
組成物および使用の方法

本発明は、疾患、障害、またはその症状(例えば、増殖障害、癌)の治療のための、植物抽出物、その組成物、および、植物抽出物から単離されるか、またはその合成手段から得られる化合物の使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明の技術分野
本発明は、植物抽出組成物、単離した活性薬剤、および癌の治療のための使用の方法に関する。
【背景技術】
【0002】
治療用途のための新しい抽出物および化合物の同定は、継続して生物医学的に重要であり、天然生成物がこの試みのために注目を集め続けている。増殖障害はヒトおよび動物の健康に問題を与え続け、このような障害に対する治療計画は、幅広い観点から未充足の必要性として残っている。増殖障害に関与する機構、信号伝達過程、および標的の変動のために、新しくかつより有効な組成物、化合物、および治療の方法の同定は、医学界ならびに一般大衆にとって大いに興味深いものである。
【0003】
オオカミノアシ(Wolf's
Foot)およびクラブモスヒカゲノカズラ科(Club Moss Lycopodiaceae)としても知られるヒカゲノカズラ(Lycopodium clavatum)は、ヒカゲノカズラ属(genus
Lycopodium)およびヒカゲノカズラ科(family Lycopodiaceae)に属する。その名前は、枝の先端が狼の鉤爪足に似ているために狼の足に由来する(lukosは狼を意味し、podosは足を意味する)。当該植物は、世界中の亜熱帯林および熱帯林内で遍在的に見つけられる。胞子体は、減数分裂の後に胞子嚢内で胞子を生産する。ヒカゲノカズラ類(Lycopodium)において、胞子体は、その中で各胞子嚢が葉状の胞子葉により保護される、円錐状の球果内に群生している。植物の胞子からの抽出物が、新規の治療活性を有する新しくかつ興味深い組成物の探索中に、分画されて単離した抽出物および化合物を産生した。
【発明の概要】
【0004】
本発明の概要
本発明は、本明細書における植物抽出物を用い、対象者における増殖障害を治療する方法に関する。本発明の1つの態様は、本明細書における植物抽出物からの単離した化合物を用い、対象者における増殖障害を治療する方法である。1つの実施形態では、増殖障害は癌である。特定の実施形態は、癌が、白血病、肺癌、肝臓癌、または結腸癌であると定めている。1つの実施形態では、癌は非小細胞肺癌である。別の実施形態では、癌は肝臓癌である。
【0005】
1つの実施形態では、当該化合物は、8−ヒドロキシ−パルミチン酸(「8−HHA」)、またはその塩、前駆薬、前駆薬塩、溶媒和物、水和物、および多形体である。別の実施形態では、本発明の方法に従って用いられる化合物は、8−ヒドロキシ−パルミチン酸のR−エナンチオマーおよびS−エナンチオマーのラセミ混合物である。1つの実施形態では、当該化合物は、8−ヒドロキシパルミチン酸のS−エナンチオマー(「(S)−8−HHA」)である。別の実施形態では、当該化合物は、8−ヒドロキシ−パルミチン酸のR−エナンチオマー(「(R)−8−HHA」)である。
【0006】
本発明の別の態様は、本明細書における植物抽出物(例えば、ヒカゲノカズラ(Lycopodium clavatum)の抽出物)を含む組成物である。別の態様は、本明細書における植物抽出物からの単離した化合物を含む組成物である。別の態様は、1つ以上の置換基を有する、本明細書における植物抽出物からの単離した化合物を含む組成物である。
【0007】
別の態様は、本明細書(または本明細書に描出されている化合物/組み合わせを用いる方法)に描出されている一つの化合物(または複数の化合物の組み合わせ)であり、そこで、当該化合物または化合物の組み合わせは、対象者(動物モデル、マウス、ラット、ウサギ、霊長類、ヒト)における抗癌活性を有することが実証されている。本明細書における例は、このような効果を決定するための代表的な方法にとって有益である。
【0008】
1つの実施形態では、本明細書に描出されている化合物(または複数の化合物の組み合わせ)は、植物からの抽出を含む手順から得られる。特定の実施形態では、化合物(または複数の化合物の組み合わせ)を得る際の使用のための手順は、植物抽出物の単離段階、蒸発段階、および分配段階の何れかをさらに含む。
【0009】
別の実施形態では、本明細書に描出されている化合物(または複数の化合物の組み合わせ)は、合成手段から得られる。
【0010】
本発明の別の態様は、本明細書における植物抽出物(例えば、BCP−21抽出物、表1または2内のBCP−21抽出物画分の何れか)、または、本明細書における植物抽出物(例えば、BCP−21抽出物、表1または2内のBCP−21抽出物画分の何れか)中に生じる化合物(例えば、8−ヒドロキシヘキサデカン酸、本明細書におけるBCP化合物1〜10の何れか)を含む医薬組成物である。1つの実施形態では、本医薬組成物は、8−HHAのラセミ混合物、またはその塩、前駆薬、前駆薬塩、溶媒和物、水和物、および多形体を含む。1つの実施形態では、本医薬組成物は、相当なエナンチオマー的純度で8−ヒドロキシ−パルミチン酸のS−エナンチオマー(「(S)−8−HHA」)を含む。別の実施形態では、本医薬組成物は、相当なエナンチオマー的純度で8−ヒドロキシ−パルミチン酸のR−エナンチオマー(「(R)−8−HHA」)を含む。
【0011】
別の態様では、このような組成物をキットに含めることができる。
【0012】
上記の組成物(および本明細書に描出されている治療の方法)を、適正な化学治療薬剤および生物治療薬剤とさらに組み合わせることができる。このような薬剤の例としては、アルデスロイキン、アレムツズマブ、アリトレチノイン、アルトレタミン、アミノレブリン酸、アナグレリド、アナストロゾール、三酸化ヒ素、アスパラギナーゼ、カルメット・ゲラン桿菌(BCG)、ベタメタゾン、ベキサロテン、ビカルタミド、ブレオマイシン、ブスルファン、カペシタビン、カルボプラチン、カルムスチン、クロラムブシル、リン酸クロムP−32、シスプラチン、クラドリビン、結合型エストロゲン類、コルチゾン、シクロホスファミド、シタラビンリポソーム、シタラビン、ara−、ダカルバジン、ダクチノマイシン、アクチノマイシンD、ダウノルビシン、クエン酸ダウノルビシンリポソーム、デニロイキンジフチトクス、デキサメタゾン、ジクロフェナク、ドセタキセル、ドキソルビシン、ドキソルビシンリポソーム、エピルビシン、エステル型エストロゲン類、エストラジオール、吉草酸エストラジオール、エストラムスチン、エストロン、エチニルエストラジオール、エトポシド、リン酸エトポシド、エキセメスタン、フロクスウリジン、リン酸フルダラビン、フルオロウラシル、フルオロウラシル、フルオロウラシル、フルオキシメステロン、フルタミド、ゲムシタビン、ゲムツズマブオゾガマイシン、酢酸ゴセレリン、グラニセトロン、ヒドロコルチゾン、ヒドロキシプロゲステロン、ヒドロキシ尿素、イブリツモマブチウキセタン、イダルビシン、イホスファミド、メシル酸イマチニブ、インターフェロンアルファ−2a、インターフェロンアルファ−2b、イリノテカン、レトロゾール、酢酸ロイプロリド、レボチロキシン、ロムスチン、メクロレタミン、メドロキシプロゲステロン、メドロキシプロゲステロン、酢酸メゲストロール、メルファラン、メルカプトプリン、メトトレキサートナトリウム、メトキサレン、メチルプレドニゾロン、メチルテストステロン、メチロシン、マイトマイシン、マイトマイシンC、ミトタン、ミトキサントロン、フェンプロピオン酸ナンドロロン、ニルタミド、酢酸オクトレオチド、オプレルベキン、オキシメトロン、ペガスパルガーゼ、ペントスタチン、プリカマイシン、ポリフェプロザン20/カルムスチン、ポルフィマーナトリウム、プレドニゾロン、プレドニゾン、プロカルバジン、プロゲステロン、リツキシマブ、サマリウム−153レキシドロナムペンタナトリウム、ヨウ化ナトリウムI−131、リン酸ナトリウムP−32、ストレプトゾシン、塩化ストロンチウム−89、滑石、クエン酸タモキシフェン、テモゾロミド、テニポシド、テストラクトン、エナント酸テストステロン、チオグアニン、チオテパ、トポテカン、クエン酸トレミフェン、トラスツズマブ、トレチノイン、トリアムシノロン、パモ酸トリプトレリン、バルルビシン、ビンブラスチン、ビンクリスチン、およびビノレルビンが挙げられるが、これらに限定されない。
【0013】
さらに別の態様では、本発明は、本明細書に明記されている対象者における疾患、障害、または症状の治療または防止のための、単一の組成物または別個の剤形の何れかとしての薬物の製造における、単独での、または1つ以上の追加の治療薬剤と合わせての、本明細書における化学式の何れかの化合物の使用を提供する。本発明の別の態様は、本明細書に描出されている対象者における疾患、障害、またはその症状の治療または防止における使用のための、(単離した天然または合成の)本明細書における化学式の化合物である。
【図面の簡単な説明】
【0014】
【図1】BCP−21抽出物から単離された化合物のNMRスペクトルである。
【図2】BCP−21抽出物から単離された化合物のNMRスペクトルである。
【図3】BCP−21抽出物から単離された化合物のNMRスペクトルである。
【図4】BCP−21抽出物から単離された化合物のMSスペクトルである。
【図5】BCP−21抽出物から単離された化合物のMSスペクトルである。
【図6】BCP−21が、用量依存的な様態でHeLa細胞のアポトーシスを誘発することを示す。
【図7】ヒカゲノカズラ(lycopodiumclavatum)の抽出物が、時間依存的な様態でPARP切断を誘発することを示す、ウエスタンブロッティングの結果である。
【図8】(S)−8−HHAが、用量依存的な様態でHOP−92の肺癌細胞株の生存を阻害したことを示す。
【図9】100μMの8−HHA(ラセミ混合物、S−エナンチオマーまたはR−エナンチオマー)が、比色MTT検定を用いて、PCL5、Hep3B、およびSNUの肝臓癌細胞株の生存を減少させることを結果的にもたらしたことを示す。
【図10】(S)−8−HHAが、用量依存的な様態でPLC−5の肝臓癌細胞株の生存を阻害したことを実証する。
【図11】100μMの(S)−8−HHAが、SNUおよびPCL5の肝臓癌細胞株内にPARP切断を結果的にもたらしたことを示す。
【図12】(S)−8−HHAの処理が、対照と比較してFAS受容体濃度の相当な減少を結果的にもたらしたことを実証する。
【図13】8−HHAが、肝臓異種移植マウスモデルにおけるPLC5の肝臓癌細胞内にて腫瘍成長を減少させることを描写する。
【図14】異種移植マウスモデルにおけるHOP92の肺癌細胞内での8−HHAの抗腫瘍効能を描写する。
【発明を実施するための形態】
【0015】
本発明の詳細な説明
定義
「寛解させる」および「治療する」という用語は、相互交換可能に用いられ、疾患(例えば、本明細書に描出されている疾患または障害)の発生または進行を減少させ、抑制し、減弱し、軽減し、阻止し、または安定させることを両方とも意味する。
【0016】
「疾患」とは、細胞、組織、または器官の正常な機能を損傷するかまたは妨げる、任意の容態または障害を意味する。
【0017】
「標識」とは、疾患または障害と関連する任意の変化を意味する。例えば、疾患または障害と関連する発現量または活性における変化を有する、任意のタンパク質またはポリヌクレオチドである。
【0018】
「癌」という用語は、一群の細胞が、非制御成長(正常限界を超える分裂)、侵襲(隣接する組織への侵入およびこれの破壊)、および時には転移(体内の他の場所への蔓延)を見せる一種の疾患を概して指す。癌の例としては、白血病、肺癌、肝臓癌、結腸癌、黒色腫、乳癌、CNS癌、卵巣癌、腎臓癌、および前立腺癌が挙げられるが、これらに限定されない。
【0019】
本開示において、「備える」、「包含する」、「含有する」、および「有する」および同様のものは、米国特許法でそれらに帰される意味を有することがあり、かつ「含める」、「含む」、および同様のものを意味することがあり、「から実質的に成る」または「実質的に成る」は、米国特許法で帰される意味を同様に有し、当該用語は変更可能であり、列挙されるものの基本的な特徴または新規の特徴が、列挙されるが先行技術の実施形態を除くものより多くの存在により変更されない限り、列挙されるものより多くの存在を考慮に入れる。
【0020】
本明細書における化合物を、本明細書における化学式の化合物の塩、前駆薬、および前駆薬塩、溶媒和物、水和物、および多形体を含む化合物の形態で利用することもできる。
【0021】
本発明の化合物の塩は、酸とアミノ官能基などの化合物の塩基性基との間に形成されるか、あるいは、塩基とカルボキシル官能基などの化合物の酸性基との間に形成される。別の好適な実施形態によれば、化合物は医薬的に許容可能な酸付加塩である。
【0022】
本明細書で用いられる際および他に示されない限り、「前駆薬」という用語は、生物学的条件下(インビトロまたはインビボ)で加水分解し、酸化し、またはそれ以外の反応を起こし、本発明の化合物を与えることができる化合物の誘導体を意味する。前駆薬は、生物学的条件下でこのような反応において活性になるのみであり得、あるいは、その未反応の形態で活性を有し得る。本明細書で意図されている前駆薬の例としては、アミド類、エステル類、カルバミン酸塩類、炭酸塩類、およびリン酸塩類似体等の生物加水分解可能な部分を含む、本明細書で開示されている化学式の何れか一つの化合物の類似体または誘導体が挙げられるが、これらに限定されない。「バーガーの医薬品化学および創薬(1995年)172〜178ページ、949〜982ページ(マンフレッド・E.ウォルフ編、第5版)(Burger's Medicinal Chemistry and Drug Discovery (1995) 172-178, 949-982
(Manfred E. Wolff ed., 5th ed))」により記載されている方法等の、十分に知られているものを用いて、前駆薬を典型的に調製することができ、「グッドマンおよびギルマンの治療学の薬理学的基礎、第8版、マグローヒル国際版、1992年、『薬物の生体変換』(Goodman and Gilman's, The Pharmacological basis of Therapeutics, 8th
ed., McGraw-Hill, Int. Ed. 1992, "Biotransformation of Drugs")」も参照されたい。
【0023】
本明細書で用いられる際および他に示されない限り、「生物加水分解可能な部分」という用語は、1)化合物の生物学的活性を破壊せず、かつ、取り込み、作用の持続時間、または作用の発現等の、インビボでの有利な特性を化合物に付与するか、あるいは、2)それ自体は生物学的に不活性であるが、インビボで生物学的に活性な化合物に転換されるか、の何れかである官能基(例えば、アミド、エステル、カルバミン酸塩、炭酸塩、またはリン酸塩)類似体を意味する。
【0024】
前駆薬塩は、酸とアミノ官能基などの前駆薬の塩基性基との間に形成されるか、あるいは、塩基とカルボキシル官能基などの前駆薬の酸性基との間に形成される化合物である。1つの実施形態では、前駆薬塩は医薬的に許容可能な塩である。
【0025】
特に好ましい前駆薬および前駆薬塩は、本発明の化合物が(例えば、経口的に投与される化合物を血液中により容易に吸収することを可能にすることにより)哺乳類に投与される際に、このような化合物の生物利用可能性を増加させるか、あるいは、親種と比較して親化合物の生物学的区画(例えば、脳または中枢神経系)への運搬を強化するものである。好適な前駆薬としては、水溶性または腸膜を通じる能動輸送を強化する基が、本明細書に記載されている化学式の構造に付加される誘導体が挙げられる。例えば、「アレクサンダー・J.他、医薬品化学誌、1988年、第31号、318〜322ページ(Alexander, J. et al. Journal of Medicinal Chemistry 1988, 31,
318-322)」、「バンガード・H.、前駆薬の設計、エルゼビア:アムステルダム、1985年、1〜92ページ(Bundgaard,
H. Design of Prodrugs; Elsevier: Amsterdam, 1985; pp 1-92)」、「バンガード・H.、ニールセン・N.M.、医薬品化学誌、1987年、第30号、451〜454ページ(Bundgaard, H.; Nielsen, N. M. Journal of Medicinal Chemistry 1987,
30, 451-454)」、「バンガード・H.、薬の設計および開発の教科書、ハーウッド・アカデミック出版:スイス、1991年、113〜191ページ(Bundgaard, H. A Textbook of Drug Design and Development; Harwood
Academic Publ.: Switzerland, 1991; pp 113-191)」、「ディゲニス・G.A.他、実験薬理学の手引書、1975年、第28号、86〜112ページ(Digenis, G. A. et al. Handbook of Experimental Pharmacology 1975,
28, 86-112)」、「フリース・G.J.、バンガード・H.、薬の設計および開発の教科書、第2版、海外出版:アムステルダム、1996年、351〜385ページ(Friis, G. J.; Bundgaard, H. A Textbook of Drug Design and
Development; 2 ed.; Overseas Publ.: Amsterdam, 1996; pp 351-385)」、「ピットマン・I.H.、医薬品研究総説、1981年、第1号、189〜214ページ(Pitman, I. H. Medicinal Research Reviews 1981, 1, 189-214)」を参照されたい。
【0026】
「医薬的に許容可能な」という用語は、本明細書で用いられる際に、健全医学的判断の範囲内において、過度の毒性、刺激、アレルギー反応、および同様のものを伴わない、ヒトおよび他の哺乳類の組織と接触する使用に適切であり、かつ、妥当な恩恵/危険比に相応である成分を指す。「医薬的に許容可能な塩」は、受容者への投与の際に、直接的にも間接的にも、本発明の化合物または化合物の前駆薬を与えることができる、任意の非毒性塩を意味する。
【0027】
医薬的に許容可能な塩を形成するために一般に使用される酸としては、重硫化水素、塩化水素酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸等の無機酸、ならびに、パラ−トルエンスルホン酸、サリチル酸、酒石酸、二酒石酸、アスコルビン酸、マレイン酸、ベシル酸、フマル酸、グルコン酸、グルクロン酸、蟻酸、グルタミン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、乳酸、蓚酸、パラ−ブロモフェニルスルホン酸、炭酸、コハク酸、クエン酸、安息香酸、および酢酸等の有機酸、および関連する無機酸と有機酸が挙げられる。このような医薬的に許容可能な塩としては、そのため、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、リン酸一水素塩、リン酸二水素塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、蟻酸塩、イソ酪酸塩、カプリン酸塩、ヘプタン酸塩、プロピオール酸塩、蓚酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオール酸塩、ヘキシン−1,6−ジオール酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、テレフタル酸塩、スルホン酸塩、キシレンスルホン酸、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、β−ヒドロキシ酪酸塩、グリコール酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スルホン酸塩、マンデル酸塩、および同様の塩が挙げられる。好適な医薬的に許容可能な酸付加塩としては、塩化水素酸および臭化水素酸等の鉱酸と形成されるもの、および、特にマレイン酸等の有機酸と形成されるものが挙げられる。
【0028】
医薬的に許容可能な塩を本発明の前駆薬の酸性官能基と形成するための好適な塩基としては、ナトリウム、カリウム、およびリチウム等のアルカリ金属の水酸化物、カルシウムおよびマグネシウム等のアルカリ土類金属の水酸化物、アルミニウムおよび亜鉛等の他の金属の水酸化物、アンモニア、および非置換またはヒドロキシ置換のモノアルキルアミン、ジアルキルアミン、およびトリアルキルアミン等の有機アミン類、ジシクロヘキシルアミン、トリブチルアミン、ピリジン、N−メチル−N−エチルアミン、ジエチルアミン、トリエチルアミン、モノ−、ビス−、またはトリス−(2−ヒドロキシエチル)アミン、2−ヒドロキシ−tert−ブチルアミン、またはトリス−(2−ヒドロキシエチル)アミン等のモノ−、ビス−、またはトリス−(2−ヒドロキシ−低級アルキルアミン)、N,N−ジメチル−N−(2−ヒドロキシエチル)アミン、またはトリ−(2−ヒドロキシエチル)アミン等のN,N−ジ−低級アルキル−N−(ヒドロキシ低級アルキル)−アミン類、N−メチル−D−グルカミン、ならびにアルギニン、リジン、および同様のもの等のアミノ酸類が挙げられるが、これらに限定されない。
【0029】
本明細書で用いられる際に、「水和物」という用語は、非共有分子間力により結合した、化学量論量または非化学量論量の水をさらに含む化合物を意味する。
【0030】
「単離した」、「精製した」、または「生物学的に純粋な」という用語は、その天然状態で見つかる際に通常はそれを伴う成分が、実質的にまたは本質的に非含有である物質を指す。純度または均質性は、ポリアクリルアミドゲル電気泳動法または高性能液体クロマトグラフィー等の分析化学技術を用いて典型的に決定される。特に、実施形態では、化合物は少なくとも純度85%、より好ましくは少なくとも純度90%、より好ましくは少なくとも純度95%、最も好ましくは少なくとも純度99%である。
【0031】
本明細書で用いられる際に、「溶媒和物」という用語は、非共有分子間力により結合した、化学量論量または非化学量論量の、水、アセトン、エタノール、メタノール、ジクロロメタン、2−プロパノール、または同様のもの等の溶媒をさらに含む化合物を意味する。
【0032】
本明細書で用いられる際に、「多形体」という用語は、例えば、X線粉末回折図形または赤外分光法等の物理的手段により特徴付けることができる、化合物またはその錯体の固体結晶形態を意味する。同じ化合物の異なる多形体は、異なる物理的特性、化学的特性、および/または分光学的特性を呈することがある。異なる物理的特性としては、安定性(例えば、熱、光、または水分に対して)、圧縮性および密度(調合および製品製造において重要)、吸湿性、可溶性、および溶解率(生物利用可能性に影響し得る)が挙げられるが、これらに限定されない。安定性における差異は、化学反応性(例えば、剤形が、1つの多形体から成る際に、別の多形体から成る際よりも急速に変色するような、酸化の違い)、または力学的特徴(例えば、動力学的に有利な多形体が熱力学的により安定した多形体に転換する時に、錠剤が貯蔵の際に砕ける)、またはその両方(例えば、1つの多形体の錠剤が、高い湿度で分解の影響をより受けやすい)における変化に起因し得る。多形体の異なる物理的特性は、加工に影響し得る。例えば、1つの多形体は、より溶媒和物を形成する可能性があり得るだろうし、あるいは、例えば、それの粒子の形状または大きさの分布に起因して、別のものよりも不純物を完全に濾過または洗浄することがより困難であり得るだろう。
【0033】
本明細書で用いられる際の「他の立体異性体が実質的に非含有の」という用語は、25%未満の他の立体異性体、好ましくは10%未満の他の立体異性体、より好ましくは5%未満の他の立体異性体、最も好ましくは2%未満の他の立体異性体、あるいは「X」%未満の他の立体異性体(式中、Xは0および100の間の全体を含む数である)が存在することを意味する。ジアステレオマーを得るかまたは合成する方法は、当該技術分野で十分に知られており、最終化合物に対して、あるいは出発物質または中間体に対して実行可能なものとして適用されてよい。他の実施形態は、化合物が単離した化合物であるものである。本明細書で用いられる際の「少なくともX%がエナンチオマー的に豊富な」という用語は、化合物の少なくともX%が単一のエナンチオマー形態であることを意味し、式中、Xは0および100の間の全体を含む数である。
【0034】
「安定化合物」という用語は、本明細書で用いられる際に、製造を可能にするのに十分な安定性を有し、かつ、本明細書に詳述されている目的(例えば、治療薬剤に応答する疾患または障害を治療する、治療製品、治療化合物の製造の際の使用のための中間体、単離可能または貯蔵可能な中間化合物への調合)のために有用であるのに十分な期間にわたり化合物の統合性を維持する化合物を指す。
【0035】
「立体異性体」は、エナンチオマーおよびジアステレオマーを両方とも指す。
【0036】
本明細書で用いられる際に、「ハロ」または「ハロゲン」という用語は、フッ素、塩素、臭素、またはヨウ素の任意のラジカルを指す。
【0037】
「アルク」または「アルキル」という用語は、1乃至12個の炭素原子、好ましくは1乃至8個の炭素原子を有する、直鎖または分枝鎖の炭化水素基を指す。「低級アルキル」という表現は、1乃至4(全体を含む)個の炭素原子のアルキル基を指す。「アリールアルキル」という用語は、その中でアルキルの水素原子がアリール基により置き換えられる部分を指す。「アルケニル」という用語は、2乃至10個の、好ましくは2乃至4個の炭素原子の、少なくとも1つの二重結合を有する直鎖または分枝鎖の炭化水素基を指す。アルケニル基が窒素原子に結合される場合には、このような基が二重結合を有する炭素を通じて直接的に結合されないことが好適である。
【0038】
「アルコキシ」という用語は、−O−アルキルラジカルを指す。「アルキレンジオキソ」という用語は、−O−R−O−という構造の二価種を指し、その中で、Rはアルキレンを表す。
【0039】
「アルキニル」という用語は、2乃至10個の、好ましくは2乃至4個の炭素原子の、少なくとも1つの三重結合を有する直鎖または分枝鎖の炭化水素基を指す。アルキニル基が窒素原子に結合される場合には、このような基が三重結合を有する炭素を通じて直接的に結合されないことが好適である。
【0040】
「アルキレン」という用語は、1乃至3個の低級アルキル基で置換することができる、単結合により接続される1乃至5個の炭素原子の二価の直鎖橋(例えば、−(CH2)x−、式中、xは1乃至5である)を指す。
【0041】
「アルケニレン」という用語は、単結合により接続され、かつ1乃至3個の低級アルキル基で置換することができる、1つまたは2つの二重結合を有する2乃至5個の炭素原子の直鎖橋を指す。例示的なアルケニレン基は、−CH=CH−CH=CH−、−CH2−CH=CH−、−CH2−CH=CH−CH2−、−C(CH3)2CH=CH−、および−CH(C2H5)−CH=CH−である。
【0042】
「アルキニレン」という用語は、その中に一つの三重結合を有し、単結合により接続され、かつ1乃至3個の低級アルキル基で置換することができる、2乃至5個の炭素原子の直鎖橋を指す。例示的なアルキニレン基は、−C≡C−、−CH2−C≡C−、−CH(CH3)−C≡C−、および−C≡C−CH(C2H5)CH2−である。
【0043】
本明細書で使用される際の「シクロアルキル」および「シクロアルケニル」という用語は、それぞれ、飽和および部分不飽和の、3乃至12個の炭素、好ましくは3乃至8個の炭素、より好ましくは3乃至6個の炭素を有する環式炭化水素基を含む。「Ar」または「アリール」という用語は、6乃至14個の炭素原子を含有する芳香族環式基(例えば、6員環の単環式環系、10員環の二環式環系、または14員環の三環式環系)を指す。例示的なアリール基としては、フェニル、ナフチル、ビフェニル、およびアントラセンが挙げられる。
【0044】
「ヘテロアリール」は、N、O、またはSから選択される1個、2個、3個、または4個の環複素原子を含有する、5乃至12個の環原子の単環式または縮合環(すなわち、隣接する一対の原子を共有する環)の基を指し、残りの環原子はCであり、かつ、加えて、完全に共役したパイ電子系を有し、そこで、各環の0個、1個、2個、3個、または4個の原子を置換基により置換することができる。ヘテロアリール基の例は、制限なく、ピロール、フラン、チオフェン、イミダゾール、オキサゾール、チアゾール、ピラゾール、ピリジン、ピリミジン、キノリン、キナゾリン、イソキノリン、プリン、およびカルバゾールである。
【0045】
「複素環」、「複素環式」、または「ヘテロシクロ」という用語は、完全飽和または部分不飽和の環式基、例えば、3乃至7員環の単環式、7乃至12員環の二環式、または10乃至15員環の三環式の環系を指し、これらは少なくとも1個の複素原子を少なくとも1個の環内に有し、そこで、各環の0個、1個、2個、または3個の原子を置換基により置換することができる。複素原子を含有する複素環式基の各環は、窒素原子、酸素原子、および/または硫黄原子から選択される1個、2個、3個、または4個の複素原子を有し得、そこで、窒素および硫黄の複素原子を随意に酸化させることができ、かつ、窒素複素原子を随意に四級化することができる。複素環式基は、環または環系の任意の複素原子または炭素原子にて結合させることができる。
【0046】
「置換基」という用語は、本明細書に描出されている任意の官能基上、例えば、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、シクロアルケニル基、アリール基、ヘテロシクリル基、またはヘテロアリール基上にて、その基の任意の原子において「置換された」基を指す。態様では、本明細書に描出されている官能基、例えば、アルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクリル、またはヘテロアリールを、一つの置換基(例えば、下記に列挙されているもの)で置換することができる。適切な置換基としては、制限なく、ハロゲン、CN、NO2、OR15、SR15、S(O)2R15、NR15R16、C1〜C2のペルフルオロアルキル、C1〜C2のペルフルオロアルコキシ、1,2−メチレンジオキシ、C(O)OR15、C(O)NR15R16、OC(O)NR15R16、NR15C(O)NR15R16、C(NR16)NR15R16、NR15C(NR16)NR15R16、S(O)2NR15R16、R17、C(O)R17、NR15C(O)R17、S(O)R17、S(O)2R17、R16、オキソ、C(O)R16、C(O)(CH2)nOH、(CH2)nOR15、(CH2)nC(O)NR15R16、NR15S(O)2R17が挙げられ、式中、nは独立して0〜6の全体を含むものである。各R15は、独立して、水素、C1〜C4のアルキル、またはC3〜C6のシクロアルキルである。各R16は、独立して、水素、アルケニル、アルキニル、C3〜C6のシクロアルキル、アリール、ヘテロシクリル、ヘテロアリール、C1〜C4のアルキル、あるいは、C3〜C6のシクロアルキル、アリール、ヘテロシクリル、またはヘテロアリールで置換されたC1〜C4のアルキルである。各R17は、独立して、C3〜C6のシクロアルキル、アリール、ヘテロシクリル、ヘテロアリール、C1〜C4のアルキル、あるいは、C3〜C6のシクロアルキル、アリール、ヘテロシクリル、またはヘテロアリールで置換されたC1〜C4のアルキルである。各R15、R16、およびR17中の各C3〜C6のシクロアルキル、アリール、ヘテロシクリル、ヘテロアリール、およびC1〜C4のアルキルを、ハロゲン、CN、C1〜C4のアルキル、OH、C1〜C4のアルコキシ、NH2、C1〜C4のアルキルアミノ、C1〜C4のジアルキルアミノ、C1〜C2のペルフルオロアルキル、C1〜C2のペルフルオロアルコキシ、または1,2−メチレンジオキシで随意に置換することができる。
