
組成物
実質的に非晶質のタキサン、担体及び界面活性剤を含む経口投与のための固体医薬タキサン組成物であって、該実質的に非晶質のタキサンは溶媒蒸発法により、及び特別には、スプレードライ法により調製される組成物。本組成物の調製方法及び本組成物の使用も又含まれる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬組成物に関する。詳細には、排他的ではないが、本発明は、腫瘍性疾患を治療するための組成物に関する。
【背景技術】
【0002】
経口形にある薬物の投与は、多数の利点を提供する。経口抗癌薬、例えば、5−フルオロウラシル(5−FU)プロドラッグ(例、カペシタビン)及びシグナル伝達経路又は血管新生プロセスを妨害する薬物のアベイラビリティは、治療が最適に効果的であるように長期的に適用されなければならない場合には重要である(非特許文献1)。さらに経口薬は、通院患者ベース又は自宅で投与することができ、便宜性及び患者のクオリティ・オブ・ライフを増加させ、入院を減少させることによってコストを低下させる可能性がある(非特許文献2)。このため、抗癌薬を経口的に投与しようとすることは有益である。
【0003】
一般に、薬物の経口投与は、便宜的且つ実際的である。しかし抗癌薬の大多数は、残念なことに低く変動性の経口バイオアベイラビリティを有する(非特許文献1)。典型的な例は、広汎に使用されているタキサン類であるドセタキセル及びパクリタキセルであり、これらは10%未満の経口バイオアベイラビリティを有する(非特許文献3、4)。より高いバイオアベイラビリティを備える数種の他の抗癌剤は、より高い変動性を示す。例には、トポイソメラーゼI阻害剤、ビンカアルカロイド類、及びミトキサントロンが含まれる(非特許文献1、5、6)。狭い治療濃度域を考えると、変動性のバイオアベイラビリティは、予測できない毒性又は治療血漿レベルが達成されない場合に低下した有効性を生じさせることがある。Hellriegel et al.は試験において、経口投与後の血漿レベルが、一般には静脈内投与後より変動性であることを証明した(非特許文献7)。適正な経口バイオアベイラビリティは、薬物曝露期間が抗癌療法の主要決定因子である場合には重要である(非特許文献8)。適正な経口バイオアベイラビリティは、局所毒性を生じる可能性がある消化管内での高い局所薬物濃度を防止するためにも又重要である。
【0004】
このため、先行技術に関連する問題は、低い変動性とともに高いバイオアベイラビリティを有するタキサンを含む経口組成物を開発することができなかった点である。経口パクリタキセルを用いた臨床試験(例、非特許文献3)及び経口ドセタキセルを用いた臨床試験(例、非特許文献9)が本発明者らによって実施されたが、これらの試験ではタキサン静脈内投与調製物(さらに賦形剤、例えばCremophor EL及びエタノール、又はポリソルベート80及びエタノールを含有する)が経口摂取された。悪心、嘔吐及び不快な味覚が患者によって頻回に報告された。
【0005】
Chen et al.(非特許文献13)は、そのバイオアベイラビリティを改善するために抗癌薬ドセタキセルの溶解度の改善を試みる実験を実施した。Chen et al.は、ドセタキセルの固体分散体を様々な担体、つまりグリセリルモノステアレート、PVP−K30又はポロキサマー188とともに使用することを試みた。Chen et al.は、ポロキサマー188が、5:95のドセタキセル 対 ポロキサマー比を使用した場合に、ドセタキセルの溶解度を20分後(標準溶解試験において)には約3.3μg/mLへ、そしては約120分後には最高約5.5μg/mLへ増加させることを見いだした(Chenの論文における図7を参照)。PVP−K30は、ドセタキセルの溶解度を20分後には約0.8μg/mLへ、そして約300分後には最高約4.2μg/mLにしか増加させなかった(図2を参照)。グリセロールモノステアレートは、ドセタキセルの溶解度を全く増加させなかった。そこで、ドセタキセルの溶解度及び溶解速度は、特に高いレベルには増加させられなかった。優れた経口バイオアベイラビリティを達成するためには、薬物は、最初の約0.5〜1.5時間以内に溶液中に吸収のために利用できる十分に高い薬物量が存在するように、相当に高い溶解度及び溶解速度を有していなければならない。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Demario MD, Ratain MJ. J Clin Oncol 1998;16:2557-2567.
【非特許文献2】Liu G, Franssen E, Fitch MI et al. J Clin Oncol 1997;15:110-115.
【非特許文献3】Meerum Terwogt JM, Beijnen JH, ten Bokkel Huinink WW et al. Lancet 1998;352:285.
【非特許文献4】Sparreboom A, van Asperen J, Mayer U et al. Proc Natl Acad Sci USA 1997;94:2031 -2035.
【非特許文献5】Kuhn J, Rizzo J, Eckhardt J et al. Proc Am Soc Clin Oncol 1995;14:474.
【非特許文献6】Schellens JHM, Creemers GJ, Beijnen JH et al. Br J Cancer 1996;73:1268-1271.
【非特許文献7】Hellriegel ET, Bjornnson TD, Hauck WW. Clin Pharmacol Ther 1996;60:601 -607.
【非特許文献8】Huizing MT, Giaccone G, van Warmerdam LJC et al. J Clin Oncol 1997;15:317-329.
【非特許文献9】Malingre MM, Richel DJ, Beijnen JH et al. J Clin Oncol 2001;19:l160-1166.
【非特許文献10】Chen J, et al. Drug Development and Industrial Pharmacy 2008;34:588-594.
【発明の概要】
【発明が解決しようとする課題】
【0007】
第1態様では、本発明は、実質的に非晶質のタキサン、親水性担体及び界面活性剤を含む経口投与のための固体医薬組成物であって、該非晶質タキサンが溶媒蒸発法により調製される固体医薬組成物を提供する。
【課題を解決するための手段】
【0008】
本発明の組成物によって提供される利点は、タキサンの溶解度、タキサンの溶解速度及び/又はタキサンが結晶化し始める前に溶液中に留まっている時間量が驚くべき程度まで増加させられることにある。これらの因子は、タキサンのバイオアベイラビリティの有意な増加を結果として生じさせる。これは、少なくとも一部には、タキサンが他の方法により製造される真に非晶質である可能性が低い疑似非晶質タキサンと比較してより非晶質状態にあることに起因すると考えられる。結晶性タキサン類は、極めて低い溶解度を有する。
【0009】
担体は、タキサンを非晶質状態に維持することに役立つ。さらに、タキサンが水性媒体中に配置されると、担体は、タキサンを溶液中で過飽和状態に維持することに役立つ。これは、タキサンが結晶化するのを停止させる、又はタキサンが溶液中で結晶化し始める前の時間の長さを増加させる。このため、タキサンの溶解度及び溶解速度は高いままとなる。さらに、担体は本組成物に優れた物理的及び化学的安定性を与える。これはタキサンの分解を防止するのに役立ち、さらに非晶質タキサンが固体状態において経時的に実質的により結晶性構造へ変化するのを防止するのにも役立つ。優れた物理的安定性は、タキサンの溶解度が高いままとなることを保証する。
【0010】
界面活性剤も又、水性媒体中に配置されるとタキサンを非晶質状態で維持することに役立ち、そして驚くべきことに、非晶質タキサン及び担体を含む組成物に比較してタキサンの溶解度を実質的に増加させる。
【0011】
用語「実質的に非晶質」は、タキサン分子の長距離に渡る位置の列が殆ど又は全くあってはならないことを意味している。分子の大多数は、ランダムに配向されていなければならない。完全に非晶質の構造は決して長距離に渡る列を有しておらず、それが一体何であれ、結晶構造を含有していない:これは結晶性固体と正反対である。しかし、一部の固体については、完全に非晶質の構造を得ることは困難なことがある。このため、多数の「非晶質」構造は完全に非晶質ではなく、所定量の長距離に渡る列又は結晶性を依然含有している。例えば、固体は主として非晶質であるが結晶性構造のポケットを有する、又は極めて小さな結晶を含有することがあるので、真の非晶質と近似している。このため、用語「実質的に非晶質」は、一部の非晶質構造を有するが、一部の結晶性構造も又同様に有する固体を含んでいる。実質的に非晶質のタキサンの結晶化度は、50%未満でなければならない。好ましくは、実質的に非晶質のタキサンの結晶化度は、40%未満、一層より好ましくは30%未満、なおより好ましくは25%未満、さらにより好ましくは20%未満、さらにより好ましくは15%未満、一層より好ましくは12.5%未満、一層より好ましくは10%未満、一層より好ましくは7.5%未満、より一層好ましくは5%未満、及び最も好ましくは2.5%未満である。結晶性タキサン類は低溶解度を有するので、実質的に非晶質のタキサンの結晶化度が低いほど、実質的に非晶質のタキサンの溶解度は改善される。
【0012】
実質的に非晶質のタキサンは、任意の適切な溶媒蒸発法を使用して調製できる。適切な溶媒蒸発法は、例えば、[18]に記載されたスプレードライ法及び真空乾燥法である。好ましくは、溶媒蒸発法は、スプレードライ法である。驚くべきことに、溶媒蒸発法、特別にはスプレードライ法を使用して非晶質のタキサンを調製する工程は、他の方法を使用して調製された組成物と比較して特別に優れた溶解度、溶解速度を有し、及び/又は結晶化を開始する前により長時間にわたり溶液中に留まる組成物を生じさせることが見いだされた。これは、他の方法に比較してより非晶質のタキサンを製造する溶媒蒸発法に起因すると考えられる。
【0013】
経口投与のための組成物は、固体形態にある。本固体組成物は、タキサンが実質的に非晶質状態にある限り、任意の適切な形態にあってよい。例えば、本組成物は、非晶質タキサン、担体及び界面活性剤の物理的混合物を含むことができる。所定の実施形態では、担体及び/又は界面活性剤も又実質的に非晶質の状態にある。好ましくは、タキサン及び担体は、固体分散体の形態にある。用語「固体分散体」は、当業者には周知であり、タキサンが担体内で部分的に分子状で分散していることを意味する。より好ましくは、タキサン及び担体は、固溶体の形態にある。用語「固溶体」は、当業者には周知であり、タキサンが担体内で実質的に完全に分子状で分散していることを意味する。固溶体は、本質的に固体分散体より非晶質であると考えられる。固体分散体及び固溶体を調製する方法は、当業者には周知である[11,12]。これらの方法を使用すると、タキサン及び担体はどちらも非晶質状態にある。タキサン及び担体が固体分散体若しくは固溶体の形態にある場合は、タキサンの溶解度及び溶解速度は非晶質タキサン及び担体の物理的混合物より大きい。タキサンが固体分散体若しくは固溶体中に場合は、タキサンは自然に疑似非晶質タキサンに比較してより非晶質状態にあると考えられる。これは改善された溶解度及び溶解を生じさせると考えられる。固体分散体若しくは固溶体の結晶化度は、50%未満でなければならない。好ましくは、固体分散体若しくは固溶体の結晶化度は、40%未満、一層より好ましくは30%未満、なおより好ましくは25%未満、さらにより好ましくは20%未満、さらにより好ましくは15%未満、一層より好ましくは12.5%未満、一層より好ましくは10%未満、一層より好ましくは7.5%未満、より一層好ましくは5%未満、及び最も好ましくは2.5%未満である。
【0014】
タキサン及び担体が固体分散体中にある場合は、界面活性剤は固体分散体若しくは固溶体との物理的混合物中にあってもよい。しかし好ましくは、本組成物は、固体分散体、又はより好ましくは固溶体の形態にあるタキサン、担体及び界面活性剤を含んでいる。固体分散体若しくは固溶体中に全3種の構成成分を有する利点は、それが溶解度及び溶解速度における同一の改善を達成するためにより少量の界面活性剤の使用を可能にすることである。好ましくは、タキサン、担体及び界面活性剤は、全部が実質的に非晶質状態にある。
【0015】
タキサン及び担体;又はタキサン、担体及び界面活性剤の固体分散体若しくは固溶体は、上述したような任意の適切な溶媒蒸発法を使用して製造することができる。好ましくは、固体分散体若しくは固溶体は、スプレードライ法により調製される。驚くべきことに、溶媒蒸発法、特別にはスプレードライ法を使用して固体分散体若しくは固溶体を調製する工程は、特別に優れた溶解度特性を有する組成物を生じさせるので、タキサンは他の方法を使用して調製された組成物と比較して優れた溶解度、溶解速度を有し、及び/又は結晶化を開始する前により長時間にわたり溶液中に留まることが見いだされた。これは、他の方法に比較してその中で全構成成分がより非晶質状態にある組成物を製造する溶媒蒸発法に起因すると考えられる。他の方法では、構成要素の1つ又はそれ以上が依然として一部の結晶の性質を有する可能性が見出され、この性質が溶液中での減少した溶解度特性及び/又は物理的安定性を有する組成物を生じさせると考えられる。
【0016】
1つの実施形態では、本組成物は、経口投与用のカプセル中に含有させることができる。カプセルは、多数の様々な方法で充填することができる。例えば、非晶質タキサンは、スプレードライをして、粉末化し、担体及び界面活性剤と組み合わせ、そして次にカプセル中に分注することにより調製することができる。
【0017】
又別の実施形態では、本組成物は、錠剤に圧縮することができる。例えば、非晶質タキサンは、スプレードライをして、粉末化し、担体及び界面活性剤(及び任意で他の賦形剤)と組み合わせ、次に適切な量を錠剤に圧縮することにより調製することができる。
【0018】
タキサン類は、イチイ属(Taxus(イチイ))の植物を起源とするジテルペン化合物である。しかし一部のタキサン類は、現在では合成的又は半合成的に製造されている。タキサン類は、細胞分裂を停止させることによって細胞増殖を阻害するので、癌の治療に使用されている。タキサン類は、微小管形成を中断させることによって細胞分裂を停止させる。タキサン類は、血管新生阻害剤としても作用できる。本明細書で使用する用語「タキサン」には、自然に生成されようと人工的に製造されようと、チューブリンに結合する全てのジテルペンタキサン類、機能的誘導体及び医薬上許容される塩若しくはエステルが含まれる。
【0019】
生理化学的特性を修飾するための基を含有するタキサン類の誘導体も又本発明の範囲内に含まれる。そこで、改善又は修飾された溶解度特性を備える、タキサン類のポリアルキレングリコール(例えば、ポリエチレングリコール)又はサッカライドコンジュゲートが含まれる。
【0020】
本組成物のタキサンは、上記に定義した任意の適切なタキサンであってよい。好ましいタキサン類は、ドセタキセル、パクリタキセル、BMS−275183、それらの機能的誘導体及びそれらの医薬上許容される塩又はエステルである。BMS−275183は、パクリタキセルのC−3’−t−ブチル−3’−N−t−ブチルオキシカルボニルアナログである[10]。より好ましくは、タキサンは、ドセタキセル、パクリタキセル、それらの機能的誘導体及びそれらの医薬上許容される塩若しくはエステルから選択される。
【0021】
本組成物の親水性担体は、pH7.4の水性媒体中に少なくとも部分的に溶解できる、及び/又はそのような水性媒体中で膨潤又はゲル化できる有機化合物である。担体は、タキサンが本組成物中で非晶質状態に留まることを保証し、タキサンの溶解度及び溶解速度を増加させる任意の適切な親水性担体であってよい。好ましくは、担体はポリマー性のものである。好ましくは、担体は:ポリビニルピロリドン(PVP);ポリエチレングリコール(PEG);ポリビニルアルコール(PVA);クロスポビドン(PVP−CL);ポリビニルピロリドン−ポリビニルアセテートコポリマー(PVP−VA);セルロース誘導体、例えばメチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルエチルセルロース、ヒドロキシプロピルメチルセルロース(HPMC)、セルロースアセテートフタレート及びヒドロキシプロピルメチルセルロースフタレート;ポリアクリレート類;ポリメタクリレート類;糖類、ポリオール類及びそれらのポリマー類、例えばマンニトール、スクロース、ソルビトール、デキストロース及びキトサン;並びにシクロデキストリン類から選択される。より好ましくは、担体は、PVP、PEG及びPVP−VA、から選択され、一層より好ましくは、担体は、PVP及びPVP−VAから選択される。1つの実施形態では、担体は、PVPである。又別の実施形態では、担体は、PVP−VAである。
【0022】
担体がPVPである場合は、担体として機能し、タキサンを非晶質状態で維持するのに役立つ任意の適切なPVP[16]であってよい。例えば、PVPは、PVP−K12、PVP−K15、PVP−K17、PVP−K25、PVP−K30、PVP−K60、PVP−K90及びPVP−K120から選択されてよい。好ましくは、PVPは、PVP−K30、PVP−K60及びPVP−K90から選択される。最も好ましくは、PVPは、PVP−K30である。
【0023】
担体がPEGである場合は、担体として機能し、タキサンを非晶質状態で維持するのに役立つ任意の適切なPEG[16]であってよい。例えば、PEGは、PEG1500、PEG6000及びPEG20000から選択することができる。好ましくは、PEGは、PEG1500及びPEG6000から選択され、最も好ましくは、PEGは、PEG1500である。
【0024】
担体がPVP−VAである場合は、担体として機能し、タキサンを非晶質状態で維持するのに役立つ任意の適切なPVP−VA[16]であってよい。例えば、PVP−VAは、PVP−VA64であってよい。
【0025】
本組成物は、非晶質タキサンと比して、担体が非晶質タキサンをその非晶質状態に維持するように任意の適切な量の担体を含有することができる。好ましくは、担体に対するタキサンの重量比は、約0.01:99.99(w/w)〜約75:25(w/w)である。より好ましくは、担体に対するタキサンの重量比は、約0.01:99.99(w/w)〜約50:50(w/w)、一層より好ましくは約0.01:99.99(w/w)〜約40:60(w/w)、より一層好ましくは約0.01:99.99(w/w)〜約30:70(w/w)、一層より好ましくは約0.1:99.9(w/w)〜約20:80(w/w)、なお一層より好ましくは約1:99(w/w)〜約20:80(w/w)、一層より好ましくは約2.5:97.5(w/w)〜約20:80(w/w)、さらにより好ましくは約2.5:97.5(w/w)〜約15:85(w/w)、一層より好ましくは約5:95(w/w)〜約15:85(w/w)であり、及び最も好ましくは約10:90(w/w)である。
【0026】
界面活性剤は、任意の適切な医薬上許容される界面活性剤であってよく、そのような界面活性剤は当業者には周知である。例えば、界面活性剤は、アニオン性、カチオン性又は非イオン性界面活性剤であってよい。好ましくは、界面活性剤は、カチオン性又はアニオン性界面活性剤である。より好ましくは、界面活性剤は、アニオン性界面活性剤である。
【0027】
1つの実施形態では、界面活性剤は、好ましくは約2より大きいHLB(親水性親油性バランス)値を有する。より好ましくは、HLB値は、約4より大きく、なおより好ましくはHLB値は約10より大きく、一層より好ましくはHLB値は約14より大きく、より好ましくはHLB値は約20より大きく、一層より好ましくはHLB値は約25より大きく、より好ましくはHLB値は約30より大きく、最も好ましくはHLB値は約35より大きい。好ましくは、HLB値は、約45未満でなければならない。
【0028】
好ましくは、界面活性剤は、トリエタノールアミン、ヒマワリ油、ステアリン酸、一塩基ナトリウムホスフェート、ナトリウムシトレートジハイドレート、プロピレングリコールアルギネート、オレイン酸、モノエタノールアミン、鉱油及びラノリンアルコール類、メチルセルロース、中鎖トリグリセリド類、レシチン、加水ラノリン、ラノリン、ヒドロキシプロピルセルロース、グリセリルモノステアレート、エチレングリコールパミトステアレート(pamitostearate)、ジエタノールアミン、ラノリンアルコール類、コレステロール、セチルアルコール、セトステアリルアルコール、ヒマシ油、ナトリウムドデシルスルフェート(SDS)、ソルビタンエステル類(ソルビタン脂肪酸エステル類)、ポリオキシエチレンステアレート類、ポリオキシエチレンソルビタン脂肪酸エステル類、ポリオキシエチレンヒマシ油誘導体、ポリオキシエチレンアルキルエーテル類、ポロキサマー、グリセリルモノオレエート、ドキュセートナトリウム、セトリミド、ベンジルベンゾエート、ベンザアルコニウムクロライド、ベンゼトニウムクロライド、ヒプロメロース、非イオン性乳化ろう、アニオン性乳化ろう及びトリエチルシトレート(これらの化合物はHandbook of Pharmaceutical Excipients (4th Edition, editors:RC Rowe, PJ Sheskey, PJ Weller)の中で乳化剤及び界面活性剤として表示されている)から選択される。より好ましくは、界面活性剤は、ナトリウムドデシルスルフェート(SDS)、ソルビタンエステル類(ソルビタン脂肪酸エステル類)、ポリオキシエチレンステアレート類、ポリオキシエチレンソルビタン脂肪酸エステル類、ポリオキシエチレンヒマシ油誘導体、ポリオキシエチレンアルキルエーテル類、ポロキサマー、グリセリルモノオレエート、ドキュセートナトリウム、セトリミド、ベンジルベンゾエート、ベンザアルコニウムクロライド、ベンゼトニウムクロライド、ヒプロメロース、非イオン性乳化ろう、アニオン性乳化ろう及びトリエチルシトレート(これらの化合物はthe Handbook of Pharmaceutical Excipients (4th Edition, editors:RC Rowe, PJ Sheskey, PJ Weller)の中で界面活性剤として表示されている)から選択される。より好ましくは、界面活性剤は、ナトリウムドデシルスルフェート(SDS)、ソルビタンエステル類(ソルビタン脂肪酸エステル)、及びポリオキシエチレンソルビタン脂肪酸エステルから選択される。1つの実施形態では、界面活性剤は、セチルピリジニウムクロライド(CPC)であってよい。又別の実施形態では、界面活性剤は、SDS、CPC、ポリオキシエチレン(20)ソルビタンモノオレート(ポリソルベート80)及びポリソルビタンモノオレエートから選択される。好ましくは、界面活性剤は、SDS、CPC及びポリソルベート80から選択される。より好ましくは、界面活性剤は、SDS及びCPCから選択される。最も好ましくは、界面活性剤は、SDSである。
【0029】
本組成物中では、タキサンの溶解度及び溶解速度を改善するために任意の適切な量の界面活性剤を使用できる。好ましくは、組み合わされたタキサンおよび担体に対する界面活性剤の重量比は、約1:99(w/w)〜約50:50(w/w)、より好ましくは約1:99(w/w)〜約44:56(w/w)、一層より好ましくは約1:99(w/w)〜約33:67(w/w)、なお一層より好ましくは約2:98(w/w)〜約33:67(w/w)、一層より好ましくは約2:98(w/w)〜約17:83(w/w)、さらにより好ましくは約5:95(w/w)〜約17:83(w/w)であり、及び最も好ましくは約9:91(w/w)である。
【0030】
又は、タキサンに対する界面活性剤の重量比は、好ましくは約1:100(w/w)〜約60:1(w/w)、より好ましくは約1:50(w/w)〜約40:1(w/w)、一層より好ましくは約1:20(w/w)〜約20:1(w/w)、なお一層より好ましくは約1:10(w/w)〜約10:1(w/w)、一層より好ましくは約1:5(w/w)〜約5:1(w/w)、さらにより好ましくは約1:3(w/w)〜約3:1(w/w)、一層より好ましくは約1:2(w/w)〜約2:1(w/w)であり、及び最も好ましくは約1:1(w/w)である。
【0031】
1つの実施形態では、本組成物は、腸溶コーティングを含んでいる。任意の適切な腸溶コーティング、例えば、セルロースアセテートフタレート、ポリビニルアセテートフタレート及び適切なアクリル誘導体、例えばポリメタクリレート類を使用できる。腸溶コーティングは、胃内でのタキサンの放出を防止し、それによりタキサンの酸媒介性分解を防止する。さらに、腸溶コーティングは、タキサンが吸収される腸へのタキサンの標的化送達を可能にし、それによってその間はタキサンが溶液中で(結晶化が始まる前に)存在する限定された時間を吸収が可能である部位でのみ過ごすことを保証する。
【0032】
1つの実施形態では、本組成物は、1つ又はそれ以上の追加の医薬上活性な成分をさらに含むことができる。好ましくは、1つ又はそれ以上の追加の医薬上活性な成分は、CYP3A4阻害剤である。適切なCYP3A4阻害剤は、グレープフルーツジュース又はセイヨウオトギリソウ(St. John’s wort)(又はいずれかの成分)、リトナビル、ロピナビル又はイミダゾール化合物、例えばケトコナゾールである。好ましくは、CYP3A4阻害剤はリトナビルである。
【0033】
本組成物が1つ又はそれ以上の追加の医薬上活性な成分を含む場合は、該医薬上活性な成分は、本組成物中に物理的混合物として含めることができる。又は、医薬上活性な成分は、非晶質形態にあってよい。医薬上活性な成分は、他の非晶質成分及び/又は非晶質ではない成分との物理的混合物中での非晶質形態にあってよい。又は、医薬上活性な成分は、タキサン;タキサン及び担体;又はタキサン、担体及び界面活性剤との固体分散体若しくは好ましくは固溶体中にあってよい。追加の医薬上活性な成分が非晶質状態にある場合、又は固体分散体若しくは固溶体中にある場合は、追加の医薬上活性な成分は、溶媒蒸発法、例えば、スプレードライ法を使用して調製されなければならない。
【0034】
本組成物が錠剤形にあり、1つ又はそれ以上の追加の医薬上活性な成分を含む場合は、該1つ又はそれ以上の追加の医薬上活性な成分は、好ましくは該非晶質タキサンと同一錠剤中に、即ち他の成分との単一錠剤中にある。
【0035】
本医薬組成物は、追加の医薬上許容される賦形剤、アジュバント及びビヒクルを含むことができ、それらは当業者には周知である。本発明の医薬組成物中で使用できる医薬上許容される賦形剤、アジュバント及びビヒクルには、イオン交換剤、アルミナ、アルミニウムステアレート、血清タンパク質、例えばヒト血清アルブミン、バッファー物質、例えばホスフェート、グリセリン、ソルビン酸、ソルベートカリウム、飽和植物性脂肪酸の部分グリセリド混合物、水、塩又は電解質、例えば、硫酸プロタミン、リン酸水素二ナトリウム、リン酸水素カリウム、ナトリウムクロライド、亜鉛塩、コロイダル・シリカ、マグネシウムトリシリケート及びウールファットが含まれるがそれらに限定されない。
【0036】
本医薬組成物は、カプセル剤、錠剤、散剤又は被覆顆粒剤を含むがそれらに限定されない任意の経口的に許容できる剤形で経口投与することができる。錠剤は、即時放出型、持続放出型、反復放出型又は徐放型であるように調製できる。錠剤はさらに、又は、発泡性、二重層及び/又はコーティング錠であってよい。カプセル剤は、即時放出型、持続放出型、反復放出型又は徐放型であるように調製できる。潤滑剤、例えばマグネシウムステアレートも又典型的に加えられる。カプセル形にある経口投与のためには、有用な希釈剤には、ラクトース及び乾燥コーンスターチが含まれる。錠剤及びカプセル剤のためには、添加できる医薬上賦形剤は、結合剤、充填剤、充填剤/結合剤、吸着剤、湿潤剤、錠剤分解剤、潤滑剤、流動促進剤などである。錠剤及びカプセル剤は、錠剤及びカプセル剤の外観又は特性を変化させるために、例えば、錠剤又はカプセル剤の味覚を変化させる、又は着色コーティングするためにコーティングすることができる。
【0037】
本組成物に添加できる他の医薬上許容される添加物は、当業者には周知である。
【0038】
本発明は、治療に使用するための上記の組成物をさらに提供する。
【0039】
さらに、本発明は、腫瘍性疾患の治療において使用するための上記の組成物を提供する。
【0040】
本発明により治療される腫瘍性疾患は、好ましくは充実性腫瘍である。充実性腫瘍は、好ましくは、乳房、肺、胃、結腸直腸、頭頸部、食道、肝臓、腎臓、膵臓、膀胱、前立腺、精巣、子宮頸部、子宮内膜、卵巣の癌及び非ホジキン(non-Hodgkin’s)リンパ腫(NHL)から選択される。充実性腫瘍は、より好ましくは、乳房、胃、卵巣、前立腺、頭頸部及び非小細胞肺癌から選択される。
【0041】
本発明により治療できる又別の腫瘍性疾患は、多発性骨髄腫、慢性骨髄単球性白血病(CMML)、急性骨髄性白血病(AML)及びカポジ(Kapsoi’s)肉腫である。さらに、疾患範囲には、骨髄異形成症候群(MDS)も含まれる。
【0042】
本発明は、腫瘍性疾患の治療方法であって、そのような治療を必要とする被験者への有効量の上記の組成物の投与を含む方法をさらに提供する。
【0043】
好ましくは、本方法は、ヒト被験者を治療するために使用される。
【0044】
本発明は、上記の組成物を調製する方法であって、
溶媒蒸発法を使用して非晶質タキサンを調製する工程と、
該組成物を生成するために該非晶質タキサンを親水性担体及び界面活性剤と組み合わせる工程と、を含む方法をさらに提供する。
【0045】
非晶質タキサンは、任意の適切な溶媒蒸発法、例えば、上述した方法により製造することができる。好ましくは、非晶質タキサンは、スプレードライ法により製造できる。
【0046】
非晶質タキサンの調製、並びにそれと担体及び/又は界面活性剤との組み合わせは、単一工程で実施することができ、例えば、タキサン及び担体及び/又は界面活性剤は一緒に(例えば固体分散体若しくは固溶体を形成するために)非晶質化処理にかけられる。
【0047】
好ましくは、本方法は、タキサン及び親水性担体を含む固体分散体を調製する工程と、該固体分散体を該界面活性剤と組み合わせる工程と、を含んでいる。
【0048】
より好ましくは、本方法は、タキサン、親水性担体、及び界面活性剤を含む固体分散体を調製する工程を含んでいる。
【0049】
又別の態様では、本発明は、実質的に非晶質のタキサン及び親水性担体を含む経口投与のための医薬組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製される医薬組成物を提供する。
【0050】
本発明は、実質的に非晶質のタキサン及び親水性担体を含む経口投与のための固体医薬組成物を調製する方法であって、
非晶質タキサンをスプレードライ法により調製する工程と、
該組成物を生成するために該非晶質タキサンを親水性担体と組み合わせる工程と、を含む方法をさらに提供する。
【0051】
本組成物によって提供される利点は、タキサンの溶解度、タキサンの溶解速度及び/又はタキサンが結晶化し始める前に溶液中に留まっている時間量が驚くべき程度まで増加させられることにある。これは、溶媒蒸発法が、タキサンが真に非晶質である可能性が低い非晶質タキサンを製造する他の方法に比較して一層より非晶質のタキサンを製造するからであると考えられる。本タキサンのより非晶質の性質が増加した溶解度特性を提供すると考えられる。
【0052】
本組成物の追加の任意の特徴は、非晶質タキサン、担体及び界面活性剤を含む組成物と同一である。例えば、実質的に非晶質のタキサン及び担体を含んでいる本組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製される組成物は、好ましくは界面活性剤をさらに含んでいる。タキサン、担体、タキサンの結晶化度、担体に対するタキサンの比率、タキサン及び担体の状態の好ましい実施形態などは、上記に規定した通りである。
【0053】
又別の態様では、本発明は、溶液中にタキサン、親水性担体及び界面活性剤を含む医薬組成物をさらに提供する。タキサン、担体及び界面活性剤の同一性、特性などに関連する上記の説明は、本発明のこの態様に同等に適合する。そのような組成物中では、本組成物の全3種の成分が溶液中にある。
【0054】
本医薬組成物は、被験者に投与するための飲料液の形態にあってよい。又は、本医薬組成物は、カプセル剤中に、例えば、該溶液を含有する液体充填カプセルを形成するためにゼラチンカプセル中に配置することができる。
【0055】
本組成物は、適切な溶媒中に上述した固体組成物を溶解させる工程により入手できる。このため、1つの実施形態では、本組成物は、上述した固体組成物を適切な溶媒、例えばトリアセチン中に溶解させる工程により入手可能である。1つの実施形態では、本溶媒は、水性溶媒である。
【0056】
又別の態様では、本発明は、実質的に非晶質のタキサン及び1つ又はそれ以上の医薬上許容される賦形剤を含む経口投与のための固体医薬組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製される固体医薬組成物を提供する。
【0057】
本組成物の特性及び好ましい特徴は、本発明の他の組成物について上述した通りである。
以下では本発明を添付の図面を参照しながら例としてのみ記載する。
【図面の簡単な説明】
【0058】
【図1】パクリタキセル固体分散体とパクリタキセルの物理的混合物との溶解試験の結果を示す図である(条件:900mLのWfI、37℃、75rpm)。
【図2】ナトリウムドデシルスルフェートを含む、及び含まないパクリタキセル(PCT)固体分散体カプセルの溶解試験の結果を示す図である(条件:900mLのWfI、37℃、75rpm)。
【図3】固体分散体中に組み込まれた、又はカプセルに加えられたナトリウムドデシルスルフェートを含むパクリタキセル固体分散体の溶解試験の結果を示す図である(条件:500mLのWfI、37℃、75rpm(カプセルに加えられたSDSについては100rpm))。
【図4】様々な担体を含むパクリタキセル固体分散体の溶解試験の結果を示す図である(条件:500mLのWfI、37℃、100rpm)。
【図5】様々な薬物−担体比を含むパクリタキセル/PVP−K17固体分散体の溶解試験の結果を示す図である(条件:25mLのWfI、37℃、7,200rpm)。
【図6】様々な媒体中でのパクリタキセル固体分散体の溶解試験の結果を示す図である(条件:500mLのFaSSIF(ライトグレー)、37℃、75rpm;又は500mLのSGFsp及び629mLのSIFsp、37℃、75rpm(ダークグレー))。
【図7】5種の異なる調製物のドセタキセル溶解度を示す図である(表13を参照)。A:無水ドセタキセル;B:非晶質ドセタキセル;C:無水ドセタキセル、PVP−K30及びSDSの物理的混合物;D:非晶質ドセタキセル、PVP−K30及びSDSの物理的混合物;E:非晶質ドセタキセル、PVP−K30及びSDSの固体分散体(溶解条件:±6mgのドセタキセル、25mLのWfI、37℃、720rpm)。
【図8】異なる担体を含む固体分散体のドセタキセル溶解度を示す図である(表13を参照)。E:非晶質ドセタキセル、PVP−K30及びSDSの固体分散体;F:非晶質ドセタキセル、HPβ−CD及びSDSの固体分散体(溶解条件:±6mgのドセタキセル、25mLのWfI、37℃、720rpm)。
【図9】様々な鎖長のPVPを含む固体分散体のドセタキセル溶解度を示す図である(表13を参照)。E:非晶質ドセタキセル、PVP−K30及びSDSの固体分散体;G:非晶質ドセタキセル、PVP−K12及びSDSの固体分散体;H:非晶質ドセタキセル、PVP−K17及びSDSの固体分散体;I:非晶質ドセタキセル、PVP−K25及びSDSの固体分散体;J:非晶質ドセタキセル、PVP−K90及びSDSの固体分散体(溶解条件:±6mgのドセタキセル、25mLのWfI、37℃、720rpm)。
【図10】様々な薬物負荷を含む固体分散体のドセタキセル溶解度を示す図である(表13を参照)。E:1/11ドセタキセル;K:5/7ドセタキセル;L:1/3ドセタキセル;M:1/6ドセタキセル;N:1/21ドセタキセル(溶解条件:±6mgのドセタキセル、25mLのWfI、37℃、720rpm)。
【図11】ドセタキセル及びPVP−K30の固体分散体についての文献データ(Chen et al,[13])に比較したドセタキセル、PVP−K30及びSDSの固体分散体の溶解した相対量のドセタキセルに関する溶解試験結果を示す図である。
【図12】ドセタキセル及びPVP−K30の固体分散体についての文献データ(Chen et al.[13])に比較したドセタキセル、PVP−K30及びSDSの固体分散体の溶解した絶対量のドセタキセルに関する溶解試験の結果を示す図である。
【図13】文献データ(Chen et al.[13])と比較したドセタキセルカプセル(PVP−K30+SDSを含む1カプセル当たり15mgのドセタキセル(DXT))の溶解試験の結果を示す図である。
【図14】ドセタキセル、PVP−K30及びSDSの固体分散体の溶解した絶対量に関する溶解試験の結果を示す図である。溶解試験は、模擬腸液サインパンクレアチン(Simulated Intestinal Fluid sine Pancreatin(SIFsp))中で実施した。
【図15】ドセタキセル、PVP−K30及びSDSの固体分散体の溶解した相対量に関する溶解試験の結果を示す図である。溶解試験は、模擬腸液サインパンクレアチン中で実施した。
【図16】第1サイクルにおいてドセタキセル及びリトナビルを同時に摂取した患者のドセタキセル薬物動態曲線を示す図である。第2サイクルでは、患者はドセタキセル及びリトナビルをt=0に同時に摂取し、次にt=4hに追加のブースター用量のリトナビルを摂取した。
【図17】ドセタキセルの液体調製物及び/又はドセタキセルを含む固体分散体(MODRAと呼ぶ)を摂取した患者4例の薬物動態曲線を示す図である。
【図18】ドセタキセルの固体経口調製物(MODRA)を摂取した患者に比較したドセタキセルの液体経口調製物を摂取した患者の薬物動態曲線を示す図である。
【図19】ドセタキセルの静脈内及び経口投与後の薬物動態曲線を示す図である。静脈内及び経口投与の両方をリトナビルの投与と組み合わせた。注意:計算バイオアベイラビリティは、投与された用量について補正されている。
【図20】異なるタイプの界面活性剤を含む5種のドセタキセル調製物の平均溶解曲線を示す図である(n=3)。
【図21】様々な量のSDS界面活性剤を含むドセタキセル調製物の平均溶解曲線を示す図である(n=3)。
【図22】様々な量の界面活性剤を含む4種のドセタキセル調製物の0〜10分間の平均溶解速度を示す図である(n=3)。
【図23】凍結乾燥及び結晶性ドセタキセルのX線粉末回折パターンを示す図である。
【図24】凍結乾燥及び結晶性ドセタキセルDSC(示差走査熱量測定)サーモグラムを示す図である。
【図25】結晶性及び非晶質ドセタキセルの物理的混合物のX線粉末回折パターンを示す図である。
【図26】結晶性及び非晶質ドセタキセルの物理的混合物のDSCサーモグラムを示す図である。
