、以下に示すHindIIIなどの制限部位を挿入した後、エピトープ及び/又はアフィニティタグを付加する。
【0161】
【0162】
糸状ファージ由来の遺伝子III及びVIII:
ファージ被殻(遺伝子VIII)又は付着(遺伝子III)タンパク質コード領域との融合物は、ファージディスプレイに最も広く使用されている。その各遺伝子セグメントとのFd(VH CH1)遺伝子セグメントの融合物を構築する。この遺伝子をPCRによってfd tetファージから単離する。遺伝子III融合物では、この遺伝子のP198からS406までのセグメントを使用する。CH1と遺伝子IIIセグメントの間のHSVエピトープタグを保持しながら、遺伝子IIIセグメントが上記のベクター内のヘキサヒスチジンタグと置き換わるように、融合物を構築する。多価のFabディスプレイとするため、遺伝子VIIIのセグメントで同様の構築物を作製する。
【0163】
ファージ分析:
標準的な条件を使用して、ファージの調製及び分析を行う。ファージミドを大腸菌株XL1 Blueにおいて増殖させる。37℃で増殖させた対数増殖期の培養物に、ヘルパーファージVCSM13を感染させ、約12時間培養する。PEG/NaCl沈殿による培養上清からファージを単離し、得られたペレットをTBS15に再懸濁させる。表面上にFabが存在するかどうか、ファージの一部をELISAで分析する。さらに、結合したファージをイムノプレートから溶出させて、ファージが表面結合sCD16 Ig又はsCD32 Ig同一の調製物と結合するかどうかを決定する。活性型抗CD16 Fabを有するファージは、前者の分子と結合するが、後者と結合しないはずである。溶液中でsCD16 Igとともに予めインキュベートして、結合を遮断するために使用する。低pH溶液(グリシンHCl、pH2.2)を用い、その後中和して、プレートからのファージミドの溶出を行う。力価を決定するため、アンピシリン含有プレート上にファージミドをXL1 Blueとともに播く。
【0164】
(Fab’)2
【0165】
Fab’発現では、以下の改変について、ヒンジCH1イントロンをHCミニ遺伝子中の適当な部位に導入後試験した(配列番号28〜31)。
【0166】
【0167】
本発明は、記載した特定の実施形態による範囲に限定されるべきでなく、この実施形態は本発明の個々の態様の例示に過ぎないものとし、機能上等価な方法及び構成要素は本発明の範囲に含まれる。実際、本明細書に示し記載したものに加えて、本発明の様々な変更形態が、前記の記載及び添付した図面から当業者に明らかとなるであろう。このような変更形態は、添付した特許請求の範囲内に含まれるものとする。
【0168】
本明細書において引用したすべての文献は、すべての目的でその全体が参照により本明細書に組み込まれる。
【図面の簡単な説明】
【0169】
【図1】非還元条件での精製Ch3G8Fab(pMGX513)のクーマシーブルー染色を示す図である。レーン1:標準タンパク質(SeeBlue(登録商標)Plus Stained;Invitrogen);レーン2:ヒトIgG(対照);レーン3:ch3G8Fab。
【図2】sCD16−Igと免疫複合体との結合阻害を示す図である。1)HuIgG1、ヒトIgG1、すなわちCh4 4 20(陰性対照として);2)ChFab;3)Ch3G8(IgG1)。
【図3】ch3G8FabとsCD16Aとの結合を示す図である。
【図4】イントロン配列の設計を示す図である。Aは、哺乳動物シグナルペプチドコード配列におけるイントロン内のlacプロモーター及び細菌シグナル配列の配置を示す(配列番号6、7、及び8)。Bは、CH1ヒンジイントロン内のTAA停止コドンの配置を示す(配列番号9〜12)。
【図5】重鎖(HC)及び軽鎖(LC)発現プラスミドの構築及び名称を示す図である。
【図6】大腸菌周辺質抽出物におけるキメラLCの検出を示す図である。
【図7】pMFX583から発現した精製IgGのクーマシーブルー染色及びウェスタンブロットを示す図である。
【図8】抗CD16のELISAによって測定された、周辺質抽出物由来のpMGX583のHu3G8 FabとsCD16Aとの結合を示す図である。
【配列表】
【技術分野】
【0001】
本発明は、細菌におけるFabフラグメントの発現及び分泌、並びに哺乳動物細胞における対応する全長IgGの発現及び分泌のための二重発現ベクター及びその使用方法を提供する。このベクターは、目的のポリペプチド、例えばIgG重鎖又は軽鎖の調節配列及びコード配列を含み、細菌プロモーター及びシグナル配列が、ポリペプチド、例えばIgG重鎖又は軽鎖遺伝子のシグナル配列内に位置する第1イントロン内に含まれ、目的のタンパク質が、例えばIg重鎖が複数のイントロンを有する場合、細菌停止コドンが、第2イントロン、例えば重鎖遺伝子のCH1ドメインとヒンジ領域の間のイントロン内に含まれる。このベクターは、哺乳動物プロモーター、細菌細胞と哺乳動物細胞の両方の複製起点、及び場合によっては、1つ又は複数の選択マーカーも含む。したがって、細菌において発現させた場合、細菌プロモーターから転写し、第2イントロン内の停止コドンにおいて終結することによって、ポリペプチドフラグメント、例えばFabフラグメントが発現する。これに対し、哺乳動物細胞においては、スプライシングによりイントロン内に位置する細菌調節配列が除去され、哺乳動物シグナル配列が生成され、これにより全長ポリペプチド、例えばIgG重鎖又は軽鎖ポリペプチドが発現する。この二重ベクター発現系は、新規なモノクローナル抗体を選択及びスクリーニングし、並びに目的の抗原分子に対する結合についてモノクローナル抗体を最適化するために用いることができる。この系を用いると、大腸菌(E. coli)でFab(又はscFv)を発現させることにより、最初のスクリーニング又は選択のステップが実現され、機能試験用に所望のアイソタイプの二価IgG分子として、得られたFab結合分子を容易に発現させることができる。
【背景技術】
【0002】
組換え発現系は、現在の抗体工学技術開発の鍵となっている。哺乳動物細胞におけるIgM又はIgGのクローン化された軽鎖及び重鎖遺伝子の共発現が示されると、ヒトの定常領域を含んでいるキメラMabが迅速に生成され、試験された(Ochi et al., 1983、Proc. Natl. Acad. Sci. U.S.A. 80:6351-6355;Oi et al., 1983、Proc. Natl. Acad. Sci. U.S.A. 80:825-829;Morrison et al., 1984、Proc. Natl. Acad. Sci. U.S.A. 81(21):6851-5)。続いて、アビディティを低下させずにヒトの配列をマウスイムノグロブリン可変領域に導入して、ヒト被験者内での潜在的な免疫原性が非常に低い抗体を得る方法が開発された(Jones et al., 1986、Nature 321:522-525;Queen et al., 1989、Proc. Natl. Acad. Sci. U.S.A. 86:10029-10033)。ヒトでの試験用及び最終的な販売用の高度に精製された物質を調製するため、CHO細胞又はマウス骨髄腫細胞内でこのような組換え抗体を大量に発現させる再現可能な方法が開発された。癌治療用のリツキサン及びハーセプチン、RSV感染防止用のシナジス、慢性関節リウマチ治療用のレミケード、並びに移植片拒絶防止用のゼナパックスを含めて(Reff et al., 1994、Blood 83:435-445;Carter et al., 1992、Proc. Natl. Acad. Sci. U.S.A. 89(10):4285-9;Johnson et al., 1997、J. Infect. Dis. 176:1215-1224;Queen et al., 1989、Proc. Natl. Acad. Sci. U.S.A. 86:10029-10033;表1を参照のこと)、現在多数のこのようなMabがヒトでの使用に承認され市販されている。
【0003】
同様に、Fv、一本鎖Fv、又はFab分子を細菌の系でうまく発現させることができたことが実証されると、この発現技術を利用して、VH及びVL配列の多様なライブラリーを利用する方法が急速に開発された(Skerra et al., 1988、Science 240:1038-1041;Bird et al., 1988、Science 242:423-426;Huston et al., 1988、Proc. Natl. Acad. Sci. U.S.A. 85:5879-5883)。VL及びVH配列のコンビナトリアルライブラリーは、最初にバクテリオファージλから発現させ、プラークリフトアッセイを用いて特定の組合せと抗原との結合についてスクリーニングしていた(Huse et al., 1989、Science 246:1275-81;Huse et al., 1992、Biotechnology 24:517-523)。糸状バクテリオファージの表面上にscFv又はFabのどちらか一方を固定することにより、パニング技術を用いて、そのゲノム内に結合領域の遺伝子を含むファージと結合するかどうかで選択することが可能になった(McCafferty et al., 1990、Nature 348:552-554;Hoogenboom et al., 1991、19: 4133-4137;Bird et al., 1988、Science 242:423-426;Kang et al., 1991、Proc. Natl. Acad. Sci. U.S.A. 88:4363-4366)。スクリーニングではなく選択することができることにより、まれな結合物を同定し単離するために開発された、個々のメンバーが109以上という大きなライブラリーに含まれる莫大な多様性が可能となった。したがって、現在大きく多様なバクテリオファージライブラリーから、ほとんどどんなタンパク質抗原でも適度なアフィニティで結合する抗体フラグメントを単離することが可能である。CDRの突然変異誘発を何ラウンドも反復実施することによって抗体フラグメントのアフィニティを改善し、抗原との改善された結合についてスクリーニング又は選択を行う方法も開発されている(Schier et al., 1996、J. Mol. Biol. 263: 551-567;Wu et al., 1998、Proc. Natl. Acad. Sci. U.S.A. 95:6037-6042)。
【0004】
Fab又はscFv発現系を用いて新規な特異性を有するものの同定及びアフィニティ成熟を行うことにおける大きな進歩があったにも関わらず、全長IgG分子はいくつかの利点を提供する。IgGの重要な特徴の1つは、その二価構造である。抗原との結合におけるIgGの2本のFab腕間の共同性により、一価のFabと比べて二価IgGのアビディティがより高くなる。抗原もまた多価である又は表面に結合している場合には、個々のFab腕のアフィニティとIgGのアビディティとの差異の程度は最も顕著である。抗原がより高い密度で存在している場合には、共同性の総計はより顕著となるが、Fab腕が高アフィニティのMabでは顕著ではなくなる。実際的な表現では、このことは、特定の抗原密度の閾値を超えると、アフィニティは高いが共同性が低いMabが、アフィニティは中程度であるが共同性が高いFab腕を有するMabと同じアビディティを有することを意味する。この後者のMabは、前者のMabと比べて抗原の密度が高い領域に対して選択的であるはずである。どちらかのMabが有利であるかという場合を想像することができる。例えば、真の癌抗原、すなわち腫瘍細胞上にしか発現していない抗原は極めて少数である。大部分は、より高密度で腫瘍細胞上に発現しているが、同様に他の細胞型にも発現している。したがって、アビディティは高いがアフィニティが中程度であるMabは、より低密度で抗原を発現している正常細胞より腫瘍細胞に対して選択的である可能性がある。同様に、ウイルス感染過程において、抗原は、ウイルス上に存在し、ウイルス感染細胞上に存在し、遊離型で分泌されている可能性がある。抗原密度がより高い領域に選択的な中和Mabは、遊離抗原、又は抗原密度が低い他の領域ではなくウイルス及び感染細胞を標的とすることができ、したがって高アフィニティMabと比べて同等又はより高い効力を有する可能性がある。Fab又はscFv単量体と連結する様々な手法を用いて、アビディティがより高いフラグメントを選択する方法が開発されている(Hudson et al., 1999、J Immunol. Methods 231:177-189)。この構築物は有用であるが、Fcを用いてFab腕を連結することによってもたらされたアビディティを正確に複製できない可能性がある。さらに、最初に特定の結合物を同定し、次いで共同性がより高いものを同定することが望まれることがある。例えば、上記の例で、ウイルスを中和するかどうかでスクリーニングしたいが、大部分の一価Fabがほとんど活性がないことが判明することがある。全長IgGに転換することにより、アビディティの増大に起因して中和活性を有するかどうかで選択を行うことが可能となる。
【0005】
他の場合では、エフェクター機能が、結合分子の最適な効力を得るのに必要となる可能性がある。イムノグロブリン分子のFc部分と特定の細胞表面受容体との相互作用により、抗原結合とエフェクター細胞機能の組合せが可能になる。
【0006】
ヒト及びげっ歯類に存在するIgGのFc受容体は3クラスあり、それらはRI、RII及びRIIIと名付けられている(Ravetch and Bolland、2001、Annu. Rev. Immunol. 19: 275 290)。RIは、単球及びマクロファージ上に存在し、単量体IgGと高いアフィニティで結合する。RIIは、B細胞、血小板、顆粒球、マクロファージ及び単球を含めた多種多様な細胞上に存在し、中程度のアフィニティで多量体IgG(免疫複合体の又は凝集したIgG)と結合する。2種の形態のRIIが発現し、これらは、受容体の細胞内部分に活性化型(ITAM)RIIaドメイン又は抑制型(ITIM)RIIbドメインが存在するかどうかによって異なる。所与の細胞上の活性化型及び抑制型受容体の相対レベルによって、免疫複合体に対する反応が決まる。B細胞は抑制型のみを発現している。RIIIは、RIIと同様に、中程度のアフィニティで多量体IgG(免疫複合体の又は凝集したIgG)と結合する。RIIIにも2種の形態が存在する。結合したγ鎖上のITAMドメインは、RIによるシグナル伝達も媒介し、RIIIa及びFcE受容体によるシグナル伝達も媒介する。シグナル伝達分子RIIIaは、NK細胞、単球、マクロファージ、及び特定のT細胞上でITAMを含むγ鎖と結合する。NK細胞上では、RIIIaによるシグナル伝達にTCRζ鎖も関与する。RIIIbは、非シグナル伝達型であり、GPI結合分子として(ヒト)好中球上で発現している。
【0007】
体内で、RI部位は、一般に単量体IgGによって占有されているが、RII及びRIII受容体は、占有されておらず、免疫複合体との相互作用に使用可能である。抗体抗原複合体によって活性化型Fc受容体が架橋されると、病原体を貪食し、直接の細胞傷害性によって外来細胞及び形質転換細胞を死滅させ、毒性物質を排除し、炎症反応を開始することができる。さらに、Fcは補体成分と相互作用する部位を含んでいる(Tao et al., 1993、J Exp Med 178:661-667)。最後に、Fcは、MHC関連FcRn受容体との特異的な相互作用によって、in vivoでIgGの半減期を長くする役割を果たしている(Ghetie and Ward、2002、Immunol Res. 25:97-113)。
【0008】
明らかに、標的が細菌又は癌細胞の場合、結合するだけでなく排除し又は死滅させる作用因子を試験すると有利であるはずである。その場合、IgGは、試験するのに好ましい分子であるりうる。例えば、キメラ抗CD20 Mabであるリツキサンは、それがヒトB細胞に対する強いADCC活性を有することに基づいて選択された(Reff et al., 1994、Blood 83:435-445)。さらに、抗HER2抗体であるハーセプチンは、腫瘍細胞上のEGF様受容体と結合し、それを介するシグナル伝達を遮断するが、最近の研究から、腫瘍の防御が主としてFcを媒介するものであることが示唆されている(Clynes et al., 2000、Nat. Med. 6:443-446)。
【0009】
scFv又はFabの系を用いた抗体の発現、選択及び改良についての関心は高い。しかし、現在使用可能な技術を用いても、さらなる試験及び開発のために、得られたscFv又はFabフラグメントを、糖鎖付加した全長Mabを発現させるためのベクター中に再クローン化しなければならない。このステップにより、この段階で試験することができるMabの数が厳格に制限される。したがって、その技術におけるこのような関心があるにも関わらず、ヒト治療薬として有用な全長Mab分子を選択し改良する有効な系は、現在まで開発されていない。
【0010】
本明細書における文献の引用又は考察を、そのようなものが本発明に対する従来技術であると認めるものと解釈すべきでない。
【発明の開示】
【0011】
本発明は、最適化されたモノクローナル抗体の選択、スクリーニング及び発現のためのベクター系、及びその使用方法を提供する。このベクター系を用いて、細菌内、好ましくは大腸菌内でFabフラグメントを、真核細胞内、好ましくは哺乳動物細胞内で対応する全長抗体(例えば、IgG)を発現させ選択することができる。この系を用いると、最初の結合分子のスクリーニング及び/又は選択を、Fab又はscFvを発現した大腸菌を用いて実現することができ、得られた結合分子を、機能試験用に所望のアイソタイプの二価抗体として迅速に発現させることができる。
【0012】
本発明は、細菌において細胞膜周辺腔へと抗体フラグメントを発現させ分泌し、哺乳動物細胞において全長IgGを発現させ分泌することができる二重発現ベクター系の出願人による発見及び開発に部分的に基づいている。この新規なベクターでは、細菌内でFabフラグメントを発現させ分泌するのに必要な調節エレメントは、哺乳動物細胞におけるIgG重鎖及び軽鎖RNA転写物の適当なプロセシングにも、IgG重鎖及び軽鎖ポリペプチドを分泌するのにも必要な配列エレメントと重複する。具体的には、細菌プロモーター及びシグナル配列は、哺乳動物IgG重鎖又は軽鎖遺伝子のシグナル配列をコードする配列内に位置するイントロン内に含まれ、細菌停止コドンは、重鎖遺伝子のCH1とヒンジ領域の間にある他のイントロンに(又は、別の実施形態ではヒンジ領域とCH2ドメインの間に位置するイントロンに)含まれる。したがって、細菌において発現させた場合、細菌プロモーターから転写し、第2イントロン内の停止コドンにおいて終結することによって、細菌の細胞膜周辺腔でFabフラグメントが発現する。これに対し哺乳動物細胞においては、スプライシングにより細菌プロモーター及びシグナル配列が除去され、哺乳動物シグナル配列が再生成され、これにより全長IgG重鎖又は軽鎖ポリペプチドが発現する。このベクターの重要な特徴は、細菌の及び哺乳動物の調節配列エレメント、すなわち哺乳動物シグナル配列及びスプライス受容部位、並びに細菌プロモーター及びシグナル配列が、この配列エレメント4種すべての機能が維持されるように、構造的にかつ機能的に重複していることである。したがって、このベクターの構築において、この重複した領域内でどんな変化が生じても、この配列エレメント4種の機能が維持されることが重要である。
【0013】
この改良系は、治療薬として使用するのに最良の機能性Mabの同定を促進し合理化する。細菌細胞において発現させた場合、細菌プロモーターは、Fab(又はscFv)の発現を制御し、それによって細菌細胞において抗原と結合するかどうかで選択しスクリーニングすることが可能になる。しかし、哺乳動物細胞において発現させた場合、スプライシングにより哺乳動物シグナル配列内のイントロン、及び重鎖のCH1とヒンジ領域との間のイントロンが除去され、したがって細菌プロモーター、細菌シグナル配列、及び停止コドンが除去され、哺乳動物シグナル配列が再構築される。哺乳動物プロモーター、例えばCMVプロモーターは、哺乳動物シグナル配列の開始部位の5’に位置し、IgG分子の重鎖及び軽鎖をコードするヌクレオチド配列の転写を誘導し、それによって全長の重鎖又は軽鎖IgG分子が哺乳動物細胞内で発現する。
【0014】
一実施形態では、本発明は、哺乳動物細胞においてIgG重鎖又は軽鎖を、細菌において重鎖又は軽鎖のFabフラグメント部分を発現させるためのベクターを包含し、前記ベクターは、(a)細菌性複製起点、(b)哺乳動物複製起点、及び(c)細胞内発現のための哺乳動物プロモーターを含み、そして前記重鎖又は前記軽鎖をコードするヌクレオチド配列と機能的に結合しており、前記ヌクレオチド配列は、(i)第1イントロンを含む哺乳動物シグナル配列であって、第1イントロンが、細菌細胞において細菌プロモーター及びシグナル配列が前記重鎖又は前記軽鎖のFabフラグメントの細胞膜周辺腔への発現及び分泌を誘導し、哺乳動物細胞において前記哺乳動物プロモーター及びシグナル配列が前記重鎖又は前記軽鎖の発現及び分泌を誘導するように、前記重鎖又は前記軽鎖のFabドメインをコードする配列と機能的に結合した細菌プロモーター及び細菌シグナル配列を含むものである哺乳動物シグナル配列と、(ii)前記ベクターが前記重鎖をコードし、第2イントロンが前記重鎖配列のCH1とヒンジ領域の間に含まれる場合、細菌における翻訳が、前記ヒンジ領域の配列の後で終わるように、好ましくはイントロンの5’末端に近接して停止コドンを含む第2イントロンとを含む。他の実施形態では、本発明は、細菌プロモーターがlacPO配列を含む、上記に記載のベクターを提供する。特定の実施形態では、本発明は、細菌シグナル配列がpelBシグナル配列であるそのようなベクターを提供する。他の特定の実施形態では、本発明は、エピトープタグ又はアフィニティ標識をコードする配列を含むように、軽鎖配列を遺伝子改変した、上記に記載のベクターを提供する。他の実施形態では、本発明は、エピトープタグがFd鎖のC末端にあるHSVタグである、上記に記載のベクターを提供する。他の実施形態では、このベクターのアフィニティタグは、Fd鎖のC末端にあるヘキサヒスチジンタグである。
【0015】
他の特定の実施形態では、このベクターは、重鎖及び軽鎖の両方をコードする配列を含み、それぞれが哺乳動物及び細菌のプロモーター及びシグナル配列と機能的に連結している。他の特定の実施形態では、重鎖又は軽鎖は、キメラ重鎖又は軽鎖である。他の特定の実施形態では、重鎖又は軽鎖配列は、ヒト又はヒト化重鎖又は軽鎖配列である。
【0016】
他の実施形態では、本発明は、上記に記載のベクターを含む細菌細胞を提供する。特定の実施形態では、その細菌細胞は、大腸菌細胞である。
【0017】
他の特定の実施形態では、本発明は、上記に記載のベクターを含む哺乳動物細胞を提供する。特定の実施形態では、その哺乳動物細胞はヒト又はネズミ細胞であり、好ましくは、骨髄腫細胞、CHO細胞、HEK細胞、NSO細胞、NS1細胞、BHK細胞、COS細胞、293細胞、又は3T3細胞である。