【0047】
「オキソ」という用語は、炭素に結合される際にはカルボニル、窒素に結合される際にはN−オキシド、および硫黄に結合される際にはスルホキシドまたはスルホンを形成する、酸素原子を指す。
【0048】
「アシル」という用語は、アルキルカルボニル、シクロアルキルカルボニル、アリールカルボニル、ヘテロシクリルカルボニル、またはヘテロアリールカルボニルの置換基を指し、これらの何れかを置換基によりさらに置換することができる。
【0049】
一可変物の任意の定義における化学基の一覧の詳述は、本明細書において、任意の単一の基または列挙されている基の組み合わせとしてのその可変物の定義を含む。一可変物についての一実施形態の詳述は、本明細書において、任意の単一の実施形態としての、あるいは、任意の他の実施形態またはその部分と組み合わせるその実施形態を含む。
【0050】
本発明の化合物は、1つ以上の不斉中心を含有することがあり、そのため、ラセミ体およびラセミ混合物、単一のエナンチオマー、各個のジアステレオマーおよびジアステレオマー混合物として生じることがある。これらの化合物の全てのこのような異性形態は、本発明に明確に含まれる。本発明の化合物を多数の互変異性形態にて表すこともでき、このような場合において、本発明は、本明細書に記載されている化合物の全ての互変異性形態を明確に含む。このような化合物の全てのこのような異性形態は、本発明に明確に含まれる。本明細書に記載されている化合物の全ての結晶形態は、本発明に明確に含まれる。
【0051】
本明細書における化学式の化合物を合成するための他の手法を、本明細書で引用されている参考文献から容易に適合させることができる。これらの手順の変化形およびそれらの最適化は、通常の実務者の技能の範囲内にある。
【0052】
上記で示されている特定の手法および化合物は、限定的であることを目的としていない。本明細書における図式内の化学構造は、同じ可変物名(例えば、R1、R2、R、R’、Xなど)により識別されるか否かにかかわらず、本明細書における化合物の化学式内の対応する位置の化学基の定義(部分、原子など)に相応に本明細書により定義される可変物を描写する。ある化合物構造内の化学基の、別の化合物構造の合成の際の使用のための適合性は、当該技術分野における通常の技能を有する者の知識の範囲内にある。本明細書における図式内に明白に示されていない経路内にあるものを含む、本明細書における化合物およびその合成前駆体を合成する追加の方法は、当該技術分野における通常の技能を有する化学者の技量の範囲内にある。反応条件を最適化し、必要であれば競合副生成物を最小化するための方法は、当該技術分野で知られている。本明細書に記載されている方法は、本明細書に具体的に記載されている段階の前でも後でも、本明細書における化合物の合成を最終的に可能にするために、適切な保護基を追加または除去する段階を追加的に含むこともある。加えて、様々な合成段階を交互の順序または順番で行い、所望の化合物を与えることができる。適用可能な化合物を合成する際に有用な、合成化学の変換および保護基の方法論(保護および脱保護)は、当該技術分野で知られており、例えば、「R.ラロック、包括的有機変換、VCH出版(1989年)(R. Larock, Comprehensive Organic Transformations, VCH Publishers
(1989))」、「T.W.グリーンおよびP.G.M.ウッツ、有機合成における保護基、第3版、ジョン・ワイリー・アンド・サンズ(1999年)(T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis,
3rd Ed., John Wiley and Sons (1999))」、「L.フィーザーおよびM.フィーザー、フィーザーおよびフィーザーの試薬または有機合成、ジョン・ワイリー・アンド・サンズ(1994年)(L. Fieser and M. Fieser, Fieser and Fieser's Reagents or Organic
Synthesis, John Wiley and Sons (1994))」、および、「L.パケット編、有機合成のための試薬の百科事典、ジョン・ワイリー・アンド・サンズ(1995年)(L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis,
John Wiley and Sons (1995))」、およびそれらの後続版に記載されているものが挙げられる。
【0053】
本明細書に記載されている合成法は、任意の図式に記載されている段階の何れかの前でも後でも、本明細書に記載されている化学式の化合物の合成を最終的に可能にするために、適切な保護基を追加または除去する段階を追加的に含むこともある。本明細書に描出されている方法は、1つの化学式の化合物を別の化学式の化合物に転換することを意図している。転換する工程は、その場で行うことができるか、あるいは中間化合物の単離を伴う、1つ以上の化学変換を指す。当該変換は、本明細書で引用されている参考文献中にあるものを含む、当該技術分野で知られている技術および実験計画法を用いて、出発化合物または中間体を追加の試薬と反応させることを含み得る。精製(例えば、濾過、蒸留、昇華、結晶化、研和、固相抽出、およびクロマトグラフィー)を伴うか否かにかかわらず、中間体を用いることができる。
【0054】
本発明により想定される置換基および可変物の組み合わせは、安定化合物の形成を結果的にもたらすもののみである。
【0055】
本発明は、有効量の本明細書における化合物、または、妥当な場合、前記化合物の医薬的に許容可能な塩、溶媒和物、水和物、多形体、もしくは前駆薬、および許容可能な担体を含む組成物も提供する。好ましくは、本発明の組成物は医薬用途のために調合され(医薬組成物)、そこで、担体は医薬的に許容可能な担体である。1つ(または複数)の担体は、調合物の他の成分と相溶性があるという意味で「許容可能」でなければならず、かつ、医薬的に許容可能な担体の場合には、薬物中に典型的に用いられる量でその受容者に有害であってはならない。
【0056】
本発明の医薬組成物において用いることができる、医薬的に許容可能な担体、補助剤、および媒介物としては、イオン交換体、アルミナ、ステアリン酸アルミニウム、レシチン、ヒト血清アルブミン等の血清タンパク質、リン酸塩類等の緩衝物質、グリシン、ソルビン酸、ソルビン酸カリウム、飽和植物脂肪酸の部分グリセリド混合物、水、硫酸プロタミン、リン酸水素ジナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩類、コロイド状シリカ、三ケイ酸マグネシウム、ポリビニルピロリドン等の塩または電解質、セルロース系物質、ポリエチレングリコール、カルボキシメチルセルロースナトリウム、ポリアクリル酸塩類、蝋、ポリエチレン−ポリオキシプロピレン−ブロック重合体、ポリエチレングリコール、および羊毛脂が挙げられるが、これらに限定されない。
【0057】
本発明の医薬組成物としては、経口、直腸、経鼻、局所(頬側および舌下を含む)、膣または非経口(皮下、筋肉内、静脈内、および皮内を含む)の投与に適したものが挙げられる。特定の実施形態では、本明細書における化学式の化合物は、(例えば、経皮貼布を用いて)経皮的に投与される。他の調合物を、単位剤形、例えば、錠剤および持続放出型カプセルで、およびリポソームで便利に提供することができ、かつ、薬学の技術分野において十分に知られている任意の方法により調製することができる。例えば、「レミントンの薬科学、マック出版社、ペンシルヴァニア州、フィラデルフィア(第17版、1985年)(Remington's Pharmaceutical Sciences, Mack Publishing Company,
Philadelphia, PA (17th ed. 1985))」を参照されたい。
【0058】
このような調製法は、1つ以上の付属成分を構成する担体等の成分を、投与されることになる分子との関連に至らせる段階を含む。一般的に、本組成物は、活性成分を、液状担体、リポソームつまり微細化した固体担体、あるいはその両方との関連に均一にかつ密接に至らせ、その後必要であれば生成物を成形することにより調製される。
【0059】
特定の好適な実施形態では、化合物は経口的に投与される。経口投与に適した本発明の組成物を、所定量の活性成分をそれぞれ含有するカプセル、小袋、または錠剤等の不連続単位として、粉末または顆粒として、水性液体または非水液体中の溶液または懸濁液として、または水中油型液状乳濁液もしくは油中水型液状乳濁液として提供することができ、あるいは、リポソーム中におよび急速静注薬などとして包装することができる。軟質のゼラチンカプセルは、化合物の吸収の速度を有益に増加させることができるような懸濁液を含有するのに有用であり得る。
【0060】
随意に1つ以上の付属成分を用いて、圧縮または成形により錠剤を作ることができる。適切な機械内で、粉末または顆粒等の自由流動形態にある活性成分を、結合剤、潤滑剤、不活性希釈剤、保存剤、表面活性剤、または分散剤と随意に混合されて圧縮することにより、圧縮錠剤を調製することができる。適切な機械内で、不活性液状希釈剤で湿らせた粉末化合物の混合物を成形することにより、成形錠剤を作ることができる。当該錠剤を随意に被覆するかまたは刻み付けることができ、かつ、その中の活性成分の徐放つまり制御放出を提供するように調合することができる。本明細書におけるものおよび当該技術分野で知られている他の化合物等の、医薬的に活性な成分のこのような徐放または制御放出の組成物を調合する方法は、当該技術分野で知られかついくつかの発行された米国特許に記載されており、その一部としては、米国特許第4,369,172号、および第4,842,866号、および当該特許で引用されている参考文献が挙げられるが、これらに限定されない。化合物の腸への運搬のために、被覆剤を用いることができる(例えば、米国特許第6,638,534号、第5,217,720号、および第6,569,457号、第6,461,631号、第6,528,080号、第6,800,663号、および当該特許で引用されている参考文献を参照されたい)。本発明の化合物のための有用な調合物は、腸溶層が酢酸コハク酸ヒドロキシプロピルメチルセルロースを含む形態の腸溶性丸薬である。
【0061】
経口用途のための錠剤の場合において、一般に用いられる担体としては、乳糖およびトウモロコシデンプンが挙げられる。ステアリン酸マグネシウム等の潤滑剤も、典型的に添加される。カプセル形態における経口投与のために、有用な希釈剤としては、乳糖および乾燥トウモロコシデンプンが挙げられる。水性懸濁液が経口的に投与される際に、活性成分は乳化剤および懸濁剤と混合される。所望であれば、特定の甘味剤および/または香味剤および/または着色剤を添加してよい。
【0062】
局所投与に適した組成物としては、通常は蔗糖およびアカシアまたはトラガカントである香味基礎中に成分を含む飴剤、ならびに、ゼラチンおよびグリセリン、または蔗糖およびアカシア等の不活性基礎中に活性成分を含む香錠が挙げられる。
【0063】
非経口投与に適した組成物としては、調合物を対象とする受容者の血液と等張にする抗酸化剤、緩衝剤、静菌剤、および溶質を含有することがある、水性および非水の無菌注入溶液、ならびに、懸濁剤および増粘剤を含むことがある、水性および非水の無菌懸濁液が挙げられる。調合物を、単位用量または多重用量の容器、例えば、封止したアンプルおよび薬瓶内で提供することができ、ならびに、使用の直前に、無菌液状担体、例えば注入用の水の添加のみを必要とする凍結乾燥(氷結乾燥)条件において貯蔵することができる。即時の注入溶液および懸濁液を、無菌の粉末、顆粒、および錠剤から調製することができる。
【0064】
このような注入溶液は、例えば、無菌注入可能な水性または油性の懸濁液の形態にあってよい。当該技術分野で知られている技術により、適切な分散剤または湿潤剤(例えば、Tween 80等)および懸濁剤を用いて、この懸濁液を調合することができる。無菌注入可能な調製物は、非毒性で非経口的に許容可能な希釈剤または溶媒中の無菌注入可能な溶液または懸濁液、例えば、1,3−ブタンジオール中の溶液としてであってもよい。使用することができる許容可能な媒介物および溶媒の中には、マンニトール、水、リンガー溶液、および等張塩化ナトリウム溶液がある。加えて、無菌の固定油は、溶媒または懸濁媒体として従来的に使用される。この目的のために、合成のモノグリセリドまたはジグリセリドを含む、任意の無刺激性の固定油を使用することができる。オレイン酸およびそのグリセリド誘導体等の脂肪酸は、注入剤の調製において有用であり、特にそのポリオキシエチル化型にある、オリーブ油またはヒマシ油等の、天然の医薬的に許容可能な油も同様である。こうした油の溶液または懸濁液は、長鎖アルコールの希釈剤または分散剤を含有することもある。
【0065】
直腸投与のための坐剤の形態で、本発明の医薬組成物を投与することができる。室温で固体であるが直腸温度で液体であり、そのため、直腸内で溶けて活性成分を放出することになる、適切な非刺激性の賦形剤と、本発明の化合物を混合することにより、こうした組成物を調製することができる。このような物質としては、ココアバター、蜜蝋、およびポリエチレングリコール類が挙げられるが、これらに限定されない。
【0066】
経鼻の噴霧または吸入により、本発明の医薬組成物を投与することができる。このような組成物は、医薬調合の技術分野で十分に知られている技術により調製され、かつ、生理食塩水中の溶液として調製し、ベンジルアルコールまたは他の適切な保存剤、生物利用可能性を高める吸収促進剤、フッ化炭素類、および/または、当該技術分野で知られている他の可溶化剤または分散剤を使用することができる。
【0067】
本発明の医薬組成物の局所投与は、所望の治療が局所適用により容易に到達可能な領域または器官に影響を及ぼす際に特に有用である。皮膚への局所的な適用のためには、担体中に懸濁または溶存した活性成分を含有する適切な軟膏を用いて、本医薬組成物を調合するべきである。本発明の化合物の局所投与のための担体としては、鉱油、液状石油、白色石油、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化蝋、および水が挙げられるが、これらに限定されない。代替的には、担体中に懸濁または溶存した活性成分を含有する適切なローションまたはクリームを用いて、本医薬組成物を調合することができる。適切な担体としては、鉱油、モノステアリン酸ソルビタン、ポリソルベート60、セチルエステル類、蝋、セテアリルアルコール、2−オクチルドデカノール、ベンジルアルコール、および水が挙げられるが、これらに限定されない。本発明の医薬組成物を、直腸坐剤調合物により、または、適切な浣腸調合物中で、下部腸管に局所的に適用することもできる。局所経皮貼布およびイオン導入投与も、本発明に含まれる。
【0068】
特に好ましい誘導体および前駆薬は、本発明の化合物が(例えば、経口的に投与される化合物を血液中により容易に吸収することを可能にすることにより)哺乳類に投与される際に、このような化合物の生物利用可能性を増加させるか、あるいは、親種と比較して親化合物の生物学的区画(例えば、脳または中枢神経系)への運搬を強化するものである。好適な前駆薬としては、水溶性または腸膜を通じる能動輸送を強化する基が、本明細書に記載されている化学式の構造に付加される誘導体が挙げられる。例えば、「アレクサンダー・J.他、医薬品化学誌、1988年、第31号、318〜322ページ(Alexander, J. et al. Journal of Medicinal Chemistry 1988, 31,
318-322)」、「バンガード・H.、前駆薬の設計、エルゼビア:アムステルダム、1985年、1〜92ページ(Bundgaard,
H. Design of Prodrugs; Elsevier: Amsterdam, 1985; pp 1-92)」、「バンガード・H.、ニールセン・N.M.、医薬品化学誌、1987年、第30号、451〜454ページ(Bundgaard, H.; Nielsen, N. M. Journal of Medicinal Chemistry 1987,
30, 451-454)」、「バンガード・H.、薬の設計および開発の教科書、ハーウッド・アカデミック出版:スイス、1991年、113〜191ページ(Bundgaard, H. A Textbook of Drug Design and Development; Harwood
Academic Publ.: Switzerland, 1991; pp 113-191)」、「ディゲニス・G.A.他、実験薬理学の手引書、1975年、第28号、86〜112ページ(Digenis, G. A. et al. Handbook of Experimental Pharmacology 1975,
28, 86-112)」、「フリース・G.J.、バンガード・H.、薬の設計および開発の教科書、第2版、海外出版:アムステルダム、1996年、351〜385ページ(Friis, G. J.; Bundgaard, H. A Textbook of Drug Design and
Development; 2 ed.; Overseas Publ.: Amsterdam, 1996; pp 351-385)」、「ピットマン・I.H.、医薬品研究総説、1981年、第1号、189〜214ページ(Pitman, I. H. Medicinal Research Reviews 1981, 1, 189-214)」を参照されたい。
【0069】
本治療法の適用は、関心部位に投与が行なわれるように局所的であり得る。関心部位に本組成物を提供するために、注入、カテーテル、套管針、発射体、プルロンゲル、ステント、持続薬物放出重合体、または内部到達を提供する他の機器の使用等の、様々な技術を用いることができる。
【0070】
別の実施形態によれば、本発明は、移植可能な薬物放出機器を本発明の化合物または組成物と接触させる段階を含む、前記薬物放出機器を含浸させる方法を提供する。移植可能な薬物放出機器としては、生分解性重合体のカプセルまたは弾丸、非生分解性で拡散性の重合体のカプセル、および生分解性重合体の薄板が挙げられるが、これらに限定されない。
【0071】
別の実施形態によれば、本発明は、化合物、または本発明の化合物を含む組成物で、前記化合物が治療的に活性であるように被覆された移植可能な医療機器を提供する。
【0072】
別の実施形態では、本発明の組成物は第2の治療薬剤をさらに含む。第2の治療薬剤は、単独で、または、本明細書における化学式の何れかの化合物と投与される際に、有利な特性を有すると知られているかまたは実証する任意の化合物または治療薬剤を含む。こうした化合物と有用に組み合わせることができるだろう薬物としては、他のキナーゼ阻害剤、および/または、上記で検討されている疾患および障害の治療のための他の化学治療薬剤が挙げられる。
【0073】
このような薬剤は、当該技術分野で詳細に説明されている。好ましくは、第2の治療薬剤は、癌から選択される疾患または容態の治療または防止において有用な薬剤である。
【0074】
さらにより好ましくは、本発明の化合物と共調合された第2の治療薬剤は、癌、免疫障害、心血管疾患、ウイルス感染、炎症、代謝/内分泌の障害、および神経学的障害等の、キナーゼ媒介の疾患/障害の治療において有用な薬剤である。
【0075】
別の実施形態では、本発明は、互いに関連する、本発明の化合物および第2の治療薬剤の別個の剤形を提供する。本明細書で用いられる際の「互いに関連する」という用語は、別個の剤形が共に包装され、またはそうでなければ、別個の剤形が(連続的または同時に、互いに24時間未満以内に)一緒に販売、かつ投与されることを目的とすることが容易に明白であるように互いに付着されることを意味する。
【0076】
本発明の医薬組成物において、本発明の化合物は有効量で存在する。本明細書で用いられる際に、「有効量」という用語は、適切な投与計画内で投与される際に、治療される障害の重症度、持続時間、または進行を低減するかまたは寛解させるのに十分であり、治療される障害の前進を防止し、治療される障害の後退を引き起こし、あるいは、別の治療の1つ(または複数)の予防効果または治療効果を強化または改善する量を指す。
【0077】
動物およびヒトに対する用量の相互関係(体表面の1平方メートル当たりのミリグラム数に基づく)は、「フライライヒ他、(1966年)癌化学療法報告書第50号:219ページ(Freireich et al., (1966) Cancer Chemother Rep 50: 219)」に記載されている。患者の身長および体重から、体表面積をおおよそ決定することができる。例えば、「科学表、ガイギー製薬、ニューヨーク州、アードリー、1970年、537ページ(Scientific Tables, Geigy Pharmaceuticals, Ardley, N. Y., 1970, 537)」を参照されたい。本発明の化合物の有効量は、約0.001mg/kgから約500mg/kgまで、より好ましくは0.01mg/kg乃至約50mg/kg、より好ましくは0.1mg/kg乃至約2.5mg/kgの範囲であり得る。有効用量も、当業者により認識されるように、治療される疾患、当該疾患の重症度、投与の経路、患者の性別、年齢、および一般的健康状態、賦形剤の利用、他の薬剤の使用等の他の治療処置との共利用の可能性、および治療する医師の判断に応じて変化することになる。
【0078】
第2の治療薬剤を含む医薬組成物について、当該第2の治療薬剤の有効量は、その薬剤のみを用いる単独治療計画で通常利用される用量の約20%および100%の間である。好ましくは、有効量は通常の単独治療用量の約70%および100%の間である。こうした第2の治療薬剤の通常の単独治療用量は、当該技術分野で十分に知られている。例えば、「ウェルズ他編、薬物治療の手引書、第2版、アップルトン・アンド・ラング、コネティカット州、スタムフォード(2000年)(Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton
and Lange, Stamford, Conn. (2000))」、「PDR薬局方、タラスコン小型薬局方2000、豪華版、タラスコン出版、カリフォルニア州、ロマ・リンダ(2000年)(PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe
Edition, Tarascon Publishing, Loma Linda, Calif. (2000))」を参照されたく、その参考文献のそれぞれは、本明細書に参照により全体的に組み込まれている。
【0079】
上記で参照されている第2の治療薬剤のいくつかは、本発明の化合物と相乗的に作用することになることが予想される。これが起こる際に、それにより、第2の治療薬剤および/または本発明の化合物の有効用量を、単独治療に必要とされるものから減らすことが可能になることになる。これは、第2の治療薬剤または本発明の化合物の何れか毒性の副作用の最小化、有効性における相乗的改善、投与または使用の改善した容易さ、および/または化合物の調製または調合の低減した全体の費用、という利点を有する。
【0080】
治療の方法
1つの態様によれば、本発明は、対象者に有効量の化合物(例えば、単離した化合物、本明細書における化合物)または本発明の組成物を投与する段階を含む、疾患または障害またはその症状(例えば、本明細書で描出されているもの)を患っているか、またはこれらの影響を受けやすい前記対象者を治療する方法を提供する。このような疾患は当該技術分野で十分に知られており、かつ本明細書でも開示されている。
【0081】
本方法は、組成物が、ヒカゲノカズラ(Lycopodium clavatum)の抽出物(例えば、BCP−21抽出物、表1または2内のBCP−21抽出物画分の何れか)、または、本明細書における植物抽出物(例えば、BCP−21抽出物、表1または2内のBCP−21抽出物画分の何れか)中に生じる単離した化合物(例えば、8−ヒドロキシヘキサデカン酸(8−HHA)、本明細書におけるBCP化合物1〜10の何れか)であるものをさらに含み得る。
【0082】
本発明の1つの実施形態では、単離した化合物は、8−ヒドロキシヘキサデカン酸(「8−HHA」)、またはその塩、前駆薬、前駆薬塩、溶媒和物、水和物、および多形体である。特定の実施形態は、当該化合物が、8−ヒドロキシ−パルミチン酸のR−エナンチオマーおよびS−エナンチオマーのラセミ混合物であることを規定している。1つの実施形態では、当該化合物は、8−ヒドロキシ−パルミチン酸のS−エナンチオマー(「(S)−8−HHA」)である。別の実施形態では、当該化合物は、8−ヒドロキシ−パルミチン酸のR−エナンチオマー(「(R)−8−HHA」)である。
【0083】
別の実施形態では、本発明の方法において用いられる化合物(または組成物)は、合成手段を通じて得られる。
【0084】
本発明の1つの態様では、当該疾患または障害は、カスパーゼ媒介の疾患または障害である。
【0085】
本発明の1つの態様では、当該疾患または障害は、カスパーゼ−3媒介の細胞死により媒介される。
【0086】
本発明の1つの態様では、当該疾患または障害は、カスパーゼ−3媒介の疾患または障害である。
【0087】
本発明の1つの態様では、当該疾患または障害は、細胞死を誘発することにより治療される。
【0088】
本発明の1つの態様では、当該疾患または障害は、カスパーゼ−3により媒介される細胞死を誘発することにより治療される。
【0089】
本発明の1つの態様では、当該疾患または障害をカスパーゼ−3により調節することができる。
【0090】
1つの態様では、治療する方法は、増殖障害である障害またはその症状の治療を伴う。これらは、癌、腫瘍、腫瘍剤が適切である任意の疾患を含む。
【0091】
本化合物、組成物、および治療の方法を用いて治療することができる癌の例としては、癌(例えば、白血病、肝臓癌、肺癌、結腸癌、CNS癌、黒色腫、腎臓癌、およびカスパーゼ−3媒介癌)、アレルギー性障害、および炎症性障害が挙げられる。増殖障害、例えば、癌を患っているヒトまたは動物の患者を、そのため、上記で定義されている通りの本発明の化合物の当該患者への投与を含む方法により治療することができる。患者の容態を、それにより、改善するかまたは寛解させることができる。本発明の方法により治療可能な疾患および容態としては、癌および炎症性障害が挙げられるが、これらに限定されない。本発明の方法により治療することができる癌としては、肝臓癌、肝細胞性、白血病、肺癌、結腸癌、CNS癌、黒色腫、腎臓癌などが挙げられるが、これらに限定されない。
【0092】
1つの実施形態は、本発明の方法により治療可能な癌が、白血病、肺癌、肝臓癌、または結腸癌であることを規定している。1つの実施形態では、当該癌は肝臓癌である。別の実施形態は、当該癌が肺癌であることを規定している。1つの実施形態では、当該肺癌は非小細胞肺癌である。特定の実例は、本発明の方法により治療可能な癌が、HOP−92媒介の疾患(例えば、非小細胞肺癌)に関することを規定している。
【0093】
さらに別の実施形態では、本発明の方法により治療することができる癌は、白血病である。
【0094】
本明細書に描出されている方法としては、対象者が特定の所定の治療を必要としていると確認されるものが挙げられる。このような治療を必要としている対象者を確認することは、対象者または健康管理専門家の判断のうちにあり得、かつ、主観的(例えば、意見)または客観的(例えば、試験または診断法により測定可能)であり得る。
【0095】
1つの実施形態では、本発明は、本明細書における化学式の何れかの1つ以上の化合物と細胞を接触させることを含む、細胞内のカスパーゼ酵素(例えば、カスパーゼ−3)活性を調節する方法を提供する。
【0096】
別の実施形態では、上記の治療の方法は、前記患者に1つ以上の第2の治療薬剤を共投与するさらなる段階を含む。第2の治療薬剤の選択を、本明細書における適応症に有用であると知られている任意の第2の治療薬剤から行うことができる。
【0097】
本明細書で用いられる際の「共投与される」という用語は、単一の剤形(上記に記載されているような本発明の化合物および第2の治療薬剤を含む、本発明の組成物等)の一部として、あるいは、別々の複数の剤形として、第2の治療薬剤を本発明の化合物と共に投与することができることを意味する。代替的には、本発明の化合物の投与より前に、これと連続して、またはこれの後に、追加の薬剤を投与することができる。このような組み合わせの治療処置では、本発明の化合物および1つ(または複数)の第2の治療薬剤の両方が、従来の方法により投与される。本発明の化合物および第2の治療薬剤を含む、本発明の組成物の対象者への投与は、治療の経過中の別の時間における、その同じ治療薬剤、任意の他の第2の治療薬剤、または本発明の任意の化合物の前記対象者への別個の投与を除外しない。
【0098】
こうした第2の治療薬剤の有効量は当業者に十分に知られており、本明細書で参照されている特許および公開特許出願、ならびに、「ウェルズ他編、薬物治療の手引書、第2版、アップルトン・アンド・ラング、コネティカット州、スタムフォード(2000年)(Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton
and Lange, Stamford, Conn. (2000))」、「PDR薬局方、タラスコン小型薬局方2000、豪華版、タラスコン出版、カリフォルニア州、ロマ・リンダ(2000年)(PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition,
Tarascon Publishing, Loma Linda, Calif. (2000))」、および他の医学書で投薬の指針を見つけることができる。しかしながら、第2の治療薬剤の最適な有効量の範囲を決定することは、十分に当業者の能力が及ぶ範囲内にある。
【0099】
第2の治療薬剤が対象者に投与される本発明の1つの実施形態では、本発明の化合物の有効量は、その有効量が当該第2の治療薬剤が投与されない場合のものより少ない。別の実施形態では、当該第2の治療薬剤の有効量は、その有効量が本発明の化合物が投与されない場合のものより少ない。このようにして、両方の薬剤の高い用量と関連する非所望の副作用を最小化することができる。他の潜在的な利点(制限なく、改善した投薬計画および/または低減した薬物費用を含む)は、当業者にとって明白であることになる。
【0100】
さらに別の態様では、本発明は、上記で明記されている対象者における疾患、障害、または症状の治療または防止のための、単一の組成物または別個の剤形の何れかとしての薬物の製造における、単独での、または、上記に記載されている第2の治療薬剤のうち1つ以上と合わせての、本明細書における化学式の何れかの化合物の使用を提供する。本発明の別の態様は、本明細書に描出されている対象者における疾患、障害、またはその症状の治療または防止における使用のための、本明細書における化学式の化合物である。