【図27】結晶性ドセタキセル含有量に対する、全熱流量サーモグラムにおける165℃でのピーク面積を示す図である。黒色の線は、0.990のR2を備える線形回帰線である。
【図28】凍結乾燥及び結晶性パクリタキセルのX線粉末回折パターンを示す図である。
【図29】凍結乾燥及び結晶性パクリタキセルのDSCサーモグラムを示す図である。
【図30】ドセタキセル、PVP−K30及びSDSのX線粉末回折パターンを示す図である。
【図31】PVP−K30、SDS及びドセタキセルのDSCサーモグラムを示す図である。
【図32】1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを備える物理的混合物及び固体分散体のX線粉末回折スペクトルを示す図である。
【図33】1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを備える物理的混合物及び固体分散体のDSCサーモグラムを示す図である。
【図34】凍結乾燥法及びスプレードライ法により製造された固体分散体のX線粉末回折スペクトルを示す図である。
【図35】凍結乾燥法及びスプレードライ法により製造された固体分散体のDSCサーモグラムを示す図である。
【図36】凍結乾燥法及びスプレードライ法により製造された固体分散体(n=4)の平均溶解スクリーニング曲線を示す図である。
【図37】PVP−K30(n=4)、PEG1500(n=2)、PEG6000(n=2)及びPEG20000(n=2)を含むドセタキセルの固体分散体の平均溶解スクリーニング曲線を示す図である。
【図38】PVP−K30(n=4)及びPVP−VA64(n=2)を含むドセタキセルの固体分散体の平均溶解スクリーニング曲線を示す図である。
【図39】20mgのドセタキセル錠剤(n=2)及び10mgのドセタキセルカプセル(n=6)の平均溶解曲線を示す図である。
【図40】20mgのドセタキセル錠剤(n=2)及び10mgのドセタキセルカプセル(n=6)の0〜10分間のドセタキセルの平均放出速度を示す図である。
【図41】非晶質(スプレードライ)及び結晶性リトナビルのDSCサーモグラムを示す図である。
【図42】ドセタキセル、リトナビル、PVP−K30及びSDSの組み合わせのスプレードライ法固体分散体粉末のDSCサーモグラムを示す図である。
【図43】37℃及び50RPMで1,000mLの0.1N HCl中におけるドセタキセル/リトナビル/PVP−K30/SDSカプセルの溶解プロファイルを示す図である。
【発明を実施するための形態】
【実施例1】
【0059】
パクリタキセルの経口調製物
1.1:固体分散体と物理的混合物
この実験では、SDSと混合されたパクリタキセル及びPVP−K17の固体分散体を含む組成物の溶解度及び溶解速度を無水パクリタキセル、PVP−K17及びSDSの物理的混合物と比較した。
【0060】
PVP−K17中のパクリタキセル固体分散体の5mgカプセル
PVP−K17中の20%パクリタキセルの固体分散体は、10mLのt−ブタノール中に100mgのパクリタキセル及び6.67mLの水中に400mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(条件については表1を参照)。25mgの20%パクリタキセル/PVP−K17固体分散体(=5mgのパクリタキセル)を125mgのラクトース、30mgのナトリウムドデシルスルフェート、及び30mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表2参照)。
【0061】
【表1】

【0062】
【表2】
【0063】
PVP−K17との物理的混合物中のパクリタキセルの5mgカプセル
物理的混合物は、5mgの無水パクリタキセルを20mgのPVP、125mgのラクトース、30mgのナトリウムドデシルスルフェート、及び30mgのクロスカルメロースナトリウムと混合することにより調製した。結果として生じた粉末混合物はカプセル充填した。
【0064】
【表3】
【0065】
溶解試験
両方のカプセル調製物は、75rpmの回転速度を備えるUSP2(パドル式)溶解装置内で37℃に維持した900mLの注射用水中で試験した。第1実験では、各調製物の1つのカプセルを使用した。第2実験では、各調製物の2つのカプセルを使用した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0066】
【表4】
【0067】
結果及び結論
結果は図1に示した。溶解したパクリタキセルの量は、ラベル記載内容(5及び10mg)に比較して表示した。パクリタキセルの溶解は、固体分散体中へのPVPの組み込みによって大きく改善されることは明白に見て取ることができる。溶解したパクリタキセルの最高量は、物理的混合物が使用された場合はラベル記載内容に比較して20%未満に留まる。固体分散体が使用された場合は、溶解度は約65%(5mgのパクリタキセル)又は70%超(10mgのパクリタキセル)である。10mgのパクリタキセル実験については、これは約8μg/mLの絶対溶解度に対応し、これは約15分間後に達成される。このため、固体分散体は溶解度を有意に増加させ、さらにどちらもバイオアベイラビリティにとって重要である迅速な溶解速度も提供する。
【0068】
固溶体又は固体分散体では、担体の非晶質状態は、担体及びタキサンの混合を通して可能になる。担体は、保管中並びに水性媒体内での溶解中の結晶化を防止する。
【0069】
1.2:カプセル調製物へのナトリウムドデシルスルフェートの添加
この実験ではカプセル中の界面活性剤SDSの存在又は非存在が溶解度に及ぼす作用を決定した。
【0070】
PVP−K17中の20%パクリタキセル固体分散体
固体分散体は、10mLのt−ブタノール中に100mgのパクリタキセル及び6.67mLの水中に400mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0071】
ナトリウムドデシルスルフェートを含まない5mgのパクリタキセルカプセル
25mgの20%パクリタキセル/PVP−K17固体分散体(=5mgのパクリタキセル)を125mgのラクトースと混合し、カプセル充填した(表5を参照)。
【0072】
【表5】
【0073】
ナトリウムドデシルスルフェートを含む5mgのパクリタキセルカプセル
25mgの20%パクリタキセル/PVP−K17固体分散体(=5mgのパクリタキセル)を125mgのラクトース、30mgのナトリウムドデシルスルフェート、及び30mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表6参照)。
【0074】
【表6】
【0075】
溶解試験
両方のカプセル調製物は、75rpmの回転速度を備えるUSP2(パドル式)溶解装置内で37℃に維持した900mLの注射用水中で試験した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0076】
結果及び結論
結果は図2に示した。溶解したパクリタキセルの量は、ラベル記載内容に比較して表示した(この場合には5mg)。凍結乾燥タキサン及び担体固体分散体の多孔性は、粉末形にある場合の迅速な溶解を保証するために十分に高かった(結果は示していない)。しかし粉末がカプセル中に圧縮されると、湿潤性は劇的に減少した。このため、カプセル又は錠剤中に圧縮される場合は、固体分散体を湿潤させるために界面活性剤が必要とされる。
【0077】
図2からパクリタキセルの溶解は、界面活性剤のナトリウムドデシルスルフェートの添加により大きく改善されることを明白に見て取ることができる。以前の実験は、クロスカルメロースナトリウム、より多くのラクトースの添加、又はより大きなカプセルの使用がカプセル調製物の増加した溶解速度を生じさせないことを示した。さらに又、この実験はSDSのような界面活性剤を用いると、約10〜15分間で最高溶解が達成されることを示している。
【0078】
1.3:固体分散体調製物へのナトリウムドデシルスルフェートの添加
この実験では、固体分散体へのSDSの添加が溶解度に及ぼす作用が決定された。
【0079】
PVP−K17中の40%パクリタキセル固体分散体
固体分散体は、60mLのt−ブタノール中に600mgのパクリタキセル及び40mLの水中に900mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0080】
PVP−K17及び10%ナトリウムドデシルスルフェート中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのPVP−K17及び62.5mgのナトリウムドデシルスルフェート(SDS)を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/ナトリウムドデシルスルフェート/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0081】
パクリタキセル/PVP−K17固体分散体の25mgパクリタキセルカプセル
62.5mgの40%パクリタキセル/PVP−K17固体分散体(=25mgのパクリタキセル)を160mgのラクトース、30mgのナトリウムドデシルスルフェート、及び10mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表7参照)。
【0082】
【表7】
【0083】
パクリタキセル/PVP−K17/ナトリウムドデシルスルフェート固体分散体の25mgパクリタキセルカプセル
68.75mgの40%パクリタキセル/PVP−K17/10%ナトリウムドデシルスルフェート固体分散体(=25mgのパクリタキセル)を160mgのラクトース及び10mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表8参照)。
【0084】
【表8】
【0085】
溶解試験
両方のカプセル調製物は、USP2(パドル式)溶解装置内で37℃に維持した500mLの注射用水中で試験した。回転速度は、パクリタキセル/PVP−K17/ナトリウムドデシルスルフェート固体分散体を含むカプセルについては75rpm及びパクリタキセル/PVP−K17固体分散体を含むカプセルについては100rpmに設定した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0086】
結果及び結論
結果は図3に示した。溶解したパクリタキセルの量は、ラベル記載内容に比較して表示した(この場合には25mg)。固体分散体中に組み込まれたナトリウムドデシルスルフェートを含むカプセルからのパクリタキセルの溶解は、カプセルに加えられたナトリウムドデシルスルフェートを含むカプセルからのパクリタキセルの溶解に匹敵することを明白に見て取ることができる。さらに6.25mgのナトリウムドデシルスルフェートしか固体分散体中に組み込むために使用されなかったが、30mgのナトリウムドデシルスルフェートはカプセル調製物への添加剤として使用された。これは、界面活性剤が固体分散体中に組み込まれる場合は類似の結果を達成するためにカプセル中に組み込まれる場合より少ない界面活性剤が必要とされることを示している。この実験からの又別の驚くべき結果は、両方の組成物が約26μg/mLの絶対パクリタキセル溶解度を提供し、このレベルは20〜30分間で達することである。この結果は、以前に達成されていたより高い溶解度及び迅速な溶解速度を提供する。
【0087】
1.4:担体が及ぼす影響
実施例1.4の実験において使用された固体分散体は、最初の実験が薬物負荷間で明白な差を示さなかった後に製造された。40%の薬物負荷を選択したのは、これらの調製物が上記で記載した実験において20%薬物負荷調製物と同等に機能し、1つの錠剤又はカプセルにおいてより多くのタキサンを送達する可能性を提供するからであった。
【0088】
PVP−K12中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのPVP−K12を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K12/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0089】
PVP−K17中の40%パクリタキセル固体分散体
固体分散体は、60mLのt−ブタノール中に600mgのパクリタキセル及び40mLの水中に900mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0090】
PVP−K30中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのPVP−K30を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K30の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0091】
HP−シクロデキストリン中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのHP−シクロデキストリンを溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらHP−シクロデキストリンの水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0092】
25mgのパクリタキセル固体分散体カプセル
62.5mgのパクリタキセル担体/固体分散体(=25mgのパクリタキセル)を160mgのラクトース、30mgのナトリウムドデシルスルフェート、及び10mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表9参照)。
【0093】
【表9】
【0094】
溶解試験
全カプセル調製物は、100rpmの回転速度を備えるUSP2(パドル式)溶解装置内で37℃に維持した500mLの注射用水中で試験した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0095】
結果及び結論
2〜3回の実験の平均結果は、図4に示した。溶解したパクリタキセルの量は、ラベル記載内容に比較して表示した(この場合には25mg)。PVP−K30固体分散体からのパクリタキセルの溶解は、PVP−K17固体分散体からのパクリタキセルの溶解と同等に迅速であることを明白に見て取ることができる。しかし、溶解したパクリタキセルの量は、PVP−K30固体分散体の症例において4時間の実験を通して高いままであった。
【0096】
ポリマー性担体の鎖長は、水性環境内で結晶化するための時間を決定する。
【0097】
1.5:薬物/担体比の影響
実施例1.5の実験において使用した固体分散体は、最初の実験が担体間で明白な差を示さなかった後に製造された。これらの最初の実験は、実施例1.4のより詳細な実験の前に実施された。結果として、PVP−K17がその後の実験のための担体として自由裁量で選択された。
【0098】
PVP−K17中の10%パクリタキセル固体分散体
固体分散体は、10mLのt−ブタノール中に100mgのパクリタキセル及び40mLの水中に900mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0099】
PVP−K17中の25%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に750mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0100】
PVP−K17中の40%パクリタキセル固体分散体
固体分散体は、60mLのt−ブタノール中に600mgのパクリタキセル及び6.67mLの水中に900mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0101】
PVP−K17中の75%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に83mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0102】
100%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセルを溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながら16.67mLの水に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0103】
溶解試験
およそ4mgのパクリタキセルと同等の量の固体分散体粉末を50mLのビーカー内に配置した。磁気攪拌棒及び25mLの水をビーカーに加えた。この溶液を720rpmで攪拌した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0104】
結果及び結論
2〜3回の実験の平均結果は、図5に示した。溶解したパクリタキセル(PCT)の量は、ラベル記載内容に比較して表示した(この場合にはおよそ4mg)。薬物/担体比が及ぼす影響は、図5から直ちに明らかである。パクリタキセルのピーク濃度値は、薬物/担体比と反比例していた。最高ピーク濃度は最低薬物/担体比(10%)の場合に達するが、最低ピーク濃度は最高薬物/担体比(100%)の場合に達する。さらに、10%薬物/担体比固体分散体のAUC値が最高であり、その後に25、40、75及び100%薬物/担体比固体分散体が続いた。
【0105】
薬物の量に比した担体の量が、水性環境内で結晶化するための時間を決定する。
【0106】
1.6:腸溶コーティングの存在
PVP−K17及びド10%ナトリウムドデシルスルフェート中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのPVP−K17及び62.5mgのナトリウムドデシルスルフェート(SDS)を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/ナトリウムドデシルスルフェート/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0107】
パクリタキセル/PVP−K17/ナトリウムドデシルスルフェート固体分散体の25mgパクリタキセルカプセル
68.75mgの20%パクリタキセル/PVP−K17/10%ナトリウムドデシルサルフェート固体分散体(=25mgのパクリタキセル)を160mgのラクトース及び10mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表10参照)。
【0108】
【表10】
【0109】
溶解試験
これらのカプセルに2種の異なる溶解試験を2回ずつ受けさせた。第1回試験は、ペプシンを含まない500mLの模擬胃液(SGFsp;表11を参照)中での2時間の溶解試験、その後にペプシンを含まない629mLの模擬胃液(SIFsp;表11を参照)中での2時間の溶解試験から構成される2段階式溶解試験であった。第2回試験は、500mLの絶食状態の模擬腸液(FaSSIF;表12を参照)媒体中で4時間にわたって実施した。
【0110】
両方の溶解試験は、37℃に維持し、パドル回転速度75rpmで維持した500mLの媒体を備えるUSP2(パドル式)溶解装置内で実施した。SGFsp媒体は、129mLのスイッチ媒体の添加によりSIFsp媒体に変更した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0111】
【表11】
【0112】
【表12】
【0113】
結果及び結論
結果は図6に示した。溶解したパクリタキセルの量は、ラベル記載内容に比較して表示した(この場合には25mg)。絶食状態の模擬腸液中でのパクリタキセルの溶解度は、模擬胃液(SGFsp)中においてよりおよそ20%高かった。SGFsp中に2時間置いた後の溶液中のパクリタキセルの量は、媒体を模擬腸液(SIFsp)に変更した場合は僅かにしか増加しなかった。
【0114】
腸溶コーティングは、胃内でのタキサンの放出を防止し、それにより活性成分の分解を防止する。さらに、腸溶コーティングは、タキサンが吸収される腸へのタキサンの標的化送達を可能にし、それによってその間はタキサンが溶液中で(結晶化が始まる前に)存在する限定された時間を吸収が可能である部位でのみ過ごすことを保証する。
【実施例2】
【0115】
ドセタキセルの経口調製物
材料及び方法
以下の実験において使用した調製物は、以下で略述する方法及び表13に記載した組成物に従って調製した。
【0116】
純粋無水ドセタキセル
無水ドセタキセルは、ScinoPharm社(台湾国)から入手したままで使用した。
【0117】
純粋非晶質ドセタキセル
ドセタキセルは、300mgのドセタキセル無水物を30mLのt−ブタノール中に溶解させることにより非晶質化した。ドセタキセル/t−ブタノール溶液は、絶え間なく攪拌しながら20mLのWfI(注射用水)に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronorm、サイズ1/9)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表14を参照)。
【0118】
物理的混合物
物理的混合物は、150mgのドセタキセル及び対応する量の担体及び界面活性剤(表13を参照)を乳鉢と乳棒で混合することにより調製した。
【0119】
固体分散体
固体分散体は、30mLのt−ブタノール中の300mgの無水ドセタキセル、及び20mLの注射用水中の対応する量の担体及び界面活性剤(表13を参照)を溶解させることにより入手した。ドセタキセル/t−ブタノール溶液は、絶え間なく攪拌しながら担体/界面活性剤/WfIの溶液に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronorm、サイズ1/9)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表14を参照)。
【表13】
【0120】
【表14】
【0121】
【表15】
【0122】
2.1:調製物のタイプ
第1回実験では、調製物のタイプがドセタキセルの溶解度に及ぼす影響を試験した。調製物A〜Eについて実施した溶解試験からのデータを比較した。結果は図7に示した。調製物Eは4回ずつ試験し、調製物A〜Dを2回ずつ試験した。
【0123】
結果
調製物A(純粋ドセタキセル無水物)は5分間の攪拌後におよそ12μg/mL(4.7%の全ドセタキセルが存在する)の最高濃度に到達し、15分間の攪拌後におよそ6μg/mL(2%)の平衡濃度に達する。
【0124】
調製物B(純粋非晶質ドセタキセル)は0.5分後に最高32μg/mL(13%)に到達し、10〜60分後の溶解度は調製物Aに匹敵する。
【0125】
調製物C(無水ドセタキセル、PVP−K30及びSDSの物理的混合物)は5分後におよそ85μg/mL(37%)の濃度に達する。15〜25分後に、ドセタキセル濃度は85μg/mL(37%)〜30μg/mL(12%)へ急速に低下し、その後にさらに60分後には20μg/mL(9%)へ低下する。
【0126】
調製物D(非晶質ドセタキセル、PVP−K30及びSDSの物理的混合物)は7.5分後におよそ172μg/mL(70%)の最高ドセタキセル濃度に達する。7.5〜20分後に、溶液中のドセタキセルの量は24μg/mL(10%)に低下する。60分後、19μg/mL(7%)の平衡濃度に達する。
【0127】
調製物E(非晶質ドセタキセル、PVP−K30及びSDSの固体分散体)は5分後に到達した213μg/mL(90%)の最高最大濃度を有する。10〜25分後に、溶液中のドセタキセルの量は迅速に低下し、結果として45分後に20μg/mL(8%)の平衡濃度を生じさせる。
【0128】
結論
全調製物は、最初はより高い溶解度を示したが、これらは45〜60分間の攪拌後に平衡溶解度へ減少する。溶解度の減少は、過飽和溶液の結果としてドセタキセルの結晶化によって生じる。過飽和度は、薬物の物理的状態に、即ち薬物が非晶質又は結晶性のいずれであるかに依存する。PVP−K30が担体である場合は、過飽和状態は、ドセタキセルの溶解度が急速に減少しないようにより長時間にわたり維持される。さらに、これらの結果は、非晶質ドセタキセルを使用することが無水結晶性ドセタキセルに比較してドセタキセルの溶解度を有意に増加させることを示している。さらに、非晶質ドセタキセルは、約5〜7.5分間でピークに達する相当に高い溶解速度を示す。
【0129】
この実験は、溶液中のドセタキセルの量が無水ドセタキセルをPVP−K30及びSDSと物理的混合することによって、及び非晶質ドセタキセルをPVP−K30及びSDSと物理的混合することによってより一層顕著に増加することを示した。しかし溶解度における最大増加は、PVP−K30及びSDSの固体分散体中にドセタキセルを組み込むことによって達成される。
【0130】
2.2:担体のタイプ
第2回実験では、担体のタイプがドセタキセルの溶解度に及ぼす影響を試験した。調製物E及びFについて実施した溶解試験からのデータを比較した。結果は図8に示した。調製物Eは4回ずつ試験し、調製物Fを2回ずつ試験した。
【0131】
結果
調製物E(非晶質ドセタキセル、PVP−K30及びSDSの固体分散体)は5分後に到達した213μg/mL(90%の全ドセタキセルが存在する)の最高最大濃度を有する。10〜25分後に、溶液中のドセタキセルの量は迅速に低下し、結果として45分後に20μg/mL(8%)の平衡濃度を生じさせる。
【0132】
調製物F(非晶質ドセタキセル、HPβ−CD及びSDSの固体分散体中)は、約2分後におよそ200μg/mL(81%)の最高ドセタキセル濃度に達する。5〜10分後に、溶液中のドセタキセルの量は16μg/mL(6%)の値に低下し、45分後に、11μg/mL(4%)の平衡濃度に達する。
【0133】
結論
この実験は、PVP−K30及びHPβ−CDがドセタキセルの溶解度を増加させることを示した。PVP−K30がHPβ−CDと比較して担体として使用された場合は、最大ドセタキセル濃度は僅かに高く、過飽和の状態は長時間維持されたので、ドセタキセルの溶解度は経時的に迅速には減少しなかった。さらに、ドセタキセルの沈降後に到達した平衡濃度は、HPβ−CDと比較してPVP−K30を用いた場合の方が高い。
【0134】
2.3:鎖長
第3回実験では、PVP鎖長がドセタキセルの溶解度に及ぼす影響を試験した。調製物E及びG〜Jについて実施した溶解試験からのデータを比較した。結果は図9に示した。調製物Eは4回ずつ試験し、調製物G〜Jを2回ずつ試験した。
【0135】
結果
調製物G(PVP−K12)は、5分後に206μg/mL(全ドセタキセルの77%が存在する)の最高ドセタキセル濃度に達する。5〜30分後に、溶液中のドセタキセルの量は20μg/mL(7%)の値に低下し、45分後には、ドセタキセル濃度は17μg/mL(6%)である。
【0136】
調製物H(PVP−K17)は5分後に200μg/mL(83%)の最高ドセタキセル濃度に達し、この濃度を攪拌10分間まで維持し、その後に溶液中のドセタキセルの量は15分後には44μg/mL(18%)へ、及び30分後には22μg/mL(9%)に迅速に低下する。45〜60分後の平衡濃度は、およそ21μg/mL(8%)であった。
【0137】
調製物I(PVP−K25)は、5分間の攪拌後に214μg/mL(88%)の最高ドセタキセル濃度に達する。溶液中のドセタキセルの量は10〜30分後に22μg/mL(9%)へ減少し、及び60分後にドセタキセルの濃度は19μg/mL(8%)である。
【0138】
調製物E(PVP−K30)は213μg/mL(90%)の最高ドセタキセル濃度を有するが、これは5分後に達する。10〜25分後に、溶液中のドセタキセルの量は迅速に低下し、結果として45分後に20μg/mL(8%)の平衡濃度を生じさせる。
【0139】
調製物J(PVP−K90)は、10分間の攪拌後に214μg/mL(88%)の最高ドセタキセル濃度に達する。15分後に、溶液中のドセタキセルの量は依然として151μg/mL(61%)である。60分後に、ドセタキセル濃度は、19μg/mL(7%)に低下した。
【0140】
結論
この実験は、PVPの鎖長が過飽和度及び過飽和が維持される期間の両方に影響を及ぼすことを示している。より長いPVP鎖長の使用はより高い最高ドセタキセル濃度及びより長い過飽和期間、そこでより長期間にわたってより高い溶解度を生じさせる。
【0141】
2.4:薬物負荷
第4回実験では、薬物負荷がドセタキセルの溶解度に及ぼす影響を試験した。調製物E及びK〜Nについて実施した溶解試験からのデータを比較した。結果は図10に示した。調製物Eは4回ずつ試験し、調製物K〜Nを2回ずつ試験した。
【0142】
調製物N(全組成物の重量で1/21のドセタキセル;5:95(w/w)のドセタキセル 対 PVP)は、10分後に197μg/mL(全ドセタキセルの79%が存在する)の最高ドセタキセル濃度に達する。15分後に、溶液中のドセタキセルの量は依然として120μg/mL(48%)であり、15〜30分後には、ドセタキセル濃度は24μg/mL(12%)へ減少する。60分後に、ドセタキセル濃度は、20μg/mL(8%)である。
【0143】
調製物E(全組成物の重量で1/11のドセタキセル;10:90(w/w)のドセタキセル 対 PVP)は、5分後に213μg/mL(90%)の最高ドセタキセル濃度を有する。10〜30分後に、溶液中のドセタキセルの量は迅速に低下し、45分後には20μg/mL(8%)の平衡濃度に達する。
【0144】
調製物M(全組成物の重量で1/6のドセタキセル;20:80(w/w)のドセタキセル 対 PVP)は、10分間の攪拌後に196μg/mL(80%)のドセタキセル濃度を有する。溶液中のドセタキセルの量は10〜30分後に25μg/mL(10%)へ減少し、60分後にドセタキセルの濃度は18μg/mL(7%)である。
【0145】
調製物L(全組成物の重量で1/3のドセタキセル;40:60(w/w)のドセタキセル 対 PVP)は、176μg/mL(71%)のドセタキセル濃度に達する。10〜15分後に、溶液中のドセタキセルの量は46μg/mL(18%)に迅速に低下し、60分後には、溶液中のドセタキセル濃度は18μg/mL(7%)である。
【0146】
調製物K(全組成物の重量で5/7のドセタキセル;75:25(w/w)のドセタキセル 対 PVP)は、5分間の攪拌後に172μg/mL(71%)の最高ドセタキセル値に達する。5〜10分後に、ドセタキセル濃度は42μg/mL(17%)に急速に低下し、60分後に、18μg/mL(7%)のドセタキセル濃度に達する。
【0147】
結論
この実験は、固体分散体中で使用したドセタキセルの量に比較したPVP−K30の量は、過飽和度及び過飽和が維持される期間の両方に影響を及ぼすことを示している。より高い薬物負荷の使用はより低い最高ドセタキセル濃度、及びより短い過飽和期間、そこで経時的により低い溶解度を生じさせる。
【0148】
2.5:先行技術組成物との溶解度の比較
この実験では、15mgのドセタキセル、135mgのPVP−K30及び15mgのSDSの固体分散体を含有する組成物を、Chen et al.[13]に開示された5mgのドセタキセル及びPVP−K30の固体分散体を含む組成物の文献データと比較した。溶解度試験結果は、Chen et al.[13]に記載された溶解試験を使用して入手し、図11及び12に表示した。溶解試験はさらに、模擬腸液中でも実施し、Chenの文献データと比較した。結果は図13に示した。
【0149】
結果
図11から、Chen et al.の組成物は、900mLの水中での組成物中の5mgのドセタキセルの最高約80%を溶解させることができる。この最高値に達するには5時間超を要した。ドセタキセル、PVP−K30及びSDS組成物は、約60分間で15mgのドセタキセルの100%を溶解させた。
【0150】
図12には、ドセタキセルの絶対濃度を表示した。Chenの組成物は、約5時間後に約4.2μg/mLの最高ドセタキセル濃度を生じさせた。ドセタキセル、PVP−K30及びSDS組成物は、約60分後に約16.7μg/mLの最高ドセタキセル濃度をもたらした。
【0151】
図13では、ドセタキセルカプセルは、28μg/mL(>90%の溶解度)の溶解度に達する。Chen et al.に記載された固体分散体(ドセタキセル+PVP−K30)は、4.2μg/mL(900mL中への溶解について試験した80%未満の5mgのドセタキセル固体分散体)の溶解度に達する。そこで本カプセル調製物は、より高い溶解速度とともに6.6倍良好な溶解度に達する(最高溶解速度はChenによる90〜120分後であるのに対して30分後に到達した)。
【0152】
結論
これらの結果から、ドセタキセル、PVP−K30及びSDS組成物は、Chenの組成物に比較してより高速の溶解速度及びより高い溶解度を生じさせた。バイオアベイラビリティについては、薬物がどのように高速で溶解するのか、及びどのような溶解度に0.5〜1.5時間で達するのかを見るのかが重要である。
【0153】
Chenの結果から、当業者は、組成物中のドセタキセルの量を増加させるとドセタキセルの絶対溶解度を増加させるであろうと考察しないであろう。Chenの組成物は900mLの水中で5mgのドセタキセル中80%(即ち、4mg)しか溶解させないので、15mgへドセタキセルの量を増加させることが4mgより多いドセタキセルが溶解することを生じさせるであろうとは予想しないであろう。そこでChenによる15mgのドセタキセル組成物は、ドセタキセル、PVP−K30及びSDS組成物についての100%に比較して最高約27%のドセタキセルを溶解させると予想されるであろう。このため、ドセタキセル、PVP−K30及びSDS組成物は、Chenに比較して驚くべき優れた結果を提供する。
【0154】
2.6:模擬腸液サインパンクレアチン(SIFsp)中での溶解試験
この実験では、ドセタキセル、PVP−K30及びSDSの固体分散体を含有するカプセルの溶解を模擬腸液サインパンクレアチン(SIFsp)中で試験した。カプセルは、調製物Eによる15mgのドセタキセルを含有していた(表13を参照)。SIFspは、USP28に従って調製した。15mgのドセタキセルを含有するカプセルは、75rpmで攪拌しながら37℃の500mLのUSP SIFsp中に溶解させた。結果は図14及び15に示した。
【0155】
図14及び15は、ほぼ100%のドセタキセルが溶解したことを示している。これは約29μg/mLの絶対ドセタキセル濃度と等価であり、約30分間で達成される。そこで、本組成物は、相当に短時間で相当に高い溶解度を提供する。
【0156】
2.7:安定性
調製物E(表13を参照)による、そして臨床試験のためのカプセル中で使用された(以下の実施例を参照)ドセタキセル、PVP−K30及びSDSの固体分散体は、4〜8℃で保管された場合に少なくとも180日間にわたって化学的(分解なし)及び物理的に(溶解特性に変化なし)の両方が安定性であることが見いだされた。
【実施例3】
【0157】
調製物を用いた臨床試験のデータ
材料及び方法
現在進行中の第I相臨床試験には患者10例が参加した。
【0158】
これらの患者には、以下の番号が与えられた:301、302、303、304、305、306、307、308、309及び310。
【0159】
これらの患者には、ドセタキセルの液体調製物又はドセタキセル、PVP−K30及びSDSの固体分散体を含む固体組成物からなる薬剤(以下ではMODRAと呼ぶ)が与えられた。
【0160】
液体調製物
ドセタキセルの用量:全患者(20mgのドセタキセルを摂取した患者306を除く)に対して30mg。30mgの用量は、以下の通りに調製した:静脈内投与用の3.0mLのTaxotere(登録商標)プレミックス(ポリソルベート80(25(v/v)%)、エタノール(10(w/w)%)、及び水)中に1mL当たり10mgのドセタキセルを含有する)を最終容量が25mLとなるように水と混合した。この溶液は、患者に100mLの水道水とともに経口摂取された。
【0161】
MODRA
ドセタキセルの用量:30mg;1カプセルに付き15mgのドセタキセルを含む2カプセルが摂取された。本臨床試験のその後の試験のためには、先行実施例からの調製物E(1/11ドセタキセル、9/11 PVP−K30及び1/11 SDS)が選択された。新規バッチの調製物Eは、120mLのt−ブタノール中の1,200mgのドセタキセル無水物、及び80mLの注射用水中の10,800mgのPVP−K30及び1,200mgのSDS(表13を参照)を溶解させることにより製造した。ドセタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K30/SDS/WfI溶液に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronorm、サイズ1/3)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表14を参照)。
【0162】
サイズ0の計60個のゼラチンカプセルに15mgのドセタキセルと同等の量の固体分散体を充填し、HPLCアッセイを使用して固体分散体1mg当たりのドセタキセルの正確な量を決定した。このアッセイは、カプセルが15mgのドセタキセルを含有することを確認した。
【0163】
患者は、100mLの水道水とともに朝の胃が空のときにこの薬剤を経口摂取した。
【0164】
患者の治療
患者301、302、303、304及び305は、液体調製物だけを摂取した。
【0165】
患者306は、第1サイクルでは液体調製物としての20mgのドセタキセル+リトナビル、及び第2サイクルではドセタキセル摂取4時間後に追加のリトナビルを摂取した以外は同一薬剤を摂取した。
【0166】
患者307、308、309及び310は、液体調製物及び/又はMODRAを摂取した。治療サイクルは、1週間間隔で投与された。
【0167】
施設内ガイドラインに従って、経口及び静注ドセタキセルの両方について、全患者が経口デキサメタゾンを用いて治療された。用量4mgのデキメタソンは、治験薬の1時間前に投与され、その後は12時間毎に4mg投与された(2回)。ドセタキセル治療の1時間前、患者は悪心及び嘔吐を予防するために1mgのグラニセトロン(Kytril(登録商標))も又摂取した。
【0168】
薬物投与後、血液サンプルを薬物動態分析のために採取した。投与前にはブランクサンプルを採取した。血液サンプルは遠心分離し、血漿を分離し、分析時まで直ちに−20℃で保管した。分析は、GLP(医薬品安全性試験実施基準)認証検査室において妥当性の確認されたHPLC法を用いて実施した[17]。
【0169】
結果
表16は、個々の薬物動態試験結果の要約を表示している。
【0170】
【表16】
【0171】
患者301、302、303、304、305、307、309及び310は、液体調製物を摂取した。AUCの平均値、及び平均値についての95%信頼区間(無限大まで補外させた):1156(+348)ng*h/mL.