【0018】
他の特定の実施形態では、本発明は、重鎖及び軽鎖の両方を発現する、上記に記載のベクターを含む細胞を提供する。本発明のこの態様の他の特定の実施形態では、重鎖及び軽鎖は、同じ細胞内で異なるベクターから発現し、その少なくとも1つ(好ましくは両方)は、上記に記載のベクターである。
【0019】
他の実施形態では、本発明は、細菌細胞においてベクターを発現させた場合にFd遺伝子VIII又はFd遺伝子III融合物が産生されるように、糸状ファージ遺伝子VIII又は遺伝子IIIタンパク質のコード領域をコードする配列と機能的に連結した(好ましくは融合した)、IgG重鎖又は軽鎖のFd(VH CH1)セグメントをコードするヌクレオチド配列を含む、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクターを提供する。他の実施形態では、このベクターは、相補的な重鎖遺伝子又は軽鎖遺伝子をコードするヌクレオチド配列を含み、これはファージ配列と機能的に連結していない。
【0020】
本発明の他の態様では、治療薬として使用するMabを同定する方法が提供される。この方法は、(a)対照IgGをコードする、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクターを含む対照細胞を用意するステップと、(b)対照IgGと比べて遺伝子改変されたIgGをコードする、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクターをそれぞれ発現する試験細胞のライブラリーと抗原とを接触させるステップと、(c)対照細胞の周辺質抽出物と前記抗原の結合アフィニティと比較して試験細胞の周辺質抽出物と前記抗原の結合アフィニティを測定するステップとを含む。この方法の特定の実施形態では、この方法はさらに、ステップ(c)の後に、(d)哺乳動物細胞においてステップ(c)の試験細胞から単離したベクターを発現させるステップと、(e)その哺乳動物細胞と前記抗原とを接触させるステップと、(f)対照IgGの結合アフィニティと比較して、その哺乳動物細胞内で発現した遺伝子改変IgGの結合アフィニティを測定するステップとを含み、試験細胞の周辺質抽出物と前記抗原の結合アフィニティが対照細胞の周辺質抽出物と前記抗原の結合アフィニティより大きい場合に治療薬として有用なMabを発現している細胞が同定される。
【0021】
本発明の他の態様では、治療薬として使用するMabを同定するファージディスプレイスクリーニング法が提供される。この方法は、(a)fd−糸状ファージ−遺伝子IIIの融合物又はfd−糸状ファージ−遺伝子VIIIの融合物をコードする対照ファージを発現する細菌細胞を用意するステップと、(b)対照ファージと比べて改変された、軽鎖及びfd(VH CH1)遺伝子III融合物又はfd(VH CH1)遺伝子VIII融合物をコードする試験ファージを産生する複数の試験細胞を含むファージライブラリーのメンバーと抗原とを接触させるステップと、(c)対照ファージと前記抗原の結合アフィニティと比較して試験ファージと前記抗原の結合アフィニティを測定するステップとを含み、試験ファージと前記抗原の結合アフィニティが対照ファージと前記抗原の結合アフィニティより大きい場合に治療薬として有用なMabを発現している細胞が同定される。この方法の特定の実施形態では、この方法はさらに、ステップ(c)の後に、(d)哺乳動物細胞においてステップ(c)の試験細胞から単離したベクターを発現させるステップと、(e)その哺乳動物細胞と前記抗原とを接触させるステップと、(f)対照IgGの結合アフィニティと比較して、その哺乳動物細胞内で発現した遺伝子改変IgGの結合アフィニティを測定するステップとを含む。
【0022】
本発明はまた、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクターを含む、Fabポリペプチドを発現する複数の細菌細胞を含む組成物をも提供する。
【0023】
さらに本発明はまた、哺乳動物細胞においてIgGを、大腸菌においてFabフラグメントを発現させるためのベクター、及びFabポリペプチドを発現する糸状ファージを含む、複数の細菌細胞を含む組成物をも提供する。特定の実施形態では、その細菌細胞は大腸菌細胞であり、その糸状ファージはfdファージである。
【0024】
他の実施形態では、本発明はさらに、複数の標的に対する受動治療薬の迅速な開発に特に有用なMabカクテルの作製を包含する。本明細書に記載のベクターを用いて、臨床的に重要なMabを単離するために、未感作の及び免疫感作されたヒト被験体に由来するFab発現大腸菌及びファージのライブラリーを作製することができる。
【0025】
さらに、細菌においてFabフラグメントを、哺乳動物細胞において全長IgGを発現させるためのベクターを設計する際に使用する原理を適用して、細菌において特定のタンパク質の一部を、哺乳動物細胞において全長タンパク質を発現させるためのベクターを作製することができる。例えば、本発明は、哺乳動物細胞において分泌型又は膜結合型ポリペプチドを、細菌において前記ポリペプチドの可溶性フラグメントを発現させるためのベクターを提供し、前記ベクターは、(a)細菌性複製起点、(b)哺乳動物複製起点、及び(c)前記分泌型又は膜結合型ポリペプチドをコードするヌクレオチド配列と機能的に結合した哺乳動物プロモーターを含み、前記ヌクレオチド配列は、イントロンを少なくとも1つ含む哺乳動物シグナル配列を含むものであり、前記イントロンは、前記細菌プロモーター及び細菌シグナル配列が細菌細胞において前記ポリペプチドの前記可溶性ドメインの細胞膜周辺腔への発現及び分泌を誘導し、及び前記哺乳動物プロモーター前記哺乳動物シグナル配列が哺乳動物細胞において前記ポリペプチドの発現及び分泌を誘導するように、前記ポリペプチドの前記可溶性ドメインをコードする配列と機能的に結合した細菌プロモーター及び細菌シグナル配列を含み、前記哺乳動物プロモーターは、前記ポリペプチドの前記可溶性ドメインをコードする前記ヌクレオチド配列と機能的に結合している。
【0026】
本明細書において、「抗体」(antibody及びantibodies)という用語は、モノクローナル抗体、ヒト化抗体、キメラ抗体、一本鎖Fv(scFv)、一本鎖抗体、Fabフラグメント、F(ab’)フラグメント、ジスルフィド結合Fv(sdFv)、及び抗イディオタイプ(抗Id)抗体(例えば、対象とする抗体に対する抗Id抗体を含む)、並びに上記のいずれかのエピトープ結合性フラグメントを指す。具体的には、抗体には、イムノグロブリン分子、及びイムノグロブリン分子の免疫活性フラグメント、すなわち抗原結合部位を含む分子が含まれる。イムノグロブリン分子は、どのタイプ(例えば、IgG、IgE、IgM、IgD、IgA及びIgY)でもよく、どのクラス(例えば、IgG1、IgG2、IgG3、IgG4、IgA1及びIgA2)でもよく、どのサブクラスでもよい。
【0027】
本明細書において、「二重発現ベクター系」という用語は、真核細胞において、好ましくは哺乳動物細胞において対象とするポリペプチドを発現させ、細菌細胞において細胞膜周辺腔へとそのポリペプチドフラグメントを発現させるベクター系を指す。好ましい実施形態では、対象とするポリペプチドは抗体鎖であり、そのフラグメント又はドメインは、Fabフラグメント又はscFvフラグメントである。最も好ましくは、対象とするポリペプチドは、IgGの重鎖又は軽鎖、及びIgGのFabフラグメントである。したがって、本明細書においてこの用語は、「抗体二重発現ベクター系」及び「IgG二重発現ベクター系」という用語も指している。
【0028】
本明細書において、「二重発現ベクターカセット」又は「二重発現ベクターポリヌクレオチドカセット」という用語は、本明細書で同義に用いられ、対象とするポリヌクレオチドのコード配列、並びに真核細胞において、好ましくは哺乳動物細胞において対象とするポリペプチドの発現及び分泌に、細菌細胞において対象とするポリペプチドのフラグメント又はドメインの細胞膜周辺腔への発現及び分泌に必要な調節配列を含むポリヌクレオチドを指す。このような調節配列は、真核生物のシグナル配列内に、好ましくは哺乳動物のシグナル配列内にイントロンを含み、この配列は、真核細胞において、好ましくは哺乳動物細胞において対象とするポリペプチドの発現及び分泌を、細菌細胞において(対象とするポリペプチドをコードする配列の第2イントロン内に停止コドンを置くことで決定する)対象とするポリペプチドのフラグメント又はドメインの細胞膜周辺腔への発現及び分泌を可能にするように、本明細書で詳細に説明するように特定の形で位置する細菌プロモーター及び細菌シグナル配列をイントロン内に含む。「抗体発現ベクターポリヌクレオチドカセット」という用語は、本発明の好ましい実施形態を指し、この場合、対象とするポリペプチドは抗体鎖であり、そのフラグメント又はドメインは、Fabフラグメント又はscFvフラグメントである。同様に、「IgG発現ベクターポリヌクレオチドカセット」という用語は、特定の実施形態を指し、この場合、対象とするポリペプチドは、IgGの重鎖又は軽鎖、及びIgGのFabフラグメントである。
【0029】
本発明のこれらの態様及び他の態様は、添付の図面及び以下の詳細な説明の項を参照することによってより理解されるであろう。
【発明を実施するための最良の形態】
【0030】
本発明は、細菌細胞、例えば大腸菌細胞においてFabフラグメントを発現させ分泌し、真核細胞、好ましくは哺乳動物細胞において、しかし例えば昆虫細胞又は鳥類細胞においてもIgG重鎖又は軽鎖ポリペプチドを発現させるための新規な二重発現ベクター系、並びに特定の結合性を有するモノクローナル抗体のスクリーニング及び最適化にそれを使用する方法に関する。上記で論じたように、本発明は、細菌において細胞膜周辺腔へと抗体フラグメントを発現させ分泌し、哺乳動物細胞において全長抗体を発現させ分泌することができる二重発現ベクター系の出願人による開発に部分的に基づいている。この新規なベクターでは、細菌プロモーター及びシグナル配列は、哺乳動物IgG重鎖又は軽鎖遺伝子のシグナル配列内に位置するイントロン内に含まれ、細菌停止コドンは、重鎖遺伝子のCH1とヒンジ領域の間にある他のイントロンに含まれる。したがって、細菌において発現させた場合、細菌プロモーターから転写し、第2イントロン内の停止コドンにおいて終結することによってFabフラグメントが発現する。これに対し哺乳動物細胞においては、スプライシングにより細菌性制御エレメントが除去され、哺乳動物シグナル配列が生成され、これにより全長IgG重鎖又は軽鎖ポリペプチドが発現する。したがって、この二重の目的のベクターは、細菌性の及び哺乳動物の調節配列エレメント、すなわち哺乳動物シグナル配列及びスプライス受容部位、並びに細菌プロモーター及びシグナル配列の構造及び機能を維持するように設計する。
【0031】
この新規な二重発現ベクター系の構築及び使用に関する組成物及び方法について以下に説明する。具体的には、第5.1節で、真核細胞において対象とするポリペプチドが、細菌においてそのフラグメントが二重に発現するように設計されたDNAカセット、二重発現ベクターポリヌクレオチドカセットを含むベクター、このようなカセット及びベクターを含む宿主細胞、並びにこのようなカセット、ベクター、及び宿主細胞を含むキットを含めた本発明の組成物について説明する。第5.2節及び第5.3節で、新規な抗体の同定にこの新規なベクター配列を使用する方法、真核細胞の系と細菌の系の両方において、最適化されたモノクローナル抗体を選択しスクリーニングする方法、並びにこの原理を適用して、対象とする任意の膜結合型又は分泌型タンパク質用の二重発現ベクター系をも構築する一般化した方法を含めた、本発明の使用方法について説明する。
【0032】
5.1.二重発現ベクター
上記で概略を述べた配列及び機能エレメントを有する二重発現ベクター系は、当技術分野で使用可能な様々な技術を用いて構築することができる。好ましい実施形態では、ベクターは、
(1)哺乳動物プロモーター、
(2)IgG重鎖又は軽鎖をコードするヌクレオチド配列であって、(a)以下の(i)及び(ii)を含む哺乳動物シグナル配列を含む上記ヌクレオチド配列:
(i)細菌プロモーター及びシグナル配列が細菌細胞において前記重鎖又は前記軽鎖のFabドメインの細胞膜周辺腔への発現及び分泌を誘導し、前記哺乳動物プロモーター及びシグナル配列が哺乳動物細胞において前記重鎖又は前記軽鎖の発現及び分泌を誘導するように、前記細菌プロモーター及びシグナル配列が第1スプライス受容部位と重複し、IgG重鎖又は軽鎖コード配列と機能的に連結するように前記細菌プロモーター及びシグナル配列を含む第1イントロンと、(ii)前記ベクターが前記重鎖をコードする場合には、CH1をコードする配列と重鎖遺伝子のヒンジ領域との間にあり、細菌停止コドンを含む第2イントロン、
(3)細菌性複製起点、並びに
(4)哺乳動物複製起点
を含む。前記の配列成分に加えて、ベクターは、細菌細胞においてベクターをクローン化し増殖させ、細菌細胞と真核細胞の両方でベクターを有する細胞を増殖させ選択するための選択マーカー、他のヌクレオチド配列を付加するためのマルチクローニング部位配列、並びに対象とする他の配列をさらに含むことができる。この配列エレメントを、本明細書において以下に詳細に説明する。
【0033】
5.1.1.二重発現ベクターカセット配列
対象とするポリペプチド、例えばIgG重鎖又は軽鎖をコードするヌクレオチド配列は、第1イントロンを含む哺乳動物シグナル配列、及びその配列が重鎖をコードする場合は第2イントロンを含むCH1からヒンジまでの領域を用いて設計する。第1及び第2イントロンは、細菌細胞においてポリペプチドの発現及び分泌を誘導し、二重発現ベクターが哺乳動物細胞において発現する場合、スプライシングによって除去される細菌調節配列を含むように設計する。二重発現ベクターの具体的な配列組成及び構造について、本明細書において詳細に説明する。
【0034】
第1イントロンは、対象とするポリペプチドの哺乳動物シグナル配列内に位置するように設計する。哺乳動物シグナル配列が天然にイントロンを有さない場合、第1イントロンは、当技術分野で知られている任意の組換えDNA法を用いて構築する。第1イントロンは、スプライス受容部位と重複する細菌プロモーター及びシグナル配列を含む。細菌プロモーター及びシグナル配列は、ポリペプチド配列と、例えばIgG重鎖配列又は軽鎖配列と「機能的に連結する」ように構築する。すなわち、細菌細胞においてFabフラグメント又はscFv配列の転写を誘導するように、細菌プロモーターを配置し、細菌細胞において細胞膜周辺腔へとポリペプチドが分泌されるようにスプライス受容部位と重複するように、細菌シグナル配列を配置する。
【0035】
細菌プロモーター、細菌シグナル配列、哺乳動物シグナル配列及びスプライス受容部位の機能を維持するため、第1イントロンのヌクレオチド配列は、当技術分野で周知であり、第6節で提示する実施例において示すプロモーターコンセンサス配列、シグナル配列コンセンサス配列及びスプライス部位コンセンサス配列を用いて設計することができる。例えば、対象とするポリペプチド、例えばIgGなどの抗体を標的として細菌の周辺質膜へと誘導するいずれのシグナル配列も使用することができる。細菌シグナル配列は、由来が天然のものでもよく、合成のものでもよい。天然に周辺質に存在することが定められているタンパク質と結合しているリーダー配列は、例えば、外来タンパク質の周辺質への分泌を誘導することが知られている(MacIntyre et al., 1990、Mol. Gen. Genet. 221:466-474)。好ましい実施形態では、シグナル配列は、pelB配列及びOmpAタンパク質リーダー配列(Hobom et al., 1995、Dev. Biol. Stand. 84:255-262)をコードする。それだけに限らないが、大腸菌PhoA(Oka et al., 1985、Proc. Natl. Acad. Sci 82:7212-16)、OmpT(Johnson et al., 1996、Protein Expression 7:104-113)、LamB及びOmpF(Hoffman & Wright、1985、Proc. Natl. Acad. Sci. USA 82:5107-5111)、β−ラクタマーゼ(Kadonaga et al., 1984、J. Biol. Chem. 259:2149-54)、エンテロトキシン(Morioka-Fujimoto et al., 1991、J. Biol. Chem. 266:1728-32)、黄色ブドウ球菌(Staphylococcus aureus)のプロテインA(Abrahmsen et al., 1986、Nucleic Acids Res. 14:7487-7500)、枯草菌(B. subtilis)のエンドグルカナーゼ(Lo et al., Appl. Environ. Microbiol. 54:2287-2292)由来のリーダー配列、並びに人工及び合成シグナル配列(MacIntyre et al., 1990、Mol. Gen. Genet. 221:466-74;Kaiser et al., 1987、Science、235:312-317)を含めた他のシグナル配列も考えられる。
【0036】
Fabフラグメント又はscFvの細菌の周辺質への分泌は、最初のベクターを作製した後にベクターの配列を変更することによって改善することができる。例えば、シグナル配列内に可変性を導入した一本鎖fv(scFv)のファージディスプレイを用いて、分泌が改善した変異体を選択することができる。さらに、他のシグナルペプチドのコード配列を改変し試験することもできる。この場合、シグナルペプチドのコード配列のデータベースを作成して所望のスプライス部位と比較することができる。次いで、最も相同なセグメントを必要なら改変して、大腸菌においてFabを分泌させ、HEK293細胞においてMabの発現及び分泌を保持することができる。
【0037】
すべての配列エレメントの機能、すなわち、細菌と哺乳動物の両方の宿主での転写、哺乳動物宿主での転写物のスプライシング、細菌での周辺質への分泌及び哺乳動物宿主での分泌が維持される条件で、個々のエレメントの機能を高めるのに適した変化を加えることにより、この領域内の個々の配列成分を最適化することができる。
【0038】
改変した原核細胞シグナルペプチドが依然として好ましいスプライス切断部位を保持しているかどうかを予測するために、神経ネットワークアルゴリズムを用いるシグナルP(SignalP)プログラムによって配列を分析することができる(Nielsen et al., 1997、Int. J. Neural Sys. 8、581 599)。バークレーショウジョウバエゲノムプロジェクト(Berkeley Drosophila Genome Project)ウェブサイトのスプライス部位予測(Splice Site Prediction)プログラムを用いて、スプライス部位の潜在的機能を評価することができる(Reese et al., J. Comput. Biol.、1997、4(3):311 23を参照のこと)。このプログラムも、ヒト遺伝子向けの神経ネットワークアルゴリズムを用いている。
【0039】
精製又はスクリーニングのプロトコルの間に、エピトープ/アフィニティタグを付加して、ポリペプチド、例えばFabフラグメント、scFv、或いは軽鎖又は重鎖の精製又は同定を改善することが望ましいこともある。確実に分泌後にタンパク質上にタグをさらすために、GGGGSなどの柔軟なリンカー配列を、機能ドメインとエピトープ/アフィニティタグ配列の間に導入する。ペプチドタグには、タグ付きのポリペプチド又はフラグメントの同定を容易にするが、タグ付きのポリペプチド又はフラグメントの機能を阻害し又はそれに干渉する可能性が低い方法/試薬が存在するタグが含まれ得る。タグは、対応する結合用試薬との結合が可能であるが、mRNAと結合しているタグ付きタンパク質を干渉しない任意の長さでよい。好ましい実施形態では、タグの長さは、約8、10、12、15、18又は20アミノ酸であり、15、20、25、30、40又は50アミノ酸以下であるが、100、150、200、300、400アミノ酸又は500アミノ酸以上でもよい。タグは、(1)対象とする細胞、又は(2)対象とするポリソーム調製物、又は(3)タグと結合する試薬と接触している、対象とするいずれの細胞分画、のどんな成分とも結合しない試薬と特異的に結合することができる。分子タグには、一例としてであり限定するものではないが、プロテインAフラグメント、mycエピトープ(Evan et al., Mol. Cell Biol. 5(12):3610-3616)、Btag(Wang et al., 1996、Gene 169(1): 53-58)及びポリヒスチジン束(Bornhorst et al., 2000、Methods Enzymol 326:245-54)が含まれ得る。他の好ましいタグには、以下のものがあるが、それだけに限らない:
(1)インフルエンザウイルス血球凝集素タンパク質の一部(Tyr−Pro−Tyr−Asp−Val−Pro−Asp−Tyr−Ala、配列番号1)。精製に用いる試薬は、タグ付きタンパク質を認識するモノクローナル抗体(12CA5)である(Wilson et al., 1984、Cell 37(3):767 78)。
【0040】
(2)ヒトc myc遺伝子の一部(Glu−Gln−Lys−Leu−Ile−Ser−Glu−Glu−Asp−Leu、配列番号2)。精製に用いる試薬は、タグ付きタンパク質を認識するモノクローナル抗体(9E10)である(Evan et al., 1985、Mol Cell Biol. 5(12):3610 6)。
【0041】
(3)ブルータングウイルス(bluetongue virus)VP7タンパク質の一部(Gln−Tyr−Pro−Ala−Leu−Thr、配列番号3)。精製に用いる試薬は、タグ付きタンパク質を認識するモノクローナル抗体(D11及び/又はF10)である(Wang et al., Gene. 1996 Feb 22;169(1):53-8)。
【0042】
(4)FLAGペプチド(例えば、Asp−Tyr−Lys−Asp−Asp−Asp−Asp−Lys、配列番号4)。精製に用いる試薬は、タグ付きタンパク質を認識するモノクローナル抗体(例えばM1及び/又はM2)(Sigma)である(Hopp et al., 「Synthesis of protein with an identification peptide」という名称の、1987年10月27日発行の米国特許第4,703,004号;Brizzard et al., 1994、Biotechniques. Apr;16(4):730-5;Knappik et al., 1994、Biotechniques 17(4):754-761)。
【0043】