【0101】
他の態様では、本明細書における方法としては、治療投与に対する対象者の応答を監視することをさらに含むものが挙げられる。このような監視は、治療計画の標識または指標としての、対象者の組織、体液、検体、細胞、タンパク質、化学標識、遺伝物質などの周期的な試料抽出を含み得る。他の方法では、当該対象者は、このような治療に対する適切性の関連する標識または指標に対する評価により、前検査されるかまたはこのような治療を必要としていると確認される。
【0102】
1つの実施形態では、本発明は治療経過を監視する方法を提供する。当該方法は、本明細書に描出されている障害またはその症状を患っているか、またはこれらの影響を受けやすい患者における、診断標識(標識)(例えば、本明細書における化合物により調節される、本明細書に描出されている任意の標的または細胞型)の濃度の決定、または診断測定(例えば、検査、検定)の段階を含み、ここで、この対象者は、疾患またはその症状を治療するのに十分な治療量の本明細書における化合物を投与されている。当該方法において決定される標識の濃度を、健常な通常の対照または他の罹患した患者の何れかにおける標識の既知の濃度と比較し、対象者の疾患状態を確証することができる。好適な実施形態では、対象者における標識の第2の濃度は、第1の濃度の決定より遅い時点で決定され、2つの濃度は、疾患の経過または治療の有効性を監視するために比較される。特定の好適な実施形態では、対象者における標識の前治療濃度は、本発明による治療を始めるより前に決定され、この標識の前治療濃度を、その後、治療を開始した後の対象者における標識の濃度と比較し、治療の有効性を決定することができる。
【0103】
特定の方法の実施形態では、対象者における標識の濃度または標識活性は、少なくとも1回決定される。標識濃度の、例えば、同じ患者、別の患者、または通常の患者から前にまたは後に得られる標識濃度の別の測定結果との比較は、本発明による治療が所望の効果を有しているかどうかを決定する際に有用であり、それにより、投薬濃度の調節を必要に応じて可能にすることができる。当該技術分野で知られているか、または本明細書に記載されている、任意の適切な試料抽出法/発現検定法を用いて、標識濃度の決定を行うことができる。好ましくは、組織または体液の試料は対象者から最初に取り出される。適切な試料の例としては、血液、尿、組織、口または頬の細胞、および毛根を含有する毛髪試料が挙げられる。他の適切な試料が、当業者に公知であろう。当該試料中のタンパク質濃度および/またはmRNA濃度(例えば、標識濃度)の決定を、酵素免疫測定法、ELISA、放射標識技術/検定技術、吸い取り法/化学発光法、実時間PCR、および同様のものを含むが、これらに限定されない、当該技術分野で知られている任意の適切な技術を用いて行うことができる。
【0104】
本発明の化合物の使用
本発明は、本明細書に描出されているものを含む、疾患、障害、またはその症状を治療する使用のためのキットも提供する。こうしたキットは、a)本明細書における化学式の何れかの1つ以上の化合物もしくはその塩、またはその前駆薬もしくはその前駆薬の塩、またはその水和物、溶媒和物、もしくは多形体を含む医薬組成物(例えば、本明細書における組成物、本明細書における任意の特定の化合物)であって、前記医薬組成物が容器内にある、医薬組成物と、b)当該医薬組成物を用いて、本明細書に描出されているものを含む、疾患、障害、またはその症状を治療する方法を説明している取扱説明書と、を備える。放射線治療の実施と逐次的に、または同時に、当該化合物/組成物を投与することができる。
【0105】
当該容器は、前記医薬組成物を保持することができる、任意の器あるいは他の封止された装置または封止可能な装置であってよい。例としては、瓶、各区分または室が単一用量の前記組成物を含む分割型または多室型の保持瓶、各区分が単一用量の前記組成物を含む分割型箔包、または、単一用量の前記組成物を分注する分注器が挙げられる。当該容器は、医薬的に許容可能な物質で作られる、当該技術分野で知られているような任意の従来の形状または形態にあり、例えば、紙または段ボール箱、ガラスまたはプラスチックの瓶または広口瓶、再封止可能な袋(例えば、異なる容器内への配置のために錠剤の「補充分」を保持する)、または、治療予定により包装から押し出すための各個の用量を有するブリスターパックであってよい。使用される容器は関連する的確な剤形に依存し得、例えば、従来の段ボール箱は一般的に液状懸濁液を保持するために用いられることはないだろう。単一の剤形を市販するために、1つより多くの容器を単一の包装内において共に用いることができることが可能である。例えば、錠剤を瓶内に包含することができ、これは同様に箱の中に包含される。好ましくは、当該容器はブリスターパックである。
【0106】
当該キットは、医師、薬剤師、または対象者のための情報および/または取扱説明書を追加的に備え得る。このような記憶補助としては、そのように規定される錠剤またはカプセルが対象者に摂取または投与されるべきである投与計画の日数と一致する投薬量を包含する各室または区分上に印刷される数、または各室または区分上に印刷される週の日数、または同じ種類の情報を含有するカードが挙げられる。1つの態様では、当該取扱説明書はさらに、対象者への放射線管理に関する。
【0107】
さらに別の態様では、本発明は、本明細書に明記されている対象者における疾患、障害、または症状の治療または防止のための、単一の組成物または別個の剤形の何れかとしての薬物の製造における、単独での、または、1つ以上の追加の治療薬剤と合わせての、本発明の一つの化合物(または複数の化合物の組み合わせ)の使用を提供する。本発明の別の態様は、本明細書に描出されている対象者における疾患、障害、またはその症状の治療または防止における使用のための、本発明の一つの化合物(または複数の化合物の組み合わせ)である。
【0108】
本明細書で引用されている全ての参考文献は、印刷媒体、電子媒体、コンピュータ可読記憶媒体、または他の形態にあるかどうかにかかわらず、参照により全体として明確に組み込まれ、要約書、記事、機関誌、刊行物、教科書、論文、技術資料集、インターネットウェブサイト、データベース、特許、特許出願、および特許公開を含むが、これらに限定されない。
【実施例】
【0109】
実施例
一般的な手順:
用いた溶媒(CDCl3)を参照したJEOL ECLIPSE 400 MHz NMRの分光計にて、NMRスペクトルを取得した。ESI−MS(陰性モード)をHitachi M−8000の質量分光計を用いて得た。Phenomenex ZB−WAXのカラム(300m×0.32mm×0.25m)およびAgilent Model 5890のガスクロマトグラフィーを有する、Agilent Model 597 IAの質量分光計(範囲:70〜550amu)を用いて分析を行った。
【0110】
HPLC検定法1(逆相法):この勾配法を用いて、抽出物およびカラムの画分を検定することができる。そのカラムは、4.6×100mm、Phenomenex Luna(2)、3μ C18カラム(PIN 00D−4251−EO)である。流速は1.0mL/分、温度は40℃、注入は10μLである。検出は、ELSDおよび200nmでのUVである。順相クロマトグラフィーの試料を蒸発させ、注入する前にアセトン中で元に戻す。
【0111】
HPLC検定法2(順相法):この勾配法を用いて、カラム画分および最終生成物を検定することができる。そのカラムは、4.6×250mm、5μ、Alltech、Adsorbosilのシリカカラム(P/N 298017)である。移動相:ヘキサン/EtOAc、流速は1.0mL/分、温度は40℃、注入は10μLである。検出はELSDである。順相クロマトグラフィーの試料を直接的に注入することができ、注入する前に固体試料をEtOAc中で元に戻す。アセトン溶液を用いれば、ブロードまたは二重ピークを与え得るだろう。
【0112】
HPLC画分のアポトーシス活性についての生物検定:100,000個のHeLa細胞/穴を、24穴の細胞培養板中に蒔いた。その細胞を、24時間にわたり細胞培養恒温器内で成長させた。24時間後に、BCP21の精製から生じた化合物でその細胞を処理した。その精製の最中にHPLCにより発生させた画分を乾燥させ、DMSO中で再懸濁させた。各画分中の3つの異なる濃度の化合物でその細胞を処理した。その細胞を24時間後に採取し、アポトーシス(細胞死)(%)を決定した。その細胞をトリパンブルーで処理し、細胞死を推定した。10%未満の細胞死を+と標識付け、一方で、100%を++++により表し、50および75%を++および+++でそれぞれ標識付けた。
【0113】
実施例1.ヒカゲノカズラ(Lycopodium
clavatum)からの抽出および単離
BCP−21の単離:ヒカゲノカズラ(Lycopodium clavatum)の胞子を、95%のエタノールを用いて抽出する。その結果として生じた抽出相を混合し、95%のエタノール中の溶液として貯蔵することができるか、あるいは濃縮することができる。
【0114】
抽出物の蒸発および分割:BCP21のエタノール溶液(1L)を45℃にて真空下で蒸発させ、少量の沈殿物を含有する懸濁液を与えた。50mLの水をその懸濁液に添加し、200mLの塩化メチレン(MC)で分割した。そのMC層を分液漏斗内で分離して蒸発乾固させ、18.9gの油性残留物を与えた。その水層を乾燥させ、2.84gの固体を与えた。BCP21のエタノール抽出物およびMC可溶性残留物のHPLC特性描写を行った。
【0115】
実施例2.40MのSilica Biotageの分画:
前述したMC可溶性残留物が活性化合物を含有していると生物検定が示したので、5gのこの物質を15mLのMC中に溶解させ、800mLのヘキサンで平衡化させていた40Mのシリカカートリッジ上に装填した。10%のアセトン/ヘキサン(A/H)(1L)、20%のA/H(1L)、25%のA/H(1.2L)、およびアセトン(0.5L)と共に、そのカラムを溶出させた。生物検定の結果に基づき、25%のA/H溶出液のF5乃至F8、およびF9乃至F11を混合して窒素流により乾燥させ、2615−182−8(43.4mg)および2615−182−9(36.3mg)をそれぞれ与えた。表1は、アポトーシス検定により導いた画分試料抽出の結果を詳記している。
【表1】

【0116】
実施例3.12MのSilica Biotageの分画:
前述した溜めた活性生成物2615−182−8(43.4mg)を、2mLの30%のEtOAc/ヘキサン中に50℃にて溶解させ、100mLの30%のEtOAc/ヘキサンで平衡化させた12MのBiotageのシリカカートリッジ上にその溶液を注入した。60mLの35%のEtOAc/ヘキサン、100mLの40%のEtOAc/ヘキサン、60mLの45%のEtOAc/ヘキサン、60mLの80%のEtOAc/ヘキサンと共にそのカラムを溶出させ、26個の画分を得た。1mLのそれぞれを乾燥後に生物検定へ送った。各画分の残りの溶液を乾燥させて重量を得た。表2は、アポトーシス検定により導いた画分試料抽出の結果を詳記している。
【表2】
【表3】
【0117】
実施例4.検定のための試験化合物試料の調製。
前述した化合物の画分の調製を、下記の通りに表4に描出しているものに従って行った:
【表4】
【0118】
実施例5.ラセミ8−ヒドロキシパルミチン酸(「8−HHA」)の合成
【化1】
ラセミ8−ヒドロキシパルミチン酸の合成についての合成図式
【0119】
2つの三つ口フラスコは、気体注入補助具、隔壁、および撹拌子を備えており、その後、フラスコをアルゴンで浄化した。スベリン酸モノメチルエステル(10.0g、53.1mmol、1当量)を、THF(380mL、0.17M)中に1つのフラスコ内で溶解させた。トリエチルアミン(0.95mL、64.1mmol、1.2当量)を徐々に(4分にわたり、気密注射器を用いて)添加し、その混合物を室温で1.25時間にわたり撹拌した。無水DCM中でPPh3Cl2(24.4g、73.2mmol、1.4当量)から別個の溶液を作り、これは後で<30℃に冷却される。そのスベリン酸溶液を、黄色のPPh3Cl2溶液に滴下して添加し、2時間にわたり−35乃至−20℃で撹拌した。そのグリニャール試薬を、その後、温度を−41乃至−7℃に維持しつつ、45分にわたり滴下して添加した。その反応混合物を1.5時間にわたり撹拌し、その後、依然として冷たい間に2NのHCl(75mL)で反応停止した。20分にわたり撹拌した後に、その黄色は薄れる。薄層クロマトグラフィー(シリカゲル60、25%のアセトン/ヘキサン類)が、2つの主生成物のスポットに加えてPPh3およびOPPh3として反応混合物を与える。
【0120】
その反応混合物を、酢酸エチル(200mL)および2NのHCl(200mL)と共に分液漏斗に添加した。その有機層を収集し、その水層を2×80mLの酢酸エチルで逆抽出した。1つにまとめた有機相を、NaHCO3(飽和溶液、2×90mL)で洗浄することにより中和し、その後塩水(90mL)で洗浄し、MgSO4(20g、30分)で乾燥させ、濾過し、25℃で回転蒸発させ、高真空下で3時間にわたり乾燥させた。酢酸エチルを除去するために、その物質を20mLのDCMと混合して再蒸発させ、33gの粗製物質を与える。
【0121】
その粗製物質を、10%のアセトン/DCM中に取り出し、シリカゲル栓(240ml)上に装填し、4CVの10%のアセトン/DCMで洗い流した。その第1の栓は10.4gのOPPh3を除去し、第2の栓は6.8gのOPPh3を除去した。フラッシュカラムクロマトグラフィーを用いて精製を継続し、その中で、5×29cmのカラムを10%のアセトン/ヘキサン類中のシリカゲル60で充填する。55%のDCM/ヘキサン類の最小体積で、その粗製物質を装填した。所望の化合物を、10%のDCM/5%のアセトン/85%のヘキサン類と共に溶出させる。1つの画分の蒸発および乾燥は、2.7gの化合物1(8−ケトパルミチン酸メチルエステル)を与え、いくつかの不純な画分上の反復カラムは、さらに2.0gの化合物1を与えた。全収率は、スベリン酸モノメチルエステルに基づいて31%であった。化合物1(8−ケトパルミチン酸メチルエステル)は、1H NMR(CDCl3中):3.65(s,3H,OCH3),2.37(m,2H,O=CCH2),2.29(t,2H,O=CCH2),1.57および1.28(br.,22H,CH2),0.87(s,3H,CH3末端)により同定した。
【0122】
化合物1(2.86g、10.1mmol)を、50mLのエタノール中に溶解させ、氷浴内で冷却した。水素化ホウ素ナトリウム(1.9g、59.5mmol)を、一部分ずつ1〜17Cで添加した。その反応をTLC(25%のアセトン/ヘキサン類中で展開した板)により監視し、そこで、Rfは、ケトンについて0.56であり、メチルエステルについて0.28であった。固体NaBH4の追加分を、出発物質を全く検出しなくなるまで2時間の期間にわたり添加した(全部で60当量を用いた)。その反応を、2NのHCl(60mL)をその冷溶液にゆっくり添加することにより停止し、分液漏斗に移し、DCM(3×35mL)で洗浄した。1つにまとめた有機層を、NaHCO3(飽和、35mL)および塩水(35mL)で洗浄し、その後MgSO4で乾燥させ(7g、30分)、濾過し、回転蒸発させて澄んだ油を与え、これは4℃で貯蔵する際に白色で蝋状の固体となった(収量は2.86g)。
【0123】
その物質を、フラッシュクロマトグラフィー(シリカゲル60、20%のアセトン/ヘプタン)により精製した。回転蒸発乾固させた後は、その化合物は油であり、高真空上で乾燥させた後は、その生成物は白色で蝋状の固体である。その1H NMRスペクトル(CDCl3)は、その優勢生成物を、少量の未知の不純物を有する化合物2として示した。化合物2(8−ヒドロキシパルミチン酸メチルエステル)は、1H NMR(CDCl3中):3.66(s,3H,OCH3),(br,1H,メチン),2.29(t,2H,O=CCH2),1.62および1.31(br.,28H,CH2),0.87(s,3H,CH3末端)により同定した。
【0124】
化合物2(1.97g、6.9mmol)をTHF(28mL)中に溶解させた。水酸化リチウムを水(Nanopure、15mL)中に溶解させ、5分撹拌して溶解した。そのLiOH溶液を、5分にわたりそのエステル溶液にゆっくり添加し、室温で撹拌した。その生成物が消え、新たなスポットが基線に現れるまで(4時間)、その反応をTLC(20%のアセトン/ヘキサン)により監視した。単離精製操作は、その反応混合物を回転蒸発させ、揮発物を除去し、豊富な白色の固体を得ることを伴った。その固体をDCMですすぎ、その後、2NのHCl(固体はほとんど不溶)およびクロロホルムと混合した。その水層をCHCl3(3×75mL)3で抽出した。1つにまとめた有機相を、塩水およびMgSO4で乾燥させ(30分)、その後濾過し、回転蒸発させ、高真空下で一晩にわたり乾燥させ、1.67gの白色の固体を最終生成物として得た(89%の収率)。
【0125】
その最終生成物の分析は、8−ヒドロキシ−パルミチン酸としての同定を、その優勢生成物として支持した。1H NMR(CDCl3中):5.5(非常に広範,1H,OH),3.58(br,1H,メチン),2.34(t,2H,O=CCH2),1.64および1.33,(br.,28H,CH2),0.87(s,3H,CH3末端)。13C NMR(CDCl3中):178.40,72.08,37.59,37.43,33.79,31.97,29.79,29.68,29.37,29.09,25.73,25.51,24.69,22.76,14.20。LC−MS(ACN−NH4OH中でESIモード)。親イオン 271.13 m/z,理論:272(100%) 273(17%)。融点:70.5℃。
【0126】
実施例6.(S)−8−ヒドロキシパルミチン酸および(R)−8−ヒドロキシパルミチン酸の両方の合成
R、S、またはラセミ1,2エポキシデカンの1,2−デカンジオールからの形成
【化2】
塩基の存在下における、ラセミ1,2−デカンジオール類の塩化p−トルエンスルホニルとの反応は、所望の第一級トシル酸塩、ビス−トシル酸塩、および少量の第二級トシル酸塩の混合物を与えることが知られている(「韓国化学会会報、2009年、第30巻、第7号、1671−4(Bull. Korean Chem. Soc. 2009 Vol. 30, No. 7, 1671-4)」)。任意の第二級トシル酸塩の発生は、2位での閉環の最中における立体中心の反転が原因で、その結果として生じるエポキシドの立体化学的純度に不利に影響し得るだろう。
【0127】
この反応では、10mlの無水ジクロロメタン中の23.9ml(0.172mol、3.0当量)のトリエチルアミンを、10.00g(0.0574mol)の1,2(S)−デカンジオール(99.5%、「S」)、13.7g(0.0717mol、1.25当量)の塩化p−トルエンスルホニル、0.70g(0.00574mol、0.10当量)かつ30mlの無水ジクロロメタンの溶液に、<10℃において、ゆっくり40分の期間にわたり添加した。20分にわたり<10℃で撹拌した後に、その反応はTLC(100%のCH2Cl2)により完了した。室温に温めた後に、200mlのMTBEおよび120mlの1MのHClの添加により、その生成物の混合物を有機相に分配した。その有機層を、1×80mlの飽和NaHCO3溶液、1×80mlの水、および1×80mlの飽和NaCl溶液で洗浄した。硫酸ナトリウムでの乾燥および濃縮は、直接的にエポキシドへの転換まで進めた21.7gの残留物を結果的にもたらした。この残留物を217mlの無水THF中に溶解させ、9.64g(0.086mol、1.30当量)のカリウムt−ブトキシドを室温で冷却することなしに添加した。30分後に、その反応はTLC(10:1のヘプタン/酢酸エチル)により完了した。その反応を100mlの水の添加により停止し、THFを真空中で除去した。その生成物の混合物を300mlのMTBE中に分配し、その水層を1×50mlのMTBEで逆抽出した。1つにまとめた有機物を、その後、1×80mlの飽和NaCl溶液で洗浄した。硫酸ナトリウムでの乾燥および濃縮は、10.4gの残留物を結果的にもたらした。その残留物を短経路蒸留し、1,2(S)−エポキシデカンを約60℃および1mmgHgで収集した。>99%のGC−MS純度、68%の回収率で、6.11gを結果的にもたらした。
【0128】
臭化アルケニルマグネシウム類のR、S、またはラセミ1,2−エポキシデカンとの反応
【化3】
グリニャール試薬の形成、およびラセミまたは不斉の1−エポキシデカンへの添加を、文献の手順に従って行った(「有機化学誌、2009年、第74号、5063〜5066ページ(J. Org. Chem. 2009, 74, 5063-5066)」)。そのグリニャール試薬を、自己連結生成物を最小化するために約0.5Mで<10℃にて調製した。そのグリニャール試薬を滴定する試みを全く行わず、それらを0.5Mであると想定した。マグネシウムの削り屑を活性化してそのグリニャール反応を開始させるために、I2の添加を必要としなかった。
【0129】
乾燥した250mlのフラスコに、N2下、THF(上記で調製した通り)中で約0.5Mの98ml(0.049mol、1.5当量)の臭化ヘプト−6−エニルマグネシウムを投入する。そのフラスコの内容物を、撹拌しながら−40℃に冷却する。1.24g(0.00653mol、0.20当量)のヨウ化銅(I)を、そのフラスコに投入する。−40℃で0.5時間後に、51mlの無水THF中の5.10g(0.0326mol)の1,2(S)−エポキシデカンの溶液を、少なくとも1時間にわたり−40±5℃でゆっくり添加する。その添加の完了後、−40℃で2時間にわたり撹拌し続け、その後、その反応は、通常、TLC(ヘプタン中で40%のEtOAc)により完了する。完了後に、500mlのMTBE、125mlの飽和塩化アンモニウム、および50mlの水を添加し、15分にわたり十分に混ぜる。層を切り、その有機物を、1部の飽和塩化アンモニウムおよび1部の水の混合物100mlで洗浄する。その有機物を、その後、3×100mlの水、1×50mlの飽和NaCl溶液で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、静置の際に凝固する油になるまで濃縮する(8.6gの結果物)。その生成物を、220gのシリカ上でクロマトグラフィーに掛け、いかなる非極性不純物をも除去するために500mlのヘプタンで最初に溶出し、その後、10:1(v:v)のヘプタン/酢酸エチルで溶出する。その生成物を含有する画分を溜めて濃縮し、8.11g(97.6%の収率)の9(S)−ヒドロキシヘプタデス−1−エンを、GC−MSで96.7%純粋である蝋状の固体として得る。
【0130】
【化4】
1.50当量の塩化t−ブチルジメチルシリルの、2.0当量のイミダゾールを塩基として有するアルケノール類のTHF溶液への添加により、アルコールの保護を達成した。概して、室温で16〜20時間後に、その反応は92〜96%完了した。追加の塩化TBSおよびイミダゾールは、その反応を完了まで完全には推進しなかった。ヘプタンへの分配を伴う酸性で水性の単離精製操作は、酸感受性のTBSにより保護したアルコールの分解なしに、イミダゾールの除去を可能にした。シリカ上のヘプタンを用いたフラッシュクロマトグラフィーは、その残余の未反応のアルコールを容易に除去し、その中間体を高い純度(GC−MSで>99%)をもって単離することを可能にした(1362−42、45、86、1341−89、1380−15に留意されたい)。
【0131】
TBSにより保護した酸へのオゾン分解/ピニック酸化の経路
【化5】
138mlのジクロロメタンおよび138mlのメタノール中の9.83g(0.0267mol)の9(S)−(t−ブチルジメチルシリルオキシ)ヘプタデス−1−エン、0.10g(0.00027mol、0.01当量)のスーダンレッド7Bの溶液を、−25±5℃にてオゾンで処理した。指示薬の色が脱色した後に、オゾンの添加をさらに6〜7分にわたり継続した。その溶液に、その後N2を少なくとも30分にわたり注入する。13.99g(0.0533mol、2.00当量)のトリフェニルホスフィンの溶液を、その後4〜5分にわたり<−15℃で添加し、その後室温に温まるようにした。30分にわたり室温で撹拌した後に、その反応をGC−MSにより検査し、1.25時間および3時間で再び検査する。全ての3回の検査は、同じ濃度のトリフェニルホスフィン、酸化トリフェニルホスフィン、および生成物を概して示し、オゾン化物が消費されたことを示す。その反応混合物を、その後糊状の固体になるまで濃縮し、(メタノールを除去するために)100mlのジクロロメタンから1回再濃縮する。その残留物を、その後、ヘプタン、続いて20:1のヘプタン/酢酸エチルと共に溶出する、300gのシリカ上でクロマトグラフィーに掛けた。その生成物を含有する画分を溜めて濃縮し、GC−MSで31%のトリフェニルホスフィンを依然として含有する13gの油を与える。その油を500gのシリカ上で再度クロマトグラフィーに掛け、そのトリフェニルホスフィン濃度はその時13%であった。この油を100mlのジクロロメタン中に溶解させた。0.46ml(0.0074mol)のヨードメタンを添加し、その混合物を一晩にわたり撹拌し、その後、トリフェニルホスフィンの全てが消費されていた(GC−MSにより監視した)。最後に、その生成物を、10:1のヘプタン/酢酸エチルと共に溶出する、200gのシリカ上でフラッシュクロマトグラフィーに掛けた。濃縮後に、8.00g(89%の収率)の8(S)−(t−ブチルジメチルシリルオキシ)ヘキサデカナールを、98.7%のGC−MS純度をもって結果的にもたらした。
【0132】
315mlのt−ブタノール中の3.79g(0.0102mol)の8(S)−(t−ブチルジメチルシリルオキシ)ヘキサデカナールの溶液に、125mlの水中の8.49g(0.0939mol、9.17当量)の亜塩素酸ナトリウムおよび8.50g(0.0708mol)のリン酸二水素ナトリウムの溶液を、室温で30分にわたり添加した。その温和な発熱を水浴により制御した。一晩にわたり撹拌した後に、その反応はTLC(1:1:0.01のヘプタン:EtOAc:HOAc)により完了した。t−ブタノールを真空中で除去した。その残留物を200mlの水および300mlのヘプタン中に溶解させ、その後、80mlの1.0MのHClを添加した。その結果として生じたヘプタン層を、3×100mlの水、1×75mlの飽和塩化ナトリウム溶液で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濃縮し、3.79g(95.7%の収率)の8(S)−(t−ブチルジメチルシリルオキシ)ヘキサデカン酸(GC−MSで99.4%の純度)を(TMSCHN2からのメチルエステルとして)得た。
【0133】
TBS保護基の除去および8(S)−ヒドロキシパルミチン酸の精製
【化6】
3.79g(0.00980mol)の8(S)−(t−ブチルジメチルシリルオキシ)ヘキサデカン酸、48mlのACN、および2.13ml(0.059mol、6当量)の48%のフッ化水素酸を混合し、1時間にわたり撹拌した。20〜30分後に、その油性の混合物は懸濁液となる。1時間後に、その出発物質を完全に消費した(TLC(1:1:0.01のヘプタン:EtOAc:HOAc)により監視した通り)。完了の際に、300mlのMTBEおよび300mlの水を添加し、徹底的に混ぜた。その水層を除去し、その有機層を、3×100mlの水、1×100mlの飽和塩化ナトリウムで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濃縮し、2.50gの固体を得た。その固体はこの時点で95.6%eeであった(1380−51に留意されたい)。その固体を40mlの熱いACN中に溶解させ、撹拌しながら冷ました。その結果として生じた懸濁液を、<10℃に30分にわたり冷却し、濾過により収集し、20mlの冷たいACNで洗浄し、真空下にて40℃で乾燥させ、2.18g(82%の収率)の8(S)−ヒドロキシパルミチン酸が得られ、それは、GC−MSで>99%であり(TMSCHN2からのメチルエステルとして)、HPLC−MSで98.4%eeであった。
【0134】
8(R)エナンチオマーを与える光延反転
【化7】
8−ヒドロキシ酸の立体中心を光延条件下で反転させるためには、カルボン酸をそのメチルエステルとして保護する必要があった。最初は小規模に、ジアゾメタンを転換の迅速で効率的な方法として用いた。ジアゾメタンの危険な性質のために、より安全な代替物、TMSジアゾメタンをより大規模に用いた。MTBE/メタノール中の酸を完全にエステル化するために、2.6当量のTMSジアゾメタンを必要とした。立体中心を光延条件下で酢酸を用いて反転させる試みは、決して理想的でないと判明した。4−ニトロ安息香酸を置換する同じ条件(「有機合成、全集第9巻、607ページ(Organic Synthesis, Collective Volume 9, page 607)」)は、8(R)−4−ニトロ安息香酸塩を清浄に提供した。THF/水中のLiOHを用いた鹸化、続いてACNからの再結晶は、HPLC−MSで100%eeである所望の8(R)−ヒドロキシパルミチン酸を提供した。
【0135】
実施例7:Alamar Blue検定によるHeLa細胞の生存能の決定のための一般的な検定手順。Alamar Blue還元法による細胞生存能の評価は、候補薬物の細胞傷害能を例解するために日常的に用いられる。Alamar blueは、細胞代謝中間体により桃色の蛍光染料のレゾルフィンに転換される、青色の非蛍光染料のレサズリンを利用することにより、細胞生存能を検出する。本検定から発生した蛍光信号は、試料中の生細胞の数に比例する。T−75フラスコ内で成長させた集密的なHeLa細胞(70〜80%)を、トリプシン−EDTAを用いて剥離した。遠心分離に続いて、その細胞の沈渣をDMEM媒体中で再懸濁させ、血球計を用いて計数した。細胞を96穴板に10,000細胞/100μl/穴の密度で播種し、37℃/5%のCO2で8時間にわたり培養した。媒体を除去し、試験化合物を100μlの体積で添加した。その板を22時間、46時間、および70時間にわたり培養し、その後、穴1つ当たり10μlのalamar blueを添加した。試験化合物での24時間、48時間、および72時間の終わりに、マイクロプレートリーダーを用いて530nmの励起および590nmの発光で蛍光を測定した。対照(%)として表現した生存能を、薬物濃度に対して描図した。テルフェナジンを実験計画法における参照化合物として用い、これは、再現可能なIC50値を有する全ての時点(24時間、48時間、および72時間)において用量依存的な阻害を示した。異なる化合物は異なる生存能を示した。
【0136】
実施例8.実施例7の検定のデータ分析。2つの反復サンプルとしての穴の平均値を算出した。対照細胞の生存能を、全ての試験濃度について100%と考えた。試験試料の細胞生存能の百分率を、
【数1】
として算出した。S字状の用量応答(変数)等式(Graph Pad Prism 4のソフトウェア)を用いて、薬物濃度に対する百分率生存能データの非線形回帰分析(曲線適合)により、IC50値を決定した。BCP−6およびBCP−7は、細胞に対する格別の影響を実証した。
質的管理:本検定を以下の質的検査について評価した:
i.参照化合物のIC50値:テルフェナジンのIC50値
ii.反復サンプル間の変動係数(%):反復サンプル間のCV(%)は許容可能限界(10%)以内であった。
【0137】
実施例9.試験化合物試料の調製。前述した化合物の画分の調製を、表5に描出したものに従って行った。
【表5】
【0138】
実施例10:HepG2細胞に対するカスパーゼ−3活性の決定のための一般的な検定手順:
疑わしい、つまり、アポトーシスを経るように誘発されている細胞を最初に溶解させ、その細胞内内容物を収集する。我々の試験系では、蛍光レポーター分子の7−アミノ−4−メチルクマリン(AMC)に共役するカスパーゼ特異的ペプチドの添加により、スタウロスポリンで処理した細胞からの溶解物をプロテアーゼ活性について試験した。そのカスパーゼによるそのペプチドの開裂は、380nmの波長で励起される際に、460nmで蛍光を発する蛍光色素を放出する。その細胞溶解物中のカスパーゼ酵素活性の程度は、蛍光マイクロプレートリーダーで検出した蛍光信号に直接的に比例する。T−75フラスコ内で成長させた集密的なHepG2細胞(70〜80%)を、トリプシン−EDTAを用いて剥離した。遠心分離に続いて、その細胞の沈渣をDMEM媒体中で再懸濁させ、血球計を用いて計数した。細胞を96穴板に40,000細胞/100μl/穴の密度で播種し、37℃/5%のCO2で8時間にわたり培養した。媒体を除去し、試験化合物を100μlの体積で添加した。30分の上記のような前処理の後に、50μLの3×濃縮のスタウロスポリン(20μMの最終濃度)、または50μLの媒体(未処理の対照)を、それぞれの穴に添加した。スタウロスポリンで16時間にわたり誘発した後、50μlの溶解緩衝液の添加、続いて30分の培養により細胞を溶解した。顕微鏡下で観察されるような完全な溶解の後に、100μlの冷やした緩衝液(検定緩衝液)を各穴に添加した。次に、DEVD−AMC(基質、最終濃度は15μM)を添加し、その反応が2時間にわたり継続することを可能にした。BMG Polarstarの蛍光マイクロプレートリーダーを用いて、380nMで励起させ、460nMで発光を捕捉することにより、その蛍光を測定した。
【0139】
実施例11:実施例10の検定のデータ分析。各標準、ブランク、および試料について、反復サンプルの相対蛍光値(RFU)を平均する。対照(阻害剤なしで処理したスタウロスポリン)の酵素活性を、100%の活性(0%の阻害)と考えた。試験化合物/参照化合物の蛍光値(RFU)を、阻害(%)を算出するためにこれに対して比較する。試験化合物/参照化合物の百分率阻害を、下記のように算出した:
【数2】
S字状の用量応答(変数)等式(Graph Pad Prism 4のソフトウェア)を用いて、薬物濃度に対する百分率生存能データの非線形回帰分析(曲線適合)により、IC50値を決定した。BCP−6およびBCP−7は、カスパーゼ3阻害に対する格別の影響を実証した。
質的管理:本検定を以下の質的検査について評価した:
(i)参照化合物のIC50値:Ac−DEVD−CHOのIC50値
(ii)反復サンプル間の変動係数(%):反復サンプル間のCV(%)は許容可能限界(10%)以内であった。
【0140】
実施例12:ヒカゲノカズラ(Lycopodium
Clavatum)の抽出物が、アポトーシスと一致する形態学的特徴を誘発する
American Type Tissue Culture(ATCC)から得られたHeLa細胞(子宮頸癌)を、48時間にわたりヒカゲノカズラ(Lycopodium Clavatum)からの抽出物で処理し、抗チューブリン抗体で染色してその細胞骨格を検査し、DNAに強く結合する蛍光染色液である、4’,6−ジアミジノ−2−フェニルインドール(DAPI)で核を染色した。媒介物で処理した細胞は、無傷の細胞質構造および核を暴露した。その抽出物で処理した細胞がその細胞質構造(丸い細胞)を失うと同時に、核中の空所として現れるDNAの喪失により明らかであるように、細胞中の核DNAは完全に喪失した。こうした形態学的変化は、BCP−21が子宮頸癌細胞株内にアポトーシスを誘発することを示唆している。
【0141】
実施例13:ヒカゲノカズラ(Lycopodium
Clavatum)の抽出物が、sub−G1集団を誘発する
ヒカゲノカズラ(Lycopodium
Clavatum)の抽出物がアポトーシスを示唆する変化を誘発するため、流動細胞計測法を用いてDNA含量を測定することによりアポトーシスを評価した。そのアポトーシス細胞は断片化によりDNAを失い、それ故、2n未満のDNA含量を有することになるだろう。流動細胞計測法の分析の際には、その細胞はG1ピークの左に延びるように見えることになり、それ故、「sub−G1ピーク」と呼ばれる。対照および様々な希釈液(1:500)および1:250のヒカゲノカズラ(Lycopodium Clavatum)の抽出物で処理したHeLa細胞を、無水エタノール中に固定し、ヨウ化プロピジウムで30分にわたりRNAseと共に染色した。ハーバード医学校、ダナ・ファーバー癌研究所(Dana Farber Cancer Institute, Harvard Medical School)(マサチューセッツ州、ボストン)の中核施設にて、その細胞を分析した。フェニックス・フロー・システムズ(Phoenix Flow Systems)(カリフォルニア州、サンディエゴ)製のMultiCycleのソフトウェアを用い、細胞DNA含量の柱状図を逆畳み込みし、sub−G1相内の細胞の百分率の定量化を得ることになる。
【0142】
ヒカゲノカズラ(Lycopodium
Clavatum)の粗製抽出物は、用量依存的な様態でその対照(6.7%)に比べてsub−G1ピーク(23%)を増加させた(図6)。そのデータは、その抽出物が用量依存的な様態でHeLa細胞のアポトーシスを誘発することを強く示している。
【0143】
実施例14:ヒカゲノカズラ(Lycopodium
Clavatum)の抽出物が、PARP切断を誘発する
50mMのトリス−HCl、7.6のpH、150mMのNaCl、30mMのEDTA、0.5%のTriton X−100中で、完全プロテアーゼ阻害剤((ロシェ)Roche)を用いて細胞を溶解させた。タンパク質試料を、適切な百分率のゲル上にSDS−PAGEにより分離した。概して、20〜60μgのタンパク質を内因性タンパク質について、10〜15μgを形質移入タンパク質について分析した。タンパク質を0.2μmのニトロセルロース膜(Bio−Rad)に、1時間にわたり4℃で1時間にわたり移した。5%の乳および0.1%のTween−20(アメリカン・バイオアナリティカル(American Bioanalytical))を含有するPBS溶液中にて、膜を45分にわたり室温(RT)でブロッキングした。そのニトロセルロース膜を、その後、RTで2時間または4℃で一晩中の何れかにわたり、1%の乳および0.1%のTween−20を含有するPBS中にて、適切な濃度で希釈した一次抗体中で培養した。その一次抗体の除去の際に、洗浄緩衝液(1%の乳および0.1%のTween−20を有する1×PBS)中にて、その膜を10分にわたりRTで3回洗浄した。その膜を、その後、洗浄緩衝液中で希釈した適切な二次抗体中にて、1時間にわたりRTで培養した。膜をその後10分にわたり3回洗浄した。製造者の取扱説明書に従い、ECL(ピアース研究所(Pierce Laboratories))を用いてウエスタンブロットを発色させた。
【0144】
その結果は、全長PARP(分子量は116kDa)が24時間後に完全に切断されたことを示した。その抽出物で処理したHeLa細胞は、時間依存的な様態でPARP切断における増加を示している(図7)。このデータは、ヒカゲノカズラ類(Lycopodium)の抽出物がアポトーシスを誘発することを強く示している。
【0145】
実施例15:ヒト腫瘍細胞株に対する8−HHAのラセミ混合物の検査
DMC中に溶解させた8−HHAの合成ラセミ混合物を、60のヒト腫瘍細胞株のパネルに対して検査するために、NIH、国立癌研究所、癌診療部(the Division of Cancer Diagnosis and Treatment, National Cancer
Institute, NIH)が受け取った。その検査は、生存細胞中のテトラゾリウム染料の有色ホルマザン生成物への代謝還元に依存する、単純な比色(MTT)検定に基づくマイクロプレート細胞毒性検定である。その化合物の検査のために修正実験計画法を用い、これは様々な腫瘍細胞株の成長の阻害を測定する。この検定において試験した8−HHAは、10μMの濃度にあった。
【0146】
その結果は、8−HHAが、40〜96%の範囲で、白血病細胞株(例えば、CCRF−CEM、K−562、SR、MOLT−4、RPMI−8226、およびHL−60(TB))の成長を著しく抑制していることを示している。それは、非小細胞肺癌細胞株(例えば、HOP−92およびNCI−H460)上における成長の顕著な阻害も実証した。また、検査した7つの結腸直腸癌細胞株のうち3つ(すなわち、HCT−116、HCT−15、およびKM12)は、成長抑制の中等度の証拠を示した。このデータは、8−HHAが、その程度は様々ではあるが、白血病、肺、およびの結腸直腸の癌細胞株等の、様々な癌細胞株に対して成長阻害活性を有することを示している。
【0147】
実施例16:癌細胞株の生存に対する8−HHAの影響
1)肺癌細胞株に対して:8−HHAのラセミ混合物の影響、および、各個のエナンチオマー、つまりSエナンチオマーおよびRエナンチオマーのそれを、HOP−92肺癌細胞株の生存に対して検査した。その細胞生存能を、比色MTT検定を用いて、対数増殖期にて100μMの8−HHAで72時間にわたり検査した。別個の検定において、20〜100μMに及ぶ、異なる濃度の8−HHAのS−エナンチオマーで72時間にわたり、HOP−92細胞を処理した。
【0148】
その結果は、8−HHAのラセミ混合物が、HOP92細胞株の生存を25%低減したことを示している。そのR−エナンチオマーは、HOP92細胞株の生存を13%低減した。対照的に、そのS−エナンチオマーは、肺癌細胞株の生存に対して最大の影響を有した(72%低減した)。このデータは、8−HHAのSエナンチオマーが、HOP92肺癌細胞の生存に対して強力な阻害効果を有することを示している。さらに、図8は、増加する濃度の(S)−8−HHAが、用量依存的な様態で肺癌細胞株の生存を阻害したことを示している。細胞生存における50%の低減(IC50)が、8−HHAのS−エナンチオマーのおおよそ90μMの用量にて観察された。このデータは、8−HHAのSエナンチオマーが、肺癌細胞株に対して用量依存的な阻害効果を有することを示している。
【0149】
2)肝臓癌細胞株に対して:8−HHAのラセミ混合物の影響、および、各個のエナンチオマー、つまりSエナンチオマーおよびRエナンチオマーのそれを、PLC/PRF/5(「PLC−5」または「PLC5」とも呼ばれる)、Hep3B、およびSNU449(「SNU」とも呼ばれる)の肝臓癌細胞株の生存に対して検査した。その細胞生存能を、比色MTT検定を用いて、対数増殖期にて100μMの8−HHAで72時間にわたり検査した。別個の検定において、20〜100μMに及ぶ、異なる濃度の8−HHAのS−エナンチオマーで72時間にわたり、PLC−5細胞を処理した。
【0150】
その結果(図9)は、8−HHAのラセミ混合物が、PLC−5およびHep3Bの細胞株の生存をそれぞれ52〜25%低減したことを示している。それは、SNU細胞株に対して全く大きな影響を有しなかった。一方、8−HHAのS−エナンチオマーは、ラセミ混合物またはR−エナンチオマーの何れかと比較して、全ての肝臓癌細胞株の生存に対して最大の影響(ほとんど75%の細胞生存の低減)を有した。このデータは、8−HHAのSエナンチオマーが、肝臓癌細胞の生存の全てに対して強力な阻害効果を有することを示している。
【0151】
さらに、図10は、増加する濃度の(S)−8−HHAが、用量依存的な様態でPLC−5肝臓癌細胞株の生存を阻害したことを実証している。細胞生存における50%の低減(IC50)が、8−HHAのS−エナンチオマーのおおよそ50μMの用量にて観察された。このデータは、8−HHAのSエナンチオマーが、肺癌細胞株に対して用量依存的な阻害効果を有することを強く示している。
【0152】
実施例17:8−HHAは、肝臓および肺の癌細胞株内でアポトーシスを誘発する
PARP切断は、アポトーシスの特異的でかつ十分に確立した標識と見なされる。アポトーシスの最中に、FAS受容体は下方調節を受け、これはアポトーシスの確立した指標と見なされる。FAS配位子に結合するFAS受容体により活性化した経路によりアポトーシスを誘発することができ、外因性機構と呼ぶ。この経路によるアポトーシスは、FAS受容体の低減を結果的にもたらす。従って、FAS受容体のタンパク質濃度における低減は、アポトーシスが外因性機構により誘発されることを示している。
【0153】
1)肝臓癌細胞株に対して:8−HHAの影響を肝臓癌細胞株内でのアポトーシスに対して検査した。図11は、100μMの8−HHAのS−エナンチオマーの処理が、SNUおよびPLC−5の肝臓癌細胞株内におけるPARP切断を結果的にもたらしたことを示している。このデータは、8−HHAが肝臓癌細胞株内でアポトーシスを誘発することを示している。
【0154】
PLC−5細胞株を、100μMの8−HHAで72時間にわたり処理した。これは、対照と比較して、(S)−8−HHAの処理を用いて、FAS受容体濃度における相当な低減を結果的にもたらした(図12)。このデータは、(S)−8−HHAが、外因性機構により肝臓癌細胞株内でアポトーシスを誘発することを示唆している。
【0155】
2)肺癌細胞株に対して:HOP−92肺癌細胞株を、100μMの(S)−8−HHAで72時間にわたり処理した。その(S)−8−HHA処理は、HOP92細胞株内におけるFASタンパク質濃度の低減を結果的にもたらした。このデータは、8−HHAが、外因性機構を通じてHOP92細胞株内でアポトーシスを誘発することを示している。
【0156】
実施例18:肺癌細胞株内のプロアポトーシスBim1に対する8−HHA処理の影響。
Bim1は、その上方調節がアポトーシスを引き起こすプロアポトーシスタンパク質である。HOP92細胞株を、100μMの8−HHAのS−エナンチオマーで72時間にわたり処理した。その結果は、Bim1が、対照試料と比較して、処理した8−HHA中で2倍に増加したことを示している。このデータは、8−HHAが肺癌細胞株内でアポトーシスを誘発することを示している。
【0157】
実施例19:インビボの動物モデルに対する、8−HHAの最大耐用量(MTD)の決定:本研究の目的は、ICRマウスにおける静脈内(IV)投与に続く、8−HHAの最大耐用量を決定することであった。8−HHAのラセミ混合物、偽薬、および媒介物を、25〜30グラムの体重を有する20匹のICR雌マウスにそれぞれ投与した。8−HHAを、ナノ脂質中にて、0、10、20、および40mg/mlの濃度で調合した。一群当たり4匹の動物がおり、それらに8−HHAの0、10、20、および40mg/kgの用量を注射した。マウスに単一の急速静注用量を、尾静脈を通じて群割り当てに従って与えた。投薬の1週間後に、マウスを屠殺した。毎日の臨床観察を、投与後15分および1時間に行った。研究8日目での屍検まで、そのマウスを任意の有害作用について毎日観察した。試験物質投与より前に、かつ試験期間中を通して1日置きに、体重を全てのマウスについて記録した。この全体の研究を、トキシコン社(Toxikon Corporation)、マサチューセッツ州、ベッドフォードで行った。その結果を表6に描写する。
【表6】
【0158】
結果および考察:6日目および8日目の体重は、投薬前の体重から変化しなかった。6日目および8日目の体重は、投薬前の体重から変化しなかった。高用量(40mg/kg)を投薬した2匹のマウスは、投薬した直後に死んだ。偽薬、10mg/kg、および20mg/kgを投薬した動物は、8HHAに十分に耐性があり、いかなる留意すべき臨床的異常も実証しなかった。20mg/kgの用量を、その後8−HHAの最大耐用量と考えた。
【0159】
実施例20:肝臓異種移植マウスモデル:
本研究の目的は、PLC5細胞、肝細胞癌細胞株を用いて、肝臓癌における8−HHAの抗腫瘍効能を決定することであった。腫瘍成長および動物の生存を主要評価項目と考えた。
【0160】
動物および飼育:生後5〜6週間でかつ16〜20gmの体重がある、30匹のBABL/cの、非妊娠で、かつ未経産の雌ヌードマウスを本研究で用いた。薬物候補の抗腫瘍効能試験のための異種移植の研究において歴史的に用いられてきたために、BALB/c nu/nuのマウスを用いた。実際の試験用と同じ条件下で、マウスを最小で5日にわたり順化させた。30〜70%の室内相対湿度、時間当たり最小で10回の交換という時間当たりの空気交換、12時間の明暗周期の光曝露、全領域蛍光灯を用いて、マウスを68±5°Fの室温で収容した。ポリカーボネートでできている換気したマイクロアイソレーターケージ内に、マウスを集団で収容した。マウスは、高圧滅菌した実験室等級の寝藁を用いており、放射線照射したペレットおよび高圧滅菌した水を自由に提供された。飼料、水、または寝藁には、試験データに干渉すると予想される既知の汚染物は全くなかった。実験室および動物室を、出入り制限施設として維持した。
【0161】
注射および投薬の経路:8−HHAを、腹腔内(IP)注射を通じて投与した。8−HHAは不十分に水溶性であり、それ故、100mMに対応する27.2mg/mlの濃度でナノ脂質分散溶液(ePharse、スイス、バーゼル)中に懸濁する。MTD検定(予備データ)は、マウスが20mg/kgという最大用量に耐えたことを示した。本研究のために、2つの用量―10mg/kgおよび100mg/kgを用いた。
【0162】
動物の準備
腫瘍誘発:細胞株PLC5をヴァナス・オンコロジー(Vanas Oncology)での推奨仕様に従って培養し、そこで細胞を成長させ、10%のウシ胎仔血清、2mMのL−グルタミン、100単位/mlのストレプトマイシン、および100単位/mlのペニシリンを含有するRPMI媒体内に保持した。血球計を用いたトリパンブルー生存能試験を用いて、細胞をトリプシン処理および計数した。血球計の象限における細胞計数を細胞/mL値に換算し、これにより、マウス1匹につき適切な数の細胞の単離が可能になることになる。腫瘍細胞(5×106細胞/マウス)の懸濁液を含有する0.2mLの50%のRPMI/50%のMatrigel(商標)の混合物を、各マウスに右側腹部内に皮下接種した。十分に確定するまで、細胞を週に2回観察した。式:腫瘍重量(mg)=(a×b2/2)(式中、「b」はミリメートルでの最小直径であり、「a」は最大直径である)を用いて、腫瘍量を算出した。
【0163】
動物の割り当て:確定した腫瘍がおおよそ50〜100mgの平均算出量に到達した時点で、一群当たりの腫瘍の大きさの変動性を低減するために、マウスを適切なソフトウェアを用いて無作為に処理群とした。
【0164】
投薬前の手順:1日目における投薬より前に、順化させた動物を秤量し、毒性の臨床的兆候について観察した。
【0165】
用量の管理:2つの用量の1および10mg/kgを、MTD検定指針に基づいて投与した。第1の投薬日が1日目である。1日目には、8−HHA、および媒介物の注射を、下記の表7における研究設計に従って投与した。
【表7】
【0166】
投薬後の手順:処理に続いて、腫瘍およびマウスの体重の測定結果を週に2回記録し、総括的観察を毎日少なくとも1回行った。触知可能でない腫瘍を有するマウスを、完全退縮と考えた。マウス死亡率の百分率および死期を、本研究における全ての群について記録することになる。下記の判定基準のうち1つ以上を満たす場合に、動物を瀕死と定義し、屠殺してよい:
・1週間の期間内における20%以上の体重の減少。
・摂食、飲水、運動性、および排尿および/または排便する能力等の、正常な生理的機能を阻害するマウス。
・測径器で測定する際に2,000mgの最大寸法を超える腫瘍。
・潰瘍化した腫瘍、あるいは出血するか滲出液を生じる腫瘍。
・過度の体重減少(>20%)に繋がる長期で過度の下痢。
・持続性喘鳴および呼吸窮迫。
・衰弱、猫背姿勢、麻痺/不全麻痺、膨隆腹、潰瘍化、膿瘍、発作、および/または出血等の、臨床観察で定義する際の長期または過度の痛みまたは苦痛。
投薬の完了後に、マウスを追加の2週間にわたり観察し、腫瘍の再成長について検査した。
【0167】
屠殺:全ての動物を本研究の終了時に屠殺することになり、その腫瘍を採取することになる。その腫瘍の一部をOCT溶液中で凍結させることになり、その他部をホルマリン中で固定することになる。凍結および固定した組織を、出資者に送ることになる。全ての動物実験を、トキシコン社(Toxikon Corporation)(マサチューセッツ州、ベッドフォード)で行った。
【0168】
結果:8−HHA処理の1日目に、マウスを一群当たり10匹のマウスを有する3つの群に無作為に分けた:媒介物のみの対照群(C1)、10mg/kg体重の8−HHA(T1)、および100mg/kg体重の8−HHA(T2)。群T1およびT2は両方とも、8−HHA処理の4日目からマウスにおける腫瘍成長を著しく阻害した。対照的に、8−HHAで処理した群(T1)において、腫瘍量は、対照群と比較して、44.5%、49.8%、72.3%、77.7%、および78.4%(それぞれ、4日目、9日目、11日目、16日目、および18日目に)減少した。T2群において、腫瘍量は、56.3%、61.1%、81.1%、87.9%、および88.3%(それぞれ、4日目、9日目、11日目、16日目、および18日目に)減少した(図13および表8を参照されたい)。
【0169】
これらの結果は、本研究で用いた2つの濃度の8−HHAが、PLC−5細胞を用いる我々のインビトロ実験から明らかなように、抗増殖効果およびプロアポトーシス効果により、肝臓癌マウスモデルにおいて腫瘍成長を減少させることを示している。8−HHAは、そのため、標準的な化学療法計画の非常に効果的で、安全で、かつ安価な補助薬であることが判明し得る。
(*S:対照群と比較して有意、N=10/群)
平均±SEMの腫瘍量(PLC−5)
【表8】
【0170】
実施例21:小細胞肺癌異種移植マウスモデル:
本研究の目的は、HOP92を用いて、肺癌における8−HHAの抗腫瘍効能を決定することであった。
【0171】
動物および飼育:生後5〜6週間でかつ16〜20gmの体重がある、30匹のBABL/cの、非妊娠で、かつ未経産の雌ヌードマウスを本研究で用いた。薬物候補の抗腫瘍効能試験のための異種移植の研究において歴史的に用いられてきたために、BALB/c nu/nuのマウスを用いた。実際の試験用と同じ条件下で、マウスを最小で5日にわたり順化させた。30〜70%の室内相対湿度、時間当たり最小で10回の交換という時間当たりの空気交換、12時間の明暗周期の光曝露、全領域蛍光灯を用いて、マウスを68±5°Fの室温で収容した。ポリカーボネートでできている換気したマイクロマイクロアイソレーターケージ内に、マウスを集団で収容した。マウスは、高圧滅菌した実験室等級の寝藁を用いており、放射線照射したペレットおよび高圧滅菌した水を自由に提供された。飼料、水、または寝藁には、試験データに干渉すると予想される既知の汚染物は全くなかった。実験室および動物室を、出入り制限施設として維持した。
【0172】
注射および投薬の経路:8−HHAを、腹腔内(IP)注射を通じて投与した。8−HHAは不十分に水溶性であり、それ故、100mMに対応する27.2mg/mlの濃度でナノ脂質分散溶液(ePharse、スイス、バーゼル)中に懸濁する。MTD検定(予備データ)は、マウスが20mg/kgという最大用量に耐えたことを示した。
【0173】
実験設計:
動物の準備
腫瘍誘発:細胞株HOP92を、実施例20にて上記で検討したように、ヴァナス・オンコロジー(Vanas Oncology)での推奨仕様に従って培養した。血球計を用いたトリパンブルー生存能試験を用いて、細胞をトリプシン処理および計数した。血球計の象限における細胞計数を細胞/mL値に換算し、これにより、マウス1匹につき適切な数の細胞の単離が可能になることになる。腫瘍細胞(5×106細胞/マウス)の懸濁液を含有する0.2mLの50%のRPMI/50%のMatrigel(商標)の混合物を、各マウスに右側腹部内に皮下接種した。十分に確定するまで、細胞を週に2回観察した。式:腫瘍重量(mg)=(a×b2/2)(式中、「b」はミリメートルでの最小直径であり、「a」は最大直径である)を用いて、腫瘍量を算出した。
【0174】
動物の割り当て:確定した腫瘍がおおよそ50〜100mgの平均算出量に到達した時点で、一群当たりの腫瘍の大きさの変動性を低減するために、マウスを適切なソフトウェアを用いて無作為に処理群とした。
【0175】
投薬前の手順:1日目における投薬より前に、順化させた動物を秤量し、毒性の臨床的兆候について観察した。
【0176】
用量の管理:2つの用量の1および10mg/kgを投与した。第1の投薬日が1日目である。1日目には、8−HHA、および媒介物の注射を、下記の表9における研究設計に従って投与した。
【表9】
【0177】
投薬後の手順:処理に続いて、腫瘍およびマウスの体重の測定結果を週に2回記録し、総括的観察を毎日少なくとも1回行った。触知可能でない腫瘍を有するマウスを、完全退縮と考えた。マウス死亡率の百分率および死期を、本研究における全ての群について記録した。下記の判定基準のうち1つ以上を満たす場合に、動物を瀕死と定義し、屠殺してよい:
・1週間の期間内における20%以上の体重の減少。
・摂食、飲水、運動性、および排尿および/または排便する能力等の、正常な生理的機能を阻害するマウス。
・測径器で測定する際に2,000mgの最大寸法を超える腫瘍。
・潰瘍化した腫瘍、あるいは出血するか滲出液を生じる腫瘍。
・過度の体重減少(>20%)に繋がる長期で過度の下痢。
・持続性喘鳴および呼吸窮迫。
・衰弱、猫背姿勢、麻痺/不全麻痺、膨隆腹、潰瘍化、膿瘍、発作、および/または出血等の、臨床観察で定義する際の長期または過度の痛みまたは苦痛。
投薬の完了後に、マウスを追加の2週間にわたり観察し、腫瘍の再成長について検査した。
【0178】
屠殺:全ての動物を本研究の終了時に屠殺することになり、その腫瘍を採取することになる。その腫瘍の一部をOCT溶液中で凍結させることになり、その他部をホルマリン中で固定することになる。凍結および固定した組織を出資者に送った。
【0179】
結果:8−HHA処理の1日目に、マウスを一群当たり10匹のマウスを有する3つの群に無作為に分けた:媒介物のみの対照群(C1)、10mg/kg体重の8−HHA(T1)、および100mg/kg体重の8−HHA(T2)。群T1およびT2は両方とも、8−HHA処理の16日目からマウスにおける腫瘍成長を著しく阻害した。腫瘍量は、対照群において、16日目および18日目にそれぞれ214.6±38.65mm3および258.4±64.96mm3であった。対照的に、8−HHAで処理した群(T1)において、腫瘍量は、16日目および18日目にそれぞれ88.78±28.61mm3および82.02±25.93mm3であり、16日目および18日目での32.63mm3の腫瘍における58.6%および68.2%の減少にそれぞれ相当し、腫瘍量における56.5%および73.1%の減少に相当した(表10および図14を参照されたい)。
【表10】
*S:対照群と比較して有意
平均±SEMの腫瘍量(HOP−92)(N=10/群)
【0180】
結論として、本研究で用いた2つの濃度の8−HHAが、HOP92細胞を用いる我々のインビトロ実験から明らかなように、抗増殖効果およびプロアポトーシス効果により、肺癌マウスモデルにおいて腫瘍成長を減少させた。8−HHAは、そのため、標準的な化学療法計画の非常に効果的で、安全で、かつ安価な補助薬であることが判明し得る。
【0181】
本発明は特定の実施形態を参照して開示されてきたが、本発明の真の趣旨および範囲を逸脱することなく、他の当業者が本発明の他の実施形態および変化形を考案することができることは明白である。
【技術分野】
【0001】
本発明の技術分野
本発明は、植物抽出組成物、単離した活性薬剤、および癌の治療のための使用の方法に関する。
【背景技術】
【0002】
治療用途のための新しい抽出物および化合物の同定は、継続して生物医学的に重要であり、天然生成物がこの試みのために注目を集め続けている。増殖障害はヒトおよび動物の健康に問題を与え続け、このような障害に対する治療計画は、幅広い観点から未充足の必要性として残っている。増殖障害に関与する機構、信号伝達過程、および標的の変動のために、新しくかつより有効な組成物、化合物、および治療の方法の同定は、医学界ならびに一般大衆にとって大いに興味深いものである。
【0003】
オオカミノアシ(Wolf's
Foot)およびクラブモスヒカゲノカズラ科(Club Moss Lycopodiaceae)としても知られるヒカゲノカズラ(Lycopodium clavatum)は、ヒカゲノカズラ属(genus
Lycopodium)およびヒカゲノカズラ科(family Lycopodiaceae)に属する。その名前は、枝の先端が狼の鉤爪足に似ているために狼の足に由来する(lukosは狼を意味し、podosは足を意味する)。当該植物は、世界中の亜熱帯林および熱帯林内で遍在的に見つけられる。胞子体は、減数分裂の後に胞子嚢内で胞子を生産する。ヒカゲノカズラ類(Lycopodium)において、胞子体は、その中で各胞子嚢が葉状の胞子葉により保護される、円錐状の球果内に群生している。植物の胞子からの抽出物が、新規の治療活性を有する新しくかつ興味深い組成物の探索中に、分画されて単離した抽出物および化合物を産生した。
【発明の概要】
【0004】
本発明の概要
本発明は、本明細書における植物抽出物を用い、対象者における増殖障害を治療する方法に関する。本発明の1つの態様は、本明細書における植物抽出物からの単離した化合物を用い、対象者における増殖障害を治療する方法である。1つの実施形態では、増殖障害は癌である。特定の実施形態は、癌が、白血病、肺癌、肝臓癌、または結腸癌であると定めている。1つの実施形態では、癌は非小細胞肺癌である。別の実施形態では、癌は肝臓癌である。
【0005】
1つの実施形態では、当該化合物は、8−ヒドロキシ−パルミチン酸(「8−HHA」)、またはその塩、前駆薬、前駆薬塩、溶媒和物、水和物、および多形体である。別の実施形態では、本発明の方法に従って用いられる化合物は、8−ヒドロキシ−パルミチン酸のR−エナンチオマーおよびS−エナンチオマーのラセミ混合物である。1つの実施形態では、当該化合物は、8−ヒドロキシパルミチン酸のS−エナンチオマー(「(S)−8−HHA」)である。別の実施形態では、当該化合物は、8−ヒドロキシ−パルミチン酸のR−エナンチオマー(「(R)−8−HHA」)である。
【0006】
本発明の別の態様は、本明細書における植物抽出物(例えば、ヒカゲノカズラ(Lycopodium clavatum)の抽出物)を含む組成物である。別の態様は、本明細書における植物抽出物からの単離した化合物を含む組成物である。別の態様は、1つ以上の置換基を有する、本明細書における植物抽出物からの単離した化合物を含む組成物である。
【0007】