個人間変動性は、85%である。
【0172】
患者306は第1サイクルにおいて100mgのリトナビルと同時に20mgのドセタキセル(液体調製物として)を摂取し、1週間後の第2サイクルでは同一の組み合わせを摂取したが、ドセタキセルの摂取4時間後には追加の100mgのリトナビルを摂取した。即ち、2用量のリトナビルを第1回はt=0、及び第2回はt=4hで摂取した。薬物動態曲線は、図16に示した。
【0173】
患者307、308、309及び310は、液体調製物及び/又はMODRAを摂取した。薬物動態曲線は、図17に示した。
【0174】
図18は、液体調製物を摂取した患者(307、309及び310)の薬物動態曲線並びにMODRAを摂取した患者4例(307、308、309及び310)の全経過(n=6)を示している。
【0175】
どちらも100mgのリトナビルと組み合わせた、液体調製物 対 MODRAの薬物動態試験結果を以下に要約した。
液体調製物(30mgのドセタキセル)
AUCinf(平均値の95%信頼区間):1156(808−1504)ng*h/mL.
個人間変動性:85%(n=8)
MODRA(30mgのドセタキセル)
AUCinf(平均値の95%信頼区間):768(568−968)ng*h/mL.
個人間変動性:29%(n=4)
個人内変動性:33%(n=2)
【0176】
MODRAの平均AUCは、4例の患者からの6本の曲線を使用して計算した。各患者に投与されたMODRAの第1用量を使用して個人間変動性を計算した。個人内変動性は、2用量のMODRAを摂取した患者307及び308からのデータに基づいている。
【0177】
結論
試験したドセタキセル液体調製物は、新規なカプセル調製物(MODRA)で投与された同一用量(30mg)よりおよそ1.5倍高いAUC値を生じさせる。
【0178】
本液体調製物の個人間変動性は高いが(85%)、カプセル調製物の個人間変動性は実質的に低い(29%)。これは新規なカプセル調製物の重要な特徴であり、はるかに優れた予測可能なドセタキセル曝露を提供する。さらに安全性の理由からも、低い個人間変動性は経口化学療法レジメンにおいて極めてはるかに望ましい。
【0179】
個人内変動性(限定されたデータ)は、個人間変動性と同一の程度にある。
【0180】
ドセタキセル投与の4時間後に摂取された100mgのリトナビルの第2ブースト投与用量は、ドセタキセルのAUCを1.5倍増加させる。
【0181】
経口カプセル調製物と静脈内投与との比較
図19は、静脈内投与(静脈内1時間注入としての20mgのドセタキセル、Taxotere(登録商標)(n=患者5例)及びドセタキセルの経口投与(30mgのドセタキセル;MODRAカプセル、上記参照)(n=患者4例;6コース)後の薬物動態曲線を示している。静脈内及び経口投与の両方を100mgリトナビル(カプセル、Norvir(登録商標))の投与と組み合わせた。施設内ガイドラインに従って、経口及び静注ドセタキセルの両方について、全患者が経口デキサメタゾンを用いて治療された。用量4mgのデキメタソンは、治験薬の1時間前に投与され、その後は12時間毎に4mg投与された(2回)。ドセタキセル治療の1時間前、患者は悪心及び嘔吐を予防するために1mgのグラニセトロン(Kytril(登録商標))も又摂取した。
【0182】
MODRAカプセルのバイオアベイラビリティは:
(AUC 30mg経口/AUC 20mg静注)×(20/30)×100%=73%(SD 18%)により計算した。
【0183】
これは、カプセルのバイオアベイラビリティが相当に高く、低い個人間変動性を伴うことを示している。
【実施例4】
【0184】
経口調製物の又別の特性解析
重要な所見:
スプレードライ法により製造された調製物は十分に非晶質であり、凍結乾燥法により製造された調製物に比較して溶解試験後に長期間の過飽和状態を有する(以下のセクション7を参照)。
【0185】
溶解試験での沈降後のドセタキセルの平衡溶解度は、PVP−K30を含む調製物(20μg/mL)及び担体を含まない調製物(7μg/mL)と比較した、PVP−VA64を含む調製物(40μg/mL)中では有意に増加する(以下のセクション8を参照)。
【0186】
結論の要約:
界面活性剤のタイプがドセタキセルの溶解に及ぼす作用(セクション1):
・試験した全界面活性剤(SDS、CPC、ポリソルベート80及びソルビタンモノオレート)は、ドセタキセルの溶解度を増加させる。
・界面活性剤がドセタキセルの溶解速度に及ぼす作用は、界面活性剤のHLB値と相関すると思われる。
・SDS及びCPCは、ドセタキセルの溶解速度における類似の増加を示す。
【0187】
SDSの量がドセタキセルの溶解に及ぼす作用(セクション2):
・少量のSDS(1/41)は、既にドセタキセルの溶解速度を増加させる。
・より多量のSDS(1/11)は、ドセタキセルの溶解速度をさらに一層増加させる。
・界面活性剤の作用は粉末の圧密レベルと相関すると思われるので、このため界面活性剤を含む粉末と含まない粉末との間のドセタキセルの溶解速度の差は、錠剤では一層大きくなると予想される。
【0188】
ドセタキセルの非晶質の性質(セクション3):
・ドセタキセルの凍結乾燥は、X線粉末回折スペクトルにおける回折ピークの消失を生じさせる。
・ドセタキセルの凍結乾燥は165℃近くで吸熱ピークの消失及び124℃近くでガラス転移の出現を生じさせる。
・165℃近くでの吸熱ピークの面積は、非晶質及び結晶性ドセタキセルの物理的混合物中の結晶性物質の量とよく相関している。
【0189】
パクリタキセルの非晶質の性質(セクション4):
・パクリタキセルの凍結乾燥は、X線粉末回折スペクトルにおける回折ピークの消失を生じさせる。
・パクリタキセルの凍結乾燥は、58、80及び163℃近くでの吸熱ピークの消失、61℃近くでの吸熱ピークの出現及び154℃近くでのガラス転移を生じさせる。
【0190】
ドセタキセルの固体分散体の特性解析(セクション5):
・ドセタキセル、PVP−K30及びSDSの混合物の凍結乾燥は、X線粉末回折パターンにおいてドセタキセルに属する回折ピークの消失を生じさせる。
・ドセタキセルの回折ピークは、固体分散体成分の物理的混合物中に存在する。
【0191】
パクリタキセルの固体分散体の特性解析(セクション6):
・パクリタキセル固体分散体系は、ドセタキセル固体分散体系に匹敵する。
【0192】
製造方法がドセタキセル固体分散体の特性及び性能に及ぼす作用(セクション7):
・ドセタキセル、PVP−K30及びSDSの混合物のスプレードライは、ドセタキセル及びSDSに属する回折ピークの消失を生じさせる。
・ドセタキセル、PVP−K30及びSDSの混合物のスプレードライは、DSCサーモグラムにおいてドセタキセル及びSDSに属する吸熱ピークの消失を生じさせる。
・ドセタキセル、PVP−K30及びSDSの混合物のスプレードライは、溶解試験後にドセタキセル、PVP−K30及びSDSの凍結乾燥混合物に比較して沈降までにより長い時間を生じさせる。
【0193】
担体のタイプがドセタキセル固体分散体の溶解性能に及ぼす作用(セクション8):
・PVP−K30の代りのポリエチレングリコール(1500、6000又は20000)の使用は、沈降までのより短い時間及び溶解試験後のドセタキセルのより低い平衡溶解度を生じさせる。
・PVP−K30に代えてのPVP−VA64の使用は、沈降までの匹敵する時間及び溶解試験後のドセタキセルの有意に高い平衡溶解度を生じさせる(各々、40μg/mL 対 20μg/mL)。
【0194】
錠剤の製造(セクション9):
・現在使用されているカプセルと同等以上に高い溶解速度を備える錠剤を製造することは実行可能である。
・現在使用されているカプセルより高いドセタキセルの含有量を備える錠剤を製造することは実行可能である。
【0195】
材料及び方法
一般
以下の実験において使用した調製物は、以下で略述する方法及び表18、表19、表20及び表17に記載した組成物に従って調製した。試験した薬物は、パクリタキセル及びドセタキセルであった。
【0196】
結晶性薬物:
結晶性薬物は供給業者から入手したままで使用した。
【0197】
非晶質薬物:
薬物は、薬物を30mLのt−ブタノール中に溶解させることにより非晶質化した。薬物/t−ブタノール溶液は、絶え間なく攪拌しながら20mLの注射用水(WfI)に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronorm、サイズ1/9)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表20を参照)。
【0198】
固体分散体の成分の物理的混合物
物理的混合物は、150mgのドセタキセル及び対応する量の担体及び界面活性剤(表17を参照)を乳鉢と乳棒で混合することにより調製した。
【0199】
【表17】
【0200】
非晶質及び結晶性ドセタキセルの物理的混合物
物理的混合物は、正確に計量した量の結晶性及び非晶質ドセタキセルを混合することにより調製した(表18を参照)。
【0201】
【表18】
【0202】
固体分散体(凍結乾燥)
固体分散体は、30mLのt−ブタノール中にドセタキセル、及び20mLの注射用水中の対応する量の担体及び界面活性剤(表19を参照)を溶解させることにより入手した。ドセタキセル/t−ブタノール溶液は、絶え間なく攪拌しながら担体/界面活性剤/WfIの溶液に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronormサイズ1/9)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表20を参照)。
【0203】
【表19】
【0204】
【表20】
【0205】
固体分散体(スプレードライ)
固体分散体は、ドセタキセルを45mLのエタノール及び5mLのWfI中に溶解させることにより入手した。薬物を完全に溶解させた後、絶え間なく攪拌しながらPVP−K30及びSDS(表21を参照)を薬物/エタノール/WfI溶液に加えた。最終混合物をフラスコに移し、その後にエタノール及び水をスプレードライ法により除去した(表22を参照)。
【0206】
【表21】
【0207】
【表22】
【0208】
カプセル(セクション1及び2において使用した)
カプセルは、10〜15mgの薬物と同等の量の凍結乾燥した固体分散体粉末(1/11ドセタキセル、9/11 PVP−K30及び1/11 SDS)を計量することにより製造した。固体分散体粉末を乳鉢及び乳棒で粉砕して微細粉末にし、サイズ0の硬質ゼラチンカプセル内に手動カプセル充填装置を用いてカプセル充填した。1カプセル当たりのドセタキセルの量は、製造後に正味カプセル重量を総カプセル重量から減じ、それに固体分散体粉末のドセタキセル比率を掛けることによって推定した(図19)。カプセルの含有量は、HPLC品質管理によって確認した。
【0209】
臨床試験用カプセル(セクション9において使用)
臨床試験用カプセルは、10mgの薬物と同等の量の凍結乾燥した固体分散体粉末(1/11ドセタキセル、9/11 PVP−K30及び1/11 SDS)、110mgのラクトースモノヒドレート及び1.1mgのコロイド状シリコンジオキシドを計量することにより製造した。全成分は、均質な混合物が得られるまで乳鉢及び乳棒を用いて混合した。この混合物は、サイズ0の硬質ゼラチンカプセル内に手動カプセル充填装置を用いてカプセル充填した。
【0210】
錠剤(セクション9において使用)
錠剤は、20mgのドセタキセルと同等の量のスプレードライ法した固体分散体粉末(1/11ドセタキセル、9/11 PVP−K30及び1/11 SDS)、110mgのラクトースモノヒドレート及び110mgの架橋ポリビニルピロリドンを計量することにより製造した。全成分は、均質な混合物が得られるまで乳鉢及び乳棒を用いて混合した。この混合物は、13mmフラット工具を装備した偏心プレス機上で手動により圧縮した。充填量は13.5mmで固定し、圧力は10.5mmに設定した。圧縮後に錠剤を計量し、ドセタキセルの量は錠剤重量に固体分散体粉末中の薬物の重量分率と錠剤中の固体分散体粉末の重量分率との積を掛けることによって推定した。錠剤の含有量は、HPLC品質管理によって確認した。
【0211】
溶解スクリーニングテスト
およそ6mgの薬物と同等の量の粉末を50mLのビーカー内に配置した。磁気攪拌棒及び25mLの水をビーカーに加えた。この溶液を720rpmで攪拌し、およそ37℃で維持した。サンプルを様々な時点で採取し、0.45μmフィルターを使用して濾過し、その後にそれらをメタノール及びアセトニトリルの1:4(v/v)混合液で希釈した。濾過及び希釈したサンプルは、その後にHPLC−UV(表23を参照)により分析した。溶解した薬物の量は、μg/mLの濃度として表示した。
【0212】
溶解試験
カプセル又は錠剤は、37℃で500mLのWfIが充填された2型(パドル式)溶解装置に配置し、パドルの回転速度は75rpmであった。サンプルを様々な時点で採取し、0.45μmフィルターを使用して濾過し、その後にそれらをメタノール及びアセトニトリルの1:4(v/v)混合液を用いて1:1に希釈した。濾過及び希釈したサンプルは、その後にHPLC−UV(表23を参照)により分析した。溶解したドセタキセルの量は、μg/mLでの濃度又はラベル記載内容に対するパーセンテージ(% RLC)のいずれかで表示した。ラベル記載内容は、製造後の各カプセル又は錠剤中に存在する薬物の推定量である。
【0213】
【表23】
【0214】
示差走査熱量(DSC)測定
DSC測定は、Q2000 DSC(TA Instruments社、米国デラウエア州ニューキャッスル)上で実施した。温度計及び熱流量計はインジウムを用いて校正した。およそ10mgの粉末のサンプルをTzeroアルミニウム製鍋(TA instruments社)に移し、密封しオートサンプラー内に配置した。表24に列挙したプログラムを全サンプルに対して使用した。
【0215】
【表24】
【0216】
X線粉末回折測定
X線粉末回折測定は、X−celeratorを装備したPhlips X’pert pro回折装置上で実施した。厚さがおよそ0.5mmのサンプルを金属製サンプルホルダー内に配置し、これを回折装置内に配置し、表25に表示した設定で走査した。
【0217】
【表25】
【0218】
実験
セクション1:界面活性剤のタイプがドセタキセルの溶解に及ぼす作用
目的
界面活性剤のタイプが及ぼす作用を決定するために、異なる界面活性剤(表26を参照)を備える5種の調製物(表19を参照、調製物1〜5)を凍結乾燥法により調製した(表20を参照)。界面活性剤の選択は、界面活性剤のクラス及びHLB値に基づいていた。選択された界面活性剤は、全3種の界面活性剤クラス(アニオン性、カチオン性及び非イオン性)並びに広範囲のHLB値(4.3〜40)を表している。各調製物は、同一量のドセタキセル、PVP−K30及び界面活性剤を有していた。各調製物から添加物を全く含めずに3個のカプセルを製造し、溶解試験にかけた。
【0219】
所見
図20は、0〜5分間にSDSを含む調製物中に溶解したドセタキセルの量は10% RLC未満であったが、CPC及びポリソルベート80を含む調製物中に溶解したドセタキセルの量は30% RLCを超えていた。この時点で、ソルビタンモノオレート調製物中のドセタキセルのおよそ17% RLCが溶解する。しかし10分後、SDS(63% RLC)、CPC(75% RLC)及びポリソルベート80(68% RLC)調製物間の溶解したドセタキセルの量の差は顕著に減少したが、ソルビタンモノオレート調製物からのドセタキセルの放出は、48% RLCと相当に低いままである。15分後、SDSを含む調製物から放出したドセタキセルの量は、CPCを含む調製物から放出したドセタキセルの量と同等である。ポリソルベート80及びソルビタンモノオレート調製物は73% RLC及び63% RLCというより低い放出したドセタキセルの量を有するが、これらの数値はそれでも界面活性剤を含まない固体分散体系からのドセタキセルの放出より依然としてはるかに高い(37% RLC)。60分後、CPC及びSDS系から放出したドセタキセルの量は、各々87% RLC及び89% RLCであるが、ポリソルベート80及びソルビタンモノオレート調製物からの放出は、各々77% RLC及び83% RLCと低い。
【0220】
結論
界面活性剤を備える全固体分散体系は、界面活性剤を備えない同一固体分散体系に比較してドセタキセルの溶解を増加させる。CPC及びSDSがドセタキセルの溶解に及ぼす作用は匹敵しており、これら2種の界面活性剤間の初期の差は、おそらくカプセル外被の溶解の変動の結果であると考えられる。これらの界面活性剤のHLB値は、固体分散体調製物の性能とよく相関すると思われる。
【0221】
さらに、パクリタキセル固体分散体系から製造されたカプセルを用いた溶解試験は、固体分散系内への界面活性剤(SDS)の組み込みに起因して溶解速度の一層より大きな変化を示した。この差は、活性成分(パクリタキセル 対 ドセタキセル)の溶解度の作用の差と関連している可能性があり、このため、種々の界面活性剤間の差は固体分散体系を含有するパクリタキセルから製造された製剤については大きくなる可能性が高い。
【0222】
【表26】
【0223】
セクション2:界面活性剤の量がドセタキセルの溶解に及ぼす作用
目的
SDSの量の影響を決定するために、ドセタキセルを含有する4種の固体分散体粉末(表19を参照、調製物4〜7)を凍結乾燥法により調製した(表20を参照)。SDSの量は4種の調製物間で変動したが、ドセタキセル及びPVP−K30の量は一定に維持された。各調製物から添加物を全く含めずに3個のカプセルを製造し、溶解試験にかけた。
【0224】
所見
図21及び図22は、溶解試験の結果を示している。0〜5分間に、ドセタキセルの溶解は、全4種の調製物中で限定される。初期の緩徐な溶解度は、カプセル外被の溶解によって生じる遅延時間に起因する。外被が溶解した後、固体分散体粉末の溶解を開始できる。界面活性剤を含まない調製物からのドセタキセルの溶解は、1/11 SDSを含む調製物からのドセタキセルの溶解より緩徐であると考えられる。
【0225】
1/11 SDS調製物は30分間以内に90% RLCの量の溶解したドセタキセルに達するが、SDSを含まない調製物は60分後に70% RLC(ラベル記載内容に比較して)の量の溶解したドセタキセルにしか到達しない。さらに、界面活性剤を含まない調製物のカプセル間のドセタキセルの放出速度における変動は、1/11 SDSを含む調製物のカプセル間の変動よりはるかに高い。
【0226】
図20及び21はさらに又、様々な量のSDS間の差も又示している。ドセタキセルの溶解は、既に固体分散体系への1/41 SDSだけの添加によって改善されるが、しかし1/41及び1/21 SDSの溶解パターン間に明確な差はない。ある量の1/11 SDSは、1/21 SDS及び1/41 SDSに比較してドセタキセルの最善の溶解パターンを生じさせる。
【0227】
結論
固体分散体系へのSDSへのより高い量の組み込みは、より高速の溶解速度を生じさせる。1/11(w/w)のSDSの組み込みは、最速の溶解速度を生じさせる。
【0228】
固体分散体粉末の圧縮は界面活性剤の使用を不可欠にする可能性が高い。錠剤の製造はカプセルの製造より一層高い圧縮を生じさせるので、界面活性剤の組み込みは、固体分散体錠剤において一層より必要になるであろう。
【0229】
固体分散体系へのSDSの組み込みは、カプセル充填及び錠剤化後に固体分散体粉末の湿潤性を改善する、界面活性剤の均質な分布を保証する。
【0230】
セクション3:ドセタキセルの非晶質の性質
目的
凍結乾燥後のドセタキセルの物理的形態は、ドセタキセルの凍結乾燥後に結晶度を決定するために示差走査熱量(DSC)(表24を参照)及びX線粉末回折測定(表25を参照)により調査した。
【0231】
所見
図23は、凍結乾燥法がドセタキセルのX線粉末回折パターンに及ぼす作用を示している。ドセタキセルの凍結乾燥前に、X線回折スペクトルは10〜40°2θ間の数多くの回折ピークを有しており、これはドセタキセルが結晶性状態にあることを示している。ドセタキセルの凍結乾燥後、X線粉末回折パターンには非晶質ハローが存在し、これはドセタキセルが非晶質形にあることを示している。
【0232】
図24は、凍結乾燥法がドセタキセルのDSCサーモグラムに及ぼす作用を示している。ドセタキセルの凍結乾燥前に、DSCサーモグラムはおそらくはドセタキセルの結晶構造の再配列によって生じた165℃での大きな吸熱ピークを示している。ドセタキセルの凍結乾燥後、DSCサーモグラムは165℃で吸熱ピークを有していない。しかし、広汎な吸熱ピークはおよそ50℃に存在しており、これは水及びt−ブタノールの蒸発法により引き起こされる。さらに、ガラス転移は124℃で観察することができ、これはドセタキセルが非晶質状態にあることを示している。
【0233】
図25は、非晶質(凍結乾燥)及び結晶性ドセタキセルの様々な混合物のX線粉末回折スペクトルを示す図である。混合物中の結晶性ドセタキセル含有量における減少は、X線粉末回折スペクトルにおける回折ピークの強度及び数の減少を生じさせる。5%結晶性ドセタキセルを含む混合物のX線回折パターンは回折ピークを有しておらず、これはX線粉末回折を伴う結晶性ドセタキセルの検出可能な最低量が5(w/w)%(純粋薬物物質)を超えることを指示している。
【0234】
図26は、非晶質(凍結乾燥)及び結晶性ドセタキセルの様々な混合物のDSCサーモグラムを示す図である。混合物中の結晶性ドセタキセルの含有量における減少は165℃での吸熱ピークのサイズの減少及びおよそ50℃での広汎な吸熱ピークの増加を生じさせる。これは0%結晶性ドセタキセル(純粋凍結乾燥ドセタキセル)及び5%結晶性ドセタキセルのサーモグラムにおける50及び165℃での吸熱ピークのサイズには差があり、これはDSCを含む結晶性ドセタキセルの検出可能な最低量が0〜5(w/w)%結晶性ドセタキセル(純粋薬物物質)であることを指示している。
【0235】
図27は、結晶性及び非晶質ドセタキセルの物理的混合物中の結晶性物質の量に対する、全熱流量サーモグラムにおける165℃でのピーク面積のプロット図を示している(表18を参照)。非晶質及び結晶性薬物中の結晶性物質の量は、各々0〜100(w/w)%であると推定される。回帰線は、0.990の決定係数及び0.995の回帰係数を有しており、これは165℃でのピークサイズと結晶度との間に強度の相関があることを確認している。
【0236】
結論
ドセタキセルの凍結乾燥は、凍結乾燥ドセタキセルのX線粉末回折スペクトルが回折ピークを示さないような、並びにDSCサーモグラムが結晶再配列と関連する吸熱ピークを示さないような程度への結晶度の減少を生じさせる。さらにDSCサーモグラムでは、ガラス転移が出現する。これは全てが、凍結乾燥後にドセタキセルが非晶質状態にあることの指標である。
【0237】
結晶再配列と関連する吸熱ピークのピーク面積は、非晶質及び結晶性ドセタキセルの物理的混合物中の結晶性ドセタキセルの含有量とよく相関する。
【0238】
セクション4:パクリタキセルの非晶質の性質
目的
凍結乾燥後のパクリタキセルの物理的形態をX線粉末回折及び示差走査熱量測定により調査し、凍結乾燥後のパクリタキセルの結晶度を決定した。
【0239】
所見
図28は、凍結乾燥がパクリタキセルのX線粉末回折パターンに及ぼす作用を示している。パクリタキセルの凍結乾燥前に、X線回折スペクトルは10〜40°2θ間の数多くの回折ピークを有しており、これはパクリタキセルが結晶性状態にあることを示している。パクリタキセルの凍結乾燥後、X線粉末回折パターンには非晶質ハローが存在し、これはパクリタキセルが非晶質形にあることを示している。
【0240】
図29は、凍結乾燥がパクリタキセルのDSCサーモグラムに及ぼす作用を示している。パクリタキセルの凍結乾燥前に、DSCサーモグラムは、58、80及び163℃で大きな吸熱ピークを示す。58及び80℃での両方のピークは、結晶格子からの水の消失により生じる。163℃での吸熱ピークは、おそらくパクリタキセルの結晶構造の再配列により生じる。パクリタキセルの凍結乾燥後、DSCサーモグラムは、61℃での広範囲の吸熱ピークの代わりに58、80又は163℃での吸熱ピークを有しておらず、154℃でのガラス転移を観察できる。
【0241】
結論
パクリタキセルの凍結乾燥は、凍結乾燥パクリタキセルのX線粉末回折スペクトルが回折ピークを示さない、並びにDSCサーモグラムが結晶再配列と関連する吸熱ピークを示さない程度への結晶度の減少を生じさせる。さらにDSCサーモグラムでは、ガラス転移が出現する。これは全てが、凍結乾燥後にパクリタキセルが非晶質状態にあることの指標である。
【0242】
セクション5:ドセタキセルを含む固体分散体の特性解析
目的
ドセタキセルの固体分散体を特性解析するために、X線粉末回折測定及びDSC測定は、ドセタキセル、PVP−K30及びSDSの物理的混合物及び固体分散体上に実施した。
【0243】
所見
図30には、固体分散体の成分であるドセタキセル、PVP−K30及びSDSのX線回折パターンを示した。ドセタキセルは結晶性であり、10〜40°の2θの数多くの回折ピークを有しており、SDSは結晶性で20〜22°の2θ間に鋭い回折ピークを有しており、PVP−K30は非晶質で回折ピークを有していない。
【0244】
図31は、固体分散体の成分であるドセタキセル、PVP−K30及びSDSのDSCサーモグラムを示している。ドセタキセルは、おそらくは結晶再配列により生じた165℃での大きな吸熱ピークを有している。PVP−K30は、76℃の近くで水の蒸発により生じた大きな吸熱ピークを有しており、ガラス状態及びゴム状態の間での熱容量における差により生じた162℃の近くでのガラス転移を有する。SDSは、おそらくは小さな非晶質分率により生じた67℃近くでの相転移、及び複数のピークを含有する80〜120℃の大きな吸熱領域を有している。これらのピークは、一部にはSDSの結晶性バルクを溶融させることによって、及び一部には未知の非可逆性吸熱事象により生じる。
【0245】
図32は、1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを含有する物理的混合物及び固体分散体系のX線粉末回折スペクトルを示す図である。ドセタキセル及びSDSの結晶性回折ピークは物理的混合物のX線粉末回折スペクトルに出現するが、ドセタキセルの回折ピークは固体分散体系のX線回折スペクトルにおいては出現しない。
【0246】
図33は、1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを含有する物理的混合物及び固体分散体系のDSCサーモグラムを示す図である。物理的混合物のサーモグラムは、おそらくはSDSにより生じた200℃近くでの吸熱下落;おそらくは結晶性ドセタキセルにより生じた162℃の近くでの小さな吸熱ピーク及びおそらくはPVP−K30により生じた164℃の近くでのガラス転移を示している。固体分散体系のサーモグラムは、おそらくはSDSにより生じた113℃の近くでの吸熱ピーク;並びに非晶質ドセタキセル及び非晶質PVP−K30の組み合わせにより生じた155℃の近くでのガラス転移を示す。
【0247】
結論
固体分散体のX線粉末回折スペクトルはドセタキセルに属する回折ピークを示さないが、ドセタキセル、PVP−K30及びSDSを含有する物理的混合物のX線粉末回折スペクトルはドセタキセルに属する回折ピークを示す。ドセタキセル、PVP−K30及びSDSの凍結乾燥混合物のDSCサーモグラムは、おそらくは非晶質ドセタキセル及びPVP−K30の分子混合により生じる155℃でのガラス転移を示すが、ドセタキセル、PVP−K30及びSDSを含有する物理的混合物のDSCサーモグラムは、おそらくはPVP−K30により生じた163℃でのガラス転移温度を示す。これに加えて162℃の近くでの吸熱ピークは物理的混合物のDSCサーモグラムにおいてのみ視認することができるが、固体分散体のDSCサーモグラムにおいては視認できない。
【0248】
SDSは固体分散体及び物理的混合物両方において結晶性状態で存在し、固体分散体及び物理的混合物両方のX線回折スペクトルにおいて20〜22°の2θ間での回折ピーク、並びに物理的混合物及び固体分散体のDSCサーモグラムにおける100〜113℃近くでの吸熱ピークを各々生じさせる。
【0249】
さらに、ドセタキセル単独の凍結乾燥は非晶質ドセタキセルを生じさせたが(セクション3を参照)、ドセタキセルを含有する混合物の凍結乾燥はさらに又非晶質ドセタキセルを生じさせる可能性が極めて高い。
【0250】
セクション6:パクリタキセルの固体分散体の特性解析
入手できるデータは、パクリタキセル固体分散体系がドセタキセル固体分散体系に匹敵する、即ち、パクリタキセルは凍結乾燥後には非晶質状態で存在するがSDSはそうではないことを指示している。
【0251】
セクション7:製造方法がドセタキセル固体分散体の特性及び性能に及ぼす作用
目的
製造方法が固体分散体特性に及ぼす影響を決定するために、凍結乾燥法及びスプレードライ法の両方を使用して1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを備える固体分散体系を製造した(表19及び表21を参照)。両方の系をX線回折、DSC及び溶解スクリーニング試験により試験した。
【0252】
所見
図34は、凍結乾燥法及びスプレードライ法により製造された固体分散体のX線粉末回折スペクトルを示している。凍結乾燥法(フリーズドライ法)により製造された固体分散体は、20〜22°2θのスペクトル内に存在する回折ピークが存在するために、一部には結晶である。これらの回折ピークはSDSに属する(図30)。スプレードライ法により製造された固体分散体は、X線粉末回折パターンにおける回折ピークの非存在から結論できるように、十分に非晶質である。
【0253】
図35は、凍結乾燥法及びスプレードライ法により製造された固体分散体のDSCサーモグラムを示している。凍結乾燥法により製造された固体分散体のサーモグラムは、おそらくはSDSにより生じた113℃の近くでの吸熱ピーク;並びに非晶質ドセタキセル及び非晶質PVP−K30の組み合わせにより生じる155℃の近くでのガラス転移を示す。スプレードライ法により製造された固体分散体は、147℃の近くでのガラス転移しか示さず、これは十分な非晶質形の指標である。
【0254】
図36は、凍結乾燥法及びスプレードライ法により製造された固体分散体の溶解スクリーニング曲線を示している。凍結乾燥法により製造された固体分散体は、5分後にピークドセタキセル濃度に到達し、沈降し始める。スプレードライ法により製造された固体分散体は、10分後にピークドセタキセル濃度に到達し、15分後に沈降し始める。
【0255】
結論
ドセタキセルの固体分散体の製造における凍結乾燥法に比較したスプレードライ法の使用は、長期間の過飽和状態に関して溶解スクリーニング試験における改善された性能を生じさせるより非晶質系をもたらす。
【0256】
さらに、スプレードライ法後に得られた粉末は低静止性であり、より一様な粒径を有するので、凍結乾燥生成物に比較してその後に加工処理するためにより適切である。
【0257】
セクション8:担体のタイプがドセタキセル固体分散体の溶解性能に及ぼす作用
目的
異なる担体がドセタキセルの固体分散体の溶解性能に及ぼす性能を試験すること。I/11ドセタキセル、9/11担体及び1/11 SDSを含有する固体分散体系の製造において様々な担体を使用した(表19を参照、調製物8〜11)。全系を溶解スクリーニング試験にかけた。
【0258】
所見