(5)Strepタグペプチド(例えば、Ala Trp Arg His Pro Gln Phe Gly Gly、配列番号5)。好ましい実施形態では、strepタグペプチドを用いる。精製に用いる試薬は、タグ付きタンパク質を認識するいくつかの最適化された形のストレプトアビジンのうちの1つ(IBA GmbH)である(Skerraら、「Fusion peptides with binding activity for streptavidin」という名称の、1996年4月9日発行の米国特許第5,506,121号;Skerra et al., 1999、Biomol. Eng. 16(14):79-86;Skerra et al., 2000、Methods Enzymol. 2000;326:271-304)。
【0044】
細菌において発現されるポリペプチドの可溶性ドメイン/フラグメントが、対象とする全長ポリペプチドと共通の末端でない場合、細菌停止コドンを含む第2イントロンを、対象とするポリペプチド内に含める。このイントロンは、可溶性ドメイン/フラグメントの終結が所望されるポリペプチドの領域内に位置する。イントロンが、対象とするタンパク質の所望の位置に天然に存在しない場合、当技術分野で周知の組換え又は合成DNA技術を用いて、所望の位置に適当なイントロン配列を導入することができる。例えば、好ましい実施形態では、IgG重鎖の配列は、CH1ドメインとヒンジ領域の間に位置する第2イントロンを含む。細菌においてこの部位で翻訳が終結する結果、Fab1フラグメントが発現する。或いは、Fab2フラグメントの産生が所望される場合、ヒンジとCH2ドメインの間のポリペプチドをコードする配列内に、停止コドンを含んでいるイントロンを含めることもできる。これによって、より大きなFab2フラグメントが産生されるはずである。
【0045】
スプライス受容部位及び細菌シグナル配列の機能が確実に維持されているかどうか、二重発現ベクターカセット配列を試験する。少なくともスクリーニングの目的について、機能に関するわずかな影響なら許容することができるが、哺乳動物細胞から分泌される全長IgGの能力に負の影響を与えるどんな配列変化も有用でないはずである。
【0046】
当技術分野で知られている標準的な方法を用いて、IgG及びFab発現配列を組み込んだ環状ベクターを構築することができる(Sambrook et al., 1989、上記;Ausubel et al., Current Protocols in Molecular Biology、Greene Publishing Associates and Wiley Interscience、New Yorkを参照のこと)。例えば、合成又は組換えDNA技術を用いることができる。一実施形態では、ポリメラーゼ連鎖反応(「PCR」)増幅によって、二重発現ベクターカセット配列を含むベクターを作製する。この方法では、その5’末端に制限酵素部位を、その3’末端にIgGの調節配列とコード配列の境界配列と相補的なPCRプライマー配列を含むようにオリゴヌクレオチドを合成する。次いでそのオリゴヌクレオチドをPCR増幅反応でプライマーとして使用して、IgGの調節配列及びコード配列の領域を増幅する。次いで、標準的な分子生物学技術を用いて、哺乳動物の及び細菌性複製起点、並びに適当な選択マーカー配列を含むベクター中にこの増幅した領域をクローン化する(例えば、Methods in Enzymology、1987、Volume 154、Academic Press;Sambrook et al., 1989、上記;及びAusubel et al., 上記を参照のこと)。次いで、増幅のためにこの環状産物で大腸菌を形質転換して、大量のベクターを得る。
【0047】
好ましくは、下記に詳細に論じるように、このベクターは、細菌性複製起点、哺乳動物複製起点、及び1つ又は複数の選択マーカーを含む。本明細書に記載の二重発現ベクター系が哺乳動物宿主と細菌宿主のどちらにも使用されるように設計されているので、様々な細菌株又は細胞系統を使用することができる。複製起点配列など特定のベクター配列の選択は、宿主の選択に依存し、対象とする特定のポリペプチド又は抗体の発現、分泌、及びスクリーニング又は選択に必要とするファクターなど様々なファクターにも依存する可能性がある。
【0048】
5.1.2.抗体の製造方法
抗原と免疫特異的に結合する抗体は、抗体を合成するための当技術分野で公知の任意の方法により、特に化学合成又は好ましくは組換え発現技術により製造することができる。
【0049】
モノクローナル抗体は、ハイブリドーマ技術、組換え技術、及びファージディスプレイ技術、又はそれらの組合せの利用を含む、当技術分野で公知の多種多様な技術を用いて調製することができる。例えば、モノクローナル抗体は、当技術分野で公知であって、例えば、Harlow et al., 「Antibodies: A Laboratory Manual」, (Cold Spring Harbor Laboratory Press, 第2版 1988);Hammerling et al., 「Monoclonal Antibodies and T-Cell Hybridomas」 563-681に収載(Elsevier, N. Y., 1981)(前記参考文献は、参照によりその全文が組み入れられる)に教示される方法をはじめとするハイブリドーマ技術を利用して製造することができる。本明細書に用いる用語「モノクローナル抗体」は、ハイブリドーマ技術によって製造される抗体に限定されるものではない。用語「モノクローナル抗体」は、それを製造する方法ではなく、任意の真核生物、原核生物、又はファージクローンを含む単一クローンから得られる抗体を意味する。
【0050】
ハイブリドーマ技術を用いて特異的抗体を製造しスクリーニングする方法は、当技術分野では日常的に利用されかつ公知である。簡単に説明すると、マウスを、非マウス抗原を用いて免疫感作することができ、そして免疫応答が一度検出されれば(例えば、その抗原に特異的な抗体がマウス血清中に検出されれば)、マウス脾臓を採取して脾細胞を単離する。次いでその脾細胞を公知の技術により任意の適当な骨髄腫細胞、例えばATCCから入手しうる培養細胞株SP20由来の細胞と、融合する。ハイブリドーマを選択し、限定希釈によりクローン化する。次いで、ハイブリドーマクローンを、当技術分野で公知の方法により、本発明のポリペプチドと結合できる抗体を分泌する細胞についてアッセイする。一般的に高レベルの抗体を含有する腹水液は、マウスを陽性ハイブリドーマクローンを用いて免疫感作することにより生成することができる。
【0051】
従って、本発明は、抗体を分泌するハイブリドーマ細胞を培養することを含むモノクローナル抗体を作製する方法、並びにその方法により製造される抗体を提供するものであり、好ましくは、上記方法におけるハイブリドーマは、非マウス抗原を用いて免疫感作したマウスから単離した脾細胞を骨髄腫細胞と融合し、次いで融合から得たハイブリドーマを、その抗原と結合しうる抗体を分泌するハイブリドーマクローンについてスクリーニングすることにより作製されるものである。
【0052】
特異的な特定のエピトープを認識する抗体フラグメントは、当業者に公知のいかなる技術により作製してもよい。例えば、Fab及びF(ab’)2フラグメントは、パパイン(Fabフラグメントを生成する)又はペプシン(F(ab’)2フラグメントを生成する)などの酵素を用いて、免疫グロブリン分子のタンパク質分解切断により生成することができる。F(ab’)2フラグメントは可変領域、軽鎖定常領域及び重鎖のCH1ドメインを含有する。さらに、本発明の抗体はまた、当技術分野で公知の様々なファージディスプレイ法を用いて作製することができる。
【0053】
ヒトにおける抗体のin vivo使用、及びin vitro検出アッセイを含む複数の用途について、ヒト又はキメラ抗体を使用することが好ましい場合がある。完全なヒト抗体は、ヒト被験者の治療のためには特に望ましい。ヒト抗体は、ヒト免疫グロブリン配列由来の抗体ライブラリーを用いる上記のファージディスプレイ法を含む、当技術分野で公知の様々な方法により作製することができる。米国特許第4,444,887号及び第4,716,111号、;並びにPCT公報WO98/46645号、WO98/50433号、WO98/24893号、W098/16654号、WO96/34096号、WO96/33735号、及びWO91/10741号も参照すること;これらはそれぞれ参照によりその全文が本明細書に組み入れられる。
【0054】
ヒト抗体はまた、機能性の内因性免疫グロブリンを発現できないがヒト免疫グロブリン遺伝子を発現できるトランスジェニックマウスを用いて製造することもできる。例えば、ヒト重鎖及び軽鎖免疫グロブリン遺伝子複合体を、無作為に又は相同組換えによりマウス胚幹細胞中に導入してもよい。あるいは、ヒト重鎖及び軽鎖遺伝子に加えて、ヒト可変領域、定常領域、及び多様性領域を、マウス胚性幹細胞中に導入してもよい。相同組換えによりヒト免疫グロブリン遺伝子座を導入して、マウス重鎖及び軽鎖免疫グロブリン遺伝子を別々に又は同時に非機能化してもよい。特に、JH領域のホモ接合性欠失は内因性抗体産生を妨げる。改変した胚幹細胞を増殖させ、胚盤胞中にマイクロインジェクションしてキメラマウスを作る。次いでキメラマウスを交配し、ヒト抗体を発現するホモ接合性子孫を作る。そのトランスジェニックマウスを、通常の方式で、選択した抗原、例えば目的のポリペプチドの全体若しくは部分を用いて、免疫感作する。その抗原に対するモノクローナル抗体を、免疫感作したトランスジェニックマウスから通常のハイブリドーマ技術を利用して得ることができる。トランスジェニックマウスによって保持されるヒト免疫グロブリントランスジーンはB細胞分化の際に再配列し、その後、クラススイッチ及び体細胞突然変異を受ける。従って、かかる技術を利用して、治療上有用なIgG、IgA、IgM及びIgE抗体を製造することが可能である。ヒト抗体を製造するためのこの技術の総括については、Lonberg and Huszar(1995, Int. Rev. Immunol. 13:65-93)を参照すること。ヒト抗体及びヒトモノクローナル抗体を製造するためのこの技術並びにかかる抗体を製造するためのプロトコルの詳細な考察については、例えば、参照によりその全文が本明細書に組み入れられる、PCT公報WO98/24893号、WO96/34096号、及びWO96/33735号;及び米国特許第5,413,923号、第5,625,126号、第5,633,425号、第5,569,825号、第5,661,016号、第5,545,806号、第5,814,318号、及び第5,939,598号を参照すること。さらに、Abgenix, Inc.(Freemont、CA)及びGenpharm(San Jose、CA)などの会社と、選択した抗原に対するヒト抗体を上記と類似の技術を利用して提供する契約を結ぶことができる。
【0055】
キメラ抗体は、抗体の異なる部分が異なる免疫グロブリンに由来する分子、例えばヒト抗体由来の可変領域及び非ヒト免疫グロブリンの定常領域を有する抗体である。キメラ抗体を製造する方法は当技術分野で公知である。例えば、Morrison, 1985, Science 229:1202;Oi et al., 1986, BioTechniques 4:214;Gillies et al., 1989, J. Immunol. Methods 125:191-202;並びに米国特許第5,807,715号、及び第4,816,567号、第4,816,397号(これらは本明細書に参照によりその全文が組み入れられる)を参照すること。ヒト種からの1以上のCDRと非ヒト免疫グロブリン分子からのフレームワーク領域を含むキメラ抗体は、当技術分野で公知の種々の技法を用いて作製しうる、例えば、CDRグラフト(欧州特許EP239,400;PCT公報WO91/09967号;及び米国特許第5,225,539号、第5,530,101号、第5,585,089号)、ベニアリング(veneering)又は再表面形成(resurfacing)(欧州特許EP592,106;EP519,596;Padlan, 1991, Molecular Immunology 28 (4/5):489-498;Studnicka et al., 1994, Protein Engineering 7 (6):805-814;及びRoguska et al., 1994, PNAS 91:969-973)、及びチェーンシャッフリング(chain shuffling)(米国特許第5,565,332号)が挙げられる。好ましい実施形態において、キメラ抗体は、表2に例示したCDR3のいずれか1つのアミノ酸配列を有するヒトCDR3と非ヒトフレームワーク領域を含む。しばしば、フレームワーク領域のフレームワーク残基を、CDRドナー抗体由来の対応する残基で置換して、抗原結合を改変し、好ましくは改良する。これらのフレームワーク置換は、当技術分野で公知の方法によって、例えば、CDRとフレームワーク残基の相互作用をモデル化して抗原結合に重要なフレームワーク残基を同定し、さらに配列比較により特定位置の異常なフレームワーク残基を同定することによって確認する(例えば、本明細書に参照によりその全文が組み入れられる、Queen et al., 米国特許第5,585,089号;及びRiechmann et al., 1988, Nature 332:323を参照)。
【0056】
さらに、目的の抗体は、続いて、当業者に周知の技術を用いて抗原を「模倣」する抗イディオタイプ抗体を作製するために用いることができる(例えば、Greenspan & Bona, 1989, FASEB J. 7(5):437-444;及びNissinoff, 1991, J. Immunol. 147(8):2429-2438参照)。例えば、目的の抗原と結合し、その宿主細胞受容体への結合を競合的に阻害する抗体(当技術分野で周知の及び上述のアッセイにより測定しうる)を用いて、目的の結合ドメインの抗原を「模倣」し、その結果として該抗原及び/又はその宿主細胞受容体と結合し、中和する抗イディオタイプを作製することができる。かかる中和抗イディオタイプ又はかかる抗イディオタイプのFabフラグメントは、抗原を中和するために治療方法において用いることができる。例えば、かかる抗イディオタイプ抗体は、目的の抗原と結合するため及び/又はその宿主細胞受容体と結合するために用いることができる。
【0057】
5.1.3.組換え発現及びタンパク質産生
目的のポリペプチドをコードするカセット配列(例えば適当に設計したイントロン配列を有する抗体分子をコードするカセット配列)を含む二重発現ベクターを構築した後、本発明の二重発現ベクターは、当技術分野で周知の技術を利用して組換えDNA技術により作製することができる。例えば、本明細書に参照によりその全文が組み入れられる、米国特許第6,331,415号を参照すること。
【0058】
例えば、好ましい実施形態においては、当業者に周知の方法を利用して、適当な転写及び翻訳制御シグナルを含有する二重発現ベクターカセットを構築することができる。これらの方法としては、例えば、in vitro組換えDNA技術、合成技術、及びin vivo遺伝子組換えが挙げられる。本発明は、従って、抗体分子、抗体の重鎖若しくは軽鎖、抗体の重鎖若しくは軽鎖可変ドメイン又はそれらの部分、あるいは重鎖若しくは軽鎖のCDRをコードし、プロモーターと機能的に連結されたヌクレオチド配列を含む発現ベクターを提供する。かかるベクターは、抗体分子の定常領域をコードするヌクレオチド配列(例えば、PCT公報WO86/05807号;PCT公報WO89/01036号;及び米国特許第5,122,464号を参照)を含んでもよく、また抗体の可変ドメインをかかるベクター中にクローニングして重鎖全体、軽鎖全体、又は重鎖全体と軽鎖全体の両方を発現させてもよい。
【0059】
二重発現ベクターを通常の技術により宿主細胞に導入し、次いでトランスフェクトした細胞を通常の技術により培養して、目的の抗体を製造する。従って、本発明は、目的の抗体又はそのフラグメント、あるいはその重鎖若しくは軽鎖又はそれらの一部分、あるいは本発明の一本鎖抗体をコードし、異種プロモーターと機能的に連結されたポリヌクレオチドを含有する宿主細胞を含む。二本鎖抗体を発現させるための好ましい実施形態においては、以下に詳しく説明するように、重鎖及び軽鎖の両方をコードするベクターを宿主細胞において共発現させ、免疫グロブリン分子全体を発現させることができる。
【0060】
様々な宿主系を利用して、本発明の二重発現ベクターを発現させることができる(例えば、米国特許第5,807,715号を参照)。かかる宿主系は、適当なヌクレオチドコード配列を用いて形質転換又はトランスフェクトすると、目的の抗体分子をin situで発現することができる細胞も意味する。これらとしては、限定されるものでないが、細菌などの微生物(例えば、大腸菌(E.coli)及び枯草菌(B.subtilis));二重発現ベクターを用いて形質転換した酵母(例えば、サッカロミセス・ピキア(Saccharomyces Pichia));組換えウイルス発現ベクター(例えば、バキュロウイルス)に感染した昆虫細胞系;二重発現カセットで形質転換した組換えウイルス発現ベクター(例えば、カリフラワーモザイクウイルス、CaMV;タバコモザイクウイルス、TMV)に感染した植物細胞系;又は、哺乳動物細胞のゲノム由来のプロモーター(例えば、メタロチオネインプロモーター)若しくは哺乳動物ウイルス由来のプロモーター(例えば、アデノウイルス後期プロモーター;ワクシニアウイルス7.5Kプロモーター)を含有する組換え発現構築物を保持する哺乳動物細胞系(例えば、COS、HEK、CHO、BHK、293、NSO、及び3T3細胞)が挙げられる。
【0061】
細菌宿主での発現のために、二重発現ベクターは、プラスミドベクターの複製及び増幅のために必要とされる複製起点を含む。大腸菌におけるクローニング及び増幅のために、大腸菌の複製起点、例えば当技術分野で周知のものを用いる(Miller, 1992, A Short Course in Bacterial Genetics, Cold Spring Harbor Laboratory Press, NY、及びそれに含まれる参考文献を参照)。容易に入手可能なプラスミドの複製起点の例としては、限定されるものではないが、ColE1由来の複製起点(Bolivar et al., 1977, Gene 2:95-113;Sambrook et al., 1989、前掲を参照)、pACYC184などのプラスミド上に存在するp15A起点(Chang and Cohen, 1978, J. Bacteriol. 134:1141-56;Miller, 1992, p.10.4-10.11も参照)、及び低コピープラスミド発現のために利用可能なpSC101起点が当技術分野で周知である。
【0062】
例えば、一実施形態において、高コピープラスミド、例えば、ColE1由来複製起点を含有するプラスミドなどに由来する複製起点を用いることができ、この例は当技術分野で周知である(Sambrook et al., 1989、前掲参照;Miller, 1992, A Short Course in Bacterial Genetics, Cold Spring Harbor Laboratory Press, NY、及びそれに含まれる参考文献も参照)。一例としては、pUC19由来の起点及びその誘導体がある(Yanisch-Perron et al., 1985, Gene 33:103-119)。pUCベクターは、細胞当たり300〜500コピーのレベルで存在し、外来遺伝子の挿入のための都合のよいクローニング部位を有している。非常に高い発現のため、λベクター、例えばλgt11(Huynh et al., 1984, "DNA Cloning Techniques: VolI: A Practical Approach", D. Glover編, pp.49-78, IRL Press, Oxford)、又はT7及びSp6ポリメラーゼ発現系を含有する細胞におけるT7若しくはSP6ファージプロモーター(Studier et al., 1990, Methods Enzymol. 185:60-89)を使用することも可能である。
【0063】
低レベルの発現が望まれる場合には、中程度又は低コピーに由来する複製起点を使用しうる。中程度コピープラスミドは当技術分野で周知であり、例えばColE1由来の複製起点を有し、細胞当たり20〜100コピーであるpBR322(Bolivar et al., 1977, Gene 2:95-113;Sambrook et al., 1989、前掲を参照)、又はpACYC100系のプラスミドの1つであり、p15A複製起点を有し、細胞当たり10〜12コピーで存在するpACYC184(Chang and Cohen, 1978, J. Bacteriol. 134:1141-56;またMiller, 1992, p.10.4-10.11参照)がある。低コピープラスミドもまた当技術分野で周知であり、例えば、pSC101起点を有し、細胞当たり約5コピーで存在するpSC101がある。pACYC及びpSC101プラスミドベクターは両方とも都合の良いクローニング部位を有し、同じ細胞においてpBR及びpUCプラスミドとして共存しうる。これは、これらが適合可能な複製起点を有し、特有の選択的抗生物質マーカーを有するためである。他の好適なプラスミド複製起点としては、λ又はファージP1レプリコンに基づくプラスミド、例えばLorist系が挙げられる(Gibson et al., 1987, Gene 53:283-286)。
【0064】
さらに低い発現が望まれる場合には、複製起点は、細菌染色体から得てもよい(Miller, 1992、前掲;Niedhardt, F.C.編、1987, Escherichia coli and Salmonella typhimurium, American Society for Microbiology, Washington, D.C.;Yarmolinsky, M.B. & Sternberg, N., 1988, pp.291-438, The Bacteriophages, Vol.1、R. Calendar編、Plenum Press, New York)。さらに、合成の複製起点、細菌プロモーター、又は細菌シグナル配列を用いることも可能である。
【0065】
哺乳動物宿主細胞において、二重発現ベクター配列は、エピソームとして哺乳動物宿主細胞中に存在するように設計してもよいし、あるいは例えばベクターを線状化して設計することなどにより、宿主のゲノムDNAへの組込みを促進し、安定な細胞系が作製されるように設計を行ってもよい。かかるベクターは当技術分野で公知である。
【0066】