別の態様は、本明細書(または本明細書に描出されている化合物/組み合わせを用いる方法)に描出されている一つの化合物(または複数の化合物の組み合わせ)であり、そこで、当該化合物または化合物の組み合わせは、対象者(動物モデル、マウス、ラット、ウサギ、霊長類、ヒト)における抗癌活性を有することが実証されている。本明細書における例は、このような効果を決定するための代表的な方法にとって有益である。
【0008】
1つの実施形態では、本明細書に描出されている化合物(または複数の化合物の組み合わせ)は、植物からの抽出を含む手順から得られる。特定の実施形態では、化合物(または複数の化合物の組み合わせ)を得る際の使用のための手順は、植物抽出物の単離段階、蒸発段階、および分配段階の何れかをさらに含む。
【0009】
別の実施形態では、本明細書に描出されている化合物(または複数の化合物の組み合わせ)は、合成手段から得られる。
【0010】
本発明の別の態様は、本明細書における植物抽出物(例えば、BCP−21抽出物、表1または2内のBCP−21抽出物画分の何れか)、または、本明細書における植物抽出物(例えば、BCP−21抽出物、表1または2内のBCP−21抽出物画分の何れか)中に生じる化合物(例えば、8−ヒドロキシヘキサデカン酸、本明細書におけるBCP化合物1〜10の何れか)を含む医薬組成物である。1つの実施形態では、本医薬組成物は、8−HHAのラセミ混合物、またはその塩、前駆薬、前駆薬塩、溶媒和物、水和物、および多形体を含む。1つの実施形態では、本医薬組成物は、相当なエナンチオマー的純度で8−ヒドロキシ−パルミチン酸のS−エナンチオマー(「(S)−8−HHA」)を含む。別の実施形態では、本医薬組成物は、相当なエナンチオマー的純度で8−ヒドロキシ−パルミチン酸のR−エナンチオマー(「(R)−8−HHA」)を含む。
【0011】
別の態様では、このような組成物をキットに含めることができる。
【0012】
上記の組成物(および本明細書に描出されている治療の方法)を、適正な化学治療薬剤および生物治療薬剤とさらに組み合わせることができる。このような薬剤の例としては、アルデスロイキン、アレムツズマブ、アリトレチノイン、アルトレタミン、アミノレブリン酸、アナグレリド、アナストロゾール、三酸化ヒ素、アスパラギナーゼ、カルメット・ゲラン桿菌(BCG)、ベタメタゾン、ベキサロテン、ビカルタミド、ブレオマイシン、ブスルファン、カペシタビン、カルボプラチン、カルムスチン、クロラムブシル、リン酸クロムP−32、シスプラチン、クラドリビン、結合型エストロゲン類、コルチゾン、シクロホスファミド、シタラビンリポソーム、シタラビン、ara−、ダカルバジン、ダクチノマイシン、アクチノマイシンD、ダウノルビシン、クエン酸ダウノルビシンリポソーム、デニロイキンジフチトクス、デキサメタゾン、ジクロフェナク、ドセタキセル、ドキソルビシン、ドキソルビシンリポソーム、エピルビシン、エステル型エストロゲン類、エストラジオール、吉草酸エストラジオール、エストラムスチン、エストロン、エチニルエストラジオール、エトポシド、リン酸エトポシド、エキセメスタン、フロクスウリジン、リン酸フルダラビン、フルオロウラシル、フルオロウラシル、フルオロウラシル、フルオキシメステロン、フルタミド、ゲムシタビン、ゲムツズマブオゾガマイシン、酢酸ゴセレリン、グラニセトロン、ヒドロコルチゾン、ヒドロキシプロゲステロン、ヒドロキシ尿素、イブリツモマブチウキセタン、イダルビシン、イホスファミド、メシル酸イマチニブ、インターフェロンアルファ−2a、インターフェロンアルファ−2b、イリノテカン、レトロゾール、酢酸ロイプロリド、レボチロキシン、ロムスチン、メクロレタミン、メドロキシプロゲステロン、メドロキシプロゲステロン、酢酸メゲストロール、メルファラン、メルカプトプリン、メトトレキサートナトリウム、メトキサレン、メチルプレドニゾロン、メチルテストステロン、メチロシン、マイトマイシン、マイトマイシンC、ミトタン、ミトキサントロン、フェンプロピオン酸ナンドロロン、ニルタミド、酢酸オクトレオチド、オプレルベキン、オキシメトロン、ペガスパルガーゼ、ペントスタチン、プリカマイシン、ポリフェプロザン20/カルムスチン、ポルフィマーナトリウム、プレドニゾロン、プレドニゾン、プロカルバジン、プロゲステロン、リツキシマブ、サマリウム−153レキシドロナムペンタナトリウム、ヨウ化ナトリウムI−131、リン酸ナトリウムP−32、ストレプトゾシン、塩化ストロンチウム−89、滑石、クエン酸タモキシフェン、テモゾロミド、テニポシド、テストラクトン、エナント酸テストステロン、チオグアニン、チオテパ、トポテカン、クエン酸トレミフェン、トラスツズマブ、トレチノイン、トリアムシノロン、パモ酸トリプトレリン、バルルビシン、ビンブラスチン、ビンクリスチン、およびビノレルビンが挙げられるが、これらに限定されない。
【0013】
さらに別の態様では、本発明は、本明細書に明記されている対象者における疾患、障害、または症状の治療または防止のための、単一の組成物または別個の剤形の何れかとしての薬物の製造における、単独での、または1つ以上の追加の治療薬剤と合わせての、本明細書における化学式の何れかの化合物の使用を提供する。本発明の別の態様は、本明細書に描出されている対象者における疾患、障害、またはその症状の治療または防止における使用のための、(単離した天然または合成の)本明細書における化学式の化合物である。
【図面の簡単な説明】
【0014】
【図1】BCP−21抽出物から単離された化合物のNMRスペクトルである。
【図2】BCP−21抽出物から単離された化合物のNMRスペクトルである。
【図3】BCP−21抽出物から単離された化合物のNMRスペクトルである。
【図4】BCP−21抽出物から単離された化合物のMSスペクトルである。
【図5】BCP−21抽出物から単離された化合物のMSスペクトルである。
【図6】BCP−21が、用量依存的な様態でHeLa細胞のアポトーシスを誘発することを示す。
【図7】ヒカゲノカズラ(lycopodiumclavatum)の抽出物が、時間依存的な様態でPARP切断を誘発することを示す、ウエスタンブロッティングの結果である。
【図8】(S)−8−HHAが、用量依存的な様態でHOP−92の肺癌細胞株の生存を阻害したことを示す。
【図9】100μMの8−HHA(ラセミ混合物、S−エナンチオマーまたはR−エナンチオマー)が、比色MTT検定を用いて、PCL5、Hep3B、およびSNUの肝臓癌細胞株の生存を減少させることを結果的にもたらしたことを示す。
【図10】(S)−8−HHAが、用量依存的な様態でPLC−5の肝臓癌細胞株の生存を阻害したことを実証する。
【図11】100μMの(S)−8−HHAが、SNUおよびPCL5の肝臓癌細胞株内にPARP切断を結果的にもたらしたことを示す。
【図12】(S)−8−HHAの処理が、対照と比較してFAS受容体濃度の相当な減少を結果的にもたらしたことを実証する。
【図13】8−HHAが、肝臓異種移植マウスモデルにおけるPLC5の肝臓癌細胞内にて腫瘍成長を減少させることを描写する。
【図14】異種移植マウスモデルにおけるHOP92の肺癌細胞内での8−HHAの抗腫瘍効能を描写する。
【発明を実施するための形態】
【0015】
本発明の詳細な説明
定義
「寛解させる」および「治療する」という用語は、相互交換可能に用いられ、疾患(例えば、本明細書に描出されている疾患または障害)の発生または進行を減少させ、抑制し、減弱し、軽減し、阻止し、または安定させることを両方とも意味する。
【0016】
「疾患」とは、細胞、組織、または器官の正常な機能を損傷するかまたは妨げる、任意の容態または障害を意味する。
【0017】
「標識」とは、疾患または障害と関連する任意の変化を意味する。例えば、疾患または障害と関連する発現量または活性における変化を有する、任意のタンパク質またはポリヌクレオチドである。
【0018】
「癌」という用語は、一群の細胞が、非制御成長(正常限界を超える分裂)、侵襲(隣接する組織への侵入およびこれの破壊)、および時には転移(体内の他の場所への蔓延)を見せる一種の疾患を概して指す。癌の例としては、白血病、肺癌、肝臓癌、結腸癌、黒色腫、乳癌、CNS癌、卵巣癌、腎臓癌、および前立腺癌が挙げられるが、これらに限定されない。
【0019】
本開示において、「備える」、「包含する」、「含有する」、および「有する」および同様のものは、米国特許法でそれらに帰される意味を有することがあり、かつ「含める」、「含む」、および同様のものを意味することがあり、「から実質的に成る」または「実質的に成る」は、米国特許法で帰される意味を同様に有し、当該用語は変更可能であり、列挙されるものの基本的な特徴または新規の特徴が、列挙されるが先行技術の実施形態を除くものより多くの存在により変更されない限り、列挙されるものより多くの存在を考慮に入れる。
【0020】
本明細書における化合物を、本明細書における化学式の化合物の塩、前駆薬、および前駆薬塩、溶媒和物、水和物、および多形体を含む化合物の形態で利用することもできる。
【0021】
本発明の化合物の塩は、酸とアミノ官能基などの化合物の塩基性基との間に形成されるか、あるいは、塩基とカルボキシル官能基などの化合物の酸性基との間に形成される。別の好適な実施形態によれば、化合物は医薬的に許容可能な酸付加塩である。
【0022】
本明細書で用いられる際および他に示されない限り、「前駆薬」という用語は、生物学的条件下(インビトロまたはインビボ)で加水分解し、酸化し、またはそれ以外の反応を起こし、本発明の化合物を与えることができる化合物の誘導体を意味する。前駆薬は、生物学的条件下でこのような反応において活性になるのみであり得、あるいは、その未反応の形態で活性を有し得る。本明細書で意図されている前駆薬の例としては、アミド類、エステル類、カルバミン酸塩類、炭酸塩類、およびリン酸塩類似体等の生物加水分解可能な部分を含む、本明細書で開示されている化学式の何れか一つの化合物の類似体または誘導体が挙げられるが、これらに限定されない。「バーガーの医薬品化学および創薬(1995年)172〜178ページ、949〜982ページ(マンフレッド・E.ウォルフ編、第5版)(Burger's Medicinal Chemistry and Drug Discovery (1995) 172-178, 949-982
(Manfred E. Wolff ed., 5th ed))」により記載されている方法等の、十分に知られているものを用いて、前駆薬を典型的に調製することができ、「グッドマンおよびギルマンの治療学の薬理学的基礎、第8版、マグローヒル国際版、1992年、『薬物の生体変換』(Goodman and Gilman's, The Pharmacological basis of Therapeutics, 8th
ed., McGraw-Hill, Int. Ed. 1992, "Biotransformation of Drugs")」も参照されたい。
【0023】
本明細書で用いられる際および他に示されない限り、「生物加水分解可能な部分」という用語は、1)化合物の生物学的活性を破壊せず、かつ、取り込み、作用の持続時間、または作用の発現等の、インビボでの有利な特性を化合物に付与するか、あるいは、2)それ自体は生物学的に不活性であるが、インビボで生物学的に活性な化合物に転換されるか、の何れかである官能基(例えば、アミド、エステル、カルバミン酸塩、炭酸塩、またはリン酸塩)類似体を意味する。
【0024】
前駆薬塩は、酸とアミノ官能基などの前駆薬の塩基性基との間に形成されるか、あるいは、塩基とカルボキシル官能基などの前駆薬の酸性基との間に形成される化合物である。1つの実施形態では、前駆薬塩は医薬的に許容可能な塩である。
【0025】
特に好ましい前駆薬および前駆薬塩は、本発明の化合物が(例えば、経口的に投与される化合物を血液中により容易に吸収することを可能にすることにより)哺乳類に投与される際に、このような化合物の生物利用可能性を増加させるか、あるいは、親種と比較して親化合物の生物学的区画(例えば、脳または中枢神経系)への運搬を強化するものである。好適な前駆薬としては、水溶性または腸膜を通じる能動輸送を強化する基が、本明細書に記載されている化学式の構造に付加される誘導体が挙げられる。例えば、「アレクサンダー・J.他、医薬品化学誌、1988年、第31号、318〜322ページ(Alexander, J. et al. Journal of Medicinal Chemistry 1988, 31,
318-322)」、「バンガード・H.、前駆薬の設計、エルゼビア:アムステルダム、1985年、1〜92ページ(Bundgaard,
H. Design of Prodrugs; Elsevier: Amsterdam, 1985; pp 1-92)」、「バンガード・H.、ニールセン・N.M.、医薬品化学誌、1987年、第30号、451〜454ページ(Bundgaard, H.; Nielsen, N. M. Journal of Medicinal Chemistry 1987,
30, 451-454)」、「バンガード・H.、薬の設計および開発の教科書、ハーウッド・アカデミック出版:スイス、1991年、113〜191ページ(Bundgaard, H. A Textbook of Drug Design and Development; Harwood
Academic Publ.: Switzerland, 1991; pp 113-191)」、「ディゲニス・G.A.他、実験薬理学の手引書、1975年、第28号、86〜112ページ(Digenis, G. A. et al. Handbook of Experimental Pharmacology 1975,
28, 86-112)」、「フリース・G.J.、バンガード・H.、薬の設計および開発の教科書、第2版、海外出版:アムステルダム、1996年、351〜385ページ(Friis, G. J.; Bundgaard, H. A Textbook of Drug Design and
Development; 2 ed.; Overseas Publ.: Amsterdam, 1996; pp 351-385)」、「ピットマン・I.H.、医薬品研究総説、1981年、第1号、189〜214ページ(Pitman, I. H. Medicinal Research Reviews 1981, 1, 189-214)」を参照されたい。
【0026】
「医薬的に許容可能な」という用語は、本明細書で用いられる際に、健全医学的判断の範囲内において、過度の毒性、刺激、アレルギー反応、および同様のものを伴わない、ヒトおよび他の哺乳類の組織と接触する使用に適切であり、かつ、妥当な恩恵/危険比に相応である成分を指す。「医薬的に許容可能な塩」は、受容者への投与の際に、直接的にも間接的にも、本発明の化合物または化合物の前駆薬を与えることができる、任意の非毒性塩を意味する。
【0027】
医薬的に許容可能な塩を形成するために一般に使用される酸としては、重硫化水素、塩化水素酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸等の無機酸、ならびに、パラ−トルエンスルホン酸、サリチル酸、酒石酸、二酒石酸、アスコルビン酸、マレイン酸、ベシル酸、フマル酸、グルコン酸、グルクロン酸、蟻酸、グルタミン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、乳酸、蓚酸、パラ−ブロモフェニルスルホン酸、炭酸、コハク酸、クエン酸、安息香酸、および酢酸等の有機酸、および関連する無機酸と有機酸が挙げられる。このような医薬的に許容可能な塩としては、そのため、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、リン酸一水素塩、リン酸二水素塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、蟻酸塩、イソ酪酸塩、カプリン酸塩、ヘプタン酸塩、プロピオール酸塩、蓚酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオール酸塩、ヘキシン−1,6−ジオール酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、テレフタル酸塩、スルホン酸塩、キシレンスルホン酸、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、β−ヒドロキシ酪酸塩、グリコール酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スルホン酸塩、マンデル酸塩、および同様の塩が挙げられる。好適な医薬的に許容可能な酸付加塩としては、塩化水素酸および臭化水素酸等の鉱酸と形成されるもの、および、特にマレイン酸等の有機酸と形成されるものが挙げられる。
【0028】
医薬的に許容可能な塩を本発明の前駆薬の酸性官能基と形成するための好適な塩基としては、ナトリウム、カリウム、およびリチウム等のアルカリ金属の水酸化物、カルシウムおよびマグネシウム等のアルカリ土類金属の水酸化物、アルミニウムおよび亜鉛等の他の金属の水酸化物、アンモニア、および非置換またはヒドロキシ置換のモノアルキルアミン、ジアルキルアミン、およびトリアルキルアミン等の有機アミン類、ジシクロヘキシルアミン、トリブチルアミン、ピリジン、N−メチル−N−エチルアミン、ジエチルアミン、トリエチルアミン、モノ−、ビス−、またはトリス−(2−ヒドロキシエチル)アミン、2−ヒドロキシ−tert−ブチルアミン、またはトリス−(2−ヒドロキシエチル)アミン等のモノ−、ビス−、またはトリス−(2−ヒドロキシ−低級アルキルアミン)、N,N−ジメチル−N−(2−ヒドロキシエチル)アミン、またはトリ−(2−ヒドロキシエチル)アミン等のN,N−ジ−低級アルキル−N−(ヒドロキシ低級アルキル)−アミン類、N−メチル−D−グルカミン、ならびにアルギニン、リジン、および同様のもの等のアミノ酸類が挙げられるが、これらに限定されない。
【0029】
本明細書で用いられる際に、「水和物」という用語は、非共有分子間力により結合した、化学量論量または非化学量論量の水をさらに含む化合物を意味する。
【0030】
「単離した」、「精製した」、または「生物学的に純粋な」という用語は、その天然状態で見つかる際に通常はそれを伴う成分が、実質的にまたは本質的に非含有である物質を指す。純度または均質性は、ポリアクリルアミドゲル電気泳動法または高性能液体クロマトグラフィー等の分析化学技術を用いて典型的に決定される。特に、実施形態では、化合物は少なくとも純度85%、より好ましくは少なくとも純度90%、より好ましくは少なくとも純度95%、最も好ましくは少なくとも純度99%である。
【0031】
本明細書で用いられる際に、「溶媒和物」という用語は、非共有分子間力により結合した、化学量論量または非化学量論量の、水、アセトン、エタノール、メタノール、ジクロロメタン、2−プロパノール、または同様のもの等の溶媒をさらに含む化合物を意味する。
【0032】
本明細書で用いられる際に、「多形体」という用語は、例えば、X線粉末回折図形または赤外分光法等の物理的手段により特徴付けることができる、化合物またはその錯体の固体結晶形態を意味する。同じ化合物の異なる多形体は、異なる物理的特性、化学的特性、および/または分光学的特性を呈することがある。異なる物理的特性としては、安定性(例えば、熱、光、または水分に対して)、圧縮性および密度(調合および製品製造において重要)、吸湿性、可溶性、および溶解率(生物利用可能性に影響し得る)が挙げられるが、これらに限定されない。安定性における差異は、化学反応性(例えば、剤形が、1つの多形体から成る際に、別の多形体から成る際よりも急速に変色するような、酸化の違い)、または力学的特徴(例えば、動力学的に有利な多形体が熱力学的により安定した多形体に転換する時に、錠剤が貯蔵の際に砕ける)、またはその両方(例えば、1つの多形体の錠剤が、高い湿度で分解の影響をより受けやすい)における変化に起因し得る。多形体の異なる物理的特性は、加工に影響し得る。例えば、1つの多形体は、より溶媒和物を形成する可能性があり得るだろうし、あるいは、例えば、それの粒子の形状または大きさの分布に起因して、別のものよりも不純物を完全に濾過または洗浄することがより困難であり得るだろう。
【0033】
本明細書で用いられる際の「他の立体異性体が実質的に非含有の」という用語は、25%未満の他の立体異性体、好ましくは10%未満の他の立体異性体、より好ましくは5%未満の他の立体異性体、最も好ましくは2%未満の他の立体異性体、あるいは「X」%未満の他の立体異性体(式中、Xは0および100の間の全体を含む数である)が存在することを意味する。ジアステレオマーを得るかまたは合成する方法は、当該技術分野で十分に知られており、最終化合物に対して、あるいは出発物質または中間体に対して実行可能なものとして適用されてよい。他の実施形態は、化合物が単離した化合物であるものである。本明細書で用いられる際の「少なくともX%がエナンチオマー的に豊富な」という用語は、化合物の少なくともX%が単一のエナンチオマー形態であることを意味し、式中、Xは0および100の間の全体を含む数である。
【0034】
「安定化合物」という用語は、本明細書で用いられる際に、製造を可能にするのに十分な安定性を有し、かつ、本明細書に詳述されている目的(例えば、治療薬剤に応答する疾患または障害を治療する、治療製品、治療化合物の製造の際の使用のための中間体、単離可能または貯蔵可能な中間化合物への調合)のために有用であるのに十分な期間にわたり化合物の統合性を維持する化合物を指す。
【0035】
「立体異性体」は、エナンチオマーおよびジアステレオマーを両方とも指す。
【0036】
本明細書で用いられる際に、「ハロ」または「ハロゲン」という用語は、フッ素、塩素、臭素、またはヨウ素の任意のラジカルを指す。
【0037】
「アルク」または「アルキル」という用語は、1乃至12個の炭素原子、好ましくは1乃至8個の炭素原子を有する、直鎖または分枝鎖の炭化水素基を指す。「低級アルキル」という表現は、1乃至4(全体を含む)個の炭素原子のアルキル基を指す。「アリールアルキル」という用語は、その中でアルキルの水素原子がアリール基により置き換えられる部分を指す。「アルケニル」という用語は、2乃至10個の、好ましくは2乃至4個の炭素原子の、少なくとも1つの二重結合を有する直鎖または分枝鎖の炭化水素基を指す。アルケニル基が窒素原子に結合される場合には、このような基が二重結合を有する炭素を通じて直接的に結合されないことが好適である。
【0038】
「アルコキシ」という用語は、−O−アルキルラジカルを指す。「アルキレンジオキソ」という用語は、−O−R−O−という構造の二価種を指し、その中で、Rはアルキレンを表す。
【0039】
「アルキニル」という用語は、2乃至10個の、好ましくは2乃至4個の炭素原子の、少なくとも1つの三重結合を有する直鎖または分枝鎖の炭化水素基を指す。アルキニル基が窒素原子に結合される場合には、このような基が三重結合を有する炭素を通じて直接的に結合されないことが好適である。
【0040】
「アルキレン」という用語は、1乃至3個の低級アルキル基で置換することができる、単結合により接続される1乃至5個の炭素原子の二価の直鎖橋(例えば、−(CH2)x−、式中、xは1乃至5である)を指す。
【0041】
「アルケニレン」という用語は、単結合により接続され、かつ1乃至3個の低級アルキル基で置換することができる、1つまたは2つの二重結合を有する2乃至5個の炭素原子の直鎖橋を指す。例示的なアルケニレン基は、−CH=CH−CH=CH−、−CH2−CH=CH−、−CH2−CH=CH−CH2−、−C(CH3)2CH=CH−、および−CH(C2H5)−CH=CH−である。
【0042】
「アルキニレン」という用語は、その中に一つの三重結合を有し、単結合により接続され、かつ1乃至3個の低級アルキル基で置換することができる、2乃至5個の炭素原子の直鎖橋を指す。例示的なアルキニレン基は、−C≡C−、−CH2−C≡C−、−CH(CH3)−C≡C−、および−C≡C−CH(C2H5)CH2−である。
【0043】
本明細書で使用される際の「シクロアルキル」および「シクロアルケニル」という用語は、それぞれ、飽和および部分不飽和の、3乃至12個の炭素、好ましくは3乃至8個の炭素、より好ましくは3乃至6個の炭素を有する環式炭化水素基を含む。「Ar」または「アリール」という用語は、6乃至14個の炭素原子を含有する芳香族環式基(例えば、6員環の単環式環系、10員環の二環式環系、または14員環の三環式環系)を指す。例示的なアリール基としては、フェニル、ナフチル、ビフェニル、およびアントラセンが挙げられる。
【0044】
「ヘテロアリール」は、N、O、またはSから選択される1個、2個、3個、または4個の環複素原子を含有する、5乃至12個の環原子の単環式または縮合環(すなわち、隣接する一対の原子を共有する環)の基を指し、残りの環原子はCであり、かつ、加えて、完全に共役したパイ電子系を有し、そこで、各環の0個、1個、2個、3個、または4個の原子を置換基により置換することができる。ヘテロアリール基の例は、制限なく、ピロール、フラン、チオフェン、イミダゾール、オキサゾール、チアゾール、ピラゾール、ピリジン、ピリミジン、キノリン、キナゾリン、イソキノリン、プリン、およびカルバゾールである。
【0045】
「複素環」、「複素環式」、または「ヘテロシクロ」という用語は、完全飽和または部分不飽和の環式基、例えば、3乃至7員環の単環式、7乃至12員環の二環式、または10乃至15員環の三環式の環系を指し、これらは少なくとも1個の複素原子を少なくとも1個の環内に有し、そこで、各環の0個、1個、2個、または3個の原子を置換基により置換することができる。複素原子を含有する複素環式基の各環は、窒素原子、酸素原子、および/または硫黄原子から選択される1個、2個、3個、または4個の複素原子を有し得、そこで、窒素および硫黄の複素原子を随意に酸化させることができ、かつ、窒素複素原子を随意に四級化することができる。複素環式基は、環または環系の任意の複素原子または炭素原子にて結合させることができる。
【0046】
「置換基」という用語は、本明細書に描出されている任意の官能基上、例えば、アルキル基、アルケニル基、アルキニル基、シクロアルキル基、シクロアルケニル基、アリール基、ヘテロシクリル基、またはヘテロアリール基上にて、その基の任意の原子において「置換された」基を指す。態様では、本明細書に描出されている官能基、例えば、アルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アリール、ヘテロシクリル、またはヘテロアリールを、一つの置換基(例えば、下記に列挙されているもの)で置換することができる。適切な置換基としては、制限なく、ハロゲン、CN、NO2、OR15、SR15、S(O)2R15、NR15R16、C1〜C2のペルフルオロアルキル、C1〜C2のペルフルオロアルコキシ、1,2−メチレンジオキシ、C(O)OR15、C(O)NR15R16、OC(O)NR15R16、NR15C(O)NR15R16、C(NR16)NR15R16、NR15C(NR16)NR15R16、S(O)2NR15R16、R17、C(O)R17、NR15C(O)R17、S(O)R17、S(O)2R17、R16、オキソ、C(O)R16、C(O)(CH2)nOH、(CH2)nOR15、(CH2)nC(O)NR15R16、NR15S(O)2R17が挙げられ、式中、nは独立して0〜6の全体を含むものである。各R15は、独立して、水素、C1〜C4のアルキル、またはC3〜C6のシクロアルキルである。各R16は、独立して、水素、アルケニル、アルキニル、C3〜C6のシクロアルキル、アリール、ヘテロシクリル、ヘテロアリール、C1〜C4のアルキル、あるいは、C3〜C6のシクロアルキル、アリール、ヘテロシクリル、またはヘテロアリールで置換されたC1〜C4のアルキルである。各R17は、独立して、C3〜C6のシクロアルキル、アリール、ヘテロシクリル、ヘテロアリール、C1〜C4のアルキル、あるいは、C3〜C6のシクロアルキル、アリール、ヘテロシクリル、またはヘテロアリールで置換されたC1〜C4のアルキルである。各R15、R16、およびR17中の各C3〜C6のシクロアルキル、アリール、ヘテロシクリル、ヘテロアリール、およびC1〜C4のアルキルを、ハロゲン、CN、C1〜C4のアルキル、OH、C1〜C4のアルコキシ、NH2、C1〜C4のアルキルアミノ、C1〜C4のジアルキルアミノ、C1〜C2のペルフルオロアルキル、C1〜C2のペルフルオロアルコキシ、または1,2−メチレンジオキシで随意に置換することができる。
【0047】
「オキソ」という用語は、炭素に結合される際にはカルボニル、窒素に結合される際にはN−オキシド、および硫黄に結合される際にはスルホキシドまたはスルホンを形成する、酸素原子を指す。
【0048】
「アシル」という用語は、アルキルカルボニル、シクロアルキルカルボニル、アリールカルボニル、ヘテロシクリルカルボニル、またはヘテロアリールカルボニルの置換基を指し、これらの何れかを置換基によりさらに置換することができる。
【0049】
一可変物の任意の定義における化学基の一覧の詳述は、本明細書において、任意の単一の基または列挙されている基の組み合わせとしてのその可変物の定義を含む。一可変物についての一実施形態の詳述は、本明細書において、任意の単一の実施形態としての、あるいは、任意の他の実施形態またはその部分と組み合わせるその実施形態を含む。
【0050】
本発明の化合物は、1つ以上の不斉中心を含有することがあり、そのため、ラセミ体およびラセミ混合物、単一のエナンチオマー、各個のジアステレオマーおよびジアステレオマー混合物として生じることがある。これらの化合物の全てのこのような異性形態は、本発明に明確に含まれる。本発明の化合物を多数の互変異性形態にて表すこともでき、このような場合において、本発明は、本明細書に記載されている化合物の全ての互変異性形態を明確に含む。このような化合物の全てのこのような異性形態は、本発明に明確に含まれる。本明細書に記載されている化合物の全ての結晶形態は、本発明に明確に含まれる。
【0051】
本明細書における化学式の化合物を合成するための他の手法を、本明細書で引用されている参考文献から容易に適合させることができる。これらの手順の変化形およびそれらの最適化は、通常の実務者の技能の範囲内にある。
【0052】
上記で示されている特定の手法および化合物は、限定的であることを目的としていない。本明細書における図式内の化学構造は、同じ可変物名(例えば、R1、R2、R、R’、Xなど)により識別されるか否かにかかわらず、本明細書における化合物の化学式内の対応する位置の化学基の定義(部分、原子など)に相応に本明細書により定義される可変物を描写する。ある化合物構造内の化学基の、別の化合物構造の合成の際の使用のための適合性は、当該技術分野における通常の技能を有する者の知識の範囲内にある。本明細書における図式内に明白に示されていない経路内にあるものを含む、本明細書における化合物およびその合成前駆体を合成する追加の方法は、当該技術分野における通常の技能を有する化学者の技量の範囲内にある。反応条件を最適化し、必要であれば競合副生成物を最小化するための方法は、当該技術分野で知られている。本明細書に記載されている方法は、本明細書に具体的に記載されている段階の前でも後でも、本明細書における化合物の合成を最終的に可能にするために、適切な保護基を追加または除去する段階を追加的に含むこともある。加えて、様々な合成段階を交互の順序または順番で行い、所望の化合物を与えることができる。適用可能な化合物を合成する際に有用な、合成化学の変換および保護基の方法論(保護および脱保護)は、当該技術分野で知られており、例えば、「R.ラロック、包括的有機変換、VCH出版(1989年)(R. Larock, Comprehensive Organic Transformations, VCH Publishers