図37は、PVP−K30、PEG1500、PEG6000又はPEG20000を含有する固体分散体系の溶解スクリーニング曲線を示している。全系は5分後には匹敵するドセタキセルのピーク濃度に達するが、全3種のPEG系についてドセタキセルの沈降は既に5分後に始まる。さらに、沈降後の溶液中のドセタキセルの量は、固体分散体系を含有するPEG中においておよそ8μg/mLであるが、PVP−K30含有系は沈降後に20μg/mLの溶液中のドセタキセルの量に達する。
【0259】
図38は、PVP−K30又はPVP−VA64を含有する固体分散体系の溶解スクリーニング曲線を示している。両方の系について、5分後に同一ドセタキセルピーク濃度に達し、両方の系についてドセタキセルはおよそ10分後に沈降し始める。しかし沈降後の溶液中のドセタキセルの量には有意な差がある:PVP−VA64(40μg/mL)及びPVP−K30(20μg/mL)。
【0260】
結論
PEGを含有する固体分散体系は、溶解スクリーニング試験においてPVP−K30を含有する固体分散体系より不良に機能する。
【0261】
PVP−VA64を含有する固体分散体系は、沈降後にPVP−K30を含有する固体分散体系よりドセタキセルの有意に高い濃度に達する。
【0262】
さらに、パクリタキセルはドセタキセルより低い溶解度を有するために、PVP−VA64の使用は、固体分散体系を含有するパクリタキセル中において特に有用な可能性がある。
【0263】
セクション9:錠剤の製造
目的
ドセタキセルの固体分散体からの錠剤製造の実行可能性を調査すること。
【0264】
所見
図39は、現在臨床試験において使用されている20mgのドセタキセル錠及び10mgのドセタキセルカプセルの平均溶解曲線を示している。図40は、0〜10分間に錠剤及びカプセルの平均溶解速度を示している。錠剤は、35μg/mLの濃度に達するまで0〜30分間の定常溶解速度を示している。カプセルは、およそ16μg/mLの濃度に達するまで0〜10分間の定常溶解速度を示している。0〜10分間に、カプセル及び錠剤両方からのドセタキセルの放出速度はおよそ0.8mg/分である。
【0265】
結論
固体分散体系を含有するドセタキセルの錠剤の製造は実行可能である。ドセタキセル錠剤の溶解速度は、現在臨床試験において使用されているドセタキセルカプセルの溶解速度に匹敵する。
【実施例5】
【0266】
ドセタキセルの固体分散体への追加の医薬上有効成分の添加
非晶質リトナビルの製造
固体成分:
300mgのリトナビル
溶媒:
45mLのエタノール
5mLの注射用水
リトナビルはエタノール/水混合物中に溶解させ、Buchi 290ミニスプレー乾燥機を用いてスプレードライした(表27を参照)。
【0267】
ドセタキセル、リトナビル、PVP及びSDSの組み合わせ生成物の製造
固体成分:
1gのドセタキセル無水物
4gのリトナビル
25gのPVP−K30
5gのSDS
溶媒:
900mLのエタノール
100mLの注射用水
全固体成分は、エタノール−水混合物中に溶解させ、Buchi 290ミニスプレー乾燥機を用いてスプレードライした(表27を参照)。これは視覚的に均質な白色粉末を生じさせた。この固体分散体粉末は、およそ12.5mgのドセタキセル及び50mgのリトナビル(1カプセル当たり)と等価であり、サイズ0の硬質ゼラチンカプセル内に手動でカプセル充填した。
【0268】
【表27】
【0269】
【表28】
【0270】
図41は、非晶質(スプレードライ)及び結晶性リトナビルのDSCサーモグラムを示している。非晶質リトナビルは、およそ25℃のTgを示し、結晶はおよそ122℃の溶融吸熱を示す。スプレードライ法は非晶質リトナビルを生じさせると結論付けた。
【0271】
図42は、ドセタキセル、リトナビル、PVP−K30及びSDSの組み合わせのスプレードライ法固体分散体粉末のDSCサーモグラムを示している。サーモグラムは、およそ117℃の単一Tgを示すが、リトナビル、ドセタキセル、PVP−K30又はSDSの溶融吸熱は示さなかった。
【0272】
図43は、37℃及び50RPMで1,000mLの0.1N HCl中におけるドセタキセル/リトナビル/PVP−K30/SDSカプセルの溶解プロファイルを示している。各カプセルは、およそ12.5mgのドセタキセル及びおよそ50mgのリトナビルを含有していた。ドセタキセル及びリトナビルはどちらも45分間以内に完全に放出される(n=3)。
【0273】
上記の実施例は、本発明の特定の実施形態を例示することを企図しているが、本発明の範囲を限定することは企図しておらず、本発明の範囲は添付の特許請求の範囲によって規定される。本明細書に言及した全文献は、全体として参照により本明細書に組み込まれる。
【0274】
参考文献
1. Demario MD, Ratain MJ. J Clin Oncol 1998;16:2557-2567.
2. Liu G, Franssen E, Fitch MI et al. J Clin Oncol 1997;15:110-115.
3. Meerum Terwogt JM, Beijnen JH, ten Bokkel Huinink WW et al. Lancet 1998;352:285.
4. Sparreboom A, van Asperen J, Mayer U et al. Proc Natl Acad Sci USA 1997;94:2031 -2035.
5. Kuhn J, Rizzo J, Eckhardt J et al. Proc Am Soc Clin Oncol 1995;14:474.
6. Schellens JHM, Creemers GJ, Beijnen JH et al. Br J Cancer 1996;73:1268-1271.
7. Hellriegel ET, Bjornnson TD, Hauck WW. Clin Pharmacol Ther 1996;60:601 -607.
8. Huizing MT, Giaccone G, van Warmerdam LJC et al. J Clin Oncol 1997;15:317-329.
9. Malingre MM, Richel DJ, Beijnen JH et al. J Clin Oncol 2001;19:l160-1166.
10. Mastalerz H, Cook D, Fairchild CR, Hansel S, Johnson W, Kadow JF, Long BH, Rose WC, Tarrant J, Wu MJ, Xue MQ, Zhang G, Zoeckler M, Vyas DM. Bioorg Med Chem. 2003; 11:4315-23.
11. Serajuddin AT. J Pharm Sci 1999;88(10):1058 1066.
12. Karanth H, Shenoy VS, Murthy RR. AAPS PharmSciTech 2006;7(4):87.
13. Chen J, et al. Drug Development and Industrial Pharmacy 2008;34:588-594.
14. Schellekens RC, Stuurman FE, van der Weert FH, Kosterink JG, Frijlink HW. Eur. J. Pharm. Sci. 2007 Jan;30(1):15-20.
15. Dressman JB, Reppas C. Eur. J. Pharm. Sci. 2000 Oct;11:S73-S80.
16. The Handbook of Pharmaceutical Excipients. Fifth Edition. Edited by Raymond Rowe, Paul Sheskey and Sian Owen. See sections on Povidone, PVP VAand PEG.
17. Kuppens IE, Van Maanen MJ, Rosing H, Schellens JHM, Beijnen JH. Biomed Chromatogr 2005;19:355-361.
18. Kawakami (2009) Journal of Pharmaceutical Sciences. 98(9) 2875-2885. Published online in Wiley Interscience (www.interscience.wiley.com) DOI 10.1002/jps.21816
【技術分野】
【0001】
本発明は、医薬組成物に関する。詳細には、排他的ではないが、本発明は、腫瘍性疾患を治療するための組成物に関する。
【背景技術】
【0002】
経口形にある薬物の投与は、多数の利点を提供する。経口抗癌薬、例えば、5−フルオロウラシル(5−FU)プロドラッグ(例、カペシタビン)及びシグナル伝達経路又は血管新生プロセスを妨害する薬物のアベイラビリティは、治療が最適に効果的であるように長期的に適用されなければならない場合には重要である(非特許文献1)。さらに経口薬は、通院患者ベース又は自宅で投与することができ、便宜性及び患者のクオリティ・オブ・ライフを増加させ、入院を減少させることによってコストを低下させる可能性がある(非特許文献2)。このため、抗癌薬を経口的に投与しようとすることは有益である。
【0003】
一般に、薬物の経口投与は、便宜的且つ実際的である。しかし抗癌薬の大多数は、残念なことに低く変動性の経口バイオアベイラビリティを有する(非特許文献1)。典型的な例は、広汎に使用されているタキサン類であるドセタキセル及びパクリタキセルであり、これらは10%未満の経口バイオアベイラビリティを有する(非特許文献3、4)。より高いバイオアベイラビリティを備える数種の他の抗癌剤は、より高い変動性を示す。例には、トポイソメラーゼI阻害剤、ビンカアルカロイド類、及びミトキサントロンが含まれる(非特許文献1、5、6)。狭い治療濃度域を考えると、変動性のバイオアベイラビリティは、予測できない毒性又は治療血漿レベルが達成されない場合に低下した有効性を生じさせることがある。Hellriegel et al.は試験において、経口投与後の血漿レベルが、一般には静脈内投与後より変動性であることを証明した(非特許文献7)。適正な経口バイオアベイラビリティは、薬物曝露期間が抗癌療法の主要決定因子である場合には重要である(非特許文献8)。適正な経口バイオアベイラビリティは、局所毒性を生じる可能性がある消化管内での高い局所薬物濃度を防止するためにも又重要である。
【0004】
このため、先行技術に関連する問題は、低い変動性とともに高いバイオアベイラビリティを有するタキサンを含む経口組成物を開発することができなかった点である。経口パクリタキセルを用いた臨床試験(例、非特許文献3)及び経口ドセタキセルを用いた臨床試験(例、非特許文献9)が本発明者らによって実施されたが、これらの試験ではタキサン静脈内投与調製物(さらに賦形剤、例えばCremophor EL及びエタノール、又はポリソルベート80及びエタノールを含有する)が経口摂取された。悪心、嘔吐及び不快な味覚が患者によって頻回に報告された。
【0005】
Chen et al.(非特許文献13)は、そのバイオアベイラビリティを改善するために抗癌薬ドセタキセルの溶解度の改善を試みる実験を実施した。Chen et al.は、ドセタキセルの固体分散体を様々な担体、つまりグリセリルモノステアレート、PVP−K30又はポロキサマー188とともに使用することを試みた。Chen et al.は、ポロキサマー188が、5:95のドセタキセル 対 ポロキサマー比を使用した場合に、ドセタキセルの溶解度を20分後(標準溶解試験において)には約3.3μg/mLへ、そしては約120分後には最高約5.5μg/mLへ増加させることを見いだした(Chenの論文における図7を参照)。PVP−K30は、ドセタキセルの溶解度を20分後には約0.8μg/mLへ、そして約300分後には最高約4.2μg/mLにしか増加させなかった(図2を参照)。グリセロールモノステアレートは、ドセタキセルの溶解度を全く増加させなかった。そこで、ドセタキセルの溶解度及び溶解速度は、特に高いレベルには増加させられなかった。優れた経口バイオアベイラビリティを達成するためには、薬物は、最初の約0.5〜1.5時間以内に溶液中に吸収のために利用できる十分に高い薬物量が存在するように、相当に高い溶解度及び溶解速度を有していなければならない。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Demario MD, Ratain MJ. J Clin Oncol 1998;16:2557-2567.
【非特許文献2】Liu G, Franssen E, Fitch MI et al. J Clin Oncol 1997;15:110-115.
【非特許文献3】Meerum Terwogt JM, Beijnen JH, ten Bokkel Huinink WW et al. Lancet 1998;352:285.
【非特許文献4】Sparreboom A, van Asperen J, Mayer U et al. Proc Natl Acad Sci USA 1997;94:2031 -2035.
【非特許文献5】Kuhn J, Rizzo J, Eckhardt J et al. Proc Am Soc Clin Oncol 1995;14:474.
【非特許文献6】Schellens JHM, Creemers GJ, Beijnen JH et al. Br J Cancer 1996;73:1268-1271.
【非特許文献7】Hellriegel ET, Bjornnson TD, Hauck WW. Clin Pharmacol Ther 1996;60:601 -607.
【非特許文献8】Huizing MT, Giaccone G, van Warmerdam LJC et al. J Clin Oncol 1997;15:317-329.
【非特許文献9】Malingre MM, Richel DJ, Beijnen JH et al. J Clin Oncol 2001;19:l160-1166.
【非特許文献10】Chen J, et al. Drug Development and Industrial Pharmacy 2008;34:588-594.
【発明の概要】
【発明が解決しようとする課題】
【0007】
第1態様では、本発明は、実質的に非晶質のタキサン、親水性担体及び界面活性剤を含む経口投与のための固体医薬組成物であって、該非晶質タキサンが溶媒蒸発法により調製される固体医薬組成物を提供する。
【課題を解決するための手段】
【0008】
本発明の組成物によって提供される利点は、タキサンの溶解度、タキサンの溶解速度及び/又はタキサンが結晶化し始める前に溶液中に留まっている時間量が驚くべき程度まで増加させられることにある。これらの因子は、タキサンのバイオアベイラビリティの有意な増加を結果として生じさせる。これは、少なくとも一部には、タキサンが他の方法により製造される真に非晶質である可能性が低い疑似非晶質タキサンと比較してより非晶質状態にあることに起因すると考えられる。結晶性タキサン類は、極めて低い溶解度を有する。
【0009】
担体は、タキサンを非晶質状態に維持することに役立つ。さらに、タキサンが水性媒体中に配置されると、担体は、タキサンを溶液中で過飽和状態に維持することに役立つ。これは、タキサンが結晶化するのを停止させる、又はタキサンが溶液中で結晶化し始める前の時間の長さを増加させる。このため、タキサンの溶解度及び溶解速度は高いままとなる。さらに、担体は本組成物に優れた物理的及び化学的安定性を与える。これはタキサンの分解を防止するのに役立ち、さらに非晶質タキサンが固体状態において経時的に実質的により結晶性構造へ変化するのを防止するのにも役立つ。優れた物理的安定性は、タキサンの溶解度が高いままとなることを保証する。
【0010】
界面活性剤も又、水性媒体中に配置されるとタキサンを非晶質状態で維持することに役立ち、そして驚くべきことに、非晶質タキサン及び担体を含む組成物に比較してタキサンの溶解度を実質的に増加させる。
【0011】
用語「実質的に非晶質」は、タキサン分子の長距離に渡る位置の列が殆ど又は全くあってはならないことを意味している。分子の大多数は、ランダムに配向されていなければならない。完全に非晶質の構造は決して長距離に渡る列を有しておらず、それが一体何であれ、結晶構造を含有していない:これは結晶性固体と正反対である。しかし、一部の固体については、完全に非晶質の構造を得ることは困難なことがある。このため、多数の「非晶質」構造は完全に非晶質ではなく、所定量の長距離に渡る列又は結晶性を依然含有している。例えば、固体は主として非晶質であるが結晶性構造のポケットを有する、又は極めて小さな結晶を含有することがあるので、真の非晶質と近似している。このため、用語「実質的に非晶質」は、一部の非晶質構造を有するが、一部の結晶性構造も又同様に有する固体を含んでいる。実質的に非晶質のタキサンの結晶化度は、50%未満でなければならない。好ましくは、実質的に非晶質のタキサンの結晶化度は、40%未満、一層より好ましくは30%未満、なおより好ましくは25%未満、さらにより好ましくは20%未満、さらにより好ましくは15%未満、一層より好ましくは12.5%未満、一層より好ましくは10%未満、一層より好ましくは7.5%未満、より一層好ましくは5%未満、及び最も好ましくは2.5%未満である。結晶性タキサン類は低溶解度を有するので、実質的に非晶質のタキサンの結晶化度が低いほど、実質的に非晶質のタキサンの溶解度は改善される。
【0012】
実質的に非晶質のタキサンは、任意の適切な溶媒蒸発法を使用して調製できる。適切な溶媒蒸発法は、例えば、[18]に記載されたスプレードライ法及び真空乾燥法である。好ましくは、溶媒蒸発法は、スプレードライ法である。驚くべきことに、溶媒蒸発法、特別にはスプレードライ法を使用して非晶質のタキサンを調製する工程は、他の方法を使用して調製された組成物と比較して特別に優れた溶解度、溶解速度を有し、及び/又は結晶化を開始する前により長時間にわたり溶液中に留まる組成物を生じさせることが見いだされた。これは、他の方法に比較してより非晶質のタキサンを製造する溶媒蒸発法に起因すると考えられる。
【0013】
経口投与のための組成物は、固体形態にある。本固体組成物は、タキサンが実質的に非晶質状態にある限り、任意の適切な形態にあってよい。例えば、本組成物は、非晶質タキサン、担体及び界面活性剤の物理的混合物を含むことができる。所定の実施形態では、担体及び/又は界面活性剤も又実質的に非晶質の状態にある。好ましくは、タキサン及び担体は、固体分散体の形態にある。用語「固体分散体」は、当業者には周知であり、タキサンが担体内で部分的に分子状で分散していることを意味する。より好ましくは、タキサン及び担体は、固溶体の形態にある。用語「固溶体」は、当業者には周知であり、タキサンが担体内で実質的に完全に分子状で分散していることを意味する。固溶体は、本質的に固体分散体より非晶質であると考えられる。固体分散体及び固溶体を調製する方法は、当業者には周知である[11,12]。これらの方法を使用すると、タキサン及び担体はどちらも非晶質状態にある。タキサン及び担体が固体分散体若しくは固溶体の形態にある場合は、タキサンの溶解度及び溶解速度は非晶質タキサン及び担体の物理的混合物より大きい。タキサンが固体分散体若しくは固溶体中に場合は、タキサンは自然に疑似非晶質タキサンに比較してより非晶質状態にあると考えられる。これは改善された溶解度及び溶解を生じさせると考えられる。固体分散体若しくは固溶体の結晶化度は、50%未満でなければならない。好ましくは、固体分散体若しくは固溶体の結晶化度は、40%未満、一層より好ましくは30%未満、なおより好ましくは25%未満、さらにより好ましくは20%未満、さらにより好ましくは15%未満、一層より好ましくは12.5%未満、一層より好ましくは10%未満、一層より好ましくは7.5%未満、より一層好ましくは5%未満、及び最も好ましくは2.5%未満である。
【0014】
タキサン及び担体が固体分散体中にある場合は、界面活性剤は固体分散体若しくは固溶体との物理的混合物中にあってもよい。しかし好ましくは、本組成物は、固体分散体、又はより好ましくは固溶体の形態にあるタキサン、担体及び界面活性剤を含んでいる。固体分散体若しくは固溶体中に全3種の構成成分を有する利点は、それが溶解度及び溶解速度における同一の改善を達成するためにより少量の界面活性剤の使用を可能にすることである。好ましくは、タキサン、担体及び界面活性剤は、全部が実質的に非晶質状態にある。
【0015】
タキサン及び担体;又はタキサン、担体及び界面活性剤の固体分散体若しくは固溶体は、上述したような任意の適切な溶媒蒸発法を使用して製造することができる。好ましくは、固体分散体若しくは固溶体は、スプレードライ法により調製される。驚くべきことに、溶媒蒸発法、特別にはスプレードライ法を使用して固体分散体若しくは固溶体を調製する工程は、特別に優れた溶解度特性を有する組成物を生じさせるので、タキサンは他の方法を使用して調製された組成物と比較して優れた溶解度、溶解速度を有し、及び/又は結晶化を開始する前により長時間にわたり溶液中に留まることが見いだされた。これは、他の方法に比較してその中で全構成成分がより非晶質状態にある組成物を製造する溶媒蒸発法に起因すると考えられる。他の方法では、構成要素の1つ又はそれ以上が依然として一部の結晶の性質を有する可能性が見出され、この性質が溶液中での減少した溶解度特性及び/又は物理的安定性を有する組成物を生じさせると考えられる。
【0016】
1つの実施形態では、本組成物は、経口投与用のカプセル中に含有させることができる。カプセルは、多数の様々な方法で充填することができる。例えば、非晶質タキサンは、スプレードライをして、粉末化し、担体及び界面活性剤と組み合わせ、そして次にカプセル中に分注することにより調製することができる。
【0017】
又別の実施形態では、本組成物は、錠剤に圧縮することができる。例えば、非晶質タキサンは、スプレードライをして、粉末化し、担体及び界面活性剤(及び任意で他の賦形剤)と組み合わせ、次に適切な量を錠剤に圧縮することにより調製することができる。
【0018】
タキサン類は、イチイ属(Taxus(イチイ))の植物を起源とするジテルペン化合物である。しかし一部のタキサン類は、現在では合成的又は半合成的に製造されている。タキサン類は、細胞分裂を停止させることによって細胞増殖を阻害するので、癌の治療に使用されている。タキサン類は、微小管形成を中断させることによって細胞分裂を停止させる。タキサン類は、血管新生阻害剤としても作用できる。本明細書で使用する用語「タキサン」には、自然に生成されようと人工的に製造されようと、チューブリンに結合する全てのジテルペンタキサン類、機能的誘導体及び医薬上許容される塩若しくはエステルが含まれる。
【0019】
生理化学的特性を修飾するための基を含有するタキサン類の誘導体も又本発明の範囲内に含まれる。そこで、改善又は修飾された溶解度特性を備える、タキサン類のポリアルキレングリコール(例えば、ポリエチレングリコール)又はサッカライドコンジュゲートが含まれる。
【0020】
本組成物のタキサンは、上記に定義した任意の適切なタキサンであってよい。好ましいタキサン類は、ドセタキセル、パクリタキセル、BMS−275183、それらの機能的誘導体及びそれらの医薬上許容される塩又はエステルである。BMS−275183は、パクリタキセルのC−3’−t−ブチル−3’−N−t−ブチルオキシカルボニルアナログである[10]。より好ましくは、タキサンは、ドセタキセル、パクリタキセル、それらの機能的誘導体及びそれらの医薬上許容される塩若しくはエステルから選択される。
【0021】
本組成物の親水性担体は、pH7.4の水性媒体中に少なくとも部分的に溶解できる、及び/又はそのような水性媒体中で膨潤又はゲル化できる有機化合物である。担体は、タキサンが本組成物中で非晶質状態に留まることを保証し、タキサンの溶解度及び溶解速度を増加させる任意の適切な親水性担体であってよい。好ましくは、担体はポリマー性のものである。好ましくは、担体は:ポリビニルピロリドン(PVP);ポリエチレングリコール(PEG);ポリビニルアルコール(PVA);クロスポビドン(PVP−CL);ポリビニルピロリドン−ポリビニルアセテートコポリマー(PVP−VA);セルロース誘導体、例えばメチルセルロース、ヒドロキシプロピルセルロース、カルボキシメチルエチルセルロース、ヒドロキシプロピルメチルセルロース(HPMC)、セルロースアセテートフタレート及びヒドロキシプロピルメチルセルロースフタレート;ポリアクリレート類;ポリメタクリレート類;糖類、ポリオール類及びそれらのポリマー類、例えばマンニトール、スクロース、ソルビトール、デキストロース及びキトサン;並びにシクロデキストリン類から選択される。より好ましくは、担体は、PVP、PEG及びPVP−VA、から選択され、一層より好ましくは、担体は、PVP及びPVP−VAから選択される。1つの実施形態では、担体は、PVPである。又別の実施形態では、担体は、PVP−VAである。
【0022】
担体がPVPである場合は、担体として機能し、タキサンを非晶質状態で維持するのに役立つ任意の適切なPVP[16]であってよい。例えば、PVPは、PVP−K12、PVP−K15、PVP−K17、PVP−K25、PVP−K30、PVP−K60、PVP−K90及びPVP−K120から選択されてよい。好ましくは、PVPは、PVP−K30、PVP−K60及びPVP−K90から選択される。最も好ましくは、PVPは、PVP−K30である。
【0023】
担体がPEGである場合は、担体として機能し、タキサンを非晶質状態で維持するのに役立つ任意の適切なPEG[16]であってよい。例えば、PEGは、PEG1500、PEG6000及びPEG20000から選択することができる。好ましくは、PEGは、PEG1500及びPEG6000から選択され、最も好ましくは、PEGは、PEG1500である。
【0024】
担体がPVP−VAである場合は、担体として機能し、タキサンを非晶質状態で維持するのに役立つ任意の適切なPVP−VA[16]であってよい。例えば、PVP−VAは、PVP−VA64であってよい。
【0025】
本組成物は、非晶質タキサンと比して、担体が非晶質タキサンをその非晶質状態に維持するように任意の適切な量の担体を含有することができる。好ましくは、担体に対するタキサンの重量比は、約0.01:99.99(w/w)〜約75:25(w/w)である。より好ましくは、担体に対するタキサンの重量比は、約0.01:99.99(w/w)〜約50:50(w/w)、一層より好ましくは約0.01:99.99(w/w)〜約40:60(w/w)、より一層好ましくは約0.01:99.99(w/w)〜約30:70(w/w)、一層より好ましくは約0.1:99.9(w/w)〜約20:80(w/w)、なお一層より好ましくは約1:99(w/w)〜約20:80(w/w)、一層より好ましくは約2.5:97.5(w/w)〜約20:80(w/w)、さらにより好ましくは約2.5:97.5(w/w)〜約15:85(w/w)、一層より好ましくは約5:95(w/w)〜約15:85(w/w)であり、及び最も好ましくは約10:90(w/w)である。
【0026】
界面活性剤は、任意の適切な医薬上許容される界面活性剤であってよく、そのような界面活性剤は当業者には周知である。例えば、界面活性剤は、アニオン性、カチオン性又は非イオン性界面活性剤であってよい。好ましくは、界面活性剤は、カチオン性又はアニオン性界面活性剤である。より好ましくは、界面活性剤は、アニオン性界面活性剤である。
【0027】
1つの実施形態では、界面活性剤は、好ましくは約2より大きいHLB(親水性親油性バランス)値を有する。より好ましくは、HLB値は、約4より大きく、なおより好ましくはHLB値は約10より大きく、一層より好ましくはHLB値は約14より大きく、より好ましくはHLB値は約20より大きく、一層より好ましくはHLB値は約25より大きく、より好ましくはHLB値は約30より大きく、最も好ましくはHLB値は約35より大きい。好ましくは、HLB値は、約45未満でなければならない。
【0028】
好ましくは、界面活性剤は、トリエタノールアミン、ヒマワリ油、ステアリン酸、一塩基ナトリウムホスフェート、ナトリウムシトレートジハイドレート、プロピレングリコールアルギネート、オレイン酸、モノエタノールアミン、鉱油及びラノリンアルコール類、メチルセルロース、中鎖トリグリセリド類、レシチン、加水ラノリン、ラノリン、ヒドロキシプロピルセルロース、グリセリルモノステアレート、エチレングリコールパミトステアレート(pamitostearate)、ジエタノールアミン、ラノリンアルコール類、コレステロール、セチルアルコール、セトステアリルアルコール、ヒマシ油、ナトリウムドデシルスルフェート(SDS)、ソルビタンエステル類(ソルビタン脂肪酸エステル類)、ポリオキシエチレンステアレート類、ポリオキシエチレンソルビタン脂肪酸エステル類、ポリオキシエチレンヒマシ油誘導体、ポリオキシエチレンアルキルエーテル類、ポロキサマー、グリセリルモノオレエート、ドキュセートナトリウム、セトリミド、ベンジルベンゾエート、ベンザアルコニウムクロライド、ベンゼトニウムクロライド、ヒプロメロース、非イオン性乳化ろう、アニオン性乳化ろう及びトリエチルシトレート(これらの化合物はHandbook of Pharmaceutical Excipients (4th Edition, editors:RC Rowe, PJ Sheskey, PJ Weller)の中で乳化剤及び界面活性剤として表示されている)から選択される。より好ましくは、界面活性剤は、ナトリウムドデシルスルフェート(SDS)、ソルビタンエステル類(ソルビタン脂肪酸エステル類)、ポリオキシエチレンステアレート類、ポリオキシエチレンソルビタン脂肪酸エステル類、ポリオキシエチレンヒマシ油誘導体、ポリオキシエチレンアルキルエーテル類、ポロキサマー、グリセリルモノオレエート、ドキュセートナトリウム、セトリミド、ベンジルベンゾエート、ベンザアルコニウムクロライド、ベンゼトニウムクロライド、ヒプロメロース、非イオン性乳化ろう、アニオン性乳化ろう及びトリエチルシトレート(これらの化合物はthe Handbook of Pharmaceutical Excipients (4th Edition, editors:RC Rowe, PJ Sheskey, PJ Weller)の中で界面活性剤として表示されている)から選択される。より好ましくは、界面活性剤は、ナトリウムドデシルスルフェート(SDS)、ソルビタンエステル類(ソルビタン脂肪酸エステル)、及びポリオキシエチレンソルビタン脂肪酸エステルから選択される。1つの実施形態では、界面活性剤は、セチルピリジニウムクロライド(CPC)であってよい。又別の実施形態では、界面活性剤は、SDS、CPC、ポリオキシエチレン(20)ソルビタンモノオレート(ポリソルベート80)及びポリソルビタンモノオレエートから選択される。好ましくは、界面活性剤は、SDS、CPC及びポリソルベート80から選択される。より好ましくは、界面活性剤は、SDS及びCPCから選択される。最も好ましくは、界面活性剤は、SDSである。
【0029】