例えば、哺乳動物宿主細胞においては、様々なウイルスに基づく発現系を使用することができる。アデノウイルスを発現ベクターとして利用する場合、二重発現ベクターのカセット配列を、アデノウイルス転写/翻訳制御複合体、例えば、後期プロモーター及びトリパータイト(tripartite)リーダー配列と連結しうる。次いでこのキメラ遺伝子を、in vitro又はin vivo組換えによりアデノウイルスゲノムに挿入することができる。ウイルスゲノムの非必須領域(例えば、領域E1又はE3)への挿入により、感染宿主において生存可能でかつIgG遺伝子産物を発現しうる組換えウイルスを得ることができる(例えば、Logan and Shenk, 1984, Proc. Natl. Acad. Sci. USA 8 1:3655-3659を参照)。特定の開始シグナルも、挿入された発現ベクターカセット配列の効率的な翻訳に必要でありうる。これらのシグナルとしてはATG開始コドン及び隣接配列が挙げられる。IgG重鎖又は軽鎖を、それ自体の開始コドン及び隣接配列を含めて、適当な発現ベクターに挿入する場合には、さらに翻訳制御シグナルは必要ない。しかしながら、IgGコード配列の一部のみを挿入する場合には、おそらくATG開始コドンを含む外因性の翻訳制御シグナルを提供する必要がある。さらに、全インサートの翻訳を保証するため開始コドンは所望のコード配列のリーディングフレームと一致している必要がある。これらの外因性翻訳制御シグナル及び開始コドンは、天然及び合成の両方の様々な起源のものであってよい。発現効率は、適当な転写エンハンサーエレメント、転写ターミネーターなど(例えば、Bittner et al., 1987, Methods in Enzymol. 153:51-544を参照)を組み入れることにより増強することができる。
【0067】
さらに、挿入された配列の発現をモジュレートするか、又は所望の特定の様式で遺伝子産物を修飾及びプロセシングする宿主細胞株を選択することができる。タンパク質産物のかかる修飾(例えば、グリコシル化)及びプロセシング(例えば、切断)は、タンパク質の機能にとって重要でありうる。異なる宿主細胞は、タンパク質及び遺伝子産物の翻訳後プロセシング及び修飾のための特有かつ特定の機構を有する。適当な細胞系又は宿主系を選んで、発現された外来タンパク質の正しい修飾とプロセシングを保証することができる。この目的のために、適切な一次転写産物のプロセシング、遺伝子産物のグリコシル化及びリン酸化のための細胞機構を持つ宿主真核細胞を使用することができる。かかる哺乳動物宿主細胞としては、限定されるものでないが、CHO、VERY、BHK、Hela、COS、MDCK、293、3T3、W138、BT483、Hs578T、HTB2、BT20及びT47D、NSO(内因的に全く免疫グロブリン鎖を産生しないマウス骨髄腫培養細胞株)が挙げられる。
【0068】
組換えタンパク質の長期間にわたる高収量の生産のためには、安定した発現が好ましい。例えば、安定して抗体分子を発現する培養細胞株を遺伝子操作によって作ることができる。ウイルスの複製起点を含有する発現ベクターを用いるのではなしに、宿主細胞を、適当な発現制御エレメント(例えば、プロモーター、エンハンサー、配列、転写ターミネーター、ポリアデニル化部位など)により制御したDNA、及び選択マーカーを用いて形質転換することができる。外来DNAの導入後、遺伝子操作した細胞を、1〜2日間富栄養培地で増殖させ、次いで選択培地に切り換えてもよい。組換えプラスミド中の選択マーカーは、選択に対する耐性を付与し、細胞がプラスミドをその染色体中に安定的に組み込み、増殖して細胞巣を形成することを可能にするので、これを次にクローニングして培養細胞株に拡大することができる。この方法を利用して抗体分子を発現する細胞系を遺伝子操作により有利に作製することができる。かかる遺伝子操作で作製した培養細胞株は、抗体分子と直接又は間接に相互作用する組成物をスクリーニングしたり評価したりするのに特に有用である。
【0069】
様々な選択系を利用することができ、限定されるものでないが、例えば単純ヘルペスウイルスチミジンキナーゼ(Wigler et al., 1977, Cell 11:223)、ヒポキサンチン−グアニン・ホスホリボシルトランスフェラーゼ(Szybalska & Szybalski, 1992, Proc. Natl. Acad. Sci. USA 48:202)、及びアデニン・ホスホリボシルトランスフェラーゼ(Lowy et al., 1980, Cell 22:8-17)遺伝子が挙げられ、これらはそれぞれtk−、hgprt−又はaprt−細胞において使用することができる。また、以下の遺伝子については代謝拮抗物質耐性を選択の基礎として利用することができる:メトトレキセート耐性を付与するdhfr(Wigler et al., 1980, Natl. Acad. Sci. USA 77:357;O'Hare et al., 1981, Proc. Natl. Acad. Sci. USA 78:1527);ミコフェノール酸耐性を付与するgpt(Mulligan & Berg, 1981, Proc. Natl. Acad. Sci. USA 78:2072);アミノグリコシドG−418耐性を付与するneo(Wu and Wu, 1991, Biotherapy 3:87-95;Tolstoshev, 1993, Ann. Rev. Pharmacol. Toxicol. 32:573-596;Mulligan, 1993, Science 260:926-932;並びにMorgan and Anderson, 1993, Ann. Rev. Biochem. 62:191-217;May, 1993, TIB TECH 11 (5):155-215);並びにハイグロマイシン耐性を付与するhygro(Santerre et al., 1984, Gene 30:147)。当技術分野で公知の組換えDNA技法を慣用的に適用して所望の組換えクローンを選択することができ、かかる方法は、例えば、Ausubel et al.(編), 「Current Protocols in Molecular Biology」, John Wiley & Sons, NY (1993);Kriegler, 「Gene Transfer and Expression, A Laboratory Manual」, Stockton Press, NY (1990);及びDracopoli et al.(編), 「Current Protocols in Human Genetics」, John Wiley & Sons, NY (1994)の第12章及び第13章;Colberre-Garapin et al., 1981, J. Mol. Biol. 150:1(これらは参照により本明細書にその全文が組み入れられる)に記載されている。
【0070】
細菌における選択のために、好ましくは抗生物質耐性マーカー、例えば、Tn903由来のカナマイシン耐性遺伝子(Friedrich and Soriano, 1991, Genes Dev. 5:1513-1523)、又は他のアミノグリコシド(限定されるものではないが、ジヒドロストレプトマイシン、ゲンタマイシン、ネオマイシン、パロマイシン及びストレプトマイシンなど)に対する耐性を付与する遺伝子、ペニシリン(限定されるものではないが、アンピシリン、カルベニシリン、メチシリン、ペニシリンN、ペニシリンO、及びペニシリンV)に対する耐性を付与するTn9由来のTEM−1 β−ラクタマーゼ遺伝子を用いる。他の選択可能な遺伝子配列としては、限定されるものではないが、ゼオシン耐性を付与するポリペプチドをコードする遺伝子配列が含まれる(Hegedus et al., 1998, Gene 207:241-249)。使用しうる他の抗生物質は、アムフェニコール類、例えばクロラムフェニコールなどに対する耐性を付与する遺伝子、例えば、クロラムフェニコールトランスアセチラーゼ(CAT)のコード配列を使用することができる(Eikmanns et al., 1991, Gene 102:93-98)。当業者により理解されているように、プラスミドの維持について選択するための他の非抗生物質方法を用いることも可能であり、例えば、種々の栄養要求性マーカーなどである(Sambrook et al., 1989、前掲;Ausubel et al.、前掲)。
【0071】
抗体分子の発現レベルは、ベクター増幅により増加させることができる(総説については、Bebbington and Hentschel, 「The use of vectors based on gene amplification for the expression of cloned genes in mammalian cells in DNA cloning」, Vol.3. (Academic Press, New York, 1987)を参照)。抗体を発現するベクター系中のマーカーが増幅可能であれば、宿主細胞培養中に存在するインヒビターレベルの増加は、マーカー遺伝子のコピー数を増加させうる。増幅される領域は、抗体遺伝子と関連しているので、抗体の産生も増加しうる(Crouse et al., 1983, Mol. Cell. Biol. 3:257)。
【0072】
宿主細胞を、本発明の2つの二重発現ベクター、すなわち、重鎖由来のポリペプチドをコードする第1ベクターと軽鎖由来のポリペプチドをコードする第2ベクターとを用いて共トランスフェクトすることができる。その2つのベクターは、哺乳動物細胞においては重鎖ポリペプチドと軽鎖ポリペプチドの、また細菌細胞においてはFab又はscFvポリペプチドの同等の発現を可能にする同一の選択マーカーを含有してもよい。あるいは、重鎖ポリペプチドと軽鎖ポリペプチドの両方をコードしかつ発現できる単一ベクターを利用してもよい。かかる状況では重鎖の前に軽鎖を配置して、毒性である遊離の重鎖が過剰になることを避けなければならない(Proudfoot, 1986, Nature 322:52;及びKohler, 1980, Proc. Natl. Acad. Sci. USA 77:2 197)。重鎖と軽鎖のコード配列はcDNA又はゲノムDNAを含んでいてもよい。
【0073】
ポリペプチドは、標準組換えDNA技術により、製造することができる。例えば、遺伝子断片のPCR増幅を、2つの連続した遺伝子断片の間を相補的に重複するようなアンカープライマーを用いて実施し、次いでそれらをアニーリングして再増幅することによりキメラ遺伝子配列を作製することができる(例えば、「Current Protocols in Molecular Biology」, Ausubel et al.編, John Wiley & Sons, 1992を参照)。さらに、生物活性分子をコードする核酸を、Fcドメイン又はそのフラグメントを含有する発現ベクター中にクローニングして、その生物活性分子がFcドメイン又はFcドメインのフラグメントとイン・フレームとなるように連結することができる。
【0074】
ポリペプチドを抗体の定常領域に融合又は複合体化する方法は、当技術分野では公知である。例えば、米国特許第5,336,603号、第5,622,929号、第5,359,046号、第5,349,053号、第5,447,851号、第5,723,125号、第5,783,181号、第5,908,626号、第5,844,095号、及び第5,112,946号;欧州特許EP307,434;EP367,166;EP394,827;PCT公報WO91/06570号、WO96/04388号、WO96/22024号、WO97/34631号、及びWO99/04813号;Ashkenazi et al., 1991, Proc. Natl. Acad. Sci. USA 88:10535-10539;Traunecker et al., 1988, Nature, 331:84-86;Zheng et al., 1995, J. Immunol. 154:5590-5600;及びVil et al., 1992, Proc. Natl. Acad. Sci. USA 89:11337-11341(これらは本明細書に参照によりその全文が組み入れられる)を参照すること。
【0075】
生物活性分子及びFcドメイン又はそのフラグメントをコードするヌクレオチド配列は、当業者が利用しうる任意の情報から(すなわち、Genbank、文献から、又は慣用的なクローニングにより)得ることができる。ポリペプチド又は融合タンパク質をコードするヌクレオチド配列を、二重発現ベクター中に挿入してもよい。
【0076】
真核細胞におけるポリペプチドの発現は、当技術分野で公知の任意のプロモーター若しくはエンハンサーエレメントにより制御することができる。融合タンパク質をコードする遺伝子の発現を制御するのに利用しうるプロモーターとしては、限定されるものでないが、SV40初期プロモーター領域(Bernoist and Chambon, 1981, Nature 290:304-310)、ラウス肉腫ウイルスの3’長末端反復配列に含有されるプロモーター(Yamamoto et al., 1980, Cell 22:787-797)、ヘルペスチミジンキナーゼプロモーター(Wagner et al., 1981, Proc. Natl. Acad. Sci. U. S. A. 78:1441-1445)、メタロチオネイン遺伝子の調節配列(Brinster et al., 1982, Nature 296:39-42)、テトラサイクリン(Tet)プロモーター(Gossen et al., 1995, Proc. Nat. Acad. Sci. USA 89:5547-5551);細菌においては、原核生物プロモーター、例えばβ−ラクタマーゼ・プロモーター(Villa-Kamaroff et al., 1978, Proc. Natl. Acad. Sci. U. S. A. 75:3727-3731)若しくはtacプロモーター(DeBoer et al., 1983, Proc. Natl. Acad. Sci. U. S. A. 80:21-25などの原核生物発現ベクター;また「Useful proteins from recombinant bacteria」, Scientific American, 1980,242: 74-94も参照)も参照;植物細胞においては、ノパリンシンセターゼプロモーター領域(Herrera-Estrella et al., Nature 303:209-213)又はカリフラワーモザイクウイルス35SRNAプロモーター(Gardner et al., 1981, Nucl. Acids Res. 9:2871)を含む植物発現ベクター、及び光合成酵素リブロース二リン酸カルボキシラーゼのプロモーター(Herrera-Estrella et al., 1984, Nature 310:115-120);Gal4プロモーターなどの酵母又は他の真菌由来のプロモーターエレメント、ADC(アルコールデヒドロゲナーゼ)プロモーター、PGK(ホスホグリセロールキナーゼ)プロモーター、及びアルカリホスファターゼプロモーターが挙げられる。
【0077】
特定の実施形態においては、ポリペプチドの発現は構成的プロモーター(CMVプロモーターなど)により調節される。他の実施形態においては、ポリペプチドの発現は誘導性プロモーターにより調節される。
【0078】
ポリペプチドをコードする遺伝子のインサートを含有する発現ベクターは、3つの一般的手法:(a)核酸ハイブリダイゼーション、(b)「マーカー」遺伝子機能の存在若しくは非存在、及び(c)挿入した配列の発現により、同定することができる。第1の手法においては、発現ベクター中のポリペプチドをコードする遺伝子の存在を、ポリペプチドをコードする挿入した遺伝子と相同的な配列を含むプローブを用いて核酸ハイブリダイゼーションを行うことにより検出することができる。第2の手法においては、組換えベクター/宿主系は、ポリペプチドをコードするヌクレオチド配列のベクター中への挿入により生じる、ある特定の「マーカー」遺伝子機能(例えば、チミジンキナーゼ活性、抗生物質耐性、形質転換表現型、バキュロウイルス中の封入体(occlusion body)形成など)の存在又は非存在に基づいて同定しかつ選択することができる。例えば、融合タンパク質をコードするヌクレオチド配列がベクターのマーカー遺伝子配列内に挿入される場合には、融合タンパク質インサートをコードする遺伝子を含有する組換え体は、マーカー遺伝子機能の不在により同定することができる。第3の手法においては、組換え発現ベクターは、組換え体が発現する遺伝子産物(例えば、融合タンパク質)をアッセイすることにより同定することができる。かかるアッセイは、例えば、in vitroアッセイ系における融合タンパク質の物理的又は機能的特性、例えば、抗生物活性分子抗体との結合に基づくものでもよい。
【0079】
DNA断片のベクターへの挿入について既に記載されている方法の任意のものを用いて、適当な転写/翻訳制御シグナル及びタンパク質コード配列を含有するキメラ遺伝子を含む発現ベクターを構築しうる。これらの方法としては、in vitro組換えDNA及び合成技法、並びにin vivo組換え体(遺伝的組換え)が含まれる。哺乳動物細胞におけるIgG重鎖又は軽鎖の発現は、IgG重鎖又は軽鎖が組換えDNA分子で形質転換された宿主において発現されるように、第2の核酸配列により調節することができる。例えば、IgG重鎖又は軽鎖の発現は、当技術分野で公知の任意のプロモーター/エンハンサーエレメントにより制御されうる。
【0080】
好ましくは、Fabフラグメント又は全長IgGの細菌発現は誘導性プロモーターにより制御される。広範囲の発現をもたらす誘導性発現は、種々の誘導性調節配列を用いることにより達成しうる。一実施形態において、例えば、lacI遺伝子及びその無償性の誘導剤IPTGを用いて、ポリペプチドをコードする配列がlacOP調節配列を介して転写される場合には、大腸菌におけるFabフラグメントの誘導性の高レベル発現を得ることができる。また、使用可能である多様な他の誘導性プロモーター系が当業者に周知である。IgG二重発現ベクター系からの発現レベルもまた種々の強度のプロモーターで変更することができる。
【0081】
使用可能である他の調節される発現系としては、限定されるものではないが、アラビノース(AraC)により誘導されうるaraCプロモーター(例えば、Schleif, 2000, Trends Genet. 16:559-565参照)、TET系(Geissendorfer and Hillen, 1990, Appl. Microbiol. Biotechnol. 33:657-663)、ファージλ温度のpLプロモーター及び誘導性λリプレッサーCI857(Pirrotta, 1975, Nature 254:114-117;Petrenko et al., 1989, Gene 78:85-91)、trpプロモーター及びtrpリプレッサー系(Bennett et al., 1976, Proc. Natl. Acad. Sci USA 73:2351-55;Wame et al., 1986, Gene 46:103-112)、lacUV5プロモーター(Gilbert and Maxam, 1973, Proc. Natl. Acad. Sci. USA 70:1559-63)、lpp(Nokamura et al., 1982, J. Mol. Appl. Gen. 1:289-299)、T7遺伝子10プロモーター、phoA(アルカリホスファターゼ)、recA(Horii et al., 1980, Proc. Natl. Acad. Sci. USA 77:313-7)、及びIPTGにより誘導されるtacプロモーター、trp−lac融合プロモーター(Amann et al., 1983, Gene 25:167-78)は全て一般的に使用される強力なプロモーターであり、その結果、各プロモーターによりそのレベルが制御されるタンパク質について約1〜10%の総細胞タンパク質のレベルが蓄積する。より強力なプロモーターが望まれる場合には、tacプロモーターはlacUV5よりも約10倍強力であるが、基底レベルの発現が高く、過剰発現が必要とされる場合にのみ使用すべきである。より弱いプロモーターが必要な場合には、例えば、マルトース、ガラクトース、又は他の所望のプロモーターなど(かかるプロモーターの配列はGenBankから入手可能である)の、他の細菌プロモーターが当技術分野で周知である(Burks et al., 1991, Nucl. Acids Res. 19:2227-2230)。
【0082】
全長IgG重鎖又は軽鎖の真核生物における発現については、ベクターは、RNAポリメラーゼの認識、結合及び転写開始に十分な特異的配列を含む、真核生物特異的複製起点及びプロモーター領域を含みうる。さらに、プロモーター領域は、RNAポリメラーゼの認識、結合及び転写開始活性をモジュレートする配列を含んでもよい。かかる配列としては、cic作用性因子であるか、又はtrans作用性因子に応答するものでありうる。調節の性質に応じて、プロモーターは構成的であるか又は調節型でありうる。TnpI発現を制御するために用い得るプロモーターとしては、限定されるものではないが、SV40初期プロモーター領域(Benoist and Chambon, 1981, Nature 290:304-310)、ラウス肉腫ウイルスの3’長末端リピートに含まれるプロモーター(Yamamoto et al., 1980, Cell 22:787-797)、ヘルペスチミジンキナーゼプロモーター(Wagner et al., 1981, Proc. Natl. Acad. Sci. U.S.A. 78:1441-1445)、メタロチオネイン遺伝子の調節配列(Brinster et al., 1982, Nature 296:39-42)、ノパリンシンテターゼプロモーター領域を含む植物発現ベクター(Herrera-Estrella et al., 1984, Nature 303:209-213)、又はカリフラワーモザイクウイルス35S RNAプロモーター(Gardner et al., 1981, Nucl. Acids Res. 9:2871)、及び光合成酵素リブロース二リン酸カルボキシラーゼのプロモーター(Herrera-Estrella et al., 1984, Nature 310:115-120)、酵母又は他の真菌由来のプロモーターエレメント、例えばGal4プロモーター、又はADC(アルコールデヒドロゲナーゼ)プロモーターが挙げられる。
【0083】
プロモーター、及びポリヌクレオチドを機能的に連結しうるクローニング部位を含有するベクターは当技術分野で周知である。かかるベクターは、in vitroで又はin vivoでRNAを転写可能であり、Stratagene(La Jolla, Calif.)及びPromega Biotech(Madison, Wis.)などの供与元から市販されている。発現及び/又はin vitro転写を最適化するために、クローン化DNAの5’及び/又は3’非翻訳部分を除去し、付加し又は改変して、さらなる潜在的な不適当な別の翻訳開始コドン、又は転写若しくは翻訳レベルのいずれかで発現を妨害若しくは低減しうるほかの配列を排除する必要がありうる。あるいは、コンセンサスリボソーム結合部位を開始コドンのすぐ5’側に挿入して発現を増強することができる(例えばKozak, 1991, J. Biol. Chem. 266:19867参照)。同様に、同じアミノ酸をコードする別のコドンをコード配列と置換して、翻訳を増強してもよい(例えば、宿主細胞のコドン最適性を採用して、G−Cリッチドメインの存在を低減する、等)。