(1989))」、「T.W.グリーンおよびP.G.M.ウッツ、有機合成における保護基、第3版、ジョン・ワイリー・アンド・サンズ(1999年)(T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis,
3rd Ed., John Wiley and Sons (1999))」、「L.フィーザーおよびM.フィーザー、フィーザーおよびフィーザーの試薬または有機合成、ジョン・ワイリー・アンド・サンズ(1994年)(L. Fieser and M. Fieser, Fieser and Fieser's Reagents or Organic
Synthesis, John Wiley and Sons (1994))」、および、「L.パケット編、有機合成のための試薬の百科事典、ジョン・ワイリー・アンド・サンズ(1995年)(L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis,
John Wiley and Sons (1995))」、およびそれらの後続版に記載されているものが挙げられる。
【0053】
本明細書に記載されている合成法は、任意の図式に記載されている段階の何れかの前でも後でも、本明細書に記載されている化学式の化合物の合成を最終的に可能にするために、適切な保護基を追加または除去する段階を追加的に含むこともある。本明細書に描出されている方法は、1つの化学式の化合物を別の化学式の化合物に転換することを意図している。転換する工程は、その場で行うことができるか、あるいは中間化合物の単離を伴う、1つ以上の化学変換を指す。当該変換は、本明細書で引用されている参考文献中にあるものを含む、当該技術分野で知られている技術および実験計画法を用いて、出発化合物または中間体を追加の試薬と反応させることを含み得る。精製(例えば、濾過、蒸留、昇華、結晶化、研和、固相抽出、およびクロマトグラフィー)を伴うか否かにかかわらず、中間体を用いることができる。
【0054】
本発明により想定される置換基および可変物の組み合わせは、安定化合物の形成を結果的にもたらすもののみである。
【0055】
本発明は、有効量の本明細書における化合物、または、妥当な場合、前記化合物の医薬的に許容可能な塩、溶媒和物、水和物、多形体、もしくは前駆薬、および許容可能な担体を含む組成物も提供する。好ましくは、本発明の組成物は医薬用途のために調合され(医薬組成物)、そこで、担体は医薬的に許容可能な担体である。1つ(または複数)の担体は、調合物の他の成分と相溶性があるという意味で「許容可能」でなければならず、かつ、医薬的に許容可能な担体の場合には、薬物中に典型的に用いられる量でその受容者に有害であってはならない。
【0056】
本発明の医薬組成物において用いることができる、医薬的に許容可能な担体、補助剤、および媒介物としては、イオン交換体、アルミナ、ステアリン酸アルミニウム、レシチン、ヒト血清アルブミン等の血清タンパク質、リン酸塩類等の緩衝物質、グリシン、ソルビン酸、ソルビン酸カリウム、飽和植物脂肪酸の部分グリセリド混合物、水、硫酸プロタミン、リン酸水素ジナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩類、コロイド状シリカ、三ケイ酸マグネシウム、ポリビニルピロリドン等の塩または電解質、セルロース系物質、ポリエチレングリコール、カルボキシメチルセルロースナトリウム、ポリアクリル酸塩類、蝋、ポリエチレン−ポリオキシプロピレン−ブロック重合体、ポリエチレングリコール、および羊毛脂が挙げられるが、これらに限定されない。
【0057】
本発明の医薬組成物としては、経口、直腸、経鼻、局所(頬側および舌下を含む)、膣または非経口(皮下、筋肉内、静脈内、および皮内を含む)の投与に適したものが挙げられる。特定の実施形態では、本明細書における化学式の化合物は、(例えば、経皮貼布を用いて)経皮的に投与される。他の調合物を、単位剤形、例えば、錠剤および持続放出型カプセルで、およびリポソームで便利に提供することができ、かつ、薬学の技術分野において十分に知られている任意の方法により調製することができる。例えば、「レミントンの薬科学、マック出版社、ペンシルヴァニア州、フィラデルフィア(第17版、1985年)(Remington's Pharmaceutical Sciences, Mack Publishing Company,
Philadelphia, PA (17th ed. 1985))」を参照されたい。
【0058】
このような調製法は、1つ以上の付属成分を構成する担体等の成分を、投与されることになる分子との関連に至らせる段階を含む。一般的に、本組成物は、活性成分を、液状担体、リポソームつまり微細化した固体担体、あるいはその両方との関連に均一にかつ密接に至らせ、その後必要であれば生成物を成形することにより調製される。
【0059】
特定の好適な実施形態では、化合物は経口的に投与される。経口投与に適した本発明の組成物を、所定量の活性成分をそれぞれ含有するカプセル、小袋、または錠剤等の不連続単位として、粉末または顆粒として、水性液体または非水液体中の溶液または懸濁液として、または水中油型液状乳濁液もしくは油中水型液状乳濁液として提供することができ、あるいは、リポソーム中におよび急速静注薬などとして包装することができる。軟質のゼラチンカプセルは、化合物の吸収の速度を有益に増加させることができるような懸濁液を含有するのに有用であり得る。
【0060】
随意に1つ以上の付属成分を用いて、圧縮または成形により錠剤を作ることができる。適切な機械内で、粉末または顆粒等の自由流動形態にある活性成分を、結合剤、潤滑剤、不活性希釈剤、保存剤、表面活性剤、または分散剤と随意に混合されて圧縮することにより、圧縮錠剤を調製することができる。適切な機械内で、不活性液状希釈剤で湿らせた粉末化合物の混合物を成形することにより、成形錠剤を作ることができる。当該錠剤を随意に被覆するかまたは刻み付けることができ、かつ、その中の活性成分の徐放つまり制御放出を提供するように調合することができる。本明細書におけるものおよび当該技術分野で知られている他の化合物等の、医薬的に活性な成分のこのような徐放または制御放出の組成物を調合する方法は、当該技術分野で知られかついくつかの発行された米国特許に記載されており、その一部としては、米国特許第4,369,172号、および第4,842,866号、および当該特許で引用されている参考文献が挙げられるが、これらに限定されない。化合物の腸への運搬のために、被覆剤を用いることができる(例えば、米国特許第6,638,534号、第5,217,720号、および第6,569,457号、第6,461,631号、第6,528,080号、第6,800,663号、および当該特許で引用されている参考文献を参照されたい)。本発明の化合物のための有用な調合物は、腸溶層が酢酸コハク酸ヒドロキシプロピルメチルセルロースを含む形態の腸溶性丸薬である。
【0061】
経口用途のための錠剤の場合において、一般に用いられる担体としては、乳糖およびトウモロコシデンプンが挙げられる。ステアリン酸マグネシウム等の潤滑剤も、典型的に添加される。カプセル形態における経口投与のために、有用な希釈剤としては、乳糖および乾燥トウモロコシデンプンが挙げられる。水性懸濁液が経口的に投与される際に、活性成分は乳化剤および懸濁剤と混合される。所望であれば、特定の甘味剤および/または香味剤および/または着色剤を添加してよい。
【0062】
局所投与に適した組成物としては、通常は蔗糖およびアカシアまたはトラガカントである香味基礎中に成分を含む飴剤、ならびに、ゼラチンおよびグリセリン、または蔗糖およびアカシア等の不活性基礎中に活性成分を含む香錠が挙げられる。
【0063】
非経口投与に適した組成物としては、調合物を対象とする受容者の血液と等張にする抗酸化剤、緩衝剤、静菌剤、および溶質を含有することがある、水性および非水の無菌注入溶液、ならびに、懸濁剤および増粘剤を含むことがある、水性および非水の無菌懸濁液が挙げられる。調合物を、単位用量または多重用量の容器、例えば、封止したアンプルおよび薬瓶内で提供することができ、ならびに、使用の直前に、無菌液状担体、例えば注入用の水の添加のみを必要とする凍結乾燥(氷結乾燥)条件において貯蔵することができる。即時の注入溶液および懸濁液を、無菌の粉末、顆粒、および錠剤から調製することができる。
【0064】
このような注入溶液は、例えば、無菌注入可能な水性または油性の懸濁液の形態にあってよい。当該技術分野で知られている技術により、適切な分散剤または湿潤剤(例えば、Tween 80等)および懸濁剤を用いて、この懸濁液を調合することができる。無菌注入可能な調製物は、非毒性で非経口的に許容可能な希釈剤または溶媒中の無菌注入可能な溶液または懸濁液、例えば、1,3−ブタンジオール中の溶液としてであってもよい。使用することができる許容可能な媒介物および溶媒の中には、マンニトール、水、リンガー溶液、および等張塩化ナトリウム溶液がある。加えて、無菌の固定油は、溶媒または懸濁媒体として従来的に使用される。この目的のために、合成のモノグリセリドまたはジグリセリドを含む、任意の無刺激性の固定油を使用することができる。オレイン酸およびそのグリセリド誘導体等の脂肪酸は、注入剤の調製において有用であり、特にそのポリオキシエチル化型にある、オリーブ油またはヒマシ油等の、天然の医薬的に許容可能な油も同様である。こうした油の溶液または懸濁液は、長鎖アルコールの希釈剤または分散剤を含有することもある。
【0065】
直腸投与のための坐剤の形態で、本発明の医薬組成物を投与することができる。室温で固体であるが直腸温度で液体であり、そのため、直腸内で溶けて活性成分を放出することになる、適切な非刺激性の賦形剤と、本発明の化合物を混合することにより、こうした組成物を調製することができる。このような物質としては、ココアバター、蜜蝋、およびポリエチレングリコール類が挙げられるが、これらに限定されない。
【0066】
経鼻の噴霧または吸入により、本発明の医薬組成物を投与することができる。このような組成物は、医薬調合の技術分野で十分に知られている技術により調製され、かつ、生理食塩水中の溶液として調製し、ベンジルアルコールまたは他の適切な保存剤、生物利用可能性を高める吸収促進剤、フッ化炭素類、および/または、当該技術分野で知られている他の可溶化剤または分散剤を使用することができる。
【0067】
本発明の医薬組成物の局所投与は、所望の治療が局所適用により容易に到達可能な領域または器官に影響を及ぼす際に特に有用である。皮膚への局所的な適用のためには、担体中に懸濁または溶存した活性成分を含有する適切な軟膏を用いて、本医薬組成物を調合するべきである。本発明の化合物の局所投与のための担体としては、鉱油、液状石油、白色石油、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化蝋、および水が挙げられるが、これらに限定されない。代替的には、担体中に懸濁または溶存した活性成分を含有する適切なローションまたはクリームを用いて、本医薬組成物を調合することができる。適切な担体としては、鉱油、モノステアリン酸ソルビタン、ポリソルベート60、セチルエステル類、蝋、セテアリルアルコール、2−オクチルドデカノール、ベンジルアルコール、および水が挙げられるが、これらに限定されない。本発明の医薬組成物を、直腸坐剤調合物により、または、適切な浣腸調合物中で、下部腸管に局所的に適用することもできる。局所経皮貼布およびイオン導入投与も、本発明に含まれる。
【0068】
特に好ましい誘導体および前駆薬は、本発明の化合物が(例えば、経口的に投与される化合物を血液中により容易に吸収することを可能にすることにより)哺乳類に投与される際に、このような化合物の生物利用可能性を増加させるか、あるいは、親種と比較して親化合物の生物学的区画(例えば、脳または中枢神経系)への運搬を強化するものである。好適な前駆薬としては、水溶性または腸膜を通じる能動輸送を強化する基が、本明細書に記載されている化学式の構造に付加される誘導体が挙げられる。例えば、「アレクサンダー・J.他、医薬品化学誌、1988年、第31号、318〜322ページ(Alexander, J. et al. Journal of Medicinal Chemistry 1988, 31,
318-322)」、「バンガード・H.、前駆薬の設計、エルゼビア:アムステルダム、1985年、1〜92ページ(Bundgaard,
H. Design of Prodrugs; Elsevier: Amsterdam, 1985; pp 1-92)」、「バンガード・H.、ニールセン・N.M.、医薬品化学誌、1987年、第30号、451〜454ページ(Bundgaard, H.; Nielsen, N. M. Journal of Medicinal Chemistry 1987,
30, 451-454)」、「バンガード・H.、薬の設計および開発の教科書、ハーウッド・アカデミック出版:スイス、1991年、113〜191ページ(Bundgaard, H. A Textbook of Drug Design and Development; Harwood
Academic Publ.: Switzerland, 1991; pp 113-191)」、「ディゲニス・G.A.他、実験薬理学の手引書、1975年、第28号、86〜112ページ(Digenis, G. A. et al. Handbook of Experimental Pharmacology 1975,
28, 86-112)」、「フリース・G.J.、バンガード・H.、薬の設計および開発の教科書、第2版、海外出版:アムステルダム、1996年、351〜385ページ(Friis, G. J.; Bundgaard, H. A Textbook of Drug Design and
Development; 2 ed.; Overseas Publ.: Amsterdam, 1996; pp 351-385)」、「ピットマン・I.H.、医薬品研究総説、1981年、第1号、189〜214ページ(Pitman, I. H. Medicinal Research Reviews 1981, 1, 189-214)」を参照されたい。
【0069】
本治療法の適用は、関心部位に投与が行なわれるように局所的であり得る。関心部位に本組成物を提供するために、注入、カテーテル、套管針、発射体、プルロンゲル、ステント、持続薬物放出重合体、または内部到達を提供する他の機器の使用等の、様々な技術を用いることができる。
【0070】
別の実施形態によれば、本発明は、移植可能な薬物放出機器を本発明の化合物または組成物と接触させる段階を含む、前記薬物放出機器を含浸させる方法を提供する。移植可能な薬物放出機器としては、生分解性重合体のカプセルまたは弾丸、非生分解性で拡散性の重合体のカプセル、および生分解性重合体の薄板が挙げられるが、これらに限定されない。
【0071】
別の実施形態によれば、本発明は、化合物、または本発明の化合物を含む組成物で、前記化合物が治療的に活性であるように被覆された移植可能な医療機器を提供する。
【0072】
別の実施形態では、本発明の組成物は第2の治療薬剤をさらに含む。第2の治療薬剤は、単独で、または、本明細書における化学式の何れかの化合物と投与される際に、有利な特性を有すると知られているかまたは実証する任意の化合物または治療薬剤を含む。こうした化合物と有用に組み合わせることができるだろう薬物としては、他のキナーゼ阻害剤、および/または、上記で検討されている疾患および障害の治療のための他の化学治療薬剤が挙げられる。
【0073】
このような薬剤は、当該技術分野で詳細に説明されている。好ましくは、第2の治療薬剤は、癌から選択される疾患または容態の治療または防止において有用な薬剤である。
【0074】
さらにより好ましくは、本発明の化合物と共調合された第2の治療薬剤は、癌、免疫障害、心血管疾患、ウイルス感染、炎症、代謝/内分泌の障害、および神経学的障害等の、キナーゼ媒介の疾患/障害の治療において有用な薬剤である。
【0075】
別の実施形態では、本発明は、互いに関連する、本発明の化合物および第2の治療薬剤の別個の剤形を提供する。本明細書で用いられる際の「互いに関連する」という用語は、別個の剤形が共に包装され、またはそうでなければ、別個の剤形が(連続的または同時に、互いに24時間未満以内に)一緒に販売、かつ投与されることを目的とすることが容易に明白であるように互いに付着されることを意味する。
【0076】
本発明の医薬組成物において、本発明の化合物は有効量で存在する。本明細書で用いられる際に、「有効量」という用語は、適切な投与計画内で投与される際に、治療される障害の重症度、持続時間、または進行を低減するかまたは寛解させるのに十分であり、治療される障害の前進を防止し、治療される障害の後退を引き起こし、あるいは、別の治療の1つ(または複数)の予防効果または治療効果を強化または改善する量を指す。
【0077】
動物およびヒトに対する用量の相互関係(体表面の1平方メートル当たりのミリグラム数に基づく)は、「フライライヒ他、(1966年)癌化学療法報告書第50号:219ページ(Freireich et al., (1966) Cancer Chemother Rep 50: 219)」に記載されている。患者の身長および体重から、体表面積をおおよそ決定することができる。例えば、「科学表、ガイギー製薬、ニューヨーク州、アードリー、1970年、537ページ(Scientific Tables, Geigy Pharmaceuticals, Ardley, N. Y., 1970, 537)」を参照されたい。本発明の化合物の有効量は、約0.001mg/kgから約500mg/kgまで、より好ましくは0.01mg/kg乃至約50mg/kg、より好ましくは0.1mg/kg乃至約2.5mg/kgの範囲であり得る。有効用量も、当業者により認識されるように、治療される疾患、当該疾患の重症度、投与の経路、患者の性別、年齢、および一般的健康状態、賦形剤の利用、他の薬剤の使用等の他の治療処置との共利用の可能性、および治療する医師の判断に応じて変化することになる。
【0078】
第2の治療薬剤を含む医薬組成物について、当該第2の治療薬剤の有効量は、その薬剤のみを用いる単独治療計画で通常利用される用量の約20%および100%の間である。好ましくは、有効量は通常の単独治療用量の約70%および100%の間である。こうした第2の治療薬剤の通常の単独治療用量は、当該技術分野で十分に知られている。例えば、「ウェルズ他編、薬物治療の手引書、第2版、アップルトン・アンド・ラング、コネティカット州、スタムフォード(2000年)(Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton
and Lange, Stamford, Conn. (2000))」、「PDR薬局方、タラスコン小型薬局方2000、豪華版、タラスコン出版、カリフォルニア州、ロマ・リンダ(2000年)(PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe
Edition, Tarascon Publishing, Loma Linda, Calif. (2000))」を参照されたく、その参考文献のそれぞれは、本明細書に参照により全体的に組み込まれている。
【0079】
上記で参照されている第2の治療薬剤のいくつかは、本発明の化合物と相乗的に作用することになることが予想される。これが起こる際に、それにより、第2の治療薬剤および/または本発明の化合物の有効用量を、単独治療に必要とされるものから減らすことが可能になることになる。これは、第2の治療薬剤または本発明の化合物の何れか毒性の副作用の最小化、有効性における相乗的改善、投与または使用の改善した容易さ、および/または化合物の調製または調合の低減した全体の費用、という利点を有する。
【0080】
治療の方法
1つの態様によれば、本発明は、対象者に有効量の化合物(例えば、単離した化合物、本明細書における化合物)または本発明の組成物を投与する段階を含む、疾患または障害またはその症状(例えば、本明細書で描出されているもの)を患っているか、またはこれらの影響を受けやすい前記対象者を治療する方法を提供する。このような疾患は当該技術分野で十分に知られており、かつ本明細書でも開示されている。
【0081】
本方法は、組成物が、ヒカゲノカズラ(Lycopodium clavatum)の抽出物(例えば、BCP−21抽出物、表1または2内のBCP−21抽出物画分の何れか)、または、本明細書における植物抽出物(例えば、BCP−21抽出物、表1または2内のBCP−21抽出物画分の何れか)中に生じる単離した化合物(例えば、8−ヒドロキシヘキサデカン酸(8−HHA)、本明細書におけるBCP化合物1〜10の何れか)であるものをさらに含み得る。
【0082】
本発明の1つの実施形態では、単離した化合物は、8−ヒドロキシヘキサデカン酸(「8−HHA」)、またはその塩、前駆薬、前駆薬塩、溶媒和物、水和物、および多形体である。特定の実施形態は、当該化合物が、8−ヒドロキシ−パルミチン酸のR−エナンチオマーおよびS−エナンチオマーのラセミ混合物であることを規定している。1つの実施形態では、当該化合物は、8−ヒドロキシ−パルミチン酸のS−エナンチオマー(「(S)−8−HHA」)である。別の実施形態では、当該化合物は、8−ヒドロキシ−パルミチン酸のR−エナンチオマー(「(R)−8−HHA」)である。
【0083】
別の実施形態では、本発明の方法において用いられる化合物(または組成物)は、合成手段を通じて得られる。
【0084】
本発明の1つの態様では、当該疾患または障害は、カスパーゼ媒介の疾患または障害である。
【0085】
本発明の1つの態様では、当該疾患または障害は、カスパーゼ−3媒介の細胞死により媒介される。
【0086】
本発明の1つの態様では、当該疾患または障害は、カスパーゼ−3媒介の疾患または障害である。
【0087】
本発明の1つの態様では、当該疾患または障害は、細胞死を誘発することにより治療される。
【0088】
本発明の1つの態様では、当該疾患または障害は、カスパーゼ−3により媒介される細胞死を誘発することにより治療される。
【0089】
本発明の1つの態様では、当該疾患または障害をカスパーゼ−3により調節することができる。
【0090】
1つの態様では、治療する方法は、増殖障害である障害またはその症状の治療を伴う。これらは、癌、腫瘍、腫瘍剤が適切である任意の疾患を含む。
【0091】
本化合物、組成物、および治療の方法を用いて治療することができる癌の例としては、癌(例えば、白血病、肝臓癌、肺癌、結腸癌、CNS癌、黒色腫、腎臓癌、およびカスパーゼ−3媒介癌)、アレルギー性障害、および炎症性障害が挙げられる。増殖障害、例えば、癌を患っているヒトまたは動物の患者を、そのため、上記で定義されている通りの本発明の化合物の当該患者への投与を含む方法により治療することができる。患者の容態を、それにより、改善するかまたは寛解させることができる。本発明の方法により治療可能な疾患および容態としては、癌および炎症性障害が挙げられるが、これらに限定されない。本発明の方法により治療することができる癌としては、肝臓癌、肝細胞性、白血病、肺癌、結腸癌、CNS癌、黒色腫、腎臓癌などが挙げられるが、これらに限定されない。
【0092】
1つの実施形態は、本発明の方法により治療可能な癌が、白血病、肺癌、肝臓癌、または結腸癌であることを規定している。1つの実施形態では、当該癌は肝臓癌である。別の実施形態は、当該癌が肺癌であることを規定している。1つの実施形態では、当該肺癌は非小細胞肺癌である。特定の実例は、本発明の方法により治療可能な癌が、HOP−92媒介の疾患(例えば、非小細胞肺癌)に関することを規定している。
【0093】
さらに別の実施形態では、本発明の方法により治療することができる癌は、白血病である。
【0094】
本明細書に描出されている方法としては、対象者が特定の所定の治療を必要としていると確認されるものが挙げられる。このような治療を必要としている対象者を確認することは、対象者または健康管理専門家の判断のうちにあり得、かつ、主観的(例えば、意見)または客観的(例えば、試験または診断法により測定可能)であり得る。
【0095】
1つの実施形態では、本発明は、本明細書における化学式の何れかの1つ以上の化合物と細胞を接触させることを含む、細胞内のカスパーゼ酵素(例えば、カスパーゼ−3)活性を調節する方法を提供する。
【0096】
別の実施形態では、上記の治療の方法は、前記患者に1つ以上の第2の治療薬剤を共投与するさらなる段階を含む。第2の治療薬剤の選択を、本明細書における適応症に有用であると知られている任意の第2の治療薬剤から行うことができる。
【0097】
本明細書で用いられる際の「共投与される」という用語は、単一の剤形(上記に記載されているような本発明の化合物および第2の治療薬剤を含む、本発明の組成物等)の一部として、あるいは、別々の複数の剤形として、第2の治療薬剤を本発明の化合物と共に投与することができることを意味する。代替的には、本発明の化合物の投与より前に、これと連続して、またはこれの後に、追加の薬剤を投与することができる。このような組み合わせの治療処置では、本発明の化合物および1つ(または複数)の第2の治療薬剤の両方が、従来の方法により投与される。本発明の化合物および第2の治療薬剤を含む、本発明の組成物の対象者への投与は、治療の経過中の別の時間における、その同じ治療薬剤、任意の他の第2の治療薬剤、または本発明の任意の化合物の前記対象者への別個の投与を除外しない。
【0098】
こうした第2の治療薬剤の有効量は当業者に十分に知られており、本明細書で参照されている特許および公開特許出願、ならびに、「ウェルズ他編、薬物治療の手引書、第2版、アップルトン・アンド・ラング、コネティカット州、スタムフォード(2000年)(Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton
and Lange, Stamford, Conn. (2000))」、「PDR薬局方、タラスコン小型薬局方2000、豪華版、タラスコン出版、カリフォルニア州、ロマ・リンダ(2000年)(PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition,
Tarascon Publishing, Loma Linda, Calif. (2000))」、および他の医学書で投薬の指針を見つけることができる。しかしながら、第2の治療薬剤の最適な有効量の範囲を決定することは、十分に当業者の能力が及ぶ範囲内にある。
【0099】
第2の治療薬剤が対象者に投与される本発明の1つの実施形態では、本発明の化合物の有効量は、その有効量が当該第2の治療薬剤が投与されない場合のものより少ない。別の実施形態では、当該第2の治療薬剤の有効量は、その有効量が本発明の化合物が投与されない場合のものより少ない。このようにして、両方の薬剤の高い用量と関連する非所望の副作用を最小化することができる。他の潜在的な利点(制限なく、改善した投薬計画および/または低減した薬物費用を含む)は、当業者にとって明白であることになる。
【0100】
さらに別の態様では、本発明は、上記で明記されている対象者における疾患、障害、または症状の治療または防止のための、単一の組成物または別個の剤形の何れかとしての薬物の製造における、単独での、または、上記に記載されている第2の治療薬剤のうち1つ以上と合わせての、本明細書における化学式の何れかの化合物の使用を提供する。本発明の別の態様は、本明細書に描出されている対象者における疾患、障害、またはその症状の治療または防止における使用のための、本明細書における化学式の化合物である。
【0101】
他の態様では、本明細書における方法としては、治療投与に対する対象者の応答を監視することをさらに含むものが挙げられる。このような監視は、治療計画の標識または指標としての、対象者の組織、体液、検体、細胞、タンパク質、化学標識、遺伝物質などの周期的な試料抽出を含み得る。他の方法では、当該対象者は、このような治療に対する適切性の関連する標識または指標に対する評価により、前検査されるかまたはこのような治療を必要としていると確認される。
【0102】
1つの実施形態では、本発明は治療経過を監視する方法を提供する。当該方法は、本明細書に描出されている障害またはその症状を患っているか、またはこれらの影響を受けやすい患者における、診断標識(標識)(例えば、本明細書における化合物により調節される、本明細書に描出されている任意の標的または細胞型)の濃度の決定、または診断測定(例えば、検査、検定)の段階を含み、ここで、この対象者は、疾患またはその症状を治療するのに十分な治療量の本明細書における化合物を投与されている。当該方法において決定される標識の濃度を、健常な通常の対照または他の罹患した患者の何れかにおける標識の既知の濃度と比較し、対象者の疾患状態を確証することができる。好適な実施形態では、対象者における標識の第2の濃度は、第1の濃度の決定より遅い時点で決定され、2つの濃度は、疾患の経過または治療の有効性を監視するために比較される。特定の好適な実施形態では、対象者における標識の前治療濃度は、本発明による治療を始めるより前に決定され、この標識の前治療濃度を、その後、治療を開始した後の対象者における標識の濃度と比較し、治療の有効性を決定することができる。
【0103】
特定の方法の実施形態では、対象者における標識の濃度または標識活性は、少なくとも1回決定される。標識濃度の、例えば、同じ患者、別の患者、または通常の患者から前にまたは後に得られる標識濃度の別の測定結果との比較は、本発明による治療が所望の効果を有しているかどうかを決定する際に有用であり、それにより、投薬濃度の調節を必要に応じて可能にすることができる。当該技術分野で知られているか、または本明細書に記載されている、任意の適切な試料抽出法/発現検定法を用いて、標識濃度の決定を行うことができる。好ましくは、組織または体液の試料は対象者から最初に取り出される。適切な試料の例としては、血液、尿、組織、口または頬の細胞、および毛根を含有する毛髪試料が挙げられる。他の適切な試料が、当業者に公知であろう。当該試料中のタンパク質濃度および/またはmRNA濃度(例えば、標識濃度)の決定を、酵素免疫測定法、ELISA、放射標識技術/検定技術、吸い取り法/化学発光法、実時間PCR、および同様のものを含むが、これらに限定されない、当該技術分野で知られている任意の適切な技術を用いて行うことができる。
【0104】
本発明の化合物の使用
本発明は、本明細書に描出されているものを含む、疾患、障害、またはその症状を治療する使用のためのキットも提供する。こうしたキットは、a)本明細書における化学式の何れかの1つ以上の化合物もしくはその塩、またはその前駆薬もしくはその前駆薬の塩、またはその水和物、溶媒和物、もしくは多形体を含む医薬組成物(例えば、本明細書における組成物、本明細書における任意の特定の化合物)であって、前記医薬組成物が容器内にある、医薬組成物と、b)当該医薬組成物を用いて、本明細書に描出されているものを含む、疾患、障害、またはその症状を治療する方法を説明している取扱説明書と、を備える。放射線治療の実施と逐次的に、または同時に、当該化合物/組成物を投与することができる。