本組成物中では、タキサンの溶解度及び溶解速度を改善するために任意の適切な量の界面活性剤を使用できる。好ましくは、組み合わされたタキサンおよび担体に対する界面活性剤の重量比は、約1:99(w/w)〜約50:50(w/w)、より好ましくは約1:99(w/w)〜約44:56(w/w)、一層より好ましくは約1:99(w/w)〜約33:67(w/w)、なお一層より好ましくは約2:98(w/w)〜約33:67(w/w)、一層より好ましくは約2:98(w/w)〜約17:83(w/w)、さらにより好ましくは約5:95(w/w)〜約17:83(w/w)であり、及び最も好ましくは約9:91(w/w)である。
【0030】
又は、タキサンに対する界面活性剤の重量比は、好ましくは約1:100(w/w)〜約60:1(w/w)、より好ましくは約1:50(w/w)〜約40:1(w/w)、一層より好ましくは約1:20(w/w)〜約20:1(w/w)、なお一層より好ましくは約1:10(w/w)〜約10:1(w/w)、一層より好ましくは約1:5(w/w)〜約5:1(w/w)、さらにより好ましくは約1:3(w/w)〜約3:1(w/w)、一層より好ましくは約1:2(w/w)〜約2:1(w/w)であり、及び最も好ましくは約1:1(w/w)である。
【0031】
1つの実施形態では、本組成物は、腸溶コーティングを含んでいる。任意の適切な腸溶コーティング、例えば、セルロースアセテートフタレート、ポリビニルアセテートフタレート及び適切なアクリル誘導体、例えばポリメタクリレート類を使用できる。腸溶コーティングは、胃内でのタキサンの放出を防止し、それによりタキサンの酸媒介性分解を防止する。さらに、腸溶コーティングは、タキサンが吸収される腸へのタキサンの標的化送達を可能にし、それによってその間はタキサンが溶液中で(結晶化が始まる前に)存在する限定された時間を吸収が可能である部位でのみ過ごすことを保証する。
【0032】
1つの実施形態では、本組成物は、1つ又はそれ以上の追加の医薬上活性な成分をさらに含むことができる。好ましくは、1つ又はそれ以上の追加の医薬上活性な成分は、CYP3A4阻害剤である。適切なCYP3A4阻害剤は、グレープフルーツジュース又はセイヨウオトギリソウ(St. John’s wort)(又はいずれかの成分)、リトナビル、ロピナビル又はイミダゾール化合物、例えばケトコナゾールである。好ましくは、CYP3A4阻害剤はリトナビルである。
【0033】
本組成物が1つ又はそれ以上の追加の医薬上活性な成分を含む場合は、該医薬上活性な成分は、本組成物中に物理的混合物として含めることができる。又は、医薬上活性な成分は、非晶質形態にあってよい。医薬上活性な成分は、他の非晶質成分及び/又は非晶質ではない成分との物理的混合物中での非晶質形態にあってよい。又は、医薬上活性な成分は、タキサン;タキサン及び担体;又はタキサン、担体及び界面活性剤との固体分散体若しくは好ましくは固溶体中にあってよい。追加の医薬上活性な成分が非晶質状態にある場合、又は固体分散体若しくは固溶体中にある場合は、追加の医薬上活性な成分は、溶媒蒸発法、例えば、スプレードライ法を使用して調製されなければならない。
【0034】
本組成物が錠剤形にあり、1つ又はそれ以上の追加の医薬上活性な成分を含む場合は、該1つ又はそれ以上の追加の医薬上活性な成分は、好ましくは該非晶質タキサンと同一錠剤中に、即ち他の成分との単一錠剤中にある。
【0035】
本医薬組成物は、追加の医薬上許容される賦形剤、アジュバント及びビヒクルを含むことができ、それらは当業者には周知である。本発明の医薬組成物中で使用できる医薬上許容される賦形剤、アジュバント及びビヒクルには、イオン交換剤、アルミナ、アルミニウムステアレート、血清タンパク質、例えばヒト血清アルブミン、バッファー物質、例えばホスフェート、グリセリン、ソルビン酸、ソルベートカリウム、飽和植物性脂肪酸の部分グリセリド混合物、水、塩又は電解質、例えば、硫酸プロタミン、リン酸水素二ナトリウム、リン酸水素カリウム、ナトリウムクロライド、亜鉛塩、コロイダル・シリカ、マグネシウムトリシリケート及びウールファットが含まれるがそれらに限定されない。
【0036】
本医薬組成物は、カプセル剤、錠剤、散剤又は被覆顆粒剤を含むがそれらに限定されない任意の経口的に許容できる剤形で経口投与することができる。錠剤は、即時放出型、持続放出型、反復放出型又は徐放型であるように調製できる。錠剤はさらに、又は、発泡性、二重層及び/又はコーティング錠であってよい。カプセル剤は、即時放出型、持続放出型、反復放出型又は徐放型であるように調製できる。潤滑剤、例えばマグネシウムステアレートも又典型的に加えられる。カプセル形にある経口投与のためには、有用な希釈剤には、ラクトース及び乾燥コーンスターチが含まれる。錠剤及びカプセル剤のためには、添加できる医薬上賦形剤は、結合剤、充填剤、充填剤/結合剤、吸着剤、湿潤剤、錠剤分解剤、潤滑剤、流動促進剤などである。錠剤及びカプセル剤は、錠剤及びカプセル剤の外観又は特性を変化させるために、例えば、錠剤又はカプセル剤の味覚を変化させる、又は着色コーティングするためにコーティングすることができる。
【0037】
本組成物に添加できる他の医薬上許容される添加物は、当業者には周知である。
【0038】
本発明は、治療に使用するための上記の組成物をさらに提供する。
【0039】
さらに、本発明は、腫瘍性疾患の治療において使用するための上記の組成物を提供する。
【0040】
本発明により治療される腫瘍性疾患は、好ましくは充実性腫瘍である。充実性腫瘍は、好ましくは、乳房、肺、胃、結腸直腸、頭頸部、食道、肝臓、腎臓、膵臓、膀胱、前立腺、精巣、子宮頸部、子宮内膜、卵巣の癌及び非ホジキン(non-Hodgkin’s)リンパ腫(NHL)から選択される。充実性腫瘍は、より好ましくは、乳房、胃、卵巣、前立腺、頭頸部及び非小細胞肺癌から選択される。
【0041】
本発明により治療できる又別の腫瘍性疾患は、多発性骨髄腫、慢性骨髄単球性白血病(CMML)、急性骨髄性白血病(AML)及びカポジ(Kapsoi’s)肉腫である。さらに、疾患範囲には、骨髄異形成症候群(MDS)も含まれる。
【0042】
本発明は、腫瘍性疾患の治療方法であって、そのような治療を必要とする被験者への有効量の上記の組成物の投与を含む方法をさらに提供する。
【0043】
好ましくは、本方法は、ヒト被験者を治療するために使用される。
【0044】
本発明は、上記の組成物を調製する方法であって、
溶媒蒸発法を使用して非晶質タキサンを調製する工程と、
該組成物を生成するために該非晶質タキサンを親水性担体及び界面活性剤と組み合わせる工程と、を含む方法をさらに提供する。
【0045】
非晶質タキサンは、任意の適切な溶媒蒸発法、例えば、上述した方法により製造することができる。好ましくは、非晶質タキサンは、スプレードライ法により製造できる。
【0046】
非晶質タキサンの調製、並びにそれと担体及び/又は界面活性剤との組み合わせは、単一工程で実施することができ、例えば、タキサン及び担体及び/又は界面活性剤は一緒に(例えば固体分散体若しくは固溶体を形成するために)非晶質化処理にかけられる。
【0047】
好ましくは、本方法は、タキサン及び親水性担体を含む固体分散体を調製する工程と、該固体分散体を該界面活性剤と組み合わせる工程と、を含んでいる。
【0048】
より好ましくは、本方法は、タキサン、親水性担体、及び界面活性剤を含む固体分散体を調製する工程を含んでいる。
【0049】
又別の態様では、本発明は、実質的に非晶質のタキサン及び親水性担体を含む経口投与のための医薬組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製される医薬組成物を提供する。
【0050】
本発明は、実質的に非晶質のタキサン及び親水性担体を含む経口投与のための固体医薬組成物を調製する方法であって、
非晶質タキサンをスプレードライ法により調製する工程と、
該組成物を生成するために該非晶質タキサンを親水性担体と組み合わせる工程と、を含む方法をさらに提供する。
【0051】
本組成物によって提供される利点は、タキサンの溶解度、タキサンの溶解速度及び/又はタキサンが結晶化し始める前に溶液中に留まっている時間量が驚くべき程度まで増加させられることにある。これは、溶媒蒸発法が、タキサンが真に非晶質である可能性が低い非晶質タキサンを製造する他の方法に比較して一層より非晶質のタキサンを製造するからであると考えられる。本タキサンのより非晶質の性質が増加した溶解度特性を提供すると考えられる。
【0052】
本組成物の追加の任意の特徴は、非晶質タキサン、担体及び界面活性剤を含む組成物と同一である。例えば、実質的に非晶質のタキサン及び担体を含んでいる本組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製される組成物は、好ましくは界面活性剤をさらに含んでいる。タキサン、担体、タキサンの結晶化度、担体に対するタキサンの比率、タキサン及び担体の状態の好ましい実施形態などは、上記に規定した通りである。
【0053】
又別の態様では、本発明は、溶液中にタキサン、親水性担体及び界面活性剤を含む医薬組成物をさらに提供する。タキサン、担体及び界面活性剤の同一性、特性などに関連する上記の説明は、本発明のこの態様に同等に適合する。そのような組成物中では、本組成物の全3種の成分が溶液中にある。
【0054】
本医薬組成物は、被験者に投与するための飲料液の形態にあってよい。又は、本医薬組成物は、カプセル剤中に、例えば、該溶液を含有する液体充填カプセルを形成するためにゼラチンカプセル中に配置することができる。
【0055】
本組成物は、適切な溶媒中に上述した固体組成物を溶解させる工程により入手できる。このため、1つの実施形態では、本組成物は、上述した固体組成物を適切な溶媒、例えばトリアセチン中に溶解させる工程により入手可能である。1つの実施形態では、本溶媒は、水性溶媒である。
【0056】
又別の態様では、本発明は、実質的に非晶質のタキサン及び1つ又はそれ以上の医薬上許容される賦形剤を含む経口投与のための固体医薬組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製される固体医薬組成物を提供する。
【0057】
本組成物の特性及び好ましい特徴は、本発明の他の組成物について上述した通りである。
以下では本発明を添付の図面を参照しながら例としてのみ記載する。
【図面の簡単な説明】
【0058】
【図1】パクリタキセル固体分散体とパクリタキセルの物理的混合物との溶解試験の結果を示す図である(条件:900mLのWfI、37℃、75rpm)。
【図2】ナトリウムドデシルスルフェートを含む、及び含まないパクリタキセル(PCT)固体分散体カプセルの溶解試験の結果を示す図である(条件:900mLのWfI、37℃、75rpm)。
【図3】固体分散体中に組み込まれた、又はカプセルに加えられたナトリウムドデシルスルフェートを含むパクリタキセル固体分散体の溶解試験の結果を示す図である(条件:500mLのWfI、37℃、75rpm(カプセルに加えられたSDSについては100rpm))。
【図4】様々な担体を含むパクリタキセル固体分散体の溶解試験の結果を示す図である(条件:500mLのWfI、37℃、100rpm)。
【図5】様々な薬物−担体比を含むパクリタキセル/PVP−K17固体分散体の溶解試験の結果を示す図である(条件:25mLのWfI、37℃、7,200rpm)。
【図6】様々な媒体中でのパクリタキセル固体分散体の溶解試験の結果を示す図である(条件:500mLのFaSSIF(ライトグレー)、37℃、75rpm;又は500mLのSGFsp及び629mLのSIFsp、37℃、75rpm(ダークグレー))。
【図7】5種の異なる調製物のドセタキセル溶解度を示す図である(表13を参照)。A:無水ドセタキセル;B:非晶質ドセタキセル;C:無水ドセタキセル、PVP−K30及びSDSの物理的混合物;D:非晶質ドセタキセル、PVP−K30及びSDSの物理的混合物;E:非晶質ドセタキセル、PVP−K30及びSDSの固体分散体(溶解条件:±6mgのドセタキセル、25mLのWfI、37℃、720rpm)。
【図8】異なる担体を含む固体分散体のドセタキセル溶解度を示す図である(表13を参照)。E:非晶質ドセタキセル、PVP−K30及びSDSの固体分散体;F:非晶質ドセタキセル、HPβ−CD及びSDSの固体分散体(溶解条件:±6mgのドセタキセル、25mLのWfI、37℃、720rpm)。
【図9】様々な鎖長のPVPを含む固体分散体のドセタキセル溶解度を示す図である(表13を参照)。E:非晶質ドセタキセル、PVP−K30及びSDSの固体分散体;G:非晶質ドセタキセル、PVP−K12及びSDSの固体分散体;H:非晶質ドセタキセル、PVP−K17及びSDSの固体分散体;I:非晶質ドセタキセル、PVP−K25及びSDSの固体分散体;J:非晶質ドセタキセル、PVP−K90及びSDSの固体分散体(溶解条件:±6mgのドセタキセル、25mLのWfI、37℃、720rpm)。
【図10】様々な薬物負荷を含む固体分散体のドセタキセル溶解度を示す図である(表13を参照)。E:1/11ドセタキセル;K:5/7ドセタキセル;L:1/3ドセタキセル;M:1/6ドセタキセル;N:1/21ドセタキセル(溶解条件:±6mgのドセタキセル、25mLのWfI、37℃、720rpm)。
【図11】ドセタキセル及びPVP−K30の固体分散体についての文献データ(Chen et al,[13])に比較したドセタキセル、PVP−K30及びSDSの固体分散体の溶解した相対量のドセタキセルに関する溶解試験結果を示す図である。
【図12】ドセタキセル及びPVP−K30の固体分散体についての文献データ(Chen et al.[13])に比較したドセタキセル、PVP−K30及びSDSの固体分散体の溶解した絶対量のドセタキセルに関する溶解試験の結果を示す図である。
【図13】文献データ(Chen et al.[13])と比較したドセタキセルカプセル(PVP−K30+SDSを含む1カプセル当たり15mgのドセタキセル(DXT))の溶解試験の結果を示す図である。
【図14】ドセタキセル、PVP−K30及びSDSの固体分散体の溶解した絶対量に関する溶解試験の結果を示す図である。溶解試験は、模擬腸液サインパンクレアチン(Simulated Intestinal Fluid sine Pancreatin(SIFsp))中で実施した。
【図15】ドセタキセル、PVP−K30及びSDSの固体分散体の溶解した相対量に関する溶解試験の結果を示す図である。溶解試験は、模擬腸液サインパンクレアチン中で実施した。
【図16】第1サイクルにおいてドセタキセル及びリトナビルを同時に摂取した患者のドセタキセル薬物動態曲線を示す図である。第2サイクルでは、患者はドセタキセル及びリトナビルをt=0に同時に摂取し、次にt=4hに追加のブースター用量のリトナビルを摂取した。
【図17】ドセタキセルの液体調製物及び/又はドセタキセルを含む固体分散体(MODRAと呼ぶ)を摂取した患者4例の薬物動態曲線を示す図である。
【図18】ドセタキセルの固体経口調製物(MODRA)を摂取した患者に比較したドセタキセルの液体経口調製物を摂取した患者の薬物動態曲線を示す図である。
【図19】ドセタキセルの静脈内及び経口投与後の薬物動態曲線を示す図である。静脈内及び経口投与の両方をリトナビルの投与と組み合わせた。注意:計算バイオアベイラビリティは、投与された用量について補正されている。
【図20】異なるタイプの界面活性剤を含む5種のドセタキセル調製物の平均溶解曲線を示す図である(n=3)。
【図21】様々な量のSDS界面活性剤を含むドセタキセル調製物の平均溶解曲線を示す図である(n=3)。
【図22】様々な量の界面活性剤を含む4種のドセタキセル調製物の0〜10分間の平均溶解速度を示す図である(n=3)。
【図23】凍結乾燥及び結晶性ドセタキセルのX線粉末回折パターンを示す図である。
【図24】凍結乾燥及び結晶性ドセタキセルDSC(示差走査熱量測定)サーモグラムを示す図である。
【図25】結晶性及び非晶質ドセタキセルの物理的混合物のX線粉末回折パターンを示す図である。
【図26】結晶性及び非晶質ドセタキセルの物理的混合物のDSCサーモグラムを示す図である。
【図27】結晶性ドセタキセル含有量に対する、全熱流量サーモグラムにおける165℃でのピーク面積を示す図である。黒色の線は、0.990のR2を備える線形回帰線である。
【図28】凍結乾燥及び結晶性パクリタキセルのX線粉末回折パターンを示す図である。
【図29】凍結乾燥及び結晶性パクリタキセルのDSCサーモグラムを示す図である。
【図30】ドセタキセル、PVP−K30及びSDSのX線粉末回折パターンを示す図である。
【図31】PVP−K30、SDS及びドセタキセルのDSCサーモグラムを示す図である。
【図32】1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを備える物理的混合物及び固体分散体のX線粉末回折スペクトルを示す図である。
【図33】1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを備える物理的混合物及び固体分散体のDSCサーモグラムを示す図である。
【図34】凍結乾燥法及びスプレードライ法により製造された固体分散体のX線粉末回折スペクトルを示す図である。
【図35】凍結乾燥法及びスプレードライ法により製造された固体分散体のDSCサーモグラムを示す図である。
【図36】凍結乾燥法及びスプレードライ法により製造された固体分散体(n=4)の平均溶解スクリーニング曲線を示す図である。
【図37】PVP−K30(n=4)、PEG1500(n=2)、PEG6000(n=2)及びPEG20000(n=2)を含むドセタキセルの固体分散体の平均溶解スクリーニング曲線を示す図である。
【図38】PVP−K30(n=4)及びPVP−VA64(n=2)を含むドセタキセルの固体分散体の平均溶解スクリーニング曲線を示す図である。
【図39】20mgのドセタキセル錠剤(n=2)及び10mgのドセタキセルカプセル(n=6)の平均溶解曲線を示す図である。
【図40】20mgのドセタキセル錠剤(n=2)及び10mgのドセタキセルカプセル(n=6)の0〜10分間のドセタキセルの平均放出速度を示す図である。
【図41】非晶質(スプレードライ)及び結晶性リトナビルのDSCサーモグラムを示す図である。
【図42】ドセタキセル、リトナビル、PVP−K30及びSDSの組み合わせのスプレードライ法固体分散体粉末のDSCサーモグラムを示す図である。
【図43】37℃及び50RPMで1,000mLの0.1N HCl中におけるドセタキセル/リトナビル/PVP−K30/SDSカプセルの溶解プロファイルを示す図である。
【発明を実施するための形態】
【実施例1】
【0059】
パクリタキセルの経口調製物
1.1:固体分散体と物理的混合物
この実験では、SDSと混合されたパクリタキセル及びPVP−K17の固体分散体を含む組成物の溶解度及び溶解速度を無水パクリタキセル、PVP−K17及びSDSの物理的混合物と比較した。
【0060】
PVP−K17中のパクリタキセル固体分散体の5mgカプセル
PVP−K17中の20%パクリタキセルの固体分散体は、10mLのt−ブタノール中に100mgのパクリタキセル及び6.67mLの水中に400mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(条件については表1を参照)。25mgの20%パクリタキセル/PVP−K17固体分散体(=5mgのパクリタキセル)を125mgのラクトース、30mgのナトリウムドデシルスルフェート、及び30mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表2参照)。
【0061】
【表1】
【0062】
【表2】
【0063】
PVP−K17との物理的混合物中のパクリタキセルの5mgカプセル
物理的混合物は、5mgの無水パクリタキセルを20mgのPVP、125mgのラクトース、30mgのナトリウムドデシルスルフェート、及び30mgのクロスカルメロースナトリウムと混合することにより調製した。結果として生じた粉末混合物はカプセル充填した。
【0064】
【表3】
【0065】
溶解試験
両方のカプセル調製物は、75rpmの回転速度を備えるUSP2(パドル式)溶解装置内で37℃に維持した900mLの注射用水中で試験した。第1実験では、各調製物の1つのカプセルを使用した。第2実験では、各調製物の2つのカプセルを使用した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0066】
【表4】
【0067】
結果及び結論
結果は図1に示した。溶解したパクリタキセルの量は、ラベル記載内容(5及び10mg)に比較して表示した。パクリタキセルの溶解は、固体分散体中へのPVPの組み込みによって大きく改善されることは明白に見て取ることができる。溶解したパクリタキセルの最高量は、物理的混合物が使用された場合はラベル記載内容に比較して20%未満に留まる。固体分散体が使用された場合は、溶解度は約65%(5mgのパクリタキセル)又は70%超(10mgのパクリタキセル)である。10mgのパクリタキセル実験については、これは約8μg/mLの絶対溶解度に対応し、これは約15分間後に達成される。このため、固体分散体は溶解度を有意に増加させ、さらにどちらもバイオアベイラビリティにとって重要である迅速な溶解速度も提供する。
【0068】
固溶体又は固体分散体では、担体の非晶質状態は、担体及びタキサンの混合を通して可能になる。担体は、保管中並びに水性媒体内での溶解中の結晶化を防止する。
【0069】
1.2:カプセル調製物へのナトリウムドデシルスルフェートの添加
この実験ではカプセル中の界面活性剤SDSの存在又は非存在が溶解度に及ぼす作用を決定した。
【0070】
PVP−K17中の20%パクリタキセル固体分散体
固体分散体は、10mLのt−ブタノール中に100mgのパクリタキセル及び6.67mLの水中に400mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0071】
ナトリウムドデシルスルフェートを含まない5mgのパクリタキセルカプセル
25mgの20%パクリタキセル/PVP−K17固体分散体(=5mgのパクリタキセル)を125mgのラクトースと混合し、カプセル充填した(表5を参照)。
【0072】
【表5】
【0073】
ナトリウムドデシルスルフェートを含む5mgのパクリタキセルカプセル
25mgの20%パクリタキセル/PVP−K17固体分散体(=5mgのパクリタキセル)を125mgのラクトース、30mgのナトリウムドデシルスルフェート、及び30mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表6参照)。
【0074】
【表6】
【0075】
溶解試験
両方のカプセル調製物は、75rpmの回転速度を備えるUSP2(パドル式)溶解装置内で37℃に維持した900mLの注射用水中で試験した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0076】
結果及び結論
結果は図2に示した。溶解したパクリタキセルの量は、ラベル記載内容に比較して表示した(この場合には5mg)。凍結乾燥タキサン及び担体固体分散体の多孔性は、粉末形にある場合の迅速な溶解を保証するために十分に高かった(結果は示していない)。しかし粉末がカプセル中に圧縮されると、湿潤性は劇的に減少した。このため、カプセル又は錠剤中に圧縮される場合は、固体分散体を湿潤させるために界面活性剤が必要とされる。
【0077】
図2からパクリタキセルの溶解は、界面活性剤のナトリウムドデシルスルフェートの添加により大きく改善されることを明白に見て取ることができる。以前の実験は、クロスカルメロースナトリウム、より多くのラクトースの添加、又はより大きなカプセルの使用がカプセル調製物の増加した溶解速度を生じさせないことを示した。さらに又、この実験はSDSのような界面活性剤を用いると、約10〜15分間で最高溶解が達成されることを示している。
【0078】
1.3:固体分散体調製物へのナトリウムドデシルスルフェートの添加
この実験では、固体分散体へのSDSの添加が溶解度に及ぼす作用が決定された。
【0079】
PVP−K17中の40%パクリタキセル固体分散体
固体分散体は、60mLのt−ブタノール中に600mgのパクリタキセル及び40mLの水中に900mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0080】
PVP−K17及び10%ナトリウムドデシルスルフェート中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのPVP−K17及び62.5mgのナトリウムドデシルスルフェート(SDS)を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/ナトリウムドデシルスルフェート/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0081】
パクリタキセル/PVP−K17固体分散体の25mgパクリタキセルカプセル
62.5mgの40%パクリタキセル/PVP−K17固体分散体(=25mgのパクリタキセル)を160mgのラクトース、30mgのナトリウムドデシルスルフェート、及び10mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表7参照)。
【0082】
【表7】
【0083】
パクリタキセル/PVP−K17/ナトリウムドデシルスルフェート固体分散体の25mgパクリタキセルカプセル
68.75mgの40%パクリタキセル/PVP−K17/10%ナトリウムドデシルスルフェート固体分散体(=25mgのパクリタキセル)を160mgのラクトース及び10mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表8参照)。
【0084】
【表8】
【0085】
溶解試験
両方のカプセル調製物は、USP2(パドル式)溶解装置内で37℃に維持した500mLの注射用水中で試験した。回転速度は、パクリタキセル/PVP−K17/ナトリウムドデシルスルフェート固体分散体を含むカプセルについては75rpm及びパクリタキセル/PVP−K17固体分散体を含むカプセルについては100rpmに設定した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0086】
結果及び結論
結果は図3に示した。溶解したパクリタキセルの量は、ラベル記載内容に比較して表示した(この場合には25mg)。固体分散体中に組み込まれたナトリウムドデシルスルフェートを含むカプセルからのパクリタキセルの溶解は、カプセルに加えられたナトリウムドデシルスルフェートを含むカプセルからのパクリタキセルの溶解に匹敵することを明白に見て取ることができる。さらに6.25mgのナトリウムドデシルスルフェートしか固体分散体中に組み込むために使用されなかったが、30mgのナトリウムドデシルスルフェートはカプセル調製物への添加剤として使用された。これは、界面活性剤が固体分散体中に組み込まれる場合は類似の結果を達成するためにカプセル中に組み込まれる場合より少ない界面活性剤が必要とされることを示している。この実験からの又別の驚くべき結果は、両方の組成物が約26μg/mLの絶対パクリタキセル溶解度を提供し、このレベルは20〜30分間で達することである。この結果は、以前に達成されていたより高い溶解度及び迅速な溶解速度を提供する。
【0087】
1.4:担体が及ぼす影響
実施例1.4の実験において使用された固体分散体は、最初の実験が薬物負荷間で明白な差を示さなかった後に製造された。40%の薬物負荷を選択したのは、これらの調製物が上記で記載した実験において20%薬物負荷調製物と同等に機能し、1つの錠剤又はカプセルにおいてより多くのタキサンを送達する可能性を提供するからであった。
【0088】
PVP−K12中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのPVP−K12を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K12/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0089】
PVP−K17中の40%パクリタキセル固体分散体
固体分散体は、60mLのt−ブタノール中に600mgのパクリタキセル及び40mLの水中に900mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0090】
PVP−K30中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのPVP−K30を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K30の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0091】
HP−シクロデキストリン中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのHP−シクロデキストリンを溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらHP−シクロデキストリンの水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0092】
25mgのパクリタキセル固体分散体カプセル
62.5mgのパクリタキセル担体/固体分散体(=25mgのパクリタキセル)を160mgのラクトース、30mgのナトリウムドデシルスルフェート、及び10mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表9参照)。
【0093】
【表9】
【0094】
溶解試験
全カプセル調製物は、100rpmの回転速度を備えるUSP2(パドル式)溶解装置内で37℃に維持した500mLの注射用水中で試験した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0095】
結果及び結論
2〜3回の実験の平均結果は、図4に示した。溶解したパクリタキセルの量は、ラベル記載内容に比較して表示した(この場合には25mg)。PVP−K30固体分散体からのパクリタキセルの溶解は、PVP−K17固体分散体からのパクリタキセルの溶解と同等に迅速であることを明白に見て取ることができる。しかし、溶解したパクリタキセルの量は、PVP−K30固体分散体の症例において4時間の実験を通して高いままであった。
【0096】
ポリマー性担体の鎖長は、水性環境内で結晶化するための時間を決定する。
【0097】
1.5:薬物/担体比の影響
実施例1.5の実験において使用した固体分散体は、最初の実験が担体間で明白な差を示さなかった後に製造された。これらの最初の実験は、実施例1.4のより詳細な実験の前に実施された。結果として、PVP−K17がその後の実験のための担体として自由裁量で選択された。
【0098】
PVP−K17中の10%パクリタキセル固体分散体
固体分散体は、10mLのt−ブタノール中に100mgのパクリタキセル及び40mLの水中に900mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0099】
PVP−K17中の25%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に750mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0100】