【0084】
ベクターはまた、タンパク質発現、操作又は挿入された標的DNAの維持のための目的のヌクレオチド配列を含み得る。例えば、プロモーター配列、エンハンサー配列、翻訳配列(例えばシャイン及びダルガーノ配列)、転写因子認識部位、Kozakコンセンサス配列、及び終結シグナルを、ベクターの適当な位置に含有させてもよい。
【0085】
ベクターはまた、天然又は合成由来のシグナル配列を含む必要がある。ポリペプチド(例えば抗体、IgGなど)を細胞膜内に標的化しうるシグナル配列を用いることができる。天然に周辺質に存在することが定められたタンパク質と結合しているリーダー配列は、例えば、外来タンパク質の周辺質への分泌を誘導することが知られている(MacIntyre et al., 1990、Mol. Gen. Genet. 221:466-474)。好ましい実施形態では、シグナル配列は、OmpAタンパク質リーダー配列(Hobom et al., 1995、Dev. Biol. Stand. 84:255-262)をコードする。他のシグナル配列もまた可能であり、限定されるものではないが、大腸菌PhoA(Oka et al., 1985、Proc. Natl. Acad. Sci 82:7212-16)、OmpT(Johnson et al., 1996、Protein Expression 7:104-113)、LamB及びOmpF(Hoffman and Wright、1985、Proc. Natl. Acad. Sci. USA 82:5107-5111)由来のリーダー、β−ラクタマーゼ(Kadonaga et al., 1984、J. Biol. Chem. 259:2149-54)、エンテロトキシン(Morioka-Fujimoto et al., 1991、J. Biol. Chem. 266:1728-32)、黄色ブドウ球菌(Staphylococcus aureus)のプロテインA(Abrahmsen et al., 1986、Nucleic Acids Res. 14:7487-7500)、枯草菌(B. subtilis)のエンドグルカナーゼ(Lo et al., Appl. Environ. Microbiol. 54:2287-2292)由来のリーダー配列、並びに人工及び合成シグナル配列(MacIntyre et al., 1990、Mol. Gen. Genet. 221:466-74;Kaiser et al., 1987、Science、235:312-317)が挙げられる。
【0086】
二重発現ベクターカセット配列を含むDNA調製物を宿主細胞に送達するための当技術分野で公知の方法の任意のものが上記方法と共に使用するのに適している。かかる方法は当技術分野で公知であり、限定されるものではないが、細胞のエレクトロポレーション、塩化カルシウム又はルビジウムを用いたコンピテント細胞の調製、及びウイルス粒子にパッケージングした標的DNAを用いたDNAの形質導入が挙げられる。真核細胞については、限定されるものではないが、エレクトロポレーション、DNAのリン酸カルシウム沈殿によるトランスフェクション、及びウイルスパッケージングなどの方法がある。好ましい実施形態においては、エレクトロポレーションを使用する。細胞は、標準的な方法によりエレクトロポレーションにコンピテントとなるように処理する(Ausubel et al., Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley Interscience, New York参照)。好ましくは、エレクトロコンピテント細胞の標準調製物約50μlを標準的な手順によりエレクトロポレーションに用いる。線状又は環状ベクターの形質転換を必要とする実験においては、好ましくは0.3g以上のベクターを使用する。IgG DNAを含有するDNA調製物の形質転換を必要とする実験においては、好ましくは0.3μg以上のベクターを使用する。共形質転換実験においては、DNAはエレクトロポレーションの前に混合することが好ましい。エレクトロポレーション後、細胞を培養培地に希釈し、約1時間半の回復期間にわたりインキュベートし、その後選択マーカー遺伝子により付与される表現型変化を同定する条件下で培養することが好ましい。
【0087】
好適には、表現型の変化は抗生物質に対する耐性であり、細胞は対応する抗生物質を含有するプレート上で培養する。この場合、抗生物質耐性コロニーは終夜培養後に出現するが、これは大部分が所望のサブクローニング産物を含有する。選択マーカーに関して、好ましくは抗生物質耐性マーカー、例えば、Tn903由来のカナマイシン耐性遺伝子(Friedrich and Soriano, 1991, Genes Dev. 5:1513-1523)、又は他のアミノグリコシド(限定されるものではないが、ジヒドロストレプトマイシン、ゲンタマイシン、ネオマイシン、パロマイシン及びストレプトマイシンなど)に対する耐性を付与する遺伝子、ペニシリン(限定されるものではないが、アンピシリン、カルベニシリン、メチシリン、ペニシリンN、ペニシリンO、及びペニシリンV)に対する耐性を付与するTn9由来のTEM−1 β−ラクタマーゼ遺伝子を用いる。他の選択可能な遺伝子配列としては、限定されるものではないが、ゼオシン耐性を付与するポリペプチドをコードする遺伝子配列が含まれる(Hegedus et al., 1998, Gene 207:241-249)。使用しうる他の抗生物質は、アムフェニコール類、例えばクロラムフェニコールなどに対する耐性を付与する遺伝子、例えば、クロラムフェニコールトランスアセチラーゼ(CAT)のコード配列を使用することができる(Eikmanns et al., 1991, Gene 102:93-98)。当業者により理解されているように、プラスミドの維持について選択するための他の非抗生物質方法を用いることも可能であり、例えば、種々の栄養要求性マーカーなどである(Sambrook et al., 1989、前掲;Ausubel et al.、前掲を参照)。
【0088】
別の実施形態において、DNAは、ファージ粒子にパッケージングされたDNAの形質導入により宿主細胞に送達する。P1又はλ形質導入及びパッケージングプロトコールは当技術分野で公知である。λパッケージング抽出物は市販されている(例えば、Promega, Madison, WI製)。
【0089】
目的の抗体分子を組換え発現により産生させた後、免疫グロブリン分子を精製するための当技術分野で公知の任意の方法、例えば、クロマトグラフィー(例えば、イオン交換、アフィニティー、特に、プロテインAの後の特異的抗原に対するアフィニティ、及びサイズ分離カラムクロマトグラフィ)により、遠心分離、溶解度差により、又はタンパク質を精製するための他の任意の標準技術により、それらを精製することができる。さらに、本発明の抗体又はそのフラグメントは、精製を容易にするために、本明細書に記載の又は当技術分野で公知の異種ポリペプチド配列と融合してもよい。
【0090】
5.2.抗体の選択及びスクリーニング
5.2.1.抗体の選択及び特性決定
本発明の全長IgG及びFabフラグメントを様々な方法で特徴決定することができる。特に、全長IgG及びFabフラグメントを目的の抗原と免疫特異的に結合する能力についてアッセイすることができる。かかるアッセイは、溶液中で(例えば、Houghten, 1992, Bio/Techniques 13:412-421)、マイクロタイターディッシュなどの固相支持体上で、ビーズ上で(Lam, 1991, Nature 354:82-84)、チップ上で(Fodor, 1993, Nature 364:555-556)、細菌上で(米国特許第5,223,409号)、胞子上で(米国特許第5,571,698号、第5,403,484号及び第5,223,409号)、プラスミド上で(Cull et al., 1992, Proc. Natl. Acad. Sci. USA 89:1865-1869)又はファージ上で(Scott and Smith, 1990, Science 249:386-390;Devlin, 1990, Science 249: 404-406;Cwirla et al., 1990, Proc. Natl. Acad. Sci. USA 87:6378-6382;並びにFelici, 1991, J. Mol. Biol. 222:301-310)(これらの参考文献はそれぞれ参照により本明細書にその全文が組み入れられる)実施することができる。目的の抗原又はその断片と免疫特異的に結合すると同定されている抗体又はそのフラグメントを、次いで目的の抗原に対する特異性とアフィニティについて試験することができる。
【0091】
目的の抗体又はそのフラグメントを、当技術分野で公知のいずれかの方法により、目的の抗原に対する免疫特異的結合及び他の抗原との交差反応性についてアッセイすることができる。免疫特異的結合と交差反応性を分析するために利用できるイムノアッセイとしては、限定されるものでないが、いくつかを列挙すれば、ウェスタンブロット、ラジオイムノアッセイ、ELISA(酵素結合免疫吸着アッセイ)、「サンドイッチ」イムノアッセイ、免疫沈降アッセイ、沈降反応、ゲル拡散沈降反応、免疫拡散アッセイ、凝集アッセイ、補体結合試験、免疫放射線測定アッセイ、蛍光イムノアッセイ、プロテインAイムノアッセイを利用する競合及び非競合アッセイ系が挙げられる。かかるアッセイは日常的に行われかつ当技術分野では公知である(例えば、Ausubel et al.編, 1994, 「Current Protocols in Molecular Biology」, Vol.1, John Wiley & Sons, Inc., New York、これは参照により本明細書にその全文が組み入れられる)。例示のイムノアッセイを簡単に以下に(限定を意図するのではないが)記載する。
【0092】
免疫沈降プロトコルは一般的に、細胞の集団を、タンパク質ホスファターゼ及び/又はプロテアーゼ阻害剤(例えば、EDTA、PMSF、アプロチニン、バナジン酸ナトリウム)を添加したRIPAバッファー(1%NP−40又はTritonX−100、1%デオキシコール酸ナトリウム、0.1%SDS、0.15M NaCl、0.01Mリン酸ナトリウム、pH7.2、1%トラジロール)などの溶解バッファーに溶解し、目的の抗体を該細胞溶解物に加え、ある時間(例えば、1〜4時間)40℃にてインキュベートし、プロテインA及び/又はプロテインGセファロースビーズを細胞溶解液に加え、約1時間以上40℃にてインキュベートし、ビーズを溶解バッファー中で洗浄し、そしてSDS/サンプルバッファーに再懸濁することを含む。目的の抗体の特定抗原と免疫沈降する能力は、例えばウェスタンブロット分析により評価することができる。当業者であれば、抗体の抗原との結合を増加しかつバックグラウンドを減少するように改変しうるパラメーター(例えば、セファロースビーズによる細胞溶解液の前清澄化)を理解しているであろう。免疫沈降プロトコルに関するさらなる考察については、例えば、Ausubel et al.編, 1994, 「Current Protocols in Molecular Biology」, Vol.1, John Wiley & Sons, Inc., New Yorkの10.16.1を参照されたい。
【0093】
ウェスタンブロット分析は一般的に、タンパク質サンプルを調製し、タンパク質サンプルをポリアクリルアミド中で電気泳動し(例えば、抗原の分子量に基づいて8%〜20%SDS−PAGE)、タンパク質サンプルをポリアクリルアミドゲルからニトロセルロース、PVDF又はナイロンなどのメンブランに移し、ブロッキング溶液(例えば、3%BSA又は無脂肪乳入りのPBS)中でメンブランをブロッキングし、メンブランを洗浄バッファー(例えば、PBS−Tween20)で洗浄し、メンブランをブロッキング溶液に希釈した一次抗体(例えば、目的の抗体)とともにインキュベートし、メンブランを洗浄バッファーで洗浄し、メンブランをブロッキング溶液に希釈した酵素基質(例えば、西洋ワサビペルオキシダーゼ又はアルカリホスファターゼ)若しくは放射性分子(例えば32P又は125I)と複合した二次抗体(一次抗体を認識するもの、例えば抗ヒト抗体)と共にインキュベートし、メンブランを洗浄バッファーで洗浄し、そして抗原の存在を検出することを含む。当業者であれば、検出されるシグナルを増加しかつバックグラウンドノイズを減少するように改変することができるパラメーターを理解しているであろう。ウェスタンブロットプロトコルに関するさらなる考察については、例えば、Ausubel et al.編, 1994, 「Current Protocols in Molecular Biology」, Vol.1, John Wiley & Sons, Inc., New Yorkの10.8.1を参照されたい。
【0094】
ELISAは、抗原を調製し、96ウエルマイクロタイタープレートのウエルを該抗原によりコーティングし、酵素基質(例えば、西洋ワサビペルオキシダーゼ又はアルカリホスファターゼ)などの検出可能な化合物と複合した目的の抗体をウエルに加え、そしてある時間インキュベートして、該抗原の存在を検出することを含む。ELISAにおいて、目的の抗体を検出可能な化合物と必ずしも複合させる必要はなく;代わりに検出可能な化合物と複合した二次抗体(目的の抗体を認識する抗体)をウエルに加えてもよい。さらに、ウエルを抗原でコーティングする代わりに、抗体をウエルにコーティングしてもよい。この場合、コーティングしたウエルに目的の抗原を加えた後、検出可能な化合物と複合した二次抗体を加えてもよい。当業者であれば、検出されるシグナルを増加するように改変することができるパラメーターだけでなく当技術分野で公知のELISAの他の変法も理解しているであろう。ウェスタンブロットプロトコルに関するさらなる考察については、例えば、Ausubel et al.編, 1994, 「Current Protocols in Molecular Biology」, Vol.1, John Wiley & Sons, Inc., New Yorkの11.2.1を参照されたい。
【0095】
抗体の抗原との結合アフィニティ及び抗体−抗原相互作用の解離速度(off-rate)は、競合結合アッセイにより測定することができる。競合結合アッセイの一例はラジオイムノアッセイであって、これは、増加する量の非標識抗原の存在のもとでの、標識(例えば、3H又は125I)した抗原を目的の抗体と共にインキュベーション、及び標識した抗原と結合した抗体の検出を含む。本発明の抗体又はそのフラグメントの目的の抗原に対するアフィニティ及び解離速度は、データからスキャッチャードプロット)分析により測定することができる。二次抗体との競合をラジオイムノアッセイを用いて測定することもできる。この場合、非標識の二次抗体の増加する量の存在のもとで、目的の抗原を標識(例えば、3H又は125I)した化合物と複合した本発明の抗体又はそのフラグメントとともにインキュベートする。
【0096】
好ましい実施形態においては、BIAcore動力学分析を用いて抗体又はそのフラグメントの目的の抗原との会合及び解離速度を決定することができる。BIAcore動力学分析は、その表面上に抗体又はそのフラグメントを固定したチップからの、目的の抗原の結合及び解離を分析することを含む(後述の実施例を参照)。
【0097】
抗体又はそのフラグメントはまた、目的の抗原のその宿主細胞受容体への結合を阻害するその能力について、当技術分野で公知の方法を用いてアッセイしうる。例えば、目的の抗原に対する受容体を発現する細胞を、抗体又はそのフラグメント(すなわちFabフラグメント)の存在下又は不在下にて抗原と接触させ、該抗体又はそのフラグメントが目的の抗原の結合を阻害する能力を、例えばフローサイトメトリー又はシンチレーションアッセイにより測定しうる。目的の抗原又は抗体若しくは抗体フラグメントは、検出可能な化合物、例えば放射性標識(例えば32P、35S、及び125I)、又は蛍光標識(例えば、フルオレセインイソチオシアネート、ローダミン、フィコエリスリン、フィコシアニン、アロフィコシアニン、o−フタルデヒド及びフルオレスカミン)で標識して、目的の抗原とその宿主細胞受容体との相互作用の検出を可能にしてもよい。あるいは、抗体又はそのフラグメントが目的の抗原のその受容体との結合を阻害する能力は、細胞非含有アッセイにおいて測定しうる。例えば、目的の抗原を抗体又はFabフラグメントと接触させ、該抗体又は抗体フラグメントが目的の抗原のその宿主細胞との結合を阻害する能力を測定しうる。好ましくは、抗体又はFabフラグメントを固相支持体に固定化し、目的の抗原を検出可能な化合物で標識する。あるいは、目的の抗原を固相支持体に固定化し、抗体又はFabフラグメントを検出可能な化合物で標識する。目的の抗原は、完全に又は部分的に精製されたものであってもよいし(例えば部分的又は完全に他のポリペプチドを含まない)、あるいは細胞溶解物の一部であってもよい。さらに、抗原は、抗原及びドメイン(結合ドメインなど)を含む融合タンパク質であってもよい。あるいは、抗原は、当業者に周知の技法を用いてビオチニル化されていてもよい(例えば、ビオチニル化キット、Pierce Chemicals, Rockford, IL)。
【0098】
目的の抗体又はそのフラグメントはまた、抗原の活性を阻害又はダウンレギュレートするその能力について、当業者に公知の技法を用いてアッセイしてもよい。本発明のベクター系により作製される抗体又はFabフラグメントは、抗原ポリペプチドの発現を阻害又はダウンレギュレートするそれらの能力についてアッセイすることも可能である。当業者に公知の技術としては、限定されるものではないが、ウエスタンブロット分析、ノーザンブロット分析、及びRT−PCRを用いてタンパク質発現を測定することができる。
【0099】
本発明のベクター系により作製される抗体又はFabフラグメントは、好ましくはin vitroで試験し、その後、ヒトに使用する前に、所望の治療的又は予防的活性についてin vivoで試験しうる。例えば、特異的抗体又は本発明の組成物の投与が適応か否かを判定するために用い得るin vitroアッセイとしては、被験体の組織サンプルを培養中で増殖させ、本発明の抗体又は組成物に曝露し又はそれを投与し、かかる本発明の抗体又は組成物の組織サンプルに対する効果を観察することを含むin vitro細胞培養アッセイが挙げられる。種々の特定の実施形態において、in vitroアッセイは、本発明の抗体又は組成物が特定の細胞型に対して所望の効果を有するか否かを判定するために行いうる。好ましくは、本発明のベクター系により作製された抗体又はFabフラグメントはまた、in vitroアッセイ及び動物モデル系においてヒトに投与する前に試験しうる。さらに、この実施形態に従って、犠牲にしたラット由来の組織を組織学的変化について試験してもよい。
【0100】
本発明においては、本発明のベクター系により作製された抗体又はFabフラグメントの予防的及び/又は治療的効力を証明するために、ヒト被験者を用いた臨床試験を行う必要はない。抗体又はそのフラグメントを用いたin vitro及び動物モデル試験からヒトについても推定することができ、またこれらの試験は該抗体又は抗体フラグメントの予防用途及び/又は治療用途を証明するために十分である。
【0101】
治療に使用するための本発明の抗体又は組成物は、適当な動物モデル系(限定されるものではないが、ラット、マウス、ウシ、サル、及びウサギなど)においてその毒性を試験することができる。抗体又は組成物の毒性のin vivo試験については、当技術分野で公知の任意の動物モデル系を用いることができる。
【0102】
ウイルス感染の治療又は予防における効力は、本発明のベクター系により作製される抗体又はFabフラグメントが、病原体の感染を阻害する、又は抗原と関連する1以上の症候を予防、改善若しくは緩和する能力を検出することにより証明することができる。例えば1以上の症候の改善、又は目的の抗体若しくは組成物の投与後の死亡率及び/若しくは疾患率の低減がある場合には、その処置は治療的と考えられる。さらに、本発明のベクター系により作製され、1以上の抗原と免疫特異的に結合する1以上の抗体又はFabフラグメントの投与後の免疫応答の増大がある場合には、その処置は治療的と考えられる。
【0103】
目的の抗体又は組成物は、IFN−α、IFN−β、IFN−γ、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、IL−8、IL−9、IL−10、IL−12及びIL−15などのサイトカインの発現を誘導する能力についてin vitro及びin vivoにおいて試験しうる。当業者に公知の技法を用いて、サイトカインの発現レベルを測定しうる。例えば、サイトカインの発現レベルは、例えばRT−PCR及びノーザンブロット分析によりサイトカインのRNAのレベルを分析することで、例えば免疫沈降の後にウエスタンブロット分析及びELISAを行うことによりサイトカインのレベルを分析することで、測定することができる。好ましい実施形態において、本発明のベクター系により作製される抗体又はFabフラグメントは、IFN−γの発現を誘導するその能力について試験する。
【0104】
目的の抗体又は組成物は、それらが免疫細胞、好ましくはヒト免疫細胞(例えばT細胞、B細胞、及びナチュラルキラー細胞)の生物学的活性をモジュレートする能力についてin vitro及びin vivoで試験しうる。目的のベクター系により作製された抗体又はFabフラグメントが免疫細胞の生物学的活性をモジュレートする能力は、抗原の発現を検出し、免疫細胞の増殖を検出し、シグナル伝達分子の活性化を検出し、免疫細胞のエフェクター機能を検出し、あるいは免疫細胞の分化を検出することにより、評価しうる。これらの活性を測定するために、当業者に公知の技法を使用することができる。例えば、細胞増殖は、3Hチミジン取り込みアッセイ及びトリパンブルー細胞計数によりアッセイしうる。抗原の発現は、例えば、限定されるものではないが、ウエスタンブロット、免疫組織化学的ラジオイムノアッセイ、ELISA(酵素結合免疫吸着アッセイ)、「サンドイッチ」イムノアッセイ、免疫沈降アッセイ、沈降素反応、ゲル拡散沈降素反応、免疫拡散アッセイ、凝集アッセイ、補体結合アッセイ、免疫放射アッセイ、蛍光イムノアッセイ、プロテインAイムノアッセイ、及びFACS分析等の技法を用いる競合及び非競合アッセイ系などのイムノアッセイによりアッセイしうる。シグナル伝達分子の活性化は、例えばキナーゼアッセイ及び電気泳動シフトアッセイ(EMSA)によりアッセイしうる。
【0105】