【0105】
当該容器は、前記医薬組成物を保持することができる、任意の器あるいは他の封止された装置または封止可能な装置であってよい。例としては、瓶、各区分または室が単一用量の前記組成物を含む分割型または多室型の保持瓶、各区分が単一用量の前記組成物を含む分割型箔包、または、単一用量の前記組成物を分注する分注器が挙げられる。当該容器は、医薬的に許容可能な物質で作られる、当該技術分野で知られているような任意の従来の形状または形態にあり、例えば、紙または段ボール箱、ガラスまたはプラスチックの瓶または広口瓶、再封止可能な袋(例えば、異なる容器内への配置のために錠剤の「補充分」を保持する)、または、治療予定により包装から押し出すための各個の用量を有するブリスターパックであってよい。使用される容器は関連する的確な剤形に依存し得、例えば、従来の段ボール箱は一般的に液状懸濁液を保持するために用いられることはないだろう。単一の剤形を市販するために、1つより多くの容器を単一の包装内において共に用いることができることが可能である。例えば、錠剤を瓶内に包含することができ、これは同様に箱の中に包含される。好ましくは、当該容器はブリスターパックである。
【0106】
当該キットは、医師、薬剤師、または対象者のための情報および/または取扱説明書を追加的に備え得る。このような記憶補助としては、そのように規定される錠剤またはカプセルが対象者に摂取または投与されるべきである投与計画の日数と一致する投薬量を包含する各室または区分上に印刷される数、または各室または区分上に印刷される週の日数、または同じ種類の情報を含有するカードが挙げられる。1つの態様では、当該取扱説明書はさらに、対象者への放射線管理に関する。
【0107】
さらに別の態様では、本発明は、本明細書に明記されている対象者における疾患、障害、または症状の治療または防止のための、単一の組成物または別個の剤形の何れかとしての薬物の製造における、単独での、または、1つ以上の追加の治療薬剤と合わせての、本発明の一つの化合物(または複数の化合物の組み合わせ)の使用を提供する。本発明の別の態様は、本明細書に描出されている対象者における疾患、障害、またはその症状の治療または防止における使用のための、本発明の一つの化合物(または複数の化合物の組み合わせ)である。
【0108】
本明細書で引用されている全ての参考文献は、印刷媒体、電子媒体、コンピュータ可読記憶媒体、または他の形態にあるかどうかにかかわらず、参照により全体として明確に組み込まれ、要約書、記事、機関誌、刊行物、教科書、論文、技術資料集、インターネットウェブサイト、データベース、特許、特許出願、および特許公開を含むが、これらに限定されない。
【実施例】
【0109】
実施例
一般的な手順:
用いた溶媒(CDCl3)を参照したJEOL ECLIPSE 400 MHz NMRの分光計にて、NMRスペクトルを取得した。ESI−MS(陰性モード)をHitachi M−8000の質量分光計を用いて得た。Phenomenex ZB−WAXのカラム(300m×0.32mm×0.25m)およびAgilent Model 5890のガスクロマトグラフィーを有する、Agilent Model 597 IAの質量分光計(範囲:70〜550amu)を用いて分析を行った。
【0110】
HPLC検定法1(逆相法):この勾配法を用いて、抽出物およびカラムの画分を検定することができる。そのカラムは、4.6×100mm、Phenomenex Luna(2)、3μ C18カラム(PIN 00D−4251−EO)である。流速は1.0mL/分、温度は40℃、注入は10μLである。検出は、ELSDおよび200nmでのUVである。順相クロマトグラフィーの試料を蒸発させ、注入する前にアセトン中で元に戻す。
【0111】
HPLC検定法2(順相法):この勾配法を用いて、カラム画分および最終生成物を検定することができる。そのカラムは、4.6×250mm、5μ、Alltech、Adsorbosilのシリカカラム(P/N 298017)である。移動相:ヘキサン/EtOAc、流速は1.0mL/分、温度は40℃、注入は10μLである。検出はELSDである。順相クロマトグラフィーの試料を直接的に注入することができ、注入する前に固体試料をEtOAc中で元に戻す。アセトン溶液を用いれば、ブロードまたは二重ピークを与え得るだろう。
【0112】
HPLC画分のアポトーシス活性についての生物検定:100,000個のHeLa細胞/穴を、24穴の細胞培養板中に蒔いた。その細胞を、24時間にわたり細胞培養恒温器内で成長させた。24時間後に、BCP21の精製から生じた化合物でその細胞を処理した。その精製の最中にHPLCにより発生させた画分を乾燥させ、DMSO中で再懸濁させた。各画分中の3つの異なる濃度の化合物でその細胞を処理した。その細胞を24時間後に採取し、アポトーシス(細胞死)(%)を決定した。その細胞をトリパンブルーで処理し、細胞死を推定した。10%未満の細胞死を+と標識付け、一方で、100%を++++により表し、50および75%を++および+++でそれぞれ標識付けた。
【0113】
実施例1.ヒカゲノカズラ(Lycopodium
clavatum)からの抽出および単離
BCP−21の単離:ヒカゲノカズラ(Lycopodium clavatum)の胞子を、95%のエタノールを用いて抽出する。その結果として生じた抽出相を混合し、95%のエタノール中の溶液として貯蔵することができるか、あるいは濃縮することができる。
【0114】
抽出物の蒸発および分割:BCP21のエタノール溶液(1L)を45℃にて真空下で蒸発させ、少量の沈殿物を含有する懸濁液を与えた。50mLの水をその懸濁液に添加し、200mLの塩化メチレン(MC)で分割した。そのMC層を分液漏斗内で分離して蒸発乾固させ、18.9gの油性残留物を与えた。その水層を乾燥させ、2.84gの固体を与えた。BCP21のエタノール抽出物およびMC可溶性残留物のHPLC特性描写を行った。
【0115】
実施例2.40MのSilica Biotageの分画:
前述したMC可溶性残留物が活性化合物を含有していると生物検定が示したので、5gのこの物質を15mLのMC中に溶解させ、800mLのヘキサンで平衡化させていた40Mのシリカカートリッジ上に装填した。10%のアセトン/ヘキサン(A/H)(1L)、20%のA/H(1L)、25%のA/H(1.2L)、およびアセトン(0.5L)と共に、そのカラムを溶出させた。生物検定の結果に基づき、25%のA/H溶出液のF5乃至F8、およびF9乃至F11を混合して窒素流により乾燥させ、2615−182−8(43.4mg)および2615−182−9(36.3mg)をそれぞれ与えた。表1は、アポトーシス検定により導いた画分試料抽出の結果を詳記している。
【表1】
【0116】
実施例3.12MのSilica Biotageの分画:
前述した溜めた活性生成物2615−182−8(43.4mg)を、2mLの30%のEtOAc/ヘキサン中に50℃にて溶解させ、100mLの30%のEtOAc/ヘキサンで平衡化させた12MのBiotageのシリカカートリッジ上にその溶液を注入した。60mLの35%のEtOAc/ヘキサン、100mLの40%のEtOAc/ヘキサン、60mLの45%のEtOAc/ヘキサン、60mLの80%のEtOAc/ヘキサンと共にそのカラムを溶出させ、26個の画分を得た。1mLのそれぞれを乾燥後に生物検定へ送った。各画分の残りの溶液を乾燥させて重量を得た。表2は、アポトーシス検定により導いた画分試料抽出の結果を詳記している。
【表2】
【表3】
【0117】
実施例4.検定のための試験化合物試料の調製。
前述した化合物の画分の調製を、下記の通りに表4に描出しているものに従って行った:
【表4】
【0118】
実施例5.ラセミ8−ヒドロキシパルミチン酸(「8−HHA」)の合成
【化1】
ラセミ8−ヒドロキシパルミチン酸の合成についての合成図式
【0119】
2つの三つ口フラスコは、気体注入補助具、隔壁、および撹拌子を備えており、その後、フラスコをアルゴンで浄化した。スベリン酸モノメチルエステル(10.0g、53.1mmol、1当量)を、THF(380mL、0.17M)中に1つのフラスコ内で溶解させた。トリエチルアミン(0.95mL、64.1mmol、1.2当量)を徐々に(4分にわたり、気密注射器を用いて)添加し、その混合物を室温で1.25時間にわたり撹拌した。無水DCM中でPPh3Cl2(24.4g、73.2mmol、1.4当量)から別個の溶液を作り、これは後で<30℃に冷却される。そのスベリン酸溶液を、黄色のPPh3Cl2溶液に滴下して添加し、2時間にわたり−35乃至−20℃で撹拌した。そのグリニャール試薬を、その後、温度を−41乃至−7℃に維持しつつ、45分にわたり滴下して添加した。その反応混合物を1.5時間にわたり撹拌し、その後、依然として冷たい間に2NのHCl(75mL)で反応停止した。20分にわたり撹拌した後に、その黄色は薄れる。薄層クロマトグラフィー(シリカゲル60、25%のアセトン/ヘキサン類)が、2つの主生成物のスポットに加えてPPh3およびOPPh3として反応混合物を与える。
【0120】
その反応混合物を、酢酸エチル(200mL)および2NのHCl(200mL)と共に分液漏斗に添加した。その有機層を収集し、その水層を2×80mLの酢酸エチルで逆抽出した。1つにまとめた有機相を、NaHCO3(飽和溶液、2×90mL)で洗浄することにより中和し、その後塩水(90mL)で洗浄し、MgSO4(20g、30分)で乾燥させ、濾過し、25℃で回転蒸発させ、高真空下で3時間にわたり乾燥させた。酢酸エチルを除去するために、その物質を20mLのDCMと混合して再蒸発させ、33gの粗製物質を与える。
【0121】
その粗製物質を、10%のアセトン/DCM中に取り出し、シリカゲル栓(240ml)上に装填し、4CVの10%のアセトン/DCMで洗い流した。その第1の栓は10.4gのOPPh3を除去し、第2の栓は6.8gのOPPh3を除去した。フラッシュカラムクロマトグラフィーを用いて精製を継続し、その中で、5×29cmのカラムを10%のアセトン/ヘキサン類中のシリカゲル60で充填する。55%のDCM/ヘキサン類の最小体積で、その粗製物質を装填した。所望の化合物を、10%のDCM/5%のアセトン/85%のヘキサン類と共に溶出させる。1つの画分の蒸発および乾燥は、2.7gの化合物1(8−ケトパルミチン酸メチルエステル)を与え、いくつかの不純な画分上の反復カラムは、さらに2.0gの化合物1を与えた。全収率は、スベリン酸モノメチルエステルに基づいて31%であった。化合物1(8−ケトパルミチン酸メチルエステル)は、1H NMR(CDCl3中):3.65(s,3H,OCH3),2.37(m,2H,O=CCH2),2.29(t,2H,O=CCH2),1.57および1.28(br.,22H,CH2),0.87(s,3H,CH3末端)により同定した。
【0122】
化合物1(2.86g、10.1mmol)を、50mLのエタノール中に溶解させ、氷浴内で冷却した。水素化ホウ素ナトリウム(1.9g、59.5mmol)を、一部分ずつ1〜17Cで添加した。その反応をTLC(25%のアセトン/ヘキサン類中で展開した板)により監視し、そこで、Rfは、ケトンについて0.56であり、メチルエステルについて0.28であった。固体NaBH4の追加分を、出発物質を全く検出しなくなるまで2時間の期間にわたり添加した(全部で60当量を用いた)。その反応を、2NのHCl(60mL)をその冷溶液にゆっくり添加することにより停止し、分液漏斗に移し、DCM(3×35mL)で洗浄した。1つにまとめた有機層を、NaHCO3(飽和、35mL)および塩水(35mL)で洗浄し、その後MgSO4で乾燥させ(7g、30分)、濾過し、回転蒸発させて澄んだ油を与え、これは4℃で貯蔵する際に白色で蝋状の固体となった(収量は2.86g)。
【0123】
その物質を、フラッシュクロマトグラフィー(シリカゲル60、20%のアセトン/ヘプタン)により精製した。回転蒸発乾固させた後は、その化合物は油であり、高真空上で乾燥させた後は、その生成物は白色で蝋状の固体である。その1H NMRスペクトル(CDCl3)は、その優勢生成物を、少量の未知の不純物を有する化合物2として示した。化合物2(8−ヒドロキシパルミチン酸メチルエステル)は、1H NMR(CDCl3中):3.66(s,3H,OCH3),(br,1H,メチン),2.29(t,2H,O=CCH2),1.62および1.31(br.,28H,CH2),0.87(s,3H,CH3末端)により同定した。
【0124】
化合物2(1.97g、6.9mmol)をTHF(28mL)中に溶解させた。水酸化リチウムを水(Nanopure、15mL)中に溶解させ、5分撹拌して溶解した。そのLiOH溶液を、5分にわたりそのエステル溶液にゆっくり添加し、室温で撹拌した。その生成物が消え、新たなスポットが基線に現れるまで(4時間)、その反応をTLC(20%のアセトン/ヘキサン)により監視した。単離精製操作は、その反応混合物を回転蒸発させ、揮発物を除去し、豊富な白色の固体を得ることを伴った。その固体をDCMですすぎ、その後、2NのHCl(固体はほとんど不溶)およびクロロホルムと混合した。その水層をCHCl3(3×75mL)3で抽出した。1つにまとめた有機相を、塩水およびMgSO4で乾燥させ(30分)、その後濾過し、回転蒸発させ、高真空下で一晩にわたり乾燥させ、1.67gの白色の固体を最終生成物として得た(89%の収率)。
【0125】
その最終生成物の分析は、8−ヒドロキシ−パルミチン酸としての同定を、その優勢生成物として支持した。1H NMR(CDCl3中):5.5(非常に広範,1H,OH),3.58(br,1H,メチン),2.34(t,2H,O=CCH2),1.64および1.33,(br.,28H,CH2),0.87(s,3H,CH3末端)。13C NMR(CDCl3中):178.40,72.08,37.59,37.43,33.79,31.97,29.79,29.68,29.37,29.09,25.73,25.51,24.69,22.76,14.20。LC−MS(ACN−NH4OH中でESIモード)。親イオン 271.13 m/z,理論:272(100%) 273(17%)。融点:70.5℃。
【0126】
実施例6.(S)−8−ヒドロキシパルミチン酸および(R)−8−ヒドロキシパルミチン酸の両方の合成
R、S、またはラセミ1,2エポキシデカンの1,2−デカンジオールからの形成
【化2】
塩基の存在下における、ラセミ1,2−デカンジオール類の塩化p−トルエンスルホニルとの反応は、所望の第一級トシル酸塩、ビス−トシル酸塩、および少量の第二級トシル酸塩の混合物を与えることが知られている(「韓国化学会会報、2009年、第30巻、第7号、1671−4(Bull. Korean Chem. Soc. 2009 Vol. 30, No. 7, 1671-4)」)。任意の第二級トシル酸塩の発生は、2位での閉環の最中における立体中心の反転が原因で、その結果として生じるエポキシドの立体化学的純度に不利に影響し得るだろう。
【0127】
この反応では、10mlの無水ジクロロメタン中の23.9ml(0.172mol、3.0当量)のトリエチルアミンを、10.00g(0.0574mol)の1,2(S)−デカンジオール(99.5%、「S」)、13.7g(0.0717mol、1.25当量)の塩化p−トルエンスルホニル、0.70g(0.00574mol、0.10当量)かつ30mlの無水ジクロロメタンの溶液に、<10℃において、ゆっくり40分の期間にわたり添加した。20分にわたり<10℃で撹拌した後に、その反応はTLC(100%のCH2Cl2)により完了した。室温に温めた後に、200mlのMTBEおよび120mlの1MのHClの添加により、その生成物の混合物を有機相に分配した。その有機層を、1×80mlの飽和NaHCO3溶液、1×80mlの水、および1×80mlの飽和NaCl溶液で洗浄した。硫酸ナトリウムでの乾燥および濃縮は、直接的にエポキシドへの転換まで進めた21.7gの残留物を結果的にもたらした。この残留物を217mlの無水THF中に溶解させ、9.64g(0.086mol、1.30当量)のカリウムt−ブトキシドを室温で冷却することなしに添加した。30分後に、その反応はTLC(10:1のヘプタン/酢酸エチル)により完了した。その反応を100mlの水の添加により停止し、THFを真空中で除去した。その生成物の混合物を300mlのMTBE中に分配し、その水層を1×50mlのMTBEで逆抽出した。1つにまとめた有機物を、その後、1×80mlの飽和NaCl溶液で洗浄した。硫酸ナトリウムでの乾燥および濃縮は、10.4gの残留物を結果的にもたらした。その残留物を短経路蒸留し、1,2(S)−エポキシデカンを約60℃および1mmgHgで収集した。>99%のGC−MS純度、68%の回収率で、6.11gを結果的にもたらした。
【0128】
臭化アルケニルマグネシウム類のR、S、またはラセミ1,2−エポキシデカンとの反応
【化3】
グリニャール試薬の形成、およびラセミまたは不斉の1−エポキシデカンへの添加を、文献の手順に従って行った(「有機化学誌、2009年、第74号、5063〜5066ページ(J. Org. Chem. 2009, 74, 5063-5066)」)。そのグリニャール試薬を、自己連結生成物を最小化するために約0.5Mで<10℃にて調製した。そのグリニャール試薬を滴定する試みを全く行わず、それらを0.5Mであると想定した。マグネシウムの削り屑を活性化してそのグリニャール反応を開始させるために、I2の添加を必要としなかった。
【0129】
乾燥した250mlのフラスコに、N2下、THF(上記で調製した通り)中で約0.5Mの98ml(0.049mol、1.5当量)の臭化ヘプト−6−エニルマグネシウムを投入する。そのフラスコの内容物を、撹拌しながら−40℃に冷却する。1.24g(0.00653mol、0.20当量)のヨウ化銅(I)を、そのフラスコに投入する。−40℃で0.5時間後に、51mlの無水THF中の5.10g(0.0326mol)の1,2(S)−エポキシデカンの溶液を、少なくとも1時間にわたり−40±5℃でゆっくり添加する。その添加の完了後、−40℃で2時間にわたり撹拌し続け、その後、その反応は、通常、TLC(ヘプタン中で40%のEtOAc)により完了する。完了後に、500mlのMTBE、125mlの飽和塩化アンモニウム、および50mlの水を添加し、15分にわたり十分に混ぜる。層を切り、その有機物を、1部の飽和塩化アンモニウムおよび1部の水の混合物100mlで洗浄する。その有機物を、その後、3×100mlの水、1×50mlの飽和NaCl溶液で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、静置の際に凝固する油になるまで濃縮する(8.6gの結果物)。その生成物を、220gのシリカ上でクロマトグラフィーに掛け、いかなる非極性不純物をも除去するために500mlのヘプタンで最初に溶出し、その後、10:1(v:v)のヘプタン/酢酸エチルで溶出する。その生成物を含有する画分を溜めて濃縮し、8.11g(97.6%の収率)の9(S)−ヒドロキシヘプタデス−1−エンを、GC−MSで96.7%純粋である蝋状の固体として得る。
【0130】
【化4】
1.50当量の塩化t−ブチルジメチルシリルの、2.0当量のイミダゾールを塩基として有するアルケノール類のTHF溶液への添加により、アルコールの保護を達成した。概して、室温で16〜20時間後に、その反応は92〜96%完了した。追加の塩化TBSおよびイミダゾールは、その反応を完了まで完全には推進しなかった。ヘプタンへの分配を伴う酸性で水性の単離精製操作は、酸感受性のTBSにより保護したアルコールの分解なしに、イミダゾールの除去を可能にした。シリカ上のヘプタンを用いたフラッシュクロマトグラフィーは、その残余の未反応のアルコールを容易に除去し、その中間体を高い純度(GC−MSで>99%)をもって単離することを可能にした(1362−42、45、86、1341−89、1380−15に留意されたい)。
【0131】
TBSにより保護した酸へのオゾン分解/ピニック酸化の経路
【化5】
138mlのジクロロメタンおよび138mlのメタノール中の9.83g(0.0267mol)の9(S)−(t−ブチルジメチルシリルオキシ)ヘプタデス−1−エン、0.10g(0.00027mol、0.01当量)のスーダンレッド7Bの溶液を、−25±5℃にてオゾンで処理した。指示薬の色が脱色した後に、オゾンの添加をさらに6〜7分にわたり継続した。その溶液に、その後N2を少なくとも30分にわたり注入する。13.99g(0.0533mol、2.00当量)のトリフェニルホスフィンの溶液を、その後4〜5分にわたり<−15℃で添加し、その後室温に温まるようにした。30分にわたり室温で撹拌した後に、その反応をGC−MSにより検査し、1.25時間および3時間で再び検査する。全ての3回の検査は、同じ濃度のトリフェニルホスフィン、酸化トリフェニルホスフィン、および生成物を概して示し、オゾン化物が消費されたことを示す。その反応混合物を、その後糊状の固体になるまで濃縮し、(メタノールを除去するために)100mlのジクロロメタンから1回再濃縮する。その残留物を、その後、ヘプタン、続いて20:1のヘプタン/酢酸エチルと共に溶出する、300gのシリカ上でクロマトグラフィーに掛けた。その生成物を含有する画分を溜めて濃縮し、GC−MSで31%のトリフェニルホスフィンを依然として含有する13gの油を与える。その油を500gのシリカ上で再度クロマトグラフィーに掛け、そのトリフェニルホスフィン濃度はその時13%であった。この油を100mlのジクロロメタン中に溶解させた。0.46ml(0.0074mol)のヨードメタンを添加し、その混合物を一晩にわたり撹拌し、その後、トリフェニルホスフィンの全てが消費されていた(GC−MSにより監視した)。最後に、その生成物を、10:1のヘプタン/酢酸エチルと共に溶出する、200gのシリカ上でフラッシュクロマトグラフィーに掛けた。濃縮後に、8.00g(89%の収率)の8(S)−(t−ブチルジメチルシリルオキシ)ヘキサデカナールを、98.7%のGC−MS純度をもって結果的にもたらした。
【0132】
315mlのt−ブタノール中の3.79g(0.0102mol)の8(S)−(t−ブチルジメチルシリルオキシ)ヘキサデカナールの溶液に、125mlの水中の8.49g(0.0939mol、9.17当量)の亜塩素酸ナトリウムおよび8.50g(0.0708mol)のリン酸二水素ナトリウムの溶液を、室温で30分にわたり添加した。その温和な発熱を水浴により制御した。一晩にわたり撹拌した後に、その反応はTLC(1:1:0.01のヘプタン:EtOAc:HOAc)により完了した。t−ブタノールを真空中で除去した。その残留物を200mlの水および300mlのヘプタン中に溶解させ、その後、80mlの1.0MのHClを添加した。その結果として生じたヘプタン層を、3×100mlの水、1×75mlの飽和塩化ナトリウム溶液で洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濃縮し、3.79g(95.7%の収率)の8(S)−(t−ブチルジメチルシリルオキシ)ヘキサデカン酸(GC−MSで99.4%の純度)を(TMSCHN2からのメチルエステルとして)得た。
【0133】
TBS保護基の除去および8(S)−ヒドロキシパルミチン酸の精製
【化6】
3.79g(0.00980mol)の8(S)−(t−ブチルジメチルシリルオキシ)ヘキサデカン酸、48mlのACN、および2.13ml(0.059mol、6当量)の48%のフッ化水素酸を混合し、1時間にわたり撹拌した。20〜30分後に、その油性の混合物は懸濁液となる。1時間後に、その出発物質を完全に消費した(TLC(1:1:0.01のヘプタン:EtOAc:HOAc)により監視した通り)。完了の際に、300mlのMTBEおよび300mlの水を添加し、徹底的に混ぜた。その水層を除去し、その有機層を、3×100mlの水、1×100mlの飽和塩化ナトリウムで洗浄し、硫酸ナトリウムで乾燥させ、濾過し、濃縮し、2.50gの固体を得た。その固体はこの時点で95.6%eeであった(1380−51に留意されたい)。その固体を40mlの熱いACN中に溶解させ、撹拌しながら冷ました。その結果として生じた懸濁液を、<10℃に30分にわたり冷却し、濾過により収集し、20mlの冷たいACNで洗浄し、真空下にて40℃で乾燥させ、2.18g(82%の収率)の8(S)−ヒドロキシパルミチン酸が得られ、それは、GC−MSで>99%であり(TMSCHN2からのメチルエステルとして)、HPLC−MSで98.4%eeであった。
【0134】
8(R)エナンチオマーを与える光延反転
【化7】
8−ヒドロキシ酸の立体中心を光延条件下で反転させるためには、カルボン酸をそのメチルエステルとして保護する必要があった。最初は小規模に、ジアゾメタンを転換の迅速で効率的な方法として用いた。ジアゾメタンの危険な性質のために、より安全な代替物、TMSジアゾメタンをより大規模に用いた。MTBE/メタノール中の酸を完全にエステル化するために、2.6当量のTMSジアゾメタンを必要とした。立体中心を光延条件下で酢酸を用いて反転させる試みは、決して理想的でないと判明した。4−ニトロ安息香酸を置換する同じ条件(「有機合成、全集第9巻、607ページ(Organic Synthesis, Collective Volume 9, page 607)」)は、8(R)−4−ニトロ安息香酸塩を清浄に提供した。THF/水中のLiOHを用いた鹸化、続いてACNからの再結晶は、HPLC−MSで100%eeである所望の8(R)−ヒドロキシパルミチン酸を提供した。
【0135】
実施例7:Alamar Blue検定によるHeLa細胞の生存能の決定のための一般的な検定手順。Alamar Blue還元法による細胞生存能の評価は、候補薬物の細胞傷害能を例解するために日常的に用いられる。Alamar blueは、細胞代謝中間体により桃色の蛍光染料のレゾルフィンに転換される、青色の非蛍光染料のレサズリンを利用することにより、細胞生存能を検出する。本検定から発生した蛍光信号は、試料中の生細胞の数に比例する。T−75フラスコ内で成長させた集密的なHeLa細胞(70〜80%)を、トリプシン−EDTAを用いて剥離した。遠心分離に続いて、その細胞の沈渣をDMEM媒体中で再懸濁させ、血球計を用いて計数した。細胞を96穴板に10,000細胞/100μl/穴の密度で播種し、37℃/5%のCO2で8時間にわたり培養した。媒体を除去し、試験化合物を100μlの体積で添加した。その板を22時間、46時間、および70時間にわたり培養し、その後、穴1つ当たり10μlのalamar blueを添加した。試験化合物での24時間、48時間、および72時間の終わりに、マイクロプレートリーダーを用いて530nmの励起および590nmの発光で蛍光を測定した。対照(%)として表現した生存能を、薬物濃度に対して描図した。テルフェナジンを実験計画法における参照化合物として用い、これは、再現可能なIC50値を有する全ての時点(24時間、48時間、および72時間)において用量依存的な阻害を示した。異なる化合物は異なる生存能を示した。
【0136】
実施例8.実施例7の検定のデータ分析。2つの反復サンプルとしての穴の平均値を算出した。対照細胞の生存能を、全ての試験濃度について100%と考えた。試験試料の細胞生存能の百分率を、
【数1】
として算出した。S字状の用量応答(変数)等式(Graph Pad Prism 4のソフトウェア)を用いて、薬物濃度に対する百分率生存能データの非線形回帰分析(曲線適合)により、IC50値を決定した。BCP−6およびBCP−7は、細胞に対する格別の影響を実証した。
質的管理:本検定を以下の質的検査について評価した:
i.参照化合物のIC50値:テルフェナジンのIC50値
ii.反復サンプル間の変動係数(%):反復サンプル間のCV(%)は許容可能限界(10%)以内であった。
【0137】
実施例9.試験化合物試料の調製。前述した化合物の画分の調製を、表5に描出したものに従って行った。
【表5】
【0138】
実施例10:HepG2細胞に対するカスパーゼ−3活性の決定のための一般的な検定手順:
疑わしい、つまり、アポトーシスを経るように誘発されている細胞を最初に溶解させ、その細胞内内容物を収集する。我々の試験系では、蛍光レポーター分子の7−アミノ−4−メチルクマリン(AMC)に共役するカスパーゼ特異的ペプチドの添加により、スタウロスポリンで処理した細胞からの溶解物をプロテアーゼ活性について試験した。そのカスパーゼによるそのペプチドの開裂は、380nmの波長で励起される際に、460nmで蛍光を発する蛍光色素を放出する。その細胞溶解物中のカスパーゼ酵素活性の程度は、蛍光マイクロプレートリーダーで検出した蛍光信号に直接的に比例する。T−75フラスコ内で成長させた集密的なHepG2細胞(70〜80%)を、トリプシン−EDTAを用いて剥離した。遠心分離に続いて、その細胞の沈渣をDMEM媒体中で再懸濁させ、血球計を用いて計数した。細胞を96穴板に40,000細胞/100μl/穴の密度で播種し、37℃/5%のCO2で8時間にわたり培養した。媒体を除去し、試験化合物を100μlの体積で添加した。30分の上記のような前処理の後に、50μLの3×濃縮のスタウロスポリン(20μMの最終濃度)、または50μLの媒体(未処理の対照)を、それぞれの穴に添加した。スタウロスポリンで16時間にわたり誘発した後、50μlの溶解緩衝液の添加、続いて30分の培養により細胞を溶解した。顕微鏡下で観察されるような完全な溶解の後に、100μlの冷やした緩衝液(検定緩衝液)を各穴に添加した。次に、DEVD−AMC(基質、最終濃度は15μM)を添加し、その反応が2時間にわたり継続することを可能にした。BMG Polarstarの蛍光マイクロプレートリーダーを用いて、380nMで励起させ、460nMで発光を捕捉することにより、その蛍光を測定した。
【0139】
実施例11:実施例10の検定のデータ分析。各標準、ブランク、および試料について、反復サンプルの相対蛍光値(RFU)を平均する。対照(阻害剤なしで処理したスタウロスポリン)の酵素活性を、100%の活性(0%の阻害)と考えた。試験化合物/参照化合物の蛍光値(RFU)を、阻害(%)を算出するためにこれに対して比較する。試験化合物/参照化合物の百分率阻害を、下記のように算出した:
【数2】
S字状の用量応答(変数)等式(Graph Pad Prism 4のソフトウェア)を用いて、薬物濃度に対する百分率生存能データの非線形回帰分析(曲線適合)により、IC50値を決定した。BCP−6およびBCP−7は、カスパーゼ3阻害に対する格別の影響を実証した。
質的管理:本検定を以下の質的検査について評価した:
(i)参照化合物のIC50値:Ac−DEVD−CHOのIC50値
(ii)反復サンプル間の変動係数(%):反復サンプル間のCV(%)は許容可能限界(10%)以内であった。
【0140】
実施例12:ヒカゲノカズラ(Lycopodium
Clavatum)の抽出物が、アポトーシスと一致する形態学的特徴を誘発する
American Type Tissue Culture(ATCC)から得られたHeLa細胞(子宮頸癌)を、48時間にわたりヒカゲノカズラ(Lycopodium Clavatum)からの抽出物で処理し、抗チューブリン抗体で染色してその細胞骨格を検査し、DNAに強く結合する蛍光染色液である、4’,6−ジアミジノ−2−フェニルインドール(DAPI)で核を染色した。媒介物で処理した細胞は、無傷の細胞質構造および核を暴露した。その抽出物で処理した細胞がその細胞質構造(丸い細胞)を失うと同時に、核中の空所として現れるDNAの喪失により明らかであるように、細胞中の核DNAは完全に喪失した。こうした形態学的変化は、BCP−21が子宮頸癌細胞株内にアポトーシスを誘発することを示唆している。