PVP−K17中の40%パクリタキセル固体分散体
固体分散体は、60mLのt−ブタノール中に600mgのパクリタキセル及び6.67mLの水中に900mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0101】
PVP−K17中の75%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に83mgのPVP−K17を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17の水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0102】
100%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセルを溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながら16.67mLの水に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0103】
溶解試験
およそ4mgのパクリタキセルと同等の量の固体分散体粉末を50mLのビーカー内に配置した。磁気攪拌棒及び25mLの水をビーカーに加えた。この溶液を720rpmで攪拌した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0104】
結果及び結論
2〜3回の実験の平均結果は、図5に示した。溶解したパクリタキセル(PCT)の量は、ラベル記載内容に比較して表示した(この場合にはおよそ4mg)。薬物/担体比が及ぼす影響は、図5から直ちに明らかである。パクリタキセルのピーク濃度値は、薬物/担体比と反比例していた。最高ピーク濃度は最低薬物/担体比(10%)の場合に達するが、最低ピーク濃度は最高薬物/担体比(100%)の場合に達する。さらに、10%薬物/担体比固体分散体のAUC値が最高であり、その後に25、40、75及び100%薬物/担体比固体分散体が続いた。
【0105】
薬物の量に比した担体の量が、水性環境内で結晶化するための時間を決定する。
【0106】
1.6:腸溶コーティングの存在
PVP−K17及びド10%ナトリウムドデシルスルフェート中の40%パクリタキセル固体分散体
固体分散体は、25mLのt−ブタノール中に250mgのパクリタキセル及び16.67mLの水中に375mgのPVP−K17及び62.5mgのナトリウムドデシルスルフェート(SDS)を溶解させることにより調製した。パクリタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K17/ナトリウムドデシルスルフェート/水の溶液に加えた。最終混合液は、最高充填レベルが2mLである8mLバイアルに移した。その後にt−ブタノール及び水を凍結乾燥法により除去した(表1を参照)。
【0107】
パクリタキセル/PVP−K17/ナトリウムドデシルスルフェート固体分散体の25mgパクリタキセルカプセル
68.75mgの20%パクリタキセル/PVP−K17/10%ナトリウムドデシルサルフェート固体分散体(=25mgのパクリタキセル)を160mgのラクトース及び10mgのクロスカルメロースナトリウムと混合した。結果として生じた粉末混合物はカプセル充填した(表10参照)。
【0108】
【表10】
【0109】
溶解試験
これらのカプセルに2種の異なる溶解試験を2回ずつ受けさせた。第1回試験は、ペプシンを含まない500mLの模擬胃液(SGFsp;表11を参照)中での2時間の溶解試験、その後にペプシンを含まない629mLの模擬胃液(SIFsp;表11を参照)中での2時間の溶解試験から構成される2段階式溶解試験であった。第2回試験は、500mLの絶食状態の模擬腸液(FaSSIF;表12を参照)媒体中で4時間にわたって実施した。
【0110】
両方の溶解試験は、37℃に維持し、パドル回転速度75rpmで維持した500mLの媒体を備えるUSP2(パドル式)溶解装置内で実施した。SGFsp媒体は、129mLのスイッチ媒体の添加によりSIFsp媒体に変更した。サンプルを様々な時点で採取し、HPLC−UVにより分析した(表4を参照)。
【0111】
【表11】
【0112】
【表12】
【0113】
結果及び結論
結果は図6に示した。溶解したパクリタキセルの量は、ラベル記載内容に比較して表示した(この場合には25mg)。絶食状態の模擬腸液中でのパクリタキセルの溶解度は、模擬胃液(SGFsp)中においてよりおよそ20%高かった。SGFsp中に2時間置いた後の溶液中のパクリタキセルの量は、媒体を模擬腸液(SIFsp)に変更した場合は僅かにしか増加しなかった。
【0114】
腸溶コーティングは、胃内でのタキサンの放出を防止し、それにより活性成分の分解を防止する。さらに、腸溶コーティングは、タキサンが吸収される腸へのタキサンの標的化送達を可能にし、それによってその間はタキサンが溶液中で(結晶化が始まる前に)存在する限定された時間を吸収が可能である部位でのみ過ごすことを保証する。
【実施例2】
【0115】
ドセタキセルの経口調製物
材料及び方法
以下の実験において使用した調製物は、以下で略述する方法及び表13に記載した組成物に従って調製した。
【0116】
純粋無水ドセタキセル
無水ドセタキセルは、ScinoPharm社(台湾国)から入手したままで使用した。
【0117】
純粋非晶質ドセタキセル
ドセタキセルは、300mgのドセタキセル無水物を30mLのt−ブタノール中に溶解させることにより非晶質化した。ドセタキセル/t−ブタノール溶液は、絶え間なく攪拌しながら20mLのWfI(注射用水)に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronorm、サイズ1/9)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表14を参照)。
【0118】
物理的混合物
物理的混合物は、150mgのドセタキセル及び対応する量の担体及び界面活性剤(表13を参照)を乳鉢と乳棒で混合することにより調製した。
【0119】
固体分散体
固体分散体は、30mLのt−ブタノール中の300mgの無水ドセタキセル、及び20mLの注射用水中の対応する量の担体及び界面活性剤(表13を参照)を溶解させることにより入手した。ドセタキセル/t−ブタノール溶液は、絶え間なく攪拌しながら担体/界面活性剤/WfIの溶液に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronorm、サイズ1/9)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表14を参照)。
【表13】
【0120】
【表14】
【0121】
【表15】
【0122】
2.1:調製物のタイプ
第1回実験では、調製物のタイプがドセタキセルの溶解度に及ぼす影響を試験した。調製物A〜Eについて実施した溶解試験からのデータを比較した。結果は図7に示した。調製物Eは4回ずつ試験し、調製物A〜Dを2回ずつ試験した。
【0123】
結果
調製物A(純粋ドセタキセル無水物)は5分間の攪拌後におよそ12μg/mL(4.7%の全ドセタキセルが存在する)の最高濃度に到達し、15分間の攪拌後におよそ6μg/mL(2%)の平衡濃度に達する。
【0124】
調製物B(純粋非晶質ドセタキセル)は0.5分後に最高32μg/mL(13%)に到達し、10〜60分後の溶解度は調製物Aに匹敵する。
【0125】
調製物C(無水ドセタキセル、PVP−K30及びSDSの物理的混合物)は5分後におよそ85μg/mL(37%)の濃度に達する。15〜25分後に、ドセタキセル濃度は85μg/mL(37%)〜30μg/mL(12%)へ急速に低下し、その後にさらに60分後には20μg/mL(9%)へ低下する。
【0126】
調製物D(非晶質ドセタキセル、PVP−K30及びSDSの物理的混合物)は7.5分後におよそ172μg/mL(70%)の最高ドセタキセル濃度に達する。7.5〜20分後に、溶液中のドセタキセルの量は24μg/mL(10%)に低下する。60分後、19μg/mL(7%)の平衡濃度に達する。
【0127】
調製物E(非晶質ドセタキセル、PVP−K30及びSDSの固体分散体)は5分後に到達した213μg/mL(90%)の最高最大濃度を有する。10〜25分後に、溶液中のドセタキセルの量は迅速に低下し、結果として45分後に20μg/mL(8%)の平衡濃度を生じさせる。
【0128】
結論
全調製物は、最初はより高い溶解度を示したが、これらは45〜60分間の攪拌後に平衡溶解度へ減少する。溶解度の減少は、過飽和溶液の結果としてドセタキセルの結晶化によって生じる。過飽和度は、薬物の物理的状態に、即ち薬物が非晶質又は結晶性のいずれであるかに依存する。PVP−K30が担体である場合は、過飽和状態は、ドセタキセルの溶解度が急速に減少しないようにより長時間にわたり維持される。さらに、これらの結果は、非晶質ドセタキセルを使用することが無水結晶性ドセタキセルに比較してドセタキセルの溶解度を有意に増加させることを示している。さらに、非晶質ドセタキセルは、約5〜7.5分間でピークに達する相当に高い溶解速度を示す。
【0129】
この実験は、溶液中のドセタキセルの量が無水ドセタキセルをPVP−K30及びSDSと物理的混合することによって、及び非晶質ドセタキセルをPVP−K30及びSDSと物理的混合することによってより一層顕著に増加することを示した。しかし溶解度における最大増加は、PVP−K30及びSDSの固体分散体中にドセタキセルを組み込むことによって達成される。
【0130】
2.2:担体のタイプ
第2回実験では、担体のタイプがドセタキセルの溶解度に及ぼす影響を試験した。調製物E及びFについて実施した溶解試験からのデータを比較した。結果は図8に示した。調製物Eは4回ずつ試験し、調製物Fを2回ずつ試験した。
【0131】
結果
調製物E(非晶質ドセタキセル、PVP−K30及びSDSの固体分散体)は5分後に到達した213μg/mL(90%の全ドセタキセルが存在する)の最高最大濃度を有する。10〜25分後に、溶液中のドセタキセルの量は迅速に低下し、結果として45分後に20μg/mL(8%)の平衡濃度を生じさせる。
【0132】
調製物F(非晶質ドセタキセル、HPβ−CD及びSDSの固体分散体中)は、約2分後におよそ200μg/mL(81%)の最高ドセタキセル濃度に達する。5〜10分後に、溶液中のドセタキセルの量は16μg/mL(6%)の値に低下し、45分後に、11μg/mL(4%)の平衡濃度に達する。
【0133】
結論
この実験は、PVP−K30及びHPβ−CDがドセタキセルの溶解度を増加させることを示した。PVP−K30がHPβ−CDと比較して担体として使用された場合は、最大ドセタキセル濃度は僅かに高く、過飽和の状態は長時間維持されたので、ドセタキセルの溶解度は経時的に迅速には減少しなかった。さらに、ドセタキセルの沈降後に到達した平衡濃度は、HPβ−CDと比較してPVP−K30を用いた場合の方が高い。
【0134】
2.3:鎖長
第3回実験では、PVP鎖長がドセタキセルの溶解度に及ぼす影響を試験した。調製物E及びG〜Jについて実施した溶解試験からのデータを比較した。結果は図9に示した。調製物Eは4回ずつ試験し、調製物G〜Jを2回ずつ試験した。
【0135】
結果
調製物G(PVP−K12)は、5分後に206μg/mL(全ドセタキセルの77%が存在する)の最高ドセタキセル濃度に達する。5〜30分後に、溶液中のドセタキセルの量は20μg/mL(7%)の値に低下し、45分後には、ドセタキセル濃度は17μg/mL(6%)である。
【0136】
調製物H(PVP−K17)は5分後に200μg/mL(83%)の最高ドセタキセル濃度に達し、この濃度を攪拌10分間まで維持し、その後に溶液中のドセタキセルの量は15分後には44μg/mL(18%)へ、及び30分後には22μg/mL(9%)に迅速に低下する。45〜60分後の平衡濃度は、およそ21μg/mL(8%)であった。
【0137】
調製物I(PVP−K25)は、5分間の攪拌後に214μg/mL(88%)の最高ドセタキセル濃度に達する。溶液中のドセタキセルの量は10〜30分後に22μg/mL(9%)へ減少し、及び60分後にドセタキセルの濃度は19μg/mL(8%)である。
【0138】
調製物E(PVP−K30)は213μg/mL(90%)の最高ドセタキセル濃度を有するが、これは5分後に達する。10〜25分後に、溶液中のドセタキセルの量は迅速に低下し、結果として45分後に20μg/mL(8%)の平衡濃度を生じさせる。
【0139】
調製物J(PVP−K90)は、10分間の攪拌後に214μg/mL(88%)の最高ドセタキセル濃度に達する。15分後に、溶液中のドセタキセルの量は依然として151μg/mL(61%)である。60分後に、ドセタキセル濃度は、19μg/mL(7%)に低下した。
【0140】
結論
この実験は、PVPの鎖長が過飽和度及び過飽和が維持される期間の両方に影響を及ぼすことを示している。より長いPVP鎖長の使用はより高い最高ドセタキセル濃度及びより長い過飽和期間、そこでより長期間にわたってより高い溶解度を生じさせる。
【0141】
2.4:薬物負荷
第4回実験では、薬物負荷がドセタキセルの溶解度に及ぼす影響を試験した。調製物E及びK〜Nについて実施した溶解試験からのデータを比較した。結果は図10に示した。調製物Eは4回ずつ試験し、調製物K〜Nを2回ずつ試験した。
【0142】
調製物N(全組成物の重量で1/21のドセタキセル;5:95(w/w)のドセタキセル 対 PVP)は、10分後に197μg/mL(全ドセタキセルの79%が存在する)の最高ドセタキセル濃度に達する。15分後に、溶液中のドセタキセルの量は依然として120μg/mL(48%)であり、15〜30分後には、ドセタキセル濃度は24μg/mL(12%)へ減少する。60分後に、ドセタキセル濃度は、20μg/mL(8%)である。
【0143】
調製物E(全組成物の重量で1/11のドセタキセル;10:90(w/w)のドセタキセル 対 PVP)は、5分後に213μg/mL(90%)の最高ドセタキセル濃度を有する。10〜30分後に、溶液中のドセタキセルの量は迅速に低下し、45分後には20μg/mL(8%)の平衡濃度に達する。
【0144】
調製物M(全組成物の重量で1/6のドセタキセル;20:80(w/w)のドセタキセル 対 PVP)は、10分間の攪拌後に196μg/mL(80%)のドセタキセル濃度を有する。溶液中のドセタキセルの量は10〜30分後に25μg/mL(10%)へ減少し、60分後にドセタキセルの濃度は18μg/mL(7%)である。
【0145】
調製物L(全組成物の重量で1/3のドセタキセル;40:60(w/w)のドセタキセル 対 PVP)は、176μg/mL(71%)のドセタキセル濃度に達する。10〜15分後に、溶液中のドセタキセルの量は46μg/mL(18%)に迅速に低下し、60分後には、溶液中のドセタキセル濃度は18μg/mL(7%)である。
【0146】
調製物K(全組成物の重量で5/7のドセタキセル;75:25(w/w)のドセタキセル 対 PVP)は、5分間の攪拌後に172μg/mL(71%)の最高ドセタキセル値に達する。5〜10分後に、ドセタキセル濃度は42μg/mL(17%)に急速に低下し、60分後に、18μg/mL(7%)のドセタキセル濃度に達する。
【0147】
結論
この実験は、固体分散体中で使用したドセタキセルの量に比較したPVP−K30の量は、過飽和度及び過飽和が維持される期間の両方に影響を及ぼすことを示している。より高い薬物負荷の使用はより低い最高ドセタキセル濃度、及びより短い過飽和期間、そこで経時的により低い溶解度を生じさせる。
【0148】
2.5:先行技術組成物との溶解度の比較
この実験では、15mgのドセタキセル、135mgのPVP−K30及び15mgのSDSの固体分散体を含有する組成物を、Chen et al.[13]に開示された5mgのドセタキセル及びPVP−K30の固体分散体を含む組成物の文献データと比較した。溶解度試験結果は、Chen et al.[13]に記載された溶解試験を使用して入手し、図11及び12に表示した。溶解試験はさらに、模擬腸液中でも実施し、Chenの文献データと比較した。結果は図13に示した。
【0149】
結果
図11から、Chen et al.の組成物は、900mLの水中での組成物中の5mgのドセタキセルの最高約80%を溶解させることができる。この最高値に達するには5時間超を要した。ドセタキセル、PVP−K30及びSDS組成物は、約60分間で15mgのドセタキセルの100%を溶解させた。
【0150】
図12には、ドセタキセルの絶対濃度を表示した。Chenの組成物は、約5時間後に約4.2μg/mLの最高ドセタキセル濃度を生じさせた。ドセタキセル、PVP−K30及びSDS組成物は、約60分後に約16.7μg/mLの最高ドセタキセル濃度をもたらした。
【0151】
図13では、ドセタキセルカプセルは、28μg/mL(>90%の溶解度)の溶解度に達する。Chen et al.に記載された固体分散体(ドセタキセル+PVP−K30)は、4.2μg/mL(900mL中への溶解について試験した80%未満の5mgのドセタキセル固体分散体)の溶解度に達する。そこで本カプセル調製物は、より高い溶解速度とともに6.6倍良好な溶解度に達する(最高溶解速度はChenによる90〜120分後であるのに対して30分後に到達した)。
【0152】
結論
これらの結果から、ドセタキセル、PVP−K30及びSDS組成物は、Chenの組成物に比較してより高速の溶解速度及びより高い溶解度を生じさせた。バイオアベイラビリティについては、薬物がどのように高速で溶解するのか、及びどのような溶解度に0.5〜1.5時間で達するのかを見るのかが重要である。
【0153】
Chenの結果から、当業者は、組成物中のドセタキセルの量を増加させるとドセタキセルの絶対溶解度を増加させるであろうと考察しないであろう。Chenの組成物は900mLの水中で5mgのドセタキセル中80%(即ち、4mg)しか溶解させないので、15mgへドセタキセルの量を増加させることが4mgより多いドセタキセルが溶解することを生じさせるであろうとは予想しないであろう。そこでChenによる15mgのドセタキセル組成物は、ドセタキセル、PVP−K30及びSDS組成物についての100%に比較して最高約27%のドセタキセルを溶解させると予想されるであろう。このため、ドセタキセル、PVP−K30及びSDS組成物は、Chenに比較して驚くべき優れた結果を提供する。
【0154】
2.6:模擬腸液サインパンクレアチン(SIFsp)中での溶解試験
この実験では、ドセタキセル、PVP−K30及びSDSの固体分散体を含有するカプセルの溶解を模擬腸液サインパンクレアチン(SIFsp)中で試験した。カプセルは、調製物Eによる15mgのドセタキセルを含有していた(表13を参照)。SIFspは、USP28に従って調製した。15mgのドセタキセルを含有するカプセルは、75rpmで攪拌しながら37℃の500mLのUSP SIFsp中に溶解させた。結果は図14及び15に示した。
【0155】
図14及び15は、ほぼ100%のドセタキセルが溶解したことを示している。これは約29μg/mLの絶対ドセタキセル濃度と等価であり、約30分間で達成される。そこで、本組成物は、相当に短時間で相当に高い溶解度を提供する。
【0156】
2.7:安定性
調製物E(表13を参照)による、そして臨床試験のためのカプセル中で使用された(以下の実施例を参照)ドセタキセル、PVP−K30及びSDSの固体分散体は、4〜8℃で保管された場合に少なくとも180日間にわたって化学的(分解なし)及び物理的に(溶解特性に変化なし)の両方が安定性であることが見いだされた。
【実施例3】
【0157】
調製物を用いた臨床試験のデータ
材料及び方法
現在進行中の第I相臨床試験には患者10例が参加した。
【0158】
これらの患者には、以下の番号が与えられた:301、302、303、304、305、306、307、308、309及び310。
【0159】
これらの患者には、ドセタキセルの液体調製物又はドセタキセル、PVP−K30及びSDSの固体分散体を含む固体組成物からなる薬剤(以下ではMODRAと呼ぶ)が与えられた。
【0160】
液体調製物
ドセタキセルの用量:全患者(20mgのドセタキセルを摂取した患者306を除く)に対して30mg。30mgの用量は、以下の通りに調製した:静脈内投与用の3.0mLのTaxotere(登録商標)プレミックス(ポリソルベート80(25(v/v)%)、エタノール(10(w/w)%)、及び水)中に1mL当たり10mgのドセタキセルを含有する)を最終容量が25mLとなるように水と混合した。この溶液は、患者に100mLの水道水とともに経口摂取された。
【0161】
MODRA
ドセタキセルの用量:30mg;1カプセルに付き15mgのドセタキセルを含む2カプセルが摂取された。本臨床試験のその後の試験のためには、先行実施例からの調製物E(1/11ドセタキセル、9/11 PVP−K30及び1/11 SDS)が選択された。新規バッチの調製物Eは、120mLのt−ブタノール中の1,200mgのドセタキセル無水物、及び80mLの注射用水中の10,800mgのPVP−K30及び1,200mgのSDS(表13を参照)を溶解させることにより製造した。ドセタキセル/t−ブタノール溶液は、絶え間なく攪拌しながらPVP−K30/SDS/WfI溶液に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronorm、サイズ1/3)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表14を参照)。
【0162】
サイズ0の計60個のゼラチンカプセルに15mgのドセタキセルと同等の量の固体分散体を充填し、HPLCアッセイを使用して固体分散体1mg当たりのドセタキセルの正確な量を決定した。このアッセイは、カプセルが15mgのドセタキセルを含有することを確認した。
【0163】
患者は、100mLの水道水とともに朝の胃が空のときにこの薬剤を経口摂取した。
【0164】
患者の治療
患者301、302、303、304及び305は、液体調製物だけを摂取した。
【0165】
患者306は、第1サイクルでは液体調製物としての20mgのドセタキセル+リトナビル、及び第2サイクルではドセタキセル摂取4時間後に追加のリトナビルを摂取した以外は同一薬剤を摂取した。
【0166】
患者307、308、309及び310は、液体調製物及び/又はMODRAを摂取した。治療サイクルは、1週間間隔で投与された。
【0167】
施設内ガイドラインに従って、経口及び静注ドセタキセルの両方について、全患者が経口デキサメタゾンを用いて治療された。用量4mgのデキメタソンは、治験薬の1時間前に投与され、その後は12時間毎に4mg投与された(2回)。ドセタキセル治療の1時間前、患者は悪心及び嘔吐を予防するために1mgのグラニセトロン(Kytril(登録商標))も又摂取した。
【0168】
薬物投与後、血液サンプルを薬物動態分析のために採取した。投与前にはブランクサンプルを採取した。血液サンプルは遠心分離し、血漿を分離し、分析時まで直ちに−20℃で保管した。分析は、GLP(医薬品安全性試験実施基準)認証検査室において妥当性の確認されたHPLC法を用いて実施した[17]。
【0169】
結果
表16は、個々の薬物動態試験結果の要約を表示している。
【0170】
【表16】
【0171】
患者301、302、303、304、305、307、309及び310は、液体調製物を摂取した。AUCの平均値、及び平均値についての95%信頼区間(無限大まで補外させた):1156(+348)ng*h/mL.
個人間変動性は、85%である。
【0172】
患者306は第1サイクルにおいて100mgのリトナビルと同時に20mgのドセタキセル(液体調製物として)を摂取し、1週間後の第2サイクルでは同一の組み合わせを摂取したが、ドセタキセルの摂取4時間後には追加の100mgのリトナビルを摂取した。即ち、2用量のリトナビルを第1回はt=0、及び第2回はt=4hで摂取した。薬物動態曲線は、図16に示した。
【0173】
患者307、308、309及び310は、液体調製物及び/又はMODRAを摂取した。薬物動態曲線は、図17に示した。
【0174】
図18は、液体調製物を摂取した患者(307、309及び310)の薬物動態曲線並びにMODRAを摂取した患者4例(307、308、309及び310)の全経過(n=6)を示している。
【0175】
どちらも100mgのリトナビルと組み合わせた、液体調製物 対 MODRAの薬物動態試験結果を以下に要約した。
液体調製物(30mgのドセタキセル)
AUCinf(平均値の95%信頼区間):1156(808−1504)ng*h/mL.
個人間変動性:85%(n=8)
MODRA(30mgのドセタキセル)
AUCinf(平均値の95%信頼区間):768(568−968)ng*h/mL.
個人間変動性:29%(n=4)
個人内変動性:33%(n=2)
【0176】
MODRAの平均AUCは、4例の患者からの6本の曲線を使用して計算した。各患者に投与されたMODRAの第1用量を使用して個人間変動性を計算した。個人内変動性は、2用量のMODRAを摂取した患者307及び308からのデータに基づいている。
【0177】
結論
試験したドセタキセル液体調製物は、新規なカプセル調製物(MODRA)で投与された同一用量(30mg)よりおよそ1.5倍高いAUC値を生じさせる。
【0178】
本液体調製物の個人間変動性は高いが(85%)、カプセル調製物の個人間変動性は実質的に低い(29%)。これは新規なカプセル調製物の重要な特徴であり、はるかに優れた予測可能なドセタキセル曝露を提供する。さらに安全性の理由からも、低い個人間変動性は経口化学療法レジメンにおいて極めてはるかに望ましい。
【0179】
個人内変動性(限定されたデータ)は、個人間変動性と同一の程度にある。
【0180】
ドセタキセル投与の4時間後に摂取された100mgのリトナビルの第2ブースト投与用量は、ドセタキセルのAUCを1.5倍増加させる。
【0181】
経口カプセル調製物と静脈内投与との比較
図19は、静脈内投与(静脈内1時間注入としての20mgのドセタキセル、Taxotere(登録商標)(n=患者5例)及びドセタキセルの経口投与(30mgのドセタキセル;MODRAカプセル、上記参照)(n=患者4例;6コース)後の薬物動態曲線を示している。静脈内及び経口投与の両方を100mgリトナビル(カプセル、Norvir(登録商標))の投与と組み合わせた。施設内ガイドラインに従って、経口及び静注ドセタキセルの両方について、全患者が経口デキサメタゾンを用いて治療された。用量4mgのデキメタソンは、治験薬の1時間前に投与され、その後は12時間毎に4mg投与された(2回)。ドセタキセル治療の1時間前、患者は悪心及び嘔吐を予防するために1mgのグラニセトロン(Kytril(登録商標))も又摂取した。
【0182】
MODRAカプセルのバイオアベイラビリティは:
(AUC 30mg経口/AUC 20mg静注)×(20/30)×100%=73%(SD 18%)により計算した。
【0183】
これは、カプセルのバイオアベイラビリティが相当に高く、低い個人間変動性を伴うことを示している。
【実施例4】
【0184】
経口調製物の又別の特性解析
重要な所見:
スプレードライ法により製造された調製物は十分に非晶質であり、凍結乾燥法により製造された調製物に比較して溶解試験後に長期間の過飽和状態を有する(以下のセクション7を参照)。
【0185】
溶解試験での沈降後のドセタキセルの平衡溶解度は、PVP−K30を含む調製物(20μg/mL)及び担体を含まない調製物(7μg/mL)と比較した、PVP−VA64を含む調製物(40μg/mL)中では有意に増加する(以下のセクション8を参照)。
【0186】
結論の要約:
界面活性剤のタイプがドセタキセルの溶解に及ぼす作用(セクション1):
・試験した全界面活性剤(SDS、CPC、ポリソルベート80及びソルビタンモノオレート)は、ドセタキセルの溶解度を増加させる。
・界面活性剤がドセタキセルの溶解速度に及ぼす作用は、界面活性剤のHLB値と相関すると思われる。
・SDS及びCPCは、ドセタキセルの溶解速度における類似の増加を示す。
【0187】
SDSの量がドセタキセルの溶解に及ぼす作用(セクション2):
・少量のSDS(1/41)は、既にドセタキセルの溶解速度を増加させる。
・より多量のSDS(1/11)は、ドセタキセルの溶解速度をさらに一層増加させる。
・界面活性剤の作用は粉末の圧密レベルと相関すると思われるので、このため界面活性剤を含む粉末と含まない粉末との間のドセタキセルの溶解速度の差は、錠剤では一層大きくなると予想される。
【0188】
ドセタキセルの非晶質の性質(セクション3):
・ドセタキセルの凍結乾燥は、X線粉末回折スペクトルにおける回折ピークの消失を生じさせる。
・ドセタキセルの凍結乾燥は165℃近くで吸熱ピークの消失及び124℃近くでガラス転移の出現を生じさせる。
・165℃近くでの吸熱ピークの面積は、非晶質及び結晶性ドセタキセルの物理的混合物中の結晶性物質の量とよく相関している。
【0189】
パクリタキセルの非晶質の性質(セクション4):
・パクリタキセルの凍結乾燥は、X線粉末回折スペクトルにおける回折ピークの消失を生じさせる。
・パクリタキセルの凍結乾燥は、58、80及び163℃近くでの吸熱ピークの消失、61℃近くでの吸熱ピークの出現及び154℃近くでのガラス転移を生じさせる。
【0190】
ドセタキセルの固体分散体の特性解析(セクション5):
・ドセタキセル、PVP−K30及びSDSの混合物の凍結乾燥は、X線粉末回折パターンにおいてドセタキセルに属する回折ピークの消失を生じさせる。
・ドセタキセルの回折ピークは、固体分散体成分の物理的混合物中に存在する。
【0191】
パクリタキセルの固体分散体の特性解析(セクション6):
・パクリタキセル固体分散体系は、ドセタキセル固体分散体系に匹敵する。
【0192】
製造方法がドセタキセル固体分散体の特性及び性能に及ぼす作用(セクション7):
・ドセタキセル、PVP−K30及びSDSの混合物のスプレードライは、ドセタキセル及びSDSに属する回折ピークの消失を生じさせる。
・ドセタキセル、PVP−K30及びSDSの混合物のスプレードライは、DSCサーモグラムにおいてドセタキセル及びSDSに属する吸熱ピークの消失を生じさせる。
・ドセタキセル、PVP−K30及びSDSの混合物のスプレードライは、溶解試験後にドセタキセル、PVP−K30及びSDSの凍結乾燥混合物に比較して沈降までにより長い時間を生じさせる。
【0193】
担体のタイプがドセタキセル固体分散体の溶解性能に及ぼす作用(セクション8):
・PVP−K30の代りのポリエチレングリコール(1500、6000又は20000)の使用は、沈降までのより短い時間及び溶解試験後のドセタキセルのより低い平衡溶解度を生じさせる。
・PVP−K30に代えてのPVP−VA64の使用は、沈降までの匹敵する時間及び溶解試験後のドセタキセルの有意に高い平衡溶解度を生じさせる(各々、40μg/mL 対 20μg/mL)。
【0194】
錠剤の製造(セクション9):
・現在使用されているカプセルと同等以上に高い溶解速度を備える錠剤を製造することは実行可能である。
・現在使用されているカプセルより高いドセタキセルの含有量を備える錠剤を製造することは実行可能である。
【0195】
材料及び方法