目的の抗体又は組成物はまた、ウイルス複製を阻害又はウイルス負荷を低減する能力について、in vitro、ex vivo及びin vivoアッセイにおいて試験しうる。本発明のベクター系により作製される抗体又はFabフラグメントはまた、感染の時間経過を低減する能力について試験しうる。本発明のベクター系により作製される抗体又はFabフラグメントはまた、感染症に罹患したヒトの生存期間を少なくとも25%、好ましくは少なくとも50%、少なくとも60%、少なくとも75%、少なくとも85%、少なくとも95%又は少なくとも99%で増大する能力について試験しうる。さらに、本発明のベクター系により作製される抗体又はFabフラグメントは、感染症に罹患したヒトの入院期間を少なくとも60%、好ましくは少なくとも75%、少なくとも85%、少なくとも95%又は少なくとも99%で低減する能力について試験しうる。当業者に公知の方法を用いて、目的の抗体又は組成物の機能をin vivoで分析することも可能である。
【0106】
5.2.2.ファージディスプレイを用いたスクリーニング方法
当業者であれば理解できるように、個々のタンパク質又は他の化合物、並びにタンパク質又は他の化合物の大きなライブラリー(例えばファージディスプレイライブラリー)をスクリーニングして、目的の特定の抗原と結合する分子を同定する方法がいくつかある。
【0107】
ファージディスプレイ法においては、機能性抗体ドメインが、同ドメインをコードするポリヌクレオチド配列を保持するファージ粒子の表面に提示される。特に、VH及びVLドメインをコードするDNA配列を、動物cDNAライブラリー(例えば、リンパ組織のヒト又はマウスcDNAライブラリー)から増幅する。VH及びVLドメインをコードするDNAをPCRによりscFvリンカーと一緒に組換えて、ファージミドベクター(例えば、pCANTAB6又はpComb3 HSS)中にクローニングする。そのベクターを大腸菌(E.coli)中にエレクトロポレーションして大腸菌(E.coli)をヘルパーファージに感染させる。これらの方法で使用されるファージは、典型的には、fd及びM13を含む繊維状ファージであり、VH及びVLドメインを通常、組換え法によりファージ遺伝子III又は遺伝子VIIIのいずれかに融合させる。目的の抗原と結合する抗原結合ドメインを発現するファージを、抗原により、例えば、標識した抗原又は固体表面若しくはビーズに結合若しくは捕獲された抗原を用いて、選択又は同定することができる。本発明の抗体を作製するために利用しうるファージディスプレイ法の例としては、Brinkman et al., 1995, J. Immunol. Methods 182:41-50;Ames et al., 1995, J. Immunol. Methods 184:177-186;Kettleborough et al., 1994, Eur. J. Immunol. 24:952-958;Persic et al., 1997, Gene 187:9-18;Burton et al., 1994, Advances in Immunology 57:191-280;PCT出願第PCT/GB91/01134号;国際公報WO90/02809号、WO91/10737号、WO92/01047号、WO92/18619号、WO93/11236号、WO95/15982号、WO95/20401号、及びW097/13844号;並びに米国特許第5,698,426号、第5,223,409号、第5,403,484号、第5,580,717号、第5,427,908号、第5,750,753号、第5,821,047号、第5,571,698号、第5
,427,908号、第5,516,637号、第5,780,225号、第5,658,727号、第5,733,743号、及び第5,969,108号に開示された方法が挙げられ、これらはそれぞれ参照によりその全文が本明細書に組み入れられる。
【0108】
ファージディスプレイライブラリの例は以下の文献に記載されている:Scott and Simith, 1990, Science 249:386-390;Devlin et al., 1990, Science, 249:404-406;Christian et al., 1992, J. Mol. Biol. 227:711-718;Lenstra, 1992, J. Immunol. Meth. 152:149-157;Kay et al., 1993, Gene 128:58-65;及びPCT公報WO94/18318(1994年8月18日出願)。
【0109】
以上の参考文献に記載の通り、ファージ選択の後、ファージからの抗体コード領域を単離し、さらにそれを使用して、ヒト抗体を含む完全抗体、又は任意の他の所望の抗原結合フラグメントを作製し、それらを、例えば以下に記載のようにして、哺乳動物細胞、昆虫細胞、植物細胞、酵母、及び細菌を含む任意の所望の宿主にて発現させることができる。組換え法によりFab、Fab'及びF(ab')2フラグメントを製造する技術はまた、PCT公報WO92/22324号;Mullinax et al., 1992, BioTechniques 12 (6):864-869;Sawai et al., 1995, AJRI 34:26-34;並びにBetter et al., 1988, Science 240:1041-1043(上記参考文献は参照によりその全文が組み入れられる)に開示された方法などの当技術分野で公知の方法を用いて、使用することができる。
【0110】
完全抗体を作製するために、VH又はVLヌクレオチド配列、制限酵素切断部位、及び制限酵素切断部位を保護するフランキング配列を含むPCRプライマーを用いて、scFvクローン中のVH又はVL配列を増幅することができる。当業者に公知のクローニング技術を利用して、PCR増幅されたVHドメインをVH定常領域、例えば、ヒトγ4定常領域を発現するベクター中にクローニングし、また、PCR増幅したVLドメインをVL定常領域、例えばヒトκ若しくはλ定常領域を発現するベクター中にクローニングすることができる。好ましくは、VH又はVLドメインを発現するベクターは、EF−1αプロモーター、分泌シグナル、可変ドメイン、定常ドメイン及びネオマイシンなどの選択マーカーに対するクローニング部位を含む。VH及びVLドメインを、必要な定常領域を発現する1つのベクター中にクローニングすることもできる。次いで、重鎖変換ベクター及び軽鎖変換ベクターを培養細胞株中に同時トランスフェクトし、当業者に公知の技術を用いて、全長抗体、例えばIgGを発現する安定した一過性の培養細胞株を作製する。
【0111】
5.2.3.抗体配列の最適化方法
新規抗体の試験及び特性決定についての上記用途に加えて、二重発現ベクターは、所望の結合特性又は治療特性について既存の抗体を最適化するために用いることが可能である。本発明のこの態様において、既知のIgG重鎖又は軽鎖配列をコードするヌクレオチド配列を二重発現ベクター系にクローニングし、化学的、合成又は遺伝的突然変異誘発を行い、そのヌクレオチド配列を改変することができる。続いて、配列変異体は、目的の特性の変化について細菌及び/又はヒト細胞においてスクリーニングしうる。
【0112】
抗体をコードするポリヌクレオチドを得て、当技術分野で公知のいずれかの方法によりヌクレオチド配列を決定することができる。所望の抗原に対して免疫特異的な抗体のヌクレオチド配列は、例えば、文献又はGenBankなどのデータベースから得ることができる。VITAXINTMのアミノ酸配列は既知であるので、この抗体をコードするヌクレオチド配列を、当技術分野で公知の方法を利用して(すなわち、特定のアミノ酸をコードすることが知られるヌクレオチドコドンを、抗体をコードする核酸を生成するように組み立てて)決定することができる。抗体をコードするかかるポリヌクレオチドは、化学合成したオリゴヌクレオチドから組み立てることができ(例えば、Kutmeier et al., 1994, BioTechniques 17:242に記載の通り)、これは、簡単に説明すると、抗体をコードする配列の一部分を含有する重複オリゴヌクレオチドの合成、これらのオリゴヌクレオチドのアニーリング及びライゲーション、次いでライゲートしたオリゴヌクレオチドのPCRによる増幅を伴う。
【0113】
あるいは、抗体をコードするポリヌクレオチドを、好適な起源由来の核酸から作製することができる。特定の抗体をコードする核酸を含有するクローンが入手できなくても、その抗体分子の配列が既知であれば、その免疫グロブリンをコードする核酸は、化学的に合成することができるし、あるいは、適当な起源(例えば、抗体cDNAライブラリー若しくはそれから作製したcDNAライブラリー、それから単離した核酸、好ましくはポリA+RNA、目的の抗体を発現するように選択されたハイブリドーマ細胞などの、抗体を発現する任意の組織若しくは細胞)からその配列の3’及び5’末端にハイブリダイズする合成プライマーを用いるPCR増幅によるか、又は、特定の遺伝子配列に特異的なオリゴヌクレオチドプローブを用いて、例えばcDNAライブラリーから前記抗体をコードするcDNAクローンを同定してクローニングすることにより、取得することができる。PCRにより作製された増幅核酸は、次いで、当技術分野で公知の方法を用いて複製可能なクローニングベクター中にクローニングすることができる。
【0114】
抗体のヌクレオチド配列が一度決定されると、抗体のヌクレオチド配列を、ヌクレオチド配列の遺伝子操作に関する当技術分野で公知の方法、例えば組換えDNA技術、位置指定突然変異、PCRなど(例えば、Sambrook et al., 1990, 「Molecular Cloning, A Laboratory Manual」, 第2版, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY、及びAusubel et al.編, 1998, 「Current Protocols in Molecular Biology」, John Wiley & Sons, NY、に記載の技術を参照、これらの文献は本明細書に参照によりその全文が組み入れられる)を用いて遺伝子操作して、異なるアミノ酸配列を有する抗体を作製し、例えばアミノ酸置換、欠失、及び/又は挿入を創出することができる。
【0115】
特定の実施形態においては、1以上のCDRを、慣用される組換えDNA技術を利用してフレームワーク領域内に挿入する。フレームワーク領域は、天然又はコンセンサスフレームワーク領域であってもよく、好ましくはヒトフレームワーク領域である(例えば、ヒトフレームワーク領域の列挙については、Chothia et al., 1998, J. Mol. Biol. 278:457-479を参照)。好ましくは、フレームワーク領域とCDRの組合せにより作製されたポリヌクレオチドは、特定の抗原と特異的に結合する抗体をコードする。好ましくは先に考察したように、1以上のアミノ酸置換をフレームワーク領域内で引き起こしてもよく、かつ好ましくはそのアミノ酸置換は抗体のその抗原との結合を改良するものである。さらに、かかる方法を利用して、鎖内ジスルフィド結合に参加する1以上の可変領域システイン残基のアミノ酸置換若しくは欠失を引き起こし、1以上の鎖内ジスルフィド結合を欠く抗体分子を作製してもよい。ポリヌクレオチドに対するその他の改変は本発明により包含されかつ当技術分野の範囲内にある。
【0116】
本発明に従って使用しうる市販の抗体の例を以下の表1に示すが、これに限定されるものではない。
【表1】
【0117】
【0118】
【0119】
【0120】
5.3.本発明と共に使用するための他の方法
5.3.1.他のポリペプチド及びその断片を発現するための二重発現ベクター系の使用
本発明の二重発現ベクター系は、本明細書に開示する抗体を発現するために記載した方法を用いて、哺乳動物細胞において目的の任意のポリペプチドを、及び細菌細胞において該ポリペプチドの断片、好ましくは可能性断片を発現するために用いることができる。目的のポリペプチドは、膜結合ポリペプチド又は分泌ポリペプチドであることが好ましく、細菌細胞の周辺腔領域に発現され分泌された場合にアッセイ可能な活性を保持する可溶性ドメインを有するものである。目的のドメインとしては、限定されるものではないが、DNA結合ドメイン、タンパク質−タンパク質相互作用ドメイン、キナーゼドメイン、又は他の酵素的若しくは機能的タンパク質ドメインが挙げられる。
【0121】
二重発現ベクターは、イントロンを全長ポリペプチドのシグナル配列に挿入することにより構築する。イントロンは、スプライス受容配列と重複し、目的のポリペプチドのコード領域と読み取り枠を合わせた細菌プロモーターとシグナル配列を含み、その結果、細菌プロモーターからの転写が細菌細胞における目的のドメインの発現を指令するように設計する。そのような配列の構築方法は、以下に示す実施例において詳細を説明する。
【0122】
かかる二重発現ベクターの構成要素としては、(a)細菌性複製起点、(b)哺乳動物複製起点、(c)上記分泌又は膜結合ポリペプチドをコードするヌクレオチド配列と機能的に結合した哺乳動物プロモーター及び/又はエンハンサー配列が含まれ、該ヌクレオチド配列は、少なくとも1つのイントロンを含む哺乳動物シグナル配列を含み、該イントロンは該ポリペプチドの可溶性ドメインをコードする配列と機能的に結合された細菌プロモーター及び細菌シグナル配列を含み、その結果、該細菌プロモーター及び細菌シグナル配列が細菌細胞における周辺腔への上記ポリペプチドの可溶性ドメインの発現及び分泌を指令し、上記哺乳動物シグナル配列は該ポリペプチドの発現及び分泌を指令し、ここで哺乳動物細胞における発現のためのプロモーターは、上記ポリペプチドの可溶性ドメインをコードする上記ヌクレオチド配列と機能的に結合している。
【0123】
目的の膜結合又は分泌ポリペプチドの例としては、限定されるものではないが、細胞表面受容体、例えば限定されるものではないが、エリスロポエチン受容体(Epo−R;Noguchi et al., 1991, Blood 78(10):2548-2556)、インスリン受容体(InsR;Ebina et al., 1985, Cell 40:747-758;Ullrich, 1985, Nature 313:756-761)、及び腫瘍壊死因子α受容体(TNFαR;Gray et al., 1990, Proc. Natl. Acad. Sci. USA 87:7380-7384);一回膜貫通チロシン受容体キナーゼ(TRK)様クラス受容体のメンバー(Ullrich & Schlessinger, 1990, Cell 61:203-12;Hunter & Cooper, 1985, Ann. Rev. Biochem. 54:897-930)が挙げられる。このクラスには以下のものが含まれる:表皮増殖因子受容体ファミリー、例えば表皮増殖因子(EGF;Ullrich et al., Nature, 1984, 309:418-25;Schector et al., Nature 278:835-38)、ワクシニア増殖因子(Brown et al., 1985, Nature 313:491-92)、アンフィレギュリン/神経鞘腫由来増殖因子(AR又はSDGF;Schoyab et al., 1989, Science 243:1074-1076)、ヘアピン結合EGF様因子(HB−EGF;Higashiyama et al., 1991, Science 251:936-939)、neu分化因子(NDF;Wen et al., 1992, Cell, 69:559-72)、及びHer2(Coussens et al., 1985, Science 230:1132-39;Santanta et al., 1994, Proc. Natl. Acad. Sci. USA 91:1711-1715)などのヒレギュリン(Holmes et al., 1992, Science 256:1205-10)、インスリン受容体ファミリー、例えばINSR(上記)及びIRR、血小板由来増殖因子(PDGF)受容体ファミリー、例えばα−PDGFR(Potts & Carrington, 1993, Dev. Dyn. 198:14-21)、β−PDGFR(Chi et al., 1997, Oncogene 15:1051-58)、CSF1−R(例えば、Waterfield et al., 1983, Nature 304:35-39)、c−Kit幹細胞因子受容体(Lemmon et al., 1997, J. Biol. Chem. 272:6311-6317)、繊維芽細胞増殖因子受容体(FGFR)、例えばCEK2(Pasquale, 1990, Proc. Natl. Acad. Sci. U.S.A. 87:5812-16)、TRK受容体ファミリー、例えばTRK及びTRK−B;並びにEPH/ECK受容体ファミリー、例えばElf−1及びEck(Cheng & Flanagan, 1994, Cell 79:157-68;Lindberg & Hunter, 1990, Mol. Cell Biol. 10:6316-24)、神経増殖因子受容体(Woo et al., 1998, Protein Sci. 7:1006-1016;Johnson et al., 1986, Cell 47:545-54)、並びにインスリン様増殖因子受容体(Ullrich et al., 1986, EMBO J. 5:2503-12;及びSepp-Lorenzino, 1998, Breast Cancer Res. Treat. 47:235-253)。TK様ファミリーの受容体の他のメンバーもまた用いることが可能である。例えば、van der Greer et al., 1997, Ann. Rev. Cell Biol. 10:251-337;Herz et al., 1997, J. Recept. Signal Transduct. Res. 17:671-776(これらは参照によりその全体を本明細書に援用する)、並びにそれらの参照文献を参照されたい。
【0124】
他の実施形態において、目的のポリペプチドは、7回膜貫通クラスの受容体(例えば、Gタンパク質共役型クラスの受容体;GPCR)のメンバーでありうる:例えば、β3アドレナリン作動性受容体(Emorine et al., 1989, Science 245:1118-21;Huang et al., 1997, J. Recept. Signal Transduct. Res. 17:599-607)、ドーパミン受容体、例えばドーパミンD2受容体(Wilkie et al., 1993, Genomics 18:175-184;Bunzow et al., 1988, Nature 336:783-7)、及びムスカリン作動性アセチルコリン受容体(Strader et al., 1994, Ann. Rev. Biochem. 63:101-32、及びそれに引用された参照文献を参照。参照によりその全体を本明細書に援用する)、イオンチャネル、例えば限定されるものではないが、Kvl.3カリウムチャネル(Kath et al., 1997, Annual Reports in Med. Chem., Hagmann編、32:181-89)、NHEI及びNHE2 Na+/H+交換体(Fafournoux & Pouysseyur, 1994, J. Biol. Chem. 269:2589-96)、電位依存性イオンチャネルファミリーの受容体、例えばK+感受性チャネル及びCa2+感受性チャネル(Hille, B, "Ionic Channels of Excitable Membranes", 1992, Sinauer Associates, Sunderland, MA;Catterall, W.A., 1991, Science 253:1499-1500及びこれらに引用された参考文献を参照。参考によりその全文を本明細書に援用する)、受容体タンパク質−チロシンホスファターゼファミリーのメンバー(すなわちR−PTP)、例えば限定されるものではないが、CD45(すなわちリンパ球共通抗原、LCA)、R−PTPα、β、γ、κ及びその他(例えば、Denu et al., 1996, Cell 87:361-64;Fashena and Zinn, 1995, Curr. Biol. 5:1367-69、それぞれ参照によりその全文を本明細書に援用する)、サイトカイン受容体ファミリーのメンバー、IL−1サイトカイン受容体ファミリー(IL−1α及びIL−1β、例えば、Vigers et al., 1997, Nature 386:190-194参照)、クラスIサイトカインファミリー、特にホモダイマー形成に関与する高度に保存されたシステインを特徴とする造血サイトカイン受容体の成長ホルモン受容体サブファミリー(Watowich et al., Proc. Nat. Acad. Sci., 89:2140-44)。このファミリーには、EPO受容体(Noguchi et al., 1991、前掲)だけではなく、成長ホルモン受容体(Cunningham et al., 1989, Science 243:1330)、プロラクチン受容体(Boutin et al., 1988, Cell 53:69)、CSF、顆粒球−コロニー刺激因子受容体(Seto et al., 1992, J. Immunol. 148(1):259-266)、ソマトトロピン受容体(Leung et al., 1987, Nature 330:537)、グリア由来神経栄養因子(GDNF)受容体、例えばGFRα3(Baloh et al., Proc. Natl. Acad. Sci. 95:5801-06)、及び他多数(Herz et al. 1997、前掲)、並びにクラスIIサイトカイン受容体(インターフェロン)ファミリーメンバー(このファミリーでは、リガンド結合はJAKキナーゼを介して二量体化及び活性化を誘導しうる)(Aguet et al., 1988, Cell 55:273-80;及びUze et al., 1990, Cell 60:225-234)。
【0125】
他の実施形態において、目的のポリペプチドは、核ホルモン受容体スーパーファミリーのメンバーでありうる(例えば、Mangelsdorf et al., 1995, Cell 83:835-39及びそれに引用された参照文献。参照によりその全体を本明細書に援用する):例えば、ステロイド受容体(Beato et al., 1995, Cell 83:851-57及びそれに引用された参照文献。参照によりその全体を本明細書に援用する)、糖質コルチコイド(Hollenberg et al., 1985, Nature, 318:635-41;またEvans, 1989, Recent Prog. Horm. Res. 45:1-22及びそれに引用された参照文献)、アンドロゲン(Tilley et al., Proc. Nat. Acad. Scie. U.S.A., 1989, 86:327-31)、アルドステロン、プロゲステロン、及びエストロゲン受容体(Greene et al., 1986, Nature 320:134-39;Tsai & O'Malley, 1994, Ann. Rev. Biochem. 63:451-86及びそれに引用された参照文献。参照によりその全体を本明細書に援用する)、ヘテロダイマー受容体、例えばチロキシン、ビタミンD、ビタミンA、レチノイド(RAR、RXR)、プロスチノイド受容体(Mangelsdorf & Evans, 1995, Cell 83:841-50及びそれに引用された参照文献。参照によりその全体を本明細書に援用する)、例えば肝核因子HNF4(Sladek et al., 1990, Genes Dev. 4:2353-65)。これらのクラス内のオーファン受容体は特に興味深い配列であり、その推定リガンドが未知であるヘテロダイマー及びホモダイマー受容体のファミリーを表すリガンドを同定するために本発明の方法の一部として用いることができる。