【0141】
実施例13:ヒカゲノカズラ(Lycopodium
Clavatum)の抽出物が、sub−G1集団を誘発する
ヒカゲノカズラ(Lycopodium
Clavatum)の抽出物がアポトーシスを示唆する変化を誘発するため、流動細胞計測法を用いてDNA含量を測定することによりアポトーシスを評価した。そのアポトーシス細胞は断片化によりDNAを失い、それ故、2n未満のDNA含量を有することになるだろう。流動細胞計測法の分析の際には、その細胞はG1ピークの左に延びるように見えることになり、それ故、「sub−G1ピーク」と呼ばれる。対照および様々な希釈液(1:500)および1:250のヒカゲノカズラ(Lycopodium Clavatum)の抽出物で処理したHeLa細胞を、無水エタノール中に固定し、ヨウ化プロピジウムで30分にわたりRNAseと共に染色した。ハーバード医学校、ダナ・ファーバー癌研究所(Dana Farber Cancer Institute, Harvard Medical School)(マサチューセッツ州、ボストン)の中核施設にて、その細胞を分析した。フェニックス・フロー・システムズ(Phoenix Flow Systems)(カリフォルニア州、サンディエゴ)製のMultiCycleのソフトウェアを用い、細胞DNA含量の柱状図を逆畳み込みし、sub−G1相内の細胞の百分率の定量化を得ることになる。
【0142】
ヒカゲノカズラ(Lycopodium
Clavatum)の粗製抽出物は、用量依存的な様態でその対照(6.7%)に比べてsub−G1ピーク(23%)を増加させた(図6)。そのデータは、その抽出物が用量依存的な様態でHeLa細胞のアポトーシスを誘発することを強く示している。
【0143】
実施例14:ヒカゲノカズラ(Lycopodium
Clavatum)の抽出物が、PARP切断を誘発する
50mMのトリス−HCl、7.6のpH、150mMのNaCl、30mMのEDTA、0.5%のTriton X−100中で、完全プロテアーゼ阻害剤((ロシェ)Roche)を用いて細胞を溶解させた。タンパク質試料を、適切な百分率のゲル上にSDS−PAGEにより分離した。概して、20〜60μgのタンパク質を内因性タンパク質について、10〜15μgを形質移入タンパク質について分析した。タンパク質を0.2μmのニトロセルロース膜(Bio−Rad)に、1時間にわたり4℃で1時間にわたり移した。5%の乳および0.1%のTween−20(アメリカン・バイオアナリティカル(American Bioanalytical))を含有するPBS溶液中にて、膜を45分にわたり室温(RT)でブロッキングした。そのニトロセルロース膜を、その後、RTで2時間または4℃で一晩中の何れかにわたり、1%の乳および0.1%のTween−20を含有するPBS中にて、適切な濃度で希釈した一次抗体中で培養した。その一次抗体の除去の際に、洗浄緩衝液(1%の乳および0.1%のTween−20を有する1×PBS)中にて、その膜を10分にわたりRTで3回洗浄した。その膜を、その後、洗浄緩衝液中で希釈した適切な二次抗体中にて、1時間にわたりRTで培養した。膜をその後10分にわたり3回洗浄した。製造者の取扱説明書に従い、ECL(ピアース研究所(Pierce Laboratories))を用いてウエスタンブロットを発色させた。
【0144】
その結果は、全長PARP(分子量は116kDa)が24時間後に完全に切断されたことを示した。その抽出物で処理したHeLa細胞は、時間依存的な様態でPARP切断における増加を示している(図7)。このデータは、ヒカゲノカズラ類(Lycopodium)の抽出物がアポトーシスを誘発することを強く示している。
【0145】
実施例15:ヒト腫瘍細胞株に対する8−HHAのラセミ混合物の検査
DMC中に溶解させた8−HHAの合成ラセミ混合物を、60のヒト腫瘍細胞株のパネルに対して検査するために、NIH、国立癌研究所、癌診療部(the Division of Cancer Diagnosis and Treatment, National Cancer
Institute, NIH)が受け取った。その検査は、生存細胞中のテトラゾリウム染料の有色ホルマザン生成物への代謝還元に依存する、単純な比色(MTT)検定に基づくマイクロプレート細胞毒性検定である。その化合物の検査のために修正実験計画法を用い、これは様々な腫瘍細胞株の成長の阻害を測定する。この検定において試験した8−HHAは、10μMの濃度にあった。
【0146】
その結果は、8−HHAが、40〜96%の範囲で、白血病細胞株(例えば、CCRF−CEM、K−562、SR、MOLT−4、RPMI−8226、およびHL−60(TB))の成長を著しく抑制していることを示している。それは、非小細胞肺癌細胞株(例えば、HOP−92およびNCI−H460)上における成長の顕著な阻害も実証した。また、検査した7つの結腸直腸癌細胞株のうち3つ(すなわち、HCT−116、HCT−15、およびKM12)は、成長抑制の中等度の証拠を示した。このデータは、8−HHAが、その程度は様々ではあるが、白血病、肺、およびの結腸直腸の癌細胞株等の、様々な癌細胞株に対して成長阻害活性を有することを示している。
【0147】
実施例16:癌細胞株の生存に対する8−HHAの影響
1)肺癌細胞株に対して:8−HHAのラセミ混合物の影響、および、各個のエナンチオマー、つまりSエナンチオマーおよびRエナンチオマーのそれを、HOP−92肺癌細胞株の生存に対して検査した。その細胞生存能を、比色MTT検定を用いて、対数増殖期にて100μMの8−HHAで72時間にわたり検査した。別個の検定において、20〜100μMに及ぶ、異なる濃度の8−HHAのS−エナンチオマーで72時間にわたり、HOP−92細胞を処理した。
【0148】
その結果は、8−HHAのラセミ混合物が、HOP92細胞株の生存を25%低減したことを示している。そのR−エナンチオマーは、HOP92細胞株の生存を13%低減した。対照的に、そのS−エナンチオマーは、肺癌細胞株の生存に対して最大の影響を有した(72%低減した)。このデータは、8−HHAのSエナンチオマーが、HOP92肺癌細胞の生存に対して強力な阻害効果を有することを示している。さらに、図8は、増加する濃度の(S)−8−HHAが、用量依存的な様態で肺癌細胞株の生存を阻害したことを示している。細胞生存における50%の低減(IC50)が、8−HHAのS−エナンチオマーのおおよそ90μMの用量にて観察された。このデータは、8−HHAのSエナンチオマーが、肺癌細胞株に対して用量依存的な阻害効果を有することを示している。
【0149】
2)肝臓癌細胞株に対して:8−HHAのラセミ混合物の影響、および、各個のエナンチオマー、つまりSエナンチオマーおよびRエナンチオマーのそれを、PLC/PRF/5(「PLC−5」または「PLC5」とも呼ばれる)、Hep3B、およびSNU449(「SNU」とも呼ばれる)の肝臓癌細胞株の生存に対して検査した。その細胞生存能を、比色MTT検定を用いて、対数増殖期にて100μMの8−HHAで72時間にわたり検査した。別個の検定において、20〜100μMに及ぶ、異なる濃度の8−HHAのS−エナンチオマーで72時間にわたり、PLC−5細胞を処理した。
【0150】
その結果(図9)は、8−HHAのラセミ混合物が、PLC−5およびHep3Bの細胞株の生存をそれぞれ52〜25%低減したことを示している。それは、SNU細胞株に対して全く大きな影響を有しなかった。一方、8−HHAのS−エナンチオマーは、ラセミ混合物またはR−エナンチオマーの何れかと比較して、全ての肝臓癌細胞株の生存に対して最大の影響(ほとんど75%の細胞生存の低減)を有した。このデータは、8−HHAのSエナンチオマーが、肝臓癌細胞の生存の全てに対して強力な阻害効果を有することを示している。
【0151】
さらに、図10は、増加する濃度の(S)−8−HHAが、用量依存的な様態でPLC−5肝臓癌細胞株の生存を阻害したことを実証している。細胞生存における50%の低減(IC50)が、8−HHAのS−エナンチオマーのおおよそ50μMの用量にて観察された。このデータは、8−HHAのSエナンチオマーが、肺癌細胞株に対して用量依存的な阻害効果を有することを強く示している。
【0152】
実施例17:8−HHAは、肝臓および肺の癌細胞株内でアポトーシスを誘発する
PARP切断は、アポトーシスの特異的でかつ十分に確立した標識と見なされる。アポトーシスの最中に、FAS受容体は下方調節を受け、これはアポトーシスの確立した指標と見なされる。FAS配位子に結合するFAS受容体により活性化した経路によりアポトーシスを誘発することができ、外因性機構と呼ぶ。この経路によるアポトーシスは、FAS受容体の低減を結果的にもたらす。従って、FAS受容体のタンパク質濃度における低減は、アポトーシスが外因性機構により誘発されることを示している。
【0153】
1)肝臓癌細胞株に対して:8−HHAの影響を肝臓癌細胞株内でのアポトーシスに対して検査した。図11は、100μMの8−HHAのS−エナンチオマーの処理が、SNUおよびPLC−5の肝臓癌細胞株内におけるPARP切断を結果的にもたらしたことを示している。このデータは、8−HHAが肝臓癌細胞株内でアポトーシスを誘発することを示している。
【0154】
PLC−5細胞株を、100μMの8−HHAで72時間にわたり処理した。これは、対照と比較して、(S)−8−HHAの処理を用いて、FAS受容体濃度における相当な低減を結果的にもたらした(図12)。このデータは、(S)−8−HHAが、外因性機構により肝臓癌細胞株内でアポトーシスを誘発することを示唆している。
【0155】
2)肺癌細胞株に対して:HOP−92肺癌細胞株を、100μMの(S)−8−HHAで72時間にわたり処理した。その(S)−8−HHA処理は、HOP92細胞株内におけるFASタンパク質濃度の低減を結果的にもたらした。このデータは、8−HHAが、外因性機構を通じてHOP92細胞株内でアポトーシスを誘発することを示している。
【0156】
実施例18:肺癌細胞株内のプロアポトーシスBim1に対する8−HHA処理の影響。
Bim1は、その上方調節がアポトーシスを引き起こすプロアポトーシスタンパク質である。HOP92細胞株を、100μMの8−HHAのS−エナンチオマーで72時間にわたり処理した。その結果は、Bim1が、対照試料と比較して、処理した8−HHA中で2倍に増加したことを示している。このデータは、8−HHAが肺癌細胞株内でアポトーシスを誘発することを示している。
【0157】
実施例19:インビボの動物モデルに対する、8−HHAの最大耐用量(MTD)の決定:本研究の目的は、ICRマウスにおける静脈内(IV)投与に続く、8−HHAの最大耐用量を決定することであった。8−HHAのラセミ混合物、偽薬、および媒介物を、25〜30グラムの体重を有する20匹のICR雌マウスにそれぞれ投与した。8−HHAを、ナノ脂質中にて、0、10、20、および40mg/mlの濃度で調合した。一群当たり4匹の動物がおり、それらに8−HHAの0、10、20、および40mg/kgの用量を注射した。マウスに単一の急速静注用量を、尾静脈を通じて群割り当てに従って与えた。投薬の1週間後に、マウスを屠殺した。毎日の臨床観察を、投与後15分および1時間に行った。研究8日目での屍検まで、そのマウスを任意の有害作用について毎日観察した。試験物質投与より前に、かつ試験期間中を通して1日置きに、体重を全てのマウスについて記録した。この全体の研究を、トキシコン社(Toxikon Corporation)、マサチューセッツ州、ベッドフォードで行った。その結果を表6に描写する。
【表6】
【0158】
結果および考察:6日目および8日目の体重は、投薬前の体重から変化しなかった。6日目および8日目の体重は、投薬前の体重から変化しなかった。高用量(40mg/kg)を投薬した2匹のマウスは、投薬した直後に死んだ。偽薬、10mg/kg、および20mg/kgを投薬した動物は、8HHAに十分に耐性があり、いかなる留意すべき臨床的異常も実証しなかった。20mg/kgの用量を、その後8−HHAの最大耐用量と考えた。
【0159】
実施例20:肝臓異種移植マウスモデル:
本研究の目的は、PLC5細胞、肝細胞癌細胞株を用いて、肝臓癌における8−HHAの抗腫瘍効能を決定することであった。腫瘍成長および動物の生存を主要評価項目と考えた。
【0160】
動物および飼育:生後5〜6週間でかつ16〜20gmの体重がある、30匹のBABL/cの、非妊娠で、かつ未経産の雌ヌードマウスを本研究で用いた。薬物候補の抗腫瘍効能試験のための異種移植の研究において歴史的に用いられてきたために、BALB/c nu/nuのマウスを用いた。実際の試験用と同じ条件下で、マウスを最小で5日にわたり順化させた。30〜70%の室内相対湿度、時間当たり最小で10回の交換という時間当たりの空気交換、12時間の明暗周期の光曝露、全領域蛍光灯を用いて、マウスを68±5°Fの室温で収容した。ポリカーボネートでできている換気したマイクロアイソレーターケージ内に、マウスを集団で収容した。マウスは、高圧滅菌した実験室等級の寝藁を用いており、放射線照射したペレットおよび高圧滅菌した水を自由に提供された。飼料、水、または寝藁には、試験データに干渉すると予想される既知の汚染物は全くなかった。実験室および動物室を、出入り制限施設として維持した。
【0161】
注射および投薬の経路:8−HHAを、腹腔内(IP)注射を通じて投与した。8−HHAは不十分に水溶性であり、それ故、100mMに対応する27.2mg/mlの濃度でナノ脂質分散溶液(ePharse、スイス、バーゼル)中に懸濁する。MTD検定(予備データ)は、マウスが20mg/kgという最大用量に耐えたことを示した。本研究のために、2つの用量―10mg/kgおよび100mg/kgを用いた。
【0162】
動物の準備
腫瘍誘発:細胞株PLC5をヴァナス・オンコロジー(Vanas Oncology)での推奨仕様に従って培養し、そこで細胞を成長させ、10%のウシ胎仔血清、2mMのL−グルタミン、100単位/mlのストレプトマイシン、および100単位/mlのペニシリンを含有するRPMI媒体内に保持した。血球計を用いたトリパンブルー生存能試験を用いて、細胞をトリプシン処理および計数した。血球計の象限における細胞計数を細胞/mL値に換算し、これにより、マウス1匹につき適切な数の細胞の単離が可能になることになる。腫瘍細胞(5×106細胞/マウス)の懸濁液を含有する0.2mLの50%のRPMI/50%のMatrigel(商標)の混合物を、各マウスに右側腹部内に皮下接種した。十分に確定するまで、細胞を週に2回観察した。式:腫瘍重量(mg)=(a×b2/2)(式中、「b」はミリメートルでの最小直径であり、「a」は最大直径である)を用いて、腫瘍量を算出した。
【0163】
動物の割り当て:確定した腫瘍がおおよそ50〜100mgの平均算出量に到達した時点で、一群当たりの腫瘍の大きさの変動性を低減するために、マウスを適切なソフトウェアを用いて無作為に処理群とした。
【0164】
投薬前の手順:1日目における投薬より前に、順化させた動物を秤量し、毒性の臨床的兆候について観察した。
【0165】
用量の管理:2つの用量の1および10mg/kgを、MTD検定指針に基づいて投与した。第1の投薬日が1日目である。1日目には、8−HHA、および媒介物の注射を、下記の表7における研究設計に従って投与した。
【表7】
【0166】
投薬後の手順:処理に続いて、腫瘍およびマウスの体重の測定結果を週に2回記録し、総括的観察を毎日少なくとも1回行った。触知可能でない腫瘍を有するマウスを、完全退縮と考えた。マウス死亡率の百分率および死期を、本研究における全ての群について記録することになる。下記の判定基準のうち1つ以上を満たす場合に、動物を瀕死と定義し、屠殺してよい:
・1週間の期間内における20%以上の体重の減少。
・摂食、飲水、運動性、および排尿および/または排便する能力等の、正常な生理的機能を阻害するマウス。
・測径器で測定する際に2,000mgの最大寸法を超える腫瘍。
・潰瘍化した腫瘍、あるいは出血するか滲出液を生じる腫瘍。
・過度の体重減少(>20%)に繋がる長期で過度の下痢。
・持続性喘鳴および呼吸窮迫。
・衰弱、猫背姿勢、麻痺/不全麻痺、膨隆腹、潰瘍化、膿瘍、発作、および/または出血等の、臨床観察で定義する際の長期または過度の痛みまたは苦痛。
投薬の完了後に、マウスを追加の2週間にわたり観察し、腫瘍の再成長について検査した。
【0167】
屠殺:全ての動物を本研究の終了時に屠殺することになり、その腫瘍を採取することになる。その腫瘍の一部をOCT溶液中で凍結させることになり、その他部をホルマリン中で固定することになる。凍結および固定した組織を、出資者に送ることになる。全ての動物実験を、トキシコン社(Toxikon Corporation)(マサチューセッツ州、ベッドフォード)で行った。
【0168】
結果:8−HHA処理の1日目に、マウスを一群当たり10匹のマウスを有する3つの群に無作為に分けた:媒介物のみの対照群(C1)、10mg/kg体重の8−HHA(T1)、および100mg/kg体重の8−HHA(T2)。群T1およびT2は両方とも、8−HHA処理の4日目からマウスにおける腫瘍成長を著しく阻害した。対照的に、8−HHAで処理した群(T1)において、腫瘍量は、対照群と比較して、44.5%、49.8%、72.3%、77.7%、および78.4%(それぞれ、4日目、9日目、11日目、16日目、および18日目に)減少した。T2群において、腫瘍量は、56.3%、61.1%、81.1%、87.9%、および88.3%(それぞれ、4日目、9日目、11日目、16日目、および18日目に)減少した(図13および表8を参照されたい)。
【0169】
これらの結果は、本研究で用いた2つの濃度の8−HHAが、PLC−5細胞を用いる我々のインビトロ実験から明らかなように、抗増殖効果およびプロアポトーシス効果により、肝臓癌マウスモデルにおいて腫瘍成長を減少させることを示している。8−HHAは、そのため、標準的な化学療法計画の非常に効果的で、安全で、かつ安価な補助薬であることが判明し得る。
(*S:対照群と比較して有意、N=10/群)
平均±SEMの腫瘍量(PLC−5)
【表8】
【0170】
実施例21:小細胞肺癌異種移植マウスモデル:
本研究の目的は、HOP92を用いて、肺癌における8−HHAの抗腫瘍効能を決定することであった。
【0171】
動物および飼育:生後5〜6週間でかつ16〜20gmの体重がある、30匹のBABL/cの、非妊娠で、かつ未経産の雌ヌードマウスを本研究で用いた。薬物候補の抗腫瘍効能試験のための異種移植の研究において歴史的に用いられてきたために、BALB/c nu/nuのマウスを用いた。実際の試験用と同じ条件下で、マウスを最小で5日にわたり順化させた。30〜70%の室内相対湿度、時間当たり最小で10回の交換という時間当たりの空気交換、12時間の明暗周期の光曝露、全領域蛍光灯を用いて、マウスを68±5°Fの室温で収容した。ポリカーボネートでできている換気したマイクロマイクロアイソレーターケージ内に、マウスを集団で収容した。マウスは、高圧滅菌した実験室等級の寝藁を用いており、放射線照射したペレットおよび高圧滅菌した水を自由に提供された。飼料、水、または寝藁には、試験データに干渉すると予想される既知の汚染物は全くなかった。実験室および動物室を、出入り制限施設として維持した。
【0172】
注射および投薬の経路:8−HHAを、腹腔内(IP)注射を通じて投与した。8−HHAは不十分に水溶性であり、それ故、100mMに対応する27.2mg/mlの濃度でナノ脂質分散溶液(ePharse、スイス、バーゼル)中に懸濁する。MTD検定(予備データ)は、マウスが20mg/kgという最大用量に耐えたことを示した。
【0173】
実験設計:
動物の準備
腫瘍誘発:細胞株HOP92を、実施例20にて上記で検討したように、ヴァナス・オンコロジー(Vanas Oncology)での推奨仕様に従って培養した。血球計を用いたトリパンブルー生存能試験を用いて、細胞をトリプシン処理および計数した。血球計の象限における細胞計数を細胞/mL値に換算し、これにより、マウス1匹につき適切な数の細胞の単離が可能になることになる。腫瘍細胞(5×106細胞/マウス)の懸濁液を含有する0.2mLの50%のRPMI/50%のMatrigel(商標)の混合物を、各マウスに右側腹部内に皮下接種した。十分に確定するまで、細胞を週に2回観察した。式:腫瘍重量(mg)=(a×b2/2)(式中、「b」はミリメートルでの最小直径であり、「a」は最大直径である)を用いて、腫瘍量を算出した。
【0174】
動物の割り当て:確定した腫瘍がおおよそ50〜100mgの平均算出量に到達した時点で、一群当たりの腫瘍の大きさの変動性を低減するために、マウスを適切なソフトウェアを用いて無作為に処理群とした。
【0175】
投薬前の手順:1日目における投薬より前に、順化させた動物を秤量し、毒性の臨床的兆候について観察した。
【0176】
用量の管理:2つの用量の1および10mg/kgを投与した。第1の投薬日が1日目である。1日目には、8−HHA、および媒介物の注射を、下記の表9における研究設計に従って投与した。
【表9】
【0177】
投薬後の手順:処理に続いて、腫瘍およびマウスの体重の測定結果を週に2回記録し、総括的観察を毎日少なくとも1回行った。触知可能でない腫瘍を有するマウスを、完全退縮と考えた。マウス死亡率の百分率および死期を、本研究における全ての群について記録した。下記の判定基準のうち1つ以上を満たす場合に、動物を瀕死と定義し、屠殺してよい:
・1週間の期間内における20%以上の体重の減少。
・摂食、飲水、運動性、および排尿および/または排便する能力等の、正常な生理的機能を阻害するマウス。
・測径器で測定する際に2,000mgの最大寸法を超える腫瘍。
・潰瘍化した腫瘍、あるいは出血するか滲出液を生じる腫瘍。
・過度の体重減少(>20%)に繋がる長期で過度の下痢。
・持続性喘鳴および呼吸窮迫。
・衰弱、猫背姿勢、麻痺/不全麻痺、膨隆腹、潰瘍化、膿瘍、発作、および/または出血等の、臨床観察で定義する際の長期または過度の痛みまたは苦痛。
投薬の完了後に、マウスを追加の2週間にわたり観察し、腫瘍の再成長について検査した。
【0178】
屠殺:全ての動物を本研究の終了時に屠殺することになり、その腫瘍を採取することになる。その腫瘍の一部をOCT溶液中で凍結させることになり、その他部をホルマリン中で固定することになる。凍結および固定した組織を出資者に送った。
【0179】
結果:8−HHA処理の1日目に、マウスを一群当たり10匹のマウスを有する3つの群に無作為に分けた:媒介物のみの対照群(C1)、10mg/kg体重の8−HHA(T1)、および100mg/kg体重の8−HHA(T2)。群T1およびT2は両方とも、8−HHA処理の16日目からマウスにおける腫瘍成長を著しく阻害した。腫瘍量は、対照群において、16日目および18日目にそれぞれ214.6±38.65mm3および258.4±64.96mm3であった。対照的に、8−HHAで処理した群(T1)において、腫瘍量は、16日目および18日目にそれぞれ88.78±28.61mm3および82.02±25.93mm3であり、16日目および18日目での32.63mm3の腫瘍における58.6%および68.2%の減少にそれぞれ相当し、腫瘍量における56.5%および73.1%の減少に相当した(表10および図14を参照されたい)。
【表10】
*S:対照群と比較して有意
平均±SEMの腫瘍量(HOP−92)(N=10/群)
【0180】
結論として、本研究で用いた2つの濃度の8−HHAが、HOP92細胞を用いる我々のインビトロ実験から明らかなように、抗増殖効果およびプロアポトーシス効果により、肺癌マウスモデルにおいて腫瘍成長を減少させた。8−HHAは、そのため、標準的な化学療法計画の非常に効果的で、安全で、かつ安価な補助薬であることが判明し得る。
【0181】
本発明は特定の実施形態を参照して開示されてきたが、本発明の真の趣旨および範囲を逸脱することなく、他の当業者が本発明の他の実施形態および変化形を考案することができることは明白である。
【特許請求の範囲】
【請求項1】
ヒカゲノカズラ(Lycopodium clavatum)からの抽出物中に生じる化合物を含む組成物の対象者への投与を含む、前記対象者における癌を治療する方法。
【請求項2】
前記組成物が、BCP−21抽出物画分を含む、請求項1に記載の方法。
【請求項3】
前記組成物が、BCP−21抽出物画分からの単離した化合物を含む、請求項1に記載の方法。
【請求項4】
前記組成物が、単離した8−ヒドロキシヘキサデカン酸を含む、請求項1に記載の方法。
【請求項5】
前記組成物が、合成の8−ヒドロキシヘキサデカン酸を含む、請求項1に記載の方法。
【請求項6】
前記組成物が、8−ヒドロキシ−パルミチン酸のS−エナンチオマーを含む、請求項1に記載の方法。
【請求項7】
追加の治療薬剤の同時投与を含む、請求項1に記載の方法。
【請求項8】
前記追加の治療薬剤が抗癌剤である、請求項7に記載の方法。
【請求項9】
前記癌がカスパーゼ3により媒介される癌である、請求項1に記載の方法。
【請求項10】
前記癌が肝細胞性である、請求項1に記載の方法。
【請求項11】
前記癌が肝臓癌である、請求項1に記載の方法。
【請求項12】
前記癌が肺癌である、請求項1に記載の方法。
【請求項13】
前記肺癌が非小細胞肺癌である、請求項12に記載の方法。
【請求項14】
前記肺癌がHOP−92癌細胞に関わる、請求項12に記載の方法。
【請求項15】
前記癌が白血病である、請求項1に記載の方法。
【請求項16】
ヒカゲノカズラ(Lycopodium clavatum)の抽出物中に生じる化合物を含むキット。
【請求項17】
前記化合物が、8−ヒドロキシヘキサデカン酸である、請求項16に記載のキット。
【請求項18】
癌治療を必要としていると確認される対象者に前記化合物を投与するための取扱説明書をさらに備える、請求項16に記載のキット。
【請求項19】
追加の抗癌剤を投与するための取扱説明書をさらに備える、請求項16に記載のキット。
【請求項20】
8−ヒドロキシヘキサデカン酸および医薬的に許容可能な担体を含む医薬組成物。
【請求項21】
対象者における癌の治療の際の使用のための、8−ヒドロキシヘキサデカン酸を含む組成物。
【請求項22】
前記癌の治療に有用な薬物の製造における、8−ヒドロキシヘキサデカン酸の使用。
【請求項23】
有効量の8−ヒドロキシヘキサデカン酸、またはその塩、前駆薬、前駆薬塩、溶媒和物、水和物、および多形体を含む組成物を対象者に投与することを含む、前記対象者における癌を治療する方法。
【請求項1】
ヒカゲノカズラ(Lycopodium clavatum)からの抽出物中に生じる化合物を含む組成物の対象者への投与を含む、前記対象者における癌を治療する方法。
【請求項2】
前記組成物が、BCP−21抽出物画分を含む、請求項1に記載の方法。
【請求項3】
前記組成物が、BCP−21抽出物画分からの単離した化合物を含む、請求項1に記載の方法。
【請求項4】
前記組成物が、単離した8−ヒドロキシヘキサデカン酸を含む、請求項1に記載の方法。
【請求項5】
前記組成物が、合成の8−ヒドロキシヘキサデカン酸を含む、請求項1に記載の方法。
【請求項6】
前記組成物が、8−ヒドロキシ−パルミチン酸のS−エナンチオマーを含む、請求項1に記載の方法。
【請求項7】
追加の治療薬剤の同時投与を含む、請求項1に記載の方法。
【請求項8】
前記追加の治療薬剤が抗癌剤である、請求項7に記載の方法。
【請求項9】
前記癌がカスパーゼ3により媒介される癌である、請求項1に記載の方法。
【請求項10】
前記癌が肝細胞性である、請求項1に記載の方法。
【請求項11】
前記癌が肝臓癌である、請求項1に記載の方法。
【請求項12】
前記癌が肺癌である、請求項1に記載の方法。
【請求項13】
前記肺癌が非小細胞肺癌である、請求項12に記載の方法。
【請求項14】
前記肺癌がHOP−92癌細胞に関わる、請求項12に記載の方法。
【請求項15】
前記癌が白血病である、請求項1に記載の方法。
【請求項16】
ヒカゲノカズラ(Lycopodium clavatum)の抽出物中に生じる化合物を含むキット。
【請求項17】
前記化合物が、8−ヒドロキシヘキサデカン酸である、請求項16に記載のキット。
【請求項18】
癌治療を必要としていると確認される対象者に前記化合物を投与するための取扱説明書をさらに備える、請求項16に記載のキット。
【請求項19】
追加の抗癌剤を投与するための取扱説明書をさらに備える、請求項16に記載のキット。
【請求項20】
8−ヒドロキシヘキサデカン酸および医薬的に許容可能な担体を含む医薬組成物。
【請求項21】
対象者における癌の治療の際の使用のための、8−ヒドロキシヘキサデカン酸を含む組成物。
【請求項22】
前記癌の治療に有用な薬物の製造における、8−ヒドロキシヘキサデカン酸の使用。
【請求項23】
有効量の8−ヒドロキシヘキサデカン酸、またはその塩、前駆薬、前駆薬塩、溶媒和物、水和物、および多形体を含む組成物を対象者に投与することを含む、前記対象者における癌を治療する方法。
【図1】

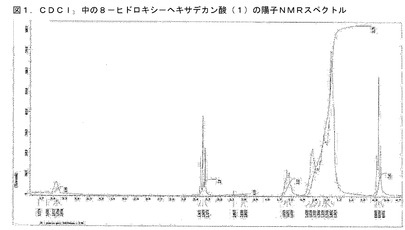
【図2】
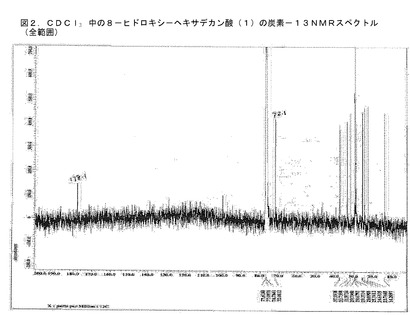
【図3】

【図4】
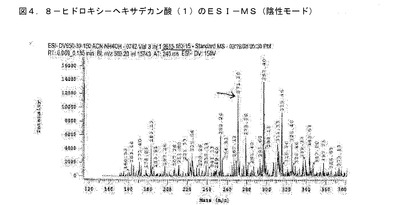
【図5】
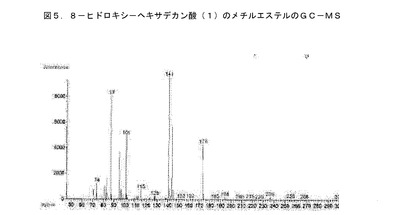
【図6】
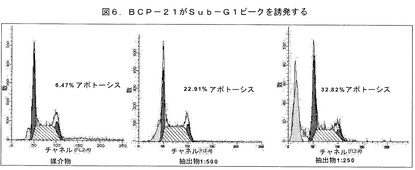
【図7】

【図8】
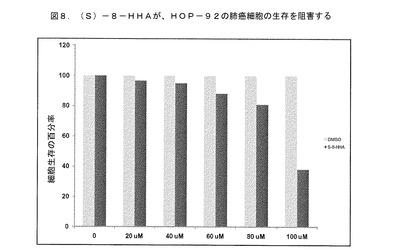
【図9】

【図10】

【図11】
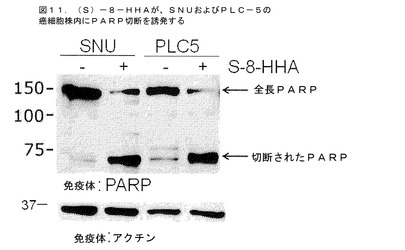
【図12】
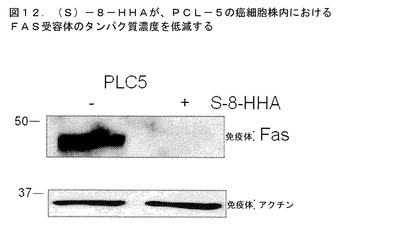
【図13】

【図14】
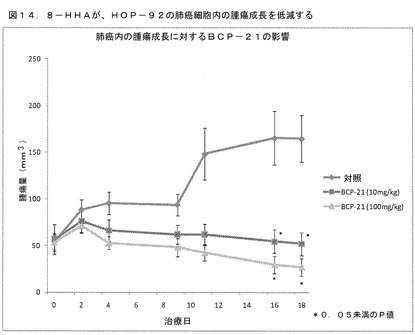
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公表番号】特表2012−515171(P2012−515171A)
【公表日】平成24年7月5日(2012.7.5)
【国際特許分類】
【出願番号】特願2011−545546(P2011−545546)
【出願日】平成22年1月14日(2010.1.14)
【国際出願番号】PCT/US2010/021046
【国際公開番号】WO2010/083311
【国際公開日】平成22年7月22日(2010.7.22)
【出願人】(511168246)
【Fターム(参考)】
【公表日】平成24年7月5日(2012.7.5)
【国際特許分類】
【出願日】平成22年1月14日(2010.1.14)
【国際出願番号】PCT/US2010/021046
【国際公開番号】WO2010/083311
【国際公開日】平成22年7月22日(2010.7.22)
【出願人】(511168246)
【Fターム(参考)】
[ Back to top ]