一般
以下の実験において使用した調製物は、以下で略述する方法及び表18、表19、表20及び表17に記載した組成物に従って調製した。試験した薬物は、パクリタキセル及びドセタキセルであった。
【0196】
結晶性薬物:
結晶性薬物は供給業者から入手したままで使用した。
【0197】
非晶質薬物:
薬物は、薬物を30mLのt−ブタノール中に溶解させることにより非晶質化した。薬物/t−ブタノール溶液は、絶え間なく攪拌しながら20mLの注射用水(WfI)に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronorm、サイズ1/9)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表20を参照)。
【0198】
固体分散体の成分の物理的混合物
物理的混合物は、150mgのドセタキセル及び対応する量の担体及び界面活性剤(表17を参照)を乳鉢と乳棒で混合することにより調製した。
【0199】
【表17】
【0200】
非晶質及び結晶性ドセタキセルの物理的混合物
物理的混合物は、正確に計量した量の結晶性及び非晶質ドセタキセルを混合することにより調製した(表18を参照)。
【0201】
【表18】
【0202】
固体分散体(凍結乾燥)
固体分散体は、30mLのt−ブタノール中にドセタキセル、及び20mLの注射用水中の対応する量の担体及び界面活性剤(表19を参照)を溶解させることにより入手した。ドセタキセル/t−ブタノール溶液は、絶え間なく攪拌しながら担体/界面活性剤/WfIの溶液に加えた。最終混合物は、ステンレススチール製凍結乾燥箱(Gastronormサイズ1/9)へ移し、その後にt−ブタノール及び水を凍結乾燥法により除去した(表20を参照)。
【0203】
【表19】
【0204】
【表20】
【0205】
固体分散体(スプレードライ)
固体分散体は、ドセタキセルを45mLのエタノール及び5mLのWfI中に溶解させることにより入手した。薬物を完全に溶解させた後、絶え間なく攪拌しながらPVP−K30及びSDS(表21を参照)を薬物/エタノール/WfI溶液に加えた。最終混合物をフラスコに移し、その後にエタノール及び水をスプレードライ法により除去した(表22を参照)。
【0206】
【表21】
【0207】
【表22】
【0208】
カプセル(セクション1及び2において使用した)
カプセルは、10〜15mgの薬物と同等の量の凍結乾燥した固体分散体粉末(1/11ドセタキセル、9/11 PVP−K30及び1/11 SDS)を計量することにより製造した。固体分散体粉末を乳鉢及び乳棒で粉砕して微細粉末にし、サイズ0の硬質ゼラチンカプセル内に手動カプセル充填装置を用いてカプセル充填した。1カプセル当たりのドセタキセルの量は、製造後に正味カプセル重量を総カプセル重量から減じ、それに固体分散体粉末のドセタキセル比率を掛けることによって推定した(図19)。カプセルの含有量は、HPLC品質管理によって確認した。
【0209】
臨床試験用カプセル(セクション9において使用)
臨床試験用カプセルは、10mgの薬物と同等の量の凍結乾燥した固体分散体粉末(1/11ドセタキセル、9/11 PVP−K30及び1/11 SDS)、110mgのラクトースモノヒドレート及び1.1mgのコロイド状シリコンジオキシドを計量することにより製造した。全成分は、均質な混合物が得られるまで乳鉢及び乳棒を用いて混合した。この混合物は、サイズ0の硬質ゼラチンカプセル内に手動カプセル充填装置を用いてカプセル充填した。
【0210】
錠剤(セクション9において使用)
錠剤は、20mgのドセタキセルと同等の量のスプレードライ法した固体分散体粉末(1/11ドセタキセル、9/11 PVP−K30及び1/11 SDS)、110mgのラクトースモノヒドレート及び110mgの架橋ポリビニルピロリドンを計量することにより製造した。全成分は、均質な混合物が得られるまで乳鉢及び乳棒を用いて混合した。この混合物は、13mmフラット工具を装備した偏心プレス機上で手動により圧縮した。充填量は13.5mmで固定し、圧力は10.5mmに設定した。圧縮後に錠剤を計量し、ドセタキセルの量は錠剤重量に固体分散体粉末中の薬物の重量分率と錠剤中の固体分散体粉末の重量分率との積を掛けることによって推定した。錠剤の含有量は、HPLC品質管理によって確認した。
【0211】
溶解スクリーニングテスト
およそ6mgの薬物と同等の量の粉末を50mLのビーカー内に配置した。磁気攪拌棒及び25mLの水をビーカーに加えた。この溶液を720rpmで攪拌し、およそ37℃で維持した。サンプルを様々な時点で採取し、0.45μmフィルターを使用して濾過し、その後にそれらをメタノール及びアセトニトリルの1:4(v/v)混合液で希釈した。濾過及び希釈したサンプルは、その後にHPLC−UV(表23を参照)により分析した。溶解した薬物の量は、μg/mLの濃度として表示した。
【0212】
溶解試験
カプセル又は錠剤は、37℃で500mLのWfIが充填された2型(パドル式)溶解装置に配置し、パドルの回転速度は75rpmであった。サンプルを様々な時点で採取し、0.45μmフィルターを使用して濾過し、その後にそれらをメタノール及びアセトニトリルの1:4(v/v)混合液を用いて1:1に希釈した。濾過及び希釈したサンプルは、その後にHPLC−UV(表23を参照)により分析した。溶解したドセタキセルの量は、μg/mLでの濃度又はラベル記載内容に対するパーセンテージ(% RLC)のいずれかで表示した。ラベル記載内容は、製造後の各カプセル又は錠剤中に存在する薬物の推定量である。
【0213】
【表23】
【0214】
示差走査熱量(DSC)測定
DSC測定は、Q2000 DSC(TA Instruments社、米国デラウエア州ニューキャッスル)上で実施した。温度計及び熱流量計はインジウムを用いて校正した。およそ10mgの粉末のサンプルをTzeroアルミニウム製鍋(TA instruments社)に移し、密封しオートサンプラー内に配置した。表24に列挙したプログラムを全サンプルに対して使用した。
【0215】
【表24】
【0216】
X線粉末回折測定
X線粉末回折測定は、X−celeratorを装備したPhlips X’pert pro回折装置上で実施した。厚さがおよそ0.5mmのサンプルを金属製サンプルホルダー内に配置し、これを回折装置内に配置し、表25に表示した設定で走査した。
【0217】
【表25】
【0218】
実験
セクション1:界面活性剤のタイプがドセタキセルの溶解に及ぼす作用
目的
界面活性剤のタイプが及ぼす作用を決定するために、異なる界面活性剤(表26を参照)を備える5種の調製物(表19を参照、調製物1〜5)を凍結乾燥法により調製した(表20を参照)。界面活性剤の選択は、界面活性剤のクラス及びHLB値に基づいていた。選択された界面活性剤は、全3種の界面活性剤クラス(アニオン性、カチオン性及び非イオン性)並びに広範囲のHLB値(4.3〜40)を表している。各調製物は、同一量のドセタキセル、PVP−K30及び界面活性剤を有していた。各調製物から添加物を全く含めずに3個のカプセルを製造し、溶解試験にかけた。
【0219】
所見
図20は、0〜5分間にSDSを含む調製物中に溶解したドセタキセルの量は10% RLC未満であったが、CPC及びポリソルベート80を含む調製物中に溶解したドセタキセルの量は30% RLCを超えていた。この時点で、ソルビタンモノオレート調製物中のドセタキセルのおよそ17% RLCが溶解する。しかし10分後、SDS(63% RLC)、CPC(75% RLC)及びポリソルベート80(68% RLC)調製物間の溶解したドセタキセルの量の差は顕著に減少したが、ソルビタンモノオレート調製物からのドセタキセルの放出は、48% RLCと相当に低いままである。15分後、SDSを含む調製物から放出したドセタキセルの量は、CPCを含む調製物から放出したドセタキセルの量と同等である。ポリソルベート80及びソルビタンモノオレート調製物は73% RLC及び63% RLCというより低い放出したドセタキセルの量を有するが、これらの数値はそれでも界面活性剤を含まない固体分散体系からのドセタキセルの放出より依然としてはるかに高い(37% RLC)。60分後、CPC及びSDS系から放出したドセタキセルの量は、各々87% RLC及び89% RLCであるが、ポリソルベート80及びソルビタンモノオレート調製物からの放出は、各々77% RLC及び83% RLCと低い。
【0220】
結論
界面活性剤を備える全固体分散体系は、界面活性剤を備えない同一固体分散体系に比較してドセタキセルの溶解を増加させる。CPC及びSDSがドセタキセルの溶解に及ぼす作用は匹敵しており、これら2種の界面活性剤間の初期の差は、おそらくカプセル外被の溶解の変動の結果であると考えられる。これらの界面活性剤のHLB値は、固体分散体調製物の性能とよく相関すると思われる。
【0221】
さらに、パクリタキセル固体分散体系から製造されたカプセルを用いた溶解試験は、固体分散系内への界面活性剤(SDS)の組み込みに起因して溶解速度の一層より大きな変化を示した。この差は、活性成分(パクリタキセル 対 ドセタキセル)の溶解度の作用の差と関連している可能性があり、このため、種々の界面活性剤間の差は固体分散体系を含有するパクリタキセルから製造された製剤については大きくなる可能性が高い。
【0222】
【表26】
【0223】
セクション2:界面活性剤の量がドセタキセルの溶解に及ぼす作用
目的
SDSの量の影響を決定するために、ドセタキセルを含有する4種の固体分散体粉末(表19を参照、調製物4〜7)を凍結乾燥法により調製した(表20を参照)。SDSの量は4種の調製物間で変動したが、ドセタキセル及びPVP−K30の量は一定に維持された。各調製物から添加物を全く含めずに3個のカプセルを製造し、溶解試験にかけた。
【0224】
所見
図21及び図22は、溶解試験の結果を示している。0〜5分間に、ドセタキセルの溶解は、全4種の調製物中で限定される。初期の緩徐な溶解度は、カプセル外被の溶解によって生じる遅延時間に起因する。外被が溶解した後、固体分散体粉末の溶解を開始できる。界面活性剤を含まない調製物からのドセタキセルの溶解は、1/11 SDSを含む調製物からのドセタキセルの溶解より緩徐であると考えられる。
【0225】
1/11 SDS調製物は30分間以内に90% RLCの量の溶解したドセタキセルに達するが、SDSを含まない調製物は60分後に70% RLC(ラベル記載内容に比較して)の量の溶解したドセタキセルにしか到達しない。さらに、界面活性剤を含まない調製物のカプセル間のドセタキセルの放出速度における変動は、1/11 SDSを含む調製物のカプセル間の変動よりはるかに高い。
【0226】
図20及び21はさらに又、様々な量のSDS間の差も又示している。ドセタキセルの溶解は、既に固体分散体系への1/41 SDSだけの添加によって改善されるが、しかし1/41及び1/21 SDSの溶解パターン間に明確な差はない。ある量の1/11 SDSは、1/21 SDS及び1/41 SDSに比較してドセタキセルの最善の溶解パターンを生じさせる。
【0227】
結論
固体分散体系へのSDSへのより高い量の組み込みは、より高速の溶解速度を生じさせる。1/11(w/w)のSDSの組み込みは、最速の溶解速度を生じさせる。
【0228】
固体分散体粉末の圧縮は界面活性剤の使用を不可欠にする可能性が高い。錠剤の製造はカプセルの製造より一層高い圧縮を生じさせるので、界面活性剤の組み込みは、固体分散体錠剤において一層より必要になるであろう。
【0229】
固体分散体系へのSDSの組み込みは、カプセル充填及び錠剤化後に固体分散体粉末の湿潤性を改善する、界面活性剤の均質な分布を保証する。
【0230】
セクション3:ドセタキセルの非晶質の性質
目的
凍結乾燥後のドセタキセルの物理的形態は、ドセタキセルの凍結乾燥後に結晶度を決定するために示差走査熱量(DSC)(表24を参照)及びX線粉末回折測定(表25を参照)により調査した。
【0231】
所見
図23は、凍結乾燥法がドセタキセルのX線粉末回折パターンに及ぼす作用を示している。ドセタキセルの凍結乾燥前に、X線回折スペクトルは10〜40°2θ間の数多くの回折ピークを有しており、これはドセタキセルが結晶性状態にあることを示している。ドセタキセルの凍結乾燥後、X線粉末回折パターンには非晶質ハローが存在し、これはドセタキセルが非晶質形にあることを示している。
【0232】
図24は、凍結乾燥法がドセタキセルのDSCサーモグラムに及ぼす作用を示している。ドセタキセルの凍結乾燥前に、DSCサーモグラムはおそらくはドセタキセルの結晶構造の再配列によって生じた165℃での大きな吸熱ピークを示している。ドセタキセルの凍結乾燥後、DSCサーモグラムは165℃で吸熱ピークを有していない。しかし、広汎な吸熱ピークはおよそ50℃に存在しており、これは水及びt−ブタノールの蒸発法により引き起こされる。さらに、ガラス転移は124℃で観察することができ、これはドセタキセルが非晶質状態にあることを示している。
【0233】
図25は、非晶質(凍結乾燥)及び結晶性ドセタキセルの様々な混合物のX線粉末回折スペクトルを示す図である。混合物中の結晶性ドセタキセル含有量における減少は、X線粉末回折スペクトルにおける回折ピークの強度及び数の減少を生じさせる。5%結晶性ドセタキセルを含む混合物のX線回折パターンは回折ピークを有しておらず、これはX線粉末回折を伴う結晶性ドセタキセルの検出可能な最低量が5(w/w)%(純粋薬物物質)を超えることを指示している。
【0234】
図26は、非晶質(凍結乾燥)及び結晶性ドセタキセルの様々な混合物のDSCサーモグラムを示す図である。混合物中の結晶性ドセタキセルの含有量における減少は165℃での吸熱ピークのサイズの減少及びおよそ50℃での広汎な吸熱ピークの増加を生じさせる。これは0%結晶性ドセタキセル(純粋凍結乾燥ドセタキセル)及び5%結晶性ドセタキセルのサーモグラムにおける50及び165℃での吸熱ピークのサイズには差があり、これはDSCを含む結晶性ドセタキセルの検出可能な最低量が0〜5(w/w)%結晶性ドセタキセル(純粋薬物物質)であることを指示している。
【0235】
図27は、結晶性及び非晶質ドセタキセルの物理的混合物中の結晶性物質の量に対する、全熱流量サーモグラムにおける165℃でのピーク面積のプロット図を示している(表18を参照)。非晶質及び結晶性薬物中の結晶性物質の量は、各々0〜100(w/w)%であると推定される。回帰線は、0.990の決定係数及び0.995の回帰係数を有しており、これは165℃でのピークサイズと結晶度との間に強度の相関があることを確認している。
【0236】
結論
ドセタキセルの凍結乾燥は、凍結乾燥ドセタキセルのX線粉末回折スペクトルが回折ピークを示さないような、並びにDSCサーモグラムが結晶再配列と関連する吸熱ピークを示さないような程度への結晶度の減少を生じさせる。さらにDSCサーモグラムでは、ガラス転移が出現する。これは全てが、凍結乾燥後にドセタキセルが非晶質状態にあることの指標である。
【0237】
結晶再配列と関連する吸熱ピークのピーク面積は、非晶質及び結晶性ドセタキセルの物理的混合物中の結晶性ドセタキセルの含有量とよく相関する。
【0238】
セクション4:パクリタキセルの非晶質の性質
目的
凍結乾燥後のパクリタキセルの物理的形態をX線粉末回折及び示差走査熱量測定により調査し、凍結乾燥後のパクリタキセルの結晶度を決定した。
【0239】
所見
図28は、凍結乾燥がパクリタキセルのX線粉末回折パターンに及ぼす作用を示している。パクリタキセルの凍結乾燥前に、X線回折スペクトルは10〜40°2θ間の数多くの回折ピークを有しており、これはパクリタキセルが結晶性状態にあることを示している。パクリタキセルの凍結乾燥後、X線粉末回折パターンには非晶質ハローが存在し、これはパクリタキセルが非晶質形にあることを示している。
【0240】
図29は、凍結乾燥がパクリタキセルのDSCサーモグラムに及ぼす作用を示している。パクリタキセルの凍結乾燥前に、DSCサーモグラムは、58、80及び163℃で大きな吸熱ピークを示す。58及び80℃での両方のピークは、結晶格子からの水の消失により生じる。163℃での吸熱ピークは、おそらくパクリタキセルの結晶構造の再配列により生じる。パクリタキセルの凍結乾燥後、DSCサーモグラムは、61℃での広範囲の吸熱ピークの代わりに58、80又は163℃での吸熱ピークを有しておらず、154℃でのガラス転移を観察できる。
【0241】
結論
パクリタキセルの凍結乾燥は、凍結乾燥パクリタキセルのX線粉末回折スペクトルが回折ピークを示さない、並びにDSCサーモグラムが結晶再配列と関連する吸熱ピークを示さない程度への結晶度の減少を生じさせる。さらにDSCサーモグラムでは、ガラス転移が出現する。これは全てが、凍結乾燥後にパクリタキセルが非晶質状態にあることの指標である。
【0242】
セクション5:ドセタキセルを含む固体分散体の特性解析
目的
ドセタキセルの固体分散体を特性解析するために、X線粉末回折測定及びDSC測定は、ドセタキセル、PVP−K30及びSDSの物理的混合物及び固体分散体上に実施した。
【0243】
所見
図30には、固体分散体の成分であるドセタキセル、PVP−K30及びSDSのX線回折パターンを示した。ドセタキセルは結晶性であり、10〜40°の2θの数多くの回折ピークを有しており、SDSは結晶性で20〜22°の2θ間に鋭い回折ピークを有しており、PVP−K30は非晶質で回折ピークを有していない。
【0244】
図31は、固体分散体の成分であるドセタキセル、PVP−K30及びSDSのDSCサーモグラムを示している。ドセタキセルは、おそらくは結晶再配列により生じた165℃での大きな吸熱ピークを有している。PVP−K30は、76℃の近くで水の蒸発により生じた大きな吸熱ピークを有しており、ガラス状態及びゴム状態の間での熱容量における差により生じた162℃の近くでのガラス転移を有する。SDSは、おそらくは小さな非晶質分率により生じた67℃近くでの相転移、及び複数のピークを含有する80〜120℃の大きな吸熱領域を有している。これらのピークは、一部にはSDSの結晶性バルクを溶融させることによって、及び一部には未知の非可逆性吸熱事象により生じる。
【0245】
図32は、1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを含有する物理的混合物及び固体分散体系のX線粉末回折スペクトルを示す図である。ドセタキセル及びSDSの結晶性回折ピークは物理的混合物のX線粉末回折スペクトルに出現するが、ドセタキセルの回折ピークは固体分散体系のX線回折スペクトルにおいては出現しない。
【0246】
図33は、1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを含有する物理的混合物及び固体分散体系のDSCサーモグラムを示す図である。物理的混合物のサーモグラムは、おそらくはSDSにより生じた200℃近くでの吸熱下落;おそらくは結晶性ドセタキセルにより生じた162℃の近くでの小さな吸熱ピーク及びおそらくはPVP−K30により生じた164℃の近くでのガラス転移を示している。固体分散体系のサーモグラムは、おそらくはSDSにより生じた113℃の近くでの吸熱ピーク;並びに非晶質ドセタキセル及び非晶質PVP−K30の組み合わせにより生じた155℃の近くでのガラス転移を示す。
【0247】
結論
固体分散体のX線粉末回折スペクトルはドセタキセルに属する回折ピークを示さないが、ドセタキセル、PVP−K30及びSDSを含有する物理的混合物のX線粉末回折スペクトルはドセタキセルに属する回折ピークを示す。ドセタキセル、PVP−K30及びSDSの凍結乾燥混合物のDSCサーモグラムは、おそらくは非晶質ドセタキセル及びPVP−K30の分子混合により生じる155℃でのガラス転移を示すが、ドセタキセル、PVP−K30及びSDSを含有する物理的混合物のDSCサーモグラムは、おそらくはPVP−K30により生じた163℃でのガラス転移温度を示す。これに加えて162℃の近くでの吸熱ピークは物理的混合物のDSCサーモグラムにおいてのみ視認することができるが、固体分散体のDSCサーモグラムにおいては視認できない。
【0248】
SDSは固体分散体及び物理的混合物両方において結晶性状態で存在し、固体分散体及び物理的混合物両方のX線回折スペクトルにおいて20〜22°の2θ間での回折ピーク、並びに物理的混合物及び固体分散体のDSCサーモグラムにおける100〜113℃近くでの吸熱ピークを各々生じさせる。
【0249】
さらに、ドセタキセル単独の凍結乾燥は非晶質ドセタキセルを生じさせたが(セクション3を参照)、ドセタキセルを含有する混合物の凍結乾燥はさらに又非晶質ドセタキセルを生じさせる可能性が極めて高い。
【0250】
セクション6:パクリタキセルの固体分散体の特性解析
入手できるデータは、パクリタキセル固体分散体系がドセタキセル固体分散体系に匹敵する、即ち、パクリタキセルは凍結乾燥後には非晶質状態で存在するがSDSはそうではないことを指示している。
【0251】
セクション7:製造方法がドセタキセル固体分散体の特性及び性能に及ぼす作用
目的
製造方法が固体分散体特性に及ぼす影響を決定するために、凍結乾燥法及びスプレードライ法の両方を使用して1/11ドセタキセル、9/11 PVP−K30及び1/11 SDSを備える固体分散体系を製造した(表19及び表21を参照)。両方の系をX線回折、DSC及び溶解スクリーニング試験により試験した。
【0252】
所見
図34は、凍結乾燥法及びスプレードライ法により製造された固体分散体のX線粉末回折スペクトルを示している。凍結乾燥法(フリーズドライ法)により製造された固体分散体は、20〜22°2θのスペクトル内に存在する回折ピークが存在するために、一部には結晶である。これらの回折ピークはSDSに属する(図30)。スプレードライ法により製造された固体分散体は、X線粉末回折パターンにおける回折ピークの非存在から結論できるように、十分に非晶質である。
【0253】
図35は、凍結乾燥法及びスプレードライ法により製造された固体分散体のDSCサーモグラムを示している。凍結乾燥法により製造された固体分散体のサーモグラムは、おそらくはSDSにより生じた113℃の近くでの吸熱ピーク;並びに非晶質ドセタキセル及び非晶質PVP−K30の組み合わせにより生じる155℃の近くでのガラス転移を示す。スプレードライ法により製造された固体分散体は、147℃の近くでのガラス転移しか示さず、これは十分な非晶質形の指標である。
【0254】
図36は、凍結乾燥法及びスプレードライ法により製造された固体分散体の溶解スクリーニング曲線を示している。凍結乾燥法により製造された固体分散体は、5分後にピークドセタキセル濃度に到達し、沈降し始める。スプレードライ法により製造された固体分散体は、10分後にピークドセタキセル濃度に到達し、15分後に沈降し始める。
【0255】
結論
ドセタキセルの固体分散体の製造における凍結乾燥法に比較したスプレードライ法の使用は、長期間の過飽和状態に関して溶解スクリーニング試験における改善された性能を生じさせるより非晶質系をもたらす。
【0256】
さらに、スプレードライ法後に得られた粉末は低静止性であり、より一様な粒径を有するので、凍結乾燥生成物に比較してその後に加工処理するためにより適切である。
【0257】
セクション8:担体のタイプがドセタキセル固体分散体の溶解性能に及ぼす作用
目的
異なる担体がドセタキセルの固体分散体の溶解性能に及ぼす性能を試験すること。I/11ドセタキセル、9/11担体及び1/11 SDSを含有する固体分散体系の製造において様々な担体を使用した(表19を参照、調製物8〜11)。全系を溶解スクリーニング試験にかけた。
【0258】
所見
図37は、PVP−K30、PEG1500、PEG6000又はPEG20000を含有する固体分散体系の溶解スクリーニング曲線を示している。全系は5分後には匹敵するドセタキセルのピーク濃度に達するが、全3種のPEG系についてドセタキセルの沈降は既に5分後に始まる。さらに、沈降後の溶液中のドセタキセルの量は、固体分散体系を含有するPEG中においておよそ8μg/mLであるが、PVP−K30含有系は沈降後に20μg/mLの溶液中のドセタキセルの量に達する。
【0259】
図38は、PVP−K30又はPVP−VA64を含有する固体分散体系の溶解スクリーニング曲線を示している。両方の系について、5分後に同一ドセタキセルピーク濃度に達し、両方の系についてドセタキセルはおよそ10分後に沈降し始める。しかし沈降後の溶液中のドセタキセルの量には有意な差がある:PVP−VA64(40μg/mL)及びPVP−K30(20μg/mL)。
【0260】
結論
PEGを含有する固体分散体系は、溶解スクリーニング試験においてPVP−K30を含有する固体分散体系より不良に機能する。
【0261】
PVP−VA64を含有する固体分散体系は、沈降後にPVP−K30を含有する固体分散体系よりドセタキセルの有意に高い濃度に達する。
【0262】
さらに、パクリタキセルはドセタキセルより低い溶解度を有するために、PVP−VA64の使用は、固体分散体系を含有するパクリタキセル中において特に有用な可能性がある。
【0263】
セクション9:錠剤の製造
目的
ドセタキセルの固体分散体からの錠剤製造の実行可能性を調査すること。
【0264】
所見
図39は、現在臨床試験において使用されている20mgのドセタキセル錠及び10mgのドセタキセルカプセルの平均溶解曲線を示している。図40は、0〜10分間に錠剤及びカプセルの平均溶解速度を示している。錠剤は、35μg/mLの濃度に達するまで0〜30分間の定常溶解速度を示している。カプセルは、およそ16μg/mLの濃度に達するまで0〜10分間の定常溶解速度を示している。0〜10分間に、カプセル及び錠剤両方からのドセタキセルの放出速度はおよそ0.8mg/分である。
【0265】
結論
固体分散体系を含有するドセタキセルの錠剤の製造は実行可能である。ドセタキセル錠剤の溶解速度は、現在臨床試験において使用されているドセタキセルカプセルの溶解速度に匹敵する。
【実施例5】
【0266】
ドセタキセルの固体分散体への追加の医薬上有効成分の添加
非晶質リトナビルの製造
固体成分:
300mgのリトナビル
溶媒:
45mLのエタノール
5mLの注射用水
リトナビルはエタノール/水混合物中に溶解させ、Buchi 290ミニスプレー乾燥機を用いてスプレードライした(表27を参照)。
【0267】
ドセタキセル、リトナビル、PVP及びSDSの組み合わせ生成物の製造
固体成分:
1gのドセタキセル無水物
4gのリトナビル
25gのPVP−K30
5gのSDS
溶媒:
900mLのエタノール
100mLの注射用水
全固体成分は、エタノール−水混合物中に溶解させ、Buchi 290ミニスプレー乾燥機を用いてスプレードライした(表27を参照)。これは視覚的に均質な白色粉末を生じさせた。この固体分散体粉末は、およそ12.5mgのドセタキセル及び50mgのリトナビル(1カプセル当たり)と等価であり、サイズ0の硬質ゼラチンカプセル内に手動でカプセル充填した。
【0268】
【表27】
【0269】
【表28】
【0270】
図41は、非晶質(スプレードライ)及び結晶性リトナビルのDSCサーモグラムを示している。非晶質リトナビルは、およそ25℃のTgを示し、結晶はおよそ122℃の溶融吸熱を示す。スプレードライ法は非晶質リトナビルを生じさせると結論付けた。
【0271】
図42は、ドセタキセル、リトナビル、PVP−K30及びSDSの組み合わせのスプレードライ法固体分散体粉末のDSCサーモグラムを示している。サーモグラムは、およそ117℃の単一Tgを示すが、リトナビル、ドセタキセル、PVP−K30又はSDSの溶融吸熱は示さなかった。
【0272】
図43は、37℃及び50RPMで1,000mLの0.1N HCl中におけるドセタキセル/リトナビル/PVP−K30/SDSカプセルの溶解プロファイルを示している。各カプセルは、およそ12.5mgのドセタキセル及びおよそ50mgのリトナビルを含有していた。ドセタキセル及びリトナビルはどちらも45分間以内に完全に放出される(n=3)。
【0273】
上記の実施例は、本発明の特定の実施形態を例示することを企図しているが、本発明の範囲を限定することは企図しておらず、本発明の範囲は添付の特許請求の範囲によって規定される。本明細書に言及した全文献は、全体として参照により本明細書に組み込まれる。
【0274】
参考文献
1. Demario MD, Ratain MJ. J Clin Oncol 1998;16:2557-2567.
2. Liu G, Franssen E, Fitch MI et al. J Clin Oncol 1997;15:110-115.
3. Meerum Terwogt JM, Beijnen JH, ten Bokkel Huinink WW et al. Lancet 1998;352:285.
4. Sparreboom A, van Asperen J, Mayer U et al. Proc Natl Acad Sci USA 1997;94:2031 -2035.
5. Kuhn J, Rizzo J, Eckhardt J et al. Proc Am Soc Clin Oncol 1995;14:474.
6. Schellens JHM, Creemers GJ, Beijnen JH et al. Br J Cancer 1996;73:1268-1271.
7. Hellriegel ET, Bjornnson TD, Hauck WW. Clin Pharmacol Ther 1996;60:601 -607.
8. Huizing MT, Giaccone G, van Warmerdam LJC et al. J Clin Oncol 1997;15:317-329.
9. Malingre MM, Richel DJ, Beijnen JH et al. J Clin Oncol 2001;19:l160-1166.
10. Mastalerz H, Cook D, Fairchild CR, Hansel S, Johnson W, Kadow JF, Long BH, Rose WC, Tarrant J, Wu MJ, Xue MQ, Zhang G, Zoeckler M, Vyas DM. Bioorg Med Chem. 2003; 11:4315-23.
11. Serajuddin AT. J Pharm Sci 1999;88(10):1058 1066.
12. Karanth H, Shenoy VS, Murthy RR. AAPS PharmSciTech 2006;7(4):87.
13. Chen J, et al. Drug Development and Industrial Pharmacy 2008;34:588-594.
14. Schellekens RC, Stuurman FE, van der Weert FH, Kosterink JG, Frijlink HW. Eur. J. Pharm. Sci. 2007 Jan;30(1):15-20.
15. Dressman JB, Reppas C. Eur. J. Pharm. Sci. 2000 Oct;11:S73-S80.
16. The Handbook of Pharmaceutical Excipients. Fifth Edition. Edited by Raymond Rowe, Paul Sheskey and Sian Owen. See sections on Povidone, PVP VAand PEG.