【0126】
別の実施形態において、目的のポリペプチドは、非膜型の非分泌型ポリペプチド、例えば核転写因子タンパク質でありうる。転写因子としては、限定されるものではないが、Fos/Jun(Bohmann et al., Science 238:1386-92;Angel et al., 1988, Nature 332:166-71)、C/EBP(Landshultz et al., 1988, Science, 240:1759-64)、GCN4(例えば、Agre et al., 1989, Science 246:922-926、また本明細書の第9節に示す実施例参照)、ヘリックスループヘリックス(HLH)ドメインタンパク質、例えばMyc(Murre et al., 1989, Cell 56:777-783)及びMyoD並びにE12/E47様タンパク質とin vivoでヘテロオリゴマー化する必要のある他の筋原性HLHタンパク質(Lasser et al., 1991, Cell 66:305-15)、並びに当技術分野で周知の他の転写因子が挙げられる。
【0127】
本明細書に例示するタンパク質に加えて、目的のポリペプチドは、公共データベース、例えばSwiss Protein Data Base(SWISS−PROT;Bairoch & Apweiler, 1998, Nucl. Acids Res. 26:38-42)などに登録された任意の膜結合又は分泌型ポリペプチドに由来するアミノ酸残基を含みうる。
【0128】
5.3.2.抗体の診断用途
本発明のベクター系により作製された抗体又はFabフラグメントを使用し、生物学的サンプル中の抗原レベルを本明細書に記載の又は当業者に公知の古典的免疫組織学的方法を用いて評価することができる(例えば、Jalkanen et al., 1985, J. Cell. Biol. 101:976-985;Jalkanen et al., 1987, J. Cell. Biol. 105:3087-3096を参照)。タンパク質遺伝子発現を検出するために有用な他の抗体に基づく方法は、イムノアッセイ、例えば、酵素結合免疫吸着アッセイ(ELISA)及びラジオイムノアッセイ(RIA)を含む。好適な抗体アッセイ標識は当技術分野で公知であり、酵素標識、例えばグルコースオキシダーゼ;放射性同位体、例えばヨウ素(125I,131I)、炭素(14C)、硫黄(35S)、トリチウム(3H)、インジウム(121In)、及びテクチニウム(99mTC);発光標識、例えばルミノール;及び蛍光標識、例えばフルオレセイン、ローダミン及びビオチンが挙げられる。
【0129】
被験者のサイズと使用するイメージングシステムが、診断イメージを作るために必要なイメージング成分の量を決定しうることは当技術分野で理解されるであろう。ヒト被験者に対する放射性同位体成分の場合、注射される放射能の量は通常約5〜20ミリキューリーの99mTcであろう。標識した抗体又は抗体フラグメントを次いで選択的に特定のタンパク質を含有する細胞の位置に蓄積させる。in vivo腫瘍イメージングは、S. W. Burchiel et al., 「Immunopharmacokinetics of Radiolabeled Antibody and Their Fragments」,Tumor Imaging :The Radiochemical Detection of Cancer, Chapt. 13, S. W. Burchiel and B. A. Rhodes, 編, Masson Publishing Inc. (1982)に記載されている。
【0130】
使用する標識のタイプ及び投与様式を含む複数の変数に依存して、標識した分子が被験者の部位に選択的に濃縮しうるためのかつ無結合の標識した分子がバックグラウンドレベルにクリアされるための投与後の時間間隔は、6〜48時間又は6〜24時間又は6〜12時間である。他の実施形態においては、投与後の間隔は5〜20日又は5〜10日である。
【0131】
被験者中の標識した分子の存在は、in vivo走査に対する当技術分野で公知の方法を用いて検出することができる。これらの方法は、使用する標識のタイプに依存する。当業者は特定標識を検出するための適当な方法を決定することができよう。本発明の診断に利用しうる方法とデバイスは、限定されるものでないが、コンピューター断層撮影(CT)、位置放出断層撮影(PET)などの全身走査、磁気共鳴イメージング(MRI)、及びソノグラフィを含む。
【0132】
特定の実施形態においては、分子を放射性同位体により標識し、患者において放射線応答外科機器(Thurston et al., 米国特許第5,441,050号)を用いて検出する。他の実施形態においては、分子を蛍光化合物により標識し、患者において蛍光応答走査機器を用いて検出する。他の実施形態においては、分子を位置放出性金属により標識し、患者においてポジトロン放出断層撮影を用いて検出する。さらに他の実施形態においては、分子を常磁性標識により標識し、患者において磁気共鳴イメージングを用いて検出する。
【実施例】
【0133】
6.1.抗CD16 Fab及びIgGの発現
大腸菌においてFabを、哺乳動物細胞においてIgGを発現させるための二重発現ベクターの設計の基礎とするため、以下の予備的研究を行った。ベクターを2種構築した。第1のものは、大腸菌においてFab分子を発現させるためのベクターであり、第2のものは、哺乳動物細胞においてIgGを発現させるためのベクターであった。キメラ型又はヒト化型抗CD16 Mabのうちいずれかの重鎖及び軽鎖cDNAを用いて、発現ベクターを検証した。
【0134】
大腸菌においてFab分子を発現させるため、lacプロモーターオペレーター(lacPO)の制御下で個々の軽鎖(LC)配列及びVH CH1(Fd)鎖配列がそれぞれpelBシグナルペプチドコード配列と融合する、Barbasにより記載のもの(Barbaset al., 1991, Proc. Natl. Acad. Sci. U.S.A. 88:7978-7982)と類似するベクターを構築した。Fdセグメント配列はまた、精製用のC末端Hisタグと同定用のHSVエピトープタグの両方をコードする配列を含む。
【0135】
このベクターを使用して、大腸菌(BL21又はXL1 Blue株)の周辺質中にキメラ又はヒト化抗CD16 Fabを約1mg/l分泌させた。この物質に結合活性があるかどうかを試験するために、周辺質抽出物からFabを精製し、サンドイッチELISAでCD16受容体の可溶性部分(sCD16)と結合できるかどうかを、又は免疫複合体とのsCD16の結合を阻害できるかどうかを分析した。図1に示すように、精製したキメラFabは、還元後25kdの単一バンドとしてSDS PAGEゲル上で移動し、又は還元しない場合50kdの単一バンドとして移動する。このことから、この産物がジスルフィド結合していることが示唆される。ウェスタンブロット上で、このバンドは、抗LC抗体にも抗HSVタグ抗体にも反応することから、LC鎖もFd鎖も存在することが示唆される。
【0136】
IgGを発現させるため、全長重鎖(γ1)又は軽鎖(K)遺伝子をコードする配列をネズミVHシグナル配列と融合させ、ベクターpCI neo(Promega)内でCMVieプロモーター/エンハンサーの下流に配置した、個々のベクターを構築した。ネズミVHシグナルペプチドコード領域は、天然に存在するイントロンを含み、多数の抗体の軽鎖配列と重鎖配列をどちらも分泌する信頼性が非常に高いことが認められた。これに対し、分泌のために発現される可変領域由来のシグナルペプチドを含むcDNA配列からは、一貫しない結果が得られた。キメラ又はヒト化抗CD16の軽鎖及び重鎖プラスミドをHEK293細胞に共トランスフェクトした。一般に、3日後に培地中に5〜10μg/ml分泌された。
【0137】
両方のベクターから精製した物質を、結合するかどうかELISAで直接試験し、又はフルオレセインBSAとヒトIgG1キメラ型抗フルオレセインMab4 4 20の間で形成された免疫複合体とsCD16との結合を阻害するかどうかについてアッセイした。免疫複合体とsCD16との結合阻害を図2に示す。このプロトコルは以下の通りである。MaxiSorpイムノプレート(Nunc F96)を、1ウエルにつき500ngの炭酸緩衝液中のBSA FITCで、4℃で一晩被覆した。0.5%BSAのPBST溶液で、このプレートを室温で30分間ブロッキングした。Ch4 4 20(ヒトIgG1)をプレートに1ウエルにつき50ng加え、室温で1時間インキュベートして免疫複合体を形成させた。精製したCh3G8Fab、(陽性対照である)Ch3G8、(陰性対照である)ヒトIgG1を、sCD16 G2ビオチンを0.5μg/ml含む0.5%BSA/PBSTで、図に示した最終濃度まで希釈し、免疫複合体を含んでいるウエルに加えた。次いで、このプレートを室温で2時間インキュベートした。sCD16 G2ビオチンが免疫複合体と結合したことを、1:5000に希釈した西洋ワサビ結合ストレプトアビジン(Pharmacia)によって検出した。室温で30分間インキュベートした後、TMB(BioFX)を検出用の基質として用いた。上記のステップの間で、1×PBS/0.1%Tween20(PBST)でプレートを常に3回洗浄した。発色させるため、このプレートを室温で5〜10分間維持した。この反応を0.18M硫酸で停止し、OD450nmを測定した。
【0138】
図3に示すように、この物質はまた、sCD16Aとの直接の結合においても活性であった。可溶性モノマーCD16は、抗CD16 Mab LNK 16で被覆したイムノプレート上に捕捉された。結合しなかったリガンドを洗浄して除いた後、キメラFabの希釈物をプレートに加え、次いでこのプレートを室温で1時間インキュベートした。次いで、上記に記載のように、ヤギ抗ヒトFab HRP結合物を用い、その後TMBで発色させて、結合したFabを検出した。このsCD16A結合アッセイの結果を図3に示す。
【0139】
6.1.1.哺乳動物細胞においてIgG軽鎖及び重鎖を、大腸菌においてFabを発現させるための発現ベクター
以下の手順を使用して、大腸菌においてFabフラグメントを、哺乳動物細胞においてIgGを発現させ、スクリーニングするための二重発現ベクターを設計し構築した。本発明のこの実施形態では、本発明に従ってどちらの系でも効率のよい発現及び分泌を得るために、2つの条件が課せられた。第1に、大腸菌における発現及び分泌では、分泌されるポリペプチドの前にシグナルペプチドが機能しなければならない。第2に、哺乳動物における発現及び分泌では、メッセージを正しくスプライシングして、シグナル配列をコードするセグメントを結合させなければならない。細菌シグナル配列をコードする領域が、哺乳動物のスプライス受容部位と重複するので、これら2つの構成要素の設計を一緒に考慮しなければならない。
【0140】
この合成セグメントの設計及び構築のための鋳型として、pET25bのpelBシグナルペプチドコード配列を使用した。このコード配列を改変して、シグナルペプチドの中心にある疎水性残基を保持しながら、コンセンサス3’スプライス供与部位との相同性を最大にした。この方法では、ロイシンコドン(CTC)で2つのアラニンコドン(GCC又はGCT)を置き換えて、一続きのピリミジン(ピリミジンストレッチ)を正確なスプライシングに十分な長さにした。さらに、このピリミジンストレッチの上流に、潜在的なスプライシング分枝点をもたらすために、Alaコドン(GCT)を、Ile(ATC)に変化させた。最後に、原核生物と哺乳動物のシグナルペプチドが共有している領域において、−1及び−2位で、どちらの系でも機能的活性を保持する可能性が最も高い残基を選択した。改変型原核生物シグナルペプチドが依然として好ましいスプライス切断部位を保持しているかどうかを予測するために、神経ネットワークアルゴリズムを用いるSignalPプログラム(Nielsen et al., 1997, Int. J. Neural Sys. 8:581-599)によって配列を分析した。バークレーショウジョウバエゲノムプロジェクトウェブサイトのスプライス部位予測プログラムを用いて、スプライス部位の潜在的機能を評価した(Reese et al., J. Comput. Biol., 1997, 4(3):311 23を参照のこと)。このプログラムも、ヒト遺伝子向けの神経ネットワークアルゴリズムを用いている。
【0141】
プラスミドベクターpMGX115は、哺乳動物発現ベクターpCI neo内にヒト化重鎖をコードするミニ遺伝子を含む。このミニ遺伝子では、唯一のイントロンがシグナルペプチドをコードする領域内にある。正確なスプライス結合部位は、Glyコドン内で、シグナルペプチド切断部位の−4位に位置する(図4A〜Bを参照のこと)。図4Aに示す設計されたセグメントを以下のように導入する。最初に、PCRによりpUC18からlacプロモーター及びオペレーター(lacPO)配列を得、BglII−XbaI断片としてこれをpET25b中に導入して、T7プロモーターを置き換え、pMGX102を生成した。次いで、pMGX102を鋳型として用いて、lacPO配列をpelBシグナル配列と一緒にPCRで増幅した。次いで、重複PCRを用いて、シグナル配列の5’側のエキソン(5’スプライス部位を含む)及び重鎖(VH Cγ1)をそれぞれ含むpMGX115由来の2つの断片間にこの断片を配置した。このプロセスの間に、成熟VHをコードするセグメントをlacPO pelB配列を含むセグメントと結合させるのに使用する重複PCRプライマー内に改変を設計することにより、シグナル配列に改変を導入した。得られた断片を発現ベクターpCI Neo中にクローン化して、pMGX121を生成した。
【0142】
イントロン、スプライス結合部位及びシグナルペプチドの改変が発現及び分泌に影響を及ぼすかどうかを決定するために、HEK293細胞においてpMGX121の発現を調べた。抗ヒトFc抗体を用いてヒトIgGを捕捉し、抗ヒト重鎖+軽鎖HRP結合物を用いて検出するELISAで測定したとき、発現に対して有害な影響は認められなかった。
【0143】
大腸菌におけるFd(VH CH1フラグメント)の発現で停止コドンをもたらすために、pMGX121中のCH1とヒンジ領域の間に第2イントロンを導入して、pMGX578を生成した(図4Bを参照のこと)。ゲノムDNAからnested PCRで天然のヒトγ1遺伝子由来のイントロンを増幅し、重複PCRで他のセグメントとこれを結合させた。次いで、部位特異的変異誘発を行ってイントロンの開始部の近くに停止コドンを導入した。さらに、スプライシングに干渉しないような配列を設計し、スプライス部位配列が保持されるかどうかを、上記のプログラムを用いて調べた。次いで、得られたプラスミドpMGX579の配列を決定し、LC(軽鎖)発現プラスミドと共トランスフェクトしたとき、これがHEK293細胞において発現することを確認した。
【0144】
類似のLC発現プラスミドを生成するために、重複PCR手順により、pMGX121由来のlacPO pelB配列を含むシグナルイントロンをヒト化軽鎖コード配列と結合させ、発現カセット全体が切り出されるようにAscI部位がCMVieプロモーターの5’末端及びSV40ポリA部位の3’末端に導入されている以外はpMGX579と同一であるpMGX581中に、この断片をクローン化した。このプラスミドをpMGX582と名付けた。
【0145】
このプラスミドからLCが発現するかどうかについて、以下の通りに試験した。0.5mM IPTGで、pMGX506又はpMGX582(LC)を有する大腸菌株XL 10goldを誘導した。3時間後、細胞を収集し、周辺質分画を単離した。この物質を1/2及び1/10に希釈し、ヤギ抗ヒトFab(Jackson)で被覆したマイクロタイタープレートに加えた。室温で約1時間インキュベートした後、結合しなかった物質を洗浄して除き、HRP結合ヤギ抗ヒトLC(Biosource,Inc.)を用いて、結合した軽鎖(LC)を検出した。室温で1時間インキュベートした後、TMB試薬を用いてプレートを発色させ、約10分後に0.18M H2SO4を用いて発色を停止した。図6に示す結果から、大腸菌においてこのプラスミドからLCが発現することが確認された。LCが大腸菌の周辺質中に明らかに分泌されたことが特に重要であり、このことからシグナル配列が機能的であったことが示される。このプラスミドを重鎖(HC)発現プラスミドpMGX115と共トランスフェクトした後にIgGが発現することも実証された。
【0146】
大腸菌におけるFab、及び哺乳動物細胞におけるIgGの発現を評価するために。プラスミドpMGX583を構築した。pMGX583は、HCとLCの発現カセットをどちらも含み、各カセットにlacPO pelBイントロン配列が含まれている。pMGX583を構築するために、AscIで消化することにより、発現カセット全体であるCMvie lacPO pelB LC SV40pAを切り出し、これを(AscI部位がCMvieプロモーターの5’末端に導入されている以外はpMGX579と同一である)pMGX580に連結した。HEK293細胞にトランスフェクトし、その後ヤギ抗ヒトFc抗体で捕捉するELISAを行うことにより、このプラスミドからIgGが発現することを確認した。プロテインGクロマトグラフィーによってIgGを精製し、SDS−PAGE及びウェスタンブロットで分析した。HEK293細胞においてpMGX583から発現した精製IgGのクーマシーブルー染色及びウェスタンブロットを行った。還元条件下で、SDS−PAGEでタンパク質を分析した。ウェスタンブロットでは、1:5000で希釈したAP結合ヤギ抗ヒトIgG(Fab’)2(Jackson)を使用し、色素原によって発色させた。この結果を図7に示す。
【0147】
大腸菌(BL21又はXL1−blue株)におけるpMGX583によるFab発現も評価した。pMGX121由来のプラスミドがlacリプレッサー遺伝子のコピーを含まないので、プラスミドpLac1(Novagen)を用意した。pLac1はクロラムフェニコール耐性プラスミドであり、lacリプレッサータンパク質をコードし、pMGX121由来のベクターと適合するp15a複製起点を有する。pLac1とpMGX583で大腸菌を共形質転換し、アンピシリン及びクロラムフェニコールでコロニーを選択した。下記のように、形質転換体を増殖させ、1mM IPTGで誘導した。図8に示すように、周辺質抽出物由来のFabをsCD16Aによって捕捉し、HRP結合ヤギ抗ヒトF(ab’)2(Jackson)で検出した。非誘導及び誘導(IPTG、1mM)の周辺質抽出物を同量使用した。市販のヒトIgG1を対照として用いた。図2に示すように、前記の構築物に由来する精製ch3G8Fabの連続希釈物を標準物質として用いた。pMGX583プラスミドによる周辺質におけるFabの推定発現量は、培養物1ml当たり約10ngである。
【0148】
【0149】
上記の予備的研究から、哺乳動物細胞において発現又は分泌を低下させずに、原核生物の転写及び翻訳シグナルをIgG重鎖又は軽鎖構築物のシグナルイントロン内に導入することができることが実証された。
【0150】
LCの及びFdフラグメントのN末端配列を得るのに十分なFabが大腸菌から調製される。非修飾のFab、並びに還元及びアルキル化された鎖に対して質量分析を行う。哺乳動物細胞において一過性に産生されるMabに対しても、同様の分析を行う。
【0151】
細菌において周辺質へのFab分泌レベルを高めるために、細菌シグナル配列領域におけるペプチドのいくつかの改変を設計し構築した。以下に示すように、多数の位置のアミノ酸をpelB配列中に含まれるものに戻した。
【0152】
【0153】
改変シグナルペプチドが依然として好ましいスプライス切断部位を保持しているかを予測するために、SignalPプログラムによって配列を分析した。個々の改変を行い、細菌細胞と哺乳動物細胞の両方の系において試験した。pMGX102由来の元のlacZ pelBセグメント(すなわち野生型pelB)とともにLC cDNAも含むベクターにおけるHC/Fd遺伝子のイントロン内に変異が導入された。このようにして、周辺質抽出物中の分泌Fabによる抗原結合について分析することにより、分泌の増大に対する変異の寄与について評価することができる。哺乳動物での発現では、第2プラスミド上のLC遺伝子を共トランスフェクトすることができる。次いで、最初の分析の結果によって定められるように、変異が組み合わされる。
【0154】
コドン利用も、シグナルペプチドの切断の成功に影響を与えることが示されている。この可能性を利用し、分泌の改善を促進することのできるどんな予測不可能な変異をも捕捉するために、この領域中に無作為な可変性が導入された大腸菌変異体のライブラリーをスクリーニングする。これは、縮重又は付加(doped)オリゴヌクレオチドを用いて構築され、コロニーリフト法及び周辺質抽出物の高速処理スクリーニングの両方によってスクリーニングされる。エピトープタグの組み込みは、このスクリーニングに特に有用である。
【0155】
材料及び方法:
基本的なクローン化法、重複PCR法及び部位特異的変異誘発(Quick change kit、Stratagene)を用いて、ベクターを改変して新規な遺伝的構成要素を導入する。新規構築物のすべてのDNA配列を決定して、構築過程で配列の中に望ましくない変異が導入されていないことを確認する。プラスミドが大腸菌内で安定であることを確認するために、受容株はlacIq+であり、また必要であれば適合するプラスミドに、又は構築物自体にlacIを供給する。さらに、CRPによるlacプロモーターの誘導を防止するため、誘導前にプラスミドを含む細胞を栄養富化培地又はブドウ糖の存在下で増殖させる。次に発現を誘導するための以下のプロトコルを行う。単一コロニー由来の細胞をLブロス(1リットル当たりバクトトリプトン10g、酵母抽出物5g、NaCl10g)中で30℃で一晩増殖させた。一晩培養物をLBで1/100に希釈し、OD600が約0.2になるまで30℃で増殖させた。この時点で、この培養物を3つのフラスコに分け、2つをそれぞれ0.1mM又は1mM IPTG(100mMストック由来)で誘導する。その他のフラスコは、非誘導対照として使用する。誘導してから3時間後、細胞を回収し、浸透圧ショックによって周辺質分画を単離する。得られた分画について、Fabが存在するかどうかELISA及びウェスタンブロットでアッセイする。ELISAアッセイでは、Fabをヤギ抗ヒト軽鎖で捕捉し、マウス抗Fdで、その後ウサギ抗マウスHRP結合物で検出する。予備的な結果についての節で記載した、又は業者から入手した精製Fabを用いて、アッセイ用の標準曲線を作成する。