17. Kuppens IE, Van Maanen MJ, Rosing H, Schellens JHM, Beijnen JH. Biomed Chromatogr 2005;19:355-361.
18. Kawakami (2009) Journal of Pharmaceutical Sciences. 98(9) 2875-2885. Published online in Wiley Interscience (www.interscience.wiley.com) DOI 10.1002/jps.21816
【特許請求の範囲】
【請求項1】
実質的に非晶質のタキサン、親水性担体及び界面活性剤を含む経口投与のための固体医薬組成物であって、該実質的に非晶質のタキサンが溶媒蒸発法により調製されるものである、前記固体医薬組成物。
【請求項2】
前記溶媒蒸発法が、スプレードライ法である、請求項1に記載の組成物。
【請求項3】
前記タキサン及び担体が、固体分散体の形態にあるものである、請求項1又は2に記載の組成物。
【請求項4】
前記タキサン、前記担体及び前記界面活性剤が、固体分散体の形態にあるものである、請求項1から3のいずれか一項に記載の組成物。
【請求項5】
前記固体分散体が、溶媒蒸発法により調製されるものである、請求項3又は4に記載の組成物。
【請求項6】
前記溶媒蒸発法が、スプレードライ法である、請求項5に記載の組成物。
【請求項7】
前記タキサンが、ドセタキセル、パクリタキセル、BMS−275183、及びそれらの機能的誘導体、並びにそれらの医薬上許容される塩若しくはエステルから選択されるものである、請求項1から6のいずれか一項に記載の組成物。
【請求項8】
前記タキサンが、ドセタキセル、パクリタキセル、及びそれらの機能的誘導体、並びにそれらの医薬上許容される塩若しくはエステルから選択されるものである、請求項7に記載の組成物。
【請求項9】
前記担体が、ポリマー性のものである、請求項1から8のいずれか一項に記載の組成物。
【請求項10】
前記担体が、PVP、PVP−VA及びPEGから選択されるものである、請求項1から9のいずれか一項に記載の組成物。
【請求項11】
前記担体が、PVPである、請求項1から10のいずれか一項に記載の組成物。
【請求項12】
前記PVPが、PVP−K12、PVP−K15、PVP−K17、PVP−K25、PVP−K30、PVP−K60及びPVP−K90から選択されるものである、請求項11に記載の組成物。
【請求項13】
前記PVPが、PVP−K30、PVP−K60及びPVP−K90から選択されるものである、請求項12に記載の組成物。
【請求項14】
前記界面活性剤が、ナトリウムドデシルスルフェート(SDS)、ソルビタンエステル類(ソルビタン脂肪酸エステル類)、ポリオキシエチレンステアレート類、ポリオキシエチレンソルビタン脂肪酸エステル類、ポリオキシエチレンヒマシ油誘導体、ポリオキシエチレンアルキルエーテル類、ポロキサマー、グリセリルモノオレエート、ドキュセートナトリウム、セトリミド、ベンジルベンゾエート、ベンザアルコニウムクロライド、ベンゼトニウムクロライド、ヒプロメロース、非イオン性乳化ろう、アニオン性乳化ろう及びトリエチルシトレートから選択されるものである、請求項1から13のいずれか一項に記載の組成物。
【請求項15】
前記界面活性剤が、SDS、ソルビタンエステル類(ソルビタン脂肪酸エステル)、ポリオキシエチレンソルビタン脂肪酸エステル及びCPCから選択されるものである、請求項1から14のいずれか一項に記載の組成物。
【請求項16】
前記界面活性剤が、SDSである、請求項14に記載の組成物。
【請求項17】
担体に対する前記タキサンの重量比が、約0.01:99.99(w/w)〜約75:25(w/w)である、請求項1から16のいずれか一項に記載の組成物。
【請求項18】
担体に対する前記タキサンの重量比が、約0.01:99.99(w/w)〜約30:70(w/w)である、請求項17に記載の組成物。
【請求項19】
組み合わされたタキサン及び担体に対する界面活性剤の重量比が、約1:99(w/w)〜約50:50(w/w)である、請求項1から18のいずれか一項に記載の組成物。
【請求項20】
組み合わされたタキサン及び担体に対する界面活性剤の重量比が、約2:98(w/w)〜約17:83(w/w)の間である、請求項19に記載の組成物。
【請求項21】
1つ又はそれ以上の追加の医薬上有効成分をさらに含む、請求項1から20のいずれか一項に記載の組成物。
【請求項22】
1つ又はそれ以上の追加の医薬上活性な成分が、CYP3A4阻害剤である、請求項21に記載の組成物。
【請求項23】
前記CYP3A4阻害剤が、リトナビルである、請求項22に記載の組成物。
【請求項24】
前記タキサン、前記担体、前記界面活性剤及び前記1つ又はそれ以上の追加の医薬上有効成分が、固体分散体の形態にあるものである、請求項21〜23のいずれか一項に記載の組成物。
【請求項25】
前記固体分散体が、溶媒蒸発法により調製されるものである、請求項24に記載の組成物。
【請求項26】
前記溶媒蒸発法が、スプレードライ法である、請求項25に記載の組成物。
【請求項27】
治療に使用するための請求項1から26のいずれか一項に記載の組成物。
【請求項28】
腫瘍性疾患の治療に使用するための請求項1〜26のいずれか一項に記載の組成物。
【請求項29】
腫瘍性疾患の治療方法であって、そのような治療を必要とする被験者に、請求項1〜26のいずれか一項に記載の組成物を有効量投与する工程を含む、前記治療方法。
【請求項30】
前記組成物を製造するために、
溶媒蒸発法を使用して非晶質タキサンを調製する工程と、
前記非晶質タキサンを親水性担体及び界面活性剤と組み合わせる工程とを含む、請求項1に記載の組成物の調製方法。
【請求項31】
実質的に非晶質のタキサン及び親水性担体を含む経口投与のための医薬組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製されるものである、前記医薬組成物。
【請求項32】
タキサン、親水性担体及び界面活性剤を溶液中に含む、医薬組成物。
【請求項33】
前記組成物が、被験者に投与するための飲料液の形態にあるものである、請求項32に記載の組成物。
【請求項34】
前記組成物が、カプセル中に含有されるものである、請求項32に記載の組成物。
【請求項35】
前記組成物が、適切な溶媒中に請求項1〜26のいずれか一項に記載の固体組成物を溶解させる工程により入手可能なものである、請求項29〜31のいずれか一項に記載の組成物。
【請求項36】
実質的に非晶質のタキサン及び1つ又はそれ以上の医薬上許容される賦形剤を含む経口投与のための固体医薬組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製されるものである、前記固体医薬組成物。
【請求項1】
実質的に非晶質のタキサン、親水性担体及び界面活性剤を含む経口投与のための固体医薬組成物であって、該実質的に非晶質のタキサンが溶媒蒸発法により調製されるものである、前記固体医薬組成物。
【請求項2】
前記溶媒蒸発法が、スプレードライ法である、請求項1に記載の組成物。
【請求項3】
前記タキサン及び担体が、固体分散体の形態にあるものである、請求項1又は2に記載の組成物。
【請求項4】
前記タキサン、前記担体及び前記界面活性剤が、固体分散体の形態にあるものである、請求項1から3のいずれか一項に記載の組成物。
【請求項5】
前記固体分散体が、溶媒蒸発法により調製されるものである、請求項3又は4に記載の組成物。
【請求項6】
前記溶媒蒸発法が、スプレードライ法である、請求項5に記載の組成物。
【請求項7】
前記タキサンが、ドセタキセル、パクリタキセル、BMS−275183、及びそれらの機能的誘導体、並びにそれらの医薬上許容される塩若しくはエステルから選択されるものである、請求項1から6のいずれか一項に記載の組成物。
【請求項8】
前記タキサンが、ドセタキセル、パクリタキセル、及びそれらの機能的誘導体、並びにそれらの医薬上許容される塩若しくはエステルから選択されるものである、請求項7に記載の組成物。
【請求項9】
前記担体が、ポリマー性のものである、請求項1から8のいずれか一項に記載の組成物。
【請求項10】
前記担体が、PVP、PVP−VA及びPEGから選択されるものである、請求項1から9のいずれか一項に記載の組成物。
【請求項11】
前記担体が、PVPである、請求項1から10のいずれか一項に記載の組成物。
【請求項12】
前記PVPが、PVP−K12、PVP−K15、PVP−K17、PVP−K25、PVP−K30、PVP−K60及びPVP−K90から選択されるものである、請求項11に記載の組成物。
【請求項13】
前記PVPが、PVP−K30、PVP−K60及びPVP−K90から選択されるものである、請求項12に記載の組成物。
【請求項14】
前記界面活性剤が、ナトリウムドデシルスルフェート(SDS)、ソルビタンエステル類(ソルビタン脂肪酸エステル類)、ポリオキシエチレンステアレート類、ポリオキシエチレンソルビタン脂肪酸エステル類、ポリオキシエチレンヒマシ油誘導体、ポリオキシエチレンアルキルエーテル類、ポロキサマー、グリセリルモノオレエート、ドキュセートナトリウム、セトリミド、ベンジルベンゾエート、ベンザアルコニウムクロライド、ベンゼトニウムクロライド、ヒプロメロース、非イオン性乳化ろう、アニオン性乳化ろう及びトリエチルシトレートから選択されるものである、請求項1から13のいずれか一項に記載の組成物。
【請求項15】
前記界面活性剤が、SDS、ソルビタンエステル類(ソルビタン脂肪酸エステル)、ポリオキシエチレンソルビタン脂肪酸エステル及びCPCから選択されるものである、請求項1から14のいずれか一項に記載の組成物。
【請求項16】
前記界面活性剤が、SDSである、請求項14に記載の組成物。
【請求項17】
担体に対する前記タキサンの重量比が、約0.01:99.99(w/w)〜約75:25(w/w)である、請求項1から16のいずれか一項に記載の組成物。
【請求項18】
担体に対する前記タキサンの重量比が、約0.01:99.99(w/w)〜約30:70(w/w)である、請求項17に記載の組成物。
【請求項19】
組み合わされたタキサン及び担体に対する界面活性剤の重量比が、約1:99(w/w)〜約50:50(w/w)である、請求項1から18のいずれか一項に記載の組成物。
【請求項20】
組み合わされたタキサン及び担体に対する界面活性剤の重量比が、約2:98(w/w)〜約17:83(w/w)の間である、請求項19に記載の組成物。
【請求項21】
1つ又はそれ以上の追加の医薬上有効成分をさらに含む、請求項1から20のいずれか一項に記載の組成物。
【請求項22】
1つ又はそれ以上の追加の医薬上活性な成分が、CYP3A4阻害剤である、請求項21に記載の組成物。
【請求項23】
前記CYP3A4阻害剤が、リトナビルである、請求項22に記載の組成物。
【請求項24】
前記タキサン、前記担体、前記界面活性剤及び前記1つ又はそれ以上の追加の医薬上有効成分が、固体分散体の形態にあるものである、請求項21〜23のいずれか一項に記載の組成物。
【請求項25】
前記固体分散体が、溶媒蒸発法により調製されるものである、請求項24に記載の組成物。
【請求項26】
前記溶媒蒸発法が、スプレードライ法である、請求項25に記載の組成物。
【請求項27】
治療に使用するための請求項1から26のいずれか一項に記載の組成物。
【請求項28】
腫瘍性疾患の治療に使用するための請求項1〜26のいずれか一項に記載の組成物。
【請求項29】
腫瘍性疾患の治療方法であって、そのような治療を必要とする被験者に、請求項1〜26のいずれか一項に記載の組成物を有効量投与する工程を含む、前記治療方法。
【請求項30】
前記組成物を製造するために、
溶媒蒸発法を使用して非晶質タキサンを調製する工程と、
前記非晶質タキサンを親水性担体及び界面活性剤と組み合わせる工程とを含む、請求項1に記載の組成物の調製方法。
【請求項31】
実質的に非晶質のタキサン及び親水性担体を含む経口投与のための医薬組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製されるものである、前記医薬組成物。
【請求項32】
タキサン、親水性担体及び界面活性剤を溶液中に含む、医薬組成物。
【請求項33】
前記組成物が、被験者に投与するための飲料液の形態にあるものである、請求項32に記載の組成物。
【請求項34】
前記組成物が、カプセル中に含有されるものである、請求項32に記載の組成物。
【請求項35】
前記組成物が、適切な溶媒中に請求項1〜26のいずれか一項に記載の固体組成物を溶解させる工程により入手可能なものである、請求項29〜31のいずれか一項に記載の組成物。
【請求項36】
実質的に非晶質のタキサン及び1つ又はそれ以上の医薬上許容される賦形剤を含む経口投与のための固体医薬組成物であって、該実質的に非晶質のタキサンがスプレードライ法により調製されるものである、前記固体医薬組成物。
【図1】

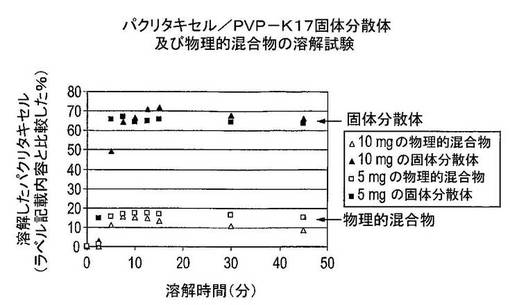
【図2】
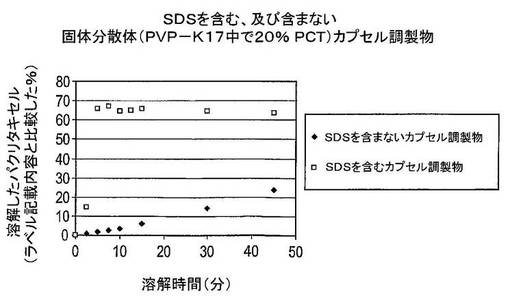
【図3】
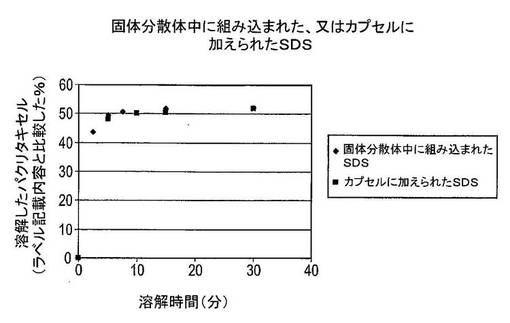
【図4】
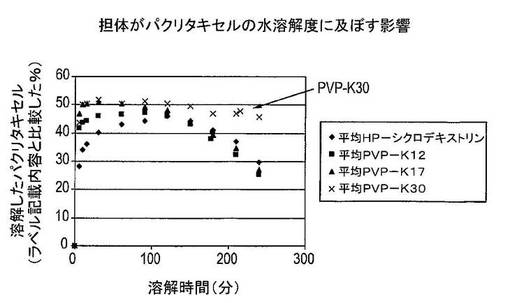
【図5】
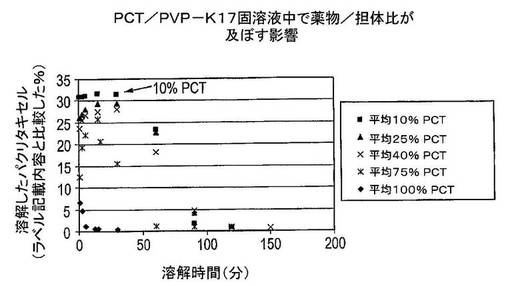
【図6】
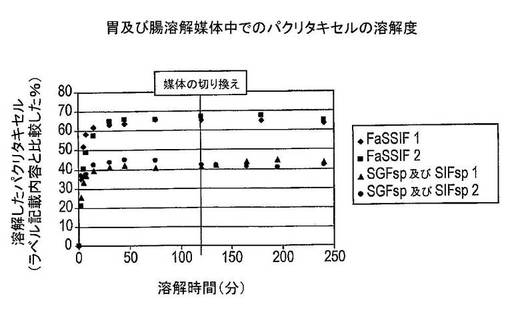
【図7】
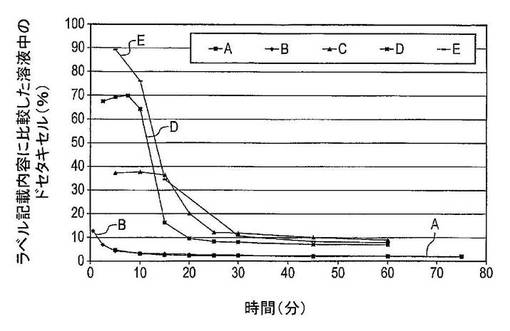
【図8】
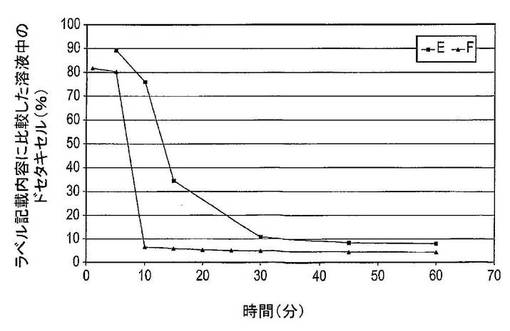
【図9】
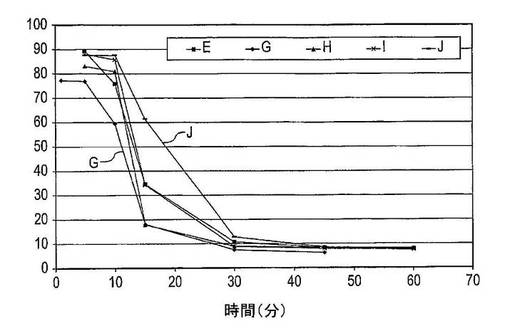
【図10】
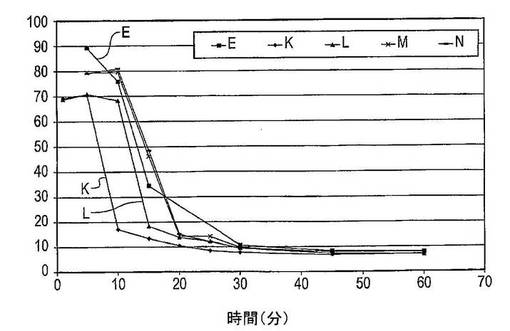
【図11】
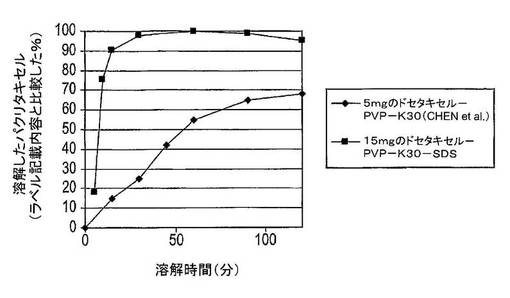
【図12】
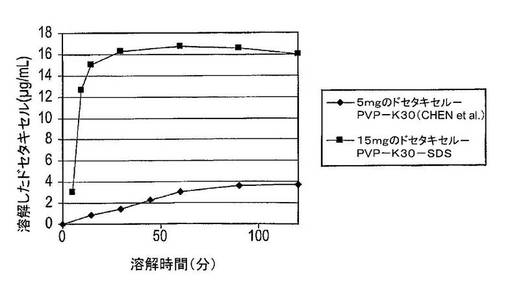
【図13】
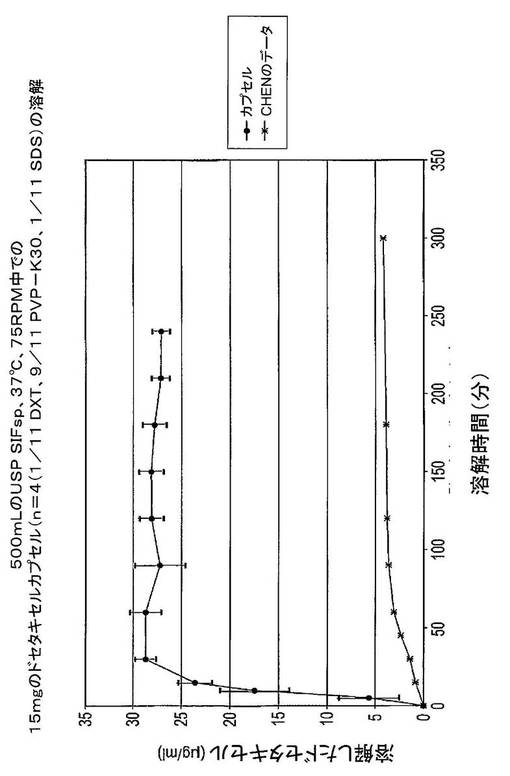
【図14】
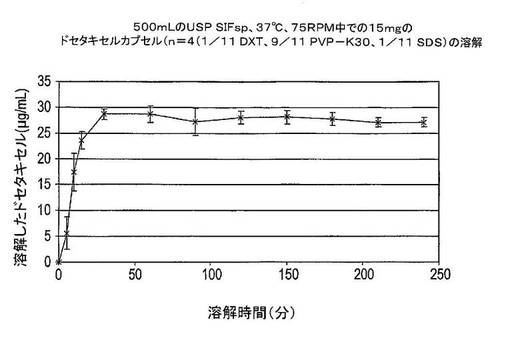
【図15】
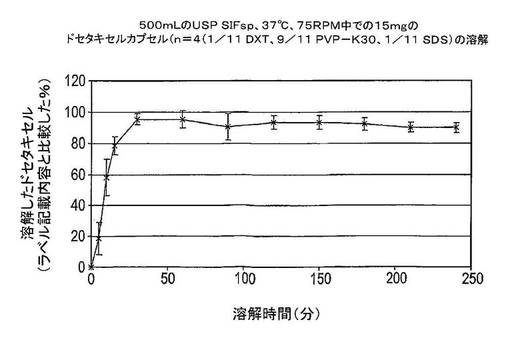
【図16】
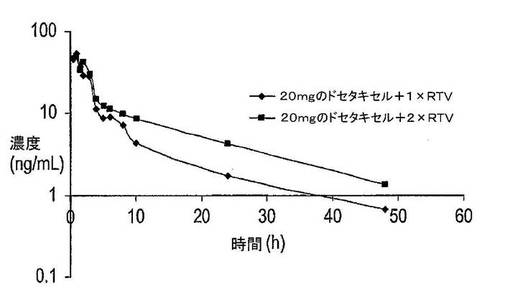
【図17】
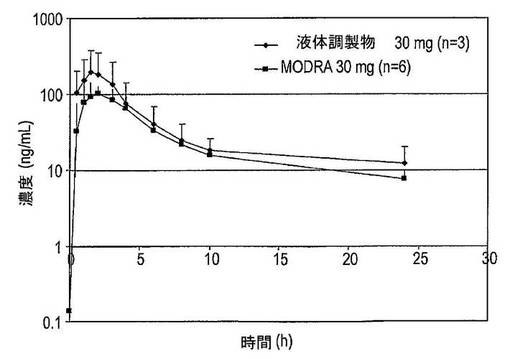
【図18】
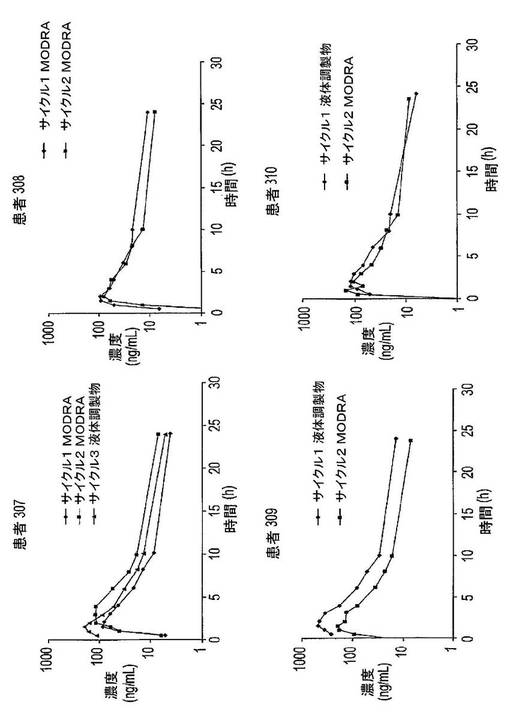
【図19】
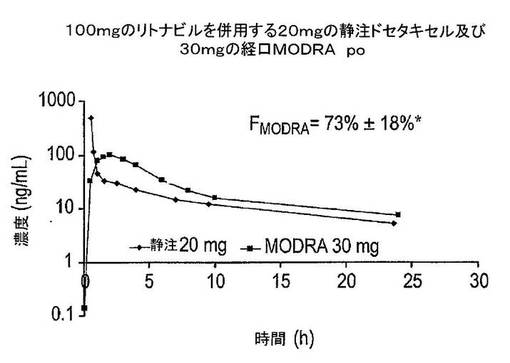
【図20】
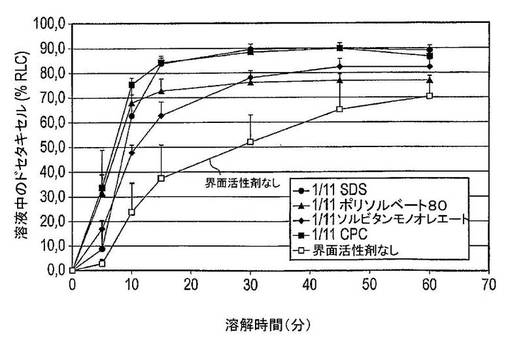
【図21】
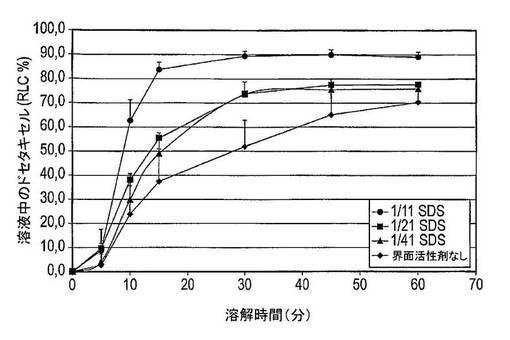
【図22】
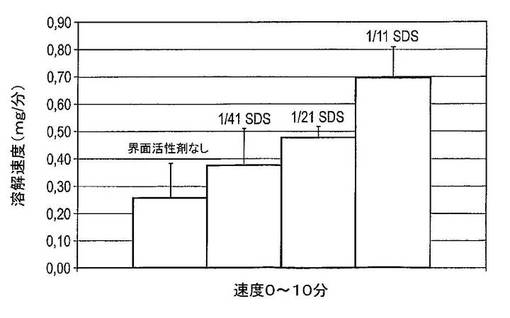
【図23】
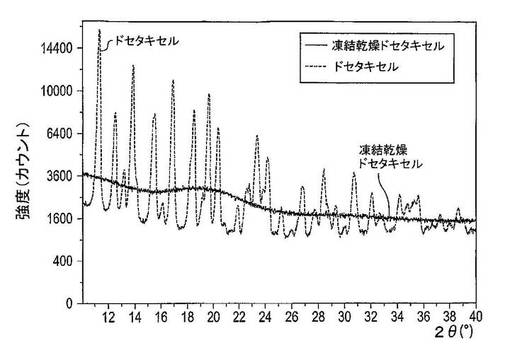
【図24】
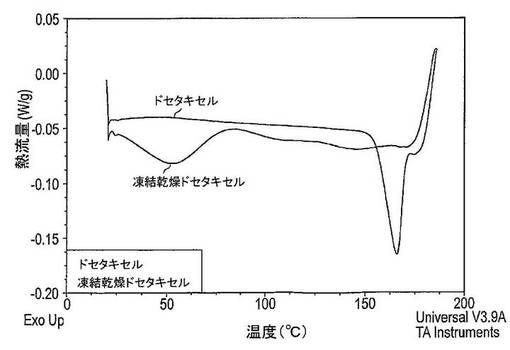
【図25】
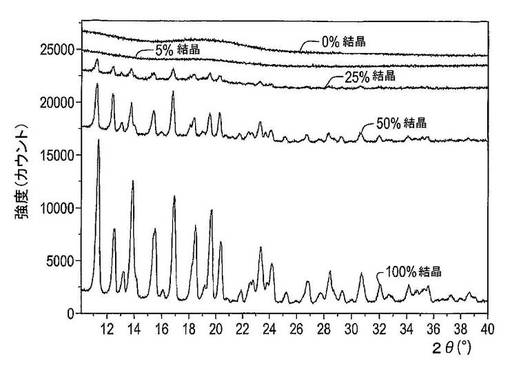
【図26】
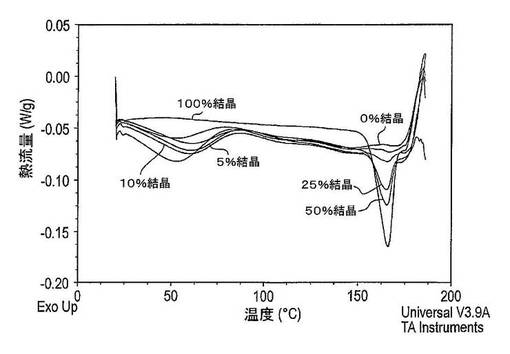
【図27】
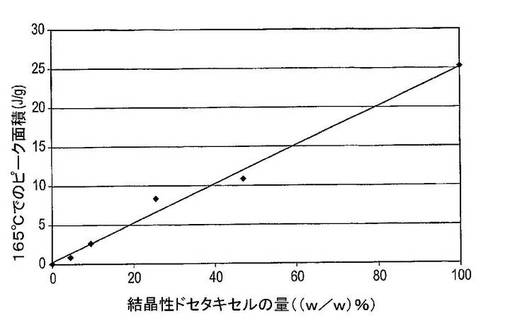
【図28】
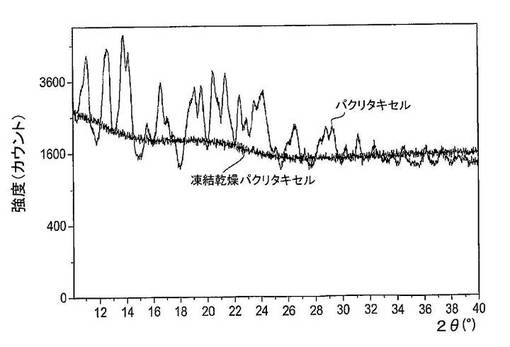
【図29】
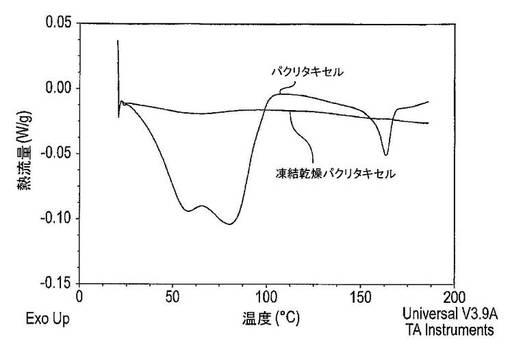
【図30】
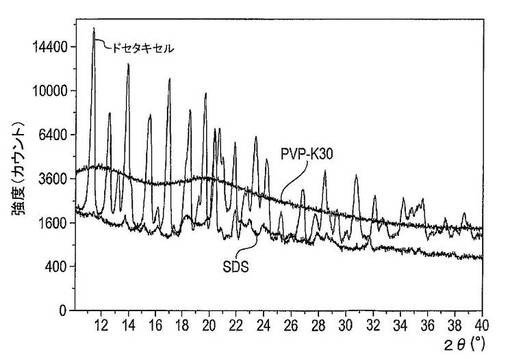
【図31】
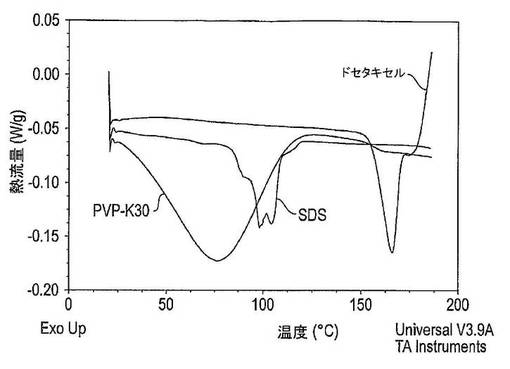
【図32】
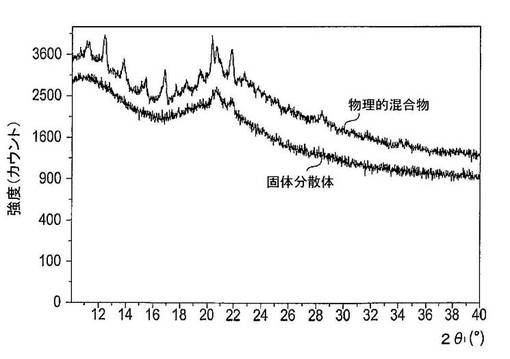
【図33】
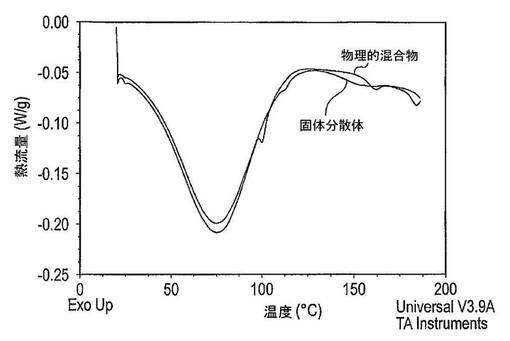
【図34】
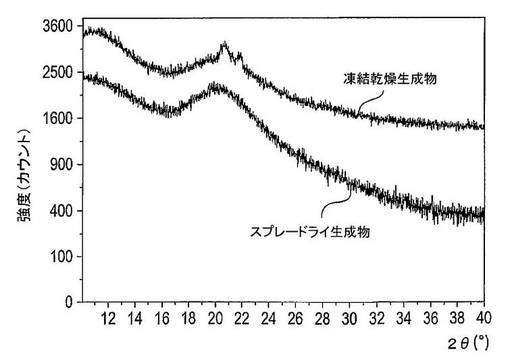
【図35】
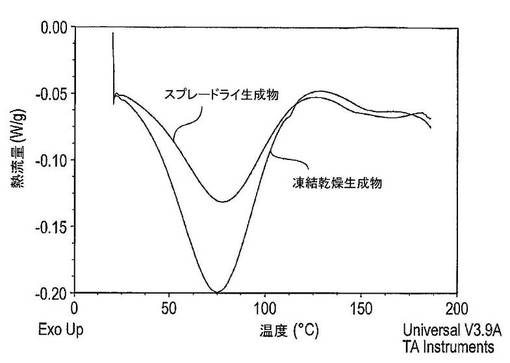
【図36】
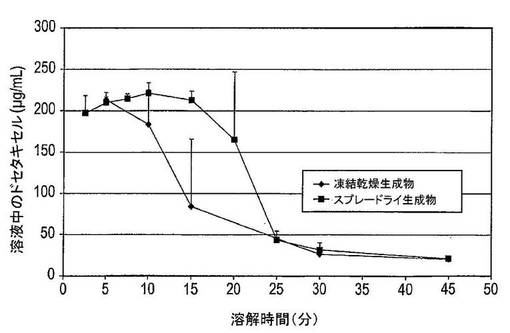
【図37】
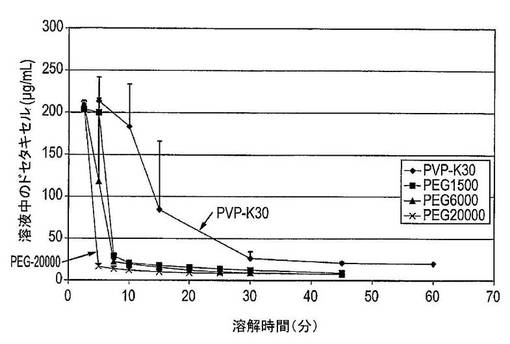
【図38】
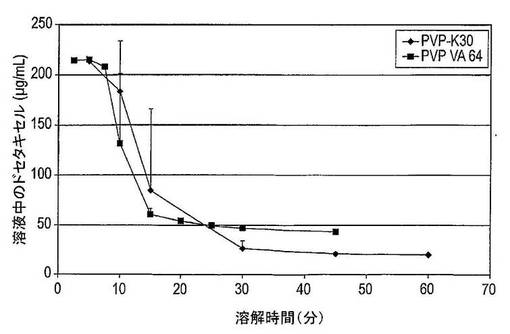
【図39】
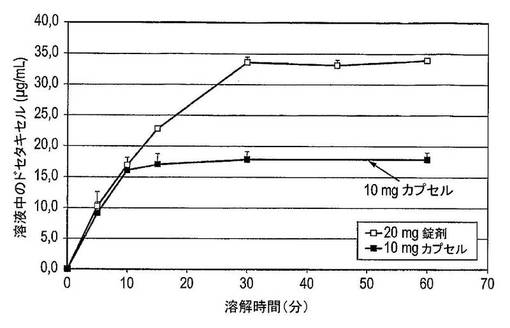
【図40】
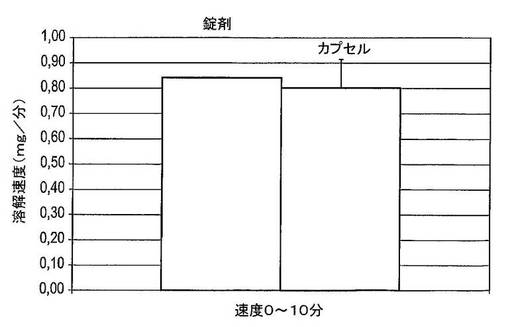
【図41】
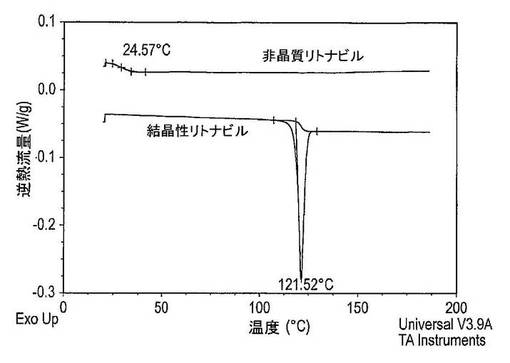
【図42】
【図43】
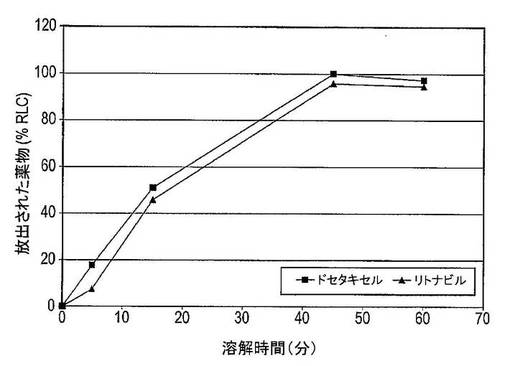
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【公表番号】特表2012−500788(P2012−500788A)
【公表日】平成24年1月12日(2012.1.12)
【国際特許分類】
【出願番号】特願2011−523454(P2011−523454)
【出願日】平成21年8月24日(2009.8.24)
【国際出願番号】PCT/GB2009/002068
【国際公開番号】WO2010/020799
【国際公開日】平成22年2月25日(2010.2.25)
【出願人】(510050188)スティヒティング ヘット ネーデルランド カンケル インスティテュート (3)
【出願人】(510050177)
【Fターム(参考)】
【公表日】平成24年1月12日(2012.1.12)
【国際特許分類】
【出願日】平成21年8月24日(2009.8.24)
【国際出願番号】PCT/GB2009/002068
【国際公開番号】WO2010/020799
【国際公開日】平成22年2月25日(2010.2.25)
【出願人】(510050188)スティヒティング ヘット ネーデルランド カンケル インスティテュート (3)
【出願人】(510050177)
【Fターム(参考)】
[ Back to top ]