【0156】
Fabの保持機能を検出するため、捕捉抗原結合ELISAアッセイを適用した。周辺質から精製したFab又は未精製周辺質抽出物をsCD16によって捕捉し、ヤギ抗ヒトF(ab’)2 HRP結合抗体(Jackson)で検出した。市販の精製ヒトIgG1を陰性対照として使用した。
【0157】
哺乳動物細胞の発現分析:
哺乳動物細胞における発現及び分泌レベルを測定するために、LipofectAMINE2000試薬(Invitrogen)を用いたトランスフェクションにより、ヒト胚性腎293細胞(HEK293)において、表1に示した個々の構築物を一過性に発現させた。トランスフェクションの前日に、ポリDリシンで予め被覆した皿(Becton Dickinson)上に1皿(100mm)当たり細胞5×106個で細胞を播いた。各皿の細胞について、総DNA18μlをOPTIMEM I還元血清培地(Invitrogen)1.4mlで希釈した。LipofectAMINE試薬(Invitrogen)54μlをOPTIMEM I還元血清培地1.4mlで希釈し、室温で5分間インキュベートした。次いで希釈したDNA及びLipofectAMINE試薬を混合し、室温で20分間インキュベートして複合体を形成させた。このDNAとLipofectAMINE試薬の複合体を細胞に直接加えた。細胞を37℃でCO2(5%)インキュベーター内で72時間インキュベートして組換えIgGを培地に分泌させた。IgG発現レベルについて、ELISA及びウェスタンブロットで条件培地(conditional medium)をアッセイする。ELISAアッセイでは、(条件培地中の)IgGをヤギ抗ヒトFc抗体(Jackson)で捕捉し、ヤギ抗ヒトIgG(軽鎖+重鎖)HRP結合物で検出する。市販の精製ヒトIgG1を標準曲線に使用した。
【0158】
6.2.ファージディスプレイ及びスクリーニング法
大腸菌によって発現されるFab及び哺乳動物によって発現されるIgGに関与するこのベクターを利用するスクリーニング法は、本発明に包含されている。改変型pET25bを使用してFabを発現させる。この場合、LCはエピトープタグが付加されておらず、Fd鎖は、C末端HSVタグと、その後にヘキサヒスチジンタグが精製用に付加されて発現した。2種類の方法で、この配列を構築物に組み込む。第1に、目下存在するオーカーコドン(TAA)の代わりにアンバー(TAG)停止コドンを使用する。これにより、大腸菌のサプレッサー(supE)株においてリードスルー翻訳が可能となる。このような構築物はファージディスプレイに特に有用であり、この手法は以前に用いられている(Hoogenboom et al., 1991, Nucleic Acids Res. 19: 4133 4137)。アンバーコドンは、XL1 blue(supE44+)などの株で抑制されるはずであり、それによってFabのファージ粒子中への組み込みが可能となるが、Fab発現に好ましい株であるBL21(sup)では抑制されない。
【0159】
【0160】
或いは、以下に示すHindIIIなどの制限部位を挿入した後、エピトープ及び/又はアフィニティタグを付加する。
【0161】
【0162】
糸状ファージ由来の遺伝子III及びVIII:
ファージ被殻(遺伝子VIII)又は付着(遺伝子III)タンパク質コード領域との融合物は、ファージディスプレイに最も広く使用されている。その各遺伝子セグメントとのFd(VH CH1)遺伝子セグメントの融合物を構築する。この遺伝子をPCRによってfd tetファージから単離する。遺伝子III融合物では、この遺伝子のP198からS406までのセグメントを使用する。CH1と遺伝子IIIセグメントの間のHSVエピトープタグを保持しながら、遺伝子IIIセグメントが上記のベクター内のヘキサヒスチジンタグと置き換わるように、融合物を構築する。多価のFabディスプレイとするため、遺伝子VIIIのセグメントで同様の構築物を作製する。
【0163】
ファージ分析:
標準的な条件を使用して、ファージの調製及び分析を行う。ファージミドを大腸菌株XL1 Blueにおいて増殖させる。37℃で増殖させた対数増殖期の培養物に、ヘルパーファージVCSM13を感染させ、約12時間培養する。PEG/NaCl沈殿による培養上清からファージを単離し、得られたペレットをTBS15に再懸濁させる。表面上にFabが存在するかどうか、ファージの一部をELISAで分析する。さらに、結合したファージをイムノプレートから溶出させて、ファージが表面結合sCD16 Ig又はsCD32 Ig同一の調製物と結合するかどうかを決定する。活性型抗CD16 Fabを有するファージは、前者の分子と結合するが、後者と結合しないはずである。溶液中でsCD16 Igとともに予めインキュベートして、結合を遮断するために使用する。低pH溶液(グリシンHCl、pH2.2)を用い、その後中和して、プレートからのファージミドの溶出を行う。力価を決定するため、アンピシリン含有プレート上にファージミドをXL1 Blueとともに播く。
【0164】
(Fab’)2
【0165】
Fab’発現では、以下の改変について、ヒンジCH1イントロンをHCミニ遺伝子中の適当な部位に導入後試験した(配列番号28〜31)。
【0166】
【0167】
本発明は、記載した特定の実施形態による範囲に限定されるべきでなく、この実施形態は本発明の個々の態様の例示に過ぎないものとし、機能上等価な方法及び構成要素は本発明の範囲に含まれる。実際、本明細書に示し記載したものに加えて、本発明の様々な変更形態が、前記の記載及び添付した図面から当業者に明らかとなるであろう。このような変更形態は、添付した特許請求の範囲内に含まれるものとする。
【0168】
本明細書において引用したすべての文献は、すべての目的でその全体が参照により本明細書に組み込まれる。
【図面の簡単な説明】
【0169】
【図1】非還元条件での精製Ch3G8Fab(pMGX513)のクーマシーブルー染色を示す図である。レーン1:標準タンパク質(SeeBlue(登録商標)Plus Stained;Invitrogen);レーン2:ヒトIgG(対照);レーン3:ch3G8Fab。
【図2】sCD16−Igと免疫複合体との結合阻害を示す図である。1)HuIgG1、ヒトIgG1、すなわちCh4 4 20(陰性対照として);2)ChFab;3)Ch3G8(IgG1)。
【図3】ch3G8FabとsCD16Aとの結合を示す図である。
【図4】イントロン配列の設計を示す図である。Aは、哺乳動物シグナルペプチドコード配列におけるイントロン内のlacプロモーター及び細菌シグナル配列の配置を示す(配列番号6、7、及び8)。Bは、CH1ヒンジイントロン内のTAA停止コドンの配置を示す(配列番号9〜12)。
【図5】重鎖(HC)及び軽鎖(LC)発現プラスミドの構築及び名称を示す図である。
【図6】大腸菌周辺質抽出物におけるキメラLCの検出を示す図である。
【図7】pMFX583から発現した精製IgGのクーマシーブルー染色及びウェスタンブロットを示す図である。
【図8】抗CD16のELISAによって測定された、周辺質抽出物由来のpMGX583のHu3G8 FabとsCD16Aとの結合を示す図である。
【配列表】
【特許請求の範囲】
【請求項1】
哺乳動物細胞においてIgG重鎖又は軽鎖を、大腸菌(E. coli)において重鎖又は軽鎖のFabフラグメント部分を発現させるためのベクターであって、前記ベクターが、
(a)細菌性複製起点と、
(b)哺乳動物複製起点と、
(c)哺乳動物プロモーター及び/又はエンハンサー配列と、
(d)前記重鎖又は前記軽鎖をコードするヌクレオチド配列であって、
(i)第1イントロンを含む哺乳動物シグナル配列であって、第1イントロンが、細菌細胞において細菌プロモーター及び細菌シグナル配列が前記重鎖又は前記軽鎖のFabドメインの細胞膜周辺腔への発現及び分泌を誘導し、哺乳動物細胞において前記哺乳動物プロモーター及び前記哺乳動物シグナル配列が前記重鎖又は前記軽鎖の発現及び分泌を誘導するように、前記重鎖又は前記軽鎖のFabドメインをコードする配列と機能的に結合した細菌プロモーター及び細菌シグナル配列を含むものである哺乳動物シグナル配列と、
(ii)前記ベクターが前記重鎖をコードする場合、停止コドンを含む、前記重鎖のCH1とヒンジ領域の間の第2イントロンと
を含む前記ヌクレオチド配列と、
を含み、哺乳動物細胞における発現のための前記プロモーターが、前記重鎖又は前記軽鎖をコードする前記ヌクレオチド配列と機能的に結合しているものである、前記ベクター。
【請求項2】
前記細菌プロモーターがlacPO配列を含む、請求項1に記載のベクター。
【請求項3】
前記重鎖又は前記軽鎖が、ヒト重鎖又は軽鎖配列、或いはヒト化重鎖又は軽鎖配列である、請求項1に記載のベクター。
【請求項4】
前記重鎖又は前記軽鎖が、キメラ重鎖又は軽鎖配列である、請求項1に記載のベクター。
【請求項5】
前記細菌シグナル配列がpelBシグナル配列である、請求項1に記載のベクター。
【請求項6】
哺乳動物細胞における発現のための前記プロモーターがCMVプロモーターである、請求項1に記載のベクター。
【請求項7】
前記ベクターが重鎖と軽鎖の両者をコードする、請求項1に記載のベクター。
【請求項8】
前記軽鎖配列が、エピトープタグ又はアフィニティ標識をコードする配列を含むように遺伝子改変されている、請求項1に記載のベクター。
【請求項9】
前記エピトープタグが、Fd鎖のC末端にあるHSVタグである、請求項8に記載のベクター。
【請求項10】
前記アフィニティタグが、Fd鎖のC末端にあるヘキサヒスチジンタグである、請求項8に記載のベクター。
【請求項11】
請求項1に記載のベクターを含む細菌細胞。
【請求項12】
大腸菌細胞である、請求項11に記載の細菌細胞。
【請求項13】
請求項1に記載のベクターを含む哺乳動物細胞。
【請求項14】
ヒト細胞又はネズミ細胞である、請求項13に記載の哺乳動物細胞。
【請求項15】
骨髄腫細胞、CHO細胞、又はHEK細胞である、請求項13に記載の哺乳動物細胞。
【請求項16】
前記ベクターが、重鎖と軽鎖の両者のヌクレオチド配列を含む、請求項11又は13に記載の細胞。
【請求項17】
重鎖を発現し、かつ軽鎖ヌクレオチド配列を含む請求項1に記載のベクターを含む細胞。
【請求項18】
軽鎖を発現し、かつ重鎖ヌクレオチド配列を含む請求項1に記載のベクターを含む細胞。
【請求項19】
前記ベクターが細菌細胞において発現する場合に、fdファージ遺伝子VIII融合物が産生されるように、IgG遺伝子のfd(VH CH1)セグメントをコードする配列が、fdファージ遺伝子VIIIタンパク質のコード領域をコードする配列と機能的に連結している、請求項1に記載のベクター。
【請求項20】
前記ベクターが細菌細胞において発現する場合に、fd遺伝子III融合物が産生されるように、IgG遺伝子のfd(VH CH1)セグメントをコードする配列が、fdファージ遺伝子IIIタンパク質のコード領域をコードする配列と機能的に連結している、請求項1に記載のベクター。
【請求項21】
治療薬として使用するMabを同定する方法であって、
(a)対照重鎖をコードする請求項1に記載のベクターと、対照軽鎖をコードする請求項1に記載のベクターとを含む対照細菌細胞を用意するステップと、
(b)試験細菌細胞のライブラリーと抗原とを接触させるステップであって、各試験細菌細胞が、対照重鎖及び軽鎖と比べて遺伝子改変された、試験重鎖をコードする請求項1に記載のベクター又は試験軽鎖をコードする請求項1に記載のベクターを発現するものである、ステップと、
(c)対照細菌細胞の周辺質抽出物と前記抗原の結合アフィニティと比較して試験細胞の周辺質抽出物と前記抗原の結合アフィニティを測定するステップと
を含み、試験細胞の周辺質抽出物と前記抗原の結合アフィニティが対照細菌細胞の周辺質抽出物と前記抗原の結合アフィニティより大きい場合に、治療薬として有用なMabを発現している細菌細胞が同定される、前記方法。
【請求項22】
ステップ(c)の後に、
(d)哺乳動物細胞においてステップ(c)の試験細胞から単離したベクターを発現させるステップと、
(e)前記哺乳動物細胞と前記抗原とを接触させるステップと、
(f)対照IgGの結合アフィニティと比較して、その哺乳動物細胞内で発現した遺伝子改変IgGの結合アフィニティを測定するステップと
をさらに含む、請求項21に記載の方法。
【請求項23】
治療薬として使用するMabを同定する方法であって、
(a)請求項18又は19に記載のFd(VH CH1)−遺伝子IIIの融合物又はFd(VH CH1)−遺伝子VIIIの融合物をコードする対照ファージを発現する細胞を用意するステップと、
(b)対照ファージと比べて改変された、Fd(VH CH1)−遺伝子IIIの融合物又はFd(VH CH1)−遺伝子VIIIの融合物をコードする試験ファージを産生する複数の試験細胞を含むファージライブラリーのメンバーと、抗原とを接触させるステップと、
(c)対照ファージと前記抗原の結合アフィニティと比較して試験ファージと前記抗原の結合アフィニティを測定するステップと
を含み、試験ファージと前記抗原の結合アフィニティが対照ファージと前記抗原の結合アフィニティより大きい場合に、治療薬として有用なMabを発現している細胞が同定される、前記方法。
【請求項24】
ステップ(c)の後に、
(a)哺乳動物細胞においてステップ(c)の試験細胞から単離したベクターを発現させるステップと、
(b)前記哺乳動物細胞と前記抗原とを接触させるステップと、
(c)対照IgGの結合アフィニティと比較して、その哺乳動物細胞内で発現した遺伝子改変IgGの結合アフィニティを測定するステップと
をさらに含む、請求項21又は23に記載の方法。
【請求項25】
請求項1に記載のベクターを含む、Fabポリペプチドを発現する複数の細菌細胞を含有する組成物。
【請求項26】
請求項1に記載のベクター、及びFabポリペプチドを発現する糸状ファージを含む複数の細菌細胞を含有する組成物。
【請求項27】
前記細菌細胞が大腸菌細胞である、請求項25又は26に記載の組成物。
【請求項28】
前記糸状ファージがfdファージである、請求項25又は26に記載の組成物。
【請求項1】
哺乳動物細胞においてIgG重鎖又は軽鎖を、大腸菌(E. coli)において重鎖又は軽鎖のFabフラグメント部分を発現させるためのベクターであって、前記ベクターが、
(a)細菌性複製起点と、
(b)哺乳動物複製起点と、
(c)哺乳動物プロモーター及び/又はエンハンサー配列と、
(d)前記重鎖又は前記軽鎖をコードするヌクレオチド配列であって、
(i)第1イントロンを含む哺乳動物シグナル配列であって、第1イントロンが、細菌細胞において細菌プロモーター及び細菌シグナル配列が前記重鎖又は前記軽鎖のFabドメインの細胞膜周辺腔への発現及び分泌を誘導し、哺乳動物細胞において前記哺乳動物プロモーター及び前記哺乳動物シグナル配列が前記重鎖又は前記軽鎖の発現及び分泌を誘導するように、前記重鎖又は前記軽鎖のFabドメインをコードする配列と機能的に結合した細菌プロモーター及び細菌シグナル配列を含むものである哺乳動物シグナル配列と、
(ii)前記ベクターが前記重鎖をコードする場合、停止コドンを含む、前記重鎖のCH1とヒンジ領域の間の第2イントロンと
を含む前記ヌクレオチド配列と、
を含み、哺乳動物細胞における発現のための前記プロモーターが、前記重鎖又は前記軽鎖をコードする前記ヌクレオチド配列と機能的に結合しているものである、前記ベクター。
【請求項2】
前記細菌プロモーターがlacPO配列を含む、請求項1に記載のベクター。
【請求項3】
前記重鎖又は前記軽鎖が、ヒト重鎖又は軽鎖配列、或いはヒト化重鎖又は軽鎖配列である、請求項1に記載のベクター。
【請求項4】
前記重鎖又は前記軽鎖が、キメラ重鎖又は軽鎖配列である、請求項1に記載のベクター。
【請求項5】
前記細菌シグナル配列がpelBシグナル配列である、請求項1に記載のベクター。
【請求項6】
哺乳動物細胞における発現のための前記プロモーターがCMVプロモーターである、請求項1に記載のベクター。
【請求項7】
前記ベクターが重鎖と軽鎖の両者をコードする、請求項1に記載のベクター。
【請求項8】
前記軽鎖配列が、エピトープタグ又はアフィニティ標識をコードする配列を含むように遺伝子改変されている、請求項1に記載のベクター。
【請求項9】
前記エピトープタグが、Fd鎖のC末端にあるHSVタグである、請求項8に記載のベクター。
【請求項10】
前記アフィニティタグが、Fd鎖のC末端にあるヘキサヒスチジンタグである、請求項8に記載のベクター。
【請求項11】
請求項1に記載のベクターを含む細菌細胞。
【請求項12】
大腸菌細胞である、請求項11に記載の細菌細胞。
【請求項13】
請求項1に記載のベクターを含む哺乳動物細胞。
【請求項14】
ヒト細胞又はネズミ細胞である、請求項13に記載の哺乳動物細胞。
【請求項15】
骨髄腫細胞、CHO細胞、又はHEK細胞である、請求項13に記載の哺乳動物細胞。
【請求項16】
前記ベクターが、重鎖と軽鎖の両者のヌクレオチド配列を含む、請求項11又は13に記載の細胞。
【請求項17】
重鎖を発現し、かつ軽鎖ヌクレオチド配列を含む請求項1に記載のベクターを含む細胞。
【請求項18】
軽鎖を発現し、かつ重鎖ヌクレオチド配列を含む請求項1に記載のベクターを含む細胞。
【請求項19】
前記ベクターが細菌細胞において発現する場合に、fdファージ遺伝子VIII融合物が産生されるように、IgG遺伝子のfd(VH CH1)セグメントをコードする配列が、fdファージ遺伝子VIIIタンパク質のコード領域をコードする配列と機能的に連結している、請求項1に記載のベクター。
【請求項20】
前記ベクターが細菌細胞において発現する場合に、fd遺伝子III融合物が産生されるように、IgG遺伝子のfd(VH CH1)セグメントをコードする配列が、fdファージ遺伝子IIIタンパク質のコード領域をコードする配列と機能的に連結している、請求項1に記載のベクター。
【請求項21】
治療薬として使用するMabを同定する方法であって、
(a)対照重鎖をコードする請求項1に記載のベクターと、対照軽鎖をコードする請求項1に記載のベクターとを含む対照細菌細胞を用意するステップと、
(b)試験細菌細胞のライブラリーと抗原とを接触させるステップであって、各試験細菌細胞が、対照重鎖及び軽鎖と比べて遺伝子改変された、試験重鎖をコードする請求項1に記載のベクター又は試験軽鎖をコードする請求項1に記載のベクターを発現するものである、ステップと、
(c)対照細菌細胞の周辺質抽出物と前記抗原の結合アフィニティと比較して試験細胞の周辺質抽出物と前記抗原の結合アフィニティを測定するステップと
を含み、試験細胞の周辺質抽出物と前記抗原の結合アフィニティが対照細菌細胞の周辺質抽出物と前記抗原の結合アフィニティより大きい場合に、治療薬として有用なMabを発現している細菌細胞が同定される、前記方法。
【請求項22】
ステップ(c)の後に、
(d)哺乳動物細胞においてステップ(c)の試験細胞から単離したベクターを発現させるステップと、
(e)前記哺乳動物細胞と前記抗原とを接触させるステップと、
(f)対照IgGの結合アフィニティと比較して、その哺乳動物細胞内で発現した遺伝子改変IgGの結合アフィニティを測定するステップと
をさらに含む、請求項21に記載の方法。
【請求項23】
治療薬として使用するMabを同定する方法であって、
(a)請求項18又は19に記載のFd(VH CH1)−遺伝子IIIの融合物又はFd(VH CH1)−遺伝子VIIIの融合物をコードする対照ファージを発現する細胞を用意するステップと、
(b)対照ファージと比べて改変された、Fd(VH CH1)−遺伝子IIIの融合物又はFd(VH CH1)−遺伝子VIIIの融合物をコードする試験ファージを産生する複数の試験細胞を含むファージライブラリーのメンバーと、抗原とを接触させるステップと、
(c)対照ファージと前記抗原の結合アフィニティと比較して試験ファージと前記抗原の結合アフィニティを測定するステップと
を含み、試験ファージと前記抗原の結合アフィニティが対照ファージと前記抗原の結合アフィニティより大きい場合に、治療薬として有用なMabを発現している細胞が同定される、前記方法。
【請求項24】
ステップ(c)の後に、
(a)哺乳動物細胞においてステップ(c)の試験細胞から単離したベクターを発現させるステップと、
(b)前記哺乳動物細胞と前記抗原とを接触させるステップと、
(c)対照IgGの結合アフィニティと比較して、その哺乳動物細胞内で発現した遺伝子改変IgGの結合アフィニティを測定するステップと
をさらに含む、請求項21又は23に記載の方法。
【請求項25】
請求項1に記載のベクターを含む、Fabポリペプチドを発現する複数の細菌細胞を含有する組成物。
【請求項26】
請求項1に記載のベクター、及びFabポリペプチドを発現する糸状ファージを含む複数の細菌細胞を含有する組成物。
【請求項27】
前記細菌細胞が大腸菌細胞である、請求項25又は26に記載の組成物。
【請求項28】
前記糸状ファージがfdファージである、請求項25又は26に記載の組成物。
【図1】

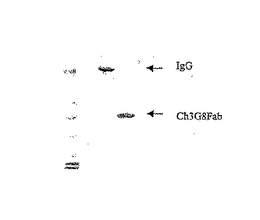
【図2】
【図3】
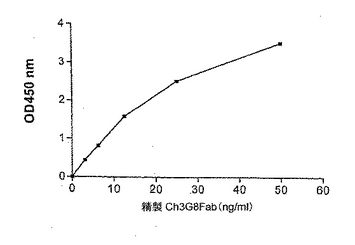
【図4】

【図5】
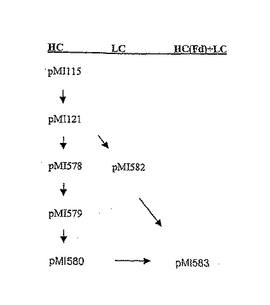
【図6】
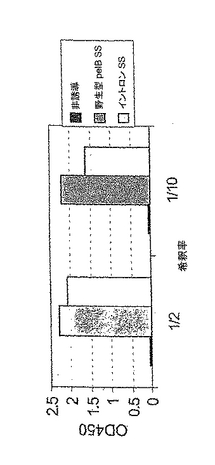
【図7】
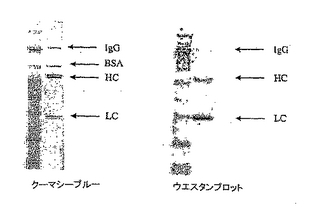
【図8】
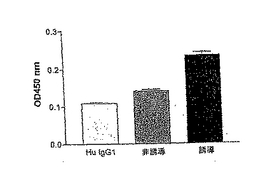
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2006−516195(P2006−516195A)
【公表日】平成18年6月29日(2006.6.29)
【国際特許分類】
【出願番号】特願2006−500864(P2006−500864)
【出願日】平成16年1月8日(2004.1.8)
【国際出願番号】PCT/US2004/000462
【国際公開番号】WO2004/063343
【国際公開日】平成16年7月29日(2004.7.29)
【出願人】(504438727)マクロジェニクス,インコーポレーテッド (23)
【Fターム(参考)】
【公表日】平成18年6月29日(2006.6.29)
【国際特許分類】
【出願日】平成16年1月8日(2004.1.8)
【国際出願番号】PCT/US2004/000462
【国際公開番号】WO2004/063343
【国際公開日】平成16年7月29日(2004.7.29)
【出願人】(504438727)マクロジェニクス,インコーポレーテッド (23)
【Fターム(参考)】
[ Back to top ]